WO2007126117A1 - フェニル 5-チオグルコシド化合物 - Google Patents
フェニル 5-チオグルコシド化合物 Download PDFInfo
- Publication number
- WO2007126117A1 WO2007126117A1 PCT/JP2007/059394 JP2007059394W WO2007126117A1 WO 2007126117 A1 WO2007126117 A1 WO 2007126117A1 JP 2007059394 W JP2007059394 W JP 2007059394W WO 2007126117 A1 WO2007126117 A1 WO 2007126117A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- mmol
- methyl
- phenol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/203—Monocyclic carbocyclic rings other than cyclohexane rings; Bicyclic carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a ferrule 5-thiodarcoside compound having an inhibitory activity on sodium-dependent glucose cotransporter 1 (SGLT1) involved in glucose reabsorption in the kidney.
- SGLT1 sodium-dependent glucose cotransporter 1
- the fasting blood glucose level is 126 mgZdL or more.
- IGT impaired glucose tolerance
- Non-Patent Document 2 administration of the a-darcosidase inhibitor acarbose, which inhibits sugar hydrolase and delays the absorption of sugar in the small intestine, suppresses the transition from IGT to type 2 diabetes. Furthermore, it has been reported that the onset of hypertension is also significantly suppressed (see Non-Patent Document 2).
- SG LT1 Sodium-dependent glucose cotransporter 1
- IGT Sodium-dependent glucose cotransporter 1
- Non-Patent Documents sodium-dependent dalcose cotransporter 2 (SGLT2) is frequently expressed in the kidney, and glucose that has been glomerularly filtered is reabsorbed via SGLT2 (Non-Patent Documents). 3). SGLT2 inhibitors promote glucose excretion in the urine and reduce blood sugar. As a result, it has come to be considered as a target molecule for new antidiabetic drugs (see Non-Patent Document 4). against this background, SGLT2 inhibitors have been studied and aryl-5 thioglycoside derivatives have been provided (see Patent Document 7).
- Patent Document 1 International Publication No. WO2002Z098893 Pamphlet
- Patent Document 2 International Publication No. WO2004Z014932 Pamphlet
- Patent Document 3 International Publication No. WO2004Z018491 Pamphlet
- Patent Document 4 International Publication No. WO2004Z019958 Pamphlet
- Patent Document 5 International Publication No. WO2005Z121161 Pamphlet
- Patent Document 6 International Publication No. WO2004Z050122 Pamphlet
- Patent Document 7 International Publication No. WO2004Z014931 Pamphlet
- Non-Patent Document 1 Pan XR, et al. Diabets Care, 20th, 534, 1997
- Non-Patent Document 2 J. Shias, et al. Lancent, 359, 2072, 2002
- Non-Patent Document 3 E. M. Wright, Am. J. Physiol. Renal. Physiol., 280, F10, 2001
- Non-Patent Document 4 G. Toggenburger, et al. Biochem. Biophys. Acta., 688, 557, 1 982
- the present invention provides a novel phenol 5-thio-13D-darcopyranoside compound capable of controlling IGT by inhibiting SGLT1 activity and suppressing glucose absorption from the gastrointestinal tract.
- the purpose is to do.
- Aspect (1) of the present invention is a phenyl 5-thiodarcosidyl compound represented by the following formula (I), a pharmaceutically acceptable salt thereof, or a hydrate thereof.
- R 1 and R 2 are the same or different and each represents a hydrogen atom, hydroxyl group, C alkyl group, C alkyl
- 1-6 1-6 is a alkoxy group or a halogen atom
- Z is —NHSO fuel, —NHSO NH, or —Y—Q,
- Y is a single bond, a C alkylene group or a C alkylene group
- 2 1-6 a 4- to 6-membered heterocycloalkyl group substituted with at least one of a kill group and an oxo group (the heterocycloalkyl group may be condensed with a phenyl group);
- R A and R B are the same or different and each represents a hydrogen atom, a C alkyl group, C cycloalkyl.
- R e is a C alkyl group substituted with 1 to 4 groups selected from the group consisting of a hydroxyl group, a carboxyl group, and —CONR ei R e2 force;
- R ei and R e2 are the same or different, I together with a hydrogen atom or it may also be substituted with a hydroxyl group V, a force is C alkyl or by bonding R ei and R e2,, Ru nitrogen atom
- a ring-constituting atom it may contain a heteroatom selected from an oxygen atom, a sulfur atom and a nitrogen atom, and a 5- to 6-membered heterocycloalkyl group (the heterocycloalkyl group is substituted with a hydroxyl group).
- C alkyl group or C alkoxy carbo May be substituted with a group ⁇ ).
- R 1 is a hydrogen atom, a hydroxyl group, a C alkyl group or a C alkyl
- the phenol of the above aspect (1) which is a alkoxy group and R 2 is a C alkyl group or a halogen atom.
- Another embodiment of the present invention is a phenyl 5 of the above embodiment (3) wherein Y is a C alkylene group.
- Z guard Y-NHCON (RA) R B (RA and Y are as defined in embodiment (1), R B is hydrogen atom) is a form (1) or (2) phenyl 5-thiodarcoside compound or a pharmaceutically acceptable salt thereof or a hydrate thereof.
- Z guard Y-CONH (R E) ( R E are as defined in embodiment (1), Y is C alkylene group or a C Aruke - an alkylene group) Embodiment (1) or (
- a phenyl 5-thiodarcoside compound of the embodiment (6) which is an alkyl group or a product thereof
- a pharmaceutically acceptable salt or a hydrate thereof is provided.
- R 1 and R 2 are the same or different and each is a hydrogen atom, a hydroxyl group, a C alkyl group, or a C alkoxy group,
- R A and R B are the same or different and each represents a hydrogen atom, a C alkyl group, C cycloalkyl.
- Y represents a C alkylene group or a C alkenylene group, and R c is substituted with —CONH.
- the phenyl 5 thiodalcoside compound according to any one of aspects (1) to (8) or a pharmaceutically acceptable salt thereof or a hydrate thereof is used as an active ingredient.
- SGLT1 sodium-dependent glucose cotransporter 1
- the phenyl 5-thiodarcoside compound or a pharmaceutically acceptable salt or hydrate thereof according to any one of aspects (1) to (8) is effective. It is a preventive or therapeutic agent for diabetes included as a component.
- the compound of the present invention has strong SGLT1 inhibitory activity, patients with borderline (pre-diabetes group) patients who have postprandial hyperglycemia even in the normal range of preprandial blood glucose obtained only by treatment of diabetic patients. It can also be treated to prevent the transition to diabetes.
- borderline pre-diabetes group
- C alkyl group means a linear or branched alkyl group having 1 to 6 carbon atoms
- a methyl group, an ethyl group, an n propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a tert butyl group, a sec butyl group, an n pentyl group, and an n xyl group can be mentioned.
- C cycloalkyl group refers to a cyclic alkyl group having 3 to 7 carbon atoms.
- Examples include chloropropyl group, cyclobutyl group, cyclopentyl group, cyclohexyl group, and cycloheptyl group. Of these, a cyclopentyl group and a cyclohexyl group are preferable.
- C alkoxy group refers to a linear or branched alkoxy having 16 carbon atoms. Means a Si group, and a C alkoxy group is preferred. Examples of the C alkoxy group include meth
- Examples thereof include a xy group, an ethoxy group, a propoxy group, an isopropoxy group, an n-butoxy group, an isobutoxy group, and a tert-butoxy group.
- halogen atom is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- ⁇ kill group '' refers to a heterocycloalkyl group having 4 to 6 ring atoms containing 1 to 2 heteroatoms with 0, S and N forces also selected, and Hydrogen atom is C
- it is a 4- to 6-membered nitrogen-containing heteroalkyl group substituted with 1 or 2 oxo groups (the heterocycloalkyl group may be substituted with a C alkyl group).
- imidazolidyl groups eg, 5,5-dimethyl-imidazolidine-1,2,4 dione and 3-yl groups
- a kill group and an oxo group 1,3 dioxo-1,3 dihydro (1) 2H isoindole-2-yl group and the like.
- Ethyl group 2-hydroxy 1,1-dimethylethyl group, 1,3 dihydroxy 2-methyl propane 2-yl group, strong rubamoylmethyl group, 2-strong rubamoylethyl group, benzyl group, phenethyl group, 3-pyridylmethyl group.
- hydroxyl group, and carboxyl group is C alkyl group was substituted with CONR ei 1 to 4 radicals selected from the group consisting of R e2 'is a hydrogen atom on that group, hydroxyl group, carboxy
- Group group consisting of a ru group and —CONR ei Re 2 represents a C alkyl group substituted by 1 to 4 (preferably 1 to 3) of at least one selected group. For example, hydroxymethyl
- hydroxyethyl group 2-hydroxy 1,1-dimethylethyl group, 1,3 dihydroxy 2-methylpropane 2-yl group, 1,3-dihydroxy 2-hydroxymethyl pro Examples include pan 2-yl group, force ruberamoylmethyl group, 2-force rubermoylethyl group, 2-force rubermoyl 2-methylethyl group.
- “It may be formed together with the nitrogen atom to which 1 and 2 are bonded, and may further include a hetero atom selected from an oxygen atom, a sulfur atom and a nitrogen atom as a ring-constituting atom.
- a hetero atom selected from an oxygen atom, a sulfur atom and a nitrogen atom as a ring-constituting atom.
- ⁇ 6-membered heterocycloalkyl group (the heterocycloalkyl group may be substituted with a C alkyl group which may be substituted with a hydroxyl group, or a C alkoxycarbonyl group) ”
- 1-pyrrolidyl group 1-piveridyl group, 1-piperazyl group, 4 morpholino group, 4-thiomorpholino group, 4-methyl-1-piperazyl group, 4 (2 hydroxy shetyl) 1-piperazyl group, 4-methoxycarbolulu 1-piperazine group and the like can be mentioned.
- C alkyl group optionally substituted with a hydroxyl group means at least one hydroxyl group
- Examples thereof include a til group, a 2-hydroxy 1,1-dimethylethyl group, a 1,3-dihydroxy 2-methylpropane-2-yl group, and a 1,3-dihydroxy 2-hydroxymethylpropane 1-2-yl group.
- the "C alkylene group” is a linear or branched divalent saturation composed of 1 to 6 carbon atoms.
- hydrocarbon It is a hydrocarbon.
- methylene, ethylene, trimethylene, tetramethylene, 2-methylpropylene, 2,2-dimethylpropylene and the like can be mentioned.
- C alk-lene group means a straight chain or branched chain having 2 to 6 carbon atoms including a double bond.
- ethylene, probelene, 2-butylene and the like can be mentioned. Of these, probelene is preferable.
- “Pharmaceutically acceptable salt” refers to a salt with an alkali metal, an alkaline earth metal, an ammonium, an alkyl ammonium, a mineral acid or an organic acid.
- Hydrate refers to a pharmaceutically acceptable hydrate of the compound of the present invention or a salt thereof.
- the compound of the present invention or a salt thereof may be exposed to air or recrystallized to absorb moisture and form adsorbed water, or may become a hydrate.
- the hydrate in the present invention includes such hydrates.
- the compounds of the present invention may have a chiral center, they exist as various diastereomeric or enantiomeric forms. Some of the compounds of the present invention also exist as keto-enol tautomers, for example. In addition, some of the compounds of the present invention also exist as geometrically different bodies (E-form, Z-form). Accordingly, the compounds of the present invention include all the individual isomers above as well as mixtures thereof.
- R 1 and R 2 are the same or different and are a hydrogen atom, a hydroxyl group, a C alkyl group, a C alkoxy group or a halogen atom, and more preferably a hydrogen atom.
- Child hydroxyl group, methyl group, methoxy group or halogen atom.
- R 2 is a cyano group or a halogen atom (especially a chlorine atom), and R 1 is a hydroxyl group, a methoxy group, or a methyl group. Furthermore, R 1 is a hydroxyl group. It is more preferable.
- Y—CONH (R e ) is more preferred in that SGLT1 inhibitory activity is stronger.
- Z is Y—NHCON (R A ) R B Y-NHCONH, Y-NHCONH-C alkyl (the alkyl is substituted with a hydroxyl group)
- Y is a C alkylene group.
- Y is a C alkylene group or C
- R e examples include a C alkyl group substituted with a hydroxyl group and 1 to 4 (more preferably 1 to 3) groups selected from the group consisting of —CO—NR ei R e2. Yes , Rei and Re2
- a hetero atom selected from an oxygen atom, a sulfur atom and a nitrogen atom as a ring atom together with the nitrogen atom to which Re and R e2 are bonded.
- a 5- to 6-membered heterocycloalkyl group (the heterocycloalkyl group is substituted with a C alkyl group or a C alkoxycarbonyl group which may be substituted with a hydroxyl group).
- the following compound groups are preferable in that they have strong SGLT1 inhibitory activity and strong SGLT2 inhibitory activity. These compounds can inhibit SGLT2 activity, which can not only be expected to have a therapeutic effect based on the SGLT1 inhibitory activity described below, thereby suppressing reabsorption of sugar and excreting excess sugar outside the body. Diabetes can be treated, hyperglycemia can be corrected without stressing pancreatic ⁇ cells, and insulin resistance can be improved.
- a compound group having strong SGLT1 inhibitory activity and strong SGLT2 inhibitory activity is ⁇ - ⁇ -CONH (R C ), and Y is a C alkylene group or a C alkkelene group (more preferably propylene.
- R C is selected from the group consisting of a hydroxyl group and —CO—NR C1 R C2 A C alkyl group substituted with 1 to 4 (more preferably 1 to 3) selected groups (the C
- 1-6 1-6 alkyl group is a tertiary carbon atom bonded to —NH—, wherein 1 and 2 are the same or different and may be substituted with a hydrogen atom or a hydroxyl group Base
- the ring member may further include a heteroatom selected from an oxygen atom, a sulfur atom and a nitrogen atom. !, May form a 5- to 6-membered heterocycloalkyl group (the heterocycloalkyl group may be substituted with a hydroxyl group or a C alkyl group).
- R 2 and preferred substitution position of Z are the same as above.
- Z is —Y—CO—NH—C alkyl (including the above).
- Alkyl is substituted with a hydroxyl group), -Y-CO-NH-C (CH) -CONH
- NH) NH or NHCON (R A ) R B can be synthesized by the following method.
- the phenol derivative (lla) can be synthesized according to International Publication WO2004Z050122.
- the 5-thioglucose derivative (III) can be synthesized according to International Publication WO2004Z014931.
- phosphines necessary as a reagent in this reaction include triphenylphosphine, tri-n-butylphosphine, tri-t-butylphosphine, tristolylphosphine and diphenyl-2-pyridylphosphine. Of these, triphenylphosphine and diphenyl-1-pyridylphosphine are preferred, and triphenylphosphine is more preferred.
- azo reagents include jetyl dicarboxylate, diisopropyl azodicarboxyloxy tert-butinoreazodicanoloxylate, 1,1'-azobis (N, N-dimethylformamide) And 1, 1 (azodicarbol) dipiperidine can be used.
- jetylazodicarboxylate (DEAD) and diisopropylazodicarboxylate are preferable.
- the solvent used in this reaction is tetrahydrofuran, dioxane, toluene, dichloromethane, chloroform, acetonitrile, ethyl acetate, dimethyl sulfoxide, N, N-dimethylformamide or the like, preferably tetrahydrofuran or toluene.
- the reaction temperature is preferably from 20 ° C to room temperature, more preferably from 5 ° C to + 5 ° C.
- the compound (IVa) obtained above is converted into an amino group by catalytic hydrogenation in a hydrogen atmosphere using a catalyst such as palladium activated carbon, palladium hydroxide, or platinum-norradium activated carbon, and the compound ( IVb) is obtained.
- a catalyst such as palladium activated carbon, palladium hydroxide, or platinum-norradium activated carbon
- palladium activated carbon and palladium hydroxide are preferred as the catalyst.
- the solvent used for this reaction include methanol, ethanol, isopropanol, ethyl acetate, and acetic acid.
- the reaction temperature is preferably room temperature, reflux temperature, or room temperature.
- Iron can also be used in the presence of tin chloride (11) monohydrate and salt ammonium.
- solvent used in this reaction include methanol, ethanol, isopropanol and the like.
- the reaction temperature is room temperature and reflux temperature.
- the compound (I) of the present invention can be produced by the following method using the compound (IVb) as an intermediate.
- R E represents a benzyloxycarbonyl group or a t-butoxycarbonyl group, and other symbols are as defined above.
- phenylsulfur chloride or isocyanide Acid chlorosulfol can be used to derive compound (IVc) or (IVd).
- Suitable bases include triethylamine, Nethyl N, N diisopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like.
- the solvent to be used include black mouth form, dichloromethane, jetyl ether, tetrahydrofuran, N, N dimethylformamide, acetonitrile, ethyl acetate and the like, or a mixed solvent thereof.
- the reaction temperature is 0 ° C force reflux temperature.
- Compound (IVb) force can also be induced to compound (IVe) using guadinodination reagent (V).
- the solvent used in this reaction include tetrahydrofuran, N, N dimethylformamide, methanol, ethanol, isopropanol, ethyl acetate, toluene and the like.
- the reaction temperature is from room temperature to reflux temperature.
- Suitable bases include triethylamine, Nethyl N, N diisopropylamine, pyridine, DBU, potassium carbonate, calcium carbonate, cesium carbonate and the like.
- the solvent used include black mouth form, dichloromethane, jetyl ether, tetrahydrofuran, N, N dimethylformamide, acetonitrile, and ethyl acetate.
- the reaction temperature is 0 ° C force and reflux temperature.
- the protecting group for 5-thioglucose is removed using a suitable base to obtain the compound (I) of the present invention.
- a suitable base sodium methoxide, sodium hydroxide, lithium hydroxide, potassium carbonate, cesium carbonate, triethylamine and the like can be used.
- Suitable solvents for the reaction are methanol, ethanol, water or a mixed solvent thereof.
- 0 ° C force is also room temperature, and room temperature is preferred.
- the protecting group R E of the guazino group is a benzyloxycarbon group, it can be removed by a catalytic hydrogenation catalyst described in Production Method 1.
- a preferred catalyst at this time is palladium hydroxide.
- the compound that is a Y-acetylene group can be produced by using, as a raw material, a compound having an amino group, which is a thioglucoside compound described in Patent Document 7, with reference to Step 5 of Production Method 1.
- Y 1 represents a single bond or a C alkylene group, and other symbols are as defined above.
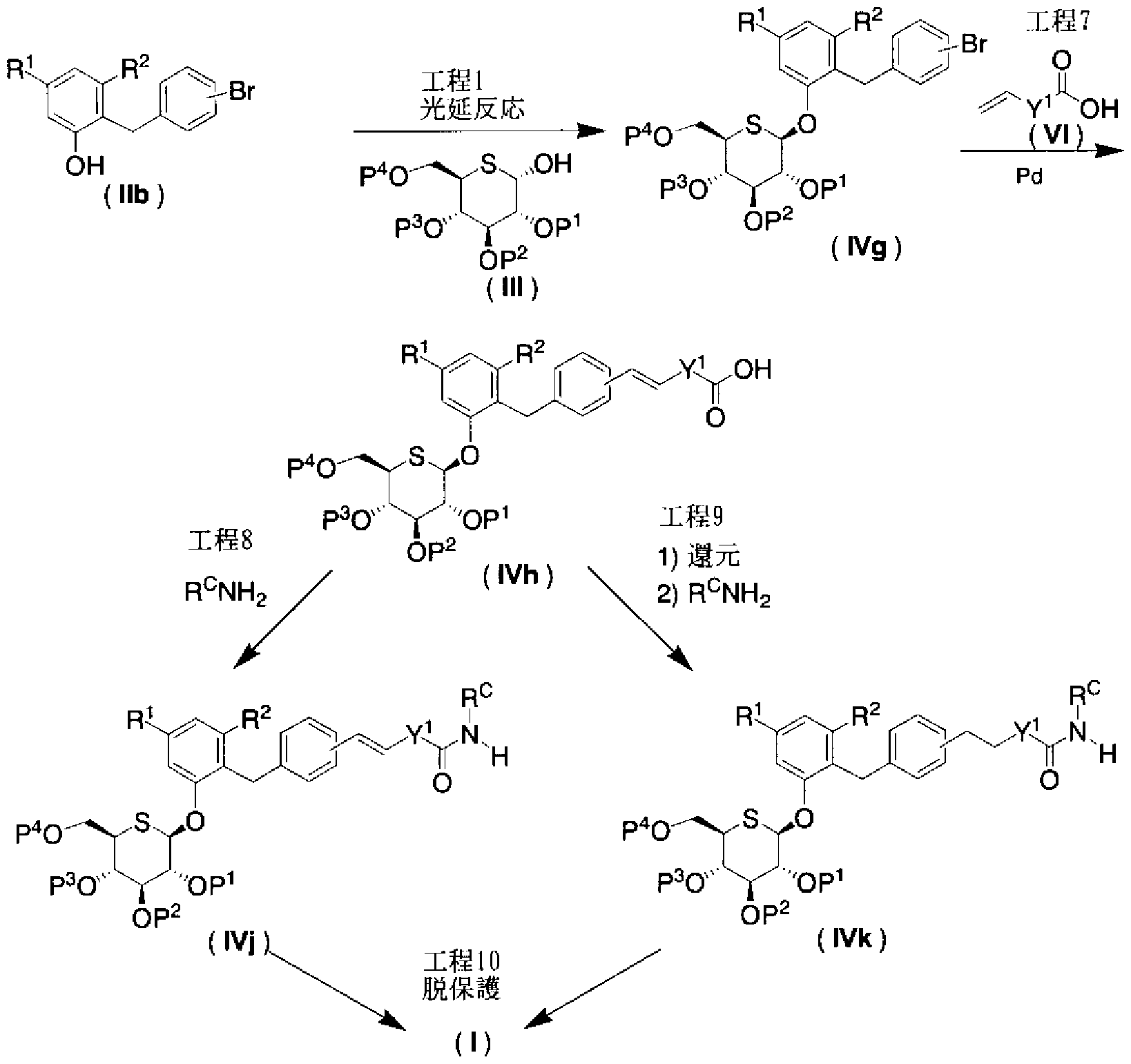
- the phenol derivative (lib) force compound (IVg) can be synthesized by the Mitsunobu reaction shown in Step 1 of Production Method 1.
- Step 7 (Heck reaction)
- Compound (IVh) can be synthesized by subjecting compound (IVg) and olefin acetic acid (VI) to Heck reaction in the presence of a palladium catalyst, a phosphine ligand, and an appropriate base.
- a palladium catalyst used at this time include palladium acetate, tetrakistriphenylphosphine palladium, dibenzylideneacetone palladium, bistriphenylphosphine paradium chloride, palladium activated carbon and the like.
- the phosphine ligand include triphenylphosphine tris (2-methylphenol) phosphine.
- triethylamine, N-ethyl-N, N-diisopropylamine, potassium carbonate, calcium carbonate, cesium carbonate, potassium t-butoxide and the like are used as the base.
- the solvent used in the reaction include acetonitrile, toluene, tetrahydrofuran and the like.
- the reaction temperature is from 0 ° C to the reflux temperature, but a microwave may be used.
- N N-dicyclocarpoimide
- N-ethyl-N N-ethyl-N
- -3 Dimethylaminopropylcarbodiimide hydrochloride
- CDI CDI
- WSC 1-hydroxybenzotriazole monohydrate, etc.
- the reaction temperature here is 0 ° C to 60 ° C.
- Compound (IVk) can also be produced by subjecting the olefin moiety of compound (IVh) to the catalytic hydrogenation shown in Step 2 of Production Method 1 and then condensing with ammine (R e NH 3).
- the compound in which Y is a single bond or a methylene group can be produced by using a compound having a carboxyl group described in Patent Document 7 as a raw material with reference to Step 8 of Production Method 2.
- Y is a C alkylene group or a C alkylene group.
- NHCON (R A ) R B or C alkyl group and oxo
- a compound which is a 4- to 6-membered heterocycloalkyl group substituted with at least one group (the heterocycloalkyl group may be condensed with a phenyl group).
- the heterocycloalkyl group may be condensed with a phenyl group
- a compound for example, a (Vic) compound
- a compound (IVm) that is a sum hydrocarbon ring (which may be condensed with a phenyl group) is converted to a compound (IVm) using the Heck reaction described in Step 7 of Production Method 2.
- the compound (I) of the present invention which is an alkyl group can be synthesized by the following method.
- RN and R NA represent a hydrogen atom or a C alkyl group, and other symbols are as defined above.
- the compound (1) of the present invention which is a 4- to 6-membered heterocycloalkyl group substituted with at least one of an alkyl group and an oxo group, can be produced.
- methanol or ethanol is preferable as the solvent preferred for sodium methoxide.
- the compound of the present invention is a disease or condition that can be ameliorated by inhibiting the activity of SGLT1, eg, for example, it can be used as an active ingredient of a medicament for preventing or treating diabetes, diabetes-related diseases and diabetic complications.
- the compound of the present invention is particularly excellent in that it inhibits SGLT1 activity, thereby suppressing absorption of sugar from the small intestine and improving IGT, thereby preventing the transition to diabetes. Yes.
- diabetes includes type 1 diabetes, type 2 diabetes, and other types of diabetes due to specific causes.
- diabetes-related disease means obesity, hyperinsulinemia, abnormal glucose metabolism, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, abnormal lipid metabolism, hypertension, congestive heart failure Edema, hyperuricemia, gout and the like.
- diabetes complications are classified into acute complications and chronic complications.
- Acute complications include hyperglycemia (eg ketoacidosis), infections (skin, soft tissue, biliary system, respiratory system, urinary tract infection, etc.).
- hyperglycemia eg ketoacidosis
- infections skin, soft tissue, biliary system, respiratory system, urinary tract infection, etc.
- Chronic complications include microangiopathy (nephropathy, retinopathy), arteriosclerosis (atherosclerosis, myocardial infarction, cerebral infarction, lower limb arterial occlusion, etc.), neuropathy (sensory nerve, Motor nerves, autonomic nerves, etc.) and foot gangrene.
- the major complications are diabetic retinopathy, diabetic nephropathy, diabetic neuropathy.
- the compound of the present invention can be administered as a medicine systemically or locally, orally or parenterally.
- the compound of the present invention is a therapeutic agent for diabetes having a different mechanism other than SGLT1 and SGLT2 activity inhibitors and diabetic complications for the purpose of enhancing the action of the compound or reducing the dose of the compound. It can be used in combination with drugs (hereinafter abbreviated as concomitant drugs) such as therapeutic agents, antihyperlipidemic agents, antihypertensive agents, antiobesity agents, diuretics and antithrombotic agents.
- concomitant drugs drugs
- the administration time of the compound of the present invention and the concomitant drug is not limited, and these may be administered simultaneously to the administration subject or may be administered with a time difference.
- the compound of the present invention and the concomitant drug may be administered as two types of preparations containing each active ingredient, or may be administered as a single preparation containing both active ingredients.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug is the subject of administration, administration route, target disease, symptoms, combination It can be appropriately selected depending on the combination.
- the concomitant drug may be used in an amount of 0.01 to LOO parts by weight per 1 part by weight of the compound of the present invention.
- diabetes therapeutic agents include insulin preparations (eg, animal insulin preparations extracted from sushi and swine spleen; human insulin preparations synthesized using E. coli or yeast and genetically engineered; Protamine insulin sub-insulin fragments or derivatives (eg, INS-1 etc.), oral insulin preparations), insulin resistance improvers (eg,
- Pioglitazone or its salt preferably hydrochloride
- rosiglitazone or its salt preferably maleate
- riboglitazone CS—Oi l
- Sipoglitazar TAK— 654
- Metaglidasen M XB- 1 0 2
- Naveglitazar LY—519818
- MX—6054 Nolaglitazone
- N—2344 Nolaglitazone
- T—131 AMG131
- PPAR y Ago-st PPAR y-antagost
- a-Darcosidase inhibitor eg, voglibose, carbolose, miglitol, emidalitate
- biguanide eg, phenformin, metformin, buformin or Their salts (eg, hydrochloride, fumarate, succinate)
- insulin secretagogues sulfururea
- Examples of the therapeutic agent for diabetic complications include aldose reductase inhibitors (eg, torles, epanolorestat, zenarestat, zoponolestat, minarerestat, fidarestat, CT-112) , Neurotrophic factor and its increasing drug (eg, NGF, NT-3, BDNF, neurotrophin production 'secretion promoter), nerve regeneration promoter (eg, Y-128), PKC inhibitor (eg, ruboxis) Taurine mesylate (LY- 333531)), AGE inhibitors (eg, ALT946, pimagedin, pyratoxatin, N-phenacyl thiazolium bromide (ALT766), ALT-711, EXO-226, pyridorin) , Pyridoxamine), active oxygen scavengers (eg, thiotate), cerebral vasodilators (eg, thiopride, mexiletine), somatostatin receptor agonist
- Antihyperlipidemic agents include, for example, statin compounds (eg, pravastatin, sympastatin, oral pastatin, atonolepastatin, flupastatin, itapastatin, rosbastatin, pitapastatin or salts thereof (eg, Sodium salts, calcium salts)), squalene synthase inhibitors (eg, TAK-475), fibrate compounds (eg, bezafibrate, clofibrate, simfibrate, clinofibrate), ACAT inhibitors (eg, abashimibe ( Avasimibe), Eflucimibe), anion exchange scab (eg, cholestyramine), probuconole, nicotinic acid drugs (eg, nicomol), niceritro Niceritrol), icosapentate ethyl, plant sterols (eg, soysterol, gamma-oryzanol), CETP inhibitors (eg,
- antihypertensive agents include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril), angiotensin II antagonists (eg, candesartan cilexetil, oral sultan, eprosartan, valsartan, telmisartan, Ilbesartan, tasosartan, azilzartan (TAK-536)), calcium antagonists (eg, iliapine, difedipine, amlodipine, efonidipine, dicanodipine), potassium channelnore openers (eg, lebucromacarim, L 27152, AL0671, NIP — 121), including clodin
- Anti-obesity agents include, for example, central anti-obesity agents (eg, dexfenfluramine, fenfluramine, phentermine, sibutramine, amphepramone, dexamphetamine, mazindol, phenol-propanolamine, clobenzolex).
- central anti-obesity agents eg, dexfenfluramine, fenfluramine, phentermine, sibutramine, amphepramone, dexamphetamine, mazindol, phenol-propanolamine, clobenzolex.
- MCH receptor antagonist eg, compound described in W 006/035967, SB—568849; SNAP—7941, T 226296); neuropeptide antagonist (eg, CP—422935); cannapinoid receptor antagonist (Eg, Rimonabant (SR—141716), SR—147778); ghrelin antagonists; 1 1 ⁇ -hydroxysteroid dehydrogenase inhibitors (eg, BVT-3498, INCB1373 9)), spleen lipase inhibitors ( Eg, orlistat, ATL—962), DGAT—1 inhibitor, / 3 3 agonist (eg, AJ—9677, AZ40140), peptidic appetite suppressant (eg, leptin, CN TF (ciliary neurotrophic) factor)) , Cholecyst kyungagost (eg, Lynch tribute, FPL-15849), antifeedant (eg, P-57).
- neuropeptide antagonist eg,
- diuretics examples include xanthine derivatives (eg, sodium theoproyl salicylate, calcium theopromin salicylate), thiazide preparations (eg, ethiazide, cyclopenthiazide, trichloromethiazide, hydrothiazide, hydroflumethiazide, benzylhydride).
- xanthine derivatives eg, sodium theoproyl salicylate, calcium theopromin salicylate
- thiazide preparations eg, ethiazide, cyclopenthiazide, trichloromethiazide, hydrothiazide, hydroflumethiazide, benzylhydride.
- Mouth-mouth mouth thiazide, penflutide, polythiazide, methiclothiazide), anti-aldosterone preparation eg, spironolatatone, triamterene
- carbonic anhydrase inhibitor eg, acetazolamide
- chlorobenzenesulfonamide preparation eg, chlorthalidone, mefluside, indapamide
- Examples include fazosemide, isosorbide, ethacrynic acid, pyrethradide, bumetide, and furosemide.
- Antithrombotic agents include, for example, heparin (eg, heparin sodium, heparin calcium, dalteparin sodium, AVE-5026), ⁇ rufarin (eg, ⁇ rufarin potassium, etc.), antithrombin Drugs (eg, argatroban, xymeragatran (Dimigatran), Dabigatran (Odiparcil, Lepirudin, bival irudin, Desirudin, ART—123, Idraparinux, SR—123781, AZD—0837, MCC—977, TGN— 255, TGN—167, RWJ—58436, LB—30870, MPC—09 20, Pegmusirudin ⁇ Org—426751, etc., thrombolytic drugs (eg, urokina se, tisokinase, anoleplase, alteplase) (natepla se), monteplase, pamiteplase,
- a pharmaceutically acceptable carrier can be blended.
- examples of such carriers include common excipients, bulking agents, binders, disintegrants, coating agents, dragees, pH adjusters, solubilizers or aqueous or non-aqueous solvents. Tablets, pills, capsules, granules, powders, powders, solutions, emulsions, suspensions, injections and the like can be prepared from the compound of the present invention and these carriers.
- the solubility of the compound of the present invention can also be improved by inclusion in a, ⁇ or ⁇ -cyclodextrin, methylicyclodextrin, or the like.
- the dose of the compound of the present invention varies depending on the disease, symptom, body weight, age, sex, route of administration, etc. S, for adults, 0.1 to 1 per person: LOOOmg / kg body weight Yes, 0.1 to 200 mg Zkg body weight is preferred 0.1 to 10 mg Zkg body weight is more preferred. This can be administered once to several times a day.
- Methyl 3 Methyl 4— (4-Trobenzyl) 5— [(2, 3, 4, 6—Tetra-1-O-cetyl 5-thio-1—Dalcopyranosyl) oxy] phenyl carbonate instead of methyl 3
- Reference Example 8 (2) The title compound was obtained in the same manner as (0. 242 g, 17%).
- Compound 34 was synthesized in the same manner as in Example 15 using N-aryl N,-(2-hydroxy-1,1-dimethylethyl) urea.
- Example 23 (4-Aminobenzil) -3 ethyl 5— [(2, 3, 4, 6—tetra-acetyl 5—thio 13 D-darcopyranosyl) oxy] phenol methyl carbonate synthesized in Reference Example 17 was used.
- the title compound (0.076 g, 82%) was obtained in the same manner as in Example 3.
- the structure, NMR data and MS data of the obtained compound are shown in Table 1.
- the drug (the compound of the present invention) is mixed with lactose monohydrate, crystalline cellulose, carboxymethyl cellulose-calcium and hydroxypropylcellulose, and this mixture is pulverized with a pulverizer.
- the pulverized mixture is mixed with a stirring granulator for 1 minute, and then granulated with water for 4-8 minutes.
- the resulting granulate is dried at 70 ° C for 40 minutes.
- the granulated dry powder after sieving and magnesium stearate are mixed at 30 rpm for 3 minutes using a V-type mixer.
- the granules for tableting obtained using a mouth-tally tablet press are compression-molded and tableted.
- a human SGLT1 sequence (NM-000343) was amplified from human small intestine-derived mRNA after reverse transcription and introduced into pCMV-tag5A (Stratagene).
- a human SGLT2 sequence (NM-003 041) was prepared from human kidney-derived mRNA by the same method and introduced into pcDNA3.1 + hygro (Invitrogen). The sequence ability of each clone was confirmed to match the reported sequence.
- Cells stably expressing human SGLT1 or human SGLT2 were used in a sodium-dependent glucose uptake activity inhibition test.
- Pretreatment buffer 140 mM choline chloride, 2 mM KC1, ImM CaCl, ImM Mg
- test compound ["C] methyl ⁇ -D-dalcopyranoside containing ⁇ -D-darcobilanoside (lmM), 145 mM NaCl, 2 mM KC1, ImM CaCl , ImM MgCl, lOmM HEPES / 5mM Tris, pH7.4
- the drug group was orally administered with a drug (lmgZkg) suspended in a 0.5% carboxymethylcellulose (CMC) aqueous solution and only a 0.5% CMC aqueous solution in the control group.
- CMC carboxymethylcellulose
- glucose solution (2 gZkg) was orally administered, and blood was collected at a total of 5 points before drug administration (Otime) and 0.25, 0.5, 1, 2 hours after oral administration.
- SGLT1 sodium-dependent glucose cotransporter expressed in the intestinal epithelium
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Description
明 細 書
フエニル 5—チォダルコシド化合物
技術分野
[0001] 本発明は、腎臓でのグルコース再吸収に関わるナトリウム依存性グルコース共輸送 体 1 (SGLT1)の阻害活性を有するフエ-ル 5—チォダルコシドィ匕合物に関する。 背景技術
[0002] 糖尿病に罹患すると、空腹時の血糖値は 126mgZdL以上を示す。また、空腹時 の血糖値が正常であっても、食事の後に 140〜200mg/dLという高い血糖値を示 す場合には、耐糖能異常(以下、 IGT (impaired glucose tolerance)という。 )と診断さ れる。 IGTから糖尿病の発症を遅らせることは、心血管障害のリスクを低減させると考 えられ、それを示す幾つかの知見が得られている。例えば、 1997年に中国で行われ た Da Qing IGT and Diabetes Studyでは、ダイエットや運動を行うことで IGTから 2型 糖尿病への移行を有意に抑制したと報告されている (非特許文献 1参照)。また、薬剤 治療が有効な例として、糖の加水分解酵素を阻害し、小腸力 の糖の吸収を遅延さ せる a—ダルコシダーゼ阻害剤ァカルボースを投与すると、 IGTから 2型糖尿病への 移行を抑制し、さらに高血圧の発症も有意に抑制することが報告されている (非特許 文献 2参照)。
[0003] この様なことから、糖尿病の発症を抑えるには、食事療法、運動及び薬物療法によ つて IGTをコント口一ルすることが重要である。
[0004] 哺乳動物の小腸上皮には高い頻度でナトリウム依存性グルコース共輸送体 1 (SG LT1)が発現している。この SGLT1は小腸において、ナトリウムに依存し、ダルコ—ス 又はガラクトースの能動輸送を司っていることが知られている。そこで、食事由来のグ ルコ—ス吸収を抑制し、 IGTの予防または治療を行うというコンセプトに基づき、 SGL T1活性を阻害するグリコシド化合物が報告されている (特許文献 1〜6参照)。
[0005] また、腎臓には高頻度にナトリウム依存性ダルコース共輸送体 2 (SGLT2)が発現 しており、糸球体でー且濾過されたグルコースは SGLT2を介して再吸収される(非 特許文献 3参照)。そして、 SGLT2阻害剤は、尿への糖排泄を促進し、血糖低下作
用を招来するので、新たな糖尿病治療薬の標的分子と考えられるようになった (非特 許文献 4参照)。このような背景から、 SGLT2阻害剤が研究されァリ—ル 5 チォグ ルコシド誘導体が提供されて ヽる (特許文献 7参照)。
[0006] し力しながら、従来 SGLT1阻害活性を有するチォダルコシド誘導体は知られてい なかった。
特許文献 1:国際公開第 WO2002Z098893号パンフレット
特許文献 2:国際公開第 WO2004Z014932号パンフレット
特許文献 3 :国際公開第 WO2004Z018491号パンフレット
特許文献 4:国際公開第 WO2004Z019958号パンフレット
特許文献 5 :国際公開第 WO2005Z121161号パンフレット
特許文献 6 :国際公開第 WO2004Z050122号パンフレット
特許文献 7 :国際公開第 WO2004Z014931号パンフレット
非特許文献 1 : Pan XR, et al. Diabets Care,第 20卷, 534項, 1997年
非特許文献 2 : J. し Chiasson, et al. Lancent,第 359卷, 2072項, 2002年
非特許文献 3 : E. M. Wright, Am. J. Physiol. Renal. Physiol. ,第 280卷, F10項, 2001 年
非特許文献 4: G. Toggenburger, et al. Biochem. Biophys. Acta.,第 688卷, 557項, 1 982年
発明の開示
発明が解決しょうとする課題
[0007] 本発明は、 SGLT1活性を阻害し、消化管からのグルコース吸収を抑制することで、 IGTをコントロールすることができる新規なフエ-ル 5—チォー 13 D—ダルコピラノ シドィ匕合物を提供することを目的とする。
課題を解決するための手段
[0008] 本発明者らは、前記課題を解決するために鋭意研究した結果、ある種の 2 べンジ ルフエノール誘導体を 5—チォダルコシル化した化合物力 今までのチォダルコシド 誘導体には見られな力つた強い SGLT1阻害活性を有することを発見し、本発明を 兀成し 7こ。
[0009] 以下に、本発明のフ -ル 5—チォダルコシド誘導体 (以下、「本発明化合物」と いう)の態様を述べる。
(1) 本発明の態様(1)は、下記式 (I)で表されるフエニル 5—チォダルコシドィ匕合 物若しくはその製薬学的に許容される塩又はそれらの水和物である。
[0010] [化 1]

