WO2007141799A1 - A process for preparing pure anastrozole - Google Patents
A process for preparing pure anastrozole Download PDFInfo
- Publication number
- WO2007141799A1 WO2007141799A1 PCT/IN2006/000338 IN2006000338W WO2007141799A1 WO 2007141799 A1 WO2007141799 A1 WO 2007141799A1 IN 2006000338 W IN2006000338 W IN 2006000338W WO 2007141799 A1 WO2007141799 A1 WO 2007141799A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- anastrozole
- triazol
- related substances
- isobutyramide
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
Definitions
- Aromatase is an enzyme, which effects aromatisation of ring A in the metabolic formation of various steroid hormones.
- Various cancers for example, breast cancer are dependent upon circulating steroid hormones, which have an aromatic ring A.
- Such cancers can be treated by removing the source of ring A aromatised steroid hormones, for example, by the combination of oophorectomy and adrenalectomy.
- An alternative way of obtaining the same effect is by administering a chemical compound, which inhibits the aromatisation of the steroid ring A.
- Anastrozole is a non-steroidal antineoplastic, claimed to inhibit the aromatase (oestrogen synthase) activity. It is useful in the treatment of advanced breast cancer in postmenopausal women. BACKGROUND OF INVENTION
- DISCRIPTION OF INVENTION Intermediate (3) undergoes condensation with 4-amino-l,2,4-triazole (4) in a suitable solvent to give 4-amino-l-[3,5-bis-(l-cyano-l-methylethyl)benzyl]-lH- [l,2,4]triazolium bromide (Q.A.-salt) (5) in good yield.
- hydrolysis of cyano groups also takes place leading to the formation of two major related substances.
- the hydrolysis products formed due to hydrolysis are characterized as 2- [3 -(cyanodimethyl-methyl)-5 - [ 1 ,2,4]triazol- 1 -ylmethyl-phenyl] -isobutyramide (6) and 2-[3 -( 1 -carbamoyl- 1 -methylethyl)-5 - [ 1 ,2,4]triazol- 1 -ylmethylphenyl] - isobutyramide (7). Both the substances are isolated and well characterized by using NMR and mass analysis.
- the HPLC chromatogram of Anastrozole shows presence of related substances (6) and (7) in 0.02 % to 1.0 % in crude product which are removed by the repeated crystallization using an alcoholic solvent with a mixture of hydrocarbon as anti-solvent.
- the removal of the related substances 2-[3-(cyanodimethyl-methyl)-5-
- [l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2-[3-(l-carbamoyl-l- methylethyl) : 5-[l,2,4]triazol-l-ylmemylphenyl]-isobutyramide (7) are accomplished by the crystallization method using various solvent systems to get Anastrozole in its purer form.
- the main embodiment of the present invention relates to the products 2- [3- (cyanodimethyl-methyl)-5-[l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2- [3 -( 1 -carbamoyl- 1 -methy lethyl)-5 -[ 1 ,2,4]triazol- 1 -ylmethylphenyl] -isobutyramide (7) as related substances in Anastrozole.
- the present invention also relates to the process for the preparation of Anastrozole with related substances 2-[3-(cyanodimethyl-methyl)-5-[l,2,4]triazol-l-ylmethyl-phenyl]- isobutyramide (6) and 2-[3-(l-carbamoyl-l-methylethyl)-5-[l,2,4]triazol-l- ylmethylphenyl] -isobutyramide (7) preferably, less than 1.0%, more preferably, 0.1% and most preferably, below quantitation limits.
- Anastrozole is obtained in its purer form but still some extent of the related substances 2-[3-(cyanodimethyl-methyl)-5- [l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2-[3-(l-carbamoyl-l- methylethyl)-5-[l,2,4]triazol-l-ylmethylphenyl]-isobutyramide (7) still remain contaminating Anastrozole, which is further purified using organic solvents preferably isopropanol, ethyl acetate or mixture of solvents preferably cyclohexane/ethyl acetate, cyclohexane/isopropanol or a mixture of solvents with water.
