WO2008077554A1 - Cycloalkylamine substituted isoquinoline derivatives - Google Patents
Cycloalkylamine substituted isoquinoline derivatives Download PDFInfo
- Publication number
- WO2008077554A1 WO2008077554A1 PCT/EP2007/011167 EP2007011167W WO2008077554A1 WO 2008077554 A1 WO2008077554 A1 WO 2008077554A1 EP 2007011167 W EP2007011167 W EP 2007011167W WO 2008077554 A1 WO2008077554 A1 WO 2008077554A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkylene
- aryl
- heterocyclyl
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*C(CCC1)C(C)(*)CC1N(*)* Chemical compound C*C(CCC1)C(C)(*)CC1N(*)* 0.000 description 2
- BCURVDYTHOJCRW-UHFFFAOYSA-N NC(CC1)CCC1Oc(ccc1c2ccnc1)c2Cl Chemical compound NC(CC1)CCC1Oc(ccc1c2ccnc1)c2Cl BCURVDYTHOJCRW-UHFFFAOYSA-N 0.000 description 1
- TVATXWYAPMOKRB-JOCQHMNTSA-N N[C@H](CC1)CC[C@@H]1Oc(c(Cl)c1)cc2c1cncc2 Chemical compound N[C@H](CC1)CC[C@@H]1Oc(c(Cl)c1)cc2c1cncc2 TVATXWYAPMOKRB-JOCQHMNTSA-N 0.000 description 1
- HJFWRLPBMPQGFY-XYPYZODXSA-N N[C@H](CC1)CC[C@@H]1Oc(c(Cl)cc1cnccc11)c1Cl Chemical compound N[C@H](CC1)CC[C@@H]1Oc(c(Cl)cc1cnccc11)c1Cl HJFWRLPBMPQGFY-XYPYZODXSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/24—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention relates to novel isoquinoline derivatives as described in the claims, their preparation and their use in the treatment and/or prevention of diseases related to the inhibition of Rho-kinase and/or of Rho-kinase mediated phosphorylation of myosin light chain phosphatase.
- Rho-kinase 2 Activation of a small GTPase RhoA upon agonist stimulation results in conversion of RhoA from the inactive GDP-bound form to the active GTP-bound form with a subsequent binding to and activation of Rho-kinase.
- Rho-kinase 1 and Rho-kinase 2 Two isoforms, Rho-kinase 1 and Rho-kinase 2, are known.
- Rho-kinase 2 is expressed in vascular smooth muscle cells and endothelial cells.
- Rho-kinase 2 Activation of Rho-kinase 2 by the active GTP-bound RhoA leads to calcium sensitization of smooth muscle cells through phosphorylation-mediated inhibition of the myosin light chain phosphatase activity and thereby up-regulation of the activity of myosin regulatory light chain (Uehata et al., Nature 1997, 389, 990-994).
- Rho-kinase is involved in vasoconstriction, including the development of myogenic tone and smooth muscle hypercontractility (Gokina et al. J. Appl. Physiol. 2005, 98, 1940-8), bronchial smooth muscle contraction (Yoshii et al. Am. J. Resp. Cell MoI. Biol. 20, 1190-1200), asthma (Setoguchi et al. Br J Pharmacol. 2001 , 132, 111 -8; Nakahara, et al.
- PAOD peripheral arterial occlusive disease
- sexual dysfunction e.g., penile erectile dysfunction (Chitaley et al. Nature Medicine 2001 , 7, 119-122), retinopathy, inflammation, immune diseases, AIDS, osteoporosis, endocrine dysfunctions, e.g. hyperaldosteronism, central nervous system disorders such as neuronal degeneration and spinal cord injury (Hara, et al. JNeurosurg 2000, 93, 94), cerebral ischemia (Uehata, et al. Nature 1997,389,990; Satoh et al. Life Sci. 2001 , 69, 1441-53; Hitomi, et al.
- a compound having inhibitory effect on Rho-kinase and/or on Rho-kinase mediated phosphorylation of myosin light chain phosphatase is useful for the treatment and/or prevention of cardiovascular and non-cardiovascular diseases involving Rho- kinase as the primary or secondary disease cause, like hypertension, pulmonary hypertension, ocular hypertension, retinopathy, and glaucoma, peripheral circulatory disorder, peripheral arterial occlusive disease (PAOD), coronary heart disease, angina pectoris, heart hypertrophy, heart failure, ischemic diseases, ischemic organ failure (end organ damage), fibroid lung, fibroid liver, liver failure, nephropathy, including hypertension-induced, non-hypertension-induced, and diabetic nephropathies, renal failure, fibroid kidney, renal glomerulosclerosis, organ hypertrophy, asthma, chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome, thrombotic disorders, stroke, cerebral
- neuropathic pain neuronal degeneration, spinal cord injury, Alzheimer's disease, premature birth, erectile dysfunction, endocrine dysfunctions, arteriosclerosis, prostatic hypertrophy, diabetes and complications of diabetes, metabolic syndrome, blood vessel restenosis, atherosclerosis, inflammation, autoimmune diseases, AIDS, osteopathy such as osteoporosis, infection of digestive tracts with bacteria, sepsis, cancer development and progression, e.g. cancers of the breast, colon, prostate, ovaries, brain and lung and their metastases.
- WO 01/64238 describes isoquinoline-5-sulfonamide derivatives optionally substituted by a -(CH 2 )i-6-0-(CH 2 )o-6-. a -(CH 2 ) ⁇ -6-S-(CH 2 ) ⁇ -6- or a -(CH 2 ) 0 _6-linked heterocyclic group useful as neuroprotective agents.
- WO 2004/106325 (Schering AG) describes prodrugs of the Rho-kinase inhibitor fasudil carrying an ether or ester group in the 1-position of the isoquinoline ring.
- JP 10087629 A describes isoquinoline derivatives useful for the treatment of diseases caused by Heliobacter pylori such as for example gastritis cancer or ulcer.
- the isoquinoline derivatives may be substituted by OH in the 1 -position and are preferably 5-substituted by X-[(C 1 -C 6 )alkylene)]o--
- I may be among others an optionally substituted isoquinolone and Ar Il may be among others an optionally substituted 03.7 monocyclic saturated heterocyclic system.
- WO 2005/030791 (Merck & Co.) generically describes as potassium channel inhibitors for the treatment of cardiac arrhythmias, stroke, congestive heart failure etc. isoquinolone derivatives which are optionally substituted in 6-position by a group
- R 43 is e.g. a (C3-C ⁇
- WO 2005/030130 (Merck & Co.) generically describes as potassium channel inhibitors for the treatment of cardiac arrhythmias, stroke, congestive heart failure etc. isoquinoline derivatives which may be substituted by hydroxyl in the 1 -position and are optionally substituted in 6-position by a group (CR e Rf)pOR 43 wherein p may be zero, and R 43 is e.g.
- R 51 and R 52 may be hydrogen, (C 1 -C 6 )alkyl etc.; or R 43 is a group R81 defined as a 4-6 membered unsaturated or saturated monocyclic heterocylic ring with 1 , 2, 3 or 4 heteroatoms; and are substituted by a directly bound optionally substituted aryl or heteroaryl ring in the 4-position.
- WO 03/053330 (Ube) generically describes isoquinolone derivatives of the formula
- Rho-kinase inhibitors As Rho-kinase inhibitors.
- An embodiment of the present invention is a compound of the formula (I)
- R2 is H, halogen or (C 1 -C 6 )SlRyI;
- R 3 is H 1 halogen
- Rg and RQ are independently of each other
- R8 is H, halogen or (C 1 -C 6 )alkyl
- n 1 , 2, 3 or 4;
- n 1 , 2 ,3 ,4 or 5
- L is O or O- (C 1 -C 6 )alkylene
- R" is (C 3 -C 8 )cycloalkyl
- residues R4, R5, RQ, RQ , RJ and Rs alkyl, alkylene or cycloalkyl can optionally be substituted one or more times by OH, OCH3, COOH, COOCH3, NH 2 , NHCH3, N(CH3)2, CONH2, CONHCH3 or CON(CH3)2 ;
- residues R 1 to Rs alkyl or alkylene can optionally be substituted one or more times by halogen
- and R3 to R ⁇ (Ce-C 1 rj)aryl and (Cs-C 10 )heterocyclyl are unsubstituted or substituted one or more times by suitable groups independently selected from halogen, OH, NO2, N 3 , CN, C(O)-(C 1 -Ce)alkyl, C(O)-(C ⁇
- Ce)alkyl-C ⁇ HC-i-Ce)alkyl N(C 1 -C 6 )alkyl-C(O)O-(C 1 -C 6 )alkyl,
- C 6 )alkyl CONH 2 , (C 1 -C 6 )alkylene-O-fC-i-Ce)alkyl, (C-i-C ⁇ alkylene-O ⁇ Ce-C 10 )aryl, or O ⁇ C-i-C ⁇ alkylene-tCe-C-icOaryl; or wherein (C 6 -C 1 o)aryl is vicinally substituted by a O-(C 1 -C4)alkylene-O group whereby a 5-8-membered ring is formed together with the carbon atoms the oxygen atoms are attached to; and wherein aryl or heterocyclyl substituents of (C 6 -C 1 o)aryl and (Cs-C 10 )heterocyclyl groups may not be further substituted by an aryl or heterocyclyl containing group;
- R 1 is H, (C-i-C ⁇ alkyl, (C 6 -C 10 )aryl, NH-(C 1 -C 6 )alkyl, NH-(C 6 -C 1 rj)aryl or Nt(C 1 -C 6 )alkyl] 2 . More preferably, R 1 is H, halogen, (C-i-C ⁇ alkyl, NH-(C 1 -C4)alkyl, Nt(C 1 -C 4 )alkyl] 2 or NH-phenyl. Most preferably, R 1 is H, (C-
- R3 is preferably H, halogen, (C-
- R4 is H, halogen or (C 1 -C 6 )alkyl. More preferred, R4 is H, halogen or (C-
- R5 is H, halogen, CN, (C 1 -C 6 )alkyl, (C2-C6)alkenyl, R', NH-(C 6 -Ci rj)aryl or (C 1 -C 6 )alkylene-R'.
- R5 is H, halogen, (C 1 -C 6 )alkyl, (C2-C6)alkenyl, R', NH-(C 6 -Ci ⁇ jaryl or (C 1 -C 6 )alkylene-R ⁇
- R5 is H, halogen, (C 1 -C 6 )alkyl, (C2-C6)alkenyl, (C 6 -Ci o)aryl, NH-(C 6 -C 10 )aryl, (C-
- R5 is H, halogen, phenyl, (C-
- R5 is H, halogen, methyl, ethyl, vinyl, phenyl, thienyl or pyridyl.
- R5 are hydrogen, fluoro, chloro, bromo, iodo, methyl, ethyl, vinyl, phenyl, thienyl or pyridyl, nitrile, nitro, (p-methoxy)-phenyl, N-aniline, benzyl, 2-propenyl, s- butenyl, cyclopropyl, tetrazol, amino, 4-methoxy-aniline or N-acetyl, preferably hydrogen, fluoro, chloro, bromo, iodo, methyl, ethyl, vinyl, phenyl, thienyl or pyridyl. More preferred, R5 is H, halogen, methyl, or ethyl, most preferred R5 is H.
- R 6 and R 6 ' are independently of each other H, (C 1 -C 6 )aIkYl 1
- R' (C 1 -C4)alkylene-(C3-C8)cycloalkyl
- RQ and RQ' are independently of each other
- Rg is H, (C 1 -C 6 )alkyl, (C3-C6)cycloalkyl or (C-] -C4)alkylene-(C3-C6)cycloalkyl, and
- RQ is H, (C 1 -C 6 )alkyl and RQ' is
- RQ and Rg' together with the N-atom to which they are attached, form a (C5-C 10 )heterocyclyl group.
- RQ is H, (C 1 -C 6 )alkyl and RQ' is H, (C 1 -Ce) 3 IkYl;
- -C4)alkylene-(C5-C 10 )heterocyclyl wherein heterocyclyl is unsubstituted or substituted one or more times, preferably one to three times, more preferabyl one or two times, by a group independently selected from (C-
- -C 4 )alkyl preferably CH3 or CF3; O-(Ci-C4)alkyl; CN, SO2-NH2; SO2-
- O-(C 1 -C4)alkyl, or (C6-C 10 )aryl is substituted once by unsubstituted phenyl, unsubstituted O-phenyl or unsubstituted (C5-C6)heterocyclyl; or RQ and RQ', together with the N-atom to which they are attached, form a
- (C5-C6)heterocyclyl group which is unsubstituted or substituted one to three times, preferably once, by (C-
- the formed heterocyclyl group is morpholino, piperidino, pyrrolidine or piperazino. More preferably the heterocyclyl group is morpholino or 4- (ethoxycarbonyl)-piperazinyl.
- R ⁇ is H, (C 1 -C 6 )alkyl and R ⁇ ' is H, (C 1 -C 6 )alky ⁇ , (C3-C8)cycloalkyl.
- R ⁇ is H and RQ' is H, unsubstituted (C-
- R ⁇ and R ⁇ ' are H.
- R6 or R6 ' are, independently from each other, hydrogen, methyl, ethyl, propyl, isopropyl, 3-methyl-butyl, 2-methyl-propyl, butyl, pentyl, 3,3,3-trifuoropropyl, 4,4,4-trifluorobutyl or a substituent selected from the group consisting of
- R 6 or R 6 ' are, independently from each other,
- R 6 , R 6 ' forming a (C 5 -C io)heterocyclyl is
- R7 is H, halogen, CN, (C 1 -C 6 )alkyl, O-(C ⁇
- R7 is H, fluoro, chloro, bromo, methyl, ethyl, methoxy, phenyl, nitrile, cyclopropyl, thienyl or vinyl, most especially preferred R7 is H, fluoro, chloro, methyl or methoxy. More particular preferred R7 is H.
- Rg is preferably H, halogen or (C-
- R2 is H, halogen or (C-
- R2 is H or (C-
- R2 may be bound to any carbon atom of the ring including the position where the linker group L is bound.
- n is 1 , 2 or 3. More preferred, n is 1 or 2. Most preferred n is 1.
- n is 2, 3 or 4. More preferred m is 3.
- the linker group L may be bound to the ring in any position via a ring carbon atom.
- m is 3 and L is attached to the 4-position of the amino cyclohexane ring
- L is attached to the 4-position of the amino cyclohexane ring.
- L is O-methylene, O-ethylene or preferably O. More preferably, m is 3 and L is O-methylene, O-ethylene or O attached to the 4- position of the amino cyclohexane ring.
- an alkyl or alkylene can optionally be substituted one or more times by halogen.
- alkyl or alkylene is substituted one to three times by halogen selected from chloro or bromo but may be substituted by fluoro once or more, e.g. being perfluorinated.
- halogen is fluor. More preferred an alkyl or alkylene is not halogenated.
- alkylene or cycloalkyl can optionally be substituted one or more times by a group selected independently from OH, OCH3, COOH, COOCH 3 , NH 2 , NHCH 3 , N(CH 3 ) 2 , CONH 2 , CONHCH 3 or CON(CH 3 ) 2 .
- the number of substituents is preferably between 1 , 2, 3 or 4, more preferably 1 or 2 with 1 being even more preferred.
- R4, R5, R7 and Re are not substituted.
- an alkylene or cycloalkyl is not substituted. More preferably an alkyl, alkylene or cycloalkyl is not substituted.
- one or more or all of the groups contained in the compounds of formula (I) can independently of each other have any of the preferred, more preferred or most preferred definitions of the groups specified above or any one or some of the specific denotations which are comprised by the definitions of the groups and specified above, all combinations of preferred definitions, more preferred or most preferred and/or specific denotations being a subject of the present invention.
- the invention includes the compounds of the formula (I) in all stereoisomeric forms and mixtures of stereoisomeric forms in all ratios, and their pharmaceutically acceptable salts.
- a preferred embodiment is a compound of the formula (I) wherein
- R 1 is H, (C-i-C ⁇ alkyl, (C 6 -C 10 )aryl, NH-(C 1 -C 6 )alkyl, NH-(C 6 -C 10 )aryl, or N[(C ⁇ C ⁇ )alkylk;
- R2 is hydrogen, halogen, or (C 1 -C 6 )alkyl
- R3 is H, halogen, (C-j-C ⁇ alkylene-R 1 , O-R" or NHR";
- F*4 is H 1 halogen or (C 1 -C 6 )alkyl;
- R 5 is H, (C 1 -C 6 ) ⁇ kYl, halogen, CN, (C 2 -C 6 )alkenyl, (C 6 -C 10 )aryl, NH-(C 6 -C 10 )aryl,
- R 6 and R 6 ' are independently of each other H, R', (C-
- R7 is H, halogen, CN, (C 1 -C 6 )alkyl, 0-(C ⁇
- R 8 is H, halogen or (C-
- n 2, 3 or 4
- n 1 , 2 or 3
- L is O, O-methylene or O-ethylene; and their pharmaceutically acceptable salts.
- a further preferred embodiment is a compound of the formula (I) wherein R 1 is H, (C 1 -C 6 )aIlCyI 1 (C 6 -C 10 )aryl, NH-(C 1 -C 6 )alkyl, NH-(C 6 -C 10 )aryl, or N[(C ⁇ Ce)alkyl ⁇ ;
- F*2 is H Or (C 1 -C ⁇ aIKyI;
- R3 is H, halogen or NHR", wherein R" is defined as above;
- R4 is H, halogen or (C-i-C ⁇ alkyl
- R5 is H, (C-i-C ⁇ )alkyl, halogen, (C2-C4)alkenyl, (C 6 -C 1 o)aryl, (C 1 -C 6 )alkylene-(C6- C 10 )aryl or (C5-C 10 )heterocyclyl;
- R 6 and R 6 ' are independently of each other H, (C ⁇ -CsJcycloalkyl, (C-
- R7 is H, halogen, CN, (C-
- R ⁇ is H, halogen or (C-
- n 1 , 2 or 3;
- L is O; and their pharmaceutically acceptable salts.
- An especially preferred embodiment is a compound of the formula (I) wherein
- R 1 is H, (Ci-C4)alkyl, NH-(C 1 -C 4 )alkyl, N[(Ci-C4)alkyl]2 or NH-phenyl;
- R 2 is H, (C-i-C 4 )alkyl;
- R3 is H, NH-(C5-C6)heteroaryl or NH-phenyl
- R4 is H, halogen or (C-
- R5 is H, (Ci-C4)alkyl, halogen, (C ⁇
- R6 is H, (C3-C6)cycloalkyl or (Ci-C4)alkyl;
- R6' is H 1 (C3-C8)cycloalkyl, (Ci-C8)alkyl, (Ci-C3)alkylene-R ⁇ C(O)O-(C 1 -C 6 )alkyl, C(O)(C 1 -C 6 )alkyl, C(O)(C3-C6)cycloalkyl, C(O)(C5-C6)heterocyclyl, C(O)(Ci-C3)alkylene-(C3-C6)cycloalkyl, C(O)(Ci-C3)alkylene-(C5-C6)heterocyclyl, or C(O)(Ci -C3)alkylene-phenyl;
- R7 is H, halogen, CN, (Ci-C4)alkyl, O(Ci-C4)alkyl, (Ci-C4)alkenyl, phenyl, cyclopropyl, (C5-C6)heteroaryl;
- R8 is H, halogen or (Ci-C4)alkyl
- n 1 ;
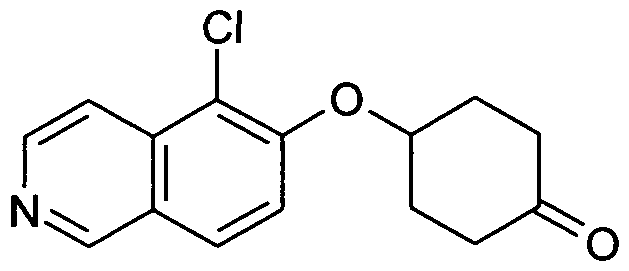
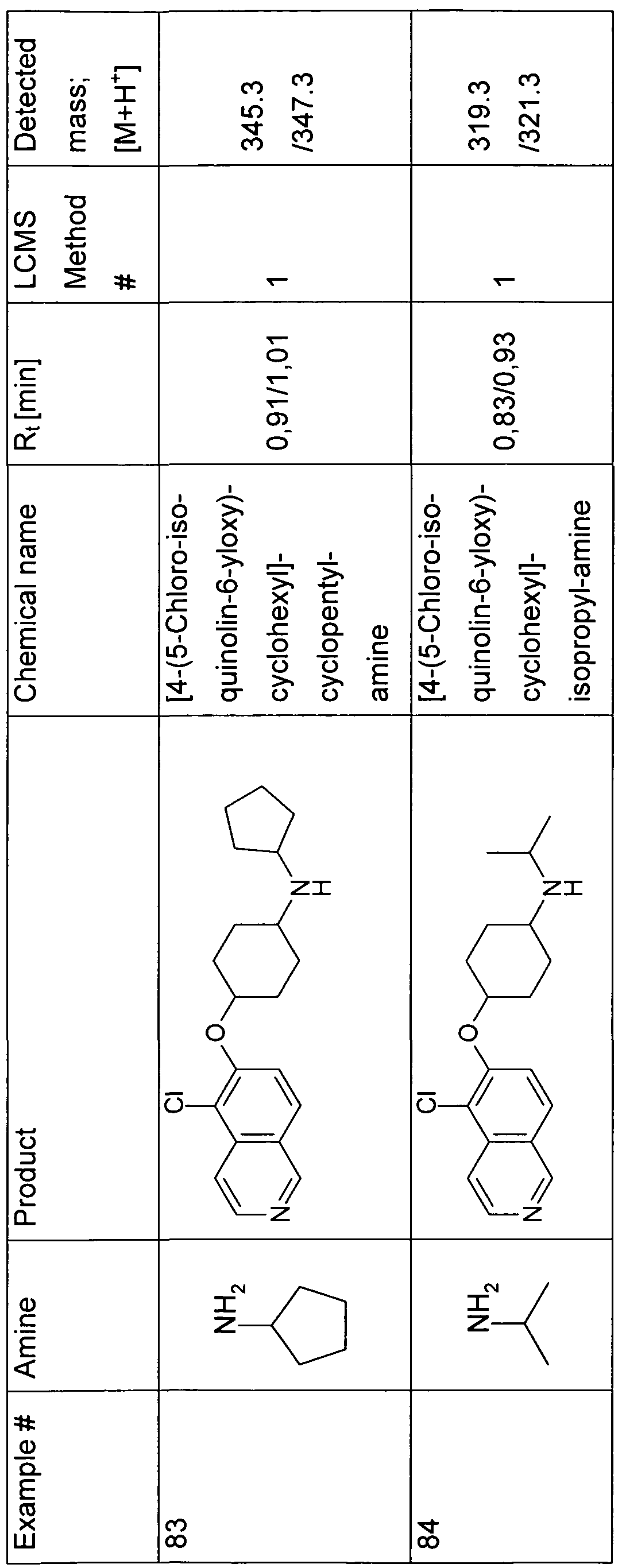
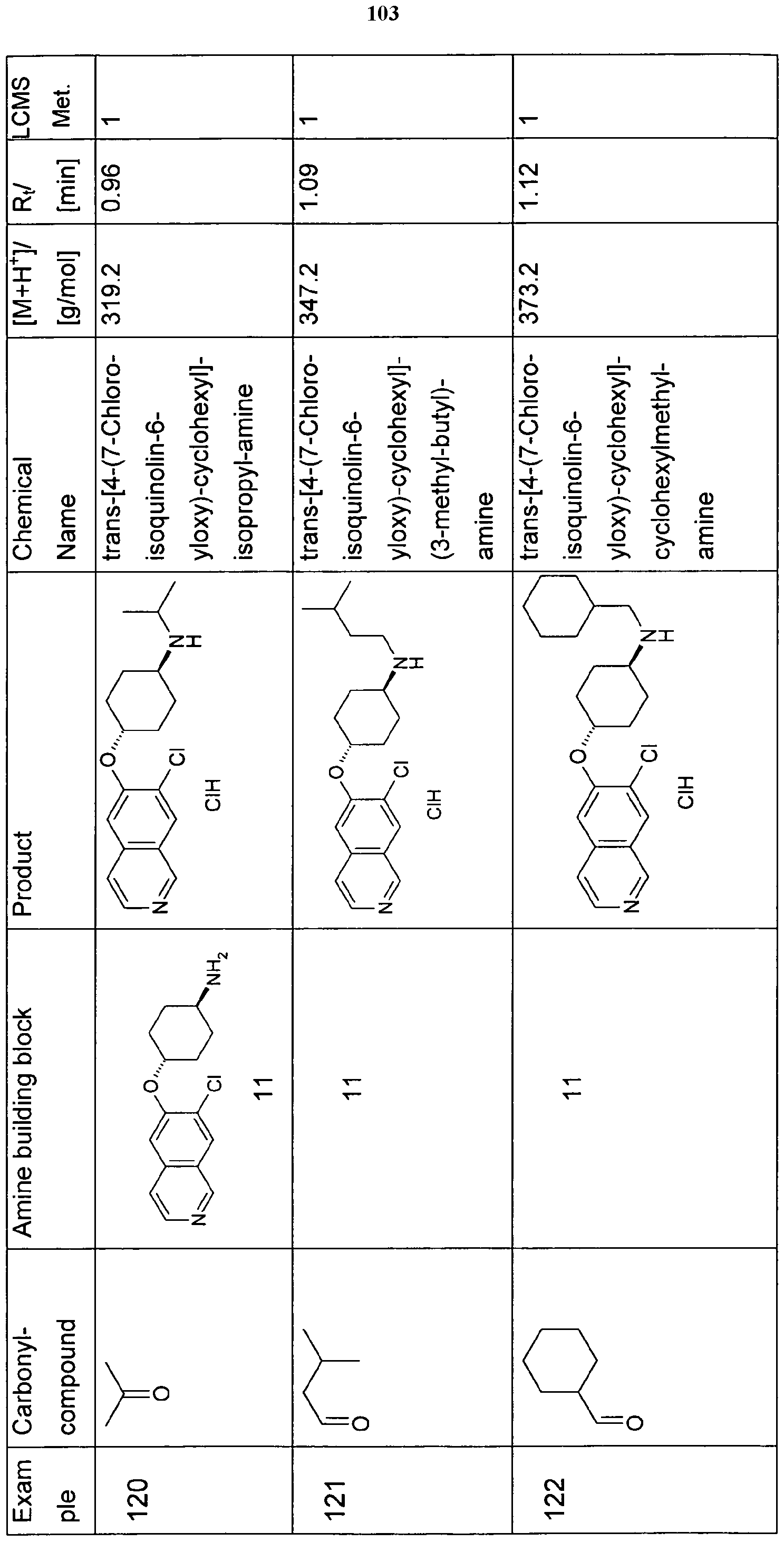
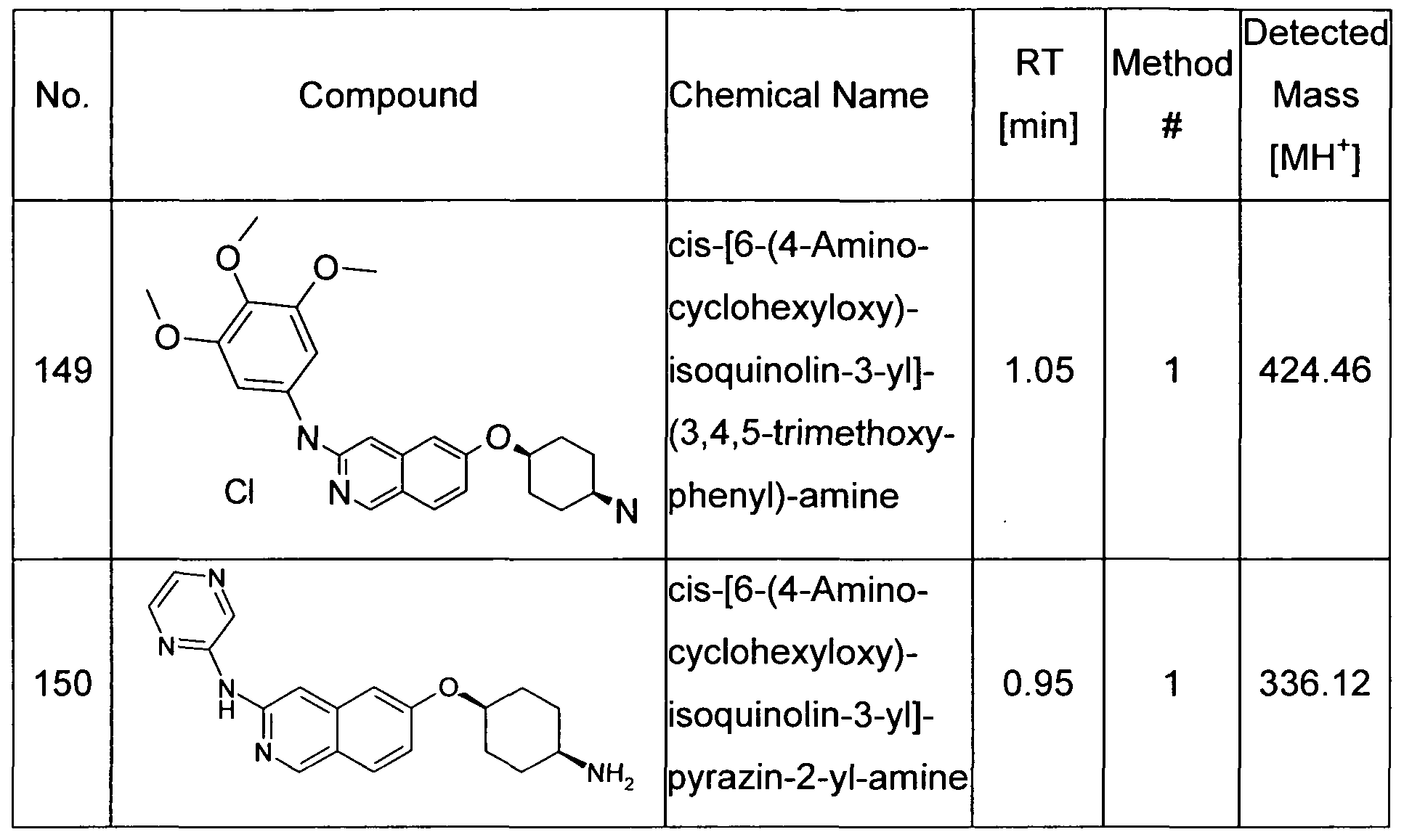
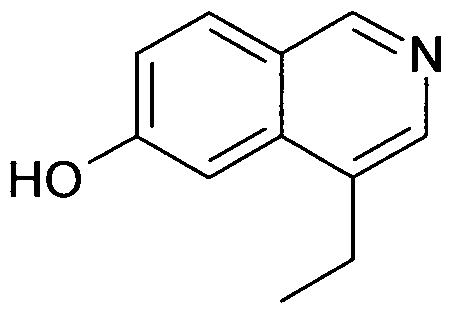
- the present invention relates to a compound of formula (I) independently selected from the group of
- one or more or all of the groups can have any of its preferred, more preferred, most preferred definitions specified above or any one or some of the specific denotations which are comprised by its definitions and are specified above.
- Isoquinoline substitution pattern is numbered according to IUPAC rules:
- compositions of the formula (I) mean both their organic and inorganic salts as described in Remington's Pharmaceutical Sciences (17th edition, page 1418 (1985)). Because of the physical and chemical stability and the solubility, preference is given for acidic groups inter alia to sodium, potassium, calcium and ammonium salts; preference is given for basic groups inter alia to salts of maleic acid, fumaric acid, succinic acid, malic acid, tartaric acid, methylsulfonic acid, hydrochloric acid, sulfuric acid, phosphoric acid or of carboxylic acids or sulfonic acids, for example as hydrochlorides, hydrobromides, phosphates, sulfates, methanesulfonates, acetates, lactates, maleates, fumarates, malates, gluconates, and salts of amino acids, of natural bases or carboxylic acids.
