WO2008087514A2 - Hdac inhibitors - Google Patents
Hdac inhibitors Download PDFInfo
- Publication number
- WO2008087514A2 WO2008087514A2 PCT/IB2008/000045 IB2008000045W WO2008087514A2 WO 2008087514 A2 WO2008087514 A2 WO 2008087514A2 IB 2008000045 W IB2008000045 W IB 2008000045W WO 2008087514 A2 WO2008087514 A2 WO 2008087514A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- piperazine
- carboxylate
- oxoprop
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*C(*)=C(*)C(N(*)*)=* Chemical compound C*C(*)=C(*)C(N(*)*)=* 0.000 description 1
- UYZKYFLIXDJKPO-FMIVXFBMSA-N CC(C(Nc1ccccc1)=O)Oc1ccc(/C=C/C(NO)=O)cc1 Chemical compound CC(C(Nc1ccccc1)=O)Oc1ccc(/C=C/C(NO)=O)cc1 UYZKYFLIXDJKPO-FMIVXFBMSA-N 0.000 description 1
- DCPLOIFDMMEBQZ-UHFFFAOYSA-N O=C(CBr)Nc1ccccc1 Chemical compound O=C(CBr)Nc1ccccc1 DCPLOIFDMMEBQZ-UHFFFAOYSA-N 0.000 description 1
- SLOZMICNVIKCGO-JXMROGBWSA-N ONC(/C=C/c(cc1)ccc1N(CC1)CCN1C(OCc1nc2ccccc2[s]1)=O)=O Chemical compound ONC(/C=C/c(cc1)ccc1N(CC1)CCN1C(OCc1nc2ccccc2[s]1)=O)=O SLOZMICNVIKCGO-JXMROGBWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/12—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D215/14—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/06—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/30—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
- C07D285/02—Thiadiazoles; Hydrogenated thiadiazoles
- C07D285/04—Thiadiazoles; Hydrogenated thiadiazoles not condensed with other rings
- C07D285/08—1,2,4-Thiadiazoles; Hydrogenated 1,2,4-thiadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/60—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/14—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems
- C07D319/16—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D319/20—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring with substituents attached to the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/22—Radicals substituted by doubly bound hetero atoms, or by two hetero atoms other than halogen singly bound to the same carbon atom
Definitions
- novel compounds of the formula (I) their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, solvates, pharmaceutically acceptable salts and compositions, metabolites and prodrugs thereof.
- the present invention more particularly provides novel compounds of the general formula (I).
- the present invention also provides a process for the preparation of the above said novel compounds of the general formula (I), their derivatives, analogs, stereoisomers, polymorphs, hydrates, solvates, pharmaceutically acceptable salts pharmaceutical compositions, metabolites and prodrugs thereof.
- the present invention relates to potentially pharmaceutical compositions and in particular to new molecules as active ingredients, that are used in particular as anticancer agents.
- Compounds of the general formula (I), or pharmaceutically acceptable salts thereof according to the present invention have an ability of inhibiting histone deacetylating enzyme and of inducing differentiation and are useful as therapeutic or ameliorating agent for diseases that are involved in cellular growth such as malignant tumors, autoimmune diseases, skin diseases, infections etc.
- novel compounds (I) of the present invention are useful for the treatment of cancer, which is one of the leading causes of death in the present society.
- a great deal of effort has been underway to treat various forms of cancer for decades and until recently, chemoprevention of cancer is receiving its due share of attention.
- Cancer may affect people at all ages, but risks tends to increase with age, due to the fact that DNA damage becomes more apparent in the aging DNA.
- Cancer is one of the principal causes of death in developed countries, more than 11 million people are diagnosed with cancer every year, and it is estimated that there will be 16 million new cases every year by 2020. Cancer causes 7 million deaths every year or 12.5% of deaths worldwide. Cancer particularly affects major portion of people in industrialized world than in the non-industrialized world.
- Cancer is a disease of multifactorial origin characterized by uncontrolled division of cells; when the cancer cell faces spatial restrictions, due to uncontrolled proliferation in an organ of the body, the ability of the cell to invade other distinct tissues occurs by a process defined as "metastasis" the stage in which cancer cells are transported through the bloodstream or the lymphatic system.
- PKI 166 is a dual inhibitor of EGF receptor (HER 1) as well as erbB (HER 2).
- EGF-receptor and PTK787 potent inhibitors of VEGF-receptor 2 (KDR) are able to suppress tumor growth via suppression of tumor angiogenesis and also these agents have entered clinical trials in tumor patients (Michal et.al., Drug Discovery Today, 10, 2005, 839-846)
- KDR potent inhibitors of VEGF-receptor 2
- Transcriptional regulation is a major event in cell differentiation, proliferation and apoptosis. Transcriptional activation of a set of genes determines cell destination and for this reason transcription is tightly regulated by a variety of factors.
- One of its regulatory mechanisms, involved in the process is an alteration in the tertiary structure of DNA, which affects transcription factors to their target DNA regiments.
- Nucleosomal integrity is regulated by the acetylating status of the core histone, with the result being permissiveness to transcription.
- the regulations of transcription factor are thought to involve by changes in the structure of chromatin. Changing its affinity of histone proteins for coiled DNA in the nuclesome alters the structure of chromatin.
- hypo acetylated histones are believed to have greater affinity to the DNA and form a tightly bound DNA-histone complex and render the DNA inaccessible to transcriptional regulation.
- the acetylating status of the histone is governed by the balance activities of the histone acetyl transferase (HAT) and histone deacetylase (HDAC).
- HAT histone acetyl transferase
- HDAC histone deacetylase
- HDACs histone deacetylases
- HDACs histone deacetylases
- RPD 3 class I includes HDAC I, 2, 3, 8, and 11
- Hda 1 class II includes HDAC 4, 6, 7, 9, and 10
- All of the HDACs have a highly conserved zinc dependent catalytic domain.
- HDAC inhibitors are promising reagents for cancer therapy as effective inducers of apoptosis.
- HDACIs structural classes of HDAC inhibitors
- Zolinza Vorinostat, SAHA
- This invention relates to immuno suppressants, process for their production, their uses and pharmaceutical compositions.
- the invention provides a novel class of compounds useful in the treatment or prevention of diseases or disorders mediated by interactions, particularly diseases associated with EDG receptor mediated signal transduction, infectious diseases and cancer.
- An alteration in the EDG receptor activity contributes to the pathology and/or symptomology of these diseases.
- molecules that themselves alter the activity of EDG receptors are useful as therapeutic agents in the treatment of such diseases; wherein, n is chosen from 0, 1 and 2; m is chosen from 1, 2 and 3; R ⁇ represents C ⁇ -ioaryl, and Cs-ioheteroaryl etc; wherein aryl or heteroaryl of Ri is optionally substituted.
- R 2 , R 3 , R 4 and R 5 are independently chosen from hydrogen, halo, hydroxy, Ci-ioalkyl etc.
- A represents -XiC(O)OR 7 .
- Xi represents B is CRgR ⁇ >; Rg and R 9 are independently hydrogen.
- E represents CR 8 or N;
- X represents -XiC(O)NR 7 X 2 -.
- Xi and X 2 represent a bond, Ci- 3 alkylene and C 2-3 alkylene; R 7 is hydrogen and Ci- ⁇ alkyl; and heteroarylene of X is optionally substituted by halo and Ci -6 alkyl.
- This invention relates to novel cross-linkable, photoactive silane derivatives of the formula I.
- silane derivative of formula I and mixtures thereof are used as orientation layers for liquid crystals and for the production of unstructured or structured optical elements and multiplayer systems; wherein, X 1 , X 2 and X 3 denote alkyl, alkoxy or halogen, but atleast one of these radicals is either alkoxy or halogen.
- L denotes a single bond or linking functional groups such as O, COO, OOC, NR 1 , NR 1 -CO-, CO-NR 1
- Z denotes -O- or -NR 4 - and R 4 denotes hydrogen or lower alkyl.
- Ring A denotes piperazin-1,4- diyl.
- K is an alkyl-OCO group having 1-20 carbon atoms, which is optionally substituted by fluorine, chlorine, cyano or nitro etc. III.
- US 2004/0077726 Al discloses certain active carbamic acid compounds which inhibit HDAC activity and which have the following formula I
- A is an aryl group
- Q 1 is a covalent bond or an aryl leader group
- R1 is a sulfonamido substituent; and
- Q 2 is an acid leader group; with the provision that if J is
- Ql is an aryl leader group; and pharmaceutically acceptable salts, solvates, amides, esters, ethers, chemically protected forms, and prodrugs thereof.
- This invention also pertains to pharmaceutical compositions comprising such compounds, and the use of such compounds and compositions, both in vitro and in vivo, to inhibit proliferative conditions, such as cancer and psoriasis.
- Q 1 is a covalent bond
- J is 2
- Q 2 is phenylene-meta-trans-ethylene
- the compounds have the following formula II, wherein R B m is as defined in the patent.
- Cancer is now believed to be the number one cause of premature death in industrialized countries.
- the market for anti-cancer agents was estimated at about US $ 85.3billion in 2010 and continues to escalate. Befause of the need and the value of these drugs, many laboratories are intensively investigating the nature and vulnerability of cancerous cells, resulting in the development of novel screens and approaches.
- Histone acetylation and deacetylation play an essential role in modifying the chromatin structure and regulating gene expression in eukaryotic cells.
- Hyperacetylated histones are generally found in transcriptionally active genes and in the transcriptionally silent regions of the genome.
- Key enzymes, which modify histone proteins and thereby regulate gene expression, are histone acetyl transferases (HATs) and histone deacetylases (HDACs).
- HATs histone acetyl transferases
- HDACs histone deacetylases
- HDAC inhibitors such as Trichostatin A (TSA), Trapoxin (TPX), Suberoylanilide hydroxamic acid (SAHA), Sodium butyrate (NaB), Sodium valproate (VPA), Cyclic hydroxamic acid containing peptides (CHAPs), Depsipeptide FK-228 and MS-275 can de-repress these genes, resulting in antiproliferative effects in vitro and antitumor effects in vivo.
- TSA Trichostatin A
- TPX Trapoxin
- SAHA Suberoylanilide hydroxamic acid
- NaB Sodium butyrate
- VPA Sodium valproate
- CHAPs Cyclic hydroxamic acid containing peptides
- Depsipeptide FK-228 and MS-275 can de-repress these genes, resulting in antiproliferative effects in vitro and antitumor effects in vivo.
- HDACs are primarily responsible for the manifestation of the disease and can selective inhibitors be developed to address these specific diseases? It is clear that somewhat selective agents can be developed that distinguishes at least the histone and nonhistone protein deacetylation process. As these more selective agents advance into clinical trials their utility will become apparent, but as discussed, their approval (efficacy in a disease state) may depend on effective combinations with other therapeutics to maximize the desired pharmacological benefit.
- novel compounds of the formula (I) their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, solvates, pharmaceutically acceptable salts and pharmaceutical compositions, metabolites and prodrugs thereof,
- Q 2 represents optionally substituted groups such as aryl, aralkyl, heterocyclyl, heteroaryl and benzofused heteroaryl groups.
- Ri represents hydrogen and alkyl.
- R 2 represents hydrogen, -CN, -COOH, -COOCH 3 , -COOEt, -
- R, R 3 and R 4 may be same or different and independently represent hydrogen, hydroxy, alkyl, alkoxy, acetyl, cycloalkyl, or optionally substituted aryl, aryloxy, benzyloxy, heteroaryl, heterocyclyl and benzofused heteroaryl groups; k, 1 and m are integers in the range of O to 2.
- A represents saturated or unsaturated heterocyclyl groups containing one or more hetero atoms selected from O, S and N such as pyrrolidinyl, thiazolidinyl, oxazolidinyl, morpholinyl, thiomorpholinyl, piperidinyl, piperazinyl, pyridyl, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, isooxazolyl, oxadiazolyl, triazolyl, thiadiazolyl, tetrazolyl, pyrimidinyl, pyrazinyl and pyridazinyl;
- Qi represents a bond, -NH, -CO,
- Suitable groups substituted on A, R, R 3 , R 4 and Q 2 may be selected from halogens such as fluorine, chlorine, bromine, iodine; hydroxy; nitro; cyano; azido; nitroso; hydrazine; formyl; alkyl; alkoxy; haloalkyl; haloalkoxy; cycloalkyl; aryl; aryloxy; acyl; acyloxy; acyloxyacyl; heterocyclyl; heteroaryl; amino; monoalkylamino; dialkylamino; acylamino; alkoxycarbonyl; aryloxycarbonyl; alkylsulfonyl; arylsulfonyl; alkylsulfinyl; arylsulflnyl; alkylthio; arylthio; sulfamoyl; alkoxyalkyl groups and carboxylic acids or its derivatives; Wherein
- R, R 3 , R 4 and Q 2 which are cyclic rings represents substituted or unsubstituted 5 to 10 membered ring systems, and also the rings may be monocyclic or bicyclic, saturated or partially saturated or aromatic containing 1 to 4 hetero atoms selected from O, S, N and the like.
- Pharmaceutically acceptable salts include base addition salts such as alkali metal salts like Li, Na, and K salts; alkaline earth metal salts like Ca and Mg, salts of organic bases such as lysine, arginine, guanidine, diethanolamine, ⁇ -phenylethylamine, benzylamine, piperidine, morpholine, pyridine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, choline and the like, ammonium or substituted ammonium salts, aluminum salts. Salts also include amino acid salts such as glycine, alanine, cystine, cysteine, lysine, arginine, phenylalanine, guanidine etc.
- Salts may include acid addition salts where appropriate, which are sulphates, nitrates, phosphates, perchlorates, borates, hydrohalides, acetates, tartrates, maleates, citrates, succinates, palmoates, methanesulphonates, tosylates, benzoates, salicylates, hydroxynaphthoates, benzenesulfonates, ascorbates, glycerophosphates, ketoglutarates and the like.
- Pharmaceutically acceptable solvates may be hydrates or comprising of other solvents of crystallization such as alcohols.
- analog includes a compound, which differs from the parent structure by one or more C, N, O or S atoms.
- a compound in which one of the N atoms in the parent structure is replaced by an S atom is an analog of the former.
- stereoisomer includes isomers that differ from one another in the way the atoms are arranged in space, but whose chemical formulas and structures are otherwise identical. Stereoisomers include enantiomers and diastereoisomers.
- tautomers include readily interconvertible isomeric forms of a compound in equilibrium.
- the enol-keto tautomerism is an example.
- polymorphs include crystallographically distinct forms of compounds with chemically identical structures.
- pharmaceutically acceptable solvates includes combinations of solvent molecules with molecules or ions of the solute compound.
- derivative refers to a compound obtained from a compound according to formula (I), an analog, tautomeric form, stereoisomer, polymorph, hydrate, pharmaceutically acceptable salt or pharmaceutically acceptable solvate thereof, by a simple chemical process converting one or more functional groups, such as, by oxidation, hydrogenation, alkylation, esterification, halogenation, and the like.
- Representative compounds include:
- the compound of the general formula (I) is prepared by the following procedure (Scheme 1): Step (I): The condensation of the compound of formula (Ia), with 1,1'carbonyl diimidazole and primary or secondary amine is carried out in the presence of solvents selected from toluene, DMF, tetrahydrofuran, chloroform, dichloromethane, dichloroethane, ethyl acetate, o-dichlorobenzene or a mixture thereof, in the presence of bases such as triethylamine, diethylamine, pyridine, DMAP and alkali carbonates such as sodium carbonate, potassium carbonate and the like to afford the compound of formula (Ib).
- the reaction is carried out at a temperature in the range of O 0 C to room temperature.
- solvents such as toluene, tetrahydrofuran, DMF, chloroform, dichloromethane, dichloroethane, ethylacetate, o- dichlorobenz
- a primary or secondary amine e.g. hydroxylamine hydrochloride, N-methoxy hydroxylamine hydrochloride, Ophenylenediamine, or 2-aminothiazole
- an acid activating reagent such as BOP, DCC, EDCI, isobutyl chloroformate and the like in the presence of solvents mentioned above to afford a compound of the formula (I).
- any reactive group in the substrate molecule may be protected according to the conventional chemical practice.
- Suitable protecting groups in any of the above-mentioned reactions are those used conventionally in the art.
- the methods of formation and removal of such protecting groups are those conventional methods appropriate to the molecule being protected.
- the pharmaceutically acceptable salts are prepared by reacting the compound of formula (I) with 1 to 10 equivalents of a base such as sodium hydroxide, sodium methoxide, sodium hydride, potassium t-butoxide, calcium hydroxide, magnesium hydroxide and the like, in solvents like ether, tetrahydrofuran, methanol, t-butanol, dioxane, isopropanol, ethanol etc.
- Organic bases such as diethanolamine, ⁇ -phenylethylamine, benzylamine, piperidine, morpholine, pyridine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, choline, guanidine and the like, ammonium or substituted ammonium salts, aluminum salts.
- Amino acids such as glycine, alanine, cystine, cysteine, lysine, arginine, phenylalanine etc may be used for the preparation of amino acid salts.
- acid addition salts wherever applicable are prepared by treatment with acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, p-toluenesulphonic acid, methanesulfonic acid, acetic acid, citric acid, maleic acid, salicylic acid, hydroxynaphthoic acid, ascorbic acid, palmitic acid, succinic acid, benzoic acid, benzenesulfonic acid, tartaric acid, oxalic acid and the like in solvents like ethyl acetate, ether, alcohols, acetone, tetrahydrofuran, dioxane etc. Mixture of solvents may also be used.
- acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, p-toluenesulphonic acid, methanesulfonic acid, acetic acid, citric acid, maleic acid, salicylic acid
- compounds of the invention may contain groups that may exist in tautomeric forms, and though one form is named, described, displayed and/or claimed herein, all the forms are intended to be inherently included in such name, description, display and/or claim.
- stereoisomers of the compounds forming part of this invention may be prepared by using reactants in their single enantiomeric form, in the process wherever possible or by conducting the reaction in the presence of reagents or catalysts in their single enantiomeric form or by resolving the mixture of stereoisomers by conventional methods.
- Some of the preferred methods include use of microbial resolution, resolving the diastereomeric salts formed with chiral acids such as mandelic acid, camphorsulfonic acid, tartaric acid, lactic acid, and the like wherever applicable or by using chiral bases such as brucine, cinchona alkaloids, their derivatives and the like.
- Prodrugs of the compounds of formula (I) are also contemplated by this invention.
- a prodrug is an active or inactive compound that is modified chemically through in-vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following administration of the prodrug to a patient.
- the suitability and techniques involved in making and using prodrugs are well known to those skilled in the art.
- polymorphs of the compounds of the general formula (I), forming part of this invention may be prepared by crystallization of the compounds of formula (I) under different conditions. For example, using different commonly used solvents, or their mixtures for recrystallization; crystallizations at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallizations. Heating or melting the compounds followed by cooling gradually or immediately, one can also obtain polymorphs.
- the presence of polymorphs may be determined by solid probe NMR spectroscopy, IR spectroscopy, differential scanning calorimetry and powder X-ray diffraction or other such techniques.
- solvates of the compounds of the formula (I) forming part of this invention may be prepared by conventional methods such as dissolving the compounds of the formula (I) in solvents such as water, methanol, ethanol, mixture of solvents such as acetone:water, dioxane:water, N,N- dimethylformamide:water and the like, preferably water and recrystallization by using different crystallization techniques
- the present invention also provides a pharmaceutical composition, containing one or more of the compounds of the general formula (I) as defined above, their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, metabolites, prodrugs, pharmaceutically acceptable salts, pharmaceutically acceptable solvates in combination with the usual pharmaceutically employed carriers, diluents and the like, useful for the treatment of cancer, psoriasis, proliferative conditions and conditions mediated by HDAC.
- a pharmaceutical composition containing one or more of the compounds of the general formula (I) as defined above, their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, metabolites, prodrugs, pharmaceutically acceptable salts, pharmaceutically acceptable solvates in combination with the usual pharmaceutically employed carriers, diluents and the like, useful for the treatment of cancer, psoriasis, proliferative conditions and conditions mediated by HDAC.
- the derivatives provided by the present invention can be employed as pharmaceutical compositions, for example, in the form of pharmaceutical compositions containing the derivatives together with appropriate, pharmaceutically acceptable carriers.
- the products in accordance with the invention can be administered, for example, perorally, such as in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions, or rectally, such as in the form of suppositories, etc.
- the compositions may be sterilized and may contain auxiliary agents generally employed in the pharmaceutical art, such as sodium hydrogen carbonate, citric acid, propylene glycol, tween 80, etc.
- the compounds can be used orally or parenterally.
- the pharmaceutical composition may be in the forms normally employed, such as tablets, capsules, powders, syrups, solutions, suspensions and the like, may contain flavorants, sweeteners etc. in suitable solid or liquid carriers or diluents, or in suitable sterile media to form injectable solutions or suspensions.
- the compositions may be prepared by processes known in the art, such as by combining the ingredients into a dosage form together with suitable, non-toxic, inert, therapeutically compatible solid or liquid carrier materials and if desired, the usual pharmaceutical adjuvants.
- the amount of the active ingredient in the composition may be less than 70% by weight.
- Such compositions typically contain from 1 to 25%, preferably 1 to 15% by ⁇ weight of active compound, the remainder of the composition being pharmaceutically acceptable carriers, diluents, excipients or solvents.
- Suitable pharmaceutically acceptable carriers include solid fillers or diluents and sterile aqueous or organic solutions.
- the active compound will be present in such pharmaceutical compositions in the amounts sufficient to provide the desired dosage in the range as described above.
- the compounds can be combined with a suitable solid or liquid carrier or diluent to form capsules, tablets, powders, syrups, solutions, suspensions and the like.
- the pharmaceutical compositions may, if desired, contain additional components such as flavorants, sweeteners, excipients and the like.
- the compounds can be combined with sterile aqueous or organic media to form injectable solutions or suspensions.
- solutions in sesame or peanut oil, aqueous propylene glycol and the like can be used, as well as aqueous solutions of water-soluble pharmaceutically-acceptable acid addition salts or alkali or alkaline earth metal salts of the compounds.
- the injectable solutions prepared in this manner can then be, administered intravenously, intraperitoneal Iy, subcutaneously, or intramuscularly, with intramuscular administration being preferred in humans.
- the effective dose for treating a particular condition in a patient may be readily determined and adjusted by the physician during treatment to alleviate the symptoms or indications of the condition or disease.
- a daily dose of active compound in the range of about 0.01 to 1000 mg/kg of body weight is appropriate for administration to obtain effective results.
- the daily dose may be administered in a single dose or divided into several doses. In some cases, depending upon the individual response, it may be necessary to deviate upwards or downwards from the initially prescribed daily dose.
- Typical pharmaceutical preparations normally contain from about 0.2 to about 500 mg of active compound of formula I and/or its pharmaceutically active salts or solvates per dose.
- the compounds of the invention can be administered as the sole active pharmaceutical agent, they can also be used in combination with one or more compounds of the invention or other agents.
- the therapeutic agents can be formulated as separate compositions that are given at the same time or different times, or the therapeutic agents can be given as a single composition.
- therapeutically effective amount or “effective amount” refers to that amount of a compound or mixture of compounds of Formula I that is sufficient to effect treatment, as defined below, when administered alone or in combination with other therapies to a mammal in need of such treatment.
- the term "animal” as used herein is meant to include all mammals, and in particular humans. Such animals are also referred to herein as subjects or patients in need of treatment.
- the therapeutically effective amount will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the particular compound of Formula I chosen, the dosing regimen to be followed, timing of administration, the manner of administration and the like, all of which can readily be determined by one of ordinary skill in the art.
- treatment means any treatment of a disease in a mammal, including: a) Preventing the disease, that is, causing the clinical symptoms of the disease not to develop; b) Inhibiting the disease, that is, slowing or arresting the development of clinical symptoms; and/or c) Relieving the disease, that is, causing the regression of clinical symptoms.
- Anti-cancer screen Experimental drugs are screened for anti-cancer activity in three cell lines for their GI 5 0, TGI and LC50 values (using five concentrations for each compound). The cell lines are maintained in DMEM containing 10% fetal bovine serum. 96 well micro titer plates are inoculated with cells in 100 ⁇ L for 24 hours at 37°C, 5% CO2, 95% air and 100% relative humidity. 5000 HCT 116 cells/well, 5000 NCIH 460 cells/well and 5000 U251 cells/well are plated. A separate plate with these cell lines is also inoculated to determine cell viability before the addition of the compounds (T 0 ). Addition of experimental drugs:
- mice Following 24-hour incubation, experimental drugs are added to the 96 well plates. Each plate contains one of the above cell lines and the following in triplicate: five different concentrations (0.01, 0.1, 1, 10 and 100 ⁇ M) of four different compounds, appropriate dilutions of a cytotoxic standard and control (untreated) wells. Compounds are dissolved in DMSO to make 20 mM stock solutions on the day of drug addition and frozen at -20 0 C. Serial dilutions of these 20 mM stock solutions are made in complete growth medium such that 100 ⁇ L of these drug solutions in medium, of final concentrations equaling 0.01, 0.1, 1, 10 and 100 ⁇ M can be added to the cells in triplicate. Standard drugs whose anti-cancer activity has been well documented and which are regularly used are doxorubicin and SAHA. End-point measurement;
- GI 50 is the concentration required to decrease % growth by 50%; TGI is the concentration required to decrease % growth by 100% and LC 50 is the concentration required to decrease % cell death by 50%.
- Histone Deacetylase (HDAC) Inhibition Assay using Boc-Lys (Ac)-AMC Substrate Inhibition of HDAC has been implicated to modulate transcription and to induce apoptosis or differentiation in cancer cells.
- the fluorometric assay provides a fast and fluorescence based method that eliminates radioactivity, extractions or chromatography, as used in traditional assays. The assay is based on two steps. First, the HDAC fluorometric substrate, which comprises an acetylated lysine side chain, is incubated with a sample containing HDAC activity (Mouse Liver Extract).
- Deacetylation of the substrate sensitizes the substrate, in the second step; treatment with the Trypsin stop solution produces a fluorophore that can be easily analyzed using fluorescence plate reader.
- Assay was done in 96 well black microplate and total volume of the assay is 100 ⁇ L.
- Mouse liver enzyme is diluted 1:6 with HDAC buffer.
- Enzyme cocktail made of 10 ⁇ L of diluted enzyme and 30 ⁇ L of HDAC buffer. 40 ⁇ L of enzyme cocktail dispensed into each well. 10 ⁇ L of different concentrations of inhibitor added in to each well, except enzyme control well. Preincubated the plate at 30 0 C for 5 minutes.
- the HDAC reaction is started by adding 50 ⁇ L of HDAC substrate ( ⁇ oc-Lys (Ac)-AMC Substrate) solution. Incubated the plate at 30 0 C for 30 minutes. Adding 100 ⁇ L of Trypsin stop solution stops the reaction. The plate is incubated again at 30 0 C for 20-30 minutes. The release of AMC is monitored by measuring the fluorescence at excitation wavelength of 365 or 360 nm and emission wavelength of 440 or 460 nm. Buffer and substrate alone kept for blank subtraction. (Dennis Wegener et al, Anal. Biochem, 321, 2003, 202-208).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Novel compounds of the formula (I), their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, solvates, pharmaceutically acceptable salts and compositions, metabolites and prodrugs thereof are described. These novel compounds can inhibit HDACs and are useful as a therapeutic or ameliorating agent for diseases that are involved in cellular growth such as malignant tumors, autoimmune diseases, skin diseases, infections, inflammation, cancer, psoriasis, proliferative conditions and conditions mediated by HDAC.
Description
HDAC INHIBITORS
The following specification particularly describes the nature of the invention and the manner in which it has to be performed;
Field
Described are novel compounds of the formula (I), their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, solvates, pharmaceutically acceptable salts and compositions, metabolites and prodrugs thereof. The present invention more particularly provides novel compounds of the general formula (I).

