Substituierte Heterozyklen und ihre Verwendung
Die Erfindung betrifft neue substituierte Heterozyklen, Verfahren zu ihrer Herstellung, ihre Verwendung zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbesondere von thromboembolischen Erkrankungen.
Die Blutgerinnung ist ein Schutzmechanismus des Organismus, mit dessen Hilfe Defekte in der Gefäßwand rasch und zuverlässig „abgedichtet" werden können. So kann ein Blutverlust vermieden bzw. minimiert werden. Die Blutstillung nach Gefäßverletzung erfolgt im wesentlichen durch das Gerinnungssystem, bei dem eine enzymatische Kaskade komplexer Reaktionen von Plasmaproteinen ausgelöst wird. Hierbei sind zahlreiche Blutgerinnungsfaktoren beteiligt, von denen jeder, sobald aktiviert, die jeweils nächste inaktive Vorstufe in ihre aktive Form überführt. Am Ende der Kaskade steht die Umwandlung des löslichen Fibrinogens in das unlösliche Fibrin, so dass es zu einem Blutgerinnsel kommt. Traditionell unterscheidet man bei der Blutgerinnung zwischen dem intrinsischen und dem extrinsischen System, die in einem abschließenden gemein- samen Reaktionsweg münden. Hierbei kommt dem Faktor Xa, der aus dem Proenzym Faktor X gebildet wird, eine Schlüsselrolle zu, da er beide Gerinnungswege verbindet. Die aktivierte Serin- protease Xa spaltet Prothrombin zu Thrombin. Das entstandene Thrombin wiederum spaltet seinerseits Fibrinogen zu Fibrin. Durch anschließende Quervernetzung der Fibrin-Monomere kommt es zur Bildung von Blutgerinnseln und damit zur Blutstillung. Darüber hinaus ist Thrombin ein potenter Auslöser der Thrombozytenaggregation, die ebenfalls einen erheblichen Beitrag bei der Hämostase leistet.
Die Hämostase unterliegt einem komplexen Regulationsmechanismus. Eine unkontrollierte Aktivierung des Gerinnungssystems oder eine defekte Hemmung der Aktivierungsprozesse kann die Bildung von lokalen Thrombosen oder Embolien in Gefäßen (Arterien, Venen, Lymphgefäßen) oder Herzhöhlen bewirken. Dies kann zu schwerwiegenden thromboembolischen Erkrankungen führen. Darüber hinaus kann eine Hyperkoagulabilität - systemisch - bei einer Verbrauchskoagulo- pathie zur disseminierten intravasalen Gerinnung führen. Thromboembolische Komplikationen treten ferner auf bei mikroangiopathischen hämolytischen Anämien, extrakorporalen Blutkreisläufen, wie Hämodialyse, sowie Herzklappenprothesen.
Thromboembolische Erkrankungen sind die häufigste Ursache von Morbidität und Mortalität in den meisten industrialisierten Ländern [Heart Disease: A Textbook of Cardiovascular Medicine, Eugene Braunwald, 5. Auflage, 1997, W.B. Saunders Company, Philadelphia].
Die aus dem Stand der Technik bekannten Antikoagulantien, d.h. Stoffe zur Hemmung oder Verhinderung der Blutgerinnung, weisen verschiedene, oftmals gravierende Nachteile auf. Eine effiziente Behandlungsmethode bzw. Prophylaxe von thromboembolischen Erkrankungen erweist sich in der Praxis deshalb als sehr schwierig und unbefriedigend.
Für die Therapie und Prophylaxe von thromboembolischen Erkrankungen findet zum einen Heparin Verwendung, das parenteral oder subkutan appliziert wird. Aufgrund günstigerer pharmakokinetischer Eigenschaften wird zwar heutzutage zunehmend niedermolekulares Heparin bevorzugt; allerdings können auch hierdurch die im folgenden geschilderten bekannten Nachteile nicht vermieden werden, die bei der Therapierung mit Heparin bestehen. So ist Heparin oral unwirksam und besitzt nur eine vergleichsweise geringe Halbwertszeit. Da Heparin gleichzeitig mehrere Faktoren der Blutgerinnungskaskade hemmt, kommt es zu einer unselektiven Wirkung. Darüber hinaus besteht ein hohes Blutungsrisiko, insbesondere können Hirnblutungen und Blutungen im Gastrointestinaltrakt auftreten, und es kann zu Thrombopenie, Alopecia medico- mentosa oder Osteoporose kommen [Pschyrembel, Klinisches Wörterbuch, 257. Auflage, 1994, Walter de Gruyter Verlag, Seite 610, Stichwort „Heparin"; Römpp Lexikon Chemie, Version 1.5, 1998, Georg Thieme Verlag Stuttgart, Stichwort „Heparin"].
Eine zweite Klasse von Antikoagulantien stellen die Vitamin K-Antagonisten dar. Hierzu gehören beispielsweise 1,3-Indandjone, vor allem aber Verbindungen wie Warfarin, Phenprocoumon, Dicumarol und andere Cumarin-Derivate, die unselektiv die Synthese verschiedener Produkte bestimmter Vitamin K-abhängiger Gerinnungsfaktoren in der Leber hemmen. Durch den Wirkmechanismus bedingt, setzt die Wirkung aber nur sehr langsam ein (Latenzzeit bis zum Wirkeintritt 36 bis 48 Stunden). Die Verbindungen können zwar oral appliziert werden, aufgrund des hohen Blutungsrisikos und des engen therapeutischen Indexes ist aber eine aufwendige individuelle Einstellung und Beobachtung des Patienten notwendig [J. Hirsh, J. Dalen, D.R. Anderson et al., „Oral anticoagulants: Mechanism of action, clinical effectiveness, and optimal therapeutic ränge" Chest 2001, 119, 8S-21S; J. Ansell, J. Hirsh, J. Dalen et al, „Managing oral anticoagulant therapy" Chest 2001, 119, 22S-38S; P.S. Wells, A.M. Holbrook, N.R. Crowther et al, „Inter- actions of warfarin with drugs and food" Ann. Intern. Med. 1994, 121, 676-683].
In jüngster Zeit ist ein neuer Therapieansatz für die Behandlung und Prophylaxe von thrombo- embolischen Erkrankungen beschrieben worden. Ziel dieses neuen Therapieansatzes ist die Inhibierung von Faktor Xa. Entsprechend der zentralen Rolle, die Faktor Xa in der Blutgerinnungskaskade spielt, stellt Faktor Xa eines der wichtigsten Targets für antikoagulatorische Wirkstoffe dar [J. Hauptmann, J. Stürzebecher, Thrombosis Research 1999, 93, 203; S.A.V. Raghavan, M. Dikshit, „Recent advances in the Status and targets of antithrombotic agents" Drugs Fut. 2002, 27,
669-683; H.A. Wieland, V. Laux, D. Kozian, M. Lorenz, „Approaches in anticoagulation: Rationales for target positioning" Curr. Opin. Investig. Drugs 2003, 4, 264-271; UJ. Ries, W. Wienen, „Serine proteases as targets for antithrombotic therapy" Drugs FuL 2003, 28, 355-370; L.-A. Linkins, J.I. Weitz, „New anticoagulant therapy" Annu. Rev. Med. 2005, 56, 63-77 (online- Publikation August 2004)].
Dabei ist gezeigt worden, dass verschiedene, sowohl peptidische wie nicht-peptidische Verbindungen in Tiermodellen als Faktor Xa-Inhibitoren wirksam sind. Eine große Anzahl von direkten Faktor Xa-Inhibitoren ist bislang bekannt [J.M. Walenga, W.P. Jeske, D. Hoppensteadt, J. Fareed, „Factor Xa Inhibitors: Today and beyond" Curr. Opin. Investig. Drugs 2003, 4, 272-281; J. Ruef, H.A. Katus, „New antithrombotic drugs on the horizon" Expert Opin. Investig. Drugs 2003, 12, 781- 797; M.L. Quan, J.M. Smallheer, „The race to an orally active Factor Xa inhibitor: Recent advances" Curr. Opin. Drug Discovery & Development 2004, 7, 460-469; A. Casimiro-Garcia et al. , „Progress in the discovery of Factor Xa inhibitors" Expert Opin. Ther. Patents 2006, 15, 119-145]. Weiterhin sind nicht-peptidische, niedermolekulare Faktor Xa-Inhibitoren beispielsweise auch in WO 06/002099 und WO 03/026652 beschrieben.
Eine Aufgabe der vorliegenden Erfindung ist die Bereitstellung neuer alternativer Verbindungen mit vergleichbarer oder verbesserter Wirkung und besserer Löslichkeit in wässrigen Lösungen, zur Bekämpfung von Erkrankungen, insbesondere von thromboembolischen Erkrankungen, bei Menschen und Tieren.