( I ) 式中、
R1及び R2は、同一または異なって、水素原子、水酸基、 C アルキル基、 C アル
1-6 1-6 コキシ基又はハロゲン原子であり、
Zは、— NHSOフエ-ル、— NHSO NH、又は— Y— Qであり、
2 2 2
Yは、単結合、 C アルキレン基又は C ァルケ-レン基であり、
1-6 2-6
Qは、一 NHC ( = NH) NH 、 一 NHCON (RA)RB、 一 CONH (RC)、又は C ァノレ
2 1-6 キル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロアルキル基( 該ヘテロシクロアルキル基はフエ-ル基と縮合してもょ 、)であり、
RA及び RBは、同一または異なって、水素原子、 C アルキル基、 C シクロアルキ
1-6 3-7
ル基、又は水酸基、 -CONH、フエニル基及びピリジル基力 なる群より選択される
2
1〜3個の基で置換された C アルキル基を示し、
1-6
Reは、水酸基、カルボキシル基及び— CONReiRe2力もなる群より選択される 1〜 4個の基で置換された C アルキル基であり、
1-6
Rei及び Re2は、同一または異なって、水素原子または水酸基で置換されても良 V、C アルキル基である力、あるいは Rei及び Re2が結合して 、る窒素原子と一緒にな
1-6
つて、さらに環構成原子として、酸素原子、硫黄原子及び窒素原子から選択されるへ テロ原子を含んでも良 、5〜6員のへテロシクロアルキル基 (該ヘテロシクロアルキル 基は、水酸基で置換されても良い C アルキル基、または C アルコキシカルボ-ル
基で置換されてもょ ヽ)を形成してもよ 、。
(2) 本発明の他の態様は、 R1が、水素原子、水酸基、 C アルキル基又は C アル
1-4 1-4 コキシ基であり、 R2が、 C アルキル基又はハロゲン原子である、上記態様(1)のフエ
1-4
-ル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又はそれらの 水和物である。
(3) 本発明の他の態様は、 Zがー Y—NHC ( = NH) NH (Yは態様(1)で定義した
2
とおりである)である上記態様(1)または(2)のフエ-ル 5—チォダルコシド化合物若 しくはその製薬学的に許容される塩又はそれらの水和物である。
(4) 本発明の他の態様は、 Yが C アルキレン基である上記態様(3)のフエニル 5
1-6
ーチォダルコシドィ匕合物若しくはその製薬学的に許容される塩又はそれらの水和物 である。
( 5) 本発明の他の態様は、 Zがー Y—NHCON (RA) RB (RA及び Yは態様(1 )で定 義したとおりであり、 RBは水素原子である)である態様(1)または(2)のフエニル 5 - チォダルコシドィ匕合物若しくはその製薬学的に許容される塩又はそれらの水和物で ある。
(6) 本発明の他の態様は、 Zがー Y—CONH (RE) (REは態様(1)で定義したとおり であり、 Yは C アルキレン基又は C ァルケ-レン基である)である態様(1)または(
1-6 2-6
2)のフエ-ル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又は それらの水和物である。
(7) 本発明の他の態様は、 が水酸基及び— CO— NRG1RE2 (REI及び 2は、態 様(1)で定義したとおりである)からなる群より選択される 1〜4個の基で置換された C
1 アルキル基である、態様(6)のフエニル 5—チォダルコシドィ匕合物若しくはその製
-6
薬学的に許容される塩又はそれらの水和物である。
(8) 本発明の他の態様は、 R1及び R2は、同一または異なって、水素原子、水酸基、 C アルキル基、又は C アルコキシ基であり、
1-6 1-6
Zは、一 NHSOフエ-ノレ、 -NHSO NH 、 一 NHC ( = NH) NH 、 一 NHCON (R
2 2 2 2
A) RB、又はメチル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロ アルキル基、又は— Y,— CONH(RC)であり、
RA及び RBは、同一または異なって、水素原子、 C アルキル基、 C シクロアルキ
1-6 3-7
ル基、又は水酸基、 -CONH、フエニル基及びピリジル基力 なる群より選択される
2
1 3個の基で置換された C アルキル基であり、
1-6
Y,は、 C アルキレン基又は C アルケニレン基を示し、 Rcは— CONHで置換さ
1-6 2-6 2 れた C アルキル基である、態様(1)のフエニル 5 チォダルコシドィ匕合物若しくは
1-6
その製薬学的に許容される塩又はそれらの水和物である。
(9) 本発明の他の態様は、態様(1)〜(8)のいずれかに記載のフエニル 5 チォ ダルコシド化合物若しくはその製薬学的に許容される塩又はそれらの水和物を有効 成分として含むナトリウム依存性グルコース共輸送体 1 (SGLT1)活性阻害剤である
(10) 本発明の他の態様は、態様(1)〜(8)のいずれかに記載のフ ニル 5 チ ォダルコシド化合物若しくはその製薬学的に許容される塩又はそれらの水和物を有 効成分として含む糖尿病の予防又は治療剤である。
発明の効果
[0011] 本発明の化合物は、強い SGLT1阻害活性を有するので、糖尿病患者の治療だけ でなぐ食前血糖が正常域であっても食後高血糖がみられる、境界型 (糖尿病予備 群)の患者の治療もすることができ、これにより糖尿病への移行予防をすることができ る。
発明を実施するための最良の形態
[0012] 本発明において使用する用語を以下に定義する。
[0013] 「C アルキル基」とは、炭素原子を 1—6個有する直鎖状又は分枝状のアルキル基
1-6
を意味する。例えば、メチル基、ェチル基、 n プロピル基、イソプロピル基、 n—ブチ ル基、イソブチル基、 tert ブチル基、 sec ブチル基、 n ペンチル基、 n キシ ル基が挙げられる。
[0014] 「C シクロアルキル基」とは、炭素数 3— 7個の環状アルキル基を示す。例えば、シ
3-7
クロプロピル基、シクロブチル基、シクロペンチル基、シクロへキシル基、シクロへプチ ル基挙げられる。中でも、シクロペンチル基、シクロへキシル基が好ましい。
[0015] 「C アルコキシ基」とは、炭素原子を 1 6個有する直鎖状又は分枝状のアルコキ
シ基を意味し、 C アルコキシ基が好ましい。 C アルコキシ基としては、例えば、メト
1-4 1-4
キシ基、エトキシ基、プロポキシ基、イソプロポキシ基、 n ブトキシ基、イソブトキシ基 、 tert ブトキシ基が挙げられる。
「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子である。
「C アルキル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロアル
1-6
キル基」とは、 0、 S及び N力も選択された 1〜2個のへテロ原子を含有する環構成原 子数 4〜6個力 なるヘテロシクロアルキル基にぉ 、て、その基上の水素原子が C
1-6 アルキル基及びォキソ基の少なくとも 1種で置換された基である。また、ヘテロシクロ アルキル基はフエ-ル基と共に縮合環を形成しても良い。
[0016] より好ましくは、 1若しくは 2個のォキソ基で置換された 4〜6員窒素含有へテロシク 口アルキル基である(該ヘテロシクロアルキル基は、 C アルキル基で置換されてよく
1-6
、また、フエ-ル基で縮合されてもよい)。例えば、 2—ォキソピロリジ -ル基、 C アル
1-6 キル基及びォキソ基で置換されてよいイミダゾリジ-ル基 (例えば、 5,5—ジメチル— イミダゾリジン一 2,4 ジオン一 3—ィル基)、 1, 3 ジォキソ一 1, 3 ジヒドロ一 2H イソインドールー 2—ィル基等が挙げられる。
[0017] 「水酸基、 -CONH、フエ-ル基及びピリジル基力 なる群より選択される 1〜3個
2
の基で置換された C アルキル基」とは、その基上の水素原子が、水酸基、 CON
1-6
H、フ ニル基及びピリジル基力 なる群力 選択される少なくとも 1種の基の 1〜3
2
個によって置換された C アルキル基を示す。例えば、ヒドロキシメチル基、ヒドロキシ
1-6
ェチル基、 2 ヒドロキシ 1,1ージメチルェチル基、 1,3 ジヒドロキシ 2 メチル プロパン 2—ィル基、力ルバモイルメチル基、 2—力ルバモイルェチル基、ベンジル 基、フエネチル基、 3—ピリジルメチル基が挙げられる。
[0018] 「水酸基、カルボキシル基及び— CONReiRe2からなる群より選択される 1〜4個の 基で置換された C アルキル基」とは、その基上の水素原子が、水酸基、カルボキシ
1-6
ル基及び― CONReiRe2からなる群力 選択される少なくとも 1種の基の 1〜4個(好 ましくは 1〜3個)によって置換された C アルキル基を示す。例えば、ヒドロキシメチ
1-6
ル基、ヒドロキシェチル基、 2 ヒドロキシ 1,1ージメチルェチル基、 1,3 ジヒドロキ シ 2—メチルプロパン 2—ィル基、 1,3-ジヒドロキシ 2—ヒドロキシメチルプロ
パン 2—ィル基、力ルバモイルメチル基、 2—力ルバモイルェチル基、 2—力ルバモ ィルー 2—メチルェチル基が挙げられる。
[0019] 「 1及び 2が結合している窒素原子と一緒になつて形成し、さらに環構成原子と して、酸素原子、硫黄原子及び窒素原子から選択されるへテロ原子を含んでも良い 5〜6員のへテロシクロアルキル基 (該ヘテロシクロアルキル基は、水酸基で置換され ても良い C アルキル基、または C アルコキシカルボ-ル基で置換されてもよい)」
1-6 1-6
の例としては、 1—ピロリジ -ル基、 1—ピベリジ-ル基、 1—ピペラジ-ル基、 4 モ ルホリノ基、 4ーチオモルホリノ基、 4ーメチルー 1ーピペラジ-ル基、 4一(2 ヒドロキ シェチル) 1ーピペラジ-ル基、 4ーメトキシカルボ-ルー 1ーピペラジ-ル基等が 挙げられる。
[0020] 「水酸基で置換されてもよい C アルキル基」とは、少なくとも 1個の水酸基によって
1-6
置換されてもよい C アルキル基を意味し、例えば、ヒドロキシメチル基、ヒドロキシェ
1-6
チル基、 2 ヒドロキシ 1,1ージメチルェチル基、 1,3 ジヒドロキシ 2—メチルプ 口パン一 2—ィル基、 1,3-ジヒドロキシ 2—ヒドロキシメチルプロパン一 2—ィル基 が挙げられる。
[0021] 「C アルキレン基」とは、炭素数 1〜6個からなる、直鎖または分岐状の 2価の飽和
1- 6
炭化水素である。例えば、メチレン、エチレン、トリメチレン、テトラメチレン、 2—メチル プロピレン、 2, 2—ジメチルプロピレン等が挙げられる。
[0022] 「C ァルケ-レン基」とは、二重結合を含む炭素数 2〜6個力もなる、直鎖または分
2- 6
岐状の 2価の不飽和炭化水素である。例えば、エチレン、プロべ-レン、 2—プチ-レ ン等が挙げられ、中でもプロべ-レンが好ましい。
[0023] 「製薬学的に許容される塩」とは、アルカリ金属類、アルカリ土類金属類、アンモ-ゥ ム、アルキルアンモ-ゥムなどとの塩、鉱酸又は有機酸との塩であり、例えば、ナトリウ ム塩、カリウム塩、カルシウム塩、アンモ-ゥム塩、アルミニウム塩、トリェチルアンモ- ゥム塩、酢酸塩、プロピオン酸塩、酪酸塩、ぎ酸塩、トリフルォロ酢酸塩、マレイン酸 塩、酒石酸塩、クェン酸塩、ステアリン酸塩、コハク酸塩、ェチルコハク酸塩、ラタトビ オン酸塩、ダルコン酸塩、ダルコヘプトン酸塩、安息香酸塩、メタンスルホン酸塩、ェ タンスルホン酸塩、 2—ヒドロキシエタンスルホン酸塩、ベンゼンスルホン酸塩、パラト
ルエンスルホン酸塩、ラウリル硫酸塩、リンゴ酸塩、ァスパラギン酸塩、グルタミン酸塩 、アジピン酸塩、システィンとの塩、 N ァセチルシスティンとの塩、塩酸塩、臭化水 素酸塩、リン酸塩、硫酸塩、よう化水素酸塩、ニコチン酸塩、シユウ酸塩、ピクリン酸塩 、チォシアン酸塩、ゥンデカン酸塩、アクリル酸ポリマーとの塩、カルボキシビュルポリ マーとの塩などが挙げられる。
[0024] 「水和物」とは、本発明の化合物又はその塩の製薬学的に許容される水和物である 。本発明の化合物又はその塩は、大気にさらされ、あるいは再結晶することなどにより 、水分を吸収し、吸着水がつく場合や、水和物となる場合がある。本発明における水 和物には、そのような水和物も含まれる。
[0025] 本発明の化合物はキラル中心を有することがあるので、種々なジァステレオマー形 又はェナンチォマー形として存在する。また、本発明の化合物の一部は、例えば、ケ ト—エノール互変異性体としても存在する。また、本発明の化合物の一部は、幾何異 性体 (E体、 Z体)としても存在する。したがって、本発明の化合物は、上記全ての個 々の異性体並びにこれらの混合物を包含する。
本発明化合物の好ましい態様を以下にあげる。
式 (I)において、 R1及び R2の好ましい例は、同一または異なって、水素原子、水酸基 、 C アルキル基、 C アルコキシ基又はハロゲン原子であり、より好ましくは、水素原
1-4 1-4
子、水酸基、メチル基、メトキシ基又はハロゲン原子である。
[0026] さらに、 R2カ チル基又はハロゲン原子 (特に、塩素原子)であり、 R1が水酸基、メト キシ基、又はメチル基であることが好ましぐさらには、 R1が水酸基であることがより好 ましい。
[0027] また、式 (I)にお 、て、 Zの置換位置がベンジル基に対してパラ位であることが好ま しい。
[0028] Zの好ましい態様を以下にあげる:
Zが、 Y—NHCON (RA)RB、 一 Y—NHC ( = NH) NH又は Y—CONH(RC)
2
であることが好ましぐ SGLT1阻害活性がより強い点で、 Y— CONH(Re)がより好 ましい。
[0029] Zが、 Y— NHCON (RA)RBである場合の好ましい態様は、
Y-NHCONH、 Y-NHCONH-C アルキル(該アルキルは水酸基で置換され
2 1-6
てもよい)、 Y— NHCONH— C シクロアルキル、 NHCONH— CH—フエ-ル、
3-7 2
— NHCONH— CH—ピリジルである。より好ましくは、 Y- NHCONH -C アルキ
2 1-6 ル (該アルキルは水酸基で置換されて 、る)である。
[0030] Zがー Y—NHC ( = NH) NHである場合の好ましい態様は、 Yが C アルキレン基
2 1-6
である場合である。
[0031] Zがー Y—CONH(Re)である場合の好ましい態様は、 Yが C アルキレン基又は C
1-6 2- ァルケ-レン基である場合であり、より好ましくは、プロピレン又はプロべ-レン(—C
6
H = CH— CH—)である。
2
[0032] 好ましい Reの例としては、水酸基及び— CO— NReiRe2からなる群より選択される 1 〜4個(より好ましくは 1〜3個)の基で置換された C アルキル基である、 Rei及び Re2
1-6
は同一または異なって、水素原子または水酸基で置換されても良い C アルキル基
1-6
であるか、あるいは Rei及び Re2が結合している窒素原子と一緒になつて、さらに環構 成原子として、酸素原子、硫黄原子及び窒素原子から選択される 1個のへテロ原子 を含んでも良 、5〜6員のへテロシクロアルキル基 (該ヘテロシクロアルキル基は、水 酸基で置換されても良い C アルキル基または C アルコキシカルボ-ル基で置換さ
1-6 1-6
れてもよ ヽ)を形成してもよ ヽ。
— Y— CONH(RC)において、 に定義される C アルキル基は、 NH—に結合す
1-6
る炭素原子が分岐して 、る場合が好ま U、。
[0033] さらに、本発明の化合物のうち、下記の化合物群は、 SGLT1阻害活性が強くかつ SGLT2阻害活性も強い点で好ましい。これらの化合物は、下記に述べる SGLT1阻 害活性に基づく治療効果が期待できるだけでなぐ SGLT2活性を阻害できるので、そ れによって、糖の再吸収を抑制し、余分な糖を体外に排泄することによって糖尿病を 治療することができ、すい臓の β細胞に負荷を与えずに高血糖を是正し、またインス リン抵抗性を改善することができる。
[0034] SGLT1阻害活性が強くかつ SGLT2阻害活性も強い化合物群は、 Ζがー Υ— CO NH(RC)であり、 Yが C アルキレン基又は C ァルケ-レン基(より好ましくは、プロピ
1-6 2-6
レン又はプロべ-レンである)、 RCが水酸基及び— CO— NRC1RC2からなる群より選
択される 1〜4個(より好ましくは 1〜3個)の基で置換された C アルキル基 (該 C ァ
1-6 1-6 ルキル基は、—NH—に結合する炭素原子が 3級である)である、ここで 1及び 2 は同一または異なって、水素原子または水酸基で置換されても良い C アルキル基
1-6 であるか、あるいは 1及び 2が結合している窒素原子と一緒になつて、さらに環構 成原子として、酸素原子、硫黄原子及び窒素原子から選択されるへテロ原子を含ん でも良!、5〜6員のへテロシクロアルキル基 (該ヘテロシクロアルキル基は、水酸基で 置換されても良 、C アルキル基で置換されてもょ 、)を形成してもよ 、。なお、 R1及
1-6
び R2の好ましい例、 Zの好ましい置換位置は、上記と同様である。
[0035] 上記の化合物群において、より好ましい Zは、—Y— CO— NH— C アルキル(該
1-6
アルキルは水酸基で置換されている)、 -Y-CO -NH-C (CH ) —CONH 、 一
3 2 2
Y-CO -NH-C (CH ) -CONH-C アルキル(該アルキルは水酸基で置換さ
3 2 1-6
れても良い)、 -Y-CO -NH-C (CH ) —CO ピペラジノ(該ピペラジノは、水酸
3 2
基で置換されても良 、C アルキル基で置換されてもよ!、)である。ここで、 Yは、プロ
1-6
ピレン又はプロべ-レン(一 CH = CH— CH—)である。
2
[0036] 以下に、本発明化合物 (I)の製造方法を例をあげて以下に詳細に説明するが、例 示されたものに特に限定されない。
[0037] 製造法 1
本発明化合物(I)において、 Zが— NHSOフエ-ル、 -NHSO NH 、— NHC ( =
2 2 2
NH) NH、又は NHCON(RA)RBである化合物は以下の方法で合成することがで
2
きる。
[0038] ただし、 ルカノィル基又

