明 細 書
ピぺリジン誘導体またはその塩
技術分野
[0001] 本発明は、医薬、特に副甲状腺機能亢進症等のカルシウム感知受容体 (Calcium s ensing rec印 tor; CaSR)の関与する疾患の治療剤として有用な新規なピぺリジン誘導 体に関する。
背景技術
[0002] 細胞外 Ca2+濃度は生命の維持をはじめとする種々の生体機能に非常に重要な役 割を果たしている。そのため、血中 Ca2+濃度は多くの調節機構により非常に狭い範囲 に厳密に制御されている。
副甲状腺ホルモン (Parathyroid hormone; PTH)は副甲状腺で産生 ·分泌されるポリ ペプチドホルモンであり、主に血中 Ca2+濃度を調節している。この PTHは骨吸収を促 進し、腎臓においてカルシウムの再吸収を促進することなどにより血中 Ca2+濃度を上 昇させる。血中 Ca2+濃度の上昇は PTHの分泌を抑制し、逆に Ca2+濃度の低下は PTH の分泌を促進させることから、言わばネガティブ ·フィ一ドバック機構により血中 Ca2+濃 度が制御されて!/、ると考えられる。
[0003] PTHの過剰分泌が持続的に生ずる副甲状腺機能亢進症には、副甲状腺自体の腺 腫 ·過形成 ·癌等によると考えられる原発性副甲状腺機能亢進症と腎機能低下等に よる二次性副甲状腺機能亢進症がある。
腎不全患者の多くが二次性副甲状腺機能亢進症を併発していることが報告されて いる。二次性副甲状腺機能亢進症は異所性石灰化などを含む腎性骨異栄養症の原 因疾患のひとつとなっており、骨折 ·骨痛などによる腎不全患者の QOL低下や心血 管系の石灰化に起因すると考えられる心血管系疾患による腎不全患者の死亡の要 因となってレ、ると考えられて!/、る。このため二次性副甲状腺機能亢進症は臨床でも 大きな問題となっている。
腎不全による二次性副甲状腺機能亢進症では、腎でのリン排泄能の低下や活性 型ビタミン D減少により血中 Ca2+濃度が低下することが引き金となって、 PTHの過剰分
泌が生じている。この PTHの過剰分泌は、さらなる腎機能の低下や副甲状腺自体の 過形成、 PTH標的臓器の抵抗性などによって持続 '増悪していると考えられている。
[0004] 副甲状腺機能亢進症の内科的治療法としては、現在ビタミン D補充療法が中心に 行われている。しかし、ビタミン D製剤は血中 Ca2+濃度を上昇させることから投与制限 があり、十分な治療ができている状態ではない。以上のことから、血中 Ca2+濃度が上 昇しな!/、有効性の高レ、副甲状腺機能亢進症治療薬の開発が望まれてレ、た。
カルシウム感知受容体 (Calcium sensing rec印 tor ; CaSR)は、当初ゥシ副甲状腺で 細胞外 Ca2+を感知する Gタンパク共役 7回膜貫通型受容体(G-protein coupled recep tor ; GPCR)としてクローニングされた(非特許文献 1)。 CaSRは細胞外の Ca2+濃度を感 知して細胞内の Ca2+濃度を変化させ、それによつて PTHに代表される Ca2+代謝調節 に関係する分子の産生を調節する機能を有する。このことを裏付ける事実として、ヒト CaSRの活性化変異や不活性化変異が家族性の高カルシウム血症や低カルシウム血 症の原因となっていることが多数報告されている。また、原発性、二次性いずれの副 甲状腺機能亢進症においても、 Ca2+に対する副甲状腺の感受性の低下が認められ ている。
[0005] CaSRの作動的な調節薬は、副甲状腺の CaSRに直接作用して Ca2+感受性を上げる ことにより、血中 Ca2+濃度の上昇なしに PTHを低下させると考えられる。最近、 CaSRの 作動的な調節薬であるシナカルセト(Cinacalcet)が、副甲状腺の CaSRに直接作用し て CaSRの Ca2+感受性を上げることにより、 PTH分泌を抑制する作用を有することが報 告された(非特許文献 2及び 3)。シナカルセトは、既存の療法であったビタミン D製剤 や高リン血症治療の目的で使用されてきた Ca2+含有リン吸着剤などとの併用が可能 な、新し!/ヽ副甲状腺機能亢進症治療薬として期待されて!ヽる。
[0006] しかしながら、シナカルセトはシトクローム p450 (Cytochrome p450; CYP)のサブタイ プのひとつである CYP2D6の強力な阻害活性を有することが報告されて!/、る。この CY P2D6は臨床で使用される種々の薬剤の代謝にも重要な役割を担っている。シナカル セトは CYP2D6を阻害することから、 CYP2D6で代謝される薬剤の代謝を遅らせること で体内動態を変化させ、薬剤相互作用 (DDI)を起こすことが懸念されている(非特許 文献 4)。以上のことから、 CYP2D6阻害のない強力な CaSR調節薬の開発が求められ
ている。
[0007] CaSRの mRNAは PTHの主分泌組織である副甲状腺に加え、腎臓 '副甲状腺をはじ めとした種々の組織に発現しており、種々の生理的役割を担っているものと考えられ ている。
CaSRを作動的もしくは拮抗的に調節する薬剤 (CaSR調節薬)は、上述の副甲状腺 機能亢進症に加え骨疾患 ·上部下部消化器疾患 (非特許文献 5及び 6) ·糖尿病 (非 特許文献 7及び 8) ·下垂体前葉機能低下/亢進症 (非特許文献 9)などを含む種々の 疾患の治療薬となることが期待されてレ、る。
[0008] CaSR調節薬としては以下の特許文献 1乃至 3の報告がある。
特許文献 1では広範な化合物を含む下記式 (A)及び式 (B)で示される化合物が開 示されている。し力、しながら、本発明化合物の具体的な開示はない。
[化 1]
(A) (B)
(式中、 Ar、 R、及び Rは以下の意味を示す。
3
Ar :疎水性物質。
R:水素、メチル、ェチル、プロピル、イソプロピル、ブチル、イソブチル、シクロペンチ ノレ、シクロへキシル、シクロへプチル、シクロォクチノレ、インデュル、インダニル、また は 2-、 3-もしくは 4ーピペリジル。
R:置換されていることもある、 5〜6の環構成原子を有する、単環式または双環式ァ
3
リールまたはシクロアルキル。
他の記号は該公報参照。 )
[0009] 特許文献 2では、下記式 (C)で示される化合物が開示されている。しかしながら、式
(C)で示される化合物は、含窒素環にアミノ基が直結しており、また、本発明化合物 の R2に相当する部分がない。
[化 2]
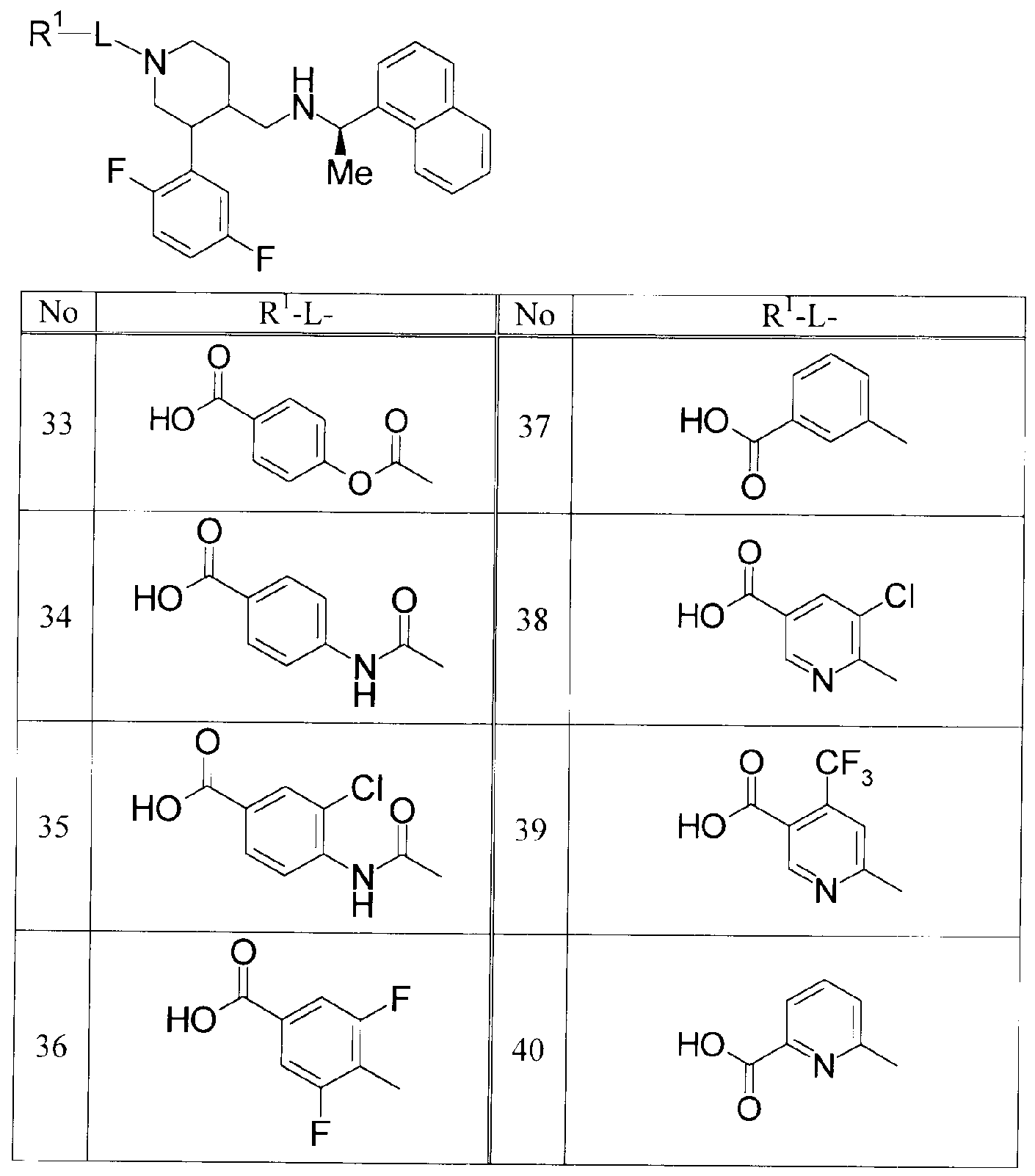
(式中の記号は該公報参照。 )
本出願人により出願され本願の優先日後に公開された特許文献 3では、下記式 (D )で示されるピロリジン誘導体が開示されている。し力もながら、 A及び Bを含む含窒素 ヘテロ環がピロリジン環に限定されて!/、る。
[化 3]
(式中の A及び Bは、 _C(R7)(R7a)_または- C(0)_を示す。他の記号は該公報参照。 ) ) また、ピぺリジン誘導体としては以下の特許文献 4及び 5が報告されている。
特許文献 4では、下記式 (E)で示されるピぺリジン誘導体が脳細胞中のカルシウム オーバーロード(overload)防止活性を有し、酸素欠乏症等の神経変性疾患に有効で あること力 S記載されている。し力もながら、化合物(E)は本発明化合物の R4に相当す る部分がない。また、 CaSR調節作用及び副甲状腺機能亢進症に対する有効性に関 する記載はない。
[化 4]
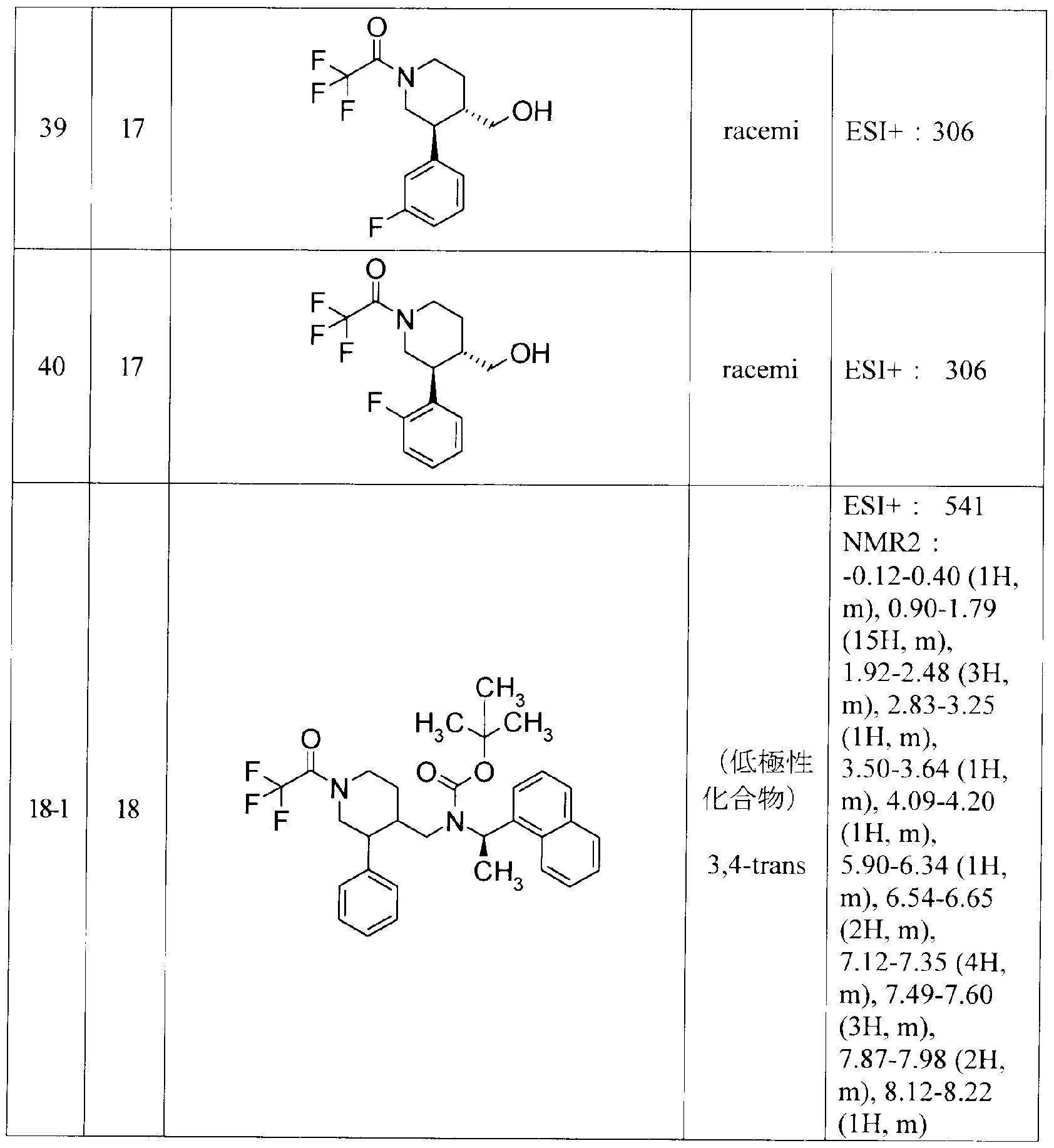
Y(CH2)nR3 (K)
(式中の Yは 0、 S又は NRを、 nは 0力、ら 4を、 R3は 3,4-メチレンジォキシフエニル、フエ ニル、ナフチル、又は 5若しくは 6員へテロ環基を意味する。他の記号は該公報参照。
)
特許文献 5では、下記式 (F)で示されるピぺリジン誘導体がタキキュン受容体拮抗 活性を有し、疼痛、炎症、アレルギー等に有効であることが記載されている。しかしな がら、化合物(F)はピペリジンの 4位の置換基がアミド又はエステルである。また、 CaS R調節作用及び副甲状腺機能亢進症に対する有効性に関する記載はなレ、。
[化 5]
(式中の Zは 0、又は N (R3)を意味する。他の記号は該公報参照。 )
非特許文献 1 :ブラウン (Brown)ら、「ネイチヤー (Nature)」、(英国)、 1993年、第 366巻
、 .575-580
非特許文献 2 :コーヘン (Cohen)ら、 「カレント 'オピニオン'イン'ファーマコロジー(Cur rent Opinion in Pharmacology)」、(オランダ)、 2002年、第 2巻、 ρ· 734-739
非特許文献 3 :ジョイ(Joy)ら、「ザ'アナルズ 'ォブ 'ファーマコセラピー (The Annals of Pharmacotherapy)」、(米国)、 2004年、第 38巻、 .1871-1880
非特許文献 4:「センシパー (登録商標)(シナカルセト '塩酸塩)錠 (SensiparTM (cinaca lcet HC1) Tablets) J , [online], 2004年、米国食品医薬局(FDA) [検索日、平成 17年 3 月 28日]、インターネット、 (URL:http:〃 www.fda.govAider/foi/label/^OCM/^ieSS— Sen sipar_lbl.pdf)。
非特許文献 5 :ジーニーンら (Jeannine)、「ザ'ジャーナル'ォブ 'クリニカル'インべステ ィゲーシヨン (The Journal of Clinical Investigation) J、 (米国)、 1997年、第 99巻、 .23 28-2333
非特許文献 6 :チェン (Cheng)ら、「ザ'アメリカン 'ジャーナル'ォブ 'フイジォロジ一一
ガストロインテスティナノレ'アンド ·リノ一 ·フィンォロン一 (The American Journal of Ph ysiology-Gastrointestinal and Liver Physiology)」、(米国)、 2002年、第 283巻、 .G24 0-G250
非特許文献 7:ブルース (Bruce)ら、「ザ ·ジャーナル ·ォブ 'バイオロジカル ·ケミストリ一 (The Journal of Biological Chemistry)」、(米国)、 1999年、第 274巻、 p.20561-20568 非特許文献 8 :ストローブ (Straub)ら、「ザ'ジャーナル'ォブ 'バイオロジカル 'ケミストリ 一 (The Journal of Biological Chemistry)」、(米国)、 2000年、第 275巻、 .18777-187 84
非特許文献 9 :エマニュエル (Emanuel)ら、 「モレキュラー 'エンドクライノロジー(Molecu lar Endocrinology)」、(米国)、 1996年、第 10巻、 .555-565
特許文献 1:国際公開第 94/18959号パンフレット
特許文献 2 :国際公開第 2005/115975号パンフレット
特許文献 3:国際公開第 2006/123725号パンフレット
特許文献 4 :国際公開第 03/101964号パンフレット
特許文献 5:国際公開第 2006/004195号パンフレット
発明の開示
発明が解決しょうとする課題
[0013] 本発明の課題は、新規な CaSR調節作用を有する医薬、特に副甲状腺機能亢進 症治療剤として有用な、新規な化合物の提供である。
課題を解決するための手段
[0014] 既存の CaSR調節薬は、有効性、安全性の面のいずれかの点で満足できるもので はないことから、有効性、安全性に優れた CaSR調節薬の提供が切望されている。こ のような状況下、本発明者等は、有効性、安全性に優れた CaSR調節薬の開発を目 的として鋭意研究した。その結果、 3及び 4位の一方に置換アミノメチル基、他方にァ リール又はへテロアリール基等を有する新規なピぺリジン誘導体が強力な CaSR作動 的調節作用を示すことを見出した。また、これらの新規なピぺリジン誘導体は薬物相 互作用を起こす懸念のある CYP2D6阻害作用との選択性が高いことも知見して本発 明を完成した。
即ち、本発明は、一般式 (I)で示されるピぺリジン誘導体またはその製薬学的に許 容される塩に関する。
[化 6]
[式中の記号は以下の意味を示す。
X及び Y:どちらか一方が- CH -であり、他方が単結合。
し単結合、 *_C(0)-、 *-OC(0)_又は *-N(R°)C(0)-。ただし、 *は R1への結合を示す。
R° : -H又は低級アルキル。
R1: -H又はそれぞれ置換されて!/、てもよ!/、C アルキル、低級アルケニル、シクロア
1-12
ルキル、シクロアルケニル、ァリール若しくはヘテロ環基。
R2: C アルキル、低級アルケニル、シクロアルキル、シクロアルケニル又はそれぞれ
1-12
置換されてレ、てもよ!/、ァリール若しくはへテロァリール。
R3:それぞれ置換されて!/、てもよ!/、ァリール又はへテロァリール。
R4 :低級アルキル。
以下同様。 ]
また、本発明は、一般式 (I)で示されるピぺリジン誘導体またはその製薬学的に許 容される塩 (以下、「式 (I)記載の化合物またはその製薬学的に許容される塩」、「化 合物 (1)」等と記すことがある。)と、製薬的に許容される担体とからなる医薬組成物、 殊に、カルシウム感知受容体調節薬、副甲状腺機能亢進症治療薬、腎性骨異栄養 症治療薬または高カルシウム血症治療薬である医薬組成物にも関する。
即ち、(1)式 (I)記載の化合物またはその製薬学的に許容される塩と、製薬学的に 許容される担体とからなる医薬組成物;
(2)カルシウム感知受容体調節薬である(1)記載の医薬組成物;
(3)副甲状腺機能亢進症治療薬である(1)記載の医薬組成物;
(4)腎性骨異栄養症治療薬である(1)記載の医薬組成物;
(5)高カルシウム血症治療薬である(1)記載の医薬組成物;
(6)カルシウム感知受容体調節薬、副甲状腺機能亢進症治療薬、腎性骨異栄養症 治療薬または高カルシウム血症治療薬の製造のための、式 (I)記載の化合物または その製薬学的に許容される塩の使用;及び、
(7)式 (I)記載の化合物またはその塩の治療有効量を患者に投与することを含む、 副甲状腺機能亢進症、腎性骨異栄養症または高カルシウム血症の治療方法、にも 関する。
発明の効果
[0017] 本発明化合物は、 CaSR受容体調節作用を有することから、副甲状腺機能亢進症 等の治療剤として有用である。
[0018] 以下、本発明を詳細に説明する。
本明細書中の定義において「アルキル」、 「ァルケ二ル」、 「アルキレン」及び「ァルケ 二レン」とは、特に断らない限り、直鎖又は分枝状の炭化水素鎖を意味する。
[0019] 「低級アルキル」として好ましくは、炭素数が 1から 6 (以後、 C と略す)のアルキル、
1-6
具体的には、メチル、ェチル、 n-プロピル、イソプロピル、 n-ブチル、イソブチル、 sec- ブチル、 tert-ブチル、 n-ペンチル、 n-へキシル基等である。より好ましくは C アルキ
1-4 ルであり、特に好ましくはメチル、ェチル、 n-プロピル、イソプロピルである。
[0020] 「低級アルケニル」として好ましくは、 C アルケニル、具体的には、ビュル、プロぺ
2-6
ニル、ブテュル、ペンテュル、 1-メチルビュル、 1-メチル -2-プロぺニル、 1,3-ブタジ ェニル、 1,3-ペンタジェニル基等である。より好ましくは C アルケニルであり、特に好
2-4
ましくはビュル、プロぺニルである。
[0021] 「低級アルキレン」として好ましくは、 C アルキレン、具体的には、メチレン、ェチレ
1-6
ン、トリメチレン、テトラメチレン、ペンタメチレン、へキサメチレン、プロピレン、メチノレメ チレン、ェチルエチレン、 1,2-ジメチルエチレン、 1, 1,2,2-テトラメチルエチレン基等で ある。より好ましくは C アルキレンであり、特に好ましくはメチレン、エチレン、トリメチ
2-4
レンである。
[0022] 「低級アルケニレン」として好ましくは、 C アルケニレン、具体的には、ビニレン、ェ
2-6
チリデン、プロぺニレン、ブテニレン、ペンテ二レン、へキセニレン、 1,3-ブタジェニレ ン、 1,3-ペンタジェ二レン基等である。より好ましくは C アルケニレンであり、特に好
ましくは、ビニレン、ェチリデン、プロぺニレンである。
[0023] 「ハロゲン」は、 F、 Cl、 Br、 Iを意味する。
「ハロゲノ低級アルキル」とは、 1個以上のハロゲンで置換された、 C アルキル、具
1-6
体的には、フルォロメチル、ジフルォロメチル、トリフルォロメチル、 2,2,2-トリフルォロ ェチル、ペンタフルォロェチル、へキサフルォロプロピル基等である。好ましくは 1〜 5個のハロゲンで置換された低級アルキルであり、より好ましくは、トリフルォロメチル である。
[0024] 「シクロアルキル」とは、 C の飽和炭化水素環基であり、架橋を有していてもよい。
3-10
具体的には、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシル、シクロ ヘプチル、シクロォクチル、ァダマンチル基等である。好ましくは C シクロアルキル
3-8
であり、さらに好ましくは C シクロアルキルであり、特に好ましくは、シクロプロピノレ、
3-6
シクロブチル、シクロペンチル、シクロへキシルである。
[0025] 「シクロアルケニル」は C シクロアルケニルであり、架橋を有していてもよぐ二重
3-15
結合部位でベンゼン環と縮合した環基を包含する。具体的には、シクロペンテュル、 シクロペンタジェニノレ、シクロへキセニノレ、シクロへキサジェニノレ、 1-テトラヒドロナフ チル、 1-インデュル、 9-フルォレニル、ノルボルネニル基等である。より好ましくは、 C
5 シクロアルケニルであり、特に好ましくは、シクロペンテュル及びシクロへキセニル
- 10
である。
[0026] 「ァリール」は C の単環乃至三環式芳香族炭化水素環基であり、 C シクロアルケ
6-14 5-8
ンとその二重結合部位で縮合した環基を包含する。具体的には、フエニル、ナフチル 、 5-テトラヒドロナフチル、 4-インデュル、 1-フルォレニル基等である。より好ましくは、 フエニル、ナフチルであり、さらに好ましくは、フエニルである。
[0027] 「ヘテロ環」基とは、 i) 0、 S及び Nから選択されるへテロ原子を 1〜4個含有する単環
3〜8員(好ましくは 5〜7員)ヘテロ環、 ii)当該単環へテロ環と、単環へテロ環、ベン ゼン環、 C シクロアルカン及び C シクロアルケンからなる群より選択される 1又は 2
5-8 5-8
個の環とが縮環し形成される、 0、 S及び Nから選択されるへテロ原子を 1〜5個含有 する二環式 8〜; 14員(好ましくは 9〜; 11員)ヘテロ環及び三環式;!;!〜 20員(好ましく は 12〜; 15員)ヘテロ環、からなる環基を意味する。環原子である S又は Nが酸化され
ォキシドゃジォキシドを形成してもよレ、。
「ヘテロ環」基として好ましくは、アジリジニル、ァゼチジル、ピロリジニル、ピペリジニ ノレ、ピぺラジュノレ、ホモピぺラジュノレ、ォキシラニノレ、ォキセタニノレ、テトラヒドロフラニ ノレ、テトラヒドロビラニノレ、 モノレホリニノレ、ホモモノレホリニノレ、ピロリノレ、 イミダゾリノレ、 トリ ァゾリル、テトラゾリル、ピリジノレ、ピリミジェノレ、ピラジュル、フリル、チェニル、ォキサ ゾリル、ォキサジァゾリル、チアゾリル、チアジアゾリル、インドリル、インドリジニル、ベ ンゾイミダゾリル、イミダゾ [l,2-a]ピリジニル、キノキサリニル、キノリル、イソキノリル、キ ナゾリノレ、 シンノニノレ、フタラジノレ、 ベンゾフラニノレ、 ベンゾチェニノレ、 ベンゾォキサゾ リル、ベンゾチアゾリル、カルバゾリル、キヌタリジニルであり、より好ましくは、ピロリジ ニル、ピペリジニル、ピぺラジュル、モルホリニル、ピロリル、テトラゾリル、ピリジノレ、ピ リミジ二ノレ、ピラジュル、フリル、チェニル、ォキサゾリル、チアゾリル、インドリル、キノリ ノレ、ベンゾフラニル、ベンゾチェニル、ベンゾォキサゾリル、ベンゾチアゾリルであり、 特〖こ好ましく ίま、ピロリジニノレ、ピペリジニノレ、ピロリノレ、テトラゾリノレ、ピリジノレ、フリノレ、 チェニル、チアゾリル、ベンゾチェニルである。
[0028] 「ヘテロァリール」は上記のへテロ環基のうち、芳香族性を有するヘテロ環基であり、 例えば、ピロリル、イミダゾリル、トリァゾリル、テトラゾリル、ピリジル、ピリミジェル、ビラ ジニル、フリル、チェニル、ォキサゾリル、ォキサジァゾリル、チアゾリル、チアジアゾリ ノレ、インドリル、インドリジニル、ベンゾイミダゾリル、イミダゾ [l,2-a]ピリジニル、キノキ サリニル、キノリノレ、イソキノリル、キナゾリル、シンノエル、フタラジル、ベンゾフラ二ノレ 、ベンゾチェニル、ベンゾォキサゾリル、ベンゾチアゾリル、カルバゾリルなどであり、 好ましくは、ピロリル、テトラゾリル、ピリジノレ、ピリミジェノレ、ピラジュル、フリル、チェ二 ノレ、ォキサゾリル、チアゾリル、インドリル、キノリノレ、ベンゾフラニル、ベンゾチェ二ノレ 、ベンゾォキサゾリル、ベンゾチアゾリルであり、特に好ましくは、ピロリル、テトラゾリル 、ピリジノレ、フリル、チェニル、チアゾリル、ベンゾチェニルである。
[0029] 「置換されていてもよい」とは、「無置換」あるいは「同一又は異なる置換基を 1〜5個 有していること」を示す。なお、複数個の置換基を有する場合、それらの置換基は同 一でも互いに異なって!/、てもよレ、。
[0030] R1におけるそれぞれ置換されていてもよい「ァリール」及び「ヘテロ環基」における置
換基として好ましくは、以下の G1群から選択される基であり、より好ましくは、低級アル キル、ハロゲン、ハロゲノ低級アルキル、 -OR°、 -CO R°及びへテロ環基からなる群か ら選択される基であり、さらに好ましくは、低級アルキル、ハロゲン、ハロゲノ低級アル キル、 -0-低級アルキル、 -CO H及びテトラゾールからなる群から選択される基であ
G1群:ノヽロゲン、ニトロ、低級アルキル、ハロゲノ低級アルキル、 - OR°、 -CO R°、 - C(0 )N(R°) 、 -C(0)N(R°)S(0) -低級アルキル、 -C(0)N(R°)S(0) -低級アルキレン- OR°、 - N(R°) 、 -NR°- C(0)R°、ァリール、ヘテロ環基、ォキソ、低級アルキレン- CO R°、低級 アルケニレン- CO R°、低級アルキレン-ァリール、低級アルキレン-ヘテロ環基、 -0- 低級アルキレン- CO R°、 -N(R°)_低級アルキレン- CO R°及び- S(O) -低級アルキレン
2 2 n
-CO R°。 (nは 0、 1又は 2を意味する。以下同様。 )
ただし、 G1群におけるァリール及びへテロ環基は、それぞれ以下の P群から選択され る基で置換されてレ、てもよ!/、。
P群:ノヽロゲン、低級アルキル、ハロゲノ低級アルキル、 -OR°、 -0-ハロゲノ低級アル キル、ォキソ及び- CO R°0
[0031] R1における置換されていてもよい「シクロアルキル」及び「シクロアルケニル」におけ る置換基として好ましくは、以下の G2群から選択される基である。
G2群:ハロゲン、低級アルキル、ヘテロ環基、 _OR°及び- CO R°。
[0032] R1における置換されていてもよい「C アルキル」及び「低級アルケニル」における
1-12
置換基として好ましくは、以下の G3群より選択される基であり、より好ましくは、 -CO R° 、又は、それぞれハロゲン、 -OR0, -CO R°及びへテロ環基からなる群から選択される 基で置換されてレ、てもよ!/、ァリール若しくはへテロ環基である。
G3群:ハロゲン、 -OR0, - 0-ァリール、 -0-ヘテロ環基、 - N(R。)、 - N(R。)-ァリール、 - N (R。)-ヘテロ環基、 - N(R°)C(0)R°、 -0-低級アルキレン- CO R°、 - N(R°)-低級アルキレ ン -CO R。、 -S(0) -低級アルキレン- CO R。、 -CO R。、 -C(0)N(R°) 、 _C(0)N(R。)-ァリ ール、 -C(0)N(R°)_ヘテロ環基、 -C(0)-ァリール、 -C(0)-ヘテロ環基、シクロアルキ ノレ、ァリール及びへテロ環基。
ただし、 G3群におけるシクロアルキルは前記 G2群から選択される基で置換されていて
もよく、ァリール及びへテロ環基は前記 G1群から選択される基で置換されて!/、てもよ い。
[0033] R2におけるそれぞれ置換されていてもよい「ァリール」及び「ヘテロァリール」におけ る置換基として好ましくは、以下の G4群より選択される基であり、より好ましくはハロゲ ン、低級アルキル又はハロゲノ低級アルキルであり、さらにより好ましくはハロゲンで ある。
G4群:ノヽロゲン、低級アルキル、ハロゲノ低級アルキル、 _OR°及び- 0-ハロゲノ低級 アルキル。
[0034] R3におけるそれぞれ置換されていてもよい「ァリール」及び「ヘテロァリール」におけ る置換基として好ましくは、前記 G4群から選択される基であり、より好ましくは- 0-低級 アルキルである。
[0035] 本発明における好まし!/、態様を以下に示す。
(a) R1として、好ましくは、 -CO H及びテトラゾールからなる群より選択される基でそれ ぞれ置換されている C アルキル、シクロアルキル、ァリール又はへテロ環基であり(
1-12
ただし、前記 C アルキル、シクロアルキル、ァリール及びへテロ環基はさらに置換さ
1-12
れていてもよい。 );より好ましくは、 -CO H及びテトラゾールからなる群より選択される 基でそれぞれ置換されている低級アルキル、ァリール又はへテロアリールであり(た だし、前記低級アルキル、ァリール及びへテロアリールはさらに置換されていてもよい 。 );さらにより好ましくは、 -CO H及びテトラゾールからなる群より選択される基でそれ ぞれ置換されているァリール又はへテロアリールであり(ただし、前記ァリール基及び ヘテロァリールは低級アルキル、ハロゲン、ハロゲノ低級アルキル及び- 0-低級アル キルからなる群より選択される基でさらに置換されていてもよい。 );特に好ましくは、 - CO H及びテトラゾールからなる群より選択される基で置換されて!/、るフエニル又はピ リジルである(ただし、前記フエニル及びピリジルは低級アルキル、ハロゲン、ハロゲノ 低級アルキル及び- 0-低級アルキルからなる群より選択される基でさらに置換されて いてもよい)。
(b) Lとして、好ましくは、単結合、 _C(0)-、 -OC(O)-又は- NHC(O)-であり、より好まし くは単結合である。
(c) R2として、好ましくは、置換されていてもよいフエニルであり、より好ましくは、ハロ ゲン、低級アルキル又はハロゲノ低級アルキルで置換されて!/、てもよ!/、フエニルであ り、さらにより好ましくは、ハロゲンで置換されていてもよいフエニルである。
(d) R3として、好ましくはそれぞれ置換されて!/、てもよ!/、ァリール又はべンゾチェニル であり、より好ましくは、 -0-低級アルキルで置換されていてもよいァリール又はベン ゾチェニルであり、さらにより好ましくは、 -0-低級アルキルで置換されていてもよいナ フチル又はべンゾチアゾールである。
(e) R4として、好ましくは、メチルである。
( 乂及ひ として、好ましくは、 Xが単結合であり、 Yが- CH -である。
さらに上記の(a)から(f )の好まし!/、基の組み合わせからなる化合物がより好まし!/、 また、一般式 (I)で示される本発明化合物における別の好ましい態様を以下に示す
(1) R4がメチルである式 (I)記載の化合物またはその塩。
(2) Xが単結合であり、 Yが- CH -である(1)記載の化合物またはその塩。
(3) R3が- 0-低級アルキルで置換されていてもよいァリール、又は、ベンゾチェニル である(2)記載の化合物またはその塩。
(4) R2が、ハロゲン、低級アルキル及びハロゲノ低級アルキルからなる群より選択され る基で置換されてレ、てもよ!/、フエニルである(3)記載の化合物またはその塩。
(5) Lが単結合である(4)記載の化合物またはその塩。
(6) が、 -CO H及びテトラゾールからなる群より選択される基でそれぞれ置換され ているァリール又はへテロアリール(ただし、前記ァリール及びへテロアリールは低級 アルキル、ハロゲン、ハロゲノ低級アルキル及び- 0-低級アルキル力、ら選択される基 でさらにそれぞれ置換されて!/、てもよ!/、)である(5)記載の化合物またはその塩。
(7) 3-[3_(2-フルオロフェニル )-4-({[(lR)-l-(lナフチノレ)ェチノレ]アミノ}メチノレ)ピペリ ジン -1-ィル]安息香酸、
5-クロ口- 6-[4-({[(lR)-l-(3-メトキシフエ二ノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリ ジン- 1-ィル]ニコチン酸、
6-[4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ピリ ジン- 2-カルボン酸、
6-[4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -4- (トリフルォロメチル)ニコチン酸、
6-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン -1-ィル] -4- (トリフルォロメチル)ニコチン酸、
6-[3_(2-フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン -1-ィル]ニコチン酸、
3-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン -1-ィル]安息香酸、
2- [4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -1,
3-チアゾール -4-カルボン酸、
4-クロ口- 6-[4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1- ィル]ニコチン酸、
6-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン -1-ィル]ピリジン- 2-カルボン酸、
6-[3_(2-フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン -1-ィル]ピリジン- 2-カルボン酸、及び、
5-クロ口- 6-[3_(2-フルオロフヱニル) -4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル) ピぺリジン- 1-ィル]ニコチン酸
力 なる群より選択される式 (I)記載の化合物、またはその製薬学的に許容される塩
〇
本発明の化合物は、置換基の種類によっては他の互変異性体や幾何異性体が存 在する場合もある。本明細書中、それら異性体の一形態のみで記載することがあるが 、本発明にはこれらの異性体も包含し、異性体の分離したもの、あるいは混合物も包 含する。
また、化合物 (I)は不斉炭素原子や軸不斉を有する場合があり、これに基づく (R)体 、(S)体などの光学異性体が存在しうる。本発明はこれらの光学異性体の混合物や単
離されたものを全て包含する。
更に、本発明には、化合物 (I)の薬理学的に許容されるプロドラッグも含まれる。薬 理学的に許容されるプロドラッグとは、加溶媒分解により又は生理学的条件下でアミ ノ基、 OH、 CO H等に変換できる基を有する化合物である。プロドラッグを形成する基 としては、例えば、 Prog. Med., 5, 2157-2161 (1985)や「医薬品の開発」(廣川書店、 1 990年)第 7巻 分子設計 163-198に記載の基が挙げられる。
[0038] また、本発明化合物は、置換基の種類によっては酸付加塩又は塩基との塩を形成 する場合もあり、力、かる塩が製薬学的に許容され得る塩である限りにおいて本発明に 包含される。具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等 の無機酸や、ギ酸、酢酸、プロピオン酸、シユウ酸、マロン酸、コハク酸、フマル酸、マ レイン酸、乳酸、リンゴ酸、酒石酸、クェン酸、メタンスルホン酸、エタンスルホン酸、 p -トルエンスルホン酸、ァスパラギン酸、又はグルタミン酸等の有機酸との酸付加塩、 ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機塩基、メチルァ ミン、ェチルァミン、エタノールァミン、リシン、オル二チン等の有機塩基との塩やアン モユウム塩等が挙げられる。
更に、本発明は、本発明化合物及びその製薬学的に許容される塩の各種の水和 物や溶媒和物、及び結晶多形の物質をも包含する。また、本発明は、種々の放射性 又は非放射性同位体でラベルされた化合物も包含する。
[0039] (製造法)
本発明化合物及びその製薬学的に許容され得る塩は、その基本骨格あるいは置 換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することが できる。その際、官能基の種類によっては、当該官能基を原料乃至中間体の段階で 適当な保護基 (容易に当該官能基に転化可能な基)に置き換えておくことが製造技 術上効果的な場合がある。このような官能基としては例えばアミノ基、水酸基、カルボ キシル基等であり、それらの保護基としては例えばグリーン (Greene)及びウッツ (Wuts) 著、 「プロテクティブ'グループス'イン'オーガニック 'シンセシス(Protective Groups i n Organic Synthesis) (第 3版、 1999年)」に記載の保護基等を挙げることができ、これら を反応条件に応じて適宜選択して用いればよい。このような方法では、当該保護基を
導入して反応を行った後、必要に応じて保護基を除去することにより、所望の化合物 を得ること力 Sでさる。
また、化合物(I)のプロドラッグは上記保護基と同様、原料乃至中間体の段階で特 定の基を導入、あるいは得られた化合物(I)を用い反応を行うことで製造できる。反応 は通常のエステル化、アミド化、脱水等、当業者により公知の方法を適用することによ り fiうこと力 Sでさる。
以下、本発明化合物の代表的な製造法を説明する。各製法は、当該説明に付した 参考文献を参照して行うこともできる。なお、本発明の製造法は以下に示した例には 限定されない。
第一製法:還元的ァミノ化 1
[化 7]
本製法は、化合物(1)と化合物(2)とを反応させ、本発明化合物 (I)を得る方法で め 0。
反応は、化合物(1)と化合物(2)とを等量若しくは一方を過剰量用い、還元剤の存 在下、反応に不活性な溶媒中、 _45°Cから加熱還流下、好ましくは 0°C〜室温におい て、通常 0.1時間〜 5日間撹拌することによって行なわれる。ここに、溶媒としては特に 限定されないが、例えば、メタノール、エタノール等のアルコール類、ジェチルエーテ ノレ、テトラヒドロフラン(THF)、ジォキサン、ジメトキシェタン等のエーテノレ類、或いは これらの混合物が挙げられる。還元剤としては、シアン化水素化ホウ素ナトリウム、トリ ァセトキシ水素化ホウ素ナトリウム、水素化ホウ素ナトリウム、または還元剤を担持した ポリスチレン樹脂、例えば MP-トリァセトキシ水素化ホウ素(MP-Triacetoxyborohydrid e) (ァルゴノートテクノロジ一社 (Argonaut Technologies)、米国)等が挙げられる。モ レキユラーシーブス等の脱水剤、又は酢酸、塩酸、チタニウム(IV)イソプロポキシド錯 体等の酸存在下に反応を行うことが好ましい場合がある。反応によっては、中間体と
して反応系内に生成するイミン体を安定に単離できる場合には、当該イミン体を得た 後、別途還元反応を行っても良い。また、前記還元剤での処理の代わりにメタノール 、エタノール、酢酸ェチル等の溶媒中、酢酸、塩酸等の酸の存在下又は非存在下で
、還元触媒 (例えば、パラジウム炭素、ラネーニッケル等)を用いて行うこともできる。こ の場合、反応を常圧から 50気圧の水素雰囲気下で、 0°Cから加熱下で行うことが好ま しい。更に反応終了後の過剰なアミンを除去する目的でイソシァネートを担持したポ リスチレン樹脂、例えば PS-イソシァネート(PS-Isocyanate) (ァルゴノートテクノロジー 社(Argonaut Technologies)、米国)等を用いることもできる。
第二製法:加水分解
[化 8]
( は- H又は保護基(好ましくは tert-ブトキシカルボニル (Boc)基)を意味する。以下 同様。)
本製法は、化合物(3)を加水分解して化合物 (4)を得る方法である。
加水分解反応は例えば、前記「プロテクティブ ·グループス ·イン ·オーガニック ·シン セシス」に記載の方法により行うことができる。
第三製法:還元的ァミノ化 2
[化 9]
(式中、 Rla及び Rlbは R1の残部を意味する。以下同様。 )
本製法は、化合物(5)と化合物(4)とを反応させ、化合物(6)を得る方法である。 反応は、前記第一製法と同様にして行うことが出来る。
[0042] 第四製法:求核置換反応
[化 10]
(式中、 L1は脱離基を示す。以下同様。 )
本製法は、化合物(7)と化合物 (4)とを反応させ、化合物(8)を得る方法である。こ こで、 L1における脱離基としては、例えばハロゲン、メタンスルホニルォキシ、 P-トルェ ンスルホニルォキシ基等が挙げられる。
反応は、化合物(7)と化合物 (4)とを等量若しくは一方を過剰量用い、反応に不活 性な溶媒中、又は無溶媒下、冷却下から加熱還流下、好ましくは 0°Cから 80°Cにおい て、通常 0.1時間〜 5日間撹拌することによって行なわれる。ここに、溶媒としては特に 限定はされないが、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、 ジェチルエーテル、テトラヒドロフラン、ジ才キサン、ジメトキシェタン等のエーテル類 、ジクロロメタン、 1,2-ジクロロェタン、クロ口ホルム等のハロゲン化炭化水素類、 N,N- ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)、酢酸ェチル、ァセトニト リル或いはこれらの混合物が挙げられる。トリエチルァミン、 N,N-ジイソプロピルェチ ルァミン若しくは N-メチルモルホリン等の有機塩基、又は炭酸カリウム、炭酸ナトリウ ム若しくは水酸化カリウム等の無機塩基の存在下に反応させるの力 S、反応を円滑に 進行させる上で有利な場合がある。
[0043] 第五製法:アミド化
[化 11]
本製法は、化合物(9)と化合物 (4)とを反応させ、化合物(3)を得る方法である。
反応は、化合物(9)と化合物 (4)とを等量若しくは一方を過剰量用い、縮合剤の存 在下、反応に不活性な溶媒中、冷却下から加熱下、好ましくは- 20°C〜60°Cにおい て、通常 0.1時間〜 5日間撹拌することによって行なわれる。ここに、溶媒としては特に 限定はされないが、例えば、ベンゼン、トルエン若しくはキシレン等の芳香族炭化水 素類、ジクロロメタン、 1,2-ジクロロェタン若しくはクロ口ホルム等のハロゲン化炭化水 素類、ジェチルエーテル、テトラヒドロフラン、ジォキサン、ジメトキシェタン等のエー テル類、 N,N-ジメチルホルムアミド、ジメチルスルホキシド、酢酸ェチル、ァセトニトリ ル又は水、或いはこれらの混合物が挙げられる。縮合剤としては、 1_(3-ジメチルアミ ノプロピル) -3-ェチルカルポジイミド(WSC)、ジシクロへキシルカルポジイミド、 1,1 ' - カルボニルジイミダゾール(CDI)、ジフエニルリン酸アジド(DPPA)、ォキシ塩化リン等 が挙げられるが、これらに限定されるものではない。または縮合剤を担持したポリスチ レン樹脂、例えば PS-カルポジイミド(PS-Carbodiimide) (ァルゴノートテクノロジ一社( Argonaut Technologies)、米国)、 PL-DCCレジン(PL-DCC Resin) (ポリマー'ラボラ トリーズ社(Polymer Laboratories)、英国)を用いることもできる。添加剤(例えば 1_ヒド ロキシベンゾトリアゾール (HOBt)、 N-ヒドロキシスクシンイミド(HONSu)等)を用いるこ とが反応に好ましい場合がある。トリェチルァミン、 N,N-ジイソプロピルェチルァミン若 しくは N-メチルモルホリン等の有機塩基、又は炭酸カリウム、炭酸ナトリウム若しくは 水酸化カリウム等の無機塩基の存在下に反応させるの力 反応を円滑に進行させる 上で有利な場合がある。また、場合によっては、更に反応終了後の過剰なアミンを除 去する目的でイソシァネートを担持したポリスチレン樹脂、例えば PS-イソシァネート( PS-Isocyanate) (ァルゴノートテクノロジ一社、米国)等を用いることが好ましい。
また、カルボン酸(9)を反応性誘導体へ導いた後にアミン化合物(4)と反応させる 方法も用いることができる。ここにカルボン酸の反応性誘導体としては、ォキシ塩化リ ン、塩化チォニル等のハロゲン化剤と反応して得られる酸ハロゲン化物、クロロギ酸 イソブチル等と反応して得られる混合酸無水物、 HOBt等と縮合して得られる活性ェ ステル等が挙げられる。これらの反応性誘導体と化合物(4)との反応は、ハロゲン化 炭化水素類、芳香族炭化水素類、エーテル類等の反応に不活性な溶媒中、冷却下 〜加熱下、好ましくは、 -20°C〜60°Cで行うことができる。
第六製法:力ルバマート化
[化 12]
(式中、 L2は、力ルバマート化剤の残部を意味する。以下同様。 )
本製法は、化合物(10)と化合物(4)とを反応させ、化合物(11)を得る方法である 反応は、例えば日本化学会編「実験化学講座 (第 4版)」 20巻(1992年) (丸善)、 P355 -365等に記載の方法、または、前記の「プロテクティブ'グループス'イン'オーガニッ ク 'シンセシス」に記載の力ルバマート化の条件が適用できる。反応はァミン化合物(4 )と力ルバマート化剤(10)とを等モルあるいは一方を過剰量用いて、ベンゼン、トル ェン、キシレン等の芳香族炭化水素類、酢酸ェチル等のエステル類、ジェチルエー テル、テトラヒドロフラン、ジォキサン、ジメトキシェタン等のエーテル類、ジクロロメタン 、 1,2-ジクロロェタン若しくはクロ口ホルム等のハロゲン化炭化水素類、アルコール類 、アセトン、メチルェチルケトン等のケトン類、 DMF、 N,N-ジメチルァセトアミド(DMA) 、 N_メチルピロリドン(NMP)、 DMSO、ァセトニトリル、ピリジン、水等の反応に不活性 な溶媒中、冷却下乃至加熱還流下に行うことができる。力ルバマート化剤(10)として は酸ハロゲン化物(クロ口ホルマート等)、酸無水物(クロ口炭酸ェチル、クロ口炭酸べ ンジル、クロ口炭酸フエニル、 P-トルエンスルホン酸、イソ吉草酸等との反応で得られ る混合酸無水物、或いは対称酸無水物)、活性エステル (ニトロ基あるいはフッ素原 子などの電子吸引基で置換していてもよいフエノール、 CDI、 HONSu等を用いて調製 できるエステル)等が挙げられる。これらの反応性誘導体は常法により製造することが できる。化合物によっては、有機塩基(トリエチルァミン、ジイソプロピルェチルァミン、 N-メチルモルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン等が好適に用いられる )、又は金属塩塩基 (炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、 水素化ナトリウム、 tert-ブトキシカリウム等が好適に用いられる)の存在下に行うことが
、反応を円滑に進行させる上で有利な場合がある。 P-ニトロフエノールまたは CDIを 用いた力ルバマート化剤の調製及び力ルバマート化は例えば、ヴァテーレ (Vatele)ら の方法(「テトラへドロン(Tetrahedron)」、 2004年、 60巻、 p4251_4260)等に準じて 行うこと力 Sできる。また、例えば HONSuを用いた力ルバマート化剤の調製及び力ルバ マート化はゴーシュ (Ghosh)らの方法(「テトラへドロン'レターズ(Tetrahedron Letters )」、 1992年、 33巻、 p2781-2784)等に準じて行うことができる。
第七製法:ウレァ化
[化 13]
(式中、 L3は、ウレァ化剤の残部を意味する。以下同様。 )
本製法は、化合物(4)と化合物(12)または化合物(13)とを 応させ、化合物(14 )を得る方法である。
反応は、例えば日本化学会編「実験化学講座 (第 4版)」 20巻(1992年) (丸善)、 Ρ355 -365等に記載の方法が適用できる。反応はァミン化合物(4)とウレァ化剤(12)又は ( 13)とを等モルあるいは一方を過剰量用いて、ベンゼン、トルエン、キシレン等の芳 香族炭化水素類、酢酸ェチル等のエステル類、ジェチルエーテル、テトラヒドロフラン 、ジォキサン、ジメトキシェタン等のエーテル類、ジクロロメタン、 1,2-ジクロロェタン若 しくはクロ口ホルム等のハロゲン化炭化水素類、メタノール、エタノール等のアルコー ル類、アセトン、メチルェチルケトン等のケトン類、 DMF、 DMA, NMP、 DMSO、ァセト 二トリル、ピリジン、水等の反応に不活性な溶媒中、冷却下乃至加熱還流下に行うこ とができる。化合物によっては、有機塩基(トリエチルァミン、ジイソプロピルェチルアミ ン、 N-メチルモルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン等が好適に用いら れる)、又は金属塩塩基 (炭酸カリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリ ゥム、水素化ナトリウム、 tert-ブトキシカリウム等が好適に用いられる)の存在下に行う ことが、反応を円滑に進行させる上で有利な場合がある。
当該ウレァ化剤(12)としては酸ノ、ロゲン化物(クロロカルバメート等)、酸無水物(ク ロロ炭酸ェチル、クロ口炭酸ベンジル、クロ口炭酸フエニル、 p-トルエンスルホン酸、ィ ソ吉草酸等との反応で得られる混合酸無水物、或いは対称酸無水物)、活性エステ ル (ニトロ基あるいはフッ素原子などの電子吸引基で置換して!/、てもよ!/、フエノール、 CDI、 HONSu等を用いて調製できるエステル)、酸アジド等が挙げられる。これらのゥ レア化剤は常法により製造することができる。例えば p -ニトロフエノールを用いたウレ ァ化剤の調製及びウレァ化はトール (Tor)らの方法(「テトラへドロン ·レターズ(Tetrah edron Letters)」、 2001年、 42巻、 pl445_1447)等に準じて行うことができる。例えば CDIを用いたウレァ化剤の調製及びはペイティ (Batey)らの方法(「テトラへドロン ·レタ ーズ(Tetrahedron Letters)」、 1998年、 39巻、 p6267_6270)、コガ (Koga)らの方法(「 バイオオーガニック'アンド'メディシナル 'ケミストリー 'レターズ(Bioorganic & Medicin al Chemistry Letters)」、 1998年、 8巻、 pl471_1476)等に準じて行うことができる。 例えば HONSuを用いたウレァ化剤の調製及びウレァ化はォグラ (Ogura)らの方法(「 テトラへドロン.レターズ(Tetrahedron Letters)」、 1983年、 24巻、 p4569_4572)等に 準じて行うこと力 Sできる。例えば酸アジドを用いたウレァ化剤の調製及びウレァ化は力 一セラー (Carceller)らの方法(「ジャーナル'ォブ 'メディシナル 'ケミストリー(Journal o f Medicinal Chemistry)」、 1996年、 39巻、 p487_493)、リング (Ryng)らの方法(「ファ ルマジ一(Pharmazie)」、 1999年、 54巻、 p359_361)等に準じて行うことができる。

[化 14]
(式中、 Rleはそれぞれ置換されていてもよいァリール又はへテロアリールを、 L4は脱 離基を意味する。以下同様。 )
本製法は、化合物(15)と化合物(4)とを反応させ、化合物(16)を得る方法である 。ここで、 L4における脱離基としては、例えばハロゲン、トリフルォロメタンスルホニル
ォキシ基等が挙げられる。
反応は、パラジウム触媒及び塩基存在下、ァミン化合物(4)と化合物(15)とを等モ ルあるいは一方を過剰量用いて、ベンゼン、トルエン、キシレン等の芳香族炭化水素 類、ジェチルエーテル、テトラヒドロフラン、ジ才キサン、ジメトキシェタン等のエーテ ノレ類、メタノール、エタノール、 tert-ブチルアルコール等のアルコール類、 DMF、 DM A、 NMP等の反応に不活性な溶媒中、室温下乃至加熱還流下に行うことができる。パ ラジウム触媒としては、ビス(トリ- tert-ブチルホスフィン)パラジウム(0)、トリス(ジベン ジリデンアセトン)パラジウム(0)、酢酸パラジウム等のパラジウム錯体とトリ- 0-トルイ ノレホスフィン、 2,2'-ビス(ジフエニルホスフイノ) -1,1'-ビナフチル、 2-ジシクロへキシル ホスフイノ- 2'-(N,N-ジメチルァミノ)ビフエニル等のホスフィンリガンドより調整されるパ ラジウム触媒等が好適に用いられる。塩基としては炭酸ナトリウム、炭酸カリウム、炭 酸セシウム、カリウム tert-ブトキシド、ナトリウム tert-ブトキシドが挙げられる。
リン酸カリウム等が好適に用いられる。
[0047] また、上記第二乃至第八製法において が保護基の場合、保護基の脱保護は当 業者が通常用いる方法により行うことが出来る。例えば、前記「プロテクティブ 'グルー プス'イン'オーガニック 'シンセシス」に記載の方法により行うことが出来る。
[0048] 第九製法 その他の製法
種々の官能基、例えばカルボキシル基、アミド基、ヒドロキシル基、アルキルアミノ基 等を有する本発明化合物は、対応するエステル基、カルボキシル基、アミノ基等を有 する本発明化合物を原料として、当業者が自明の方法または公知の方法、あるいは その変法を用いることによって製造することができる。
[0049] (原料合成)
本発明化合物 (I)の製造に使用する原料化合物は、例えば下記の方法、公知の方 法、あるいはその変法を用いて製造することができる。
(原料合成 1)
[化 15]
(式中、 Rは低級アルキルを意味する。以下同様。 )
化合物(17)と化合物(18)をマイケル (Michael)付加することにより、化合物(19)を 得ること力 Sできる。マイケル付加反応は、例えば、水素化ナトリウム等の塩基存在下に 反応を fiうこと力できる。
化合物(19)のシァノ基を還元し分子内環化することにより、化合物(20)を得ること ができる。シァノ基の還元は、例えば、塩化コバルト存在下、水素化ホウ素ナトリウム を還元剤として用いて行うことができる。
化合物(20)のエステル基、アミド基を還元して、化合物(21)を得ることができる。 エステル基及びアミド基の還元は、例えば、水素化アルミニウムリチウムの還元剤を 用いて行うことができる。
(原料合成 2)
[化 16]
化合物(22)から、還元的ァミノ化、求核置換反応、アミド化、力ルバマート化又はゥ レア化により、化合物(23)を得ることができる。求核置換反応、アミド化、力ルバマー ト化、ウレァ化は、それぞれ第三乃至第七製法と同様にして行うことができる。
化合物(23)を酸化して、アルデヒド化合物(1)を得ることができる。酸化反応は、例
えばスワン (Swem)酸化又はデス.マー 酸化を用いることができる c
(原料合成 3)
[化 17]
化合物(I)を Boc化して化合物(24)を得ることができる。 Boc化は、例えば前記「プ ロテクティブ'グループス'イン'オーガニック 'シンセシス」に記載の方法により行うこと ができる。
(原料合成 4)
[化 18]
(式中、 R はァリール又はへテロアリールを、 Tfはトリフルォロメチルスルホニル基を、 R1Qは同一又は互いに異なって- H又は低級アルキル、或いは、 2つの R1Qがー体となつ て低級アルキレンを意味する。以下同様。 )
化合物(25)をトリフルォロメチルスルホニル化して、化合物(26)を得ること力 Sできる 。反応は、例えば、水素化ナトリウム等の塩基存在下、
無水物をトリフルォロメチルスルホニル化剤として用いて行うことができる。
化合物(26)と化合物(27)をカップリングして、化合物(28)を得ることができる。力 ップリング反応は、塩基及びパラジウム触媒の存在下に行うことができる。塩基として は、炭酸ナトリウム、炭酸カリウム、水酸化ナトリウム等の無機塩基が好ましい。また、 パラジウム触媒としては、テトラキス(トリフエュルホスフィン)パラジウム、ジクロロビス( トリフエニルホスフィン)パラジウム、塩化パラジウム- 1,1'-ビス(ジフエニルホスフイノ) フエ口セン等が好ましい。
化合物(28)を水素添加して、化合物(29)を得ることができる。反応は、例えば、パ ラジウム炭素、酸化白金、ラネーニッケル等の触媒存在下、水素雰囲気下に行うこと ができる。
化合物(29)を Boc化して、化合物(30)を得ることができる。 Boc化は、例えば前記「 プロテクティブ.グループス.イン.オーガニック.シンセシス」に記載の方法により行う こと力 Sでさる。
化合物(30)を異性化して、化合物(31)を得ることができる。反応は、例えば、ナトリ ゥムェトキシドを塩基として用いて行うことができる。
化合物(31)を脱 Boc化して、化合物(32)を得ることができる。反応は、例えば前記 「プロテクティブ.グループス.イン.オーガニック.シンセシス」に記載の方法により行う こと力 Sでさる。
化合物(32)を還元して、化合物(33)を得ることができる。反応は、例えば、水素化 アルミニウムリチウムを還元剤として用いて行うことができる
化合物(33)をトリフルォロアセチル化して、化合物(34)を得ることができる。反応 は、例えば、第五製法のアミド化と同様にして得ることができる。
本発明化合物は、遊離化合物、その製薬学的に許容される塩、水和物、溶媒和物 、あるいは結晶多形の物質として単離され、精製される。本発明化合物 (I)の製薬学 的に許容される塩は、常法の造塩反応に付すことにより製造することもできる。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等通常の化学操作 を適用して行われる。
各種の異性体は、適当な原料化合物を選択することにより、あるいは異性体間の物
理化学的性質の差を利用して分離することができる。例えば、光学異性体は一般的 な光学分割法 (例えば、光学活性な塩基又は酸とのジァステレオマー塩に導く分別 結晶化やキラルカラム等を用いたクロマトグラフィー等)により、立体化学的に純粋な 異性体に導くことができる。また、適当な光学活性な原料化合物より製造することもで きる。
本発明化合物(I)の優れた CaSR作動的調節作用は以下の試験により確認した。 試験 1.ヒトカルシウム感知受容体 (CaSR)作動性試験
1)ヒト CaSR発現ベクターの作製
ヒト CaSRをコードする DNA断片は定法によりクローニングした。詳細には、 NM_000388 の 203-2387を DNA断片 D4として、及び 2210-3633を断片 B2としてヒト腎臓 cDNA (イン ビトロジェン (Invitrogen)社製)をテンプレートとして DNAポリメラーゼ (登録商標: Pyrob est,タカラバイオ(Takara Bio)社製)を用いて増幅し、それぞれ pCR2. l-Topoベクタ 一 (インビトロジェン社製)を用いて PCR2.1ベクターにクローニングした。次に pCR2.1- D4を Spe I及び Xba Iで切断した DNA断片を pcDNA3.1/Zeo(+)ベクターの同サイトに 揷入した。さらに pCR2.1-B2を Sac I及び Xba Iで切断した断片を、先に作製した pcDN A3.1/Zeo(+)-D4(Spe I-Xba I)の Sac I及び Xba Iサイトに導入し、 pcDNA3.1/Zeo(+)ベ クタ一にヒト CaSRオープンリーディングフレーム (ORF)を含む、ヒト CaSR発現ベクター である pcDNA3. l/Zeo(+)_hCaSRを得た。
2)ヒト CaSR発現細胞の作製
ヒト CaSR発現ベクターをトランスフエクシヨン試薬(登録商標: FuGene 6、ロシュ'ダイ ァグノスティックス (Roche Diagnostics)社製)を用いて HEK293細胞に導入した。遺伝 子導入後 40 g/mLゼォシン (Ze0Cin、登録商標)(インビトロジェン社製)および 10%ゥ シ胎児血清を含む DMEM (インビトロジェン社製)培地中で 37°C、 5% CO存在下で 2 週間培養し、ゼォシン(Zeocin)耐性クローンを得た。これらのクローンの中からヒト Ca SRを安定発現する HEK293細胞を、細胞外 Ca2+に対する反応性を指標に選択し取得 した。
3)ヒト CaSR作動性試験
ヒト CaSR安定発現 HEK293細胞をポリ- D-リジン (poly-D-lysine)コートブラッククリア
一底 96ゥエルプレート(BDバイオサイエンス (BD biosciences)社製)に播種した。 20m M Hepes (2-[4_(2_ヒドロキシェチル) _1_ピぺラジュル]エタンスルホン酸)緩衝液 (pH7 • 4)、 2.5mMプロべネシド(Probenecid) (シグマ (Sigma)社製)、 0.1%ゥシ血清アルブミ ン (BSA)を含むハンクス平衡緩衝塩溶液 (HBSS) (Ca2+(_)、 Mg2+(_);インビトロジェン 社製)を、洗浄用緩衝液として作成した。播種一晩後培地を除き、 1ゥエルあたり 100 Lの ImM CaClおよび 10 M Fluo-3 AM (商品名、同仁化学(DOJINDO)社製)を含 む洗浄用緩衝液を加え、 37°C、 5%C0存在下で 1時間インキュベートした。洗浄用緩 衝液 200 H Lで 2回洗浄した後 100 H Lの 0.5mM CaClを含む洗浄用緩衝液に置換し 1 0分静置した後、蛍光測定画像解析用プレートリーダー(登録商標: FLIPR、モレキュ ラー ·デバイス(Molecular Devices)社)を用いて評価化合物に対する反応を検出した 。なお、評価化合物は 0.5mM Ca2+を含む洗浄用緩衝液に適宜希釈して用いた。 評価化合物のヒト CaSR作動活性強度は、溶媒群を 0%とし、最終濃度 2mMの Ca2+を 100%として算出し、 50%活性を示す化合物濃度 (EC )を濃度活性曲線から最小二乗 法により算出した。
その結果、本発明化合物は強いヒト CaSR作動活性を有することが明らかとなった。 本発明の代表的化合物の活性強度は表 1に示す。ただし、 Exは後記実施例番号を 示す (以下同様。)。
[表 1]
Ex EC50 (nM)
3 1.8
13 2.9
25 13
30 5.5
31 5.0
32 7.5
33 20
34 8.1
35 7.6
39 2.6
52 13
53 3.8
87 1 1
98 17
104 14
[0056] 試験 2.ラット血漿カルシウム濃度および血漿 PTH濃度の測定
本発明の化合物をラットに投与し、血漿カルシウム濃度および血漿 PTH濃度に及 ぼす影響を検討した。試験は、本発明の化合物および対照化合物を正常雄性ラット もしくは腎不全雄性ラット各 5-6匹に経口で単回投与することにより行った。
Vehicle群として 0.5%メチルセルロース(MC)溶液を 5mL/kgで投与した。比較対 照としては cinacalcetを MC溶液に溶解して、 3mg/kgの用量で投与した。本発明化合 物は MC溶液に溶解もしくは懸濁し、 1、 3もしくは 10mg/kgの用量で投与した。
投与前,および投与 2時間, 4時間,場合により 8時間後にエーテル麻酔下にて眼窩 静脈叢より採血し,血漿カルシウム濃度はカルシウム E—テストヮコー(和光純薬)を 用いて,また血漿 PTH濃度はラットインタタト PTH ELISAキット(ィムトピックス (Immutop ics)社)を用いて測定した。
その結果、本発明の化合物は in vivo試験において血漿カルシウムおよび血漿 PTH のレベルを低下させる作用があることが確認できた。正常雄性ラットにおける本発明 の代表的化合物の結果を表 2に示す。
[0057] [表 2]
投与 2時間後、 ラッ ト血漿
Ex
カルシウム濃度低下率(%) (3mg/kg)
25 17
31 24
32 34
33 29
35 33
39 23
52 26
53 20
[0058] 試験 3. ヒト CYP2D6阻害試験
CYP2D6に対する阻害活性評価は、文献(「ドラッグ ·メタボリズム 'アンド'デイスポジ シヨン(Drug Metabolism and Disposition)」、 2001年、第 29巻、 ρ· 1196-1200)に概ね 従って測定した。
酵素反応液中の試薬の最終濃度は、それぞれ、 CYP2D6 = 7.5 pmol/mL (BDジェ ンテスト(BD Gentest)社、カタログ番号: 456217)、還元型ニコチンアミドアデニンジヌ クレオチドリン酸(NADPH)再生系 (0.0081 mMニコチンアミドアデニンジヌクレオチド リン酸(NADP+) , 0.41 mMグルコース- 6-リン酸(glucose-6- phosphate)、 0.41 mM M gCl、 0.4 units/mLグルコース- 6_リン酸脱水素酵素(glucose-6-phosphate dehydro genase))、蛍光基質 AMMC = 1.5 μ Μ、 100 mMリン酸カリウム緩衝液 (ρΗ7·4)とした。 化合物は 50%ァセトニトリル溶液として、酵素反応溶液中に添加した(ァセトニトリル最 終濃度は 2.5%)。酵素反応は 37°C下にて 30分間実施し、反応停止液 (0.1Mトリス (ヒド 口キシメチル)ァミノメタン(Tris-base) :ァセトニトリル = 20: 80)により反応停止後、蛍 光強度を測定した。得られた蛍光強度から、化合物無添加時の酵素活性を 100%と して 50%阻害を示す濃度 (IC )を算出した。
その結果本発明化合物はヒト CYP2D6阻害活性が弱いことが明らかとなった。本発 明の代表化合物の CYP2D6阻害強度は表 3に示す。
[0059] [表 3]
Ex
3 ≥20
13 ≥20
25 ≥20
30 ≥20
34 ≥20
35 ≥20
52 ≥20
87 ≥20
98 ≥20
[0060] 上記の各試験の結果、本発明化合物は CaSR作動活性を有すること、また、薬物相 互作用を起こす懸念のある CYP2D6の阻害作用との選択性に優れることが明らかとな つた。このこと力、ら、本発明化合物は CaSRが関与する疾患、殊に副甲状腺機能亢進 症、腎性骨異栄養症、高カルシウム血症等の治療剤として有用であることは明らかで ある。
[0061] 本発明化合物(I)又はその塩の 1種又は 2種以上を有効成分として含有する製剤 は、当分野において通常用いられている薬剤用担体、賦形剤等を用いて通常使用さ れてレヽる方法によって調製することができる。
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関 節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、 経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれ の形態であってもよい。
[0062] 本発明による経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用 いられる。このような固体組成物においては、 1種又は 2種以上の有効成分を、少なく とも 1種の不活性な賦形剤、例えば乳糖、マンニトール、ブドウ糖、ヒドロキシプロピル セルロース、微結晶セルロース、デンプン、ポリビュルピロリドン、及び/又はメタケイ 酸アルミン酸マグネシウム等と混合される。組成物は、常法に従って、不活性な添カロ 剤、例えばステアリン酸マグネシウムのような滑沢剤やカルボキシメチルスターチナト リウム等のような崩壊剤、安定化剤、溶解補助剤を含有していてもよい。錠剤又は丸 剤は必要により糖衣又は胃溶性若しくは腸溶性物質のフィルムで被膜してもよい。
経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、 シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例え ば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化 剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有してい てもよい。
非経口投与のための注射剤は、無菌の水性又は非水性の溶液剤、懸濁剤又は乳 濁剤を含有する。水性の溶剤としては、例えば注射用蒸留水又は生理食塩液が含ま れる。非水溶性の溶剤としては、例えばプロピレングリコール、ポリエチレングリコール 又はオリーブ油のような植物油、エタノールのようなアルコール類、又はポリソルベー ト 80 (局方名)等がある。このような組成物は、さらに等張化剤、防腐剤、湿潤剤、乳 化剤、分散剤、安定化剤、又は溶解補助剤を含んでもよい。これらは例えばバタテリ ァ保留フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。また、 これらは無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶 解又は懸濁して使用することもできる。
外用剤としては、軟膏剤、硬膏剤、クリーム剤、ゼリー剤、パップ剤、噴霧剤、ローシ ヨン剤、点眼剤、眼軟膏等を包含する。一般に用いられる軟膏基剤、ローション基剤 、水性又は非水性の液剤、懸濁剤、乳剤等を含有する。例えば、軟膏又はローション 基剤としては、ポリエチレングリコール、プロピレングリコール、 白色ワセリン、サラシミ ッロウ、ポリオキシエチレン硬化ヒマシ油、モノステアリン酸グリセリン、ステアリルアル コール、セチルアルコール、ラウロマクロゴール、セスキォレイン酸ソルビタン等が挙 げられる。
吸入剤や経鼻剤等の経粘膜剤は固体、液体又は半固体状のものが用いられ、従 来公知の方法に従って製造することができる。例えば公知の賦形剤や、更に、 pH調 整剤、防腐剤、界面活性剤、滑沢剤、安定剤や増粘剤等が適宜添加されていてもよ い。投与は、適当な吸入又は吹送のためのデバイスを使用することができる。例えば 、計量投与吸入デバイス等の公知のデバイスや噴霧器を使用して、化合物を単独で 又は処方された混合物の粉末として、もしくは医薬的に許容し得る担体と組み合わせ て溶液又は懸濁液として投与することができる。乾燥粉末吸入器等は、単回又は多
数回の投与用のものであってもよぐ乾燥粉末又は粉末含有カプセルを利用すること 力できる。あるいは、適当な駆出剤、例えば、クロロフノレォロアルカン、ヒドロフルォロ アルカン又は二酸化炭素等の好適な気体を使用した加圧エアゾールスプレー等の 形態であってもよい。
[0064] 通常経口投与の場合、 1日の投与量は、体重当たり約 0.001〜100 mg/kg、好ましく は 0.1〜30 mg/kg、更に好ましくは 0· 1〜10 mg/kgが適当であり、これを 1回であるい は 2乃至 4回に分けて投与する。静脈内投与される場合は、 1日の投与量は、体重当 たり約 0.0001〜10 mg/kgが適当で、 1日 1回乃至複数回に分けて投与する。また、経 粘膜剤としては、体重当たり約 0.001〜100 mg/kgを 1日 1回乃至複数回に分けて投与 する。投与量は症状、年令、性別等を考慮して個々の場合に応じて適宜決定される
〇
[0065] 本発明化合物は、前述の本発明化合物が有効と考えられる疾患の種々の治療又 は予防剤と併用することができる。当該併用は、同時投与、或いは別個に連続しても しくは所望の時間間隔をおいて投与してもよい。同時投与製剤は、配合剤であっても 別個に製剤化されて!/、てもよレ、。
実施例
[0066] 以下、実施例に基づき本発明化合物(I)の製法を更に詳細に説明する。本発明化 合物は下記実施例に記載の化合物に限定されるものではない。また原料化合物の 製法を製造例に示す。
また、実施例、製造例及び後記表中以下の略号を用いる。
PEx :製造例、 Ex :実施例、 No :化合物番号、 MS :質量分析における m/z値 (EI : EI-M S、(特に断らない限り、(M)+を示す); FAB : FAB-MS、 ESI : ESI_MS、 API : AH-MS (ィ オン化法のうしろの +は陽イオンをを示し、一は陰イオンを示す。特に断らない限り、 陽イオンの場合は (M+H)+を示し、陰イオンの場合は (M— H)—を示す。))、 NMR1 : DMS O-d中の1 H NMRにおける δ (ppm)、 NMR2 : CDC1中の1 H NMRにおける δ (ppm)、 D
811 : 1,8-ジァザビシクロ[5.4.0]-7_ゥンデセン、 Me :メチル、 Syn :製造法(数字は、そ の番号を実施例番号として有する実施例化合物と同様にして、対応する原料を用い て製造したことを示す。数字が複数ある場合は、順次同様にして反応を行い製造した
ことを示す。)。 PSyn :製造法 (数字は、その番号を製造例番号として有する製造例化 合物と同様にして、製造したことを示す。数字が複数ある場合は、順次同様にして反 応を行い製造したことを示す。)。尚、構造式中の HC1はその化合物が塩酸塩である ことを示す。 Note :備考(racemiはその化合物がラセミ体であることを意味する。 diaster eo mixtureはその化合物がジァステレオ混合物であることを意味する。 3,4-transはピ ペリジンの 3位と 4位がトランス配置であることを意味する。また、 l',2'-cisのように数字 にダッシュがある場合は、ピぺリジンの 1位のシクロアルキルの立体を表している。例 えば、 l',2'-cisはピペリジンの 1位のシクロアルキルの立体がシス配置であることを意 味する。低極性化合物及び高極性化合物はその化合物が対応するジァステレオマ 一に対して TLC (メルク社、シリカゲル(Silica Gel) 60F )で低極性画分であるか高
254
極性画分であるかを意味する。 )
[0067] 製造例 1
シァノ酢酸メチル 5g、メタノール 20 mL、およびトルエン 20 mLの混合液に氷冷下、 水素化ナトリウム(55%オイル 'デイスパージヨン) 1.61gを加えた。室温にて 30分撹拌し た後、反応混合物に桂皮酸メチル 3.24 mLを加え、 66°Cにて 15時間撹拌した。反応 混合物を室温まで冷却後、 1M塩酸を加えて、溶液の pHを約 7に調整した。酢酸ェチ ルで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し て得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)により 精製し、 2-シァノ -3-フエ二ルペンタンニ酸ジメチル 5.19 gを得た。
[0068] 製造例 2
酢酸メチル 22.38 gのトルエン 24.2 mL溶液にナトリウムメトキシド(28wt%メタノール 溶液) 11.66 gを室温にて加えた。得られた白色懸濁液に 2-フルォロベンズアルデヒド 5.00 gを室温にて加えた。室温で 2時間撹拌した後、シァノ酢酸メチルとナトリウムメト キシド(28wt%メタノール溶液) 7.77 gをさらに加え、 65 °Cにて終夜撹拌した。反応混 合物を室温まで冷却後、 1M塩酸(70 mL)、飽和食塩水を順次加え、酢酸ェチルで 抽出した。有機層を無水硫酸ナトリウムにより乾燥し、ろ過後、ろ液を減圧下濃縮して 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン一酢酸ェチル)により精 製し、 2-シァノ -3_(2-フルオロフヱニル)ペンタンニ酸ジメチル 4.66 gを淡黄色油状物
として得た。
[0069] 製造例 3
2-シァノ -3-フエ二ルペンタンニ酸ジメチル 20.0 gと塩化コバルト (Π)六水和物 36.4 g のメタノール 1.0 L溶液に、氷冷下、水素化ホウ素ナトリウム 17.4 gを注意深く加えた。 室温で 30分撹拌した後、 1M塩酸(1.0 L)を加えて、室温でさらに 30分撹拌した。反 応混合物を半分量程度まで減圧下濃縮した後、酢酸ェチルで抽出した。有機層を 飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧下濃 縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム-メタノール)で 精製し、 re卜 (3R,4S)-6-ォキソ -4-フエ二ルビペリジン- 3-カルボン酸メチル 3.84 gを白 色固体として得た。
[0070] 製造例 4
(1)水素化アルミニウムリチウム 1.8 gのトルエン 40 mL-THF 40mL懸濁液に re卜 (3R, 4S)_6-ォキソ -4-フエ二ルビペリジン- 3-カルボン酸メチル 6.3 gを室温にて加えた。反 応混合物を 75 °Cにて 35時間撹拌した。反応終了後、室温まで冷却し、水 1.0 mL、 1 M水酸化ナトリウム水溶液 1.0 mL、 水 1.0 mLを順次に加えて、 10分間撹拌した。こ の混合物を無水硫酸ナトリウムで乾燥し、不溶物を濾過にて除去した後、ろ液を減圧 下濃縮して白色固体 reH(3R,4S)_4-フエ二ルビペリジン- 3-ィル]メタノール 4.37 gを 粗生成物として得た。 ESI+: 192
(2)得られた粗 reH(3R,4S)-4-フエ二ルビペリジン- 3-ィル]メタノール 4.34 g、トリェチ ルァミン 9.40 mL、およびトルエン 150 mLの混合物に氷冷下トリフルォロ酢酸無水物 4 .80 mLを滴下した。反応混合物を 30分間室温にて撹拌した後、 THF100 mLと飽和炭 酸水素ナトリウム水溶液 100 mLを加えて、更に 30分間激しく撹拌した。酢酸ェチルで 抽出した後、有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後
、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 一酢酸ェチル)により精製し、 reH(3R,4S)-4-フエニル -1- (トリフルォロアセチノレ)ピぺ リジン- 3-ィル]メタノール 3.69 gを得た。
[0071] 製造例 5
(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-ィル]
メチノレ }エタナミン 1.42 gの THF 15 mL溶液に、室温でジ炭酸-ジ -tert-ブチル 1.82 g の THF 15 mL溶液とトリェチルァミン 1.75 mLを加え、 60 °Cで終夜撹拌した。反応混 合物を室温に冷ました後、メタノール 22 mLと 1M水酸化ナトリウム水溶液 50 mLを加え 、室温で 30分間撹拌した。酢酸ェチルで抽出した後、有機層を飽和食塩水で洗浄し 、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリ 力ゲルカラムクロマトグラフィー(クロ口ホルム メタノール)にて精製し、 tert-ブチル [( 1R)-1-(1-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチル]力ルバメート 864 mgを得た。
[0072] 製造例 6
(lR)-N-{[4-(2-フルオロフェニル )-1- (トリフルォロアセチル)ピぺリジン- 3-ィル]メチ ル}-1-(1-ナフチル)エタナミン 1.24 gの THF20 mL溶液に、室温でジ炭酸-ジ -tert-ブ チル 2.36 gの THF10 mL溶液とトリェチルァミン 2.26 mLをカロえ、 60 °Cに加熱して終夜 撹拌した。反応混合物を室温に冷ました後、減圧下濃縮して得た残渣をシリカゲル力 ラムクロマトグラフィー(へキサン一酢酸ェチル)にて精製し、 tert-ブチル {[4-(2-フル オロフェニル )-1- (トリフルォロアセチル)ピぺリジン- 3-ィル]メチル }[(1R)-1-(1-ナフチ ノレ)ェチル]力ルバメート 703 mg (製造例 6— 1、低極性分画、 Rf値 0.29 (展開溶媒:へ キサン/酢酸ェチル = 7/1) )と tert-ブチル {[4-(2-フルオロフェニル )-1- (トリフルォロ ァセチル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフチル)ェチル]力ルバメート 700 m g (製造例 6— 2、高極性分画、 Rf値 0.21 (展開溶媒:へキサン/酢酸ェチル = 7/1) )を 各々無色泡状物質として得た。
[0073] 製造例 7
2,6-ジクロロニコチン酸ェチル l.OOgを塩化メチレン 10mLに溶解し、室温にてナトリ ゥムメトキシド(28wt%メタノール溶液) 1.05gを加えた。室温にて 2日間撹拌した後、反 応混合物を濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン-酢 酸ェチル)で精製し、 6-クロ口- 2-メトキシニコチン酸メチル 511mgを白色固体として 得た。
[0074] 製造例 8
tert-ブチル {[4-(2-フルオロフェニル )-1- (トリフルォロアセチル)ピぺリジン- 3-ィル]
メチル }[(1R)-1_(1-ナフチノレ)ェチノレ]力ルバメート 695mgを THF5 mL—メタノール 2 m Uこ溶解し、 1M水酸化ナトリウム水溶液 lmLを加え、室温で 30分間撹拌した。反応混 合物を減圧下濃縮し、得られた残渣にクロ口ホルムを加え、無水硫酸ナトリウムで乾 燥した。ろ過後、ろ液を減圧下濃縮して、 tert-ブチル {[4-(2-フルオロフェニル)ピペリ ジン- 3-ィル]メチル }[(1R)-1_(1-ナフチル)ェチル]力ルバメート 636 mgを無色泡状物 質を粗生成物として得た。
[0075] 製造例 9
re卜 [(3R,4S)-4-(4-フルオロフェニル)ピぺリジン- 3-ィル]メタノール(ァスタテック(As tatech, Inc)、米国) 382 mgをジクロロメタン 5 mLに溶解し、水 5 mLおよび炭酸ナトリウ ム 755 mgを加えて 0°Cに冷却した後、 4- [(クロ口カルボニル)ォキシ]安息香酸メチル( フル力(Fluka社)) 785 mgを少量ずつ加え、 1時間攪拌した。反応混合物をクロロホ ルムにて抽出し、飽和炭酸ナトリウム水溶液にて洗浄した後有機層を無水硫酸ナトリ ゥムで乾燥し、ろ過後、ろ液を減圧下濃縮した。得られた残渣をジイソプロピルエー テル中攪拌洗浄し、 re卜 (3R,4S)-4-(4-フルオロフェニル )-3- (ヒドロキシメチノレ)ピペリ ジン- 1-カルボン酸 4- (メトキシカルボニル)フエニル 490 mgを白色粉末として得た。
[0076] 製造例 10
4- (メトキシカルボニル)フエニル 4-(4-フルオロフヱニル) -3-({[(lR)-l_(l-ナフチル) ェチノレ]アミノ}メチノレ)ピぺリジン- 1-カルボキシレート 888 mgのジクロロメタン 9 mL溶 液に、トリエチルァミン 0.7 mL及び炭酸ジ tert-ブチル 868 mgを加え、室温にて 37時 間攪拌した。反応混合物を減圧下濃縮し、得られた残渣をシリカゲルクロマトグラフィ 一(へキサン-酢酸ェチル)にて精製し、 4- (メトキシカルボニル)フエニル 3_({(tert-ブ トキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ) -4-(4-フルオロフェニル )ピペリジン- 1-カルボキシレート 503 mgを淡黄色泡状物質として得た。
[0077] 製造例 11
氷冷下クロロギ酸 4-ニトロフエニル 5.43 gのジクロロメタン 100 mL溶液に、ピリジン 2 .4 mLを加え、得られた混合物に 4-ァミノ- 3-クロ口安息香酸メチル 5 gを加えた。室温 にて終夜撹拌した。反応混合物を減圧下濃縮した後、残渣を酢酸ェチルを加え 1M 塩酸、水、飽和炭酸水素ナトリウム水溶液、水、飽和塩化ナトリウム水溶液で順次洗
浄した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後、ろ液を減圧下濃縮した。得ら れた固体をトルエン 200 mLに加熱溶解させた室温まで放冷した後、さらに氷浴で冷 却した。析出した固体をろ過により採取し、減圧乾燥して、 3-クロ口- 4-{[(4-ニトロフエ ノキシ)カルボニル]アミノ}安息香酸メチル 5.59 gを白色固体として得た。
製造例 12
(1)触媒の調製:国際特許公開 W01998/42643号公報の方法に準じて不斉ロジウム 触媒を調製した。(lR,2S)-(+)-cis_l-ァミノ- 2-インダノール 75.3 mgとジクロ口(ペンタ メチルシクロペンタジェニル)ロジウム (III)ジクロライド,二量体(ストリームケミカルス(S trem Chemicals, Inc.)、米国) 156 mgの混合物をイソプロパノール 63 mLに溶解させ た。この溶液の脱気を行い、アルゴン雰囲気に置換した。得られた懸濁液を 40。Cに て 2時間撹拌した後、室温まで冷却し、オレンジ色の不斉ロジウム触媒溶液を調製し た。
(2) 1-(1-ベンゾチェン- 3-ィル)エタノン 4.45 gのイソプロパノール 95 mL溶液に上述 のように調製した不斉ロジウム触媒溶液を加えた。減圧下 (約 6600Pa)、ナトリウムイソ プロポキシドのイソプロパノール溶液(0.1M、 10.0 mL)を加えた。更に約 3700Paまで 減圧し、室温で 2時間撹拌した。反応混合物に酢酸 (2 mL)を加え反応を停止した後
、減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸 ェチル)で精製し、(lS)-l-(l-ベンゾチェン- 3-ィル)エタノール(4.48 g、 94%ee)を淡 黄色油状物として得た。 (HPLC分析条件:ダイセル化学工業株式会社製 CHIRALC EL OD-Hカラム、展開溶媒へキサン/イソプロパノール = 90/10、流速 1.0 mLmin— 保持時間: 8.1 min (S体), 12.6 min (R体)) EI: 178
(3) Thompsonらの方法(J. Org. Chem. 1993, 58, 5886)を参考に行った。アルゴン雰 囲気下、(lS)-l-(l-ベンゾチェン- 3-ィル)エタノール 5.50 gと DPPA 7.98 gの混合物 をトルエンに溶解し、氷浴下 DBU 5.54 mLを加えた。氷冷下 30分間撹拌した後、室 温で更に 15時間撹拌した。反応混合物を水、 1M塩酸で順次洗浄した後、無水硫酸 ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラ ムクロマトグラフィー(へキサン 酢酸ェチル)で精製し (lR)-l-(l-ベンゾチェン- 3-ィ ノレ)ェチルアジド 5.86 gを無色油状物として得た。 EI: 203
(4) (1R)-1-(1-ベンゾチェン- 3-ィル)ェチルアジド 5.57 g、 10%パラジウム/炭素 557 mg、および酢酸ェチル 250 mLを混合し、常圧水素ガス雰囲気下で 2時間激しく撹拌 した。アルゴンで置換した後、不溶物をセライト層にてろ過し、ろ液を減圧下で濃縮し た。得られた残渣をエタノール 20 mLに溶解し、 4M塩化水素 /1,4 -ジォキサン溶液 7. 0 mLを室温で加え 5分間撹拌した後、再び減圧濃縮した。エタノール溶媒から 2回結 晶化を繰り返して、(lR)-l-(l-ベンゾチェン- 3-ィル)エタナミン塩酸塩(2.35 g、 96% ee)を白色固体として得た。 (HPLC分析条件:ダイセル化学工業株式会社製 CHIRA LCEL OD-Hカラム,展開溶媒:へキサン/イソプロパノール = 90/10、流速 1.0 mLmi n— 保持時間: 10.4 min (R体), 12.2 min (S体))
製造例 13
(1)水素化ナトリウム(60%オイル 'デイスパージヨン) 11.8 gのジェチルエーテル 600 m L懸濁液を 0 °Cに冷却した後、 1-ベンジル -3-ォキソピペリジン -4-カルボン酸ェチル 塩酸塩 40.0 gを加え、 0 °Cにて 30分間撹拌した。反応混合物にトリフルォロメタンス ルホン酸無水物 26.4 mLを加え、さらに 0 °Cで 1時間撹拌した。飽和塩化アンモニゥム 水溶液を加え反応を停止した後、有機層を分離し、水層を酢酸ェチルで抽出した。 混合した有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ 液を減圧下濃縮、乾燥し 1-ベンジル _5-{[(トリフルォロメチル)スルホニル]ォキシ }-1,2 ,3,6-テトラヒドロピリジン- 4-カルボン酸ェチルを粗生成物(濃茶色油状物、 64.3 g)と して得た。 FAB + : 394
(2)得られた 1-ベンジル -5_{[(トリフルォロメチル)スルホニル]ォキシト 1,2,3,6-テトラヒ ドロピリジン- 4-カルボン酸ェチルの粗生成物 64.3 g、フエ二ルポロン酸 19.7 g、炭酸 カリウム 22.3 g、ジメトキシェタン 600 mL及びテトラキス(トリフエニルホスフィン)パラジ ゥム 4.66 gの混合物を 90 °Cにて終夜攪拌した。室温まで冷却後、不溶物をセライト層 にてろ過した。ろ液を減圧下濃縮して得られた残渣にクロ口ホルムを加え、水で洗浄 した後、有機層を無水硫酸ナトリウムにて乾燥した。ろ過後、ろ液を減圧下濃縮して 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)で精製し 1-ベンジル -5-フエニル -1,2,3,6-テトラヒドロピリジン- 4-カルボン酸ェチル 35.9 gを 黄色油状物として得た。
[0080] 製造例 14
(1) 1-ベンジル -5-フエニル -1,2,3,6-テトラヒドロピリジン- 4-カルボン酸ェチル 9.98 g 、ギ酸アンモニゥム 10.31 g及びメタノール 90 mLの混合物に 10%パラジウム/炭素 1.49 gを水 10 mLとメタノール 10 mL懸濁させながら加えた。反応混合物を 60 °Cにて 2時間 激しく撹拌した。室温まで冷却後、不溶物をセライト層にてろ過し、ろ液を減圧下濃 縮して得られた残渣にクロ口ホルムを加え、飽和重層水で洗浄した。有機層を無水硫 酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して re卜 (3R,4S)-3-フエ二 ルビペリジン- 4-カルボン酸ェチル 7.09 gを淡黄色油状物、粗生成物として得た。 ES I+: 234
(2)粗生成物として得られた rel-(3R,4S)_3-フエ二ルビペリジン- 4-カルボン酸ェチル 5.58 gを THF55 mLに溶解し、トリエチノレアミン 5.0 mL及びジ炭酸-ジ- tert-ブチル 6. 26 gを加え、室温で 2日間撹拌した。反応混合物を減圧下濃縮し、得られた残渣をシ リカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)により精製して、 re卜 (3R,4S )-3-フエ二ルビペリジン- 1,4-ジカルボン酸 1-tert-ブチル 4-ェチル 7.06 gを淡黄色油 状物として得た。
[0081] 製造例 15
re卜 (3R,4S)-3-フエ二ルビペリジン- 1,4-ジカルボン酸 1-tert-ブチル 4-ェチル 26.5 gのトルエン 200 mL溶液にカリウム t-ブトキシド 2.94 gを室温で加え、加熱還流下終 夜撹拌した。反応混合物を室温まで冷却後、 1M塩酸 27 mLを加え中和し、酢酸ェチ ルを加え有機層を分取した。有機層を水及び飽和食塩水で順次洗浄し、無水硫酸 ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して rd-(3R,4R)-3-フエニル ピぺリジン- 1,4-ジカルボン酸 1-tert-ブチル 4-ェチル 25.8 gを白色蠟状固体として 得た。
[0082] 製造例 16
re卜 (3R,4R)-3-フエ二ルビペリジン- 1,4-ジカルボン酸 1-tert-ブチル 4-ェチル 1.40 gに 4M塩化水素 /1,4-ジォキサン溶液 10 mLを加え、室温にて終夜撹拌した。反応 混合物に酢酸ェチルを加え、析出固体を懸濁させた後、ろ過により単離し、減圧下 乾燥して re卜 (3S,4S)-3-フエ二ルビペリジン- 4-カルボン酸ェチル塩酸塩 1.05 gを白
色固体として得た。
[0083] 製造例 17
(1)水素化リチウムアルミニウム 6.00gのトルエン 115 mL-THF115 mL懸濁液に reH 3S,4S)-3-フエ二ルビペリジン- 4-カルボン酸ェチル塩酸塩 17.7 gを室温にて注意深 く加え、室温で 5時間撹拌した。氷冷下、反応混合物にメタノール 20 mLを加え反応を 停止した後、水 6 mL、 15w%水酸化ナトリウム水溶液 18 mL,水 18 mLを順次加えて 10 分間撹拌した。不溶物を濾過にて除去した後、ろ液を減圧下濃縮して reH(3R,4R)-3 -フエ二ルビペリジン- 4-ィル]メタノール 12.9 gを白色固体、粗生成物として得た。 ESI +: 192
(2)粗 re卜 [(3R,4R)-3-フエ二ルビペリジン- 4-ィル]メタノール 3.59 gとトリエチノレアミン 10.46 mLのトルエン 70 mL溶液に氷冷下トリフルォロ酢酸無水物 5.30 mLを加えた。 室温にて 30分間撹拌した後、反応混合物に飽和炭酸水素ナトリウム水溶液 50 mLと T HF50 mLを加え、更に 30分間激しく撹拌した。有機層を分取し、飽和塩化ナトリウム 水溶液で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得 られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)で精製し rel _[(3R,4R)-3-フエニル -1- (トリフルォロアセチノレ)ピぺリジン- 4-ィル]メタノール 5.143 g を淡黄色油状物として得た。
[0084] 製造例 18
(lR)-l-(l-ナフチル) -N-{[3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4-ィル] メチル }エタナミン 2.15 gの THF 43.0 mL溶液にトリェチルァミン 2.72 mL及びジ炭酸- ジ -tert-ブチル 2.13 gを室温にて加え、 60 °Cて終夜撹拌した。室温まで冷却後、反 応混合物を減圧下で濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(へ キサン 酢酸ェチル)で精製し、 2つのジァステレオマーとして低極性分画 tert-ブチ ノレ [(lR)-l-(l-ナフチル)ェチル ]{[3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4- ィル]メチル }力ルバメート(製造例 18-1、 Rf値 0.57 (へキサン/酢酸ェチル = 7/1)) 1.21 g及び高極性分画 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ]{[3-フエニル -1- (トリフ ルォロアセチル)ピぺリジン- 4-ィル]メチル }力ルバメート(製造例 18-2、 Rf値 0.42 (へキ サン/酢酸ェチル = 7/1)) 1.20 gを各々無色泡状物質として得た。
[0085] 製造例 19
(1) re卜 [(3R,4R)-3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4-ィル]メタノール 500 mgに THF8.0 mL、メタノール 4.0 mL及び 1M水酸化ナトリウム水溶液 4.0 mLをカロ え、室温にて終夜撹拌した。反応混合物を減圧下濃縮し、残渣を酢酸ェチルで抽出 した。有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥し 得られた粗生成物 376 mg、 2,6-ジクロロ- 5-フルォロニコチン酸メチル 429 mg、およ び炭酸カリウム 289 mgの混合物に DMSO 10 mLを室温にて加え、混合液を室温にて 5時間撹拌した。混合物に酢酸ェチルを加え、不溶物をろ過により除いた後、ろ液を 減圧下で濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)で精製し、 re卜 2-クロ口- 5-フルォ口- 6-[(3R,4R)-4- (ヒドロキシメチル) -3- フエ二ルビペリジン- 1-ィル]ニコチン酸メチル 493 mgを無色泡状物質として得た。 ES I+: 379
(2) re卜 2-クロ口- 5-フルォ口- 6-[(3R,4R)-4- (ヒドロキシメチル) -3-フエ二ルビペリジン- 1-ィノレ]ニコチン酸メチル 488 mg、ギ酸アンモニゥム 428 mg及びメタノール 12 mLの 混合物に 10%パラジウム/炭素 70 mgを水 1 mLとメタノール 6 mL懸濁させながら加えた 。反応混合物を 60 °Cにて 2時間激しく撹拌した。室温まで冷却後、不溶物をセライト 層にてろ過し、ろ液を減圧下濃縮して得られた残渣にクロ口ホルムを加え、飽和重層 水で洗浄した。有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮 し、得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル)で精 製し、 re卜 5-フルォ口- 6-[(3R,4R)-4- (ヒドロキシメチル) -3-フエ二ルビペリジン- 1-ィ ノレ]ニコチン酸メチル 283 mgを無色泡状物質として得た。
[0086] 製造例 20
製造例 18で分取した低極性分画ジァステレオマー tert-ブチル [(lR)-l-(l-ナフチ ノレ)ェチル ]{[3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4-ィル]メチル }力ルバメ ート 5.00 g、 THF 40 mL及びメタノール 40mLの混合液に、 1M水酸化ナトリウム水溶 液 18.5mLを加えた。室温で 3日間撹拌した後、反応混合物を減圧下濃縮した。残渣 に水を加え、クロ口ホルムで抽出した後、有機層を無水硫酸ナトリウムで乾燥した。ろ 過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロ
口ホルム メタノール)で精製し tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二 ルビペリジン- 4-ィル)メチル]力ルバメート 3.78gを無色泡状物質として得た。
[0087] 製造例 21
製造例 18で分取したジァステレオマー tert-ブチル [(lR)-l-(l-ナフチル)ェチル] {[ 3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4-ィル]メチル }力ルバメートの低極性 分画 900 mgと高極性分画 900 mgを混合し、 THF5 mL、メタノール 2.5 mL及び 1M水 酸化ナトリウム水溶液 2.5 mLを加えた。室温で 3時間撹拌した後、反応混合物を減圧 下濃縮した。残渣に水を加え、クロ口ホルムで抽出した後、有機層を無水硫酸ナトリウ ムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して tert-ブチル [(lR)-l-(l-ナフチ ノレ)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチル]力ルバメート 1.55 gを無色泡状物 質として得た。
[0088] 製造例 22
4,6-ジクロロニコチン酸メチル 500mg及び THF 5.0 mLの混合物を氷浴で冷却下、 ナトリウムメトキシド(28wt%メタノール溶液) 157mgを滴下した。室温に昇温し、終夜撹 拌した。反応混合物を減圧下濃縮後、残渣をシリカゲルカラムクロマトグラフィーによ り精製し、 6-クロ口- 4-メトキシニコチン酸メチルを白色固体として 162mg得た。
[0089] 製造例 23
メタノール 10mLを氷浴で冷却下、チォユルク口ライド 2.5mLを 30分間かけて滴下し た。反応混合物に 3,5-ジフルォロピリジン- 2-カルボン酸 500mgを加え、室温に昇温 して 3日間撹拌した。反応混合物を減圧下濃縮し、飽和炭酸水素ナトリウム水溶液を くわえ、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄した後、無水硫酸ナトリ ゥムで乾燥した。ろ過後、減圧下濃縮して、 3,5-ジフルォロピリジン- 2_カルボン酸メ チルを 496mg得た。
[0090] 製造例 1〜23の方法と同様にして、後記表に示す製造例 24〜52の化合物を製造 した。製造例化合物の構造、製造法及び物理化学的データを表 4〜; 14にそれぞれ 示す。
[0091] 実施例 1
(l) DMSO 1.37 mLのジクロロメタン 20 mL溶液を- 78 °Cに冷却し、ォキサリルクロリド
0.845 mLを滴下した(内部温度- 60 °C以下)。同温度で 20分間攪拌した後、 rd_[(3R, 4S)_4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-ィル]メタノール 1.46 gのジク ロロメタン 20 mL溶液を滴下し、さらに 20分間攪拌した。トリェチルァミン 4.40 mLを滴 下した後、反応温度を- 30 °Cに昇温し、さらに 15分撹拌した。反応混合物に飽和塩 化アンモニゥム水溶液を加えて反応を停止し、室温に昇温した後、クロ口ホルムで抽 出した。有機層を水で洗浄後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧 下濃縮し、 re卜 (3R,4S)-4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-カルボア ルデヒドを粗生成物 1 · 597gとして得た。
(2) (1R)-1-(1-ナフチル)エタナミン 868 mg、酢酸 0.073 mL、トリァセトキシ水素化ホウ 素ナトリウム 1.29 g及び 1, 2-ジクロロェタン 75 mLの混合物に、粗 reト (3R,4S)_4-フエ ニル -1- (トリフルォロアセチル)ピぺリジン- 3-カルボアルデヒド 1.446 gの 1, 2-ジクロロ ェタン 25 mL溶液を室温にて滴下し、終夜撹拌した。反応混合物に飽和炭酸水素ナ トリウム水溶液を加え 10分間激しく撹拌した後、クロ口ホルムにて抽出し、有機層を無 水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲ ルカラムクロマトグラフィー(クロ口ホルム一メタノール)で精製し (lR)-l-(l-ナフチル) - N-{[4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-ィル]メチル }エタナミン 1.84 g を無色泡状物質として得た。
(3) (1R)-1_(1-ナフチル) -N-{[4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-ィ ル]メチノレ }エタナミン 200 mgをエタノール 4.0 mLに溶解し、 4M塩化水素 /1,4_ジォキ サン溶液 300 ^ Lを室温にて加えた。 5分間撹拌した後、反応混合物を減圧下濃縮 し、得られた残渣をイソプロパノールから結晶化して (lR)-l-(l-ナフチル) -N-{[4-フエ ニル -1- (トリフルォロアセチノレ)ピぺリジン- 3-ィル]メチル }エタナミン塩酸塩 173 mgを 白色粉末として得た。
実施例 2
ジァステレオマーの混合物である(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフル ォロアセチル)ピぺリジン- 3-ィル]メチル }エタナミン 220 mgを高速液体クロマトグラフィ 一(関東化学株式会社製カラム, Mightysil, RP-18, GP 250-20, 5 m、展開溶媒: ァセトニトリル一水)により処理し、(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフルォ
ロアセチル)ピぺリジン- 3-ィル]メチル }エタナミン(低極性分画、 HPLC保持時間: 12mi n)と(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフルォロアセチル)ピぺリジン- 3-ィ ノレ]メチル }エタナミン(高極性分画: HPLC保持時間: 8.7min)を分取した(分取高速液 体クロマトグラフィー条件、カラム: TSK-GEL, ODS-80TM (東ソ一(TOSO)社、内径 4· 6mm、長さ 150mm)、流速: lml/min、 0.01Mリン酸二水素カリウム水溶液/ァセト 二トリル =30/70)。それぞれ別々に 4M塩化水素 /1,4-ジォキサン溶液で処理した後、 イソプロパノールから固体化し、(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフルォ ロアセチル)ピぺリジン- 3-ィル]メチル }エタナミン塩酸塩(実施例 2— 1、低極性分画よ り) 69 mgと(lR)-l-(l-ナフチル) -N-{[4-フエニル -1- (トリフルォロアセチル)ピぺリジン -3-ィル]メチル }エタナミン塩酸塩 (実施例 2— 2、高極性分画より) 78 mgを白色粉末 として得た。
[0093] 実施例 3
3-メトキシ _4-({[3-({[(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-4-フエ二ルビペリジ ン -1-ィル]カルボ二ノレ }ァミノ)安息香酸メチル 159mgを THF 6.0mL_メタノーノレ 3.0mL に溶解し、室温で 1M水酸化ナトリウム水溶液 3.0mLを加え終夜撹拌した。反応混合 物に 1M塩酸 3.0 mLを加えて中和した後、減圧下濃縮した。残渣に水を加えクロロホ ルムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、濾液を濃縮して得 られた残渣をシリカゲルカラムクロマトグラフィー(メタノール-クロ口ホルム)で精製し、 3-メトキシ _4-({[3-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン -1-ィル]カルボ二ル}ァミノ)安息香酸メチルを無色アモルファスとして 144 mg得た。得 られたアモルファスをクロ口ホルムに溶解した後、へキサンを加え固体化した。析出物 をろ取し、減圧下乾燥して 3-メトキシ _4-({[3-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メ チル) -4-フエ二ルビペリジン -1-ィル]カルボ二ル}ァミノ)安息香酸を白色固体として 10 2mg得た。
[0094] 実施例 4
実施例 2で分取した低極性分画ジァステレオマー(lR)-l-(l-ナフチル) -N-{[4-フエ ニル -1- (トリフルォロアセチル)ピペリジン- 3-ィル]メチル }エタナミン塩酸塩(実施例 2 -1) 610 mgを THF4 mLとメタノーノレ 2 mLに懸濁させ、 1M水酸化ナトリウム水溶液 3 m
Lを加えた。室温で 2時間撹拌した後、反応混合物を減圧下濃縮した。残渣に水を加 え酢酸ェチルで抽出した後、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液 を減圧下濃縮して、(lR)-l-(l-ナフチル) -N-K4-フエュルピペリジン -3-ィル)メチル] エタナミン 475 mgを得た。
[0095] 実施例 5
( 1 )ォキサリルクロリド 0.3 mLのジクロロメタン 5 mL溶液に、 DMSO 0.5 mLを- 78。Cに て滴下し、 10分間攪拌した。反応混合物に rd-(3R,4S)_4-(4-フルオロフヱニル) -3- (ヒ ドロキシメチル)ピぺリジン- 1-カルボン酸 4- (メトキシカルボニル)フエニル 664 mgのジ クロロメタン 5 mL溶液を- 78 °Cにて滴下し、 15分間攪拌後、ジイソプロピルェチルアミ ン 1.8 mLのジクロロメタン 3 mL溶液を滴下し、 2時間かけて室温まで昇温した。反応 混合物を減圧下濃縮し、残渣にジェチルエーテルを加え飽和食塩水にて洗浄し、無 水硫酸ナトリウムにて乾燥した。ろ過後、ろ液を減圧下濃縮し、 rd-(3R,4S)_4-(4-フル オロフェニル )-3-ホルミノレビペリジン- 1-カルボン酸 4- (メトキシカルボニル)フエニルを 粗生成物として得た。 ESI +: 386
(2)得られた粗 re卜 (3R,4S)-4-(4-フルオロフェニル )-3-ホルミルピぺリジン- 1-カルボ ン酸 4- (メトキシカルボ二ノレ)フエニルの 1 , 2-ジクロロェタン 4 mL溶液を、(1R)_1_(1-ナ フチル)エタナミン 311 mg、酢酸 0.030 mL、トリァセトキシ水素化ホウ素ナトリウム 1.09 g及び 1 , 2-ジクロロェタン 6 mLの混合物に加え、室温にて終夜撹拌した。反応混合 物が中性になるまで飽和炭酸水素ナトリウム水溶液を加えた後、クロ口ホルムにて抽 出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後、ろ液を減圧下濃縮した。得ら れた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム-メタノール)で精製し 4-( メトキシカルボ二ノレ)フエニル 4-(4-フルオロフェニル )-3-({[(lR)-l_(l-ナフチノレ)ェチ ノレ]アミノ}メチル)ピぺリジン- 1-カルボキシレート 888 mgを淡黄色泡状物質として得た
〇
[0096] 実施例 6
(lR)-l-(l-ナフチル) -N-K4-フエ二ルビペリジン- 3-ィル)メチル]エタナミンの粗生成 物 175 mgとトリエチルァミン 142 μ Lの THF3.5 mL溶液に、氷冷下 4- [(クロ口カルボ二 ノレ)ォキシ]安息香酸メチル (フル力(Fluka社)) 120 mgを加えた。室温にて 30分間撹
拌した後、反応混合物を減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトダラ フィー(へキサン—酢酸ェチル)で精製して 4- (メトキシカルボニル)フエニル 3-({[(lR)_ 1-(1-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-カルボキシレート 237 mgを得た。
[0097] 実施例 7
(lR)-l-(l-ナフチル) -N-K4-フエ二ルビペリジン- 3-ィル)メチル]エタナミン 158 mg とトリエチルァミン 0.100 mLのトルエン 3.0 mL溶液に 2-イソシアナト安息香酸メチル 8 3 mgを室温にて加えた。 90 °Cにて終夜撹拌した後、室温まで冷却した。反応混合物 を減圧下濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルムーメ タノール)で精製し 2-({[3-({[(lR)-l_(l-ナフチル)ェチル]アミノ}メチル )_4-フエ二ルビ ペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸メチル 27.3 mgを無色泡状物質として得 た。
[0098] 実施例 8
(lR)-l-(l-ナフチル) -N-K4-フエ二ルビペリジン- 3-ィル)メチル]エタナミンの粗生成 物 217 mgとトリエチルァミン 0.175 mLの THF4.0 mL溶液に 3_クロ口 _4-{[(4_ニトロフエ ノキシ)カルボニル]アミノ}安息香酸メチル 243 mgを加えた。室温で終夜撹拌した後、 反応混合物を減圧下濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー( へキサン 酢酸ェチル)で精製し、 3-クロ口- 4-({[3-({[(lR)-l_(l-ナフチル)ェチル]ァ ミノ }メチル )-4-フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸メチル 214 mg を得た。
[0099] 実施例 9
(l) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチノレ] 力ルバメートの粗生成物 150 mgとトリエチルァミン 0.094 mLの THF3 mL溶液に 2,2-ジ メチルダルタル酸無水物 62.3 mgを室温にて加えた。室温で終夜撹拌した後、反応 混合物を減圧下濃縮した。残渣を酢酸ェチルに溶解した後、 1M塩酸で洗浄し、有機 層を無水硫酸ナトリウムで乾燥した。ろ過、ろ液を減圧下濃縮し、 5-[3-({(tert-ブトキ シカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィ ノレ] -2,2-ジメチル -5-ォキソペンタン酸を粗生成物(無色泡状物質、 225 mg)として得
た。 ESI+: 587
(2)粗 5-[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4 -フエ二ルビペリジン- 1-ィル] -2,2-ジメチル -5-ォキソペンタン酸 225 mgに 4M塩化水 素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 1時間撹拌した。反応混合物を減 圧下濃縮した後、残渣を THFに溶解した。これにジイソプロピルエーテルを滴下し、 析出物をろ過により単離した。これを減圧下 50 °Cで乾燥し、 2,2-ジメチル -5-[3_({[(1 R)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル] -5-ォキソぺ ンタン酸塩酸塩 170 mgを白色固体として得た。
実施例 10
(1)粗 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチ ル]力ルバメート 238 mgの THF3 mL溶液に炭酸水素ナトリウム 200 mgを水 2 mLに懸 濁させながら加えた。反応混合物に 4- [(クロ口カルボニル)ォキシ]安息香酸メチル (フ ルカ(Fluka社)) 170 mgを加えた。室温にて終夜撹拌した後、混合液を酢酸ェチル で抽出した。有機層を水及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥 した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィ 一(へキサン 酢酸ェチル)で精製し 4- (メトキシカルボニル)フエニル 3_({(tert-ブトキ シカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1- カルボキシレートを粗生成物(353mg,無色泡状物質)として得た。 ESI+: 623
(2)得られた 4- (メトキシカルボニル)フエニル 3_({(tert-ブトキシカルボニル) [(lR)-l-( 1-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-カルボキシレートの粗生 成物 333 mgに THF3.0 mL、メタノーノレ 3.0 mL及び 1M水酸化ナトリウム水溶液 1.5 mL を加え、室温にて終夜撹拌した。 1M塩酸 1.6mLを加えて中和した後、酢酸ェチルで 抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得 られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム一メタノール)にて精製 し、 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 260 mgを淡黄色泡状物質 として得た。 ESI+: 609
(3) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-
フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 255 mgに 4M塩化水素 /1,4 -ジォキサン溶液 3.0 mLを加え、室温にて 1時間撹拌した。反応混合物を減圧下濃 縮した後、残渣を THFに溶解した。これにジイソプロピルエーテルを滴下し、析出をろ 過により単離した。これを減圧下 50 °Cで乾燥し、 4-({[3-({[(lR)-l_(l-ナフチル)ェチ ノレ]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸塩酸塩 159 mgを白色固体として得た。
[0101] 実施例 11
(1) tert-ブチノレ {[4-(3_フルオロフェニル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ チノレ)ェチノレ]力ルバメート 300 mgの THF2 mL溶液にトリェチルァミン 0.136 mLと 4- [( クロ口カルボニル)ォキシ]安息香酸メチル(フル力(Fluka社)) 167 mgを加え、室温に て 2時間で撹拌した。水を加え、酢酸ェチルで抽出した後、有機層を飽和食塩水で 洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して、得られた残 渣に THF8 mL、イソプロパノール 8 mL、及び 1M水酸化ナトリウム水溶液 4 mLを加え
、室温で 2日間撹拌した。 1M塩酸 4.1 mLを加えて中和した後、反応混合物を減圧下 濃縮した。得られた残渣をクロ口ホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥 した。ろ過後、ろ液を減圧下濃縮し得られた残渣をシリカゲルカラムクロマトグラフィー (クロ口ホルム一メタノール)により精製し、 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-( 1-ナフチル)ェチル]アミノ}メチル )-4-(3_フルオロフェニル)ピぺリジン- 1-ィル]カルボ 二ル}ォキシ)安息香酸 387 mgを無色泡状物質として得た。 ESI+: 627
(2) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- (3-フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 385 mgに 4M 塩化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 2時間撹拌した。反応混合 物を減圧下濃縮した後、残渣にイソプロパノールを加え、加熱し溶解させた。これに ジイソプロピルエーテルを滴下し析出物をろ過により単離し、減圧下乾燥し、 4-({[4-( 3-フルオロフェニル )-3-({[(lR)-l_(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン- 1-ィ ノレ]カルボ二ル}ォキシ)安息香酸塩酸塩 178 mgを白色固体として得た。
[0102] 実施例 12
(l) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチノレ]
力ルバメート 150 mgとトリエチルァミン 0.140 mLの THF3.0 mL溶液に製造例 11と同様 にして調製した 3-メトキシ _4-{[(4-ニトロフエノキシ)カルボニル]アミノ}安息香酸メチル 155 mgを加え、室温で終夜撹拌した。反応混合物を減圧下濃縮して得られた残渣を シリカゲルカラムクロマトグラフィー(へキサン一酢酸ェチル)で精製し 4-({[3-({(tert- ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジ ン- 1-ィル]カルボ二ル}ァミノ) -3-メトキシ安息香酸メチルを粗生成物(黄色泡状物質 、 297 mg)として得た。 ESI+: 652
(2)得られた 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミ メ チル) -4-フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ) -3-メトキシ安息香酸メチルの 粗生成物 220 mgに THF3 mL、メタノール 3 mL、及び 1M水酸化ナトリウム水溶液 1.5 mLを加え、室温にて終夜撹拌した。 1M塩酸 1.6mLを加えて中和した後、酢酸ェチル で抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し得 られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム メタノール)で精製し 、 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-4-フ ェニルビペリジン- 1-ィル]カルボ二ル}ァミノ) -3-メトキシ安息香酸 203 mgを淡黄色泡 状物質として得た。 ESI+: 638
(3) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- フエ二ルビペリジン -1-ィル]カルボ二ル}ァミノ) -3-メトキシ安息香酸 197 mgに 4M塩化 水素 /1,4-ジォキサン溶液 3.0 mLを加え、室温にて 1時間撹拌した。反応混合物を 減圧下濃縮した後、残渣を THFに溶解した。これにジイソプロピルエーテルを滴下し 、析出物をろ過により単離し、減圧下 50 °Cで乾燥して、 3-メトキシ _4-({[3-({[(lR)-l-( 1-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ)安 息香酸塩酸塩 88 mgを白色固体として得た。
実施例 13
(l) tert-ブチノレ {[4-(3_フルオロフェニル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ チノレ)ェチノレ]力ルバメート 300 mgのトノレェン 3.0 mL溶液にトリェチルァミン 0.136 mL と 4-イソシアナト安息香酸ェチル 149 mgを室温にて加え、 100 °Cにて 2日間撹拌した 。反応混合物を室温まで冷却後、水を加え、酢酸ェチルで抽出した。有機層を飽和
食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、得ら れた残渣に THF8 mL、メタノーノレ 4 mL及び 1M水酸化ナトリウム水溶液 4 mLを加え、 室温にて 24時間撹拌した。反応混合物に 1M塩酸 4.1 mLを加え中和した後、減圧下 濃縮し、クロ口ホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥し、ろ過後、ろ 液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム —メタノール)により精製し、 4-({[3-({(tert_ブトキシカルボニル) [(lR)-l-(l-ナフチル) ェチノレ]アミノ}メチル )-4-(3_フルオロフェニノレ)ピぺリジン- 1-ィル]カルボ二ノレ }ァミノ) 安息香酸 356 mgを無色泡状物質として得た。 ESI+: 626
(2) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- (3-フルオロフェニノレ)ピぺリジン- 1-ィル]カルボ二ノレ }ァミノ)安息香酸 354 mgに 4M塩 化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 2時間撹拌した。反応混合物 を減圧下濃縮した後、残渣にイソプロパノールを加え、加熱、溶解させた。これにジィ ソプロピルエーテルを滴下し、析出物をろ過により単離し、減圧下乾燥して、 4-({[4-( 3-フルオロフェニル )-3-({[(lR)-l_(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン- 1-ィ ノレ]カルボ二ル}ァミノ)安息香酸塩酸塩 180 mgを白色固体として得た。
実施例 14
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチノレ] 力ルバメート 150 mg、 3,4,5-トリフルォロ安息香酸メチル 96.2 mg、炭酸カリウム 93.3m g、および DMSO1.0 mLの混合物を 110 °Cにて 1時間撹拌した。室温まで冷却後、反 応混合物に水を加え、酢酸ェチルで抽出した。有機層を水、飽和食塩水で順次洗 浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣 をシリカゲルカラムクロマトグラフィー(へキサン一酢酸ェチル)で精製し、 4-[3-({(tert -ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジ ン -1-ィル] -3,5-ジフルォロ安息香酸メチル 127 mgを無色泡状物質として得た。 ESI+ : 615
(2) 4-[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸メチル 122 mgに、 THF2.0 mL 、メタノーノレ 2.0 mL、および 1M水酸化ナトリウム水溶液 1.0 mLをカロえ、室温にて終夜
撹拌した。 1M塩酸 1.1 mLを加え、中和した後、酢酸ェチルで抽出し、有機層を無水 硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲノレ カラムクロマトグラフィー(クロ口ホルム一メタノール)で精製し、 4-[3-({(tert-ブトキシカ ルポニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル] - 3,5-ジフルォロ安息香酸 115 mgを淡黄色油状物として得た。 ESI+: 601
(3) 4-[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-4- フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸 127 mgに 4M塩化水素 /1,4-ジ ォキサン溶液 3.0 mLを加え、室温にて 1時間撹拌した。反応混合物を減圧下濃縮し た後、残渣に THF、イソプロパノール、およびジイソプロピルエーテルを加え析出した 固体をろ過により単離し、減圧下乾燥して、 3,5-ジフルォ口- 4-[3-({[(lR)-Hl-ナフ チル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル]安息香酸塩酸塩 78 mgを 淡ピンク色固体として得た。
実施例 15
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチノレ] 力ルバメート 200 mgと 3,4,5-トリフルォロベンゾニトリノレ 212 mg、及び炭酸カリウム 18 7 mgの DMSO4.0 mL溶液を 110 °Cにて 2時間加熱した。反応混合物を室温まで冷却 後、水を加え、酢酸ェチルで抽出した。有機層を水、飽和食塩水で順次洗浄し、無 水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲ ルカラムクロマトグラフィー(へキサン—酢酸ェチル)で精製し、 tert-ブチル {[1_(4-シ ァノ -2,6-ジフルオロフェニル )-4-フエ二ルビペリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ チル)ェチル]力ルバメート 191 mgを無色泡状物質として得た。 ESI+: 582
(2) tert-ブチル {[1-(4-シァノ -2,6-ジフルオロフェニル )-4-フエ二ルビペリジン- 3-ィ ノレ]メチル }[(1R)-1-(1-ナフチノレ)ェチノレ]力ルバメート 185 mg、アジ化ナトリウム 414 mg 、トリェチルァミン塩酸塩 876 mg及び DMF 4.0 mLの混合物を 120 °Cにて終夜撹拌し た。反応混合物を室温まで冷却後、水を加え、酢酸ェチルで抽出した。有機層を水
、飽和塩化ナトリウム水溶液で順次洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、 ろ液を減圧下濃縮して、 tert-ブチル({1-[2,6_ジフルォ口- 4-( -テトラゾール- 5-ィ ノレ)フエニル] -4-フエ二ルビペリジン- 3-ィル }メチル) [(lR)-l-(l-ナフチノレ)ェチノレ]カル
バメートを粗生成物(ベージュ色泡状物質、 208 mg)として得た。 ESI+: 625
(3) tert_ブチル({1-[2,6_ジフルォ口- 4-(1Η-テトラゾール -5-ィル)フエニル] -4-フエ 二ルビペリジン- 3-ィル }メチル) [(lR)-l-(l-ナフチル)ェチル]力ルバメートの粗生成物 206 mgに 1,4-ジォキサン 2.0 mLと 4M塩化水素 /1,4_ジォキサン溶液 2.0 mLをカロえ、 室温にて 2日間撹拌した。反応混合物を減圧下濃縮後、残渣に酢酸ェチルと少量の エタノールを加えて加熱溶解させた。これにへキサンを加え、析出物をろ過により単 離し、減圧下乾燥して、(lR)-N-({l-[2,6-ジフルォ口- 4-( -テトラゾール -5-ィル)フ ェニル ]-4-フエ二ルビペリジン- 3-ィル }メチル )-1-(1_ナフチル)エタナミン塩酸塩 153 mgをベージュ色固体として得た。
[0106] 実施例 16
4- (メトキシカルボニル)フエニル 3-({[(lR)-l_(l-ナフチノレ)ェチノレ]アミノ}メチル )-4- フエ二ルビペリジン- 1-カルボキシレート 232 mgに THF6.0 mL、メタノーノレ 3.0 mL、お よび 1M水酸化ナトリウム水溶液 3.0 mLを加え、室温にて終夜撹拌した。 1M塩酸 3.0 mLを加えて中和した後、反応混合物を減圧下濃縮した。残渣に水を加え、クロロホ ルムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮 して得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム メタノール)で 精製した。これを少量のクロ口ホルムに溶解した後へキサンを加えて、析出物をろ過 にて単離し、減圧下 50 °Cで乾燥して 4-({[3-({[(lR)-l_(l-ナフチル)ェチル]アミノ}メチ ル) -4-フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 66 mgを白色固体と して得た。
[0107] 実施例 17
(l) tert-ブチル {[4-(2-フルオロフェニル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ チル)ェチル]力ルバメート 192 mgと炭酸水素ナトリウム 157 mgの混合物に THF3 mL と水 1.5 mLを加えた後、 4- [(クロ口カルボニル)ォキシ]安息香酸メチル(フル力(Fluka 社)) 133 mgを加えた。室温にて 1時間撹拌した後、酢酸ェチルで抽出した。有機層 を水及び飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を 減圧下濃縮し得られた残渣 327 mgに THF4 mL、メタノール 2 mL、及び 1M水酸化ナト リウム水溶液 2.5 mLを加え、室温で終夜撹拌した。反応混合物に 1M塩酸 2.6 mLを
加え中和した後、減圧下濃縮し、酢酸ェチルで抽出した。有機層を無水硫酸ナトリウ ムで乾燥した後、ろ過し、ろ液を減圧下濃縮した。得られた残渣をシリカゲルカラムク 口マトグラフィー(クロ口ホルム メタノール)で精製し、 4-({[3-({(tert_ブトキシカルボ二 ル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-4_(2-フルオロフェニノレ)ピぺリジン- 1- ィル]カルボ二ル}ォキシ)安息香酸 228 mgを無色泡状物質として得た。 ESI+: 627 (2) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- (2-フルオロフェニノレ)ピぺリジン- 1-ィノレ]カルボ二ノレ }ォキシ)安息香酸 213 mgに 4M 塩化水素 /1,4-ジォキサン溶液 3.0 mLを加え、室温にて終夜撹拌した。反応混合物 を減圧下濃縮した後、得られた残渣にクロ口ホルムを加え、飽和炭酸水素ナトリウム 水溶液(1.0 mL)で洗浄した。有機層を無水硫酸ナトリウムで乾燥した後、ろ過し、ろ 液を減圧下濃縮した。得られた残渣を酢酸ェチルに溶解した後へキサンを加えて、 析出物をろ過により単離した。減圧下乾燥して、 4-({[4-(2-フルオロフェニル )-3_({[(1 R)-l-(l-ナフチル)ェチル]アミノ}メチル)ピペリジン- 1-ィル]カルボ二ル}ォキシ)安息 香酸 135 mgを白色固体として得た。
実施例 18
(1)アルゴン雰囲気下、ドライアイス-アセトン浴で冷却した DMSO 1.62 mLのジクロ口 メタン 40 mL溶液に,内温を -70 °C以下に保ちながらォキサリルクロリド 0.996 mLを滴 下した。 10分撹拌後、 reH(3R,4R)_3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4 -ィル]メタノール 1.64 gのジクロロメタン 25 mL溶液を 20分間かけて滴下した。 -70 °C 以下を保ったまま 10分間撹拌後、トリェチルァミン 4.77 mLを 15分間かけて滴下し、 反応系を- 30 °Cまで昇温し、 15分間撹拌した。反応混合物に飽和塩化アンモニゥム 水溶液を加えた後、室温に昇温した。クロ口ホルムで抽出した後、有機層を水で洗浄 し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し re卜 (3R,4R)-3-フエ ニル -1- (トリフルォロアセチル)ピぺリジン- 4-カルボアルデヒドを粗生成物(淡黄色油 状物、 1.78 g)として得た。
(2) (1R)-1-(1-ナフチル)エタナミン 1.03 g、酢酸 0.327 mL、トリァセトキシ水素化ホウ 素ナトリウム 3.63 g及び 1, 2-ジクロロェタン 70 mLの混合物に得られた粗 re卜 (3R,4R)_ 3-フエニル -1- (トリフルォロアセチル)ピぺリジン- 4-カルボアルデヒド 1.78 gの 1, 2-ジ
クロロェタン 30 mL溶液を室温にて滴下し、終夜撹拌した。飽和炭酸水素ナトリウム水 溶液を加え 10分間撹拌した後、クロ口ホルムにて抽出し、有機層を無水硫酸ナトリウ ムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマ トグラフィー(へキサン 酢酸ェチル)で精製し (lR)-l-(l-ナフチル) -N-{[3-フエ二ル- 1- (トリフルォロアセチル)ピぺリジン- 4-ィル]メチル }エタナミン 2.19 gを無色泡状物質 として得た。
[0109] 実施例 19
(1) re卜 [(3R,4R)-3-(3-フルオロフェニル )-1- (トリフルォロアセチル)ピぺリジン- 4-ィル ]メタノール 1.50 gのジクロロメタン 15 mL溶液に、室温にてデス一マーチンペルョー ジナン 2.29 gを加え、 1.5時間撹拌した。反応混合物に飽和チォ硫酸ナトリウム水溶 液および飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出した。有機層を 水および飽和食塩水で順次洗浄し、硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧 下濃縮し、粗製の re卜 (3R,4R)-3-(3-フルオロフヱニル) -1- (トリフルォロアセチル)ピぺ リジン- 4-カルボアルデヒド 1.83 gを橙色油状化合物として得た。
(2)得られた粗 re卜 (3R,4R)-3-(3-フルオロフヱニル) -1- (トリフルォロアセチル)ピペリ ジン- 4-カルボアルデヒドおよび (lR)-l-(l-ナフチル)エタナミン 882 mgのジクロ口メタ ン 20 mL溶液に、室温にてトリァセトキシ水素化ホウ素ナトリウム 3.12 gを加え、 1時間 撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出 した。有機層を水および飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥した。 ろ過後、ろ液を減圧下濃縮し得られた残渣を NHシリカゲル(富士シリシァ化学、 日本 )を用いたカラムクロマトグラフィー(へキサン一酢酸ェチル)により精製し、(lR)-N-{[3 -(3-フルオロフェニル )- 1- (トリフルォロァセチル)ピペリジン- 4-ィル]メチル }- 1-( 1-ナ フチル)エタナミン 1.64 gを無色泡状物質として得た。
[0110] 実施例 20
(l) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mgとトリエチルァミン 0.038 mLの THF3 mL溶液に 4- (クロ口カルボ二 ノレ)安息香酸メチル 46.9 mgを加え、室温にて終夜撹拌した。反応混合物に飽和炭 酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出し、有機層を無水硫酸ナトリウム
で乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマト グラフィー(へキサン—酢酸ェチル)で精製し 4-{[4-({(tert-ブトキシカルボニル) [(1R)- 1-(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息 香酸メチル 56.0 mgを無色泡状物質として得た。 FAB+: 607
(2) 4-{[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィノレ]カルボ二ノレ }安息香酸メチル 55 mgに THF2.0 mL、メタノ ール 1.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mLを加え、室温で終夜撹拌した。 1M 塩酸を 1.1 mLを加えて中和した後、クロ口ホルムで抽出し、有機層を無水硫酸ナトリ ゥムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して、 4-{[4-({(tert-ブトキシカルボ ニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カル ボニル }安息香酸を粗生成物(無色泡状物質、 57.5 mg)として得た。
(3)得られた 4-{[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミ メ チル) -3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息香酸の粗生成物 57.5 mgに 4M 塩化水素 /1,4-ジォキサン溶液 2.0 mLを加えた。室温にて 2時間撹拌した後、反応 混合物を減圧下濃縮した。得られた残渣にエタノール 酢酸ェチルを加え、結晶化 を行い、 4-{[4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1- ィル]カルボ二ル}安息香酸塩酸塩 15.6 mgを白色固体として得た。
実施例 21
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mg、イソフタル酸モノメチル 44.6 mg、および HOBt 51.7 mgの混合 物をジクロロメタン 1.0 mLに溶解した後、 WSO塩酸塩 36.5 mgを加え、室温で 2日間 撹拌した。反応混合物を減圧下濃縮し、得られた残渣をシリカゲルカラムクロマトダラ フィー(へキサン 酢酸ェチル)で精製し 3-{[4-({(tert-ブトキシカルボニル) [(1R)-1-(1 -ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息香酸 メチル 122 mgを無色泡状物質として得た。
(2)得られた 3-{[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミ メ チル) -3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息香酸メチル 122 mgに THF2.0 mL、メタノール 1.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mLをカロえ、室温にて終夜
撹拌した。 1M塩酸 1.1 mLを加えて中和した後、酢酸ェチルで抽出し、有機層を無 水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して、 3_{[4-({(tert-ブ トキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息香酸を粗生成物(無色泡状物質、 121 mg)として得た。
(3)得られた粗 3-{[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ} メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ二ル}安息香酸 118 mgに 4M塩化水素/ 1,4-ジォキサン溶液 2.0 mLを加えた。室温にて終夜撹拌した後、反応混合物を減圧 下濃縮した。得られた残渣をイソプロパノールに加熱溶解し、ジイソプロピルエーテ ルを加えて目的物の粗沈殿を析出させた。析出物をろ過して得られた白色固体に飽 和炭酸水素ナトリウム水溶液を加え、クロ口ホルムで抽出し、有機層を無水硫酸ナトリ ゥムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣に 4M塩化水素 /1,4- ジォキサン溶液 0.5 mLを加えた後、濃縮した。得られた白色固体をイソプロパノール で洗浄して、乾燥し、 3-{[4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエニル ピペリジン- 1-ィル]カルボ二ル}安息香酸塩酸塩 93 mgを白色固体として得た。
実施例 22
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mg、 2,2'-[(tert-ブトキシカルボニル)ィミノ]ジ酢酸(フル力(Fluka社) ) 57.7 mg、および HOBt 51.7 mgの混合物をジクロロメタン 1.0 mLに溶解した後、 WS C '塩酸塩 36.5 mgを加え、室温にて 2日間撹拌した。反応混合物を減圧下濃縮し、得 られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム メタノール)で精製し [ (tert-ブトキシカルボニル) {2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェ チル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -2-ォキソェチル }ァミノ]酢酸 139 mgを無色泡状物質として得た。
(2) [ 6 -ブトキシカルボニル){2-[4-({ 6 -ブトキシカルボニル)[(11¾-1-(1-ナフチル )ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -2-ォキソェチル }ァミノ]酢酸 13 8 mgに 4M塩化水素 /1,4-ジォキサン溶液 2.0 mLを加えた。室温で 2時間撹拌した後 、反応混合物を減圧下濃縮し、得られた残渣にイソプロパノールとジイソプロピルェ 一テルを加え析出した固体を濾取し、白色固体として ({2-[4-({[(lR)-l-(l-ナフチル)
ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -2-ォキソェチル }ァミノ)酢酸塩 酸塩 83.9 mgを得た。
[0113] 実施例 23
(1) tert-ブチル {[3-(3_フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チル)ェチル]力ルバメート 139 mg、 4 クロロー 4 ォキソブタン酸ェチル 54 mg及び THF 4.0 mLの混合物に室温にてトリエチルァミン 0.084 mLをカロえ 3日間撹拌した。反 応混合物に飽和塩化アンモユウム水溶液を加え酢酸ェチルで抽出し、有機層を水 および飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減 圧下濃縮し、無色油状化合物として 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナ フチノレ)ェチノレ]アミノ}メチノレ) -3-(3_フルオロフェニノレ)ピぺリジン- 1-ィル] -4-ォキソブ タン酸ェチノレ 177 mgを得た。 ESI+: 591
(2) 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタン酸ェチル 177 mgのエタノール 2.0 mL溶液に、室温にて 1M水酸化ナトリウム水溶液 1.00 mLを加え、 2時間撹拌した
。 1M塩酸 1.00 mLを加え中和した。酢酸ェチルで抽出し、有機層を飽和食塩水で洗 浄した後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、無色油状 化合物として 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミ メ チル) -3-(3_フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタン酸 174 mgを得た。 E SI+: 563
(3) 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタン酸 174 mgの酢酸ェチル 1.0 m L溶液に室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え 2時間撹拌した。ジィ ソプロピルエーテルを加え、生じた析出物をろ取した後、減圧下乾燥し、白色固体と して 4-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピペリ ジン- 1-ィル] -4-ォキソブタン酸塩酸塩 100 mgを得た。
[0114] 実施例 24
(l) 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタン酸 169 mg、 4ーァミノ安息香
酸メチル 54 mg、 WSO塩酸塩 61 mg、 HOBt 45 mgおよび DMF 2.00 mLの混合液を、 室温にて 2時間攪拌した後、飽和炭酸水素ナトリウム水溶液を加えた。酢酸ェチルで 抽出し、有機層を水および飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過 後、ろ液を減圧下濃縮し、淡褐色油状化合物として 4-({4-[4-({(tert-ブトキシカルボ ニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )_3-(3-フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタノィル }ァミノ)安息香酸メチル 208 mgを得た。 ESI+: 696
(2) 4-({4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル) - 3-(3_フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタノィル }ァミノ)安息香酸メチ ル 208 mgの THF 2.0 mLおよびメタノール 1.0 mL溶液に室温にて 1M水酸化ナトリウ ム水溶液 1.00 mLを加えた後、 60 °Cにて 2時間撹拌した。反応混合物を室温まで冷 却後、酢酸ェチル及び水を加え分液操作を行った。得られた水層に、 1M塩酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄後、無 水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラ ムクロマトグラフィー(クロ口ホルム メタノール)により精製し、黄色油状化合物として 4 _({4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-(3_ フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタノィル }ァミノ)安息香酸 107 mgを 得た。 ESI-: 680
(3) 4-({4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル) - 3-(3_フルオロフェニル)ピぺリジン- 1-ィル] -4-ォキソブタノィル }ァミノ)安息香酸 107 mgの酢酸ェチル 2.0 mL溶液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを 加え、 2時間撹拌した。ジイソプロピルエーテルを加え、生じた析出物をろ取、減圧下 乾燥し、淡黄色固体として 4-({4-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチル) ェチル]アミノ}メチル)ピペリジン- 1-ィル ]_4-ォキソブタノィル }ァミノ)安息香酸塩酸塩
28 mgを得た。
実施例 25
(1)粗 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチ ル]力ルバメート 220 mgとトリエチルァミン 0.104 mLの THF4.0 mL溶液に、室温下 4_[( クロ口カルボニル)ォキシ]安息香酸メチル(フル力(Fluka社)) 127 mgを加えた。室温
にて 1時間撹拌した後、反応混合物を減圧下濃縮し、得られた残渣をシリカゲルカラ ムクロマトグラフィー(へキサン 酢酸ェチル)で精製して 4- (メトキシカルボニル)フエ ニル 4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フ ェニルビペリジン- 1-カルボキシレートを無色アモルファス状の粗生成物 264 mgとして 得た。 ESI+: 623
(2)得られた 4- (メトキシカルボニル)フエニル 4-({(tert-ブトキシカルボニル) [(lR)-l-( 1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-カルボキシレートの粗生 成物 260 mgに THF4.0 mL、メタノーノレ 2.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mL を加え、室温にて終夜撹拌した。 1M塩酸 l. lmLを加えて中和した後、クロ口ホルムで 抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得 られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム一メタノール)にて精製 し、 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 197 mgを淡黄色泡状物質 として得た。 ESI+: 609
(3) 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 193 mgに 1,4-ジォキサン 2. 0 mLおよび 4M塩化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 5時間撹拌し た。反応混合物を減圧下濃縮した後、残渣をイソプロパノールに加熱還流した後、混 合物を室温に放冷した。析出物を濾取し、減圧下乾燥して 4-({[4-({[(lR)-l-(l-ナフ チル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ二ル}ォキシ)安息香 酸塩酸塩 117 mgを白色固体として得た。
実施例 26
(1) 3—ヒドロキシ安息香酸メチル 5.000 g、クロロギ酸ー4一二トロフエニル 6.955 gおよ びトルエン 100 mLの懸濁液に、室温にてトリエチノレアミン 4.81 mLをカロえ、終夜攪拌し た。不溶物を濾過にて除去した後、減圧下溶媒を留去し、淡黄色固体として 3-{[(4_ ニトロフエノキシ)カルボ二ノレ]ォキシ }安息香酸メチル 5.948 gを得た。 3_{[(4-ニトロフエ ノキシ)カルボニル]ォキシ }安息香酸メチル 100 mg、 tert-ブチル {[3-(3_フルオロフェ 二ノレ)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチル)ェチル]力ルバメート 139 mgお
よび THF 4 ml混合物に室温にてトリエチルァミン 0.084 mLを加え終夜攪拌した。飽 和炭酸水素ナトリウム水溶液を加えた後、酢酸ェチルで抽出した。有機層を水およ び飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し 、淡黄色油状化合物として 3- (メトキシカルボニル)フエニル 4-({(tert-ブトキシカルボ二 ル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )_3-(3-フルオロフェニノレ)ピぺリジン- 1- カルボン酸 196 mgを得た。
(2) 3-(メトキシカルボニノレ)フェニル4-({ 6 -ブトキシカルボニル)[(11¾-1-(1-ナフチ ノレ)ェチノレ]アミノ}メチノレ) -3-(3_フルオロフェニノレ)ピぺリジン- 1-カルボン酸 196 mgの THF 2.0 mLおよびメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム水溶液 1.0 0 mLを加え、 1時間撹拌した。反応混合物に酢酸ェチル及び水を加え分液操作を行 つた。得られた水層に 1M塩酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。 有機層を飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下 濃縮し、 3-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-(3_フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 88 mgを得 た。 ESト: 625
(3) 3-({[4-({ 6 -ブトキシカルボニル)[(11¾-1-(1-ナフチル)ェチル]ァミノ}メチル)-3- (3-フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ォキシ)安息香酸 88 mgの酢酸 ェチル 2.0 mL溶液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを力 Pえ、 2時 間撹拌した。生じた析出物をろ取、減圧下乾燥し、白色固体として 3-({[3-(3_フルォ 口フエニル) -4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン- 1-ィル]カルボ 二ル}ォキシ)安息香酸塩酸塩 22 mgを得た。
実施例 27
(1)粗 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチ ノレ]力ルバメート 181 mgとトリエチルァミン 0.114 mLの THF2.7 mL溶液に 4-イソシアナ ト安息香酸ェチル 117 mgを室温にて加えた。反応容器を密閉し、 100 °Cにて終夜撹 拌した。反応混合物を室温まで冷却した後、飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧 下濃縮して、 4-({[4-({(tert_ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メ
チル) -3-フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸ェチル 357 mgを粗 生成物として得た。 ESI+: 636
(2)得られた 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メ チル) -3-フエ二ルビペリジン -1-ィル]カルボ二ル}ァミノ)安息香酸ェチルの粗生成物 3 55 mgに THF4.0 mL、メタノーノレ 2.0 mL及び 1M水酸化ナトリウム水溶液 2.0 mLを加え 、室温にて終夜撹拌した。 1M塩酸 2.2mLを加えて中和した後、クロ口ホルムで抽出し 、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた 残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム一メタノール)にて精製し、 4-({
[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二 ルビペリジン -1-ィル]カルボ二ル}ァミノ)安息香酸の粗生成物 291 mgを得た。 ESI+: 6 08
(3) 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸の粗生成物 289 mgに 4M塩 化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 2時間撹拌した。反応混合物 を減圧下濃縮した後、残渣をイソプロパノールに加熱還流下、溶解した。これにジィ ソプロピルエーテルを滴下し、析出物をろ過により単離し、減圧下で乾燥して、 4-({[4 -({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ 二ル}ァミノ)安息香酸塩酸塩 159 mgを白色固体として得た。
実施例 28
(1)粗 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチ ル]力ルバメート 181 mgおよびトリェチルァミン 0.114 mLの THF3.0 mL溶液に、室温に て 3-クロ口- 4-{[(4-ニトロフエノキシ)カルボニル]アミノ}安息香酸メチル 171 mgを加え
、終夜攪拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルム で抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 4 _({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ 二ルビペリジン -1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸メチルの粗生成物 352 mgを得た。 ESI+: 656
(2)得られた 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メ
チル) -3-フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸メチルの粗 生成物 347 mgに THF4.0 mL、メタノーノレ 2.0 mL及び 1M水酸化ナトリウム水溶液 2.0 m Lをカロえ、室温にて終夜撹拌した。 1M塩酸 2.2mLを加えて中和した後、クロ口ホルム で抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して 得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム メタノール)にて精 製し、 4-({[4-({(tert-ブトキシカルボニル) [( 1 R)_ 1 _( 1 -ナフチル)ェチル]アミ メチル) -
3-フエ二ルビペリジン -1-ィル]カルボ二ル}ァミノ )_3-クロ口安息香酸の粗生成物 306 mgを得た。 ESI+: 642
(3) 4-({[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸の粗生成物 302 mg に 4M塩化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 2時間撹拌した。反応 混合物を減圧下濃縮した後、残渣にイソプロパノールを加え、加熱還流した。混合物 を室温に冷却した後、析出物をろ過により単離し、減圧下で乾燥して、 3-クロ口- 4-({[
4- ({[(lR)-l_(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ 二ル}ァミノ)安息香酸塩酸塩 147 mgを白色固体として得た。
実施例 29
(1) tert-ブチル {[3-(3_フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チノレ)ェチノレ]力ルバメート 231 mgおよび 3-クロ口- 4-{[(4-ニトロフエノキシ)カルボニル ]ァミノ }安息香酸メチル 210 mgの THF4.0 mL溶液に、室温にてトリエチルァミン 0.139 mLを加え、終夜攪拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、酢 酸ェチルで抽出した。有機層を水および飽和食塩水で順次洗浄し、無水硫酸ナトリ ゥムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 4-({[4-({(tert-ブトキシカルボニル) [(1 R)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-(3_フルオロフェニノレ)ピぺリジン- 1-ィル] カルボ二ル}ァミノ) -3-クロ口安息香酸メチルの粗生成物を淡黄色泡状物質として 394 mg得た。 ESI+: 674
( 2)粗 4-({[4-({(tert-ブトキシカルボニル) [( 1 R)_ 1 _( 1 -ナフチル)ェチル]アミノ}メチル) - 3-(3_フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸メチ ル 241 mgを 4M塩化水素 /1,4 ジォキサン 4.0 mL溶液に室温にて溶解し、終夜攪拌
した。反応混合物を減圧下濃縮後、残渣をシリカゲルカラムクロマトグラフィー(クロ口 ホルム-メタノール)で精製し、 3-クロ口- 4-({[4-(3_フルオロフヱニル) _3-({[(lR)-l-(l- ナフチル)ェチル]アミノ}メチル)ピペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸メチル の粗生成物を黄色油状化合物として得た。
(3)得られた粗 3-クロ口- 4-({[4-(3_フルオロフヱニル) _3-({[(lR)-l-(l-ナフチル)ェチ ノレ]アミノ}メチル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸メチルとメタノール 2.0 mLおよび THF2.0 mLの混合液に、室温にて 1M水酸化ナトリウム水溶液 2.00 mLを 加えた後、 65 °Cにて 2.5時間撹拌した。反応混合物を室温まで冷却後、水層を酢酸 ェチルにて洗浄後、水層に 1M塩酸を加え中和し、酢酸ェチルで抽出した。有機層を 飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し 、 3-クロ口- 4-({[3-(3_フルオロフヱニル) -4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチ ノレ)ピぺリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸を粗生成物として得た。
(4)得られた粗 3-クロ口- 4-({[3-(3_フルオロフヱニル) -4-({[(lR)-l-(l-ナフチル)ェチ ノレ]アミノ}メチル)ピペリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸とメタノール 2.0 mLお よび THF2.0 mLの混合液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え 、 3時間撹拌した。反応混合物を減圧下濃縮し、残渣を酢酸ェチルおよび THFで洗 浄し、ろ取により単離した。減圧下乾燥し 3-クロ口- 4-({[3-(3_フルオロフェニル )_4-({[( 1 R)_ 1 -( 1 -ナフチル)ェチル]アミノ}メチル)ピペリジン- 1 -ィル]カルボ二ル}アミノ)安息 香酸塩酸塩 92 mgを白色固体として得た。
実施例 30
(l) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mg、 6-クロ口ピリジン- 2-カルボン酸ェチル 46 mg、炭酸カリウム 37 m gおよび DMSO 2.0 mLの混合物を、 100 °Cにて終夜攪拌した。反応混合物を室温ま で冷却後、水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄、無水硫 酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロ マトグラフィー(へキサン 酢酸ェチル)により精製し、無色樹脂状化合物として 6_[4- ({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビ ペリジン- 1-ィル]ピリジン- 2-カルボン酸ェチル 75 mgを得た。 ESI + : 594
(2) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィノレ]ピリジン- 2-カルボン酸ェチル 75 mgの THF 2.0 mLおよ びメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム水溶液 1.00 mLをカロえ、終 夜攪拌した。 1M塩酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。有機層を 飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 無色樹脂状化合物化合物として 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチ ル)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ピリジン- 2-カルボン酸 75 mg を粗生成物として得た。 ESI+ : 566
(3)粗 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -フエ二ルビペリジン- 1-ィル]ピリジン- 2-カルボン酸 75 mgの酢酸ェチル 2.0 mL溶液 に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え 2時間撹拌した。反応混 合物にジイソプロピルエーテルを加え、生じた析出物をろ取、減圧下乾燥し、淡黄色 固体として 6-[4-({[(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ピリジン- 2-カルボン酸塩酸塩 50 mgを得た。
実施例 31
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mg、 5,6-ジクロロニコチン酸ェチル 59.4 mg、および炭酸カリウム 3 7.3 mgの混合物に DMSO 2.0 mLを室温にて加え、混合液を 100 °Cにて終夜撹拌し た。室温まで冷却後、水を加えた。酢酸ェチルで抽出した後、有機層を無水硫酸ナト リウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムク 口マトグラフィー(へキサン一酢酸ェチル)で精製し 6-[4-({(tert-ブトキシカルボニル) [( 1R)-1-(1-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5-クロロニ コチン酸ェチル 128 mgを無色泡状物質として得た。 FAB+: 628
(2) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -5-クロ口ニコチン酸ェチル 126 mgに THF2.0 mL、メタノ ール 1.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mLを加え、室温にて 2日間撹拌した 。 1M塩酸 1.1 mLを加えて中和した後、酢酸ェチルで抽出し、有機層を無水硫酸ナト リウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して、 6_[4-({(tert-ブトキシカル
ボニル )[(1R)-1_(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5- クロ口ニコチン酸を粗生成物(無色泡状物質、 142 mg)として得た。
(3) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -5-クロ口ニコチン酸の粗生成物 141 mgに 4M塩化水素 /1 ,4-ジォキサン溶液 2.0 mLを加えた。室温で 2時間撹拌した後、反応混合物を減圧 下濃縮し、得られた残渣にエタノールを加え、結晶化を行い、 5-クロ口- 6_[4-({[(lR)_ 1-(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ニコチン酸塩酸 塩 49.8 mgを白色固体として得た。
実施例 32
(1) tert-ブチル {[3_(2-フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チル)ェチル]力ルバメート 116 mg、 5,6—ジクロ口ニコチン酸ェチル 66 mg、炭酸カリ ゥム 42 mgおよび DMSO 2.0 mLの混合物を 100 °Cで終夜攪拌した。室温にまで放冷 し、反応混合物に水を加え、生じた析出物をろ取した。減圧下乾燥し、 6-[4-({(tert- ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ) -3_(2-フルオロフェニ ル)ピぺリジン- 1-ィル] -5-クロ口ニコチン酸ェチルの粗生成物 110 mgを白色固体とし て得た。 ESI+: 646
(2)前述の反応で得られた粗 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル) ェチノレ]アミノ}メチノレ) -3_(2-フルオロフェニノレ)ピぺリジン- 1-ィル] -5-クロ口ニコチン酸 ェチルの THF 2.0 mLおよびメタノール 1.0 mL溶液に、 1M水酸化ナトリウム水溶液 1· 00 mLを力 Pえ、室温にて終夜攪拌した。 1M塩酸 1.00 mLを加え中和し、酢酸ェチル で抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。ろ過後 、濾液を減圧下濃縮し、 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチ ノレ]アミノ}メチノレ) -3_(2-フルオロフェニノレ)ピぺリジン- 1-ィル] -5-クロ口ニコチン酸の 粗生成物を淡黄色非晶状化合物として 115 mgを得た。 ESI+: 618
(3)前述の反応で得られた粗 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル) ェチノレ]アミノ}メチノレ) -3_(2-フルオロフェニノレ)ピぺリジン- 1-ィル] -5-クロ口ニコチン酸 の酢酸ェチル 2.0 mL溶液に、 4M塩化水素/酢酸ェチル溶液 1.00 mLをカロえ、室温 にて 2時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣を逆相シリカゲル力
ラムクロマトグラフィー(ァセトニトリル- 0.01M塩酸)により精製し、白色固体として 5-ク ロロ- 6-[3_(2-フルオロフヱニル) -4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル)ピぺ リジン- 1-ィル]ニコチン酸塩酸塩 56 mgを得た。
実施例 33
(1)粗 tert-ブチル [(lR)-l-(3-メトキシフエニル)ェチル ][(3-フエ二ルビペリジン- 4-ィ ル)メチル]力ルバメート 119 mg、 5,6-ジクロロニコチン酸ェチル 62 mg、および炭酸 カリウム 39 mgの混合物に DMSO 1.0 mLを室温にて加え、混合液を 100 °Cにて終夜 撹拌した。室温まで冷却後、反応混合物に水を加え、酢酸ェチルで抽出した後、有 機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣 をシリカゲルカラムクロマトグラフィー(へキサン一酢酸ェチル)で精製し 6-[4-({(tert- ブトキシカルボニル) [(lR)-l-(3-メトキシフエ二ノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビ ペリジン- 1-ィル] -5-クロ口ニコチン酸ェチル 132 mgを無色泡状物質として得た。 FA B+: 608
(2) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(3-メトキシフエニル)ェチル]アミノ}メチ ル) -3-フエ二ルビペリジン- 1-ィル] -5-クロ口ニコチン酸ェチル 131 mgに THF2.0 mL 、メタノール 1.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mLをカロえ、室温にて 2日間撹 拌した。 1M塩酸 1.1 mLを加えて中和した後、酢酸ェチルで抽出し、有機層を無水硫 酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮、乾燥して、 6_[4-({(tert-ブトキシ カルボニル) [(lR)-l-(3-メトキシフエ二ノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビペリジン -1-ィル] -5-クロ口ニコチン酸の粗生成物を無色泡状物質として 143 mg得た。 ESI+: 5 80
(3) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(3-メトキシフエニル)ェチル]アミノ}メチ ル) -3-フエ二ルビペリジン- 1-ィル] -5-クロ口ニコチン酸の粗生成物 142 mgに 4M塩化 水素 /1,4-ジォキサン溶液 2.0 mLを加えた。室温で 2時間撹拌した後、反応混合物 を減圧下濃縮し、得られた残渣にエタノールと酢酸ェチルを加え、加熱還流した後、 室温まで放冷し、析出物を濾取し、減圧下乾燥して 5-クロ口- 6-[4-({[(lR)-l-(3-メトキ シフエニル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ニコチン酸塩酸塩 7 8 mgを白色固体として得た。
[0124] 実施例 34
(1)トリス (ジベンジリデンアセトン)ジパラジウム (0) 2.3 mg、 2- (ジシクロへキシルホスフ イノ) -2'- (ジメチルァミノ)ビフエニル 1.9 mg、および 1,4-ジォキサン 0.5 mLの混合物に 、 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチル]力 ルバメート 100 mg、 3—ョード安息香酸ェチル 83.8 mg、炭酸セシウム 105 mg、 1,4-ジ ォキサン 1.0 mLおよび t-ブタノール 1.5 mLを加え 100 °Cにて終夜撹拌した。室温ま で冷却後、反応混合物に酢酸ェチルを加え、不溶物を除き溶媒を減圧留去した。残 渣をシリカゲルカラムクロマトグラフィー(へキサン一酢酸ェチル)で精製し 3-[4-({(tert -ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジ ン -1-ィル]安息香酸ェチル 108 mgを淡黄色泡状物質として得た。 ESI+: 593
(2) 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィノレ]安息香酸ェチル 107 mgに THF2.0 mL、メタノール 1.0 m L及び 1M水酸化ナトリウム水溶液 1.0 mLを加え、室温で 2日間撹拌した。 1M塩酸 1.1 mLを加えて中和した後、酢酸ェチルで抽出し、有機層を無水硫酸ナトリウムで乾燥 した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィ 一(クロ口ホルム一メタノール)により精製し 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-( 1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]安息香酸 100 mgを 無色泡状物質として得た。 ESI+: 565
(3) 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル]安息香酸 99 mgに塩酸の 4M塩化水素 /1,4-ジォキサン 溶液 2.0 mLを加えた。室温で 2時間撹拌した後、反応混合物を減圧下濃縮し、得ら れた残渣に THFとイソプロパノールを加え、固体化を行い、 3-[4-({[(lR)-l-(l-ナフチ ル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]安息香酸塩酸塩 76 mgを白 色固体として得た。
[0125] 実施例 35
(1) tert-ブチル {[3_(2-フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チル)ェチル]力ルバメート 100 mg、 2—ジシクロへキシルホスフィノー 2'— (N,N—ジメ チルァミノ)ビフエニル 2 mg、トリス(ジベンジリデンアセトン)パラジウム(0) 2 mg、炭酸
セシウム 106 mg、 3—ョード安息香酸ェチル 84 mg、 t—ブチルアルコール 1.50 mLお よび 1,4—ジォキサン 1.50 mLの混合物を、 100 °Cにて終夜攪拌した。反応混合物を 室温まで冷却後、酢酸ェチルで希釈し、不溶物を濾過にて除去した。ろ液を減圧下 濃縮し、淡黄色油状化合物として 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフ チル)ェチル]アミノ}メチル )-3-(2-フルオロフェニル)ピぺリジン- 1-ィル]安息香酸ェチ ル 172 mgを粗生成物として得た。 ESI+: 611
(2)粗 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -(2-フルオロフェニノレ)ピぺリジン- 1-ィノレ]安息香酸ェチル 172 mgの THF 2.0 mLお よびメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム水溶液 1.00 mLを加え、 終夜攪拌した。 85 °Cにて 1時間撹拌した。 1M水酸化ナトリウム水溶液 0.50 mLを加え 、 85 °Cにて 1時間撹拌した。反応混合物を室温まで冷却後、 1M塩酸 1.50 mLを加え 中和した後、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸ナトリウ ムで乾燥した。ろ過後、ろ液を減圧下濃縮し、淡黄色油状化合物として 3-[4-({(tert_ ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ) -3_(2-フルオロフェニ ル)ピぺリジン- 1-ィル]安息香酸 161 mgを粗生成物として得た。 ESI+: 583
(3)粗 3-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -(2-フルオロフェニル)ピぺリジン- 1-ィル]安息香酸 161 mgの酢酸ェチル 2.0 mL溶 液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え、 1時間撹拌した。減圧 下溶媒を留去した。残渣を逆相シリカゲルカラムクロマトグラフィー(ァセトニトリル- 0.0 01M塩酸)により精製し、白色固体として 3-[3_(2-フルオロフヱニル) -4-({[(lR)-l-(l- ナフチル)ェチル]アミノ}メチル)ピぺリジン- 1-ィル]安息香酸塩酸塩 58 mgを得た。 実施例 36
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチノレ] 力ルバメート 100 mg、 2,6—ジクロ口イソニコチン酸メチル 56 mg、リン酸カリウム 95 mg 、ビス (トリ- tert-ブチルホスフィン)パラジウム(0) 6 mgおよびジメチルァセトアミド 2.0 m Lの混合物を、 100 °Cにて終夜攪拌した。反応混合物を室温まで冷却後、水を加え、 酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥し た。ろ過後、ろ液を減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(へキ
サン—酢酸ェチル)により精製し、淡黄色油状化合物として 2-[4-({(tert-ブトキシカル ボニル )[(1R)-1_(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -6- クロ口イソニコチン酸メチル 66 mgを得た。 ESI+: 614
(2) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -6-クロ口イソニコチン酸メチル 66 mgの THF 2.0 mlおよ びメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム水溶液 1.00 mLをカロえ、終 夜攪拌した。 1M塩酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。有機層を 飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 淡黄色樹脂状化合物として 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェ チル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -6-クロ口イソニコチン酸 68 mgを 得た。 ESI+: 600
(3) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -6-クロ口イソニコチン酸 68 mgの 1,4—ジォキサン 1.0 mL 溶液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え、 2時間撹拌した。減 圧下溶媒を留去した。残渣をジイソピロピルエーテルで洗浄、減圧下乾燥し、淡黄色 固体として 2-クロ口- 6-[4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3-フエ二ルビ ペリジン -1-ィル]イソニコチン酸塩酸塩 29 mgを得た。
実施例 37
(1) tert-ブチル {[3-(3_フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チノレ)ェチル]力ルバメート 216 mg、ベンゾィルイソチオシアナート 80 mg及びトノレエン 2.00 mLの混合物を、室温にて 2時間撹拌した。減圧下溶媒を留去した。残渣にメタノ 一ノレ 2.0 mLを加えた後、室温にてメチルァミン 9.8Mメタノール溶液 0.24 mLをカロえ、 2 日間撹拌した。減圧下溶媒を留去し、 tert-ブチル {[1- (ァミノカルボノチオイル) -3-(3_ フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチル)ェチル]カルバメー ト 303 mgを淡橙色非晶状化合物の粗生成物として得た。 ESI+: 522
(2)粗 tert-ブチル {[1- (ァミノカルボノチオイル) -3-(3_フルオロフェニル)ピぺリジン- 4 -ィル]メチル }[(1R)-1-(1-ナフチル)ェチル]力ルバメート 303 mgおよびブロモピルビン 酸ェチル 182 mgのエタノール 2.0 ml溶液に、室温にて 1M水酸化ナトリウム水溶液 2.8
mLを加えた。 85 °Cにて 3時間撹拌した。反応混合物を室温まで冷却後、減圧下濃 縮した。残渣に水およびジェチルエーテルを加え、分液操作を行った。得られた水 層に、 1M塩酸を加え酸性とし、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄 、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、橙色油状化合物とし て 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3-(3_ フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸 198 mgを粗生 成物として得た。 ESI+: 590
(3)粗 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -(3-フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸 198 mgの THF 2.0 ml溶液に、室温にて 4M塩化水素酢酸ェチル溶液 1.00 mlを加え、 1時間撹 拌した。生じた析出物をろ取、減圧下乾燥し、白色固体として 2-[3-(3_フルオロフェ ニル) -4-({[(lR)-l_(l-ナフチノレ)ェチル]アミノ}メチノレ)ピぺリジン- 1-ィル] -1,3-チアゾ ール -4-カルボン酸塩酸塩 20 mgを得た。
実施例 38
(1)実施例 37(1)で得られた粗 tert-ブチル {[1- (ァミノカルボノチオイル) -3-(3_フルォ 口フエ二ノレ)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチノレ)ェチノレ]力ルバメート 125 mg、硫酸マグネシウム 50 mg、 2—クロロー 3—ォキソプロパン酸ェチル(5%ベンゼン懸 濁液) 2000 mgおよびアセトン 5.0 mLの混合物を、 65 °Cにて 4時間撹拌した。反応混 合物を室温まで冷却後、不溶物をろ別、ろ液を減圧下濃縮し、淡黄色油状化合物と して 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3_( 3-フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -5-カルボン酸ェチル 223 mgを粗生成物として得た。 ESI+: 618
(2)粗 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -(3-フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -5-カルボン酸ェチル 223 mgの THF 2.0 mLおよびメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム水 溶液 1.50 mLを加えた。 85 °Cにて 1時間撹拌した。反応混合物を室温まで冷却後、 1 M塩酸 1.50 mLを加え中和した後、酢酸ェチルで抽出した。有機層を飽和食塩水で 洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、淡黄色油状化
合物として 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチ ノレ) -3-(3_フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -5-カルボン酸 172 mgを粗生成物として得た。 ESI+: 590
(3)粗 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -(3-フルオロフェニル)ピぺリジン- 1-ィル] -1,3-チアゾール -5-カルボン酸 172 mgの THF 2.0 mL溶液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLをカロえ、 1時間 撹拌した。減圧下溶媒を留去した。残渣を逆相シリカゲルカラムクロマトグラフィー(ァ セトニトリル- 0.001M塩酸)により精製し、白色固体として 2-[3-(3_フルオロフェニル )-4 -({[(lR)-l-(l-ナフチル)ェチル]アミ メチル)ピぺリジン- 1-ィル] -1,3-チアゾール -5- カルボン酸塩酸塩 19 mgを得た。
実施例 39
(1)粗 tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチ ノレ]力ルバメート 136 mg、 2-ブロモ -1,3-チアゾール -4-カルボン酸メチル 64 mg、お よび炭酸カリウム 38 mgの混合物に DMSO 1.5 mLを室温にて加え、混合液を室温に て終夜撹拌した。反応混合物に水を加え、酢酸ェチルで抽出した後、有機層を水、 飽和食塩水で順に洗浄した後、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧 下濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチ ノレ)で精製し 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチ ル) -3-フエ二ルビペリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸メチル 113 mgを 無色泡状物質として得た。 ESI+: 586
(2) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸メチル 112 mgに THF2.0 mL、メタノール 1.0 mL及び 1M水酸化ナトリウム水溶液 1.0 mLをカロえ、 60 °Cにて終夜 撹拌した。混合物に 1M塩酸 1.1 mLを加えた後、減圧下濃縮し、残渣にクロ口ホルムと 無水硫酸ナトリウムを加えた。ろ過後、ろ液を減圧下濃縮、乾燥して、 2-[4-({(tert-ブ トキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸の粗生成物を無色泡状物質として 116 mg得 た。
(3) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -1,3-チアゾール -4-カルボン酸の粗生成物 116 mgに 4M 塩化水素 /1,4-ジォキサン溶液 2.0 mLを加えた。室温で 2時間撹拌した後、反応混 合物を減圧下濃縮し、得られた残渣にイソプロパノール及び酢酸ェチルを加え、カロ 熱還流した後、室温まで放冷した。析出物を濾取し、減圧下乾燥して 2-[4-({[(lR)-l- (1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -1,3-チアゾール -4 -カルボン酸塩酸塩 67 mgを白色固体として得た。
実施例 40
(1) 3—アミノー 4ービドロキシ安息香酸メチル 2.000 g、ェチルキサントゲン酸カリウム 1.918 gおよびエタノール 20.0 mlの混合物を加熱還流下 3日間撹拌し、減圧下溶媒を 留去した。残渣を水に溶解し酢酸を加え酸性とし、析出物をろ取した。減圧下乾燥し 、 2-スルファニル -1,3-ベンズォキサゾール -5-カルボン酸メチル 2.049 gを得た。 2— スルファニルー 1,3 ベンゾォキサゾールー 6 力ルボン酸メチル 200 mg、五塩化リン 239 mg、ォキシ塩化リン 0.78 mlおよびジクロロメタン 1 mLの混合物を、室温にて 3時 間撹拌した。減圧下溶媒を留去した。残渣に飽和炭酸水素ナトリウム水溶液を加え p H8とし、クロ口ホルムで抽出した。有機層を水および飽和食塩水で洗浄、無水硫酸ナ トリウムで乾燥した。減圧下溶媒を留去し、橙褐色固体として 2-クロ口- 1,3-ベンズォ キサゾール -5-カルボン酸メチル 191 mgを得た。 ESI+: 212
(2) tert_ブチノレ {[3-(3_フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チル)ェチル]力ルバメート 185 mgの DMSO 2.00 mL溶液に、室温にて水素化ナトリウ ム(55%デイスパージヨン) 18 mgを加え、 10分間撹拌した。 2-クロ口- 1,3_ベンズォキサ ゾール -5-カルボン酸メチル 89 mgを加え、 100 °Cにて終夜攪拌した。 2-クロ口- 1,3_ ベンズォキサゾール -5-カルボン酸メチル 55 mgを更に加え、 100 °Cにて終夜攪拌し た。反応混合物を室温まで冷却後、水を加え、酢酸ェチルで抽出した。有機層を飽 和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮した。 残渣をシリカゲルカラムクロマトグラフィー(へキサン 酢酸ェチル及びクロ口ホルム —メタノール)により精製し、無色樹脂状化合物として 2-[4-({(tert-ブトキシカルボ二 ル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )_3-(3-フルオロフェニノレ)ピぺリジン- 1-
ィル] -1,3-ベンズォキサゾール -5-カルボン酸メチル 33 mgを得た。 ESI+: 638
(3) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル] -1,3-ベンズォキサゾール -5-カルボン酸メチ ル 33 mgの THF 2.0 mLおよびメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウ ム水溶液 1.00 mLを加え、終夜攪拌した。 1M塩酸 1.00 mLを加え中和した後、酢酸ェ チルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過 後、ろ液を減圧下濃縮し、無色樹脂状化合物として 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-(3_フルオロフェニル)ピぺリジン- 1-ィ ル] -1,3-ベンズォキサゾール -5-カルボン酸 35 mgを得た。 ESI+: 624
(4) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3_(3 -フルオロフェニノレ)ピぺリジン- 1-ィル] -1,3-ベンズォキサゾール -5-カルボン酸 35 m gの 1 ,4 ジォキサン 1.0 mL溶液に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mL を加え、 1時間撹拌した。ジイソプロピルエーテルを加え、生じた析出物をろ取、減圧 下乾燥し、 2-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )ピペリジン- 1-ィル] -1,3-ベンズォキサゾール -5-カルボン酸塩酸塩 14 mgを得た。 実施例 41
(1) 2 ォキソー2,3 ジヒドロー 1H べンズイミダゾールー 5 力ルボン酸 228 mg、ォ キシ塩化リン 2.00 mLおよび濃塩酸 1滴の混合物を、 100 °Cにて終夜攪拌した。減圧 下溶媒を留去した。残渣に、室温にてメタノール 4.0 mLを加え、 1時間撹拌した。水 および酢酸ェチルで希釈し、炭酸カリウムを pH8になるまで加えた。不溶物をろ別し た後、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄、無水硫酸ナトリウムで 乾燥した。ろ過後、ろ液を減圧下濃縮し、淡褐色固体として 2-クロ口- 1H-ベンズイミ ダゾール -6-カルボン酸メチノレ 313 mgを得た。 ESI+: 211
(2) 2-クロ口- 1H-ベンズイミダゾール -6-カルボン酸メチル 313 mgおよび tert-ブチル {[3-(3_フルオロフェニノレ)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチノレ)ェチノレ]カル バメート 100 mgの DMSO 2.00 mL溶液を、 130 °Cにて 9時間攪拌した。反応混合物を 室温まで冷却後、水を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄、 無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮した。残渣をシリカゲル力
ラムクロマトグラフィー(へキサン 酢酸ェチル)により精製し、淡黄色固体として 2-[4_ ({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-(3_フルォ 口フエ二ノレ)ピぺリジン- 1-ィノレ] -1H-ベンズイミダゾール -6-カルボン酸メチル 48 mgを 得た。 ESI+: 637
(3) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル] -1H-ベンズイミダゾール -6-カルボン酸メチル 48 mgの THF 2.0 mLおよびメタノール 1.0 mL溶液に、室温にて 1M水酸化ナトリウム 水溶液 l.OOmLを加え、終夜攪拌した。 80 °Cにて 6時間撹拌した。反応混合物を室温 まで冷却後、 1M塩酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。有機層を 飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 淡黄色非晶状化合物として 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェ チノレ]アミノ}メチノレ) -3-(3_フルオロフェニノレ)ピぺリジン- 1-ィノレ] -1H-ベンズイミダゾー ル -6-カルボン酸 19 mgを得た。 ESI+: 623
(4) 2-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3_(3 -フルオロフェニノレ)ピぺリジン- 1-ィノレ] -1H-ベンズイミダゾール -6-カルボン酸 19 mg の 4M塩化水素 /1,4 ジォキサン 1.00 mL溶液を、室温にて 2時間撹拌した。ジイソプ 口ピルエーテルを加え、生じた析出物をろ取、減圧下乾燥し、 2-[3-(3_フルオロフェ ニル) -4-({[(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチノレ)ピぺリジン- 1-ィル] -1H-ベンズ イミダゾール -6-カルボン酸二塩酸塩 15 mgを得た。
実施例 42
re卜 5-フルォ口- 6-[(3R,4R)-4- (ヒドロキシメチル) -3-フエ二ルビペリジン- 1-ィノレ]ニコ チン酸メチル 282 mgのジクロロメタン 3.0 mL溶液に、室温にてデス一マーチンペルョ ージナン 382 mgを加え、 1.5時間撹拌した。この反応混合物を 1,2-ジクロロェタンで洗 いこみながら滴下ロートに移し、(lR)-l-(l-ナフチル)エタナミン 147 mg、トリァセトキ シ水素化ホウ素ナトリウム 521 mg、酢酸 49 mg及び 1,2-ジクロロェタン 10 mLの混合物 に室温にて滴下した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、 10分間 激しく撹拌した後、クロ口ホルムで抽出した。有機層を水、飽和食塩水で順次洗浄し 、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し得られた残渣を NHシ
リカゲル(富士シリシァ化学、 日本)を用いたカラムクロマトグラフィー(へキサン 酢 酸ェチル)により精製し、 5-フノレオ口- 6_[4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチ ル) -3-フエ二ルビペリジン- 1-ィル]ニコチン酸メチル 314 mgを無色泡状物質として得 た。
実施例 43
(1) 5-フルォ口- 6-[4-({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリ ジン- 1-ィル]ニコチン酸メチル 312 mgの THF5 mL溶液に、室温でトリェチルァミン 0· 35 mL及びジ炭酸-ジ- tert-ブチル 274 mgを加え、混合物を 60 °Cで終夜撹拌した。 反応混合物を室温に冷ました後、減圧下濃縮して得た残渣をシリカゲルカラムクロマ トグラフィー(へキサン 酢酸ェチル)にて精製し、 6-[4-({(tert-ブトキシカルボニル) [( 1R)-1-(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5-フルォロ ニコチン酸メチル 174 mg (低極性分画、 Rf値 0.23 (展開溶媒:へキサン/酢酸ェチル =
7/1)、 FAB+: 598)と 6_[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル] アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5-フルォロニコチン酸メチル 176 mg (高 極性分画、 Rf値 0.15 (展開溶媒:へキサン/酢酸ェチル = 7/1)、 FAB+: 598)を各々 無色泡状物質として得た。
(2)分取した低極性分画ジァステレオマー 6-[4-({(tert-ブトキシカルボニル) [(1R)-1- (1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5-フルォロニコチ ン酸メチノレ 173 mgに THF2.0 mL、メタノーノレ 1.0 mL及び 1M水酸化ナトリウム水溶液 1 .0 mLを加え、室温にて 2日間撹拌した。 1M塩酸 1.1 mLを加えて中和した後、酢酸ェ チルで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮
、乾燥して、 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチ ル) -3-フエ二ルビペリジン- 1-ィル] -5-フルォロニコチン酸の粗生成物をオフホワイト 色泡状物質として 188 mg得た。
(3) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -5-フルォロニコチン酸の粗生成物 187 mgに 4M塩化水 素 /1,4-ジォキサン溶液 2.0 mLを加えた。室温で 2時間撹拌した後、反応混合物を 減圧下濃縮し、得られた残渣にイソプロパノール及び酢酸ェチルを加え、加熱還流
した後、室温まで放冷した。析出物を濾取し、減圧下乾燥して 5-フルォ口- 6_[4-({[(1 R)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]ニコチン酸 塩酸塩 106 mgを白色固体として得た。
[0134] 実施例 44
(1) tert-ブチル {[3-(3_フルオロフェニル)ピぺリジン- 4-ィル]メチル }[(1R)-1-(1-ナフ チル)ェチル]力ルバメート 139 mgおよび 6-ォキソへキサン酸メチル 48 mgのジクロロメ タン 4.0 mL溶液に、室温にてトリァセトキシ水素化ホウ素ナトリウム 191 mgを加え、終 夜撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、クロ口ホルムを用 いて抽出した。有機層を水および飽和食塩水で洗浄、無水硫酸ナトリウムで乾燥した 。ろ過後、ろ液を減圧下濃縮し、無色油状化合物として 6-[4-({(tert-ブトキシカルボ ニル) [(lR)-l-(l-ナフチノレ)ェチノレ]アミノ}メチル )_3-(3-フルオロフェニル)ピぺリジン- 1-ィル]へキサン酸メチル 180 mgを得た。 ESI+: 591
(2) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニノレ)ピぺリジン- 1-ィノレ]へキサン酸メチル 180 mgのメタノール 2.0 mL 溶液に、室温にて 1M水酸化ナトリウム水溶液 1.00 mLを加え、 1時間撹拌した。 1M塩 酸 1.00 mLを加え中和した後、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄
、無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し、 6_[4-({(tert-ブトキ シカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-(3_フルオロフェニル)ピ ペリジン- 1-ィル]へキサン酸 168 mgを得た。 ESI+: 577
(3) 6-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )-3_(3 -フルオロフェニル)ピぺリジン- 1-ィル]へキサン酸 168 mgの酢酸ェチル 2.0 mL溶液 に、室温にて 4M塩化水素/酢酸ェチル溶液 1.00 mLを加え、 2時間撹拌した。減圧下 溶媒を留去した。残渣を逆相シリカゲルカラムクロマトグラフィー(ァセトニトリル- 0.001 M塩酸)により精製し、淡黄色固体として 6-[3-(3_フルオロフェニル )-4-({[(lR)-l-(l- ナフチル)ェチル]アミノ}メチル)ピぺリジン- 1-ィル]へキサン酸二塩酸塩 70 mgを得 た。
[0135] 実施例 45
(l) tert-ブチル {[4-(2-フルオロフェニル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ
チル)ェチル]力ルバメート 191mg、トリェチルァミン 0.115mL及び THF 3.0mLの混合 物に 3-クロ口- 4-{[(4-ニトロフエノキシ)カルボニル]アミノ}安息香酸メチル 175mgを室 温にて加えた。室温にて終夜撹拌した後、反応混合物を減圧下濃縮した。残渣をシ リカゲルカラムクロマトグラフィー(へキサン-酢酸ェチル)で精製し、粗生成物として 4 _({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )_4-(2-フ ルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸メチルを 330 mg得た。 ESI+: 674
(2)粗 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル) - 4-(2-フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸メチ ル 330mg及び THF lOmL-メタノール 5. OmLの混合物に 1M水酸化ナトリウム水溶液 5 • OmLを室温にて滴下し、終夜撹拌した。 1M塩酸を 5.2 mL加えて中和した後、酢酸ェ チルで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過後、、ろ液を減圧下濃 縮して得られた残渣をシリカゲルカラムクロマトグラフィー(メタノール-クロ口ホルム)に て精製し 4-({[3-({(tert_ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-(2-フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸を 淡黄色アモルファスとして 274mg得た。 ESI+: 660
(3) 4-({[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4- (2-フルオロフェニル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ) -3-クロ口安息香酸 274mg に 4M塩化水素 /1,4-ジォキサン溶液 3.0mLを加え、室温で終夜撹拌した。反応混合 物を減圧下濃縮し、クロ口ホルム及び飽和炭酸水素ナトリウム水溶液を加え撹拌した 後、分離し無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮し得られた残 渣をシリカゲルカラムクロマトグラフィー(メタノール-クロ口ホルム)により精製し、 3-クロ 口- 4-({[4-(2-フルオロフヱニル) -3-({[(lR)-l_(l-ナフチル)ェチル]アミノ}メチル)ピペリ ジン- 1-ィル]カルボ二ル}ァミノ)安息香酸を無色アモルファスとして 157 mg得た。得ら れたアモルファスをクロ口ホルムに溶解した後、へキサンを加えて白色固体を析出さ せ、ろ過取し、 3-クロ口- 4-({[4-(2-フルオロフヱニル) -3-({[(lR)-l_(l-ナフチル)ェチ ノレ]アミノ}メチル)ピぺリジン- 1-ィル]カルボ二ル}ァミノ)安息香酸を白色固体として 136 mgを得た。
[0136] 実施例 46
tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチル]力 ノレバメート 13.3 mg、 4-メトキシカルボニル安息香酸 6.3 mg、 HOBt 4.7 mg及び DMF 1 mLの混合物に室温にて PS-カルポジイミド(PS-Carbodiimide) (ァルゴノートテクノロ ジ一社、米国) 100 mgを加え終夜撹拌した。反応混合物に室温にて MP-カルボネー ト(MP-Carbonate) (ァルゴノートテクノロジ一社、米国) 50 mg、 PS-イソシァネート(PS -Isocyanate) (ァルゴノートテクノロジ一社、米国) 50 mg及び DMF 0.5 mLを加え 4時 間攪拌し、反応混合物を濾過した。濾液を減圧下濃縮し粗生成物として 4_{[3-({(tert -ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジ ン- 1-ィル]カルボ二ル}安息香酸メチルを得た。得られた粗生成物の 1,4-ジォキサン 溶液 0.5 mLに室温にて 4M塩化水素/酢酸ェチル溶液 0.5 mLを加え終夜撹拌した。 反応混合物を減圧下濃縮し、粗生成物として 4-{[3-({[(lR)-l_(l-ナフチル)ェチル]ァ ミノ }メチル )_4-フエ二ルビペリジン -1-ィル]カルボ二ル}安息香酸メチルを得た。得ら れた粗生成物の THF溶液 0.5 mLに室温にてメタノール 0.5 mL及び 1M水酸化ナトリウ ム水溶液 0.5 mLを加え終夜撹拌した。反応混合物に 1M塩酸を 0.5 mL加え減圧下濃 縮した。得られた残渣を分取高速液体クロマトグラフィー(カラム:登録商標サンフアイ ァ(SunFire、ウォーターズ(Waters)社、粒径 5 m、内径 19mm、長さ 100mm)、 流速: 25ml/min、カラム温度: 20 °C、メタノール 0.1%ギ酸水溶液)で精製し 4_{[3 -({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-4-フエ二ルビペリジン- 1-ィル]カルボ 二ル}安息香酸 8.4 mgを得た。
[0137] 実施例 47
tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチル]力 ノレバメート 13.3 mg、 4-メトキシカルボニル安息香酸 6.3 mg、 HOBt 4.7 mg及び DMF 1 mLの混合物に室温にて PS-カルポジイミド(PS-Carbodiimide) (ァルゴノートテクノロ ジ一社、米国) 100 mgを加え終夜撹拌した。反応混合物に室温にて MP-カルボネー ト(MP-Carbonate) (ァルゴノートテクノロジ一社、米国) 50 mg、 PS-イソシァネート(PS -Isocyanate) (ァルゴノートテクノロジ一社、米国) 50 mg及び DMF 0.5 mLを加え 4時 間攪拌し、反応混合物を濾過した。濾液を減圧下濃縮し粗生成物として 4_{[4-({(tert
-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジ ン- 1-ィル]カルボ二ル}安息香酸メチルを得た。得られた粗生成物の 1,4-ジォキサン 溶液 0.5 mLに室温にて 4M塩化水素/酢酸ェチル溶液 0.5 mLを加え 6時間撹拌した。 反応混合物を減圧下濃縮し、粗生成物として 4-{[4-({[(lR)-l-(l-ナフチル)ェチル]ァ ミノ }メチル )_3-フエ二ルビペリジン -1-ィル]カルボ二ル}安息香酸メチルを得た。得ら れた粗生成物の THF溶液 0.5 mLに室温にてメタノール 0.5 mL及び 1M水酸化ナトリウ ム水溶液 0.5 mLを加え終夜撹拌した。反応混合物に 1M塩酸を 0.5 mL加え減圧下濃 縮した。得られた残渣を分取高速液体クロマトグラフィー(カラム:登録商標サンフアイ ァ(SunFire、ウォーターズ(Waters)社、粒径 5 m、内径 19mm、長さ 100mm)、 流速: 25ml/min、カラム温度: 20 °C、メタノールー0.1%ギ酸水溶液)で精製し4_{[4 -({[(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]カルボ 二ル}安息香酸 7.7 mgを得た。
[0138] 実施例 48
tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(4-フエ二ルビペリジン- 3-ィル)メチル]力 ノレバメート 13.3 mg、 3-フルアルデヒド 3.4 mg、酢酸 0.050 mL及び DMF 0.5 mLの混 合物に室温にて MP-トリァセトキシ水素化ホウ素(MP-Triacetoxyborohydride) (アル ゴノートテクノロジ一社、米国) 75mgを加え終夜撹拌した。反応混合物に室温にて PS -イソシァネート(PS-Isocyanate) (ァルゴノートテクノロジ一社、米国) 50 mgを加え 2時 間攪拌し、反応混合物を濾過した。濾液を減圧下濃縮し粗生成物として tert-ブチル {
[1-(3_フリルメチル )-4-フエ二ルビペリジン- 3-ィル]メチル }[(1R)-1_(1-ナフチル)ェチ ル]力ルバメートを得た。得られた粗生成物のメタノール溶液 0.5 mLに室温にて 4M塩 化水素/酢酸ェチル溶液 0.5 mLを加え 4時間撹拌した。反応混合物を減圧下濃縮し 、得られた残渣を分取高速液体クロマトグラフィー(カラム:登録商標サンファイア(Su nFire、ウォーターズ(Waters)社、粒径 5 m、内径 19mm、長さ 100mm)、流速: 2 5ml/min、カラム温度: 20 °C、メタノールー0.1%ギ酸水溶液)で精製し(11¾,-{[1-( 3-フリルメチル )-4-フエ二ルビペリジン- 3-ィル]メチル }-1-(1_ナフチル)エタナミン 4.9 mgを得た。
[0139] 実施例 49
tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチル]力 ノレバメート 13.3 mg、 3-フルアルデヒド 3.4 mg、酢酸 0.050 mL及び DMF 0.5 mLの混 合物に室温にて MP-トリァセトキシ水素化ホウ素(MP-Triacetoxyborohydride) (アル ゴノートテクノロジ一社、米国) 75mgを加え終夜撹拌した。反応混合物に室温にて PS -イソシァネート(PS-Isocyanate) (ァルゴノートテクノロジ一社、米国) 50 mgを加え 2時 間攪拌し、反応混合物を濾過した。濾液を減圧下濃縮し粗生成物として tert-ブチル {
[1-(3_フリルメチル )-3-フエ二ルビペリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチル)ェチ ル]力ルバメートを得た。得られた粗生成物のメタノール溶液 0.5 mLに室温にて 4M塩 化水素/酢酸ェチル溶液 0.5 mLを加え 4時間撹拌した。反応混合物を減圧下濃縮し 、得られた残渣を分取高速液体クロマトグラフィー(カラム:登録商標サンファイア(Su nFire、ウォーターズ(Waters)社、粒径 5 m、内径 19mm、長さ 100mm)、流速: 2 5ml/min、カラム温度: 20 °C、メタノールー0.1%ギ酸水溶液)で精製し(11¾,-{[1-( 3-フリルメチル )-3-フエ二ルビペリジン- 4-ィル]メチル }-1-(1-ナフチル)エタナミン 8.6 mgを得た。
実施例 50
(1) tert-ブチル {[4-(2-フルオロフェニル)ピぺリジン- 3-ィル]メチル }[(1R)-1_(1-ナフ チル)ェチル]力ルバメート 191mgと 3,4,5-トリフルォロ安息香酸メチル 118mg、炭酸力 リウム 114mg及び THF 2.0mL-DMSO 2.0mL混合物を 100 °Cにて終夜撹拌した。冷 却後、反応混合物に水を加え、酢酸ェチルで抽出した後、無水硫酸ナトリウムで乾 燥した。ろ過後、ろ液を減圧下濃縮し得られた残渣をシリカゲルカラムクロマトグラフィ 一(へキサン-酢酸ェチル)で精製し、 4-[(3R,4S)-3-({(tert-ブトキシカルボニル) [(1R) -1-(1-ナフチル)ェチル]アミノ}メチル )-4-(2-フルオロフェニル)ピぺリジン- 1-ィル] -3, 5-ジフルォロ安息香酸メチルを無色アモルファスとして 147mg得た。
(2) 4-[(3R,4S)-3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチ ル) -4-(2-フルオロフェニル)ピぺリジン- 1-ィル] -3,5-ジフルォロ安息香酸メチル 143 mgを THF 4.0mL-メタノール 2.0mLに溶解し、室温で 1M水酸化ナトリウム水溶液 2.0 mLを力 Pえた。室温で 3日間撹拌したのち、 1M塩酸 2.1 mLを加えて中和した。反応 混合物を減圧下濃縮した後、酢酸ェチルで抽出し、有機層を無水硫酸ナトリウムで
乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリカゲルカラムクロマトグ ラフィー(メタノール-クロ口ホルム)で精製し、 4-[3-({(tert_ブトキシカルボニル) [(1R)-1 -(1-ナフチノレ)ェチノレ]アミノ}メチノレ) -4-(2-フルオロフェニノレ)ピぺリジン- 1-ィル] -3,5- ジフルォロ安息香酸を 138mg得た。 ESI+: 619
(3) 4-[3-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチノレ)ェチル]アミノ}メチル )_4-(2 -フルオロフェニル)ピぺリジン- 1-ィル] -3,5-ジフルォロ安息香酸 135mgに 4M塩化水 素 /1,4-ジォキサン溶液 3.0mLを加え、室温で終夜撹拌した。反応混合物を減圧下 濃縮し、得られた残渣にクロ口ホルム及び飽和炭酸水素ナトリウム水溶液を加え撹拌 した後、有機層を分離し無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮 し得られた残渣をシリカゲルカラムクロマトグラフィー(メタノール-クロ口ホルム)により 精製し、 3,5-ジフルォロ _4-[4-(2-フルオロフヱニル) -3-({[(lR)-l_(l-ナフチル)ェチ ル]アミノ}メチル)ピペリジン- 1-ィル]安息香酸を無色アモルファスとして 11 lmg得た。 得られたアモルファスをクロ口ホルムに溶解した後、へキサンを加えて白色固体を析 出させ、ろ取して 3,5-ジフルォロ _4-[4-(2-フルオロフェニル )-3-({[(lR)-l_(l-ナフチ ノレ)ェチル]アミノ}メチル)ピペリジン- 1-ィル]安息香酸 93mgを白色固体として得た。 実施例 51
(1) tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィル)メチル] 力ルバメート 181mg、トリェチルァミン 0.114mL及び THF 3.0mLの混合物に 3,3_ジメ チルダルタル酸無水物 64mgを室温にて加え、室温で終夜撹拌した。反応混合物に 飽和炭酸水素ナトリウム水溶液を加え撹拌した後、クロ口ホルムで抽出し、無水硫酸 ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して、 5-[4-({(tert-ブトキシカルボ二 ル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -3,3-ジ メチル -5-ォキソペンタン酸を粗生成物として 266 mg得た。 ESI+: 587
(2)粗 5-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -フエ二ルビペリジン- 1-ィル] -3,3-ジメチル -5-ォキソペンタン酸 265mgに 4M塩化水 素 /1,4-ジォキサン溶液 2.0 mLを加え室温で 2時間撹拌した。反応混合物を減圧下 濃縮後得られた残渣をイソプロパノールで加熱中懸濁化した後、放冷した。析出物を ろ過により単離し、減圧下乾燥して、 3,3-ジメチル _5-[4-({[(lR)-l-(l-ナフチル)ェチ
ル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -5-ォキソペンタン酸塩酸塩を白色 固体として 189mg得た。
実施例 52
(1) 3,4,5-トリフルォロ安息香酸メチル 116mg、炭酸カリウム 113mg及び DMSO 2.0m Lの混合物に tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィ ル)メチノレ]力ルバメート 181mgの THF 3.0mL溶液を室温にて加えた。 100°Cにて終夜 撹拌した後、冷却した。反応混合物に水を加え酢酸ェチルで抽出した後、有機層を 水、飽和食塩水で順次洗浄し、無水硫酸ナトリウムにより乾燥した。ろ過後、ろ液を減 圧下濃縮し、 4-[4-({(tert_ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メ チル) -3-フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸メチルを粗生成物とし て 248 mg得た。 FAB+: 615
(2)粗 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3 -フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸メチル 247mg、 THF 4.0mL及 びメタノール 2.0mLの混合物に 1M水酸化ナトリウム水溶液を室温にて滴下し、終夜 撹拌した。反応混合物に 1M塩酸を加えて中和した後、酢酸ェチルで抽出し、有機層 を無水硫酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣をシリ 力ゲルカラムクロマトグラフィー(メタノール-クロ口ホルム)にて精製し、 4-[4-({(tert-ブ トキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸を白色固体として 233mg得た。 FAB+: 601
(3) 4-[4-({(tert-ブトキシカルボニル) [(lR)-l-(l-ナフチル)ェチル]アミノ}メチル )-3- フエ二ルビペリジン- 1-ィル] -3,5-ジフルォロ安息香酸 230mgに 4M塩化水素 /1,4-ジ ォキサン溶液 1.5mLを室温にて加えた。室温にて 2時間撹拌した後、反応混合物を 減圧下濃縮した。得られた残渣をイソプロパノール及びジイソプロピルエーテルで固 体化した後、ろ取した。得られた固体をシリカゲルカラムクロマトグラフィー(メタノール -クロ口ホルム)で精製した後、 4M塩化水素 /1,4-ジォキサン溶液を加えた。混合物を 減圧下濃縮し、得られた個体をイソプロパノールで洗浄し、 3,5-ジフルォ口- 4_[4-({[( 1R)-1-(1-ナフチル)ェチル]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]安息香酸 塩酸塩を 140mg得た。
実施例 53
(1) 3,4,5-トリフルォロベンゾニトリノレ 96 mg、炭酸カリウム 113 mg、及び DMSO3.0 m Lの混合物に tert-ブチル [(lR)-l-(l-ナフチル)ェチル ][(3-フエ二ルビペリジン- 4-ィ ル)メチル]力ルバメートの粗生成物 181 mgの THF3.0 mL溶液を加え、反応容器を密 栓し、混合物を 100 °Cにて終夜加熱した。反応混合物を室温まで冷却した後、水を 加え、酢酸ェチルで抽出した。有機層を水、飽和食塩水で順次洗浄し、無水硫酸ナ トリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して、 tert-ブチル {[1-(4_シァノ -2,6- ジフルオロフェニル )-3-フエ二ルビペリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチル)ェチ ル]力ルバメートを粗生成物 232 mgとして得た。 FAB+: 582
(2) tert-ブチル {[1-(4-シァノ -2,6-ジフルオロフェニル )-3-フエ二ルビペリジン- 4-ィ ル]メチル }[(1R)-1-(1-ナフチル)ェチル]力ルバメートの粗生成物 232 mg、アジ化ナトリ ゥム 518 mg、トリェチルァミン塩酸塩 1.10 g及び DMF 4.6 mLの混合物を 120 °Cにて 終夜撹拌した。反応混合物を室温まで冷却後、水を加え、酢酸ェチルで抽出した。 有機層を水、飽和塩化ナトリウム水溶液で順次洗浄し、無水硫酸ナトリウムで乾燥し た。これをろ過した後、ろ液を減圧下濃縮して、得られた残渣をシリカゲルカラムクロ マトグラフィー(クロ口ホルム/メタノール)で精製し、 tert-ブチル ({1-[2,6_ジフルォロ -4 -(1H-テトラゾール -5-ィノレ)フエニル] -3-フエ二ルビペリジン- 4-ィル }メチル) [(lR)-l-( 1-ナフチル)ェチル]力ルバメート 208 mgを黄色油状物として得た。 FAB+: 625
(3) tert-ブチル({1-[2,6_ジフルォ口- 4-(1Η-テトラゾール -5-ィル)フエニル] -3-フエ 二ルビペリジン- 4-ィル }メチル) [(lR)-l-(l-ナフチル)ェチル]力ルバメート 205 mgに 4 M塩化水素 /1,4-ジォキサン溶液 2.0 mLを加え、室温にて 2時間撹拌した。反応混 合物を減圧下濃縮した後、残渣に飽和炭酸水素ナトリウム水溶液を加え、クロロホノレ ムで抽出し、有機層を無水硫酸ナトリウムで乾燥した。ろ過した後、ろ液を減圧下で 濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(クロ口ホルム/メタノール) で精製し、(lR)-N-({l-[2,6-ジフルォロ- 4-(1Η -テトラゾール- 5-ィル)フエニル] -3-フ ェニルビペリジン- 4-ィル }メチル )-1-(1-ナフチノレ)エタナミン 158 mgを得た。これに塩 化水素 /1,4-ジォキサン溶液 2.0 mLを加え、減圧下で濃縮した後、得られた残渣に イソプロパノールを約 3 mLを加え、加熱した。混合物を室温に放冷した後、析出物を
濾取し、減圧下乾燥して、(lR)-N-({l-[2,6-ジフルォロ _4-( -テトラゾール -5-ィル) フエニル] -3-フエ二ルビペリジン- 4-ィル }メチル )-1-(1-ナフチル)エタナミン塩酸塩 1 53 mgを白色固体として得た。
実施例 54
(1)実施例 53 (1)で得られた tert-ブチル {[1_(4-シァノ -2,6-ジフルオロフヱニル) -3- フエ二ルビペリジン- 4-ィル]メチル }[(1R)-1-(1-ナフチル)ェチル]力ルバメート 183 mg 、ヒドロキシルァミン塩酸塩 44 mg、エタノール 2 mLの混合物にトリェチルァミン 0.088 mLを加え、混合物を加熱還流下、 4時間撹拌した。混合物を室温に放冷した後、水 を加え、酢酸ェチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウム で乾燥した。これをろ過した後、ろ液を減圧下濃縮し、残渣 (無色泡状物質、 233 mg) を得た。この残渣に DMF 2 mL、ピリジン 0.033 mLを加え、混合物を氷浴で冷却した 後、クロロギ酸 2-ェチルへキシル 0.061mLを加え、氷冷のまま 1時間撹拌した。反応 混合物を酢酸ェチルで希釈し、水、飽和食塩水で順次洗浄した後、有機層を無水硫 酸ナトリウムで乾燥した。ろ過後、ろ液を減圧下濃縮して得られた残渣にキシレン 2 m Lを加え、混合物を加熱還流下、 5時間撹拌した。これを室温まで放冷した後、シリカ ゲルカラムクロマトグラフィー(クロ口ホルム/メタノール)により精製し、 tert-ブチル
2,6-ジフルォロ -4-(5_ォキソ -4,5-ジヒドロ- 1,2,4-ォキサジァゾール -3-ィル)フエ二ノレ] -3-フエ二ルビペリジン- 4-ィル }メチル) [(lR)-l-(l-ナフチル)ェチル]力ルバメート 184 mgを黄色泡状物質として得た。 ESI+: 641
(2) 16 -ブチル({1-[2,6-ジフルォロ-4-(5-ォキソ-4,5-ジヒドロ-1,2,4-ォキサジァゾー ル -3-ィル)フエニル] -3-フエ二ルビペリジン- 4-ィル }メチル) [(lR)-l-(l-ナフチル)ェチ ル]力ルバメート 183 mgに 4M塩化水素 /1,4-ジォキサン溶液 1.5 mLをカロえた。室温 で 2時間撹拌した後、反応混合物を減圧下濃縮し、得られた残渣をシリカゲルカラム クロマトグラフィー(クロ口ホルム/メタノール)で精製しした後、 4M塩化水素 /1,4 -ジォ キサン溶液 0.5 mLを加え、再び減圧下濃縮した。得られた残渣にエタノール、酢酸 ェチルを加え、加熱還流した後、室温に放冷した。析出物を濾取し、減圧下乾燥して 、イソプロパノール、酢酸ェチルを加え、加熱還流した後、室温まで放冷した。析出 物を濾取し、減圧下乾燥して 3-{3,5-ジフルォ口- 4-[4-({[(lR)-l-(l-ナフチル)ェチル
]アミノ}メチル )-3-フエ二ルビペリジン- 1-ィル]フエ二ル}_1,2,4-ォキサジァゾール -5(4
H)-オン塩酸塩 57 mgを白色固体として得た。
[0145] 実施例 1〜54の方法と同様にして、後記表に示す実施例 55〜297の化合物を、そ れぞれ対応する原料を使用して製造した。各実施例化合物の構造を表 15〜74に、 物理化学的データ及び製造法を表 75〜89に示す。
[0146] また、表 90〜97に本発明の別の化合物の構造を示す。これらは、上記の製造法や 実施例に記載の方法及び当業者にとって自明である方法、又はこれらの変法を用い ることにより、容易に合成することカでさる。
[0147] [表 4]
[0150] [表 7]
//:i O ε90ί/-0/-00ί1£ooiAV
¾星ΐ9
[0156] [表 13]
[0161] [表 18]
,4 -trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0162] [表 19]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans
3,4-trans
3,4-trans
301
£90ZL0/L0QZdr/13d
90 o-o/ozl:>d
3,4-trans
3,4-trans
3,4-trans
3,4-trans
[0171] [表 28]
3,4-trans
3,4-trans
3,4 -trans
3,4- trans
3,4-trans
zvv
£90ZL0/L00Zdr/13d 17S86S0/800Z; OAV
[0174] [表 31]
3,4-trans
3,4-trans
3,4-trans
3,4-trans
3,4-trans
[0175] [表 32]
4-trans
3,4-trans
3,4-trans
3,4- trans
3,4-trans
3,4-trans
3,4-trans
3,4-trans
3,4-trans
3,4-trans
[0178] [表 35]
6]
4 -trans
3,4-trans
3,4-trans
3,4-trans
3,4-trans
[0180] [表 37]
[8ε挲] [Ϊ8ΐ0]
031- 10/ LOOZdT/lDd 17S86S0/800Z OAV
9]
,4-trans
3,4-trans
40]
[0184] [表 41]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3.4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans
1,,2,-cis diastereo mixture
[0186] [表 43]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3.4 -trans diastereo mixture
3,4-trans diastereo mixture
[0187] [表 44]
45]
3,4-trans
1 ',3,-cis diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0189] [表 46]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans 2,,3,-cis-en do diastereo mixture
[0190] [表 47]
3 ,4-trans diastereo mixture
3.4-trans diastereo mixture
3, 4 -trans diastereo mixture
3.4-trans diastereo mixture
[0191] [表 48]
131
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0194] [表 51]
3,4-trans diastereo mixture
I 3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0195] [表 52]
]
]
]
]
7]
]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0202] [表 59]
]
3.4-trans diastereo mixture
3,4-trans diasterco mixture
3,4-trans diasterco mixture
3.4-trans diastereo mixture
3,4-trans diastereo mixture
[0204] [表 61]
2]
3,4-trans diastereo mixture
3.4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0207] [表 64]
3.4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0208] [表 65]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0209] [表 66]
j,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
[0210] [表 67]
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3,4-trans diastereo mixture
3.4-trans
[0211] [表 68]
j,4-trans diastereo mixture
3.4-trans diastereo mixture
3, 4 -trans diastereo mixture
3,4-trans diastereo mixture
[0212] [表 69]
]
]
]
]
]
]
:2
〔〕
〔〕
ε90/ζ/-0/-00ί/:12s8/6so800i OAV.
〔〔星
OAV
[98挲] [6ZZ0]
: +ISH Li' 181
S : +IS3 LP 081
£ : +isa it 6L\
Ιί : +ISH 8Z.I
: +IS3 9 LL\ i : +ISH 9P 9L\
: +IS3 9 ςι\ ξ9ζ : +IS3 9 VL\
9£ : +IS3 9 ill し : +ISH ZL\ ,
: +IS3 9t \L\ ξ^ς : +ISH 0ん I
: +ISH 9^ 691
Sii : +ISH 891
9ίζ ·' +IS3 9t L9\
6 : +ISH 9 991 ιζς : +IS3 9 S91
ΖΖξ : +IS3 9 t9l
ΙΖξ : +IS3 9f £91
615 : +IS3 9i7 Z9\
61 : +IS3 9 191
9\ς : +IS3 9 091
CIS : +ISH 9V 651
60 ς ·' +ISH 9^ 8SI
60S : +IS3 Lil
Z.0S : +IS3 91/ 9SI
COS : +IS3 9 SSI
ΙΟξ : ^IS3 9
66t : +IS3 9fr ίςι
66 : +ISH 9 Ζξ\
66^ : +ISH 9t7 191
66fr : +ISH 9f oci
66^ : +IS3 9 6W
乙 6 : +IS3 9fr 8セ1
: +IS3 9 L l
: +ISH 9V 9Π
891-
£90ZL0/L00Zd£/13d t^S86S0/800Z OAV
69レ
£90ZL0/L00Zd£/13d 17S86S0/800Z OAV
217 47 ESI+: 565
218 47 ESト : 576
219 47 ESI : 443
220 47 ES1+ : 482
221 48 ESI十 : 435
222 48 ESI+ : 387
223 48 ESI+ : 401
224 48 ES1+ : 403
225 48 FSI+ : 403
48 48 ESI+ : 425
226 48 ES1+ : 436
227 48 ESI+ . 436
228 48 ESI+ • 438
229 48 ESI+ 441
230 48 ESI+ 449
231 48 ESI+ 451
232 48 ESI+ 451
233 48 ESI+ 451
234 48 ESI+ 453
235 48 ESI+ 461
236 48 ESI+ 463
237 48 ESI+ 465
238 48 ESI+ 465
239 48 ESI+ 469
240 48 ESI+ 469
241 48 ES1+ 469
242 48 ESI+ 469
243 48 ESI+ 478 44 48 ESI+ 485 45 48 ESI+ 485 46 48 ESK 492 47 48 ESI+ 503 48 48 ESI+ 509 49 48 ESI+ 509 50 48 ESI+ · 511 51 48 HSI+: 424 88]
252 48 ESI+ 425
253 48 ESI+ 441
254 48 ESI 442
255 48 ESI- 453
256 48 ESI+ 453
257 48 ESI+ 465
258 48 ESI+ 479
259 48 ES1+ 479
260 48 ES1+ 485
261 48 ESI+ 505
262 49 ESI+ 387
263 49 ESI+ 403
264 49 ESI+ 403
265 49 ESI+ • 425
49 49 ESI+ 425
266 49 ESI+ 436
267 49 ESI+ 438
268 49 ESI+ 441
269 49 ESI+ 441
270 49 ESI+ 442
271 49 ESI+ 449
272 49 ESI+ 451
273 49 ESI 451
274 49 ESI+ 451
275 49 ESI 453
276 49 ES1+ 453
277 49 ESI+ 461 78 49 ESI+ 463 79 49 ESI+ 465 80 49 ESI+ 465 81 49 ESI+ 465 82 49 ESI+ 469 83 49 ESI+ . 469 84 49 ESI+ 469 85 49 ESI+ 469 86 49 ESI+ 478 9]
287 49 ESI+ 479
288 49 HSI+ 479
289 49 ESI+ 485
290 49 ESI+ 485
291 49 ESI+ 485
292 49 ESI+ 492
293 49 F.SK 503
294 49 ESI+ 505
295 49 ESI+ 509
296 49 ESI+ 509
297 49 ESI+ 511 0]
£90110/ LOOZdT/lDd 17S86S0/800Z OAV
[0236] [表 93]
[0238] [表 95]
本発明化合物は、 CaSR作動的調節作用に優れ、また、薬物相互作用を起こす懸 念のある CYP2D6の阻害作用との選択性に優れることから、 CaSRが関与する疾患(副 甲状腺機能亢進症、腎性骨異栄養症、高カルシウム血症等)の治療剤として有用で ある。