WO2007113218A1 - Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) - Google Patents
Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) Download PDFInfo
- Publication number
- WO2007113218A1 WO2007113218A1 PCT/EP2007/053050 EP2007053050W WO2007113218A1 WO 2007113218 A1 WO2007113218 A1 WO 2007113218A1 EP 2007053050 W EP2007053050 W EP 2007053050W WO 2007113218 A1 WO2007113218 A1 WO 2007113218A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dimethyl
- propyl
- palladium
- methyl
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/30—1,4-Oxazines; Hydrogenated 1,4-oxazines not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/10—Preparation of halogenated hydrocarbons by replacement by halogens of hydrogen atoms
- C07C17/12—Preparation of halogenated hydrocarbons by replacement by halogens of hydrogen atoms in the ring of aromatic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C25/00—Compounds containing at least one halogen atom bound to a six-membered aromatic ring
- C07C25/02—Monocyclic aromatic halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
- C07C45/512—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups the singly bound functional group being a free hydroxyl group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/20—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms
- C07C47/228—Unsaturated compounds having —CHO groups bound to acyclic carbon atoms containing six-membered aromatic rings, e.g. phenylacetaldehyde
Definitions
- the present invention relates to a new process for the synthesis of 3-[4-(l,l-dimethyl- propyl)-phenyl]-2-methyl-propionaldehyde and cis-4- ⁇ 3- [4-( 1 , 1 -dimethyl-propyl)-phenyl] -2- methyl-propyl ⁇ -2,6-dimethyl-morpholine or its salts.
- Amorolfine, cis-4- ⁇ 3- [4-( 1 , 1 -dimethyl-propyl)-phenyl] -2-methyl-propyl-2,6-dimethyl- morpholine of formula (I) is a potent antifungal drug exhibiting a large spectrum of in vitro activity. It belongs to a new chemical class of antimycotics and exhibits a broad spectrum of antifungal activity against dermatophytes, dimorphic fungi, Candida albicans, Cryptococus neoformans and some dematiaceae. It finds a major use as the active ingredient in nail lacquer as a topical antifungal in treatment of onychomycosis and as topical antimycotic indicated for the treatment of dermatomycosis.
- Zhixiang teaches a multi-step synthesis of Amorolfine from tert-pentylbenzene (F. Zhixiang & coll., Zhongguo Yaowu Huaxue Zazhi, H)(I), 64-65, (200O)).
- tert-pentylbenzene F. Zhixiang & coll., Zhongguo Yaowu Huaxue Zazhi, H)(I), 64-65, (200O)
- 4-bromomethyl-tert- pentylbenzene is prepared. It is coupled with methyl malonic ester, hydrolyzed and decarboxylated to afford 2-methyl-3-(4-tert-pentylphenyl) propionic acid.
- This intermediate is amidated with c/5'-2,6-dimethylmorpholine and reduced to afford Amorolfine of formula (I).
- Hoffmann-La Roche patent (FR 2,463,767 or EP 24,334) disclose other synthetic pathways for the production of Amorolfine of formula (I) or its hydrochloride salt of formula (Ia).
- Amorolfine is produced as a racemic mixture of the cis isomers by tree different synthetic pathways.
- a second synthetic pathway discloses a reduction of a compound of the formula to produce Amorolfine of formula (I)
- a third synthetic pathway discloses an amino reduction between (E)-2-Methyl-3- phenyl-propenal and ds ⁇ -dimethyl morpholine to produce c/s-2,6-dimethyl-4-(2-methyl-3- phenyl-propyl)-morpholine followed by a Friedel-Craft reaction with 2-methyl-2-butanol to produce Amorolfine of formula (I)
- Fenpropimorph 4-[3-(4-fert-butylphenyl)-2-methylpropyl]-2,6- dimethylmorpholine, which is a pesticide, specifically categorised as a morpholine fungicide, has been recently synthesised by Forsyth in a two steps one pot process (S.A.Forsyth & coll; Organic Process Research & Development, 10, 94-102, (2006)).
- This process teaches a Heck reaction between 4-te/t-butyl-iodobenzene and 2-methyl- prop-2-en-l-ol catalyzed by palladium chloride (PdCl 2 ) in ionic liquid solvent followed by an amino reduction in presence of c/5'-2,6-dimethyl morpholine catalyzed by palladium on carbon (Pd/C) under hydrogen pressure in ionic liquid solvent.
- Amorolfine or its salts can be produced in a two steps one pot process involving a Heck reaction between (l,l-dimethyl-propyl)-4-iodo-benzene and 2- methyl-prop-2-en-l-ol catalyzed by palladium catalyst such as palladium acetate (PdOAc 2 ) in N,N-dimethylformamide (DMF) followed by an amino reduction in alcohol in presence of c/5'-2,6-dimethyl morpholine and a reducing agent such as hydrogen gas/palladium catalyst or a mixed metal hydrides, in alcoholic solvents.
- palladium catalyst such as palladium acetate (PdOAc 2 ) in N,N-dimethylformamide (DMF)
- a reducing agent such as hydrogen gas/palladium catalyst or a mixed metal hydrides
- the solvent used in the invention for the Heck reaction is DMF, polar protic or non polar organic solvents.
- the solvent used in the invention for the amino reduction reaction is alcohol. With such a two steps one pot process, the production of Amorolfine is optimized.
