WO2009010416A2 - Inhibitors of 11b-hydroxysteroid dehydrogenase - Google Patents
Inhibitors of 11b-hydroxysteroid dehydrogenase Download PDFInfo
- Publication number
- WO2009010416A2 WO2009010416A2 PCT/EP2008/058804 EP2008058804W WO2009010416A2 WO 2009010416 A2 WO2009010416 A2 WO 2009010416A2 EP 2008058804 W EP2008058804 W EP 2008058804W WO 2009010416 A2 WO2009010416 A2 WO 2009010416A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- pyrazole
- carboxylic acid
- ylamide
- acid adamantan
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCOC(c(cn[n]1*)c1Cl)=O Chemical compound CCOC(c(cn[n]1*)c1Cl)=O 0.000 description 4
- SWLSIHINYKHSSA-UHFFFAOYSA-N O=C(c(cn[n]1-c2ccccc2)c1Cl)NC1C2CC(C3)CC1CC3C2 Chemical compound O=C(c(cn[n]1-c2ccccc2)c1Cl)NC1C2CC(C3)CC1CC3C2 SWLSIHINYKHSSA-UHFFFAOYSA-N 0.000 description 4
- YKQJFCFMBSCWCQ-UHFFFAOYSA-M CCOC(c(cn1)c(N)[n]1[AlH2])=O Chemical compound CCOC(c(cn1)c(N)[n]1[AlH2])=O YKQJFCFMBSCWCQ-UHFFFAOYSA-M 0.000 description 2
- DXYHZBCAXZUFHB-UHFFFAOYSA-M C=[Al][n]1ncc(C(O)=O)c1Cl Chemical compound C=[Al][n]1ncc(C(O)=O)c1Cl DXYHZBCAXZUFHB-UHFFFAOYSA-M 0.000 description 1
- TZOAXKVIPKNHQL-UHFFFAOYSA-N CC(NC1(CC2CC(C3)C1)CC3C2NC(OCc1ccccc1)=O)=O Chemical compound CC(NC1(CC2CC(C3)C1)CC3C2NC(OCc1ccccc1)=O)=O TZOAXKVIPKNHQL-UHFFFAOYSA-N 0.000 description 1
- KTMGNAIGXYODKQ-VOTSOKGWSA-N CCO/C=C(/C(OCC)=O)\C#N Chemical compound CCO/C=C(/C(OCC)=O)\C#N KTMGNAIGXYODKQ-VOTSOKGWSA-N 0.000 description 1
- WKIXDVMMTUXWJG-UHFFFAOYSA-N CCOC(C(C(OCC)=O)=CNN[AlH2])=O Chemical compound CCOC(C(C(OCC)=O)=CNN[AlH2])=O WKIXDVMMTUXWJG-UHFFFAOYSA-N 0.000 description 1
- IQVGOYIZHXLXFL-UHFFFAOYSA-M CCOC(c(cn[n]1[AlH2])c1Cl)=O Chemical compound CCOC(c(cn[n]1[AlH2])c1Cl)=O IQVGOYIZHXLXFL-UHFFFAOYSA-M 0.000 description 1
- MDUDRENNDMFIIA-UHFFFAOYSA-M CCOC(c(cn[n]1[AlH2])c1O)=O Chemical compound CCOC(c(cn[n]1[AlH2])c1O)=O MDUDRENNDMFIIA-UHFFFAOYSA-M 0.000 description 1
- HFUORKXQSYEZET-UHFFFAOYSA-N O=C(c1c(N2CCCC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 Chemical compound O=C(c1c(N2CCCC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 HFUORKXQSYEZET-UHFFFAOYSA-N 0.000 description 1
- OETYWRAYEGRZOY-UHFFFAOYSA-N O=C(c1c(NCC2CC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 Chemical compound O=C(c1c(NCC2CC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 OETYWRAYEGRZOY-UHFFFAOYSA-N 0.000 description 1
- YAWQHBSZDSKOHL-UHFFFAOYSA-N O=C(c1c(NCC2OCCC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 Chemical compound O=C(c1c(NCC2OCCC2)[n](-c2ccccc2)nc1)NC1C2CC(C3)CC1CC3C2 YAWQHBSZDSKOHL-UHFFFAOYSA-N 0.000 description 1
- ZBSIEFINRFWWBJ-UHFFFAOYSA-N OC(CCC1)CN1c([n](-c1ccccc1)nc1)c1C(NC1C2CC(C3)CC1CC3C2)=O Chemical compound OC(CCC1)CN1c([n](-c1ccccc1)nc1)c1C(NC1C2CC(C3)CC1CC3C2)=O ZBSIEFINRFWWBJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/38—Nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the invention relates to inhibitors of 11 ⁇ -hydroxysteroid dehydrogenase.
- the inhibitors include, for example, amino- l-aryl-pyrazole-3-carboxylic acid adamantan-2-yl amides, and derivatives thereof, and are useful for the treatment of diseases such as type II diabetes mellitus and metabolic syndrome.
- the invention relates to a compound of formula (I):
- Ri is hydrogen or lower alkyl
- R 2 is lower alkyl, -(CH 2 ) n -cycloalkyl, -(CH 2 ) n -heterocycloalkyl, -(CH 2 ) n -aryl, -(CH 2 ) n -heteroaryl, -(CH 2 ) n OH, -(CH 2 ) n CH(CH 3 )OH or -(CH 2 ) n OCH 3;
- Ri and R 2 together with the N atom to which they are attached, form a 5- to 7- membered monocyclic ring, which contains the N atom to which Ri and R 2 are attached, and optionally another hetero atom which is selected from O and S, wherein the 5- to 7-membered monocyclic ring is unsubstituted or mono- or bi- substituted with substituents independently selected from hydroxy, lower alkyl and -(CH 2 ) n OH;
- R 3 is one or more substituents independently selected from H, halogen, lower alkyl and lower alkoxy;
- n 1, 2, 3 or 4; and pharmaceutically acceptable salts thereof.
- Diabetes mellitus is a serious illness that affects an increasing number of people across the world.
- a recent press release by the International Diabetes Federation suggests that by 2025, there will be a total of 380 million people worldwide suffering from diabetes.
- the incidence of diabetes in many countries is escalating in parallel with an upward trend in obesity.
- Serious consequences of diabetes include increased risk of stroke, heart disease, kidney damage, blindness, and amputation.
- Cardiovascular diseases are the cause of death of more than 70% of patients with Type 2 diabetes mellitus (T2DM) [B. Pourcet et al. Expert Opin. Emerging Drugs 2006, 11, 379-401.]
- Diabetes is characterized by decreased insulin secretion and/or an impaired ability of peripheral tissues to respond to insulin, resulting in increased plasma glucose levels.
- diabetes There are two forms of diabetes: insulin-dependent and non-insulin-dependent, with the great majority of diabetics suffering from the non-insulin-dependent form of the disease, known as type 2 diabetes or non-insulin-dependent diabetes mellitus (NIDDM). Because of the serious consequences, there is an urgent need to control diabetes.
- NIDDM non-insulin-dependent diabetes mellitus
- the metabolic syndrome is a condition where patients exhibit more than two of the following symptoms: obesity, hypertriglyceridemia, low levels of HDL-cholesterol, high blood pressure, and elevated fasting glucose levels [R. H. Eckel Proc. Nutr. Soc. 2007, 66, 82-95; J.-P. despres and I. Lemieux Nature 2006, 444, 881-887; E. Ratto et al. /. Am. Soc. Nephrol. 2006, 17, S120-S122; A. M. McNeill et al. Diabetes Care 2005, 28, 385-390].
- This syndrome is often a precursor of type 2 diabetes, and has high prevalence in the United States, estimated at 24% [E. S. Ford et al. JAMA 2002, 287, 356].
- a therapeutic agent that ameliorates the metabolic syndrome would be useful in potentially slowing or stopping the progression to type 2 diabetes.
- HbAi c test tests for the levels of glycosylated hemoglobin in the red blood cells [D. R. McCane et al. BMJ 1994, 308, 1323-1328; R. J. McCarter et al. Diabetes Care 2006, 29, 352-355].
- Red blood cells have a normal life-span of 120 days in the body, and they contain hemoglobin which becomes progressively glycosylated, with the level of glycosylation correlating with the average levels of blood glucose.
- the HbAi c levels give an indication of the average levels of blood glucose over the preceding 3-4 months, and they do not fluctuate during the course of the day.
- the level of HbAi c in normal blood is approximately 5%, and the level in poorly controlled diabetic patients is 8% or above.
- the current guideline from the American Diabetes Association is to maintain the HbAi c level below 7%. This level corresponds to a mean plasma glucose level of approximately 170 mg/dL [D. E. Goldstein et al. Diabetes Care 2004, 27, 1761-1773].
- NIDDM NIDDM-induced diabetes fibrosis .
- Treatment of NIDDM generally starts with weight loss, a healthy diet and an exercise program. These factors are especially important in addressing the increased cardiovascular risks associated with diabetes, but they are generally ineffective in controlling the disease itself.
- drug treatments available including insulin, metformin, sulfonylureas, acarbose, thiazolidinediones, GLP-I analogues, and DPP IV inhibitors.
- these treatments have disadvantages, and there is an ongoing need for new drugs to treat diabetes.
- metformin is an effective agent that reduces fasting plasma glucose levels and enhances the insulin sensitivity of peripheral tissue.
- Metformin has a number of effects in vivo, including an increase in the synthesis of glycogen, the polymeric form in which glucose is stored [R. A. De Fronzo Drugs 1999, 58 Suppl. 1, 29].
- Metformin also has beneficial effects on lipid profile, with favorable results on cardiovascular health. Treatment with metformin leads to reductions in the levels of LDL cholesterol and triglycerides [S. E. Inzucchi/ ⁇ M ⁇ 2002, 287, 360].
- metformin loses its effectiveness [R. C. Turner et al. JAMA 1999, 281, 2005] and there is consequently a need for new treatments for diabetes.
- Thiazolidinediones are activators of the nuclear receptor peroxisome-proliferator activated receptor- gamma (PPAR ⁇ ). They are effective in reducing blood glucose levels, and their efficacy has been attributed primarily to decreasing insulin resistance in skeletal muscle [M. Tadayyon and S. A. Smith Expert Opin. Investig. Drugs 2003, 12, 307]. Three thiazolidinediones have been approved for use in the United States for the treatment of diabetes but one was subsequently withdrawn because of hepato toxicity issues. The two currently approved drugs, pioglitazone and rosiglitazone, are effective in reducing blood sugar and HbAi c levels in diabetic patients [G. Boden and M. Zhang Expert Opin.
- PPAR ⁇ nuclear receptor peroxisome-proliferator activated receptor- gamma
- Sulfonylureas bind to the sulfonylurea receptor on pancreatic beta cells, stimulate insulin secretion, and consequently reduce blood glucose levels. Weight gain is also associated with the use of sulfonylureas [S. E. Inzucchi JAMA 2002, 287, 360] and, like metformin, they lose efficacy over time [R. C. Turner et al. JAMA 1999, 281, 2005].
- a further problem - A - often encountered in patients treated with sulfonylureas is hypoglycemia [M. Salas J. J. and Caro Adv. Drug React. Tox. Rev. 2002, 21, 205-217].
- Acarbose is an inhibitor of the enzyme alpha- glucosidase, which breaks down disaccharides and complex carbohydrates in the intestine. It has lower efficacy than metformin or the sulfonylureas, and it causes intestinal discomfort and diarrhea which often lead to the discontinuation of its use [S. E. Inzucchi/ ⁇ M ⁇ 2002, 287, 360]
- 1 l ⁇ -hydroxysteroid dehydrogenase type I 1 l ⁇ -HSDl
- 1 l ⁇ -HSDl is an enzyme that catalyzes the reduction of cortisone to Cortisol (or dehydrocorticosterone to corticosterone in rodents).
- Cortisol is a corticosteroid hormone produced in the adrenal gland, and it has been shown to increase levels of glucose production, mostly by increasing gluconeogenesis [S. Khani and J. A. Tayek Clinical. ScL 2001, 101, 739-747].
- a second enzyme, 1 l ⁇ -hydroxysteroid dehydrogenase type II (l l ⁇ -HSD2) is responsible for the oxidation of Cortisol to cortisone.
- the enzymes have low homology and are expressed in different tissues.
- 1 l ⁇ -HSDl is highly expressed in a number of tissues including liver, adipose tissue, and brain, while 1 l ⁇ -HSD2 is highly expressed in mineralocorticoid target tissues, such as kidney and colon.
- 1 l ⁇ -HSD2 prevents the binding of Cortisol to the mineralocorticoid receptor, and defects in this enzyme have been found to be associated with the syndrome of apparent mineralocorticoid excess (AME).
- mice demonstrate that modulation of the activity of 1 l ⁇ -HSDl could have beneficial therapeutic effects in diabetes and in the metabolic syndrome.
- 1 l ⁇ -HSDl gene when the 1 l ⁇ -HSDl gene is knocked out in mice, fasting does not lead to the normal increase in levels of G6Pase and PEPCK, and the animals are not susceptible to stress- or obesity-related hyperglycemia.
- knockout animals which are rendered obese on a high-fat diet have significantly lower fasting glucose levels than weight- matched controls (Y. Kotolevtsev et al. Proc. Natl. Acad. Sd. USA 1997, 94, 14924).
- mice have also been found to have improved lipid profile, insulin sensitivity, and glucose tolerance (N. M. Morton et al. /. Biol. Chem. 2001, 276, 41293).
- the effect of overexpressing the 1 l ⁇ -HSDl gene in mice has also been studied.
- These transgenic mice displayed increased 1 l ⁇ -HSDl activity in adipose tissue, and they also exhibit visceral obesity which is associated with the metabolic syndrome. Levels of the corticosterone were increased in adipose tissue, but not in serum, and the mice had increased levels of obesity, especially when on a high-fat diet. Mice fed on low- fat diets were hyperglycemic and hyperinsulinemic, and also showed glucose intolerance and insulin resistance (H. Masuzaki et al. Science, 2001, 294, 2166).
- carbenoxolone was found to lead to an increase in whole body insulin sensitivity, and this increase was attributed to a decrease in hepatic glucose production (B. R. Walker et al. /. Clin. Endocrinol. Metab. 1995, 80, 3155).
- carbenoxolone was found to improve cognitive function in healthy elderly men and also in type 2 diabetics (T. C. Sandeep et al. Proc. Natl. Acad. Sci USA 2004, 101, 6734).
- a number of non-specific inhibitors of 1 l ⁇ -HSDl and 1 l ⁇ -HSD2 have been identified, including glycyrrhetinic acid, abietic acid, and carbenoxolone.
- a number of selective inhibitors of 1 l ⁇ -HSDl have been found, including chenodeoxycholic acid, flavanone and 2'-hydroxyflavanone (S. Diederich et al. Eur. J. Endocrinol. 2000, 142, 200 and R. A. S. Schweizer et al. MoI. Cell. Endocrinol. 2003, 212, Al).
- 1 l ⁇ -HSDl inhibitors that have efficacy for the treatment of diseases such as, for example, type II diabetes mellitus and metabolic syndrome. Further, a need exists in the art for 1 l ⁇ -HSDl inhibitors having IC50 values less than about 1 ⁇ M.
- a pharmaceutical composition comprising a therapeutically effective amount of a compound according to formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- a method for treating diabetes comprising the step of administering a therapeutically effective amount of a compound according to formula (I), or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- the present invention is directed to inhibitors of ll ⁇ -HSDl.
- the invention provides for pharmaceutical compositions comprising compounds of the formula (I) as well as pharmaceutically acceptable salts thereof, that are useful as inhibitors of l l ⁇ -HSDl .
- alkyl means, for example, a branched or unbranched, cyclic ("cycloalkyl") or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl radical which may be substituted or unsubstituted.
- cyclic the alkyl group is preferably C3 to C12, more preferably C4 to C10, more preferably C4 to C7.
- the alkyl group is preferably Ci to C10, more preferably Ci to Ce, more preferably methyl, ethyl, propyl (n-propyl or isopropyl), butyl (n-butyl, isobutyl or tertiary-butyl) or pentyl ( including n-pentyl and isopentyl), more preferably methyl.
- alkyl as used herein includes alkyl (branched or unbranched), substituted alkyl (branched or unbranched), alkenyl (branched or unbranched), substituted alkenyl (branched or unbranched), alkynyl (branched or unbranched), substituted alkynyl (branched or unbranched), cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, cycloalkynyl and substituted cycloalkynyl.
- a preferred example of cycloalkyl includes cycloalkenyl.
- the "cycloalkyl" moieties can optionally be substituted with one, two, three or four substituents, wherein each substituent is independently, for example, hydroxy, alkyl, alkoxy, halogen or amino, unless otherwise specifically indicated.
- substituents include, but are not limited to, optionally substituted cyclopropyl, optionally substituted cyclobutyl, optionally substituted cyclopentyl, optionally substituted cyclopentenyl, optionally substituted cyclohexyl, optionally substituted cyclohexylenyl, optionally substituted cycloheptyl, and the like or those which are specifically exemplified herein.
- Preferred cycloalkyl are cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. Further preferred cycloalkyl are cyclopropyl, cyclobutyl and cyclohexyl.
- heterocycloalkyl denotes a cyclic alkyl ring, wherein one, two or three of the carbon ring atoms are replaced by a heteroatom such as N, O or S.
- heterocycloalkyl groups include, but are not limited to, morpholinyl, thiomorpholinyl, piperazinyl, piperidinyl, pyrrolidinyl, azepanyl, tetrahydrofuranyl and the like.
- the heterocycloalkyl groups may be unsubstituted or substituted with one, two or three substituents independently selected from methyl, hydroxyl and hydroymethyl.
- Preferred heterocycloalkyl groups are morpholinyl, piperidinyl, pyrrolidinyl, azepanyl and tetrahydrofuranyl, optionally substituted with one, two or three substituents independently selected from methyl, hydroxyl and hydroymethyl.
- Further preferred heterocycloalkyl are morpholinyl, dimethylmorpholinyl, methylpiperidinyl, hydroxypiperidinyl, hydroxymethylpiperidinyl, pyrrolidinyl, methylpyrrolidinyl, dimethylpyrrolidinyl, hydroxymethylpyrrolidinyl, azepanyl and tetrahydrofuranyl.
- lower alkyl means, for example, a branched or unbranched, cyclic or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl radical wherein said cyclic lower alkyl group is C3, Q, C5, Ce or C7, and wherein said acyclic lower alkyl group is C 1 , C 2 , C3 or Q, and is preferably selected from methyl, ethyl, propyl (n-propyl or isopropyl) or butyl (n-butyl, isobutyl or tertiary-butyl).
- lower alkyl as used herein includes, for example, lower alkyl (branched or unbranched), lower alkenyl (branched or unbranched), lower alkynyl (branched or unbranched), cycloloweralkyl, cycloloweralkenyl and cycloloweralkynyl.
- the lower alkyl can optionally be substituted with hydroxy.
- Particularly preferred examples of lower alkyl are methyl, isopropyl, hydroxymethyl, hydroxyethyl, hydroxypropyl and hydroxybutyl.
- aryl means, for example, a substituted or unsubstituted carbocyclic aromatic group.
- aryl groups are phenyl, naphthyl and the like.
- a preferred aryl groups is phenyl.
- heteroaryl alone or in combination with other groups, means a monocyclic or bicyclic radical of 5 to 12 ring atoms having at least one aromatic ring having one, two, or three ring heteroatoms selected from N, O, and S, the remaining ring atoms being C.
- One or two ring carbon atoms of the heteroaryl group may be replaced with a carbonyl group.
- the heteroaryl group described above may be substituted independently with one, two, or three substituents, preferably one or two substituents such as, for example, halogen, hydroxy, C 1 ⁇ alkyl, halo C 1 ⁇ alkyl, d-6 alkoxy, C 1 ⁇ alkyl sulfonyl, C 1 ⁇ alkyl sulfinyl, d-6 alkylthio, amino, amino Ci-6 alkyl, mono- or di-substituted amino-Ci-6 alkyl, nitro, cyano, acyl, carbamoyl, mono- or di-substituted amino, aminocarbonyl, mono- or di-substituted amino-carbonyl, aminocarbonyl Ci-6 alkoxy, mono- or di- substituted amino-carbonyl-Ci-6 alkoxy, hydroxy- C 1 ⁇ alkyl, carboxyl, C 1 ⁇ alkoxy carbonyl, aryl d-6 alkoxy, hetero
- heteroaryl examples include thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, oxadiazolyl, thiadiazolyl, aziridinyl, azetidinyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, imidazolinyl, pyrazolidinyl, tetrahydrofuranyl, pyranyl, pyronyl, pyridyl, pyrazinyl, pyridazinyl, piperidyl, hexahydroazepinyl, piperazinyl, morpholinyl, thianaphthyl, benzofuranyl, isobenzofuranyl, indolyl, oxyindolyl, isoindolyl, indazolyl, indolinyl, 7-
- alkyl and aryl groups may be substituted or unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substituents present, preferably 1 substituent.
- Substituents may include, for example: carbon-containing groups such as alkyl, aryl, arylalkyl (e.g. substituted and unsubstituted phenyl, substituted and unsubstituted benzyl); halogen atoms and halogen-containing groups such as haloalkyl (e.g. trifluoromethyl); oxygen-containing groups such as alcohols (e.g. hydroxyl, hydroxyalkyl, aryl(hydroxyl)alkyl), ethers (e.g.
- aminocarbonyl mono- or di-alkylaminocarbonyl, aminocarbonylalkyl, mono-or di-alkylaminocarbonylalkyl, arylaminocarbonyl
- carbamates e.g. alkoxycarbonylamino, arloxycarbonylamino, aminocarbonyloxy, mono- or di-alkylaminocarbonyloxy, arylminocarbonloxy
- ureas e.g. mono- or di- alkylaminocarbonylamino or arylaminocarbonylamino
- nitrogen-containing groups such as amines (e.g.
- the lower alkyl groups may be substituted or unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substitutents present, preferably 1 substituent.
- alkoxy means, for example, alkyl-O- and "alkoyl” means, for example, alkyl-CO-.
- Alkoxy substituent groups or alkoxy- containing substituent groups may be substituted by, for example, one or more alkyl groups.
- Preferred alkoxy substituents are methoxy, ethoxy, propyloxy and butyloxy. Particularly preferred is methoxy.
- halogen means, for example, a fluorine, chlorine, bromine or iodine radical, preferably a fluorine, chlorine or bromine radical, and more preferably a fluorine or chlorine radical.
- Pharmaceutically acceptable salt refers to conventional acid-addition salts or base- addition salts that retain the biological effectiveness and properties of the compounds of formula (I) and are formed from suitable organic or inorganic acids or organic or inorganic bases.
- Sample acid-addition salts include those derived from inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, phosphoric acid and nitric acid, and those derived from organic acids such as p- toluenesulfonic acid, salicylic acid, methanesulfonic acid, oxalic acid, succinic acid, citric acid, malic acid, lactic acid, fumaric acid, and the like.
- Sample base-addition salts include those derived from ammonium, potassium, sodium and, quaternary ammonium hydroxides, such as for example, tetramethylammonium hydroxide.
- “Pharmaceutically acceptable ester” refers to a conventionally esterified compound of formula I having a carboxyl group, which esters retain the biological effectiveness and properties of the compounds of formula I and are cleaved in vivo (in the organism) to the corresponding active carboxylic acid.
- ester groups which are cleaved (in this case hydrolyzed) in vivo to the corresponding carboxylic acids are those in which the hydrogen is replaced with lower alkyl which is optionally substituted, e.g., with heterocycle, cycloalkyl, etc.
- substituted lower alkyl esters are those in which lower alkyl is substituted with pyrrolidine, piperidine, morpholine, N-methylpiperazine, etc.
- the group which is cleaved in vivo may be, for example, ethyl, morpholino ethyl, and diethylamino ethyl.
- -CONH 2 is also considered an ester, as the -NH 2 may be cleaved in vivo and replaced with a hydroxy group, to form the corresponding carboxylic acid.
- Preferred is a compound of formula (I), wherein Ri is methyl.
- a compound of formula (I) wherein Ri and R 2 , together with the N atom to which they are attached, form an unsubstituted 5- to 7-membered monocyclic ring, which contains the N atom to which Ri and R 2 are attached.
- a compound of formula (I) wherein Ri and R 2 , together with the N atom to which they are attached, form piperidinyl, pyrrolidinyl or azepanyl.
- a compound of formula (I) wherein Ri and R 2 , together with the N atom to which they are attached, form an unsubstituted 5- to 7-membered monocyclic ring, which contains the N atom to which Ri and R 2 are attached and another hetero atom which is selected from O and S.
- a compound of formula (I) wherein Ri and R 2 , together with the N atom to which they are attached, form morpholinyl.
- Ri and R 2 together with the N atom to which they are attached, form methylpiperidinyl, hydroxypiperidinyl, hydroxymethylpiperidinyl, ethylpyrrolidinyl, dimethylpyrrolidinyl or hydroxymethylpyrrolidinyl.
- a compound of formula (I) wherein Ri and R 2 , together with the N atom to which they are attached, form a 5- to 7-membered monocyclic ring, which contains the N atom to which Ri and R 2 are attached, and another hetero atom which is selected from O and S, wherein the 5- to 7-membered monocyclic ring is mono- or bi- substituted with hydroxy, lower alkyl or -(CH 2 ) n OH.
- Particularly preferred is a compound of formula (I), wherein Ri and R 2 , together with the N atom to which they are attached, form dimethylmorpholinyl.
- R3 can represent one to five substituents, preferably three, more preferably two and more preferably one.
- Preferred is a compound of formula (I), wherein R 3 is hydrogen or halogen and particularly preferred is a compound of formula (I), wherein R 3 is hydrogen.
- Preferred is a compound of formula (I), wherein R 4 is hydrogen, -OH, or -NHC( O)CH 3 and particularly preferred is a compound of formula (I), wherein R 4 is hydrogen.
- the present invention also relates to a process for the preparation of a compound of formula (I) comprising the reaction of a compound according to formula (II)
- R 1 , R 2 , R3 and R4 are as defined above.
- the present invention also relates to a compound of formula (I) for use as therapeutically active substance.
- the present invention also relates to a compound of formula (I) for the preparation of medicaments for the prophylaxis or therapy of illnesses which are caused by disorders associated with the enzyme 1 lbeta-hydroxysteroid dehydrogenase 1.
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) and a therapeutically inert carrier.
- the present invention also relates to the use of a compound of formula (I) for the preparation of medicaments for the treatment or prophylaxis of diabetes, obesity, eating disorders or dyslipidemia.
- the present invention also relates to a compound according of formula (I) for use as medicament for the treatment or prophylaxis of diabetes, obesity, eating disorders or dyslipidemia.
- the present invention also relates to the use of a compound of formula (I) for the preparation of medicaments for the treatment or prophylaxis of diabetes Type II.
- the present invention also relates to a compound of formula (I) for use as medicament for the treatment or prophylaxis of diabetes Type II.
- the present invention also relates to the use of a compound of formula (I), when manufactured according to a process according to the present invention.
- the present invention also relates to a method for the treatment and prophylaxis of diabetes, obesity, eating disorders, or dyslipidemia, which method comprises administering an effective amount of a compound of formula (I).
- the present invention also relates to a method for the treatment or prophylaxis of diabetes Type II, which method comprises administering an effective amount of a compound of formula (I).
- an effective amount of any one of the compounds of this invention or a combination of any of the compounds of this invention or a pharmaceutically acceptable salt or ester thereof is administered via any of the usual and acceptable methods known in the art, either singly or in combination.
- the compounds or compositions can thus be administered orally (e.g., buccal cavity), sublingually, parenterally (e.g., intramuscularly, intravenously, or subcutaneously), rectally (e.g., by suppositories or washings), transdermally (e.g., skin electroporation) or by inhalation (e.g., by aerosol), and in the form or solid, liquid or gaseous dosages, including tablets and suspensions.
- buccal cavity e.g., buccal cavity
- parenterally e.g., intramuscularly, intravenously, or subcutaneously
- rectally e.g., by suppositories or washings
- transdermally e.g., skin electroporation
- the administration can be conducted in a single unit dosage form with continuous therapy or in a single dose therapy ad libitum.
- the therapeutic composition can also be in the form of an oil emulsion or dispersion in conjunction with a lipophilic salt such as pamoic acid, or in the form of a biodegradable sustained- release composition for subcutaneous or intramuscular administration.
- Useful pharmaceutical carriers for the preparation of the compositions hereof can be solids, liquids or gases; thus, the compositions can take the form of tablets, pills, capsules, suppositories, powders, enterically coated or other protected formulations (e.g. binding on ion-exchange resins or packaging in lipid-protein vesicles), sustained release formulations, solutions, suspensions, elixirs, aerosols, and the like.
- the carrier can be selected from the various oils including those of petroleum, animal, vegetable or synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, and the like.
- formulations for intravenous administration comprise sterile aqueous solutions of the active ingredient(s) which are prepared by dissolving solid active ingredient(s) in water to produce an aqueous solution, and rendering the solution sterile.
- suitable pharmaceutical excipients include starch, cellulose, glucose, lactose, talc, gelatin, malt, rice, flour, chalk, silica, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, dried skim milk, glycerol, propylene glycol, water, ethanol, and the like.
- compositions may be subjected to conventional pharmaceutical additives such as preservatives, stabilizing agents, wetting or emulsifying agents, salts for adjusting osmotic pressure, buffers and the like.
- suitable pharmaceutical carriers and their formulation are described in Remington's Pharmaceutical Sciences by E. W. Martin. Such compositions will, in any event, contain an effective amount of the active compound together with a suitable carrier so as to prepare the proper dosage form for proper administration to the recipient.
- the pharmaceutical preparations can also contain preserving agents, solubilizing agents, stabilizing agents, wetting agents, emulsifying agents, sweetening agents, coloring agents, flavoring agents, salts for varying the osmotic pressure, buffers, coating agents or antioxidants. They can also contain other therapeutically valuable substances, including additional active ingredients other than those of formula I.
- the "therapeutically effective amount” or “dosage” of a compound according to this invention can vary within wide limits and may be determined in a manner known in the art. Such dosage will be adjusted to the individual requirements in each particular case including the specific compound(s) being administered, the route of administration, the condition being treated, as well as the patient being treated. In general, in the case of oral or parenteral administration to adult humans weighing approximately 70 kg, a daily dosage of from about 0.01 mg/kg to about 50 mg/kg should be appropriate, although the upper limit may be exceeded when indicated. The dosage is preferably from about 0.3 mg/kg to about 10 mg/kg per day. A preferred dosage may be from about 0.70 mg/kg to about 3.5 mg/kg per day. The daily dosage can be administered as a single dose or in divided doses, or for parenteral administration it may be given as continuous infusion.
- the compounds of the present invention can be prepared by any conventional manner. Suitable processes for synthesizing these compounds are provided in the examples.
- a l-aryl-5-(substituted amino) -pyrazole-4-carboxamide derivative of formula 1 can be prepared starting from ethyl (ethoxymethylene)cyanoacetate of formula 2 (which is available from Aldrich) by the following sequence of reactions:
- the first reaction in the sequence can be conveniently carried out by treating ethyl (ethoxymethylene)cyanoacetate of formula 2 with a hydrazine of formula ArNHNH 2 in an inert solvent such as ethanol at the reflux temperature.
- Conditions suitable for this reaction can be found in the literature, for example in A. Costanzo et al. /. Heterocycl. Chem. 1994, 31, 1369-1376; in M. Kopp et al. /. Heterocycl. Chem. 2001, 38, 1045-1050; A. Costanzo et al. /. Heterocycl. Chem. 1992, 29, 1499-1505; in N. P. Peet et al. /. Med. Chem.
- the Sandmeyer-type reaction of the intermediate of formula 3 involves diazotization of the amino group in the presence of a chlorinating agent such as copper(I) chloride, or copper(II) chloride, or nitrosyl chloride.
- a chlorinating agent such as copper(I) chloride, or copper(II) chloride, or nitrosyl chloride.
- the reaction is conveniently carried out by treating the compound of formula 3 with an alkyl nitrite such as tert-butyl nitrite or isoamyl nitrite in an inert solvent such as acetonitrile or a halogenated hydrocarbon (for example, carbon tetrachloride) at a temperature between about 50 degrees and about 65 degrees, in the presence of a chlorine source such as copper(I) chloride.
- the reaction can be carried out by treating the compound of formula 3 with sodium nitrite in the presence of aqueous hydrochloric acid and a chlorinating agent such as copper(II) chloride initially at a temperature preferably below 10 degrees and most preferably at about 0 degrees, and then at about 40 degrees.
- the conversion of the amino-pyrazole of formula 3 to the chloro- pyrazole of formula 4 may be carried out by treating a solution of the compound of formula 3 in an inert solvent such as a chlorinated hydrocarbon (e.g., chloroform) with hydrogen chloride, and then with liquid nitrosyl chloride at a temperature below about 10 degrees and then at about room temperature.
- an inert solvent such as a chlorinated hydrocarbon (e.g., chloroform)
- the cleavage of a compound of formula 4 to the corresponding carboxylic acid of formula 5 is carried out using reaction conditions that are well known in the field of organic synthesis, many of which are outlined in "Protective Groups in Organic Synthesis" [T. W. Greene and P. G. M. Wuts, 2nd Edition, John Wiley & Sons, N.Y. 1991].
- the reaction can be conveniently effected by treating the compound of formula 4 with one equivalent of an alkali metal hydroxide, such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide, in a suitable solvent, such as a mixture of tetrahydrofuran, methanol, and water.
- an alkali metal hydroxide such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide
- the reaction can be carried out at a temperature between about 0 0 C and about room temperature, preferably at about room temperature.
- the ester may be treated with a strong inorganic acid, for example a hydrohalic acid such as hydrogen chloride or hydrogen bromide, in aqueous solution, preferably at the reflux temperature.
- the carboxylic acid of formula 5 can be converted conveniently to the amide of formula 7 by treating the carboxylic acid of structure 5 with the hydrochloride of the adamantane derivative of formula 6 in the presence of an appropriate base, such as diisopropylethylamine, a coupling agent such as O-(benzotriazol-l-yl)-l, 1,3,3- tetramethyluronium hexafluorophosphate, and in the optional additional presence of a substance that increases the rate of the reaction, such as 1-hydroxybenzotriazole or 1- hydroxy-7-azabenzotriazole, in an inert solvent, such as a chlorinated hydrocarbon (e.g., dichloromethane) or N,N-dimethylformamide or N-methylpyrrolidinone, at a temperature between about 0 degrees and about room temperature, preferably at about room temperature.
- an appropriate base such as diisopropylethylamine
- a coupling agent such as O-(benzo
- reaction can be carried out by converting the carboxylic acid of formula 5 to an activated ester derivative, such as the N-hydroxysuccinimide ester, and subsequently reacting this with the adamantane derivative of formula 6 or a corresponding acid addition salt.
- an activated ester derivative such as the N-hydroxysuccinimide ester
- This reaction sequence can be carried out by reacting the carboxylic acid of formula 5 with N- hydroxysuccinimide in the presence of a coupling agent such as N,N'-dicyclohexyl- carbodiimide in an inert solvent such as tetrahydrofuran at a temperature between about 0 degrees and about room temperature.
- a coupling agent such as N,N'-dicyclohexyl- carbodiimide
- an inert solvent such as tetrahydrofuran
- N-hydroxysuccinimide ester is then treated with the adamantane derivative of formula 6 or a corresponding acid addition salt, in the presence of a base, such as an organic base (e.g., triethylamine or diisopropylethylamine or the like) in a suitable inert solvent such as N,N- dimethylformamide at around room temperature to give the l-aryl-5-chloro-pyrazole-4- carboxamide of formula 7.
- a base such as an organic base (e.g., triethylamine or diisopropylethylamine or the like) in a suitable inert solvent such as N,N- dimethylformamide at around room temperature to give the l-aryl-5-chloro-pyrazole-4- carboxamide of formula 7.
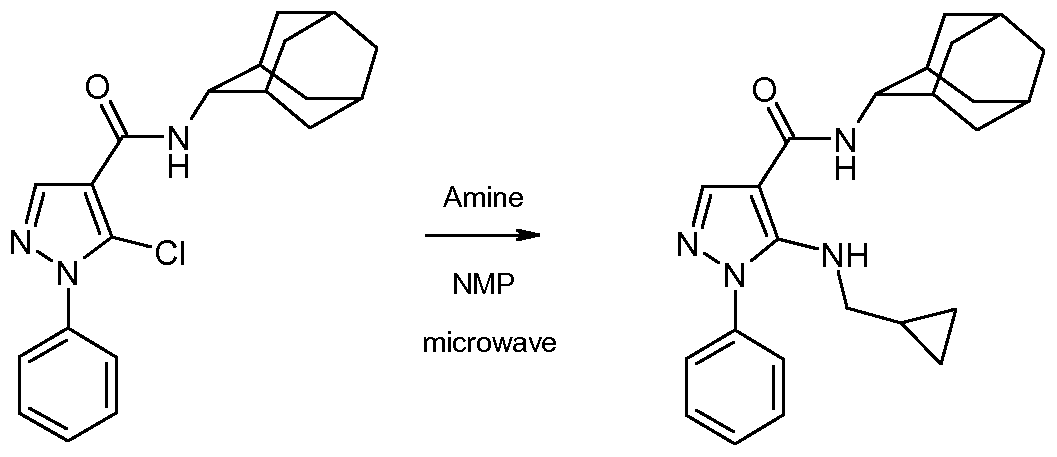
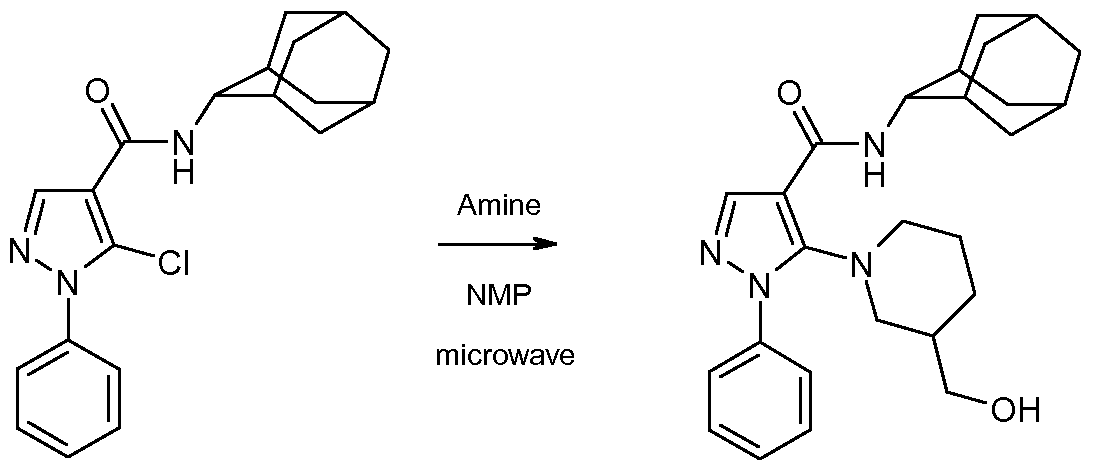
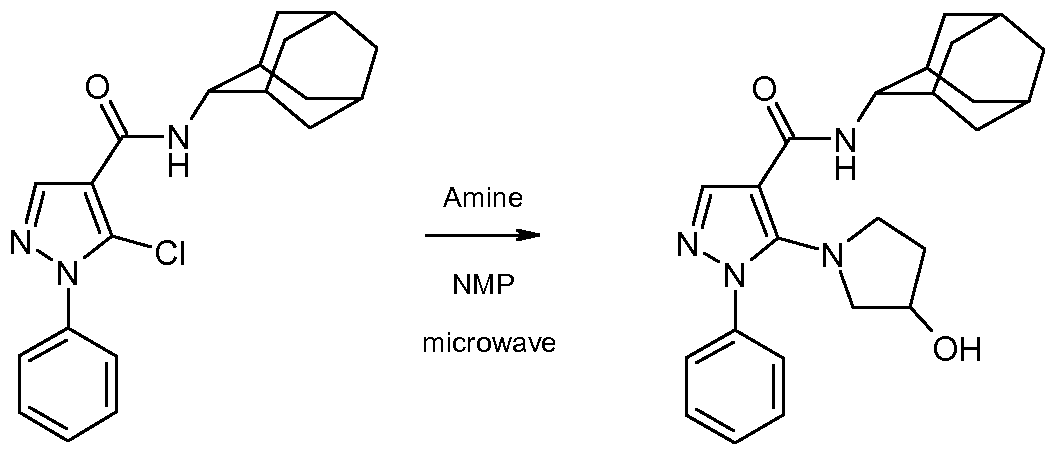
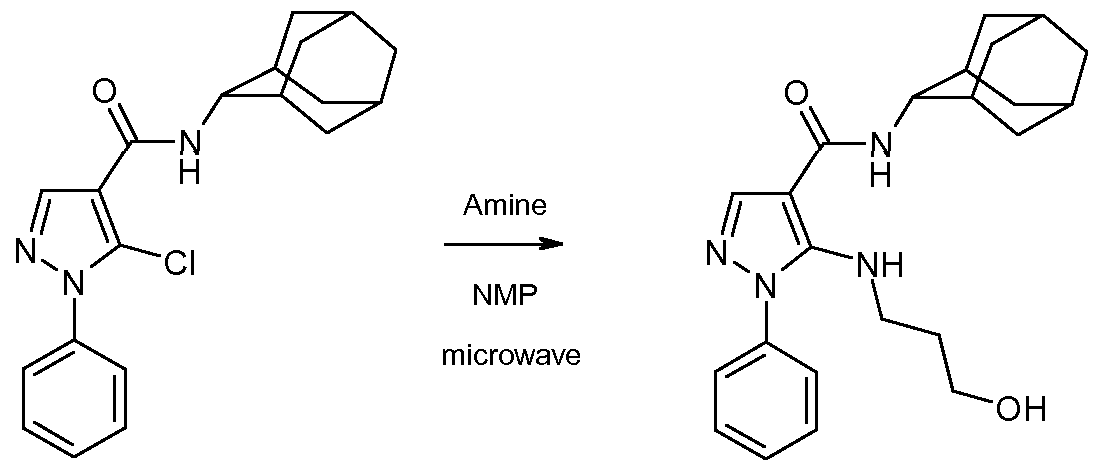
- the l-aryl-5-chloro-pyrazole-4-carboxamide of formula 7 can then be converted to the compound of the invention of formula 1 by heating it with an amine of formula HRiR 2 in an inert solvent such as N-methylpyrrolidinone at a temperature about 250 degrees, under microwave irradiation.
- an inert solvent such as N-methylpyrrolidinone
- a second alternative procedure for the preparation of compounds of formula 4 is shown in Scheme 3.
- the reaction of commercially available diethyl ethoxymethylenemalonate of formula 9 with an arylhydrazine of formula ArNHNH 2 can be carried out under a variety of conditions.
- the compound of formula 9 can be reacted with an arylhydrazine or the acid addition salt of an arylhydrazine in an inert solvent such as an alcohol (for example, ethanol).
- an acid addition salt of the arylhydrazine is used, then the reaction is carried out in the additional presence of a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- the reaction is conveniently carried out at a temperature between about -20 degrees and about 80 degrees. Examples of conditions for this reaction can be found in the literature, for example, in R. Gehring et al. US 4,804,398; in W. Holzer and E. Schmid/. Heterocycl. Chem. 1995, 32, 1341-1349.
- the intermediate of formula 10 is then heated to approximately 170 degrees with the evolved ethanol being removed by distillation. This process gives the 5-hydroxy-pyrazole of formula 11.
- Conditions for this reaction can be found in the literature, for example in R. Gehring et al. US 4,804,398.
- the intermediate of formula 10 can be heated at reflux in ethanol in the presence of a base such as potassium carbonate to give the 5-hydroxy- pyrazole of formula 11.
- the 5- hydroxy-pyrazole of formula 11 can then be converted into the chloro-pyrazole of formula 4 through a chlorination reaction.
- the reaction can conveniently be carried out by heating the 5-hydroxy-pyrazole of formula 11 with a chlorinating agent such as phosphorus oxychloride in the absence of additional solvents at a temperature about 100 degrees.
- a chlorinating agent such as phosphorus oxychloride
- Precise conditions for such a reaction can be found in the literature, for example in W. Holzer and K. Hahn /. Heterocycl. Chem. 2003, 40, 303-308; in H. A. DeWaId et al. /. Med. Chem. 1981, 24, 982-987.
- the N-arylglycine derivative of formula 13 is then nitrosated to give the N-nitroso derivative of formula 14 by treatment with sodium nitrite in aqueous hydrochloric acid at a temperature about 0 degrees (for details, see D. L. Hammick and D. J. Voaden /. Chem. Soc. 1961, 3303-3308 or F. Dumitra ⁇ cu et al. ARKIVOC 2002, 80-86).
- the compound of formula 14 is then treated with acetic acid and pyridine to give the sydnone of formula 15.
- the sydnone is then chlorinated to give the chloro-sydnone of formula 16.
- the chlorination reaction can be carried out by treating the sydnone of formula 15 with chlorine in a mixture of sodium acetate and acetic acid at a temperature about room temperature (see F. Dumitra ⁇ cu et al. ARKIVOC 2002, 80-86); by treating the sydnone of formula 15 with iodobenzene dichloride in a mixture of triethylamine and dichloromethane (see S. Ito and K. Turnbull Synth. Commun. 1996, 26, 1441-1446); or by treating the sydnone of formula 15 with N- chlorosuccinimide in an inert solvent such as dimethylformamide at a temperature about room temperature (see K. Turnbull et al. /.
- the chloro-sydnone of formula 16 can then be treated with excess dimethyl acetylenedicarboxylate in ethylene glycol at 120 degrees to give the [3+2] dipolar cycloaddition product 17.
- the compound of formula 17 can then be treated with 20% aqueous hydrochloric acid at reflux to effect hydrolysis to the dicarboxylate which undergoes selective decarboxylation upon heating to about 250 degrees (bath temperature) to give the monocarboxylic acid of formula 5.
- Specific conditions for the reactions that lead from the chloro-sydnone of formula 16 to the monocarboxylic acid of formula 5 can be found in the literature, for example in H. Dickopp Chem. Ber. 1974, 107, 3036-3042.
- a compound of the invention where R2 represents hydrogen can be prepared in four steps from the l-aryl-5-amino-pyrazole-4-carboxylate ester of formula 3 by converting the amino substituent to a carbamate which can then be alkylated to give the intermediate of formula 19. Deprotection of the carbamate and ethyl ester then gives intermediate 21 which can be coupled with an adamantanamine of formula 6 to give the product of formula 1.
- the conversion of the amine of formula 3 to the carbamate of formula 18 can be effected through any conventional means, several of which will be apparent to one of average skill in the art of organic synthesis.
- the amine may be treated with a loweralkyl chloroformate (such as ethyl chloroformate) in the presence of a base such as sodium hydride in an inert solvent such as dimethylformamide or tetrahydrofuran.
- a base such as sodium hydride
- an inert solvent such as dimethylformamide or tetrahydrofuran.
- the amine of formula 3 can be treated with an excess of phenyl chloroformate in the presence of a base such as sodium hydride in an inert solvent such as dimethylformamide to give the bis(phenoxycarbonyl) amino derivative, as described in L. R. Hatton et al.
- the reaction can also be carried out using pyridine as base and chloroform as solvent. In this case, the reaction is preferably carried out at low temperature such as at about 0 degrees.
- Conditions for this transformation can be found in the literature, for example in L. R. Hatton et al. US 4,629,495.
- This bis(phenoxycarbonyl) amino derivative can then be treated with ferf-butanol at the reflux temperature to give the intermediate of formula 18 where R' represents tert-butyl.
- Conditions for this transformation can be found in the literature, for example in L. R. Hatton et al. US 4,629,495.
- the alkylation of the carbamate of formula 18 with an alkylating agent of formula RiX can be effected using a variety of different procedures which are well known.
- the leaving group X can be a halogen (e.g., bromine or iodine) or it can be a sulfonate ester (e.g., mesylate, tosylate, or nosylate) etc.
- the reaction is conveniently effected by treatment of the carbamate with a base such as sodium hydride in an inert solvent such as tetrahydrofuran at a temperature between about room temperature and the reflux temperature of the solvent, depending on the reactivity of the alkylating agent.
- the carbamate protective group is then removed from the carbamate of formula 19 to give the amine of formula 20 using conditions well known in the art for this transformation, which may be specific to the nature of the R' group. Many examples of appropriate conditions are outlined in the book "Protective Groups in Organic Synthesis” [T. W. Greene and P. G. M. Wuts, 2nd Edition, John Wiley & Sons, N.Y. 1991].
- the protective group may be removed by treating the compound of formula 20 with an acid such as tifluoroacetic acid in an inert solvent such as a halogenated hydrocarbon (e.g., dichloromethane) at about room temperature.
- the protective group may be removed by heating the compound of formula 19 with potassium hydroxide in ethylene glycol at about 100 degrees. Conditions for this reaction may be found in the literature, for example in K. Matsushita et al. EP 885890.
- the ethyl ester in the compound of formula 20 can then be removed hydrolytically under conditions well known in the field of organic synthesis.
- the compound of formula 20 may be treated with one equivalent of an alkali metal hydroxide, such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide, in a suitable solvent, such as a mixture of tetrahydrofuran, methanol, and water.
- an alkali metal hydroxide such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide
- a suitable solvent such as a mixture of tetrahydrofuran, methanol, and water.
- the reaction can be carried out at a temperature between about 0 degrees and about room temperature, preferably at about room temperature.
- the carbamate and ester may be removed in one process from the compound of formula 19 by subjecting the compound of formula 19 to acidic conditions, for example by heating in dilute aqueous hydrochloric acid at the reflux temperature.
- the compound of the invention of formula 1 can then be prepared by reaction of the carboxylic acid of structure 21 or of an appropriate derivative thereof such as an activated ester, with an adamantane derivative of formula 6 or a corresponding acid addition salt (e.g., the hydrochloride salt) in the presence, if necessary, of a coupling agent, many examples of which are well known per se in peptide chemistry.
- the reaction is conveniently carried out by treating the carboxylic acid of structure 21 with the hydrochloride of the adamantane derivative of formula 6 in the presence of an appropriate base, such as diisopropylethylamine, a coupling agent such as O- (benzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate, and in the optional additional presence of a substance that increases the rate of the reaction, such as 1-hydroxybenzotriazole or l-hydroxy-7-azabenzotriazole, in an inert solvent, such as a chlorinated hydrocarbon (e.g., dichloromethane) or N,N-dimethylformamide or N- methylpyrrolidinone, at a temperature between about 0 degrees and about room temperature, preferably at about room temperature.
- an appropriate base such as diisopropylethylamine
- a coupling agent such as O- (benzotriazol-l-yl)-l
- reaction can be carried out by converting the carboxylic acid of formula 21 to an activated ester derivative, such as the N-hydroxysuccinimide ester, and subsequently reacting this with the adamantane derivative of formula 6 or a corresponding acid addition salt.
- This reaction sequence can be carried out by reacting the carboxylic acid of formula 21 with N-hydroxysuccinimide in the presence of a coupling agent such as N,N'- dicyclohexylcarbodiimide in an inert solvent such as tetrahydrofuran at a temperature between about 0 degrees and about room temperature.
- N- hydroxysuccinimide ester is then treated with the adamantane derivative of formula 6 or a corresponding acid addition salt, in the presence of a base, such as an organic base (e.g., triethylamine or diisopropylethylamine or the like) in a suitable inert solvent such as N,N-dimethylformamide at around room temperature.
- a base such as an organic base (e.g., triethylamine or diisopropylethylamine or the like) in a suitable inert solvent such as N,N-dimethylformamide at around room temperature.
- Examples of processes useful for the preparation of aryl-hydrazines include diazotization of an aniline followed by reduction of the diazonium salt (P. Barraja et al. Bioorg. Med.
- 2-Adamantanamine hydrochloride is available from Aldrich.
- 2-Amino-5-hydroxy-adamantane (formula 22) can be prepared by hydrogenation of the imine derived from 5-hydroxy-2-adamantanone and 1-S- ⁇ -methylbenzylamine according to the procedure described in L. Jaraskova et al. Tetrahedron Lett. 2006, 47, 8063-8067.
- 2-amino-5-acetamido-adamantane (of formula 25) can be prepared starting from 2-amino-5-hydroxy-adamantane (22).
- the Cbz-protected compound of formula 23 is prepared conveniently from 2-amino-5-hydroxy-adamantane by treatment with benzyl chloroformate in the presence of a base such as triethylamine in an inert solvent such as dichloromethane at about room temperature.
- the alcohol of formula 23 is then treated with an inorganic acid such as sulfuric acid in acetonitrile at room temperature in a reaction known as the Ritter reaction. Conditions for this reaction may be found in L. Jaroskova et al. WO 2006024627; in B.
- the carbobenzyloxy protective group is then removed from the compound of formula 24 using hydrogenation under noble metal catalysis to give the amine of formula 25.
- the compound of formula 24 may be hydrogenated at approximately 50 psi in the presence of a catalytic amount of 5% palladium-on-carbon in an alcoholic solvent (such as ethanol) at room temperature.
- the alcohol of formula 23 can be subjected to Ritter reaction conditions with chloroacetonitrile, in the presence of sulfuric acid at about room temperature, to give the chloroacetyl derivative of formula 26.
- This compound then undergoes reaction with thiourea in the presence of acetic acid in ethanol at a temperature between about 50 degrees and about 120 degrees to give the amine of formula 27.
- Conditions for the Ritter reaction and the deprotection of the chloroacetamide can be found in the literature, for example in I. R. Gladwell WO 2007010356; in B. Gopalan et al. WO2006090244; and in A. Jirgensons et al. Synthesis 2000, 1709-1712.
- the amine of formula 27 can then be treated with methanesulfonyl chloride in the presence of a base such as triethylamine or diisopropylethylamine in an inert solvent such as dichloromethane at about room temperature to give the sulfonamide of formula 28.
- a base such as triethylamine or diisopropylethylamine in an inert solvent such as dichloromethane at about room temperature
- the carbobenzyloxy protective group is then removed from the compound of formula 28 using hydrogenation under noble metal catalysis to give the amine of formula 29.
- the compound of formula 28 may be hydrogenated at approximately 50 psi in the presence of a catalytic amount of 5% palladium-on-carbon in an alcholic solvent (such as ethanol) at room temperature.
- a l-aryl-pyrazole-4-carboxylate ester of formula 11 can be conveniently prepared by the reaction of (ethoxycarbonyl)-malondialdehyde with an arylhydrazine.
- the synthesis of (ethoxycarbonyl)-malondialdehyde is described in two steps from ethyl propiolate in S. H. Bertz et al. /. Org. Chem. 1982, 47, 2216-2217.
- the compound of formula 11 is conveniently prepared by treating (ethoxycarbonyl)- malondialdehyde with an arylhydrazine in an inert solvent such as a lower alcohol (e.g., ethanol) at room temperature. Conditions suitable for this reaction can be found in the literature, for example in W. Holzer and G. Seringer /. Heterocycl. Chem. 1993, 30, 865- 872.
- a l-aryl-pyrazole-4-carboxylate ester of formula 11 can be conveniently prepared in two steps from the commercially available l,3-dimethyluracil-5- carboxaldehyde (of formula 30).
- the aldehyde is treated with an arylhydrazine of formula ArNHNH 2 in water in the presence of acetic acid at about 100 degrees to give the hydrazone of formula 31.
- This is then heated in the presence of sodium methoxide in methanol at the reflux temperature to give the l-aryl-pyrazole-4-carboxylate ester of formula 11.
- Conditions suitable for this reaction can be found in the literature, for example in K. Hirota et al. /. Chem. Soc. Perkin Trans. 1 1983, 1293-1297.
- a l-aryl-pyrazole-4-carboxylate ester of formula 11 can be isolated as the minor product of a [3+2] dipolar cycloaddition reaction of a sydnone of formula 15 (prepared as described above) with a loweralkyl propiolate (e.g., methyl propiolate).
- the reaction is conveniently carried out by treating the sydnone of formula 15 with methyl propiolate in an inert solvent such as 1,2-dichlorobenzene, isobutyl alcohol, p-xylene, or dimethylformamide at the reflux temperature.
- an inert solvent such as 1,2-dichlorobenzene, isobutyl alcohol, p-xylene, or dimethylformamide
- reaction mixture was stirred at room temperature for 1 hour then at 65 0 C for 1 hour. Following complete consumption of starting material (monitored by TLC), the reaction mixture was poured into 6N hydrochloric acid (200 mL) and extracted with dichloromethane (3 x 300 mL). The combined organic layers were dried over magnesium sulfate and purified by column chromatography (eluting with heptane then 20% ethyl acetate in heptane) to give 5-chloro-l-phenyl-lH-pyrazole-4-carboxylic acid ethyl ester (7.3 g, 66%), which NMR and HPLC analysis indicated was 78% pure. This material was used directly in the next step without further purification.
- reaction mixture was stirred at 70 0 C for ⁇ 2 hours (reaction progress monitored by TLC and LCMS).
- the reaction mixture was dissolved in dichloromethane (300 mL), washed with IN hydrochloric acid (2 x 200 mL), and brine (200 mL), then dried over magnesium sulfate, filtered, evaporated, and purified by column chromatography (eluting with 20% ethyl acetate in heptane) to give 5-chloro-l-phenyl-lH-pyrazole-4-carboxylic acid adamantan-2-ylamide (4.2 g, 11.8 mmol, 45%), which HPLC analysis indicated was 90% pure. This material was used directly in the next step without further purification.
- the compounds were purified by chromatography, eluting with the following solvent sequence: heptane; 10% ethyl acetate in heptane; 20% ethyl acetate in heptane; and 50% ethyl acetate in heptane.
- Example 17 5- ( (S) -2-Hydroxymethyl-pyrrolidin- 1 -yl) - 1 -phenyl- lH-pyrazole-4- carboxylic acid adamantan-2-ylamide
- Example 25 l-Phenyl-5-[(tetrahydro-furan-2-ylmethyl)-amino]-lH-pyrazole-4- carboxylic acid adamantan-2-ylamide
- H4IIE cells stably transfected with full-length human 1 lbetaHSDl cDNA were propagated and expanded in DMEM high glucose media (Invitrogen Cat# 11995-065), supplemented with 10% FCS (Invitrogen Cat# 10082-147), 100 units/mL and 100 ⁇ g/mL pen/ strep (Invitrogen Cat#15140-122), and geneticin (800 ⁇ g/mL).
- FCS Invitrogen Cat# 10082-147
- pen/ strep Invitrogen Cat#15140-122
- geneticin 800 ⁇ g/mL
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description























































Claims




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008800247172A CN101743226B (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11beta-hydroxysteroid dehydrogenase |
| EP08774850.5A EP2178845B1 (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| CA2693457A CA2693457C (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| BRPI0814825-2A2A BRPI0814825A2 (en) | 2007-07-17 | 2008-07-07 | COMPOUND, PROCESS FOR THE PREPARATION OF COMPOUND, PHARMACEUTICAL COMPOSITION UNDERSTANDING ITS USE AND METHODS FOR TREATMENT OR PROPHYLAXY OF DIABETES, OBESITY, DISORDERS OF FOOD OR DISLIPIMEDIA AND TYPE II DIABETES |
| AU2008277783A AU2008277783B2 (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11beta-hydroxysteroid dehydrogenase |
| ES08774850T ES2423181T3 (en) | 2007-07-17 | 2008-07-07 | 11ß-hydroxysteroid dehydrogenase inhibitors |
| JP2010516456A JP5189165B2 (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11β-hydroxysteroid dehydrogenase |
| KR1020107003122A KR101158191B1 (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11b-hydroxysteroid dehydrogenase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US95016807P | 2007-07-17 | 2007-07-17 | |
| US60/950,168 | 2007-07-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009010416A2 true WO2009010416A2 (en) | 2009-01-22 |
| WO2009010416A3 WO2009010416A3 (en) | 2009-03-05 |
Family
ID=40019292
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/058804 Ceased WO2009010416A2 (en) | 2007-07-17 | 2008-07-07 | Inhibitors of 11b-hydroxysteroid dehydrogenase |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US7790711B2 (en) |
| EP (1) | EP2178845B1 (en) |
| JP (1) | JP5189165B2 (en) |
| KR (1) | KR101158191B1 (en) |
| CN (1) | CN101743226B (en) |
| AR (1) | AR067538A1 (en) |
| AU (1) | AU2008277783B2 (en) |
| BR (1) | BRPI0814825A2 (en) |
| CA (1) | CA2693457C (en) |
| CL (1) | CL2008002052A1 (en) |
| ES (1) | ES2423181T3 (en) |
| PE (1) | PE20090813A1 (en) |
| RU (1) | RU2440989C2 (en) |
| TW (1) | TWI372623B (en) |
| WO (1) | WO2009010416A2 (en) |
| ZA (1) | ZA201000318B (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7816391B2 (en) | 2007-02-12 | 2010-10-19 | Astrazeneca Ab | Chemical compounds |
| US7951833B2 (en) | 2008-02-04 | 2011-05-31 | Astrazeneca Ab | Crystalline forms of 4-[4-(2-adamantylcarbamoyl)-5-tert-butyl-pyrazol-1-yl]Benzoic acid 471 |
| US7964618B2 (en) | 2006-11-03 | 2011-06-21 | Astrazeneca Ab | Chemical compounds |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8324265B2 (en) | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| US8383622B2 (en) | 2007-05-18 | 2013-02-26 | Shionogi & Co., Ltd. | Nitrogen-containing heterocyclic derivative having 11β-hydroxysteroid dehydrogenase type I inhibitory activity |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Families Citing this family (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2604591A4 (en) * | 2010-08-09 | 2015-07-22 | Shionogi & Co | Process for preparing aminoadamantyl carbamate derivatives |
| CN103649066B (en) * | 2011-06-10 | 2016-08-17 | 爱思开生物制药株式会社 | 5-carbamyl-diamantane (obsolete)-2-yl amide derivatives, its pharmaceutically acceptable salt and preparation method thereof |
| ES2643403T3 (en) | 2011-12-28 | 2017-11-22 | Global Blood Therapeutics, Inc. | Substituted benzaldehyde compounds and methods for use in increasing tissue oxygenation |
| HK1203412A1 (en) | 2011-12-28 | 2015-10-30 | Global Blood Therapeutics, Inc. | Substituted heteroaryl aldehyde compounds and methods for their use in increasing tissue oxygenation |
| MX379235B (en) | 2013-03-15 | 2025-03-11 | Global Blood Therapeutics Inc | COMPOUNDS AND THEIR USES TO MODULATE HEMOGLOBIN. |
| WO2014150258A1 (en) * | 2013-03-15 | 2014-09-25 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US9802900B2 (en) | 2013-03-15 | 2017-10-31 | Global Blood Therapeutics, Inc. | Bicyclic heteroaryl compounds and uses thereof for the modulation of hemoglobin |
| US9422279B2 (en) | 2013-03-15 | 2016-08-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10100043B2 (en) | 2013-03-15 | 2018-10-16 | Global Blood Therapeutics, Inc. | Substituted aldehyde compounds and methods for their use in increasing tissue oxygenation |
| US9604999B2 (en) | 2013-03-15 | 2017-03-28 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US8952171B2 (en) | 2013-03-15 | 2015-02-10 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| US10266551B2 (en) | 2013-03-15 | 2019-04-23 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| CN105073728A (en) | 2013-03-15 | 2015-11-18 | 全球血液疗法股份有限公司 | Compounds and their use for regulating hemoglobin |
| US9458139B2 (en) | 2013-03-15 | 2016-10-04 | Global Blood Therapeutics, Inc. | Compounds and uses thereof for the modulation of hemoglobin |
| CA2926095A1 (en) | 2013-10-17 | 2015-04-23 | Dow Agrosciences Llc | Processes for the preparation of pesticidal compounds |
| CA2925953C (en) | 2013-10-17 | 2021-11-02 | Dow Agrosciences Llc | Processes for the preparation of pesticidal compounds |
| US9029554B1 (en) | 2013-10-17 | 2015-05-12 | Dow Agrosciences Llc | Processes for the preparation of pesticidal compounds |
| CA2925952A1 (en) | 2013-10-17 | 2015-04-23 | Dow Agrosciences Llc | Processes for the preparation of pesticidal compounds |
| MX2016004941A (en) | 2013-10-17 | 2016-06-28 | Dow Agrosciences Llc | Processes for the preparation of pesticidal compounds. |
| EP3057429A4 (en) | 2013-10-17 | 2017-08-09 | Dow AgroSciences LLC | Processes for the preparation of pesticidal compounds |
| JP2016539092A (en) | 2013-10-17 | 2016-12-15 | ダウ アグロサイエンシィズ エルエルシー | Method for producing pest control compound |
| EA201992707A1 (en) | 2013-11-18 | 2020-06-30 | Глобал Блад Терапьютикс, Инк. | COMPOUNDS AND THEIR APPLICATIONS FOR HEMOGLOBIN MODULATION |
| CN114181195A (en) | 2014-02-07 | 2022-03-15 | 全球血液疗法股份有限公司 | Crystalline polymorph of a compound |
| JP2017523168A (en) | 2014-07-31 | 2017-08-17 | ダウ アグロサイエンシィズ エルエルシー | Method for producing 3- (3-chloro-1H-pyrazol-1-yl) pyridine |
| BR112017000293A2 (en) | 2014-07-31 | 2017-10-31 | Dow Agrosciences Llc | Process for the preparation of 3- (3-chloro-1h-pyrazol-1-yl) pyridine |
| US9249122B1 (en) | 2014-07-31 | 2016-02-02 | Dow Agrosciences Llc | Process for the preparation of 3-(3-chloro-1H-pyrazol-1-yl)pyridine |
| JP2017525703A (en) | 2014-08-19 | 2017-09-07 | ダウ アグロサイエンシィズ エルエルシー | Method for preparing 3- (3-chloro-1H-pyrazol-1-yl) pyridine |
| AR098113A1 (en) | 2014-09-12 | 2016-05-04 | Dow Agrosciences Llc | PROCESS FOR THE PREPARATION OF 3- (3-CHLORINE-1H-PIRAZOL-1-IL) PIRIDINE |
| KR101759874B1 (en) | 2015-08-07 | 2017-07-21 | 코스맥스 주식회사 | Skin external composition or Cosmetics composition for anti-wrinkle comprising compound of 11β-hydroxysteroid dehydrogenase type 1 inhibitor |
| US10324166B2 (en) * | 2015-09-28 | 2019-06-18 | Rockwell Collins, Inc. | Affordable combined pulsed/FMCW radar AESA |
| SG11201804647TA (en) | 2015-12-04 | 2018-06-28 | Global Blood Therapeutics Inc | Dosing regimens for 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| TWI663160B (en) | 2016-05-12 | 2019-06-21 | 全球血液治療公司 | Process for synthesizing 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)-pyridin-3-yl)methoxy)benzaldehyde |
| EP3258288B1 (en) * | 2016-06-14 | 2024-07-31 | Rohde & Schwarz GmbH & Co. KG | Method for testing the transmission and reflection properties of an automotive radome body as well as apparatus for testing the transmission and reflection properties of an automotive radome body |
| TW202332423A (en) | 2016-10-12 | 2023-08-16 | 美商全球血液治療公司 | Tablets comprising 2-hydroxy-6-((2-(1-isopropyl-1h-pyrazol-5-yl)pyridin-3-yl)methoxy)benzaldehyde |
| JP2020503336A (en) | 2016-12-29 | 2020-01-30 | ダウ アグロサイエンシィズ エルエルシー | Method for preparing pesticidal compounds |
| CN110325036B (en) | 2016-12-29 | 2021-10-26 | 美国陶氏益农公司 | Process for preparing pesticidal compounds |
| US11014884B2 (en) | 2018-10-01 | 2021-05-25 | Global Blood Therapeutics, Inc. | Modulators of hemoglobin |
| US12479816B2 (en) | 2019-02-08 | 2025-11-25 | University of Pittsburgh—of the Commonwealth System of Higher Education | 20-HETE formation inhibitors |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2732967B1 (en) * | 1995-04-11 | 1997-07-04 | Sanofi Sa | 1-PHENYLPYRAZOLE-3-CARBOXAMIDES SUBSTITUTED, ACTIVE IN NEUROTENSIN, THEIR PREPARATION, THE PHARMACEUTICAL COMPOSITIONS CONTAINING IT |
| EP0979228A4 (en) * | 1997-03-18 | 2000-05-03 | Smithkline Beecham Corp | Novel cannabinoid receptor agonists |
| WO2004056744A1 (en) | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| ATE467616T1 (en) | 2003-04-11 | 2010-05-15 | High Point Pharmaceuticals Llc | COMPOUNDS WITH ACTIVITY AT 11BETA-HYDROXASTEROID DEHYDROGENASE |
| JP2006522750A (en) | 2003-04-11 | 2006-10-05 | ノボ ノルディスク アクティーゼルスカブ | Combination therapy using 11β-hydroxysteroid dehydrogenase type 1 inhibitors and antihypertensive agents to treat metabolic syndrome and related diseases and disorders |
| EP1615667A2 (en) | 2003-04-11 | 2006-01-18 | Novo Nordisk A/S | Combinations of an 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor and a glucocorticoid receptor agonist |
| DK1618092T3 (en) | 2003-05-01 | 2011-01-31 | Bristol Myers Squibb Co | Aryl-substituted pyrazole-amide compounds useful as kinase inhibitors |
| US7659408B2 (en) | 2003-08-07 | 2010-02-09 | Merck Sharp & Dhome Corp. | Pyrazole carboxamides as inhibitors of 11-β-hydroxysteroid dehydrogenase-1 |
| US7880001B2 (en) | 2004-04-29 | 2011-02-01 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US20050261302A1 (en) | 2004-04-29 | 2005-11-24 | Hoff Ethan D | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme and their therapeutic application |
| US20050245533A1 (en) | 2004-04-29 | 2005-11-03 | Hoff Ethan D | Inhibitors of the 11-beta-hydroxysteroid dehydrogenaseType 1 enzyme and their therapeutic application |
| US20050245534A1 (en) | 2004-04-29 | 2005-11-03 | Link James T | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US20050245532A1 (en) | 2004-04-29 | 2005-11-03 | Hoff Ethan D | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme and their therapeutic application |
| CA2565843A1 (en) | 2004-05-06 | 2005-11-17 | Pfizer Inc. | Novel compounds of proline and morpholine derivatives |
| NZ551076A (en) | 2004-05-07 | 2009-05-31 | Janssen Pharmaceutica Nv | Adamantyl pyrrolidin-2-one derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| CN1993320A (en) | 2004-08-06 | 2007-07-04 | 默克公司 | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| BRPI0515121A (en) | 2004-08-30 | 2008-07-08 | Janssen Pharmaceutica Nv | n-2 adamantyl-2-phenoxy acetamide derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| MY141198A (en) | 2004-08-30 | 2010-03-31 | Janssen Pharmaceutica Nv | Tricyclic adamantylamide derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| US7713979B2 (en) | 2004-10-29 | 2010-05-11 | Eli Lilly And Company | Cycloalkyl lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| JP2008518903A (en) | 2004-11-02 | 2008-06-05 | ファイザー・インク | New compounds of substituted and unsubstituted adamantylamides |
| EP1659113A1 (en) | 2004-11-08 | 2006-05-24 | Evotec AG | Inhibitors of 11beta-hydroxy steroid dehydrogenase type 1 (11beta-HSD1) |
| US20060148871A1 (en) | 2005-01-05 | 2006-07-06 | Rohde Jeffrey J | Metabolic stabilization of substituted adamantane |
| JP5133702B2 (en) | 2005-01-05 | 2013-01-30 | アボット・ラボラトリーズ | Inhibitors of 11-β-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2006074244A2 (en) | 2005-01-05 | 2006-07-13 | Abbott Laboratories | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| JP2008537939A (en) | 2005-03-31 | 2008-10-02 | エクソンモービル・ケミカル・パテンツ・インク | Production of multi-phase alkyl aromatics |
| US20090264650A1 (en) | 2005-03-31 | 2009-10-22 | Nobuo Cho | Prophylactic/Therapeutic Agent for Diabetes |
| DK1928840T3 (en) * | 2005-04-05 | 2011-09-12 | Hoffmann La Roche | 1H-pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors |
| EP1894919B1 (en) * | 2005-06-07 | 2012-03-28 | Shionogi & Co., Ltd. | Heterocyclic compound having type i 11 beta hydroxysteroid dehydrogenase inhibitory activity |
| US8324265B2 (en) * | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| EP1999114B1 (en) * | 2006-03-22 | 2015-07-22 | F. Hoffmann-La Roche AG | Pyrazoles as 11-beta-hsd-1 |
| TW200836719A (en) | 2007-02-12 | 2008-09-16 | Astrazeneca Ab | Chemical compounds |
| WO2011068927A2 (en) * | 2009-12-04 | 2011-06-09 | Abbott Laboratories | 11-β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 (11B-HSD1) INHIBITORS AND USES THEREOF |
-
2008
- 2008-07-07 AU AU2008277783A patent/AU2008277783B2/en not_active Ceased
- 2008-07-07 RU RU2010105299/04A patent/RU2440989C2/en not_active IP Right Cessation
- 2008-07-07 JP JP2010516456A patent/JP5189165B2/en not_active Expired - Fee Related
- 2008-07-07 EP EP08774850.5A patent/EP2178845B1/en not_active Not-in-force
- 2008-07-07 BR BRPI0814825-2A2A patent/BRPI0814825A2/en not_active IP Right Cessation
- 2008-07-07 ES ES08774850T patent/ES2423181T3/en active Active
- 2008-07-07 WO PCT/EP2008/058804 patent/WO2009010416A2/en not_active Ceased
- 2008-07-07 CN CN2008800247172A patent/CN101743226B/en not_active Expired - Fee Related
- 2008-07-07 CA CA2693457A patent/CA2693457C/en not_active Expired - Fee Related
- 2008-07-07 KR KR1020107003122A patent/KR101158191B1/en not_active Expired - Fee Related
- 2008-07-14 US US12/172,389 patent/US7790711B2/en not_active Expired - Fee Related
- 2008-07-14 CL CL2008002052A patent/CL2008002052A1/en unknown
- 2008-07-14 AR ARP080103024A patent/AR067538A1/en not_active Application Discontinuation
- 2008-07-15 PE PE2008001191A patent/PE20090813A1/en not_active Application Discontinuation
- 2008-07-16 TW TW097127011A patent/TWI372623B/en not_active IP Right Cessation
-
2010
- 2010-01-15 ZA ZA201000318A patent/ZA201000318B/en unknown
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8324265B2 (en) | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| US7964618B2 (en) | 2006-11-03 | 2011-06-21 | Astrazeneca Ab | Chemical compounds |
| US8673938B2 (en) | 2006-11-03 | 2014-03-18 | Astrazeneca Ab | Chemical compounds |
| US7816391B2 (en) | 2007-02-12 | 2010-10-19 | Astrazeneca Ab | Chemical compounds |
| US8344016B2 (en) | 2007-02-12 | 2013-01-01 | Astrazeneca Ab | Pyrazole derivatives as 11-beta-HSD1 inhibitors |
| US8383622B2 (en) | 2007-05-18 | 2013-02-26 | Shionogi & Co., Ltd. | Nitrogen-containing heterocyclic derivative having 11β-hydroxysteroid dehydrogenase type I inhibitory activity |
| US7951833B2 (en) | 2008-02-04 | 2011-05-31 | Astrazeneca Ab | Crystalline forms of 4-[4-(2-adamantylcarbamoyl)-5-tert-butyl-pyrazol-1-yl]Benzoic acid 471 |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2440989C2 (en) | 2012-01-27 |
| CN101743226A (en) | 2010-06-16 |
| CN101743226B (en) | 2012-10-10 |
| ES2423181T3 (en) | 2013-09-18 |
| BRPI0814825A2 (en) | 2015-02-03 |
| AR067538A1 (en) | 2009-10-14 |
| AU2008277783A1 (en) | 2009-01-22 |
| TW200913992A (en) | 2009-04-01 |
| US7790711B2 (en) | 2010-09-07 |
| TWI372623B (en) | 2012-09-21 |
| WO2009010416A3 (en) | 2009-03-05 |
| JP5189165B2 (en) | 2013-04-24 |
| JP2010533670A (en) | 2010-10-28 |
| CA2693457A1 (en) | 2009-01-22 |
| CA2693457C (en) | 2012-10-23 |
| PE20090813A1 (en) | 2009-06-27 |
| ZA201000318B (en) | 2010-10-27 |
| CL2008002052A1 (en) | 2009-05-22 |
| EP2178845A2 (en) | 2010-04-28 |
| RU2010105299A (en) | 2011-08-27 |
| AU2008277783B2 (en) | 2012-09-20 |
| US20090023709A1 (en) | 2009-01-22 |
| KR101158191B1 (en) | 2012-06-19 |
| KR20100034763A (en) | 2010-04-01 |
| EP2178845B1 (en) | 2013-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2178845B1 (en) | Inhibitors of 11b-hydroxysteroid dehydrogenase | |
| AU2007228887B2 (en) | Pyrazoles as 11-beta-HSD-1 | |
| EP1928840B1 (en) | 1H-Pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors | |
| EP1924563B1 (en) | 11-beta-hydroxysteroid dehydrogenase-1-inhibitor-diabetes-type 2-1 | |
| US9840470B2 (en) | Targeting GLI proteins in human cancer by small molecules |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880024717.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08774850 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008774850 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2693457 Country of ref document: CA Ref document number: 2008277783 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12010500084 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010516456 Country of ref document: JP Ref document number: MX/A/2010/000639 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 325/DELNP/2010 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2008277783 Country of ref document: AU Date of ref document: 20080707 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20107003122 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010105299 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0814825 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100114 |