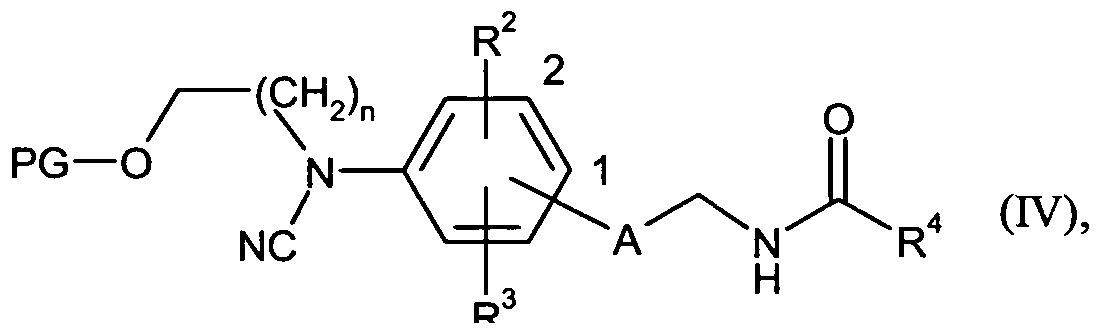
Gegenstand der Erfindung sind Verbindungen der Formel
in welcher
n für die Zahl 1, 2 oder 3 steht,
A für ein 5-gliedriges Heteroaryl oder ein 5-gliedriges Heterocyclyl steht,
wobei Heteroaryl und Heterocyclyl in 1 oder 2 Position an den Phenyl-Ring gebunden sind und Heteroaryl und Heterocyclyl selber eine 1,3 -Verknüpfung mit dem Phenyl-Ring und der Carbonylaminomethyl-Gruppe aufweisen,
und
wobei Heteroaryl und Heterocyclyl substituiert sein können mit einem Substituenten R8,
wobei R8 am Nachbaratom des Atoms gebunden ist, an das die Carbonylamino- methyl-Gruppe gebunden ist, und eine 1,4-Verknüpfung zum Phenyl-Ring aufweist
und
wobei das Atom, an das R8 gebunden ist, ein Stickstoff- oder Kohlenstoffatom ist
und
wobei R8 für Halogen, Hydroxy, Amino, Ci-C4-Alkyl, Q-C4-AIkOXy, Ci-C4-Al- kylamino, Hydroxycarbonyl, Aminocarbonyl, Ci-C4-Alkoxycarbonyl, Ci-C4-Alkyl- aminocarbonyl, Aminosulfonyl, C1-C4-Alkylaminosulfonyl oder Ci-C4-Alkyl- sulfonyl steht,
worin Alkyl, Alkylamino und Alkylaminosulfonyl substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus Hydroxy, Amino, Ci-C4-Alkoxy, Cj-Q-Alkylamino, Hydroxycarbonyl, Aminocarbonyl, Ci-C4-Alkoxycarbonyl, Ci-C4-Alkyl- aminocarbonyl und über ein Stickstoffatom gebundenes 5- oder 6- gliedriges Heterocyclyl,
und
worin Alkylaminocarbonyl substituiert sein kann mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus
Hydroxy, Amino, Ci-C4-Alkylamino und über ein Stickstoffatom gebundenes 5- oder 6-gliedriges Heterocyclyl,
R1 für Wasserstoff, Cyano, Hydroxy, CrC4-Alkyl, CrC4-Alkylcarbonyl, C3-C7- Cycloalkylcarbonyl, Phenylcarbonyl, 4- bis 7-gliedriges Heterocyclylcarbonyl oder 5- oder 6-gliedriges Heteroarylcarbonyl steht,
R2 für Wasserstoff, Fluor, Chlor, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, CrC4-Alkyl, CrC4-Alkoxy, CrC4-Alkoxymethyl, Ci-C4-Alkylamino, C3-C6-Cycloalkyl, Aminocarbonyl, Ci-C4-Alkoxycarbonyl oder Cj -C4- Alkylaminocarbonyl steht,
R3 für Wasserstoff, Fluor, Chlor, Cyano, Hydroxy, Amino, Trifluormethyl, Trifluormethoxy, CrC4-Alkyl, Ci-C4-Alkoxy, C1-C4-AIkOXyTnCtIIyI, CrC4-Alkylamino, C3-C6-Cycloalkyl, Aminocarbonyl, Ci-C4-Alkoxycarbonyl oder Ci-C4-Alkylaminocarbonyl steht,
R4 für eine Gruppe der Formel
steht,
wobei
die Anknüpfstelle an die Carbonylgruppe ist,
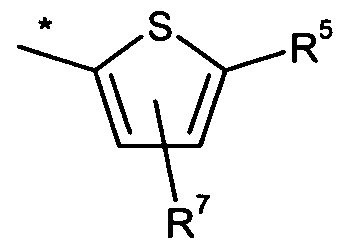
R5 für Wasserstoff, Fluor, Chlor, Cyano, Ethinyl, CrC4-Alkyl, CrC4-Alkoxy oder C3- C6-Cycloalkyl steht,
R6 für Wasserstoff, Amino, Ci-C4-Alkyl, Ci-C4-Alkylamino oder Ca-Cβ-Cycloalkyl steht,
und
R7 für Wasserstoff, Fluor, Chlor, Amino oder Ci-C4-Alkyl steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Erfϊndungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formehl und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische An- Wendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasser- stoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kalium- salze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" umfasst Verbindungen, welche selbst biologisch aktiv oder
inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl per se und "Alk" und "Alkyl" in Alkoxy, Alkylamino. Alkoxycarbonyl. Alkylaminocarbonyl, Alkylaminosulfonyl und Alkylsulfonyl steht für einen linearen oder verzweigten Alkylrest mit in der Regel 1 bis 4, bevorzugt 1 oder 2 Kohlenstoffatomen, beispielhaft und vorzugsweise für Methyl, Ethyl, n-Propyl, Isopropyl und tert-Butyl.
Alkoxy steht beispielhaft und vorzugsweise für Methoxy, Ethoxy, n-Propoxy, Isopropoxy und tert- Butoxy.
Alkylamino steht für einen Alkylaminorest mit einem oder zwei (unabhängig voneinander gewählten) Alkylsubstituenten, beispielhaft und vorzugsweise für Methylamino, Ethylamino, n- Propylamino, Isopropylamino, tert-Butylamino, N,N-Dimethylamino, NN-Diethylamino, N-Ethyl-N- methylamino, N-Methyl-N-n-propylamino, N-Isopropyl-N-n-propylamino und N-tert-Butyl-N-methyl- amino. Ci-C3-Alkylamino steht beispielsweise für einen Monoalkylaminorest mit 1 bis 3 Kohlenstoffatomen oder für einen Dialkylaminorest mit jeweils 1 bis 3 Kohlenstoffatomen pro Alkylsubstituent.
Alkoxycarbonyl steht beispielhaft und vorzugsweise für Methoxycarbonyl, Ethoxycarbonyl, n-Prop- oxycarbonyl, Isopropoxycarbonyl und tert-Butoxycarbonyl.
Alkylaminocarbonyl steht für einen Alkylaminocarbonylrest mit einem oder zwei (unabhängig voneinander gewählten) Alkylsubstituenten, beispielhaft und vorzugsweise für Methylaminocarbonyl, Ethylaminocarbonyl, n-Propylaminocarbonyl, iso-Propylaminocarbonyl, tert-Butylaminocarbonyl, N,N-Dimethylaminocarbonyl, NN-Diethylaminocarbonyl, N-Ethyl-N-methylaminocarbonyl, N- Methyl-N-n-propylaminocarbonyl, N-iso-Propyl-N-n-propylaminocarbonyl und N-tert-Butyl-N- methylaminocarbonyl. Ci-Cs-Alkylaminocarbonyl steht beispielsweise für einen Monoalkylamino- carbonylrest mit 1 bis 3 Kohlenstoffatomen oder für einen Dialkylaminocarbonylrest mit jeweils 1 bis 3 Kohlenstoffatomen pro Alkylsubstituent.
Alkylaminosulfonyl steht für einen Alkylaminosulfonylrest mit einem oder zwei (unabhängig voneinander gewählten) Alkylsubstituenten, beispielhaft und vorzugsweise für Methylaminosulfonyl, Ethylaminosulfonyl, n-Propylaminosulfonyl, iso-Propylaminosulfonyl, tert-Butylaminosulfonyl, NN- Dimethylaminosulfonyl, NN-Diethylaminosulfonyl, N-Ethyl-N-methylaminosulfonyl, N-Methyl-N-n- propylaminosulfonyl, N-iso-Propyl-N-n-propylaminosulfonyl und N-tert-Butyl-N-methylamino-
sulfonyl. Ci-C3-Alkylaminosulfonyl steht beispielsweise für einen Monoalkylaminosulfonylrest mit 1 bis 3 Kohlenstoffatomen oder für einen Dialkylaminosulfonylrest mit jeweils 1 bis 3 Kohlenstoffatomen pro Alkylsubstituent.
Alkylsulfonyl steht beispielhaft und vorzugsweise für Methylsulfonyl, Ethylsulfonyl, n-Propyl- sulfonyl, Isopropylsulfonyl und tert-Butylsulfonyl.
Cycloalkyl steht für eine Cycloalkylgruppe mit in der Regel 3 bis 7 Kohlenstoffatomen, bevorzugt mit 3 bis 5 Kohlenstoffatomen, beispielhaft und vorzugsweise für Cyclopropyl, Cyclobutyl, Cyclo- pentyl, Cyclohexyl und Cycloheptyl.
Heterocvclyl steht für einen monocyclischen Rest mit 5 oder 6 Ringatomen und bis zu 3, vorzugsweise bis zu 2 Heteroatomen und/oder Heterogruppen aus der Reihe N, O, S, SO, SO2. Die Heterocyclyl-Reste können gesättigt oder teilweise ungesättigt sein. Bevorzugt sind Hetero- cyclylreste mit bis zu zwei Heteroatomen aus der Reihe O, N und S, wie beispielhaft und vorzugsweise Tetrahydrofuranyl, Pyrrolidinyl, Pyrrolinyl, Isoxazolinyl und Morpholinyl.
Heteroaryl steht für einen aromatischen, monocyclischen Rest mit 5 Ringatomen und bis zu 4 Heteroatomen aus der Reihe S, O und N, beispielhaft und vorzugsweise für Thienyl, Furyl, Pyrrolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Imidazolyl und Pyrazolyl.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
In den Formern der Gruppe, die für R4 stehen kann, steht der Endpunkt der Linie, neben der jeweils ein * steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Gruppe sondern ist Bestandteil der Bindung zu dem Atom, an das R4 gebunden ist.
In den Formeln der Gruppe, die für A stehen kann, steht der Endpunkt der Linie, neben der jeweils ein #1 oder #2 steht, nicht für ein Kohlenstoffatom beziehungsweise eine CH2-Gruppe sondern ist Bestandteil der Bindung zu dem Atom, an das A gebunden ist.