- This invention is a new process for preparing 3-[4-(l,l-dimethyl-propyl)-phenyl]-2- methyl-propionaldehyde of formula (II): characterized in that a compound of general formula (VI)
- X can be an halide such as bromine, chlorine, iodine or fluorine, or a trifluoromethane sulfonate radical (OSO 2 CF 3 ) is reacting in Heck reaction with 2-methyl- prop-2-en-l-ol of formula (VII)
- This invention extends to a new process of producing Amorolfine of formula (I)
- X is an halide such as bromine, chlorine, iodine or fluorine or a trifluoromethane sulfonate radical (OSO 2 CF 3 ) is subjected to Heck coupling conditions with 2-methyl-prop-2- en-l-ol of formula (VII)
- This compound can be further converted to a corresponding salt of c/,s-4- ⁇ 3-[4-(l,l-dimethyl- propyl)-phenyl]-2-methyl-propyl ⁇ -2,6-dimethyl-morpholine.
- the salification can be performed thanks to the addition of an acid, like hydrochloric acid.
- the preferred salt of c/s-4- ⁇ 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propyl ⁇ -2,6- dimethyl-morpholine is the hydrochloride salt of formula (Ia)
- the palladium catalyst used in the Heck reaction conditions of the invention is chosen among palladium(II)acetate, palladium(II)chloride, tetrakis (triphenylphosphine)-palladium(O), palladium on activated carbon, dichloro [1,1'- bis(diphenylphosphino)ferrocene] palladium(II), dichloro-bis-triphenylphosphino palladium(II).
- a phosphinic ligand can be used in combination with the palladium catalyst.
- phosphinic ligands used are for example triphenylphosphine, tri-o-tolylphosphine, tri-m-tolylphosphine or tri-p-tolylphosphine.
- the organic solvents used are N,N- dimethylformamide (DMF), polar pro tic solvent as for example methanol, ethanol, n- propanol, i-propanol, n-butanol, i-butanol, t-butanol or appropriate mixture of these solvents with water or non polar solvent as for example tetrahydrofurane, ethyl acetate, toluene, o- xylene, m- xylene, p-xylene.
- DMF N,N- dimethylformamide
- the reaction is carried out in the presence of a base such as a tertiary amine, as for example triethylamine, tripropylamine, tributylamine, diisopropylethylamine, a metal carbonates, as for example sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate or a metal acetates as for example potassium acetate or sodium acetate.
- a base such as a tertiary amine, as for example triethylamine, tripropylamine, tributylamine, diisopropylethylamine
- a metal carbonates as for example sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate or a metal acetates as for example potassium acetate or sodium acetate.
- the reaction is carried out in an inert atmosphere, such as under nitrogen or argon gas in a suitable reaction vessel.
- the temperatures are hold between about 60° to about 150 0 C during about 30 min to 24 h.
- the preferred conditions for the Heck reaction are DMF as solvent, an homogeneous palladium catalyst such as palladium(II) acetate, a base such as sodium carbonate, a range time between 8 hours and 10 hours, for example 9 hours, and a temperature between 80° and HO 0 C.
- the preferred compound of general formula (VI) is the (1,1- dimethyl-propyl)-4-iodo-benzene of formula (Via)
- a new process for iodination reaction of compound of formula (VIII) is achieved by using sodium metaperiodate and iodine in a mixture of acetic acid and acetic anhydride followed by the addition of sulfuric acid.
- (l,l-dimethyl-propyl)-4-iodo- benzene of formula (Via) is obtained in excellent yield (98%)
- the amino reduction step is performed with reducing agent such as : hydrogen gas in the presence of a palladium catalyst such as for example palladium on activated carbon, palladium on activated carbon in the presence of metal salts like Ba(OH) 2 , Ca(OH) 2 , CaCO 3 , BaCO 3 or Perlmans catalyst Pd(OH) 2 . or mixed metal hydrides like metal borohydride such as for example sodium borohydride (NaBH 4) and lithium borohydride (LiBH 4 ), or like metal cyano borohydride such as for example sodium cyano borohydride (NaCNBH 3 ) and lithium cyanoborohydride (LiCNBH 3 ).
- a palladium catalyst such as for example palladium on activated carbon
- metal salts like Ba(OH) 2 , Ca(OH) 2 , CaCO 3 , BaCO 3 or Perlmans catalyst Pd(OH) 2 .
- mixed metal hydrides like metal borohydride such as for example sodium
- the solvent in which the amino reduction is performed is N,N-dimethylformamide, a polar protic solvent such as methanol, ethanol, propanol, i-propanol, butanol, iso-butanol, tert- butanol or a non polar organic solvent such as toluene, tetrahydrofurane, ethyl acetate.
- a polar protic solvent such as methanol, ethanol, propanol, i-propanol, butanol, iso-butanol, tert- butanol or a non polar organic solvent such as toluene, tetrahydrofurane, ethyl acetate.
- the temperature for this amino reduction may typically be set at no more than 45°C, preferably between 20° and 45°C, and more preferably between 20° and 3O 0 C in the case of metal catalyst such as palladium catalyst, and preferably between O 0 C and 3O 0 C in the case of mixed metal hydride.
- the preferred conditions are to a) first react (l,l-dimethyl-propyl)-4-iodo-benzene of formula (Via) with 2- methyl-prop-2-en-l-ol of formula (VII) in DMF as solvent and in the presence of palladium acetate (PdOAc 2 ) as catalyst and sodium bicarbonate
- Example 1 Two steps synthesis of cis-4- ⁇ 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propyl ⁇ -2,6-dimethyl-morpholine hydrochloride (Ia) using NaBH 4 as reducing agent
- Example 3 Two steps synthesis of cis-4- ⁇ 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propyl ⁇ -2,6-dimethyl-morpholine hydrochloride (Ia) using hydrogen and palladium on carbon as reducing agent
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
The present invention discloses a new process for the preparation of 3-[4-(l,l- dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and for the preparation of c/5'-4-{3-[4- (1,1 -dimethyl-propyl)-phenyl] -2-methyl-propyl } -2,6-dimethyl-morpholine (Amorolfine) .