- organic solvents preferably isopropanol, ethyl acetate or mixture of solvents preferably cyclohexane/ethyl acetate, cyclohexane/is
- Another embodiment of the present invention relates to the process for the preparation of Anastrozole free from related substances (6) and (7) by crystallization of crude Anastrozole using alcohols preferably selected from Cl to ClO alcohols and hydrocarbons, preferably selected from aliphatic hydrocarbons preferably Cl to ClO.
- Alcohols preferably selected from Cl to ClO alcohols and hydrocarbons, preferably selected from aliphatic hydrocarbons preferably Cl to ClO.
- Example - 1 2,2'-[5-(lH-l,2,4-Triazol-l-ylmethyl)-l,3-phenylene]di(2-methylpropiononitrile) (1), Anastrozole
- reaction mixture was quenched by the addition of a solution of urea (4.5 g) in water (15 mL). Toluene (700 mL) was added to the reaction mixture and the heterogeneous solution was further cooled down to 0 - 5 0 C. The solution was basified by the addition of liquor ammonia (365 mL) slowly in 4 hours at 5 - 25 0 C. Organic layer was separated and further washed with water (200 mL). Aqueous layer was removed and a solution of cone. HCl (140 mL) in water (140 mL) was added to the organic layer slowly in 30 minutes at 25 - 30 0 C and reaction mass was heated at 60 - 65 0 C for 30 minutes.
- the lower aqueous layer (280 - 300 mL), containing product was collected in a conical flask maintaining at 50 0 C.
- the aqueous part was again washed with toluene (700 mL) at 60 - 65 0 C for 30 minutes.
- the lower aqueous layer, containing product was charged in a separating funnel and again washed with fresh toluene (700 mL).
- the aqueous layer, containing product was transferred in a R.B. flask and ethyl acetate (350 mL) was added to it.
- the heterogeneous solution was cooled to 0 - 5 0 C basified by the slow addition of liquor ammonia (280 mL) in 2 - 3 hours at 5 - 25 0 C.
- the solution was stirred for one hour at 25 - 35 0 C, and the upper organic layer (360 - 375 mL), containing product was separated and filtered through hyflow super cell bed.
- Solvent was distilled out below 50 0 C under vacuum leaving approximately 100 mL ethyl acetate in the flask.
- the content of the flask was cooled down to 25 - 35 0 C and cyclohexane (500 mL) was added to the solution slowly in 30 minutes.
- Anastrozole (33 g) from example - 2 was dissolved in isopropanol (100 mL) at 45 - 50 0 C. The solution was cooled down to 25 - 35 0 C and cyclohexane (100 mL) was added drop wise in 30 minutes. The solution was stirred at 25 - 35 0 C for 2 hours; the precipitated solid product was filtered and washed with fresh cyclohexane (30 mL x 2) and dried at 50 0 C to get 23 g of pure Anastrozole contaminated with related substance (6) as 0.09% and with related substance (7) below detection limit.
- Example - 3
- Pure Anastrozole (11 g) from Example - 2 was further purified by dissolving in isopropanol (33 mL) at 45 - 50 0 C. The solution was cooled down to 25 - 35 0 C and cyclohexane (33 mL) was added drop wise in 30 minutes. The solution was stirred at 25 - 35 0 C for 2 hours; the precipitated solid product was filtered and washed with fresh cyclohexane (30 mL x 2) and dried at 50 0 C to get 8.9 g of pure Anastrozole containing with 0.03% of (6) as related substance and another related substance (7) below detection limit. Related substance (6) can be further removed below detection limit by repeating the same process.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The present invention discloses two new related substances (6) and (7) of Anastrozole synthesis from Q.A. Salt (5) as in Scheme - 1 and purification procedures to get Anastrozole (1) free from (6) and (7).
Description
A PROCESS FOR PREPARING PURE ANASTROZOLE
FIELD OF INVENTION
Aromatase is an enzyme, which effects aromatisation of ring A in the metabolic formation of various steroid hormones. Various cancers, for example, breast cancer are dependent upon circulating steroid hormones, which have an aromatic ring A. Such cancers can be treated by removing the source of ring A aromatised steroid hormones, for example, by the combination of oophorectomy and adrenalectomy. An alternative way of obtaining the same effect is by administering a chemical compound, which inhibits the aromatisation of the steroid ring A.
