WO2007132478A2 - Process for the preparation of pure risedronic acid or salts - Google Patents
Process for the preparation of pure risedronic acid or salts Download PDFInfo
- Publication number
- WO2007132478A2 WO2007132478A2 PCT/IN2007/000187 IN2007000187W WO2007132478A2 WO 2007132478 A2 WO2007132478 A2 WO 2007132478A2 IN 2007000187 W IN2007000187 W IN 2007000187W WO 2007132478 A2 WO2007132478 A2 WO 2007132478A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phosphorous
- acid
- formula
- risedronic acid
- reaction mass
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- WGNUNYPERJMVRM-UHFFFAOYSA-N OC(Cc1cccnc1)=O Chemical compound OC(Cc1cccnc1)=O WGNUNYPERJMVRM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/55—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/58—Pyridine rings
Definitions
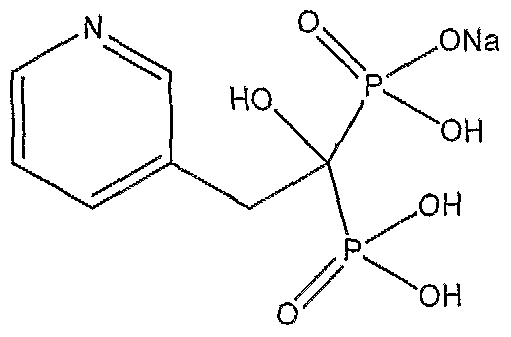
- the present invention relates to a process for the preparation of bisphosphonic acid in particular risedronic acid or its pharmaceutically acceptable salt, useful in the treatment of bone disorders. More particularly, the present invention relates to a novel method for the preparation of risedronic acid, namely, [l-hydiOxy-2(3-pyridinyl)ethylidene] bisphosphonic acid having Formula-I or its salts in high purity and high yield.
- the bisphosponates which are salts of bisphosphonic acids, are an important class of medicaments useful in the treatment of bone disorders such as Paget's disease and osteoporosis.
- the bisphosphonates for example etidronate, pamidronate, and risedronate are used in the form of various non-toxic and pharmaceutically acceptable esters, alkali metal salts and salts of alkaline-earth metals and their various hydrates.
- the form of the substance can have a fundamental influence on its solubility and its biological availability.
- the preferred forms of risedronate are the sodium and calcium salts.
- Risedronic acid chemically known as [l-hydiOxy-2(3-pyridinyl)ethylidene] bisphosphonic acid, presently marketed as risedronate sodium under the tradename Actonel, is an important active pharmaceutical ingredient for the treatment of osteoporosis.
- reaction when the reaction is carried out in chlorobenzene as a diluent, it does not solubilize the reaction components.
- the reaction starts as a two phase system, in which the melt gradually thickens into a non-stirrable mass. This semisolid sticky mass finally turns into a hard, rigid material coated on the walls of the reaction vessel which is preventing smooth heat transfer.
- the process might be suitable for laboratory preparation of gram quantities of the product; however, for industrial production it is not acceptable and is not reasonable even for a modest scale up.
- US Patent 6,562,974 discloses a process for the preparation of geminal bisphosphonate using pyridine hydrochloride, morpholine hydrochloride & phosphoric acid at 7O 0 C.
- US Patent application 2004/0043967A1 discloses a process for the preparation of risedronate comprising the use of aromatic hydrocarbon or a silicone fluid optionally with poly-alkene glycol.
- these solvents have a high cost and are difficult to eliminate from the finished product
- PCT application WO 03/93282 Al describes a process for the preparation of risedronate & its monovalent cation using ionic liquid (ti ⁇ -butyl ammonium chloride) as solvent at 15-120 0 C.
- ionic liquid ti ⁇ -butyl ammonium chloride
- the disclosed invention uses a solvent which is an expensive reagent, difficult to recover. Also, the yields reported are very low.
- PCT application WO 05/044831A2 describes a process for the preparation of risedronic acid by using sulfolane as reaction solvent. Quenching of the reaction mixture containing sulfolane. with water causes high exothermicity and reaction becomes uncontrollable, hence is difficult to handle.
- PCT application WO05/63779A2 describes a process for the preparation of risedronate using a mixture of phosphorous acid & phosphorous chloride in the absence of solvent. Requirement of more reaction time and large quantities of reactants for the reaction makes the process inefficient and expensive for use on a large scale. Also, the yields obtained are very low.
- the present invention provides an industrially advantageous process for preparation of risedronic acid or salts, chemically known as [l-hydroxy-2(3 ⁇ pyridinyl)ethylidene] bisphosphonic acid, of formula-! in high purity and high yields.
- the present invention provides improved processes for preparing risedronic acid and risedronic acid monosodium salt.
- risedronic acid or salts preferably monosodium salt can be prepared by one pot process comprising reacting carboxylic acid compound in particular 3-pyridyl acetic acid with phosphorous acid in the presence of phosphorous halide in a water miscible neutral solvent such as acetonitrile, optionally distilling the solvent, quenching the reaction mixture with water, adjusting the pH using sodium source and isolating pure risedronic acid monosodium salt in high yield and purity.
- a water miscible neutral solvent such as acetonitrile
- the present invention provides an efficient, economic and also environmentally friendly process which comprises obtaining the risedronic acid by reacting carboxylic acid compound in particular 3-pyridyl acetic acid with phosphorous acid in the presence of phosphorous halide in a water miscible neutral solvent such as acetonitrile, optionally distilling the solvent, quenching the reaction mixture with water. Thereafter risedronic acid is converted to risedronic acid mono sodium salt by the methods reported in the prior art.
- Yet another embodiment of the present invention provides a process for the preparation of risedronic acid using chlorobenzene, avoiding decantation and isolation of the pure product by a simple layer separation method.
- the present invention provides a process for the preparation of risedronic acid or salts of formula- ⁇ ,
- the present invention relates to a safe mode of preparing risedronic acid, in high yields and high purity.
- the present invention uses acetonitrile, which is a water miscible neutral solvent, relatively safe and inexpensive.
- acetonitrile which is a water miscible neutral solvent, relatively safe and inexpensive.
- 3-pyridyl acetic acid of formula II and the phosphorous acid in acetonitrile are reacted with phosphorous halide at temperature 40-80°C, and preferably at about 70-75 0 C till phosphonylation is complete.
- Phosphorous halide can be selected from the group comprising phosphorous trichloride, phosphorous tribromide, phosphorous pentachloride, phosphorous pentabromide, phosphorous oxychloride, phosphorous oxybromide and the like.
- 3-pyridyl acetic acid of formula II and the phosphorous acid in acetonitrile are reacted with phosphorous trichloride.
- phosphorous trichloride is added in small portions.
- the reaction mixture is refluxed to a temperature of 70 ⁇ 5°C for a period of about 1 to about 12 hours till phosphonylation is complete.
- acetonitrile is distilled off completely. The acetonitrile so recovered by distillation can be reused, thus making the process more economical and cost effective.
- the reaction mixture is quenched at ambient temperature with demineralized water.
- the substrate is effective to treat the substrate with an adsorbent, preferably with active charcoal.
- the filtered mass is further refluxed for a period of about 1 to about 12 hours at a temperature of 85 ⁇ 5°C till complete hydrolysis.
- the reaction mixture is cooled to 0-5°C and stirred for a period of 2-3 hours.
- risedronic acid is filtered and washed with demineralized water.
- Risedronic acid prepared can be optionally purified by acid base treatment like risedronic acid in water is treated with base to adjust pH above seven and further treatment with mineral acid to bring pH at 1-2.
- risedronic acid in high yields and better quality as compared to any of the prior art processes. Thereafter risedronic acid is converted to risedronic acid monosodium salt by the methods reported in the prior art.
- the process for preparing risedronic acid monosodium salt comprises suspending the risedronic acid formed in demineralized water and pH is suitably adjusted to 4.2-4.5 with a base selected from alkali carbonates, alkali hydroxides or alkali bicarbonates.
- a base selected from alkali carbonates, alkali hydroxides or alkali bicarbonates.
- the base employed is sodium hydroxide, preferably 50% sodium hydroxide.
- the precipitated salt may be isolated by a manner well known in art.
- the present invention provides a one pot process for the preparation of risedronic acid mono sodium salt of formula-TIT,
- 3-pyridyl acetic acid of formula TT and the phosphorous acid are reacted in a suitable solvent like acetonitrile at reflux temperature.
- Phosphorous halide is slowly added to the above reaction mixture at a temperature of 40-100 0 C, preferably at about 70 ⁇ 5 'J C till phosphonylation is complete.
- Phosphorous halide can be selected from amongst phosphorous trichloride, phosphorous tribromide, phosphorous pentachloride, phosphorous pentabromide, phosphorous oxychloride, phosphorous oxybromide and the like and preferably phosphorous trichloride is used.
