WO2008056827A1 - Procédé pour la production d'une bétaïne - Google Patents
Procédé pour la production d'une bétaïne Download PDFInfo
- Publication number
- WO2008056827A1 WO2008056827A1 PCT/JP2007/072237 JP2007072237W WO2008056827A1 WO 2008056827 A1 WO2008056827 A1 WO 2008056827A1 JP 2007072237 W JP2007072237 W JP 2007072237W WO 2008056827 A1 WO2008056827 A1 WO 2008056827A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- genus
- reaction
- represented
- halo
- halogen atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/12—Formation of amino and carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/06—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/007—Carnitine; Butyrobetaine; Crotonobetaine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/02—Amides, e.g. chloramphenicol or polyamides; Imides or polyimides; Urethanes, i.e. compounds comprising N-C=O structural element or polyurethanes
Definitions
- the present invention relates to a method for producing betaine, in particular calcutin.
- L-Carrotin a kind of betaine, is also called vitamin B T and is an important compound involved in the metabolism of fatty acids in vivo.
- Heart disease therapeutic agent Japanese Patent Laid-Open No. 5-4-
- hyperlipidemia treatment agent Japanese Patent Laid-Open No. 54-13-1340 9
- venous disease treatment agent Japanese Patent Laid-Open No. 5-8-8 8 3 1 2
- Japanese Patent Application Laid-Open No. Sho 5 7-1 6 5 3 5 2 describes a method for obtaining L-carnitine from D-mannitol as a raw material.
- 7 2 9 8 3 includes (R, S) 1, 3, 4-epoxybutyric acid ester as a starting material and (R) — 3, 4-epoxybutyric acid ester.
- the step of introducing a quaternary amino group using trimethylamine into the C 4 skeleton in the carnitine production process is an unavoidable important step, and the yield of this step is the overall yield. Often has a large impact on rate.
- Japanese Patent Laid-Open No. 2-1 4 2 7 5 8 describes 4 halogenated 3- A method is shown in which methylbutyrate is quaternized with trimethylamine in a ketone solvent and the resulting butyrate derivative is hydrolyzed.
- This quaternized amination reaction uses alcohol trimethylamine as an alcohol solvent and is allowed to react in an autoclave at a temperature as high as 80 ° C.
- the dehydration of 4-hydroxygeno-3-hydroxybutyrate proceeds. Therefore, the selectivity is extremely low, about 30-40%, and the reaction time is as long as 20 hours or more. Although it is described that the selectivity is improved by using a ketone solvent, although the selectivity is greatly improved, the reaction time is further increased, and even if it exceeds 50 hours, 4-halogeno 3-hydroxybutyric acid is used. The conversion rate of methyl is only about 80%. When ether type, toluene or the like is used as the solvent, the reaction time is further delayed and the selectivity is not high.
- Japanese Patent Application Laid-Open No. 2-27995 describes a method of adding quaternary amination by adding 30% trimethylamine aqueous solution to ⁇ -chloro-) 3-hydroxybutyronitrile. After standing overnight at 4 ° C, the solid obtained by filtering the reaction solution under reduced pressure is only about 75% yield of carnitine nitryl chloride with a purity of 100%. I can't say that.
- JP-A-60-2 5 848 7 4-chloro-3-hydroxybutyronitrile is added with a large excess of anhydrous trimethylamine and reacted in an autoclave at 100 ° C. without solvent.
- the quaternary amination process with trialkylamine which significantly affects the overall yield in the production process of carnitine and other betaines, is based on the conventional 4-monohalogeno 3-methylhydroxybutyrate ⁇ -chloro-hydroxyl
- side reactions often proceed, making it difficult to obtain the desired quaternary amination product in high yield. Disclosure of the invention
- the present invention aims to prevent side reactions significantly in the quaternary amination process, which is important in the production process of betaine such as carcin, and to obtain betaine such as cartine with high yield. To do.
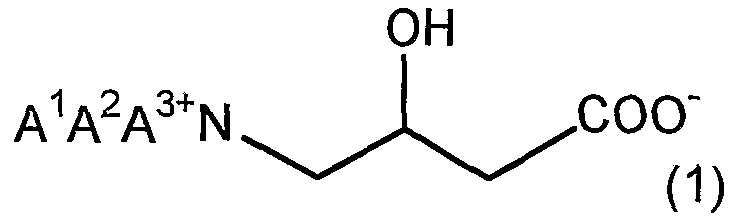
- AA 2 and A 3 are each independently the same or different and may have a substituent; ⁇ Hydrocarbon group, X 1 is a halogen atom. ]
- a process for producing a 4-trialkylamino hydride represented by the formula 3-hydroxybutane halide (2) The method according to (1), wherein the trialkylamine is selected from the group consisting of trimethylamine, triethylamine, and triptylamamine.
- a 1 A 2 and A 3 are each independently the same or different and may have a substituent.
- X 1 is a halogen atom.
- a 1 A 2 and A 3 are each, independently of one another, identical or different, Ji ⁇ which may have a substituent 2. It is a hydrocarbon group.
- the nitrile hydratase is a genus of Achr omo bacter, genus Acido V ora X, genus Agrobacteri um, genus Arthrobacter, The genus Bacillus (B aci 1 1 us), the genus Brevibacterium, the genus Burkholderia, the genus C andida, the genus Caseobacter, the comamonas ( Comamo nas), Corynebacteri um, Dietzia, En terobacter, Erwinia (E rw 1 n 1 a) 3 ⁇ 4, N.
- X 1 has the above-mentioned meaning.
- X 2 is the same or different haguchigen atom independently of X 1 .
- the halohydrin epoxidase is a microorganism belonging to the genus Corynebacterium, Microbacteiiumj, Agronocterium, Mycobacterium, or Arthrobacter.
- trialkylamine which is important in the production process of betaine such as carcin is used.
- 4-halogenated 3-hydroxybutane amide as a substrate, the production of crotonic acid derivatives, which are the main by-product, can be greatly suppressed compared to the conventional method. It becomes possible to produce a carnitine or other betaine at a high rate.
- AA 2 and A 3 are each independently the same or different, and are optionally substituted C 1 to C 2 Q hydrocarbon groups, and X 1 is a halogen atom. is there.
- AA 2 and A 3 are each, independently of one another, identical or different, ⁇ may have a substituent ⁇ 2. It is a hydrocarbon group, and X 1 is a halogen atom. ]
- ⁇ ⁇ 2 and ⁇ 3 are each independently the same or different and may have a substituent. It is a hydrocarbon group.
- a method for producing betaine or a salt of betaine is provided.
- a process for producing a bein represented by the following formula (1) comprising a step of quaternizing an amide represented by the following formula (2) and a step of hydrolyzing an amide group.
- the present invention provides a method for producing betaine represented by the following formula (1), which comprises a step of quaternizing an amide represented by the following formula (2).
- AA 2 and A 3 are each independently Ci Cs, which may be the same or different and may have a substituent. It is a hydrocarbon group.
- hydrocarbon group of “hydrocarbon group” may be a sum or unsaturated acyclic group, or a saturated or unsaturated cyclic group. C 1 ⁇ c 2. When the hydrocarbon group is acyclic, it may be linear or branched. “Ci to C 2. Hydrocarbon group” includes C i to C 2 . Alkyl groups, C 2 ⁇ C 2. Alkenyl group, c 2 ⁇ c 2. Alkynyl group, c 4 ⁇ c 2. Alkyl Jenny Le group, c 6 to c 18 Ariru group, C 7 -C 20 alkyl ⁇ aryl group, C 7 ⁇ C 2. Aryl alkyl group, C 3 to C 20 cyclic alkyl group, C 4 to C 2 . A cycloalkenyl group, (Cg Ci. Cycloalkyl) C-dialkyl group, and the like.