Anastrozole is a non-steroidal antineoplastic, claimed to inhibit the aromatase (oestrogen synthase) activity. It is useful in the treatment of advanced breast cancer in postmenopausal women. BACKGROUND OF INVENTION
Synthesis of Anastrozole is reported in US 4,935,437 and European Patent Application EP 0,296,749. The synthetic route mentioned in the said patents suffers a major disadvantage of the formation of Anastrozole regioisomer (2).

To overcome the formation of regioisomer (2), another synthetic route is reported in US Patent No. 4,935,437; in which compound (3) is reacted with 4-amino-
1,2,4-triazole (4) to form quaternary ammonium salt (5), which further undergoes diazotisation reaction to give Anastrozole (1) free from regioisomeric impurity (2)
(Scheme - 1).
-l-

It has been observed that the cyano groups undergo hydrolysis in various conditions to form hydrolysed related compounds. OBJECTS OF THE INVENTION
It is an object of the present invention to provide an improved process for the preparation of pure Anastrozole (1) free from impurities arising due to hydrolysis of cyano groups during the course of the preparation of Anastrozole (1). DISCRIPTION OF INVENTION Intermediate (3) undergoes condensation with 4-amino-l,2,4-triazole (4) in a suitable solvent to give 4-amino-l-[3,5-bis-(l-cyano-l-methylethyl)benzyl]-lH- [l,2,4]triazolium bromide (Q.A.-salt) (5) in good yield.
It has been further observed that during the preparation of Anastrozole, hydrolysis of cyano groups also takes place leading to the formation of two major related substances. The hydrolysis products formed due to hydrolysis are characterized as 2- [3 -(cyanodimethyl-methyl)-5 - [ 1 ,2,4]triazol- 1 -ylmethyl-phenyl] -isobutyramide (6) and 2-[3 -( 1 -carbamoyl- 1 -methylethyl)-5 - [ 1 ,2,4]triazol- 1 -ylmethylphenyl] - isobutyramide (7). Both the substances are isolated and well characterized by using NMR and mass analysis. The 1H-NMR, 13C-NMR and mass analysis of the isolated products 2-[3 -(cyanodimethyl-methyl)-5 - [ 1 ,2,4]triazol- 1 -ylmethyl-phenyl] - isobutyramide (6) and 2-[3-(l-carbamoyl-l-methylethyl)-5-[l,2,4]triazol-l- ylmethylphenyl] -isobutyramide (7) are in accordance with the chemical structure. 1H- NMR of compound (6) shows three singlets at δ 7.4, 7.3 and 7.24 for three protons in aromatic ring, and two protons at δ 6.95 for amide group. However four methyl groups appear at δ 1.65 and 1.41, each for six protons. The 13C-NMR of compound (6) shows a quaternary peak at δ 177.4 for amide carbonyl carbon, three tertiary aromatic carbons at δ 125.0, 122.7 and 122.2; two aliphatic quaternary carbons at δ 52.1 and 46.1 and two peaks for methyl carbons at δ 28.4 and 26.7. Further, the structure is also confirmed by the mass analysis of compound (6).

(6) 2-[3-(Cyano-dimethyl-methyl)-5-[1 ,2,4]triazol-1-ylmethyl-phenyl]-isobutyramide
The IH-NMR of compound (7) shows peaks at δ 6.86 for amide protons and its 13C-NMR shows amide carbonyl carbon at δ 179.6. Further, the structure is also confirmed by the mass analysis of compound (7).
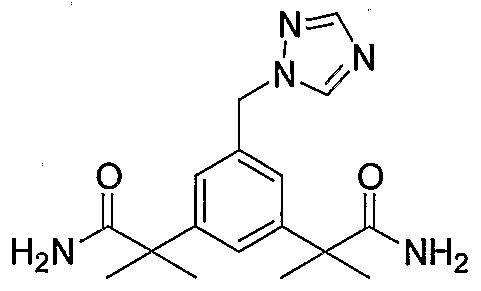
(7) 2-[3-(1-Carbamoyl-1-methylethyl)-5-[1 ,2,4]triazol-1-ylmethylphenyl]-isobutyramide
The HPLC chromatogram of Anastrozole shows presence of related substances (6) and (7) in 0.02 % to 1.0 % in crude product which are removed by the repeated crystallization using an alcoholic solvent with a mixture of hydrocarbon as anti-solvent. The removal of the related substances 2-[3-(cyanodimethyl-methyl)-5-
[l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2-[3-(l-carbamoyl-l- methylethyl):5-[l,2,4]triazol-l-ylmemylphenyl]-isobutyramide (7) are accomplished by the crystallization method using various solvent systems to get Anastrozole in its purer form. Thus, the main embodiment of the present invention relates to the products 2- [3- (cyanodimethyl-methyl)-5-[l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2- [3 -( 1 -carbamoyl- 1 -methy lethyl)-5 -[ 1 ,2,4]triazol- 1 -ylmethylphenyl] -isobutyramide (7) as related substances in Anastrozole. According to another embodiment, the present invention also relates to the process for the preparation of Anastrozole with related substances 2-[3-(cyanodimethyl-methyl)-5-[l,2,4]triazol-l-ylmethyl-phenyl]- isobutyramide (6) and 2-[3-(l-carbamoyl-l-methylethyl)-5-[l,2,4]triazol-l- ylmethylphenyl] -isobutyramide (7) preferably, less than 1.0%, more preferably, 0.1% and most preferably, below quantitation limits.
NMR data of Anastrozole (1) and related substances (6) and (7)

Following the procedures as per Scheme - 1 Anastrozole is obtained in its purer form but still some extent of the related substances 2-[3-(cyanodimethyl-methyl)-5- [l,2,4]triazol-l-ylmethyl-phenyl]-isobutyramide (6) and 2-[3-(l-carbamoyl-l- methylethyl)-5-[l,2,4]triazol-l-ylmethylphenyl]-isobutyramide (7) still remain contaminating Anastrozole, which is further purified using organic solvents preferably isopropanol, ethyl acetate or mixture of solvents preferably cyclohexane/ethyl acetate, cyclohexane/isopropanol or a mixture of solvents with water. Thus another embodiment of the present invention relates to the process for the preparation of Anastrozole free from related substances (6) and (7) by crystallization of crude Anastrozole using alcohols preferably selected from Cl to ClO alcohols and hydrocarbons, preferably selected from aliphatic hydrocarbons preferably Cl to ClO. Example - 1 2,2'-[5-(lH-l,2,4-Triazol-l-ylmethyl)-l,3-phenylene]di(2-methylpropiononitrile) (1), Anastrozole
4- Amino- 1 -[3 , 5 -bis-( 1 -cyano- 1 -methylethyl)benzyl] -IH-[1 ,2,4]triazolium bromide (5) (70 g) was dissolved in cone. HCl (280 mL) in a 5 L R.B. flask and cooled to -5 0C. A solution of sodium nitrite (15 g) in water (70 mL) was slowly added to the reaction mixture at 0 - 5 0C in 4 hrs and the reaction mixture was stirred for one hour at 0 - 5 0C and further at 10 - 20 0C for next 3 hours. The reaction mixture was quenched by the addition of a solution of urea (4.5 g) in water (15 mL). Toluene (700 mL) was
added to the reaction mixture and the heterogeneous solution was further cooled down to 0 - 5 0C. The solution was basified by the addition of liquor ammonia (365 mL) slowly in 4 hours at 5 - 25 0C. Organic layer was separated and further washed with water (200 mL). Aqueous layer was removed and a solution of cone. HCl (140 mL) in water (140 mL) was added to the organic layer slowly in 30 minutes at 25 - 30 0C and reaction mass was heated at 60 - 65 0C for 30 minutes. The lower aqueous layer (280 - 300 mL), containing product was collected in a conical flask maintaining at 50 0C. The aqueous part was again washed with toluene (700 mL) at 60 - 65 0C for 30 minutes. The lower aqueous layer, containing product was charged in a separating funnel and again washed with fresh toluene (700 mL). The aqueous layer, containing product was transferred in a R.B. flask and ethyl acetate (350 mL) was added to it. The heterogeneous solution was cooled to 0 - 5 0C basified by the slow addition of liquor ammonia (280 mL) in 2 - 3 hours at 5 - 25 0C. The solution was stirred for one hour at 25 - 35 0C, and the upper organic layer (360 - 375 mL), containing product was separated and filtered through hyflow super cell bed. Solvent was distilled out below 50 0C under vacuum leaving approximately 100 mL ethyl acetate in the flask. The content of the flask was cooled down to 25 - 35 0C and cyclohexane (500 mL) was added to the solution slowly in 30 minutes. The precipitated solid product was filtered and washed with fresh cyclohexane (20 mL x 2). The product was dried at 45 - 50 0C to get crude Anastrozole (44 g) with more than 98% HPLC purity contaminated with related substance (6) as 0.36% and with related substance (7) as 0.05%. Example - 2 Removal of Related substances (6) and (7) from Anastrozole
Anastrozole (33 g) from example - 2 was dissolved in isopropanol (100 mL) at 45 - 50 0C. The solution was cooled down to 25 - 35 0C and cyclohexane (100 mL) was added drop wise in 30 minutes. The solution was stirred at 25 - 35 0C for 2 hours; the precipitated solid product was filtered and washed with fresh cyclohexane (30 mL x 2) and dried at 50 0C to get 23 g of pure Anastrozole contaminated with related substance (6) as 0.09% and with related substance (7) below detection limit. Example - 3
Purification of Anastrozole
Pure Anastrozole (11 g) from Example - 2 was further purified by dissolving in isopropanol (33 mL) at 45 - 50 0C. The solution was cooled down to 25 - 35 0C and cyclohexane (33 mL) was added drop wise in 30 minutes. The solution was stirred at 25
- 35 0C for 2 hours; the precipitated solid product was filtered and washed with fresh cyclohexane (30 mL x 2) and dried at 50 0C to get 8.9 g of pure Anastrozole containing with 0.03% of (6) as related substance and another related substance (7) below detection limit. Related substance (6) can be further removed below detection limit by repeating the same process.
Claims
1. Compounds (6) and (7) as related substances in Anastrozole

2. Crude Anastrozole with compounds of structural formulae (6) and (7)

as impurities, present preferably in amounts less than 1.0% more preferably, less than 0.1% and most preferably below quantitation limits.
3. A process for producing pure Anastrozole by the removal of related substances of structural formula (6) and (7) from Anastrozole, comprising: a) dissolving Anastrozole in an alcoholic solvent b) adding hydrocarbon to the alcoholic solution and c) isolating pure Anastrozole.
4. A process as claimed in claim 3 wherein the alcoholic solvents used are selected from Cl - C6 straight chain, branched or cyclic alcohols.
