WO2008006113A2 - Novel belactosin derivatives as therapeutic agents/biological probes and their synthesis - Google Patents
Novel belactosin derivatives as therapeutic agents/biological probes and their synthesis Download PDFInfo
- Publication number
- WO2008006113A2 WO2008006113A2 PCT/US2007/073077 US2007073077W WO2008006113A2 WO 2008006113 A2 WO2008006113 A2 WO 2008006113A2 US 2007073077 W US2007073077 W US 2007073077W WO 2008006113 A2 WO2008006113 A2 WO 2008006113A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- alkyl
- aryl
- protecting group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*(CC(C)(C)C(*)N(C)*)N(C)* Chemical compound C*(CC(C)(C)C(*)N(C)*)N(C)* 0.000 description 6
- VDFHYRKHUXAZGV-CGXNFDGLSA-N CC1(C)O[C@@H]([C@@H](c2ccccc2)OC(C=O)=O)[C@H]([C@@H](c2ccccc2)OC)O1 Chemical compound CC1(C)O[C@@H]([C@@H](c2ccccc2)OC(C=O)=O)[C@H]([C@@H](c2ccccc2)OC)O1 VDFHYRKHUXAZGV-CGXNFDGLSA-N 0.000 description 1
- ISNIYCPYAVEALF-ZRNYENFQSA-N CC1(C)O[C@@H]([C@@H](c2ccccc2)OC)[C@H]([C@@H](c2ccccc2)OC)O1 Chemical compound CC1(C)O[C@@H]([C@@H](c2ccccc2)OC)[C@H]([C@@H](c2ccccc2)OC)O1 ISNIYCPYAVEALF-ZRNYENFQSA-N 0.000 description 1
- KWIWIYBHHYUBQH-VIFPVBQESA-N CC[C@H](C)CC(Sc1ccccn1)=O Chemical compound CC[C@H](C)CC(Sc1ccccn1)=O KWIWIYBHHYUBQH-VIFPVBQESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/02—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D305/10—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having one or more double bonds between ring members or between ring members and non-ring members
- C07D305/12—Beta-lactones
Definitions
- the present invention relates generally to derivatives of belactosin and their synthesis.
- Certain belactosin derivatives of the present invention are inhibitors of both fatty acid synthase and the proteasome.
- these derivatives in certain embodiments, are candidates as anticancer and/or auto-immune therapeutics, antivirals, or antibiotics.
- the synthesis of these compounds generally comprises a concise, single step formation of a common beta-lactone (2-oxetanone) moiety.
- Belactosin and its derivatives feature a beta-lactone ring, and this moiety is pivotal for bioactivity. Kumaraswamy et ah, 2006. Thus, synthetic derivatives of belactosin featuring this moiety are potential therapeutic targets of the proteasome.
- Orlistat a drug approved for treating obesity, also features such a beta-lactone ring moiety.
- This drug has recently been shown to be a potent inhibitor of human fatty acid synthase (FAS); further, this natural product derivative is cytotoxic and cytostatic to tumor cells in vitro and can inhibit tumor growth in vivo.
- FAS is responsible for the cellular synthesis of palmitate and is attracting great interest as a drug target in oncology because it is up-regulated in most solid tumors, including those of the breast, prostate and ovary.
- orlistat has poor solubility and poor bioavailability, so there is a need to develop new beta-lactones that overcome these problems and that can be deployed as potential antitumor drugs.
- simplified derivatives that are readily prepared would also ultimately be attractive from the standpoint of process development.
- the present invention overcomes deficiencies in the art by providing novel derivatives of belactosin comprising a beta-lactone ring structure.
- a particularly attractive feature of certain compounds of the present invention is their ability, in certain embodiments, to act as inhibitors of the proteasome and/or FAS. This inhibitory activity significantly increases the therapeutic potential of these compounds.
- certain compounds of the present invention are low nanomolar inhibitors of the 2OS proteasome. As such, these compounds could be used in treatments for cancer as well as malaria, tuberculosis and other indications. Indeed, any protein that recognizes the beta-lactone moiety as a substrate may recognize a compound as described herein.
- the present invention contemplates, in certain embodiments, a concise synthesis of these compounds that allows access to a variety of beta-lactones in a diastereoselective manner, often in fewer steps than disclosed in previous methodologies.
- the synthetic steps comprise a tandem Mukaiyama aldol lactonization (TMAL) reaction.
- TMAL Mukaiyama aldol lactonization
- Other compounds comprising a beta-lactone moiety besides belactosins include the vegetal poison anisatin and the antibiotic 1233 A (Kumaraswamy and Markondaiah, 2007): such compounds may also be accessed via methods of the present invention.
- Xo is a chiral auxiliary, such as
- Ro may be selected from a group consisting of H, alkyl, an amine protecting group and
- R 5 and Rg may each independently be selected from the group consisting of H, alkyl, aryl, an amine protecting group and
- m may range in length from 1-5, depending on the desired length of this flexible linker, and X may comprise a fluorophore.
- linkers or spacers known to those of skill in the art may also be employed. Linkers or spacers typically are of low reactivity and are internally flexible moieties that allow one moiety to be joined with another at a desired distance. Fluorophores and methods of incorporation into various compounds are well known to those of skill in the art. See, e.g., FluoProbes® BioDirectory of Fluorescence; Molecular Probes, The Handbook — A Guide to Fluorescent Probes and Labeling Technologies, 10 th Ed. (2005), both of which are herein incorporated by reference in their entirety.
- Compounds of the present invention comprising a fluorophore may be useful as tools (e.g., biological probes) for studying proteins, in vivo or in vitro, such as FAS, the proteasome, and various enzymes and other proteins that recognize beta-lactone rings of the present invention.
- the fluorophore may be selected from the group consisting of:
- R 7 may be selected from the group consisting of H, alkyl and aryl.
- Ri and R 3 may each independently be selected from the group consisting of H, alkyl, aryl and an amine protecting group;
- R 2 may be selected from the group consisting of H, aryl, alkyl, -CO 2 W, wherein W is H, alkyl, aryl or a carboxylic acid protecting group, and
- Zi and Z2 are each independently selected from the group consisting of H, -OH, -NH 2 , -NHCH 3 and -CO 2 H; and R 4 may equal
- R 9 may each independently be selected from the group consisting of H, alkyl, aryl, -CH(OH)alkyl, -CH(OH)aryl, -ORi 0 and -NRnRi 2 , wherein R 1 0-R 12 may each independently be selected from the group consisting of H, alkyl, aryl, acetyl, -SiRoRi 4 Ri S , an alcohol protecting group and an amine protecting group, wherein R 13 -R 15 may each independently be selected from the group consisting of alkyl and aryl; and n may equal 1-7, depending on the need for greater chain length, flexibility and/or distance between the two termini of the overall compound.
- provisos such as the following may apply: if R 0 equals
- R 2 equals H and n equals 3, then R 4 cannot be
- R 5 is, in certain preferred embodiments, an amine protecting group.
- Re is preferably H.
- R 7 is preferably -CH3.
- R 5 is an amine protecting group, Re is H and R 7 is -CH3.
- the amine protecting group may be Cbz, in certain embodiments.
- R 7 is -CH3 and R 5 is
- m is 1 and wherein, in certain preferred embodiments, X is
- Ri is H.
- R 2 is alkyl and in certain other embodiments, R 2 is a carboxylic acid or a protected carboxylic acid.
- the protected carboxylic acid may, in certain embodiments, be protected by a benzyl protecting group.
- R 3 is H.
- R 4 is
- R 4 is
- Rg is -OR 1 0 and Rg is H.
- Ri 0 may be, in certain preferred embodiments, selected from the group consisting of H, an alcohol protecting group and - SR 13 R 14 R 15 .
- R 13 -R 15 may independently be, in certain embodiments, H, alky or aryl, with ethyl groups comprising a preferred embodiment.
- Rs is either alkyl or aryl and R 9 is H.
- Rg is -CH(OH)alkyl or -CH(OH)aryl and R 9 is H.
- R 8 may be phenyl, in certain embodiments, and may be substituted with any of the substituents as described herein.
- Rg is -NR 11 R 12 and R9 is H.
- Rn and R 12 may each independently be selected from the group consisting of H, acetyl and an amine protecting group, in preferred embodiments. Preferably, n equals 1-5 and even more preferably, n equals 3.
- Yi is selected from the group consisting of H, Cbz, Fmoc, PMB, BOM and
- a compound of the following formula is contemplated:
- R A is selected from the group consisting of H, Bn and PMB and R B is selected from the group consisting of H, Bn, PMB, -C(O)CH 3 , Cbz and Boc.
- Ys is selected from the group consisting of H, Cbz, Fmoc, PMB, BOM and
- Zi and Z 2 are each independently selected from the group consisting of H, -OH, -NH 2 , -NHCH 3 and - CO 2 H;
- Y 8 is selected from the group consisting of -CH 3 , -C 6 Hi 3 , -CH 2 Cy, -CH(OH)Cy, phenyl, -CH 2 C 6 H 5 and -CH(OH)C 6 H 5 ;
- r is 0 or 1; and s is 1-5.
- Rc is selected from the group consisting of H, Bn and PMB and R D is selected from the group consisting of H, Bn, PMB, -C(O)CH 3 , Cbz and Boc.
- Another general aspect of the present invention contemplates a method of synthesizing a compound of formula (I):
- the first compound may comprise, for example, a dipeptide or a tripeptide.
- the reaction conditions may comprise, in certain embodiments, a Lewis acid.
- Lewis acids are well-known to those of skill in the art. Non-limiting examples of Lewis acids include ZnCl 2 , Zn(OTf) 2 and SnCl 4 .
- a chosen solvent for this reaction may comprise any type as described herein and in preferred embodiments, the solvent is methylene chloride. Reaction conditions typically take place at room temperature for periods of 8-36 hours, with 12-24 hours being more preferable. Reactions may be monitored by any means known to those of skill in the art, such as thin-layer chromatography and HPLC. Purification can take place via any means known to those of skill in the art, with silica gel column chromatography comprising a preferred embodiment.
- Rs and R 9 may each independently be selected from the group consisting of H, alkyl, aryl, -CH(OH)alkyl, -CH(OH)aryl, -OR 22 and -NR 2 3R 24 , wherein R 22 -R 24 may each independently be selected from the group consisting of H, alkyl, aryl, acetyl, -SiR 2 sR 2 6R 2 7, an alcohol protecting group and an amine protecting group, wherein R 2 5-R 2 7 niay each independently be selected from the group consisting of H, alkyl and aryl; and n may equal 1-7, depending on the need or wish for greater chain length, flexibility and/or distance between the two termini of the overall compound.
- R 2 5-R 2 7 are each ethyl.
- Ri 8 -R 2 O may comprise, independently, H, alkyl or aryl, with ethyl groups being more preferred.
- R 2 i may comprise, in some embodiments, H, alkyl or aryl, with aryl groups being preferable, and pyridyl and phenyl groups being more preferable, and pyridyl groups being even more preferable.
- reaction conditions may comprise a coupling agent, as described herein.
- a chosen solvent for this reaction may comprise any type described herein and in preferred embodiments, the solvent comprises ethyl acetate/water.
- Reaction conditions typically take place at room temperature for periods under 24 hours. Reactions may be monitored by any means known to those of skill in the art, such as thin-layer chromatography and HPLC. Purification can take place via any means known to those of skill in the art, with silica gel column chromatography comprising a preferred embodiment.
- Ro may be selected from the group consisting of H, alkyl, an amine protecting group and
- R 5 and Re may each independently be selected from the group consisting of H, alkyl, aryl, an amine protecting group and
- n may range in length from 1-5, depending on the desired length of this flexible linker
- X may comprise a fluorophore as described herein.
- the fluorophore may be selected from the group consisting of:
- R 7 may be selected from the group consisting of H, alkyl and aryl.
- Ri and R 3 may each independently be selected from the group consisting of H, alkyl, aryl and an amine protecting group; and R2 may be selected from the group consisting of H, aryl, alkyl, -CO 2 W, wherein W is H, alkyl, aryl or a carboxylic acid protecting group, and
- Ri 6 may be selected from the group consisting of H, -C(O)CHO and -C(O)C(O)Rn, wherein R ⁇ 7 may be selected from the group consisting of alkyl and aryl.
- the chain length represented by n may range from 1-7, depending on the need or wish for greater chain length, flexibility and/or distance between the two termini of the overall compound. Further, R 4 , as generated via this method, may equal
- Rg and R 9 may be as described above.
- Ro H and Ri equals an amine protecting group, such as Cbz. In other preferred embodiments, Ro equals
- R5 is an amine protecting group.
- R 6 is H.
- R 7 is CH 3 .
- R5 is an amine protecting group, Re is H and R 7 is CH3.
- the amine protecting group may be, in certain embodiments, Cbz.
- R 7 is CH3 and R 5 is
- n 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 10 or 11 or 12 or 13 or 14 or 14 or 14 or 15 or 16 or 16 or 17 or 18 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or 20 or 19 or any other linker or spacer known to those of skill in the art. In certain preferred embodiments, m is 1. In certain preferred embodiments, X is
- Ri is H.
- R 2 is alkyl and in other preferred embodiments, R 2 is a carboxylic acid protecting group, such as Bn.
- R3 is H.
- the R 4 group generated by this method comprises
- the R 4 group generated by this method comprises
- Rg or Rg is H.
- Rs is either alkyl or aryl and R 9 is H.
- Rs is -OR10 and R9 is H.
- Rs is -CH(OH)alkyl or - CH(OH)aryl and R9 is H.
- Rio is selected from the group consisting of H, an alcohol protecting group and SR 13 R 14 R 15 .
- R 13 -R 15 may comprise, for example, H, alkyl or aryl, with ethyl groups being more preferred.
- Rs is phenyl.
- Rs is -NR 11 R 12 and R9 is H.
- Rn and R 12 in preferred embodiments, may each independently be selected from the group consisting of H, acetyl and an amine protecting group.
- n equaling 1-5 contemplates n equaling 3 being a more preferred embodiment.
- Yet another preferred embodiment comprises Rn as Bn.
- provisos such as the following may apply: when formula (II) comprises
- R 28 is -OH or
- Ph and R 29 is selected from the group consisting of H, alkyl, aryl, -CH 2 Cy, -CH(OH)alkyl, -CH(OH)aryl, -OR 22 and -NR 23 R 24 , wherein R 22 -R 24 are each independently selected from the group consisting of H, alkyl, aryl, acetyl, -SiR 2 5R 2 6R27, an alcohol protecting group and an amine protecting group, wherein R 2S -R 27 are each independently selected from the group consisting of alkyl and aryl; and optical isomers thereof.
- Certain aspects of the present invention contemplate a method of inhibiting the 2OS proteasome, comprising contacting a cell with an effective amount of a compound as described herein.
- One skilled in the art can purify and measure the activity of the proteasome using approaches such as those described in the following citations: Hirano et al., 2005; Akaishi et al., 1996; Ugai et al., 1993; Adams et al., 1999; and Mellgren, 1997, each of which is incorporated herein by reference in its entirety. The method may take place in vitro or in vivo.
- Compounds of the present invention may also be used to treat proteasome -related conditions, such as cancer, Alzheimer's disease, malaria, tuberculosis, eye disorders and asthma. Accordingly, methods of treatment of these conditions are also contemplated. Compounds used in these methods may be comprised in a pharmaceutically acceptable excipient, diluent, or vehicle.
- Certain aspects of the present invention contemplate a method of inhibiting fatty acid synthase, comprising contacting a cell with an effective amount of a compound as described herein.
- compounds may be screened for their ability to inhibit the thioesterase domain of fatty acid synthase, which liberates palmitate, the natural substrate, from the enzyme.
- One of ordinary skill in the art could express and purify the recombinant thioesterase using procedures described in, e.g., Chakravarty et al., 2004 and Kridel et al., 2004.
- the thioesterase domain of fatty acid synthase was PCR amplified using the following primers: 5_ ATG ACG CCC AAG GAG GAT GGT CTG GCC CAG CAG (corresponds to nucleotides 6727-6756) and 3_ GCC CTC CCG CAC GCT CAC GCG TGG CT (corresponds to nucleotides 7625-7650).
- the recombinant thioesterase domain was cloned into pTrcHis (Invitrogen) and expressed in Escheria coli.
- the recombinant protein corresponds to residues 2202 through 2509 of FAS.
- the thioesterase was purified by Ni- affinity chromatography.
- the method may take place in vitro or in vivo.
- Compounds of the present invention may also be used to treat fatty acid-synthase-related conditions, which generally includes diseases characterized by hyperproliferation of cells such as inflammation, angiogenesis and cancer. Such compounds may also be of use in treating obesity as fatty acid synthase is the only enzyme that converts dietary carbohydrate to fat. Accordingly, methods of treatment of these conditions are also contemplated.
- Compounds used in these methods may be comprised in a pharmaceutically acceptable excipient, diluent, or vehicle.
- the inhibition of fatty acid synthase in cells can be measured by, for example, directly determining the amount of palmitate synthesized by the cell using methods described in Browne et al, 2006, Kridel et al., 2004 and Pizer et al, 1996.
- Another general aspect of the present invention contemplates a method of treating cancer comprising administering to a subject an effective amount of a compound as described herein.
- the subject may be a mammal, such as a human.
- the cancer may be any cancer treatable by administration of a compound described herein.
- the cancer may be breast, prostate, ovarian, brain hepatocarcinoma, melanoma, colorectal, liver, lymphoma, lung, oral, head, neck, spleen, lymph node, small intestine, large intestine, blood cells, stomach, endometrium, testicle, skin, esophagus, bone marrow, blood, cervical, bladder, Ewing's sarcoma, thyroid, and/or gastrointestinal.
- contact when applied to a cell, is used herein to describe the process by which a compound of the invention is delivered to a target cell or is placed in direct juxtaposition with the target cell.
- an effective amount means adequate to accomplish a desired, expected, or intended result.
- an “effective amount” may be an amount of a compound sufficient to produce a therapeutic benefit (e.g., effective to reproducibly inhibit decrease, reduce, inhibit or otherwise abrogate the growth of a cancer cell).
- Effective amounts or a “therapeutically relevant amount” are those amounts of a compound sufficient to produce a therapeutic benefit (e.g., effective to reproducibly inhibit decrease, reduce, inhibit or otherwise abrogate the growth of a cancer cell).
- An effective amount in the context of treating a subject, is sufficient to produce a therapeutic benefit.
- a "suitable solvent” or a “chosen solvent” is a solvent that will facilitate, or at least not significantly impede, the reaction that takes place within that solvent.
- a suitable solvent choice may depend on, for example, which one(s) will facilitate the solubilizing of all the reagents, or, for example, which one(s) will best facilitate the desired reaction (particularly if the mechanism of the reaction is known). However, a suitable solvent need not completely solubilize each reagent and may actually impede the desired reaction to some degree.
- Suitable solvents for the methods of the present invention will be known to one of ordinary skill in the art. More than one solvent may be chosen for any particular reaction. Water may also be admixed into any solvent choice, particularly when improvements in solubility of the reagents are required.
- Suitable solvents may include, for example, polar solvents and non-polar solvents. Suitable solvents include, but are not limited to, dimethylformamide, dimethylsulfoxide, dioxane, methanol, ethanol, hexane, ethyl acetate, methylene chloride, tetrahydrofuran and acetonitrile.
- suitable solvents include methylene chloride, tetrahydrofuran and ethyl acetate/water.
- a "coupling agent” is a reagent used to facilitate the coupling of two compounds.
- a coupling agent facilitates the coupling of an NH 2 - containing compound with a -CChH-containing compound to form an amide bond between the two compounds.
- Coupling agents are well known to those of ordinary skill in the art and may be employed in certain embodiments of methods of the present invention.
- coupling agents include, but are not limited to, l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC) and dicyclohexylcarbodiimide (DCC), optionally in conjunction with catalytic amounts of dimethylaminopyridine (DMAP) as a promoter.
- Other carbodiimides are also envisioned as coupling agents.
- Coupling agents are discussed, for example, in Bodansky, 1993 and Grant, 1992. These coupling agents may be used singly or in combination with each other or other agents to facilitate conjugation.
- a urea by-product is typically formed.
- the urea by-product may be removed by filtration.
- the conjugated product may then be purified by, for example, silica gel column chromatography or HPLC.
- a "chiral auxiliary" refers to an easily removable chiral group that is capable of influencing the direction of nucleophilic attack. Chiral auxiliaries typically control the diastereoselectivity of a reaction. Persons of skill in the art are familiar with such compounds, and many are commercially available. Known and unknown equivalents of the specific compounds, agents, and active ingredients discussed throughout this specification can be used with the compositions and methods of the present invention. The equivalents can be used as substitutes for the specific compounds, agents, and active components. The equivalents can also be used to add to the methods and compositions of the present invention. A person of ordinary skill in the art would be able to recognize and identify acceptable known and unknown equivalents to the specific compounds, agents, and active ingredients without undue experimentation.
- compounds of the present invention can generally be purified at any step, including the purification of intermediates as well as purification of the final products.
- purification of a compound does not remove all impurities. In some embodiments, such impurities can be identified.
- Non-limiting examples of purification methods include gel filtration, size exclusion chromatography (also called gel filtration chromatography, gel permeation chromatography or molecular exclusion), dialysis, distillation, crystallization, recrystallization, reprecipitation, sublimation, electrophoresis, prep thin-layer chromatography, silica gel column chromatography and high-performance liquid chromatography (HPLC), including normal-phase HPLC and reverse-phase HPLC.
- purification comprises silica gel column chromatography.
- Methods of determining the purity of compounds are well known to those of skill in the art and include, in non-limiting examples, autoradiography, mass spectroscopy, melting point determination, ultra violet analysis, colorimetric analysis, (HPLC), thin-layer chromatography and nuclear magnetic resonance (NMR) analysis (including, but not limited to, H and C NMR).
- purity is determined via NMR.
- Software available on various instruments e.g., spectrophotometers, HPLCs, NMRs
- Prodrugs and solvates of the compounds of the present invention are also contemplated herein. Any compound described herein may be a prodrug.
- the term "prodrug” as used herein is understood as being a compound which, upon administration to a subject, such as a mammal, undergoes chemical conversion by metabolic or chemical processes to yield a compound any of the formulas herein, or a salt and/or solvate thereof (Bundgaard, 1991; Bundgaard, 1985).
- Solvates of the compounds of the present invention are preferably hydrates.
- “predominantly one enantiomer” or “substantially free” from other optical isomers means that the compound contains at least about 95% of one enantiomer, or more preferably at least about 98% of one enantiomer, or most preferably at least about 99% of one enantiomer. Any compound described herein may, in certain embodiments, be present as predominantly one enantiomer.
- substantially pure compounds are contemplated. That is, any compound as described herein may be a substantially pure compound.
- substantially pure refers to compounds that are at least about 95% pure, or more preferably at least about 98% pure, or most preferably at least about 99% pure.
- the term "about” is used to indicate that a value includes the inherent variation of error for the device, the method being employed to determine the value, or the variation that exists among the study subjects.
- “about” can be within 10%, preferably within 5%, more preferably within 1%, and most preferably within
- FIG. 2 General synthetic method for belactosin C and derivatives: Synthesis of dipeptide and beta-lactone separately, followed by coupling.
- FIG. 3 Proteasome 2OS inhibition assays.
- Known and novel belactosin derivatives inhibit the turnover of a fluorogenic substrate by the 2OS proteasome.
- the 2OS proteasome (5 nM) was incubated with (A) 1.25 ⁇ M or (B) 50 ⁇ M test compound and 100 mM suc-LLVY-
- AMC substrate Fluorescence measurements were taken every ten minutes at 380/460 nm.
- Results shown are the average of (A) triplicate or (B) duplicate data points.
- belactosin and its derivatives feature a beta-lactone ring, and this moiety is pivotal for bioactivity.
- belactosin derivatives of the present invention may function as inhibitors of human fatty acid synthase
- Orlistat a drug approved for treating obesity, also features a beta-lactone ring moiety.
- This drug has recently been shown to be a potent inhibitor of FAS; further, this natural product derivative is cytotoxic and cytostatic to tumor cells in vitro and can inhibit tumor growth in vivo.
- FAS is responsible for the cellular synthesis of palmitate and is attracting great interest as a drug target in oncology because it is up-regulated in most solid tumors, including those of the breast, prostate and ovary.
- Beta-lactone inhibitors of FAS typically function by acylating the active site serine residue, leading to inhibition of thioesterase (TE) activity and ultimately apoptosis — that is, cell death. Purohit et al, 2006. Similarly, beta-lactone inhibitors of the proteasome typically function by acylation of the active site threonine (chymotrypsin activity of the proteasome). Simon et al, 2006; Maier et al, 2006.
- Beta-lactone moieties similar to those found in such agents as the belactosins and orlistat are capable, in certain embodiments, of acylating the active site serine of the FAS-TE and also the threonine of the proteasome, thus typically leading to dual inhibition. Accordingly, in certain embodiments, compounds of the present invention are attractive anticancer agents for study. Such compounds may also be useful for as substrates for other proteins ⁇ e.g., enzymes) which recognize the beta-lactone moiety.
- a facile means of accessing compounds of the present invention comprises, in preferred embodiments, a tandem Mukaiyama aldol lactonization (TMAL) reaction.
- TMAL Mukaiyama aldol lactonization
- the synthetic methodology of the present invention offers, in certain embodiments, access to these biologically interesting beta-lactones in fewer steps than many methodologies previously attempted.
- any compound comprising a beta-lactone moiety may be prepared using methods disclosed herein.
- alkyl includes straight-chain alkyl, branched chain alkyl, cycloalkyl (Cy) (alicyclic), cyclic alkyl (e.g., -CH 2 -cyclopropyl), heteroatom-unsubstituted alkyl, heteroatom- substituted alkyl, heteroatom-unsubstituted C n -alkyl, and heteroatom-substituted C n -alkyl.
- Alkyl groups may also optionally contain alkene or alkyne C-C bonds (but not such that aromatic compounds result). In certain embodiments, lower alkyls are contemplated.
- lower alkyl refers to alkyls of 1-6 carbon atoms (that is, 1, 2, 3, 4, 5 or 6 carbon atoms).
- heteroatom-unsubstituted C n -alkyl refers to a radical, having a linear or branched, cyclic or acyclic structure, further having no carbon-carbon double or triple bonds, further having a total of n carbon atoms, all of which are nonaromatic, 3 or more hydrogen atoms, and no heteroatoms.
- a heteroatom-unsubstituted Ci-Cio-alkyl has 1 to 10 carbon atoms.
- heteroatom-substituted C n -alkyl refers to a radical, having a single saturated carbon atom as the point of attachment, no carbon-carbon double or triple bonds, further having a linear or branched, cyclic or acyclic structure, further having a total of n carbon atoms, all of which are nonaromatic, 0, 1, or more than one hydrogen atom, at least one heteroatom, wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S.
- a heteroatom- substituted Ci-Cio-alkyl has 1 to 10 carbon atoms.
- "lower alkyl” groups are contemplated. As used herein, "lower alkyl" groups
- amino alone or in combination, is used interchangeably with “amine” and may refer to any one or more of the following: a primary (e.g., -NH 2 ), secondary (e.g., alkyl- NH-), tertiary (e.g., (alkyl) 2 -N-), or quarternary (e.g., (alkyl) 3 — N(+)-) amine radical.
- a primary e.g., -NH 2
- secondary e.g., alkyl- NH-
- tertiary e.g., (alkyl) 2 -N-
- quarternary e.g., (alkyl) 3 — N(+)-
- aryl includes heteroatom-unsubstituted aryl, heteroatom-substituted aryl, heteroatom-unsubstituted C n -aryl, heteroatom-substituted C n -aryl, heteroaryl, heterocyclic aryl groups, carbocyclic aryl groups, biaryl groups, and radicals derived from polycyclic fused hydrocarbons (PAHs).
- PAHs polycyclic fused hydrocarbons
- heteroatom-unsubstituted C n -aryl refers to a radical, having a single carbon atom as a point of attachment, wherein the carbon atom is part of an aromatic ring structure containing only carbon atoms, further having a total of n carbon atoms, 5 or more hydrogen atoms, and no heteroatoms.
- a heteroatom- unsubstituted C ⁇ -Cio-aryl has 6 to 10 carbon atoms.
- heteroatom-substituted C n -aryl refers to a radical, having either a single aromatic carbon atom or a single aromatic heteroatom as the point of attachment, further having a total of n carbon atoms, at least one hydrogen atom, and at least one heteroatom, further wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S.
- a heteroatom-unsubstituted Ci- Cio-heteroaryl has 1 to 10 carbon atoms.
- Non-limiting examples of heteroatom-substituted aryl groups include the groups: -C 6 H 4 F, -C 6 H 4 Cl, -C 6 H 4 Br, -C 6 H 4 I, -C 6 H 4 OH, -C 6 H 4 OCH 3 , -C 6 H 4 OCH 2 CH 3 , -C 6 H 4 OC(O)CH 3 , -C 6 H 4 NH 2 , -C 6 H 4 NHCH 3 , -C 6 H 4 N(CH 3 ) 2 , -C 6 H 4 CH 2 OH, -C 6 H 4 CH 2 OC(O)CH 3 , -C 6 H 4 CH 2 NH 2 , -C 6 H 4 CF 3 , -C 6 H 4 CN, -C 6 H 4 CHO, -C 6 H 4 CHO, -C 6 H 4 C(O)CH 3 , -C 6 H 4 C(O)C 6 H 5 , -C 6 H 4
- aralkyl includes heteroatom-unsubstituted aralkyl, heteroatom-substituted aralkyl, heteroatom-unsubstituted C n -aralkyl, heteroatom-substituted C n -aralkyl, heteroaralkyl, and heterocyclic aralkyl groups. In certain embodiments, lower aralkyls are contemplated.
- Aralkyls generally refer to radicals comprising the formula -alkyl-aryl.
- lower aralkyl refers to aralkyls of 7-12 carbon atoms (that is, 7, 8, 9, 10, 11 or 12 carbon atoms).
- heteroatom-unsubstituted C n -aralkyl refers to a radical, having a single saturated carbon atom as the point of attachment, further having a total of n carbon atoms, wherein at least 6 of the carbon atoms form an aromatic ring structure containing only carbon atoms, 7 or more hydrogen atoms, and no heteroatoms.
- a heteroatom- unsubstituted C 7 -Ci i-aralkyl has 7 to 11 carbon atoms.
- heteroatom- unsubstituted aralkyls are: 2,3-dihydro-lH-indenyl, 1,2,3,4-tetrahydronaphthalenyl, phenylmethyl (benzyl, Bn) and phenylethyl.
- heteroatom-substituted C n -aralkyl refers to a radical, having a single saturated carbon atom as the point of attachment, further having a total of n carbon atoms, 0, 1, or more than one hydrogen atom, and at least one heteroatom, wherein at least one of the carbon atoms is incorporated an aromatic ring structures, further wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S.
- a heteroatom-substituted C 2 -CiO- heteroaralkyl has 2 to 10 carbon atoms.
- heteroatom-substituted C n -aralkyls include indolinyl, benzofuranyl and benzothiophenyl. In certain embodiments, certain aryl groups may be specifically excluded.
- halide refers to fluoro, chloro, bromo or iodo. In certain embodiments, certain halides may be specifically excluded.
- Compounds as described herein may contain one or more asymmetric centers and thus can occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. All possible stereoisomers of the all the compounds described herein, unless otherwise noted, are contemplated as being within the scope of the present invention. In preferred embodiments, a single diastereomer is contemplated.
- the chiral centers of the compounds of the present invention can have the S- or the R-configuration, as defined by the IUPAC 1974 Recommendations. The present invention is meant to comprehend all such isomeric forms of the compounds of the invention.
- the claimed invention is also intended to encompass salts of any of the synthesized compounds of the present invention.
- salt(s) as used herein, is understood as being acidic and/or basic salts formed with inorganic and/or organic acids and bases.
- Zwitterions are understood as being included within the term “salt(s)” as used herein, as are quaternary ammonium salts such as alkylammonium salts.
- Nontoxic, pharmaceutically acceptable salts are preferred as described below, although other salts may be useful, as for example in isolation or purification steps.
- Non-limiting examples of acid addition salts include but are not limited to acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate
- Non-limiting examples of basic salts include but are not limited to ammonium salts; alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; salts comprising organic bases such as amines (e.g., dicyclohexylamine, alkylamines such as z-butylamine and f-amylamine, substituted alkylamines, aryl-alkylamines such as benzylamine, dialkylamines, substituted dialkylamines such as N-methyl glucamine (especially N-methyl D-glucamine), trialkylamines, and substituted trialkylamines); and salts comprising amino acids such as arginine, lysine and so forth.
- amines e.g., dicyclohexylamine, alkylamines such as z-butylamine and f-amylamine, substituted alkylamines, aryl-alkyl
- the basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myrtistyl and stearyl chlorides, bromides and iodides), arylalkyl halides (e.g., benzyl and phenethyl bromides) and others known in the art.
- lower alkyl halides e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates e.g., dimethyl, diethyl, dibutyl, and dia
- a “protecting group,” as used herein, is defined as a group used for the purpose of this temporary blockage.
- protecting groups or protecting agents
- use of the phrases "protected carboxylic acid,” “protected amine” or “protected hydroxy,” for example, does not mean that every functional group available to be protected is protected.
- Functional groups necessary for the desired transformation, for example, should be unprotected.
- the function of a protecting group is to protect one or more functionalities (e.g.,
- the same protecting group may be used to protect one or more of the same or different functional group(s).
- different protecting groups can be used to protect the same type of functional group within a compound of the present invention in multiple steps. There are a number of methods well known to those skilled in the art for accomplishing such a step. For protecting agents, their reactivity, installation and use, see, e.g., Greene & Wuts, 1999.
- a protecting group When a protecting group is no longer needed, it is removed by methods well known to those skilled in the art.
- Agents used to remove the protecting group are sometimes called deprotecting agents.
- Protecting groups must be readily removable (as is known to those skilled in the art) by methods employing deprotecting agents that are well known to those skilled in the art. It is well known that certain deprotecting agents remove some protective groups and not others, while other deprotecting agents remove several types of protecting groups from several types of functional groups. Thus, a first deprotecting agent may be used to remove one type of protecting group, followed by the use of a second deprotecting agent to remove a second type of protecting group, and so on.
- the deprotecting agent is the use of gaseous H 2 in the presence of a palladium catalyst dispersed on carbon in order to remove a benzyl (Bn) protecting group and reveal a free carboxylic acid.
- a palladium catalyst dispersed on carbon in order to remove a benzyl (Bn) protecting group and reveal a free carboxylic acid.
- Bn benzyl
- Persons of ordinary skill in the art will be familiar with the proper ordering of protective group removal using deprotecting agents. See e.g., Greene & Wuts, 1999. Particular non-limiting examples of protecting groups are discussed below.
- amino protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 7.
- the amino protecting group may be a carbamate.
- a suitable amino protecting group may be selected from the group consisting of t- butoxycarbonyl, benzyloxycarbonyl, formyl, trityl, acetyl, trichloroacetyl, dichloroacetyl, chloroacetyl, trifluoroacetyl, difluoroacetyl, fluoroacetyl, benzyl chloroformate, 4- phenylbenzyloxycarbonyl, 2-methylbenzyloxycarbonyl, 4-ethoxybenzyloxycarbonyl, 4- fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2- chlorobenzyloxycarbonyl, 2,4-dichlorobenzyloxycarbonyl, 4-brom
- triphenylphosphino ethoxycarbonyl, fluorenylmethoxycarbonyl, 2- (trimethylsilyl)ethoxycarbonyl, allyloxycarbonyl, l-(trimethylsilylmethyl)prop-l- enyloxycarbonyl, 5-benzisoxalylmethoxycarbonyl, 4-acetoxybenzyloxycarbonyl, 2,2,2- trichloroethoxycarbonyl, 2-ethynyl-2-propoxycarbonyl, cyclopropylmethoxycarbonyl, 4- (decyloxyl)benzyloxycarbonyl, isobomyloxycarbonyl, 1 -piperidyloxycarbonyl and 9- fluorenylmethylcarbonyl, for example.
- the amine protecting group is Z-butoxycarbonyl (Boc), benzyloxycarbonyl (CBz), paramethoxybenzyl (PMB), or 9-fluorenylmethylcarbonyl (Fmoc).
- Thiol protecting groups are well known to those skilled in the art. See, for example,
- a suitable thiol protecting group may be selected from the group consisting of acetamidomethyl, benzamidomethyl, 1 -ethoxyethyl, benzoyl, triphenylmethyl, z-butyl, benzyl, adamantyl, cyanoethyl, acetyl, and trifluoroacetyl, for example.
- Alcohol protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 2.
- a suitable alcohol protecting group may be selected from the group consisting of methoxymethyl, paramethoxybenzyl, (phenyldimethylsilyl)methoxymethyl, triethylsilyl, benzyloxymethyl, t-butoxymethyl, and tetrahydropyranyl, for example.
- the an alcohol protecting group is benzyl (Bn), paramethoxybenzyl (PMB), or triethylsilyl (TES).
- Carboxylic acid protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 5.
- a suitable carboxylic acid protecting group may be selected from the group consisting of dimethylacetal, methoxymethylester, phenylacetoxymethyl ester and tetrahydropyranyl ester, for example.
- the carboxylic acid protecting group is benzyl (Bn).
- a "protected carboxylic acid” refers to a carboxylic acid group protected by a carboxylic acid protecting group.
- the compounds, agents, and active ingredients e.g., solvents, catalysts, bases used in reactions, and other compounds, agents, and active ingredients described herein
- the compounds, agents, and active ingredients can be obtained by any means known to a person of ordinary skill in the art.
- the compounds, agents, and active ingredients can be isolated by obtaining the source of such compounds, agents, and active ingredients.
- the compounds, agents, and active ingredients are commercially available (e.g., Sigma- Aldrich, Milwaukee, WI). Modifications or derivatives of the compounds, agents, and active ingredients disclosed throughout this specification are contemplated as being useful with the methods and compositions of the present invention.
- Derivatives may be prepared and the properties of such derivatives may be assayed for their desired properties by any method known to those of skill in the art.
- “derivative” refers to a chemically modified compound that still retains the desired effects of the compound prior to the chemical modification. Such derivatives may have the addition, removal, or substitution of one or more chemical moieties on the parent molecule.
- Non-limiting examples of the types modifications that can be made to the compounds and structures disclosed herein include the addition or removal of lower alkanes such as methyl, ethyl, propyl, or substituted lower alkanes such as hydroxymethyl or aminomethyl groups; carboxyl groups and carbonyl groups; hydroxyls; nitro, amino, amide, and azo groups; sulfate, sulfonate, sulfono, sulfhydryl, sulfonyl, sulfoxido, phosphate, phosphono, phosphoryl groups, and halide substituents.
- lower alkanes such as methyl, ethyl, propyl, or substituted lower alkanes
- carboxyl groups and carbonyl groups hydroxyls; nitro, amino, amide, and azo groups
- sulfate, sulfonate, sulfono, sulfhydryl, sulfonyl s
- Additional modifications can include an addition or a deletion of one or more atoms of the atomic framework, for example, substitution of an ethyl by a propyl; substitution of a phenyl by a larger or smaller aromatic group.
- hetero atoms such as N, S, or O can be substituted into the structure instead of a carbon atom.
- compositions of the present invention comprise an effective amount of one or more candidate substance or additional agent dissolved or dispersed in a pharmaceutically acceptable carrier.
- pharmaceutically acceptable refers to molecular entities and compositions that do not produce an adverse, allergic or other untoward reaction when administered to an animal, such as, for example, a human, as appropriate.
- the preparation of a pharmaceutical composition that contains at least one candidate substance or additional active ingredient will be known to those of skill in the art in light of the present disclosure, as exemplified by Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, incorporated herein by reference.
- pharmaceutically acceptable carrier includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drugs, drug stabilizers, gels, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, such like materials and combinations thereof, as would be known to one of ordinary skill in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
- the candidate substance may comprise different types of carriers depending on whether it is to be administered in solid, liquid or aerosol form, and whether it need to be sterile for such routes of administration as injection.
- the present invention can be administered intravenously, intradermally, intraarterially, intraperitoneally, intralesionally, intracranially, intraarticularly, intraprostaticaly, intrapleurally, intratracheally, intranasally, intravitreally, intravaginally, intrarectally, topically, intratumorally, intramuscularly, subcutaneously, subconjunctival, intravesicularlly, mucosally, intrapericardially, intraumbilically, intraocularally, orally, locally, via inhalation (e.g., aerosol inhalation), via injection, via infusion, via continuous infusion, via localized perfusion bathing target cells directly, via a catheter, via a lavage, in cremes, in lipid compositions (e.g., liposomes), or by other method or any combination
- compositions of the present invention administered to an animal patient can be determined by physical and physiological factors such as body weight, severity of condition, the type of disease being treated, previous or concurrent therapeutic interventions, idiopathy of the patient and on the route of administration.
- the practitioner responsible for administration will, in any event, determine the concentration of active ingredient(s) in a composition and appropriate dose(s) for the individual subject.
- pharmaceutical compositions may comprise, for example, at least about 0.1% of a compound of the present invention.
- the compound may comprise between about 2% to about 75% of the weight of the unit, or between about 25% to about 60%, for example, and any range derivable therein.
- a dose may also comprise from about 1 microgram/kg/body weight, about 5 microgram/kg/body weight, about 10 microgram/kg/body weight, about 50 microgram/kg/body weight, about 100 microgram/kg/body weight, about 200 microgram/kg/body weight, about 350 microgram/kg/body weight, about 500 microgram/kg/body weight, about 1 milligram/kg/body weight, about 5 milligram/kg/body weight, about 10 milligram/kg/body weight, about 50 milligram/kg/body weight, about 100 milligram/kg/body weight, about 200 milligram/kg/body weight, about 350 milligram/kg/body weight, about 500 milligram/kg/body weight, to about 1000 mg/kg/body weight or more per administration, and any range derivable therein.
- a range of about 5 mg/kg/body weight to about 100 mg/kg/body weight, about 5 microgram/kg/body weight to about 500 milligram/kg/body weight, etc. can be administered, based on the numbers described above.
- the composition may comprise various antioxidants to retard oxidation of one or more component.
- the prevention of the action of microorganisms can be brought about by preservatives such as various antibacterial and antifungal agents, including but not limited to parabens (e.g., methylparabens, propylparabens), chlorobutanol, phenol, sorbic acid, thimerosal, or combinations thereof.
- parabens e.g., methylparabens, propylparabens
- chlorobutanol phenol
- sorbic acid thimerosal, or combinations thereof.
- the candidate substance may be formulated into a composition in a free base, neutral or salt form.
- Pharmaceutically acceptable salts include the acid addition salts, e.g., those formed with the free amino groups of a proteinaceous composition, or which are formed with inorganic acids such as for example, hydrochloric or phosphoric acids, or such organic acids as acetic, oxalic, tartaric or mandelic acid. Salts formed with the free carboxyl groups can also be derived from inorganic bases such as for example, sodium, potassium, ammonium, calcium or ferric hydroxides; or such organic bases as isopropylamine, trimethylamine, histidine, or procaine.
- a carrier can be a solvent or dispersion medium comprising but not limited to, water, ethanol, polyol (e.g., glycerol, propylene glycol, liquid polyethylene glycol, etc.), lipids (e.g., triglycerides, vegetable oils, liposomes) and combinations thereof.
- the proper fluidity can be maintained, for example, by the use of a coating, such as lecithin; by the maintenance of the required particle size by dispersion in carriers such as, for example liquid polyol or lipids; by the use of surfactants such as, for example hydroxypropylcellulose; or combinations thereof such methods.
- nasal solutions are usually aqueous solutions designed to be administered to the nasal passages in drops or sprays.
- Nasal solutions are prepared so that they are similar in many respects to nasal secretions, so that normal ciliary action is maintained.
- the aqueous nasal solutions usually are isotonic or slightly buffered to maintain a pH of about 5.5 to about 6.5.
- antimicrobial preservatives similar to those used in ophthalmic preparations, drugs, or appropriate drug stabilizers, if required, may be included in the formulation.
- various commercial nasal preparations are known and include drugs such as antibiotics or antihistamines.
- the candidate substance is prepared for administration by such routes as oral ingestion.
- the solid composition may comprise, for example, solutions, suspensions, emulsions, tablets, pills, capsules (e.g., hard or soft shelled gelatin capsules), sustained release formulations, buccal compositions, troches, elixirs, suspensions, syrups, wafers, or combinations thereof.
- Oral compositions may be incorporated directly with the food of the diet.
- carriers for oral administration comprise inert diluents, assimilable edible carriers or combinations thereof.
- the oral composition may be prepared as a syrup or elixir.
- a syrup or elixir and may comprise, for example, at least one active agent, a sweetening agent, a preservative, a flavoring agent, a dye, a preservative, or combinations thereof.
- an oral composition may comprise one or more binders, excipients, disintegration agents, lubricants, flavoring agents, and combinations thereof.
- a composition may comprise one or more of the following: a binder, such as, for example, gum tragacanth, acacia, cornstarch, gelatin or combinations thereof; an excipient, such as, for example, dicalcium phosphate, mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate or combinations thereof; a disintegrating agent, such as, for example, corn starch, potato starch, alginic acid or combinations thereof; a lubricant, such as, for example, magnesium stearate; a sweetening agent, such as, for example, sucrose, lactose, saccharin or combinations thereof; a flavoring agent, such as, for example peppermint, oil of wintergreen, cherry flavoring, orange flavoring, etc.; or combinations thereof the fore
- the dosage unit form When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, carriers such as a liquid carrier. Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar, or both.
- suppositories are solid dosage forms of various weights and shapes, usually medicated, for insertion into the rectum, vagina, or urethra. After insertion, suppositories soften, melt or dissolve in the cavity fluids.
- traditional carriers may include, for example, polyalkylene glycols, triglycerides, or combinations thereof.
- suppositories may be formed from mixtures containing, for example, the active ingredient in the range of about 0.5% to about 10%, and preferably about 1% to about 2%.
- Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and/or the other ingredients.
- certain methods of preparation may include vacuum-drying or freeze-drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered liquid medium thereof.
- the liquid medium should be suitably buffered if necessary and the liquid diluent first rendered isotonic prior to injection with sufficient saline or glucose.
- the preparation of highly concentrated compositions for direct injection is also contemplated, where the use of DMSO as solvent is envisioned to result in extremely rapid penetration, delivering high concentrations of the active agents to a small area.
- the composition must be stable under the conditions of manufacture and storage, and preserved against the contaminating action of microorganisms, such as bacteria and fungi. It will be appreciated that endotoxin contamination should be kept minimally at a safe level, for example, less that 0.5 ng/mg protein.
- prolonged absorption of an injectable composition can be brought about by the use in the compositions of agents delaying absorption, such as, for example, aluminum monostearate, gelatin, or combinations thereof.
- the compound may be combined with traditional drugs. It is contemplated that this type of combination therapy may be used in vitro or in vivo.
- an anticancer agent may be used in combination with a compound.
- An anti-viral or antibiotic agent may be used in combination with a compound, for example.
- agents of the present invention may be provided in a combined amount with an effective amount of an anti-cancer agent. This process may involve contacting the cell(s) with the agents at the same time or within a period of time wherein separate administration of the substances produces a desired therapeutic benefit. This may be achieved by contacting the cell, tissue or organism with a single composition or pharmacological formulation that includes two or more agents, or by contacting the cell with two or more distinct compositions or formulations, wherein one composition includes one agent and the other includes another.
- the compounds of the present invention may precede, be co-current with and/or follow the other agents by intervals ranging from minutes to weeks.
- the agents are applied separately to a cell, tissue or organism, one would generally ensure that a significant period of time did not expire between the time of each delivery, such that the agents would still be able to exert an advantageously combined effect on the cell, tissue or organism.
- one may contact the cell, tissue or organism with two, three, four or more modalities substantially simultaneously (i.e., within less than about a minute) as the candidate substance.
- one or more agents may be administered within of from substantially simultaneously, about 1 minute, about 5 minutes, about 10 minutes, about 20 minutes about 30 minutes, about 45 minutes, about 60 minutes, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 22 hours, about 23 hours, about 24 hours, about 25 hours, about 26 hours, about 27 hours, about 28 hours, about 29 hours, about 30 hours, about 31 hours, about 32 hours, about 33 hours, about 34 hours, about 35 hours, about 36 hours, about 37 hours, about 38 hours, about 39 hours, about 40 hours, about 41 hours, about 42 hours, about 43 hours, about 44 hours, about 45 hours, about 46 hours, about 47 hours, about 48 hours, about 1 day, about 2 days, about 3 days, about 4 days, about 5 days, about 2 hours
- an "anti-cancer” agent is capable of negatively affecting cancer in a subject, for example, by killing one or more cancer cells, inducing apoptosis in one or more cancer cells, reducing the growth rate of one or more cancer cells, reducing the incidence or number of metastases, reducing tumor size, inhibiting tumor growth, reducing the blood supply to a tumor or one or more cancer cells, promoting an immune response against one or more cancer cells or a tumor, preventing or inhibiting the progression of a cancer, or increasing the lifespan of a subject with a cancer.
- Anti-cancer agents are well-known in the art and include, for example, chemotherapy agents (chemotherapy), radiotherapy agents (radiotherapy), a surgical procedure, immune therapy agents (immunotherapy), genetic therapy agents (gene therapy), reoviral therapy, hormonal therapy, other biological agents (biotherapy), and/or alternative therapies.
- FIGs. 1 and 2 represent, in certain embodiments, general synthetic methods for accessing compounds of the present invention.
- Protected ornithine and alanine were purchased from Acros and used as received. Other reagents were purchased from Aldrich (Milwaukee, WI) and used as received.
- the fluorogenic peptide substrate Suc-LL VY-AMC was purchased from Biomol International, Inc. (Plymouth Meeting, PA). The reaction mixture consisted of approximately 5 nM 2OS proteasome in buffer (50 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 % v/v DMSO, 5 mM MgCl 2 , and 0.02 % SDS) and 1 ⁇ L test compounds dissolved in DMSO at final concentrations of 0.4-50 ⁇ M along with 100 ⁇ M Suc-LLVY-AMC. The resulting fluorescence from liberated AMC was measured every ten minutes at 380/460 nm for 2-3 hours. Suc-LLVY-Amc incubated without proteasome served as a background control. Results are the average of triplicate time points. The results are shown below and in FIG. 3:
- Known and novel belactosin derivatives inhibit the turnover of a fluorogenic substrate by fatty acid synthase thioesterase.
- the thioesterase (230 nM) was incubated with a range of test compound concentrations (100 ⁇ M to 0.16 ⁇ M) and 120 ⁇ M 4-MU-heptanoate substrate, approximately 3.5x K m . Fluorescence measurements were taken every five minutes at 350/450 nm. Results shown are the calculated IC 5O value and corresponding 95% confidence intervals from a dose-response curve fit of the data in triplicate. See Example 13 for compounds tested.
- compositions and/or methods disclosed and claimed in this specification can be made and executed without undue experimentation in light of the present disclosure. While the compositions and methods of this invention have been described in terms of preferred embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and/or methods and in the steps or in the sequence of steps of the method described herein without departing from the concept, spirit and scope of the invention. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the invention as defined by the appended claims.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Derivatives of belactosin and their synthesis are disclosed. In certain embodiments, compounds of the present invention exhibit anti-cancer, antiviral, antibiotic, and/or autoimmune therapeutic abilities. In general, methods of synthesis disclosed herein allow for introduction of a variety of substituents at numerous positions as well as the facile introduction of a beta-lactone ring moiety. The synthetic steps comprise, in preferred embodiments, a tandem Mukaiyama aldol lactonization reaction. Data demonstrating the utility of some of the derivatives as proteasome inhibitors is also disclosed.
Description
DESCRIPTION
NOVEL BELACTOSIN DERIVATIVES AS THERAPEUTIC AGENTS/BIOLOGICAL PROBES AND THEIR SYNTHESIS
BACKGROUND OF THE INVENTION
This application claims the benefit of U.S. Provisional Application No. 60/819,213 filed July 7, 2006, the contents of which are incorporated herein in their entirety.
This invention was made with government support under CHE-0416260 awarded by the National Science Foundation (NSF). The government has certain rights in the invention.
A. Field of the Invention
The present invention relates generally to derivatives of belactosin and their synthesis. Certain belactosin derivatives of the present invention are inhibitors of both fatty acid synthase and the proteasome. As such, these derivatives, in certain embodiments, are candidates as anticancer and/or auto-immune therapeutics, antivirals, or antibiotics. The synthesis of these compounds generally comprises a concise, single step formation of a common beta-lactone (2-oxetanone) moiety.
B. Background of the Invention
Belactosin and its derivatives have recently garnered interest in the scientific community due to their potential anti-cancer activities. Initial studies of the belactosins revealed that these molecules could represent good lead compounds for cancer treatment by regulating the ubiquitin-proteasome pathways. Mizukami et al, 1997; Asai et al, 2004. The ubiquitin-proteasome pathway generates peptide products with a narrow length distribution centered around 8-12 mers, a size suitable for binding to MHC class I molecules. Groll et al, 2005; Michalek et al, 1993. This process allows CD8+ T lymphocytes to identify and eliminate cells that are synthesizing abnormal or "foreign" proteins, as may arise through mutations or infection by viruses. Cresswell et al, 1999. The 2OS proteasome is the central component of this degradation system. Kumaraswamy et al, 2006. Certain belactosins have been shown to exhibit behavior similar to that of lactacystin, an inhibitor of the 2OS proteasome. Kumaraswamy et al, 2006. Proteasome inhibitors represent a novel anticancer
therapy. Adams et al, 2000; Murray et al, 2000; Adams et al, 1999; Adams et al, 2002a; Adams et al, 2002b. It has been shown that belactosins A and C arrest cell-cycle progression at the G2/M phase. Asai et al, 2000. Belactosin and its derivatives feature a beta-lactone ring, and this moiety is pivotal for bioactivity. Kumaraswamy et ah, 2006. Thus, synthetic derivatives of belactosin featuring this moiety are potential therapeutic targets of the proteasome.
(-)-belatctosin C

