WO2009055749A1 - 2-aminooxazole derivatives as trpvl antagonists useful for treating pain - Google Patents
2-aminooxazole derivatives as trpvl antagonists useful for treating pain Download PDFInfo
- Publication number
- WO2009055749A1 WO2009055749A1 PCT/US2008/081229 US2008081229W WO2009055749A1 WO 2009055749 A1 WO2009055749 A1 WO 2009055749A1 US 2008081229 W US2008081229 W US 2008081229W WO 2009055749 A1 WO2009055749 A1 WO 2009055749A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tetrahydronaphthalen
- amino
- oxazol
- methyl
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCOC(c1c(*)[o]cn1)=O Chemical compound CCOC(c1c(*)[o]cn1)=O 0.000 description 7
- NTNZTEQNFHNYBC-UHFFFAOYSA-N CCOC(CN)=O Chemical compound CCOC(CN)=O NTNZTEQNFHNYBC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/48—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention generally relates to oxazole containing compounds, compositions comprising such compounds, and methods of treating conditions and disorders using such compounds and compositions.
- Nociceptors are primary sensory afferent (C and A ⁇ fibers) neurons that are activated by a wide variety of noxious stimuli including chemical, mechanical, thermal, and proton (pH ⁇ 6) modalities.
- the lipophilic vanilloid, capsaicin activates primary sensory fibers via a specific cell surface capsaicin receptor, cloned as the transient receptor potential vanilloid- 1 (TRPVl).
- TRPVl is also known as vanilloid receptor- 1 (VRl).
- the intradermal administration of capsaicin is characterized by an initial burning or hot sensation followed by a prolonged period of analgesia.
- TRPVl receptor activation is mediated by a capsaicin-induced desensitization of the primary sensory afferent terminal.
- capsaicin analogs as analgesic agents.
- capsazepine a capsaicin receptor antagonist can reduce inflammation-induced hyperalgesia in animal models.
- TRPVl receptors are also localized on sensory afferents, which innervate the bladder. Capsaicin or resiniferatoxin has been shown to ameliorate incontinence symptoms upon injection into the bladder.
- the TRPVl receptor has been called a "polymodal detector" of noxious stimuli since it can be activated in several ways.
- the receptor channel is activated by capsaicin and other vanilloids, and thus is classified as a ligand-gated ion channel.
- TRPVl receptor activation by capsaicin can be blocked by the competitive TRPVl receptor antagonist, capsazepine.
- Protons acidic pH
- the affinity of capsaicin for the receptor is increased, whereas at pH ⁇ 6, direct activation of the channel occurs.
- membrane temperature reaches 43 0 C, the channel is opened. Thus heat can directly gate the channel in the absence of ligand.
- the capsaicin analog, capsazepine which is a competitive antagonist of capsaicin, blocks activation of the channel in response to capsaicin, acid, or heat.
- the channel is a nonspecific cation conductor. Both extracellular sodium and calcium enter through the channel pore, resulting in cell membrane depolarization. This depolarization increases neuronal excitability, leading to action potential firing and transmission of a noxious nerve impulse to the spinal cord.
- depolarization of the peripheral terminal can lead to release of inflammatory peptides such as, but not limited to, substance P and CGRP, leading to enhanced peripheral sensitization of tissue.
- TRPVl knockout mice lacking the TRPVl receptor. Electrophysiological studies of sensory neurons (dorsal root ganglia) from these animals revealed a marked absence of responses evoked by noxious stimuli including capsaicin, heat, and reduced pH. These animals did not display any overt signs of behavioral impairment and showed no differences in responses to acute non-noxious thermal and mechanical stimulation relative to wild-type mice. The TRPVl (-/-) mice also did not show reduced sensitivity to nerve injury-induced mechanical or thermal nociception. However, the TRPVl knockout mice were insensitive to the noxious effects of intradermal capsaicin, exposure to intense heat (50-55 0 C), and failed to develop thermal hyperalgesia following the intradermal administration of carrageenan.
- TRPVl receptors have been found in both the peripheral nervous system and the central nervous system (CNS). In the brain, TRPVl receptors have been identified in various regions known for their role in pain transmission or modulation (Merzey et al., Proc. Natl. Acad. Sci. USA 97:3655-3660 (2000); Szabo et al., MoI. Brain Res. 98:51-57 (2002); Roberts et al., Brain Res. 995:176-183 (2004)). Using TRPVl antagonists with similar in vitro potency but different CNS penetration, studies by Cui, M.; Honore, P. et al. J.
- TRPVl antagonists are discussed in U.S. Publication No. 2006/0281799 with unknown CNS penetration. Accordingly, the need exists to develop TRPVl antagonists that exhibit good CNS penetration. Such antagonists may provide full efficacy across a wide array of pain states.
- the present invention generally provides oxazole containing compounds and pharmaceutical compositions and methods for the treatment of disorders using these compounds and pharmaceutical compositions.
- One aspect of the invention is directed towards compounds of formula (I), or pharmaceutically acceptable salts, prodrugs, salts of prodrugs, or combinations thereof,
- W is CH 2 or O
- R 1 is phenyl, a monocyclic heteroaryl, or a monocyclic cycloalkenyl, a monocyclic cycloalkyl, each of which is independently unsubstituted or substituted with 1, 2, 3, 4, or 5 substituents as represented by R 3 , wherein each R 3 is independently alkyl, alkenyl, alkynyl, -NO 2 , -CN, halogen, -0R a , -OC(O)R a , -SR a , -SF 5 , -S(O)R b , -S(O) 2 R b , -S(O) 2 N(R a )(R c ), -N(R a )(R c ), -N(R c )C(0)R a , -N(R c )S(O) 2 R b , -N(R c )C(0)N(R a )(
- R a at each occurrence, is independently hydrogen, alkyl, or haloalkyl
- R b at each occurrence, is independently alkyl or haloalkyl
- R c at each occurrence, is independently hydrogen, alkyl, or haloalkyl
- R e and R f are each independently hydrogen, alkyl, or haloalkyl; m is 1, 2, or 3;
- R is hydrogen or alkyl
- R 4 is methyl, ethyl, Ci-C 2 haloalkyl, or -CN.
- Another aspect of the inventions relates to a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, and one or more pharmaceutically acceptable carriers.
- Another aspect of the inventions relates to a pharmaceutical composition comprising a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof, one or more pharmaceutically acceptable carriers, and one or more analgesic agents, wherein the analgesic agent is a nonsteroidal anti-inflammatory drug (NSAID), or acetaminophen, or a combination thereof.
- NSAID nonsteroidal anti-inflammatory drug
- Yet other aspect provides methods for treating diseases or disorders as defined herein.
- Said methods comprise the step of administering therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described herein, to a subject in need thereof.
- NSAID nonsteroidal anti-inflammatory drug
- R 1 , R 2 , R 4 , and W are defined above in the Summary of the Invention and below in the Detailed Description.
- compounds of the invention are TRPVl antagonists that exhibit CNS penetration.
- compositions comprising such compounds and methods for treating conditions and disorders using such compounds and compositions are also disclosed.
- the present invention provides at least one variable that occurs more than one time in any substituent or in the compound of the invention or any other formulae herein.
- Definition of a variable on each occurrence is independent of its definition at another occurrence. Further, combinations of substituents are permissible only if such combinations result in stable compounds.
- Stable compounds are compounds, which can be isolated from a reaction mixture. a. Definition
- alkenyl as used herein, means a straight or branched chain hydrocarbon containing from 2 to 10 carbons and containing at least one carbon-carbon double bond.
- Representative examples of alkenyl include, but are not limited to, ethenyl, 2-propenyl, 2-methyl- 2-propenyl, 3-butenyl, 4-pentenyl, 5-hexenyl, 2-heptenyl, 2-methyl-l-heptenyl, and 3-decenyl.
- C 2 -C 3 alkenyl means a straight or branched chain hydrocarbon containing from 2 or 3 carbons and containing at least one carbon-carbon double bond.
- alkyl as used herein, means a straight or branched chain saturated hydrocarbon containing from 1 to 10 carbon atoms, for example, from 1 to 6 carbon atoms.
- Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, 3- methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, 3,3-dimethylbutyl, n-heptyl, n-octyl, n- nonyl, and n-decyl.
- Ci-C 3 alkyl as used herein, means a straight or branched chain saturated hydrocarbon containing 1, 2, or 3 carbon atoms.
- C1-C4 alkyl as used herein, means a straight or branched chain saturated hydrocarbon containing 1, 2, or 4 carbon atoms.
- Ci-C 2 alkyl as used herein, means methyl and ethyl.
- alkynyl as used herein, means a straight or branched chain hydrocarbon group containing from 2 to 10 carbon atoms and containing at least one carbon-carbon triple bond.
- Representative examples of alkynyl include, but are not limited, to acetylenyl, 1-propynyl, 2- propynyl, 3-butynyl, 2-pentynyl, and 1-butynyl.
- halo or halogen as used herein, means -Cl, -Br, -I or -F.
- haloalkyl means at least one halogen (for example, one to six halogens), as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of haloalkyl include, but are not limited to, chloromethyl, difluoromethyl, 2-fluoroethyl, trifluoromethyl, pentafluoroethyl, and 2-chloro-3- fluoropentyl.
- C 1 -C 2 haloalkyl as used herein, means at least one halogen (for example, one to six halogens), as defined herein, appended to the parent molecular moiety through a Ci-C 2 alkyl group, as defined herein.
- monocyclic cycloalkenyl as used herein, means an optionally substituted monocyclic ring system containing three-, four-, five-, six-, seven- or eight carbon atoms and zero heteroatoms.
- the three or four-membered ring systems have one double bond, the five-or six- membered ring systems have one or two double bonds, and the seven- or eight-membered ring systems have one, two or three double bonds.
- monocyclic cycloalkenyls include, but are not limited to, cyclohex-1-en-l-yl, 2-cyclohexen-l-yl, 3- cyclohexen- 1 -yl, 2,4-cyclohexadien-l-yl and 3-cyclopenten-l-yl.
- monocyclic cycloalkyl as used herein, means an optionally substituted monocyclic carbocyclic ring system containing 3, 4, 5, 6, 7, or 8 carbon atoms and zero heteroatoms as ring atoms, and zero double bonds.
- monocyclic cycloalkyls include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- monocyclic heteroaryl as used herein, means an optionally substituted 5-or 6-membered ring containing at least one heteroatom independently selected from the group consisting of O, N, and S.
- the 5-membered ring contains two double bonds and one, two, three, or four heteroatoms.
- the 6-membered ring contains three double bonds and one, two, three or four heteroatoms.
- monocyclic heteroaryls include, but are not limited to, furanyl (including furan-2-yl), imidazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, oxazolyl, pyridinyl (including pyridine-3-yl, pyridine-2-yl), pyridazinyl, pyrimidinyl, pyrazinyl, pyrazolyl, pyrrolyl, tetrazolyl, thiadiazolyl, thiazolyl, thienyl (including thien-2-yl and thien-3-yl), triazolyl, and triazinyl.
- the monocyclic heteroaryl groups are connected to the parent molecular moiety through any substitutable carbon atom or any substitutable nitrogen atom contained within the groups.
- the nitrogen and sulfur heteroatoms of the rings may optionally be oxidized, and are contemplated within the scope of the invention.
- variable groups in compounds of formula (I) are as follows. Such values may be used where appropriate with any of the other values, definitions, claims or embodiments defined hereinbefore or hereinafter.
- R 1 is phenyl, a monocyclic heteroaryl, or a monocyclic cycloalkenyl, a monocyclic cycloalkyl, each of which is optionally substituted as described in the Summary.
- R 1 is optionally substituted phenyl.
- R 1 is substituted phenyl.
- R 1 is phenyl, substituted with 1, 2, or 3 substituents as represented by R 3 .
- R 1 is a substituted phenyl, it is preferred that at least one substituent is located at the fourth carbon atom of the phenyl ring relative to the point of attachment of the phenyl ring to the parent moiety.
- examples of compounds of the invention include, but are not limited to, those represented by formula (Ia)
- R 1 is optionally substituted monocyclic heteroaryl.
- R 1 is monocyclic heteroaryl, optionally substituted with 1, 2, or 3 substituents as represented by R 3 .
- Examples of the monocyclic heteroaryl include, but are not limited to, furanyl (including furan-2-yl), thienyl (including thien-2-yl and thien-3-yl) and pyridinyl (including pyridin-2-yl, pyridin-3-yl), each of which is optionally substituted.
- R 1 is optionally substituted monocyclic cycloalkenyl.
- An example of the cycloalkenyl includes, but is not limited to, optionally substituted cyclohexenyl (e.g. cyclohex-1-en-yl).
- R 3 examples include, but are not limited to, alkyl, alkenyl, -CN, halogen, -OR a , -SF 5 , -S(O) 2 R b , haloalkyl, -(CR e R f ) m -CN, and an optionally substituted cycloalkyl; wherein m, R a , R b , R e , and R f are as described in the Summary.
- R a and R b are each independently alkyl (e.g. methyl) or haloalkyl (e.g. trifluoromethyl).
- R e and R f are each independently hydrogen or alkyl (e.g. methyl).
- substituents of R 1 include, but are not limited to, Ci-C 4 alkyl (e.g. methyl, ethyl, tert-butyl), C 2 -C 3 alkenyl (e.g. 2-methyl-2-propenyl), -CN, halogen (e.g. F, Cl, Br), -OCH 3 , -OCF 3 , -SF 5 , -S(O) 2 (CF 3 ), trifluoromethyl, difluoromethyl, -C(CH 3 ) 2 -CN, and an optionally substituted cyclopropyl
- Examples of compounds of formula (I) or (Ia) include those wherein R 2 is hydrogen or alkyl.
- R is hydrogen or Ci-C 3 alkyl such as, but not limited to, methyl.
- R 2 is hydrogen.
- R 4 has values as described generally in the Summary. In certain embodiments, R 4 is methyl or ethyl. In other embodiments, R 4 is Ci-C 2 haloalkyl such as, but not limited to, difluoromethyl. In yet other embodiments, R 4 is -CN. Examples of compounds of formula (I) and (Ia) include, but not limited to, those wherein R 4 is methyl, ethyl, -CN, or difluoromethyl.
- Compounds of the present invention contain one or more asymmetrically substituted carbon atoms in the cycloalkene ring of formula (I) and (Ia).
- Compounds of formula (I) and (Ia) can have stereoisomers including, but not limited to, those shown below: wherein R 1 , R 2 , R 3 , R 4 , and W are as disclosed in the Summary and the Detailed Description sections. It is understood that embodiments for R 1 , R 2 , R 3 , R 4 , and W, and combinations of embodiments, including particular, and more particular embodiments as described for formula (I), are also contemplated for compounds of formula (I-i), (I-ii), (Ia-i), and (Ia-ii).
- the present invention contemplates various individual stereoisomers (including enantiomers and diastereomers) and mixtures thereof.
- Individual stereoisomers of compounds of the present invention may be prepared synthetically from commercially available starting materials that contain asymmetric or chiral centers or by preparation of racemic mixtures followed by resolution of the individual stereoisomer using methods that are known to those of ordinary skill in the art. Examples of resolution are, for example, (i) attachment of a mixture of enantiomers to a chiral auxiliary, separation of the resulting mixture of diastereomers by recrystallization or chromatography, followed by liberation of the optically pure product; or (ii) separation of the mixture of enantiomers or diastereomers on chiral chromatographic columns.
- the invention also contemplates the various geometric isomers and mixtures thereof resulting from the disposition of substituents around a carbon-carbon double bond, a carbon- nitrogen double bond, a cycloalkyl group, or a heterocycle group.
- substituents around a carbon- carbon double bond or a carbon-nitrogen double bond are designated as being of Z or E configuration and substituents around a cycloalkyl or heterocycle are designated as being of cis or trans configuration.
- one aspect of the invention relates to a group of compounds of formula (I), (Ia), (I-i), (I-ii), (Ia-i), or (Ia-ii) wherein W is O or CH 2 .
- Another aspect of the invention is directed to a group of compounds of formula (I), (Ia), (I-i), (I-ii), (Ia-i), or (Ia-ii) wherein W is CH 2 .
- a further aspect of the invention relates to a group of compounds of formula (I), (Ia), (I- i), (I-ii), (Ia-i), or (Ia-ii) wherein W is O.
- R 1 , R 2 , R 3 , and R 4 are as disclosed in the Summary and in specific embodiments herein.
- examples of a subgroup include those having formula (I), (Ia), (I-i), (I- ⁇ ), (Ia-i), or (Ia-ii) wherein R 1 is optionally substituted phenyl and R 2 is hydrogen, and the optional substituents of the phenyl group have meanings as disclosed in the Summary and in specific embodiments herein.
- the phenyl group is substituted, preferably with 1, 2, or 3 substituents as described herein above.
- Examples of another subgroup include those having formula (I), (Ia), (I-i), (I-ii), (Ia-i), or (Ia-ii) wherein R 1 is an optionally substituted monocyclic heteroaryl and R 2 is hydrogen, and the optional substituents of the monocyclic heteroaryl have meanings as disclosed in the Summary and in specific embodiments herein.
- Furanyl including furan-2-yl
- thienyl including thien-2-yl and thien-3-yl
- pyridinyl including pyridin-2-yl, pyridin-3-yl
- Examples of yet another subgroup include those having formula (I), (Ia), (I-i), (I-ii), (Ia- i), or (Ia-ii) wherein R 1 is optionally substituted monocyclic cycloalkenyl and R 2 is hydrogen.
- R 1 is optionally substituted monocyclic cycloalkenyl and R 2 is hydrogen.
- An example of the cycloalkenyl includes, but is not limited to, optionally substituted cyclohexenyl (e.g. cyclohex-1-en-yl).
- R 3 and R 4 have values as disclosed in the Summary and the Detailed Description.
- R 3 is alkyl, alkenyl, -CN, halogen, -OR a , -SF 5 , -S(O) 2 R b , haloalkyl, -(CR e R f ) m -CN, and an optionally substituted cycloalkyl; wherein m, R a , R b , R e , and R f are as described in the Summary.
- R a and R are each independently alkyl (e.g. methyl) or haloalkyl (e.g. trifluoromethyl).
- R e and R f are each independently hydrogen or alkyl (e.g. methyl).
- Particular examples of the substituents of R 1 include, but are not limited to, Ci-C 4 alkyl (e.g. methyl, ethyl, tert-butyl), C 2 -C 3 alkenyl (e.g. 2- methyl-2-propenyl), -CN, halogen (e.g.
- R 4 is methyl, ethyl, -CN, or difluoromethyl.
- R 4 is methyl or ethyl.
- R is Ci-C 2 haloalkyl such as, but not limited to, difluoromethyl.
- R 4 is -CN.
- Exemplary compounds of the present invention include, but are not limited to:
- D-MEM Dulbecco's modified Eagle medium
- D-PBS Dulbecco's phosphate-buffered saline
- L-glutamine L-glutamine
- hygromycin B Lipofectamine®
- G418 sulfate was obtained from Calbiochem-Novabiochem Corp. (San Diego, Calif.).
- Fluo-4 AM N-[4-[6-[(acetyloxy)methoxy]-2,7-difluoro-3- oxo-3H-xanthen-9-yl]-2-[2-[2-[bis[2 -[(acetyl oxy)methoxy]-2-oxyethyl]amino]-5-methy lphenoxy] ethoxy] phenyl] -N- [2- [(acetyloxy)methoxy]-2-oxyethyl] -glycine, (acetyl oxy)methyl ester) was purchased from Molecular Probes (Eugene, Oreg.).
- the cDNAs for the human TRPVl receptor were isolated by reverse transcriptase- polymerase chain reaction (RT-PCR) from human small intestine poly A+RNA supplied by Clontech (Palo Alto, Calif.) using primers designed surrounding the initiation and termination codons identical to the published sequences (Hayes et al. Pain Vol. 88 pages 205-215, 2000).
- the resulting cDNA PCR products were subcloned into pCIneo mammalian expression vector (Promega) and fully sequenced using fluorescent dye-terminator reagents (Prism, Perkin-Elmer Applied Biosystems Division) and a Perkin-Elmer Applied Biosystems Model 373 DNA sequencer or Model 310 genetic analyzer.
- Expression plasmids encoding the hTRPVl cDNA were transfected individually into 1321N1 human astrocytoma cells using Lipofectamine®. Forty-eight hours after transfection, the neomycin-resistant cells were selected with growth medium containing 800 ⁇ g/mL Geneticin (Gibco BRL). Surviving individual colonies were isolated and screened for TRPVl receptor activity. Cells expressing recombinant homomeric TRPVl receptors were maintained at 37° C. in D-MEM containing 4 mM L-glutamine, 300 ⁇ g/mL G418 (Cal-biochem) and 10% fetal bovine serum under a humidified 5% CO 2 atmosphere.
- the functional activity of compounds at the TRPVl receptor was determined with a Ca 2+ influx assay and measurement of intracellular Ca + levels ([Ca J 1 ). All compounds were tested over an 11 -point half-log concentration range.
- Compound solutions were prepared in D-PBS (4 ⁇ final concentration), and diluted serially across 96-well v-bottom tissue culture plates using a Biomek 2000 robotic automation workstation (B eckman- Coulter, Inc., Fullerton, Calif.).
- a 0.2 ⁇ M solution of the TRPVl agonist capsaicin was also prepared in D-PBS.
- the fluorescent Ca + chelating dye Fluo-4 AM was used as an indicator of the relative levels of [Ca J 1 in a 96-well format using a Fluorescence Imaging Plate Reader (FLIPR)(Molecular Devices, Sunnyvale, Calif.). Cells were grown to confluency in 96-well black-walled tissue culture plates. Then, prior to the assay, the cells were loaded with 100 ⁇ L per well of Fluo-4 AM (2 ⁇ M, in D-PBS) for 1-2 hours at 23° C. Washing of the cells was performed to remove extracellular Fluo-4 AM (2 ⁇ l mL D-PBS per well), and afterward, the cells were placed in the reading chamber of the FLIPR instrument.
- FLIPR Fluorescence Imaging Plate Reader
- TRPVl antagonists with IC 50 values from about 10 ⁇ M to about 10 nM, for example, from about 1 ⁇ M to about 10 nM, and preferably, from about 200 nM to about 10 nM.
- the compounds were prepared as a solution in a 10% DMSO/poly(ethylene glycol)-400 (v/v). Blood and brain samples were obtained from each animal two hours after dosing.
- Rat brain tissue was homogenized with two parts water (by weight).
- Test compound was selectively extracted from plasma and brain homogenate using liquid-liquid extraction with tert- butylmethylether.
- the compounds of interest were separated from co-extracted contaminants on a 50 x 3 mm, Luna CN column (Phenomenex), with an acetonitrile:0.1% aqueous trifluoroacetic acid mobile phase at a flow rate of 0.35 ml/min.
- Plasma and tissue concentrations were determined by HPLC-MS/MS on an API2000 with Turbo Ion Spray interface, with MRM detection in the positive ionization mode. To calibrate compound concentration, a separate portion of each tissue sample was spiked with a known quantity of test compound and analyzed simultaneously with the samples. The results are shown in Table 1.
- Compound A is 8-( ⁇ 5-[4-(trifluoromethyl)phenyl]-l,3-oxazol-2-yl ⁇ amino)- l,2,3,4-tetrahydronaphthalen-2-ol, a compound that is disclosed as Example 2 in the U.S. Patent
- One embodiment of the present invention provides a method for treating a disorder that may be ameliorated by inhibiting vanilloid receptor subtype 1 (TRPVl) receptor in a host mammal in need of such treatment.
- the method comprises administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Another embodiment of the present invention provides a method for treating pain in a mammal in need of such treatment.
- This method comprises administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof.
- Yet another embodiment of the present invention provides a method of treating ischemia including acute cerebral ischemia, pain including chronic pain, neuropathic pain, nociceptive pain, allodynia, inflammatory pain, inflammatory hyperalgesia, post herpetic neuralgia, neuropathies, neuralgia, diabetic neuropathy, HlV-related neuropathy, nerve injury, rheumatoid arthritic pain, osteoarthritic pain, burns, back pain, visceral pain, cancer pain, dental pain, headache, migraine, carpal tunnel syndrome, fibromyalgia, neuritis, sciatica, pelvic hypersensitivity, pelvic pain, menstrual pain, bladder disease, such as incontinence and bladder overactivity, micturition disorder, renal colic; and cystitis; inflammation such as burns, rheumatoid arthritis and osteoarthritis; neurodegenerative disease such as stroke, post stroke pain and multiple sclerosis; pulmonary disease such as asthma, cough, chronic obstructive pulmonary disease (COP
- the compounds of the invention are useful for the treatment of pain, particularly nociceptive and inflammatory pain.
- This method comprises the step of administering a therapeutically effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof, to a subject in need thereof.
- Compounds of the present invention can be used to treat pain as demonstrated by Nolano, M. et al., Pain 81:135 (1999); Caterina, M. and Julius, D., Annu. Rev. Neurosci. 24:487-517 (2001); Caterina, M. et al., Science 288:306-313 (2000); Caterina, M. et al., Nature 389:816-824 (1997); and Cui et al., J. of Neuroscience, 26:9385-9393 (2006).
- Compounds of the present invention can be used to treat bladder overactivity and/ or urinary incontinence as demonstrated by Fowler, C. Urology 55:60 (2000).
- Compounds of the present invention can be used as for the treatment of anxiety -related disorders as demonstrated by Marsch, R. et al., J. Neuroscience 27:832-839 (2007).
- Compounds of the present invention can be used as for the treatment of disorders associated with hyperdopaminergia such as psychosis, attention deficit hyperactivity disorder and schizophrenia as demonstrated by Tzavara, E. et al., Biol. Psychiatry 59:508-515 (2006).
- Compounds of the invention may be administered alone, or in combination with one or more other compounds of the invention, or in combination (i.e. co-administered) with one or more additional pharmaceutical agents.
- a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof may be administered in combination with one or more analgesic agents selected from the group consisting of nonsteroidal anti-inflammatory drug (NSAID) and acetaminophen, or a combination thereof.
- NSAID nonsteroidal anti-inflammatory drug
- acetaminophen or a combination thereof.
- nonsteroidal antiinflammatory drug examples include, but are not limited to, aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, meloxicam, nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin and zomepirac.
- NSAID nonsteroidal antiinflammatory drug
- Combination therapy includes administration of a single pharmaceutical dosage formulation containing one or more of the compounds of invention and one or more additional pharmaceutical agents, as well as administration of the compounds of the invention and each additional pharmaceutical agent, each in its own separate pharmaceutical dosage formulation.
- a compound of formula (I) and one or more additional pharmaceutical agent may be administered to the patient together, in a single oral dosage composition having a fixed ratio of each active ingredient, such as a tablet or capsule; or each agent may be administered in separate oral dosage formulations.
- compounds of the invention and one or more additional pharmaceutical agents may be administered at essentially the same time (e.g., concurrently) or at separately staggered times (e.g., sequentially).
- Actual dosage levels of active ingredients in the pharmaceutical compositions of this invention can be varied so as to obtain an amount of the active compound(s) that is effective to achieve the desired therapeutic response for a particular patient, compositions and mode of administration.
- the selected dosage level will depend upon the activity of the particular compound, the route of administration, the severity of the condition being treated and the condition and prior medical history of the patient being treated. However, it is within the skill of the art to start doses of the compound at levels lower than required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved.
- Compounds of the invention can also be administered as a pharmaceutical composition comprising the compounds of interest in combination with one or more pharmaceutically acceptable carriers.
- therapeutically effective amount means a sufficient amount of the compound to treat disorders, at a reasonable benefit/risk ratio applicable to any medical treatment. It will be understood, however, that the total daily usage of the compounds and compositions of the invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific therapeutically effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed; and like factors well-known in the medical arts. For example, it is well within the skill of the art to start doses of the compound at levels lower than required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved.
- the total daily dose of the compounds of this invention administered to a human or lower animal range from about 0.10 ⁇ g/kg body weight to about 50 mg/kg body weight. More preferable doses can be in the range of from about 0.10 ⁇ g/kg body weight to about 3 mg/kg body weight. (Please verify the ranges) If desired, the effective daily dose can be divided into multiple doses for purposes of administration. Consequently, single dose compositions may contain such amounts or submultiples thereof to make up the daily dose. e. Pharmaceutical Compositions
- the present invention further provides pharmaceutical compositions that comprise compounds of the present invention or a pharmaceutically acceptable salt or solvate thereof.
- the pharmaceutical compositions comprise compounds of the present invention that may be formulated together with one or more non-toxic pharmaceutically acceptable carriers.
- Another aspect of the present invention is a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and one or more pharmaceutically acceptable carriers, alone or in combination with one or more nonsteroidal antiinflammatory drug (NSAID).
- NSAID nonsteroidal antiinflammatory drug
- compositions of the invention can be used for the treatment of the disorders as described herein in mammals including human.
- compositions of this invention can be administered to humans and other mammals orally, rectally, parenterally, intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments or drops), bucally or as an oral or nasal spray.
- parenterally refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection and infusion.
- pharmaceutically acceptable carrier means a non-toxic, inert solid, semi-solid or liquid filler, diluent, encapsulating material or formulation auxiliary of any type.
- materials which can serve as pharmaceutically acceptable carriers are sugars such as, but not limited to, lactose, glucose and sucrose; starches such as, but not limited to, corn starch and potato starch; cellulose and its derivatives such as, but not limited to, sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc; excipients such as, but not limited to, cocoa butter and suppository waxes; oils such as, but not limited to, peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; glycols; such a propylene glycol; esters such as, but not limited to, ethyl oleate and
- compositions of this invention for parenteral injection comprise pharmaceutically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions as well as sterile powders for reconstitution into sterile injectable solutions or dispersions just prior to use.
- suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol and the like), vegetable oils (such as olive oil), injectable organic esters (such as ethyl oleate) and suitable mixtures thereof.
- Proper fluidity can be maintained, for example, by the use of coating materials such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
- compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents.
- adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents.
- Prevention of the action of microorganisms can be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid and the like. It may also be desirable to include isotonic agents such as sugars, sodium chloride and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the inclusion of agents, which delay absorption such as aluminum monostearate and gelatin.
- the absorption of the drug in order to prolong the effect of the drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This can be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally -administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
- Injectable depot forms are made by forming microencapsule matrices of the drug in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio of drug to polymer and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions which are compatible with body tissues.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium just prior to use.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules.
- the active compound may be mixed with at least one inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol and silicic acid; b) binders such as carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; c) humectants such as glycerol; d) disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates and sodium carbonate; e) solution retarding agents such as paraffin; f) absorption accelerators such as quaternary ammonium compounds; g) wetting agents such as cetyl alcohol and glycerol monostearate; h)
- compositions of a similar type may also be employed as fillers in soft and hard- filled gelatin capsules using such carriers as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills and granules can be prepared with coatings and shells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition such that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- coatings and shells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition such that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- embedding compositions which can be used include polymeric substances and waxes.
- the active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned carriers.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3- butylene glycol, dimethyl formamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan and mixtures thereof.
- inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as
- the oral compositions may also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
- Suspensions in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, tragacanth and mixtures thereof.
- suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, tragacanth and mixtures thereof.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating carriers or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating carriers or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multi-lamellar hydrated liquid crystals which are dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and metabolizable lipid capable of forming liposomes can be used.
- the present compositions in liposome form can contain, in addition to a compound of the present invention, stabilizers, preservatives, excipients and the like.
- the preferred lipids are natural and synthetic phospholipids and phosphatidyl cholines (lecithins) used separately or together.
- Dosage forms for topical administration of a compound of this invention include powders, sprays, ointments and inhalants.
- the active compound may be mixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives, buffers or propellants which may be required.
- Opthalmic formulations, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- the compounds of the present invention can be used in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salt means those salts which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like and are commensurate with a reasonable benefit/risk ratio.
- the compounds of the invention may contain either a basic or an acidic functionality, or both, and can be converted to a pharmaceutically acceptable salt, when desired, by using a suitable acid or base.
- the salts can be prepared in situ during the final isolation and purification of the compounds of the invention.
- Representative acid addition salts include, but are not limited to acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate, digluconate, glycerophosphate, hemisulfate, heptanoate, hexanoate, fumarate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethansulfonate (isothionate), lactate, malate, maleate, methanesulfonate, nicotinate, 2-naphthalenesulfonate, oxalate, palmitoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, phosphate, glutamate, bicarbonate, p-toluenesulfonate and undecan
- the basic nitrogen-containing groups can be quaternized with such agents as lower alkyl halides such as, but not limited to, methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates; long chain halides such as, but not limited to, decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides; arylalkyl halides like benzyl and phenethyl bromides and others. Water or oil-soluble or dispersible products are thereby obtained.
- lower alkyl halides such as, but not limited to, methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl and diamyl
- acids which can be employed to form pharmaceutically acceptable acid addition salts include such inorganic acids as hydrochloric acid, hydrobromic acid, sulfuric acid, and phosphoric acid and such organic acids as acetic acid, fumaric acid, maleic acid, A- methylbenzenesulfonic acid, succinic acid and citric acid.
- Basic addition salts can be prepared in situ during the final isolation and purification of compounds of this invention by reacting a carboxylic acid-containing moiety with a suitable base such as, but not limited to, the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine.
- a suitable base such as, but not limited to, the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine.
- Pharmaceutically acceptable salts include, but are not limited to, cations based on alkali metals or alkaline earth metals such as, but not limited to, lithium, sodium, potassium, calcium, magnesium and aluminum salts and the like and nontoxic quaternary ammonia and amine cations including ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, diethylamine, ethylamine and the like.
- Other representative organic amines useful for the formation of base addition salts include ethyl enediamine, ethanolamine, diethanolamine, piperidine, piperazine and the like.
- prodrug or “prodrug”as used herein, represents those prodrugs of the compounds of the present invention which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use.
- the present invention contemplates compounds of formula (I) formed by synthetic means or formed by in vivo biotransformation of a prodrug.
- the compounds of the invention can exist in unsolvated as well as solvated forms, including hydrated forms, such as hemi-hydrates.
- the solvated forms with pharmaceutically acceptable solvents such as water and ethanol among others are equivalent to the unsolvated forms for the purposes of the invention.
- This invention is intended to encompass compounds of the invention when prepared by synthetic processes or by metabolic processes. Preparation of the compounds by metabolic processes includes those occurring in the human or animal body (in vivo) or processes occurring in vitro.
- the compounds of the invention may be prepared by a variety of processes well known for the preparation of compounds of this class.
- the compounds of the invention wherein the groups W, R 1 , R 2 , R 3 and R 4 have the meanings as set forth in the summary section and Detailed description unless otherwise noted, can be synthesized as shown in Schemes 1-4.
- BINAP for 2,2'-bis(diphenylphosphino)-l,r- binaphthyl, BuOH for n-butanol, dppf for l ,r-bis(diphenylphosphino)ferrocene, Et 2 O for diethyl ether, EtOH for ethanol, EtOAc for ethyl acetate, Et 3 N for triethyl amine, Et for ethyl, LAH for lithium aluminum hydride, mCPBA for 3-chloroperbenzoic acid, NaHMDS for sodium bis(trimethylsilyl)amide, MeOH for methanol, Pd(PPh 3 ) 4 for tetrakis(triphenylphosphine)palladium(0), Ph for phenyl, for TBS for tert-butyldimethylsilyl, THF for tetra
- reaction of producing compound (3) from compound (1) wherein P is a hydrogen atom or a hydroxy protecting group such as, but not limited to, TBS or tert-butyldiphenylsilyl, and (2) is generally conducted in a solvent such as n-butanol or isopropyl alcohol, at a temperature ranging from about 50 0 C to about 120 0 C.
- a solvent such as n-butanol or isopropyl alcohol
- coupling of (1) and (2) can be carried out in acetonitrile under microwave condition at a temperature ranging from about 100 0 C to about 150 0 C.
- the presence of a catalytic amount of acid such as, but not limited to, hydrochloric acid, trifluoroacetic acid, and p- toluenesulfonic acid is beneficial in selectively coupling the amino functionality of (1) to (2). It may sometimes be necessary to cool the reaction mixture after the coupling and then subject the reaction mixture to an acid such as, but not limited to, hydrochloric acid or hydrogen fluoride- triethylamine complex to facilitate complete removal of the hydroxy protecting group.
- Single enantiomers of (3) can be separated by chiral HPLC using a chiral column such as, but not limited to, a Chiralpak AD-H column and using methanol/CO 2 as eluant.
- the single enantiomer of compounds of formula (3) can be prepared by coupling substantially pure single enantiomer of (1) with compound (2).
- Preparations of the enantiomers of (1) wherein P is a hydrogen atom or a tert-butyldiphenylsilyl group are shown in Examples 24, 25, and 26.
- Compound (6) wherein X is methyl or ethyl can be obtained from the reaction of compound (4) with aldehyde (5) in the presence of a base such as potassium carbonate, and in a solvent such as methanol, at a temperature ranging from about 50 0 C to about 100 0 C.
- a base such as potassium carbonate
- a solvent such as methanol
- Step (a) is generally carried out in a solvent mixture such as tetrahydrofuran/l,3-dimethyl-3,4,5,6- tetrahydro-2(l//)-pyrimidinone, at a temperature ranging from about —78 0 C to about room temperature.
- Step (b) is generally conducted in a solvent such as dioxane at a temperature ranging from about 50 0 C to about 100 0 C.
- 2-chloro-oxazoles of formula (13) can be prepared from chlorides of formula R 1 C(O)Cl and ethyl-2-isocyanoacetate utilizing general procedures as illustrated in Scheme 3.
- (11) When subjected to an acid such as, but not limited to, oxalic acid, (11) can be transformed to the corresponding aldehydes (12).
- Intermediates (13) can be obtained by reacting (12) with [bis(2-methoxyethyl)amino]sulfur trifluoride in a suitable solvent such as, but not limited to, dichloromethane, at about 0 0 C.
- reaction conditions and reaction times for each individual step may vary depending on the particular reactants employed and substituents present in the reactants used. Unless otherwise specified, solvents, temperatures and other reaction conditions may be readily selected by one of ordinary skill in the art. Specific procedures are provided in the Synthetic Examples section. Reactions may be worked up in the conventional manner, e.g. by eliminating the solvent from the residue and further purified according to methodologies generally known in the art such as, but not limited to, crystallization, distillation, extraction, trituration and chromatography. Unless otherwise described, the starting materials and reagents are either commercially available or may be prepared by one skilled in the art from commercially available materials using methods described in the chemical literature.
- an optically active form of a compound of the invention When an optically active form of a compound of the invention is required, it may be obtained by carrying out one of the procedures described herein using an optically active starting material (prepared, for example, by asymmetric induction of a suitable reaction step), or by resolution of a mixture of the stereoisomers of the compound or intermediates using a standard procedure (such as chromatographic separation, recrystallization or enzymatic resolution).
- a pure geometric isomer of a compound of the invention it may be obtained by carrying out one of the above procedures using a pure geometric isomer as a starting material, or by resolution of a mixture of the geometric isomers of the compound or intermediates using a standard procedure such as chromatographic separation.
- the oil was dissolved in CH 2 Cl 2 (600 mL) and was treated with imidazole (65.9 g, 969 mmol) and tert-butylchlorodimethylsilane (59.0 g, 392 mmol).
- the reaction mixture was stirred at room temperature for 18 hours, and washed with water and brine.
- the organic solution was dried over Na 2 S ⁇ 4 and evaporated.
- the residue was chromatographed on silica gel (9:1 to 4:1 hexane-ethyl acetate, eluant) to afford the title compound as a dark purple oil, 44.22 g (49%).
- Example 1C (0.78 g, 2.79 mmol) and Example IA (0.774 g, 2.79 mmol) were heated to reflux in n-butanol (14 mL) for 2 hours, cooled to room temperature and concentrated in vacuo. The residue was chromatographed on silica gel (3 to 20% methanol- CH 2 Cl 2 , eluant) to afford the title compound as an off-white solid (485 mg, 43%).
- Example 1 The title compound was prepared according to the procedure of Example 1, substituting 3,4-bis(trifluoromethyl)benzaldehyde for 3-fluoro-4-(trifluoromethyl)benzaldehyde used in Example IB.
- Example 3B (0.648 g, 1.97 mmol) and Example IA in n-butanol (15 mL) was heated to reflux. After 2 hours the reaction mixture was cooled to room temperature and stirred for 1 h with 15 mL of aqueous IN HCl. After this time, saturated NaHCO 3 was added to increase the basicity to pH 8-9, then the aqueous mixture was extracted with ethyl acetate. The solvent was evaporated and the residue was chromatographed on silica gel (3 to 6% methanol- CH 2 Cl 2 , eluant) to afford a tan solid that was then triturated to give the title compound as an off- white solid (307 mg, 34%).
- Example 5A A solution of Example 5A (6.46 g, 27.9 mmol) in tetrahydrofuran (48 mL) and 1,3- dimethyl-3,4,5,6-tetrahydro-2(lH)-pyrimidinone (36 mL) was cooled to -78 0 C, and then treated slowly with lithium bis(trimethylsilyl)amide (IM in tetrahydrofuran, 34 mL, 34 mmol). The reaction was stirred for 1 hour and then treated dropwise with bromine (1.43 ml, 27.9 mmol). The reaction stirred for another hour at -78 0 C, and then was poured into 900 mL ether and 270 mL 10% Na 2 SO 3 solution.
- IM lithium bis(trimethylsilyl)amide
- Example 5 C 5-[2-fluoro-4-(tafluoromethyl)phenyl]-4-methyl-l,3-oxazole
- a mixture of Example 5B (4.58 g, 14.8 mmol) and [1,1'- bis(diphenylphosphino)ferrocene]dichloropalladium (II), 1 :1 complex with CH 2 Cl 2 (0.362 g, 0.443 mmol) in dioxane (49 mL) was treated carefully with dimethyl zinc (2M in toluene, 14.8 mL, 29.6 mmol). The reaction was heated to reflux for 2 hours, then cooled to room temperature and carefully quenched with a few milliliters of methanol.
- Example 5D (1.32 g, 4.72 mmol) and Example IA (1.31 g, 4.72 mmol) in acetonitrile (16 mL) was heated at 15O 0 C in a microwave reactor for 20 minutes. The solution was cooled and concentrated, and the crude material was chromatographed on silica gel (1 to 20% methanol-CH 2 Cl 2 eluant). The resulting crude product was further purified by trituration with 1 : 1 hexane-ether, which yielded 0.278 g (14 % yield) of the title compound as an off-white solid.
- Example 6 8-( ⁇ 5- [2,3 -difluoro-4-(trifluoromethyl)phenyl] -4-methyl- 1 ,3 -oxazol-2-yl ⁇ amino)- 1,2,3,4- tetrahydronaphthalen-2-ol
- Example 7D 0.425 g, 2.13 mmol
- Example IA 0.591 g, 2.13 mmol
- acetonitrile 10 mL
- the reaction mixture was cooled to room temperature and evaporated.
- the residue was taken up in tetrahydrofuran (36 mL) and stirred with 6N HCl (3.6 mL) for several hours.
- Example 7A The title compound was prepared according to the procedure of Example 7, substituting 4-(trifluoromethoxy)benzaldehyde for thiophene-3-carbaldehyde used in Example 7A.
- Lithium bis(trimethylsilyl)amide (1 M in tetrahydrofuran, 19.4 mL, 19.4 mmol) was added dropwise to a solution of Example 12A (4.0 g, 17.6 mmol) in tetrahydrofuran (80 mL) at - 78 0 C. After stirring 30 min, solid hexachloroethane (8.34 g, 35.2 mmol) was added in one portion, and the reaction was allowed to gradually warm to ambient temperature. After 16 hours the reaction was quenched with half-saturated NH 4 CI solution.
- Example 12C 8-( ⁇ 4-methyl-5-[4-(trifluoromethyl)phenyl]-l,3-oxazol-2-yl ⁇ amino)-l,2,3,4- tetrahydronaphthalen-2-ol
- Example IA (1.08 g, 3.89 mmol) and Example 12B (1.02 g, 3.89 mmol) in 15 mL n-butanol was heated to reflux for 1.5 hours.
- the reaction mixture was cooled to ambient temperature and partitioned between ethyl acetate and saturated aqueous NaHC ⁇ 3 solution.
- the separated organic phase was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated at the rotary evaporator to give a dark oil. Hexanes was added to the residue, and the resulting solid was collected by suction filtration, washed with hexanes and dried under vacuum. The result was 0.89 g of the title compound as a white solid.
- Example IA 3.12 g, 11.2 mmol
- Example 12B 2.94 g, 11.2 mmol
- hexane was added to precipitate the product.
- the result was 2.52 g that was combined with the material from the first reaction to afford a total of 3.41 g (58% yield) of the title compound as a white solid.
- Example 13 8- ⁇ [5-(4-chlorophenyl)-4-methyl- 1 ,3-oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 13E 8- ⁇ [5-(4-chlorophenyl)-4-methyl- 1 ,3-oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 13D (540 mg, 2.37 mmol) and Example IA (657 mg, 2.37 mmol) in acetonitrile (6 mL) was heated in a microwave reactor at 150 0 C for 20 minutes. The reaction was cooled and the solvent was evaporated. The residue was dissolved in tetrahydrofuran (20 mL) and 10 mL 6N HCl and the mixture was stirred several hours at ambient temperature. The mixture was then diluted with ethyl acetate, and the organic phase was washed with water, brine, dried over Na 2 SO 4 , filtered, and evaporated.
- Example 14 8- ⁇ [5-(4-methoxyphenyl)-4-methyl- 1 ,3-oxazol-2-yl]amino ⁇ - 1 ,2,3,4-tetrahydronaphthalen-2-ol
- Example 14A
- Example 14D (0.840 g, 3.76 mmol) and Example IA (1.04 g, 3.76 mmol) in acetonitrile (19 mL) was heated in a microwave reactor at 150 0 C for 20 minutes. Two drops of aqueous concentrated HCl were added and the reaction was reheated in the microwave to 150 0 C for 20 min. The volatiles were evaporated and the crude material was dissolved in tetrahydrofuran (20 mL), followed by addition of 10 mL 6N HCl. The mixture was stirred overnight at ambient temperature, then diluted with ethyl acetate. The organic phase was washed with water, brine, dried over Na 2 SC> 4 , and concentrated in vacuo.
- Example 15A A solution of Example 15A (1.70 g, 9.23 mmol) in tetrahydrofuran (37 ml) was cooled to -78°C, followed by addition of lithium bis(trimethylsilyl)amide (IM, in tetrahydrofuran, 10.2 mL, 10.2 mmol). After 30 min hexchloroethane (4.37 g, 18.5 mmol) was added as a solid in one portion. The reaction was stirred overnight being allowed to warm to room temperature, and then quenched with brine, and diluted with ethyl acetate. Water was added and the product was extracted with ethyl acetate.
- IM lithium bis(trimethylsilyl)amide
- Example 15B (1.36 g, 6.22 mmol) and Example IA (1.73 g, 6.22 mmol) in 2-propanol (63 mL) was heated to reflux overnight. The reaction was cooled and the solvent evaporated. The residue was triturated with 1 :1:1 hexanes: ether: CH 2 Cl 2 , followed by recrystallization from ethyl acetate and collection of the solid by filtration. The mother liquor was evaporated and the resulting solid was recrystallized from ethyl acetate. All of the solids were combined and triturated with ether and collected by filtration to give 0.986 g (46% yield) of the title compound as a light gray cotton like material.
- Example 16 8- ⁇ [5-(4-bromophenyl)-4-methyl- 1 ,3-oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 16C 8- ⁇ [5-(4-bromophenyl)-4-methyl- 1 ,3-oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 13E The title compound was prepared according to the procedure of Example 13E, substituting Example 16B for Example 13D.
- MS (ESI) m/z 346 (M+H) + .
- Example 17 8- ⁇ [5-(3 ,4-dichlorophenyl)-4-methyl- 1 ,3 -oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 17C 8- ⁇ [5-(3 ,4-dichlorophenyl)-4-methyl- 1 ,3 -oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-2-ol
- Example 13E The title compound was prepared according to the procedure of Example 13E, substituting Example 17B for Example 13D.
- MS (ESI) m/z 388 (M+H) + .
- Example 18B 500 mg, 2.36 mmol
- Example IA 656 mg, 2.36 mmol
- n-butanol 12 mL
- the reaction was cooled to room temperature, followed by addition of 12 mL IM HCl and continued stirring for less than 1 hour.
- the mixture was diluted with water and ethyl acetate, and the organic phase was washed with water, dried, filtered, and evaporated.
- the crude product was triturated with CH 2 Cl 2 (with sonication), and the solid was collected by filtration and washed with diethyl ether to give 192 mg (24% yield) of the title compound as a white solid.
- Example IA A mixture of Example IA (516 mg, 1.86 mmol), Example 19B (450 mg, 1.86 mmol), and 1 drop aqueous concentrated HCl in n-butanol (9.3 mL) was heated to reflux for 2 hours. The reaction was cooled and concentrated, and the residue was partitioned between water and ethyl acetate. The organic phase was washed with water and brine, dried over Na 2 S ⁇ 4 , filtered and evaporated.
- Example 2OB 200 mg, 0.688 mmol
- Example IA (191 mg, 0.688 mmol) in n-butanol (10 mL) was heated to reflux. After 1.5 hours 1 drop aqueous concentrated HCl was added and the solution stirred an additional 1.5 hour. The reaction was cooled to room temperature, and two drops of 6M HCl were added and stirring was continued 15 minutes. The n-butanol was evaporated and the residue was partitioned between water and ethyl acetate. The organic layer was washed with water and brine, dried over Na 2 SC> 4 , filtered, and evaporated.
- Example 12C (3.3 g) was dissolved in ethanol (17 mg/mL after sonication), loaded on a Chiralpak AD-H (3 cm ID x 25 cm) preparative chiral HPLC column (4.5 mL/injection), and eluted with 40% methanol in supercritical CO 2 (120 bar) under supercritical fluid chromatography (SFC) conditions at 40 0 C with a flow rate of 40 gram/min. The early eluting peak was collected and the solvent evaporated to afford 1.46 g (44% yield) of the title compound as a pale gray solid.
- SFC supercritical fluid chromatography
- Example 24E 133 mg, 0.815 mmol
- Example 12B 213 mg, 0.815 mmol
- n-butanol 3 mL
- the reaction mixture was cooled to ambient temperature, diluted with ethyl acetate, and quenched with aqueous saturated NaHCO 3 solution.
- the separated organic phase was washed with brine, dried (Na 2 SO/i), filtered, and concentrated in vacuo to about 2 mL.
- Example 26F (71 mg, 0.18 mmol) and Example 12B (50 mg, 0.19 mmol) was heated to reflux in n-butanol (1 mL) for 2 hours, and cooled to room temperature. The solvent was evaporated in vacuo, and the residue was dissolved in 4 mL tetrahydrofuran and treated with hydrogen fluoride -triethylamine complex (1.0 mL). The reaction mixture was heated at 45 0 C for 4 hours, after which time the reaction mixture was diluted with ethyl acetate, and washed with water and brine.
- Example 12C (3.3 g) was dissolved in ethanol (17 mg/mL after sonication), loaded on a Chiralpak AD-H (3 cm ID x 25 cm) preparative chiral HPLC column (4.5 mL/injection), and eluted with 40% methanol in supercritical CO 2 (120 bar) under supercritical fluid chromatography (SFC) conditions at 40 0 C with a flow rate of 40 gram/min. The late eluting peak was collected and the solvent evaporated to afford 1.55 g (47% yield) of the title compound as a pale gray solid.
- SFC supercritical fluid chromatography
- Example 25B 145 mg, 0.888 mmol
- Example 12B 232 mg, 0.888 mmol
- n-butanol 3 mL
- 6-(trifluoromethyl)nicotinaldehyde (7 g, 40 mmol), l-(l-isocyanoethylsulfonyl)-4- methylbenzene (8.41 g, 40.0 mmol) and K 2 CO 3 (6.63 g, 48.0 mmol) were heated to reflux in methanol (200 mL) overnight. The solution was cooled and the solvents removed in vacuo. The orange semi-solid was partitioned between water and ethyl acetate. The organic phase was washed with water and brine, dried over Na 2 SO/!, filtered, and the solvents removed in vacuo.
- Example 23 A (8.05 g, 35.3 mmol) was dissolved in tetrahydrofuran (118 mL) and cooled to -78 0 C. Lithium bis(trimethylsilyl)amide (1 M in tetrahydrofuran, 38.8 mL, 38.8 mmol) was added dropwise, and the resulting solution stirred 1/2 hour at -78°C. Hexachloroethane (9.19 g, 38.8 mmol) was then added in one portion. The mixture was stirred overnight while warming to room temperature.
- the reaction mixture was diluted with water and extracted with ethyl acetate.
- the organic phase was washed with water, brine, dried over Na 2 S ⁇ 4 , filtered, and the solvents removed in vacuo.
- the resulting solid was chromatographed on a SiO 2 column using two column volumes of hexane, followed by 10%-40% ethyl acetate/hexanes to afford 7.89 g (85% yield) of the title compound as a yellow solid.
- Example IA A solution of Example IA (506 mg, 1.82 mmol) and Example 23B (479 mg, 1.82 mmol) in n-butanol (18 mL) was heated to 110 0 C for 1.5 hour. The reaction mixture was cooled to ambient temperature, aqueous IM HCl (6 mL) was added, and stirring was continued 1 hour. The reaction was then diluted with water, basified with saturated aq NaHC ⁇ 3 solution, and the product was extracted with ethyl acetate. The organic phase was washed with water, brine, dried over Na 2 SO 4 , and evaporated.
- a gradient of acetonitrile (A) and 0.1% trifluoroacetic acid in H 2 O (B) was used at a flow rate of 50 mL/min (0-0.5 min 10% A, 0.5-12 min linear gradient 10%-95% A, 12-15 min 95% A, 15-17 min linear gradient 95-10% A) to afford 73 mg of the title compound as a tan solid.
- Ethanol (1 liter) was added to 8-amino-2-naphthol (100 g, 0.61 mol), Raney nickel (4Og water wet), and sodium hydroxide (4.Og 8 mol% aqueous) in a stirred reactor.
- the reactor was sealed and sparged with hydrogen.
- the reaction mixture was stirred for 13 hours at 85 0 C and then an additional 8 hours at 100 0 C.
- the mixture was then filtered through a pad of Celite.
- the resulting solution was treated with Darco G-60 (35 grams) and heated to reflux for 1 hour, then cooled to ambient temperature and stirred an additional 3 hours. This mixture was filtered through Celite (350grams), and the pad washed with ethyl acetate (1.5 liters).
- Example 24B A mixture of Example 24B (1.03 g, 3.46 mmol), (R)-2-methoxy-2-phenylacetic acid (0.633 g, 3.81 mmol), 1,3-dicyclohexylcarbodiimide (1.14 g, 5.54 mmol), and 4- dimethylaminopyridine (0.846 g, 6.93 mmol) in dichloromethane (35 mL) was stirred at ambient temperature for 18 hr. The reaction mixture was diluted with CH 2 Cl 2 and washed with saturated aqueous NH 4 Cl solution, brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 24D A mixture of Example 24D (3.05 g, 9.80 mmol) in tetrahydrofuran (40 mL) and lithium hydroxide (1 M aq, 30 mL, 30.0 mmol) was stirred vigorously at ambient temperature. After 1.5 hours the reaction mixture was partitioned between 100 mL ethyl acetate:H 2 O (1:1). The separated organic phase was washed with aq saturated NaHCO 3 and brine, dried over Na 2 SO 4 , and filtered. The solvent was removed at the rotary evaporator to yield 1.31 g (82% yield) of the title compound as a tan solid.
- Example 24A was dissolved in ethanol, loaded on a Chiralpak AD-H (4.6 mm ID x 250 mm) chiral HPLC column (0.1 mL/injection), and eluted with 12% ethanol + 1% isopropylamine in supercritical CO 2 (200 bar) under supercritical fluid chromatography (SFC) conditions at 40 0 C with a flow rate of 3 mL/min. The earlier eluting peak was collected and the solvent evaporated to afford the title compound as an off-white solid in >99% ee.
- SFC supercritical fluid chromatography
- Example 25A A mixture of Example 25A (300 mg, 0.963 mmol) in tetrahydrofuran (4 mL) and aqueous lithium hydroxide (I M aq, 3 mL, 3.00 mmol) was stirred at ambient temperature for 1.5 hour. The reaction was diluted with ethyl acetate and poured into aqueous saturated NaHCO 3 solution. The separated organic phase was washed with brine, dried over Na 2 SO 4 , and concentrated in vacuo. The result was 145 mg (92% yield) of the title compound as a tan solid.
- Example 24A was dissolved in ethanol, loaded on a Chiralpak AD-H (4.6 mm ID x 250 mm) chiral HPLC column (0.1 mL/injection), and eluted with 12% ethanol + 1% isopropylamine in supercritical CO 2 (200 bar) under supercritical fluid chromatography (SFC) conditions at 40 0 C with a flow rate of 3 mL/min. The later eluting peak was collected and the solvent evaporated to afford the title compound as an off-white solid in >99% ee.
- SFC supercritical fluid chromatography
- N-((lR,2R)-2-amino-l,2-diphenylethyl)-4-methylbenzenesulfonamide (0.333 g, 0.908 mmol) and benzene ruthenium(II) chloride dimer (0.113 g, 0.226 mmol) in 80 mL isopropanol were stirred at 80 0 C for 30 minutes, then cooled to room temperature.
- a solution of 8-methoxy- 2-tetralone (4.0 g, 23 mmol) in isopropanol (400 mL) was added, followed by potassium hydroxide (0.038M in isopropanol, 120 mL, 4.56 mmol).
- the reaction mixture was stirred at 50 0 C for 1.5 hours, at which time the starting material had been consumed.
- the isopropanol was evaporated, the residue was taken up in ethyl acetate and washed with water and brine.
- the organic layer was dried over Na 2 SO 4 , filtered, and concentrated.
- the residue was chromatographed on silica gel (15 to 30% ethyl acetate -hexane, eluant) to afford 3.0 g (74% yield) of the title compound as a fluffy off-white solid.
- Example 26B tert-butyl ⁇ [(27?)-8-methoxy- 1 ,2,3 ,4-tetrahydronaphthalen-2-yl] oxy ⁇ diphenylsilane
- Example 26A (3 g, 16.8 mmol), ter ⁇ -butylchlorodiphenylsilane (4.4 mL, 17 mmol), and imidazole (3.44 g, 50.5 mmol) were stirred overnight in CH 2 CI 2 (70 mL) at room temperature. The mixture was diluted with more CH 2 Cl 2 and washed with water and brine.
- Example 26D [IR)-I- ⁇ [tert-butyl(diphenyl)silyl]oxy ⁇ -5,6,7,8-tetrahydronaphthalen- 1 -yl trifluoroacetate
- a solution of Example 26C (190 mg, 0.472 mmol) in dichloromethane (3 rnL) was treated with triethylamine (0.48 mL, 3.44 mmol) and a small amount of 4- (dimethylamino)pyridine, and cooled to -10 0 C.
- trifluoroacetic anhydride (0.14 mL, 0.83 mmol) was slowly added, and the reaction was stirred at -10 0 C for 1 hour, and at room temperature for 2 hours.
- Example 26D A mixture of Example 26D (291 mg, 0.544 mmol), benzophenone imine (0.18 mL, 1.1 mmol), cesium carbonate (390 mg, 1.20 mmol), tris(dibenzylideneacetone)dipalladium(0) (14 mg, 0.015 mmol), and 2,2'-bis(diphenylphosphino)-l,l'-binaphthyl (22 mg, 0.035 mmol) in dimethoxyethane (2.2 mL) was heated at 150 0 C for 25 minutes in a microwave reactor. The mixture was cooled to room temperature, and the solution was pipetted away from the residual solid and concentrated.
- Example 27B (6.34 g, 22.7 mmol) and Example 24E (3.7 g, 23 mmol) were dissolved in n-butanol (227 mL) and heated at 110 0 C for 4 hours. Two drops aqueous concentrated HCl were added and heating was continued 2 hours additional, after which the solvent was evaporated in vacuo. The resulting solid was chromatographed (Si ⁇ 2 , 5% to 40% methanol/methylene chloride as eluent) to give a solid that was triturated with hexanes: ethyl ether (1 :1), collected by filtration and air-dried to afford 5.00 g (54% yield) of the title compound as a gray solid.
- Example IA (191 mg, 0.688 mmol) and Example 29B (220 mg, 0.757 mmol) in BuOH (3 mL) was heated at 120 0 C for 1 hr.
- the reaction was cooled to ambient temperature, diluted with EtOAc, washed with aqueous saturated NaHCO 3 solution and brine, dried over Na2SO4, and filtered.
- the solution was concentrated to approximately 5 mL, followed by addition of hexanes.
- the resulting precipitate was collected by filtration, washed with EtOAc, CH 2 Cl 2 and Et 2 O, and air-dried. The result was 176 mg (61%) of the title compound as a light grey solid.
- Example 30 8- ⁇ [5 -(3 -fluoro-4-methylphenyl)-4-methyl- 1 ,3 -oxazol-2-yl]amino ⁇ - 1 ,2,3 ,4-tetrahydronaphthalen-
- Example 3OA A solution of Example 3OA (1.56 g, 8.16 mmol) in THF (36 mL) was cooled to -78°C, followed by dropwise addition of LiHMDS (1 M in THF, 10.2 mL, 10.2 mmol). The reaction was stirred 30 min and then hexachloroethane (3.86 g, 16.3 mmol) was added in one portion. The reaction was allowed to slowly warm to room temperature and stir over the weekend. The reaction was then quenched with 50 mL of 1:1 water: saturated aqueous NH 4 Cl solution, and extracted with Et 2 O (2X). The combined organic layer was dried with MgSO 4 , filtered, and concentrated in vacuo.
- Example IA 400 mg, 1.44 mmol
- Example 3OB 325 mg, 1.44 mmol
- BuOH 14 mL
- the reaction was cooled to ambient temperature, followed by addition of 10 mL IM HCl.
- the mixture was stirred approximately 0.5 hour. Water was added, the layers were separated, and the organic mixture was diluted with EtOAc.
- Example 31A To a solution of Example 31A (0.62 g, 2.8 mmol) in THF (20 mL) at -78°C was added LiHMDS (1 M in THF, 3.4 mL, 3.4 mmol) dropwise over a period of 6-8 min. After 30 min hexachloroethane (1.35 g, 5.68 mmol) was added at once as a solid and the mixture was left to stir overnight warming to ambient temperature. The mixture was diluted with Et2 ⁇ and washed with aqueous NH 4 CI solution and water.
- Example 3 IB 200 mg, 0.795 mmol
- Example IA 221 mg, 0.795 mmol
- BuOH 5 mL
- the reaction was cooled to room temperature, diluted with EtOAc, and washed 3X with NaHCO 3 solution and water, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo.
- the residue was treated with approximately 25 mL hexane, causing precipitation of a pale-grey solid.
- Example 32A (R)-4-methyl-5-(4-(prop-l-en-2-yl)cyclohex-l-enyl)oxazole
- (R)-4-(prop-l-en-2-yl)cyclohex-l-enecarbaldehyde 0.574 g, 3.82 mmol
- l-(l-isocyanoethylsulfonyl)-4-methylbenzene (0.80 g, 3.82 mmol)
- K 2 CO 3 (1.06 g, 7.65 mmol) in MeOH (25 mL) was heated at reflux for 2 h.
- the reaction mixture was cooled to ambient temperature, concentrated in vacuo, and diluted with EtOAc.
- Example 32A To a solution of Example 32A (0.55 g, 2.7 mmol) in THF (20 mL) at -78°C was added LiHMDS (1 M in THF, 3.25 mL, 3.25 mmol) dropwise over a period of 6-8 min. After 30 min hexachloroethane (1.28 g, 5.41 mmol) was added at once as a solid and the mixture was left to stir overnight warming to ambient temperature. The mixture was diluted with Et 2 O and washed with aqueous NH 4 Cl solution and water.
- Example 32B 300 mg, 1.26 mmol
- Example IA 350 mg, 1.26 mmol
- BuOH 12 mL
- Example 33A A solution of Example 33A (0.94 g, 4.8 mmol) in THF (20 mL) was cooled to -78°C, then treated slowly with LiHMDS (IM in THF, 6.3 mL, 6.3 mmol). The reaction was stirred at - 78°C for 20 min and then treated all at once with solid hexachloroethane (2.31 g, 9.75 mmol). The flask was removed from the cold bath, and the reaction mixture was allowed to stir overnight at room temperature. After this time, it was poured into water and extracted with Et 2 O.
- Example 33B 8-(5-(5-ethylthiophen-2-yl)-4-methyloxazol-2-ylamino)-l,2,3,4-tetrahydronaphthalen-2-ol
- BuOH 2.4 mL
- Example IA 100 mg, 0.36 mmol
- the reaction was then cooled to room temperature and allowed to stand overnight.
- the reaction mixture was then stirred with 2.4 mL of IN HCl for 1 h, and then diluted with EtOAc.
- the reaction mixture was washed with saturated NaHCO 3 solution (3X), water and brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo.
- Example 34A NaBH 4 (0.369 g, 9.75 mmol) was added to Example 34A (Ig, 5 mmol) in EtOH (40 mL). The suspension was heated at reflux overnight. The reaction was cooled to ambient temperature and quenched with saturated NH 4 Cl solution. The mixture was diluted with water and extracted with EtOAc. The separated organic layer was washed with water and brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo. The crude product was chromatographed on silica gel (Analogix® 25x40 column, 30%-50% EtOAc/hexanes eluant) to afford 0.62 g (72%) of the title compound as a colorless oil.
- Example 34B A solution of Example 34B (3.89 g, 22.0 mmol) in CH 2 Cl 2 (10 mL) was added to a deoxygenated solution of Dess-Martin periodinane (l,l,l-tris(acetyloxy)-l,l-dihydro-l,2- benziodoxol-3-(l//)-one, 10.3 g, 24.2 mmol) in CH 2 Cl 2 (73 mL) in one portion. The reaction was stirred under nitrogen at ambient temperature for about 20 minutes after which a solid formed. The reaction was diluted with 50 mL CH 2 Cl 2 and carefully quenched with approximately 30 mL of saturated NaHCO 3 solution.
- Example 34D A solution of Example 34D (3.37 g, 14.8 mmol) in THF (50 mL) was cooled to -78°C, followed by addition of LiHMDS (IM in THF, 16.3 ml, 16.3 mmol). After 0.5 hr at -78°C, hexachloroethane (3.50 g, 14.8 mmol) was added in one portion. The reaction was allowed to stir overnight with gradual warming to room temperature. The reaction was quenched with water and extracted with EtOAc. The combined organic extract was washed with water and brine, and then filtered through celite filter aid. The filtrate was dried over Na 2 SO 4 , filtered, and concentrated in vacuo.
- Example 24E (1.24 g, 7.62 mmol) and Example 34E (2.20 g, 8.38 mmol) were taken up in isopropanol (31 mL), followed by addition of 2 drops of trifluoroacetic acid. The reaction was heated at reflux for 3 hr. The solution was cooled to ambient temperature and concentrated in vacuo, and the residue was dissolved in CH 2 Cl 2 ZEtOAc. The organic solution was washed with saturated NaHCO 3 solution, water and brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo. The crude product was triturated with 1:1 Et 2 O:hexanes and collected by filtration, then triturated again with CH 2 Cl 2 /Et0Ac/Me0H.
- Example 35C 2-(4-(2-chloro-4-methyloxazol-5-yl)phenyl)-2-methylpropanenitrile
- LiHMDS 1.0 M in THF, 11.4 ml, 11.4 mmol
- the solution was stirred 45 min, and then solid hexachloroethane (3.69 g, 15.6 mmol) was added in one portion.
- the reaction was allowed to proceed for 16 hr, gradually warming to ambient temperature.
- the reaction was quenched with half-saturated aq NH4CI solution, and the product was extracted with Et 2 O.
- Example 24A (277 mg, 1.70 mmol) and Example 35D (442 mg, 1.70 mmol) were combined in BuOH (7 mL) and heated to 110 0 C. After 1 hr the solution was cooled, diluted with EtOAc, and poured into saturated aq NaHC ⁇ 3 solution. The separated organic layer was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo. Hexane was added to the residue, and the resulting solid was collected by vacuum filtration and air-dried. The result was 494 mg (75%) of the title compound as a pale pink solid.
- Example 36A A mixture of Example 36A (870 mg, 3.40 mmol), Pd(OAc) 2 (38.1 mg, 0.170 mmol), tricyclohexylphosphine (95 mg, 0.34 mmol), K 3 PO 4 (2524 mg, 11.9 mmol), and cyclopropylboronic acid (379 mg, 4.42 mmol) in toluene (16 mL) and water (0.8 mL) was heated in a microwave reactor at 100 0 C for 1.5 hr. A second, identical reaction was performed. The two reactions were combined, and then filtered, rinsed with EtOAc, and concentrated to an orange oil.
- Example 36B (1.25 g, 5.75 mmol) was dissolved in THF (30 mL) and cooled to -75°C for 10 min, followed by addition of LiHMDS (1.0 M in THF, 7.6 mL, 7.6 mmol) in portions. The orange solution was stirred at -75°C for 25 min. Hexachloroethane (2.74 g, 11.6 mmol) was added and the reaction mixture was stirred overnight at ambient temperature. Saturated NH 4 Cl solution (200 mL) was added and the product was extracted with 1:1 EtOAc:hexanes (2 x 200 mL). The combined organic layer was washed with brine and dried overNa 2 SO 4 , filtered, and concentrated to a yellow oil.
- LiHMDS 1.0 M in THF, 7.6 mL, 7.6 mmol
- Example 36D 8-(5-(4-cyclopropyl-3-fluorophenyl)-4-methyloxazol-2-ylamino)-l,2,3,4-tetrahydronaphthalen-2- ol
- Example 36C 200 mg, 0.795 mmol
- Example 24A 130 mg, 0.795 mmol
- BuOH 8 mL
- One drop of concentrated aqueous HCl was added and heating was continued 0.5 hr.
- the reaction was cooled to ambient temperature and concentrated in vacuo.
- the crude product was purified by chromatography on silica gel (Analogix® 45x115 column, 5% to 40% MeOH/CH 2 Cl 2 ) which yielded 132 mg (44%) of the title compound as a light tan solid.
- Example 37B 2-chloro-5-(4-chloro-3-fluorophenyl)-4-methyloxazole
- Example 37A (1.22 g, 5.78 mmol) was dissolved in THF (30 mL) and cooled to -75°C for 10 min.
- LiHMDS 1.0 M in THF, 7.8 mL, 7.8 mmol
- Hexachloroethane (2.77 g, 11.7 mmol) was then added and the reaction stirred overnight at ambient temperature.
- Saturated NH 4 CI solution 200 mL was added and the product was extracted with 1:1 EtOAc:hexanes (2 x 200 mL).
- Example 24A 400 mg, 2.45 mmol
- Example 37B (603 mg, 2.45 mmol) were taken up in BuOH (25 mL) and the mixture was heated at reflux for 1.5 hr. The reaction was cooled to ambient temperature and concentrated to a purple oil. Hexane ( ⁇ 60 mL) and BuOH ( ⁇ 5 mL) were added, and the mixture was stirred overnight. The resulting gray solid was collected by filtration, rinsed with hexane, and air dried.
- This material was triturated with 1:1 Et 2 O:hexane, collected by filtration and rinsed with 1 : 1 Et 2 O:hexane, and vacuum dried overnight to afford product that was contaminated with BuOH.
- This material was then chromatographed on silica gel (Analogix® SF25-40G column, 0%-50% MeOH/CH 2 Cl 2 , 30 mL/min) to yield a purple foam that was triturated with Et 2 O, then collected by filtration and rinsed with Et 2 O, and vacuum dried overnight. The result was 530 mg, (58%) of the title compound as a gray powder.
- Example 38A A solution of LAH (1.0 M in THF, 31 mL, 31 mmol) was added dropwise to a solution of Example 38A (5.82 g, 31.3 mmol) in THF (100 mL) at 0 0 C. The reaction was stirred for 1 hr, then quenched by careful addition of solid Na 2 SO 4 - 1 OH2O. The mixture was warmed to ambient temperature and stirred 30 min. Celite filter aid was added and the mixture was filtered. The filtrate was concentrated in vacuo to yield 4.02 g (81%) of the title compound as a colorless oil.
- Example 38C A mixture of Example 38C (3.68 g, 23.6 mmol), l-(l-isocyanoethylsulfonyl)-4- methylbenzene (4.93 g, 23.6 mmol), and K 2 CO 3 (3.26 g, 23.6 mmol) in MeOH (110 mL) was heated to reflux. After 1 hr the reaction mixture was cooled to ambient temperature and the volatiles evaporated at reduced pressure. The residue was partitioned between Et 2 O and H 2 O. The separated aqueous phase was extracted with Et 2 O, and the combined organic extract was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 38D To a solution of Example 38D (4.39 g, 21.0 mmol) in THF (100 mL) at -78°C was added LiHMDS (1.0 M in THF, 23 ml, 23 mmol) slowly via syringe. The solution was stirred 45 min, and then solid hexachloroethane (5.46 g, 23.1 mmol) was added in one portion. The reaction was allowed to proceed for 24 hr, gradually warming to ambient temperature. The reaction was quenched with half-saturated aq NH 4 Cl solution, and the product was extracted with Et 2 O. The combined organic extract was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- LiHMDS 1.0 M in THF, 23 ml, 23 mmol
- Example 24A (181 mg, 1.11 mmol) and Example 38E (270 mg, 1.11 mmol) in BuOH (5 mL) was heated at 110 0 C for 1 hr.
- the reaction was cooled to ambient temperature, diluted with EtOAc, and poured into aq saturated NaHCO 3 solution.
- the separated organic layer was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo to approximately 3 mL. Hexane was added to precipitate the product, and the resulting solid was collected by vacuum filtration, washed with hexane and minimal Et 2 O, and dried in the vacuum oven at 50 0 C for 30 min.
- the result was 312 mg (76%) of the title compound as a pale pink solid.
- Example 24E (2.40 g, 14.7 mmol) and Example 38E (3.58 g, 14.7 mmol) in BuOH (70 mL) was heated at 120 0 C for 1 hr. One drop of trifluoroacetic acid was added and the red solution was heated at reflux for 30 min. The reaction was cooled to ambient temperature and diluted with EtOAc (500 mL). The organic mixture was washed with saturated NaHCO 3 solution, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo to a give brown slurry.
- the crude product was supported on silica gel and chromatographed (Analogix® SF40-115G, 50%-100% EtOAc/CH 2 Cl 2 eluant, 70 mL/min) which yielded both pure and impure lots.
- the chromatography was repeated on the impure material (Analogix® SF25-60G column, 50%-100% EtOAc/CH 2 Cl 2 eluant, 35 mL/min) with similar results.
- the remaining impure material was triturated with 1 : 1 Et 2 O/hexanes in a sonicator, and the resulting solid was collected by filtration, washed with 1 : 1 Et 2 O/hexanes, and vacuum dried.
- Example 4OB 117 mg, 0.41 mmol
- THF 4 mL
- LiHMDS 1.0 M in THF, 0.56 mL, 0.56 mmol
- Solid hexachloroethane 200 mg, 0.845 mmol
- a solution of saturated aqueous NH4CI solution 50 mL was added and the product was extracted with 50 mL EtOAc. The separated organic layer was washed with brine and dried overNa 2 S ⁇ 4 , filtered, and concentrated to a light yellow residue.
- Example 24A 44 mg, 0.27 mmol
- Example 4OC 82 mg, 0.26 mmol
- BuOH 3 mL
- the reaction was cooled to ambient temperature, diluted with 50 mL EtOAc, and washed with saturated NaHCO 3 solution, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo.
- Example 24E (2.97 g, 18.2 mmol) and Example 37B (4.47 g, 18.2 mmol) in BuOH (90 mL) was heated to reflux. After 1 hr the reaction mixture was cooled to ambient temperature and diluted with EtOAc (500 mL). The organic solution was washed with saturated NaHCO 3 solution, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo to a beige solid.
- Example 29C (2.7 g) was dissolved in (50/50) ethanol/hexane (1 g/200 mL after addition of minimal Et 2 NH and sonication), loaded on a Chiralpak AD-H SFC (3 cm ID x 25 cm) preparative chiral HPLC column (20 mL/injection), and eluted with ethanol/hexane (50/50) at 40 0 C with a flow rate of 30 mL/min. The early eluting peak was collected and the solvent evaporated to afford 1.18 g (87%) of the title compound as a pale gray solid.
- Example 43A LiHMDS (1.0 M in THF, 26 mL, 26 mmol) was added dropwise to a solution of Example 43A (5.75 g, 23.8 mmol) in THF (100 mL) at -78°C. After 30 min solid hexachloroethane (6.21 g, 26.2 mmol) was added in one portion, and the reaction mixture was allowed to proceed for 18 hr with gradual warming to ambient temperature. The reaction was quenched with saturated aq NH 4 Cl solution (50 mL), and the mixture was partitioned between H 2 O and Et 2 O.
- the aqueous phase was extracted once with Et 2 O, and the combined organic extract was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- the crude product was purified on silica gel (Analogix® Intelliflash 280TM; SF40-150g column; 100% hexane 7 min, then 0% to 20% EtOAc/hexanes eluant, 23 min) which yielded 6.23 g (95%) of the title compound as a pale yellow oil.
- Example 24E (3.69 g, 22.6 mmol) and Example 43B (6.23 g, 22.6 mmol) in BuOH (75 mL) was heated at 110 0 C for 2 hr.
- p-Toluenesulfonic acid monohydrate (0.430 g, 2.26 mmol) was then added and heating was continued for 1.5 hr.
- the reaction mixture was cooled to ambient temperature, diluted with EtOAc, and quenched with aq saturated NaHCO 3 solution.
- Example 44A 5-(5-bromopyridin-2-yl)-4-ethyloxazole A mixture of l-(l-isocyanopropylsulfonyl)-4-methylbenzene (1.98 g, 8.87 mmol), 5- bromopicolinaldehyde (1.65 g, 8.87 mmol), and K 2 CO 3 (1.47 g, 10.6 mmol) in MeOH (45 mL) was heated at reflux overnight. The reaction was cooled to ambient temperature and concentrated in vacuo. Water was added to the residue and the product was extracted with EtOAc. The combined organic extract was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 24A (0.401 g, 2.46 mmol) and Example 44B (0.777 g, 2.70 mmol) in BuOH (10 mL) was heated at reflux for 3 hr. The solvent was evaporated at reduced pressure and the residue was dissolved in CH 2 Cl 2 /Et0Ac. The organic solution was washed with saturated NaHCO 3 solution resulting in the formation of a yellow precipitate. The solid was collected by filtration, washed with Et 2 O, and air-dried. The result was 392 mg (39%) of the title compound.
- Example 45A A mixture of Example 45A (2.84 g, 13.6 mmol), l-(l-isocyanoethylsulfonyl)-4- methylbenzene (2.85 g, 13.6 mmol), and K 2 CO 3 (2.26 g, 16.3 mmol) in MeOH (70 mL) was heated to reflux. After 2 hr the reaction mixture was cooled to ambient temperature, and the volatiles were evaporated at reduced pressure. The residue was partitioned between Et 2 O and H 2 O. The separated aqueous layer was extracted with Et 2 O, and the combined organic phase was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 45D 8-(5-(3-chloro-4-(trifluoromethyl)phenyl)-4-methyloxazol-2-ylamino)-l,2,3,4- tetrahydronaphthalen-2-ol
- Example 24A 136 mg, 0.834 mmol
- Example 45C (247 mg, 0.834 mmol) in BuOH (3.5 mL) was heated to 110 0 C. After 1 hr the solution was cooled to ambient temperature, diluted with EtOAc, and washed with saturated aq NaHCO3 solution and brine, dried (Na 2 SO 4 ), filtered, and concentrated to approximately 2 mL during which the product precipitatee. Hexanes (10-15 mL) was added to complete the product precipitation, which was collected by vacuum filtration. The solid was washed with hexanes and minimal Et 2 O, and dried under vacuum at 50 0 C overnight to provide 224 mg (64%) of the title compound as a light grey solid.
- Example 46B 2-chloro-4-ethyl-5-(2-fluoro-4-(trifluoromethyl)phenyl)oxazole LiHMDS (1.0 M in THF, 6.1 mL, 6.1 mmol) was added to Example 46A (1.44 g, 5.56 mmol) in THF (19 mL) at -78°C. After 30 min solid hexachloroethane (1.32 g, 5.56 mmol) was added in one portion and the reaction mixture was stirred while warming to ambient temperature overnight. The reaction was then diluted with water and the product was extracted with EtOAc. The organic extract was washed with water and brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 24A (0.207 g, 1.27 mmol) and Example 46B (0.410 g, 1.40 mmol) were taken up in BuOH (5 mL), followed by addition of 1 drop of trifluoroacetic acid. The reaction was heated at 110 0 C for several hr, and then 1 drop of concentrated HCl was added. After heating at 110 0 C an additional 3 hrs the reaction mixture was cooled to ambient temperature and concentrated in vacuo. The residue was triturated with EtOAc and the solid filtered. The filtrate was then concentrated in vacuo and the residue was sonicated with Et 2 O, and the resulting solid was collected by filtration to afford 172 mg (32%) of the title compound as a white solid.
- Example 24A 0.627 g, 3.84 mmol
- Example 47B (1.21 g, 4.22 mmol) in BuOH (15 mL) was heated at 110 0 C for 2.5 hr. The reaction was cooled to ambient temperature and the solvent evaporated. The resulting solid was dissolved in EtOAcZCH 2 Cl 2 and washed with saturated NaHCO 3 solution and water, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo until a slurry formed.
- Example 46C (140 mg) was dissolved in methanol and loaded on a Chiralpak AD (21.1 mm x 250 mm) preparative chiral HPLC column (1.5 mL/injection, 35 mg/mL), and eluted in supercritical CO 2 (100 bar) with methanol as the modifier (gradient: 10% for 1 min, ramp 2.8%/min to 50%, hold 4 min) under supercritical fluid chromatography (SFC) conditions at 35°C with a flow rate of 40 mL/min. Pure fractions of the late eluting peak were pooled and the solvent evaporated to afford the title compound (30 mg) as a white solid.
- SFC supercritical fluid chromatography
- Example 25B (2.22 g, 13.6 mmol), Example 43B (3.75 g, 13.6 mmol), and p-toluenesulfonic acid (0.129 g, 0.680 mmol) in BuOH (45 mL) was heated at 110 0 C for 2 hr.
- the reaction was cooled to ambient temperature, diluted with ethyl acetate, and washed with saturated NaHCO 3 solution and water, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo to a volume of approximately 25 mL resulting in a thick slurry.
- the mixture was dilute with 1 : 1 ether/hexanes and sonicated.
- Example 5 IA 500 mg, 2.35 mmol
- triethylamine (0.50 mL, 3.52 mmol
- acetic anhydride (0.27 mL, 2.82 mmol) in CH 2 Cl 2 (5 mL) was stirred at room temperature.
- the reaction mixture was diluted with CH 2 Cl 2 , washed with 2N aq HCl (2 x 10 mL) and saturated NaHCO 3 solution (10 mL), dried over Na 2 SO 4 , filtered, and concentrated in vacuo.
- the crude product (599 mg, 2.35 mmol) was dissolved in CHCl 3 (6 mL) and mCPBA (789 mg, 3.52 mmol) was added.
- Example 51B A mixture of Example 51B (607 mg, 2.24 mmol) and sodium iodide (168 mg, 1.12 mmol) in acetone (3 mL) was heated to 55°C. After 2h, the reaction mixture was cooled to room temperature and water and CH 2 Cl 2 were added. The separated aqueous phase was extracted with CH 2 Cl 2 , and the combined organic extract was dried over Na 2 S ⁇ 4 , filtered, and concentrated in vacuo. Flash chromatography (0%-25% EtOAc/hexanes) gave 415 mg (68%) of the title compound as a clear oil.
- Example 51C (404 mg, 1.49 mmol), Pd(OAc) 2 (16.7 mg, 0.075 mmol), BINAP (186 mg, 0.298 mmol), and Cs 2 CO 3 (1214 mg, 3.73 mmol) were combined in toluene (4 mL). While bubbling N 2 through the mixture for 10 min, benzophenone imine (0.50 mL, 2.98 mmol) was added and the red mixture was heated to 100 0 C. After 6hr, the reaction mixture was cooled to ambient temperature, filtered, and the filter cake washed with CH 2 Cl 2 .
- Example 5 ID A solution of Example 5 ID (504 mg, 1.36 mmol) in THF (5 mL) was stirred at room temperature, followed by addition of 6N aq HCl (3.4 mL, 6.8 mmol). After 10 min water (20 mL) and CH 2 Cl 2 (10 mL) were added and the layers separated. The aqueous layer was washed with CH 2 Cl 2 (5 mL). The aqueous layer was then neutralized with saturated NaHCO 3 solution and extracted with CH 2 Cl 2 (3 x 20 mL). The combined organic layers were dried (Na 2 SO 4 ), filtered, and concentrated in vacuo. The residue was taken up in MeOH (5 mL) and K 2 CO 3 (938 mg, 6.78 mmol) was added.
- Example 51E 143 mg, 0.866 mmol
- Example 12B (272 mg, 1.04 mmol)
- p-toluenesulfonic acid monohydrate 21 mg, 0.11 mmol
- isopropanol 3.5 mL
- Example 52A A solution of Example 52A (2.08 g, 7.29 mmol) in THF (20 mL) was cooled to -78°C.
- Diisobutylaluminum hydride (1.0 M in toluene, 14.6 mL, 14.6 mmol) was added slowly via syringe, and the reaction mixture was allowed to proceed 1 hr, then warmed to -2O 0 C. After an additional 1 hr the reaction mixture was quenched with 1 N aq HCl, and the mixture was partitioned between Et 2 O and saturated aq NH 4 Cl solution.
- Example 52B A solution of Example 52B (1.26 g, 5.22 mmol) in CH 2 Cl 2 (5 mL) was cooled to -78°C after which the starting material was observed to precipitate.
- Methoxytrimethylsilane (1.44 mL, 10.5 mmol) and trimethylsilyltrifluoromethane sulfonate (0.047 mL, 0.261 mmol) were added and the reaction mixture was allowed to proceed for 24 hr with gradual warming to ambient temperature.
- the reaction was quenched with saturated aq NaHC ⁇ 3 solution (10 mL), then diluted with EtOAc (50 mL) and H 2 O (30 mL).
- Example 52D A mixture of Example 52D (1.59 g, 4.94 mmol) and oxalic acid (0.71 g, 7.9 mmol) in THF (16 mL)/water (8 mL) was stirred at ambient temperature for 2 hr. Additional oxalic acid (0.71 g, 7.9 mmol) and THF (16 mL) were added and the reaction mixture was stirred for 2 additional hr. HCl (4 N in dioxane, 5 mL) was then added and the reaction mixture was stirred 16 hr at ambient temperature, and then quenched by addition of saturated aq Na 2 CO 3 solution (10 mL).
- Example 52E To a solution of Example 52E (1.13 g, 4.10 mmol) in CH 2 Cl 2 (12 mL) at 0 0 C was added [bis(2-methoxyethyl)amino]sulfur trifluoride (1.1 mL, 6.2 mmol). The reaction was allowed to proceed for 3 hr, then H 2 O (25 mL) was added and the phases were separated. The aqueous phase was extracted with CH 2 Cl 2 , and the combined organic phase was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 25B (253 mg, 1.55 mmol), Example 52F (461 mg, 1.55 mmol), and p-toluenesulfonic acid monohydrate (29 mg, 0.16 mmol) in 2-propanol (5 mL) was heated at 100 0 C for 1 hr, then the heat was increased to 12O 0 C, and stirring was continued for 7 hr.
- the reaction was cooled to ambient temperature, diluted with EtOAc (30 mL), and washed with 1 N aq HCl, saturated aq NaHCO 3 solution, brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo to approximately 5 mL.
- Example 24E (303 mg, 1.86 mmol), Example 52F (552 mg, 1.86 mmol), and p- toluenesulfonic acid monohydrate (35 mg, 0.18 mmol) were combined in BuOH (5 mL), and the reaction mixture was heated to 120 0 C. After 5 hr the mixture was cooled to ambient temperature, diluted with EtOAc, and washed with 1 M aq HCl and saturated aq NaHCO 3 solution. The separated organic phase was washed with brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo to a wet yellow solid. Hexane ( ⁇ 40 mL) was added and the solid was collected by filtration.
- Example 24E (1.27 g, 7.76 mmol), Example 54B (2.00 g, 7.76 mmol), and p-toluenesulfonic acid monohydrate (0.074 g, 0.388 mmol) in BuOH (25 mL) was heated at 110 0 C for 1.5 hr. The reaction was cooled to ambient temperature, and washed with saturated NaHCO 3 solution and water, and then brine during which a solid formed. The solid was collected by filtration, washed with water and hexane:Et 2 O (1:1), and air-dried to yield 1.82 g (61%) of the title compound as an off-white solid.
- Example 25B (1.44 g, 8.81 mmol), Example 54B (2.27 g, 8.81 mmol), and p-toluenesulfonic acid monohydrate (0.084 g, 0.440 mmol) in BuOH (29 mL) was heated at 110 0 C for 1.5 hours.
- the reaction was cooled to ambient temperature, and washed with saturated NaHCO 3 solution and water, and then brine during which a solid formed.
- the solid was collected by filtration, washed with water and hexane:Et 2 O (1:1), and air-dried to provide 1.14 g (34%) of the title compound as an off-white solid.
- Example IB The title compound was prepared according to the procedure of Example IB, substituting 4-(trifluoromethyl)benzaldehyde for 3-fluoro-4-(trifluoromethyl)benzaldehyde and substituting p- toluenesulfonylmethyl isocyanide for l-(l-isocyanoethylsulfonyl)-4-methylbenzene.
- Example 56B 500 mg, 1.71 mmol, dicyanozinc (221 mg, 1.88 mmol), and Pd(Ph 3 P) 4 (101 mg, 0.087 mmol) in DMF (3 mL) was heated in a microwave reactor to 160 0 C for 6 min.
- the reaction mixture was cooled to ambient temperature, diluted with water (50 mL) and extracted with EtOAc.
- the combined organic extracts were filtered through celite, and the filtrate was washed with water and brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- Example 56D 2-chloro-5-(4-(trifluoromethyl)phenyl)oxazole-4-carbonitrile LiHMDS (1.0 M in THF, 4.5 mL, 4.5 mmol) was added dropwise to a solution of Example 56C (0.980 g, 4.11 mmol) in THF (13 mL) at -78°C. After 30 min solid hexachloroethane (0.974 g, 4.11 mmol) was added in one portion. The reaction was allowed to warm to room temperature while stirring overnight during which a solid formed. The mixture was diluted with hexanes and water, and filtered. The filtrate was diluted with EtOAc, and the phases separated.
- LiHMDS 2-chloro-5-(4-(trifluoromethyl)phenyl)oxazole-4-carbonitrile LiHMDS (1.0 M in THF, 4.5 mL, 4.5 mmol) was added dropwise to a solution of Example 56C (0.980 g, 4.11
- Example 24E (0.305 g, 1.87 mmol), Example 56D (0.51 g, 1.87 mmol), and p-toluenesulfonic acid monohydrate (0.018 g, 0.094 mmol) in isopropanol (6 mL) was heated at reflux for 2 hr.
- the reaction was cooled to ambient temperature, diluted with EtOAc, and washed with saturated NaHCO 3 solution and water, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated in vacuo.
- the crude product was triturated with Et 2 O.
- Example 57A A solution of Example 57A (7.32 g, 30.6 mmol) in THF (150 mL) was cooled to -78°C, and treated carefully with LiHMDS (1 M in THF, 37 mL, 37 mmol). The mixture was stirred at - 78°C for 20 minutes, then was treated all at once with hexachloroethane (14.5 g, 61.2 mmol). The flask was removed from the cold bath, and the reaction mixture was stirred overnight at room temperature. After this time, the mixture was diluted with Et 2 O and washed with saturated NH 4 Cl solution and water, dried over Na 2 SO 4 , filtered, and concentrated in vacuo.
- Example 24A (3.58 g, 21.9 mmol) and Example 57B (6.0 g, 21.9 mmol) in BuOH (75 mL) was heated at 110 0 C for 2 hr. The mixture was cooled to room temperature and diluted with EtOAc. The organic solution was washed three times with saturated NaHCO 3 solution and once with water and once with brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo. The crude product was triturated with hexane, and the solid was collected by filtration, washed with additional hexane, and air-dried to afford 6.86 g (78%) of the title compound.
- Example 57C (8.4 g) was dissolved in ethanol, loaded on a Chiralpak AD-H SFC (3 cm ID x 25 cm) preparative chiral HPLC column (4.5 mL/injection), and eluted with 40% ethanol (containing 1% isopropylamine) in supercritical CO 2 (120 bar) under supercritical fluid chromatography (SFC) conditions at 40 0 C with a flow rate of 40 gram/min. The early eluting peak was collected and the solvent evaporated to afford 1.98 g (24%) of the title compound as a brown solid.
- SFC supercritical fluid chromatography
- Example 58A (7.22 g, 27.4 mmol) was dissolved in THF (130 mL) and cooled to -75°C for 15 min. LiHMDS (1.0 M in THF, 36 mL, 36 mmol) was added in portions, and the orange solution was stirred at -75°C for 30 min. Solid hexachloroethane (13.0 g, 54.9 mmol) was added, and the reaction mixture was stirred overnight while warming to ambient temperature. Water (500 mL) was added and the product was extracted with 1:1 EtOAc/hexanes (2 x 500 mL).
- Example 24E A mixture of Example 24E (3.76 g, 23.0 mmol) and Example 58B (6.83 g, 22.9 mmol) in BuOH (115 mL) was heated at reflux for 70 min. The reaction was cooled to ambient temperature and combined with the reaction mixture from an identical reaction that employed Example 24E (494 mg, 3.03 mmol) and Example 58B (898 mg, 3.02 mmol) in BuOH (15 mL). The combined reaction mixture was diluted with EtOAc (700 mL), and washed with saturated NaHCO 3 solution, washed with brine and dried overNa 2 SO 4 , filtered, and concentrated to a brown solid.
- EtOAc 700 mL
- Example 47C (425 mg) was dissolved in methanol and loaded on a Chiralpak AD (21.1 mm x 250 mm) preparative chiral HPLC column (1.5 mL/injection, 35 mg/mL), and eluted in supercritical CO 2 (100 bar) with methanol as the modifier (gradient: 10% for 1 min, ramp 4%/min to 50%, total runtime 20 min) under supercritical fluid chromatography (SFC) conditions at 35°C with a flow rate of 40 mL/min. Pure fractions of the early eluting component were pooled and concentrated in vacuo to give 36 mg (17%) of the title compound as a white solid.
- SFC supercritical fluid chromatography
- Example 47C (425 mg) was dissolved in methanol and loaded on a Chiralpak AD (21.1 mm x 250 mm) preparative chiral HPLC column (1.5 mL/injection, 35 mg/mL), and eluted in supercritical CO 2 (100 bar) with methanol as the modifier (gradient: 10% for 1 min, ramp 4%/min to 50%, total runtime 20 min) under supercritical fluid chromatography (SFC) conditions at 35°C with a flow rate of 40 mL/min. Pure fractions of the late eluting component were pooled and concentrated in vacuo to give 36 mg (17%) of the title compound as a white solid.
- SFC supercritical fluid chromatography
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pain & Pain Management (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Child & Adolescent Psychology (AREA)
- Vascular Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Hospice & Palliative Care (AREA)
- Otolaryngology (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description








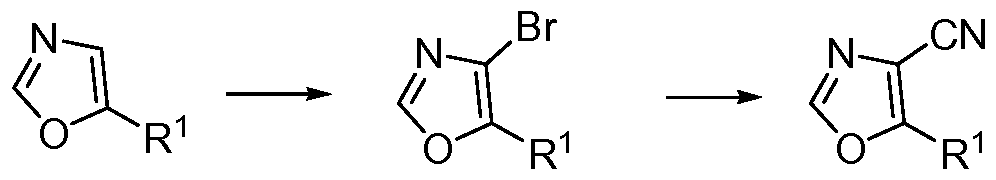
Claims


Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200880113257A CN101835762A (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as TRPV antagonists useful for the treatment of pain |
| JP2010531302A JP2011500852A (en) | 2007-10-25 | 2008-10-25 | 2-Aminooxazole derivatives as TRPVL antagonists useful for the treatment of pain |
| AT08841479T ATE518846T1 (en) | 2007-10-25 | 2008-10-25 | 2-AMINOOXAZOLE DERIVATIVES AS TRPVL ANTAGONISTS SUITABLE FOR THE TREATMENT OF PAIN |
| MX2010004495A MX2010004495A (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as trpvl antagonists useful for treating pain. |
| HK11101672.0A HK1147493B (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as trpvl antagonists useful for treating pain |
| EP08841479A EP2220059B1 (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as trpvl antagonists useful for treating pain |
| CA2702976A CA2702976A1 (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as trpv1 antagonists useful for treating pain |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US98249007P | 2007-10-25 | 2007-10-25 | |
| US60/982,490 | 2007-10-25 | ||
| US12/256,931 US7998993B2 (en) | 2007-10-25 | 2008-10-23 | TRPV1 antagonists |
| US12/256,931 | 2008-10-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009055749A1 true WO2009055749A1 (en) | 2009-04-30 |
Family
ID=40227493
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2008/081229 Ceased WO2009055749A1 (en) | 2007-10-25 | 2008-10-25 | 2-aminooxazole derivatives as trpvl antagonists useful for treating pain |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7998993B2 (en) |
| EP (2) | EP2412372A1 (en) |
| JP (1) | JP2011500852A (en) |
| CN (1) | CN101835762A (en) |
| AT (1) | ATE518846T1 (en) |
| CA (1) | CA2702976A1 (en) |
| MX (1) | MX2010004495A (en) |
| TW (1) | TW200934487A (en) |
| WO (1) | WO2009055749A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8722964B2 (en) | 2009-04-23 | 2014-05-13 | Transposagen Biopharmaceuticals, Inc. | Genetically engineered or transgenic rats exhibiting a cancer phenotype due to a disruption of germline tumor suppressor genes |
| US10131634B2 (en) | 2011-12-16 | 2018-11-20 | Poseida Therapeutics, Inc. | Method of treating pain |
| WO2021039023A1 (en) | 2019-08-23 | 2021-03-04 | 持田製薬株式会社 | Method for producing heterocyclidene acetamide derivatives |
| US11089765B2 (en) | 2009-07-01 | 2021-08-17 | Hera Testing Laboratories, Inc. | Genetically modified rat models for severe combined immunodeficiency (SCID) |
| KR20220051168A (en) | 2019-08-23 | 2022-04-26 | 모찌다 세이야쿠 가부시끼가이샤 | Method for preparing heterocyclideneacetamide derivatives |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100210682A1 (en) * | 2009-02-19 | 2010-08-19 | Abbott Laboratories | Repeated Dosing of TRPV1 Antagonists |
| EP2460016A2 (en) * | 2009-07-30 | 2012-06-06 | Transposagen Biopharmaceuticals, Inc. | Genetically modified rat models for pharmacokinetics |
| WO2011017518A2 (en) * | 2009-08-05 | 2011-02-10 | Transposagen Biopharmaceuticals, Inc. | Genetically modified rat models for drug metabolism |
| US8602579B2 (en) | 2009-09-25 | 2013-12-10 | Cree, Inc. | Lighting devices including thermally conductive housings and related structures |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5151440A (en) * | 1990-02-28 | 1992-09-29 | Allergan, Inc. | Method for reducing or maintaining intraocular pressure in the mammalian eye by administering pharmaceutical compositions containing 2-(2-alkylphenylamino)-oxazolines, 2-(2-alkylphenyl-amino)-thiazolines and 2-(2-alkylphenylamino)-imidazolines |
| WO2006122250A2 (en) * | 2005-05-11 | 2006-11-16 | Abbott Laboratories | Antagonists of the vanilloid receptor subtype 1 (vr1) and uses thereof |
-
2008
- 2008-10-23 US US12/256,931 patent/US7998993B2/en not_active Expired - Fee Related
- 2008-10-24 TW TW097141042A patent/TW200934487A/en unknown
- 2008-10-25 CA CA2702976A patent/CA2702976A1/en not_active Abandoned
- 2008-10-25 AT AT08841479T patent/ATE518846T1/en not_active IP Right Cessation
- 2008-10-25 EP EP11176290A patent/EP2412372A1/en not_active Withdrawn
- 2008-10-25 EP EP08841479A patent/EP2220059B1/en active Active
- 2008-10-25 WO PCT/US2008/081229 patent/WO2009055749A1/en not_active Ceased
- 2008-10-25 JP JP2010531302A patent/JP2011500852A/en active Pending
- 2008-10-25 MX MX2010004495A patent/MX2010004495A/en unknown
- 2008-10-25 CN CN200880113257A patent/CN101835762A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5151440A (en) * | 1990-02-28 | 1992-09-29 | Allergan, Inc. | Method for reducing or maintaining intraocular pressure in the mammalian eye by administering pharmaceutical compositions containing 2-(2-alkylphenylamino)-oxazolines, 2-(2-alkylphenyl-amino)-thiazolines and 2-(2-alkylphenylamino)-imidazolines |
| WO2006122250A2 (en) * | 2005-05-11 | 2006-11-16 | Abbott Laboratories | Antagonists of the vanilloid receptor subtype 1 (vr1) and uses thereof |
Non-Patent Citations (1)
| Title |
|---|
| SUSAN M. WESTAWAY: "The potential of transient receptor potential vanilloid type 1 channel modulators for the treatment of pain", JOURNAL OF MEDICINAL CHEMISTRY, vol. 50, 2007, pages 2589 - 2596, XP002452089, ISSN: 0022-2623 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8722964B2 (en) | 2009-04-23 | 2014-05-13 | Transposagen Biopharmaceuticals, Inc. | Genetically engineered or transgenic rats exhibiting a cancer phenotype due to a disruption of germline tumor suppressor genes |
| US11089765B2 (en) | 2009-07-01 | 2021-08-17 | Hera Testing Laboratories, Inc. | Genetically modified rat models for severe combined immunodeficiency (SCID) |
| US11849709B2 (en) | 2009-07-01 | 2023-12-26 | Hera Testing Laboratories, Inc. | Genetically modified rat models for severe combined immunodeficiency (SCID) |
| US12274247B2 (en) | 2009-07-01 | 2025-04-15 | Hera Testing Laboratories, Inc. | Genetically modified rat models for severe combined immunodeficiency (SCID) |
| US10131634B2 (en) | 2011-12-16 | 2018-11-20 | Poseida Therapeutics, Inc. | Method of treating pain |
| WO2021039023A1 (en) | 2019-08-23 | 2021-03-04 | 持田製薬株式会社 | Method for producing heterocyclidene acetamide derivatives |
| KR20220051168A (en) | 2019-08-23 | 2022-04-26 | 모찌다 세이야쿠 가부시끼가이샤 | Method for preparing heterocyclideneacetamide derivatives |
| EP4410989A2 (en) | 2019-08-23 | 2024-08-07 | Mochida Pharmaceutical Co., Ltd. | Method for producing heterocyclidene acetamide derivatives |
| US12286414B2 (en) | 2019-08-23 | 2025-04-29 | Mochida Pharmaceutical Co., Ltd. | Method for producing heterocyclidene acetamide derivative |
| KR20250166332A (en) | 2019-08-23 | 2025-11-27 | 모찌다 세이야쿠 가부시끼가이샤 | Method for producing heterocyclidene acetamide derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2220059B1 (en) | 2011-08-03 |
| US20090124666A1 (en) | 2009-05-14 |
| JP2011500852A (en) | 2011-01-06 |
| US7998993B2 (en) | 2011-08-16 |
| HK1147493A1 (en) | 2011-08-12 |
| TW200934487A (en) | 2009-08-16 |
| CN101835762A (en) | 2010-09-15 |
| ATE518846T1 (en) | 2011-08-15 |
| EP2220059A1 (en) | 2010-08-25 |
| EP2412372A1 (en) | 2012-02-01 |
| CA2702976A1 (en) | 2009-04-30 |
| MX2010004495A (en) | 2010-05-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7998993B2 (en) | TRPV1 antagonists | |
| KR102203552B1 (en) | Allosteric modulator of nicotinic acid acetylcholine receptor | |
| CN103635458B (en) | TRPV1 antagonist | |
| CN101421279B (en) | Imidazo compounds | |
| US7998982B2 (en) | Amide derivatives as TRPV1 antagonists | |
| EP2300472B1 (en) | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof | |
| US8338603B2 (en) | TRPV1 antagonists | |
| US20100010055A1 (en) | Antagonists of the vanilloid receptor subtype 1 (vr1) and use thereof | |
| CA2942871A1 (en) | Ror-gamma modulators and uses thereof | |
| EP3557998A1 (en) | Compouns, compositions and methods of use | |
| CA3129492A1 (en) | Substituted bicyclic compounds as farnesoid x receptor modulators | |
| CA3129533A1 (en) | Substituted bicyclic compounds as farnesoid x receptor modulators | |
| HK1147493B (en) | 2-aminooxazole derivatives as trpvl antagonists useful for treating pain | |
| EP3924329A2 (en) | Substituted amide compounds useful as farnesoid x receptor modulators | |
| CA3129619A1 (en) | Substituted amide compounds useful as farnesoid x receptor modulators | |
| HK1259808B (en) | Allosteric modulators of nicotinic acetylcholine receptors | |
| HK1114837B (en) | Antagonists of the vanilloid receptor subtype 1 (vr1) and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880113257.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08841479 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2702976 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010531302 Country of ref document: JP Ref document number: MX/A/2010/004495 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008841479 Country of ref document: EP |