WO2007137962A1 - Thiazole derivatives - Google Patents
Thiazole derivatives Download PDFInfo
- Publication number
- WO2007137962A1 WO2007137962A1 PCT/EP2007/054908 EP2007054908W WO2007137962A1 WO 2007137962 A1 WO2007137962 A1 WO 2007137962A1 EP 2007054908 W EP2007054908 W EP 2007054908W WO 2007137962 A1 WO2007137962 A1 WO 2007137962A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbamoyl
- bromo
- thiazol
- methyl
- sulfonamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 **(C=C1)C=CC(S(Cl)(=O)=O)=C*1S Chemical compound **(C=C1)C=CC(S(Cl)(=O)=O)=C*1S 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/48—Acylated amino or imino radicals by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof, e.g. carbonylguanidines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/54—Nitrogen and either oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/58—Nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/68—Benzothiazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D277/82—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to novel compounds useful as FBPase inhibitors.
- the invention is concerned particularly with compounds of formula
- R 1 is hydrogen, alkyl, cycloalkyl, halogen, haloalkyl, heterocyclyl, heterocyclylalkyl, aralkyl, -S-R 4 , -O-R 4 or nitro;
- R 2 is hydrogen, alkyl, cycloalkyl, halogen, haloalkyl, alkoxyalkyl, hydroxyalkyl or alkoxycarbonyl;
- R 3 is phenyl, thiazolyl, thiophenyl, pyridinyl, pyrimidinyl, pyradizinyl, oxazoyl or isoxazoyl, wherein phenyl, thiazolyl, thiophenyl, pyridinyl, pyrimidinyl, pyradizinyl, oxazoyl and isoxazoyl are optionally substituted with one to three substituents independently selected from alkyl, cycloalkyl, alkoxy, hydroxy, halogen, haloalkyl, haloalkoxy, aryl, aryloxy, heterocyclyl, heterocyclyloxy, amino, nitro, alkoxyalkyl, hydroxyalkyl, alkoxyalkoxy and hydroxyalkoxy;
- R 4 is alkyl, cycloalkyl, heterocyclylalkyl, aralkyl or haloalkyl;
- R 1 and R 2 are not both at the same time hydrogen and, wherein N-((2-benzothiazolylamino)carbonyl)-4-methyl-benzenesulfonamide;
- the present invention relates to new chemical compounds that are inhibitors of Fructose- 1,6-bisphosphatase (FBPase), a rate-limiting enzyme of gluconeogenesis that is allosterically regulated by AMP and responsible for the hydrolysis of Fructose- 1,6- bisphosphate to Fructose-6-phosphate.
- FBPase Fructose- 1,6-bisphosphatase
- Compounds of the present invention represent novel FBPase AMP site inhibitors and have valuable pharmacological properties suitable in both human and veterinary medicine.
- compounds of the present invention are hypoglycaemic agents and are indicated for the treatment and/or the prophylaxis of disorders of glucose homeostasis, such as Diabetes Mellitus, in particular Type II and Type I Diabetes Mellitus (NIDDM and IDDM), Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT) , and for the prevention of the progression of disorders of the Metabolic Syndrome (MetS, also described as Syndrome X or Insulin Resistance Syndrome) which most important components are insulin resistance (with or without IGT), obesity, dyslipidemia, hypertension, prothrombic and proinflammatory states.
- disorders of glucose homeostasis such as Diabetes Mellitus, in particular Type II and Type I Diabetes Mellitus (NIDDM and IDDM), Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT)
- IGT Impaired Glucose Tolerance
- MetS also described as Syndrome X or Insulin Resistance Syndrome
- compounds of the present invention are also indicated for the prevention and/or the treatment of diabetic complications or diabetic- associated diseases such as cardiomyopathy, macrovascular atherosclerotic disorders, including coronary, cerebrovascular and peripheral artery diseases, microvascular diseases including retinopathy, cataracts, blindness and nephropathy, neuropathy (peripheral neuropathy and sympathetic nerve disorders), diabetic necrosis, infection or depression, and so forth.
- diabetic complications or diabetic- associated diseases such as cardiomyopathy, macrovascular atherosclerotic disorders, including coronary, cerebrovascular and peripheral artery diseases, microvascular diseases including retinopathy, cataracts, blindness and nephropathy, neuropathy (peripheral neuropathy and sympathetic nerve disorders), diabetic necrosis, infection or depression, and so forth.
- compounds of the present invention have cytoprotective effects as anti-ischaemic agents and are useful for preventing ischaemia- induced tissue damage. Therefore, compounds of the present invention can be used in a variety of ischaemic and inflammatory conditions where acute management of tissue injury could be beneficial such as surgical trauma, myocardial infarction, congestive heart failure, stroke, sickle cell disease, and so forth, and have further utility in cardioprotection, in improving cardiac function and tolerance to exercise, in improving red-blood cells and pulmonary endothelial functions, in organ preservation in transplants, and so forth. As such, compounds of the present invention can also be used to treat asthma attacks, hypertension, atheriosclerosis and so forth, and in the management of certain excess glycogen storage diseases such as McArdle disease (GSD-Type V) and others.
- McArdle disease GSD-Type V
- compounds of the present invention can have further utility in the prevention and/or the management of a large set of diseases including obesity, atherosclerosis, inflammation, Alzheimer disease, cancer or respiratory disorders such as excess mucus production and allergic asthma, excess surfactant synthesis, cystic fibrosis, and so forth.
- compounds of the present invention can be used in any disease, syndrome, symptom or organ malfunction found associated with increased expression and/or activity of one or another FBPase isoform, at the obvious exception of certain deficiencies where FBPase upregulation might be beneficial for ensuring normal body function, e.g. certain glycogen storage diseases, such as GSD-Type 0 (glycogen synthase deficiency) .
- GSD-Type 0 glycogen storage diseases
- the compounds of formula I and their pharmaceutically acceptable salts and esters are novel and have valuable pharmacological properties.
- they are FBPase inhibitors and can be used in the prophylaxis and/or treatment of Diabetes Mellitus such as Type I, Type II and Type III Diabetes, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), Metabolic Syndrome, insulin resistance, dyslipidemia, obesity, hypertension, atherosclerosis, diabetic complications, inflammation, respiratory diseases or ischaemia.
- IGF Impaired Fasting Glucose
- ITT Impaired Glucose Tolerance
- Metabolic Syndrome insulin resistance, dyslipidemia, obesity, hypertension, atherosclerosis, diabetic complications, inflammation, respiratory diseases or ischaemia.
- Preferred is the prophylaxis and/or prevention of progression and/or treatment of Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III,
- Impaired Fasting Glucose IGF
- Impaired Glucose Tolerance ITT
- Metabolic Syndrome diabetic complications and ischaemia.
- prophylaxis and/or treatment of Diabetes Mellitus Type II and Diabetes Mellitus Type I is particularly preferred.
- Objects of the present invention are the compounds of formula I and their aforementioned salts and esters per se and their use as therapeutically active substances, a process for the manufacture of the said compounds, intermediates, pharmaceutical compositions, medicaments containing the said compounds, their pharmaceutically acceptable salts and esters, the use of the said compounds, esters and salts for the prophylaxis and/or therapy of illnesses, especially in the treatment and/or prophylaxis of Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia, particularly Diabetes Mellitus Type II and Diabetes Mellitus Type I
- the compounds of the present invention can be combined with one or more additional active substances indicated for the management of human and veterinary homeostasis in any suitable ratio.
- Such substances may be insulin sensitizers such as peroxisome proliferator- activated receptor modulators (PPAR alpha, gamma, delta agonists, particularly with thiazolinediones such as rosiglitazone and pioglitazone), insulin secretagogues (sulfonylureas such as glyburide, glimepiride and glipizide, and non- sulfonylurea secretagogues such as the meglitinides repaglinide and nateglinide), insulin, metformin, alpha- glucosidase inhibitors (e.g.
- PPAR alpha, gamma, delta agonists particularly with thiazolinediones such as rosiglitazone and pioglitazone
- insulin secretagogues sulfon
- acarbose miglitol
- GLP-I glucagon-like peptide
- DPP-IV dipeptidyl peptidase- IV
- glycogen phosphorylase inhibitors glycogen synthase kinase-3 inhibitors
- ll ⁇ -hydroxysteroid dehydrogenase- 1 inhibitors glycogen synthase kinase-3 inhibitors
- ll ⁇ -hydroxysteroid dehydrogenase- 1 inhibitors ll ⁇ -hydroxysteroid dehydrogenase- 1 inhibitors
- carnitine palmitoyltranferase- 1 inhibitors glucocorticoid receptor antagonists
- glucagon receptor antagonists Adenosine (A 2B ) receptor agonists
- amylin agonists e.g. pramlintide
- lipase inhibitor e.g. orlistat
- alkyl signifies a straight-chain or branched-chain alkyl group with 1 to 8 carbon atoms, preferably a straight or branched-chain alkyl group with 1 to 6 carbon atoms and particularly preferred a straight or branched-chain alkyl group with 1 to 4 carbon atoms
- straight- chain and branched Ci-Cs alkyl groups are methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert.
- cycloalkyl signifies a cycloalkyl ring with 3 to 8 carbon atoms and preferably a cycloalkyl ring with 3 to 6 carbon atoms.
- Examples of C 3 -Cs cycloalkyl are cyclopropyl, methyl-cyclopropyl, dimethylcyclopropyl, cyclobutyl, methyl- cyclobutyl, cyclopentyl, methyl-cyclopentyl, cyclohexyl, methyl-cyclohexyl, dimethyl- cyclohexyl, cycloheptyl and cyclooctyl, preferably cyclopropyl.
- alkoxy signifies a group of the formula alkyl-O- in which the term “alkyl” has the previously given significance, such as methoxy, ethoxy, n- propoxy, isopropoxy, n-butoxy, isobutoxy, sec. butoxy and tert.butoxy, preferably methoxy and ethoxy and most preferred methoxy.
- haloalkyl signifies an alkyl group as previously defined, wherein one to five hydrogen atoms are substituted by halogen, preferably fluoro.
- halogen preferably fluoro.
- Preferred examples are pentafluoroethyl and particularly trifluoromethyl and difluoromethyl.
- haloalkoxy alone or in combination, signifies a group of the formula haloalkyl-O- in which the term “haloalkyl” is defined as beofore.
- hydroxyalkyl signifies an alkyl group as defined before, wherein one or more hydrogen atoms, preferably one hydrogen atom is replaced by a hydroxy group.
- hydroxyalkyl are hydroxymethyl and hydroxyethyl.
- aryl signifies a phenyl or naphthyl group, preferably a phenyl group which optionally carries one or more substituents, preferably one to three, each independently selected from halogen, trifluoromethyl, trifluoromethoxy, amino, alkyl, alkoxy, alkylcarbonyl, cyano, carbamoyl, alkoxycarbamoyl, methylendioxy, carboxy, alkoxycarbonyl, aminocarbonyl, alkyaminocarbonyl, dialkylaminocarbonyl, hydroxy, nitro, alkyl-SO2-, amino-SO2-, cycloalkyl and the like.
- Examples are phenyl or naphthyl, particularly phenyl optionally substituted with one to three, preferably one or two substituents independently selected from alkyl, halogen, alkoxy, trifluoromethoxy, nitro and trifluoromethyl.
- Preferred examples are phenyl or phenyl substituted with one to three, preferably one or two substituents independently selected from alkyl, halogen and alkoxy.
- aryloxy alone or in combination, signifies an aryl-O- group in which the term “aryl” has the previously given significance.
- alkoxyalkyl signifies an alkyl group as defined before, wherein one or more hydrogen atoms, preferably one hydrogen atom is replaced by an alkoxy group as defined before.
- alkoxyalkyl are methoxymethyl and methoxyethyl.
- alkoxycarbonyl alone or in combination, signifies an alkoxy-CO- group in which the term “alkoxy” has the previously given significance.
- alkoxyalkoxy signifies an alkoxy group as defined before, wherein one or more hydrogen atoms, preferably one hydrogen atom is replaced by an alkoxy group as defined before.
- alkoxyalkoxy are methoxymethoxy and methoxyethoxy.
- hydroxyalkoxy signifies an alkoxy group as defined before, wherein one or more hydrogen atoms, preferably one hydrogen atom is replaced by an hydroxy group.
- An example of hydroxyalkoxy is hydroxyethoxy.
- heterocyclyl alone or in combination signifies a saturated, partially unsaturated or aromatic 5- to 10-membered heterocycle which contains one or more hetero atoms selected from nitrogen, oxygen and sulphur. If desired, it can be substituted on one or more carbon atoms e.g. by halogen, alkyl, alkoxy, oxo etc. and/or on a secondary nitrogen atom (i.e.
- alkyl, cycloalkyl, aralkoxycarbonyl, alkanoyl, phenyl or phenylalkyl or on a tertiary nitrogen atom (i.e. N-) by oxido, with halogen, alkyl, cycloalkyl and alkoxy being preferred.
- heterocyclyl groups are pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, pyrazoyl, imidazoyl (e.g.
- Preferred are oxazolyl, thienyl, pyrazolyl, thiazolyl, 1,2,3-thiadiazolyl and pyrrolidinyl, wherein oxazolyl, thienyl, pyrazolyl, thiazolyl, 1,2,3-thiadiazolyl and pyrrolidinyl are optionally substituted with one to three substituents, preferably one or two substituents independently selected from alkyl, halogen and cyclalkyl, particularly cyclohexyl.
- heterocyclylalkyl signifies the heterocyclyl- alkyl group, wherein the terms “heterocyclyl” and “alkyl” are as priviously defined.
- amino signifies a primary, secondary or tertiary amino group bonded via the nitrogen atom, with the secondary amino group carrying an alkyl or cycloalkyl substituent and the tertiary amino group carrying two similar or different alkyl or cycloalkyl substituents or the two nitrogen substitutents together forming a ring, such as, for example, -NH 2, methylamino, ethylamino, dimethylamino, diethylamino, methyl-ethylamino, pyrrolidin-1-yl or piperidino etc., preferably primary amino, dimethylamino and diethylamino and particularly dimethylamino.
- halogen alone or in combination, signifies fluorine, chlorine, bromine or iodine and preferably fluorine, chlorine or bromine.
- carbonyl alone or in combination, signifies the -C(O)- group.
- aralkyl alone or in combination, signifies the aryl-alkyl group, wherein the terms “aryl” and “alkyl” are as priviously defined. Preferred is benzyl.
- nitro alone or in combination signifies the -NO 2 group.
- salts refers to those salts which retain the biological effectiveness and properties of the free bases or free acids, which are not biologically or otherwise undesirable.
- the salts are formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, preferably hydrochloric acid, and organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxylic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, N-acetylcystein and the like.
- salts derived from an inorganic base include, but are not limited to, the sodium, potassium, lithium, ammonium, calcium, magnesium salts and the like.
- Salts derived from organic bases include, but are not limited to salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, lysine, arginine, N-ethylpiperidine, piperidine, polymine resins and the like.
- the compound of formula I can also be present in the form of zwitterions.
- a preferred pharmaceutically acceptable salt of compounds of formula I is the sodium salt.
- the compounds of formula I can also be solvated, e.g. hydrated.
- the solvation can be effected in the course of the manufacturing process or can take place e.g. as a consequence of hygroscopic properties of an initially anhydrous compound of formula I (hydration).
- pharmaceutically acceptable salts also includes physiologically acceptable solvates.
- “Pharmaceutically acceptable esters” means that compounds of general formula (I) may be derivatised at functional groups to provide derivatives which are capable of conversion back to the parent compounds in vivo. Examples of such compounds include physiologically acceptable and metabolically labile ester derivatives, such as methoxymethyl esters, methylthiomethyl esters and pivaloyloxymethyl esters. Additionally, any physiologically acceptable equivalents of the compounds of general formula (I) , similar to the metabolically labile esters, which are capable of producing the parent compounds of general formula (I) in vivo, are within the scope of this invention.
- the compounds of formula I can contain several asymmetric centers and can be present in the form of optically pure enantiomers, mixtures of enantiomers such as, for example, racemates, optically pure diastereioisomers, mixtures of diastereoisomers, diastereoisomeric racemates or mixtures of diastereoisomeric racemates.
- R 1 is hydrogen, alkyl, cycloalkyl, halogen, haloalkyl, heterocyclyl, heterocyclylalkyl, aralkyl, -S-R 4 or -O-R 4 ;
- R 2 is hydrogen, alkyl, cycloalkyl, halogen or haloalkyl
- R 3 is phenyl, thiazolyl, thiophenyl, pyridinyl, pyrimidinyl, pyradizinyl, oxazoyl or isoxazoyl, wherein phenyl, thiazolyl, thiophenyl, pyridinyl, pyrimidinyl, pyradizinyl, oxazoyl and isoxazoyl are optionally substituted with one to three substituents independently selected from alkyl, cycloalkyl, alkoxy, hydroxy, halogen, haloalkyl, haloalkoxy, aryl, aryloxy, heterocyclyl, heterocyclyloxy, amino and nitro;
- R 4 is alkyl, cycloalkyl, heterocyclylalkyl or aralkyl
- R 1 is hydrogen, methyl, halogen, thienyl or -S-R 4 .
- Particularly preferred are those compounds of formula I, wherein R 1 is bromo.
- R 2 is hydrogen, methyl, or halogen. Particularly preferred are those, wherein R is hydrogen. Further preferred are those compounds of formula I, wherein R 2 alkoxyalkyl, hydroxyalkyl or alkoxycarbonyl.
- R 3 is phenyl, thiophenyl or pyridinyl, wherein phenyl, thiophenyl and pyridinyl are optionally substituted with one to three substituents, preferably one or two substituents independently selected from alkyl, cycloalkyl, halogen, haloalkoxy, aryloxy, dichloro-methyl-lH-pyrazolyl)oxy, methylthiazolyl, cyclohexyl-methyl-oxazolyl, oxazolyl, thiadiazolyl, methyloxazolyl, methylpyrrolidinyl, (methoxymethyl)-pyrrolidinyl, (methylethyl)-pyrrolidinyl and methoxypyridinyl.
- R 3 is phenyl, thiophenyl or pyridinyl, wherein phenyl, thiophenyl and pyridinyl are optionally substituted with one to three substituents, particularly one or two substituents independently selected from alkyl, cycloalkyl, halogen, haloalkoxy, aryloxy, 3,4-dichloro- 1- methyl- lH-pyrazol-5-yl)oxy, 2-methyl-4-thiazolyl, 4-cyclohexyl-2-methyl-5-oxazolyl, oxazolyl, l,2,3-thiadiazol-4-yl, 4-methyl-5-oxazolyl, 2-methyl-l-pyrrolidinyl, 2- (methoxymethyl)-l-pyrrolidinyl, 2-(l-methylethyl)-l-pyrrolidinyl and 6-methoxypyridin- 3-yl.
- R 3 is phenyl, thiophenyl or pyridinyl, wherein phenyl, thiophenyl and pyridinyl are optionally substituted with one to three substituents independently selected from alkyl, cyclopropyl, halogen, haloalkoxy, phenyl, difluorophenyl, methylpenyl, methoxyphenyl, methyloxazolyl and methoxypyridinyl.
- R 3 is phenyl, thiophenyl, pyridinyl or thiazolyl, wherein phenyl, thiophenyl, pyridinyl and thiazolyl are optionally substituted with one to three substituents independently selected from alkyl, cycloalkyl, halogen, haloalkoxy, alkoxyalkyl, alkoxyalkoxy, aryloxy, dichloro-methyl- IH- pyrazolyl)oxy, methylthiazolyl, cyclohexyl-methyl-oxazolyl, oxazolyl, thiadiazolyl, methyloxazolyl, methylpyrrolidinyl, (methoxymethyl)-pyrrolidinyl, (methylethyl)- pyrrolidinyl, methoxypyridinyl, 6-methoxy-4-methyl-pyridin-3-yl, alkoxy-alkyl-phen
- R 3 is phenyl, thiophenyl, pyridinyl or thiazolyl, wherein phenyl, thiophenyl, pyridinyl and thiazolyl are optionally substituted with one to three substituents independently selected from alkyl, cycloalkyl, halogen, haloalkoxy, alkoxyalkyl, alkoxyalkoxy, aryloxy, 3,4-dichloro-l-methyl-lH- pyrazol-5-yl)oxy, 2-methyl-4-thiazolyl, 4-cyclohexyl-2-methyl-5-oxazolyl, oxazolyl, 1,2,3- thiadiazol-4-yl, 4-methyl-5-oxazolyl, 2-methyl-l-pyrrolidinyl, 2-(methoxymethyl)-l- pyrrolidinyl, 2-(l-methylethyl)-l-pyrrolidinyl, 6-
- R 4 is alkyl, alkyloxazolylmethyl or phenylmethyl. Particularly preferred are those, wherein R 4 is methyl or ((l,l-dimethylethyl)-2-oxazolyl)methyl.
- Preferred compounds of formula I are those, wherein R 4 is haloalkyl.
- Examples of particularly preferred compounds of formula (I) are: N- [(5-bromo- 1,3-thiazo 1- 2- yl)carbamoyl]-3-(difluoromethoxy)benzenesulfonamide; 4-bromo-N- [(5-bromo- 1,3-thiazo 1-2- yl)carbamoyl]-5-chlorothiophene- 2- sulfonamide;
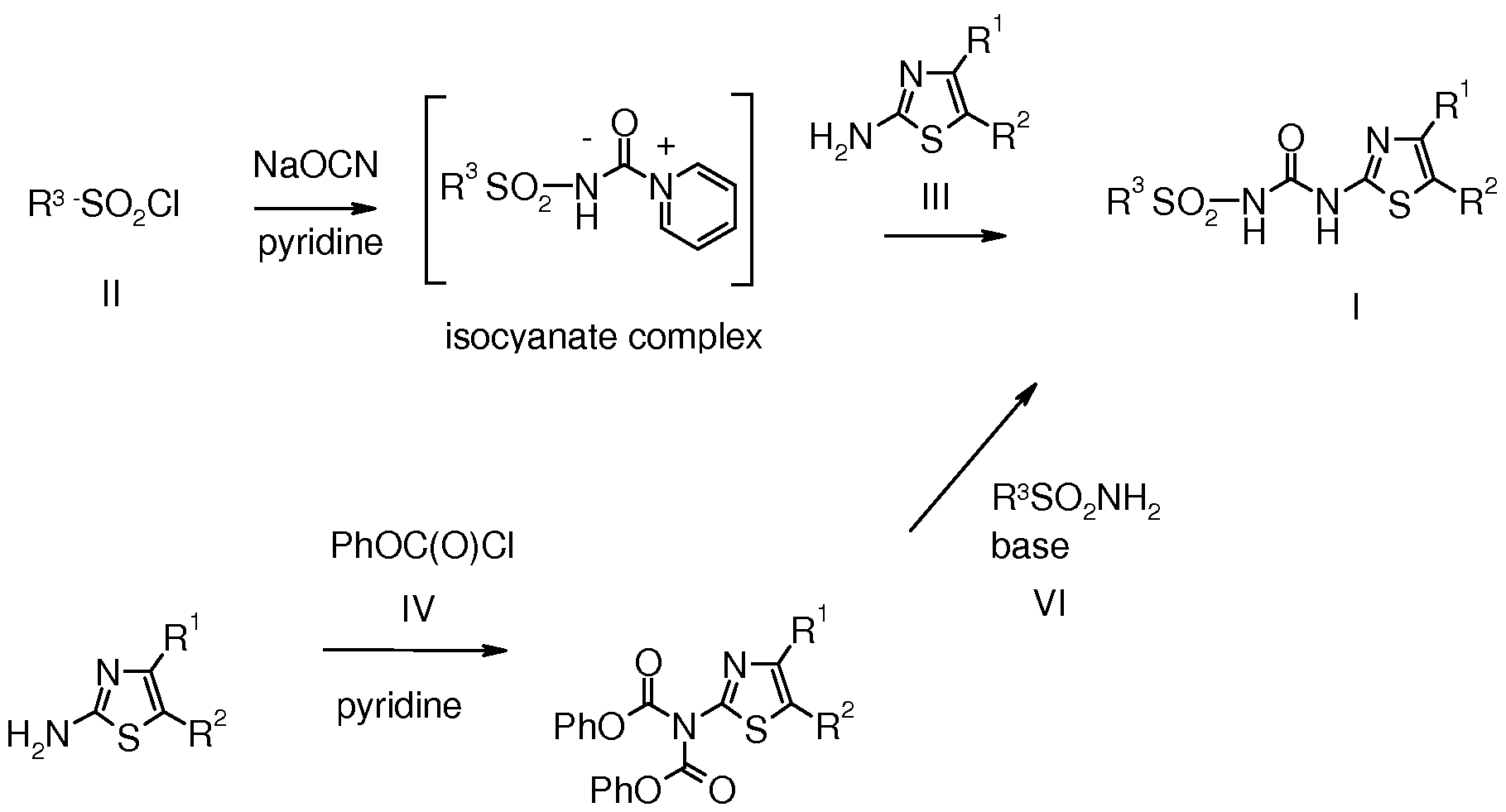
- aminothiazole III was reacted with phenyl chloroformate IV in the presence of pyridine to prepare the bis-carbamate derivative V (lit.: DE 19501174) .
- An aryl or heteroaryl sulfonamide VI was then reacted with V in the presence of base such as DBU to yield compounds of type I.
- Phenyl sulfonyl chlorides HA were prepared from the corresponding bromide VII by first, lithium halogen exchange at low temperature in an inert solvent such as THF and trapping the lithiated species with SO 2 gas. The resultant intermediate was reacted with a chlorinating reagent such as sulfuryl chloride or N-chlorosuccinimide to prepare HA. Alternatively, an aryl amine VIII underwent diazotisation with sodium nitrite. The diazonium salt intermediate, in the presence of SO 2 gas, copper (I) chloride and in an acidic solution, underwent the Meerwein reaction to yield HA. A third method employed was the direct reaction of activated phenyl IX and chlorosulfonic acid.
- Thiophene sulfonyl chlorides HB were prepared from thiophene prucursors X by reacting with a preformed DMF-SO 2 Cl 2 complex. Alternatively, lithiation with butyl lithium at low temperature and subsequent reaction with sulfuryl chloride also furnished compounds of type HB (Scheme 2). Scheme 2
- a further object of the invention are the compounds according to formula I for the preparation of medicaments for the prophylaxis and/or therapy of illnesses which are caused by disorders associated with the enzyme Fructose- 1,6-bisphosphatase, preferrably Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia.
- Fructose- 1,6-bisphosphatase preferrably Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia.
- composition comprising a compound of formula I as described and a therapeutically inert carrier.
- a further preferred embodiment of the invention is the use of a compound according to formula I as described for the preparation of medicaments for the treatment and/or prophylaxis of Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia and particularly preferred for the treatment and/or prophylaxis of Diabetes Mellitus Type II or Diabetes Mellitus Type I.
- IGF Impaired Fasting Glucose
- ITT Impaired Glucose Tolerance
- a further object of the present invention is a compound according to formula I, when manufactured according to any one of the described processes.
- a method for the treatment and/or prophylaxis of Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia which method comprises administering an effective amount of a compound of formula I as described.
- Human liver FBPase cDNA (NM_000507) was purchased from Origene Technologies, Inc, subcloned in a vector for expression in ECoIi., and sequenced.
- Recombinant human liver FBPase (hlFBPase) was purified according to the following protocol that uses heat denaturation similarly to that described by El-Maghrabi et. al. [El- Maghrabi,M.R. et al. "Isolation of a human liver fructose- 1,6-bisphosphatase cDNA and expression of the protein in Escherichia coli.” J Biol Chem 268:9466-9472, 1993.] .
- E coli cells transiently expressing very high levels of soluble and active human liver FBPase, were suspended in 20 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM DTT and were lysed by French press.
- the soluble extract was heat denatured at 65 0 C for 5 min, and insoluble, denatured proteins were removed by centrifugation.
- the extract was then applied to a BioRad Macro-Prep High Q column equilibrated with 20 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM DTT and the flow-through (containing FBPase activity) was collected and applied to a BioRad Macro-Prep HS column equilibrated with 20 mM HEPES pH 7.2, 1 mM DTT. A gradient of increasing NaCl concentration was then applied to the HS column and fractions were collected.
- Fractions containing active FBPase were pooled and further purified by size exclusion chromatography on a Sephacryl S200 column equilibrated in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM DTT. Purity of the enzyme preparation was > 90% as assessed by Mass spectrometry.
- in vitro activity Recombinant human liver FBPase (hlFBPase) activity was assayed through measuring the inorganic phosphate release that results from the hydrolysis of Fructose- 1,6- bisphosphate by the enzyme.
- inorganic phosphate can be readily quantified by spectrophotometry at 620 nm after complexation with ammonium molybdate/malachite green reagent.
- Enzymatic reaction was carried out with modifications of the procedure described by Wright S.W. et al. [Wright S.W. et al., "Anilinoquinazoline inhibitors of Fructose- 1,6-bisphosphatase bind to a novel allosteric site: synthesis, in vitro characterization , and X-ray crystallography". J.Med.Chem. 45:3865- 3877, 2002] . Specifically, the reaction was carried out in 96 well plates in a final volume of 100 ⁇ l in the presence or in the absence of allosteric inhibitors.
- Reaction was started adding 25 ng of hlFBPase to the reaction mixture containing 50 rnM HEPES-KOH buffer pH 7.2, 2mM MgCl 2 , 2mM EDTA, ImM DTT, 50 ⁇ M fructose- 1,6-bisphosphate and 1% DMSO.
- the phosphate released was allowed to form a colored complex for 10 min by adding 150 ⁇ l of ammonium molybdate/malachite green reagent containing 0.03% malachite green, 0.2% ammonium molybdate, 0.05% Triton X-100 and 0.7 M H 2 SO 4 in water that was stirred for 30 min at room temperature and filtered through 0.2 ⁇ m filter. Under these conditions, the assay was linear with time and able to detect FBPase inhibition after spectrophotometric read-out at 620 nm.
- Glucose lowering activity of representative compounds of the present invention was demonstrated after acute treatment in male adult and diabetic db/db mice, db/db mice ( 12-20 weeks of age) were purchased from Jackson laboratories and time- course effect of compounds on blood glucose levels was measured from tail vein samplings using fluorometric method (Glucotrend systems (Roche AG)).
- the compounds of formula I and their pharmaceutically acceptable salts and esters can be used as medicaments (e.g. in the form of pharmaceutical preparations).
- the pharmaceutical preparations can be administered internally, such as orally (e.g. in the form of tablets, coated tablets, dragees, hard and soft gelatin capsules, solutions, emulsions or suspensions), nasally (e.g. in the form of nasal sprays), as aerosol formulations or rectally (e.g. in the form of suppositories).
- the administration can also be effected parentally, such as intramuscularly or intravenously (e.g. in the form of injection solutions).
- the compounds of formula I and their pharmaceutically acceptable salts and esters can be processed with pharmaceutically inert, inorganic or organic adjuvants for the production of tablets, coated tablets, dragees and hard gelatin capsules.
- Lactose, corn starch or derivatives thereof, talc, stearic acid or its salts etc. can be used, for example, as such adjuvants for tablets, dragees and hard gelatin capsules.
- Suitable adjuvants for soft gelatin capsules are, for example, vegetable oils, waxes, fats, semi-solid substances and liquid polyols, etc.
- Suitable adjuvants for the production of solutions and syrups are, for example, water, polyols, saccharose, invert sugar, etc.
- Suitable adjuvants for injection solutions are, for example, water, alcohols, polyols, glycerol, vegetable oils, etc.
- Suitable adjuvants for suppositories are, for example, natural or hardened oils, waxes, fats, semi-solid or liquid polyols, etc.
- the pharmaceutical preparations can contain preservatives, solubilizers, viscosity- increasing substances, stabilizers, wetting agents, emulsif ⁇ ers, sweeteners, colorants, flavorants, salts for varying the osmotic pressure, buffers, masking agents or antioxidants. They can also contain still other therapeutically valuable substances.
- the compounds of formula I and their pharmaceutically acceptable salts can be used e.g. for the prophylaxis and/or treatment of Diabetes Mellitus Type II, Diabetes Mellitus Type I, Diabetes Mellitus Type III, Impaired Fasting Glucose (IFG), Impaired Glucose Tolerance (IGT), diabetic complications or ischaemia.
- the dosage can vary in wide limits and will, of course, be fitted to the individual requirements in each particular case.
- a daily dosage of about 0.1 mg to 100 mg per kg body weight, preferably about 0.5 mg to 10 mg per kg body weight (e.g. about 300 mg per person), divided into preferably 1-3 individual doses, which can consist, for example, of the same amounts, should be appropriate. It will, however, be clear that the upper limit given above can be exceeded when this is shown to be indicated.
- Phenyl chloroformate (1.4 ml, 11.0 mmol) was slowly added to a suspension of 5-bromo- thiazol-2-ylamine hydrobromide ( 1.3 g, 5.0 mmol) in 20 ml pyridine under an argon atmosphere. The reaction mixture was stirred for 3 h at room temperature and then concentrated under reduced pressure. The residue was suspended in water and acidified using cone. HCl to yield light brown crystals (2.3 g, 93%), m/e 419.2 (MH + ).
- 2,6-Dimethylbiphenyl-4-sulfonyl chloride (0.147 g, 0.53 mmol, 1.4 equiv.) and pyridine (0.152 ml, 1.87 mmol, 5.0 equiv.) were added to a stirred suspension of sodium cyanate (56 mg, 0.86 mmol, 2.3 equiv.) in dry acetonitrile (1.5 ml) and the mixture stirred at room temperature for 3 hours.
- 5-Bromo-thiazol-2-ylamine hydrobromide (97 mg, 0.38 mmol, 1.0 equiv.) was added and the reaction stirred for 1 hour. Water (2 ml) and acetic acid (3 drops) were added.
- N-[(5-Bromo-l,3-thiazol-2-yl)carbamoyl]- 5,6-dichloropyridine-3-sulfonamide (20 mg, 0.05 mmol) was dissolved in DMF (1 ml) and reacted with 2-methylpyrrolidine (0.10 ml, 1 mmol) at 5O 0 C for 2.5 hours.
- the reaction mixture was diluted with ethyl acetate (30 ml) and washed with water, NaHCO 3 (half sat. 3x) , 5% KHSO 4 / 10% K 2 SO 4 , NaCl sat., dried (MgSO 4 .2H 2 O), filtered and concentrated under reduced pressure.
- the crude was purified by preparative RP-HPLC: 6 mg, MS: m/e 477.9 (MH " ) .
- 3-Ethyl-phenylamine (2.4 g, 20 mmol) was added dropwise to a stirred mixture containing HCl cone. (37%, 12 ml) and acetic acid (5 ml) at 5 0 C (ice-salt bath).
- a solution of sodium nitrite ( 1.5 g, 22 mmol) in water (3 ml) was added dropwise at O 0 C and the resultant black slurry was further stirred at the low temperature for 30 min.
- a copper (I) chloride (0.5 g, 5 mmol) solution in acetic acid (50 ml) was saturated with SO 2 gas .
- N-chlorosuccinimide (1.1 g, 8.2 mmol, 1.1 equiv.) was added in one portion. After two hours stirring, the mixture is filtered and the filtrate was concentrated under reduced pressure. The residue was dissolved in acetone and an excess of 25% aq. ammonium hydroxide solution was added and the mixture was stirred for 30 min. at room temp. The solvent was evaporated and the remaining oil was dissolved in ethyla ceteate and extracted with water, washed with brine, dried over magnesiumsulfate dihydrate, filtered and evaporated. After purification on silica- gel with ethyl acetate/n-heptane, 210 mg of the title compound were obtained as a light brown solid. MS (ES): m/e 206.0 (M-H)
- 1,3-Dibromoacetone (15 g, 52 mmol) and thiourea (4.05 g, 53.2 mmol, 1.02 equiv.) in methanol were refluxed overnight.
- the reaction mixture was diluted with water and the pH was adjusted to 1 with HCl 25%, followed by extraction with ethyl acetate.
- the pH of the aqueous layer was adjusted to 8-9 using solid sodium carbonate.
- the aqueous layer was extracted with ethyl acetate, and the combined organic extracts were washed with brine, dried over magnesiumsulfat dihydrate, filtered and evaporated.
- a compound of formula I can be used in a manner known per se as the active ingredient for the production of tablets of the following composition:
- a compound of formula I can be used in a manner known per se as the active ingredient for the production of capsules of the following composition:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Abstract
Description











Claims




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007267191A AU2007267191B2 (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives |
| JP2009512545A JP2009538860A (en) | 2006-06-01 | 2007-05-22 | Thiazole derivative |
| EP07729351A EP2032548A1 (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives |
| MX2008015143A MX2008015143A (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives. |
| BRPI0712144-0A BRPI0712144A2 (en) | 2006-06-01 | 2007-05-22 | compounds, process for their preparation, pharmaceutical composition comprising them, their use and method for the treatment and / or prophylaxis of diseases |
| CA002652162A CA2652162A1 (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives |
| KR1020087029281A KR101156367B1 (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives |
| IL195007A IL195007A0 (en) | 2006-06-01 | 2008-10-30 | Thiazole derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06114848 | 2006-06-01 | ||
| EP06114848.2 | 2006-06-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007137962A1 true WO2007137962A1 (en) | 2007-12-06 |
Family
ID=38370786
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/054908 Ceased WO2007137962A1 (en) | 2006-06-01 | 2007-05-22 | Thiazole derivatives |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US7868030B2 (en) |
| EP (1) | EP2032548A1 (en) |
| JP (1) | JP2009538860A (en) |
| KR (1) | KR101156367B1 (en) |
| CN (1) | CN101454301A (en) |
| AR (1) | AR061220A1 (en) |
| AU (1) | AU2007267191B2 (en) |
| BR (1) | BRPI0712144A2 (en) |
| CA (1) | CA2652162A1 (en) |
| CL (1) | CL2007001530A1 (en) |
| IL (1) | IL195007A0 (en) |
| MX (1) | MX2008015143A (en) |
| TW (1) | TW200815380A (en) |
| WO (1) | WO2007137962A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009068467A1 (en) * | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Aminothiazole derivatives |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2058198A1 (en) * | 1991-01-04 | 1992-07-05 | Adalbert Wagner | Azole derivatives, process for their preparation, and their use |
| CA2396713A1 (en) | 1999-12-22 | 2001-07-05 | Metabasis Therapeutics, Inc. | Novel bisamidate phosphonate prodrugs |
| US6906063B2 (en) * | 2000-02-04 | 2005-06-14 | Portola Pharmaceuticals, Inc. | Platelet ADP receptor inhibitors |
| AU2003208757A1 (en) * | 2002-03-06 | 2003-09-16 | Ciba Specialty Chemicals Holding Inc. | Heat sensitive recording material |
| JP2004244409A (en) | 2002-07-23 | 2004-09-02 | Sankyo Co Ltd | Agent for preventing diabetes onset |
| NZ550114A (en) * | 2004-04-20 | 2011-02-25 | Transtech Pharma Inc | Substituted thiazole and pyrimidine derivatives as melanocortin receptor modulators |
-
2007
- 2007-05-22 CA CA002652162A patent/CA2652162A1/en not_active Abandoned
- 2007-05-22 BR BRPI0712144-0A patent/BRPI0712144A2/en not_active IP Right Cessation
- 2007-05-22 KR KR1020087029281A patent/KR101156367B1/en not_active Expired - Fee Related
- 2007-05-22 AU AU2007267191A patent/AU2007267191B2/en not_active Ceased
- 2007-05-22 WO PCT/EP2007/054908 patent/WO2007137962A1/en not_active Ceased