5. A process as claimed in claim 3, wherein the hydrocarbons used as anti-solvent are selected from aromatic or aliphatic hydrocarbons; preferably selected from Cl - ClO, straight chain, branched or cyclic hydrocarbons.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06842740A EP2029557A1 (en) | 2006-06-05 | 2006-09-04 | A process for preparing pure anastrozole |
| US12/303,235 US8058302B2 (en) | 2006-06-05 | 2006-09-04 | Process for preparing pure anastrozole |
| US12/952,707 US20110065927A1 (en) | 2006-06-05 | 2010-11-23 | Process for preparing pure anastrozole |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN865-MUM-2006 | 2006-06-05 | ||
| IN865MU2006 | 2006-06-05 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/303,235 A-371-Of-International US8058302B2 (en) | 2006-06-05 | 2006-09-04 | Process for preparing pure anastrozole |
| US12/952,707 Division US20110065927A1 (en) | 2006-06-05 | 2010-11-23 | Process for preparing pure anastrozole |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007141799A1 true WO2007141799A1 (en) | 2007-12-13 |
Family
ID=38068935
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2006/000338 Ceased WO2007141799A1 (en) | 2006-06-05 | 2006-09-04 | A process for preparing pure anastrozole |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US8058302B2 (en) |
| EP (1) | EP2029557A1 (en) |
| WO (1) | WO2007141799A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7692025B2 (en) | 2005-04-06 | 2010-04-06 | Sicor, Inc. | Process for the preparation of anticancer drugs |
| WO2009010991A3 (en) * | 2007-07-17 | 2010-11-11 | Ind-Swift Laboratories Limited | Purification process to prepare highly pure anastrozole |
| CN106083748A (en) * | 2016-06-21 | 2016-11-09 | 扬子江药业集团江苏海慈生物药业有限公司 | A kind of preparation method of Anastrozole |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL304863B2 (en) | 2017-09-11 | 2025-08-01 | Atossa Therapeutics Inc | Methods for preparing and using endoxifen |
| JP7662204B2 (en) | 2019-07-03 | 2025-04-15 | アトッサ・セラピューティクス・インコーポレイテッド | Sustained release compositions of endoxifen |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296749A1 (en) * | 1987-06-16 | 1988-12-28 | Zeneca Limited | (Substituted aralkyl) heterocyclic compounds |
| WO2005105762A1 (en) * | 2004-05-05 | 2005-11-10 | Natco Pharma Limited | Improved process for the preparation of high purity anastrozole |
| US20060035950A1 (en) * | 2004-08-09 | 2006-02-16 | Mohammed Alnabari | Novel processes for preparing substantially pure anastrozole |
| US20060189670A1 (en) * | 2005-02-22 | 2006-08-24 | Glenmark Pharmaceuticals Limited | Process for the preparation of anastrozole and intermediates thereof |
| WO2006108155A2 (en) * | 2005-04-06 | 2006-10-12 | Sicor, Inc. | Process for the preparation of anastrozole |
| WO2007002720A2 (en) * | 2005-06-27 | 2007-01-04 | Sicor, Inc. | An impurity of anastrozole intermediate, and uses thereof |
-
2006
- 2006-09-04 EP EP06842740A patent/EP2029557A1/en not_active Withdrawn
- 2006-09-04 US US12/303,235 patent/US8058302B2/en not_active Expired - Fee Related
- 2006-09-04 WO PCT/IN2006/000338 patent/WO2007141799A1/en not_active Ceased
-
2010
- 2010-11-23 US US12/952,707 patent/US20110065927A1/en not_active Abandoned
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296749A1 (en) * | 1987-06-16 | 1988-12-28 | Zeneca Limited | (Substituted aralkyl) heterocyclic compounds |
| WO2005105762A1 (en) * | 2004-05-05 | 2005-11-10 | Natco Pharma Limited | Improved process for the preparation of high purity anastrozole |
| US20060035950A1 (en) * | 2004-08-09 | 2006-02-16 | Mohammed Alnabari | Novel processes for preparing substantially pure anastrozole |
| US20060189670A1 (en) * | 2005-02-22 | 2006-08-24 | Glenmark Pharmaceuticals Limited | Process for the preparation of anastrozole and intermediates thereof |
| WO2006108155A2 (en) * | 2005-04-06 | 2006-10-12 | Sicor, Inc. | Process for the preparation of anastrozole |
