WO2008093960A1 - Novel dipeptidyl peptidase-iv inhibitors - Google Patents
Novel dipeptidyl peptidase-iv inhibitors Download PDFInfo
- Publication number
- WO2008093960A1 WO2008093960A1 PCT/KR2008/000428 KR2008000428W WO2008093960A1 WO 2008093960 A1 WO2008093960 A1 WO 2008093960A1 KR 2008000428 W KR2008000428 W KR 2008000428W WO 2008093960 A1 WO2008093960 A1 WO 2008093960A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- alkyl
- preparation
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 BCC(CCN1C(*)CC(C(C)C)C(*)*1)N Chemical compound BCC(CCN1C(*)CC(C(C)C)C(*)*1)N 0.000 description 3
- MQZBPVCYZIMAOS-UHFFFAOYSA-N BCC(CC(O)=O)NP Chemical compound BCC(CC(O)=O)NP MQZBPVCYZIMAOS-UHFFFAOYSA-N 0.000 description 1
- AMURTXUSCGARMK-UHFFFAOYSA-N BCC(CCO)NP Chemical compound BCC(CCO)NP AMURTXUSCGARMK-UHFFFAOYSA-N 0.000 description 1
- IDKFIYARQSBVSM-UHFFFAOYSA-N CN(C)C(CCCF)=O Chemical compound CN(C)C(CCCF)=O IDKFIYARQSBVSM-UHFFFAOYSA-N 0.000 description 1
- NXUMMUGQBVXMFH-YCPHGPKFSA-N C[C@H](C1)CN(C[C@H](CC(N(CCC2)[C@@H]2c2nc(-c3ccccn3)n[o]2)=O)N)C1=O Chemical compound C[C@H](C1)CN(C[C@H](CC(N(CCC2)[C@@H]2c2nc(-c3ccccn3)n[o]2)=O)N)C1=O NXUMMUGQBVXMFH-YCPHGPKFSA-N 0.000 description 1
- KGXKNDXCFMAOJM-YNTUMEFWSA-N C[C@H](CC1)CN(C[C@H](CC(N(CCC2)[C@@H]2C2ON=C(c3ccccc3)N2)=O)N)C1=O Chemical compound C[C@H](CC1)CN(C[C@H](CC(N(CCC2)[C@@H]2C2ON=C(c3ccccc3)N2)=O)N)C1=O KGXKNDXCFMAOJM-YNTUMEFWSA-N 0.000 description 1
- FDKCUZOFVNFZSM-KBPBESRZSA-N C[N](CCC1)([C@@H]1c1nc(C2CC2)n[o]1)C(C[C@@H](CN(CC(CC1)(F)F)C1=O)N)=O Chemical compound C[N](CCC1)([C@@H]1c1nc(C2CC2)n[o]1)C(C[C@@H](CN(CC(CC1)(F)F)C1=O)N)=O FDKCUZOFVNFZSM-KBPBESRZSA-N 0.000 description 1
- WNMMJPHGUXDYQV-WMZOPIPTSA-N N[C@@H](CC(N(CCC1)[C@@H]1c1nc(-c2ccccc2)c[o]1)=O)CN(CC(CC1)(F)F)C1=O Chemical compound N[C@@H](CC(N(CCC1)[C@@H]1c1nc(-c2ccccc2)c[o]1)=O)CN(CC(CC1)(F)F)C1=O WNMMJPHGUXDYQV-WMZOPIPTSA-N 0.000 description 1
- GXEXMPYRKFZRFS-STQMWFEESA-N N[C@@H](CC(N(CCC1)[C@@H]1c1nc(C2CC2)n[o]1)=O)CN(CC(CC1)(F)F)C1=O Chemical compound N[C@@H](CC(N(CCC1)[C@@H]1c1nc(C2CC2)n[o]1)=O)CN(CC(CC1)(F)F)C1=O GXEXMPYRKFZRFS-STQMWFEESA-N 0.000 description 1
- CHVXJQGIFIXMKK-KBPBESRZSA-N N[C@@H](CC(N(CCC1)[C@@H]1c1nc(C2CCC2)n[o]1)=O)CN(CC(CC1)(F)F)C1=O Chemical compound N[C@@H](CC(N(CCC1)[C@@H]1c1nc(C2CCC2)n[o]1)=O)CN(CC(CC1)(F)F)C1=O CHVXJQGIFIXMKK-KBPBESRZSA-N 0.000 description 1
- FUJWQXZVBZTPAA-VXKWHMMOSA-N N[C@@H](CC(N1[C@H](CNC(c2ccccc2)=O)Cc2ccccc2C1)=O)CN(CC(CC1)(F)F)C1=O Chemical compound N[C@@H](CC(N1[C@H](CNC(c2ccccc2)=O)Cc2ccccc2C1)=O)CN(CC(CC1)(F)F)C1=O FUJWQXZVBZTPAA-VXKWHMMOSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4453—Non condensed piperidines, e.g. piperocaine only substituted in position 1, e.g. propipocaine, diperodon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/45—Non condensed piperidines, e.g. piperocaine having oxo groups directly attached to the heterocyclic ring, e.g. cycloheximide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to compounds of novel structure, having good inhibition activity versus Dipeptidyl Peptidase-IV (DPP-IV), methods of preparing the same and pharmaceutical compositions containing the same as an active agent.
- DPP-IV Dipeptidyl Peptidase-IV
- Diabetes mellitus has serious effects on people's health and accompanies various complications.
- type I diabetes mellitus characterized by little or no insulin secretory capacity due to the destruction of pancreatic cells
- type II diabetes mellitus characterized by insulin deficiency and insulin resistance due to other causes.
- the prevalence of type II diabetes mellitus is 90% or more of total patients with diabetes mellitus.
- Representative examples of complications accompanying diabetes include hyperlipidemia, hypertension, retinopathy and renal insufficiency (Paul Zimmer, et al., Nature, 2001, 414, 782).
- Sulfonylureas (stimulating insulin secretion in pancreatic cells), biguanides (inhibiting glucose production in the liver), ⁇ -glucosidase inhibitors (inhibiting glucose absorption in the intestines), etc. have been used as agents to treat diabetes.
- peroxisome pro- liferator- activated receptor gamma (PP AR ⁇ ) accelerators Thiazolidinediones, increasing insulin sensitivity
- these drugs have side effects such as hypoglycemia, weight gain and the like (David E. Moller, Nature, 2001, 414, 821). Accordingly, there is a strong need to develope diabetes therapeutic agents with decreased side effects, in particular without inducing hypoglycemia and weight gain.
- DPP-IV dipeptidyl peptidase-IV
- GLP-I glucagon-like protein 1
- pancreatic ⁇ -cells pancreatic ⁇ -cells in vivo and plays an important role in the production and secretion of insulin.
- GLP- 1 is inactivated by DPP-IV, and DPP-IV inhibitors have been reported to increase insulin secretion by means of inhibiting said inactivation mechanism.
- DPP-IV inhibitors are also being developed as agents for treatment of obesity because they lead to satiety in rats and slow down digestion of foods in the intestines, resulting in weight loss. Further, many investigators have also shown that DPP-IV inhibitors control blood glucose and lipid levels in animal experiments (Pospislik J. A., et al, Diabetes, (2002) 51, 943-950). In this regard, DPP-IV inhibitors can be considered as potentially useful agents for treatement of diabetes.
- DPP-IV inhibitors [4] To date, many candidate materials as DPP-IV inhibitors have been on clinical trials, and Sitagliptin of Merck Company had gained FDA acceptance. Much research for developing DPP-IV inhibotors has focused on materials in which cyano group is bonded to pyrrolidine ring. Representative examples of these DPP-IV inhibitors, series of cy- anopyrrolidine, are WO00/34241, WO04/064778, WO03/004498 and WO03/082817. Other examples of DPP-IV inhibitors, series of xanthine not distributed with peptide bond, are WO02/062764, WO03/068757 and WO04/087053.
- DPP-IV inhibitors using ⁇ -amino acid derivatives have been recently reported including WO03/004498.
- WO03/082817, WO05/120494, WO05/120494 and WO 05/056003 have also been reported.
- These DPP-IV inhibitors set forth in the above documents are based upon cyclic compounds having rings connected with amide bond, which is similar to that of the present invention; however, the molecular structures substituted with phenyl groups, which are represented as Ar or Z in the above documents, are entirely different from the structures substituted with saturated or unsaturated, 5-membered or 6-membered cyclic moiety in the present invention.
- DPP-IV inhibitors according to the present invention having the molecular structure in which a lactam ring is substituted at the phenyl group position, and the method for perparation thereof have not been disclosed in the prior art.
- n 0, 1 or 2;
- a 1 , A 2 and A 3 are each independently selected from the group consisting of
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen, or linear or branched Ci-C 7 alkyl
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen, linear or branched Ci-C 7 alkyl, or substituted or unsubstituted phenyl; [28] (9) substituted or unsubstituted phenyl,
- group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, OH, CN, COOH and
- group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, OH, CN, COOH and
- m is 1 or 2
- X is -NH-, -O- , -S-, -CONR- or -CO-
- Y is each independently hydrogen, -SO 2 -R, -CH 2 -R, -CO-R, -CONH-R, substituted or unsubstituted het- erocycle, or halogen-substituted or unsubstituted phenyl
- R is each independently hydrogen, substituted or unsubstituted Ci-Ci 0 alkyl, or halogen-substituted or unsubstituted phenyl;
- D) B is selected from the Formula (II) or Formula (III) groups below:
- n 0, 1 or 2;
- D is selected from the group consisting of sulfur(S), oxygen (O), NR' and CRR';
- R and R' are each independently selected from hydrogen or linear or branched Ci-C 7 alkyl
- R 1 , R 2 , R 3 , R 4 and R 5 are selected from the group consisting of,
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen, or linear or branched Ci-C 7 alkyl
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen, or linear or branched Ci-C 7 alkyl
- group(s) for substitution is/are one or more than two each independently selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, OH, CN, COOH and NH 2 ;
- group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, OH, CN, COOH and NH 2 ;
- group(s) for substitution is/are each independently one or more selected from the group consisting of halogen, OR', NR'R", substituted or unsubstituted linear or branched Ci-Ci 0 alkyl, CN, COOR', CONR'R", substituted or unsubstituted phenyl, substituted or unsubstituted C 3 -C 6 cycloalky, or substituted or unsubstituted heterocycle, where R' and R" are each independently hydrogen or linear or branched Ci-C 7 alkyl.
- Ai, A 2 and A 3 may be each independently hydrogen, halogen, substituted or unsubstituted Ci-C 4 alkyl, substituted or unsubstituted Ci-C 4 alkoxy, substituted or unsubstituted heterocycle, CONR'R" or -CH 2 -X-Y, where R' and R" are each independently hydrogen, linear or branched Ci-C 7 alkyl, or substituted or unsubstituted phenyl; X is - NH-, -O-, -S-, or -CO-; and Y is substituted or unsubstituted phenyl, -SO 2 -R, -CO-R, - CONH-R, or substituted or unsubstituted heterocycle, wherein R is substituted or unsubstituted Ci-Cio alkyl or phenyl, and the phenyl is preferably halogen-substituted or unsubstituted phenyl
- Z is -CH 2 -, Ai, A 2 and A 3 are each independently hydrogen, halogen, C 3 -C 6 cycloalkyl-substituted or unsubstituted heterocycle, or -CH 2 - X-Y, wherein X is -NH-, or -O-; and Y is each independently -SO 2 -R, -CO-R, or halogen-substituted or unsubstituted phenyl, wherein R is substituted or unsubstituted Ci-Cio alkyl, or halogen-substituted or unsubstituted phenyl.
- R is preferably halogen-substituted or unsubstituted phenyl.
- a 2 and A 3 may be interconnected to form a cyclic structure (E), E may be selected from the group consisting of substituted or unsubstituted phenyl, substituted or unsubstituted heterocycle, and substituted or unsubstituted C 3 -C 6 cycloalkyl.
- E is preferably phenyl.
- group(s) for substitution may be each independently substituted or unsubstituted linear or branched Ci-C 4 alkyl, substituted or unsubstituted C 3 -C 6 cycloalkyl, substituted or unsubstituted phenyl, or substituted or unsubstituted heterocycle, and the heterocycle is one selected from the group consisting of furan, thiophene, pyrrol, pyrrolidine, imidazole, pyrazole, pyrazoline, oxazole, oxazoline, isoxazole, isoxazoline, thiazole, thiazoline, isothiazole, isothiazolidine, thiadiazole, thiadiazoline, tetrahydrofuran, tetrahydrothiophene, im- idazolidine, pyrazolidine, oxazolidine, isoxazolidine, thia
- Ri, R 2 , R 3 , R 4 and R 5 are preferably, each independently, hydrogen, halogen, or substituted or unsubstituted linear or branched Ci-Ci 0 alkyl.
- the compound of formula (I) is a compound as represented by Formula (Ia) or pharmaceutically acceptable salt below:
- n is 0 or l
- a 4 and A 5 are each independently selected from the following groups,
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen or linear or branched Ci-C 7 alkyl; [80] (5) substituted or unsubstituted linear or branched Ci-C 7 alkyl
- R' is linear or branched Ci-C 7 alkyl
- R' and R" are each independently hydrogen or linear or branched Ci-C 7 alkyl
- group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, CN, OH, COOH and
- group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C 6 alkyl, Ci-C 6 alkoxy, halogen, CN, OH, COOH and
- the compound according to the present invention may be a compound as represented by Formula (Ib) below wherein Z is -CH 2 -, and the compound can also include isomers thereof.
- preferable is the compound in which the carbon substituted with an amine group (-NH 2 ) forms a stereo center, as seen in the structure of Formula (Ib) below.
- the compound according to the present invention may be a compound as represented by Formula (Ic) below wherein Z is -CO-, and the compound can also include isomers thereof.
- Z is -CO-
- preferable is the compound in which the carbon substituted with an amine group (-NH 2 ) forms a stereo center, as seen in the structure of Formula (Ic) below.
- Formula (Ic) can be diastereomers.
- the compounds of Formula 1 according to the present invention are compounds as defined below, but are not limited to the following compounds: [101] l-[(2S)-2-amino-4- ⁇ (2S)-2-[(3-fluorophenoxy)methyl]pyrrolidine-l-yl ⁇ butyl]-5,5-dif luoropiperidine-2-one [102] N-( ⁇ (2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]pyrrolidine-2-y l ⁇ methyl)me thane sulfonamide [103] l- ⁇ (2S)-2-amino-4-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidine-7(6
- the compound of the present invention may form an acid adduct with a pharmaceutically acceptable acid.
- the term "pharmaceutically acceptable salt” includes inorganic salts, organic salts, amino acid salts, etc., and more specifically, salts with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid; salts with organic carboxylic acids such as acetic acid, trifluoroacetic acid, citric acid, formic acid, maleic acid, oxalic acid, succinic acid, benzoic acid, tartaric acid, fumaric acid, mandelic acid, ascorbic acid, malic acid and the like; salts with methanesulfonic acid, p-toluenesulfonic acid and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid
- organic carboxylic acids such as acetic acid, trifluoroacetic acid, citric acid, formic acid, maleic acid, oxalic acid, succinic acid, benzoic acid, tartaric acid, fum
- the compound of the present invention or the pharmaceutically acceptable salts thereof can be present in a form of hydrate or solvate.
- the present invention is also directed to processes for preparation of the compound of Formula (I), and the compound of Formula (I) wherein Z is -CH 2 - can be prepared from the following important intermediates.
- One of the intermediates is an amino acid derivative represented by the following compound of Formula (IV), and the other is a cycloamine or its corresponding salt (representatively, hydrochloric acid salt or trifluoroacetic acid salt) represented by the following Formula (V).
- the compound of Formula (I) wherein Z is -CO- can be prepared from an amino acid derivative represented by the compound of Formula (IV) below and a cycloamine or its corresponding salt represented by the following Formula (V).
- n, A 1 , A 2 , A 3 and B are as defined in the above;
- Pi is an amine-protecting group such as t-butoxycarbonyl (Boc), benzyl oxycarbonyl
- Pi is as defined in the above;
- P 2 is a linear or branched Ci-C 7 alkyl, preferably t-butyl or isopropyl.
- Intermediates (1) and (V) being aspartic acid derivatives are commercially obtainable starting materials.
- the intermediates (1) and (V) are firstly reacted with isobutylchlo- roformate and N-methylmorpholine in the presence of anhydrous tetrahydrofuran (THF), followed by being reduced with sodiumborohydride (NaBH 4 ) to prepare intermediates (2) and (2') are prepared.
- THF anhydrous tetrahydrofuran
- NaBH 4 sodiumborohydride
- the intermediate (2) is oxidized to give the aldehyde intermediates (3) and (3') by Swern oxidation method using dimethylsul- furoxide (DMSO) and oxalic acid in the presence of methylenechloride solvent or by using 2,2,6,6-tetramethyl-l-piperidinyloxy free radical (TEMPO), lithium bromide and sodiumbicarbonate.
- Cycloamides (4) and (4') can be prepared from the aldehyde intermediates (3) and (3') by using an amine 'T-NH 2 ' or its corresponding salt (for examples, hydrochloric acid salt or trifluoroacetic acid salt) and sodiumtriacetoxy- borohydride (NaBH(OAc) 3 ).
- Amine'T-NH 2 ' or its corresponding salt as a start material for synthesizing the intermediate (4) in Reaction Scheme 1 and 1-1 can be represented by Fomula (Ha) and Fomula (Ilia).
- the compounds of the following Fomula (Ha) and Fomula (Ilia) are important intermediates for preparing the compounds of the Fomula (II) and Fomula (III).
- Intermediate (5) can be commercially obtainable, or prepared by introducing amine- protecting group (P 1 ) from an amine which is commercially obtainable, in which the amine-protecting group (P 1 ) generally means t-butoxycarbonyl group (Boc).
- the intermediate (5) is first reacted with an amine base (for examples, triethylamine or N,N'-diisopropylethylamine) and isobutylchloroformate, followed by preparing a thiazoketone intermediate using diazomethane, and silverbenzoate and amine base are added thereto in the presence of methanol solvent, then the amino acid intermediate (5a) is obtained by sonication method. 4N hydrochloric acid-dioxane solution or tri- fluoroacetic acid is added to the thus prepared intermediate (5a) to give the compound of Fomula (Ha).
- an amine base for examples, triethylamine or N,N'-diisopropylethylamine
- isobutylchloroformate followed by preparing a thiazoketone intermediate using diazomethane, and silverbenzoate and amine base are added thereto in the presence of methanol solvent, then the amino acid intermediate (5a) is obtained by
- the intermediate (IV) can be prepared by the following Reaction Scheme 3 using a commercially obtainable intermediate (IV).
- the compound of Fomula (IV) can be prepared by reacting isobutylchloroformate and N- methylmorpholine in the presence of tetrahydrofuran (THF), and then reducing the resulting compound with sodiumborohydride (NaBH 4 ), in the same reduction manner as in the starting material (1) in Reaction Scheme 1.
- An amine or its corresponding salt as another important intermediate represented by the following Fomula (V) may be, for example, the compounds of Fomulas (Va), (Vb) and (Vc) below.
- Intermediate (6) can be commercially obtainable, or prepared by introducing an amine-protecting group (Pi) from a commercially obtainable amine, in which the amine-protecting group (Pi) generally means t-butoxycarbonyl group (Boc).
- Intermediate (7) can be prepared from the intermediate (6) with triphenylphosphine (TPP), diphenylphosphorylazaide (DPPA) and diethylazodicarboxylate (DEAD) in anhydrous tetrahydrofuran (THF) as solvent.
- TPP triphenylphosphine
- DPPA diphenylphosphorylazaide
- DEAD diethylazodicarboxylate
- THF anhydrous tetrahydrofuran
- the intermediate (7) is converted to amine intermediate (8) by palladium-activated carbon in alcohol as solvent, and the amine intermediate (8) thus obtained is reacted with an acid derivative or amine receptor to give intermediate (9).
- the reactant is reacted under the same condition as the condition using EDC, HOBT and amine base, as described in the above.
- Intermediate (Vb) is obtained by removing an amine- protecting group (P) from the intermediate (9) obtained by polymerization.
- amine-protecting group (P) is t-butoxycarbonyl (Boc)
- the amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid (TFA).
- Amine or its corresponding salt represented by Fomula (V) may be the compounds of the following Fomulas (Vd), (Ve) and (Vf), where Ai in Formula (I) is oxadiazole or oxazolidine as heterocycle.
- intermediate (11) is reacted with carbodiimidazole and alkyl- amidoxim (12) to give intermediate (13).
- An amine-protecting group (P) of the intermediate (13) thus obtained is removed to give intermediate (Vd).
- the amine- protecting group is t-butoxycarbonyl group (B oc)
- amine-protecting group is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid.
- intermediate (14) is reacted with acid derivative (15) and N,N-dimethylformamide (DMF) in the same condition as in Reaction Scheme 5 to give intermediate (16), and an amine-protecting group (P) of the intermediate (16) is removed to give intermediate (Ve).
- the amine-protecting group is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid in which the amine-protecting group is t-butoxycarbonyl (B oc).
- intermediate (11) and amino alcohol starting material (17) are used to give intermediate (18) by EDC coupling reaction.
- Intermediate (19) is prepared from the intermediate (18) using diethylaminosulfur trifluoride (DAST).
- DAST diethylaminosulfur trifluoride
- An amine- protecting group of the intermediate (19) is removed to give intermediate (Vf).
- the amine-protecting group is t-butoxycarbonyl (B oc)
- the amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid.
- a 4 , A 5 and n are as defined in the above above, and an amine- protecting group (P) is t-butoxycarbonyl (Boc), benzyl (Bn), benzylbenzyloxycarbonyl and the like, preferably t-butoxycarbonyl (Boc).
- Dicarbonyl intermediate (20) is reacted with amidine (21) to form heterocycle, i.e., intermediate (22) in the presence of pyridine solvent.
- amine-protecting group (P) is t-butoxycarbonyl group (Boc)
- an amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid to give amine or its corresponding salt (Vg).
- the detailed preparation method is also described in Korean Patent Application No. 2006-29138.
- an aldehyde intermediate (23) is obtained by oxidation using 2,2,6,6-tetramethyl-l-piperidinyloxy free radical (TEMPO), lithium bromide, sodi- umbicarbonate, and alcohol (IV) as a starting material obtained from Reaction Scheme 3, or by Swern oxidation method using dimethylsulfuroxide (DMSO) and oxalic acid in the presence of methylenechloride solvent.
- Intermediate (24) can be obtained by reducing aminization reaction of the aldehyde intermediate (23) and the compound of Fomula (V).
- an amine-protecting group of the intermediate (24) is removed by 4N hydrochloric acid- dioxane solution or trifluoroacetic acid/methylene chloride to give Compound (I) in which the substituent Z is CH 2 .
- the compound of Fomula (I) in which the substituent Z is -CO- can be converted to Compound (I 1 ), by the following Reaction Scheme 10, which can is prepared from the compounds of Fomula (V) and Fomula (IV) obtained by the above method.
- Intermediate (25) can be prepared by a general amino acid synthesis method; for example, l-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 1-hydroxybenzotriazole (HOBT) and amine base (for example, N,N-diisopropylethylamine) are reacted in the presence of N,N-dimethylformamide (DMF) or ethylene chloride solution.
- EDC l-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBT 1-hydroxybenzotriazole
- amine base for example, N,N-diisopropylethylamine
- Compound (T) in which the substituent Z is -CO- can be obtained from the intermediate (25) by treatment with 4N hydrochloric acid-dioxane solution or trifluoroacetic acid/methylene chloride in the presence of methylenechloride solvent.
- a compound of Formula (I) can be isolated and purified from the reaction product by means of conventional methods such as recrystallization, ion electrophoresis, silica gel column chromatography or ion exchange resin chromatography and the like.
- the present invention also provides a pharmaceutical composition for inhibiting
- DPP-IV comprising the compound of Formula (I) or a pharmaceutically acceptable salt thereof.
- composition can include a mixture of a compound of the invention with other chemical components, such as diluents or carriers. Accordingly, carriers, diluents, excipient(s), or their combination can be included in the pharmaceutical composition, if necessary.
- the pharmaceutical composition facilitates administration of the compound to an organism. Multiple techniques of administering a compound exist in the art including, but are not limited to oral, injection, aerosol, parenteral, and topical administrations.
- the compound of the present invention can be administered in various pharmaceutical dosage forms in accordance with intended use.
- an active agent more specifically the compound of Formula (I) may be mixed with one or more pharmaceutically acceptable carriers which can be selected depending on the dosage form to be prepared.
- the pharmaceutical composition according to the present invention can be formulated into dosage forms suitable for injection or oral administration.
- the compound of Formula (I) may be formulated in a conventional manner using known pharmaceutically acceptable carriers and excipients and presented in unit dosage form or in multidose containers.
- the formulations may take such forms as solutions, suspensions or emulsions in oily or aqueous vehicles, and may contain conventional dispersing, suspending or stabilizing agents.
- the active ingredient may be in the powder form for reconstitution with sterile pyrogen-free water, before use.
- the compound of Formula (I) may also be formulated into suppositories containing conventional suppository bases such as cocoa butter or other glycerides.
- Solid dosage forms for oral administration include capsule, tablet, pill, powder and granule. Preferable dosage forms are capsule and tablet.
- the solid dosage forms for oral administration may be obtained by mixing the compound of Formula (I) as an active agent with inactive diluents such as sucrose, lactose, starch and the like and carriers such as lubricant, for example magnesium stearate, including disintegrator, binder and the like.
- inactive diluents such as sucrose, lactose, starch and the like
- carriers such as lubricant, for example magnesium stearate, including disintegrator, binder and the like.
- the compound of Formula (I) and compositions comprising the same according to the present invention may be administrated in combination with other pharmaceutical agents, for example, other diabetes treating agents.
- the compound of Formula (I) as an active agent can be preferably contained in an amount of about 0.1 ⁇ 1,500 mg unit dosage.
- the dosage amount of the compound of Formula (I) will be dependent on the subject's weight and age, the nature and severity of the affliction and the judgment of the prescribing physician.
- the dosage amount required will be about in the range of 1 to 500 mg a day depending on the frequency and strength of the dosage.
- a total dosage amount of about 5 ⁇ 300 mg a day will be sufficient. In some patients, the dosage amount in a day will be higher than that.
- the present invention provides the method for treatment or prevention of diseases involving inappropriate activity of DPP-IV by using the compound of Formula (I) as defined in claim 1 in effective dose.
- diseases caused by inappropriate levels of DPP-IV include, but are in no way limited to, diabetes mellitus, obesity and the like as described above.
- the present invention isuseful to treat and prevent type II diabetes mellitus. Best Mode for Carrying Out the Invention
- T-butyl (3S)-3-(hydroxyketyl)-3,4-dihydroisoquinoline-2(lH)-carboxylate obtained in PREPARATION 4(1) was dissolved in 40 mL of anhydrous tetrahydrofuran (THF), and cooled to O 0 C. 3.05 g (116 mmol) of triphenylphosphine was added thereto. To this solution were added dropwise in sequence 2.5 mL (11.6 mmol) of diphenylphos- phorylazide and 1.83 mL (11.6 mmol) of diethylazodicarboxylate (DEAD).
- THF anhydrous tetrahydrofuran
- PREPARATION 7 Synthesis of t- butyll ( 1 S V 3-hydroxy- 1 - r(4-methyl-2-oxo-2.5-dihydro- lH-pyrrole- 1 -yPmethyllpropyl ) carbamate
- TEMPO 2,2,6,6-tetramethyl-l-piperidinyloxy preradical
- (2S)-l-(3-fluorophenoxy)-N-methylpentane-2-amine chloric acid salt obtained by referring to WO05/ 120494, was added to filterated solution, obtained by drying over anhydrous magnesium sulfate and filtering the methylene chloride layer at room temperature. After 5 minutes, 60 mg (0.285 mmol) of sodium triacetoxyborohydride was added therero. After stirring the suspension solution for 18 hours, the organic layer was separated by extracting with ethylacetate, dried over anhydrous magnesiunsulfate, and then concentrated under reduced pressure. The thus obtained concentrate was isolated and purified with prep-TLC to give 18 mg of the title compound in a yield of 19%.
- PREPARATION 11 Synthesis of t- butyl( ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-r(2SV2-(r(methylsulfonvD amino!
- PREPARATION 12 Synthesis of t- butyl( ⁇ SV3-r2.4-bis ⁇ rifluoromethylV5.8-dihvdropyridor3.4-dlpyrimidine-7(6HVyll- 1 - IY5.5-difruoro-2-oxopiperidine- 1 -yPmethyllpropyl ) carbamate
- PREPARATION 13 Synthesis of t- butvK ⁇ SVS-r ⁇ S ⁇ -G-cvclopropyl-l ⁇ -oxadiazole-S-vDpyrrolidine-l-yll-l-rrS.S-di fluoro-2-oxopiperidine- 1 yPmethyllpropyl ) carbamate
- EXAMPLE 4 Synthesis of l-lflSVl-amino- ⁇ rflSVl-G-cvclopropyl-l.l ⁇ -oxadiazole-S-vD pyrrolidine-l-yll butyl)-5.5-difluoropiperidine2-one [376] o N ⁇
- PREPARATION 14 Synthesis of t- butyl( ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-lvDmethyll-3-r(2SV2-(r ⁇ henylsulfonvDa minol methyl ⁇ pyrrolidine- 1 -yll propyl ⁇ carbamate
- PREPARATION 16 Synthesis of t- butyl( ⁇ SVl-3-r(2SV2-G-cvclobutyl-1.2.4-oxadiazole-5-vDpiperidine-l-yll-l-r(5.5-dif luoro-2-oxopiperidine-lyl s )methyllpropyl) carbamate [404] 38 mg of the title compound was obtained in a yield of 32%, in the same manner as in PREPARATION 10, except that 72.2 mg (0.224 mmol) of t- butyl ⁇ (lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl ⁇ carbamate obtained in PREPARATION 9 and 57 mg (0.235 mmol) of
- PREPARATION 17 Synthesis of t- butyl((lS s )-l-[(5.5-difluoro-2-oxopiperidine-l-yl s )methyll-3-[2-(3-furyl s )-4-(trifluorome thyl s )-5.8-dihvdropyrido[3.4-dlpyrimidine-7(6H s )-yllpropyl)carbamate [415] 173 mg of the title compound was obtained in a yield of 58%, in the same manner as in PREPARATION 10, except that 168 mg (0.52 mmol) of t- butyl ⁇ (lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)
- PREPARATION 18 Synthesis of t- butylf ⁇ SV3-rGSV3-r ⁇ ?enzoylamino)metfayl1-34-dihvdroisoquinoline-2flHVyl1-l-r(5
- PREPARATION 19 Synthesis of t-butylf flSV3-f (2SV2-r ⁇ ?enzoylami no ⁇ methyllpyrrolidine- 1 yl ) - 1 - [(5.5-difluoro-2-oxopiperidine- 1 - vDmethyll propyl ) carba mate
- 66 mg of the title compound was obtained in a yield of 25%, in the same manner as in PREPARATION 10, except that 168 mg (0.52 mmol) of t- butyl ⁇ (lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl ⁇ carbamate obtained in PREPARATION 9 and 138 mg (0.57 mmol) of N-
- PREPARATION 20 Synthesis of t- butvir ⁇ SV3-r(2S.4SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v ll-l-(r(4RV4-methyl-2-oxopyrrolidine-l-yllmethyl)propyllcarbamate
- PREPARATION 21 Synthesis of t- butvir ⁇ SV3-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v 11-1-1 r(5R)-5-methyl-2-oxopiperidine- 1 -yllmethyl Ipropyllcarbamate
- PREPARATION 22 Synthesis of t- butyl( ⁇ SV3-r(2S.4SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v 11-1 - r(4-methyl-2-oxo-2.5-dihydro- lH-pyrrole- 1 -yPmethyllpropyl ) carbamate
- EXAMPLE 13 Synthesis of l-((2SV2-amino-4-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5ylV4- fluoropyrrolidine- 1 -yllbutyl ) -4-methyl- 1.5-dihvdro-2H-pyrrole-2-one
- (2S,4S)-4-fluoropyrrolidine-l,2-dicarboxylate obtained by referring to WO 03/002553 was dissolved in 16 mL of tetrahydrofuran and 4 mL of distilled water. Thereafter, 250 mg (6.06 mmol) of lithium hydroxide was added thereto, then stirred for 15 hours at room temperature. The solvent was distilled off under reduced pressure, and ethylacetoacetate was added thereto. The resulting solution was neutralized with IN hydrochloric acid, then dried over magnesium sulfate. The solvent was distilled off under reduced pressure to give 330 mg of the title compound (yield: 70%).
- (2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-(l-ethoxyethoxy)pyrrolidine-l-carb oxylate obtained in PREPARATION 24(3) was dissolved in 30 mL of anhydrous ethanol, and then 135 mg (0.536 mmol) of pyridinium p-toluenesulfonate (PPTS) was added thereto. After the solution was heated to 5O 0 C and then cooled to room temperature, the ethanol is removed under reduced pressure to give 1.52 g of the title compound without further purification (yield: 96%).
- PPTS pyridinium p-toluenesulfonate
- (2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-hydroxypyrrolidine-l-carboxylate obtained in PREPARATION 24(4) was dissolved in 10 rnL of anhydrous tetrahy- drofuran (THF), then 34 mg (0.837 mmol) of sodium hydride (55% dispersion in mineral oil) was added thereto in ice bath and stirred for 5 minutes. Thereafter, 0.065 mL (1.05 mmol) of idiomethane as a reaction solution was added thereto, the icebath was removed.
- THF anhydrous tetrahy- drofuran
- PREPARATION 26 Synthesis of (2SVN-phenylpiperidine-2-carboxamide hy- drochloric acid salt
- EXAMPLE 14 Synthesis of l-K2S s )-2-amino-4-[(2S s )-2-(hvdroxymethyl s )pyrrolidine-l-yll-4-oxobutyl)-5.5-difluoro piperidine-2-one
- PREPARATION 31 Synthesis of t- butvirriS ⁇ -l-rrS.S-difluoro-l-oxopiperidine-l-vDmethyll-S-oxo-S-KlS'l-l-r ⁇ yrimidin e-2-ylthio)methv ⁇ pyrrolidine- 1 -yl Ipropyllcarbamate [588] 41.8 mg of the title compound (yield: 19%) was obtained, in the same manner as in
- PREPARATION 35 Synthesis of t- butyl(QSV3-r(2SV2-(anilinomethyl s )pyrrolidine-l-yll-l-r(5.5-difluoro-2-oxopiperidine -l-yDmethyll-S-oxopropyDcarbamate
- EXAMPLE 22 Synthesis of l-((2SV2-amino-4-r(2SV2-(anilinomethyl s )pyrrolidine-l-yll-4-oxobutyl)-5.5-difluorop iperidine-2-one
- PREPARATION 37 Synthesis of t- butylKlS s )-l-[(5.5-difluoro-2-oxopiperidine-l-yl s )methyll-3-[(3S s )-3-(morpholine-4-ylc arbonvD-S ⁇ -dihvdroisoquinoline-iriH ⁇ -yll-S-oxopropyllcarbamate
- (3S)-3-(morpholine-4-ylcarbonyl)-l,2,3,4-tetrahydroisoquinoline hydrochloric acid salt obtained by referring to WO 05/056003 and 60.0 mg (0.180 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
- EXAMPLE 24 Synthesis of l-((2SV2-amino-4-r(3SV3-(morpholine-4-ylcarbonylV3.4-dihvdroisoquinoline-2 ⁇ HV yll -4-oxobutyl ) -5.5-difluoropiperidine-2-one
- PREPARATION 38 Synthesis of t- butyl( ⁇ SV3-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-r(5.5-di fluoro-2-oxopiperidine- 1 -yPmethyll -3-oxopropyl lcarbamate
- EXAMPLE 25 Synthesis of l-((2SV2-amino-4-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-vD pyrrolidine- l-yll-4-oxobutyl)-5.5-difluoropiperidine-2-one
- PREPARATION 39 Synthesis of t-butyl( ⁇ SV3-r(2SV2-(5-cvclopropyl-1.2.4- ox- adiazole-S-yDpyrrolidine- 1 -yll - 1 - IY5.5-difruoro-2-oxopiperidine- 1 -yPmethyll -3-oxopr opyl) carbamate
- PREPARATION 40 Synthesis of t- butvir ⁇ SV3-r(2SV2-G-cvclopropyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-(r(5RV 5-methyl-2-oxopiperidine- l-yllmethyl)-3- oxopropyllcarbamate
- PREPARATION 41 Synthesis of t- butyll QSV l-r(5.5-difluoro-2-oxopiperidine- l-vDmethyll-3-oxo-3-r(2SV2-G-phenyl- 1 .2.4-oxadiazole-5-yl)pyrrolidine-l-yllpropyl)carbamate
- PREPARATION 43 Synthesis of t- butyll (IS)- l-[(5.5-difluoro-2-oxopiperidine- 1-yl) methyll-3-oxo-3-[(2S)-2-(3-pyridine-2-yl-1.2.4-oxadiazole-5-yl)pyrrolidine-l-yll propyl ) carbamate
- PREPARATION 44 Synthesis of t- butylf flSVl-f rf5RV5-methyl-2-oxopiperidine-l-yl1methyl)-3-oxo-3-r(2SV2-f3-pyridi ne-2-yl- 1.2.4-oxadiazole-5-yl)pyrrolidine- 1 -yllpropyl ) carbamate
- PREPARATION 2 except that 3.44 g (16.0 mmol) of N-(t-butoxycarbonyl)-L-proline and 1.06 g (8.27 mmol) of N'-hydroxycyclopentanecarboximidamide obtained in PREPARATION 45(1) were used.
- PREPARATION 46 Synthesis of t- butyl( ⁇ SV3-r(2SV2-(3-cvclopentyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-r(5.5-dif luoro-2-oxopiperidine-l-yl s )methyll-3-oxopropyl)carbamate
- EXAMPLE 32 l-((2SV2-amino-4-r(2SV2-(3-cvclopentyl-1.2.4-oxadiazole-5-vD pyrrolidine- l-yll-4-oxobutyl)-5.5-difluoropiperidine-2- one
- PREPARATION 50 Synthesis of t- butyl( ⁇ SVl-(r(4RV4-methyl-2-oxopyrrolidine-l-yllmethyl)-3-oxo-3-r(2SV2-G-pyrid ine-2-yl-1.2.4-oxadiazole-5-yl)pyrroridine-l-v ⁇ propyl) carbamate [802] 27.0 mg of the title compound (yield: 55%) was obtained, in the same manner as in
- PREPARATION 51 Synthesis of t- butyl( ⁇ SV3-r(2SV2-r3-(cvclopropylmethylV1.2.4-oxadiazole-5-yllpyrrolidine-l-yll-l -r(5.5-difluoro-2-oxopiperidine-l-yl s )methyll-3- oxopropyl) carbamate
- EXAMPLE 37 Synthesis of l-((2SV2-amino-4-r( ' 2SV2-G-cvclobutyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-4-ox obutyl)-5.5-difluoropiperidine-2-one
- (2S)-2-[(4S)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-carboxylate obtained by referring to WO 05/121131 was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 mL). Thereafter, the resulting solution was stirred for 20 minutes and concentrated under reduced pressure, then the residue was purified with prep-TLC (6:1 CH 2 Cl 2 MeOH) to give 130 mg of the title compound (yield: 82%).
- PREPARATION 54 Synthesis of t- butvir ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-oxo-3-((2SV2-r(4SV4-phe nyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)propyllcarbamate
- PREPARATION 56 Synthesis of t- butvir ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-((2S.4SV4-fluoro-2-r(4SV 4-phenyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)-3-oxopropyllcarbamate
- EXAMPLE 39 Synthesis of l-r(2SV2-amino-4-oxo-4-((2S.4SV4-fluoro-2-r(4SV4-phenyl-4.5-dihvdro-1.3-oxazole- 2-yllpyrrolidine-l-yl)-4-oxobutyll-5.5-difluoropiperidine-2-one
- PREPARATION 58 Synthesis of t- butvir ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-((2S.4RV4-hvdroxy-2-r(4 SV4-phenyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)-3-oxopropyllcarbamate
- PREPARATION 59 Synthesis of 4-phenyl-2-r(2SVpyrrolidine-2-yll-1.3-oxazole hy- drochloric acid salt
- (2S)-2-( ⁇ [(lS)-2-hydroxy- 1-phenylethyl] amino ⁇ carbonyl)pyrrolidine- 1-carboxylate obtained by referring to WO 05/121131 was dissolved in 10 mL of methylenechloride, and 0.65 g (1.54 mmol) of Dess-martin periodinane was added thereto. The resulting solution was stirred for 3 hours, filtered by Cellite and concentrated under reduced pressure. The next step was preceded without further purification.
- (2S)-2-(4-phenyl-l,3-oxazole-2-yl)pyrrolidine-l-carboxylate obtained in PREPARATION 58(1) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). The resulting solution was stirred for 20 minutes and concentrated under reduced pressure, and the residue was purified with diethylether to give 140 mg of the title compound (yield: 97%).
- PREPARATION 60 Synthesis of t- butylf f ISV l-r(5.5-difluoro-2-oxopiperidine- l-yl)methyl1-3-oxo-3-r(2SV2-f4-phenyl- 1 .3-oxazole-2-yl)pyrrolidine-l-yllpropyl)carbamate
- PREPARATION 62 Synthesis of t- butyl( ⁇ SVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-oxo-3-r(2SV2-(5-ri-(triflu oromethyDcyclopropyll - 1.2.4-oxadiazole-3-yl Ipyrrolidine- 1 -yllpropyl ) carbamate
- EXAMPLE 42 Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-(5-ri-(trifluoromethyl s )cvclopropyll-1.2.4-oxadiazol e-3-yl Ipyrrolidine- 1 -yllbutyl ) -5.5-difluoropiperidine-2-one
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The present invention relates to novel compounds exhibiting excellent inhibitory activity versus Dipeptidyl Peptidase-IV (DPP-IV), methods of preparing the same and pharmaceutical compositions containing the same as an active agent.
Description
Description
NOVEL DIPEPTIDYL PEPTIDASE-IV INHIBITORS
Technical Field
[1] The present invention relates to compounds of novel structure, having good inhibition activity versus Dipeptidyl Peptidase-IV (DPP-IV), methods of preparing the same and pharmaceutical compositions containing the same as an active agent. Background Art
[2] Diabetes mellitus has serious effects on people's health and accompanies various complications. There are two major types of diabetes mellitus: type I diabetes mellitus characterized by little or no insulin secretory capacity due to the destruction of pancreatic cells, and type II diabetes mellitus characterized by insulin deficiency and insulin resistance due to other causes. The prevalence of type II diabetes mellitus is 90% or more of total patients with diabetes mellitus. Representative examples of complications accompanying diabetes include hyperlipidemia, hypertension, retinopathy and renal insufficiency (Paul Zimmer, et al., Nature, 2001, 414, 782). Sulfonylureas (stimulating insulin secretion in pancreatic cells), biguanides (inhibiting glucose production in the liver), α-glucosidase inhibitors (inhibiting glucose absorption in the intestines), etc. have been used as agents to treat diabetes. Recently, peroxisome pro- liferator- activated receptor gamma (PP ARγ) accelerators (Thiazolidinediones, increasing insulin sensitivity) have drawn attention as therapeutic agents for diabetes. However, these drugs have side effects such as hypoglycemia, weight gain and the like (David E. Moller, Nature, 2001, 414, 821). Accordingly, there is a strong need to develope diabetes therapeutic agents with decreased side effects, in particular without inducing hypoglycemia and weight gain.
[3] Recently, it has been found that dipeptidyl peptidase-IV (DPP-IV) deficient mice maintained glucagon-like protein 1 (GLP-I) activity and high insulin levels, resulting in decreased blood glucose levels, which suggested the possibility of it being used as a therapeutic agent for diabetes (Marguet D. et al, Natl. Acad. ScL USA, (2000) 97, 6874-6879). GLP-I induces differentiation and growth of pancreatic β-cells in vivo and plays an important role in the production and secretion of insulin. GLP- 1 is inactivated by DPP-IV, and DPP-IV inhibitors have been reported to increase insulin secretion by means of inhibiting said inactivation mechanism. DPP-IV inhibitors are also being developed as agents for treatment of obesity because they lead to satiety in rats and slow down digestion of foods in the intestines, resulting in weight loss. Further, many investigators have also shown that DPP-IV inhibitors control blood glucose and lipid levels in animal experiments (Pospislik J. A., et al, Diabetes, (2002) 51, 943-950). In
this regard, DPP-IV inhibitors can be considered as potentially useful agents for treatement of diabetes.
[4] To date, many candidate materials as DPP-IV inhibitors have been on clinical trials, and Sitagliptin of Merck Company had gained FDA acceptance. Much research for developing DPP-IV inhibotors has focused on materials in which cyano group is bonded to pyrrolidine ring. Representative examples of these DPP-IV inhibitors, series of cy- anopyrrolidine, are WO00/34241, WO04/064778, WO03/004498 and WO03/082817. Other examples of DPP-IV inhibitors, series of xanthine not distributed with peptide bond, are WO02/062764, WO03/068757 and WO04/087053.
[5] In addition, many DPP-IV inhibitors using β-amino acid derivatives have been recently reported including WO03/004498. In this connection, WO03/082817, WO05/120494, WO05/120494 and WO 05/056003 have also been reported. These DPP-IV inhibitors set forth in the above documents are based upon cyclic compounds having rings connected with amide bond, which is similar to that of the present invention; however, the molecular structures substituted with phenyl groups, which are represented as Ar or Z in the above documents, are entirely different from the structures substituted with saturated or unsaturated, 5-membered or 6-membered cyclic moiety in the present invention. Moreover, DPP-IV inhibitors according to the present invention, having the molecular structure in which a lactam ring is substituted at the phenyl group position, and the method for perparation thereof have not been disclosed in the prior art.
Disclosure of Invention
Technical Problem
[6] The inventors of the present invention, while carrying out extensive research and many experiments to develop compounds effective in DPP-IV inhibition, found that compounds having an optionally substituted lactam ring structure exhibit excellent inhibitory activity versus DPP-IV. The present invention was accomplished on the basis of such finding.
[7] It is therefore an object of the invention to provide novel compounds of an optionally substituted lactam ring structure having inhibitory activity versus DPP-IV.
[8] It is a further object of the present invention to provide processes for preparation of such compounds.
[9] It is another object of the present invention to provide pharmaceutical compositions for inhibiting DPP-IV activity comprising a pharmceutically effective amount of these compounds as an active agent, and also provide methods for treating or preventing diseases caused by inappropriate activity of DPP-IV by the use of the compounds of the present invention.
[10] Other objects and advantages of the present invention will become apparent to those skilled in the art from the following detailed description.
Technical Solution [11] According to the present invention, there is provided the compound of the following
Formula (I) or pharmaceutically acceptable salt thereof:

(D
[13] wherein
[14] A) n is 0, 1 or 2;
[15] B) A1, A2 and A3 are each independently selected from the group consisting of
[16] (1) hydrogen;
[17] (2) halogen;
[18] (3) OH or OR',
[19] wherein R' is linear or branched Ci-C7 alkyl;
[20] (4) substituted or unsubstituted linear or branched Ci-C7 alkyl;
[21] (5) NR1R"
[22] wherein R' and R" are each independently hydrogen, or linear or branched Ci-C7 alkyl;
[23] (6) CN;
[24] (7) COOH or COOR',
[25] wherein R' is linear or branched Ci-C7 alkyl;
[26] (8) CONR'R",
[27] wherein R' and R" are each independently hydrogen, linear or branched Ci-C7 alkyl, or substituted or unsubstituted phenyl; [28] (9) substituted or unsubstituted phenyl,
[29] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, OH, CN, COOH and
NH2;
[30] (10) substituted or unsubstituted C3-C6 cycloalkyl,
[31] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, OH, CN, COOH and
NH2; [32] (11) substituted or unsubstituted heterocycle,
[33] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of halogen, OR', NR'R", substituted or unsubstituted linear or branched Ci-Ci0 alkyl, CN, COOR', CONR'R", substituted or unsubstituted phenyl, substituted or unsubstituted C3-C6 cycloalkyl, or substituted or unsubstituted het- erocycle, where R' and R" are each independently hydrogen, or linear or branched Ci -C7 alkyl; and
[34] (12) -(CH2)m-X-Y
[35] wherein m is 1 or 2, X is -NH-, -O-, -S-, -CONR- or -CO-, Y is each independently hydrogen, -SO2-R, -CH2-R, -CO-R, -CONH-R, substituted or unsubstituted het- erocycle, or halogen-substituted or unsubstituted phenyl, where R is each independently hydrogen, substituted or unsubstituted Ci-Ci0 alkyl, or halogen-substituted or unsubstituted phenyl;
[36] wherein A2 and A3 are interconnected to make a ring (E), and where Z is -CO-, E is phenyl
[37] C) Z is -CH2- or -CO-;
[38] D) B is selected from the Formula (II) or Formula (III) groups below:

(III)
[41] wherein
[42] (a) n is 0, 1 or 2;
[43] (b) D is selected from the group consisting of sulfur(S), oxygen (O), NR' and CRR';
[44] wherein R and R' are each independently selected from hydrogen or linear or branched Ci-C7 alkyl,
[45] (c) R1, R2, R3, R4 and R5 are selected from the group consisting of,
[46] (1) hydrogen;
[47] (2) halogen;
[48] (3) OH or OR',
[49] wherein R' is linear or branched Ci-C7 alkyl;
[50] (4) substituted or unsubstituted linear or branched Cj-C7 alkyl; (5) NR1R"
[51] wherein R' and R" are each independently hydrogen, or linear or branched Ci-C7 alkyl;
[52] (6) CN;
[53] (7) COOH or COOR'
[54] wherein R' is linear or branched Ci-C7 alkyl;
[55] (8) CONR'R"
[56] wherein R' and R" are each independently hydrogen, or linear or branched Ci-C7 alkyl;
[57] (9) substituted or unsubstituted phenyl,
[58] wherein group(s) for substitution is/are one or more than two each independently selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, OH, CN, COOH and NH2;
[59] (10) substituted or unsubstituted C3-C6 cycloalkyl,
[60] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, OH, CN, COOH and NH2;
[61] (11) substituted or unsubstituted heterocycle,
[62] wherein the group(s) for substitution is/are each independently one or more selected from the group consisting of halogen, OR', NR'R", substituted or unsubstituted linear or branched Ci-Ci0 alkyl, CN, COOR', CONR'R", substituted or unsubstituted phenyl, substituted or unsubstituted C3-C6 cycloalky, or substituted or unsubstituted heterocycle, where R' and R" are each independently hydrogen or linear or branched Ci-C 7 alkyl.
[63] Ai, A2 and A3 may be each independently hydrogen, halogen, substituted or unsubstituted Ci-C4 alkyl, substituted or unsubstituted Ci-C4 alkoxy, substituted or unsubstituted heterocycle, CONR'R" or -CH2-X-Y, where R' and R" are each independently hydrogen, linear or branched Ci-C7 alkyl, or substituted or unsubstituted phenyl; X is - NH-, -O-, -S-, or -CO-; and Y is substituted or unsubstituted phenyl, -SO2-R, -CO-R, - CONH-R, or substituted or unsubstituted heterocycle, wherein R is substituted or unsubstituted Ci-Cio alkyl or phenyl, and the phenyl is preferably halogen-substituted or unsubstituted phenyl.
[64] In a preferable embodiment, where Z is -CH2-, Ai, A2 and A3 are each independently hydrogen, halogen, C3-C6 cycloalkyl-substituted or unsubstituted heterocycle, or -CH2 - X-Y, wherein X is -NH-, or -O-; and Y is each independently -SO2-R, -CO-R, or halogen-substituted or unsubstituted phenyl, wherein R is substituted or unsubstituted
Ci-Cio alkyl, or halogen-substituted or unsubstituted phenyl.
[65] In the above, R is preferably halogen-substituted or unsubstituted phenyl.
[66] A2 and A3 may be interconnected to form a cyclic structure (E), E may be selected from the group consisting of substituted or unsubstituted phenyl, substituted or unsubstituted heterocycle, and substituted or unsubstituted C3-C6 cycloalkyl. When Z is - CO-, E is preferably phenyl.
[67] In case of the heterocycles having one or more substituent, group(s) for substitution may be each independently substituted or unsubstituted linear or branched Ci-C4 alkyl, substituted or unsubstituted C3-C6 cycloalkyl, substituted or unsubstituted phenyl, or substituted or unsubstituted heterocycle, and the heterocycle is one selected from the group consisting of furan, thiophene, pyrrol, pyrrolidine, imidazole, pyrazole, pyrazoline, oxazole, oxazoline, isoxazole, isoxazoline, thiazole, thiazoline, isothiazole, isothiazolidine, thiadiazole, thiadiazoline, tetrahydrofuran, tetrahydrothiophene, im- idazolidine, pyrazolidine, oxazolidine, isoxazolidine, thiazolidine, isothiazolidine, thi- adiazolidine, sulforan, pyran, dihydropyran, tetrahydropyran, pyridine, pyridinone, pyridazine, pyrazine, pyrimidine, piperidine, piperazine, morpholine, pyridazinone, tetrazole, triazole, triazolidine and azepine.
[68] In the substituent B, Ri, R2, R3, R4 and R5 are preferably, each independently, hydrogen, halogen, or substituted or unsubstituted linear or branched Ci-Ci0 alkyl.
[69] In a preferable embodiment, the compound of formula (I) is a compound as represented by Formula (Ia) or pharmaceutically acceptable salt below:

(Ia)
[71] wherein,
[72] A) n is 0 or l;
[73] B) A4 and A5 are each independently selected from the following groups,
[74] (1) hydrogen;
[75] (2) halogen;
[76] (3) OH or OR',
[77] wherein R' is linear or branched Ci-C7 alkyl;
[78] (4) NR1R",
[79] wherein R' and R" are each independently hydrogen or linear or branched Ci-C7 alkyl;
[80] (5) substituted or unsubstituted linear or branched Ci-C7 alkyl
[81] (6) CN;
[82] (7) COOH or COOR'
[83] wherein R' is linear or branched Ci-C7 alkyl;
[84] (8) CONR'R"
[85] wherein R' and R" are each independently hydrogen or linear or branched Ci-C7 alkyl;
[86] (9) substituted or unsubstituted phenyl,
[87] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, CN, OH, COOH and
NH2;
[88] (10) substituted or unsubstituted C3-C6 cycloalkyl,
[89] wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, CN, OH, COOH and
NH2;
[90] (11) substituted or unsubstituted heterocycle,
[91] wherein the group(s) for substitution is/are each independently halogen, OR', NR'R", substituted or unsubstituted linear or branched Ci-Ci0 alkyl, CN, COOR', CONR'R", substituted or unsubstituted phenyl, substituted or unsubstituted C3-C6 cycloalkyl, or substituted or unsubstituted heterocycle, wherein R' and R" are each independently hydrogen, or linear or branched Ci-C7 alkyl. [92] When any substituent(s) is/are expressed to be "substituted" without any exceptional explanation in the speciation, it means that all of the various substitution structures described in the above examples are possible. [93] In a preferable embodiment, the compound according to the present invention may be a compound as represented by Formula (Ib) below wherein Z is -CH2-, and the compound can also include isomers thereof. In this case, preferable is the compound in which the carbon substituted with an amine group (-NH2) forms a stereo center, as seen in the structure of Formula (Ib) below.

(Ib)
[95] wherein,
[96] n, Ai, A2, A3 and B are as defined in Formula (I).
[97] In another preferable embodiment, the compound according to the present invention may be a compound as represented by Formula (Ic) below wherein Z is -CO-, and the compound can also include isomers thereof. In this case, preferable is the compound in which the carbon substituted with an amine group (-NH2) forms a stereo center, as seen in the structure of Formula (Ic) below.

(Ic) [99] When the substituents A1, A2, A3, R1, R2, R3, R4 and R5 in Formulas (I), (II) and (III) are substituted with two different substituents, the compounds of Formula (Ib) and
Formula (Ic) can be diastereomers. [100] In a particularly preferred embodiment, the compounds of Formula 1 according to the present invention are compounds as defined below, but are not limited to the following compounds: [101] l-[(2S)-2-amino-4-{(2S)-2-[(3-fluorophenoxy)methyl]pyrrolidine-l-yl}butyl]-5,5-dif luoropiperidine-2-one [102] N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]pyrrolidine-2-y l}methyl)me thane sulfonamide [103] l-{(2S)-2-amino-4-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidine-7(6
H)-yl]butyl}-5,5-difluoropiperidine-2-one [104] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl] butyl}-5,5-difluoropiperidine2-one [105] N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]pyrrolidine-2-y
1 } methyl)benzenesulf onamide [106] l-{(2S)-2-amino-4-[(3S)-3-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)-3,4-dihydroisoquino line-2(lH)-yl]butyl}-5,5-difluoropiperidine-2-one [107] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine-l-yl]butyl
}-5,5- difluoropiperidine-2-one [108] l-{(2S)-2-amino-4-[2-(3-furyl)-4-(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidi ne-7(6H)-yl]}butyl}-5,5-difluoropiperidine-2-one [109] N-({(3S)-2-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]-l,2,3,4-tetrais oquinoline- 3 -yl } methyl)benzamide [110] N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]pyrrolidine-2-y
1 } methyl)benzamide [111] (4R)-l-{(2S)-2-amino-4-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluorop
yrrolidine- 1 -yl]butyl } -4-methylpyrrolidine-2-one [112] (5R)-l-{(2S)-2-amino-4-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluorop yrrolidine- 1 -yl]butyl } -5-methylpiperidine-2-one [113] l-{(2S)-2-amino-4-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5yl)-4- fluoropyrrolidine- 1 -yl]butyl } -4-methyl- 1 ,5-dihydro-2H-pyrrole-2-one [114] l-{(2S)-2-amino-4-[(2S)-2-(hydroxymethyl)pyrrolidine-l-yl]-4-oxobutyl}-5,5-difluo ropiperidine-2-one [115] l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(phenoxymethyl)pyrrolidine-l-yl]butyl}-5,5-difluo ropiperidine-2-one [116] N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]pyrrolidine
-2-yl } methyl)benzenesulfonamide [117] N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]pyrrolidine
-2-yl } methyl)benzamide [118] l-[(2S)-2-amino-4-oxo-4-{(2S)-2-[(pyrimidine-2-ylthio)methyl]pyrrolidine-l-yl}but yl]-5,5- difluoropiperidine-2-one [119] l-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]pyrrolidine-
2-yl}methyl)-3-phenylurea [120] l-[(2S)-2-amino-4-oxo-4-{(2S)-2-[(phenylthio)methyl]pyrrolidine-l-yl}butyl]-5,5-di fluoropiperidine-2-one [121] (3S)-2-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]-N-benzyl-l,2,3
,4-tetrahydroisoquinoline-3-carboxamide [122] l-{(2S)-2-amino-4-[(2S)-2-(anilinomethyl)pyrrolidine-l-yl]-4-oxobutyl}-5,5-difluor opiperidine-2-one [123] (3S)-2-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]-l,2,3,4-tetrahy droisoquinoline- 3 -carboxamide [124] l-{(2S)-2-amino-4-[(3S)-3-(morpholine-4-ylcarbonyl)-3,4-dihydroisoquinoline-2(lH
)-yl] -4-oxobutyl } -5,5-difluoropiperidine-2-one [125] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-4- oxobutyl}-5,5-difluoropiperidine-2-one [126] l-{(2S)-2-amino-4-[(2S)-2-(5-cyclopropyl-l,2,4-oxadiazole-3-yl)pyrrolidine-l-yl]-4- oxobutyl}-5,5-difluoropiperidine-2-one [127] (5R)-l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l- yl] -4-oxobutyl } -5-methylpiperidine-2-one [128] l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-phenyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]b utyl}-5,5-difluoropiperidine-2-one [129] (5R)-l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-phenyl-l,2,4-oxadiazole-5-yl)pyrrolidine-
1 -yl]butyl } -5-methylpiperidine-2-one [130] l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-pyridine-2-yl-l,2,4-oxadiazole-5-yl)pyrrolidine-
1 -yl]butyl } -5,5-difluoropiperidine-2-one [131] (5R)-l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-pyridine-2-yl-l,2,4-oxadiazole-5-yl)pyrrol idine- 1 -yl]butyl } -5-methylpiperidine-2-one [132] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopentyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-4- oxobutyl}-5,5-difluoropiperidine-2- one [133] l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-piperidine-l-yl-l,2,4-oxadiazole-5-yl)pyrrolidin e- 1 -yl]butyl } -5,5-difluoropiperidine-2-one [134] (4R)-l-{(2S)-2-amino-4-[(2S)-2-(anilinomethyl)pyrrolidine-l-yl]-4-oxobutyl}-4-met hylpyrrolidine-2-one [135] (4R)-l-{(2S)-2-amino-4-oxo-4-[(2S)-2-(3-pyridine-2-yl-l,2,4-oxadiazole-5-yl)pyrrol idine- 1 -yl]butyl } -4-methylpyrrolidine-2-one [136] l-[(2S)-2-amino-4-{(2S)-2-[3-(cyclopropylmethyl)-l,2,4-oxadiazole-5-yl]pyrrolidine
-l-yl}-4-oxobutyl]-5,5-difluoropiperidine-2-one [137] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-4-o xobutyl}-5,5-difluoropiperidine-2-one [138] l-[(2S)-2-amino-4-oxo-4-{(2S)-2-[(4S)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrro lidine- 1 -yl } butyl] -5,5 -difluoropiperidine-2-one [139] l-[(2S)-2-amino-4-oxo-4-{(2S,4S)-4-fluoro-2-[(4S)-4-phenyl-4,5-dihydro-l,3-oxazol e-2-yl]pyrrolidine- 1 -yl } -4-oxobutyl] -5,5-difluoropiperidine-2-one [140] l-[(2S)-2-amino-4-{(2S,4R)-4-hydroxy-2-[(4S)-4-phenyl-4,5-dihydro-l,3-oxazole-2- yl]pyrrolidine-l-yl}-4-oxobutyl]-5,5-difluoropiperidine-2-one [141] 1 - { (2S)-2-amino-4-oxo-4- [(2S)-2-(4-phenyl- 1 ,3-oxazole-2-yl)pyrrolidine- 1 -yl]butyl }
-5,5 -difluoropiperidine-2-one [142] l-{(2S)-2-amino-4-oxo-4-[(2S)-2-{5-[l-(trifluoromethyl)cyclopropyl]-l,2,4-oxadiaz ole-3-yl}pyrrolidine-l-yl]butyl}-5,5-difluoropiperidine-2-one [143] l-{(2S)-2-amino-4-[(2S)-2-(4,5-dihydro-l,3-oxazole-2-yl)pyrrolidine-l-yl]-4-oxobut yl } -5,5-difluoropiperidine-2-one [144] l-[(2S)-2-amino-4-{(2S)-2-[(4S)-4-isopropyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidi ne- 1 -yl } -4-oxobutyl] -5,5-difluoropiperidine-2-one [145] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-4- oxobutyl } -4-methyl- 1 ,5-dihydro-2H-pyrrole-2-one [146] N-({(2S)-l-[(3S)-3-amino-4-(4-methyl-2-oxo-2,5-dihydro-lH-pyrrole-l-yl)butanoyl] pyrrolidine-2-yl } methyl)benzenesulfonamide [147] N-({(3S)-2-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]-l,2,3,4-tet rahydroisoquinoline-3-yl}methyl)benzamide [148] l-[(2S)-2-amino-4-{(2S)-2-[3-(cyclobutyl)-l,2,4-oxadiazole-5-yl]piperidine-l-yl}-4- oxobutyl]-5,5-difluoropiperidine-2-one [149] l-{(2S)-2-amino-4-[(3S)-3-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)-3,4-dihydroisoquino
line-2( 1 H)-yl] -4-oxobutyl } -5 ,5-difluoropiperidine-2-one
[150] l-[(2S)-2-amino-4-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolid ine- 1 -yl] -4-oxobutyl] -5,5-difluoropiperidine-2-one
[151] l-{(2S)-2-amino-4-[(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-hydroxypyrr olidine- 1 -yl] -4-oxobutyl } -5,5-difluoropiperidine-2-one
[152] l-{(2S)-2-amino-4-[(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-metoxypyrro lidine- 1 -yl] -4-oxobutyl } -5,5-difluoropiperidine-2-one
[153] l-{(2S)-2-amino-4-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4,4-difluoropyrroli dine- 1 -yl] -4-oxobutyl } -5,5-difluoropiperidine-2-one
[154] (3S)-2-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butanoyl]-N-phenyl-l,2,3 ,4-tetraisoquinoline-3-carboxamide
[155] (2S ) - 1 - [ (3 S ) -3 - amino-4- (5,5 -difluoro-2-oxopiperidine- 1 -yl)butanoyl] -N-phenylpiperi dine-2-carboxamide.
[156] The compound of the present invention may form an acid adduct with a pharmaceutically acceptable acid.
[157] As used herein, the term "pharmaceutically acceptable salt" includes inorganic salts, organic salts, amino acid salts, etc., and more specifically, salts with inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid; salts with organic carboxylic acids such as acetic acid, trifluoroacetic acid, citric acid, formic acid, maleic acid, oxalic acid, succinic acid, benzoic acid, tartaric acid, fumaric acid, mandelic acid, ascorbic acid, malic acid and the like; salts with methanesulfonic acid, p-toluenesulfonic acid and the like.
[158] The compound of the present invention or the pharmaceutically acceptable salts thereof can be present in a form of hydrate or solvate.
[159] The present invention is also directed to processes for preparation of the compound of Formula (I), and the compound of Formula (I) wherein Z is -CH2- can be prepared from the following important intermediates. One of the intermediates is an amino acid derivative represented by the following compound of Formula (IV), and the other is a cycloamine or its corresponding salt (representatively, hydrochloric acid salt or trifluoroacetic acid salt) represented by the following Formula (V).