- 2007-05-22 JP JP2009512545A patent/JP2009538860A/en active Pending
- 2007-05-22 CN CNA2007800195065A patent/CN101454301A/en active Pending
- 2007-05-22 MX MX2008015143A patent/MX2008015143A/en active IP Right Grant
- 2007-05-22 EP EP07729351A patent/EP2032548A1/en not_active Withdrawn
- 2007-05-29 CL CL2007001530A patent/CL2007001530A1/en unknown
- 2007-05-29 TW TW096119170A patent/TW200815380A/en unknown
- 2007-05-30 US US11/809,043 patent/US7868030B2/en not_active Expired - Fee Related
- 2007-05-30 AR ARP070102328A patent/AR061220A1/en not_active Application Discontinuation
-
2008
- 2008-10-30 IL IL195007A patent/IL195007A0/en unknown
Non-Patent Citations (17)
| Title |
|---|
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 11 March 2001 (2001-03-11), INTERBIOSCREEN, XP002451597 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 15 September 2004 (2004-09-15), TIMTEC, XP002451586 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 2 May 2004 (2004-05-02), TIMTEC, INC, XP002451589 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 2 May 2004 (2004-05-02), TIMTEC, INC., XP002451588 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 20 April 2004 (2004-04-20), TIMTEC, INC., XP002451591 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 22 April 2004 (2004-04-22), TIMTEC, INC, XP002451596 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 23 April 2004 (2004-04-23), TIMTEC, INC, XP002451595 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 26 April 2004 (2004-04-26), TIMTEC, INC., XP002451594 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 27 April 2004 (2004-04-27), TIMTEC, INC., XP002451593 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 29 April 2004 (2004-04-29), TIMTEC, INC., XP002451592 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 30 April 2004 (2004-04-30), TIMTEC, INC., XP002451590 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 5 March 2001 (2001-03-05), OAK SAMPLES LTD., XP002451598 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; 5 May 2004 (2004-05-05), TIMTEC, INC., XP002451587 * |
| DATABASE CHEMCATS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO; SCIENTIFIC EXCHANGE, XP002451585 * |
| HOF H ET AL: "Therapeutic activities of nitrothiazoles against trichomonads", ARZNEIMITTEL FORSCHUNG. DRUG RESEARCH, ECV EDITIO CANTOR VERLAG, AULENDORF, DE, vol. 37, no. 3, 1987, pages 306 - 309, XP002128088, ISSN: 0004-4172 * |
| HUSAIN ET AL: "Synthesis of some new N1-(5-arylazo-4-phenyl-2-thiazolyl)-N-arylsulf onyl ureas as oral hypoglycemic agents", INDIAN DRUGS, vol. 24, no. 1, 1986, pages 21 - 23, XP009088826 * |
| RUSCHING H ET AL: "New Orally effective blood sugar reducing compounds Neue peroral wirksame blutzckersenkende Substanzen", ARZNEIMITTEL FORSCHUNG / DRUG RESEARCH, VERLAG FUER NATURWISSENSCHAFTEN GMBH, AULENDORF, DE, vol. 8, no. 7a, 1958, pages 448 - 454, XP009088824, ISSN: 0004-4172 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009068467A1 (en) * | 2007-11-30 | 2009-06-04 | F. Hoffmann-La Roche Ag | Aminothiazole derivatives |
| US7973051B2 (en) | 2007-11-30 | 2011-07-05 | Hoffman-La Roche Inc. | Aminothiazoles as FBPase inhibitors for diabetes |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2008015143A (en) | 2008-12-10 |
| AR061220A1 (en) | 2008-08-13 |
| AU2007267191B2 (en) | 2010-08-26 |
| KR20090005406A (en) | 2009-01-13 |
| BRPI0712144A2 (en) | 2012-01-24 |
| IL195007A0 (en) | 2009-08-03 |
| US20070281979A1 (en) | 2007-12-06 |
| EP2032548A1 (en) | 2009-03-11 |
| AU2007267191A1 (en) | 2007-12-06 |
| KR101156367B1 (en) | 2012-06-13 |
| CA2652162A1 (en) | 2007-12-06 |
| TW200815380A (en) | 2008-04-01 |
| US7868030B2 (en) | 2011-01-11 |
| JP2009538860A (en) | 2009-11-12 |
| CL2007001530A1 (en) | 2008-01-04 |
| CN101454301A (en) | 2009-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7868030B2 (en) | FBPase inhibitors for diabetes | |
| AU2008328955B2 (en) | Aminothiazoles as FBPase inhibitors for diabetes | |
| RU2330030C2 (en) | Derivatives of heteroarylcarbamoylbenzene | |
| CA2551324C (en) | Heteroaryl-ureas and their use as glucokinase activators | |
| AU2004218239B2 (en) | 5-phenylthiazole derivatives and their use as P13 kinase inhibitors | |
| EP1709019B1 (en) | Thiazole derivatives and use thereof | |
| US8163778B2 (en) | Pyridines as FBPase inhibitors for treatment of diabetes | |
| CA2488161A1 (en) | Novel aminobenzamide derivative | |
| EP1658288B1 (en) | 2-amino-5-benzoylthiazole npy antagonists | |
| AU2007203160B2 (en) | 5-phenylthiazole derivatives and use as P13 kinase inhibitors | |
| HK1103720B (en) | Thiazole derivatives and use thereof | |
| MXPA06007934A (en) | Thiazole derivatives and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780019506.5 Country of ref document: CN |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07729351 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007729351 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 195007 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007267191 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2652162 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2007267191 Country of ref document: AU Date of ref document: 20070522 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/015143 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087029281 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009512545 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10416/DELNP/2008 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0712144 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081201 |