Orlistat, a drug approved for treating obesity, also features such a beta-lactone ring moiety. Purohit et al., 2006; Kridel et al., 2004; Knowles et al., 2004. This drug has recently been shown to be a potent inhibitor of human fatty acid synthase (FAS); further, this natural product derivative is cytotoxic and cytostatic to tumor cells in vitro and can inhibit tumor growth in vivo. Kridel et al., 2004; Knowles et al., 2004. FAS is responsible for the cellular synthesis of palmitate and is attracting great interest as a drug target in oncology because it is up-regulated in most solid tumors, including those of the breast, prostate and ovary. Purohit et al, 2006; AIo et al, 1996; Swinnen et al, 2002; Rossi et al, 2003; Pizer et al, 1996a; Pizer et al, 1996b; Pizer et al, 2000; Pizer et al, 2001; Gansler et al, 1997. Furthermore, a number of studies show that a pharmacologic blockade of FAS can be cytostatic and cytotoxic to tumor cells. Kuhajda et al, 2000; Kuhajda et al, 1994; Pizer et al, 1998; Funabashi et al. 1989; Pizer et al, 2000. However, orlistat has poor solubility and poor bioavailability, so there is a need to develop new beta-lactones that overcome these problems and that can be deployed as potential antitumor drugs. In addition, simplified derivatives that are readily prepared would also ultimately be attractive from the standpoint of process development.
Previous syntheses of belactosin and derivatives have relied on multi-step syntheses of the beta-lactone moiety involving, for example, aldol chemistry followed by a subsequent lactonization step. In addition, syntheses to date have focused on synthesis of appropriate beta-lactones followed by coupling to a dipeptide. To date, there have been three known reports of syntheses of belactosin and derivatives. Armstrong and Scutt, 2004; Larionov and
de Meijere, 2004; Kumaraswamy et ah, 2006. Improved methods of preparing belactosin and derivatives are needed, such as methods involving fewer synthetic steps.
SUMMARY OF THE INVENTION
The present invention overcomes deficiencies in the art by providing novel derivatives of belactosin comprising a beta-lactone ring structure. A particularly attractive feature of certain compounds of the present invention is their ability, in certain embodiments, to act as inhibitors of the proteasome and/or FAS. This inhibitory activity significantly increases the therapeutic potential of these compounds. For example, certain compounds of the present invention are low nanomolar inhibitors of the 2OS proteasome. As such, these compounds could be used in treatments for cancer as well as malaria, tuberculosis and other indications. Indeed, any protein that recognizes the beta-lactone moiety as a substrate may recognize a compound as described herein.
Further, the present invention contemplates, in certain embodiments, a concise synthesis of these compounds that allows access to a variety of beta-lactones in a diastereoselective manner, often in fewer steps than disclosed in previous methodologies. In preferred embodiments, the synthetic steps comprise a tandem Mukaiyama aldol lactonization (TMAL) reaction. Other compounds comprising a beta-lactone moiety besides belactosins include the vegetal poison anisatin and the antibiotic 1233 A (Kumaraswamy and Markondaiah, 2007): such compounds may also be accessed via methods of the present invention.
Accordingly, compounds of the present invention are generally drawn to compounds of formula (X):