はベンゾィル基を示し、その他の記号は前記と同義である。
[0039] [化 2]

ここに、フ ノール誘導体 (lla)は、国際公開 WO2004Z050122号に準拠して合 成することができる。 5—チォグルコース誘導体 (III)は、国際公開 WO2004Z0149 31号に準拠して合成することができる。
(1)工程 1 (光延反応)
光延反応(国際公開 WO2004Z089966号)によって、フエノール誘導体 (lla)と 5 —チォグルコース誘導体(III)力らフヱ-ル 5—チォ— β—D—ダルコシド誘導体(I Va)を選択的に製造することができる。この反応における試薬として必要なホスフィン 類の例としては、トリフエ-ルホスフィン、トリー n—ブチルホスフィン、トリー t—ブチル ホスフィン、トリストリルホスフィンゃジフエ-ル一 2—ピリジルホスフィンが挙げられる。 中でもトリフエ-ルホスフィン、ジフエ-ル一 2—ピリジルホスフィンが好ましぐトリフエ -ルホスフィンがより好まし 、。
[0040] ァゾ試薬の例としては、ジェチルァゾジカルボキシレート、ジイソプロピルァゾジカル ボキシレートゃジー tert—ブチノレアゾジカノレボキシレート、 1, 1 'ーァゾビス(N, N— ジメチルホルムアミド)や 1, 1 (ァゾジカルボ-ル)ジピペリジンを用いることができ る。中でも、ジェチルァゾジカルボキシレート(DEAD)、ジイソプロピルァゾジカルボ キシレートが好ましい。
[0041] 本反応に用いる溶媒はテトラヒドロフラン、ジォキサン、トルエン、ジクロロメタン、クロ 口ホルム、ァセトニトリル、酢酸ェチル、ジメチルスルホキシド、 N, N—ジメチルホルム アミド等であり、好ましくはテトラヒドロフラン、トルエンである。
[0042] 反応温度は 20°Cから室温が好ましぐ 5°Cから + 5°Cがより好ましい。
(2)工程 2 トロ基の還元)
上記で得られた化合物 (IVa)をパラジウム活性炭、水酸化パラジウム、又は白金 ノ ラジウム活性炭等の触媒を用いて水素雰囲気下にて接触水素添加することにより ニトロ基をァミノ基に変換し、化合物 (IVb)が得られる。中でもパラジウム活性炭、水 酸化パラジウムが触媒として好ましい。この反応に使用する溶媒としては、メタノール 、エタノール、イソプロパノール、酢酸ェチル、酢酸等が挙げられる。反応温度は室 温力 還流温度である力 室温が好ましい。
[0043] また、塩化スズ (11)1水和物、塩ィ匕アンモ-ゥムの存在下、鉄を用いることもできる。こ の反応に使用する溶媒としては、メタノール、エタノール、イソプロパノール等が挙げ られる。反応温度は室温力 還流温度である。
[0044] 引き続いて、化合物 (IVb)を中間体として以下に示す方法で本発明化合物 (I)を製 造することができる。
[0045] REはべンジルォキシカルボ-ル基又は t ブトキシカルボ-ル基を示し、その他の 記号は前記と同義である。
[0046] [化 3]

( IVf )
(3)工程 3 (スルホ-ル基の導入)
化合物(IVb)から適当な塩基の存在下、フエ-ルスルホユルクロリド又はイソシアン
酸クロロスルホ -ルを用い、化合物(IVc)又は(IVd)に誘導することができる。適当な 塩基としては、トリェチルァミン、 N ェチル N, N ジイソプロピルァミン、ピリジン 、 DBU、炭酸カリウム、炭酸カルシウム、炭酸セシウム等が挙げられる。また、使用す る溶媒としては、クロ口ホルム、ジクロロメタン、ジェチルエーテル、テトラヒドロフラン、 N, N ジメチルホルムアミド、ァセトニトリル、酢酸ェチル等、又はそれらの混合溶媒 が挙げられる。反応温度は 0°C力 還流温度である。
(4)工程 4 (グァニジノ基の導入)
化合物(IVb)力もグァ-ジノ化試薬 (V)を用いて化合物(IVe)に誘導することができ る。この反応に使用する溶媒としては、テトラヒドロフラン、 N, N ジメチルホルムアミ ド、メタノール、エタノール、イソプロパノール、酢酸ェチル、トルエン等が挙げられる。 反応温度は室温から還流温度である。
(5)工程 5 (ウレイド基の構築)
化合物(IVb)から適当な塩基の存在下、 p -トロフエ-ルクロロホルメート、 N, N —カルボ-ルジイミダゾール(CDI)やトリホスゲンを用いて、ァミン RARBNHと縮合す ることにより、化合物(IVf)に誘導することができる。適当な塩基としては、トリェチルァ ミン、 N ェチル N, N ジイソプロピルァミン、ピリジン、 DBU、炭酸カリウム、炭酸 カルシウム、炭酸セシウム等が挙げられる。また、使用する溶媒としては、クロ口ホルム 、ジクロロメタン、ジェチルエーテル、テトラヒドロフラン、 N, N ジメチルホルムアミド 、ァセトニトリル、酢酸ェチル等が挙げられる。反応温度は 0°C力も還流温度である。
(6)工程 6
 P4及び REの脱保護)
P4及び REの脱保護)
5—チォグルコースの保護基を適当な塩基を用いて除去し、本発明化合物 (I)が得 られる。この反応に使用する塩基として、ナトリウムメトキシド、水酸化ナトリウム、水酸 化リチウム、炭酸カリウム、炭酸セシウム、トリェチルアミン等を用いることができる。反 応に適当な溶媒はメタノール、エタノール、水又はこれらの混合溶媒である。反応温 度は 0°C力も室温であり、室温が好ましい。
また、グァ -ジノ基の保護基 REがべンジルォキシカルボ-ル基である場合は、製造 法 1に記載された接触水素添カ卩にて除去することもできる。このときの好ましい触媒は 水酸化パラジウムである。
[0048] さらに、 Yカ チレン基である化合物は、特許文献 7に記載されたチォグルコシドィ匕 合物でアミノ基を有する化合物を原料に用い、製造法 1の工程 5を参考に製造できる
[0049] 製造法 2
本発明の化合物(I)において、 Ζがー Υ— CONH (Re)である化合物は以下の方法 で合成できる。
[0050] ただし、 Y1は単結合又は C アルキレン基を示し、その他の記号は前記と同義であ
1-4
る。
[0051] [化 4]
まず、製造法 1の工程 1に示した光延反応により、フ ノール誘導体 (lib)力 化合 物(IVg)を合成することができる。
(7)工程 7 (Heck反応)
化合物(IVg)とォレフイン酢酸 (VI)をパラジウム触媒とホスフィンリガンド、及び適当 な塩基の存在下、 Heck反応を行うことによりィ匕合物(IVh)を合成することができる。こ のとき用いるパラジウム触媒としては、酢酸パラジウム、テトラキストリフエ-ルホスフィ ンパラジウム、ジベンジリデンアセトンパラジウム、ビストリフエニルホスフィンパラジゥ ムクロリド、パラジウム活性炭等が挙げられる。ホスフィンリガンドとしてはトリフエ-ルホ スフインゃトリス (2—メチルフエ-ル)ホスフィン等が挙げられる。また、塩基にはトリエ チルァミン、 N—ェチル— N, N—ジイソプロピルァミン、炭酸カリウム、炭酸カルシゥ ム、炭酸セシウム、カリウム t—ブトキシド等が用いられる。反応に用いられる溶媒とし ては、ァセトニトリル、トルエン、テトラヒドロフラン等が挙げられる。反応温度は 0°Cか ら還流温度であるが、マイクロウエーブを用いることもある。
(8)工程 8 (アミド基への変換)
化合物(IVh)とァミン (ReNH )にて脱水縮合し、化合物(IVj)が得られる。この反応
2
に使用する溶媒としては、クロ口ホルム、ジクロロメタン、 N, N—ジメチルホルムアミド 等が好ましぐ脱水縮合剤としては、 N, N—ジシクロカルポジイミド(DCC)、 N—ェ チルー N,ー3—ジメチルァミノプロピルカルボジィミド塩酸(WSC)、CDI、 WSC/1 ーヒドロキシベンゾトリアゾール 1水和物等が好まし 、。ここでの反応温度は 0°C〜60 °Cである。
(9)工程 9
化合物(IVh)のォレフイン部分を製造法 1の工程 2に示した接触水素添加を行った 後に、ァミン (ReNH )と縮合することで、化合物 (IVk)を製造することもできる。
2
( 10)工程 10 (脱保護)
最後に、
 P2、 P3及び P4を製造法 1の工程 6に示した方法で脱保護し、本発明化 合物 (I)が得られる。
P2、 P3及び P4を製造法 1の工程 6に示した方法で脱保護し、本発明化 合物 (I)が得られる。
[0052] さらに、 Yが単結合またはメチレン基である化合物は、特許文献 7に記載されたチォ ダルコシド化合物でカルボキシル基を有する化合物を原料に用い、製造法 2の工程 8を参考に製造できる。
[0053] 製造法 3
本発明の化合物(I)にお 、て、 Yが C アルキレン基又は C ァルケ-レン基であり
、 Qがー NHC ( = NH) NH 、 一 NHCON (RA)RB、又は C アルキル基及びォキソ
2 1-6
基の少なくとも 1種で置換された 4〜6員へテロシクロアルキル基 (該ヘテロシクロアル キル基はフエニル基と縮合してもよ!ヽ)である化合物は以下の方法で合成できる。
[化 5]

(11)工程 11 (Heck反応)
化合物(IVg)とァリルアミン誘導体、式 (Via)化合物、式 (Vlb)化合物、、又は 1若し くは 2個のォキシ基で置換され、 C アルキル基で置換されてもよい 4〜6員へテロ飽
1-6
和炭化水素環 (該環はフ ニル基と縮合してもょ 、)である化合物(例えば、(Vic)化 合物)を製造法 2の工程 7で記載した Heck反応を用いて化合物 (IVm)を製造すること ができる。
(12)工程 12
化合物(IVm)のォレフイン部分を製造法 1の工程 2に示した接触水素添加を行った 後に、
 P3及び P4を製造法 1の工程 6に示した方法で脱保護することで、 Yが C アルキレン基である本発明化合物 (I)が得られる。
P3及び P4を製造法 1の工程 6に示した方法で脱保護することで、 Yが C アルキレン基である本発明化合物 (I)が得られる。
2-6
(13)工程 13 (脱保護)
また、化合物(IVm)の P1 P2、 P3及び P4を製造法 1の工程 6に示した方法で脱保護 することで、 Yが C ァルケ-レン基である、本発明化合物 (I)が得られる。
2-6
[0055] 製造法 4
Zが C アルキル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロ
1-6
アルキル基である本発明化合物 (I)は以下の方法で合成できる。
ただし、 RN及び RNAは水素原子または C アルキル基を示し、その他の記号は前記と
1-6
同義である。
[0056] [化 6]

(14)工程 14 (ウレイド基の構築)
製造法 1の工程 5に記載の方法で、 RARBNHとして、アミノ酸 (例えば、 2—ァミノイソ プチリックアシッド)を用いて、化合物(IVb)と縮合することにより、例えば、カルボキシ ル基を有する化合物(IVn)を合成することができる。
(15)工程 15 (塩基性条件での脱保護)
化合物 (IVn)を塩基性条件にて、 P1— P4の脱保護を行うと同時に、化合物 (IVn)中の — NHCONHCRN(RNA) COOHが分子内環化し、 C アルキル基 (好ましくは、メチ
1-6
ル基)及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロアルキル基で ある、本発明化合物(1)を製造することができる。このときに用いる塩基としてはナトリ ゥムメトキシドが好ましぐ溶媒はメタノールまたはエタノールが良い。
[0057] 本発明の化合物は、 SGLT1の活性を阻害することで改善しうる疾患又は状態、例
えば、糖尿病、糖尿病関連疾患及び糖尿病合併症を予防又は治療するための医薬 の有効成分として用いることができる。 本発明化合物は、 SGLT1活性を阻害するこ とによって、小腸からの糖の吸収を抑制して、 IGTを改善することができ、これにより 糖尿病への移行予防をすることができる点で特に優れている。
[0058] ここで、「糖尿病」とは、 1型糖尿病、 2型糖尿病、特定の原因によるその他の型の糖 尿病を包含する。
[0059] ここで、「糖尿病関連疾患」とは、肥満、高インスリン血症、糖代謝異常、高脂質血 症、高コレステロール血症、高トリグリセリド血症、脂質代謝異常、高血圧、うつ血性心 不全、浮腫、高尿酸血症、痛風などが挙げられる。
[0060] ここで、「糖尿病合併症」は、急性合併症及び慢性合併症に分類される。
「急性合併症」には、高血糖 (ケトアシドーシスなど)、感染症 (皮膚、軟部組織、胆道 系、呼吸系、尿路感染など)などが挙げられる。
[0061] 「慢性合併症」には、細小血管症(腎症、網膜症)、動脈硬化症 (ァテローム性動脈 硬化症、心筋梗塞、脳梗塞、下肢動脈閉塞など)、神経障害 (感覚神経、運動神経、 自律神経など)、足壊疽などが挙げられる。
[0062] 主要な合併症は、糖尿病網膜症、糖尿病腎症、糖尿病神経障害である。
[0063] 本発明の化合物は、医薬として、全身的又は局所的に、経口投与又は非経口投与 することができる。
[0064] 本発明化合物は、該化合物の作用の増強または該化合物の投与量の低減などを 目的として、 SGLT1及び SGLT2活性阻害薬以外のことなった作用機序の糖尿病 治療剤、糖尿病性合併症治療剤、抗高脂血症剤、降圧剤、抗肥満剤、利尿剤、抗 血栓剤などの薬剤(以下、併用薬剤と略記する)と組み合わせて用いることができる 。この際、本発明化合物と併用薬剤の投与時期は限定されず、これらを投与対象に 対し、同時に投与してもよいし、時間差をおいて投与してもよい。さらに、本発明化合 物と併用薬剤とは、それぞれの活性成分を含む 2種類の製剤として投与されてもょ ヽ し、両方の活性成分を含む単一の製剤として投与されてもよい。併用薬剤の投与量 は、臨床上用いられている用量を基準として適宜選択することができる。また、本発 明化合物と併用薬剤の配合比は、投与対象、投与ルート、対象疾患、症状、組み合
わせなどにより適宜選択することができる。例えば、投与対象がヒトである場合、本発 明化合物 1重量部に対し、併用薬剤を 0. 01〜: LOO重量部用いればよい。
なお、糖尿病治療剤としては、例えばインスリン製剤(例、ゥシ、ブタの脾臓力ゝら抽 出された動物インスリン製剤;大腸菌またはイーストを用い、遺伝子工学的に合成し たヒトインスリン製剤;インスリン亜 プロタミンインスリン亜 インスリンのフラグメン トまたは誘導体 (例、 INS— 1等)、経口インスリン製剤)、インスリン抵抗性改善剤 (例
、ピオグリタゾンまたはその塩 (好ましくは塩酸塩)、ロシグリタゾンまたはその塩 (好ま しくはマレイン酸塩) 、リボグリタゾン (Rivoglitazone) (CS— Oi l) (R— 119702)、 シポグリタザール(Sipoglitazar) (TAK—654)、メタグリダセン (Metaglidasen) (M XB- 1 0 2)、ナベグリタザール (Naveglitazar) (LY— 519818)、 MX— 6054、ノ ラグリタゾン (Balaglitazone) (NN— 2344)、 T— 131 (AMG131)、 PPAR y ァゴ -スト、 PPAR yアンタゴ-スト、 PPAR y / aデュアルァゴ-スト、 a—ダルコシダー ゼ阻害剤(例、ボグリボース、ァカルボース、ミグリトール、エミダリテート)、ビグアナィ ド剤(例、フェンホルミン、メトホルミン、ブホルミンまたはそれらの塩 (例、塩酸塩、フマ ル酸塩、コハク酸塩))、インスリン分泌促進剤 (スルホ-ルゥレア剤(例、トルプタミド、 ダリベンクラミド、ダリクラジド、クロルプロパミド、トラザミド、ァセトへキサミド、ダリクロピ ラミド、グリメピリド、ダリピザイド、ダリブゾール等)、レパグリニド、セナグリニド、ナテグ リニド、ミチグリニドまたはそのカルシウム塩水和物) 、 GPR40ァゴ-スト、 GPR40ァ ンタゴ-スト、 GLP— 1受容体ァゴ-スト(例、 GLP— 1、 GLP— 1MR剤、リラグルチ ド(Liraglutide) (NN— 2211)、 Exenatide(AC - 2993) (exendin— 4)、 Exenati de LAR、: BIM— 51077、 A i b (8, 35) hGLP— 1(7, 37) NH2、 CJC— 1131、 A VE0010、 GSK— 716155)、アミリンァゴニスト(例、プラムリンチド) 、フォスフォチ口 シンフォスファターゼ阻害剤(例、バナジン酸ナトリウム) 、ジぺプチジルぺプチダー ゼ I V阻害剤(例、 WO02/038541に記載のィ匕合物、 NVP— DPP— 278、 PT— 100、 P32/98,ビルダグジプチン (Vildagliptin) (LAF— 237)、 P93Z01、シタグ リプチン (SitagliptinXMK— 431)、サクサグリプチン(Saxagliptin) (BMS— 4771 18)、 SYR— 322、 MP— 513、 T— 6666、 GRC— 8200等)、 j8 3ァゴ-スト(例、 A J 9677、 AZ40140等) 、糖新生阻害剤(例、グリコーゲンホスホリラーゼ阻害剤、
グルコース一 6 ホスファターゼ阻害剤、グルカゴン拮抗剤、フルクトース一 1, 6 ビ スホスファターゼ阻害剤)、 SGLT (sodium-glucose cotransporter) 阻害剤( ί列、 WO04/014931, WO04/089967, WO06/073197に記載のィ匕合物、 Τ 1 095、 Sergliflozin (GSK- 869682)、 GSK— 189075、 KGT— 1251、 KGT— 1 681、 KGA— 2727、 BMS— 512148、 AVE2268、 SAR7226等)、 1 I j8—ヒドロ キシステロイドデヒドロゲナーゼ阻害薬(例、 WO06/51662に記載のィ匕合物、 BVT — 3498、 INCB13739)、 GPR119ァゴ-スト(例、 PSN— 632408、 APD— 668) 、アディポネクチンまたはその作動薬、 IKK阻害薬(例、 A S— 2868)、 AMPK活性 化薬、レブチン抵抗性改善薬、ソマトスタチン受容体作動薬、ダルコキナーゼ活性化 薬(例、 Ro— 28— 1675)、脾リパーゼ阻害薬(例、オルリスタツト、 ATL—962)、 D GAT— 1阻害薬が挙げられる。
[0066] 糖尿病性合併症治療剤としては、例えばアルドース還元酵素阻害剤(例、トルレス タツト、ェパノレレスタツト、ゼナレスタツト、ゾポノレレスタツト、ミナノレレスタツト、フィダレス タツト、 CT— 112)、神経栄養因子およびその増加薬(例、 NGF、 NT— 3、 BDNF、 ニューロトロフィン産生'分泌促進剤)、神経再生促進薬 (例、 Y—128)、 PKC阻害 剤(例、ルボキシスタウリンメシレート (ruboxistaurin mesylate; LY— 333531))、 AGE阻害剤(例、 ALT946、ピマゲジン、ピラトキサチン、 N—フエナシルチアゾリゥ ムブロマイド(ALT766)、 ALT— 711、 EXO— 226、ピリドリン(Pyridorin)、ピリド キサミン)、活性酸素消去薬 (例、チォタト酸)、脳血管拡張剤 (例、チアプリド、メキシ レチン)、ソマトスタチン受容体作動薬(例、 BIM23190)、アポトーシスシグナルレギ ユレ一ティングキナーゼ 1 (ASK— 1) 阻害薬が挙げられる。
[0067] 抗高脂血症剤としては、例えばスタチン系化合物(例、プラバスタチン、シンパスタ チン、口パスタチン、アトノレパスタチン、フルパスタチン、イタパスタチン、ロスバスタチ ン、ピタパスタチンまたはそれらの塩(例、ナトリウム塩、カルシウム塩)) 、スクアレン 合成酵素阻害剤(例、 TAK— 475)、フイブラート系化合物 (例、ベザフイブラート、 クロフイブラート、シムフイブラート、クリノフイブラート)、 AC AT阻害剤(例、アバシマ イブ(Avasimibe)、エフルシマイブ(Eflucimibe) )、陰イオン交換榭脂(例、コレス チラミン)、プロブコーノレ、ニコチン酸系薬剤(例、ニコモーノレ (nicomol)、ニセリトロー
ル (niceritrol))、ィコサペント酸ェチル、植物ステロール(例、ソイステロール (soyste rol)、ガンマオリザノール(γ—oryzanol))、 CETP阻害薬(例、 Torcetrapib、 JTT 705、JTT 302、 FN— VP4等)、コレステロール吸収抑制薬(例、ェゼチミブ(E zetimibe)等)が上げられる。
[0068] 降圧剤としては、例えばアンジォテンシン変換酵素阻害剤(例、カプトプリル、ェナ ラプリル、デラプリル) 、アンジォテンシン II拮抗剤(例、カンデサルタン シレキセチ ル、口サルタン、ェプロサルタン、バルサルタン、テルミサルタン、ィルベサルタン、タ ソサルタン、アジルザルタン(TAK— 536) )、カルシウム拮抗剤(例、マ-ジピン、二 フエジピン、アムロジピン、エホニジピン、二カノレジピン)、カリウムチャンネノレ開口薬( 例、レブクロマカリム、 L 27152、 AL0671、 NIP— 121)、クロ-ジンが挙げられる
[0069] 抗肥満剤としては、例えば中枢性抗肥満薬 (例、デキスフェンフルラミン、フェンフ ルラミン、フェンテルミン、シブトラミン、アンフエプラモン、デキサンフエタミン、マジン ドール、フエ-ルプロパノールァミン、クロべンゾレックス; MCH受容体拮抗薬(例、 W 006/035967に記載のィ匕合物、 SB— 568849 ; SNAP— 7941、 T 226296); ニューロペプチド 拮抗薬 (例、 CP— 422935);カンナピノイド受容体拮抗薬(例、 リモナノ ン卜(Rimonabant) (SR— 141716)、 SR— 147778) ;グレリン拮抗薬; 1 1 β—ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、 BVT— 3498、 INCB1373 9) )、脾リパーゼ阻害薬 (例、オルリスタツト、 ATL— 962)、 DGAT—1阻害薬、 /3 3 ァゴ-スト(例、 AJ— 9677、 AZ40140)、ペプチド性食欲抑制薬(例、レプチン、 CN TF (毛様体神経栄養因子))、コレシストキュンァゴ-スト(例、リンチトリブト、 FPL— 15849)、摂食抑制薬 (例、 P— 57)が挙げられる。
[0070] 利尿剤としては、例えば、キサンチン誘導体(例、サリチル酸ナトリウムテオプロミン 、サリチル酸カルシウムテオプロミン)、チアジド系製剤(例、ェチアジド、シクロペンチ アジド、トリクロルメチアジド、ヒドロクロ口チアジド、ヒドロフルメチアジド、ベンチルヒド 口クロ口チアジド、ペンフルチジド、ポリチアジド、メチクロチアジド)、抗アルドステロン 製剤 (例、スピロノラタトン、トリアムテレン)、炭酸脱水酵素阻害剤 (例、ァセタゾラミド) 、クロルベンゼンスルホンアミド系製剤(例、クロルタリドン、メフルシド、インダパミド)、
ァゾセミド、イソソルビド、エタクリン酸、ピレタ -ド、ブメタ-ド、フロセミドが挙げられる
[0071] 抗血栓剤としては、例えば、へパリン (例、へパリンナトリウム、へパリンカルシウム、 ダルテパリンナトリウム (dalteparin sodium), AVE - 5026) 、ヮルフアリン(例、ヮ ルフアリンカリウムなど) 、抗トロンビン薬(例、アルガトロバン (argatroban)、キシメラ ガトフン (Ximelagatran入ダビ'ガトフン (Dabigatran入 Odiparcil、 Lepirudin、 bival irudin、 Desirudin、 ART— 123、 Idraparinux、 SR— 123781、 AZD— 0837、 M CC— 977、 TGN— 255、 TGN— 167、 RWJ— 58436、 LB— 30870、 MPC— 09 20、 Pegmusirudinゝ Org— 426751等)、血栓溶解薬(例、ゥロキナーゼ (urokina se)、チソキナーゼ (tisokinase)、ァノレテプラーゼ (alteplase)、ナテプラーゼ (natepla se)、モンテプラーゼ (monteplase)、パミテプラーゼ (pamiteplase)等)、血小板凝集 抑制薬(例、塩酸チクロピジン (ticlepidine hydrochloride),シロスタゾール(cilost azol)、ィコサペント酸ェチノレ、ベラプロストナトリウム (beraprost sodium),塩酸サノレ ポグレフート (sarpogrelate hydrochloride)など)、 Xa阻害薬 (例、 Fondaparinu x、 BAY— 59— 7939、 DU— 176b、 YM— 150、 SR— 126517、 Apixaban, Raz axaban, LY— 517717、 MLN— 102、 Octaparine、 Otamixaban, EMD— 503 982、 TC— 10、 CS— 3030、 AVE— 3247、 GSK— 813893、 KFA— 1982等)、 血漿中カルボキシぺプチターゼ B (または活性型 thrombin—activatable fibrinol ysis inhibitor [TAFIa]としても知られている)阻害薬(例、 AZD— 9684、 EF— 62 65、 MN— 462)などが挙げられる。
[0072] 本発明の化合物を医薬として提供する場合、固形剤、液剤等の種々の態様の製剤 形態を適宜に採択することができる。その際、製薬学的に許容される担体を配合する ことも可能である。そのような担体の例としては、一般的な賦形剤、増量剤、結合剤、 崩壊剤、被覆剤、糖衣剤、 pH調整剤、溶解剤又は水性若しくは非水性溶媒などが 挙げられる。本発明の化合物とこれらの担体から、錠剤、丸剤、カプセル剤、顆粒剤 、粉剤、散剤、液剤、乳剤、懸濁剤、注射剤等を調製することができる。
[0073] また、本発明の化合物は、 a、 β若しくは γ—シクロデキストリン又はメチルイ匕シクロ デキストリン等に包接させて、その溶解性を改善することも可能である。
[0074] 本発明の化合物の投与量は、疾患、症状、体重、年齢、性別、投与経路等により異 なってくる力 S、成人に対し、 1曰当たり 0. 1〜: LOOOmg/kg体重であり、 0. 1〜200 mgZkg体重が好ましぐ 0. l〜10mgZkg体重がより好ましい。これを 1日 1回から 数回に分けて投与することができる。
» 列
以下に、本発明化合物を製造するための中間体の製造例を参考例によって示す。
[0075] 参考例 1
5—メトキシ 3—メチル - 2- (4 -トロベンジル)フエノールの合成
[0076] [化 7]

(1) 3, 5 ジメトキシトルエン(3. 83mL、 26. 2mmol)と 4 -トロべンゾイルクロリド (5. 35g、 28. 8mmol)のクロ口ホルム(75mL)溶液に氷冷下塩化アルミニウム(3. 67g、 27. 5mmol)を加えて 30分攪拌した後、室温にて 4時間攪拌した。反応混合 物に氷水、 1M塩酸を加えた後、クロ口ホルムで抽出した。有機層を 1M塩酸、 1M水 酸ィ匕ナトリウム水溶液及び飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥 し、溶媒を減圧下に留去した。残渣をシリカゲルクロマトグラフィー(へキサン:酢酸ェ チル= 10 : 1→3 : 1)にて精製し、 (2, 4 ジメトキシ— 6—メチルフエ-ル)(4 ニトロ フエ-ル)メタノンを得た。
(2)次に、(2, 4 ジメトキシ一 6—メチルフエ-ル)(4 -トロフエ-ル)メタノンをクロ 口ホルム(30mL)に溶解し、氷冷下 1. 0M三塩化ホウ素のヘプタン溶液(10. 7mL
、 10. 7mmol)を滴下して、室温にて 15時間攪拌した。反応混合物に氷水、 10%塩 酸を加えた後、クロ口ホルムで抽出した。有機層を 10%塩酸、飽和炭酸水素ナトリウ ム水溶液及び飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を 減圧下留去した。残渣をシリカゲルクロマトグラフィー(へキサン:酢酸ェチル =4 : 1 →2 : 1)にて精製し、(2 ヒドロキシ一 4—メトキシ一 6—メチルフエ-ル)(4 -トロフ ェ -ル)メタノンを得た。
(3)次に、(2 ヒドロキシ一 4—メトキシ一 6—メチルフエ-ル)(4 -トロフエ-ル)メ タノンをテトラヒドロフラン(26mL)に溶解しトリェチルァミン(1. 39mL、 9. 96mmol) を加え、この溶液に氷冷下クロロギ酸メチル(0. 67mL、 8. 76mmol)をカ卩えて、室 温で 21時間攪拌した。反応混合物に水を加え、酢酸ェチルで抽出した。有機層を飽 和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣 をシリカゲルクロマトグラフィー(へキサン:酢酸ェチル = 5 : 1)にて精製して 5—メトキ シ— 3—メチル—2— (4— -トロべンゾィル)フエ-ル メチル カルボナートを得た。
(4)次に、 5—メトキシー3—メチルー 2—(4 -トロベンゾィル)フエ-ル メチル 力 ルボナートをテトラヒドロフラン(26mL)と水(26mL)に懸濁させ、氷冷下水素化ホウ 素ナトリウム(1. 20g、 31. 8mmoL)をカ卩えて、室温で 2. 5時間攪拌した。反応混合 物に飽和塩ィ匕アンモ-ゥム水溶液をカ卩え、酢酸ェチルで抽出した。有機層を飽和食 塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリ 力ゲルクロマトグラフィー(へキサン:酢酸ェチル =4 : 1)にて精製し、淡黄色粉末の 表題化合物(2. 0g、 28%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 2.20 (s, 3 H) 3.77 (s, 3 H) 4.07 (s, 2 H) 4.75 - 4.77 (m, 1 H) 6.26 (d, J=2.49 Hz, 1 H) 6.39 (d, J=2.49 Hz, 1 H) 7.27 - 7.33 (m, 2 H) 8.07 - 8.13 (m, 2 H).
ESI m/z = 272 (M- H).
参考例 2
3, 5 ジメチルー 2—(4 -トロベンジル)フエノールの合成
[化 8]

(1) 3, 5 ジメチルフエノール(8. 0g、0. 066mol)とトリェチルァミン(9. 9mL)のク ロロホルム溶液に 4°Cにて 4 -トロべンゾイルクロリド(12. 0g、0. 065mol)を加え て、室温にて 1. 5時間攪拌した。反応混合物をクロ口ホルムで希釈し、有機層を水、 飽和炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄後、無水硫酸マグネシウム
で乾燥し、溶媒を減圧下留去した。残渣をへキサン:酢酸ェチル = 10 : 1の混合溶媒 で洗浄し、無色粉末状の 3, 5 ジメチルフエ-ル 4 -トロべンゾアート(14. 9g、 8 5%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 2.36 (s, 6 H) 6.85 (s, 2 H) 6.95 (s, 1 H) 8.36 (s, 4 H).
(2)次に、 3, 5 ジメチルフエ-ル 4 -トロべンゾアート(9. 9g、 36. 5mmol)に 塩ィ匕アルミニウム(6. 3g、 47. 4mmol)をカロえ、 140°Cにて 1時間攪拌した。反応混 合物を室温まで冷却した後、 1M塩酸を力卩ぇジェチルエーテルで抽出した。有機層 を 5%炭酸水素ナトリウム水溶液、 5%炭酸ナトリウム水溶液で洗浄し、 2%水酸化ナ トリウム水溶液を加えて水層を分取した。氷冷下、水層に濃塩酸を加えて酸性にし、 析出した結晶を濾過して黄色粉末状の(2 ヒドロキシー 4, 6 ジメチルフエニル)(4 —ニトロフエ-ル)メタノン(3. 39g、 34%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.91 (s, 3 H) 2.34 (s, 3 H) 6.59 (s, 1 H) 6.74 (s, 1 H) 7.76 - 7.83 (m, 2 H) 8.27 - 8.35 (m, 2 H) 9.60 (s, 1 H).
ESI m/z = 270 (M- H).
(3)次に、(2 ヒドロキシ一 4, 6 ジメチルフエ-ル)(4 -トロフエ-ル)メタノン(3 . 39g、 12. 5mmol)及びトリェチルァミン(2. 17mL、 13. 7mmol)のテトラヒドロフ ラン(40mL)溶液に氷冷下クロロギ酸メチル(1. 06mL、 13. 7mmol)を加え、室温 で 16時間攪拌した。反応混合物に水を加え、酢酸ェチルで抽出した。有機層を飽和 食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を シリカゲルクロマトグラフィー(へキサン:酢酸ェチル = 5 : 1)にて精製して 3, 5 ジメ チルー 2—(4 -トロベンゾィル)フエ-ル メチル カルボナートを得た。これをテト ラヒドロフラン (40mL)と水(40mL)に懸濁し、氷冷下水素化ホウ素ナトリウム(1. 89 g、 50. Ommol)をカ卩えて、室温で 2. 5時間攪拌した。反応混合物に飽和塩化アン モ -ゥム水溶液を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄後、無 水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルクロマトダラ フィー(へキサン:酢酸ェチル =4 : 1)にて精製し、淡黄色粉末の 3, 5 ジメチル— 2 一 (4一-トロベンジル)フエノール(2. 55g、 79%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.17 (s, 3 H) 2.41 (s, 3 H) 3.67 (s 3 H) 6.97 (s, 1 H) 7.03 (s, 1 H) 7.91 - 7.98 (m, 2 H) 8.25 - 8.32 (m, 2 H).
ESI m/z = 256 (M- H).
参考例 3
3—メチルー 2—(4 -トロベンジル)フエノールの合成
[化 9]

(1) 3 メチルフエノールと 4 -トロベンゾイルクロリドから参考例 2の(1)と同様の方 法で、 3—メチルフエ-ル 4 -トロベンゾアートを得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.41 (s, 3 H) 7.00 - 7.07 (m, 2 H) 7.13 (d, J=6.84 Hz, 1 H) 7.34 (t, J=7.85 Hz, 1 H) 8.37 (s, 4 H).
(2)次に、 3, 5 ジメチルフエ-ル 4 -トロベンゾアートの代わりに 3 メチルフエ -ル 4 -トロベンゾアート(15. 0g、0. 058mol)を用いて参考例 2の(2)と同様の Fries転移を用いて、(2 ヒドロキシ一 6—メチルフエ-ル)(4 -トロフエ-ル)メタノ ン (0. 42g)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.99 (s, 3 H) 6.80 (d, J=6.84 Hz, 1 H) 6.90 (d, J=8.86 Hz, 1 H) 7.30 - 7.41 (m, 1 H) 7.77 - 7.90 (m, 2 H) 8.27 - 8.36 ( m, 2 H) 8.62 - 8.71 (m, 1 H).
また、(2 ヒドロキシ一 4—メチルフエ-ル)(4 -トロフエ-ル)メタノン(1. 32g)を 得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.40 (s, 3 H) 6.71 (d, J=8.24 Hz, 1 H) 6.92 (s, 1 H) 7.32 (d, J=8.24 Hz, 1 H) 7.81 (d, J=8.86 Hz, 2 H) 8.37 (d, J=8.86 Hz, 2 H) 11.86 (s, 1 H).さらに、(4 ヒドロキシ一 2—メチルフエ-ル)(4 -トロフエ -ル)メタノン(1. 31g)を得た。
(3)次に、(2 ヒドロキシ一 4, 6 ジメチルフエ-ル)(4 -トロフエ-ル)メタノンの 代わりに(2 ヒドロキシ 6 メチルフエ-ル)(4 -トロフエ-ル)メタノンを用 V、て
参考例 2の(3)と同様の方法で 3—メチルー 2—(4 -トロベンジル)フエノールを得 た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.24 (s, 3 H) 4.15 (s, 2 H) 4.68 (s, 1 H) 6.67 (d, J=8.08 Hz, 1 H) 6.82 (d, J=7.31 Hz, 1 H) 7.07 (t, J=7.77 Hz, 1 H) 7.2
8 - 7.35 (m, 2 H) 8.07 - 8.14 (m, 2 H).
ESI m/z = 242 (M- H).
参考例 4
5—メチルー 2—(4 -トロベンジル)フエノールの合成
[0079] [化 10]

(2 ヒドロキシ一 4, 6 ジメチルフエ-ル)(4 -トロフエ-ル)メタノンの代わりに( 2 -ヒドロキシ - 4 メチルフエ-ル)(4 -トロフエ-ル)メタノンを用 V、て参考例 2の (3)と同様の方法で表題ィ匕合物を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.29 (s, 3 H) 4.03 (s, 2 H) 4.57 (s, 1 H) 6.59 (s, 1 H) 6.73 (d, J=8.39 Hz, 1 H) 7.00 (d, J=7.62 Hz, 1 H) 7.33 - 7.41 (m
, 2 H) 8.07 - 8.16 (m, 2 H).
ESI m/z = 242 (M- H).
参考例 5
3—ヒドロキシ一 5—メチル 4— (4—二トロベンジル)フエ-ル メチル カルボナ ートの合成
[0080] [化 11]

3, 5 ジメトキシトルエンと 4 -トロべンゾイルクロリドより、国際公開 WO2004Z0 50122号に準拠して合成した。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.25 (s, 3 H) 3.91 (s, 3 H) 4.08 (s, 2 H) 5.29 (s, 1 H) 6.53 - 6.56 (m, 1 H) 6.63 - 6.65 (m, 1 H) 7.35 - 7.49 (m, 2 H) 7
.99 - 8.07 (m, 2 H).
ESI m/z = 316 (M- H).
参考例 6
3—ヒドロキシ一 4— (4—二トロベンジル)フエ-ル メチル カルボナートの合成
[0081] [化 12]

3, 5 ジメトキシベンゼンと 4 -トロべンゾイルクロリドより国際公開 WO2004Z0 50122号に準拠して合成した。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 3.90 (s, 3 H) 4.04 (s, 2 H) 6.65 (d, J=2.29 Hz, 1 H) 6.73 (dd, J=8.36, 2.29 Hz, 1 H) 7.09 (d, J=8.36 Hz, 1 H) 7.33 - 7. 39 (m, 2 H) 8.09 - 8.15 (m, 2 H).
ESI m/z = 302 (M- H).
参考例 7
4一(4一ブロモベンジル)一 3—ヒドロキシ - 5—メチルフエニル メチル カルボナ ートの合成
[0082] [化 13]

3, 5 ジメトキシトルエンと 4 ブロモベンゾイルクロリドより国際特許公開 WO2004 Z050122号に準拠して合成した。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 2.23 (s, 3 H) 3.90 (s, 3 H) 3.94 (s, 2 H) 4.97 (s, 1 H) 6.52 - 6.55 (m, 1 H) 6.61 - 6.65 (m, 1 H) 6.98 - 7.04 (m, 2 H) 7 .32 - 7.39 (m, 2 H).
ESI m/z = 374 (M+Na).
参考例 8
N— [4— [4—ヒドロキシ一 2—メチル 6— [ (5—チォ
ォキシ]ベンジル]フエ-ル]メタンスルホンアミドの合成
[化 14]