- reaction mass After distillation, the filtered mass is cooled to ambient temperature preferably 25 ⁇ 2°C and reaction mass is quenched with water. The resultant reaction mass is then charcoalised to decolorize the reaction mass.
- reaction mass is refluxed for a period of about 1 to 12 hours to complete hydrolysis.
- risedronic acid is converted to monosodium salt.
- the preparation of sodium salt is pH dependent, from about pH 3.0 to about pH 12.0.
- the pH is adjusted to 4.3 to obtain the mono sodium salt using a suitable base.
- the base can be selected from alkali carbonates, alkali hydroxides or alkali bicarbonates.
- the base employed is sodium hydroxide and more preferably 50% sodium hydroxide.
- reaction mass is directly basified with sodium hydroxide at a pH of 4.2-4.5.
- reaction mass is first basified with sodium hydroxide at a pH of 8-9 and then acidified with concentrated hydrochloric acid to bring the pH to 4.2-4.5.
- the reaction mass is initially cooled to ambient temperature and finally to a temperature of below 5 0 C. Thereafter the product can be isolated from the reaction mixture by the methods known in prior art such as filtration.
- reaction mixture After completion of reaction, the reaction mixture is cooled to a temperature below 50°C and demineralized water is added slowly. The reaction mass is stirred for a period of about 30 minutes and allow the layers to settle and separate. The organic layer is then extracted with demineralized water. The combined aqueous layer is charcoalised to decolorize the reaction mass. The filtered mass is refluxed azeotropically for a period of about 1 to 12 hours to complete hydrolysis and removal of traces of chlorobenzene. The reaction mass is then cooled to ambient temperature. Thereafter the product can be isolated from the reaction mixture by the methods known in prior art such as Filtration. The product obtained is highly pure having purity greater than 97% by high performance liquid chromatography (HPLC) and yield is above 80%.
- HPLC high performance liquid chromatography
- the reaction mass was cooled to ambient temperature and then to 0-5 0 C and stirred further for 2.0 hour.
- the solid was filtered, washed with 20% aqueous ethyl alcohol to get the pure risedronate sodium (33 g ) having purity of 99.24% by HPLC.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
Abstract
Description






Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009508675A JP2009536639A (en) | 2006-05-11 | 2007-05-09 | Process for preparing pure risedronic acid or salt |
| MX2008014248A MX2008014248A (en) | 2006-05-11 | 2007-05-09 | Process for the preparation of pure risedronic acid or salts. |
| BRPI0710421-9A BRPI0710421A2 (en) | 2006-05-11 | 2007-05-09 | process for the preparation of pure risedronic acid or salts |
| US12/299,615 US8076483B2 (en) | 2006-05-11 | 2007-05-09 | Process for the preparation of pure risedronic acid or salts |
| EP07736604A EP2016084A2 (en) | 2006-05-11 | 2007-05-09 | Process for the preparation of pure risedronic acid or salts |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1177/DEL/2006 | 2006-05-11 | ||
| IN1177DE2006 | 2006-05-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007132478A2 true WO2007132478A2 (en) | 2007-11-22 |
| WO2007132478A3 WO2007132478A3 (en) | 2009-06-04 |
Family
ID=38694309
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2007/000187 Ceased WO2007132478A2 (en) | 2006-05-11 | 2007-05-09 | Process for the preparation of pure risedronic acid or salts |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8076483B2 (en) |
| EP (1) | EP2016084A2 (en) |
| JP (1) | JP2009536639A (en) |
| KR (1) | KR20090005206A (en) |
| BR (1) | BRPI0710421A2 (en) |
| MX (1) | MX2008014248A (en) |