(I)
The present invention also provides a process for the preparation of the above said novel compounds of the general formula (I), their derivatives, analogs, stereoisomers, polymorphs, hydrates, solvates, pharmaceutically acceptable salts pharmaceutical compositions, metabolites and prodrugs thereof.
Background
The present invention relates to potentially pharmaceutical compositions and in particular to new molecules as active ingredients, that are used in particular as anticancer agents. Compounds of the general formula (I), or pharmaceutically acceptable salts thereof according to the present invention have an ability of inhibiting histone deacetylating enzyme and of inducing differentiation and are useful as therapeutic or ameliorating agent for diseases that are involved in cellular growth such as malignant tumors, autoimmune diseases, skin diseases, infections etc.
The novel compounds (I) of the present invention are useful for the treatment of cancer, which is one of the leading causes of death in the present society. A great deal of effort has been underway to treat various forms of cancer for decades and until recently, chemoprevention of cancer is receiving its due share of attention. Cancer may affect people at all ages, but risks tends to increase with age, due to the fact that DNA damage becomes more apparent in the aging DNA. Cancer is one of
the principal causes of death in developed countries, more than 11 million people are diagnosed with cancer every year, and it is estimated that there will be 16 million new cases every year by 2020. Cancer causes 7 million deaths every year or 12.5% of deaths worldwide. Cancer particularly affects major portion of people in industrialized world than in the non-industrialized world. From a total of 58 million deaths worldwide in 2005, cancer accounts for 7.6 million (or 13%) of all deaths. The main types of cancer leading to overall cancer mortality are Lung (1.3 million deaths/year), Stomach (almost 1 million deaths/year), Liver (662,000 deaths/year), Colon (655,000 deaths/year) and Breast (502,000 deaths/year). Deaths from cancer in the world are projected to continue rising, with an estimated 9 million people dying from cancer in 2015 and 11.4 million dying in 2030.
Every cell constantly faces decisions. Should it divide? Or should it differentiate? Or should it die (Apoptosis)? proper development and tissue homeostasis rely on the correct balance between division and apoptosis. Too much apoptosis leads to tissue atrophy such as in Alzheimer's disease. Too much proliferation or too little apoptosis leads to cancer. Cancer is a disease of multifactorial origin characterized by uncontrolled division of cells; when the cancer cell faces spatial restrictions, due to uncontrolled proliferation in an organ of the body, the ability of the cell to invade other distinct tissues occurs by a process defined as "metastasis" the stage in which cancer cells are transported through the bloodstream or the lymphatic system.
The most common treatment for easily accessible cancer is surgical removal of diseased tissues and radiation. The choice of treatment for in-accessible tumors is chemotherapy. Also chemotherapy is given as additional insurance for most cancers as it is difficult to access the extent of metastasis. Most clinically revelant anticancer drugs currently used in the clinics, interfere with cell division and hence are not highly selective to cancer cells and there are potential chances, that chemotherapy can lead to secondary cancers in due course of time. Also the quality of life is hampered in the patients upon chemotherapy, hence there is an unmet medical need for treating cancer patients without affecting the quality of life.
The cell cycle deregulation and the molecular basis of cancer cell growth has been thoroughly exploited in the recent years. Inhibition of signal transduction has become a viable and attractive avenue in biomedical cancer research based on the
discovery of a large number of somatic mutations in many different types of cancers that lead to deregulated growth signal transduction and subsequent aberrant growth, invasion, tumor-derived angiogenesis and metastasis. Most of the noncytotoxic drugs that have been recently developed include protein kinase inhibitors such as Gleevec, Iressa and Tarceva. Glivec™ (STI571), is an inhibitor of the bcr-abl kinase and CML. PKI 166, on the other hand, is a dual inhibitor of EGF receptor (HER 1) as well as erbB (HER 2). EGF-receptor and PTK787, potent inhibitors of VEGF-receptor 2 (KDR) are able to suppress tumor growth via suppression of tumor angiogenesis and also these agents have entered clinical trials in tumor patients (Michal et.al., Drug Discovery Today, 10, 2005, 839-846) These types of orally active and relatively well-tolerated compounds can be used in the clinics; either as single agents or in combination with other well established cytotoxic agents.
Transcriptional regulation is a major event in cell differentiation, proliferation and apoptosis. Transcriptional activation of a set of genes determines cell destination and for this reason transcription is tightly regulated by a variety of factors. One of its regulatory mechanisms, involved in the process is an alteration in the tertiary structure of DNA, which affects transcription factors to their target DNA regiments. Nucleosomal integrity is regulated by the acetylating status of the core histone, with the result being permissiveness to transcription. The regulations of transcription factor are thought to involve by changes in the structure of chromatin. Changing its affinity of histone proteins for coiled DNA in the nuclesome alters the structure of chromatin. Hypo acetylated histones are believed to have greater affinity to the DNA and form a tightly bound DNA-histone complex and render the DNA inaccessible to transcriptional regulation. The acetylating status of the histone is governed by the balance activities of the histone acetyl transferase (HAT) and histone deacetylase (HDAC).
The first isolation of histone deacetylase was described in 1964 from crude nuclear extracts of cells, but the molecular characterization of isoforms of the enzyme has been achieved only recently. Inhibitors of histone deacetylase (HDACs) are zinc hydrolase's responsible for the deacetylation of N-acetyl lysine residues of histone and nonhistone protein substrates. Human HDACs are classified into two distinct classes, the HDACs and sirtuins. The HDACs are devised into two subclasses based on their similarity to yeast histone deacetylases, RPD 3 (class I includes HDAC I, 2, 3, 8, and
11) and Hda 1 (class II includes HDAC 4, 6, 7, 9, and 10). All of the HDACs have a highly conserved zinc dependent catalytic domain. There is growing evidence that the acetylation state of proteins and thus the HDAC enzyme family plays a crucial role in the modulation of a number of biological processes, including transcription and cell cycle.
Given that apoptosis is a crucial factor for cancer progression, HDAC inhibitors are promising reagents for cancer therapy as effective inducers of apoptosis. Several structural classes of HDAC inhibitors (HDACIs) have been identified and are reviewed in Marks, P.A. et al., J. Natl. Cancer Inst., 92, (2000), 1210-1215. More specifically the patents WO 98/55449 and US 5,369,108 report alkanoyl hydroxamates with HDAC inhibitory activity. HDACIs currently in clinical development cover pan-HDACIs (Vorinostat, Belinostat, and LBH589) and somewhat isotype selective agents (Romidepsin, MS-275 and MGCDO 103) With the approval of Zolinza (Vorinostat, SAHA) by the FDA on October 2006 for the treatment of CTCL and with other histone deacetylase inhibitors awaiting approval for various cancers, this will hopefully prompt the investigation of histone deacetylase inhibitors into a broader range of disease states where altered chromatin function may play a role in their pathophysiology.
Prior art:
I. WO 2005082089 A2, discloses compounds of the formula I:

This invention relates to immuno suppressants, process for their production, their uses and pharmaceutical compositions. The invention provides a novel class of compounds useful in the treatment or prevention of diseases or disorders mediated by interactions, particularly diseases associated with EDG receptor mediated signal transduction, infectious diseases and cancer. An alteration in the EDG receptor activity contributes to the pathology and/or symptomology of these diseases. Accordingly,
molecules that themselves alter the activity of EDG receptors are useful as therapeutic agents in the treatment of such diseases; wherein, n is chosen from 0, 1 and 2; m is chosen from 1, 2 and 3; R\ represents Cβ-ioaryl, and Cs-ioheteroaryl etc; wherein aryl or heteroaryl of Ri is optionally substituted. R2, R3, R4 and R5 are independently chosen from hydrogen, halo, hydroxy, Ci-ioalkyl etc. A represents -XiC(O)OR7. Xi represents
 B is CRgRς>; Rg and R9 are independently hydrogen. E represents CR8 or N; X represents -XiC(O)NR7X2-. Xi and X2 represent a bond, Ci-3alkylene and C2-3 alkylene; R7 is hydrogen and Ci-βalkyl; and heteroarylene of X is optionally substituted by halo and Ci-6 alkyl. II. US 6277502 Bl, discloses compounds of the formula I:
B is CRgRς>; Rg and R9 are independently hydrogen. E represents CR8 or N; X represents -XiC(O)NR7X2-. Xi and X2 represent a bond, Ci-3alkylene and C2-3 alkylene; R7 is hydrogen and Ci-βalkyl; and heteroarylene of X is optionally substituted by halo and Ci-6 alkyl. II. US 6277502 Bl, discloses compounds of the formula I:
This invention relates to novel cross-linkable, photoactive silane derivatives of the formula I.
X3

O Il A— Q1 — J — Q2— C — NH - OH
I wherein A is an aryl group; Q1 is a covalent bond or an aryl leader group; J is a Sulfonamide linkage selected from: ~S(=°)2 NR1— and — NR1S(=O)2— . R1 is a sulfonamido substituent; and Q2 is an acid leader group; with the provision that if J is
^~ '2 , then Ql is an aryl leader group; and pharmaceutically acceptable salts, solvates, amides, esters, ethers, chemically protected forms, and prodrugs thereof.
This invention also pertains to pharmaceutical compositions comprising such compounds, and the use of such compounds and compositions, both in vitro and in vivo, to inhibit proliferative conditions, such as cancer and psoriasis.
In one preferred embodiment, Q1 is a covalent bond, J is 2 , Q2 is phenylene-meta-trans-ethylene, and the compounds have the following formula II, wherein RB m is as defined in the patent.

Objectives
Cancer is now believed to be the number one cause of premature death in industrialized nations. The market for anti-cancer agents was estimated at about US $ 85.3billion in 2010 and continues to escalate. Befause of the need and the value of these drugs, many laboratories are intensively investigating the nature and vulnerability of cancerous cells, resulting in the development of novel screens and approaches.
Our sustained efforts have resulted in the novel anticancer agents of the general formula (I). Histone acetylation and deacetylation play an essential role in modifying the chromatin structure and regulating gene expression in eukaryotic cells.
Hyperacetylated histones are generally found in transcriptionally active genes and in
the transcriptionally silent regions of the genome. Key enzymes, which modify histone proteins and thereby regulate gene expression, are histone acetyl transferases (HATs) and histone deacetylases (HDACs). Compounds able to inhibit HDAC activity i.e. HDAC inhibitors such as Trichostatin A (TSA), Trapoxin (TPX), Suberoylanilide hydroxamic acid (SAHA), Sodium butyrate (NaB), Sodium valproate (VPA), Cyclic hydroxamic acid containing peptides (CHAPs), Depsipeptide FK-228 and MS-275 can de-repress these genes, resulting in antiproliferative effects in vitro and antitumor effects in vivo.
With the approval of Vorinostat for the treatment of CTCL and PTCL, the application of epigenetic regulation as an avenue in treatment has expanded, not only for hematological malignancies, but also to a much broader range of cancers. The response rates in CTCL are impressive and the side effects are manageable. The greatest utility of these epigenetic modulators will be in combination with other therapeutics that synergize with the regulation being controlled by the epigenetic modulator. Only in this manner of combination will there be a sufficient response rate in solid tumors. These experiments are now ongoing in clinical trials of Vorinostat, Romidepsin, Belinostat and LBH589.
One of the major issues still remaining is, which of the HDACs are primarily responsible for the manifestation of the disease and can selective inhibitors be developed to address these specific diseases? It is clear that somewhat selective agents can be developed that distinguishes at least the histone and nonhistone protein deacetylation process. As these more selective agents advance into clinical trials their utility will become apparent, but as discussed, their approval (efficacy in a disease state) may depend on effective combinations with other therapeutics to maximize the desired pharmacological benefit.
Summary
Described are novel compounds of the formula (I), their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, solvates, pharmaceutically acceptable salts and pharmaceutical compositions, metabolites and prodrugs thereof,