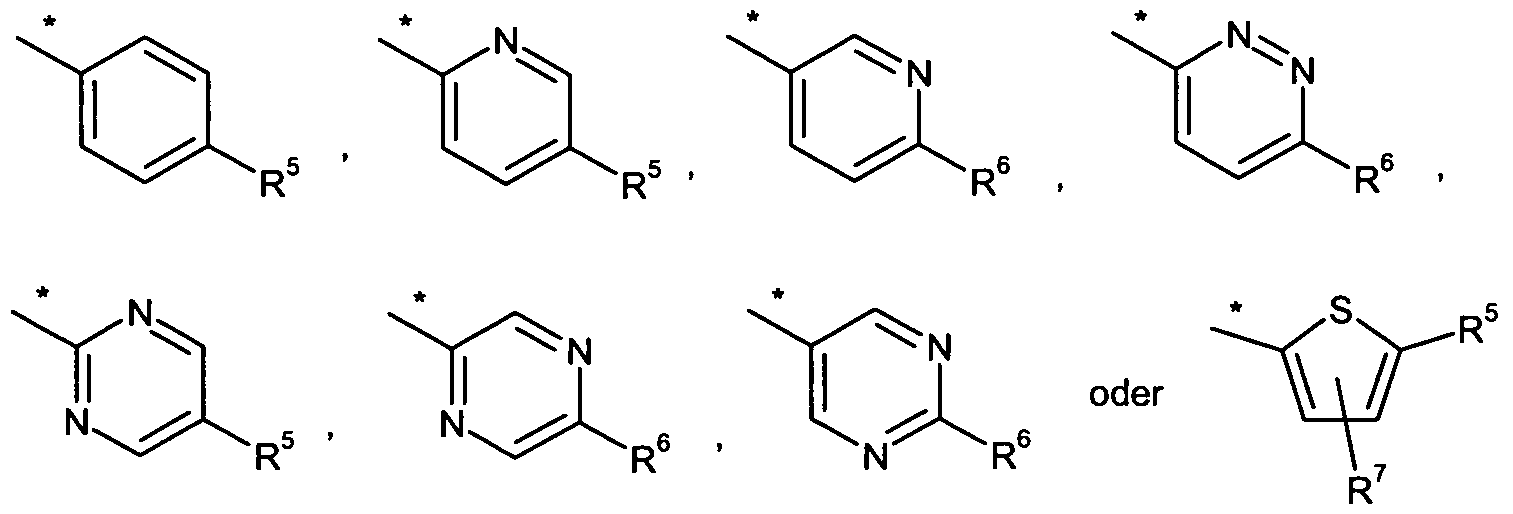
Bevorzugt sind Verbindungen der Formel (I), in welcher
n für die Zahl 1 , 2 oder 3 steht,
A für ein 5-gliedriges Heteroaryl oder teilweise ungesättigtes 5-gliedriges Heterocyclyl steht,
wobei Heteroaryl und Heterocyclyl in 1 oder 2 Position an den Phenyl-Ring gebunden sind und Heteroaryl und Heterocyclyl selber eine 1,3-Verknüpfung mit dem Phenyl-Ring und der Carbonylaminomethyl-Gruppe aufweisen,
und
wobei Heteroaryl und Heterocyclyl substituiert sein können mit einem Substituenten R8,
wobei R8 am Nachbaratom des Atoms gebunden ist, an das die Carbonylaminomethyl-Gruppe gebunden ist, und eine 1 ,4-Verknüprung zum Phenyl-Ring aufweist
und
wobei das Atom, an das R8 gebunden ist, ein Stickstoff- oder Kohlenstoffatom ist
und
wobei R8 für Amino, CrC4-Alkyl, CrC4-Alkoxy, CrC4-Alkoxymethyl, CpC4- Alkylamino, Ci-C4-Alkylaminomethyl, Hydroxycarbonyl, Hydroxycarbonyl- methyl, Hydroxycarbonylethyl, Aminocarbonyl, Aminocarbonylmethyl, Amino- carbonylethyl, C]-C4-Alkoxycarbonyl, Ci-C4-Alkoxycarbonylmethyl, Ci-C4-AIk- oxycarbonylethyl, Ci-C4-Alkylaminocarbonyl, Ci-C4-Alkylaminocarbonylmethyl, Ci-C4-Alkylaminocarbonylethyl, Aminosulfonyl, Ci-C4-Alkylaminosulfonyl oder Ci-C4-Alkylsulfonyl steht,
worin Alkyl substituiert sein kann mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus Hydroxy und
Amino,
und
worin Ethylaminocarbonyl und Propylaminocarbonyl substituiert sein können mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus Hydroxy, Amino und Ci-C4-Alkylamino,
R1 für Wasserstoff, Cyano, Hydroxy oder d-C4-Alkyl steht,
R2 für Wasserstoff, Fluor, Chlor, Cyano, CrC4-Alkyl oder CrC4-Alkoxy steht,
R3 für Wasserstoff, Fluor, Chlor, Cyano, Hydroxy, C,-C4-Alkyl, C,-C4-Alkoxy, C1-C4-AIk- oxymethyl, Cyclopropyl, Aminocarbonyl, CrC4-Alkoxycarbonyl oder CrC4-Alkylamino- carbonyl steht,
R4 für eine Gruppe der Formel
steht,
wobei
die Anknüpfstelle an die Carbonylgruppe ist,
R5 für Fluor, Chlor, Ethinyl, Methyl oder Methoxy steht,
und
R7 für Wasserstoff steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher
n für die Zahl 1 oder 2 steht,
A für eine Gruppe der Formel
steht,
wobei
#1 die Anknüpfstelle an den Phenyl-Ring ist, und in 1 Position an den Phenyl-Ring gebunden ist,
#2 die Anknüpfstelle an die Carbonylaminomethyl-Gruppe ist,
R8 für Wasserstoff, CrC4-Alkyl, Ci-C4-Alkoxy, Ci-C4-Alkoxymethyl, CrC4-Alkyl- amino, Ci-C4-Alkylaminomethyl, Hydroxycarbonyl, Hydroxycarbonylmethyl,
Aminocarbonyl, Aminocarbonylmethyl, C]-C4-Alkoxycarbonyl, CrC4-Alkoxy- carbonylmethyl, Ci-C4-Alkylaminocarbonyl oder Ci-C4-Alkylaminocarbonyl- methyl steht,
worin Alkyl substituiert sein kann mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus Hydroxy und Amino,
und
worin Ethylaminocarbonyl substituiert sein kann mit einem Substituenten, wobei der Substituent ausgewählt wird aus der Gruppe bestehen aus Hydroxy, Amino und Ci-C4-Alkylamino,
R1 für Wasserstoff steht,
R2 für Wasserstoff oder Fluor steht,
R3 für Wasserstoff, Fluor, Chlor, Cyano, Methyl, Ethyl, n-Propyl, iso-Propyl, Methoxy, Ethoxy, Methoxymethyl oder Cyclopropyl steht,
R4 für eine Gruppe der Formel
steht,
wobei
* die Anknüpfstelle an die Carbonylgruppe ist,
R5 für Fluor, Chlor oder Methyl steht,
und
R7 für Wasserstoff steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher
n für die Zahl 1 steht,
A für eine Gruppe der Formel
steht,
wobei
#1 die Anknüpfstelle an den Phenyl-Ring ist, und in 1 Position an den Phenyl-Ring gebunden ist,
#2 die Anknüpfstelle an die Carbonylaminomethyl-Gruppe ist,
R8 für Wasserstoff, Hydroxymethyl, Aminomethyl, Ci-C4-Alkyl, Ci-C4-Alkoxy, Ci-C4-Alkoxymethyl, Ci-C4-Alkylaminomethyl, Hydroxycarbonyl, Aminocarb- onyl, C]-C4-Alkoxycarbonyl, Ci-C4-Alkylaminocarbonyl, Hydroxyethylaminocarb- onyl oder C ] -C4-Alkylaminoethylaminocarbonyl steht,
R1 für Wasserstoff steht,
R2 für Wasserstoff oder Fluor steht,
R3 für Wasserstoff, Fluor, Chlor, Methyl oder Methoxy steht,
R4 für eine Gruppe der Formel
steht,
wobei
* die Anknüpfstelle an die Carbonylgruppe ist,
R5 für Chlor steht,
und
R7 für Wasserstoff steht,
und ihre Salze, ihre Solvate und die Solvate ihrer Salze.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher nfür die Zahl 1 steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher A für eine Gruppe der Formel
steht,
wobei
#1 die Anknüpf stelle an den Phenyl-Ring ist, und in 1 Position an den Phenyl-Ring gebunden ist,
#2 die Anknüpfstelle an die Carbonylaminomethyl-Gruppe ist,
und
R8 für Wasserstoff, Hydroxymethyl, Aminomethyl, Hydroxycarbonyl, Aminocarb- onyl, Ci-C4-Alkoxycarbonyl oder Ci-C4-Alkylaminocarbonyl steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R2 für Wasserstoff steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R3 für Wasserstoff, Fluor, Chlor, Methyl oder Methoxy steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R3 für Wasserstoff steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R2 und R3 für Wasserstoff stehen.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R2 für Wasserstoff und R3 für Fluor steht.
Bevorzugt sind auch Verbindungen der Formel (I), in welcher R4 für eine Gruppe der Formel
steht, wobei * die Anknüpfstelle an die Carbonylgruppe ist, R5 für Chlor steht und R7 für Wasserstoff steht.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombina- tionen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Gegenstand der Erfindung ist weiterhin ein Verfahren zur Herstellung der Verbindungen der Formel (I), oder ihrer Salze, ihrer Solvate oder der Solvate ihrer Salze, wobei
[A] die Verbindungen der Formel
in welcher n, A, R2, R3 und R4 die oben angegebene Bedeutung haben,
in einem inerten Lösungsmittel in Gegenwart einer Säure mit Bromcyan zu Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht, umgesetzt werden,
oder
[B] die Verbindungen der Formel
in welcher n, A, R2, R3 und R4 die oben angegebene Bedeutung haben, und
PG für eine Hydroxy-Schutzgruppe, vorzugsweise für Trimethylsilyl oder tert.-Butyldimethyl- silyl, steht,
in einem dreistufigen Verfahren zuerst in einem inerten Lösungsmittel mit Bromcyan, vorzugsweise in Gegenwart einer Base, zu Verbindungen der Formel
in welcher n, A, R2, R3 und R4 die oben angegebene Bedeutung haben, und
PG für eine Hydroxy-Schutzgruppe, vorzugsweise für Trimethylsilyl oder tert.-Butyldimethyl- silyl, steht,
und anschließend durch Abspaltung der Schutzgruppe PG zu Verbindungen der Formel
in welcher n, A, R2, R3 und R4 die oben angegebene Bedeutung haben,
umgesetzt werden und in der dritten Stufe die Verbindungen der Formel (V) in einem inerten Lösungsmittel in Gegenwart einer Säure zu Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht, cyclisiert werden, wobei die Abspaltung der Schutzgruppe und die Cyclisierung bevorzugt in einem Reaktionsschritt erfolgen,
oder
[C] die Verbindungen der Formel (II) in der ersten Stufe mit Verbindungen der Formel
N S
R** (VI),
in welcher
R1 für CrC4-Alkyl, CrC4-Alkylcarbonyl, C3-C7-Cycloalkylcarbonyl, Phenylcarbonyl, 4- bis 7- gliedriges Heterocyclylcarbonyl oder 5- oder 6-gliedriges Heteroarylcarbonyl steht,
umgesetzt werden und in der zweiten Stufe cyclisiert werden,
oder
[D] die Verbindungen der Formel (II) mit Verbindungen der Formel
in welcher
R1 für Cyano oder Ci-C4-Alkyl steht, und
G für eine Abgangsgruppe, bevorzugt Phenoxy oder Methylthio, steht,
umgesetzt werden,
oder
[E] die Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht, mit Hydroxylamin- Hydrochlorid zu Verbindungen der Formel (I), in welcher R1 für Hydroxy steht, umgesetzt werden.
Die Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht, können gegebenenfalls mit den entsprechenden Lösungsmitteln und/oder Basen oder Säuren zu ihren Salzen, ihren Solvaten und/oder den Solvaten ihrer Salze umgesetzt werden.
Die freie Base der Salze kann zum Beispiel durch Chromatographie an einer Reversed Phase Säule mit einem Acetonitril-Wasser-Gradienten unter Zusatz einer Base erhalten werden, insbesondere durch Verwendung einer RP 18 Phenomenex Luna C 18(2) Säule und Diethylamin als Base, oder
durch Lösen der Salze in einem organischen Lösungsmittel und Ausschütteln mit wässrigen Lösungen von basischen Salzen wie Natriumhydrogencarbonat.
In einem alternativen Verfahren werden die Salze in Wasser gelöst und durch Zugabe von Natriumhydrogencabonat-Lösung wird die Base ausgefällt.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der Verbindungen der Formel (I) oder ihrer Solvate, bei dem Salze der Verbindungen oder Solvate der Salze der Verbindungen durch Chromatographie unter Zusatz einer Base in die Verbindungen überführt werden.
Die Umsetzung nach Verfahren [A] erfolgt im Allgemeinen in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von -200C bis 5O0C bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Tetrahydrofuran, Dichlormethan oder Acetonitril oder Gemische dieser Lösungsmittel.
Säuren sind beispielsweise starke anorganische oder organische Säuren wie Fluorwasserstoff, Chlorwasserstoff, Bromwasserstoff, Methansulfonsäure, Trifluormethansulfonsäure oder Trifluor- essigsaure.
Die Umsetzung der ersten Stufe nach Verfahren [B] erfolgt im Allgemeinen in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von -200C bis 500C bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Tetrahydrofuran, Dichlormethan oder Acetonitril oder Gemische dieser Lösungsmittel.
Basen sind beispielsweise anorganische Basen wie Alkali- oder Erdalkalicarbonate oder -hydrogencarbonate wie Lithium-, Natrium-, Kalium-, Calcium- oder Cäsiumcarbonat oder Natrium- oder Kaliumhydrogencarbonat, oder Alkalihydride wie Natriumhydrid.
Die Abspaltung von Trimethylsilyl oder tert.-Butyldimethylsilyl als bevorzugt verwendete Hydroxy-Schutzgruppen (PG) in der zweiten Stufe nach Verfahren [B] erfolgt im Allgemeinen in Tetrahydrofuran als Lösungsmittel, vorzugsweise mit Hilfe von Tetra-n-butylammoniumfluorid (TBAF), bevorzugt in einem Temperaturbereich von 00C bis 400C bei Normaldruck.
