明 細 書
ゥレア化合物又はその塩
技術分野
[0001] 本発明は、医薬、殊に頻尿、尿失禁、及び/又は過活動膀胱の治療剤として有用な ゥレア化合物又はその製薬学的に許容される塩に関する。
背景技術
[0002] 過活動膀胱は失禁の有無にかかわらず尿意切迫感を訴える病態であり、通常、頻 尿及び/又は夜間頻尿を伴う(非特許文献 1)。現在その治療には主に抗コリン薬が 使用され、一定の治療成績を示している。しかし、一方でロ渴、便秘、かすみ目とい つた副作用の発現も知られているほか、尿閉の危険性もあるため、前立腺肥大患者 や高齢者には使いづらいことが報告されている。また、抗コリン治療で有効性を示さ ない患者の存在も知られている。以上のことから、過活動膀胱に対する新規機序の 薬剤への期待は大きい。
[0003] 脂肪酸アミド加水分解酵素 (Fatty acid amide hydrolase;FAAH)は、エンドカンナビノ イドを加水分解することで、その活性を消失させることが知られている (非特許文献 2- 5)。エンドカンナビノイド (endocannabinoid)とは、カンナビノイド受容体に作用して生 理作用を発揮する生体内物質の総称である。代表的なエンドカンナビノイドとしてァ ナンダミド、パルミトイルエタノールアミド、ォレアミド、 2-ァラキドン酸グリセロールがあ り、 FAAHによって加水分解を受け活性が消失することが知られている。また、大麻( マリファナ)の活性成分であると考えられて!/、る Δ 9-テトラヒドロカンナビノールは、力 ンナビノイド受容体を活性化することが知られてレ、る(非特許文献 6)。
哺乳動物にはこれまで 2種類のカンナビノイド受容体 CB1、 CB2が知られている。 C B1は中枢及び末梢神経系に発現しており、その活性化により精神作用及び鎮痛作 用等が惹起される。 CB2は免疫系組織に発現し、その活性化により抗炎症作用及び 鎮痛 (炎症性)作用等が惹起される。
一方、ラット膀胱炎モデルにおいて、カンナビノイド受容体作動薬は膀胱容量及び 排尿閾値を増大させること (非特許文献 7及び非特許文献 8)、また、カンナビノイド受
容体作動薬を動物に投与した場合に観察される幻覚、妄想、頻脈、起立性低血圧等 の副作用が、 FAAH阻害剤を投与した場合には観察されないこと (非特許文献 9)から 、 FAAH阻害剤は大麻様の副作用や常用性の懸念が少ない新規な頻尿、尿失禁、 及び/又は過活動膀胱の治療剤として期待される。
FAAH阻害活性を有するピぺラジュルゥレア誘導体又はピペリジルゥレア誘導体と しては下記化合物が報告されて!/、る。
例えば、特許文献 1には下記化合物が
[化 1コ
R1 a Z
1 I I I
R— N-C-R-R-R
(式中、 R1は置換されていてもよいァリール又は置換されていてもよい複素環基、 Rla は H又は炭化水素基等、 Zは 0又は S、 R2は置換されていてもよいピぺリジン- 1,4-ジィ ル又は置換されていてもよいピぺラジン- 1,4-ジィル、 R3は- CO-、 - CO-O-、 -CONH -又は複素環基、 R4は置換されて!/、てもよ!/、炭化水素基又は置換されて!/、てもよ!/ヽ複 素環基であるが、 R3が- CO_、 -CO-O-又は- CONH-のとき、 R4はべンゾイソォキサゾ リル、をそれぞれ示す。詳細は当該公報参照。)、
特許文献 2には下記化合物が
[化 2]
(式中、 Ar1は 1又は 2個の Raで置換されていてもよい、 2-チアゾリル、 2-ピリジル、 4-ピ リジノレ、 2-ピリミジニノレ、 4-ピリミジニノレ、 5-ピリミジニル又はフエニル、 R1は H又は C
1-4 アルキル、 Zは N又は CH、 Ar2は、それぞれ特定の基で置換又は縮環していてもよい
、フエニル又は複素環基等をそれぞれ示す。詳細は当該公報参照。)、
それぞれ開示されている。し力、しながら、いずれも本願化合物の具体的開示は無い。
また、 FAAH阻害活性を有する力ルバメート誘導体として、例えば、特許文献 3及び 4、並びに、本願の優先日後に公開となった特許文献 5が報告されている。
[0005] 特許文献 1:国際公開 WO2006/054652号パンフレット
特許文献 2:国際公開 WO2006/074025号パンフレット
特許文献 3:国際公開 WO2003/065989号パンフレット
特許文献 4:国際公開 WO2004/033422号パンフレット
特許文献 5:国際公開 WO2006/088075号パンフレット
非特許文献 1:「ニューロウロロジ一'アンド'ゥロダイナミクス(Neurourology and Urody namics) J、 (英国)、 2002年、第 21巻、 ρ· 167-78
非特許文献 2 :「プロスタグランディンズ 'ロイコトリェンズ 'アンド 'エッセンシャル'ファ アイ · /ンッズ (Prostaglandins Leukotrienes and Essential Fatty Acids)」、、 国)、 20 02年、第 66巻、 .143-160
非特許文献 3 :「ブリティッシュ 'ジャーナル'ォブ 'ファーマコロジー (British Journal of
Pharmacology)]、(英国)、 2004年、第 141巻、 p.253-262
非特許文献 4 :「ネイチヤー(Nature)」、(英国)、 1996年、第 384巻、 p.83-87 非特許文献 5:「バイオケミカル 'ファーマコロジー(Biochemical Pharmacology)」、(米 国)、 2001年、第 62巻、 p.517-526
非特許文献 6 :「カレント'メディシナノレ 'ケミストリー(Current Medicinal Chemistry)」、 (米国)、 1999年、第 6巻、 .635-664
非特許文献 7 :「ザ'ジャーナル'ォブ 'ニューロサイエンス(The Journal of Neuroscien ce) J、 2002年、第 22巻、 p.7147-7153
非特許文献 8 :「ペイン(Pain)」、 1998年、第 76巻、 p.189-199
非特許文献 9 :「ネィチヤ一 ·メデイシン(Nature Medicine)」、(英国)、 2003年、第 9巻 、 .76-81
発明の開示
発明が解決しょうとする課題
[0006] 本発明の課題は、 FAAH阻害作用を有する医薬、特に頻尿、尿失禁、及び/又は過 活動膀胱の治療剤として有用な化合物の提供である。
課題を解決するための手段
[0007] 上述のように、 FAAH阻害剤は、抗コリン薬に見られるロ渴、尿閉等の副作用が少 なぐ安全性の高い頻尿、尿失禁、及び/又は過活動膀胱の治療剤となることが期待 できる。そこで本発明者等は頻尿、尿失禁、及び/又は過活動膀胱の治療に有用な 化合物を提供することを目的として、 FAAH阻害活性を有する化合物につき鋭意検 討した結果、本発明のゥレア化合物が良好な FAAH阻害作用を有することを知見し、 更に、 FAAH阻害活性を有する化合物がシクロフォスフアミド(Cyclophosphamide;CP A)で誘発した頻尿ラットにおいて、有効膀胱容量を増加させることを初めて見出し、 本発明を完成した。
[0008] 即ち、本発明は、以下に関する。
[1]式 (I)で示されるゥレア化合物又はその製薬学的に許容される塩。
[化 3]
[式中の記号は以下の意味を示す。
R' I H,ァリール、ァリール-〇-、ァリール-低級アルキレン-、ァリール-低級アルケニ レン-、ァリール-低級アルキレン-〇-、ァリール-低級アルキレン _NR°-、ァリール- N R。-低級アルキレン-、ァリール- C(0)-NR。-、ァリール- SO - NR。-、ヘテロァリール 、ヘテロァリール-低級アルキレン-〇-、シクロアルキル-低級アルキレン-、シクロアル キル-低級アルキレン- 0-、シクロアルキル-低級アルキレン- NR°-、環の構成原子で ある Nに結合手を有する含窒素へテロ環基、又は含酸素飽和へテロ環基-低級アル キレン-〇-、
ここで、 R1における各ァリール、各へテロアリールは下記 G1群から選択される基で 置換されていてもよぐ
G1群:ノヽロゲン、低級アルキル、 -0-低級アルキル、 -0-ベンジル、ハロゲノ低級アル
キル、 -O-ハロゲノ低級アルキル、 -CN、 -N(R°) 、 -C(0)-OR。、 -C(0)-N(R°)、及 びフエニル、
R° :同一又は異なって、 H、又は低級アルキル、
A:下記 G2群から選択される基でそれぞれ置換されて!/、てもよ!/、ベンゼン環、又はへ テロ環、
G2群:ノヽロゲン、低級アルキル、 -0-C アルキル、 _OH、 -NO 、 _C(0)-OR°、及び
1-8 2
-C(0)-N(R°) 、
X : N、又は CH、
L :低級アルキレン、低級アルケニレン、 -〇-、 -0-低級アルキレン-、 -S(O) -、 -低級 m アルキレン- s(o) -、 -c(o)-、 -低級アルキレン- c(o)-、 -低級アルケニレン- c(o)- m
、 -NR°-、 -C(0)-NR°-、 -NR°-C(0)-、 -O-低級アルキレン- C(0)-、 -低級アルキレ ン -0-C(0)-、又は-低級アルキレン- NR°-C(0)-、
(但し、 Xが Nの場合、 Lは-〇-、 - S -、 - NR。-、 - C(0)-NR。-ではない。 )、
m :同一又は異なって、 0、 1、又は 2、
R2、及び R3 :同一又は異なって、 H、又は低級アルキル、
n : 0、又は 1、
B : (i) n= lの場合、単結合、或いは、下記 G3群から選択される基でそれぞれ置換さ れていてもよいベンゼン環、又は芳香族へテロ環、(ii)n = 0の場合、単結合、
G3群:ノヽロゲン、低級アルキル、 -0-低級アルキル、ハロゲノ低級アルキル、 -0-ハロ ゲノ低級アルキル、 -OH、 -0-ベンジル、 -O-C(O)-低級アルキル、 -低級アルキレ ン -OR°、 -低級アルキレン- O-C(O)-低級アルキル、 _CN、 -NO 、 _C(0)-OR°、 _C( 0)-N(R°) 、 -N(R°) 、 -NR°-C(0)_低級アルキル、 -NR°_SO -低級アルキル、及び -S(O) -低級アルキル、
m
R4 : (i) n= l、かつ、 B =単結合の場合、
-c(o)-z、又は- S(O) -z、
m
[ここで、
z :低級アルキル、シクロアルキル、ァリーノレ、ヘテロァリール、 -低級アルキレン- 0- 低級アルキル、又は-低級アルキレン- 0-ベンジル、 ]
(ii) n= l、かつ、 B =単結合以外の場合、
H、下記 G4群から選択される基でそれぞれ置換されていてもよいフエニル、含窒素へ テロ環基、 _c(o)-含窒素へテロ環基、又は- W -低級アルキレン- Y、
W : _低級アルキレン- C(0)-NR°-、 -C(0)-NR°-、 -0-、又は単結合、
Y : - OH、 - N(R。)、 - C(O)- OR。、又は- C(O)- N(R。)、 ]
(iii) n = 0の場合、
下記 G4群から選択される基で置換されて!/、てもよ!/、、環の構成原子である Nに結合 手を有する含窒素へテロ環基、
G4群:低級アルキル、 _OH、 _N(R°)、 _C(0)-OR°、 _C(0)-N(R°)、 -低級アルキレ ン -C(0)-OR°、及び-低級アルキレン- C(0)-N(R°) 、
但し、 Lカ C(O)-又は-低級アルキレン- C(0)_、 Xが N、 n= l、かつ、 、
及び R
4が!/、ずれも Hである場合、 Bは置換されて!/、な!/、ベンゾイソォキサゾールを除 く。以下同様。 ]
[2]式 (I-A)で示される [1]記載のゥレア化合物又はその製薬学的に許容される塩。
[化 4]
[式中の記号は以下の意味を示す。
Rla :ァリール-低級アルキレン-、ァリール-低級アルケニレン-、ァリール-低級アルキ レン-〇-、ァリール-低級アルキレン- NR°-、ヘテロァリール-低級アルキレン- 0-、シ クロアルキル-低級アルキレン-、シクロアルキル-低級アルキレン-〇-、シクロアルキ ノレ-低級アルキレン- NR°-、又は含酸素飽和へテロ環基-低級アルキレン-〇-、 ここで、 Rlaにおける各ァリール、各へテロァリールは下記 G1群から選択される基で 置換されていてもよぐ
G1群:ノヽロゲン、低級アルキル、 -0-低級アルキル、 -0-ベンジル、ハロゲノ低級アル キル、 -0-ハロゲノ低級アルキル、 _CN、 -N(R°) 、 -C(0)-OR。、 -C(0)-N(R°)、及 びフエニル、
R° :同一又は異なって、 H、又は低級アルキル、
A1:下記 G2群から選択される基でそれぞれ置換されて!/、てもよ!/、ベンゼン環、又は 芳香族へテロ環、
G2群:ノヽロゲン、低級アルキル、 - 0- C アルキル、 - OH、 -NO 、 - C(0)-OR°、及び
1-8 2
-C(0)-N(R°) 、
X : N、又は CH、
L1 :メチレン、 - 0-、 - S(O) -、 - C(0)_、又は- NR0-、
m
(但し、 Xが Nの場合、 L1はメチレン、 -S(O) -、又は _C(0)_である。 )、
m :同一又は異なって、 0、 1、又は 2、
R2 : H、又は低級アルキル、
n : 0、又は 1、
B1 : © n= lの場合、単結合、或いは、下記 G3群から選択される基で置換されていて もよい 5又は 6員芳香族含窒素へテロ環、(ii)n = 0の場合、単結合、
G3群:ノヽロゲン、低級アルキル、 -0-低級アルキル、ハロゲノ低級アルキル、 -0-ハロ ゲノ低級アルキル、 -OH、 -0-ベンジル、 -O-C(O)-低級アルキル、 -低級アルキレ ン -OR°、 -低級アルキレン- O-C(O)-低級アルキル、 _CN、 -NO 、 _C(0)-OR°、 _C( 0)-N(R°) 、 -N(R°) 、 -NR°-C(0)_低級アルキル、 -NR°_SO -低級アルキル、及び -S(O) -低級アルキル、
m
R4a : (i) n = l、かつ、 単結合の場合、
-c(o)-z、又は- S(O) -z、
m
[ここで、
z :低級アルキル、シクロアルキル、ァリーノレ、ヘテロァリール、 -低級アルキレン- 0- 低級アルキル、又は-低級アルキレン- 0-ベンジル、 ]
(ii) n= l、かつ、 上記 G3群から選択される基で置換されていてもよい 5又は 6員 芳香族含窒素へテロ環の場合、
H、下記 G4群から選択される基でそれぞれ置換されていてもよいフエニル、含窒素へ テロ環基、 _c(o)-含窒素へテロ環基、又は- W -低級アルキレン- Y、
[ここで、
W : _低級アルキレン- C(0)-NR°-、 -C(0)-NR°-、 -0-、又は単結合、
Y : - OH、 - N(R。)、 - C(O)- OR。、又は- C(O)- N(R。)、 ]
(iii) n = 0の場合、
下記 G4群から選択される基で置換されて!/、てもよ!/、、環の構成原子である Nに結合 手を有する 5員芳香族含窒素へテロ環基、
G4群:低級アルキル、 _OH、 _N(R°)、 _C(0)-OR°、 _C(0)-N(R°)、 -低級アルキレ ン -C(〇)-OR°、及び-低級アルキレン- C(〇)-N(R°)2 。以下同様。 ]
[3]式 (I-B)で示される [2]記載のゥレア化合物又はその製薬学的に許容される塩。
[化 5]
[式中の記号は以下の意味を示す。
Rla :ァリール-低級アルキレン-、ァリール-低級アルケニレン-、ァリール-低級アルキ レン-〇-、ァリール-低級アルキレン- NR°-、ヘテロァリール-低級アルキレン- 0-、シ クロアルキル-低級アルキレン-、シクロアルキル-低級アルキレン-〇-、シクロアルキ ノレ-低級アルキレン- NR°-、又は含酸素飽和へテロ環基-低級アルキレン-〇-、 ここで、 Rlaにおける各ァリール、各へテロァリールは下記 G1群から選択される基で 置換されていてもよぐ
G1群:ノヽロゲン、低級アルキル、 -0-低級アルキル、 -0-ベンジル、ハロゲノ低級アル キル、 -0-ハロゲノ低級アルキル、 _CN、 -N(R°)、 -C(0)-OR。、 -C(0)-N(R°)、及 びフエニル、
R° :同一又は異なって、 H、又は低級アルキル、
A1:下記 G2群から選択される基でそれぞれ置換されて!/、てもよ!/、ベンゼン環、又は 芳香族へテロ環、
G2群:ノヽロゲン、低級アルキル、 -0-C アルキル、 _OH、 -NO 、 _C(0)-OR°、及び
1-8 2
-C(0)-N(R°) 、
X : N、又は CH、
L1 :メチレン、 - 0-、 - S(O) -、 - C(0)_、又は- NR0-、
m
(但し、 Xが Nの場合、 L1はメチレン、 -S(O) -、又は _C(0)_である。 )、
m :同一又は異なって、 0、 1、又は 2、
R2 : H、又は低級アルキル、
B2:下記 G3群から選択される基で置換されて!/、てもよい 5又は 6員芳香族含窒素へテ 口環、
G3群:ノヽロゲン、低級アルキル、 -0-低級アルキル、ハロゲノ低級アルキル、 -0-ハロ ゲノ低級アルキル、 -OH、 -0-ベンジル、 -O-C(O)-低級アルキル、 -低級アルキレ ン -OR°、 -低級アルキレン- O-C(O)-低級アルキル、 _CN、 -NO 、 _C(0)-OR°、 _C( 0)-N(R°) 、 -N(R°) 、 -NR°-C(0)_低級アルキル、 -NR°_SO -低級アルキル、及び -S(O) -低級アルキル、
m
R4b : H、下記 G4群から選択される基でそれぞれ置換されていてもよいフエニル、含窒 素へテロ環基、 -C(O)-含窒素へテロ環基、又は- W-低級アルキレン- Y、
[ここで、
W : _低級アルキレン- C(0)-NR°-、 -C(0)-NR°-、 -0-、又は単結合、
Y : - OH、 - N(R。)、 - C(O)- OR。、又は- C(O)- N(R。)、 ]
G4群:低級アルキル、 _OH、 _N(R°) 、 _C(0)-OR°、 _C(0)-N(R°)、 -低級アルキレ ン -C(〇)-OR°、及び-低級アルキレン- C(〇)-N(R°)2 。以下同様。 ]
[4] A1がベンゼン環、或いは 5又は 6員芳香族へテロ環である [3]記載のゥレア化合物 又はその製薬学的に許容される塩。
[5] Rlaがァリール-低級アルキレン-〇-、ァリール-低級アルキレン- NR°-、ヘテロァリ ール-低級アルキレン-〇-、シクロアルキル-低級アルキレン- o-、シクロアルキル-低 級アルキレン- NR°-、又は含酸素飽和へテロ環基-低級アルキレン- 0-である [4]記
載のウレァ化合物又はその製薬学的に許容される塩。
[6] X=Nで、かつ、 L1が- C(0)_、又はメチレンである [5]記載のゥレア化合物又はそ の製薬学的に許容される塩。
[7] X=CHで、かつ、 L1が- 0-である [5]記載のゥレア化合物又はその製薬学的に許 容される塩。
[8]式 (I-C)で示される [2]記載のゥレア化合物又はその製薬学的に許容される塩。
[化 6]
[式中の記号は以下の意味を示す。
Rla :ァリール-低級アルキレン-、ァリール-低級アルケニレン-、ァリール-低級アルキ レン-〇-、ァリール-低級アルキレン- NR°-、ヘテロァリール-低級アルキレン- 0-、シ クロアルキル-低級アルキレン-、シクロアルキル-低級アルキレン-〇-、シクロアルキ ノレ-低級アルキレン- NR°-、又は含酸素飽和へテロ環基-低級アルキレン-〇-、 ここで、 Rlaにおける各ァリール、各へテロァリールは下記 G1群から選択される基で 置換されていてもよぐ
G1群:ノヽロゲン、低級アルキル、 -0-低級アルキル、 -0-ベンジル、ハロゲノ低級アル キル、 -0-ハロゲノ低級アルキル、 _CN、 -N(R°) 、 -C(0)-OR。、 -C(0)-N(R°)、及 びフエニル、
R° :同一又は異なって、 H、又は低級アルキル、
A2 :下記 G2群から選択される基でそれぞれ置換されていてもよいベンゼン環、又は 5 又は 6員芳香族へテロ環、
G2群:ノヽロゲン、低級アルキル、 -0-C アルキル、 _OH、 -NO 、 _C(0)-OR°、及び
1-8 2
-C(0)-N(R°) 、
X : N、又は CH、
L1 :メチレン、 - 0-、 - S(O) -、 - C(0)_、又は- NR0-、
(但し、 Xが Nの場合、 L1はメチレン、 -S(O) -、又は _C(0)_である。 )、
m :同一又は異なって、 0、 1、又は 2、
R2 : H、又は低級アルキル、
R4c : -C(0)-Z,又は- S(O) -Z、
m
[ここで、
z:低級アルキル、シクロアルキル、ァリール、ヘテロァリール、 -低級アルキレン- 0- 低級アルキル、又は-低級アルキレン- 0-ベンジル 。以下同様。 ]
[9]式 (I-D)で示される [2]記載のゥレア化合物又はその製薬学的に許容される塩。
[化 7]
[式中の記号は以下の意味を示す。
R :ァリール-低級アルキレン-、ァリール-低級アルケニレン-、ァリール-低級アルキ レン-〇-、ァリール-低級アルキレン- NR°-、ヘテロァリール-低級アルキレン- 0-、シ クロアルキル-低級アルキレン-、シクロアルキル-低級アルキレン-〇-、シクロアルキ ノレ-低級アルキレン- NR°-、又は含酸素飽和へテロ環基-低級アルキレン-〇-、 ここで、 Rlaにおける各ァリール、各へテロァリールは下記 G1群から選択される基で 置換されていてもよぐ
G1群:ノヽロゲン、低級アルキル、 -0-低級アルキル、 -0-ベンジル、ハロゲノ低級アル キル、 -0-ハロゲノ低級アルキル、 _CN、 -N(R°) 、 -C(0)-OR。、 -C(0)-N(R°)、及 びフエニル、
R° :同一又は異なって、 H、又は低級アルキル、
A2 :下記 G2群から選択される基でそれぞれ置換されていてもよいベンゼン環、又は 5 又は 6員芳香族へテロ環、
G2群:ノヽロゲン、低級アルキル、 - 0- C アルキル、 - OH、 -NO 、 - C(0)-OR°、及び
1-8 2
-C(0)-N(R°) 、
X : N、又は CH、
L1 :メチレン、 - 0-、 - S(O) -、 - C(0)_、又は- NR0-、
m
(但し、 Xが Nの場合、 L1はメチレン、 -S(O) -、又は _C(0)_である。 )、
m : 0、 1、又は 2、
R2 : H、又は低級アルキル、
R4d :下記 G4群から選択される基で置換されていてもよい、環の構成原子である Nに 結合手を有する 5員芳香族含窒素へテロ環基、
G4群:低級アルキル、 _OH、 _N(R°)、 _C(0)-OR°、 _C(0)-N(R°)、 -低級アルキレ ン -C(〇)-OR°、及び-低級アルキレン- C(〇)-N(R°)2 。以下同様。 ]
[10] 4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }-N-ピリジン- 3-ィルピペリジン- 1- カルボキサミド、
4_{4-[(3-フルォロベンジル)ォキシ]ベンジル }-N-ピリジン- 3-ィルピペラジン- 1-カル ボキサミド、
4-{4-[(2-シクロへキシルェチル) (メチル)ァミノ]ベンゾィル }-N-ピリジン- 3-ィルピペラ ジン- 1-カルボキサミド、
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-ピリミジン- 2-ィルピペラジン- 1-カル ボキサミド、
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-ピリダジン- 3-ィルピペラジン- 1-力 ルポキサミド、
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-ピラジン- 2-ィルピペラジン- 1-カル ボキサミド、
4-[2-(2-シクロへキシルエトキシ)イソニコチノィル] -N-ピラジン- 2-ィルピペラジン- 1- カルボキサミド、
4-[3_(2-シクロへキシルエトキシ)ベンジル] -N-ピリジン- 3-ィルピペラジン- 1-カルボ キサミド、
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-ピリミジン- 5-ィルピペラジン- 1-カル ボキサミド、
N-(6-アミノビリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンゾィノレ]ピぺラジン
-1-カノレポキサミド、
4-[3-(2-シクロへキシルエトキシ)ベンジル] -N-ピリダジン- 3-ィルピペラジン- 1-カル ボキサミド、
4-{3-[(2,3-ジフルォロベンジル)ォキシ]ベンジル }-N-ピリジン- 3-ィルピペラジン- 1- カルボキサミド、
4-{[2-(2-シクロへキシルエトキシ)ピリジン- 4-ィル]メチル }-N-ピリジン- 3-ィルピペラ ジン- 1-カルボキサミド、
4-{3-[2-(3_フルオロフェニノレ)エトキシ]ベンゾィル }-N-ピラジン- 2-ィルピペラジン- 1- カルボキサミド、
4-{[5_(2-シクロへキシルエトキシ)ピリジン- 2-ィノレ]カルボ二ル}-N-ピラジン- 2-ィルピ ペラジン- 1-カルボキサミド、
4-[3_(2-シクロへキシルエトキシ) -5-フルォロベンゾィル] -N-ピラジン- 2-ィルピペラ ジン- 1-カルボキサミド、
4-[3_(2-シクロへキシルエトキシ) -4-フルォロベンゾィル] -N-ピラジン- 2-ィルピペラ ジン- 1-カルボキサミド、
4-{2-[2-(2-フルオロフェニノレ)エトキシ]イソニコチノィル }-N-ピラジン- 2-ィルピペラジ ン -1-カルボキサミド、
N-(3-クロロビラジン- 2-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンゾィノレ]ピぺラジン -1-カノレポキサミド、
N-(3-クロロビラジン- 2-ィル) -4-[3_(2-シクロへキシルエトキシ)フエノキシ]ピぺリジン- 1-カノレポキサミド、
4-[5_(2-シクロへキシルエトキシ) -2-フルォロベンゾィル] -N-ピラジン- 2-ィルピペラ ジン- 1-カルボキサミド、
N-(3-クロロビラジン- 2-ィル) -4-[2-(2-シクロへキシルエトキシ)イソニコチノィノレ]ピぺ ラジン- 1-カルボキサミド、
N-(3-クロロビラジン- 2-ィル) -4-{[2-(2-シクロへキシルエトキシ)ピリジン- 4-ィル]メチ ル}ピペラジン- 1-カルボキサミド、
4-{[2-(2-シクロへキシルエトキシ)ピリジン- 4-ィル]メチル }-N-ピラジン- 2-ィルピペラ
ジン- 1-カルボキサミド、
N-(3-クロロビラジン- 2-ィノレ) -4-{2-[2-(3-フルオロフェニノレ)エトキシ]イソニコチノィル }ピペラジン- 1-カルボキサミド、
4-{2-[2-(2-クロ口フエ二ノレ)エトキシ]イソニコチノィルト N-(3-クロロビラジン -2-ィノレ)ピ ペラジン- 1-カルボキサミド、
N-(3-クロロビラジン- 2-ィル) -4-(2-{2-[2- (トリフルォロメチノレ)フエ二ノレ]エトキシ }イソ二 コチノィル)ピぺラジン- 1-カルボキサミド、
N-(3-クロロビラジン- 2-ィノレ) -4-{2-[2-(2-フルオロフェニノレ)エトキシ]イソニコチノィル }ピペラジン- 1-カルボキサミド、
4_{4-[(3-フルォロベンジル)ォキシ]フエノキシ }-1-(1Η-イミダゾール -1-ィルカルボ二 ル)ピぺリジン、
1-{4-[(3-フルォロベンジル)ォキシ]ベンゾィル }-4-(1Η-イミダゾール -1-ィルカルボ二 ノレ)ピぺラジン、
4_({4-[3-(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カルボニル) -4H- 1,2,4-トリァゾール -3-ァミン、
1_({4-[3-(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カルボニル) -1H- ピラゾール -5-ァミン、及び
4-[3_(2-シクロへキシルエトキシ)ベンジル] -N- (メトキシァセチル)ピぺラジン- 1-カル ボキサミド
からなる群から選択される [1]記載の化合物又はその製薬学的に許容される塩。
[11] [1]記載の化合物又はその製薬学的に許容される塩を有効成分として含有する 医薬組成物。
[12] FAAH阻害剤である、 [11]記載の医薬組成物。
[13]頻尿、尿失禁、及び/又は過活動膀胱の治療剤である、 [11]記載の医薬組成物
[14]頻尿、尿失禁、及び/又は過活動膀胱の治療剤の製造のための、 [1]記載の化 合物又はその製薬学的に許容される塩の使用。
[15] [1]記載の化合物又はその製薬学的に許容される塩の有効量を患者に投与する
ことを含む、頻尿、尿失禁、及び/又は過活動膀胱の治療方法。
発明の効果
[0009] 本発明化合物は、後記試験例 1〜3において良好な FAAH阻害作用を有すること、 及び後記試験例 4において有効膀胱容量を増加させ、頻尿状態を改善することが確 認されたことから、頻尿、尿失禁、及び/又は過活動膀胱の治療剤として有用である。 発明を実施するための最良の形態
[0010] 以下、本発明を詳細に説明する。
本明細書中、「低級」なる語は、特に断らない限り炭素数 1〜6個(以下、 C と略す)
1-6 の直鎖または分枝状の炭化水素鎖を意味する。
[0011] 「低級アルキル」とは、 C のアルキルを意味する。具体的には、メチル、ェチル、ノ
1-6
