WO2008104875A1 - Oxazolidinones as cholesterol absorption inhibitors - Google Patents
Oxazolidinones as cholesterol absorption inhibitors Download PDFInfo
- Publication number
- WO2008104875A1 WO2008104875A1 PCT/IB2008/000523 IB2008000523W WO2008104875A1 WO 2008104875 A1 WO2008104875 A1 WO 2008104875A1 IB 2008000523 W IB2008000523 W IB 2008000523W WO 2008104875 A1 WO2008104875 A1 WO 2008104875A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- hydroxy
- fluoro
- oxazolidin
- ethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 O=C1OC(*c2ccccc2)C(c2ccccc2)N1c1ccccc1 Chemical compound O=C1OC(*c2ccccc2)C(c2ccccc2)N1c1ccccc1 0.000 description 4
- DLYSSBVKGUWJDX-NYJWAPBASA-N CCOC(CN(CC([C@H]([C@@H](c(cc1)ccc1O)N1c(cc2)ccc2F)OC1=O)O)c(cc1)ccc1F)=O Chemical compound CCOC(CN(CC([C@H]([C@@H](c(cc1)ccc1O)N1c(cc2)ccc2F)OC1=O)O)c(cc1)ccc1F)=O DLYSSBVKGUWJDX-NYJWAPBASA-N 0.000 description 1
- UURCHUTWJHPNQX-DOTOQJQBSA-N COc1ccc([C@H]([C@H](CC2=O)C(O)=O)N2c(cc2)ccc2F)cc1 Chemical compound COc1ccc([C@H]([C@H](CC2=O)C(O)=O)N2c(cc2)ccc2F)cc1 UURCHUTWJHPNQX-DOTOQJQBSA-N 0.000 description 1
- ORDWQKDFZGBZRA-MRAPNPIVSA-N C[C@H]([C@H]([C@@H]([C@H]1OC(C)=O)OC(C)=O)OC(C)=O)OC1C(C(Cl)(Cl)Cl)=N Chemical compound C[C@H]([C@H]([C@@H]([C@H]1OC(C)=O)OC(C)=O)OC(C)=O)OC1C(C(Cl)(Cl)Cl)=N ORDWQKDFZGBZRA-MRAPNPIVSA-N 0.000 description 1
- JBVQNUIDYBNYRN-NRWPOFLRSA-N N[C@](CO1)(C(Cc(cc2)ccc2O)N(c(cc2)ccc2F)C1=O)C(NCc1ccccc1)=O Chemical compound N[C@](CO1)(C(Cc(cc2)ccc2O)N(c(cc2)ccc2F)C1=O)C(NCc1ccccc1)=O JBVQNUIDYBNYRN-NRWPOFLRSA-N 0.000 description 1
- KWDRNYHTESKZCF-RBZQAINGSA-N Oc1ccc([C@H]([C@@H]([C@H](C2)OCCN2c(cc2)ccc2F)OC2=O)N2c(cc2)ccc2F)cc1 Chemical compound Oc1ccc([C@H]([C@@H]([C@H](C2)OCCN2c(cc2)ccc2F)OC2=O)N2c(cc2)ccc2F)cc1 KWDRNYHTESKZCF-RBZQAINGSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/16—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/18—Oxygen atoms
- C07D263/20—Oxygen atoms attached in position 2
- C07D263/24—Oxygen atoms attached in position 2 with hydrocarbon radicals, substituted by oxygen atoms, attached to other ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- This invention relates to a group of novel oxazolidinones. These compounds inhibit the cholesterol transporter NPC1 L1 and thus are useful as hypocholesterolemic agents and in the treatment and prevention of atherosclerosis.
- Atherosclerotic coronary heart disease represents the major cause of death and cardiovascular morbidity in the western world. Risk factors for atherosclerotic coronary heart disease include hypertension, diabetes mellitus, family history, maleness, smoking and elevated plasma cholesterol. Elevated plasma cholesterol and lipoprotein are significant atherosclerotic risk factors. Thus, a causative link between elevated plasma cholesterol levels, atherosclerosis, and coronary heart disease has been firmly established. Harwood et al., 34 J. Lipid Research 377-378 (1993). More specifically, a total cholesterol level in excess of 225-250 mg/dl is associated with significant elevation of risk.
- LDL low density lipoprotein
- VLDL very low-density lipoprotein
- RE37721 describes 2-azetidinone compounds wherein the 3-position substituent is an arylalkylene group substituted in the alkylene portion by a hydroxy group
- US 2003/0105028 describes glucose-derived conjugates of 2-azetidinone compounds wherein the 1-position substituent is a hydroxyl-substituted phenyl group and the 4- position substituent is a hydroxyphenyl group
- U.S. Pat. No. 5,756,470 discloses 2- azetidinones having an aryl group at the 4-position which is substituted with a hydroxyl and a glucuronide group.
- WO 2006/102674 discloses certain substituted azetidinones, as well as certain substituted oxazolidinones, useful as cholesterol absorption inhibitors.
- At least one substituted azetidinone, ezetimibe is currently commercially available for the treatment of hypercholesterolemia.
- the effectiveness of available antilipidemic therapies is limited, in part because of poor patient compliance due to unacceptable side effects and tolerability as well as minimal efficacy or potency.
- the present invention provides a compound having a Formula (I),
- Ar 1 and Ar 2 are each independently aryl or heteroaryl, optionally substituted; Y 3 is alky!, aryl, aralkyl, heteroalkyl or heteroaralkyl; optionally substituted; Z is -O-CR"R"'CH(OR')-; -NR'-CR"R"'-CH(OR')-; -CR"R"'-CR"R"'-CH(OR')-;
- Vv/w-" indicates the points of attachment;
- R' is H; or lower alkyl, optionally substituted;
- R" and R"' are each independently H; lower alkyl, optionally substituted, or flourine;
- W is O or NR'; and
- n is 0, 1 or 2.
- the present invention further provides a compound having a Formula (Ia), -A-
- R 1 , R 2 and R 3 are each independently halo; -OR', -COR', -COOR', -CONR'R"; CH 2 NR 1 R"; CH 2 NR 1 C(O)R"; C 1 -C 12 alkyl, aryl, or heteroaryl; optionally substituted; S(O) n R', P(O) n R', OG, CR'R"G, S(O) n G, NR 1 G or SG; G is is selected from the group consisting of hydrogen,
- R 5 , R 6 , R 7 , R 8 , R 9 , and R 10 are each independently selected from the group consisting of hydrogen, Ci-C 6 alkyl, Ci-C 6 aralkyl, -C(O)Ci-C 6 alkyl, -C(O)aryl, and aryl; and R 11 is selected from the group consisting of hydrogen, hydroxy, Ci.C ⁇ alkyl, -OC L C 6 alkyl, and NR'R"; R' is H; or lower alkyl, optionally substituted; R" and R'" are each independently H; lower alkyl, optionally substituted, or flourine; W is O or NR'; and n is 0, 1 or 2.
- the present invention further provides inter alia the following compounds: 3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one;
- Acetic acid 1 [3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl- propyl ester;
- Acetic acid 1 S-[3,4-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester; 5S-(1 R-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one;
- the present invention provides a compound having a Formula (I),
- the present invention further provides a compound having a Formula (Ia),
- a method of inhibiting cholesterol absorption in a mammal requiring inhibition comprising administering to the mammal a therapeutically effective amount of the compound or the pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof. Further provided is a method of treating, preventing or controlling hyperlipidemia in a mammal.
- a combination comprising the above-described compound and a pharmaceutically active agent.
- said pharmaceutically active agent is a CETP inhibitor, a PPAR- activator, an MTP/Apo B secretion inhibitor, HDL- cholesterol raising agent, HMG-CoA reductase inhibitor, triglyceride lowering agent, a cholesterol synthesis inhibitor, a cholesterol modulating agent, a fibrate, niacin, an ion- exchange resin, an antioxidant, an ACAT inhibitor, bile acid sequestrant, an anti- hypertensive agent, or an acetylcholine esterase inhibitor.
- HMG-CoA reductase inhibitor is a statin.
- composition comprising the above combination and a pharmaceutically acceptable carrier, diluent, solvent or vehicle.
- a pharmaceutically acceptable carrier diluent, solvent or vehicle.
- present invention further encompasses each of the title compounds set forth in the Examples herein.
- the present invention further includes each of the title compounds set forth in the Examples herein.
- alkyl refers to both the singular and plural form of the object to which it refers.
- the following definitions apply regardless of whether a term is used by itself or in combination with other terms, unless otherwise indicated. Therefore, the definition of “alkyl” applies to “alkyl” as well as the “alkyl” portions of “hydroxyalkyl”, “haloalkyl”, “alkoxy”, “aralkyl”, etc.
- aryl applies to “aryl” as well as the “aryl” portions of “heteroaryl", “aralkyl”, “arylthio”, etc.
- alkyl refers to a linear or branched hydrocarbon of from 1 to 20 carbon atoms.
- Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyi, n-decyl, tetradecyl, and the like.
- lower alkyl refers to a subset of alkyl which means a linear or branched hydrocarbon radical having from 1 to 6 carbon atoms. Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert- butyl, n-pentyl, n-hexyl, and the like. Alternatively, lower alkyl is referred to as "C 1 -C 6 alkyl.”
- the lower alkyl group can also be substituted with at least one to three of the substituents as previously recited for the term alkyl.
- alkoxy refers to an alkyl-O- group in which the alkyl group is as previously defined. Useful alkoxy groups can comprise 1 to 12 carbon atoms.
- lower alkoxy means an alkyl-O- group in which the alkyl group comprises 1 to 6 carbon atoms. Non-limiting examples of a lower alkoxy include methoxy, ethoxy, isopropoxy, and the like.
- the alkyl group of the alkoxy is linked to an adjacent moiety through the ether oxygen.
- alkenyl as used herein means a linear or branched hydrocarbon radical from 2 to 12 carbon atoms having at least one carbon-carbon double bond.
- alkenyl include ethenyl, 1-propenyl, 1-butenyl, 2-butenyl, 2- pentenyl, 3-methyl-3-butenyl, 1-hexenyl, 3-heptenyl, l-octenyl, 1-nonenyl, 1-decenyl, 1- undecenyl, 1-dodecenyl, and the like.
- the alkenyl group may be optionally substituted with at least one to three of the substituents as previously recited for the term alkyl.
- alkynyl as used herein means a linear or branched hydrocarbon radical from 2 to 12 carbon atoms having at least one carbon-carbon triple bond. Non- limiting examples include 3-propynyl,
- alkynyl group may be optionally substituted with at least one to three of the substituents as previously recited for the term alkyl.
- aryl refers to a C5-C 14 mono-, bi- or polycarbocyclic aromatic ring system which is optionally substituted by at least one substituent selected from alkyl, lower alkoxy, lower thioalkoxy, halogen, -CO2H, -002(Ci-C 6 ) alkyl, - C(O)C 1 -C 6 alkyl, -OSO 3 H, -OPO 3 H, Or -OC 1 -C 6 alkyl,-0(CH 2 )o-2CF 3 , -O-aryl, -OSO 2 R', nitro, cyano -OH, -SH, -CF 3 , -NR 1 R", -NR 1 SO 2 R", -NR 1 C(O)NR 1 R", -S(O) 1 .
- R 1 , and R" are independently hydrogen, C r C 6 alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, or heteroaralkyl, or N, R' and R" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S.
- aryl include phenyl, naphthyl, indenyl, 2-chlorophenyl, 3-chlorophenyl, 4- chlorophenyl, 2-methylphenyl, 3-methylphenyl, 4-methylphenyl, 2-methoxyphenyl, 3- methoxyphenyl, 4-methoxyphenyl, 2-chloro-3-methylphenyl, 2-chloro-4-methylphenyl, 2- chloro-5-methylphenyl, 3-chloro-2-methylphenyl, 3-chloro-4-methylphenyl, 4-chloro-2- methylphenyl, 4-chloro-3-methylphenyl, 5-chloro-2-methylphenyl, 2,3-dichlorophenyl, 2,5-dichlorophenyl, 3,4-dichlorophenyl, 2,3-dimethylphenyl, 3,4-dimethylphenyl, and the like.
- the aryl group may be optionally substituted with at least one to three "ring system
- aralkyl as used herein means an aryl-alkyl group, in which the aryl and alkyl groups are as previously defined. Linkage to the rest of the molecule may be through either the aryl or alkyl portion of the aralkyl moiety.
- the aralkyl group may be optionally substituted by at least one to three substituents as recited above for alkyl and aryl.
- Non-limiting examples of aralkyl include benzyl, phenethyl, naphthlenylmethyl, tolyl, and the like.
- aralkenyl as used herein means an aryl-alkenyl group in which the aryl and alkenyl groups are as previously defined.
- the aralkenyl group may be optionally substituted with one to three substituents as recited above for aryl and alkenyl.
- Non-limiting examples of aralkenyl include 2-phenethenyl, 2-naphthylethenyl, and the like.
- alkylene refers to a divalent group derived from a linear or branched chain saturated hydrocarbon having from 1 to 10 carbon atoms by the removal of two hydrogen atoms.
- the preferred alkylene refers to a linear or branched hydrocarbon chain diradical having from 1 to 3 carbon atoms.
- Useful alkylene groups have from 1 to 6 carbon atoms (Ci-C ⁇ alkylene).
- alkylene include methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), propylene (-(CH 2 ) 3 -), and the like.
- aroyl means an aryl-C(O)- group in which the aryl group is as previously defined.
- Non-limiting examples of aroyl include benzoyl, 1-naphthoyl, 2- naphthoyl, and the like.
- acyl as used herein means an HC(O)- or alkyl-C(O)- in which the alkyl group is as previously defined. Preferred acyls contain a lower alkyl.
- Non-limiting examples of acyl include formyl, acetyl, propanoyl, 2-methylpropanoyl, butanoyl, and the like.
- aryloxy as used herein means an aryl-O- in which the aryl group is as previously defined. Non-limitng examples of aryloxy include phenoxy, naphthoxy, and the like.
- arylthio as used herein means an aryl-S- in which the aryl group is as previously described.
- Non-limiting examples of arylthio include phenylthio, heptylthio, and the like.
- aralkylthio as used herein means an aralkyl-S- group in which the aralkyl is as previously defined.
- Non-limiting examples of aralkylthio include benzylthio, 2-phenyl-ethanethiol, and the like.
- alkoxycarbonyl as used herein means an alkoxy-C(O)- in which the alkoxy is as previously defined.
- Non-limiting examples of alkoxycarbonyl include methoxycarbonyl, ethoxycarbonyl, and the like.
- aryloxycarbonyl as used herein means an aryl-O-C(O)- group in which the aryl group is as previously described.
- Non-limiting examples of aryloxycarbonyl include phenoxycarbonyl, naphthoxycarbonyl, and the like.
- aralkoxycarbonyl as used herein means an aralkyl-O-C(O)- group in which the aralkyl group is as previously defined.
- Non-limiting examples of aralkoxycarbonyl include benzyloxycarbonyl, and the like
- alkylsulfonyl as used herein means an alkyl-S(O) 2 - in which the alkyl group is as previously defined. Preferred groups are those in which the alkyl group is lower alkyl.
- alkylsulfinyl as used herein means an alkyl-S(O)- group. Preferred groups are those in which the alkyl group is lower alkyl.
- arylsulfonyl as used herein means an aryl-S(O) 2 - group.
- arylsulfinyl as used herein means an aryl-S(O)- group.
- cycloalkyl refers to a saturated cyclic C 3 -Ci 2 alkyl group, where alkyl is as previously defined.
- Non-limitng examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cycloctyl, decalinyl, norpinanyl, or adamantyl.
- the cycloalkyl group may be optionally substituted with at least one of those substituents recited above for alkyl or alkylene.
- Non-limiting examples of substituted cycloalkyl groups include fluorocyclopropyl, 2-iodocyclobutyl, 2,3-dimethylcyclopentyl, 2,2-dimethoxycyclohexyl, 3-phenylcyclopentyl, and the like.
- cycloalkenyl refers to a saturated cyclic C3-C 12 alke ⁇ yl group having at least one carbon-carbon double bond, where alkenyl is as previously defined.
- Nonlimiting examples of cycloalkenyl include cyclopropene, cyclopentene, cyclopenta-1-3-diene, cyclohexene, cycloheptene, cyclohepta-i-4-diene, and the like.
- hydrocarbon chain refers to a linear hydrocarbon of from 1 to 12 carbon atoms.
- halogen or halo, as used herein means fluorine or fluoro, chlorine or chloro, bromine or bromo or iodine or iodo.
- heteroatom means oxygen (O), nitrogen (N), or sulfur (S) as well as sulfoxyl or sulfonyl (S(O) or SO 2 ) unless otherwise indicated.
- heteroaryl as used herein means an aryl group, as previously defined, containing one or more heteroatoms, as previously defined.
- the heteroaryl may be optionally substituted with at least one of the substituents previously recited for "aryl”.
- heteroaryl examples include thienyl, benzothienyl (2-benzothienyl, 3-benzothienyl, and the like), indolizinyl, pyrazinyl, furanyl, benzofuranyl, pyrrolyl, pyridyl, pyrimidinyl (2-pyrimidinyl, 4-pyrimidinyl, and the like), imidazolyl (1-imidazolyl, 2- imidazolyl, and the like), benzimidazolyl (l-benzimidazolyl, 2-benzimidazolyl, and the like), triazolyl (1-triazolyl, 3-triazolyl, and the like), isothiazolyl, pyrazolyl (l-pyrazolyl, 3- pyrazolyl, 4-pyrazolyl, and the like), oxazolyl (2-oxazolyl, 4-oxazolyl, and the like), benzoxazolyl (2-benzoxazolyl,
- heterocycle means a saturated mono-, bi- or polycyclic ring containing one or more heteroatoms selected from N, O, and S.
- the heterocycle may be optionally substituted with at least one of those substituents recited above for alkyl.
- Non-limiting examples of heterocycle include piperidinyl, pyrrolidinyl, I- piperazinyl, 2-piperazinyl, 2-morpholinyl, 4-morpholinyl, piperazinyl, azetidinyl, aziridinyl, thietanyl, and the like.
- heterocyclenyl means a non-aromatic monocyclic or multicyclic ring system of about 3 to about 12 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is/are element(s) other than carbon, for example nitrogen, oxygen, or sulfur atoms, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond.
- aza, oxa, or thia before heterocyclenyl means that at least a nitrogen, oxygen, or sulfur atom, respectively, is present as a ring atom.
- Non-limiting examples of heterocyclenyl include 1 ,2,3,4-tetrahydropyridine, 2-pyrrolinyl, 2-imidazolinyl, 1 ,2-dihydropyridyl, and the like.
- heteroarylkyl means heteroaryl-alkyl, in which heteroaryl and alkyl are both as previously defined. Linkage to the rest of the molecule can be either through the heteroaryl or the alkyl portion of the heteroaralkyl moiety.
- the heteroaralkyl may be optionally substituted with at least one of those substituents previously recited for alkyl and heteroaryl.
- Nonlimiting examples of heteroarylalkyl include 2-propyl-pyridine, 3,4-methyl-1H-pyrrole, and the like.
- heteroarylkenyl means heteroaryl-alkenyl, in which heteroaryl and alkenyl are both as previously defined. Linkage to the rest of the molecule can be either through the heteroaryl or the alkenyl portion of the heteroaralkenyl moiety.
- the heteroaralkenyl may be optionally substituted with at least one of those substituents previously recited for alkenyl and heteroaryl.
- Non-limiting examples of heteroaralkenyl include 2-(pyrid-3-yl)ethenyl, 2-(quinolin-3-yl)ethenyl, and the like.
- heterocycloalkyl as used herein means heterocycle-alkyl, in which the heterocycle and the alkyl are both as previously defined. Linkage to the rest of the molecule can be either through the heterocycle or the alkyl portion of the heterocycloalkyl moiety.
- the heterocycloalkyl may be optionally substituted with at least one of those substituents recited above for alkyl and heterocycle.
- Non-limiting examples of heterocycloalkyl include 2-methyl piperidine, 2-ethyl-5-methyl-pyrrolidine, and the like.
- thioalkyl or “alkylthio” means an alkyl-S- in which the alkyl group is a previously defined.
- the alkyl is linked to an adjacent moiety through the sulfinyl moiety.
- thioalkyl include methylthio, ethylthio, isopropylthio, and the like.
- thioalkoxy means an alkoxy-S- in which the alkoxy group is a s previously defined. The alkoxy is linked to an adjacent moiety throught the sulfinyl moiety.
- the term “lower thioalkoxy” means an alkyl-O-S- group in which the alkyl group comprises 1 to 6 carbon atoms.
- thioalkoxy include methoxysulfanyl, ethoxysulfanyl, and the like.
- ring as used herein includes heteroaryl, heterocycle, cycloalkyl and aryl, each as previously defined, and further includes fused, monocyclic, bicyclic, and polycyclic permutations thereof.
- Ring system substituent means a substituent attached to an aromatic or non- aromatic ring system which, for example, replaces hydrogen on the ring system.
- Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, aryl, heteroaryl, aralkyl, aralkenyl, heteroaralkyl, heteroaralkenyl, hydroxy, alkoxy, aryloxy, aralkoxy, acyl, halo, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, aroyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio
- stereoisomer refers to both geometric (e.g., cis and trans isomers) and/or optical isomers (e.g., R and S enantiomers) of a compound of the invention. Racemic, enatiomeric, diastereomeric, and epimeric mixtures of isomers are contemplated by the present invention.
- Compounds of formula 1 , 2, or 3 containing one or more asymmetric carbon atom can exist as two or more stereoisomers. Where a compound of formula 1 , 2, or 3 contains an alkenyl or alkenylene group, geometric cis/trans isomers are possible.
- tautomeric isomerism ('tautomerism') can occur.
- This can take the form of proton tautomerism in compounds of formula 1 , 2, or 3 containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism. Accordingly, included within the scope of the present invention are all stereoisomers and tautomeric forms of the compounds of formula 1 , 2, or 3, including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallization.
- enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
- HPLC high pressure liquid chromatography
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula 1 , 2, or 3 contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula 1 , 2, or 3 contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantio
- Chiral compounds of the invention may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- racemate as used herein, is meant to include both the racemic compound wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts and the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each containing the single enantiomer.
- Such mixtures may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994).
- compound of the invention or “compounds of the invention” includes the compound itself as well as pharmaceutically acceptable salts, esters, amides, hydrates, or stereoisomers thereof.
- patient or “subject” means all animals and mammals, including humans. Examples of patients or subjects include humans, cows, dogs, cats, goats, sheep, pigs, and rabbits.
- phrases "effective amount” and "therapeutically effective amount” mean that amount of a compound of Formula 1 , 2, or 3, and other pharmacological or therapeutic agents described below, that will elicit a biological or medical response in a tissue, system, animal, or mammal that is being sought by the administrator (such as a researcher, doctor, or veterinarian) which includes alleviation of the symptoms of the condition or disease being treated and the prevention, slowing or halting of progression of one or more conditions, for example vascular conditions such as hyperlipidemia, atherosclerosis, hypercholesterolemia, hypertriglyceridemia, sitosterolemia, vascular inflammation, and the like.
- vascular conditions such as hyperlipidemia, atherosclerosis, hypercholesterolemia, hypertriglyceridemia, sitosterolemia, vascular inflammation, and the like.
- a "therapeutically effective amount” will vary from subject to subject and will be determined on a case by case basis. Factors to consider include, but are not limited to, the subject being treated, weight, health, and
- a pharmaceutically acceptable salt, ester, amide, hydrate, or stereoisomer refers to those acid addition salts, base addition salts, esters, amides, hydrates, and stereoisomers (optical, geometric, and tautomeric) of the compounds of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of patients without undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compounds of the invention.
- a pharmaceutically acceptable salt refers to the relatively nontoxic, inorganic and organic acid addition or base salts of compounds of the invention.
- salts can be prepared in situ during the final isolation and purification of the compounds or by separately reacting the purified compound in its free form with a suitable organic or inorganic acid or base and isolating the salt thus formed.
- anionic or acid addition salts include acetate, aspartate, besylate, bicarbonate, carbonate, camysylate, citrate, edisylate, fumarate, gluconate, hydrobromide, bromide, hydrochloride, chloride, D-lactate, L-lactate, malate, mesylate, pamoate, phosphate, succinate, sulphate, D-tartrate, L-tartrate, benzoate, gluceptate, glucuronate, hibenzate, isethionate, malonate, methylsulphate, 2-napsylate, nicotinate, nitrate, orotate, stearate, tosylate, adipate, arabogalactanesulphate, ascorbate
- the free base form may be regenerated by contacting the salt form with a base. While the free base may differ from the salt form in terms of physical properties, such as solubility, the salts are equivalent to their respective free bases for the purposes of the present invention.
- Representative cationic or base salts include calcium, choline, magnesium, potassium, sodium, aluminum, ammonium, quaternary ammonium, and amine cations including tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, and the like, arginine, benzathine, diethylamide, diolamine, glycine, lysine, meglumine, olamine, tromethamine (Tris), 2- amino-2-methylpropan-1-ol, benethamine, erbumine (tert-butylamine), epolamine (hydroxyethylpyrrolidine), ethylenediamine, hydrabamine, morpholine, piperazine, procaine, silver, trolamine, zinc, adenine, arginine, cytosine, glucosamine, guanidine, guanine, nic
- esters of the compounds of the invention include C 1 -C 6 alkyl esters wherein the alkyl group is a linear or branched chain. Acceptable esters also include C 5 -C 7 cycloalkyl esters as well as aralkyl esters such as, but not limited to, benzyl. C 1 -C4 alkyl esters are preferred. Esters of the compounds of the present invention may be prepared according to conventional methods.
- Examples of pharmaceutically acceptable, non-toxic amides of the compounds of the invention include amides derived from ammonia, primary (Ci-Ce)alkyl amines and secondary di-(CrC6)alkyl amines wherein the alkyl groups are linear or branched chain.
- the amine may also be in the form of a 5- or 6-membered heterocycle containing one nitrogen atom.
- Amides derived from ammonia, CrC 3 alkyl primary amines and C 1 -C 2 dialkyl secondary amines are preferred.
- Amides of the compounds of the invention may be prepared according to conventional methods. Certain compounds of the present invention can exist in unsolvated form as well as solvated form including hydrated form. In general, the solvated form including hydrated form is equivalent to the unsolvated form and is intended to be encompassed within the scope of the present invention.
- prodrugs are intended to include any covalently bonded carrier which releases the active parent drug according to Formula 1 , 2, or 3, in vivo. Further, the term “prodrug” refers to compounds that are transformed in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood.
- prodrug refers to compounds that are transformed in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood.
- prodrugs include acetates, formates, benzoate derivatives of alcohols, and amines present in compounds of Formula 1, 2, or 3.
- the compounds of the present invention are suitable to be administered to a patient or subject for the treatment of hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, and atherosclerosis.
- the compounds of the present invention can be administered to a patient/subject alone, or with another compound of the invention, or as part of a pharmaceutical composition.
- a pharmaceutical composition of the invention contains at least one compound of the invention and at least one pharmaceutically acceptable carrier, diluent, solvent or vehicle.
- the pharmaceutically acceptable carrier, diluent, solvent or vehicle may be any such carrier known in the art including those described in, for example, Remington's Pharmaceutical Sciences, Mack Publishing Co., (A. R. Gennaro edit. 1985).
- a pharmaceutical composition of the invention may be prepared by conventional means known in the art including, for example, mixing at least one compound of the invention with a pharmaceutically acceptable carrier.
- the compounds, compositions, and treatments of the present invention can be administered by any suitable means which produce contact of these compounds with the site of action in the body, for example, in the plasma, liver, rectum, or small intestine of an animal or mammal. Compositions of compounds of the invention are contemplated herein.
- a composition of the invention can be administered to a patient/subject either orally, rectally, parenterally (intravenously, intramuscularly, or subcutaneously), intracisternally, intravaginally, intraperitoneally, intravesically, locally (powders, ointments, or drops), or as a buccal or nasal spray.
- compositions suitable for parenteral injection may comprise physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions.
- suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil), and injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
- compositions may also contain additives such as preserving, wetting, emulsifying, and dispensing agents.
- Prevention of the action of microorganisms can be ensured by various antibacterial and antifungal agents, for example, parabens, chlorob ⁇ tanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example sugars, sodium chloride, and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is admixed with at least one (a) inert customary excipient (or carrier) such as sodium citrate or dicalcium phosphate; (b) fillers or extenders, as for example, starches, lactose, sucrose, glucose, mannitol, and silicic acid; (c) binders, as for example, carboxymethylcellulose, alignates, gelatin, polyvinylpyrrolidone, sucrose, and acacia; (d) humectants, as for example, glycerol; (e) disintegrating agents, as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (f) solution retarders, as for example paraffin; (g) absorption accelerators, as for example, quaternary ammonium compounds; (h) wetting agents, as for example, cetyl alcohol and glycerol monostearate; (a) inert
- compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethyleneglycols, and the like.
- Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells, such as enteric coatings and others well-known in the art. They may contain opacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedding compositions which can be used are polymeric substances and waxes. The active compounds can also be in micro- encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-butyleneglycol, dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil and sesame oil, glycerol, tetrahydrofurfuryl alcohol, polyethyleneglycols and fatty acid esters of sorbitan or mixtures of these substances, and the like.
- inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate,
- compositions include additives, such as, for example, wetting agents, emulsifying and the pending agents, sweetening, flavoring, and perfuming agents, or mixtures thereof.
- Suspensions in addition to the active compounds, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances.
- compositions for rectal administrations are preferably suppositories which can be prepared by mixing the compounds of the present invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethyleneglycol, or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore, melt in the rectum or vaginal cavity and release the active component.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethyleneglycol, or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore, melt in the rectum or vaginal cavity and release the active component.
- Dosage forms for topical administration of a compound of this invention include ointments, powders, sprays, and inhalants.
- the active component is admixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants as may be required.
- Ophthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
- the compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 2,000 mg per day, preferably about 5 to about 250 mg. per day.
- dosage levels in the range of about 0.1 to about 2,000 mg per day, preferably about 5 to about 250 mg. per day.
- a dosage in the range of about 0.01 to about 100 mg per kilogram of body weight per day is preferable.
- the specific dosage used can vary from patient to patient.
- the dosage can depend on a numbers of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used. The determination of optimum dosages for a particular patient is well known to those skilled in the art.
- treating refers to curative, palliative and prophylactic treatment, including reversing, ameliorating, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- the compounds of the invention may be used either alone or in combination with another pharmaceutically active agent described herein, in the treatment of the following diseases/conditions: dyslipidemia, hypercholesterolemia, hypertriglyceridemia, atherosclerosis, peripheral vascular disease, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, diabetes and vascular complications of diabetes, obesity, unstable angina pectoris, Alzheimer's Disease, BPH, osteoporosis, cerebrovascular disease, coronary artery disease, ventricular dysfunction, cardiac arrhythmia, pulmonary vascular disease, renal-vascular disease, renal disease, vascular hemostatic disease, autoimmune disorders, pulmonary disease, sexual dysfunction, cognitive dysfunction, cancer, organ transplant rejection, psoriasis, endometriosis, and macular degeneration.
- a combination of the invention may be part of a pharmaceutical composition further containing a pharmaceutically active carrier, diluent, solvent or vehicle, each