IV V
[161] Further, the compound of Formula (I) wherein Z is -CO- can be prepared from an amino acid derivative represented by the compound of Formula (IV) below and a
cycloamine or its corresponding salt represented by the following Formula (V).
[162]
HO.
B
HN-
IV
[163] wherein,
[164] n, A1, A2, A3 and B are as defined in the above;
[165] Pi is an amine-protecting group such as t-butoxycarbonyl (Boc), benzyl oxycarbonyl
(Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), preferably t-butoxycarbonyl (Boc). [166] The intermediates IVa and IV can be prepared from Reaction Scheme 1 and
Reaction Scheme 1-1 below. [167] [Reaction Scheme 1] [168]

T-NH2. NaBH(OAc)1 P,O
[169] [170] [Reaction Scheme 1-1] [171]

T-NH1, NaBH(OAc),

4' IV-a
[172] wherein,
[173] Pi is as defined in the above;
[174] P2 is a linear or branched Ci-C7 alkyl, preferably t-butyl or isopropyl. [175] Intermediates (1) and (V) being aspartic acid derivatives are commercially obtainable starting materials. The intermediates (1) and (V) are firstly reacted with isobutylchlo- roformate and N-methylmorpholine in the presence of anhydrous tetrahydrofuran (THF), followed by being reduced with sodiumborohydride (NaBH4) to prepare intermediates (2) and (2') are prepared. The intermediate (2) is oxidized to give the aldehyde intermediates (3) and (3') by Swern oxidation method using dimethylsul- furoxide (DMSO) and oxalic acid in the presence of methylenechloride solvent or by using 2,2,6,6-tetramethyl-l-piperidinyloxy free radical (TEMPO), lithium bromide and sodiumbicarbonate. Cycloamides (4) and (4') can be prepared from the aldehyde intermediates (3) and (3') by using an amine 'T-NH2' or its corresponding salt (for examples, hydrochloric acid salt or trifluoroacetic acid salt) and sodiumtriacetoxy- borohydride (NaBH(OAc)3). Where P1 is t-butoxycarbonyl (Boc), an amine-protecting group is removed from trifluoroacetic acid/methylene chloride, and then the intermediates (4) and (4') are reacted with di-t-butyldicarbonate (BoC2O) and sodium- hydroxide solution in the presence of water and t-butanol to give Fomula (IVa) and Fomula (IVa).
[176] Amine'T-NH2' or its corresponding salt as a start material for synthesizing the intermediate (4) in Reaction Scheme 1 and 1-1 can be represented by Fomula (Ha) and Fomula (Ilia). The compounds of the following Fomula (Ha) and Fomula (Ilia) are important intermediates for preparing the compounds of the Fomula (II) and Fomula (III).

[178] wherein, [179] Ri, R2, R3, R4, R5 and D are as defined in the above; [180] R6 and R7 are linear or non-linear Ci-C5 alkyl. [181] The compound of Fomula (Ha) can be commercially obtainable or prepared by the same method as in Korean Patent Application No. 2006-29138. Also, the compound can be prepared by the following Reaction Scheme 2 using the method set forth in WO 04/007468.
[182] [Reaction Scheme 2] [183]

[184] Intermediate (5) can be commercially obtainable, or prepared by introducing amine- protecting group (P1) from an amine which is commercially obtainable, in which the amine-protecting group (P1) generally means t-butoxycarbonyl group (Boc).
[185] The intermediate (5) is first reacted with an amine base (for examples, triethylamine or N,N'-diisopropylethylamine) and isobutylchloroformate, followed by preparing a thiazoketone intermediate using diazomethane, and silverbenzoate and amine base are added thereto in the presence of methanol solvent, then the amino acid intermediate (5a) is obtained by sonication method. 4N hydrochloric acid-dioxane solution or tri- fluoroacetic acid is added to the thus prepared intermediate (5a) to give the compound of Fomula (Ha).
[186] In addition, the intermediate (IV) can be prepared by the following Reaction Scheme 3 using a commercially obtainable intermediate (IV). In Reaction Scheme 3, the compound of Fomula (IV) can be prepared by reacting isobutylchloroformate and N- methylmorpholine in the presence of tetrahydrofuran (THF), and then reducing the resulting compound with sodiumborohydride (NaBH4), in the same reduction manner as in the starting material (1) in Reaction Scheme 1.
[187] [Reaction Scheme 3] [188]

[189] An amine or its corresponding salt as another important intermediate represented by the following Fomula (V) may be, for example, the compounds of Fomulas (Va), (Vb) and (Vc) below.

Va Vb Vc
[191] The preparation methods of the compounds of Fomulas (Va), (Vb) and (Vc) are
disclosed in WO 05/0560138 and many articles. For example, the compound of Fomula (Vb) can be prepared by the following Reaction Scheme 4. Herein, each sub- stituent, namely -OR', -NHR', -CONR'R", is included within the scope of A1 defined in the Fomula (I).


9 10
[194] Intermediate (6) can be commercially obtainable, or prepared by introducing an amine-protecting group (Pi) from a commercially obtainable amine, in which the amine-protecting group (Pi) generally means t-butoxycarbonyl group (Boc). Intermediate (7) can be prepared from the intermediate (6) with triphenylphosphine (TPP), diphenylphosphorylazaide (DPPA) and diethylazodicarboxylate (DEAD) in anhydrous tetrahydrofuran (THF) as solvent. The intermediate (7) is converted to amine intermediate (8) by palladium-activated carbon in alcohol as solvent, and the amine intermediate (8) thus obtained is reacted with an acid derivative or amine receptor to give intermediate (9). Where reacted with an acid derivative, the reactant is reacted under the same condition as the condition using EDC, HOBT and amine base, as described in the above. Intermediate (Vb) is obtained by removing an amine- protecting group (P) from the intermediate (9) obtained by polymerization. Where amine-protecting group (P) is t-butoxycarbonyl (Boc), the amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid (TFA).
[195] Amine or its corresponding salt represented by Fomula (V) may be the compounds of the following Fomulas (Vd), (Ve) and (Vf), where Ai in Formula (I) is oxadiazole or oxazolidine as heterocycle.
[196]

Vd Ve Vf
[197] The amine or its corresponding salt represented by Fomula (Vd) is prepared by the following reaction scheme 5.
[198] [Reaction Scheme 5]

12
[200] In Reaction Scheme 5, intermediate (11) is reacted with carbodiimidazole and alkyl- amidoxim (12) to give intermediate (13). An amine-protecting group (P) of the intermediate (13) thus obtained is removed to give intermediate (Vd). Where the amine- protecting group is t-butoxycarbonyl group (B oc), amine-protecting group is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid.
[201] The amine or its corresponding salt, represented by Fomula (Ve) below, is prepared by the following Reaction Scheme 6.
[202] [Reaction Scheme 6] [203]

15
[204] In Reaction Scheme 6, intermediate (14) is reacted with acid derivative (15) and
N,N-dimethylformamide (DMF) in the same condition as in Reaction Scheme 5 to give intermediate (16), and an amine-protecting group (P) of the intermediate (16) is removed to give intermediate (Ve). The amine-protecting group is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid in which the amine-protecting group is t-butoxycarbonyl (B oc).
[205] The amine or its corresponding salt, represented by Fomula (Vf) below, is prepared by the following Reaction Scheme 7.
[206] [Reaction Scheme 7]

11 17 18
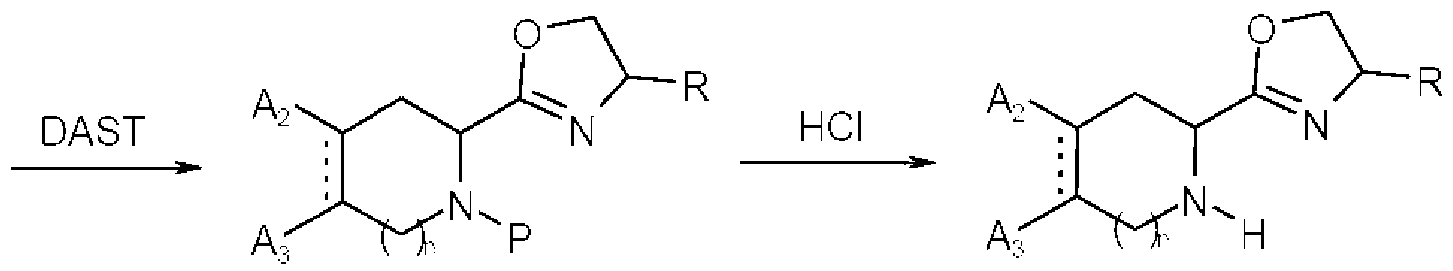
19 Vf
[208] In Reaction Scheme 7, intermediate (11) and amino alcohol starting material (17) are used to give intermediate (18) by EDC coupling reaction. Intermediate (19) is prepared from the intermediate (18) using diethylaminosulfur trifluoride (DAST). An amine- protecting group of the intermediate (19) is removed to give intermediate (Vf). Where the amine-protecting group is t-butoxycarbonyl (B oc), the amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid.
[209] The amine or its corresponding salt (for examples, hydrochloricacid salt or trifluoro- aceticacid salt), represented by Fomula (Ia) below, is prepared by the following Reaction Scheme 8.
[210] [Reaction Scheme 8] [211]

21
[212] In Reaction Scheme 8, A4, A5 and n are as defined in the above above, and an amine- protecting group (P) is t-butoxycarbonyl (Boc), benzyl (Bn), benzylbenzyloxycarbonyl and the like, preferably t-butoxycarbonyl (Boc). Dicarbonyl intermediate (20) is reacted with amidine (21) to form heterocycle, i.e., intermediate (22) in the presence of pyridine solvent. Where the amine-protecting group (P) is t-butoxycarbonyl group (Boc), an amine-protecting group (P) is removed by 4N hydrochloric acid-dioxane solution or trifluoroacetic acid to give amine or its corresponding salt (Vg). The detailed preparation method is also described in Korean Patent Application No. 2006-29138.
[213] Compound (I) in which Z is -CH- can be prepared by the following Reaction Scheme 9 using the compounds of Fomulas (IV) and (V) obtained in the above.
[214] [215] [Reaction Scheme 9]

IV 23

[217] In Reaction Scheme 9, an aldehyde intermediate (23) is obtained by oxidation using
2,2,6,6-tetramethyl-l-piperidinyloxy free radical (TEMPO), lithium bromide, sodi- umbicarbonate, and alcohol (IV) as a starting material obtained from Reaction Scheme 3, or by Swern oxidation method using dimethylsulfuroxide (DMSO) and oxalic acid in the presence of methylenechloride solvent. Intermediate (24) can be obtained by reducing aminization reaction of the aldehyde intermediate (23) and the compound of Fomula (V). Where the amine-protecting group is t-butoxycarbonyl group (Boc), an amine-protecting group of the intermediate (24) is removed by 4N hydrochloric acid- dioxane solution or trifluoroacetic acid/methylene chloride to give Compound (I) in which the substituent Z is CH2.
[218] Further, the compound of Fomula (I) in which the substituent Z is -CO- can be converted to Compound (I1), by the following Reaction Scheme 10, which can is prepared from the compounds of Fomula (V) and Fomula (IV) obtained by the above method.
[219] [Reaction Scheme 10]

[221] Intermediate (25) can be prepared by a general amino acid synthesis method; for example, l-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 1-hydroxybenzotriazole (HOBT) and amine base (for example, N,N-diisopropylethylamine) are reacted in the presence of N,N-dimethylformamide (DMF) or ethylene chloride solution. Where an amine-protecting group (P1) is t- butoxycarbonyl group (Boc), Compound (T) in which the substituent Z is -CO- can be obtained from the intermediate (25) by treatment with 4N hydrochloric acid-dioxane solution or trifluoroacetic acid/methylene chloride in the presence of methylenechloride solvent.
[222] When the preparation methods for any compounds as starting materials are not
explained in the present invention, these compounds are well known or can be synthesized by a known method or similar methods.
[223] A compound of Formula (I) can be isolated and purified from the reaction product by means of conventional methods such as recrystallization, ion electrophoresis, silica gel column chromatography or ion exchange resin chromatography and the like.
[224] As described above, the compounds according to the present invention, starting materials for preparation thereof and intermediates can be synthesized by various methods, thus those methods should be interpreted to be included within the scope of the present invention in view of preparation of the compound of Formula (I).
[225] The present invention also provides a pharmaceutical composition for inhibiting
DPP-IV comprising the compound of Formula (I) or a pharmaceutically acceptable salt thereof.
[226] The term "pharmaceutical composition" as used herein can include a mixture of a compound of the invention with other chemical components, such as diluents or carriers. Accordingly, carriers, diluents, excipient(s), or their combination can be included in the pharmaceutical composition, if necessary. The pharmaceutical composition facilitates administration of the compound to an organism. Multiple techniques of administering a compound exist in the art including, but are not limited to oral, injection, aerosol, parenteral, and topical administrations.
[227] The compound of the present invention can be administered in various pharmaceutical dosage forms in accordance with intended use. In the preparation of pharmaceutical compositions in accordance with the present invention, an active agent, more specifically the compound of Formula (I) may be mixed with one or more pharmaceutically acceptable carriers which can be selected depending on the dosage form to be prepared. For example, the pharmaceutical composition according to the present invention can be formulated into dosage forms suitable for injection or oral administration.
[228] The compound of Formula (I) may be formulated in a conventional manner using known pharmaceutically acceptable carriers and excipients and presented in unit dosage form or in multidose containers. The formulations may take such forms as solutions, suspensions or emulsions in oily or aqueous vehicles, and may contain conventional dispersing, suspending or stabilizing agents. Alternatively, the active ingredient may be in the powder form for reconstitution with sterile pyrogen-free water, before use. The compound of Formula (I) may also be formulated into suppositories containing conventional suppository bases such as cocoa butter or other glycerides. Solid dosage forms for oral administration include capsule, tablet, pill, powder and granule. Preferable dosage forms are capsule and tablet. It is preferable that tablets and pills be coated. The solid dosage forms for oral administration may be obtained by
mixing the compound of Formula (I) as an active agent with inactive diluents such as sucrose, lactose, starch and the like and carriers such as lubricant, for example magnesium stearate, including disintegrator, binder and the like.
[229] If necessary, the compound of Formula (I) and compositions comprising the same according to the present invention may be administrated in combination with other pharmaceutical agents, for example, other diabetes treating agents.
[230] When the formulation is presented in the unit dosage form, the compound of Formula (I) as an active agent can be preferably contained in an amount of about 0.1 ~ 1,500 mg unit dosage. The dosage amount of the compound of Formula (I) will be dependent on the subject's weight and age, the nature and severity of the affliction and the judgment of the prescribing physician. For adult administration, the dosage amount required will be about in the range of 1 to 500 mg a day depending on the frequency and strength of the dosage. For intramuscular or intravenous administration to adults, a total dosage amount of about 5 ~ 300 mg a day will be sufficient. In some patients, the dosage amount in a day will be higher than that.
[231] Further, the present invention provides the method for treatment or prevention of diseases involving inappropriate activity of DPP-IV by using the compound of Formula (I) as defined in claim 1 in effective dose. Representative examples of the diseases caused by inappropriate levels of DPP-IV include, but are in no way limited to, diabetes mellitus, obesity and the like as described above. Among diabetes mellitus, the present invention isuseful to treat and prevent type II diabetes mellitus. Best Mode for Carrying Out the Invention
[232] The present invention will now be illustrated in more detail by the following preparations and examples. However, it will be understood that the present invention is not limited to these specific preparations and examples.
[233]
[234] PREPARATION 1: Synthesis of
GSVN-phenyl-1.2.3.4-tetrahydroisoquinoline-3-carboxamide hydrochloric acid salt
[235] mSvnthesis of t-butyl
GSV3-(anilinocarbonylV3.4-dihydroisoquinoline-2-QHVcarboxylate
[236] 118 mg (0.43 mmol) of
(3S)-2-(tbutoxycarbonyl)-l,2,3,4-tetraisoquinoline-3-carboxylic acid was dissolved in 20 mL of methylene chloride, and then 86 mg (0.64 mmol) of 1-hydroxybenzotriazole, 98 mg (0.51 mmol) of EDC and 41.6 mg (0.45 mmol) of toluene were added thereto at room temperature in sequence. After stirring for 5 minutes, 0.22 mL (1.28 mmol) of diisopropyl ethylamine was dropwise added at room temperature to the solution. After further stirring for 18 hours, the solution was extracted with ethylacetate and 0.5N HCl
aqueous solutions. The organic layer was washed with sodiumbicarbonate aqueous solutions, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The residue was isolated and purified with Prep- TLC to give 67 mg of the title compound in a yield of 45%.
[237] 1H NMR (CDCl3) bill -123 (9H, m), 4.64 (2H, brs), 3.36 (IH, brs), 3.14 (IH, brs), 1.50 (9H, brs)
[238] Mass (m/e) 353 (M+l)
[239]
[240] (2)Svnthesis of (3S)-N-phenyl- 1 ^.SΛ-tetrahvdroisoquinoline-S-carboxamide hy- drochloric acid salt
[241] 67 mg (0.190 mmol) of t-butyl (3S)-3-(anilinocarbonyl)-3, 4-dihydroiso quinoline- 2-(lH)-carboxylate obtained in PREPARATION 1(1) was dissolved in 1 mL of methylene chloride at room temperature, then 4 mL of 4N hydrochloric acid- 1,4-dioxane solution was added thereto. After stirring for 15 minutes, 2.5 mL of 4N hydrochloric acid-l,4-dioxane solution was added further to the solution, followed by stirring for 20 minutes further, then the resulting solution was concentrated under reduced pressure to give 50 mg of the title compound in a yield of 91%.
[242] 1H NMR (CD3OD) δ 7.68-7.66 (2H, m), 7.39-7.29 (6H, m), 7.19-7.16 (IH, m), 4.41-4.37 (IH, m), 3.77-3.69 (2H, m), 3.61-3.48 (IH, m), 3.36-3.27 (IH, m)
[243] Mass (m/e) 253 (M+l)
[244]
[245] PREPARATION 2: Synthesis of
(2S)-2-(3-cvclobutyl-1.2.4-oxadiazole-5-yl)piperidine hydrochloric acid salt
[246] mSvnthesis of t-butyl
(2S)-2-(3-cvclobutyl-1.2.4-oxadiazole-5-yl)piperidine-l-carboxylate
[247] 200 mg (0.872 mmol) of (2S)-l-(tbutoxycarbonyl)piperidine-2-carboxylic acid was dissolved in 10 mL of N,N-dimethylformaide, and 170 mg (1.05 mmol) of 1,1-carbodimide was added thereto at room temperature. After stirring for 30 minutes, 100 mg (0.872 mmol) of N'-hydroxycyclobutanecarboximide was added thereto, and the reaction solution was heated to 12O0C, followed by stirring for 18 hours. After cooling to room temperature, the solvent was removed under reduced pressure. The residue was isolated and purified with column chromatography to give 204 mg of the title compound in a yield of 76%.
[248] H NMR (CDCl3) δ 5.57-5.44 (IH, m), 4.03 (IH, brs), 3.65-3.58 (IH, m), 2.93 (IH, brs), 2.39-2.26 (6H, m), 2.10-1.94 (2H, m), 1.90-1.80 (2H, m), 1.71-1.60 (2H, m), 1.43 (9H, s)
[249] Mass (m/e) 308 (M+l)
[250]
[251] (2)Svnthesis of (2S)-2-G-cvclobutyl-1.2.4-oxadiazole-5-yl)piperidine hydrochloric acid salt [252] 17 rnL of 4N hydrochloric acid-l,4-dioxane solution was added to 204 mg (0.664 mmol) of t-butyl (2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine-l-carboxylate obtained in PREPARATION 2(1) at room temperature. After stirring for 20 minutes, the reaction solution was concentrated under reduced pressure to give 153 mg of the title compound in a yield of 95%. [253] H NMR (CD3OD) δ 4.86 (IH, dd, J=I 1.6 Hz, J=3.4 Hz), 3.78-3.67 (IH, m),
3.62-3.54 (IH, m), 3.28-3.22 (IH, m), 2.45-2.39 (5H, m), 2.21-2.14 (IH, m), 2.06-1.96
(4H, m), 1.85-1.82 (2H, m) [254] Mass (m/e) 208 (M+ 1) [255] [256] PREPARATION 3: Synthesis of
GS V 3-G-cyclobutyl- 1.2.4-oxadiazole-5-ylV 1.2.3.4-tetraisoquinoline hydrochloric acid salt [257] m Synthesis of t-butyl
GSV3-G-cyclobutyl-1.2.4-oxadiazole-5-ylV3.4-dihydroisoquinoline-2QHVcarboxylat e [258] 203 mg of the title compound was obtained in a yield of 63%, in the same manner as in PREPARATION 2(1), except that 250 mg (0.902 mmol) of
(3S)-2-(tbutoxycarbonyl)-l,2,3,4-tetraisoquinoline-3-carboxylic acid and 103 mg
(0.902 mmol) of N'-hydroxycyclobutanecarboximidamide were used. [259] H NMR (CDC13) 67.26-7.09 (4H, m), 5.89 (IH, brs), 5.49 (IH, brs), 4.86-4.79 (IH, m), 4.56 (IH, brs), 3.55 (IH, brs), 3.38-3.30 (2H, m), 2.28 (4H, brs), 2.04-1.96 (IH, m), 1.54-1.23 (9H, m) [260] Mass (m/e) 356 (M+ 1) [261] [262] (2) Synthesis of GS)-3-G-cvclobutyl-1.2.4-oxadiazole-5-yl)-1.2.3.4-trisoquinoline hydrochloric acid salt [263] 159 mg of the title compound was obtained in a yield of 95%, in the same manner as in PREPARATION 2(2), except that 203 mg (0.571 mmol) of
(3S)-3-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)-3,4-dihydroisoquinoline-2(lH)-carboxylat e obtained in PREPARATION 3(1) was used. [264] H NMR (CDCl3) 67.38-7.32 (4H, m), 5.32 (IH, dd, J=10.4 Hz, 5.2 Hz), 4.87 (3H, brs), 4.68-4.59 (2H, m), 3.82-3.74 (IH, m), 3.69-3.60 (IH, s), 3.51-3.44 (IH, m),
2.46-2.40 (4H, m), 2.24-2.15 (IH, m), 2.13-2.03 (IH, m) [265] Mass (m/e) 256 (M+ 1) [266]
[267] PREPARATION 4: Synthesis of N-
[(3S)- 1.l.SΛ-tetrahvdroisoquinoline-S-ylmethyllbenzamide
[268] (I) Synthesis of t-butyl
(3Ss)-3-(hvdroxyketyls)-3.4-dihvdroisoquinoline-2(lHs)-carboxylate
[269] 2.34 g (8.44 mmol) of
(3S)-2-(t-butoxyCarbonyl)-l,2,3,4-tetraisoquinoline-3-carboxylic acid was dissolved in 30 niL of anhydrous tetrahydrofuran(THF), and the resulting solution was cooled to - 150C. To this solution were added dropwise in sequence 0.97 rnL (8.86 mmol) of N- methylmorpholine and 1.16 mL (8.86 mmol) of isobutylchloroformate at the same temperature. After 50 minutes, suspended reaction solution was added to 492 mg (13.0 mmol) of sodiumborohydride dissolved in 10 mL of water for 20 minutes, at the same temperature. After completion of dropwise addition, the temperature of the reaction solution was raised to O0C, followed by stirring for 2 hours. When the reaction is over, the resulting solution was neutralized with ammoniumchloride aqueous solution, and then the organic layer, obtained by extracting with both IN HCl solution and ethylacetate, was washed with sodiumbicarbonate solution, dried over anhydrous magnesium sulfate, filtered and concentrated by eliminating solvents under reduced pressure. The residue was purified with column chromatography to give 2.05 g of the title compound in a yield of 92%.
[270] Mass (m/e) 264 (M+ 1)
[271]
[272] (2) Synthesis of t-butyl
(3S)-3-(azidomethyl)-3.4-dihvdroisoquinoline-2(lH)-carboxylate
[273] T-butyl (3S)-3-(hydroxyketyl)-3,4-dihydroisoquinoline-2(lH)-carboxylate obtained in PREPARATION 4(1) was dissolved in 40 mL of anhydrous tetrahydrofuran (THF), and cooled to O0C. 3.05 g (116 mmol) of triphenylphosphine was added thereto. To this solution were added dropwise in sequence 2.5 mL (11.6 mmol) of diphenylphos- phorylazide and 1.83 mL (11.6 mmol) of diethylazodicarboxylate (DEAD). After stirring for 2 hours while raising the reaction temperature to room temperature, the resulting solution was extracted with sodium bicarbonate aqueous solution and ethylacetate. The organic layer thus obtained was dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The residue was purified with column chromatography to give 1.85 g of the title compound in a yield of 83%.
[274] Mass (m/e) 289 (M+ 1)
[275]
[276] (3) Synthesis of t-butyl
(3S)-3-(aminomethyl)-3.4-dihydroisoquinoline-2(lH)-carboxylate
[277] 1.85 g (6.40 mmol) of t-butyl
(3S)-3-(azidomethyl)-3,4-dihydroisoquinoline-2(lH)-carboxylate obtained in PREPARATION 4(2) was dissolved in 50 niL of methanol, then 400 mg of activated carbon containing 10% of palladium, was added thereto, followed by stirring for 18 hours under hydrogen atmosphere. The reaction solution was filtered by Cellite and concentrated under reduced pressure without further purification, to give 1.68 g of the title compound in a yield of 100%
[278] Mass (m/e) 263 (M+l)
[279]
[280] (4) Synthesis of t-butyl
(3S)-3-[(benzoylamino)methyll-3Λ-dihvdroisoquinorine-2(lH)- carboxylate
[281] 280 mg (2.10 mmol) of 1-hydroxybenzotriazole and 400 mg (2.10 mmol) of EDC were added in sequence to the solution composed of 500 mg (1.91 mmol) of t-butyl (3S)-3-(aminomethyl)-3,4-dihydroisoquinoline-2(lH)-carboxylate obtained in PREPARATION 4(3) dissolved in 10 mL of methylene chloride. After stirring for 10 minutes, 230 mg (1.91 mmol) benzoic acid which is dissolved in 5 mL of methylene chloride was added thereto. After stirring for 10 minutes further at room temperature, 0.50 mL (2.87 mmol) of diisopropylethylamine was added thereto. After stirring for 12 hours at room temperature, the impure title compound, obtained by concentrating under reduced pressure, was purified with column chromatography (1:10 methanol: methylene chloride) to give 577 mg of the title compound in a yield of 83%.
[282] Mass (m/e) 367 (M+l)
[283]
[284] (5) Synthesis of N-[(3S)-1.2.3.4-tetrahvdroisoquinoline-3-ylmethyllbenzamide
[285] 577 mg (1.57 mmol) of t-butyl
(3S)-3-[(benzoylamino)methyl]-3,4-dihydroisoquinoline-2(lH)-carboxylate obtained in PREPARATION 4(4) was dissolved in 7.0 ml of 4N 1.4-dioaxane hydrochloric aqueous solution. The resulting solution was stirred for 20 minutes, concentrated under reduced pressure. The residue was purified with diethylether to give 300 mg of the title compound in a yield of 64%.
[286] 1H NMR (CD3OD) δ 7.91-7.89 (2H, m), 7.70-7.40 (3H, m), 7.29-7.19 (4H, m), 3.85-3.72 (3H, m), 3.19-3.06 (5H, m),
[287] Mass (m/e) 267 (M+l)
[288]
[289] PREPARATION 5: Synthesis of
GSV3-r(butoxycarbonyls)aminol-4-r(4RV4-methyl-2-oxopyrrolidine-l-yllbutyric acid
[290] (1) Synthesis of t-butvir(2SV4-diazo-2-methyl-3-oxobutyllcarbamate
[291] 1.30 g (5.98 mmol) of methyl
(2S)-3-[(t-butoxycarbonyl)amino]-2-methylpropanoate, obtained by referring to
Korean Patent Application No. 2006-29138, was dissolved in 16 rnL of tetrahy- drofuran and 4 rnL of distilled water, followed by addition of 0.75g (18.0 mmol) of lithium hydroxide thereto and stirred for 15 hours at room temperature. The solvent was distilled off under reduced pressure, and ethylacetate was added thereto, then the resulting solution was neutralized by IN HCl and dried over magnesium sulfate. Thereafter, the solvent was distilled off, and the residue was purified with column chromatography to give 1.05 g of the compound (yield: 87%).
[292] This solution was dissolved in 20 mL of diethylether, cooled to -3O0C. 0.82 mL (5.90 mmol) of triethylamine and 0.78 mL (5.90 mmol) of isobutylchloroformate were added thereto. After stirring for 15 minutes at -3O0C, diazomethane(excess amount) dissolved in dithylether was added slowly thereto. After stirring for 2 hours, acetic acid and ethylacetate were added in sequence thereto, followed by washing with sodiumbi- carbonate and water. The residue was purified with column chromatography to give 0.3 Ig of the title compound (yield: 28%).
[293] 1H NMR (CDCl3) δ 5.35 (IH, s), 5.01 (IH, s), 3.28 (2H, t, J=6Hz), 2.71 (IH, m), 1.45 (9H, s), 1.15 (3H, d, J=6.8Hz),
[294] Mass (m/e) 228 (M+ 1)
[295]
[296] (2) Synthesis of methyl GRV4-r(t-butoxycarbonyls)aminol-3- methylbutanoate
[297] 310 mg (1.36 mmol) of t-butyl[(2S)-4-diazo-2-methyl-3-oxobutyl]carbamate obtained in PREPARATION 5(1) was dissolved in 10 mL of methanol, followed by adding 0.71 mL (4.08 mmol) of diisoprpylethylamine and 62 mg (0.27 mmol) of sil- verbenzoate at -3O0C. After stirring for 2.5 hours at room temperature, the solvent was distilled off under reduced pressure, and the residue was diluted with methylene chloride and filtered by Cellite. Thereafter, the filtrated solution was distilled off under reduced pressure and then purified withcolumn chromatography to give 105 mg of the title compound (yield: 32%).
[298] 1H NMR (CDCl3) δ 4.68 (IH, s), 3.71 (3H, s), 3.11-3.09 (2H, m), 2.41-2.36 (IH, m), 2.24-2.08 (2H, m), 1.47 (9H, s), 1.00 (3H, d, J=6.8Hz)
[299] Mass (m/e) 232(M+1)
[300]
[301] G) Synthesis of methyl GRV4-amino-3- methylbutanoate hydrochloric acid salt
[302] 330 mg of the title compound (yield: 88%) was obtained in the same manner as in PREPARATION 1(2), except that 367 mg (1.60 mmol) of
(3R)-4-[(t-butoxycarbonyl)amino]-3-methylbutanoate obtained in PREPARATION 5(2) was used.
[303] 1H NMR (CD3OD) δ 3.69 (3H, s), 2.92-2.82 (2H, m), 2.51-2.30 (3H, m), 1.07 (3H, d, J=6.8Hz)
[304] Mass (m/e) 132 (M+ 1)
[305]
[306] (4) Synthesis of t-butyl
(3Ss)-3-[(t-butoxycarbonyls)aminol-4-[(4Rs)-4-methyl-2-oxopyrrolidine-l-yllbutanoate
[307] 20 niL of methylene chloride and 5 niL of distilled water were added to 480 mg (1.76 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-hydroxy butanoate. 197 mg (1.94 mmol) of sodiumbromide and TEMPO were added in an amount of catalyst at O0C. The solution composed of 4% sodiumperchloride (2.11 mmol), 428 mg (5.10 mmol) of sodiumbicarbonate and water, prepared in advance, was slowly added thereto. After stirring for 30 minutes, the resulting solution was diluted with methylene chloride, washed with water. The organic layer was dried over magnesiumsulfate. The filtrated aldehyde solution obtained by filtering under reduced pressure was used in next reaction without further purification process.
[308] 330 mg of (3R)-4-amino-3-methylbutanoate hydrochloric acid salt was added to the solution obtained in PREPARATION 5(3) at O0C. After stirring for 20 minutes at the same temperature, 445 mg (2.1 mmol) of sodium triacetoxyborohydride was added thereto. After stirring for 2 hours, stirring was processed further for about 18 hours with raising the reaction temperature to room temperature. Thereafter, the reaction solution was diluted with methylene chloride, and washed with IN Hydrochloric acid and salt water in sequence. The organic layer obtained was dried over anhydrous sulfate magnesium. The impure title compound, obtained by concentrating under reduced pressure, was isolated and purified with column chromatography (3:1 → 1:1 hexane: aceticacid ethyl) to give 367 mg of the title compound in a yield of 77% (2 steps).
[309] 1H NMR (CDCl3) 65.18-5.17 (IH, m), 3.75-3.49 (2H, m), 3.22-3.18 (2H, m),
2.59-2.39 (4H, m), 2.07-2.04 (IH, m), 1.49-1.45 (18H, m), 1.14 (3H, d, J=6.8Hz),
[310] Mass (m/e) 357 (M+ 1)
[311]
[312] (5) Synthesis of
(3S)-3-[(t-butoxycarbonyl)aminol-4-[(4R)-4-methyl-2-oxopyrrolidine-l-yllbutyric acid
[313] 367 mg (1.06 mmol) of t-butyl
(3S)-3-[(t-butoxycarbonyl)amino]-4-[(4R)-4-methyl-2-oxopyrrolidine-l-yl]butanoate obtained in PREPARATION 5(4) was dissolved in 5 mL of dichloromethane/tri- fluoroacetic acid (4/1) solution and stirred for 7 hours at room temperature. Excess tri- fluoroaceticacid and dichloromethane were removed under reduced pressure. This concentrate was dissolved in 4 mL of t-butanol solvent, followed by adding 3 mL of 1 N sodium hydroxide, and 350 mg (1.59 mmol) of di-t-butyldicarbonate (BoC2O) at room
temperature thereto. The resulting solution was stirred for 1 hour at room temperature, and diluted with methylene chloride. The organic layer was washed with salt water, followed by drying over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give 280 mg of the title compound (yield: 88%).
[314] 1H NMR (CDCl3) δl l.26 (IH, s), 5.51 (IH, d, J=4Hz), 4.22-4.15 (IH, m), 3.76-3.56 (2H, m), 3.29-3.27 (2H, m), 2.73-2.50 (4H, m), 2.17-2.12 (IH, m), 1.55-1.49 (9H, m), 1.16 (IH, d, J=4Hz)
[315] Mass (m/e) 301 (M+l)
[316]
[317] PREPARATION 6: Synthesis of t-butyl
( 1 S V 3-hydroxy- 1-1 [5Rl -5-mehtyl-2-oxopiperidine- 1 -yl) methyl lpropylcarbamate
[318] (3S)-3-[(t-butoxycarbonyl)amino]-4-[(5R)-5-mehtyl-2-oxopiperidine-l-yl]butri cacid, obtained by referring to Korean Patent Application No. 2006-29138, was dissolved in 14 mL of tetrahydrofuran, 0.84 mL (7.63 mmol) of N-methylmorpholine was added thereto at -1O0C, and stirred for 5 minutes. After 0.99 mL (7.63 mmol) of isobutylchloroformate was added thereto, stirring was processed further for 30 minutes. The resulting solution was filtered by Cellite, and the temperature was dropped to -1O0C, 0.39 g (10.4 mmol) of sodiumborohydride dissolved in 40 mL of water in advance was added thereto. After the reaction temperature was raised to room temperature, the resulting solution was stirred for 1 hour and diluted with ethylacetoacetate, followed by washing with IN hydrochloric acid, water and sodi- umbicarbonate. The organic solution layer thus obtained was dried over anhydrous magnesium sulfate. The impure title compound, obtained by concentrating under reduced pressure, was isolated and purified with column chromatography (1:1 → 1:2 hexane: aceticacid ethyl) to give 951 mg of the title compound (yield: 46%).
[319] 1H NMR (CDCl3) δ 5.38 (IH, d, J=8 Hz), 4.12-3.92 (2H, m), 3.67-3.65 (3H, m),
3.24-3.20 (IH, m), 3.07-2.95 (2H, m), 2.50-2.32 (2H, m), 2.04-1.98(1H, m), 1.84-1.80 (IH, m), 1.81-1.71 (IH, m), 1.49-1.34 (1OH, m), 1.01 (3H, d, J=6.4 Hz)
[320] Mass (m/e) 301 (M+l)
[321]
[322] PREPARATION 7: Synthesis of t- butyll ( 1 S V 3-hydroxy- 1 - r(4-methyl-2-oxo-2.5-dihydro- lH-pyrrole- 1 -yPmethyllpropyl ) carbamate
[323] 245 mg of the title compound (yield: 42%) was obtained, in the same manner as in PREPARATION 6, except that 620 mg (2.08 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-[4-methyl-2-oxo-2,5-dihydro-lH-pyrrol-l-yl]buty ric acid, obtained by referring to Korean Patent Application No. 2006-29138, was used.
[324] 1H NMR (CDCl3) δ 5.85-5.84 (IH, m), 5.35 (IH, d, J=8.8Hz), 4.02-3.97 (2H, m),
3.86-3.82 (IH, m), 3.68-3.62 (3H, m), 3.50-3.31 (2H, m), 2.06 (3H, s), 1.79-1.74 (IH, m), 1.41-1.39 (9H, m) [325] Mass (m/e) 285 (M+ 1) [326] [327] PREPARATION 8: Synthesis of t- butyl((lSs)-3-hvdroxy-l-([(4Rs)-4-methyl-2-oxopyrrolidine-l-yllmethyl)propyl)carba mate [328] 150 mg of the title compound (yield: 56%) was obtained, in the same manner as in
PREPARATION 6, except that 280 mg (0.930 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-[(4R)-4-methyl-2-oxo pyrrolidine- l-yl]butyric acid obtained in PREPARATION 5 was used. [329] 1H NMR (CDCl3) 65.11 (IH, d, J=8.8Hz), 4.12-3.99 (IH, m), 3.69-3.60 (3H, m),
3.08-3.00 (2H, m), 2.59-2.42 (2H, m), 2.04-1.98 (IH, m), 1.72-1.70 (4H, m), 1.57 (9H, s), 1.09 (3H, d, J=6.8) [330] Mass (m/e) 287 (M+ 1) [331] [332] PREPARATION 9: Synthesis of t- butyl(QSVl-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-hydroxypropyl)carbamate [333] 400 mg of the title compound (yield: 86%) was obtained, in the same manner as in
PREPARATION 6, except that 460 mg (1.380 mmol)
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid, obtained by referring to Korean Patent Application No. 2006-29138, was used. [334] 1H NMR (CDCl3) 65.10 (IH, d, J=8.6Hz), 4.08-3.99 (IH, m), 3.98-3.90 (IH, m),
3.80-3.67 (2H, m), 3.60-3.51 (IH, m), 3.27 (IH, s), 3.01 (IH, dd), 2.59-2.58 (2H, m),
2.28-2.26(2H, m ), 1.75-1.72 (IH, m), 1.43-1.41(9H, m) [335] Mass (m/e) 323 (M+ 1) [336] [337] PREPARATION 10: Synthesis of t- butyl[(lS)-l-[(5.5-difluoro-2-oxopiperidine-l-yl)methyll-3-((2S)-2-[(2-fluorophenoxy
^methyllpyrrolidine- 1 -yl lpropyllcarbamate [338] 3 mL of distilled water and 5 mL of methylene chloride were added to 62 mg (0.190 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9, and then 22 mg (0.209 mmol) of sodium bromide and
0.3 mg of TEMPO (2,2,6,6-tetramethyl-l-piperidinyloxy preradical) were added thereto in icebath sequently. 47 mg (0.550 mmol) of sodiumbicarbonate dissolved in 2 mL of distilled water in advance was dropwise added to the reaction solution in ice
bath. After 20 minutes, 45 mg (0.194 mmol) of
(2S)-l-(3-fluorophenoxy)-N-methylpentane-2-amine chloric acid salt, obtained by referring to WO05/ 120494, was added to filterated solution, obtained by drying over anhydrous magnesium sulfate and filtering the methylene chloride layer at room temperature. After 5 minutes, 60 mg (0.285 mmol) of sodium triacetoxyborohydride was added therero. After stirring the suspension solution for 18 hours, the organic layer was separated by extracting with ethylacetate, dried over anhydrous magnesiunsulfate, and then concentrated under reduced pressure. The thus obtained concentrate was isolated and purified with prep-TLC to give 18 mg of the title compound in a yield of 19%.
[339] 1H NMR (CDCl3) 67.21-7.17 (IH, m), 6.72-6.70 (IH, m), 6.67-6.62 (2H, m), 5.85 (IH, d, J=7.95 Hz), 3.95 (IH, brs), 3.97-3.83 (IH, m), 3.74-3.66 (IH, m), 3.54-3.29 (2H, m), 3.20-3.14 (2H, m), 2.84 (IH, brs), 2.56-2.45 (3H, m), 2.26-2.17 (3H, m), 2.01-1.95 (lH,m), 1.81-1.72 (4H, m), 1.53-1.51 (IH, m), 1.41-1.39 (9H, m)
[340] Mass (m/e) 500 (M+ 1)
[341]
[342] EXAMPLE 1: Synthesis of l-r(2SV2-amino-4-((2SV2-r(3-fluorophenox ys)methyllpyrrolidine-l-yl)butyll-5.5-difluoropiperidine-2-one
[343] . , F
N N
J —
NH2 O
[344] 18 mg (0.036 mmol) of
[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-{(2S)-2-[(2-fluorophenoxy)met hyl]pyrrolidine-l-yl}propyl]carbamate obtained in PREPARATION 10 was dissolved in 1 mL of methylene chloride, and then 1 mL of 4N hydrochloric acid-l,4-dioxane solution was added thereto at room temperature. After 15 minutes, the residual solvent and hydrochloric acid were removed by concentrating under reduced pressure to give 16.9 mg of the title compound in a yield of 100%.
[345] 1H NMR (CD3OD) δ 7.32-7.28 (IH, m), 6.89-6.84 (2H, m), 6.75-6.74 (IH, m),
4.39-4.29 (2H, m), 4.04 (IH, brs), 3.87-3.78 (3H, brs), 3.73-3.63 (5H, m), 3.48-3.45 (IH, m), 3.32-3.28 (IH, m), 2.62-2.58 (2H, m), 2.42-2.33 (3H, m), 2.23-2.13 (3H, m), 2.05-2.02 (IH, m)
[346] Mass (m/e) 400 (M++ 1)
[347]
[348] PREPARATION 11: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-r(2SV2-(r(methylsulfonvD amino! methyl ) pyrrolidine- 1 - yll propyl ) carbamate
[349] 12 mg of the title compound was obtained in a yield of 13%, in the same manner as in PREPARATION 10, except that 62 mg (0.190 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 42 mg (0.194 mmol) of N-
[(2S)-pyrrolidine-2-ylmethyl] methanesulfonamide hydrochloric acid salt obtained by referring to WO05/ 120494 were used. [350] 1H NMR (CDCl3) δ 5.98 (IH, d, J=8.6 Hz), 5.83 (IH, brs), 4.13-4.08 (IH, m),
4.03-3.95 (2H, m), 3.52-3.45 (IH, m), 3.30-3.20 (IH, m), 3.16-3.15 (IH, m), 3.10-3.04
(2H, m), 2.97 (3H, s), 2.84-2.81 (IH, m), 2.62-2.51 (3H, m), 2.34-2.20 (3H, m),
2.13-2.08 (lH,m), 1.87-1.71 (6H, m), 1.40 (9H, s) [351] Mass (m/e) 483 (M+ 1) [352] [353] EXAMPLE 2: Synthesis of N-
(((2SVl-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-yls)butyll pyrrolidine-
2-yl)methyls)methane sulfonamide [354] o O
N N
1 —
N-I2 O
[355] 16.9 mg of the title compound was obtained in a yield of 100%, in the same manner as in EXAMPLE 1, except that 2 mg (0.025 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[(2S)-2-{[(methylsulfonyl) amino]methyl}pyrrolidine-l-yl}propyl]carbamate obtained in PREPARATION 11 and 42 mg (0.194 mmol) of N-[(2S)-pyrrolidine-2-ylmethyl] methanesulfonamide hydrochloric acid salt obtained by referring to WO05/ 120494 were used.
[356] 1H NMR (CD3OD) δ 3.87-3.77 (4H, m), 3.71-3.60 (4H, m), 3.48-3.45 (2H, m),
3.29-3.25 (2H, m), 3.05 (3H, s), 2.61-2.59 (2H, m), 2.41-2.33 (2H, m), 2.28-2.23 (IH, m), 2.16-2.13 (3H, m), 2.08-2.05 (lH,m), 1.92-1.87 (IH, m)
[357] Mass (m/e) 383 (M++l)
[358]
[359] PREPARATION 12: Synthesis of t- butyl(αSV3-r2.4-bisαrifluoromethylV5.8-dihvdropyridor3.4-dlpyrimidine-7(6HVyll- 1 - IY5.5-difruoro-2-oxopiperidine- 1 -yPmethyllpropyl ) carbamate
[360] 59 mg of the title compound was obtained in a yield of 54%, in the same manner as in PREPARATION 10, except that 61.5 mg (0.190 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 60 mg (0.194 mmol) of 2,4-bis(trifluoromethyl)-5,6,7,8-tetrahydro[3,4-d]pyrimidine synthesized by referring
to Korean Patent Application No. 2006-0029138 were used. [361] 1H NMR (CDCl3) δ 5.16 (IH, d, J=8.8 Hz), 4.00-3.90 (IH, m), 3.88-3.75 (4H, m),
3.60-3.51 (IH, m), 3.16-3.15 (3H, m), 2.93-2.87 (IH, m), 2.82-2.75 (2H, m), 2.69-2.63
(IH, m), 2.60-2.53 (IH, m), 2.31-2.20 (2H,m), 1.86-1.77 (IH, m), 1.66-1.55 (IH, m),
1.45-1.40 (1OH, m) [362] Mass (m/e) 576 (M+ 1) [363] [364] EXAMPLE 3: Synthesis of l-((2SV2-amino-4-r2.4-bisαrifluoromethylV5.8-dihvdropyridor3.4-dlpyrimidine-7(6H
)-yllbutyl)-5.5-difluoropiperidine-2-one [365] F F F
_. N N
Fs H 1 F