Die Umsetzung der dritten Stufe nach Verfahren [B] erfolgt im Allgemeinen in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von -200C bis 500C bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Tetrahydrofuran, Dichlormethan oder Acetonitril oder Gemische dieser Lösungsmittel.
Säuren sind beispielsweise starke anorganische oder organische Säuren wie Fluorwasserstoff, Chlorwasserstoff, Bromwasserstoff, Methansulfonsäure, Trifluormethansulfonsäure oder Trifluor- essigsaure.
Die Umsetzung der zweiten und dritten Stufe nach Verfahren [B] erfolgt besonders bevorzugt unter Verwendung einer säurelabilen Hydroxy-Schutzgruppe, wie beispielsweise Trimethylsilyl oder tert.-Butyldimethylsilyl, in Gegenwart eines Überschusses einer Säure als Eintopf-Reaktion, in inerten Lösungsmitteln, bevorzugt in einem Temperaturbereich von -200C bis 500C bei Normaldruck, ohne Isolierung der Zwischenstufe der Verbindungen der Formel (V).
Inerte Lösungsmittel sind beispielsweise Tetrahydrofuran, Dichlormethan oder Acetonitril oder Gemische dieser Lösungsmittel.
Säuren sind beispielsweise starke anorganische oder organische Säuren wie Fluorwasserstoff, Chlorwasserstoff, Bromwasserstoff, Methansulfonsäure, Trifluormethansulfonsäure oder Trifluor- essigsaure.
Die Umsetzung der ersten Stufe nach Verfahren [C] erfolgt im Allgemeinen in Analogie zu literaturbekannten Verfahren, wie beschrieben in z. B. A. Hetenyi, et al., J. Org. Chem. 2003, 68, 2175-2182, D. Douglass, J. Amer. Chem. Soc. 1934, 56, 719, F.B. Dains et al, J. Amer. Chem. Soc. 1925, 47, 1981-1989 oder F.B. Dains et al., J. Amer. Chem. Soc. 1922, 44, 2637-2643.
Die Umsetzung der zweiten Stufe nach Verfahren [C] erfolgt im Allgemeinen in Analogie zu literaturbekannten Verfahren, wie beschrieben in z. B. T. Shibanuma, M. Shiono, T. Mukaiyama, Chem. Lett. 1977, 575-576.
Die Umsetzung nach Verfahren [D] erfolgt im Allgemeinen in Analogie zu literaturbekannten Verfahren, wie beschrieben in z. B. N. Maezaki, A. Furusawa, S. Uchida, T. Tanaka, Tetrahedron 2001, 57, 9309-9316, G. Berecz, J. Reiter, G. Argay, A. Kaiman, J. Heterocycl. Chem. 2002, 39, 319-326, R. Evers, M. Michalik, J. Prakt. Chem. 1991, 333, 699-710, R. Mohr, A. Buschauer, W. Schunack, Arch. Pharm. (Weinheim Ger.) 1988, 321, 221-227, P. J. Garratt, et al., Tetrahedron 1989, 45, 829-834 oder V.A. Vaillancourt et al., J. Med. Chem. 2001, 44, 1231-1248.
Die Umsetzung nach Verfahren [E] erfolgt im Allgemeinen in Analogie zu literaturbekannten Ver- fahren, wie beschrieben in z. B. G. Zinner, G. Nebel, Arch. Pharm. Ber. Dtsch. Ges. 1970, 303, 385-390.
Die Verbindungen der Formeln (VI) und (VE) sind bekannt oder lassen sich nach bekannten Verfahren aus den entsprechenden Ausgangsverbindungen synthetisieren.
Die Verbindungen der Formel (IH) sind bekannt oder können hergestellt werden, aus den Verbindungen der Formel (II) durch Einführung der Schutzgruppe PG nach dem Fachmann bekannten Bedingungen.
Die Einführung von Trimethylsilyl oder tert.-Butyldimethylsilyl als bevorzugt verwendete Hydroxy-Schutzgruppen (PG) erfolgt im Allgemeinen durch Umsetzung mit Trimethylsilylchlorid, tert.-Butyldimethylsilylchlorid oder tert.-Butyldimethylsilyl-trifluormethansulfonat in Tetrahydro- furan, Dimethylformamid oder Dichlormethan als Lösungsmittel, vorzugsweise in Gegenwart von Imidazol oder 2,6-Dimethylpyridin, bevorzugt in einem Temperaturbereich von 00C bis 400C bei Normaldruck.
Die Verbindungen der Formel (IT) sind bekannt oder können hergestellt werden, indem Verbindungen der Formel
in welcher
A, R2, R3 und R4 die oben angegebene Bedeutung haben, und
X1 für Brom oder Iod steht,
mit Verbindungen der Formel
■(CH2)n
HO (K), NH,
in welcher n die oben angegebene Bedeutung hat,
umgesetzt werden.
Die Umsetzung erfolgt im Allgemeinen in inerten Lösungsmitteln unter Zugabe eines Kupfer(I)- Salzes, einer Base und eines Diamin-Liganden, bevorzugt in einem Temperaturbereich von 600C bis zum Rückfluss der Lösungsmittels bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise aprotische Lösungsmittel wie Toluol, Dioxan, Tetrahy- drofuran oder Dimethylformamid, bevorzugt ist Dioxan.
Kupfer(I)-Salze sind beispielsweise Kupfer(I)-iodid, Kupfer(I)-chlorid oder Kupfer(I)-oxid, bevorzugt ist Kupfer(I)-iodid.
Basen sind beispielsweise Kaliumphosphat, Kaliumcarbonat oder Cäsiumcarbonat, bevorzugt ist Kaliumphosphat.
Diamin-Liganden sind beispielsweise 1 ,2-Diamine wie NN'-Dimethylethylendiamin.
Die Verbindungen der Formel (VIII) sind bekannt oder lassen sich nach dem Fachmann bekannten Verfahren zum Aufbau des Heterozyklus A aus den entsprechenden Ausgangsverbindungen synthetisieren.
Die Verbindungen der Formel (EX) sind bekannt oder lassen sich nach bekannten Verfahren aus den entsprechenden Ausgangsverbindungen synthetisieren.
Der Stickstoff des Amides in Verbindungen der Formeln (II), (ffl), (IV), (V) und (VHf) kann gegebenenfalls während der Umsetzung mit einer dem Fachmann bekannten Schutzgruppe geschützt sein, bevorzugt ist eine 2,4-Dimethoxybenzyl-Gruppe, die unter den Bedingungen der letzten Stufe der Synthese der Verbindungen der Formel (I) abgespalten wird.
Die Herstellung der erfϊndungsgemäßen Verbindungen kann durch die folgenden Syntheseschemata veranschaulicht werden:
Schema 1
BiCN NaHCO3
Die erfindungsgemäßen Verbindungen zeigen ein nicht vorhersehbares, wertvolles pharmakolo- gisches Wirkspektrum.
Sie eignen sich daher zur Verwendung als Arzneimittel zur Behandlung und/oder Prophylaxe von Krankheiten bei Menschen und Tieren.
Bei den erfindungsgemäßen Verbindungen handelt es sich um selektive Inhibitoren des Blutgerinnungsfaktors Xa, die insbesondere als Antikoagulantien wirken.
Darüber hinaus verfugen die erfindungsgemäßen Verbindungen über günstige physikochemische Eigenschaften, wie beispielsweise eine gute Löslichkeit in Wasser und physiologischen Medien, was für ihre therapeutische Anwendung von Vorteil ist.
Weiterer Gegenstand der vorliegenden Erfindung ist der Einsatz der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, vorzugsweise von thromboembo- lischen Erkrankungen und/oder thromboembolischen Komplikationen.
Zu den „thromboembolischen Erkrankungen" im Sinne der vorliegenden Erfindung zählen insbesondere Erkrankungen wie Herzinfarkt mit ST-Segment-Erhöhung (STEMI) und ohne ST- Segment-Erhöhung (non-STEMI), stabile Angina Pectoris, instabile Angina Pectoris, Reokklusio- nen und Restenosen nach Koronarinterventionen wie Angioplastie oder aortokoronarem Bypass, periphere arterielle Verschlusskrankheiten, Lungenembolien, tiefe venöse Thrombosen und
Nierenvenenthrombosen, transitorische ischämische Attacken sowie thrombotischer und thrombo- embolischer Hirnschlag.
Die Substanzen eignen sich daher auch zur Prävention und Behandlung von kardiogenen Thrombo- embolien, wie beispielsweise Hirn-Ischämien, Schlaganfall und systemischen Thromboembolien und Ischämien, bei Patienten mit akuten, intermittierenden oder persistierenden Herzarrhythmien, wie beispielsweise Vorhofflimmern, und solchen, die sich einer Kardioversion unterziehen, ferner bei Patienten mit Herzklappen-Erkrankungen oder mit künstlichen Herzklappen. Darüber hinaus sind die erfindungsgemäßen Verbindungen zur Behandlung der disseminierten intravasalen Gerinnung (DIC) geeignet.
Thromboembolische Komplikationen treten ferner auf bei mikroangiopathischen hämolytischen Anämien, extrakorporalen Blutkreisläufen, wie Hämodialyse, sowie Herzklappenprothesen.
Außerdem kommen die erfindungsgemäßen Verbindungen auch für die Prophylaxe und/oder Behandlung von atherosklerotischen Gefäßerkrankungen und entzündlichen Erkrankungen wie rheumatische Erkrankungen des Bewegungsapparats in Betracht, darüber hinaus ebenso für die Prophylaxe und/oder Behandlung der Alzheimer'schen Erkrankung. Außerdem können die erfindungsgemäßen Verbindungen zur Inhibition des Tumorwachstums und der Metastasenbildung, bei Mikroangiopathien, altersbedingter Makula-Degeneration, diabetischer Retinopathie, diabetischer Nephropathie und anderen mikrovaskulären Erkrankungen sowie zur Prävention und Behandlung thromboembolischer Komplikationen, wie beispielsweise venöser Thromboembolien, bei Tumorpatienten, insbesondere solchen, die sich größeren chirurgischen Eingriffen oder einer Chemo- oder Radiotherapie unterziehen, eingesetzt werden.