Within formula (X), Ro may be selected from a group consisting of H, alkyl, an amine protecting group and

wherein R5 and Rg may each independently be selected from the group consisting of H, alkyl, aryl, an amine protecting group and


Further, R7 may be selected from the group consisting of H, alkyl and aryl.
Also within formula (I), Ri and R3 may each independently be selected from the group consisting of H, alkyl, aryl and an amine protecting group; R2 may be selected from the group consisting of H, aryl, alkyl, -CO2W, wherein W is H, alkyl, aryl or a carboxylic acid protecting group, and


wherein Rs and R9 may each independently be selected from the group consisting of H, alkyl, aryl, -CH(OH)alkyl, -CH(OH)aryl, -ORi0 and -NRnRi2, wherein R10-R12 may each independently be selected from the group consisting of H, alkyl, aryl, acetyl, -SiRoRi4RiS, an alcohol protecting group and an amine protecting group, wherein R13-R15 may each
independently be selected from the group consisting of alkyl and aryl; and n may equal 1-7, depending on the need for greater chain length, flexibility and/or distance between the two termini of the overall compound.
When a compound of formula (I) is contemplated, provisos such as the following may apply: if R0 equals
CBzHN

Me , R2 equals Bn and n equals 3, then R4 cannot be
; and if Ro equals

, R2 equals H and n equals 3, then R4 cannot be

In certain preferred embodiments of the compound comprising formula (I), Ro equals
H and Ri equals an amine protecting group.
In other preferred embodiments, Ro equals