ノレマノレプロピノレ、イソプロピノレ、ノノレマノレブチノレ、イソブチノレ、 sec-ブチノレ、 tert-ブチ ル、ノルマルペンチル、ノルマルへキシル等が挙げられる。好ましくは、炭素数;!〜 3 のものであり、より好ましくは、メチル、ェチル、又はイソプロピルである。
「低級アルキレン」とは、上記「低級アルキル」の任意の位置の水素を 1個除去して なる 2価基を意味する。具体的にはメチレン、エチレン、メチルメチレン、ジメチルメチ レン、トリメチレン等が挙げられる。好ましくは、メチレン、エチレン、又はトリメチレンで あり、より好ましくは、メチレン、又はエチレンである。
「低級アルケニレン」とは、上記「低級アルキレン」の任意の位置に少なくとも 1つの 二重結合を有する 2価基を意味する。具体的にはビニレン、プロぺニレン、 1—ブテ 二レン、 2—ブテニレン等が挙げられる。好ましくは、ビニレンである。
「シクロアルキル」とは、 C の飽和炭化水素環を意味し、架橋環を形成してレ、ても
3 - 10
よい。具体的には、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、 シクロへプチル、シクロオタチル、ァダマンチル、ノルボルニル等が挙げられる。好ま しくは、架橋を有していない単環式の C シクロアルキルであり、より好ましくは、シクロ
3-8
へキシルである。
[0012] 「ハロゲン」とは、 F、 Cl、 Br、 Iを意味する。好ましくは F、 Cl、又は Brである。
「ハロゲノ低級アルキル」とは、前記「低級アルキル」の 1個以上の任意の水素原子 力 同一又は異なって前記「ハロゲン」で置換された基を意味する。具体的には、フ
ノレオロメチル、ジフルォロメチル、トリフルォロメチル、 2,2,2-トリフルォロェチル、ペン タフルォロェチル等が挙げられる。好ましくは、トリフルォロメチルである。
「ァリール」とは、 C の単環式、二環式、又は三環式芳香族炭化水素環基を意味
6-14
し、 C シクロアルケン環と縮合した環基を包含する。但し、 C シクロアルケン環が縮
5-7 5-7
環している場合、結合手は芳香環上にある。具体的には、フエニル、ナフチル、イン ダニル、テトラヒドロナフチル、フルォレニル等が挙げられる。好ましくは、フエニルで ある。
[0013] 「ヘテロ環」とは、〇、 S及び Nから選択されるへテロ原子を 1〜4個含有する 4〜12 員の、単環式、又は 2環式の飽和又は不飽和環である。不飽和環には芳香族へテロ 環を含む。また、環原子である S又は Nが酸化されて、ォキシドゃジォキシドを形成し ていてもよい。具体的には、単環式としては、ァゼチジン、ピロリジン、ピぺリジン、ピ ペラジン、モノレホリン、チォモノレホリン、ァゼパン、ジァゼパン、ォキセタン、テトラヒド 口フラン、テトラヒドロピラン、 1,3-ジォキソール、 2,3-ジヒドロ- 1,4-ジォキシン、ビラゾリ ジン、フラン、チォフェン、ピロ一ノレ、イミダゾーノレ、ピラゾーノレ、チアゾーノレ、ォキサゾ 一ノレ、イソチアゾーノレ、イソォキサゾーノレ、トリァゾーノレ、テトラゾール、チアジアゾー ル、ォキサジァゾ一ノレ、ピリジン、ピラジン、ピリミジン、ピリダジン、トリアジン、 2,3-ジヒ ドロ- 1,3-ォキサゾール等力 二環式としては、 1,3-ベンゾジォキソール、 2,3-ジヒドロ -1,4-ベンゾジォキシン、インドーノレ、ベンゾフラン、ベンゾチォフェン、ベンゾ才キサ ゾーノレ、ベンゾイソォキサゾ一ノレ、ベンゾチアゾーノレ、ベンゾイソチアゾーノレ、ベンゾ イミダゾール、インダゾール、ベンゾトリァゾール、キノリン、イソキノリン、 1,2,3,4-テトラ ヒドロキノリン、 1,2,3,4-テトラヒドロイソキノリン、キノキサリン、キナゾリン、フタラジン等 が挙げられる。好ましくは単環式のへテロ環である。 「ヘテロ環基」とは、上記のへテロ 環で構成される環基を意味する。
[0014] 「芳香族へテロ環」とは、上記「ヘテロ環」のうち、 i) 0、 S及び Nから選択されるへテ 口原子を 1〜4個含有する単環式 5又は 6員芳香族へテロ環、 ii)上記 i)の芳香族へテ 口環が縮環した 2環式へテロ環 (但し、縮合する 2つの芳香族へテロ環は互いに同一 でも異なっていてもよい)、及び iii)上記 i)の芳香族へテロ環とベンゼン環又は 5〜7 員シクロアルカンが縮合した 2環式へテロ環、から選択される環を意味する。具体的
には、 i)ピリジン、ピラジン、ピリミジン、ピリダジン、トリアジン、ピロール、フラン、チォ フェン、イミダゾーノレ、ピラゾーノレ、 トリァゾーノレ、テトラゾーノレ、ォキサゾーノレ、イソォ キサゾ一ノレ、才キサジァゾ一ノレ、チアゾーノレ、イソチアゾーノレ、チアジアゾーノレ、 ii)ナ フチリジン、イミダゾピリジン、ピロ口ピリミジン、チェノビリジン、チエノピロリン、 iii)ベン ゾイミ ゾーノレ ベン、,フラン ベ.、ノゾ千才フェン ベン、/チア、ソァ、一ノレ ベンゾ千ァ ゾーノレ、ベンゾイソチアゾーノレ、ベンゾォキサゾーノレ、ベンゾイソォキサゾ一ノレ、キノリ ン、イソキノリン、 5,6,7,8-テトラヒドロキノリン、 5,6,7,8-テトラヒドロイソキノリン、キナゾリ ン、キノキサリン、フタラジン、インドール、イソインドール、テトラヒドロべンゾイミダゾー ノレ、クロマン、インダゾール等が挙げられる。好ましくは、上記 i)又は iii)であり、より好ま しくは、上記の i)単環式 5又は 6員芳香族へテロ環である。「ヘテロァリール」とは、上 記の芳香族へテロ環で構成される環基を意味する。
[0015] 「含窒素へテロ環」とは、上記「ヘテロ環」のうち、環の構成元素として少なくとも 1つ 以上の Nを必ず含有するへテロ環を意味する。具体的には、ピロリジン、ピぺリジン、 ピぺラジン、モノレホリン、チォモノレホリン、ァゼパン、ジァゼパン、 1,2,3,4-テトラヒドロ キノリン、 1,2,3,4-テトラヒドロイソキノリン、ピリジン、ピラジン、ピリミジン、ピリダジン、ト リアジン、ピロ一ノレ、イミダゾーノレ、ピラゾール、トリァゾーノレ、テトラゾーノレ、ォキサゾ 一ノレ、イソ才キサゾーノレ、才キサジァゾ一ノレ、チアゾーノレ、イソチアゾーノレ、チアジア ゾーノレ、ベンゾイミダゾーノレ、ベンゾチアジアゾーノレ、ベンゾチアゾーノレ、ベンゾイソ チアゾーノレ、ベンゾォキサゾーノレ、ベンゾイソォキサゾ一ノレ、キノリン、イソキノリン、 5, 6,7,8-テトラヒドロキノリン、 5,6,7,8-テトラヒドロイソキノリン、キナゾリン、キノキサリン、 フタラジン、インドーノレ、イソインドーノレ、テトラヒドロべンゾイミダゾーノレ、インダゾール 等が挙げられる。「含窒素へテロ環基」とは、上記の含窒素へテロ環で構成される環 基を意味する。
[0016] 「5又は 6員芳香族含窒素へテロ環」とは、上記「含窒素へテロ環」のうち、芳香族の 単環式 5又は 6員環を意味する。具体的には、ピリジン、ピラジン、ピリミジン、ピリダジ ン、トリアジン、ピロール、イミダゾール、ピラゾール、トリァゾール、テトラゾール、ォキ サゾーノレ、イソ才キサゾーノレ、才キサジァゾ一ノレ、チアゾーノレ、イソチアゾーノレ、チア ジァゾール等が挙げられる。
「環の構成原子である Nに結合手を有する含窒素へテロ環基」とは、上記「含窒素 ヘテロ環基」のうち、例えば、 1H-イミダゾール -1-ィル、 4H-1,2,4-トリァゾール -4-ィ ノレ、 1H-ピラゾール -1-ィル、 1-ピロリジニノレ、 1-ピペリジル、 1-ピぺラジュル、 4-モノレ ホリニル等のように、必ず結合手が環の構成原子である Nにあるものを意味する。
「環の構成原子である Nに結合手を有する 5員芳香族含窒素へテロ環基」とは、上 記「環の構成原子である Nに結合手を有する含窒素へテロ環基」のうち、芳香族の単 環式 5員環で構成される環基を意味する。具体的には、 1H-イミダゾール -1-ィル、 4 H-1, 2,4-トリァゾール -4-ィル、 1H-ピラゾール -1-ィル等が挙げられる。
[0017] 「含酸素飽和へテロ環」とは、上記「ヘテロ環」のうち、環の構成元素として少なくとも
1つ以上の Oを必ず有し、更に〇、 S及び Nから選択されるへテロ原子を有してもよい 飽和へテロ環を意味する。好ましくは、ォキセタン、テトラヒドロフラン、テトラヒドロビラ ン、ォキセパン、 1,4-ジォキサン等のように環の構成元素が C及び Oのみの飽和へテ 口環である。 「含酸素飽和へテロ環基」とは、上記の含酸素飽和へテロ環で構成され る環基を意味する。
[0018] 「置換されていてもよい」とは、「置換されていない」、又は「同一又は異なる 1〜5個 の、好ましくは 1〜2個の置換基で置換された」ことを意味する。
また、例えば、 -N(R°)における R°のように複数個の基が存在する場合、それぞれ の基(この場合は R°)は互いに同一であっても異なって!/、てもよ!/、。
[0019] 本発明化合物 (I)における好ましい態様を以下に示す。
(1) R1としては、好ましくは Rlaであり、より好ましくはァリール-低級アルキレン-〇-、ァ リール-低級アルキレン- NR°-、ヘテロァリール-低級アルキレン- 0-、シクロアルキル -低級アルキレン-〇-、シクロアルキル-低級アルキレン- NR°-、又は含酸素飽和へテ 口環基-低級アルキレン- 0-であり、更に好ましくは、ァリール-低級アルキレン-〇-、 ァリール-低級アルキレン- NR°-、シクロアルキル-低級アルキレン- 0-、又はシクロア ルキル-低級アルキレン- NR°-であり、より更に好ましくは、ァリール-低級アルキレン- 〇-、ァリール-低級アルキレン- NR°-、又はシクロアルキル-低級アルキレン- 0-であ る。 R1における各ァリール、及び各へテロァリールは前記 G1群から選択される基で置 換されていてもよいが、好ましくは、無置換である場合、或いはハロゲン、低級アルキ
ル、 -o-低級アルキル、及びハロゲノ低級アルキルからなる群から選択される同一又 は異なる 1又は 2個の置換基で置換されている場合であり、より好ましくは、無置換で ある場合、或いはハロゲン及びノヽロゲノ低級アルキルからなる群から選択される同一 又は異なる 1又は 2個の置換基で置換されている場合である。 R1におけるァリールと しては、好ましくはフエニルであり、またシクロアルキルとしては、好ましくは 5〜7員シ クロアルキルであり、より好ましくはシクロへキシルである。更に、 R1における低級アル キレンとしては、好ましくはメチレン、エチレン、又はトリメチレンであり、より好ましくは エチレンである。
(2) R°としては、好ましくは H、又はメチルである。
(3) Aとしては、好ましくは A1であり、より好ましくはそれぞれ置換されていてもよいべ ンゼン環、或いは 5又は 6員芳香族へテロ環であり、更に好ましくはベンゼン環、又は 6員芳香族へテロ環であり、より更に好ましくはベンゼン環、又はピリジン環である。 A におけるベンゼン環、及びへテロ環は前記 G2群から選択される基で置換されていて もよいが、好ましくは、無置換である場合、或いは、ハロゲン、低級アルキル、及び- O -C アルキルからなる群から選択される同一又は異なる 1又は 2個の置換基で置換
1-8
されている場合であり、より好ましくは無置換である場合、又は 1個のハロゲンで置換 されている場合である。
(4) Lとしては、好ましくは L1であり、より好ましくはメチレン、 -〇-、又は- C(O)-であり、 更に好ましくは、(i)Xが CHの場合、 -〇-、(ii)Xが Nである場合、メチレン、又は- C(O)
-である。
(5) R2としては、好ましくは H、又はメチルであり、より好ましくは Hである。
(6) Bとしては、好ましくは B1であり、より好ましくは単結合、又は 6員芳香族含窒素へ テロ環であり、更に好ましくは単結合、ピリジン環、ピラジン環、ピリミジン環、ピリダジ ン環、又はトリアジン環である。 Bにおけるベンゼン環、及び芳香族へテロ環は前記 G 3群から選択される基で置換されていてもよいが、好ましくは、無置換である場合、或 いは、ハロゲン、低級アルキル、 -0-低級アルキル、 -OH、 -低級アルキレン- OR°、 - CN、 -C(0)-N(R°)、 -N(R°)、及び _NR°_C(0)-低級アルキルからなる群から選択さ れる 1個の置換基で置換されている場合であり、より好ましくは無置換である場合、又
は 1個のハロゲンで置換されている場合である。
(7) R4としては、好ましくは R4aであり、更に
(i) n= l、かつ、 B =単結合の場合、
より好ましくは- c(o)-zである。ここにおいて、 Zとしては、好ましくは低級アルキル、 シクロアルキル、ヘテロァリール、又は-低級アルキレン- 0-低級アルキルであり、より 好ましくは低級アルキル、ヘテロァリール、又は-低級アルキレン- 0-低級アルキルで あり、更に好ましくはメチル、ピリジル、又はメトキシメチルである。
(ii) n= l、かつ、 B =上記 G3群から選択される基で置換されていてもよい 5又は 6員 芳香族含窒素へテロ環の場合、
より好ましくは H、又は含窒素へテロ環基であり、更に好ましくは Hである。
(iii) n = 0の場合、
より好ましくは無置換又は- NHで置換された、環の構成原子である Nに結合手を 有する 5員芳香族含窒素へテロ環基であり、更に好ましくは 1H-イミダゾール -1-ィル 、 3-ァミノ- 4H-1, 2,4-トリァゾール -4-ィル、又は 5-ァミノ- 1H-ピラゾール -1-ィルであ 本発明化合物 (I)の特に好ましい態様としては、上記 (1)〜 )に記載の各好ましい基 の組合わせからなる化合物である。また、本発明化合物 (I)の別の好ましい態様として は、式 (I-A)、式 (Ι-Β)、式 (I_C)、又は式 (I-D)で示される化合物である。
本発明の化合物は、置換基の種類によっては他の互変異性体や幾何異性体が存 在する場合もある。本明細書中、それら異性体の一形態のみで記載することがあるが 、本発明にはこれらの異性体も包含し、異性体の分離したもの、あるいは混合物も包 含する。
また、化合物 0)は不斉炭素原子や軸不斉を有する場合があり、これに基づく (R)体、 (S)体などの光学異性体が存在しうる。本発明はこれらの光学異性体の混合物や単 離されたものを全て包含する。
更に、本発明には、化合物 0)の薬理学的に許容されるプロドラッグも含まれる。薬 理学的に許容されるプロドラッグとは、加溶媒分解により又は生理学的条件下でアミ ノ基、 OH、 CO H等に変換できる基を有する化合物である。プロドラッグを形成する基
としては、例えば、 Prog. Med., 5, 2157-2161 (1985)や「医薬品の開発」(廣川書店、 1 990年)第 7巻 分子設計 163-198に記載の基が挙げられる。
[0021] また、本発明化合物は、置換基の種類によって、酸付加塩又は塩基との塩を形成 する場合もあり、力、かる塩が製薬学的に許容され得る塩である限りにおいて本発明に 包含される。具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等 の無機酸や、ギ酸、酢酸、プロピオン酸、シユウ酸、マロン酸、コハク酸、フマル酸、マ レイン酸、乳酸、リンゴ酸、酒石酸、クェン酸、メタンスルホン酸、エタンスルホン酸、 p -トルエンスルホン酸、ァスパラギン酸、又はグルタミン酸等の有機酸との酸付加塩、 ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機塩基、メチルァ ミン、ェチルァミン、エタノールァミン、リシン、オル二チン等の有機塩基との塩やアン モユウム塩等が挙げられる。
更に、本発明は、本発明化合物及びその製薬学的に許容される塩の各種の水和 物や溶媒和物、及び結晶多形の物質をも包含する。また、本発明は、種々の放射性 又は非放射性同位体でラベルされた化合物も包含する。
[0022] 本明細書における「頻尿」とは、排尿回数が正常範囲を越えて増加した状態のこと をいう。また前記「尿失禁」とは、社会的、衛生的に問題となる不随意の尿漏出状態 のことをいう。
本明細書における「過活動膀胱」とは、頻尿及び/又は尿意切迫感といった自覚症 状によって診断される症候群のことをいう(非特許文献 1)。その発症原因として神経 障害 (例えば神経因性膀胱、脳梗塞に起因するもの)、下部尿路閉塞 (例えば前立 腺肥大)、加齢などがあり、これらに共通する発症メカニズムとして、カブサイシン感受 性求心性神経の活動亢進が考えられて!/、る。
頻尿、尿失禁、尿意切迫感などの症状を改善することにより、過活動膀胱を治療す ること力 Sできる。それは、例えば抗コリン薬である塩酸ォキシプチニン(日本標準商品 分類番号 87259 ;アベンテイスファーマ株式会社)を過活動膀胱の患者に 1日 3回、 1 回 2〜3 mg投与することにより頻尿、尿失禁、尿意切迫感などの症状を改善し、過活 動膀胱を治療できることからも明らかである。
[0023] 頻尿、尿失禁、及び/又は過活動膀胱の治療効果があることの確認は、当業者に
公知の方法、あるいはそれを改良した方法を用いることにより実施することができる。 例えば、ラット、モルモット、ィヌ等にシクロフォスフアミド(CPA)を 50〜200 mg投与す ることにより誘発する病態モデルが当分野においては非常によく用いられる(Ozawaら 、 The Journal of Urology,第 162巻、第 2211-2216頁、 1999年; Boucherら、 The Journ al of Urology,第 164巻、第 203-208頁、 2000年)。このモデルは出血性膀胱炎に伴う 病態モデルである力 S、この頻尿発症機序にカブサイシン感受性求心性神経が関与 することから、本モデルは神経因性膀胱を含む各種過活動膀胱に即した病態モデル で ¾?·0と考えられる (Carlo Alberto Maggiら、 Journal of the Autonomic Nervous Syste m、第 38巻、第 201-208頁、 1992年)。頻尿状態は有効膀胱容量の減少により確認す ることができる。この病態モデル動物に対し、有効用量の医薬組成物を経口、腹腔内 又は静脈内投与で、単回又は反復投与することにより、有効膀胱容量の増加をもつ て頻尿、尿失禁、及び/又は過活動膀胱治療効果を確認することができる。
(製造法)
本発明化合物及びその製薬学的に許容され得る塩は、その基本骨格あるいは置 換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することが できる。その際、官能基の種類によっては、当該官能基を原料乃至中間体の段階で 適当な保護基 (容易に当該官能基に転化可能な基)に置き換えておくことが製造技 術上効果的な場合がある。このような官能基としては例えばアミノ基、水酸基、カルボ キシル基等であり、それらの保護基としては例えばグリーン (Greene)及びウッツ (Wuts) 著、「Protective Groups in Organic Synthesis (第 3版、 1999年)」に記載の保護基等を 挙げること力 Sでき、これらを反応条件に応じて適宜選択して用いればよい。このような 方法では、当該保護基を導入して反応を行った後、必要に応じて保護基を除去する ことにより、所望の化合物を得ることができる。
また、化合物 0)のプロドラッグは上記保護基と同様、原料乃至中間体の段階で特定 の基を導入、あるいは得られた化合物 (I)を用い反応を行うことで製造できる。反応は 通常のエステル化、アミド化、脱水等、当業者により公知の方法を適用することにより fiうこと力 Sでさる。
以下、本発明化合物の代表的な製造法を説明する。なお、本発明の製造法は以
下に示した例には限定されない。
(文章中の記号は次の通りである。
DMF: N,N-ジメチルホルムアミド; DMSO:ジメチルスルホキシド; THF:テトラヒドロフラン ; Tol:トルエン; EtOAc:酢酸ェチル; TEA:トリェチルァミン。以下同様。 )
また、本発明化合物中に同様な置換基が当該製造法の反応式中以外の位置に存 在している場合は置換基修飾反応により、容易に本発明に包含される化合物を製造 できる。
(製法 1)
[化 8]
D
(l-a)
(式中、 Dは本反応において有利な脱離基又は- OHを示す。以下同様。 ) 本製法は、カルボン酸誘導体 (Π)からイソシァネート (III)を調製し、次いで、環状アミ ン誘導体 (IV)を作用させることにより、本発明化合物 (I)において、 n= lで、かつ、 R3 が H、かつ、 Bがそれぞれ置換されていてもよいベンゼン環又は芳香族へテロ環であ るゥレア化合物 (I-a)を製造する方法である。 Dの脱離基としては、ハロゲン等が挙げ られる。イソシァネート (III)の調製は、 Dがハロゲン等である場合、例えば、ァセトニトリ ルのような反応に不活性な溶媒中、アジ化ナトリウムを冷却下〜室温下作用させ、一 且、対応する酸アジドを形成させた後、分液操作により過剰のアジ化ナトリウムを除去 し、溶媒を Tolのような反応に不活性な溶媒に置換し、加熱 (好ましくは、 110°C前後) することで行われる。イソシァネート (III)の調製は、 Dカ OHである遊離のカルボン酸 を原料として、ジフエニルホスホリルアジド (DPPA)を TEAのような有機塩基の存在下、 Tolのような反応に不活性な溶媒中で冷却下〜室温下作用させ、次いで上記と同様 にして行うこともできる。イソシァネート (III)と環状アミン誘導体 (IV)との反応は上記のよ
うに調整したイソシァネート (III)の Tol溶液を用いて、 THF等の反応に不活性な溶媒 中で冷却下〜加熱還流下に行うことができる。また、イソシァネート (III)が市販されて いる場合は、これを使用して行うこともできる。
[0026] (製法 2)
[化 9]
(V) (I)
(式中、 Eは本反応において有利な脱離基を示す。)
本製法は、含窒素化合物に TEA、ピリジン等の有機塩基の存在下又は非存在下、 ジクロロメタン、 THF等の反応に不活性な溶媒中、冷却下〜室温下で、トリホスゲン、 1,1'-カルボニルジイミダゾール(CDI)、クロロギ酸フエニル、又はクロ口ギ酸ェチルを 作用させて調製される脱離基を有する化合物 (V)と環状アミン誘導体 (IV)を反応させ 、本発明化合物 (I)を製造する方法である。化合物 (V)は単離して反応に用いる場合も 、反応系中で調整して単離することなく反応に用いる場合もあるが、 Eの脱離基として は、クロ口、イミダゾリル、フエノキシ、又はエトキシが挙げられる。反応は、ジクロロメタ ン、 DMF、 DMSO、 THF、ジォキサンのような反応に不活性な溶媒中、冷却下〜加熱 還流下で行われる。
[0027] (製法 3)
[化 10]
(1 )
(1)本製法は、環状アミン誘導体 (VI)とカルボン酸 (VII)又はその反応性誘導体を反 応させ、本発明化合物 (I)において、 Xが Nで、かつ、 Lが- C(O)-である化合物 (I-b)を 製造する方法である。反応は縮合剤(例えば、ジシクロへキシルカルポジイミド (DCC
)、ジイソプロピルカルポジイミド(DIPC)、 1-ェチル -3-(3_ジメチルァミノプロピル)カル ポジイミド (WSC)、 1,1'-カルボニルジイミダゾール(CDI)等)、場合によっては、更に 添加剤(例えば、 N-ヒドロキシスクシンイミド(HONSu)、 1-ヒドロキシベンゾトリァゾー ノレ(HOBt)、 4-(N,N-ジメチルァミノ)ピリジン(DMAP)や TEAのような有機塩基等)の 存在下行うことができる。カルボン酸の反応性誘導体としては、酸ノヽライド、酸無水物 、活性エステル等が使用できる。反応は、例えば日本化学会編「実験化学講座 (第 4 版)」 22巻(1992年)(丸善)等に記載の方法により行うこともできる。
(2)本製法は、環状アミン誘導体 (VI)とスルホユルクロリド (VIII)を反応させ、本発明化 合物 (I)において、 Xが Nで、かつ、 Lカ S(O) -である化合物 (I-c)を製造する方法で ある。反応は、例えば、前記の「プロテクティブ'グループス'イン'オーガニック 'シン セシス(Protective Groups in Organic Synthesis)」に記載のスノレホニノレ化の条件が適 用できる。具体的には、 THF、ジクロロメタン、ァセトニトリル等の溶媒中、必要により T EA、ピリジン等の塩基の存在下、冷却下〜加熱還流下にて行うことができる。
(製法 4)
本製法は、環状アミン誘導体 (VI)とアルデヒド (IX)で還元的アルキル化して、本発明 化合物 (I)において、 Xが Nで、かつ、 L力 Sメチレンである化合物 (I-d)を製造する方法 である。反応は、例えば日本化学会編「実験化学講座 (第 4版)」 20巻(1992年)(丸善 )
P.300等に記載の方法が適用できる。具体的には、無溶媒中またはジクロロメタン、 1 ,2-ジクロロェタン、クロ口ホルム等のハロゲン化炭化水素類、ベンゼン、 Tol、キシレン 等の芳香族炭化水素類、 EtOAc等のエステル類、ジェチルエーテル、 THF、ジォキ サン等のエーテル類、メタノール、エタノール等のアルコール類、酢酸等の反応に不 活性な溶媒中、水素化ホウ素ナトリウム (NaBH )、ナトリウムシァノポロヒドリド (NaBH C
4 3
N)、トリァセトキシ水素化ホウ素ナトリウム (NaBH(OAc) )等の還元剤を用い冷却下、冷
3
却下〜加熱還流下に行うのが好適である。化合物によっては硫酸、塩酸、臭化水素 酸等の鉱酸、ギ酸、酢酸等の有機酸、又は四塩化チタン等のルイス酸存在下反応を 行うことが有利な場合がある。また、例えば触媒として、パラジウム炭素、ラネ一二ッケ ル、白金等を用い、常圧乃至加圧の水素雰囲気下、前述の芳香族炭化水素類、ェ ステル類、エーテル類、ハロゲン化炭化水素類、 DMF、 N,N-ジメチルァセトアミド(D MA)、 N-メチルピロリドン (NMP)、ァセトニトリル、酢酸等反応に不活性な溶媒中、室 温下〜加熱還流下に行うことができる。
(製法 5)
[化 12]
(l-e)
(式中、 R1Aは前記 G1群で置換されていてもよいァリール-低級アルキレン-、及びシク 口アルキル-低級アルキレン-を示す。)
本製法は、フエノール誘導体又は水酸基を有する芳香族へテロ環誘導体 (X)を脱 離基を有する化合物 (XI)を用いてアルキル化することにより、本発明化合物 (I)にお V、て、 R1が前記 G1群で置換されて!/、てもよ!/、ァリール-低級アルキレン- 0-又はシク 口アルキル-低級アルキレン- 0-である化合物 (I-e)を製造する方法である。 Dの脱離 基は、求核置換反応において常用される脱離基であればいずれでもよぐクロ口、ブ ロモ等のハロゲン、メタンスルホニルォキシ、 p-トルエンスルホニルォキシ、トリフノレオ ロメタンスルホニルォキシ等のスルホニルォキシ、低級アルキルスルホニル、ァリール スルホニル等のスルホニル等が好適に用いられる。本工程のアルキル化反応は当業 者が通常用いうるアルキル化を採用することができる。例えば、ベンゼン、 Tol、キシレ ン等の芳香族炭化水素類、 EtOAc等のエステル類、ジェチルエーテル、 THF、ジォ キサン等のエーテノレ類、ジクロロメタン、 1,2-ジクロロェタン、クロロホノレム等のハロゲ ン化炭化水素類、 DMF、 DMA, NMP、 DMSO、ァセトニトリル等の反応に不活性な溶 媒、あるいはアルコール類等の溶媒中、室温〜加熱還流下に行うことができる。化合 物によっては、有機塩基(TEA、ジイソプロピルェチルァミン、 N-メチノレモノレホリン、ピ リジン、 DMAP等が好適に用いられる)、又は金属塩塩基 (炭酸カリウム、炭酸セシゥ ム、水酸化ナトリウム、水酸化カリウム、水素化ナトリウム、 tert-ブトキシカリウム等が好 適に用いられる)の存在下に行うことが、反応を円滑に進行させる上で有利な場合が ある。
[0030] また、式 (XI)において Dカ OHである化合物を用いて、 THF、ジォキサン、ジェチ ルエーテル等のエーテル類、塩化メチレン、クロ口ホルム等のハロゲン化炭化水素類
、 Tol、ベンゼン等の芳香族炭化水素類、 DMF等の溶媒中、トリフエニルホスフィン (Ph P)、トリ- n-ブチルホスフィン等のホスフィン類、及びジェチルァゾジカルボキシレート(
、冷却下〜室温下にて行うこともできる。
[0031] (原料合成 1)
[化 13]
(I
(xiii) (xiv) (XVII) (XVIII) L=CO, X=N L=0, X=CH L=NH, X=CH
(式中、 Pはァミノ基の保護基を示し好ましくは Boc基である。また、 D2は脱離基を示し 好ましくはメタンスルホニルォキシ基である。以下同様。 )
環状アミン誘導体 (IV)は L及び Xの態様によって種々の方法によって製造すること ができる。例えば、アルデヒド誘導体(IX)及びアミン (XII)の還元的ァミノ化、カルボン 酸 (VII)又はその反応性誘導体及びアミン (XII)のアミド化、化合物(XIII)及びアルコ ール(XIV)の光延反応、化合物(XV)及び化合物(XVI)の S-アルキル化、並びに化 合物 (XVII)及びケトン (XVIII)の還元的ァミノ化等によって製造することができる。
(A. R. Katritzky及び R. J. K. Taylor 、「Comprehensive Organic Functional roup Transrormations II」、 2 、 Elsevier Perg腿 on、 2005牛
日本化学会編「実験化学講座 (第 5版)」 14巻 (2005年) (丸善)