Die erfindungsgemäßen Verbindungen können darüber hinaus auch zur Verhinderung von Koagulation ex vivo eingesetzt werden, z.B. zur Konservierung von Blut- und Plasmaprodukten, zur Reinigung/Vorbehandlung von Kathetern und anderen medizinischen Hilfsmitteln und Geräten, zur Beschichtung künstlicher Oberflächen von in vivo oder ex vivo eingesetzten medizinischen Hilfsmitteln und Geräten oder bei biologischen Proben, die Faktor Xa enthalten.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer antikoagulatorisch wirksamen Menge der erfindungsgemäßen Verbindung.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Verhinderung der Blutkoagulation in vitro, insbesondere bei Blutkonserven oder biologischen Proben, die Faktor Xa enthalten, das dadurch gekennzeichnet ist, dass eine antikoagulatorisch wirksame Menge der erfindungsgemäßen Verbindung zugegeben wird.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend eine erfindungsgemäße Verbindung und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Erkrankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• Lipidsenker, insbesondere HMG-CoA-(3-Hydroxy-3-methylglutaryl-Coenzym A)-Reduktase- Inhibitoren;
• Koronartherapeutika/Vasodilatatoren, insbesondere ACE-(Angiotensin-Converting-Enzyme)- Inhibitoren; AII-(Angiotensin II)-Rezeptor- Antagonisten; ß-Adrenozeptor- Antagonisten; alpha- 1-Adrenozeptor- Antagonisten; Diuretika; Calciumkanal-Blocker; Substanzen, die eine Erhöhung von cyclischem Guanosinmonophosphat (cGMP) bewirken, wie beispielsweise Stimulatoren der löslichen Guanylatcyclase;
• Plasminogen-Aktivatoren (Thrombolytika/Fibrinolytika) und die Thrombolyse/Fibrinolyse steigernde Verbindungen wie Inhibitoren des Plasminogen-Aktivator-Inhibitors (P AI-Inhibitoren) oder Inhibitoren des Thrombin-aktivierten Fibrinolyse-Inhibitors (TAFI-Inhibitoren);
• antikoagulatorisch wirksame Substanzen (Antikoagulantien);
• plättchenaggregationshemmende Substanzen (Plättchenaggregationshemmer, Thrombozytenaggregationshemmer) ;
• Fibrinogen-Rezeptor-Antagonisten (Glycoprotein-IIb/πia-Antagonisten);
• sowie Antiarrhythmika.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapsem (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräpara- tionen, Vaginalkapseln, wässrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspen- sionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharma- zeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl-
sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Abkürzungen
DC Dünnschicht-Chromatographie
DCI direkte chemische Ionisation (bei MS)
DMF NN-Dimethylformamid
DMSO Dimethylsulfoxid d Tag(e) d. Th. der Theorie (bei Ausbeute) ee Enantiomerenüberschuss eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS) h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
LC-MS Flüssigchromatographie-gekoppelte Massenspektroskopie min Minute(n)
MS Massenspektroskopie
NMR Kernresonanzspektroskopie
RP reverse phase (bei HPLC)
RT Raumtemperatur
R, Retentionszeit (bei HPLC)
TBTU 0-(Benzotriazol-l-yl)-N,NN',N'-tetramethyluronium-tetrafluoroborat
THF Tetrahydrofuran
LC-MS- und HPLC-Methoden
Methode 1 : Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml Perchlorsäure (70%-ig) / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 6.5 min 90% B → 6.7 min 2% B → 7.5 min 2% B; Fluss: 0.75 ml/min; Säulentemperatur: 300C; UV-Detektion: 210 nm.
Methode 2: Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml Perchlorsäure (70%-ig) / 1 Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 9 min 0% B → 9.2 min 2% B → 10 min 2% B; Fluss: 0.75 ml/min; Säulentemperatur: 300C; UV-Detektion: 210 nm.
Methode 3: Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%- ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 4: Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%- ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 5: Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%- ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min, 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 6: Säule: GROM-SIL 120 ODS-4 HE, 10 μM, 250 mm x 30 mm; Laufinittel und Gradientenprogramm: Acetonitril/0.1% wässrige Ameisensäure 10:90 (0-3 min), Acetonitril/0.1% wässrige Ameisensäure 10:90 — » 95:5 (3-27 min), Acetonitril/0. f% wässrige Ameisensäure 95T5" (27-34 min), Acetonitril/0.1% wässrige Ameisensäure 10:90 (34-38 min); Fluss: 50 ml/min; Temperatur: 22°C; UV-Detektion: 254 nm.
Ausgangsverbindungen
Beispiel IA
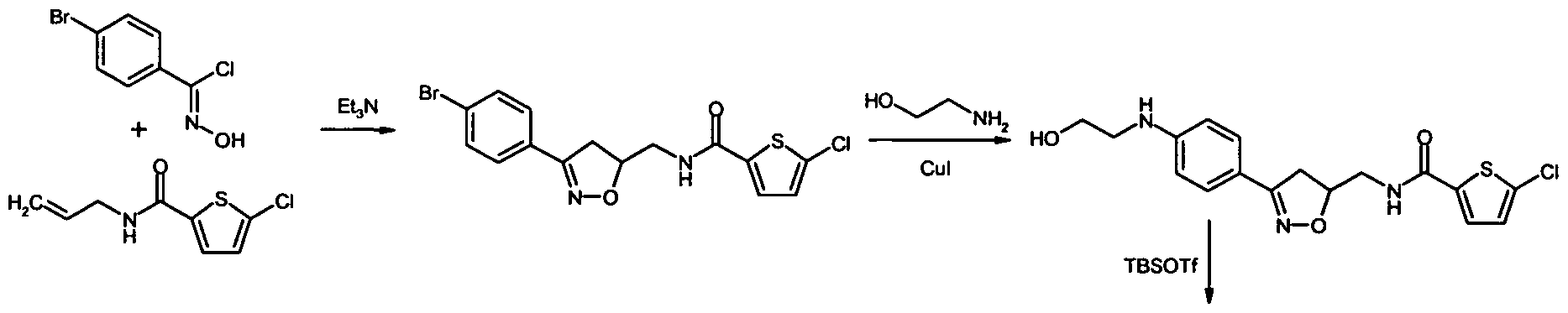
N-{[3-(4-Bromphenyl)-4,5-dihydroisoxazol-5-yl]methyl}-5-chlorthiophen-2-carbonsäureamid
Eine Lösung von 748 mg (3.708 mmol) 4-Brom-N-hydroxybenzimidoylchlorid (M.R. Barbachyn et al., J. Med. Chem. 46(2), 284-302 (2003)) und 1.0 g (4.265 mmol) N-Allyl-5-chlorthiophen- carbonsäureamid in 30 ml wasserfreiem Dichlormethan wird bei 00C tropfenweise mit 594 μl (4.265 mmol) Triethylamin versetzt. Das Reaktionsgemisch wird 15 Stunden bei Raumtemperatur gerührt. Anschließend wird der Ansatz am Rotationsverdampfer zur Trockene eingeengt. Der erhaltene Rückstand wird mit einem Gemisch aus Acetonitril und Wasser im Volumenverhältnis 1 : 1 verrührt. Das darin unlösliche Produkt wird abgesaugt, mit Acetonitril nachgewaschen und im Hochvakuum getrocknet. Es werden 1.1 g (71% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSO-Cl6, δ/ppm): 8.90 (t, IH), 7.67 (2 d, zusammen 3H), 7.60 (d, 2H), 7.19 (d, IH), 4.90-4.84 (m, IH), 3.52 (dd, IH), 3.45-3.42 (m, 2H), 3.21 (dd, IH).
HPLC (Methode 3): R, = 2.36 min.
MS (ESIpos, m/z): 399/401/403 (7W1Br, 35C1/37C1) (M+H)+.
Beispiel 2A
5-Chlor-N-[(3-{4-[(2-hydroxyethyl)amino]phenyl}-4,5-dihydroisoxazol-5-yl)methyl]thiophen-2- carbonsäureamid
1.09 g (2.737 mmol) des Produktes aus Beispiel IA werden in 20 ml wasserfreiem Dioxan gelöst und nacheinander mit 397 μl (6.569 mmol) Aminoethanol, 104 mg (0.547 mmol) Kupfer(I)-jodid, 2.32 g (10.95 mmol) Kaliumphosphat und 175 μl (1.642 mmol) N,N'-Dimethylethylendiamin
versetzt. Die Rückflußapparatur wird durch wiederholtes Anlegen eines leichten Vakuums und Begasen mit Argon inertisiert. Das Reaktionsgemisch wird 15 Stunden zum Rückfluß erhitzt. Da der Umsatz nach dieser Zeit etwa 50% beträgt, läßt man das Gemisch auf RT kommen und versetzt erneut mit den gleichen Mengen an Aminoethanol, Kupfer(I)-jodid, Kaliumphosphat und NN- Dimethylethylendiamin. Es wird nach Inertisieren weitere 20 Stunden zum Rückfluß erhitzt. Nach dieser Zeit läßt man auf RT abkühlen. Es wird mit Wasser versetzt und mit Ethylacetat extrahiert. Der organische Extrakt wird nacheinander mit Wasser und gesättigter Kochsalz-Lösung gewaschen. Es wird über wasserfreiem Magnesiumsulfat getrocknet, filtriert und das Filtrat im Vakuum vom Lösemittel befreit. Der Rückstand wird mittels präparativer HPLC (Methode 6) gereinigt. Die erhaltene Produktfraktion wird mit einem Gemisch aus Acetonitril und NN- Dimethylformamid verrührt. Der Feststoff wird abgesaugt, mit Acetonitril gewaschen und im Hochvakuum getrocknet. Es werden 152 mg (15% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSO-Cl6, δ/ppm): 8.88 (t, IH), 7.68 (d, IH), 7.37 (d, 2H), 7.19 (d, IH), 6.61 (d, 2H), 6.09 (t, IH), 4.75-4.70 (m, IH), 4.72 (t, IH), 3.54 (dt, 2H), 3.42-3.35 (m, 3H), 3.17-3.08 (m, 3H).
HPLC (Methode 1): R, = 3.62 min.
MS (DCI, NH3, m/z): 380/382 (35C1/37C1) (M+H)+, 397/399 (M+NH»)+.
Beispiel 3A
N-[(3-{4-[(2-{[tert.-Butyl(dimethyl)silyl]oxy}ethyl)amino]phenyl}-4,5-dihydroisoxazol-5- yl)methyl]-5-chlorthiophen-2-carbonsäureamid
Eine Suspension von 146 mg (0.384 mmol) des Produktes aus Beispiel 2A und 67 μl (0.577 mmol) 2,6-Dimethylpyridin in 15 ml wasserfreiem Dichlormethan wird bei -500C mit 93 μl (0.404 mmol) ter£-Butyl(dimethyl)silyl-trifluormethansulfonat versetzt. Das Reaktionsgemisch wird 15 Stunden bei Raumtemperatur gerührt. Dann werden ca. 30 ml Wasser zugesetzt und mit Dichlormethan extrahiert. Der Extrakt wird mit Wasser gewaschen, über wasserfreiem Νatriumsulfat getrocknet, filtriert und am Rotationsverdampfer vom Lösemittel befreit. Der Rückstand wird mittels
präparativer HPLC (Methode 6) gereinigt. Es werden 136 mg (72% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSOd6, δfpprn): 8.86 (t, IH), 7.66 (d, IH), 7.33 (d, 2H), 7.17 (d, IH), 6.58 (d, 2H), 6.08 (t, IH), 4.73-4.67 (m, IH), 3.67 (t, 2H), 3.39-3.33 (m, 3H), 3.17 (dt, 2H), 3.08 (dd, IH), 0.83 (s, 9H), 0.01 (s, 6H).
HPLC (Methode 3): R, = 2.99 min.
MS (ESIpos, m/z): 494/496 (35C1/37C1) (M+H)+.
