WO2009016191A1 - Process for preparing a leukotriene antagonist and an intermediate thereof - Google Patents
Process for preparing a leukotriene antagonist and an intermediate thereof Download PDFInfo
- Publication number
- WO2009016191A1 WO2009016191A1 PCT/EP2008/059965 EP2008059965W WO2009016191A1 WO 2009016191 A1 WO2009016191 A1 WO 2009016191A1 EP 2008059965 W EP2008059965 W EP 2008059965W WO 2009016191 A1 WO2009016191 A1 WO 2009016191A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- salt
- preparation according
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- DYLOVNSFPNMSRY-OTVRWNPNSA-N CC(c1c(CC[C@H](c2cc(/C=C/c(cc3)nc4c3ccc(Cl)c4)ccc2)SCC2(CC(O)=O)CC2)cccc1)=O Chemical compound CC(c1c(CC[C@H](c2cc(/C=C/c(cc3)nc4c3ccc(Cl)c4)ccc2)SCC2(CC(O)=O)CC2)cccc1)=O DYLOVNSFPNMSRY-OTVRWNPNSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/18—Halogen atoms or nitro radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
Definitions
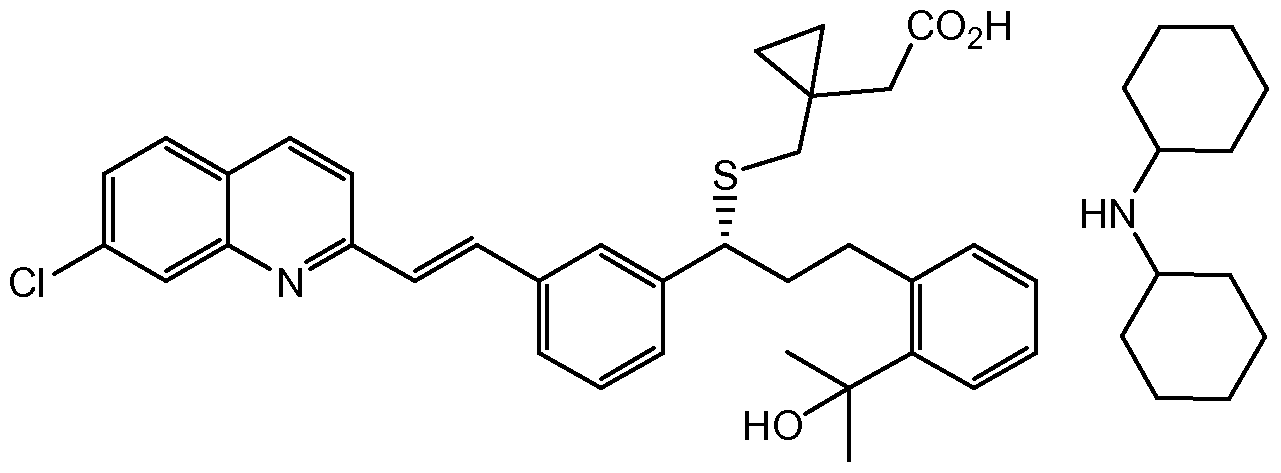
- the present invention relates to a process for preparing montelukast, and novel intermediates used in such process.
- Montelukast is the International Non-proprietary Name (INN) for (R)-(E)-I -(((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-(1 -hydroxy-1 - methylethyl)phenyl)propyl)thio)methyl)cyclopropane acetic acid, and CAS RN 158966-92-8.
- INN International Non-proprietary Name
- European Patent Application EP 737186 discloses a process to prepare a crystalline form of montelukast sodium from the dicyclohexylamine salt of montelukast. This process is hardly applicable on an industrial scale since a highly flammable base (n-butyl lithium) and reaction temperatures below -1 O 0 C are used. In addition, an impurity, which is derived from dehydration of the te/t-butanol group, is formed during the preparation of montelukast and its removal becomes very difficult.
- the purification steps via the dicyclohexylamine salt are very time consuming and comprise seeding of montulekast and dicyclohexylamine solution in toluene or ethyl acetate, and subsequent addition of heptane or hexane respectively.
- the dicyclohexylamine salt of formula Ma is much more insoluble in polar solvents than the known dicyclohexylamine salts of montelukast and its methyl ester intermediate. Due to its different solubility, this salt shows the advantage of being easily crystallizable in said polar solvents, and the duration of salt formation becomes considerably shorter. Likewise, seeding of the solution to start crystallization is not necessary, which facilitates -in practice- the industrial-scale operating conditions since procedures that may be dangerous due to the flammability of such solvents are avoided. Additionally, the product is obtained in high yield and with a high purity.
- Another advantage of the use of the salt of formula Ma is that it can be obtained by using polar solvents without the need for mixtures of non-polar solvents, which facilitates the isolation of the product. Furthermore, the use of one sole solvent for crystallization facilitates its recovery and reuse in an industrial process.
- purification of the salt of formula Ma may be carried out in the same solvent as that used for crystallization.
- a first aspect of the invention relates to a compound of formula Ma,
- Another aspect of the invention relates to a process for the preparation of a compound of formula Ma as defined above, which comprises reaction of a compound
- Another aspect of the invention relates to a process for preparing a compound of formula I,
- Montelukast (compound of formula I) and its intermediates may be prepared by the processes hereinafter described.
- the most appropriate conditions under which the process is carried out may vary depending on different parameters considered by those skilled in the art, such as the concentration of starting material, temperature, solvent used and the like. These parameters may be easily determined by those skilled in the art through routine testing and using the teachings in the examples of the present specification.
- an aqueous treatment is utilized in an acid medium for isolation of the reaction product.
- said acid may be an organic acid.
- organic acids include, among others, acetic acid, methanesulfonic acid, thfluoromethanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid or p-toluenesulfonic acid.
- the acid used is acetic acid.
- the compound of formula I may be prepared by reaction of a compound of formula Il with a compound of formula III, wherein X is halogen, as depicted on the following scheme:
- This reaction is optionally carried out in the presence of a Lewis acid in a suitable solvent, such us tetrahydrofuran, and at a suitable temperature, preferably at O 0 C, and a subsequent aqueous treatment in an acid medium is performed to yield the compound of formula I.
- a Lewis acid such as tetrahydrofuran
- Lewis acid is used herein to refer to a substance which can accept an electron pair.
- Lewis acids include, among others, AICI 3 , FeCI 3 , ZnCI 2 and CeCI 3 .
- in the compound of formula III X is Cl and the reaction is carried out in the presence of a Lewis acid.
- in the compound of formula III X is Cl and the Lewis acid is CeCI 3 .
- the compound of formula Il is obtained by aqueous treatment of the salt of formula Ma in an acid medium, preferably in acetic acid, in a suitable solvent, such as a mixture of toluene and water, and at a suitable temperature, preferably room temperature.
- a suitable solvent such as a mixture of toluene and water
- the process for preparing a compound of formula Ma comprises reaction of a compound of formula IV, wherein LG represents a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl, with a compound of formula V, wherein R represents an alkaline metal, such as Na, Li or K, followed by treatment with dicyclohexylamine (DCHA) in a polar solvent, as depicted on the following synthetic scheme:
- LG represents a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl
- R represents an alkaline metal, such as Na, Li or K
- DCHA dicyclohexylamine
- the first step is carried out preferably in a solvent system such as dimethylformamide, acetone and toluene, at room temperature.
- a solvent system such as dimethylformamide, acetone and toluene, at room temperature.
- LG represents methanesulfonyloxyl.
- R represents Na.
- the second step comprises reaction of the compound in free acid form, which is obtained in the first step after an aqueous treatment in an acid medium, with dicyclohexylamine to yield, within a few minutes and without the need for seeding, the salt of formula Ma.
- This reaction is carried out in a polar solvent.
- polar solvent refers to a solvent in whose molecules there is either a permanent separation of positive and negative charges, or the centres of positive and negative charges do not coincide.
- polar solvents include ketones; alcohols, such as isopropanol or ethanol; esters; halogenated hydrocarbons, such as chloroform or methylene chloride; ethers, such as isopropyl ether or methyl te/t-butyl ether; and polar aromatic hydrocarbons.
- aromatic hydrocarbon is used herein to refer to a mono- or di- substituted benzene, wherein the substituent is selected from halogen or methyl.
- aromatic hydrocarbons of the invention include toluene, xylene or chlorobenzene.
- the polar solvent is selected from the group consisting of a ketone of formula RCORi, an ester of formula RCO 2 Ri and toluene; wherein R and Ri may be the same or different and represent (Ci-C 4 )alkyl.
- (Ci-C 4 )alkyl represents a saturated straight or branched hydrocarbon chain having from 1 to 4 carbon atoms. Examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and te/t-buthyl groups.
- ketones of formula RCORi include, among others, acetone, methyl ethyl ketone, diethyl ketone or methyl isobutyl ketone; and examples of esters of formula RCO 2 Ri include, among others, ethyl acetate or isopropyl acetate.
- the solvent is acetone, isopropyl acetate or toluene.
- the solvent is acetone.
- crystallization is carried out at room temperature. If necessary, the obtained salt may be purified by using the same solvent.
- the compound of formula Ma thus obtained is in a crystalline form.
- the compound of formula Ma thus obtained is the crystalline form of the (R)-(E)-I -(((1 -(3-(2-(7-chloro-2- quinolinyl)ethenyl)phenyl)-3-(2-acetylphenyl)propyl)thio)methyl)cyclopropane acetic acid dicyclohexylamine salt, whose X-ray diffractogram is substantially the same as that shown in FIGURE 1.
- the compounds of formula V may be obtained from mercaptomethylcyclopropylacetic acid by reaction with a base, for example NaH, in a suitable solvent such as dimethylformamide, and preferably upon cooling.
- a base for example NaH
- a suitable solvent such as dimethylformamide
- the compounds of formula IV may be prepared in a two-step sequence.
- the compound of formula VM is reacted with a compound of formula Ml wherein X is halogen, preferably chloro, to give, after aqueous treatment, a compound of formula Vl.
- This reaction is carried out in the presence of lithium bis(trimethylsilyl)amide in a suitable solvent, such as a mixture of tetrahydrofuran and toluene, and at a suitable temperature, preferably upon cooling.
- the hydroxyl group of the compound of formula Vl is converted into a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl.
- LG represents methanesulfonyloxyl.
- This reaction is carried out in the presence of a sulfonyl halide, such as methanesulfonyl chloride or p- toluenesulfonyl chloride, in the presence of a base, such as pyridine or triethylamine, in a suitable solvent such as toluene, and at a suitable temperature, preferably upon cooling.
- the present invention also relates to salts of the compounds of formula I.
- a preferred embodiment relates to montelukast sodium salt of formula Ia.
- Said salt may be prepared from a compound of formula I or a salt thereof in the presence of NaOH in a suitable polar solvent, such as toluene or isopropyl acetate, by using a suitable non-polar solvent, such as n-heptane, to precipitate the product, and at an appropriate temperature, preferably at 15- 2O 0 C.
- FIGURE 1 depicts the X-ray diffractogram of the crystalline form of (R)-(E)-I - (((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-acetylphenyl)propyl)thio) methyl )cyclopropane acetic acid dicyclohexylamine salt.
- HPLC spectra were recorded using a Waters Alliance 2695 system equipped with UV Waters 2487 detector.
- reaction mixture was maintained for 15 hours under N 2 atmosphere at a temperature of 0-5 0 C.
- the reaction mixture was added to an aqueous solution containing 400 ml of water, 75 g of sodium chloride, 70 ml of glacial acetic acid and 60 g of ammonium acetate, at a temperature below 25 0 C.
- the aqueous phase was separated and the organic phase was washed with two 100-ml portions of 5% aqueous sodium bicarbonate solution.
- the solvent from the resulting organic phase was removed by distillation under reduced pressure and the obtained residue was dissolved in 90 ml of acetone at 2O 0 C. Then, 6 ml of water were added to afford a suspension.
- the enantiomeric excess of the starting compound VII was 96.5%, which led to compound Vl with an enantiomeric excess higher than 99%, due to the precipitation of the latter in the mixture of acetone-water.
- compound Vl is obtained with an enantiomeric excess less than 99%, it may be purified by a recrystallization in 4 volumes of acetone and 0.5 volumes of water referred to the weight of compound Vl.
- the solution obtained when heating the initial solution is cooled slowly to 15-20 0 C in order to crystallize the product.
- the isolated solid by filtration shows an enantiomeric excess higher than 99.5%. This methodology avoids the dependency on the enantiomeric purity of the starting hydroxyester VII.
- Example 2 20 g of the compound obtained in Example 1 (4% water, Vl) were dissolved in 200 ml of toluene. The solution was heated at reflux and water was separated by azeotropic distillation. The solution was then concentrated to a volume of approximately 50 ml.
- Previous solution containing the mesyl derivative from step a) was quickly added to the suspension of mercaptomethylcyclopropylacetic acid disodium salt from step b) and the temperature was kept at 5-10 0 C. After the addition was completed, the temperature was adjusted to 20-25 0 C and kept within this range for about 15 hours. Then, the reaction mixture was cooled to 0-5 0 C and 100 ml of an aqueous solution of sodium chloride were added. Acetone was removed by vacuum distillation and then 100 ml of isopropyl acetate were added. The aqueous phase was separated and the organic phase was washed with one 100-ml portion of aqueous sodium chloride solution.
- the product thus obtained may be purified using a suspension in 15 volumes of acetone at reflux temperature. After cooling at a temperature of 20-25 0 C, the solid was filtered off to afford a product with 99.0% purity by HPLC analysis and a purification yield higher than 98%. The titled compound was obtained with an enantiomeric excess higher than 99.8%.
- Example 2 In a light-protected flask under N 2 atmosphere, 10 g of the compound obtained in Example 2, 100 ml of toluene and 50 ml of water were placed. 1 g of glacial acetic acid was added and the mixture was kept under stirring until obtaining a solution. The aqueous phase was separated and the organic phase was washed with two 50-ml portions of water. Water was removed from the resulting solution by azeotropic distillation and the solution was concentrated until obtaining a residue which was dissolved in 50 ml of tetrahydrofuran.
- the aqueous phase was separated and the organic phase was washed with 100 ml of water. Thereafter, the resulting organic phase was treated with an aqueous solution containing 1.75 g of 30% sodium hydroxide and 100 ml of a 20% aqueous sodium chloride solution. The aqueous phase was separated and the organic phase was repeatedly treated with two portions (100 and 50 ml) of water. To the combined aqueous phases, 100 ml of toluene and 30 g of sodium chloride were added, and the mixture was kept under stirring until complete dissolution of sodium chloride. The aqueous phase was separated and tetrahydrofuran and water were removed by azeotropic distillation.
- the solution obtained was treated with 0.5 g active charcoal. After filtration, the resulting solution was concentrated and the residue obtained was added to 50 ml of n-heptane at the temperature of 20° C. The resulting mixture was concentrated by distillation and 35 ml of n-heptane were added. The resulting mixture was kept under stirring for 2 hours at the temperature of 15-20° C. Finally, the precipitated solid was filtered off, washed with n-heptane and dried at the temperature of 60-80° C, to yield 6.4 g of amorphous solid form of montelukast sodium salt (>99.0% purity by HPLC analysis, 81.0% yield). The title compound was obtained with an enantiomeric excess higher than 99.8%.
- Table 1 illustrates the starting material from which the dicyclohexilamine salt is formed, and crystallization conditions: solvent, time and if seeding is necessary. In all cases, temperature is 20-25°C. Table 1
- Comparative solubilities of montelukast dicyclohexilamine salts, the methyl ester intermediate and the compound of the invention Ma are shown in Table 2. In particular, the amounts of solvents needed to dissolve each compound at reflux are provided.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pulmonology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Dicyclohexylamine salt of formula (IIa) or a pharmaceutically acceptable solvate thereof, including a hydrate, and its process for preparation. Process for the preparation of montelukast of formula (I) or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including a hydrate, comprising the conversion of a salt of formula (IIa) into its form of free acid, followed by reaction with a compound of formula (III) CH3MgX, wherein X is halogen, in a suitable solvent, optionally in the presence of a Lewis acid.
Description
Process for preparing a leukotriene antagonist and an intermediate thereof
The present invention relates to a process for preparing montelukast, and novel intermediates used in such process.
BACKGROUND OF THE INVENTION
Montelukast is the International Non-proprietary Name (INN) for (R)-(E)-I -(((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-(1 -hydroxy-1 - methylethyl)phenyl)propyl)thio)methyl)cyclopropane acetic acid, and CAS RN 158966-92-8.
Montelukast monosodium salt (CAS RN 151767-02-1 ) of formula Ia
Ia
belongs to a group of drugs known as leukotriene antagonists. It is an orally active compound, which binds with high affinity and selectivity to CysLTI receptor. Montelukast monosodium salt is currently used in the treatment of asthma, inflammation, angina, cerebral spasm, glomerular nephritis, hepatitis, endotoxemia, uveitis and allograft rejection.
The preparation of montelukast sodium salt was first described in EP 480717 (Example 161 ). Later on, an alternative process was described for the preparation of a montelukast intermediate in EP 500360. However, these processes are not particularly suitable for industrial-scale application due to the use of solvents such as dichloromethane, reagents such as hydrazine, and temperatures below -4O0C. In addition, such processes require tedious chromatographic purifications of some intermediates and/or final products, and yields of final product are low.
Among the strategies for the preparation of montelukast, processes comprising the formation of amine salts of montelukast which are subsequently converted into its sodium salt have also been described. Some of these processes, as described below, comprise the purification of the dicyclohexylamine salt of montelukast:
Thus, for example, European Patent Application EP 737186 discloses a process to prepare a crystalline form of montelukast sodium from the dicyclohexylamine salt of montelukast. This process is hardly applicable on an industrial scale since a highly flammable base (n-butyl lithium) and reaction temperatures below -1 O0C are used. In addition, an impurity, which is derived from dehydration of the te/t-butanol group, is formed during the preparation of montelukast and its removal becomes very difficult. Moreover, the purification steps via the dicyclohexylamine salt are very time consuming and comprise seeding of montulekast and dicyclohexylamine solution in toluene or ethyl acetate, and subsequent addition of heptane or hexane respectively.
In document WO 06/008751 another process is described for obtaining montelukast sodium salt which comprises converting montelukast into its dicyclohexylamine salt. However, this process also requires seeding of the solution, and a very long period of time (more than one day) is needed for crystallization of the dicyclohexylamine salt.
Moreover, purification via the dicyclohexylamine salts has also been applied to intermediates which are subsequently converted into montelukast.
Thus, documents WO 06/008751 and WO 07/004237 describe the preparation of montelukast through the formation of a dicyclohexylamine salt of the methyl ester intermediate of formula:

However, these processes show the same drawbacks as those described for the formation of dicyclohexylamine salts over montelukast acid.
Therefore, due to the difficulty in purifying montelukast and its intermediates, the provision of alternative processes for preparation of montelukast, it is of great interest, particularly if they are easily industrializable.
SUMMARY OF THE INVENTION
The inventors have found that the preparation of a dicyclohexylamine salt of formula Ma

allows to obtain montelukast, as well as its salts and solvates, including hydrates, by an easily scalable process which overcomes the prior art drawbacks.
The dicyclohexylamine salt of formula Ma, as illustrated by the examples, is much more insoluble in polar solvents than the known dicyclohexylamine salts of montelukast and its methyl ester intermediate. Due to its different solubility, this salt shows the advantage of being easily crystallizable in said
polar solvents, and the duration of salt formation becomes considerably shorter. Likewise, seeding of the solution to start crystallization is not necessary, which facilitates -in practice- the industrial-scale operating conditions since procedures that may be dangerous due to the flammability of such solvents are avoided. Additionally, the product is obtained in high yield and with a high purity.
Another advantage of the use of the salt of formula Ma is that it can be obtained by using polar solvents without the need for mixtures of non-polar solvents, which facilitates the isolation of the product. Furthermore, the use of one sole solvent for crystallization facilitates its recovery and reuse in an industrial process.
Moreover, if desired, purification of the salt of formula Ma may be carried out in the same solvent as that used for crystallization.
Therefore, a first aspect of the invention relates to a compound of formula Ma,

Another aspect of the invention relates to a process for the preparation of a compound of formula Ma as defined above, which comprises reaction of a compound

Another aspect of the invention relates to a process for preparing a compound of formula I,

I
or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including a hydrate; which comprises conversion of a compound of formula Ma


Montelukast (compound of formula I) and its intermediates may be prepared by the processes hereinafter described. The most appropriate conditions under which the process is carried out may vary depending on different parameters considered by those skilled in the art, such as the concentration of starting material, temperature, solvent used and the like. These parameters may be easily determined by those skilled in the art through routine testing and using the teachings in the examples of the present specification.
In some reactions of the present invention, an aqueous treatment is utilized in an acid medium for isolation of the reaction product. Generally, said acid may be an organic acid. Examples or organic acids include, among others, acetic acid, methanesulfonic acid, thfluoromethanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid or p-toluenesulfonic acid. In a particular embodiment the acid used is acetic acid.
As mentioned above, the compound of formula I may be prepared by reaction of a compound of formula Il with a compound of formula III, wherein X is halogen, as depicted on the following scheme:

This reaction is optionally carried out in the presence of a Lewis acid in a suitable solvent, such us tetrahydrofuran, and at a suitable temperature, preferably at O0C, and a subsequent aqueous treatment in an acid medium is performed to yield the compound of formula I.
Throughout the specification, the term Lewis acid is used herein to refer to a substance which can accept an electron pair. Examples of Lewis acids include, among others, AICI3, FeCI3, ZnCI2 and CeCI3.
In a preferred embodiment, in the compound of formula III X is Cl and the reaction is carried out in the presence of a Lewis acid. In another preferred reaction, in the compound of formula III X is Cl and the Lewis acid is CeCI3.
The compound of formula Il is obtained by aqueous treatment of the salt of formula Ma in an acid medium, preferably in acetic acid, in a suitable solvent, such as a mixture of toluene and water, and at a suitable temperature,
preferably room temperature.

The process for preparing a compound of formula Ma comprises reaction of a compound of formula IV, wherein LG represents a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl, with a compound of formula V, wherein R represents an alkaline metal, such as Na, Li or K, followed by treatment with dicyclohexylamine (DCHA) in a polar solvent, as depicted on the following synthetic scheme:

The second step comprises reaction of the compound in free acid form, which is obtained in the first step after an aqueous treatment in an acid medium, with dicyclohexylamine to yield, within a few minutes and without the need for seeding, the salt of formula Ma. This reaction is carried out in a polar solvent.
The term polar solvent, as used in the present invention, refers to a solvent in whose molecules there is either a permanent separation of positive and negative charges, or the centres of positive and negative charges do not coincide. Examples of polar solvents include ketones; alcohols, such as isopropanol or ethanol; esters; halogenated hydrocarbons, such as chloroform or methylene chloride; ethers, such as isopropyl ether or methyl te/t-butyl ether; and polar aromatic hydrocarbons.
The term aromatic hydrocarbon is used herein to refer to a mono- or di- substituted benzene, wherein the substituent is selected from halogen or methyl. Examples of aromatic hydrocarbons of the invention include toluene, xylene or chlorobenzene.
In a preferred embodiment, the polar solvent is selected from the group consisting of a ketone of formula RCORi, an ester of formula RCO2Ri and toluene; wherein R and Ri may be the same or different and represent (Ci-C4)alkyl.
The term (Ci-C4)alkyl represents a saturated straight or branched hydrocarbon chain having from 1 to 4 carbon atoms. Examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and te/t-buthyl groups. Thus, examples of ketones of formula RCORi include, among others, acetone, methyl ethyl ketone, diethyl ketone or methyl isobutyl ketone; and examples of esters of formula RCO2Ri include, among others, ethyl acetate or isopropyl acetate.
In a yet more preferred embodiment, the solvent is acetone, isopropyl acetate or toluene. In the most preferred embodiment the solvent is acetone. Preferably, crystallization is carried out at room temperature. If necessary, the obtained salt may be purified by using the same solvent.
Preferably, the compound of formula Ma thus obtained is in a crystalline form. In a particular embodiment, the compound of formula Ma thus obtained is the crystalline form of the (R)-(E)-I -(((1 -(3-(2-(7-chloro-2- quinolinyl)ethenyl)phenyl)-3-(2-acetylphenyl)propyl)thio)methyl)cyclopropane acetic acid dicyclohexylamine salt, whose X-ray diffractogram is substantially the same as that shown in FIGURE 1.
The compounds of formula V may be obtained from mercaptomethylcyclopropylacetic acid by reaction with a base, for example NaH, in a suitable solvent such as dimethylformamide, and preferably upon cooling.
The compounds of formula IV may be prepared in a two-step sequence. In the first step, the compound of formula VM is reacted with a compound of formula Ml wherein X is halogen, preferably chloro, to give, after aqueous treatment, a compound of formula Vl. This reaction is carried out in the presence of lithium bis(trimethylsilyl)amide in a suitable solvent, such as a mixture of tetrahydrofuran and toluene, and at a suitable temperature, preferably upon cooling.
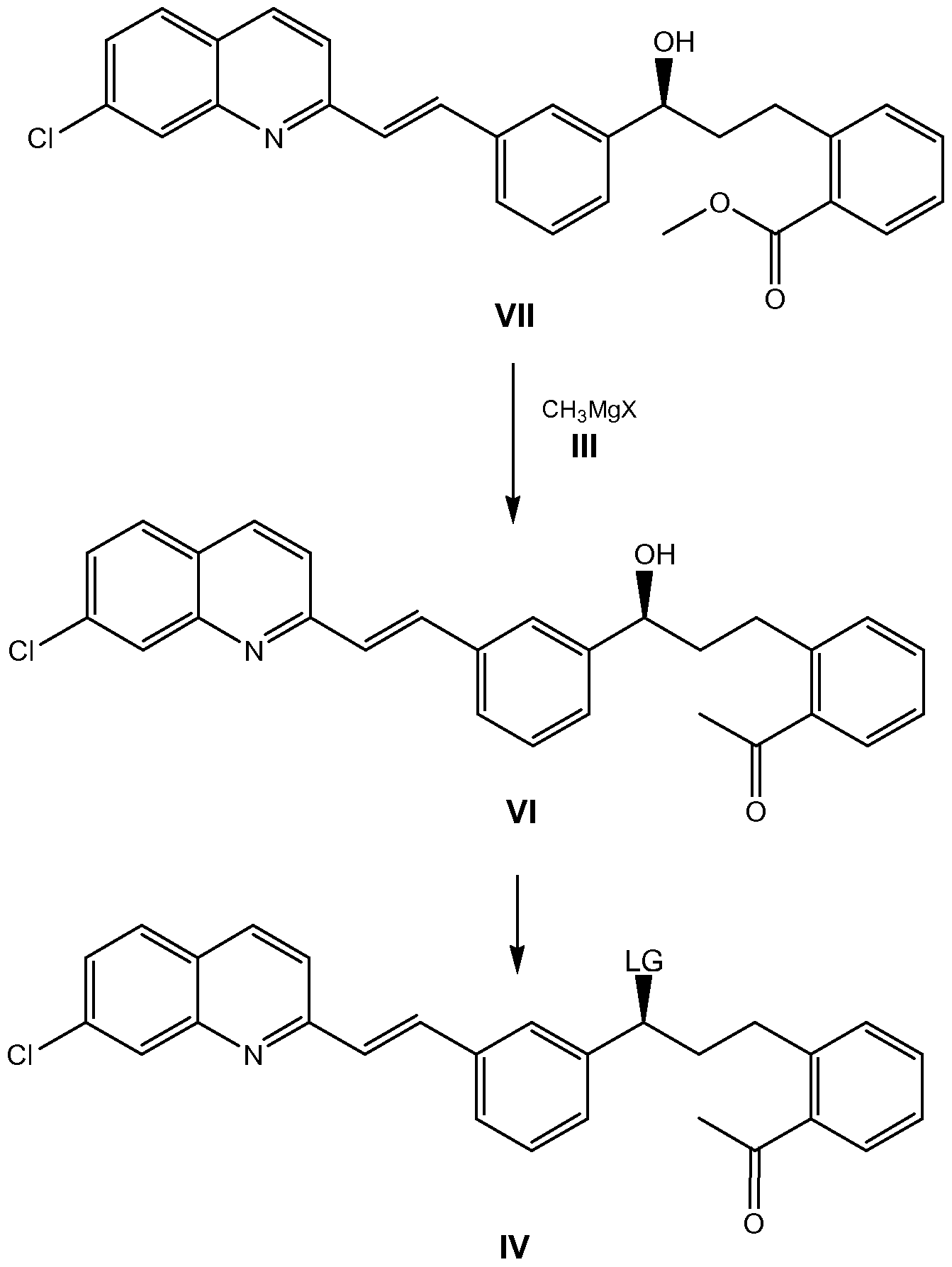
In the second step, the hydroxyl group of the compound of formula Vl is converted into a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl. In a preferred embodiment, LG represents methanesulfonyloxyl. This reaction is carried out in the presence of a sulfonyl halide, such as methanesulfonyl chloride or p- toluenesulfonyl chloride, in the presence of a base, such as pyridine or triethylamine, in a suitable solvent such as toluene, and at a suitable temperature, preferably upon cooling.
The compounds of formula IV, wherein LG is p-toluenesulfonyloxyl, are new
and form also part of the invention.
The present invention also relates to salts of the compounds of formula I. A preferred embodiment relates to montelukast sodium salt of formula Ia. Said salt may be prepared from a compound of formula I or a salt thereof in the presence of NaOH in a suitable polar solvent, such as toluene or isopropyl acetate, by using a suitable non-polar solvent, such as n-heptane, to precipitate the product, and at an appropriate temperature, preferably at 15- 2O0C.
Throughout the specification and claims the term "comprising" and its variations are not meant to exclude other technical characteristics, additives, components or steps. Other objectives, advantages and characteristics of the invention will become apparent to those skilled in the art in part from the description and partly from the practice of the invention. The following examples are provided for illustrative purposes and are not meant to be limiting of the present invention
BRIEF DESCRIPTION OF THE DRAWINGS
FIGURE 1 depicts the X-ray diffractogram of the crystalline form of (R)-(E)-I - (((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-acetylphenyl)propyl)thio) methyl )cyclopropane acetic acid dicyclohexylamine salt.
EXAMPLES
1H-NMR and 13C-NMR spectra were recorded at room temperature on a Mercury 400 MHz spectrometer.
HPLC spectra were recorded using a Waters Alliance 2695 system equipped with UV Waters 2487 detector.
X-ray diffractogram was obtained at room temperature using a Siemens D500 diffractometer equipped with a Cu anode (λ= 1.54056 A).
Example 1. (S)-(E)-I -(2-(1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)- hydroxypropyl)phenyl)ethanone monohydrate (Vl)
To a 1 M solution (390 ml) of lithium bis(trimethylsilyl)amide in tetrahydrofuran, 65 ml of a 3M solution of methylmagnesium chloride in tetrahydrofuran were slowly added under N2 atmosphere at a temperature of O0C. The solution obtained was maintained for 30 minutes at 0-50C under stirring. 25 g of methyl (S)-(E)-2-(3-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)- hydroxypropyl) benzoate monohydrate (3.8% of water, VII, EP 480717, example 146, step 2) were dissolved in 250 ml of toluene. The solution was heated at reflux and water was separated by azeotropic disuntilation at a temperature above 11 O0C. The solution was concentrated to a volume of approximately 130 ml and the initially prepared reducing solution was slowly added under N2 atmosphere at a temperature from O0C to -5 0C. After the addition was completed, the reaction mixture was maintained for 15 hours under N2 atmosphere at a temperature of 0-50C. The reaction mixture was added to an aqueous solution containing 400 ml of water, 75 g of sodium chloride, 70 ml of glacial acetic acid and 60 g of ammonium acetate, at a temperature below 250C. The aqueous phase was separated and the organic phase was washed with two 100-ml portions of 5% aqueous sodium bicarbonate solution. The solvent from the resulting organic phase was removed by distillation under reduced pressure and the obtained residue was dissolved in 90 ml of acetone at 2O0C. Then, 6 ml of water were added to afford a suspension. This suspension was maintained for 2 hours at a temperature of 15-2O0C and then filtered to afford a solid which was washed with methyl-te/t-butylether. The isolated solid was dried at a temperature of 45-5O0C to afford 18.1 g of the title compound (99% purity by HPLC analysis and 74.0% yield).
1H-NMR (400 MHz, CD3OD): 1.97-2.03 (2H, m); 2.53 (3H, s); 2.80-2.98 (2H, m); 4.66-4.69 (1 H, t); 7.24-7.45 (7H, m); 751 -7.53 (1 H, dt); 7.63 (1 H,s); 7.68- 7.72 (2H, m);7.76-7.78 (1 H, d); 7.78-7.80 (1 H, d); 7.91 -7.92 (1 H, d); 8.18-8.20 (1 H, d).
In this example, the enantiomeric excess of the starting compound VII was 96.5%, which led to compound Vl with an enantiomeric excess higher than 99%, due to the precipitation of the latter in the mixture of acetone-water. When compound Vl is obtained with an enantiomeric excess less than 99%, it may be purified by a recrystallization in 4 volumes of acetone and 0.5 volumes of water referred to the weight of compound Vl. Thus, the solution
obtained when heating the initial solution is cooled slowly to 15-20 0C in order to crystallize the product. The isolated solid by filtration shows an enantiomeric excess higher than 99.5%. This methodology avoids the dependency on the enantiomeric purity of the starting hydroxyester VII.
Example 2. (R)-(E)-I -(((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2- acetylphenyl)propyl)thio)methyl)cyclopropane acetic acid dicyclohexylamine salt (Ma)
a) (S)-(E)-I -(2-(1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-1 - (methanesulfonyl-oxy)propyl)phenyl)ethanone (IV.1 )
20 g of the compound obtained in Example 1 (4% water, Vl) were dissolved in 200 ml of toluene. The solution was heated at reflux and water was separated by azeotropic distillation. The solution was then concentrated to a volume of approximately 50 ml.
To the solution thus obtained, 8.4 g of triethylamine were added and the mixture was cooled to a temperature from -5 to -1 O0C. While maintaining this temperature, 7.1 g of mesyl chloride were slowly added and the mixture was stirred for 1 hour at the temperature from -5 to -10 0C under N2 atmosphere. Thereafter, 200 ml of previously cooled acetone were added with stirring for 1 further hour at the temperature from 0 to -5 0C. The solid present in the reaction mixture was separated by filtration and the resultant solution containing the mesyl derivative was kept at the temperature of -5 0C.
b) Mercaptomethylcyclopropylacetic acid disodium salt (V.1)
In a light-protected flask and under N2 atmosphere, 4.4 g of 60% sodium hydride and 88 ml of dimethylformamide were mixed. To this mixture, a solution containing 8.1 g of mercaptomethylcyclopropylacetic acid and 20 ml of dimethylformamide was added while maintaining the temperature at 0-50C. The reaction mixture was kept at this temperature for 1 hour.
c) Title compound
Previous solution containing the mesyl derivative from step a) was quickly added to the suspension of mercaptomethylcyclopropylacetic acid disodium salt from step b) and the temperature was kept at 5-100C. After the addition was completed, the temperature was adjusted to 20-250C and kept within this range for about 15 hours.
Then, the reaction mixture was cooled to 0-5 0C and 100 ml of an aqueous solution of sodium chloride were added. Acetone was removed by vacuum distillation and then 100 ml of isopropyl acetate were added. The aqueous phase was separated and the organic phase was washed with one 100-ml portion of aqueous sodium chloride solution. 100 ml of water were added and pH was adjusted to 4.5-5.5 with glacial acetic acid. The aqueous phase was separated and the solvent from the organic phase was removed by vacuum distillation, and the resulting residue was dissolved in 260 ml of acetone in a light-protected flask under N2 atmosphere and the temperature was kept at 20-25 0C. 7.9 g of dicyclohexylamine were obtained to afford within 5 minutes a white precipitate. The suspension was heated at reflux for 30 minutes. Then the suspension was cooled to 20-250C and kept within this temperature range for 2 hours. The resulting suspension was filtered off to give a white solid which was washed with acetone. The isolated solid was dried at the temperature of 45-5O0C to afford 24.6 g of the title compound (98.0% purity by HPLC analysis and 74.6% yield).
If necessary, the product thus obtained may be purified using a suspension in 15 volumes of acetone at reflux temperature. After cooling at a temperature of 20-250C, the solid was filtered off to afford a product with 99.0% purity by HPLC analysis and a purification yield higher than 98%. The titled compound was obtained with an enantiomeric excess higher than 99.8%.
1H-NMR (400 MHz, CD3OD): 0.28-0.51 (4H, m); 1.15-1.39 (12H, m); 1.65-1.69 (2H, dd); 1.81 -1.84 (4H, dd); 2.01 -2.03 (4H, d); 2.11 -2.15 (2H, m); 2.25-2.39 (2H, q); 2.46-2.50 (1 H, d); 2.51 (3H, s); 2.60-2.63 (1 H, d); 2.77-2.80 (1 H, m); 2.88-2.90 (1 H, m); 3.08-3.13 (2H, m); 3.90-3.94 (1 H, t); 7.23-7.30 (2H, m); 7.35-7.42 (4H, m); 7.48-7.51 (1 H, dd); 7.54-7.55 (1 H, dd); 7.64 (1 H, s); 7.70- 7.72 (1 H, dd); 7.76-7.80 (1 H, d); 7.85-7.89 (2H, dd); 7.97-7.98 (1 H, d); 8.26- 8.28 (1 H, d). 13C-NMR (400 MHz, CD3OD): 12.85, 13.31 , 18.48, 25.54, 26.20, 29.93, 30.75, 33.42, 39.82, 41 .01 , 44.53, 50.86, 54.38, 120.90, 126.93, 127.24, 127.30, 127.97, 128.17, 128.32, 128.87, 130.05, 130.51 , 130.55, 132.43, 132.74, 136.84, 137.40, 137.73, 138.17, 139.18, 142.74, 145.34, 149.38, 158.83, 180.30, 204.22.
Example 3. (R)-(E)-I -(((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2- (1-hydroxy-1-methylethyl)phenyl)propyl)thio)methyl)-cyclopropane acetic acid sodium salt (Montelukast sodium salt, Ia)
In a light-protected flask under N2 atmosphere, 10 g of the compound obtained in Example 2, 100 ml of toluene and 50 ml of water were placed. 1 g of glacial acetic acid was added and the mixture was kept under stirring until obtaining a solution. The aqueous phase was separated and the organic phase was washed with two 50-ml portions of water. Water was removed from the resulting solution by azeotropic distillation and the solution was concentrated until obtaining a residue which was dissolved in 50 ml of tetrahydrofuran.
In parallel, a mixture of 2.9 g of anhydrous cerium (III) chloride and 50 ml of tetrahydrofuran was prepared and kept at reflux temperature for 1 hour. The mixture was then cooled to 0-5 0C and 17.5 ml of a 3M solution of methylmagnesium chloride in tetrahydrofuran was added while maintaining previous temperature range and under N2 atmosphere. The resulting mixture was kept under stirring for 1 hour. Then, the previously prepared solution in tetrahydrofuran was slowly added at a temperature below 1 O0C. The reaction mixture was allowed to develop for 30 minutes at the temperature of 10-150C. 56 ml of an aqueous solution containing 6 ml of glacial acetic acid and 50 ml of 10% aqueous acetic acid-sodium chloride solution were slowly added while maintaining the temperature below 20° C. After adding 100 ml of toluene, the aqueous phase was separated and the organic phase was washed with 100 ml of water. Water was removed from the resulting solution by azeotropic distillation and the solution was concentrated by distillation to afford a residue which was dissolved in 50 ml of anhydrous tetrahydrofuran. Following the above described treatment, a mixture of 0.16 g of anhydrous cerium (III) chloride and 10 ml of a 3M solution of methylmagnesium chloride in tetrahydrofuran was prepared. The previously obtained tetrahydrofuran solution was slowly added and the temperature was kept below 10 0C. The reaction mixture was allowed to develop for 30 minutes at the temperature of 10-150C 56 ml of an aqueous solution containing 6 ml of glacial acetic acid and 50 ml of 10% aqueous acetic acid-sodium chloride solution were slowly added while maintaining the temperature below 20° C. After adding 100 ml of toluene, the aqueous phase was separated and the organic phase was washed with 100
ml of water. Thereafter, the resulting organic phase was treated with an aqueous solution containing 1.75 g of 30% sodium hydroxide and 100 ml of a 20% aqueous sodium chloride solution. The aqueous phase was separated and the organic phase was repeatedly treated with two portions (100 and 50 ml) of water. To the combined aqueous phases, 100 ml of toluene and 30 g of sodium chloride were added, and the mixture was kept under stirring until complete dissolution of sodium chloride. The aqueous phase was separated and tetrahydrofuran and water were removed by azeotropic distillation. The solution obtained was treated with 0.5 g active charcoal. After filtration, the resulting solution was concentrated and the residue obtained was added to 50 ml of n-heptane at the temperature of 20° C. The resulting mixture was concentrated by distillation and 35 ml of n-heptane were added. The resulting mixture was kept under stirring for 2 hours at the temperature of 15-20° C. Finally, the precipitated solid was filtered off, washed with n-heptane and dried at the temperature of 60-80° C, to yield 6.4 g of amorphous solid form of montelukast sodium salt (>99.0% purity by HPLC analysis, 81.0% yield). The title compound was obtained with an enantiomeric excess higher than 99.8%.
Example 4. Comparison among crystallization procedures of dicyclohexylamine salt
Comparison of crystallization procedures among montelukast dicyclohexilamine salt and methyl ester intermediate (as described in prior art) and the compound of the invention Ma is shown in Table 1.
Table 1 illustrates the starting material from which the dicyclohexilamine salt is formed, and crystallization conditions: solvent, time and if seeding is necessary. In all cases, temperature is 20-25°C.
Table 1

It has been observed that unlike prior art compounds which even require the addition of large quantities of non-polar solvents to perform crystallization, the dicyclohexylamine salt of the compound of the invention Ma was obtained much more readily under the same temperature and without the need for addition of non-polar solvents.
Example 5. Solubility of dicyclohexylamine salts
Comparative solubilities of montelukast dicyclohexilamine salts, the methyl ester intermediate and the compound of the invention Ma are shown in Table 2. In particular, the amounts of solvents needed to dissolve each compound at reflux are provided.
The amounts of solvents needed to dissolve the salts are approximate. These amounts might slightly change depending on the quantity and purity of
product or whether a crystalline or amorphous form is used. In the present example, the solubility of the same quantity of each compound was tested, and their purities were substantially similar. The results obtained show that the difference in solubility of the compound of the invention Ma in contrast to that of prior art compounds is significant.
Table 2

Claims
1. A compound of formula Ma,

2. Crystalline form of the (R)-(E)-I -(((1 -(3-(2-(7-chloro-2-quinolinyl)ethenyl) phenyl)-3-(2-acetylphenyl)propyl)thio)methyl)cyclopropane acetic acid dicyclohexylamine salt according to claim 1 , characterized in that its X-ray diffractogram is substantially the same as that shown in FIGURE 1.
3. A process for the preparation of a compound of formula Ma as defined in any one of claims 1 and 2, comprising the reaction of a compound of formula

with dicyclohexylamine in the presence of a polar solvent.
4. The process of preparation according to claim 3, wherein the polar solvent is selected from the group consisting of a ketone of formula RCORi, an ester of formula RCO2Ri wherein R and Ri may be the same or different and are (C-|-C4)alkyl, and an aromatic hydrocarbon which is a mono or disubstituted benzene, wherein the substituent is selected from halogen or methyl.
5. The process of preparation according to claim 4, wherein the polar solvent is acetone.
6. The process of preparation according to any one of claims 3-5, wherein a compound of formula IV

wherein LG represents a leaving group selected from the group consisting of methanesulfonyloxyl and p-toluenesulfonyloxyl; is previously reacted with a compound of formula V 
V
wherein R represents an alkaline metal, in a suitable solvent, followed by aqueous treatment in an acid medium, to afford the compounds of formula II.
7. The process of preparation according to claim 6 wherein LG represents methanesulfonyloxyl .
8. The process of preparation according to any one of claims 6 and 7 wherein R represents Na.
9. A process for the preparation of a compound of formula I,

I
or a pharmaceutically acceptable salt thereof, or a pharmaceutically acceptable solvate thereof, including a hydrate; comprising the conversion of a compound of formula Ma


followed by reaction with a compound of formula III CH3MgX, wherein X is halogen, in a suitable solvent, optionally in the presence of a Lewis acid; and subsequent aqueous treatment in an acid medium to afford the compound of formula I; and, optionally, the compound of formula I is converted into a pharmaceutically acceptable salt thereof by treatment with the corresponding base, or a salt of the compound of formula I is converted into another salt of the compound of formula I by ion exchange, or the compound of formula I is converted into a pharmaceutically acceptable solvate thereof, including a hydrate, by crystallization/precipitation in a suitable solvent.
10. A process of preparation according to claim 9, wherein X is Cl in the presence of a Lewis acid.
11. A process of preparation according to claim 10, wherein the Lewis acid is CeCI3.
12. A process of preparation according to any one of claims 9-11 , wherein a compound of formula Il

is previously reacted with dicyclohexylamine in the presence of a polar solvent to afford a compound of formula Ma.
13. A process of preparation according to claim 12, wherein a compound of formula IV


wherein R represents an alkaline metal, in a suitable solvent, followed by aqueous treatment in an acid medium, to afford the compound of formula
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/670,530 US8163924B2 (en) | 2007-07-31 | 2008-07-30 | Process for preparing a leukotriene antagonist and an intermediate thereof |
| EP08786600A EP2185517B1 (en) | 2007-07-31 | 2008-07-30 | Process for preparing a leukotriene antagonist and an intermediate thereof |
| ES08786600T ES2395439T3 (en) | 2007-07-31 | 2008-07-30 | Procedure for preparing a leukotriene antagonist and an intermediate thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ESP200702220 | 2007-07-31 | ||
| ES200702220A ES2320077B1 (en) | 2007-07-31 | 2007-07-31 | PROCESS OF PREPARATION OF AN ANTIGONIST OF LEUCOTRIENS AND AN INTERMEDIATE OF THE SAME. |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009016191A1 true WO2009016191A1 (en) | 2009-02-05 |
Family
ID=39963083
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/059965 Ceased WO2009016191A1 (en) | 2007-07-31 | 2008-07-30 | Process for preparing a leukotriene antagonist and an intermediate thereof |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8163924B2 (en) |
| EP (1) | EP2185517B1 (en) |
| ES (2) | ES2320077B1 (en) |
| WO (1) | WO2009016191A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011121091A1 (en) * | 2010-03-31 | 2011-10-06 | Krka, D.D., Novo Mesto | Efficient synthesis for the preparation of montelukast and novel crystalline form of intermediates therein |
| WO2012020271A1 (en) | 2010-08-11 | 2012-02-16 | Richter Gedeon Nyrt. | Process for the preparation of montelukast sodium |
| CN111170939A (en) * | 2019-12-20 | 2020-05-19 | 牡丹江恒远药业股份有限公司 | Preparation method of high-purity montelukast sodium and intermediate thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995018107A1 (en) * | 1993-12-28 | 1995-07-06 | Merck & Co., Inc. | Process for the preparation of leukotriene antagonists |
| WO2006008751A2 (en) * | 2004-07-19 | 2006-01-26 | Matrix Laboratories Ltd | Process for the preparation of montelukast and its salts |
| WO2007004237A2 (en) * | 2005-07-05 | 2007-01-11 | Matrix Laboratories Ltd | A process for the preparation of montelukast |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE165088T1 (en) | 1990-10-12 | 1998-05-15 | Merck Frosst Canada Inc | UNSATURATED HYDROXYALKYLQUINOLINIC ACIDS AS LEUCOTRIEN ANTAGONISTS |
| IE920499A1 (en) * | 1991-02-21 | 1992-08-26 | Merck Frosst Canada Inc | Quinoline-containing ketoacids as leukotriene antagonists |
| US20050107612A1 (en) * | 2002-12-30 | 2005-05-19 | Dr. Reddy's Laboratories Limited | Process for preparation of montelukast and its salts |
| AR057908A1 (en) * | 2005-11-18 | 2007-12-26 | Synthon Bv | PROCESS TO PREPARE MONTELUKAST AND INTERMEDIARIES OF THE SAME |
| KR20090080038A (en) * | 2006-09-15 | 2009-07-23 | 씨아이피엘에이 엘티디. | Method for preparing montelukast and intermediates thereof |
-
2007
- 2007-07-31 ES ES200702220A patent/ES2320077B1/en not_active Expired - Fee Related
-
2008
- 2008-07-30 EP EP08786600A patent/EP2185517B1/en not_active Not-in-force
- 2008-07-30 WO PCT/EP2008/059965 patent/WO2009016191A1/en not_active Ceased
- 2008-07-30 US US12/670,530 patent/US8163924B2/en not_active Expired - Fee Related
- 2008-07-30 ES ES08786600T patent/ES2395439T3/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995018107A1 (en) * | 1993-12-28 | 1995-07-06 | Merck & Co., Inc. | Process for the preparation of leukotriene antagonists |
| WO2006008751A2 (en) * | 2004-07-19 | 2006-01-26 | Matrix Laboratories Ltd | Process for the preparation of montelukast and its salts |
| WO2007004237A2 (en) * | 2005-07-05 | 2007-01-11 | Matrix Laboratories Ltd | A process for the preparation of montelukast |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011121091A1 (en) * | 2010-03-31 | 2011-10-06 | Krka, D.D., Novo Mesto | Efficient synthesis for the preparation of montelukast and novel crystalline form of intermediates therein |
| WO2012020271A1 (en) | 2010-08-11 | 2012-02-16 | Richter Gedeon Nyrt. | Process for the preparation of montelukast sodium |
| CN111170939A (en) * | 2019-12-20 | 2020-05-19 | 牡丹江恒远药业股份有限公司 | Preparation method of high-purity montelukast sodium and intermediate thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100204476A1 (en) | 2010-08-12 |
| EP2185517A1 (en) | 2010-05-19 |
| ES2395439T3 (en) | 2013-02-12 |
| ES2320077A1 (en) | 2009-05-18 |
| US8163924B2 (en) | 2012-04-24 |
| ES2320077B1 (en) | 2010-02-26 |
| EP2185517B1 (en) | 2012-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8188285B2 (en) | Purification process of Montelukast and its amine salts | |
| WO2008058118A2 (en) | Preparation of montelukast and its salts | |
| CZ2007455A3 (en) | Method of isolation and purification of montelukast | |
| WO2006058545A1 (en) | New process for the preparation of a leukotriene antagonist | |
| JP2003506425A (en) | Method for producing nitroxyalkyl ester of naproxen | |
| US7417149B2 (en) | Process for the preparation of Montelukast | |
| EP2066638B1 (en) | Process for the purification of montelukast | |
| EP2185517B1 (en) | Process for preparing a leukotriene antagonist and an intermediate thereof | |
| JP2009515922A (en) | Process for producing montelukast and intermediates therefor | |
| PL205444B1 (en) | The manner of production of salt of 1-(((1(R)-(3-(2-(7--chloro-2- chinolinylo)-ethenylo)phenylo)-3-(2-(1-hydroxy-1- methyloethylo)phenylo)propylo)sulphanylo)methylo)-cyclopropaiacetic acid | |
| RU2402532C2 (en) | Method of producing montelukast and compounds for realising said method | |
| WO2009098271A1 (en) | Process for the purification of montelukast by the preparation of acid addition salts and tert-amylamine salt | |
| JP6059157B2 (en) | Montelukast Intermediate Camphorsulfonate | |
| CN101356157B (en) | Process for making montelukast and intermediates therefor | |
| JP3523874B2 (en) | Quinolone disulfide as an intermediate | |
| WO2023126376A2 (en) | Process for the preparation of cysteamine bitartrate and product so obtained | |
| JP2010521475A (en) | Processes and intermediates for the preparation of arzoxifene | |
| EP2287154A1 (en) | Efficient synthesis for the preparation of montelukast |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08786600 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12670530 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008786600 Country of ref document: EP |