R. sandier及び W. Karo 、「 Organic functional Group Preparations」、 版、 ^ 1巻、 Academic Press Inc.、 1991年)
(原料合成 2)
[化 14]
(V)
(VI)
環状アミン誘導体 (VI)は化合物 (V)及びアミン (XIX)を反応させ、次!/、でアミノ基を 脱保護することにより製造すること力でさる。
[0033] さらに、式 (I)で示されるいくつかの化合物は、以上のように製造された本発明化合 物から、後述の実施例に記載した方法及びそれらの変法、又は公知のアルキル化、 ァシル化、置換反応、酸化、還元、加水分解、脱保護等、当業者が通常採用しうるェ 程を任意に組み合わせることにより製造することもできる。
[0034] 本発明化合物は、遊離化合物、その製薬学的に許容される塩、水和物、溶媒和物 、あるいは結晶多形の物質として単離され、精製される。本発明化合物 (I)の製薬学 的に許容される塩は、常法の造塩反応に付すことにより製造することもできる。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等通常の化学操作 を適用して行われる。
各種の異性体は、適当な原料化合物を選択することにより、あるいは異性体間の物 理化学的性質の差を利用して分離することができる。例えば、光学異性体は一般的 な光学分割法 (例えば、光学活性な塩基又は酸とのジァステレオマー塩に導く分別 結晶化やキラルカラム等を用いたクロマトグラフィー等)により、立体化学的に純粋な 異性体に導くことができる。また、適当な光学活性な原料化合物より製造することもで きる。
[0035] 本発明化合物の薬理活性は以下の試験により確認した。
試験例 1 ラット脳破砕液を用レ、た FAAH活性を阻害する物質のスクリ一ユング
(1)ラット脳破砕液の調製
10週齢の SD系雄性ラット(日本 SLC社)をエーテル麻酔下にて断頭し、大脳を摘出 し重量を測定した。重量の 5倍容の氷冷した緩衝液(50mM Tris-HCl (pH7.4)、 0.32 M Sucrose)を加え、氷中でホモジナイザーにより均一な懸濁液になるまで摩砕した。 遠心分離 (1500 X g、 4°C、 15分間)後,その上清をさらに遠心分離(15000 X g、 4°C、 20分間)し沈殿を得た。さらに超音波発生機 (UR-20P ;トミー精ェ社)により、 5秒間、 超音波破砕 (Power dial 4)した。得られた破砕液の蛋白質濃度を色素結合法 (プロ ティンアツセィ CBB溶液;ナカライテスタ社)により測定した。緩衝液(50mM Tris-HCl (pH 8.0) , ImM EDTA、 O. lmg/ml BSA、 lOOmM NaCl)を用いて、蛋白質の濃度が 60
μ g/mlになるようにラット脳破砕液を希釈し酵素液を調製した。
(2) FAAH活性を阻害する物質のスクリーニング
2 μ Ci/ml放射標識アナンダミド(Anandamide [ethanolamine l- ] (American Radio labeled Chemical社))、 8 Mアナンダミド(フナコシ社)、 50mM Tris_HCl (pH 8.0)、 ImM EDTA、 O. lmg/ml BSA及び lOOmM NaCU:りなる基質液を調製した。 1ηΜ〜100 11 Mになるように DMSOに溶解した試験物質溶液を調製した。 50 1の酵素液に 50 1 の上記基質液、 1 1の試験物質溶液を加え、 1時間放置した。また、コントロールとし ては DMSOを試験物質溶液の代わりに添加した。これに 200 1のクロ口ホルムとメタノ ールの 1: 1 (容量比)溶液を加え攪拌した。遠心分離(15000回転/分、 2分間)すること により、上層(水/メタノール層)に分解産物のエタノールァミン(ethanolamine 1_3H) 、下層(クロ口ホルム層)に未反応の放射標識アナンダミド(Anandamide [ethanolam ine 1-3H])が分離された。上層の 30 1を 96ゥエルの有機溶媒耐性白色マイクロプレ ート(PicoPlate-96; PerkinElmer社)に移し、 150 μ 1のマイクロシンチ 20 (PerkinElmer 社)を加えマイクロプレートシンチレーシヨンカウンター(TopCount™; Beckman社)に て測定した。コントロールと比較して測定値を減少させる物質を、 FAAH活性を阻害 する物質として選択した。
(3) FAAH活性阻害物質の IC 値の測定
化合物を lnM lOO Mになるように DMSOに溶解して試験物質溶液を調製し、上 記に記載の方法で、 FAAH活性に及ぼす影響を調べた。コントロールとして DMSOを 用いた。各測定値より、酵素液の代わりに緩衝液(50mM Tris-HCl (pH 8.0)、 ImM E DTA、 O. lmg/ml BSA、 lOOmM NaCl)を用いて反応させた場合の測定値を差し引い た。コントロールの測定値を 100%として IC 値を求めた。上記測定結果を表 1に示す。 なお、 Exは後記実施例番号を示す。
[表 1]
Ex I C50 (nM)
30 30
38 5. 0
155 25
210 3. 3
334 42
439 25 試験例 2 ヒト膀胱上皮癌由来細胞を用レ、た FAAH活性を阻害する物質のスクリ一二 ング
(1) FAAH活性を阻害する物質のスクリーニング
ヒト膀胱上皮癌由来細胞株 5637細胞(HTB-9; ATCC)を 48ゥエルの細胞培養プレ ートに 1ゥエルあたり 1X105個、 10%ゥシ胎児血清(HyClone社)を含有する RPMI1640培 地(Invitrogen社)を用いて播種した。 37°Cで 12時間以上培養した後、細胞を 1ゥエル あたり 400〃 1の緩衝液(Hank' s Balanced Salt Solution, 20mM Hepes-NaOH (pH 7.4 ) )で洗浄した。基質液(3 Ci/ml放射標識アナンダミド(Anandamide [ethanolamine 1-3H])、 10 μ Μアナンダミドを含む前記緩衝液)に DMSOに溶解した試験物質を 0.00 3ηΜ〜30ηΜになるように添加した。コントロールとして DMSOのみを添加した。上記細 胞に 1ゥエルあたり 100 1の基質液を加え、 COインキュベーター内で、 37°Cで 30分 間インキュベートした。その後、細胞培養プレートを氷上に移し、基質液を吸引除去 し、 1ゥエルあたり 75 1の氷冷した細胞溶解用の溶液(0.5% TritonX-100, 10 \の? AAH阻害活十生を するィ匕合物 cyclohexylcarbamic acid 3 -carbamoylbiphenyl-3-yl e ster (URB597 ; Cayman chemical社; Kathuriaら、 Nature Med. ,第 9巻、第 76-81頁、 20 03年)を含む前記緩衝液)を加え攪拌した。得られた細胞溶解液をゥエルごとに 1.5 m 1容のサンプルチューブに移し、 150 1のクロ口ホルムとメタノールの 1 : 1 (容量比)溶液 を加え攪拌した。遠心分離(15000回転/分、 2分間)すると、上層(水/メタノール層) に分解産物のエタノールァミン(ethanolamine 1_3H)力 下層(クロ口ホルム層)に未 反応の放射標識アナンダミドが分離される。上層の 25 1を 96ゥエルの有機溶媒耐性 白色マイクロプレート(PicoPlate_96 ; PerkinElmer社)に移し、 150 μ 1のマイクロシンチ 20 (PerkinElmer社)を加えマイクロプレートシンチレーシヨンカウンター(TopCount™;
Beckman社)にて測定した。コントロールと比較して測定値を減少させる物質を、 FAA H活性を阻害する物質として選択した。
(2) FAAH活性阻害物質の IC 値の測定
DMSOに 10mMになるように溶解した化合物を 0·003ηΜ〜30ηΜになるように、基質 液に加え、上記に記載の方法で FAAH活性に及ぼす影響を調べた。ネガティブコント ロールとして DMSOを、ポジティブコントロールとして URB597を 10 Μになるように基 質液に添加し、ポジティブコントロールの測定値を 0%、ネガティブコントロールの測定 値を 100%として IC 値を求めた。上記測定結果を表 2に示す。なお、 Exは後記実施例 番号を示す。
[表 2]
試験例 3 試験物質を投与したラットの組織破砕液を用レ、た FAAH活性を阻害する物 質のスクリーニング
(1)ラットへの投与、及び組織破砕液の調製
2匹の 9週齢の Wistar系雄性ラット(日本 SLC社)に 0.5%メチルセルロース(MC)溶液 に懸濁した試験物質を l〜3mg/kgで経口投与した。コントロールとして 2匹のラットに は 0.5% MC溶液を経口投与した。 30分後に、エーテル麻酔下にて下大動脈から血液 を採取した。その後、断頭し、大脳を採取した。
採取した血液 3mlを等量の生理食塩水で希釈し、遠心チューブ内の 3mlの血球分 離剤(Nycopl印; AXIS-SHIELD社)の上に静かに重層した。遠心分離(400 X g、 20分
間)し、単球層を採取した。得られた単球を生理食塩水で 2回洗浄し、測定まで- 20°C で凍結保存した。
採取したラット脳に、重量の 5倍容の氷冷した緩衝液(50mM Tris-HCl (pH 8.0)、 lm M EDTA)を加え、氷中でホモジナイザーにより、均一な溶液になるまで摩砕した。さ らに超音波発生機(UR-20P (Power dial 4) ;トミー精ェ社)により、 5秒間、超音波破砕 した。前記の凍結保存した単球には、氷冷した緩衝液(50mM Tris-HCl (pH 8.0)、 1 mM EDTA)を 100 1加え、超音波発生機(UR_20P(Power dial 4) ;トミー精ェ社)によ り 5秒間、超音波処理した。脳、単球の破砕液について、色素結合法(プロテインアツ セィ CBB溶液;ナカライテスタ社)により蛋白質濃度を測定した。脳、単球の破砕液を 緩衝液(50mM Tris-HCl (pH 8.0) , ImM EDTA, 0. lmg/ml BSA、 lOOmM NaCl)を用 いて、蛋白質濃度がそれぞれ 80 g/ml、 400 g/mlになるように希釈し酵素液とした
〇
(2) FAAHの活性の測定
50 ,1 1の酵素液に 50 ,1 1の基質液(2 ,1 Ci/ml放射標識アナンダミド (Anandamide [et hanolamine 1-°H] (American Radiolabeled Chemical社) )、 8 μ Mアナンダミド (フナコ シ社)、 50mM Tris-HCl (pH8.0)、 ImM EDTA)を加え室温にて 1時間反応させた。 20 0 1のクロ口ホルムとメタノールの 1 : 1 (容量比)溶液を加え攪拌した。遠心分離(12000
X g、 2分間)により、上層(水/メタノール層)に分解産物のエタノールァミン(ethanola mine が、下層(クロ口ホルム層)に未反応の放射標識アナンダミド (Anandamide
[ethanolamine が分離された。上層の 25 1を 96ゥエルの有機溶媒耐性白色マ イク口プレート(PicoPlate- 96; PerkinElmer社)に移し、 150 μ 1のマイクロシンチ 20 (Per kinElmer社)を加えマイクロプレートシンチレーシヨンカウンター(TopCount™; Beckma n社)にて測定した。
試験物質を投与してレ、な!/、コントロールラットの脳、単球の破砕液における FAAH 活性を 100%とし、組織破砕液を含まない緩衝液(50mM Tris-HCl (pH 8.0) , ImM ED TA、 0.1 mg/ml BSA、 lOOmM NaCl)の FAAH活性を 0%とし、試験物質を投与したラッ ト組織破砕液の FAAH活性の相対値 (%)を求めた。 FAAH活性の相対値を低下させ る物質を、 FAAH活性を阻害する物質として選択した。
以上の結果より、試験物質を実験動物に投与した後、摘出した組織の破砕液にお ける試験物質依存的な FAAH活性の変化を測定することにより、 FAAH活性を阻害す る物質をスクリーニングできることが示された。
[0040] 試験例 4 シクロフォスフアミド(CPA)誘発頻尿ラットに対する化合物の作用
化合物の膀胱刺激症状改善作用を病態モデルを用いて検討した。シクロフォスファ ミド(CPA)は全身投与により代謝物であるァクロレインに変換され、尿中から膀胱粘 膜を傷害することが知られている。ラットにおいては、 CPA投与により出血性膀胱炎に 伴う膀胱痛あるいは頻尿状態が誘発されるため、これら症状に対する薬効評価が可 能である。実験には 9週齢の Wistar系雌性ラット(チヤ一ルスリバ一社)を用いた。 CPA (100mg/kg)を腹腔内投与し、その 2日後に実験を行った。化合物を経口投与 (ρ·ο·) し 15分後に、蒸留水(30ml/kg)を強制的に経口投与した。ラットを代謝ケージに入れ 、排尿重量を 1時間連続的に測定した。総排尿量を総排尿回数で割ることにより、有 効膀胱容量を算出した。その結果、溶媒である 0.5%メチルセルロース(MC)投与群 においては有効膀胱容量が減少し、頻尿状態が認められた。有効経口投与量は、 例えば、実施例 155、及び 210の化合物ではそれぞれ 10mg/kg、及び 3mg/kgであり 、減少した有効膀胱容量を増加させ、頻尿状態を改善した。
[0041] 以上の各試験の結果より、本発明化合物は、良好な FAAH阻害作用を有すること、 及び減少した有効膀胱容量を増加させ、頻尿状態を改善することが確認された。こ のことから、本発明化合物は、 FAAHが関与する疾患、特に頻尿、尿失禁、及び/又 は過活動膀胱の治療剤として有用であることは明らかである。
更に、本発明化合物は良好な FAAH阻害作用を有するため、(1)精神神経疾患( 不安 *鬱*てんかん等)、(2)脳傷害,神経変性疾患 (頭部外傷 ·脳虚血 ·認知症 (痴 呆)等)、(3)疼痛、 (4)免疫,炎症性疾患、(5)嘔吐、(6)摂食障害、(7)過敏性腸 症候群 ·潰瘍性大腸炎、(8)高血圧、(9)緑内障、又は(10)睡眠障害の治療剤とし ても有用である。
[0042] 本発明化合物 (I)又はその塩の 1種又は 2種以上を有効成分として含有する製剤は 、当分野において通常用いられている薬剤用担体、賦形剤等を用いて通常使用され てレ、る方法によって調製することができる。
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関 節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、 経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれ の形態であってもよい。
本発明による経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用 いられる。このような固体組成物においては、 1種又は 2種以上の有効成分を、少なく とも 1種の不活性な賦形剤、例えば乳糖、マンニトール、ブドウ糖、ヒドロキシプロピル セルロース、微結晶セルロース、デンプン、ポリビュルピロリドン、及び/又はメタケイ 酸アルミン酸マグネシウム等と混合される。組成物は、常法に従って、不活性な添カロ 剤、例えばステアリン酸マグネシウムのような潤滑剤やカルボキシメチルスターチナト リウム等のような崩壊剤、安定化剤、溶解補助剤を含有していてもよい。錠剤又は丸 剤は必要により糖衣又は胃溶性若しくは腸溶性物質のフィルムで被膜してもよい。 経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、 シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例え ば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化 剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有してい てもよい。
非経口投与のための注射剤は、無菌の水性又は非水性の溶液剤、懸濁剤又は乳 濁剤を含有する。水性の溶剤としては、例えば注射用蒸留水又は生理食塩液が含ま れる。非水溶性の溶剤としては、例えばプロピレングリコール、ポリエチレングリコール 又はオリーブ油のような植物油、エタノールのようなアルコール類、又はポリソルベー ト 80 (局方名)等がある。このような組成物は、さらに等張化剤、防腐剤、湿潤剤、乳 化剤、分散剤、安定化剤、又は溶解補助剤を含んでもよい。これらは例えばバタテリ ァ保留フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。また、 これらは無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶 解又は懸濁して使用することもできる。
外用剤としては、軟膏剤、硬膏剤、クリーム剤、ゼリー剤、パップ剤、噴霧剤、ローシ ヨン剤、点眼剤、眼軟膏等を包含する。一般に用いられる軟膏基剤、ローション基剤
、水性又は非水性の液剤、懸濁剤、乳剤等を含有する。例えば、軟膏又はローション 基剤としては、ポリエチレングリコール、プロピレングリコール、白色ワセリン、サラシミ ッロウ、ポリオキシエチレン硬化ヒマシ油、モノステアリン酸グリセリン、ステアリルアル コール、セチルアルコール、ラウロマクロゴール、セスキォレイン酸ソルビタン等が挙 げられる。
吸入剤や経鼻剤等の経粘膜剤は固体、液体又は半固体状のものが用いられ、従 来公知の方法に従って製造することができる。例えば公知の賦形剤や、更に、 pH調 整剤、防腐剤、界面活性剤、滑沢剤、安定剤や増粘剤等が適宜添加されていてもよ い。投与は、適当な吸入又は吹送のためのデバイスを使用することができる。例えば 、計量投与吸入デバイス等の公知のデバイスや噴霧器を使用して、化合物を単独で 又は処方された混合物の粉末として、もしくは医薬的に許容し得る担体と組み合わせ て溶液又は懸濁液として投与することができる。乾燥粉末吸入器等は、単回又は多 数回の投与用のものであってもよぐ乾燥粉末又は粉末含有カプセルを利用すること 力できる。あるいは、適当な駆出剤、例えば、クロロフノレォロアルカン、ヒドロフルォロ アルカン又は二酸化炭素等の好適な気体を使用した加圧エアゾールスプレー等の 形態であってもよい。
[0044] 通常経口投与の場合、 1日の投与量は、体重当たり約 0.001〜100mg/kg、好ましく は 0.1〜30mg/kg、更に好ましくは 0.1〜10mg/kgが適当であり、これを 1回であるいは 2 乃至 4回に分けて投与する。静脈内投与される場合は、 1日の投与量は、体重当たり 約 0.0001〜10mg/kgが適当で、 1日 1回乃至複数回に分けて投与する。また、経粘膜 剤としては、体重当たり約 0.001〜100mg/kgを 1日 1回乃至複数回に分けて投与する 。投与量は症状、年令、性別等を考慮して個々の場合に応じて適宜決定される。
[0045] 本発明化合物は、前述の本発明化合物が有効と考えられる疾患の種々の治療又 は予防剤と併用することができる。当該併用は、同時投与、或いは別個に連続しても しくは所望の時間間隔をおいて投与してもよい。同時投与製剤は、配合剤であっても 別個に製剤化されて!/、てもよレ、。
実施例
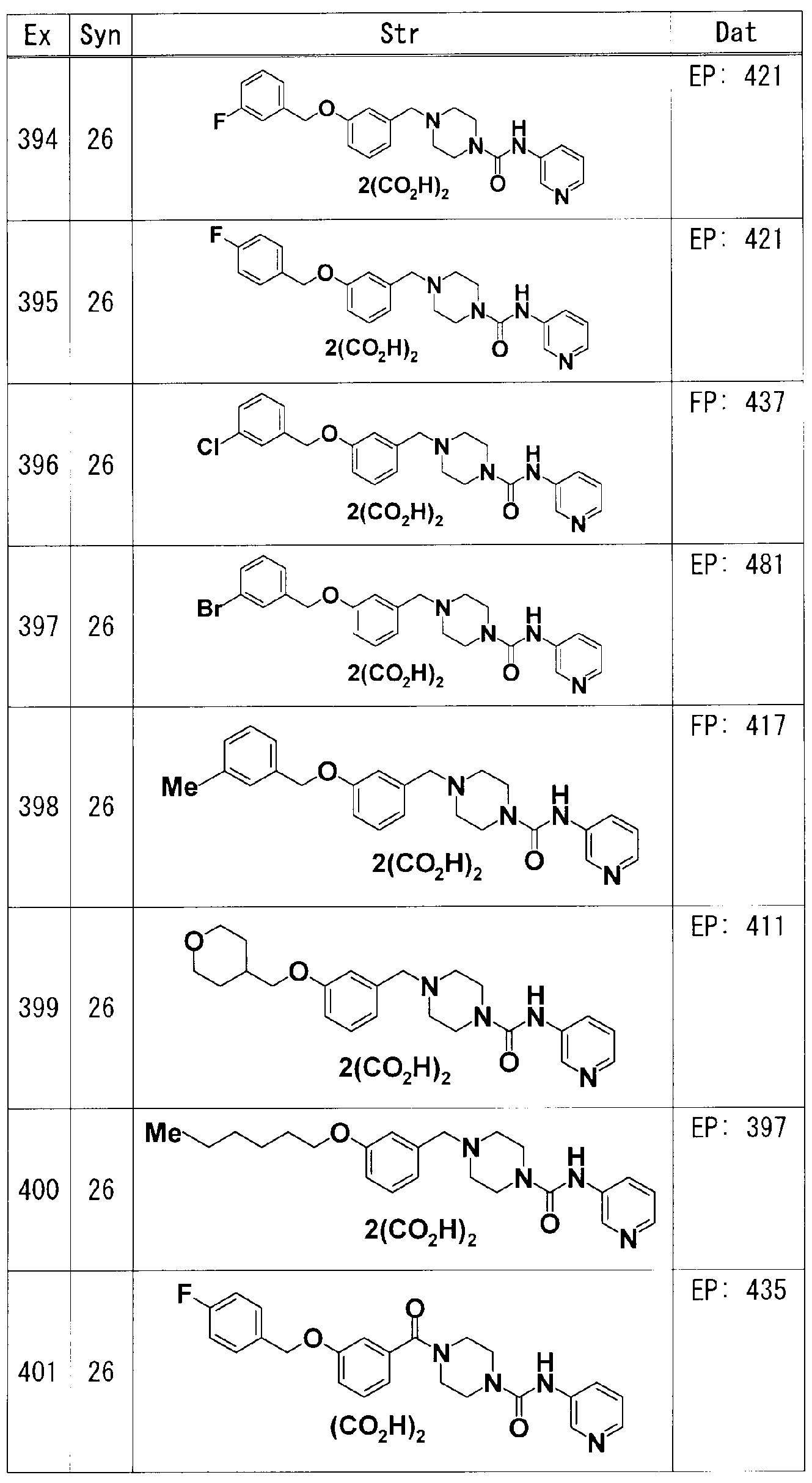
[0046] 以下、実施例に基づき本発明化合物 (I)の製法を更に詳細に説明する。本発明化
合物は下記実施例に記載の化合物に限定されるものではない。また原料化合物の 製法を参考例に示す。
また、実施例、参考例及び後記表中以下の略号を用いる。
Rf:参考例番号、 Ex :実施例番号、 Str :構造式、 Syn :製造方法 (数字は、その番号を 実施例番号として有する実施例化合物と同様に、対応する原料を用いて製造したこ とを示す。数字の前に Rが付!/、て!/、る場合はその番号を参考例番号として有する参 考例化合物と同様に、対応する原料を用いて製造したことを示す。)、 Dat :物理化学 的データ(EI:EI-MS ([M]+); EP: ESI-MS (Pos) (無記載である場合は [M+H]+); EN: ESI -MS (Neg) ([M-H]— ); ?? : ?八8_\ 5 (?03) (無記載でぁる場合は[\ + +); FN: FAB-M S (Neg) (無記載である場合は [M-H]— )); NMRl : DMSO-d中の1 H-NMRにおけるピー クの δ (ppm); NMR2 : CDC1中の1 H-NMRにおけるピークの δ (ppm); DIBAL:ジイソブ チルアルミニウムヒドリド; Pd(PPh ):テトラキス(トリフエニルホスフィン)パラジウム; pTs
OH:p-トノレエンスルホン酸; Me:メチル; Et:ェチル。
参考例 1
tert-ブチル 4-ヒドロキシピペリジン- 1-カルボキシレート(7.85g)の THF (60ml)溶液 に、 PPh (10.2g)を加え、更に氷冷下、 4-ベンジルォキシフエノール(6.01g)及び DEA
D (6.14ml、 40%Tol溶液)の THF (50ml)溶液を滴下し、室温にて 20時間攪拌した。反 応液を減圧濃縮し、得られた残渣に水酸化ナトリウム水溶液を加え、 EtOAcにて抽出 した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮した。残渣をシリカゲル力 ラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精製して、 tert-ブチ ル 4-[4- (ベンジルォキシ)フエノキシ]ピぺリジン- 1-カルボキシレート(9.13g)を得た。 得られた tert-ブチル 4-[(4-ベンジルォキシ)フエノキシ]ピぺリジン- 1-カルボキシレ ート(9.13g)のメタノール溶液に、 10%パラジウム炭素(0.913g)を加え、水素雰囲気下 、室温にて 3時間攪拌した。不溶物を濾去した後、濾液を減圧濃縮した。得られた残 渣をへキサン/ EtOAcから再結晶して、 tert-ブチル 4-(4-ヒドロキシフエノキシ)ピペリ ジン- 1-カルボキシレート(5.04g)を得た。
得られた tert-ブチル 4-(4-ヒドロキシフエノキシ)ピぺリジン- 1-カルボキシレート(3.8 2g)のァセトニトリル(50ml)溶液に、炭酸カリウム(2.70g)及び 3-フルォロベンジルブ口
ミド(2.95g)を加え、 80°Cにて 3時間攪拌した。反応液を室温まで冷却後、水を加え、 クロ口ホルムにて抽出した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮した 。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) ) で精製して、 tert-ブチル 4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1 -カルボキシレート(5.77g)を得た。
得られた tert-ブチル 4-[4-(3_フルォロベンジルォキシ)フエノキシ]ピぺリジン- 1-力 ルポキシレート(5.77g)の EtOAc (20ml)溶液に、 4M塩化水素/ EtOAc溶液(19.5ml) を加え、室温にて 5時間攪拌した。析出した固体を濾取し、 EtOAcで洗浄した。得ら れた固体を水に溶解させ、水酸化ナトリウム水溶液を加えアルカリ性とし、クロロホノレ ムにて抽出した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮して、 4-{4-[(3 -フルォロベンジル)ォキシ]フエノキシ }ピペリジン(3.56g)を得た。
参考例 2
3-ブロモ安息香酸ェチル(2.29g)及び (3-ヒドロキシフエニル)ボロン酸(1.66g)のァ セトニトリル(40ml)溶液に 0.5M炭酸ナトリウム水溶液(40ml)、 Pd(PPh ) (578mg)を順 次添加し、 90°Cにて 1時間撹拌した。反応液を室温まで冷却後、不溶物をセライトで 濾去した。濾液に水を加えて、 EtOAcにて抽出した。有機層を飽和食塩水にて洗浄 後、無水硫酸ナトリウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、淡黄色油状物(2.62g) を得た。
tert-ブチル -4-ヒドロキシピペリジン- 1 -カルボキシレート( 3 · 00g)及び PPh ( 3 · 91 g) の THF (20ml)溶液に、氷冷下、上記で得られた化合物(2.61g)及び DEAD (2.59g、 4 0%Tol溶液)の THF (lOml)溶液を滴下した後、室温にて 20時間攪拌した。反応液に 水酸化ナトリウム水溶液を加えて、 EtOAcにて抽出した。有機層を飽和食塩水にて洗 浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精製して、無色油状物(4.77g)を 得た。
得られた化合物(4.75g)の EtOAc (20ml)溶液に、 4M塩化水素/ EtOAc溶液(19.8ml )を加え、室温にて 6時間攪拌した。反応液を減圧濃縮し、得られた残渣を EtOAc/ァ
セトニトリル/エタノール混合溶媒から結晶化させて、無色粉末のェチル 3' -(ピペリジ ン- 4-ィルォキシ)ビフエニル -3-カルボキシレート塩酸塩(2.81g)を得た。
[0049] 参考例 3
4-ヒドロキシチォフエノール(1.26g)の DMA(15ml)溶液に、炭酸セシウム(3.91g)及 び tert-ブチル -4- [(メタンスルホニル)ォキシ]ピぺリジン- 1-カルボキシレート(3.35g) を添加した後、 50°Cにて 2時間加熱した。反応液を室温まで冷却後、水を加えて EtO Acにて抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減 圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2 : 1 (V/V) )で精製して、無色油状物(2.28g)を得た。
得られた化合物(1.24g)のァセトニトリル(20ml)溶液に、炭酸カリウム(829mg)及び 1 - (ブロモメチル) -3-フルォロベンゼン(832mg)を添加した後、 80°Cにて 3時間撹拌し た。反応液を室温まで冷却後、水酸化ナトリウム水溶液を加え、クロ口ホルムにて抽 出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮し た。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精製して、無色油状物(1.86g)を得た。
得られた油状物(1.85g)の EtOAc (lOml)溶液に、 4M塩化水素/ EtOAc溶液(4.94ml) を加え、室温にて 4時間攪拌した。析出した固体を濾取して、加熱乾燥し、無色粉末 の 4-({4-[(3_フルォロベンジル)ォキシ]フエ二ル}スルファニル)ピぺリジン塩酸塩(1.20 g)を得た。
[0050] 参考例 4
4-ァミノフエノール(1.09g)の DMF(40ml)溶液に、氷冷下、 tert-ブトキシカリウム(1. 35g)、 1- (ブロモメチル) -3-フルォロベンゼン(1.89g)を順次添加した後、室温にて 1 時間攪拌した。反応液に氷冷下、水を加え EtOAcにて抽出した。有機層を飽和食塩 水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラム クロマトグラフィー(溶出液;へキサン: EtOAc= 1: 1 (V/V) )で精製して、淡黄色油状 物(1.87g)を得た。
得られた油状物(1.05g)の酢酸(15ml)溶液に、 l-(tert-ブトキシカルボニル) -4-ピ ペリドン (996mg)、 NaBH(OAc) (1.27g)を順次添加した後、室温にて 3時間攪拌した。
減圧濃縮して酢酸を留去した後、得られた残渣に 5M水酸化ナトリウム水溶液を加え 、クロ口ホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮した 。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2 : l (V/V) ) で精製して、黄色油状物(1.96g)を得た。
得られた油状物( 1.96g)の酢酸(10ml)溶液に、 35%ホルマリン水溶液(2.10g)、 NaB H(OAc) (2.07g)を順次添加した後、室温にて 16時間攪拌した。減圧濃縮して酢酸を 留去した後、得られた残渣に 5M水酸化ナトリウム水溶液を加え、クロ口ホルムで抽出 した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮した。残渣をシリカゲル力 ラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、黄色油 状物(1.88g)を得た。
得られた油状物(1.88g)の EtOAc (lOml)溶液に、 4M塩化水素/ EtOAc溶液(9.05ml )を加え、室温にて 5時間攪拌した。反応液を減圧濃縮し、得られた残渣をァセトニトリ ル /メタノールから結晶化させて、淡黄色粉末の N- -K3-フルォロベンジル)ォキシ] フエ二ル},-メチルビペリジン- 4 -ァミン二塩酸塩( 1 · 29g)を得た。