Description
PROCESS FOR PRODUCING 3-[4-(1,1-DIMETHYL-PROPYL)-PHENYL]^-
METHYL-PROPIONALDEHYDE AND cis-4-{3-[4-(l,l-DIMETHYL-PROPYL)-
PHENYL]-2-METHYL-PROPYL}-2,6-DIMETHYL-MORPHOLINE (AMOROLFINE)
The present invention relates to a new process for the synthesis of 3-[4-(l,l-dimethyl- propyl)-phenyl]-2-methyl-propionaldehyde and cis-4- { 3- [4-( 1 , 1 -dimethyl-propyl)-phenyl] -2- methyl-propyl}-2,6-dimethyl-morpholine or its salts.
Amorolfine, cis-4- { 3- [4-( 1 , 1 -dimethyl-propyl)-phenyl] -2-methyl-propyl-2,6-dimethyl- morpholine of formula (I) is a potent antifungal drug exhibiting a large spectrum of in vitro activity. It belongs to a new chemical class of antimycotics and exhibits a broad spectrum of antifungal activity against dermatophytes, dimorphic fungi, Candida albicans, Cryptococus neoformans and some dematiaceae. It finds a major use as the active ingredient in nail lacquer as a topical antifungal in treatment of onychomycosis and as topical antimycotic indicated for the treatment of dermatomycosis.
Zhixiang teaches a multi-step synthesis of Amorolfine from tert-pentylbenzene (F. Zhixiang & coll., Zhongguo Yaowu Huaxue Zazhi, H)(I), 64-65, (200O)). By hydroxymethylation and bromination from tert-pentylbenzene, 4-bromomethyl-tert- pentylbenzene is prepared. It is coupled with methyl malonic ester, hydrolyzed and decarboxylated to afford 2-methyl-3-(4-tert-pentylphenyl) propionic acid. This intermediate is amidated with c/5'-2,6-dimethylmorpholine and reduced to afford Amorolfine of formula (I).
Hoffmann-La Roche patent (FR 2,463,767 or EP 24,334) disclose other synthetic pathways for the production of Amorolfine of formula (I) or its hydrochloride salt of formula (Ia). In the Hoffmann-La Roche patent, Amorolfine is produced as a racemic mixture of the cis isomers by tree different synthetic pathways.
A first synthetic pathway discloses a nucleophilic substitution on l-(3-halo-2-methyl- propyl)-4-(l,l-dimethyl-propyl)-benzene (halo=chlorine, bromine or iodine) by cis-2,6- dimethyl morpholine to produce Amorolfine of formula (I)
A second synthetic pathway discloses a reduction of a compound of the formula
 to produce Amorolfine of formula (I)
to produce Amorolfine of formula (I)
A third synthetic pathway discloses an amino reduction between (E)-2-Methyl-3- phenyl-propenal and ds^ό-dimethyl morpholine to produce c/s-2,6-dimethyl-4-(2-methyl-3- phenyl-propyl)-morpholine followed by a Friedel-Craft reaction with 2-methyl-2-butanol to produce Amorolfine of formula (I)
On another side, Fenpropimorph, 4-[3-(4-fert-butylphenyl)-2-methylpropyl]-2,6- dimethylmorpholine, which is a pesticide, specifically categorised as a morpholine fungicide, has been recently synthesised by Forsyth in a two steps one pot process (S.A.Forsyth & coll; Organic Process Research & Development, 10, 94-102, (2006)).
This process teaches a Heck reaction between 4-te/t-butyl-iodobenzene and 2-methyl- prop-2-en-l-ol catalyzed by palladium chloride (PdCl2) in ionic liquid solvent followed by an amino reduction in presence of c/5'-2,6-dimethyl morpholine catalyzed by palladium on carbon (Pd/C) under hydrogen pressure in ionic liquid solvent.
We have now found that Amorolfine or its salts can be produced in a two steps one pot process involving a Heck reaction between (l,l-dimethyl-propyl)-4-iodo-benzene and 2- methyl-prop-2-en-l-ol catalyzed by palladium catalyst such as palladium acetate (PdOAc2) in N,N-dimethylformamide (DMF) followed by an amino reduction in alcohol in presence of c/5'-2,6-dimethyl morpholine and a reducing agent such as hydrogen gas/palladium catalyst or a mixed metal hydrides, in alcoholic solvents.
This new process has never been used in the prior art to synthesize Amorolfine. The solvent used in the invention for the Heck reaction is DMF, polar protic or non polar organic solvents.
Likewise, the solvent used in the invention for the amino reduction reaction is alcohol. With such a two steps one pot process, the production of Amorolfine is optimized.
This invention is a new process for preparing 3-[4-(l,l-dimethyl-propyl)-phenyl]-2- methyl-propionaldehyde of formula (II):
 characterized in that a compound of general formula (VI)
characterized in that a compound of general formula (VI)

in the presence of palladium catalyst and a base in a solvent chosen from N,N- dimethylformamide (DMF), polar protic and non polar organic solvents, at temperature between 60° to 15O0C
This invention extends to a new process of producing Amorolfine of formula (I)

or its salts characterized in that in the first step, compound of general formula (VI)

in the presence of palladium catalyst and a base in a solvent chosen from N,N- dimethylformamide (DMF), polar protic and non polar organic solvents, at temperature
between 60° and 15O0C, to provide 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propionaldehyde of formula (II):

and in the second step (amino reduction step), 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propionaldehyde of formula (II) is reacted with ds^ό-dimethyl morpholine of formula (IV)


This compound can be further converted to a corresponding salt of c/,s-4-{3-[4-(l,l-dimethyl- propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine. The salification can be performed thanks to the addition of an acid, like hydrochloric acid.