| WO (1) | WO2007132478A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2041148A1 (en) * | 2006-07-03 | 2009-04-01 | Generics Ýuk¨Limited | Novel process for the preparation of bisphosphonic acids |
| JP2009269867A (en) * | 2008-05-08 | 2009-11-19 | Daito Kk | Method for industrially producing risedronic acid |
| CN104418886A (en) * | 2013-09-02 | 2015-03-18 | 河南天方药业股份有限公司 | Risedronic acid synthesized by one-pot process |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009050731A2 (en) * | 2007-06-20 | 2009-04-23 | Alkem Laboratories Ltd | Novel process for preparing risedronic acid |
Family Cites Families (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3016289A1 (en) * | 1980-04-28 | 1981-10-29 | Henkel KGaA, 4000 Düsseldorf | METHOD FOR PRODUCING OMEGA-AMINO-1-HYDROXYALKYLIDEN-1,1-BIS-PHOSPHONIC ACIDS |
| IL77243A (en) | 1984-12-21 | 1996-11-14 | Procter & Gamble | Pharmaceutical compositions containing geminal diphosphonic acid compounds and certain such novel compounds |
| US5019651A (en) * | 1990-06-20 | 1991-05-28 | Merck & Co., Inc. | Process for preparing 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid (ABP) or salts thereof |
| TW257765B (en) | 1993-08-25 | 1995-09-21 | Merck & Co Inc | |
| US5449819A (en) * | 1994-06-06 | 1995-09-12 | Merck & Co., Inc. | Process for removing waste pox, alendronate and its by products |
| CA2197267C (en) | 1997-02-11 | 2000-02-08 | Yong Tao | Process for the production of 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid or salts thereof |
| ES2153794B1 (en) * | 1999-08-06 | 2001-10-16 | Medichem Sa | PROCEDURE FOR OBTAINING THE 4-AMINO-1-HYDROXIBUTILIDEN-1,1-BISPHOSPHONIC ACID AND ITS TRIHYDRATED MONOSODIC SALT. |
| US6562974B2 (en) | 2000-02-01 | 2003-05-13 | The Procter & Gamble Company | Process for making geminal bisphosphonates |
| PE20011061A1 (en) | 2000-02-01 | 2001-11-20 | Procter & Gamble | SELECTIVE CRYSTALLIZATION OF 3-PYRIDYL-1-HYDROXY-ETHYLIDEN-1,1-BISPHOSPHONIC SODIUM ACID AS HEMIPENTAHYDRATE OR MONOHYDRATE |
| GB0115824D0 (en) * | 2001-06-28 | 2001-08-22 | Rhodia Cons Spec Ltd | Improved solvent systems |
| HRP20041051A2 (en) | 2002-04-11 | 2005-02-28 | Teva Pharmaceutical Industries Ltd. | Novel polymorphs and pseudopolymorphs of risedronate sodium |
| ITMI20020908A1 (en) | 2002-04-29 | 2003-10-29 | Chemi Spa | ALENDRONATE SODIUM PREPARATION PROCESS |
| EP1476451B1 (en) * | 2002-05-17 | 2010-03-17 | Teva Pharmaceutical Industries Ltd. | Use of certain diluents for making bisphosphonic acids |
| CZ293349B6 (en) | 2002-10-25 | 2004-04-14 | Léčiva, A.S. | Novel crystalline form of 3-pyridyl-1-hydroxyethylidene-1,1-bisphopshonoc acid sodium salt |
| ES2311142T3 (en) | 2003-01-17 | 2009-02-01 | Teva Pharmaceutical Industries Limited | PROCEDURE TO REDUCE THE IRON CONTENT OF SODIUM RISEDRONATE. |
| HUP0300227A2 (en) | 2003-01-28 | 2004-09-28 | Richter Gedeon Vegyészeti Gyár Rt. | Process for preparing 2-substituted-1-hidroxyetylidene-1,1-bisphosphonic acids and their salts with high purity |
| BRPI0413067A (en) | 2003-07-30 | 2006-10-17 | Procter & Gamble | process to control the crystal structure of risedronate |
| JP4642762B2 (en) | 2003-08-21 | 2011-03-02 | サン・ファーマシューティカル・インダストリーズ・リミテッド | Method for producing bisphosphonic acid compound |
| DE602004032577D1 (en) | 2003-12-23 | 2011-06-16 | Alchymars S P A | Amorphous form the sodium salt of ibandronic acid |
| WO2005066190A1 (en) | 2004-01-02 | 2005-07-21 | Hexal A/S | New risedronate salts |
| CZ298639B6 (en) | 2004-02-05 | 2007-12-05 | Zentiva, A. S. | Crystalline form of monosodium risedronate |
| US7618953B2 (en) | 2004-02-26 | 2009-11-17 | Zentiva, A.S. | Amorphous forms of risedronate monosodium |
| DE602005001873T2 (en) | 2004-03-03 | 2008-04-24 | Chemi S.P.A., Cinisello Balsamo | Amorphous 3-pyridyl-1-hydroxyethylidene-1,1-bisphosphonic acid monosodium salt and process for its preparation |