参考例 5
4-(4-メトキシベンゾィル)ピぺリジン塩酸塩(1.05g)を 48%臭化水素酸水溶液(20ml) 中に加え、 19時間加熱還流した。反応液を室温まで冷却後、減圧濃縮し、淡黄色固 体を得た。
得られた固体のジォキサン(10ml) /水(5ml)混合溶液に、 TEA(1.24g)、ジ (tert-ブチ ル)ジカルボネート(984mg)のジォキサン(5ml)溶液を順次添加した後、室温にて 2時 間攪拌した。反応液に 5%クェン酸水溶液を加えて、 EtOAcにて抽出した。有機層を 飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカ ゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =24: 1 (V/V) )で精製 して、淡黄色油状物(1.36g)を得た。
得られた油状物(1.34g)のァセトニトリル(15ml)溶液に、炭酸カリウム(840mg)、 1_( ブロモメチル )-3-フルォロベンゼン(843mg)を順次添加した後、 80°Cにて 3時間撹拌 した。反応液を室温まで冷却後、水酸化ナトリウム水溶液を加えて、 EtOAcにて抽出 した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥して、減圧濃縮し
た。残渣をへキサン/ EtOAcから結晶化させて、無色粉末(1.25g)を得た。
得られた粉末(1.22g)の EtOAc (lOml)溶液に、 4M塩化水素/ EtOAc溶液(7.38ml) を加えた後、室温にて 10時間攪拌した。析出した固体を濾取して、無色粉末の {4-[(3 -フルォロベンジル)ォキシ]フエニル }(ピぺリジン- 4-ィル)メタンオン塩酸塩(l.Olg)を 得た。
参考例 6
4-ヒドロキシ安息香酸メチル (4.56g)のァセトニトリル(50ml)溶液に、炭酸カリウム(4. 98g)、 1- (ブロモメチル) -3-フルォロベンゼン(6.24g)を順次添加した後、 80°Cにて 2 時間撹拌した。反応液を室温まで冷却後、水を加えてクロ口ホルムにて抽出した。有 機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣を へキサンにて洗浄し、無色粉末の 4-[(3-フルォロベンジル)ォキシ]安息香酸メチル(6 • 92g)を得た。
4-[(3-フルォロベンジル)ォキシ]安息香酸メチル(6.92g)の THF (30ml) /メタノーノレ( 30ml)混合溶液に、 1M水酸化ナトリウム水溶液(39.9ml)を加え、 50°Cにて 2時間撹拌 した。反応液を室温まで冷却後、減圧濃縮した。残渣に 1M塩酸水溶液を加えて酸性 とし、析出した固体を濾取して無色粉末の 4-[(3-フルォロベンジル)ォキシ]安息香酸 (6.66g)を得た。
l-(tert-ブトキシカルボニル) -4-ピぺリドン(1.99g)のクロ口ホルム(30ml)溶液に、酢 酸(1.80g)、 40%メチルァミン含有メタノール溶液(2.33g)、 NaBH(OAc) (3.18g)を順次 添加した後、室温にて 3時間攪拌した。反応液に 1M水酸化ナトリウム水溶液を加えて アルカリ性とした後、クロ口ホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥し た後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム: メタノール = 21 : 4 (V/V) )で精製して、黄色油状物の tert-ブチル 4- (メチルァミノ)ピ ペリジン- 1-カルボキシレート(2.01g)を得た。
tert-ブチル 4- (メチルァミノ)ピぺリジン- 1-カルボキシレート(536mg)の THF (10ml) 溶液に、 4-[(3-フルォロベンジル)ォキシ]安息香酸(616mg)、 HOBt (169mg)、 WSC ( 527mg)を順次添加した後、室温にて 18時間攪拌した。反応液に塩酸水溶液を加え E tOAcにて抽出した。有機層を水酸化ナトリウム水溶液と水で洗浄後、無水硫酸ナトリ
ゥムで乾燥し、減圧濃縮した。残渣をへキサン/ EtOAcから結晶化させて、無色粉末 の化合物(940mg)を得た。
得られた化合物(930mg)の EtOAc (lOml)溶液に、 4M塩化水素/ EtOAc溶液(5.25 ml)を加え、室温にて 14時間攪拌した。析出した固体を濾取して、無色粉末の 4-[(3- フルォロベンジル)ォキシ] -N-メチル -N-ピぺリジン- 4-ィルベンズアミド塩酸塩(674m g)を得た。
参考例 7
4-(4-メトキシベンゾィル)ピぺリジン塩酸塩(1.05g)のジォキサン(10ml)溶液に、 TE A (1.06g)、ジ (tert-ブチル)ジカルボネート(1.09g)のジォキサン(10ml)溶液を順次添 加した後、室温にて 1時間攪拌した。反応液に 5%クェン酸水溶液を加えて EtOAcにて 抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮 して、淡黄色油状物(1.89g)を得た。
得られた油状物(1.89g)のメタノール(20ml)溶液に、 NaBH (284mg)を添加し、室
4
温にて 30分間攪拌した。反応液に水を加えた後、クロ口ホルムで抽出した。有機層を 無水硫酸ナトリウムで乾燥した後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフ ィー(溶出液;へキサン: EtOAc= 1: 1 (V/V) )で精製して、黄色油状物(1.65g)を得 た。
得られた油状物(1.63g)のエタノール(30ml)溶液に、 10%パラジウム炭素(796mg)を 添加した後、 3kgf/cm2の水素ガス雰囲気下、室温にて 17時間攪拌した。触媒を濾去 し、濾液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 4: 1 (V/V) )で精製して、無色油状物( 1 · 31 g)を得た。
得られた油状物(1.29g)を 48%臭化水素酸水溶液(20ml)に溶解させ、 24時間加熱 還流した。反応液を室温まで冷却後、減圧濃縮し、淡黄色固体を得た。
得られた固体のジォキサン(10ml) /水(5ml)混合溶液に、 TEA (1.29g)、ジ (tert-ブ チル)ジカルボネート(l.Olg)のジォキサン(5ml)溶液を順次添加した後、室温にて 30 分間攪拌した。反応液に 5%クェン酸水溶液を加えて EtOAcにて抽出した。有機層を 飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカ ゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、無
色油状物(1.05g)を得た。
得られた油状物(1.03g)のァセトニトリル(15ml)溶液に、炭酸カリウム(733mg)、 1_( ブロモメチル )-3-フルォロベンゼン(733mg)を順次添加した後、 80°Cにて 14時間撹 拌した。反応液を室温まで冷却後、水を加えてクロ口ホルムにて抽出した。有機層を 水洗後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマト グラフィー(溶出液;へキサン: EtOAc = 4 : l (V/V) )で精製して、無色油状物(1.49g) を得た。
得られた油状物(1.47g)の EtOAc (lOml)溶液に、 4M塩化水素/ EtOAc溶液(8.70ml )を添加した後、室温にて 4時間攪拌した。生成した固体を濾取して、無色粉末の 4-{ 4-[(3-フルォロベンジル)ォキシ]ベンジル }ピペリジン塩酸塩(802mg)を得た。
[0054] 参考例 8
4-[(3-フルォロベンジル)ォキシ]ァニリン塩酸塩(761mg)の THF(lOml)溶液に、 TE A (304mg)、 l-(tert-ブトキシカルボニル)ピぺリジン- 4-カルボン酸(688mg)、 HOBt (2 03mg)、 WSC (633mg)を順次添加した後、室温にて 16時間攪拌した。反応液に塩酸 水溶液を加え、 EtOAcにて抽出した。有機層を水酸化ナトリウム水溶液と飽和食塩水 で洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をへキサン/ EtOAcから 結晶化させて、無色粉末の化合物(880mg)を得た。
得られた粉末(855mg)の DMF (lOml)溶液に、氷冷下、 60%水素化ナトリウム(96mg) を加えた後、室温にて 20分間攪拌した。反応液に、ヨウ化メチル (566mg)を添加した 後、 40°Cにて 15分間撹拌した。反応液を室温まで冷却後、水を加えて EtOAcにて抽 出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮し た。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =24: 1 (V/V) )で精製して、淡黄色油状物(1.03g)を得た。
得られた油状物(l.Olg)の EtOAc (10ml)溶液に、 4M塩化水素/ EtOAc溶液(4.85ml )を加え、室温にて 4時間攪拌した。反応液を減圧濃縮し、残渣をァセトニトリル/ジェ チルエーテルから結晶化させて、無色粉末の N- -K3-フルォロベンジル)ォキシ]フ ェニル }-N-メチルビペリジン- 4-カルボキサミド塩酸塩(590mg)を得た。
[0055] 参考例 9
シクロへキシルメタノール(514mg)及び PPh (1.18g)の THF (5ml)溶液に、氷冷下、
4-ヒドロキシ安息香酸メチル(456mg)及び DEAD (784mg、 40%Tol溶液)の THF (5ml) 溶液を滴下した後、室温にて 21時間攪拌した。反応液に水酸化ナトリウム水溶液を 加えて EtOAcにて抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウム で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 4: 1 (V/V) )で精製して、無色固体(934mg)を得た。
得られた化合物(920mg)のメタノール(5ml) /THF (3ml)混合溶液に、 1M水酸化ナト リウム水溶液 (4.43ml)を加え、 50°Cにて 6時間撹拌した。反応液を室温まで冷却した 後、水酸化ナトリウム水溶液を加えて、水層を EtOAcにて洗浄した。水層を濃塩酸に て酸性とした後、クロ口ホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥した後 、減圧濃縮し、残渣をへキサン/ EtOAcから結晶化させて、無色粉末の 4- (シクロへキ シルメトキシ)安息香酸 (603mg)を得た。
[0056] 参考例 10
2,2-ジメチルプロパノール(397mg)の DMF (5ml)溶液に、氷冷下、 tert-ブトキシカリ ゥム(505mg)を加え、室温にて 30分間攪拌した。反応液に、氷冷下、 4-フルォロベン ゾニトリル(363mg)の DMF (5ml)溶液を加え、室温にて 3時間攪拌した。反応液に水 を加えて EtOAcにて抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウム で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 4: 1 (V/V) )で精製して、無色油状物(587mg)を得た。
得られた化合物(575mg)のエタノール(5ml)溶液に、 5M水酸化ナトリウム水溶液(5. 90ml)を加え、 100°Cにて 23時間撹拌した。反応液を室温まで冷却した後、減圧濃縮 した。残渣に水と 1M塩酸水溶液を加え、析出した固体を濾取し、加熱乾燥して、無 色粉末の 4-(2,2-ジメチルプロボキシ)安息香酸(560mg)を得た。
[0057] 参考例 11
3-ヒドロキシ安息香酸メチル (610mg)のァセトニトリル(10ml)溶液に、炭酸カリウム( l. l lg)、ブロモメチルシクロへキサン(1.06g)を順次添加した後、 80°Cにて 28時間撹 拌した。反応液を冷却した後、水酸化ナトリウム水溶液を加えて、クロ口ホルムにて抽 出した。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮し、残渣をシリカゲル力
ラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精製して、無色油 状物(475mg)を得た。
得られた化合物(465mg)のメタノール(4ml) /THF (4ml)混合溶液に、 1M水酸化ナト リウム水溶液(3.75ml)を加え、 50°Cにて 3時間撹拌した。反応液を室温まで冷却した 後、減圧濃縮し、得られた残渣を水に溶解させた。 1M塩酸水溶液を加えて酸性とし 、析出した固体を濾取して、加熱乾燥し、無色粉末の 3- (シクロへキシルメトキシ)安息 香酸 (371mg)を得た。
[0058] 参考例 12
4-ァミノ安息香酸ェチル(1.65g)のクロ口ホルム(30ml)溶液に、酢酸(3.00g)、 3_フ ルォ口べンズアルデヒド(1.30g)、 NaBH(OAc) (2.54g)を順次添加した後、室温にて 1
7時間攪拌した。反応液に 1M水酸化ナトリウム水溶液を加え、クロ口ホルムで抽出し た。有機層を無水硫酸ナトリウムで乾燥した後、減圧濃縮し、残渣をシリカゲルカラム クロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、淡黄色固体( 1.68g)を得た。
得られた化合物(683mg)のエタノール(5ml) /THF (5ml)混合溶液に、 1M水酸化ナ トリウム水溶液(12.5ml)を加え、 50°Cにて 24時間撹拌した。反応液を室温まで冷却し た後、減圧濃縮し、得られた残渣を水に溶解させた。 1M塩酸水溶液を加えて酸性と し、析出した固体を濾取して、加熱乾燥し、無色粉末の 4-[(3-フルォロベンジル)アミ ノ]安息香酸 (574mg)を得た。
[0059] 参考例 13
シクロへキシルメタノール(685mg)の DMF (lOml)溶液に、氷冷下、 tert-ブトキシカリ ゥム(673mg)を加え、室温にて 30分間攪拌した。反応液に、氷冷下、 2-クロ口- 4-シァ ノビリジン (693mg)の DMF (2ml)溶液を加え、室温にて 2時間攪拌した。反応液に水 を加え、 EtOAcにて抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウム で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 4: 1 (V/V) )で精製して、無色油状物(807mg)を得た。
得られた化合物(792mg)のエタノール(5ml)溶液に、 5M水酸化ナトリウム水溶液(7. 32ml)を加え、 100°Cにて 2時間撹拌した。反応液を室温まで冷却した後、減圧濃縮し
、得られた残渣を水に溶解させた。 1M塩酸水溶液を加えて酸性とし、析出した固体 を濾取して、加熱乾燥し、淡黄色粉末の 2- (シクロへキシルメトキシ)イソニコチン酸 (8 64mg)を得た。
[0060] 参考例 14
6-クロ口ニコチノ二トリノレ(508mg)及び TEA(1.02ml)のァセトニトリノレ(10ml)溶液に( シクロへキシルメチル)メチルァミン塩酸塩 (600mg)を加え 90°Cで終夜攪拌した。反応 液を減圧濃縮後、 EtOAcで希釈し、有機層を飽和炭酸水素ナトリウム水溶液で洗浄 後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣を塩基性シリカゲルカラム クロマトグラフィー(溶出液;へキサン: EtOAc= 10: 1 (V/V) )で精製し、得られた油状 物をエタノール(5ml)に溶解後、 5M水酸化ナトリウム水溶液(5ml)を加え、 100°Cで 6 時間攪拌した。反応液を室温まで冷却後、 1M塩酸を加え中和し、析出した結晶を濾 取し、加熱乾燥して、 6- [(シクロへキシルメチル )(メチル)ァミノ]ニコチン酸(530mg)を 得た。
参考例 15
シクロへキシルメタノール(594mg)及び PPh (1.36g)の THF (lOml)溶液に、氷冷下
、 5-ヒドロキシニコチン酸メチル(613mg)及び DEAD (2.26g、 40%Tol溶液)の THF (30 ml)溶液を滴下した後、室温にて 18時間攪拌した。反応液に水酸化ナトリウム水溶液 を加え、 EtOAcにて抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウム で乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 2 : 1 (V/V) )で精製して、淡黄色油状物の 5- (シクロへキシルメトキシ)ニコ チン酸メチル( 1 · 17g)を得た。
[0061] 参考例 16
2-シクロへキシルエタノール(l.OOg)の DMF (20ml)溶液に、氷冷下で tert-ブトキシ カリウム(0.972g)を加え、室温にて 30分間攪拌した。反応液に 2-クロ口イソニコチノ二 トリル(1.1 lg)を加え、室温で 3日間攪拌した。反応液に水を加え、 EtOAcで抽出した 。有機層を飽和食塩水にて洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した 。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) ) で精製して、 2-(2-シクロへキシルエトキシ)イソニコチノ二トリル(1.27g)を得た。
2-(2-シクロへキシルエトキシ)イソニコチノ二トリル(1.25g)の Tol (40ml)溶液に、 -78 °Cで DIBAL (0.543ml、 1M Tol溶液)を加え、 2時間攪拌した。反応液に DIBAL (0.109 ml、 1M Tol溶液)を追加し、さらに 2時間攪拌した。反応液に飽和塩化アンモニゥム水 溶液を加え、 EtOAcで抽出後、有機層を無水硫酸マグネシウムで乾燥し、減圧濃縮 した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/ V) )で精製して、 2-(2-シクロへキシルエトキシ)イソニコチンアルデヒド(338mg)を無色 油状物として得た。
[0062] 参考例 17
tert-ブチルピぺラジン- 1-カルボキシレート(931mg)の THF (15ml)溶液に、 4_[(3_ フルォロベンジル)ォキシ]安息香酸(1.23g)、 HOBt (338mg)、 WSC (1.05g)を順次添 加した後、室温にて 7時間攪拌した。反応液に塩酸水溶液を加え、 EtOAcにて抽出し た。有機層を水酸化ナトリウム水溶液、水で順次洗浄後、無水硫酸ナトリウムで乾燥 し、減圧濃縮した。残渣をへキサン/ EtOAcから結晶化させて、無色粉末の化合物(1 • 79g)を得た。
得られた化合物(1.75g)の EtOAc (10ml)溶液に、 4M塩化水素/ EtOAc溶液(10.6ml )を加え、室温にて 7時間攪拌した。減圧濃縮後、残渣を EtOAcから結晶化させて、 無色粉末の 1-{4-[(3-フルォロベンジル)ォキシ]ベンゾィル }ピペラジン塩酸塩(1.47g) を得た。
[0063] 参考例 18
tert-ブチルピぺラジン- 1-カルボキシレート塩酸塩(12.4g)の DMF (200ml)溶液に 、 3-ァセチル安息香酸(10g)、 HOBt (9.0g)、 WSC (10.3g)を順次添加した後、室温 にて終夜攪拌した。反応液に塩酸水溶液を加え、 EtOAcにて抽出した。有機層を飽 和炭酸水素ナトリウム水溶液と飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥 し、減圧濃縮した。残渣をへキサン/ EtOAcから結晶化させて、白色粉末の tert-プチ ル 4-(3_ァセトキシベンゾィル)ピぺラジン- 1-カルボキシレート(16.5g)を得た。
tert-ブチル 4-(3_ァセトキシベンゾィノレ)ピぺラジン- 1-カルボキシレート(8.0g)の T HF(lOOml)溶液に、 1M水酸化ナトリウム水溶液(45.9ml)を加え、室温にて 1時間撹 拌した。反応液を減圧濃縮した後、 1M塩酸水溶液を加えて中和し、析出した固体を
濾取し、加熱乾燥して、白色粉末の tert-ブチル 4-(3_ヒドロキシベンゾィル)ピペラジ ン -1-カルボキシレート(7.0g)を得た。
PPh (3.85g)の THF (60ml)溶液に、氷冷下、 DEAD (2.26g、 40%Tol溶液)を滴下した 後、室温にて 1時間攪拌した。氷冷下、反応液に、 3-フエニル -1-プロパノール (2.00 ml)、 tert-ブチル 4-(3_ヒドロキシベンゾィノレ)ピぺラジン- 1-カルボキシレート(3.0g)を 順次加え、室温にて終夜撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、 EtOAcにて抽出した。有機層を水洗後、無水硫酸ナトリウムで乾燥し、減圧濃縮した 。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 3 : l (V/V) ) で精製して、淡黄色油状物の tert-ブチル 4-[3-(3_フエニルプロポキシ)ベンゾィル] ピペラジン- 1-カルボキシレート(3.60g)を得た。
tert-ブチル 4-[3-(3_フエニルプロポキシ)ベンゾィル]ピぺラジン- 1-カルボキシレー ト(3.60g)の EtOAc (15ml)溶液に、 4M塩化水素/ EtOAc溶液(10.6ml)を加え、室温 にて 2時間攪拌した。減圧濃縮後、残渣をジイソプロピルエーテルから結晶化させて 、白色粉末の 1_[3-(3-フエニルプロポキシ)ベンゾィル]ピぺラジン塩酸塩(2.40g)を得 た。
参考例 19
4-フルォ口- 3-ヒドロキシ安息香酸(1.0g)、 tert-ブチルピぺラジン- 1-カルボキシレ 一ト塩酸塩( 1 · 79g)及び HOBt ( 1 · 04g)の DMF (20ml)溶液に WSC ( 1 · 47g)を加え、室 温で終夜攪拌した。反応液を EtOAcで希釈後、飽和炭酸水素ナトリウム水溶液で洗 浄し、無水硫酸マグネシウムで乾燥して、減圧濃縮した。得られた固体をへキサン/ E tOAcで洗浄後、加熱乾燥し、 tert-ブチル 4-(4-フルォ口- 3-ヒドロキシベンゾィル)ピ ペラジン- 1 -カルボキシレート( 1.70g)を白色固体として得た。
tert-ブチル 4-(4-フルォ口- 3-ヒドロキシベンゾィル)ピぺラジン- 1-カルボキシレート (400mg)、 2-シクロへキシノレエタノーノレ(240mg)及びトリブチノレホスフィン(374mg)の THF ( 13ml)溶液に、氷冷下で 1, 1 '- (ァゾジカルボニル)ジピペリジン(466mg)を加え、 室温で終夜攪拌した。反応液を EtOAcで希釈し、不溶物を濾過した後、濾液を減圧 濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2 : 1 (V/V) )で精製し、得られた固体をへキサン/ EtOAcで洗浄後、加熱乾燥し、 tert-ブ
チル 4-[3-(2-シクロへキシルエトキシ) -4-フルォロベンゾィル]ピぺラジン- 1-カルボキ シレート(255mg)を白色固体として得た。
tert-ブチル 4-[3_(2-シクロへキシルエトキシ) -4-フルォロベンゾィル]ピぺラジン- 1- カルボキシレート(240mg)の EtOAc (3ml)溶液に 4M塩化水素/ EtOAc溶液(2ml)を加 え、室温で終夜攪拌した。析出した固体を濾取し、 EtOAcで洗浄後、加熱乾燥して、 1-[3_(2-シクロへキシルエトキシ) -4-フルォロベンゾィル]ピぺラジン塩酸塩(193mg) を白色固体として得た。
[0065] 参考例 20
2-メチルビペラジン(1.38g)の THF (40ml)溶液に、氷冷下、ジ (tert-ブチル)ジカル ボネート(2.00g)を加え、室温で終夜撹拌した。反応液を減圧濃縮し、得られた残渣 をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =85: 15 (V/V) )で精製して、 tert-ブチル -3-メチルビペラジン- 1-カルボキシレート(966mg)を無色 油状物として得た。
3_(2-シクロへキシルエトキシ)安息香酸(300mg)、 tert-ブチル -3-メチルビペラジン -1-カルボキシレート(371mg)、 HOBt (202mg)及び WSC (287mg)のジクロロメタン (4. 5ml)懸濁液を室温で 2日間攪拌した。反応液を水、飽和炭酸水素ナトリウム水溶液、 飽和食塩水で順次洗浄し、無水硫酸マグネシウムで乾燥、減圧濃縮した。残渣をシ リカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =97: 3 (V/V) )で 精製して、 tert-ブチル 4-[3_(2-シクロへキシルエトキシ)ベンゾィル ]-2-メチルビペラ ジン- 1-カルボキシレート(601mg)を無色油状物として得た。
tert-ブチル 4-[3_(2-シクロへキシルエトキシ)ベンゾィル ]-3-メチルピペラジン- 1- カルボキシレート(522mg)に 4M塩化水素/ EtOAc溶液を加え、室温で 1時間撹拌した 。減圧濃縮して、 4-[3_(2-シクロへキシルエトキシ)ベンゾィル ]-2-メチルビペラジン塩 酸塩 (444mg)を無色油状物として得た。
[0066] 参考例 21
3_(2-シクロへキシルエトキシ)安息香酸(350mg)、 HOBt (190mg)及び WSC (270mg )のジクロロメタン懸濁液を室温で 2時間撹拌した後、 2-メチルビペラジンを加えて終 夜撹拌した。反応液を飽和炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄し、
無水硫酸マグネシウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;クロ口ホルム:メタノール =93: 7 (V/V) )で精製し、 1-[3_(2-シクロへ キシルエトキシ)ベンゾィル ]-3-メチルビペラジン(466mg)を無色油状物として得た。
[0067] 参考例 22
3- (ベンジルォキシ)ベンズアルデヒド(8.00g)の THF (150ml)溶液に、氷冷下、 tert- ブチルピぺラジン- 1-カルボキシレート塩酸塩(7.72g)、酢酸(2· 16ml)及びNaBH(OA c) (9.59g)を加え、室温で終夜攪拌した。反応液に 1M水酸化ナトリウム水溶液をカロえ アルカリ性にし、クロ口ホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥後、 減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4 : 1 (V/V) )で精製して、 tert-ブチル 4-[3_ (ベンジルォキシ)ベンジル]ピぺラジン- 1-カルボキシレート(13.4g)を淡黄色油状物として得た。
tert-ブチル 4-[3_ (ベンジルォキシ)ベンジル]ピぺラジン- 1-カルボキシレート(13.4 g)の EtOAc (200ml)溶液に、 10%パラジウム炭素(1.12g)を加え、水素雰囲気下室温 で 10時間攪拌した。反応液をセライト濾過し、濾液を減圧濃縮して、 tert-ブチル 4-(3 -ヒドロキシベンジル)ピぺラジン- 1-カルボキシレート(9.36g)を得た。
tert-ブチル 4-(3_ヒドロキシベンジル)ピぺラジン- 1-カルボキシレート(10.6g)、 2-シ クロへキシルエタノール(5.54ml)及び PPh (14.2g)の THF (140ml)溶液に、氷冷下で
DEAD (24.6ml、 40%Tol溶液)を加え、室温で 2日間攪拌した。減圧濃縮後、得られた 残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で 精製し、更に再度シリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム)で精製し て、 tert-ブチル 4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキシ レート(5.06g)を淡黄色油状物として得た。
tert-ブチル 4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキシレ ート(5.06g)の EtOAc (31.4ml)溶液に、 4M塩化水素/ EtOAc溶液(31.4ml)を加え、室 温で終夜攪拌した。析出した固形物を濾取し、加熱乾燥して、 1-[3_(2-シクロへキシ ルエトキシ)ベンジル]ピぺラジン二塩酸塩 (3.45g)を無色粉末として得た。
[0068] 参考例 23
6-メチル -3-ピリジノール(2.00g)、 2-シクロへキシルエタノール(3.1ml)及び PPh (5.