The two steps of the present invention as described above can be realized advantageously in a "one pot" process without isolation of the aldehyde intermediate of formula (II).
The preferred salt of c/s-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6- dimethyl-morpholine is the hydrochloride salt of formula (Ia)

Particularly, the palladium catalyst used in the Heck reaction conditions of the
invention is chosen among palladium(II)acetate, palladium(II)chloride, tetrakis (triphenylphosphine)-palladium(O), palladium on activated carbon, dichloro [1,1'- bis(diphenylphosphino)ferrocene] palladium(II), dichloro-bis-triphenylphosphino palladium(II).
Notably, a phosphinic ligand can be used in combination with the palladium catalyst. Commonly phosphinic ligands used are for example triphenylphosphine, tri-o-tolylphosphine, tri-m-tolylphosphine or tri-p-tolylphosphine.
In this first step, the organic solvents used, according to the invention, are N,N- dimethylformamide (DMF), polar pro tic solvent as for example methanol, ethanol, n- propanol, i-propanol, n-butanol, i-butanol, t-butanol or appropriate mixture of these solvents with water or non polar solvent as for exemple tetrahydrofurane, ethyl acetate, toluene, o- xylene, m- xylene, p-xylene. The reaction is carried out in the presence of a base such as a tertiary amine, as for example triethylamine, tripropylamine, tributylamine, diisopropylethylamine, a metal carbonates, as for example sodium carbonate, sodium bicarbonate, potassium carbonate, potassium bicarbonate or a metal acetates as for example potassium acetate or sodium acetate.
Advantageously, the reaction is carried out in an inert atmosphere, such as under nitrogen or argon gas in a suitable reaction vessel.
The temperatures are hold between about 60° to about 1500C during about 30 min to 24 h.
Advantageously, the molar ratio between the two starting materials (VI)/(VII) range from (VI)/(VII) =1/1 to (VI)/(VII) =1/2. In the field of the invention, the preferred conditions for the Heck reaction are DMF as solvent, an homogeneous palladium catalyst such as palladium(II) acetate, a base such as sodium carbonate, a range time between 8 hours and 10 hours, for example 9 hours, and a temperature between 80° and HO0C.
As starting material, the preferred compound of general formula (VI) is the (1,1- dimethyl-propyl)-4-iodo-benzene of formula (Via)
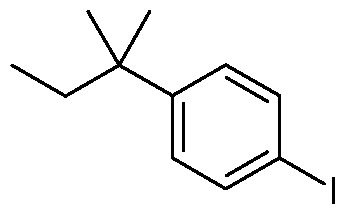

A new process for iodination reaction of compound of formula (VIII) is achieved by using sodium metaperiodate and iodine in a mixture of acetic acid and acetic anhydride followed by the addition of sulfuric acid. In those conditions, (l,l-dimethyl-propyl)-4-iodo- benzene of formula (Via) is obtained in excellent yield (98%)
The amino reduction step is performed with reducing agent such as : hydrogen gas in the presence of a palladium catalyst such as for example palladium on activated carbon, palladium on activated carbon in the presence of metal salts like Ba(OH)2, Ca(OH)2, CaCO3, BaCO3 or Perlmans catalyst Pd(OH)2. or mixed metal hydrides like metal borohydride such as for example sodium borohydride (NaBH4) and lithium borohydride (LiBH4), or like metal cyano borohydride such as for example sodium cyano borohydride (NaCNBH3) and lithium cyanoborohydride (LiCNBH3).
The solvent in which the amino reduction is performed is N,N-dimethylformamide, a polar protic solvent such as methanol, ethanol, propanol, i-propanol, butanol, iso-butanol, tert- butanol or a non polar organic solvent such as toluene, tetrahydrofurane, ethyl acetate.
The temperature for this amino reduction may typically be set at no more than 45°C, preferably between 20° and 45°C, and more preferably between 20° and 3O0C in the case of metal catalyst such as palladium catalyst, and preferably between O0C and 3O0C in the case of mixed metal hydride.
When the two steps of the present invention as described above are realized
advantageously in a "one pot" process without isolation of the aldehyde intermediate of formula (II), the preferred conditions are to a) first react (l,l-dimethyl-propyl)-4-iodo-benzene of formula (Via) with 2- methyl-prop-2-en-l-ol of formula (VII) in DMF as solvent and in the presence of palladium acetate (PdOAc2) as catalyst and sodium bicarbonate
(NaHCO3) as base, at 10O0C during 9h. b) filtrate the catalyst through Celite, then add ethanol, acetic acid and cis-2,6- dimethylmorpholine of formula (IV). c) Cool the mixture to -50C, then add sodium borohydride (NaBH4) d) After work-up, dissolve the crude product obtained from step c) in diisopropyl oxide (iPr2O) and add a solution of hydrochloric acid gas (HCl) in ethyl acetate (EtOAc) e) isolate cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6- dimethyl-morpholine hydrochloride of formula (Ia) obtained by filtration.
The present invention will now be described by way of example. The following examples are not intended to be limiting on the invention.
Example 1: Two steps synthesis of cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propyl}-2,6-dimethyl-morpholine hydrochloride (Ia) using NaBH4 as reducing agent
First step: a) (l,l-dimethyl-propyl)-4-iodo-benzene (Via)

To a mixture of AcOH (1188 mL) and Ac2O (594 mL) was added sodium periodate (79.83 g, 0.373 mol,) and iodine (280.94 g, 1.107 mol,). Then the mixture was cooled in ice- bath and concentrated, sulfuric acid (184 mL, 3.71 mol) was added drop-wise during 20 min so that temperature did not exceed 12°C. After the addition was complete, (1,1-dimethyl- propyl) -benzene (256.76 g, 1.732 mol, 1.0 eq) ) was added at once and the stirring continued for 24 h. The reaction was then partitioned between heptane-10% EtOAc (IL) and water (2L).
The organic phase was separated and was washed with water (2L) containing Na2SO3 (50 g). Then the organic phase was dried over MgSO4 and the solvents were evaporated.
457.87 g of (l,l-dimethyl-propyl)-4-iodo-benzene were isolated as a liquid (96% yield). This material was pure enough to be used in the next step without purification.
1U NMR (400 MHz, CDCl3) δ: 0.73 (3H, t, J = 7.4 Hz), 1.31 (6H, s), 1.67 (2H, q, J = 7.4 Hz), 7.13 (2H, d, J = 8.56 Hz), 7.66 (2H, d, J = 8.56 Hz);
13C NMR (100 MHz, CDCl3) δ: 9.59 (CH3), 28.78 (2CH3), 37.16 (CH2), 38.3 (CAi), 91.12 (CAT), 128.72 (CHA1), 137.46 (CHA1), 149.62 (CAr).
b) 3-[4-(l,l-Dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde (II)

Through a mixture of (l,l-dimethyl-propyl)-4-iodo-benzene (137.07 g, 0.50 mol), palladium(II) acetat (1.122 g, 5 mmol) and sodium bicarbonate (50.40 g, 0.60 mol) in dry DMF (500 mL) was bubbled nitrogen for 10 min. Then 2-methyl-prop-2-en-l-ol (54.083 g, 0.75 mol) was added and nitrogen was bubbled for another ten minutes. The reaction mixture was heated under nitrogen for 9 h at 1000C. The reaction was cooled and was filtered through a thin layer of Celite. The Celite was then washed with DMF (300 mL). The reaction was then partitioned between H2O (2 L) and heptane/EtOAc (9/1) (2x1 L). The combined organic layers were washed with H2O (1 L) and were dried over MgSO4. The solvents were evaporated. 109.63 g of 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde were isolated as crude product. This intermediate was purified by distillation at 112°C under 0.06 mbar to afford 84 g of pure 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde (77% yield)..
1H NMR (400 MHz, CDCl3) δ: 0.69 (3H, t, J=7.45), 1.11 (3H, d, J=6.87), 1.29 (6H, s), 1.65 (2H, q, J=7.43), 2.60 (1HAB, dd, J= 8.18, 13.52 Hz), 2.69 (IH, sex, J=7.06), 3.08 (IH, dd, J = 5.87, 13.54 Hz), 7.12 (2H, d, J = 8.27), 7.27 (2H, d, J = 8.27), 9.75 (IH, s);
13C NMR (100 MHz, CDCl3) δ: 9.6 (CH3), 13.7 (CH3), 25.8 (CH3), 36.6 (CH2), 37.3 (CH2), 38.0 (C), 48.5 (CH), 126.4 (CHAr), 128.6 (CHAr), 136.0 (CAr), 148.0 (CAr), 205.1 (C=O).
Second step:
c) cis-4-{3-[4-(l,l-Dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl- morpholine hydrochloride (Ia)

3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde (109.63 g ; 0.50 mol) obtained at the previous step were dissolved in absolute ethanol (500 rnL). To the ethanol solution was added at 00C (ice/water) AcOH (30 mL, 0.5 mol) and c/5'-2,6-dimethyl- morpholine (69.108 g, 0.60 mol) and the reaction was stirred at RT for 30 min. It was then cooled to (-150C) and NaBH4 (15.93 g, 0.42 mol) was added in portions during 1 h. Stirring was continued at 00C for 2 h and the reaction was quenched by drop-wise addition of NaOH solution in water (30 g, 0.75 mol, /100 mL H2O) to pH=12. The reaction was then partitioned between H2O (2 L) and hept/EtOAc (9/1) (2x1 L). The organic phases were combined, washed with the H2O (IL), and dried over MgSO4. After evaporation of the solvents, the residue (147.14 g) was dissolved in iPr2O (500 mL) and cooled to 00C. To this solution was added the solution of HCl gas in EtOAc (150 ml, ~4M) drop-wise (30 min) and with stirring. White precipitate that was formed during HCl addition was filtered 30 min after completion of the addition. It was washed with iPr2O (300 mL) and was dried in vacuo at 400C during 48 h providing 118.79 g of cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6- dimethyl-morpholine hydrochloride as white powder. (67% yield) (Melting Point = 205.90C)
1U NMR (400 MHz, CDCl3) δ: 0.63 (3H, t, J = 7.39 Hz), 1.12 (3H, d, J = 6.30 Hz), 1.13 (3H, d, J = 6.32 Hz), 1.24 (6H, s), 1.26 (3H, d, J = 6.65 Hz), 1.59 (2H, q, J = 7.49 Hz), 2.05 (IH, q, J = 10.77 Hz), 2.26 (IH, q, J = 10.80 Hz), 2.27-2.35 (IH, m), 2.56-2.66 (2H, m), 2.76-2.89 (2H, m), 3.22 (2H, t, J = 13.28 Hz), 4.36-4.44 (IH, m), 4.46-4.54 (IH, m), 7.06 (2H, d, J = 8.22 Hz), 7.23 (2H, d, J = 8.22 Hz), 12.55 (IH, b)
Example 2: One pot synthesis of cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propyl}-2,6-dimethyl-morpholine hydrochloride (Ia) using NaBH4 as reducing agent Through a mixture of (l,l-dimethyl-propyl)-4-iodo-benzene (137.07 g, 050 mol), palladium(II) acetat (1.122 g, 5 mmol) and sodium bicarbonate (50.40 g, 0.60 mol) in dry DMF (500 mL) was bubbled nitrogen for 10 min. Then 2-methyl-prop-2-en-l-ol (54.083 g, 0.75 mol) was added and nitrogen bubbled for another ten minutes. The reaction mixture was heated under nitrogen for 9 h at 1000C. The reaction was cooled (0°C)and was filtered through a thin layer of Celite. The Celite was then washed with cold DMF (300 mL). To DMF solution was added EtOH (abs. 700 mL) followed by