WO2008061900A1 - Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand - Google Patents
Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand Download PDFInfo
- Publication number
- WO2008061900A1 WO2008061900A1 PCT/EP2007/062111 EP2007062111W WO2008061900A1 WO 2008061900 A1 WO2008061900 A1 WO 2008061900A1 EP 2007062111 W EP2007062111 W EP 2007062111W WO 2008061900 A1 WO2008061900 A1 WO 2008061900A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ligand
- unsubstituted
- substituted
- groups
- deprotonated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C1*(*)N2*3c(cccc4)c4ccc3CC1C(*)=*2* Chemical compound *C1*(*)N2*3c(cccc4)c4ccc3CC1C(*)=*2* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/18—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms
- B01J31/1805—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes containing nitrogen, phosphorus, arsenic or antimony as complexing atoms, e.g. in pyridine ligands, or in resonance therewith, e.g. in isocyanide ligands C=N-R or as complexed central atoms the ligands containing nitrogen
- B01J31/181—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine
- B01J31/1815—Cyclic ligands, including e.g. non-condensed polycyclic ligands, comprising at least one complexing nitrogen atom as ring member, e.g. pyridine with more than one complexing nitrogen atom, e.g. bipyridyl, 2-aminopyridine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/02—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups
- C07C251/04—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups having carbon atoms of imino groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C251/10—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups having carbon atoms of imino groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of an unsaturated carbon skeleton
- C07C251/16—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton containing imino groups having carbon atoms of imino groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of an unsaturated carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/44—Radicals substituted by doubly-bound oxygen, sulfur, or nitrogen atoms, or by two such atoms singly-bound to the same carbon atom
- C07D213/53—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/12—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F11/00—Compounds containing elements of Groups 6 or 16 of the Periodic Table
- C07F11/005—Compounds containing elements of Groups 6 or 16 of the Periodic Table compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/003—Compounds containing elements of Groups 4 or 14 of the Periodic Table without C-Metal linkages
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2231/00—Catalytic reactions performed with catalysts classified in B01J31/00
- B01J2231/10—Polymerisation reactions involving at least dual use catalysts, e.g. for both oligomerisation and polymerisation
- B01J2231/12—Olefin polymerisation or copolymerisation
- B01J2231/122—Cationic (co)polymerisation, e.g. single-site or Ziegler-Natta type
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0238—Complexes comprising multidentate ligands, i.e. more than 2 ionic or coordinative bonds from the central metal to the ligand, the latter having at least two donor atoms, e.g. N, O, S, P
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/40—Complexes comprising metals of Group IV (IVA or IVB) as the central metal
- B01J2531/46—Titanium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/40—Complexes comprising metals of Group IV (IVA or IVB) as the central metal
- B01J2531/48—Zirconium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/50—Complexes comprising metals of Group V (VA or VB) as the central metal
- B01J2531/56—Vanadium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/60—Complexes comprising metals of Group VI (VIA or VIB) as the central metal
- B01J2531/62—Chromium
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/02—Ethene
Definitions
- the present invention related to the field of single site catalyst systems having anionic scorpion-like three dimensional structure that are suitable for oligomerising or polymerising ethylene and alpha-olefins.
- the present invention discloses a ligand of general formula I,
- R1 , R2, R3, R4, R6, R7, R8, R9 and R10 are each independently selected from hydrogen, unsubstituted or substituted hydrocarbyl, or inert functional group, wherein two or more of those groups can be linked together to form one or more rings, with the restriction that R1 and R3 and/or R2 and R4 and/or R9 and R10, cannot be simultaneously oxazoline, wherein Z is selected from groups 15 or 16 of the Periodic Table and wherein m is the valence of Z minus one.
- inert functional group is meant a group, other than hydrocarbyl or substituted hydrocarbyl, that is inert under the complexation conditions to which the compound containing said group is subjected.
- They can be selected for example from halo, ester, ether, amino, imino, nitro, cyano, carboxyl, phosphate, phosphonite, phosphine, phosphinite, thioether and amide.
- halo such as chloro, bromo, fluoro and iodo, or ether of formula - OR * wherein R * is unsubstituted or substituted hydrocarbyl.
- Ligand I results from the reaction between a beta-diimine Il and a compound of formula III wherein X is a leaving group, preferably halogen for example Br.
- R5 is hydrogen
- R1 and R2 are the same or different and are unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups, more preferably, they are unsubstituted or substituted phenyl groups and if they are substituted, the substituents may be joined to form a closed structure. If the phenyls are substituted, the substituents preferably occupy 2 and 6 positions.
- R3 and R4 are the same or different, hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups, more preferably, they are unsubstituted or substituted alkyl groups.
- R3 and R4 may also be linked together to form a cyclohexyl ring.
- R1 with R3 and/or R2 with R4 are linked together to form a ring.
- Z is selected from N, P, O or S.
- R6, R7, R8, R9 and R10 are the same or different, hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups.
- R8 and R9 and/or R9 and R10 can be linked together to form a ring, for example a pyridine, a quinoline, an isoquinoline, a pyrrolyl, a furyl or a thiophenyl group.
- Examples of formula III include 2-(bromomethyl)-5-nitrofuran, 2-(bromomethyl)-1 ,3- dioxalane, 2-(bromomethyl)tetrahydro-2H-pyran, 2-(bromomethyl)-5- thfluoromethyl)furan, 3(-bromomethyl)pyridazine, 2-bromomethylpyridine, 1 -bromo- 2-ethoxyethane, 2-bromoethylacetate, 1-bromo-2-(2-methoxyethoxy)ethane, [(2- bromoethoxy)methyl]benzene and 3-(bromomethyl)-2,4,10-thoxaadamantane, 2- bromo-N,N-dimethylaniline.
- the invention also discloses a catalyst component of formula IV:
- the base used for deprotonating ligand I can be selected for example from butyl lithium, methyl lithium, sodium hydride. More preferably it is butyl lithium.
- M is Ti, Zr, Hf, V, Cr, Mn, Fe, Co, Ni, Pd or rare earths. More preferably, it is Cr or Ti.
- X' is halogen, more preferably it is chlorine.
- the metal is complexed with the two nitrogen atoms of the starting beta-diimine and during the complexation reaction, the complex folds around in order to coordinate heteroatom Z to the metal to form a three dimensional scorpion-like structure.
- the solvent may be selected from dichloromethane or tetrahydrofuran and the complexation reaction is carried out at room temperature.
- the present invention also discloses an active catalyst system comprising the single site catalyst component of formula IV and an activating agent having an ionising action.
- the activating agent can be an aluminium alkyl represented by formula AIR + n X3 -n wherein R + is an alkyl having from 1 to 20 carbon atoms and X is a halogen.
- the preferred aluminium alkyls are triisobutyl aluminium (TIBAL) or triethyl aluminium (TEAL).
- it can be aluminoxane and comprise oligomeric linear and/or cyclic alkyl aluminoxanes represented by formula
- n is 1 -40, preferably 1-20, m is 3-40, preferably 3-20 and R * is a Ci-Cs alkyl group and preferably methyl or isobutyl.
- the activating agent is selected from methylaluminoxane (MAO) or tetra- isobutyldialuminoxane (IBAO), more preferably, it is MAO.
- MAO methylaluminoxane
- IBAO tetra- isobutyldialuminoxane
- the amount of activating agent is selected to give an Al/M ratio of from 100 to 3000, preferably of from 500 to 2000.
- the amount of activating agent depends upon its nature, the preferred Al/M ratio is of about 2000
- Suitable boron-containing activating agents may comprise a triphenylcarbenium boronate such as tetrakis-pentafluorophenyl-borato-triphenylcarbenium as described in EP-A-0427696, or those of the general formula [L'-H] + [B An Ar 2 X3 X 4 ]- as described in EP-A-0277004 (page 6, line 30 to page 7, line 7).
- triphenylcarbenium boronate such as tetrakis-pentafluorophenyl-borato-triphenylcarbenium as described in EP-A-0427696, or those of the general formula [L'-H] + [B An Ar 2 X3 X 4 ]- as described in EP-A-0277004 (page 6, line 30 to page 7, line 7).
- the amount of boron-containing activating agent is selected to give a B/M ratio of from 0.5 to 5, preferably of about 1.
- the single site catalyst component of formula IV may be deposited on a conventional support.
- the conventional support is silica impregnated with MAO.
- the support may also be an activating support such as fluohnated alumina silica.
- the present invention further discloses a method for preparing an active catalyst system that comprises the steps of:
- step f) catalyst component IV is deposited on a support impregnated with an activating agent or on an activating support containing fluor.
- the cocatalyst may be selected from thethylaluminium, triisobutylaluminum, tris-n- octylaluminium, tetraisobutyldialuminoxane or diethyl zinc.
- the active catalyst system is used in the oligomerisation and in the polymerisation of ethylene and alpha-olefins.
- the present invention discloses a method for the oligomerisation or the homo- or co- polymerisation of ethylene and alpha-olefins that comprises the steps of: a) injecting the active catalyst system into the reactor; b) injecting the monomer and optional comonomer either before or after or simultaneously with step a); c) maintaining under polymerisation conditions; d) retrieving the oligomers and/or polymer.
- the pressure in the reactor can vary from 0.5 to 50 bars, preferably from 5 to 25 bars.
- the polymerisation temperature can range from 10 to 100 0 C, preferably from 50 to 85°C.
- the preferred monomer and optional comonomer can be selected from ethylene, propylene or 1 -hexene.
- the optional comonomer can be a polar functionalised alpha-olefin.
- the polymer formed is characterised by a melting point comprised between 130 and 135 0 C as measured by Differential Scanning Calohmetry (DSC) method. They also have a high weight average molecular weight Mw.
- the molecular weight distribution is measured by the polydispersity index D defined as the ratio Mw/Mn of the weight average molecular weight Mw over the number average molecular weight Mn.
- Molecular weights are measured by Gel Permeation Chromatography (GPC)
- the beta-diimine was synthesised according to the procedure published by Feldman and al. (Organometallics 1997, 16, p. 1514).
- Step 3 1.26 g (3 mmol) of beta-diimine were dissolved in 15 mL of dry THF under argon. The solution was cooled to a temperature of -20 0 C and 2 ml_ (3.15 mmol) of n-BuLi (1.6 M in hexane) were added dropwise. The colourless solution turned immediately bright yellow and was stirred at room temperature for 30 minutes. The solution was cooled to a temperature of -20 0 C and a solution of 2-bromomethylpyhdine in 10 ml_ of dry THF was added by canula. The solution was allowed to warm to room temperature and was stirred overnight, before being heated at a temperature of 80°C for 6 hours under reflux.
- ligand L2 was obtained with a yield of 61 %.
- ligand L3 was obtained with a yield of 41 %.
- ligand L4 was obtained with a yield of 23%.
- ligand L5 was obtained with a yield of 57%.
- ligand L7 was obtained with a yield of 40%.
- ligand L9 was obtained with a yield of 69%.
- ligand L11 was obtained with a yield of 76%.
- 0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of titanium (IV) tetrachloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -78°C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
- 0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of zirconium (IV) tetrachloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of - 78°C. The mixture was stirred under argon overnight at 70 0 C. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
- 0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of vanadium (III) trichloride tetrahydrofuran complex dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -78°C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
- 0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -20 0 C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of iron (III) trichloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -20 0 C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
- Ti(IV) complex 4 was obtained as a brown solid with a yield of 94%.
- Ti(IV) complex 6 was obtained as a brown solid with a yield of 75%.
- Ti(IV) complex 8 was obtained as a orange solid with a yield of 56%.
- V(III) complex 10 was obtained as a green solid with a yield of 76%.
- Fe(III) complex 11 was obtained as a brown solid with a yield of 63%.
- Ti(IV) complex 14 was obtained as a clear brown solid with a yield of 70%.
- Ti(IV) complex 15 was obtained as a black solid with a yield of 44%.
- Ti(IV) complex 16 was obtained as a red solid with a yield of 19%.
- Ti(IV) complex 17 was obtained as a yellow solid with a yield of 81 %.
- Ti(IV) complex 18 was obtained as an orange solid with a yield of 34%.
- Ti(IV) complex 19 was obtained as a clear brown-orange solid with a yield of 52%.
- Zr(IV) complex 20 was obtained as a yellow solid with a yield of 63%.
- Ethylene polymerisation reactions were performed in a 20 ml_ stainless steel autoclave containing a glass insert, fitted with mechanical stirring, external thermocouple and pressure gauge and controlled by computer.
- 4 ml_ of dry solvent toluene or n-heptane
- the temperature and ethylene pressure were raised to the desired values.
- Ethylene was fed continuously.
- an argon-filled glove box about 5 ⁇ mol of the appropriate catalyst were weighted.
- Activities are expressed in kg of polyethylene per mol Cr per hour.
- Activities are expressed in kg of polyethylene per mol Zr per hour.
- Activities are expressed in kg of polyethylene per mol V per hour.
- Activity is expressed as g of polyethylene per g of supported catalyst per hour.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Abstract
The present invention discloses single site catalyst systems having anionic scorpion-like three dimensional structure, that are suitable for oligomerising or polymerising ethylene and alpha-olefins.
Description
POLYMERISATION OF ETHYLENE AND ALPHA OLEFINS WITH SINGLE SITE CATALYSTS HAVING AN ANIONIC SCORPION-LIKE LIGAND
The present invention related to the field of single site catalyst systems having anionic scorpion-like three dimensional structure that are suitable for oligomerising or polymerising ethylene and alpha-olefins.
There exists a multitude of catalyst systems available for polymerising or oligomerising ethylene and alpha-olefins, but there is a growing need for finding new systems capable to tailor polymers with very specific properties. More and more post- metallocene catalyst components based on early or late transition metals from Groups 3 to 10 of the Periodic Table have recently been investigated such as for example those disclosed in Gibson and al. review (Gibson, V.C.; Spitzmesser, S. K., Chem. Rev. 2003, 103, p. 283). But there is still a need to improve either the specificities or the performances of these systems.
It is an aim of the present invention to provide a new single site catalyst component based on beta-diimine ligands with a chelating pendant arm.
It is also an aim of the present invention to provide single site catalyst components having a scorpion-like spatial organisation.
It is another aim of the present invention to provide active catalyst systems based on these catalyst components.
It is a further aim of the present invention to provide a process for polymerising or for oligomerising ethylene and alpha-olefins with these new catalyst systems.
It is yet a further aim of the present invention to provide a polyethylene by polymerising ethylene with these new catalyst systems.
Accordingly, the present invention discloses a ligand of general formula I,

wherein R1 , R2, R3, R4, R6, R7, R8, R9 and R10 are each independently selected from hydrogen, unsubstituted or substituted hydrocarbyl, or inert functional group, wherein two or more of those groups can be linked together to form one or more rings, with the restriction that R1 and R3 and/or R2 and R4 and/or R9 and R10, cannot be simultaneously oxazoline, wherein Z is selected from groups 15 or 16 of the Periodic Table and wherein m is the valence of Z minus one.
By inert functional group, is meant a group, other than hydrocarbyl or substituted hydrocarbyl, that is inert under the complexation conditions to which the compound containing said group is subjected. They can be selected for example from halo, ester, ether, amino, imino, nitro, cyano, carboxyl, phosphate, phosphonite, phosphine, phosphinite, thioether and amide. Preferably, they are selected from halo, such as chloro, bromo, fluoro and iodo, or ether of formula - OR* wherein R* is unsubstituted or substituted hydrocarbyl. After metallation of the ligand, an inert functional group must not coordinate to the metal more strongly than the groups organised to coordinate to the metal and thereby displace the desired coordinating group.
Ligand I results from the reaction between a beta-diimine Il and a compound of formula III wherein X is a leaving group, preferably halogen for example Br.

wherein R5 is hydrogen.
Preferably, R1 and R2 are the same or different and are unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups, more preferably, they are unsubstituted or substituted phenyl groups and if they are substituted, the substituents may be joined to form a closed structure. If the phenyls are substituted, the substituents preferably occupy 2 and 6 positions.
Preferably, R3 and R4 are the same or different, hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups, more preferably, they are unsubstituted or substituted alkyl groups. Optionally R3 and R4 may also be linked together to form a cyclohexyl ring.
In another embodiment according to the present invention, R1 with R3 and/or R2 with R4 are linked together to form a ring.
Preferably, Z is selected from N, P, O or S.
Preferably, R6, R7, R8, R9 and R10 are the same or different, hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups. R8 and R9 and/or R9 and R10 can be
linked together to form a ring, for example a pyridine, a quinoline, an isoquinoline, a pyrrolyl, a furyl or a thiophenyl group.
Examples of formula III include 2-(bromomethyl)-5-nitrofuran, 2-(bromomethyl)-1 ,3- dioxalane, 2-(bromomethyl)tetrahydro-2H-pyran, 2-(bromomethyl)-5- thfluoromethyl)furan, 3(-bromomethyl)pyridazine, 2-bromomethylpyridine, 1 -bromo- 2-ethoxyethane, 2-bromoethylacetate, 1-bromo-2-(2-methoxyethoxy)ethane, [(2- bromoethoxy)methyl]benzene and 3-(bromomethyl)-2,4,10-thoxaadamantane, 2- bromo-N,N-dimethylaniline.
The invention also discloses a catalyst component of formula IV:

IV
resulting from the complexation of deprotonated ligand I with the metallic salt MX'n+i in a solvent, wherein M is a metal Group 3 to 10 of the periodic Table, wherein X' is the same or different and can be a halogen, alcoholate, or substituted or unsubstituted hydrocarbyl, wherein n+1 is the valence of M and wherein said ligand I is deprotonated by a base and characterised in that pending arm

is folding to coordinate heteroatom Z to metal M.
The base used for deprotonating ligand I can be selected for example from butyl lithium, methyl lithium, sodium hydride. More preferably it is butyl lithium.
Preferably, M is Ti, Zr, Hf, V, Cr, Mn, Fe, Co, Ni, Pd or rare earths. More preferably, it is Cr or Ti.
Preferably, X' is halogen, more preferably it is chlorine.
The metal is complexed with the two nitrogen atoms of the starting beta-diimine and during the complexation reaction, the complex folds around in order to coordinate heteroatom Z to the metal to form a three dimensional scorpion-like structure.
The solvent may be selected from dichloromethane or tetrahydrofuran and the complexation reaction is carried out at room temperature.
The present invention also discloses an active catalyst system comprising the single site catalyst component of formula IV and an activating agent having an ionising action.
Suitable activating agents are well known in the art. The activating agent can be an aluminium alkyl represented by formula AIR+ nX3-n wherein R+ is an alkyl having from 1 to 20 carbon atoms and X is a halogen. The preferred aluminium alkyls are triisobutyl aluminium (TIBAL) or triethyl aluminium (TEAL).
Alternatively, it can be aluminoxane and comprise oligomeric linear and/or cyclic alkyl aluminoxanes represented by formula
R - (AI -O)n-Al R*2
R*
for oligomeric, linear aluminoxanes and by formula
R*
for oligomeric, cyclic aluminoxane,
wherein n is 1 -40, preferably 1-20, m is 3-40, preferably 3-20 and R* is a Ci-Cs alkyl group and preferably methyl or isobutyl.
Preferably, the activating agent is selected from methylaluminoxane (MAO) or tetra- isobutyldialuminoxane (IBAO), more preferably, it is MAO.
The amount of activating agent is selected to give an Al/M ratio of from 100 to 3000, preferably of from 500 to 2000. The amount of activating agent depends upon its nature, the preferred Al/M ratio is of about 2000
Suitable boron-containing activating agents may comprise a triphenylcarbenium boronate such as tetrakis-pentafluorophenyl-borato-triphenylcarbenium as described in EP-A-0427696, or those of the general formula [L'-H] + [B An Ar2 X3 X4]- as described in EP-A-0277004 (page 6, line 30 to page 7, line 7).
The amount of boron-containing activating agent is selected to give a B/M ratio of from 0.5 to 5, preferably of about 1.
In another embodiment, according to the present invention, the single site catalyst component of formula IV may be deposited on a conventional support. Preferably, the conventional support is silica impregnated with MAO. Alternatively the support may also be an activating support such as fluohnated alumina silica.
The present invention further discloses a method for preparing an active catalyst system that comprises the steps of:
a) providing a beta-diimine ligand precursor of formula II;
b) reacting the beta-diimine ligand precursor of formula Il with compound III to obtain a ligand; c) deprotonating the ligand of step b) with a base; d) complexing the deprotonated ligand of step c) with a metallic salt MX'n+i; e) retrieving a catalyst component of formula IV; f) activating the catalyst component of step e) with an activating agent having an ionising action; g) optionally adding a cocatalyst; h) retrieving an active oligomerisation or polymerisation catalyst system.
Alternatively, in step f) catalyst component IV is deposited on a support impregnated with an activating agent or on an activating support containing fluor.
The cocatalyst may be selected from thethylaluminium, triisobutylaluminum, tris-n- octylaluminium, tetraisobutyldialuminoxane or diethyl zinc.
The active catalyst system is used in the oligomerisation and in the polymerisation of ethylene and alpha-olefins.
The present invention discloses a method for the oligomerisation or the homo- or co- polymerisation of ethylene and alpha-olefins that comprises the steps of: a) injecting the active catalyst system into the reactor; b) injecting the monomer and optional comonomer either before or after or simultaneously with step a); c) maintaining under polymerisation conditions; d) retrieving the oligomers and/or polymer.
The pressure in the reactor can vary from 0.5 to 50 bars, preferably from 5 to 25 bars.
The polymerisation temperature can range from 10 to 1000C, preferably from 50 to 85°C.
The preferred monomer and optional comonomer can be selected from ethylene, propylene or 1 -hexene. Alternatively, the optional comonomer can be a polar functionalised alpha-olefin.
With a catalyst activated by MAO, the polymer formed is characterised by a melting point comprised between 130 and 135 0C as measured by Differential Scanning Calohmetry (DSC) method. They also have a high weight average molecular weight Mw. The molecular weight distribution is measured by the polydispersity index D defined as the ratio Mw/Mn of the weight average molecular weight Mw over the number average molecular weight Mn. Molecular weights are measured by Gel Permeation Chromatography (GPC)
Examples-
Preparation of catalyst components-
Synthesis of ligand L1.
Step 1.
The beta-diimine was synthesised according to the procedure published by Feldman and al. (Organometallics 1997, 16, p. 1514).
Step 2.
760 mg (3 mmol) of 2-bromomethylpyridine.HBr and 436 mg (3.15 mmol) of potassium carbonate were degassed under vacuum for 1 hour. 10 ml_ of dry acetone were added and the mixture was stirred under argon for 6 hours at room temperature (about 25 0C). The solvent was removed and the 2-bromomethylpyhdine was extracted with 3 x 10 ml_ of diethyl ether under inert atmosphere. The solvent was removed to afford a pink oil with quantitative yield.
Step 3.
1.26 g (3 mmol) of beta-diimine were dissolved in 15 mL of dry THF under argon. The solution was cooled to a temperature of -200C and 2 ml_ (3.15 mmol) of n-BuLi (1.6 M in hexane) were added dropwise. The colourless solution turned immediately bright yellow and was stirred at room temperature for 30 minutes. The solution was cooled to a temperature of -200C and a solution of 2-bromomethylpyhdine in 10 ml_ of dry THF was added by canula. The solution was allowed to warm to room temperature and was stirred overnight, before being heated at a temperature of 80°C for 6 hours under reflux. After that time, the solvent was evaporated to dryness. The residue was extracted with 10 ml_ of dichloromethane and filtered over neutral alumina. The solution was evaporated to afford a yellow oil that was purified by column chromatography (SiO2, pentane:diethyl ether 95:5 to 80:20). 910 mg of the expected product were obtained as pale yellow oil containing isomers 1a and 1 b, with a yield of 60 %.
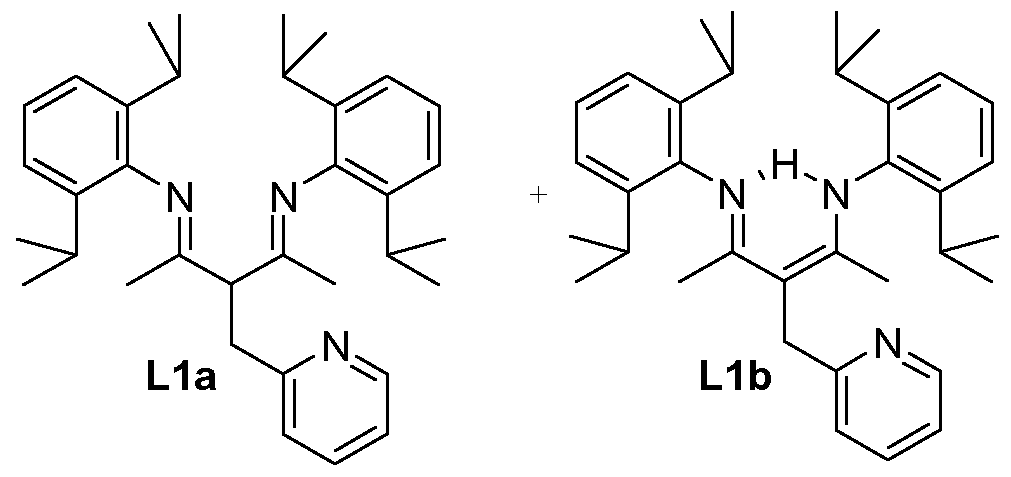
The isomers were characterised as follows:
C3SH47N3
M=509.77 g.mol"1
1H NMR (500 MHz, CDCI3) results:
Isomer 1a: δ = 1.10 (m, 12H1 CH3 ZPr), 1.21 (dd, 12H, J=6.8 Hz, CH3 /Pr), 1.92 (s, 6H,
CH3CN), 2.53 (sept, 2H, J=6.8 Hz, CH /Pr), 2.60 (sept, 2H, J=6.8 Hz, CH /Pr), 3.65
(d, 2H, J=7.5 Hz, CH2), 4.70 (t, 1 H, J=7.5 Hz, CH), 7.11 (m, 2H, CH para Ph), 7.17
(br s, 4H, CH meta Ph), 7.22 (m, 1 H, H5 pyr), 7.38 (d, 1 H, J=7.5 Hz, H3 pyr), 7.66
(td, 1 H, J=7.5 Hz, J=1.8 Hz, H4 pyr), 8.66 (d, 1 H, J=5 Hz, H6 pyr).
Isomer 1b: δ = 1.07 (m, 12H1 CH3 ZPr), 1.13 (d, 12H, J=6.9 Hz, CH3 /Pr), 1.78 (s, 6H,
CH3CN), 3.20 (sept, 4H, J=6.9 Hz, CH /Pr), 4.06 (s, 2H, CH2), 7.09 (m, 2H, CH para
Ph), 7.13 (m, 4H, CH meta Ph), 7.18 (m, 1 H, H5 pyr), 7.33 (d, 1 H, J=7.8 Hz, H3 pyr),
7.68 (m, 1 H, H4 pyr), 8.61 (d, 1 H, J=5 Hz, H6 pyr).
Synthesis of ligand L2
Using the same procedure as that used to prepare ligand 1 (steps 1 , 2 and 3), ligand L2 was obtained with a yield of 61 %.

1H NMR (500 MHz, CDCI3) : δ = 1.26 (s, 18H, CH3 ffiu), 2.00 (s, 6H, CH3CN), 3.63 (d, 2H, J=7.5 Hz, CH2), 4.44 (t, 1 H, J=7.5 Hz, CH), 6.25 (d, 2H, J=7.5 Hz), 7.05 (m, 6H), 7.34 (t, 2H, J=7.5 Hz), 7.62 (t, 1 H, J=7.5 Hz), 8.59 (d, 1 H, J=5 H).
Synthesis of ligand L3
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L3 was obtained with a yield of 41 %.

1H NMR (500 MHz, CDCI3) : δ = 1.80 (s, 6H, CH3), δ = 1.84 (s, 12H, CH3), 2.25 (s, 6H, CH3), 3.60 (d, 2H, J=7.5 Hz, CH2), 4.53 (t, 1 H, J=7.5 Hz, CH), 6.90 (s, 4H), 7.1 - 7.25 (m, 1 H), 7.30-7.80 (m, 3H), 8.58 (m, 1 H).
Synthesis of ligand L4
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3) except that 2-bromomethylquinoline.HCI was used instead of 2- bromomethylpyridine.HBr, ligand L4 was obtained with a yield of 23%.

1H NMR (300 MHz, CDCI3) : δ =1.22 (s, 18H, 2*tBu), 2.10 (s, 6H, 2*CH3), 3.86 (d, 2H, J=7.2 Hz, CH2), 4.68 (t, 1 H, J=7.2 Hz, CH), 6.31 (d, 2H, J=7.5 Hz,2*CH Ph), 7.05 (m, 4H, 4*CH Ph), 7.35 (d, 2H, J=7.5 Hz, 2*CH Ph), 7.46 (d, 1 H, J=8.4 Hz, CH quin), 7.53 (t, 1 H, J=7.5 Hz, CH quin), 7.72 (t, 1 H, J=7.6 Hz, CH quin), 7.81 (d, 1 H, J=8.1 Hz, CH quin), 809 (d, 2H, J=8.4 Hz, 2*CH quin).
Synthesis of ligand L5
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), except that 2-methoxy-5-nitrobenzyl bromide was used instead of 2- bromomethylpyridine.HBr, ligand L5 was obtained with a yield of 57%.

1H NMR (300 MHz, CDCI3) : δ =1.29 (s, 18H, 2*tBu), 1.99 (s, 6H, 2*CH3), 3.52 (d, 2H, J=7.5 Hz, CH2); 4.00 (s, 3H, OCH3), 4.17 (t, 1 H, J=7.5 Hz, CH), 6.27 (dd, 2H), 7.05 (m, 6H), 7.37 (dd, 2H), 8.20 (s, 1 H).
Synthesis of ligand L6
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L6 was obtained with a yield of 67%.

1H NMR (300 MHz, CDCI3) : δ = 1.98 (s, 6H, CH3), 3.52 (d, 2H, J=7.5 Hz, CH2), 4.27 (t, 1 H, J=7.5 Hz, CH), 7.03 (s, 4H), 7.21 (m, 1 H), 7.28 (d, 1 H, J=7.8 Hz), 7.68 (t, 1 H, J=7.6 Hz), 8.61 (m, 1 H).
Synthesis of ligand L7
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L7 was obtained with a yield of 40%.

1H NMR (300 MHz, CDCI3) : δ = 1 ,94 (s, 6H, CH3) ; 3,56 (d, 2H, d, J=7.5 Hz, CH2) ; 4,42 (t, 1 H, t, J=7.5 Hz, CH) ; 6,68 (t, JHF=8.3 Hz, 4H) ; 7,14 (m, 1 H), 7,26 (d, 1 H, J=7.5 Hz), 7,61 (t, 1 H, J=7.7 Hz), 8,56 (d, 1 H, J=3.9 Hz).
Synthesis of ligand L8
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L8 was obtained with a yield of 46%.
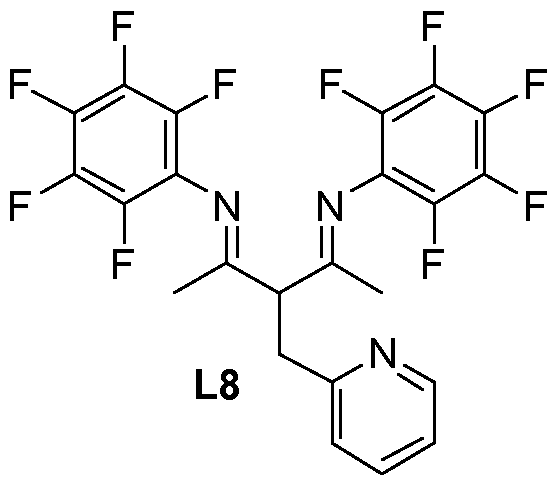
1H NMR (300 MHz, CDCI3) : δ = 1.99 (s, 6H, CH3), 3.56 (d, 2H, J=7.5 Hz, CH2), 4.53 (t, 1 H, J=7.5 Hz, CH), 7.18 (m, 1 H), 7.26 (d, 1 H, J=7.8 Hz), 7.65 (t, 1 H, J=7.5 Hz), 8.56 (m, 1 H).
Synthesis of ligand L9
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L9 was obtained with a yield of 69%.

1H NMR (300 MHz, CDCI3) : δ = 2.00 (s, 6H, CH3), 3.65 (d, 2H, J=7.8 Hz, CH2), 4.67 (t, 1 H, J=7.8 Hz, CH), 6.92 (t, 2H, J=8.1 Hz), 7.14 (m, 1 H), 7.26 (m, 4H), 7.28 (m, 1 H), 7.61 (t, 1 H, J=7.6 Hz), 8.60 (m, 1 H).
Synthesis of ligand L10
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L10 was obtained with a yield of 39%.

1H NMR (300 MHz, CDCI3) : δ = 1 ,25 (s, 18H, tBu), 2,05 (s, 6H, CH3), 2,27 (s, 3H, CH3 pyr), 2,31 (s, 3H, CH3 pyr), 3,56 (d, 2H, J=7.2 Hz, CH2), 3,75 (s, 3H, OCH3), 4,58 (t, 1 H, J=7.2 Hz, CH), 6,33 (d, J=7.6 Hz, 2H), 7,01 (t, J=7.5 Hz, 2H), 7,10 (t, J=7.4 Hz, 2H), 7,35 (d, J=7.8 Hz, 2H), 8,21 (s, 1 H).
Synthesis of ligand L11
Using the same procedure as that used to prepare ligand L1 (steps 1 , 2 and 3), ligand L11 was obtained with a yield of 76%.

1H NMR (300 MHz, CDCI3) : δ = 1 ,37 (s, 18H, tBu), 1 ,97 (s, 6H, CH3), 2,41 (q, 2H, J=6.6 Hz, CH2), 3,42 (s, 3H, OCH3), 3,62 (t, 2H, J=6.4 Hz, CH2O), 3,77 (t, 1 H, J=7.1 Hz, CH), 6,41 (d, 2H, J=7.6 Hz), 7,04 (t, 2H, J=7.5 Hz), 7,14 (t, 2H, J=7.5 Hz), 7,38 (d, 2H, J=7.8 Hz).
Method 1 Preparation of Cr(III) complexes
0.5 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.5 mmol of n-butyl lithium were added dropwise at a temperature of -15°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.5 mmol of chromium (III) chloride tetrahydrofuran complex dissolved in 5 ml_ of dry tetrahydrofuran. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 5 ml_ of pentane and dried under vacuum.
Method 2: Preparation of Ti(IV) complexes
0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of titanium (IV) tetrachloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -78°C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
Method 3: Preparation of Zr(IV) complexes
0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of zirconium (IV) tetrachloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of - 78°C. The mixture was stirred under argon overnight at 700C. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
Method 4: Preparation of V(III) complexes
0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -78°C. The solution
was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of vanadium (III) trichloride tetrahydrofuran complex dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -78°C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
Method 5: Preparation of Fe(III) complexes
0.35 mmol of the appropriate ligand were dissolved in 5 ml_ of tetrahydrofuran. 0.35 mmol of n-butyl lithium were added dropwise at a temperature of -200C. The solution was allowed to warm to room temperature and was stirred for 30 minutes. The solution was added dropwise to a solution of 0.35 mmol of iron (III) trichloride dissolved in 5 ml_ of dry tetrahydrofuran cooled at a temperature of -200C. The mixture was stirred under argon overnight at room temperature. The solution was concentrated to approximately 2 ml_ and 10 ml_ of pentane were added. The solid was filtered off, washed twice with 10 ml_ of pentane and dried under vacuum.
Using method 1 , Cr(III) complex 1 was obtained as a pale grey solid with a yield of 74%.

Using method 1 , Cr(III) complex 2 was obtained as a pale green solid with a yield of 75%.

Using method 1 , Cr(III) complex 3 was obtained as a pale green solid with a yield of 48%.
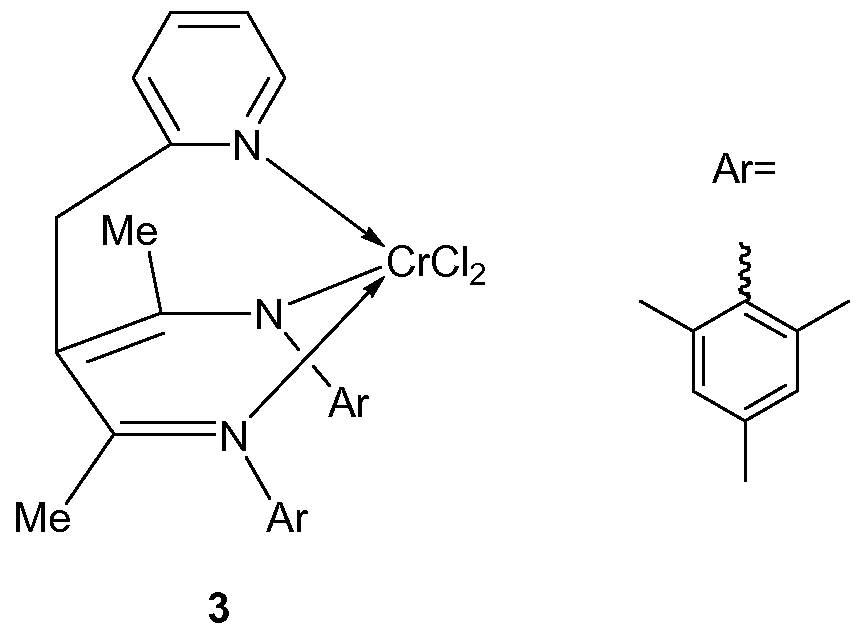
Using method 2, Ti(IV) complex 4 was obtained as a brown solid with a yield of 94%.

Using method 2, Ti(IV) complex 5 was obtained as a brown solid with a yield of 94%.

Using method 2, Ti(IV) complex 6 was obtained as a brown solid with a yield of 75%.

Using method 1 , Cr(III) complex 7 was obtained.

Using method 2, Ti(IV) complex 8 was obtained as a orange solid with a yield of 56%.

Using method 3, Zr(IV) complex 9 was obtained.

Using method 4, V(III) complex 10 was obtained as a green solid with a yield of 76%.

Using method 5, Fe(III) complex 11 was obtained as a brown solid with a yield of 63%.

11
Using method 1 , Cr(III) complex 12 was obtained as a green solid with a yield of
34%.

12
Using method 2, Ti(IV) complex 13 was obtained as a dark brown solid with a yield of
50%.

Using method 2, Ti(IV) complex 14 was obtained as a clear brown solid with a yield of 70%.

14
Using method 2, Ti(IV) complex 15 was obtained as a black solid with a yield of 44%.

15
Using method 2, Ti(IV) complex 16 was obtained as a red solid with a yield of 19%.

16
Using method 2, Ti(IV) complex 17 was obtained as a yellow solid with a yield of 81 %.

17
Using method 2, Ti(IV) complex 18 was obtained as an orange solid with a yield of 34%.

18
Using method 2, Ti(IV) complex 19 was obtained as a clear brown-orange solid with a yield of 52%.

19
Using method 3, Zr(IV) complex 20 was obtained as a yellow solid with a yield of 63%.

20
High pressure polymerisation of ethylene.
Ethylene polymerisation reactions were performed in a 20 ml_ stainless steel autoclave containing a glass insert, fitted with mechanical stirring, external thermocouple and pressure gauge and controlled by computer. In a typical reaction run, 4 ml_ of dry solvent (toluene or n-heptane) were introduced into the reactor and the temperature and ethylene pressure were raised to the desired values. Ethylene was fed continuously. In an argon-filled glove box, about 5 μmol of the appropriate catalyst were weighted. It was activated with methylaluminoxane (MAO 30 % wt in toluene) in an amount appropriate to obtain a ratio [AI]:[M] of 2000 and it was diluted with toluene to a final volume of 2 ml_. 200 μl_ of the solution of activated catalyst were placed inside the reactor. The injection loop was rinsed with 800 μl_ of solvent. After 1 hour or after an ethylene consumption of 12 mmol, the reaction was quenched with isopropanol and an aliquot was analysed by gas chromatography. The gas chromatographic analysis of the reaction products were performed on a Trace GC apparatus with a Petrocol capillary column (methyl silicone, 100 m long, i.d. 0.25 mm and film thickness of 0.5 μm) working at a temperature of 35°C for 15 min and then heated to a temperature of 250 0C at a heating rate of 5°C/min. The remaining reaction mixture was quenched with MeOH / HCI and the polymer was filtered, washed with methanol and dried at a temperature of 500C, under vacuum, for a period of time of 24 hours.
The results are displayed in Table I and in Table II.
TABLE I. Results with the Cr(III) complexes
m PE Activity DSC
Run Complex P (bar) T (0C) (mg) (kg/mol/h) Tm (0C) ΔH (J.g 1)
1 1 15 50 47 93 165.5 2 2 15 50 45 86 178.5 3 3 15 50 40 78 177.8
4 7 15 50 36 70 130 170.9
5 12 15 50 41 81 131 170.6
6 1 19 80 27 53 131 202.5
7 2 19 80 26 52 131 209.1
8 3 19 80 32 63 130 183.5
All reactions were performed with 0.5 μmol of catalyst dissolved in 5 ml_ of n-heptane during 1 hour.
MAO was added to give a [AI]:[Cr] ratio of 2000.
Activities are expressed in kg of polyethylene per mol Cr per hour.
TABLE II. Results with the Ti(IV) complexes
Time m PE Activity
Run Complex P (bar) DSC
T (0C) (min) (mg) (kg/mol/h) Tm (0C) ΔH (J.g 1)
9 4 15 50 60 552 1111 135 133,3
10 5 15 50 60 630 1198 135 122,1
1 1 6 15 50 60 583 1027 133 138,6
12 4 19 80 51 812 1909 134 136,9
13 5 19 80 48 862 2094 135 145,7
14 6 19 80 52 955 2187 136 150,4
15 8 19 80 60 257 512 134 120.0
16 13 19 80 60 274 549 134 125.0
17 14 19 80 60 408 1646 136 108.2
18 15 19 80 60 301 1209 139 119.1
19 16 19 80 60 705 1399 132 98.7
20 17 19 80 60 387 765 133 129.7
21 18 19 80 60 260 514 133 96.6
22 19 19 80 60 383 749 138 109.2
All reactions were performed with 0.5 μmol of catalyst dissolved in 5 ml_ of n-heptane during 1 hour.
MAO was added to give a [AI]:[Ti] ratio of 2000.
Activities are expressed in kg of polyethylene per mol Ti per hour.
TABLE III. Results with the Zr(IV) complex
Run Con^ P (bar) T fC, ^ , *£» , Tm ,χ, "^ ,,,-,,
23 9 15 50 395 778 133 130.1
24 9 19 80 473 932 134 148.9
25 20 19 80 249 480 138 137.7
All reactions were performed with 0.5 μmol of catalyst dissolved in 5 ml_ of n-heptane during 1 hour.
MAO was added to give a [AI]:[Zr] ratio of 2000.
Activities are expressed in kg of polyethylene per mol Zr per hour.
TABLE IV. Results with the V(III) complex
Run Convex P,bar, T1-C, ^ ,*£», " ,
25 10 15 50 52 103 133 139.1 27 10 19 80 63 125 133 146.8
All reactions were performed with 0.5 μmol of catalyst dissolved in 5 ml_ of n-heptane during 1 hour.
MAO was added to give a [AI]:[V] ratio of 2000.
Activities are expressed in kg of polyethylene per mol V per hour.
Polymerisation of ethylene with a supported catalyst
Complex 5 was deposited on a MAO impregnated silica (Sylopol 952X1836), with 2 wt % of complex based on the total weight of the obtained supported catalyst. This supported catalyst was used for the polymerisation of ethylene. Ethylene polymerisation reactions were carried out in a 130 ml stainless steel autoclave equipped with mechanical stirring and a stainless steel injection cylinder. In a typical reaction run, the reactor was first dried under nitrogen flow at 1000C during 10 min. Then it was cooled down to the reaction temperature (85°C) and 35 ml of
isobutane were introduced into the reactor with a syringe pump. The pressure was adjusted to the desired value (23.8 bar) with ethylene. In an argon-filled glove box, 301 mg of the supported catalyst, 0.6 ml of TiBAI and 0.5 ml of n-heptane were placed into the injection cylinder. The valve was closed and the cylinder was connected to the reactor under nitrogen flow. The active catalyst mixture was then introduced into the reactor with 40 ml of isobutane. After 1 hour at 750 rpm, the reactor was cooled down to room temperature and slowly depressurised. The polymer was recovered and characterised by DSC. The polymerisation results are displayed in Table V.
TABLE V: Polymerisation results with supported Ti(IV) complex 5

* Activity is expressed as g of polyethylene per g of supported catalyst per hour.
Due to the high molecular weight of the obtained polymers, GPC and 13C NMR analysis were not possible, even in TCB at 135°C.
Claims
1. A deprotonated ligand of general formula I,
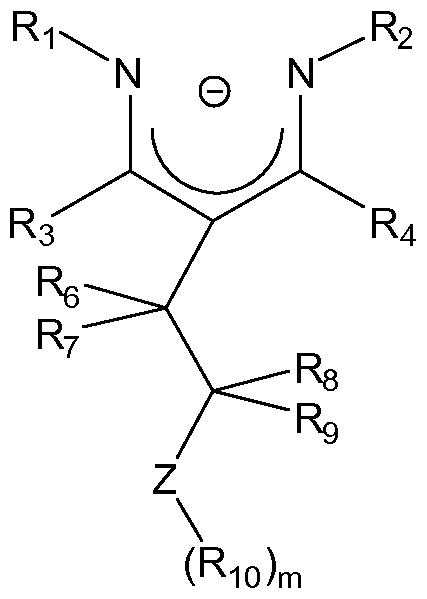
wherein R1 , R2, R3, R4, R6, R7, R8, R9 and R10 are each independently selected from hydrogen, unsubstituted or substituted hydrocarbyl, or inert functional group and wherein two or more of said R's can be linked together to form one or more rings, with the restriction that R1 and R3, R2 and R4, and R9 and R10, cannot all be simultaneously oxazoline, wherein Z is selected from groups 15 or 16 of the Periodic Table and m is the valence of Z minus one.
2. The deprotonated ligand of claim 1 wherein R1 and R2 are the same or different and are unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups.
3. The deprotonated ligand of claim 2 wherein R1 and R2 are the same and are unsubstituted or substituted phenyl groups.
4. The deprotonated ligand of any one of claims 1 to 3 wherein R3 and R4 are the same or different, and are hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups.
5. The deprotonated ligand of claim 4 wherein R3 and R4 are unsubstituted or substituted alkyl groups or are linked together to form a cyclohexyl ring.
6. The deprotonated ligand of any one of the preceding claims wherein R1 with R3 and/or R2 with R4 are linked together to form a ring.
7. The deprotonated ligand of any one of the preceding claims wherein Z is selected from N, P, O or S.
8. The deprotonated ligand of any one of the preceding claims wherein R6, R7, R8, R9 and R10 are the same or different, and are hydrogen, unsubstituted or substituted alkyl groups, unsubstituted or substituted aryl groups, or unsubstituted or substituted cycloalkyl groups.
9. A catalyst component of formula IV:

IV
resulting from the complexation of deprotonated ligand I of any one of claims 1 to 9 with the metallic salt MX'n+i in a solvent, wherein M is a metal Group 3 to 10 of the periodic Table, X' is the same or different and is halogen, alcoholate, or substituted or unsubstituted hydrocarbyl and wherein n+1 is the valence of M and characterised in that pending arm

is folding to coordinate heteroatom Z to metal M.
10. The metallic complex of claim 10 wherein M is Ti, Zr, Hf, V, Cr, Mn, Fe, Co, Ni, Pd.
11. An active catalyst system comprising the metallic complex of claim 10 or claim 11 and an activating agent having an ionising action.
12. A method for preparing an active catalysts system that comprises the steps of:
a) providing a beta-diimine ligand precursor of formula II; b) reacting the beta-diimine ligand precursor of formula Il with compound III to obtain a ligand; c) deprotonating the ligand of step b) by subtracting the R5 hydrogen with a base; d) complexing the deprotonated ligand of step c) with a metallic salt  e) retrieving a catalyst component of formula IV; f) activating the catalyst component of step e) with an activating agent having an ionising action; g) optionally adding a cocatalyst; h) retrieving an active oligomerisation or polymerisation catalyst system.
e) retrieving a catalyst component of formula IV; f) activating the catalyst component of step e) with an activating agent having an ionising action; g) optionally adding a cocatalyst; h) retrieving an active oligomerisation or polymerisation catalyst system.
13. A method for the oligomerisation or for the homo- or co-polymerisation of ethylene and alpha-olefins that comprises the steps of: a) injecting the active catalyst system of claim 13 into the reactor; b) injecting the monomer and optional comonomer either before or after or simultaneously with step a); c) maintaining under polymerisation conditions; d) retrieving the oligomers and/or polymer.
14. The method of claim 14 wherein the monomer and optional comonomer are selected from ethylene, propylene and 1 -hexene.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07857204A EP2089399B1 (en) | 2006-11-24 | 2007-11-09 | Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand |
| US12/516,203 US20100298122A1 (en) | 2006-11-24 | 2007-11-09 | Polymerisation of Ethylene and Alpha Olefins with Single Site Catalysts having an Anionic Scorpion-Like Ligand |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06124706A EP1925620A1 (en) | 2006-11-24 | 2006-11-24 | Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand |
| EP06124706.0 | 2006-11-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008061900A1 true WO2008061900A1 (en) | 2008-05-29 |
Family
ID=37909331
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/062111 Ceased WO2008061900A1 (en) | 2006-11-24 | 2007-11-09 | Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20100298122A1 (en) |
| EP (2) | EP1925620A1 (en) |
| WO (1) | WO2008061900A1 (en) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102020741B (en) * | 2010-10-20 | 2012-11-14 | 中南民族大学 | Method for preparing linear low-density polyethylene and bifunctional catalyst system |
| CN112920300B (en) * | 2021-02-01 | 2021-12-24 | 中国科学院长春应用化学研究所 | Large steric hindrance alpha-diimine ligand, nickel catalyst, preparation method and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001034665A1 (en) | 1999-11-12 | 2001-05-17 | E.I. Du Pont De Nemours And Company | Chain transfer agents for olefin polymerization |
| EP1754723A1 (en) * | 2005-07-07 | 2007-02-21 | Total Petrochemicals Research Feluy | Single site catalyst systems having a scorpion-like structure |
-
2006
- 2006-11-24 EP EP06124706A patent/EP1925620A1/en not_active Withdrawn
-
2007
- 2007-11-09 EP EP07857204A patent/EP2089399B1/en not_active Not-in-force
- 2007-11-09 US US12/516,203 patent/US20100298122A1/en not_active Abandoned
- 2007-11-09 WO PCT/EP2007/062111 patent/WO2008061900A1/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001034665A1 (en) | 1999-11-12 | 2001-05-17 | E.I. Du Pont De Nemours And Company | Chain transfer agents for olefin polymerization |
| EP1754723A1 (en) * | 2005-07-07 | 2007-02-21 | Total Petrochemicals Research Feluy | Single site catalyst systems having a scorpion-like structure |
Non-Patent Citations (1)
| Title |
|---|
| GIBSON, V.C., SPITZMESSER, S.K., CHEM. REV., vol. 103, 2003, pages 283 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100298122A1 (en) | 2010-11-25 |
| EP2089399B1 (en) | 2012-08-08 |
| EP1925620A1 (en) | 2008-05-28 |
| EP2089399A1 (en) | 2009-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008061901A1 (en) | Polymerisation of ethylene and alpha-olefins with pyrroliminophenol complexes. | |
| EP2010550B1 (en) | Polymerisation of ethylene and alpha-olefins with pyridino-iminophenol complexes | |
| EP1994006B1 (en) | Polymerisation of ethylene and alpha-olefins with imino-quinolinol complexes | |
| US7754835B2 (en) | Polymerisation of ethylene and alpha-olefins with phosphino-iminophenol complexes | |
| US8519070B2 (en) | Post-metallocene complexes based on bis(naphthoxy)pyridine and bis(naphthoxy)thiophene ligands for the polymerisation of ethylene and alpha-olefins | |
| EP2089399B1 (en) | Polymerisation of ethylene and alpha olefins with single site catalysts having an anionic scorpion-like ligand | |
| US7786232B2 (en) | Single site catalyst systems having a scorpion-like structure | |
| US20090111958A1 (en) | Single catalyst systems having a scorpion-like structure | |
| US8420756B2 (en) | Polymerisation of ethylene and alpha-olefins with catalyst systems based on binam derived ligands | |
| CN119462720A (en) | A kind of aminophenoloxy transition metal complex and its preparation method and application |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07857204 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007857204 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12516203 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |