明 細 書
ヘテロァセン誘導体、テトラハロターフェニル誘導体及びそれらの製造方 法
技術分野
[0001] 本発明は、有機半導体等の電子材料への展開が可能なヘテロァセン誘導体、その 用途及びその製造方法に関する。さらに本発明は、該ヘテロァセン誘導体の前駆化 合物であるテトラハロターフェニル誘導体及びその製造方法に関する。
背景技術
[0002] 有機薄膜トランジスタに代表される有機半導体デバイスは、省エネルギー、低コスト 及びフレキシブルといった無機半導体デバイスにはない特徴を有することから近年 注目されるようになった。この有機半導体デバイスは有機半導体活性相、基板、絶縁 相、電極等数種類の材料から構成されるが、中でも電荷のキャリアー移動を担う有機 半導体活性相は該デバイスの中心的な役割を有している。この有機半導体活性相を 構成する有機材料のキャリアー移動能により有機半導体デバイス性能が左右される 有機半導体活性相を作製する方法としては一般的に、高温真空下、有機材料を気 化させて実施する真空蒸着法及び有機材料を適当な溶媒に溶解させその溶液を塗 布する塗布法が知られている。塗布法においては、塗布は高温高真空条件を用いる ことなく印刷技術を用いても実施することができる。そのため、塗布法は印刷によりデ バイス作製の大幅な製造コストの削減を図ることができることから、経済的に好ましい プロセスである。しかし、従来、有機半導体デバイスとして性能が高い材料ほど塗布 法で有機半導体活性相を形成することが困難になるという問題があった。
[0003] 例えば、ペンタセン等の結晶性材料はアモルファスシリコン並みの高いキャリアー 移動度を有し、優れた有機半導体デバイス特性を発現することが報告されてレ、る (例 えば、非特許文献 1参照)。又、ペンタセン等のポリアセンを溶解させ塗布法で有機 半導体デバイスを製造する試みも報告されている (例えば、特許文献 1参照)。しかし ながら、ペンタセンはその強い凝集性のため溶解性が低ぐ塗布法を適用するため
には高温加熱等の条件が必要とされ、さらにペンタセンの溶液は極めて容易に空気 酸化されることから、塗布法の適用はプロセス的、経済的に困難を伴うものであった。 また、ポリ一(3—へキシルチオフェン)等の自己組織化材料は溶媒に可溶であり、塗 布法による有機半導体デバイス作製が報告されてはいる力 S、キャリアー移動度が結 晶性化合物より 1桁低いことから (例えば、非特許文献 2参照)、得られた有機半導体 デバイスの特性が低!/、とレ、う問題があった。
またチォフェン環が縮環したペンタチェノアセンはペンタセンに比べ耐酸化性が向 上して!/、る力 キャリアー移動度が低!/、こと及びその合成に多工程を必要とすること 力 (例えば、非特許文献 3参照)実用上好まし!/、材料ではなかった。
非特許文献 1 :「ジャーナル ォブ アプライドフィジックス」、(米国)、 2002年、 92巻 、 5259— 5263頁
非特許文献 2 :「サイエンス」、(米国)、 1998年、 280巻、 1741— 1744頁 非特許文献 3 :「ジャーナル ォブ アメリカン ケミカル ソサイエティー」、(米国)、 2
005年、 127巻、 13281— 13286頁
特許文献 1: WO2003/016599号
発明の開示
発明が解決しょうとする課題
[0004] そこで、本発明は上記の従来技術が有する問題点に鑑み、優れた耐酸化性を有し
、塗布法による有機半導体活性相形成が可能な、ヘテロァセン誘導体及びそれを用 いた耐酸化性有機半導体材料並びに有機薄膜を提供することを目的とする。さらに
、本発明は該ヘテロァセン誘導体の前駆体として有用なテトラハロターフェニル誘導 体及びその製造方法を提供することをも目的とする。
課題を解決するための手段
[0005] 本発明者らは上記課題を解決するため鋭意検討の結果、本発明の新規なヘテロァ セン誘導体を見出した。加えて、該ヘテロァセン誘導体が耐酸化性に優れ、塗布法 の適用が可能であるため結晶性の薄膜を容易に安定して作製することができることか ら、該ヘテロァセン誘導体を含む耐酸化性有機半導体材料及びその有機薄膜を見 出し、本発明を完成するに到った。
さらに本発明者らは、該ヘテロァセン誘導体を効率的に製造することができる新規 な前駆化合物、即ち特定のテトラハロターフェニル誘導体を見出し、且つ係るテトラ ハロターフェニル誘導体を効率的に製造する方法を見出し本発明を完成するに到つ た。
即ち、本発明は以下の構成である。
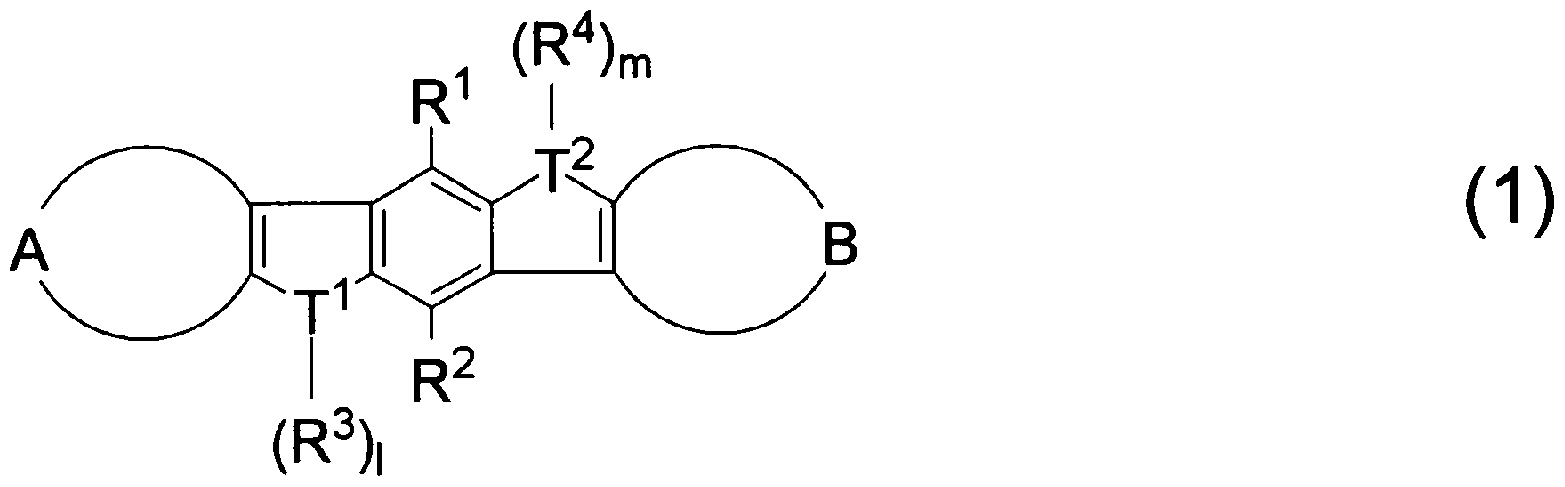
1. 下記一般式(1)で示されることを特徴とするヘテロァセン誘導体。
[化 1]
[0007] [ (ここで、置換基 I^〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、 炭素数 4〜30のァリール基、炭素数 3〜20のアルキル基、炭素数 1〜20のハロゲン 化アルキル基を示し、 T1及び T2は同一又は異なって、硫黄、セレン、テルル、酸素、 リン、ホウ素、アルミニウムを示し、 1及び mは、各々 0又は 1の整数であり、環 A及び B は同一又は異なって、下記一般式 (A— 1)又は (A— 2)で示される構造を有する。 )
[0008] [化 2]
[0010] (ここで、置換基 R5〜RUは同一又は異なって、水素原子、フッ素原子、塩素原子、 炭素数 4〜30のァリール基、炭素数 3〜20のアルキル基、炭素数 1〜20のハロゲン 化アルキル基を示す。なお、置換基 R5〜R6及び R8〜RUは、それぞれに、各置換基 内の任意の 2つ以上の置換基が互いに結合し、置換基を有して!/、てもよ!/、ベンゼン 環、置換基を有していてもよいピリジン環、置換基を有していてもよいピラジン環を形 成すること力 Sでき、置換基 T3は、硫黄、セレン、テルル、酸素、リン、ホウ素を示し、 n は 0又は 1の整数である。但し、 T1及び T2が硫黄である場合、環 A及び Bは、 (A- 1) 又は置換基を有する (A— 2)で示される環である。 ) ) ]
2. 1及び mが各々 0であり、且つ T1及び T2は同一又は異なって、硫黄、セレン、テ ルル、酸素であることを特徴とする上記 1に記載のへテロアセン誘導体。
3. 1及び mが各々 1であり、且つ T1及び T2は同一又は異なって、リン、ホウ素、アル ミニゥムであることを特徴とする上記 1に記載のへテロアセン誘導体。
4. nが 0であり、且つ T3は硫黄、セレン、テルル、酸素であることを特徴とする上記 1 〜3の!/、ずれかに記載のへテロアセン誘導体。
5. 一般式(2)で示されることを特徴とするテトラハロターフェニル誘導体。
[0011] [化 4]
[0012] (ここで、置換基 xi〜X
4は臭素原子、ヨウ素原子、塩素原子を示し、置換基 R
1及び R 2並びに環 A及び Bは上記 1に記載の一般式(1)で示される置換基及び環と同意義 を示す。 )
[0013] 6. 一般式(2)において、環 A及び Bが (A—1)で示される環であることを特徴とする 上記 5に記載のテトラハロターフェニル誘導体。
7. nが 0で、且つ T3は硫黄、セレン、テルル、酸素であることを特徴とする上記 5又 は 6に記載のテトラハロターフェニル誘導体。
8. 上記 5〜7のいずれかに記載の一般式(2)で示されるテトラハロターフェニル誘 導体をメタル化剤を用いてテトラメタル化し、下記一般式(3)及び下記一般式 (4)で 示される反応剤と反応させることを特徴とする上記 1〜4のいずれかに記載のへテロ ァセン誘導体の製造方法。
(R4) T2 (L2) (4)
m q
(ここで、置換基丁
1、 T
2、
R
4及び記号 1と mは上記 1に記載の一般式(1)で示され る置換基及び記号と同意義を示し、置換基 ΐΛ L
2は塩素原子、臭素原子、ヨウ素原 子、炭素数 1〜20のォキシ基、ァセトキシ基、ァリールスルホニル基を示し、 ρ及び q は 0又は 2の整数を示す。 )
[0014] 9. メタル化剤としてアルキルリチウムを用いることを特徴とする上記 8に記載のへテ ロアセン誘導体の製造方法。
10. 下記一般式(5)で示されるテトラハロベンゼンと下記一般式(6)及び下記一般 式(7)で示される 2—ハロアリール金属試薬をパラジウム及び/又はニッケル触媒存 在下で反応させることを特徴とする上記 5〜7のいずれかに記載のテトラハロターフェ ニル誘導体の製造方法。
[0015] [化 5]
[0016] (ここで、置換基 X
5及び X
6は臭素原子、ヨウ素原子、塩素原子を示す。
R
、 X2及び X3は上記 8に記載の一般式 (2)で示される置換基と同意義を示す。 ) [0017] [化 6]
[0018] (ここで、 M1はマグネシウム、ホウ素、亜鉛、錫、ケィ素のハロゲン化物;ノ、イドロォキ サイド;アルコキサイド;アルキル化物を示し、置換基 X1並びに環 Aは、上記 8に記載 の一般式 (2)で示される置換基並びに環と同意義を示す。 )
[0019] [化 7]
(ここで、 M2はマグネシウム、ホウ素、亜鉛、錫、ケィ素のハロゲン化物;ノヽイドロォキ サイド;アルコキサイド;アルキル化物を示し、置換基 X4並びに B環は、上記 8に記載 の一般式 (2)で示される置換基並びに環と同意義を示す。 )
11. 一般式(5)で示されるテトラハロベンゼンにおいて、 X5及び X6がヨウ素原子で あり、 X2及び X3が臭素原子及び/又は塩素原子であることを特徴とする上記 10に記 載のテトラハロターフェニル誘導体の製造方法。
[0021] 12. 一般式(6)、 であることを特徴とする上
記 10又は 11に記載のテトラハロターフェニル誘導体の製造方法。
13. 用いる触媒がテトラキス(トリフエニルホスフィン)パラジウムであることを特徴と する上記 10〜; 12のいずれかに記載のテトラハロターフェニル誘導体の製造方法。
14. 上記 1〜4のいずれかに記載のへテロアセン誘導体を含むことを特徴とする耐 酸化性有機半導体材料。
15. 上記 14に記載の耐酸化性有機半導体材料を用いることを特徴とする有機薄 膜。
16. 有機薄膜が基板上に形成されることを特徴とする上記 15に記載の有機薄膜。 発明の効果
[0022] 優れた耐酸化性を有し、塗布法による有機半導体活性相形成が可能な、ヘテロァ セン誘導体及びその用途を提供する。さらに、該ヘテロァセン誘導体の前駆化合物 であるテトラハロターフェニル誘導体及びその製造方法をも提供する。
発明を実施するための最良の形態
[0023] 以下に本発明を詳細に説明する。説明はへテロァセン誘導体及びその製造方法、 該ヘテロァセン誘導体の前駆体であるテトラハロターフェニル誘導体及びその製造 方法、並びに該ヘテロァセン誘導体を含む耐酸化性有機半導体材料及びその有機 薄膜について述べる。
[0024] (ヘテロァセン誘導体)
本発明のへテロアセン誘導体は下記一般式(1)で示される。
[0026] [ (ここで、置換基 I^〜R4は同一又は異なって、水素原子、フッ素原子、塩素原子、 炭素数 4〜30のァリール基、炭素数 3〜20のアルキル基、炭素数 1〜20のハロゲン 化アルキル基を示し、 T1及び T2は同一又は異なって、硫黄、セレン、テルル、酸素、 リン、ホウ素、アルミニウムを示し、 1及び mは、各々 0又は 1の整数であり、環 A及び B は同一又は異なって、下記一般式 (A— 1)又は (A— 2)で示される構造を有する。 )
[0027] [化 9]
[0028] [化 10]
(A-2)
[0029] (ここで、置換基 R
5〜R
Uは同一又は異なって、水素原子、フッ素原子、塩素原子、 炭素数 4〜30のァリール基、炭素数 3〜20のアルキル基、炭素数 1〜20のハロゲン 化アルキル基を示す。なお、置換基 R
5〜R
6及び R
8〜R
Uは、それぞれに、各置換基 内の任意の 2つ以上の置換基が互いに結合し、置換基を有して!/、てもよ!/、ベンゼン 環、置換基を有していてもよいピリジン環、置換基を有していてもよいピラジン環を形 成すること力 Sでき、置換基 T
3は、硫黄、セレン、テルル、酸素、リン、ホウ素を示し、 n は 0又は 1の整数である。但し、 T
1及び T
2が硫黄である場合、環 A及び Bは、 (A- 1) 又は置換基を有する (A— 2)で示される環である。 ) ]
[0030] 本発明の一般式(1)で示されるヘテロァセン誘導体の置換基について述べる。
置換基 Ri〜R4における炭素数 4〜30のァリール基は、特に限定はなぐ例えばフ ェニノレ基、 p トリノレ基、 p— (n へキシル)フエニル基、 p— (n ォクチノレ)フエニル 基、 p— (シクロへキシノレ)フエニル基、 m— (n ォクチノレ)フエニル基、 p—フルオロフ ェニノレ基、ペンタフルオロフェニル基、 p (トリフルォロメチル)フエニル基、 p— (n- ノ ーフルォロォクチル)フエニル基、 2 チェニル基、 5—(n へキシル)ー2 チェ 二ノレ基、 2, 2,一ビチェ二ルー 5 基、ビフエ二ル基、パーフルォロビフエニル基、 1 ナフチル基、 2—ナフチル基、 1 パーフルォロナフチル基、アントラセニル基、 2 フルォレニル基、 9, 9 ジメチルー 2 フルォレニル基、 1ービフエニレノ基、 2— ビフエユレノ基、ターフェニル基、 2—ピリジル基、テトラフルォロピリジル基、ビビリジ ル基、(ジフエニルァミノ)フエニル基、(ジフエニルァミノ)ビフエ二ル基等を挙げること ができ、好ましくはフエニル基、 p— (n ォクチル)フエニル基、 p— (n—パーフルォ ロォクチル)フエニル基、 5—(n へキシル) 2 チェニル基等である。
置換基 Ri〜R4における炭素数 3〜20のアルキル基は、特に限定はなぐ例えばプ 口ピル基、 n ブチル基、イソブチル基、 t ブチル基、ネオペンチル基、へキシル基 、ォクチル基、ドデシル基、ォクタデシル基、シクロへキシル基、シクロォクチル基、 2 ェチルへキシル基等を挙げることができる。
[0031] 置換基 I^〜R4における炭素数 1〜20のハロゲン化アルキル基は、特に限定はなく 、例えばトリフルォロメチル基、トリフルォロェチル基、パーフルォロォクチル基、パー フルォロドデシル基、パーフルォロォクタデシル基、パーフルォロシクロへキシル基、
パーフルォロシクロォクチル基等のパーフルォロアルキル基;あるいはペンタデカフ ノレォロォクチル基、ォクタデカフルォロデシル基等の一部の水素がフッ素に置換され たハロゲン化アルキル基を挙げることができ、好ましくはパーフルォロアルキル基であ り、特に好ましくはパーフルォロォクチル基、パーフルォロドデシル基である。
これらの置換基 Ri〜R4の中でも、特に水素原子、炭素数 4〜30のァリール基が好 ましぐさらに水素原子、フエニル基が好ましい。
置換基 T1及び T2は、硫黄、セレン、テルル、酸素、リン、ホウ素、アルミニウムであり 、その中でも好ましくは硫黄、セレン、リン、ホウ素であり、さらに好ましくは硫黄、リン、 ホウ素である。また、 T1及び T2が硫黄である時、環 A及び Bは、(A— 1)又は置換基 を有する (A— 2)で示される環であることが好まし!/、。
1及び mは、各々 0又は 1の整数である。ただし、置換基丁1、 T2が、硫黄、セレン、テ ノレル、酸素の場合は、 1、 mは 0であり、置換基丁1、 T2が、リン、ホウ素、アルミニウムの 場合は、 1、 mは 1である。
次に、一般式 (A—1 )及び (A—2)で示される、環 A及び Bについて述べる。
本発明のへテロアセン誘導体は、環 A、 Bを有する誘導体であり、環 A, Bは一般式 (A— 1)又は (A— 2)で示される構造を有するものである。
置換基 R5〜RUにおける炭素数 4〜30のァリール基は、特に限定はなぐ例えばフ ェニノレ基、 p トリノレ基、 p— (n へキシル)フエニル基、 p— (n ォクチノレ)フエニル 基、 p— (シクロへキシノレ)フエニル基、 m— (n ォクチノレ)フエニル基、 p—フルオロフ ェニノレ基、ペンタフルオロフェニル基、 p (トリフルォロメチル)フエニル基、 p— (n- ノ ーフルォロォクチル)フエニル基、 2 チェニル基、 5—(n へキシル)ー2 チェ 二ノレ基、 2, 2,一ビチェ二ルー 5 基、ビフエ二ル基、パーフルォロビフエニル基、 1 ナフチル基、 2—ナフチル基、 1 パーフルォロナフチル基、アントラセニル基、 2 フルォレニル基、 9, 9 ジメチルー 2 フルォレニル基、 1ービフエニレノ基、 2— ビフエユレノ基、ターフェニル基、 2—ピリジル基、テトラフルォロピリジル基、ビビリジ ル基、(ジフエニルァミノ)フエニル基、(ジフエニルァミノ)ビフエ二ル基等を挙げること ができ、好ましくはフエニル基、 p— (n ォクチル)フエニル基、 p— (n—パーフルォ ロォクチル)フエニル基、 5—(n へキシル) 2 チェニル基等である。
置換基 R5〜RUにおける炭素数 3〜20のアルキル基は、特に限定はなぐ例えば プロピル基、 n ブチル基、イソブチル基、 t ブチル基、ネオペンチル基、へキシノレ 基、ォクチル基、ドデシル基、シクロへキシル基、シクロォクチル基、 2—ェチルへキ シノレ基等を挙げることカできる。
置換基 R5〜RUにおける炭素数 1〜20のハロゲン化アルキル基は、特に限定はな く、例えばトリフルォロメチル基、トリフノレオロェチノレ基、パーフルォロォクチル基、ノ 一フルォロシクロへキシル基、パーフルォロシクロォクチル基等を挙げることができ、 好ましくはパーフルォロォクチル基である。
また、環 A及び Bの置換基群 R5〜R6及び R8〜RUは、それぞれに、各置換基群内 の任意の二以上の置換基が互いに結合し、置換基を有して!/、てもよ!/、ベンゼン環、 置換基を有してレ、てもよ!/、ピリジン環、置換基を有して!/、てもよ!/、ピラジン環を形成す ること力 Sでき、好ましくは置換基を有していてもよいベンゼン環である。置換基を有し てもよいベンゼン環は特に限定はなぐ例えばベンゼン環、メチルベンゼン環、(n— へキシル)ベンゼン環、(n ォクチル)ベンゼン環、ジメチルベンゼン環、ジ(n へキ シル)ベンゼン環、ジフエニルベンゼン環、ナフタレン環、メチルナフタレン環、ジメチ ルナフタレン環、ジ (n へキシル)ナフタレン環、ジ(n ォクチル)ナフタレン環、ジ( n ドデシル)ナフタレン環、ジ(n ォクタデシル)ナフタレン環、ジ(2—ェチルへキ シル)ナフタレン環、ジ(n パーフルォ口へキシル)ナフタレン環、ジ(n パーフルォ
環、ジ (n ォクタデカフルォロドデシル)ナフタレン環、フエ二ルナフタレン環、アント ラセン環、トリフエ二レン環、キノリン環等を挙げることができ、置換基を有していてもよ いピリジン環は特に限定はなぐ例えばピリジン環、メチルピリジン環、(n へキシル) ピリジン環、フエ二ルビリジン環等を挙げることができ、置換基を有していてもよいビラ ジン環は特に限定はなぐ例えばピラジン環、メチルビラジン環、ジメチルビラジン環 、(n へキシル)ピラジン環、フエ二ルビラジン環等を挙げることができ、置換基を有 していてもよいベンゼン環が好ましぐさらにジ (n ドデシル)ナフタレン環、ジ (n— パーフルォロドデシル)ナフタレン環が特に好ましい。また、置換基 R8〜RUの内、 R9
と R1Uが互いに結合し、置換基を有して!/、てもよ!/、ベンゼン環を形成することが好まし い。
これらの置換基 R5〜R6及び R8〜RUの中でも、特に水素原子、フッ素原子、置換 基を有していても良いベンゼン環が好ましぐさらに水素原子、フッ素原子、ベンゼン 環が好ましい。
置換基 T3は、硫黄、セレン、テルル、酸素、リン、ホウ素であり、その中でも好ましく は硫黄、セレン、リン、ホウ素であり、さらに好ましくは硫黄、リン、ホウ素である。 nは 0又は 1の整数であり、 nが 0の場合、 T3は硫黄、セレン、テルル、酸素であり、 n 力 aの場合、 T3はリン、ホウ素である。
[0034] これらの中でも本発明の一般式(1)で示されるヘテロァセン誘導体は、該ヘテロァ セン誘導体及び該ヘテロァセン誘導体を含む耐酸化性有機半導体材料及びその有 機薄膜が、高い耐酸化性及びキャリアー移動度を発現することから、以下の化合物 が好ましぐ
[0035] [化 11]
Κΐ¾] [9εοο]
89990/L00Zd /13d S V Ζ099蘭 ΟΟΖ OAV
89990/L00ZdT/13d Ζ099蘭 ΟΟΖ OAV
89990/L00ZdT/13d 91- Z099蘭 OOZ OAV
[9ΐ¾] [6C00]
[η^ΐ [8εοο] 89990/L00ZdT/13d 9 1- Ζ099蘭 ΟΟΖ OAV
トラチェノアセン、 P, P—ジフエニルベンゾホスホロジベンゾホスホール、 B
, B ジフエニノレベンゾボロリノレジべンゾボローノレ、テトラフノレォロジチェノアセン、テト ラフェニルジチェノアセン、ジベンゾジチェノアセン等が好ましレ、。
[0041] (テトラハロターフェニル誘導体)
次に、本発明の本発明の一般式(1)で示されるヘテロァセン誘導体の前駆化合物 であるテトラハロターフェニル誘導体につ!/、て述べる。
本発明の本発明の一般式(1)で示されるヘテロァセン誘導体の前駆化合物である テトラハロターフェニル誘導体は下記一般式(2)で示される。
[0042] [化 16]
[0043] (ここで、置換基 xi〜X4は臭素原子、ヨウ素原子、塩素原子を示し、置換基 R1及び2 並びに環 A及び Bは一般式(1)で示される置換基及び環と同意義を示す。 )
置換基 〜X4は臭素原子、ヨウ素原子、塩素原子を示し、好ましくは臭素原子、ョ ゥ素原子であり、特に好ましくはいずれも臭素原子である。
置換基 R1及び R2は、一般式(1)で示される置換基と同意義を示し、その中でも特 に水素原子が好ましい。
環 A及び Bは一般式(1)で示される環と同意義を示す。すなわち、一般式 (A— 1) 又は一般式 (A— 2)と同意義を示す。そして、その中でも一般式 (A— 1)にお!/、ては 、 T3が硫黄、 R5及び R6が結合し環状のベンゼン環であることが好ましぐ一般式 (A 2)においては、 R8〜RUが水素原子、フッ素原子、環状のベンゼン環であることが 好ましい。
[0044] 本発明の一般式(2)で示されるテトラハロターフェニル誘導体としては、以下の化 合物が好ましぐ
/ i89990/-00iv:fcl26ί S99/ OS80SAV. 皇g
[8ΐ¾] [9W)0] 89990/L00Zd /13d 03 Z099蘭 00Z OAV
l789990/Z.00Zdf/X3d Z099Z0/800Z OAV
[0047] 特に、 { 1 , 4 ビス(3 ブロモベンゾチェニル) 2, 5 ジブ口モ}ベンゼン、 2, 2, , 5,, 2" テトラフ、、口モー 1 , 1 ' , 4 ' , 1" ターフェ二ノレ、 4, 5, 4" , 5" テトラフノレ才 口 2, 2,, 5,, 2,, テトラブロモ— 1 , 1 ' , 4 ' , 1,,—ターフェ二ノレ、 4, 5, 4" , 5,,— テトラフエ二ノレ一 2, 2,, 5,, 2"—テトラブロモー 1 , 1 ' , 4,, 1"—ターフェ二ノレ、 2, 2 ,, 5,, 2"—テトラブロモー 1 , 1,, 4,, 1 "ージベンゾターフェニル等が好ましい。
(ヘテロァセン誘導体の製造方法)
本発明の一般式(1)で示されるヘテロァセン誘導体の製造方法について述べる。 本発明の一般式(1)で示されるヘテロァセン誘導体は、一般式(2)で示されるテト ラハロターフェニル誘導体をメタル化剤を用いてテトラメタル化し、下記一般式(3)及 び下記一般式 (4)で示される反応剤と反応させることにより製造することができる。な お、一般式 (3)、一般式 (4)で示される反応剤が同じ化合物であっても良い。
(R4) T2 (L2) (4)
m q
[0048] (ここで、置換基丁
1、 T
2、
R
4及び記号 1と mは一般式(1)で示される置換基及び記 号と同意義を示し、置換基 L
1 L
2は塩素原子、臭素原子、ヨウ素原子、炭素数 1〜20 のォキシ基、ァセトキシ基、フエニルスルホニル基を示し、 p及び qは 0又は 2の整数を 示す。)
[0049] なお、ここでテトラメタル化とは、一般式(2)における xi〜X4をそれぞれメタルに置 換することを意味する。
一般式(2)で示されるテトラハロターフェニル誘導体をテトラメタル化する場合、用 いるメタル化剤は、一般式(2)における xi〜X4をメタルに置換することができるもので ある限り特に限定はなぐ例えば n ブチルリチウム、 sec ブチルリチウム、 tert ブ チルリチウム、メチルリチウム、へキシルリチウム等のアルキルリチウム;フエ二ルリチウ ム、 p tert ブチノレフエニノレリチウム、 p メトキシフエニノレリチウム、 p フノレオロフ ェニルリチウム等のァリールリチウム;リチウムジイソプロピルアミド、リチウムへキサメチ ルジシラジド等のリチウムアミド;リチウムパウダー等のリチウム金属;メチルマグネシゥ
試薬;マグネシウム金属;亜鉛金属等を挙げることができ、好ましくはアルキルリチウム であり、特に好ましくは sec—ブチルリチウムである。
該メタル化剤の使用量は一般式(2)のテトラハロターフェニル誘導体 1当量に対し、 3〜20当量が好ましぐ特に好ましくは 4〜; 15当量、さらに好ましくは 5〜; 10当量の範 囲で使用することができる。
該テトラメタル化は、好ましくは溶媒中で実施する。用いる溶媒は特に限定はなぐ 例えばテトラヒドロフラン(以下、 THFと略す)、ジェチルエーテル、メチルー tert—ブ チノレエーテノレ、エチレングリコーノレジメチノレエーテノレ、ジ才キサン、トノレェン、へキサ ン、シクロへキサン等であり、特に好ましくは THFである。又、これら溶剤は 1種若しく は 2種以上の混合物を用いても良い。該テトラメタル化の温度は— 100〜50°Cで行う こと力 S好ましく、特に好ましくは一 90〜20°Cである。反応時間は 1〜; 120分が好ましく 、特に好ましくは 5〜60分である。なお、テトラメタル化の進行は、反応液の一部を取 り出し、水で反応を停止させた後、ガスクロマトグラフィーで分析することで監視するこ と力 Sできる。
該テトラメタル化により生成したテトラメタル塩は、次いで一般式(3)及び一般式 (4) で示される反応剤と反応させることにより、一般式(1)で示されるヘテロァセン誘導体 が得られるものである。係る反応剤との反応は、前記テトラメタル化により生成したテト ラメタル塩を含む反応混合物に前記反応剤を直接用いて反応させる方法、生成した テトラメタル塩を一度単離した後、前記反応剤と反応させる方法のいずれを用いても よい。
ここで、一般式(3)、一般式 (4)における置換基丁
1、 T
2、
R
4及び記号 1と mは一 般式(1)で示される置換基及び記号と同意義を示す。その中でも一般式 (3)、一般 式(4)としては、ビス(フエニルスルホニノレ)スルフイド、ジクロロフェニルホスフィン、ジ クロ口フエ二ルポラン等が好ましレ、。
また、置換基 ΐ L2は塩素原子、臭素原子、ヨウ素原子、炭素数 1〜20のォキシ基 、ァセトキシ基、ァリールスルホニル基を示し、好ましくは塩素原子、臭素原子、炭素 数;!〜 20のォキシ基、ァリールスルホニル基である。炭素数 1〜20のォキシ基は特 に限定はなぐ例えばメトキシ基、エトキシ基、 η—ブトキシ基、フエノキシ基、(2—メト
キシ)フエノキシ基等を挙げることができ、ァリールスルホニル基は特に限定はなぐ 例えばフエニルスルホニル基、 p—トリルスルホニル基等を挙げることができる。これら の中でも特にフエニルスルホニル基が好ましい。
[0051] そして、具体的な一般式(3)、一般式 (4)で示される反応剤としては、例えば 2塩化 硫黄; 2臭化硫黄;ビス(フエニルスルホニノレ)スノレフイド、ビス(p -トリノレスノレホニノレ)ス ルフイド等のビス(ァリールスルホニル)スルフイド類;硫黄; 2塩化セレン;セレン; 2塩 化テノレル;テノレル;ジクロロフェニルホスフィン、ジメトキシフエニルホスフィン、ジフエノ キシフエニルホスフィン、ジクロロ {4— (n—ォクチノレ)フエ二ノレ }ホスフィン等のァリー ノレホスフィン類;ジクロロ(n—へキシノレ)ホスフィン、ジクロロ(n—ォクチノレ)ホスフィン 、ジメトキシ(n -へキシル)ホスフィン等のアルキルホスフィン類;ジクロロフェニルボラ ン、ジメトキシフエ二ルポラン、ジメトキシ {4一(n—へキシル)フエ二ノレ }ボラン、ジフエ ノキシフエニルボラン、ジクロロ {4一(n—ォクチノレ)フエ二ノレ }ボラン等のァリールボラ ン類;ジクロロ(n—へキシル)ボラン、ジクロロ(n—ォクチル)ボラン、ジメトキシ(n—へ キシル)ボラン等のアルキルボラン類;ジクロロフェニルアルミニウム、ジメトキシフエ二 ノレアノレミニゥム、ジメトキシ {4— (n—へキシル)フエ二ノレ }アルミニウム、ジフエノキシフ ェニルアルミニウム、ジクロロ {4一(n—ォクチノレ)フエ二ノレ }アルミニウム等のァリール アルミニウム類;ジクロ口(n—へキシル)ァノレミニゥム、ジクロロ(n -ォクチル)アルミ二 ゥム、ジメトキシ(n—へキシル)アルミユウム等のアルキルアルミユウム類等を挙げるこ とができ、好ましくはビス(フエニルスルホニノレ)スルフイド、ジクロロフェニルホスフィン 、ジクロロフエ二ルポラン等である。
[0052] テトラメタル化により生成したテトラメタル塩と一般式(3)及び一般式 (4)で示される 反応剤と反応させる際には、好ましくは溶媒中で実施する。用いる溶媒は特に限定 はなく、例えば THF、ジェチルエーテル、メチルー tert—ブチルエーテル、エチレン グリコールジメチルエーテル、ジグライム、ジ才キサン、トルエン、へキサン、シクロへ キサン等であり、好ましくは THFである。用いる反応剤の量は、一般式(2)のテトラハ 口ターフェニル誘導体 1当量に対し、 1. 2〜; 10当量が好ましぐ特に好ましくは 2〜8 当量である。該反応剤との反応温度は— 100〜50°Cが好ましぐ特に好ましくは— 9 0〜30°Cであり、反応時間は 0. 5〜30時間が好ましぐ特に好ましくは 1〜; 18時間で
本発明の一般式(1)のへテロァセン誘導体の製造は、好ましくは窒素又はアルゴン 等の不活性雰囲気下で実施する。
本発明の一般式(1)のへテロァセン誘導体の製造方法では、一般式(2)のテトラハ 口ターフェニル誘導体をテトラメタル化した後、塩化マグネシウムと反応させ、その後 に一般式(3)及び一般式 (4)で示される反応剤で処理することもできる。
力、くして得られた、本発明の一般式(1)で示されるヘテロァセン誘導体は、さらに精 製すること力 Sできる。精製する方法は特に限定はなぐ例えばカラムクロマトグラフィー 、再結晶化、あるいは昇華による方法を挙げること力 Sできる。
[0053] (テトラハロターフェニル誘導体の製造方法)
次に、本発明の一般式(1)で示されるヘテロァセン誘導体の前駆体として用いられ る一般式(2)で示されるテトラハロターフェニル誘導体の製造方法について述べる。 本発明の一般式(2)で示されるテトラハロターフェニル誘導体は下記一般式(5)で 示されるテトラハロベンゼンと下記一般式(6)及び下記一般式(7)で示される 2—ハ ロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させることに より製造すること力 Sできる。なお、一般式 (6)、一般式(7)で示される反応剤が同じ化 合物であっても良い。
[0054] [化 19]
[0055] (ここで、置換基 及び X
6は臭素原子、ヨウ素原子、塩素原子を示す。
R
、 X2及び X3は一般式 (2)で示される置換基と同意義を示す。 )
[0056] [化 20]
[0057] (ここで、 M1はマグネシウム、ホウ素、亜鉛、錫、ケィ素のハロゲン化物;ノ、イドロォキ サイド;アルコキサイド;アルキル化物を示し、置換基 X1並びに環 Aは、一般式(2)で 示される置換基並びに環と同意義を示す。 )
[0058] [化 21]
[0059] (ここで、 M2はマグネシウム、ホウ素、亜鉛、錫、ケィ素のハロゲン化物;ノ、イドロォキ サイド;アルコキサイド;アルキル化物を示し、置換基 X4並びに B環は、一般式(2)で 示される置換基並びに環と同意義を示す。 )
[0060] 本発明の一般式(5)、 (6)及び(7)について、さらに述べる。
一般式(5)の置換基 X5及び X6は、臭素原子、ヨウ素原子、塩素原子を示し、好まし くは臭素原子及びヨウ素原子であり、さらに好ましくはヨウ素原子である。
R
2、 X
2及び X
3は、一般式(2)で示される置換基と同意義を示す。
そして、具体的な一般式(5)で示される化合物としては、 1 , 4—ジブロモ一 2, 5- ジョードベンゼンが挙げられる。
一般式(6)、 (7)
M
2はマグネシウム、ホウ素、亜鉛、錫、ケィ素のハロ ゲン化物;ハイド口オキサイド;アルコキサイド;アルキル化物であり、上記のパラジウム 及び/又はニッケル触媒により脱離され、パラジウム及び/又はニッケルと置換でき
る基である限り特に限定はなぐ例えば MgCl MgBr B (OH) B (OMe) 、テトラ
2 2 メチノレジオキサボロラニノレ基、 ZnCl ZnBr Znl Sn (Bu— n) Si (Bu— n) 等を
3 3 挙げること力 Sでき、好ましくは ZnCl Β (ΟΗ) である。
2
置換基 X1 X4並びに環 Α Βは、一般式(2)で示される置換基並びに環と同意義を 示す。
[0061] そして、具体的な一般式(6)、一般式(7)で示される化合物としては、例えば 3—ブ ロモベンゾチェ二ルー 2 ジンククロライド、 2 ブロモー 4, 5 ジフルオロフェニルマ グネシゥムブロマイド、 2 ブロモナフチルー 3 マグネシウムブロマイド、 2 ブロモ フエ二ルポロン酸等が挙げられる。
なお、一般式(6)、一般式(7)で示される 2 ハロアリール金属試薬は、例えば、そ れらの原料となるァリールジハロゲン置換体をイソプロピルマグネシウムブロマイド等 のグリニャール試薬あるいは η ブチルリチウム等の有機リチウム試薬によりハロゲン /金属交換反応を行った後、塩化亜鉛、トリメトキシボラン等と反応させることで好適 に調製すること力できる。なお、グリニャール試薬によるハロゲン/金属交換反応は、 ί列免 ίί「ジャーナノレ 才フ、、 才ノレガニック ゲミストリィー」、 2000年、 65巻、 4618— 4 634頁」に記載されている方法、有機リチウム試薬によるハロゲン/金属交換反応は 、例えば「ジャーナル ォブ ケミカノレ リサーチ シノプシス」、 1981年、 185頁に記 載されてレ、る方法を用いることもできる。
[0062] 一般式(5)で示されるテトラハロベンゼンと一般式(6)及び一般式(7)で示される 2
ロアリール金属試薬の反応に用いる触媒はパラジウム及び/又はニッケル触媒 であれば特に限定はなぐ例えばテトラキス(トリフエニルホスフィン)パラジウム、トリス (ジベンジリデンアセトン)ジパラジウム/トリフエニルホスフィン混合物、ジクロロビス( トリフエニルホスフィン)パラジウム、ビス(トリー tert ブチルホスフィン)パラジウム、ジ ァセタトビス(トリフエニルホスフィン)パラジウム、ジクロロ(1 , 2—ビス(ジフエニルホス フイノ)ェタン)パラジウム、酢酸パラジウム/トリフエニルホスフィン混合物、酢酸パラ ジゥム /トリー tert ブチルホスフィン混合物、酢酸パラジウム /2—(ジシクロへキシ ルホスフイノ) 1 , 1,ービフエニル混合物、ジクロロ(エチレンジァミン)パラジウム、ジ クロ口(N, N, Ν' , Ν,一テトラメチルエチレンジァミン)パラジウム、ジクロロ(Ν, Ν, Ν
' , N'—テトラメチルエチレンジァミン)パラジウム/トリフエニルホスフィン混合物等の パラジウム触媒;ジクロロビス(トリフエニルホスフィン)ニッケル、ジクロロ(1 , 2—ビス( ジフエニルホスフイノ)ェタン)ニッケル、ジクロロ(エチレンジァミン)ニッケル、ジクロロ (N, N, Ν' , Ν,一テトラメチルエチレンジァミン)ニッケル、ジクロロ(Ν, Ν, Ν' , Ν,
—テトラメチルエチレンジァミン)ニッケル/トリフエニルホスフィン混合物、ビス(1 , 5 —シクロォクタジェン)ニッケル/トリフエニルホスフィン混合物等のニッケル触媒;を 挙げること力 Sできる。中でも、好ましい触媒は 0価のパラジウム化合物であり、特に好 ましい触媒はテトラキス(トリフエニルホスフィン)パラジウムである。又、これら触媒は 1 種若しくは 2種以上の混合物を用いても良い。
[0063] 一般式(5)で示されるテトラハロベンゼンと一般式(6)及び一般式(7)で示される 2 ーハロアリール金属試薬をパラジウム及び/又はニッケル触媒存在下で反応させる 際には、好ましくは溶媒中で実施する。用いる溶媒に特に限定はなぐ例えば THF、 ジェチノレエーテノレ、メチノレー tert—ブチノレエーテノレ、ジ才キサン、エチレングリコー ノレジメチルエーテル、トルエン、キシレン、へキサン、シクロへキサン、エタノール、水 、 N, N—ジメチルホルムアミド、 N—メチルピロリドン、トリエチルァミン、ピぺリジン、ピ 口リジン、ジイソプロピルアミン等を挙げることができ、又、これら溶剤は 1種若しくは 2 種以上の混合物を用いても良ぐ例えばトルエン/水、トルエン/エタノール/水の ような 2乃至 3成分系でも使用することができる。
ノ ラジウム触媒、ニッケル触媒の使用量は一般式(5)のテトラハロベンゼン 1モルに 対し、 0. ;!〜 20モル0 /0が好ましぐ特に好ましくは 1〜; 10モル0 /0の範囲である。 一般式 (6)、一般式(7)の 2—ハロアリール金属試薬の使用量は一般式(5)のテト ラハロベンゼン 1当量に対し、 0. 8〜3. 2当量が好ましぐ特に好ましくは 1. 0〜2. 8 当量、さらに好ましくは 1. ;!〜 2. 5当量の範囲で使用することができる。
反応の際の温度は 10〜; 120°Cが好ましぐ特に好ましくは 30〜; 100°C、さらに好ま しくは 40〜90°Cであり、反応時間は;!〜 48時間が好ましぐ特に好ましくは 2〜30時 間の範囲で好適に実施することができる。
[0064] なお、反応系中に塩基を存在させることもできる。この場合の塩基の種類としては特 に限定はなぐ例えば炭酸ナトリウム、炭酸水素ナトリウム、炭酸カリウム、炭酸セシゥ
ム、りん酸カリウム、りん酸ナトリウム、ナトリウム tert ブトキサイド、フッ化カリウム等の 無機塩基;トリェチルァミン、トリメチルァミン、トリブチルァミン、エチレンジァミン、 N, N, N' , N'—テトラメチルエチレンジァミン、ジイソプロピルァミン、ピリジン等の有機 塩基を好適なものとして挙げること力 Sできる。これらの塩基の使用量は一般式(5)の テトラハロベンゼン 1当量に対し、 0. 5〜10. 0当量が好ましぐ特に好ましくは 2. 0 〜8. 0当量の範囲で使用することができる。さらにこれらの塩基と併用し、相間移動 触媒を用いることもできる。相間移動触媒の種類は特に限定はなぐ例えばトリオクチ ルメチルアンモニゥムクロライド、テトラプチルアンモニゥムクロライド、セチルピリジニ ゥムクロライド等を好適なものとして挙げること力 Sできる。これらの相間移動触媒の使 用量は一般式(5)のテトラハロベンゼン 1当量に対し、 0· ;!〜 1 · 5当量が好ましぐ特 に好ましくは 0. 2〜0. 8当量の範囲である。
さらに反応系中にトリフエニルホスフィン等のホスフィンを存在させることもできる。こ れらのホスフィンの使用量は、該パラジウム及び/又はニッケル触媒 1当量に対し、 0 . 9〜8. 0当量が好ましぐ特に好ましくは 1. 0〜3. 0当量の範囲で使用することが できる。
なお、反応系中に銅化合物を存在させることもできる。該銅化合物しては特に限定 はなぐ例えば塩化銅 (1)、臭化銅 (1)、ヨウ化銅 (1)、酢酸銅 (I)等の 1価銅;塩化銅 (I I)、臭化銅 (II)、ヨウ化銅 (II)、酢酸銅 (II)、ァセチルァセトナート銅 (II)等の 2価銅 等を挙げること力 Sできる。その中でも好ましくは 1価銅であり、特に好ましくはヨウ化銅( I)である。これらの銅化合物の使用量は該パラジウム及び/又はニッケル触媒 1当量 に対し、 0. 3-10. 0当量が好ましぐ特に好ましくは 0. 6〜6. 0当量の範囲で使用 すること力 Sでさる。
また、一般式(5)で示されるテトラハロベンゼンと一般式(6)及び(7)の 2 ハロアリ ール金属試薬の反応により炭素 炭素結合が形成される位置はハロゲンの種類に より制卸すること力 Sでさる。
即ち、ヨウ素原子の反応性が最も高ぐ臭素原子、塩素原子の順に反応性が低下 することから、これらハロゲンの種類の反応性を利用することで反応する位置を任意 に決めることができる。そのため、一般式(2)のテトロハ口ターフェニル誘導体の製造
は、例えば一般式(5)の X5及び X6をヨウ素原子とし、 X2及び X3を臭素原子及び/又 は塩素原子とすることにより、製造すること力 Sできる。
力、くして得られた、本発明の一般式(2)で示されるテトラハロターフェニル誘導体は 、さらに精製することができる。精製する方法は特に限定はなぐ例えばカラムクロマト グラフィー、再結晶化、あるいは昇華による方法を挙げること力 Sできる。
[0066] (耐酸化性有機半導体材料)
次に、本発明の一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機半 導体材料について述べる。該耐酸化性有機半導体材料は溶剤への溶解性、耐酸化 性に優れ、好適な塗布性を有する。該耐酸化性有機半導体材料は本発明の一般式 (1)で示されるヘテロァセン誘導体を溶剤に溶解することにより製造することができる
〇
本発明の一般式(1)で示されるヘテロァセン誘導体の溶解に用いる溶剤は、特に 限定はなぐ例えば o ジクロロベンゼン、クロ口ベンゼン、 1 , 2—ジクロロェタン、 1 , 1 , 2, 2—テトラクロロェタン、クロ口ホルム等のハロゲン系溶剤; THF、ジォキサン等 のエーテル系溶剤;トルエン、キシレン、メシチレン等の芳香族化合物の炭化水素系 溶剤;酢酸ェチル、 Ί ブチロラタトン等のエステル系溶剤; N, N ジメチルホルム アミド、 N メチルピロリドン等のアミド系溶剤;等が挙げられる。又、これら溶剤は 1種 若しくは 2種以上の混合物を用いても良い。中でも、好ましくはクロ口ベンゼン、トルェ ン等である。
上記に挙げた溶剤と一般式(1)で示されるヘテロァセン誘導体を混合攪拌すること により、一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機半導体材料 となるものである。混合攪拌する際の温度は 10〜200°Cが好ましぐ特に好ましくは 2 0〜190°Cである。混合攪拌する際の一般式(1)で示されるヘテロァセン誘導体の濃 度は、溶剤及び温度により変えることができ、 0. 0;!〜 10. 0重量%であることが好ま しい。溶液の調製は空気中でも実施することができる力 好ましくは窒素、アルゴン等 の不活性雰囲気下で調製する。
[0067] 一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機半導体材料の耐酸 化性の評価は、該溶液を所定時間、空気と接触させる方法で実施することができる。
まず用いる溶剤は予め脱気しておき、溶存酸素を除去する。空気との接触時間は、 温度により適宜選択することができ、 0. 5分〜 3時間が好適である。酸化の進行は、 溶液の色の変化並びにガスクロマトグラフィー及びガスクロマトグラフィー(GC)—マ ススペクトル (GCMS)分析による酸化物の検出により行うことができる。
本発明の一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機半導体材 料は、用いられる一般式(1)で示されるヘテロァセン誘導体自体が適度の凝集性を 有することから比較的に低温で溶剤へ溶解でき、且つ耐酸化性があることから、塗布 法による有機薄膜の製造に好適に適用できる。即ち、雰囲気から厳密に空気を除く 必要がないことから塗布工程を簡略化することができる。塗布は空気中でも実施でき るが、好ましくは溶剤の乾燥を考慮して窒素気流下で行う。なお、好適な塗布性を得 るために、本発明の一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機 半導体材料の粘度は、 0. 005〜20ポアズの範囲にあることが好ましい。
(有機薄膜)
次に本発明の一般式(1)で示されるヘテロァセン誘導体を含む耐酸化性有機半導 体材料を用いた有機薄膜につ!/、て述べる。係る有機薄膜は上記の耐酸化性有機半 導体材料 (溶液)の再結晶化若しくは基板への塗布により製造することができ、特に 基板への塗布により製造することが好ましい。そして、基板への塗布により製造するこ とにより、基板上に形成される有機薄膜となるものである。
再結晶化による薄膜は、前記耐酸化性有機半導体材料を冷却することで形成する ことができる。有機薄膜を製造する時の雰囲気は、窒素、アルゴン等の不活性ガス又 は空気下で行うことが好ましぐ特に窒素、アルゴン等の不活性ガス下で行うことが好 ましい。該溶液中の一般式(1)で示されるヘテロァセン誘導体の濃度は、特に限定 はなぐ例えば 0. 01-10. 0重量%である。冷却は 60〜200°Cの温度から 20〜 60°Cが好ましぐ特に好ましくは 10°C〜40°Cの間に冷却することにより好適に実 施すること力 Sできる。またこのようにして製造した結晶状の有機薄膜を適当な基板の 上に張り合わせる、即ちラミネーシヨン等により基板上に製造することもできる。再結 晶化により得られる有機薄膜の膜厚は特に限定はなぐ好ましくは 50nm〜2mm、特 に好ましくは;!〜 500 μ mである。
基板への塗布による有機薄膜の製造は、前記耐酸化性有機半導体材料を基板上 に塗布した後、加熱、気流及び自然乾燥等の方法により溶剤を気化させることで実 施すること力 Sできる。該溶液中の一般式(1)で示されるヘテロァセン誘導体の濃度は 、特に限定はなぐ例えば 0. 0;!〜 10. 0重量%であることが好ましい。塗布温度は特 に限定はなぐ例えば 20〜200°Cの間で好適に実施することができる。塗布の具体 的方法は特に限定はなぐ公知の方法、例えばスピンコート、キャストコート及びディ ップコート等を用いること力 Sできる。さらにスクリーン印局 I インクジェット印局 I グラビア 印刷等の印刷技術を用いても作製することが可能である。使用する基板の材料は特 に限定はなぐ結晶性、非結晶性の種々の材料を用いることができる。基板の具体例 としては、例えばポリエチレンテレフタレート、ポリメチルメタタリレート、ポリエチレン、 ポリプロピレン、ポリスチレン、環状ポリオレフイン、ポリイミド、ポリカーボネート、ポリビ ユルフェノール、ポリビュルアルコール、ポリ(ジイソプロピルフマル酸)、ポリ(ジェチ ルフマル酸)、ポリ(ジイソプロピルマレイン酸)等のプラスチック基板;ガラス、石英、 酸化アルミニウム、シリコン、酸化シリコン、二酸化タンタル、五酸化タンタル、インジゥ ム錫酸化物等の無機材料基板;金、銅、クロム、チタン等の金属基板を好適に用いる こと力 Sできる。またこれらの基板の表面は、例えばォクタデシルトリクロロシラン、ォクタ デシノレトリメトキシシラン等のシラン類;へキサメチノレジシラザン等のシリノレアミン類で 修飾処理したものであっても使用することができる。さらに、基板は絶縁性あるいは誘 電性を有する材料であっても良い。塗布した後の溶剤の乾燥は、常圧若しくは減圧 で除去することができる、又、加熱、窒素気流により乾燥してもよい。さらに、溶剤の気 化速度を調節することで本発明の一般式(1)で示されるヘテロァセン誘導体の結晶 成長を制御すること力 Sできる。基板への塗布により得られる有機薄膜の膜厚は特に 限定はなぐ好ましくは lnm〜; 100 μ m、特に好ましくは 10nm〜20 μ mである。 本発明の一般式(1)で示されるヘテロァセン誘導体は平面剛直性の高い分子構造 を有すること力 、優れた半導体特性を与えることが期待できる。又、該ヘテロァセン 誘導体はトルエンあるいはクロ口ベンゼン等の溶媒に溶解し、溶液状態にあっても容 易に空気酸化されることはない。従って、塗布法により半導体薄膜を容易に作成でき る。したがって、本発明の一般式(1)で示されるヘテロァセン誘導体は、電子ぺーパ
一、有機 ELディスプレイ、液晶ディスプレイ、 ICタグ用等のトランジスタの有機半導体 活性相用途;有機 ELディスプレイ材料;有機半導体レーザー材料;有機薄膜太陽電 池材料;フォトニック結晶材料等の電子材料に利用することができる。
実施例
[0070] 以下、実施例により本発明をさらに詳細に説明するが、本発明はこれら実施例に限 定されるものではない。
生成物の同定には1 H— NMRスペクトル及びマススペクトルを用いた。なお、 ifi— NMRスペクトルは日本電子 tiJEOL GSX— 270WB (270MHz)を用いた。マスス ぺクトル (MS)は日本電子 ti!EOL JMS— 700を用いて、試料を直接導入し、電子 衝突(EI)法(70エレクトロンボルト)あるいは FAB法(6キロエレクトロンボルト、キセノ ンガス、マトリックス(ジチオスレィトール:ジチォエリスリトール = 3 : 1) ) (FABMS)で 測定した。
反応の進行の確認等はガスクロマトグラフィー(GC)及びガスクロマトグラフィ マ ススぺクトノレ(GCMS)分析を用いた。
ガスクロマトグラフィー分析
装置 島津 GC14B
カラム J&Wサイエンティフィック社製、 DB—1 , 30m
ガスクロマトグラフィ^——マススペクトル分析
装置 パーキンエルマ一オートシステム XL (MS部;ターボマスゴールド) カラム J&Wサイエンティフィック社製、 DB—1 , 30
反応用の試薬及び溶媒は、断りのない限り市販品を用いた。なお、グリニャール試 薬あるいはブチルリチウム等の有機金属試薬を用いた場合は、市販の脱水溶媒をそ のまま用いた。
[0071] 合成例 1 (1 , 4 ジブ口モー 2, 5 ジョードベンゼンの合成)
1 , 4 ジブ口モー 2, 5—ジョードベンゼンはジャーナル ォブ アメリカン ケミカル ソサイエティー、 1997年、 119巻、 4578— 4593頁 ίこ記載されてレヽる方法を参考 に合成を行った。
メカニカルスターラー付き 11の三口フラスコに過ヨウ素酸 16· 7g (73. Ommol)及び
硫酸 525mlを加えた。過ヨウ素酸が溶解した後、ヨウ化カリウム 36. 4g (219mmol) を少しずつ添加した。その内容物の温度を— 30°Cに冷却し、 1 , 4 ジブロモベンゼ ン 34. 5g (146mmol)を 5分間かけて添加した。得られた混合物を— 25°Cで 36時間 撹拌した。反応混合物を氷(2Kg)中へ注いだ後、濾過し固体を取り出した。その固 体をクロ口ホルムに溶解させ、 5%苛性ソーダ水溶液及び水で洗浄し、有機相を無水 硫酸マグネシウムで乾燥した。減圧濃縮後、残渣をクロ口ホルムから再結晶化し、白 色結晶を得た(36. Og、収率 50%)。
'H-NMR CCDCl , 21°C): δ = 8. 02 (s, 2H)。
3
ifi— NMRスペクトルが文献値と一致したことより、 1 , 4 ジブ口モー 2, 5—ジョー ドベンゼンが得られたことを確認した。
合成例 2 (2 フエ二ルー 5 ブロモー 4ービフエ二ルポロン酸の合成)
1) 1 , 2 ジブ口モー 4, 5 ジョードベンゼンの合成
1 , 2 ジブ口モー 4, 5 ジョードベンゼンを「シンレット」、 2003年、 29— 34頁に 従い合成した。
メカニカルスターラー付き 11の三口フラスコに過ヨウ素酸 36. 9g (162mmol)及び 硫酸 150mlをカロえた。過ヨウ素酸が溶解した後、ヨウ化カリウム 80. 7g (486mmol) を少しずつ添加した。その内容物の温度を 0°Cに冷却し、 1 , 2 ジブロモベンゼン 7 5. 0g (318mmol)を添加した。得られた混合物を 0°Cで 30分間撹拌した。反応混合 物を氷へ注いだ後、濾過し固体を取り出した。その固体を THF/メタノールから 2回 再結晶化し、白色結晶を得た(76. 2g、収率 49%)。
'H-NMR CCDCl , 21°C): δ = 8. 03 (s, 2H)。
3
ifi— NMR測定より、 1 , 2 ジブ口モー 4, 5 ジョードベンゼンが得られたことを確 した。
2) 1 , 2 ジブ口モー 4, 5 ジフエニルベンゼンの合成
窒素雰囲気下、 200mトンュレンク反応容器に 1)で合成した 1 , 2 ジブ口モー 4, 5 —ジョードベンゼン 3· 074g (6. 30mmol)、テトラキス(トリフエニルホスフィン)パラジ ゥム(東京化成工業製) 600mg (0. 519mmol)及びフエ二ルポロン酸(和光純薬ェ 業製) 1. 920g (15. 7mmol)を添カロした。さらにトノレエン 50ml、エタノーノレ 13ml及
び炭酸ナトリウム 4. 007g (37. 8mmol)と水 16πύからなる水溶液を添加した。 82°C に加熱し、 24時間撹拌した。室温まで冷却後、トルエン及び水を添加し分相した。有 機相を濃縮し、得られた残渣をトルエン 26mlに溶解後、 70%tert ブチルノヽイド口 パーオキサイド溶液 (和光純薬工業製) 1. Omlを添加し、室温で 2時間撹拌した。こ のトルエン溶液を水で 2回洗浄後、有機相を減圧濃縮し、得られた残渣をシリカゲル カラムクロマトグラフィーで精製後(溶媒、へキサン)、白色固体を得た(1. 953g、収 率 80% )。
'H-NMR CCDCl , 21°C): δ = 7. 67 (s, 2H) , 7. 24— 7. 13 (m, 6Η) , 7. 12
3
- 6. 90 (m, 4Η)。
MS m/z : 388 (M+, 100%) , 308 (M+— Br, 23) , 228 (M+— 2Br, 53)。
丄^1 NMR及び MS測定より、 1 , 2 ジブ口モー 4, 5 ジフエ二ルベンゼンが得ら れたことを確認した。
3) 2—フエ二ノレ一 5 -ブロモ 4 ビフエニルボロン酸の合成
窒素雰囲気下、 100mトンュレンク反応容器に 2)で合成した 1 , 2 ジブ口モー 4, 5 ージフエニルベンゼン 755mg (l . 95mmol)及び THF12mlを添加した。この溶液 を— 100°Cに冷却し、 n ブチルリチウム(関東化学製、 1 · 59M)のへキサン溶液 1 . 3ml (2. lmmol)を滴下した。 30分間熟成後、その温度でホウ酸トリイソプロピル( 東京化成工業製) 472mg (2. 51mmol)を滴下した。徐々に室温まで昇温した後、 3 N塩酸を添加し、分相した。有機相を減圧濃縮し、 770mgの白色固体(2 フエニル 5—ブロモー 4 ビフエ二ルポロン酸)を得た。
合成例 3 (2 ブロモー 3 ョードナフタレンの合成)
2 ブロモー 3 ョードナフタレンはシンセティック コミュニュケーシヨンズ、 2003年 、 33巻、 2751— 2756頁に記載されている方法を参考に合成を行った。なお、原料 の 2—ブロモ ビス (へキサクロロシクロペンタジェン)ナフタレンはシグマ ァノレドリッ チから購入したものをそのまま使用した。
窒素雰囲気下、 500mlの 3口フラスコ反応容器にメタンスルホン酸 200ml及びオル ト過ヨウ素酸 1. 31g (5. 74mmol)をカロえた。 30分間撹拌後、ヨウ素 4. 36g (17. 2 mmol)を加えた。この混合物を 2時間撹拌した後、 2—ブロモービス(へキサクロロシ
クロペンタジェン)ナフタレン 30· lg (40. Ommol)を少しずつ加えた。この混合物を 30°Cで 3日間撹拌した。反応混合物を氷水中に注ぎ、生成した固体を濾過した。さら にこの固体を水で洗浄し、減圧乾燥した後、 2 ブロモー 3 ョードービス(へキサク ロロシクロペンタジェン)ナフタレンの白色粉体を得た(34· 8g、収率 99%)。
ガラス製昇華管の末端に上記で得た 2 ブロモ 3 ョードービス(へキサクロロシ クロペンタジェン)ナフタレン 8· 05g (9. 16mmol)をカロえた。末端を 210°Cに加熱し 、 1. 5パスカルに減圧した。発生した 2—ブロモー 3—ョードナフタレンは減圧側のガ ラス管に付着し、へキサクロロシクロペンタジェンは減圧側の底に溜まった。 1時間後 昇華操作を中断し、ガラス管の付着物を取り出し、再度同じ操作を繰り返した。 1時間 の昇華操作を行った(2. 29g、収率 75%)。
'H-NMR CCDCl , 21°C): δ = 8. 41 (s, 1H) , 8. 14 (s, 1H) , 7. 75— 7. 65 (
3
m, 2H) , 7. 54- 7. 45 (m, 2H)。
XH NMRスペクトルが文献値と一致したことより、 2 ブロモ 3 ョードナフタレ ンが得られたことを確認した。
実施例 1 ({ 1 , 4 ビス(3 ブロモベンゾチェニル) 2, 5 ジブ口モ}ベンゼン( テトラハロターフェニル誘導体)の合成)
窒素雰囲気下、 lOOmhンュレンク反応容器に 2, 3 ジブロモベンゾチォフェン(シ グマ—アルドリッチ製) 886mg (3. 03mmol)及び THF8mlを添加した。この溶液を — 30°Cに冷却し、イソプロピルマグネシウムブロマイド (東京化成工業製、 0. 80M) の THF溶液 3. 8ml (3. Ommol)を滴下した。 30分間熟成後、 50°Cに冷却し、そ の温度で塩化亜鉛(シグマ アルドリッチ製、 1. OM)のジェチルエーテル溶液 3. 0 ml (3. Ommol)を滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を 減圧濃縮した。得られた白色固体 [ (3 ブロモベンゾチェニル 2 ジンククロライド ) (一般式(6)及び(7)の化合物)]に、合成例 1で合成した 1 , 4 ジブ口モー 2, 5— ジョードベンゼン 492mg (l . Olmmol) (一般式(5)の化合物)、触媒としてテトラキ ス(トリフエニルホスフィン)パラジウム(東京化成工業製) 91 · 7mg (0. 079mmol)及 び THF8mlを添加した。 63°Cで 10時間反応を実施した後、容器を水冷し 1N塩酸 4 mlを添加することで反応を停止させた。トルエンを添加し、得られた懸濁液を濾過し
、濾板上の固体をトルエン及び水で洗浄した。固体を減圧乾燥し、白色固体 292mg を得た。一方、濾液を分相し有機相を水洗した。有機相を減圧濃縮し、溶媒を留去し た。得られた固体をへキサン洗浄し(10ml)、残渣をトルエンから再結晶化した。析出 した結晶を減圧乾燥後、 206mgの白色固体を得た。先の濾過後の白色固体と合わ せ、収率 75%で目的物を得た。
'H-NMRCCDCl , 21°C): δ =7.95— 7.84 (m, 4H) , 7.81 (s, 2Η) , 7.58
3
-7.44 (m, 4Η)。
MS m/z: 658 (M+, 44%), 498(M+— 2Br, 34), 338(M+— 4Br, 100), 3 06(M+-4Br-S), 9), 169(M+-4Br)/2, 66)。
NMR及び MS測定より、 {1, 4 ビス(3 ブロモベンゾチェ二ル)一 2, 5 ジ ブロモ }ベンゼンが得られたことを確認した。なお、その構造式を下記に示す。
[化 22]
実施例 2 (テトラチェノアセン (ヘテロァセン誘導体)の合成)
窒素雰囲気下、 lOOmhンュレンク反応容器に、実施例 1で合成した {1, 4—ビス(3 ブロモベンゾチェニル)—2, 5 ジブ口モ}ベンゼン 422mg(0.641mmol)及び THF30mlを添加した。この溶液を— 75°Cに冷却し、メタル化剤として sec ブチルリ チウム(関東化学製 1· 0M)のシクロへキサン/へキサン溶液 5· lml (5. lmmol)を 滴下し、テトラメタル化を行った。 40分間撹拌後、 70°Cで反応剤としてビス(フエ二 ルスルホニル)スルフイド(ァクロス製)(一般式(3)及び(4)の化合物) 730mg(2.32 mmol)を一気に投入した。ー晚かけて室温まで温度を上げた。飽和食塩水を添加し た後、分相し、さらに有機相を飽和食塩水で洗浄した。有機相は黄色懸濁液であつ たことから濾過し、黄色固体を取り出し、真空乾燥し、 123mgの黄色固体を得た。生
成した固体を濾過した。さらにこの得られた固体を o ジクロ口ベンゼンで抽出(50°C )した後、減圧乾燥し残渣を 60°Cでトルエンにて洗浄し、残渣を減圧乾燥することで 黄色固体を得た(71mg、収率 28%)。
MS m/z: 402 (M+, 100%), 201(M+/2, 14)。
MS測定より、テトラチェノアセンが得られたことを確認した。なお、その構造式を下 記に示す。
[化 23]
実施例 3 (2, 2,, 5,, 2"—テトラブロモー 1, 1,, 4,, 1 "—ターフェ二ノレ(テトラノヽ 口ターフェニル誘導体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に合成例 1で合成した 1, 4 ジブロモ 2, 5 ジョードベンゼン 4· 39g(9. OOmmol) (一般式(5)の化合物)、触媒として テトラキス(トリフエニルホスフィン)パラジウム(東京化成工業製) 974mg (0.84mmo 1)及び 2 ブロモフエ二ルポロン酸(シグマ アルドリッチ製) 4· 16g (一般式(6)及 び(7)の化合物) (20.7mmol)を添加した。さらにトルエン 72ml、エタノール 18ml 及び炭酸ナトリウム 5.72g(54. Ommol)と水 22πύからなる水溶液を添加した。 85 °Cのオイルバスに浸し、 15時間撹拌した。室温まで冷却後、ジクロロメタン及び飽和 食塩水を添加し分相した。有機相を減圧濃縮し、残渣をトルエンから再結晶化し、白 色針状晶を得た(3.68g、収率 75%)。
融点: 230— 231°C。
'H-NMRCCDCl , 21°C): δ =7.70 (d, J = 8. OHz, 2H) , 7.55(d, J=l.5H
3
z, 2H), 7.45-7.23 (m, 6H)。
MS m/z: 546 (M+, 92%), 466(M+-Br, 45), 386(M+-2Br, 53), 226
(M+-4Br, 100)。
ifi— NMR及び MS測定より、 2, 2,, 5,, 2"—テトラブロモー 1, 1', 4', ,一ター フエニルが得られたことを確認した。なお、その構造式を下記に示す。
[0079] [化 24]
[0080] 実施例 4 (P, P ジフエニルベンゾホスホロジベンゾホスホール(ヘテロァセン誘 導体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に実施例 3で合成した 2, 2', 5', 2"— テトラブロモー 1, 1', 4', 1"—ターフェニル 410mg(0.752mmol)及び THF30ml を添加した。この溶液を一 80°Cに冷却し、メタル化剤として sec ブチルリチウム(関 東化学製 1.0M)のシクロへキサン/へキサン溶液 6. Oml (6. Ommol)を滴下し、 テトラメタル化を行った。溶液の色が薄黄色から真緑色へ変化した。 20分間撹拌後、 — 75°Cで反応化剤としてジクロロフヱニルホスフィン (東京化成工業製) 452mg (2. 52mmol) (一般式(3)及び(4)の化合物)を添加し、ー晚かけて室温まで温度を上 げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄し た。減圧濃縮し、得られた残渣にへキサンを添加し撹拌後静置し、上澄み液を取り除 き、減圧乾燥した。残渣をトルエンから再結晶化し、淡黄色の結晶を得た(101mg、 収率 30%)。
'H-NMRCCDCl , 21°C): δ =8.26 (s, 2H) , 7.94 (d, J = 7.8Hz, 2H) , 7.
3
69(d, J = 7. 1Hz, 2H), 7.44(t, J = 7.8Hz, 2H) , 7.41— 7.10 (m, 12H)。 MS m/z: 442 (M+, 100%), 364(M+— Ph— 1, 38), 288(M+— 2Ph, 19) , 221(M+/2, 10)。
ェ^1 NMR及び MS測定より、 P, P ジフエニルベンゾホスホロジベンゾホスホール
が得られたことを確認した。なお、その構造式を下記に示す。
[0081] [化 25]
[0082] 実施例 5 (B, B—シフエニルベンゾボロリルジベンゾボロール(ヘテロァセン誘導 体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に実施例 3で合成した 2, 2 ' , 5 ' , 2"— テトラブロモー 1 , 1 ' , 4 ' , 1"—ターフェニル 425mg (0. 778mmol)及び THF30ml を添加した。この溶液を一 80°Cに冷却し、メタル化剤として sec—ブチルリチウム(関 東化学製 1. 0M)のシクロへキサン/へキサン溶液 6. 2ml (6. 2mmol)を滴下し、 テトラメタル化を行った。溶液の色が薄黄色から真緑色へ変化した。 20分間撹拌後、 — 75°Cで反応化剤としてジクロロフエ二ルポラン(シグマ—アルドリッチ製) 410mg (2 . 58mmol) (一般式(3)及び(4)の化合物)を添加し、ー晚かけて室温まで温度を上 げた。飽和食塩水を添加した後分相し、さらに有機相を炭酸カリウム水溶液で洗浄し た。減圧濃縮し、得られた残渣にへキサンを添加し撹拌後静置し、上澄み液を取り除 き、減圧乾燥した。残渣をトルエンから再結晶化し、淡黄色の結晶を得た(78mg、収 率 25%)。
MS m/z : 402 (M+, 100%) , 201 (M+/2, 14)。
MS測定より、 B, B—ジフエニルベンゾポロリルジベンゾボロールが得られたことを 確認した。なお、その構造式を下記に示す。
[0083] [化 26]
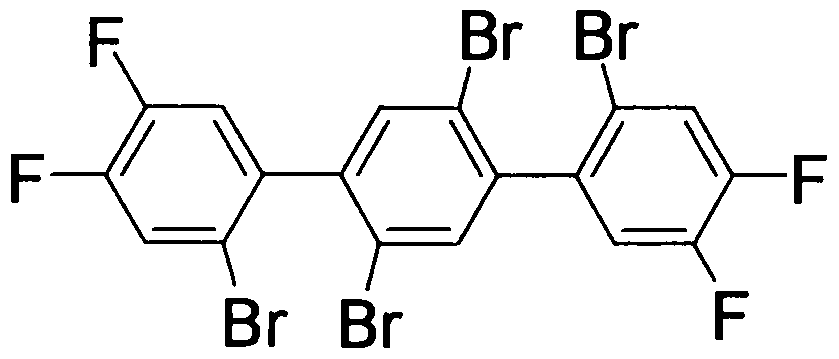
実施例 6 (4, 5, 4" , 5"—テトラフルオロー 2, 2,, 5,, 2"—テトラブロモー 1 , 1 ' , 4,, 1" ターフェニル(テトラハロターフェニル誘導体)の合成)
窒素雰囲気下、 100mトンュレンク反応容器に 1 , 2 ジブ口モー 4, 5 ジフルォロ ベンゼン(和光純薬工業製) 2. 53g (9. 30mmol)及び THF15mlを添加した。この 溶液を— 40°Cに冷却し、イソプロピルマグネシウムブロマイド(関東化学製、 0. 65M )の1¾?溶液1 51111 (9. 7mmol)を滴下した。 30分間熟成後、その温度で塩化亜鉛 (シグマ アルドリッチ製、 1. 0M)のジェチルエーテル溶液 9· 8ml (9. 8mmol)を 滴下した。徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得 られた白色固体(2 ブロモー 4, 5 ジフルオロフェニルマグネシウムブロマイド)(一 般式(6)及び(7)の化合物)に、合成例 1で合成した 1 , 4 ジブ口モー 2, 5 ジョー ドベンゼン 2· 15g (4. 41mmol) (一般式(5)の化合物)、テトラキス(トリフエニルホス フィン)パラジウム(東京化成工業製) 408mg (0. 353mmol)及び THF30mlを添加 した。 60°Cで 6時間反応を実施した後、容器を水冷し 3N塩酸(8ml)を添加すること で反応を停止させた。トルエン及び食塩を添加後、分相し、有機相を食塩水で洗浄 した。有機相を減圧濃縮し溶媒を留去した。この得られた残渣をトルエン 10mlに溶 解させ、 70%tert—プチルノヽイド口パーオキサイド溶液(和光純薬工業製)(0· 5ml) を添加し、室温で 2時間撹拌した。この溶液を水洗浄し、有機相を減圧濃縮した。有 機相をトルエン:へキサン = 1: 1に溶解させ、シリカゲルを充填したカラムを通過させ た。溶出液を減圧濃縮し、得られた固体をへキサン:トルエン = 3 : 1の混合溶媒を用 いて再結晶化を行い、白色固体を得た(1. 48g,収率 54%)。
H-NMR(CDC1 , 21°C): δ =7.58— 7.45 (m, 2H) , 7.53(s, 2H) , 7.23
3
-7.09 (m, 2H)。
MS m/z: 618(M+, 73%), 538(M+— Br, 32), 458(M+— 2Br, 45), 378 (M+-3Br, 4), 298(M+-4Br, 100)。
ifi— NMR及び MS測定より、 4, 5, 4", 5"—テトラフノレォロ一 2, 2,, 5,, 2"—テ トラブロモー 1, 1', 4', 1 "—ターフェニルが得られたことを確認した。なお、その構 造式を下記に示す。
[化 27]
実施例 7 (テトラフルォロジチェノアセン (ヘテロァセン誘導体)の合成)) 窒素雰囲気下、 lOOmhンュレンク反応容器に、実施例 6で合成した 4, 5, 4", 5"— テトラフノレオロー 2, 2,, 5,, 2"—テトラブロモー 1, 1', 4', 1"—ターフェ二ノレ 506m g(0.818mmol)及び THF28mlを添加した。この懸濁溶液を— 80°Cに冷却し、メタ ル化剤として sec—ブチルリチウム(関東化学製 1. OM)のシクロへキサン/へキサン 溶液 5.9ml (5.9mmol)を滴下し、テトラメタル化を行った。 20分間撹拌後、—75 。Cでビス(フエニルスルホニル)スルフイド(ァクロス製) 900mg (2· 86mmol) (一般式 (3)及び (4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室温まで反応 温度を上げた。飽和食塩水及びトルエンを添加した後、分相し、さらに有機相を飽和 食塩水で洗浄した。減圧濃縮し、得られた残渣にへキサンを添加し撹拌後、静置し、 上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化した(77mg,収率 26%)。
H-NMR(CDC1 , 21°C): δ =8.46 (s, 2H) , 8. 10 (m, 2H) , 7.81 (m, 2H)
MS m/z: 362(M+, 100%), 181(M+/2, 18)。
丄^1 NMR及び MS測定より、テトラフルォロジチェノアセンが得られたことを確認し た。なお、その構造式を下記に示す。
[化 28]
実施例 8 (4, 5, 4", 5"—テトラフエ二ノレ一 2, 2,, 5,, 2"—テトラブロモ一 1, 1', 4,, 1" ターフェニル(テトラハロターフェニル誘導体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に合成例 2で合成した 2 フエニル— 5 ーブロモー 4ービフエ二ルポロン酸(一般式(6)及び(7)の化合物) 770mg、合成例 1で合成した 1, 4 ジブ口モー 2, 5 ジョードベンゼン 476mg(0.976mmol) (一 般式(5)の化合物)、テトラキス(トリフエニルホスフィン)パラジウム (東京化成工業製) 90. lmg(0.078mmol)、 トノレェン 7.6ml及びエタノーノレ 1.8mlを添カロした。さら に炭酸ナトリウム 625mg(5.90mmol)と水 2.3mlからなる溶液を添加し、この混合 物を 85°Cで 30時間反応を実施した。室温まで冷却させた後、トルエン及び食塩水を 添加分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を留去した。得 られた固体をトルエン:へキサン = 7: 3の混合溶媒を用いて再結晶化を行い、白色固 体を得た(467mg、収率 56%)。
'H-NMRCCDCl , 21°C): δ =7.77(s, 0.85H), 7.76 (s, 1.15H), 7.69(
3
s, 2H), 7.42(s, 1.15H), 7.35(s, 0.85H), 7.28— 7. 13(m, 20H)。
FABMS m/z: 850(M+, 100%), 770(M+— Br, 71)。
ifi— NMR及び FABMS測定より、 4, 5, 4", 5,,一テトラフエ二ノレ一 2, 2,, 5,, 2" ーテトラブロモー 1, 1', 4', 1 "—ターフェニルが得られたことを確認した。なお、その 構造式を下記に示す。
[化 29]
実施例 9 (テトラフエニルジチェノアセン (ヘテロァセン誘導体)の合成) 窒素雰囲気下、 lOOmhンュレンク反応容器に、実施例 8で合成した 4, 5, 4", 5"— テトラフエ二ノレ一 2, 2,, 5,, 2"—テトラブロモー 1, 1', 4,, 1"—ターフェニル 416m g(0.489mmol)及び THF30mlを添加した。この懸濁溶液を— 80°Cに冷却し、メタ ル化剤として sec—ブチルリチウム(関東化学製 1.0M)のシクロへキサン/へキサン 溶液 3.9ml (3.9mmol)を滴下し、テトラメタル化を行った。 20分間撹拌後、—75 °Cでビス(フエニルスルホニル)スルフイド(ァクロス製) 507mg(l.61mmol) (一般式 (3)及び (4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室温まで反応 温度を上げた。飽和食塩水及びトルエンを添加した後、分相し、さらに有機相を飽和 食塩水で洗浄した。減圧濃縮し、得られた残渣にへキサンを添加し撹拌後、静置し、 上澄み液を取り除き、減圧乾燥した。残渣をトルエンから再結晶化し、結晶を得た(9 Omg,収率 31%)。
MS m/z: 594 (M+, 100%), 297(M+/2, 15)。
MS測定より、テトラフエニルジチェノアセンが得られたことを確認した。なお、その 構造式を下記に示す。
[化 30]
実施例 10 (2, 2', 5', 2" テトラブロモー 1, 1', 4', 1"ージベンゾターフェニル (テトラハロターフェニル誘導体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に合成例 3で合成した 2 ブロモ 3— ョードナフタレン 2· 03g(6. lOmmol)及び THF12mlを添加した。この溶液を一 65 °Cに冷却し、イソプロピルマグネシウムブロマイド(関東化学製、 0· 65M)の THF溶 液 9.9ml (6.4mmol)を滴下した。 30分間熟成後、その温度で塩化亜鉛(シグマ アルドリッチ製、 1. OM)のジェチルエーテル溶液 6.4ml (6.4mmol)を滴下した。 徐々に室温まで昇温した後、生成した白色スラリー液を減圧濃縮した。得られた白色 固体(2 ブロモナフチルー 3 マグネシウムブロマイド)(一般式(6)及び(7)の化合 物)に、合成例 1で合成した 1, 4 ジブ口モー 2, 5 ジョードベンゼン 1· 41g(2.88 mmol) (一般式(5)の化合物)、テトラキス(トリフエニルホスフィン)パラジウム(東京化 成工業製) 285mg(0.247mmol)及び THF31mlを添加した。 60°Cで 4時間反応 を実施した後、容器を水冷し 3N塩酸 4mlを添加することで反応を停止させた。全体 を減圧濃縮し、溶媒を留去した。析出した固体を濾液が中性になるまで水で洗浄し、 さらにクロ口ホルムと THFで洗浄した。得られた結晶を減圧乾燥後、結晶を得た(1. 20g,収率 64%)。
'H-NMRCCDCl , 60°C): δ =8.22(s, 2H) , 7.90— 7.75 (m, 4Η) , 7.85(
3
s, 2Η), 7.67(s, 2Η), 7.60— 7.48 (m, 4Η)。
MS m/z: 646 (M+, 64%), 566(M+-Br, 8), 486(M+-2Br, 34), 406 (
M -3Br, 6), 326(M -4Br, 92), 163((M -4Br)/2, 100)。 ifi— NMR及び MS測定より、 2, 2,, 5,, 2"—テトラブロモー 1, 1', 4', ,一ジ ベンゾターフェニルが得られたことを確認した。なお、その構造式を下記に示す。
[0093] [化 31]
[0094] 実施例 11 (ジベンゾジチェノアセン (ヘテロァセン誘導体)の合成)
窒素雰囲気下、 100mlシュレンク反応容器に、実施例 10で合成した 2, 2', 5', 2" —テトラブロモー 1, 1', 4', 1"—ジベンゾターフェニル 388mg(0.601mmol)及び THF27mlを添加した。この懸濁溶液を— 80°Cに冷却し、メタル化剤として sec—ブ チルリチウム(関東化学製 1.0M)のシクロへキサン/へキサン溶液 4.8ml (4.8m mol)を滴下し、テトラメタル化を行った。 20分間撹拌後、— 75°Cでビス(フエニルス ルホニル)スルフイド(ァクロス製) 660mg(2. lOmmol) (一般式(3)及び(4)の化合 物)を一気に投入した。徐々に昇温し、一晩かけて室温まで反応温度を上げた。飽 和食塩水を添加した後、分相し、さらに有機相を飽和食塩水で洗浄した。減圧濃縮 し、得られた残渣にトルエンを添加し撹拌後、静置し、上澄み液を取り除き、減圧乾 燥し結晶を得た(59mg,収率 25%)。
MS m/z: 390(M+, 100%), 195(M+/2, 11)。
MS測定より、ジベンゾジチェノアセンが得られたことを確認した。なお、その構造式 を下記に示す。
[0095] [化 32]
合成例 4 (2 ブロモー 3 ョードー 6, 7 -
1) 1 , 2—ジドデシルベンゼンの合成
1 , 2 ジドデシルベンゼンは、「シンセシス」、 1993年、 387— 390頁の方法を参 考に 1 , 2—ジクロ口ベンゼンと n ドデシルマグネシウムブロマイドから次のように合 成した。
窒素雰囲気下、 200mトンュレンク反応容器に 1 , 2 ジクロ口ベンゼン 2. 7ml (24. Ommol)、塩ィ匕二ッケノレ {ビス (ンフエニノレホスフイノ)プロノヽン } 66mg (0. 12mmol) 、及びジェチルエーテル 18mlを添加した。 0°Cに冷却し、 n ドデシルマグネシウム ブロマイド(シグマ アルドリッチ製、 1. OM)のジェチルエーテル溶液 65ml (65mm ol)を滴下した。 35°Cで 11時間反応後、 3N塩酸を加えて反応を停止させた。ジェチ ルエーテルで抽出し、有機相を水及び飽和炭酸水素ナトリウム水溶液で洗浄した。 塩化カルシウムで乾燥し、溶媒を減圧濃縮した。残渣を 170°Cで真空乾燥し(20Pa) 、 1 , 2 ジドデシルベンゼンの液体を得た(8 · 36g、収率 84%)。
2) 4 -ブロモ 5 ョード無水フタル酸の合成
4ーブロモー 5—ョード無水フタル酸は「ジャーナル ォブ オルガニック ケミストリ ィー」(米国)、 1951年、 16巻、 1577— 1581頁 ίこ記載されてレヽる方法を参考 ίこ 4 ブロモ無水フタル酸を原料に用いて次のように合成を行った。
100mlの三口フラスコに 4 ブロモ無水フタル酸(東京化成工業製) 6· 42g (28. 3 mmol)、 10%発煙硫酸 25ml、及びヨウ素 3. 60g (14. 2mmol)を加えた。 110°C に加熱し、 4時間反応を行った。室温に冷却後、反応物を氷中へ注いでタエンチした 。冷 20%水酸化ナトリウム水溶液で処理した後、塩酸を添加し、溶液の pHを 6〜7と
した。不溶物を濾紙を用いて除去し、さらに塩酸を少しずつ添加し、 pHを 1以下にし た。得られたスラリー液を一晩撹拌した後、生成した沈殿物を濾別し、乾燥した。得ら れた固体をトルエンで洗浄し、残渣を冷 20%水酸化ナトリウム水溶液で処理し溶解さ せた。酢酸を用いて溶液の pHを 3. 5とし生成した沈殿物を濾別した。この沈殿物を 塩酸で処理した後、さらにトルエンと無水酢酸で処理することで 1. 10gの 4 ブロモ 5—ョード無水フタル酸を得た(収率 11 % )。
[0097] 3) 2 ブロモー 3—ョードー 6, 7 ジドデシルアントラキノンの合成
2 ブロモ 3 ョード 6, 7 ジドデシルアントラキノンは「ベリヒテ」(独国)、 193 3年、 66B巻、 1876— 1891頁に記載されている方法を参考に次のように合成を行 つた。
100mlの三口フラスコに上記で合成した 4 -ブロモー 5—ョード無水フタル酸 1. 00 g (2. 83mmol)、 1 , 2 ジドデシノレベンゼン 1. 29g (3. 1 lmmol)、及びテトラクロ口 ェタン 4mlを加えた。そこへ塩化アルミニウム 0· 82g (6. 15mmol)を添加し、室温で 3時間撹拌した。得られた反応混合物に氷を少しずつ加えてタエンチした後、トルェ ンで抽出した。減圧濃縮し、 2. 5gの粘調物を得た。この粘調物に硫酸 8mlを添加し 、 80°Cで 2時間撹拌した。得られた反応混合物を室温まで冷却し、氷を加えた。トノレ ェンで抽出し、有機相を硫酸ナトリウムで乾燥後、濾過、減圧濃縮し、 678mgの 2— ブロモー 3—ョードー 6, 7—ジドデシルアントラキノンを得た(収率 35 % )。
4) 2 -ブロモ 3 ョードー 6, 7 ジドデシルアントラセンの合成
上記で得た 2 ブロモー 3 ョードー 6, 7 ジドデシルアントラキノン 678mgに TH F 14mlを加え溶解させた後、水素化ジイソプロピルアルミニウム(関東化学製、 0. 99 M)トルエン溶液 2. 7ml (2. 7mmol)を添加し、室温で 2時間撹拌した。氷冷後、 6N 塩酸 5mlを添加した後、 65°Cに加熱し、 4時間反応を行った。トルエン及び食塩水を 添加し、分相した。さらに食塩水で洗浄し、有機相を減圧濃縮及び真空乾燥した。得 られた残渣に再度、水素化ジイソプロピルアルミニウムを用いた還元、 6N塩酸による 脱水操作を繰り返した。粗生成物をトルエンから再結晶精製し、 469mgの薄黄色固 体である 2 ブロモー 3 ョードー 6 , 7 ジドデシルアントラセンを得た(収率 72%)。
[0098] 実施例 12 (3, 2,, 5,, 3" テトラブロモー 6, 7, 6" , 7" (テトラドデシル) 2,
1', 4,, 2"—ジナフトターフェニル(テトラハロターフェニル誘導体)の合成) 窒素雰囲気下、 100mlシュレンク反応容器に合成例 4で合成した 2—ブロモ— 3— ョードー 6, 7—ジドデシルアントラセン 461mg(0. 640mmol)及び THF8mlを添加 した。この溶液を— 40°Cに冷却し、イソプロピルマグネシウムブロマイド(関東化学製 、 0. 65M)の THF溶液 1.0ml (0. 65mmol)を滴下した。 30分間熟成後、— 78。C に冷却し、塩化亜鉛(シグマ—アルドリッチ製、 1.0M)のジェチルエーテル溶液 0. 65ml (0. 65mmol)を滴下した。徐々に室温まで昇温した後、得られた反応液を減 圧濃縮した。得られた残渣に、合成例 1で合成した 1, 4—ジブロモ— 2, 5—ジョード ベンゼン 145mg(0. 298mmol)、テトラキス(トリフエニルホスフィン)パラジウム(東 京化成工業製) 27. 5mg(0. 0238mmol)及び THF8mlを添加した。 60°Cで 7時間 反応を実施した後、容器を水冷し 3N塩酸 3mlを添加することで反応を停止させた。ト ルェンを添加後、分相し、有機相を食塩水で洗浄した。有機相を減圧濃縮し溶媒を 留去し、さらに真空乾燥した。得られた残渣にトルエンを添加し、 70%tert—ブチル ノ、イド口パーオキサイド溶液(和光純薬工業製)(0· 06ml)を添加し、室温で 2時間 撹拌した。この溶液を水洗浄し、有機相を減圧濃縮析出した。残渣をシリカゲルを充 填したカラムクロマトグラフィーで濾過し (溶媒;へキサン:クロ口ホルム = 5:2)、濾液 を減圧濃縮した。得られた残渣をへキサンで洗浄、真空乾燥後、 254mgの黄色固体 を得た (収率 60%)。
MS m/z: 1419(M+, 100%), 1339(M+— Br, 8), 1108(M+-2C H , 1
11 23
5).
MS測定より、 3, 2,, 5,, 3"—テトラブロモー 6, 7, 6", 7"— (テトラドデシル)一 2, 1', 4', 2"—ジナフトターフェニルが得られたことを確認した。なお、その構造式を下 記に示す。
[0100] 実施例 13 (テトラドデシルジナフトジチェノアセン (ヘテロァセン誘導体)の合成) 窒素雰囲気下、 100mlシュレンク反応容器に、実施例 12で合成した 3, 2', 5', 3" ーテトラブロモー 6, 7, 6", 7"—(テトラドデシル) 2, 1', 4', 2" ジナフトターフ ェニル 122mg(0.086mmol)及び THF6mlを添加した。この懸濁溶液を 75°Cに 冷却し、メタル化剤として sec ブチルリチウム(関東化学製 1.0M)のシクロへキサン /へキサン溶液 0. 7ml (0. 7mmol)を滴下し、テトラメタル化を行った。 20分間撹拌 後、 80。Cでビス(フエニルスルホニノレ)スルフイド(ァクロス製) 108mg(0. 344mm ol) (一般式(3)及び (4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室 温まで反応温度を上げた。トルエン及び飽和食塩水を添加した後、分相し、さらに有 機相を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣 をトルエンから再結晶精製し、 25mgの固体を得た(収率 25%)。
MS m/z: 1164(M+, 100%), 582(M+/2, 14).
MS測定より、テトラドデシルジナフトジチェノアセンが得られたことを確認した。なお 、その構造式を下記に示す。
[0102] 実施例 14 (テトラドデシルジナフトジチェノアセン (ヘテロァセン誘導体)の合成) 窒素雰囲気下、 100mトンュレンク反応容器を— 75°Cに冷却し、 THF6ml及びメタ ル化剤として sec ブチルリチウム(関東化学製 1. 0M)のシクロへキサン/へキサン 溶液 1. Oml (1. Ommol)を添加した。 75°C下で実施例 12で合成した 3, 2 ' , 5 ' , 3" テトラブロモー 6, 7, 6" , 7"—(テトラドデシル) 2, 1 ' , 4 ' , 2" ジナフトター フエニル 132mg (0. 093mmol)を投入し、テトラメタル化を行った。 20分間撹拌後、 — 80。Cでビス(フエニルスルホニル)スルフイド(ァクロス製) 117mg (0. 372mmol) ( 一般式(3)及び (4)の化合物)を一気に投入した。徐々に昇温し、一晩かけて室温ま で反応温度を上げた。トルエン及び飽和食塩水を添加した後、分相し、さらに有機相 を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、減圧濃縮した。得られた残渣をトル ェンから再結晶精製し、 40mgのテトラドデシルジナフトジチェノアセンを得た (収率 3 7%) o
[0103] 実施例 15 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、 100mトンュレンク容器にクロ口ベンゼン 5. 4gを添加し、凍結(液体 窒素) 減圧 窒素置換 融解から成るサイクルを 3回繰り返すことで溶存酸素を除 去した。そこへ実施例 2で得られたテトラチェノアセンの固体 5. lmgを添加し、 50°C に加熱し溶解させ、テトラチェノアセンを含む耐酸化性有機半導体材料を合成した( 山吹色溶液)。次に、このシュレンク容器の上部の栓を開け、 1分間、外気に接触させ ることで空気を導入(耐酸化性評価)し、さらに 50°Cで撹拌した力 色の変化は見ら れな力 た。したがって、色の変化が見られなかったことから、耐酸化性に優れるもの であった。
[0104] 実施例 16 (有機薄膜の作成)
窒素雰囲気下、実施例 2で得られたテトラチェノアセン 2. 5mgをクロ口ベンゼン 25 gと混合し、 70°Cで 1時間撹拌し、テトラチェノアセンの山吹色溶液を調製した (テトラ チェノアセンを含む耐酸化性有機半導体材料の合成)。
窒素雰囲気下、凹面のあるガラス基板を 70°Cに加熱し、この基板上に上記の溶液 をスポイトを用いて塗布し常圧下で乾燥し、膜厚 280nmの有機薄膜を作製した。
[0105] 実施例 17 (耐酸化性有機半導体材料の合成及びその耐酸化性評価)
窒素雰囲気下、 100mトンュレンク容器にクロ口ベンゼン 5. 4gを添加し、凍結(液体 窒素) 減圧 窒素置換 融解から成るサイクルを 3回繰り返すことで溶存酸素を除 去した。そこへ実施例 14で得られたテトラドデシルジナフトジチェノアセンの固体 7. 2mgを添加し、 70°Cに加熱し溶解させ、テトラドデシルジナフトジチェノアセンを含む 耐酸化性有機半導体材料を合成した (黄橙色溶液)。次に、このシュレンク容器の上 部の栓を開け、 1分間、外気に接触させることで空気を導入 (耐酸化性評価)し、さら に 70°Cで撹拌した力 色の変化は見られなかった。したがって、色の変化が見られ なかったことから、耐酸化性に優れるものであった。
さらにこの溶液を 70°C。 1時間、撹拌下で空気と接触させても溶液の色の変化は見 られず、耐酸化性に優れるものであった。
[0106] 実施例 18 (有機薄膜の作成)
窒素雰囲気下、実施例 14で得られたテトラドデシルジナフトジチェノアセン 4. 7mg をクロ口ベンゼン 15gと混合し、 70°Cで 1時間撹拌し、テトラドデシルジナフトジチエノ ァセンの黄橙色溶液を調製した (テトラドデシルジナフトジチェノアセンを含む耐酸化 性有機半導体材料の合成)。
窒素雰囲気下、凹面のあるガラス基板を 70°Cに加熱し、この基板上に上記の溶液 をスポイトを用いて塗布し常圧下で乾燥し、膜厚 220nmの有機薄膜を作製した。
[0107] 比較例 1 (耐酸化性評価)
ペンタセンを用いて耐酸化性を評価した。
窒素雰囲気下、 20mトンュレンク容器に o ジクロロベンゼン 2. 9gを添加し、凍結( 液体窒素) 減圧 窒素置換 融解から成るサイクルを 3回繰り返すことで溶存酸 素を除去した。そこへペンタセン (東京化成工業製) 2. 5mgを添加し、 120°Cに加熱
し溶解させると赤紫色溶液となった。次にこのシュレンク容器の上部の栓を開け、 1分 間、外気に接触させることで空気を導入し、さらに 120°Cで撹拌した。ガスクロマトダラ フィー及びガスクロマトグラフィ^——マススペクトル(GCMS)分析から、 6, 13—ペン タセンキノンが生成して!/、ることがわかった。
さらにこの溶液を 120°C、 1時間、撹拌下で空気と接触させると溶液の色が黄に変 化していた。ガスクロマトグラフィー分析から、 6, 13—ペンタセンキノンの生成が増加 していることがわかった。
したがって、溶液の色の変化及び 6, 13—ペンタセンキノンが生成していることから 、酸化が進行しており、耐酸化性に劣るものであった。
[0108] 本発明を詳細にまた特定の実施態様を参照して説明したが、本発明の精神と範囲 を逸脱することなく様々な変更や修正を加えることができることは当業者にとって明ら 力、である。
本出願 (ま、 2006年 8月 28曰出願の曰本特許出願(特願 2006— 231082) ίこ基づ くものであり、その内容はここに参照として取り込まれる。
産業上の利用可能性
[0109] 本発明によれば、優れた耐酸化性を有し、塗布法による有機半導体活性相形成が 可能な、ヘテロァセン誘導体及びその用途を提供することができる。更に、該ヘテロ ァセン誘導体の前駆化合物であるテトラハロターフェニル誘導体及びその製造方法 をあ提供すること力でさる。