Beispiel 4A
N-[(3- {4-[(2- {[tert. -Butyl(dimethyl)silyl]oxy} ethyl)(cyano)amino]phenyl} -4,5-dihydroisoxazol-5- yl)methyl]-5-chlorthiophen-2-carbonsäureamid
In einem dickwandigen Glasröhrchen mit Schraubverschluss wird ein Gemisch aus 106 mg (0.215 mmol) des Produktes aus Beispiel 3A, 54 mg (0.644 mmol) Νatriumhydrogencarbonat und 86 μl (0.257 mmol) einer 3-molaren Lösung von Bromcyan in Dichlormethan in 5 ml Tetrahydrofuran insgesamt 5 Tage auf 40-50°C erwärmt. Dabei wird jeweils nach Ablauf des ersten bis vierten Tages das Reaktionsgefäß bei Raumtemperatur geöffnet und nochmals die gleichen Mengen Bromcyanlösung und Νatriumhydrogencarbonat hinzugefügt. Nach Ablauf der fünf Tage wird das Reaktionsgemisch mit Dichlormethan verdünnt und nacheinander mit Wasser, gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen über wasserfreiem Natriumsulfat, filtrieren und Entfernen des Lösemittels am Rotationsverdampfer wird der Rückstand in Acetonitril gelöst und mit dem gleichen Volumen Wasser versetzt. Dabei fällt das Produkt aus. Es wird abgesaugt und im Hochvakuum getrocknet. Es werden 71 mg (64% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSOd6, δ/ppm): 8.87 (t, IH), 7.68 (d, 2H), 7.67 (d, IH), 7.25 (d, 2H), 7.17 (d, IH), 4.87-4.81 (m, IH), 3.88-3.83 (m, 4H), 3.48 (dd, IH), 3.45-3.40 (m, 2H), 3.18 (dd, IH), 0.82 (s, 9H), -0.05 (s, 6H).
HPLC (Methode 2): R, = 5.33 min.
MS (DCI, NH3, m/z): 519/521 (35Cy37Cl) (M+H)+, 536/538 (M+NHO*.
Beispiel 5A
Acetophenon-(4-jodphenyl)hydrazon
Eine Lösung von 2.0 g (8.546 mmol) 4-Jodphenylhydrazin in 30 ml 50%-iger Essigsäure wird mit einer Lösung von 1.54 g (12.82 mmol) Acetophenon in 10 ml des gleichen Lösemittels versetzt. Es wird bei Raumtemperatur gerührt, wobei ein Niederschlag ausfällt. Nach 30 Minuten wird der Niederschlag abfϊltriert und nacheinander mit Wasser und Cyclohexan gut gewaschen. Der Rückstand wird im Hochvakuum getrocknet. Es werden 1.95 g (68% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSO-(I6, δ/ppm): 7.78 (d, 2H), 7.51 (d, 2H), 7.38 (dd, 2H), 7.30 (dd, IH), 7O7 (d, 2H); 2.25 (s, 3H):
HPLC (Methode 4): R, = 3.22 min.
MS (ESIpos, m/z): 337 (M+H)+.
Beispiel 6A
1 -(4- Jodphenyl)-3 -phenyl-7H-pyrazol-4-carbaldehyd
Bei 00C werden 1.08 ml (11.58 mmol) Phosphorylchlorid (POCI3) langsam zu 10 ml wasserfreiem N,N-Dimethylformamid zugetropft. Nach 30 Minuten bei 0°C wird eine Lösung von 1.95 g (5.792
mmol) des Produktes aus Beispiel 5 A in 10 ml N.N-Dimethylformamid zugetropft und das Reaktionsgemisch eine weitere Stunde bei 00C gerührt. Dann lässt man auf Raumtemperatur erwärmen, rührt eine weitere Stunde, bevor man auf 600C erwärmt. Das Reaktionsgemisch wird 15 Stunden bei dieser Temperatur gerührt. Anschließend lässt man auf Raumtemperatur abkühlen, versetzt mit 80 ml gesättigter Νatriumhydrogencarbonat-Lösung und extrahiert mit Ethylacetat. Der organische Extrakt wird nacheinander mit Wasser und gesättigter Kochsalz-Lösung gewaschen. Nach Trocknen über wasserfreiem Natriumsulfat wird filtriert und das Lösemittel am Rotationsverdampfer entfernt. Der erhaltene Rückstand wird mit Diisopropylether verrieben. Der Feststoff wird abgesaugt, mit Diisopropylether gewaschen und im Hochvakuum getrocknet. Es werden 1.34 g (62% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSO-d6, δ/ppm): 9.98 (s, IH), 9.36 (s, IH), 7.95-7.90 (m, 4H), 7.82 (d, 2H), 7.53-7.48 (m, 3H).
HPLC (Methode 4): R, = 3.08 min.
MS (ESIpos, m/z): 375 (M+H)+.
Beispiel 7A
1 -(2,4-Dimethoxyphenyl)-N- { [ 1 -(4-j odphenyl)-3 -phenyl-/H-pyrazol-4-yl]methyl } methanamin
1.34 g (3.581 mmol) des Produktes aus Beispiel 6A und 538 μl (3.581 mmol) 2,4- Dimethoxybenzylamin werden in 40 ml Dichlorethan gelöst und eine Stunde bei Raumtemperatur gerührt. Dann werden 1.52 g (7.162 mmol) Νatriumtriacetoxyborhydrid und 820 μl (14.33 mmol) Eisessig zugefügt. Das Reaktionsgemisch wird 15 Stunden bei Raumtemperatur gerührt. Anschließend wird gesättigte Νatriumhydrogencarbonat-Lösung zugesetzt und das Produkt mit Dichlormethan extrahiert. Der organische Extrakt wird mit Wasser gewaschen und über wasserfreiem Νatriumsulfat getrocknet. Nach Filtration wird das Lösemittel am Rotationsverdampfer entfernt. Das Rohprodukt wird im Hochvakuum getrocknet und ohne weitere Reinigung in der nächsten Reaktion eingesetzt. Es werden 1.89 g der Titelverbindung erhalten.
HPLC (Methode 5): R, = 2.10 min (60%).
MS (ESIpos, m/z): 526 (M+H)+.
Beispiel 8A
5-Chlor-N-(2,4-dimethoxybenzyl)-N-{[l-(4-jodphenyl)-3-phenyl-7H-pyrazol-4-yl]methyl}- thiophen-2-carbonsäureamid
Eine Lösung von 1.89 g (3.597 mmol) des Produktes aus Beispiel 7A und 1.25 ml Diisopropylethylamin (Ηünig-Base) in 40 ml wasserfreiem Tetrahydrofuran werden mit einer Lösung von 651 mg (3.597 mmol) 5-Chlorthiophen-2-carbonsäurechlorid in 10 ml wasserfreiem Tetrahydrofuran versetzt. Das Reaktionsgemisch wird 15 Stunden bei Raumtemperatur gerührt. Anschließend wird das Lösemittel am Rotationsverdampfer entfernt, der Rückstand in Dichlor- methan aufgenommen und nacheinander mit gesättigter Νatriumhydrogencarbonat-Lösung und Wasser gewaschen. Nach Trocknen über wasserfreiem Natriumsulfat wird filtriert, eingedampft und der Rückstand mittels präparativer ΗPLC (Methode 6) gereinigt. Es werden 1.05 g (43% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSOd6, δ/ppm): 8.53 (breit, IH), 7.85 (d, 2H), 7.76 (d, 2H), 7.61 (d, 2H), 7.43-7.38 (m, 3H), 7.15 (breit, IH), 7.08 (d, IH), 7.01 (breit, IH), 6.48-6.43 (m, 2H), 4.70 (breit, 2H), 4.58 (s, breit, 2H), 3.71 (s, 3H), 3.57 (breit, 3H).
HPLC (Methode 2): Rt = 6.47 min.
MS (ESIpos, m/z): 670/672 (35CV37Cl) (M+H)+.
Beispiel 9A
5-Chlor-N-(2,4-dimethoxybenzyl)-N-[( 1 - {4-[(2-hydroxyethyl)amino]phenyl} -3-phenyl-/H-pyrazol- 4-yl)methyl]thiophen-2-carbonsäureamid
372 mg (0.555 mmol) der Verbindung aus Beispiel 8A werden wie unter Beispiel 2A beschrieben mit Aminoethanol umgesetzt. Nach Reinigung über präparative ΗPLC (Methode 6) werden 158 mg (47% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, DMSOd6, δ/ppm): 8.18 (s, breit, IH), 7.57-7.54 (m, 4H), 7.41-7.32 (m, 3H), 7.16 (breit, IH), 7.07 (d, IH), 7.00 (breit, IH), 6.68 (d, 2H), 6.49-6.43 (m, 2H), 5.73 (t, IH), 4.72- - 4:67 (m, 2H)r4.56 (S, breit, 2H),-3.72 (s, 3H), 3τ60-3.54 (m, 5H), 3.-13 (dt, 2H).
HPLC (Methode 1): R, = 4.74 min.
MS (ESIpos, m/z): 603/605 (35C1/37C1) (M+H)+.
Beispiel IQA
N-[( 1 -{4-[(2- {[tert. -Butyl(dimethyl)silyl]oxy} ethyl)amino]phenyl} -3-phenyl-7H-pyrazol-4- yl)methyl]-5-chlor-N-(2,4-dimethoxybenzyl)thiophen-2-carbonsäureamid
210 mg (0.350 mmol) der Verbindung aus Beispiel 9A werden analog zu dem unter Beispiel 3A beschriebenen Verfahren zu 194 mg (78% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (500 MHz, DMSO-d6, δ/ppm): 8.20 (s, breit, IH), 7.57-7.54 (m, 4H), 7.41-7.32 (m, 3H), 7.15 (breit, IH), 7.08 (d, IH), 7.00 (breit, IH), 6.68 (d, 2H), 6.49-6.43 (m, 2H), 5.77 (t, IH), 4.70 (breit, 2H), 4.56 (s, breit, 2H), 3.73 (t, 2H), 3.71 (s, 3H), 3.58 (breit, 3H), 3.19 (dt, 2H).
HPLC (Methode 2): R, = 5.86 min.
MS (ESIpos, m/z): 717/719 (35C1/37C1) (M+H)+.
Beispiel IIA
N-[O- {4-[(2- { [tert. -Butyl(dimethyl)silyl]oxy} ethyl)(cyano)amino]phenyl} -3-phenyl-/H-pyrazol-4- yl)methyl]-5-chlor-N-(2,4-dimethoxybenzyl)thiophen-2-carbonsäureamid
192 mg (0.268 mmol) der Verbindung aus Beispiel 10A werden analog zu dem unter Beispiel 4A beschriebenen Verfahren zu 173 mg (87% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (500 MHz, DMSO-(I6, δ/ppm): 8.47 (breit, IH), 7.97 (d, 2H), 7.61 (d, 2H), 7.46-7.38 (m, 3H), 7.33 (d, 2H), 7.17 (breit, IH), 7.10 (d, IH), 7.03 (breit, IH), 6.50-6.45 (m, 2H), 4.73 (breit, 2H), 4.60 (s, breit, 2H), 3.91-3.87 (m, 4H), 3.73 (s, 3H), 3.59 (breit, 3H).
HPLC (Methode 3): R, = 3.41 min.
MS (ESIpos, m/z): 1A2I1AA (35C1/37C1) (M+H)+.
Beispiel 12A
1,1,1 -Trifluoraceton-(4-jodphenyl)hydrazon
Analog zu dem unter Beispiel 5 A beschriebenen Verfahren werden 2.5 g (10.68 mmol) 4- Jodphenylhydrazon und 2.28 ml (16.02 mmol) Trifluoraceton zu 2.18 g (62% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (400 MHz, DMSOd6, δ/ppm): 7.57 (d, 2H), 7.01 (d, 2H), 2.05 (s, 3H).
HPLC (Methode 3): R, = 2.74 min.
MS (ESIneg, m/z): 327 (M-H)+.