[366] 53 mg of the title compound was obtained in a yield of 95%, in the same manner as in EXAMPLE 1, except that 59 mg (0.103 mmol) of t- butyl{(lS)-3-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-yl]- l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]propyl}carbamate obtained in PREPARATION 12 was used.
[367] 1H NMR (CD3OD) δ 4.75 (2H, brs), 3.93-3.88 (2H, m), 3.85-3.72 (4H, m), 3.59-3.53 (3H, m), 3.47-3.44 (IH, m), 2.63-2.61 (2H, m), 2.40-2.34 (3H, m), 2.28-2.25 (2H,m)
[368] Mass(EI) 476 (M++ 1)
[369]
[370] PREPARATION 13: Synthesis of t- butvKαSVS-rαS^^-G-cvclopropyl-l^^-oxadiazole-S-vDpyrrolidine-l-yll-l-rrS.S-di fluoro-2-oxopiperidine- 1 yPmethyllpropyl ) carbamate
[371] 50 mg of the title compound was obtained in a yield of 54%, in the same manner as in PREPARATION 10, except that 62 mg (0.190 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 49 mg (0.194 mmol) of 3-cyclopropyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/120494 were used.
[372] 1H NMR (CDCl3) δ 5.46 (IH, d, J=8.4 Hz), 3.84-3.74 (3H, m), 3.65-3.50 (2H, m),
3.36-3.32 (IH, m), 3.23-3.19 (IH, m), 2.85-2.78 (IH, m), 2.61-2.53 (2H, m), 2.50-2.41 (2H, m), 2.30-2.21 (3H,m), 2.12-1.96 (4H, m), 1.92-1.89 (IH, m), 1.69-1.63 (IH, m), 1.60-1.57 (IH, m), 1.41 (9H, s), 1.06-1.03 (4H, m)
[373] Mass (m/e) 484 (M+ 1)
[374]
[375] EXAMPLE 4: Synthesis of l-lflSVl-amino-^rflSVl-G-cvclopropyl-l.l^-oxadiazole-S-vD pyrrolidine-l-yll butyl)-5.5-difluoropiperidine2-one [376] o N \
N -N
I —
NH2 O
[377] 46 mg of the title compound was obtained in a yield of 98%, in the same manner as in EXAMPLE 1, except that 50 mg (0.103 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-[(5,5-di fluoro-2-oxopiperidine-lyl)methyl]propyl]carbamate obtained in PREPARATION 13 was used.
[378] 1H NMR (CD3OD) δ 5.08 (IH, brs), 3.86-3.71 (5H, brs), 3.68-3.60 (2H, m),
3.47-3.40 (3H, m), 2.70-2.58 (3H, m), 2.40-2.30 (5H, m), 2.18-2.10 (2H, m), 1.14-1.10 (2H, m), 1.06-1.02 (2H, m)
[379] Mass (m/e) 384 (M++l)
[380]
[381] PREPARATION 14: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxopiperidine-lvDmethyll-3-r(2SV2-(rφhenylsulfonvDa minol methyl } pyrrolidine- 1 -yll propyl } carbamate
[382] 36 mg of the title compound was obtained in a yield of 30%, in the same manner as in PREPARATION 10, except that 72.2 mg (0.224 mmol) of
{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[(2S)-2-{[(methylsulfonyl)amin o]methyl}pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION 9 and 65 mg (0.235 mmol) of N-[(2S)-pyrrolidine-2-ylmethyl] methanesulfonamide hydrochloric acid salt obtained by referring to WO 05/120494 were used.
[383] H NMR (CDCl3) δ 7.97-7.95 (2H, m), 7.60-7.51 (3H, m), 6.21 (IH, brs), 5.84 (IH, d, J=8.4 Hz), 4.09-3.95 (3H, m), 3.58-3.50 (IH, m), 3.18-3.16 (IH, m), 3.05-2.96 (3H, m), 2.90-2.87 (IH, m), 2.70-2.57 (3H, m), 2.35-2.24 (3H, m), 2.17-2.07 (IH, m), 1.90-1.72 (5H, m), 1.49-1.46 (IH, m), 1.41 (9H, s)
[384] Mass (m/e) 545 (M+l)
[385]
[386] EXAMPLE 5: Synthesis of N-
(I (2S)- l-[(3S)-3-amino-4-(5.5-difluoro-2-oxopiperidine- l-yl)butyllpyrrolidine-2-yl)m ethvDbenzenesulfonamide
[387]
N -N
I
NH2 O [388] 28 mg of the title compound was obtained in a yield of 99%, in the same manner as in EXAMPLE 1, except that 36 mg (0.066 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-lyl)methyl]-3-[(2S)-2-{[(phenylsulfonyl)a mino]methyl}pyrrolidine-l-yl]propyl}carbamate 36 mg (0.066 mmol) obtained in
PREPARATION 14 was used. [389] H NMR (CD3OD) δ 7.96 (2H, d, J=7.6 Hz), 7.72-7.62 (3H, m), 3.98-3.69 (7H, m),
3.61-3.50 (IH, m), 3.41-3.27 (4H, m), 2.68-2.64 (2H, m), 2.46-2.36 (2H, m), 2.30-2.05
(5H, m), 2.00-1.90 (IH, m) [390] Mass (m/e) 445 (M+ 1) [391] [392] PREPARATION 15: Synthesis of t- butyl(αSVl-3-rGSV3-G-cvclobutyl-1.2.4-oxadiazole-5-ylV3.4-dihvdroisoquinoline-2
( lH)-yll- l-r(5.5-difluoro-2-oxopiperidine-lyl)methyllpropyl)carbamate [393] 36 mg of the title compound was obtained in a yield of 43%, in the same manner as in PREPARATION 10, except that 48 mg (0.149 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 68.5 mg (0.225 mmol) of
(3S)-3-(3-cyclobutyl- 1 ,2,4-oxadiazole-5-yl)- 1 ,2,3,4-tetra isoquinoline hydrochloric acid salt obtained in PREPARATION 3 were used. [394] H NMR (CDCl3) δ 7.19-7.14 (3H, m), 7.07-7.03 (IH, m), 5.29 (IH, brs), 4.48-4.45
(IH, m), 3.95-3.85 (3H, m), 3.70-3.57 (3H, m), 3.37-3.35 (IH, m), 3.27-3.22 (2H, m),
2.81 (IH, brs), 2.59-2.55 (2H, m), 2.39-2.24 (6H, m), 2.07-1.99 (3H, m), 1.80-1.68
(2H, m), 1.40 (9H, s)
[395] Mass (m/e) 560 (M+ 1), 582 (M+Na) [396] [397] EXAMPLE 6: Synthesis of l-((2S)-2-amino-4-r(3S)-3-(3-cvclobutyl-1.2.4-oxadiazole-5-yl)-3.4-dihvdroisoquinoli ne-2(lH)-yllbutyl)-5.5-difluoropiperidine-2-one

(lH)-yl]-l-[(5,5-difluoro-2-oxopiperidine-lyl)methyl]propyl}carbamate obtained in
PREPARATION 15 was used. [400] H NMR (CD3OD) δ 7.24-7.20 (4H, m), 5.44-2.17 (IH, m), 4.62-4.46 (2H, m),
3.82-3.59 (5H, m), 3.55-3.33 (6H, m), 2.56-2.44 (2H, m), 2.34-2.21 (7H, m), 2.13-2.02
(IH, m), 1.99-1.85 (IH, m) [401] Mass (m/e) 460 (M+ 1) [402] [403] PREPARATION 16: Synthesis of t- butyl(αSVl-3-r(2SV2-G-cvclobutyl-1.2.4-oxadiazole-5-vDpiperidine-l-yll-l-r(5.5-dif luoro-2-oxopiperidine-lyls)methyllpropyl) carbamate [404] 38 mg of the title compound was obtained in a yield of 32%, in the same manner as in PREPARATION 10, except that 72.2 mg (0.224 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 57 mg (0.235 mmol) of
(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine hydrochloric acid salt obtained in PREPARATION 2 were used. [405] H NMR (CDCl3) δ 5.58-5.56 (IH, m), 4.05 (2H, dd, J=10.0 Hz, 3.2 Hz), 3.88-3.76
(3H, m), 3.71-3.53 (5H, m), 3.32-3.28 (IH, m), 3.19-3.15 (2H, m), 3.06-3.03 (IH, m),
2.83-2.77 (2H, m), 2.58-2.51 (3H, m), 2.45-2.23 (7H, m), 2.12-2.00 (2H, m), 1.90-1.86
(IH, m), 1.77-1.53 (2H, m), 1.42 (9H, s) [406] Mass (m/e) 512 (M+ 1), 534 (M+Na) [407] [408] Example 7: Synthesis of l-((2SV2-amino-4-r(2SV2-G-cvclobutyl-1.2.4-oxadiazole-5-yls)piperidine-l-yllbutyl)-
5.5- difluoropiperidine-2-one

[410] 36 mg of the title compound was obtained in a yield of 99%, in the same manner as in EXAMPLE 1, except that 38 mg (0.074 mmol) of t- butyl{(lS)-l-3-[(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine-l-yl]-l-[(5,5-dif luoro-2-oxopiperidine-lyl)methyl]propyl} carbamate obtained in PREPARATION 16 was used.
[411] H NMR (CD3OD) δ 4.74-4.70 (IH, m), 3.74-3.62 (4H, m), 3.50-3.43 (2H, m),
3.37-3.31 (2H, m), 3.14-3.09 (IH, m), 2.53-2.50 (IH, m), 2.35-2.25 (8H, m), 2.12-2.03
(3H, m), 1.96-1.80 (5H, m), 1.75-1.67 (2H, m) [412] Mass (m/e) 412 (M+l) [413] [414] PREPARATION 17: Synthesis of t- butyl((lSs)-l-[(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-[2-(3-furyls)-4-(trifluorome thyls)-5.8-dihvdropyrido[3.4-dlpyrimidine-7(6Hs)-yllpropyl)carbamate [415] 173 mg of the title compound was obtained in a yield of 58%, in the same manner as in PREPARATION 10, except that 168 mg (0.52 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 175 mg (0.57 mmol) of
2-(3-furyl)-4-(trifluoromethyl)-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidine hydrochloric acid salt obtained by referring to Korean Patent Application No. 2006-29138 were used. [416] H NMR (CDCl3) δ 8.24 (IH, s), 7.49 (IH, m), 7.05 (IH, d, J=0.4 Hz), 5.43 (IH, d,
J=0.8 Hz), 3.96 (IH, brs), 3.91-3.69 (3H, m), 3.61-3.52 (IH, m), 3.22-3.17 (IH, m),
3.04-3.02 (2H, m), 2.90-2.85 (IH, m), 2.79-2.73 (2H, m), 2.69-2.57 (3H, m), 2.31-2.20
(2H, m), 1.86-1.82 (IH, m), 1.67-1.62 (IH, m), 1.28 (9H, s) [417] Mass (m/e) 574 (M+l) [418] [419] EXAMPLE 8: Synthesis of l-((2SV2-amino-4-r2-G-furylV4-αrifluoromethylV5.8-dihvdropyridor3.4-dlpyrimidi ne-7(6H)-yll)butyl)-5.5-difluoropiperidine-2-one [420] j, O
c F N jl N 1 F
F
F N -NN
NH2 o7
[421] 122 mg of the title compound was obtained in a yield of 74%, in the same manner as in EXAMPLE 1, except that 173 mg (0.302 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[2-(3-furyl)-4-(trifluorome thyl)-5,8-dihydropyrido[3,4-d]pyrimidine-7(6H)-yl]propyl}carbamate obtained in
PREPARATION 17 was used. [422] H NMR (CD3OD) δ 8.39 (IH, d, J=0.4 Hz), 7.67 (IH, m), 7.09 (IH, d, J=2.0 Hz),
4.72 (2H, brs), 3.99-3.82 (5H, m), 3.73-3.60 (3H, m), 3.55-3.46 (3H, m), 2.71-2.60
(2H, m), 2.47-2.36 (4H, m)
[423] Mass (m/e) 474 (M+ 1)
[424]
[425] PREPARATION 18: Synthesis of t- butylf αSV3-rGSV3-rα?enzoylamino)metfayl1-34-dihvdroisoquinoline-2flHVyl1-l-r(5
.5-difluoro-2-oxopiperidine-l-yls)methyllpropyl)carbamate [426] 159 mg of the title compound was obtained in a yield of 54%, in the same manner as in PREPARATION 10, except that 168 mg (0.52 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 173 mg (0.57 mmol) of N-
[(3S)-l,2,3,4-tetrahydroisoquinoline-3-ylmethyl]benzamide hydrochloric acid salt obtained in PREPARATION 4 were used. [427] H NMR (CDCl3) δ 7.96-7.92 (2H, m), 7.48-7.37 (4H, m), 7.14-7.10 (2H, m),
7.01-7.06 (IH, m), 7.01-6.99 (IH, m), 5.09 (IH, d, J=9.6 Hz), 4.21-4.19 (IH, m),
3.94-3.87 (2H, m), 3.77-3.50 (3H, m), 3.47-3.35 (2H, m), 3.23-3.20 (IH, m), 3.08-3.02
(IH, m), 2.95-2.91 (IH, m), 2.75-2.67 (2H, m), 2.60-2.50 (3H, m), 2.29-2.19 (2H, m),
1.78-1.73 (IH, m), 1.37 (9H, s) [428] Mass (m/e) 571 (M+ 1) [429] [430] EXAMPLE 9: Synthesis of N- α(3SV2-r(3SV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-vDbutyll-1.2.3.4-tetraisoqui noline- 3 - yl ) methvDbenzamide [431]

[432] 112 mg of the title compound was obtained in a yield of 74%, in the same manner as in EXAMPLE 1, except that 159 mg (0.28 mmol) of t- butyl{(lS)-3-[(3S)-3-[(benzoylamino)methyl]-3,4-dihydroisoquinoline-2(lH)-yl]-l-[(5 ,5-difluoro-2-oxopiperidine-l-yl)methyl]propyl}carbamate obtained in PREPARATION 18 was used.
[433] H NMR (CD3OD) δ 7.90-7.82 (2H, m), 7.55-7.54 (IH, m), 7.47 (2H, brs), 7.35-7.30 (4H, m), 4.70-4.38 (2H, m), 4.13 (IH, brs), 4.01 (IH, brs), 3.87-3.69 (6H, m), 3.64-3.55 (2H, m), 3.45-3.33 (2H, m), 3.20-3.15 (IH, m), 2.64-2.53 (2H, m), 2.40-2.30 (5H, m)
[434] Mass (m/e) 471 (M+ 1)
[435]
[436] PREPARATION 19: Synthesis of t-butylf flSV3-f (2SV2-rα?enzoylami no^methyllpyrrolidine- 1 yl ) - 1 - [(5.5-difluoro-2-oxopiperidine- 1 - vDmethyll propyl ) carba mate [437] 66 mg of the title compound was obtained in a yield of 25%, in the same manner as in PREPARATION 10, except that 168 mg (0.52 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-hydroxypropyl}carbamate obtained in PREPARATION 9 and 138 mg (0.57 mmol) of N-
[(2S)-pyrrolidine-2-ylmethyl] methanesulfonamide hydrochloric acid salt obtained by referring to WO 05/120494 were used. [438] H NMR (CDCl3) δ 7.99 (2H, d, d=7.2 Hz), 7.60 (IH, brs), 7.49-7.38 (3H, m), 5.44
(IH, d, J=9.6 Hz), 4.22-4.19 (IH, m), 4.15-4.11 91H, m), 4.09-3.93 (2H, m), 3.56-3.48
(IH, m), 3.15-3.12 (2H, m), 3.08-3.01 (IH, m), 2.80 (IH, dd, J=12.0 Hz, 4.0 Hz),
2.62-2.50 (3H, m), 2.31-2.20 (3H, m), 2.10-2.02 (IH, m), 1.93-1.85 (IH, m), 1.83-1.64
(4H, m), 1.48-1.42 (IH, m), 1.33 (9H, m) [439] Mass (m/e) 509 (M+ 1) [440] [441] EXAMPLE 10: Synthesis of N-
({ (2SV l-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine- l-yls)butyllpyrrolidine-2-yl)m ethyPbenzamide [442]
I F _
^ N O \ F
N - N
NH2 O
[443] 55 mg of the title compound was obtained in a yield of 98%, in the same manner as in Example 1, except that 66 mg (0.13 mmol) of t- butyl{(lS)-3-{(2S)-2-[(benzoylamino)methyl]pyrrolidine-lyl}-l-[(5,5-difluoro-2-oxop iperidine-l-yl)methyl]propyl}carbamate obtained in PREPARATION 19 was used.
[444] H NMR (CD3OD) δ 7.98 (2H, d, J=7.2 Hz), 7.62-7.58 (IH, m), 7.53-7.50 (2H, m),
4.01-3.75 (8H, m), 3.71-3.66 (IH, m), 3.61-3.59 (IH, m), 3.55-3.52 (IH, m), 3.43 (IH, brs), 3.30-3.22 (IH, m), 2.72-2.59 (2H, m), 2.45-2.19 (6H, m), 2.06-2.02 (2H, m)
[445] Mass (m/e) 409 (M+ 1), 431 (M+Na)
[446]
[447] PREPARATION 20: Synthesis of t- butvirαSV3-r(2S.4SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v ll-l-(r(4RV4-methyl-2-oxopyrrolidine-l-yllmethyl)propyllcarbamate
[448] 60 mg of the title compound was obtained in a yield of 31%, in the same manner as
in PREPARATION 10, except that 120 mg (0.419 mmol) of t- butyl{(lS)-3-hydroxy-l-{[(4R)-4-methyl-2-oxopyrrolidine-l-yl]methyl}propyl}carba mate obtained in PREPARATION 8 was used. [449] H NMR (CDCl3) δ 5.41 (IH, d, J=12.0 Hz), 5.28-5.12 (IH, m), 3.81-3.78 (2H, m),
3.51-3.47 (2H, m), 3.34-3.30 (IH, m), 3.10-2.94 (2H, m), 2.69-2.37 (5H, m), 2.11-1.95
(3H, m), 1.72-1.54 (2H, m), 1.41 (9H, s), 1.10 (3H, d, J=6.8 Hz), 1.08-1.02 (5H, m) [450] Mass (m/e) 466 (M+ 1), 488 (M+Na) [451] [452] EXAMPLE 11: Synthesis of
(4RVl-((2SV2-amino-4-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyr rolidine- 1 -yllbutyl ) -4-methylpyrrolidine-2-one [453] o N /\
r N N i t-
NH2 O
[454] 54.8 mg of the title compound was obtained in a yield of 83%, in the same manner as in EXAMPLE 1, except that 60 mg (0.13 mmol) of t- butyl[(lS)-3-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolidine-l-y l]-l-{[(4R)-4-methyl-2-oxopyrrolidine-l-yl]methyl}propyl]carbamate obtained in PREPARATION 20 was used.
[455] H NMR (CD3OD) δ 5.58-5.48 (IH, m), 5.26-5.17 (IH, m), 4.13-4.08 (IH, m),
3.74-3.70 (IH, m), 3.67-3.46 (6H, m), 3.19-3.10 (2H, m), 2.80-2.70 (IH, m), 2.58-2.52 (2H, m), 2.16-2.07 (4H, m), 1.51-1.10 (5H, m), 1.06-0.99 (2H, m)
[456] Mass (m/e) 366 (M+l), 388 (M+Na)
[457]
[458] PREPARATION 21: Synthesis of t- butvirαSV3-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v 11-1-1 r(5R)-5-methyl-2-oxopiperidine- 1 -yllmethyl Ipropyllcarbamate
[459] 124 mg of the title compound was obtained in a yield of 62%, in the same manner as in PREPARATION 10, except that 126 mg (0.419 mmol) of t-butyl ( 1 S)-3-hydroxy- 1 - { [5R] -5-mehtyl-2-oxopiperidine- 1 -yljmethyl jpropylcarbamate obtained in PREPARATION 6 was used.
[460] H NMR (CDCl3) δ 5.53 (IH, d, J=8.4 Hz), 5.27-5.14 (IH, m), 3.81-3.77 (IH, m),
3.56-3.48 (2H, m), 3.35-3.23 (2H, m), 3.06-2.92 (2H, m), 2.69-2.53 (2H, m), 2.49-2.29 (5H, m), 2.13-2.06 (IH, m), 1.93 (IH, brs), 1.80 (IH, brs), 1.67-1.58 (2H, m), 1.41 (9H, s), 1.07-1.04 (5H, m), 1.00 (3H, d, J=6.4 Hz)
[461] Mass (m/e) 480 (M+l)
[462]
[463] EXAMPLE 12: Synthesis of
(SRVl-iαS^^-amino^-rαS^S^^-G-cvclopropyl-l^^-oxadiazole-S-vD^-fluoropyr rolidine- 1 -yllbutyl ) -5-methylpiperidine-2-one [464]

[465] 76.2 mg of the title compound in a yield of 56% was obtained, in the same manner as in EXAMPLE 1, except that 124 mg (0.26 mmol) of t- butyl[(lS)-3-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolidine-l-y l]-l-{[(5R)-5-methyl-2-oxopiperidine-l-yl]methyl}propyl]carbamate obtained in PREPARATION 21 was used.
[466] H NMR (CD3OD) δ 5.58-5.48 (IH, m), 5.18-5.14 (IH, m), 4.12-4.06 (IH, m),
3.73-3.63 (2H, m), 3.56-3.52 (2H, m), 3.48-3.34 (IH, m), 3.20-3.04 (2H, m), 2.80-2.69 (IH, m), 2.45-2.36 (2H, m), 2.17-2.10 (2H, m), 2.04 (IH, brs), 1.86-1.83 (IH, m), 1.56-1.48 (IH, m), 1.17-1.11 (2H, m), 1.05-1.03 (4H, m)
[467] Mass (m/e) 380 (M+ 1), 402 (M+Na)
[468]
[469] PREPARATION 22: Synthesis of t- butyl(αSV3-r(2S.4SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v 11-1 - r(4-methyl-2-oxo-2.5-dihydro- lH-pyrrole- 1 -yPmethyllpropyl ) carbamate
[470] 89 mg of the title compound was obtained in a yield of 46%, in the same manner as in PREPARATION 10, except that 119 mg (0.419 mmol) of t- butyl{ ( 1 S)-3-hydroxy- 1 - [(4-methyl-2-oxo-2,5-dihydro- lH-pyrrole- 1 -yl)methyl]propyl } carbamate obtained in PREPARATION 7 was used.
[471] H NMR (CDCl3) δ 5.83-5.78 (IH, m), 5.49-5.48 (IH, m), 5.29-5.23 (IH, m),
5.18-5.12 (IH, m), 4.40-4.38 (IH, m), 3.98-3.74 (4H, m), 3.50-3.41 (2H, m), 3.08-2.98 (IH, m), 2.66-2.32 (5H, m), 2.10-2.02 (5H, m), 1.67-1.55 (IH, m), 1.41-1.35 (2H, m), 1.08-1.01 (9H, m)
[472] Mass (m/e) 464 (M+ 1), 486 (M+Na)
[473]
[474] EXAMPLE 13: Synthesis of l-((2SV2-amino-4-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5ylV4- fluoropyrrolidine- 1 -yllbutyl ) -4-methyl- 1.5-dihvdro-2H-pyrrole-2-one
[475]

[476] 87.4 mg of the title compound was obtained in a yield of 89%, in the same manner as in EXAMPLE 1, except that 89 mg (0.19 mmol) of t- butyl{(lS)-3-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolidine-l-y I]-I - [(4-methyl-2-oxo-2,5-dihydro- lH-pyrrole- 1 -yl)methyl]propyl } carbamate obtained in PREPARATION 22 was used.
[477] H NMR (CD3OD) δ 5.64-5.51 (IH, m), 5.31-5.29 (IH, m), 4.20-4.10 (IH, m),
3.89-3.60 (7H, m), 3.08-2.92 (2H, m), 2.85-2.75 (2H, m), 2.19-2.15 (5H, m), 1.16-1.14 (3H, m), 1.08-1.04 (2H, m)
[478] Mass (m/e) 364 (M+ 1), 386 (M+Na)
[479]
[480] PREPARATION 23: Synthesis of
3-cvclopropyl-5-[(2S.4Ss)-4-fluoropyrrolidine-2-yll-1.2.4- oxadiazole hydrochloric acid salt
[481] (DSynthesis of (4SVl-(t-butoxycarbonylV4-fluoro-L-proline
[482] 500 mg (2.02 mmol) of 1-t-butyl 2-methyl
(2S,4S)-4-fluoropyrrolidine-l,2-dicarboxylate obtained by referring to WO 03/002553 was dissolved in 16 mL of tetrahydrofuran and 4 mL of distilled water. Thereafter, 250 mg (6.06 mmol) of lithium hydroxide was added thereto, then stirred for 15 hours at room temperature. The solvent was distilled off under reduced pressure, and ethylacetoacetate was added thereto. The resulting solution was neutralized with IN hydrochloric acid, then dried over magnesium sulfate. The solvent was distilled off under reduced pressure to give 330 mg of the title compound (yield: 70%).
[483] Mass (m/e) 234 (M+ 1)
[484]
[485] (^Synthesis of t-butyl (2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4- fluoropyrrolidine- 1 -carboxylate
[486] 699 mg of the title compound was obtained in a yield of 61%, in the same manner as in PREPARATION 2(1), except that 900 mg (3.86 mmol) of
(4S)-l-(t-butoxycarbonyl)-4-fluoro-L-proline obtained in PREPARATION 23(1) and 386 mg (3.86 mmol) of N'-hydroxycyclopropanecarboximidamide were used.
[487] 1H NMR (CDCl3) δ 5.31-5.12 (2H, m), 3.97-3.67 (2H, m), 2.59-2.52 (2H, m), 2.07-2.00 (IH, m), 1.48-1.35 (9H, m), 1.02-1.01 (4H, m)
[488] Mass (m/e) 298 (M+l)
[489]
[490] G^Svnthesis of 3-cvcloporpyl-5-r('2S.4SV4-fluoropyrrolidine-2-yll-1.2.4-oxadiazole hydrochloric acid salt
[491] 450 mg of the title compound (yield: 82%) was obtained, in the same manner as in PREPARATION 1(2), except that 699 mg (2.35 mmol) of t-butyl (2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolidine-l-carboxylate obtained in PREPARATION 23(2) was used.
[492] 1H NMR (CD3OD) δ 5.66-5.51 (IH, m), 5.34-5.31 (IH, m), 3.89-3.74 (IH, m), 3.09-2.93 (IH, m), 2.85-2.73 (IH, m), 2.21-2.14 (IH, m), 1.21-0.97 (4H, m)
[493] Mass (m/e) 198 (M+l)
[494]
[495] PREPARATION 24: Synthesis of
GR.SSVS-G-cyclopropyl-l^^-oxadiazole-S-yDpyrrolidine-S-ol hydrochloric acid salt
[496] mSvnthesis of 1 -t-butyl 2-methyl
(2S.4RV4-Q-ethoxyethoxys)pyrrolidine-1.2-dicarboxylate
[497] 5.0 g (0.0204 mol) of trans -L-hydroxyproline methylester was dissolved in 100 mL of methylenechloride, and to this solution were added 2.34 mL (0.0245 mol) of ethylv- inylester and 513 mg (2.04 mmol) of pyridinium p-toluenesulfonate in sequence. After stirring for 2 hours at room temperature, the solvent was removed under reduced pressure. The resulting solution was extracted with 100 mL of diethylester and dilute sodiumbicarbonate solution. Thereafter, the organic layer thus obtained was dried over anhydrous magnesium sulfate and filtered. The residue was concentrated under reduced pressure to give 6.30 g of the title compound in a yield of 97% without further purification.
[498] 1H NMR (CDCl3) 64.76-4.71 (IH, m), 4.43-4.31 (2H, m), 3.73 (3H, d, J=4.0 Hz),
3.71-3.42 (4H, m), 2.36-2.22 (IH, m), 2.13-2.04 (IH, m), 1.46-1.41 (9H, m), 1.31-1.28 (3H, m), 1.22-1.76 (3H, m)
[499] Mass (m/e) 318 (M+l)
[500]
[501] (2)Svnthesis of (4R)-l-(t-butoxycarbonyl)-4-(l-ethoxyethoxy)-L-proline
[502] 6.30 g (19.8 mmol) of 1-t-butyl 2-methyl
(2S,4R)-4-(l-ethoxyethoxy)pyrrolidine-l,2-dicarboxylate obtained in PREPARATION 24(1) was dissolved in 200 mL of the solvent composed of tetrahydrofuran(THF) : water : methanol (1 : 1 : 1), then 1.71 g (40.8 mmol) of lithiumhydoxide was added thereto and stirred for 15 hours at room temperature. The solvent was distilled off under reduced pressure, and ethylacetoacetate was added thereto. The resulting solution was neutralized with IN hydrochloric acid, and then dried over magnesium sulfate. The solvent was distilled off under reduced pressure to give 6.0 g of the title
compound (yield: 100%).
[503] 1H NMR (CDCl3) δ 4.75-4.73 (IH, m), 4.49-4.34 (2H, m), 3.65-3.44 (4H, m), 2.46-2.11 (2H, m), 1.49-1.44 (9H, m), 1.32-1.20 (6H, m)
[504] Mass (m/e) 304 (M+ 1)
[505]
[506] G^Svnthesis of t-butyl
(2S.4R)-2-(3-cvclopropyl-1.2.4-oxadiazole-5-yl)-4-(l-ethoxyethoxy)pyrrolidine-l-carb oxylate
[507] 425 mg of the title compound (yield: 17%) was obtained, in the same manner as in PREPARATION 2(1), except that 2.07 g (6.82 mmol) of
(4R)-l-(t-butoxycarbonyl)-4-(l-ethoxyethoxy)-L-proline obtained in PREPARATION 24(2) and 683 mg (6.82 mmol) of hydroxyl cyclopropanecarboximidamide were used.
[508] 1H NMR (CDCl3) 65.13-5.08 (IH, m), 5.01-4.96 (IH, m), 4.75-4.68 (IH, m),
4.45-4.38 (IH, m), 3.74-3.65 (IH, m), 3.62-3.54 (3H, m), 3.46-3.40 (2H, m), 2.42-2.31 (IH, m), 2.21-2.12 (IH, m), 2.08-2.00 (IH, m), 1.30-1.20 (9H, m), 1.18-1.13 (3H, m), 1.03-0.94 (3H, m)
[509] Mass (m/e) 368 (M+ 1)
[510]
[511] Synthesis of t-butyl
(2S.4RV2-G-cyclopropyl-1.2.4-oxadiazole-5-ylV4-hydroxypyrrolidine-l-carboxylate
[512] 1.97 g (5.36 mmol) of t-butyl
(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-(l-ethoxyethoxy)pyrrolidine-l-carb oxylate obtained in PREPARATION 24(3) was dissolved in 30 mL of anhydrous ethanol, and then 135 mg (0.536 mmol) of pyridinium p-toluenesulfonate (PPTS) was added thereto. After the solution was heated to 5O0C and then cooled to room temperature, the ethanol is removed under reduced pressure to give 1.52 g of the title compound without further purification (yield: 96%).
[513] 1H NMR (CDCl3) δ 5.11-5.07 (IH, m), 3.75-3.71 (IH, m), 3.64-3.62 (IH, m),
2.57-2.50 (IH, m), 2.42-2.37 (IH, m), 2.22-2.16 (IH, m), 2.07-2.04 (IH, m), 1.28-1.26 (9H, m), 1.12-1.01 (4H, m)
[514] Mass (m/e) 296 (M+l)
[515]
[516] (^Synthesis of GR. SSyS-G-cyclopropyl-l^^-oxadiazole-S-yDpyrrolidineG-ol hy- drochloric acid salt
[517] 40 mg of the title compound (yield: 94%) was obtained, in the same manner as in PREPARATION 1(2), except that 54 mg (0.183 mmol) of
(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-hydroxypyrrolidine-l-carboxylate obtained in PREPARATION 24(4) was used.
[518] 1H NMR (CD3OD) δ 5.20-5.18 (IH, m), 3.65-3.64 (IH, m), 3.56-3.53 (IH, m),
3.29-3.28 (IH, m), 2.58-2.54 (IH, m), 2.43-2.37 (IH, m), 2.16-2.11 (IH, m), 1.13-1.08 (2H, m), 1.12-0.98 (2H, m)
[519] Mass (m/e) 196 (M+l)
[520]
[521] PREPARATION 25: Synthesis of
3-cvclopropyl-5-[(2S.4Rs)-4-methoxypyrrolidine-2-yll-1.2.4-oxadiazole hydrochloric acid salt
[522] mSvnthesis of t-butyl
(2S.4Rs)-2-(3-cvclopropyl-1.2.4-oxadiazole-5-yls)-4-methoxypyrrolidine-l-carboxylate
[523] 206 mg (0.698 mmol) of
(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-hydroxypyrrolidine-l-carboxylate obtained in PREPARATION 24(4) was dissolved in 10 rnL of anhydrous tetrahy- drofuran (THF), then 34 mg (0.837 mmol) of sodium hydride (55% dispersion in mineral oil) was added thereto in ice bath and stirred for 5 minutes. Thereafter, 0.065 mL (1.05 mmol) of idiomethane as a reaction solution was added thereto, the icebath was removed. After 1 hour, the resulting solution was extracted with ammonium chloride solution and ethylacetate, and the organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified with column chromatography to give 152 mg (yield: 70%).
[524] 1H NMR (CDCl3) 65.13-5.00 (IH, m), 4.06-3.99 (IH, m), 3.78-3.53 (2H, m), 3.36 (3H, s), 2.50-2.34 (IH, m), 2.22-2.06 (2H, m), 1.35-1.26 (9H, m), 1.07-1.01 (4H, m)
[525] Mass (m/e) 310 (M+l)
[526]
[527] (^Synthesis of
3-cvclopropyl-5-[(2S.4R)-4-methoxypyrrolidine-2-yll-1.2.4-oxadiazole hydro chloric acid
[528] 110 mg of the title compound (yield: 91%) was obtained, in the same manner as in PREPARATION 1(2), except that 152 mg (0.491 mmol) of
(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-methoxypyrrolidine-l-carboxylate obtained in PREPARATION 25(1) was used.
[529] 1H NMR (CD3OD) δ 5.19-5.12 (IH, m), 4.33-4.28 (IH, m), 3.77-3.51 (2H, m), 3.41 (3H, s), 2.83-2.67 (IH, m), 2.43-2.36 (IH, m), 2.21-2.15 (IH, m), 1.28-1.02 (4H, m)
[530] Mass (m/e) 210 (M+l)
[531]
[532] PREPARATION 26: Synthesis of (2SVN-phenylpiperidine-2-carboxamide hy- drochloric acid salt
[533] (DSynthesis of t-butyl (2SV2-(anilinocarbonyls)piperidine-l-carboxylate
[534] 46.0 mg of the title compound (yield: 35%) was obtained, in the same manner as in
PREPARATION 1(1), except that 100 mg (0.436 mmol) of
(2S)-l-(t-butoxycarbonyl)piperidine-2-carboxylic acid and 42.6 mg (0.458 mmol) of toluene were used. [535] 1H NMR (CDCl3) 68.14-8.03 (IH, brs), 7.69-7.67 (2H, m), 7.59-7.27 (2H, m),
7.16-7.08 (IH, m), 4.86 (IH, s), 4.18-4.13 (2H, m), 2.90-2.82 (IH, m ), 2.36-2.33 (IH, m), 1.87-1.35 (13H, m) [536] Mass (m/e) 305 (M+ 1) [537]
[538] (2)Svnthesis of (2S)-N-phenylpiperidine-2-carboxamide hydrochloric acid salt [539] 24.5 mg of the title compound (yield: 67%) was obtained, in the same manner as in
PREPARATION 1(2), except that 46.0 mg (0.151 mmol) of t-butyl
(2S)-2-(anilinocarbonyl)piperidine-l-carboxylate obtained in PREPARATION 26(1) was used. [540] 1H NMR (CD3OD) δ 10.08 (IH, brs), 7.56 (2H, d, J=7.35), 7.33-7.30 (IH, m), 7.12
(IH, m), 3.90-3.88 (IH, m), 3.44-3.41 (IH, m), 3.10-3.00 (IH, m), 2.32-2.29 (IH, m),
2.00-1.90 (2H, m), 1.88-1.67 (3H, m), [541] Mass (m/e) 205 (M+ 1) [542] [543] PREPARATION 27: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-r(2SV2-(hvdroxymethyls)p yrrolidine- 1 -yll -3-oxopropyl lcarbamate [544] 280 mg (2.07 mmol) of 1-hydroxybenzotriazole and 320 mg (1.66 mmol) of EDC were added to 190 mg (1.38 mmol) of (2S)-pyrrolidine-2-yl methanol hydrochloric acid salt dissolved in 7 mL of methylenechloride at room temperature in sequence.
After stirring for 10 minutes, 460 mg (1.38 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperridine-l-yl)butyric acid, obtained by referring to Korean Patent Application No. 2006-29138, dissolved in 6 mL of methylenechloride was added thereto slowly. After stirring for 10 minutes at room temperature, 0.72 mL (4.14 mmol) of diisopropylethylamine was added thereto. After further stirring for 12 hours at room temperature, the impure title compound, obtained by concentrating under reduced pressure, was purified with column chromatography
(1:10 methanol: methylene chloride) to give 540 mg of the title compound (yield:
94%). [545] 1H NMR (CDCl3) 65.74-5.72 (IH, m), 4.75-4.73 (IH, m), 4.23-4.20 (2H, m),
3.78-3.43 (6H, m), 2.48-2.67 (3H, m), 2.32-2.30 (2H, m), 2.07-1.90 (4H, m), 1.66-1.62
(IH, m), 1.46-1.42 (9H, m) [546] Mass (m/e) 420 (M+ 1)
[547]
[548] EXAMPLE 14: Synthesis of l-K2Ss)-2-amino-4-[(2Ss)-2-(hvdroxymethyls)pyrrolidine-l-yll-4-oxobutyl)-5.5-difluoro piperidine-2-one

[550] 540 mg (1.30 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[(2S)-2-(hydroxymethyl)p yrrolidine-l-yl]-3-oxopropyl}carbamate obtained in PREPARATION 1 was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). Thereafter, the resulting solution was stirred for 20 minutes, and then concentrated under reduced pressure. The residue was purified with prep-TLC (6: 1 CH2Cl2: MeOH) to give 450 mg of the title compound (yield: 97%). [551] 1H NMR (CD3OD) δ 4.15-4.02 (IH, m), 3.92-3.72 (5H, m), 3.64-3.44 (4H, m),
2.92-2.67 (2H, m), 2.63-2.56 (2H, m), 2.42-2.34 (2H, m), 2.09-1.93 (4H, m) [552] Mass (m/e) 320 (M+ 1) [553] [554] PREPARATION 28: Synthesis of t- butyl((lS)-l-[(5.5-difluoro-2-oxopiperidine-l-yl)methyll-3-oxo-3-[(2S)-2-(phenoxym ethvDpyrrolidine- 1 - yllpropyl ) carbamate [555] 73.0 mg of the title compound (yield: 52%) was obtained, in the same manner as in
PREPARATION 27, except that 56.0 mg (0.280 mmol) of
(2S)-2-(phenoxymethyl)pyrrolidine hydrochloric acid salt obtained by referring to WO
05/056003 and 93.0 mg (0.280 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [556] 1H NMR (CDCl3) δ 7.38-7.27 (2H, m), 7.01-6.92 (3H, m), 4.48-4.42 (IH, m),
4.25-4.18 (IH, m), 4.15-4.05 (2H, m), 3.81-3.44 (5H, m), 2.60-2.54 (2H, m), 2.32-2.10
(5H, m), 2.08-1.95 (4H, m), 1.45-1.43 (9H, m) [557] Mass (m/e) 496 (M+ 1) [558] [559] EXAMPLE 15: Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-φhenoxymethvDpyrrolidine-l-yllbutyl)-5.5-difluor opiperidine-2-one [560]
 [561] 32.0 mg of the title compound (yield: 50%) was obtained, in the same manner as in
[561] 32.0 mg of the title compound (yield: 50%) was obtained, in the same manner as in
EXAMPLE 14, except that 73 mg (0.150 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-(phenoxym ethyl)pyrrolidin-l-yl]propyl} carbamate obtained in PREPARATION 28 was used. [562] 1H NMR (CD3OD) δ 7.30-7.23 (2H, m), 6.97-6.90 (3H, m), 4.43-4.40 (IH, m),
4.12-4.10 (IH, m), 3.86-3.79 (4H, m), 3.76-3.67 (5H, m), 2.75-2.61 (3H, m), 2.59-2.05
(6H, m),
[563] Mass (m/e) 396 (M+ 1) [564] [565] PREPARATION 29: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-oxo-3-r(2SV2-(rφhenylsu
If onyDaminol methyl ) pyrrolidine- 1 -yll propyl ) carbamate [566] 108 mg of the title compound (yield: 60%) was obtained, in the same manner as in
PREPARATION 27, except that 89.0 mg (0.320 mmol) of N-[(2S)- pyrrolidine-
2-ylmethyl]benzenesulfonamide hydrochloric acid salt obtained by referring to WO
05/056003 and 110 mg (0.320 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [567] 1H NMR (CDCl3) δ 7.95-7.80 (2H, m), 7.61-7.46 (3H, m), 4.20-4.10 (IH, m),
3.81-3.55 (5H, m), 3.55-3.18 (5H, m), 3.05-2.94 (IH, m), 2.65-2.50 (2H, m), 2.38-2.20
(2H, m), 2.07-1.75 (4H, m), 1.42-1.41 (9H, m) [568] Mass (m/e) 559 (M+ 1) [569] [570] EXAMPLE 16: Synthesis of N-
({ (2SV l-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine- l-yls)butanoyllpyrrolidine-2-yl
) methyPbenzenesulfonamide

[572] 74.0 mg of the title compound (yield: 69%) was obtained, in the same manner as in EXAMPLE 14, except that 108 mg (0.19 mmol) of t-
butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-{[(phenylsu lfonyl)amino]methyl}pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION
29 was used. [573] 1H NMR (CD3OD) δ 7.88-7.82 (2H, m), 7.62-7.54 (3H, m), 4.15-4.05 (IH, m),
3.90-3.71 (5H, m), 3.54-3.42 (4H, m), 2.98-2.92 (IH, m), 2.62-2.59 (3H, m), 2.42-2.32
(2H, m), 1.99-1.90 (3H, m) [574] Mass (m/e) 459 (M+ 1) [575] [576] PREPARATION 30: Synthesis of t- butyll QSV3-( (2SV2-r(benzoylaminos)methyllpyrrolidine- 1-yll- l-IY5.5-difluoro-2-oxo piperidine-l-yDmethyll-S-oxopropyl lcarbamate [577] 80.5 mg of the title compound (yield: 35%) was obtained, in the same manner as in
PREPARATION 27, except that 106 mg (0.440 mmol) of N-
[(2S)-pyrrolidine-2-ylmethyl]benzamide hydrochloric acid salt obtained by referring to
WO 05/056003 and 150 mg (0.440 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-0029138 were used. [578] 1H NMR (CDCl3) δ 8.30 (IH, s), 7.87-7.85 (2H, d, J=8Hz), 7.46-7.42 (2H, m),
4.48-4.46 (2H, m), 4.21-4.15 (2H, m), 3.80-3.35 (9H, m), 2.56-2.53 (IH, t, J=6.7Hz),
2.29-2.23 (2H, m), 2.10-2.03 (3H, m), 1.90-1.80 (IH, m), 1.42-1.39 (9H, m) [579] Mass (m/e) 523 (M+ 1) [580] [581] EXAMPLE 17: Synthesis of N-
(I (2S)- l-[(3S)-3-amino-4-(5.5-difluoro-2-oxopiperidine- l-yl)butanoyllpyrrolidine-2-yl
) methvDbenzamide

[583] 43.5 mg of the title compound (yield: 56%) was obtained, in the same manner as in
EXAMPLE 14, except that 85.0 mg (0.160 mmol) of t- butyl[(lS)-3-{(2S)-2-[(benzoylamino)methyl]pyrrolidine-l-yl}-l-[(5,5-difluoro-2-oxo piperidine-l-yl)methyl]-3-oxopropyl]carbamate obtained in PREPARATION 30 was used. [584] 1H NMR (CD3OD) δ 7.87-7.83 (2H, m), 7.58-7.44 (3H, m), 3.99-3.84 (4H, m),
3.71-3.63 (2H, m), 3.58-3.49 (4H, m), 2.82-2.67 (2H, m), 2.62-2.58 (2H, m), 2.40-2.35
(2H, m), 2.10-1.95 (4H, m) [585] Mass (m/e) 423 (M+l)
[586]
[587] PREPARATION 31: Synthesis of t- butvirriS^-l-rrS.S-difluoro-l-oxopiperidine-l-vDmethyll-S-oxo-S-KlS'l-l-rφyrimidin e-2-ylthio)methvπpyrrolidine- 1 -yl Ipropyllcarbamate [588] 41.8 mg of the title compound (yield: 19%) was obtained, in the same manner as in
PREPARATION 27, except that 100 mg (0.430 mmol) of 2- ([(2S)- pyrrolidine-
2-ylmethyl]thio}pyrimidine hydrochloric acid salt obtained by referring to WO
05/056003 and 150 mg (0.430 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [589] 1H NMR (CDCl3) δ 8.61 (IH, d, J=4.9Hz), 8.50 (IH, d, J=4.9Hz), 7.05-6.95 (IH, m),
6.09-5.90 (IH, m), 4.43-4.07 (2H, m), 3.89-3.42 (7H, m), 2.72-2.54 (4H, m), 2.27-1.95
(6H, m), 1.49-1.38 (9H, m) [590] Mass (m/e) 514 (M+l) [591] [592] EXAMPLE 18: Synthesis of l-r(2SV2-amino-4-oxo-4-((2SV2-rφyrimidine-2-ylthios)methyll pyrrolidine- l-yl)butyll-5.5- difluoropiperidine-2-one

EXAMPLE 14, except that 41.8 mg (0.810 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-{(2S)-2-[(pyrimidin e-2-ylthio)methyl]pyrrolidine-l-yl}propyl]carbamate obtained in PREPARATION 31 was used. [595] 1H NMR (CD3OD) δ 7.40-7.38 (IH, m), 7.28-7.21 (IH, m), 7.02-6.97 (IH, m),
3.90-3.86 (IH, m), 3.82-3.72 (IH, m), 3.53-3.39 (5H, m), 3.35-3.17 (5H, m), 2.36-2.31
(IH, m), 2.19-2.00(5H, m), 1.90-1.79 (2H, m) [596] Mass (m/e) 414 (M+l) [597] [598] PREPARATION 32: Synthesis of t- butyl ( Q SV 3 - IY2S V 2- ( T (anilinocarbonyPaminol methyl ) pyrrolidine- l-yll-l-IY5.5 -diflu oro-2-oxopiperidine- 1 -yPemthyll -3-oxopripyl ) carbamate [599] 24.0 mg of the title compound (yield: 10%) was obtained, in the same manner as in
PREPARATION 27, except that 100 mg (0.400 mmol) of
l-phenyl-3-[(2S)-pyrrolidine-2-ylmethyl]urea hydrochloric acid salt obtained by referring to WO 05/056003 and 130 mg (0.400 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [600] 1H NMR (CDCl3) δ 7.45-7.35 (2H, m), 7.27-7.24 (2H, m), 7.00-6.97 (IH, m),
4.22-4.09 (IH, m), 3.74-3.57(3H, m), 3.50-3.22 (6H, m), 2.58-2.53 (3H, m), 2.32-2.26
(2H, m), 2.03-1.85 (5H, m), 1.52-1.42 (9H, m) [601] Mass (m/e) 538 (M+l) [602] [603] EXAMPLE 19: Synthesis of l-(((2SVl-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-yls)butanoyllpyrrolidine-2- yl ) methyl V 3 -phenylurea

EXAMPLE 14, except that 24.0 mg (0.040 mmol) of l-[(2S)-2-amino-4-oxo-4-{(2S)-2-[(pyrimidine-2-ylthio)methyl]pyrrolidine-l-yl}butyl]
-5,5-difluoropiperidine-2-one obtained in PREPARATION 32 was used. [606] 1H NMR (CD3OD) δ 7.36-7.33 (2H, m), 7.28-7.22 (2H, m), 7.00-6.98 (IH, m),
3.90-3.71 (6H, m), 3.59-3.40 (6H, m), 2.65-2.59 (2H, m), 2.42-2.39 (2H, m), 2.05-1.92
(4H, m)
[607] Mass (m/e) 438 (M+l) [608] [609] PREPARATION 33: Synthesis of t- butyl[(lS)-l-[(5.5-difluoro-2-oxopiperidine-l-yl)methyll-3-oxo-3-((2S)-2-[(phenylthio
)methyllpyrrolidine- 1 -yl Ipropyllcarbamate [610] 79.7 mg of the title compound (yield: 58%) was obtained, in the same manner as in
PREPARATION 1, except that 62.0 mg (0.270 mmol) of
(2S)-2-[(phenylthio)methyl]pyrrolidine hydrochloric acid salt obtained by referring to
WO 05/056003 and 90.0 mg (0.270 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [611] 1H NMR (CDCl3) δ 7.46-7.17 (5H, m), 5.87-5.82(1H, m), 4.33-4.15 (2H, m),
3.73-3.39 (5H, m), 2.95-3.02 (IH, m), 2.58-2.52 (3H, m), 2.26-2.15 (4H, m),
2.03-1.90(5H, m), 1.46-1.44 (9H, m) [612] Mass (m/e) 512 (M+l) [613] [614] EXAMPLE 20: Synthesis of l-r('2SV2-amino-4-oxo-4-(('2SV2-r('phenylthioN)methyllpyrrolidine-l-yl)butyll-5.5-difl uoropiperidine-2-one

EXAMPLE 14, except that 79.7 mg (0.155 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-{(2S)-2-[(phenylthio
)methyl]pyrrolidine-l-yl}propyl]carbamate obtained in PREPARATION 33 was used. [617] 1H NMR (CD3OD) δ 7.48-7.44 (2H, m), 7.30-7.26 (2H, m), 7.18-7.15 (IH, m),
4.30-4.27 (IH, m), 3.89-3.69 (4H, m), 3.51-3.37 (4H, m), 3.01-2.95 (IH, m), 2.63-2.59
(4H, m), 2.42-2.31 (2H, m), 2.12-1.96(4H, m) [618] Mass (m/e) 412 (M+l) [619] [620] PREPARATION 34: Synthesis of t- butyl((lS)-3-[(3S)-3-[(benzylamino)carbonyll-3.4- dihydroisoquinoline-
2( lH)-yll - 1 - [(5.5-difluoro-2-oxopiperidine- 1 -vDmethyll -3-oxopropyl lcarbamate [621] 27.0 mg of the title compound (yield: 47%) was obtained, in the same manner as in
PREPARATION 27, except that 30.0 mg (0.100 mmol) of
(3S)-N-benzyl-l,2,3,4-tetrahydroisoquinoline-3-carboxamide hydrochloric acid salt obtained by referring to WO 05/056003 and 33.0 mg (0.100 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [622] 1H NMR (CDCl3) δ 7.35-6.81 (9H, m), 4.86 (IH, d, J=17Hz), 4.68-4.45 (3H, m),
4.30-3.90 (3H, m), 3.81-3.42 (4H, m), 3.35-3.22 (IH, m), 3.10-3.02 (2H, m), 2.72-2.07
(5H, m), 1.45 (9H, s) [623] Mass (m/e) 585 (M+l) [624] [625] EXAMPLE 21: Synthesis of
(3SV2-r(3SV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-vDbutanoyll-N-benzyl-1.2.3.4
-tetrahydroisoquinoline-3-carboxamide [626]

[627] 18.2 mg of the title compound (yield: 83%) was obtained, in the same manner as in EXAMPLE 14, except that 27.0 mg (0.046 mmol) of t- butyl{(lS)-3-[(3S)-3-[(benzylamino)carbonyl]-3,4-dihydroisoquinoline-2(lH)-yl]-l-[( 5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 34 was used.
[628] 1H NMR (CD3OD) δ 7.65-7.15 (7H, m), 6.89-6.80(2H, m), 5.10 (IH, t, J=5.6Hz),
4.79-4.65 (2H, m), 4.38-4.16 (2H, m), 3.97-3.73 (5H, m), 3.48-3.46 (IH, m), 3.30-3.28 (IH, m), 3.19-3.09 (2H, m), 2.87-2.81 (IH, m), 2.63-2.53(2H, m), 2.42-2.32(2H, m)
[629] Mass (m/e) 485 (M+ 1)
[630]
[631] PREPARATION 35: Synthesis of t- butyl(QSV3-r(2SV2-(anilinomethyls)pyrrolidine-l-yll-l-r(5.5-difluoro-2-oxopiperidine -l-yDmethyll-S-oxopropyDcarbamate
[632] 39 mg of the title compound (yield: 39%) was obtained, in the same manner as in PREPARATION 27, except that 43.0 mg (0.200 mmol) of N- [(2S)-pyrrolidine-2-ylmethyl] aniline hydrochloric acid salt obtained by referring to WO 05/056003 and 68.0 mg (0.20 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[633] 1H NMR (CDCl3) δ 7.19-7.15 (2H, m), 6.68-6.63 (3H, m), 5.85-5.80 (IH, m),
4.55-4.48 (IH, m), 4.28-4.15 (IH, m), 3.85-3.50 (6H, m), 3.48-3.15 (2H, m), 2.80-2.20 (6H, m), 2.15-1.92 (4H, m), 1.45 (9H, s)
[634] Mass (m/e) 495 (M+ 1)
[635]
[636] EXAMPLE 22: Synthesis of l-((2SV2-amino-4-r(2SV2-(anilinomethyls)pyrrolidine-l-yll-4-oxobutyl)-5.5-difluorop iperidine-2-one

EXAMPLE 14, except that 39.0 mg (0.079 mmol) of t- butyl{(lS)-3-[(2S)-2-(anilinomethyl)pyrrolidine-l-yl]-l-[(5,5-difluoro-2-oxopiperidine
-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 35 was used. [639] 1H NMR (CD3OD) δ 7.70-7.38 (5H, m), 4.48-4.38 (IH, m), 3.96-3.83 (4H, m),
3.66-3.54 (5H, m), 2.91-2.66 (2H, m), 2.64-2.58 (2H, m), 2.41-2.38 (2H, m), 2.22-2.15
(IH, m), 2.08-1.99 (2H, m), 1.88-1.85 (IH, m) [640] Mass (m/e) 395 (M+ 1) [641] [642] PREPARATION 36: Synthesis of t- butyl(αSV3-r(3SV3-(aminos)carbonyl-3.4-dihvdroisoquinoline-2αHVyll-l-r(5.5-diflu oro-2-oxopiperidine- 1 -yPmethyll -3-oxopropyl ) carbamate [643] 39.0 mg of the title compound (yield: 57%) was obtained, in the same manner as in
PREPARATION 27, except that 30.0 mg (0.140 mmol) of
(3S)-l,2,3,4-tetrahydroisoquinoline-3-carboxamide hydrochloric acid salt obtained by referring to WO 05/056003 and 48.0 mg (0.140 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [644] 1H NMR (CDCl3) δ 7.25-7.14 (4H, m), 5.35-5.12 (3H, m), 4.70-4.57 (IH, m),
4.45-4.27 (IH, m), 4.15-4.05 (IH, m), 3.82-3.48 (6H, m), 3.18-3.16 (IH, m), 3.08-2.90
(IH, m), 2.61-2.52 (2H, m), 2.35-2.22 (2H, m), 1.46-1.44 (9H, m) [645] Mass (m/e) 495 (M+ 1) [646] [647] EXAMPLE 23: Synthesis of
GSV2-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-yls)butanoyll-1.2.3.4-tetrahvdr oisoquinoline-3-carboxamide

[649] 20.0 mg of the title compound (yield: 59%) was obtained, in the same manner as in EXAMPLE 14, except that 39.0 mg (0.079 mmol) of t- butyl{(lS)-3-[(3S)-3-(amino)carbonyl-3,4-dihydroisoquinoline-2(lH)-yl]-l-[(5,5-diflu oro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 36 was used.
[650] 1H NMR (CD3OD) δ 7.25-7.21 (4H, m), 5.04 (IH, t, J=4Hz), 4.71-4.69 (IH, m),
3.96-3.70 (5H, m), 3.54-3.47 (IH, m), 3.27-3.08 (3H, m), 2.91-2.81 (IH, m), 2.65-2.55 (2H, m), 2.41-2.32 (2H, m)
[651] Mass (m/e) 395 (M+l)
[652]
[653] PREPARATION 37: Synthesis of t- butylKlSs)-l-[(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-[(3Ss)-3-(morpholine-4-ylc arbonvD-SΛ-dihvdroisoquinoline-iriH^-yll-S-oxopropyllcarbamate
[654] 69.0 mg of the title compound (yield: 69%) was obtained, in the same manner as in PREPARATION 27, except that 50.0 mg (0.180 mmol) of
(3S)-3-(morpholine-4-ylcarbonyl)-l,2,3,4-tetrahydroisoquinoline hydrochloric acid salt obtained by referring to WO 05/056003 and 60.0 mg (0.180 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[655] 1H NMR (CDCl3) δ 7.25-7.20 (4H, m), 4.73-4.62 (2H, m), 4.18-4.17 (IH, m), 3.84-3.48 (HH, m), 3.45-3.38(1H, m), 3.12-2.98 (2H, m), 2.86-2.84 (IH, m), 2.64-2.52 (4H, m), 2.30-2.22 (2H, m), 1.43 (9H, s)
[656] Mass (m/e) 565 (M+l)
[657]
[658] EXAMPLE 24: Synthesis of l-((2SV2-amino-4-r(3SV3-(morpholine-4-ylcarbonylV3.4-dihvdroisoquinoline-2αHV yll -4-oxobutyl ) -5.5-difluoropiperidine-2-one

[660] 20.0 mg of the title compound (yield: 33%) was obtained, in the same manner as in EXAMPLE 14, except that 69.0 mg (0.120 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[(3S)-3-(morpholine-4-ylc arbonyl)-3,4-dihydroisoquinoline-2(lH)-yl]-3-oxopropyl}carbamate obtained in PREPARATION 37 was used.
[661] 1H NMR (CD3OD) δ 7.25-7.22 (4H, m), 5.26 (IH, t, J=6.4Hz), 4.76-4.65 (2H, m), 3.93-3.46 (12H, m), 3.25-3.19 (2H, m), 3.10-2.98 (2H, m), 2.87-2.81 (IH, m), 2.64-2.60 (2H, m), 2.42-2.32 (2H, m)
[662] Mass (m/e) 465 (M+l)
[663]
[664] PREPARATION 38: Synthesis of t- butyl(αSV3-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-r(5.5-di fluoro-2-oxopiperidine- 1 -yPmethyll -3-oxopropyl lcarbamate
[665] 58.0 mg of the title compound (yield: 53%) was obtained, in the same manner as in
PREPARATION 27, except that 50.0 mg (0.230 mmol) of 3-cyclopropyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 78.0 mg (0.230 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[666] 1H NMR (CDCl3) δ 5.92-5.91 (IH, m), 5.29-5.27 (IH, m), 4.17-4.13 (IH, m),
3.78-3.68 (3H, m), 3.58-3.53 (3H, m), 2.59-2.51 (4H, m), 2.32-2.24 (3H, m), 2.10-2.06 (4H, m), 1.44 (9H, s), 1.05 (4H, d, J=6.4Hz)
[667] Mass (m/e) 498 (M+ 1)
[668]
[669] EXAMPLE 25: Synthesis of l-((2SV2-amino-4-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-vD pyrrolidine- l-yll-4-oxobutyl)-5.5-difluoropiperidine-2-one

[671] 20.0 mg of the title compound (yield: 38%) was obtained, in the same manner as in EXAMPLE 14, except that 58.0 mg (0.120 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-[(5,5-di fluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 38 was used.
[672] 1H NMR (CD3OD) δ 5.25-5.22 (IH, m), 3.85-3.71 (5H, m), 3.49-3.46 (IH, m),
2.86-2.77 (2H, m), 2.63-2.59 (2H, m), 2.38-2.32 (4H, m), 2.17-2.03 (4H, m), 1.08-0.93 (4H, m)
[673] Mass (m/e) 398 (M+ 1)
[674]
[675] PREPARATION 39: Synthesis of t-butyl(αSV3-r(2SV2-(5-cvclopropyl-1.2.4- ox- adiazole-S-yDpyrrolidine- 1 -yll - 1 - IY5.5-difruoro-2-oxopiperidine- 1 -yPmethyll -3-oxopr opyl) carbamate
[676] 90.0 mg of the title compound (yield: 87%) was obtained, in the same manner as in PREPARATION 27, except that 50.0 mg (0.198 mmol) of 5-cyclopropyl-3-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 70.0 mg (0.208 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[677] 1H NMR (CDCl3) δ 6.00-5.72 (IH, m), 5.23-4.94 (IH, m), 4.20-4.11 (IH, m),
3.81-3.64 (3H, m), 3.60-3.51 (3H, m), 2.71-2.48 (4H, m), 2.30-2.10 (5H, m), 1.41 (9H, s), 1.28-1.11 (6H, m)
[678] Mass (m/e) 498 (M+ 1), 520 (M+Na) [679] [680] EXAMPLE 26: Synthesis of l-((2SV2-amino-4-r(2SV2-(5-cvclopropyl-1.2.4-oxadiazole-3-yls)pyrrolidine-l-yll-4-o xobutyl)-5.5-difluoropiperidine-2-one

[682] 72.0 mg of the title compound (yield: 92%) was obtained, in the same manner as in EXAMPLE 14, except that 40.0 mg (0.181 mmol) of t- butyl{(lS)-3-[(2S)-2-(5-cyclopropyl-l,2,4-oxadiazole-3-yl)pyrrolidine-l-yl]-l-[(5,5-di fluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 39 was used.
[683] 1H NMR (CD3OD) δ 5.21-5.16 (IH, m), 3.89-3.60 (6H, m), 3.52-3.46 (IH, m),
2.96-2.72 (2H, m), 2.65-2.62 (2H, m), 2.52-2.23 (4H, m), 2.14-2.04 (3H, m), 1.32-1.26 (2H, m), 1.22-1.15 (2H, m)
[684] Mass (m/e) 398 (M+ 1)
[685]
[686] PREPARATION 40: Synthesis of t- butvirαSV3-r(2SV2-G-cvclopropyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-(r(5RV 5-methyl-2-oxopiperidine- l-yllmethyl)-3- oxopropyllcarbamate
[687] 30.0 mg of the title compound (yield: 28%) was obtained, in the same manner as in PREPARATION 1, except that 50.0 mg (0.230 mmol) of 3-cyclopropyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 73.0 mg (0.230 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[(5R)-5-methyl-2-oxopiperidine-l-yl]butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[688] 1H NMR (CDCl3) δ 6.05-6.03 (IH, m), 5.32-5.28 (IH, m), 4.20-4.15 (IH, m),
3.69-3.51 (4H, m), 3.53-3.32 (IH, m), 3.05-3.02 (IH, m), 2.43-2.06 (5H, m), 1.92-1.85 (4H, m), 1.84-1.65 (3H, m), 1.44 (9H, s), 1.06-1.00 (7H, m)
[689] Mass (m/e) 476 (M+ 1)
[690]
[691] EXAMPLE 27: Synthesis of
(5RVl-((2SV2-amino-4-r(2SV2-G-cvclopropyl-1.2.4-oxadiazole-5-yls)pyrrolidine-l-yl 1 -4-oxobutyl ) -5-methylpiperidine-2-one

[693] 20.0 mg of the title compound (yield: 83%) was obtained, in the same manner as in EXAMPLE 14, except that 30 mg (0.060 mmol) of t- butyl[(lS)-3-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-{[(5R)- 5-methyl-2-oxopiperidine-l-yl]methyl}-3-oxopropyl]carbamate obtained in PREPARATION 40 was used.
[694] 1H NMR (CD3OD) δ 5.22-5.20 (IH, m), 4.87-4.82 (IH, m), 3.80-3.71 (3H, m),
3.70-3.62 (3H, m), 3.55-3.40 (3H, m), 3.07-3.02 (IH, m), 2.86-2.81 (IH, m), 2.70-2.68 (IH, m), 2.62-2.55 (IH, m), 2.30-2.02 (5H, m), 1.02-1.01 (7H, m)
[695] Mass (m/e) 376 (M+ 1)
[696]
[697] PREPARATION 41: Synthesis of t- butyll QSV l-r(5.5-difluoro-2-oxopiperidine- l-vDmethyll-3-oxo-3-r(2SV2-G-phenyl- 1 .2.4-oxadiazole-5-yl)pyrrolidine-l-yllpropyl)carbamate
[698] 35.0 mg of the title compound (yield: 55%) was obtained, in the same manner as in PREPARATION 27, except that 30 mg (0.240 mmol) of
3-phenyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 80.0 mg (0.240 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[699] 1H NMR (CDCl3) δ 8.09-8.07 (2H, m), 7.50-7.48 (3H, m), 5.43-5.41 (IH, m),
4.22-4.15 (IH, m), 3.77-3.62 (6H, m), 2.74-2.39 (4H, m), 2.38-2.05 (7H, m), 1.43 (9H, s)
[700] Mass (m/e) 534 (M+ 1) [701] [702] EXAMPLE 28: Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-G-phenyl-1.2.4-oxadiazole-5-yls)pyrrolidine-l-yllbut yl ) -5.5-difluoropiperidine-2-one [703]

EXAMPLE 14, except that 35.0 mg (0.06 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-(3-phenyl-l
,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION
41 was used. [705] 1H NMR (CD3OD) δ 8.07-8.00 (2H, m), 7.56-7.48 (3H, m), 5.39-5.37 (IH, m),
3.87-3.65 (7H, m), 3.53-3.49 (IH, m), 2.92-2.83 (2H, m), 2.55-2.10 (7H, m), [706] Mass (m/e) 434 (M+ 1) [707] [708] PREPARATION 42: Synthesis of t- butyl(αSVl-(r(5RV5-methyl-2-oxopiperidine-l-yllmethyl)-3-oxo-3-r(2SV2-(3-phenv l-l^^-oxadiazole-S-yDpyrrolidine-l-yllpropyl) carbamate [709] 130 mg of the title compound (yield: 72%) was obtained, in the same manner as in
PREPARATION 1, except that 90.0 mg (0.240 mmol) of
3-phenyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 110 mg (0.240 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-[(5R)-5-methyl-2-oxopiperidine-l-yl]butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [710] 1H NMR (CDCl3) δ 8.05-8.03 (2H, m), 7.49-7.44 (3H, m), 6.02-6.00 (IH, m),
5.40-5.38 (2H, m), 4.13-4.11 (IH, m), 3.65-3.63 (IH, m), 3.62-3.51 (5H, m), 3.27-3.25
(IH, m), 3.01-2.99 (IH, m), 2.79-2.75 (IH, m), 2.50-2.13 (7H, m), 1.39-1.35 (9H, m),
0.95-0.91 (3H, m) [711] Mass (m/e) 512 (M+l) [712] [713] EXAMPLE 29:
(5RVl-((2SV2-amino-4-oxo-4-r(2SV2-G-phenyl-1.2.4-oxadiazole-5- vDpyrrolidine- 1 -yllbutyl ) -5-methylpiperidine-2-one

[715] 35 mg of the title compound (yield: 29%) was obtained, in the same manner as in EXAMPLE 14, except that 132 mg (0.26 mmol) of t- butyl{(lS)-l-{[(5R)-5-methyl-2-oxopiperidine-l-yl]methyl}-3-oxo-3-[(2S)-2-(3-pheny l-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]propyl} carbamate obtained in PREPARATION 42 was used.
[716] 1H NMR (CD3OD) δ 8.05-8.00 (2H, m), 7.60-7.48 (3H, m), 5.38-5.26 (IH, m),
3.85-3.79 (2H, m), 3.75-3.71 (2H, m), 3.60-3.55 (IH, m), 3.31-3.24 (IH, m), 3.07-2.85 (3H, m), 2.47-2.38 (3H, m), 2.28-2.17 (3H, m), 1.92-1.90 (IH, m), 1.78-1.74 (IH, m), 1.49-1.37 (IH, m), 0.95-0.92 (3H, m)
[717] Mass (m/e) 412 (M+l)
[718]
[719] PREPARATION 43: Synthesis of t- butyll (IS)- l-[(5.5-difluoro-2-oxopiperidine- 1-yl) methyll-3-oxo-3-[(2S)-2-(3-pyridine-2-yl-1.2.4-oxadiazole-5-yl)pyrrolidine-l-yll propyl ) carbamate
[720] 37.3 mg of the title compound (yield: 35%) was obtained, in the same manner as in PREPARATION 27, except that 50.0 mg (0.200 mmol) of 2-{5-[(2S)-pyrrolidine-yl]-l,2,4-oxadiazole-3-yl}pyridine hydrochloric acid salt obtained by referring to WO 05/121131 and 67.0 mg (0.200 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[721] 1H NMR (CDCl3) δ 8.78-8.77 (IH, m), 8.13-8.09 (IH, m), 7.84-7.82 (IH, m), 7.42-7.41 (IH, m), 5.89(1H, d, J=7.9Hz), 5.41-5.39 (IH, m), 4.15-4.11 (IH, m), 3.78-3.58 (5H, m), 2.60-2.48 (4H, m), 2.42-2.34 (IH, m), 2.30-2.18 (5H, m), 2.17-2.10 (IH, m), 1.44-1.35 (9H, m)
[722] Mass (m/e) 535 (M+l)
[723]
[724] EXAMPLE 30: Synthesis of
1-1 (2S)-2-amino-4-oxo-4-[(2S)-2-(3-pyridine-2-yl- 1.2.4-oxadiazole-5-yl)pyrrolidine- 1 -yllbutyl)-5.5-difluoropiperidine-2-one

[726] 26.0 mg of the title compound (yield: 79%) was obtained, in the same manner as in EXAMPLE 14, except that 37.0 mg (0.070 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-(3-pyridine- 2-yl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION 43 was used.
[727] 1H NMR (CD3OD) δ 8.84-8.82 (IH, m), 8.38-8.31 (2H, m), 7.88-7.85 (IH, m),
5.48-5.43 (IH, m), 3.89-3.71 (6H, m), 3.51-3.48 (IH, m), 2.97-2.78 (2H, m), 2.62-2.55
(2H, m), 2.53-2.46 (IH, m), 2.39-2.20 (5H, m)
[728] Mass (m/e) 435 (M+ 1)
[729]
[730] PREPARATION 44: Synthesis of t- butylf flSVl-f rf5RV5-methyl-2-oxopiperidine-l-yl1methyl)-3-oxo-3-r(2SV2-f3-pyridi ne-2-yl- 1.2.4-oxadiazole-5-yl)pyrrolidine- 1 -yllpropyl ) carbamate
[731] 27.0 m of the title compound (yield: 26%) was obtained, in the same manner as in PREPARATION 27, except that 50.0 mg (0.200 mmol) of
2-{5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole-3-yl}pyridine hydrochloric acid salt obtained by referring to WO 05/121131 and 63.0 mg (0.200 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-[(5R)-5-methyl-2-oxopiperidine-l-yl]butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[732] 1H NMR (CDCl3) δ 8.78-8.77 (lH,m), 8.12-8.09 (IH, m), 7.83-7.82 (IH, m),
7.41-7.40 (IH, m), 6.02-6.00 (IH, m), 5.42-5.40 (IH, m), 3.78-3.72 (IH, m), 3.64-3.60 (2H, m), 3.56-3.50 (3H, m), 3.30-3.27 (IH, m), 3.02-2.98 (IH, m), 2.79-2.76 (IH, m), 2.48-2.28 (4H, m), 2.27-2.09 (3H, m), 1.93-1.87 (IH, m), 1.80-1.77 (IH, m), 1.40 (9H, s), 0.94 (3H, J=6.7Hz)
[733] Mass (m/e) 513 (M+l)
[734]
[735] EXAMPLE 31: Synthesis of
(5RVl-((2SV2-amino-4-oxo-4-r(2SV2-G-pyridine-2-yl-1.2.4-oxadiazole-5-yls)pyrrolid ine- 1 -yllbutyl ) -5-methylpiperidine-2-one

[737] 26.0 mg of the title compound (yield: 79%) was obtained, in the same manner as in EXAMPLE 14, except that 37.0 mg (0.070 mmol) of t- butyl{(lS)-l-{[(5R)-5-methyl-2-oxopiperidine-l-yl]methyl}-3-oxo-3-[(2S)-2-(3-pyridi ne-2-yl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION 44 was used.
[738] 1H NMR (CD3OD) δ 8.79-8.78 (IH, m), 8.29-8.19 (2H, m), 1.11-1.16 (IH, m),
5.44-5.42 (IH, m), 3.84-3.66 (6H, m), 3.59-3.50 (IH, m), 3.08-2.91 (2H, m) 2.81-2.77 (IH, m), 2.50-2.42 (2H, m), 2.38-2.19 (3H, m), 2.05-1.90 (IH, m), 1.85-1.80 (IH, m), 1.52-1.48 (IH, m), 0.98 (3H, d, J=6.4Hz)
[739] Mass (m/e) 413 (M+l)
[740]
[741] PREPARATION 45: Synthesis of
3-cvclopentyl-5-[(2S)-pyrrolidine-2-yll-1.2.4-oxadiazole hydrochloric acid salt
[742] (l)Svnthesis of N'-hvdroxycvclopentanecarboximidamide
[743] 2.07 g (15.0 mmol) of potassium carbonate and 320 mg (1.66 mmol) of hydroxylam- inehydrochloride dissolved in 12 mL of methanol were gradually added to 1.14 g (10.0 mmol) of cyclopentanecarbonitrile dissolved in 4 mL of methanol. The reaction solution was heated to 8O0C and stirred for 5 hours. After the reaction solution was cooled to room temperature, the methanol was removed. The resulting solution was concentrated, filtered by Cellite, then distilled off under reduced pressure to give 1.06 g of the title compound in a yield of 145% without purification, and the next step was preceded.
[744] Mass (m/e) 129 (M+l)
[745]
[746] (^Synthesis of t-butyl (2SV2-G-cyclopentyl-1.2.4-oxadiazole-5-yls)pyrrolidine-l- carboxylate
[747] 0.24 g of the title compound (yield: 10%) was obtained, in the same manner as in
PREPARATION 2, except that 3.44 g (16.0 mmol) of N-(t-butoxycarbonyl)-L-proline and 1.06 g (8.27 mmol) of N'-hydroxycyclopentanecarboximidamide obtained in PREPARATION 45(1) were used.
[748] 1H NMR (CDCl3) 65.05-4.95 (IH, m), 3.68-3.48 (2H, m), 3.28-3.18 (IH, m), 2.37-2.18 (IH, m), 2.17-1.86 (5H, m), 1.84-1.59 (6H, m), 1.31-1.28 (9H, s)
[749] Mass (m/e) 308 (M+l)
[750]
[751] (3)Svnthesis of 3-cvclopentyl-5-[(2S)-pyrrolidine-2-yll-1.2.4-oxadiazole hy- drochloric acid salt
[752] 240 mg (0.780 mmol) of t-butyl
(2S)-2-(3-cyclopentyl- 1 ,2,4-oxadiazole-5-yl)pyrrolidine- 1 -carboxylate obtained in PREPARATION 45(2) was dissolved in 4 M of dioxane/hydrochloric acid, and the resulting solution was stirred for about 30 minutes and concentrated. The residue was purified with prep-TLC (10:1 CH2Cl2:Me0H) to give 170 mg of the title compound (yield: 89%).
[753] Mass (m/e) 208 (M+l)
[754]
[755] PREPARATION 46: Synthesis of t- butyl(αSV3-r(2SV2-(3-cvclopentyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-l-r(5.5-dif luoro-2-oxopiperidine-l-yls)methyll-3-oxopropyl)carbamate
[756] 51.0 mg of the title compound (yield: 81%) was obtained, in the same manner as in
PREPARATION 27, except that 30.0 mg (0.120 mmol) of 3-cyclopentyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained in PREPARATION 45 and 41.0 mg (0.120 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[757] 1H NMR (CDCl3) δ5.90 (IH, d, J=7Hz), 5.29 (IH, m), 4.25-4.13 (IH, m), 3.80-3.51 (6H, m), 3.18-3.15 (IH, m), 2.59-2.51 (3H, m), 2.45-2.22 (4H, m), 2.21-2.02 (4H, m), 1.85-1.68 (4H, m), 1.66-1.54 (3H, m), 1.40 (9H, m)
[758] Mass (m/e) 526 (M+l)
[759]
[760] EXAMPLE 32: l-((2SV2-amino-4-r(2SV2-(3-cvclopentyl-1.2.4-oxadiazole-5-vD pyrrolidine- l-yll-4-oxobutyl)-5.5-difluoropiperidine-2- one

EXAMPLE 14, except that 51.0 mg (0.097 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclopentyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-[(5,5-dif luoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 46 was used. [763] 1H NMR (CD3OD) δ 5.29-5.26 (IH, m), 3.86-3.63 (6H, m), 3.48-3.46 (IH, m),
3.20-3.18 (IH, m), 2.91-2.76 (2H, m), 2.63-2.60 (2H, m), 2.40-2.32 (3H, m), 2.16-2.02
(5H, m), 1.85-1.70 (6H, m) [764] Mass (m/e) 426 (M+l) [765] [766] PREPARATION 47: Synthesis of l-(5-r(2SVpyrrolidine-2-yll-1.2.4-oxadiazole-3-yl)piperidine hydrochloric acid salt [767] (DSynthesis of N'-hydroxypiperidine-l-carboximidamide [768] 1.79 g of the title compound (yield: 118%) prepared from 1.22 g (10.0 mmol) of piperidine-1-carbonitrile, in the same manner as in PREPARATION 45(1), was used in the next reaction without further purification. [769] Mass (m/) 144 (M+l) [770] [771] (^Synthesis of t-butyl
(2S)-2-(3-piperidine-l-yl-1.2.4-oxadiazole-5-yl)pyrrolidine-l-carboxvlate
[772] 0.47g of the title compound (yield: 12%) was obtained, in the same manner as in
PREPARATION 42, except that 1.79 g (3.28 mmol) of
N'-hydroxypiperidine-l-carboximidamide was used. [773] 1H NMR (CDCl3) δ 4.52-4.77 (IH, m), 4.33-4.24 (IH, m), 3.71-3.41 (4H, m),
3.72-3.68 (IH, m), 2.25-2.20 (IH, m), 2.19-2.06 (IH, m), 1.97-1.80 (4H, m), 1.65-1.59
(2H, m), 1.48-1.43 (9H, m), 1.36-1.30 (2H, m) [774] Mass (m/e) 323 (M+ 1) [775] [776] (3)Svnthesis of l-(5-[(2S)-pyrrolidine-2-yll-1.2.4-oxadiazole-3-yl)piperidine hy- drochloric acid salt [777] 350 mg of the title compound (yield: 92%) was obtained, in the same manner as in
PREPARATION 45(3), except that 470 mg (1.46 mmol) of t-butyl
(2S)-2-(3-piperidine- 1-yl- l,2,4-oxadiazole-5-yl)pyrrolidine- 1-carboxylate was used. [778] Mass (m/e) 223 (M+l) [779] [780] PREPARATION 48: Synthesis of t- butyl(QSVl-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-oxo-3-r(2SV2-G-piperidin e- 1 -yl- 1 ^^-oxadiazole-S-yDpyrrolidine- 1 -yll propyl ) carbamate [781] 42.0 mg of the title compound (yield: 65%) was obtained, in the same manner as in
PREPARATION 27, except that 30.0 mg (0.120 mmol) of l-{5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole-3-yl}piperidine hydrochloric acid salt obtained in PREPARATION 47 and 39.0 mg (0.120 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [782] 1H NMR (CDCl3) δ 5.95-5.90 (IH, m), 5.17-5.15 (IH, m), 4.48-4.46 (IH, m),
4.30-4.12 (2H, m), 3.76-3.47 (6H, m), 2.75-2.50 (4H, m), 2.45-1.99 (5H, m), 1.48-1.40
(9H, m)
[783] Mass (m/e) 541 (M+l) [784] [785] EXAMPLE 33: Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-(3-piperidine-l-yl-1.2.4-oxadiazole-5-vDpyrrolidine-
1 -yllbutyl ) -5.5-difluoropiperidine-2-one [786]

EXAMPLE 14, except that 42.0 mg (0.078 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-(3-piperidin e-l-yl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl] propyl} carbamate obtained in PREPARATION 48 was used. [788] 1H NMR (CD3OD) δ 5.15-5.13 (IH, m), 4.49-4.47 (IH, m), 3.87-3.72 (7H, m),
3.67-3.30 (7H, m), 2.85-2.64 (2H, m), 2.61-2.59 (2H, m), 2.41-2.32 (4H, m), 2.06-2.03
(2H, m), 1.63-1.59 (2H, m) [789] Mass (m/e) 441 (M+l) [790] [791] PREPARATION 49: Synthesis of t- butyl(αSV3-r(2SV2-(anilinomethyls)pyrrolidine-l-yll-l-(r(4RV4-methyl-2-oxopyrroli dine- 1 -yll methyl } -3-oxopropyl } carbamate [792] 15 mg of the title compound (yield: 33%) was obtained, in the same manner as in
PREPARATION 27, except that 21.0 mg (0.100 mmol) of N-[(2S)- pyrrolidine-
2-ylmethyl] aniline hydrochloric acid salt obtained by referring to WO 05/056003 and
30.0 mg (0.10 mmol) of
(3S)-3-[(butoxycarbonyl)amino]-4-[(4R)-4-methyl-2-oxopyrrolidine-l-yl]butyric acid obtained in PREPARATION 5 were used. [793] Mass (m/e) 459 (M+l) [794] [795] EXAMPLE 34: Synthesis of
(4RV 1-1 (2SV2-amino-4-r(2SV2-(anilinomethyls)pyrrolidine- l-yll-4-oxobutyl)-4-meth ylpyrrolidine-2-one [796]

EXAMPLE 14, except that 15.0 mg (0.033 mmol) of t- butyl{(lS)-3-[(2S)-2-(anilinomethyl)pyrrolidine-l-yl]-l-{[(4R)-4-methyl-2-oxopyrroli dine-l-yl]methyl}-3-oxopropyl}carbamate obtained in PREPARATION 49 was used. [798] 1H NMR (CD3OD) δ 7.55-7.47 (5H, m), 4.39-4.37 (IH, m), 3.89-3.83 (IH, m),
3.71-3.57 (6H, m), 3.27-3.23 (IH, m), 2.88-2.74 (2H, m), 2.55-2.53 (2H, m), 2.18-1.98
(5H, m), 1.97-1.84 (IH, m), 1.14-1.09 (3H, m) [799] Mass (m/e) 359 (M+l) [800]
[801] PREPARATION 50: Synthesis of t- butyl(αSVl-(r(4RV4-methyl-2-oxopyrrolidine-l-yllmethyl)-3-oxo-3-r(2SV2-G-pyrid ine-2-yl-1.2.4-oxadiazole-5-yl)pyrroridine-l-vπ propyl) carbamate [802] 27.0 mg of the title compound (yield: 55%) was obtained, in the same manner as in
PREPARATION 27, except that 25.0 mg (0.100 mmol) of 2-{5-[(2S)- pyrrolidine-
2-yl]-l,2,4-oxadiazole-3-yl}pyridine hydrochloric acid salt obtained by referring to
WO 05/121131 and 30.0 mg (0.100 mmol) of
(3S)-3-[(butoxycarbonyl)amino]-4-[(4R)-4-methyl-2-oxopyrrolidine-l-yl]butyric acid obtained in PREPARATION 5 were used. [803] Mass (m/e) 499 (M+ 1) [804] [805] EXAMPLE 35: Synthesis of
(4RVl-((2SV2-amino-4-oxo-4-r(2SV2-(3-pyridine-2-yl-1.2.4- oxadiazole-
S-yDpyrrolidine- 1 -yllbutyl ) -4-methylpyrrolidine-2-one

[807] 21.0 mg of the title compound (yield: 84%) was obtained, in the same manner as in EXAMPLE 14, except that 27.0 mg (0.054 mmol) of t- butyl{(lS)-l-{[(4R)-4-methyl-2-oxopyrrolidine-l-yl]methyl}-3-oxo-3-[(2S)-2-(3-pyrid ine-2-yl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl] propyl} carbamate obtained in PREPARATION 50 was used.
[808] 1H NMR (CD3OD) δ 8.94-8.93 (IH, m), 8.63-8.56 (2H, m), 8.12-8.09 (IH, m),
5.48-5.46 (IH, m), 3.86-3.55 (6H, m), 3.13-3.11 (IH, m), 2.92-2.86 (2H, m) 2.56-2.52 (3H, m), 2.26-2.06 (4H, m), 1.15-1.11 (3H, m)
[809] Mass (m/e) 399 (M+ 1)
[810]
[811] PREPARATION 51: Synthesis of t- butyl(αSV3-r(2SV2-r3-(cvclopropylmethylV1.2.4-oxadiazole-5-yllpyrrolidine-l-yll-l -r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3- oxopropyl) carbamate
[812] 17.0 mg of the title compound (yield: 32%) was obtained, in the same manner as in PREPARATION 27, except that 30.0 mg (0.130 mmol) of
3-cyclopropylmethyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 44.0 mg (0.130 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid
obtained by referring to Korean Patent Application No. 2006-29138 were used. [813] 1H NMR (CDCl3) δ 5.97-5.95 (IH, m), 5.35-5.32 (IH, m), 4.22-4.19 (IH, m),
3.79-3.54 (7H, m), 2.72-2.56 (4H, m), 2.32-1.93 (6H, m), 1.43 (9H, s ), 1.39-1.30 (IH, m), 1.28-1.14 (IH, m), 0.59-0.57 (2H, m), 0.28-0.25 (2H, m) [814] Mass (m/e) 512 (M+l) [815] [816] EXAMPLE 36: Synthesis of l-r(2SV2-amino-4-(('2SV2-r3-('cvclopropylmethylV1.2.4-oxadiazole-5-yllpyrrolidine- l-yl)-4-oxobutyll-5.5-difluoropiperidine-2-one

EXAMPLE 14, except that 17.0 mg (0.041 mmol) of t- butyl{(lS)-3-[(2S)-2-[3-(cyclopropylmethyl)-l,2,4-oxadiazole-5-yl]pyrrolidine-l-yl]-l
-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3- oxopropyl} carbamate obtained in
PREPARATION 51 was used. [819] 1H NMR (CD3OD) δ 5.31-5.28 (IH, m), 3.85-3.66 (5H, m), 3.49-3.46 (2H, m), 2.90-
2.59 (7H, m), 2.40-2.33 (3H, m), 2.17-2.10 (2H, m), 1.15-1.05 (IH, m), 0.57-0.54 (2H, m), 0.24-0.21 (2H, m) [820] Mass (m/e) 412 (M+l) [821] [822] PREPARATION 52: Synthesis of t-butyl(αSV3-r(2SV2-G-cvclobutyl-1.2.4- ox- adiazole-S-yDpyrrolidine- 1 -yll - 1 - IY5.5-difruoro-2-oxopiperidine- 1 -yPmethyll -3-oxopr opyl) carbamate [823] 23.7 mg of the title compound (yield: 45%) was obtained, in the same manner as in
PREPARATION 1, except that 30.0 mg (0.130 mmol) of
3-cyclobutyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 44.0 mg (0.130 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [824] 1H NMR (CDCl3) δ 5.94-5.93 (IH, m), 5.34-5.32 (IH, m), 4.19-4.13 (IH, m),
3.76-3.53 (1OH, m), 2.59-2.56 (IH, m), 2.57-2.53 (3H, m), 2.40-2.06 (9H, m),
1.44-1.43 (9H, m) [825] Mass (m/e) 512 (M+l)
[826]
[827] EXAMPLE 37: Synthesis of l-((2SV2-amino-4-r('2SV2-G-cvclobutyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-4-ox obutyl)-5.5-difluoropiperidine-2-one

[829] 18.0 mg of the title compound (yield: 90%) was obtained, in the same manner as in EXAMPLE 14, except that 23.7 mg (0.046 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-[(5,5-difl uoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 52 was used.
[830] 1H NMR (CD3OD) δ 5.30-5.28 (IH, m), 3.86-3.61 (7H, m), 3.48-3.45 (IH, m), 2.91- 2.74 (2H, m), 2.63-2.60 (2H, m), 2.40-2.32 (7H, m), 2.19-2.00 (5H, m)
[831] Mass (m/e) 412 (M+l)
[832]
[833] PREPARATION 53: Synthesis of
(4SV4-phenyl-2-r(2SVpyrrolidine-2-yll-4.5-dihydro-1.3-oxazole hydrochloric acid salt
[834] 200 mg (0.630 mmol) of t-butyl
(2S)-2-[(4S)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-carboxylate obtained by referring to WO 05/121131 was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 mL). Thereafter, the resulting solution was stirred for 20 minutes and concentrated under reduced pressure, then the residue was purified with prep-TLC (6:1 CH2Cl2 MeOH) to give 130 mg of the title compound (yield: 82%).
[835] 1H NMR (CD3OD) δ 7.80-7.38 (5H, m), 5.25-5.19 (IH, m), 4.41-4.38 (IH, m), 3.92-3.78 (2H, m), 3.66 (2H, s), 2.52-2.47 (IH, m), 2.20-2.00 (3H, m)
[836] Mass (m/e) 217 (M+l)
[837]
[838] PREPARATION 54: Synthesis of t- butvirαSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-oxo-3-((2SV2-r(4SV4-phe nyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)propyllcarbamate
[839] 35.0 mg of the title compound (yield: 41%) was obtained, in the same manner as in PREPARATION 27, except that 40.0 mg (0.160 mmol) of
(4S)-4-phenyl-2-[(2S)-pyrrolidine-2-yl]-4,5-dihydro-l,3-oxazole hydrochloric acid salt obtained in PREPARATION 53 and 53.0 mg (0.160 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [840] Mass (m/e) 535 (M+ 1) [841] [842] EXAMPLE 38: Synthesis of l-r(2SV2-amino-4-oxo-4-((2SV2-r(4SV4-phenyl-4.5-dirivdro-1.3-oxazole-2-yllpyrroli dine-l-yl)butyll-5.5-difluoropiperidine-2-one

EXAMPLE 14, except that 35.0 mg (0.070 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-{(2S)-2-[(4S)-4-phe nyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-yl}propyl]carbamate obtained in PREPARATION 54 was used. [845] 1H NMR (CD3OD) δ 8.78 (IH, d, J=8.4Hz), 7.50-7.26 (5H, m), 5.22-5.16 (IH, m),
4.55-4.53 (IH, m), 3.90-3.40 (8H, m), 2.81-2.57 (4H, m), 2.36-2.27 (3H, m), 2.09-1.93
(3H, m)
[846] Mass (m/e) 435 (M+ 1) [847] [848] PREPARATION 55: Synthesis of
(4SV2-r(2S.4SV4-fluoropyrrolidine-2-yll-4-phenyl-4.5-dihvdro-1.3-oxazole hy- drochloric acid salt [849] ( ^Synthesis of t-butyl (2S.4S V4-fluoro-2-α IY 1 S V 2-hvdroxy- 1 -phenylethyll amino ) carbonyDpyrrolidine- 1 -carboxylate [850] 282 mg of the title compound (yield: 56%) was obtained, in the same manner as in
PREPARATION 1, except that 330 mg (1.41 mmol) of
(4S)-l-(t-butoxycarbonyl)4-fluoro-L-proline obtained in PREPARATION 23(1) and
190 mg (1.41 mmol) of (2S)-2-amino-2-phenylethanol were used, isolated and purified. [851] 1H NMR (CDCl3) 67.35-7.26 (5H, m), 5.30-5.28 (IH, m), 5.15-5.07 (3H, m),
4.48-4.45 (2H, m), 3.94-3.72 (5H, m), 1.49-1.47 (9H, m) [852] Mass (m/e) 353 (M+ 1) [853] [854] (^Synthesis of t-butyl
(2S.4SV4-fluoro-2-r(4RV4-phenyl-4.5-dihvdro-1.3-oxazole-2-yllpyrrolidine-l-carbox ylate
[855] 282 mg of t-butyl
(2S ,4S)-4-fluoro-2- ( { [( 1 S)-2-hydroxy- 1 -phenylethyl] amino } carbonyl)pyrrolidine- 1 -ca rboxylate obtained in PREPARATION 55(1) was dissolved in 5 mL of tetrahy- drofuran, and the solution, composed of 0.11 mL of diethylaminosulfur trifluoride (DAST) dissolved in 3mL of tetrahydrofuran in advance, was gradually added thereto. The resulting solution was heated to 7O0C and stirred for 90 minutes. The impure title compound, obtained by concentrating under reduced pressure, was isolated and purified to give 220 mg of the title compound (yield: 81%).
[856] 1H NMR (CDCl3) δ 7.31-7.24 (5H, m), 5.33-5.20 (2H, m), 4.78-4.64 (2H, m), 4.18-4.16 (IH, m), 3.91-3.72 (2H, m), 2.61-2.40 (2H, m), 1.48-1.46 (9H, m)
[857] Mass (m/e) 335 (M+ 1)
[858]
[859] (3) Synthesis of
(4SV2-r(2S.4SV4-fluoropyrrolidine-2-yll-4-phenyl-4.5-dihvdro-1.3-oxazole hy- drochloric acid salt
[860] 220 mg (0.650 mmol) of t-butyl
(2S,4S)-4-fluoro-2-[(4R)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-carbox ylate obtained in PREPARATION 55(2) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). The resulting solution was stirred for 20 minutes, concentrated under reduced pressure, and the residue was purified with diethylether to give 170 mg of the title compound (yield: 74%).
[861] 1H NMR (CD3OD) δ 7.38-7.31 (5H, m), 5.49-5.36 (IH, m), 5.25-5.22 (IH, m), 4.49-4.47 (IH, m), 3.91-3.77 (2H, m), 3.62-3.58 (2H, m), 2.87-2.80 (IH, m), 2.57-2.45(1H, m)
[862] Mass (m/e) 235 (M+ 1)
[863]
[864] PREPARATION 56: Synthesis of t- butvirαSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-((2S.4SV4-fluoro-2-r(4SV 4-phenyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)-3-oxopropyllcarbamate
[865] 140 mg of the title compound (yield: 40%) was obtained, in the same manner as in PREPARATION 27, except that 170 mg (0.630 mmol) of (4S)-2-[(2S,4S)-4-fluoropyrrolidine-2-yl]-4-phenyl-4,5-dihydro-l,3-oxazole hydrochloric acid salt obtained in PREPARATION 55 and 210 mg (0.630 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[866] 1H NMR (CDCl3) δ 7.40-7.25 (5H, m), 6.20-6.19 (IH, m), 5.66-5.64 (IH, m),
5.45-5.42 (IH, m), 5.33-5.30 (2H, m), 4.21-4.10 (2H, m), 4.00-3.90 (IH, m ), 3.88-3.58 (5H, m), 2.68-2.52 (4H, m), 2.49-2.15 (3H, m), 1.45-1.42 (9H, m)
[867] Mass (m/e) 553 (M+ 1)
[868]
[869] EXAMPLE 39: Synthesis of l-r(2SV2-amino-4-oxo-4-((2S.4SV4-fluoro-2-r(4SV4-phenyl-4.5-dihvdro-1.3-oxazole- 2-yllpyrrolidine-l-yl)-4-oxobutyll-5.5-difluoropiperidine-2-one

EXAMPLE 14, except that 140 mg (0.250 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-{(2S,4S)-4-fluoro-2-[(4S)-
4-phenyl-4,5-dihydro- 1 ,3-oxazole-2-yl]pyrrolidine- 1 -yl } -3-oxopropyl]carbamate obtained in PREPARATION 56 was used. [872] 1H NMR (CD3OD) δ 7.41-7.22 (5H, m), 5.38-5.15 (2H, m), 4.70-4.68 (IH, m),
3.94-3.75 (8H, m), 3.47-3.42 (IH, m), 2.81-2.28 (8H, m) [873] Mass (m/e) 453 (M+ 1) [874] [875] PREPARATION 57: Synthesis of
GR.5SV5-r(4RV4-phenyl-4.5-dihvdro-1.3-oxazole-2-yllpyrrolidine-3-ol hydrochloric acid salt [876] mSvnthesis of t-butyl
(2S.4RV 4-( 1 -etoxyetoxy )-2-( { \(l S V 2-hydroxy- 1 -phenylethyll amino IcarbonyDpyrroli dine- 1 -carboxylate [877] 330 mg of the title compound (yield: 63%) was obtained, in the same manner as in
PREPARATION 1, except that 253 mg (1.24 mmol) of
(4R)-l-(t-butoxycarbonyl)-4-(l-etoxyetoxy)-L-proline obtained in PREPARATION
24(2) and 170 mg (1.24 mmol) of (2S)-2-amino-2-phenylethanol 170 mg (1.24 mmol) were used. [878] 1H NMR (CDCl3) δ 7.31-7.27 (5H, m), 5.09-5.05 (IH, m), 4.74-4.70 (IH, m),
4.49-4.32 (2H, m), 3.91-3.89 (2H, m), 3.62-3.44 (3H, m), 2.71-2.69 (IH, m), 2.53-2.50
(IH, m), 2.22-2.05 (IH, m), 1.46-1.43 (9H, m), 1.32-1.18 (6H, m) [879] Mass (m/e) 423 (M+ 1) [880]
[881] (2) Synthesis of t-butyl
(2S.4RV4-α-etoxyetoxyV2-r(4RV4-phenyl-4.5-dihvdro-1.3-oxazole-2-yllpyrrolidine- 1-carboxylate
[882] 240 mg of t-butyl
(2S,4R)-4-( 1 -etoxyetoxy)-2-( { [( 1 S)-2-hydroxy- 1 -phenylethyl] amino }carbonyl)pyrroli dine- 1-carboxy late obtained in PREPARATION 57(1) was dissolved in 5 niL of tet- rahydrofuran and the solution, composed of 0.077 mL of diethylaminosulfur trifluoride (DAST) dissolved in 3 mL of tetrahydrofuran in advance, was gradually added thereto. The resulting solution was heated to 7O0C and stirred for 90 minutes. The impure title compound, obtained by concentrating under reduced pressure, was isolated and purified with column chromatography (2:1 → 1:1 hexane: aceticacid ethyl) to give 120 mg of the title compound (yield: 52%).
[883] 1H NMR (CDCl3) δ 7.38-7.30 (5H, m), 5.25-5.23 (IH, m), 4.80-4.78 (IH, m),
4.71-4.65 (2H, m), 4.50-4.41 (IH, m), 4.20-4.16 (IH, m), 3.85-3.66 (2H, m), 3.54-3.46 (2H, m), 2.34-2.26 (2H, m), 1.51-1.49 (9H, m), 1.36-1.30 (3H, m), 1.26-1.20 (3H, m)
[884] Mass (m/e) 405 (M+l)
[885]
[886] GVSvnthesis of
GR.5SV5-r(4RV4-phenyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-3-ol hydrochloric acid salt
[887] 117 mg (0.350 mmol) of t-butyl
(2S,4R)-4-(l-etoxyetoxy)-2-[(4R)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine- 1-carboxylate obtained in PREPARATION 57(2) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). The resulting solution was stirred for 20 minutes, concentrated under reduced pressure, and the residue was purified with diethylether to give 91.0 mg of the title compound (yield: 97%).
[888] 1H NMR (CD3OD) δ 7.70-7.68 (IH, m), 7.42-7.21 (4H, m), 4.72-4.53 (4H, m), 3.85-3.82 (IH, m), 3.38-3.29 (2H, m), 2.47-2.43 (IH, m)
[889] Mass (m/e) 233 (M+l)
[890]
[891] PREPARATION 58: Synthesis of t- butvirαSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-((2S.4RV4-hvdroxy-2-r(4 SV4-phenyl-4.5-dihydro-1.3-oxazole-2-yllpyrrolidine-l-yl)-3-oxopropyllcarbamate
[892] 65.0 mg of the title compound (yield: 35%) was obtained, in the same manner as in PREPARATION 27, except that 91.0 mg (0.340 mmol) of
(3R,5S)-5-[(4R)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-3-ol hydrochloric acid salt obtained in PREPARATION 57 and 110 mg (0.340 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid
obtained by referring to Korean Patent Application No. 2006-29138 were used. [893] 1H NMR (CDCl3) δ 7.69-7.56 (2H, m), 7.42-7.27 (3H, m), 6.28-6.22 (IH, m),
4.79-4.76 (IH, m), 4.46-4.42 (IH, m), 4.18-4.12 (IH, m), 3.83-3.53 (7H, m ),
3.10-3.08 (IH, m), 2.57-2.21 (9H, m), 1.45-1.42 (9H, m) [894] Mass (m/e) 551 (M+ 1) [895] [896] EXAMPLE 40: Synthesis of l-r(2SV2-amino-4-((2S.4RV4-hvdroxy-2-r(4SV4-phenyl-4.5-dihvdro-1.3-oxazole-2-yl lpyrrolidine-l-yl)-4-oxobutyll-5.5-difluoropiperidine-2-one

[898] 25.0 mg of the title compound (yield: 44%) was obtained, in the same manner as in EXAMPLE 14, except that 65.0 mg (0.118 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-{(2S,4R)-4-hydroxy-2-[(4 S)-4-phenyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-yl}-3-oxopropyl]carbamate obtained in PREPARATION 58 was used.
[899] 1H NMR (CD3OD) δ 7.42-7.26 (5H, m), 5.18-5.17 (IH, m), 4.63-4.61 (IH, m), 4.59-4.51 (IH, m), 3.89-3.31 (9H, m), 2.73-2.56 (6H, m), 2.34-2.29 (2H, m)
[900] Mass (m/e) 451 (M+ 1)
[901]
[902] PREPARATION 59: Synthesis of 4-phenyl-2-r(2SVpyrrolidine-2-yll-1.3-oxazole hy- drochloric acid salt
[903] mSvnthesis of t-butyl (2SV2-(4-phenyl-1.3-oxazole-2-vDpyrrolidine-l-carboxylate
[904] 500 mg ( 1.50 mmol) of t-butyl
(2S)-2-({ [(lS)-2-hydroxy- 1-phenylethyl] amino } carbonyl)pyrrolidine- 1-carboxylate obtained by referring to WO 05/121131 was dissolved in 10 mL of methylenechloride, and 0.65 g (1.54 mmol) of Dess-martin periodinane was added thereto. The resulting solution was stirred for 3 hours, filtered by Cellite and concentrated under reduced pressure. The next step was preceded without further purification.
[905] The solution, composed of 1.29 g (4.90 mmol) of triphenylphosphine, 1.02 g (4.00 mmol) of iodine, 1.12 mL (8.00 mmol) of triethylamine and 30 mL of tetrahydrofuran, was cooled to -78.O0C, and aldehyde dissolved in 15 mL of tetrahydrofuran was gradually added thereto. At this temperature, the resulting solution was stirred for 3 hours. After diluting with ethyl acetoacetate, an organic layer was washed with 10%
sodium thiosulfinate, 2M hydrochloric acid, and salt water. The organic layer thus obtained was dried over anhydrous magnesium sulfate and filtered. The impure title compound, obtained by concentrating under reduced pressure, was isolated and purified with column chromatography (5:1 → 2:1 hexane: aceticacid ethyl) to give 170 mg of the title compound (yield: 26%).
[906] 1H NMR (CDCl3) δ7.82 (IH, s), 7.73-7.71 (2H, m), 7.40-7.27 (3H, m), 5.11-4.90
(IH, m), 3.67-3.56 (2H, m), 2.33-2.05 (3H, m), 2.04-1.91 (IH, m), 1.29-1.24 (9H, m)
[907] Mass (m/e) 315 (M+l)
[908]
[909] (2)Svnthesis of 4-phenyl-2-[(2S)-pyrrolidine-2-yll-1.3-oxazole hydrochloric acid salt
[910] 170 mg (0.520 mmol) of t-butyl
(2S)-2-(4-phenyl-l,3-oxazole-2-yl)pyrrolidine-l-carboxylate obtained in PREPARATION 58(1) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). The resulting solution was stirred for 20 minutes and concentrated under reduced pressure, and the residue was purified with diethylether to give 140 mg of the title compound (yield: 97%).
[911] 1H NMR (CD3OD) 8.43 (IH, s), 7.86-7.84 (2H, m), 7.48-7.39 (3H, m), 3.53-3.37 (3H, m), 2.49-2.10 (4H, m)
[912] Mass (m/e) 215 (M+l)
[913]
[914] PREPARATION 60: Synthesis of t- butylf f ISV l-r(5.5-difluoro-2-oxopiperidine- l-yl)methyl1-3-oxo-3-r(2SV2-f4-phenyl- 1 .3-oxazole-2-yl)pyrrolidine-l-yllpropyl)carbamate
[915] 162 mg of the title compound (yield: 62%) was obtained, in the same manner as in PREPARATION 27, except that 140 mg (0.520 mmol) of
4-phenyl-2-[(2S)-pyrrolidine-2-yl]-l,3-oxazole hydrochloric acid salt obtained in PREPARATION 59 and 170 mg (0.520 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[916] 1H NMR (CDCl3) δ 7.83 (IH, s), 7.74-7.72 (2H, m), 7.42-7.31 (3H, m), 5.32-5.30 (2H, m), 4.25-4.23 (IH, m), 3.76-3.44 (6H, m), 2.69-2.07 (9H, m), 1.57 (9H, s)
[917] Mass (m/e) 533 (M+l)
[918]
[919] EXAMPLE 41:Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-(4-phenyl-1.3-oxazole-2-vDpyrrolidine-l-yllbutyl)- 5.5-difluoropiperidine-2-one
[920]

[921] 110 mg of the title compound (yield: 78%) was obtained, in the same manner as in EXAMPLE 14, except that 160 mg (0.301 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-(4-phenyl-l ,3-oxazole-2-yl)pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION 60 was used.
[922] 1H NMR (CD3OD) δ 8.15 (IH, s), 7.74-7.67 (2H, m), 7.40-7.27 (3H, m), 5.28-5.20 (IH, m), 3.86-3.64 (6H, m), 3.47-3.44 (IH, m), 2.87-2.75 (2H, m), 2.59-2.55 (2H, m), 2.35-2.12 (6H, m)
[923] Mass (m/e) 433 (M+ 1)
[924]
[925] PREPARATION 61: Synthesis of
3- [(2S)-pyrrolidine-2-yll -5- [ 1 -(trifluoromethyl)cvclopropyll - 1.2.4-oxadiazole hy- drochloric acid salt
[926] (I) Synthesis of t-butyl
(2SV2- ( 5-[ 1 -(trifluoromethvDcvclopropyll - 1.2.4-oxadiazole-3-yl Ipyrrolidine- 1 -carbox ylate
[927] 270 mg of l-(trifluoromethyl)cyclopropanecarboxylic acid was dissolved in 5 mL of dimethylformamide (DMF), and 4.70 g (29.0 mmol) of l,l'-carbonyldiimidazole was added thereto. The resulting solution was stirred for 20 minutes, and 200 mg of t-butyl (2S)-2- [(Z)-amino(hydroxyimino)methyl]pyrrolidine- 1 -carboxylate obtained by referring to WO 05/121131 was added thereto. The reaction solution was heated to 12O0C and stirred for 2 hours. The solvent was distilled off under reduced pressure, and 20 mL of ethylacetoacetate was added thereto. An organic layer was washed with water and dried over magnesium sulfate. After the solvent was distilled off, the residue was purified with column chromatography to give 140 mg of the title compound (yield: 23%).
[928] 1H NMR (CDCl3) δ 5.05-4.90 (IH, m), 3.62-3.40 (2H, m), 2.36-2.20 (IH, m), 2.10-1.90 (3H, m), 1.68-1.60 (4H, m), 1.48-1.30 (9H, m)
[929] Mass (m/e) 294 (M+ 1)
[930]
[931] (^Synthesis of
3- [(2S)-pyrrolidine-2-yll -5- [ 1 -(trifluoromethvDcvclopropyll - 1.2.4-oxadiazole hy-
drochloric acid salt
[932] 140 mg (0.400 mmol) of t-butyl
(2S)-2- { 5-[ 1 -(trifluoromethyl)cyclopropyl] - 1 ,2,4-oxadiazole-3-yl jpyrrolidine- 1 -carbox ylate obtained in PREPARATION 61(1) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml). The resulting solution was stirred for 20 minutes and concentrated under reduced pressure, and the residue was purified with diethylether to give 110 mg of the title compound (yield: 97%).
[933] 1H NMR (CD3OD) δ 4.94-4.90 (IH, m), 3.53-3.48 (2H, m), 2.58-2.52 (IH, m), 2.34-2.15 (3H, m), 1.82-1.73 (4H, m)
[934] Mass (m/e) 194 (M+l)
[935]
[936] PREPARATION 62: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-oxo-3-r(2SV2-(5-ri-(triflu oromethyDcyclopropyll - 1.2.4-oxadiazole-3-yl Ipyrrolidine- 1 -yllpropyl ) carbamate
[937] 150 mg of the title compound (yield: 68%) was obtained, in the same manner as in PREPARATION 27, except that 110 mg (0.389 mmol) of
3- [(2S)-pyrrolidine-2-yl] -5- [ 1 -(trifluoromethyl)cyclopropyl] - 1 ,2,4-oxadiazole hydrochloric acid salt obtained in PREPARATION 61 and 180 mg (0.550 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[938] 1H NMR (CDCl3) δ 5.88-5.86 (IH, m), 5.26-5.24 (IH, m), 4.19-4.17 (IH, m), 3.77-3.51 (6H, m), 2.57-2.46 (3H, m), 2.27-2.23 (3H, m), 2.08-2.04 (3H, m ), 1.69-1.57 (4H, m), 1.41 (9H, s)
[939] Mass (m/e) 512 (M+l)
[940]
[941] EXAMPLE 42: Synthesis of l-((2SV2-amino-4-oxo-4-r(2SV2-(5-ri-(trifluoromethyls)cvclopropyll-1.2.4-oxadiazol e-3-yl Ipyrrolidine- 1 -yllbutyl ) -5.5-difluoropiperidine-2-one

[943] 100 mg of the title compound (yield: 78%) was obtained, in the same manner as in EXAMPLE 14, except that 148 mg (0.257 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxo-3-[(2S)-2-{5-[l-(triflu oromethyl)cyclopropyl] - 1 ,2,4-oxadiazole-3-yl jpyrrolidine- 1 -yl]propyl } carbamate
obtained in PREPARATION 62 was used. [944] 1H NMR (CD3OD) δ 5.22-5.16 (IH, m), 3.82-3.55 (6H, m), 3.44-3.40 (IH, m),
2.79-2.60 (2H, m), 2.58-2.55 (2H, m), 2.34-2.27 (3H, m), 2.10-1.96 (3H, m), 1.72-1.62
(4H, m)
[945] Mass (m/e) 412 (M+l) [946] [947] PREPARATION 63: Synthesis of 2-r(2SVpyrrolidine-2-yll-4.5-dihvdro-1.3-oxazole hydrochloric acid salt [948] mSvnthesis of t-butyl
(2Ss)-2-(r(2-hvdroxyehtyls)aminolcarbonyl)pyrrolidine-l-carboxylate [949] 510 mg of the title compound (yield: 85%) was obtained, in the same manner as in
PREPARATION 1, except that 500 mg (2.32 mmol) of N-
(t-butoxycarbonyl)-L-proline and 140 mg (2.32 mmol) of 2-aminoethanol were used. [950] Mass (m/e) 259 (M+l) [951] [952] ^Synthesis of t-butyl
(2SV2-(4.5-dihydro-1.3-oxazole-2-yls)pyrrolidine-l-carboxylate [953] 84.2 mg of the title compound (yield: 18%) was obtained, in the same manner as in
PREPARATION 55(2), except that 510 mg (1.98 mmol) of t-butyl
(2S)-2-{[(2-hydroxy ethyl)amino]carbonyl}pyrrolidine-l-carboxylate obtained in
PREPARATION 63(1) was used. [954] 1H NMR (CDCl3) δ 4.50-4.41 (IH, m), 4.28-4.09 (2H, m), 3.86-3.81 (2H, m),
3.50-3.41 (2H, m), 2.17-1.85 (4H, m), 1.44-1.40 (9H, m) [955] Mass (m/e) 241 (M+l) [956] [957] (3)Svnthesis of 2-r(2S)-pyrrolidine-2-yll-4.5-dihvdro-1.3-oxazole hydrochloric acid salt [958] 84.0 mg (0.350 mmol) of t-butyl
(2S)-2-(4,5-dihydro-l,3-oxazole-2-yl)pyrrolidine-l-carboxylate obtained in PREPARATION 63(2) was dissolved in 4N 1,4-dioxane hydrochloric acid solution (5.0 ml), stirred for 20 minutes and concentrated under reduced pressure. The residue was purified with diethylether to give 79.4 mg of the title compound (yield: 98%). [959] 1H NMR (CD3OD) δ 4.26-4.22 (IH, m), 3.60-3.52 (3H, m), 3.39-3.28 (3H, m),
2.45-2.37 (IH, m), 2.07-1.99 (3H, m) [960] Mass (m/e) 141 (M+l) [961] [962] PREPARATION 64: Synthesis of t- butyl(αSVl-r(5.5-difluoro-2-oxoDiDeridine-l-vDmethyll-3-r(2SV2-(4.5-dihvdro-1.3-o
xazole-2-yl)pyrrolidine- 1 -yll -3-oxopropyl lcarbamate [963] 119 mg of the title compound (yield: 58%) was obtained, in the same manner as in
PREPARATION 27, except that 79.4 mg (0.450 mmol) of
2-[(2S)-pyrrolidine-2-yl]-4,5-dihydro-l,3-oxazole hydrochloric acid salt obtained in
PREPARATION 63 and 150 mg (0.450 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [964] Mass (m/e) 459 (M+ 1) [965] [966] EXAMPLE 43: Synthesis of l-((2SV2-amino-4-r(2SV2-(4.5-dihvdro-1.3-oxazole-2-vDpyrrolidine-l-yll-4-oxobutyl
)-5.5-difluoropiperidine-2-one

EXAMPLE 14, except that 110 mg (0.260 mmol) of t- butyl{(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-[(2S)-2-(4,5-dihydro-l,3-o xazole-2-yl)pyrrolidine-l-yl]-3-oxopropyl}carbamate obtained in PREPARATION 64 was used. [969] 1H NMR (CD3OD) δ 4.36-4.33 (IH, m), 3.86-3.61 (4H, m), 3.56-3.41 (5H, m),
2.81-2.56 (6H, m), 2.33-2.18 (5H, m), 2.08-1.94 (IH, m) [970] Mass (m/e) 359 (M+ 1) [971] [972] PREPARATION 65: Synthesis of
(4S)-4-isopropyl-2-[(2S)-pyrrolidine-2-yll-4.5-dihvdro- 1.3-oxazole hydrochloric acid salt [973] mSvnthesis of t-butyl
(2SV2-((rQSVl-(hydroxymethylV2-methylpropyllamino)carbonyls)pyrrolidine-l-carb oxylate [974] 740 mg of the title compound (yield: 73%) was obtained, in the same manner as in
PREPARATION 1, except that 730 mg (3.39 mmol) of N-
(t-butoxycarbonyl)-L-proline and 350 mg (3.39 mmol) of
(2S)-2-amino-3-methylbutane-l-ol were used. [975] 1H NMR (CDCl3) 66.95-6.88 (IH, m), 4.34-4.28 (IH, m), 3.77-3.30 (5H, m),
2.91-2.75 (IH, m), 2.48-2.10 (2H, m), 2.00-1.80 (3H, m), 1.55-1.52 (9H, m), 0.98-0.91
(6H, m)
[976] Mass (m/e) 301 (M+ 1)
[977]
[978] (^Synthesis of t-butyl
(2S)-2-[(4S)-4-isopropyl-4.5-dihvdro-13-oxazole-2-yllpyrroridine-l-carboxylate [979] 660 mg of the title compound (yield: 90%) was obtained, in the same manner as in
PREPARATION 55(2), except that 740 mg (2.47 mmol) of t-butyl
(2S)-2-({[(lS)-l-(hydroxymethyl)-2-methylpropyl]amino}carbonyl)pyrrolidine-l-carb oxylate obtained in PREPARATION 65(1) was used. [980] Mass (m/e) 283 (M+ 1) [981] [982] GVSvnthesis of (4SV4-isoprop yl-2-IY2S Vp yrrolidine-2-yll-4.5-dihvdro-1.3-oxazole hydrochloric acid salt [983] 480 mg of the title compound (yield: 94%) was obtained, in the same manner as in
PREPARATION 63(3), except that 661 mg (2.34 mmol) of t-butyl
(2S)-2-[(4S)-4-isopropyl-4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-carboxylate obtained in PREPARATION 65(2) was used. [984] 1H NMR (CD3OD) δ 3.89-3.88 (IH, m), 3.74-3.61 (2H, m), 3.43-3.37 (IH, m),
2.50-2.40 (2H, m), 2.11-1.89 (5H, m), 0.97-0.95(6H, m) [985] Mass (m/e) 183 (M+ 1) [986] [987] PREPARATION 66: Synthesis of t- butvirαSVl-r(5.5-difluoro-2-oxopiperidine-l-vDmethyll-3-((2SV2-r(4SV4-isopropyl-
4.5-dihvdro-1.3-oxazole-2-yllpyrrolidine-l-yl)-3-oxopropyllcarbamate [988] 20.0 mg of the title compound (yield: 17%) was obtained, in the same manner as in
PREPARATION 27, except that 50.0 mg (0.230 mmol) of
(4S)-4-isopropyl-2-[(2S)-pyrrolidine-2-yl]-4,5-dihydro- 1 ,3-oxazole hydrochloric acid salt obtained in PREPARATION 65 and 77.0 mg (0.230 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [989] 1H NMR (CDCl3) δ 4.12-4.05 (IH, m), 4.00-3.90 (IH, m), 3.70-3.62 (5H, m),
2.55-2.49 (4H, m), 2.25-1.80 (HH, m), 1.47-1.44 (9H, m), 1.21-1.19 (6H, m ) [990] Mass (m/e) 501 (M+ 1) [991] [992] EXAMPLE 44: Synthesis of l-r(2SV2-amino-4-((2SV2-r(4SV4-isopropyl-4.5-dihvdro-1.3-oxazole-2-yllpyrrolidine
-l-yl)-4-oxobutyll-5.5-difluoropiperidine-2-one [993]
 [994] 14.0 mg the title compound (yield: 82%) was obtained, in the same manner as in
[994] 14.0 mg the title compound (yield: 82%) was obtained, in the same manner as in
EXAMPLE 14, except that 20.0 mg (0.040 mmol) of t- butyl[(lS)-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-{(2S)-2-[(4S)-4-isopropyl-
4,5-dihydro-l,3-oxazole-2-yl]pyrrolidine-l-yl}-3-oxopropyl]carbamate obtained in
PREPARATION 66 was used. [995] 1H NMR (CD3OD) δ 4.71-4.68 (IH, m), 3.92-3.56 (9H, m), 3.42-3.29 (2H, m),
2.60-2.45 (3H, m), 2.34-2.21 (3H, m), 2.05-1.89 (4H, m), 0.96-0.94(6H, m) [996] Mass (m/e) 401 (M+ 1) [997] [998] PREPARATION 67: Synthesis of t- butyl(αSV3-r(2SV2-G-cvclopropyl-1.2.4-oxadiazole-5-yls)pyrrolidine-l-yll-l-r(4-met hyl-2-oxo-2.5-dihvdro-lH-pyrrole-l-yl)methyll-3-oxopropyl)carbamate [999] 58.0 mg of the title compound (yield: 55%) was obtained, in the same manner as in
PREPARATION 27, except that
3-cyclopropyl-5-[(2S)-pyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt 50.0 mg (0.230 mmol) obtained by referring to WO 05/121131 and 68.6 mg (0.230 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-(4-methyl-2-oxo-2,5-dihydro-lH-pyrrole-l-yl)but yric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [1000] 1H NMR (CDCl3) δ 6.22-6.20 (IH, m), 5.81(1H, s), 5.26-5.23 (IH, m), 4.26-4.23
(IH, m), 4.10-3.90 (2H, m), 3.70-3.58 (5H, m), 2.75-2.71 (IH, m), 2.49-2.27 (5H, m),
2.13-2.01 (3H, s), 1.41 (9H, s), 1.06-1.00 (4H, m) [1001] Mass (m/e) 460 (M+l) [1002] [1003] EXAMPLE 45: Synthesis of l-((2SV2-amino-4-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-vDpyrrolidine-l-yll-4-o xobutyl ) -4-methyl- 1.5-dihvdro-2H-pyrrole-2-one

EXAMPLE 14, except that 58.0 mg (0.120 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-l-yl]-l-[(4-met hyl-2-oxo-2,5-dihydro-lH-pyrrole-l-yl)methyl]-3-oxopropyl}carbamate obtained in
PREPARATION 67 was used. [1006] 1H NMR (CD3OD) δ 5.85 (IH, s), 5.32-5.27 (IH, m), 4.07-3.63 (8H, m), 3.30-3.17
(IH, m), 3.00-2.86 (IH, m), 2.45-2.32 (IH, m), 2.20-2.03 (6H, m), 1.13-1.02 (4H, m) [1007] Mass (m/e) 360 (M+l) [1008] [1009] PREPARATION 68: Synthesis of t- butvK QSV l-r(4-methyl-2-oxo-2.5-dihvdro- lH-pyrrole- l-vDmethyll-3-oxo-3-r(2SV2-
([(phenylsulfonyDaminolmethyDpyrrolidine-l-yllpropyDcarbamate [1010] 108 mg of the title compound (yield: 65%) was obtained, in the same manner as in
PREPARATION 27, except that 89.0 mg (0.320 mmol) of N-
[(2S)-pyrrolidine-2-ylmethyl]benzenesulfonamide hydrochloric acid salt obtained by referring to WO 05/056003 and 95 mg (0.320 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [1011] 1H NMR (CDCl3) δ 7.89-7.84 (2H, m), 7.56-7.47 (3H, m), 6.67-6.66 (IH, m),
5.84-5.76 (2H, m), 4.12-3.93 (4H, m), 3.64-3.59 (2H, m), 3.56-3.39 (2H, m), 3.35-3.16
(IH, m), 3.01-2.95 (IH, m), 2.55-2.32 (2H, m), 2.00-1.84(5H, m), 1.41-1.40(9H, m) [1012] Mass (m/e) 521 (M+l) [1013] [1014] EXAMPLE 46: Synthesis of N- α(2SVl-rGSV3-amino-4-(4-methyl-2-oxo-2.5-dihvdro-lH-pyrrole-l-yls)butanoyllpyrr olidine-2-yl ) methvDbenzenesulfonamide

[1016] 74.0 mg of the title compound (yield: 78%) was obtained, in the same manner as in EXAMPLE 14, except that 108 mg (0.210 mmol) of t- butyl{(lS)-l-[(4-methyl-2-oxo-2,5-dihydro-lH-pyrrole-l-yl)methyl]-3-oxo-3-[(2S)-2- {[(phenylsulfonyl)amino]methyl}pyrrolidine-l-yl]propyl}carbamate obtained in PREPARATION 68 was used.
[1017] 1H NMR (CD3OD) δ 7.85-7.80 (2H, m), 7.65-7.60 (3H, m), 5.78-5.73 (IH, m),
4.02-3.91 (3H, m), 3.64-3.36 (8H, m), 3.02-2.97 (IH, m), 2.80-2.72 (IH, m), 2.58-2.51
(2H, m), 2.02-1.86 (4H, m)
[1018] Mass (m/e) 421 (M+l)
[1019]
[1020] PREPARATION 69: Synthesis of t- butylf αSV3-rGSV3-rα?enzoylamino)metfayl1-34-dihvdroisoquinoline-2flHVyl1-l-r(5 .5-difluoro-2-oxopiperidine- 1 -vDmethvil -3-oxopropyl lcarbamate
[1021] 80.5 mg of the title compound (yield: 35%) was obtained, in the same manner as in PREPARATION 27, except that 106 mg (0.398 mmol) of N- [(3S)-l,2,3,4-tetrahydroisoquinoline-3-ylmethyl]benzamide hydrochloric acid salt obtained in PREPARATION 4 and 150 mg (0.440 mmol) of (3S)-3-[(t-butoxycarbonyl)amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1022] 1H NMR (CDCl3) δ 7.82-7.77 (2H, m), 7.50-7.42 (3H, m), 7.30-7.18 (4H, m),
5.12-5.11 (IH, m), 4.67-4.51 (2H, m), 3.76-3.37 (5H, m), 3.21-3.12 (2H, m), 2.91-2.83 (2H, m), 2.57-2.47 (3H, m), 2.29-2.22 (2H, m), 2.07-2.01 (IH, m), 1.44-1.43 (9H, m)
[1023] Mass (m/e) 585 (M+l)
[1024]
[1025] EXAMPLE 47: Synthesis of N- α(3SV2-r(3SV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-vDbutanoyll-1.2.3.4-tetrahv droisoquinoline-S-yDmethyDbenzamide

[1027] 43.5 mg of the title compound (yield: 61%) was obtained, in the same manner as in EXAMPLE 14, except that 80.5 mg (0.137 mmol) of t- butyl{(lS)-3-[(3S)-3-[(benzoylamino)methyl]-3,4-dihydroisoquinoline-2(lH)-yl]-l-[(5 ,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 69 was used.
[1028] 1H NMR (CD3OD) δ 7.87-7.86 (IH, m), 1.11-1.16 (IH, m), 7.50-7.39 (3H, m),
7.22-7.21 (4H, m), 5.10-5.06 (IH, m), 4.65-4.43 (2H, m), 3.93-3.61 (6H, m), 3.51-2.91 (5H, m), 2.58-2.03 (4H, m)
[1029] Mass (m/e) 485 (M+l)
[1030]
[1031] PREPARATION 70: Synthesis of t- butvKαSVS-rαS^^-G-cvclobutyl-l^^-oxadiazole-S-vDpiperidine-l-yll-l-rrS.S-diflu oro-2-oxopiperidine- 1 - vDmethyll -3-oxopropyl ) carbamate
[1032] 46.0 mg of the title compound (yield: 40%) was obtained, in the same manner as in
PREPARATION 27, except that 53.0 mg (0.217 mmol of
(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine hydrochloric acid salt) obtained in PREPARATION 2 and 73.0 mg (0.217 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [1033] 1H NMR (CDCl3) δ 6.11-6.10 (IH, m), 5.81-5.79 (IH, m), 4.28-4.26 (IH, m),
3.83-3.61 (6H, m), 3.35-3.27 (IH, m), 2.90-2.82 (IH, m), 2.64-2.58 (3H, m), 2.43-2.28
(7H, m), 2.20-2.00 (2H, m), 1.99-1.72 (4H,m), 1.49-1.45 (1OH, m) [1034] Mass (m/e) 526 (M+l) [1035] [1036] EXAMPLE 48: Synthesis of l-r(2SV2-amino-4-((2SV2-r3-(cvclobutylV1.2.4-oxadiazole-5-yllpiperidine-l-yl)-4-o xobutyl)-5.5-difluoropiperidine-2-one

[1038] 37.5 mg of the title compound (yield: 93%) was obtained, in the same manner as in EXAMPLE 14, except that 46.0 mg (0.088 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)piperidine-l-yl]-l-[(5,5-diflu oro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 70 was used.
[1039] 1H NMR (CD3OD) δ 6.09-6.07 (IH, m), 3.93-3.61 (6H, m), 3.60-3.51 (IH, m), 2.95- 2.91 (2H, m), 2.66-2.63 (2H, m), 2.42-2.34 (8H, m), 2.21-2.09 (IH, m), 2.05-1.89 (2H, m), 1.84-1.71 (2H, m), 1.68-1.48 (2H, m)
[1040] Mass (m/e) 426 (M+l)
[1041]
[1042] PREPARATION 71: Synthesis of t- butyl(αSV3-rGSV3-G-cvclobutyl-1.2.4-oxadiazole-5-ylV3.4-dihvdroisoquinoline-2α HVyIl- l-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-oxopropyl)carbamate
[1043] 20.0 mg of the title compound (yield: 22%) was obtained, in the same manner as in PREPARATION 27, except that 45.5 mg (0.156 mmol) of
(3S)-3-(3-cyclobutyl- 1 ,2,4-oxadiazole-5-yl)- 1 ,2,3,4-tetraisoquinoline hydrochloric acid salt obtained in PREPARATION 3 and 53.5 mg (0.159 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1044] 1H NMR (CDCl3) δ 7.22-7.09 (4H, m), 6.22-6.20 (IH, m), 5.81-5.79 (IH, m), 4.80-4.67 (2H, m), 4.29-4.28 (IH, m), 3.75-3.35 (7H, m), 3.00-2.89 (IH, m ), 2.73-2.67 (IH, m), 2.59-2.50 (2H, m), 2.30-2.22 (6H,m), 2.07-1.89 (2H, m), 1.42-1.41(9H, m)
[1045] Mass (m/e) 574 (M+l)
[1046]
[1047] EXAMPLE 49: Synthesis of l-((2SV2-amino-4-rGSV3-G-cvclobutyl-1.2.4-oxadiazole-5-ylV3.4-dihvdroisoquinoli ne-2(lHs)-yll-4-oxobutyl)-5.5-difluoropiperidine-2-one

[1049] 16.6 mg of the title compound (yield: 93%) was obtained, in the same manner as in EXAMPLE 14, except that 20.0 mg (0.035 mmol) of t- butyl{(lS)-3-[(3S)-3-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)-3,4-dihydroisoquinoline-2(l H)-yl]- l-[(5,5-difluoro-2-oxopiperidine- l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 71 was used.
[1050] 1H NMR (CD3OD) δ 7.14-7.06 (IH, m), 6.03-6.02 (IH, m), 3.83-3.62 (4H, m), 3.58- 3.54 (2H, m), 3.49-3.31 (4H, m), 3.06-3.02 (IH, m), 2.88-2.80 (IH, m), 2.55-2.51(2H, m), 2.31-2.07 (6H, m), 2.01-1.92(1H, m), 1.82-1.76 (IH, m)
[1051] Mass (m/e) 474 (M+l)
[1052]
[1053] PREPARATION 72: Synthesis of t- butyl(αSV3-r(2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidine-l-v ll-l-[(5.5-difluoro-2-oxopiperidine-l-yl)methyll-3-oxopropyl)carbamate
[1054] 56 mg of the title compound (yield: 51%) was obtained, in the same manner as in PREPARATION 27, except that 50.0 mg (0.214 mmol) of
3-cyclopropyl-5-[(2S,4S)-4-fluoropyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained in PREPARATION 23 and 72.0 mg (0.214 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1055] 1H NMR (CDCl3) δ 6.02-6.00 (IH, m), 5.45-5.43 (IH, m), 5.38-5.36 (IH, m),
5.27-5.26 (IH, m), 4.19-4.12 (IH, m), 3.95-3.66 (3H, m), 3.62-3.53 (IH, m), 3.51-3.45 (IH, m), 2.68-2.42 (6H, m), 2.28-2.20 (2H,m), 2.08-2.00 (IH, m), 1.39 (9H, s), 1.05-0.95 (4H, m)
[1056] Mass (m/e) 532 (M+l)
[1057]
[1058] EXAMPLE 50: Synthesis of l-r(2SV2-amino-4-r('2S.4SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-fluoropyrrolidi ne-l-yll-4-oxobutyll-5.5-difluoropiperidine-2-one

[1060] 46.0 mg of the title compound (yield: 94%) was obtained, in the same manner as in EXAMPLE 14, except that 56.0 mg (0.109 mmol) of t- butyl{(lS)-3-[(2S,4S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-fluoropyrrolidine-l-y l]-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 72 was used.
[1061] 1H NMR (CD3OD) δ 5.62-5.30 (IH, m), 4.07-3.74 (7H, m), 3.61-3.54 (IH, m), 3.04- 2.94 (IH, m), 2.86-2.49 (4H, m), 2.44-2.34 (2H, m), 2.13-2.03 (IH, m), 1.11-0.92 (4H, m), Mass (m/e) 432 (M+l)
[1062]
[1063] PREPARATION 73: Synthesis of t- butyl(αSV3-r(2S.4RV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-hvdroxypyrrolidine- l-yll-l-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-oxopropyl)carbamate
[1064] 43.0 mg of the title compound (yield: 48%) was obtained, in the same manner as in PREPARATION 27, except that 40.0 mg (0.173 mmol) of (3R, 5S)-5-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)pyrrolidine-3-ol hydrochloric acid salt obtained in PREPARATION 24 and 59.0 mg (0.176 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1065] 1H NMR (CDCl3) 66.40-6.38 (IH, m), 5.35-5.32 (IH, m), 4.50 (IH, m), 4.08-4.07
(IH, m), 3.85-3.68 (5H, m), 3.21-3.19 (IH, m), 2.72-2.69 (IH, m ), 2.57-2.39 (4H, m), 2.29-2.22 (2H, m), 2.12-2.03 (3H,m), 1.40 (9H, s), 1.13-0.98 (4H, m)
[1066] Mass (m/e) 530 (M+l)
[1067]
[1068] EXAMPLE 51: Synthesis of l-((2SV2-amino-4-r(2S.4RV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-hvdroxypyrroli dine- 1 -yll -4-oxobutyl ) -5.5-difluoropiperidine-2-one
[1069]

[1070] 34.2 mg of the title compound (yield: 91%) was obtained, in the same manner as in EXAMPLE 14, except that 43.0 mg (0.084 mmol) of t- butyl{(lS)-3-[(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-hydroxypyrrolidine- 1-yl]- l-[(5,5-difluoro-2-oxopiperidine- l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 73 was used.
[1071] 1H NMR (CD3OD) δ 5.29-5.26 (IH, m), 4.60-4.59 (IH, m), 3.90-3.61 (6H, m), 3.58- 3.52 (IH, m), 2.84-2.80 (2H, m), 2.65-2.61 (2H, m), 2.44-2.32 (IH, m), 2.22-2.04 (2H, m), 1.12-0.94 (4H, m)
[1072] Mass (m/e) 430 (M+l)
[1073]
[1074] PREPARATION 74: Synthesis of t- butyl(αSV3-r(2S.4RV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-methoxypyrrolidine- l-yll-l-[(5.5-difluoro-2-oxopiperidine-l-yl)methyll-3-oxopropyl)carbamate
[1075] 50.0 mg of the title compound (yield: 45%) was obtained, in the same manner as in PREPARATION 27, except that 52.0 mg (0.211 mmol) of
3-cyclopropyl-5-[(2S,4R)-4-methoxypyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained in PREPARATION 25 and 71 mg (0.211 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1076] 1H NMR (CDCl3) δ 5.71-5.69 (IH, m), 5.25-5.22 (IH, m), 4.22-4.05 (2H, m),
3.80-3.41 (5H, m), 3.33 (3H, s), 2.72-2.69 (IH, m), 2.61-2.39 (4H, m ), 2.30-2.19 (2H, m), 2.15-2.11 (IH, m), 2.06-2.02 (lH,m), 1.39 (9H, s), 1.07-0.95 (4H, m)
[1077] Mass (m/e) 528 (M+l)
[1078]
[1079] EXAMPLE 52: Synthesis of l-((2SV2-amino-4-r(2S.4RV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4-metoxypyrroli dine- 1 -yll -4-oxobutyl ) -5.5-difluoropiperidine-2-one
[1080]

[1082] 1H NMR (CD3OD) δ 5.19-5.16 (IH, m), 4.19-4.18 (IH, m), 3.87-3.63 (5H, m), 3.57- 3.56 (IH, m), 3.36 (3H, m), 2.84-2.72 (2H, m), 2.60-2.50 (4H, m), 2.38-2.28 (2H, m), 2.17-2.12 (IH, m), 2.05-2.02 (IH, m), 1.06-1.03 (2H, m), 0.93-0.91(2H, m)
[1083] Mass (m/e) 428 (M+l)
[1084]
[1085] PREPARATION 75: Synthesis of t- butyl(αSV3-r(2SV2-(3-cvclopropyl-1.2.4-oxadiazole-5-ylV4.4-difluoropyrrolidine-l- yll-l-r(5.5-difluoro-2-oxopiperidine-l-yls)methyll-3-oxopropyl)carbamate
[1086] 26.0 mg of the title compound (yield: 20%) was obtained, in the same manner as in PREPARATION 27, except that 59.9 mg (0.238 mmol) of
3-cyclopropyl-5-[(2S)-4,4-difluoropyrrolidine-2-yl]-l,2,4-oxadiazole hydrochloric acid salt obtained by referring to WO 05/121131 and 80.0 mg (0.238 mmol) of (3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1087] 1H NMR (CDCl3) δ 5.81-5.80 (IH, m), 5.47-5.26 (IH, m), 4.27-4.10 (2H, m), 4.02-3.96 (IH, m), 3.75-3.49 (4H, m), 3.28-3.24 (IH, m), 2.86-2.80 (IH, m ), 2.66-2.44 (3H, m), 2.28-2.20 (3H, m), 2.08-2.01 (lH,m), 1.40 (9H, s), 1.08-0.95 (4H, m)
[1088] Mass (m/e) 534 (M+l)
[1089]
[1090] EXAMPLE 53: Synthesis of l-((2SV2-amino-4-r(2SV2-G-cvclopropyl-1.2.4-oxadiazole-5-ylV4.4-difluoropyrrolidi ne-l-yll-4-oxobutyl)-5.5-difluoropiperidine-2-one

[1092] 22.5 mg of the title compound (yield: 98%) was obtained, in the same manner as in EXAMPLE 14, except that 26.0 mg (0.049 mmol) of t- butyl{(lS)-3-[(2S)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4,4-difluoropyrrolidine-l- yl]-l-[(5,5-difluoro-2-oxopiperidine-l-yl)methyl]-3-oxopropyl}carbamate obtained in
PREPARATION 75 was used. [1093] 1H NMR (CD3OD) δ 5.58-5.53 (IH, m), 4.36-4.33 (IH, m), 4.20-4.14 (IH, m),
3.91-3.69 (4H, m), 3.61- 3.59 (IH, m), 3.50-3.48 (IH, m), 2.84-2.80 (2H, m),
2.68-2.55 (3H, m), 2.38-2.29 (2H, m), 2.13-2.07 (IH, m), 1.13-0.95 (4H, m) [1094] Mass (m/e) 434 (M+l) [1095] [1096] PREPARATION 76: Synthesis of t- butyl(αSV3-rGSV3-(anilinocarbonylV3.4-dihvdroisoquinoline-2αHVyll-l-r(5.5-difl uoro-2-oxopiperidine- 1 - vDmethyll -3-oxopropyl ) carbamate [1097] 90.0 mg of the title compound (yield: 80%) was obtained, in the same manner as in
PREPARATION 27, except that 54.0 mg (0.187 mmol) of
(3S)-N-phenyl-l,2,3,4-tetrahydroisoquinoline-3-carboxamide hydrochloric acid salt obtained in PREPARATION 1 and 66.0 mg (0.196 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used. [1098] 1H NMR (CDCl3) δ 9.17 (IH, s), 8.92 (IH, s), 7.58-7.51 (2H, m), 7.25-7.12 (4H, m),
7.10-7.00 (3H, m), 6.46-6.44 (IH, m), 5.39-5.36 (IH, m ), 4.65-4.53 (2H, m),
4.35-4.29 (IH, m), 3.76-3.48 (4H, m), 3.02-2.97 (IH, m), 2.82-2.74 (IH, m), 2.52-2.49
(2H, m), 2.30-2.14 (3H, m), 1.42-1.40 (9H, m) [1099] Mass (m/e) 571 (M+l) [1100] [1101] EXAMPLE 54: Synthesis of
GSV2-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-yls)butanoyll-N-phenyl-1.2.3.4
-tetraisoquinoline-3-carboxamide

[1103] 73.2 mg of the title compound (yield: 91%) was obtained, in the same manner as in EXAMPLE 14, except that 90.0 mg (0.158 mmol) of t- butyl{(lS)-3-[(3S)-3-(3-cyclobutyl-l,2,4-oxadiazole-5-yl)-3,4-dihydroisoquinoline-2(l H)-yl]- l-[(5,5-difluoro-2-oxopiperidine- l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 76 was used.
[1104] 1H NMR (CD3OD) δ 7.43-7.38 (2H, m), 7.29-7.19 (6H, m), 7.08-7.03 (IH, m),
4.99-4.97 (IH, m), 3.99-3.70 (5H, m), 3.64-3.60 (2H, m), 3.55-3.46 (IH, m), 3.27-3.24 (IH, m), 3.18-3.11 (IH, m), 2.92-2.84 (IH, m), 2.62-2.58 (2H, m), 2.40-2.30 (2H, m)
[1105] Mass (m/e) 471 (M+l)
[1106]
[1107] PREPARATION 77: Synthesis of t- butylKlS^-S-rriS^-l-ranilinocarbonvDpiperidine-l-yll-l-rrS.S-difluoro-l-oxopiperidin e- 1 - vDmethyll -3-oxopropyl ) carbamate
[1108] 35.0 mg of the title compound (yield: 64%) was obtained, in the same manner as in PREPARATION 27, except that 24.5 mg (0.102 mmol) of (2S)-N-phenylpiperidine-2-carboxamide hydrochloric acid salt obtained in PREPARATION 26 and 35.0 mg (0.104 mmol) of
(3S)-3-[(t-butoxycarbonyl)amino]-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyric acid obtained by referring to Korean Patent Application No. 2006-29138 were used.
[1109] 1H NMR (CDCl3) δ 9.38 (IH, s), 7.79-7.71 (IH, m), 7.64-7.62 (IH, m), 7.31-7.21
(2H, m), 7.10-7.07 (IH, m), 6.64-6.62 (IH, m), 5.35-5.22 (IH, m ), 4.64-4.62 (IH, m), 4.31-4.26 (IH, m), 3.83-3.66 (3H,m), 3.61-3.50 (IH, m), 3.19-3.05 (IH, m), 2.76-2.10 (7H, m), 1.80-1.56 (3H, m), 1.50-1.34(1 IH, m)
[1110] Mass (m/e) 523 (M+l)
[1111]
[1112] EXAMPLE 55: Synthesis of
(2SVl-rGSV3-amino-4-(5.5-difluoro-2-oxopiperidine-l-yls)butanoyll-N-phenylpiperidi ne-2-carboxamide

[1114] 30.6 mg of the title compound (yield: 100%) was obtained, in the same manner as in EXAMPLE 14, except that 35.0 mg (0.067 mmol) of t- butyl{(lS)-3-[(2S)-2-(anilinocarbonyl)piperidine-l-yl]-l-[(5,5-difluoro-2-oxopiperidin e-l-yl)methyl]-3-oxopropyl}carbamate obtained in PREPARATION 77 was used.
[1115] 1H NMR (CD3OD) δ 7.54-7.46 (3H, m), 7.41-7.38 (2H, m), 5.25-5.22 (IH, m), 3.97-3.55 (9H, m), 2.98-2.77 (IH, m), 2.65-2.54(2H, m), 2.40-2.24 (3H, m), 1.76-1.61(3H, m), 1.52-1.32 (2H, m)
[1116] Mass (m/e) 423 (M+l)
[1117]
[1118] EXPERIMENT: Measurement of DPP-IV activity-inhibiting ability
[1119] Dipeptidyl Peptidase-IV (DPP-IV), known as serine protease, was obtained by a modification of the known method (Tanaka T. et al, Proc. Natl. Acad. Sci. USA, (1994) 91, 3082-3086), which comprises cloning, purification by use of Baculo- Virus and activation steps. DPP-IV was used to test the pharmaceutical efficacy of candidate
inhibitors as follows. The cloned DPP-IV was expressed in Baculo- Virus, followed by purifying with nickel column and then subjected to dialysis. The inhibitors synthesized in Examples were tested to determine the inhibiting activity thereof using a fluorescent substrate, Ac-Gly-Pro-AFC. Enzyme reactions were conducted for various concentrations of inhibitors, using 100 μM Ac-Gly-Pro-AFC at 250C in a buffer solution containing 50 mmol HEPES (pH 7.4), with the concentration of DPP-IV being 7.1 nM. The inhibitor's IC50 value was determined by measuring the amount of fluorescence emitted in a fluorescent spectrometer after allowing enzyme reaction for 1 hour, and then calculating the concentration of inhibitors exhibiting 50% inhibition of the total enzyme reaction. As the fluorescent spectrometer, Spectra MAX GeminiXS fluorescent spectrometer from Molecular Device Co. was used, and the excitation frequency and emission frequency were set to 400 nm and 505 nm, respectively. The result is summarized in TABLE 1 below.
[1120] IT ABLE 11
[1121]

[1122] * DPP-IV inhibition activity (IC50) was classified in 4 parts according to the below groups:
[ 1123] - Classification A: below 100 nM [1124] - Classification B: from 101 nM to 1.000 M [1125] - Classification C: from 1.001 M to 2.499 M [1126] - Classification D: above 2.5 M
Industrial Applicability
[1127] From the above results, it is clear that the compounds according to the present invention are very effective in inhibiting DPP-IV enzyme activity to be used for treating and preventing diabetes mellitus (especially, type II diabetes mellitus), obesity and the like.
[1128] However, it will be understood that the present invention is not limited to these specific preparations and examples, but is subject to various modifications that will be recognized by one skilled in the art to which the present invention pertains.
Claims
(1) hydrogen;
(2) halogen;
(3) OH or OR', wherein R' is linear or branched Ci-C7 alkyl;
(4) NR1R", wherein R' and R" are each independently hydrogen or linear or branched Ci-C7 alkyl;
(5) substituted or unsubstituted linear or branched Ci-C7 alkyl
(6) CN;
(7) COOH or COOR' wherein R' is linear or branched Ci-C7 alkyl;
(8) CONR'R" wherein R' and R" are each independently hydrogen or linear or branched Ci-C7 alkyl;
(9) substituted or unsubstituted phenyl, wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, CN, OH, COOH and NH2;
(10) substituted or unsubstituted C3-C6 cycloalkyl, wherein group(s) for substitution is/are each independently one or more selected from the group consisting of Ci-C6 alkyl, Ci-C6 alkoxy, halogen, CN, OH, COOH and NH2;
(11) substituted or unsubstituted heterocycle, wherein group(s) for substitution is/are each independently halogen, OR', NR'R", substituted or unsubstituted linear or branched Ci-Ci0 alkyl, CN, COOR', CONR'R", substituted or unsubstituted phenyl, substituted or unsubstituted C3-C 6 cycloalkyl, or substituted or unsubstituted heterocycle, wherein R' and R" are each independently hydrogen, or linear or branched Ci-C7 alkyl.
[8] The compound according to claim 1, or pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is a compound as represented by Formula (Ib) below:

(Ib) wherein Ai, A2, A3 and B are defined as in Formula (I).
[9] The compound according to claim 1, or pharmaceutically acceptable salt thereof, wherein the compound of formula (I) is a compound as represented by Formula
(Ic) below:

(Ic) wherein Ai, A2, A3 and B are defined as in Formula (I).
[10] The compound according to claim 1, or pharmaceutically acceptable salt thereof, wherein when the heterocycle is substituted, group(s) for substitution is/are each independently substituted or unsubstituted linear or branched Ci-C4 alkyl, sub-
stituted or unsubstituted C3-C6 cycloalkyl, substituted or unsubstituted phenyl, or substituted or unsubstituted heterocycle.
[11] The compound according to claim 10, or pharmaceutically acceptable salt thereof, wherein the heterocycle is selected from the group consisting of furan, thiophene, pyrrol, pyrrolidine, imidazole, pyrazole, pyrazoline, oxazole, oxazoline, isoxazole, isoxazoline, thiazole, thiazoline, isothiazole, iso- thiazolidine, thiadiazole, thiadiazoline, tetrahydrofuran, tetrahydrothiophene, im- idazolidine, pyrazolidine, oxazolidine, isoxazolidine, thiazolidine, iso- thiazolidine, thiadiazolidine, sulforan, pyran, dihydropyran, tetrahydropyran, pyridine, pyridinone, pyridazine, pyrazine, pyrimidine, piperidine, piperazine, morpholine, pyridazinone, tetrazole, triazole, triazolidine and azepine.
[12] The compound according to claim 1, or pharmaceutically acceptable salt thereof, wherein the compound is one of the below compounds: l-{(2S)-2-amino-4-[2,4-bis(trifluoromethyl)-5,8-dihydropyrido[3,4-d]pyrimidine -7(6H)-yl]butyl}-5,5-difluoropiperidine-2-one;
N-({(2S)-l-[(3S)-3-amino-4-(5,5-difluoro-2-oxopiperidine-l-yl)butyl]pyrrolidine -2-yl } methyl)benzenesulfonamide;
(4R)-l-{(2S)-2-amino-4-[(2S)-2-(anilinomethyl)pyrrolidine-l-yl]-4-oxobutyl}-4- methylpyrrolidine-2-one; l-[(2S)-2-amino-4-{(2S)-2-[3-(cyclobutyl)-l,2,4-oxadiazole-5-yl]piperidine-l-yl ]-4-oxobutyl}-5,5-difluoropiperidine-2-one; l-{(2S)-2-amino-4-[(2S,4R)-2-(3-cyclopropyl-l,2,4-oxadiazole-5-yl)-4-methoxy pyrrolidine- 1 -yl] -4-oxobutyl } -5,5-difluoropiperidine-2-one.
[13] A pharmaceutical composition for inhibiting Dipeptidyl Peptidase-IV (DPP-IV) comprising the compound of Formula (I) as defined in claim 1 or a pharmaceutically acceptable salt thereof.
[14] The pharmaceutical composition according to claim 13, wherein the pharmaceutical composition is used for treating or preventing diseases caused by DPP- IV.
[15] The pharmaceutical composition according to claim 14, wherein the diseases caused by DPP-IV are diabetes mellitus or obesity.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20070009315 | 2007-01-30 | ||
| KR20070009298 | 2007-01-30 | ||
| KR10-2007-0009298 | 2007-01-30 | ||
| KR10-2007-0009315 | 2007-01-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008093960A1 true WO2008093960A1 (en) | 2008-08-07 |
Family
ID=39674227
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2008/000428 Ceased WO2008093960A1 (en) | 2007-01-30 | 2008-01-23 | Novel dipeptidyl peptidase-iv inhibitors |
Country Status (2)
| Country | Link |
|---|---|
| KR (1) | KR20080071476A (en) |
| WO (1) | WO2008093960A1 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| EP2404909A4 (en) * | 2008-12-16 | 2012-05-16 | Tianjin Inst Pharm Research | Amide thiazole derivative, preparation method and uses thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| JP2013542245A (en) * | 2010-11-11 | 2013-11-21 | レッドエックス ファーマ リミテッド | Drug derivative |
| JP2013543484A (en) * | 2010-09-03 | 2013-12-05 | エルジー・ライフ・サイエンシーズ・リミテッド | Method for producing intermediate compound for pharmaceutical synthesis |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| EP3565543A4 (en) * | 2017-01-06 | 2020-10-14 | The Regents of The University of California | MU-OPIOID RECEPTOR MODULATORS |
| US11352316B2 (en) | 2018-04-04 | 2022-06-07 | Epiodyne, Inc. | Opioid receptor modulators and products and methods related thereto |
| US11634396B2 (en) | 2021-04-05 | 2023-04-25 | Epiodyne, Inc. | Opioid receptor modulators |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101555033B1 (en) * | 2014-07-28 | 2015-09-23 | 충남대학교산학협력단 | Novel methanone derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating blindness related diseases containing the same as an active ingredient |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003004498A1 (en) * | 2001-07-06 | 2003-01-16 | Merck & Co., Inc. | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2005056003A1 (en) * | 2003-12-09 | 2005-06-23 | Santhera Pharmaceuticals (Schweiz) Gmbh | Dpp-iv inhibitors |
| WO2005120494A1 (en) * | 2004-06-08 | 2005-12-22 | Santhera Pharmaceuticals (Schweiz) Ag | 1-`(3r)-amino-4-(2-fluoro-phenyl)-butyl !-pyrrolidine-(2r)-carboxilic acid-benzyl amine derivatives and related compounds as dipeptidyl-peptidase iv (dpp-iv) inhibitors for the treatment of type 2 diabetes mellitus |
-
2007
- 2007-10-23 KR KR1020070106402A patent/KR20080071476A/en not_active Ceased
-
2008
- 2008-01-23 WO PCT/KR2008/000428 patent/WO2008093960A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003004498A1 (en) * | 2001-07-06 | 2003-01-16 | Merck & Co., Inc. | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2005056003A1 (en) * | 2003-12-09 | 2005-06-23 | Santhera Pharmaceuticals (Schweiz) Gmbh | Dpp-iv inhibitors |
| WO2005120494A1 (en) * | 2004-06-08 | 2005-12-22 | Santhera Pharmaceuticals (Schweiz) Ag | 1-`(3r)-amino-4-(2-fluoro-phenyl)-butyl !-pyrrolidine-(2r)-carboxilic acid-benzyl amine derivatives and related compounds as dipeptidyl-peptidase iv (dpp-iv) inhibitors for the treatment of type 2 diabetes mellitus |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2404909A4 (en) * | 2008-12-16 | 2012-05-16 | Tianjin Inst Pharm Research | Amide thiazole derivative, preparation method and uses thereof |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| JP2013543484A (en) * | 2010-09-03 | 2013-12-05 | エルジー・ライフ・サイエンシーズ・リミテッド | Method for producing intermediate compound for pharmaceutical synthesis |
| JP2013542245A (en) * | 2010-11-11 | 2013-11-21 | レッドエックス ファーマ リミテッド | Drug derivative |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| EP3565543A4 (en) * | 2017-01-06 | 2020-10-14 | The Regents of The University of California | MU-OPIOID RECEPTOR MODULATORS |
| US11352316B2 (en) | 2018-04-04 | 2022-06-07 | Epiodyne, Inc. | Opioid receptor modulators and products and methods related thereto |
| US11634396B2 (en) | 2021-04-05 | 2023-04-25 | Epiodyne, Inc. | Opioid receptor modulators |
| US12168648B2 (en) | 2021-04-05 | 2024-12-17 | Epiodyne, Inc. | Opioid receptor modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20080071476A (en) | 2008-08-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008093960A1 (en) | Novel dipeptidyl peptidase-iv inhibitors | |
| AU2018202000B2 (en) | Carboxamide derivatives and the use thereof as medicaments for the treatment of Hepatitis B | |
| JP4101053B2 (en) | Proline derivative and its pharmaceutical use | |
| AU2013268415B2 (en) | Novel beta-lactamase inhibitor and method for producing same | |
| JP4977685B2 (en) | Dipeptidyl peptidase-IV inhibitory compound, method for producing the same, and pharmaceutical composition comprising the same as an active substance | |
| JP5324574B2 (en) | Method for the synthesis of compounds useful for the treatment of hepatitis C | |
| EP2226322B1 (en) | Imidazole carbonyl compound | |
| US10167281B2 (en) | Substituted thiazole or oxazole P2X7 receptor antagonists | |
| AU2010237750A1 (en) | Derivatives of N-acyl-N'-phenylpiperazine useful (inter alia) for the prophylaxis or treatment of diabetes | |
| JPWO2002014271A1 (en) | Proline derivatives and their medical uses | |
| KR20050037973A (en) | Novel inhibitors of dpp-iv, methods of preparing the same, and pharmaceutical compositions containing the same as an active agent | |
| US5547978A (en) | Derivatives of pyrrolidin-2-ylcarbonylheterocyclic compounds | |
| KR20200115550A (en) | Aminopyrrolotriazine as a kinase inhibitor | |
| JP2011026305A (en) | Pharmaceutical composition comprising imidazole carbonyl compound | |
| CN110724142B (en) | Amide or sulfonamide substituted hydrazine derivatives as JAK kinase inhibitors | |
| JP5560287B2 (en) | Alkyl-heterocyclic carbamate derivatives, their preparation and their use | |
| EP1961750B1 (en) | Vla-4 inhibitory drug | |
| CN115151544B (en) | Beta-lactamase inhibitor and preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08704935 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08704935 Country of ref document: EP Kind code of ref document: A1 |