- rc c ⁇ alkyl group means C Ci. It is preferably an alkyl group, Re is more preferably a C 1 -C 6 alkyl group.
- ⁇ / kill groups include, but are not limited to, methyl, ethyl, propyl, isopropylene, n-butinole, sec-butyl, tert-butyl, pentinole, hexyl, dodecanyl, etc. .
- alkenyl group is C 2 -C 1 . It is preferably an alkenyl group, and more preferably a 0 2 to 6 alkyl group. Examples of alkenyl groups include, but are not limited to, vinyl, 1-propenyl, 2-propenyl, iso-propyl, 2-butenyl and the like.
- C 2 -C 2. Alkyl group is Cs Ci. It is preferably an alkynyl group, more preferably a C 2 to C 6 alkynyl group. Examples of alkyl groups include, but are not limited to, ethynyl, probule, and petityl.
- alkylgenyl It is preferably a genyl group, and more preferably a C 4 -C 6 alkyl genyl group.
- alkylgenyl groups include, but are not limited to, 1,
- C 6 -C 18 aryl group is preferably a C 6 -C 12 aryl group.
- aryl groups include, but are not limited to, phenyl, 1-naphthyl, 2-naphthyl, indul, biphenylyl, anthryl, phenanthryl, and the like.
- Alkylaryl group is preferably a c 7 to c 12 alkylaryl group.
- alkylaryl groups include, but are not limited to: o-trinole, m-trinole, ⁇ -trinole, 2,3-xylyl, 2,4-xylyl, 2,5-xylinole, o-cumenore, m-Kume-Nole, p-Tameninole, mesityl and the like.
- Arylalkyl group is preferably a c 7 -c 12 aryl alkyl group.
- arylalkyl groups include, but are not limited to, benzyl, phenethylenore, dipheninolemethyl, tripheninolemethinole, 1-naphthylmethinole, 2-naphthinoremethinole, 2,2-diphenenoreethino. Les, 3-phenylpropyl, 4-phenylbutyl, 5-phenylpentyl, and the like.
- Cycloalkyl group is Cs Ci.
- a cycloalkyl group is preferred. Examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like.
- C 4 -C 20 cycloalkenyl group is preferably a C 4 -C 10 cycloalkenyl group.
- cycloalkenyl groups include, but are not limited to, cycloprobes, cyclobutyrs, cyclopentenyls, cyclohexenyls, and the like.
- a substituent may be introduced into the “hydrocarbon group”.
- This substituent for example, Anorekokishi group (e.g., main butoxy, ethoxy, propoxy, butoxy, etc.), C 6 ⁇ 2 Ariruokishi group (e.g., Fueniruokishi, Nafuchiruokishi, biphenyl Okishi etc.), an amino group, a hydroxyl group, a halogen atom (e.g., fluorine, chlorine, bromine, Iodine) or a silyl group.
- one or more substituents may be introduced at substitutable positions, and preferably 1 to 4 substituents may be introduced. When the number of substituents is 2 or more, each substituent may be the same or different.
- AA 2 and A 3 are each independently the same or different and are each a C 1 to C 20 alkyl group, a C 3 to C 20 cycloalkyl group, or a C 6 to C 18 aryl group.
- it is a group.
- a salt of betaine is a salt of betaine with a mineral acid such as hydrochloric acid, sulfuric acid or nitric acid, a salt with an organic acid such as fumaric acid or tartaric acid, a base such as sodium hydroxide or potassium hydroxide.
- a mineral acid such as hydrochloric acid, sulfuric acid or nitric acid
- a salt with an organic acid such as fumaric acid or tartaric acid
- a base such as sodium hydroxide or potassium hydroxide.
- X 1 represents a halogen element, and is a force showing fluorine, chlorine, bromine or iodine, and is more preferably chlorine or bromine from the viewpoint of availability.
- the aqueous solvent means water or a mixture of water and an organic solvent. is there.
- the water and organic solvent may be a two-phase system.
- alcohol solvents such as methanol, ethanol, propanol, isopropanol and propanol
- keton solvents such as acetone and methylisobutylketone, ethyl acetate, ethyl propionate
- metathalic acid Ester solvents such as methyl, hydrocarbon solvents such as pentane, hexane and heptane, aromatic solvents such as benzene, toluene and xylene, chlorine solvents such as dichloromethane and chloroform, acetonitrile and dimethylform Examples thereof include amide, tetrahydrofuran, dimethyl sulfoxide, and the like, and a mixed solvent thereof may be used.
- an alcohol solvent having high solubility in water acetonitrile, dimethylformamide, or dimethyl sulfoxide. It is more preferable to use an organic solvent as long as it is soluble in water, and it is particularly preferable to use water alone because the reaction rate increases when the amount of water is large.
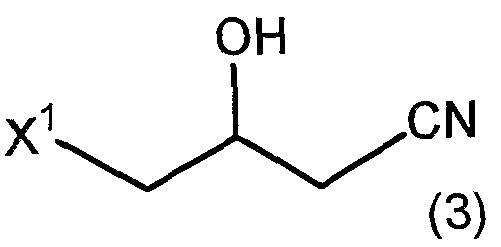
- the production method of the present invention includes a step of passing through 4-halo-3-hydroxybutane amide represented by the following formula (2).
- “via” means that when an amide represented by the following formula (2) is used as a starting material, and other substances are used as a starting material, but an intermediate represented by the following formula (2): Meaning both via the indicated amide.
- the production method of 4-halo-3-hydroxybutane amide represented by the above formula (2) is particularly there is no limit.
- a method obtained by chemical reaction a method of converting to an amide using an ester and ammonia, a method of converting to an amide using an acid halide and ammonia, and a conversion of -tolyl to an amide using an acid or base as a catalyst. Any method can be used.
- a method for converting nitrile to amide a method using mineral acid such as hydrochloric acid, a method using hydrogen peroxide and an alkali, a method using manganese dioxide, etc. are known. It may be used.
- the 4-halo-3-hydroxyptyramide represented by the above formula (2) is produced by amidating the 4-halo-3-hydroxypropylonitrile represented by the above formula (3) by the action of nitrile hydratase. You can also. Since this reaction can be carried out under mild conditions, it is possible to suppress the progress of side reactions when using an acid / base catalyst that requires strict conditions or complicated operations.
- the method for producing 4-halo-3-hydroxypetit-tolyl which is a precursor of the amide represented by the above formula (2).
- epihalohydrin represented by the following formula (4) A method of synthesizing from hydrocyanic acid (shown in Japanese Patent Laid-Open No. 2 0 0 2 — 2 4 1 3 5 7), a method of synthesizing from Epihaguchi hydrin and cyanate (Japanese Patent Laid-Open No. 2 0 0 4 — 1 8 2 6 0 7),
- X 1 has the above-mentioned meaning.
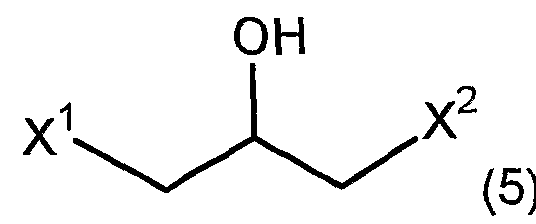
- X 2 is independently the same as X 1 and is the same or different halogen atom.
- cyanate it is preferable to use an alkali metal salt of hydrocyanic acid, and it is more preferable to use 'NaCN, KCN, or LiCN.
- an optically active epihalohydrin represented by the formula (4) it is also possible to produce an optically active 4-monohalo-3-hydroxypetit nitrile.
- the halohydrin epoxidase is prepared from the 1,3-dihalo-2-propanol represented by the above formula (5) from the 1,3-dihalo-2-propanol represented by the above formula (4).
- the 1,3-dihalo-2-propanol represented by the above formula (4) There is no particular limitation as long as it has a function of generating nitrile.
- Microorganisms producing halohydrin epoxidase include the genus Corynebacterium, the genus Microbacterium, the agropa It is a microorganism belonging to the genus Cterium, the genus Mycobacterium and the genus Arthrobacter.
- microorganisms include Corynebacterium sp.N-1074 (FEEM BP-2643), Microbacterium sp.N-4701 (FERM BP-2644), Agrobacteriu radiobacter AD 1, Mycobacterium sp.GPl, Arthrobacter sp.AD2, and the like.
- Particularly preferred microorganisms are Corynebacterium sp. N-1074 (FERM BP-2634), and Microbacterium sp. N-4701 (FERM BP-2644).
- N-4701 shares have been deposited with the National Institute of Advanced Industrial Science and Technology (National Institute of Advanced Industrial Science and Technology), Biological Depositary Center (1-1-1 Tsukuba, Tsukuba 1-1-1) on April 19th, 1999.
- the accession numbers are FERM BP-2643 and FERM BP-2644.
- microorganisms obtained by cloning and transforming (introducing) the halohydrin epoxidase gene of the above microorganism are also included as microorganisms producing the above halohydrin epoxidase.
- halohydrin epoxidase gene is published in, for example, GenBank, and the accession number of the halohydrin epoxidase gene (hheB) derived from Corynebacterium sp. (Corynebacterim) N-1074 is D90350.
- the halohydrin epoxidase according to the present invention can be obtained by culturing a microorganism having halohydrin epoxidase activity prepared by the above-described method and collecting it from the culture.
- a medium for culturing microorganisms having halohydrin epoxidase activity a medium that contains a carbon source, a nitrogen source, inorganic salts, etc. that can be assimilated by microorganisms and that can efficiently culture transformants. If so, either a natural medium or a synthetic medium may be used.
- the carbon source include carbohydrates such as gnolecose, fructose, sucrose, and starch, organic acids such as acetic acid and propionic acid, and alcohols such as ethanol and propanol.
- Nitrogen sources include inorganic acids such as ammonia, ammonium chloride, ammonium sulfate, ammonium acetate, and ammonium phosphate.
- Ammonium salt of acid or other nitrogen-containing compounds peptone, meat extract, corn steep liquor and the like.
- the inorganic substance include potassium phosphate, potassium phosphate, magnesium phosphate, magnesium sulfate, sodium chloride, ferrous sulfate, manganese sulfate, copper sulfate, or calcium carbonate.
- the above microorganisms may be cultured by a conventional method, for example, aerobically cultured at a pH of 4 to 10 and a temperature of 10 to 45 ° C. for 10 to 180 hours. Culturing can be performed by either liquid culture or solid culture.
- the transformant is preferably cultured at 30 to 40 ° C. under aerobic conditions such as shaking culture or aeration and agitation culture.
- antibiotics such as ampicillin or kana machine and inducers may be added to the medium as needed.
- the culture obtained by culturing the halohydrin epoxidase prepared by the above method or its treated product is an epihalohydrin represented by the above formula (4) or 1,3-dihalo-2-propanol represented by the above formula (5). From 4 1-halo 3-hydroxy ptyronitrile can be produced.
- “Processed product” means a microbial cell extract, such as a crushed cell product, a microbial cell treated with a drug, an immobilized microbial cell, or a crude enzyme / purified enzyme.
- the solvent for the reaction solution is not particularly limited, but the aqueous solvent is generally used.
- water is preferable, and water or a buffer solution having an optimum enzyme activity pH of about 4 to 1 ° is used.
- a buffer solution composed of a salt such as phosphoric acid, boric acid, citrate, dartaric acid, malic acid, malonic acid, 0-phthalic acid, succinic acid or acetic acid, ris buffer or Good
- a buffer solution or the like is preferable.
- the reaction temperature is preferably 5 to 50 ° C., and the reaction pH is preferably 4 to 10.
- the reaction temperature is more preferably 10 to 40 ° C.
- the reaction pH is more preferably pH 6-9.
- the reaction time is selected as appropriate depending on the concentration of the substrate, etc., the bacterial cell concentration, or other reaction conditions, but it is preferable to set the conditions so that the reaction is completed in 1 to 120 hours. In this reaction, the reaction can proceed more smoothly by neutralizing the chlorine ions produced as the reaction proceeds with an appropriate strength.
- Cyanide compounds include hydrogen cyanide, potassium cyanide, and sodium cyanide.
- a compound that generates cyanide ion (CN-) or hydrogen cyanide or a solution thereof when added to a reaction solution such as trium, cyanic acid, or acetone cyanhydrin can be used.
- the substrate concentration in the reaction solution is preferably from 0.01 to 20 (W / V)%, particularly preferably from 0.01 to L0%, from the viewpoint of enzyme stability. .
- the amount of cyanide used is preferably 1 to 3 times the amount of the substrate (mole) from the viewpoint of enzyme stability.
- the 4-halo-3-hydroxypropylonitrile represented by the above formula (3) produced by the method as described above can be collected and purified using a known method. For example, after removing the cells from the reaction solution using a method such as centrifugation, extraction with a solvent such as ethyl acetate and removal of the solvent under reduced pressure results in 4 hello 3-hydroxypropylonitrile. You can get syrup. Coloring often occurs and the reaction solution turns brown, but decolorization may be performed using activated carbon or the like. Further, these syrups can be further purified by distillation under reduced pressure.
- the optical purity of the reaction is mild under the conditions, so that the optical purity can be reduced to betaine without lowering the optical purity. It is possible to manufacture.
- nitrile hydratase is not particularly limited, but is an enzyme that catalyzes a reaction for converting nitrile of 4-halo-3-hydroxypropylonitrile represented by the above formula (3) to amide. According to international enzyme classification, it is an enzyme belonging to lyase.
- Microorganisms having nitrile hydratase used in the present invention include, for example, the genus Achromobacter, the genus Acidovorax, the genus Agrobacterium, the genus Arthrobacter, the batinoles.
- Microbial strains assigned ATCC numbers can be easily obtained from the American Type Culture Collection (ATCC).
- Microorganisms with I FO numbers are listed in “List of Cultures, 8th edition, 1st edition (1 98 8) J” published by Fermentation Research Institute (IFO). Is available from the Genetic Resource Conservation Division, Biological Resources Division, Biotechnology Headquarters, National Institute of Technology and Technology, I.
- Microorganisms with AM numbers can be obtained from the Institute of Applied Microbiology, University of Tokyo J Numbering These microorganisms are described in the “Catalog of strains 4th edition (1 9 8 9)” issued by the Institute of Physical and Microbiology, RIKEN, and can be obtained from the Institute of Microorganisms of RIKEN.
- Microorganisms with HUT numbers are listed in “Catalogue of Cultures, 4th edition (1 9 8 7) j” issued by the Japan Microbial Conservation Federation (J FCC) and can be obtained from the Faculty of Engineering, Hiroshima University. The attached microorganism can be obtained from the Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology.
- nitrile hydratase for use in the present invention, further Yamada et al Rhodococcus rhodochrous J one 1 CFERM No. BP-1478 isolated from soil], ⁇ beauty ground port Pakuta S p. SK 1 0 3 [FERMP- 11300), Caseopactor sp. BC 2 3 [FERM P-11261], Syudumonas sp. BC 1 5-2 FERMBP-3320], Syudumonas s p. SK 3 1 [FERMP-11310], Syudomonas s p. S K8 7 [FEBM P-11311], Syudomonas sp.
- microorganisms cloned and transformed (introduced) by cloning the nitrile hydratase gene of the above-mentioned microorganisms are also included as microorganisms producing the above-mentioned utriyl hydratase.
- Escherichia coli M 10822 strain (FERM BP-5785)
- the medium composition for culturing the microorganism producing nitrinole hydratase used in the present invention is not particularly limited, and any medium can be used as long as these microorganisms can normally grow.
- sugars such as glucose, fructose, sucrose, and maltose as carbon sources
- organic acids such as acetic acid and citrate
- alcohols such as ethanol and glycerol
- peptone meat extract, yeast extract, and protein hydrolysate
- various inorganic and organic acid ammonium salts can be used.
- inorganic salts, trace metals, vitamins, etc. are used as needed.
- various nitrile compounds such as 4-chloro-3-hydroxypropylonitrile, propionitol, isobutyric-tolyl, benzyl cyanide, etc.
- various amide compounds such as hydroxybutamide, propionamide, and isobutaneamide to the medium.
- the microorganisms may be cultured by a conventional method.
- the microorganisms are aerobically cultured at a pH of 4 to 10 and a temperature of 10 to 45 ° C. for 10 to 180 hours. Culturing can be performed by either liquid culture or solid culture.
- an enzyme is allowed to act on 4-halo-3-hydroxypropylonitrile.
- the method for obtaining 4-halo-3-hydroxybutane amide is not particularly limited, but was obtained by culturing the microorganisms that produced nitrile hydratase obtained as described above or by centrifugation.
- a method for adding a substrate to a suspension of bacterial cells, a substrate for a suspension of the above-mentioned microorganism treated product for example, crushed bacterial cells, bacterial cell extract, etc.
- reaction solution adding a substrate to the culture solution when cultivating microorganisms, and simultaneously with the culture Method of performing reaction, microbial culture solution obtained by preparing an aqueous solution of the substrate as described above, microbial cells, processed microbial cells, nitrile hydratase crude enzyme, purified enzyme, or immobilizing them thing
- the amidation reaction is an exothermic reaction, and the reaction rate is increased by adding a suspension of bacterial cells to the substrate aqueous solution to prevent runaway reaction due to the exotherm. It is preferable to control.
- the following amidation reaction with nitrile hydratase may be performed.
- the cyanide compound remaining in the system often deactivates the nitrile hydratase.
- the amidation reaction of 4-halo-3-hydroxybutyronitrile does not proceed rapidly even when the concentration of hydrocyanic acid in the system is 1 ppm or less, heat-treat the 4-halo-3-hydroxybutyronitrile or its solution. By doing so, the amidation reaction may proceed rapidly.
- the heating method is not particularly limited as long as the object of the present invention is achieved, and any of pressurization, decompression, and normal pressure conditions can be selected.
- the solution may be an organic solvent, an aqueous solution, or a mixture thereof.
- the amidation reaction is usually performed in an aqueous solvent, it is possible to perform the reaction continuously.
- Heat treatment is preferably performed in an aqueous solvent.
- the calorie heat time is about 0.1 to 100 hours, preferably 0.5 to 50 hours, and more preferably 1 to 10 hours.
- the heating temperature can be in a range from about 40 ° C. to a temperature at which 4 1-halo 3-hydroxyptyronitrile does not decompose, and is preferably 60 to: 150 ° C. 4
- the pH is preferably in the neutral to acidic range, and more preferably in the pH range from 1 to 7, in order to prevent premature rectification.
- the cyanide ion contained in a very small amount in 4-halo-3-hydroxybutyronitrile is reduced to a metal complex, thereby allowing the amidation reaction to proceed rapidly.
- a metal that reacts with hydrocyanic acid to form a metal cyanide complex is converted into a solution containing the nitrile or the -tolyl as a metal salt.
- cyanide is converted to a metal cyanated complex by addition.
- the metal cobalt, nickel, and zinc are preferable, and the form of the salt is not particularly limited, but examples thereof include nitrate, chloride, sulfate, and carboxylate. A hydrate may also be used.
- the conditions for the amidation reaction with nitrile hydratase in the present invention are not particularly limited, and the reaction temperature is appropriately determined depending on the enzyme used. In general, it is the temperature at which the reaction solution does not freeze ⁇ 50 ° C. The temperature at which the reaction solution does not freeze depends on the reaction solution in which the amidation reaction is performed. If the reaction is a dilute solution, the temperature can be calculated from the molar concentration (mol / kg) of the solute dissolved in the reaction solution and the value of the molar freezing point depression of the solvent.
- the amidation reaction of 4-halo-3-hydroxyptyronitrile to 4-halo-3-hydroxybutanamide is carried out in order to suppress the progress of side reactions.
- the temperature at which the reaction solution does not freeze is preferably about 30 ° C., and the temperature at which the reaction solution does not freeze is particularly preferably about 5 ° C.
- 4-Halo 3-Hydroxyptyronitrile concentration is not particularly limited, but is usually about 0.11 to 50 (W / V)%, and about 0.1 to 40% is more preferable.
- the pH of the reaction solution is preferably in the range of 4 to 10, more preferably 6 to 9.
- reaction time varies depending on the substrate concentration, bacterial cell concentration, and other reaction conditions
- the 4-halo-3-hydroxybutanamide synthesized as described above can be isolated and purified according to conventional methods such as extraction, column separation, and recrystallization. When a microorganism having nitrile hydratase is used, the cells may be filtered after completion of the reaction. However, 4-halo-3-hydroxybutanamide is unstable in an aqueous solution and should be filtered at a low temperature. There is a need to do. It is more preferable to operate at about 0 to 10 ° C.
- 4-halo-3-hydroxybutanamide is also an optically active substance. If one of them is excessive, the optical purity can be improved by performing a recrystallization operation of 4-halo-3-hydroxybutane amide.
- racemic 4-halo-3-hydroxybutanamide is produced, optically active 4-halo-3-hydroxybutanamide can be obtained using an optical resolving agent.
- the 4-halo-3-hydroxybutaneamide represented by the above formula (2) used in the present invention may be a purified one, or may be in the state of an aqueous solution after the action of etryl hydratase. .
- isolation and purification may cause further reaction and decrease yield.
- isolation and purification by column purification, etc. is complicated and has an isolation and purification yield of about 80%, and considering the total yield up to the final product, nitrinorehydratase is effective. It is preferable to use the aqueous solution after the treatment as it is.
- 4-halo-3-hydroxybutane amide is unstable in aqueous solution, so in order to suppress side reactions, within 48 hours after the production of 4-halo-3-hydroxybutanamide is completed. It is more preferable to proceed to a quaternary amination reaction with trialkylamine, particularly preferably within 10 hours. It is preferable to keep the temperature of the reaction-terminated liquid at 0 to 10 ° C. until contact with trial alkylamine.
- the method for producing betaine represented by the following formula (1) preferably includes a step of quaternizing an amide represented by the following formula (2) and a step of hydrolyzing the amide group. . Since various side reactions occur in the hydrolysis reaction of compound (2), the 4-quaternary amination step is performed first from the viewpoint of yield improvement, and 4-trialkylamino-3-hydroxybutane represented by compound (2 ') is used. Produce amide halide followed by hydrolysis step.
- X 1 , A 1 , A 2 and A 3 have the above-mentioned meanings.
- a trialkylamine such as trimethyl / reamine, triethylamine, or triptylamin in the step of quaternizing the amide represented by the above formula (2).
- the trialkylamine used in the present invention may be in an anhydrous state or an aqueous solution.
- the solvent is not particularly limited, but the aqueous solvent is used. It is preferable.
- the trialkylamine used in this reaction may be anhydrous trialkylamine. Either one of these aqueous solutions may be used. However, when an organic solvent compatible with water is used as the solvent for the 4-halo-3-hydroxybutane amide solution, it is better to use a trialkylamine aqueous solution. And the reaction rate is higher, and it is more preferable to perform the reaction using water alone, so that a higher yield and a higher reaction rate can be obtained.
- the concentration of 4-halo 3-hydroxybutane amide in the quaternary amination reaction is not particularly limited, but is usually about 0.1 to 50%, more preferably about 1 to 20 ° / 0. .
- the amount of trialkylamine used is not particularly limited, but is usually 1.0 to 20.0 equivalents (mole) based on 4 1-halo 3-hydroxybutanamide, and 1.1 to About 8.0 equivalent is more preferable.
- the reaction temperature is not particularly limited, but is usually about 10 ° C to 60 ° C, more preferably about 0 ° C to 50 ° C.
- the quaternary amination reaction it may be colored brown.
- a decolorization operation with activated carbon or the like may be performed.
- the 4-trialkylamino 1-3-hydroxybutane amide halide represented by the above formula (2 ′) synthesized as described above is subjected to conventional methods such as extraction, column separation, recrystallization, electrodialysis, and ion exchange. It can be isolated and purified.
- the optically active form of 4-halo-3-hydroxybutyronitrile or 4-nobutaguchi 3-hydroxybutaneamide is used as a precursor or substrate for quaternary amination
- the above formula (2 ') The 4-trialkylamino-3-hydroxybutane amide halide represented by) is also an optically active substance.
- the 4-trialkyl represented by the above formula (2 ') The optical purity can be improved by performing a recrystallization operation of the killamino-3-hydroxybutane amide halide.
- the quaternary amination reaction consists mainly of a reaction in which a dehydrochlorination reaction occurs from a halohydrin compound to produce an epoxy compound, followed by a nucleophilic attack of trialkylamine to produce a quaternary amination product.
- the isomerization reaction to the acid derivative is a side reaction.
- the susceptibility of this crotonic acid derivative to the isomerization reaction determines the yield of the quaternary amination product, and it is the electron withdrawing property of the functional group that determines the ease of the isomerization. It is expected to be. Amides have lower electron-withdrawing properties than functional groups such as nitrinole and esters, and isomerization to crotonic acid derivatives is unlikely to occur. For this reason, the ratio of the isomerization reaction from the epoxy compound to the cutotonic acid derivative decreases, and the ratio of nucleophilic attack of the trialkylamine increases accordingly, resulting in a high yield of quaternary amination. It is assumed.
- the method of the hydrolysis reaction is not particularly limited, and an acid catalyst, Various known methods using a base catalyst can be used.
- Japanese Patent Publication No. Sho 4 3-2 6 8 4 9 Japanese Patent Application Laid-Open No. Sho 5 5-1 3 2 9 9 has a hydrolysis reaction using oxalic acid
- Japanese Patent Publication No. Sho 4 3-2 6 8 5 0 has A method of hydrolysis using n-butyl nitrite, glacial acetic acid and hydrochloric acid gas
- alkali metal hydroxides are alkaline earths from carnitine nitrile chloride.
- a method of hydrolysis to nitrile ⁇ amide ⁇ carboxylic acid using a base catalyst such as metal hydroxide is shown.
- JP-A-4-3 2 0 6 7 9 discloses a method using an enzyme that converts rutile amide to L-carnitine. It is limited to any method of acid catalyst, base catalyst and enzyme catalyst. Is not to be done.
- the main by-product in the hydrolysis reaction of 4-trialkylaminos 3-hydroxybutane amides is crotonobetaine, a dehydrated form.
- the basic substances used are alkali metal hydroxides, alkaline earth metal hydroxides, alkaline earth metal carbonates or bicarbonates, tertiary amines, quaternary ammonium hydroxys. de, include such basic anion exchange resin, in particular NaOH, KOH, C a (OH ) 2, Na 2 C_ ⁇ 3, K 2 C0 3, triethyl Chiruamin, NH 4 OH, an anion exchange resin IRA-400. These may be used alone or in combination of two or more. In particular, Na OH and KOH are preferable because the reaction can proceed efficiently even at room temperature and there are few by-products of crotonobetaine.
- the amount of acid and base used may be added so as to be equimolar or more with respect to the 4-monoalkylamino-3-hydroxybutanamide halide represented by the above formula (2 ′). 1.1-5. About 0 equivalent is more preferable.
- the concentration of the 4-trialkylamino-3-hydroxybutane amide halide represented by the above formula (2 ′) is not particularly limited, but is usually about 1 to 50%, more preferably about 5 to 30%.
- the reaction temperature is not particularly limited, but usually 5 to 100 ° C is preferable. When an acid catalyst is used, the production of crotonobetaine is extremely small, so 60 to 100 ° C. is particularly preferable from the viewpoint of reaction time.
- aqueous solvent is preferable and it is especially preferable to use water alone as a solvent from a viewpoint of reaction rate and side reaction suppression. Further, when the quaternary amino reaction is carried out with an aqueous solvent, it is more preferred to carry out the hydrolysis reaction with an aqueous solvent as it is.
- the hydrolysis reaction may be colored pale brown. It may also be colored after neutralization after completion of the hydrolysis reaction. For this decolorization, you can also perform a decoloring operation with activated charcoal.
- Betaine obtained as described above can be isolated and purified according to conventional methods such as extraction, column separation, recrystallization, electrodialysis, and ion exchange.
- Halo 3-hydroxybutane amide, 4 trialkynoleamino 1-hydroxybutanamide halide, and betaine can be used to improve optical purity by recrystallization operation, etc. Although it is possible, by-products are generated in each reaction step, and it is more preferable to perform the optical purity improving operation collectively by recrystallization operation that also serves as a purification after producing betaine.
- an optically active epihalohydrin, 4-halo-3-hydroxybutyronitrile risole, 4-halo-3-hydroxybutane amide, 4-trianoloxylamino 3-hydroxybutane amide halide are produced.
- betaine the corresponding optically active bein can be produced without degrading the optical purity. That is, the present invention is useful as a method for producing optically active betaine, particularly L-carcin.
- DC P 1,3-dichloro-2-propanol
- ECH Epoxychlorohydrin
- CHBN 4-chloro-3-hydroxybutyronitrile
- CHBA 4-chloro-3-hydroxybutanamide
- HCAm 4-hydroxycrotonamide
- C a r Carnitine-tolyl chloride
- C a r Car-tin
- C a r Carnitine
- CB Crotonobetaine
- HP LC system used in this analysis is as follows.
- Kahum--Bun temperature 40 ° C
- optical purity of L-carnitine is expressed by the following formula from the ARE A of D, L-caretine.
- a closed glass flask equipped with a stirrer, dropping funnel, thermometer and pH meter was charged with 262.3 g of epichlorohydrin and 700 g of water under a nitrogen atmosphere, and reacted at 40 ° C under stirring. While controlling the pH of the solution to be 8, adjust 32.6 wt% sodium cyanate aqueous solution 432.3 g (2.7 mol) and 65 wt% sulfuric acid aqueous solution 203.7 g (1.35 mol 1) for 3 hours. It was dripped at the same time. After the dropwise addition, the mixture was reacted at 40 ° C for 5 hours with stirring. Next, with the glass flask sealed, ethyl acetate 25 Om 1 was added and extraction was performed at 40 ° C.
- the contents were transferred to a separatory funnel to obtain an organic layer.
- the obtained organic layer was concentrated under reduced pressure at 50 ° C., and further distilled under reduced pressure to obtain 190.6 g of CHBN.
- the actual yield of the obtained CHBN was 58.1%.
- Escherichia coli with halohydrin epoxidase activity JM109 / p ST 1 1 1 (F ERM ABP-10922, see Japanese Patent Laid-Open No. 5-31 7066) is added to LB medium (1% bacto tryptone, 0.5% Bacterial toast extract, 0.5% NaCl, 1 mM I PTG, 50 ⁇ g / m1 ampicillin) in 50 OmL Erlenmeyer flasks each containing 1 O OmL, inoculated into 20 each, 37 ° Cultured with shaking at C for 20 hours.
- LB medium 1% bacto tryptone, 0.5% Bacterial toast extract, 0.5% NaCl, 1 mM I PTG, 50 ⁇ g / m1 ampicillin
- PSTll is a BamH I— P st I 1.1 Kb fragment containing the halohydrin epoxidase gene (hhe B) of Corynebacterium sp. It is a joined plasmid.
- P ST 1 1 1 is described in Japanese Patent Application Laid-Open No. 5-3 17066.
- JM109 / p ST lll is a receipt number F ERM ABP-10922. It has been deposited internationally on March 1, 1991 at the Deposit Center (Tsukuba Sakai Higashi Ibaraki Pref. 1 1 1 1 Central 6).
- a cell suspension 20.O 2 Og having halohydrin epoxidase activity prepared by the method i) was added, and the reaction was started at 20 ° C.
- the pH controller was set so that 30% NaOH was added, and 1,3-dichloro- 2 at a ratio of approximately equimolar to the input NaOH.
- the concentration of 1,3-dichloro-2-propanol in the system should not exceed 0.5mo 1 / kg, and the cyanide ion concentration in the system should be reduced. 1. Not to exceed 1 mo 1 / kg.
- the nitrile hydratase used in the present invention was prepared as follows.
- Mouth dococcus with nitrile hydracuse activity (Rhodococcus rhodochrous) J 1 1 (FEEM BP-1478), 30 L jar fermenter (Takasugi 2 wt% glucose at Seisakusho) urea 1 wt%, peptone 0.5% by weight, including yeast extract 0.3 mass 0/0, salts Ihikobaruto 0.05 mass 0/0, 20 medium (H7 0) and aerobically cultured at a temperature of 30 ° C for 60 hours. Bacteria obtained by culturing by the above method are collected by centrifugation, washed twice with 5 OmM phosphate buffer (pH 7.7) in the same amount, suspended, and used for reaction. It was.
- 5 OmM phosphate buffer pH 7.7
- CHBN 20 mM phosphate buffer
- Example 1 2 reaction end solution 1 6. 37 g (containing 2.59 g of Car amide) added 4 8% NaOHa q 2.2 g (26.4 mmo l) at 30 ° C After 8 hours, when the conversion rate of Car-amide reached 80%, the temperature was raised to 40 ° C and the reaction was continued for another 6 hours. After neutralizing the system with HC 1 aq and quantifying (L) -Car and CB in the reaction solution, (L) —C ar was 2.09 g ( Yield 98.5%), and the same 8 was found to be 0.015 g (vs. Car O. 7%) A part of this aqueous solution was taken and dried, When optical purity was measured, it was 94.2% e. E.
- Example 1 2 reaction finished solution 1 6.37 g (containing 2.59 g of Car-amide) 3 4.01 g (39.6 mmol) of 6% HC laq was added, and the reaction was started at 80 ° C. After 8 hours, the conversion rate of Car-amide reached 100%, and the reaction was completed. After neutralizing the system with NaOH aq, the amount of (L) -Car and CB in the reaction mixture was quantified. (L) -Car 2.11 g (yield 99.4%), CB 0 001 g rate 0. 05%). A part of this aqueous solution was collected and dried, and the optical purity of Car was measured. As a result, it was 94.5% e. E. Industrial applicability
- a method for producing betaine is provided.
- the present invention is useful in that side reactions can be prevented much more than before, and betaine such as carnitine can be obtained in high yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Description


































Claims








Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07831967A EP2096103A4 (en) | 2006-11-09 | 2007-11-09 | PROCESS FOR PREPARING A BETAIN |
| US12/514,000 US8334132B2 (en) | 2006-11-09 | 2007-11-09 | Process for production of a betaine such as carnitine |
| JP2007555405A JP5214249B2 (ja) | 2006-11-09 | 2007-11-09 | ベタインの製造方法 |
| CN200780041539XA CN101573326B (zh) | 2006-11-09 | 2007-11-09 | 甜菜碱的制造方法 |
| KR1020097011837A KR101097006B1 (ko) | 2006-11-09 | 2007-11-09 | 베타인의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-303972 | 2006-11-09 | ||
| JP2006303972 | 2006-11-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008056827A1 true WO2008056827A1 (fr) | 2008-05-15 |
Family
ID=39364629
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/072237 Ceased WO2008056827A1 (fr) | 2006-11-09 | 2007-11-09 | Procédé pour la production d'une bétaïne |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8334132B2 (ja) |
| EP (1) | EP2096103A4 (ja) |
| JP (1) | JP5214249B2 (ja) |
| KR (1) | KR101097006B1 (ja) |
| CN (1) | CN101573326B (ja) |
| WO (1) | WO2008056827A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008133198A (ja) * | 2006-11-27 | 2008-06-12 | Mitsubishi Rayon Co Ltd | L−カルニチンの製造方法 |
| JP2010143857A (ja) * | 2008-12-18 | 2010-07-01 | Mitsubishi Rayon Co Ltd | カルニチンの精製方法 |
| JP2011503132A (ja) * | 2007-11-16 | 2011-01-27 | ロンザ リミテッド | ベタインの製造方法 |
| CN119345743A (zh) * | 2024-12-23 | 2025-01-24 | 山东瑞弘生物科技股份有限公司 | 一种甜菜碱磷酸盐提纯装置和提纯方法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104768919B (zh) | 2012-11-16 | 2018-05-18 | 隆萨有限公司 | 使包含甜菜碱的组合物脱色的方法 |
| CN103012177B (zh) * | 2012-12-17 | 2015-04-08 | 苏州浩波科技股份有限公司 | 左旋肉碱的制备方法 |
Citations (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4911828A (ja) * | 1972-05-11 | 1974-02-01 | ||
| JPS5476830A (en) | 1977-11-03 | 1979-06-19 | Sigma Tau Ind Farmaceuti | Cardial disease treating agent |
| JPS54113409A (en) | 1978-02-03 | 1979-09-05 | Sigma Tau Ind Farmaceuti | Treating agent for hyperlipoid disease |
| JPS5513299A (en) | 1978-07-10 | 1980-01-30 | Cavazza Claudio | Manufacture of camphoric acid carnitine amide |
| JPS57165352A (en) | 1981-03-18 | 1982-10-12 | Anic Spa | Manufacture of l-carnitine |
| JPS5888312A (ja) | 1981-11-06 | 1983-05-26 | シグマ―タウ・インダストリエ・ファルマシゥティシェ・リウニテ・ソシエタ・ペル・アチオーニ | カルニチンまたは低級アシルカルニチンからなる静脈疾患治療剤 |
| JPS60258487A (ja) | 1984-05-26 | 1985-12-20 | バスフ アクチェン ゲゼルシャフト | カルニチン又はカルニチンアミドの水溶液の製法 |
| JPS62272983A (ja) | 1986-03-14 | 1987-11-27 | イステイトウト・グイド・ドネガニ・ソチエタ・ペル・アツイオニ | 3,4−エポキシ酪酸エステルから出発するl−(−)−カルニチンクロライドの製造方法 |
| JPH01287065A (ja) | 1988-05-13 | 1989-11-17 | Kanegafuchi Chem Ind Co Ltd | カルニチンの製造方法 |
| JPH0227995A (ja) | 1988-07-15 | 1990-01-30 | Earth Chem Corp Ltd | l‐カルニチンクロライドの製造法 |
| JPH02142758A (ja) | 1988-11-24 | 1990-05-31 | Takasago Internatl Corp | カルニチンメチルエステルハライドの製造方法 |
| JPH0314171B2 (ja) | 1982-04-13 | 1991-02-26 | Mitsubishi Electric Corp | |
| JPH04211379A (ja) | 1990-02-28 | 1992-08-03 | Teruhiko Beppu | ニトリルヒドラターゼ活性を有するポリペプチドをコードする遺伝子dna、これを含有する形質転換体 |
| JPH04278089A (ja) * | 1991-03-04 | 1992-10-02 | Nitto Chem Ind Co Ltd | ハロヒドリンエポキシダ−ゼ遺伝子を有する組換え体プラ スミドおよび該プラスミドにより形質転換された微生物 |
| JPH04320679A (ja) | 1991-02-28 | 1992-11-11 | Degussa Ag | L‐カルニチン‐アミダーゼを生産する微生物、微生物学的に生産されたl‐カルニチン‐アミダーゼ、その取得方法およびl‐カルニチンへのdl‐および/またはカルニチンアミドの酵素的変換方法 |
| JPH04365491A (ja) * | 1991-06-06 | 1992-12-17 | Nitto Chem Ind Co Ltd | 4−ハロ−3−ヒドロキシブチルアミドの製造法 |
| JPH05219965A (ja) | 1992-02-10 | 1993-08-31 | Nitto Chem Ind Co Ltd | 光学活性ハロヒドリンの製造法 |
| JPH05317066A (ja) | 1991-03-04 | 1993-12-03 | Hideaki Yamada | ハロヒドリンエポキシダ−ゼ遺伝子を有する組換え体プラ スミドにより形質転換された微生物による3−ヒドロキシ ニトリル化合物の製造法 |
| JPH08266277A (ja) | 1995-03-29 | 1996-10-15 | Mitsui Toatsu Chem Inc | 形質転換体を用いたアミド化合物の製造法 |
| US5807730A (en) | 1996-02-14 | 1998-09-15 | Mitsui Chemicals, Inc. | Nitrile hydratase |
| JP2002241357A (ja) | 2001-02-19 | 2002-08-28 | Mitsubishi Rayon Co Ltd | 4−クロロ−3−ヒドロキシブチロニトリルの製造方法 |
| JP2002529528A (ja) | 1998-11-16 | 2002-09-10 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | L−カルニチンの製造のための工業的方法 |
| JP2002544252A (ja) | 1999-05-18 | 2002-12-24 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | S−(−)−クロロコハク酸またはその誘導体からr−(−)−カルニチンを調製するための方法 |
| JP2004182607A (ja) | 2002-11-29 | 2004-07-02 | Mitsubishi Rayon Co Ltd | 4−クロロ−3−ヒドロキシブチロニトリルの製造方法 |
| JP2006303972A (ja) | 2005-04-21 | 2006-11-02 | Toshiba Digital Media Engineering Corp | 課金システム |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE659194A (ja) * | 1965-02-03 | 1965-08-03 | ||
| JPS5920379B2 (ja) | 1976-07-23 | 1984-05-12 | 三菱重工業株式会社 | 粉体の充填方法 |
| IE51711B1 (en) | 1977-11-03 | 1987-03-04 | Sigma Tau Ind Farmaceuti | Carnitine derivatives |
| JP2840722B2 (ja) * | 1989-07-20 | 1998-12-24 | 日東化学工業株式会社 | 4‐ハロ‐3‐ヒドロキシブチロニトリルの製造法 |
-
2007
- 2007-11-09 KR KR1020097011837A patent/KR101097006B1/ko not_active Expired - Fee Related
- 2007-11-09 JP JP2007555405A patent/JP5214249B2/ja not_active Expired - Fee Related
- 2007-11-09 EP EP07831967A patent/EP2096103A4/en not_active Withdrawn
- 2007-11-09 US US12/514,000 patent/US8334132B2/en not_active Expired - Fee Related
- 2007-11-09 WO PCT/JP2007/072237 patent/WO2008056827A1/ja not_active Ceased
- 2007-11-09 CN CN200780041539XA patent/CN101573326B/zh not_active Expired - Fee Related
Patent Citations (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4911828A (ja) * | 1972-05-11 | 1974-02-01 | ||
| JPS5313611B2 (ja) | 1972-05-11 | 1978-05-11 | ||
| JPS5476830A (en) | 1977-11-03 | 1979-06-19 | Sigma Tau Ind Farmaceuti | Cardial disease treating agent |
| JPS54113409A (en) | 1978-02-03 | 1979-09-05 | Sigma Tau Ind Farmaceuti | Treating agent for hyperlipoid disease |
| JPS5513299A (en) | 1978-07-10 | 1980-01-30 | Cavazza Claudio | Manufacture of camphoric acid carnitine amide |
| JPS57165352A (en) | 1981-03-18 | 1982-10-12 | Anic Spa | Manufacture of l-carnitine |
| JPS5888312A (ja) | 1981-11-06 | 1983-05-26 | シグマ―タウ・インダストリエ・ファルマシゥティシェ・リウニテ・ソシエタ・ペル・アチオーニ | カルニチンまたは低級アシルカルニチンからなる静脈疾患治療剤 |
| JPH0314171B2 (ja) | 1982-04-13 | 1991-02-26 | Mitsubishi Electric Corp | |
| JPS60258487A (ja) | 1984-05-26 | 1985-12-20 | バスフ アクチェン ゲゼルシャフト | カルニチン又はカルニチンアミドの水溶液の製法 |
| JPS62272983A (ja) | 1986-03-14 | 1987-11-27 | イステイトウト・グイド・ドネガニ・ソチエタ・ペル・アツイオニ | 3,4−エポキシ酪酸エステルから出発するl−(−)−カルニチンクロライドの製造方法 |
| JPH01287065A (ja) | 1988-05-13 | 1989-11-17 | Kanegafuchi Chem Ind Co Ltd | カルニチンの製造方法 |
| JPH0227995A (ja) | 1988-07-15 | 1990-01-30 | Earth Chem Corp Ltd | l‐カルニチンクロライドの製造法 |
| JPH02142758A (ja) | 1988-11-24 | 1990-05-31 | Takasago Internatl Corp | カルニチンメチルエステルハライドの製造方法 |
| JPH04211379A (ja) | 1990-02-28 | 1992-08-03 | Teruhiko Beppu | ニトリルヒドラターゼ活性を有するポリペプチドをコードする遺伝子dna、これを含有する形質転換体 |
| JPH04320679A (ja) | 1991-02-28 | 1992-11-11 | Degussa Ag | L‐カルニチン‐アミダーゼを生産する微生物、微生物学的に生産されたl‐カルニチン‐アミダーゼ、その取得方法およびl‐カルニチンへのdl‐および/またはカルニチンアミドの酵素的変換方法 |
| JPH05317066A (ja) | 1991-03-04 | 1993-12-03 | Hideaki Yamada | ハロヒドリンエポキシダ−ゼ遺伝子を有する組換え体プラ スミドにより形質転換された微生物による3−ヒドロキシ ニトリル化合物の製造法 |
| JPH04278089A (ja) * | 1991-03-04 | 1992-10-02 | Nitto Chem Ind Co Ltd | ハロヒドリンエポキシダ−ゼ遺伝子を有する組換え体プラ スミドおよび該プラスミドにより形質転換された微生物 |
| JPH04365491A (ja) * | 1991-06-06 | 1992-12-17 | Nitto Chem Ind Co Ltd | 4−ハロ−3−ヒドロキシブチルアミドの製造法 |
| JPH05219965A (ja) | 1992-02-10 | 1993-08-31 | Nitto Chem Ind Co Ltd | 光学活性ハロヒドリンの製造法 |
| JPH08266277A (ja) | 1995-03-29 | 1996-10-15 | Mitsui Toatsu Chem Inc | 形質転換体を用いたアミド化合物の製造法 |
| US5807730A (en) | 1996-02-14 | 1998-09-15 | Mitsui Chemicals, Inc. | Nitrile hydratase |
| JP2002529528A (ja) | 1998-11-16 | 2002-09-10 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | L−カルニチンの製造のための工業的方法 |
| JP2002544252A (ja) | 1999-05-18 | 2002-12-24 | シグマ−タウ・インドゥストリエ・ファルマチェウチケ・リウニテ・ソシエタ・ペル・アチオニ | S−(−)−クロロコハク酸またはその誘導体からr−(−)−カルニチンを調製するための方法 |
| JP2002241357A (ja) | 2001-02-19 | 2002-08-28 | Mitsubishi Rayon Co Ltd | 4−クロロ−3−ヒドロキシブチロニトリルの製造方法 |
| JP2004182607A (ja) | 2002-11-29 | 2004-07-02 | Mitsubishi Rayon Co Ltd | 4−クロロ−3−ヒドロキシブチロニトリルの製造方法 |
| JP2006303972A (ja) | 2005-04-21 | 2006-11-02 | Toshiba Digital Media Engineering Corp | 課金システム |
Non-Patent Citations (4)
| Title |
|---|
| "Microorganisms with IFO number are listed in the List of Cultures", vol. 1, 1988, INSTITUTE FOR FENNENTATION |
| HETEROCYCLES, vol. 53, no. 1, 2000 |
| J. PHARM. BIO. ANAL., vol. 14, 1996, pages 1579 - 1584 |
| See also references of EP2096103A4 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008133198A (ja) * | 2006-11-27 | 2008-06-12 | Mitsubishi Rayon Co Ltd | L−カルニチンの製造方法 |
| JP2011503132A (ja) * | 2007-11-16 | 2011-01-27 | ロンザ リミテッド | ベタインの製造方法 |
| JP2010143857A (ja) * | 2008-12-18 | 2010-07-01 | Mitsubishi Rayon Co Ltd | カルニチンの精製方法 |
| CN119345743A (zh) * | 2024-12-23 | 2025-01-24 | 山东瑞弘生物科技股份有限公司 | 一种甜菜碱磷酸盐提纯装置和提纯方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2096103A1 (en) | 2009-09-02 |
| KR101097006B1 (ko) | 2011-12-20 |
| US20090325246A1 (en) | 2009-12-31 |
| CN101573326B (zh) | 2013-05-01 |
| EP2096103A4 (en) | 2012-11-21 |
| JPWO2008056827A1 (ja) | 2010-02-25 |
| US8334132B2 (en) | 2012-12-18 |
| CN101573326A (zh) | 2009-11-04 |
| JP5214249B2 (ja) | 2013-06-19 |
| KR20090090329A (ko) | 2009-08-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI304095B (en) | Stereoselective bioconversion of aliphatic dinitriles into cyano carboxylic acids | |
| WO2008056827A1 (fr) | Procédé pour la production d'une bétaïne | |
| WO2013127354A1 (zh) | 一种合成左旋吡喹酮的方法 | |
| CN113234698B (zh) | 一种氰基还原酶和加巴喷丁的制备方法 | |
| CN101701222A (zh) | 一种编码产碱杆菌腈水解酶的基因及其用于制备扁桃酸单一对映体的方法 | |
| WO2023184791A1 (zh) | 一种酶法合成布瓦西坦手性中间体的方法 | |
| US6781009B2 (en) | Method for producing 4-cyano-3-oxobutanoate and 4-ctano-3-hydroxybutanoate | |
| CN113388600A (zh) | 一种醛肟脱水酶及其在催化合成芳香腈类化合物中的应用 | |
| JP5001725B2 (ja) | 4−ハロ−3−ヒドロキシブチロニトリルの製造方法 | |
| JP2008133198A (ja) | L−カルニチンの製造方法 | |
| WO2007097336A1 (ja) | (2r,3r)および(2s,3s)-3-フェニルイソセリン誘導体の製造法 | |
| JP2008067626A (ja) | 4−ハロ−3−ヒドロキシブチルアミドの製造方法 | |
| JP4608216B2 (ja) | ラクトン類の製造方法 | |
| KR100598475B1 (ko) | 아미도 그룹 함유 구아니딘 유도체 및 이의 염의 제조방법 | |
| JP2011072303A (ja) | 4−ハロ−3−ヒドロキシブチルアミドの製造方法 | |
| JP2011045361A (ja) | 4−ハロ−3−ヒドロキシブチルアミドの製造方法 | |
| JP5001631B2 (ja) | 4−ハロ−3−ヒドロキシブチロニトリルの工業的製造方法 | |
| CN121628937A (zh) | 一种高产d-泛酸的基因工程菌及其构建方法、应用 | |
| JP5165393B2 (ja) | グリコール酸又はグリコール酸アンモニウムの製造方法 | |
| EP0339618A2 (en) | Method for preparing optically active 3,4-dihydroxy butyric acid derivatives | |
| JPWO2006101266A1 (ja) | 光学活性なヒドロキシメチル置換フェニルアラニンの製造方法 | |
| JP2008148637A (ja) | 4−ハロ−3−ヒドロキシブチルアミドの製造方法 | |
| JP2003040853A (ja) | アミド基含有グアニジン誘導体およびその塩の製造方法 | |
| WO2000065079A1 (en) | Processes for producing s,s-2-hydroxypropylenediamine-n,n'-disuccinic acid | |
| US20080241896A1 (en) | Process for production of optically active hydroxymethyl-substituted phenylalanine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780041539.X Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007555405 Country of ref document: JP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07831967 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007831967 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3216/CHENP/2009 Country of ref document: IN Ref document number: 1020097011837 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12514000 Country of ref document: US |