AcOH (30 mL) and cis-2,6- dimethylmorpholine (73.9 mL). After 15 min stirring at RT, the reaction was cooled to -5°C and NaBH4 (15.93 g, 0.42 mol) was added in portions over Ih 30 min so that temperature did not exceed 2-3°C. Stirring was continued at 00C for 1 h and the reaction was quenched by drop-wise addition of NaOH solution in water at 00C (30 g, 0.75 mol, /100 mL H2O) to pH=12. The reaction was then partitioned between H2O (2 L) and Hept/EtOAc (9/1) (2x1 L). The organic phases were combined, washed with the H2O (IL), and dried over MgSO4. After evaporation of the solvents, the residue (147.14 g) was dissolved in iPr2O (500 mL) and cooled to 00C. To this solution was added the solution of HCl gas in EtOAc (150 ml, ~4M) drop-wise (30 min) and with stirring. White precipitate was formed during HCl addition and was filtered 30 min after completion of the addition. It was washed with iPr2O (300 mL) and was dried in vacuo at 400C during 48 h providing 112.34 g of cis-4-{3-[4-(l,l-dimethyl- propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine hydrochloride as white powder. (64% yield) (Melting Point = 205.80C)
1U NMR (400 MHz, CDCl3) δ: 0.63 (3H, t, J = 7.39 Hz), 1.12 (3H, d, J = 6.30 Hz), 1.13 (3H, d, J = 6.32 Hz), 1.24 (6H, s), 1.26 (3H, d, J = 6.65 Hz), 1.59 (2H, q, J = 7.49 Hz), 2.05 (IH, q, J = 10.77 Hz), 2.26 (IH, q, J = 10.80 Hz), 2.27-2.35 (IH, m), 2.56-2.66 (2H, m), 2.76-2.89 (2H, m), 3.22 (2H, t, J = 13.28 Hz), 4.36-4.44 (IH, m), 4.46-4.54 (IH, m), 7.06 (2H, d, J = 8.22 Hz), 7.23 (2H, d, J = 8.22 Hz), 12.55 (IH, b)
Example 3: Two steps synthesis of cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propyl}-2,6-dimethyl-morpholine hydrochloride (Ia) using hydrogen and palladium on carbon as reducing agent
First step:
Same as example 1
Second step:
3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde (100.56 g, 0,46 mol) obtained as described in example 1-b) was dissolved in absolute methanol (500 mL). To the methanol solution cooled at 00C (ice/water) was added AcOH (30 mL, 0.50 mol) and cis-2,6- dimethylmorpholine (69.108, 0.60 mol) and the reaction was stirred at room temperature for 30 min. It was then placed under nitrogen and Pd/C 10% (5.00 g) added to it. nitrogen was replaced with hydrogen and the reaction was heated at 37°C under hydrogen pressure. At the end of the reduction, the reaction mixture was filtered through Celite. The Celite was washed with MeOH (200 mL), then aqueous NaOH solution was added to the methanolic reaction mixture until pH=12. The reaction mixture was partitioned with Hept/EtOAc (9/1) (2x1 L), the organic phase was washed with water (1 L) and was dried over MgSO4. After evaporation of the solvents, the residue (144.32 g) was dissolved in iPr2O (500 mL) and cooled to 00C. To this solution was added a solution of HCl gas in EtOAc (150 ml, ~4M) drop- wise (30 min) and with stirring. White precipitate formed during HCl in EtOAc addition was filtered 30 min after completion of the addition. It was washed with iPr2O (300 mL) and was dried in vacuo at 400C during 48 h providing 112.65 g of cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2- methyl-propyl}-2,6-dimethyl-morpholine hydrochloride as white powder. (64% yield) (Melting Point = 205.70C)
1U NMR (400 MHz, CDCl3) δ: 0.63 (3H, t, J = 7.39 Hz), 1.12 (3H, d, J = 6.30 Hz), 1.13 (3H, d, J = 6.32 Hz), 1.24 (6H, s), 1.26 (3H, d, J = 6.65 Hz), 1.59 (2H, q, J = 7.49 Hz), 2.05 (IH, q, J = 10.77 Hz), 2.26 (IH, q, J = 10.80 Hz), 2.27-2.35 (IH, m), 2.56-2.66 (2H, m), 2.76-2.89 (2H, m), 3.22 (2H, t, J = 13.28 Hz), 4.36-4.44 (IH, m), 4.46-4.54 (IH, m), 7.06 (2H, d, J = 8.22 Hz), 7.23 (2H, d, J = 8.22 Hz), 12.55 (IH, b)
Claims
1 Process for preparing 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propionaldehyde of formula (II):


J^°" (VII)
in the presence of palladium catalyst and a base in a solvent chosen from N,N- dimethylformamide, polar protic and non polar organic solvents at temperature between 60° to 15O0C.
Process of producing Amorolfine of formula (I)

or its salts, characterized in that it comprises, after the process according to claim 1, an amino reduction step consisting in reacting 3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl- propionaldehyde of formula (II) with c/5'-2,6-dimethyl morpholine of formula (IV)

4 Process as claimed in one of claims 1 to 3 characterized in that the palladium catalyst used in the Heck reaction is selected from the group consisting of palladium(II)acetate, palladium(II)chloride, tetrakis (triphenylphosphine)-palladium(O), palladium on activated carbon, dichloro [l,l'-bis(diphenylphosphino)ferrocene] palladium(II) and dichloro-bis- triphenylphosphino palladium(II).
5 Process as claimed in one of claims 1 to 4 characterized in that a phosphinic ligand is used in combination with the palladium catalyst.
6 Process as claimed in claim 5 characterized in that the phosphinic ligand is selected from the group consisting of triphenylphosphine, tri-o-tolylphosphine, tri-m-tolylphosphine and tri-p-tolylphosphine
7 Process as claimed in one of claims 1 to 6 characterized in that the polar protic solvent used in the Heck reaction is selected from the group consisting of methanol, ethanol, n- propanol, i-propanol, n-butanol, i-butanol, t-butanol and appropriate mixture of these solvents with water.
8 Process as claimed in one of claims 1 to 7 characterized in that the non polar organic solvent used in the Heck reaction is selected from the group consisting of tetrahydrofurane, ethyl acetate, toluene, o- xylene, m- xylene and p-xylene.
9 Process as claimed in one of claims 1 to 8 characterized in that the base used in the Heck reaction is selected from the group consisting of a tertiary amine, a metal carbonate, and a metal acetate.
10 Process as claimed in one of claims 2 to 9 characterized in that the reducing agent used in the amino reduction step is selected from the group consisting of hydrogen gas in the presence of a palladium catalyst and mixed metal hydrides.
11 Process as claimed in claim 10 characterized in that the palladium catalyst used in
the amino reduction step is selected from the group consisting of palladium on activated carbon, palladium on activated carbon in the presence of metal salts or Perlmans catalyst.
12 Process as claimed in claim 10 characterized in that the mixed metal hydride used in the amino reduction step is selected from the group consisting of sodium borohydride, lithium borohydride, sodium cyano borohydride and lithium cyanoborohydride.
13. Process as claimed in one of claims 2 to 12 characterized in that the polar protic solvent used in the amino reduction step is selected from the group consisting of methanol, ethanol, propanol, i-propanol, butanol, iso-butanol and tert-butanol.
14 Process as claimed in one of claims 2 to 13 characterized in that the non polar solvent used in the amino reduction step is selected from the group consisting of toluene, tetrahydrofurane and ethyl acetate.
15 Process as claimed in claim 11 characterized in that the temperature in the amino reduction step is no more than 45°C, preferably between 20° and 45°C, and more preferably between 20° and 3O0C.
16 Process as claimed in claim 12 characterized in that the temperature in the amino reduction step is between O0C and 3O0C.
17. Process as claimed in one of claims 2 to 16 characterized in that the two steps are performed in a one pot process without isolation of the aldehyde intermediate of formula (II).
18. Process as claimed in claim 17 characterized in that it comprises the following steps: a) first react (l,l-dimethyl-propyl)-4-iodo-benzene with 2-methyl-prop-2-en-l-ol in N,N-dimethylformamide and in the presence of palladium acetate and sodium bicarbonate, at 1000C during 9h; b) filtrate the catalyst through Celite, then add ethanol, acetic acid and cis-2,6- dimethylmorpholine ; c) Cool the mixture to -50C, then add sodium borohydride ; d) After work-up, dissolve the crude product obtained from step c) in diisopropyl
oxide and add a solution of hydrochloric acid gas in ethyl acetate; e) isolate cis-4-{3-[4-(l,l-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl- morpholine hydrochloride by filtration.
19. Process for preparing (l,l-dimethyl-propyl)-4-iodo-benzene of formula (Via)

with sodium metaperiodate and iodine in a mixture of acetic acid and acetic anhydride followed by addition of sulfuric acid.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07727522A EP2007742A1 (en) | 2006-04-03 | 2007-03-29 | Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) |
| CA002647586A CA2647586A1 (en) | 2006-04-03 | 2007-03-29 | Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) |
| US12/244,788 US20090131660A1 (en) | 2006-04-03 | 2008-10-03 | SYNTHESIS OF 3-[4-(1,1-DIMETHYL-PROPYL)-PHENYL]-2-METHYL-PROPIONALDEHYDE AND cis-4--2,6-DIMETHYL-MORPHOLINE (AMOROLFINE) |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06290532A EP1842848A1 (en) | 2006-04-03 | 2006-04-03 | Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]2-methyl-propionaldehyde and cis-4{3-[4-(1,1-dimethyl-propyl)-phenyl]2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) |
| EP06290532.8 | 2006-04-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007113218A1 true WO2007113218A1 (en) | 2007-10-11 |
Family
ID=37012060
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/053050 Ceased WO2007113218A1 (en) | 2006-04-03 | 2007-03-29 | Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20090131660A1 (en) |
| EP (2) | EP1842848A1 (en) |
| AR (1) | AR060341A1 (en) |
| CA (1) | CA2647586A1 (en) |
| WO (1) | WO2007113218A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013097629A1 (en) * | 2011-12-30 | 2013-07-04 | 浙江海翔药业股份有限公司 | Preparation method of amorolfine hydrochloride |
| CN115093309A (en) * | 2022-06-20 | 2022-09-23 | 浙江海翔药业股份有限公司 | Amorolfine hydrochloride intermediate impurity and preparation method thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109232458A (en) * | 2018-06-15 | 2019-01-18 | 南通常佑药业科技有限公司 | A kind of preparation method of chirality morpholinium compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0024334A1 (en) * | 1979-08-17 | 1981-03-04 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Morpholine derivatives, process for their production, pharmaceutical preparations containing these derivatives |
| EP0447947A1 (en) * | 1990-03-23 | 1991-09-25 | BASF Aktiengesellschaft | N-(3-Phenyl-2-methylpropyl and -methyl-prop-2-enyl)-azaheterocycles |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1548595A (en) | 1921-07-14 | 1925-08-04 | Susannah M Freileweh | Suitcase |
| AT354187B (en) | 1976-11-22 | 1979-12-27 | Hoffmann La Roche | FUNGICIDE AGENT |
-
2006
- 2006-04-03 EP EP06290532A patent/EP1842848A1/en not_active Withdrawn
-
2007
- 2007-03-29 EP EP07727522A patent/EP2007742A1/en not_active Withdrawn
- 2007-03-29 CA CA002647586A patent/CA2647586A1/en not_active Abandoned
- 2007-03-29 WO PCT/EP2007/053050 patent/WO2007113218A1/en not_active Ceased
- 2007-04-03 AR ARP070101392A patent/AR060341A1/en not_active Application Discontinuation
-
2008
- 2008-10-03 US US12/244,788 patent/US20090131660A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0024334A1 (en) * | 1979-08-17 | 1981-03-04 | F. HOFFMANN-LA ROCHE & CO. Aktiengesellschaft | Morpholine derivatives, process for their production, pharmaceutical preparations containing these derivatives |
| EP0447947A1 (en) * | 1990-03-23 | 1991-09-25 | BASF Aktiengesellschaft | N-(3-Phenyl-2-methylpropyl and -methyl-prop-2-enyl)-azaheterocycles |
Non-Patent Citations (4)
| Title |
|---|
| FORSYTH, S.A. ET AL.: "One-Pot Multistep Synthetic Strategies for the Production of Fenpropimorph Using an Ionic Liquid Solvent", ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 10, no. 1, 30 November 2005 (2005-11-30), pages 94 - 102, XP002401290 * |
| M.E. BOEDTKER: "Sur quelques homologues du diphényle", BULLETIN DE LA SOCIETE CHIMIQUE DE FRANCE, vol. 45, 1929, pages 645 - 650, XP009073169 * |
| P. LULINSKI, L. SKULSKI: "Iodination of Both Deactivated and ActivatedArenes with Sodium Periodate or Sodium Iodate as the Oxidants", BULLETIN OF THE CHEMICAL SOCIETY OF JAPAN, vol. 73, no. 4, 2000, pages 951 - 956, XP002401584 * |
| See also references of EP2007742A1 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013097629A1 (en) * | 2011-12-30 | 2013-07-04 | 浙江海翔药业股份有限公司 | Preparation method of amorolfine hydrochloride |
| CN115093309A (en) * | 2022-06-20 | 2022-09-23 | 浙江海翔药业股份有限公司 | Amorolfine hydrochloride intermediate impurity and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2647586A1 (en) | 2007-10-11 |
| US20090131660A1 (en) | 2009-05-21 |
| AR060341A1 (en) | 2008-06-11 |
| EP2007742A1 (en) | 2008-12-31 |
| EP1842848A1 (en) | 2007-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2699549B2 (en) | Method for producing 4-benzoyl-5-hydroxypyrazoles | |
| CN105218378A (en) | Prepare the method for substituted biphenyl | |
| US10239831B2 (en) | Method for producing bis(3-aminophenyl)disulfides and 3-aminothiols | |
| WO2007113218A1 (en) | Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine) | |
| CN111689852B (en) | Preparation method of 2,3,5,6-tetrachlorobenzoyl chloride | |
| CN107428648B (en) | Process for the preparation of compounds such as 3-arylbutyraldehyde useful for the synthesis of medetomidine | |
| CN113548982B (en) | Preparation method of 4-cyano-2-fluorobenzyl alcohol | |
| CN114409600A (en) | Synthetic method of enilconazole | |
| JP3522310B2 (en) | Method for producing 2-chloro-4-methylsulfonylbenzoic acid | |
| JPWO2006016510A1 (en) | Process for producing 2-amino-5-iodobenzoic acid | |
| WO2001074799A1 (en) | Fluorinated oxetane derivatives and production process thereof | |
| JP4649733B2 (en) | Method for producing acetophenone compound containing trifluoromethyl group | |
| JPH07300445A (en) | Preparation of 4-halo-2'-nitrobutyrophenone compound | |
| CN112745244B (en) | Intermediate of simotimod and synthesis method thereof | |
| NO20025652L (en) | Process for the preparation of trifluoroethoxy-substituted benzoic acids | |
| JP5448572B2 (en) | Acetyl compound, method for producing the acetyl compound, and method for producing a naphthol compound using the acetyl compound | |
| CN114181097A (en) | Synthetic method of methoxamine hydrochloride | |
| FR2602766A1 (en) | DICHLOROTRIFLUOROMETHYLNITROTOLUENE COMPOUNDS AND PROCESS FOR THE PREPARATION OF AMINOTRIFLUOROMETHYLTOLUENES THEREFROM | |
| CN113603604B (en) | Preparation method of tert-butyl glycinate | |
| CN116425623B (en) | Method for synthesizing 3,5-dichloro-4-methylbenzoic acid by one-pot method | |
| CN1318379C (en) | Process for roducing alpha-methyl-ss-keto ester | |
| CN111217709A (en) | Preparation method of (1-fluorocyclopropyl) methylamine hydrochloride | |
| CN101993413B (en) | Method for preparing 4-pyridine propyl alcohol | |
| CN116239468A (en) | The preparation method of 2-(2,6-diethyl-4-methylphenyl)-malonic acid dimethyl ester | |
| GB2336155A (en) | Process for producing 1-bromo-4-phenylbutane |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07727522 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007727522 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2647586 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |