WO2008036022A1 - Tetrahydro-lh-pyrido[3,4-b] indole derivatives as cb1 receptor ligands - Google Patents
Tetrahydro-lh-pyrido[3,4-b] indole derivatives as cb1 receptor ligands Download PDFInfo
- Publication number
- WO2008036022A1 WO2008036022A1 PCT/SE2007/000822 SE2007000822W WO2008036022A1 WO 2008036022 A1 WO2008036022 A1 WO 2008036022A1 SE 2007000822 W SE2007000822 W SE 2007000822W WO 2008036022 A1 WO2008036022 A1 WO 2008036022A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- tetrahydro
- pyrido
- alkyl
- oxobutyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)(C)OC(N(CC1)Cc(c2c3)c1[n]c2ccc3C(*)=O)=O Chemical compound CC(C)(C)OC(N(CC1)Cc(c2c3)c1[n]c2ccc3C(*)=O)=O 0.000 description 3
- NCRWBLSVIRVJJQ-UHFFFAOYSA-N COC(c1ccc2[nH]c(CCNC3)c3c2c1)=O Chemical compound COC(c1ccc2[nH]c(CCNC3)c3c2c1)=O NCRWBLSVIRVJJQ-UHFFFAOYSA-N 0.000 description 2
- DGOOWCRAFJNCIH-UHFFFAOYSA-N CCS([n](c(CCN(C1)C2CCOCC2)c1c1c2)c1ccc2C(OC)=O)(=O)=O Chemical compound CCS([n](c(CCN(C1)C2CCOCC2)c1c1c2)c1ccc2C(OC)=O)(=O)=O DGOOWCRAFJNCIH-UHFFFAOYSA-N 0.000 description 1
- LCKVZLSFTPNJSJ-UHFFFAOYSA-N CCS([n](c(CCNC1)c1c1c2)c1ccc2C(OC)=O)(=O)=O Chemical compound CCS([n](c(CCNC1)c1c1c2)c1ccc2C(OC)=O)(=O)=O LCKVZLSFTPNJSJ-UHFFFAOYSA-N 0.000 description 1
- ZMLGETIZAKBAJF-UHFFFAOYSA-N COC(c1ccc2[nH]c(CCN(C3)C4CCCC4)c3c2c1)=O Chemical compound COC(c1ccc2[nH]c(CCN(C3)C4CCCC4)c3c2c1)=O ZMLGETIZAKBAJF-UHFFFAOYSA-N 0.000 description 1
- IJKQHGCIYZFPRF-UHFFFAOYSA-N CS([n](c(CCN(C1)C2CCCC2)c1c1c2)c1ccc2C(O)=O)(=O)=O Chemical compound CS([n](c(CCN(C1)C2CCCC2)c1c1c2)c1ccc2C(O)=O)(=O)=O IJKQHGCIYZFPRF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the invention is related to therapeutic compounds, pharmaceutical compositions containing these compounds, manufacturing processes thereof and uses thereof.
- the present invention is related to compounds that may be effective in treating pain, cancer, multiple sclerosis, Parkinson's disease, Huntington's chorea, Alzheimer's disease, anxiety disorders, gastrointestinal disorders and/or cardiovascular disorders.
- cannabinoid receptor e.g., CBi receptor, CB 2 receptor
- CBi receptors e.g., CBi receptor, CB 2 receptor
- agonists, antagonists and inverse agonists produce relief of pain in a variety of animal models by interacting with CBi and/or CB 2 receptors.
- CBi receptors are located predominately in the central nervous system
- CB 2 receptors are located primarily in the periphery and are primarily restricted to the cells and tissues derived from the immune system.
- CBi receptor agonists such as ⁇ 9 -tetrahydrocannabinol ( ⁇ 9 -THC) and anadamide
- CNS side-effects e.g., psychoactive side effects, the abuse potential, drug dependence and tolerance, etc.
- CBi receptors located in CNS There are lines of evidence, however, suggesting that CBi agonists acting at peripheral sites or with limited CNS exposure can manage pain in humans or animals with much improved overall in vivo profile. Therefore, there is a need for new CBi receptor ligands such as agonists that may be useful in managing pain or treating other related symptoms or diseases with reduced or minimal undesirable CNS side-effects.
- the present invention provides CBi receptor ligands which may be useful in treating pain and/or other related symptoms or diseases.
- C m . n or "C m . n group” refers to any group having m to n carbon atoms.
- alkyl refers to a saturated monovalent straight or branched chain hydrocarbon radical comprising 1 to about 12 carbon atoms.
- alky Is include, but are not limited to, groups, such as methyl, ethyl, propyl, isopropyl, 2-methyl-l -propyl, 2-methyl-2-propyl, 2-methyl-l -butyl, 3 -methyl- 1 -butyl, 2-methyl-3-butyl, 2,2-dimethyl-l -propyl, 2-methyl-l -pentyl, 3-methyl-l- ⁇ enryl, 4- methyl-1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 2,2- dimethyl-1-butyl, 3,3-dirnethyl-l-butyl, 2-ethyI-l -butyl, butyl, isobutyl, t-butyl, pentyl, isopentyl, neopentyl, and hexyl, and longer alkyl groups, such as heptyl,
- alkyl can be unsubstituted or substituted with one or two suitable substituents.
- cycloalkyl refers to a saturated monovalent ring-containing hydrocarbon radical comprising at least 3 up to about 12 carbon atoms.
- Examples of cycloalkyls include, but are not limited to, C 3 _ 7 cycloalkyl groups, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl, and saturated cyclic and bicyclic terpenes.
- a cycloalkyl can be unsubstituted or substituted by one or two suitable substituents.
- the cycloalkyl is a monocyclic ring or bicyclic ring.
- cycloalkenyl refers to a monovalent ring-containing hydrocarbon radical having at least one carbon-carbon double bond and comprising at least 3 up to about 12 carbon atoms.
- aryl refers to a monovalent hydrocarbon radical having one or more polyunsaturated carbon rings having aromatic character, (e.g. ⁇ 4n + 2 delocalized electrons) and comprising 5 up to about 14 carbon atoms.
- heterocyclyc ⁇ e refers to a ring-containing structure or molecule having one or more multivalent heteroatoms, independently selected from N, O, P and S, as a part of the ring structure and including at least 3 and up to about 20 atoms in the ring(s).
- Heterocycle may be saturated or unsaturated, containing one or more double bonds, and heterocycle may contain more than one ring. When a heterocycle contains more than one ring, the rings may be fused or unfused. Fused rings generally refer to at least two rings share two atoms therebetween.
- Heterocycle may have aromatic character or may not have aromatic character.
- heteromatic refers to a ring-containing structure or molecule having one or more multivalent heteroatoms, independently selected from N, O, P and S, as a part of the ring structure and including at least 3 and up to about 20 atoms in the ring(s), wherein the ring-containing structure or molecule has an aromatic character (e.g., 4n + 2 delocalized electrons).
- heterocyclic group refers to a radical derived from a heterocycle by removing one or more hydrogens therefrom.
- heterocyclyl refers a monovalent radical derived from a heterocycle by removing one hydrogen therefrom.
- heterocyclylene refers to a divalent radical derived from a heterocycle by removing two hydrogens therefrom, which serves to links two structures together.
- heteroaryl refers to a heterocyclyl having aromatic character.
- heterocycloalkyl refers to a monocyclic or polycyclic ring comprising carbon and hydrogen atoms and at least one heteroatom, preferably, 1 to 3 heteroatoms selected from nitrogen, oxygen, and sulfur, and having no unsaturation.
- heterocycloalkyl groups include pyrrolidinyl, pyrrolidino, piperidinyl, piperidino, piperazinyl, piperazino, morpholinyl, morpholino, thiomorpholinyl, thiomorpholino, and pyranyl.
- a heterocycloalkyl group can be unsubstituted or substituted with one or two suitable substiruents.
- the heterocycloalkyl group is a monocyclic or bicyclic ring, more preferably, a monocyclic ring, wherein the ring comprises from 3 to 6 carbon atoms and form 1 to 3 heteroatoms, referred to herein as Cs- ⁇ heterocycloalkyl.
- ix-membered refers to a group having a ring that contains six ring atoms.
- five-membered refers to a group having a ring that contains five ring atoms.
- a five-membered ring heteroaryl is a heteroaryl with a ring having five ring atoms wherein 1, 2 or 3 ring atoms are independently selected from N, O and S.
- Exemplary five-membered ring heteroaryls are thienyl, fliryl, pyrrolyl, imidazolyl, thiazolyl, oxazolyl, pyrazolyl, isothiazolyl, isoxazolyl, 1,2,3-triazolyl, tetrazolyl, 1,2,3-thiadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-triazolyl, 1,2,4-thiadiazolyl, 1,2,4-oxadiazolyl, 1,3,4-triazolyl, 1,3,4-thiadiazolyl, and 1,3,4- oxadiazolyl.
- a six-membered ring heteroaryl is a heteroaryl with a ring having six ring atoms
- Exemplary six-membered ring heteroaryls are pyridyl, pyrazinyl, pyrimidinyl, triazinyl and pyridazinyl.
- Heterocyclyl includes, for example, monocyclic heterocyclyls, such as: aziridinyl, oxiranyl, thiiranyl, azetidinyl, oxetanyl, thietanyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, pyrazolidinyl, pyrazolinyl, dioxolanyl, sulfolanyl, 2,3-dihydrofuranyl, 2,5-dihydrofuranyl, tetrahydrofuranyl, thiophanyl, piperidinyl, 1,2,3,6-tetrahydro- pyridinyl, piperazinyl, mo ⁇ holinyl, thiomorpholinyl, pyranyl, thiopyranyl, 2,3- dihydropyranyl, tetrahydropyranyl, 1,4-dihydropyridinyl, 1,4-d
- heterocyclyl includes aromatic heterocyclyls or heteroaryl, for example, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl, furyl, furazanyl, pyrrolyl, imidazolyl, thiazolyl, oxazolyl, pyrazolyl, isothiazolyl, isoxazolyl, 1,2,3- triazolyl, tetrazolyl, 1,2,3-thiadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-triazolyl, 1,2,4- thiadiazolyl, 1,2,4-oxadiazolyl, 1,3,4-triazolyl, 1,3,4-thiadiazolyl, and 1,3,4 oxadiazolyl.
- heterocyclyl encompasses polycyclic heterocyclyls (including both aromatic or non-aromatic), for example, indolyl, indolinyl, isoindolinyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, 1,4- benzodioxanyl, coumarinyl, dihydrocoumarinyl, benzofuranyl, 2,3- dihydrobenzofuranyl, isobenzofuranyl, chromenyl, chromanyl, isochromanyl, xanthenyl, phenoxathiinyl, thianthrenyl, indolizinyl, isoindolyl, indazolyl, purinyl, phthalazinyl, naphthyridinyl, quinoxalinyl, quinazolinyl, cinnolmyl, pteri
- heterocyclyl includes polycyclic heterocyclyls wherein the ring fusion between two or more rings includes more than one bond common to both rings and more than two atoms common to both rings.
- bridged heterocycles include quinuclidinyl, diazabicyclo[2.2.1]heptyl; and 7-oxabicyclo[2.2.1]heptyl.
- alkoxy refers to radicals of the general formula -O-R, wherein R is selected from a hydrocarbon radical.
- exemplary alkoxy includes methoxy, ethoxy, propoxy, isopropoxy, butoxy, t-butoxy, isobutoxy, cyclopropylmethoxy, allyloxy, and propargyloxy.
- Halogen includes fluorine, chlorine, bromine and iodine.
- RT room temperature
- an embodiment of the invention provides a compound of Formula I, a pharmaceutically acceptable salt thereof, diastereomers, enantiomers, or mixtures thereof:
- A is -(CHk) n - optionally substituted with one or more groups selected from methyl, ethyl, phenyl, benzyl and halogen, wherein n is 2, 3 or 4;
- R 1 is selected from Chalky!, halogenated Ci ⁇ alkyl and Cs ⁇ cycloalkyl;
- R 9 and R 10 are independently selected from -H, Q-galkyl, Ce-io ⁇ ryl, C ⁇ -ioaryl- C].4alkyl, Cs- ⁇ heterocyclyl, Cs- ⁇ heterocyclyl-C M alkyl, C 2 - 6 alkenyI, C 3-6 cycloalkyl, and C 3-6 cycloalkyl-Ci. 4 alkyl; NjN-diCCi ⁇ alkyOamido-Ci-ealkyl, hydroxy-Ci- ⁇ alkyl and Ci-ealkoxy-Ci- ⁇ alkyl.
- A is -(CHa) 3 -.
- A is -(CHa) 2 -.
- A is -(CH 2 V.
- A is -CH 2 -CH 2 -CH(-Ph)-.
- A is -CH 2 -CH 2 -CH(-CH 2 Ph)-.
- R 1 is selected from methyl, ethyl, 2-fluoroethyl, isopropyl, tert-butyl and cyclopropyl.
- R 2 is selected from methyl, methylsulfonyl and ethylsulfonyl.
- R 3 is selected from ethyl, isopropyl, propyl, 2-methy- propyl, 1 -butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 2-pyrimidinyl, 1- iminoethyl, 2-pyridinyl, 3,4,5,6-teteahydropyrdin-2-yl, 3,4-dihydro-2H-pyrrol-5-yl, 2- pyridinyl-methyl, 3-pyridiny
- R 3 is selected from ethyl, isopropyl, propyl, 2- methy-propyl, 1-butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 1-iminoethyl, 3,4,5,6-tetrahydropyrdin-2-yl, 3,4-dihydro-2H-pyrrol-5-yl, tetrahydrofuran-3- yhnethyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, l-methyl-4-piperidinyl, 2- (tetrahydrohydrofuran
- R 3 is selected from cyclopentyl and 4- tetrahydro-2H-pyranyl.
- the compounds of the invention may exist in, and be isolated as, enantiomeric or diastereomeric forms, or as a racemic mixture.
- the present invention includes any possible enantiomers, diastereomers, racemates or mixtures thereof, of a compound of Formula I.
- the optically active forms of the compound of the invention may be prepared, for example, by chiral chromatographic separation of aracemate, by synthesis from optically active starting materials or by asymmetric synthesis based on the procedures described thereafter.
- certain compounds of the present invention may exist as geometrical isomers, for example E and Z isomers of alkenes.
- the present invention includes any geometrical isomer of a compound of Formula I. It will further be understood that the present invention encompasses tautomers of the compounds of the Formula I.
- salts of the compounds of the Formula I are also salts of the compounds of the Formula I.
- pharmaceutically acceptable salts of compounds of the present invention may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound, for example an alkyl amine with a suitable acid, for example, HCl or acetic acid, to afford a physiologically acceptable anion.
- a corresponding alkali metal such as sodium, potassium, or lithium
- an alkaline earth metal such as a calcium
- a compound of the present invention having a suitably acidic proton, such as a carboxylic acid or a phenol with one equivalent of an alkali metal or alkaline earth metal hydroxide or alkoxide (such as the ethoxide or methoxide), or a suitably basic organic amine (such as choline or meglumine) in an aqueous medium, followed by conventional purification techniques.
- a suitably acidic proton such as a carboxylic acid or a phenol
- an alkali metal or alkaline earth metal hydroxide or alkoxide such as the ethoxide or methoxide
- a suitably basic organic amine such as choline or meglumine
- the compound of Formula I above may be converted to a pharmaceutically acceptable salt or solvate thereof, particularly, an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or/7-toluenesulphonate.
- an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or/7-toluenesulphonate.
- the compounds of the invention exhibit selective activity as agonist of the CBi receptors and are useful in therapy, especially for relief of various pain conditions such as chronic pain, neuropathic pain, acute pain, cancer pain, pain caused by rheumatoid arthritis, migraine, visceral pain etc.
- various pain conditions such as chronic pain, neuropathic pain, acute pain, cancer pain, pain caused by rheumatoid arthritis, migraine, visceral pain etc.
- compounds of the present invention are useful in other disease states in which dysfunction of CBi receptors is present or implicated.
- the compounds of the invention may be used to treat cancer, multiple sclerosis, Parkinson's disease, Huntington's chorea, Alzheimer's disease, anxiety disorders, obesity, gastrointestinal disorders and cardiovascular disorders. Even furthermore, the compounds of the invention may be useful in enhancing smoking cessation.
- Compounds of the invention are useful as immunomodulators, especially for autoimmune diseases, such as arthritis, for skin grafts, organ transplants and similar surgical needs, for collagen diseases, various allergies
- Compounds of the invention are useful in disease states where degeneration or dysfunction of cannabinoid receptors is present or implicated in that paradigm. This may involve the use of isotopically labelled versions of the compounds of the invention in diagnostic techniques and imaging applications such as positron emission tomography (PET).
- PET positron emission tomography
- Compounds of the invention are useful for the treatment of diarrhoea, depression, anxiety and stress-related disorders such as post-traumatic stress disorders, panic disorder, generalized anxiety disorder, social phobia, and obsessive compulsive disorder, urinary incontinence, premature ejaculation, various mental illnesses, cough, lung oedema, various gastro-intestinal disorders, e.g. constipation, functional gastrointestinal disorders such as Irritable Bowel Syndrome and Functional Dyspepsia, Parkinson's disease and other motor disorders, traumatic brain injury, stroke, cardioprotection following miocardial infarction, obesity, spinal injury and drug addiction, including the treatment of alcohol, nicotine, opioid and other drug abuse and for disorders of the sympathetic nervous system for example hypertension.
- stress-related disorders such as post-traumatic stress disorders, panic disorder, generalized anxiety disorder, social phobia, and obsessive compulsive disorder, urinary incontinence, premature ejaculation, various mental illnesses, cough, lung oedema
- Compounds of the invention are useful as an analgesic agent for use during general anaesthesia and monitored anaesthesia care.
- Combinations of agents with different properties are often used to achieve a balance of effects needed to maintain the anaesthetic state (e.g. amnesia, analgesia, muscle relaxation and sedation). Included in this combination are inhaled anaesthetics, hypnotics, anxiolytics, neuromuscular blockers and opioids.
- a further aspect of the invention is a method for the treatment of a subject suffering from any of the conditions discussed above, whereby an effective amount of a compound according to the Formula I above, is administered to a patient in need of such treatment.
- the invention provides a compound of Formula I or pharmaceutically acceptable salt or solvate thereof, as hereinbefore defined for use in therapy.
- the present invention provides the use of a compound of Formula I or a pharmaceutically acceptable salt or solvate thereof, as hereinbefore defined in the manufacture of a medicament for use in therapy.
- the term “therapy” also includes “prophylaxis” unless there are specific indications to the contrary.
- the term “therapeutic” and “therapeutically” should be contrued accordingly.
- the term “therapy” within the context of the present invention further encompasses to administer an effective amount of a compound of the present invention, to mitigate either a pre-existing disease state, acute or chronic, or a recurring condition. This definition also encompasses prophylactic therapies for prevention of recurring conditions and continued therapy for chronic disorders.
- the compounds of the present invention are useful in therapy, especially for the therapy of various pain conditions including, but not limited to: acute pain, chronic pain, neuropathic pain, back pain, cancer pain, and visceral pain.
- the compound of the invention may be administered in the form of a conventional pharmaceutical composition by any route including orally, intramuscularly, subcutaneously, topically, intranasally, intraperitoneally, intrathoracially, intravenously, epidurally, intxathecally, transdermally, intracerebroventricularly and by injection into the joints.
- the route of administration may be oral, intravenous or intramuscular.
- the dosage will depend on the route ' of administration, the severity of the disease, age and weight of the patient and other factors normally considered by the attending physician, when determining the individual regimen and dosage level at the most appropriate for a particular patient.
- a solid carrier can be one or more substances, which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, or table disintegrating agents; it can also be an encapsulating material.
- the carrier is a finely divided solid, which is in a mixture with the finely divided compound of the invention, or the active component.
- the active component is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- a low-melting wax such as a mixture of fatty acid glycerides and cocoa butter is first melted and the active ingredient is dispersed therein by, for example, stirring. The molten homogeneous mixture in then poured into convenient sized moulds and allowed to cool and solidify.
- Suitable carriers are magnesium carbonate, magnesium stearate, talc, lactose, sugar, pectin, dextrin, starch, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, a low-melting wax, cocoa butter, and the like.
- composition is also intended to include the formulation of the active component with encapsulating material as a carrier providing a capsule in which the active component (with or without other carriers) is surrounded by a carrier which is thus in association with it. Similarly, cachets are included.
- Liquid form compositions include solutions, suspensions, and emulsions.
- sterile water or water propylene glycol solutions of the active compounds may be liquid preparations suitable for parenteral administration.
- Liquid compositions can also be formulated in solution in aqueous polyethylene glycol solution.
- Aqueous solutions for oral administration can be prepared by dissolving the active component in water and adding suitable colorants, flavoring agents, stabilizers, and thickening agents as desired.
- Aqueous suspensions for oral use can be made by dispersing the finely divided active component in water together with a viscous material such as natural synthetic gums, resins, methyl cellulose, sodium carboxymethyl cellulose, and other suspending agents known to the pharmaceutical formulation art.
- the pharmaceutical composition will preferably include from 0.05% to 99%w (per cent by weight), more preferably from 0.10 to 50%w, of the compound of the invention, all percentages by weight being based on total composition.
- a therapeutically effective amount for the practice of the present invention may be determined, by the use of known criteria including the age, weight and response of the individual patient, and interpreted within the context of the disease which is being treated or which is being prevented, by one of ordinary skills in the art.
- the use of any compound of Formula I as defined above for the manufacture of a medicament is the use of any compound of Formula I as defined above for the manufacture of a medicament.
- any compound of Formula I for the manufacture of a medicament for the therapy of pain. Additionally provided is the use of any compound according to Formula I for the manufacture of a medicament for the therapy of various pain conditions including, but not limited to: acute pain, chronic pain, neuropathic pain, back pain, cancer pain, and visceral pain.
- a further aspect of the invention is a method for therapy of a subject suffering from any of the conditions discussed above, whereby an effective amount of a compound according to the Formula I above, is administered to a patient in need of such therapy.
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier.
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier for therapy, more particularly for therapy of pain.
- composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier use in any of the conditions discussed above.
- IiCB 1 and hCB? receptor binding Human CBi receptor from Receptor Biology (hCBi) or human CB 2 receptor from BioSignal (I1CB 2 ) membranes are thawed at 37 0 C, passed 3 times through a 25- gauge blunt-end needle, diluted in the cannabinoid binding buffer (50 mM Tris, 2.5 mM EDTA, 5 mM MgCl 2 , and 0.5 mg/mL BSA fatty acid free, pH 7.4) and aliquots containing the appropriate amount of protein are distributed in 96-well plates.
- cannabinoid binding buffer 50 mM Tris, 2.5 mM EDTA, 5 mM MgCl 2 , and 0.5 mg/mL BSA fatty acid free, pH 7.4
- the IC5 0 of the compounds of the invention at hCBi and I1CB 2 are evaluated from 10-point dose-response curves done with 3 H-CP55,940 at 20000 to 25000 dpm per well (0.17- 0.21 nM) in a final volume of 300 ⁇ l.
- the total and non-specific binding are determined in the absence and presence of 0.2 ⁇ M of HU210 respectively.
- the plates are vortexed and incubated for 60 minutes at room temperature, filtered through Unifilters GF/B (presoaked in 0.1% polyethyleneimine) with the Tomtec or Packard harvester using 3 mL of wash buffer (50 mM Tris, 5 mM MgCl 2 , 0.5 mg BSA pH 7.0). The filters are dried for 1 hour at 55 °C.
- the radioactivity (cpm) is counted in a TopCount (Packard) after adding 65 ⁇ l/well of MS-20 scintillation liquid.
- Human CBj receptor from Receptor Biology (hCBi) or human CB 2 receptor membranes (BioSignal) are thawed at 37 0 C, passed 3 times through a 25-gauge blunt-end needle and diluted in the GTP ⁇ S binding buffer (50 mM Hepes, 20 mM NaOH, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl 2 , pH 7.4, 0.1% BSA).
- the EC 50 and E max of the compounds of the invention are evaluated from 10-point dose- response curves done in 300 ⁇ l with the appropriate amount of membrane protein and 100000-130000 dpm of GTPg 35 S per well (0.11 -0.14 nM).
- the basal and maximal stimulated binding is determined in absence and presence of 1 ⁇ M (hCB 2 ) or 10 ⁇ M (hCBi) Win 55,212-2 respectively.
- the membranes are pre-incubated for 5 minutes with 56.25 ⁇ M (hCB2) or 112.5 ⁇ M (hCBi) GDP prior to distribution in plates (15 ⁇ M (TiCB 2 ) or 30 ⁇ M (hCBi) GDP final).
- the plates are vortexed and incubated for 60 minutes at room temperature, filtered on Unifilters GFfB (presoaked in water) with the Tomtec or Packard harvester using 3 ml of wash buffer (50 mM Tris, 5 mM MgCl 2 , 50 mM NaCl, pH 7.0). The filters are dried for 1 hour at 55 0 C. The radioactivity (cpm) is counted in a TopCount (Packard) after adding 65 ⁇ l/well of MS-20 scintillation liquid.
- wash buffer 50 mM Tris, 5 mM MgCl 2 , 50 mM NaCl, pH 7.0.
- IC 50 is the concentration of the compound of the invention at which 50% displacement has been observed
- [rad] is a standard or reference radioactive ligand concentration at that moment; and Kd is the dissociation constant of the radioactive ligand towards the particular receptor.
- the compounds of the invention are found to be active towards human CBi receptors.
- Step A ⁇ ir -[4-(Cyclopropylamino)-4-oxobutyl]-iV,5-dimethyl-2-(tetrahydro-2J3 r - pyran-4-yI)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indoIe-8-carboxamide
- HATU(146 mg, 0.38 mmol) was added portionwise into a mixture of 5-methyl-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3- ⁇ ]indole-8-carboxylic acid (100 mg, 0.32 mmol) (see following steps B, C, D, E, F, G, and ⁇ for its preparation), 4-(methylamino) butyric acid hydrochloride (50 mg, 0.32 mmol) and NN-diisopropylethylamine (0.3 uL, 1.3 mmol) in dry DMF (5 mL) at 0 0 C.
- the carboxylic acid (87.7 g) was mixed with methanol in a 3L round bottom flask and cooled in an ice bath. Acetyl chloride (100 mL) was slowly added and the ice bath was removed. The mixture was heated at reflux for 3.5 hours. After cooling at room temperature, the mixture was concentrated by evaporation of methanol and the solid was collected by filtration to provide final product (87.3 g, 111 % for two steps, still containing ammonium chloride).
- Step E 2-fert-Butyl 8-methyl 5-methyl-l,3,4,5-tetrahydro-2H-pyrido[4,3- ⁇ ]indole-2,8-dicarboxyIate
- Step F Methyl 5-methyl-2,3,4,5-tetrahydro-lia r -pyrido[4,3-6]indole-8- carboxylate
- Step G Methyl 5-methyl-2-(tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH- pyrido [4,3-6] indole-8-carboxylate
- Step H 5-Methyl-2-(tetrahydro-2fr-pyran-4-yl)-2,3,4,5-tetrahydro-lJ3 r - pyrido [4,3-6]indole-8-carboxylic acid
- HATU (0.21 g, 0.56 mmol) was added to a solution of 4-[ ⁇ [2-cyclopentyl-5- (methylsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3- ⁇ ]indol-8- yl]carbonyl ⁇ (methyl)amino]butanoic acid (0.20 g, 0.43 mmol), DIPEA (0.18 roL, 1.0 mmol) and methylamine (2M in T ⁇ F, 0.25 mL, 0.52 mmol) in DMF (15 mL). The reaction mixture was stirred for 3 hrs. and was then concentrated.
- Step E 2-(4-Tetrahydropyranyl)-5-raetliylsulfone-2,3,4,5-tetrahydro-li ⁇ - pyrido[4-3b]indole-8-carboxylic acid
- the crude methyl ester was diluted in hot denatured ethanol (350 mL) and 1,2 M lithium hydroxide solution (60 mL) was added. The mixture was heated at reflux for 4 hours. After cooling to room temperature, ethanol was evaporated and water (60 mL) was added. Acetic acid was slowly added to obtain a pH of 5-6. Then the product precipitated as a thick solid that gave to the mixture a creamy texture. After filtration under vacuum, the final product was obtained as a beige solid (free amine).
- Step F Methyl 4-[ ⁇ [2-cyclopentyl-5-(methylsuIfonyl)-2,3,4,5-tetrahydro-lH- pyrido[4,3-AJindol-8-yl]carbonyl ⁇ (methyl)amino]butanoate
- the reaction mixture was slowly added to a mixture OfNaHCO 3 (0.40 g, 4.8 mmol) and MeOH (80 mL) at -78°C. The resulting mixture was allowed to warm to ambient temperature and the solvent was concentrated. CH 2 CI 2 (20 mL) was added and the precipitated salts were filtered. The filtrate was concentrated and the product was purified by preparative reverse-phase HPLC to provide the TFA salt of the title compound as a white solid (60 mg, 20 %).
- Step G 2-Cyclopentyl- ⁇ '-[4-(cyclopropylamino)-4-oxobutyl]-iV-methyl-2,3,4,5- tetrahydro ⁇ lH-pyrido[4,3-Z>]indole-8-carboxamide
- Step A iV-[4-(Cyclopropylamino)-4-oxobutyl]-5-(ethyIsulfonyl)-iV-methyl-2- (tetrahydro-2J3 r -pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indole-8- carboxamide
- Step D Methyl 5-(ethylsulfonyl)-2-(tetrahydro-2H-pyraa-4-yl)-2,3,4,5- tetrahydro-lH-pyrido [4,3-b]indoIe-8-carboxylate
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides tetrahydro-1H-pyrido [3,4- b] indole derivatives, processes for their preparation, pharmaceutical compositions containing them and their use in therapy. The compounds act as cannabinoid receptor ligands (CB1) and thus may be used in the treatment of pain, cancer, multiple sclerosis, Parkinson's disease, Huntington's chorea, Alzheimer's disease, anxiety disorders, gastrointestinal disorders and/or cardiovascular disorders.
Description
Tetrahydro- lH-pyrido [3 , 4 -b] indole derivatives as CBl receptor ligands
BACKGROUND OF THE INVENTION 1. Field of the invention
The invention is related to therapeutic compounds, pharmaceutical compositions containing these compounds, manufacturing processes thereof and uses thereof. Particularly, the present invention is related to compounds that may be effective in treating pain, cancer, multiple sclerosis, Parkinson's disease, Huntington's chorea, Alzheimer's disease, anxiety disorders, gastrointestinal disorders and/or cardiovascular disorders.
2. Discussion of Relevant Technology
Pain management has been studied for many years. It is known that cannabinoid receptor (e.g., CBi receptor, CB2 receptor) ligands including agonists, antagonists and inverse agonists produce relief of pain in a variety of animal models by interacting with CBi and/or CB2 receptors. Generally, CBi receptors are located predominately in the central nervous system, whereas CB2 receptors are located primarily in the periphery and are primarily restricted to the cells and tissues derived from the immune system.
While CBi receptor agonists, such as Δ9-tetrahydrocannabinol (Δ9-THC) and anadamide, are useful in anti-nociception models in animals, they tend to exert undesired CNS side-effects, e.g., psychoactive side effects, the abuse potential, drug dependence and tolerance, etc. These undesired side effects are known to be mediated by the CBi receptors located in CNS. There are lines of evidence, however, suggesting that CBi agonists acting at peripheral sites or with limited CNS exposure can manage pain in humans or animals with much improved overall in vivo profile. Therefore, there is a need for new CBi receptor ligands such as agonists that may be useful in managing pain or treating other related symptoms or diseases with reduced or minimal undesirable CNS side-effects.
DESCRIPTION OF THE EMBODIMENTS
The present invention provides CBi receptor ligands which may be useful in treating pain and/or other related symptoms or diseases.
The term "Cm.n" or "Cm.n group" refers to any group having m to n carbon atoms. The term "alkyl" refers to a saturated monovalent straight or branched chain hydrocarbon radical comprising 1 to about 12 carbon atoms. Illustrative examples of alky Is include, but are not limited to,
 groups, such as methyl, ethyl, propyl, isopropyl, 2-methyl-l -propyl, 2-methyl-2-propyl, 2-methyl-l -butyl, 3 -methyl- 1 -butyl, 2-methyl-3-butyl, 2,2-dimethyl-l -propyl, 2-methyl-l -pentyl, 3-methyl-l-ρenryl, 4- methyl-1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 2,2- dimethyl-1-butyl, 3,3-dirnethyl-l-butyl, 2-ethyI-l -butyl, butyl, isobutyl, t-butyl, pentyl, isopentyl, neopentyl, and hexyl, and longer alkyl groups, such as heptyl, and octyl. An alkyl can be unsubstituted or substituted with one or two suitable substituents. The term "cycloalkyl" refers to a saturated monovalent ring-containing hydrocarbon radical comprising at least 3 up to about 12 carbon atoms. Examples of cycloalkyls include, but are not limited to, C3_7cycloalkyl groups, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl, and saturated cyclic and bicyclic terpenes. A cycloalkyl can be unsubstituted or substituted by one or two suitable substituents. Preferably, the cycloalkyl is a monocyclic ring or bicyclic ring. The term "cycloalkenyl" refers to a monovalent ring-containing hydrocarbon radical having at least one carbon-carbon double bond and comprising at least 3 up to about 12 carbon atoms.
groups, such as methyl, ethyl, propyl, isopropyl, 2-methyl-l -propyl, 2-methyl-2-propyl, 2-methyl-l -butyl, 3 -methyl- 1 -butyl, 2-methyl-3-butyl, 2,2-dimethyl-l -propyl, 2-methyl-l -pentyl, 3-methyl-l-ρenryl, 4- methyl-1-pentyl, 2-methyl-2-pentyl, 3-methyl-2-pentyl, 4-methyl-2-pentyl, 2,2- dimethyl-1-butyl, 3,3-dirnethyl-l-butyl, 2-ethyI-l -butyl, butyl, isobutyl, t-butyl, pentyl, isopentyl, neopentyl, and hexyl, and longer alkyl groups, such as heptyl, and octyl. An alkyl can be unsubstituted or substituted with one or two suitable substituents. The term "cycloalkyl" refers to a saturated monovalent ring-containing hydrocarbon radical comprising at least 3 up to about 12 carbon atoms. Examples of cycloalkyls include, but are not limited to, C3_7cycloalkyl groups, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl, and saturated cyclic and bicyclic terpenes. A cycloalkyl can be unsubstituted or substituted by one or two suitable substituents. Preferably, the cycloalkyl is a monocyclic ring or bicyclic ring. The term "cycloalkenyl" refers to a monovalent ring-containing hydrocarbon radical having at least one carbon-carbon double bond and comprising at least 3 up to about 12 carbon atoms.
The term "aryl" refers to a monovalent hydrocarbon radical having one or more polyunsaturated carbon rings having aromatic character, (e.g. ^ 4n + 2 delocalized electrons) and comprising 5 up to about 14 carbon atoms.
The term "heterocycϊe" refers to a ring-containing structure or molecule having one or more multivalent heteroatoms, independently selected from N, O, P and S, as a part of the ring structure and including at least 3 and up to about 20 atoms in the ring(s). Heterocycle may be saturated or unsaturated, containing one or more double bonds, and heterocycle may contain more than one ring. When a heterocycle contains more than one ring, the rings may be fused or unfused. Fused rings generally
refer to at least two rings share two atoms therebetween. Heterocycle may have aromatic character or may not have aromatic character.
The term "heteroaromatic" refers to a ring-containing structure or molecule having one or more multivalent heteroatoms, independently selected from N, O, P and S, as a part of the ring structure and including at least 3 and up to about 20 atoms in the ring(s), wherein the ring-containing structure or molecule has an aromatic character (e.g., 4n + 2 delocalized electrons).
The term "heterocyclic group," "heterocyclic moiety," "heterocyclic," or "heterocyclo" refers to a radical derived from a heterocycle by removing one or more hydrogens therefrom.
The term "heterocyclyl" refers a monovalent radical derived from a heterocycle by removing one hydrogen therefrom.
The term "heterocyclylene" refers to a divalent radical derived from a heterocycle by removing two hydrogens therefrom, which serves to links two structures together.
The term "heteroaryl" refers to a heterocyclyl having aromatic character.
The term "heterocylcoalkyl" refers to a monocyclic or polycyclic ring comprising carbon and hydrogen atoms and at least one heteroatom, preferably, 1 to 3 heteroatoms selected from nitrogen, oxygen, and sulfur, and having no unsaturation. Examples of heterocycloalkyl groups include pyrrolidinyl, pyrrolidino, piperidinyl, piperidino, piperazinyl, piperazino, morpholinyl, morpholino, thiomorpholinyl, thiomorpholino, and pyranyl. A heterocycloalkyl group can be unsubstituted or substituted with one or two suitable substiruents. Preferably, the heterocycloalkyl group is a monocyclic or bicyclic ring, more preferably, a monocyclic ring, wherein the ring comprises from 3 to 6 carbon atoms and form 1 to 3 heteroatoms, referred to herein as Cs-βheterocycloalkyl.
The term "six-membered" refers to a group having a ring that contains six ring atoms.
The term "five-membered" refers to a group having a ring that contains five ring atoms.
A five-membered ring heteroaryl is a heteroaryl with a ring having five ring atoms wherein 1, 2 or 3 ring atoms are independently selected from N, O and S.
Exemplary five-membered ring heteroaryls are thienyl, fliryl, pyrrolyl, imidazolyl, thiazolyl, oxazolyl, pyrazolyl, isothiazolyl, isoxazolyl, 1,2,3-triazolyl, tetrazolyl, 1,2,3-thiadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-triazolyl, 1,2,4-thiadiazolyl, 1,2,4-oxadiazolyl, 1,3,4-triazolyl, 1,3,4-thiadiazolyl, and 1,3,4- oxadiazolyl. A six-membered ring heteroaryl is a heteroaryl with a ring having six ring atoms wherein 1, 2 or 3 ring atoms are independently selected from N, O and S.
Exemplary six-membered ring heteroaryls are pyridyl, pyrazinyl, pyrimidinyl, triazinyl and pyridazinyl.
Heterocyclyl includes, for example, monocyclic heterocyclyls, such as: aziridinyl, oxiranyl, thiiranyl, azetidinyl, oxetanyl, thietanyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, pyrazolidinyl, pyrazolinyl, dioxolanyl, sulfolanyl, 2,3-dihydrofuranyl, 2,5-dihydrofuranyl, tetrahydrofuranyl, thiophanyl, piperidinyl, 1,2,3,6-tetrahydro- pyridinyl, piperazinyl, moφholinyl, thiomorpholinyl, pyranyl, thiopyranyl, 2,3- dihydropyranyl, tetrahydropyranyl, 1,4-dihydropyridinyl, 1,4-dioxanyl, 1,3-dioxanyl, dioxanyl, homopiperidinyl, 2,3,4,7-tetrahydro-lH-azepinyl, homopiperazinyl, 1,3- dioxepanyl, 4,7-dihydro-l,3-dioxepinyl, and hexamethylene oxidyl.
In addition, heterocyclyl includes aromatic heterocyclyls or heteroaryl, for example, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl, furyl, furazanyl, pyrrolyl, imidazolyl, thiazolyl, oxazolyl, pyrazolyl, isothiazolyl, isoxazolyl, 1,2,3- triazolyl, tetrazolyl, 1,2,3-thiadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-triazolyl, 1,2,4- thiadiazolyl, 1,2,4-oxadiazolyl, 1,3,4-triazolyl, 1,3,4-thiadiazolyl, and 1,3,4 oxadiazolyl.
Additionally, heterocyclyl encompasses polycyclic heterocyclyls (including both aromatic or non-aromatic), for example, indolyl, indolinyl, isoindolinyl, quinolinyl, tetrahydroquinolinyl, isoquinolinyl, tetrahydroisoquinolinyl, 1,4- benzodioxanyl, coumarinyl, dihydrocoumarinyl, benzofuranyl, 2,3- dihydrobenzofuranyl, isobenzofuranyl, chromenyl, chromanyl, isochromanyl, xanthenyl, phenoxathiinyl, thianthrenyl, indolizinyl, isoindolyl, indazolyl, purinyl, phthalazinyl, naphthyridinyl, quinoxalinyl, quinazolinyl, cinnolmyl, pteridinyl, phenanthridinyl, perimidinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxazinyl, 1,2-benzisoxazolyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benzimidazolyl, benztriazolyl, thioxanthinyl, carbazolyl, carbolinyl, acridinyl, pyrolizidinyl, and quinolizidinyl.
In addition to the polycyclic heterocyclyls described above, heterocyclyl includes polycyclic heterocyclyls wherein the ring fusion between two or more rings includes more than one bond common to both rings and more than two atoms common to both rings. Examples of such bridged heterocycles include quinuclidinyl, diazabicyclo[2.2.1]heptyl; and 7-oxabicyclo[2.2.1]heptyl.
The term "alkoxy" refers to radicals of the general formula -O-R, wherein R is selected from a hydrocarbon radical. Exemplary alkoxy includes methoxy, ethoxy, propoxy, isopropoxy, butoxy, t-butoxy, isobutoxy, cyclopropylmethoxy, allyloxy, and propargyloxy.
Halogen includes fluorine, chlorine, bromine and iodine.
"RT" or "rt" means room temperature.
In one aspect, an embodiment of the invention provides a compound of Formula I, a pharmaceutically acceptable salt thereof, diastereomers, enantiomers, or mixtures thereof:

wherein
A is -(CHk)n- optionally substituted with one or more groups selected from methyl, ethyl, phenyl, benzyl and halogen, wherein n is 2, 3 or 4;
R1 is selected from Chalky!, halogenated Ci^alkyl and Cs^cycloalkyl;
R2 is selected from -H, Cwalkyl, C2-6alkenyl, -C(=O)-NR9R10, -S(O)2- NR9R10, -S(=O)2-Ci-6alkyl, -S(=0)2-C6-ioaryl, -S(=O)2-C3-5heteroaryl, -C(=0)- Ci-βalkyl;
 and
and
 wherein said Ci-βalkyl, C2.6alkenyl, -S(=O)2-C1-6alkyl, -S(=O)2-C6-10aryl, -S(=O)2-C3-5heteroaryl, -C(=0)- Ci-6alkyl;
wherein said Ci-βalkyl, C2.6alkenyl, -S(=O)2-C1-6alkyl, -S(=O)2-C6-10aryl, -S(=O)2-C3-5heteroaryl, -C(=0)- Ci-6alkyl;
 and
and
 used in defining R2 is optionally substituted with one or more group selected from -OR, R, -CO2H, -CO2-R; -SO2-R; halogen, -NO2, -OH, -NH2, -NHR, -C(=0)-NH2, and -C(=0)-NHR;
R3 is selected from C^eheterocycloalkyl, Cs-βheterocycloalkyl-Ci^alkyl, C3-6cycloalkyl, C3.6cycloalkyl-Ci-4alkyl, Q.ealkyl, C2-6alkenyl,
used in defining R2 is optionally substituted with one or more group selected from -OR, R, -CO2H, -CO2-R; -SO2-R; halogen, -NO2, -OH, -NH2, -NHR, -C(=0)-NH2, and -C(=0)-NHR;
R3 is selected from C^eheterocycloalkyl, Cs-βheterocycloalkyl-Ci^alkyl, C3-6cycloalkyl, C3.6cycloalkyl-Ci-4alkyl, Q.ealkyl, C2-6alkenyl,
 C3- 6heteroaryl-CMallcyl, -C(=O)-Ci-6alkyl, -C(=O)-C3-6cycloalkyl and -Ct=NH)-C1- 6alkyl, wherein said C^eheterocycloalkyl, C3-6heterocycloalkyI-Ci.4alkyl, C3-6cycloalkyl, C3-6cycloalkyl-Ci.4alkyl, Ci^alkyl, C2-6alkenyI, C6-ioaryl-Ci.4alkyl, C3- 6heteroaryl-Ci-4alkyl, -C(=O)-Ci-6alkyl, -C(=O)-C3-6cycloalkyl and
C3- 6heteroaryl-CMallcyl, -C(=O)-Ci-6alkyl, -C(=O)-C3-6cycloalkyl and -Ct=NH)-C1- 6alkyl, wherein said C^eheterocycloalkyl, C3-6heterocycloalkyI-Ci.4alkyl, C3-6cycloalkyl, C3-6cycloalkyl-Ci.4alkyl, Ci^alkyl, C2-6alkenyI, C6-ioaryl-Ci.4alkyl, C3- 6heteroaryl-Ci-4alkyl, -C(=O)-Ci-6alkyl, -C(=O)-C3-6cycloalkyl and
 βalkyl used in defining R3 is optionally substituted with one or more groups selected from -OR, R, NO2, -CO2H, -CO2-R; -SO2-R; halogen; -OH; -NH2; -NHR, -C(=0)- NH2, and -C(=0)-NHR; R is C1-6alkyl; and
βalkyl used in defining R3 is optionally substituted with one or more groups selected from -OR, R, NO2, -CO2H, -CO2-R; -SO2-R; halogen; -OH; -NH2; -NHR, -C(=0)- NH2, and -C(=0)-NHR; R is C1-6alkyl; and
R9 and R10 are independently selected from -H, Q-galkyl, Ce-io^ryl, Cδ-ioaryl- C].4alkyl, Cs-βheterocyclyl, Cs-βheterocyclyl-CMalkyl, C2-6alkenyI, C3-6cycloalkyl, and C3-6cycloalkyl-Ci.4alkyl; NjN-diCCi^alkyOamido-Ci-ealkyl, hydroxy-Ci-βalkyl and Ci-ealkoxy-Ci-βalkyl. In another embodiment, A is -(CHa)3-.
In a further embodiment, A is -(CHa)2-.
In an even further embodiment, A is -(CH2V.
In another embodiment, A is -CH2-CH2-CH(-Ph)-.
In another embodiment, A is -CH2-CH2-CH(-CH2Ph)-. In another embodiment, R1 is selected from methyl, ethyl, 2-fluoroethyl, isopropyl, tert-butyl and cyclopropyl.
In a further embodiment, R2 is selected from methyl, ethyl, 1 -propyl, 2-propyl, 1-butyl, 2-butyl, t-butyl, aUyl, -S(=O)2-CH3, -S(=O)2-CH2CH3, 2-methoxyethyl, tetrahydropyran-4-yl-methyl, 1-propylsulfonyl, methylsulfonyl, ethylsulfonyl, cyclopropylsulfonyl, phenyl, phenylsulfonyl, 2-(methoxycarbonyl)-phenylsulfonyl; 2- (hydroxycarbonyl)-phenylsulfonyl, l-methyl-lH-imidazol-4-yl-sulfonyI, IH- imidazol-1-yl-sulfonyl, furylsulfonyl, (5-methylisoxazol-4-yl)sulfonyl, morpholin-4- ylcarbonyl, 4-amino-phenyl, -CH2-C(=O)-N(CH3)2, -C(=O)-N(CH3)2, -S(^O)2- N(CHs)2, -S(O)2-NHCH2CH3, -C(=O)-CH2CH2CH3, -CH2-C(=O)-OCH3, -CH2- C(=O)-OCH2CH3, -CH2-CO2H, ben∑yl, 4-aminobenzyl, 4-nitrobenzyl, 4- methylsulfonyl-benzyl, 4-methylthio-benzyl, 4-acetylamino-benzyl, 4-methoxy- benzyl, 4-ethoxy-benzyl, 2,6-difluorobenzyl, (6-chloro-l,3-benzodioxol-5-yl)methyl,
(5-ethoxycarbonyl)-fur-2-yl-methyl, (2-methyl-l,3-thiazol-4-yl)-memyl, (5-methyl- isoxazol-4-yl)-methyl, pyridin-2-ylmethyl, cyclobutylmethyl, and cyclopropylmethyl.
In another embodiment, R2 is selected from methyl, methylsulfonyl and ethylsulfonyl. In another embodiment, R3 is selected from ethyl, isopropyl, propyl, 2-methy- propyl, 1 -butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 2-pyrimidinyl, 1- iminoethyl, 2-pyridinyl, 3,4,5,6-teteahydropyrdin-2-yl, 3,4-dihydro-2H-pyrrol-5-yl, 2- pyridinyl-methyl, 3-pyridinylmethyl, 4-pyridinyhnethyl, l-methyl-4-piρeridinyl, A- piperidinyl, (6-methyl-pyridin-2-yl)methyl, (2-ethyl-4-methyl- lH-imidazol-5- yl)methyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrofuran-3-ylmethyl, 1- ethyl-lH-pyrazol-4-yl, l,3-dimethyl-lH-pyrazol-5-yl, (3-methylpyridin-4-yl)methyl, l,3-oxazol-2-ylmethyl, l,3-oxazol-5-ylmethyl, 2-(tetrahydro-2H-pyran-4-yl)ethyl, tetrahydro-2H-pyran-4-ylmethyl, 2-phenylethyl, 2-methoxybenzyl, 3,3,3- trifluoropropyl, 2,2-difluoroethyl, 2-hydroxycyclopentyl, (l-ethyl-3-methyl-lH- pyrazol-5-yl)methyl, 2,l,3-benzoxadiazol-5-ylmethyl, 3-thienylmetb.yl, 2- trifluoromethyl-benzyl, 3-methylbutyl, cyclohex-3-en-l-ylmethyl, 2-fluoro-6- methoxybenzyl, 2-phenyl-propyl, 2-ethyl-butyl, cyclobutylcarbonyl, 2,2- difluoropropanoyl, cyclopentylcarbonyl, tetrahydro-2H-pyran-4-ylcarbonyl, cyclopropylcarbonyl, propylcarbonyl, N-ethylaminocarbonyl, N- isopropylaminocarbonyl, cyclopropylsulfonyl, and ethylsulfonyl.
In a further embodiment, R3 is selected from ethyl, isopropyl, propyl, 2- methy-propyl, 1-butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 1-iminoethyl, 3,4,5,6-tetrahydropyrdin-2-yl, 3,4-dihydro-2H-pyrrol-5-yl, tetrahydrofuran-3- yhnethyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, l-methyl-4-piperidinyl, 2- (tetrahydro-2H-pyran-4-yl)ethyI, tetrahydro-2H-pyran-4-ylmethyl, 3,3,3- trifluoropropyl, 2,2-difluoroethyl, 2-hydroxycyclopentyl, 3-methylbutyl, cyclohex-3- en-1-ylmethyl, and 2-ethyl-butyl.
In an even further embodiment, R3 is selected from cyclopentyl and 4- tetrahydro-2H-pyranyl.
It will be understood that when compounds of the present invention contain one or more chiral centers, the compounds of the invention may exist in, and be isolated as, enantiomeric or diastereomeric forms, or as a racemic mixture. The present invention includes any possible enantiomers, diastereomers, racemates or mixtures thereof, of a compound of Formula I. The optically active forms of the compound of the invention may be prepared, for example, by chiral chromatographic separation of aracemate, by synthesis from optically active starting materials or by asymmetric synthesis based on the procedures described thereafter.
It will also be appreciated that certain compounds of the present invention may exist as geometrical isomers, for example E and Z isomers of alkenes. The present invention includes any geometrical isomer of a compound of Formula I. It will further be understood that the present invention encompasses tautomers of the compounds of the Formula I.
It will also be understood that certain compounds of the present invention may exist in solvated, for example hydrated, as well as unsolvated forms. It will further be understood that the present invention encompasses all such solvated forms of the compounds of the Formula I.
Within the scope of the invention are also salts of the compounds of the Formula I. Generally, pharmaceutically acceptable salts of compounds of the present invention may be obtained using standard procedures well known in the art, for example by reacting a sufficiently basic compound, for example an alkyl amine with a suitable acid, for example, HCl or acetic acid, to afford a physiologically acceptable anion. It may also be possible to make a corresponding alkali metal (such as sodium, potassium, or lithium) or an alkaline earth metal (such as a calcium) salt by treating a compound of the present invention having a suitably acidic proton, such as a carboxylic acid or a phenol with one equivalent of an alkali metal or alkaline earth metal hydroxide or alkoxide (such as the ethoxide or methoxide), or a suitably basic organic amine (such as choline or meglumine) in an aqueous medium, followed by conventional purification techniques. In one embodiment, the compound of Formula I above may be converted to a pharmaceutically acceptable salt or solvate thereof, particularly, an acid addition salt such as a hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartrate, citrate, methanesulphonate or/7-toluenesulphonate.
We have now found that the compounds of the invention have activity as pharmaceuticals, in particular as modulators or ligands such as agonists, partial agonists, inverse agonist or antagonists of CBi receptors. More particularly, the compounds of the invention exhibit selective activity as agonist of the CBi receptors and are useful in therapy, especially for relief of various pain conditions such as chronic pain, neuropathic pain, acute pain, cancer pain, pain caused by rheumatoid arthritis, migraine, visceral pain etc. This list should however not be interpreted as exhaustive. Additionally, compounds of the present invention are useful in other disease states in which dysfunction of CBi receptors is present or implicated. Furthermore, the compounds of the invention may be used to treat cancer, multiple sclerosis, Parkinson's disease, Huntington's chorea, Alzheimer's disease, anxiety disorders, obesity, gastrointestinal disorders and cardiovascular disorders. Even furthermore, the compounds of the invention may be useful in enhancing smoking cessation. Compounds of the invention are useful as immunomodulators, especially for autoimmune diseases, such as arthritis, for skin grafts, organ transplants and similar surgical needs, for collagen diseases, various allergies, for use as anti-tumour agents and anti viral agents.
Compounds of the invention are useful in disease states where degeneration or dysfunction of cannabinoid receptors is present or implicated in that paradigm. This may involve the use of isotopically labelled versions of the compounds of the invention in diagnostic techniques and imaging applications such as positron emission tomography (PET).
Compounds of the invention are useful for the treatment of diarrhoea, depression, anxiety and stress-related disorders such as post-traumatic stress disorders, panic disorder, generalized anxiety disorder, social phobia, and obsessive compulsive disorder, urinary incontinence, premature ejaculation, various mental illnesses, cough, lung oedema, various gastro-intestinal disorders, e.g. constipation, functional gastrointestinal disorders such as Irritable Bowel Syndrome and Functional Dyspepsia, Parkinson's disease and other motor disorders, traumatic brain injury, stroke, cardioprotection following miocardial infarction, obesity, spinal injury and drug addiction, including the treatment of alcohol, nicotine, opioid and other drug abuse and for disorders of the sympathetic nervous system for example hypertension.
Compounds of the invention are useful as an analgesic agent for use during general anaesthesia and monitored anaesthesia care. Combinations of agents with different properties are often used to achieve a balance of effects needed to maintain the anaesthetic state (e.g. amnesia, analgesia, muscle relaxation and sedation). Included in this combination are inhaled anaesthetics, hypnotics, anxiolytics, neuromuscular blockers and opioids.
Also within the scope of the invention is the use of any of the compounds according to the Formula I above, for the manufacture of a medicament for the treatment of any of the conditions discussed above. A further aspect of the invention is a method for the treatment of a subject suffering from any of the conditions discussed above, whereby an effective amount of a compound according to the Formula I above, is administered to a patient in need of such treatment.
Thus, the invention provides a compound of Formula I or pharmaceutically acceptable salt or solvate thereof, as hereinbefore defined for use in therapy.
In a further aspect, the present invention provides the use of a compound of Formula I or a pharmaceutically acceptable salt or solvate thereof, as hereinbefore defined in the manufacture of a medicament for use in therapy.
In the context of the present specification, the term "therapy" also includes "prophylaxis" unless there are specific indications to the contrary. The term "therapeutic" and "therapeutically" should be contrued accordingly. The term "therapy" within the context of the present invention further encompasses to administer an effective amount of a compound of the present invention, to mitigate either a pre-existing disease state, acute or chronic, or a recurring condition. This definition also encompasses prophylactic therapies for prevention of recurring conditions and continued therapy for chronic disorders.
The compounds of the present invention are useful in therapy, especially for the therapy of various pain conditions including, but not limited to: acute pain, chronic pain, neuropathic pain, back pain, cancer pain, and visceral pain. In use for therapy in a warm-blooded animal such as a human, the compound of the invention may be administered in the form of a conventional pharmaceutical composition by any route including orally, intramuscularly, subcutaneously, topically,
intranasally, intraperitoneally, intrathoracially, intravenously, epidurally, intxathecally, transdermally, intracerebroventricularly and by injection into the joints. In one embodiment of the invention, the route of administration may be oral, intravenous or intramuscular. The dosage will depend on the route' of administration, the severity of the disease, age and weight of the patient and other factors normally considered by the attending physician, when determining the individual regimen and dosage level at the most appropriate for a particular patient.
For preparing pharmaceutical compositions from the compounds of this invention, inert, pharmaceutically acceptable carriers can be either solid and liquid. Solid form preparations include powders, tablets, dispersible granules, capsules, cachets, and suppositories.
A solid carrier can be one or more substances, which may also act as diluents, flavoring agents, solubilizers, lubricants, suspending agents, binders, or table disintegrating agents; it can also be an encapsulating material. hi powders, the carrier is a finely divided solid, which is in a mixture with the finely divided compound of the invention, or the active component. In tablets, the active component is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired. For preparing suppository compositions, a low-melting wax such as a mixture of fatty acid glycerides and cocoa butter is first melted and the active ingredient is dispersed therein by, for example, stirring. The molten homogeneous mixture in then poured into convenient sized moulds and allowed to cool and solidify.
Suitable carriers are magnesium carbonate, magnesium stearate, talc, lactose, sugar, pectin, dextrin, starch, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, a low-melting wax, cocoa butter, and the like.
The term composition is also intended to include the formulation of the active component with encapsulating material as a carrier providing a capsule in which the active component (with or without other carriers) is surrounded by a carrier which is thus in association with it. Similarly, cachets are included.
Tablets, powders, cachets, and capsules can be used as solid dosage forms suitable for oral administration.
Liquid form compositions include solutions, suspensions, and emulsions. For example, sterile water or water propylene glycol solutions of the active compounds may be liquid preparations suitable for parenteral administration. Liquid compositions can also be formulated in solution in aqueous polyethylene glycol solution.
Aqueous solutions for oral administration can be prepared by dissolving the active component in water and adding suitable colorants, flavoring agents, stabilizers, and thickening agents as desired. Aqueous suspensions for oral use can be made by dispersing the finely divided active component in water together with a viscous material such as natural synthetic gums, resins, methyl cellulose, sodium carboxymethyl cellulose, and other suspending agents known to the pharmaceutical formulation art.
Depending on the mode of administration, the pharmaceutical composition will preferably include from 0.05% to 99%w (per cent by weight), more preferably from 0.10 to 50%w, of the compound of the invention, all percentages by weight being based on total composition.
A therapeutically effective amount for the practice of the present invention may be determined, by the use of known criteria including the age, weight and response of the individual patient, and interpreted within the context of the disease which is being treated or which is being prevented, by one of ordinary skills in the art. Within the scope of the invention is the use of any compound of Formula I as defined above for the manufacture of a medicament.
Also within the scope of the invention is the use of any compound of Formula I for the manufacture of a medicament for the therapy of pain. Additionally provided is the use of any compound according to Formula I for the manufacture of a medicament for the therapy of various pain conditions including, but not limited to: acute pain, chronic pain, neuropathic pain, back pain, cancer pain, and visceral pain.
A further aspect of the invention is a method for therapy of a subject suffering from any of the conditions discussed above, whereby an effective amount of a compound according to the Formula I above, is administered to a patient in need of such therapy.
Additionally, there is provided a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier.
Particularly, there is provided a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier for therapy, more particularly for therapy of pain.
Further, there is provided a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier use in any of the conditions discussed above.
Biological Evaluation
IiCB1 and hCB?. receptor binding Human CBi receptor from Receptor Biology (hCBi) or human CB2 receptor from BioSignal (I1CB2) membranes are thawed at 37 0C, passed 3 times through a 25- gauge blunt-end needle, diluted in the cannabinoid binding buffer (50 mM Tris, 2.5 mM EDTA, 5 mM MgCl2, and 0.5 mg/mL BSA fatty acid free, pH 7.4) and aliquots containing the appropriate amount of protein are distributed in 96-well plates. The IC50 of the compounds of the invention at hCBi and I1CB2 are evaluated from 10-point dose-response curves done with 3H-CP55,940 at 20000 to 25000 dpm per well (0.17- 0.21 nM) in a final volume of 300 μl. The total and non-specific binding are determined in the absence and presence of 0.2 μM of HU210 respectively. The plates are vortexed and incubated for 60 minutes at room temperature, filtered through Unifilters GF/B (presoaked in 0.1% polyethyleneimine) with the Tomtec or Packard harvester using 3 mL of wash buffer (50 mM Tris, 5 mM MgCl2, 0.5 mg BSA pH 7.0). The filters are dried for 1 hour at 55 °C. The radioactivity (cpm) is counted in a TopCount (Packard) after adding 65 μl/well of MS-20 scintillation liquid.
!1CB1 and hCB? GTPγS binding
Human CBj receptor from Receptor Biology (hCBi) or human CB2 receptor membranes (BioSignal) are thawed at 37 0C, passed 3 times through a 25-gauge blunt-end needle and diluted in the GTPγS binding buffer (50 mM Hepes, 20 mM
NaOH, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, pH 7.4, 0.1% BSA). The EC50 and Emax of the compounds of the invention are evaluated from 10-point dose- response curves done in 300μl with the appropriate amount of membrane protein and 100000-130000 dpm of GTPg35S per well (0.11 -0.14 nM). The basal and maximal stimulated binding is determined in absence and presence of 1 μM (hCB2) or 10 μM (hCBi) Win 55,212-2 respectively. The membranes are pre-incubated for 5 minutes with 56.25 μM (hCB2) or 112.5 μM (hCBi) GDP prior to distribution in plates (15 μM (TiCB2) or 30 μM (hCBi) GDP final). The plates are vortexed and incubated for 60 minutes at room temperature, filtered on Unifilters GFfB (presoaked in water) with the Tomtec or Packard harvester using 3 ml of wash buffer (50 mM Tris, 5 mM MgCl2, 50 mM NaCl, pH 7.0). The filters are dried for 1 hour at 55 0C. The radioactivity (cpm) is counted in a TopCount (Packard) after adding 65 μl/well of MS-20 scintillation liquid. Antagonist reversal studies are done in the same way except that (a) an agonist dose-response curve is done in the presence of a constant concentration of antagonist, or (b) an antagonist dose-response curve is done in the presence of a constant concentration of agonist.
Based on the above assays, the dissociation constant (KLi) for a particular compound of the invention towards a particular receptor is determined using the following equation: Ki = IC50/(l+[rad]/Kd),
Wherein IC50 is the concentration of the compound of the invention at which 50% displacement has been observed;
[rad] is a standard or reference radioactive ligand concentration at that moment; and Kd is the dissociation constant of the radioactive ligand towards the particular receptor.
Using the above-mentioned assays, the compounds of the invention are found to be active towards human CBi receptors.
EXAMPLES
The invention will further be described in more detail by the following Examples which describe methods whereby compounds of the present invention may
be prepared, purified, analyzed and biologically tested, and which are not to be construed as limiting the invention.
Example 1 N- [4-(Cyclopropylamino)-4-oxobutyl] -iV^-dimethyl^-^etrahydro-lH-pyran-^ yl)-2β,4,5-tetrahydro-lH-pyrido[4,3-£]indole-8-carboxamide

Step A: Λir-[4-(Cyclopropylamino)-4-oxobutyl]-iV,5-dimethyl-2-(tetrahydro-2J3r- pyran-4-yI)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indoIe-8-carboxamide

HATU(146 mg, 0.38 mmol) was added portionwise into a mixture of 5-methyl-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indole-8-carboxylic acid (100 mg, 0.32 mmol) (see following steps B, C, D, E, F, G, and Η for its preparation), 4-(methylamino) butyric acid hydrochloride (50 mg, 0.32 mmol) and NN-diisopropylethylamine (0.3 uL, 1.3 mmol) in dry DMF (5 mL) at 00C. The reaction was stirred at room temperature for 2 hours, followed by addition of cyclopropylamine (37 mg, 0.64 mmol) and ΗATU (146 mg, 0.38 mmol). After stirred overnight, the reaction mixture was concentrated, extracted with EtOAc, washed with water then brine. The crude product was purified by LCMS using high pΗ column 40-65% acetonitrile gradient to give the title compound as a white solid (TFA salt, 20 mg, 37%). 1H ΝMR (400 MHz, METHAΝOL-D4) δ 0.09 - 0.19 (m, 2 H) 0.35 - 0.41 (m, 2 H) 1.73 - 1.93 (m, 6 H) 2.05 - 2.13 (m, 1 H) 2.89 - 3.02 (m, 3 H) 3.12 - 3.19 (m, 3 H) 3.35 - 3.53 (m, 6 H) 3.64 (s, 6 H) 4.03 (dd, J=I 1.72, 4.30 Hz, 4 H) 7.37 (s, 2 H) 7.40 (s, 1 H); MS (APPI) (M+H)+=453.3
Step B.2,3,4,5-Tetrahydro-lH-pyrido[4-3b]indole-8-carboxylic acid

In a 3 L round bottom flask with a mechanical stirrer, 3-hydrazinoic acid hydrochloride (55.3 g, 0.29 mol) and 4-piperidone hydrochloride monohydrate (45 g, 0.29 mol) were heated at reflux with dioxane (IL) and hydrochloric acid 12N (100 mL) for 17 hours. After cooling at room temperature, the dioxane was removed by evaporation and ethanol (100 mL) was added. The suspension was cooled in an ice bath and the solid was collected by filtration and washed with ethanol to give a crude product (87.7 g, 118%), which contained ammonium chloride as the impurity, and was used in the next step without further purification. 1H-NMR (300 MHz, DMSO-fife) δ 12.44 (s, IH), 11.63 (s, IH), 8.13 (s, IH), 7.70 (dd, IH, J= 1.6, 8.5 Hz), 7.38 (d, IH, J= 9.2 Hz), 4.32 (s, 2H), 3.45-3.38 (m, 2H), 3.03 (t, 2H, J= 5.7 Hz).
Step C. 2,3,4,5-Tetrahydro-lH-pyrido[4-3b]indoIe-8-methyl carboxylate

The carboxylic acid (87.7 g) was mixed with methanol in a 3L round bottom flask and cooled in an ice bath. Acetyl chloride (100 mL) was slowly added and the ice bath was removed. The mixture was heated at reflux for 3.5 hours. After cooling at room temperature, the mixture was concentrated by evaporation of methanol and the solid was collected by filtration to provide final product (87.3 g, 111 % for two steps, still containing ammonium chloride). 1H-NMR (300 MHz, DMSO-ύfe) δ 11.69 (s, IH), 9.66 (s, IH), 8.16 (s, IH), 7.71 (dd, IH, J= 1.6, 8.6 Hz), 7.41 (dd, IH, J= 0.5, 8.5 Hz), 4.33 (s, 2H), 3.82 (s, 3H), 3.43 (s, 2H), 3.03 (t, 2H, J= 5.9 Hz).
Step D.2-Boc-2,3,4,5-tetrahydro-lH-pyrido[4-3b]indole-8-methyl carboxylate

The crude amine (87.3 g) was stirred in methanol (1,5 L) in an ice bath. Then 5N sodium hydroxide solution (60 mL) was slowly added, followed by addition of Boc anhydride (80 g 0.37 mol). The mixture was stirred at room temperature for 2.5 hours. The starting material then disappeared by TLC (AcOEt). The mixture was concentrated by evaporation of methanol and the solid was collected by filtration to give a pink solid (97.3 g, quantitative yield for 3 steps). 1H-NMR (300 MHz, DMSO- d6) δ 11.39 (s, IH), 8.06 (s, IH), 7.67 (dd, IH, J= 1.7, 8.5 Hz), 7.36 (d, IH3 J= 8.5 Hz), 4.55 (s, 2H), 3.81 (s, 3H), 3.69 (t, 2H, J= 5.7 Hz)3 2.77 (t, 2H3 J= 5.5Hz)3 1.42 (S3 9H).
Step E: 2-fert-Butyl 8-methyl 5-methyl-l,3,4,5-tetrahydro-2H-pyrido[4,3- έ]indole-2,8-dicarboxyIate

Step F: Methyl 5-methyl-2,3,4,5-tetrahydro-liar-pyrido[4,3-6]indole-8- carboxylate

2-tert-Butyl 8-methyl 5-methyl-l,3,4,5-tetrahydro-2H-pyrido[4,3-i]indole-2,8- dicarboxylate (3.4 g, 10 mmol) was dissolved in (15 mL) CHzCl2, TFA (15 rciL) was added and the reaction mixture was stirred at room temperature for 2 hours. The mixture was concentrated and lyophilized to yield 3.4 g of crude product as TFA salt, which was carried over to the next step. 1H NMR (400 MHz, METHANOL-D4) δ 3.13 - 3.21 (m, 2 H) 3.66 (t, J=6.25 Hz, 2 H) 3.74 (s, 4 H) 3.90 (s, 4 H) 4.48 (dd, 1 H) 7.47 (dd, J=8.79, 0.59 Hz, 1 H) 7.89 (dd, J=8.69, 1.66 Hz, 1 H) 8.21 (d, J=1.56 Hz, 1 H).
Step G: Methyl 5-methyl-2-(tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH- pyrido [4,3-6] indole-8-carboxylate

Methyl 5-methyl-2,3,4,5'tetrahydro-lH-pyrido[4,3-έ]indole-8-carboxylate (1.5 g, 4.2 mmol) was dissolved in a mixture of CEbCtaMeOH (1:1, 40 mL), triethylamine (1.2 mL, 8.4 mmol) was added, followed by the addition of tetrahydro-4H-pyran-4-one (0.8 mL, 8.4 mmol) and NaBHsCN (528 mg, 8.4 mmol). The reaction mixture was stirred at 50 0C overnight, concentrated and extracted with CH2CI2, washed with NaHCO3 then brine. The crude product (1.7g) was carried over to the next step. MS (APPI) (M+H)+= 329.10.
Step H: 5-Methyl-2-(tetrahydro-2fr-pyran-4-yl)-2,3,4,5-tetrahydro-lJ3r- pyrido [4,3-6]indole-8-carboxylic acid

Methyl 5-methyl-2-(tetxahydro-2H-pyran-4-yl)-2,3,4,5-tetraliydro-lH-pyrido[4,3- ό]indole-8-carboxylate(1.7 g, 5 mmol) was dissolved in a mixture of THF/MeOH/H2O (1:1:1), KOH (1.4 g, 25 mmol) was added and the reaction was stirred at room temperature for 3 hours. The reaction mixture was neutralized with 5N HCl to pH ~ 5.0, concentrated and used as crude product (1.5 g) for the next step. MS (APPI) (M+H)+= 315.11.
Example 2
2-Cyclopentyl-iV-methyl-iV-[4-(methylamino)-4-oxobutyl]-5-(methylsulfonyl)-
2,3,4,5-tetrahydro-lH-pyrido[4,3-&]indole-8-carboxamide

Step A. 2-Cyclopentyl-iV-inethyl-Λ/-[4-(methylamino)-4-oxobutyl]-5- (methylsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indoIe-8-carboxamide

HATU (0.21 g, 0.56 mmol) was added to a solution of 4-[{[2-cyclopentyl-5- (methylsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indol-8- yl]carbonyl}(methyl)amino]butanoic acid (0.20 g, 0.43 mmol), DIPEA (0.18 roL, 1.0 mmol) and methylamine (2M in TΗF, 0.25 mL, 0.52 mmol) in DMF (15 mL). The reaction mixture was stirred for 3 hrs. and was then concentrated. The product was
purified by preparative reverse-phase HPLC to provide the TFA salt of the title compound as white solid (93 mg, 36 %). 1H NMR (400 MHz, CD3OD) δ 1.65 - 2.11 (m, 12 H), 2.21 - 2.41 (m, 4 H), 2.44 - 2.56 (m, 1 H), 2.63 - 2.79 (m, 2 H), 2.94 - 3.04 (m, 2 H), 3.09 (s, 1 H), 3.34 - 3.66 (m, 4 H), 3.75 - 3.89 (m, 1 H), 3.90 - 4.06 (m, 1 H), 4.33 - 4.51 (m, 1 H), 4.71 - 4.86 (m, J=13.48, 13.48 Hz, 1 H), 7.43 (dd, J=17.19, 8.79 Hz, 1 H), 7.65 (d, /=18.36 Hz, 1 H), 8.04 (d, J=8.59 Hz, 1 H); MS (ESI) (MfH)+ 475.3.
Step B. 2-Boc-5-methylsulfone-2,3,4,5-tetrahydro-lH-pyrido[4-3b]indole-8- methyl carboxylate

The amine (20 g, 60.5 mmol) was stirred in anhydrous THF (220 mL) with mechanical stirring under a dry nitrogen atmosphere. Sodium hydride (60 %, 6.1 g, 151 mmol, 2.5 eq.) was added to the solution at 00C. The mixture became thick after warmed to room temperature. Methanesulfonyl chloride (7 mL, 1.5 eq.) was slowly added at O0C and the mixture was stirred for 1 hour at room temperature. After completion of the reaction by TLC (AcOEt/MeOH 4/6), water (50 mL) was added at 00C and THF was evaporated. The solid was collected and washed with water to give a yellow powder (27.8 g, with water as the impurity). 1H-NMR (300 MHz, DMSO-Cf6) ppm : 8.15 (s, IH), 7.94 (td, IH, J= 5.2, 8.8 Hz), 4.57 (s, 2H), 3.87 (s, 3H), 3.70 (t, 2H, J= 5.6Hz), 3.44 (s, 3H), 3.00 (t, 2H, J= 5.4Hz), 1.43 (s, 9H).
Step C. 5-Methylsulfone-2,3,4,5-tetrahydro-LI?-pyrido[4-3b]indoIe-8-methyl carboxylate

The protected amine (24.7 g, 60.5 mmol) was heated at reflux for 1 hour in methanol (300 mL) and 5N hydrochloric acid in isopropanol (15 mL). After completion of the
reaction (TLC MeOH/AcOEt 4/6), the mixture was concentrated by evaporation of methanol and the solid was collected by filtration to give the desired product as its hydrochloride salt (17.7 g, 85 % for 2 steps). The HCl was neutralized by stirring the salt in MeOH and triethylamine and evaporating the solution to dryness. The crude product was used for the next step without any further purification.
Step D. 2-(4-TetrahydropyranyI)-5-methylsulfone-2,3,4,5-tetrahydro-lH- pyrido[4-3b]indoIe-8-methyl carboxylate

Sodium triacetoxyborohydride (13.5 g, 63.5 mmol, 1.1 eq.) was added into a mixture of cyclopentanone (5.6 mL, 64.5 mmol, 1.1 eq.) and crude methyϊsulfone-2,3,4,5- tetrahydro-lH-pyrido[4-3b]indole-8-methyl carboxylate (17.7 g) in TΗF (400 mL), followed by addition of acetic acid (6.6 mL, 115.4 mmol, 2 eq.). The mixture was stirred for 16 hours at room temperature. More cyclopentanone (0.5 mL, 0.1 eq.) and sodium triacetoxyborohydride (1.2 g, 0.1 eq.) were then added and the mixture was stirred for an additional 5 hours. After completion of the reaction, saturated sodium carbonate solution (300 mL) was added and TΗF was evaporated by vacuum. Then the product precipitated as a thick gum. The aqueous phase was decanted and the crude product was used for the next step without any further purification.
Step E.2-(4-Tetrahydropyranyl)-5-raetliylsulfone-2,3,4,5-tetrahydro-liϊ- pyrido[4-3b]indole-8-carboxylic acid

Step F. Methyl 4-[{[2-cyclopentyl-5-(methylsuIfonyl)-2,3,4,5-tetrahydro-lH- pyrido[4,3-AJindol-8-yl]carbonyl}(methyl)amino]butanoate

2-Cyclopentyl-5-(methylsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indole-8- carboxylic acid (3.0 g, 7.5 mmol) and SOCl2 (20 mL) were mixed together and heated to 850C for 3 hrs. The reaction mixture was evaporated to dryness and the solid residue was added to a solution of the HCl salt of methyl 4-(methylamino)butanoate (3.8 g, 22 mmol) and DIPEA (13 mL, 75 mmol) in DCM (280 mL) at 00C. The reaction mixture was stirred for 2 hrs. and the solvent was concentrated. The product was purified by normal-phase MPLC using 2% Et3N, 5% MeOH and 10% acetone in DCM to provide the title compound as a yellow solid (2.59 g, 72 %). 1H NMR (400 MHz, CD3OD) δ 1.46 - 1.71 (m, 4 H), 1.72 - 1.92 (m, 3 H), 1.93 - 2.14 (m, 3 H), 2.14 - 2.23 (m, ./=6.64, 6.64 Hz, 1 H), 2.41 - 2.52 (m, 1 H), 2.84 - 2.94 (m, /=8.11, 8.11 Hz, 1 H), 2.94 - 3.03 (m, 4 H), 3.03 - 3.14 (m, 4 H), 3.16 (s, 3 H), 3.34 - 3.41 (m, 1
H), 3.56 - 3.63 (m, J=6.45, 6.45 Hz, 1 H), 3.68 (s, 2 H), 3.73 - 3.83 (m, 2 H), 7.24 - 7.43 (m, J=9.37, 9.37 Hz, 1 H), 7.49 - 7.62 (m, J=8.20 Hz, 1 H), 8.02 (d, J=8.59 Hz, 1 H); MS (ESI) (M+H)+ 476.2.
Step G. 4-[{[2-CyclopentyI-5-(methylsulfonyI)-2,3,4,5-tetrahydro-lJ?-pyrido[4,3- δ]indoI-8-yI]carbonyI}(methyl)amino]butanoic acid

MeOH (4 mL) was added to a mixture of methyl 4-[{[2-cyclopentyl-5- (methylsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indol-8- yl]carbonyl}(methyl)amino]butanoate (2.4 g, 5.1 mmol), LiOH 2M (4 mL) and water (40 mL). The reaction mixture was stirred for 5 hrs. and concentrated to 40 mL. The solution was neutralized at 00C to pΗ 6 using HCl (2M). The solvent was evaporated to dryness by azeotropic co-evaporation with EtOH to provide the title compound as a white solid, which was used in the next step without further purification. MS (ESI) (M+Η)+ 462.2.
Example 3
2-Cyclopentyl-iV-[4-(ethylamiiio)-4-oxobutyl]-iV:-methyl-5-(methyIsulfonyI)-
2,3,4,5-tetrahydro-li7-pyrido[4,3~A]iiidoIe-8-carboxamide

Following the procedure for Step A in Example 2, using ethylamine in THF (2M, 0.25 mL, 0.52 mmol) provided the TFA salt of the title compound as a white solid (77 mg, 29 %). 1H NMR (400 MHz, CD3OD) 50.94 (t, 7=7.13 Hz, 1 H), 1.12 (t, J=7.23 Hz, 1
H), 1.65 - 2.11 (m, 12 H), 2.19 - 2.44 (m, 4 H), 3.00 (s, 3 H), 3.11 (s, 2 H), 3.21 (q, J=7.23 Hz, 1 H), 3.35 - 3.67 (m, 4 H), 3.84 (t, J=7.91 Hz, 1 H), 3.92 - 4.07 (m, ./=5.27 Hz, 1 H), 4.33 - 4.50 (m, J=I 8.55 Hz, 1 H), 4.70 - 4.87 (m, 1 H), 7.44 (dd, J=17.28, 9.08 Hz, 1 H), 7.66 (d, J=I 8.55 Hz, 1 H), 8.06 (d, J=8.59 Hz, 1 H); MS (ESI) (M+H)+ 489.3.
Example 4
2-Cyclopentyl-iV-[4-(isopropyIamino)-4-oxobutyl]-Λ'-methyl-5-(raethylsulfonyI)-
2,3,4,5-tetrahydro~lff-pyrido[4,3-£]indoIe-8-carboxamide

Following the procedure for Step A in Example 2, using isopropylamine (0.03 g, 0.52 mmol) provided the TFA salt of the title compound as a white solid (73 mg, 27 %). 1H NMR (400 MHz, CD3OD) δ 0.95 (d, J=6.44 Hz, 3 H), 1.13 (d, J=6.45 Hz, 3 H), 1.65 - 2.10 (m, 13 H), 2.15 - 2.44 (m, 4 H), 2.99 (s, 2 H), 3.10 (s, 1 H), 3.34 - 3.64 (m, 4 H), 3.83 (t, J=7.81 Hz, 1 H), 3.90 - 4.07 (m, 1 H), 4.32 - 4.51 (m, J=9.96, 9.96 Hz, 1 H), 4.71 - 4.85 (m, J=13.28, 13.28 Hz, 1 H), 7.43 (dd, J=19.82, 8.50 Hz, 1 H), 7.66 (d, J=20.12 Hz, 1 H), 8.04 (d, J=8.59 Hz, 1 H); MS (ESI) (MH-H)+ 503.3.
Example 5
2-Cyclopentyl-iV-[4-(cyclopropylamino)-4-oxobutyl]-iV-methyl-5-
(methyIsulfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-Z>lindole-8-carboxamide

Following the procedure for Step A in Example 2, using cyclopropylamine (0.03 g,
0.52 mmol) provided the TFA salt of the title compound as a white solid (58 mg, 21 %). 1H NMR (400 MHz, CD3OD) δ 0.16 - 0.33 (m, 1 H), 0.42 - 0.54 (m, 1 H), 0.58
(d, /=6.25 Hz, 1 H), 0.71 (d, J=6.84 Hz, 1 H),- 1.66 - 2.10 (m, 12 H), 2.19 - 2.39 (m, 4 H), 3.00 (s, 2 H), 3.10 (s, 1 H), 3.30 - 3.34 (m, /=1.17 Hz, 4 H), 3.58 (t, ./=6.93 Hz, 1 H), 3.84 (t, /=7.91 Hz, 1 H), 3.92 - 4.07 (m, 1 H), 4.33 - 4.53 (m, 1 H), 4.71 - 4.86 (m, 1 H), 7.44 (dd, /=17.67, 8.89 Hz, 1 H), 7.66 (d, /=18.36 Hz, 1 H), 8.06 (dd, /=8.69, 0.68 Hz, 1 H); MS (ESI) (M+H)+ 501.3.
Example 6 iV-[4-(tert-Butylamino)-4-oxobutyl]-2-cyclopentyl-iV-methyl-5-(methylsuIfonyl)-
2,3,4,5-tetrahydro-lH-pyτido[4,3-#]indole-8-carboxamide

Following the procedure for Step A in Example 2, using fer^-butylamine (0.04 g, 0.52 mmol) provided the TFA salt of the title compound as a white solid (99 mg, 36 %). 1H NMR (400 MHz, CD3OD) δ 1.14 (s, 4 H), 1.33 (s, 5 H), 1.67 - 2.05 (m, 12 H), 2.21 (t, /=7.81 Hz, 1 H), 2.26 - 2.42 (m, 3 H), 2.94 - 3.04 (m, 2 H), 3.05 - 3.15 (m, 1 H), 3.33 - 3.66 (m, 4 H), 3.75 - 3.91 (m, 1 H)5 3.91 - 4.08 (m, 1 H), 4.33 - 4.53 (m, 1 H), 4.67 - 4.84 (m, 1 H), 7.44 (dd, /=19.53, 8.98 Hz, 1 H), 7.66 (d, /=19.33 Hz, 1 H), 8.05 (d, /=8.79 Hz, 1 H); MS (ESI) (M+H)+ 517.3.
Example 7
2-CyclopentyI-iV-{4-[(2-fluoroethyl)amino]-4-oxobutyl}-iV-methyl-5-
(methyIsuIfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indole-8-carboxamide

Following the procedure for Step A in Example 2, using 2-fluoroethylamine (0.05 g,
0.52 mmol) provided the TFA salt of the title compound as a white solid (80 mg, 29 %). 1H NMR (400 MHz, CD3OD) δ 1.63 - 2.19 (m, 12 H), 2.22 - 2.47 (m, 4 H), 3.01
(s, 2 H), 3.11 (s, 1 H), 3.35 - 3.69 (m, 8 H), 3.84 (t, J=7.62 Hz, 1 H), 4.14 - 4.36 (m, 1 H), 4.36 - 4.57 (m, 1 H), 4.72 - 4.87 (m, 1 H), 7.44 (dd, J=16.99, 9.18 Hz, 1 H), 7.67 (d, J=16.80 Hz, 1 H), 8.06 (d, J=8.40 Hz, 1 H); MS (ESI) (M+H)+ 507.3.
Example 8
2-Cyclopentyl-iV-[4-(cyclopropylamino)-4-oxobutyl]-5-(ethylsulfonyl)-Λr-methyl- 2,3,4,5-tetrahydro-lH-pyrido[4,3-£]indole-8-carboxamide

Step A.2-Cyclopentyl-Λr-[4-(cyclopropylamino)-4-oxobutyl]-5-(ethylsulfonyl)-Λ':- methyl-2,3,4,5-tetrahydro-lH-pyrido[4,3-A]indole-8-carboxamide

NaH (0.18 g, 4.7 mmol) was added to a solution of cyclopentyl-N-[4- (cyclopropylamino)-4-oxobutyl]-N-methyl-2,3,4,5-tetrahydro-lH-pyrido[4,3- Z>]indole-8-carboxamide (0.20 g, 0.47 mmol) in DMF (20 mL) under nitrogen atmosphere. The reaction mixture was stirred for 3 hrs. Ethyl sulfonyl chloride (0.44 mL, 4.7 mmol) was added and the reaction mixture was stirred for 2 hrs. The reaction mixture was slowly added to a mixture OfNaHCO3 (0.40 g, 4.8 mmol) and MeOH (80 mL) at -78°C. The resulting mixture was allowed to warm to ambient temperature and the solvent was concentrated. CH2CI2 (20 mL) was added and the precipitated salts were filtered. The filtrate was concentrated and the product was purified by preparative reverse-phase HPLC to provide the TFA salt of the title compound as a white solid (60 mg, 20 %). 1H NMR (400 MHz, CD3OD) δ 0.17 - 0.52 (m, /=89.06 Hz, 2 H), 0.53 - 0.78 (m, 2 H), 1.23 (t, J=6.84 Hz, 3 H), 1.64 - 2.10 (m, 12 H), 2.16 -
2.43 (m, 4 H), 3.00 (s, 2 H), 3.10 (s, 1 H), 3.41 - 3.67 (m, 4 H), 3.74 - 3.90 (m, 1 H), 3.95 - 4.06 (m, J=13.87 Hz, 1 H), 4.33 - 4.54 (m, 7=13.38, 13.38 Hz, 1 H), 4.74 - 4.87 (m, 1 H), 7.44 (dd, ./=17.87, 9.86 Hz, 1 H), 7.67 (d, /=18.16 Hz, 1 H), 8.05 (d, J=9.37 Hz, 1 H); MS (ESI) (M+H)+ 515.3.
Step B. Methyl 2,3,4,5-tetrahydro-lH-pyrido[4,3-i>]indole-8-carboxylate

2-tert-Butyl 8-methyl l,3,4,5-tetrahydro-2H-pyrido[4,3-ό]indole-2,8-dicarboxylate (5.4 g, 16 mmol) was added to TFA (40 mL) in CH2Cl2 (40 mL). The solution was stirred for 1 hr. and concentrated to dryness. The residue was recovered in water (50 mL), stirred at 00C for 1 hr. and filtered. The solid was thoroughly washed with ether (2 x 150 mL), filtered and dried to provide the pure TFA salt of the title compound as a pink solid (5.6 g, 99 %). MS (ESI) (M+H)+ 231.0.
Step C. Methyl l-cyclopentyl^S^S-tetrahydro-lH-pyrido^S-^indole-S- carboxylate

Cyclopentanone (5.6 mL, 63 mmol) was added to a solution of methyl 2,3,4,5- tetrahydro-lH-pyrido[4,3-£]indole-8-carboxylate (5.6 g, 16 mmol) in EtOH (80 mL). The solution was stirred for 1 hr. and NaBH(OAc)3 (5.3 g, 25 mmol) was added. The reaction mixture was stirred for 4 hrs. and the solvent was concentrated. The product was purified by normal-phase MPLC using 2% Et3N, 5% MeOH and 10% acetone in DCM to provide the title compound as a pale-yellow solid (4.56 g, 94 %). MS (ESI) (M+H)+ 299.1.
Step D.2-Cyclopentyl-2,3,4,5-tetrahydro-lH-pyrido [4,3-έ]indole-8-carboxylic acid

MeOH (15 rαL) was added to a mixture of methyl 2-cyclopentyl-2,3,4,5-tetrahydro- lH-pyrido[4,3-έ]indole-8-carboxylate (4.5 g, 15 mmol) and NaOΗ 2M (50 mL). The reaction mixture was heated to 85°C upon clear solution obtained and was concentrated to 50 mL. The solution was neutralized with concentrated HCl at 00C. The precipitate was filtered, washed with ether and dried to provide the pure title compound as apink solid (4.25 g, 98 %). MS (ESI) (M+H)+ 285.1.
Step E. Methyl 4-[[(2-cyclopentyl-2,3,4,5-tetrahydro-lH-pyrido[4,3-*]indol-8- yl)carbonyl](methyl)amino]butanoate
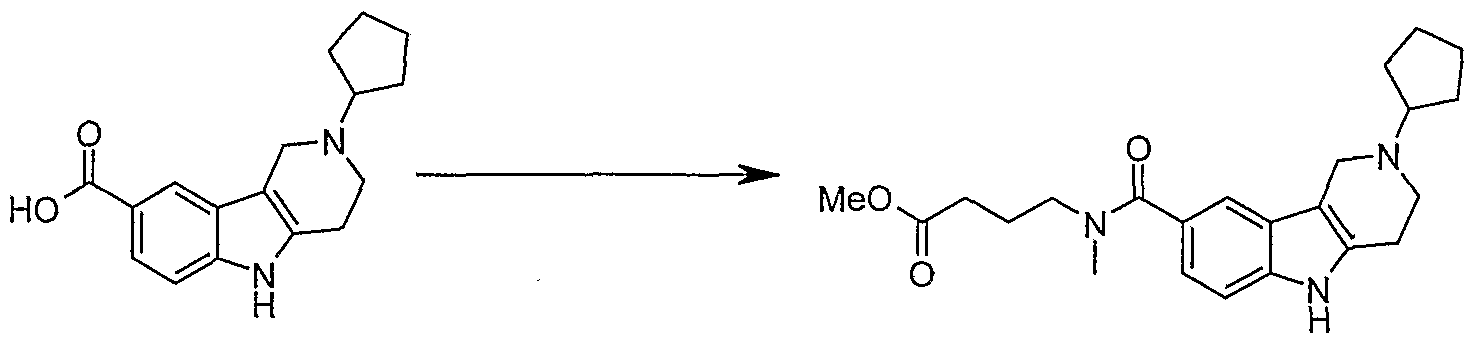
Following the procedure for Step F in Example 2, using 2-cyclopentyl-2,3,4,5- tetrahydro-lH-pyrido[4,3-Z>]indole-8-carboxylic acid (1.0 g, 3.6 mmol) provided the title compound as a colorless oil (0.72 g, 49 %). 1H NMR (400 MHz, CD3OD) δ 1.48 - 1.70 (m, 5 H), 1.70 - 1.82 (m, 2 H), 1.81 - 2.11 (m, 5 H), 2.84 - 2.94 (m, 3 H), 2.94 - 2.99 (m, J=5.37, 5.37 Hz, 2 H), 3.00 - 3.12 (m, 3 H), 3.36 - 3.73 (m, 5 H), 3.76 - 3.84 (m, 2 H), 7.03 - 7.15 (m, J=7.03 Hz, 1 H), 7.32 (d, /=8.20 Hz, 1 H), 7.42 - 7.52 (m, 1 H); MS (ESI) (M+H)+ 398.2.
Step F^-fp-Cyclopentyl-lβ^^-tetrahydro-lH-pyridoHjS-^indol-S- yl)carbonyl](methyl)amino]butanoic acid

MeOH (4 niL) was added to a mixture of methyl 4-[[(2-cyclopentyl-2,3,4,5- tetrahydro-lH-pyrido[4,3-έ]indol-8-yl)carbonyl](methyl)amino]butanoate (0.70 g, 1.7 mmol) and NaOH (2M, 20 mL). The reaction mixture was heated to 600C upon a clear solution, and was then concentrated to 20 mL. The solution was neutralized with concentrated HCl at O0C. The precipitate was filtered, washed with ether and dried to provide the pure title compound as a pink solid (0.66 g, 97 %). MS (ESI) (M+H)+ 384.2.
Step G. 2-Cyclopentyl-Λ'-[4-(cyclopropylamino)-4-oxobutyl]-iV-methyl-2,3,4,5- tetrahydro~lH-pyrido[4,3-Z>]indole-8-carboxamide

Following the procedure for Step A in Example 2, using 4-[[(2-cyclopentyl-2,3,4,5- tetrahydro-lH-pyrido[4,3-Z)]mdol-8-yl)carbonyl](methyl)amino]butanoic acid (0.65 g, 1.7 mmol) and cyclopropylamine (0.14 g, 2.5 mmol) provided the title compound as a white solid (0.70 g, 97 %). 1HNMR (400 MHz, CD3OD) δ 0.15 - 0.31 (m, J=3.71 Hz, 2 H), 0.42 - 0.53 (m, 2 H), 0.62 - 0.79 (m, J=7.52, 7.52 Hz, 2 H), 1.68 - 2.06 (m, 12 H), 2.15 - 2.43 (m, 4 H), 2.97 - 3.13 (m, J=19.33 Hz, 2 H), 3.13 - 3.28 (m, 1 H), 3.46 - 3.66 (m, 1 H), 3.75 - 3.88 (m, J=7.71, 7.71 Hz, 1 H), 3.89 - 4.02 (m, /=13.09 Hz, 1
H), 4.31 - 4.48 (m, 1 H), 4.69 - 4.85 (m, J=15.82 Hz, 1 H), 7.16 - 7.29 (m, 1 H), 7.35 - 7.47 (m, J=8.40, 0.59 Hz, 1 H), 7.53 - 7.64 (m, 1 H); MS (ESI) (M+H)+ 423.3.
Example 9
ΛL[4-(Cyclopropylamino)-4-oxobutyl]-5-(ethylsulfonyl)-Λr-methyl-2-(tetrahydro- 2J3r-pyran-4-yl)-2,3,4,5-tetrahydro-lZr-pyrido[4,3-6]indole-8-carboxamide

Step A. iV-[4-(Cyclopropylamino)-4-oxobutyl]-5-(ethyIsulfonyl)-iV-methyl-2- (tetrahydro-2J3r-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indole-8- carboxamide

Following the procedure for Step A in Example 2, using 4-[{[5-(ethylsulfonyl)-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indol-8- yl]carbonyl}(methyl)amino]butanoic acid hydrochloride (107 mg, 0.20 mmol), cyclopropylamine (23 mg, 0.40 mmol), N^V-diisopropylethylamine (77 mg, 104 uL, 0.60 mmol) and ΗATU (99 mg, 0.26 mmol) in DMF (5 mL). The crude product was purified by reverse-phase ΗPLC using high pΗ column 20-40% acetom'trile gradient to give the title compound as a white solid (63 mg, 60%). 1H ΝMR (400 MHz, METHAΝOL-D4) δ -0.09 - 0.06 (m, 1 H)5 0.16 - 0.27 (m, 1 H), 0.28 - 0.36 (m, 1 H), 0.41 - 0.50 (m, 1 H), 0.97 (t, 7=7.03 Hz, 3 H), 1.55 - 1.79 (m, 5 H), 1.86 - 2.05 (m, 3 H), 2.10 - 2.24 (m, 0.5 H), 2.35 - 2.44 (m, 0.5 H), 2.75 (s, 1.5 H), 2.84 (s, 1.5 H), 3.12 - 3.38 (m, 10 H), 3.41 - 3.55 (m, 1 H), 3.66 - 3.82 (m, 1 H), 3.88 (dd, J=11.52, 4.30 Hz, 2 H), 4.14 - 4.57 (m, 2 H), 7.19 (m, 1 H), 7.38 (s, 0.5 H), 7.43 (s, 0.5 H), 7.79 (d, J=8.79 Hz, 1 H); MS (APPI) (M+H)+= 531.2.
Step B.2-tert-Butyl-8-methyl-5-(ethylsulfonyl)-3,4-dihydro-lH-pyrido [4,3- b]indole-2,8(5H)-dicarboxylate

In a three-neck round bottom flask, equipped with a mechanical stirrer and under a nitrogen atmosphere, the indole (40.0 g, 121mmol) was dissolved in dry THF (400 mL). The solution was cooled at 0 0C and NaH (7.03 g, 145 mmol) was added portionwise over 20 minutes. The ice bath was then removed and the mixture was stirred at room temperature for 30 minutes. After 30 minutes, the mixture was cooled at 0 0C and ethanesulfonyl chloride (17 mL, 179 mmol) was added slowly. The mixture was then stirred for one hour at 0 0C and then at room temperature for 3 hours. The solution was cooled at 0 0C and water was slowly added to quench excess of sodium hydride. THF was removed under reduced pressure. Water (200 mL) and ethyl acetate (300 mL) was added to the residue. A solid precipitated out, was removed by filtration and set aside. The phases were separated. The organic phase was washed with NaHCC>3 (5%), brine, dried over Na2SO4 and then concentrated to give a beige solid. The solid collected previously by filtration contained salts. It was dissolved in a mixture of EtOAc (800 mL) and water (200 mL). The phases were separated. The organic portion was washed with brine, dried over Na2SO4 and concentrated to give a white solid. Both solids were reunited and triturated in ether, recovered by filtration and dried under vacuum to yield the titled compound (35.5 g,
Step C. Methyl 5-(ethyIsulfonyl)-2,3,4,5-tetrahydro-lfr-pyrido[4,3-b]indole-8- carboxylate

Step D. Methyl 5-(ethylsulfonyl)-2-(tetrahydro-2H-pyraa-4-yl)-2,3,4,5- tetrahydro-lH-pyrido [4,3-b]indoIe-8-carboxylate

The amine (16.0 g, 44.6 mmol) was suspended in THF (400 mL). Triethylamine (7.0 mL, 50 mmol) was added and the mixture was stirred for 10 minutes at room temperature. Ketone (5.0 mL, 54 mmol), NaBH(OAc)3 (10.4 g, 49.0 mmol) and acetic acid (3.0 mL, 52 mmol) were then added. The mixture was stirred overnight at room temperature. After 16 hours, TLC (95/5/0.1 CH2Cl2MeOHZNH4OH) showed some starting material left. The ketone (1 mL) and NaBH(O Ac)3 (1.02 g) were added and the mixture was stirred at room temperature for 8 hours. After that time, there was still some starting material left. The ketone (0.5 mL), NaBH(OAc)3 (1.9 g), Et3N (0.7 mL) and AcOH (2.5 mL) were added and the mixture was stirred overnight. The solid was then filtered off and discarded and the solvent was evaporated. The residue was taken up in dichloromethane and the organic phase was washed with a saturated solution OfNa2CO3, dried over Na2SO4 and concentrated to give the titled compound which was used without any further purification for the next step (19.1 g).
Step E. 5-(Ethylsulfonyl)-2-(tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH- pyrido[4,3-b]ϊndole-8-carboxylic acid

The methyl ester (18.1 g , a wet solid, 44 mmol) was dissolved in a mixture of THF (70 mL) and methanol (40 mL). An aqueous solution of NaOH (20 mL, 5N, 100 mmol) was added and the mixture was stirred at room temperature overnight. The solvents were then concentrated under reduced pressure. The remaining aqueous solution was diluted with water and acetic acid was added until the pH reached 5. The mixture was cooled at 0 0C and the material came out as a sticky gum. Water was decanted and set aside. The gum was triturated in methanol and a solid formed. This solid was recovered by filtration. The aqueous solution was concentrated under vacuum until a solid came out. This solid was recovered by filtration and added to the solid previously isolated. The beige solid was then suspended in methanol and a solution of 5M HCl in isopropanol was added until all the material went into solution. The solution was stirred for 10 minutes and cooled down at 0 0C and a precipitate formed. This precipitate was collected by filtration. The solid was triturated in ether and dried under vacuum to yield the titled compound (10.4 g, 55 %).
Step F. Methyl 4-[{[5-(ethylsulfonyl)-2-(tetrahydro-2H-pyran-4-yl)-2,3,4,5- tetrahydro-lH-pyrido [4,3-Λ]indol-8-yl] carbonyl} (methyl)amino]butanoate
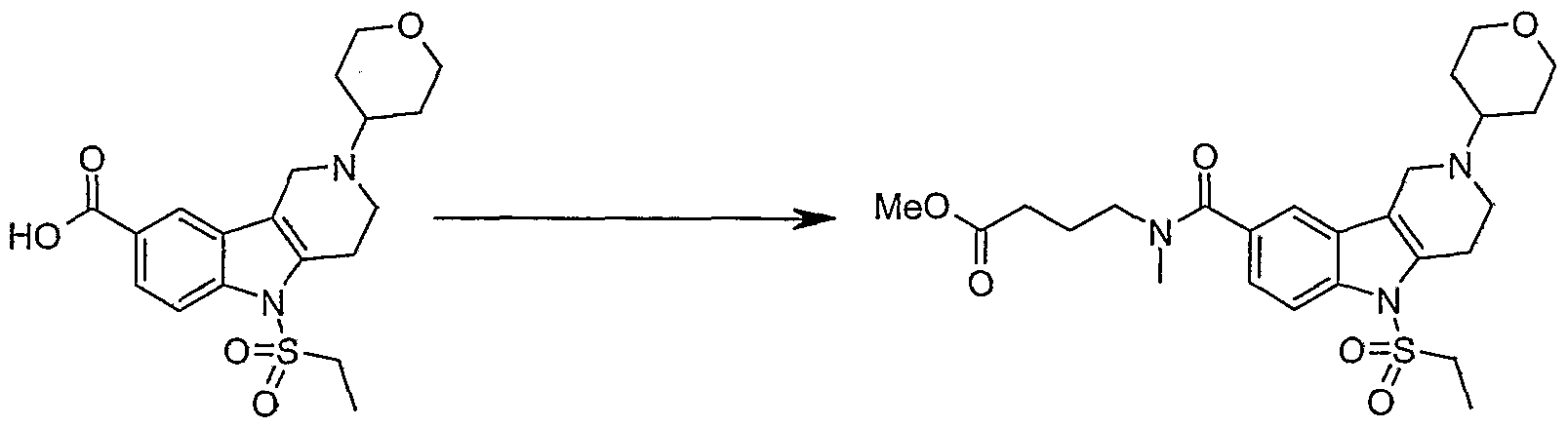
Following the procedure for Step F in Example 2, using 5-(ethylsulfonyl)-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[453-&]indole-δ-carboxylic
acid hydrochloride (4.29 g, 10 mmol), methyl 4-(methylamino)butanoate (1.97 g, 15 mmol), NN-diisopropylethylamine (3.88 g, 5.23 mL, 30 mmol) and HATU (4.94 g, 13 mmol) in DMF (40 mL). The crude product was purified by MPLC on silica gel using CH2Cl2MeOH (20:1) to give the title compound as a white solid (1.91 g, 38 %). 1H ΝMR (400 MHz, METHAΝOL-D4) δ 1.21 (t, /=7.32 Hz, 3 H), 1.83 - 2.03 (m, 4 H), 2.10 - 2.23 (m, 4 H), 2.44 (t, 7=6.93 Hz, 1 H), 2.99 (s, 1.5 H), 3.08 (s, 1.5 H), 3.31 - 3.39 (m, 1 H), 3.42 - 3.55 (m, 7 H), 3.60 (t, J=7.23 Hz, 2 H), 3.65 - 3.77 (m, 3 H), 4.12 (dd, J=I 1.72, 4.10 Hz, 2 H), 4.36 - 4.83 (m, 2 H), 7.37 - 7.48 (m, 1 H), 7.66 (s, 1 H), 8.03 (d, /=8.79 Hz, 1 H); MS (APPI) (M+H)+= 506.2.
Step G^-tftS-CEthylsuIfony^-l-^etrahydro^H-pyran^-y^-lβ^jS-tetrahydro- lH-pyrido[4,3-i]indol-8-yl]carbonyl}(methyl)amino]butanoic acid

Lithium hydroxide (0.18 g, 7.55 mmol) was added to a solution of methyl 4-[{[5- (ethylsulfonyl)-2-(tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3- έ]indol-8-yl]carbonyl}(methyl)amino]butanoate (1.91 g, 3.78 mmol) in 70 mL of THF-H2O (7:3). Stirring for 1.5 h at room temperature, the reaction mixture was neutralized by 2N HCl until pH= 5-6. After concentration and dried in vacuo, a white solid was obtained as a hydrochloride salt (2.01 g, 99%). The crude product was used directly for the next step without further purification. MS (APPI) (M+H)+= 492.3.
Example 10
5-(EthylsuIfonyl)-Λr-methyl-iV-[4-(methylanimo)-4-oxobutyl]-2-(tetrahydro-2H- pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-ό]indoIe-8-carboxamide

Following the procedure for Step A in Example 2, using 4-[{[5-(ethylsulfonyl)-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-o]indol-8- yl]carbonyl}(methyl)amino]butanoic acid hydrochloride (107 mg, 0.20 mmol), methylamine in TΗF (2.0 M, 0.2 mL, 0.40 mmol),NN-diisopropylethylamine (77 mg, 104 uL, 0.60 mmol) and ΗATU (99 mg5 0.26 mmol) in DMF (5 mL). The crude product was purified by reverse-phase ΗPLC using high pΗ column 20-40% acetonitrile gradient to give the title compound as white solid (50 mg, 50 %). 1H ΝMR (400 MHz, METHAΝOL-D4) δ 1.19 (t, J=7.42 Hz, 3 H), 1.81 - 2.05 (m, 6 H), 2.10 - 2.21 (m, 2 H), 2.26 (t, J=6.84 Hz, 1 H), 2.48 (s, 1.5 H), 2.69 (s, 1.5 H), 2.97 (s, 1.5 H), 3.07 (s, 1.5 H), 3.48 (m, 6 H), 3.53 - 3.62 (m, 2 H), 3.64 - 3.76 (m, 2 H), 3.89 - 4.05 (m, 1 H), 4.10 (dd, J=I 1.33, 4.49 Hz, 2 H), 4.41 - 4.80 (m, 2 H), 7.36 - 7.46 (m, 1 H), 7.60 (s, 0.5 H), 7.65 (s, 0.5 H), 8.01 (d, J=8.79 Hz, 1 H); MS (APPI) (M+H)+= 505.3.
Example 11
Λr-[4-(EthyIamino)-4-oxobutyl]-5-(ethylsulfonyl)-Λ'-methyl-2-(tetrahydro-2H- pyran-4-yl)-2,3,4,5-tetrahydro-L-ar-pyrido[4,3-δ]indole-8-carboxamide
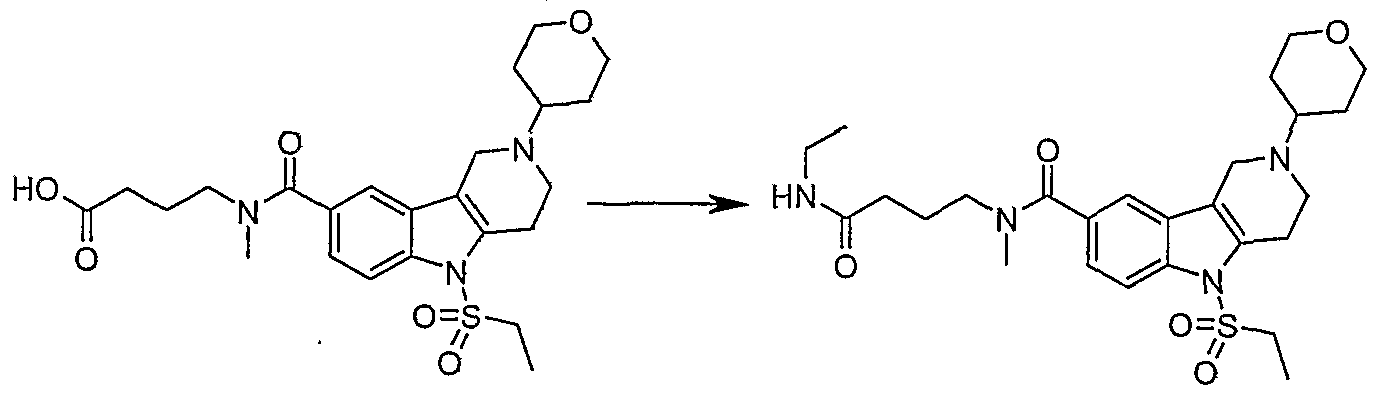
Example 12
5-(EthylsuIfonyI)-iV-[4-(isopropylamino)-4-oxobutyl]-iV-methyl-2-(tetrahydro- 2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-i]indole-8-carboxamide

Following the procedure for Step A in Example 2, using 4-[{[5-(ethylsuIfonyl)-2- (tetrahydro-2H-pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indol-8- yl]carbonyl}(methyl)amino]butanoic acid hydrochloride (107 mg, 0.20 mmol), isopropylamine (24 mg, 0.40 mmol), N,N-diisopropylethylamine (77 mg, 104 uL, 0.60 mmol) and ΗATU (99 mg, 0.26 mmol) in DMF (5 mL). The crude product was purified by reverse-phase ΗPLC using high pΗ column 20-40% acetonitrile gradient to give the title compound as white solid (65 mg, 55 %). 1H ΝMR (400 MHz,
METHAΝOL-D4) 5 0.92 (d, J=6.05 Hz, 3 H), 1.10 (d, /=6.44 Hz, 3 H), 1.16 - 1.23 (m, 3 H), 1.76 - 2.03 (m, 6 H), 2.09 - 2.19 (m, 2 H), 2.22 (t, J=7.03 Hz, 1 H), 2.97 (s, 1.5 H), 3.07 (s, 1.5 H), 3.48 (m, 7 H), 3.55 (t, J=6.25 Hz, 2 H), 3.64 - 3.77 (m, 2 H), 3.85 - 4.03 (m, 1 H), 4.10 (dd, 7=11.13, 4.10 Hz, 2 H), 4.38 - 4.81 (m, 2 H), 7.33 - 7.48 (m, 1 H), 7.61 (s, 0.5 H), 7.65 (s, 0.5 H), 8.01 (d, 7=8.79 Hz, 1 H); MS (APPI) (M+H)+= 533.3.
Claims
1. A compound of formula I, a pharmaceutically acceptable salt thereof, a diastereomer, an enantiomer, or a mixture thereof:

wherein
A is -(CBk)n- optionally substituted with one or more groups selected from methyl, ethyl, phenyl, benzyl and halogen, wherein n is 2, 3 or 4;
R1 is selected from  and C3-6cycloalkyl; R2 is selected from -H, Ci-6alkyl, C2-6alkenyl, -C(=O)-NR9R10, -S(O)2-
and C3-6cycloalkyl; R2 is selected from -H, Ci-6alkyl, C2-6alkenyl, -C(=O)-NR9R10, -S(O)2-
NR9R10, -S(=O)2-Ci-6alkyl, -S(=0)2-C6-ioaryl, -S(=O)2-C3-sheteroaryl, -C(O)- Ci-βalkyl;  and
and  wherein said Ci-βalkyl, C2-6alkenyl, -S(=O)2-C1-6alkyl, -S(=O)2-C6-i0aryl, -S(=O)2-C3.5heteroaryl, -C(O)- Ci-βalkyl;
wherein said Ci-βalkyl, C2-6alkenyl, -S(=O)2-C1-6alkyl, -S(=O)2-C6-i0aryl, -S(=O)2-C3.5heteroaryl, -C(O)- Ci-βalkyl;  and
and  used in defining R2 is optionally substituted with one or more group selected from -OR, R, -CO2H, -CO2-R; -SO2-R; halogen, -NO2, -OH, -NH2, -NHR, -C(O)-NH2, and -C(O)-NHR;
used in defining R2 is optionally substituted with one or more group selected from -OR, R, -CO2H, -CO2-R; -SO2-R; halogen, -NO2, -OH, -NH2, -NHR, -C(O)-NH2, and -C(O)-NHR;
R3 is selected from Cs-βheterocycloalkyl, Cs-βheterocycloalkyl-Ci^alkyl, C3-6cycloalkyl, C3.6cycloalkyl-C1.4aU.yl, Chalky!, C2-6alkenyl,  C3. eheteroaryl-Ci^alkyl, -C(=O)-C1-6alkyl, -C(=O)-C3.6cycloalkyl and -Ct=NH)-C1-
C3. eheteroaryl-Ci^alkyl, -C(=O)-C1-6alkyl, -C(=O)-C3.6cycloalkyl and -Ct=NH)-C1-  C3-6heterocycloalkyl-Ci.4alkyl,
C3-6heterocycloalkyl-Ci.4alkyl,
C3.6cycloalkyl,  C3- 6heteroaryl-C1-4alkyl, -C(=O)-d-6alkyl, -C(=O)-C3-6cycloalkyl and -C(=NH)-Ci- βalkyl used in defining R3 is optionally substituted with one or more groups selected from -OR, R, NO2, -CO2H, -CO2-R; -SO2-R; halogen; -OH; -NH2; -NHR, -C(O)- NH2, and -C(O)-NHR;
C3- 6heteroaryl-C1-4alkyl, -C(=O)-d-6alkyl, -C(=O)-C3-6cycloalkyl and -C(=NH)-Ci- βalkyl used in defining R3 is optionally substituted with one or more groups selected from -OR, R, NO2, -CO2H, -CO2-R; -SO2-R; halogen; -OH; -NH2; -NHR, -C(O)- NH2, and -C(O)-NHR;
R is Ci-βalkyl; and
R9 and R10 are independently selected from -H, Ci-ealkyl, Cβ-ioaryl, Cβ-ioaryl- Ci-4alkyl, C3.6heterocyclyl, Cs-eheterocyclyl-Ci^alkyl, C2.6alkenyl, C3-6cycloalkyl, and  N,N-di(Ci-4alkyl)amido-Ci.6alkyl7 hydroxy-Cμβalkyl and Ci-6alkoxy-Ci-6alkyL
N,N-di(Ci-4alkyl)amido-Ci.6alkyl7 hydroxy-Cμβalkyl and Ci-6alkoxy-Ci-6alkyL
2. A compound as claimed in claim 1, wherein R2 is selected from methyl, ethyl, 1 -propyl, 2-propyl, 1 -butyl, 2-butyl, t-butyl,' allyl, -S(=O)2-CH3, -S(=O)2-CH2CH3, 2-methoxyethyl, tetrahydropyran-4-yl-methyl, 1-propylsulfonyl, methylsulfonyl, ethylsulfonyl, cyclopropylsulfonyl, phenyl, phenylsulfonyl, 2-(methoxycarbonyl)-phenylsulfonyl; 2-(hydroxycarbonyl)- phenylsulfonyl, 1 -methyl-1 H-imidazol-4-yl-sulfonyl, 1 H-imidazol- 1 -yl-sulfonyl, furylsulfonyl, (5-methylisoxazol-4-yl)sulfonyl, morpholin-4-ylcarbonyl, 4-amino- phenyl, -CH2-C(=O)-N(CH3)2, -C(=O)-N(CH3)2, -S(=O)2-N(CH3)2, -S(=O)2- NHCH2CH3, -CC=O)-CH2CH2CH3, -CH2-C(O)-OCH3, -CH2-C(=O)-OCH2CH3, - CH2-CO2H, benzyl, 4-aminobenzyl, 4-nitroben2yl, 4-methylsulfonyl-benzyl, 4- methylthio-benzyl, 4-acetylamino-benzyl, 4-methoxy-benzyl, 4-ethoxy-benzyl, 2,6- difluorobenzyl, (6-chloro-l,3-benzodioxol-5-yl)methyl, (5-ethoxycarbonyl)-fur-2-yl- methyl, (2-methyl-l,3-thiazol-4-yl)-methyl, (5-methyl-isoxazol-4-yl)-methyl, pyridin- 2-ylmethyl, cyclobutylmethyl, and cyclopropylmethyl;
R3 is selected from ethyl, isopropyl, propyl, 2-methy-propyl, 1 -butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutyhnethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 2-pyrimidinyl, 1-iminoethyl, 2-pyridiαyI, 3,4,5,6- tetrahydropyrdin-2-yl, 3,4-dihydro-2H-pyrrol-5-yl, 2-pyridinyl-methyl, 3- pyridinylmethyl, 4-pyridinyhnethyl, l-methyl-4-piperidinyl, 4-piperidinyl, (6-methyl- pyridin-2-yl)methyl, (2-ethyl-4-methyl- 1 H-imidazol-5 -yl)methyl, tetrahydrofuran-2- yl, tetrahydrofuran-3-yl, tetrahydrofuran-3-yhnethyl, l-ethyl-lH-pyrazol-4-yl, 1,3- dimethyl-lH-pyrazol-5-yl, (3-methylpyridin-4-yl)methyl, l,3-oxazol-2-ylmethyl, 1,3- oxazol-5 -ylmethyl, 2-(tetrahydro-2H-pyran-4-yl)ethyl, tetrahydro-2H-pyran-4- ylmethyl, 2-phenylethyl, 2-methoxybenzyl, 3,3,3-trifluoropropyl, 2,2-difluoroethyl, 2- hydroxycyclopentyl, (l-ethyl-3-methyl-lH-pyrazol-5-yl)methyl, 2,1,3-benzoxadiazol- 5-ylmethyl, 3-thienylmethyl, 2-trifluoromethyl-benzyl, 3-methylbutyl, cyclohex-3-en- 1 -ylmethyl, 2-fluoro-6-methoxybenzyl, 2-phenyl-propyl, 2-ethyl-butyl, cyclobutylcarbonyl, 2,2-difluoropropanoyl, cyclopentylcarbonyl, tetrahydro-2H- pyran-4-ylcarbonyl, cyclopropylcarbonyl, propylcarbonyl, N-ethylaminocarbonyl, N- isopropylaminocarbonyl, cyclopropylsulfonyl, and ethylsulfonyl.
3. A compound as claimed in claim 1, wherein R1 is selected from methyl, ethyl, 2-fluoroethyl, isopropyl, tert-butyl and cyclopropyl;
R2 is selected from methyl, methylsulfonyl and ethylsulfonyl; and R3 is selected from ethyl, isopropyl, propyl, 2-methy-propyI, 1-butyl, 1-pentyl, l-acetyl-piperidin-4-yl, tetrahydrothien-3-yl, cyclopropylmethyl, cyclobutylmethyl, cyclopentylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 4-tetrahydro-2H-pyranyl, tetrahydro-thiopyran-4-yl, 1-iminoethyl, 3,4,5,6-tetrahydropyrdin-2-yl, 3,4-dihydro- 2H-pyrrol-5-yl, tetrahydrofuran-3-yhnethyl, tetrahydrofuran-2-yl, tetrahydrofuran-3- yl, l-methyl-4-piperidinyl, 2-(tetrahydro-2H-pyran-4-yl)ethyl, tetrahydro-2H-pyran-4- ylmethyl, 3,3,3-trifluoropropyl, 2,2-difluoroethyl, 2-hydroxycyclopentyl, 3- methylbutyl, cyclohex-3-en-l-ylmethyl, and 2-ethyl-butyl.
4. A compound as claimed in claim 1, wherein A is -(CH2)S--
5. A compound as claimed in claim 1, wherein A is -(CHO2-.
6. A compound as claimed in claim 1, wherein A is -(CH2)4-.
7. A compound as claimed in claim 1, wherein A is -CH2-CH2-CH(-Ph)-.
8. A compound as claimed in claim 1, wherein A is -CH2-CH2-CH(-CH2Ph)-.
9. A compound selected from
N-[4-(Cyclopropylarnino)-4-oxobutyl]-N,5-dimethyl-2-(tetrahydro-2H-pyran-4-yl)- 2,3,4,5-tetrahydro-lH-pyrido[4,3-Z>]mdole-8-carboxamide; 2-Cyclopentyl-N-methyl-N-[4-(methylamino)-4-oxobutyl]-5-(methylsulfonyl)- 2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]indole-8-carboxamide;
2-Cyclopentyl-N-[4-(ethylamino)-4-oxobutyl]-N-methyl-5-(methylsulfonyl)-2,3,4,5- tetrahydro-lH-pyrido[4,3-δ]indole-8-carboxamide; 2-Cyclopen1yl-N-[4-(isopropylairiino)-4-oxobutyl]-N-methyl-5-(methylsulfonyl)-
2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]mdole-8-carboxamide;
2-Cyclopentyl-N-[4-(cyclopropylamino)-4-oxobutyl]-N-methyl-5-(methylsulfonyl)-
2,3,4,5-tetrahydro-lH-pyrMo[4,3-Z>]indole-8-carboxamide; N-[4-(fe^Bu1ylamino)-4-oxobutyl]-2-cyclopentyl-N-methyl-5-(methylsulfonyl)-
2,3,4,5-tetrab.ydro-lH-pyrido[4,3-έ]indole-8-carboxamide;
2-Cyclopentyl-N-{4-[(2-fluoroethyl)amino]-4-oxobutyl}-N-methyl-5-
(methylsuIfonyl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-Z?]indole-8-carboxamide;
2-Cyclopentyl-N-[4-(cyclopropylamino)-4-oxobutyl]-5-(ethylsulfonyl)-N-methyl- 2,3,4,5-tetrahydro-lH-pyrido[4,3-6]indole-8-carboxamide;
N-[4-(Cyclopropylamino)-4-oxobutyl]-5-(ethylsulfonyl)-N-methyl-2-(tetrahydro-2H- pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-Z>]indoIe-8-carboxamide;
5-(Ethylsulfonyl)-N-methyl-N-[4-(metb.ylamino)-4-oxobutyl]-2-(tetrahydro-2H- pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-έ]mdole-8-carboxarriide; N-[4-(Ethylaimno)-4-oxobutyl]-5-(etiiylsulfonyl)-N-methyl-2-(tetrahydro-2H-pyran-
4-yl)-2,3,4,5-tetrahydro4H-pyrido[4,3-έ]mdole-8-carboxamide;
5-(Ethylsulfonyl)-N-[4-(isopropylamino)-4-oxobutyl]-N-methyl-2-(tetrahydro-2H- pyran-4-yl)-2,3,4,5-tetrahydro-lH-pyrido[4,3-o]indole-8-carboxamide; and pharmaceutically acceptable salts thereof.
10. A compound according to any one of claims 1-9 for use as a medicament.
11. The use of a compound according to any one of claims 1-9 in the manufacture of a medicament for the therapy of pain.
12. The use of a compound according to any one of claims 1-9 in the manufacture of a medicament for the treatment of anxiety disorders.
13. The use of a compound according to any one of claims 1-9 in the manufacture of a medicament for the treatment of cancer, multiple sclerosis, Parkinson' s disease,
Ηuntington's chorea, Alzheimer's disease, gastrointestinal disorders and cardiovascular disorders.
14. A pharmaceutical composition comprising a compound according to any one of claims 1-9 and a pharmaceutically acceptable carrier.
15. A method for the therapy of pain in a warm-blooded animal, comprising the step of administering to said animal in need of such therapy a therapeutically effective amount of a compound according to any one of claims 1-9.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82625006P | 2006-09-20 | 2006-09-20 | |
| US60/826,250 | 2006-09-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008036022A1 true WO2008036022A1 (en) | 2008-03-27 |
Family
ID=39200762
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2007/000822 Ceased WO2008036022A1 (en) | 2006-09-20 | 2007-09-19 | Tetrahydro-lh-pyrido[3,4-b] indole derivatives as cb1 receptor ligands |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008036022A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP1863810A4 (en) * | 2005-03-22 | 2010-03-31 | Astrazeneca Ab | NOVEL TETRAHYDRO-1H-PYRIDO [4,3-b] INDOLE DERIVATIVES AS CB1' RECEPTOR LIGANDS |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040023947A1 (en) * | 2002-05-24 | 2004-02-05 | X-Ceptor Therapeutics Inc. | Azepinoindole and pyridoindole derivatives as pharmaceutical agents |
| JP2005162657A (en) * | 2003-12-02 | 2005-06-23 | Takeda Chem Ind Ltd | Cannabinoid receptor regulator |
| WO2006101434A1 (en) * | 2005-03-22 | 2006-09-28 | Astrazeneca Ab | NOVEL TETRAHYDRO-1H-PYRIDO [4,3-b] INDOLE DERIVATIVES AS CB1’ RECEPTOR LIGANDS |
-
2007
- 2007-09-19 WO PCT/SE2007/000822 patent/WO2008036022A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040023947A1 (en) * | 2002-05-24 | 2004-02-05 | X-Ceptor Therapeutics Inc. | Azepinoindole and pyridoindole derivatives as pharmaceutical agents |
| JP2005162657A (en) * | 2003-12-02 | 2005-06-23 | Takeda Chem Ind Ltd | Cannabinoid receptor regulator |
| WO2006101434A1 (en) * | 2005-03-22 | 2006-09-28 | Astrazeneca Ab | NOVEL TETRAHYDRO-1H-PYRIDO [4,3-b] INDOLE DERIVATIVES AS CB1’ RECEPTOR LIGANDS |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1863810A4 (en) * | 2005-03-22 | 2010-03-31 | Astrazeneca Ab | NOVEL TETRAHYDRO-1H-PYRIDO [4,3-b] INDOLE DERIVATIVES AS CB1' RECEPTOR LIGANDS |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7615642B2 (en) | Therapeutic compounds | |
| US20100113502A1 (en) | Novel Tetrahydro-1H-Pyrido[4,3-b] Indole Derivatives as CB1 Receptor Ligands | |
| RU2346938C2 (en) | Derivatives of benzimidazol, compositions containing them, obtaining and application | |
| US20090062251A1 (en) | Novel Compounds 002 | |
| ES2285409T3 (en) | PIRROL DERIVATIVES (2,3-B) PIRIDINE SUBSTITUTED WITH HETEROARILO AS ANALOGONISTS OF THE CRF RECEIVER. | |
| JP2008519833A (en) | Indazolesulfonamide derivatives | |
| US7501447B2 (en) | Benzimidazole derivatives, compositions containing them, preparation thereof and uses thereof | |
| WO2004087704A1 (en) | Azaindole derivatives, preparations thereof, uses thereof and compositions containing them | |
| MXPA05003901A (en) | Novel compounds. | |
| JP2006527247A (en) | Benzimidazole derivatives, compositions containing them, their production and their use | |
| US20070072853A1 (en) | Benzimidazole derivatives compositions containing them, preparation thereof and uses thereof | |
| WO2008036021A1 (en) | Tetrahydro-lh-pyrido [3,4 -b] indole derivatives as cbl receptor ligands | |
| EP1670770B1 (en) | Benzimidazole derivatives, compositions containing them, preparation therof and uses thereof | |
| SK8542002A3 (en) | Substituted homopiperidinyl benzimidazole analogues as fundic relaxants | |
| WO2008036022A1 (en) | Tetrahydro-lh-pyrido[3,4-b] indole derivatives as cb1 receptor ligands | |
| US20070161619A1 (en) | Pyrroloquinoline and piperidoquinoline derivatives, preparation thereof, compositions containing them and uses thereof | |
| AU2005287429A1 (en) | Compounds, compositions containing them, preparations thereof and uses thereof II | |
| AU2017329111B2 (en) | Compositions for the treatment of hypertension and/or fibrosis | |
| US20110086853A1 (en) | Therapeutic Compounds | |
| EP1572691A1 (en) | Pyrazole compounds as integrin receptor antagonists derivatives | |
| MXPA06008941A (en) | Pyrroloquinoline and piperidoquinoline derivatives, preparation thereof, compositions containing them and uses thereof | |
| JP2008534641A (en) | Method for preparing bicyclic compounds | |
| KR20070057856A (en) | Compounds, compositions containing them, preparation methods thereof and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07808831 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07808831 Country of ref document: EP Kind code of ref document: A1 |