WO2007144363A2 - Process for the preparation of 6-alpha, 9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl)oxy)-11-beta -hydroxy-16-alpha -methyl-s-oxo-androsta-1,4-diene-17-beta- -carbothioic acid s-fluoromethyl - Google Patents
Process for the preparation of 6-alpha, 9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl)oxy)-11-beta -hydroxy-16-alpha -methyl-s-oxo-androsta-1,4-diene-17-beta- -carbothioic acid s-fluoromethyl Download PDFInfo
- Publication number
- WO2007144363A2 WO2007144363A2 PCT/EP2007/055805 EP2007055805W WO2007144363A2 WO 2007144363 A2 WO2007144363 A2 WO 2007144363A2 EP 2007055805 W EP2007055805 W EP 2007055805W WO 2007144363 A2 WO2007144363 A2 WO 2007144363A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- process according
- salt
- alpha
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
Definitions
- the present invention relates to a novel process for preparing a glucocorticoid.
- Glucocorticoids which have anti-inflammatory properties are known and are widely used for the treatment of inflammatory disorders or diseases such as asthma and rhinitis.
- US Patent 4,335,121 discloses 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -(1- oxopropoxy)-1 1 ⁇ -hydroxy-16 ⁇ -methyl-3-oxo-androsta-1 ,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester (known by the generic name of fluticasone propionate) and derivatives thereof.
- fluticasone propionate known by the generic name of fluticasone propionate
- glucocorticoids include suppression of the Hypothalamic- Pituitary-Adrenal (HPA) axis, effects on bone growth in children and on bone density in the elderly, ocular complications (cataract formation and glaucoma) and skin atrophy.
- HPA Hypothalamic- Pituitary-Adrenal
- Certain glucocorticoid compounds also have complex paths of metabolism wherein the production of active metabolites may make the pharmacodynamics and pharmacokinetics of such compounds difficult to understand. Whilst the modern steroids are very much safer than those originally introduced, it remains an object of research to produce new molecules which have excellent anti-inflammatory properties, with predictable pharmacokinetic and pharmacodynamic properties, with an attractive side effect profile, and with a convenient treatment regime.
- WO02/08243 discloses processes for preparing intermediates useful in the preparation of fluticasone propionate and a compound of formula (I).
- the object of the present invention is principally to provide a process for preparing a compound of formula (I) without isolating any intermediates.
- the process may be performed in a homogeneous solution.
- a process for the preparation of compounds of formula (III) and its precursors In order to perform the process of synthesis of the compound of formula (I) from the compound of formula (II) without isolating any intermediates it is necessary to undertake the reactions in a solvent which is acceptable for all stages of the process.
- suitable solvents may include pentan-2-one, methylethylketone (MEK) and mixtures thereof.
- MEK methylethylketone
- MEK methylethylketone
- the conversion of a compound of formula (II) to a compound of formula (III) may be performed by employing a deprotecting reagent such as an amine base, thiol or alcohol, for example a primary or secondary amine or a molecule containing both secondary and tertiary amine bases, for example, N-methyl piperazine.
- a deprotecting reagent such as an amine base, thiol or alcohol, for example a primary or secondary amine or a molecule containing both secondary and tertiary amine bases, for example, N-methyl piperazine.
- N-methyl piperazine is that the N-methyl piperazine-furoyl amide which is formed as a result of the process is readily soluble in water (especially as its hydrochloride (HCI) salt) and can therefore be removed from the reaction mixture during an aqueous work-up at the end of the process.
- the deprotection reaction is suitably performed at a temperature in the range of -10 to 10 0
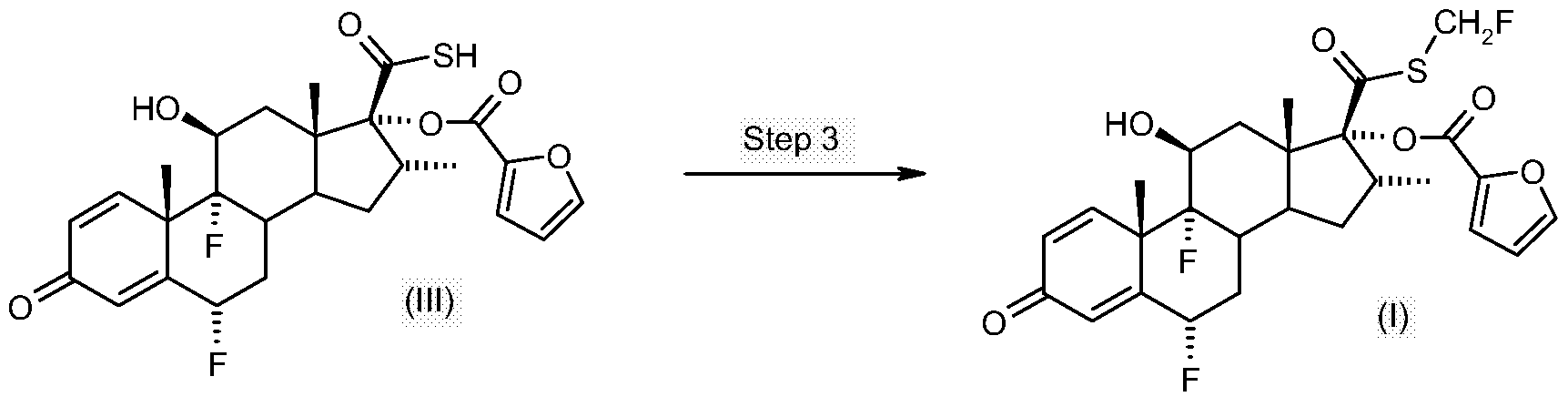
- the latter compound may be further converted to the compound of formula (I) by reacting the compound of formula (III) with a fluoromethylating agent such as chlorofluoromethane (CFM) or bromofluoromethane (BFM), particularly bromofluoromethane (BFM).
- a fluoromethylating agent such as chlorofluoromethane (CFM) or bromofluoromethane (BFM), particularly bromofluoromethane (BFM).
- CFM chlorofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- BFM bromofluoromethane
- the BFM is added at a low temperature, for example 0 0 C, and the reaction mixture is then warmed to 15 to 60 0 C, for example 20 to 22 0 C.
- the reaction time is relatively short, for example less than 30 minutes.
- the reaction is considerably slower, for example taking 5 hours, but a slight improvement in quality is achieved by reducing the level of alkylation on the carbonyl oxygen rather than on sulphur.
- the compound of formula (III) may be employed as a salt, such as an organic amine thiolate salt, for example a trialkylamine salt wherein the trialkylamine group is represented by R 1 R 2 R 3 N wherein each of R 1 , R 2 and R 3 independently represents a C3-6 straight or branched alkyl group.
- the organic amine thiolate salt of the compound of formula (III) is the tripropylamine or the tributylamine salt.
- the excess reagent may be quenched or removed .
- the fluoroalkylating agent is quenched with a chemical quenching agent i.e. a chemical reagent that reacts with the fluoroalkylating agent to produce an unreactive substance.
- a chemical quenching agent i.e. a chemical reagent that reacts with the fluoroalkylating agent to produce an unreactive substance.
- compounds having strongly nucleophilic functionality for example, thiol compounds are suitable.
- N,N-diethylaminoethane thiol is a suitable reagent for quenching BFM.
- the excess reagent can be removed by distillation.
- the compound of formula (I) resulting from the aforesaid process can be purified using conventional extraction processes.
- the compound of formula (I) is extracted into a solvent in which it has adequate solubility when blended with MEK and, more importantly, which is relatively immiscible with aqueous media, for example, dilute acids and bases, with which it may be washed to extract water soluble impurities.
- a particularly suitable extraction solvent for use in the process of the invention is methylisobutylketone (MIBK).
- MIBK methylisobutylketone
- the solution of the compound of formula (I) resulting from the aforesaid process may be diluted with an excess quantity of MIBK thereby to extract the compound of formula (I) into MIBK.
- This solution may then be worked up and washed in a conventional manner with successive washes of aqueous components, such as, aqueous acid, for example, dilute aqueous HCI, aqueous base, for example, dilute aqueous potassium carbonate and water.
- aqueous acid for example, dilute aqueous HCI
- aqueous base for example, dilute aqueous potassium carbonate and water.
- the compound of formula (I) in solid form may be prepared by precipitating the solid from a solution by addition of an anti-solvent.
- a suitable solvent is a mixture of MEK/MIBK, for example in the ratio of 1 :9 v/v, and a suitable anti-solvent is n- heptane.
- the solvent may be evaporated from the previously formed solution to yield a solid and a solution of the correct composition made up again (e.g. made up again in a mixture of MEK/MIBK 1 :9 v/v).
- the distillation process previously mentioned may be concluded at the stage when the ratio of MEK/MIBK reaches the appropriate level e.g. 1 :9 v/v.
- n-heptane as anti- solvent dropwise over an extended period, for example 2 hours, at ambient temperature or slightly above, for example a temperature of approximately 30 to 35 0 C, leads to precipitation of the compound of formula (I).
- the suspension may then be cooled and the product collected by filtration.
- the precipitation is suitably initiated by seeding with one or more crystals of the compound of formula (I).
- the above mentioned ratio of MEK/MIBK 1 :9 v/v is advantageous since it reflects a balance between having a sufficient proportion of MEK to enhance solubility of the compound of formula (I) in the solvent and not having too high a proportion which would lead to generation of an MEK solvate of the compound of formula (I) upon crystallisation.
- the compound of formula (II) may be prepared by a process which comprises reacting a compound of formula (IV)
- F or a salt thereof, for example the thiolate salt, with an activated 2-furoic acid derivative may be performed in a homogeneous solution.
- activated 2-furoic acid derivatives include halides and mixed anhydrides formed from 2-furoic acid.
- the reagent is 2-furoyl chloride (hereinafter "furoyl chloride"). This reagent may be employed without additional solvent.
- Suitable solvents for this reaction may include ethyl acetate (EtOAc), MEK, pentan-2- one and MIBK, for example, MEK, pentan-2-one and mixtures thereof.
- EtOAc ethyl acetate
- MEK ethyl acetate
- MIBK MIBK
- the 17- ⁇ -furoyl ester of the compound of formula (III) is formed via a kinetically favoured 5-exo-trigonal intramolecular S-O acyl transfer, which then goes on to react with a further mole of the furoic acid derivative to produce the compound of formula (II) (the difuroate).
- more than 2 molar equivalents of the activated 2-furoic acid derivative are employed per mole of compound of formula (IV), for example, around 2.2 molar equivalents.
- the reaction may be performed below 0 0 C, such as in the range of -10 to 0 0 C, for example -5 to 0 0 C.
- DMAP dimethylaminopyridine
- the compound of formula (II) may be prepared from the compound of formula (IV) i.e. the compound of formula (II) is not isolated before ongoing processing to the compound of formula (I) via the compound of formula (III).
- the compound of formula (IV) may be employed in the reaction in the form of a thiolate salt which is more reactive than the parent thioacid.
- Suitable salts are salts formed with organic amines, for example, tertiary amines especially tripropylamine.
- the salt of compound of formula (IV) with tripropylamine (TPA) is very soluble in MEK.
- tripropylamine hydrochloride (TPA. HCI) which is formed as a result of the reaction of the compound of formula (IV) with furoyl chloride is also very soluble in MEK.
- the salt of the compound of formula (IV) is the TPA salt.
- Salts of the compound of formula (IV) may be produced by reacting the compound of formula (IV) with the base, for example the organic amine such as TPA, in the prevailing solvent for example, MEK. This may typically be performed between 5 0 C and ambient temperature.
- the base for example the organic amine such as TPA
- MEK prevailing solvent
- the compound of formula (IV) may be employed as a salt, such as an organic amine thiolate salt, such as a trialkylamine salt wherein the trialkylamine group is represented by R 1 R 2 R 3 N wherein each of R 1 , R 2 and R 3 independently represents a C3-6 straight or branched alkyl group.
- the organic amine thiolate salt of the compound of formula (IV) is the tripropylamine or the tributylamine salt.
- HPLC HPLC
- This technique may be used to ensure that the reaction has gone to completion and that the level of impurities generated conforms to specification.
- HPLC techniques may be performed in a conventional manner.
- Control of temperature where heating or cooling is required, for example where reactions are exothermic, may be achieved through appropriate jacketing and heat exchange.
- the overall conversion of a compound of formula (IV) to a compound of formula (I) can be performed in a very efficient process. All stages from the compound of formula (IV) to the compound of formula (I) may be performed as a batch process.
- Step 1 6 ⁇ ,9 ⁇ -difluoro-1 1 ⁇ ,17 ⁇ -dihydroxy-16 ⁇ -methyl-3-oxo-androsta-1 ,4-diene-17 ⁇ - carbothioic acid (compound of formula (IV), the thioacid) (1 Og) and DMAP (0.296g, 0.1 eq wrt the thioacid) were dissolved in MEK (120ml, 8% w/v) by stirring at 20-22 0 C under nitrogen for 10 minutes. Tripropylamine (14.3ml, 3.1 eq wrt the thioacid) was then added as a single charge and the resulting solution cooled to between -8 to - 5°C.
- Neat furoyl chloride (3.59 ml, 1.5 eq wrt the thioacid) was then added dropwise over 2-3 minutes at -5 to 0°C and the reaction mixture stirred for a total of 15 minutes at -5 to 0°C (HPLC indicated that ⁇ 0.5% of the thioacid of formula (IV) remained).
- Step 2 A solution of N-methylpiperazine (1.62ml, 0.6eq wrt the thioacid) in water (4.8ml, 30.5% w/v) was then added dropwise over 2-3 minutes at -5 to 0°C and the reaction mixture stirred for a total of 10 minutes at -5 to 0°C (HPLC indicated that ⁇ 0.1 % of the difuroate (compound of formula (M)) remained).
- Step 3 A solution of bromofluoromethane (3.28g, 1.2eq wrt the thioacid) in MEK (10ml, 32.8% w/v) was then added rapidly as a single charge at 0°C. The solution was then warmed rapidly to 20-22°C and stirred for a total of 5 hours at 20-22°C (HPLC indicated that no thioacid furoate (compound of formula (III)) remained).
- reaction mixture was then diluted with MIBK (230ml) and washed subsequently with aqueous 2M hydrochloric acid (2 x 50ml); water (1 x 50 ml); aqueous potassium carbonate (4% w/v, 1 x 30ml) and then water (1 x 30ml).
- aqueous 2M hydrochloric acid (2 x 50ml); water (1 x 50 ml); aqueous potassium carbonate (4% w/v, 1 x 30ml) and then water (1 x 30ml).
- the final organic phase was then concentrated under reduced pressure to give a fine off-white solid (13.01g, 99.3% theoretical yield after correction for MIBK, 97.43% purity).
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pulmonology (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Immunology (AREA)
- Steroid Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Catalysts (AREA)
- Saccharide Compounds (AREA)
Abstract
Description









Claims



Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07730112A EP2044098B1 (en) | 2006-06-16 | 2007-06-13 | Process for the preparation of 6-alpha ,9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl)oxy)-11-beta -hydroxy-16-alpha -methyl-3-oxo-androsta-1,4-diene-17-beta- -carbothioic acid s-fluoromethyl ester |
| ES07730112T ES2379016T3 (en) | 2006-06-16 | 2007-06-13 | Process for the preparation of S-fluoromethyl ester of 6-alpha, 9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl) oxy) -11-beta-hydroxy-16-alpha-methyl-3-oxo- androsta-1,4-diene-17-beta-carbothioic |
| JP2009514789A JP5568299B2 (en) | 2006-06-16 | 2007-06-13 | Preparation of 6α, 9α-difluoro-17α-[(2-furanylcarbonyl) oxy] -11β-hydroxy-16α-methyl-S-oxo-androst-1,4-diene-17β-carbothioic acid S-fluoromethyl Method |
| AT07730112T ATE546458T1 (en) | 2006-06-16 | 2007-06-13 | METHOD FOR PRODUCING 6-ALPHA, 9-ALPHA |
| US12/304,839 US8309713B2 (en) | 2006-06-16 | 2007-06-13 | Process for the preparation of 6-α,9-α-difluoro-17-α-((2-furanylcarbonyl)oxy)-11-β-hydroxy-16-α-methy1-3-oxo-androsta-1,4-diene-17-β- -carbothioic acid S-fluoromethyl |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0612027.3 | 2006-06-16 | ||
| GBGB0612027.3A GB0612027D0 (en) | 2006-06-16 | 2006-06-16 | Novel process |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007144363A2 true WO2007144363A2 (en) | 2007-12-21 |
| WO2007144363A3 WO2007144363A3 (en) | 2008-05-02 |
Family
ID=36775829
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/055805 Ceased WO2007144363A2 (en) | 2006-06-16 | 2007-06-13 | Process for the preparation of 6-alpha, 9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl)oxy)-11-beta -hydroxy-16-alpha -methyl-s-oxo-androsta-1,4-diene-17-beta- -carbothioic acid s-fluoromethyl |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8309713B2 (en) |
| EP (1) | EP2044098B1 (en) |
| JP (2) | JP5568299B2 (en) |
| AT (1) | ATE546458T1 (en) |
| ES (1) | ES2379016T3 (en) |
| GB (1) | GB0612027D0 (en) |
| WO (1) | WO2007144363A2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012029077A2 (en) | 2010-09-01 | 2012-03-08 | Cadila Healthcare Limited | Process for preparing fluticasone propionate/furoate |
| WO2012079275A1 (en) * | 2010-12-14 | 2012-06-21 | 浙江省天台县奥锐特药业有限公司 | Method for preparing fluticasone furoate |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE887518A (en) * | 1980-02-15 | 1981-08-13 | Glaxo Group Ltd | ANDROSTAN CARTOTHIOATES |
| GB0017988D0 (en) | 2000-07-21 | 2000-09-13 | Glaxo Group Ltd | Novel process |
| SI1775305T1 (en) | 2000-08-05 | 2015-01-30 | Glaxo Group Limited | 6.Alpha.,9.alpha.-difluoro-17.alpha.-'(2-furanylcarboxyl) oxyĆ-11.beta.-hydroxy-16.alpha.-methyl-3-oxo-androst-1,4,-diene-17-carbothiotic acid s-fluoromethyl ester as an anti-inflammatory agent |
-
2006
- 2006-06-16 GB GBGB0612027.3A patent/GB0612027D0/en not_active Ceased
-
2007
- 2007-06-13 US US12/304,839 patent/US8309713B2/en active Active
- 2007-06-13 JP JP2009514789A patent/JP5568299B2/en active Active
- 2007-06-13 AT AT07730112T patent/ATE546458T1/en active
- 2007-06-13 ES ES07730112T patent/ES2379016T3/en active Active
- 2007-06-13 WO PCT/EP2007/055805 patent/WO2007144363A2/en not_active Ceased
- 2007-06-13 EP EP07730112A patent/EP2044098B1/en active Active
-
2014
- 2014-05-20 JP JP2014103872A patent/JP2014177482A/en active Pending
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012029077A2 (en) | 2010-09-01 | 2012-03-08 | Cadila Healthcare Limited | Process for preparing fluticasone propionate/furoate |
| WO2012079275A1 (en) * | 2010-12-14 | 2012-06-21 | 浙江省天台县奥锐特药业有限公司 | Method for preparing fluticasone furoate |
| CN102558273A (en) * | 2010-12-14 | 2012-07-11 | 浙江省天台县奥锐特药业有限公司 | Method for preparing fluticasone furoate |
| CN102558273B (en) * | 2010-12-14 | 2014-07-02 | 浙江省天台县奥锐特药业有限公司 | Method for preparing fluticasone furoate |
| US8969547B2 (en) | 2010-12-14 | 2015-03-03 | Zhejiang Tiantai Aurisco Pharmaceuticals Co. Ltd. | Method for preparing fluticasone furoate |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2044098B1 (en) | 2012-02-22 |
| ATE546458T1 (en) | 2012-03-15 |
| JP5568299B2 (en) | 2014-08-06 |
| EP2044098A2 (en) | 2009-04-08 |
| US8309713B2 (en) | 2012-11-13 |
| JP2014177482A (en) | 2014-09-25 |
| JP2009539934A (en) | 2009-11-19 |
| GB0612027D0 (en) | 2006-07-26 |
| ES2379016T3 (en) | 2012-04-19 |
| US20090118495A1 (en) | 2009-05-07 |
| WO2007144363A3 (en) | 2008-05-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6108944B2 (en) | Enzymatic method for obtaining 17α-monoester of cortexolone and / or its 9,11-dehydro derivative | |
| EP3490973B1 (en) | Polymorphic forms of belinostat and processes for preparation thereof | |
| US8148353B2 (en) | Polymorphs of fluticasone furoate and process for preparation thereof | |
| WO2014083512A1 (en) | Process for preparation of abiraterone acetate | |
| US8969547B2 (en) | Method for preparing fluticasone furoate | |
| RU2208616C2 (en) | Method for preparing mometasone furoate | |
| EP2044098B1 (en) | Process for the preparation of 6-alpha ,9-alpha-difluoro-17-alpha - ((2-furanylcarbonyl)oxy)-11-beta -hydroxy-16-alpha -methyl-3-oxo-androsta-1,4-diene-17-beta- -carbothioic acid s-fluoromethyl ester | |
| CN112028956A (en) | Method for synthesizing 21-hydroxy-17- (1-oxopropoxy) pregn-4-ene-3,20-dione | |
| CN105732754B (en) | Synthesis method of alkyl acid testosterone compound | |
| KR20240048007A (en) | Δ9,11 steroid synthesis | |
| JP7583398B2 (en) | Method for mass production of sodium taurodeoxycholate | |
| CN108239040B (en) | Preparation method of nitric acid 2- (4-methylthiazol-5-yl) ethyl ester hydrochloride | |
| WO2014188445A1 (en) | PROCESS FOR THE PREPARATION OF (3β)-17-(3-PYRIDINYL)ANDROSTA-5,16-DIEN-3-YL ACETATE AND POLYMORPH THEREOF | |
| CN119504830A (en) | A key intermediate of monepantel and its splitting method | |
| CN104804055B (en) | A kind of method adopting silylating reagent purification 7-Ketolithocholsaeure | |
| CH719319B1 (en) | PROCESS FOR THE PREPARATION OF 21-(ACETYLOXI)-17-(1-OXOPROPOXI)-PREGN-4-ENE-3,20-DION | |
| RU2351605C2 (en) | Method of thiocarboxylic acid etherification | |
| WO2025105347A1 (en) | Composition, method for producing fluticasone furancarboxylic acid ester, method for purifying fluticasone furancarboxylic acid ester, and method for producing labeled compound | |
| EP2275410A1 (en) | Process for production of compound having antagonistic activity on npyy5 receptor, and useful crystal | |
| JP5192807B2 (en) | Stable crystals of protected pseudouridine | |
| HK40003591B (en) | Polymorphic forms of belinostat and processes for preparation thereof | |
| KR20000017955A (en) | Process for preparing thiosalicylic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 12304839 Country of ref document: US Ref document number: 2009514789 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007730112 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07730112 Country of ref document: EP Kind code of ref document: A2 |