WO2008101857A2 - Coryneform bacteria with formate-thf-synthetase and/or glycine cleavage activity - Google Patents
Coryneform bacteria with formate-thf-synthetase and/or glycine cleavage activity Download PDFInfo
- Publication number
- WO2008101857A2 WO2008101857A2 PCT/EP2008/051795 EP2008051795W WO2008101857A2 WO 2008101857 A2 WO2008101857 A2 WO 2008101857A2 EP 2008051795 W EP2008051795 W EP 2008051795W WO 2008101857 A2 WO2008101857 A2 WO 2008101857A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- microorganism
- activity
- thf
- increased
- amount
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
- C12P13/12—Methionine; Cysteine; Cystine
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
Definitions
- the present invention relates to microorganisms and methods for producing L- methionine.
- the present invention relates to Coryneform bacteria which display formate-THF-synthetase activity and/or a functional glycine cleavage system.
- Methionine is the first limiting amino acid in livestock of poultry and feed and, due to this, mainly applied as feed supplement.
- methionine is almost exclusively applied as a racemate of D- and L-methionine which is produced by chemical synthesis.
- Fermentative production of fine chemicals such as amino acids, aromatic compounds, vitamins and cofactors is today typically carried out in microorganisms such as Corynebacterium glutamicum (C. glutamicum), Escherichia coli (E.coli), Saccharomyces cerevisiae (S. cerevisiae), Schizzosaccharomycs pombe (S. pombe), Pichia pastoris (P. pastoris), Aspergillus niger, Bacillus subtilis, Ashbya gossypii, Kluyveromyces lactis, Kluyveromyces marxianus or Gluconobacter oxydans.
- C. glutamicum Corynebacterium glutamicum
- Escherichia coli E.coli
- Saccharomyces cerevisiae Saccharomyces cerevisiae
- Schizzosaccharomycs pombe S. pombe
- Pichia pastoris P. pastoris
- Amino acids such as glutamate are thus produced using fermentation methods.
- certain microorganisms such as Escherichia coli (E. col ⁇ ) and Cory neb acterium glutamicum (C. glutamicum) have proven to be particularly suitable.
- the production of amino acids by fermentation also has inter alia the advantage that only L-amino acids are produced and that environmentally problematic chemicals such as solvents as they are typically used in chemical synthesis are avoided.
- a method for producing L- methionine is preferred, in a microorganism is provided which comprises the step of culturing a microorganism that is derived by genetic modification from a starting organism such that said microorganism produces more N 5 , N 10 -methylene- tetrahydrofolate (THF) compared to the starting organism.
- THF N 10 -methylene- tetrahydrofolate
- the method uses a microorganism that is selected from the group comprising microorganisms of the genus Enterobacteria, Corynebacterium, Bacillus and Streptomyces. Use of the species Corynebacterium glutamicum is particularly preferred.
- the method comprises the step of culturing a microorganism that is derived from a genetic modification from a starting organism such that the amount and/or activity of formate-THF- synthetase is increased compared to the starting organism.
- a preferred aspect of this latter embodiment of the invention relates to the cultivation of microorganisms in which additionally the amount and/or activity of formate-THF- deformylase is decreased compared to the starting organism and/or in which the amount and/or activity of N 5 , N 10 -methenyl-THF-cyclosynthetase, N 5 , N 10 -methenyl- THF-reductase and/or N 5 , N 10 -methylene-THF-reductase is increased compared to the starting organism.
- One of the other preferred embodiments of the present invention relates to methods in which a microorganism is cultivated that is derived by genetic modification from a starting organism such that the enzymatic activity of a glycine cleavage system (GCS) is increased compared to the starting organism.
- GCS glycine cleavage system
- PLP-dependent glycine decarboxylase gcvP, P-protein
- lipoamide-containing aminomethyl transferase gcvH, H-protein
- gcvT methylene-THF-synthesizing enzyme
- the microorganisms may be further genetically modified such that they display an increased amount and/or biological activity of lipoic acid synthase (HpA), lipoyl tansferase (HpE), and/or lipoic acid synthetase (IpIA).
- HpA lipoic acid synthase
- HpE lipoyl tansferase
- IpIA lipoic acid synthetase
- the cultivated microorganisms may additionally or alternatively be genetically modified to display an increased amount and/or activity of an NAD -dependent, FAD-requiring lipoamide dehydrogenase (Ipd).
- the method allows one to produce L-methionine by cultivating microorganisms which have been genetically modified such that the amount and/or biological activity of formate-THF-synthetase is increased and such that a functional glycine cleavage system has been established.
- microorganisms will typically display an increased amount and/or biological activity of formate-THF-synthetase, gcvH, gcvP and gcvT (gcvHPT).
- these latter microorganisms will be cultivated in the presence of lipoic acid and/or lipoamide.
- the microorganisms may be further genetically modified to display an increased amount and/or activity of HpA, HpB and/or IpI A.
- the microorganisms may also display an increased amount and/or biological activity of Ipd.
- the above-described embodiments of methods in accordance with the invention are preferably undertaken by cultivating microorganisms of the species C. glutamicum.
- the above described genetic modifications may be introduced into a C. glutamicum wild-type strain.
- these genetic alterations are introduced into a C. glutamicum strain that already is considered to be a methionine -producing strain.
- the coding sequences for the above-mentioned formate-THF-synthetase, gcvP, gcvT, gcvH, IpI A, lip A and HpB are preferably derived from C.jeikeium or E.coli.
- Sequences of C. jeikeium are particularly to be considered in case that the method is undertaken by cultivating C. glutamicum strains.
- the present invention relates to microorganisms which have been derived by genetic modification from a starting microorganism to produce more N 5 , N 10 -methylene-THF in comparison to the starting organism.
- the microorganisms can again be selected from the group comprising the genus Enterobacteria, Coryneform bacteriua, Bacillus and Streptomyces with Coryneform bacteria and particularly the species C. glutamicum being preferred.
- the microorganism is derived by genetic modification from a starting organism such that the amount and/or activity of formate-THF-synthetase is increased compared to the starting organism.
- Further elaborations of this latter embodiment of the invention comprise microorganisms with a decrease in the amount and/or activity of formate-THF-deformylase and/or with an increase in any of the activities of N 5 , N 10 -methenyl-THF-cyclosynthetase, N 5 , N 10 -methenyl-THF-reductase and/or N 5 , N 10 -methylene-THF-reductase.
- Another aspect of the invention relates to microorganisms which are derived by genetic modification from a starting organism such that the enzymatic activity of a glycine cleavage system is increased in said organism compared to the starting organism.
- the microorganism may be genetically altered in order to display an increased amount and/or activity of gcvP, gcvT and gcvH.
- the microorganism may be genetically further modified to display an increased capacity for uptake of externally provided lipoic acid and/or lipoamide and/or for synthesizing endogenously lipoic acid.
- the amount and/or activity of IpIA, lip A and/or HpB may be increased in said microorganisms.
- the amount and/or activity of lpd may also be increased compared to the starting organism.
- the microorganism is preferably derived from the species of C. glutamicum.
- the genetic alterations can be introduced in a wild-type strain of C. glutamicum or in a strain which is already considered to be a methionine-producing strain.
- a preferred aspect of the present invention relates to microorganisms in which the amount and/or biological activity of formate -THF-synthetase, gcvP, gcvT and gcvH are increased compared to the starting organism.
- the amount and/or activity o ⁇ lplA, lip A and/or HpB can be increased compared to the starting organism.
- the amount and/or activity of lpd can be increased.
- the microorganism is preferably derived from the species of C. glutamicum.
- the genetic alterations can be introduced in a wild-type strain of C. glutamicum or in a strain which is already considered to be a methionine-producing strain.
- Figure 1 shows a sequence comparison of the amino acid sequence of lpd of C. jeiekeum and C. glutamicum.
- the present invention relates to a method of producing L-methionine, comprising the step of cultivating a genetically modified microorganism and optionally isolating methionine.
- the present invention also relates to a genetically modified microorganism which is capable of producing L-methionine.
- the invention is based on the finding that an efficient production of L-methionine (also designated as methionine) can be achieved in microorganisms if such organisms have been genetically modified from a starting organism such that the resulting microorganism produces more N 5 , N 10 -methylene-tetrahydrofolate (THF) compared to the starting organism.
- L-methionine also designated as methionine
- a gene name or a protein name for example, but not limited to, formate-THF- synthetase, gcvPTH, HpA, HpB, IpI A, lpd and any other gene or protein name contained herein, shall refer to either or both the gene and/or the protein or enzyme encoded by said gene, depending on the context in which the name is used.
- microorganism for the purposes of the present invention refers to prokaryotes and lower eukaryotes.
- the organisms of the present invention thus comprise microorganisms as they are known in the art to be useful for production of fine chemicals such as amino acids, vitamins, emzyme cofactors etc. They can be selected from the genus of
- Corynebacterium with a particular focus on Corynebacterium glutamicum the genus of Enterobacteria with a particular focus on Escherichia coli, the genus of Bacillus, with a particular focus on Bacillus subtilis, the genus of actinobacteria, the genus of cyanobacteria, the genus of proteobacteria, the genus of halobacteria, the genus of methanococci, the genus of mycobacteria, the genus of salmonella, the genus of shigella and the genus of streptomycetaceae.
- Yeasts such as S. pombe, S. cerevisiae, K. lactis, K. marxianus, Ashbya gosypii, and Pichia pastoris are also understood to be encompassed by the term "microorganism”.
- the present invention is primarily concerned with microorganisms that have been genetically modified in order to display an increased amount and/or activity of certain enzymes.
- genetic modification and “genetic alteration” as well as their grammatical variations within the meaning of the present invention are intended to mean that a microorganism has been modified by means of gene technology to express an altered amount of one or more proteins which can be naturally present in the respective microorganism, one or more proteins which are not naturally present in the respective microorganism, or one or more proteins with an altered activity in comparison to the proteins of the respective non-modified microorganism.
- a non- modified microorganism is considered to be a "starting organism", the genetic alteration of which results in a microorganism in accordance with the present invention.
- starting organism therefore can refer to the wild-type of an organism. In the case of C. glutamicum, this may e.g. be ATCC 13032. However, the term “starting organism” for the purposes of the present invention may also refer to an organism which already carries genetic alterations in comparison to the wild-type organism of the respective species, but which is then further genetically modified in order to yield an organism in accordance with the present invention.
- the starting organism may thus be a wild-type C. glutamicum strain such as ATCC 13032.
- the starting organism may preferably also be e.g. a C. glutamicum strain which has already been engineered for production of methionine.
- Such a methionine -producing starting organism can e.g. be derived from a wild type Coryneform bacterium and preferably from a wild type C. glutamicum bacterium which contains genetic alterations in at least one one of the following genes: ask ⁇ ' ' , and metH wherein the genetic alterations lead to overexpression of any of these genes, thereby resulting in increased production of methionine relative to methionine produced in the absence of the genetic alterations.
- such a methionine producing starter organism will contain genetic alterations simulatenously in askf 1 " ' , and metH thereby resulting in increased production of methionine relative to methionine produced in the absence of the genetic alterations.
- C. glutamicum strain which includes these genetic alterations is e.g. C. glutamicum DSM 17322.
- the person skilled in the art will be aware that alternative genetic alterations to those being described below for generation of C. glutamicum DSM 17322 can be used to also achieve overexpression of ask fbr , hom fbr and metH.
- a methionine-producing starting organism can be derived from a wild type Coryneform bacterium and preferably from a wild type C. glutamicum bacterium which contains genetic alterations in at least one one of the following genes: asl ⁇ r , hom*" ⁇ , metH, metA (also referred to as metX), metY (also referred to as metZ), and hsk mut ⁇ ted .
- such a methionine producing starter organism will contain genetic alterations simulatenously in ask ® ' ' , metH, metA (also referred to as metX), metY (also referred to as metZ), and hsk mut ⁇ ted thereby resulting in increased production of methionine relative to methionine produced in the absence of the genetic alterations.
- C. glutamicum M2014 A C. glutamicum strain which includes these genetic alterations is e.g. C. glutamicum M2014.
- the person skilled in the art will be aware that alternative genetic alterations to those being described below specifically for generation of C. glutamicum M2014 can be used to also achieve overexpression of asl ⁇ r , hom*" ⁇ , metH, metA (also referred to as metX), metY (also referred to as metZ), and hsk mutated .
- metA denotes a homoserine succinyltransferase e.g. from E. coli.
- MetY denotes a O-Acetylhomoserine sulfhydrylase.
- ff s ⁇ mutated denotes a homoserine kinase which has been mutated to show reduced enzymatic activity. This may be achieved by exchanging threonine with serine or alanine at a position corresponding to T 190 of hsk of C. glutamicum ATCC13032 with Genbank accession no. CgIl 184. Alternatively or additionally one may replace the ATG start codon with a TTG start codon. Such mutations lead to a reduction in enzymatic activity of the resulting hsk protein compared the non- mutated hsk gene.
- a methionine-producing starting organism can be derived from a wild type Coryneform bacterium and preferably from a wild type C.
- glutamicum bacterium which contains genetic alterations in at least one of the following genes: metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mut ⁇ ted and metF wherein the genetic alterations lead to overexpression of any of these genes, in combination with a genetic alterations in one of the following genes: serA wherein the genetic alterations decrease expression of this gene where the combination results in increased methionine production by the microorganism relative to methionine production in absence of the combination.
- C. glutamicum strain which includes these genetic alterations is e.g. C. glutamicum OM264C.
- C. glutamicum OM264C The person skilled in the art will be aware that alternative genetic alterations to those being described below specifically for generation of C. glutamicum OM264C can be used to also achieve overexpression of as ⁇ , metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mutated and metF and reduced expression of serA.
- serA denotes 3-phosphoglycerate dehydrogenase (see Table 1)
- a methionine-producing starting organism can be derived from a wild type Coryneform bacterium and preferably from a wild type C.
- glutamicum bacterium which contains genetic alterations in at least one of the following genes: metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mutated and metF wherein the genetic alterations lead to overexpression of any of these genes, in combination with genetic alterations in at least one of the following genes : mcbR and metQ wherein the genetic alterations decrease expression of any of these genes where the combination results in increased methionine production by the microorganism relative to methionine production in absence of the combination.
- such a methionine producing starter organism will contain genetic alterations simulatenously in asli br , honi hr , metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mutated and metF wherein the genetic alterations lead to overexpression of any of these genes, in combination with genetic alterations in mcbR and metQ wherein the genetic alterations decrease expression of any of these genes where the combination results in increased methionine production by the microorganism relative to methionine production in absence of the combination.
- C. glutamicum strain which includes these genetic alterations is e.g. C. glutamicum OM469.
- C. glutamicum OM469 The person skilled in the art will be aware that alternative genetic alterations to those being described below specifically for generation of C. glutamicum OM469 can be used to also achieve overexpression metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mut ⁇ ted and metF and reduced expression of mcbR and metQ.
- metF denotes a N5,10-methylene- tetrahydrofolate reductase (EC 1 .5 . 1 .20).
- McbR denotes a TetR-type transcriptional regulator of sulfur metabolism (Genbank accession no: AAP45010).
- MetQ denotes a D-methionine binding lipoprotein which functiony in methionine import.
- a methionine-producing starting organism can be derived from a wild type Coryneform bacterium and preferably from a wild type C.
- glutamicum bacterium which contains genetic alterations in at least one of the following genes: metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mut ⁇ ted , metF, tkt, t ⁇ l, zwf and 6pgl wherein the genetic alterations lead to overexpression of any of these genes, in combination with genetic alterations in at least one of the following genes : mcbR, metQ and sd ⁇ wherein the genetic alterations decrease expression of any of these genes where the combination results in increased methionine production by the microorganism relative to methionine production in absence of the combination.
- such a methionine producing starter organism will contain genetic alterations simulatenously in metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mut ⁇ ted , metF, tkt, t ⁇ l, zwf and 6pgl wherein the genetic alterations lead to overexpression of any of these genes, in combination with genetic alterations in mcbR, metQ and sd ⁇ wherein the genetic alterations decrease expression of any of these genes where the combination results in increased methionine production by the microorganism relative to methionine production in absence of the combination.
- a C. glutamicum strain which includes these genetic alterations is e.g. C. glutamicum GK1259.
- C. glutamicum GK1259 can be used to also achieve overexpression of a sk ⁇ r , metH, metA (also referred to as metX), metY (also referred to as metZ), hsk mutated , metF, tkt, tal, zwf ' and 6pgl and reduced expression of mcbR, metQ and sda.
- tkt denotes transketolase
- tal denotes transaldolase
- zwf denotes glucose-6-phosphate-dehydrogenase
- 6pgl denotes 6- phospho-glucono-lactonase
- sda denotes serine deaminase (see Table 1).
- the person skilled in the art understands that for increasing the amount and/or activity of zwf one will also increase the amount and/or activity of opca which serves as a structural scaffolding protein fo zwf. In GK1259, this is achieved by the use of the P SOD promoter which simutenously increases transcription of the pentose phosphate operon comprising tkt, tal, zwf and 6pgl.
- the genetically modified microorganisms of the present invention are characterized in that the amount of N 5 , N 10 -methylene-THF is increased.
- the amount of N 5 , N 10 -methylene-THF will be increased in the microorganism in accordance with the present invention compared to the respective starting organism by at least about 2%, at least about 5%, at least about 10%, or at least about 20%. In other preferred embodiments, the amount of N 5 , N 10 -methylene- THF will be increased by at least about 30%, by at least about 50% or by at least about 75%. Even more preferred embodiments relate to microorganisms in which the amount of N 5 , N 10 -methylene-THF is increased by at least about factor 2, at least about factor 5 or at least about factor 10.
- the methods and microorganisms in accordance with the present invention can be used to produce more methionine compared to a situation where the respective starting organism,which has not been genetically modified as outlined below, is cultivated.
- the microorganisms and methods of the present invention can also be used to increase the efficiency of methionine synthesis.
- efficiency of methionine synthesis describes the carbon yield of methionine. This efficiency is calculated as a percentage of the energy input which entered the system in the form of a carbon substrate. Throughout the invention this value is given in percent values ((mol methionine) (mol carbon substrate ( -1 x 100).
- increase efficiency of methionine synthesis thus relates to a comparison between the starting organism and the actual Coryneform bacterium in which the amount and/or activity of at least one of the below mentioned enzymes has been increased.
- Preferred carbon sources according to the present invention are sugars such as mono- , di- or polysaccharides.
- sugars selected from the group comprising glucose, fructose, hanose, galactose, ribose, sorbose, lactose, maltose, sucrose, raffinose, starch or cellulose may serve as particularly preferred carbon sources.
- the methods and Coryneform bacteria in accordance with the invention may also be used to produce more methionine compared to the starting organism.
- the methods and Coryneform bacteria in accordance with the invention may also be used to produce methionine at a faster rate compared to the starting organism. If, for example, a typical production period is considered, the methods and Coryneform bacteria will allow to produce methionine at a faster rate, i.e. the same amount methionine will be produced at an earlier point in time compared to the starting organism. This particularly applies for the logarithmic growth phase.
- Methods and Coryneform bacteria in accordance with the invention allow to produce at least about 3 g methionine/1 culture volume if the strain is incubated in shake flask incubations.
- a titer of at least about 4g methionine/1 culture volume, at least about 5g methionine/1 culture volume or at least about 7g methionine/1 culture volume can be preferred if the strain is incubated in shake flask incubations.
- a more preferred value amounts to at least about 1Og methionine/1 culture volume and even more preferably to at least about 20 g methionine/1 cell mass if the strain is incubated in shake flask incubations.
- Methods and Coryneform bacteria in accordance with the invention allow to produce at least about 25 g methionine/1 culture volume if the strain is incubated in fermentation experiments using a stirred and carbon source fed fermentor.
- An titer of at least about 3Og methionine/1 culture volume, at least about 35g methionine/1 culture volume or at least about 4Og methionine/1 culture volume can be preferred if the strain is incubated in fermentation experiments using a stirred and carbon source fed fermentor.
- a more preferred value amounts to at least about 5Og methionine/1 culture volume and even more preferably to at least about 60 g methionine/1 cell mass if the strain is incubated in fermentation experiments using a stirred and carbon source fed fermentor.
- the methods and microorganisms of the invention allow to increase the efficiency of methionine synthesis and/or the amount of methionine and/or the titer and/or the rate of methionine synthesis in comparison to the starting organism by at least about 2%, at least about 5%, at least about 10% or at least about 20%.
- the efficiency of methionine synthesis and/or the amount of methionine and/or the titer and/or the rated is increased compared to the starting organism by at least about 30%, at least about 40%, or at least about 50%. Even more preferred is an increase of at least about factor 2, at least about factor 3, at least about factor 5 and at least about factor 10.
- metabolite refers to chemical compounds that are used in the metabolic pathways of organisms as precursors, intermediates and/or end products. Such metabolites may not only serve as chemical building units, but may also exert a regulatory activity on enzymes and their catalytic activity. It is known from the literature that such metabolites may inhibit or stimulate the activity of enzymes (Stryer, Biochemistry (2002) W.H. Freeman & Co., New York, New York).
- standard conditions refers to the cultivation of a microorganism in a standard medium which is not enriched with respect to a particular compound. The temperature, pH and incubation time can vary, as will be described in more detail below.
- Minimal media are media that contain only the necessities for the growth of wild- type or mutant cells, i.e. inorganic salts, a carbon source and water.
- a minimal medium can contain one or more additives of substantially pure chemical compounds to allow growth of mutant cells that are deficient in production of such chemical(s).
- enriched media are designed to fulfill all growth requirements of a specific organism, i.e. in addition to the contents of the minimal media, they contain, e.g. amino acids, growth factors, enzyme co-factors, etc.
- increasing the amount of at least one protein such as formate-THF- synthetase compared to a starting organism in the context of the present invention means that a starting micororganism is genetically modified to express a higher amount of e.g. one of the above-mentioned enzymes. It is to be understood that increasing the amount of e.g. one enzyme refers to a situation where the amount of functional enzyme is increased.
- An enzyme such as formate-THF-synthetase in the context of the present invention is considered to be functional if it is capable of catalysing the respective reaction.
- the term "increasing the activity" of at least one protein refers to the situation that at least one mutation is introduced into the respective wild-type sequences of the protein which leads to production of more methionine compared to a situation where the same amount of wild-type protein is expressed. This may achieved by e.g. using enzymes which carry specific mutations that allow for an increased activity of the enzyme. Such mutations may e.g. inactivate the regions of the enzymes that are responsible for feedback inhibition. By mutating these positions by e.g. introducing non-conservative point mutations, the enzyme does not provide for feedback regulation any more and thus the activity of the enzyme is not down-regulated if e.g. more product molecules are produced.
- the activity of an enzyme can be increased by introducing mutations which increase the catalytic turnover of an enzyme.
- mutations may be either introduced into the endogenous copy of the gene encoding for the respective enzyme, or they may be provided by over- expressing a corresponding mutant from the exogenous nucleic acid sequences encoding such an enzyme.
- Such mutations may comprise point mutations, deletions or insertions. Point mutations may be conservative (replacement of an amino acid with an amino acid of comparable biochemical and physical-chemical properties) or non-conservative (replacement of an amino acid with another which is not comparable in terms of biochemical and physical-chemical properties).
- the deletions may comprise only two or three amino acids up to complete domains of the respective protein.
- the term "increasing the activity" of at least one enzyme refers to the situation where mutations are introduced into the respective wild-type sequence to reduce negative regulatory mechanisms such as feedback-inhibition and/or to increase catalytic turnover of the enzyme.
- An increase of the amount and/or activity of a protein such as an enzyme may be achieved by different routes, e.g. by switching off inhibitory regulatory mechanisms at the transcriptional, translational or protein level, and/or by increasing gene expression of a nucleic acid encoding for this protein in comparison with the starting organism, e.g. by inducing the endogenous gene or by introducing nucleic acid sequences coding for the protein.
- aprotein such as an enzyme
- the nucleic acid sequences encoding for a protein such as an enzyme may be of endogenous or exogenous origin.
- a protein such as an enzyme may for example increase the amount of a protein such as an enzyme by either increasing expression of nucleic acid sequences that naturally occur within the respective starting microorganism by e.g. chromosomal integration of additional nucleic acid sequences, or by using a strong promoter in front of the endogenous gene.
- one may also increase the amount of a protein such as an enzyme by expressing the nucleic acid sequence encoding for a homo log of this enzyme from another organism. Examples for this latter scenario will be put forward below.
- endogenous enzymes as the endogenous coding sequence of e.g. C. glutamicum are already optimized with respect to its codon usage for expression in C. glutamicum.
- Reduction of the amount and/or activity of a protein such as an enzyme may be achieved by partially or completely deleting the nucleic acid sequences encoding the respective protein, by inhibiting transcription by e.g. introducing weak promoters, by inhibiting translation by amending the codon usage accordingly, by introducing mutations into the nucleic acid sequences encoding the respective proteins which render the proteins non- functional and/or combinations thereof.
- functional homo log for the purposes of the present invention relates to the fact that a certain enzymatic activity may not only be provided by a specific protein of defined amino acid sequence, but also by proteins of similar sequence from other (un)related organisms.
- formate-THF-synthetase For example, the activity of formate-THF-synthetase can be established in
- C. glutamicum by expressing nucleic acid sequences which encode for the formate- THF-synthetase of C.jeikeium (SEQ ID NO. 1 : nucleic acid sequence, SEQ ID NO. 2: amino acid sequence, gene bank accession number (NP 939608)) or by functional homologs thereof.
- Homologues of a protein from other organisms can be easily identified by the skilled person by homology analysis. This can be done by determining similarity, i.e. percent identity between amino acid or nucleic acid sequences for putative homologs and the sequences for the genes or proteins encoded by them (e.g., nucleic acid sequences for formate-THF-synthetase, gcvH, gcvP, gcvT, Ipd, IpI A, HpA, HpA).
- Percent identity may be determined, for example, by visual inspection or by using algorithm-based homology.
- the algorithm will align the sequences for optimal comparison purposes (e.g., gaps can be introduced in the amino acid sequence of one protein for optimal alignment with the amino acid sequence of another protein).
- the amino acid residues at corresponding amino acid positions are then compared. When a position in one sequence is occupied by the same amino acid residue as the corresponding position in the other, then the molecules are identical at that position.
- % identity # of identical positions/total # of positions multiplied by 100).
- percent identity of two nucleic acid or amino acid sequences can be determined by comparing sequence information using the GAP computer program described by Devereux et al. (1984) Nucl. Acids. Res., 12:387 and available from the University of Wisconsin Genetics Computer Group (UWGCG). Percent identity can also be determined by aligning two nucleic acid or amino acid sequences using the Basic Local Alignment Search Tool (BLASTTM) program (as described by Tatusova et al. (1999) FEMS Microbiol. Lett., 174:247.
- BLASTTM Basic Local Alignment Search Tool
- a standard software package providing the BLAST programme can be found on the BLAST website of the NCBI (http://www.ncbi.nlm.nih.gov/BLAST/). For example, if one uses any of the aforementioned SEQ IDs, one can either perform a nucleic acid sequence- or amino sequence-based BLAST search and identify closely related homo logs of the respective enzymes in e.g. E.coli, S. cervisiae, Bacillus subtilis, etc. For example, for nucleic acid sequence alignments using the BLASTTM program, the default settings are as follows: reward for match is 2, penalty for mismatch is -2, open gap and extension gap penalties are 5 and 2 respectively, gap .times. dropoff is 50, expect is 10, word size is 11, and filter is OFF.
- Comparable sequence searches and analysis can be performed at the EMBL database (http://www.embl.org) or the Expasy homepage (http://www.expasy.org/). All of the above sequences searches are typically performed with the default parameters as they are pre-installed by the database providers at the filing date of the present application. Homology searches may also routinely be performed using software programmes such as the laser gene software of DNA Star, Inc., Madison, Winconsin, USA, which uses the CLUSTAL method (Higgins et al. (1989), Comput. Appl. Biosci., 5(2) 151). The skilled person understands that two proteins will likely perform the same function (e.g. provide the same enzymatic activity) if they share a certain degree of identity as described above. A typical lower limit on the amino acid level is typically at least about 25% identity. On the nucleic acid level, the lower limit is typically at least 50%.
- Preferred identity grades for both type of sequences are at least about 50%, at least about 60% or least about 70%. More preferred identity levels are at least about 80%, at least about 90% or at least about 95%. These identity levels are considered to be significant.
- homologues are not limited to designate proteins having a theoretical common genetic ancestor, but includes proteins which may be genetically unrelated that have, none the less, evolved to perform similar functions and/or have similar structures.
- the requirement that the homologues should be functional means that the homologues herein described encompasse proteins that have substantially the same activity as the reference protein.
- proteins to have functional homology it is not necessarily required that they have significant identity in their amino acid sequences, but, rather, proteins having functional homology are so defined by having similar or identical activities, e.g., enzymatic activities.
- an enzyme from another organism than e.g. the host Coryneform bacteria will be considered to be a functional homo log if it shows at least significant similarity, i.e. about 50% sequence identity on the amino acid level, and catalyses the same reaction as its counterpart in the Coryneform bacterium.
- Functional homologues which provide the same enzymatic activity and share a higher degree of identity such as at least about 60%, at least about 70%, at least about 80% or at least about 90% sequence identity on the amino acid level are further preferred functional homolgues.
- fragments or mutated versions of the aforementioned enzymes from Corynefrom bacteria and of their functional homologues in other organisms as long as these fragments and mutated versions display the same type of functional activity.
- Typical functionally active fragments will display N-terminal and/or C -terminal deletions while mutated versions typically comprise deletions, insertions or point mutations.
- a sequence of E. coli will be considered to encode for a functional homo log of C. jeikeum formate-THF-synthetase if it displays the above- mentioned identity levels on the amino acid level to SEQ ID NO. 2 and displays the same enzymatic activity. Examples can be taken from Table 1. One can also use fragments or e.g. point mutants of these sequences as long as the resulting proteins still catalyse the same type of reaction as the full-length enzymes.
- One preferred aspect of the present invention relates to microorganisms in which a starting organism is genetically manipulated such that the amount and/or activity of formate-THF-synthetase is increased in the resulting microorganism compared to the starting organism.
- the present invention also relates to methods of producing methionine in microorganisms comprising the step of cultivating the aforementioned microorganism.
- microorganisms such as E.coli and C.jeikeium sequences for formate-THF- synthetase are known.
- increasing the amount and/or activity of formate-THF-synthetase will require raising the amount and/or activity of this enzyme above the level of the respective starting organism by e.g. over- expressing nucleic acid sequences encoding for this enzymatic activity.
- C. glutamicum which is the preferred host organism of the present invention
- a formate-THF-synthetase is not known.
- the following passages describe how a formate-THF-synthetase activity can be established in C. glutamicum.
- the person skilled in the art will nevertheless be aware how the amount and/or activity of a formate-THF-synthetase can be increased in other microorganisms such as C. jeikeium and E.coli.
- the present invention thus relates inter alia to a C. glutamicum microorganism in which the amount and/or activity of formate-THF-synthetase is increased and the use of such a microorganism for producing methionine.
- This can be achieved by e.g. increasing the copy number of nucleic acid sequences encoding for a formate-THF- synthetase, increasing transcription and/or translation of sequences encoding a formate-THF-synthetase or a combination thereof.
- a formate-THF-synthetase from C.jeikeium.
- the nucleic acid sequence of this formate-THF-synthetase is depicted in SEQ ID NO. 1, while the amino acid sequence is depicted in SEQ ID NO. 2.
- the gene bank accession number is YP 250663.1. This sequence is derived from the strain C.jeikeium NCTC Kl 1915 which is also designated as K411.
- a formate-THF-synthetase can also be obtained from the strain strain DSMZ 7171.
- the nucleic acid sequence is depicted by SED ID No: 51 and the amino acid sequence is depicted by SEQ ID No. 52.
- the copy number of nucleic acid sequences encoding formate-THF-synthetase can be increased in a microorganism and preferably in C. glutamicum by e.g. either expressing the sequence from autonomously replicating plasmids or by integrating additional copies of the respective nucleic acid sequences into the genome of the microorganism and preferably of C. glutamicum.
- Typical vectors for expressing polypeptides and enzymes such as formate-THF-synthetase in C. glutamicum include pCliK, pB and pEKO as described in Bott, M. and Eggeling, L., eds. Handbook o ⁇ Cory neb acterium glutamicum. CRC Press LLC, Boca Raton, FL; Deb, J.K. et al. (FEMS Microbiol. Lett. (1999), 175(1), 11-20), Kirchner O. et al. (J. Biotechnol. (2003), 104 (1-3), 287- 299), WO2006069711 and in WO2007012078.
- nucleic acid sequences encoding a polypeptide in a Coryneform bacterium In another approach for increasing the copy number of nucleic acid sequences encoding a polypeptide in a Coryneform bacterium, one can integrate additional copies of nucleic acid sequences encoding such polypeptides into the chromosome of C. glutamicum. Chromosomal integration can e.g. take place at the locus where the endogenous copy of the respective polypeptide is localized. Additionally and/or alternatively, chromosomal multiplication of polypeptide encoding nucleic acid sequences can take place at other loci in the genome of a Coryneform bacterium. In case of C. glutamicum, there are various methods known to the person skilled in the art for increasing the gene copy number by chromosomal integration.
- vectors for chromosomal integration of polypeptide-encoding nucleic acid sequences include or pCLIK int sacB as described in WO2005059093 and WO2007011845.
- Another preferred approach for increasing the amount and/or activity of formate- THF-synthetase in microorganisms and particularly in C. glutamicum is to increase transcription of the coding sequences by use of a strong promoter.
- strong promoter means that transcription from the newly introduced promoter is stronger than from the naturally occurring endogenous promoter.
- a promoter can be used which is known to provide strong expression of endogenous genes of C. glutamicum.
- Preferred promoters in this context are the promoters P SO D (SEQ ID NO. 3), P gr0 ES (SEQ ID No 4), P EFTu (SEQ ID No 5), phage SPOl promoter Pi 5 (SEQ ID No 42), and XP R (SEQ ID No 6), also sometimes referred to as lambdaPR.
- the XP R promoter can be stronger than the P SOD promoter.
- the P SOD promoter can be stronger than the P gr0 E S promoter, and the P gr0 E S promoter can be weaker than the P EFTU promoter or the P15 promoter.
- the P EFTU promoter can be stronger than the P SOD promoter.
- the strength of a promoter in any organism is not necessarily an inherent property of the promoter, since promoter strength can vary widely depending on the context in which the promoter is placed by the genetic engineering.
- formate -THF-synthetase in microorganisms and particularly in C. glutamicum will allow the microorganisms to grow on media comprising formate as the carbon source. Furthermore, the use of formate which also occurs as a metabolite during various biosynthetic pathways will also allow to increase the production of N 5 , N 10 -methylene-THF. An increased level of N 5 , N 10 -methylene-THF will lead to an increased production of methyl-THFand production of methionine.
- the present invention therefore also relates to a method which comprises culturing the above-described microorganisms and optionally isolating methionine.
- the amount of N 5 , N 10 -methylene- THF can be further increased by decreasing the amount and/or activity of formyl- THF-deformylase.
- a nucleic acid sequence for formyl-THF-deformylase is depicted in SEQ ID NO. 7, the amino acid sequence is depicted in SEQ ID NO. 8.
- Table 1 provides gene bank accession numbers for this enzymatic activity.
- the amount and/or activity of N 5 , N 10 -methenyl-THF- cyclosynthetase, N 5 , N 10 -methenyl-THF-reductase and/or N 5 , N 10 -methylene-THF- reductase are increased compared to the starting organism.
- the amount and/or activity of formate-THF-synthetase is increased, the amount and/or activity of formyl-THF-deformylase is decreased and the amount and/or activity of N 5 N 10 -methenyl-THF-cyclosynthetase, N 5 , N 10 -methenyl-THF- reductase and N 5 , N 10 -methenyl-reductase are increased compared to the starting organism.
- enzymatic activities are present in microorganisms and also in C.
- glutamicum (with the exception of formate-THF- synthetase), it can be preferred to use the endogenous nucleic acid sequences for increasing and/or decreasing the amount and/or activity of the respective enzymatic activities in the microorganisms in accordance with the present invention and preferably in C. glutamicum.
- a preferred embodiment relates to C. glutamicum microorganisms which express formate THF-synthetase.
- the present invention also relates preferably to the use of these C. glutamicum organisms in the production of methionine.
- These strains may show additionally the above mentioned genetic alterations discussed for formyl- THF-deformylase, N 5 , N 10 -methenyl-THF-cyclosynthetase, N 5 , N 10 -methenyl-THF- reductase and/or N 5 , N 10 -methylene-THF-reductase.
- a typical C. glutamicum strain that can be used as a starting organism will be a wild- type strain such as ATCC 13032. However, it can be preferred to use a starting organism which has already been genetically modified to ensure increased methionine production. Such an organism may display the characteristics of DSM 17323 and thus display an increased amount and/or activity and metH.
- a preferred starting strain may also have the characteristics of M2014 and display an increased amount and/or activity of asl/ br , hon/ br , metH, met A, metY, and hsk mutated .
- Other preferred starting organisms may have the characteristics of OM469 and display an increased amount and/or activity of asli br , metH, met A, , metY, hsk mutated and metF and display a reduced amount and/or activity of mcbR and metQ.
- Yet other preferred starting organisms may have the characteristics of GK1259 and display an increased amount and/or activity metH, met A, , metY, hsk mutated , tkt (and optionally g ⁇ pdh, zwfa and 6pg ⁇ ) and metF and display a reduced amount and/or activity of mcbR, metQ and sda or of M2616 and display an increased amount and/or activity oiasli hr , honf" ' , metH, met A, , metY, hsk mutated , tkt (and optionally g ⁇ pdh, zwfa and 6pg ⁇ ) and metF and display a reduced amount and/or activity of mcbR, metQ and serA .
- the inventors further found that production of methionine can be further stimulated if one cultivates the above-described microorganisms which display an increased amount and/or activity of formate-THF-synthetase, in a medium containing increased amounts of formate.
- This embodiment of the methods in accordance with the invention where formate is purposively added to the culture medium, is particularly preferably performed with strains of C. glutamicum that have been genetically modified to display the above activities of formate-THF-synthetase and the other genetic alterations. Again, it will be preferred to increase activity of formate-THF-synthetase by expressing the corresponding sequences of C. jeikeium in C. glutamicum or functional homo logs and fragments thereof. Microorganisms with increased amount and/or activity of the glycine cleavage system
- the present invention in one aspect relates to microorganisms and preferably
- the present invention also relates to methods which make use of these microorganisms for the production of methionine by cultivating said microorganisms and optionally isolating methionine.
- microorganisms such as E.coli or C. glutamicumcan produce glycine as by-product.
- the present invention makes use of this by-product by providing microorganisms that display an increased activity of the glycine cleavage system.
- the glycine cleavage system of microorganisms is typically comprised of 4 to 5 subunits.
- the first subunit is a PLP-dependent glycine decarboxylase (GcvP, also named simply P-protein).
- the second subunit is a lipoamide-containing amino methyl transferase (GcvH, also names H-protein).
- the third subunit is a N 5 , N 10 -methylene- THF synthesizing enzyme (GcvT). These three factors are sometimes also designated as gcvPTH.
- the fourth subunit is a NAD + -dependent, FAD-requiring lipoamide dehydrogenase (Ipd, also named simply L-protein).
- the corresponding genes are names gcvP, gcvT, gcvH and Ipd, respectively.
- GCS GCS-subunit
- the Ipd subunit is typically also shared by at least two other multi-subunit enzymes, namely pyruvate dehydrogenase and ⁇ - ketoglutarate dehydrogenase. If the enzymatic activity of the GCS-system is increased, glycine in excess of that required for cell mass will be preferably metabolised into NH 4 + , CO 2 and N 5 , N 10 - methylene-THF, which can then be used e.g. for increased methionine synthesis.
- some microorganisms such as e.g. C. glutamicum lack a native GCS- system. Nevertheless, such organisms will usually have an lpd gene that encodes the subunit for use in the aforementioned two enzyme systems.
- the native Lpd protein in C. glutamicum is able to function together with a non-native GCS such that only the gcvP, gcvT and gcvH genes need to be installed and expressed to obtain an active GCS function in C. glutamicum. It can however be preferred to also over-express a non-native lpd gene, since this gene may be more capable of specifically and efficiently interacting with the gcvP, gcvT and gcvH factors.
- the glycine cleavage system is comprised of five subunits.
- the P-subunit is e.g. divided into two polypeptides, sometimes named Pl and P2, which are encoded by two genes, sometimes called gcvPl and gcvP2.
- the present invention relates in a preferred embodiment to microorganisms that have been genetically modified to display an increased glycine cleavage system activity.
- Such an increased glycine cleavage system activity can be attained by increasing the amount and/or activity of the enzymatic activities encoded by gcvP, gcvH and gcvT. It will be addressed below how an increased glycine cleavage system activity can be established in C. glutamicum, as this represents a preferred embodiment of the present invention. Nevertheless, the person skilled in the art will be clearly aware how an increased glycine cleavage system activity may also be attained in other organisms such as E.coli or C.jeikeium.
- One aspect of the present invention relates to microorganisms, and particularly C. glutamicum, in which the amount and/or activity of the enzymatic activities encoded by gcvP, gcvH and gcvT (collectively called gcvPHT) is increased by genetic alteration compared to the starting organism.
- the nucleic acid sequence of gcvP is depicted in SEQ ID NO. 9, the amino acid sequence is depicted in SEQ ID NO. 10.
- Genbank accession number is C AB 6361.1.
- the nucleic acid sequence of gcvH of C. jeikeium is depicted in SEQ ID NO. 11.
- the amino acid sequence is depicted in SEQ ID NO. 12.
- the Genbank accession number is CAI36363.1.
- the nucleic acid sequence of gcvT of C. jeikeium is depicted in SEQ ID NO. 13.
- the amino acid sequence is depicted in SEQ ID NO. 14.
- the gene bank accession number is CAI36362.1.
- the activity of a glycine cleavage system in a microorganism and particularly in C. glutamicum can be increased by expressing, and preferably over-expressing, the aforementioned sequences, either alone or in combination, with the latter being a particularly preferred embodiment of the present invention.
- the present invention particularly relates to C. glutamicum microorganisms in which the enzymatic activities of gcvPHT are concomitantly increased. This may be attained by over-expressing the sequences of gcvP, gcvH and gcvT as depicted by SEQ ID NOS. 9-14, functional fragments thereof, or functional homo logs thereof.
- gcvP, gcvH and gcvT factors smoothly and efficiently interact with the lpd of C. glutamicum.
- the amount and/or activity of gcvP, gcvH and gcvT can be increased in microorganisms and preferably in C. glutamicum by the methods mentioned above in the context of formate-THF-synthetase.
- a functional unit which comprises the coding sequences of gcvP, gcvH and gcvT and increase the copy number of the nucleic acid sequence comprising this unit by using e.g. autonomously replicating plasmids or plasmids which integrate into the genome of the microorganism and preferably into the genome of C. glutamicum.
- a promoter in front of this operon which ensures strong transcription of the coding sequences for gcvP, gcvH and gcvT.
- a promoter may preferably be selected from the group of the PEFTU, PgroES, P SO D, PIS and ⁇ PR promoter.
- the lpd factor In principle, it is not necessary to increase the amount and/or activity of the lpd factor. This factor will typically be present in sufficiently abundant amounts by the host microorganism, which is genetically manipulated in order to increase the amount and/or activity of gcvP, gcvH and gcvT. Nonetheless, in some embodiments of the invention, it can be preferred to also increase the amount and/or activity of lpd. To this end, the endogenous sequences of lpd may be over-expressed by any of the above-described methods which are put forward in some more detail below.
- lpd of the starting organism Depending on the similarity of the lpd of the starting organism and the lpd of the from which gcvP, gcvH and gcvT factors are taken, it can be preferred to increase the amount and/or activity of lpd by increasing expression of endogenous or exogenous lpd. In this case, one will preferably select the lpd of that organism from which the other factors of the glycine cleavage system are taken.
- one embodiment of the present invention relates to microorganisms which are derived from the starting organism such that the resulting microorganism displays an increased amount and/or activity of the factors gcvP, gcvH and gcvT.
- the invention relates also to methods which use these microorganisms for production of methionine by cultivating the microorganisms and optionally isolating methionine.
- a preferred embodiment relates to C. glutamicum microorganisms which express the gcvP, gcvH and gcvT factors of C.jeikeium as depicted in SEQ ID NOS. 9-14, or functional homo logs and functional fragments thereof.
- the present invention also relates preferably to the use of these C. glutamicum organisms in the production of methionine.
- a typical C. glutamicum strain that can be used as a starting organism will be a wild- type strain such as ATCC 13032. However, it can be preferred to use a starting organism which has already been genetically modified to ensure increased methionine production. Such an organism may display the characteristics of DSM 17323 and thus display an increased amount and/or activity of asli br , hont * and metH.
- a preferred starting strain may also have the characteristics of M2014 and display an increased amount and/or activity of asli hr , honi hr , metH, met A, metY, and hsk mutated .
- Other preferred starting organisms may have the characteristics of OM469 and display an increased amount and/or activity o ⁇ as ⁇ , horn ⁇ 1 ⁇ , metH, met A, , metY, hsk mutated and metF and display a reduced amount and/or activity of mcbR and metQ.
- the starting microorganism which preferably is one of the aforementioned C. glutamicum strains displays further genetic modifications with respect to enzymes that are involved in the serine biosynthetic pathway.
- Seren biosynthetic pathway is art-recognized and describes a series of reactions which take place in a wild-type organism and lead to the biosynthesis of serine.
- the pathway may vary from organism to organism. The details of an organism-specific pathway can be taken from textbooks and the scientific literature which is listed e.g. on the website http://www.genome.jp.
- Serine is synthesized from the glycolytic intermediate 3 -phosphogly cerate which is first oxidized to phosphohydroxypyruvate by the action of 3-phosphoglycerate dehydrogenase (SerA).
- Serine is synthesized from the glycolytic intermediate 3 -phosphogly cerate which is first oxidized to phosphohydroxypyruvate by the action of 3-phosphoglycerate dehydrogenase (SerA).
- Serine is synthesized from the glycolytic intermediate 3 -phosphogly cerate which is first oxidized to phosphohydroxypyruvate by the action of 3-phosphoglycerate dehydrogenase (SerA).
- Serine is synthesized from the glycolytic intermediate 3 -phosphogly cerate which is first oxidized to phosphohydroxypyruvate by the action of 3-phosphoglycerate dehydrogenase (SerA).
- Serine is synthesized from the glycolytic intermediate 3 -phosphogly cerate which is first oxidized to phosphohydroxypyr
- L-serine can be converted to pyruvate by the serine dehydratase (sdaA) and to glycine and methylene tetrahydrofolate by serine hydroxymethyltransferase (SHMT or glyA).
- sdaA serine dehydratase
- SHMT serine hydroxymethyltransferase
- the starting organism in addition to the above-described genetic modifications which aim to introduce an improved glycine cleavage system and to ensure improved use of lipoic acid and/or lipoamide, the amount and/or activity of enzymes selected from the group consisting of D-3- phosphoglycerate dehydrogenase (SerA), phosphoserine phosphotase (SerB), phosphoserine aminotransferase (SerC) and serine hydroxy methyl transferase (SHMT) are increased. Alternatively or additionally, the amount and/or activity of serine deaminase (SdaA) may be reduced.
- SerA D-3- phosphoglycerate dehydrogenase
- SerB phosphoserine phosphotase
- SerC phosphoserine aminotransferase
- SHMT serine hydroxy methyl transferase
- a preferred starting strainfor may thus have the characteristics of OM264C and display an increased amount and/or activity of asl/ br , hon/ br , metH, met A, , metY, and hsk mutated and display a reduced amount and/or activity of serA.
- Another preferred starting strain for methionine production may have the characteristics of GK1259 and display an increased amount and/or activity of asli br , honi hr , metH, met A, , metY, hsk mutated , metF, tkt, tal, zwf and 6pgl and display a reduced amount and/or activity of mcbR, metQ and sd ⁇ .
- microorganisms can be particularly preferably be used for the production of methionine in methods in accordance with the invention.
- the present inventors furthermore found that N 5 , N 10 -methylene-THF and methionine production can be increased in microorganisms that display an increased activity of the glycine cleavage system as described above, if the microorganisms are (i) provided with external lipoic acid and/or lipoamide and/or (ii) are further genetically modified to produce more lipoic acid and/or lipoamide than the starting organism.
- the present invention thus in one aspect relates to methods which make use of the above-described microorganisms that display an increased amount and/or activity of the gcvP, gcvH and gcvT factors of the glycine cleavage system (and optionally of the lpd factor) and which are cultivated in a medium containing increased amounts of lipoic acid and/or lipoamide.
- lipoic acid and/or lipoamide may be added to the medium up to a final concentration of at least about 0,1 mg/1, at least about 1 mg/ml and preferably at least about 10 mg/L.
- lipoamide sometimes called lipoic acid amide
- can substitute for lipoic acid for the lipoylation of enzymes in some organisms (Reed, LJ. et al. (1958) J. Biol. Chem. 232, 143-158).
- feeding of lipoamide can substitute for feeding of lipoic acid in the invention disclosed herein.
- lipoamide which is commercially available from the same supplier as lipoic acid in its various forms (for example, Sigma-Aldrich catalog numbers T 5875, T 5625, T 1395, T 8260) can also be used to stimulate or increase glycine cleavage activity in organisms and methods of the invention disclosed herein. Both the oxidized and the reduced forms of these two compounds can be used, as well as various salt and esters of any of these forms.
- the source of the lipoyl group can vary.
- This embodiment of the methods in accordance with the invention, where lipoic acid and/or lipoamide is purposively added to the culture medium, is particularly preferably performed with strains of C. glutamicum that have been genetically modified to display the activities of the gcvP, gcvH and gcvT factors of a glycine cleavage system. Again, it will be preferred to increase activity of the glycine cleavage system by expressing the corresponding sequences of C. jeikeium in C. glutamicum or functional homologs and fragments thereof. In a further elaboration of this aspect of the invention, the amount and/or activity of the lpd factor may be increased by using either the endogenous C. glutamicum sequences or exogenous sequences (see Table 1).
- the final concentration of lipoic acid and/or lipoamide in the medium be at least about 0,1 mg/1, at least about 1 mg/1 and preferably at least about 10 mg/1.
- microorganisms in accordance with the present invention that display increased activity in the glycine cleavage system resulting from an increased amount and/or activity of the gcvP, gcvH and gcvT factors can be further genetically modified to display an increased amount of internally synthesized lipoic acid and/or lipoamide.
- the IpIA gene encodes forlipoyl synthetase (LpIA protein). This enzyme activates lipoic acid with ATP and subsequently attaches lipoyl-AMP to gcvH.
- the HpB dependent pathway comprises two enzymes(Morris et al. (1995) J. Bacteriol., 177, 1-10).
- LipA encodes for lipoic acid synthase (LipA protein).
- the HpB gene encodes for lipoyl transferase (LipB protein).
- LipA converts octanoyl-ACP to lipoyl-ACP.
- HpB attaches the lipoyl moiety to the lipoyl domain of gcvH and other lipoylated proteins.
- an increase in the amount and/or activity of IpIA allows for better incorporation of externally added lipoic acid, while an increase in the amount, type and/or activity of HpA and/or HpB increases the amount of internally synthesized lipoic acidthat becomes transferred to GcvH.
- a nucleic acid sequence o ⁇ lplA o ⁇ E. col is depicted in SEQ ID NO. 15.
- the amino acid sequence is depicted in SEQ ID NO. 16.
- the gene bank accession number is EGl 1796.
- the coding sequence for HpA of C. jeikeium is depicted in SEQ ID NO. 17.
- the amino acid sequence is depicted in SEQ ID NO. 18.
- the gene bank accession number is GeneID:3433570.
- the nucleic acid sequence for HpB of C. jeikeium is depicted in SEQ ID NO. 19.
- the amino acid sequence is depicted in SEQ ID NO. 20.
- the gene bank accession number is GeneID:3433571.
- a microorganism in accordance with the present invention which displays an increased amount and/or activity of the glycine cleavage system factors gcvP, gcvH and gcvT can thus be further optimized with respect to N 5 , N 10 -methylene-THF and methionine synthesis by increasing the amount and/or activity o ⁇ lplA.
- Such a microorganism will show a better incorporation of externally added lipoic acid and/or lipoamide and may thus particularly be suitable for those methods in accordance with the present invention in which the microorganisms are cultivated in the medium being supplemented with lipoic acid and/or lipoamide.
- the present invention relates to a microorganism which, in addition to the increase in the amount and/or activity of the glycine cleavage system factors gcvP, gcvH and gcvT displays an increased amount and/or activity for HpA, HpB or lip A and HpB.
- a microorganism that in addition to an increased amount and/or activity of gcvP, gcvH and gcvT displays an increased amount and/or activity of HpA and HpB is particularly preferred.
- microorganisms may show a better formation and accommodation of endogenously synthesized lipoic acid and/or lipoamide and will thus contribute to the production of N 5 , N 10 -methylene-THF and methionine.
- these microorganisms can also be used in the methods in accordance with the present invention which pertain to the cultivation of genetically modified organisms with increased glycine cleavage system activity in medium supplemented with lipoic acid and/or lipoamide.
- microorganisms which in addition to an increased amount and/or activity of gcvP, gcvH and gcvT show an increased amount and/or activity of IpI A, HpA and HpB.
- a particularly preferred embodiment of the present invention again relates to C. glutamicum microorganisms which by way of genetic modification of a starting C. glutamicum organism display an increased amount and/or activity of the glycine cleavage system factors gcvP, gcvH and gcvT and which display improved accommodation of the externally provided lipoic acid and/or lipoamide and/or improved formation and accommodation of internally produced lipoic acid and/or lipoamide by being genetically modified in order to display an increased amount and/or activity o ⁇ lplA, lip A and/or HpB.
- Preferred embodiments of the present invention thus relate to C.
- glutamicum microorganisms in which the amount and/or activity of gcvP, gcvH and gcvT and IpI A is increased compared to the starting organism.
- the C. glutamicum microorganism displays an increased amount and/or activity of gcvP, gcvH and gcvT and HpA or HpB.
- a C. glutamicum microorganism which displays an increased amount and/or activity of gcvP, gcvH and gcvT, HpA and HpB.
- a C. glutamicum microorganism which displays an increased amount and/or activity of gcvP, gcvH and gcvT, IpI, HpA and HpB is can be particularly preferred.
- the C. glutamicum starting organism which is used for introducing the above-mentioned genetic modification may be a wild-type strain such as ATCC 13032. However, it can be preferred to use a starting organism which has already been genetically modified to ensure increased methionine production. Such an organism may display the characteristics of DSM 17323 and thus display an increased amount and/or activity of asl/ br , hon/ br and metH. A preferred starting strain may also have the characteristics of M2014 and display an increased amount and/or activity of askf 1 " ' , metH, met A, metY, and hsk mutated .
- Other preferred starting organisms may have the characteristics of OM469 and display an increased amount and/or activity of asli br , honi hr , metH, met A, , metY, hsk mutated and metF and display a reduced amount and/or activity of mcbR and metQ.
- the starting microorganism which preferably is one of the aforementioned C. glutamicum strains displays further genetic modifications with respect to enzymes that are involved in the serine biosynthetic pathway as described above.
- a preferred starting strain may thus have the characteristics of OM264C and display an increased amount and/or activity of metH, met A, , metY, and hsk mutated and display a reduced amount and/or activity of serA.
- Another preferred starting strain may have the characteristics of GK1259 and display an increased amount and/or activity oiasli hr , honf" ' , metH, met A, , metY, hsk mutated , metF, tkt, tal, zwf and 6pgl and display a reduced amount and/or activity of mcbR, metQ and sd ⁇ .
- Microorganisms with increased amount and/or activity of formate-THF- synthetase and of the glycine cleavage system are produced by Microorganisms with increased amount and/or activity of formate-THF- synthetase and of the glycine cleavage system
- Another preferred embodiment of the present invention refers to microorganisms which combine the properties of the above-mentioned organisms, i.e. increasing the amount and/or activity of formate-THF-synthetase and an increased glycine cleavage system activity. It is understood that the above-described particularly preferred embodiments are also to be combined for this aspect of the present invention.
- a microorganism in accordance with the invention will be genetically modified such that it will display an increased amount and/or activity of formate-THF- synthetase, gcvP, gcvH and gcvT.
- the microorganism will be further genetically modified to display an increased amount and/or activity o ⁇ lplA.
- a preferred embodiment of the present invention also relates to a microorganism which displays an increased activity of formate-THF-synthetase, gcvP, gcvH and gcvT and HpA or HpB. Even more preferred are microorganisms which display an increased amount and/or activity of a formate-THF-synthetase, gcvP, gcvH and gcvT, HpA and HpB.
- Another preferred embodiment relates to a microorganism that displays an increased amount and/or activity of formate-THF-synthetase, gcvP, gcvH and gcvT, IpIA, HpA and HpB.
- the microorganisms may of course also display an increased amount and/or activity of Ip d.
- Such a C. glutamicum strain may be a wild-type strain such as ATCC 13032.
- a starting organism which has already been genetically modified to ensure increased methionine production.
- Such an organism may display the characteristics of DSM 17323 and thus display an increased amount and/or activity of asl/ br , hon/ br and metH.
- a preferred starting strain may also have the characteristics of M2014 and display an increased amount and/or activity metH, met A, metY, and hsk mutated .
- Other preferred starting organisms may have the characteristics of OM469 and display an increased amount and/or activity of asl/ br , hon/ br , metH, met A, , metY, hsk mutated and metF and display a reduced amount and/or activity of mcbR and metQ or of M2616 and display an increased amount and/or activity of asli br , honi hr , metH, met A, , metY, hsk mutated , tkt (and optionally g ⁇ pdh, zwfa and 6pgl) and metF and display a reduced amount and/or activity of mcbR, metQ and serA.
- the starting microorganism which preferably is one of the aforementioned C.
- glutamicum strains displays further genetic modifications with respect to enzymes that are involved in the serine biosynthetic pathway as described above and/or enzymes involved in the metabolisation of formyl such as formyl-THF-deformylase, N 5 , N 10 -methenyl-THF-cyclosynthetase, N 5 , N 10 -methenyl-THF-reductase and/or N 5 , N 10 -methylene-THF-reductase .
- a preferred starting strain may thus have the characteristics of OM264C and display an increased amount and/or activity o ⁇ as ⁇ , horn ⁇ 1 ' , metH, met A, , metY, and hsk mutated and display a reduced amount and/or activity of serA.
- Another preferred starting strain may have the characteristics of GK1259 and display an increased amount and/or activity oiasli hr , hom fh ' , metH, met A, , metY, hsk mutated , metF, tkt, tal, zw/ " and 6pgl and display a reduced amount and/or activity of mcbR, metQ and sda.
- microorganisms and particularly C. glutamicum microorganisms in accordance with the invention can be used in the production of methionine by culturing them and optionally isolating methionine.
- the modified organisms may be cultivated in a medium supplemented with increased amounts of formate and lipoic acid and/or lipoamide.
- rrnAC2696 MJ1018, TTE2613, amb3193, AF0813, MK0297, DET0599, CYA_1354, Synpcc7942_1501, syc2486_c, Saro_2680, ELI_01970, MM1753, cbdb_A580, BR1685, MTH970, Mbar_A1431, SPO3355, BruAbl_1670, BAB1 1697, BMEI0349, SYNW0533, Syncc9605_2150, Ava_3759, MA0592, alrl890, Mhun_3063, Syncc9902_0527, RPB_1315, glr2139, RPD 3905, Nwi_2968, RPA4308, SYN_00123, ABC1843, Nham_1119, STH9, W17401, slll908, CTC00694, BH1602, GK2247, RPC_410
- accession numbers are the official accession numbers of Genbank or are synonyms for accession numbers which have cross-references at Genbank. These numbers can be searched and found at http://www.ncbi.nlm.nih.gov/.
- the amount of the enzyme is increased by expression of an exogenous version of the respective protein.
- expression of the endogenous protein is increased by influencing the activity of e.g. the promoter and/or enhancers element and/or other regulatory activities that regulate the activities of the respective proteins either on a transcriptional, translational or post- translational level.
- the increase of the activity and the amount of a protein may be achieved via different routes, e.g. by switching off inhibitory regulatory mechanisms at the transcriptional, translational, and protein level or by increase of gene expression of a nucleic acid coding for these proteins in comparison with the starting organism, e.g. by inducing endogenous transketolase by a strong promoter and/ or by introducing nucleic acids encoding for transketolase.
- the increase of the amount and/or activity of the enzymes of Table 1 is achieved by introducing nucleic acids encoding the enzymes of Table 1 into microorganism such as C. glutamicum and E. coli.
- any protein of different organisms with an enzymatic activity of the proteins listed in Table 1 can be used.
- genomic nucleic acid sequences of such enzymes from eukaryotic sources containing introns already processed nucleic acid sequences like the corresponding cDNAs are to be used in the case as the host organism is not capable or cannot be made capable of splicing the corresponding mRNAs.
- All nucleic acids mentioned in the description can be, e.g., an RNA, DNA or cDNA sequence.
- increasing or introducing the amount of a protein typically comprises the following steps:
- a promoter sequence functional in an organism of the invention operatively linked thereto a DNA sequence coding for a protein of e.g. Table 1, functional homologues, functional fragments or functional mutated versions thereof optionally, a termination sequence functional in the organisms of the invention
- step b) transfer of the vector from step a) to an organisms of the invention such as C. glutamicum and, optionally, integration into the respective genomes.
- functional fragments relate to fragments of nucleic acid sequences coding for enzymes of e.g. Table 1, the expression of which still leads to proteins having the enzymatic activity substantially similar to that of the respective full length protein.
- the above-mentioned method can be used for increasing the expression of DNA sequences coding for enzymes of e.g. Table 1 or functional fragments thereof.
- the use of such vectors comprising regulatory sequences, like promoter and termination sequences are, is known to the person skilled in the art.
- the person skilled in the art knows how a vector from step a) can be transferred to organisms such as C. glutamicum or E. coli and which properties a vector must have to be able to be integrated into their genomes.
- the enzyme content in an organism such as C. glutamicum is increased by transferring a nucleic acid coding for an enzyme from another organism, like e.g. E. coli, it is advisable to transfer the amino acid sequence encoded by the nucleic acid sequence e.g. from E. coli by back-translation of the polypeptide sequence according to the genetic code into a nucleic acid sequence comprising mainly those codons, which are used more often due to the organism- specific codon usage.
- the codon usage can be determined by means of computer evaluations of other known genes of the relevant organisms.
- an increase of the gene expression of a nucleic acid encoding an enzyme of Table 1 is also understood to be the manipulation of the expression of the endogenous respective endogenous enzymes of an organism, in particular of C. glutamicum. This can be achieved, e.g., by altering the promoter DNA sequence for genes encoding these enzymes. Such an alteration, which causes an altered, preferably increased, expression rate of these enzymes can be achieved by replacement with strong promoters and by deletion and/or insertion of DNA sequences.
- An alteration of the promoter sequence of endogenous genes usually causes an alteration of the expressed amount of the gene and therefore also an alteration of the activity detectable in the cell or in the organism.
- an altered and increased expression, respectively, of an endogenous gene can be achieved by a regulatory protein, which does not occur or has been deleted in the transformed organism, and which interacts with the promoter of these genes.
- a regulatory protein which does not occur or has been deleted in the transformed organism, and which interacts with the promoter of these genes.
- a regulator can be a chimeric protein consisting of a DNA binding domain and a transcription activator domain, as e.g. described in WO 96/06166.
- a further possibility for increasing the activity and the content of endogenous genes is to up-regulate transcription factors involved in the transcription of the endogenous genes, e.g. by means of overexpression.
- the measures for overexpression of transcription factors are known to the person skilled in the art.
- the expression of endogenous enzymes such as those of Table 1 can e.g. be regulated via the expression of aptamers specifically binding to the promoter sequences of the genes. Depending on the aptamer binding to stimulating or repressing promoter regions, the amount of the enzymes of Table 1 can e.g. be increased.
- an alteration of the activity of endogenous genes can be achieved by targeted mutagenesis of the endogenous gene copies.
- An alteration of the endogenous genes coding for the enzymes of e.g. Table 1 can also be achieved by influencing the post-translational modifications of the enzymes. This can happen e.g. by regulating the activity of enzymes like kinases or phosphatases involved in the post-translational modification of the enzymes by means of corresponding measures like overexpression or gene silencing.
- an enzyme may be improved in efficiency, or its allosteric control region destroyed such that feedback inhibition of production of the compound is prevented.
- a degradative enzyme may be deleted or modified by substitution, deletion, or addition such that its degradative activity is lessened for the desired enzyme of Table 1 without impairing the viability of the cell. In each case, the overall yield, rate of production or amount of methionine be increased.
- C. glutamicum one may, for example, downregulate the activity of metQ.
- the expression of endogenous enzymes such as those of Table 1 can e.g. be regulated via the expression of aptamers specifically binding to the promoter sequences of the genes. Depending on the aptamer binding to stimulating or repressing promoter regions, the amount and thus, in this case, the activity of the enzymes of Table 1 can e.g. be reduced.
- Aptamers can also be designed in a way as to specifically bind to the enzymes themselves and to reduce the activity of the enzymes by e.g. binding to the catalytic center of the respective enzymes.
- the expression of aptamers is usually achieved by vector-based overexpression (see above) and is, as well as the design and the selection of aptamers, well known to the person skilled in the art (Famulok et al., (1999) Curr Top Microbiol Immunol, 243,123-36).
- a decrease of the amount and the activity of the endogenous enzymes of Table 1 can be achieved by means of various experimental measures, which are well known to the person skilled in the art. These measures are usually summarized under the term "gene silencing".
- an endogenous gene can be silenced by transferring an above-mentioned vector, which has a DNA sequence coding for the enzyme or parts thereof in antisense order, to organisms such as C. glutamicum.
- an RNA which can hybridize with the mRNA transcribed by the endogenous gene and therefore prevents its translation.
- the antisense strategy can be coupled with a ribozyme method.
- Ribozymes are catalytically active RNA sequences, which, if coupled to the antisense sequences, cleave the target sequences catalytically (Tanner et al, (1999) FEMS Microbiol Rev. 23 (3), 251 -IS). This can enhance the efficiency of an antisense strategy.
- a vector is prepared which contains at least a portion of gene coding for an enzyme of Table 1 into which a deletion, addition or substitution has been introduced to thereby alter, e.g., functionally disrupt, the endogenous gene.
- the vector is designed such that, upon homologous recombination, the endogenous gene is functionally disrupted (i. e., no longer encodes a functional protein).
- the vector can be designed such that, upon homologous recombination, the endogenous gene is mutated or otherwise altered but still encodes functional protein, e.g., the upstream regulatory region can be altered to thereby alter the expression of the endogenous enzymes of Table 1.
- This approach can have the advantage that expression of an enzyme is not completely abolished, but reduced to the required minimum level.
- the skilled person knows which vectors can be used to replace or delete endogenous sequences. A specific description for disrupting chromosomal sequences in C. glutamicum is provided below.
- Factors inhibiting the target protein itself can also be introduced into a cell.
- the protein-binding factors may e.g. be the above-mentioned aptamers (Famulok et al., (1999) Curr Top Microbiol Immunol. 243, 123-36).
- enzyme-specific antibodies may be considered.
- the production of recombinant enzyme-specific antibodies such as single chain antibodies is known in the art.
- the expression of antibodies is also known from the literature (Fiedler et al., (1997) Immunotechnology 3, 205-216; Maynard and Georgiou (2000) Annu. Rev. Biomed. Eng. 2, 339-76).
- nucleic acid constructs used for e.g. antisense methods must have and which complementarity, homology or identity, the respective nucleic acid sequences must have.
- complementarity, homology, and identity are known to the person skilled in the art.
- complementarity describes the capability of a nucleic acid molecule to hybridize with another nucleic acid molecule due to hydrogen bonds between two complementary bases.
- the person skilled in the art knows that two nucleic acid molecules do not have to display a complementarity of 100% in order to be able to hybridize with each other.
- a nucleic acid sequence, which is to hybridize with another nucleic acid sequence is preferably at least 30%, at least 40%, at least 50%, at least 60%, preferably at least 70%, particularly preferred at least 80%, also particularly preferred at least 90%, in particular preferred at least 95% and most preferably at least 98 or 100%, respectively, complementary with said other nucleic acid sequence.
- hybridization of an antisense sequence with an endogenous mRNA sequence typically occurs in vivo under cellular conditions or in vitro. According to the present invention, hybridization is carried out in vivo or in vitro under conditions that are stringent enough to ensure a specific hybridization.
- stringent conditions therefore refers to conditions, under which a nucleic acid sequence preferentially binds to a target sequence, but not, or at least to a significantly reduced extent, to other sequences.
- Stringent conditions are dependent on the circumstances. Longer sequences specifically hybridize at higher temperatures. In general, stringent conditions are chosen in such a way that the hybridization temperature lies about 5°C below the melting point (Tm) of the specific sequence with a defined ionic strength and a defined pH value. Tm is the temperature (with a defined pH value, a defined ionic strength and a defined nucleic acid concentration), at which 50% of the molecules, which are complementary to a target sequence, hybridize with said target sequence.
- stringent conditions comprise salt concentrations between 0.01 and 1.0 M sodium ions (or ions of another salt) and a pH value between 7.0 and 8.3. The temperature is at least 30 0 C for short molecules (e.g. for such molecules comprising between 10 and 50 nucleic acids).
- stringent conditions can comprise the addition of destabilizing agents like e.g. form amide.
- Typical hybridization and washing buffers are of the following composition.
- Hybridization solution Pre-hybridization solution
- Pre-hybridization at least 2 h at 50-55 0 C
- nucleic acids For antisense purposes complementarity over sequence lengths of 100 nucleic acids, 80 nucleic acids, 60 nucleic acids, 40 nucleic acids and 20 nucleic acids may suffice. Longer nucleic acid lengths will certainly also suffice. A combined application of the above-mentioned methods is also conceivable.
- vectors can, in general, be constructed, which, after the transfer to the organism's cells, allow the overexpression of the coding sequence or cause the suppression or competition and blockage of endogenous nucleic acid sequences and the proteins expressed there from, respectively.
- the activity of a particular enzyme may also be reduced by over-expressing a nonfunctional mutant thereof in the organism.
- a non- functional mutant which is not able to catalyze the reaction in question, but that is able to bind e.g. the substrate or co-factor, can, by way of over-expression out-compete the endogenous enzyme and therefore inhibit the reaction.
- Further methods in order to reduce the amount and/or activity of an enzyme in a host cell are well known to the person skilled in the art.
- non-functional enzymes have essentially the same nucleic acid sequences and amino acid sequences, respectively, as functional enzymes and functionally fragments thereof, but have, at some positions, point mutations, insertions or deletions of nucleic acids or amino acids, which have the effect that the non- functional enzyme are not, or only to a very limited extent, capable of catalyzing the respective reaction.
- These non-functional enzymes may not be intermixed with enzymes that still are capable of catalyzing the respective reaction, but which are not feedback regulated anymore.
- the term "non-functional enzyme” does not comprise such proteins having no substantial sequence homology to the respective functional enzymes at the amino acid level and nucleic acid level, respectively.
- Non-functional enzymes are, within the scope of the present invention, also referred to as inactivated or inactive enzymes.
- non-functional enzymes of e.g.Table 1 according to the present invention bearing the above-mentioned point mutations, insertions, and/or deletions are characterized by an substantial sequence homology to the wild type enzymes of e.g. Table 1 according to the present invention or functionally equivalent parts thereof.
- the above describded identity grades are to applied.
- vectors preferably expression vectors, containing a modified nucleic acid sequences as mentioned above.
- vector refers to a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked.
- vector refers to a circular double stranded DNA loop into which additional DNA segments can be ligated.
- viral vector Another type of vector, wherein additional DNA segments can be ligated into the viral genome.
- Certain vectors are capable of autonomous replication in a host cell into which they are introduced (e.g., bacterial vectors having a bacterial origin of replication and episomal mammalian vectors). Other vectors (e. g., non-episomal mammalian vectors) are integrated into the genome of a host cell upon introduction into the host cell, and thereby are replicated along with the host genome. Moreover, certain vectors are capable of directing the expression of genes to which they are operative Iy linked.
- expression vectors Such vectors are referred to herein as "expression vectors”.
- expression vectors of utility in recombinant DNA techniques are often in the form of plasmids.
- plasmid and “vector” can be used interchangeably as the plasmid is the most commonly used form of vector.
- the invention is intended to include such other forms of expression vectors, such as viral vectors (e. g., replication defective retroviruses, adenoviruses and adeno-associated viruses), which serve equivalent functions.
- the recombinant expression vectors of the invention may comprise a modified nucleic acid as mentioned above in a form suitable for expression of the respective nucleic acid in a host cell, which means that the recombinant expression vectors include one or more regulatory sequences, selected on the basis of the host cells to be used for expression, which is operatively linked to the nucleic acid sequence to be expressed.
- operably linked is intended to mean that the nucleic acid sequence of interest is linked to the regulatory sequence (s) in a manner which allows for expression of the nucleic acid sequence (e.g., in an in vitro transcription/translation system or in a host cell when the vector is introduced into the host cell).
- regulatory sequence is intended to include promoters, repressor binding sites, activator binding sites, enhancers and other expression control elements (e.g., terminators, polyadenylation signals, or other elements of mRNA secondary structure). Such regulatory sequences are described, for example, in Goeddel; Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990).
- regulatory sequences include those which direct constitutive expression of a nucleic acid sequence in many types of host cell and those which direct expression of the nucleic acid sequence only in certain host cells.
- Preferred regulatory sequences are, for example, promoters such as cos-, tac-, trp-, tet-, trp-, tet-, lpp-, lac-, lpp-lac-, laclq-, T7-, T5-, T3-, gal-, trc-, ara-, SP6-, arny, SP02,phage lambdaP R , phage lambdaP L , phage SPOl Pi 5 , phage SPOl P 26 , pSOD, EFTu, EFTs, GroEL, MetZ (last 5 from C.
- promoters from yeasts and fungi such as ADCl, MFa, AC, P-60, CYCl, GAPDH, TEF, rp28, ADH, ENO2, promoters from plants such as CaMV/35S, SSU, OCS, Iib4, usp, STLSl, B33, nos or ubiquitin-or phaseolin-promoters. It is also possible to use artificial promoters. It will be appreciated by one of ordinary skill in the art that the design of the expression vector can depend on such factors as the choice of the host cell to be transformed, the level of expression of protein desired, etc.
- the expression vectors of the invention can be introduced into host cells to thereby produce proteins or peptides, including fusion proteins or peptides, encoded by the above-mentioned modified nucleic acid sequences.
- Fusion vectors add a number of amino acids to a protein encoded therein, usually to the amino terminus of the recombinant protein but also to the C-terminus or fused within suitable regions in the proteins. Such fusion vectors typically serve four purposes: 1) to increase expression of recombinant protein; 2) to increase the solubility of the recombinant protein; and 3) to aid in the purification of the recombinant protein by acting as a ligand in affinity purification 4) to provide a "tag" for later detection of the protein.
- a proteolytic cleavage site is introduced at the junction of the fusion moiety and the recombinant protein to enable separation of the recombinant protein from the fusion moiety subsequent to purification of the fusion protein.
- enzymes, and their cognate recognition sequences include Factor Xa, thrombin and enterokinase.
- Typical fusion expression vectors include pGEX (Pharmacia Biotech Inc; Smith, D. B. and Johnson, K. S. (1988) Gene 67: 31-40), pMAL (New England Biolabs, Beverly, MA) and pRIT5 (Pharmacia, Piscataway, NJ) which fuse glutathione S- transferase (GST), maltose E binding protein, or protein A, respectively.
- Suitable inducible non- fusion E. coli expression vectors include pTrc (Amann et al, (1988) Gene 69: 301-315), pLG338, pACYC184, pBR322,pUC18, pUC19, pKC30, pRep4,pHSl, pHS2, P PLc236, pMBL24, pLG200, pUR290,pIN- IIIl 13-Bl, egtll, pBdCl, and pET Hd (Studier etal, Gene Expression Technology : Methods in Enzymology 185, Academic Press, San Diego, California (1990) 60-89; and Pouwels et al, eds.
- Target gene expression from the pTrc vector relies on host RNA polymerase transcription from a hybrid trp-lac fusion promoter.
- Target gene expression from the pET Hd vector relies on transcription from a T7 gnlO-lac fusion promoter mediated by a coexpressed viral RNA polymerase (T7gnl). This viral polymerase is supplied by host strains BL21 (DE3) or HMS 174 (DE3) from a resident X prophage harboring a T7gnl gene under the transcriptional control of the lacUV 5 promoter. For transformation of other varieties of bacteria, appropriate vectors may be selected.
- the plasmids pIJlOl, pIJ364, pIJ702 and pIJ361 are known to be useful in transforming Streptomyces, while plasmidspUBl 10, pC194 or pBD214 are suited for transformation of Bacillus species.
- plasmidspUBl 10, pC194 or pBD214 are suited for transformation of Bacillus species.
- plasmids of use in the transfer of genetic information into Corynebacterium include pHM1519, pBLl, pSA77 or pAJ667 (Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York IBSN 0 444 904018).
- C. glutamicum and E coli shuttle vectors are e.g. pClik5aMCS (WO2005059093; or can be found in Eikmanns et al ⁇ Gene. (1991) 102, 93-8).
- E. coli - C. glutamicum shuttle vectors (table 23.1), a list of E. coli - C. glutamicum shuttle expression vectors (table 23.2), a list of vectors which can be used for the integration of DNA into the C. glutamicum chromosome (table 23.3), a list of expression vectors for integration into the C. glutamicum chromosome (table 23.4.) as well as a list of vectors for site-specific integration into the C. glutamicum chromosome (table 23.6).
- the protein expression vector is a yeast expression vector.
- yeast expression vectors for expression in yeast S. cerevisiae include pYepSecl (Baldari, et al, (1987) Embo J. 6: 229-234),, 2i, pAG-1, Yep6, Yepl3, P EMBLYe23, pMFa (Kurjan and Herskowitz, (1982) Cell 30: 933-943), pJRY88 (Schultz et al., (1987) Gene 54: 113-123), and pYES2 (Invitrogen Corporation, San Diego, CA).
- Vectors and methods for the construction of vectors appropriate for use in other fungi, such as the filamentous fungi include those detailed in: van den Hondel, C. A. M. J. J. & Punt,P. J. (1991) in: Applied Molecular Genetics of Fungi, J. F. Peberdy, et al., eds., p. 1-28, Cambridge University Press: Cambridge, and Pouwels et al., eds. (1985) Cloning Vectors. Elsevier: New York (IBSN 0 444 904018).
- an operative link is understood to be the sequential arrangement of promoter, coding sequence, terminator and, optionally, further regulatory elements in such a way that each of the regulatory elements can fulfill its function, according to its determination, when expressing the coding sequence.
- Vector DNA can be introduced into prokaryotic via conventional transformation or trans fection techniques.
- transformation and “transfection”, “conjugation” and “transduction” are intended to refer to a variety of art-recognized techniques for introducing foreign nucleic acid (e. g., linear DNA or RNA (e.
- a linearized vector or a gene construct alone without a vector or nucleic acid in the form of a vector (e.g., a plasmid, phage, phasmid, phagemid, transposon or other DNA into a host cell, including calcium phosphate or calcium chloride co- precipitation, DEAE-dextran-mediated transfection, lipofection, natural competence, chemical-mediated transfer, or electroporation.
- a vector e.g., a plasmid, phage, phasmid, phagemid, transposon or other DNA into a host cell, including calcium phosphate or calcium chloride co- precipitation, DEAE-dextran-mediated transfection, lipofection, natural competence, chemical-mediated transfer, or electroporation.
- Suitable methods for transforming or transfecting host cells can be found in Sambrook, et al. (Molecular Cloning : A Laboratory Manual. 3rd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring
- a gene that encodes a selectable marker is generally introduced into the host cells along with the gene of interest.
- selectable markers include those which confer resistance to drugs, such as, but not limited to, G418, hygromycin , kanamycine, tetracycline, neomycineampicillin (and other pencillins), cephalosporins, fluoroquinones, naladixic a id, chloramphenicol, spectinomyin, ertythromycin, streptomycin and methotrexate.
- Other selectable markers include wild type genes that can complement mutated versions of the equivalent gene in a host or starting strain.
- an essential gene for growth on a minimal medium such as serA
- a minimal medium such as serA
- a vector containing a wild type or other functional copy of a serA gene can be used toselect for transformants or integrants.
- Nucleic acid encoding a selectable marker can be introduced into a host cell on the same vector as that encoding the above-mentioned modified nucleic acid sequences or can be introduced on a separate vector. Cells stably transfected with the introduced nucleic acid can be identified by drug selection (e. g., cells that have incorporated the selectable marker gene will survive, while the other cells die).
- plasmids without an origin of replication and two different marker genes e.g. pClik int sacB
- pClik int sacB When plasmids without an origin of replication and two different marker genes are used (e.g. pClik int sacB), it is also possible to generate marker-free strains which have part of the insert inserted into the genome. This is achieved by two consecutive events of homologous recombination (see also Becker et al., Applied and Environmental Microbilogy, 71 (12), p. 8587-8596).
- the sequence of plasmid pClik int sacB can be found in WO2005059093; SEQ ID 24; the plasmid is called pCIS in this document.
- recombinant microorganisms can be produced which contain selected systems which allow for regulated expression of the introduced gene. For example, inclusion of one of the above-mentioned nucleic acid sequences on a vector placing it under control of the lac operon permits expression of the gene only in the presence of IPTG.
- Such regulatory systems are well known in the art.
- Another aspect of the invention pertains to organisms or host cells into which a recombinant expression vector of the invention has been introduced.
- host cell and “recombinant host cell” are used interchangeably herein. It is understood that such terms refer not only to the particular subject cell but also to the progeny or potential progeny of such a cell. Because certain modifications may occur in succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent cell, but are still included within the scope of the term as used herein.
- E. coli strains are routinely grown in MB and LB broth, respectively (Follettie et al. (1993) J. Bacteriol. 175, 4096-4103).
- Minimal Several minimal media for bacteria, including E. coli and C glutamicum are well knoiwn in the art .
- Minimal media for E. coli isinclude,but are not limited to, E medium, M9medium and modified MCGC (Yoshihama et al. (1985) J. Bacteriol. 162,591-507), respectively.
- Glucose may be added at a final concentration of between about 0.2% and 1%.
- Antibiotics may be added in the following amounts (micrograms per millilitre): ampicillin, 5 to 1000; kanamycin, 25; nalidixic acid, 25; chloramphenicol, 5 to 120, spectinomycin 50 to 100, tetracyline 5 to 120.
- Amino acids, vitamins, and other supplements may be added, for example, in the following amounts: methionine, 9.3 mM; arginine, 9.3 mM; histidine, 9.3 mM; thiamine, 0.05 mM.
- E. coli cells are routinely grown at 18 to 37 44° C, respectivelydepending on the particular experiment or proceddure being performed.
- Corynebacteria are typically cultured in synthetic or natural growth media.
- a number of different growth media for Corynebacteria are both well-known and readily available (Lieb et al. (1989) Appl. Microbiol. Biotechnol., 32: 205-210; von der Osten et al. (1998) Biotechnology Letters, 11 : 11-16; Patent DE 4,120,867; Liebl(1992) "The Genus Corynebacterium, in: The Procaryotes, Volume II, Balows, A. et al., eds. Springer- Verlag). Instructions can also be found in the Handbook of Corynebacterium (edited by Eggeling and Bott, ISBN 0-8493-1821-1, 2005).
- These media consist of one or more carbon sources, nitrogen sources, inorganic salts, vitamins and trace elements.
- Preferred carbon sources are sugars, such as mono-, di-, or polysaccharides. For example, glucose, fructose, mannose, galactose, ribose, sorbose, ribose, lactose, maltose, sucrose, glycerol, raffmose, starch or cellulose serve as very good carbon sources.
- Nitrogen sources are usually organic or inorganic nitrogen compounds, or materials which contain these compounds. Exemplary nitrogen sources include ammonia gas or ammonia salts, such as NH 4 C 1 or (NH 4 ) 2 S0 4 , NH 4 OH, nitrates, urea, amino acids or complex nitrogen sources like corn steep liquor, soy bean flour, soy bean protein, yeast extract, meat extract and others.
- the overproduction of methionine is possible using different sulfur sources.
- Sulfates, thiosulfates, sulfites and also more reduced sulfur sources like H 2 S and sulfides and derivatives can be used.
- organic sulfur sources like methyl mercaptan, thioglycolates, thiocyanates, and thiourea, sulfur containing amino acids like cysteine and other sulfur containing compounds can be used to achieve efficient methionine production.
- Formate may also be possible as a supplement as are other Cl sources such as methanol or formaldehyde.
- Inorganic salt compounds which may be included in the media include the chloride-, phosphorous-or sulfate-salts of calcium, magnesium, sodium, cobalt, molybdenum, potassium, manganese, zinc, copper and iron.
- Chelating compounds can be added to the medium to keep the metal ions in solution.
- Particularly useful chelating compounds include dihydroxyphenols, like catechol or protocatechuate, or organic acids, such as citric acid. It is typical for the media to also contain other growth factors, such as vitamins or growth promoters, examples of which include cyanocobalamin (or other form of vitamin B 12), biotin, riboflavin, thiamine, folic acid, nicotinic acid, pantothenate and pyridoxine.
- the exact composition of the media compounds depends strongly on the immediate experiment and is individually decided for each specific case. Information about media optimization is available in the textbook "Applied Microbiol. Physiology, A Practical Approach (Eds. P. M. Rhodes, P.F. Stanbury, IRL Press (1997) pp. 53-73, ISBN 0 19 963577 3). It is also possible to select growth media from commercial suppliers, like standard 1 (Merck) or BHI (grain heart infusion, DIFCO) or others.
- All medium components should be sterilized, either by heat (20 minutes at 1.5 bar andl21 C) or by sterile filtration.
- the components can either be sterilized together or, if necessary, separately.
- All media components may be present at the beginning of growth, or they can optionally be added continuously or batch wise. Culture conditions are defined separately for each experiment.
- the temperature should be is usually in a range between 15°C and 45°C, but the range may be higher, up to 105 0 C for thermophilic oraganisms.
- the temperature can be kept constant or can be altered during the experiment.
- the pH of the medium may be in the range of 5 to 8.5, preferably around 7.0, and can be maintained by the addition of buffers to the media.
- An exemplary buffer for this purpose is a potassium phosphate buffer. Synthetic buffers such as MOPS, HEPES, ACES and others can alternatively or simultaneously be used. It is also possible to maintain a constant culture pH through the addition of an acid or base, such as acetic acid, sulfuric acid, phosphoric acid, NaOH, KOH or NH 4 OH during growth.
- the pH can also be controlled using gaseous ammonia.
- the incubation time is usually in a range from several hours to several days. This time is selected in order to permit the maximal amount of product to accumulate in the broth.
- the disclosed growth experiments can be carried out in a variety of vessels, such as microtiter plates, glass tubes, glass flasks or glass or metal fermentors of different sizes.
- the microorganisms should be cultured in microtiter plates, glass tubes or shake flasks, either with or without baffles.
- 100 ml or 250 shake flasks are used, filled with about 10% (by volume) of the required growth medium.
- the flasks should be shaken on a rotary shaker (amplitudeabout 25 mm) using a speed-range of about 100- 300 'rpm. Evaporation losses can be diminished by the maintenance of a humid atmosphere; alternatively, a mathematical correction for evaporation losses should be performed.
- the medium is inoculated to an OD600 of 0.5-1.5 using cells grown on agar plates, such as CM plates (lOg/1 glucose, 2,5g/l NaCl, 2g/l urea, lOg/1 polypeptone, 5g/l yeast extract, 5g/l meat extract, 22g/l (NFLi) 2 SO 4 , 2g/l urea, lOg/1 polypeptone, 5g/l yeast extract, 5g/l meat extract, 22g/l agar, pH about 6.8 to 7.2 with 2M NaOH) that had been incubated at 30° C. Inoculation of the media is accomplished by either introduction of a saline suspension of C. glutamicum cells from CM plates or addition of a liquid preculture of this bacterium.
- Strains can be taken e.g. for example, but not limited to, from the following list: Corynebacterium glutamicum ATCC 13032,
- Brevibacterium divaricatum ATCC 14020 or strains which have been derived therefrom such as Corynebacterium glutamicum KFCC 10065, DSM 17322 or
- the following gradient is applied: Start 0% B; 39 min 39 % B; 70 min 64 % B; 100 % B for 3.5 min; 2 min 0 % B for equilibration.
- Derivatization at room temperature is automated as described below. Initially 0.5 ⁇ l of 0.5% 2-MCE in bicine (0.5M, pH 8.5) are mixed with 0.5 ⁇ l cell extract.
- Detection is performed by a fluorescence detector (340 nm excitation, emission 450 nm, Agilent, Waldbronn, Germany).
- ⁇ -amino butyric acid (ABA) is used as internal standard
- “Campbell in,” as used herein, refers to a transformant of an original host cell in which an entire circular double stranded DNA molecule (for example a plasmid being based on pCLIK int sacB has integrated into a chromosome by a single homologous recombination event (a cross-in event), and that effectively results in the insertion of a linearized version of said circular DNA molecule into a first DNA sequence of the chromosome that is homologous to a first DNA sequence of the said circular DNA molecule.
- “Campbelled in” refers to the linearized DNA sequence that has been integrated into the chromosome of a "Campbell in” transformant.
- a "Campbell in” contains a duplication of the first homologous DNA sequence, each copy of which includes and surrounds a copy of the homologous recombination crossover point.
- the name comes from Professor Alan Campbell, who first proposed this kind of recombination.
- “Campbell out,” as used herein, refers to a cell descending from a "Campbell in” transformant, in which a second homologous recombination event (a cross out event) has occurred between a second DNA sequence that is contained on the linearized inserted DNA of the "Campbelled in” DNA, and a second DNA sequence of chromosomal origin, which is homologous to the second DNA sequence of said linearized insert, the second recombination event resulting in the deletion (jettisoning) of a portion of the integrated DNA sequence, but, importantly, also resulting in a portion (this can be as little as a single base) of the integrated Campbelled in DNA remaining in the chromosome, such that compared to the original host cell, the "Campbell out” cell contains one or more intentional changes in the chromosome (for example, a single base substitution, multiple base substitutions, insertion of a heterologous gene or DNA sequence, insertion of an additional copy or copies of a homologous gene or a modified homologous
- a "Campbell out” cell or strain is usually, but not necessarily, obtained by a counter- selection against a gene that is contained in a portion (the portion that is desired to be jettisoned) of the "Campbelled in” DNA sequence, for example the Bacillus subtilis sacB gene, which is lethal when expressed in a cell that is grown in the presence of about 5% to 10% sucrose.
- a desired "Campbell out” cell can be obtained or identified by screening for the desired cell, using any screenable phenotype, such as, but not limited to, colony morphology, colony color, presence or absence of antibiotic resistance, presence or absence of a given DNA sequence by polymerase chain reaction, presence or absence of an auxotrophy, presence or absence of an enzyme, colony nucleic acid hybridization, antibody screening, etc.
- the term "Campbell in” and “Campbell out” can also be used as verbs in various tenses to refer to the method or process described above.
- the homologous recombination events that leads to a "Campbell in” or “Campbell out” can occur over a range of DNA bases within the homologous DNA sequence, and since the homologous sequences will be identical to each other for at least part of this range, it is not usually possible to specify exactly where the crossover event occurred. In other words, it is not possible to specify precisely which sequence was originally from the inserted DNA, and which was originally from the chromosomal DNA.
- the first homologous DNA sequence and the second homologous DNA sequence are usually separated by a region of partial non-homo logy, and it is this region of non- homo logy that remains deposited in a chromosome of the "Campbell out" cell.
- typical first and second homologous DNA sequence are at least about 200 base pairs in length, and can be up to several thousand base pairs in length, however, the procedure can be made to work with shorter or longer sequences.
- a length for the first and second homologous sequences can range from about 500 to 2000 bases, and the obtaining of a "Campbell out" from a "Campbell in” is facilitated by arranging the first and second homologous sequences to be approximately the same length, preferably with a difference of less than 200 base pairs and most preferably with the shorter of the two being at least 70% of the length of the longer in base pairs.
- the "Campbell In and -Out-method" is described in WO2007012078
- Molasses Medium contained in one liter of medium: 40 g glucose; 60 g molasses; 20 g (NH 4 ) 2 SO 4 ; 0.4 g MgSO 4 *7H 2 O; 0.6 g KH 2 PO 4 ; 10 g yeast extract (DIFCO); 5 ml of 400 mM threonine; 2 mgFeSO 4 .7H 2 O; 2 mg of MnSO 4 -H 2 O; and 50 g CaCO 3 (Riedel-de Haen), with the volume made up with ddH 2 O. The pH was adjusted to 7.8 with 20% NH 4 OH.
- the filtrates were assayed using HPLC for the concentrations of methionine, glycine plus homoserine, O- acetylhomoserine, threonine, isoleucine, lysine, and other indicated amino acids.
- filtered supernatants were diluted 1 : 100 with 0.45 ⁇ m filtered 1 mM Na 2 EDTA and 1 ⁇ l of the solution was derivatized with OPA reagent (AGILENT) in Borate buffer (80 mM NaBO 3 , 2.5 mM EDTA, pH 10.2) and injected onto a 200 x 4.1 mm Hypersil 5 ⁇ AA-ODS column run on an Agilent 1100 series HPLC equipped with a G1321A fluorescence detector (AGILENT). The excitation wavelength was 338 nm and the monitored emission wavelength was 425 nm. Amino acid standard solutions were chromatographed and used to determine the retention times and standard peak areas for the various amino acids. Chem Station, the accompanying software package provided by Agilent, was used for instrument control, data acquisition and data manipulation. The hardware was an HP Pentium 4 computer that supports Microsoft Windows NT 4.0 updated with a Microsoft Service Pack (SP6a).
- SP6a Microsoft Service Pack
- the strain M440 was subsequently transformed with DNA B (also referred to as pH373) (SEQ ID NO: 22) to yield a "Campbell in” strain.
- the "Campbell in” strain were then “Campbelled out” to yield a "Campbell out” strain, M603, which contains a gene encoding a feedback resistant aspartate kinase enzyme (Ask ⁇ 1 ) (encoded by lysC).
- Ask ⁇ 1 a feedback resistant aspartate kinase enzyme
- T311 was changed to 1311 (referred to as LysC T311I).
- the strain M603 produced about 17.4 mM lysine, while the ATCC 13032 strain produced no measurable amount of lysine. Additionally, the M603 strain produced about 0.5 mM homoserine, compared to no measurable amount produced by the ATCC 13032 strain, as summarized in Table 2.
- Table 2 Amounts of homoserine, O-acetylhomoserine, methionine and lysine produced by strains ATCC 13032 and M603
- the strain M603 was transformed with DNA C (also referred to as pH304) (SEQ ID NO:23) to yield a "Campbell in” strain, which was then "Campbelled out” to yield a "Campbell out” strain, M690.
- the M690 strain contained a PgroES promoter upstream of the metH gene (referred to as P497 metH). The sequence of the P497 promoter is depicted in SEQ ID NO: 4.
- the M690 strain produced about 77.2 mM lysine and about 41.6 mM homoserine, as shown below in Table 3.
- Table 3 Amounts of homoserine, O-acetyl homoserine, methionine and lysine produced by the strains M603 and M690
- the M690 strain was subsequently mutagenized as follows: an overnight culture of M603, grown in BHI medium (BECTON DICKINSON), was washed in 5OmM citrate buffer pH 5.5, treated for 20 min at 30 0 C with N-methyl-N-nitrosoguanidine (10 mg/ml in 5OmM citrate pH 5.5).
- the cells were again washed in 50 mM citrate buffer pH 5.5 and plated on a medium containing the following ingredients: (all mentioned amounts are calculated for 500 ml medium) 1Og (NH 4 ) 2 SO 4 ; 0.5g KH 2 PO 4 ; 0.5g K 2 HPO 4 ; 0.125g MgSO 4 *7H 2 O; 21g MOPS; 50 mg CaCl 2 ; 15 mg protocatechuic acid; 0.5 mg biotin; 1 mg thiamine; and 5 g/1 D,L- ethionine (SIGMA CHEMICALS, CATALOG #E5139), adjusted to pH 7.0 with KOH.
- 1Og (NH 4 ) 2 SO 4 0.5g KH 2 PO 4 ; 0.5g K 2 HPO 4 ; 0.125g MgSO 4 *7H 2 O; 21g MOPS
- 50 mg CaCl 2 15 mg protocatechuic acid; 0.5 mg biotin; 1 mg thiamine; and 5 g/1 D,L- e
- the medium contained 0.5 ml of a trace metal solution composed of: 10 g/1 FeSO 4 *7H 2 O; 1 g/1 MnSO 4 *H 2 O; 0.1 g/1 ZnSO 4 *7H 2 O; 0.02 g/1 CuSO 4 ; and 0.002 g/1 MC1 2 *6H 2 O, all dissolved in 0.1 M HCl.
- the final medium was sterilized by filtration and to the medium, 40 mis of sterile 50% glucose solution (40 ml) and sterile agar to a final concentration of 1.5 % were added.
- the final agar containing medium was poured to agar plates and was labeled as minimal-ethionine medium.
- the mutagenized strains were spread on the plates (minimal-ethionine) and incubated for 3-7 days at 30 0 C. Clones that grew on the medium were isolated and restreaked on the same minimal-ethionine medium. Several clones were selected for methionine production analysis. Methionine production was analyzed as follows.
- Strains were grown on CM-agar medium for two days at 30 0 C, which contained: 10 g/1 D-glucose, 2.5 g/1 NaCl; 2 g/1 urea; 10 g/1 Bacto Peptone (DIFCO); 5 g/1 Yeast Extract (DIFCO); 5 g/1 Beef Extract (DIFCO); 22 g/1 Agar (DIFCO); and which was autoclaved for 20 min at about 12FC.
- DIFCO Bacto Peptone
- DIFCO 5 g/1 Yeast Extract
- DIFCO 5 g/1 Beef Extract
- DIFCO 22 g/1 Agar
- Medium II contained: 40 g/1 sucrose; 60 g/1 total sugar from molasses (calculated for the sugar content); 10 g/1 (NH 4 ) 2 SO 4 ; 0.4 g/1 MgSO 4 *7H 2 O; 0.6 g/1 KH 2 PO 4 ; 0.3 mg/1 thiamine*HCl; 1 mg/1 biotin; 2 mg/1 FeSO 4 ; and 2 mg/1 MnSO 4 .
- the medium was adjusted to pH 7.8 with NH 4 OH and autoclaved at about 121°C for about 20 min). After autoclaving and cooling, vitamin Bi 2 (cyanocobalamine) (SIGMA CHEMICALS) was added from a filter sterile stock solution (200 ⁇ g/ml) to a final concentration of 100 ⁇ g/1.
- Table 4 Amounts of homoserine, O-acetylhomoserine, methionine and lysine produced by strains M690 and M 1197
- the strain Ml 197 was transformed with DNA F (also referred to as pH399, SEQ ID NO: 24) to yield a "Campbell in” strain, which was subsequently "Campbelled out” to yield strain M 1494.
- This strain contains a mutation in the gene for the homoserine kinase, which results in an amino acid change in the resulting homoserine kinase enzyme from T 190 to A 190 (referred to as HskT190A).
- Amino acid production by the strain M 1494 was compared to the production by strain Ml 197, as summarized below in Table 5.
- Table 5 Amounts of homoserine, O-acetylhomoserine, methionine and lysine produced by strains Ml 197 and M 1494
- the strain M 1494 was transformed with DNA D (also referred to as pH484, SEQ ID NO:25) to yield a "Campbell in” strain, which was subsequently "Campbelled out” to yield the M 1990 strain.
- the M 1990 strain overexpresses a metY allele using both a groES-promoter and an EFTU (elongation factor Tu)-promoter (referred to as P 497 Pi284 metY).
- the sequence of P497P1284 promoter is set forth in SEQ ID NO:26 Amino acid production by the strain M 1494 was compared to the production by strain M 1990, as summarized below in Table 6.
- Table 6 Amounts of homoserine, O-acetylhomoserine, methionine and lysine produced by strains M 1494 and M 1990
- the strain M 1990 was transformed with DNA E (also referred to as pH 491, SEQ ID NO: 27) to yield a "Campbell in” strain, which was then "Campbelled out” to yield a "Campbell out” strain M2014.
- the M2014 strain overexpresses a metA allele using a superoxide dismutase promoter (referred to as P3119 metA).
- P3119 metA The sequence of P3119 promoter is set forth in SEQ ID NO: 3. Amino acid production by the strain M2014 was compared to the production by strain M 1990, as summarized below in Table 7
- Table 7 Amounts of homoserine, O-acetylhomoserine, methionine and lysine produced by strains M 1494 and M 1990
- Plasmid pH429 containing an RXA00655 deletion (SEQ ID NO:28) was used to introduce the mcbR deletion into C. glutamicum via integration and excision (see WO 2004/050694 Al). Plasmid pH429 was transformed into the M2014 strain with selection for kanamycin resistance (Campbell in). Using sacB counter-selection, kanamycin-sensitive derivatives of the transformed strain were isolated which presumably had lost the integrated plasmid by excision (Campbell out). The transformed strain produced kanamycin-sensitive derivatives that made small colonies and larger colonies. Colonies of both sizes were screened by PCR to detect the presence ofmcbR deletion. None of the larger colonies contained the deletion, whereas 60-70% of the smaller colonies contained the expected mcbR deletion.
- Table 9 Amino acid production by two isolates of OM403 in shake flask cultures inoculated with freshly grown cells.
- OM403-8 In order to decrease the import of methionine in OM403-8, the promoter and 5' portion of the metQ gene were deleted.
- the metQ gene encodes a subunit of a methionine import complex that is required for the complex to function. This was accomplished using the standard Campbelling in and Campbelling out technique with plasmid pH449 (SEQ ID NO: 29).
- OM403-8 and OM456-2 were assayed for methionine production in shake flask assays. The results (Table 10) show that OM456-2 produced more methionine than OM403-8. Cultures were grown for 48 hours in standard molasses medium. Table 10: Shake flask assays of OM456-2
- OM469 A strain referred to as OM469 was constructed which included both deletion o ⁇ metQ and overexpression of metF by replacing the metF promoter with the phage XP R promoter in OM456-2. This was accomplished using the standard Campbelling in and Campbelling out technique with plasmid pOM427 (SEQ ID NO 30). Four isolates of OM469 were assayed for methionine production in shake flask culture assays where they all produced more methionine than OM456-2, as shown in Table 11. Cultures were grown for 48 hours in standard molasses medium containing 2 mM threonine.
- Table 11 Shake flask assays of OM469, a derivative of OM456-2 containing the phage lambda P R promoter in place of the metF promoter.
- the strain OM469-2 was transformed by electroporation with the plasmid pCLIK5 A PSOD TKT as depicted in SEQ ID NO. 31. This was accomplished using the standard Campbelling in and Campbelling out technique.
- sda serine deaminase
- Plasmid pOM253 (SEQ ID No. 33) was used to delete serA and substitute it with spectinomycin resistance in C glutamicum strain M2014.
- OM264C is a serine auxotroph. Since it also lacks a functional GCS, OM264C cannot grow on a minimal medium lacking serine but containing glycine. If, however, a functional GCS system is installed in OM264C, then it will gain the ability to grow on minimal medium containing glycine.
- Lipoic acid (when added) 1 g/1 in 20 niM potassium I mI phosphate, pH 7.0
- Micronutrient solution amount for 1 liter
- C.jeikeium does contain a GCS.
- the gcvP,T, and H genes are clustered together in an operon (Tauch et al., 2005, J. Bacteriol., vol 187, pp4671-4682). This cluster was cloned in four overlapping subset pieces by polymerase chain reaction (PCR) using C.jeikeium strain K411 chromosomal DNA as the template.
- the necessary DNA was obtained by dividing the sequence into four smaller regions and obtaining four independent smaller PCR fragments.
- the four pieces were amplified with four sets of primers.
- An artificial Xba ⁇ site and an artificial ribosome binding site were engineered just upstream from the gcvP start codon in the respective sense primer, and an artificial BamHI site was engineered just downstream from the gcvH stop codon in the respective antisense primer. This allowed the coding sequences of the gcvPT ⁇ cluster to be reconstituted and carried on an Xbal to BamHI fragment.
- the resulting Xbal to BamHI fragment containing a reconstituted gcvPTH cluster was next cloned into C glutamicum replicating plasmids designed to express genes from the C glutamicum groESL promoter, herein named P4 9 7, or a B. subtilis phage SPOl promoter, herein named P 15 (SEQ ID No. 42).
- the resulting plasmids were named pOM615 (SEQ ID No. 34) and pOM616 (SEQ ID No. 35), respectively.
- the P 497 and Pi 5 promoters were used because they promote constitutive expression of genes situated downstream.
- promoters are not significantly regulated by any glycine related metabolite, such as glycine, serine, methionine, thymidine, purine etc.
- glycine related metabolite such as glycine, serine, methionine, thymidine, purine etc.
- Plasmids pOM615, pOM616, and empty vector pCLIK were each separately transformed into the tester strain C. glutamicum strain OM264C and the methionine producer C. glutamicum strain GK 1259 using selection on Brain Heart Infusion (formerly Difco, now Becton Dickenson) agar plates containing kanamycin sulfate (25 mg/1). The OM264C trans formants were streaked on minimal plates lacking serine but containing glycine. Lipoic acid was added to the medium to give a final concentration of 1 mg/1 to ensure that a sufficient amounts of this co factor of GcvH was present.
- C. glutamicum is a lipoic acid prototroph, and C. glutamicum presumably has a lipoyl synthetase, since pyruvate dehydrogenase and ⁇ -ketoglutarate dehydrogenase are active.
- Plasmids pOM620AF and pOM621AR were each transformed into strain OM264C resulting in OM264C(pOM620AF) and OM264C(pOM621AR) respectively and into methionine producing strain GK1259 resulting in GK1259(pOM620AF) and
- the GK1259 transformants of pOM615, pOM616, pOM620AF, pOM621AR, and pCLIK were tested for methionine and glycine production in shake flasks using molasses medium, without lipoic acid or with lipoic acid added to a final concentration of about 10 mg/ml.
- the results are shown in Tables 13 and 14 below.
- Table 13 Methionine and glycine production by GK1259 transformed with various plasmids designed to express C. jeikeium gcvPTH with lipBA and grown in shake flasks in molasses medium.
- Table 14 Methionine and glycine production by O264C transformed with various plasmids designed to express C. jeikeium gcvPTH with lipBA and grown in shake flasks in molasses medium.
- Methionine and glycine titers are averages of duplicate samples, except for the pOM616 plus lipoic acid sample, which is a single sample.
- the gcv and lip genes were installed on plasmids that replicate in the C. glutamicum.
- these genes can also be installed through an integrating vector.
- the C. jeikeium gcvPTH operon expressed from promoter P ;j has been ligated into an integrating vector to give pOM627 (SEQ ID No. 38), which is designed to integrate at the bioB locus of C. glutamicum.
- pOM627 can be "Campbelled in” and “Campbelled out” of strains OM469 and GK1259. As in the above example, one observes improved methionine production and reduced glycine accumulation as is shown in Table 15. Table 15: Methionine and glycine production by transformants of OM469 and GK1259 using pOM627 designed to express C. jeikeium gcvPTH from P 15 after integration at bioB, and grown in shake flasks in molasses medium with or without 10 m /1 li oic acid added.
- the effect from the integrating plasmid pOM627 on glycine and methionine production can be improved by installing multiple copies or by increasing the promoter strength.
- the C. jeikeium HpBA operon can also be added (either on replicating or integrating vectors).
- An example of an integrating plasmid that expresses HpBA from promoter P 497 after integrating at the bio AD locus of C. glutamicum is pOM180 (SEQ ID No 39).
- the E. coli lpd gene was amplified by PCR and installed in an integrating plasmid to give pOM331 (SEQ ID No 40).
- the integration site is a gene herein named metE2, a gene that is homologous to a portion of metE, but which does not appear to be essential for growth or methionine production in C. glutamicum.
- pOM331 the E. coli lpd gene is expressed from the P 4 9 7 promoter.
- pOM331 was transformed into and Campbelled out of OM264Cto give new strain OM 197, which was confirmed by an appropriate diagnostic PCR to contain the integrated P4 9 7 lpd cassette.
- the E. coli gcvTHP operon was amplified by PCR and ligated just downstream from the P 497 promoter in a replicating vector to give pOM344 (SEQ ID No. 41).
- pOM344 and pCLIK were each separately transformed into strain OM 197, and the resulting strains were streaked on minimal glycine plates containing glycine and lipoic acid, but lacking serine (see above).
- genes encoding GCS subunits P, T and H, and genes encoding enzymes that catalyze lipoic acid ligation or synthesis, cloned from either of two "donor" organisms (which are relatively unrelated to each other) are each capable of functioning to give measurable GCS activity in C. glutamicum, either by showing complementation of a serine auxotrophy on plates containing glycine but no serine or by showing a decrease in glycine production by a methionine producing strain compared to the glycine production of the relevant parent or precursor strain transformed with an empty vector as a control.
- Plasmid pHF96 (SEQ ID 43) is an integrating plasmid designed to create a deletion- substitution in the serA gene of C. glutamicum strains related to C. glutamicum ATCC 13032. Plasmid pHF96 int sacB delta serA was used to delete sera in M2543. The plasmid was transformed in to C. glutamicum M2543 cby electroporation and kanamycin resistant clones were isolated.
- M2616 is a serine auxotroph when assayed on minimal medium. Also as expected, since it lacks a functional formate THF synthetase, M2616 cannot grow on a minimal medium lacking serine but containing glycine and formate (see Example for recipe for minimal (chemically defined) medium). If a functional formate THF synthetase system is installed in M2616, then it will gain the ability to grow on minimal medium containing glycine.
- Corynebacterium jeikeium does contain a formyl-THF synthetase protein with the accession number NP 939608 and a coresponding gene with the accession number GenelD: 2649808.
- primers HS 1302 SEQ ID No. 46
- HS 1303 SEQ ID No. 47
- primers HS 1302 SEQ ID No. 46
- HS 1303 SEQ ID No. 47
- Both fragments were added in a third PCR in which the primers HS 13032 and HS 1305 were added.
- the third PCR was performed with limiting amounts of the PCR fragments I and II at sufficient amounts of the end to end primers and was a fused using the end-to end primers.
- HS1302+HS1305 were used to amplify a fusion construct of the PCR fragments resulting from primers HS1302+HS1303 and HS1304+HS1305.
- Pwo polymerase from Roche Mannheim was used at the following conditions: Annealing at 55°C for 30" and elongation at 72°C for 120" yielded a PCR fragment of about 1900Bp.
- the fragment was purified with the GFX PCR purification kit and was digested using the restriction enzymes MIuI and Xbal. Positive plasmids containing the insert of the formyl-THF synthetase gene were sequenced.
- the gene derived from C jeikeium NCTC Kl 1915 was sequenced. Sequencing of this gene revealed the sequence to be as expected.
- the gene derived from C jeikeium DSMZ 7171 was sequenced. Sequencing of this gene revealed a sequence as described in the plasmid sequence pH657. Plasmid pH655 (SEQ ID No. 48) comprising formate-THF-synthetase from NCTC Kl 1915 and pH657 (SEQ ID No. 49) comprising formate-THF-synthetase from DSMZ7171 were transformed into the strain M2616 lacking the functional serA gene by electroporation.
- strains M2616(pH655) and M2616(pH657) as well as the strain lacking a plasmid were streaked on minimal medium containing 1OmM threonine, 2OmM Na- formate and 2OmM glycine +- 1OmM serine.
- Strains containing the plasmids pH655 and pH657 but not pCLIK5a formed colonies on minimal medium containing formate and glycine, while even after prolonged incubation the strain M2616 did not produce colonies on this medium lacking serine.
- Na-formate and glycine but with added serine (2OmM) all strains formed colonies including M2616.
- Plasmids pH655 and pH657 were transformed into the strain GK1259 by electroporation.
- the resulting strains GK1259(pH655) and GK1259(pH657) as well as the strain containing no plasmid were incubated in shake flask assays as previoulsy described.
- 2OmM formate was added to the growth medium. It was observed that expression of the formate THF synthetase gene improved the methionine productivity of the strain over the strain lacking the formate THF synthetase gene. The data is found in table 16.
- Table 16 Shake flask assays of GK1259, and GK1259(pH655) overexpressing formate-THF s nthetase
- C. glutamicum contains a gene, which has been annotated as a formyl-THF deformylase (Accession Ncgl 0371 ) .
- This gene has been annotated as coding for an enzyme with the enzymatic activity of formyl-THF deformylase, cleaving formate from the metabolite formyl- tetrahydro folate (Annual review of plant physiology and plant molecular biology
- Knockout of the formyl-THF deformylase was performed by cloning a DNA fragment in which the upstream region of the gene coding for Ncgl 0371 (approximately 500Bp long) and its downstream region (approximately 500Bp long) of the same gene were amplified, fused by fusion-PCR and cloned into a vector resulting in pH670 int sacB delta deformylase (SEQ ID No. 50). The plasmid is transformed into the strain M2543 to yield first recombinants ("Campbell in step").
- the strain GKl 546 is transformed with the plasmids pH655 and pH657. Resulting strains GK1546(pH655) and GK1546(pH657) show significantly improved growth behaviour when they are grown on a minimal medium containing formate, glycine and threonine but no serine over the strain M2543.
- Strain M2616 was transformed with pH655 (formate-THF-synthetase NCTC 11915) and pH657 (formate-THF-synthetase DSMZ 7171). The resulting strains M2616(pH655) and M2616(pH657) were analyzed in shake flask assays as described previously.
- the medium was supplied with 2OmM glycine as well as 2OmM Na- formate in addition to the normal composition described above. Shake flasks were incubated at 30 0 C with a shaking rate of 200RPM. After 48h methionine was determined. It was found that the parental strain M2616 without the formate-THF synthetase expressing plasmid did not grow to measurable OD and it did not utilize the given carbon source. Supernatants were assayed for formate in the supernatant by HPLC. Strains containing the plasmids pH655 and pH657 show an utilisation of all formate added to the medium.
Landscapes
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description











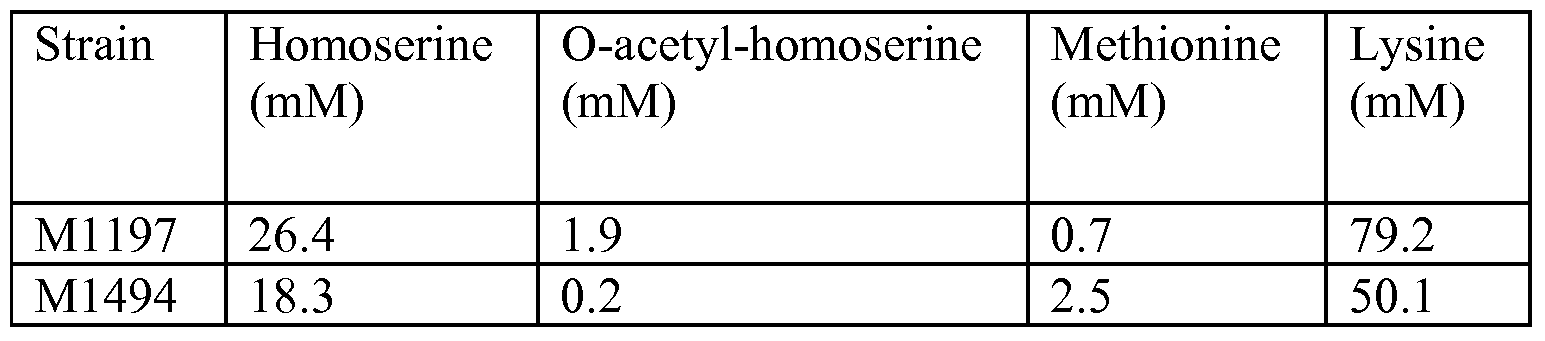








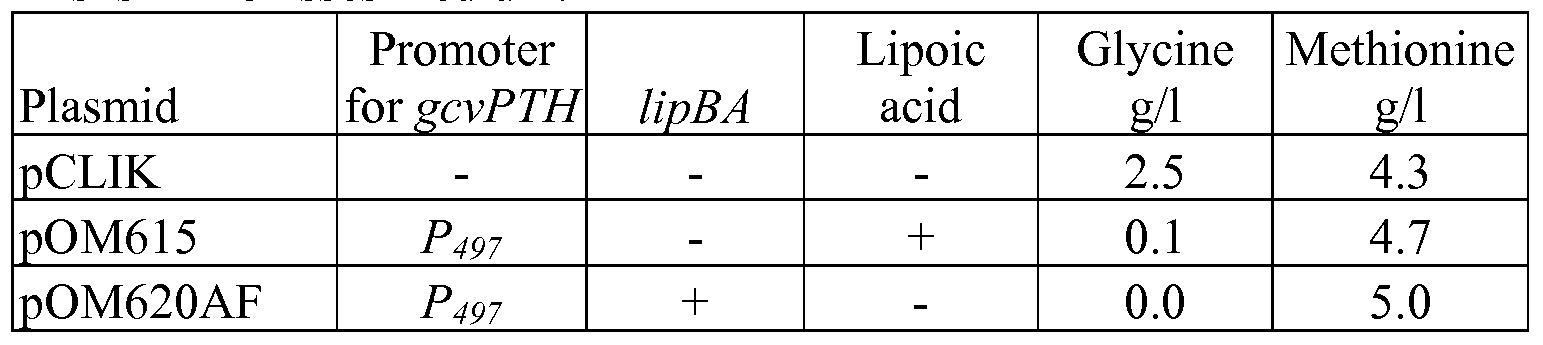





Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0807760-6A2A BRPI0807760A2 (en) | 2007-02-19 | 2008-02-14 | CORINEFORM BACTERIA WITH FORMAT-THF SYNTETASE ACTIVITY AND / OR GLYCIN CLIVING ACTIVITY |
| CN200880005502A CN101849016A (en) | 2007-02-19 | 2008-02-14 | Coryneform bacteria having formyl-THF-synthetase and/or glycine cleavage activity |
| EP08716849A EP2121954A2 (en) | 2007-02-19 | 2008-02-14 | Coryneform bacteria with formate-thf-synthetase and/or glycine cleavage activity |
| JP2009550695A JP2010518837A (en) | 2007-02-19 | 2008-02-14 | Coryneform bacterium having formate-THF-synthetase and / or glycine cleavage activity |
| US12/519,909 US20100003727A1 (en) | 2007-02-19 | 2008-02-14 | Coryneform Bacteria with Formate-THF-Synthetase and/or Glycine Cleavage Activity |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07102652.0 | 2007-02-19 | ||
| EP07102652 | 2007-02-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008101857A2 true WO2008101857A2 (en) | 2008-08-28 |
| WO2008101857A3 WO2008101857A3 (en) | 2008-12-11 |
Family
ID=39523537
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/051795 Ceased WO2008101857A2 (en) | 2007-02-19 | 2008-02-14 | Coryneform bacteria with formate-thf-synthetase and/or glycine cleavage activity |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20100003727A1 (en) |
| EP (2) | EP2431476B1 (en) |
| JP (1) | JP2010518837A (en) |
| CN (1) | CN101849016A (en) |
| BR (1) | BRPI0807760A2 (en) |
| ES (1) | ES2517394T3 (en) |
| RU (1) | RU2009134796A (en) |
| WO (1) | WO2008101857A2 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009043803A3 (en) * | 2007-10-02 | 2009-06-18 | Metabolic Explorer Sa | Increasing methionine yield |
| WO2011080301A3 (en) * | 2009-12-30 | 2012-04-12 | Metabolic Explorer | Strains and method for the production of methionine |
| US8906653B2 (en) | 2008-01-23 | 2014-12-09 | Basf Se | Method for fermentatively producing 1,5-diaminopentane |
| WO2015118126A1 (en) * | 2014-02-07 | 2015-08-13 | Dsm Ip Assets B.V. | Improved bacillus host |
| EP2940039A1 (en) | 2014-04-30 | 2015-11-04 | Evonik Degussa GmbH | Method for the production of l-amino acids in coryne bacteria using a glycine splitting system |
| WO2016146633A1 (en) * | 2015-03-18 | 2016-09-22 | Basf Se | Recombinant microorganism for improved production of fine chemicals |
| CN112301050A (en) * | 2019-07-26 | 2021-02-02 | 浙江大学 | Method for constructing high-yield glutathione recombinant strain and application |
| US11214816B2 (en) | 2016-12-27 | 2022-01-04 | Korea Advanced Institute Of Science And Technology | Recombinant microorganism having heterologous genes introduced thereto and method for producing useful material from formic acid and carbon dioxide using same microorganism |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8647642B2 (en) | 2008-09-18 | 2014-02-11 | Aviex Technologies, Llc | Live bacterial vaccines resistant to carbon dioxide (CO2), acidic PH and/or osmolarity for viral infection prophylaxis or treatment |
| EP3145482A1 (en) * | 2014-05-23 | 2017-03-29 | Evonik Degussa GmbH | Biosynthetic production of acyl amino acids |
| KR20160065504A (en) * | 2014-12-01 | 2016-06-09 | 엘지전자 주식회사 | Multimedia device and method for controlling the same |
| US10676723B2 (en) | 2015-05-11 | 2020-06-09 | David Gordon Bermudes | Chimeric protein toxins for expression by therapeutic bacteria |
| WO2017214417A1 (en) | 2016-06-10 | 2017-12-14 | Takara Bio Usa, Inc. | Methods and compositions employing blocked primers |
| US11129906B1 (en) | 2016-12-07 | 2021-09-28 | David Gordon Bermudes | Chimeric protein toxins for expression by therapeutic bacteria |
| US11180535B1 (en) | 2016-12-07 | 2021-11-23 | David Gordon Bermudes | Saccharide binding, tumor penetration, and cytotoxic antitumor chimeric peptides from therapeutic bacteria |
| KR102204917B1 (en) * | 2019-04-22 | 2021-01-20 | 씨제이제일제당 주식회사 | Microorganisms with enhanced ability to produce L-histidine and methods for producing L-histidine using the same |
| CN116284282B (en) * | 2023-03-17 | 2025-09-16 | 黑龙江伊品生物科技有限公司 | Tkt gene mutant and application thereof in preparation of L-lysine |
| WO2025002383A1 (en) * | 2023-06-28 | 2025-01-02 | Westlake University | Regulation of cellular energy metabolism by lipoate protein ligase and its applications |
| CN120796161B (en) * | 2025-09-09 | 2026-01-09 | 西湖大学 | Streptomyces fermentation method for improving yield of secondary metabolites by genetically modified streptomycete |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4601893A (en) | 1984-02-08 | 1986-07-22 | Pfizer Inc. | Laminate device for controlled and prolonged release of substances to an ambient environment and method of use |
| DE4027453A1 (en) | 1990-08-30 | 1992-03-05 | Degussa | NEW PLASMIDES FROM CORYNEBACTERIUM GLUTAMICUM AND DERIVED PLASMIDE VECTORS |
| DE4120867A1 (en) | 1991-06-25 | 1993-01-07 | Agfa Gevaert Ag | PHOTOGRAPHIC PROCESSING METHOD AND DEVICE |
| ATE524545T1 (en) | 1994-08-20 | 2011-09-15 | Gendaq Ltd | IMPROVEMENTS IN OR RELATED TO BINDING PROTEINS FOR DNA DETECTION |
| DE4440118C1 (en) | 1994-11-11 | 1995-11-09 | Forschungszentrum Juelich Gmbh | Gene expression in coryneform bacteria regulating DNA |
| JPH10229891A (en) | 1997-02-20 | 1998-09-02 | Mitsubishi Rayon Co Ltd | Method for producing malonic acid derivative |
| WO2002010209A1 (en) | 2000-08-02 | 2002-02-07 | Degussa Ag | Nucleotide sequences which code for the meth gene |
| AU2002368410A1 (en) | 2002-11-29 | 2004-06-23 | Basf Aktiengesellschaft | Methods for the production of methionine |
| DE10359594A1 (en) | 2003-12-18 | 2005-07-28 | Basf Ag | PEF TU-expression units |
| DE102004035065A1 (en) | 2004-07-20 | 2006-02-16 | Basf Ag | P-ET-TS expression units |
| DE102004061846A1 (en) * | 2004-12-22 | 2006-07-13 | Basf Ag | Multiple promoters |
| US20070026505A1 (en) * | 2005-06-17 | 2007-02-01 | Madden Kevin T | Amino acid and metabolite biosynthesis |
| BRPI0613660A2 (en) | 2005-07-18 | 2012-11-06 | Basf Ag | microorganism, metl expression cassette, vector, methods for producing methionine, lycopene, incremented levels of a desired carotenoid, at least two compounds in a fermentation process, a desired carotenoid compound, at least two compounds in a fermentation process, one carotenoid compound, and a sulfur-containing fine chemical, and to increase methionine production capacity in a microorganism, and, dna sequence |
| CA2615416A1 (en) * | 2005-07-18 | 2007-01-25 | Basf Aktiengesellschaft | Methionine producing recombinant microorganisms |
| KR20080039906A (en) * | 2005-08-18 | 2008-05-07 | 바스프 에스이 | Microorganisms with Increased Methionine Synthesis Efficiency |
| GB0602339D0 (en) * | 2006-02-06 | 2006-03-15 | Glaxo Group Ltd | Novel process |
| WO2009043372A1 (en) * | 2007-10-02 | 2009-04-09 | Metabolic Explorer | Increasing methionine yield |
-
2008
- 2008-02-14 JP JP2009550695A patent/JP2010518837A/en active Pending
- 2008-02-14 CN CN200880005502A patent/CN101849016A/en active Pending
- 2008-02-14 ES ES11193520.1T patent/ES2517394T3/en active Active
- 2008-02-14 EP EP11193520.1A patent/EP2431476B1/en not_active Not-in-force
- 2008-02-14 EP EP08716849A patent/EP2121954A2/en not_active Withdrawn
- 2008-02-14 WO PCT/EP2008/051795 patent/WO2008101857A2/en not_active Ceased
- 2008-02-14 US US12/519,909 patent/US20100003727A1/en not_active Abandoned
- 2008-02-14 RU RU2009134796/10A patent/RU2009134796A/en unknown
- 2008-02-14 BR BRPI0807760-6A2A patent/BRPI0807760A2/en not_active Application Discontinuation
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10858679B2 (en) | 2007-10-02 | 2020-12-08 | Evonik Degussa Gmbh | Increasing methionine yield |
| EP2573189A1 (en) * | 2007-10-02 | 2013-03-27 | Metabolic Explorer | Increasing methionine yield |
| WO2009043803A3 (en) * | 2007-10-02 | 2009-06-18 | Metabolic Explorer Sa | Increasing methionine yield |
| US8906653B2 (en) | 2008-01-23 | 2014-12-09 | Basf Se | Method for fermentatively producing 1,5-diaminopentane |
| US9382561B2 (en) | 2009-12-30 | 2016-07-05 | Metabolic Explorer | Strains and method for the production of methionine |
| WO2011080301A3 (en) * | 2009-12-30 | 2012-04-12 | Metabolic Explorer | Strains and method for the production of methionine |
| JP2013516162A (en) * | 2009-12-30 | 2013-05-13 | メタボリック エクスプローラー | Strains and methods for the production of methionine |
| US10287592B2 (en) | 2014-02-07 | 2019-05-14 | Dsm Ip Assets B.V. | Bacillus host |
| WO2015118126A1 (en) * | 2014-02-07 | 2015-08-13 | Dsm Ip Assets B.V. | Improved bacillus host |
| WO2015165740A3 (en) * | 2014-04-30 | 2016-02-25 | Evonik Degussa Gmbh | Method for producing l-amino acids in corynebacteria using a glycine cleavage system |
| EP2940039A1 (en) | 2014-04-30 | 2015-11-04 | Evonik Degussa GmbH | Method for the production of l-amino acids in coryne bacteria using a glycine splitting system |
| KR20160148006A (en) * | 2014-04-30 | 2016-12-23 | 에보닉 데구사 게엠베하 | Method for producing l-amino acids in corynebacteria using a glycine cleavage system |
| RU2713298C2 (en) * | 2014-04-30 | 2020-02-04 | Эвоник Оперейшенс ГмбХ | Method for producing l-amino acids using corynebacteria using glycine cleavage system |
| KR102260001B1 (en) * | 2014-04-30 | 2021-06-02 | 에보닉 오퍼레이션스 게엠베하 | Method for producing l-amino acids in corynebacteria using a glycine cleavage system |
| WO2016146633A1 (en) * | 2015-03-18 | 2016-09-22 | Basf Se | Recombinant microorganism for improved production of fine chemicals |
| US10717998B2 (en) | 2015-03-18 | 2020-07-21 | Basf Se | Recombinant microorganism for improved production of fine chemicals |
| US11214816B2 (en) | 2016-12-27 | 2022-01-04 | Korea Advanced Institute Of Science And Technology | Recombinant microorganism having heterologous genes introduced thereto and method for producing useful material from formic acid and carbon dioxide using same microorganism |
| CN112301050A (en) * | 2019-07-26 | 2021-02-02 | 浙江大学 | Method for constructing high-yield glutathione recombinant strain and application |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2431476A1 (en) | 2012-03-21 |
| EP2431476B1 (en) | 2014-08-20 |
| CN101849016A (en) | 2010-09-29 |
| RU2009134796A (en) | 2011-03-27 |
| EP2121954A2 (en) | 2009-11-25 |
| WO2008101857A3 (en) | 2008-12-11 |
| ES2517394T3 (en) | 2014-11-03 |
| US20100003727A1 (en) | 2010-01-07 |
| BRPI0807760A2 (en) | 2014-06-17 |
| JP2010518837A (en) | 2010-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2431476B1 (en) | Coryneform bacteria with glycine cleavage activity | |
| US8163532B2 (en) | Microorganisms with a reactivation system for cob(I)alamin-dependent methionine synthase | |
| US9169502B2 (en) | Method of producing L-lysine using a Corynebacterium glutamicum microorganism | |
| US20100047881A1 (en) | Microorganisms with Deregulated Vitamin B12 System | |
| US8252555B2 (en) | Nucleic acid encoding a cobalamin-dependent methionine synthase polypeptide | |
| US20090298136A1 (en) | Methionine producing recombinant microorganisms | |
| US20120288901A1 (en) | Method of Producing Methionine in Corynebacteria by Over-Expressing Enzymes of the Pentose Phosphate Pathway | |
| JP4648947B2 (en) | Microorganisms for producing sulfur-containing compounds | |
| EP2283142A1 (en) | Production process for fine chemicals using microorganisms with reduced isocitrate dehydrogenase activity | |
| RU2723714C2 (en) | Method and microorganism for enzymatic production of methionine with improved output of methionine | |
| CA2922597A1 (en) | Microorganism for methionine production with improved methionine synthase activity and methionine efflux | |
| WO2007020295A2 (en) | Microorganisms with increased efficiency for methionine synthesis | |
| CN101291590A (en) | Microorganisms and methods for producing L-methionine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880005502.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08716849 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008716849 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009550695 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12519909 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3244/KOLNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009134796 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0807760 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090818 |