. When R0 is so chosen, R5 is, in certain preferred embodiments, an amine protecting group. When Ro is so chosen, Re is preferably H. When Ro is so chosen, R7 is preferably -CH3. In another preferred embodiment, R5 is an amine protecting group, Re is H and R7 is -CH3. The amine protecting group may be Cbz, in certain embodiments. In other preferred embodiments, R7 is -CH3 and R5 is


In other preferred embodiments of the compound comprising formula (I), Ri is H. In certain embodiments, R2 is alkyl and in certain other embodiments, R2 is a carboxylic acid or a protected carboxylic acid. The protected carboxylic acid may, in certain embodiments, be protected by a benzyl protecting group. In certain embodiments, R3 is H. In certain embodiments, R4 is


. In some preferred embodiments, either Re or R9 is H, whereas in other preferred embodiments, Rg is -OR10 and Rg is H. Ri0 may be, in certain preferred embodiments, selected from the group consisting of H, an alcohol protecting group and - SR13R14R15. R13-R15 may independently be, in certain embodiments, H, alky or aryl, with ethyl groups comprising a preferred embodiment. In certain embodiments, Rs is either alkyl or aryl and R9 is H. In certain embodiments, Rg is -CH(OH)alkyl or -CH(OH)aryl and R9 is H. R8 may be phenyl, in certain embodiments, and may be substituted with any of the substituents as described herein. In certain embodiments, Rg is -NR11R12 and R9 is H. Rn and R12 may each independently be selected from the group consisting of H, acetyl and an amine protecting group, in preferred embodiments. Preferably, n equals 1-5 and even more preferably, n equals 3.
In certain embodiments, a compound of the following formula is contemplated:

wherein: Yi is selected from the group consisting of H, Cbz, Fmoc, PMB, BOM and
CbzHN
^e H t H , wherein t is 1-2; Y2 is H or -CH3; Y3 is selected from the group
consisting of H, Bn, Z
 and , wherein Zi and Z2 are each independently selected from the group consisting of H, -OH, -NH2, -NHCH3 and -CO2H; Y4 is selected from the group consisting of -CH3, -COHI3, -CH2Cy, -CH(OH)Cy, phenyl, - CH2C6H5 and -CH(OH)C6H5; p is 0 or 1; and q is 1-5.
In certain embodiments, a compound of the following formula is contemplated:
and , wherein Zi and Z2 are each independently selected from the group consisting of H, -OH, -NH2, -NHCH3 and -CO2H; Y4 is selected from the group consisting of -CH3, -COHI3, -CH2Cy, -CH(OH)Cy, phenyl, - CH2C6H5 and -CH(OH)C6H5; p is 0 or 1; and q is 1-5.
In certain embodiments, a compound of the following formula is contemplated:

wherein RA is selected from the group consisting of H, Bn and PMB and RB is selected from the group consisting of H, Bn, PMB, -C(O)CH3, Cbz and Boc.
In certain embodiments, a compound of the following formula is contemplated:

wherein:
Ys is selected from the group consisting of H, Cbz, Fmoc, PMB, BOM and
CbzHN
 wherein t is 1-2; Y6 is H or -CH3; Y7 is selected from the
wherein t is 1-2; Y6 is H or -CH3; Y7 is selected from the
group consisting of H, Bn,
 and
and
 , wherein Zi and Z2 are each independently selected from the group consisting of H, -OH, -NH2, -NHCH3 and - CO2H; Y8 is selected from the group consisting of -CH3, -C6Hi3, -CH2Cy, -CH(OH)Cy, phenyl, -CH2C6H5 and -CH(OH)C6H5; r is 0 or 1; and s is 1-5.
, wherein Zi and Z2 are each independently selected from the group consisting of H, -OH, -NH2, -NHCH3 and - CO2H; Y8 is selected from the group consisting of -CH3, -C6Hi3, -CH2Cy, -CH(OH)Cy, phenyl, -CH2C6H5 and -CH(OH)C6H5; r is 0 or 1; and s is 1-5.
In certain embodiments, a compound of the following formula is contemplated:

wherein Rc is selected from the group consisting of H, Bn and PMB and RD is selected from the group consisting of H, Bn, PMB, -C(O)CH3, Cbz and Boc.
In specific embodiments, the following compound is contemplated:

In specific embodiments, the following compound is contemplated:

Another general aspect of the present invention contemplates a method of synthesizing a compound of formula (I):

comprising obtaining a first compound of formula (II):
(II), and admixing it with a second compound of formula (III)


(III) or (IV). Preparations of these compounds may be by any method known to those of skill in the art, and exemplary methods are described herein. The first compound may comprise, for example, a dipeptide or a tripeptide.
When a compound of formula (III) is chosen as the second compound, the reaction conditions may comprise, in certain embodiments, a Lewis acid. Lewis acids are well-known to those of skill in the art. Non-limiting examples of Lewis acids include ZnCl2, Zn(OTf)2 and SnCl4. A chosen solvent for this reaction may comprise any type as described herein and in preferred embodiments, the solvent is methylene chloride. Reaction conditions typically take place at room temperature for periods of 8-36 hours, with 12-24 hours being more preferable. Reactions may be monitored by any means known to those of skill in the art, such as thin-layer chromatography and HPLC. Purification can take place via any means known to those of skill in the art, with silica gel column chromatography comprising a preferred embodiment.
Within formula (III) or (IV), Rs and R9 may each independently be selected from the group consisting of H, alkyl, aryl, -CH(OH)alkyl, -CH(OH)aryl, -OR22 and -NR23R24, wherein R22-R24 may each independently be selected from the group consisting of H, alkyl, aryl, acetyl, -SiR2sR26R27, an alcohol protecting group and an amine protecting group, wherein R25-R27 niay each independently be selected from the group consisting of H, alkyl and aryl; and n may equal 1-7, depending on the need or wish for greater chain length, flexibility and/or distance between the two termini of the overall compound. In certain preferred embodiments, R25-R27 are each ethyl. Ri8-R2O may comprise, independently, H,
alkyl or aryl, with ethyl groups being more preferred. R2i may comprise, in some embodiments, H, alkyl or aryl, with aryl groups being preferable, and pyridyl and phenyl groups being more preferable, and pyridyl groups being even more preferable.
When a compound of formula (IV) is chosen as the second compound, the reaction conditions may comprise a coupling agent, as described herein. A chosen solvent for this reaction may comprise any type described herein and in preferred embodiments, the solvent comprises ethyl acetate/water. Reaction conditions typically take place at room temperature for periods under 24 hours. Reactions may be monitored by any means known to those of skill in the art, such as thin-layer chromatography and HPLC. Purification can take place via any means known to those of skill in the art, with silica gel column chromatography comprising a preferred embodiment.
When employing this method, Ro may be selected from the group consisting of H, alkyl, an amine protecting group and

, wherein R5 and Re may each independently be selected from the group consisting of H, alkyl, aryl, an amine protecting group and


Further, R7 may be selected from the group consisting of H, alkyl and aryl. Also within this method, Ri and R3 may each independently be selected from the group consisting of H, alkyl, aryl and an amine protecting group; and R2 may be selected from the group consisting of H, aryl, alkyl, -CO2W, wherein W is H, alkyl, aryl or a carboxylic acid protecting group, and


In certain preferred embodiments of this method, Ro equals H and Ri equals an amine protecting group, such as Cbz. In other preferred embodiments, Ro equals

. In certain embodiments, R5 is an amine protecting group. In certain embodiments, R6 is H. In certain embodiments, R7 is CH3. In a certain preferred embodiment, R5 is an amine protecting group, Re is H and R7 is CH3. The amine protecting group may be, in certain embodiments, Cbz. In some preferred embodiments, R7 is CH3 and R5 is


In certain embodiments of the method, the R4 group generated by this method comprises

, whereas in other embodiments, the R4 group generated by this method comprises

In certain preferred embodiments of this method, either Rg or Rg is H. In other preferred embodiments, Rs is either alkyl or aryl and R9 is H. In other preferred embodiments, Rs is -OR10 and R9 is H. In certain embodiments, Rs is -CH(OH)alkyl or - CH(OH)aryl and R9 is H. In certain preferred embodiments, Rio is selected from the group consisting of H, an alcohol protecting group and SR13R14R15. R13-R15 may comprise, for example, H, alkyl or aryl, with ethyl groups being more preferred. In certain preferred embodiments, Rs is phenyl. In other preferred embodiments, Rs is -NR11R12 and R9 is H. Rn and R12, in preferred embodiments, may each independently be selected from the group consisting of H, acetyl and an amine protecting group.
Another preferred embodiment of this method contemplates n equaling 1-5, with n equaling 3 being a more preferred embodiment. Yet another preferred embodiment comprises Rn as Bn.
When employing the method of the present invention, provisos such as the following may apply: when formula (II) comprises

Formula (IV), then in the structure of Formula (IV), Rs and R9 together may not be H and propyl, respectively, or H and 2-methylpropyl, respectively.
In certain methods of the present invention, a compound of formula (XI) is formed as an intermediate:

wherein R28 is -OH or

It is specifically contemplated that in certain embodiments, compounds disclosed in WO 00/043000 and/or U.S. Pat. 7,223,745 are not encompassed by the present invention. Compounds disclosed in Arionov and De Meijere, 2004, are also excluded, in certain embodiments. Each of these three references are specifically incorporated herein in their entireties. For example, the following compounds may be excluded, in certain embodiments:

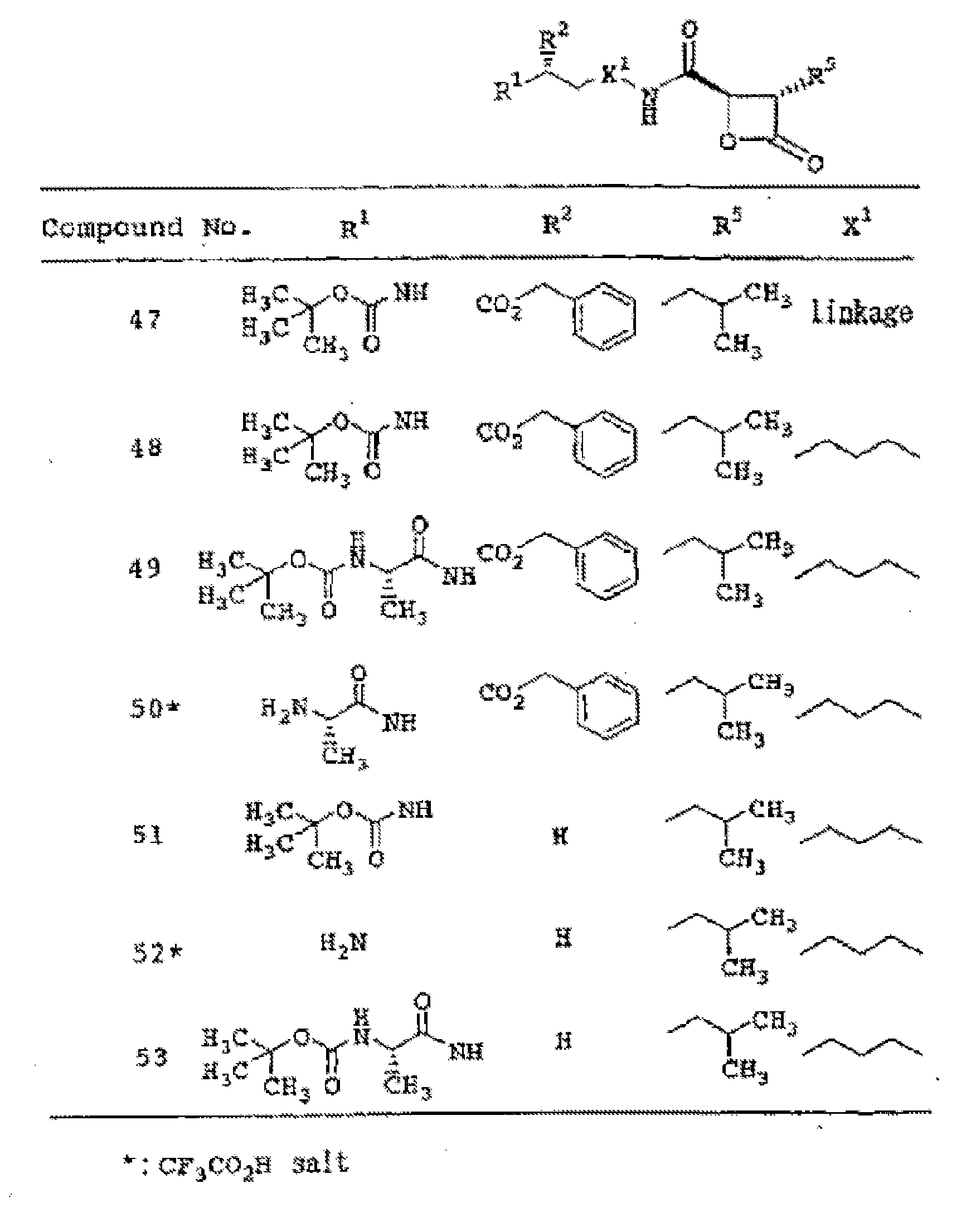




and

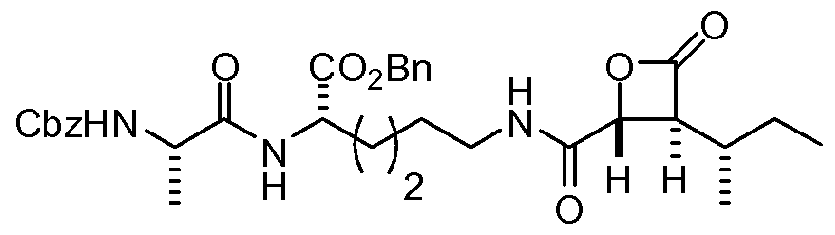

Certain aspects of the present invention contemplate a method of inhibiting the 2OS proteasome, comprising contacting a cell with an effective amount of a compound as described herein. One skilled in the art can purify and measure the activity of the proteasome using approaches such as those described in the following citations: Hirano et al., 2005; Akaishi et al., 1996; Ugai et al., 1993; Adams et al., 1999; and Mellgren, 1997, each of which is incorporated herein by reference in its entirety. The method may take place in vitro or in vivo. Compounds of the present invention may also be used to treat proteasome -related conditions, such as cancer, Alzheimer's disease, malaria, tuberculosis, eye disorders and asthma. Accordingly, methods of treatment of these conditions are also contemplated. Compounds used in these methods may be comprised in a pharmaceutically acceptable excipient, diluent, or vehicle.
Certain aspects of the present invention contemplate a method of inhibiting fatty acid synthase, comprising contacting a cell with an effective amount of a compound as described herein. For example, compounds may be screened for their ability to inhibit the thioesterase domain of fatty acid synthase, which liberates palmitate, the natural substrate, from the enzyme. One of ordinary skill in the art could express and purify the recombinant thioesterase using procedures described in, e.g., Chakravarty et al., 2004 and Kridel et al., 2004. In this study the thioesterase domain of fatty acid synthase was PCR amplified using the following primers: 5_ ATG ACG CCC AAG GAG GAT GGT CTG GCC CAG CAG (corresponds to nucleotides 6727-6756) and 3_ GCC CTC CCG CAC GCT CAC GCG TGG CT (corresponds to nucleotides 7625-7650). The recombinant thioesterase domain was cloned into pTrcHis (Invitrogen) and expressed in Escheria coli. The recombinant protein corresponds to residues 2202 through 2509 of FAS. The thioesterase was purified by Ni- affinity chromatography. The method may take place in vitro or in vivo. Compounds of the present invention may also be used to treat fatty acid-synthase-related conditions, which generally includes diseases characterized by hyperproliferation of cells such as inflammation,
angiogenesis and cancer. Such compounds may also be of use in treating obesity as fatty acid synthase is the only enzyme that converts dietary carbohydrate to fat. Accordingly, methods of treatment of these conditions are also contemplated. Compounds used in these methods may be comprised in a pharmaceutically acceptable excipient, diluent, or vehicle. The inhibition of fatty acid synthase in cells can be measured by, for example, directly determining the amount of palmitate synthesized by the cell using methods described in Browne et al, 2006, Kridel et al., 2004 and Pizer et al, 1996.
Another general aspect of the present invention contemplates a method of treating cancer comprising administering to a subject an effective amount of a compound as described herein. The subject may be a mammal, such as a human. The cancer may be any cancer treatable by administration of a compound described herein. For example, the cancer may be breast, prostate, ovarian, brain hepatocarcinoma, melanoma, colorectal, liver, lymphoma, lung, oral, head, neck, spleen, lymph node, small intestine, large intestine, blood cells, stomach, endometrium, testicle, skin, esophagus, bone marrow, blood, cervical, bladder, Ewing's sarcoma, thyroid, and/or gastrointestinal.
The terms "inhibiting," "reducing," or "prevention," or any variation of these terms, when used in the claims and/or the specification includes any measurable decrease or complete inhibition to achieve a desired result.
The term "contact," when applied to a cell, is used herein to describe the process by which a compound of the invention is delivered to a target cell or is placed in direct juxtaposition with the target cell.
As used herein, the term "effective" (e.g., "an effective amount") means adequate to accomplish a desired, expected, or intended result. For example, an "effective amount" may be an amount of a compound sufficient to produce a therapeutic benefit (e.g., effective to reproducibly inhibit decrease, reduce, inhibit or otherwise abrogate the growth of a cancer cell). "Effective amounts" or a "therapeutically relevant amount" are those amounts of a compound sufficient to produce a therapeutic benefit (e.g., effective to reproducibly inhibit decrease, reduce, inhibit or otherwise abrogate the growth of a cancer cell). An effective amount, in the context of treating a subject, is sufficient to produce a therapeutic benefit. The term "therapeutic benefit" as used herein refers to anything that promotes or enhances the well-being of the subject with respect to the medical treatment of the subject's condition.
As used herein, a "suitable solvent" or a "chosen solvent" is a solvent that will facilitate, or at least not significantly impede, the reaction that takes place within that solvent. A suitable solvent choice may depend on, for example, which one(s) will facilitate the solubilizing of all the reagents, or, for example, which one(s) will best facilitate the desired reaction (particularly if the mechanism of the reaction is known). However, a suitable solvent need not completely solubilize each reagent and may actually impede the desired reaction to some degree. Suitable solvents for the methods of the present invention will be known to one of ordinary skill in the art. More than one solvent may be chosen for any particular reaction. Water may also be admixed into any solvent choice, particularly when improvements in solubility of the reagents are required. Suitable solvents may include, for example, polar solvents and non-polar solvents. Suitable solvents include, but are not limited to, dimethylformamide, dimethylsulfoxide, dioxane, methanol, ethanol, hexane, ethyl acetate, methylene chloride, tetrahydrofuran and acetonitrile. In some preferred embodiments, suitable solvents include methylene chloride, tetrahydrofuran and ethyl acetate/water. As used herein a "coupling agent" is a reagent used to facilitate the coupling of two compounds. In preferred embodiments, a coupling agent facilitates the coupling of an NH2- containing compound with a -CChH-containing compound to form an amide bond between the two compounds. Coupling agents are well known to those of ordinary skill in the art and may be employed in certain embodiments of methods of the present invention. Examples of coupling agents include, but are not limited to, l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC) and dicyclohexylcarbodiimide (DCC), optionally in conjunction with catalytic amounts of dimethylaminopyridine (DMAP) as a promoter. Other carbodiimides are also envisioned as coupling agents. Coupling agents are discussed, for example, in Bodansky, 1993 and Grant, 1992. These coupling agents may be used singly or in combination with each other or other agents to facilitate conjugation. Once the two substrates are conjugated using coupling agents, a urea by-product is typically formed. The urea by-product may be removed by filtration. The conjugated product may then be purified by, for example, silica gel column chromatography or HPLC.
As used herein, a "chiral auxiliary" refers to an easily removable chiral group that is capable of influencing the direction of nucleophilic attack. Chiral auxiliaries typically control the diastereoselectivity of a reaction. Persons of skill in the art are familiar with such compounds, and many are commercially available.
Known and unknown equivalents of the specific compounds, agents, and active ingredients discussed throughout this specification can be used with the compositions and methods of the present invention. The equivalents can be used as substitutes for the specific compounds, agents, and active components. The equivalents can also be used to add to the methods and compositions of the present invention. A person of ordinary skill in the art would be able to recognize and identify acceptable known and unknown equivalents to the specific compounds, agents, and active ingredients without undue experimentation.
Persons of ordinary skill in the art will be familiar with methods of purifying compounds of the present invention. One of ordinary skill in the art will understand that compounds of the present invention can generally be purified at any step, including the purification of intermediates as well as purification of the final products. In certain embodiments of the present invention, purification of a compound does not remove all impurities. In some embodiments, such impurities can be identified. Non-limiting examples of purification methods include gel filtration, size exclusion chromatography (also called gel filtration chromatography, gel permeation chromatography or molecular exclusion), dialysis, distillation, crystallization, recrystallization, reprecipitation, sublimation, electrophoresis, prep thin-layer chromatography, silica gel column chromatography and high-performance liquid chromatography (HPLC), including normal-phase HPLC and reverse-phase HPLC. In preferred embodiments, purification comprises silica gel column chromatography. Methods of determining the purity of compounds are well known to those of skill in the art and include, in non-limiting examples, autoradiography, mass spectroscopy, melting point determination, ultra violet analysis, colorimetric analysis, (HPLC), thin-layer chromatography and nuclear magnetic resonance (NMR) analysis (including, but not limited to, H and C NMR). In preferred embodiments, purity is determined via NMR. Software available on various instruments (e.g., spectrophotometers, HPLCs, NMRs) can aid one of skill in the art in making these determinations, as well as other means known to those of skill in the art.
Prodrugs and solvates of the compounds of the present invention are also contemplated herein. Any compound described herein may be a prodrug. The term "prodrug" as used herein, is understood as being a compound which, upon administration to a subject, such as a mammal, undergoes chemical conversion by metabolic or chemical processes to yield a compound any of the formulas herein, or a salt and/or solvate thereof
(Bundgaard, 1991; Bundgaard, 1985). Solvates of the compounds of the present invention are preferably hydrates.
As used herein, "predominantly one enantiomer" or "substantially free" from other optical isomers means that the compound contains at least about 95% of one enantiomer, or more preferably at least about 98% of one enantiomer, or most preferably at least about 99% of one enantiomer. Any compound described herein may, in certain embodiments, be present as predominantly one enantiomer.
In certain embodiments, "substantially pure" compounds are contemplated. That is, any compound as described herein may be a substantially pure compound. As used herein, the term "substantially pure" refers to compounds that are at least about 95% pure, or more preferably at least about 98% pure, or most preferably at least about 99% pure.
It is contemplated that any embodiment discussed in this specification can be implemented with respect to any method, compound or composition of the invention, and vice versa. Furthermore, compounds and compositions of the invention can be used to achieve methods of the invention.
The use of the word "a" or "an" when used in conjunction with the term "comprising" in the claims and/or the specification may mean "one," but it is also consistent with the meaning of "one or more," "at least one," and "one or more than one."
Throughout this application, the term "about" is used to indicate that a value includes the inherent variation of error for the device, the method being employed to determine the value, or the variation that exists among the study subjects. For example, "about" can be within 10%, preferably within 5%, more preferably within 1%, and most preferably within
0.5%.
The use of the term "or" in the claims is used to mean "and/or" unless explicitly indicated to refer to alternatives only or the alternatives are mutually exclusive, although the disclosure supports a definition that refers to only alternatives and "and/or."
As used in this specification and claim(s), the words "comprising" (and any form of comprising, such as "comprise" and "comprises"), "having" (and any form of having, such as
"have" and "has"), "including" (and any form of including, such as "includes" and "include") or "containing" (and any form of containing, such as "contains" and "contain") are inclusive or open-ended and do not exclude additional, unrecited elements or method steps.
Other objects, features and advantages of the present invention will become apparent from the following detailed description. It should be understood, however, that the detailed description and the specific examples, while indicating preferred embodiments of the invention, are given by way of illustration only, since various changes and modifications within the spirit and scope of the invention will become apparent to those skilled in the art from this detailed description.
BRIEF DESCRIPTION OF THE DRAWINGS
FIG. 1. General synthetic method for belactosin C and derivatives: Synthesis of aldehyde dipeptide and direct synthesis of belactosin via the tandem Mukaiyama aldol lactonization (TMAL) process. Note: 4 D M. S = 4 A molecular sieves.
FIG. 2. General synthetic method for belactosin C and derivatives: Synthesis of dipeptide and beta-lactone separately, followed by coupling.
FIG. 3. Proteasome 2OS inhibition assays. Known and novel belactosin derivatives inhibit the turnover of a fluorogenic substrate by the 2OS proteasome. The 2OS proteasome (5 nM) was incubated with (A) 1.25 μM or (B) 50 μM test compound and 100 mM suc-LLVY-
AMC substrate. Fluorescence measurements were taken every ten minutes at 380/460 nm.
Results shown are the average of (A) triplicate or (B) duplicate data points.
DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
The present invention overcomes the deficiencies of the prior art by providing novel derivatives of belactosin as well as a facile means of synthesizing said derivatives. As discussed above, belactosin and its derivatives feature a beta-lactone ring, and this moiety is pivotal for bioactivity. Kumaraswamy et al, 2006. In some embodiments, belactosin derivatives of the present invention may function as inhibitors of human fatty acid synthase
(FAS) and/or the proteasome. Since some proteasome inhibitors represent potential novel anticancer therapy, synthetic derivatives of belactosin featuring the beta-lactone moiety are potential therapeutic targets of the proteasome. Adams et al, 2000; Murray et al, 2000; Adams et al, 1999; Adams et al, 2002a; Adams et al, 2002b.
Orlistat, a drug approved for treating obesity, also features a beta-lactone ring moiety. Purohit et al, 2006; Kridel et al, 2004; Knowles et al, 2004. This drug has recently been shown to be a potent inhibitor of FAS; further, this natural product derivative is cytotoxic and cytostatic to tumor cells in vitro and can inhibit tumor growth in vivo. Kridel et al., 2004; Knowles et al, 2004. FAS is responsible for the cellular synthesis of palmitate and is attracting great interest as a drug target in oncology because it is up-regulated in most solid tumors, including those of the breast, prostate and ovary. Purohit et al, 2006; AIo et al, 1996; Swinnen et al, 2002; Rossi et al, 2003; Pizer et al, 1996a; Pizer et al, 1996b; Pizer et al, 2000; Pizer et al, 2001; Gansler et al, 1997 '. Furthermore, a number of studies show that a pharmacologic blockade of FAS can be cytostatic and cytotoxic to tumor cells. Kuhajda et al, 2000; Kuhajda et al, 1994; Pizer et al, 1998; Funabashi et al 1989; Pizer et al, 2000.
Beta-lactone inhibitors of FAS typically function by acylating the active site serine residue, leading to inhibition of thioesterase (TE) activity and ultimately apoptosis — that is, cell death. Purohit et al, 2006. Similarly, beta-lactone inhibitors of the proteasome typically function by acylation of the active site threonine (chymotrypsin activity of the proteasome). Simon et al, 2006; Maier et al, 2006. Compounds of the present invention, which typically comprise beta-lactone moieties similar to those found in such agents as the belactosins and orlistat, are capable, in certain embodiments, of acylating the active site serine of the FAS-TE and also the threonine of the proteasome, thus typically leading to dual inhibition. Accordingly, in certain embodiments, compounds of the present invention are attractive anticancer agents for study. Such compounds may also be useful for as substrates for other proteins {e.g., enzymes) which recognize the beta-lactone moiety.
Further, a facile means of accessing compounds of the present invention comprises, in preferred embodiments, a tandem Mukaiyama aldol lactonization (TMAL) reaction. The synthetic methodology of the present invention offers, in certain embodiments, access to these biologically interesting beta-lactones in fewer steps than many methodologies previously attempted. Moreover, any compound comprising a beta-lactone moiety may be prepared using methods disclosed herein.
1. Chemical Definitions
The term "alkyl" includes straight-chain alkyl, branched chain alkyl, cycloalkyl (Cy) (alicyclic), cyclic alkyl (e.g., -CH2-cyclopropyl), heteroatom-unsubstituted alkyl, heteroatom- substituted alkyl, heteroatom-unsubstituted Cn-alkyl, and heteroatom-substituted Cn-alkyl. Alkyl groups may also optionally contain alkene or alkyne C-C bonds (but not such that aromatic compounds result). In certain embodiments, lower alkyls are contemplated. The term "lower alkyl" refers to alkyls of 1-6 carbon atoms (that is, 1, 2, 3, 4, 5 or 6 carbon atoms). The term "heteroatom-unsubstituted Cn-alkyl" refers to a radical, having a linear or branched, cyclic or acyclic structure, further having no carbon-carbon double or triple bonds, further having a total of n carbon atoms, all of which are nonaromatic, 3 or more hydrogen atoms, and no heteroatoms. For example, a heteroatom-unsubstituted Ci-Cio-alkyl has 1 to 10 carbon atoms. The groups -CH3 (Me), -CH2CH3 (Et), -CH2CH2CH3 (/j-Pr), -CH(CH3)2 (ώo-Pr), -CH2CH2CH2CH3 (IΪ-BU), -CH(CH3)CH2CH3 (sec-butyl), -CH2CH(CH3)2 (iso- butyl), -C(CH3)3 (tert-butyl), -CH2C(CH3)3 (neo-pentyl), -CH(CH3)CH2CH3, hexyl,
cyclohexenyl, cyclohexadienyl, and -CHCH2CH2 are aγγ non-limiting examples of heteroatom-unsubstituted alkyl groups. The term "heteroatom-substituted Cn-alkyl" refers to a radical, having a single saturated carbon atom as the point of attachment, no carbon-carbon double or triple bonds, further having a linear or branched, cyclic or acyclic structure, further having a total of n carbon atoms, all of which are nonaromatic, 0, 1, or more than one hydrogen atom, at least one heteroatom, wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S. For example, a heteroatom- substituted Ci-Cio-alkyl has 1 to 10 carbon atoms. The following groups are all non-limiting examples of heteroatom-substituted alkyl groups: trifluoromethyl, -CH2F, -CH2Cl, -CH2Br, -CH2OH, -CH2OCH3, -CH2OCH2CF3, -CH2OC(O)CH3, -CH2NH2, -CH2NHCH3, -CH2N(CH3)2, -CH2CH2Cl, -CH2CH2OH, CH2CH2OC(O)CH3, -CH2CH2NHCO2C(CH3)3, - C(=N-OH)CH(CH3)2 and -CH2Si(CH3)3. In certain embodiments, "lower alkyl" groups are
contemplated. As used herein, "lower alkyl" refers to an alkyl containing 1-6 carbon atoms. In certain embodiments, certain alkyl groups may be excluded.
The term "amino," alone or in combination, is used interchangeably with "amine" and may refer to any one or more of the following: a primary (e.g., -NH2), secondary (e.g., alkyl- NH-), tertiary (e.g., (alkyl)2-N-), or quarternary (e.g., (alkyl)3 — N(+)-) amine radical.
The term "aryl" includes heteroatom-unsubstituted aryl, heteroatom-substituted aryl, heteroatom-unsubstituted Cn-aryl, heteroatom-substituted Cn-aryl, heteroaryl, heterocyclic aryl groups, carbocyclic aryl groups, biaryl groups, and radicals derived from polycyclic fused hydrocarbons (PAHs). The term "heteroatom-unsubstituted Cn-aryl" refers to a radical, having a single carbon atom as a point of attachment, wherein the carbon atom is part of an aromatic ring structure containing only carbon atoms, further having a total of n carbon atoms, 5 or more hydrogen atoms, and no heteroatoms. For example, a heteroatom- unsubstituted Cβ-Cio-aryl has 6 to 10 carbon atoms. Non-limiting examples of heteroatom- unsubstituted aryl groups include methylphenyl, (dimethyl)phenyl, -C6H4CH2CH3, -C6H4CH2CH2CH3, -C6H4CH(CH3)2, -C6H4CH(CH2)2, -C6H3(CH3)CH2CH3, -C6H4CH=CH2, -C6H4CH=CHCH3, -C6H4C=CH, -C6H4C=CCH3, naphthyl, and the radical derived from biphenyl. The term "heteroatom-substituted Cn-aryl" refers to a radical, having either a single aromatic carbon atom or a single aromatic heteroatom as the point of attachment, further having a total of n carbon atoms, at least one hydrogen atom, and at least one heteroatom, further wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S. For example, a heteroatom-unsubstituted Ci- Cio-heteroaryl has 1 to 10 carbon atoms. Non-limiting examples of heteroatom-substituted aryl groups include the groups: -C6H4F, -C6H4Cl, -C6H4Br, -C6H4I, -C6H4OH, -C6H4OCH3, -C6H4OCH2CH3, -C6H4OC(O)CH3, -C6H4NH2, -C6H4NHCH3, -C6H4N(CH3)2, -C6H4CH2OH, -C6H4CH2OC(O)CH3, -C6H4CH2NH2, -C6H4CF3, -C6H4CN, -C6H4CHO, -C6H4CHO, -C6H4C(O)CH3, -C6H4C(O)C6H5, -C6H4CO2H, -C6H4CO2CH3, -C6H4CONH2, -C6H4CONHCH3, -C6H4CON(CH3)2, furanyl, thienyl, pyridyl, pyrrolyl, pyrimidyl, pyrazinyl, indolyl, quinolyl, and imidazoyl.
The term "aralkyl" includes heteroatom-unsubstituted aralkyl, heteroatom-substituted aralkyl, heteroatom-unsubstituted Cn-aralkyl, heteroatom-substituted Cn-aralkyl, heteroaralkyl, and heterocyclic aralkyl groups. In certain embodiments, lower aralkyls are contemplated. Aralkyls generally refer to radicals comprising the formula -alkyl-aryl. The term "lower aralkyl" refers to aralkyls of 7-12 carbon atoms (that is, 7, 8, 9, 10, 11 or 12
carbon atoms). The term "heteroatom-unsubstituted Cn-aralkyl" refers to a radical, having a single saturated carbon atom as the point of attachment, further having a total of n carbon atoms, wherein at least 6 of the carbon atoms form an aromatic ring structure containing only carbon atoms, 7 or more hydrogen atoms, and no heteroatoms. For example, a heteroatom- unsubstituted C7-Ci i-aralkyl has 7 to 11 carbon atoms. Non-limiting examples of heteroatom- unsubstituted aralkyls are: 2,3-dihydro-lH-indenyl, 1,2,3,4-tetrahydronaphthalenyl, phenylmethyl (benzyl, Bn) and phenylethyl. The term "heteroatom-substituted Cn-aralkyl" refers to a radical, having a single saturated carbon atom as the point of attachment, further having a total of n carbon atoms, 0, 1, or more than one hydrogen atom, and at least one heteroatom, wherein at least one of the carbon atoms is incorporated an aromatic ring structures, further wherein each heteroatom is independently selected from the group consisting of N, O, F, Cl, Br, I, Si, P, and S. For example, a heteroatom-substituted C2-CiO- heteroaralkyl has 2 to 10 carbon atoms. Examples of heteroatom-substituted Cn-aralkyls include indolinyl, benzofuranyl and benzothiophenyl. In certain embodiments, certain aryl groups may be specifically excluded.
As used herein, a "halide" (or "halo") refers to fluoro, chloro, bromo or iodo. In certain embodiments, certain halides may be specifically excluded.
Compounds as described herein may contain one or more asymmetric centers and thus can occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. All possible stereoisomers of the all the compounds described herein, unless otherwise noted, are contemplated as being within the scope of the present invention. In preferred embodiments, a single diastereomer is contemplated. The chiral centers of the compounds of the present invention can have the S- or the R-configuration, as defined by the IUPAC 1974 Recommendations. The present invention is meant to comprehend all such isomeric forms of the compounds of the invention.
The claimed invention is also intended to encompass salts of any of the synthesized compounds of the present invention. The term "salt(s)" as used herein, is understood as being acidic and/or basic salts formed with inorganic and/or organic acids and bases. Zwitterions (internal or inner salts) are understood as being included within the term "salt(s)" as used herein, as are quaternary ammonium salts such as alkylammonium salts. Nontoxic, pharmaceutically acceptable salts are preferred as described below, although other salts may be useful, as for example in isolation or purification steps.
Non-limiting examples of acid addition salts include but are not limited to acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, oxalate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, tosylate and undecanoate.
Non-limiting examples of basic salts include but are not limited to ammonium salts; alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; salts comprising organic bases such as amines (e.g., dicyclohexylamine, alkylamines such as z-butylamine and f-amylamine, substituted alkylamines, aryl-alkylamines such as benzylamine, dialkylamines, substituted dialkylamines such as N-methyl glucamine (especially N-methyl D-glucamine), trialkylamines, and substituted trialkylamines); and salts comprising amino acids such as arginine, lysine and so forth. The basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myrtistyl and stearyl chlorides, bromides and iodides), arylalkyl halides (e.g., benzyl and phenethyl bromides) and others known in the art.
When a chemical reaction is to be carried out selectively at one reactive site in a multifunctional compound, other reactive sites must be temporarily blocked. A "protecting group," as used herein, is defined as a group used for the purpose of this temporary blockage. During the synthesis of the compounds of the present invention, various functional groups must be protected using protecting groups (or protecting agents) at various stages of the synthesis. However, use of the phrases "protected carboxylic acid," "protected amine" or "protected hydroxy," for example, does not mean that every functional group available to be protected is protected. Functional groups necessary for the desired transformation, for example, should be unprotected. Thus, the function of a protecting group is to protect one or more functionalities (e.g.,
-NH2, -SH, -COOH) during subsequent reactions which would not proceed well, either because the free (in other words, unprotected) functional group would react and be functionalized in a way that is inconsistent with its need to be free for subsequent reactions,
or the free functional group would interfere in the reaction. The same protecting group may be used to protect one or more of the same or different functional group(s). Also, different protecting groups can be used to protect the same type of functional group within a compound of the present invention in multiple steps. There are a number of methods well known to those skilled in the art for accomplishing such a step. For protecting agents, their reactivity, installation and use, see, e.g., Greene & Wuts, 1999.
When a protecting group is no longer needed, it is removed by methods well known to those skilled in the art. For deprotecting agents and their use, see, e.g., Greene & Wuts, 1999. Agents used to remove the protecting group are sometimes called deprotecting agents. Protecting groups must be readily removable (as is known to those skilled in the art) by methods employing deprotecting agents that are well known to those skilled in the art. It is well known that certain deprotecting agents remove some protective groups and not others, while other deprotecting agents remove several types of protecting groups from several types of functional groups. Thus, a first deprotecting agent may be used to remove one type of protecting group, followed by the use of a second deprotecting agent to remove a second type of protecting group, and so on.
In one embodiment of the present invention, the deprotecting agent is the use of gaseous H2 in the presence of a palladium catalyst dispersed on carbon in order to remove a benzyl (Bn) protecting group and reveal a free carboxylic acid. Persons of ordinary skill in the art will be familiar with the proper ordering of protective group removal using deprotecting agents. See e.g., Greene & Wuts, 1999. Particular non-limiting examples of protecting groups are discussed below.
Amino protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 7. The amino protecting group may be a carbamate. A suitable amino protecting group may be selected from the group consisting of t- butoxycarbonyl, benzyloxycarbonyl, formyl, trityl, acetyl, trichloroacetyl, dichloroacetyl, chloroacetyl, trifluoroacetyl, difluoroacetyl, fluoroacetyl, benzyl chloroformate, 4- phenylbenzyloxycarbonyl, 2-methylbenzyloxycarbonyl, 4-ethoxybenzyloxycarbonyl, 4- fluorobenzyloxycarbonyl, 4-chlorobenzyloxycarbonyl, 3-chlorobenzyloxycarbonyl, 2- chlorobenzyloxycarbonyl, 2,4-dichlorobenzyloxycarbonyl, 4-bromobenzyloxycarbonyl, 3- bromobenzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, 4-cyanobenzyloxycarbonyl, 2-(4- xenyl)isopropoxycarbonyl, 1 , 1 -diphenyleth- 1 -yloxycarbonyl, 1 , 1 -diphenylprop- 1 - yloxycarbonyl, 2-phenylprop-2-yloxycarbonyl, 2-(p-toluyl)prop-2 -yloxycarbonyl,
cyclopentanyloxycarbonyl, l-methylcyclopentanyloxycarbonyl, cyclohexanyloxycarbonyl, 1- methylcyclohexanyloxycabonyl, 2-methylcyclohexanyloxycarbonyl, 2-(4- toluylsulfonyl)ethoxycarbonyl, 2-(methylsulfonyl)ethoxycarbonyl, 2-
(triphenylphosphino)ethoxycarbonyl, fluorenylmethoxycarbonyl, 2- (trimethylsilyl)ethoxycarbonyl, allyloxycarbonyl, l-(trimethylsilylmethyl)prop-l- enyloxycarbonyl, 5-benzisoxalylmethoxycarbonyl, 4-acetoxybenzyloxycarbonyl, 2,2,2- trichloroethoxycarbonyl, 2-ethynyl-2-propoxycarbonyl, cyclopropylmethoxycarbonyl, 4- (decyloxyl)benzyloxycarbonyl, isobomyloxycarbonyl, 1 -piperidyloxycarbonyl and 9- fluorenylmethylcarbonyl, for example. In certain embodiments of the present invention, the amine protecting group is Z-butoxycarbonyl (Boc), benzyloxycarbonyl (CBz), paramethoxybenzyl (PMB), or 9-fluorenylmethylcarbonyl (Fmoc).
Thiol protecting groups are well known to those skilled in the art. See, for example,
Greene & Wuts, 1999, Chapter 6. A suitable thiol protecting group may be selected from the group consisting of acetamidomethyl, benzamidomethyl, 1 -ethoxyethyl, benzoyl, triphenylmethyl, z-butyl, benzyl, adamantyl, cyanoethyl, acetyl, and trifluoroacetyl, for example.
Alcohol protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 2. A suitable alcohol protecting group may be selected from the group consisting of methoxymethyl, paramethoxybenzyl, (phenyldimethylsilyl)methoxymethyl, triethylsilyl, benzyloxymethyl, t-butoxymethyl, and tetrahydropyranyl, for example. In certain embodiments of the present invention, the an alcohol protecting group is benzyl (Bn), paramethoxybenzyl (PMB), or triethylsilyl (TES).
Carboxylic acid protecting groups are well known to those skilled in the art. See, for example, Greene & Wuts, 1999, Chapter 5. A suitable carboxylic acid protecting group may be selected from the group consisting of dimethylacetal, methoxymethylester, phenylacetoxymethyl ester and tetrahydropyranyl ester, for example. In certain embodiments of the present invention, the carboxylic acid protecting group is benzyl (Bn). As used herein, a "protected carboxylic acid" refers to a carboxylic acid group protected by a carboxylic acid protecting group. The compounds, agents, and active ingredients (e.g., solvents, catalysts, bases used in reactions, and other compounds, agents, and active ingredients described herein) that are described in the claims and specification can be obtained by any means known to a person of
ordinary skill in the art. In a non-limiting embodiment, for example, the compounds, agents, and active ingredients can be isolated by obtaining the source of such compounds, agents, and active ingredients. In many instances, the compounds, agents, and active ingredients are commercially available (e.g., Sigma- Aldrich, Milwaukee, WI). Modifications or derivatives of the compounds, agents, and active ingredients disclosed throughout this specification are contemplated as being useful with the methods and compositions of the present invention. Derivatives may be prepared and the properties of such derivatives may be assayed for their desired properties by any method known to those of skill in the art. In certain aspects, "derivative" refers to a chemically modified compound that still retains the desired effects of the compound prior to the chemical modification. Such derivatives may have the addition, removal, or substitution of one or more chemical moieties on the parent molecule. Non-limiting examples of the types modifications that can be made to the compounds and structures disclosed herein include the addition or removal of lower alkanes such as methyl, ethyl, propyl, or substituted lower alkanes such as hydroxymethyl or aminomethyl groups; carboxyl groups and carbonyl groups; hydroxyls; nitro, amino, amide, and azo groups; sulfate, sulfonate, sulfono, sulfhydryl, sulfonyl, sulfoxido, phosphate, phosphono, phosphoryl groups, and halide substituents. Additional modifications can include an addition or a deletion of one or more atoms of the atomic framework, for example, substitution of an ethyl by a propyl; substitution of a phenyl by a larger or smaller aromatic group. Alternatively, in a cyclic or bicyclic structure, hetero atoms such as N, S, or O can be substituted into the structure instead of a carbon atom.
2. Pharmaceutical Formulations and Administration Thereof
A. Pharmaceutical Formulations and Routes for Administration to Subjects Pharmaceutical compositions of the present invention comprise an effective amount of one or more candidate substance or additional agent dissolved or dispersed in a pharmaceutically acceptable carrier. The phrases "pharmaceutical or pharmacologically acceptable" refers to molecular entities and compositions that do not produce an adverse, allergic or other untoward reaction when administered to an animal, such as, for example, a human, as appropriate. The preparation of a pharmaceutical composition that contains at least one candidate substance or additional active ingredient will be known to those of skill in the art in light of the present disclosure, as exemplified by Remington's Pharmaceutical
Sciences, 18th Ed. Mack Printing Company, 1990, incorporated herein by reference. Moreover, for animal (e.g., human) administration, it will be understood that preparations should meet sterility, pyrogenicity, general safety and purity standards as required by FDA Office of Biological Standards. As used herein, "pharmaceutically acceptable carrier" includes any and all solvents, dispersion media, coatings, surfactants, antioxidants, preservatives (e.g., antibacterial agents, antifungal agents), isotonic agents, absorption delaying agents, salts, preservatives, drugs, drug stabilizers, gels, binders, excipients, disintegration agents, lubricants, sweetening agents, flavoring agents, dyes, such like materials and combinations thereof, as would be known to one of ordinary skill in the art (see, for example, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). Except insofar as any conventional carrier is incompatible with the active ingredient, its use in the therapeutic or pharmaceutical compositions is contemplated.
The candidate substance may comprise different types of carriers depending on whether it is to be administered in solid, liquid or aerosol form, and whether it need to be sterile for such routes of administration as injection. The present invention can be administered intravenously, intradermally, intraarterially, intraperitoneally, intralesionally, intracranially, intraarticularly, intraprostaticaly, intrapleurally, intratracheally, intranasally, intravitreally, intravaginally, intrarectally, topically, intratumorally, intramuscularly, subcutaneously, subconjunctival, intravesicularlly, mucosally, intrapericardially, intraumbilically, intraocularally, orally, locally, via inhalation (e.g., aerosol inhalation), via injection, via infusion, via continuous infusion, via localized perfusion bathing target cells directly, via a catheter, via a lavage, in cremes, in lipid compositions (e.g., liposomes), or by other method or any combination of the foregoing as would be known to one of ordinary skill in the art (see, for example, Remington's Pharmaceutical Sciences, 1990).
The actual dosage amount of a composition of the present invention administered to an animal patient can be determined by physical and physiological factors such as body weight, severity of condition, the type of disease being treated, previous or concurrent therapeutic interventions, idiopathy of the patient and on the route of administration. The practitioner responsible for administration will, in any event, determine the concentration of active ingredient(s) in a composition and appropriate dose(s) for the individual subject.
In certain embodiments, pharmaceutical compositions may comprise, for example, at least about 0.1% of a compound of the present invention. In other embodiments, the compound may comprise between about 2% to about 75% of the weight of the unit, or between about 25% to about 60%, for example, and any range derivable therein. In other non-limiting examples, a dose may also comprise from about 1 microgram/kg/body weight, about 5 microgram/kg/body weight, about 10 microgram/kg/body weight, about 50 microgram/kg/body weight, about 100 microgram/kg/body weight, about 200 microgram/kg/body weight, about 350 microgram/kg/body weight, about 500 microgram/kg/body weight, about 1 milligram/kg/body weight, about 5 milligram/kg/body weight, about 10 milligram/kg/body weight, about 50 milligram/kg/body weight, about 100 milligram/kg/body weight, about 200 milligram/kg/body weight, about 350 milligram/kg/body weight, about 500 milligram/kg/body weight, to about 1000 mg/kg/body weight or more per administration, and any range derivable therein. In non-limiting examples of a derivable range from the numbers listed herein, a range of about 5 mg/kg/body weight to about 100 mg/kg/body weight, about 5 microgram/kg/body weight to about 500 milligram/kg/body weight, etc., can be administered, based on the numbers described above.
In any case, the composition may comprise various antioxidants to retard oxidation of one or more component. Additionally, the prevention of the action of microorganisms can be brought about by preservatives such as various antibacterial and antifungal agents, including but not limited to parabens (e.g., methylparabens, propylparabens), chlorobutanol, phenol, sorbic acid, thimerosal, or combinations thereof.
The candidate substance may be formulated into a composition in a free base, neutral or salt form. Pharmaceutically acceptable salts, include the acid addition salts, e.g., those formed with the free amino groups of a proteinaceous composition, or which are formed with inorganic acids such as for example, hydrochloric or phosphoric acids, or such organic acids as acetic, oxalic, tartaric or mandelic acid. Salts formed with the free carboxyl groups can also be derived from inorganic bases such as for example, sodium, potassium, ammonium, calcium or ferric hydroxides; or such organic bases as isopropylamine, trimethylamine, histidine, or procaine. In embodiments where the composition is in a liquid form, a carrier can be a solvent or dispersion medium comprising but not limited to, water, ethanol, polyol (e.g., glycerol, propylene glycol, liquid polyethylene glycol, etc.), lipids (e.g., triglycerides, vegetable oils, liposomes) and combinations thereof. The proper fluidity can be maintained, for example, by
the use of a coating, such as lecithin; by the maintenance of the required particle size by dispersion in carriers such as, for example liquid polyol or lipids; by the use of surfactants such as, for example hydroxypropylcellulose; or combinations thereof such methods. It may be preferable to include isotonic agents, such as, for example, sugars, sodium chloride or combinations thereof.
In other embodiments, one may use eye drops, nasal solutions or sprays, aerosols or inhalants in the present invention. Such compositions are generally designed to be compatible with the target tissue type. In a non-limiting example, nasal solutions are usually aqueous solutions designed to be administered to the nasal passages in drops or sprays. Nasal solutions are prepared so that they are similar in many respects to nasal secretions, so that normal ciliary action is maintained. Thus, in certain embodiments the aqueous nasal solutions usually are isotonic or slightly buffered to maintain a pH of about 5.5 to about 6.5. In addition, antimicrobial preservatives, similar to those used in ophthalmic preparations, drugs, or appropriate drug stabilizers, if required, may be included in the formulation. For example, various commercial nasal preparations are known and include drugs such as antibiotics or antihistamines.
In certain embodiments the candidate substance is prepared for administration by such routes as oral ingestion. In these embodiments, the solid composition may comprise, for example, solutions, suspensions, emulsions, tablets, pills, capsules (e.g., hard or soft shelled gelatin capsules), sustained release formulations, buccal compositions, troches, elixirs, suspensions, syrups, wafers, or combinations thereof. Oral compositions may be incorporated directly with the food of the diet. In certain embodiments, carriers for oral administration comprise inert diluents, assimilable edible carriers or combinations thereof. In other aspects of the invention, the oral composition may be prepared as a syrup or elixir. A syrup or elixir, and may comprise, for example, at least one active agent, a sweetening agent, a preservative, a flavoring agent, a dye, a preservative, or combinations thereof.
In certain embodiments an oral composition may comprise one or more binders, excipients, disintegration agents, lubricants, flavoring agents, and combinations thereof. In certain embodiments, a composition may comprise one or more of the following: a binder, such as, for example, gum tragacanth, acacia, cornstarch, gelatin or combinations thereof; an excipient, such as, for example, dicalcium phosphate, mannitol, lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium carbonate or combinations thereof; a disintegrating agent, such as, for example, corn starch, potato starch, alginic acid or
combinations thereof; a lubricant, such as, for example, magnesium stearate; a sweetening agent, such as, for example, sucrose, lactose, saccharin or combinations thereof; a flavoring agent, such as, for example peppermint, oil of wintergreen, cherry flavoring, orange flavoring, etc.; or combinations thereof the foregoing. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, carriers such as a liquid carrier. Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar, or both.
Additional formulations which are suitable for other modes of administration include suppositories. Suppositories are solid dosage forms of various weights and shapes, usually medicated, for insertion into the rectum, vagina, or urethra. After insertion, suppositories soften, melt or dissolve in the cavity fluids. In general, for suppositories, traditional carriers may include, for example, polyalkylene glycols, triglycerides, or combinations thereof. In certain embodiments, suppositories may be formed from mixtures containing, for example, the active ingredient in the range of about 0.5% to about 10%, and preferably about 1% to about 2%.
Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and/or the other ingredients. In the case of sterile powders for the preparation of sterile injectable solutions, suspensions or emulsion, certain methods of preparation may include vacuum-drying or freeze-drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered liquid medium thereof. The liquid medium should be suitably buffered if necessary and the liquid diluent first rendered isotonic prior to injection with sufficient saline or glucose. The preparation of highly concentrated compositions for direct injection is also contemplated, where the use of DMSO as solvent is envisioned to result in extremely rapid penetration, delivering high concentrations of the active agents to a small area. The composition must be stable under the conditions of manufacture and storage, and preserved against the contaminating action of microorganisms, such as bacteria and fungi. It will be appreciated that endotoxin contamination should be kept minimally at a safe level, for example, less that 0.5 ng/mg protein.
In particular embodiments, prolonged absorption of an injectable composition can be brought about by the use in the compositions of agents delaying absorption, such as, for example, aluminum monostearate, gelatin, or combinations thereof.
B. Combination Therapy In order to increase the effectiveness of a compound of the present invention, the compound may be combined with traditional drugs. It is contemplated that this type of combination therapy may be used in vitro or in vivo. In a non-limiting example, an anticancer agent may be used in combination with a compound. An anti-viral or antibiotic agent may be used in combination with a compound, for example. More generally, agents of the present invention may be provided in a combined amount with an effective amount of an anti-cancer agent. This process may involve contacting the cell(s) with the agents at the same time or within a period of time wherein separate administration of the substances produces a desired therapeutic benefit. This may be achieved by contacting the cell, tissue or organism with a single composition or pharmacological formulation that includes two or more agents, or by contacting the cell with two or more distinct compositions or formulations, wherein one composition includes one agent and the other includes another.
The compounds of the present invention may precede, be co-current with and/or follow the other agents by intervals ranging from minutes to weeks. In embodiments where the agents are applied separately to a cell, tissue or organism, one would generally ensure that a significant period of time did not expire between the time of each delivery, such that the agents would still be able to exert an advantageously combined effect on the cell, tissue or organism. For example, in such instances, it is contemplated that one may contact the cell, tissue or organism with two, three, four or more modalities substantially simultaneously (i.e., within less than about a minute) as the candidate substance. In other aspects, one or more agents may be administered within of from substantially simultaneously, about 1 minute, about 5 minutes, about 10 minutes, about 20 minutes about 30 minutes, about 45 minutes, about 60 minutes, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours about 8 hours, about 9 hours, about 10 hours, about 11 hours, about 12 hours, about 13 hours, about 14 hours, about 15 hours, about 16 hours, about 17 hours, about 18 hours, about 19 hours, about 20 hours, about 21 hours, about 22 hours, about 22 hours, about 23 hours, about 24 hours, about 25 hours, about 26 hours, about 27 hours, about 28 hours,
about 29 hours, about 30 hours, about 31 hours, about 32 hours, about 33 hours, about 34 hours, about 35 hours, about 36 hours, about 37 hours, about 38 hours, about 39 hours, about 40 hours, about 41 hours, about 42 hours, about 43 hours, about 44 hours, about 45 hours, about 46 hours, about 47 hours, about 48 hours, about 1 day, about 2 days, about 3 days, about 4 days, about 5 days, about 6 days, about 7 days, about 8 days, about 9 days, about 10 days, about 11 days, about 12 days, about 13 days, about 14 days, about 15 days, about 16 days, about 17 days, about 18 days, about 19 days, about 20 days, about 21 days, about 1, about 2, about 3, about 4, about 5, about 6, about 7 or about 8 weeks or more, and any range derivable therein, prior to and/or after administering the candidate substance. Various combination regimens of the agents may be employed. Non-limiting examples of such combinations are shown below, wherein a compound is "A" and a second agent, such as an anti-cancer agent, is "B":
A/B/A B/A/B B/B/A A/A/B A/B/B B/A/A A/B/B/B B/A/B/B
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A
An "anti-cancer" agent is capable of negatively affecting cancer in a subject, for example, by killing one or more cancer cells, inducing apoptosis in one or more cancer cells, reducing the growth rate of one or more cancer cells, reducing the incidence or number of metastases, reducing tumor size, inhibiting tumor growth, reducing the blood supply to a tumor or one or more cancer cells, promoting an immune response against one or more cancer cells or a tumor, preventing or inhibiting the progression of a cancer, or increasing the lifespan of a subject with a cancer. Anti-cancer agents are well-known in the art and include, for example, chemotherapy agents (chemotherapy), radiotherapy agents (radiotherapy), a surgical procedure, immune therapy agents (immunotherapy), genetic therapy agents (gene therapy), reoviral therapy, hormonal therapy, other biological agents (biotherapy), and/or alternative therapies.
2. Examples
The following examples are included to demonstrate certain non-limiting aspects of the invention. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques discovered by the inventor to function well in the practice of the invention. However, those of skill in the art should, in
light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the invention.
General FIGs. 1 and 2 represent, in certain embodiments, general synthetic methods for accessing compounds of the present invention.
Protected ornithine and alanine were purchased from Acros and used as received. Other reagents were purchased from Aldrich (Milwaukee, WI) and used as received.
Example 1 Synthesis of a Vinyl Amide