(I) wherein X represents oxygen or sulphur; A represents saturated or unsaturated heterocyclyl groups containing one or more hetero atoms selected from O, S and N. Qi represents a bond, -NH, -CO, -CH2-, -CONH-, -CO-CH2-O-, -NHCO-CH(CH3)-O-, NH-
CO-CH2-O-; wherein Q2 represents optionally substituted groups such as aryl, aralkyl, heterocyclyl, heteroaryl and benzofused heteroaryl groups. Wherein Ri represents hydrogen and alkyl. R2 represents hydrogen, -CN, -COOH, -COOCH3, -COOEt, -
COCH3, -CONH2. R, R3 and R4 may be same or different and independently represent hydrogen, hydroxy, alkyl, alkoxy, acetyl, cycloalkyl, or optionally substituted aryl, aryloxy, benzyloxy, heteroaryl, heterocyclyl and benzofused heteroaryl groups; k, 1 and m are integers in the range of O to 2.
Detailed Description Novel compounds of the general formula (I),

(I) their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, solvates, pharmaceutically acceptable salts and compositions metabolites and prodrugs thereof, wherein X represents oxygen or sulphur; A represents saturated or unsaturated heterocyclyl groups containing one or more hetero atoms selected from O, S and N such as pyrrolidinyl, thiazolidinyl, oxazolidinyl, morpholinyl, thiomorpholinyl, piperidinyl, piperazinyl, pyridyl, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, isooxazolyl, oxadiazolyl, triazolyl, thiadiazolyl, tetrazolyl, pyrimidinyl, pyrazinyl and pyridazinyl; Qi represents a bond, -NH, -CO, -CH2-, -CONH-, -CO-CH2-O-, -NHCO- CH(CH3)-0- and -NH-CO-CH2-O-; wherein Q2 represents optionally substituted aryl groups such as phenyl, naphthyl and the like; aralkyl groups such as benzyl and the like; heteroaryl groups such as pyridyl, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl,
imidazolyl, isooxazolyl, oxadiazolyl, triazolyl, thiadiazolyl, tetrazolyl, pyrimidinyl, pyrazinyl, pyridazinyl and the like; heterocyclyl groups such as pyrrolidinyl, thiazolidinyl, oxazolidinyl, morpholinyl, thiomoφholinyl, piperidinyl, piperazinyl, and the like; benzofused heteroaryl groups such as indolyl, indolinyl, benzothiazolyl, quinoline, quinoxaline, acridine, phenazine, 1,3-benzodioxole, 2,3dihydro-l,4- benzodioxine and the like; Wherein R1 represents hydrogen, alkyl groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t-butyl and the like; R2 represents such as hydrogen, -CN, -COOH, -COOCH3, -COOEt, -COCH3, and -CONH2; R, R3 and R4 may be same or different and independently represent a groups like hydrogen, hydroxy, alkyl, alkoxy groups such as methoxy, ethoxy, propoxy, n-butoxy, isobutoxy, t-butoxy and the like; acetyl; or optionally substituted cycloalkyl groups such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl and the like; aryl, aryloxy groups such as phenoxy and naphthoxy; benzyloxy; aralkyl groups; heteroaryl; heterocyclyl; benzofused heteroaryl groups. The terms alkyl, aryl, heteroaryl, heterocyclyl, and benzofused heteroaryl groups are as defined above.
Suitable groups substituted on A, R, R3, R4 and Q2 may be selected from halogens such as fluorine, chlorine, bromine, iodine; hydroxy; nitro; cyano; azido; nitroso; hydrazine; formyl; alkyl; alkoxy; haloalkyl; haloalkoxy; cycloalkyl; aryl; aryloxy; acyl; acyloxy; acyloxyacyl; heterocyclyl; heteroaryl; amino; monoalkylamino; dialkylamino; acylamino; alkoxycarbonyl; aryloxycarbonyl; alkylsulfonyl; arylsulfonyl; alkylsulfinyl; arylsulflnyl; alkylthio; arylthio; sulfamoyl; alkoxyalkyl groups and carboxylic acids or its derivatives; Wherein k, 1 and m are integers in the range of O to 2.
Furthermore A, R, R3, R4 and Q2 which are cyclic rings represents substituted or unsubstituted 5 to 10 membered ring systems, and also the rings may be monocyclic or bicyclic, saturated or partially saturated or aromatic containing 1 to 4 hetero atoms selected from O, S, N and the like.
Pharmaceutically acceptable salts include base addition salts such as alkali metal salts like Li, Na, and K salts; alkaline earth metal salts like Ca and Mg, salts of organic bases such as lysine, arginine, guanidine, diethanolamine, α-phenylethylamine, benzylamine, piperidine, morpholine, pyridine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, choline and the like, ammonium or substituted ammonium salts, aluminum salts. Salts also include amino acid salts such as glycine, alanine,
cystine, cysteine, lysine, arginine, phenylalanine, guanidine etc. Salts may include acid addition salts where appropriate, which are sulphates, nitrates, phosphates, perchlorates, borates, hydrohalides, acetates, tartrates, maleates, citrates, succinates, palmoates, methanesulphonates, tosylates, benzoates, salicylates, hydroxynaphthoates, benzenesulfonates, ascorbates, glycerophosphates, ketoglutarates and the like. Pharmaceutically acceptable solvates may be hydrates or comprising of other solvents of crystallization such as alcohols.
The term analog includes a compound, which differs from the parent structure by one or more C, N, O or S atoms. Hence, a compound in which one of the N atoms in the parent structure is replaced by an S atom is an analog of the former.
The term stereoisomer includes isomers that differ from one another in the way the atoms are arranged in space, but whose chemical formulas and structures are otherwise identical. Stereoisomers include enantiomers and diastereoisomers.
The term tautomers include readily interconvertible isomeric forms of a compound in equilibrium. The enol-keto tautomerism is an example.
The term polymorphs include crystallographically distinct forms of compounds with chemically identical structures.
The term pharmaceutically acceptable solvates includes combinations of solvent molecules with molecules or ions of the solute compound. The term derivative refers to a compound obtained from a compound according to formula (I), an analog, tautomeric form, stereoisomer, polymorph, hydrate, pharmaceutically acceptable salt or pharmaceutically acceptable solvate thereof, by a simple chemical process converting one or more functional groups, such as, by oxidation, hydrogenation, alkylation, esterification, halogenation, and the like. A term once described, the same meaning applies for it, throught the patent
Representative compounds include:
1. Benzothiazol-2-yl methyl 4-{4-[(l.E)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate; 2. Pyridin-3-yl methyl 4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
3. Thiophen-2-yl methyl 4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
4.Thiophen-3-yl methyl 4- {4-[( 12s)-3-(hydroxyamino)-3 -oxoprop- 1 -en- 1 -yl] phenyl } piperazine- 1 -carboxy late; 5. Thiazol-2-yl methyl 4-{4-[(12T)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl } piperazine- 1 -carboxy late; 6. 3,4-(Methylenedioxy)benzyl-4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l- yl] phenyl } piperazine- 1 -carboxylate;
7. 4-Methoxybenzyl-4-{4-[(liT)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
8. Thiophen-3-ylmethyl-4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] 2- fluorophenyl} piperazine- 1 -carboxylate;
9. Thiophen-2-yl methyl-4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] 2- fluorophenyl}piperazine-l-carboxylate;
10. 4-Methoxybenzyl- 4-{4-[( l.E)-3-(hydroxyamino)-3-oxoprop- 1 -en- 1 -yl] - 2-fluorophenyl } piperazine- 1 -carboxylate; 11. (2£)-2-Cyano-3-(4-{4-[(benzothiazol-2-yl methoxy)carbonyl]piperazin-l
-yl}phenyl)-N-hydroxy acrylamide;
12. (2£)-2-Cyano-3-(4-{4-[(4-methoxybenzyl methoxy)carbonyl]piperazin-l
-yl}phenyl)-N-hydroxy acrylamide;
13. (2£)-N-(2-Aminophenyl)-3-{4-(4-[([l,3]benzothiazol-2-yl methoxy) carbonyl] piperazin-1-yl) phenyl} acrylamide;
14.4-Methoxybenzyl-4-{4-[(lE)-3-(5-cyclopropyl-[l,3,4] thiadizol-2-yl amino)-3-oxo prop-1-en-l-yl] phenyl}piperazine-l-carboxylate;
15. 4-Methoxybenzyl-4-(4-{(lE)-3-[(2-aminophenyl)amino]-3-oxoprop-l- en- 1 -yl } -2-fluorophenyl)piperazine- 1 -carboxylate; 16. (2£)-N-(2-Amino phenyl)-3-{4-(4-[(2-thienyl methoxy) carbonyl] piperazin-1-yl) phenyl}acrylamide;
17. (2£)-N-(Thiazol-2-yl)-3-{4-(4-[(benzo thiazol-2-yl methoxy) carbonyl] piperazin- 1 -yl) phenyl} aery lamide;
18. 2-Thienylmethyl-4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenoxy } acety l)piperazine- 1 -carboxylate;
19. 2-Benzothiazolylmethyl-4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l- yl] phenoxy} acetyl)piperazine-l -carboxylate;
20. 3-Quinolinylmethyl -4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl]
phenoxy } acetyl)piperazine- 1 -carboxylate; 21. 3 -Thienylmethyl-4-( { 4-[( 1 E)-3 -(hydroxyamino)-3 -oxoprop- 1 -en- 1 -yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
22. 3,5 -Dimethoxybenzy l-4-( {4- [( 1 E)-3 -(hydroxyamino)-3 -oxoprop- 1 -en- 1 -yl] phenoxy } acetyl)piperazine- 1 -carboxylate;
23. 2,3-Dihydro-l,4-benzodioxin-2-yl methyl-4({4-[(lE)-3-[(2-aminophenyl) amino]-3-oxo prop-1-en-l-yl] phenoxy} acetyl) piperazine-1- carboxylate;
24. 2,3-Dihydro-l,4-benzodioxin-2-yl methyl-4-({4-[(l£)-3- (hydroxyamino)-3-oxoprop- 1 -en- 1 -yljphenoxy } acetyl)piperazine- 1 - carboxylate;
25. 4-Nitrobenzyl-4-({4-[( l£)-3-(hydroxyamino)-3-oxoprop- 1 -en- 1 -yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
26. 4-Trifluoromethylbenzyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -yl]phenoxy } acetyl)piperazine- 1 -carboxylate ;
27. 2-Benzothiazolylmethyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
28. 2-Quinolinylmethyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -yl] phenoxy } acetyl)piperazine- 1 -carboxylate; 29. (2£)-3-[4-(2-anilino-2-oxoethoxy)phenyl]-N-hydroxyacrylamide;
30. (2is)-N-(2-aminophenyl)-3-[4-(2-anilino-2-oxoethoxy)phenyl] acrylamide;
31. (2£)-3 -(4- { 2- [(4-fluorobenzy l)amino] -2-oxoethoxy } pheny l)-N-hydroxy acrylamide;
32. (2£)-3-(4-{2-[(3,4,5-trimethoxyphenyl)amino]-2-oxoethoxy}phenyl)-N -hydroxyacrylamide;
33. (2£)-3-(4-{2-[(3,4,5-trimethoxybenzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide;
34. (2£)-3-(4-{2-[(3-chloro-4-fluorobenzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide; 35. (2£)-3-(4-{2-[(4-trifluoromethoxy benzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide;
36. (2£)-3-[4-(2-anilino-l-methyl-2-oxoethoxy)phenyl]-N-hydroxyacrylamide;
37. (2£)-3-(4-{2-[(4-methoxy benzyl)amino]-2-oxo ethoxy}phenyl)-N-hydroxy
acrylamide;
38. (2£)-3-(4-{2-[(2,4-dimethoxy benzyl)amino]-2-oxo ethoxy}phenyl)-N -hydroxy acrylamide;
39. (2£)-3-(4-{2-[(3,5-difluorophenyl)amino]-2-oxoethoxy}phenyl)-N -hydroxyacrylamide;
40. (2£)-3-(4-{2-[(4-fluorophenyl)amino]-2-oxoethoxy}phenyl)-N -hydroxyacrylamide;
41. (22s)-N-hydroxy-3-[4-(2-{[2-(2-methoxyphenoxy)ethyl]amino}-2 -oxoethoxy)phenyl]acrylamide; 42. (22ϊ)-3-(4-{2-[(2-aminophenyl)amino]-2-oxoethoxy}phenyl)-N
-hydroxyacrylamide;
43. (2£)-N-(2-aminophenyl)-3-[4-(2-[(3,4,5-trimethoxybenzyl)amino]-2 -oxoethoxy)phenyl] acrylamide and
44. (2E)-3-[4-(2-{[4-(dimethylamino)phenyl]amino}-2-oxoethoxy)phenyl]- Ν-hydroxy prop-2-enamide.
According to another feature of the present invention, there is provided a process as shown in the following schemes, for the preparation of compounds of the formula (I), wherein all the groups are as defined earlier. a) By condensing the compound of formula (Ia) with 1,1'carbonyl diimidazole and a secondary amine to yield a compound of the formula (Ib), wherein R, k, 1, A and X are as defined earlier. b) By reacting the compound of the formula (Ib) with (Ic) to yield a compound of the general formula (Id), wherein L represents a group selected from halides, alkyl halide and k, X, 1, m, A and Q2 are as defined earlier. c) By reacting the compound of formula (Id) with an active methylene compound to yield a compound of the general formula (Ie), wherein k, X, 1, m, A, Q2, and R2 are as defined earlier. d) By reacting the compound of formula (Ie) with a primary or secondary amine to yield a compound of the general formula (I), wherein R3 and R4 are as defined earlier.
*

Formula ( I ) Scheme 1
The compound of the general formula (I) is prepared by the following procedure (Scheme 1): Step (I): The condensation of the compound of formula (Ia), with 1,1'carbonyl diimidazole and primary or secondary amine is carried out in the presence of solvents selected from toluene, DMF, tetrahydrofuran, chloroform, dichloromethane, dichloroethane, ethyl acetate, o-dichlorobenzene or a mixture thereof, in the presence of bases such as triethylamine, diethylamine, pyridine, DMAP and alkali carbonates such as sodium carbonate, potassium carbonate and the like to afford the compound of formula (Ib). The reaction is carried out at a temperature in the range of O0C to room temperature.
Step (II): The compound of formula (Ib) is reacted with the compound of formula (Ic) which include substituted 4-fluorobenzaldehyde, or 4-bromomethylbenzaldehyde in the presence of solvents such as toluene, tetrahydrofuran, DMF, chloroform, dichloromethane, dichloroethane, ethylacetate, o- dichlorobenzene or a mixture thereof in the presence of a base such as triethylamine, diethylamine, diisopropyl ethylamine, pyridine, DMAP (N,N dimethylaminopyridine), alkali carbonates such as sodium carbonate, potassium carbonate and the like to produce a compound of the formula (Id).
Step (III): The compound of formula (Id) is reacted with active methylene derivatives which include ethyl cyanoacetate, ethyl acetoacetate, or ethyl 4-chloro acetoacetate in the presence of solvents such as toluene, tetrahydrofuran, DMF, chloroform, dichloromethane, dichloroethane, ethylacetate, o-dichlorobenzene, pyridine or a mixture thereof in the presence of a base such as triethylamine, diethylamine, diisopropylethylamine, pyridine, piperidine, DMAP, alkali carbonates such as sodium carbonate, potassium carbonate and the like to produce a compound of the formula
(Ie).
Step (IV): The compound of formula (Ie) is reacted with a primary or secondary amine e.g. hydroxylamine hydrochloride, N-methoxy hydroxylamine hydrochloride, Ophenylenediamine, or 2-aminothiazole and an acid activating reagent such as BOP, DCC, EDCI, isobutyl chloroformate and the like in the presence of solvents mentioned above to afford a compound of the formula (I).
It is appreciated that in any of the above-mentioned reactions, any reactive group in the substrate molecule may be protected according to the conventional chemical practice. Suitable protecting groups in any of the above-mentioned reactions are those used conventionally in the art. The methods of formation and removal of such protecting groups are those conventional methods appropriate to the molecule being protected. The pharmaceutically acceptable salts are prepared by reacting the compound of formula (I) with 1 to 10 equivalents of a base such as sodium hydroxide, sodium methoxide, sodium hydride, potassium t-butoxide, calcium hydroxide, magnesium hydroxide and the like, in solvents like ether, tetrahydrofuran, methanol, t-butanol, dioxane, isopropanol, ethanol etc. Mixture of solvents may also be used. Organic bases such as diethanolamine, α-phenylethylamine, benzylamine, piperidine, morpholine, pyridine, hydroxyethylpyrrolidine, hydroxyethylpiperidine, choline, guanidine and the like, ammonium or substituted ammonium salts, aluminum salts. Amino acids such as glycine, alanine, cystine, cysteine, lysine, arginine, phenylalanine etc may be used for the preparation of amino acid salts. Alternatively, acid addition salts wherever applicable are prepared by treatment with acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, p-toluenesulphonic acid, methanesulfonic acid, acetic acid, citric acid, maleic acid, salicylic acid, hydroxynaphthoic acid, ascorbic acid, palmitic acid, succinic acid, benzoic acid,
benzenesulfonic acid, tartaric acid, oxalic acid and the like in solvents like ethyl acetate, ether, alcohols, acetone, tetrahydrofuran, dioxane etc. Mixture of solvents may also be used.
It should be noted that compounds of the invention may contain groups that may exist in tautomeric forms, and though one form is named, described, displayed and/or claimed herein, all the forms are intended to be inherently included in such name, description, display and/or claim.
The stereoisomers of the compounds forming part of this invention may be prepared by using reactants in their single enantiomeric form, in the process wherever possible or by conducting the reaction in the presence of reagents or catalysts in their single enantiomeric form or by resolving the mixture of stereoisomers by conventional methods. Some of the preferred methods include use of microbial resolution, resolving the diastereomeric salts formed with chiral acids such as mandelic acid, camphorsulfonic acid, tartaric acid, lactic acid, and the like wherever applicable or by using chiral bases such as brucine, cinchona alkaloids, their derivatives and the like.
Prodrugs of the compounds of formula (I) are also contemplated by this invention. A prodrug is an active or inactive compound that is modified chemically through in-vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following administration of the prodrug to a patient. The suitability and techniques involved in making and using prodrugs are well known to those skilled in the art.
Various polymorphs of the compounds of the general formula (I), forming part of this invention may be prepared by crystallization of the compounds of formula (I) under different conditions. For example, using different commonly used solvents, or their mixtures for recrystallization; crystallizations at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallizations. Heating or melting the compounds followed by cooling gradually or immediately, one can also obtain polymorphs. The presence of polymorphs may be determined by solid probe NMR spectroscopy, IR spectroscopy, differential scanning calorimetry and powder X-ray diffraction or other such techniques.
Pharmaceutically acceptable solvates of the compounds of the formula (I) forming part of this invention may be prepared by conventional methods such as dissolving the compounds of the formula (I) in solvents such as water, methanol,
ethanol, mixture of solvents such as acetone:water, dioxane:water, N,N- dimethylformamide:water and the like, preferably water and recrystallization by using different crystallization techniques
The present invention also provides a pharmaceutical composition, containing one or more of the compounds of the general formula (I) as defined above, their derivatives, analogs, tautomeric forms, stereoisomers, polymorphs, hydrates, metabolites, prodrugs, pharmaceutically acceptable salts, pharmaceutically acceptable solvates in combination with the usual pharmaceutically employed carriers, diluents and the like, useful for the treatment of cancer, psoriasis, proliferative conditions and conditions mediated by HDAC.
The derivatives provided by the present invention can be employed as pharmaceutical compositions, for example, in the form of pharmaceutical compositions containing the derivatives together with appropriate, pharmaceutically acceptable carriers. The products in accordance with the invention can be administered, for example, perorally, such as in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions, or rectally, such as in the form of suppositories, etc. The compositions may be sterilized and may contain auxiliary agents generally employed in the pharmaceutical art, such as sodium hydrogen carbonate, citric acid, propylene glycol, tween 80, etc. The compounds can be used orally or parenterally.
The pharmaceutical composition may be in the forms normally employed, such as tablets, capsules, powders, syrups, solutions, suspensions and the like, may contain flavorants, sweeteners etc. in suitable solid or liquid carriers or diluents, or in suitable sterile media to form injectable solutions or suspensions. The compositions may be prepared by processes known in the art, such as by combining the ingredients into a dosage form together with suitable, non-toxic, inert, therapeutically compatible solid or liquid carrier materials and if desired, the usual pharmaceutical adjuvants. The amount of the active ingredient in the composition may be less than 70% by weight. Such compositions typically contain from 1 to 25%, preferably 1 to 15% by~ weight of active compound, the remainder of the composition being pharmaceutically acceptable carriers, diluents, excipients or solvents.
Suitable pharmaceutically acceptable carriers include solid fillers or diluents and sterile aqueous or organic solutions. The active compound will be present in such
pharmaceutical compositions in the amounts sufficient to provide the desired dosage in the range as described above. Thus, for oral administration, the compounds can be combined with a suitable solid or liquid carrier or diluent to form capsules, tablets, powders, syrups, solutions, suspensions and the like. The pharmaceutical compositions, may, if desired, contain additional components such as flavorants, sweeteners, excipients and the like. For parenteral administration, the compounds can be combined with sterile aqueous or organic media to form injectable solutions or suspensions. For example, solutions in sesame or peanut oil, aqueous propylene glycol and the like can be used, as well as aqueous solutions of water-soluble pharmaceutically-acceptable acid addition salts or alkali or alkaline earth metal salts of the compounds. The injectable solutions prepared in this manner can then be, administered intravenously, intraperitoneal Iy, subcutaneously, or intramuscularly, with intramuscular administration being preferred in humans.
Generally, the effective dose for treating a particular condition in a patient may be readily determined and adjusted by the physician during treatment to alleviate the symptoms or indications of the condition or disease. Generally, a daily dose of active compound in the range of about 0.01 to 1000 mg/kg of body weight is appropriate for administration to obtain effective results. The daily dose may be administered in a single dose or divided into several doses. In some cases, depending upon the individual response, it may be necessary to deviate upwards or downwards from the initially prescribed daily dose. Typical pharmaceutical preparations normally contain from about 0.2 to about 500 mg of active compound of formula I and/or its pharmaceutically active salts or solvates per dose.
While the compounds of the invention can be administered as the sole active pharmaceutical agent, they can also be used in combination with one or more compounds of the invention or other agents. When administered as a combination, the therapeutic agents can be formulated as separate compositions that are given at the same time or different times, or the therapeutic agents can be given as a single composition. The term "therapeutically effective amount" or "effective amount" refers to that amount of a compound or mixture of compounds of Formula I that is sufficient to effect treatment, as defined below, when administered alone or in combination with other therapies to a mammal in need of such treatment.
The term "animal" as used herein is meant to include all mammals, and in particular humans. Such animals are also referred to herein as subjects or patients in need of treatment. The therapeutically effective amount will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the particular compound of Formula I chosen, the dosing regimen to be followed, timing of administration, the manner of administration and the like, all of which can readily be determined by one of ordinary skill in the art.
The term "treatment" or "treating" means any treatment of a disease in a mammal, including: a) Preventing the disease, that is, causing the clinical symptoms of the disease not to develop; b) Inhibiting the disease, that is, slowing or arresting the development of clinical symptoms; and/or c) Relieving the disease, that is, causing the regression of clinical symptoms.
From the foregoing description, one skilled in the art can easily ascertain the essential characteristics of this invention, and without departing from the spirit and scope thereof, make various changes and modifications of the invention to adapt it to various usages and conditions. The present invention is provided by the examples given below, which are provided by the way of illustration only, and should not be considered to limit the scope of the invention. Variation and changes, which are obvious to one skilled in the art, are intended to be within the scope and nature of the invention, which are defined in the appended claims. Example: 1
Synthesis of benzothiazol-2-ylmethyl 4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l- en-l-yl]phenyl}piperazine-l-carboxylate.

Preparation of benzothiazol-2-yImethyl piperazine-1-carboxylate.

Preparation of benzothiazol-2-ylmethyl 4-(4-formyIphenyl)piperazine-l- carboxylate.

Preparation of (2£)-3-(4-{4-[(benzothiazol-2-ylmethoxy)carbonyl]piperazin-l- yl}phenyl)acrylic acid

(0.76g, 2mmol) followed by piperidine (0.6mL, 6mmol). The reaction mixture was stirred for 2 hours at 130 0C. After completion of the reaction, the mixture was first added to cold water (10OmL) and then acidified with concentrated hydrochloric acid to pH-4. The yellow coloured solid that precipitated out was filtered and dried under vacuum to afford the title compound Ic (0.5Og, 59.5% yield). Stage-IV
Preparation of benzothiazol-2-ylmethyl 4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l- en-l-yl]phenyl}piperazine-l-carboxylate.

1.3mmol), HOBT (0.15g, l.lmol), hydroxylamine hydrochloride (0.09g, 1.3mmol) followed by diisopropylethylamine (0.6mL, 3.3mmol). The reaction mixture was stirred for 2 hours. After which, the mixture was added to cold water (2OmL). The solid precipitated was filtered, dried under vacuum and then triturated with chloroform (2OmL), followed by diethyl ether (4OmL) to afford the title compound 1 (0.1 Ig, 23% yield). 1H NMR (DMSO-d6) δ (ppm): 3.33 (4H, bs, -CH2), 3.56 (4H, d, -CH2), 5.52 (2H, s, -CH2), 6.24-6.28 (IH, d, =CH), 6.97-6.99 (2H, d, Ar-H), 7.34-7.38 (IH, d, =CH), 7.41-7.55 (4H, m, Ar-H), 7.99-8.01(1H, d, Ar-H), 8.11-8.13 (IH, d, Ar-H), 8.92(1H, s, D2O exchangeable), 1O.59(1H, s, D2O exchangeable). MS m/z: 439.1 (M++!). M.p: 176.9-179.1 0C
The following compounds were prepared according to the procedure given in Example: 1
 s, -
s, -
- d,
t,
d,
s, -


Synthesis of (2E)-Λr-(2-ainino phenyl)-3-{4-(4-[(benzo thiazoI-2-yl methoxy) carbonyl] piperazin-1-yl) phenyl} acrylamide.
Stage-I
Preparation of (2£)-N-(2-amino phenyl)-3-{4-(4-[(benzo thiazol-2-yl methoxy) carbonyl] piperazin-1-yl) phenyljacrylamide.

To a suspension of (2£)-3-(4-{4-[(benzothiazol-2-ylmethoxy) carbonyl]piperazin-l-yl}phenyl)acrylic acid (Ic) (0.2Og, 0.47mmol, prepared according to the procedure described in example 1, stage III) in THF (5 mL) was added EDCI
(0.16g, 0.85mmol), HOBT (0.06g, 0.47mmol), o-phenylenediamine (0.05g, 0.43mmol), followed by triethylamine (0.2mL, 1.4mmol). The reaction mixture was stirred for 4 hours after which the mixture was added to cold water (2OmL). The yellow solid precipitated was filtered, dried under vacuum and finally triturated with diethyl ether
(2OmL) to afford the title compound (14) (0.05g, 21%yield). 1H NMR (DMSO-d6) δ
(ppm): 3.34 (4H, t, -CH2), 3.63 (4H, t, -CH2), 4.92 (2H, s, D2O exchangeable), 6.53
(2H, s, CH2), 6.58 (IH, d, =CH), 6.74 (2H, d, Ar-H), 6.89 (IH, d, Ar-H), 7.01 (2H, d,
Ar-H), 7.33 (IH, d, =CH), 7.51 (5H, m, Ar-H), 8.01 (IH, d, Ar-H), 8.13 (lH,d, Ar-H), 9.25 (IH, s, D2O exchangeable). MS m/z: 514.2 (M++!). m.p: 202.2-205.2 0C.
The following compounds were prepared according to the procedure given in Example: 13
m, - d,
s,
s,


Example: 18
Synthesis of 2-thienylmethyI-4-({4- [(12i)-3-(hydroxyamino)-3-oxoprop-l-en-l- yl]phenoxy}acetyl)piperazine-l-carboxylate.

Stage-I Preparation of 2-thieny lmethyl 4-(bromoacetyl) piperazine-1-carboxylate.

To a solution of 2-thienylmethyl piperazine-1-carboxylate (0.6Og, 2.7 mmol, prepared according to the procedure of example-1, stage-I) in DCM (15mL) at 50C was added bromoacetyl bromide (0.64g, 3.2mmol), drop-by-drop and was allowed to stir for 5 minutes. The reaction mixture was further stirred for 15 minutes at room temperature, subsequently it was diluted with ethyl acetate (15OmL), the organic layer washed with water (2 x 75mL), followed by brine solution (10OmL) and finally dried over anhydrous sodium sulfate followed by evaporation to dryness to afford the title compound 18a as a yellow solid (0.7Og, 71% yield).
Stage-II
Preparation of 2-thienylmethyl-4-({4-[(l£)-3-ethoxy-3-oxoprop-l-en-l-yl] phenoxy}acetyl)piperazine-l-carboxylate.

To a suspension of NaH (0.05g, 2.3mmol) in DMF (0.5mL) at 0 0C was added ethyl (2£)-3-(4-hydroxyphenyl)acrylate (18a) (0.34g, 1.8 mmol) in DMF (0.5mL) drop by drop with stirring. The stirring was continued for half an hour at 0 0C. 2- Thienylmethyl-4-(bromoacetyl)piperazine-l-carboxylate (0.60g,1.6 mmol) in DMF (ImL), catalytic amount of 18-crown-6 was then added slowly to the above reaction mixture at 0 0C. The reaction mixture was allowed to stir for three hours at room temperature. Subsequently it was quenched with methanol (3mL), diluted with water (15OmL) and extracted with ethyl acetate (3 x 10OmL). The organic layer was washed with water (2 x 10OmL), brine solution (10OmL), dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by column chromatography using silica gel and 0.2% methanol in DCM as an eluent to give 2-thienylmethyl-4-({4- [( 1 £)-3 -ethoxy-3-oxoprop- 1 -en- 1 -yl]phenoxy} acetyl)piperazine- 1 -carboxylate (18b) as a yellow sticky mass (0.4Og, 32% yield). Stage-Ill Preparation of (2£)-3-(4-{2-[4-(2-thienylmethoxycarbonyl)piperazin-l-yl]-2- oxoethoxy}phenyl)acrylic acid.

To a solution of 2-thienylmethyl- 4-({4-[(lE)-3-ethoxy-3-oxoprop-l-en-l- yl]phenoxy}acetyl)piperazine-l -carboxylate 18b (0.4Og, 0.87mmol) in ethanol (10 mL) was added, a solution of NaOH (0.1 Ig, 2.8mmol) in water (0.5mL). The reaction mixture was stirred for 2 hours at 70 0C. The solvent was completely removed by evaporation and the pasty mass obtained was dissolved in water and adjusted to a pH of
6.0-6.5 using aqueous HCl. The solid that precipitated out was filtered and dried under vacuum to give the product (18c) as an off-white solid (0.25g, 66% yield).
Stage-IV
Preparation of 2-thienylmethyl-4-({4-[(l£)-3-(hydroxyainino)-3-oxoprop-l-en-l- yl]phenoxy}acetyl)piperazine-l-carboxylate.

To a suspension of 18c (0.5Og, 1.2mmol) in THF (1OmL) was added BOP (0.62g, 1.4mmol), HOBT (0.16g, 1.2mmol), hydroxylamine hydrochloride (0.097 g, 1.4mmol) followed by diisopropylethylamine (0.6mL, 3.5mmol). The reaction mixture was stirred for 2 hours after which the mixture was added to water (10OmL) and extracted with ethyl acetate (2 x 10OmL). The organic layer was washed with water (2 x 10OmL) and brine solution (10OmL), dried over anhydrous sodium sulfate and evaporated to dryness. The crude material was purified by column chromatography using silica gel and 1.2% methanol in DCM as an eluent to afford title compound (18). 1H NMR (DMSO) δ(ppm): 3.06-3.17 (4H, m, -CH2), 3.34-3.45 (4H, m, -CH2), 4.89 (2H, s, -CH2), 5.27 (2H, s, -CH2), 6.31(1H, d, =CH), 6.93-6.96 (2H, d, Ar-H), 7.01-7.03 (IH, t, Ar-H), 7.16 (IH, s, Ar-H), 7.40 (IH, d, =CH), 7.47-7.55 (3H, m, Ar-H), 8.98 (IH, s, D2O exchangeable), 10.67 (IH, s, D2O exchangeable), MS m/z: 446.1 (M++!). The following compounds were prepared according to the procedure given in Example: 18

t,
s, - d,
s, - - d,
d,
 m, d, s, -
m, d, s, -


28 1H NMR (DMSOd6) δ (ppm): 3.50 (8H, bs, CH2), 4.92-4.94 (4H, d, -CH2&-NH2),
 5.34 (2H, s, -CH2), 6.57 (IH, t, =CH), 6.74- 6.78 (2H, t, Ar-H), 6.91-7.00 (3H, m, Ar- H), 7.32-7.34 (IH, d, Ar-H), 7.48-7.56 (3H, q, Ar-H&=CH), 7.64-7.66 (IH, d, Ar-H), 7.77-7.79 (IH, d, Ar-H) 8.01-8.05 (2H, t, Ar-H), 8.37 (IH, s, Ar-H), 8.94-8.95 (LH, d, Ar-H) 9.33 (IH, s, - D2O exchangeable). MS m/z: 566.2 (M++!).
5.34 (2H, s, -CH2), 6.57 (IH, t, =CH), 6.74- 6.78 (2H, t, Ar-H), 6.91-7.00 (3H, m, Ar- H), 7.32-7.34 (IH, d, Ar-H), 7.48-7.56 (3H, q, Ar-H&=CH), 7.64-7.66 (IH, d, Ar-H), 7.77-7.79 (IH, d, Ar-H) 8.01-8.05 (2H, t, Ar-H), 8.37 (IH, s, Ar-H), 8.94-8.95 (LH, d, Ar-H) 9.33 (IH, s, - D2O exchangeable). MS m/z: 566.2 (M++!).
Example: 29
Synthesis of (2£)-3-[4-(2-anilino-2-oxoethoxy)phenyl]-Λr-hydroxyacrylamide

Sta2e-I
Preparation of 2-bromo-7V-phenylacetamide.

29a
To a solution of bromo acetyl bromide (3.3g, lόmmol) in DCM (15mL) was added aniline (1.5g, lόmmol) in the DCM (5mL) at 5 0C and the mixture was stirred at room temperature for 0.5 hour. After stirring for 0.5 hour at room temperature the mixture was evaporated to remove the DCM and was then diluted with water (10OmL). The pale brown solid that precipitated out was filtered, washed with water (50OmL) and dried to afford the title compound 29a (2.7Og, 79 % yield). Stage-II Preparation of methyl (2£)-3-[4-(2-anilino-2-oxoethoxy)phenyl]acrylate.

To a suspension of sodium hydride (60% w/w, 0.43g, 18mmol) in DMF (5 mL) was added methyl-4-hydroxy cinnamate (2.3g, 13mmol) in DMF (6mL) at 5 0C and the reaction mixture was stirred for 30 minutes. A solution of 29a (2.72g, 13mmol) in DMF (5mL) was added drop wise to the above reaction mixture at 0 0C and it was allowed to warm up to room temperature and was stirred at room temperature for 4 hours. The reaction mixture was diluted with ice water (50OmL and the precipitated solid was filtered and washed with water (20OmL), dried under vacuum to afford the title compound (29b) as pale brown colored solid (3.6g, 91% yield). Stage-Ill Preparation of (2£)-3-[4-(2-anilino-2-oxoethoxy)phenyl]-Λ'-hydroxy acrylamide.

Hydroxylamine hydrochloride (2g, 29mmol) in methanol (3mL) was mixed with KOH (1.6g, 29mmol) in methanol (2mL) at 0 0C, and the reaction mixture was sonicated for 2 minutes. Subsequently the white precipitate formed was filtered; the filtrate was added to 29b (0.5g, 1.6 mmol) in DCM (5mL) and the mixture was stirred at 30 0C for 30 minutes. The reaction mixture was then diluted with water (20OmL)
and extracted with ethyl acetate (2 x 15OmL), the ethyl acetate layer was dried over anhydrous Na2SO4 and concentrated to obtain a solid compound, which was triturated with DCM (15mL). The pale brown solid obtained was filtered and washed with DCM (thrice) to afford the title compound 29 (0.2g, 40% yield). 1H NMR (DMSOd6) δ (ppm): 4.75 (2H, s, -CH2), 6.31-6.35 (IH, d, =CH), 7.02-7.10 (3H, m, Ar-H), 7.30-7.34 (IH, t, Ar-H), 7.39-7.43 (IH, d, =CH), 7.52-7.54 (2H, d, Ar-H), 7.62-7.64 (2H, d, Ar- H), 8.98 (IH, s, - D2O exchangeable), 10.12 (IH, s, - D2O exchangeable), 10.68 (IH, s, - D2O exchangeable). MS m/z: 313.1 (M++!). Stage-IV Preparation of acid.

To a solution of 29b (0.7g, 2.3mmol) in methanol (1OmL) was added, a solution of NaOH (0.13g, 3.2mmol) in water (0.5 mL). The reaction mixture was stirred for 1 hour at 70 0C. The solvent was completely removed by evaporation, diluted with water (50 mL) and extracted with ethyl acetate (2 X 5OmL). The aqueous layer was acidified to pH 2 with dilute aqueous HCl (1:1) and was allowed to stand at 4 0C for 30 minutes, the solid precipitated out was filtered and dried under vacuum to give 29c as a white solid (0.35g, 52% yield). Example 30 (Stage- V)
Synthesis of (2£)-7V-(2-aminophenyl)-3-[4-(2-anilino-2-oxoethoxy)phenyl] acrylamide.
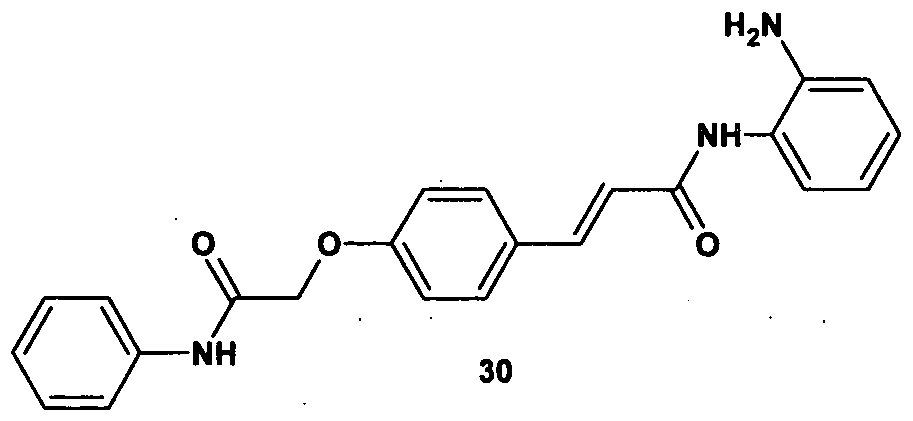
To a solution of 29c (0.15g, 0.5mmol) in DMF (8mL) was added EDCI (0.19g, 1 mmol), HOBt (0.07g, 0.5mmol), o-phenylenediamine (0.05g, 0.45mmol), followed by triethylamine (0.23mL, 1.5mmol). The reaction mixture was stirred for 1 hour after
which the mixture was added to cold water (2OmL). The aqueous layer was extracted with ethyl acetate (1 x 15OmL), washed with water (2 x 50 mL), brine (1 x 10OmL). The organic layer was dried over anhydrous Na2SO4 and concentrated to obtain the crude compound, which was triturated with ethyl acetate (20 mL) to afford title compound (29) as a yellow solid (0.05g, 26% yield). 1H NMR (DMSO-d6) δ (ppm): 4.79 (2H, s, -CH2), 4.94 (2H, s, -NH2), 6.58 (IH, t, =CH), 6.74-6.76 (2H, t, Ar-H), 6.9 (IH, t, Ar-H) 7.06-7.09 (3H, q, Ar-H), 7.31-7.35 (3H, q, Ar-H), 7.49-7.53 (IH, d, =CH), 7.58-7.65 (4H, m, Ar-H), 9.33 (IH, s, D2O exchangeable), 10.13 (IH, s, - D2O exchangeable). MS m/z: 388.2 (M++!).
The following compounds were prepared according to the procedure given in Example: 29 ,
(2H,
s, s, -


1H NMR (DMSO-de) δ (ppm): 4.31-4.33 (2H, d, -CH2), 4.61 (2H, s, -CH2), 6.30-
 6.34 (IH, d, =CH), 6.98-7.00 (2H, d, Ar- H), 7.26-7.27 (IH, d, Ar-H), 7.32-7.42 (3H, m, Ar-H &=CH), 7.51-7.53 (2H, d, Ar-H), 8.72 (IH, s, - D2O exchangeable), 8.92 (IH, t, - D2O exchangeable), 10.62 (IH, s, - D2O exchangeable). MS m/z: 379.1 (M++!).
6.34 (IH, d, =CH), 6.98-7.00 (2H, d, Ar- H), 7.26-7.27 (IH, d, Ar-H), 7.32-7.42 (3H, m, Ar-H &=CH), 7.51-7.53 (2H, d, Ar-H), 8.72 (IH, s, - D2O exchangeable), 8.92 (IH, t, - D2O exchangeable), 10.62 (IH, s, - D2O exchangeable). MS m/z: 379.1 (M++!).
1H NMR (DMSO-d6) δ (ppm): 4.36-4.37 (2H, d, -CH2), 4.60 (2H, s, -CH2), 6.31-
 6.35 (IH, d, =CH), 6.94-7.01 (2H, d, Ar- H), 7.29-7.31 (2H, d, Ar-H), 7.36-7.42 (3H, m, Ar-H &=CH), 7.50-7.52 (2H, d, Ar-H), 8.73 (IH, s, - D2O exchangeable), 10.02 (IH, s, - D2O exchangeable), MS m/z: 411.1 (M++!).
6.35 (IH, d, =CH), 6.94-7.01 (2H, d, Ar- H), 7.29-7.31 (2H, d, Ar-H), 7.36-7.42 (3H, m, Ar-H &=CH), 7.50-7.52 (2H, d, Ar-H), 8.73 (IH, s, - D2O exchangeable), 10.02 (IH, s, - D2O exchangeable), MS m/z: 411.1 (M++!).
1H NMR (DMSO-d6) δ (ppm): 1.55-1.57 (3H, d, -CH3), 4.91-4.95 (IH, q, -CH), 6.29-6.33 (IH, d, =CH), 6.97-6.99 (2H, d,
 Ar-H), 7.05-7.09 (IH, t, Ar-H), 7.29-7.31 (2H, m, Ar-H), 7.36-7.40 (IH, d, =CH), 7.50-7.52 (2H, d, Ar-H), 7.58-7.62 (2H, t,
-
Ar-H), 7.05-7.09 (IH, t, Ar-H), 7.29-7.31 (2H, m, Ar-H), 7.36-7.40 (IH, d, =CH), 7.50-7.52 (2H, d, Ar-H), 7.58-7.62 (2H, t,
-
s, (2H,
s,
s,
s,
d,
 s,
s,
s, - d, - -


Anti-cancer experimental methods
Anti-cancer screen: Experimental drugs are screened for anti-cancer activity in three cell lines for their GI50, TGI and LC50 values (using five concentrations for each compound). The cell lines are maintained in DMEM containing 10% fetal bovine serum. 96 well micro titer plates are inoculated with cells in 100 μL for 24 hours at 37°C, 5% CO2, 95% air and 100% relative humidity. 5000 HCT 116 cells/well, 5000 NCIH 460 cells/well and 5000 U251 cells/well are plated. A separate plate with these cell lines is also inoculated to determine cell viability before the addition of the compounds (T0). Addition of experimental drugs:
Following 24-hour incubation, experimental drugs are added to the 96 well plates. Each plate contains one of the above cell lines and the following in triplicate: five different concentrations (0.01, 0.1, 1, 10 and 100 μM) of four different compounds, appropriate dilutions of a cytotoxic standard and control (untreated) wells. Compounds are dissolved in DMSO to make 20 mM stock solutions on the day of drug addition and frozen at -200C. Serial dilutions of these 20 mM stock solutions are made in complete growth medium such that 100 μL of these drug solutions in medium, of final concentrations equaling 0.01, 0.1, 1, 10 and 100 μM can be added to the cells in triplicate. Standard drugs whose anti-cancer activity has been well documented and which are regularly used are doxorubicin and SAHA.
End-point measurement;
For To measurement, 24 hours after seeding the cells, 10 μL of 3-(4,5-dimethyl- 2-thiazolyl)-2,5-diphenyl-2H-tetrazolium (MTT) solution per well is added and incubation carried out for 3 hours at 37°C, 5% CO2, 95% air and 100% relative humidity, protected from light. Cells incubated with compounds for 48 hours are treated similarly except with the addition of 20μL MTT solution per well and a subsequent incubation under the same conditions. After 3 hours of MTT incubation, well contents are aspirated carefully followed by addition of 150 μL DMSO per well. Plates are agitated to ensure solution of the formazan crystals in DMSO and absorbance read at 570 nm.
Calculation of GIsn. TGI and LCsn:
Percent growth is calculated for each compound's concentration relative to the control and zero measurement wells (To; viability right before compound addition). If a test well's O.D. value is greater than the T0 measurement for that cell line % Growth = (test - zero) / (control - zero) X 100
If a test well's O.D. value is lower than the T0 measurement for that cell line, then, % Growth = (test - zero) / zero X 100
Plotting % growth versus experimental drug concentration, GI50 is the concentration required to decrease % growth by 50%; TGI is the concentration required to decrease % growth by 100% and LC50 is the concentration required to decrease % cell death by 50%. HDAC Activity screening:
Histone Deacetylase (HDAC) Inhibition Assay using Boc-Lys (Ac)-AMC Substrate: Inhibition of HDAC has been implicated to modulate transcription and to induce apoptosis or differentiation in cancer cells. The fluorometric assay provides a fast and fluorescence based method that eliminates radioactivity, extractions or chromatography, as used in traditional assays. The assay is based on two steps. First, the HDAC fluorometric substrate, which comprises an acetylated lysine side chain, is incubated with a sample containing HDAC activity (Mouse Liver Extract). Deacetylation of the substrate sensitizes the substrate, in the second step; treatment with the Trypsin stop solution produces a fluorophore that can be easily analyzed using fluorescence plate reader.
Assay was done in 96 well black microplate and total volume of the assay is 100 μL. Mouse liver enzyme is diluted 1:6 with HDAC buffer. Enzyme cocktail made of 10 μL of diluted enzyme and 30 μL of HDAC buffer. 40 μL of enzyme cocktail dispensed into each well. 10 μL of different concentrations of inhibitor added in to each well, except enzyme control well. Preincubated the plate at 30 0C for 5 minutes. The HDAC reaction is started by adding 50 μL of HDAC substrate (βoc-Lys (Ac)-AMC Substrate) solution. Incubated the plate at 30 0C for 30 minutes. Adding 100 μL of Trypsin stop solution stops the reaction. The plate is incubated again at 30 0C for 20-30 minutes. The release of AMC is monitored by measuring the fluorescence at excitation wavelength of 365 or 360 nm and emission wavelength of 440 or 460 nm. Buffer and substrate alone kept for blank subtraction. (Dennis Wegener et al, Anal. Biochem, 321, 2003, 202-208).
Representative results of growth are shown below in the Table I. Table- 1:


Claims
1. Novel compounds of the general formula (I),

A represents saturated or unsaturated heterocyclyl groups containing one or more hetero atoms selected from O, S and N comprising pyrrolidinyl, thiazolidinyl, oxazolidinyl, morpholinyl, thiomorpholinyl, piperidinyl, piperazinyl, pyridyl, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, isooxazolyl, oxadiazolyl, triazolyl, thiadiazolyl, tetrazolyl, pyrimidinyl, pyrazinyl and pyridazinyl;
Q1 represents a bond, -NH, -CO, -CH2-, -CONH-, -CO-CH2-O-, -NHCO- CH(CH3)-O- and -NH-CO-CH2-O-;
Q2 represents optionally substituted aryl groups comprising phenyl and naphthyl; aralkyl groups comprising benzyl; heteroaryl groups comprising pyridyl, thienyl, furyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, isooxazolyl, oxadiazolyl, triazolyl, thiadiazolyl, tetrazolyl, pyrimidinyl, pyrazinyl and pyridazinyl; heterocyclyl groups comprising pyrrolidinyl, thiazolidinyl, oxazolidinyl, morpholinyl, thiomorpholinyl, piperidinyl and piperazinyl; benzofused heteroaryl groups comprising indolyl, indolinyl, benzothiazolyl, quinoline, quinoxaline, acridine, phenazine, 1,3- benzodioxole and 2,3dihydro-l,4-benzodioxine;
Ri represents hydrogen, alkyl groups comprising methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl and t-butyl;
R2 represents hydrogen, -CN, -COOH, -COOCH3, -COOEt, -COCH3, and - CONH2;
R, R3 and R4 are same or different and independently represent hydrogen; hydroxy; alkyl; alkoxy groups comprising methoxy, ethoxy, propoxy, n-butoxy, isobutoxy and t-butoxy; acetyl; optionally substituted cycloalkyl groups comprising cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl; aryl; aryloxy groups comprising phenoxy and naphthoxy; benzyloxy; aralkyl groups; heteroaryl; heterocyclyl; benzofused heteroaryl groups; the terms alkyl, aryl, heteroaryl, heterocyclyl, and benzofused heteroaryl groups are as defined above; k, 1 and m are integers in the range of 0 to 2; suitable groups substituted on A, R, R3, R4 and Q2 are selected from halogens comprising fluorine, chlorine, bromine, iodine; hydroxy; nitro; cyano; azido; nitroso; hydrazine; formyl; alkyl; alkoxy; haloalkyl; haloalkoxy; cycloalkyl; aryl; aryloxy; acyl; acyloxy; acyloxyacyl; heterocyclyl; heteroaryl; amino; monoalkylamino; dialkylamino; acylamino; alkoxycarbonyl; aryloxycarbonyl; alkylsulfonyl; arylsulfonyl; alkylsulfinyl; arylsulfinyl; alkylthio; arylthio; sulfamoyl; alkoxyalkyl groups; carboxylic acids and its derivatives;
A, R, R3, R4 and Q2 are cyclic rings, that are substituted or unsubstituted 5 to 10 membered ring systems, monocyclic or bicyclic, saturated, partially saturated or aromatic, containing 1 to 4 hetero atoms selected from O, S and N.
2. A compound of formula (I) as defined in claim 1 selected from the group consisting of:
Benzothiazol-2-yl methyl 4- {4-[( 1 £)-3-(hydroxyamino)-3-oxoprop- 1 -en- 1 -yl] phenyl } piperazine- 1 -carboxylate ; Pyridin-3-yl methyl 4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
Thiophen-2-yl methyl 4- {4-[( lE)-3-(hydroxyamino)-3-oxoprop- 1 -en- 1 -yl] phenyl }piperazine-l -carboxylate;
Thiophen-3-yl methyl 4-{4-[(lis)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
Thiazol-2-yl methyl 4-{4-[(l.E)-3-(hydroxyarnino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
3,4-(Methylenedioxy)benzyl-4-{4-[(12i)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl } piperazine- 1 -carboxylate; 4-Methoxybenzyl-4-{4-[(ljE)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenyl} piperazine- 1 -carboxylate;
Thiophen-3 -y lmethyl-4- { 4- [( 1 £)-3-(hydroxyam ino)-3 -oxoprop- 1 -en- 1 -yl] 2- fluorophenyl } piperazine- 1 -carboxylate; Thiophen-2-ylmethyl-4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl]2- fluorophenyl}piperazine-l-carboxylate;
4-Methoxybenzyl-4-{4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl]-2- fluorophenyl } piperazine- 1 -carboxylate ; (2£)-2-Cyano-3-(4- {4-[(benzothiazol-2-ylmethoxy)carbonyl]piperazin- 1 - yl}phenyl)-N-hydroxy acrylamide;
(2£)-2-Cyano-3-(4-{4-[(4-methoxybenzylmethoxy)carbonyl]piperazin-l- yl}phenyl)-N-hydroxy acrylamide;
4-Methoxybenzyl-4-{4-[(lE)-3-(5-cyclopropyl-[l,3,4] thiadizol-2-yl amino)-3-oxo prop-l-en-l-yl] phenyl}piperazine-l-carboxylate;
(2^-N-(2-Aminophenyl)-3-{4-(4-[([l,3]benzothiazol-2-yl methoxy) carbonyl] plperazin-1-yl) phenyl} acrylamide;
4-Methoxybenzyl-4-(4- { ( 1 E)-3-[(2-aminophenyl)amino]-3-oxoprop- 1 - en- 1 -yl } -2-fluorophenyl)piperazine- 1 -carboxylate; (2£)-N-(2-Amino phenyl)-3-{4-(4-[(2-thienyl methoxy) carbonyl] piperazin-1-yl) phenyl}acrylamide;
(2£)-N-(Thiazol-2-yl)-3-{4-(4-[(benzo thiazol-2-yl methoxy) carbonyl] piperazin-1-yl) phenyl} acrylamide;
2-Thienylmethyl-4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
2-Benzothiazolylmethyl-4-( {4-[( 1 £)-3-(hydroxyamino)-3-oxoprop- 1 -en- 1 - yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
3-Quinolinylmethyl -4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenoxy} acetyl)piperazine- 1 -carboxylate ; 3-Thienylmethyl-4-({4-[(l£)-3-(hydroxyamino)-3-oxoprop-l-en-l-yl] phenoxy} acetyl)piperazine- 1 -carboxylate ;
3 ,5 -Dimethoxybenzy l-4-( {4-[( 1 £)-3 -(hydroxyamino)-3 -oxoprop- 1 -en- 1 -yl] phenoxy } acetyl)piperazine- 1 -carboxylate;
2,3 -Dihydro- 1 ,4-benzodioxin-2-ylmethyl-4( {4-[( 1 E)-3-[(2-aminophenyl) amino]-3-oxo prop-1-en-l-yl] phenoxyjacetyl) piperazine- 1 -carboxylate;
2,3-Dihydro-l,4-benzodioxin-2-ylmethyl-4-({4-[(l£)-3-
(hydroxyamino)-3-oxoprop- 1 -en- 1 -yl]phenoxy } acetyl)piperazine- 1 - carboxylate; 4-Nitrobenzyl-4-( {4-[( 1 E)-3 -(hydroxyamino)-3 -oxoprop- 1 -en- 1 -y 1] phenoxy } acety l)piperazine- 1 -carboxylate ;
4-Trifluoromethylbenzyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -yl]phenoxy} acetyl)piperazine- 1 -carboxylate; 2-Benzothiazolylmethyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -yl] phenoxy} acetyl)piperazine- 1 -carboxylate;
2-Quinolinylmethyl-4-({4-[(lE)-3-[(2-aminophenyl)amino]-3- oxoprop- 1 -en- 1 -y 1] phenoxy} acetyl)piperazine- 1 -carboxylate ;
(2£)-3-[4-(2-anilino-2-oxoethoxy)phenyl]-N-hydroxyacrylamide; (2£)-N-(2-aminophenyl)-3-[4-(2-anilino-2-oxoethoxy)phenyl] acrylamide;
(2£)-3-(4-{2-[(4-fluorobenzyl)amino]-2-oxoethoxy}phenyl)-N-hydroxy acrylamide;
(2£)-3-(4-{2-[(3,4,5-trimethoxyphenyl)amino]-2-oxoethoxy}phenyl)-N- hydroxyacrylamide; (2£)-3-(4-{2-[(3,4,5-trimethoxybenzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide;
(2£)-3-(4-{2-[(3-chloro-4-fluorobenzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide;
(2£)-3-(4-{2-[(4-trifluoromethoxy benzyl)amino]-2-oxoethoxy}phenyl)-N- hydroxy acrylamide;
(2£)-3-[4-(2-anilino-l-methyl-2-oxoethoxy)phenyl]-N-hydroxyacrylamide;
(2^)-3-(4-{2-[(4-methoxy benzyl)amino]-2-oxo ethoxy}phenyl)-N-hydroxy acrylamide;
(2£)-3-(4-{2-[(2,4-dimethoxy benzyl)amino]-2-oxo ethoxy}pheny I)-N- hydroxy acrylamide;
(2£)-3-(4-{2-[(3,5-difluorophenyl)amino]-2-oxoethoxy}phenyl)-N- hydroxyacrylamide;
(2£)-3-(4-{2-[(4-fluorophenyl)amino]-2-oxoethoxy}phenyl)-N hydroxyacrylamide; (2JE)-N-hydroxy-3-[4-(2-{[2-(2-methoxyphenoxy)ethyl]amino}-2- oxoethoxy)phenyl]acrylamide;
(2£)-3-(4-{2-[(2-aminophenyl)amino]-2-oxoethoxy}phenyl)-N- hydroxyacrylamide; (2£)-N-(2-aminophenyl)-3-[4-(2-[(3,4,5-trimethoxyberizyl)amino]-2- oxoethoxy)phenyl] acrylamide and
(2E)-3-[4-(2-{[4-(dimethylamino)phenyl]amino}-2-oxoethoxy)phenyl]- Ν-hydroxy prop-2-enamide.
3. A pharmaceutical composition comprising a compound of formula (I) as claimed in claim 1, as an active ingredient along with a pharmaceutically acceptable carrier, diluent, excipient or solvate.
4. The pharmaceutical composition according to claim 3, wherein the said composition is in the form of a tablet, capsule, powder, syrup, solution, aerosol or suspension.
5. The pharmaceutical composition as claimed in claim 3, wherein the amount of the compound of claim 1 in the composition is less than 70 % by weight.
6. A method of inhibiting HDAC in a cell comprising treating said cell with an effective amount of a compound according to claim 1.
7. A method for the treatment of a condition mediated by HDAC comprising administering to a subject suffering from a condition mediated by HDAC a therapeutically effective amount of a compound according to the claim 1.
8. A method for the treatment of a proliferative condition comprising administering to a subject suffering from a proliferative condition a therapeutically effective amount of a compound according to claim 1, to the mammal in need thereof.
9. A method for the treatment and/or prevention of cancer comprising administering to a subject suffering from cancer a therapeutically effective amount of a compound according to claim 1, to the mammal in need thereof.
10. A method for the treatment of psoriasis comprising administering to a subject suffering from psoriasis a therapeutically effective amount of a compound according to claim 1, to the mammal in need thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN92CH2007 | 2007-01-17 | ||
| IN92/CHE/2007 | 2007-01-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008087514A2 true WO2008087514A2 (en) | 2008-07-24 |
| WO2008087514A3 WO2008087514A3 (en) | 2009-02-19 |
Family
ID=39636436
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2008/000045 Ceased WO2008087514A2 (en) | 2007-01-17 | 2008-01-10 | Hdac inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008087514A2 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2236503A1 (en) * | 2009-04-03 | 2010-10-06 | NatureWise Biotech & Medicals Corporation | Cinamic compounds and derivatives therefrom for the inhibition of histone deacetylase |
| CN101857554A (en) * | 2009-04-03 | 2010-10-13 | 彦臣生技药品股份有限公司 | Cinnamon compounds and derivatives thereof inhibiting histone deacetylase |
| JP2011526936A (en) * | 2008-09-17 | 2011-10-20 | エフ.ホフマン−ラ ロシュ アーゲー | Novel ortho-aminoanilides for the treatment of cancer |
| WO2013154870A1 (en) | 2012-04-10 | 2013-10-17 | Annji Pharmaceutical Co., Ltd. | Histone deacetylases (hdacs) inhibitors |
| US9745253B2 (en) | 2015-03-13 | 2017-08-29 | Forma Therapeutics, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| WO2018213364A1 (en) | 2017-05-16 | 2018-11-22 | Annji Pharmaceutical Co., Ltd. | Histone deacetylases (hdacs) inhibitors |
| WO2021252475A1 (en) | 2020-06-08 | 2021-12-16 | Annji Pharmaceutical Co., Ltd. | Quinazoline derivatives useful as selective hdac6 inhibitors |
| CN116375698A (en) * | 2023-06-05 | 2023-07-04 | 中国林业科学研究院林产化学工业研究所 | Novel urushiol-based hydroxamic acid type HDAC inhibitor with targeted antitumor activity and preparation method and application thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4894386A (en) * | 1987-04-15 | 1990-01-16 | Ici Americas Inc. | Aliphatic carboxamides |
| US20040180889A1 (en) * | 2002-03-01 | 2004-09-16 | Pintex Pharmaceuticals, Inc. | Pin1-modulating compounds and methods of use thereof |
| CN101001851B (en) * | 2004-08-09 | 2011-04-20 | 安斯泰来制药有限公司 | Hydroxyamide compounds having activity as inhibitors of histone deacetylase (HDAC) |
-
2008
- 2008-01-10 WO PCT/IB2008/000045 patent/WO2008087514A2/en not_active Ceased
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011526936A (en) * | 2008-09-17 | 2011-10-20 | エフ.ホフマン−ラ ロシュ アーゲー | Novel ortho-aminoanilides for the treatment of cancer |
| EP2236503A1 (en) * | 2009-04-03 | 2010-10-06 | NatureWise Biotech & Medicals Corporation | Cinamic compounds and derivatives therefrom for the inhibition of histone deacetylase |
| CN101857554A (en) * | 2009-04-03 | 2010-10-13 | 彦臣生技药品股份有限公司 | Cinnamon compounds and derivatives thereof inhibiting histone deacetylase |
| JP2011020999A (en) * | 2009-04-03 | 2011-02-03 | Genshin Seigi Yakuhin Kofun Yugenkoshi | Synamic compounds and their derivatives for inhibiting histone deacetylase |
| WO2013154870A1 (en) | 2012-04-10 | 2013-10-17 | Annji Pharmaceutical Co., Ltd. | Histone deacetylases (hdacs) inhibitors |
| US9387209B2 (en) | 2012-04-10 | 2016-07-12 | Annji Pharmaceutical Co., Ltd. | Histone deacetylases (HDACs) inhibitors |
| US9745253B2 (en) | 2015-03-13 | 2017-08-29 | Forma Therapeutics, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| US10266487B2 (en) | 2015-03-13 | 2019-04-23 | Forma Therapeutics, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| US10508077B2 (en) | 2015-03-13 | 2019-12-17 | Forma Therapeutics, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| US10988441B2 (en) | 2015-03-13 | 2021-04-27 | Valo Early Discovery, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| US11919839B2 (en) | 2015-03-13 | 2024-03-05 | Valo Health, Inc. | Alpha-cinnamide compounds and compositions as HDAC8 inhibitors |
| WO2018213364A1 (en) | 2017-05-16 | 2018-11-22 | Annji Pharmaceutical Co., Ltd. | Histone deacetylases (hdacs) inhibitors |
| WO2021252475A1 (en) | 2020-06-08 | 2021-12-16 | Annji Pharmaceutical Co., Ltd. | Quinazoline derivatives useful as selective hdac6 inhibitors |
| CN116375698A (en) * | 2023-06-05 | 2023-07-04 | 中国林业科学研究院林产化学工业研究所 | Novel urushiol-based hydroxamic acid type HDAC inhibitor with targeted antitumor activity and preparation method and application thereof |
| CN116375698B (en) * | 2023-06-05 | 2023-08-01 | 中国林业科学研究院林产化学工业研究所 | Urushiol-based hydroxamic acid-type HDAC inhibitor with targeted anti-tumor activity, preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008087514A3 (en) | 2009-02-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2453536C2 (en) | Histone deacetylase inhibitors | |
| EP1786773B1 (en) | Isoindolin-1-one derivatives | |
| WO2008087514A2 (en) | Hdac inhibitors | |
| EA011087B1 (en) | Compounds and pharmaceutical compositions for treating inflammation diseases | |
| PT1995242E (en) | Novel 1,2,3,4-tetrahydroquinoxaline derivative having glucocorticoid receptor binding activity | |
| WO2008113255A1 (en) | Benzamide derivatives with anti-proliferative activity, pharmaceutical preparations thereof | |
| JP2009242437A (en) | Sulfonamide derivative | |
| GB2441396A (en) | Indoline sulfonamide compounds | |
| US20050209195A1 (en) | Indolinone derivatives | |
| WO2017045271A1 (en) | Benzofuran derivatives, preparation method therefor and therapeutic use thereof | |
| BRPI0915513B1 (en) | 6-substituted phenoxy-chroman carboxylic acid derivative compounds, pharmaceutical compositions comprising said compounds, use thereof, and process for the preparation thereof | |
| AU2020424661B2 (en) | Disubstituted adamantyl derivative or pharmaceutically acceptable salt thereof, and pharmaceutical composition for suppressing cancer growth comprising same as active ingredient | |
| CN103848795A (en) | I,2,5-oxadiazole-2-oxide histone deacetylase inhibitor as well as preparation method and application thereof | |
| WO2007017728A2 (en) | Novel heterocyclic compounds | |
| CA2663943A1 (en) | 3-amino-pyridine derivatives for the treatment of metabolic disorders | |
| US8263044B2 (en) | Stilbene like compounds as novel HDAC inhibitors | |
| JP2011519966A (en) | Novel N- (2-amino-phenyl) -acrylamide | |
| Reddy et al. | Design, synthesis and evaluation of (E)-α-benzylthio chalcones as novel inhibitors of BCR-ABL kinase | |
| WO2007045962A2 (en) | Novel hdac inhibitors | |
| CA2787860C (en) | Substituted 2-imidazolidones and analogs and their use against cancer | |
| TW201934547A (en) | A pyrimidine compound and the preparation method and medical use thereof | |
| CN114644560A (en) | A kind of phenylacrylic acid compound and its preparation method and application | |
| WO2022021683A1 (en) | Indole alkaloid compound, preparation method therefor, and application thereof | |
| JP2007501833A (en) | Novel oxazole, its production method and its use as a drug | |
| WO2007113644A2 (en) | New hdac inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08702209 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08702209 Country of ref document: EP Kind code of ref document: A2 |