Beispiel 13A
1 -(4- Jodphenyl)-3 -(trifluormethyl)-/H-pyrazol-4-carbaldehyd
Analog zu dem unter Beispiel 6A beschriebenen Verfahren werden 2.18 g (6.64 mmol) der Verbindung aus Beispiel 12A zu 2.46 g (100% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (400 MHz, DMSO-d6, δ/ppm): 9.97 (s, IH), 9.50 (s, IH), 7.97 (d, 2H), 7.50 (d, 2H).
HPLC (Methode 4): R, = 2.89 min.
Beispiel 14A
1 -(2,4-Dimethoxyphenyl)-N- { [ 1 -(4-j odphenyl)-3-(trifluormethyl)-7H-pyrazol-4-yl]methyl} - methanamin
Analog zu dem unter Beispiel 7A beschriebenen Verfahren werden 2.43 g (6.64 mmol) der Verbindung aus Beispiel 13A zu 3.46 g (100% d. Th.) der Titelverbindung umgesetzt.
ΗPLC (Methode 3): R, = 1.87 min.
MS (ESIpos, m/z): 518 (M+Η)+.
Beispiel 15A
5-Chlor-N-(2,4-dimethoxybenzyl)-N- { [ 1 -(4-j odphenyl)-3-(trifluormethyl)-/H-pyrazol-4- yl]methyl}thiophen-2-carbonsäureamid
Analog zu dem unter Beispiel 8A beschriebenen Verfahren werden 3.43 g (6.64 mmol) der Verbindung aus Beispiel 14A zu 987 mg (22% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (400 MHz, DMSOd6, δ/ppm): 8.63 (s, breit, IH), 7.90 (d, 2H), 7.71 (d, 2H), 7.21 (d, 2H), 7.11 (d, IH), 7.09 (breit, IH), 6.54-6.49 (m, 2H), 4.68 (s, breit, 2H), 4.58 (s, breit, 2H), 3.73 (s, 3H), 3.70 (s, 3H).
HPLC (Methode 2): R, = 6.25 min.
Beispiel 16A
Acetaldehyd-(4-jodphenyl)hydrazon
Analog zu dem unter Beispiel 5A beschriebenen Verfahren werden 17.5 g (74.77 mmol) 4- Jodphenylhydrazon und 6.27 ml (112.2 mmol) Acetaldehyd zu 12.5 g (64% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (400 MHz, DMSOd6, δ/ppm): 9.18 (s, breit, IH), 7.46 (d, 2H), 6.91 (d, 2H), 6.57 (quart, IH), 1.83 (d, 3H).
HPLC (Methode 1): R, = 4.59 min.
MS (ESIpos, m/z): 261 (M+H)+.
Beispiel 17A
l-(4-Jodphenyl)-/H-pyrazol-4-carbaldehyd
Analog zu dem unter Beispiel 6A beschriebenen Verfahren werden 12.5 g (48.06 mmol) der Verbindung aus Beispiel 16A zu 6.37 g (40% d. Th., bezogen auf 90% Reinheit) der Titelverbindung umgesetzt. Anstelle von 600C wird der Ansatz bei 800C gerührt.
1H-NMR (400 MHz, DMSO-Cl6, δ/ppm): 9.91 (s, IH), 9.27 (s, IH), 8.29 (s, IH), 7.91 (d, 2H), 7.73 (d, 2H).
HPLC (Methode 1): R, = 4.44 min.
MS (ESIpos, m/z): 299 (M+H)+.
Beispiel 18A
1 -(2 ,4-Dimethoxyphenyl)-N- { [ 1 -(4-j odphenyl)-/H-pyrazol-4-yl]methyl } methanamin
Analog zu dem unter Beispiel 7A beschriebenen Verfahren werden 6.30 g (21.14 mmol) der Verbindung aus Beispiel 17A zu 9.5 g (62% d. Th., bezogen auf 62% Reinheit) der Titelverbindung umgesetzt.
HPLC (Methode 5): R, = 1.70 min.
MS (ESIpos, m/z): 450 (M+H)+.
Beispiel 19A
5-Chlor-N-(2,4-dimethoxybenzyl)-N-{[l-(4-jodphenyl)-7H-pyrazol-4-yl]methyl}thiophen-2- carbonsäureamid
Analog zu dem unter Beispiel 8 A beschriebenen Verfahren werden 9.5 g (21.13 mmol) der Verbindung aus Beispiel 18A zu 5.14 g (37% d. Th.) der Titelverbindung umgesetzt.
1H-NMR (500 MHz, DMSOd6, δ/ppm): 8.33 (breit, IH), 7.82 (d, 2H), 7.63-7.60 (m, 3H), 7.17 (breit, IH), 7.11-7.10 (m, 2H), 6.53-6.51 (m, 2H), 4.62 (breit, 2H), 4.46 (breit, 2H), 3.73 (s, 3H), 3.71 (s, 3H).
HPLC (Methode 3): R, = 3.08 min.
MS (ESIpos, m/z): 594/596 (35C1/37C1) (M+H)+.
Ausführungsbeispiele
Beispiel 1
5-Chlor-N-({3-[4-(2-imino-l,3-oxazolidin-3-yl)phenyl]-4,5-dihydroisoxazol-5-yl}methyl)thiophen- 2-carbonsäureamid
Eine Suspension von 71 mg (0.137 mmol) der Verbindung aus Beispiel 4A und 19 μl (0.287 mmol) Methansulfonsäure in 10 ml wasserfreiem Acetonitril wird bei Raumtemperatur 15 Stunden lang gerührt. Dabei entsteht eine klare Lösung, die zur Trockene am Rotationsverdampfer eingedampft wird. Der Rückstand wird mit 2 ml Wasser aufgenommen und mit 0.6 ml gesättigter Νatriumhydrogencarbonat-Lösung versetzt. Dabei fällt das Produkt aus. Der Feststoff wird abgesaugt, mit Wasser gewaschen und im Hochvakuum getrocknet. Es werden 48 mg (87% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSOd6, δ/ppm): 8.90 (t, IH), 7.90 (d, 2H), 7.68 (d, IH), 7.62 (d, 2H), 7.19 (d, IH), 6.33 (s, breit, IH), 4.85-4.79 (m, IH), 4.35 (t, 2H), 4.02 (t, 2H), 3.49 (dd, IH), 3.43-3.40 (m, 2H), 3.20 (dd, IH).
HPLC (Methode 1): R4 = 3.75 min.
MS (ESIpos, m/z): 405/407 (35C1/37C1) (M+H)+.
Beispiel 2
5-Chlor-N-( { 1 -[4-(2-imino-l ,3-oxazolidin-3-yl)phenyl]-3-phenyl-/H-pyrazol-4-yl}methyl)- thiophen-2-carbonsäureamid-Ηydrochlorid
172 mg (0.232 mmol) der Verbindung aus Beispiel I IA wird analog zu dem unter Beispiel 1 beschriebenen Verfahren umgesetzt. Abweichend davon wird das Reaktionsgemisch vor dem Eindampfen zur Trockene noch mit 0.5 ml Trifluoressigsäure versetzt und 30 Minuten bei 40 0C gerührt. Dann werden alle flüchtigen Bestandteile am Rotationsverdampfer entfernt. Der erhaltene Rückstand wird in wenig Acetonitril aufgenommen und über Celite filtriert. Das Filtrat wird eingeengt, in Methanol gelöst und mit 1 ml 1 -molarer Salzsäure versetzt. Es wird erneut eingedampft. Das Lösen in Methanol und Eindampfen nach Salzsäure-Zusatz wird noch einmal wiederholt. Es werden 110 mg (88% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, DMSO-(I6, δ/ppm): 9.64 (s, breit, IH), 9.07 (t, IH), 8.90 (s, breit, IH), 8.57 (s, IH), 8.09 (d, 2H), 7.77 (d, 2H)? 7.69 (d, IH), 7.66 (d, 2H), 7.51-7.48 (m, 2H), 7.43-7.41 (m, IH), 7.19 (d, IH), 4.77 (t, 2H), 4.53 (d, 2H), 4.27 (t, 2H).
HPLC (Methode 1): R, = 4.32 min.
MS (ESIpos, m/z): 478/480 (35C1/37C1) (M+H)+.
Beispiel 3
5-Chlor-N-({l-[4-(2-imino-l,3-oxazolidin-3-yl)phenyl]-3-(trifluoromethyl)-/H-pyrazol-4- yl } methyl)thiophen-2-carbonsäureamid-Ηydrochlorid
Die Titelverbindung wird aus der Verbindung aus Beispiel 15A analog zu den unter den Beispielen 9A, 10A, I IA und 2 beschriebenen Verfahren gewonnen.
Beispiel 4
5-Chlor-N-({l-[4-(2-imino-l,3-oxazolidin-3-yl)phenyl]-7H-pyrazol-4-yl}methyl)thiophen-2- carbonsäureamid-Ηydrochlorid
Die Titelverbindung wird aus der Verbindung aus Beispiel 19A analog zu den unter den Beispielen 9 A, 10A, I IA und 2 beschriebenen Verfahren gewonnen.
B. Bewertung der pharmakologischen Wirksamkeit
Die erfϊndungsgemäßen Verbindungen wirken insbesondere als selektive Inhibitoren des Blutgerinnungsfaktors Xa und hemmen nicht oder erst bei deutlich höheren Konzentrationen auch andere Serinproteasen wie Plasmin oder Trypsin.
Als „selektiv" werden solche Inhibitoren des Blutgerinnungsfaktors Xa bezeichnet, bei denen die IC5o-Werte für die Faktor Xa-Inhibierung gegenüber den IC50- Werten für die Inhibierung anderer Serinproteasen, insbesondere Plasmin und Trypsin, um mindestens das 100-fache kleiner sind, wobei bezüglich der Testmethoden für die Selektivität Bezug genommen wird auf die im folgenden beschriebenen Testmethoden der Beispiele B.a.l) und B.a.2).
Die vorteilhaften pharmakologischen Eigenschaften der erfindungsgemäßen Verbindungen können durch folgende Methoden festgestellt werden:
a) Testbeschreibungen (in vitro)
a. I) Messung der Faktor Xa-Hemmung
Zur Bestimmung der Faktor Xa-Hemmung der oben aufgeführten Substanzen wird ein biochemisches Testsystem aufgebaut, in dem die Umsetzung eines Faktor Xa-Substrates zur Ermittlung der enzymätischen Aktivität von humanem Faktor Xa benutzt wird. Dabei spaltet Faktor Xa aus dem peptischen Substrat Aminomethylcoumarin ab, das fluoreszent gemessen wird. Die Bestimmungen werden in Mikrotiterplatten durchgeführt.
Zu testende Substanzen werden in unterschiedlichen Konzentrationen in Dimethylsulfoxid gelöst und 15 min mit humanem Faktor Xa (1.3 nmol/1 gelöst in 50 mmol/1 Tris-Puffer [C,C,C-
Tris(hydroxymethyl)-aminomethan], 100 mmol/1 NaCl, 0.1% BSA [bovines Serumalbumin], pH
7.4) bei 22°C inkubiert. Anschließend wird das Substrat (5 μmol/1 Boc-Ile-Glu-Gly-Arg-AMC von der Firma Bachern) hinzugefügt. Nach einer Inkubation von 30 min wird die Probe bei einer
Wellenlänge von 360 nm angeregt und die Emission bei 460 nm gemessen. Die gemessenen Emissionen der Testansätze mit Prüfsubstanz werden mit den Kontrollansätzen ohne Prüfsubstanz
(ausschließlich Dimethylsulfoxid anstatt Prüfsubstanz in Dimethylsulfoxid) verglichen und aus den Konzentrations-Wirkungs-Beziehungen IC50- Werte berechnet.
Repräsentative Wirkdaten aus diesem Test sind in der folgenden Tabelle 1 aufgeführt:
Tabelle 1
a.2) Bestimmung der Selektivität
Zum Nachweis der Selektivität der Substanzen bezüglich Faktor Xa -Hemmung werden die Prüfsubstanzen auf ihre Hemmung anderer humaner Serinproteasen wie Trypsin und Plasmin hin untersucht. Zur Bestimmung der enzymatischen Aktivität von Trypsin (83 mU/ml von Sigma) und Plasmin (0.1 μg/ml von Kordia) werden diese Enzyme gelöst (50 mmol/1 Tris-Puffer [C,C,C- Tris(hydroxymethyl)-aminomethan], 100 mmol/1 NaCl, 0.1% BSA [bovines Serumalbumin], 5 mmol/1 Calciumchlorid, pH 7.4) und für 15 min mit Prüfsubstanz in verschiedenen Konzentrationen in Dimethylsulfoxid sowie mit Dimethylsulfoxid ohne Prüfsubstanz inkubiert. Anschließend wird die enzymatische Reaktion durch Zugabe der entsprechenden Substrate gestartet (5 μmol/1 Boc-Ile-Glu-Gly-Arg-AMC von Bachern für Trypsin, 50 μmol/1 MeOSuc-Ala- Phe-Lys-AMC von Bachern für Plasmin). Nach einer Inkubationszeit von 30 min bei 22°C wird die Fluoreszenz gemessen (Anregung: 360 nm, Emission: 460 nm). Die gemessenen Emissionen der Testansätze mit Prüfsubstanz werden mit den Kontrollansätzen ohne Prüfsubstanz (ausschließlich Dimethylsulfoxid anstatt Prüfsubstanz in Dimethylsulfoxid) verglichen und aus den Konzentrations-Wirkungs-Beziehungen IC50-Werte berechnet.
a.3) Bestimmung der antikoagulatorischen Wirkung:
Die antikoagulatorische Wirkung der Prüfsubstanzen wird in vitro in Human- und Kaninchen- plasma bestimmt. Dazu wird Blut unter Verwendung einer 0.11 molaren Natriumcitrat-Lösung als Vorlage in einem Mischungsverhältnis Natriumcitrat/Blut 1 :9 abgenommen. Das Blut wird unmittelbar nach der Abnahme gut gemischt und 10 Minuten bei ca. 2500 g zentrifugiert. Der Überstand wird abpipettiert. Die Prothrombinzeit (PT, Synonyme: Thromboplastinzeit, Quick-Test) wird in Gegenwart variierender Konzentrationen an Prüfsubstanz oder dem entsprechenden Lösungsmittel mit einem handelsüblichen Testkit (Hemoliance® RecombiPlastin, Fa. Instrumentation Laboratory) bestimmt. Die Testverbindungen werden 3 Minuten bei 37°C mit dem Plasma inkubiert. Anschließend wird durch Zugabe von Thromboplastin die Gerinnung ausgelöst und der Zeitpunkt des Gerinnungseintritts bestimmt. Es wird die Konzentration an Prüfsubstanz ermittelt, die eine Verdoppelung der Prothrombinzeit bewirkt.
b) Bestimmung der antithrombotischen Wirkung (in vivo)
b.1) Arteriovenöses Shunt-Modell (Kaninchen):
Nüchterne Kaninchen (Stamm: Esd: NZW) werden durch intramuskuläre Gabe einer Rompun/ Ketavet-Lösung narkotisiert (5 mg/kg bzw. 40 mg/kg). Die Thrombusbildung wird in einem arteriovenösen Shunt in Anlehnung an die von CN. Berry et al. [Semin. Thromb. Hemost. 1996, 22, 233-241] beschriebene Methode ausgelöst. Dazu werden die linke Vena jugularis und die rechte Arteria carotis freipräpariert. Ein extracorporaler Shunt wird mittels eines 10 cm langen Venenkatheders zwischen den beiden Gefäßen gelegt. Dieser Katheder ist in der Mitte in einen weiteren, 4 cm langen Polyethylenschlauch (PE 160, Becton Dickenson), der zur Erzeugung einer thrombogenen Oberfläche einen aufgerauhten und zu einer Schlinge gelegten Nylonfaden enthält, eingebunden. Der extrakorporale Kreislauf wird 15 Minuten lang aufrechterhalten. Dann wird der Shunt entfernt und der Nylonfaden mit dem Thrombus sofort gewogen. Das Leergewicht des Nylonfadens ist vor Versuchsbeginn ermittelt worden. Die Prüfsubstanzen werden vor Anlegung des extrakorporalen Kreislaufs entweder intravenös über eine Ohrvene oder oral mittels Schlund- sonde verabreicht.
c) Löslichkeitsassav
Benötigte Reagenzien:
• PBS-Puffer pH 7.4: 90.00 g NaCl p.a. (z.B. Merck Art. Nr. 1.06404.1000), 13.61 g KH2PO4 p.a. (z.B. Merck Art. Nr. 1.04873.1000) und 83.35 g IN NaOH (z.B. Bernd Kraft GmbH Art. Nr. 01030.4000) in einen 1 1 Messkolben einwiegen, mit Wasser auffüllen und ca. 1 Stunde rühren.
• Acetatpuffer pH 4.6: 5.4 g Natriumacetat x 3 H2O p.a. (z.B. Merck Art. Nr. 1.06267.0500) in einen 100 ml Messkolben einwiegen, in 50 ml Wasser lösen, mit 2.4 g Eisessig versetzen, auf 100 ml mit Wasser auffüllen, pH-Wert überprüfen und falls notwendig auf pH 4.6 einstellen.
• Dimethylsulfoxid (z.B. Baker Art. Nr. 7157.2500)
• destilliertes Wasser
Herstellung der Kalibrierlösungen:
Herstellung der Ausgangslösung für Kalibrierlösungen (Stammlösung): In ein 2 ml Eppendorf- Safe-Lock Tube (Eppendorf Art. Nr. 0030 120.094) werden ca. 0.5 mg des Wirkstoffes genau eingewogen, zu einer Konzentration von 600 μg/ml mit DMSO versetzt (z.B. 0.5 mg Wirkstoff + 833 μl DMSO) und bis zur vollständigen Lösung mittels eines Vortexers geschüttelt.
Kalibήerlösung 1 (20 μg/ml): 34.4 μl der Stammlösung werden mit 1000 μl DMSO versetzt und homogenisiert.
Kalibrierlösung 2 (2.5 μg/ml): 100 μl der Kalibrierlösung 1 werden mit 700 μl DMSO versetzt und homogenisiert.
Herstellung der Probenlösuneen:
Probenlösung fύr Löslichkeit bis 10 g/l in PBS-Puffer pH 7.4: In ein 2 ml Eppendorf-Safe-Lock Tube (Eppendorf Art. Nr. 0030 120.094) werden ca. 5 mg des Wirkstoffes genau eingewogen und zu einer Konzentration von 5 g/l mit PBS-Puffer pH 7.4 versetzt (z.B. 5 mg Wirkstoff + 500 μl PBS-Puffer pH 7.4).
Probenlösung fύr Löslichkeit bis 10 g/l in Acetatpuffer pH 4.6: In ein 2 ml Eppendorf-Safe-Lock Tube (Eppendorf Art. Nr. 0030 120.094) werden ca. 5 mg des Wirkstoffes genau eingewogen und zu einer Konzentration von 5 g/l mit Acetatpuffer pH 4.6 versetzt (z.B. 5 mg Wirkstoff + 500 μl Acetatpuffer pH 4.6).
Probenlösung fiir Löslichkeit bis 10 g/l in Wasser: In ein 2 ml Eppendorf-Safe-Lock Tube (Eppendorf Art. Nr. 0030 120.094) werden ca. 5 mg des Wirkstoffes genau eingewogen und zu einer Konzentration von 5 g/l mit Wasser versetzt (z.B. 5 mg Wirkstoff + 500 μl Wasser).
Durchfuhrung:
Die so hergestellten Probenlösungen werden 24 Stunden bei 1400 rpm mittels eines temperierbaren Schüttlers (z.B. Eppendorf Thermomixer comfort Art. Nr. 5355 000.011 mit Wechselblock Art. Nr. 5362.000.019) bei 200C geschüttelt. Von diesen Lösungen werden jeweils 180 μl abgenommen und in Beckman Polyallomer Centrifuge Tubes (Art. Nr. 343621) überfuhrt. Diese Lösungen werden 1 Stunde mit ca. 223.000 *g zentrifugiert (z.B. Beckman Optima L-90K Ultracentrifuge mit Type 42.2 Ti Rotor bei 42.000 rpm). Von jeder Probenlösung werden 100 μl des Überstandes abgenommen und 1:5, 1:100 und 1:1000 mit dem jeweils verwendeten Lösungsmittel (Wasser, PBS-Puffer 7.4 oder Acetatpuffer pH 4.6) verdünnt. Es wird von jeder Verdünnung eine Abfüllung in ein geeignetes Gefäß für die HPLC-Analytik vorgenommen.
Analytik:
Die Proben werden mittels RP-HPLC analysiert. Quantifiziert wird über eine Zwei-Punkt- Kalibrationskurve der Testverbindung in DMSO. Die Löslichkeit wird in mg/1 ausgedrückt.
Analysensequenz:
1. Kallibrierlösung 2.5 mg/ml
2. Kallibrierlösung 20 μg/ml
3. Probenlösung 1 :5
4. Probenlösung 1:100
5. Probenlösung 1: 1000
HPLC-Methode für Säuren:
Agilent 1100 mit DAD (Gl 315A), quat. Pumpe (Gl 31 IA), Autosampier CTC HTS PAL, Degaser (G1322A) and Säulenthermostat (G1316A); Säule: Phenomenex Gemini C18, 50 x 2 mm, 5 μ; Temperatur: 400C; Eluent A: Wasser/Phosphorsäure pH 2; Eluent B: Acetonitril; Flussrate: 0.7 ml/min; Gradient: 0-0.5 min 85% A, 15% B; Rampe: 0.5-3 min 10% A, 90% B; 3-3.5 min 10% A, 90% B; Rampe: 3.5-4 min 85% A, 15% B; 4-5 min 85% A, 15% B.
HPLC-Methode für Basen:
Agilent 1100 mit DAD (Gl 315A), quat. Pumpe (Gl 31 IA), Autosampier CTC HTS PAL, Degaser (G1322A) and Säulenthermostat (G1316A); Säule: VDSoptilab Kromasil 100 C18, 60 x 2.1 mm, 3.5 μ; Temperatur: 300C; Eluent A: Wasser + 5 ml Perchlorsäure/l; Eluent B: Acetonitril; Flussrate: 0.75 ml/min; Gradient: 0-0.5 min 98% A, 2% B; Rampe: 0.5-4.5 min 10% A, 90% B; 4.5-6 min 10% A, 90% B; Rampe: 6.5-6.7 min 98% A, 2% B; 6.7-7.5 min 98% A, 2% B.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfϊndungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfϊndungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.