To a stirred solution of the dipeptide of Example 2 (28.00 g, 53.07 mmol) and Et3SiH (25.71 g, 159.21 mmol) in Ethyl acetate (300 mL) was added HCl (10.47 mL, 106.14 mmol). The reaction mixture was stirred for 16 h at room temperature, and then the solvent was evaporated. To the resulting residue was poured hexane (500 mL), and stirred to give a white solid. The resulting free amine solid was filtered and can be used for next step without further purification. To a stirred solution of free amine (25.00 g, 53.89 mmol) and TMP (22.74 g, 134.7 mmol) in CH2Cl2 (300 mL) was added acryloyl chloride (5.25 g, 64.7 mmol) at 0 0C. The reaction mixture was stirred for 4 h at 0 0C and then quenched by slow additional IN HCl (100 mL) and neutralized by washing with saturated ΝaHCC>3 solution (100 mL x 2). The organic layer was separated and evaporated to give a crude solid. The crude compound was triturated by pentane to give the acrylamide peptide (24.0 g, 92.5 %). IR (thin film) 2937, 1796, 1728, 1665, 1536, 1403 cm 4; 1H NMR (500 MHz, CDCl3) δ 7.32-7.43 (m, 10H), 6.99 (s, IH), 6.26 (d, J= 4.2 Hz, IH), 6.04 (dd, J= 9.9, 16.6 Hz, 2H), 5.12 (m, 2H), 4.60 (m, IH), 4.28 (m, IH), 3.27 (m, 2H), 1.82-1.96 (m, IH), 1.61-1.73 (m, IH), 1.42-1.58 (m, 2H), 1.41
(d, J= 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.6, 171.7, 165.8, 136.1, 135.1, 130.8, 128.7, 128.6, 128.5, 128.4, 128.2, 128.0, 126.5, 67.4, 67.1, 52.1, 50.6, 38.7, 29.4, 25.4, 18.5; LRMS (ESI) Calcd. for C26H32N3O6 [M+H]+ : 482. Found : 482.
Example 2 Synthesis of an Oxoaldehyde