(1)メチル 3—メチル 4— (4 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O —ァセチル一 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カルボナート の合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(3. 46g、 9. 50mmol)、 3 ヒドロキシ一 5—メチル 4— (4 -トロベンジル)フエ-ル メチル カルボナート(2. 19g、 6. 33mmol)及びトリフエ-ルホスフィン(2. 49g、 9. 50mm ol)のトルエン(16mL)溶液に食塩を含む氷冷下、ジイソプロピルァゾジカルボキシ レート(40%トルエン溶液; 5. 16mL、 9. 50mmol)をゆっくりと滴下した。氷冷下で 3 0分間攪拌し、室温で一晩攪拌した。反応液を減圧下留去し、得られた残渣をシリカ ゲルカラムクロマトグラフィー(へキサン:酢酸ェチル = 2 : 1〜1: 1)にて精製し、褐色 ガム状物質として表題ィ匕合物(1. 89g、 45%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.83 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.22 (s, 3 H) 3.21 - 3.31 (m, 1 H) 3.87 - 4.18 (m, 6 H) 4.28 (dd, J=12.05, 5.21 Hz, 1 H) 5.08 - 5.54 (m, 4 H) 6.78 - 6.81 (m, 1 H) 6.88 - 6.91 (m, 1 H) 7.17 - 7.24 (m, 2 H) 8.06 - 8.14 (m, 2 H).
ESI m/z = 686 (M+Na).
(2) 4— (4 ァミノベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの 合成
メチル 3—メチル 4— (4 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O ァ セチルー 5 チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル カルボナート(1. 8 9g、 2. 85mmol)のメタノール(30mL)溶液にパラジウム 活性炭素(10%) (0. 63 g)を加え、水素雰囲気下、室温にて 2. 5日間攪拌した。反応液をセライトろ過後、減 圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン:酢酸ェチ ル = 2 : 1〜1 : 10)にて精製し、淡黄色粉末として表題ィ匕合物(1. 13g、 63%)を得 た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.97 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.06 (s, 3 H) 2.19 (s, 3 H) 3.20 - 3.31 (m, 1 H) 3.70 - 4.18 (m, 6 H) 4.25 - 4. 36 (m, 1 H) 5.06 - 5.40 (m, 3 H) 5.49 - 5.62 (m, 1 H) 6.51 - 6.60 (m, 2 H) 6.71 - 6 .95 (m, 4 H).
ESI m/z = 656 (M+Na).
(3) N [4— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 j8—D—ダルコビラノシ ル)ォキシ]ベンジル]フエ-ル]メタンスルホンアミドの合成
4— (4 ァミノベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチル一
5—チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200 mg、0. 316mmol)のクロ口ホルム(10mL)溶液にピリジン(0. 061mL、 0. 758m mol)とメタンスルホユルクロリド(0. 049mL、 0. 632mmol)を加え、室温でー晚攪 拌した。反応液を水にあけ、酢酸ェチルで抽出した。有機層を水、飽和食塩水で洗 浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を炉別後、溶媒を減圧下留去 し、得られた残渣にメタノール(2mL)とナトリウムメトキシド(25%メタノール溶液; 0. 1 4mL、 0. 632mmol)を加え、室温で 1. 5時間攪拌した。反応液にドライアイスを加 え中和し、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー( 酢酸ェチル:エタノール:水 = 20 : 2 : 1〜5 : 2: 1で精製し、褐色粉末として表題化合 物(125mg、 81%)を得た。
1H NMR (600 MHz, METHANOL— d ) δ ppm 2.12 (s, 3 H) 2.81 - 2.94 (m, 4 H) 3.2
4
2 - 3.28 (m, 1 H) 3.49 - 3.56 (m, 1 H) 3.68 - 3.80 (m, 2 H) 3.86 - 3.94 (m, 2 H) 4. 02 (d, J=15.59 Hz, 1 H) 5.08 (d, J=8.71 Hz, 1 H) 6.31 - 6.35 (m, 1 H) 6.65 - 6.69 (
m, 1 H) 7.03 - 7.12 (m, 4 H). ESI m/z = 508 (M+Na).
参考例 9
N- [4 [4ーヒドロキシー2—メチルー 6— [ (5 チォ
)ォキシ]ベンジル]フエ-ル]ァセトアミドの合成
[0084] [化 15]

参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナー卜(200mg、 0. 316mmol)のピリジン(0. 060mL)溶液に無水 酢酸 (0. 061mL)を加え室温で 1. 5時間攪拌した。反応液を濃縮後、得られた残渣 にメタノール(2mL)とナトリウムメトキシド(25%メタノール溶液; 0. 27mL)を加え、室 温で 4時間攪拌した。反応液にドライアイスを加え中和し、溶媒を減圧留去した。得ら れた残渣をシリカゲルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20: 2: 1〜5: 2 : 1で精製し、褐色粉末として表題ィ匕合物(106mg、 75%)を得た。
1H NMR (600 MHz, METHANOL— d ) δ ppm 2.08 (s, 3 H) 2.10 (s, 3 H) 2.85 - 2.9
4
0 (m, 1 H) 3.23 - 3.28 (m, 1 H) 3.50 - 3.57 (m, 1 H) 3.71 - 3.79 (m, 2 H) 3.87 - 3. 94 (m, 2 H) 4.01 (d, J=15.59 Hz, 1 H) 5.08 (d, J=8.71 Hz, 1 H) 6.32 (d, J=2.29 Hz, 1 H) 6.67 (d, J=2.29 Hz, 1 H) 7.03 - 7.07 (m, 2 H) 7.33 - 7.36 (m, 2 H). ESI m/z = 472 (M+Na).
参考例 10
2- (4 ァミノベンジル) 5 ヒドロキシ一 3—メチルフエ-ル 5 チォ一 β—D —ダルコビラノシドの合成
[0085] [化 16]

参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200mg、 0. 316mmol)のメタノール(2mL)溶液にナトリウム メトキシド(25%メタノール溶液、 0. 27mL、 1. 26mmol)をカ卩え、室温で 4時間攪拌 した。反応液にドライアイスを加え中和し、溶媒を減圧留去した。得られた残渣をシリ 力ゲルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20 : 2 : 1〜5 : 2 : 1)で 精製し、無色粉末として表題ィ匕合物(94mg、 73%)を得た。
1H NMR (600 MHz, METHANOL— d ) δ ppm 2.10 (s, 3 H) 2.84 - 2.89 (m, 1 H) 3.2
4
5 (t, J=9.17 Hz, 1 H) 3.54 (dd, J=10.09, 9.17 Hz, 1 H) 3.70 - 3.83 (m, 3 H) 3.88 - 3.94 (m, 2 H) 5.05 (d, J=8.71 Hz, 1 H) 6.29 - 6.33 (m, 1 H) 6.59 - 6.62 (m, 2 H) 6. 64 - 6.66 (m, 1 H) 6.83 - 6.88 (m, 2 H). ESI m/z = 430 (M+Na).
参考例 11
4— [4— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 β—D ダルコビラノシル
[化 17]

(1) 4— (4 ブロモベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの 合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(15. 4g、 42 . 3mmol)と 4— (4—ブロモベンジル) 3—ヒドロキシ一 5—メチルフエ-ル メチル
カルボナート(9. 92g、 28. 2mmol)、トリフエ-ルホスフィン(11. lg、 42. 3mmol )、ジイソプロピルァゾジカルボキシレート(40%トルエン溶液; 23. OmL、 42. 3mm ol)を用い、参考例 8の(1)に示す方法と同様の方法で表題化合物 (4. 24g、 22%) を合成した。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.77 (s, 3 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.19 (s, 3 H) 3.21 - 3.33 (m, 1 H) 3.75 - 3.82 (m, 2 H) 3.93 (s,
3 H) 4.06 - 4.15 (m, 1 H) 4.25 - 4.34 (m, 1 H) 5.07 - 5.41 (m, 3 H) 5.48 - 5.59 (m, 1 H) 6.71 - 7.05 (m, 4 H) 7.29 - 7.38 (m, 2 H).
ESI m/z = 719 (M+Na).
(2) (3E)— 4 [4 [4 [ (メトキシカルボニル)ォキシ] 2—メチルー 6— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジ ル]フ -ル]ブター 3—エノイツクアシッドの合成
4— (4 ブロモベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチル 5—チォー j8—D ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(1. 08g、 1. 55mmol)のァセトニトリル(16mL)溶液にビュル酢酸(321mg、 3. 73mm ol)、酢酸パラジウム(II) (35mg、 0. 155mmol)、トリ— O トリルホスフィン(94mg、
0. 310mmol)、トリェチルァミン(1. 08mL、 7. 75mmol)を加え、 biotage社製マイ クロウ ーブを用いて 120°C、 20分間反応を行った。反応液を減圧下留去し、得られ た残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム:メタノール = 100 : 1〜30:
1、へキサン:酢酸ェチル= 1 :4〜0 : 1)にて精製し、淡黄色粉末として表題化合物( 990mg、 91%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.96 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.05 (s, 3 H) 2.18 (s, 3 H) 3.21 - 3.31 (m, 2 H) 3.79 - 4.17 (m, 7 H) 4.25 - 4. 34 (m, 1 H) 5.06 - 5.38 (m, 3 H) 5.49 - 5.60 (m, 1 H) 6.13 - 6.26 (m, 1 H) 6.40 - 6 .50 (m, 1 H) 6.74 - 6.77 (m, 1 H) 6.88 - 6.92 (m, 1 H) 6.94 - 6.99 (m, 2 H) 7.19 - 7.25 (m, 2 H).
ESI m/z = 725 (M+Na).
(3) 4— [4— [4 [ (メトキシカルボ-ル)ォキシ ]ー2—メチルー 6— [ (2, 3, 4, 6—
テトラ一 O ァセチルー 5—チォ一 β—D—ダルコピラノシル)ォキシ]ベンジル]フエ

(3E)—4 [4 [4ー[ (メトキシカルボ-ル)ォキシ]ー2—メチルー6—[ (2, 3, 4 , 6—テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル ]フエ-ル]ブタ一 3 エノイツクアシッド(1. 99g、 2. 83mmol)のメタノール(28mL) 溶液にパラジウム 活性炭素(10%) (660mg) を加え、水素雰囲気下、室温にて 2時間攪拌した。反応液をセライトろ過後、減圧下留去し、得られた残渣をシリカゲル カラムクロマトグラフィ—(クロ口ホルム:メタノール = 50: 1〜30: 1)にて精製し、無色 粉末として表題化合物(1. 81g、 91%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.74 (s, 3 H) 1.85 - 1.94 (m, 2 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.19 (s, 3 H) 2.30 - 2.38 (m, 2 H) 2.55 - 2. 64 (m, 2 H) 3.21 - 3.31 (m, 1 H) 3.77 - 4.17 (m, 6 H) 4.30 (dd, J=11.89, 5.21 Hz, 1 H) 5.06 - 5.39 (m, 3 H) 5.49 - 5.59 (m, 1 H) 6.68 - 7.09 (m, 6 H).
ESI m/z = 727 (M+Na).
(4) 4— [4— [4—ヒドロキシ一 2—メチル 6— [ ( 5—チォ— β — Ό—ダルコビラノシ ル)ォキシ]ベンジル]フエ-ル]ブタノイツクアシッドの合成
4 [4 [4ー[ (メトキシカルボニル)ォキシ]ー2—メチルー6—[ (2, 3, 4, 6—テト ラ一 Ο ァセチノレ一 5—チォ一 β—D—ダルコピラノシル)ォキシ]ベンジル]フエ- ル]ブタノイツクアシッド(160mg、 0. 227mmol)のメタノール溶液に、ナトリウムメトキ シド(25%メタノール溶液; 0. 20mL、 0. 908mmol)をカ卩え、室温で 2時間攪拌した 。反応液にドライアイスを加え中和し、溶媒を減圧留去した。得られた残渣をシリカゲ ルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20 : 2 : 1〜 5 : 2 : 1)で精製 し、表題ィ匕合物(51mg、 47%)を合成した。
1H NMR (600 MHz, METHANOL— d ) δ ppm 1.82 - 1.89 (m, 2 H) 2.10 (s, 3 H) 2.2
4
2 - 2.28 (m, 2 H) 2.54 - 2.60 (m, 2 H) 2.85 - 2.90 (m, 1 H) 3.22 - 3.27 (m, 1 H) 3. 51 - 3.57 (m, 1 H) 3.70 - 3.78 (m, 2 H) 3.82 - 3.94 (m, 2 H) 3.97 - 4.03 (m, 1 H) 5 .06 (d, J=8.71 Hz, 1 H) 6.32 (d, J=2.29 Hz, 1 H) 6.67 (d, J=2.29 Hz, 1 H) 6.98 - 7. 04 (m, 4 H). ESI m/z = 501 (M+Na).
参考例 12
4— [4— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 13—D ダルコビラノシル )ォキシ]ベンジル]フエ-ル]ブタンアミドの合成
[化 18]

( 1) 4— [4— (4 アミノー 4ーォキソブチル)ベンジル ]ー3—メチルー 5— [ (2, 3, 4 , 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの合成
参考例 11の(3)で得られた 4 [4 [4 [ (メトキシカルボニル)ォキシ ] 2—メチ ルー 6— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 j8—D—ダルコビラノシ ル)ォキシ]ベンジル]フエ-ル]ブタノイツクアシッド(180mg、 0. 255mmol)のクロ口 ホルム(3mL)の溶液に、塩化アンモ-ゥム(27mg、 0. 511mmol)、 1ーヒドロキシ ベンゾトリアゾール(43mg、 0. 281mmol)、 1ーェチルー 3—(3 ジメチルアミノプ 口ピル)カルボジイミド塩酸塩(68mg、 0. 357mmol)、トリェチルァミン(0. 18mL、 1 . 28mmol)を加え、室温にて 3時間攪拌した。反応液に水を加え、クロ口ホルムで抽 出した。有機層を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。乾 燥剤を炉別後、溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフ ィ—(クロ口ホルム:メタノール = 50 : 1〜10 : 1)で精製し、表題化合物(140mg、 78 %)を合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.78 (s, 3 H) 1.88 - 1.96 (m, 2 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.10 - 2.19 (m, 2 H) 2.20 (s, 3 H) 2.55 - 2. 66 (m, 2 H) 3.20 - 3.33 (m, 1 H) 3.78 - 4.17 (m, 6 H) 4.25 - 4.34 (m, 1 H) 5.06 - 5 .60 (m, 6 H) 6.68 - 7.09 (m, 6 H).
ESI m/z = 726 (M+Na).
(2) 4— [4— [4 ヒドロキシ一 2—メチル 6— [ ( 5 チォ— β— Ό—ダルコビラノシ
ル)ォキシ]ベンジル]フエ-ル]ブタンアミドの合成
4— [4— (4 アミノー 4—ォキソブチル)ベンジル]—3—メチル 5— [ (2, 3, 4, 6 —テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メ チル カルボナート(140mg、 0. 199mmol)のメタノール溶液に、ナトリウムメトキシ ド(25%メタノール溶液; 0. 17mL、 0. 796mmol)を加え、室温で 2時間攪拌した。 反応液にドライアイスを加え中和し、溶媒を減圧留去した。得られた残渣をシリカゲル カラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20 : 2 : 1〜5 : 2 : 1)で精製し、 表題ィ匕合物(66mg、 69%)を合成した。
1H NMR (600 MHz, METHANOL- d ) δ ppm 1.83 - 1.90 (m, 2 H) 2.11 (s, 3 H) 2.1
4
4 - 2.19 (m, 2 H) 2.54 - 2.59 (m, 2 H) 2.84 - 2.90 (m, 1 H) 3.21 - 3.26 (m, 1 H) 3. 50 - 3.56 (m, 1 H) 3.69 - 3.78 (m, 2 H) 3.80 - 4.04 (m, 3 H) 5.05 (d, J=8.71 Hz, 1 H) 6.32 (d, J=2.29 Hz, 1 H) 6.67 (d, J=2.29 Hz, 1 H) 6.99 - 7.05 (m, 4 H). ESI m/z = 500 (M+Na).
参考例 13
ジベンジル [(Z) - (ァリルァミノ)メチルイリデン]ビスカルバメ―トの合成
[化 19]
H
^ ,N. ,NHCbz
NCbz
ァリルアミン(250mg、 4. 38mmol)のテトラヒドロフラン(4. 3mL)溶液に N, N, 一 ビス—ベンジルォキシカルボ-ルー 1—グァ -ルピラゾール(1. 98g、 5. 25mmol) を加え、室温にて一晩攪拌した。反応液を減圧下留去し得られた残渣をシリカゲル力 ラムクロマトグラフィ—(へキサン:酢酸ェチル =4 : 1)にて精製し、無色粉末として表 題化合物(1. 45g、 90%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 4.03 - 4.12 (m, 2 H) 5.11 - 5.28 (m, 6 H) 5.81 5.96 (m, 1 H) 7.23 7.43 (m, 10 H) 8.35 8.45 (m, 1 H) 11.76 (s, 1 H).
ESI m/z = 368(M+H).
参考例 14
N—ァリル— N, - (2—ヒドロキシ— 1, 1—ジメチルェチル)ゥレアの合成
[0089] [化 20]

ァリルァミン(1. 5g、 26. 3mmol)のクロ口ホルム(60mL)溶液にトリェチルアミン( 4. 9mL、 35. 5mmol)をカ卩え、 4°Cにて 4—二トロフエ-ル クロ口ホルメ一ト(6. 09g 、 30. 2mmol)をカロえ 1時間攪拌した。この反応液に同温にて 2—ァミノ一 2—メチル プロパノール(2. 58g、 28. 9mmol)のクロ口ホルム(3mL)溶液を加えて、室温で一 晚攪拌した。反応溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトダラ フィ—(酢酸ェチル)にて精製し、黄色油状ィ匕合物として表題ィ匕合物 (4. Og、 88%) を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.26 (s, 6 H) 3.55 (s, 2 H) 3.71 - 3 .80 (m, 2 H) 4.85 - 5.08 (m, 2 H) 5.08 - 5.24 (m, 2 H) 5.77 - 5.91 (m, 1 H).
ESI m/z = 195 (M+Na).
参考例 15
1ーェチルー 3, 5—ジメトキシベンゼンの合成
[0090] [化 21]

(1) 1, 3—ジメトキシー 5—ビュルベンゼンの合成
メチルトリフエ-ルホスホ-ゥムブロミド(43. 0g、 120mmol)のテトラヒドロフラン(15 OmL)溶液に、— 78。Cで、 n—ブチルリチウム(2. 67Mへキサン溶液、 45mL、 120 mmol)を滴下した。室温で 30分攪拌後、反応液を 0°Cに冷却し、 3, 5—ジメトキシべ ンズアルデヒド(9. 93g、 60. 2mmol)のテトラヒドロフラン(50mL)溶液を滴下した。 室温に昇温後、反応液に水を加え、酢酸ェチルで 2回抽出した。有機層を飽和食塩 水で洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を炉別後、溶媒を減圧 下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(n—へキサン:酢酸ェ
チル = 9: 1)で精製し、表題化合物(8. 2g、 83%)を合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 3.79 (s, 6 H) 5.24 (dd, J=10.88, 0.
93 Hz, 1 H) 5.72 (dd, J=17.56, 0.93 Hz, 1 H) 6.38 (t, J=2.25 Hz, 1 H) 6.56 (s, 1 H)
6.57 (s, 1 H) 6.64 (dd, J=17.56, 10.88 Hz, 1 H).
(2) 1—ェチルー 3, 5 ジメトキシベンゼンの合成
1, 3 ジメトキシー 5 ビュルベンゼン(8. 2g)のメタノール(200mL)溶液に 10% パラジウム—活性炭素 (0. 82g)を加え、水素雰囲気下で一晩攪拌した。反応液をセ ライト濾過後、溶媒を減圧下留去し、得られた残渣をシリカゲルを通して濾過し (n— へキサン:酢酸ェチル =4 : 1)、減圧濃縮した。残渣を同様にメタノール(200mL)に 溶解させ、 10%パラジウム—活性炭素 (0. 82g)を加え、水素雰囲気下で一晩攪拌 した。反応液をセライト濾過後、溶媒を減圧下留去した。得られた残渣をシリカゲルを 通して濾過し (n—へキサン:酢酸ェチル =4 : 1)、減圧濃縮し、無色液体として表題 化合物(7. 2g、 87%)を得た。
[0091] 1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.23 (t, J=7.62 Hz, 3 H) 2.60 (q, J =7.62 Hz, 2 H) 3.78 (s, 6 H) 6.30 (t, J=2.25 Hz, 1 H) 6.37 (d, J=2.25 Hz, 2 H). 参考例 16
3—ェチル 5—ヒドロキシ一 4— (4— -トロベンジル)フエ-ル メチル カーボナー トの合成
[0092] [化 22]

1ーェチルー 3, 5 ジメトキシベンゼンと 4 -トロベンゾイルクロリドより、国際公開 WO2004Z050122号に準拠して合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.11 (t, J=7.54 Hz, 3 H) 2.56 (q, J =7.54 Hz, 2 H) 3.92 (s, 3 H) 4.07 (s, 2 H) 5.80 (s, 1 H) 6.54 (d, J=2.33 Hz, 1 H) 6. 65 (d, J=2.33 Hz, 1 H) 7.24 (d, J=8.24 Hz, 2 H) 8.05 (d, J=8.24 Hz, 2 H).
参考例 17
4— (4 ァミノベンジル) 3 ェチル 5— [ (2, 3, 4, 6—テトラ一 0 ァセチル一
5—チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カーボナートの合成 [化 23]

(1)メチル 3 ェチル 4— (4 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O— ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カーボナートの 合成
3—ヒドロキシ一 5—メチル 4— (4— -トロベンジル)フエ-ル メチル カルボナート の代わりに 3—ェチル 5—ヒドロキシ一 4— (4— -トロベンジル)フエ-ル メチル カーボナートを用い、参考例 8 (1)と同様の方法で表題ィ匕合物を得た(1. 47g、 31% )
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.12 (t, J=7.36 Hz, 3 H) 1.83 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.04 (s, 3 H) 2.56 (q, J=7.36 Hz, 2 H) 3.21 - 3.29 (m, 1 H) 3.94 (s, 3 H) 3.96 - 4.16 (m, 3 H) 4.26 (dd, J=11.97, 5.28 Hz, 1 H) 5.12 (t, J= 9.33 Hz, 1 H) 5.23 - 5.33 (m, 2 H) 5.41 - 5.50 (m, 1 H) 6.83 (d, J=2.33 Hz, 1 H) 6. 89 (d, J=2.33 Hz, 1 H) 7.20 (d, J=8.86 Hz, 2 H) 8.10 (d, J=8.86 Hz, 2 H).
(2) 4— (4 ァミノベンジル) 3 ェチル 5— [ (2, 3, 4, 6—テトラ一 O ァセ チルー 5—チォー j8—D ダルコピラノシル)ォキシ]フエ-ル メチル カーボナー ト の合成
メチル 3—メチル 4— (4 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O ァ セチル 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カルボナートの代 わりに、メチル 3 ェチル 4— (4 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カーボナー トを用い、参考例 8 (2)と同様の方法で表題ィ匕合物を得た (0. 242g、 17%)。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.09 (t, J=7.46 Hz, 3 H) 1.77 (s, 3 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.55 (q, J=7.46 Hz, 2 H) 3.20 - 3.29 (m,
1 H) 3.74 - 3.82 (m, 1 H) 3.89 - 3.98 (m, 4 H) 4.07 - 4.17 (m, 1 H) 4.30 (dd, J=ll. 89, 5.21 Hz, 1 H) 5.11 (t, J=9.25 Hz, 1 H) 5.24 (d, J=8.55 Hz, 1 H) 5.29 - 5.39 (m,
1 H) 5.54 (t, J=8.55 Hz, 1 H) 6.55 (d, J=8.39 Hz, 2 H) 6.75 - 6.83 (m, 3 H) 6.89 (d , J=2.18 Hz, 1 H).
実施例
[0094] 以下に実施例及び試験例をあげて本発明をさらに詳しく説明するが、本発明はこ れらの記載によって限定的に解釈されるものではない。
[0095] 実施例 1
N— (2 ヒドロキシェチル) Ν'— [4— [4 ヒドロキシ一 2—メチル 6— [ (5 チ ォ一 13—D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ゥレアの合成
[0096] [化 24]

参考例 8の(2)で合成した、 4一(4ーァミノベンジル) 3—メチルー 5— [ (2, 3, 4 , 6—テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200mg、 0. 316mmol)のクロ口ホルム(8mL)溶液にピリジ ン(0. 034mL、 0. 426mmol)、クロロギ酸 4 -トロフエ-ル(73mgゝ 0. 363mm ol)を加え、室温で 1時間攪拌した。トリェチルァミン(0. 088mL、 0. 631mmol)と 2 —アミノエタノール(21mg、 0. 347mmol)をカ卩え、室温で 1時間攪拌した。反応液を 減圧下留去し、得られた残渣にメタノール(2mL)とナトリウムメトキシド(25%メタノー ル溶液、 0. 30mL、 1. 38mmol)をカ卩え、室温で 4時間攪拌した。反応液にドライア イスを加え中和し、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグ ラフィ (酢酸ェチル:エタノール:水 = 20 : 2 : 1〜 5 : 2 : 1)で精製し、酢酸ェチルで 洗浄し、無色粉末として表題化合物 (71mg、 45%)を得た。得られた化合物の構
造、 NMRデータ及び MSデータを表 1に示す。
[0097] 実施例 2 ウレイド誘導体の合成
参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートとそれぞれ対応するァミン、すなわち、 3—アミノメチルビリジン、 2—アミノー 2—メチルー 1 プロパノール、グリシンアミド塩酸塩、 j8—ァラニンアミド 塩酸塩、ベンジルァミン、イソプロピルァミン、ジメチルァミン、 n—へキシルァミン、シ クロへキシルァミンを用い、実施例 1に示した方法と同様の方法で"ィ匕合物 2"乃至" 化合物 5"または"化合物 7"乃至"化合物 11"を合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0098] 実施例 3
N— [4— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 13—D—ダルコビラノシル )ォキシ]ベンジノレ]フエ二ノレ]ゥレアの合成
[0099] [化 25]