| US7361761B2 (en) | 2004-09-28 | 2008-04-22 | Orchid Chemicals & Pharmaceuticals Ltd. | Process for the preparation of bisphosphonic acid |
| WO2006051553A1 (en) | 2004-11-09 | 2006-05-18 | Jubilant Organosys Limited | Process for preparing a pure polymorphic form of 3-pyridyl-1-hydroxyethylidine-1,1-bisphosphonic acid sodium salt |
| PL199215B1 (en) | 2004-12-28 | 2008-08-29 | Politechnika Gdanska | Method for the manufacture of [1-hydroxy-2-(3-pyridyl) ethylidene bis-phosphonic] acid and its 2 and a half aqueous monosodium salt |
| US20080194525A1 (en) | 2005-05-06 | 2008-08-14 | Jordi Puig Serrano | Process of Making Geminal Bisphosphonic Acids and Pharmaceutically Acceptable Salts and/or Hydrates Thereof |
| EP1888606A2 (en) | 2005-05-28 | 2008-02-20 | PLIVA HRVATSKA d.o.o. | Process and novel salt |
| US7872144B2 (en) | 2005-06-13 | 2011-01-18 | Jubilant Organosys Limited | Process for producing biphosphonic acids and forms thereof |
| WO2007026379A2 (en) | 2005-08-30 | 2007-03-08 | Natco Pharma Limited | Novel crystalline forms of risedronate monosodium |
| GB0519891D0 (en) | 2005-09-30 | 2005-11-09 | Pliva Hrvatska D O O | Pharmaceutically acceptable salts and hydrates |
| WO2007042048A2 (en) | 2005-10-11 | 2007-04-19 | Sandoz A/S | Method for preparing crystalline sodium risedronate |
| US8003820B2 (en) | 2005-10-20 | 2011-08-23 | Dr. Reddy's Laboratories Limited | Process for preparing bisphosphonic acids |
| WO2007083240A2 (en) | 2006-01-20 | 2007-07-26 | Aurobindo Pharma Limited | An improved process for the preparation of bisphosphonic acids |
| WO2007083243A2 (en) | 2006-01-20 | 2007-07-26 | Aurobindo Pharma Limited | An improved process for the preparation of risedronate sodium hemi-pentahydrate |
| WO2007096896A1 (en) | 2006-02-20 | 2007-08-30 | Alembic Limited | An improved process for the preparation of biphosphonic derivatives |
| GB0609465D0 (en) | 2006-05-12 | 2006-06-21 | Pliva Hrvatska D O O | Pharmaceutically acceptable salts and polymorphic forms |
| WO2008004000A1 (en) | 2006-07-03 | 2008-01-10 | Generics [Uk] Limited | Novel process for the preparation of bisphosphonic acids |
| WO2008065542A2 (en) | 2006-09-22 | 2008-06-05 | Orchid Chemicals & Pharmaceuticals Ltd. | An improved process for the preparation of risedronate sodium |
| WO2008044245A2 (en) | 2006-10-10 | 2008-04-17 | Matrix Laboratories Ltd | A process for the preparation of risedronate sodium hemipentahydrate |
| PT103600B (en) | 2006-11-06 | 2009-01-30 | Hovione Farmaciencia Sa | PROCESS FOR THE PREPARATION OF BIOSPHONIC ACIDS AND THEIR PHARMACEUTICALLY ACCEPTABLE SALTS |
| KR100775440B1 (en) | 2006-12-20 | 2007-11-12 | 동우신테크 주식회사 | Method for preparing risedronate sodium hemipentahydrate |
| WO2008152518A2 (en) | 2007-05-16 | 2008-12-18 | Actavis Group Ptc Ehf | Process for the preparation of risedronic acid or risedronate sodium |
| WO2009050731A2 (en) | 2007-06-20 | 2009-04-23 | Alkem Laboratories Ltd | Novel process for preparing risedronic acid |
| WO2009003001A2 (en) | 2007-06-27 | 2008-12-31 | Dr. Reddy's Laboratories Ltd. | Preparation of risedronate sodium hemi-pentahydrate |
| WO2009034580A1 (en) | 2007-09-11 | 2009-03-19 | Fleming Laboratories Limited | Improved process for the preparation of risedronate sodium hemipentahydrate |
| KR100925835B1 (en) | 2007-12-07 | 2009-11-06 | 동우신테크 주식회사 | Process for preparing risedronate sodium anhydride and hydrate |
| US8026388B2 (en) | 2008-07-11 | 2011-09-27 | Synthon Bv | Process for making 1-hydroxyalkylidene-1,1-biphosphonic acids |
| EA201270328A1 (en) | 2009-08-28 | 2012-09-28 | Синтон Б. В. | METHOD OF OBTAINING 1-HYDROXYLKYLIDENE-1,1-DIPHOSPHONE ACIDS |
-
2007
- 2007-05-09 JP JP2009508675A patent/JP2009536639A/en active Pending
- 2007-05-09 KR KR1020087028495A patent/KR20090005206A/en not_active Withdrawn
- 2007-05-09 US US12/299,615 patent/US8076483B2/en not_active Expired - Fee Related