77g)の THF (50ml)溶液に DEAD (10ml, 40%Tol溶液)を滴下し、室温で終夜攪拌した 。減圧濃縮後、得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロロホ ルム:メタノール = 20 : 1 (V/V) )により精製し、無色油状の 5_(2-シクロへキシルェタノ ール) -2-メチルピリジン(3.93g)を得た。
5_(2-シクロへキシルエタノール) -2-メチルピリジン(3.90g)のクロ口ホルム(50ml)溶 液に m-クロ口過安息香酸(3.38g)を加え、室温にて 3時間攪拌した。反応液に 10%チ ォ硫酸ナトリウム水溶液(50ml)を加えて 5分間攪拌した後、 EtOAcで抽出した。有機 層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄し、無水硫酸マグネシ ゥムで乾燥して減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へ キサン: EtOAc = 2 : 1 (V/V) )により精製し、無色結晶の 5_(2-シクロへキシルエトキシ) -2-メチルピリジン 1-ォキシド (4· 1 lg)を得た。
5_(2-シクロへキシルエトキシ) -2-メチルピリジン- 1-ォキシド(3.9g)と無水酢酸(1.69 g)の混合物を 100°Cで 2時間攪拌した。反応液に Tolを加え減圧濃縮した。 EtOAcに て希釈後、飽和炭酸水素ナトリウム水溶液で洗浄した。無水硫酸マグネシウムで乾燥 後、減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサ ン: EtOAc = 4: 1 (V/V) )で精製し、 [5_(2-シクロへキシルエトキシ)ピリジン- 2-ィル]メ チルアセテート(3.9g)を油状物として得た。
[5_(2-シクロへキシルエトキシ)ピリジン- 2-ィル]メチルアセテート(3.9g)のメタノー ル(70ml)溶液に 1M水酸化ナトリウム水溶液(30ml)を加え、室温で終夜攪拌した。反 応液に 1M塩酸水溶液(30ml)を加えた後、 EtOAcで抽出した。有機層を水で洗浄後 、無水硫酸マグネシウムで乾燥し、減圧濃縮して、 [5_(2-シクロへキシルエトキシ)ピリ ジン- 2-ィル]メタノール(3.26g)を油状物として得た。
[5_(2-シクロへキシルエトキシ)ピリジン- 2-ィル]メタノール(l.Og)のクロ口ホルム(30 ml)溶液に二酸化マンガン (3.70g)を加え、 65°Cで 1時間攪拌した。セライト濾過した 後、濾液を濃縮し、 5_(2-シクロへキシルエトキシ)ピリジン- 2-カルボアルデヒド(840m g)を油状物として得た。
5_(2-シクロへキシルエトキシ)ピリジン- 2-カルボアルデヒド(830mg)、酢酸(430mg) 及び tert-ブチル 1-ピぺラジンカルボキシレート(660mg)のジクロロメタン(16ml)溶液
に NaBH(OAc) (1.13g)を加え、室温で 2時間攪拌した。反応液をジクロロメタンで希 釈し、飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥して、 減圧濃縮した。得られた固体をへキサン/ EtOAcで洗浄し、加熱乾燥して、 tert-プチ ル 4-{[5_(2-シクロへキシルエトキシ)ピリジン- 2-ィル]メチノレ }ピペラジン- 1-カルボキ シレート(830mg)を白色固体として得た。
tert-ブチル 4-{[5_(2-シクロへキシルエトキシ)ピリジン- 2-ィル]メチル }ピペラジン- 1 -カルボキシレート(800mg)のメタノール(10ml)溶液に 4M塩化水素/ジォキサン溶液 (4ml)を加え、室温で終夜攪拌した。減圧濃縮後得られた固体を EtOAcで洗浄し、加 熱乾燥して、 1_{[5-(2-シクロへキシルエトキシ)ピリジン -2-ィル]メチル }ピペラジン三 塩酸塩(730mg)を白色固体として得た。
参考例 24
5- (ベンジルォキシ) -1H-インドール- 2-カルボン酸(3.0g)、 tert-ブチルピぺラジン -1-カルボキシレート塩酸塩(2.51g)及び HOBt (1.82g)の DMF (30ml)溶液に WSC (2. 58g)を加え、室温で 2時間攪拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え 、析出した固体を濾取した。得られた固体を水で洗浄後、加熱乾燥して、 tert-ブチル 4_{[5- (ベンジルォキシ) -1H-インドール- 2-ィノレ]カルボ二ノレ }ピペラジン- 1-カルボキ シレート(4.48g)を白色固体として得た。
tert-ブチル 4-{[5_ (ベンジルォキシ) -1H-インドール- 2-ィル]カルボ二ル}ピペラジ ン- 1-カルボキシレート(1.0g)、 1,4-ジァザビシクロ [2.2.2]オクタン(25mg)、炭酸ジメ チル(10ml)及び DMF (lml)の混合物を 100°Cで終夜攪拌した。反応液を EtOAcで希 釈し、水で洗浄後、無水硫酸マグネシウムで乾燥した。減圧濃縮後、得られた固体を へキサン/ EtOAcで洗浄し、加熱乾燥して、 tert-ブチル 4-{[5_ (ベンジルォキシ) -1-メ チル -1H-インドール- 2-ィル]カルボ二ル}ピペラジン- 1-カルボキシレート(940mg)を 白色固体として得た。
tert-ブチル 4-{[5_ (ベンジルォキシ) -1-メチル -1H-インドール- 2-ィル]カルボ二ノレ } ピぺラジン- 1-カルボキシレート(630mg)の THF (15ml) /エタノール(9ml)混合溶液に 10%パラジウム炭素(30mg)を加え、水素雰囲気下室温で 5時間攪拌した。触媒をセラ イトで濾去し、濾液を減圧濃縮して、 tert-ブチル 4-[(5-ヒドロキシ -1-メチル -1H -イン
ドール- 2-ィル)カルボニル]ピぺラジン- 1-カルボキシレート(500mg)を無色ァモルフ ァスとして得た。
tert-ブチル 4-[(5-ヒドロキシ -1-メチル -1H-インドール- 2-ィル)カルボニル]ピペラ ジン- 1-カルボキシレート(490mg)、 PPh (540mg)及びシクロへキシルメタノール(233
3
mg)の THF (lOml)溶液に、 DEAD (0.93ml、 40%Tol溶液)を氷冷下で加えた後、室温 で終夜攪拌した。反応液を EtOAcで希釈し、水、飽和炭酸水素ナトリウム水溶液で順 次洗浄した後、無水硫酸マグネシウムで乾燥した。減圧濃縮後、得られた残渣をシリ 力ゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製し、 ter t-ブチノレ 4-{[5_ (シクロへキシルメトキシ) -1-メチル -1H-インドール- 2-ィノレ]カルボ二 ル}ピペリジン- 1-カルボキシレート(310mg)を得た。
tert-ブチル 4-{[5_ (シクロへキシルメトキシ) -1-メチル -1H-インドール- 2-ィル]カル ボニル }ピペリジン- 1-カルボキシレート(300mg)の EtOAc ( 10ml)溶液に 4M塩化水素/ EtOAc溶液(3ml)を加え、室温で 6時間攪拌した。析出した固体を濾取し、 EtOAcで 洗浄後加熱乾燥して、 5- (シクロへキシルメトキシ) -1-メチル -2- (ピペラジン- 1-ィルカ ルポニル) - -インドール塩酸塩(236mg)を白色固体として得た。
参考例 25
メチル 3-(3_シクロへキシルプロピル)ベンゾエート(1.50g)の THF (20ml)溶液に、 _7 8°Cにて DIBAL (13ml、 1M Tol溶液)を滴下した後、 0°Cにて 1時間攪拌した。反応液 に飽和 (土) -酒石酸ナトリウムカリウム水溶液(30ml)及び EtOAcを加えて 30分間激しく 攪拌し、 EtOAcで抽出した。有機層を無水硫酸マグネシウムで乾燥し、減圧濃縮した 。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc =3 : 1 (V/V) ) にて精製し、無色油状の [3-(3_シクロへキシルプロピル)フエニル]メタノール(1.30g) を得た。
[3-(3_シクロへキシルプロピル)フエニル]メタノール( 1.30g)のクロ口ホルム(30ml)溶 液に二酸化マンガン (25.0g)をカロえ、室温にて 1時間攪拌した。セライト濾過により二 酸化マンガンを濾去し、濾液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィ 一 (溶出液;へキサン: EtOAc=4: 1 (V/V) )にて精製し、無色油状の 3-(3_シクロへキシ ルプロピル)ベンズアルデヒド(1· 10g)を得た。
3_(3-シクロへキシルプロピル)ベンズアルデヒド(600mg)の THF (30ml)溶液に、氷 冷下、 tert-ブチルピぺラジン- 1-カルボキシレート塩酸塩(583mg)、酢酸(0.30ml)及 び NaBH(OAc) (1.1 lg)を加えて、室温にて 2時間撹拌した。反応液を 1M水酸化ナトリ ゥム水溶液でアルカリ性とし、クロ口ホルムで抽出した。有機層を無水硫酸マグネシゥ ムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキ サン: EtOAc = 3 : 1 (V/V) )にて精製して、無色油状の tert-ブチル 4-[3_(3_シクロへ キシルプロピル)ベンジル]ピぺラジン- 1-カルボキシレート(800mg)を得た。
tert-ブチル 4-[3-(3_シクロへキシルプロピル)ベンジル]ピぺラジン- 1-カルボキシレ ート(800mg)の EtOAc (10ml)溶液に 4M塩化水素/ EtOAc溶液(2.0ml)を加え、室温 にて終夜攪拌した。析出した固体を EtOAcで洗浄し、加熱乾燥して、白色固体の 1_[ 3-(3_シクロへキシル)ベンジル]ピぺラジン(530mg)を得た。
[0071] 参考例 26
tert-ブチル(5-ブロモ -3-ピリジル)力ルバメート(300mg)、酢酸パラジウム(II) (12.3 mg)、 2- (ジ -tert-ブチルホスフイノ)ビフエニル(32.8mg)、モルホリン(0.287ml)及びリ ン酸カリウム(699mg)のジォキサン(10ml)懸濁液を、 100°Cで終夜攪拌した。反応液 を室温まで冷却し、さらに酢酸パラジウム(II) (12.3mg)、 2- (ジ -tert-ブチルホスフイノ) ビフエニル(32.8mg)及びモルホリン(0.287ml)を追加し、 100°Cで 24時間攪拌した。 反応液を室温まで冷却後、セライト濾過し、濾液を減圧濃縮した。残渣をシリカゲル カラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製し、 tert-ブチ ル(5-モルホリン- 4-ィルピリジン- 3-ィル)力ルバメート(147mg)を無色油状物として得 た。
tert-ブチル(5-モルホリン- 4-ィルピリジン- 3-ィノレ)力ルバメート(147mg)のジォキサ ン (4.0ml)溶液に、 4M塩化水素/ジォキサン溶液 (4.0ml)を加え、終夜攪拌した。反 応液を減圧濃縮し、 5-モルホリン- 4-ィルピリジン- 3アミンニ塩酸塩(132mg)を無色ァ モルファス状物として得た。
[0072] 参考例 27
ニコチン酸クロリド塩酸塩(3.03g)のァセトニトリル (40ml)懸濁液に、氷冷下、アジ化 ナトリウム(2.76g)、 TEA (5.16g)を順次添加した後、氷冷下にて 1時間攪拌した。反応
液に塩酸水溶液を加えて EtOAcにて抽出した。有機層を水酸化ナトリウム水溶液と 水で洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮して、褐色固体(2.08g)を得た。 得られた固体(2.08g)の Tol (20ml)溶液を 110°Cにて 40分間撹拌した。反応液を冷 却して、褐色の溶液を得た。
tert-ブチルピぺラジン- 1-カルボキシレート塩酸塩(1.86g)の THF (20ml)溶液に、 氷冷下、上記で得られた褐色の溶液を添加した後、室温にて 15時間攪拌した。反応 液に水酸化ナトリウム水溶液を加えて、 EtOAcにて抽出した。有機層を水洗後、無水 硫酸ナトリウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー( 溶出液;クロ口ホルム:メタノール = 23: 2 (V/V) )で精製して、淡黄色油状物(3.64g)を 得た。
得られた油状物(3.62g)の EtOAc (20ml)溶液に、 4M塩化水素/ EtOAc溶液(19.9ml )を加えた後、室温にて 5時間攪拌した。析出した固体を濾取し、加熱乾燥して、褐色 粉末の N-ピリジン- 3-ィルピペリジン- 1-カルボキサミドニ塩酸塩(2.57g)を得た。 参考例 28
60%水素化ナトリウム(4.62g)の THF (200ml)懸濁液に、氷冷下、 2-アミノビラジン(10 • 0g)を加え、室温にて 1時間撹拌した。反応液に、氷冷下、ジフエ二ルカルポネート(2 2.5g)を加え、 45°Cにて終夜加熱した。反応液に、氷冷下、 1M塩酸水溶液を加えた 後、 EtOAcにて抽出した。有機層を水洗後、無水硫酸ナトリウムで乾燥し、減圧濃縮 した。得られた固体をへキサンにて洗浄して、ベージュ色粉末のフエ二ルビラジン- 2- ィルカルバメート(20.5g)を得た。
tert-ブチルピぺラジン- 1-カルボキシレート塩酸塩(10.0g)のァセトニトリル(200ml) 溶液に、フエ二ルビラジン- 2-ィルカルバメート(11.6g)及び TEA (6.3ml)を加え、室温 にて終夜撹拌した。反応液に飽和炭酸水素ナトリウム水溶液を加えて、 EtOAcにて 抽出した。有機層を水洗後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリ 力ゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、 白色粉末 tert-ブチル 4- (ピラジン- 2-ィルカルバモイル)ピぺラジン- 1-カルボキシレ ート(12.0g)を得た。
tert-ブチル 4- (ピラジン- 2-ィルカルバモイル)ピぺラジン- 1-カルボキシレート(12.0
g)のジォキサン(30ml) /メタノール(10ml)混合溶液に、 4M塩化水素/ジォキサン溶液 (48.8ml)を加えた後、室温にて 3時間攪拌した。析出した固体を EtOAcに懸濁させて 濾取して、加熱乾燥し、淡黄色粉末の N-ピラジン- 2-ィルピペラジン- 1-カルボキサミ ドニ塩酸塩 (9.50g)を得た。
[0074] 参考例 29
1-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(200mg)のジクロロメ タン懸濁液にジイソプロピルェチルアミン( 126mg)及びトリホスゲン(84mg)を加え室 温にて 3時間攪拌した。さらに、 TEA (0.4ml)及び塩化アンモニゥム(152mg)を加え 15 時間攪拌した。反応液に水を加え、クロ口ホルムで抽出し、有機層を無水硫酸マグネ シゥムで乾燥した。減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー (溶出液; クロ口ホルム:メタノール = 10 : 1 (V/V) )で精製し、 EtOAcから結晶化して、 4-[3_(2-シ クロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-カルボキサミド( 117mg)を白色固体と して得た。
[0075] 参考例 96
N- 2-ピラジュル -1-ピぺラジンカルボキサミドニ塩酸塩(210mg)、 5_ヒドロキシ _1_ベ ンゾチォフェン- 2_カルボン酸( 5mg)、 WSC ( 172mg)及び HOBt (121mg)の DMF (5 ml)溶液に、 TEA (0.31ml)を加え、室温で終夜攪拌した。反応液に水を加え、 EtOAc で抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸マグネシ ゥムで乾燥し、減圧濃縮した。得られた固体をへキサン/ EtOAcで洗浄後、加熱乾燥 して、 4-(5_ヒドロキシ -1-ベンゾチォフェン- 2-ィノレ)カルボニル] -N-ピラジン- 2-ィルピ ペラジン- 1-カルボキサミド(216mg)を白色固体として得た。
参考例 97
5-フエ二ルチオフェン- 2-アルデヒド(640mg)及び酢酸(0.39ml)のジクロロメタン(13 ml)溶液に tert-ブチルビペラジン- 1-カルボキシレート(696mg)を加え、室温で 1時間 攪拌した。更に、 NaBH(OAc) (1.08g)を加え終夜攪拌した。反応液に水を加え、クロ 口ホルムで抽出した。有機層を飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸 マグネシウムで乾燥し、減圧濃縮した。残渣を EtOAc (15ml)に溶解させ、 4M塩化水 素/ EtOAc溶液(5ml)を加え終夜攪拌した。析出した固体を濾取後、 EtOAcで洗浄し
、加熱乾燥して 1-K5-フエニル -2-チェニル)メチル]ピぺラジン二塩酸塩(1.02g)を白 色固体として得た。
[0076] 参考例 98
60%水素化ナトリウム(4.62g)の THF (200ml)懸濁液に、氷冷下アミノビラジン(10g) を加え、室温にて 1時間撹拌した。更に氷冷下、炭酸ジフエニル (22.5g)を少しずつ 添加し、 45°Cにて 4時間撹拌した。反応溶に 1M塩酸水溶液(120ml)を加え、 EtOAc にて抽出した。有機層を飽和炭酸水素ナトリウム水溶液にて洗浄し、無水硫酸マグネ シゥムにて乾燥後、減圧濃縮した。得られた固形物をへキサンにて洗浄後加熱乾燥 して、フエニルピラジン- 2-ィルカルバメート(12.4g)を得た。
[0077] 参考例 99
2-ァミノ- 3-クロロビラジン(500mg)のピリジン(5ml)溶液に、氷冷下クロロギ酸フエ二 ノレ(1.1ml)を加え、室温で 2時間攪拌した。反応液にトルエンを加え減圧濃縮した。残 渣を水で洗浄後、加熱乾燥し、ジフエニル(3-クロロビラジン- 2-ィル)イミドジカルボネ ート(1.41g)を褐色固体として得た。
参考例 100
ジフエニル(3-クロロビラジン- 2-ィル)イミドジカルボネート(3.5g)及び tert-ブチルビ ペラジン- 1-カルボキシレート(3.52g)の DMF (35ml)溶液に、 TEA (2.64ml)を加え、 80 °Cで 1時間攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗 浄後、無水硫酸マグネシウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロ マトグラフィー(溶出液;へキサン: EtOAc= l : l (V/V) )で精製し、 tert-ブチル 4_[(3_ クロロビラジン- 2-ィノレ)力ルバモイノレ]ピぺラジン- 1-カルボキシレート(2.28g)を白色 固体として得た。
[0078] 参考例 101
tert-ブチル 4-[(3-クロロビラジン- 2-ィル)力ルバモイル]ピぺラジン- 1-カルボキシレ ート(2.28g)のイソプロパノール(30ml)溶液に、 4M塩化水素/ EtOAc (15ml)を加え、 室温で終夜攪拌した。反応液を減圧濃縮し、得られた固体をイソプロパノールで洗浄 後加熱乾燥して、 N-(3-クロロビラジン -2-ィル)ピペラジン- 1-カルボキサミドニ塩酸塩 (1.13g)を得た。
参考例 102
2-クロ口イソニコチノ二トリル(l.OOg)のジメトキシェタン(20ml)溶液に、 [3- (ベンジル ォキシ)フエニル]ボロン酸(1.81g)、 Pd(PPh ) (417mg)、炭酸ナトリウム(1.53g)及び水
3 4
(10ml)を加え、 80°Cにて 3時間撹拌した。反応液を室温まで冷却した後、 EtOAcにて 抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸マグネシウムにて乾燥し、減 圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 6 : 1 (V/V) )にて精製して、 2-[3_ (ベンジルォキシ)フエニル]イソニコチノ二トリル(1.50g )を白色粉末として得た。
2-[3_ (ベンジルォキシ)フエ二ノレ]イソニコチノ二トリノレ(1· 15g)のエタノール(23ml)溶 液に、 4M水酸化ナトリウム水溶液(10ml)を加え、 80°Cにて 4時間撹拌した。反応液を 室温まで冷却した後、エタノールを留去し、塩酸水溶液にて中和した。析出した固形 物を濾取し、水で洗浄した後、加熱乾燥して、 2-[3_ (ベンジルォキシ)フエニル]イソ二 コチン酸( 1.20g)を白色粉末として得た。
[0079] 参考例 103
2-クロ口イソニコチノ二トリル(l.OOg)のトルエン(30ml)溶液に、 2,5_ジフルォロベン ジルァミン(1.10g)、酢酸パラジウム(81mg)、 2,2'-ビス (ジフエニルホスフイノ) -1,1'-ビ ナフチル(224mg)及び tert-ブトキシナトリウム(762mg)を加え、 50°Cにて 2時間撹拌し た。反応液を室温まで冷却した後、水を加え、 EtOAcにて抽出した。有機層を飽和食 塩水にて洗浄後、無水硫酸マグネシウムにて乾燥し、減圧濃縮した。残渣をシリカゲ ルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 6 : l (V/V) )にて精製して、 2- [(2,5_フノレオ口ベンジル)ァミノ]イソニコチノ二トリル (800mg)を白色粉末として得た。
2- [(2,5-フルォロベンジル)ァミノ]イソニコチノ二トリル(800mg)のエタノール(16ml) 溶液に、 4M水酸化ナトリウム水溶液 (8ml)を加え、 80°Cにて 4時間撹拌した。反応液 を室温まで冷却した後、エタノールを留去し、塩酸水溶液にて中和した。析出した固 形物を濾取し、水で洗浄した後、加熱乾燥して、 2-[(2,5-ジフルォロベンジル)ァミノ] イソニコチン酸 (240mg)を淡黄色粉末として得た。
[0080] 参考例 104
3- (ベンジルォキシ)フエノール(5.00g)、 tert-ブチル 4-ヒドロキシピペリジン- 1-カル
ボキシレート(7.53g)及び PPh (9.82g)の THF (250ml)溶液に、氷冷下 DEAD (17ml、 4
0%Tol溶液)を加え、室温で 48時間攪拌した。反応液を減圧濃縮した後、 EtOAcを加 え、 1M水酸化ナトリウム水溶液、水にて順次洗浄後、無水硫酸マグネシウムで乾燥し た。減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOA c = 4: 1 (V/V) )で精製して、 tert-ブチル 4-[3_ (ベンジルォキシ)フエノキシ]ピぺリジン -1-カルボキシレート(7.67g)を無色油状物として得た。
参考例 105
tert-ブチル 4-[3_ (ベンジルォキシ)フエノキシ]ピぺリジン- 1-カルボキシレート(4.67 g)のエタノール溶液に、 10%パラジウム炭素(500mg)を加え、水素雰囲気下、室温に て終夜攪拌した。触媒をセライト濾過で除去し、濾液を減圧濃縮した。得られた固体 をへキサン/ EtOAcで洗浄後、加熱乾燥して、 tert-ブチル 4-(3_ヒドロキシフエノキシ) ピぺリジン- 1-カルボキシレート(3.08g)を白色固体として得た。
参考例 106
tert-ブチル 4-(3_ヒドロキシフエノキシ)ピぺリジン- 1-カルボキシレート(400mg)、 2- シクロへキシルエタノール(262mg)及びトリブチルホスフィン(413mg)の THF (15ml)溶 液に、氷冷下 1,1'_ (ァゾジカルボニル)ジピペリジン(516mg)を加え、室温で終夜攪拌 した。反応液を減圧濃縮した後、 EtOAcを加え、 1M水酸化ナトリウム水溶液で洗浄後 、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;へキサン: EtOAc = 7 : 3 (V/V) )で精製し、油状物を得た。得られた 油状物をイソプロパノール/ EtOAc混合溶媒に溶解させ、 4M塩化水素/ EtOAcを加え 、室温で終夜攪拌した。反応液を減圧濃縮し、得られた固体をへキサン/ EtOAcで洗 浄後、加熱乾燥して、 4-[3_(2-シクロへキシルエトキシ)フエノキシ]ピぺリジン塩酸塩( 252mg)を白色固体として得た。
参考例 107
4-[3_ (ベンジルォキシ)フエノキシ]ピぺリジン塩酸塩(2.16g)、フエニルピラジン- 2- ィルカルバメート(1.45g)及び TEA(1.88ml)のァセトニトリル(43ml)溶液を 80°Cで 1時 間攪拌した。反応液を減圧濃縮した後、 EtOAcを加え、飽和炭酸水素ナトリウム水溶 液で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラ
ムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 1 (V/V) )で精製し、油状物を得 た。得られた油状物をへキサン/ EtOAcから結晶化させ、加熱乾燥して、 4-[3_ (ベン ジルォキシ)フエノキシ] -N-ピラジン- 2-ィルピペラジン -1-カルボキサミド(2.3g)を白 色固体として得た。
[0082] 参考例 108
4-[3_ (ベンジルォキシ)フエノキシ] -N-ピラジン- 2-ィルピペラジン- 1-カルボキサミド (2.2g)のエタノール(50ml)溶液に 10%パラジウム炭素(200mg)を加え,、水素雰囲気 下、室温で終夜攪拌した。触媒をセライトで濾去した後、濾液を減圧濃縮した。残渣 をへキサン/ EtOAcから結晶化させ、加熱乾燥して、 4-(3_ヒドロキフエノキシ) -N-ビラ ジン -2-ィルピペリジン -1-カルボキサミド(1.67g)を白色固体として得た。
参考例 109
2-クロ口- 4-ニトロピリジン(lg)及び tert-ブチル 4-ヒドロキシピペリジン- 1-カルボキ シレート(1.40g)の DMF (30ml)溶液に、氷冷下 60%水素化ナトリウム(277mg)を加え、 室温で 2時間攪拌した。反応液に水を加え、析出した固体を水、へキサンで順次洗 浄後、加熱乾燥し、 tert-ブチル 4-[(2-クロ口ピリジン- 4-ィル)ォキシ]ピぺリジン- 1-力 ルポキシレート(1.56g)を白色固体として得た。
[0083] 参考例 136
4-[(3-フルォロベンジル)ォキシ]ベンズアルデヒド(1.40g)の THF (20ml)溶液に、氷 冷下、 t-ブチルピぺラジン- 1-カルボキシレート(1.23g)、酢酸(0.343ml)、 NaBH(OA c) (1.53g)を順次加え、室温にて 19時間攪拌した。反応液に氷冷下、 1M水酸化ナト リウム水溶液を加えてアルカリ性とした後、クロ口ホルムで抽出した。有機層を無水硫 酸ナトリウムで乾燥した後、減圧濃縮した。得られた残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;クロ口ホルム:メタノール = 96 : 4 (V/V) )で精製して、 t-ブチル 4-{4- [(3-フルォロベンジル)ォキシ]ベンジル }ピペラジン- 1-カルボキシレート(2.41g)を淡 黄色油状物として得た。
t-ブチノレ 4-{4-[(3_フルォロベンジル)ォキシ]ベンジル }ピペラジン- 1-カルボキシレ ート(2.39g)のメタノール(10ml)溶液に、氷冷下、 4M塩化水素/ EtOAc溶液(11.9ml) を加え、室温にて 5時間攪拌した。反応液を減圧濃縮し、得られた残渣をァセトニトリ
ルに懸濁させて濾取して、 1-{4-[(3-フルォロベンジル)ォキシ]ベンジル }ピぺラジン 二塩酸塩(1.71g)を無色固体として得た。
[0084] 上記参考例 1〜29、 96〜; 109、及び 136の方法と同様にして、後記表 3〜; 17に示 す参考例化合物 1〜 137をそれぞれ対応する原料を使用して製造した。表 3〜; 17に の参考例化合物の構造、製造法及び物理化学的データを示す。
[0085] 実施例 1
ニコチン酸クロリド塩酸塩(356mg)をァセトニトリル(10ml)に懸濁し、これに氷冷下、 アジ化ナトリウム(325mg)及び TEA(607mg)を加え、氷冷下 1時間攪拌した。反応液 に水を加え、 EtOAcにて抽出した。有機層を無水硫酸ナトリウムで乾燥した後、減圧 濃縮し、得られた残渣を Tol ( 10ml)に溶解し、 110°Cにて 30分間攪拌した。反応液を 冷却後、次反応に用いた。
4_{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペリジン(301mg)の THF(5ml)溶液 に、氷冷下、上記の Tol溶液を加え、室温にて 3時間攪拌した。反応液に水酸化ナトリ ゥム水溶液を加え、 EtOAcにて抽出し、有機層を無水硫酸ナトリウムで乾燥した後、 減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノ ール =24: 1 (V/V) )で精製した。得られた油状物を EtOAc (5ml)に溶解し、氷冷下、 4 M塩化水素/ EtOAc溶液(0.30ml)を加え、減圧濃縮した。得られた残渣をァセトニトリ ノレ/メタノール混合溶媒から再結晶して、 4-{4-[(3-フルォロベンジル)ォキシ]フエノキ シト N- (ピリジン- 3-ィル)ピぺリジン- 1-カルボキサミド塩酸塩 (325mg)を得た。
[0086] 実施例 2
5-アミノビリジン- 2-カルボ二トリル(l.Og)のピリジン(10ml)溶液にクロロギ酸フエ二 ル(1.05ml)を加え、室温で 2時間攪拌した。反応液を減圧濃縮後、得られた残渣を 飽和炭酸水素ナトリウム水溶液、ァセトニトリルで順次洗浄後、加熱乾燥して、フエ二 ル(6-シァノピリジン- 3-ィル)力ルバメート(l.Olg)を白色固体として得た。
4-{4-[(3-フルォロべンジル)ォキシ]フェノキシ}ピぺリジン(594mg)及びTEA(0.30ml )の DMF (lOml)溶液に、フエ二ノレ(6-シァノピリジン- 3_ィノレ)力ルバメート(519mg)を 加え、 100°Cにて 2時間攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム 水溶液で洗浄した。無水硫酸マグネシウムで乾燥後、減圧濃縮し、残渣をシリカゲル
カラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =95: 5 (V/V) )で精製した 。得られた固体をへキサン/ EtOAcで洗浄し、 N-(6-シァノピリジン- 3-ィル) -4-{4-[(3- フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1-カルボキサミド(530mg)を無色粉 末として得た。
[0087] 実施例 3
5-ブロモニコチン酸(1.21g)及び TEA(lml)の Tol (20ml)溶液に氷冷下、ジフエ二ル ホスホリルアジド(1.82g)を加え室温で 2日攪拌した。反応液を EtOAcで希釈し、飽和 炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾 燥し、減圧濃縮した。得られた残渣を Tol (30ml)に溶解し、 110°Cで 2時間攪拌した。 反応液に氷冷下、 4-[4-(3_フルォロベンジルォキシ)フエノキシ]ピぺリジン塩酸塩(1. 5g)の THF (30ml)溶液を加え室温で 5時間攪拌した。反応液を EtOAcで希釈し、 1M 水酸化ナトリウム水溶液、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減 圧濃縮した。得られた残渣を EtOAc/ァセトニトリルから再結晶し、 N-(5-ブロモピリジ ン -3-ィル) -4-{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1-カルボキサミ ド(1.73g)を白色固体として得た。
[0088] 実施例 4
N-(5-ブロモピリジン- 3-ィル) -4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリ ジン- 1-カルボキサミド(720mg)、ジシァノジンク(lOlmg)、トリス (ジベンジリデンァセト ン)ジパラジウム(7.0mg)及び 1,1'-ビス (ジフエニルホスフイノ)フエ口セン(10mg)の DM F懸濁液を 120°Cで 15時間攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリ ゥム水溶液、飽和食塩水で洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮し、残 渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= l:;!〜 1 : 9 (V/V ) )で精製して、 N-(5-シァノピリジン -3-ィル) -4-{4-[(3_フルォロベンジル)ォキシ]フエ ノキシ }ピペリジン- 1-カルボキサミド(500mg)を得た。
N-(5-シァノピリジン- 3-ィル) -4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリ ジン- 1-カルボキサミド(500mg)のエタノール溶液に 10M水酸化ナトリウム水溶液(0.6 7ml)を加え 80°Cで 15時間攪拌した。反応液を減圧濃縮後、残渣に水(20ml)、メタノ ール(5ml)を加え不溶物を濾去した。濾液に氷冷下、 1M塩酸水溶液を加え、析出し
た結晶を濾収後、加熱乾燥し、 5-{[(4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピ ペリジン -1-ィル)カルボニル]アミノ}ニコチン酸(129mg)を白色固体として得た。
[0089] 実施例 5
メチル 4-ァミノべンゾエート(2.0g)のピリジン溶液に、氷冷下クロロギ酸フエ二ルを加 え、室温で 3時間攪拌した。反応液を減圧濃縮した後、 EtOAcを加え、有機層を水、 飽和炭酸水素ナトリウム水溶液、飽和塩化ナトリウム水溶液で順次洗浄した。無水硫 酸マグネシウムで乾燥した後、減圧濃縮して、メチル 4- [(フエノキシカルボニル)ァミノ ]ベンゾェート(4.23g)を白色固体として得た。
メチル 4- [(フエノキシカルボニル)ァミノ]ベンゾエート(1.0g)、 4-[4- (ベンジルォキシ )フエノキシ]ピぺリジン塩酸塩(1.2g)及び TEA (0.96g)のァセトニトリル溶液を 60°Cで 3 時間攪拌した。反応液を減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー (溶 出液;へキサン^10八0 = 2 : 1〜1 : 10 /¥) )で精製して、メチル 4-[({4-[4- (ベンジル ォキシ)フエノキシ]ピペリジン- 1-ィル }カルボ二ノレ)ァミノ]ベンゾエート(1.25g)を白色 固体として得た。
メチル 4-[({4-[4- (ベンジルォキシ)フエノキシ]ピペリジン- 1-ィル }カルボニル)ァミノ] ベンゾエート(800mg)の THF (16ml) /メタノール(8ml)混合溶液に 1M水酸化ナトリウム 水溶液を加え 70°Cで 15時間攪拌した。反応液を減圧濃縮後、残渣に水を加え溶解 し、 1M塩酸水溶液を加え酸性とした。析出した結晶を濾収した後、加熱乾燥して、 4- [({4_[4- (ベンジルォキシ)フヱノキシ]ピペリジン- 1-ィル }カルボニル)ァミノ]安息香酸( 690mg)を白色固体として得た。
[0090] 実施例 6
3_[({4-[4- (ベンジルォキシ)フエノキシ]ピぺリジン- 1-ィル }カルボニル)ァミノ]安息香 酸(103mg)の DMF (2.0ml)溶液に、 WSC (53mg)、 HOBt (46mg)、ピぺリジン(29mg) を順次添加した後、室温にて 12時間攪拌した。反応液に飽和炭酸水素ナトリウム水 溶液を加え、 EtOAcで抽出し、有機層を飽和食塩水にて洗浄後、無水硫酸ナトリウム で乾燥した。減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー (溶出液;へキサ ン: EtOAc = 1: 1 (V/V) )で精製した。ジイソプロピルエーテルにて結晶化し、加熱乾 燥して、 4-[4- (ベンジルォキシ)フエノキシ] -N-[3- (ピペリジン- 1-ィルカルボ二ノレ)フエ
ニル]ピぺリジン- 1-カルボキサミド(63mg)を得た。
[0091] 実施例 7
4_{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペラジン(400mg)の THF (8ml)溶 液に、ェチル 3-イソシァネートべンゾエート(305mg)を加え、室温にて 24時間攪拌し た。反応液を減圧濃縮し、クロ口ホルムで希釈後、飽和炭酸水素ナトリウム水溶液で 洗浄した。無水硫酸マグネシウムで乾燥後、減圧濃縮し、残渣をシリカゲルカラムクロ マトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製した。得られた固体を へキサン/ EtOAcで再結晶し、ェチル 3-{[(4-{4-[(3_フルォロベンジル)ォキシ]フエノ キシ }ピペリジン- 1-ィル)カルボニル]アミノ}ベンゾエート(562mg)を無色粉末として得 た。
[0092] 実施例 8
ェチル 3-{[(4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1-ィル)カル ボニル]アミノ}ベンゾエート(400mg)の THF (5ml) /メタノール(3ml)の混合溶液に、 1M 水酸化ナトリウム水溶液(3ml)を加え、室温にて 10時間攪拌した。反応液に 1M塩酸 水溶液(3ml)を加え、析出した固体を濾取し、水で洗浄後、加熱乾燥し、 3-{[(4-{4-[( 3-フルォロベンジル)ォキシ]フエノキシ }ピペリジン -1-ィル)カルボニル]アミノ}安息香 酸 (36 lmg)を無色粉末として得た。
実施例 9
3_{[(4-{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1-ィル)カルボニル] アミノ}安息香酸(253mg)及び HOBt (88mg)の DMF (5ml)溶液に WSC (124mg)を加え 、室温で 2時間攪拌した。反応液に濃アンモニア水(0.5ml)を加え、室温で 1時間攪 拌した。反応液に水を加え、析出した固体を濾取し、エタノールで再結晶して、 N-(3- 力ルバモイルフエニル) -4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1- カルボキサミド(163mg)を無色粉末として得た。
[0093] 実施例 10
N-(6-シァノピリジン- 3-ィル) -4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }ピペリ ジン- 1-カルボキサミド(285mg)及び炭酸カリウム(88mg)の DMSO (3ml)懸濁液に、 氷冷下 31%過酸化水素水(0.3ml)を加え室温で 1時間攪拌した。反応液に水を加え、
析出した固体を濾取した。固体を水で洗浄後、加熱乾燥し、 5-{[(4-{4-[(3_フルォロ ベンジル)ォキシ]フエノキシ }ピペリジン- 1-ィル)カルボニル]アミノ}ピリジン- 2-カルボ キサミド(272mg)を無色粉末として得た。
実施例 11
3_{[(4-{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペリジン- 1-ィル)カルボニル] アミノ}フエニルアセテート(l.Og)のメタノール(10ml)溶液に、 1M水酸化ナトリウム水溶 液(5ml)を加え、室温にて 4時間攪拌した。 1M塩酸水溶液(5ml)を加え、析出した固 体を濾取し、水で洗浄後加熱乾燥し、 4-{4-[(3-フルォロベンジル)ォキシ]フエノキシ } -N-(3-ヒドロキシフエニル)ピぺリジン- 1-カルボキサミド(0.85g)を無色粉末として得た
[0094] 実施例 12
4_{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペラジン(500mg)のジクロロメタン( 5ml)溶液に CDI (269mg)を氷冷下加え、室温で終夜攪拌した。反応液を濃縮後、ク ロロホルムで希釈し、水で洗浄した。減圧濃縮後、得られた残渣をへキサン/ EtOAc にて結晶化し、へキサン/ EtOAcで再結晶して、 4-{4-[(3-フルォロベンジル)ォキシ] フエノキシ }-1-(1Η-イミダゾール -1-ィルカルボニル)ピぺリジン(370mg)を無色粉末と して得た。
実施例 13
4-{4-[(3_フルォロベンジル)ォキシ]フエノキシ }-N-ピリジン- 3-ィルピペリジン- 1-力 ルポキサミドのクロ口ホルム(8ml)溶液に m-クロ口過安息香酸(281mg)を加え室温で 2 時間攪拌した。反応液をクロ口ホルムで希釈し、飽和炭酸水素ナトリウム水溶液で洗 浄後、無水硫酸マグネシウムで乾燥した。減圧濃縮後、得られた固体をへキサン/ Et OAcで洗浄し、エタノール/ EtOAcで再結晶して、 4-{4-[(3_フルォロベンジル)ォキシ] フエノキシ }-Ν-(1-ォキシドピリジン- 3-ィル)ピぺリジン- 1-カルボキサミド(185mg)を無 色粉末として得た。
[0095] 実施例 14
1H-ピラゾール -3-ァミン(2.0g)及び TEA (3.7ml)の THF (40ml)溶液にクロ口ギ酸フ ェニル (3.3ml)を加え、室温で終夜攪拌した。反応液を減圧濃縮後、得られた残渣を
EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄した。無水硫酸マグネシウム で乾燥後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン : EtOAc = 5 :ト 0: 1 (V/V) )で精製し、白色固体 (2. lg)を得た。
トリホスゲン(118mg)の THF (5ml)溶液に、上記の方法で得られた固体(203ml)及 び TEA (0.20ml)の THF (3ml)溶液を氷冷下滴下し、室温で 30分攪拌した。反応液に 4_{4-[(3-フルォロベンジル)ォキシ]フエノキシ }ピペラジン(250mg)及び TEA (0.14ml) の THF (2ml)溶液を氷冷下滴下し、室温で 2時間攪拌した。反応液を減圧濃縮後、 Et OAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄した。無水硫酸マグネシウムで 乾燥後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: E tOAc = 5 : 1 (V/V) )で精製し、固体を得た。得られた固体をへキサン/ EtOAcで洗浄 後加熱乾燥し、白色固体(318mg)を得た。
上記の方法で得られた固体(310mg)のァセトニトリル(5ml)溶液に水酸化リチウム一 水和物(245mg)を加え、 50°Cで 3時間攪拌した。反応液に水を加え、析出した固体を 水で洗浄後、 EtOAcで再結晶し、 4-{4-[(3-フルォロベンジル)ォキシ]フエノキシト N-1 H -ピラゾール -3-ィルピペリジン -1-カルボキサミド(168mg)を無色粉末として得た。 実施例 15
3_[({4-[4- (ベンジルォキシ)フエノキシ]ピぺリジン- 1-ィル }カルボニル)ァミノ]安息香 酸(300mg)の DMF (6 · 7ml)溶液に、 WSC ( 193mg)、 HOBt ( 136mg)、 N-メチル - 1, 2-ベ ンゼンジァミン (410mg)、 TEA (340mg)を順次添加した後、室温にて 12時間攪拌した 。反応液に水と飽和炭酸水素ナトリウム水溶液を加え、 EtOAcで抽出し、有機層を飽 和食塩水にて洗浄後、無水硫酸ナトリウムで乾燥した。減圧濃縮後、残渣をシリカゲ ルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 2 (V/V) )で精製して、 4-[4 - (ベンジルォキシ)フエノキシ] -N-(3-{[2- (メチルァミノ)フエニル]力ルバモイル}フエ二 ル)ピぺリジン- 1-カルボキサミド(234mg)を得た。
4-[4- (ベンジルォキシ)フエノキシ] -N-(3-{[2- (メチルァミノ)フエニル]力ルバモイル} フエニル)ピぺリジン- 1-カルボキサミド(230mg)の酢酸(6.0ml)溶液を、 80°Cで 3時間 攪拌後、減圧濃縮した。残渣を塩基性シリカゲルカラムクロマトグラフィー (溶出液;へ キサン: EtOAc = 1: 2 (V/V) )で精製し、ジイソプロピルエーテルにて結晶化後、加熱
乾燥して、 4-[4- (ベンジルォキシ)フエノキシ] -N-[3-(l-メチル -1H-ベンズイミダゾー ル -2-ィル)フエニル]ピぺリジン- 1-カルボキサミド(lOOmg)を得た。
[0097] 実施例 16
3- (ベンジルォキシ)安息香酸(3.50g)、 N-ピリジン- 3-ィルピペラジン- 1-カルボキサ ミドニ塩酸塩(4.29g)、 WSC (4.41g)及び HOBt (3.1 lg)の DMF (100ml)溶液に、 TEA ( 6.5ml)を滴下し、室温にて終夜攪拌した。反応液に水(100ml)を加え、 EtOAcにて抽 出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水にて順次洗浄し、無 水硫酸マグネシウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロマトダラ フィー (溶出液;クロ口ホルム:メタノール =20: 1 (V/V) )により精製し、無色油状物とし て、 4-[3_ (ベンジルォキシ)ベンゾィル] -N-ピリジン- 3-ィルピペラジン- 1-カルボキサ ミド(5.80g)を得た。得られた油状物(230mg)をエタノール(10ml)に溶解させ、シユウ 酸(99mg)を加え、 4-[3_ (ベンジルォキシ)ベンゾィル] -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミドシユウ酸塩(202mg)を得た。
[0098] 実施例 17
5- (シクロへキシルメトキシ)ニコチン酸メチル(303mg)のメタノール(5ml)溶液に、 1M 水酸化ナトリウム水溶液(1.56ml)を加え、室温にて 2時間攪拌した。反応液を 1M塩酸 水溶液を加えて酸性とした後、減圧濃縮し、残渣を次の反応に用いた。
N-ピリジン- 3-ィルピペラジン- 1-カルボキサミドニ塩酸塩(223mg)の DMF (5ml)溶 液に、 TEA(162mg)、上記で得られた残渣の DMF (3ml)溶液、 HOBt (108mg)、 WSC ( 230mg)を順次添加した後、室温にて 4時間攪拌した。反応液に水酸化ナトリウム水溶 液を加え、クロ口ホルムにて抽出した。有機層を無水硫酸ナトリウムで乾燥した後、減 圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノー ル =22: 3 (V/V) )で精製して、淡黄色油状物(332mg)を得た。得られた油状物のメタ ノール(5ml)溶液に、シユウ酸(141mg)を添加した後、ァセトニトリルを加え結晶化さ せて、淡黄色粉末の 4-{[5_ (シクロへキシルメトキシ)ピリジン- 3-ィル]カルボ二ル}-N- ピリジン -3-ィルピペラジン -1-カルボキサミドシユウ酸塩(351mg)を得た。
[0099] 実施例 18
N-ピリジン- 3-ィルピペリジン- 1-カルボキサミドニ塩酸塩(280mg)の DMF (15ml)溶
液に、 TEA (0.42ml)と 4-ペンチルベンゼン- 1-スルホユルクロリド(370mg)を加え、室 温にて終夜撹拌した。反応液に水を加え、 EtOAcで抽出した。有機層を水洗後、無 水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をへキサン/ EtOAcから結晶化させて 、淡橙色粉末の 4-[(4-ペンチルフエニル)スルホニル] -N-ピリジン- 3-ィルピペリジン- 1-カルボキサミド(244mg)を得た。
実施例 19
3,5-ビス (ァセチルォキシ)安息香酸(202mg)及び TEA (103mg)の Tol (5ml)溶液に、 ジフエニルホスホリルアジド(257mg)を加え 3時間攪拌した。反応液に水を加え EtOAc で抽出し、有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。減圧 濃縮して得られた残渣を Tolに溶解し、 100°Cで 4時間攪拌した。反応液を室温まで冷 却後、 1_[3-(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(200mg)及び T EA (58mg)のァセトニトリル(10ml)溶液に加え 2時間攪拌した。反応液を EtOAcで希 釈後、水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧濃縮後、残 渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 1〜1: 2 (V/V) )で精製し、 5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カル ボニノレ)ァミノ] -1,3-フエ二レンジアセテート(260mg)をアモルファスとして得た。
5_[({4-[3-(2-シクロへキシルエトキシ)ベンゾィノレ]ピぺラジン- 1-イノレ}カルボ二ノレ)ァ ミノ] -1,3-フエ二レンジアセテート(260mg)のイソプロパノール(5ml)溶液に、 1M水酸 化ナトリウム水溶液(2.4ml)を加え、 3時間攪拌した。反応液に 1M塩酸水溶液を加え 酸性にした後、 EtOAcにて抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグ ネシゥムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー (溶出液 ;クロ口ホルム:メタノール = 10: 0〜10: 1 (V/V) )で精製後、再度シリカゲルカラムクロ マトグラフィー(溶出液;へキサン: EtOAc = 2: 1〜1: 2 (V/V) )で精製し、 4-[3_(2_シク 口へキシルエトキシ)ベンゾィル] -N-(3,5-ジヒドロキシフエ二ノレ)ピぺラジン- 1-カルボ キサミド(54mg)を白色固体として得た。
実施例 20
メチル 5- (アジドカルボニル)ニコチネート(1.2g)の Tol溶液(20ml)を 120°Cで 1時間 攪拌した。この反応液を 1_[3-(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸
塩 (700mg)及び TEA (241mg)の THF (20ml)溶液に氷冷下加え、室温で 10時間攪拌 した。反応液を EtOAcにて希釈後、水、飽和炭酸水素ナトリウム水溶液、飽和食塩水 で順次洗浄し、無水硫酸マグネシウムで乾燥して、減圧濃縮した。残渣をシリカゲル カラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 10: 0〜5: 1 (V/V) )で精 製し、メチル 5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピペラジン- 1-ィル }力 ルポニル)ァミノ]ニコチネート(l.Og)をアモルファスとして得た。
メチル 5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カルボ ニル)ァミノ]ニコチネート(lOOmg)の THF (5ml) /エタノール(lml)混合溶液に水素化 ホウ素リチウム (44mg)を加え、 3時間攪拌した。反応液に飽和炭酸水素ナトリウム水 溶液を加え 10分間攪拌した。 EtOAcにて抽出後、有機層を飽和食塩水で洗浄し、無 水硫酸マグネシウムで乾燥して、減圧濃縮した。残渣をシリカゲルカラムクロマトダラ フィー(溶出液;クロ口ホルム:メタノール =5: 1 (V/V) )で精製し、 4-[3_(2-シクロへキ シルエトキシ)ベンゾィル] -N-[5- (ヒドロキシメチノレ)ピリジン- 3-ィノレ]ピぺラジン- 1-力 ルポキサミド(38mg)をアモルファスとして得た。
4- [3_(2-シクロへキシルエトキシ)ベンゾィル] -N-[5- (ヒドロキシメチル)ピリジン- 3-ィ ノレ]ピぺラジン- 1-カルボキサミド(38mg)の EtOAc (2.0ml)溶液に、 4M塩化水素/ EtO Ac溶液 (0.1ml)を加え、室温にて 3時間攪拌した。反応液を減圧濃縮して、 4-[3-(2- シクロへキシルエトキシ)ベンゾィル] -N-[5- (ヒドロキシメチノレ)ピリジン- 3-ィノレ]ピペラ ジン- 1-カルボキサミド塩酸塩(30mg)をアモルファスとして得た。
実施例 21
5-ブロモニコチン酸(20.0g)の Tol (300ml)溶液に、 TEA(12.0g)を加え、続いて氷冷 下ジフエニルホスホリルアジド(30.0g)を加えた。室温で 3時間攪拌した後、反応液を E tOAcで希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄した。有機 層を無水硫酸マグネシウムで乾燥後、減圧濃縮し、 5-ブロモニコチノィルアジド(26.5 g)を白色固体として得た。
5-ブロモニコチノィルアジド(2.95g)の Tol (30ml)溶液を 110°Cで 1時間攪拌した。反 応液に、氷冷下 1_[3-(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(3.0g )及び TEA(1.72g)の THF (50ml)溶液を加え、室温で 3時間攪拌した。反応液を減圧
濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 10 : 0〜10: 1 (V/V) )で精製し、 N-(5-ブロモピリジン- 3-ィル) -4-[3-(2-シクロへキ シルエトキシ)ベンゾィル]ピぺラジン- 1-カルボキサミド(3.57g)をアモルファスとして得 た。
N-(5-ブロモピリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピペラジ ン -1-カルボキサミド(150mg)、(3-ァミノカルボユルフェ二ノレ)ボロン酸(96mg)及び Pd(PPh ) (67mg)の Tol (7.5ml)懸濁液に、 2M炭酸ナトリウム水溶液(0.44ml)を加え、
3 4
100°Cで 3時間攪拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、 EtOAcで 抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃 縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 20: 1 (V/V) )で精製し、 N-[5-(3-力ルバモイルフエニル)ピリジン- 3-ィル] -4-[3_(2-シ クロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-カルボキサミド(66mg)を白色固体と して得た。
実施例 22
5-ヒドロキシニコチン酸メチル(5.0g)のアセトン(150ml)溶液に、炭酸カリウム(13.5g )及び臭化べンジル (9.71ml)を加え、 5時間加熱還流した。反応液を室温まで冷却後 、不溶物を濾去し、濾液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー( 溶出液; EtOAc:メタノール = 10 : 1 (V/V) )で精製して、 5-ベンジルォキシニコチン酸 メチル( 1.70g)を淡黄色結晶として得た。
5-ベンジルォキシニコチン酸メチル(1.70g)のメタノール(40ml) /THF(lOml)混合 溶液に、氷冷下 1M水酸化ナトリウム水溶液(7.34ml)を加え、室温で終夜攪拌した。 反応液を減圧濃縮後、析出した結晶を濾取し、 EtOAcで洗浄して、 5-ベンジルォキ シニコチン酸ナトリウム(1.66g)を得た。
5-ベンジルォキシニコチン酸ナトリウム(1.66g)の DMF (26ml)溶液に、室温で TEA( 1.10ml)を加え、さらに氷冷下でジフエ二ルホスホリルアジド(1.56ml)を加え、室温で 5 時間攪拌した。反応液に水を加え、 EtOAcで抽出し、有機層を飽和炭酸水素ナトリウ ム水溶液、飽和食塩水で順次洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮して 5 - (ベンジルォキシ)ニコチノィルアジドを得た。
5- (ベンジルォキシ)ニコチノィルアジド(1.68g)の Tol (20ml)溶液を 120°Cで 2時間攪 拌した。この溶液を 1-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(1. 55g)及び TEA (1.23ml)の THF (20ml)溶液に、氷冷下で加え、 2時間攪拌し、さらに 室温で 10時間攪拌した。反応液に水を加え、 EtOAcで抽出し、有機層を飽和炭酸水 素ナトリウム水溶液、飽和食塩水で順に洗浄後、無水硫酸ナトリウムで乾燥した。減 圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 2 : 1 (V/V) )で精製して、 N-[5- (ベンジルォキシ)ピリジン- 3-ィル] -4-[3_(2-シクロへキシ ルエトキシ)ベンゾィル]ピぺラジン- 1-カルボキサミド(1.78g)を淡黄色アモルファス状 物として得た。
N-[5- (ベンジルォキシ)ピリジン- 3-ィル] -4-[3_(2-シクロへキシルエトキシ)ベンゾィ ル]ピぺラジン- 1-カルボキサミド(1.78g)のメタノール(20ml)溶液に、 5%パラジウム炭 素 (698mg)を加え、水素雰囲気下室温で終夜攪拌した。触媒をセライトで濾去し、濾 液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム: メタノール = 10: 1 (V/V) )で精製して、 4-[3_(2-シクロへキシルエトキシ)ベンゾィル] - N-(5-ヒドロキシピリジン- 3-ィル)ピぺラジン- 1-カルボキサミド(1.15g)を淡黄色ァモル ファス状物として得た。
実施例 23
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-(5-ヒドロキシピリジン- 3-ィノレ)ピぺ ラジン- 1-カルボキサミド(200mg)、 2- (ベンジルォキシ)エタノール(0.126ml)及び PPh
(232mg)の THF (4.0ml)溶液に、氷冷下で DEAD (203mg、 40%Tol溶液)を加え、室温 で終夜攪拌した。反応液に水を加え、 EtOAcで抽出し、有機層を飽和食塩水で洗浄 後、無水硫酸ナトリウムで乾燥した。減圧濃縮後、残渣をシリカゲルカラムクロマトダラ フィー(溶出液;クロロホノレム:メタノーノレ = 10: 1 (V/V) )で精製して、 N-{5-[2- (ベンジ ルォキシ)エトキシ]ピリジン- 3-ィル }-4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピ ペラジン- 1-カルボキサミド(94mg)を褐色油状物として得た。
N-{5-[2- (ベンジルォキシ)エトキシ]ピリジン- 3-ィル }-4-[3_(2-シクロへキシルェトキ シ)ベンゾィル]ピぺラジン- 1-カルボキサミド(94mg)の EtOAc (5.0ml)溶液に、 5%パラ ジゥム炭素(34mg)を加え、水素雰囲気下で終夜攪拌した。さらに 5%パラジウム炭素
(lOOmg)を追加し、水素雰囲気下で 24時間攪拌した。触媒をセライトで濾去し、濾液 を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メ タノール = 10: 1 (V/V) )で精製して 4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-[ 5_(2-ヒドロキシエトキシピリジン- 3-ィル]ピぺラジン- 1-カルボキサミド(33mg)を淡黄 色アモルファス状物として得た。
[0104] 実施例 24
インドーノレ( 116mg)のァセトニトリノレ(10ml)溶液に、 CDI (169mg)と触媒量の DMAP を加え、 80°Cにて 6時間撹拌した。反応液を室温まで冷却後、 1-[3_(2-シクロへキシ ルエトキシ)ベンゾィル]ピぺラジン塩酸塩(350mg)の DMF (5ml)溶液を加え、 80°Cに て 10時間加熱した。反応液を室温まで冷却後、水を加えて EtOAcにて抽出した。有 機層を水洗後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラム クロマトグラフィー(溶出液;へキサン: EtOAc = 2: 1 (V/V) )で精製して、白色粉末の 1 -({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カルボニル) -1H- インドーノレ(288mg)を得た。
[0105] 実施例 25
4-[3_ (ベンジルォキシ)ベンゾィル] -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミド (5.4g)のエタノール(100ml)溶液に、 10%パラジウム炭素(500mg)を加え、水素雰囲 気下にて 5時間攪拌した。触媒をセライトで濾去し、濾液を減圧濃縮した。残渣を EtO Acから結晶化し、 4-(3_ヒドロキシベンゾィル) -N-ピリジン- 3-ィルピペラジン- 1-カル ボキサミド(3.9g)を白色固体として得た。
[0106] 実施例 26
4-(3_ヒドロキシベンゾィル) -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミド(300mg )、 2-フルォロベンジルアルコール(0.10ml)及び PPh (362mg)の THF (lOml)溶液に、
DEAD (0.63ml、 40%Tol溶液)を滴下し、室温で終夜攪拌した。反応液を減圧濃縮後 、残渣をシリカゲルカラムクロマトグラフィー (溶出液;クロ口ホルム:メタノール = 20: 1)に て精製して、無色油状の 4-{3_[(2-フルォロベンジル)ォキシ]ベンゾィル }-Ν-ピリジン- 3-ィルピペラジン- 1-カルボキサミド(298mg)を得た。得られた化合物をエタノール(1 0ml)に溶解させ、シユウ酸(186mg)を加えて室温にて 30分間攪拌した。析出した固
体を濾取し、エタノールで洗浄、加熱乾燥して、白色粉末の 4-{3_[(2-フルォロベンジ ル)ォキシ]ベンゾィル }-N-ピリジン- 3-ィルピペラジン- 1-カルボキサミドシユウ酸塩(1 40mg)を得た。
[0107] 実施例 27
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-(5-ヒドロキシピリジン- 3-ィノレ)ピぺ ラジン- 1-カルボキサミド(150mg)、ブロモ酢酸ェチル(0.077ml)及び炭酸カリウム(48 mg)の DMF (2.0ml)懸濁液を、室温で終夜攪拌した。反応液に水を加え、 EtOAcで抽 出し、有機層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥した。減圧濃縮後、 残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 10: 1 (V /V) )で精製して、ェチル({5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピペラジ ン- 1-ィル }カルボニル)ァミノ]ピリジン -3-ィル }ォキシ)アセテート(66mg)を褐色油状物 として得た。
ェチル({5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カル ボニノレ)ァミノ]ピリジン -3-ィル }ォキシ)アセテート(66mg)の THF (5.0ml) /メタノール(1 .0ml)混合溶液に、 1M水酸化ナトリウム水溶液(0.130ml)を加え、室温で終夜攪拌し た。反応液を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液 ;クロ口ホルム:メタノール = 10: 1 (V/V) )で精製し、({5-[({4-[3_(2-シクロへキシルエト キシ)ベンゾィル]ピぺラジン- 1-ィル }カルボニル)ァミノ]ピリジン- 3-ィル }ォキシ)酢酸( 38mg)を褐色アモルファス状物として得た。
[0108] 実施例 28
3- (ベンジルォキシ)ベンズアルデヒド(15.0g)の THF (250ml)溶液に、氷冷下、 tert- ブチルピぺラジン- 1-カルボキシレート塩酸塩(19.8g)、酢酸(6.1ml)及びNaBH(OAc ) (24.0g)を加えて室温にて 2時間撹拌した。反応液に 1M水酸化ナトリウム水溶液を 加えアルカリ性とし、クロ口ホルムで抽出した。有機層を無水硫酸マグネシウムで乾燥 し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー (溶出液;へキサン: EtO Ac = 3 : 1 (V/V) )にて精製して、無色油状の tert-ブチル 4_[3_ (ベンジルォキシ)ベン ジル]ピぺラジン- 1-カルボキシレート(26.4g)を得た。
tert-ブチル 4-[3_ (ベンジルォキシ)ベンジル]ピぺラジン- 1-カルボキシレート(24.0
g)の EtOAc溶液に、 4M塩化水素/ EtOAc溶液(70ml)を加え、室温にて 5時間攪拌し た。析出した固体を濾取し、 EtOAcで洗浄して、白色固体の 1-[3_ (ベンジルォキシ) ベンジル]ピぺラジン塩酸塩(18.8g)を得た。
1-[3_ (ベンジルォキシ)ベンジル]ピぺラジン塩酸塩(3.00g)及びフエニル 3-ピリジン 力ルバメート(2.02g)の DMF (45ml)溶液に TEA (3.3ml)を加え、 80°Cで 1時間撹拌し た。反応液を室温まで冷却後、飽和炭酸水素ナトリウム水溶液を加え、 EtOAcで抽出 した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥して減圧濃縮し た。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =20 : 1 (V/V) )にて精製し、淡褐色アモルファス状物として 4-[3_ (ベンジルォキシ)ベンジル] - N-ピリジン- 3-ィルピペラジン- 1-カルボキサミド(1.76g)を得た。
4-[3_ (ベンジルォキシ)ベンジル] -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミド( 200mg)をエタノール(10ml)に溶解させ、シユウ酸(90.0mg)を加えて室温にて 30分間 攪拌した。析出した固体を濾取し、エタノールで洗浄後、加熱乾燥して、白色粉末の 4-[3_ (ベンジルォキシ)ベンジル] -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミドシ ユウ酸塩(135mg)を得た。
実施例 29
水(5ml) /THF (5ml)混合溶媒に OXONE (登録商標、 376mg)を懸濁させ、室温で 4- [3_(2-シクロへキシルエトキシ)ベンゾィル] -N-[5- (メチルチオ) -1,3,4-チアジアゾー ル -2-ィル]ピぺラジン- 1-カルボキサミド(200mg)の THF溶液(5ml)を滴下し、 4時間 撹拌した。反応液に水を加え、 EtOAcで抽出し、有機層を水、飽和食塩水で洗浄後 、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;クロ口ホルム:メタノール = 20: 1 (V/V) )で精製し、へキサンにて結 晶化して、 4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-[5- (メチルスルホニル) -1, 3,4-チアジアゾール -2-ィル]ピぺラジン- 1-カルボキサミド(209mg)を無色結晶として 得た。
実施例 30
N-3-ピリジニル -1-ピぺラジンカルボキサミドニ塩酸塩(227mg)の THF (3.8ml)溶液 に、室温で TEA (0.114ml)を加え、 1時間攪拌した。反応液に 2_(2_シクロへキシルエト
キシ)イソニコチンアルデヒド(190mg)、 NaBH(OAc) (207mg)及び酢酸(0.0470ml)を 加え、終夜攪拌した。反応液に 1M水酸化ナトリウム水溶液を加えてアルカリ性にした 後、クロ口ホルムで抽出し、有機層を無水硫酸マグネシウムで乾燥した。減圧濃縮後 、残渣をシリカゲルカラムクロマトグラフィー(溶出液; EtOA メタノール = 10 : 1 (V/V) )で精製して、無色油状物を得た。この油状物を EtOAc (5.0ml)に溶解させ、 4M塩化 水素/ EtOAc溶液 (0.36ml)を加え、室温で 2時間攪拌した。析出した結晶を濾取し、 4 -{[2-(2-シクロへキシルエトキシ)ピリジン- 4-ィル]メチル }-N-ピリジン- 3-ィルピペラジ ン -1-カルボキサミド三塩酸塩 (338mg)を無色粉末として得た。
[0110] 実施例 31
5-[(tert-ブトキシカルボニル)ァミノ]ニコチン酸(900mg)の Tol (10ml)溶液に、 TEA ( 459mg)及びジフエニルホスホリルアジド(1.25g)を加え 1時間攪拌した。反応液を EtO Acにて希釈し、飽和炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄して、無水 硫酸マグネシウムで乾燥した。減圧濃縮後、得られた残渣を Tol (10ml)に溶解し、 10 0°Cで 3時間攪拌した。反応液を室温まで冷却後、 TEA(382mg)及び 1-[3_(2_シクロ へキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(1.33g)の THF(lOml)溶液に加え、 1 5時間攪拌した。反応液を減圧濃縮後、得られた残渣をシリカゲルカラムクロマトダラ フィー(溶出液;へキサン: EtOAc = 2 : 1 (V/V) )で精製し、 tert-ブチル {5_[({4-[3-(2_ シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル }カルボニル)ァミノ]ピリジン- 3- ィル }力ルバメート(1.31g)を白色固体として得た。
tert-ブチル {5-[({4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-ィル } カルボニル)ァミノ]ピリジン- 3-ィル }力ルバメート(1.31g)の EtOAc (20ml)溶液に、 4M 塩化水素/ EtOAc溶液(20ml)を加え、室温にて 3時間攪拌した。析出した固体を濾取 し、加熱乾燥して、 N-(5-アミノビリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベ ンゾィル]ピぺラジン- 1-カルボキサミド塩酸塩(956mg)を淡黄色固体として得た。
[0111] 実施例 32
N-(5-アミノビリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピペラジ ン -1-カルボキサミド塩酸塩(150mg)のジクロロメタン(5ml)溶液に、氷冷下 TEA(78m g)及びメタンスルホユルクロリド(39mg)を加え 3時間攪拌した。反応液に飽和炭酸水
素ナトリウム水溶液を加えクロ口ホルムで抽出した。有機層を無水硫酸マグネシウム で乾燥した後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口 ホルム:メタノール = 10: 0〜20: 1 (V/V) )で精製して、 4-[3_(2-シクロへキシルェトキ シ)ベンゾィル] -N-{5- [(メチルスルホニノレ)ァミノ]ピリジン- 3-イノレ}ピペラジン- 1-カルボ キサミド(63mg)を白色固体として得た。
実施例 33
6-クロ口ニコチノ二トリル(272mg)及び炭酸カリウム(544mg)の DMF (10ml)懸濁液に (2-シクロへキシルェチル)メチルァミン塩酸塩(350mg)を加え、 120°Cで 2時間攪拌し た。反応液を室温まで冷却後、 EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗 浄して、無水硫酸マグネシウムで乾燥した。減圧濃縮後、得られた残渣をエタノール (5ml)に溶解させ、 5M水酸化ナトリウム水溶液 (5ml)を加え、 100°Cで 6時間攪拌した。 反応液を室温まで冷却後、 1M塩酸水溶液を加え中和し、析出した結晶を濾取後、 加熱乾燥して、褐色固体を得た。得られた褐色固体、 N-ピリジン- 3-ィルピペラジン- 1-カルボキサミドニ塩酸塩(159mg)、 HOBt (92mg)及び TEA (0.16ml)の DMF (3ml) 溶液に WSC (131mg)を加え、室温で終夜攪拌した。反応液を EtOAcで希釈後、飽和 炭酸水素ナトリウム水溶液で洗浄し、無水硫酸マグネシウムで乾燥した。減圧濃縮後 、得られた残渣を分取 HPLC (ODSカラム、溶出液;水:ァセトニトリル = 60 : 40〜5: 95 ( V/V) )により精製し、固体を得た。得られた固体をへキサン/ EtOAcで洗浄後、加熱 乾燥して、 4-({6_[(2-シクロへキシルェチル )(メチル)ァミノ]ピリジン -3-ィル }カルボ二 ル)- N-ピリジン -3-ィルピペラジン -1-カルボキサミド(157mg)を無色粉末として得た。 実施例 34
メチル 3- (ブロモメチル)ベンゾエート(821mg)及び N-ピリジン- 3-ィルピペラジン- 1- カルボキサミドニ塩酸塩 ( l.OOg)の DMF (20ml)溶液に炭酸力リウム( 1.49g)を加え、 室温にて 2時間攪拌した。反応液に飽和炭酸水素ナトリウム水溶液を加え、 EtOAcで 抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃 縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =2 0 : 1 (V/V) )にて精製し、無色油状のメチル 3-({4- [(ピリジン -3-ィルァミノ)カルボニル ]ピペラジン- 1-ィル }メチル)ベンゾエート(995mg)を得た。
メチル 3_({4- [(ピリジン- 3-ィルァミノ)カルボニル]ピぺラジン- 1-ィル }メチル)ベンゾ エート(995mg)の THF (20ml)溶液に 1M水酸化ナトリウム水溶液(2.9ml)を加え、室温 にて終夜撹拌した。反応液を減圧濃縮し、残渣を Tol (50ml)で共沸して脱水した。 Et OAcを加え加熱還流後、析出した固体を濾取し、ピンク色粉末として、 3_({4- [(ピリジ ン -3-ィルァミノ)カルボニル]ピぺラジン- 1-ィル }メチル)安息香酸ナトリウム(995mg)を 得た。
実施例 35
3-ヒドロキシベンズアルデヒド(5.0g)、 2-シクロへキシルエタノール(6.82g)及び PPh
(14.0g)の THF (lOOml)溶液に、氷冷下 DEAD (21ml、 40%Tol溶液)を加え、室温で 5 時間攪拌した。反応液を減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー (溶 出液;へキサン: EtOAc = 10 : 1 (V/V) )で精製し、 3_(2-シクロへキシルエトキシ)ベン ズアルデヒド(8.81 g)を褐色油状物として得た。
3_(2-シクロへキシルエトキシ)ベンズアルデヒド(8.81g)の THF (100ml)溶液に、氷 冷下、 tert-ブチルピぺラジン- 1-カルボキシレート塩酸塩(7. lg)、酢酸(4.3ml)及び NaBH(OAc) (16. lg)を加えて室温で 2時間撹拌した。反応液に 1M水酸化ナトリウム 水溶液を加えアルカリ性とした後、 EtOAcで抽出した。有機層を飽和食塩水で洗浄し 、無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;へキサン: EtOAc = 3 : l (V/V) )で精製し、 tert-ブチル 4-[3_(2_シ クロへキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキシレート(11.6g)を無色油状 物として得た。
tert-ブチル 4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキシレ ート(11.6g)の EtOAc (100ml)溶液に、 4M塩化水素/ EtOAc溶液(100ml)を加え 3時 間攪拌した。反応液にジェチルエーテル(100ml)を加え 10分間攪拌した。析出した 固体を濾収し、加熱乾燥して、 1_[3-(2-シクロへキシルエトキシ)ベンジル]ピぺラジン 二塩酸塩 (9.67g)を白色固体として得た。
1-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン二塩酸塩(2.04g)及びシアン 酸カリウム(880mg)のメタノール(50ml)溶液に、 1M塩酸水溶液(11ml)を加え 1時間 攪拌した。反応液に 1M水酸化ナトリウム水溶液を加えアルカリ性とした後、 EtOAcで
抽出し、有機層を水、飽和食塩水で順次洗浄後、無水硫酸マグネシウムで乾燥した 。減圧濃縮後、得られた固体をへキサン/ EtOAcから再結晶し、 4-[3_(2-シクロへキシ ルエトキシ)ベンジル]ピペラジン- 1-カルボキサミド(1.41g)を白色固体として得た。
4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキサミド(200mg)の ジクロロメタン溶液に、 1,8-ジァザビシクロ [5·4·0]-7-ゥンデセン(530mg)及びァセチ ルクロリド(272mg)を加え室温で 3時間攪拌した。反応液に氷冷下、水を加え、 EtOAc で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧 濃縮した。得られた残渣をエタノールに溶解し、氷冷下、 1M水酸化ナトリウム水溶液( 0.6ml)を加え 30分攪拌した。反応液を減圧濃縮した後、水を加え、 EtOAcで抽出した 。有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。 残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= 10 : 1〜1: 1 (V /V) )で精製した。得られた油状物(142mg)をアセトンに溶解し、シユウ酸(10mg)をカロ え 10分攪拌した。析出した固体を濾収し、加熱乾燥して、 N-ァセチル -4_[3-(2-シク 口へキシルエトキシ)ベンジル]ピぺラジン- 1-カルボキサミドシユウ酸塩(122mg)を白 色固体として得た。
実施例 36
N-(6-ァセトアミドピリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピ ペラジン- 1-カルボキサミド(276mg)のメタノール(5ml) /THF (3ml)の混合溶液に 8M 水酸化ナトリウム水溶液(0.124ml)を加え、 60°Cで終夜攪拌した。反応液を EtOAcで 希釈し、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残 渣をシリカゲルカラムクロマトグラフィー(溶出液;クロロホノレム:メタノーノレ = 10: 1 (V/V ) )で精製し、 EtOAc/イソプロパノールから再結晶して、 N-(6-アミノビリジン -3-ィル) - 4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1-カルボキサミド(22mg)を 淡黄色固体として得た。
実施例 37
l-[3-(2-シクロへキシルェトキシ)べンゾィル]ピぺラジン塩酸塩(300mg)とTEA (0· 12 ml)をァセトニトリル(4.5ml)中に懸濁させ、室温で CDI (137mg)を加えて、透明溶液 になるまで撹拌した。反応液にメタンスルホンアミド(243mg)と 1,8-ジァザビシクロ [5.4.
0]-7_ゥンデセン (0.38ml)を加え、 60°Cで終夜撹拌した。反応液を室温まで冷却後減 圧濃縮し、 EtOAcを加えて水、飽和食塩水で順次洗浄した。無水硫酸マグネシウム で乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー (溶出液;クロ口 ホルム:メタノール =8: 2 (V/V) )で精製し、得られた油状物をエタノール/水から結晶 化して、加熱乾燥し、 4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N- (メチルスルホ ニル)ピペラジン- 1-カルボキサミド(343mg)を白色結晶として得た。
実施例 38
2-シクロへキシルエタノール(6.03ml)の DMF (75ml)溶液に、氷冷下で tert-ブトキシ カリウム(4.86g)を加え、室温にて 30分間攪拌した。反応液に 2-クロ口イソニコチノニト リノレ(5.00g)を加え、室温で 3日間攪拌した。反応液に水を加え、 EtOAcで抽出し、有 機層を飽和食塩水にて洗浄後、無水硫酸マグネシウムで乾燥した。減圧濃縮後、残 渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精 製して、 2-(2-シクロへキシルエトキシ)イソニコチノ二トリル(6.97g)を無色粉末として 得た。
2-(2-シクロへキシルエトキシ)イソニコチノ二トリル(6.97g)のエタノール(80ml)溶液 に、 5M水酸化ナトリウム水溶液(60ml)を加え、 100°Cで終夜攪拌した。反応液を室温 まで冷却後、反応液に 1M塩酸水溶液を加え酸性とした。析出した固体を濾取し、加 熱乾燥して、 2-(2-シクロへキシルエトキシ)イソニコチン酸(6.90g)を無色粉末として 得た。
2-(2-シクロへキシルエトキシ)イソニコチン酸(3.00g)、 WSC (2.77g)及び HOBt (1.95 g)の DMF (60ml)溶液を 30分間攪拌後、 tert-ブチルピぺラジン- 1-カルボキシレート 塩酸塩(2.24g)を加え、 4時間攪拌した。反応液に水を加え、 EtOAcで抽出し、有機 層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。減圧濃縮後、残渣を シリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 4: 1 (V/V) )で精製し 、 tert-ブチル 4-[2-(2-シクロへキシルエトキシ)イソニコチノィル]ピぺラジン- 1-カル ボキシレート(5.14g)を無色アモルファス状物として得た。
tert-ブチル 4-[2-(2-シクロへキシルエトキシ)イソニコチノィル]ピぺラジン- 1-カル ボキシレート(5.14g)の EtOAc (50ml)溶液に、 4M塩化水素/ EtOAc溶液(50ml)を加
え、終夜攪拌した。生じた固体を濾取し、加熱乾燥して、 1_[2-(2-シクロへキシルエト キシ)イソニコチノィル]ピぺラジン二塩酸塩 (4.78g)を無色粉末として得た。
2-アミノビラジン(5.00g)のピリジン(50ml)溶液に、氷冷下でクロロギ酸フエニル(6.9 7ml)を加え、室温で 4時間攪拌した。反応液を減圧濃縮後、残渣に飽和炭酸水素ナ トリウム水溶液を加え、 EtOAcで抽出し、有機層を飽和食塩水で洗浄後、無水硫酸ナ トリウムで乾燥した。減圧濃縮後、残渣をシリカゲルカラムクロマトグラフィー (溶出液; へキサン: EtOAc = 2 : l (V/V) )にて精製し、フエニルピラジン- 2-ィルカルバメート(2 . l lg)を無色粉末として得た。
1-[2-(2-シクロへキシルエトキシ)イソニコチノィル]ピぺラジン二塩酸塩(362mg)及 びフエ二ノレピラジン- 2-ィルカルバメート(200mg)のァセトニトリノレ(10ml)溶液に、 TE A (0.259ml)を加え、終夜攪拌した。反応液に、飽和炭酸水素ナトリウム水溶液を加え 、 EtOAcで抽出し、有機層を飽和食塩水で洗浄後、無水硫酸マグネシウムにて乾燥 した。減圧濃縮後、残留物をシリカゲルカラムクロマトグラフィー (溶出液; EtOAc)で 精製し、 4-[2-(2-シクロへキシルエトキシ)イソニコチノィル] -N-ピラジン- 2-ィルピペラ ジン- 1-カルボキサミド(136mg)を無色アモルファス状物として得た。
実施例 39
4-(3_ヒドロキシベンジル) -N-ピリジン- 3-ィルピペラジン- 1-カルボキサミド(600mg) 、メチル 3- (ヒドロキシメチル)ベンゾエート(479mg)及び PPh (756mg)の THF (20ml) 溶液に DEAD (1.4ml、 40%Tol溶液)を滴下し、室温で終夜攪拌した。反応液を減圧濃 縮し、得られた残渣をシリカゲルカラムクロマトグラフィー (溶出液;クロ口ホルム:メタノ ール = 20 : 1 (V/V) )により精製し、無色油状のメチル 3-{[3_({4- [(ピリジン -3-ィルアミ ノ)カルボニル]ピぺラジン- 1-ィル }メチル)フエノキシ]メチル }ベンゾェート(618mg)を得 た。
メチル 3-{[3_({4- [(ピリジン- 3-ィルァミノ)カルボニル]ピぺラジン- 1-ィル }メチル)フエ ノキシ]メチノレ }ベンゾェート(615mg)の THF (15ml) /メタノール(1.5ml)混合溶液に 1M 水酸化ナトリウム水溶液(1.4ml)を加え、 50°Cにて 1時間攪拌した。反応液を減圧濃 縮し、残渣に Tol (50ml)を加え共沸して脱水した。残渣に EtOAcを加え、 30分間加熱 還流して生じた固体を濾取し、白色固体として、 3-{[3_({4- [(ピリジン -3-ィルァミノ)力
ルポ二ノレ]ピぺラジン- 1-ィル }メチル)フエノキシ]メチノレ }安息香酸ナトリウム(608mg)を 得た。
[0117] 実施例 40
3_{[3-({4- [(ピリジン -3-ィルァミノ)カルボニル]ピペラジン- 1-ィル }メチル)フエノキシ] メチル }安息香酸ナトリウム(320mg)、塩化アンモニゥム(l lOmg)、 WSC (197mg)及び HOBt (139mg)の DMF (15ml)溶液を室温にて終夜攪拌した。反応液に飽和炭酸水 素ナトリウム水溶液を加え、 EtOAcにて抽出した。有機層を飽和食塩水で洗浄後、無 水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィ 一 (溶出液;クロ口ホルム:メタノール = 10: 1 (V/V) )により精製し、無色油状の 4-(3_{[3 - (ァミノカルボニル)ベンジル]ォキシ }ベンジル) -N-ピリジン- 3-ィルピペラジン- 1-カル ボキサミド(267mg)を得た。
4_(3-{[3- (ァミノカルボニル)ベンジル]ォキシ }ベンジル) -N-ピリジン- 3-ィルピペラジ ン -1-カルボキサミド(265mg)のエタノール(10ml)溶液にシユウ酸(161mg)を加え、室 温にて 30分間撹拌した。析出した固体を濾取した後、エタノールで洗浄して、白色粉 末の 4-(3-{[3_ (ァミノカルボニル)ベンジル]ォキシ }ベンジル) -N-ピリジン- 3-ィルピぺ ラジン- 1-カルボキサミドニシュゥ酸塩(201mg)を得た。
[0118] 実施例 41
[3- (メトキシカルボニル)ベンジル] (トリフエニル)ホスホニゥムブロミド(1.42g)とシクロ へキシルァセトアルデヒド(5.50g)の DMF (80ml)溶液に氷冷下 60%水素化ナトリウム(5 13mg)を加え、室温にて終夜攪拌した。反応液に水(100ml)を加え、 EtOAcにて抽出 した。有機層を飽和食塩水にて洗浄し、無水硫酸マグネシウムで乾燥して減圧濃縮 した。残渣にジェチルエーテル(100ml)、へキサン(200ml)を加えて不溶物を濾去し た。濾液を減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 9 : 1 (V/V) )により精製し、無色油状のメチノレ 3-[3_シクロへキシル -1-プロ ペン- 1-ィル]ベンゾエート(2.29g)を得た。
メチル 3-[3_シクロへキシル -1-プロペン- 1-ィル]ベンゾエート(2.28g)のエタノーノレ 溶液に 10%パラジウム炭素(500mg)を加え、水素雰囲気下にて 5時間攪拌した。触媒 をセライトで濾去し、濾液を減圧濃縮して、無色油状のメチル 3-(3_シクロへキシルプ
口ピル)ベンゾエート(2.29g)を得た。
メチル 3-(3_シクロへキシルプロピル)ベンゾエート(720mg)のメタノール(10ml)溶液 に 1M水酸化ナトリウム水溶液(5.0ml)を加え、 50°Cにて 2時間攪拌した。反応液を室 温まで冷却後、 1M塩酸水溶液を加えて酸性とし、 EtOAcにて抽出した。有機層を無 水硫酸マグネシウムで乾燥して、減圧濃縮し、白色結晶の 3-(3_シクロへキシルプロ ピル)安息香酸 (660mg)を得た。
3- (3_シクロへキシルプロピル)安息香酸(300mg)、 N-ピリジン- 3-ィルピペラジン- 1- カルボキサミドニ塩酸塩(374mg)、 WSC (257mg)及び HOBt (182mg)の DMF (lOml) 溶液に、 TEA (0.40ml)を加え、室温にて終夜攪拌した。反応液に水(20ml)を加え、 E tOAcにて抽出した。有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水にて順次 洗浄し、無水硫酸マグネシウムで乾燥して減圧濃縮した。残渣をシリカゲルカラムクロ マトグラフィー(溶出液;クロ口ホルム:メタノール =20: 1 (V/V) )により精製し、無色油 状の 4-[3-(3_シクロへキシルプロピル)ベンゾィル] -N-ピリジン- 3-ィルピペラジン- 1- カルボキサミド(185mg)を得た。
4- [3-(3_シクロへキシルプロピル)ベンゾィル] -N-ピリジン- 3-ィルピペラジン- 1-力 ルポキサミド(180mg)のエタノール(15ml)溶液に、シユウ酸(112mg)を加えて室温に て 30分間攪拌した。析出した固体を濾取し、エタノールで洗浄後、加熱乾燥して、白 色粉末の 4-[3-(3_シクロへキシルプロピル)ベンゾィル] -N-ピリジン- 3-ィルピペラジ ン -1-カルボキサミドシユウ酸塩(158mg)を得た。
実施例 42
6-クロ口ニコチノ二トリノレ(2.00g)と 2-シクロへキシルエタノール(2.23g)の DMF (30ml )溶液に、氷冷下 tert-ブトキシカリウム(1.95g)を加え、室温にて終夜攪拌した。反応 液に水を加え、 EtOAcで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸マグネ シゥムで乾燥して減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー (溶出液; へキサン: EtOAc = 4: 1 (V/V) )により精製し、無色油状の 6_(2-シクロへキシルェトキ シ)ニコチノ二トリル(2.86g)を得た。
6_(2-シクロへキシルエトキシ)ニコチノ二トリル(2.00g)の Tol (150ml)溶液に- 78°Cに て DIBAL (8.7ml、 1M Tol溶液)を滴下し、 _78°Cにて 1時間攪拌した。反応液に飽和
塩化アンモニゥム水溶液(30ml)及び EtOAcを加えて 30分間攪拌し、 EtOAcで抽出し た。有機層を無水硫酸マグネシウムで乾燥して減圧濃縮した。残渣をシリカゲルカラ ムクロマトグラフィー(溶出液;へキサン: EtOAc = 3 : l (V/V) )にて精製し、無色油状 の 6_(2-シクロへキシルエトキシ)ニコチンアルデヒド(1.62g)を得た。
6_(2-シクロへキシルエトキシ)ニコチンアルデヒド(200mg)の THF (20ml)懸濁液に T EA (0.12ml)を加え、室温にて 1時間攪拌した。これに N-ピリジン- 3_ィルピペラジン- 1 -カルボキサミド(240mg)、酢酸(50ul)、 NaBH(OAc) (219mg)を順次加えて室温にて 終夜攪拌した。反応液を 1M水酸化ナトリウム水溶液でアルカリ性とし、クロ口ホルムで 抽出した。有機層を無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲ ルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 3: 1 (V/V) )にて精製して、無 色油状の 4-{[6_(2-シクロへキシルエトキシ)ピリジン- 3-ィル]メチル }-N-ピリジン- 3-ィ ルビペラジン- 1-カルボキサミド(297mg)を得た。
4-{[6_(2-シクロへキシルエトキシ)ピリジン- 3-ィル]メチル }-N-ピリジン- 3-ィルピペラ ジン- 1-カルボキサミド(297mg)のエタノール(15ml)溶液に、シユウ酸(190mg)を加え て室温にて 30分間攪拌した。析出した固体を濾取し、エタノールで洗浄後、加熱乾 燥して、白色粉末の 4-{[6_(2-シクロへキシルエトキシ)ピリジン -3-ィル]メチルト N-ピリ ジン- 3-ィルピペラジン- 1-カルボキサミドニシュゥ酸塩(390mg)を得た。
実施例 43
ベンジルアルコール(72ml)に氷冷下で 60%水素化ナトリウム(2.0g)を加え、 15分間 攪拌した。反応液に 6-クロ口- 3-ピリダジンァミン (3.0g)を加え、室温で 2時間、 50°Cで 2時間、 80°Cで終夜攪拌した。反応液を室温まで冷却し、水を加え、 EtOAcで抽出し た。有機層を無水硫酸ナトリウムで乾燥した。減圧濃縮後、残渣をシリカゲルカラムク 口マトグラフィー(溶出液; EtOAc:メタノーノレ = 10: 1 (V/V) )で精製し、 6- (ベンジルォ キシ)ピリダジン- 3 -ァミン(1.78g)を褐色粉末として得た。
6- (ベンジルォキシ)ピリダジン- 3-ァミン(867mg)のピリジン(10ml)溶液に、氷冷下ク ロロギ酸フヱニル (0.571ml)を加え 1時間撹拌後、室温で 2時間攪拌した。反応液を減 圧濃縮した後、残渣に EtOAcを加えて析出した固体をジイソプロピルエーテル、水で 順次洗浄し、加熱乾燥して、フエニル [6- (ベンジルォキシ)ピリダジン- 3-ィル]力ルバ
メート(1.05g)を褐色粉末として得た。
フエニル [6- (ベンジルォキシ)ピリダジン- 3-ィル]力ルバメート(500mg)及び 1-[3-(2 -シクロへキシルエトキシ)ベンゾィル]ピぺラジン塩酸塩(500mg)のァセトニトリル(10m 1)溶液に、 TEA (0.197ml)を加え、終夜攪拌した。反応液に EtOAcを加え、生じた不 溶物を濾去し、濾液を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出 液; EtOAc)で精製し、ジイソプロピルエーテルにて結晶化させて、 N-[6- (ベンジルォ キシ)ピリダジン- 3-ィル] -4-[3_(2-シクロへキシルエトキシ)ベンゾィル]ピぺラジン- 1- カルボキサミド(716mg)を無色粉末として得た。
[0121] 実施例 44
N-[6- (ベンジルォキシ)ピリダジン- 3-ィル] -4-[3_(2-シクロへキシルエトキシ)ベンゾ ィノレ]ピぺラジン- 1-カルボキサミド(448mg)のエタノール(40ml) /メタノーノレ(20ml)混 合溶液に、 10%パラジウム炭素 (44mg)を加え、水素雰囲気下で終夜攪拌した。触媒 をセライトで濾去し、濾液を減圧濃縮した。得られた固体をへキサンで洗浄し、 4-[3-( 2-シクロへキシルエトキシ)ベンゾィル] -N-(6-ヒドロキシピリダジン- 3-ィル)ピぺラジン -1-カルボキサミド(299mg)を灰白色粉末として得た。
実施例 45
4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N- (メチルスルホニル)ピぺラジン- 1- カルボキサミド(l lOmg)と炭酸カリウム(105mg)の DMF (1.6ml)懸濁液にヨウ化メチル (0.08ml)を加え、室温で 3日間撹拌した。反応液に EtOAcを加えて水、飽和炭酸水 素ナトリウム水溶液、飽和食塩水で順次洗浄した。無水硫酸マグネシウムで乾燥後、 減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノ ール =97: 3 (V/V) )で精製し、 4-[3_(2-シクロへキシルエトキシ)ベンゾィル] -N-メチ ル -N- (メチルスルホニル)ピペラジン- 1-カルボキサミド(57mg)を白色結晶として得た
[0122] 実施例 46
N-(5-ァミノ- 2-ピリジル)ァセトアミド(810mg)のピリジン(20ml)溶液に、氷冷下でクロ ロギ酸フエニル (0.744ml)を加え、室温で 1時間攪拌した。反応液を減圧濃縮後、残 渣に水を加え、 EtOAcで抽出した。有機層を飽和食塩水にて洗浄後、無水硫酸マグ
ネシゥムで乾燥し、減圧濃縮して、フエニル(6-ァセトアミドピリジン- 3-ィル)カルバメ ート(1800mg)を淡黄色粉末として得た。
フエ二ノレ(6-ァセトアミドピリジン- 3-ィル)力ルバメート(291mg)と 1-[3_(2-シクロへキ シルエトキシ)ベンジル]ピぺラジン二塩酸塩(383mg)のァセトニトリル(20ml)溶液に、 TEA (0.284ml)を加え、 1日間攪拌した。析出した固体を濾取し、シリカゲルカラムクロ マトグラフィー(溶出液;クロ口ホルム:メタノール = 10: 1 (V/V) )で精製して、 N-(6-ァ セトアミドピリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺラジン- 1- カルボキサミド(353mg)を無色アモルファス状物として得た。
N-(6-ァセトアミドピリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ)ベンジル]ピぺ ラジン- 1-カルボキサミド(353mg)の THF (5.0ml) /メタノール(3.0ml)混合溶液に、室 温で 8M水酸化ナトリウム水溶液(0.28ml)を加え、 70°Cで 2日間攪拌した。反応液に 水を加え、 EtOAcで抽出した。有機層を飽和食塩水で洗浄後、無水硫酸マグネシゥ ムで乾燥し、減圧濃縮した。残渣を塩基性シリカゲルクロマトグラフィー(溶出液; EtO A メタノール = 10 : 1 (V/V) )で精製し、得られた淡黄色油状物をジイソプロピルエー テルにて結晶化させ、 N-(6-アミノビリジン- 3-ィル) -4-[3_(2-シクロへキシルエトキシ) ベンジル]ピぺラジン- 1-カルボキサミド(96mg)を淡黄色粉末として得た。
[0123] 実施例 441
4-(5_ヒドロキシ -1-ベンゾチォフェン- 2-ィノレ)カルボニル] -N-ピラジン- 2-ィルピぺ ラジン- 1-カルボキサミド(188mg)、 3-フルォロベンジルアルコール(93mg)及び PPh (
192mg)の THF (5ml)溶液に、氷冷下 DEAD (0.33ml、 40%Tol溶液)を加え、室温で終 夜攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄後、無 水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィ 一(溶出液;クロ口ホルム:メタノール = 20 : 1 (V/V) )で精製し、固体を得た。得られた 固体をへキサン/ EtOAcで洗浄後、加熱乾燥して、 4-({5-[(3_フルォロベンジル)ォキ シ] -1-ベンゾチォフェン- 2-ィル }カルボニル) -N-ピラジン- 2-ィルピペラジン- 1-カル ボキサミド(71mg)を白色粉末として得た。
[0124] 実施例 442
1-[(5-フエニル -2-チェニル)メチル]ピぺラジン二塩酸塩(0.30g)及びフエニルビラ
ジン- 2-ィルカルバメート(390mg)の DMF (6ml)溶液に、 TEA (0.38ml)を加え、 80°Cで 2時間攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄後 、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグ ラフィー(溶出液;クロ口ホルム:メタノール = 20: 1 (V/V) )、更に塩基性シリカゲルカラ ムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 1 (V/V) )で精製し、 4-[(5-フエ二 ル -2-チェニル)メチル] -N-ピラジン- 2-ィルピペラジン- 1-カルボキサミド(143mg)を 白色粉末として得た。
実施例 443
4-フエ二ルチオフェン- 2-アルデヒド(134mg)と N-2-ピラジュル -1-ピぺラジンカルボ キサミドニ塩酸塩(200mg)の THF (5ml)溶液に、酢酸(43mg)と DMF (1.25ml)を加え、 室温にて 1時間撹拌した。反応液に NaBH(OAc) (302mg)を加え、室温にて終夜撹 拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫 酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー( 溶出液;クロ口ホルム:メタノール = 20 : 1 (V/V) )で精製し、固体を得た。得られた固 体をへキサン/ EtOAcで洗浄後、加熱乾燥して、 4-[(4-フエニル -2-チェニル)メチル] -N-ピラジン- 2-ィルピペラジン -1-カルボキサミド(65mg)を白色固体として得た。 実施例 444
ジフエニル(3-クロロビラジン- 2-ィル)イミドジカルボネート(284mg)及び 1-[2_(2_シ クロへキシルエトキシ)イソニコチノィル]ピぺラジン二塩酸塩(300mg)のァセトニトリル( 10ml)溶液に TEA (0.43ml)を加え、 80°Cで 1時間攪拌した。反応液を EtOAcで希釈し 、飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃 縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 20 : 1 (V/V) )で精製し、固体を得た。得られた固体をへキサン/ EtOAcで洗浄後、カロ 熱乾燥して、 N-(3-クロロビラジン- 2-ィル) -4-[2-(2-シクロへキシルエトキシ)イソニコ チノィル]ピぺラジン- 1 -カルボキサミド( 133mg)を白色固体として得た。
実施例 445
4- (メチルァミノ) -N-ピラジン- 2-ィルピペリジン- 1-カルボキサミドニ塩酸塩(314mg) 及び 5-クロ口- 3-フエニル -1,2,4-チアジアゾール(200mg)の DMF (5ml)溶液に TEA (0
.57ml)を加え、室温で終夜攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリ ゥム水溶液で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカ ゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 1 (V/V) )で精製した。得 られた固体をへキサン/ EtOAcで洗浄後、加熱乾燥して、 4- [メチル (3-フエニル -1,2,4 -チアジアゾール -5-ィル)ァミノ] -N-ピラジン- 2-ィルピペリジン- 1-カルボキサミド(25 4mg)を白色固体として得た。
[0126] 実施例 446
N-(3-クロロビラジン- 2-ィル)ピぺラジン- 1-カルボキサミドニ塩酸塩(lOOmg)、 2_[2_ (2-クロ口フエニル)エトキシ]イソニコチン酸(97mg)、 WSC (76mg)及び HOBt (54mg)の DMF (3ml)溶液に、 TEA (0.088ml)を加え、室温にて終夜攪拌した。反応液を EtOAc で希釈し、飽和炭酸水素ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥し 、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;クロ口ホルム:メタ ノール = 20: 1 (V/V) )で精製した。得られた固体をへキサン/ EtOAcで洗浄後、加熱 乾燥して、 4-{2-[2-(2-クロ口フエニル)エトキシ]イソニコチノィルト N-(3-クロロビラジン -2-ィル)ピぺラジン- 1-カルボキサミド(89mg)を白色固体として得た。
実施例 447
N-(3-クロロビラジン- 2-ィル)ピぺラジン- 1-カルボキサミドニ塩酸塩(150mg)、 2_{[2 -(3-フルオロフェニノレ)ェチノレ]アミノ}イソニコチン酸(124mg)及び 0-(7_ァザべンゾトリ ァゾール-1-ィル)- , -テトラメチルゥロニゥムへキサフルォロホスフェート(2 71mg)の DMF (5ml)溶液に、ジイソプロピルェチルァミン(0.25ml)を加え、室温にて 終夜攪拌した。反応液を EtOAcで希釈し、飽和炭酸水素ナトリウム水溶液で洗浄後、 無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトダラ フィー(溶出液;クロ口ホルム:メタノール = 10: 1 (V/V) )で精製し、得られた固体をへ キサン/ EtOAcで洗浄後、加熱乾燥して、 N-(3-クロロビラジン- 2-ィル) -4-(2-{[2-(3_ フルオロフェニノレ)ェチノレ]アミノ}イソニコチノィル)ピぺラジン- 1-カルボキサミド(22mg )を白色固体として得た。
[0127] 実施例 448
4-(3_ヒドロキフエノキシ) -N-ピラジン- 2-ィルピペリジン- 1-カルボキサミド(150mg)、
2-シクロへキシルエタノール(91mg)及び PPh (187mg)の THF (5ml)溶液に、 DEAD (
0.33ml、 40%Tol溶液)を加え、室温にて終夜攪拌した。反応液を EtOAcで希釈し、 1M 水酸化ナトリウム水溶液、水にて順次洗浄後、無水硫酸マグネシウムで乾燥し、減圧 濃縮した。残渣をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc= 1: 2 (V/V) )で精製し、 4-[3_(2-シクロへキシル)フエノキシ] -N-ピラジン- 2-ィルピペリジン -1-カルボキサミド(72mg)をアモルファスとして得た。
[0128] 実施例 449,及び 450
tert-ブチル 4-[(2-クロ口ピリジン- 4-ィル)ォキシ]ピぺリジン- 1-カルボキシレート(50 Omg)及び 2-シクロへキシルエタノール(307mg)の DMF (lOml)溶液に、 60%水素化ナ トリウム(95mg)を加え 80°Cで 2時間攪拌した。反応液を EtOAcで希釈し、飽和炭酸水 素ナトリウム水溶液で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣 をシリカゲルカラムクロマトグラフィー(溶出液;へキサン: EtOAc = 3: 1 (V/V) )で精製 し、油状物を得た。得られた油状物を EtOAc (5mL)に溶解し、 4M塩化水素/ EtOAc ( 5mL)を加え室温で終夜攪拌した。反応液を減圧濃縮後、フエニルピラジン- 2-ィル 力ルバメート(129mg)、丁5八(0.1½11)及び0\^ (3011)を加え 80°Cで 1時間攪拌した。 反応液を EtOAcで希釈し、水、飽和炭酸水素ナトリウム水溶液で順次洗浄後、無水 硫酸マグネシウムで乾燥し、減圧濃縮した。残渣を分取 HPLC (ODSカラム、溶出液; 水:ァセトニトリル = 1: 1)で精製し、 4-{[2-(2-シクロへキシルエトキシ)ピリジン -4-ィル] ォキシ }-N-ピラジン- 2-ィルピペラジン- 1-カルボキサミド(43mg:実施例 449)、 4_{[4_ (2-シクロへキシルエトキシ)ピリジン- 2-ィル]ォキシ }-N-ピラジン- 2-ィルピペラジン- 1 -カルボキサミド(55mg:実施例 450)をそれぞれ白色固体として得た。
[0129] 実施例 451
60%水素化ナトリウム(95mg)の DMF (12ml)懸濁液に、 4_(1H_インドール- 3_ィルメ チル) -N-ピラジン- 2-ィルピペラジン- 1-カルボキサミド(400mg)を加え、室温にて 30 分間撹拌した。反応液に 1- (ブロモメチル) -3-フルォロベンゼン(247mg)を加え、室 温にて終夜撹拌した。反応液に水を加え、 EtOAcにて抽出した。有機層を飽和食塩 水にて洗浄後、無水硫酸マグネシウムにて乾燥し、減圧濃縮した。残渣をシリカゲル カラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール = 10: 1 (V/V) )にて精製し
て、淡黄色油状物を得た。得られた油状物を EtOAc (5ml)に溶解させ、 4M塩化水素/ EtOAc溶液(lml)を加え、析出した固体を濾取し、加熱乾燥して、淡黄色粉末の 4-{[ 1-(3_フルォロベンジル) -1H-インドール- 3-ィル]メチル }-N-ピラジン- 2-ィルピペラジ ン- 1 -カルボキサミドニ塩酸塩 (315mg)を得た。
実施例 452
1-(2-フエニルェチル)ピぺラジン二塩酸塩(400mg)の DMF(8ml)溶液に、ベンゾィ ルイソシァネート(245mg)と TEA (0.42ml)を加え、室温にて終夜撹拌した。反応液に 飽和炭酸水素ナトリウム水溶液を加え、 EtOAcにて抽出した。有機層を飽和食塩水 にて洗浄後、無水硫酸マグネシウムにて乾燥し、減圧濃縮した。残渣をシリカゲル力 ラムクロマトグラフィー(溶出液;クロ口ホルム:メタノール =95: 5 (V/V) )にて精製して 、 白色粉末の N-ベンゾィル -4-(2-フエニルェチル)ピぺラジン- 1-カルボキサミド(512 mg)を得た。
[0130] 上記実施例 1〜46,及び 44;!〜 452の方法と同様にして、後記表 18〜76に示す 実施例化合物 1〜492をそれぞれ対応する原料を使用して製造した。表 18〜 76に 実施例化合物の製造法及び物理化学的データを示す。また、いくつかの実施例化 合物の NMRデータを後記表 77〜81に示す。
[0131] [表 3]
9Cl990/.00idf/13d 06
[ i [εετο]
9ei990/-00Zdf/I3d !■6
[9挲] [ ΐΟ]
9CZ990/.00Zdf/X3d 36 OZ 蘭 00Z OAV
9CZ990/.00Zdf/X3d 86 OZ 蘭 00Z OAV
[8挲] [9C TO]
9£Z990/L00ZdT/13d V6 蘭 00Z OAV
9CZ990/.00Zdf/X3d 96 OZ 蘭 00Z OAV
10]
11]
9£Z990/L00Zd£/13d 86 OZ 蘭 00Z OAV
9£Z990/L00ZdT/13d 001- OZ 00Z OAV
15]
16]
7]
[81 ¾ [9W0]
9£Z990/L00Zd£/13d V0\. OAV
9]
901- OZ 蘭 00Z OAV
1]
22]
3]
24]
5]
]
27]
28]
9]
30]
1]
32]
33]
]
5]
d 0UZ0/800Z OAV
7]
[8ε挲] [99ΐ0]
d OZ 蘭 00Z OAV
[6ε挲] [ 9ΐ0]
931- OZ 蘭 00Z OAV
0]
1]
42]
43]
44]
5]
46]
47]
[晦] [9 Ϊ0]
13d 881- OZ 蘭 00Z OAV
d 6m OZ蘭 00Z OAV
64]
65]
66]
67]
]
[69挲] [ 6Ϊ0]
d 991- ο ε蘭 οοζ Ο
70]
71]
72]
73]
4]
〕 D¾〔07702
S D¾80082
¾8
Ex 瞧
NMR1:0.86-0.90 (3H, m), 1.29-1.34 (4H, m), 1.38-1.44 (2H, m) .1.68-1.74 (2H, m)
400 , 2.87-2.92 (4H, m) , 3.63-3.66 (4H, m) , 3.95-4.00 (2H, m), 6.93-7.03 (3H, m) , 7.2 8-7.33 (2H, m), 7.88 (1 H,m),8.17 (1H, m) , 8.64 (1H, m) , 8.91 (1 H, s) 画 :3.09-3.48 (8H, m) , 5.25 (2H, s) , 7.13-7.29 (3H, m) , 7.38-7.50 (4H, m) , 1.92
409
(1 H, m) , 8.50 (1 H, m) , 8.63 (1 H,m),9.13 (1 H, m), 10.35 (1 H, s)
NMR1 :3.08-3.47 (8H, m), 4.34 (2H, s) , 5.15 (2H, s) , 7.13-7.23 (3H, m) , 7.39-7.45
412 (2H,m),7.51-7.59(2H,m),7.91 (1H, m) , 8.49(1H, m) , 8.62 (1H, m) , 9.11 (1H, m) , 1 0.32(1H, s)
NMR1: 2.76-2.83 (4H, m) , 3.05-3.09 (2H, m) , 3.57-3.63 (4H, m) , 3.90 (2H, s) , 4.19
415 -4.23 (2H, m) , 6.93-7.08 (4H, m) , 7.17-7.21 (2H, m) , 7.27-7.39 (3H, m) , 7.87 (1 H, m),8.17(1H,m),8.63 (1H, m) , 8.85 (1H, s)
NMR1 :3.08-3.47 (8H, m) , 4.33 (2H, s) , 5.21 (2H, s) , 7.09-7.23 (5H, m) , 7.37-7.46
416
(2H, m) , 7.92 (1H, m) , 8.49(1H, m) 8.65(1H, m) ,9.13(1H,m), 10.38(1H, s) 隱 1: 1.99-2.05 (2H, m) , 2.73-2.77 (2H, m) , 2.86-2.90 (4H, m) , 3.61 -3.66 (4H, m)
424 , 3.96-4.00 (2H, m) , 6.94-7.04 (3H, m) , 7.17-7.34 (7H, m) , 7.88 (1H, m), 8.17(1 H, m),8.65(1H, m) , 8.90(1H, s)
NMR1: 3.04 (2H, t, J=6.8Hz) , 3.33-3.72 (8H, m), 4.24 (2H, t, J=6.8Hz) , 6.94-6.98
426 (2H, m) , 7.00-7.05 (1 H, m) , 7.19-7.38 (7H, m) , 7.83-7.89 (1H, m) , 8.13-8.17(1H, m),8.63(1H,d, J=2.4Hz),8.78(1H, s)
N R2:0.91-1.04(2H,m), 1.10-1.33(3H,m), 1.44-1.56 (1H, m), 1.61-1.81 (7H, m)
435 , 3.32-3.96 (8H, br) , 4.00 (2H, t, J=5. OHz) , 6.89-7.01 (3H, m) , 7.27-7.35 (1 H, m) 7.38-7.46 (1 H, m), 8.13-8.27 (1 H, m) , 8.73-8.85 (1H. br) , 8.99-9.23 (2H, br)
NMR1: 0.90-1.01 (2H, m) , 1.07-1.27 (3H, m) , 1.40-1.52(1H, m) , 1.58-1.78 (7H, m) , 2.91 -3.02 (2H. m) , 3.25-3.35 (4H, m) , 3.39 (3H, s) , 4.00-4.10 (4H, m) , 4.14 (2H,
439
s) , 4.28 (2H, d, J=2.8Hz) , 6.98-7.03 (1 H,m), 7.11 (1 H. d, J=5.6Hz) , 7.28 (1 H, s) , 7.35(1H, t, J=5.6Hz), 10.0K1H, s) 産業上の利用可能性
[0210] 本発明化合物は、優れた FAAH阻害活性を有することから、 FAAHが関与する疾患 、特に頻尿、尿失禁、及び/又は過活動膀胱の治療に有用である。
配列表フリーテキスト
[0211] 以下の配列表の配列番号 1の数字見出し < 223〉に、発明者を掲記する