参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200mg、 0. 316mmol)のクロ口ホルム(8mL)溶液にピリジン (0. 034mL、 0. 426mmol)、クロロギ酸 4 -トロフエ-ル(73mg、 0. 363mmol )を加え、室温で 30分間攪拌した。アンモニア(2Mメタノール溶液; 1. 58mL、 3. 1 6mmol)を加え、室温で 1時間攪拌した。反応液を減圧下留去し、得られた残渣にメ タノール(2mL)とナトリウムメトキシド(25%メタノール溶液; 0. 27mL、 1. 26mmol) を加え、室温で 3時間攪拌した。反応液にドライアイスを加え中和し、溶媒を減圧留 去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸ェチル:エタノール: 水 = 20 : 2 : 1〜5: 2 : 1)で精製し、酢酸ェチルで洗浄し、無色粉末として表題化合物
(85mg 60%)を得た。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0100] 実施例 ο s 4
N— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 13—D グノレコピラノシノレ )ォキシ]ベンジル]フエ-ル]ベンゼンスルホンアミドの合成
[0101] [化 26]
HN o
Ό
Hひ
HO 参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートとベンゼンスルホユルク口リドを用い、参考例 8 (3)に示した方法 と同様の方法でィ匕合物 14を合成した。得られたィ匕合物の構造、 NMRデータ及び Μ Sデータを表 1に示す。
[0102] 実施例 5
Ν— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5 チォ一 13—D グノレコピラノシノレ )ォキシ]ベンジル]フエ-ル]スルフアミドの合成
[0103] [化 27]

イソシアン酸 クロロスルホニル(0. 083mL 0. 948mmol)のァセトニトリル(4mL )溶液に水(0. 02mL)を加え、室温で 10分間攪拌した。この反応混合物を、参考例 8の(2)で得られた 4— (4 ァミノベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ —O ァセチルー 5—チォ一 j8—D—ダルコピラノシル)ォキシ]フエ-ル メチル 力 ルボナート(200mg 0. 316mmol)および、トリェチルァミン(0. 20mL 1. 42mm
ol)のクロ口ホルム(8mL)溶液にカ卩え、室温で 2. 5日間攪拌した。反応液を水にあけ 、クロ口ホルムで抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸マグネシゥ ムで乾燥した。乾燥剤を炉別後、溶媒を減圧下留去し、得られた残渣にメタノール (2 mL)とナトリウムメトキシド(25%メタノール溶液、 0. 27mL、 1. 26mmol)を加え、室 温で 2時間攪拌した。反応液にドライアイスを加え中和し、溶媒を減圧留去した。得ら れた残渣をシリカゲルカラムクロマトグラフィ (酢酸ェチル:エタノール:水 = 20: 2: 1〜5 : 2 : 1)で精製し、無色粉末として表題ィ匕合物(71mg、 46%)を得た。得られた 化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0104] 実施例 6
N— [4— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 β—D—ダルコビラノシル )ォキシ]ベンジル]フエ-ル]グァ-ジンの合成
[0105] [化 28]

参考例 8の(2)で得られた 4 (4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(187mg、 0. 295mmol)のテトラヒドロフラン(7mL)溶液に N, N, -ビス(ベンジルォキシカルボ-ル)— 1H—ピラゾール— 1—カルボキサミジン( 1 23mg、 0. 325mmol)を加え、室温で 3時間、 60°Cで 4時間攪拌し、 8時間加熱還 流した。反応液を減圧下留去後、得られた残渣をシリカゲルカラムクロマトグラフィー( へキサン:酢酸ェチル = 1: 1)で精製し、無色ガム状物質を得た。これにメタノール(2 mL)とナトリウムメトキシド(25%メタノール溶液、 0. 26mL、 1. 18mmol)を加え、室 温でー晚攪拌した。ナトリウムメトキシド(25%メタノール溶液、 0. 52mL、 2. 36mm ol)を加え、室温で一晩攪拌した。さらにナトリウムメトキシド(25%メタノール溶液、 0. 52mL、 2. 36mmol)を加え、室温で 3日間攪拌した。反応液にドライアイスを加え中 和し、溶媒を減圧留去した。得られた残渣を合成吸着剤 HP20 (メタノール:水 = 1 : 1
〜1 : 0)で精製し、酢酸ェチルで洗浄後、淡褐色粉末として表題化合物(24mg、 18 %)を得た。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0106] 実施例 7
N— [3— [4 ヒドロキシ一 2—メチル 6— [ (5—チォ一 β—D—ダルコビラノシル )ォキシ]ベンジノレ]フエ二ノレ]ゥレアの合成
[0107] [化 29]

(1)メチル 3—メチル 4— (3 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 Ο— ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カルボナートの 合成
2, 3, 4, 6—テトラ一 Ο ァセチルー 5 チォ一 D—ダルコビラノース(3. 4g、 9. 4 6mmol)と 3 ヒドロキシ一 5—メチル 4— (3 -トロベンジル)フエ-ル メチル 力 ルボナート(2. Og、 6. 30mmol)、トリフエ-ルホスフィン(2. 5g、 9. 46mmol)、ジィ ソプロピルァゾジカルボキシレート(40%トルエン溶液; 5. lmL、 9. 46mmol)を用 い、参考例 8の(1)に示した方法と同様の方法で表題ィ匕合物(1. 61g、 39%)を合成 した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.85 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.24 (s, 3 H) 3.22 - 3.31 (m, 1 H) 3.93 (s, 3 H) 3.99 - 4.15 (m, 3 H) 4.27 (dd, J=11.89, 5.36 Hz, 1 H) 5.09 - 5.17 (m, 1 H) 5.25 - 5.39 (m, 2 H) 5.4 6 - 5.55 (m, 1 H) 6.78 - 6.81 (m, 1 H) 6.89 - 6.93 (m, 1 H) 7.37 - 7.43 (m, 2 H) 7. 90 - 7.95 (m, 1 H) 7.98 - 8.09 (m, 1 H).
ESI m/z = 686 (M+Na).
(2) 4— (3 ァミノベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの 合成
メチル 3—メチル 4— (3 -トロベンジル) 5— [ (2, 3, 4, 6—テトラ一 O ァ セチルー 5 チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル カルボナート(1. 5 9g、 2. 40mmol)とパラジウム—活性炭素(10%) (0. 76g)を用い、参考例 8の(2) に示した方法と同様の方法で表題ィ匕合物(660mg、 43%)を合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.78 (s, 3 H) 1.97 (s, 3 H) 2.04 (s,
3 H) 2.06 (s, 3 H) 2.21 (s, 3 H) 3.21 - 3.31 (m, 1 H) 3.73 - 3.81 (m, 1 H) 3.92 (s, 3 H) 4.07 - 4.18 (m, 2 H) 4.30 (dd, J=11.97, 5.13 Hz, 1 H) 5.08 - 5.40 (m, 3 H) 5.5 2 - 5.61 (m, 1 H) 6.34 - 6.38 (m, 1 H) 6.42 - 6.50 (m, 2 H) 6.73 - 6.76 (m, 1 H) 6. 88 - 6.91 (m, 1 H) 6.96 - 7.03 (m, 1 H).
(3) N- [3— [4 ヒドロキシ一 2—メチル 6— [ (5 チォ一 j8—D—ダルコビラノシ ル)ォキシ]ベンジル]フエニル]ゥレアの合成
4— (3 ァミノベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチル 5 チォー j8—D ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(20 Omg、 0. 316mmol)とピリジン(0. 034mレ 0. 426mmol)、クロロギ酸 4 -卜口 フエ-ル(73mg、 0. 363mmol)、アンモニア(2Mメタノール溶液; 1. 58mL、 3. 16 mmol)、ナトリウムメトキシド(25%メタノール溶液)を用い、実施例 3に示した方法と 同様の方法で表題ィ匕合物(110mg、 77%)を合成した。得られたィ匕合物の構造、 N MRデータ及び MSデータを表 1に示す。
[0108] 実施例 8
—ヒドロキシ一 2— [ (5—チォ一 β—D—ダルコピラノシル)ォキシ]ベ ル]ゥレア合成
[0109] [化 30]

(1)メチル 4— (4 -トロベンジル) 3— [ (2, 3, 4, 6—テトラ一 Ο ァセチル一 5 チォー β D—ダルコピラノシル)ォキシ]フエ-ル カルボナートの合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(5. 39g、 14 . 8mmol)と 3 ヒドロキシ一 4— (4 -トロベンジル)フエ-ル メチル カルボナート (3. 27g、 9. 85mmol)、トリフエ-ルホスフィン(3. 88g、 14. 8mmol)、ジイソプロピ ルァゾジカルボキシレート(40%トルエン溶液; 8. 04mL、 14. 8mmol)を用い、参 考例 8の(1)に示した方法と同様の方法で表題ィ匕合物(0. 95g、 15%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.94 (s, 3 H) 2.02 (s, 3 H) 2.04 (s,
3 H) 2.05 (s, 3 H) 3.23 - 3.31 (m, 1 H) 3.88 - 4.03 (m, 4 H) 4.06 - 4.17 (m, 2 H) 4 .29 (dd, J=11.97, 5.28 Hz, 1 H) 5.10 - 5.58 (m, 4 H) 6.87 (dd, J=8.24, 2.26 Hz, 1 H ) 6.98 (d, J=2.26 Hz, 1 H) 7.13 (d, J=8.24 Hz, 1 H) 7.29 - 7.35 (m, 2 H) 8.10 - 8.16
(m, 2 H).
ESI m/z = 672 (M+Na).
(2) 4— (4 ァミノベンジル) 3— [ (2, 3, 4, 6—テトラ一 O ァセチル一 5 チォ — j8—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの合成 メチル 4— (4 -トロベンジル) 3— [ (2, 3, 4, 6—テトラ一 O ァセチル一 5— チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル カルボナート(0. 95g、 1. 46m mol)とパラジウム—活性炭素(10%) (0. 48g)を用い、参考例 8の(2)に示した方法 と同様の方法で表題ィ匕合物(0. 51g、 56%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.95 (s, 3 H) 2.02 (s, 3 H) 2.05 (s, 3 H) 2.07 (s, 3 H) 3.23 - 3.32 (m, 1 H) 3.73 - 3.78 (m, 1 H) 3.92 (s, 3 H) 4.07 - 4.
17 (m, 2 H) 4.32 (dd, J=11.97, 5.28 Hz, 1 H) 5.11 - 5.43 (m, 3 H) 5.57 - 5.66 (m, 1 H) 6.60 - 6.66 (m, 2 H) 6.77 - 6.82 (m, 1 H) 6.91 - 7.06 (m, 4 H).
ESI m/z = 642 (M+Na).
(3) N— [4— [4 ヒドロキシ一 2— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ] ベンジル]フエ-ル]ゥレアの合成
4— (4 ァミノベンジル) 3— [ (2, 3, 4, 6—テトラ一 O ァセチル一 5 チォ一 j8—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200mg、 0. 323 mmol)とピリジン(0. 035mL、 0. 436mmol)、クロロギ酸 4 -トロフエ-ル(75m g、 0. 371mmol)、アンモニア(2Mメタノール溶液; 1. 62mL、 3. 23mmol)、ナトリ
ゥムメトキシド(25%メタノール溶液; 0. 28mL、 1. 29mmol)を用い、実施例 3に示 した方法と同様の方法で表題ィ匕合物(89mg、 63%)を合成した。得られたィ匕合物の 構造、 NMRデータ及び MSデータを表 1に示す。
[0110] 実施例 9
N— [4— [4—メトキシ一 2—メチル 6— [ (5—チォ一 β—D—ダルコピラノシル) ォキシ]ベンジノレ]フエ二ノレ]ゥレアの合成
[0111] [化 31]

(1) 5—メトキシ一 3—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ —Ο ァセチル一 5—チォ一 β—D—ダルコビラノシドの合成
2, 3, 4, 6—テトラ一 Ο ァセチルー 5 チォ一 D—ダルコビラノース(2. Og、 5. 4 8mmol)と 5—メトキシ一 3—メチル 2— (4 -トロベンジル)フエノール(1. Og、 3. 66mmol)、トリフエ-ルホスフィン(1. 4g、 5. 48mmol)、ジイソプロピルァゾジカル ボキシレート(2. 8mL、 5. 48mmol)を用い、参考例 8の(1)に示した方法と同様の 方法で表題ィ匕合物 (0. 71g、 31%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.82 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.04 (s, 3 H) 2.19 (s, 3 H) 3.21 - 3.29 (m, 1 H) 3.82 (s, 3 H) 3.90 - 4.12 (m,
3 H) 4.28 (dd, J=11.97, 5.13 Hz, 1 H) 5.10 - 5.17 (m, 1 H) 5.26 - 5.36 (m, 2 H) 5.4
5 - 5.52 (m, 1 H) 6.48 (d, J=2.33 Hz, 1 H) 6.61 (d, J=2.49 Hz, 1 H) 7.17 - 7.23 (m, 2 H) 8.06 - 8.12 (m, 2 H).
ESI m/z = 642 (M+Na).
(2) 2— (4 ァミノベンジル) 5—メトキシ一 3—メチルフエ-ル 2, 3, 4, 6—テトラ —O ァセチル一 5—チォ一 β—D—ダルコビラノシドの合成
5—メトキシ一 3—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 Ο ァセチルー 5 チォ一 j8—D—ダルコビラノシド(0. 71g、 1. 15mmol)とパラジ
ゥム—活性炭素(10%) (0. 3g)を用い、参考例 8の(2)に示した方法と同様の方法 で表題ィ匕合物(540mg、 80%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.77 (s, 3 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.17 (s, 3 H) 3.18 - 3.26 (m, 1 H) 3.69 - 3.77 (m, 1 H) 3.80 (s, 3 H) 3.84 - 3.92 (m, 1 H) 4.11 (dd, J=12.12, 3.89 Hz, 1 H) 4.30 (dd, J=12.05, 5.05 Hz, 1 H) 5.11 (t, J=9.25 Hz, 1 H) 5.25 (d, J=8.70 Hz, 1 H) 5.34 (dd, J=10.41, 9.48 Hz, 1 H) 5.55 (t, J=8.86 Hz, 1 H) 6.45 (d, J=2.33 Hz, 1 H) 6.59 (d, J=8.39 Hz, 2 H) 6.62 (d, J=2.18 Hz, 1 H) 6.82 (d, J=8.55 Hz, 2 H).
ESI m/z = 612 (M+Na).
(3) N- [4— [4—メトキシ一 2—メチル 6— [ (5—チォ一 j8—D—ダルコビラノシル )ォキシ]ベンジル]フエ-ル]ゥレア
2— (4 ァミノベンジル) 5—メトキシ一 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチノレー 5 チォー 13 D—ダルコビラノシド(200mg、 0. 34mmol)とピリジ ン(0. 037mL、 0. 46mmol)、クロロギ酸 4 -トロフエ-ル(79mg、 0. 39mmol) 、アンモニア(2M メタノール溶液、 1. 69mL、 3. 39mmol)、ナトリウムメトキシド(2 5% メタノール溶液、 0. 3mL、 1. 36mmol)を用い、実施例 3に示した方法と同様 の方法で表題ィ匕合物(88mg、 56%)を得た。得られたィ匕合物の構造、 NMRデータ 及び MSデータを表 1に示す。
[0112] 実施例 10
N— [4— [2, 4 ジメチルー 6— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ] ベンジル]フエ-ル]ゥレアの合成
[0113] [化 32]

(1) 3, 5 ジメチル一 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O— ァセチルー 5—チォ一 β—D—ダルコビラノシドの合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(2. 12g、 5. 83mmol)と 3, 5 ジメチルー 2— (4 -トロベンジル)フエノール(1. Ogゝ 3. 89m mol)、トリフエ-ルホスフィン(1. 53g、 5. 83mmol)、ジイソプロピルァゾジカルボキ シレート(2. 9mL、 5. 83mmol)を用い、参考例 8の(1)に示した方法と同様の方法 で表題ィ匕合物 (442mg、 19%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.84 (s, 3 H) 1.99 (s, 3 H) 2.04 (d, J=2.49 Hz, 6 H) 2.17 (s, 3 H) 2.35 (s, 3 H) 3.22 - 3.31 (m, 1 H) 3.92 - 4.14 (m, 3 H) 4.29 (dd, J=12.05, 5.05 Hz, 1 H) 5.10 - 5.19 (m, 1 H) 5.28 - 5.37 (m, 2 H) 5.45 - 5.53 (m, 1 H) 6.76 (s, 1 H) 6.82 (s, 1 H) 7.18 - 7.24 (m, 2 H) 8.05 - 8.12 (m, 2 H ).
ESI m/z = 626 (M+Na).
(2) 2— (4 ァミノベンジル) 3, 5 ジメチルフエ-ル 2, 3, 4, 6—テトラ一 O ァ セチル 5—チォ一 13—D—ダルコビラノシドの合成
3, 5 ジメチル一 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O ァセ チル一 5 チォ一 13— D—ダルコビラノシド(442mg、 0. 73mmol)とパラジウム一 活性炭素(10%) (221mg)を用い、参考例 8の(2)に示した方法と同様の方法で表 題ィ匕合物(300mg、 71%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.79 (s, 3 H) 1.98 (s, 3 H) 2.05 (d, J=8.55 Hz, 6 H) 2.15 (s, 3 H) 2.33 (s, 3 H) 3.18 - 3.29 (m, 1 H) 3.72 - 3.82 (m, 1 H) 3.85 - 3.95 (m, 1 H) 4.07 - 4.16 (m, 1 H) 4.31 (dd, J=11.89, 5.05 Hz, 1 H) 5.12 (t, J=9.25 Hz, 1 H) 5.24 - 5.41 (m, 2 H) 5.55 (t, J=8.86 Hz, 1 H) 6.53 - 6.61 (m, 2 H) 6.72 (s, 1 H) 6.78 - 6.86 (m, 3 H).
ESI m/z = 596 (M+Na).
(3) N- [4- [2, 4 ジメチルー 6— [ (5—チォ一 j8—D—ダルコピラノシル)ォキシ ]ベンジル]フエ二ノレ]ゥレア
2—(4ーァミノベンジル) 3, 5 ジメチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセ チル一 5 チォ一 13— D—ダルコビラノシド(289mg、 0. 50mmol)とピリジン(0. 05 5mL、 0. 68mmol)、クロロギ酸 4 -卜口フエ-ル(117mg、 0. 579mmol)、アン
モ-ァ(2M メタノール溶液、 1. 5mL、 3. Ommol)、ナトリウムメトキシド(25% メタ ノール溶液、 0. 44mL、 2. Olmmol)を用い、実施例 3に示した方法と同様の方法 で表題ィ匕合物(179mg、 79%)を得た。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0114] 実施例 11
N— [4— [2—メチルー 6—[ (5—チォー 13 D ダルコピラノシル)ォキシ]ベンジ ル]フエ-ル]ゥレアの合成
[0115] [化 33]

(1) 3—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー 13 D—ダルコビラノシドの合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(667mg、 1. 83mmol)と 3—メチル 2— (4 -トロベンジル)フエノール(297mgゝ 1. 22mmol )、トリフエ-ルホスフィン(480mg、 1. 83mmol)、ジイソプロピルァゾジカルボキシレ 一ト(0. 93mL、 1. 83mmol)を用い、参考例 8の(1)に示した方法と同様の方法で 表題化合物を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.83 (s, 3 H) 1.99 (s, 3 H) 2.03 (s, 3 H) 2.04 (s, 3 H) 2.22 (s, 3 H) 3.22 - 3.30 (m, 1 H) 3.98 - 4.16 (m, 3 H) 4.28 (dd, J=12.05, 5.21 Hz, 1 H) 5.11 - 5.19 (m, 1 H) 5.28 - 5.37 (m, 2 H) 5.46 - 5.54 (m, 1 H) 6.94 (d, J=7.15 Hz, 1 H) 7.02 (d, J=8.39 Hz, 1 H) 7.18 - 7.25 (m, 3 H) 8.06 - 8
.12 (m, 2 H).
ESI m/z = 588 (M- H).
(2) 2— (4 ァミノベンジル) 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー 13 D—ダルコビラノシドの合成
3—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O ァセチル
—5 チォ一 β— D—ダルコビラノシド(195mg、 0. 33mmol)とパラジウム一活性 炭素(10%) (98mg)を用い、参考例 8の(2)に示した方法と同様の方法で表題ィ匕合 物(71mg、 10%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.78 (s, 3 H) 1.98 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.19 (s, 3 H) 3.19 - 3.28 (m, 1 H) 3.82 (d, J=15.70 Hz, 1 H) 3.9 6 (d, J=15.70 Hz, 1 H) 4.08 - 4.15 (m, 1 H) 4.30 (dd, J=11.89, 5.36 Hz, 1 H) 5.13 ( t, J=9.25 Hz, 1 H) 5.27 - 5.40 (m, 2 H) 5.56 (t, J=8.94 Hz, 1 H) 6.59 - 6.72 (m, 2 H) 6.81 - 6.92 (m, 3 H) 7.02 (d, J=7.31 Hz, 1 H) 7.10 - 7.17 (m, 1 H).
ESI m/z = 582 (M+Na).
(3) N— [4— [2—メチル 6— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ]ベン ジル]フ -ル]ゥレアの合成
2— (4 ァミノベンジル) 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル —5 チォ一 13— D—ダルコビラノシド(71mg、 0. 126mmol)とピリジン(0. 014m L、0. 171mmol)、クロロギ酸 4 -トロフエ-ル(29mg、 0. 146mmol)、アンモ ユア(2M メタノール溶液、 0. 63mL、 1. 26mmol)、ナトリウムメトキシド(25% メタ ノール溶液、 0. l lmL、 0. 507mmol)を用い、実施例 3に示した方法と同様の方法 で表題ィ匕合物(23mg、 42%)を得た。得られた化合物の構造、 NMRデータ及び M Sデータを表 1に示す。
[0116] 実施例 12
N— [4— [4—メチルー 2—[ (5—チォー 13 D ダルコピラノシル)ォキシ]ベンジ ル]フエ-ル]ゥレアの合成
[0117] [化 34]

(1) 5—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー β D—ダルコビラノシドの合成
2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 D—ダルコビラノース(2. 24g、 6. 16mmol)と 5—メチル 2— (4 -トロベンジル)フエノール(1. 0g、 4. l lmmol)、 トリフエ-ルホスフィン(1. 62g、 6. 16mmol)、ジイソプロピルァゾジカルボキシレー ト(3. l lmL、 6. 16mmol)を用い、参考例 8の(1)に示した方法と同様の方法で表 題化合物(0. 56g、 23%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.93 (s, 3 H) 2.02 (s, 3 H) 2.04 (s, 6 H) 2.37 (s, 3 H) 3.23 - 3.31 (m, 1 H) 3.94 (d, J=3.42 Hz, 2 H) 4.09 (dd, J=12.12, 3.73 Hz, 1 H) 4.29 (dd, J=11.97, 5.13 Hz, 1 H) 5.12 - 5.21 (m, 1 H) 5.31 - 5.40 ( m, 2 H) 5.49 - 5.56 (m, 1 H) 6.84 (d, J=8.55 Hz, 1 H) 6.90 (s, 1 H) 7.01 (d, J=7.77
Hz, 1 H) 7.29 - 7.34 (m, 2 H) 8.08 - 8.14 (m, 2 H).
ESI m/z = 612 (M+Na).
(2) 2— (4 ァミノベンジル) 5—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチ ルー 5—チォー 13 D—ダルコビラノシドの合成
5—メチル 2— (4 -トロベンジル)フエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォー 13 D—ダルコビラノシド(0. 56g、 0. 949mmol)とパラジウム 活性 炭素(10%) (0. 28g)を用い、参考例 8の(2)に示した方法と同様の方法で表題ィ匕 合物(171mg、 32%)を得た。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.94 (s, 3 H) 2.02 (s, 3 H) 2.04 (s, 3 H) 2.06 (s, 3 H) 2.33 (s, 3 H) 3.22 - 3.31 (m, 1 H) 3.76 (s, 2 H) 4.13 (dd, J=11.8
9, 3.50 Hz, 1 H) 4.32 (dd, J=11.81, 5.13 Hz, 1 H) 5.16 (t, J=9.25 Hz, 1 H) 5.29 - 5
.44 (m, 2 H) 5.60 (t, J=8.86 Hz, 1 H) 6.64 (d, J=8.39 Hz, 2 H) 6.78 (d, J=7.62 Hz, 1 H) 6.87 - 7.00 (m, 4 H).
ESI m/z = 582 (M+Na).
(3) N— [4— [4—メチル 2— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ]ベン ジル]フ -ル]ゥレアの合成
2— (4 ァミノベンジル) 5—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル —5 チォ一 13— D—ダルコビラノシド(171mg、 0. 305mmol)とピリジン(0. 033 mLゝ 0. 412mmol)、クロロギ酸 4 トロフエ-ル(71mgゝ 0. 351mmol)、アンモ
ユア(2M メタノール溶液)(1. 53mL、 3. 06mmol)、ナトリウムメトキシド(25% メ タノール溶液、 0. 26mL、 1. 22mmol)を用い、実施例 3に示した方法と同様の方法 で表題ィ匕合物(67mg、 50%)を得た。得られた化合物の構造、 NMRデータ及び M Sデータを表 1に示す。
[0118] 実施例 13
(3E)— N— (3 アミノー 3—ォキソプロピル) 4— [4— [4 ヒドロキシ一 2—メチ ルー 6— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタ一 3—ェンアミドの合成
[0119] [化 35]

(1) 4— [4— [ (1E)— 4— [ (3 ァミノ一 3—ォキソプロピル)ァミノ] 4 ォキソブタ — 1—ェン一 1—ィル }ベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセ チルー 5—チォー j8—D ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート の合成
参考例 11の(2)で得られた(3E)— 4 [4 [4 [ (メトキシカルボニル)ォキシ] 2—メチル 6— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 j8—D—ダルコ ピラノシル)ォキシ]ベンジル]フエ-ル]ブタ一 3 エノイツクアシッド(360mg、 0. 51 2mmol)のクロ口ホルム(5mL)溶液に j8—ァラニンアミド塩酸塩(128mg、 1. 02m mol)、 1—ヒドロキシベンゾトリアゾール(86mg、 0. 564mmol)、 1—ェチル 3— ( 3 ジメチルァミノプロピル)カルボジイミド塩酸塩(137mg、 0. 717mmol)、トリェチ ルァミン(0. 36mL、 2. 56mmol)をカ卩え、ー晚攪拌した。反応液に水を加え、クロ口 ホルムで抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾 燥した。乾燥剤を炉別後、溶媒を減圧下留去し、得られた残渣をシリカゲルカラムク 口マトグラフィ—(クロ口ホルム:メタノール = 50 : 1〜: L0 : 1)で精製し、無色粉末として 表題ィ匕合物(240mg、 61%)を得た。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.75 (s, 3 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.19 (s, 3 H) 2.42 - 2.50 (m, 2 H) 3.06 - 3.14 (m, 2 H) 3.21 - 3.
31 (m, 1 H) 3.49 - 3.59 (m, 2 H) 3.81 - 4.06 (m, 5 H) 4.11 (dd, J=12.16, 4.04 Hz, 1 H) 4.29 (dd, J=12.16, 5.52 Hz, 1 H) 5.07 - 5.39 (m, 3 H) 5.48 - 5.59 (m, 1 H) 5.67 (brs, 1 H) 6.11 - 6.25 (m, 1 H) 6.34 (brs, 1 H) 6.41 - 6.51 (m, 1 H) 6.74 - 6.78 (m, 1 H) 6.88 - 6.92 (m, 1 H) 6.94 - 7.00 (m, 2 H) 7.19 - 7.25 (m, 2 H).
ESI m/z = 795 (M+Na).
(2) (3E)— N— (3 ァミノ一 3—ォキソプロピル) 4— [4— [4 ヒドロキシ一 2—メ チル 6— [ ( 5—チォ— β — Ό—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタ 3—ェンアミドの合成
4— [4— [ ( 1E)— 4— [ (3 ァミノ一 3—ォキソプロピル)ァミノ] 4 ォキソブタ 1—ェン一 1—ィル }ベンジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 Ο ァセチ ルー 5 チォー j8— D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(1 20mg、0. 155mmol)とナトリウムメトキシド(25%メタノーノレ溶液; 0. 14mL、 0. 62 lmmol)を用い、実施例 3に示す方法と同様の方法で表題ィ匕合物(55mg、 65%)を 合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 13と同様な方法で、対応するァミンを用い、 "化合物 25"乃至"ィ匕合物 29"を 合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0120] 実施例 14
N— (3 アミノー 3—ォキソプロピル) 4— [4— [4 ヒドロキシ一 2 メチル 6— [ (5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミドの合 成
[0121] [化 36]

( 1) 4— [4— [4— [ (3 ァミノ一 3—ォキソプロピル)ァミノ]—4—ォキソブチル }ベン
ジル) 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチル一 5—チォ一 j8— D— ダルコピラノシル)ォキシ]フエ-ル メチル カルボナートの合成
実施例 13の( 1)で得られた 4—[4—[ (1Ε)—4—[ (3 ァミノ 3 ォキソプロピル )ァミノ] 4 ォキソブタ 1—ェン一 1—ィル]ベンジル] 3 メチル 5— [ ( 2, 3 , 4, 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ- ル メチル カルボナート(120mg、 0. 155mmol)とパラジウム 活性炭素(10%) ( 40mg)を用い、参考例 11の(3)に示した方法と同様の方法で表題ィ匕合物(80mg、 67%)を合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.76 (s, 3 H) 1.84 - 1.94 (m, 2 H) 1.97 (s, 3 H) 2.03 (s, 3 H) 2.06 (s, 3 H) 2.13 (t, J=7.38 Hz, 2 H) 2.20 (s, 3 H) 2.39 - 2.49 (m, 2 H) 2.57 (t, J=7.38 Hz, 2 H) 3.18 - 3.33 (m, 1 H) 3.44 - 3.58 (m, 2 H) 3.79 - 4.18 (m, 6 H) 4.25 - 4.34 (m, 1 H) 5.06 - 5.38 (m, 3 H) 5.49 - 5.57 (m, 1 H) 5.73 (brs, 1 H) 6.20 (brs, 1 H) 6.74 - 6.77 (m, 1 H) 6.88 - 6.91 (m, 1 H) 6.91 - 6.9 6 (m, 2 H) 6.98 - 7.04 (m, 2 H).
ESI m/z = 797 (M+Na).
(2) N— (3 アミノー 3—ォキソプロピル) 4— [4— [4 ヒドロキシ一 2 メチル 6 — [ (5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミドの 合成
4 [4 [4一 [ (3—アミノー 3—ォキソプロピル)ァミノ]—4—ォキソブチル]ベンジ ル]— 3—メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチル一 5—チォ一 j8—D—グ ルコピラノシル)ォキシ]フエ-ル メチル カルボナート(80mg、 0. 103mmol)とナト リウムメトキシド(25%メタノール溶液; 0. 090mL、 0. 412mmol)を用い、実施例 3 に示した方法と同様の方法で表題ィ匕合物(55mg、 97%)を合成した。得られた化合 物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 14と同様な方法で、対応するァミンを用い、 "ィ匕合物 30"乃至"ィ匕合物 32"を 合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 15
2— [4— [ (1E)— 3— [1, 3 ジォキソ一 1, 3 ジヒドロ一 2H—イソインドール一 2
—ィル]プロパ 1 ェン一 1—ィル]ベンジル] 5 -ヒドロキシ一 3 メチルフエ-ル 5 チォー β D—ダルコビラノシドの合成
[化 37]

(1) 4— [4— [ (1E)— 3— (1, 3 ジォキソ一 1, 3 ジヒドロ一 2Η—イソインドール —2—ィル)プロパー 1—ェン— 1—ィル]ベンジル]—3—メチル—5— [ (2, 3, 4, 6 —テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ルメチ ルカルボナートの合成
参考例 11の(1)で得られた 4— (4 ブロモベンジル)—3—メチル—5— [ (2, 3, 4 , 6—テトラ一 Ο ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(180mg、 0. 258mmol)、 2 ァリル— 1H—イソインドール — 1, 3 (2H)—ジオン(116mg、 0. 619mmol)、酢酸パラジウム(Π) (12mg、 0. 05 16mmol)、トリ— O トリルホスフィン(31mg、 0. 103mmol)、トリェチルァミン(0. 1 8mL、 1. 29mmol)を用い、参考例 11の(2)に示した方法と同様の方法で表題化 合物(190mg、 92%)を合成した。
1H NMR (300 MHz, CHLOROFORM— d) δ ppm 1.71 (s, 3 H) 1.95 (s, 3 H) 2.02 (s, 3 H) 2.05 (s, 3 H) 2.15 (s, 3 H) 3.20 - 3.31 (m, 1 H) 3.79 - 4.04 (m, 5 H) 4.10 (dd, J=11.89, 3.65 Hz, 1 H) 4.29 (dd, J=11.89, 5.21 Hz, 1 H) 4.39 - 4.45 (m, 2 H) 5.06 - 5.15 (m, 1 H) 5.21 - 5.39 (m, 2 H) 5.48 - 5.57 (m, 1 H) 6.11 - 6.24 (m, 1 H) 6.55 - 6.64 (m, 1 H) 6.72 - 6.76 (m, 1 H) 6.87 - 6.97 (m, 3 H) 7.17 - 7.24 (m, 2 H) 7.6 9 - 7.74 (m, 2 H) 7.83 - 7.88 (m, 2 H).
ESI m/z = 826 (M+Na).
(2) 2— [4— [ (IE)— 3— (1, 3 ジォキソ一 1, 3 ジヒドロ一 2H—イソインドール - 2 ィル)プロパ 1 ェン 1—ィル]ベンジル] 5 -ヒドロキシ - 3 メチルフエ- ル 5—チォー β D ダルコビラノシドの合成
4-[4-[(1Ε)-3-(1, 3 ジォキソ一 1, 3 ジヒドロ一 2H—イソインドーノレ一 2 —ィル)プロパー 1—ェン— 1—ィル]ベンジル]—3—メチル—5— [(2, 3, 4, 6—テ トラ一 O ァセチル一 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ルメチル カルボナート(190mg、 0.236mmol)とナトリウムメトキシド(25%メタノール溶液; 0 .21mL、0.945mmol)を用い、参考例 11の(4)に示した方法と同様の方法で表題 化合物(75mg、 55%)を合成した。得られた化合物の構造、 NMRデータ及び MS データを表 1に示す。
また、 N ァリル N, - (2—ヒドロキシ— 1, 1—ジメチルェチル)ウレァを用いて、実 施例 15と同様な方法で、化合物 34を合成した。
ジベンジル [(Z) (ァリルァミノ)メチルイリデン]ビス力ルバメ―トを用いて、実施例 15 ( 1)の Heck反応につづいて、参考例 11 (3)の接触水素添カ卩により、化合物 35を合 成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 16
N— [2— [ ( 2 ヒドロキシェチル)ァミノ] 1 , 1 ジメチル -2-ォキソェチル] - 4 — [4— [4 ヒドロキシ一 2—メチル 6— [(5—チォ一 13—D—ダルコピラノシル)ォ キシ]ベンジル]フエ-ル]ブタンアミドの合成
[化 38]

(1) 2-[[4-[(1Ε)-4-[[1, 1—ジメチル一 2—ォキソ 2—(フエ-ルメトキシ) ェチル]ァミノ]—4—ォキソ—1—ブテュル]フエ-ル]メチル ]—5 [(メトキシカルボ -ル)ォキシ ]—3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォ一 β -D-ダルコビラノシドの合成
参考例 11の(2)で得られた(3Ε)—4 [4 [4 [(メトキシカルボニル)ォキシ] 2—メチル 6— [(2, 3, 4, 6—テトラ一 Ο ァセチルー 5 チォ一 j8—D—ダルコ ピラノシル)ォキシ]ベンジノレ]フエ-ノレ]ブタ一 3 エノイツクアシッド(1.15g、 1.63
mmol)とべンジル 2—メチルァラ-ナート(629mg、 3. 26mmol)、 1—ヒドロキシべ ンゾトリアゾール(374mg、 2. 44mmol)、 1—ェチル—3— (3 ジメチルァミノプロピ ル)カルボジイミド塩酸塩(53 lmg、 2. 77mmol)、トリェチルァミン(1. 13mL、 8. 1 4mmol)を用い、実施例 13の(1)に示した方法と同様の方法で表題ィ匕合物(980m g、 69%)を合成した。
IH NMR (300 MHz, CHLOROFORM- d) δ ppm 1.56 (s, 6 H) 1.75 (s, 3 H) 1.96 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.19 (s, 3 H) 3.06 - 3.12 (m, 2 H) 3.21 - 3.30 (m,
1 H) 3.82 - 3.90 (m, 1 H) 3.93 (s, 3 H) 3.96 - 4.06 (m, 1 H) 4.11 (dd, J=11.97, 3.65 Hz, 1 H) 4.30 (dd, J=11.97, 5.21 Hz, 1 H) 5.08 - 5.12 (m, 1 H) 5.15 (s, 2 H) 5.23
- 5.38 (m, 2 H) 5.55 (t, J=8.86 Hz, 1 H) 6.12 - 6.24 (m, 2 H) 6.46 (d, J=16.32 Hz,
1 H) 6.76 (d, J=2.33 Hz, 1 H) 6.91 (d, J=2.33 Hz, 1 H) 6.95 - 7.00 (m, 2 H) 7.18 -
7.24 (m, 2 H) 7.31 - 7.34 (m, 5 H).
ESI m/z = 900 (M+Na).
(2) 2— [ [4 [4 [ ( 1 カルボキシ 1 メチルェチル)ァミノ] 4 ォキソブチル ]フエ-ル]メチル] 5 [ (メトキシカルボ-ル)ォキシ] 3 メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5—チォ一 j8—D—ダルコビラノシドの合成
2— [ [4— [ (1E)— 4— [ [1, 1—ジメチルー 2—ォキソ 2— (フエ-ルメトキシ)ェ チル]ァミノ]ー4 ォキソ 1ーブテュル]フエ-ル]メチル ] 5 [ (メトキシカルボ- ル)ォキシ ] 3 メチルフエ-ル 2, 3, 4, 6—テトラー O ァセチル 5 チォ j8 D ダルコビラノシド(980mg、 1. 12mmol)とパラジウム 活性炭素(10%) (24 5mg)を用い、参考例 11の(3)に示した方法と同様の方法で表題ィ匕合物(850mg、 96%)を合成した。
IH NMR (300 MHz, CHLOROFORM— d) δ ppm 1.55 (s, 3 H) 1.56 (s, 3 H) 1.81 (s, 3 H) 1.90 - 1.95 (m, 2 H) 1.97 (s, 3 H) 2.04 (s, 3 H) 2.05 (s, 3 H) 2.16 (t, J=7.38
Hz, 2 H) 2.22 (s, 3 H) 2.60 (t, J=7.31 Hz, 2 H) 3.21 - 3.29 (m, 1 H) 3.83 - 3.91 (m, 1 H) 3.93 (s, 3 H) 3.93 - 4.01 (m, 1 H) 4.08 - 4.15 (m, 1 H) 4.24 - 4.33 (m, 1 H) 5
.11 (t, J=9.17 Hz, 1 H) 5.22 - 5.39 (m, 2 H) 5.51 (t, J=8.78 Hz, 1 H) 6.03 (brs, 1 H
) 6.76 (d, J=2.33 Hz, 1 H) 6.88 (d, J=2.33 Hz, 1 H) 6.93 - 7.05 (m, 4 H).
ESI m/z = 812 (M+Na).
(3) N— [2— [ (2 ヒドロキシェチル)ァミノ] 1, 1 ジメチル - 2-ォキソェチル] - 4 [4 [4ーヒドロキシー 2—メチノレー 6— [ (2, 3, 4, 6—テトラー O ァセチノレー 5 チォー 13 D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミドの合成
2— [ [4— [4— [ ( 1 力ノレボキシ 1 メチノレエチノレ)ァミノ] 4 ォキソブチル] フエ-ル]メチル] 5 [ (メトキシカルボ-ル)ォキシ] 3 メチルフエ-ル 2, 3, 4 , 6—テトラ一 O ァセチノレ 5 チォ一 j8— D—ダルコビラノシド(208mg、 0. 263 mmol)の N, N ジメチルホルムアミド(1. 3mL)溶液にエタノールァミン(0, 032m L、0. 526mmol)、 1—ヒドロキシベンゾトリアゾール(80mg、 0. 526mmol)、 1—ェ チルー 3—(3 ジメチルァミノプロピル)カルボジイミド塩酸塩(101mg、 0. 526mm ol)を加え、一晩攪拌した。反応液に水を加え、クロ口ホルムで抽出した。有機層を水 、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を炉別後、 溶媒を減圧下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホル ム:メタノール= 20 : 1〜5 : 1)で精製し、無色粉末として表題ィ匕合物(64mg、 31%) を合成した。
1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.50 (s, 6 H) 1.76 (s, 3 H) 1.80 - 1.94 (m, 2 H) 1.96 (s, 3 H) 2.03 (s, 3 H) 2.04 (s, 3 H) 2.08 - 2.16 (m, 5 H) 2.54 (t, J=7.15 Hz, 2 H) 3.25 - 3.42 (m, 3 H) 3.69 (t, J=4.90 Hz, 2 H) 3.74 - 3.83 (m, 1 H) 3.86 - 3.95 (m, 1 H) 4.10 (dd, J=11.85, 3.81 Hz, 1 H) 4.28 (dd, J=11.85, 5.13 Hz, 1 H) 5.14 (t, J=9.17 Hz, 1 H) 5.28 - 5.38 (m, 2 H) 5.50 (t, J=8.70 Hz, 1 H) 6.18 (s, 1 H) 6.41 (d, J=2.18 Hz, 1 H) 6.56 - 6.63 (m, 2 H) 6.72 (t, J=6.06 Hz, 1 H) 6.90 - 7. 00 (m, 4 H).
ESI m/z = 775 (M+H).
(4) N— [2— [ ( 2 ヒドロキシェチル)ァミノ] 1 , 1 ジメチル - 2-ォキソェチル] - 4— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 13—D グノレコピラノシノレ)ォ キシ]ベンジル]フエ-ル]ブタンアミドの合成
Ν— [2— [ ( 2 ヒドロキシェチル)ァミノ] 1 , 1 ジメチル - 2-ォキソェチル] - 4 — [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (2, 3, 4, 6—テトラ一 Ο ァセチノレ一 5—
チォ一 β—D—ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミド(64mg、 0 . 0826mmol)とナトリウムメトキシド(25%メタノーノレ溶液; 0. 072mL, 0. 330mmo 1)を用い、参考例 11の (4)に示した方法と同様の方法で表題ィ匕合物(50mg、 99%) を合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。 また、実施例 16 (2)で得た 2— [ [4— [4 [ (1一力ノレボキシ 1ーメチノレエチノレ)ァ ミノ ] 4 ォキソブチル]フエ-ル]メチル] 5— [ (メトキシカルボ-ル)ォキシ] 3 —メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォ一 j8— D—グルコピ ラノシドを参考例 11の (4)に示した方法と同様の方法で脱保護し、化合物 37を得た o得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 17
N— [4— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 β—D グノレコピラノ シル)ォキシ]ベンジル]フエ-ル]ブタノィル] 2—メチルァラ-ルグリシンアミドの合 成
[化 39]

(1) N— [4 [4 [4 [ (メトキシカルボニル)ォキシ] 2—メチルー 6— [ (2, 3, 4 , 6—テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル }フエ-ル)ブタノィル] 2—メチルァラ-ルグリシンアミドの合成
実施例 16の(2)で得られた 2— [ [4— [4 [ (1一力ノレボキシ 1ーメチノレエチノレ) ァミノ]ー4ーォキソブチル]フエ-ル]メチル ] 5— [ (メトキシカルボ-ル)ォキシ] 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォ一 j8— D—ダルコ ビラノシド(208mg、 0. 263mmol)の N, N ジメチルホルムアミド(1. 3mL)溶液に グリシンアミド塩酸塩(58mg、 0. 526mmol)、 1—ヒドロキシベンゾトリアゾール(80 mg、0. 526mmol)、 1—ェチル—3— (3 ジメチルァミノプロピル)カルボジイミド塩 酸塩(101mg、 0. 526mmol)、トリェチルァミン(0. l lmL、 0. 789mmol)を加え、
一晩攪拌した。反応液に水を加え、クロ口ホルムで抽出した。有機層を水、飽和食塩 水で洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を炉別後、溶媒を減圧 下留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム:メタノー ル= 20 : 1〜10 : 1)で精製し、無色粉末として表題ィ匕合物(110mg、 49%)を合成し た。
1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.49 (s, 6 H) 1.84 (s, 3 H) 1.86 - 1.94 (m, 2 H) 1.96 (s, 3 H) 2.04 (s, 3 H) 2.05 (s, 3 H) 2.14 (t, J=7.62 Hz, 2 H) 2.22 (s, 3 H) 2.58 (t, J=7.15 Hz, 2 H) 3.21 - 3.30 (m, 1 H) 3.82 - 4.00 (m, 7 H) 4.08 - 4 .15 (m, 1 H) 4.27 (dd, J=11.97, 5.44 Hz, 1 H) 5.07 - 5.15 (m, 1 H) 5.23 - 5.37 (m, 3 H) 5.50 (t, J=8.55 Hz, 1 H) 6.01 (brs, 1 H) 6.54 - 6.61 (m, 1 H) 6.76 (d, J=2.10 H z, 1 H) 6.87 (d, J=2.10 Hz, 1 H) 6.93 - 7.04 (m, 4 H) 7.21 (brs, 1 H).
ESI m/z = 868 (M+Na).
(2) N— [4— [4— [4 ヒドロキシ一 2—メチル 6— [ (5 チォ一 j8—D—ダルコビラ ノシル)ォキシ]ベンジル]フエ-ル]ブタノィル] 2—メチルァラ-ルグリシンアミドの 合成
^^ [4 [4 [4ー[ (メトキシカルボ-ル)ォキシ]ー2—メチルー6—[ (2, 3, 4, 6 —テトラ一 O ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]ベンジル]フ ェ -ル]ブタノィル]—2—メチルァラ-ルグリシンアミド(110mg、 0. 130mmol)とナト リウムメトキシド(25%メタノール溶液; 0. 113mL、 0. 520mmol)を用い、参考例 11 の(4)に示した方法と同様の方法で表題ィ匕合物(74mg、 92%)を合成した。得られ た化合物の構造、 NMRデータ及び MSデータを表 1に示す。
[0126] 実施例 18
メチノレ 4— [N— [4— [4— [4—ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 13— D— ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタノィル] 2—メチルァラ -ル]ピペラ ジン 1 カルボキシラートの合成
[0127] [化 40]

(1) 4— [4— [4— [ (1, 1 -ジメチル - 2-ォキソ 2 ピぺラジン 1 ィルェチル) ァミノ] —4—ォキソブチル]ベンジル]—3—メチル 5— [ (2, 3, 4, 6—テトラ一 O —ァセチル一 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ルメチルカルボ ナートの合成
実施例 16の(2)で得られた 2— [ [4— [4 [ (1一力ノレボキシ 1ーメチノレエチノレ) ァミノ]ー4ーォキソブチル]フエ-ル]メチル ] 5— [ (メトキシカルボ-ル)ォキシ] 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォ一 j8— D—ダルコ ビラノシド(186mg、 0. 235mmol)とピペラジン無水物(41mg、 0. 471mmol)、 1 —ヒドロキシベンゾトリアゾール(72mg、 0. 471mmol)、 1—ェチル—3— (3 ジメ チルァミノプロピル)カルボジイミド塩酸塩(90mg、 0. 471mmol)、トリェチルアミン( 0. 098mL、 0. 705mmol)を用い、実施例 17の(1)に示した方法と同様の方法で 表題ィ匕合物(30mg、 15%)を合成した。
1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.55 (s, 6 H) 1.77 (s, 3 H) 1.82 - 1.94 (m, 2 H) 1.96 (s, 3 H) 2.03 (s, 3 H) 2.04 (s, 3 H) 2.07 - 2.17 (m, 5 H) 2.56 (t, J=7.07 Hz, 2 H) 3.25 - 3.35 (m, 1 H) 3.41 - 3.48 (m, 4 H) 3.58 - 3.67 (m, 4 H) 3.7 1 (s, 3 H) 3.75 - 3.84 (m, 1 H) 3.86 - 3.95 (m, 1 H) 4.10 (dd, J=11.93, 3.73 Hz, 1 H) 4.29 (dd, J=11.93, 5.21 Hz, 1 H) 5.15 (t, J=9.17 Hz, 1 H) 5.29 - 5.40 (m, 2 H) 5 .52 (t, J=8.78 Hz, 1 H) 6.43 (d, J=2.10 Hz, 1 H) 6.48 (s, 1 H) 6.62 (d, J=2.10 Hz, 1 H) 6.90 - 7.02 (m, 4 H).
ESI m/z = 880 (M+Na).
(2)メチル 4— [N— [4— [4— [4 ヒドロキシ一 2—メチル 6— [ (5 チォ一 j8— D -ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタノィル] 2—メチルァラ -ル]ピ ペラジン 1 カルボキシラートの合成
4— [4— [4— [ (1, 1—ジメチル - 2-ォキソ 2 ピぺラジン 1 ィルェチル)ァ
ミノ] —4—ォキソブチル]ベンジル ]— 3—メチル—5— [ (2, 3, 4, 6—テトラ一 O ァ セチル 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ルメチルカルボナート (30mg、 0. 0350mmol)とナトリウムメ卜キシド(25%メタノール溶液; 0. 030mL、 0 . 140mmol)を用い、参考例 11の(4)に示した方法と同様の方法で表題化合物(20 mg、 83%)を合成した。得られた化合物の構造、 NMRデータ及び MSデータを表 1 に示す。
[0128] 実施例 19
N— [2— [4— (2 ヒドロキシェチル)ピぺラジン 1 ィル] 1, 1 ジメチル - 2 —ォキソェチノレ]— 4— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 13— D— ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミドの合成
[0129] [化 41]