- a suitable pharmaceutically active agent examples include a CETP inhibitor, a PPAR- activator, an MTP/Apo B secretion inhibitor, HDL-cholesterol raising agent, triglyceride lowering agent, a cholesterol synthesis inhibitor, a cholesterol modulating agent, a fibrate, niacin, an ion-exchange resin, an antioxidant, an ACAT inhibitor, or bile acid sequestrant; an anti-hypertensive agent; an acetylcholine esterase inhibitor, an anti-diabetic compound, an anti-obesity compound, a thyromimetic agent, an anti- resorptive agent, an anti-osteoporosis agent, an antihypertensive agent, or a drug for the treatment of Alzheimer's disease.
- Specific examples of each of these agents include those known in the art as well as those specified below.
- both the compounds of the invention and the other drug therapies are administered to mammals by conventional methods.
- the following discussion more specifically describes the various combination aspects of this invention.
- CETP cholesterol ester transfer protein
- HDL high density lipoprotein
- LDL low density lipoprotein
- VLDL very low density lipoprotein
- chylomicrons may be used.
- the effect of a CETP inhibitor on lipoprotein profile is believed to be antiatherogenic. Such inhibition may be determined by means known in the art (e.g., Crook et al. Arteriosclerosis 10, 625, 1990; U.S. Pat. No. 6,140,343).
- suitable CETP inhibitors include, but are not limited to, those described in U.S. Patent Nos. 6,197,786, 6,723,752 and 6,723,753.
- CETP inhibitors include the following compounds: [2R, 4S]4-[(3,5- bis-trifluoromethyl-benzyl)-methoxycarbonyl-amino]-2-ethyl-6-trifluoromethyl-3,4- dihydroxycarbonyl-amino]-2-ethyl-6-trifluoromethyl-3,4-dihydro-2H-quinoline-1- carboxylic acid ethyl ester (TorcetrapibTM), and 3- ⁇ [3-(4-Chloro-3-ethyl-phenoxy)- phenyl]-[3-(1 ,1 ,2,2-tetrafluoro-ethoxy)-benzyl]-amino ⁇ -1 ,1 ,1-trifluoro-propan-2-ol.
- an appropriate dosage form such as one comprising (1) a solid amorphous dispersion comprising a cholesteryl ester transfer protein (CETP) inhibitor and an acidic concentration-enhancing polymer; and (2) an acid-sensitive HMG-CoA reductase inhibitor, may be necessary.
- This dosage form is more fully described in USSN 10/739,567.
- PPAR peroxisome proliferator activated receptor
- PPAR-alpha Three mammalian PPARs have been isolated and termed PPAR-alpha, PPAR-gamma, and PPAR-beta (also known as NUC1 or PPAR-delta). These PPARs regulate expression of target genes by binding to DNA sequence elements, termed PPAR response elements. These elements have been identified in the enhancers of a number of genes encoding proteins that regulate lipid metabolism suggesting that PPARs play a pivotal role in the adipogenic signaling cascade and lipid homeostasis.
- PPAR-gamma receptors are associated with regulation of insulin sensitivity and blood glucose levels.
- PPAR- ⁇ activators are associated with lowering plasma triglycerides and LDL cholesterol.
- PPAR- ⁇ activators have been reported to both increase HDL-C levels and to decrease LDL-C levels.
- activation of PPAR- ⁇ alone, or in combination with the simultaneous activation of PPAR- ⁇ and/or PPAR-gamma may be desirable in formulating a treatment for dyslipidemia in which HDL is increased and LDL lowered.
- PPAR-activation is readily determined by those skilled in the art by the standard assays (e.g. US 2003/0225158 and US 2004/0157885).
- Suitable PPAR-activator compounds include, but are not limited to, those described in US 2003/0171377, US 2003/0225158, US 2004/0157885, and U.S. Pat. No. 6,710,063. Additional examples of useful PPAR-activator compounds include the following compounds: [5-Methoxy-2- methly-4-(4'-trifluoromethly-biphenyl-4ylmethylsulfanyl)-phenoxy]-acetic acid; [5-Methoxy-2- methly-4-(4'-trifluoromethly-biphenyl-4ylmethylsulfanyl)-phenoxy]-acetic acid; [5-
- MTP/Apo B secretion inhibitor microsomal triglyceride transfer protein and/or0 apolipoprotein B secretion
- Any MTP/Apo B secretion inhibitor known in the art which inhibits the secretion of triglycerides, cholesteryl ester and phospholipids may be used. Such inhibition may be readily determined according to standard assays (e.g., Wetterau, J. R. 1992; Science 258:999).
- suitable a MTP/Apo B secretion inhibitor include, but are not limited to, imputapride (Bayer) as well as those described in WO 96/40640 and WO5 98/23593.
- Any ACAT inhibitor known in the art that inhibits the intracellular esterification of dietary cholesterol by the enzyme acyl CoA; cholesterol acyltransferase may be used. Such inhibition may be determined readily according to standard assays, such as the method of Heider et al. described in Journal of Lipid Research. 24:1127 (1983).
- suitable ACAT inhibitors include, but are not limited to, those described in U.S. Pat. No. 5,510,379 (carboxysulfonates),WO 96/26948 and WO 96/10559 (urea derivatives). Additional examples include Avasimibe (Pfizer), CS-505 (Sankyo) and Eflucimibe (EIi Lilly and Pierre Fabre).
- Any lipase inhibitor e.g., pancreatic lipase inhibitor, a gastric lipase inhibitor
- Any lipase inhibitor known in the art that inhibits the metabolic cleavage of dietary triglycerides into free fatty acids and monoglycerides may be used.
- lipase inhibition activity may be readily determined according to standard assays (e.g., Methods Enzymol. 286: 190-231).
- Examples of a suitable lipase inhibitor include, but are not limited to, lipstatin,
- Additional examples include N-3- trifluoromethylphenyl-N'-- 3-chloro-4'-trifluoromethylphenylurea, and the various urea derivatives related thereto, U.S. Pat. No. 4,405,644; esteracin (U.S. Pat. Nos. 4,189,438 and 4,242,453); and cyclo-O, O'-[(1 ,6-hexanediy!)-bis-(iminoc- arbonyl)]dioxime, and the various bis(iminocarbonyl)dioximes related thereto (Petersen et al., Liebig's Annalen, 562, 205-229 (1949).
- Any bile acid sequestrant known in the art may be used.
- suitable bile acid sequestrants include, but are not limited to, Welchol ® , Colestid ® , LoCholest ® , Questran ® and fibric acid derivatives, such as Atromid ® , Lopid ® and Tricor ® '
- a compound of the invention can be used in combination with an anti-diabetic compound, i.e. any compound (e.g. insulin) used in the treating diabetes (especially Type II), insulin resistance, impaired glucose tolerance, or the like, or any of the diabetic complications such as neuropathy, nephropathy, retinopathy or cataracts.
- an anti-diabetic compound examples include, but are not limited to, a glycogen phosphorylase inhibitor, an aldose reductase inhibitor, a sorbitol dehydrogenase inhibitor, a glucosidase inhibitor, and an amylase inhibitor.
- glycogen phosphorylase inhibitor known in the art that inhibits the bioconversion of glycogen to glucose-1 -phosphate which is catalyzed by the enzyme glycogen phosphorylase may be used. Such glycogen phosphorylase inhibition activity may be readily determined according to standard assays (e.g., J. Med. Chem. 41 (1998) 2934-2938). A variety of glycogen phosphorylase inhibitors are known to those skilled in the art including those described in WO 96/39384 and WO 96/39385. Any aldose reductase inhibitor known in the art that inhibits the bioconversion of glucose to sorbitol catalyzed by the enzyme aldose reductase.
- Aldose reductase inhibition may be readily determined according to standard assays (e.g., J. Malone, Diabetes, 29:861-864 (1980). "Red Cell Sorbitol, an Indicator of Diabetic Control”). Any sorbitol dehydrogenase inhibitor known in the art that inhibits the bioconversion of sorbitol to fructose catalyzed by the enzyme sorbitol dehydrogenase may be used. Such sorbitol dehydrogenase inhibitor activity may be readily determined according to standard assays (e.g., Analyt. Biochem (2000) 280: 329-331).
- a suitable sorbitol dehydrogenase inhibitor examples include, but are not limited to, those described in U.S. Patent Nos. 5,728,704 and 5,866,578.
- Such glucosidase inhibition activity may be readily determined by those skilled in the art according to standard assays (e.g., Biochemistry (1969) 8: 4214).
- a generally preferred glucosidase inhibitor includes an amylase inhibitor.
- amylase inhibitor known in the art that inhibits the enzymatic degradation of starch or glycogen into maltose may be used. Such amylase inhibition activity may be readily determined by those skilled in the art according to standard assays (e.g., Methods Enzymol. (1955) 1 : 149).
- Other preferred glucosidase inhibitors include, but are not limited to, acarbose and the various amino sugar derivatives related thereto (U.S. Pat. Nos. 4,062,950 and 4,174,439); adiposine (U.S. Pat. No.
- Examples include, but are not limited to, tendamistat and the various cyclic peptides related thereto (U.S. Pat. No. 4,451 ,455); AI-3688 and the various cyclic polypeptides related thereto (U.S. Pat. No. 4,623,714); and trestatin, consisting of a mixture of trestatin A, trestatin B and trestatin C and the various trehalose-containing aminosugars related thereto, (U.S. Pat. No. 4,273,765).
- biguanides e.g., metformin
- insulin secretagogues e.g., sulfonylureas and glinides
- glitazones e.g., non-glitazone PPAR. gamma
- agonists PPAR.beta.
- agonists agonists, inhibitors of DPP-IV, inhibitors of PDE5, inhibitors of GSK-3, glucagon antagonists, inhibitors of f-1 ,6-BPase (Metabasis/Sankyo), GLP-1/analogs (AC 2993, also known as exendin-4), insulin and insulin mimetics (Merck natural products), PKC-beta inhibitors, and AGE breakers.
- a compound of the invention can be used in combination with any anti-obesity agent known in the art.
- Anti-obesity activity may be readily determined according to standard assays known in the art.
- suitable anti-obesity agents include, but are not limited to, phenylpropanolamine, ephedrine, pseudoephedrine, phentermine, .beta..sub.3 adrenergic receptor agonists, apolipoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, MCR-4 agonists, cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (e.g., sibutramine - U.S.
- bombesin agonists e.g., a bombesin agonist
- anorectic agents e.g., a bombesin agonist
- Neuropeptide-Y antagonists thyroxine, thyromimetic agents, dehydroepiandrosterones or analogs thereof, glucocorticoid receptor agonists or antagonists, orexin receptor antagonists, urocortin binding protein antagonists, glucagon-like peptide-1 receptor agonists, ciliary neurotrophic factors (e.g., Axokine.TM.), human agouti-related proteins (AGRP), ghrelin receptor antagonists, histamine 3 receptor antagonists or inverse agonists, neuromedin U receptor agonists, and the like.
- bombesin agonists e.g., a bombesin agonist
- Neuropeptide-Y antagonists e.g., thyroxine, thyromimetic agents, dehydroe
- thyromimetic agent known in the art may also be used in combination with a compound of the invention. Thyromimetic activity may be readily determined according to standard assays (e.g., Atherosclerosis (1996) 126: 53-63). Examples of suitable thyromimetic agents include, but are not limited to, those described in U.S. Pat. Nos. 4,766,121; 4,826,876; 4,910,305; 5,061 ,798; 5,284,971 ; 5,401 ,772; 5,654,468; and 5,569,674.
- a compound of the invention may further be used in combination with an anti- resorptive agent (e.g., progestins, polyphosphonates, bisphosphonate(s), estrogen agonists/antagonists, estrogen, estrogen/progestin combinations, Premarin.RTM., estrone, estriol or 17. alpha.- or 17.beta.-ethynyl estradiol).
- an anti- resorptive agent e.g., progestins, polyphosphonates, bisphosphonate(s), estrogen agonists/antagonists, estrogen, estrogen/progestin combinations, Premarin.RTM., estrone, estriol or 17. alpha.- or 17.beta.-ethynyl estradiol.
- progestins are available from commercial sources and include, but are not limited to: algestone acetophenide, altrenogest, amadinone acetate, anagestone acetate, chlormadinone acetate, cingestol, clogestone acetate, clomegestone acetate, delmadinone acetate, desogestrel, dimethisterone, dydrogesterone, ethynerone, ethynodiol diacetate, etonogestrel, flurogestone acetate, gestaclone, gestodene, gestonorone caproate, gestrinone, haloprogesterone, hydroxyprogesterone caproate, levonorgestrel, lynestrenol, medrogestone, medroxyprogesterone acetate, melengestrol acetate, methynodiol diacetate, norethindrone, no
- Exemplary bone resorption inhibiting polyphosphates include polyphosphonates of the type described in U.S. Pat. No. 3,683,080.
- Preferred polyphosphonates are geminal diphosphonates (also referred to as bis-phosphonates), 6-amino-1-hydroxy-hexylidene-bisphosphonic acid and 1-hydroxy- 3(methylpentylamino)-propylidene-bisphosphonic acid.
- Tiludronate disodium, ibandronic acid, alendronate, resindronate, and zoledronic acid are each especially preferred polyphosphonates.
- the polyphosphonates may be administered in the form of the acid, or of a soluble alkali metal salt or alkaline earth metal salt.
- Hydrolyzable esters of the polyphosphonates are likewise included. Specific examples include, but are not limited to, ethane-1 -hydroxy 1 ,1-diphosphonic acid, methane diphosphonic acid, pentane-1-hydroxy-1 ,1-diphosphonic acid, methane dichloro diphosphonic acid, methane hydroxy diphosphonic acid, ethane-1-amino-1 ,1-diphosphonic acid, ethane-2- amino-1 ,1 -diphosphonic acid, propane-3-amino-1-hydroxy-1 ,1 -diphosphonic acid, propane-N,N-dimethyl-3-amino-1-hydroxy-1,1 -diphosphonic acid, propane-3,3-dimethyl- 3-amino-1-hydroxy-1 ,1 -diphosphonic acid, phenyl amino methane diphosphonic acid, N,N-dimethylamino methane diphosphonic acid, N(2-hydroxyethyl) amino methane di
- an estrogen agonist/antagonist known in the art which bind with the estrogen receptor, inhibit bone turnover and/or prevent bone loss may be used in a combination of the invention. More specifically, an estrogen agonist may be any chemical compound capable of binding to the estrogen receptor sites in mammalian tissue, and mimicking the actions of estrogen in one or more tissue.
- An estrogen antagonist may be any chemical compound capable of binding to the estrogen receptor sites in mammalian tissue, and blocking the actions of estrogen in one or more tissues. Such activities may be readily determined according to standard assays, including estrogen receptor binding assays, and standard bone histomorphometric and densitometer methods (Eriksen E. F. et al., Bone Histomo ⁇ hometry, Raven Press, New York, 1994, pages 1- 74; Grier S.
- Examples of a suitable estrogen agonist/antagonist is 3-(4-(1 ,2-diphenyl-but-1-enyl)-phenyl)-acrylic acid (see Willson et al., Endocrinology, 1997, 138, 3901-3911); tamoxifen (ethanamine, 2-(-4-(1 ,2-diphenyl-1-butenyl)phenoxy)- N.N-dimethyl, (Z)-2-, 2-hydroxy-1 ,2,3-propanetricarboxylate (1 :1)) and related compounds (U.S. Pat. No. 4,536,516); 4-hydroxy tamoxifen (U.S. Pat. No.
- raloxifene (methanone, (6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl)(4-(2-(1- piperidinyl)eth- oxy)phenyl)-hydrochloride)(U.S. Pat. No. 4,418,068); toremifene (ethanamine, 2-(4-(4-chloro-1 ,2-diphenyl-1-butenyl)phenoxy)-N,N-dimethyl- , (Z)-, 2- hydroxy-1,2,3-propanetricarboxylate (1 :1) (U.S. Pat. No.
- centchroman (1-(2- ((4-(-methoxy-2,2, dimethyl-3-phenyl-chroman-4-yl)-phenoxy)-ethyl)-p- yrrolidine)(U.S. Pat. No. 3,822,287); levormeloxifene; idoxifene ((E)-I -(2-(4-(1-(4-iodo-phenyl)-2-phenyl- but-1-enyl)-phenoxy)-ethyl)-pyrro- lidinone (U.S. Pat. No.
- Especially preferred estrogen agonist/antagonists described in U.S. Pat. No. 5,552,412 are: cis-6-(4-fluoro-phenyl)-5-(4-(2-piperidin-1-yl-ethoxy)-phenyl)-5,6,- 7,8- tetrahydro-naphthalene-2-ol; (-)-cis-6-phenyl-5-(4-(2-pyrrolidin-1-yl-ethoxy)-phenyl)- 5,6,7,8-te- trahydro-naphthalene-2-ol (also known as lasofoxifene); cis-6 ⁇ phenyl-5-(4-(2- pyrrolidin-1-yl-ethoxy)-phenyl)-5,6,7,8-tetrah- ydro-naphthalene-2-ol; cis-1-(6'- pyrrolodinoethoxy-3'-pyridyl)-2-
- any anti-osteoporosis agent known in the art may be used in a combination of the invention.
- examples include, but are not limited to, parathyroid hormone (PTH) (a bone anabolic agent); parathyroid hormone (PTH) secretagogues (see, e.g., U.S. Pat. No. 6,132,774), particularly calcium receptor antagonists; calcitonin; and vitamin D and vitamin D analogs.
- antihypertensive agent known in the art may be used in a combination of the invention.
- Antihypertensive activity may be determined according to standard tests (e.g. blood pressure measurements).
- suitable antihypertensive agents include, but are not limited to, (a) amlodipine and related dihydropyridine compounds (US Pat. Nos. 4,572,909 and 5,155,120) such as, but not limited to, amlodipine benzenesulfonate salt (also termed amlodipine besylate (Norvasc ® ))(U.S. Pat. No. 4,879,303) and other pharmaceutically acceptable acid addition salts of amlodipine (U.S. Pat. No.
- calcium channel blockers such as, but not limited to, bepridil (U.S. Pat. No. 3,962, 238 or U.S. Reissue No. 30,577), clentiazem (U.S. Pat. No. 4,567,175), diltiazem (U.S. Pat. No. 3,562), fendiline (U.S. Pat. No. 3,262,977), gallopamil (U.S. Pat. No.
- Inhibitors such as, but not limited to, alacepril (U.S. Pat. No. 4,248,883), benazepril (U.S. Pat. No. 4,410,520), captopril, ceronapril, delapril, enalapril, fosinopril, imadapril, lisinopril, moveltopril, perindopril, quinapril, ramipril, spirapril, temocapril, and trandolapril; (d) angiotensin-ll receptor antagonists such as, but not limited to, candesartan (U.S. Pat. No.
- eprosartan U.S. Pat. No. 5,185,351
- irbesartan irbesartan
- losartan irbesartan
- valsartan irbesartan
- beta-adrenergic receptor blockers beta- or ⁇ - blockers
- acebutolol U.S. Pat. No. 3,857,952
- alprenolol amosulalol
- alpha-adrenergic receptor blockers such as, but not limited to, amosulalol (U.S. Pat. No. 4,217,307), arotinolol (U.S. Pat. No.
- HMGCoA reductase activity may be determined according to standard tests (e.g. blood plasma low density lipoprotein cholesterol (LDL- C) measurements).
- HMGCoA reductase inhibitor agents include, but are not limited to, atorvastatin, simvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, simvastatin, and cerivastatin.
- atorvastatin examples include: U.S. Pat. Nos. 4,681,893, 5,273,995 and 5,969,156.
- rosuvastatin include: U.S. Pat. Nos. 5,260,440 (RE37314), 6,858,618, and 6,894,058.
- a number of patents have issued disclosing cerivastatin and include: U.S. Pat Nos.
- the present invention contains compounds that can be synthesized in a number of ways familiar to one skilled in organic synthesis.
- the following non-limiting reaction schemes illustrate the preparation of the compounds of the present invention. Unless otherwise indicated, all variables in the reaction schemes and the discussions that follow are as defined above. As would be understood by one of skill in the art, individual compounds may require manipulation of the conditions in order to accommodate various functional groups. A variety of protecting groups known to one skilled in the art may be required. Purification, if necessary, may be accomplished on a silica gel column eluted with the appropriate organic solvent system. Also, reverse phase HPLC or recrystallization may be employed.
- Schemes 1- 4 illustrate general methods for the preparation of compounds of the present invention. These illustrations are non-limiting and may be adapted by those skilled in the art for the preparation of compounds not explicitly described. The specific naming of the stereochemistry of the final compounds (e.g. R 1 S etc) will depend on the substituents selected for the variables shown, as those skilled in the art will understand. The use of solid and hashed wedges in the structures depicted will have the meaning well understood by those skilled in the art.
- Scheme 1 illustrates the preparation of variations of formula (1) where Z is an optionally substituted carbon side chain. As shown, aldehydes of structure (1) can undergo hydroxymethylation by treatment with formaldehyde, N-ethyl-benzothiazole bromide and a suitable base to provide compounds of structure (2).
- Scheme 2 illustrates the preparation of variations of formula (1) where Z contains an amine or ether linkage.
- Z contains an amine or ether linkage.
- condensation of hydroxyl acetone (8) with a suitable aldehyde (3) and a suitable amine (4) in the presence of a suitable chiral catalyst such as L-proline afford amino-alcohols of structure (9).
- a suitable chiral catalyst such as L-proline
- Scheme 3 illustrates a variation of Scheme 2 for the construction of structures of formula (I) wherein Z contains a ring system.
- epoxide (16) is treated with amine Ar 2 NH Z tO afford amino alcohol (17) which can then be treated with an electrophile of type (18) to provide structure (19).
- treatment of structure (19) with a suitable base affords structure (20).
- Scheme 4 illustrates the preparation of variations of formula (1) where Z contains an amide linkage.
- oxazolidine (10) from Scheme 2, is subjected to a haloform reaction using sodium bromate (generated in-situ from NaOH and Br 2 ) or a suitable equivalent to provide a carboxylic acid of structure (21).
- Structure (21) can be reacted with various amines in the presence of a suitable coupling reagent such as EDCI to afford structure (22).
- structure (21) can be treated with a suitable reducing agent such as borane to afford alcohols of structure (23).
- Conversion of structure (23) to amines of structure (24) followed by acylation with suitable acid chlorides provides amides of general structure (25).
- Hydrocinnamaldehyde (50.0 g, 0.37moles), paraformaldehyde (11.0 g, 0.37moles), N- ethyl-benzothiazole bromide (16.0 g, 0.067moles) and triethylamine (9.0 ml, 0.067moles) were dissolved in ethanol (500 ml) under nitrogen and heated at 70 0 C overnight. The mixture was cooled to 25 0 C and evaporated, yielding a brown oil. The crude material was purified using column chromatography (10-30% EtOAc/hexane) yielding 1-hydroxy-4-phenyl-butan-2-one (31.0 g, 51%) as an oil which crystallized on standing.
- Example 2 Acetic acid 1-[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3- phenyl-propyl ester
- the resulting compound was purified using column chromatography (30-50% EtOAc:hexane) to provide 5-(1-hydroxy-3-phenyl-propyl)-3,4-bis-(4-methoxy-phenyl)-oxazolidin-2-one (1.4g, 66%) as a mixture of diastereomers at the hydroxyl position.
- Acetic acid 1 R-[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl-propyl ester
- Examples 13 - 27 were prepared using procedures analogous to those described for Examples 10 and 11 above.
- reaction mixture was then was cooled to 25 0 C and adsorbed directly onto silica and purified by column chromatography (10-40% EtOAc:hexane) to afford 4-(4-benzyloxy-phenyl)-5-(1-bromo- 3-phenyl-propyl)-3-(4-fluoro-phenyl)-oxazolidin-2-one (1.4 g, 78%).
- reaction was then cooled to 25 0 C and adsorbed onto silica which was then purified by column chromatography on a 10% w/w KF/Silica column (20% EtOAc: hexane) to afford 4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-(3-phenyl-propyl)- oxazolidin-2-one (0.70 g, 58%).
- Example 28 Prepared according to the method of Example 28 starting from 5S-(1 R-hydroxy-3- phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one [from Example 4].
- 1 H- NMR 500 MHz, d-CDCb) ⁇ 7.4-7.1 (m, 9H), 6.8 (d, 2H), 6.7 (d, 2H), 4.7 (d, 1H), 4.3 (m, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 2.65 (m, 2H), 2.0-1.7 (m, 4H).
- Acetic acid 1-[4-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-3- phenyl-propyl ester 400 mg, 0.74 mmol was dissolved in a mixture of ethanol (6ml) and ethyl acetate (6ml) followed by the addition of cyclohexene (365mg, 4.4mmol) and Pd/C catalyst (40mg). The mixture was refluxed overnight under nitrogen. The reaction was cooled to 25 C and filtered through a pad of celite.
- Step B ⁇ 4-[5-(1-Acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-4-yl]-phenoxy ⁇ - acetic acid methyl ester
- Acetic acid 1 [4-[4-(4-chloromethyl-benzyloxy)-phenyl]-3-(4-fluoro-phenyl)-2-oxo- oxazolidin-5-yl]-3-phenyl-propyl ester
- Step B 3,4,5-Triacetoxy-6-(2,2,2-trichloro-1-imino-ethyl)-tetrahydro-pyran-2-carboxylic acid methyl ester
- reaction mixture was stirred at 0 0 C for 1 hr as and then was concentrated under reduced pressure to a brown oil that was purified by column chromatography (40% ethyl acetate/59.9% hexane/0.1% triethylamine) to provide 3,4,5- triacetoxy-6-(2,2,2-trichloro-1-imino-ethyl)-tetrahydro-pyran-2-carboxylic acid methyl ester (2.57 g, 49 %).
- Step D 6- ⁇ 4-[3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]- phenoxy ⁇ -3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid
- 6- ⁇ 4- [3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]-phenoxy ⁇ - 3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid (0.085 g, 31 %) was obtained as a light yellow foam.
- the oil was redissolved in 100 mL ethanol, cooled to O 0 C, then added sodium borohydride (0.34 g, 9.1mmoles). Stirred at 0 0 C for 60 minutes. Poured solution into 200 mL of 1M aqueous hydrogen chloride at 0 0 C. Extracted with ethyl acetate, washed with brine, dried with magnesium sulfate, filtered and concentrated to an oil. The oil was dissolved in 60 mL anhydrous tetrahydrofuran under a nitrogen atmosphere, added sodium terf-butoxide (2.7 g, 28mmoles) and stirred at ambient temperature for 1 hour.
- Step B 3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxa2olidinyl-2,2'-dione
- Example 37 3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxazolidiny!-2,2'- dione.
- Step B 4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[4-(4-fluoro-phenyl)-morpholin-2-yl]- oxazolidin-2-one
- Example 40 4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one
- Step A 1 -(4-Benzyloxy-phenyl)-2-hydroxy-1 -(4-methoxy-phenylamino)-5-phenyl-pentan-3-one
- Step B ((4-fluoro-phenyl)- ⁇ 2-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5S-yl]- 2-hydroxy-ethyl ⁇ -amino)-acetic acid ethyl ester
- Example 46 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phenyl-propionyl)- pyrrolidin-2-one
- Step B 1-(4-Fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carboxylic acid
- Step D 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phe ⁇ yl-propionyl)-pyrrolidin-2-one
- Example 47 1-(4-fluoro-phenyl)-4R-(1-hydroxy-3-phenyl-propyl)-5S-(4-methoxy- phenyl)-pyrrolidin-2-one
- Example 48 1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide
- Examples 51 - 82 were prepared as a mixture of diastereomers using procedures analogous to those described for Examples 33 and 34 above.
- Example 84 3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S- carboxylic acid (4-fluoro-benzyl)-methyl-amide
- Step B 3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid (4- fluoro-benzyl)-methyl-amide
- Selected compounds were also evaluated for in vivo inhibition of cholesterol synthesis as described below.
- Male Sprague-Dawley rats (200-400 gm) are maintained in a room with a 12 hour light cycle/12 hour dark cycle for at least one week prior to testing. On the test day the rats are fasted for 8 hours prior to dosing to synchronize initiation of eating once food is presented.
- Test drug or vehicle is administered by oral gavage approximately 1 hour prior to the start of the dark cycle.
- One group of animals is dosed with vehicle and given standard chow (chow control), one group is dosed with vehicle and given the same diet supplemented with 5.5% peanut oil, 1.5% cholesterol, and 0.4% cholic acid (PCC diet; PCC control), and the remaining animals are dosed with test agents in vehicle and are given the PCC diet.
- Animals are given access to their assigned diet ad libitum starting 30 minutes after dosing until study termination 16 hours after drug administration. Animals are euthanized with CO 2 , and blood is collected by cardiac puncture for plasma total cholesterol analysis.
- Total plasma cholesterol concentrations are ⁇ 60-90 mg/dL in chow controls and increase to ⁇ 175-240 mg/dL in PCC control animals.
- the difference in plasma cholesterol between the chow control group and the PCC control group is the elevation caused by the PCC diet.
- the dose that reduces by 50% the elevation in plasma cholesterol in animals on the PCC diet is the ED50.
- the compounds of the present invention including those exemplified herein and all compounds of Formulas l-la-lai, hereafter referred to as "compound(s)" can be administered alone or in combination with one or more therapeutic agents. These include, for example, other agents for treating, preventing or controlling dyslipidemia, non-insulin dependent diabetes mellitus, obesity, hyperglycemia, hypercholesteremia, hyperlipidemia, atherosclerosis, hypertriglyceridemia, or hyperinsulinemia.
- the ingredients are combined via melting and then poured into molds containing 2.5 g total weight.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Novel oxazolidinones and pharmaceutical compositions are described, as are methods of using such compounds and compositions to treat subjects, including humans, suffering from hyperlipidemia, hypercholeserolemia, and atherosclerosis.
Description
OXAZOLIDINONES AS CHOLESTEROL ABSORPTION INHIBITORS
FIELD OF THE INVENTION
This invention relates to a group of novel oxazolidinones. These compounds inhibit the cholesterol transporter NPC1 L1 and thus are useful as hypocholesterolemic agents and in the treatment and prevention of atherosclerosis.
BACKGROUND OF THE INVENTION
Atherosclerotic coronary heart disease represents the major cause of death and cardiovascular morbidity in the western world. Risk factors for atherosclerotic coronary heart disease include hypertension, diabetes mellitus, family history, maleness, smoking and elevated plasma cholesterol. Elevated plasma cholesterol and lipoprotein are significant atherosclerotic risk factors. Thus, a causative link between elevated plasma cholesterol levels, atherosclerosis, and coronary heart disease has been firmly established. Harwood et al., 34 J. Lipid Research 377-378 (1993). More specifically, a total cholesterol level in excess of 225-250 mg/dl is associated with significant elevation of risk.
An increase in low density lipoprotein (LDL) concentration is correlated with increased atherosclerosis. The liver is the major organ responsible for cholesterol biosynthesis and catabolism and the site of synthesis and secretion of very low-density lipoprotein (VLDL) which are subsequently metabolized to LDL. When cholesterol absorption in the intestines is reduced, by whatever means, less cholesterol is delivered to the liver. The consequence of this action is decreased hepatic lipoprotein (VLDL) production and an increase in the hepatic clearance of plasma cholesterol, mostly as LDL. Thus, the net effect of an inhibition of intestinal cholesterol absorption is a decrease in plasma cholesterol levels.
Several 2-azetidinone compounds have been reported as being useful in lowering cholesterol and/or in inhibiting the formation of cholesterol-containing lesions in mammalian arterial walls: U.S. Patent No. 5,688,785 describes 2-azetidinone compounds wherein the 3-position substituent is arylalkylene or arylalkenylene wherein the alkylene or alkenylene portion is interrupted by a hetero atom, phenylene or cycloalkylene; U.S. Patent No. 5,698,548 describes 2-azetidinone compounds wherein the 3-position substituent is an arylalkylspirocyclic group; U.S. Patent Reissue No. RE37721 describes 2-azetidinone compounds wherein the 3-position substituent is an
arylalkylene group substituted in the alkylene portion by a hydroxy group; US 2003/0105028 describes glucose-derived conjugates of 2-azetidinone compounds wherein the 1-position substituent is a hydroxyl-substituted phenyl group and the 4- position substituent is a hydroxyphenyl group; and U.S. Pat. No. 5,756,470 discloses 2- azetidinones having an aryl group at the 4-position which is substituted with a hydroxyl and a glucuronide group. WO 2006/102674 discloses certain substituted azetidinones, as well as certain substituted oxazolidinones, useful as cholesterol absorption inhibitors. Kvcernø et al. Angew. Chem. 43 Int. Ed., pp 4653-4656 (2004), discloses certain substituted oxazolidinones useful as cholesterol absorption inhibitors. At least one substituted azetidinone, ezetimibe, is currently commercially available for the treatment of hypercholesterolemia. The effectiveness of available antilipidemic therapies is limited, in part because of poor patient compliance due to unacceptable side effects and tolerability as well as minimal efficacy or potency. Furthermore, certain drug products may not be advantageous to all patients because of genetic polymorphisms regarding cholesterol biosynthesis. Furthermore, potential side effects of absorption of certain azetidinones may be detrimental. For these reasons, there is a continuing need for novel antilipidemic agents that may be used alone or in combination with other agents that provide increased efficacy and tolerability with decreased toxicity. This invention relates to novel chemical compounds having pharmacological activity, pharmaceutical compositions including these compounds and pharmaceutical methods of treatment using the compounds.
SUMMARY OF THE INVENTION
The present invention provides a compound having a Formula (I),
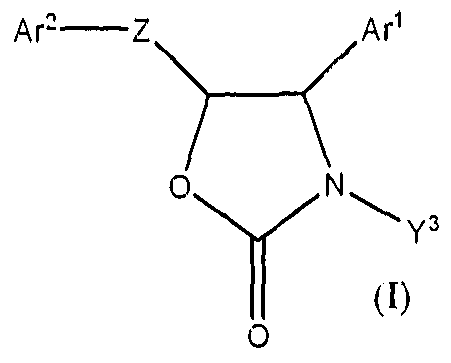
or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein Ar1 and Ar2 are each independently aryl or heteroaryl, optionally substituted; Y3 is alky!, aryl, aralkyl, heteroalkyl or heteroaralkyl; optionally substituted; Z is -O-CR"R"'CH(OR')-; -NR'-CR"R"'-CH(OR')-; -CR"R"'-CR"R"'-CH(OR')-;
-CR"R"'-NR'-C-; or is Il
O selected from

wherein Vv/w-" indicates the points of attachment; R' is H; or lower alkyl, optionally substituted; R" and R"' are each independently H; lower alkyl, optionally substituted, or flourine; W is O or NR'; and n is 0, 1 or 2.
The present invention further provides a compound having a Formula (Ia),
-A-

or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein Z is -O-CR"R"'CH(OR')-; -NR'-CR"Rm-CH(OR>;
-CR"R"'-CR"R"'-CH(OR')- -CR"R"'-NR'-C-; or is
O selected from

R1, R2 and R3 are each independently halo; -OR', -COR', -COOR', -CONR'R"; CH2NR1R"; CH2NR1C(O)R"; C1-C12 alkyl, aryl, or heteroaryl; optionally substituted; S(O)nR', P(O)nR', OG, CR'R"G, S(O)nG, NR1G or SG; G is is selected from the group consisting of hydrogen,


wherein "ΛW" indicates the point of attachment and wherein R5, R6, R7, R8, R9, and R10 are each independently selected from the group consisting of hydrogen, Ci-C6 alkyl, Ci-C6 aralkyl, -C(O)Ci-C6 alkyl, -C(O)aryl, and aryl; and R11 is selected from the group consisting of hydrogen, hydroxy, Ci.Cβ alkyl, -OCLC6 alkyl, and NR'R"; R' is H; or lower alkyl, optionally substituted; R" and R'" are each independently H; lower alkyl, optionally substituted, or flourine; W is O or NR'; and n is 0, 1 or 2.
The present invention further provides inter alia the following compounds: 3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one;
Acetic acid 1 -[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl- propyl ester;
Acetic acid 1 S-[3,4-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester; 5S-(1 R-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one;
5S-(1S-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one; 4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin- 2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2- one;
4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one; 3-(4-fIuoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-pheny)-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- benzoic acid methyl ester;
4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-pheπyl-propyl)-2-oxo-oxazolidin-4R-yl]- benzoic acid;
4-[3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- benzoic acid methyl ester; 4R-(2,4-dihydroxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl-propyl)- oxazo)idin-2-one;
4R-(2,4-dihydroxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1-hydroxy-3-phenyl-propyl)-3-(3-trifluoromethyl- phenyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(2-hydroxy-phenyl)-5S-(1R-hydroxy-3-pheny!-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(2-hydroxy-4-methoxy-phenyl)-5S-(1R-hydroxy-3-phenyl- propyl)-oxazolidin-2-one; 3-(4-f luoro-phenyl)-4R-(2-hyd roxy-4-methoxy-pheny l)-5S-( 1 S-hydroxy-3-phenyl- propyl)-oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-3-(4-trifluoromethyl- phenyl)-oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1S-hydroxy-3-phenyl-propyi)-3-(4-trifluoromethyl- phenyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(3-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(3-hydroxy-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(3-fluoro-pheny!)-4R-(4-hydroxy-pheny!)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one; 3-(3-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one;
3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one;
{4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- phenoxyj-acetic acid;
1-(4-{4-[5S-(1R-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-
4R-yl]-phenoxymethyl}-benzyl)-1-azonia-bicyclo[2.2.2]octane chloride;
6-{4-[3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]- phenoxy}-3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid; 5S-[2-(4-fluoro-phenoxy)-1 R-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
5S-[2-(4-fluoro-phenoxy)-1S-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-5S-[2-(4-fluoro-phenylamino)-1-hydroxy-ethyl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-t5,5']bioxazolidinyl-2,2'-dione;
3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy- ρhenyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one;
4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one; 5S-[1-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
5S-[1S-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fIuoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
5S-(1 ,1-difluoro-3-phenyl-propyl)-4R-(4-hydroxy-phenyl)-3-(4-methoxy-phenyl)- oxazolidin-2-oπe;
((4-flυoro-phenyl)-{2-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin-
5S-yl]-2-hydroxy-ethyl}-amino)-acetic acid ethyl ester; 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phenyl-propionyl)-pyrrolidin-2- one;
1-(4-fluoro-phenyl)-4R-(1-hydroxy-3-phenyl-propyl)-5S-(4-methoxy-phenyl)- pyrro!idin-2-one;
1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide;
1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide;
3-(4-Fluoro-phenyl)-4R-(3'-hydroxy-biphenyl-4-yl)-5S-(1-hydroxy-3-phenyl- propyl)-oxazolidin-2-one; 3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(3-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxyj-benzoic acid methyl ester;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(2-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(2-methanesulfonyl-phenoxy)-ethyl]-4-(4- hydroxy-phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1-hydroxy-2-(4-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one; 5-[2-(2,4-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3-Fluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(2,3-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3,4-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(4-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-t2-(4-Chloro-phenoxy)-1-hydroxy-ethyl]-3-(4-fiuoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one; 5-[2-(4-tert-Butyl-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3-tert-Butyl-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyi)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1-hydroxy-2-(3-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one;
2-{2-[3-(4-Flυoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzoic acid methyl ester;
5-[2-(3,4-Dichloro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxa∑olidin-2-one; 3-{2-(3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-y)]-2-hydroxy- ethoxy}-benzonitrile;
3-(4-Flυoro-phenyl)-5-[1-hydroxy-2-(naphthalen-1-yloxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(4-isopropyl-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3,5-Diflυoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-pheπyl)-4-(4-hydroxy-phenyl)-5-(1-hydroxy-2-p-tolyloxy-ethyl)- oxazolidin-2-one; 5-[2-(Biphenyl-3-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oxazolidin-2-one;
5-[2-(Biphenyl-2-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oxazolidin-2-one;
5-[2-(Biphenyl-4-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oχazolidin-2-one;
2-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzonitrile;
5-[2-(2-Fluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzonitrile; 3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1-hydroxy-2-(2-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one;
5-[2-(2-Ch!oro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(naphthalen-2-yloxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-(1-hydroxy-2-o-tolyloxy-ethyl)- oxazolidin-2-one;
3-{2-[3-(4-F)uoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzoic acid methyl ester; 3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-2-oxo-oxazolidine-5- carboxylic acid (4-fluoro-benzyl)-methyl-amide;
5-(3,4-Dihydro-1H-isoquinoline-2-carbonyl)-3-(3-fluoro-phenyl)-4-(2-hydroxy-4- methoxy-phenyl)-oxazolidin-2-one;
3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-5-(1-hydroxy-3-phenyl- propyl)-oxazolidiπ-2-one;
And pharmaceutically acceptable salts, esters, amides and hydrates thereof.
DETAILED DESCRIPTION The present invention provides a compound having a Formula (I),

or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein Ar1, Ar2 , Y3 and Z are as defined above.
The present invention further provides a compound having a Formula (Ia),

Further provided is the above compound having a Formula (lai),

(lai) or a pharmaceutically acceptable salt, ester, amide or hydrate thereof, wherein Z, R1, R2 and R3 are as defined above.
Further provided is the above-described compound wherein R1, R2, and R3 are each independently F or -OR'; and R is H or lower alkyl, optionally substituted. Further provided is a pharmaceutical composition comprising the above compound, the pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer or mixtures thereof; and a pharmaceutically acceptable carrier, diluent or vehicle.
Further provided is a method of inhibiting cholesterol absorption in a mammal requiring inhibition comprising administering to the mammal a therapeutically effective amount of the compound or the pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof. Further provided is a method of treating, preventing or controlling hyperlipidemia in a mammal.
Further provided is a method of treating, preventing or controlling hypercholesterolemia in a mammal.
Further provided is a method of treating, preventing or controlling hypertriglyceridemia in a mammal.
Further provided is a method of treating, preventing or controlling atherosclerosis ina mammal.
Further provided is a combination comprising the above-described compound and a pharmaceutically active agent. Further provided is the combination wherein said pharmaceutically active agent is a CETP inhibitor, a PPAR- activator, an MTP/Apo B secretion inhibitor, HDL- cholesterol raising agent, HMG-CoA reductase inhibitor, triglyceride lowering agent, a cholesterol synthesis inhibitor, a cholesterol modulating agent, a fibrate, niacin, an ion- exchange resin, an antioxidant, an ACAT inhibitor, bile acid sequestrant, an anti- hypertensive agent, or an acetylcholine esterase inhibitor.
Further provided is the combination wherein said' HMG-CoA reductase inhibitor is a statin.
Further provided is a pharmaceutical composition comprising the above combination and a pharmaceutically acceptable carrier, diluent, solvent or vehicle. The present invention further encompasses each of the title compounds set forth in the Examples herein.
The present invention further includes each of the title compounds set forth in the Examples herein.
As used above, and throughout the specification, the following terms, unless otherwise indicated, shall be understood to have the following meanings.
The article "a" or "an" as used herein refers to both the singular and plural form of the object to which it refers. The following definitions apply regardless of whether a term is used by itself or in combination with other terms, unless otherwise indicated. Therefore, the definition of "alkyl" applies to "alkyl" as well as the "alkyl" portions of "hydroxyalkyl", "haloalkyl", "alkoxy", "aralkyl", etc. The definition of "aryl" applies to "aryl" as well as the "aryl" portions of "heteroaryl", "aralkyl", "arylthio", etc. The term "alkyl" as used herein refers to a linear or branched hydrocarbon of from 1 to 20 carbon atoms. Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyi, n-decyl, tetradecyl, and the like. The alkyl can be optionally substituted and the term "substituted alkyl" means that the alkyl group is substituted by one or more substituents independently selected from the group consisting of halo, aryl, cycloalkyl, nitro, cyano, hydroxy, lower alkoxy, lower thioalkoxy, amino, -C(O)CrC6 alkyl, -C-OH, C1-C6 alkyl, -OSO3H, - OPO3H, -OC1-C6 alkyl, -O-aryl, =0, =S, -SH, -CO2H, -CO2C1-C6 alkyl, -NR'R", - N+R'R"R"T, -NR1SO2R", -R1C(O)NR1R", or -C(O)NR1R", where R', R", and R'" are each independently hydrogen, CrCβ alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, heteroaralkyl, where N, R' and R", or N, R', and R"', or N, R" and R'", or N, R', R", and R'" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S that can also be optionally substituted with at least one to three of the substituents recited for the term alkyl; where T is a representative counter anion forming a pharmaceutically acceptable salt, such as for example, bromide, chloride, sulfate, nitrate, bisulfate, acetate, oxalate, benzoate, tartrate, fumarate, and the like.
The term "lower alkyl" as used herein refers to a subset of alkyl which means a linear or branched hydrocarbon radical having from 1 to 6 carbon atoms. Non-limiting examples include methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert- butyl, n-pentyl, n-hexyl, and the like. Alternatively, lower alkyl is referred to as "C1-C6 alkyl." The lower alkyl group can also be substituted with at least one to three of the substituents as previously recited for the term alkyl.
The term "alkoxy" as used herein refers to an alkyl-O- group in which the alkyl group is as previously defined. Useful alkoxy groups can comprise 1 to 12 carbon atoms. The term "lower alkoxy" means an alkyl-O- group in which the alkyl group comprises 1 to 6 carbon atoms. Non-limiting examples of a lower alkoxy include methoxy, ethoxy, isopropoxy, and the like. The alkyl group of the alkoxy is linked to an adjacent moiety through the ether oxygen.
The term "alkenyl" as used herein means a linear or branched hydrocarbon radical from 2 to 12 carbon atoms having at least one carbon-carbon double bond. Non-limiting examples of an alkenyl include ethenyl, 1-propenyl, 1-butenyl, 2-butenyl, 2- pentenyl, 3-methyl-3-butenyl, 1-hexenyl, 3-heptenyl, l-octenyl, 1-nonenyl, 1-decenyl, 1- undecenyl, 1-dodecenyl, and the like. The alkenyl group may be optionally substituted with at least one to three of the substituents as previously recited for the term alkyl. The term "alkynyl" as used herein means a linear or branched hydrocarbon radical from 2 to 12 carbon atoms having at least one carbon-carbon triple bond. Non- limiting examples include 3-propynyl,
1-butynyl, 3-pentynyl, 3-methyl-3-butynyl, 1-hexynyl, 3-heptynyl, l-octynyl, 1-nonynyl, 1- decynyl, and the like. The alkynyl group may be optionally substituted with at least one to three of the substituents as previously recited for the term alkyl.
The term "aryl" as used herein refers to a C5-C14 mono-, bi- or polycarbocyclic aromatic ring system which is optionally substituted by at least one substituent selected from alkyl, lower alkoxy, lower thioalkoxy, halogen, -CO2H, -002(Ci-C6) alkyl, - C(O)C1-C6 alkyl, -OSO3H, -OPO3H, Or -OC1-C6 alkyl,-0(CH2)o-2CF3, -O-aryl, -OSO2R', nitro, cyano -OH, -SH, -CF3, -NR1R", -NR1SO2R", -NR1C(O)NR1R", -S(O)1.2alkyl, S(O)1. 2aryl, -SO2NR1R", or -C(O)NR1R", where R1, and R" are independently hydrogen, CrC6 alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, or heteroaralkyl, or N, R' and R" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S. Non-limiting examples of aryl include phenyl, naphthyl, indenyl, 2-chlorophenyl, 3-chlorophenyl, 4- chlorophenyl, 2-methylphenyl, 3-methylphenyl, 4-methylphenyl, 2-methoxyphenyl, 3- methoxyphenyl, 4-methoxyphenyl, 2-chloro-3-methylphenyl, 2-chloro-4-methylphenyl, 2- chloro-5-methylphenyl, 3-chloro-2-methylphenyl, 3-chloro-4-methylphenyl, 4-chloro-2- methylphenyl, 4-chloro-3-methylphenyl, 5-chloro-2-methylphenyl, 2,3-dichlorophenyl, 2,5-dichlorophenyl, 3,4-dichlorophenyl, 2,3-dimethylphenyl, 3,4-dimethylphenyl, and the
like. The aryl group may be optionally substituted with at least one to three "ring system substituents" which may be the same or different, and are as defined below.
The term "aralkyl" as used herein means an aryl-alkyl group, in which the aryl and alkyl groups are as previously defined. Linkage to the rest of the molecule may be through either the aryl or alkyl portion of the aralkyl moiety. The aralkyl group may be optionally substituted by at least one to three substituents as recited above for alkyl and aryl. Non-limiting examples of aralkyl include benzyl, phenethyl, naphthlenylmethyl, tolyl, and the like.
The term "aralkenyl" as used herein means an aryl-alkenyl group in which the aryl and alkenyl groups are as previously defined. The aralkenyl group may be optionally substituted with one to three substituents as recited above for aryl and alkenyl. Non-limiting examples of aralkenyl include 2-phenethenyl, 2-naphthylethenyl, and the like.
The term "alkylene" as used herein refers to a divalent group derived from a linear or branched chain saturated hydrocarbon having from 1 to 10 carbon atoms by the removal of two hydrogen atoms. The preferred alkylene refers to a linear or branched hydrocarbon chain diradical having from 1 to 3 carbon atoms. The alkylene group may be optionally substituted with one or more of the substituents recited for the term alkyl, and selected from lower alkoxy, lower thioalkoxy,
 halo, nitro, cyano, =0, =S, -OH, -SH, -CF3, -CO2H, -CO2CrC6 alkyl, -NR'R", or -C(O)NR1R", where R' and R" are independently hydrogen, CrCe alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, heteroaralkyl, or N, R' and R" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S. Useful alkylene groups have from 1 to 6 carbon atoms (Ci-Cε alkylene). Non-limiting examples of alkylene include methylene (-CH2-), ethylene (-CH2CH2-), propylene (-(CH2)3-), and the like.
halo, nitro, cyano, =0, =S, -OH, -SH, -CF3, -CO2H, -CO2CrC6 alkyl, -NR'R", or -C(O)NR1R", where R' and R" are independently hydrogen, CrCe alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, heteroaralkyl, or N, R' and R" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S. Useful alkylene groups have from 1 to 6 carbon atoms (Ci-Cε alkylene). Non-limiting examples of alkylene include methylene (-CH2-), ethylene (-CH2CH2-), propylene (-(CH2)3-), and the like.
The term "aroyl" means an aryl-C(O)- group in which the aryl group is as previously defined. Non-limiting examples of aroyl include benzoyl, 1-naphthoyl, 2- naphthoyl, and the like. The term "acyl", as used herein means an HC(O)- or alkyl-C(O)- in which the alkyl group is as previously defined. Preferred acyls contain a lower alkyl. Non-limiting examples of acyl include formyl, acetyl, propanoyl, 2-methylpropanoyl, butanoyl, and the like.
The term "aryloxy", as used herein means an aryl-O- in which the aryl group is as previously defined. Non-limitng examples of aryloxy include phenoxy, naphthoxy, and the like.
The term "arylthio", as used herein means an aryl-S- in which the aryl group is as previously described. Non-limiting examples of arylthio include phenylthio, heptylthio, and the like.
The term "aralkylthio" as used herein means an aralkyl-S- group in which the aralkyl is as previously defined. Non-limiting examples of aralkylthio include benzylthio, 2-phenyl-ethanethiol, and the like. The term "alkoxycarbonyl", as used herein means an alkoxy-C(O)- in which the alkoxy is as previously defined. Non-limiting examples of alkoxycarbonyl include methoxycarbonyl, ethoxycarbonyl, and the like.
The term "aryloxycarbonyl", as used herein means an aryl-O-C(O)- group in which the aryl group is as previously described. Non-limiting examples of aryloxycarbonyl include phenoxycarbonyl, naphthoxycarbonyl, and the like.
The term "aralkoxycarbonyl", as used herein means an aralkyl-O-C(O)- group in which the aralkyl group is as previously defined. Non-limiting examples of aralkoxycarbonyl include benzyloxycarbonyl, and the like
The term "alkylsulfonyl", as used herein means an alkyl-S(O)2- in which the alkyl group is as previously defined. Preferred groups are those in which the alkyl group is lower alkyl.
The term "alkylsulfinyl", as used herein means an alkyl-S(O)- group. Preferred groups are those in which the alkyl group is lower alkyl.
The term "arylsulfonyl", as used herein means an aryl-S(O)2- group. The term "arylsulfinyl", as used herein means an aryl-S(O)- group.
The term "cycloalkyl", as used herein refers to a saturated cyclic C3-Ci2 alkyl group, where alkyl is as previously defined. Non-limitng examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cycloctyl, decalinyl, norpinanyl, or adamantyl. The cycloalkyl group may be optionally substituted with at least one of those substituents recited above for alkyl or alkylene. Non-limiting examples of substituted cycloalkyl groups include fluorocyclopropyl, 2-iodocyclobutyl, 2,3-dimethylcyclopentyl, 2,2-dimethoxycyclohexyl, 3-phenylcyclopentyl, and the like.
The term "cycloalkenyl", as used herein refers to a saturated cyclic C3-C12 alkeπyl group having at least one carbon-carbon double bond, where alkenyl is as previously defined. Nonlimiting examples of cycloalkenyl include cyclopropene, cyclopentene, cyclopenta-1-3-diene, cyclohexene, cycloheptene, cyclohepta-i-4-diene, and the like. The term "hydrocarbon chain", as used herein refers to a linear hydrocarbon of from 1 to 12 carbon atoms. The hydrocarbon chain is optionally substituted with one or more substituents selected from alky!, alkoxy, thioalkoxy, -0(CH2)o-2CF3, halogen, nitro, cyano, =0, =S, -OH, -SH, -CF3, -CO2H, -CO2C1-C6 alkyl, -NR1R", -C(O)NR1R", - N+R'R"R"T, -NR1S(O)2R", or -R1C(O)NR1R", where R', R", and R"1 are independently hydrogen, CrC6 alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, heteroaralkyl, or N, R1 and R", or N, R1, and R1", or N, R" and R"1 or N, R', R", and R1" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S.
The term "halogen" or "halo", as used herein means fluorine or fluoro, chlorine or chloro, bromine or bromo or iodine or iodo.
The term "heteroatom", as used herein means oxygen (O), nitrogen (N), or sulfur (S) as well as sulfoxyl or sulfonyl (S(O) or SO2) unless otherwise indicated.
The term "heteroaryl", as used herein means an aryl group, as previously defined, containing one or more heteroatoms, as previously defined. The heteroaryl may be optionally substituted with at least one of the substituents previously recited for "aryl". Non-limiting examples of heteroaryl include thienyl, benzothienyl (2-benzothienyl, 3-benzothienyl, and the like), indolizinyl, pyrazinyl, furanyl, benzofuranyl, pyrrolyl, pyridyl, pyrimidinyl (2-pyrimidinyl, 4-pyrimidinyl, and the like), imidazolyl (1-imidazolyl, 2- imidazolyl, and the like), benzimidazolyl (l-benzimidazolyl, 2-benzimidazolyl, and the like), triazolyl (1-triazolyl, 3-triazolyl, and the like), isothiazolyl, pyrazolyl (l-pyrazolyl, 3- pyrazolyl, 4-pyrazolyl, and the like), oxazolyl (2-oxazolyl, 4-oxazolyl, and the like), benzoxazolyl (2-benzoxazolyl, 4-benzoxazolyl and the like), tetrazolyl (l-tetrazolyl, 3- tetrazolyl, and the like), thiazolyl (2-thiazolyl, 4- thiazolyl.and the like), indolyl (l-indolyl, 2-indolyl, and the like), isoindolyl (l-isoindolyl, 2-isoindolyl, and the like), quinazolinyl, quinolinyl (2-quinolinyl, 3-quinolinyl, and the like), isoquinolinyl (3-isoquinolinyl, 5- isoquinolinyl, and the like).
The term "heterocycle", as used herein means a saturated mono-, bi- or polycyclic ring containing one or more heteroatoms selected from N, O, and S. The
heterocycle may be optionally substituted with at least one of those substituents recited above for alkyl. Non-limiting examples of heterocycle include piperidinyl, pyrrolidinyl, I- piperazinyl, 2-piperazinyl, 2-morpholinyl, 4-morpholinyl, piperazinyl, azetidinyl, aziridinyl, thietanyl, and the like. The term "heterocyclenyl", as used herein means a non-aromatic monocyclic or multicyclic ring system of about 3 to about 12 ring atoms, preferably about 5 to about 10 ring atoms, in which one or more of the atoms in the ring system is/are element(s) other than carbon, for example nitrogen, oxygen, or sulfur atoms, and which contains at least one carbon-carbon double bond or carbon-nitrogen double bond. The prefix aza, oxa, or thia before heterocyclenyl means that at least a nitrogen, oxygen, or sulfur atom, respectively, is present as a ring atom. Non-limiting examples of heterocyclenyl include 1 ,2,3,4-tetrahydropyridine, 2-pyrrolinyl, 2-imidazolinyl, 1 ,2-dihydropyridyl, and the like.
The term "heteroaralkyl", as used herein means heteroaryl-alkyl, in which heteroaryl and alkyl are both as previously defined. Linkage to the rest of the molecule can be either through the heteroaryl or the alkyl portion of the heteroaralkyl moiety. The heteroaralkyl may be optionally substituted with at least one of those substituents previously recited for alkyl and heteroaryl. Nonlimiting examples of heteroarylalkyl include 2-propyl-pyridine, 3,4-methyl-1H-pyrrole, and the like.
The term "heteroaralkenyl", as used herein means heteroaryl-alkenyl, in which heteroaryl and alkenyl are both as previously defined. Linkage to the rest of the molecule can be either through the heteroaryl or the alkenyl portion of the heteroaralkenyl moiety. The heteroaralkenyl may be optionally substituted with at least one of those substituents previously recited for alkenyl and heteroaryl. Non-limiting examples of heteroaralkenyl include 2-(pyrid-3-yl)ethenyl, 2-(quinolin-3-yl)ethenyl, and the like.
The term "heterocycloalkyl", as used herein means heterocycle-alkyl, in which the heterocycle and the alkyl are both as previously defined. Linkage to the rest of the molecule can be either through the heterocycle or the alkyl portion of the heterocycloalkyl moiety. The heterocycloalkyl may be optionally substituted with at least one of those substituents recited above for alkyl and heterocycle. Non-limiting examples of heterocycloalkyl include 2-methyl piperidine, 2-ethyl-5-methyl-pyrrolidine, and the like.
The term "thioalkyl" or "alkylthio" means an alkyl-S- in which the alkyl group is a previously defined. The alkyl is linked to an adjacent moiety through the sulfinyl moiety. Non-limiting examples of thioalkyl include methylthio, ethylthio, isopropylthio, and the like. The term "thioalkoxy" means an alkoxy-S- in which the alkoxy group is a s previously defined. The alkoxy is linked to an adjacent moiety throught the sulfinyl moiety. The term "lower thioalkoxy" means an alkyl-O-S- group in which the alkyl group comprises 1 to 6 carbon atoms. Non-limiting examples of thioalkoxy include methoxysulfanyl, ethoxysulfanyl, and the like. The term "ring" as used herein includes heteroaryl, heterocycle, cycloalkyl and aryl, each as previously defined, and further includes fused, monocyclic, bicyclic, and polycyclic permutations thereof.
"Ring system substituent" means a substituent attached to an aromatic or non- aromatic ring system which, for example, replaces hydrogen on the ring system. Ring system substituents may be the same or different, each being independently selected from the group consisting of alkyl, aryl, heteroaryl, aralkyl, aralkenyl, heteroaralkyl, heteroaralkenyl, hydroxy, alkoxy, aryloxy, aralkoxy, acyl, halo, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, aroyl, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio, cycloalkyl, cycloalkenyl, heterocyclo, heterocyclenyl, heterocycloalkyl, and NR'R", wherein R' and R" are each independently H, Ci-C6 alkyl, cycloalkyl, alkenyl, alkynyl, aryl, aralkyl, heteroaryl, or heteroaralkyl, or N, R', and R" may be joined together to form a 4-7 member monocyclic or bicyclic ring optionally containing at least one additional heteroatom selected from N, O and S. The term "stereoisomer" as used herein refers to both geometric (e.g., cis and trans isomers) and/or optical isomers (e.g., R and S enantiomers) of a compound of the invention. Racemic, enatiomeric, diastereomeric, and epimeric mixtures of isomers are contemplated by the present invention. Compounds of formula 1 , 2, or 3 containing one or more asymmetric carbon atom can exist as two or more stereoisomers. Where a compound of formula 1 , 2, or 3 contains an alkenyl or alkenylene group, geometric cis/trans isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in compounds of formula 1 , 2, or 3 containing, for example, an
imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism. Accordingly, included within the scope of the present invention are all stereoisomers and tautomeric forms of the compounds of formula 1 , 2, or 3, including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, d-lactate or l-lysine, or racemic, for example, dl-tartrate or dl-arginine. Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallization.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC). Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula 1 , 2, or 3 contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
The term "racemate" as used herein, is meant to include both the racemic compound wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts and the racemic mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each containing the single enantiomer. Such mixtures may be separated by conventional techniques known to
those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, New York, 1994).
When a bond to a substituent is shown to cross the bond(s) connecting 2 atoms in a ring, then such substituent may be bonded to any atom in the ring, provided the atom will accept the substituent without violating its valency. When there appears to be several atoms of the substituent that may bond to the ring atom, then it is the first atom of the listed substituent that is attached to the ring, unless indicated otherwise.
Unless indicated otherwise, "compound of the invention" or "compounds of the invention" includes the compound itself as well as pharmaceutically acceptable salts, esters, amides, hydrates, or stereoisomers thereof.
The term "patient" or "subject" means all animals and mammals, including humans. Examples of patients or subjects include humans, cows, dogs, cats, goats, sheep, pigs, and rabbits.
The phrases "effective amount" and "therapeutically effective amount" mean that amount of a compound of Formula 1 , 2, or 3, and other pharmacological or therapeutic agents described below, that will elicit a biological or medical response in a tissue, system, animal, or mammal that is being sought by the administrator (such as a researcher, doctor, or veterinarian) which includes alleviation of the symptoms of the condition or disease being treated and the prevention, slowing or halting of progression of one or more conditions, for example vascular conditions such as hyperlipidemia, atherosclerosis, hypercholesterolemia, hypertriglyceridemia, sitosterolemia, vascular inflammation, and the like. As would be understood by a skilled artisan, a "therapeutically effective amount" will vary from subject to subject and will be determined on a case by case basis. Factors to consider include, but are not limited to, the subject being treated, weight, health, and compound administered.
The term "a pharmaceutically acceptable salt, ester, amide, hydrate, or stereoisomer" as used herein refers to those acid addition salts, base addition salts, esters, amides, hydrates, and stereoisomers (optical, geometric, and tautomeric) of the compounds of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of patients without undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compounds of the invention.
Further, the term "a pharmaceutically acceptable salt" refers to the relatively nontoxic, inorganic and organic acid addition or base salts of compounds of the invention. These salts can be prepared in situ during the final isolation and purification of the compounds or by separately reacting the purified compound in its free form with a suitable organic or inorganic acid or base and isolating the salt thus formed. Representative anionic or acid addition salts include acetate, aspartate, besylate, bicarbonate, carbonate, camysylate, citrate, edisylate, fumarate, gluconate, hydrobromide, bromide, hydrochloride, chloride, D-lactate, L-lactate, malate, mesylate, pamoate, phosphate, succinate, sulphate, D-tartrate, L-tartrate, benzoate, gluceptate, glucuronate, hibenzate, isethionate, malonate, methylsulphate, 2-napsylate, nicotinate, nitrate, orotate, stearate, tosylate, adipate, arabogalactanesulphate, ascorbate, estolate, galacturonate, glutamate, hippurate, 3-hydroxy-2-naphthoate, 1 -hydroxy- 2-naphthoate, iodide, lactobionate, maleate, mandelate, mucate, napadisylate, oleate, oxalate, saccharate, salicylate, sulphosalicylate, cholate, and tryptophanate. (See, for example, Berge S. M., et al., "Pharmaceutical Salts," J. Pharm. ScL, 1977;66:1-19, which is incorporated herein by reference.) The free base form may be regenerated by contacting the salt form with a base. While the free base may differ from the salt form in terms of physical properties, such as solubility, the salts are equivalent to their respective free bases for the purposes of the present invention. Representative cationic or base salts include calcium, choline, magnesium, potassium, sodium, aluminum, ammonium, quaternary ammonium, and amine cations including tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, and the like, arginine, benzathine, diethylamide, diolamine, glycine, lysine, meglumine, olamine, tromethamine (Tris), 2- amino-2-methylpropan-1-ol, benethamine, erbumine (tert-butylamine), epolamine (hydroxyethylpyrrolidine), ethylenediamine, hydrabamine, morpholine, piperazine, procaine, silver, trolamine, zinc, adenine, arginine, cytosine, glucosamine, guanidine, guanine, nicotinamide, ornithine, praline, pyridoxine, serine, tyrosine, and valine. Hemisalts, for example, hemicalcium may also be formed. Examples of pharmaceutically acceptable, non-toxic esters of the compounds of the invention include C1-C6 alkyl esters wherein the alkyl group is a linear or branched chain. Acceptable esters also include C5-C7 cycloalkyl esters as well as aralkyl esters such as, but not limited to, benzyl. C1-C4 alkyl esters are preferred. Esters of the
compounds of the present invention may be prepared according to conventional methods.
Examples of pharmaceutically acceptable, non-toxic amides of the compounds of the invention include amides derived from ammonia, primary (Ci-Ce)alkyl amines and secondary di-(CrC6)alkyl amines wherein the alkyl groups are linear or branched chain. In the case of secondary amines, the amine may also be in the form of a 5- or 6-membered heterocycle containing one nitrogen atom. Amides derived from ammonia, CrC3 alkyl primary amines and C1-C2 dialkyl secondary amines are preferred. Amides of the compounds of the invention may be prepared according to conventional methods. Certain compounds of the present invention can exist in unsolvated form as well as solvated form including hydrated form. In general, the solvated form including hydrated form is equivalent to the unsolvated form and is intended to be encompassed within the scope of the present invention.
The use of prodrugs is contemplated by the present invention. "Prodrugs" are intended to include any covalently bonded carrier which releases the active parent drug according to Formula 1 , 2, or 3, in vivo. Further, the term "prodrug" refers to compounds that are transformed in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood. A thorough discussion is provided in T. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A. C. S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, both of which are hereby incorporated by reference. Examples of prodrugs include acetates, formates, benzoate derivatives of alcohols, and amines present in compounds of Formula 1, 2, or 3. The compounds of the present invention are suitable to be administered to a patient or subject for the treatment of hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, and atherosclerosis. The compounds of the present invention can be administered to a patient/subject alone, or with another compound of the invention, or as part of a pharmaceutical composition. A pharmaceutical composition of the invention contains at least one compound of the invention and at least one pharmaceutically acceptable carrier, diluent, solvent or vehicle. The pharmaceutically acceptable carrier, diluent, solvent or vehicle may be any such carrier known in the art including those described in, for example, Remington's
Pharmaceutical Sciences, Mack Publishing Co., (A. R. Gennaro edit. 1985). A pharmaceutical composition of the invention may be prepared by conventional means known in the art including, for example, mixing at least one compound of the invention with a pharmaceutically acceptable carrier. The compounds, compositions, and treatments of the present invention can be administered by any suitable means which produce contact of these compounds with the site of action in the body, for example, in the plasma, liver, rectum, or small intestine of an animal or mammal. Compositions of compounds of the invention are contemplated herein. A composition of the invention can be administered to a patient/subject either orally, rectally, parenterally (intravenously, intramuscularly, or subcutaneously), intracisternally, intravaginally, intraperitoneally, intravesically, locally (powders, ointments, or drops), or as a buccal or nasal spray.
Compositions suitable for parenteral injection may comprise physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions. Examples of suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil), and injectable organic esters such as ethyl oleate. Proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
These compositions may also contain additives such as preserving, wetting, emulsifying, and dispensing agents. Prevention of the action of microorganisms can be ensured by various antibacterial and antifungal agents, for example, parabens, chlorobυtanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example sugars, sodium chloride, and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin. Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules. In such solid dosage forms, the active compound is admixed with at least one (a) inert customary excipient (or carrier) such as sodium citrate or dicalcium phosphate; (b) fillers or extenders, as for example, starches, lactose, sucrose, glucose, mannitol, and silicic acid; (c) binders, as for example, carboxymethylcellulose,
alignates, gelatin, polyvinylpyrrolidone, sucrose, and acacia; (d) humectants, as for example, glycerol; (e) disintegrating agents, as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (f) solution retarders, as for example paraffin; (g) absorption accelerators, as for example, quaternary ammonium compounds; (h) wetting agents, as for example, cetyl alcohol and glycerol monostearate; (i) adsorbents, as for example, kaolin and bentonite; and (j) lubricants, as for example, talc, calcium stearate, magnesium stearate, solid polyethylene glycols, sodium lauryl sulfate, or mixtures thereof. In the case of capsules, tablets, and pills, the dosage forms may also comprise buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethyleneglycols, and the like.
Solid dosage forms such as tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells, such as enteric coatings and others well-known in the art. They may contain opacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedding compositions which can be used are polymeric substances and waxes. The active compounds can also be in micro- encapsulated form, if appropriate, with one or more of the above-mentioned excipients. Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs. In addition to the active compounds, the liquid dosage forms may contain inert diluents commonly used in the art, such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-butyleneglycol, dimethylformamide, oils, in particular, cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil and sesame oil, glycerol, tetrahydrofurfuryl alcohol, polyethyleneglycols and fatty acid esters of sorbitan or mixtures of these substances, and the like. Besides inert diluents, compositions include additives, such as, for example, wetting agents, emulsifying and the pending agents, sweetening, flavoring, and perfuming agents, or mixtures thereof. Suspensions, in addition to the active compounds, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters,
microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances.
Compositions for rectal administrations are preferably suppositories which can be prepared by mixing the compounds of the present invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethyleneglycol, or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore, melt in the rectum or vaginal cavity and release the active component.
Dosage forms for topical administration of a compound of this invention include ointments, powders, sprays, and inhalants. The active component is admixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants as may be required. Ophthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
The compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 2,000 mg per day, preferably about 5 to about 250 mg. per day. For a normal human adult having a body weight of about 70 kilograms, a dosage in the range of about 0.01 to about 100 mg per kilogram of body weight per day is preferable. The specific dosage used, however, can vary from patient to patient. For example, the dosage can depend on a numbers of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used. The determination of optimum dosages for a particular patient is well known to those skilled in the art.
The term "treating" or "treatment" refers to curative, palliative and prophylactic treatment, including reversing, ameliorating, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
The compounds of the invention, as described herein, may be used either alone or in combination with another pharmaceutically active agent described herein, in the treatment of the following diseases/conditions: dyslipidemia, hypercholesterolemia, hypertriglyceridemia, atherosclerosis, peripheral vascular disease, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, diabetes and vascular complications of diabetes, obesity, unstable angina pectoris, Alzheimer's Disease, BPH, osteoporosis,
cerebrovascular disease, coronary artery disease, ventricular dysfunction, cardiac arrhythmia, pulmonary vascular disease, renal-vascular disease, renal disease, vascular hemostatic disease, autoimmune disorders, pulmonary disease, sexual dysfunction, cognitive dysfunction, cancer, organ transplant rejection, psoriasis, endometriosis, and macular degeneration. A combination of the invention may be part of a pharmaceutical composition further containing a pharmaceutically active carrier, diluent, solvent or vehicle, each as described herein.
Examples of a suitable pharmaceutically active agent include a CETP inhibitor, a PPAR- activator, an MTP/Apo B secretion inhibitor, HDL-cholesterol raising agent, triglyceride lowering agent, a cholesterol synthesis inhibitor, a cholesterol modulating agent, a fibrate, niacin, an ion-exchange resin, an antioxidant, an ACAT inhibitor, or bile acid sequestrant; an anti-hypertensive agent; an acetylcholine esterase inhibitor, an anti-diabetic compound, an anti-obesity compound, a thyromimetic agent, an anti- resorptive agent, an anti-osteoporosis agent, an antihypertensive agent, or a drug for the treatment of Alzheimer's disease. Specific examples of each of these agents include those known in the art as well as those specified below.
In combination therapy treatment, both the compounds of the invention and the other drug therapies are administered to mammals by conventional methods. The following discussion more specifically describes the various combination aspects of this invention.
Any cholesterol ester transfer protein ("CETP") inhibitor known in the art that inhibits the transfer of cholesteryl ester and triglyceride between lipoprotein particles, including high density lipoprotein (HDL), low density lipoprotein (LDL), very low density lipoprotein (VLDL), and chylomicrons may be used. The effect of a CETP inhibitor on lipoprotein profile is believed to be antiatherogenic. Such inhibition may be determined by means known in the art (e.g., Crook et al. Arteriosclerosis 10, 625, 1990; U.S. Pat. No. 6,140,343). Examples of suitable CETP inhibitors include, but are not limited to, those described in U.S. Patent Nos. 6,197,786, 6,723,752 and 6,723,753. Additional examples of useful CETP inhibitors include the following compounds: [2R, 4S]4-[(3,5- bis-trifluoromethyl-benzyl)-methoxycarbonyl-amino]-2-ethyl-6-trifluoromethyl-3,4- dihydroxycarbonyl-amino]-2-ethyl-6-trifluoromethyl-3,4-dihydro-2H-quinoline-1- carboxylic acid ethyl ester (Torcetrapib™), and 3-{[3-(4-Chloro-3-ethyl-phenoxy)- phenyl]-[3-(1 ,1 ,2,2-tetrafluoro-ethoxy)-benzyl]-amino}-1 ,1 ,1-trifluoro-propan-2-ol. To
address the poor solubility of many of the CETP inhibitors, an appropriate dosage form such as one comprising (1) a solid amorphous dispersion comprising a cholesteryl ester transfer protein (CETP) inhibitor and an acidic concentration-enhancing polymer; and (2) an acid-sensitive HMG-CoA reductase inhibitor, may be necessary. This dosage form is more fully described in USSN 10/739,567.
Any peroxisome proliferator activated receptor ("PPAR") activator known in the art that activates or otherwise interacts with a human PPAR may be used. Three mammalian PPARs have been isolated and termed PPAR-alpha, PPAR-gamma, and PPAR-beta (also known as NUC1 or PPAR-delta). These PPARs regulate expression of target genes by binding to DNA sequence elements, termed PPAR response elements. These elements have been identified in the enhancers of a number of genes encoding proteins that regulate lipid metabolism suggesting that PPARs play a pivotal role in the adipogenic signaling cascade and lipid homeostasis. PPAR-gamma receptors are associated with regulation of insulin sensitivity and blood glucose levels. PPAR-α activators are associated with lowering plasma triglycerides and LDL cholesterol. PPAR-β activators have been reported to both increase HDL-C levels and to decrease LDL-C levels. Thus, activation of PPAR-β alone, or in combination with the simultaneous activation of PPAR-α and/or PPAR-gamma may be desirable in formulating a treatment for dyslipidemia in which HDL is increased and LDL lowered. PPAR-activation is readily determined by those skilled in the art by the standard assays (e.g. US 2003/0225158 and US 2004/0157885). Examples of suitable PPAR-activator compounds include, but are not limited to, those described in US 2003/0171377, US 2003/0225158, US 2004/0157885, and U.S. Pat. No. 6,710,063. Additional examples of useful PPAR-activator compounds include the following compounds: [5-Methoxy-2- methly-4-(4'-trifluoromethly-biphenyl-4ylmethylsulfanyl)-phenoxy]-acetic acid; [5-
Methoxy-2-methyl-4-(3'-trifloromethly-biphenyl-4-ylmethylsulfanyl)-phenoxy]-acetic acid; [4-(4'Fluoro-biphenyl-4-ylmethylsulfanyl)-5-methoxy-2methyl-phenoxy]-acetic acid; {5- Methoxy-2methyl-4-[4-(4-trifluoromethyl-benzyloxy)-benzylsulfanyl]-phenoxy}-acetic acid; {{5-Methoxy-2-methyl-4-[4-(5-trifluoromethyl-pryidin-2-yl)-benzylsulfanyl]- phenoxyj-acetic acid; (4-{4-[2-(3-Fluoro-phenyl)-vinyl]-benzylsulfanyl}-5-methoxy-2- methyl-phenoxy)-acetic acid; [5-Methoxy-2-methyl-4-(3-methyl-4'-trifluoromethyl- biphenyl-4-ylmethylsulfanyl)-phenoxy]-acetic acid; [5-Methoxy-2-methyl-4-(4'- trifluoromethyl-biphenyl-3-ylmethylsulfanyl)-phenoxy]- acetic acid; {5-Methoxy-2-methyl-
4-[2-(4-trifluoromethyl-benzyloxy)-benzylsulfanyl]-phenoxy}acetic acid; 3-{5-[2-(-5- Methyl-2 phenyl-oxazol-4-yl-ethoxy] -indol- 1-yl} -propionic acid; 3-{4[2-(5-methyl-2- phenyl-1,3-oxazol-4-yl)ethoxy- 1H-indazol-1yl}propanoic acid; 2-Methyl-2-{3-[({2-(5- methyl-2-phenyl-1 ,3-oxazol-4-yl)ethoxy]carbonyl}amino)methyl] phenoxy}propionic acid; 5 1 -{3'-[2-5-Methyl-2-phenyl-1 ,3-oxazol-4-y]-1 , 1 ' -biphenyl-3-y l}oxy)cyclobutanecarboxylic acid; 3-[3-(1-Carboxy-1-methyl-ethoxy)-phenyl]-piperidine-1-carboxylic acid 3- trifluoromethyl-benzyl ester; 2-{2-methyl-4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3- thiazol-5-yl}me thyl)sulfanyl]phenoxy}acetic acid; 2-{2-methyl-4-[({4-methyl-2-[4- (trifluoromethyl )phenyl]-1 ,3-oxazol-5-yl}methyl)sulfanyl]phenoxy}acetic acid; methyl 2- o {4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl}methyl)sul fanyl]phenoxy}acetate;
2-{4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl}methyl)sulf anyl]phenoxy}acetic acid; (E)-3-[2-methyl-4-({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3- thiazol-5-yl }methoxy)phenyl]-2-propenoic acid; 5 2-{3-chloro-4-[({4-methyl-2-[4-(trifluoromethyl)phenyl]-1 ,3-thiazol-5-yl}me thyl)sulfanyl]phenyl}acetic acid; 2-{2-methyl-4-[({4-methyl-2-[3-fluoro-4- (trifluoromethyl)phenyl]-1 ,3-thiazo l-5-yl}methyl)sulfanyl]phenoxy}acetic acid; and pharmaceutically acceptable salts thereof.
Any MTP/Apo B secretion (microsomal triglyceride transfer protein and/or0 apolipoprotein B secretion) inhibitor known in the art which inhibits the secretion of triglycerides, cholesteryl ester and phospholipids may be used. Such inhibition may be readily determined according to standard assays (e.g., Wetterau, J. R. 1992; Science 258:999). Examples of suitable a MTP/Apo B secretion inhibitor include, but are not limited to, imputapride (Bayer) as well as those described in WO 96/40640 and WO5 98/23593.
Any ACAT inhibitor known in the art that inhibits the intracellular esterification of dietary cholesterol by the enzyme acyl CoA; cholesterol acyltransferase may be used. Such inhibition may be determined readily according to standard assays, such as the method of Heider et al. described in Journal of Lipid Research. 24:1127 (1983). 0 Examples of suitable ACAT inhibitors include, but are not limited to, those described in U.S. Pat. No. 5,510,379 (carboxysulfonates),WO 96/26948 and WO 96/10559 (urea derivatives). Additional examples include Avasimibe (Pfizer), CS-505 (Sankyo) and Eflucimibe (EIi Lilly and Pierre Fabre).
Any lipase inhibitor (e.g., pancreatic lipase inhibitor, a gastric lipase inhibitor) known in the art that inhibits the metabolic cleavage of dietary triglycerides into free fatty acids and monoglycerides may be used. Such lipase inhibition activity may be readily determined according to standard assays (e.g., Methods Enzymol. 286: 190-231). Examples of a suitable lipase inhibitor include, but are not limited to, lipstatin,
(2S,3S,5S,7Z,10Z)-5-[(S)-2-formamido-4-methyl-valeryloxy]-2-hexyl-3-hydro- xy-7,10- hexadecanoic acid lactone, and tetrahydrolipstatin (orlistat), (2S,3S,5S)-5-[(S)-2- formamido-4-methyl-valeryloxy)-2-hexyl-3-hydroxy-hexa- decanoic 1 ,3 acid lactone, and the variously substituted N-formylleucine derivatives and stereoisomers thereof (U.S. Pat. No. 4,598,089); tetrahydrolipstatin U.S. Pat. Nos. 5,274,143; 5,420,305; 5,540,917; and 5,643,874; FL-386, 1-[4-(2-methylpropyl)cyclohexyl]-2-[- (phenylsulfonyl)oxy]- ethanone, and the variously substituted sulfonate derivatives related thereto (U.S. Pat. No. 4,452,813); WAY-121898, 4-phenoxyphenyl-4-methylpipe- ridin-1-yl-carboxylate, and the various carbamate esters and pharmaceutically acceptable salts related thereto (U.S. Pat. Nos. 5,512,565; 5,391,571 and 5,602,151); valilactone, and a process for the preparation thereof by the microbial cultivation of Actinomycetes strain MG147-CF2 (Kitahara, et al., J. Antibiotics, 40 (11), 1647-1650 (1987)); esterastin; ebelactone A and ebelactone B, and a process for the preparation thereof by the microbial cultivation of Actinomycetes strain MG7-G1 (Umezawa, et al., J. Antibiotics, 33, 1594-1596 (1980); Japanese Kokai 08-143457, published Jun. 4, 1996). The compound tetrahydrolipstatin is especially preferred. Additional examples include N-3- trifluoromethylphenyl-N'-- 3-chloro-4'-trifluoromethylphenylurea, and the various urea derivatives related thereto, U.S. Pat. No. 4,405,644; esteracin (U.S. Pat. Nos. 4,189,438 and 4,242,453); and cyclo-O, O'-[(1 ,6-hexanediy!)-bis-(iminoc- arbonyl)]dioxime, and the various bis(iminocarbonyl)dioximes related thereto (Petersen et al., Liebig's Annalen, 562, 205-229 (1949).
Any bile acid sequestrant known in the art may be used. Examples of suitable bile acid sequestrants include, but are not limited to, Welchol®, Colestid®, LoCholest®, Questran® and fibric acid derivatives, such as Atromid®, Lopid® and Tricor®' A compound of the invention can be used in combination with an anti-diabetic compound, i.e. any compound (e.g. insulin) used in the treating diabetes (especially Type II), insulin resistance, impaired glucose tolerance, or the like, or any of the diabetic complications such as neuropathy, nephropathy, retinopathy or cataracts. Additional
examples of an anti-diabetic compound include, but are not limited to, a glycogen phosphorylase inhibitor, an aldose reductase inhibitor, a sorbitol dehydrogenase inhibitor, a glucosidase inhibitor, and an amylase inhibitor.
Any glycogen phosphorylase inhibitor known in the art that inhibits the bioconversion of glycogen to glucose-1 -phosphate which is catalyzed by the enzyme glycogen phosphorylase may be used. Such glycogen phosphorylase inhibition activity may be readily determined according to standard assays (e.g., J. Med. Chem. 41 (1998) 2934-2938). A variety of glycogen phosphorylase inhibitors are known to those skilled in the art including those described in WO 96/39384 and WO 96/39385. Any aldose reductase inhibitor known in the art that inhibits the bioconversion of glucose to sorbitol catalyzed by the enzyme aldose reductase. Aldose reductase inhibition may be readily determined according to standard assays (e.g., J. Malone, Diabetes, 29:861-864 (1980). "Red Cell Sorbitol, an Indicator of Diabetic Control"). Any sorbitol dehydrogenase inhibitor known in the art that inhibits the bioconversion of sorbitol to fructose catalyzed by the enzyme sorbitol dehydrogenase may be used. Such sorbitol dehydrogenase inhibitor activity may be readily determined according to standard assays (e.g., Analyt. Biochem (2000) 280: 329-331). Examples of a suitable sorbitol dehydrogenase inhibitor include, but are not limited to, those described in U.S. Patent Nos. 5,728,704 and 5,866,578. Any glucosidase inhibitor known in the art that inhibits the enzymatic hydrolysis of complex carbohydrates by glycoside hydrolases, for example amylase or maltase, into bioavailable simple sugars, for example, glucose. Such glucosidase inhibition activity may be readily determined by those skilled in the art according to standard assays (e.g., Biochemistry (1969) 8: 4214). A generally preferred glucosidase inhibitor includes an amylase inhibitor. Any amylase inhibitor known in the art that inhibits the enzymatic degradation of starch or glycogen into maltose may be used. Such amylase inhibition activity may be readily determined by those skilled in the art according to standard assays (e.g., Methods Enzymol. (1955) 1 : 149). Other preferred glucosidase inhibitors include, but are not limited to, acarbose and the various amino sugar derivatives related thereto (U.S. Pat. Nos. 4,062,950 and 4,174,439); adiposine (U.S. Pat. No. 4,254,256); voglibose, 3,4-dideoxy-4-[[2-hydroxy- 1-(hydroxymethyl)ethyl]amino]-2-C-(hydroxymethyl)-D-epi-inositol, and the various N-
substituted pseudo-aminosugars related thereto (U.S. Pat. No. 4,701 ,559); miglitol, (2R,3R,4R,5S)-1-(2-hydroxyethyl)-2-(hydroxymethyl)-3,4,5-piperidinetriol, and the various 3,4,5-trihydroxypiperidines related thereto (U.S. Pat. No. 4,639,436); emiglitate, ethyl p-[2-[(2R,3R,4R,5S)-3,4?5-trihydroxy-2-(hydroxymethyl)piperidino]ethoxy]- benzoate, the various derivatives related thereto and pharmaceutically acceptable acid addition salts thereof (U.S. Pat. No. 5,192,772); MDL-25637, 2,6-dideoxy-7-O-.beta.-D- glucopyrano-syl^δ-imino-D-glycero-L-gluco-heptitol, the various homodisaccharides related thereto and the pharmaceutically acceptable acid addition salts thereof (U.S. Pat. No. 4,634,765); camiglibose, methyl 6-deoxy-6-[(2R,3R,4R,5S)-3,4,5-trihydroxy-2- (hydroxy-methyl)piperidinoJ-.alpha.-D-glucopyranoside sesquihydrate, the deoxy- nojirimycin derivatives related thereto, the various pharmaceutically acceptable salts thereof and synthetic methods for the preparation thereof (U.S. Pat. Nos. 5,157,116 and 5,504,078); pradimicin-Q; and salbostatin and the various pseudosaccharides related thereto (U.S. Pat. No. 5,091 ,524). Any amylase inhibitor known in the art may be used. Examples include, but are not limited to, tendamistat and the various cyclic peptides related thereto (U.S. Pat. No. 4,451 ,455); AI-3688 and the various cyclic polypeptides related thereto (U.S. Pat. No. 4,623,714); and trestatin, consisting of a mixture of trestatin A, trestatin B and trestatin C and the various trehalose-containing aminosugars related thereto, (U.S. Pat. No. 4,273,765).
Additional examples of an anti-diabetic compound for use in a combination of the invention include: biguanides (e.g., metformin), insulin secretagogues (e.g., sulfonylureas and glinides), glitazones, non-glitazone PPAR. gamma, agonists, PPAR.beta. agonists, inhibitors of DPP-IV, inhibitors of PDE5, inhibitors of GSK-3, glucagon antagonists, inhibitors of f-1 ,6-BPase (Metabasis/Sankyo), GLP-1/analogs (AC 2993, also known as exendin-4), insulin and insulin mimetics (Merck natural products), PKC-beta inhibitors, and AGE breakers.
A compound of the invention can be used in combination with any anti-obesity agent known in the art. Anti-obesity activity may be readily determined according to standard assays known in the art. Examples of suitable anti-obesity agents include, but are not limited to, phenylpropanolamine, ephedrine, pseudoephedrine, phentermine, .beta..sub.3 adrenergic receptor agonists, apolipoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, MCR-4 agonists, cholecystokinin-A
(CCK-A) agonists, monoamine reuptake inhibitors (e.g., sibutramine - U.S. Pat. No. 4,929,629), sympathomimetic agents, serotoninergic agents, cannabinoid receptor antagonists (e.g., rimonabant (SR-141 ,716A)), dopamine agonists (e.g., bromocriptine - U.S. Pat. Nos. 3,752,814 and 3,752,888), melanocyte-stimulating hormone receptor analogs, 5HT2c agonists, melanin concentrating hormone antagonists, leptin (the OB protein), leptin analogs, leptin receptor agonists, galanin antagonists, lipase inhibitors (e.g., tetrahydrolipstatin, i.e. orlistat), bombesin agonists, anorectic agents (e.g., a bombesin agonist), Neuropeptide-Y antagonists, thyroxine, thyromimetic agents, dehydroepiandrosterones or analogs thereof, glucocorticoid receptor agonists or antagonists, orexin receptor antagonists, urocortin binding protein antagonists, glucagon-like peptide-1 receptor agonists, ciliary neurotrophic factors (e.g., Axokine.TM.), human agouti-related proteins (AGRP), ghrelin receptor antagonists, histamine 3 receptor antagonists or inverse agonists, neuromedin U receptor agonists, and the like. Any thyromimetic agent known in the art may also be used in combination with a compound of the invention. Thyromimetic activity may be readily determined according to standard assays (e.g., Atherosclerosis (1996) 126: 53-63). Examples of suitable thyromimetic agents include, but are not limited to, those described in U.S. Pat. Nos. 4,766,121; 4,826,876; 4,910,305; 5,061 ,798; 5,284,971 ; 5,401 ,772; 5,654,468; and 5,569,674.
A compound of the invention may further be used in combination with an anti- resorptive agent (e.g., progestins, polyphosphonates, bisphosphonate(s), estrogen agonists/antagonists, estrogen, estrogen/progestin combinations, Premarin.RTM., estrone, estriol or 17. alpha.- or 17.beta.-ethynyl estradiol). Exemplary progestins are available from commercial sources and include, but are not limited to: algestone acetophenide, altrenogest, amadinone acetate, anagestone acetate, chlormadinone acetate, cingestol, clogestone acetate, clomegestone acetate, delmadinone acetate, desogestrel, dimethisterone, dydrogesterone, ethynerone, ethynodiol diacetate, etonogestrel, flurogestone acetate, gestaclone, gestodene, gestonorone caproate, gestrinone, haloprogesterone, hydroxyprogesterone caproate, levonorgestrel, lynestrenol, medrogestone, medroxyprogesterone acetate, melengestrol acetate, methynodiol diacetate, norethindrone, norethindrone acetate, norethynodrel, norgestimate, norgestomet, norgestrel, Ogestone phenpropionate, progesterone,
quingestanol acetate, quingestrone, and tigestol. Preferred progestins are medroxyprogestrone, norethindrone and norethynodrel.
Exemplary bone resorption inhibiting polyphosphates include polyphosphonates of the type described in U.S. Pat. No. 3,683,080. Preferred polyphosphonates are geminal diphosphonates (also referred to as bis-phosphonates), 6-amino-1-hydroxy-hexylidene-bisphosphonic acid and 1-hydroxy- 3(methylpentylamino)-propylidene-bisphosphonic acid. Tiludronate disodium, ibandronic acid, alendronate, resindronate, and zoledronic acid are each especially preferred polyphosphonates. The polyphosphonates may be administered in the form of the acid, or of a soluble alkali metal salt or alkaline earth metal salt. Hydrolyzable esters of the polyphosphonates are likewise included. Specific examples include, but are not limited to, ethane-1 -hydroxy 1 ,1-diphosphonic acid, methane diphosphonic acid, pentane-1-hydroxy-1 ,1-diphosphonic acid, methane dichloro diphosphonic acid, methane hydroxy diphosphonic acid, ethane-1-amino-1 ,1-diphosphonic acid, ethane-2- amino-1 ,1 -diphosphonic acid, propane-3-amino-1-hydroxy-1 ,1 -diphosphonic acid, propane-N,N-dimethyl-3-amino-1-hydroxy-1,1 -diphosphonic acid, propane-3,3-dimethyl- 3-amino-1-hydroxy-1 ,1 -diphosphonic acid, phenyl amino methane diphosphonic acid, N,N-dimethylamino methane diphosphonic acid, N(2-hydroxyethyl) amino methane diphosphonic acid, butane-4-amino-1-hydroxy-1 ,1 -diphosphonic acid, pentane-5-amino- 1 -hydroxy- -1,1-diphosphonic acid, hexane-6-amino-1-hydroxy-1 ,1 -diphosphonic acid and pharmaceutically acceptable esters and salts thereof.
Any estrogen agonist/antagonist known in the art which bind with the estrogen receptor, inhibit bone turnover and/or prevent bone loss may be used in a combination of the invention. More specifically, an estrogen agonist may be any chemical compound capable of binding to the estrogen receptor sites in mammalian tissue, and mimicking the actions of estrogen in one or more tissue. An estrogen antagonist may be any chemical compound capable of binding to the estrogen receptor sites in mammalian tissue, and blocking the actions of estrogen in one or more tissues. Such activities may be readily determined according to standard assays, including estrogen receptor binding assays, and standard bone histomorphometric and densitometer methods (Eriksen E. F. et al., Bone Histomoφhometry, Raven Press, New York, 1994, pages 1- 74; Grier S. J. et. al., "The Use of Dual-Energy X-Ray Absorptiometry In Animals", Inv. Radiol., 1996, 31(1):50-62; Wahner H. W. and Fogelman I., The Evaluation of
Osteoporosis: Dual Energy X-Ray Absorptiometry in Clinical Practice., Martin Dunitz Ltd., London 1994, pages 1-296). Examples of a suitable estrogen agonist/antagonist is 3-(4-(1 ,2-diphenyl-but-1-enyl)-phenyl)-acrylic acid (see Willson et al., Endocrinology, 1997, 138, 3901-3911); tamoxifen (ethanamine, 2-(-4-(1 ,2-diphenyl-1-butenyl)phenoxy)- N.N-dimethyl, (Z)-2-, 2-hydroxy-1 ,2,3-propanetricarboxylate (1 :1)) and related compounds (U.S. Pat. No. 4,536,516); 4-hydroxy tamoxifen (U.S. Pat. No. 4,623,660); raloxifene (methanone, (6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl)(4-(2-(1- piperidinyl)eth- oxy)phenyl)-hydrochloride)(U.S. Pat. No. 4,418,068); toremifene (ethanamine, 2-(4-(4-chloro-1 ,2-diphenyl-1-butenyl)phenoxy)-N,N-dimethyl- , (Z)-, 2- hydroxy-1,2,3-propanetricarboxylate (1 :1) (U.S. Pat. No. 4,996,225); centchroman (1-(2- ((4-(-methoxy-2,2, dimethyl-3-phenyl-chroman-4-yl)-phenoxy)-ethyl)-p- yrrolidine)(U.S. Pat. No. 3,822,287); levormeloxifene; idoxifene ((E)-I -(2-(4-(1-(4-iodo-phenyl)-2-phenyl- but-1-enyl)-phenoxy)-ethyl)-pyrro- lidinone (U.S. Pat. No. 4,839,155); 2-(4-methoxy- phenyl)-3-[4-(2-piperidin-1-yl-ethoxy)-phenoxy]-benzo[b]thio- phen-6-ol (U.S. Pat. No. 5,488,058); 6-(4-hydroxy-phenyl)-5-(4-(2-piperidin-1-yl-ethoxy)-benzyl)-naphthalen-2— ol (U.S. Pat. No. 5,484,795); (4-(2-(2-aza-bicyclo[2.2.1]hept-2-yl)-ethoxy)-phenyl)-(6- hydroxy-2-(4-hyd- roxy-phenyl)-benzo[b]thiophen-3-yl)-methanone (WO 95/10513 assigned to Pfizer Inc.); TSE-424 (Wyeth-Ayerst Laboratories); arazoxifene; derivatives of 2-phenyl-3-aroyl-benzoth- iophene and 2-pheny!-3-aroylbenzothiophene-1-oxide(U.S. Pat. No. 4,133,814); estrogen agonist/antagonists described in U.S. Pat. No. 4,133,814; and estrogen agonist/antagonists described in commonly assigned U.S. Pat. No. 5,552,412.
Especially preferred estrogen agonist/antagonists described in U.S. Pat. No. 5,552,412 are: cis-6-(4-fluoro-phenyl)-5-(4-(2-piperidin-1-yl-ethoxy)-phenyl)-5,6,- 7,8- tetrahydro-naphthalene-2-ol; (-)-cis-6-phenyl-5-(4-(2-pyrrolidin-1-yl-ethoxy)-phenyl)- 5,6,7,8-te- trahydro-naphthalene-2-ol (also known as lasofoxifene); cis-6~phenyl-5-(4-(2- pyrrolidin-1-yl-ethoxy)-phenyl)-5,6,7,8-tetrah- ydro-naphthalene-2-ol; cis-1-(6'- pyrrolodinoethoxy-3'-pyridyl)-2-phenyl-6-hydroxy-1 ,2,3,4- tetrahydronaphthalene; 1-(4'- pyrrolidinoethoxyphenyl)-2-(4"-fluorophenyl)-6-hydroxy-1 ,2,3,- 4-tetrahydroisoquinoline; cis-6-(4-hydroxyphenyl)-5-(4-(2-piperidin-1-yl-ethoxy)-phenyl)-5,6,- 7,8-tetrahydro- naphthalene-2-ol; and 1-(4'-pyrrolidinolethoxyphenyl)-2-phenyl-6-hydroxy-1 ,2,3,4- tetrahyd- roisoquinoline.
Any anti-osteoporosis agent known in the art may be used in a combination of the invention. Examples include, but are not limited to, parathyroid hormone (PTH) (a bone anabolic agent); parathyroid hormone (PTH) secretagogues (see, e.g., U.S. Pat. No. 6,132,774), particularly calcium receptor antagonists; calcitonin; and vitamin D and vitamin D analogs.
Any antihypertensive agent known in the art may be used in a combination of the invention. Antihypertensive activity may be determined according to standard tests (e.g. blood pressure measurements). Examples of suitable antihypertensive agents include, but are not limited to, (a) amlodipine and related dihydropyridine compounds (US Pat. Nos. 4,572,909 and 5,155,120) such as, but not limited to, amlodipine benzenesulfonate salt (also termed amlodipine besylate (Norvasc®))(U.S. Pat. No. 4,879,303) and other pharmaceutically acceptable acid addition salts of amlodipine (U.S. Pat. No. 5,155,120); (b) calcium channel blockers such as, but not limited to, bepridil (U.S. Pat. No. 3,962, 238 or U.S. Reissue No. 30,577), clentiazem (U.S. Pat. No. 4,567,175), diltiazem (U.S. Pat. No. 3,562), fendiline (U.S. Pat. No. 3,262,977), gallopamil (U.S. Pat. No. 3,261 ,859); mibefradil, prenylamine, semotiadil, terodiline, verapamil, aranipine, barnidipine, benidipine, cilnidipine, efonidipine, elgodipine, felodipine, isradipine, lacidipine, lercanidipine, manidipine, nicardipine, nifedipine, nilvadipine, nimodipine, nisoldipine, nitrendipine, cinnarizine, flunarizine, lidoflazine, lomerizine, bencyclane, etafenone, and perhexiline; (c) angiotensin converting enzyme inhibitors ("ACE-
Inhibitors") such as, but not limited to, alacepril (U.S. Pat. No. 4,248,883), benazepril (U.S. Pat. No. 4,410,520), captopril, ceronapril, delapril, enalapril, fosinopril, imadapril, lisinopril, moveltopril, perindopril, quinapril, ramipril, spirapril, temocapril, and trandolapril; (d) angiotensin-ll receptor antagonists such as, but not limited to, candesartan (U.S. Pat. No. 5,196,444), eprosartan (U.S. Pat. No. 5,185,351), irbesartan, losartan, and valsartan; (e) beta-adrenergic receptor blockers (beta- or β- blockers) such as, but not limited to, acebutolol (U.S. Pat. No. 3,857,952), alprenolol, amosulalol (U.S. Pat. No. 4,217,305), arotinolol, atenolol, befunolol, betaxolol; and (f) alpha-adrenergic receptor blockers (alpha- or α-blockers) such as, but not limited to, amosulalol (U.S. Pat. No. 4,217,307), arotinolol (U.S. Pat. No. 3,932,400), dapiprazole, doxazosin, fenspiride, indoramin, labetolol, naftopidil, nicergoline, prazosin, tamsulosin, tolazoline, trimazosin, and yohimbine, which may be isolated from natural sources according to methods well known to those skilled in the art.
Any HMGCoA reductase inhibitor agent known in the art may be used in a combination of the invention. HMGCoA reductase activity may be determined according to standard tests (e.g. blood plasma low density lipoprotein cholesterol (LDL- C) measurements). Examples of suitable HMGCoA reductase inhibitor agents include, but are not limited to, atorvastatin, simvastatin, fluvastatin, lovastatin, pravastatin, rosuvastatin, simvastatin, and cerivastatin. A number of patents have issued disclosing atorvastatin and include: U.S. Pat. Nos. 4,681,893, 5,273,995 and 5,969,156. A number of patents have issued disclosing rosuvastatin and include: U.S. Pat. Nos. 5,260,440 (RE37314), 6,858,618, and 6,894,058. A number of patents have issued disclosing cerivastatin and include: U.S. Pat Nos. 5,006,530, 5,169,857, and 5,401 ,746. A number of patents have issued disclosing fluvastatin and include: U.S. Patent Nos. 4,739,073 and 5,354,772. A number of patents have issued disclosing lovastatin and include: U.S. Patent Nos. 4,231 ,938, 4,294,926, and 4,319,039. A number of patents have issued disclosing pravastatin and include: U.S. Pat. Nos. 4,346,227, 4,410,629, and 4,448,979.
The present invention contains compounds that can be synthesized in a number of ways familiar to one skilled in organic synthesis. The following non-limiting reaction schemes illustrate the preparation of the compounds of the present invention. Unless otherwise indicated, all variables in the reaction schemes and the discussions that follow are as defined above. As would be understood by one of skill in the art, individual compounds may require manipulation of the conditions in order to accommodate various functional groups. A variety of protecting groups known to one skilled in the art may be required. Purification, if necessary, may be accomplished on a silica gel column eluted with the appropriate organic solvent system. Also, reverse phase HPLC or recrystallization may be employed.
Schemes 1- 4 illustrate general methods for the preparation of compounds of the present invention. These illustrations are non-limiting and may be adapted by those skilled in the art for the preparation of compounds not explicitly described. The specific naming of the stereochemistry of the final compounds (e.g. R1S etc) will depend on the substituents selected for the variables shown, as those skilled in the art will understand. The use of solid and hashed wedges in the structures depicted will have the meaning well understood by those skilled in the art.
Scheme 1 illustrates the preparation of variations of formula (1) where Z is an optionally substituted carbon side chain. As shown, aldehydes of structure (1) can undergo hydroxymethylation by treatment with formaldehyde, N-ethyl-benzothiazole bromide and a suitable base to provide compounds of structure (2). Reaction of structure (2) with a suitable aldehyde (3) and suitable amine (4) in the presence of a suitable chiral catalyst such as L-proline affords amino-alcohols of structure (5). Treatment of structure (5) with triphosgene or a suitable equivalent and a base provides oxazolidinones of structure (6) which can be then be converted to structure (7) via the use of a suitable reducing agent such as sodium borohydride.
Scheme 1. Synthesis of a compounds of formula (I) where Y1 = O and Y2 = N

Tnphosgene base
Reduction


Scheme 2 illustrates the preparation of variations of formula (1) where Z contains an amine or ether linkage. As illustrated, condensation of hydroxyl acetone (8) with a suitable aldehyde (3) and a suitable amine (4) in the presence of a suitable chiral catalyst such as L-proline afford amino-alcohols of structure (9). Treatment of structure
(9) with triphosgene or a suitable equivalent and a base provides oxazolidinones of structure (10) which can then be D-brominated using phenyltrimethylammonium tribromide or an equivalent brominating agent to afford structures of type (11).
Reduction of structure (11) with sodium borohydride or a suitable equivalent provides structure (12) which can be treated with base to afford epoxides of structure (13).
Reaction of structure (13) with alcohols or amines in the presence or a suitable base provides structures (14) and (15) respectively.
S amine linkage.


ArNHR' Base

Scheme 3 illustrates a variation of Scheme 2 for the construction of structures of formula (I) wherein Z contains a ring system. Hence, epoxide (16) is treated with amine Ar2NHZtO afford amino alcohol (17) which can then be treated with an electrophile of type (18) to provide structure (19). Treatment of structure (19) with a suitable base affords structure (20).
Scheme 3. Synthesis of a compounds of formula (I) where Z contains a ring system.

Scheme 4 illustrates the preparation of variations of formula (1) where Z contains an amide linkage. As illustrated, oxazolidine (10), from Scheme 2, is subjected to a haloform reaction using sodium bromate (generated in-situ from NaOH and Br2) or a suitable equivalent to provide a carboxylic acid of structure (21). Structure (21) can be reacted with various amines in the presence of a suitable coupling reagent such as EDCI to afford structure (22). Alternatively, structure (21) can be treated with a suitable reducing agent such as borane to afford alcohols of structure (23). Conversion of
structure (23) to amines of structure (24) followed by acylation with suitable acid chlorides provides amides of general structure (25).
Scheme 4. Synthesis of a compounds of formula (I)where Z contains an amide.

EXAMPLES
The following non-limiting examples show how to carry out the present invention. The synthetic route to compounds of the present invention is not limited to the methods outlined below. One skilled in the art will be able to use the schemes outlined above to synthesize various compounds claimed in this invention.
Example 1 : 3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one

Step A
1 -Hydroxy-4-phenyl-butan-2-one

Hydrocinnamaldehyde (50.0 g, 0.37moles), paraformaldehyde (11.0 g, 0.37moles), N- ethyl-benzothiazole bromide (16.0 g, 0.067moles) and triethylamine (9.0 ml, 0.067moles) were dissolved in ethanol (500 ml) under nitrogen and heated at 70 0C overnight. The mixture was cooled to 25 0C and evaporated, yielding a brown oil. The crude material was purified using column chromatography (10-30% EtOAc/hexane) yielding 1-hydroxy-4-phenyl-butan-2-one (31.0 g, 51%) as an oil which crystallized on standing. 1H-NMR (400 MHz, d-CDCI3) δ 7.29-7.24 (m, 2 H), 7.21-7.14 (m, 3 H), 4.17 (s, 2 H), 2.95 (t, 2 H), 2.72 (t, 2 H).
Step B
2-Hydroxy-1-(4-methoxy-phenyl)-1-(4-methoxy-phenylamino)-5-phenyl-pentan-3-one

1-Hydroxy-4-phenyl-butan-2-one (2.0 g, 0.012moles), p-anisidine (0.95g, 0.0079moles), p-anisialdehyde (0.95 g, 0.0071 moles) and L-proline (0.13g, 0.0014moles) were dissolved in DMSO (12ml) under nitrogen and stirred for six days at 25 0C. The reaction mixture was extracted with water (50ml) and EtOAc (2 x 50ml). The organic layers were dried with MgSO4, filtered and evaporated to dryness. The residue was then purified using column chromatography with a (10% ether:CH2CI2) to afford 2-hydroxy-1-(4- methoxy-phenyl)-1-(4-methoxy-phenylamino)-5-phenyl-pentan-3-one (1.5g, 52%) as an oil.
Step C
3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one

2-Hydroxy-1-(4-methoxy-phenyl)-1-(4-methoxy-phenylamino)-5-phenyl-pentan-3-one (1.5g, 3.7 mmol), was dissolved in CH2CI2 (60ml) under nitrogen followed by the addition of triethylamine (1.9 g, 18 mmol) and chilled to -20 0C. A solution of triphosgene (1.1g, 3.7 mmol) dissolved in CH2CI2 (10ml) was then added drop-wise over 10 min. The reaction mixture was stirred overnight allowing it to warm to 25 0C over this period. The reaction was quenched by the addition of a saturated solution of ammonium chloride (60ml). The layers were separated, organic layer dried with magnesium sulfate, filtered and evaporated to dryness, yielding a brown oily residue which was purified by column chromatography (20% EtOAc:hexane) to provide 3,4R- bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one (450mg, 28%) as a
brown oil. 1H-NMR (500 MHz, d-CDCI3) δ 7.5-7.1 (m, 9H), 6.8 (d, 2H), 6.7 (d, 2H), 5.1 (d, 1H), 4.5 (d, 1H), 3.8 (s, 3H), 3.7 (s, 3H), 3.2-2.9 (m, 4H).
Example 2: Acetic acid 1-[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3- phenyl-propyl ester

Step A
5-(1-Hydroxy-3-phenyl-propyl)-3,4-bis-(4-methoxy-phenyl)-oxazolidin-2-one

3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one [from Example 1] (2.1 g, 4.8 mmol) was dissolved in a mixture of methanol (6ml) and THF (6ml) and cooled to 0 0C under nitrogen. Sodium borohydride (0.18g, 4.8 mmol) was then added portion-wise to the reaction and the mixture allowed to warm to 25 0C. 2M HCI was then added to the reaction and extracted with EtOAc, the resulting layers separated, organic dried with magnesium sulfate, filtered and evaporated to dryness. The resulting compound was purified using column chromatography (30-50% EtOAc:hexane) to provide 5-(1-hydroxy-3-phenyl-propyl)-3,4-bis-(4-methoxy-phenyl)-oxazolidin-2-one (1.4g, 66%) as a mixture of diastereomers at the hydroxyl position.
Step B
Acetic acid 1 R-[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl-propyl ester

5-(1-Hydroxy-3-phenyl-propyl)-3,4-bis-(4-methoxy-phenyl)-oxazolidin-2-one (1.0 g, 2.3 mmol) was dissolved in acetonitrile (20 ml) followed by the addition of pyridine (1.8 g, 23 mmol) and acetic anhydride (2.3 g, 23 mmol) under nitrogen and the reaction mixture stirred at 25 0C overnight. Reaction mixture was then washed with 1 N HCI (50ml) and extracted with EtOAc (2 x 50ml). The resulting layers were separated, organic layer dried with magnesium sulfate, filtered and evaporated to dryness yielding a yellow oil which was purified using column (0.2-0.5% THF.OH2CI2) affording acetic acid 1R-[3,4R- bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl-propyl ester (500 mg) as the lower Rf fraction. 1H-NMR (500 MHz, d-CDCI3) δ 7.5-7.0 (m, 9H), 6.85 (d, 2H), 6.77 (d, 2H), 5.30 (m, 1H), 4.99 (d, 1H), 4.42 (t, 1 H), 3.77 (s, 1H), 3.72 (s, 1H), 2.7-2.5 (m, 2H), 2.06 (m, 4H). The higher Rf fraction from this reaction was also isolated and is reported as Example 3 below.
Example 3: Acetic acid 1S-[3,4-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl- propyl ester

Acetic acid 1S-[3,4-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester (320mg) was isolated as the higher Rf fraction from the procedure of Example 2 (Step B) as a white solid. 1H-NMR (500 MHz, d-CDCI3) δ 7.5-7.0 (m, 9H), 6.87 (d, 2H), 6.77 (d, 2H), 5.24 (m, 1 H), 4.88 (d, 1H), 4.38 (d, 1 H), 3.76 (s, 3H), 3.71 (s, 3H), 2.65 (t, 2H), 2.4-1.9 (m, 5H).
Example 4: 5S-(1 R-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2- one

Acetic acid 1 R-[3,4R-bιs-(4-methoxy-phenyl)-2-oxo-oxazolιdιn-5S-yl]-3-phenyl-propyl ester [from Example 2] (289mg, O 61 mmol) was dissolved in a methanol (7 ml) at 25 0C and potassium carbonate (168 mg, 1 2 mmol) added The mixture was stirred under nitrogen The reaction was concentrated under vacuum, water (50ml) was added and extracted into ethyl acetate (2 x 50 ml) The organic layer was separated, dried, filtered and evaporated to dryness yielding the desired product as an oil which was triturated in hexane and dried under high vacuum 5S-(1R-hydroxy-3-phenyl-propyl)-3,4R-bιs-(4~ methoxy-phenyl)-oxazolιdιn-2-one (198mg, 76%) was obtained as an off-white solid 1H-NMR (500 MHz, d-CDCI3) 5 7 5-7 0 (m, 9H)1 6 84 (d, 2H), 6 76 (d, 2H), 5 24 (d, 1H), 4 32 (m, 1 H), 3 99 (m, 1 H), 3 77 (s, 3H), 3 72 (s, 3H), 2 84 (m, 1 H), 2 63 (m, 1 H), 2 17 (bs, 1H), 1 8-1 6 (m, 2H)
Example 5 5S-(1 S-hydroxy-3-phenyl-propyl)-3,4R-bιs-(4-methoxy-phenyl)-oxazolιdιn-2- one

Acetic acid 1S-[3,4-bιs-(4-methoxy-phenyl)-2-oxo-oxazolιdιn-5-yl]-3-phenyl-propyl ester
[from Example 3] (298mg, 0 63mmol) was treated according to the procedure of Example 4 to afford 5 5S-(1S-hydroxy-3-phenyl-propyl)-3,4R-bιs-(4-methoxy-phenyl)- oxazohdιn-2-one (250mg, 92%) 1H-NMR (500 MHz, d-CDCI3) 5 7 3-7 0 (m, 9H), 7 68 (d, 2H), 6 77 (d, 2H), 5 14 (d, 1H), 4 27 (m, 1H), 3 80 (s, 3H), 3 70 (s, 3H), 2 4-2 2 (m, 2H), 2 1-1 8 (3H)
Example 6 4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propιonyl)- oxazolιdιn-2-one

Step A
1-(4-Benzyloxy-phenyl)-1-(4-fluoro-phenylamino)-2-hydroxy-5-phenyl-pentan-3-one

1-Hydroxy-4-phenyl-butan-2-one [from Example 1 , Step A] (2.0 g, 0.012moles), 4- fluoroaniline (0.87 g, 0.0078moles), 4-benzyloxybenzaldehyde (1.5 g, 0.0071 moles) and L-proline (0.16g, 0.0014moles) were dissolved in DMSO (12ml) under nitrogen and stirred for 48 hrs at 25 0C. The reaction mixture was extracted with water (50 ml) and EtOAc (2 x 50ml). The organic layer was dried with MgSO4, filtered and evaporated to dryness. The residue was the purified by column chromatography (20-30% EtOAc:hexane)to afford 1-(4-benzyloxy-phenyl)-1-(4-fluoro-phenylamino)-2-hydroxy-5- phenyl-pentan-3-one (0.90 g, 27%).
Step B
4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one

Example 7: 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propionyl)- oxazolidin-2-one

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one [from Example 6] (510mg, LOmmol) was dissolved in a mixture of ethanol (6ml) and ethyl acetate (6 ml) followed by the addition of cyclohexene (0.63 ml, 6.0 mmol) and 10%Pd/C catalyst (50 mg). The mixture was refluxed overnight under nitrogen. The reaction was then cooled to 25 °C and filtered through a pad of celite. The resulting filtrate was then evaporated to dryness yielding the desired product as a white solid, which was slurried in hexane, filtered and dried to provide 3-(4-fluoro-phenyl)-4R-(4- hydroxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one (450mg, 77%). 1H-NMR (500 MHz, d-CDCIa) δ 7.4-7.0 (m, 9H), 6.9 (t, 2H), 6.7 (d, 2H), 5.2 (d, 1 H), 5.1 (bs, 1H), 4.55 (el, 1H), 3.2-2.9 (m, 4H).
Example 8: 4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl- propyl)-oxazolidin-2-one

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one [from Example 6] was converted 4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1R- hydroxy-3-phenyl-propyl)-oxazolidin-2-one using the reduction, acetylation, chromatographic separation and deacetylation procedures described for Examples 2 (Steps A and B) and Example 4. 1H-NMR (500 MHz, d-CDCI3) δ 7.5-7.0 (m, 14H), 6.9 (m, 4H), 5.18 (d, 1 H), 5.02 (s, 2H), 4.27 (m, 1H), 3.70 (m, 1H), 2.79 (m, 1 H), 2.70 (m, 1H), 2.04 (m, 1H), 1.93 (m, 2H).
Example 9: 4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1 S-hydroxy-3-phenyl- propyl)-oxazolidin-2-one

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one [from Example 6] was converted 4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S- hydroxy-3-phenyl-propyl)-oxazolidin-2-one using the reduction, acetylation, chromatographic separation and deacetylation procedures described for Examples 2 (Steps A and B) and Example 4.
Example 10: 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1 R-hydroxy-3-phenyl- propyl)-oxazolidin-2-one

4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one [from Example 8] (400mg, 0.8mmol) was dissolved in a mixture of ethanol (6ml) and ethyl acetate (6ml) followed by the addition of cyclohexene (396mg, 4.8mmol) and Pd/C catalyst (40mg). The mixture was refluxed overnight under nitrogen. The reaction was cooled to 25 0C and filtered through a pad of celite. The resulting filtrate was then evaporated to dryness, slurried in hexane, filtered and dried yielding 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one (240mg, 74%) as a white solid. 1H-NMR (500 MHz, d-CDCI3) δ 7.4-7.0 (m, 9H), 6.9 (t, 2H), 6.7 (d, 2H), 5.1 (d, 1H), 4.8 (d, 1 H), 4.2 (m, 1H), 3.7 (m, 1H), 2.9-2.6 (m, 2H), 2.1-1.8 (m, 3H).
Example 11: 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1 R-hydroxy-3-phenyl- propyl)-oxazolidin-2-one

4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one [from Example 9] (600 mg, 1.2 mmol) was dissolved in a mixture of ethanol (7 ml) and ethyl acetate (7 ml) followed by the addition of cyclohexene (595mg, 7.2 mmol) and Pd/C catalyst (60 mg). The mixture was refluxed overnight under nitrogen. The reaction was cooled to 25 0C and filtered through a pad of celite. The resulting filtrate was then evaporated to dryness, slurried in hexane, filtered and dried yielding 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-
oxazolidin-2-one (450mg, 92%) as a white solid. 1H-NMR (500 MHz, d-CDCI3) δ 7.3-7.0 (m, 14H), 7.9 (t, 2H), 6.7 (d, 2H), 5.2 (d, 1H), 4.8 (s, 1H), 4.3 (m, 1H), 3.9 (m, 1H), 2.1 (d, 1H), 1.8-1.5 (m, 2H). Example 13 -27:

Examples 13 - 27 were prepared using procedures analogous to those described for Examples 10 and 11 above.

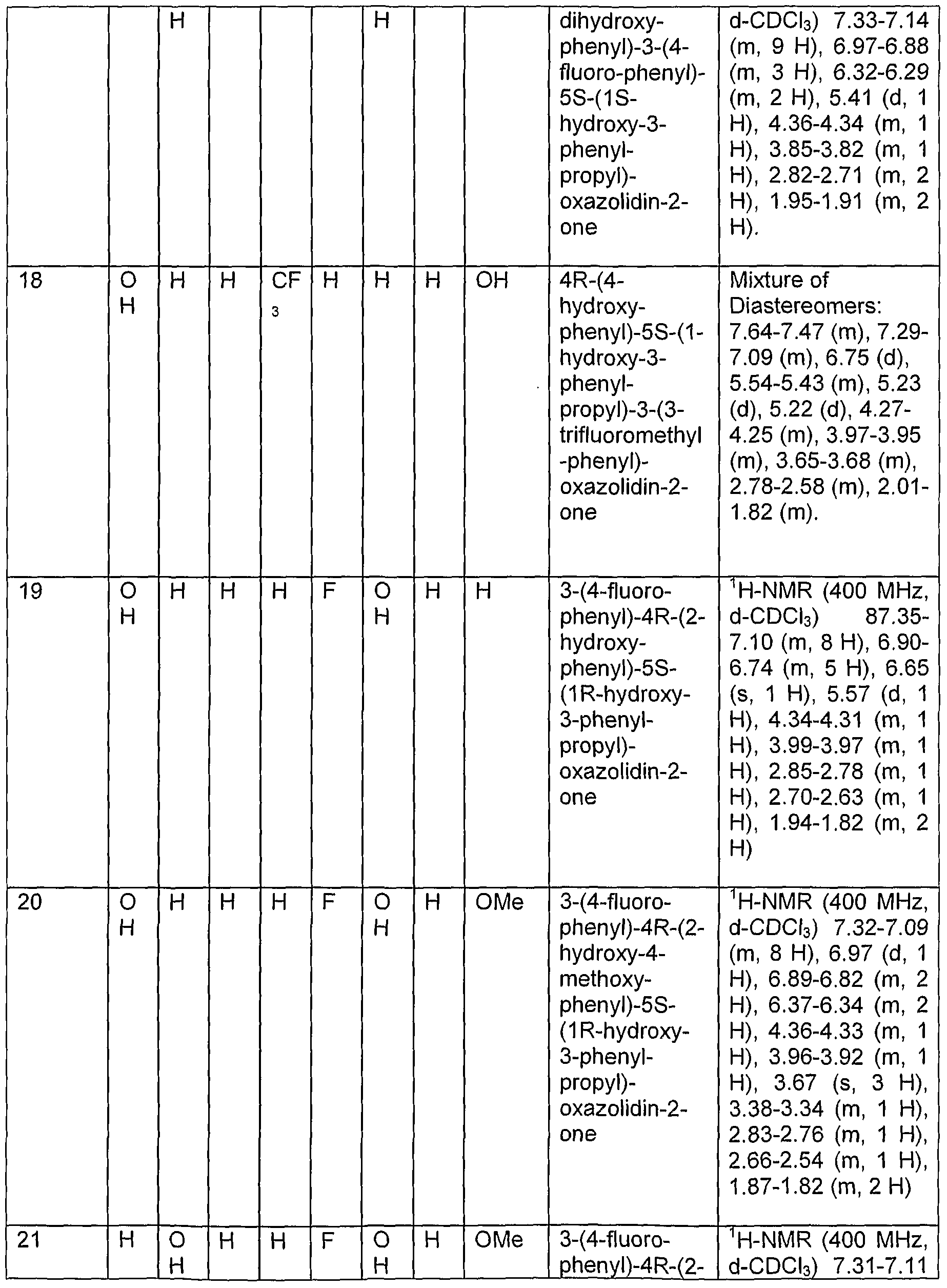


Example 28: 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin- 2-one

4-(4-Benzyloxy-phenyl)-5-(1-bromo-3-phenyl-propyl)-3-(4-fluoro-phenyl)-oxazolidin-2- one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-oxazolidin-2- one [from Example 8] (1.6g, 3.2mmol) was dissolved in CHoCI2 (40 ml) followed by the addition of carbon tetrabromide (6.3 g, 19mmol) and triphenylphosphine (1.7 g, 6.4 mmol) under nitrogen. The mixture was refluxed overnight. The reaction mixture was then was cooled to 25 0C and adsorbed directly onto silica and purified by column chromatography (10-40% EtOAc:hexane) to afford 4-(4-benzyloxy-phenyl)-5-(1-bromo- 3-phenyl-propyl)-3-(4-fluoro-phenyl)-oxazolidin-2-one (1.4 g, 78%).
Step B
4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-(3-phenyl-propyl)-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-5-(1-bromo-3-phenyl-propyl)-3-(4-fluoro-phenyl)-oxazolidin-2- one (1.4 g, 2.5 mmol) was dissolved in CH2CI2 (40ml) followed by the addition of AIBN (41 mg, 0.25 mmol). The mixture was refluxed for 10 min under nitrogen. Tributyltin hydride (0.87 mg, 3 mmol) was then added and the reaction mixture was heated to reflux overnight. The reaction was then cooled to 25 0C and adsorbed onto silica which was then purified by column chromatography on a 10% w/w KF/Silica column (20%
EtOAc: hexane) to afford 4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-(3-phenyl-propyl)- oxazolidin-2-one (0.70 g, 58%).
Step C 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-(3-phenyl-propyl)-oxazolidin-2-one (0. 50 g, 1.0 mmol) was dissolved in a mixture of ethanol (10ml) and ethyl acetate (10 ml) followed by the addition of cyclohexene (512 mg, 6 mmol) and Pd/C catalyst (50 mg). The mixture was refluxed overnight under nitrogen. The reaction mixture was then cooled to 25 0C and filtered through a pad of celite. The resulting filtrate was then evaporated to dryness yielding the desired product as a white solid which was slurried in hexane filtered and dried, to provide 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3- phenyl-propyl)-oxazolidin-2-one (0.30 g, 76%). 1H-NMR (500 MHz, d-CDCI3) δ 7.5-7.0 (m, 9H), 6.9 (t, 2H), 6.8 (d, 2H), 5.0 (s, 1 H), 4.7 (d, 1H), 4.3 (m, 1H), 2.6 (m, 2H), 1.9-1.6 (m, 4H).
Example 29: 3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one

Prepared according to the method of Example 28 starting from 5S-(1 R-hydroxy-3- phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one [from Example 4]. 1H-
NMR (500 MHz, d-CDCb) δ 7.4-7.1 (m, 9H), 6.8 (d, 2H), 6.7 (d, 2H), 4.7 (d, 1H), 4.3 (m, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 2.65 (m, 2H), 2.0-1.7 (m, 4H).
Example 30: {4-[3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin- 4R-yl]-phenoxy}-acetic acid

Step A Acetic acid 1-[3-(4-fluoro-phenyl)-4-(4~hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl- propyl ester

Acetic acid 1-[4-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-3- phenyl-propyl ester [from Example 8] (400 mg, 0.74 mmol) was dissolved in a mixture of ethanol (6ml) and ethyl acetate (6ml) followed by the addition of cyclohexene (365mg, 4.4mmol) and Pd/C catalyst (40mg). The mixture was refluxed overnight under nitrogen. The reaction was cooled to 25 C and filtered through a pad of celite. The resulting filtrate was then evaporated to dryness, slurried in hexane, filtered and dried under vacuum at 500C yielding acetic acid 1-[3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- 2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester (230mg, 69%) as a white solid.
Step B
{4-[5-(1-Acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-4-yl]-phenoxy}- acetic acid methyl ester

To a solution of acetic acid 1-[3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5-yl]-3-phenyl-propyl ester (0.50 g, 1.11 mmol) in DMF (15 ml_) at 25 C was added powdered K2CO3 (0.23 g, 1.67 mmol) followed by methyl bromoacetate (0.20 g, 1.33 mmol). The reaction mixture was stirred at 25 C for 14 hrs. Subsequently, ethyl acetate (50 ml.) and water (50 ml.) were added and the organic layer was separated, washed with brine, dried and concentrated to an oil that was purified by column chromatography (50% EtOAc/Hex) to provide {4-[5-(1-Acetoxy-3-phenyl-propyl)-3-(4- fluoro-phenyl)-2-oxo-oxazolidin-4-yl]-phenoxy}-acetic acid methyl ester (0.45 g, 77%). 1H-NMR (400 MHz, d-CDCI3) δ 7.25-7.11 (m, 9 H), 6.94-6.83 (m, 4 H), 5.29-5.25 (m, 1 H), 5.00 (d, 1 H), 4.58 (s, 2 H), 4.37 (t, 1 H), 3.77 (s, 3 H), 2.68-2.56 (m, 2 H), 2.02 (s, 3 H).
Step C
{4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- phenoxy}-acetic acid

Example 31 : 1-(4-{4-[5S-(1R-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo- oxazolidin-4R-yl]-phenoxymethyl}-benzyl)-1-azonia-bicyclo[2.2.2]octane chloride

Step A
Acetic acid 1 -[4-[4-(4-chloromethyl-benzyloxy)-phenyl]-3-(4-fluoro-phenyl)-2-oxo- oxazolidin-5-yl]-3-phenyl-propyl ester

Step B
1-(4-{4-[5S-(1R-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-4R-yl]- phenoxymethyl}-benzyl)-1-azonia-bicyclo[2.2.2]octane chloride

To a solution of acetic acid 1-[4-[4-(4-chloromethyl-benzyloxy)-phenyl]-3-(4-fluoro- phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester (0.36 g, 0.61 mmol) in MeCN (15 mL) at 25 C was added a solution of quinuclidine (0.067 g, 0.61 mmol). The reaction mixture was stirred at 25 0C for 16 hrs and then was concentrated to provide 1-(4-{4- [5S-(1R-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-4R-yl]-
phenoxymethyl}-benzyl)-1-azonia-bicyclo[2.2.2]octane chloride (0.42 g, 99%) as a white foam. 1H-NMR (40 MHz, d-DMSO) δ 7.52-7.41 (m, 6 H), 7.31 (d, 2 H), 7.23 (t, 2 H), 7.16-7.09 (m, 5 H), 6.97 (d, 2 H), 5.52 (d, 1 H), 5.18-5.15 (m, 1 H), 5.05 (s, 2 H), 4.54- 4.51 (m, 1 H), 4.36 (s, 2 H)1 3.37-3.33 (m, 6 H), 2.65-2.53 (m, 2 H), 2.04-1.80 (m, 10 H).
Example 32: 6-{4-[3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin- 4-yl]-phenoxy}-3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid

Step A
3,4,5-Triacetoxy-6-hydroxy-tetrahydro-pyran-2-carboxylic acid methyl ester

To a solution of SAδ-Ttriacetoxy-e-bromo-tetrahydro-pyran^-carboxylic acid methyl ester [Sigma Aldrich] (5.0 g, 12.6 mmol) and CdCO3 (2.28 g, 13.2 mmol) in acetonitrile (10 mL) was added water (1.6 mL) and the reaction mixture was heated to 70 0C for 4 hrs . After cooling to 25 0C, the reaction mixture was filtered through a pad of celite and the filtrate was concentrated and purified by column chromatography (60% ethyl acetate/hexane).to afford 3,4,5-Triacetoxy-6-hydroxy-tetrahydro-pyran-2-carboxylic acid methyl ester (3.26 g, 77%) as a mixture of QD-anomers.
Step B
3,4,5-Triacetoxy-6-(2,2,2-trichloro-1-imino-ethyl)-tetrahydro-pyran-2-carboxylic acid methyl ester

To a solution of SAS-triacetoxy-δ-hydroxy-tetrahydro-pyran^-carboxylic acid methyl ester (3.64 g, 10.9 mmol) in dichloroethane (100 mL) at 0 0C was added trichloroacetonitrile (15.7 g, 108.9 mmol) followed by 1 ,8-diazabicyclo[5.4.0]undec-7- ene (0.46, 3.0 mmol). The reaction mixture was stirred at 0 0C for 1 hr as and then was concentrated under reduced pressure to a brown oil that was purified by column chromatography (40% ethyl acetate/59.9% hexane/0.1% triethylamine) to provide 3,4,5- triacetoxy-6-(2,2,2-trichloro-1-imino-ethyl)-tetrahydro-pyran-2-carboxylic acid methyl ester (2.57 g, 49 %). 1H-NMR (400 MHz, d-CDCI3) δ 8.71 (s, 1 H), 6.61 (d, 1 H), 5.59 (t, 1 H), 5.27 (t, 1 H), 5.12 (dd, 1 H), 4.47 (d, 1 H), 3.74 (s, 3 H), 2.09-2.02 (m, 9 H).
Step C
3,4,5-Triacetoxy-6-{4-[5-(1-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo- oxazolidin-4-yl]-phenoxy}-tetrahydro-pyran-2-carboxylic acid methyl ester

To a solution of acetic acid 1-[3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo~ oxazolidin-5-yl]-3-phenyl-propyl ester [from Example 30] (0.94 g, 2.1 mmol) and 3,4,5- triacetoxy-6-(2,2,2-trichloro-1-imino-ethyl)-tetrahydro-pyran-2-carboxylic acid methyl ester (1.0 g, 2.1 mmol) in CH2CI2-THF (10:1 , 125 mL) at -20 0C was added BF3 OEt2
(0.03 g, 0.21 mmol). The reaction mixture was stirred at -20 0C for 1 hr and then at 25 0C for 1.5 hr. The reaction mixture was quenched by addition of saturated ammonium chloride and additional CH2CI2 (50 mL) was added. The organic layer was separated, dried and concentrated to an oil that was purified by column chromatography (25-75%, ethyl acetate/hexane) to afford 3,4,5-Triacetoxy-6-{4-[5-(1-acetoxy-3-phenyl-propyl)-3- (4-fluoro-phenyl)-2-oxo-oxazolidin-4-yl]-phenoxy}-tetrahydro-pyran-2-carboxylic acid methyl ester. APCI MS (M+1): 766.4.
Step D 6-{4-[3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]- phenoxy}-3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid

To a solution of 3,4,5-Triacetoxy-6-{4-[5-(1-acetoxy-3-phenyl-propyl)-3-(4-fluoro- phenyl)-2-oxo-oxazolidin-4-yl]-phenoxy}-tetrahydro-pyran-2-carboxylic acid methyl ester (0.36 g, 0.47 mmol) in MeOH (10 mL) and triethylamine (10 mL) at 25 0C was added (drop wise) water (18 mL) over 10 min. The reaction mixture was stirred at 25 0C for 16 hrs. Subsequently, the MeOH and triethylamine were removed under reduced pressure and ethyl acetate (50 mL) and 1N HCI (15 mL) were added. The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated. The crude product was purified by preparative TLC (5% AcOH/15% MeOH/80 % DCM). The product was recovered from the silica scrapings by washing (3 x) with MeOH: CH2CI2 (1 :1) followed each time by filtration. After concentration of the combined filtrates, 6-{4- [3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]-phenoxy}- 3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid (0.085 g, 31 %) was obtained as a
light yellow foam. 1H-NMR (400 MHz, d-MeOH) δ 7.39-6.93 (m, 13 H), 5.36 (d, 1 H), 5.01-4.85 (m, 2 H), 4.86-4.83 (m, 1 H), 3.89-3.85 (m, 1 H)1 3.29-3.21 (m, 2 H), 2.81-2.76 (m, 1 H), 2.58-2.55 (m, 1 H), 2.11-2.01 (m, 1 H), 1.76-1.71 (m, 2 H).
Example 33: 5S-[2-(4-fluoro-phenoxy)-1R-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4- hydroxy-phenyl)-oxazolidin-2-one

Step A 4-(4-Benzyloxy-phenyl)-4-(4-fluoro-phenylamino)-3-hydroxy-butan-2-one

A solution of hydroxyacetone (36 ml_, 0.5 mole), 4-benzyloxybenzaldehyde (21.2 g, O.imoles), 4-fluoroaniline (10.4 ml_, 0.11 mole), and L-proline (2.3 g, 0.02 mole) in 250 ml_ dimethylsulfoxide was stirred at ambient temperature for 4 days. The solution was poured into 2000 mL of sat. ammonium chloride, then extracted with a 3:2 of ethyl acetate:hexanes (2 x 1000 mL). Washed combined organics with brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography, eluting with a gradient of 10-70% ethyl acetate in hexanes. Combined and concentrated fractions to yield title compound as an oil. (29.6 g, 78%) APCI MS (M+1): 380.22.
Step B 5-Acetyl-4-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-4-(4-fluoro-phenylamino)-3-hydroxy-butan-2-one (29.6 g, 78mmols) was dissolved in 400 mL dichloromethane, cooled to -780C, added triethylamine (54.4 mL, 0.39moles), followed by the drop wise addition of a solution of triphosgene (23.2 g, 78mmoles) in 100 mL dichloromethane. Allowed the solution to gradually warm to ambient temperature over the course of several hours. Stirred at ambient temperature overnight. The solution was transferred to a separatory funnel and washed with water (3 x 300 mL), brine, dried with magnesium sulfate, filtered and concentrated in vacuo. Purified by column chromatography eluting with a gradient of 20-100% ethyl acetate in hexanes. Combined and concentrated fractions that contained desired product. Repurified by column chromatography eluting with 100% dichloromethane, then a gradient of 0-20% methanol in dichloromethane. Combined and concentrated desired fractions to yield title compound as an oil. (11.3 g, 36%) APCI MS (M+1): 406.16.
Step C
4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-oxiranyl-oxazolidin-2-one

5-Acetyl-4-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-oxazolidin-2-one (8.2 g, 20mmoles) was dissolved in 60 mL anhydrous tetrahydrofuran under a nitrogen atmosphere, cooled to O0C, then added phenyltrimethylammonium tribromide (8.2 g, 21.2mmoles) in portions. Solution was stirred at O0C for 5 minutes. Stirred at ambient temperature overnight. Added 150 mL diethyl ether and filtered off a white solid, washing with more diethyl ether. Concentrated filtrates to a brown oil. The oil was redissolved in 100 mL ethanol, cooled to O0C, then added sodium borohydride (0.34 g, 9.1mmoles). Stirred at 00C for 60 minutes. Poured solution into 200 mL of 1M aqueous hydrogen chloride at 00C. Extracted with ethyl acetate, washed with brine, dried with magnesium sulfate, filtered and concentrated to an oil. The oil was dissolved in 60 mL anhydrous tetrahydrofuran under a nitrogen atmosphere, added sodium terf-butoxide (2.7 g, 28mmoles) and stirred at ambient temperature for 1 hour. The solution was then partitioned between ethyl acetate and water, washed with brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 10-80% ethyl acetate in heptanes. Combined and concentrated fractions to an oil. Repurified by column chromatography eluting with a gradient of 10-80% ethyl acetate in heptanes. Concentrated in vacuo to yield title compound. (4.03 g, 50%) APCI MS (M+1): 406.08.
Step D
4-(4-Benzyloxy-phenyl)-5-[2-(4-fluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)- oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-oxiranyl-oxazolidin-2-one (1.6 g,
3.9mmoles) was dissolved in 20 ml_ tetrahydrofuran, added 4-fluorophenol (0.66 g, 5.9mmoles) and sodium fert-butoxide (0.57 g, 5.9mmoles). Stirred at ambient temperature for 1 hour. The solution was partitioned between ethyl acetate and water, washed with brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 0-80% ethyl acetate in heptanes.
Combined and concentrated desired fractions to yield title compound. (0.71 g, 35%) APCI MS (M+1): 518.19.
Step E
5S-[2-(4-fluoro-phenoxy)-1R-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)- oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-5-[2-(4-fluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)- oxazolidin-2-one was dissolved in 20 mL of ethyl acetate:ethanol (1:1), added cyclohexene (0.67 mL, 6.86mmols), followed by 10% palladium on carbon (0.15 g, 0.14mmols). Heated suspension to reflux for 2 hours. Filtered through a pad of celite, washing with ethyl acetate. Concentrated in vacuo. Purified by column chromatography eluting with a gradient of 10-80% ethyl acetate in hexanes. Combined
and concentrated pure fractions to a white sticky solid. Redissolved in 6 mL acetonitrile and added water slowly while stirring (total volume was 40 mL). Slurried at ambient temperature overnight. Filtered off solids, washing with several portions of water to yield title compound (360 mg, 61%) APCI MS (M+1): 428.11 ; Analytical HPLC (Method A) Retention time = 17.0 min. (>99%).
Example 34: 5S-[2-(4-fluoro-phenoxy)-1 S-hydroxy-ethyr]-3-(4-fluoro-phenyl)-4R-(4- hydroxy-phenyl)-oxazolidin-2-one

5S-[2-(4-fluoro-phenoxy)-1S-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)- oxazolidin-2-one was synthesized using procedures analogous to Example 33 described above. APCI MS (M+1): 428.10; Analytical HPLC (Method A) Retention time = 16.8 min. (94.5%).
Example 35: 3-(4-fluoro-phenyl)-5S-[2-(4-fluoro-phenylarnino)-1 -hydro xy-ethyl]-4R-(4- hydroxy-phenyl)-oxazolidin-2-one

Step A
4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[2-(4-fluoro-phenylamino)-1-hydroxy- ethyl]-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-oxiranyl-oxa2olidin-2-one (0.79 g ,
1.95mmoles) was dissolved in 10 mL diethyl ether, added lithium perchlorate (1.0 g, 9.7mmols) and 4-fluoroanline (0.22 mL, 2.3mmols) and stirred at ambient temperature overnight. The solution was diluted with 50 mL diethyl ether, washed with water, brine, dried with magnesium sulfate, filtered and concentrated in vacuo. Purified by column chromatography eluting with a gradient of 0-80% ethyl acetate in heptanes. Combined and concentrated pure fractions to yield title compound. (0.65 g, 65%) APCI MS (M+1): 517.25.
Step B
3-(4-fluoro-phenyl)-5S-[2-(4-fluoro-phenylamino)-1-hydroxy-ethyl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[2-(4-fluoro-phenylamino)-1~hydroxy- ethyl]-oxazolidin-2-one (0.15 g, 0.29mmols) was dissolved in 10 mL of ethyl acetate:ethanol (1:1), added cyclohexene (0.14 mL, 1.45mmols), followed by 10% palladium on carbon (31 mg, 0.029mmols). Heated suspension to reflux for 2 hours. Filtered through a pad of celite, washing with ethyl acetate. Concentrated in vacuo. Purified by column chromatography eluting with a gradient of 0-80% ethyl acetate in
hexanes. Combined and concentrated pure fractions to yield title compound as a mixture of isomers (130 mg, quant, yield) APCI MS (M+1): 427.18; Analytical HPLC (Method A) Retention time = 14.4 min. and 14.7 min. (36% and 64% respectively).
Example 36: 3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxazσlidinyl-2,2'- dione

Step A
4-(4-Benzyloxy-phenyl)-3,3'-bis-(4-fluoro-phenyl)-[5,5']bioxazolidinyl-2,2'-dione

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[2-(4-fluoro-phenylamino)-1 -hydroxy- ethyl]-oxazolidin-2-one (0.5 g, 0.97mmoles) was dissolved in 10 mL anhydrous dichloromethane under a nitrogen atmosphere, cooled to -78°C, added triethylamine (0.67 mL, 4.84mmoles) followed by triphosgene (0.14 g, 0.48mmoles). Stirred at -780C for 1 hour, then allowed to warm to ambient temperature. Stirred at ambient temperature for 1 hour then partitioned between dichloromethane and water. Dried organic layer with magnesium sulfate, filtered and concentrated in vacuo. Dissolved in approximately 15 mL tetrahydrofuran with 5% methanol with heating. Let sit at ambient
temperature overnight. A solid formed which was filtered off, washing with ethyl acetate to yield title compound. (160 mg, 30%) APCI MS (M+1): 543.1.
Step B 3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxa2olidinyl-2,2'-dione

4-(4-Benzyloxy-phenyl)-3,3'-bis-(4-fluoro-phenylH5,5']bioxazolidinyl-2,2'-dione (0.16 g, 0.295mmoles) was suspended in 10 mL of ethyl acetate:ethanol (1 :1) at reflux under nitrogen. Added 2 mL of anhydrous tetrahydrofuran. Added cyclohexene (0.14 mL, 1.5mmoles), followed by 10% palladium on carbon (31 mg, 0.029mmoles). Heated suspension to reflux for 2 hours. The solution was filtered hot through a pad of celite, washing with tetrahydrofuran. Concentrated in vacuo to yield title compound. (137 mg, quant, yield) APCI MS (M+1): 453.18; Analytical HPLC (Method A) Retention time = 17.5 min. (>99%).
Example 37: 3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxazolidiny!-2,2'- dione.

S.S'-bis^-fluoro-phenyO^R^-hydroxy-phenyO-lδ.δ'lbioxazolidinyl^^'-dione was synthesized using procedures analogous to Example 36 described above. APCI MS (M+1): 453.15; Analytical HPLC (Method A) Retention time = 17.4 min. (97%).
Example 38 : 3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one

Step A
4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-{1-hydroxy-2-[(2-hydroxy-ethyl)-phenyl- amino]-ethyl}-oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-oxiranyl-oxazolidin-2-one (0.25 g, 0.62mmoles) was dissolved in 5 mL diethyl ether, added lithium perchlorate (0.33 g, 3.1mmoles) and 2-anilinoethanol (0.093 mL, 0.74mmoles) and stirred at ambient temperature overnight. The solution was diluted with 50 mL ethyl acetate, washed with water, brine, dried with magnesium sulfate, filtered and concentrated to yield title compound. (0.33 g, quant, yield) APCI MS (M+1): 543.22.
Step B 4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[4-(4-fluoro-phenyl)-morpholin-2-yl]- oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-{1-hydroxy-2-[(2-hydroxy-ethyl)-phenyl- amino]-ethyl}-oxazolidin-2-one (0.4 g, 0.74mmoles) was dissolved in 10 mL anhydrous dichloromethane under nitrogen, added triethylamine (0.21 mL, 1.48 mmol) and methanesulfonic anhydride (0.18 g, 1.0 mmol) and stirred at ambient temperature. After 3 hours the solution was concentrated. Redissolved in 5 mL N,N-dimethylformamide, added cesium carbonate (0.48 g, 1.5mmoles) and stirred at ambient temperature overnight. The solution was diluted with ethyl acetate, washed with water, brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 0-50% ethyl acetate in heptanes. Combined and concentrated desired fractions to yield title compound (118 mg, 30%) APCI MS (M+1): 525.24.
Step C 3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy-phenyl)- oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-[4-(4-fluoro-phenyl)-morpholin-2-y!]- oxazolidin-2-one (0.115 g, 0.22mmoles) was dissolved in 6 mL of ethyl acetate:ethanol (1:1) under nitrogen. Added cyclohexene (0.11 mL, 1.1mmoles), followed by 10% palladium on carbon (23 mg, 0.022mmoles). Heated at reflux for 2 hours. The solution
was filtered hot through a pad of celite, washing with ethyl acetate. Concentrated in vacuo to yield title compound. (95 mg, quant, yield) APCI MS (M+1): 435.18; Analytical HPLC (Method A) Retention time = 17.9 min. (>99%).
Example 39 : 3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one

3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy-phenyl)- oxazolidin-2-one was synthesized using procedures analogous to Example 38 described above. APCI MS (M+1): 453.14; Analytical HPLC (Method A) Retention time = 17.4 min. (89%).
Example 40 : 4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one

Step A
6-f4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-4-(4-fluoro-phenyl)- morpholin-3-one

3-(4-fluoro-phenyl)-5S-[2-(4-fluoro-phenylamino)-1-hydroxy-ethyl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one (0.23 g, 0.45mmoles) was dissolved in 5 mL dichloromethane, added triethylamine (0.12 mL, 0.89mmoles), cooled to O0C, then added chloroacetyl chloride (0.042 mL, 0.53mmoles). Removed cooling bath and stirred at ambient temperature for 1.5 hours. The solution was concentrated in vacuo. Redissolved in 5 mL N,N-dimethylformamide, added cesium carbonate (0.29 g, 0.89mmoles) and stirred at ambient temperature over the weekend. The solution was diluted with ethyl acetate, washed with water, brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 10-100% ethyl acetate in heptanes. Combined and concentrated desired fractions to yield title compound. (80 mg, 32%) APCI MS (M+1): 557.23.
Step B
4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5S- yl]-morpholin-3-one

6-[4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-4-(4-fluoro-phenyl)- morpholin-3-one (80 mg, 0.144mmoles) was dissolved in 2 mL of ethyl acetate:ethanol (1:1) under nitrogen. Added cyclohexene (0.073 mL, 0.72mmoles), followed by 10%
palladium on carbon (15 mg, 0.014mmoles). Heated at reflux for 2 hours. The solution was filtered hot through a pad of celite, washing with ethyl acetate. Concentrated in vacuo to yield title compound. (35 mg, 52%) APCI MS (M+1): 467.19; Analytical HPLC (Method A) Retention time = 16.3 min. (97.3%).
Example 41 : 4-(4~fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one

4-(4-fluoro-phenyl)~6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5S- yl]-morpholin-3-one was synthesized using procedures analogous to Example 40 described above. APCI MS (M+1): 467.1; Analytical HPLC (Method A) Retention time 16.2 min. (>99%).
Example 42: 5S-[1-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4- hydroxy-phenyl)-oxazolidin-2-one

5S-[2-(4-fluoro-phenoxy)-1S-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)- oxazolidin-2-one (90 mg, 0.21mmoles) was suspended in 2 mL anhydrous dichloromethane under nitrogen, cooled to O0C then added (diethylamino)sulfur
trifluoride (0.14 noL, 1.05mmoles). Allowed the solution to gradually warm to ambient temperature with stirring. Stirred at ambient temperature for 1 hour. The solution was purified by column chromatography eluting with a gradient of 0-100% ethyl acetate in heptane. Combined and concentrated pure fractions to yield title compound. (45 mg, 50%) APCI MS (M+1): 430.16; Analytical HPLC (Method A) Retention time = 18.6 min. (98%).
Example 43: 5S-[1 S-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4- hydroxy-phenyl)-oxazolidin-2-one

5S-[1S-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)- oxazolidin-2-one was synthesized using procedures analogous to Example 42 described above. APCI MS (M+1): 430.09; Analytical HPLC (Method A) Retention time = 18.5 min. (96.9%).
Example 44: 5S-(1,1-difluoro-3-phenyl-propyl)-4R-(4-hydroxy-phenyl)-3~(4-methoxy- phenyl)-oxazolidin-2-one

Step A 1 -(4-Benzyloxy-phenyl)-2-hydroxy-1 -(4-methoxy-phenylamino)-5-phenyl-pentan-3-one
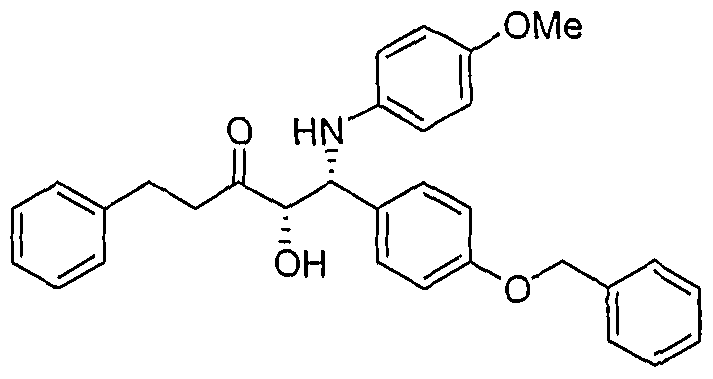
1-Hydroxy-4-phenyl-butan-2-one (8.9 g, 54mmoles), 4-benzyloxybenzaldehyde (7.7 g,
36mmoles), p-methoxyaniline (5.4 g, 44mmoles), and L-proline (1.25 g, 10.9mmoles) were combined in 150 mL dimethyl sulfoxide under nitrogen and stirred at ambient temperature for 7 days. The solution was poured into 300 mL sat. aqueous ammonium chloride, diluted with 300 mL water, extracted with 500 mL of ethyl acetate, washed with water, brine, dried with magnesium sulfate, filtered and concentrated in vacuo. Purified by column chromatography eluting with a gradient of 0-70% ethyl acetate in heptanes. Combined and concentrated fractions to yield title compound. (8.75 g, 50%) APCI MS
(M+1): 482.25.
Step B
4-(4-Benzyloxy-phenyl)-3-(4-methoxy-phenyl)-5-(3-phenyl-propionyl)-oxazolidin-2-one

1-(4-Benzyloxy-phenyl)-2-hydroxy-1-(4-methoxy-phenylamino)-5-phenyl-pentan-3-one (8.75 g, 18.2mmoles) was dissolved in 85 mL anhydrous dichloromethane under nitrogen, cooled to -78°C, added triethylamine (12.7 mL, 90.9mmoles), followed by the slow addition of triphosgene (2.7 g, 9.1 mmoles) dissolved in 15 mL dichloromethane. Stirred at -78°C for 2 hours, then allowed to gradually warm to ambient temperature where the solution was stirred overnight. The solution was partitioned between
dichloromethane and water, dried organics with magnesium sulfate, filtered and concentrated in vacuo. Purified by column chromatography eluting with a gradient of Q- 70% ethyl acetate in heptanes. Combined and concentrated fractions to yield title compound (2.46 g, 27%) APCI MS (M+1): 508.31.
Step C
4-(4-Benzyloxy-phenyl)-5-(1 , 1 -difluoro-3-phenyl-propyl)-3-(4-methoxy-phenyl)- oxazolidin-2-one

4-(4-Benzyloxy-phenyl)-3-(4-methoxy-phenyl)-5-(3-phenyl-propionyl)-oxazolidin-2-one (0.33 g, 0.65mmoles) was dissolved in 5 mL anhydrous dichloromethane under nitrogen, cooled to 00C, added (diethylamino)sulfur trifluoride (0.34 mL, 2.6mmoles). Stirred at O0C for one hour, then at ambient temperature for one hour. Added (diethylamino)sulfur trifluoride (0.34 mL, 2.6mmoles) at ambient temperature and stirred there overnight. The solution was partitioned between dichloromethane and water, separated, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 0-60% ethyl acetate in heptane. Combined and concentrated desired fractions to yield title compound (250 mg, 73%) APCI MS (M+1): 530.27
Step D
5S-(1 , 1 -difluoro-3-phenyl-propyl)-4R-(4-hydroxy-phenyl)-3-(4-methoxy-phenyl)- oxazolidin-2-one
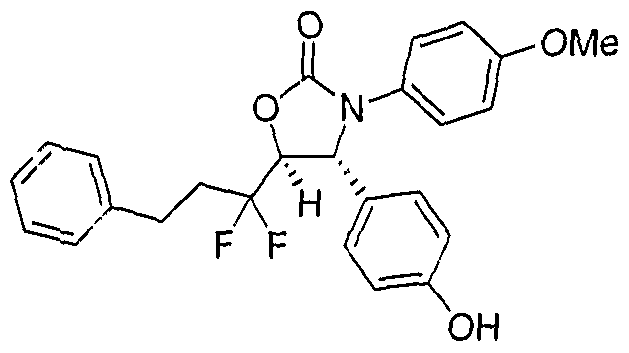
4-(4-Benzyloxy-phenyl)-5-( 1 , 1 -difluoro-3-phenyl-propyl)-3-(4-methoxy-phenyl)- oxazolidin-2-one (100 mg, 0.19mmoles) was dissolved in 2 ml_ of ethyl acetate:ethanol (1:1) under nitrogen. Added cyclohexene (0.096 mL, 0.94mmoles), followed by 10% palladium on carbon (20 mg, 0.019mmoles). Heated at reflux for 3 hours. The cooled solution was filtered through a pad of celite, washing with ethyl acetate. Concentrated in vacuo. Purified by column chromatography eluting with a gradient of 0-70% ethyl acetate in heptanes. Combined and concentrated appropriate fractions to yield title compound. (34 mg, 41%) APCI MS (M+1): 440.21 ; Analytical HPLC (Method A) Retention time = 19.4 min. (98%).
Example 45: ((4-fluoro-phenyl)-{2-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-2-hydroxy-ethyl}-amino)-acetic acid ethyl ester

Step A
[{2-[4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy-ethyl}- (4-fluoro-phenyl)-amino]-acetic acid ethyl ester

4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5-oxiranyl-oxazolidin-2-one (0.49 g, 1.21mmoles), lithium perchlorate (0.64 g, β.Ommoles), and N-phenylglycine ethyl ester (0.26 g, 1.5mmoles) were combined in 10 mL diethyl ether and stirred at RT overnight. Added additional N-phenylglycine ethyl ester (0.26 g, 1.5mmoles) and lithium perchlorate (0.64 g, δ.Ommoles) and continued stirring for another 24 hours. The solution was partitioned between ethyl acetate and water, washed with brine, dried with magnesium sulfate, filtered and concentrated. Purified by column chromatography eluting with a gradient of 0-100% ethyl acetate in heptanes. Combined and concentrated appropriate fractions to yield title compound. (310 mg, 44%) APCI MS (M+1): 585.29.
Step B ((4-fluoro-phenyl)-{2-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5S-yl]- 2-hydroxy-ethyl}-amino)-acetic acid ethyl ester

t{2-f4-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy-ethyl}- (4-fluoro-phenyl)-amino]-acetic acid ethyl ester (98 mg, 0.168mmoles) was dissolved in 6 mL of ethyl acetate:ethanol (1 :1) under nitrogen. Added cyclohexene (0.085 mL, 0.84mmσles), followed by 10% palladium on carbon (18 mg, 0.017mmoles). Heated at
reflux for 3 hours. The cooled solution was filtered through a pad of celite, washing with ethyl acetate. Concentrated in vacuo to yield title compound. (80 mg, 97%) APCI MS (M+1): 495.23; Analytical HPLC (Method A) Retention time = 18.0 min. (92.2%).
Example 46: 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phenyl-propionyl)- pyrrolidin-2-one

Step A
(4-Fluoro-phenyl)-(4-methoxy-benzylidene)-amine

A solution of p-anisaldehyde (10.0 g, 73.4 mmol) and 4-fluoroaniline (8.16 g, 73.4 mmol) in toluene (250 mL) was heated to reflux with a Dean Stark apparatus in place to remove water for 3 hrs. Reaction was then cooled to 25 C and the solvent was removed under reduced pressure to afford (4-fluoro-phenyl)-(4-methoxy-benzylidene)- amine (16.5 g, 98%) as an orange oil that was utilized without purification. 1H-NMR (400 MHz, d-CDCI3) δ 8.36 (s, 1 H), 7.82 (d, 2 H), 7.22-6.95 (m, 6 H), 3.85 (s, 3 H).
Step B 1-(4-Fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carboxylic acid

A solution of (4-fluoro-phenyl)-(4-methoxy-benzylidene)-amine (12.9 g, 56.1 mmol) And succinic anhydride (5.6 g, 56.1 mmol) in xylenes (100 ml_) was heated to reflux for 18 hrs. The reaction mixture was then slowly cooled to 25 C and extracted with saturated sodium bicarbonate (3 x 50 ml_). The aqueous extracts were washed with hexane and then made acidic by addition of concentrated hydrochloric acid resulting in the formation of a white precipitate that was subsequently extracted into ether. The ether layer was dried and concentrated to a white-pink powder that was taken up in CH2CI2/Me0H and purified by column chromatography (2-5% MeOH/ CH2Cl2 + 1% AcOH) to afford 1-(4-fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carboxylic acid (2.51 g, 14%) as a racemic mixture of trans isomers. 1H-NMR (400 MHz, d-CDCI3) δ 7.28-7.24 (m, 2 H), 7.12 (d, 2 H), 6.91 (t, 2 H), 6.81 (d, 2 H), 5.40 (d, 1 H), 3.74 (s, 3 H), 3.18-2.93 (m, 3 H).
Step C 1-(4-Fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carbonyl chloride

To a solution of 1-(4-fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carboxylic acid (1.0 g, 3.0 mmol) in CH2CI2 at 25 C was added oxalyl chloride (0.42 g, 3.3 mmol) followed by dimethylformamide (25 uL). Reaction was stirred at 25 C for 24 hrs. Reaction solvent was then evaporated and CH2CI2 (50 ml_) was added and evaporated a second time to provide 1-(4-fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrro!idine-3- carbonyl chloride (1.05 g, 99%) which was utilized without purification. 1H-NMR (400
MHz, d-CDCI3) δ 7.33-7.27 (m, 2 H), 7.18 (d, 2 H), 6.94 (t, 2 H), 6.85 (d, 2 H), 5.43 (d, 1 H), 3.75 (s, 3 H), 3.58-3.52 (m, 1 H), 3.15-2.88 (m, 2 H).
Step D 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-pheπyl-propionyl)-pyrrolidin-2-one

A solution of phenylethylmagnesium chloride (3.6 mL of 1.0 M solution in THF, 3.62 mmol) was added to THF (25 mL) at -78 C followed by the addition of Fe(acac)3 (0.053 g, 0.15 mmol). Reaction was stirred at -78 C for 2 min and then a solution of 1-(4- fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carbonyl chloride (1.05 g, 3.0 mmol) in THF (25 mL) was slowly added. The reaction mixture was stirred at -78 C for
25 min and then quenched by addition of saturated ammonium chloride. The reaction mixture was then warmed to 25 C and water (50 mL) and ethyl acetate (50 mL) were added. The organic layer was separated, washed with brine, dried and concentrated to an oil that was purified by column chromatography (15-35% EtO Ac/Hex) to afford 1-(4- fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phenyl-propionyl)-pyrrolidin-2-one (0.382 g,
30%). 1H-NMR (400 MHz, d-CDCI3) δ 7.26-7.15 (m, 5 H), 7.11-7.09 (m, 2 H), 7.03-7.00 (m, 2 H), 6.92-6.87 (m, 2 H), 6.79-6.75 (d, 2 H), 5.19 (d, 2 H), 3.74 (s, 3 H), 3.22-3.19
(m, 1 H), 2.91-2.71 (m, 6 H).
Example 47: 1-(4-fluoro-phenyl)-4R-(1-hydroxy-3-phenyl-propyl)-5S-(4-methoxy- phenyl)-pyrrolidin-2-one

To a solution of 1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)~4R-(3-phenyl-propionyl)- pyrrolidin-2-one [from Example 46] (0.34 g, 0.81 mmol) in MeOH.THF (1:1, 20 ml_) at 0
C was added NaBH4 (0.034 g, 0.90 mmol). The reaction mixture was stirred at 0 C for
30 min and then at 25 C for 1.5 hrs. The reaction mixture was quenched by addition of
1 N HCI (15 ml_), extracted with EtOAc and the organic layer was dried and concentrated to an oil that was purified by column chromatography (15-30% EtOAc/Hex) to afford 1-(4-fluoro-phenyl)-4R-(1-hydroxy-3-phenyl-propyl)-5S-(4- methoxy-phenyl)-pyrrolidin-2-one (0.27 g, 79%) as a mixture of two diastereomeric pairs of enantiomers. MS (APCI+) M+H 420.2.
Example 48: 1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide

To a solution of 1-(4-fluoro-phenyl)-2-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3-carboxylic acid [from Example 46, Step B] (0.40 g, 1.21 mmol) in CH2CI2 (100 mL) at 25 C was added EDCI (0.35 g, 1.82 mmol) followed by HOBT mono hydrate(0.26 g, 1.70 mmol) and the reaction mixture was stirred for 5 min at 25 C. Subsequently, benzyl amine (0.14 g, 1.34 mmol) was added and the resulting reaction mixture was stirred for an additional 4 hrs at 25 C. The reaction was then transferred to a separatory funnel and the organic layer was washed with 1 N HCI, saturated sodium bicarbonate and brine.
After drying and concentration, the product was purified by column chromatography (30- 50% EtOAc/Hex) to provide 1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo- pyrrolidine-3S-carboxylic acid benzylamide (0.30 g, 58%): 1H-NMR (400 MHz, d-CDCI3) δ 7.32-7.14 (m, 7 H), 7.04-7.01 (m, 2 H), 6.91-6.86 (m, 2 H), 6.88 (d, 2 H), 5.71 (t, 1 H), 5.27 (d, 2 H), 4.47 (dd, 1 H), 4.36 (dd, 1 H), 3.71 (s, 3 H), 3.09-3.01 (m, 1 H), 2.88-2.77 (m, 2 H).
Example 49: 1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide

Prepared according to the method of Example 48 using phenethylamine in place of benzyl amine to afford 1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S- carboxylic acid benzylamide. 1H-NMR (400 MHz, d-CDCI3) δ 7.26-7.16 (m, 5 H), 7.04- 7.00 (m, 4 H), 6.91-6.87 (m, 2 H), 6.75 (d, 2 H), 5.27 (t, 1 H), 5.22 (d, 2 H), 3.73 (s, 3 H), 3.67-3.55 (m, 1 H), 3.46-3.39 (m, 1 H), 3.03-2.96 (m, 1 H), 2.81-2.69 (m, 4 H).
Example 50: 3-(4-Fluoro-phenyl)-4R-(3'-hydroxy-biphenyl-4-yl)-5S-(1-hydroxy-3-phenyl- propyl)-oxazolidin-2-one

Examples 51-82:

Examples 51 - 82 were prepared as a mixture of diastereomers using procedures analogous to those described for Examples 33 and 34 above.





Example 83: 3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S- carboxylic acid benzylamide

Step A 4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid

5S-Acetyl-4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-oxazolidin-2-one (2.16 g, 5.33mmoles, Example 33, step B) was dissolved in 18 mL 1 ,4-dioxane and 2 ml_ water, cooled to O0C, then added a solution of sodium hypobromide (made by dissolving sodium hydroxide (2.98 g, 74.6mmo!s) in 10 mL water, cooling to O0C, adding 3 mL 1 ,4- dioxane, followed by dropwise addition of bromine (1.09 mL, 21.3mmoles)) dropwise. Stirred at O0C for 1 hour. Added 25 mL of 5% aqueous sodium sulfite. Extracted with Et20, dried with magnesium sulfate, filtered, and concentrated in vacuo. Purified by column chromatography eluting with a gradient of 100% ethyl acetate (with 1% acetic acid added) for 4 minutes, then 0-40% methanol in ethyl acetate (with 1% acetic acid added) for the next 20 minutes. Combined and concentrated fractions. Repurified by column chromatography eluting with a gradient of 100% dichloromethane for 4 minutes, then 0-40% methanol (with 1% acetic acid) in
dichloromethane for the next 20 minutes. Combined and concentrated desired fractions to yield title compound. (1.06 g, 49%) APCI MS (M+1): 408.2; Analytical HPLC (Method A) Retention time = 18.6 min. (84%).
Step B
4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid benzylamide

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid (0.35 g, 0.86mmols) was dissolved in 10 mL dichloromethane, added 1-hydroxybenzotriazole hydrate (0.20 g, 1.29mmoles), N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (0.25 g, 1.29mmoles), followed by benzylamine (0.15 mL, 1.29mmoles). Stirred at ambient temperature. After 1 hour the solution was diluted with 100 mL ethyl acetate, washed with sat. sodium bicarbonate (3 x 75 mL), 1M hydrogen chloride (2 x 50 mL), brine, dried with magnesium sulfate, filtered and concentrated in vacuo. Purified by column chromatography eluting with a gradient of 0-60% ethyl acetate in heptanes. Combined and concentrated fractions to yield title compound (180 mg, 39%) APCI MS (M+1): 497.2; Analytical HPLC (Method A) Retention time = 20.7 min. (93%).
Step C
3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid benzylamide

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid benzylamide (0.18 g, 0.36mmoles) was dissolved in 2 mL ethyl acetate and 2 mL ethanol under nitrogen, added cyclohexene (0.18 mL, 1.8mmoles) followed by 10% palladium on carbon (39 mg, 0.036mmoles). Refluxed for 1.5 hours, cooled, filtered through celite, washing with ethyl acetate. Concentrated filtrates. Purified by column chromatography eluting with a gradient of 10-80% ethyl acetate in heptanes. Combined and concentrated desired fractions in vacuo to yield title compound (90 mg, 61%) APCI MS (M+1): 407.07; Analytical HPLC (Method A) Retention time = 16.7 min. (97.1%).
Example 84: 3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S- carboxylic acid (4-fluoro-benzyl)-methyl-amide
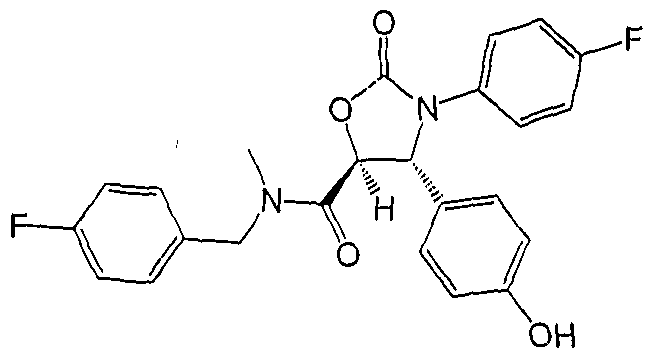
Step A
4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid (4- fluoro-benzyl)-methyl-amide

4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-pheny))-2-oxo-oxazolidine-5S-carboxylic acid (4- fluoro-benzyl)-methyl-amide was synthesized using analogous conditions to Example 83 step B substituting (4-fluoro-benzyl)-methyl-amine for benzylamine. (205 mg, 83%) APCI MS (M+1): 529.17; Analytical HPLC (Method A) Retention time = 21.2 min. (>99%).
Step B 3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid (4- fluoro-benzyl)-methyl-amide

3-(4-Fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidine-5S-carboxylic acid (4- fluoro-benzyl)-methyl-amide was synthesized using conditions found in Example 83 step C. (0.14 g, 84%) APCI MS (M+1): 439.10; Analytical HPLC (Method A) Retention time = 17.4 min. (99%).
Example 85: 3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-2-oxo-oxazolidine-5- carboxylic acid (4-fluoro-benzyl)-methyl-amide

Prepared according to the method of Example 84. APCI MS (M+1): 469.15; Analytical HPLC (Method A) Retention time = 18.5 min. (>99%).
Example 86: 5-(3,4-Dihydro~1 H-isoquinoline-2-carbonyl)-3-(3-fluoro-phenyl)-4-(2- hydroxy-4-methoxy-phenyl)-oxazolidin-2-one

Prepared according to the method of Example 84. APCI MS (M+1): 463.15; Analytical
HPLC (Method A) Retention time = 18.5 min. (>99%).
Example 87: 3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-5-(1-hydroxy-3- phenyl-propyl)-oxazolidin-2-one

Prepared according to the method of Example 10. APCI MS (M+1): 437.16.
In Vitro Method for Biological Evaluation of Cholesterol Absorption Inhibitors
Compounds of the present invention were evaluated for binding to Niemann Pick C1- Like 1 (NPC1 L1) protein according to a previously described method (Garcia-Calvo, M; Lisnock, J.; Bull, H.G; et. al Proc. Nat. Acad. Sci 2005, 102, 8132-8137). Examples 1- 87 were evaluated for binding to NPC1L1 with the results reported below. Preferred compounds have an ICs0 of less than 20 uM.


Selected compounds were also evaluated for in vivo inhibition of cholesterol synthesis as described below. Male Sprague-Dawley rats (200-400 gm) are maintained in a room with a 12 hour light cycle/12 hour dark cycle for at least one week prior to testing. On the test day the rats are fasted for 8 hours prior to dosing to synchronize initiation of eating once food is presented. Test drug or vehicle is administered by oral gavage approximately 1 hour prior to the start of the dark cycle. One group of animals is dosed with vehicle and given standard chow (chow control), one group is dosed with vehicle and given the same diet supplemented with 5.5% peanut oil, 1.5% cholesterol, and 0.4% cholic acid (PCC diet; PCC control), and the remaining animals are dosed with test agents in vehicle and are given the PCC diet. Animals are given access to their assigned diet ad libitum starting 30 minutes after dosing until study termination 16 hours after drug administration. Animals are euthanized with CO2, and blood is collected by cardiac puncture for plasma total cholesterol analysis.
Data analysis. Total plasma cholesterol concentrations are ~ 60-90 mg/dL in chow controls and increase to ~ 175-240 mg/dL in PCC control animals. The difference in plasma cholesterol between the chow control group and the PCC control group is the elevation caused by the PCC diet. The dose that reduces by 50% the elevation in plasma cholesterol in animals on the PCC diet is the ED50.
FORMULATIONS The compounds of the present invention including those exemplified herein and all compounds of Formulas l-la-lai, hereafter referred to as "compound(s)" can be administered alone or in combination with one or more therapeutic agents. These include, for example, other agents for treating, preventing or controlling dyslipidemia, non-insulin dependent diabetes mellitus, obesity, hyperglycemia, hypercholesteremia, hyperlipidemia, atherosclerosis, hypertriglyceridemia, or hyperinsulinemia.
The compounds are thus well suited to formulation for convenient administration to mammals for the prevention and treatment of such disorders.
The following examples further illustrate typical formulations of the compounds provided by the invention. Formulation 1

The above ingredients are mixed and dissolved in the saline for IV administration to a patient. Formulation 2

The ingredients are blended to uniformity and pressed into a tablet that is well suited for oral administration to a patient. Formulation 3

The ingredients are combined and milled to afford material suitable for filling hard gelatin capsules administered to a patient. Formulation 4


The ingredients are combined via melting and then poured into molds containing 2.5 g total weight.
While embodiments of the invention have been illustrated and described, it is not intended that these embodiments illustrate and describe all possible forms of the invention. Rather, the words used in the specification are words of description rather than limitation, and it is understood that various changes may be made without departing from the spirit and scope of the invention.
Claims
What is claimed is:
1. A compound having a Formula (I),

or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein Ar1 and Ar2 are each independently aryl or heteroaryl, optionally substituted; Y3 is alkyl, aryl, aralkyl, heteroalkyl, or heteroaralkyl; optionally substituted; 2 is -O-CR"R"'CH(OR>; -NR'-CR"R"'-CH(OR')-; -CR"R"'-CR"R'"-CH(OR')-; -CR"R'"-NR'-C-; or is
O selected from

wherein Vwvr" indicates the points of attachment; R' is H; or lower alkyl, optionally substituted; R" and R'" are each independently H; lower alkyl, optionally substituted, or flourine; W is O or NR'; and n is 0, 1 or 2.
2. A compound having a Formula (Ia),

or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein Z is -O-CR"R"'CH(OR')-; -NR'-CR"R"'-CH(OR')-; -CR"R'"-CR"R'"-CH(OR')-; -CR"R"'-NR'-C-; or is
O selected from

R1, R2 and R3 are each independently halo; -OR', -COR', -COOR', -CONR'R";
CH2NR1R"; CH2NR1C(O)R"; C1-Ci2 alkyl, aryl, or heteroaryl; optionally substituted;
S(O)nR', P(O)nR', OG, CR'R"G, S(O)nG, NR'G or SG;
G is is selected from the group consisting of hydrogen,


wherein 'Ww^" indicates the point of attachment and wherein R5, R6, R7, R8, R9, and R10 are each independently selected from the group consisting of hydrogen, CLC6 alkyl, Ci-C6 aralkyl, -C(O)CLC6 alkyl, -C(O)aryl, and aryl; and R11 is selected from the group consisting of hydrogen, hydroxy, Ci-C6 alkyl, -OCi- C6 alkyl, and NR1R"; R' is H; or lower alkyl, optionally substituted; R" and R"' are each independently H; lower alkyl, optionally substituted, or flourine; W is O or NR'; and n is 0, 1 or 2.
The compound of claim 2 or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein R1, R2, and R3 are each independently F or -OR'; and R is H or lower alkyl, optionally substituted.
The compound of claim 2 having a Formula (lai),

(lai) or a pharmaceutically acceptable salt, ester, amide or hydrate thereof, wherein 2, R1, R2 and R3 are as defined in claim 2.
5. The compound of claim 4 or a pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer thereof, wherein R1, R2, and R3 are each independently F or -OR'; and R is H or lower alkyl, optionally substituted.
6. A pharmaceutical composition comprising a compound of claim 1 or claim 2, the pharmaceutically acceptable salt, ester, hydrate, amide or stereoisomer or mixtures thereof; and a pharmaceutically acceptable carrier, diluent or vehicle.
7. A method of inhibiting cholesterol absorption in a mammal requiring inhibition comprising administering to the mammal a therapeutically effective amount of a compound of claim 1 or the pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof.
8. A method of treating, preventing or controlling hyperlipidemia in a mammal comprising administering to the mammal in need thereof a therapeutically effective amount of a compound of claim 1 or the pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof.
9. A method of treating, preventing or controlling hypercholesterolemia in a mammal comprising administering to the mammal in need thereof a therapeutically effective amount of a compound of claim 1 or the
pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof.
10. A method of treating, preventing or controlling hypertriglyceridemia in a mammal comprising administering to the mammal in need there of a therapeutically effective amount of a compound of claim 1 or the pharmaceutically acceptable salt, ester, hydrate, amide, or stereoisomer or mixtures thereof.
11. A method of treating, preventing or controlling atherosclerosis in a mammal comprising administering to the mammal in need thereof a therapeutically effective amount of a compound of claim 1 or the pharmaceutically acceptable salt, ester, amide, or stereoisomer or mixtures thereof.
12. A combination comprising a compound of claim 1 or claim 2 and a pharmaceutically active agent.
13. The combination of claim 12, wherein said pharmaceutically active agent is a CETP inhibitor, a PPAR- activator, an MTP/Apo B secretion inhibitor, HDL- cholesterol raising agent, HMG-CoA reductase inhibitor, triglyceride lowering agent, a cholesterol synthesis inhibitor, a cholesterol modulating agent, a fibrate, niacin, an ion-exchange resin, an antioxidant, an ACAT inhibitor, bile acid sequestrant, an anti-hypertensive agent, or an acetylcholine esterase inhibitor.
14. The combination of claim 13, wherein said HMG-CoA reductase inhibitor is a statin.
15. A pharmaceutical composition comprising the combination of claim 14 and a pharmaceutically acceptable carrier, diluent, solvent or vehicle.
16. A compound selected from:
3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2-one;
Acetic acid 1-[3,4R-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5S-yl]-3-phenyl- propyl ester;
Acetic acid 1 S-[3,4-bis-(4-methoxy-phenyl)-2-oxo-oxazolidin-5-yl]-3-phenyl-propyl ester;
5S-(1 R-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one; 5S-(1S-hydroxy-3-phenyl-propyl)-3,4R-bis-(4-methoxy-phenyl)-oxazolidin-2-one; 4R-(4-Benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin- 2-one; 3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propionyl)-oxazolidin-2- one;
4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4R-(4-benzyloxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-y!]- benzoic acid methyl ester;
4-[3-(4-fluoro-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- benzoic acid; 4-[3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- benzoic acid methyl ester;
4R-(2,4-dihydroxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
4R-(2,4-dihydroxy-phenyl)-3-(4-fluoro-phenyl)-5S-(1S-hydroxy-3-phenyl-propy!)- oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1-hydroxy-3-phenyl-propyl)-3-(3-trifluoromethyl- phenyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(2-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one; 3-(4-fluoro-phenyl)-4R-(2-hydroxy-4-methoxy-phenyl)-5S-(1R-hydroxy-3-phenyl- propyl)-oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(2-hydroxy-4-methoxy-phenyl)-5S-(1S-hydroxy-3-phenyl- propyl)-oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)-3-(4-trifluoromethyl- phenyl)-oxazolidin-2-one;
4R-(4-hydroxy-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)-3-(4-trifluoromethyl- phenyl)-oxazolidin-2-one; 3-(4-fluoro-phenyl)-4R-(3-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propy!)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(3-hydroxy-phenyl)-5S-(1S-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(3-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(3-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(1R-hydroxy-3-phenyl-propyl)- oxazolidin-2-one;
3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one;
3,4R-bis-(4-methoxy-phenyl)-5S-(3-phenyl-propyl)-oxazolidin-2-one; {4-[3-(4-fluoro-phenyl)-5S-(1 R-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4R-yl]- phenoxyj-acetic acid;
1-(4-{4-[5S-(1 R-acetoxy-3-phenyl-propyl)-3-(4-fluoro-phenyl)-2-oxo-oxazolidin-
4R-yl]-phenoxymethyl}-benzyl)-1-azonia-bicyc!o[2.2.2]octane chloride;
6-{4-[3-(4-Fluoro-phenyl)-5-(1-hydroxy-3-phenyl-propyl)-2-oxo-oxazolidin-4-yl]- phenoxy}-3,4,5-trihydroxy-tetrahydro-pyran-2-carboxylic acid;
5S-[2-(4-fluoro-phenoxy)-1R-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
5S-[2-(4-fluoro-phenoxy)-1S-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one; 3-(4-fluoro-pheny))-5S-[2-(4-flυoro-phenylamino)-1-hydroxy-ethyl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
3,3'-bis-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-[5,5']bioxazolidinyl-2,2'-dione;
3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-yl]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one; 3-(4-fluoro-phenyl)-5S-[4-(4-fluoro-phenyl)-morpholin-2-y!]-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one;
4-(4-fluoro-phenyl)-6-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo- oxazolidin-5S-yl]-morpholin-3-one;
5S-[1-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one; 5S-[1 S-fluoro-2-(4-fluoro-phenoxy)-ethyl]-3-(4-fluoro-phenyl)-4R-(4-hydroxy- phenyl)-oxazolidin-2-one;
5S-(1 ,1-difluoro-3-phenyl-propyl)-4R-(4-hydroxy-phenyl)-3-(4-methoxy-phenyl)- oxazolidin-2-one;
((4-fluoro-phenyl)-{2-[3-(4-fluoro-phenyl)-4R-(4-hydroxy-phenyl)-2-oxo-oxazolidin- 5S-yl]-2-hydroxy-ethyl}-amino)-acetic acid ethyl ester;
1-(4-fluoro-phenyl)-5S-(4-methoxy-phenyl)-4R-(3-phenyl-propionyl)-pyrrolidin-2- one;
1-(4-fluoro-phenyl)-4R-(1-hydroxy-3-phenyl-propyl)-5S-(4-methoxy-phenyl)- pyrrolidin-2-one; 1 -(4-f luoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxylic acid benzylamide;
1-(4-fluoro-phenyl)-2R-(4-methoxy-phenyl)-5-oxo-pyrrolidine-3S-carboxy)ic acid benzylamide;
3-(4-Fluoro-phenyl)-4R-(3'-hydroxy-biphenyl-4-yl)-5S-(1-hydroxy-3-phenyl- propyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(3-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzoic acid methyl ester; 3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(2-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(2-methanesulfonyl-phenoxy)-ethyl]-4-(4- hydroxy-phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1-hydroxy-2-(4-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one;
5-[2-(2,4-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3-Fluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(2,3-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one; 5-[2-(3,4-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-5~[1-hydroxy-2-(4-methoxy-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(4-Chloro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(4-tert-Butyl-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3-tert-Butyl-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one; 3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1 -hydroxy- 2-(3-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one;
2-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxyj-benzoic acid methyl ester;
5-[2-(3,4-Dichloro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzonitrile;
3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(naphthalen-1-yloxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one; 3-(4-Fluoro-phenyl)-5-[1-hydroxy-2-(4-isopropyl-phenoxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
5-[2-(3,5-Difluoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-(1-hydroxy-2-p-tolyloxy-ethyl)- oxazolidin-2-one;
5-[2-(Biphenyl-3-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oxazolidin-2-one;
5-[2-(Biphenyl-2-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oxazolidin-2-one;
5-[2-(Biphenyl-4-yloxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy-phenyl)- oxazolidin-2-oπe; 2-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxyj-benzonitrile;
5-[2-(2-Flυoro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
4-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzonitrile;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-[1-hydroxy-2-(2-trifluoromethyl- phenoxy)-ethyl]-oxazolidin-2-one;
5-[2-(2-Chloro-phenoxy)-1-hydroxy-ethyl]-3-(4-fluoro-phenyl)-4-(4-hydroxy- phenyl)-oxazolidin-2-one; 3-(4-Fluoro-phenyl)-5-[1 -hydroxy-2-(naphthalen-2-yloxy)-ethyl]-4-(4-hydroxy- phenyl)-oxazolidin-2-one;
3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-5-(1-hydroxy-2-o-tolyloxy-ethyl)- oxazolidin-2-one;
3-{2-[3-(4-Fluoro-phenyl)-4-(4-hydroxy-phenyl)-2-oxo-oxazolidin-5-yl]-2-hydroxy- ethoxy}-benzoic acid methyl ester;
3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-2-oxo-oxazolidine-5- carboxylic acid (4-fluoro-benzyl)-methyl-amide;
5-(3,4-Dihydro-1 H-isoquinoline-2-carbonyl)-3-(3-fluoro-phenyl)-4-(2-hydroxy-4- methoxy-phenyl)-oxazolidin-2-one; 3-(3-Fluoro-phenyl)-4-(2-hydroxy-4-methoxy-phenyl)-5-(1-hydroxy-3-phenyl- propyl)-oxazolidin-2-one;
And pharmaceutically acceptable salts, esters, amides and hydrates thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US89235807P | 2007-03-01 | 2007-03-01 | |
| US60/892,358 | 2007-03-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008104875A1 true WO2008104875A1 (en) | 2008-09-04 |
Family
ID=39522024
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2008/000523 Ceased WO2008104875A1 (en) | 2007-03-01 | 2008-02-20 | Oxazolidinones as cholesterol absorption inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008104875A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013157651A1 (en) | 2012-04-17 | 2013-10-24 | Sumitomo Chemical Company, Limited | Process for producing alpha-hydroxyketone compound |
| WO2014050134A1 (en) | 2012-09-27 | 2014-04-03 | 興和株式会社 | Therapeutic agent for dyslipidemia |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0720599B1 (en) * | 1993-09-21 | 1999-05-19 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
-
2008
- 2008-02-20 WO PCT/IB2008/000523 patent/WO2008104875A1/en not_active Ceased
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0720599B1 (en) * | 1993-09-21 | 1999-05-19 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
Non-Patent Citations (3)
| Title |
|---|
| DUGAR S ET AL: "Gamma-lactams and related compounds as cholesterol absorption inhibitors: homologs of the beta-lactam cholesterol absorption inhibitor SCH 48461", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 5, no. 24, 21 December 1995 (1995-12-21), pages 2947 - 2952, XP004135136, ISSN: 0960-894X * |
| KVAERNO L ET AL: "An In Vitro Assay for Evaluation of Small-Molecule Inhibitors of Cholesterol Absorption", ANGEWANDTE CHEMIE. INTERNATIONAL EDITION, WILEY VCH VERLAG, WEINHEIM, vol. 43, 1 January 2004 (2004-01-01), pages 4653 - 4655, XP002362656, ISSN: 1433-7851 * |
| RITTER T ET AL: "Heterocyclic ring scaffolds as small-molecule cholesterol absorption inhibitors", ORGANIC AND BIOMOLECULAR CHEMISTRY, ROYAL SOCIETY OF CHEMISTRY, CAMBRIDGE, GB, vol. 3, 1 January 2005 (2005-01-01), pages 3514 - 3523, XP002362655, ISSN: 1477-0520 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013157651A1 (en) | 2012-04-17 | 2013-10-24 | Sumitomo Chemical Company, Limited | Process for producing alpha-hydroxyketone compound |
| WO2014050134A1 (en) | 2012-09-27 | 2014-04-03 | 興和株式会社 | Therapeutic agent for dyslipidemia |
| KR20150063035A (en) | 2012-09-27 | 2015-06-08 | 교와 가부시키가이샤 | Therapeutic agent for dyslipidemia |
| US9572798B2 (en) | 2012-09-27 | 2017-02-21 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US9931321B2 (en) | 2012-09-27 | 2018-04-03 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US10258609B2 (en) | 2012-09-27 | 2019-04-16 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
| US11013722B2 (en) | 2012-09-27 | 2021-05-25 | Kowa Company, Ltd. | Therapeutic agent for dyslipidemia |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090170852A1 (en) | NOVEL PYRAZOLE-BASED HMG CoA REDUCTASE INHIBITORS | |
| HUT71347A (en) | Novel process for preparing chiral {[(2-bromoethyl)-amino]-methyl}-2-nitro-1h-imidazol-1-ethanol and related compounds | |
| US20080227974A1 (en) | Novel Substituted Azetidinones | |
| KR20060133013A (en) | New imidazole | |
| WO2008104875A1 (en) | Oxazolidinones as cholesterol absorption inhibitors | |
| CA2338305A1 (en) | Imidazole compounds | |
| JP5075843B2 (en) | Dibenzylamine compounds and derivatives | |
| US20050154042A1 (en) | N-alkyl pyrroles as HMG-CoA reductase inhibitors | |
| WO2005079790A1 (en) | Imidazole-based hmg-coa reductase inhibitors | |
| WO2009027785A2 (en) | 1, 3-oxazole derivatives as cetp inhibitors | |
| US20070088069A1 (en) | Novel imidazoles | |
| FR2726558A1 (en) | NOVEL 5- (ARYLOXYMETHYL) OXAZOLINES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM | |
| WO2006087630A2 (en) | OXYPYRAZOLE HMG Co-A REDUCTASE INHIBITORS | |
| CN1411442A (en) | Farnesyl transferase inhibitors | |
| WO2006059210A2 (en) | Fused bicyclic pyrrols as hmg-coa reductase inhibitors | |
| CN1942448A (en) | Novel imidazole | |
| AU4799699A (en) | Imidazole compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08709888 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08709888 Country of ref document: EP Kind code of ref document: A1 |