- 2007-05-09 WO PCT/IN2007/000187 patent/WO2007132478A2/en not_active Ceased
- 2007-05-09 BR BRPI0710421-9A patent/BRPI0710421A2/en not_active IP Right Cessation
- 2007-05-09 EP EP07736604A patent/EP2016084A2/en not_active Withdrawn
- 2007-05-09 MX MX2008014248A patent/MX2008014248A/en unknown
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2041148A1 (en) * | 2006-07-03 | 2009-04-01 | Generics Ýuk¨Limited | Novel process for the preparation of bisphosphonic acids |
| JP2009269867A (en) * | 2008-05-08 | 2009-11-19 | Daito Kk | Method for industrially producing risedronic acid |
| CN104418886A (en) * | 2013-09-02 | 2015-03-18 | 河南天方药业股份有限公司 | Risedronic acid synthesized by one-pot process |
| CN104418886B (en) * | 2013-09-02 | 2017-04-19 | 天方药业有限公司 | Risedronic acid synthesized by one-pot process |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090182147A1 (en) | 2009-07-16 |
| US8076483B2 (en) | 2011-12-13 |
| WO2007132478A3 (en) | 2009-06-04 |
| MX2008014248A (en) | 2009-02-23 |
| KR20090005206A (en) | 2009-01-12 |
| JP2009536639A (en) | 2009-10-15 |
| BRPI0710421A2 (en) | 2011-08-09 |
| EP2016084A2 (en) | 2009-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7528280B2 (en) | Process for the preparation of biphosphonic acids | |
| EP1504012B9 (en) | Preparation of biphosphonic acids and salts thereof | |
| WO2007096896A1 (en) | An improved process for the preparation of biphosphonic derivatives | |
| US8076483B2 (en) | Process for the preparation of pure risedronic acid or salts | |
| JP5015006B2 (en) | [1-Hydroxy-2- (3-pyridinyl) ethylidene] bisphosphonic acid and its 2.5 hydrate monosodium salt | |
| KR20020024320A (en) | Process for producing 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid and its trihydrated monosodium salt | |
| CN105175446B (en) | Preparation method of minodronic acid for treating osteoporosis | |
| CA2656053A1 (en) | Novel process for the preparation of bisphosphonic acids | |
| JP5220843B2 (en) | Multi-step synthesis of ibandronate | |
| WO2008152518A2 (en) | Process for the preparation of risedronic acid or risedronate sodium | |
| PL213599B1 (en) | Method of obtaining of [1-hydroxy-2-(1H-imidazole-1-yl)-ethylidene] bisphosphonic acid | |
| JP3901321B2 (en) | Method for producing riboflavin-5'-phosphate or a sodium salt thereof | |
| EP1798236A1 (en) | Process for the preparation of 3-pyridyl-1-hydroxyethylidene-1,1- biphosphonic acid and hydrated forms thereof | |
| US7414151B2 (en) | Process for manufacture of 4-amino-hydroxybutylidene-1, 1-bisphosphonic acid and its salts | |
| EP2609101B1 (en) | Process for the preparation of 3-(n-methyl-n-pentyl)amino-1-hydroxypropane-1,1-diphosphonic acid salt or derivatives thereof | |
| JPH11269184A (en) | New production of heterocyclic bis(phosphonic acid) derivative | |
| WO2013109198A1 (en) | Processes for the preparation of sodium ibandronate monohydrate polymorphs a, b and mixture of polymorphs a with b | |
| WO2007010556A1 (en) | Process for the production of 4-amino-1-hydroxybutylidene-1,1-bisphosphonic acid or salts thereof | |
| JP2022528292A (en) | Method for producing polymorph F of sodium neridronate | |
| WO2016024287A1 (en) | An improved process for preparing pure minodronic acid and intermediates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07736604 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12299615 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009508675 Country of ref document: JP Ref document number: MX/A/2008/014248 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087028495 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007736604 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08131644 Country of ref document: CO |
|
| ENP | Entry into the national phase |
Ref document number: PI0710421 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081107 |