( 1) 4一 [4一 [4一 [[2— [4一 (2 ヒドロキシェチル)ピぺラジン一 1一ィル] —1, 1一 ジメチル 2 ォキソェチル]ァミノ] 4 ォキソブチル]ベンジル] 3 メチル 5 — [ (2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 13—D—ダルコピラノシル)ォキ シ]フエニルメチルカルボナートの合成
実施例 16の(2)で得られた 2— [ [4— [4 [ (1一力ノレボキシ 1ーメチノレエチノレ) ァミノ]ー4ーォキソブチル]フエ-ル]メチル ] 5— [ (メトキシカルボ-ル)ォキシ] 3—メチルフエ-ル 2, 3, 4, 6—テトラ一 O ァセチル 5 チォ一 j8— D—ダルコ ビラノシド(186mg、 0. 235mmol)と 1—ピぺラジンエタノール(61mg、 0. 471mm ol)、 1—ヒドロキシベンゾトリアゾール(72mgゝ 0. 471mmol)、 1—ェチル 3— (3 ージメチルァミノプロピル)カルボジイミド塩酸塩(90mg、 0. 471mmol)、トリェチル ァミン(0. 098mL、 0. 705mmol)を用い、実施例 17の(1)に示した方法と同様の 方法で表題化合物(106mg、 50%)を合成した。
1H NMR (300 MHz, CHLOROFORM- d) δ ppm 1.60 (s, 6 H) 1.75 (s, 3 H) 1.82 -
1.95 (m, 2 H) 1.96 (s, 3 H) 2.03 (s, 3 H) 2.05 (s, 3 H) 2.08 - 2.17 (m, 5 H) 2.43 - 2
.62 (m, 8 H) 3.22 - 3.32 (m, 1 H) 3.49 (s, 3 H) 3.59 - 3.73 (m, 6 H) 3.74 - 3.83 (m, 1 H) 3.88 - 3.97 (m, 1 H) 4.12 (dd, J=11.93, 3.81 Hz, 1 H) 4.30 (dd, J=11.93, 4.97 Hz, 1 H) 5.13 (t, J=9.17 Hz, 1 H) 5.26 - 5.40 (m, 2 H) 5.53 (t, J=8.78 Hz, 1 H) 6.4
0 (d, J=2.26 Hz, 1 H) 6.57 (s, 1 H) 6.60 (d, J=2.26 Hz, 1 H) 6.89 - 7.03 (m, 4 H).
ESI m/z = 902 (M+H).
(2) N— [2— [4— (2 ヒドロキシェチル)ピぺラジン 1—ィル] 1, 1 ジメチル - 2—ォキソェチノレ]— 4— [4— [4 ヒドロキシ一 2—メチノレ一 6— [ (5—チォ一 13— D -ダルコピラノシル)ォキシ]ベンジル]フエ-ル]ブタンアミドの合成
4 [4 [4 [[2— [4一(2 ヒドロキシェチル)ピぺラジン 1 ィル] —1, 1ージ メチル 2 ォキソェチル]ァミノ] 4 ォキソブチル]ベンジル] 3 メチル 5— [ (2, 3, 4, 6—テトラ一 O ァセチルー 5 チォ一 13—D—ダルコピラノシル)ォキシ] フエ-ルメチルカルボナート(106mg、 0. 118mmol)とナトリウムメトキシド(25%メ タノール溶液; 0. 103mL、0. 472mmol)を用い、参考例 11の(4)に示した方法と 同様の方法で表題ィ匕合物(67mg、 84%)を合成した。得られたィ匕合物の構造、 NM Rデータ及び MSデータを表 1に示す。
実施例 22
N— [4— [2 ェチノレ一 4 ヒドロキシ一 6— [ (5—チォ一 β—D グノレコピラノシ ル)ォキシ]ベンジル]フエ-ル]尿素の合成
[化 42]

参考例 17で合成した 4 (4ーァミノべンジル)ー3 ェチルー 5—[ (2, 3, 4, 6— テトラー Ο ァセチルー 5—チォー 13 D—ダルコピラノシル)ォキシ]フエ-ル メチ ル カーボナートを用い、実施例 3同様の方法で表題ィ匕合物(0. 076g、 82%)を得 た。得られた化合物の構造、 NMRデータ及び MSデータを表 1に示す。
実施例 23
2- [4- (4, 4—ジメチル一 2, 5—ジォキソイミダゾリジン一 1—ィル)ベンジル]—5 —ヒドロキシ一 3—メチルフエ-ル 5—チォ一 β—D—ダルコビラノシドの合成
[0131] [化 43]

参考例 8の(2)で得られた 4ー(4ーァミノべンジル)ー3—メチルー 5— [ (2, 3, 4, 6—テトラ一 Ο—ァセチルー 5—チォ一 13—D—ダルコピラノシル)ォキシ]フエ-ル メチル カルボナート(200mg、 0. 316mmol)のクロ口ホルム(8mL)溶液にピリジン (0. 034mL、 0. 426mmol)、クロロギ酸 4— -トロフエ-ル(73mg、 0. 363mmol )を加え、室温で 30分間攪拌した。トリェチルァミン(0. 88mL、 0. 631mmol)と 2— ァミノイソプチリックアシッド(35mg)を加え、室温で 2時間攪拌した。反応液を減圧下 留去し、得られた残渣にメタノール(2mL)とナトリウムメトキシド(25%メタノール溶液 ;0. 27mL、 1. 26mmol)をカ卩え、室温で 2時間攪拌した。反応液にドライアイスを加 え中和し、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー( 酢酸ェチル:エタノール:水 = 20 : 2 : 1〜5: 2 : 1)で精製し、酢酸ェチルで洗浄し、無 色粉末として表題化合物 (78mg、 48%)を得た。得られたィ匕合物の構造、 NMRデ ータ及び MSデータを表 1に示す。
[0132] [表 1-1]



uεΐ εΐοι
 H]
H]

/ £6so/-ooifcv:l20Z-U O9zi-0SAV

/ £6so/-ooifcv:l>d-SU O9Z/-00ZAV
s I


£6S0.0v:0zfcl£ εΖ /-Π9ίϊ/ O£00ίAV


/寸 6£6so/-oov:zfcl£寸 Z-Π9/ Oίϊ/-00ίAV
〔〔〕ΐ0H0-

1-10]


[0143] [表 2] 薬物含量 lOOmg錠剤の処方:
1錠中含量:
薬物 108.35 mg
乳糖一水和物 38.65 mg
結晶セルロース 22.00 mg
カルポキシメチルセルロースカルシウム 20.00 mg
ヒドロキシプロピルセルロース 10.00 mg
ステアリン酸マグネシウム 1-00 mg
製造方法
薬物 (本発明化合物)を乳糖一水和物、結晶セルロース、カルボキシメチルセル口 —スカルシウム及びヒドロキシプロピルセルロースと混合し、この混合物を粉砕機で粉 砕する。粉砕された混合物を撹拌造粒機で 1分間混合し、その後、水で 4〜8分間造 粒する。得られた造粒物を 70°C、 40分間乾燥する。造粒乾燥末を 500 mの篩で 篩過する。篩過後の造粒乾燥末とステアリン酸マグネシウムを、 V型混合機を用いて 30rpmで 3分間混合する。口—タリ—式打錠機を用いて得られた打錠用顆粒を圧縮 成形し製錠する。
[0144] [表 3] 錠剤重量: 200mg
錠径 : 8 mm, 円形 試験例 1
(1)ヒト SGLT1とヒト SGLT2のクローユングと発現ベクターへの導入
ヒト小腸由来 mRNAからヒト SGLT1配列(NM— 000343)を逆転写の後増幅し、 pCMV— tag5A (ストラタジーン社)に導入した。また、ヒト SGLT2配列(NM— 003 041)はヒト腎由来 mRNAから同様な方法で調製し、 pcDNA3. 1 +hygro (インビト ロジェン社)に導入した。それぞれのクローンの配列力 報告されている配列と一致 することを確認した。
(2)ヒト SGLT1及びヒト SGLT2を安定に発現する CHO— kl細胞の作成
ヒト SGLT1およびヒト SGLT2発現ベクターを、リポフエクトァミン 2000 (インビトロジ ェン社)を用いて CHO—K1細胞へトランスフエクシヨンした。 SGLT発現細胞は、 50 0 μ gZmLの濃度のジエネティシン(SGLT1)またはハイグロマイシン B (SGLT2)の 存在下で培養し耐性株を選択し、下記に示す系により糖取り込み比活性を指標に取 得した。
(3)細胞におけるナトリウム依存的糖取り込み阻害試験
ヒト SGLT1又はヒト SGLT2を安定に発現する細胞をナトリウム依存的グルコース取り 込み活性阻害試験に用いた。
[0145] 前処理用緩衝液(140mM 塩化コリン、 2mM KC1、 ImM CaCl、 ImM Mg
2
Cl、 10mM HEPES/5mM Tris、 pH7. 4) 200 Lをヒト SGLT1発現細胞に
2
又は 2mLをヒト SGLT2発現細胞にカ卩え、 20分間インキュベーションした。前処理用 緩衝液を除去し、試験化合物を含む取り込み用緩衝液 ( ["C]メチル a— D—ダル コピラノシドを含むメチル α— D—ダルコビラノシド(lmM)、 145mM NaCl、 2m M KC1、 ImM CaCl、 ImM MgCl、 lOmM HEPES/5mM Tris、 pH7. 4
2 2
)を 75 L (SGLT1の場合)又は 200 μ L (SGLT2の場合)加え、 37°Cにて 30分 (S GLT1の場合)又は 1時間(SGLT2の場合)取り込み反応を行った。反応後細胞を 洗浄用緩衝液(lOmM メチル α— D—ダルコビラノシド、 140mM 塩化コリン 2m M KC1、 ImM CaCl、 ImM MgCl、 lOmM HEPES/5mM Tris、 pH7. 4
2 2
) 200 し(301/11の場合)又は2111し(301^2の場合)で2回洗浄し、0. 2M NaO H溶液 75 μ L (SGLT1の場合)又は 400 μ L (SGLT2の場合)に溶かした。液体シ ンチレーター(パーキンエルマ一社)を加えよく混和した後、 microBETA (SGLTlの 場合)又は液体シンチレーシヨンカウンター(SGLT2の場合)(ベックマンコールター 社)で放射活性を測定した。対照群として試験化合物を含まな!/ヽ取り込み用緩衝液 を調製した。また基礎取り込み用として NaClに代えて塩ィ匕コリンを含む取り込み用緩 衝液を調製した。
[0146] IC 値を求めるにあたり、適当な 6濃度の試験化合物を用い、対照群の糖取り込み
50
量(100%)に対し、糖取り込み量が 50%阻害される試験化合物濃度 (IC 値)を算
50 出した。試験結果を表 4に示す。
[表 4]

試験例 2
ストレブトゾトシン糖尿病モデルラットにおける血糖値上昇抑制作用確認試験 (1)糖尿病モデルラットの作製
7週齢の SDZIGSラット(日本チヤ一ルスリバ一株式会社,雄性)について約 16時 間の絶食後、エーテル麻酔下でストレブトゾトシン (STZ) 50mgZkgを尾静脈内投
与し、糖尿病モデルラットを作製した.同様にエーテル麻酔下, 1. 25mmolZLタエ ン酸生理食塩液 lmLZkgを尾静脈内投与し、正常対照ラットを作製した。 STZまた は 1. 25mmolZLクェン酸生理食塩液投与 1週後(8週齢)、経口グルコース負荷試 験に供した。
(2)経口グルコース負荷試験
ラットを約 16時間の絶食後、薬物投与群には、 0. 5%カルボキシメチルセルロース (CMC)水溶液に懸濁した薬物(lmgZkg)を,対照群には 0. 5%CMC水溶液の み経口投与した。薬物投与 5分後に、グルコース溶液 (2gZkg)を経口投与し、薬物 投与前 (Otime)、及び、経口投与 0. 25、 0. 5、 1, 2時間後の計 5点で採血した。
[0148] 採血は、エーテル麻酔下でラット眼窩静脈洞よりへノ^ンコート採血管を用いて行 い、遠心分離後、血漿を分取した。血漿中グルコース濃度の定量は、グルコース CII テストヮコー (和光純薬株式会社)を用いて測定した。血糖値上昇抑制作用強度は、 各薬物投与群の 0から 1時間までの血糖値より台形法を用いて血糖値-時間曲線下 面積 (AUC)を算出し、 basalを差し引いた血糖増加面積 ( AAUC)として表記し、対照 群のそれに対する降下の割合で表記した。結果を表 5に示す。
[0149] [表 5]

本発明により、小腸上皮に発現する SGLT1 (ナトリウム依存性グルコース共輸送体
1)を阻害し、小腸からの糖吸収を阻害することによって、 IGT (耐糖能異常)を制御 するフ ニル 5—チォダルコシドィ匕合物を有効成分として含有する糖尿病の予防又 は治療剤を提供することが期待される。
Claims
[化 1]

( I ) 式中、
R1及び R2は、同一または異なって、水素原子、水酸基、 C アルキル基、 C アル
1-6 1-6 コキシ基又はハロゲン原子であり、
Zは、 NHSOフエ-ル、 NHSO NH、又は— Y— Qであり、
2 2 2
Yは、単結合、 C アルキレン基又は C ァルケ-レン基であり、
1-6 2-6
Qは、一 NHC ( = NH) NH 、 一 NHCON (RA)RB、 一 CONH (RC)、又は C ァノレ
2 1-6 キル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロアルキル基( 該ヘテロシクロアルキル基はフエ-ル基と縮合してもょ 、)であり、
RA及び RBは、同一または異なって、水素原子、 C アルキル基、 C シクロアルキ
1-6 3-7
ル基、又は水酸基、 -CONH、フエニル基及びピリジル基力 なる群より選択される
2
1〜3個の基で置換された C アルキル基を示し、
1-6
Reは、水酸基、カルボキシル基及び— CONReiRe2力もなる群より選択される 1〜 4個の基で置換された C アルキル基であり、
1-6
Rei及び Re2は、同一または異なって、水素原子または水酸基で置換されても良 V、C アルキル基である力、あるいは Rei及び Re2が結合して 、る窒素原子と一緒にな
1-6
つて、さらに環構成原子として、酸素原子、硫黄原子及び窒素原子から選択されるへ テロ原子を含んでも良 、5〜6員のへテロシクロアルキル基 (該ヘテロシクロアルキル 基は、水酸基で置換されても良い C アルキル基、または C アルコキシカルボ-ル
基で置換されてもょ ヽ)を形成してもよ 、。
[2] R1が、水素原子、水酸基、 C アルキル基又は C アルコキシ基であり、 R2が、 C
1-4 1-4 1-4 アルキル基又はハロゲン原子である、請求項 1記載のフエ-ル 5 チォダルコシド 化合物若しくはその製薬学的に許容される塩又はそれらの水和物。
[3] Zが Y— NHC ( = NH) NH (Yは請求項 1で定義したとおりである)である請求項
2
1または 2記載のフエ-ル 5 チォダルコシド化合物若しくはその製薬学的に許容さ れる塩又はそれらの水和物。
[4] Yが C アルキレン基である請求項 3記載のフエ-ル 5—チォダルコシド化合物若
1-6
しくはその製薬学的に許容される塩又はそれらの水和物。
[5] Zが— Y— NHCON (RA) RB (RA及び Yは請求項 1で定義したとおりであり、 RBは水 素原子である)である請求項 1または 2記載のフエ-ル 5 チォダルコシドィ匕合物若 しくはその製薬学的に許容される塩又はそれらの水和物である。
[6] Zが— Y— CONH (RC) (RCは請求項 1で定義したとおりであり、 Yは C アルキレン
1-6 基又は C ァルケ-レン基である)である請求項 1または 2記載のフエ-ル 5 チォ
2-6
ダルコシド化合物若しくはその製薬学的に許容される塩又はそれらの水和物。
[7] Reが水酸基及び— CO— NReiRe2 (Rei及び Re2は請求項 1で定義したとおりである )からなる群より選択される 1〜4個の基で置換された C アルキル基である、請求項 6
1-6
記載のフエ-ル 5—チォダルコシド化合物若しくはその製薬学的に許容される塩又 はそれらの水和物。
[8] R1及び R2は、同一または異なって、水素原子、水酸基、 C アルキル基、又は C
1-6 1-6 アルコキシ基であり、
Zは、一 NHSOフエ二ノレ、 -NHSO NH 、 一 NHC ( = NH) NH、 -NHCON (R
2 2 2 2
A) RB、又はメチル基及びォキソ基の少なくとも 1種で置換された 4〜6員へテロシクロ アルキル基、又は Υ,— CONH(RC)であり、
RA及び RBは、同一または異なって、水素原子、 C アルキル基、 C シクロアルキ
1-6 3-7
ル基、又は水酸基、 -CONH、フエニル基及びピリジル基力 なる群より選択される
2
1〜3個の基で置換された C アルキル基であり、
1-6
Y,は、 C アルキレン基又は C アルケニレン基を示し、 RCは— CONHで置換さ
れた C アルキル基である、請求項 1記載のフエニル 5—チォダルコシドィ匕合物若し
1-6
くはその製薬学的に許容される塩又はそれらの水和物。
[9] 請求項 1〜8のいずれか 1項に記載のフエニル 5—チォダルコシドィ匕合物若しくは その製薬学的に許容される塩又はそれらの水和物を有効成分として含むナトリウム 依存性グルコース共輸送体 1 (SGLT1)活性阻害剤。
[10] 請求項 1〜8のいずれか 1項に記載のフエニル 5—チォダルコシドィ匕合物若しくは その製薬学的に許容される塩又はそれらの水和物を有効成分として含む糖尿病の 予防又は治療剤。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-128680 | 2006-05-02 | ||
| JP2006128680A JP2009167104A (ja) | 2006-05-02 | 2006-05-02 | フェニル5−チオグリコシド化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007126117A1 true WO2007126117A1 (ja) | 2007-11-08 |
Family
ID=38655635
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/059394 Ceased WO2007126117A1 (ja) | 2006-05-02 | 2007-05-02 | フェニル 5-チオグルコシド化合物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2009167104A (ja) |
| WO (1) | WO2007126117A1 (ja) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2011070592A2 (en) | 2009-12-09 | 2011-06-16 | Panacea Biotec Ltd. | Novel sugar derivatives |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| US8080580B2 (en) | 2008-08-28 | 2011-12-20 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| JP2012518631A (ja) * | 2009-02-23 | 2012-08-16 | 大正製薬株式会社 | Sglt1阻害剤としての4−イソプロピルフェニルグルシトール化合物 |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8669380B2 (en) | 2009-11-02 | 2014-03-11 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9061060B2 (en) | 2008-07-15 | 2015-06-23 | Theracos Inc. | Deuterated benzylbenzene derivatives and methods of use |
| US11084842B2 (en) | 2018-01-23 | 2021-08-10 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivative and use thereof |
| US11186602B2 (en) | 2018-01-31 | 2021-11-30 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivative and use thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014931A1 (ja) * | 2002-08-09 | 2004-02-19 | Taisho Pharmaceutical Co., Ltd. | アリール5−チオ−β−D−グルコピラノシド誘導体及びそれを含有する糖尿病治療薬 |
| JP2005247834A (ja) * | 2004-02-04 | 2005-09-15 | Taisho Pharmaceut Co Ltd | ナトリウム依存性グルコース供輸送体2の活性阻害剤 |
-
2006
- 2006-05-02 JP JP2006128680A patent/JP2009167104A/ja active Pending
-
2007
- 2007-05-02 WO PCT/JP2007/059394 patent/WO2007126117A1/ja not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014931A1 (ja) * | 2002-08-09 | 2004-02-19 | Taisho Pharmaceutical Co., Ltd. | アリール5−チオ−β−D−グルコピラノシド誘導体及びそれを含有する糖尿病治療薬 |
| JP2005247834A (ja) * | 2004-02-04 | 2005-09-15 | Taisho Pharmaceut Co Ltd | ナトリウム依存性グルコース供輸送体2の活性阻害剤 |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US9061060B2 (en) | 2008-07-15 | 2015-06-23 | Theracos Inc. | Deuterated benzylbenzene derivatives and methods of use |
| US8080580B2 (en) | 2008-08-28 | 2011-12-20 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| JP2012518631A (ja) * | 2009-02-23 | 2012-08-16 | 大正製薬株式会社 | Sglt1阻害剤としての4−イソプロピルフェニルグルシトール化合物 |
| US8669380B2 (en) | 2009-11-02 | 2014-03-11 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9439902B2 (en) | 2009-11-02 | 2016-09-13 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9439901B2 (en) | 2009-11-02 | 2016-09-13 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US9308204B2 (en) | 2009-11-02 | 2016-04-12 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| WO2011070592A2 (en) | 2009-12-09 | 2011-06-16 | Panacea Biotec Ltd. | Novel sugar derivatives |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US11084842B2 (en) | 2018-01-23 | 2021-08-10 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivative and use thereof |
| US11186602B2 (en) | 2018-01-31 | 2021-11-30 | Sunshine Lake Pharma Co., Ltd. | Glucopyranosyl derivative and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009167104A (ja) | 2009-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007126117A1 (ja) | フェニル 5-チオグルコシド化合物 | |
| JP5067644B2 (ja) | C−フェニルグリシト−ル化合物 | |
| JP5152519B2 (ja) | C−フェニル1−チオグルシト−ル化合物 | |
| WO2007129668A1 (ja) | ピラゾリル 5-チオグルコシド化合物 | |
| EP2598486B1 (en) | Bicyclic acetyl-coa carboxylase inhibitors | |
| CN106458994B (zh) | 联芳激酶抑制剂 | |
| IL300855A (en) | A nitrile derivative acting as an inhibitor of DIPEPTIDYL PEPTIDASE 1 and its use | |
| CA3094820A1 (en) | Modulators of g-protein coupled receptors | |
| JPWO2008072726A1 (ja) | 1−フェニル 1−チオ−d−グルシト−ル誘導体 | |
| WO2016040223A1 (en) | Cyclobutane containing carboxylic acid gpr120 modulators | |
| WO2007007588A1 (ja) | 平面性を有する環状基を母核とする化合物 | |
| TW202239756A (zh) | 作為組蛋白去乙醯酶6抑制劑之1,3,4-㗁二唑硫羰基化合物及包含該等化合物之醫藥組合物 | |
| JP2009120553A (ja) | C−フェニルグルシト−ル化合物を有効成分とする糖尿病治療剤 | |
| TWI464164B (zh) | 醯胺衍生物 | |
| JP2009107948A (ja) | フェニル5−チオグルコシド化合物を有効成分とする糖尿病治療剤 | |
| JP2009107947A (ja) | ピラゾリル5−チオグルコシド化合物を有効成分とする糖尿病治療剤 | |
| JP2009155212A (ja) | C−フェニル1−チオグルシト−ル化合物を有効成分とする糖尿病治療剤 | |
| JP2010018611A (ja) | 1−フェニル1−チオ−d−グルシト−ル誘導体を有効成分とする糖尿病、糖尿病関連疾患又は糖尿病性合併症の予防又は治療剤。 | |
| WO2023015236A2 (en) | Composition and method for treatment of gram negative bacterial infection | |
| HK40121290A (en) | Nitrile derivative that acts as inhibitor of dipeptidyl peptidase 1 and use thereof | |
| EA051684B1 (ru) | Модуляторы сопряженных с g-белком рецепторов | |
| TW202540100A (zh) | 作為parp1抑制劑之化合物及包含其之醫藥組合物 | |
| HK40012941A (en) | Macrocyclic broad spectrum antibiotics | |
| HK40012941B (en) | Macrocyclic broad spectrum antibiotics | |
| HK1130472B (zh) | C-苯基1-硫代山梨醇化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07742829 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07742829 Country of ref document: EP Kind code of ref document: A1 |