- acidic groups inter alia to sodium, potassium, calcium and ammonium salts
- basic groups inter alia to salts of maleic acid
- the compounds of the formula (I) form stable alkali metal, alkaline earth metal or optionally substituted ammonium salts with basic reagents such as hydroxides, carbonates, bicarbonates, alcoholates and ammonia or organic bases, for example trimethyl- or triethylamine, ethanolamine, diethanolamine or triethanolamine, trometamol or else basic amino acids, for example lysine, ornithine or arginine.
- basic reagents such as hydroxides, carbonates, bicarbonates, alcoholates and ammonia or organic bases, for example trimethyl- or triethylamine, ethanolamine, diethanolamine or triethanolamine, trometamol or else basic amino acids, for example lysine, ornithine or arginine.
- stable acid addition salts can also be prepared with strong acids.
- Suitable pharmaceutically acceptable acid addition salts of the compounds of the invention are salts of inorganic acids such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acid, and of organic acids such as, for example, acetic acid, benzenesulfonic, benzoic, citric, ethanesulfonic, fumaric, gluconic, glycolic, isethionic, lactic, lactobionic, maleic, malic, methanesulfonic, succinic, p-toluenesulfonic and tartaric acid.
- inorganic acids such as hydrochloric acid, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric acid
- organic acids such as, for example, acetic acid, benzenesulfonic, benzoic, citric, ethanesulfonic, fumaric, gluconic, glycolic, isethionic, lactic, lactobionic, maleic
- Salts with a pharmaceutically unacceptable anion such as, for example, trifluoroacetate likewise belong within the framework of the invention as useful intermediates for the preparation or purification of pharmaceutically acceptable salts and/or for use in nontherapeutic, for example in vitro, applications.
- physiologically functional derivative refers to any physiologically tolerated derivative of a compound of the formula (I) of the invention, for example an N-oxide, which on administration to a mammal such as, for example, a human is able to form (directly or indirectly) a compound of the formula (I) or an active metabolite thereof.
- Physiologically functional derivatives include prodrugs of the compounds of the invention, as described, for example, in H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Such prodrugs can be metabolized in vivo to a compound of the invention. These prodrugs may themselves be active or not.
- the invention relates to compounds of the formula (I) in the form of their stereoisomeric forms, which include racemates, racemic mixtures, pure enantiomers and diastereomers and mixtures thereof.
- the compounds of the invention may also exist in various polymorphous forms, for example as amorphous and crystalline polymorphous forms. All polymorphous forms of the compounds of the invention belong within the framework of the invention and are a further aspect of the invention.
- radicals or substituents may occur more than once in the compounds of the formula (I), they may all, independently of one another, have the stated meaning and be identical or different.
- -C8)alkyl and the corresposponding alkylene substituents are understood as a hydrocarbon residue which can be linear, i.e. straight-chain, or branched and has 1 , 2, 3, 4, 5, 6, 7 or 8 carbon atoms, respectively. This also applies if an alkyl group occurs as a substituent on another group, for example in an alkoxy group (O-alkyl), S-alkyl or a -O(C-
- alkyl groups are methyl, ethyl, propyl, butyl, pentyl or hexyl, the n-isomers of all these groups, isopropyl, isobutyl, 1-methylbutyl, isopentyl, neopentyl, 2,2-dimethylbutyl, 2- methylpentyl, 3-methylpentyl, isohexyl, sec-butyl, tert-butyl or tert-pentyl.
- Alkyl or alkylene groups may - if not otherwise stated - be halogenated once or more, e.g. alkyl groups may be fluorinated, e.g. perfluorinated.
- alkyl groups may be fluorinated, e.g. perfluorinated.
- halogenated alkyl groups are CF3 and CH2CF3, OCF3, SCF3, or -O-(CF2)2-O-.
- Halogen means fluoro, chloro, bromo or iodo.
- (C3-C8)cycloalkyl groups are cyclic alkyl groups containing 3, 4, 5, 6, 7 or 8 ring carbon atoms like cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cyclooctyl, which can also be substituted and/or contain 1 or 2 double bounds (unsaturated cycloalkyl groups) like, for example, cyclopentenyl or cyclohexenyl can be bonded via any carbon atom.
- a (C ⁇ -C 10 )aryl group means an aromatic ring or a ring system which comprises two aromatic rings which are fused or otherwise linked, for example a phenyl, naphthyl, biphenyl, tetrahydronaphthyl, alpha- or beta-tetralon-, indanyl- or indan-1-on-yl group.
- a preferred (Ce-C 10 )aryl group is phenyl.
- a (C5-C 10 )heterocyclyl group means a mono- or bicyclic ring system in which one or more carbon atoms can be replaced by one or more heteroatoms such as, for example, 1 , 2 or 3 nitrogen atoms, 1 or 2 oxygen atoms, 1 or 2 sulfur atoms or combinations of different hetero atoms.
- the heterocyclyl residues can be bound at any positions, for example on the 1-position, 2-position, 3-position, 4-position, 5-position, 6- position, 7-position or 8-position.
- Suitable (C5-C 10 )heterocyclyl group include acridinyl, azocinyl, benzimidazolyl, benzofuryl, benzomorpholinyl, benzothienyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, carbazolyl, 4aH-carbazolyl, carbolinyl, furanyl, quinazolinyl, quinolinyl, 4H-quinolizinyl, quinoxalinyl, quinuclidinyl, chromanyl, chromenyl, chromen-2-onyl, cinnolinyl, decahydroquinolinyl, 2H,6H-1 ,5,2-dithiazinyl, dihydrofuro[2,3-b]-tetrahydrofuran, fu
- Pyridyl stands both for 2-, 3- and 4-pyridyl.
- Thienyl stands both for 2- and 3-thienyl.
- Furyl stands both for 2- and 3-furyl.
- N-oxides of these compounds for example, 1-oxy-2-, 3- or 4-pyridyl.
- substitutions in (C5-C 10 )heterocyclyl residues can occur on free carbon atoms or on nitrogen atoms.
- Preferred examples of (C5-C 10 )heterocyclyl residues are pyrazinyl, pyridyl, pyrimidinyl, pyrazolyl, morpholinyl, pyrrolidinyl, piperazinyl, piperidinyl, thienyl, benzofuryl, quinolinyl, tetrazolyl and triazolyl.
- (C 6 -C 1 rj)aryl anc l (C 5 -C 1 rj)heterocyclyl groups are unsubstituted or, if not stated otherwise, substituted one or more times, preferably one to three times, by suitable groups independently selected from halogen, OH, NO2, N3, CN, C(O)-(C 1 -C 6 )alkyl,
- Preferred substituents for (C ⁇ -C 10 )aryl groups are (C-
- -C4)alkyl, O-phenyl, phenyl, C(O)O-(Ci -C ⁇ )alkyl, C(O)OH, C(O)-(Ci-C 4 )alkyl, halogen, NO2, SO2NH2, CN, SO 2 -(Ci -C 4 )alkyl, SO 2 -N CH-N[(C 1 -C 6 )alkyl] 2 , NH-SO 2 -(Ci- C4)alkyl, NH 2 , NH-C(O)-(Ci -C4)alkyl, (C3-C8)cycloalkyl, (Ci-C4)alkyl-OH, C(O)N[(Ci-C 4 )alkyl] 2 , CONH(C 1 -C 6 )alkyl, C(O
- the substituent can be located in the 2-position, the 3-position or the 4-position, with the 3-position and the 4-position being preferred. If a phenyl group carries two substituents, they can be located in 2, 3-position, 2,4-position, 2,5-position, 2,6-position, 3,4-position or 3,5-position. In phenyl groups carrying three substituents the substituents can be located in 2, 3,4-position, 2, 3,5-position, 2,3,6- position, 2,4,5-position, 2,4,6-position, or 3,4,5-position.
- phenyl groups correspondingly apply to divalent groups derived from phenyl groups, i.e. phenylene which can be unsubstituted or substituted 1 ,2-phenylene, 1 ,3-phenylene or 1 ,4-phenylene.
- the above statements also correspondingly apply to the aryl subgroup in arylalkylene groups.
- arylalkylene groups which can also be unsubstituted or substituted in the aryl subgroup as well as in the alkylene subgroup, are benzyl, 1-phenylethylene, 2-phenylethylene, 3- phenylpropylene, 4-phenylbutylene, 1-methyl-3-phenyl-propylene.
- Preferred substituents for (C5-Cirj)neterocyclyl groups are (C-
- substituents for (C5-C 10 )heterocyclyl groups are (C-
- the present invention therefore also relates to the compounds of the formula (I) and/or their pharmaceutically acceptable salts and/or their prodrugs for use as pharmaceuticals (or medicaments), to the use of the compounds of the formula (I) and/or their pharmaceutically acceptable salts and/or their prodrugs for the production of pharmaceuticals for the treatment and/or prevention of diseases associated with Rho-kinase and/or Rho-kinase mediated phosphorylation of myosin light chain phosphatase, i.e.
- hypertension for the treatment and/or prevention of hypertension, pulmonary hypertension, ocular hypertension, retinopathy, and glaucoma, peripheral circulatory disorder, peripheral arterial occlusive disease (PAOD), coronary heart disease, angina pectoris, heart hypertrophy, heart failure, ischemic diseases, ischemic organ failure (end organ damage), fibroid lung, fibroid liver, liver failure, nephropathy, including hypertension-induced, non-hypertension-induced, and diabetic nephropathies, renal failure, fibroid kidney, renal glomerulosclerosis, organ hypertrophy, asthma, chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome, thrombotic disorders, stroke, cerebral vasospasm, cerebral ischemia, pain, e.g.
- PAOD peripheral arterial occlusive disease
- COPD chronic obstructive pulmonary disease
- neuropathic pain neuronal degeneration, spinal cord injury, Alzheimer's disease, premature birth, erectile dysfunction, endocrine dysfunctions, arteriosclerosis, prostatic hypertrophy, diabetes and complications of diabetes, metabolic syndrome, blood vessel restenosis, atherosclerosis, inflammation, autoimmune diseases, AIDS, osteopathy such as osteoporosis, infection of digestive tracts with bacteria, sepsis, cancer development and progression, e.g. cancers of the breast, colon, prostate, ovaries, brain and lung and their metastases.
- the present invention furthermore relates to pharmaceutical preparations (or pharmaceutical compositions) which contain an effective amount of at least one compound of the formula (I) and/or its pharmaceutically acceptable salts and a pharmaceutically acceptable carrier, i. e. one or more pharmaceutically acceptable carrier substances (or vehicles) and/or additives (or excipients).
- a pharmaceutically acceptable carrier i. e. one or more pharmaceutically acceptable carrier substances (or vehicles) and/or additives (or excipients).
- the pharmaceuticals can be administered orally, for example in the form of pills, tablets, lacquered tablets, coated tablets, granules, hard and soft gelatin capsules, solutions, syrups, emulsions, suspensions or aerosol mixtures.
- Administration can also be carried out rectally, for example in the form of suppositories, or parenterally, for example intravenously, intramuscularly or subcutaneously, in the form of injection solutions or infusion solutions, microcapsules, implants or rods, or percutaneously or topically, for example in the form of ointments, solutions or tinctures, or in other ways, for example in the form of aerosols or nasal sprays.
- compositions according to the invention are prepared in a manner known per se and familiar to one skilled in the art, pharmaceutically acceptable inert inorganic and/or organic carrier substances and/or additives being used in addition to the compound(s) of the formula (I) and/or its (their) pharmaceutically acceptable salts and/or its (their) prodrugs.
- pharmaceutically acceptable inert inorganic and/or organic carrier substances and/or additives being used in addition to the compound(s) of the formula (I) and/or its (their) pharmaceutically acceptable salts and/or its (their) prodrugs.
- pharmaceutically acceptable inert inorganic and/or organic carrier substances and/or additives being used in addition to the compound(s) of the formula (I) and/or its (their) pharmaceutically acceptable salts and/or its (their) prodrugs.
- for the production of pills, tablets, coated tablets and hard gelatin capsules it is possible to use, for example, lactose, corn starch or derivatives thereof, talc
- Suitable carrier substances for the production of solutions for example injection solutions, or of emulsions or syrups are, for example, water, saline, alcohols, glycerol, polyols, sucrose, invert sugar, glucose, vegetable oils, etc.
- Suitable carrier substances for microcapsules, implants or rods are, for example, copolymers of glycolic acid and lactic acid.
- the pharmaceutical preparations normally contain about 0.5 to about 90 % by weight of the compounds of the formula (I) and/or their pharmaceutically acceptable salts and/or their prodrugs.
- the amount of the active ingredient of the formula (I) and/or its pharmaceutically acceptable salts and/or its prodrugs in the pharmaceutical preparations normally is from about 0.5 to about 1000 mg, preferably from about 1 to about 500 mg.
- the pharmaceutical preparations can contain one or more additives such as, for example, fillers, disintegrants, binders, lubricants, wetting agents, stabilizers, emulsifiers, preservatives, sweeteners, colorants, flavorings, aromatizers, thickeners, diluents, buffer substances, solvents, solubilizers, agents for achieving a depot effect, salts for altering the osmotic pressure, coating agents or antioxidants. They can also contain two or more compounds of the formula (I) and/or their pharmaceutically acceptable salts.
- a pharmaceutical preparation contains two or more compounds of the formula (I) the selection of the individual compounds can aim at a specific overall pharmacological profile of the pharmaceutical preparation. For example, a highly potent compound with a shorter duration of action may be combined with a long-acting compound of lower potency.
- the flexibility permitted with respect to the choice of substituents in the compounds of the formula (I) allows a great deal of control over the biological and physico-chemical properties of the compounds and thus allows the selection of such desired compounds.
- the pharmaceutical preparations can also contain one or more other therapeutically or prophylactically active ingredients.
- the dose can vary within wide limits and, as is customary and is known to the physician, is to be suited to the individual conditions in each individual case. It depends, for example, on the specific compound employed, on the nature and severity of the disease to be treated, on the mode and the schedule of administration, or on whether an acute or chronic condition is treated or whether prophylaxis is carried out.
- An appropriate dosage can be established using clinical approaches well known in the medical art.
- the daily dose for achieving the desired results in an adult weighing about 75 kg is from about 0.01 to about 100 mg/kg, preferably from about 0.1 to about 50 mg/kg, in particular from about 0.1 to about 10 mg/kg, (in each case in mg per kg of body weight).
- the daily dose can be divided, in particular in the case of the administration of relatively large amounts, into several, for example 2, 3 or 4, part administrations. As usual, depending on individual behavior it may be necessary to deviate upwards or downwards from the daily dose indicated.
- the compounds of the formula (I) can be used as synthesis intermediates for the preparation of other compounds, in particular of other pharmaceutical active ingredients, which are obtainable from the compounds of the formula I, for example by introduction of substituents or modification of functional groups.
- protective groups that may still be present in the products obtained in the coupling reaction are then removed by standard procedures.
- tert-butyl protecting groups in particular a tert-butoxycarbonyl group which is a protection form of an amino group
- tert-butoxycarbonyl group which is a protection form of an amino group
- functional groups can be generated from suitable precursor groups.
- a conversion into a pharmaceutically acceptable salt or a prodrug of a compound of the formulae (I) can then be carried out by known processes.
- a reaction mixture containing a final compound of the formula (I) or (I 1 ) or an intermediate is worked up and, if desired, the product is then purified by customary processes known to those skilled in the art.
- a synthesized compound can be purified using well known methods such as crystallization, chromatography or reverse phase-high performance liquid chromatography (RP-HPLC) or other methods of separation based, for example, on the size, charge or hydrophobicity of the compound.
- RP-HPLC reverse phase-high performance liquid chromatography
- well known methods such as amino acid sequence analysis, NMR, IR and mass spectrometry (MS) can be used for characterizing a compound of the invention.
- Isoquinolines can by synthesized via a variety of methods.
- the following general schemes illustrate some of the possible ways to access isoquinolines, but do not limit the present invention.
- a suitably substituted aldehyde for example substituted by X or Y being independently from each other hydrogen, alkyl, alkoxy or halide attached in a suitable position, can be reacted with a suitable compound such as for example an acetal of aminoacetaldehyde for example in a solvent like THF, chloroform or toluene under acid catalysis by toluene sulfonic acid or another appropriate acid to give imine (ii) wherein Q' can be for instance methyl or ethyl, which in turn can be cyclized by different methods to the isoquinoline (iii).
- a suitable compound such as for example an acetal of aminoacetaldehyde for example in a solvent like THF, chloroform or toluene under acid catalysis by toluene sulfonic acid or another appropriate acid to give imine (ii) wherein Q' can be for instance methyl or ethyl, which in turn can be cyclized by
- this can be done by Lewis acid catalysis by suitable Lewis acids like titanium tetrachloride, ferrous halides, aluminium halides etc. at temperatures ranging from ambient to 100 °C or by reducing the imine to the corresponding amine by action of a suitable reducing agent like sodium borohydride, converting the amine into an amide or sulphonamide by reaction with a suitable acid chloride and subsequent cyclization to the isoquinoline by action of an appropriate lewis acid.
- suitable Lewis acids like titanium tetrachloride, ferrous halides, aluminium halides etc.
- a suitable reducing agent like sodium borohydride
- converting the amine into an amide or sulphonamide by reaction with a suitable acid chloride and subsequent cyclization to the isoquinoline by action of an appropriate lewis acid.
- the products like (iv) obtained via this method can then either be liberated or, if a suitable amino functionality is present, be reacted with suitable aldehydes or ketones in the presence of a reducing agent like sodium triacetoxy borohydride, sodium borohydride or sodium cyanoborohydride in a suitable solvent and in the presence of a water withdrawing agent like molecular sieves or a suitable ortho ester.
- a reducing agent like sodium triacetoxy borohydride, sodium borohydride or sodium cyanoborohydride in a suitable solvent and in the presence of a water withdrawing agent like molecular sieves or a suitable ortho ester.
- This amino group may have to be liberated in an initial step like for example acidic removal of Boc-groups.
- Isoquinoline derivatives like (iv) can be obtained as free bases or as various salts like for example hydrochlorides, hydrobromides, phosphates, trifluoroacetates, sulfates or fumarates.
- the salts obtained can be converted into the corresponding free base by either subjecting them to ion exchange chromatography or for example by alkaline aqueous treatment and subsequent extraction with suitable organic solvents like for example methyl tert. butyl ether, chloroform, ethyl acetate or isopropanol / dichloromethane mixtures and subsequent evaporation to dryness.
- the starting material was dissolved in 2M hydrochloric acid and stirred overnight.
- methanol or dioxane was added until a homogenous solution was obtained.
- 4M hydrochloric acid in isopropanol was used to deprotect the compound.
- the reaction mixture was lyophilised and the deprotected product was obtained as the corresponding hydrochloride of the free amine.
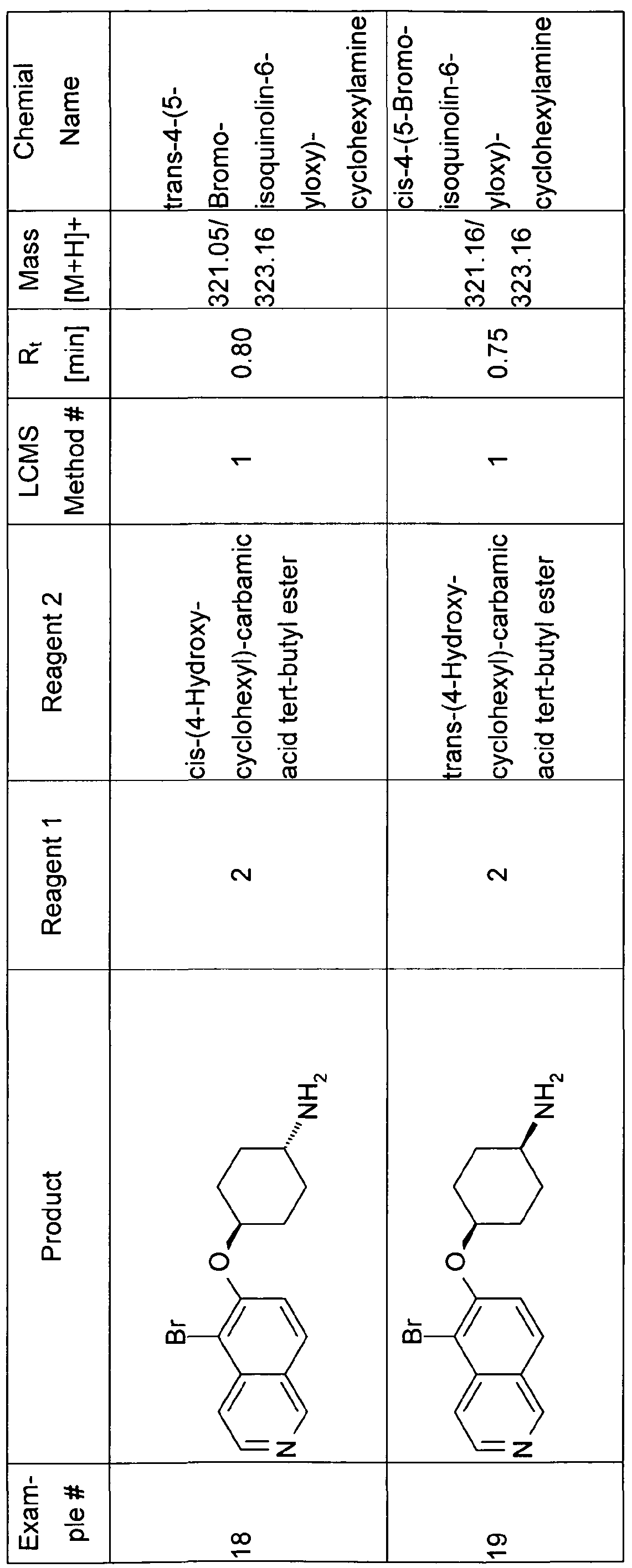
- the examples 16 and 17 were also synthesized using a similar method as described for the synthesis of 12 and 13.
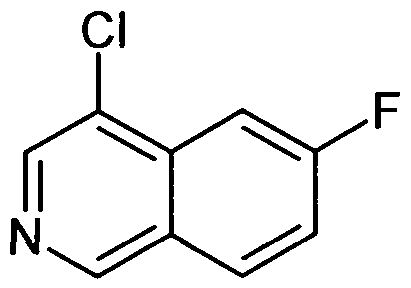
- the respective starting materials are 6-fluoro isoquinoline (5) and either cis or trans 4-amino-cyclohexanol hydrochloride.
- the crude material (2.2 g) was dissolved in 200 mL acetone and 10 mL water. 1.5 g (7.9 mmol) of para-toluene sulfonic acid were added and the reaction was heated to reflux temperature for 6 h. Then the solvents were distilled off. The remainder was dissolved in dichloromethane and was extracted with aqueous Na 2 CO 3 solution. After drying over Na 2 SO 4 , filtration and removal of the solvents the crude product was purified by flash chromatography to yield 1.19 g of 27 as a white solid.
- the Boc group was removed by dissolving the intermediate in isopropanol and addition of 5-6 N HCI in isopropanol. The precipitate was isolated by filtration.
- the Boc group was removed by dissolving the intermediate in isopropanol and addition of 5-6 N HCI in isopropanol. The precipitate was isolated by filtration.
- amine building block 0.25 mmol of the amine building block (hydrochloride) was weighted into the reaction tube. 3 ml trimethyl orthoformiate was added, then 0.25 mmol of the carbonyl compound (in 0.2 ml THF or solid), followed by 1.5 mmol (2.5 mmol in case of dihydrochlorides) Et 3 N. The mixture was stirred for 1h at room temperature, then cooled to -10 0 C. 1.5 ml of a freshly prepared solution of NaHB(OAc) 3 (1.25 mmol) in DMF was added, followed by 1.225 mmol acetic acid. The mixture was stirred for 30 min in the cold, then allowed to reach room temperature. Stirring was continued over night at room temperature.
- Step i
- the solution was allowed to stir for an additional 3 hours to reach room temperature, then the solvent was removed partially and the precipitated product was collected by filtration.
- Step 4 16.5 g of (cis-4-hydroxy-cyclohexyl)-carbamic acid tert-butyl ester were dissolved in 210 ml of diglyme and treated with 4.1g 50% NaH under nitrogen. The resulting mixture was stirred for 1 h at room temperature, then 14.8 g of the product from Step 3 were added. The mixture was allowed to stir for 1 day at room temperature, then 100 ml of toluene were added and the resulting mixture was washed with water 3 times. The organic phases were collected and the solvent was removed in vacuo.
- Step 6 The products of Step 5 are dissolved in 5 ml of ethanol saturated with gaseous HCI. After stirring for 5h the desired product is isolated as its hydrochloride by removal of the solvent in vacuo.
- the following examples were synthesized as hydrochlorides following this general procedure (Table 9):
- the aqueous layer was lyophilized and purified by HPLC, the resulting product was converted into its HCI salt by taking it up in 0.1 M HCI and subsequent lyophilisation.
- IC50 values were determined according to the following protocol:
- Buffer 25mM Tris pH7,5; 0,02% BSA; 5% Glycerol; 0,008% Triton X100; 2% DMSO,
- Biotinylated substrate diluted to 0.25 ⁇ M with buffer described above (without ATP)
- PIC50 negative decadal logarithm of the IC50
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Cardiology (AREA)
- Neurosurgery (AREA)
- Endocrinology (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Reproductive Health (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Physical Education & Sports Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Ophthalmology & Optometry (AREA)
- Virology (AREA)
- Rheumatology (AREA)
- Pregnancy & Childbirth (AREA)
- Hospice & Palliative Care (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Epidemiology (AREA)
Abstract
Description



































































































Claims

Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007338410A AU2007338410B2 (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| CA2673920A CA2673920C (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| MX2009005964A MX2009005964A (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives. |
| EP07856890A EP2125746B1 (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| BRPI0721180-5A BRPI0721180A2 (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine-Substituted Isoquinoline Derivatives |
| JP2009543375A JP5405316B2 (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| AT07856890T ATE554072T1 (en) | 2006-12-27 | 2007-12-19 | CYCLOALKYLAMINE SUBSTITUTED ISOQUINOLINE DERIVATIVES |
| CN200780048527XA CN101611012B (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| HK10106146.8A HK1140189B (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
| US12/487,386 US8710228B2 (en) | 2006-12-27 | 2009-06-18 | Cycloalkylamine substituted isoquinoline derivatives |
| IL199539A IL199539A (en) | 2006-12-27 | 2009-06-24 | Cycloalkylamine substituted isoquinoline derivatives, uses thereof and medicaments comprising the same |
| NO20092432A NO20092432L (en) | 2006-12-27 | 2009-06-26 | Cycloalkylamine substituted isoquinoline derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06026894 | 2006-12-27 | ||
| EP06026894.3 | 2006-12-27 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/487,386 Continuation US8710228B2 (en) | 2006-12-27 | 2009-06-18 | Cycloalkylamine substituted isoquinoline derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008077554A1 true WO2008077554A1 (en) | 2008-07-03 |
Family
ID=38055425
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/011167 Ceased WO2008077554A1 (en) | 2006-12-27 | 2007-12-19 | Cycloalkylamine substituted isoquinoline derivatives |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US8710228B2 (en) |
| EP (1) | EP2125746B1 (en) |
| JP (1) | JP5405316B2 (en) |
| KR (1) | KR20090092303A (en) |
| CN (1) | CN101611012B (en) |
| AT (1) | ATE554072T1 (en) |
| AU (1) | AU2007338410B2 (en) |
| BR (1) | BRPI0721180A2 (en) |
| CA (1) | CA2673920C (en) |
| IL (1) | IL199539A (en) |
| MX (1) | MX2009005964A (en) |
| MY (1) | MY151953A (en) |
| NO (1) | NO20092432L (en) |
| SV (1) | SV2009003317A (en) |
| WO (1) | WO2008077554A1 (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7470787B2 (en) | 2005-07-11 | 2008-12-30 | Aerie Pharmaceuticals, Inc. | Isoquinoline compounds |
| WO2009103966A1 (en) * | 2008-02-19 | 2009-08-27 | Cancer Research Technology Limited | Bicyclylaryl-aryl-amine compounds and their use |
| US8058045B2 (en) | 2007-10-05 | 2011-11-15 | Cancer Research Technology Limited | Pyrazin-2-yl-pyridin-2-yl-amine and pyrazin-2-yl-pyrimidin-4-yl-amine compounds and their use |
| US8357699B2 (en) | 2007-01-10 | 2013-01-22 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8394826B2 (en) | 2009-05-01 | 2013-03-12 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US8871757B2 (en) | 2008-01-17 | 2014-10-28 | Aerie Pharmaceuticals, Inc. | 6-and 7-amino isoquinoline compounds and methods for making and using the same |
| US8951997B2 (en) | 2009-06-19 | 2015-02-10 | D. Western Therapeutics Institute, Inc. | Substituted isoquinoline derivative |
| US9040540B2 (en) | 2011-11-09 | 2015-05-26 | Cancer Research Technology Limited | 5-(pyridin-2-yl-amino)-pyrazine-2-carbonitrile compounds and their therapeutic use |
| US9096569B2 (en) | 2008-07-25 | 2015-08-04 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| WO2015124877A1 (en) | 2014-02-21 | 2015-08-27 | Les Laboratoires Servier | Derivatives of 5-benzylisoquinoline for the treatment of cardiovascular diseases |
| US9415043B2 (en) | 2013-03-15 | 2016-08-16 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9643927B1 (en) | 2015-11-17 | 2017-05-09 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US9663503B2 (en) | 2012-05-15 | 2017-05-30 | Cancer Research Technology Limited | 5-[[4-[[morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2-pyridyl]amino]pyrazine-2-carbonitrile and therapeutic uses thereof |
| EP3215142A4 (en) * | 2014-11-05 | 2018-09-05 | Flexus Biosciences, Inc. | Immunoregulatory agents |
| US10550087B2 (en) | 2015-11-17 | 2020-02-04 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US10624882B2 (en) | 2006-09-20 | 2020-04-21 | Aerie Pharmaceuticals, Inc. | Rho kinase inhibitors |
| US10858339B2 (en) | 2017-03-31 | 2020-12-08 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11351149B2 (en) | 2020-09-03 | 2022-06-07 | Pfizer Inc. | Nitrile-containing antiviral compounds |
| US11389441B2 (en) | 2016-08-31 | 2022-07-19 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11427563B2 (en) | 2018-09-14 | 2022-08-30 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11566003B2 (en) | 2017-03-30 | 2023-01-31 | Genentech, Inc. | Isoquinolines as inhibitors of HPK1 |
| US11612606B2 (en) | 2018-10-03 | 2023-03-28 | Genentech, Inc. | 8-aminoisoquinoline compounds and uses thereof |
| US12378249B2 (en) | 2018-07-24 | 2025-08-05 | Genentech, Inc. | Isoquinoline compounds and uses thereof |
| US12473301B2 (en) | 2018-10-02 | 2025-11-18 | Genentech, Inc. | Isoquinoline compounds for the treatment of cancer |
| US12577240B2 (en) | 2020-07-11 | 2026-03-17 | Pfizer Inc. | Antiviral heteroaryl ketone derivatives |
| US12606550B2 (en) | 2020-04-07 | 2026-04-21 | Crt Pioneer Fund Lp | Methods for synthesis of CHK1 inhibitors |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY153756A (en) * | 2008-06-24 | 2015-03-13 | Sanofi Aventis | Bi-and polycyclic substituted isoquinoline and isoquinoline derivatives as rho kinase inhibitors |
| CA2728137C (en) * | 2008-06-24 | 2016-10-18 | Sanofi-Aventis | Substituted isoquinolines and isoquinolinones as rho kinase inhibitors |
| MY153615A (en) * | 2008-06-24 | 2015-02-27 | Sanofi Aventis | 6-substituted isoquinolines and isoquinolinones |
| CN108997222A (en) * | 2018-07-23 | 2018-12-14 | 山东省农药科学研究院 | A kind of synthetic method of the chloro- 5-FU of fluoxastrobin intermediate 4,6- bis- |
| CN111574413B (en) * | 2020-06-08 | 2022-04-05 | 杭州尚合生物医药科技有限公司 | A kind of preparation method of sulfonyl amidine with 2-aminomethylpyridine and DMF-DMA as amine source |
| CN115572262B (en) * | 2022-10-27 | 2024-08-27 | 厦门沃克沃德医药科技有限公司 | Isoquinoline derivative and preparation method thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005030791A2 (en) * | 2003-09-23 | 2005-04-07 | Merck & Co., Inc. | Isoquinolinone potassium channel inhibitors |
| EP1541559A1 (en) * | 2002-07-22 | 2005-06-15 | Asahi Kasei Pharma Corporation | 5-substituted isoquinoline derivative |
| WO2007000240A1 (en) * | 2005-06-28 | 2007-01-04 | Sanofi-Aventis | Isoquinoline derivatives as inhibitors of rho-kinase |
| WO2007012422A1 (en) * | 2005-07-26 | 2007-02-01 | Sanofi-Aventis | Cyclohexylamin isoquinolone derivatives as rho-kinase inhibitors |
Family Cites Families (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2485537B2 (en) | 1977-04-13 | 1986-05-16 | Anvar | DIPYRIDO (4,3-B) (3,4-F) INDOLES, PROCESS FOR OBTAINING IT, THERAPEUTIC APPLICATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| EP0541559A1 (en) | 1990-07-31 | 1993-05-19 | E.I. Du Pont De Nemours And Company | Catalytic equilibration of selected halocarbons |
| US5480883A (en) * | 1991-05-10 | 1996-01-02 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Bis mono- and bicyclic aryl and heteroaryl compounds which inhibit EGF and/or PDGF receptor tyrosine kinase |
| GB9516709D0 (en) | 1995-08-15 | 1995-10-18 | Zeneca Ltd | Medicament |
| ZA9610741B (en) | 1995-12-22 | 1997-06-24 | Warner Lambert Co | 4-Substituted piperidine analogs and their use as subtype selective nmda receptor antagonists |
| BR9711154A (en) | 1996-08-12 | 1999-08-17 | Yoshitomi Pharmaceutical | Pharmaceutical agent containing rho kinase inhibitor |
| JPH1087629A (en) | 1996-09-18 | 1998-04-07 | Fujisawa Pharmaceut Co Ltd | New isoquinoline derivative, and its medicinal use |
| EP1007525A1 (en) | 1997-08-29 | 2000-06-14 | Zeneca Limited | Aminometyl oxooxazolidinyl benzene derivatives |
| TW575567B (en) | 1998-10-23 | 2004-02-11 | Akzo Nobel Nv | Serine protease inhibitor |
| GB9912701D0 (en) | 1999-06-01 | 1999-08-04 | Smithkline Beecham Plc | Novel compounds |
| US6541456B1 (en) | 1999-12-01 | 2003-04-01 | Isis Pharmaceuticals, Inc. | Antimicrobial 2-deoxystreptamine compounds |
| BR0107732A (en) | 2000-01-20 | 2003-03-11 | Eisai Ltd | Method to prevent or treat a disease |
| US7217722B2 (en) * | 2000-02-01 | 2007-05-15 | Kirin Beer Kabushiki Kaisha | Nitrogen-containing compounds having kinase inhibitory activity and drugs containing the same |
| AU2001239947A1 (en) | 2000-02-29 | 2001-09-12 | Curis, Inc. | Methods and compositions for regulating adipocytes |
| GB0004887D0 (en) | 2000-03-01 | 2000-04-19 | Astrazeneca Uk Ltd | Chemical compounds |
| AR033517A1 (en) | 2000-04-08 | 2003-12-26 | Astrazeneca Ab | PIPERIDINE DERIVATIVES, PROCESS FOR THE PREPARATION AND USE OF THESE DERIVATIVES IN THE MANUFACTURE OF MEDICINES |
| GB0013060D0 (en) | 2000-05-31 | 2000-07-19 | Astrazeneca Ab | Chemical compounds |
| AU2001296008A1 (en) | 2000-10-27 | 2002-05-06 | Takeda Chemical Industries Ltd. | Process for preparing substituted aromatic compounds and intermediates therefor |
| JP2004520347A (en) | 2001-01-15 | 2004-07-08 | グラクソ グループ リミテッド | Arylpiperidine and piperazine derivatives as inducers of LDL-receptor expression |
| SE0101038D0 (en) | 2001-03-23 | 2001-03-23 | Astrazeneca Ab | Novel compounds |
| EP1389194A2 (en) | 2001-04-27 | 2004-02-18 | Vertex Pharmaceuticals Incorporated | Inhibitors of bace |
| WO2002100833A1 (en) * | 2001-06-12 | 2002-12-19 | Sumitomo Pharmaceuticals Company, Limited | Rho KINASE INHIBITORS |
| GB0117899D0 (en) | 2001-07-23 | 2001-09-12 | Astrazeneca Ab | Chemical compounds |
| WO2003024450A1 (en) | 2001-09-20 | 2003-03-27 | Eisai Co., Ltd. | Methods for treating prion diseases |
| SE0104340D0 (en) | 2001-12-20 | 2001-12-20 | Astrazeneca Ab | New compounds |
| AU2003264427A1 (en) | 2002-09-12 | 2004-04-30 | Kirin Beer Kabushiki Kaisha | Isoquinoline derivatives having kinasae inhibitory activity and drugs containing the same |
| WO2004105757A2 (en) | 2003-05-29 | 2004-12-09 | Schering Aktiengesellschaft | Use of rho-kinase inhibitors in the treatment of aneurysm and cardiac hypertrophy |
| WO2004113297A2 (en) | 2003-06-24 | 2004-12-29 | Neurosearch A/S | Aza-ring derivatives and their use as monoamine neurotransmitter re-uptake inhibitors |
| US20050067037A1 (en) | 2003-09-30 | 2005-03-31 | Conocophillips Company | Collapse resistant composite riser |
| GB0323238D0 (en) | 2003-10-03 | 2003-11-05 | Domantis Ltd | Synthetic LG binding domains of protein L |
| US20070060595A1 (en) * | 2003-10-10 | 2007-03-15 | Toshio Yoshizawa | Novel fused heterocyclic compound and use thereof |
| US7449477B2 (en) | 2003-11-25 | 2008-11-11 | Eli Lilly And Company | 7-phenyl-isoquinoline-5-sulfonylamino derivatives as inhibitors of akt (protein kinase B) |
| JP2005232175A (en) * | 2004-01-21 | 2005-09-02 | Asahi Kasei Pharma Kk | 5-substituted isoquinoline as medicine |
| WO2005074535A2 (en) | 2004-01-30 | 2005-08-18 | Eisai Co., Ltd. | Cholinesterase inhibitors for spinal cord disorders |
| EP1729761A4 (en) | 2004-03-05 | 2008-09-03 | Eisai Co Ltd | Cadasil treatment with cholinesterase inhibitors |
| SE0400850D0 (en) | 2004-03-30 | 2004-03-31 | Astrazeneca Ab | Novel Compounds |
| US7517991B2 (en) * | 2004-10-12 | 2009-04-14 | Bristol-Myers Squibb Company | N-sulfonylpiperidine cannabinoid receptor 1 antagonists |
| CA2615577C (en) | 2005-07-26 | 2014-09-09 | Sanofi-Aventis | Piperidinyl-substituted isoquinolone derivatives as rho-kinase inhibitors |
| TW200745101A (en) | 2005-09-30 | 2007-12-16 | Organon Nv | 9-Azabicyclo[3.3.1]nonane derivatives |
| TW200738682A (en) | 2005-12-08 | 2007-10-16 | Organon Nv | Isoquinoline derivatives |
| US7618985B2 (en) * | 2005-12-08 | 2009-11-17 | N.V. Organon | Isoquinoline derivatives |
| US7893088B2 (en) * | 2006-08-18 | 2011-02-22 | N.V. Organon | 6-substituted isoquinoline derivatives |
| MY148902A (en) | 2006-12-27 | 2013-06-14 | Sanofi Aventis | Substituted isoquinolines and their use as rho-kinase inhibitors |
| RU2468011C2 (en) | 2006-12-27 | 2012-11-27 | Санофи-Авентис | Cycloalkylamine-substituted isoquinoline and isoquinolinone derivatives |
-
2007
- 2007-12-19 MX MX2009005964A patent/MX2009005964A/en active IP Right Grant
- 2007-12-19 KR KR1020097013325A patent/KR20090092303A/en not_active Abandoned
- 2007-12-19 EP EP07856890A patent/EP2125746B1/en not_active Not-in-force
- 2007-12-19 WO PCT/EP2007/011167 patent/WO2008077554A1/en not_active Ceased
- 2007-12-19 BR BRPI0721180-5A patent/BRPI0721180A2/en not_active IP Right Cessation
- 2007-12-19 CN CN200780048527XA patent/CN101611012B/en not_active Expired - Fee Related
- 2007-12-19 JP JP2009543375A patent/JP5405316B2/en not_active Expired - Fee Related
- 2007-12-19 AT AT07856890T patent/ATE554072T1/en active
- 2007-12-19 AU AU2007338410A patent/AU2007338410B2/en not_active Ceased
- 2007-12-19 CA CA2673920A patent/CA2673920C/en not_active Expired - Fee Related
- 2007-12-19 MY MYPI20092541 patent/MY151953A/en unknown
-
2009
- 2009-06-18 US US12/487,386 patent/US8710228B2/en not_active Expired - Fee Related
- 2009-06-24 IL IL199539A patent/IL199539A/en not_active IP Right Cessation
- 2009-06-24 SV SV2009003317A patent/SV2009003317A/en unknown
- 2009-06-26 NO NO20092432A patent/NO20092432L/en not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1541559A1 (en) * | 2002-07-22 | 2005-06-15 | Asahi Kasei Pharma Corporation | 5-substituted isoquinoline derivative |
| WO2005030791A2 (en) * | 2003-09-23 | 2005-04-07 | Merck & Co., Inc. | Isoquinolinone potassium channel inhibitors |
| WO2007000240A1 (en) * | 2005-06-28 | 2007-01-04 | Sanofi-Aventis | Isoquinoline derivatives as inhibitors of rho-kinase |
| WO2007012422A1 (en) * | 2005-07-26 | 2007-02-01 | Sanofi-Aventis | Cyclohexylamin isoquinolone derivatives as rho-kinase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| TAMURA ET AL: "Development of specific Rho-kinase inhibitors and their clinical application", BIOCHIMICA ET BIOPHYSICA ACTA (BBA) - PROTEINS & PROTEOMICS, ELSEVIER, vol. 1754, no. 1-2, 30 December 2005 (2005-12-30), pages 245 - 252, XP005214213, ISSN: 1570-9639 * |
Cited By (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7470787B2 (en) | 2005-07-11 | 2008-12-30 | Aerie Pharmaceuticals, Inc. | Isoquinoline compounds |
| US7671205B2 (en) | 2005-07-11 | 2010-03-02 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8034943B2 (en) | 2005-07-11 | 2011-10-11 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US10624882B2 (en) | 2006-09-20 | 2020-04-21 | Aerie Pharmaceuticals, Inc. | Rho kinase inhibitors |
| US9890123B2 (en) | 2007-01-10 | 2018-02-13 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8357699B2 (en) | 2007-01-10 | 2013-01-22 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US10472327B2 (en) | 2007-01-10 | 2019-11-12 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8455513B2 (en) | 2007-01-10 | 2013-06-04 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8921392B2 (en) | 2007-01-10 | 2014-12-30 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US9365518B2 (en) | 2007-01-10 | 2016-06-14 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US10899714B2 (en) | 2007-01-10 | 2021-01-26 | Aerie Pharmaceuticals, Inc. | 6-aminoisoquinoline compounds |
| US8367658B2 (en) | 2007-10-05 | 2013-02-05 | Cancer Research Technology Limited | Pyrazin-2-yl-pyridin-2-yl-amine and pyrazin-2-yl-pyrimidin-4-yl-amine compounds and their use |
| US8058045B2 (en) | 2007-10-05 | 2011-11-15 | Cancer Research Technology Limited | Pyrazin-2-yl-pyridin-2-yl-amine and pyrazin-2-yl-pyrimidin-4-yl-amine compounds and their use |
| US8871757B2 (en) | 2008-01-17 | 2014-10-28 | Aerie Pharmaceuticals, Inc. | 6-and 7-amino isoquinoline compounds and methods for making and using the same |
| US8530468B2 (en) | 2008-02-19 | 2013-09-10 | Cancer Research Technology Limited | Bicyclylaryl-aryl-amine compounds and their use |
| WO2009103966A1 (en) * | 2008-02-19 | 2009-08-27 | Cancer Research Technology Limited | Bicyclylaryl-aryl-amine compounds and their use |
| US10882840B2 (en) | 2008-07-25 | 2021-01-05 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US10532993B2 (en) | 2008-07-25 | 2020-01-14 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US11021456B2 (en) | 2008-07-25 | 2021-06-01 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US9512101B2 (en) | 2008-07-25 | 2016-12-06 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US9096569B2 (en) | 2008-07-25 | 2015-08-04 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US10112920B2 (en) | 2008-07-25 | 2018-10-30 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US9884840B2 (en) | 2008-07-25 | 2018-02-06 | Aerie Pharmaceuticals, Inc. | Beta- and gamma-amino-isoquinoline amide compounds and substituted benzamide compounds |
| US11618748B2 (en) | 2009-05-01 | 2023-04-04 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10654844B2 (en) | 2009-05-01 | 2020-05-19 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US11028081B2 (en) | 2009-05-01 | 2021-06-08 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US9951059B2 (en) | 2009-05-01 | 2018-04-24 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US8394826B2 (en) | 2009-05-01 | 2013-03-12 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10316029B2 (en) | 2009-05-01 | 2019-06-11 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US10174017B2 (en) | 2009-05-01 | 2019-01-08 | Aerie Pharmaceuticals, Inc. | Dual mechanism inhibitors for the treatment of disease |
| US8951997B2 (en) | 2009-06-19 | 2015-02-10 | D. Western Therapeutics Institute, Inc. | Substituted isoquinoline derivative |
| US9403797B2 (en) | 2011-11-09 | 2016-08-02 | Cancer Research Technology Limited | 5-(pyridin-2-yl-amino)-pyrazine-2-carbonitrile compounds and their therapeutic use |
| US9040540B2 (en) | 2011-11-09 | 2015-05-26 | Cancer Research Technology Limited | 5-(pyridin-2-yl-amino)-pyrazine-2-carbonitrile compounds and their therapeutic use |
| US9765059B2 (en) | 2011-11-09 | 2017-09-19 | Cancer Research Technology Limited | 5-(Pyridin-2-yl-amino)-pyrazine-2-carbonitrile compounds and their therapeutic use |
| US10259806B2 (en) | 2012-05-15 | 2019-04-16 | Cancer Research Technology Limited | 5-[[4-[[morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2 pyridyl]amino]pyrazine-2-carbonitrile and therapeutic uses thereof |
| US9663503B2 (en) | 2012-05-15 | 2017-05-30 | Cancer Research Technology Limited | 5-[[4-[[morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2-pyridyl]amino]pyrazine-2-carbonitrile and therapeutic uses thereof |
| US11787792B2 (en) | 2012-05-15 | 2023-10-17 | Cancer Research Technology Limited | 5-[[4-[[morpholin-2-yl]methylamino]-5-(trifluoromethyl)-2 Pyridyl]Amino]Pyrazine-2-carbonitrile and therapeutic uses thereof |
| US11197853B2 (en) | 2013-03-15 | 2021-12-14 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US11020385B2 (en) | 2013-03-15 | 2021-06-01 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US10588901B2 (en) | 2013-03-15 | 2020-03-17 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9849122B2 (en) | 2013-03-15 | 2017-12-26 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US11185538B2 (en) | 2013-03-15 | 2021-11-30 | Aerie Pharmaceuticals, Inc. | Compositions for treating glaucoma or reducing intraocular pressure |
| US9931336B2 (en) | 2013-03-15 | 2018-04-03 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9993470B2 (en) | 2013-03-15 | 2018-06-12 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US10568878B2 (en) | 2013-03-15 | 2020-02-25 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| US9415043B2 (en) | 2013-03-15 | 2016-08-16 | Aerie Pharmaceuticals, Inc. | Combination therapy |
| WO2015124877A1 (en) | 2014-02-21 | 2015-08-27 | Les Laboratoires Servier | Derivatives of 5-benzylisoquinoline for the treatment of cardiovascular diseases |
| EP3215142A4 (en) * | 2014-11-05 | 2018-09-05 | Flexus Biosciences, Inc. | Immunoregulatory agents |
| US10206893B2 (en) | 2014-11-05 | 2019-02-19 | Flexus Biosciences, Inc. | Immunoregulatory agents |
| US9643927B1 (en) | 2015-11-17 | 2017-05-09 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US10550087B2 (en) | 2015-11-17 | 2020-02-04 | Aerie Pharmaceuticals, Inc. | Process for the preparation of kinase inhibitors and intermediates thereof |
| US11707460B2 (en) | 2016-08-31 | 2023-07-25 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11389441B2 (en) | 2016-08-31 | 2022-07-19 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11590123B2 (en) | 2016-08-31 | 2023-02-28 | Aerie Pharmaceuticals, Inc. | Ophthalmic compositions |
| US11566003B2 (en) | 2017-03-30 | 2023-01-31 | Genentech, Inc. | Isoquinolines as inhibitors of HPK1 |
| US11312700B2 (en) | 2017-03-31 | 2022-04-26 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US10858339B2 (en) | 2017-03-31 | 2020-12-08 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US12018012B2 (en) | 2017-03-31 | 2024-06-25 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US12378249B2 (en) | 2018-07-24 | 2025-08-05 | Genentech, Inc. | Isoquinoline compounds and uses thereof |
| US11427563B2 (en) | 2018-09-14 | 2022-08-30 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US11891376B2 (en) | 2018-09-14 | 2024-02-06 | Aerie Pharmaceuticals, Inc. | Aryl cyclopropyl-amino-isoquinolinyl amide compounds |
| US12473301B2 (en) | 2018-10-02 | 2025-11-18 | Genentech, Inc. | Isoquinoline compounds for the treatment of cancer |
| US11612606B2 (en) | 2018-10-03 | 2023-03-28 | Genentech, Inc. | 8-aminoisoquinoline compounds and uses thereof |
| US12606550B2 (en) | 2020-04-07 | 2026-04-21 | Crt Pioneer Fund Lp | Methods for synthesis of CHK1 inhibitors |
| US12577240B2 (en) | 2020-07-11 | 2026-03-17 | Pfizer Inc. | Antiviral heteroaryl ketone derivatives |
| US11541034B2 (en) | 2020-09-03 | 2023-01-03 | Pfizer Inc. | Nitrile-containing antiviral compounds |
| US11452711B2 (en) | 2020-09-03 | 2022-09-27 | Pfizer Inc. | Nitrile-containing antiviral compounds |
| US11351149B2 (en) | 2020-09-03 | 2022-06-07 | Pfizer Inc. | Nitrile-containing antiviral compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20090092303A (en) | 2009-08-31 |
| MX2009005964A (en) | 2009-06-15 |
| HK1140189A1 (en) | 2010-10-08 |
| AU2007338410B2 (en) | 2012-08-16 |
| JP5405316B2 (en) | 2014-02-05 |
| AU2007338410A1 (en) | 2008-07-03 |
| MY151953A (en) | 2014-07-31 |
| EP2125746A1 (en) | 2009-12-02 |
| ATE554072T1 (en) | 2012-05-15 |
| IL199539A (en) | 2014-07-31 |
| SV2009003317A (en) | 2010-02-05 |
| US8710228B2 (en) | 2014-04-29 |
| US20100081671A1 (en) | 2010-04-01 |
| CN101611012B (en) | 2012-11-14 |
| CA2673920C (en) | 2015-03-24 |
| BRPI0721180A2 (en) | 2014-03-18 |
| CN101611012A (en) | 2009-12-23 |
| JP2010514719A (en) | 2010-05-06 |
| NO20092432L (en) | 2009-09-17 |
| CA2673920A1 (en) | 2008-07-03 |
| EP2125746B1 (en) | 2012-04-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2125746B1 (en) | Cycloalkylamine substituted isoquinoline derivatives | |
| AU2007338407B2 (en) | Cycloalkylamine substituted isoquinolone derivatives | |
| CA2673919C (en) | Cycloalkylamine substituted isoquinolone and isoquinolinone derivatives | |
| EP1910333B1 (en) | Piperidinyl-substituted isoquinolone derivatives as rho-kinase inhibitors | |
| EP2102187B1 (en) | Substituted isoquinoline and isoquinolinone derivatives as inhibitors of rho-kinase | |
| AU2006274246B2 (en) | Cyclohexylamin isoquinolone derivatives as Rho-kinase inhibitors | |
| EP2102164A1 (en) | Cycloalkylamine substituted isoquinoline and isoquinolinone derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780048527.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07856890 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2009/005964 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007856890 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3728/CHENP/2009 Country of ref document: IN Ref document number: 1020097013325 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2009543375 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2673920 Country of ref document: CA Ref document number: 2007338410 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007338410 Country of ref document: AU Date of ref document: 20071219 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0721180 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090626 |