To a stirred solution of acrylamide (5.00 g, 10.4 mmol) in CH2Cl2 (200 niL) and MeOH (1 mL) was bubbled ozone at 78 °C until the blue color persisted, and the remaining ozone was removed bubbling nitrogen through the solution. Dimethyl sulfide (3.05 mL, 41.6 mmol) was added and the mixture was left to attain 25 0C for 12 h. The solvent was evaporated under reduced vacuum and the resulting oil was purified by flash chromatography on SiO2 (CH2Cl2/acetone= 1/1) to afford a mixture of hydrate and glyoxylate (3.02 g, 60 %) as a white solid. The resulting mixture of hydrate and glyoxylate was dissolved into CH2Cl2 with 4 A Molecular sieve and stirred for 24 h at room temperature to give glyoxylate. The resulting glyoxylate was used directly in the next step without further purification.
Example 3 Synthesis of a Ketene Acetal

To a stirred solution of acid (1.24 g, 10.7 mmol) in CH2Cl2 (30 mL) was added oxalyl chloride (1.38 mL, 16.0 mmol) at 25 0C. The reaction mixture was stirred for 2 h at 25 0C. The solvent was evaporated under reduced pressure. To a stirred solution of the oil residue was added 2-thiopyridine (1.42 g, 12.8 mmol) in CH2Cl2 (30 mL) followed by Et3N (2.98 mL, 21.3 mmol) at 0 0C. The reaction mixture was stirred for 2 h at 25 0C and then quenched with IN HCl (30 mL) and neutralized by washing with saturated NaHCO3 solution (30 mL x 2). The organic layers were combined and evaporated, then resulting yellow oil was purified
by flash chromatography on SiO2 (20% EtOAc/Hexane) to afford acid ester (2.19 g, 98 %) as a pure liquid : [α]D 20 +8.2 (c= 0.027, CHCl3). IR (thin film) 2953, 1702, 1440, 1418cm"1; 1H NMR (300 MHz, CDCl3) δ 8.64 (m, IH), 7.75 (m , IH), 7.64 (m , IH), 7.31 (m , IH), 2.73 (dd, J= 6.3, 14.7 Hz, IH), 2.54 (dd, J= 7.8, 14.7 Hz, IH), 1.96-2.10 (m, IH), 1.42-1.49 (m, IH), 1.29-1.35(m, IH), 0.97 (d, J= 6.3 Hz, 3H), 0.91 (t, J= 7.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 196.4, 151.9, 150.5, 137.4, 130.4, 123.7, 51.3, 32.8, 29.5, 19.4, 11.5; LRMS (ESI) Calcd. for CHHI6NOS [M+H]+ : 210. Found : 210.
Example 4 (5VE)-2-(3-methyl-l-(triethylsiryloxy)pent-l-enylthio)pyridine
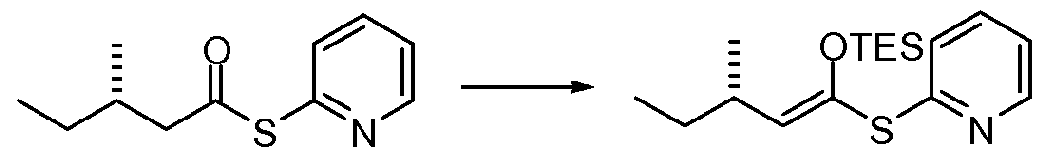
To a stirred solution of thioester (8.00 g, 35.8 mmol), DMF (3.14 g, 43.0 mmol) and
Et3N (6.04g, 43.0 mmol) in CH2Cl2 (240 mL) was added LiHMDS (1.0M, 82.6 mL, 82.6 mmol) at 78 0C. The reaction mixture was stirred for 30 min at 78 °C and TESCl (12.02 mL, 71.6 mmol) was added dropwise to the reaction mixture at 78 0C. The reaction mixture was stirred for 2 h at this temperature and then quenched with pH 7 buffer solution (40 mL x 2). The organic layer was separated, dried over Na2SO4 and evaporated. The resulting yellowish oil was purified by flash chromatography on SiO2 (10% EtOAc/Hexane) to afford ketene acetal (11.1 g, 96.2 %) as a yellow oil: [α]D 20 +1.8 (c= 0.034, CHCl3). IR (thin film) cm"1; 2950, 2873, 1624, 1568 cm"1 ; 1H NMR (300 MHz, CDCl3) δ 8.40 (m, IH), 7.53 (m, IH), 7.32 (m, IH), 6.97 (m, IH), 5.18 (d, J= 9.6, IH), 2.58 (m, IH), 1.33-1.40(m, 2H), 1.00 (d, J= 6.6 Hz, 3H), 0.93 (t, J= 7.5 Hz, 3H), 0.84-0.89 (m, 9H), 0.60-0.68(m, 6H); 13C NMR (125 MHz, CDCl3) δ 160.9, 149.6, 138.7, 136.8, 130.5, 122.3, 121.5, 119.9, 119.8, 33.5, 30.1, 20.4, 12.3, 6.8; LRMS (ESI) Calcd. for CnHi6NOS[M+H]+ : 324. Found : 324.
General Procedure for the Tandem Mukaiyama Aldol Lactonization (TMAL) Reaction
Under Normal Conditions (GPl)
Anhydrous ZnCl2 (1.2-2.0 equiv) was freshly fused at ~0.5 mm Hg and after cooling to ambient temperature, CH2Cl2 (appropriate volume to make final concentration of aldehyde in CH2Cl2 ~0.2 M) was added. The aldehyde (1.0 equiv) was then added neat or as a CH2Cl2 solution at room temperature followed by thiopyridyllketene acetal (1.1-1.2 equiv) neat. The
suspension was stirred for the appropriate time, and then quenched by pH 7 buffer, filtered. The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford the crude product which was purified by flash chromatography
Example 5 Synthesis of a beta-lactone

To a stirred solution of ketene acetal (96.74 mg, 0.30 mmol) and fused ZnCl2 (81.57 mg, 0.60 mmol) in CH2CI2 (5 mL), was added via canula activated Molecular sieve 4A (100 mg) and oxoaldehyde (100 mg, 0.20 mmol) in CH2CI2 (5 mL). The solution was stirred for
36 h at room temperature, and then quenched by pH 7 buffer (5 mL), filtered. The organic layer was separated and evaporated under reduced pressure and the residue, an oily solid, was purified by flash chromatography on SiC>2 (100% ethyl acetate) to afford beta-lactone (30 mg, 15%) as a colorless oil.
Example 6
Diastereoselective TMAL via a Chiral Auxiliary Approach: Synthesis of Belactosin C and All Diastereomers of the beta-Lactone Moiety

Example 7 Mono alcohol

To a stirred solution of diol (4.0 g, 12.7 mmol) and K2CO3 (5.3 g, 38.2 mmol) in acetone (100 mL) was added MeI (7.9 g, 127.2 mmol) at 25 0C. The reaction was refluxed for 24 h and then cooled to 25 0C. The reaction mixture was filtered and evaporated to give a crude oily solid (3.8 g, 90 %). The crude compound was purified by flash chromatography: [α]D 20 11.9 (c=0.16, CHCl3); IR (thin film) 3446, 2984, 2937, 1456 cm"1; 1H NMR (500 MHz, CDCl3) δ 7.11-7.40 (m, 10H), 4.26 (dd, J= 6.5, 7.5 Hz, IH), 4.04 (dd, J= 4.0, 7.5 Hz, IH), 3.94 (d, J= 6.5 Hz, IH), 3.87 (dd, J= 3.5, 7.5 Hz, IH), 3.24 (s, 3H), 3.01 (d, J= 7.5 Hz, IH), 1.51 (s, 3H), 1.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 141.3, 137.6, 128.9, 128.9, 128.6, 128.1, 128.0, 126.7, 110.4, 85.0, 81.4, 80.7, 72.9, 57.1, 27.7, 27.6; LRMS (ESI) Calcd. for CnHi5NOSLi [M+Li]+ : 335. Found : 335.
Example 8 Vinyl ester

To a stirred solution of alcohol (16.00 g, 28.7 mmol) and Hunig's base (6.02 g, 34.5 mmol) in CH2Cl2 (300 mL) was added acryloyl chloride (2.79 g, 34.5 mmol) at 0 0C. The reaction mixture was stirred for 12 h at 25 0C and then quenched with IN HCl (100 mL), neutralized by washing with saturated NaHCO3 solution (100 mL x 2). The organic layer was separated and evaporated to give an oily solid, which was purified by flash chromatography give a white solid (10. Ig, 83.3 %) : [α]D 2σ 15.4 (c= 0.25, CHCl3); IR (thin film) 2978 , 2925,
1727 cm"1; 1H NMR (300 MHz, CDCl3) δ 7.11-7.41 (m, 10H), 6.46 (dd, J= 1.5, 17.4 Hz, IH), 6.17 (dd, J= 10.5, 20.1 Hz, IH), 5.87 (dd, J= 1.5, 10.5 Hz, IH), 5.03 (d, J= 3.9 Hz, IH), 4.16 (m, 2H), 3.86 (d, J= 6.0 Hz, IH), 3.22 (s, 3H), 1.52 (s, 3H), 1.49 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 165.2, 137.6, 137..4, 131.5, 129.0, 128.9, 128.6, 128.5, 128.0, 127.4, 110.9, 84.9, 80.7, 80.0, 75.1, 57.0, 27.9, 27.5; LRMS (ESI) Calcd. for CnHi5NOSLi [M+Lif : 389. Found : 389.
Example 9 Glyoxylate

To a stirred solution of acrylate (2.70 g, 7.06 mmol) in CH2Cl2 (60 mL)/H2O(40 mL) was added OsO4 (0.03g, 0.35 mmol) and NaIO4 (8.39 g, 28.2 mmol) at 0 0C. The reaction mixture was stirred for 2 h at 0 0C, then water was added and the mixture was extracted with Et2O. The organic layer was separated and evaporated to give an oily solid, which was purified by flash chromatography to afford a mixture of hydrate and desired glyoxaldehyde (2.67 g, 98 %). The resulting mixture of hydrate and glyoxaldehyde was dissolved into CH2Cl2 with 4 A Molecular sieve and stirred for 24 h at room temperature to give glyoxylate. The resulting glyoxylate was used directly in the next step without further purification.
Example 10 beta-Lactone
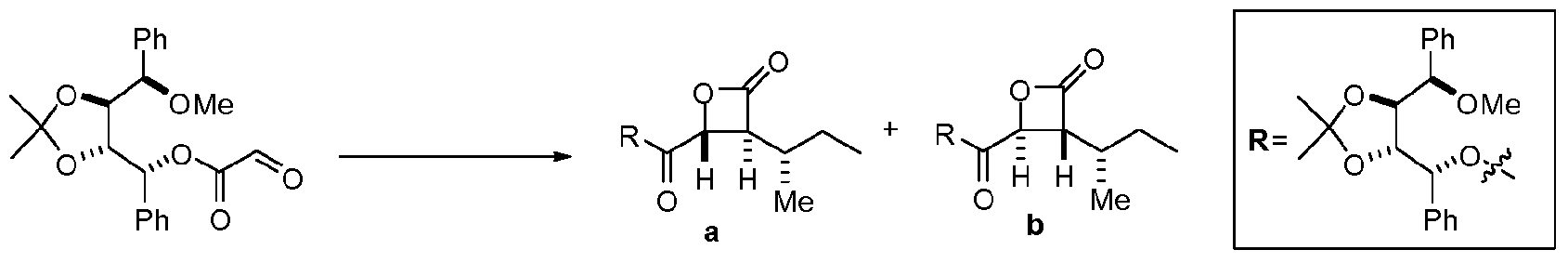
The beta-lactone a (5.0 mg, 5 %) was prepared from glyoxylate (79.7 mg, 0.20 mmol) according to GP 1 as a white solid.: [α]D 2σ 71.2 (c= 0.38, EtOAc); IR (thin film) 2917, 1843, 1733 cm 1J 1H NMR (500 MHz, CDCl3) δ 7.14-7.37 (m, 10H), 5.20 (d, J= 5.5 Hz, IH), 4.67 (d, J= 4.5 Hz, IH), 4.19 (dd, J= 6.0, 7.5 Hz, IH), 4.03 (dd, J= 6.0, 6.0 Hz, IH), 3.69 (d, 5.5 Hz, IH), 3.65 (dd, J= 4.5, 7.5 Hz, IH), 3.16 (s, 3H), 2.00 (m, IH), 1.62 (m, IH), 1.48 (s, 3H), 1.45 (s, 3H), 1.33 (m, IH), 1.07 (d, 7.0 Hz, 3H), 0.92 (t, 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 169.0, 167.6, 137.5, 136.1, 129.2, 128.9, 128.5, 127.8, 127.7, 111.0, 84.2, 80.8, 79.4, 69.3, 62.9, 57.1, 33.8, 27.8, 27.5, 27.0, 16.6, 11.2; LRMS (ESI) Calcd. for CnHi5NOSNa [M+Na]+ : 505. Found : 505.
The beta-lactone b (30.0 mg, 30 %) was prepared from glyoxylate 14 (79.7 mg, 0.20 mmol) according to GP 1 as a white solid.: [α]D 2σ51.6 (c= 0.56, EtOAc). IR (thin film) 2917 , 1843, 1733 cm"1; 1H NMR (500 MHz, CDCl3) δ 7.16-7.39 (m, 10H), 5.17 (d, J= 5.5 Hz, IH), 4.68 (d, J= 4.5 Hz, IH), 4.19 (dd, J= 5.0, 6.5 Hz, IH), 4.06 (dd, J= 6.0, 6.0 Hz, IH), 3.75 (d, 6.0 Hz, IH), 3.49 (dd, J= 4.5, 9.0 Hz, IH), 3.16 (s, 3H), 1.98 (m, IH), 1.59 (m, IH), 1.48 (s, 3H), 1.47 (s, 3H), 1.22 (m, IH), 1.10 (d, 7.0 Hz, 3H), 0.94 (t, 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.7, 167.5, 137.4, 136.1, 129.2, 129.0, 128.9, 128.9, 127.9,127.6, 111.1, 84.5, 80.9, 79.5, 77.1, 70.3, 63.4, 57.1, 34.4, 27.9, 27.6, 27.5, 16.4, 11.1; LRMS (ESI) Calcd. for CnH15NOSNa [M+Na]+ : 505. Found : 505.
Example 11 beta-Lactone acid

To a solution of beta- lactone a (80.3 mg, 0.16 mmol) in THF (3.0 mL) was added Pd/C and HCO2H (2.0 mL). The mixture was stirred under at atmosphere of H2 (rubber balloon) for 17 h. The catalyst was filtered off through a pad of cotton wool and the solvent was removed under reduced pressure to give an oily liquid, which was purified by flash chromatography give beta-lactone acid (21.2 mg, 77.0 % yield) was obtained as an oily liquid: [α]o20 3.1 (c= 0.59, CHCI3). All other data matched that previously reported (Kumaraswamy, 2006).
Example 12 N-CBZ-O-Bn protected belactosin C

Coupling of dipeptide (34.8 mg, 0.075 mmol) and beta-lactone acid 3 (15.2 mg, 0.088 mmol) was accomplished according to the published procedure (Kumaraswamy, 2006) employing DCC (24.8 mg, 0.12 mmol) and HOBT (16.2 mg, 0.12 mmol) in 1.0 mL EtOAc/H2O (1 :1 ratio) to provide amide 15 (23.4 mg, 53.7% ) as a colorless solid: [α]D 20 +3.2 (c= 0.72, CHCI3). All other data matched that previously reported (Kumaraswamy, 2006).
Example 13
Biological activity of belactosin derivatives: fluorogenic assay for detection of 2OS proteasome
The fluorogenic peptide substrate Suc-LL VY-AMC was purchased from Biomol International, Inc. (Plymouth Meeting, PA). The reaction mixture consisted of approximately 5 nM 2OS proteasome in buffer (50 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 % v/v DMSO, 5 mM MgCl2, and 0.02 % SDS) and 1 μL test compounds dissolved in DMSO at final concentrations of 0.4-50 μM along with 100 μM Suc-LLVY-AMC. The resulting fluorescence from liberated AMC was measured every ten minutes at 380/460 nm for 2-3 hours. Suc-LLVY-Amc incubated without proteasome served as a background control. Results are the average of triplicate time points. The results are shown below and in FIG. 3:

N-Cbz, O-Bn n-Propyl belactosin C (IC50 25 nM)

N-Cbz, O-Bn Methyl belactosin C (IC50 249 nM)

DR32 (1:1.7 mixture) known protected belactosin C

DR33 (1:1.2 mixture) belactosin C and isomer

DR53 (1 :1 mixture) known derivative

DR59 (1 :1 mixture)

DR60 (1 :1 mixture)
Example 14 Inhibition of Fatty Acid Synthase thioesterase domain
Known and novel belactosin derivatives inhibit the turnover of a fluorogenic substrate by fatty acid synthase thioesterase. The thioesterase (230 nM) was incubated with a range of test compound concentrations (100 μM to 0.16 μM) and 120 μM 4-MU-heptanoate substrate, approximately 3.5x Km. Fluorescence measurements were taken every five minutes at 350/450 nm. Results shown are the calculated IC5O value and corresponding 95% confidence intervals from a dose-response curve fit of the data in triplicate. See Example 13 for compounds tested.
Table 1. Inhibition of the Fatty Acid Synthase Thioesterase Domain