| WO2007002720A2 (en) * | 2005-06-27 | 2007-01-04 | Sicor, Inc. | An impurity of anastrozole intermediate, and uses thereof |
Non-Patent Citations (1)
| Title |
|---|
| TANG GU PING ET AL: "2-Ä3-(2-CYANO-2-PROPYL)-5-(1,2,4-TRIAZOL-1-YLMETHYL)PHENYLÜ-2- METHYLPROPIONONITRILE", ACTA CRYSTALLOGRAPHICA, SECTION E: STRUCTURE REPORTS ONLINE, XX, XX, vol. E61, no. 8, 2005, pages O2330 - O2331, XP008074863, ISSN: 1600-5368 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7692025B2 (en) | 2005-04-06 | 2010-04-06 | Sicor, Inc. | Process for the preparation of anticancer drugs |
| WO2009010991A3 (en) * | 2007-07-17 | 2010-11-11 | Ind-Swift Laboratories Limited | Purification process to prepare highly pure anastrozole |
| CN106083748A (en) * | 2016-06-21 | 2016-11-09 | 扬子江药业集团江苏海慈生物药业有限公司 | A kind of preparation method of Anastrozole |
| CN106083748B (en) * | 2016-06-21 | 2019-08-16 | 扬子江药业集团江苏海慈生物药业有限公司 | A kind of preparation method of Anastrozole |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2029557A1 (en) | 2009-03-04 |
| US20090247765A1 (en) | 2009-10-01 |
| US8058302B2 (en) | 2011-11-15 |
| US20110065927A1 (en) | 2011-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12049457B2 (en) | Highly purified pharmaceutical grade tasimelteon | |
| EP0676409A1 (en) | Erythromycin derivatives, their process for preparation and their application as medicaments | |
| WO2016016766A2 (en) | A process for the preparation of isavuconazonium or its salt thereof | |
| US20110065927A1 (en) | Process for preparing pure anastrozole | |
| WO2012146980A2 (en) | Preparation of fingolimod and its salts | |
| AU2016261273A1 (en) | Process for the preparation of efinaconazole | |
| US9273010B2 (en) | Process for bendamustine hydrochloride | |
| CA2800946C (en) | Crystal delta form of arginine perindopril salt, its preparation process and the pharmaceutical compounds containing same | |
| EP2240436A1 (en) | Novel process for the preparation of vorinostat | |
| EP3436439B1 (en) | Process for the preparation of diphenylpyrazine derivatives | |
| CN105085524A (en) | Preparation method of high purity valganciclovir hydrochloride | |
| CN104961787B (en) | Synthetic method of cordycepin | |
| CN120209051A (en) | Targeted compounds for delivery and their uses | |
| EP4303211B1 (en) | Industrial process for the preparation of hexanoic acid, 6(nitrooxy)-,(1s,2e)-3-[(1r,2r,3s,5r)-2-[(2z)-7-(ethylamino)-7-oxo-2-hepten-1-yl]3,5-dihydroxycyclopentyl]-1-(2-phenyl ethyl)-2-propen-1-yl ester and high pure product | |
| FR2749585A1 (en) | NOVEL AROMATIC DERIVATIVES SUBSTITUTED BY RIBOSIS, PROCESS FOR THEIR PREPARATION AND THEIR APPLICATION AS MEDICAMENTS | |
| EP1940806A2 (en) | A process for the preparation of pure anastrozole | |
| CN110698335A (en) | Synthesis method of terbutaline intermediate | |
| CN1878741A (en) | Novel polymorphs of atovaquone and process of preparation thereof | |
| CN113004202B (en) | A kind of preparation method of high-purity tolvaptan | |
| MD4303211T2 (en) | Industrial process for the preparation of hexanoic acid, 6(nitrooxy)-, (1S,2E)-3-[(1R,2R,3S,5R)-2-[(2Z)-7-(ethylamino)-7-oxo-2-hepten-1-yl]3,5-dihydroxycyclopentyl]-1-(2-phenyl ethyl)-2-propen-1-yl ester and high pure product | |
| EP4524138A1 (en) | Method for preparing glucopyranosyl-containing compound | |
| CN114573557A (en) | Preparation method of ultraconazole | |
| CN112142804A (en) | Preparation method of decitabine | |
| EA044433B1 (en) | SYNTHESIS OF A 1:1:1 CO-CRYSTAL OF 1-CYANO-2-(4-CYCLOPROPYLBENZYL)-4-(β-D-Glucopyranose-1-YL)BENZENE, L-PROLINE AND WATER | |
| WO2008107777A2 (en) | Improved method for the preparation of desloratadine with reduced levels of organic solvents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 06842740 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006842740 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12303235 Country of ref document: US |