* * * * * * * * * * * * * *
All of the compositions and/or methods disclosed and claimed in this specification can be made and executed without undue experimentation in light of the present disclosure. While the compositions and methods of this invention have been described in terms of preferred embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and/or methods and in the steps or in the sequence of steps of the method described herein without departing from the concept, spirit and scope of the invention. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the invention as defined by the appended claims.
REFERENCES
The following references, to the extent that they provide exemplary procedural or other details supplementary to those set forth herein, are specifically incorporated herein by reference.
U.S. Provisional Application entitled, "Cyclic-fused beta-Lactones and Their Synthesis," by
Daniel Romo, Huda Henry-Riyad, Gil Ma, Changsuk Lee, Henry Nguyen, Seongho
Oh and Vikram C. Purohit, filed July 9, 2007.
PCT International Pub. No. WO 04/007506 PCT International Pub. No. WO 00/043000
U.S. Pat. No. 7,223,745
Adams et al, Cancer Res. 59:2615-22, 1999.
Adams et al, Invest. New Drugs 18:109-21, 2000.
Adams et al, Curr. Opin. Chem. Biol 4:493-500, 2002a.
Adams et al, Trends MoI. Med. 8:S49-54, 2002b.
Akaishi et al, Brain Res., 722:139-144, 1996. AIo et al, Cancer, 11 Al A, 1996.
Arionov and De Meijere, Org. Lett., 6:2153, 2004.
Armstrong and Scutt, Chem. Commun., 7(5):510-511, 2004.
Asai et al, Biochem. Pharm. 67:227-34, 2004.
Asai et al, J. Antibiot, 53:81-83, 2000. Bodansky, "Peptide Chemistry," 2nd ed. , Springer- Verlag, New York, 1993.
Bundgaard, H., Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985.
Bundgaard, Drugs of the Future, 16:443-458, 1991.
Browne, FASEB J., 20:2027-35, 2006.
Chakravarty et al, Proc. Natl Acad. ScL USA, 101 :15567-72, 2004. Cresswell et al, Immunol Rev., 172:21-28, 1999.
Cho and Romo, Org. Lett, 9: 1537-1540, 2007.
FluoProbes® BioDirectory of Fluorescence.
Funabashi et al, J. Biochem., 105:751, 1989.
Ganzler et al, Hum. Pathol, 28:686, 1997.
Garcia-Echeverria, Mini Rev. Med. Chem., 2:247-259, 2002.
Garcia-Echeverria, Int. J. Pep. Res. Ther., 12:49-64, 2006.
Grant, In: Synthetic Peptides, Freeman & Co., New York, 1992. Greene & Wuts, In: Protective Groups in Organic Synthesis, 3rd ed., John Wiley & Sons, New York, N.Y., 1999.
Groll et al, Chembiochem, 6:222-256, 2005.
Groll et al., Proc. Natl. Acad. ScL USA, 103:4576-79, 2006.
Hirano et al, Methods Enzymol. 399:227-240, 2005. Knowles et al, J. Biol. Chem., 79:30540, 2004.
Kridel et al, Cancer, 64:2070-5, 2004.
Kuhajda et al, Proc. Natl Acad. ScL USA, 91 :6383, 2004.
Kuhajda et al, Proc. Natl Acad. ScL USA, 97:3450, 2000.
Kumaraswamy et al, J. Org. Chem. 71 :337-340, 2006. Kumaraswamy and Markondaiah, Tet. Lett., 48:1707-1709, 2007.
Larionov and de Meijere, Org. Lett., 6:2153, 2004.
Maier et al, Science, 311 :1258, 2006.
Mellgren, J. Biol Chem., 272:29899-903, 1997.
Michalek et al, Nature, 363:552-554, 1993. Mizukami et al, Chem. Abstr., 126:338840, 1997.
Molecular Probes, The Handbook — A Guide to Fluorescent Probes and Labeling Technologies, 10th Ed., 2005.
Murray et al., Anticancer Drugs 11 :407-417, 2000.
Pizer et al, Cancer Res., 56:745-51, 1996. Pizer et al, Cancer Res., 56: 1189-1193, 1996a.
Pizer et al, Cancer Res., 56:2745-2747, 1996b.
Pizer et al, Cancer Res., 58:4611-4615, 1998.
Pizer et al, Cancer Res., 60:213-218, 2000.
Pizer et al, Prostate, 47:102-110, 2001. Prasad and Chandrakumar, Tetrahedron: Asymmetry 1 : 1897, 2005.
Vmohit et al., J. Org. Chem. 71 :4549-58, 2006.
Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990.
Rossi et al, MoI. Cancer Res., 1 :707, 2003.
Simon et al, Science, 311 : 1263, 2006.
Swinnen et al, Int. J. Cancer, 98:19, 2002.
Ugai et al., J. Biochem. (Tokyo), 113:754-768, 1993.
Claims
A compound of the following formula:
wherein X0 is either


wherein R5 and Re are each independently selected from the group consisting of H, alkyl, aryl, an amine protecting group and


R4 equals

wherein Rg and R9 are each independently selected from the group consisting of H, alkyl, aryl, -CH2Cy, -CH(OH)alkyl, -CH(OH)aryl, -ORi0 and - NR11Ri2, wherein R10-R12 are each independently selected from the group consisting of H, alkyl, aryl, acetyl, -S1R13R14R15, an alcohol protecting group and an amine protecting group, wherein R13- Ri5 are each independently selected from the group consisting of alkyl and aryl; and n equals 1-7; and optical isomers thereof; with the proviso that when Ro equals
CBzHN 
Me , R2 equals -CO2Bn and n equals 3, then R4 cannot be 
with the proviso that if Ro equals

, R2 equals CO2H and n equals 3, then R4 cannot be

2. The compound of claim 1 , wherein the compound is not any of the following compounds:







3. The compound of claim 1 , wherein RQ equals H and Ri equals an amine protecting group.
4. The compound of claim 1 , wherein Ro equals

5. The compound of claim 4, wherein R5 is an amine protecting group.
6. The compound of claim 4, wherein Rg is H.
7. The compound of claim 4, wherein R7 is -CH3.
The compound of claim 1 , wherein R5 is 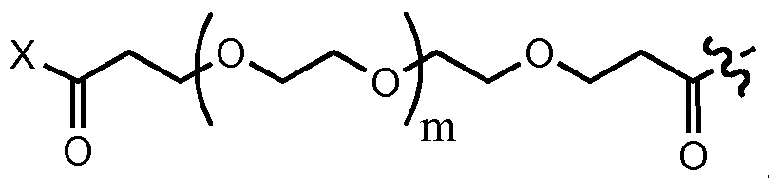
9. The compound of claim 8, wherein m is 1.
10. The compound of claim 8, wherein X is selected from the group consisting of:


11. The compound of claim 1 , wherein Ri is H.
12. The compound of claim 1, wherein R2 is alkyl or a protected carboxylic acid.
13. The compound of claim 1 , wherein R3 is H.
14. The compound of claim 1 , wherein R4 is

15. The compound of claim 1 , wherein R4 is 
16. The compound of claim 1 , wherein either R8 or R9 is H.
17. The compound of claim 1, wherein R% is -ORio and Rg is H.
18. The compound of claim 17, wherein Rio is selected from the group consisting of H, an alcohol protecting group and -SR13R14R15.
19. The compound of claim 1 , wherein R% is either alkyl or aryl and R9 is H.
20. The compound of claim 19, wherein Rg is phenyl.
21. The compound of claim 1 , wherein R« is -NRi 1R12 and R9 is H.
The compound of claim 1, wherein R« is -CH(OH)alkyl or -CH(OH)aryl and R9 is H.
23. The compound of claim 21, wherein Rn and R12 are each independently selected from the group consisting of H, acetyl and an amine protecting group.
24. The compound of claim 1, wherein n equals 1-5.
25. The compound of claim 1, having the formula:

wherein:
Yi is selected from the group consisting of H, Cbz, Fmoc, PMB and BOM; Y2 is H or -CH3; Y3 is H or Bn;
Y4 is selected from the group consisting of -CH3, -C6H13 and phenyl; p is 0 or 1; and q is 1-5.
26. The compound of claim 1, having the formula:

wherein RA is selected from the group consisting of H, Bn and PMB and RB is selected from the group consisting of H, Bn, PMB, -C(O)CH3, Cbz and Boc.
27. The compound of claim 1 , having the formula:

wherein:
Y5 is selected from the group consisting of H, Cbz, Fmoc, PMB, BOM and

Y6 is H or -CH3;
Y7 is selected from the group consisting of H, Bn,

Ye is selected from the group consisting of -CH3, -C6H13, -CH2Cy, - CH(OH)Cy, phenyl, -CH2C6H5 and -CH(OH)C6H5; r is 0 or 1 ; and s is 1-5.
28. The compound of claim 1 , having the formula:

wherein Rc is selected from the group consisting of H, Bn and PMB and RD is selected from the group consisting of H, Bn, PMB, -C(O)CH3, Cbz and Boc.
29. The compound of claim 1 , having the formula:

wherein R is alkyl or aryl.
30. The compound of claim 1 , having the formula: * 
wherein R is alkyl or aryl.
31. The compound of claim 1 , wherein the compound is comprised in a pharmaceutically acceptable excipient, diluent, or vehicle.
32. A method of synthesizing a compound of the following formula:

compπsmg: a) obtaining a first compound of formula

R1
wherein Ro is selected from the group consisting of H, alkyl, an amine protecting group and


R2 is selected from the group consisting of H, aryl, alkyl, -CO2W, wherein W is H, alkyl, aryl or a carboxylic acid protecting group, and

Ri6 is selected from the group consisting of H, -C(O)C(O)H and - C(O)C(O)Rn, wherein Rn is selected from the group consisting of alkyl and aryl; and n equals 1-7; and b) reacting it with a second compound of formula

wherein Rg and R9 are each independently selected from the group consisting of H, alkyl, aryl, -CH2Cy, -CH(OH)alkyl, -CH(OH)aryl, -OR22 and - NR23R24, wherein R22-R24 are each independently selected from the group consisting of H, alkyl, aryl, acetyl, -SiR2sR26R27, an alcohol protecting group and an amine protecting group, wherein R25-R27 are each independently selected from the group consisting of alkyl and aryl; R18-R20 are each independently selected from the group consisting of alkyl and aryl; and R21 is aryl; wherein R0-R3 and n are defined as described in step a); and R4 equals

wherein R8 and R9 are as described as in step b).
33. The method of claim 32, wherein Ro equals H and Ri equals an amine protecting group.
34. The method of claim 32, wherein Ro equals

35. The method of claim 34, wherein R5 is an amine protecting group.
36. The method of claim 34, wherein Re is H.
37. The method of claim 34, wherein R7 is -CH3.
38. The method of claim 34, wherein R5 is an amine protecting group, Ke is H and R7 is CH3.
39. The method of claim 32, wherein R5 is 
40. The method of claim 39, wherein m is 1.
41. The method of claim 39, wherein X is selected from the group consisting of:

42. The method of claim 32, wherein Ri is H.
43. The method of claim 32, wherein R2 is alkyl or a protected carboxylic acid.
44. The method of claim 32, wherein R3 is H.
45. The method of claim 32, wherein R4 is 
46. The method of claim 32, wherein R4 is

47. The method of claim 32, wherein either Rg or R9 is H.
48. The method of claim 32, wherein R8 is -OR10 and Rg is H.
49. The method of claim 48, wherein Rio is selected from the group consisting of H, an alcohol protecting group and -SR13R14R15.
50. The method of claim 32, wherein Rg is either alkyl or aryl and R9 is H.
51. The method of claim 50, wherein R8 is phenyl.
52. The method of claim 32, wherein Rg is -NR11Ri2 and R9 is H.
53. The method of claim 52, wherein Rn and R12 are each independently selected from the group consisting of H, acetyl and an amine protecting group.
54. The method of claim 32, wherein R8 is -CH(OH)alkyl or -CH(OH)aryl and R9 is H.
55. The method of claim 54, wherein Rn and R12 are each independently selected from the group consisting of H, acetyl and an amine protecting group.
56. The method of claim 1, wherein n equals 1-5.
57. The method of claim 32, wherein the first compound is reacted with a second compound of formula 
and optical isomers thereof.
58. The method of claim 57, wherein R18-R20 are each an alkyl group.
59. The method of claim 57, wherein R21 is phenyl or pyridyl.
60. The method of claim 32, wherein the first compound is reacted with a second compound of formula

and optical isomers thereof.
61. The method of claim 32, wherein a compound of formula (XI) is formed as an intermediate:

wherein R28 is -OH or 
R29 is selected from the group consisting of H, alkyl, aryl, -CH2Cy, -CH(OH)alkyl, - CH(OH)aryl, -OR22 and -NR23R24, wherein R22-R24 are each independently selected from the group consisting of H, alkyl, aryl, acetyl, -SiR2sR26R27, an alcohol protecting group and an amine protecting group, wherein R25-R27 are each independently selected from the group consisting of alkyl and aryl;
and optical isomers thereof.
62. A method of inhibiting the 2OS proteasome, comprising contacting a cell with an effective amount of a compound of claim 1.
63. A method of inhibiting fatty acid synthase, comprising contacting a cell with an effective amount of a compound of claim 1.
64. A method of treating cancer comprising administering to a subject an effective amount of a compound of claim 1.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002657103A CA2657103A1 (en) | 2006-07-07 | 2007-07-09 | Novel belactosin derivatives as therapeutic agents/biological probes and their synthesis |
| EP07799417A EP2059514A2 (en) | 2006-07-07 | 2007-07-09 | Novel belactosin derivatives as therapeutic agents/biological probes and their synthesis |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81921306P | 2006-07-07 | 2006-07-07 | |
| US60/819,213 | 2006-07-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008006113A2 true WO2008006113A2 (en) | 2008-01-10 |
| WO2008006113A3 WO2008006113A3 (en) | 2008-02-28 |
Family
ID=38716321
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/073077 Ceased WO2008006113A2 (en) | 2006-07-07 | 2007-07-09 | Novel belactosin derivatives as therapeutic agents/biological probes and their synthesis |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8119653B2 (en) |
| EP (1) | EP2059514A2 (en) |
| CA (1) | CA2657103A1 (en) |
| WO (1) | WO2008006113A2 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009106211A1 (en) * | 2008-02-26 | 2009-09-03 | Ludwig-Maximilians-Universität München | Beta-lactones as antibacterial agents |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP4180035A1 (en) | 2021-11-15 | 2023-05-17 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Novel beta-lactone inhibitors of hydrolytic enzymes and their medical and non medical uses |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3159331A1 (en) | 2010-05-05 | 2017-04-26 | Infinity Pharmaceuticals, Inc. | Tetrazolones as inhibitors of fatty acid synthase |
| US8450350B2 (en) | 2010-05-05 | 2013-05-28 | Infinity Pharmaceuticals, Inc. | Triazoles as inhibitors of fatty acid synthase |
| KR102844773B1 (en) | 2015-12-23 | 2025-08-13 | 암젠 인크 | Method for treating or improving metabolic disorders by using a binding protein for the gastric inhibitory peptide receptor (GIPR) in combination with a GLP-1 agonist |
| BR112019014588A2 (en) | 2017-01-17 | 2020-02-18 | Amgen Inc. | METHOD OF TREATING OR AMENIZING METABOLIC DISORDERS USING GLP-1 RECEPTOR AGONISTS CONJUGATED TO ANTAGONISTS FOR GASTRIC INHIBITOR PEPTIDE RECEPTOR (GIPR) |
| CN110831969B (en) | 2017-06-20 | 2024-06-21 | 安进公司 | Method for treating or improving metabolic disorders using a combination of gastric inhibitory peptide receptor (GIPR) binding protein and GLP-1 agonist |
| MX2019015544A (en) | 2017-06-21 | 2020-07-28 | Amgen Inc | Method of treating or ameliorating metabolic disorders using antagonistic binding proteins for gastric inhibitory peptide receptor (gipr)/glp-1 receptor agonist fusion proteins. |
| US11396497B2 (en) | 2018-05-04 | 2022-07-26 | New Mexico Tech University Research Park Corporation | Proteasome inhibitors |
| CN119019209A (en) * | 2024-08-07 | 2024-11-26 | 云南大学 | A kind of alpha-amino ketone compound and preparation method thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69601451T2 (en) * | 1995-10-16 | 1999-06-10 | Kyowa Hakko Kogyo Co., Ltd., Tokio/Tokyo | UCK14 connections |
| CA2359561A1 (en) * | 1999-01-20 | 2000-07-27 | Shiro Akinaga | Proteasome inhibitors |
| AU2002318538A1 (en) | 2002-07-15 | 2004-02-02 | Nippon Soda Co., Ltd. | Ruthenium compounds, diamine ligands, and process for preparation of optically active alcohols |
| US7223745B2 (en) * | 2003-08-14 | 2007-05-29 | Cephalon, Inc. | Proteasome inhibitors and methods of using the same |
-
2007
- 2007-07-09 US US11/775,154 patent/US8119653B2/en not_active Expired - Fee Related
- 2007-07-09 EP EP07799417A patent/EP2059514A2/en not_active Withdrawn
- 2007-07-09 WO PCT/US2007/073077 patent/WO2008006113A2/en not_active Ceased
- 2007-07-09 CA CA002657103A patent/CA2657103A1/en not_active Abandoned
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009106211A1 (en) * | 2008-02-26 | 2009-09-03 | Ludwig-Maximilians-Universität München | Beta-lactones as antibacterial agents |
| US8669283B2 (en) | 2008-02-26 | 2014-03-11 | Ludwig-Maximilians-Universitat Munchen | Beta-lactones as antibacterial agents |
| US9521845B2 (en) | 2008-02-26 | 2016-12-20 | Ludwig-Maximilians-Universität-München | Beta-lactones as antibacterial agents |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| EP4180035A1 (en) | 2021-11-15 | 2023-05-17 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Novel beta-lactone inhibitors of hydrolytic enzymes and their medical and non medical uses |
| WO2023084124A1 (en) | 2021-11-15 | 2023-05-19 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Novel beta-lactone inhibitors of hydrolytic enzymes and their medical and non medical uses |
Also Published As
| Publication number | Publication date |
|---|---|
| US8119653B2 (en) | 2012-02-21 |
| EP2059514A2 (en) | 2009-05-20 |
| CA2657103A1 (en) | 2008-01-10 |
| WO2008006113A3 (en) | 2008-02-28 |
| US20090042922A1 (en) | 2009-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8119653B2 (en) | Belactosin derivatives as therapeutic agents/biological probes and their synthesis | |
| US8088923B2 (en) | Cyclic-fused beta-lactones and their synthesis | |
| US6747021B2 (en) | Cryptophycin compound | |
| KR100628013B1 (en) | Tricyclic lactams and sulfam derivatives, and their use as histone deacetylase inhibitors | |
| HUP9901344A2 (en) | Radicicol derivatives and pharmaceutical compositions thereof | |
| CN113698401B (en) | β-elemene macrocyclic derivatives and their preparation methods and applications | |
| TW201625313A (en) | Novel glutamic acid derivative and use thereof | |
| JP2016513103A (en) | Albicidine derivatives, their use and synthesis | |
| US20090124681A1 (en) | Beta-lactone compounds | |
| WO2023160039A1 (en) | β-ELEMENE DERIVATIVE WITH HDACI PHARMACOPHORE, AND PREPARATION METHOD THEREFOR AND USE THEREOF | |
| CN112585142B (en) | Novel antimalarial compounds, methods for their preparation and their use in drug-resistant malaria | |
| JP6149043B2 (en) | 2-Aminated methylene or 2-esterified methylene tanshinone derivatives and methods for their preparation and use | |
| KR102452412B1 (en) | C14-hydroxyl esterified amino acid derivatives of triptolide, and methods for their preparation and uses | |
| US20050272727A1 (en) | Leptomycin compounds | |
| EP3470410A1 (en) | Crystalline form of compound suppressing protein kinase activity, and application thereof | |
| CN102844307A (en) | Triazole compounds as ksp inhibitors | |
| EP0393923A1 (en) | 6-Fluoroshikimic acid derivatives | |
| TWI293302B (en) | New derivatives of mikanolide, their preparation and their therapeutic uses | |
| US12486231B2 (en) | Targeted nitroxide compounds and their use in treating ferroptosis-related diseases | |
| CN103450164B (en) | Geldanamycin derivatives as well as preparation methods and uses thereof | |
| WO2008035222A2 (en) | Synthesis of carbohydrate-templated amino acids and methods of using same | |
| EP3075723A1 (en) | Highly soluble L-DOPA glycerol esters | |
| US9469656B2 (en) | HDAC inhibitors as anti-cancer agents | |
| CN103946217B (en) | 2-aminated methylene or 2-esterified methylene tanshinone derivatives, and preparation method and application thereof | |
| CN104803911A (en) | Pleuromutilin compounds, drug composition, synthesis method and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07799417 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2657103 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007799417 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |