WO2009065298A1 - Piperazine derivatives, preparation process and pharmaceutical use thereof - Google Patents
Piperazine derivatives, preparation process and pharmaceutical use thereof Download PDFInfo
- Publication number
- WO2009065298A1 WO2009065298A1 PCT/CN2008/001795 CN2008001795W WO2009065298A1 WO 2009065298 A1 WO2009065298 A1 WO 2009065298A1 CN 2008001795 W CN2008001795 W CN 2008001795W WO 2009065298 A1 WO2009065298 A1 WO 2009065298A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- imidazo
- trifluoromethyl
- mmol
- group
- piperazin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to a novel piperazine derivative of the formula (I), a process for the preparation thereof, and a pharmaceutical composition containing the same, and as a therapeutic agent, in particular as a dipeptidyl peptidase IV inhibitor use.
- Diabetes is a multi-pathogenic metabolic disease characterized by chronic hyperglycemia accompanied by disorders of sugar, fat and protein metabolism caused by defects in insulin secretion and/or function. Diabetes is a very old disease. It is caused by the absolute or relative lack of insulin in the human body. The concentration of grape vines in the blood is increased, and then the sugar is discharged from the urine, and there are polydipsia, polyuria, polyphagia, and consumption. , dizziness, fatigue and other symptoms.
- insulin-dependent diabetes mellitus a hormone used in the body to regulate glucose utilization.
- IPDDM insulin-independent diabetes mellitus
- NIDDM insulin-independent diabetes mellitus
- Insulin resistance is mainly caused by a decrease in the number of insulin receptors, as well as insulin receptor defects, which have not been understood so far.
- the resistance to insulin responsiveness causes insulin to fail to activate glucose uptake, oxidation, and storage in muscle tissue, and is ineffective in inhibiting adipose tissue lipolysis and production and secretion of hepatic glucose.
- DPPIV Dipeptidyl peptidase-IV
- a serine protease that cleaves N-terminal dipeptidase in a peptide chain containing a proline residue at the sub-end, although DPPIV has no physiological effects on mammals. It is fully confirmed, but it plays an important role in neuroenzyme metabolism, T-cell activation, cancer cell metastasis into the endothelium and HIV virus entry into lymphoid cells (W098/19998).
- GLP-1 glucagon-like peptide
- DPPIV can prevent the secretion of glucagon-like peptide (GLP)-1, in particular, it
- GLP-1 glucagon-like peptide
- the N-terminal group-propadipeptide enzyme in GLP-1 can be cleaved from the active form of GLP-1(7-36)NH 2 to inactive GLP-1 (9-36) N3 ⁇ 4 (Endocrinology, 1999, 140: 5356 ⁇ 5363). Due to physiological conditions, the half-life of intact GLP-1 in circulating blood is very short, and the inactive metabolites after DPPIV degrades GLP-1 can bind to GLP-1 receptor antagonistic activity GLP-1, thereby shortening the physiology of GLP-1. reaction.
- DPPIV inhibitors can completely protect endogenous and even exogenous GLP-1 from being inactivated by DPPIV, greatly increasing the physiological activity of GLP-1 (5 to 10 times) due to GLP-1 secretion of pancreatic insulin. It is an important buffer and can directly affect the distribution of glucose. DPPIV inhibitors play a very good role in the treatment of non-insulin-dependent diabetes mellitus (NIDDM) (US6110949).
- NIDDM non-insulin-dependent diabetes mellitus
- DPP-IV inhibitors have been disclosed (US 5,462,928, US 5,543,396, WO 9515309, WO2003004498, WO2003082817, WO2004032836, WO2004085661), in which Merck Corporation MK-0431 is a listed structure.
- piperazines of the formula (I) and their tautomers, enantiomers, diastereomers, Swirls and pharmaceutically acceptable salts, as well as metabolites and metabolic precursors or prodrugs.
- Ar is a phenyl group which is unsubstituted or further substituted with 1 to 5 R 7 ;
- R 1 is selected from a hydrogen atom, an alkyl group, a trifluoromethyl group, a cycloheterocyclyl group, a heterocyclic fluorenyl group, an aryl group or a heteroaryl group, wherein alkyl group, cycloalkyl group, heterocycloalkyl group, aryl group, heteroaryl group Further substituted by one or more substituents selected from halogen, cyano, aryl, hydroxy or amino, preferably trifluoromethyl;
- R 2 is selected from the group consisting of a hydrogen atom, a halogen, a cyano group, an amino group, a fluorenyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group,
- cyclodecyl, heterocycloalkyl, aryl, heteroaryl group is further selected from one or more Halogen, amino, cyano, nitro, hydroxy, alkyl, cyclodecyl, decyloxy, heteroaryl, trihaloalkyl, -NR 3 R 4 , -NR 3 C(O)R ⁇ -C(O Substituting a substituent of NR 3 R 4 , —NC(0)NR 3 R 4 , —COR 5 or —S0 2 R 6 ;
- R 3 and R 4 are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group or -S0 2 6 , wherein a fluorenyl group, a cycloalkyl group, a heterocycloalkyl group,
- the aryl or heteroaryl group is further selected from one or more selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, aryl, heterocycloalkyl, -S0 2 R 6 , -NR 3 R 4 , carboxylic acid or Substituted by a substituent of a carboxylic acid ester;
- R 3 and R 4 together form a 4 to 8 membered heterocyclic group, wherein the 4 to 8 membered heterocyclic ring contains one or more N, 0, and S atoms, and the 4 to 8 membered heterocyclic ring is further subject
- R 5 is selected from a hydrogen atom or an alkyl group
- R 6 is selected from an alkyl group or an aryl group, wherein the aryl group is further substituted with one or more alkyl groups.
- R 7 is selected from halogen, cyano, hydroxy, alkyl or alkoxy, wherein the alkyl or alkoxy group is unsubstituted or further substituted by one or more halogens.
- Typical compounds of the invention include, but are not limited to:
- the salt is a salt of the above compound with an acid selected from the group consisting of phosphate, malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, acetic acid or trifluorocarbon.
- an acid selected from the group consisting of phosphate, malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, acetic acid or trifluorocarbon.
- an acid selected from the group consisting of phosphate, malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, acetic acid or trifluorocarbon.
- Acetic acid selected from the group consisting of phosphate, malic acid, lactic acid, maleic acid, hydrochloric
- Another aspect of the invention relates to the compound of the formula (IA) or the compound of the formula (IB) which are intermediates for the synthesis of the compound of the formula (I):
- X is selected from halogen
- R 1 is selected from a hydrogen atom, an alkyl group, a trifluoromethyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group or a heteroaryl group, wherein an alkyl group, a cycloalkyl group, a heterocyclic fluorenyl group, an aryl group, a heteroaryl group Further substituted by one or more substituents selected from halogen, cyano, aryl, hydroxy or amino, preferably trifluoromethyl;
- R 2 is selected from a hydrogen atom, a halogen, a cyano group, an amino group, an alkyl group, a cyclodecyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group, -NR 3 R 4 , -NR 3 C(0)R 4 or - NC(0)NR 3 R 4 , wherein the cycloalkyl, heterocycloalkyl, aryl, heteroaryl group is further further selected from one or more selected from the group consisting of halogen, amino, cyano, nitro, hydroxy, alkyl, cycloalkane Alkyl, anthracenyloxy, heteroaryl, trihaloalkyl, -NR 3 R 4 , -NR 3 C(0)R 4 , -C(0)NR 3 R 4 , -NC(0)NR 3 R 4 , Substituted by a substituent of -COR 5 or -S0 2 R 6 ;
- R 3 and R 4 are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group or -S0 2 R 6 wherein alkyl, cycloalkyl, heterocycloalkyl Or an aryl or heteroaryl group further selected from one or more selected from the group consisting of halogen, hydroxy, amino, alkoxy, decyl, aryl, heterocycloalkyl, -S0 2 R 6 , -NR 3 R 4 , carboxylic acid Or substituted with a substituent of a carboxylic acid ester;
- R 3 and R 4 together form a 4 to 8 membered heterocyclic group, wherein the 4 to 8 membered heterocyclic ring contains one or more N, 0, and S atoms, and the 4 to 8 membered heterocyclic ring is further subjected to a Or a plurality of substituents selected from the group consisting of halogen, alkyl, aryl, heteroaryl, hydroxy, carbonyl, cyano, alkoxy, hydroxymethyl, heterocycloalkyl or -NR 3 R 4 ;
- R 5 is selected from a hydrogen atom or an alkyl group.
- R 6 is selected from a mercapto group or an aryl group, wherein the aryl group is further substituted with one or more alkyl groups.
- One aspect of the present invention relates to a process for the preparation of a compound of the formula (IA) and a compound of the formula (IB), comprising the steps of: a process for the preparation of a compound of the formula (IA), the process comprising the steps of:
- the raw material H 2 -substituted pyridazine 2-methylamine is reacted with an acid anhydride, and the resulting amide product is mixed with phosphorus oxychloride at room temperature, and then phosphorus pentoxide is added to form imidazo[1,5-a]pyridinium.
- the azine ring is then hydrogenated to reduce the compound of formula (IA) under Pd/C catalysis.
- the raw pyrazine 2-methylamine is reacted with an acid anhydride, and the resulting amide product is mixed with phosphorus oxychloride at room temperature, and then phosphorus pentoxide is added to form an imidazo[1,5-a]pyrazine ring.
- hydrogen is reduced and reacted with di-tert-butyl dicarbonate to form a t-butoxycarbonyl-protected imidazo[1,5-a] piperazine product, followed by N-halosuccinimide.
- the reaction is carried out under the conditions to obtain the compound of the formula (IB).
- the compound of the formula (IB) is reacted with a boric acid or a boric acid ester under microwaves under the catalysis of a palladium reagent.
- the compound of the formula (IA) and 3-tert-butoxycarbonylamino-4-aryl-butyric acid are carried out under the conditions of a condensation reagent bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- the resulting product is condensed and the amino protecting group is removed under acidic conditions to give the compound of the formula (1).
- the compound (IB) is deprotected with 3-tert-butoxycarbonylamino-4-aryl-butyric acid under the conditions of a condensation reagent bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- the reaction is carried out, and the obtained condensation product is catalyzed by a palladium reagent.
- reacting with boric acid or boric acid ester under microwave to carry out Suzuki coupling LAm. Chem. Soc., 2007, 129, 3358-3366; Chem. Soc. Rev., 2001, 30, 145-157
- the condensation product may also be subjected to Buchwald coupling with a substituted amine under catalytic conditions (J.
- a compound of the formula (I) is obtained; the condensation product can also be reacted with an alcohol in an oil bath with octacarbonylcobalt and ethyl chloroacetate as a catalyst under a carbon monoxide atmosphere to obtain a substituted carboxylic acid.
- the ester compound is further hydrolyzed to a carboxylic acid, and an amide formed by reacting with ammonium carbonate is further dehydrated to form a compound of the formula (1) wherein R 2 is a cyano group.
- One aspect of the present invention is that the compound of the formula (I) is purified and reacted in an acid solution of methanol, dichloromethane or ethyl acetate to obtain an acid addition product salt.
- the acid described therein is phosphate, malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, acetic acid or trifluoroacetic acid.
- One aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- One aspect of the invention is a method of inhibiting the catalytic activity of dipeptidyl peptidase IV, which comprises contacting said dipeptidyl peptidase IV with a compound or salt of any one of formula (I).
- Another aspect of the invention is the use of a compound, salt or pharmaceutical composition according to any one of the formula (I) for the treatment of a disease such as type II diabetes, hyperglycemia, obesity or insulin resistance.
- a disease such as type II diabetes, hyperglycemia, obesity or insulin resistance.
- One aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient, for the treatment of type 2 diabetes, hyperglycemia Use in drugs for obesity or insulin resistance.
- Alkyl means a saturated aliphatic hydrocarbon group including straight chain and branched chain groups of 1 to 20 carbon atoms. Preference is given to alkyl groups having 1 to 10 carbon atoms, such as methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl, tert-butyl, pentyl and the like. More preferred are lower alkyl groups having 1 to 4 carbon atoms, such as methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl or t-butyl groups and the like.
- the alkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, amino, cyano, hydroxy, decyl, cyclodecyl, heterocycloalkyl, Aryl, alkoxy, heteroaryl, trihaloalkyl, -S0 2 R 6 , -NR 3 R 4 , -NR 3 C(0)R 4 , -C(0)NR 3 R 4 or -NC( 0) NR 3 R 4 .
- Cycloalkyl means a 3 to 8 membered all carbon monocyclic, all carbon 5/6 or 6/6 fused or polycyclic fused ring ("fused" ring system means each in the system The rings share an adjacent pair of carbon atoms) with other rings in the system, wherein one or more of the rings may contain one or more double bonds, but none of the rings have a fully conjugated pi-electron system.
- cycloalkyl group examples include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclopentene group, a cyclohexanyl group, a cyclohexadiene group, an adamantane group, a cycloheptadene group, a cycloheptatriene group and the like.
- the cycloalkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, amino, cyano, hydroxy, alkyl, cycloalkyl, heterocycloalkyl, Aryl, alkoxy, heteroaryl, trihaloalkyl, carboxylic acid, carboxylic acid ester, -COR 5 , -S0 2 R 6 , -NR 3 R 4 , -NR 3 C(0)R 4 , -C(0)NR 3 R 4 or -NC(0)NR 3 R 4 .
- Aryl means a group having at least one aromatic ring structure, that is, an aromatic ring having a conjugated ⁇ -electron system, including a carbocyclic aryl group, a heteroaryl group, and a biaryl group.
- the alkynyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, amino, cyano, hydroxy, alkyl, cycloalkyl, heterocycloalkyl, Aryl, alkoxy, heteroaryl, trihaloalkyl, carboxylic acid, carboxylic acid ester, -COR 5 , -S0 2 R 6 , -NR 3 R 4 , -NR 3 C(0)R 4 , -C (0) NR 3 R 4 or -NC(0)NR 3 R 4 .
- Heteroaryl means an aryl group having from 1 to 3 heteroatoms as ring atoms, the remaining ring atoms being carbon, and heteroatoms including oxygen, sulfur and nitrogen.
- the ring may be a 5- or 6-membered ring.
- Examples of the heterocyclic aryl group include a furyl group, a thienyl group, a pyridyl group, a pyrrole, an N-alkylpyrrolyl group, a pyrimidinyl group, a pyrazinyl group, an imidazolyl group and the like.
- the heteroaryl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, amino, cyano, hydroxy, alkyl, cycloalkyl, heterocycloalkyl. , aryl, alkoxy, heteroaryl, trihaloalkyl, carboxylic acid, carboxylic acid ester, -CORK -S0 2 R 6 , -NR 3 R 4 , -NR 3 C(0)R 4 , -C( 0) NR 3 R 4 or -NC(0)NR 3 R 4 .
- Heterocycloalkyl means a monocyclic or fused ring radical having from 5 to 9 ring atoms in the ring wherein one or two ring atoms are selected from nitrogen, oxygen or S(0)n (where n is an integer) From 0 to 2), the remaining ring atoms are carbon. These rings may also have one or more double bonds. However, these rings do not have a fully conjugated ⁇ -electron system.
- Unsubstituted heterocycloalkyl includes, but is not limited to, pyrrolidinyl, piperidino, piperazino, morpholinyl, thiomorpholinyl, homopiperazine, etc., heterocycloalkyl can be substituted or Unsubstituted.
- the alkynyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, amino, cyano, hydroxy, decyl, cycloalkyl, heterocycloalkyl, Aryl, alkoxy, heteroaryl, trihaloalkyl, carboxylic acid, carboxylic acid ester, -COR 5 , -S0 2 R 6 , -NR 3 R 4 , -NR 3 C(0)R 4 , -C (0)NR 3 R 4 or -NC(0)NR 3 R 4 0
- Haldroxy means an -OH group.
- Alkoxy means -0-(alkyl) and -0-(unsubstituted cycloalkyl). Representative examples include, but are not limited to, methoxy, ethoxy, propoxy, butoxy, cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy and the like.
- Halogen means fluoro, chloro, bromo or iodo, preferably fluoro or chloro.
- Trifluoromethyl means -C.
- Amino means -NH 2 .
- Neitro means -N0 2 .
- “Pharmaceutical composition” means a mixture of one or more of the compounds described herein, or a physiologically/pharmaceutically acceptable salt or prodrug thereof, with other chemical components, such as a physiological/pharmaceutically acceptable carrier. And excipients.
- the purpose of the pharmaceutical composition is to facilitate the administration of the compound to the organism.
- Method for synthesizing the compound of the present invention In order to accomplish the object of the present invention, the present invention adopts the following technical scheme - a method for preparing the compound of the formula (IA) of the present invention, comprising the following steps:
- the raw material R 2 -substituted pyrazine 2-methylamine is reacted with an acid anhydride, and the resulting amide product is mixed with phosphorus oxychloride at room temperature, and then phosphorus pentoxide is added to form imidazo[1,5-a]pyridinium.
- the azine ring is then chlorinated to a compound of formula (IA:) under Pd/C catalysis.
- the preparation method of the compound (IB) of the present invention comprises the following steps:
- the preparation method of the compound (IA) of the present invention comprises the following steps:
- the compound of the formula (IB) is reacted with a boric acid or a boric acid ester under microwaves under the catalysis of a palladium reagent.
- the preparation method of the compound of the formula 0 of the present invention comprises the following steps:
- the compound of the formula (IA) and 3-tert-butoxycarbonylamino-4-aryl-butyric acid are carried out under the conditions of a condensation reagent bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- the resulting product is condensed and the amino protecting group is removed under acidic conditions to give the compound of the formula (1).
- the preparation method of the compound of the general formula (I) of the present invention comprises the following steps -
- the compound (IB) is deprotected with 3-tert-butoxycarbonylamino-4-aryl-butyric acid under the conditions of a condensation reagent bis(2-oxo-3-oxazolidinyl)phosphinic chloride
- the reaction is carried out, and the obtained condensation product is further catalyzed by a palladium reagent to react with boric acid or a boric acid ester under microwave to carry out Suzuki coupling
- a palladium reagent to react with boric acid or a boric acid ester under microwave to carry out Suzuki coupling
- the condensation product can also be Buchwald coupled with a substituted amine under catalytic conditions (J. Am. Chem.
- the condensation product can also be used in an oil bath with octacarbonylcobalt and ethyl chloroacetate as The catalyst is reacted with an alcohol in a carbon monoxide atmosphere, and the obtained substituted carboxylate compound is further hydrolyzed to a carboxylic acid, and an amide formed by reacting with ammonium carbonate is further dehydrated to form a compound of the formula (1) wherein R 2 is a cyano group.
- the compound of the formula (I) is purified and reacted in an acid solution of methanol, dichloromethane or ethyl acetate to obtain an acid addition product salt.
- the structure of the example compounds was determined by nuclear magnetic resonance (NMR) or mass spectrometry (MS).
- the NMR shift ( ⁇ ) is given in parts per million (ppm).
- the NMR was measured by a Bruker AVANCE-400 nuclear magnetic instrument.
- the solvent was deuterated methanol (CD 3 OD), deuterated chloroform (CDC1 3 ), and hexamethyl dimethyl sulfoxide (DMSO-d6) was labeled as the top three.
- Silane (TMS) chemical shift is given in units of l (T 6 (ppm);
- MS FINNIGAN LCQAd (ESI) mass spectrometer manufactured by Therm, model: Finnigan LCQ advantage MAX;
- the IC 50 value was determined using a NovoStar plate reader (BMG, Germany);
- DMSO-d6 hexamethylene dimethyl sulfoxide
- CD3OD deuterated methanol
- Ethyl 3-amino-4-(2,4,5-trifluoro-phenyl)-but-2-enoate 3-methoxy-4-(2,4,5-trifluoro-phenyl)- Ethyl butyrate lb (24.6 g, 94.5 mmol) was dissolved in 240 mL of methanol, ammonium acetate (36.4 g, 473 mmol) was added, and the mixture was heated under reflux for 3 hours. After TLC was followed to disappear, the solvent was evaporated and evaporated. The mixture was extracted with ethyl acetate (200 mL ⁇ 3). EtOAc was evaporated.
- 3-cyclopentyl-N*2*-ethylidene-N* 1 *-methylene-propene-1,2,3-triamine dissolves 2-cyanopyrazine 3a (6.3 g, 0.06 mol)
- 2-cyanopyrazine 3a 6.3 g, 0.06 mol
- cyclopentylmagnesium bromide 33 mL, 66 mmol
- 40 mL of isopropanol was added dropwise to the reaction solution.
- N*2*-Ethylene-N*l*-methylene-3-phenyl-propene-1,2,3-triamine Dissolves 2-cyanopyrazine 3a (3.15 g, 0.03 mol) in In 100 mL of toluene, cool the solution to -10 Torr, slowly add phenylmagnesium bromide (11 mL, 33 mmol), and add the mixture after 40 minutes. After stirring for 1 hour, add 40 mL of isopropanol to the reaction solution.
- 2,2,2-Trifluoro-N-(phenylpyrazin-2-methyl)-trifluoroacetamide 4b (940 mg, 3.5 mmol) was placed in a reaction flask under ice cooling, to which 10 mL of phosphorus oxychloride was added dropwise, and phosphorus pentoxide (994 mg, 7 mmol) was quickly added. After heating for 4 hours, the reaction was completed, and the reaction solution was concentrated under reduced pressure. 5 mL of water was added, and pH was adjusted with concentrated aqueous ammonia. The organic phase was extracted with ethyl acetate (150 mL ⁇ 4), and the organic phase was washed with 20 mL of saturated sodium chloride.
- N'*2*-Ethylene-N'*l*-methylene-but-1-ene-1,2,3-triamine 2-cyanopyrazine 3a (1.05 g, 10 mmol Dissolve in 30 mL of toluene, cool the solution to -10 ° C, slowly add methyl magnesium bromide (7.9 mL, 11 mmol), add dropwise after 30 minutes, stir the reaction for 30 minutes, then drop into the reaction solution. After adding 12 mL of ethanol, sodium borohydride (530 mg, 14 mmol) was further added thereto with stirring, and the mixture was stirred at room temperature overnight, and the reaction mixture was quenched with acetone, methanol and water until no bubbles were formed.
- 2,2,2-Trifluoropyrazin-2-ethyl)-acetamide 5b (1.8 g, 8.2 mmol) was placed in a reaction flask under ice cooling, and 20 mL of trichloroox was added dropwise thereto.
- 1-(4-Nitrophenyl)-3-trifluoromethyl-imidazo[1,5-a]pyrazine was added to a 50 mL flask with 2 mL of concentrated nitric acid, 2 mL in an ice bath. Concentrated sulfuric acid and 1-phenyl-3-trifluoromethyl-imidazo[1,5-a]pyrazine 4c (220 mg, 0.836 mmol), stirred for 1 hour in an ice bath, the reaction was completed, and the reaction was added dropwise. The mixture was extracted with ethyl acetate (25 mL ⁇ 3). Phenylphenyl)-3-trifluoromethyl-imidazo[1,5-a]pyrazine 6a (240 mg, yellow solid) was taken to the next step without isolation.
- reaction mixture was purified to silica gel elut elut elut elut elut eluting And [1,5-a] piperazin-7-yl]-4-(2,4,5-trifluorophenyl)-butan-1-one hydrochloride 15 (100 mg, white solid), yield : 50%.
- reaction was traced by thin layer chromatography, and the starting material disappeared.
- the reaction mixture was concentrated and purified by silica gel column chromatography toiel -7-yl)-3-oxo-small (2,4,5-trifluorobenzyl)-propyl]-carbamic acid tert-butyl ester 22c (200 mg, pale yellow oil), yield: 84%.
- 1-(2,4,5-trifluorobenzyl)-propyl ⁇ -carbamic acid tert-butyl ester 24a (100 mg, 0.156 mmol) was dissolved in 2 mL ethyl acetate. The solution was precipitated as a white solid. After stirring at room temperature for 10 minutes, 2 mL of 2N aqueous ethyl hydrogen chloride solution was added and the reaction was continued for 1 hour.
- the filtrate was purified by silica gel column chromatography toielield l,5-a] piperazin-7-yl]-3-oxo-1-(2,4,5-trifluorobenzyl)-propyl ⁇ -tert-butyl carbamate 30a (120 mg, light yellow Solid), Yield: 55 %.
- reaction mixture was evaporated to drynessjjjjjjjjjjjjjjjjj -yl)-4-(2,4,5-trifluoro-phenyl)-butan-1-one hydrochloride 45 (0.25 g, pale yellow solid), yield: 73.5%.
- N-(3-Trifluoromethyl-imidazo[1,5-a]piperazine small group)-acetamide trifluoroacetate salt 1-acetylamino-3-trifluoromethyl-imidazo[1, 5-a] piperazine-7-carboxylic acid tert-butyl ester 50a (190 mg, 0.545 mmol) was dissolved in 5 mL of dichloromethane, and trifluoroacetic acid (1.26 mL, 16.36 mmol) was slowly added dropwise. The reaction was traced by thin-layer chromatography, and the material was evaporated. The residue was evaporated. - acetamide trifluoroacetate 50b, the next reaction is carried out without isolation.
- N-(3-trifluoromethyl-imidazo[1 ,5-a]piperazine-1-yl)-carboxamide hydrochloride 1-methoxylylamino-3-trifluoromethyl-imidazo[1 , 5-a] piperazine-7-carboxylic acid tert-butyl ester 51a (190 mg, 0.57 mmol) was dissolved in 4 mL of 2.4 N aqueous hydrogen chloride solution, stirred at room temperature for 2 hours, and the reaction was traced by thin layer chromatography. The reaction mixture was concentrated under reduced pressure to give the title product N-(3-trifluoromethyl-imidazo[l,5-a]piperazin-1-yl)-carboxamide hydrochloride 51b.
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-carboxamide hydrochloride 51b (154 mg, 0.57 mmol) was dissolved in 5 under nitrogen. Triethylamine (0.38 mL, 1.54 mmol), (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid (187 mg, 0.58 mmol) and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (216 mg, 0.85 mmol), stirred at room temperature overnight, the reaction was traced by thin-layer chromatography, and the starting material disappeared.
- 1-Bromo-3-trifluoromethyl-imidazo[1 ,5-a]piperazine-7-carboxylic acid tert-butyl ester 7b 500 mg, 1.35 mmol
- 1,1-dimethylurea 143 Mg, 1.62 mmol
- cuprous iodide 51.3 mg, 0.27 mmol
- trans-indole ⁇ '-dimethylcyclohexane-1,2-diamine
- potassium carbonate 347 mg, 2.71 mmol
- 5 mL of xylene was poured into a 20 mL microwave reaction tube under argon gas protection at 135 ° C for 2 hours in the microwave.
- U-dimethyl-3-(3-trifluoromethyl-imidazo[1,5-a]piperazine small)-urea hydrochloride will be 1-(3,3-dimethyl-ureido) 3-trifluoromethyl-imidazo[1,5-a]piperazine-7-carboxylic acid tert-butyl ester 53a (77 mg, 0.2 mmol) was dissolved in 5 mL of ethyl acetate and 5 mL of 2.4N hydrogen chloride was added.
- N-methyl-N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-acetamide hydrochloride 1-(acetylmethylamino)-3- Trifluoromethyl-imidazo[1,5-a]piperazine-7-carboxylic acid tert-butyl ester 54a (206 mg, 0.57 mmol) was dissolved in 4 mL of 2.4N aqueous hydrogen chloride solution and stirred at room temperature 2 The reaction was followed by thin layer chromatography, the material was evaporated, and the mixture was evaporated to give the title product N-methyl-N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl. - acetamide hydrochloride 54b, the next reaction is carried out without isolation.
- N-methyl-N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-acetamide hydrochloride 54b (171 mg, 0.57 mmol) Dissolve in 5 mL of dichloromethane, and add triethylamine (0.39 mL, 2.83 mmol), (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-benzene).
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazine small group)-methanesulfonamide hydrochloride 1-methylsulfonylamino-3-trifluoromethyl-imidazo[1] , 5-a] piperazine-7-carboxylic acid tert-butyl ester 55a (158 mg, 0.41 mmol) was dissolved in 4 mL of 2.4 N aqueous hydrogen chloride solution, stirred at room temperature for 2 hours, and the reaction was traced by thin layer chromatography.
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-methanesulfonamide hydrochloride 55b (131 mg, 0.41 mmol) was dissolved in a nitrogen atmosphere.
- triethylamine (0.28 mL, 2.03 mmol)
- (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid were added in sequence.
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-benzenesulfonamide hydrochloride 56b (218 mg, 0.57 mmol) was dissolved in a stirred nitrogen atmosphere In 5 mL of dichloromethane, triethylamine (0.39 mL, 2.83 mmol), (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid were added in sequence.
- reaction mixture was concentrated under reduced pressure and purified tolululululululululululululu -yl)-3-oxo-1-(2,4,5-trifluorobenzyl)-propyl]-carbamic acid tert-butyl ester 57c (1 ll mg, colourless oily), yield: 75 %.
- reaction mixture was purified to silica gel column chromatography toiel oxazin-7-yl) -4- (2, 4, 5 - trifluoromethyl phenyl) - but-1-one hydrochloride 57 (27 mg, white solid), yield: 30%.
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-benzamide hydrochloride salt 1-benzoylamino-3-trifluoromethyl-imidazolium [1,5-a] piperazine-7-carboxylic acid tert-butyl ester 59a (90 mg, 0.27 mmol) was dissolved in 4 mL of 2.1N aqueous hydrogen chloride solution and stirred at room temperature overnight, thin layer chromatography The reaction was followed, the starting material was evaporated, and the reaction mixture was evaporated.
- N-(3-trifluoromethyl-imidazo[1,5-a]piperazin-1-yl)-benzamide hydrochloride 59b (140 mg, 0.4 mmol) was dissolved in vacuo.
- Triethylamine (204 mg, 2.02 mmol) and (R)-3-tert-butoxycarbonylamino-4-(2,4,5-trifluoro-phenyl)-butyric acid were added sequentially to 10 mL of dichloromethane.
- Le(206 mg, 0.81 mmol) after stirring for 10 min, bis(2-oxo-3-oxazolidinyl)phosphinic acid chloride (152 mg, 0.6 mmol) was added and stirred at room temperature overnight.
- 60b (250 mg, 0.63 mmol) was dissolved in 10 mL of dichloromethane with stirring, followed by triethylamine (0.2 mL, 1.26 mmol), (R)-3-tert-butoxycarbonylamino-4-(2,4 ,5-trifluoro-phenyl)-butyric acid le (210 mg, 0.63 mmol) and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (240 mg, 0.95 mmol), stirred at room temperature 3 The reaction was followed by thin-layer chromatography, and the title material was evaporated.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
哌嗪类衍生物, 其制备方法及其在医药上的应用
技术领域
本发明涉及一种通式 (I)所示新的哌嗪类衍生物、 其制备方法以及含有该衍 生物的药物组合物、 以及其作为治疗剂特别是作为二肽基肽酶 IV抑制剂的用途。 背景技术
糖尿病是一种多病因的代谢疾病, 特点是慢性高血糖, 伴随因胰岛素分泌及 / 或作用缺陷引起的糖、 脂肪和蛋白质代谢紊乱。 糖尿病是一种非常古老的疾病, 是由于人体内胰岛素绝对或相对缺乏而引起的血中葡萄糠浓度升高, 进而糖大量 从尿中排出, 并出现多饮、 多尿、 多食、 消痩、 头晕、 乏力等症状。
永久性的或不受控制的高血糖症导致发病率与死亡率的增加。 通常糖稳态异 常直接或间接地与脂质、 脂蛋白质、 脱辅基脂蛋白新陈代谢的变更或其他的代谢 和血液动力疾病有关。 II型糖尿病患者患有大多孔脂质体及微血管综合症, 如冠状 心脏病、 中风、 外周血管性疾病、 高血压、 肾病、 神经病和视网膜病等疾病危险 性显著增加。 因此, 对糖稳态、 脂类代谢、 高血压等疾病进行治疗控制, 对于临 床上治疗糖尿病是极其重要的。
通常来说,有两种类型的糖尿病。 1型糖尿病人,即胰岛素依赖型糖尿病 (IDDM) 患者自身产生的胰岛素很少或几乎没有。 胰岛素是体内用来调节葡萄糖利用的一 种荷尔蒙。 II型糖尿病人, 即胰岛素非依赖型糖尿病 (NIDDM)患者与非糖尿病患者 的血浆内胰岛素水平是相同的或者更高, 然而, 此类患者却对胰岛素产生抵抗力, 这些胰岛素对于主要的胰岛素敏感的组织细胞, 如肌肉、 肝脏、 25个脂肪组织等 的葡萄糖和脂类代谢起着剌激作用。 即使血浆胰岛素水平提高, 也无法克服患者 对于胰岛素显著的抵抗力。
胰岛素抵抗力并主要是因为胰岛素受体数量的减少而产生的, 还有胰岛素受 体缺陷, 到目前为止此机制还未能理解。 胰岛素应答性的抵抗力导致胰岛素无法 在肌肉组织中, 对葡萄糖摄取、 氧化、 存储进行激活, 无法有效抑制脂肪组织脂 解作用, 和肝脏葡萄糖的产生和分泌。
二肽基肽酶 -IV(DPPIV)是一种丝氨酸蛋白酶, 它可以在次末端含有一个脯氨 酸残基的肽链里裂解 N-末端二肽酶,尽管 DPPIV对哺乳动物的生理作用还没有得 到完全的证实, 但其在神经酶代谢, T-细胞激活, 癌细胞转移入内皮及 HIV病毒 进入淋巴样细胞过程中都起到重要的作用 (W098/19998)。
最近, 有研究表明 DPPIV可以阻止胰升糖素样肽 (GLP)-l的分泌, 尤其, 它
可以裂解 GLP-1中 N-末端的组 -丙二肽酶, 使其从活性形式的 GLP-1(7-36)NH2降 解为无活性的 GLP-l(9-36)N¾(Endocrinology, 1999, 140: 5356〜5363)。 由于生 理情况下, 循环血中完整 GLP-1的半衰期很短, DPPIV降解 GLP- 1后的无活性代 谢物能与 GLP-1受体结合拮抗活性 GLP-1从而缩短了对 GLP-1 的生理反应。 而 DPPIV抑制剂能完全保护内源性甚至外源性的 GLP-1不被 DPPIV灭活,极大的提 高 GLP-1的生理活性 (5〜10倍),由于 GLP-1对胰腺胰岛素的分泌是一个重要的剌 激器并能直接影响葡萄糖的分配, DPPIV抑制剂对非胰岛素依赖型糖尿病 (NIDDM) 的治疗起到很好的作用 (US6110949)。
目前一些 DPP-IV抑制剂已被公开(US5462928、 US5543396、 WO9515309、 WO2003004498 WO2003082817、 WO2004032836> WO2004085661 ),其中 Merck 公司 MK-0431为已上市结构。
本发明的目的是提供一种具有抑制 DPPIV活性并且可用于糖尿病或类似疾病 的治疗或缓解性药物的化合物。 发明内容
为了克服现有技术的不足之处, 本发明的目的在于提供一种通式 (I)所示的哌 嗪类化合物, 以及它们的互变异构体、 对映体、 非对映体、 消旋体和药学上可接 受的盐, 以及代谢产物和代谢前体或前药。

其中:
Ar是苯基, 该苯基是未取代的或者进一步被 1〜5个 R7所取代;
R1选自氢原子、烷基、三氟甲基、环綜基、 杂环垸基、 芳基或杂芳基, 其中烷 基、 环烷基、 杂环烷基、 芳基、 杂芳基进一步被一个或多个选自卤素、 氰基、 芳 基、 羟基或氨基的取代基所取代, 优选为三氟甲基;
R2选自氢原子、 卤素、氰基、氨基、垸基、环烷基、 杂环烷基、 芳基、杂芳基、
-NR3R4、 -NR3C(0)R4或 -NC(0)NR3R4, 其中环垸基、 杂环烷基、 芳基、 杂芳基进 一步被一个或多个选自卤素、 氨基、 氰基、 硝基、 羟基、 烷基、 环垸基、 垸氧基、 杂芳基、三卤代烷基、 -NR3R4、 -NR3C(O)R\ -C(O)NR3R4、 -NC(0)NR3R4、 -COR5 或 -S02R6的取代基所取代;
R3和 R4各自独立地选自氢原子、 烷基、 环烷基、 杂环烷基、 芳基、 杂芳基或 -S02 6, 其中垸基、 环烷基、 杂环烷基、 芳基或者杂芳基进一步被一个或多个选 自卤素、 羟基、 氨基、 烷氧基、 烷基、 芳基、 杂环烷基、 -S02R6、 - NR3R4、 羧酸 或羧酸酯的取代基所取代;
或者, R3和 R4—起形成一个 4〜8元杂环基, 其中 4〜8元杂环内含有一个或 多个 N、 0、 S原子, 并且 4〜8元杂环上进一步被一个或多个选自卤素、 烷基、 芳基、 杂芳基、 羟基、 羰基、 氰基、 烷氧基、 羟烷基、 杂环烷基或 -NR3R4的取代 基所取代;
R5选自氢原子或烷基;
R6选自烷基或芳基, 其中芳基进一步被一个或多个烷基取代。
R7选自卤素、 氰基、羟基、 烷基或者烷氧基, 其中烷基或者烷氧基是未取代的 或者进一步被一个或多个卤素取代。 本发明的典型化合物包括, 但不限于:










其中, 所述的盐为上述化合物与选自以下的酸形成的盐: 磷酸盐、 苹果酸、 乳酸、 马来酸、 盐酸、 甲磺酸、 硫酸、 磷酸、 柠檬酸、 酒石酸、 乙酸或三氟乙酸。
本发明另一方面涉及通式化合物 (IA)或通式化合物 (IB), 它们为合成通式化合 物 (I)的中间体:

其中:
X选自卤素;
R1选自氢原子、 烷基、 三氟甲基、 环烷基、 杂环烷基、 芳基或杂芳基, 其中烷 基、 环烷基、 杂环垸基、 芳基、 杂芳基进一步被一个或多个选自卤素、 氰基、 芳 基、 羟基或氨基的取代基所取代, 优选为三氟甲基;
R2选自氢原子、 卤素、 氰基、 氨基、 烷基、 环垸基、 杂环烷基、 芳基、 杂芳基、 -NR3R4、 -NR3C(0)R4或 -NC(0)NR3R4, 其中环烷基、 杂环烷基、 芳基、 杂芳基进 一步被一个或多个选自卤素、 氨基、 氰基、 硝基、 羟基、 烷基、 环烷基、 垸氧基、 杂芳基、 三卤代烷基、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4、 -NC(0)NR3R4、 -COR5 或 -S02R6的取代基所取代;
R3和 R4各自独立地选自氢原子、 烷基、 环烷基、 杂环烷基、 芳基、 杂芳基或 -S02R6, 其中烷基、 环烷基、 杂环烷基、 芳基或者杂芳基进一步被一个或多个选 自卤素、 羟基、 氨基、 烷氧基、 焼基、 芳基、 杂环烷基、 -S02R6、 - NR3R4、 羧酸 或羧酸酯的取代基所取代;
或者, R3和 R4—起形成一个 4〜8元杂环基, 其中 4〜8元杂环内含有一个或 多个 N、 0、 S原子, 并且 4~8元杂环上进一步被一个或多个选自卤素、 烷基、 芳基、 杂芳基、 羟基、 羰基、 氰基、 烷氧基、 羟垸基、 杂环烷基或 -NR3R4的取代 基所取代;
R5选自氢原子或烷基。
R6选自垸基或芳基, 其中芳基进一步被一个或多个烷基取代。
本发明的一个方面是涉及通式化合物 (IA)和通式化合物 (IB)的制备方法,包括以 下步骤 - 一种通式化合物 (IA)的制备方法, 该方法包括以下步骤:

OA)
将原料 H2取代的毗嗪 2-甲胺与酸酐进行反应, 生成的酰胺产物与三氯氧磷在 室温下混合搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催 化下, 氢化还原生成通式化合物 (IA)。
通式化合物 (IB)的制备方法, 该方法包括以下步骤-
 将原料吡嗪 2-甲胺与酸酐迸行反应,生成的酰胺产物与三氯氧磷在室温下混合 搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催化下, 氢气 还原后与二碳酸二叔丁酯反应生成叔丁氧基羰基保护的咪唑并 [1,5-a]哌嗪产物,然 后在 N-卤代琥珀酰亚胺条件下反应, 得到通式化合物 (IB)。
将原料吡嗪 2-甲胺与酸酐迸行反应,生成的酰胺产物与三氯氧磷在室温下混合 搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催化下, 氢气 还原后与二碳酸二叔丁酯反应生成叔丁氧基羰基保护的咪唑并 [1,5-a]哌嗪产物,然 后在 N-卤代琥珀酰亚胺条件下反应, 得到通式化合物 (IB)。
一种通式化合物 (IA)的制备方法, 该方法包括以下步骤:

将通式化合物 (IB)在钯类试剂催化下, 与硼酸或硼酸酯在微波下反应, 进行
Suzuki 偶 联 (J.Am.Chem.Soc., 2007,129, 3358-3366 ; C em. Soc. Rev., 2001,30,145-157),或与取代胺类在催化条件下进行 BuchwaW偶联 (J.Am.Chem.Soc., 2002,124, 7421-7428; J.Am.Chem.Soc, 2001,123, 7727-7429),得到产物在酸性条件 下脱掉氨基的保护基, 得到通式化合物 (IA)。
在本发明的一个方面是通式 (I)化合物的制备方法, 该方法包括以下步骤-

通式化合物 (IA)与 3-叔丁氧基羰基氨基 -4-芳基 -丁酸 (WO03004498)在缩合试 剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行缩合,得到的产物在酸性条件下脱 掉氨基保护基, 得到通式化合物 (1)。

通式化合物 (IB)脱保护基后与 3-叔丁氧基羰基氨基 -4-芳基 -丁酸在缩合试剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行反应,得到的缩合产物在钯类试剂催化
下, 与硼酸或硼酸酯在微波下反应, 进行 Suzuki偶联 (LAm.Chem.Soc., 2007,129, 3358-3366; Chem. Soc. Rev., 2001,30,145-157), 得到通式化合物 (I); 缩合产物也可 以与取代胺类在催化条件下, 进行 Buchwald 偶联 (J.Am.Chem.Soc., 2002,124, 7421-7428; J.Am.Chem.Soc, 2001,123, 7727-7429), 得到通式化合物 (I); 縮合产物 也可以在油浴中, 以八羰基二钴和氯乙酸乙酯作为催化剂, 在一氧化碳气氛下与 醇反应, 得到的取代羧酸酯化合物进一步水解成羧酸, 与碳酸铵反应生成的酰胺, 进一步脱水生成 R2为氰基的通式化合物 (1)。
本发明的一方面是通式化合物 (I)经纯化后在酸的甲醇、 二氯甲烷或乙酸乙酯 溶液中反应, 得到其酸加成产物盐。 其中所述的酸为磷酸盐、 苹果酸、 乳酸、 马 来酸、 盐酸、 甲磺酸、 硫酸、 磷酸、 柠檬酸、 酒石酸、 乙酸或三氟乙酸。
本发明的一方面是一种药用组合物, 其含有治疗有效剂量通式 (I)化合物或其 药学上可接受的盐, 及药学上可以接受的载体或赋形剂。
本发明的一方面是抑制二肽基肽酶 IV催化活性的方法, 其中包括将所述的二 肽基肽酶 IV与通式 (I)中任何一个所述的化合物或盐接触。
本发明的另一方面是通式 (I)中任何一个所述化合物、 盐或药物组合物用于治 疗 II型糖尿病、 高血糖症、 肥胖症或胰岛素抵抗症等疾病中的用途。
本发明的一方面是含有治疗有效剂量通式 (I)化合物或其药学上可接受的盐, 及药学上可以接受的载体或赋形剂的药物组合物在制备治疗 II型糖尿病、 高血糖 症、 肥胖症或胰岛素抵抗症的药物中的用途。 发明的详细说明
除非有相反陈述, 下列用在说明书和权利要求书中的术语具有下述含义。
"烷基"指饱和的脂族烃基团, 包括 1至 20个碳原子的直链和支链基团。 优 选含有 1至 10个碳原子的烷基, 例如甲基、 乙基、 丙基、 2-丙基、 正丁基、 异丁 基、 叔丁基、 戊基等。 更优选的是含有 1至 4个碳原子的低级烷基, 例如甲基、 乙基、丙基、 2-丙基、正丁基、异丁基或叔丁基等。烷基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自卤素、 氨基、 氰基、 羟基、 垸基、环焼基、杂环烷基、芳基、烷氧基、杂芳基、三卤代烷基、 -S02R6、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4或 -NC(0)NR3R4。
"环烷基"指 3至 8元全碳单环、全碳 5元 /6元或 6元 /6元稠合环或多环稠合 环("稠合"环系意味着系统中的每个环与体系中的其他环共享毗邻的一对碳原子) 基团, 其中一个或多个环可以含有一个或多个双键, 但没有一个环具有完全共轭 的 π电子系统。 环烷基的实例有环丙基、 环丁基、 环戊基、 环戊烯、 环己垸、 环 己二烯、 金刚烷、 环庚垸、 环庚三烯等。 环垸基可以是取代或未取代的, 当被取 代时, 取代基优选为一个或多个, 独立地选自卤素、 氨基、 氰基、 羟基、 烷基、 环烷基、杂环烷基、 芳基、烷氧基、杂芳基、三卤代烷基、羧酸、羧酸酯、 -COR5、
-S02R6、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4或 -NC(0)NR3R4。
"芳基"指具有至少一个芳环结构的基团, 即具有共轭的 π电子体系的芳环, 包括碳环芳基、 杂芳基和联芳基。 炔基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自卤素、 氨基、 氰基、 羟基、 烷基、 环烷基、 杂环烷基、 芳基、烷氧基、杂芳基、三卤代烷基、羧酸、羧酸酯、 -COR5、 -S02R6、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4或 -NC(0)NR3R4。
"杂芳基"指具有 1至 3个杂原子作为环原子, 其余的环原子为碳的芳基, 杂 原子包括氧、 硫和氮。 所述环可以是 5元或 6元环。 杂环芳基基团的实例包括呋 喃基、 噻吩基、 吡啶基、 吡咯、 N-烷基吡咯基、 嘧啶基、 吡嗪基、 咪唑基等。 杂 芳基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地 选自卤素、 氨基、 氰基、 羟基、 烷基、 环烷基、 杂环烷基、 芳基、 烷氧基、 杂芳 基、三卤代烷基、羧酸、羧酸酯、 -CORK -S02R6、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4 或 -NC(0)NR3R4。
"杂环烷基"指单环或稠环基团, 在环中, 具有 5至 9个环原子, 其中一个 或两个环原子选自氮、 氧或 S(0)n (其中 n是整数 0至 2) 的杂原子, 其余环原子 为碳。 这些环还可以具有一个或多个双键。 不过, 这些环不具有完全共轭的 π电 子系统。 未取代的杂环烷基包括但不限于吡咯烷基、 哌啶子基、 哌嗪子基、 吗啉 基、 硫代吗啉基、 高哌嗪其等、 杂环烷基可以是取代的或未取代的。 炔基可以是 取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自卤素、 氨基、 氰基、 羟基、 垸基、 环烷基、 杂环烷基、 芳基、 烷氧基、 杂芳基、 三卤代 烷基、 羧酸、 羧酸酯、 -COR5、 -S02R6、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4或 -NC(0)NR3R4 0
"羟基 "指 -OH基团。
"烷氧基"指 -0- (烷基)和 -0- (未取代的环烷基)。 代表性实例包括但不限 于甲氧基、 乙氧基、 丙氧基、 丁氧基、 环丙氧基、 环丁氧基、 环戊氧基、 环己氧 基等。
"卤素"指氟、 氯、 溴或碘, 优选氟或氯。
"三氟甲基"指 -C 。
"氨基"指 -NH2。
"氰基"指 -CN。
"硝基"指 -N02。
"羧酸"指 (垸基) C(=0)OH。
"羧酸酯"指 (垸基) C(=0)0 (垸基)。
"药物组合物 "表示一种或多种本文所述化合物或其生理学上 /药学上可接受 的盐或前体药物与其他化学组分的混合物, 其他组分例如生理学 /药学上可接受的 载体和赋形剂。 药物组合物的目的是促进化合物对生物体的给药。
本发明化合物的合成方法 为了完成本发明的目的, 本发明采用如下技术方案 - 本发明的通式化合物 (IA)的制备方法, 包括以下步骤:

(IA)
将原料 R2取代的吡嗪 2-甲胺与酸酐进行反应, 生成的酰胺产物与三氯氧磷在 室温下混合搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催 化下, 氯化还原生成通式化合物 (IA:)。
本发明的通式化合物 (IB)的制备方法, 包括以下步骤:

(IB) 将原料吡嗪 2-甲胺与酸酐进行反应,生成的酰胺产物与三氯氧磷在室温下混合 搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催化下, 氢气 还原后与二碳酸二叔丁酯反应生成叔丁氧基羰基保护的咪唑并 [1,5-a]哌嗪产物,然 后在 N-卤代琥珀酰亚胺条件下反应, 得到通式化合物 (IB)。
本发明的通式化合物 (IA)的制备方法, 包括以下步骤:

(旧) (IA)
将通式化合物 (IB)在钯类试剂催化下, 与硼酸或硼酸酯在微波下反应, 进行
Suzuki 偶 联(J.Am.Chem.Soc., 2007,129, 3358-3366 ; Chem. Soc. Rev., 2001,30,145-157),或与取代胺类在催化条件下进行 Buchwald偶联 (J.Am.Chem.Soc., 2002,124, 7421-7428; J.Am.Chem.Soc, 2001,123, 7727-7429),得到产物在酸性条件 下脱掉氨基的保护基, 得到通式化合物 (IA:)。
本发明的通式 0化合物的制备方法, 包括以下步骤:

通式化合物 (IA)与 3-叔丁氧基羰基氨基 -4-芳基 -丁酸 (WO03004498)在缩合试 剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行缩合,得到的产物在酸性条件下脱 掉氨基保护基, 得到通式化合物 (1)。
本发明的通式 (I)化合物的制备方法, 包括以下步骤-

通式化合物 (IB)脱保护基后与 3-叔丁氧基羰基氨基 -4-芳基 -丁酸在缩合试剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行反应,得到的缩合产物在钯类试剂催化 下, 进一步与硼酸或硼酸酯在微波下反应, 进行 Suzuki 偶联 (J.Am.Chem.Soc., 2007,129, 3358-3366; Chem. Soc. Rev., 2001,30,145-157),得到通式化合物 (I); 缩合 产物也可以与取代胺类在催化条件下, 进行 Buchwald 偶联 (J.Am.Chem.Soc., 2002,124, 7421-7428; J.Am.Chem.Soc, 2001,123, 7727-7429), 得到通式化合物 (I); 缩合产物也可以在油浴中, 以八羰基二钴和氯乙酸乙酯作为催化剂, 在一氧化碳 气氛下与醇反应, 得到的取代羧酸酯化合物进一步水解成羧酸, 与碳酸铵反应生 成的酰胺, 进一步脱水生成 R2为氰基的通式化合物 (1)。

将原料咪唑并 [1,5-a]吡嗪环硝化得到硝基取代的咪唑并 [l,5-a]吡嗪环, 然后在 Pd/C催化下, 氢气还原生成氨基取代的咪唑并 [1,5-a]哌嗪产物, 然后与 3-叔丁氧 羰基氨基 -4-芳基 -丁酸在缩合试剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下, 室温 搅拌过夜得到 R2为氨基的通式化合物 (1)。
通式化合物 (I)经纯化后在酸的甲醇、 二氯甲烷或乙酸乙酯溶液中反应, 得到 其酸加成产物盐。 实施例 化合物的结构是通过核磁共振 (NMR)或质谱 (MS)来确定的。 NMR位移 (δ)以百 万分之一 (ppm)的单位给出。 NMR的测定是用 Bruker AVANCE-400核磁仪, 测定 溶剂为氘代甲醇 (CD3OD)、 氘代氯仿 (CDC13), 六氘代二甲基亚砜 (DMSO-d6)内标 为三甲基硅烷 (TMS), 化学位移是以 l(T6(ppm)作为单位给出;
MS的测定用 FINNIGAN LCQAd (ESI)质谱仪 (生产商: Therm, 型号: Finnigan LCQ advantage MAX;
IC50值的测定用 NovoStar酶标仪 (德国 BMG公司);
薄层硅胶使用烟台黄海 HSGF254或青岛 GF254硅胶板;
柱层析一般使用烟台黄海硅胶 200~300目硅胶为载体;
DMSO-d6: 六氘代二甲基亚砜;
CD3OD: 氘代甲醇;
CDC13: 氘代氯仿;
Suzuki偶联参考文献: J.Am.Chem.Soc, 2007,129, 3358-3366; Chem. Soc. Rev. 2001,30,145-157;
Buc wald 偶联参考文献: J.Am.Chem.Soc., 2002,124, 7421-7428 ; J.Am.Chem.Soc, 2001,123, 7727-7429
制备实施例:
实施例 1
(R)-3-氨基小(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁小酮盐酸

第一步
2,2-二甲基 -5-[2-(2,4,5-三氟-苯基) -乙酰基 H ]二噁烷 -4,6-二酮
将 2,2-二甲基 -[1,3]二噁烷 -4,6-二酮 (5.69 g, 39.5 mmol)搅拌下溶解于 400 mL二 氯甲垸中, 在冰浴冷却下, 加入 2,4,5-三氟苯乙酸 (7.15 g, 37.6 mmol)和对二甲氨基 吡啶 (7.35 g, 60.2 mmol),缓慢滴加 250 mL 1 -(3-二甲基氨基-丙基) -3-乙基-碳二亚胺 盐酸盐 (8.28 g, 43.2 mmol)的二氯甲烷溶液, 室温下搅拌 36小时后, 用 5%硫酸氢 钠溶液 (250 ml 7)和饱和氯化钠溶液洗涤反应液,无水硫酸镁干燥,抽滤,减压浓 缩滤液, 得到标题产物 2,2-二甲基 -5-[2-(2,4,5-三氟-苯基) -乙酰基] -[1,3]二噁垸 -4,6- 二酮 la(11.4 g, 白色固体), 收率: 96%。
MS m/z (ESI): 315.5[M-l] o
笛— -Φ
3-氧代 -4-(2,4,5-三氟-苯基) -丁酸乙酯
将 2,2-二甲基 -5-[2-(2,4,5-三氟-苯基) -乙酰基] -[1,3]二噁烷 -4,6-二酮 la(15.72 g, 49.6 mmol)搅拌下溶解于 280 mL乙醇中, 油浴 70°C下搅拌过夜。 冷却, 减压浓縮 除去反应液中溶剂, 用硅胶柱色谱法纯化, 得到标题产物 3-氧代 -4-(2,4,5-三氟-苯 基) -丁酸乙酯 lb(12 g,黄色液体), 收率: 88%。
MS m/z (ESI): 259[M-1]。
第三步
3-氨基 -4-(2,4,5-三氟-苯基) -丁 -2-烯酸乙酯 将 3-氧代 -4-(2,4,5-三氟-苯基) -丁酸乙酯 lb(24.6 g, 94.5 mmol)溶解于 240 mL甲 醇中,加入醋酸铵 (36.4 g, 473 mmol),加热回流 3小时后, TLC跟踪反应至原料消 失后, 蒸干溶剂, 加入 lOO mL水, 用乙酸乙酯 (200 mLx3)萃取, 合并有机相, 用 200 mL饱和氯化钠溶液洗涤, 无水硫酸镁干燥, 过滤, 滤液减压浓缩得到的淡黄 色固体, 加入 50 mL乙酸乙酯, 在 80°C下溶解, 加入 50 mL正己烷, 晶种, 冷却 至室温下, 0.5小时后, 加入 lOO mL正己烷, 置于冰箱中过夜, 抽滤, 得到标题 产品 3-氨基 -4-(2,4,5-三氟-苯基) -丁 -2-烯酸乙酯 lc(19.5 g, 白色固体), 收率: 80%。 MS m/z (ESI): 260.1 [M+1]
第四歩
(R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸乙酯 将 3-氧代 -4-(2,4,5-三氟-苯基) -丁酸乙酯 lc(4.1 g, 15.8 mmol)加入高压釜中, 再 加入 70 mL甲醇 (通氮气 1小时后使用),二碳酸二叔丁酯 (3.8 g, 17.4 mmol),氯 (1,5- 环辛二烯)铑 (I)二聚体 (32 mg, 0.0632 mmol)和 (R)小 [(S)-2- (二苯基膦基)二茂铁基] - 乙基-叔丁基膦 (68 mg, 0.126 mmol), 在 30°C下, 90Psi的氢气中反应 24小时, 过 滤, 蒸干溶剂, 在 50°C下加入 34 mL甲醇, 完全溶解后加入 12 mL水, 在冰箱中 过夜, 过滤, 用甲醇 /水 (v:v=3:2)混合溶剂洗涤固体产品, 抽干溶剂, 得到标题产 物 (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸乙酯 ld(4 g,淡黄色固体),收率: 70%。
MS m/z (ESI): 362.4[M+1]。
第五步 -
(R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 将 (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸乙酯 ld(10 g, 27.7 mmol)和 氢氧化钠 (3.32 g, 83 mmol)搅拌下溶解于 100 mL甲醇和 50 mL水的混合溶剂中, 在 30°C下反应 4小时后, 减压浓缩除去部分溶剂, 加入少量水, 在冰浴下加入 10 %的硫酸氢钠溶液调节溶液 pH为 2.5, 用乙酸乙酯 (200 mLx3)萃取, 合并有机相, 用 200 mL饱和氯化钠溶液洗涤, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用乙
酸乙酯 /正己烷重结晶 2 次, 得到标题产物 (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯 基) -丁酸 le(9.7 g, 白色固体), 直接用于下一步反应。
MS m/z (ESI): 332.3[M- l] o
第六步
2,2,2-三氟 -N-吡嗪 -2-甲基-甲酰胺
将 C-吡嗪 -2-甲胺 lf(1.0 g, 9.2 mmol)加入到反应瓶中, 冰浴冷却至 0。C, 滴加 10 mL三氟乙酸酐, 室温下搅拌 2小时, 薄层色谱跟踪反应, 原料消失, 反应液减 压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 2,2,2-三氟 吡嗪 -2-甲基-甲酰胺 lg(2.0 g,棕色油状物), 收率: 69 %。
MS Jz (ESI): 206· 1 [Μ+1]。
第七步
3-三氟甲基-咪唑并 [1 ,5-a]吡嗪
室温条件下, 将 2,2,2-三氟 -N-吡嗪 -2-甲基-甲酰胺 lg (21.0 g, 100 mmol)加入 反应瓶中,加入 100 mL三氯氧憐,室温搅拌 30分钟后,加入五氧化二磷 (17.8 g, 125 mmol),加热回流反应 5小时,薄层色谱跟踪反应至原料消失,除去溶剂三氯化磷, 反应体系用去离子水慢慢淬灭反应, 再用 20 %氢氧化钠溶液在冰浴中调节 pH为 5-6, 用乙酸乙酯 (250 mLx4)萃取, 合并有机相, 用无水硫酸镁干燥, 抽滤, 减压 浓缩滤液, 用硅胶柱色谱法纯化, 得到标题产物 3-三氟甲基-咪唑并 [1,5-a]吡嗪 lh(12.0 g, 黄色固体), 收率: 65 %。
MS mlz (ESI): 188.0[M+1] .
第八步
3-三氟甲基-咪唑并 [1,5-a]哌嗉
将 3-三氟甲基-咪唑并 [1,5-a]吡嗪 lh (12.0 g, 64.2 mmol)搅拌下溶解于 150 mL 无水乙醇中, 加入 500 mg 10%钯 /碳, 抽换气三次, 在氢气氛下搅拌过夜。 用粗硅 胶将反应液过滤, 除去催化剂, 减压蒸干滤液, 得到标题产物 3-三氟甲基-咪唑并 [l,5-a]哌嗪 li(12.2 g, 棕色固体), 收率: 99%。
第九步
(R)-[3-氧代 -1-(2,4,5-三氟苄基) -3-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -丙基] - 氨基甲酸叔丁酯
氮气氛下,将 3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(8.6 g, 45 mmol) (根 据 WO03004498制备而得)、 9.4 mL三乙胺搅拌下溶解于 300 mL二氯甲烷中, 室 温下搅拌 5分钟后, 再分别依次加入 3-三氟甲基-咪唑并 [1,5-a]哌嗪 li(15.0 g, 45 mmol)及双 (2-氧代 -3-噁唑烷基)次膦酰氯 (17.1 g, 67.3 mmol), 室温下反应 2小时, 薄层色谱跟踪反应原料消失, 旋蒸反应液, 除去溶剂, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (RM3-氧代 -1 -(2,4,5-三氟苄基) -3-(3-三氟甲基-咪唑并 [1,5-a] 哌嗪 -7-基) -丙基] - 基甲酸叔丁酯 lj (20.0 g, 白色固体), 收率: 88 %。
第十步
(R) 3-氨基 -l-(3-三氟甲基-咪唑并 [1,5-a]脈嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸 将 (R)- [3-氧代小 (2,4,5-三氟苄基) -3-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -丙 基] -氨基甲酸叔丁酯 lj(300 mg, 0.59 nmiol)搅拌下溶解于 10 mL的 43N氯化氢甲醇 溶液, 室温下搅拌过夜, 薄层色谱跟踪反应, 原料消失, 蒸干溶剂, 用硅胶柱色 谱法纯化, 得到标题产物 (R)-3-氨基 -1-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基) _ (2,4,5-三氟苯基)—丁小酮盐酸盐 1(260 mg, 白色固体), 收率: 99%。
!HNMR (400MHz, CD3OD): δ 7.08(s, 1Η), 7.06(s, 1Η), 6.80(t, 1H), 4.51-4.59 (m, 2H), 4.09(s, 1H), 3.97(s, 1H), 3.70(d, 2H), 3.56(s, 1H), 2.75-2.78(m, 2H), 2.59-2.64(d, 2H) 实施例 2
(R)-3-氨基 -1-(1-氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1- 酮二盐酸盐

第一步
1-硝基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪
3-三氟甲基-咪唑并 [1,5-a]吡嗪 lh(2 g, 10.7 mmol)加入到 25 mL的单口烧瓶 中, 搅拌下加入 5 mL发烟硝酸和 5 mL浓硫酸, 所得的混合物加热至 110°C, 搅 拌反应 1.5小时后反应完毕,将反应液冷却至室温后,倒入冰浴冷却的 50 mL浓氨 水中, 搅拌均匀后用乙酸乙酯萃取(250 mLx3), 合并的有机相, 用无水硫酸镁干 燥, 过滤, 减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-硝基 -3- ' 三氟甲基-咪唑并 [1,5-a]吡嗪 2a (250 mg,棕色油状液体), 收率: 10%。
MS m z (ESI): 233[M+1]
第二步
3-三氟甲基-咪唑 [1,5-a]哌嗪小胺
将 1-硝基 -3-三氟甲基-咪唑并 [1,5- a]吡嗪 2a(250 mg, 1.05 mmol)搅拌下溶解于 30 mL无水乙醇中, 加入 100 mg 10%Pd/C, 抽换气三次, 在氢气氛下室温搅拌过 夜, 用硅藻土将反应液过滤, 减压蒸干滤液, 用硅胶柱色谱法纯化, 得到标题产 物 3-三氟甲基 -咪唑 [1,5-a]哌嗪 -1-胺 2b(65 mg, 白色固体), 收率: 31.6%。
MS m/z (ESI): 207[M+1]
第三步
(R)- [3-(l-氣基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -l-(2,4,5-三氟苄基) -丙基
1:!-氨基甲酸叔丁酯
氮气氛下, 将 3-三氟甲基 -咪唑 [1,5-a]哌嗪- 1-胺 2b(60 mg, 0.29 mmol)搅拌下溶 解于 20 mL二氯甲烷中,室温下依次加入二异丙基乙胺 (85 L, 0.486 mmol), (R)-3- 叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(81 mg, 0.243 mmol)和双 (2-氧代 -3-噁 唑烷基)次膦酰氯(92 1¾,0.36 11^101)。 室温下搅拌过夜, 薄层色谱跟踪反应, 原料 消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-[3-(l-氨基 -3- 三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基 1]-氨基甲酸叔丁 酯 2c(125 mg, 白色固体), 收率: 98%。
MS m/z (ESI): 522[M+1]
第四步
(R)-3-氨基 氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1- 酮二盐酸盐
将 (R)-[3-(l-氨基 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄 基) -丙基 1]-氨基甲酸叔丁酯 2c(125 mg, 0.24 mmol)搅拌下溶解于 10 mL 4.3N氯化氢 甲醇溶液, 室温下搅拌过夜, 薄层色谱跟踪反应, 原料消失, 蒸干溶剂, 用硅胶 柱色谱法纯化, 得到标题产物 (R)-3-氨基 -1-(1-氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1-酮二盐酸盐 2(117 mg, 白色固体), 收率: 98%。 MS m/z (ESI): 422[M+1]
'HNMR (400MHz, CD3OD): δ 7.35(m, 1H), 7.18(m, 1H), 5.15(m, 2H), 4.25(m, 2H), 4.05(m, 2H), 3.91(s, 1H), 3.05(s, 2H), 2.92(m, 2H) 实施例 3
(R)-3-氨基 -1-(1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-)-4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐


3-环戊基 - N*2*-亚乙基 -N* 1 *-亚甲基 -丙烯 -1 ,2,3-三胺 将 2-氰基吡嗪 3a(6.3 g, 0.06 mol)溶解于 80 mL甲苯中, 冷却溶液至 -10°C, 缓 慢滴加环戊基溴化镁(33 mL, 66 mmol), 滴加完毕搅拌 30分钟后, 向反应液中滴 加 40 mL异丙醇, 搅拌 30分钟, 继续滴加 40 mL乙醇, 0°C下, 向反应液中加入 硼氢化钠 (3.18 g, 84 mmol), 室温下搅拌过夜, 向反应液中加入丙酮、 甲醇和水猝 灭反应, 直至无气泡产生, 减压浓缩蒸掉大部分有机溶剂, 水相用乙酸乙酯萃取 (250 mLx3 ), 合并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩, 用硅胶柱色谱 法纯化所得残余物,得到标题产物 3-环戊基 -N*2*-亚乙基 -N*l*-亚甲基 -丙烯 -1,2,3- 三胺 3b ( 1.5 g,棕色油状液体), 收率: 15 %。
MS m/z (ESI): 178[M+1]
第二步
N- (环戊基吡嗪 -2-甲基) -2,2,2-三氟乙酰胺
将 3-环戊基 -N*2*-亚乙基 -N*l*-亚甲基 -丙烯 -1,2,3-三胺 3b (5.2 g, 29 mmol) 加入到反应瓶中, 冰浴冷却至 0Ό, 滴加 40 mL三氟乙酸酐, 室温下搅拌 2小时, 薄层色谱跟踪反应, 原料消失, 反应液减压浓缩, 得到的黑色油状物 N- (环戊基吡 嗉 -2-甲基) -2,2,2_三氟乙酰胺 3c, 不经分离直接进行下一步反应。
第三步
1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪
在冰浴冷却下, 将 N- (环戊基吡嗪 -2-甲基) -2,2,2-三氟乙酰胺 3c滴加入 10 mL 三氯氧磷中, 迅速加入五氧化二磷 (2 g, 14.6 mmol), 加热回流 4小时后反应完毕, 反应液减压浓缩, 加入 5 mL水, 用浓氨水调 pH=9, 用乙酸乙酯 (150 ml 4)萃取, 合并有机相, 用 20 mL饱和氯化钠溶液洗涤, 用无水硫酸镁干燥, 抽滤, 减压浓 缩滤液, 用硅胶柱色谱法纯化, 得到标题产物 1-环戊基 -3-三氟甲基-咪唑并 [1,5-a] 吡嗪 3d(166 mg,红色油状物), 收率: 25.7%。
MS m/z (ESI): 256[M+1]。
第四步
1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪
将 1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 3d ( 166 mg, 0.65 mmol)搅拌下溶 解于 10 mL无水乙醇中, 加入 40 mg 10%Pd/C, 抽换气三次, 在氢气氛下室温搅 拌过夜, 用硅藻土将反应液过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余 物, 得到标题产物 1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 3e(110 mg, 红色油状液 体), 收率: 65 %。
MS m/z (ESI): 260[M+1]
第五步
(R)-[3-(l-环戊基 1-3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯
氮气氛下, 将 1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 3e(156 mg, 0.47 mmol) 搅拌下溶解于 20 mL二氯甲垸中, 室温下依次加入二异丙基乙胺 (0.29 mL, 0.63 mmol), (R)-3-叔丁氧羰基氮基 -4-(2,4,5-三氟-苯基) -丁酸 le(156 mg, 0.47 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (160 mg, 0.63 mmol), 室温下搅拌过夜, 薄层色谱跟 踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到 标题产物 (R)-[3-(l-环戊基 1-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 3f(254 mg, 白色固体), 收率: 94%。
MS m/z (ESI): 575[M+1]
第六步
(R)-3-氨基小 (1-环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-)-4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐
将 (R)-[3-(l-环戊基 1-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟 苄基) -丙基] -氨基甲酸叔丁酯 3f(254 mg, 0.44 mmol)搅拌下溶解于 10 mL 4.3 氯化 氢甲醇溶液中, 室温下搅拌 2小时后反应完毕, 蒸干溶剂, 用硅胶柱色谱法纯化 所得残余物, 得到标题产物 (R)-3-氨基 环戊基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-)-4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 3(216 mg, 白色固体), 收率: 96%。
MS m/z (ESI): 475[M+1]
1HNMR (400MHz, CD3OD): δ 7.39(q, 1H), 7.18-7.23(m, 1H), 4.27-4.37(d, 2H), 3.66-4.05 (m, 5H), 3.07-3.12(m, 3H), 2.89-2.96(m, 2H), 2.06(s, 2H), 1.86(s, 2H), 1.71(s: 4H) 实施例 4
(R)-3-氨基小 (1-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁小 酮盐酸盐


N*2*-亚乙基 -N*l*-亚甲基 -3-苯基 -丙烯 -1,2,3-三胺 将 2-氰基吡嗪 3a(3.15 g, 0.03 mol)溶解于 100 mL甲苯中, 冷却溶液至 -10Ό, 缓慢滴加苯基溴化镁( 11 mL, 33 mmol), 40分钟后滴加完毕, 搅拌 1小时后,向反 应液中滴加 40 mL异丙醇, 搅拌下继续加入硼氢化钠 (1.59 g, 42 mmol), 室温下搅 拌过夜, 向反应液中加入丙酮、 甲醇和水猝灭反应, 直至无气泡产生, 减压浓缩 蒸掉大部分有机溶剂, 水相用乙酸乙酯萃取(250 mLx3), 合并有机相, 用无水硫 酸镁干燥,过滤,减压浓缩,用硅胶柱色谱法纯化所得残余物,得到标题产物 N*2*- 亚乙基 -N*l *-亚甲基 -3-苯基 -丙烯 -1,2,3-三胺 4a ( 1.38 g,棕色油状物), 收率- 25 %。
MS m/z (ESI): 186[M+1]
第二步
2,2,2-三氟 (苯基吡嗪 -2-甲基) -三氟乙酰胺 将 N*2*-亚乙基 -N*l*-亚甲基 -3-苯基 -丙烯 -1,2,3-三胺 4a (650 mg, 3.5 mmol) 加入到反应瓶中, 冰浴冷却至 0Ό , 滴加 5 mL三氟乙酸酐, 室温下搅拌 1小时, 薄层色谱跟踪反应, 原料消失, 反应液减压浓缩, 得到标题产物 2,2,2-三氟 (苯 基吡嗪 -2-甲基) -三氟乙酰胺 4b(950 mg,棕色固体), 不经分离直接进行下一步反 应。
第三步
1-苯基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪
在冰浴冷却下, 将 2,2,2-三氟 -N- (苯基吡嗪 -2-甲基) -三氟乙酰胺 4b(940 mg, 3.5 mmol)放入反应瓶中,向其中滴加 10 mL三氯氧磷,迅速加入五氧化二磷 (994 mg, 7 mmol), 加热回流 4小时后反应完毕, 反应液减压浓縮, 加入 5 mL水, 用浓氨水 调 pH=9, 用乙酸乙酯 (150 mLx4)萃取, 合并有机相, 用 20 mL饱和氯化钠溶液洗 涤, 收集有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色谱法纯 化所得残余物, 得到标题产物 1-苯基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 4c(700 mg,黄 色固体), 收率: 76%。
MS m/z (ESI): 264[M+l ] c
第四步
1-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪
将 1-苯基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 4c(300 mg, 1.14 mmol)搅拌下溶解于 5 mL无水乙醇中, 加入 90 mg lO%Pd/C, 抽换气三次, 在氢气氛下室温搅拌过夜, 用硅藻土将反应液过滤, 减压蒸干滤液, 得到粗品标题产物 1-苯基 -3-三氟甲基-咪 唑并 [1,5-a]哌嗪 4d(290 mg, 白色固体), 不经分离直接进行下一歩反应。
第五步
(R)-[3-氧代 -3-(l-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 )小(2,4,5-三氟苄基) -丙 基] -氨基甲酸叔丁酯
氮气氛下,将 1-苯基 -3-三氟甲基-咪唑并 [1 ,5- a]哌嗪 4d(150 mg,0.5 mmol)搅拌 下溶解于 5 mL二氯甲烷中,依次加入二异丙基乙胺 (0.35 mL, 2 mmol),(R)-3-叔丁 氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(167 mg, 0.5 mmol)和双 (2-氧代 -3-噁唑烷基) 次膦酰氯 (191 mg, 0.75 mmol), 室温下搅拌过夜, 薄层色谱跟踪反应, 原料消失, 将反应液减压浓缩, 得到粗品标题产物 (R)-[3-氧代 -3-(1-苯基 -3-三氟甲基-咪唑并 [l,5-a;|哌嗉 -7-基)小(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 4e(310 mg, 白色固体), 不经分离直接进行下一步反应。
MS m/z (ESI): 583[M+1]
第六步
(R)-3-氨基 -1-(1-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐
将 (R [3-氧代 -3-(1-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 )小(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 4e(300 mg, 0.52 mmol)搅拌下溶解于 10 mL 4.3N氯化氢 甲醇溶液中, 室温下搅拌 2小时后反应完毕, 蒸干溶剂, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R)-3-氨基 苯基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基) (2,4,5-三氟苯基) -丁小酮盐酸盐 4(200 mg, 白色固体), 收率: 74.3 %。
MS m/z (ESI): 483 [M+l]
1HNMR (400MHz, CD3OD): δ 7.61 (d, 2H), 7.45(m, 2H), 7.36(m, 2H), 7.08-7.22 (m, 1H), 4.96-5.05(m, 2H), 4.27-4.36 (m, 2H), 3.90-4.10(m, 3H), 2.90-3.09(m, 4H) 实施例 5
(R)-3-氨基小 (1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐

N'*2*-亚乙基 - N'*l*-亚甲基-丁 -1-烯 -1,2,3-三胺 将 2-氰基吡嗪 3a(l .05 g, 10 mmol)溶解于 30 mL甲苯中,冷却溶液至 -10°C,缓 慢滴加甲基溴化镁 (7.9 mL, 11 mmol), 30分钟后滴加完毕, 搅拌反应 30分钟后, 向反应液中滴加 12 mL乙醇,搅拌下继续加入硼氢化钠 (530 mg, 14 mmol),室温下 搅拌过夜, 在反应液中加入丙酮、 甲醇和水猝灭反应, 直至无气泡产生。 减压下 蒸掉大部分有机溶剂, 水相用二氯甲烷萃取 (50 mLx3), 合并的有机相用无水硫酸 镁干燥, 过滤, 减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 N*2*- 亚乙基 -N*l*-亚甲基-丁 -1-烯 -1,2,3-三胺 5a(290 mg,黄色油状物), 收率: 25%。 MS m/z (ESI): 124[M+1]
第二步
2,2,2-三氟 -N-(l-吡嗪 -2-乙基)-乙酰胺
将 N*2*-亚乙基 -N*l*-亚甲基-丁小烯 -1,2,3-三胺 5a (2.6 g; 21.1 mmol)加入 到反应瓶中, 冰浴冷却至 0°C, 滴加 20 mL三氟乙酸酐, 室温下搅拌反应 3小时, 薄层色谱跟踪反应, 原料消失, 反应液减压浓缩, 用硅胶柱色谱法纯化所得残余 物, 得到标题产物 2,2,2-三氟 -N-(l-P比嗪 -2-乙基) -乙酰胺 5b(1.8 g,棕红色油状液 体), 收率: 38.9%。
MS m/z (ESI): 220[M+1]
第三步 '
1-甲基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪
在冰浴冷却下,将 2,2,2-三氟 吡嗪 -2-乙基) -乙酰胺 5b(1.8 g, 8.2 mmol)放入 到反应瓶中,向其中滴加 20 mL三氯氧磷,迅速加入五氧化二磷 (2.3 g, 16.4 mmol), 加热回流 5小时后反应完毕, 反应液减压浓缩, 加入 5 mL水, 用浓氨水调 pH-9, 用乙酸乙酯 (50 mLx5)萃取, 合并有机相, 用 20 mL饱和氯化钠溶液洗涤, 收集有 机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余
物, 得到标题产物 1-甲基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 5c(130 mg,棕色油状液 体), 收率: 76%
MS m/z (ESI): 202[M+1]。
第四步
1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪
将 1-甲基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 5c(170 mg, 0.85 mmol)搅拌下溶解于 10 mL无水乙醇中, 加入 35 mg 10%Pd/C, 抽换气三次, 在氢气氛下室温搅拌反应 过夜, 用硅藻土将反应液过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 5d(40 mg, 白色固体), 收率: 23.1 %。
MS m/z (ESI): 206[M+1]
第五步
(R)-[3-(l-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟苄基) -丙 基] -氨基甲酸叔丁酯
氮气氛下, 将 1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 5d(40 mg, 0.2 mmol)搅拌 下溶解于 5 mL二氯甲烷中, 室温下依次加入二异丙基乙胺 (0.07 mL, 0.4 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(100 mg, 0.3 mmol)和双 (2-氧代 -3-噁唑垸基)次膦酰氯 (100 mg, 0.4 mmol), 室温下搅拌 2小时反应完毕, 将反应液 减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-(l-甲基 -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 5e(310 mg,黄色油状液体), 收率: 76.9%。
MS m/z (ESI): 521[M+1]
第六步
(R)-3-氨基小(1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐
将 (R)-[3-(l-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 5e(80 mg, 0.15 mmol)搅拌下溶解于 5 mL 4.3N氯化氢甲 醇溶液中, 室温下搅拌 2小时后反应完毕, 蒸干溶剂, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (R)-3-氨基 -1-(1-甲基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基) -4-(2,4,5-三氟苯基)-丁小酮盐酸盐 5(30 mg, 白色固体), 收率: 43.9%。
MS m/z (ESI): 421 [M+1]
'HNMR (400MHZ, CD3OD): δ 7.39(m, 1H), 7.20(m, 1H), 4.82(m, 2H), 4.40(s, 1H), 4.3 l(s, 1H), 3.91-4.06(t, 3H), 3.10(m, 2H), 2.94(m, 2H), 2.3 l(m, 3H) 实施例 6
(R)-3-氨基 -l-[l-(4-氨基苯基)- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯
基) -丁 -1-酮二盐酸盐

第一步
1-(4-硝基苯基) -3-三氟甲基-咪唑并 [1,5-a]吡嗪 在冰浴冷却下, 在 50 mL的烧瓶中依次加入 2 mL浓硝酸, 2 mL浓硫酸和 1- 苯基 -3-三氟甲基-咪唑并 [1,5-a]吡嗪 4c(220 mg, 0.836 mmol), 冰浴下搅拌 1小时后 反应完毕, 将反应液滴加至冰浴冷却下的 100 mL浓氨水中, 用乙酸乙酯萃取 (25 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩滤液, 得到粗品标题产 物 1-(4-硝基苯基) -3-三氟甲基-咪唑并 [1,5-a]吡嗪 6a(240 mg,黄色固体), 不经分离 直接进行下一步反应。
MS m/z (ESI): 309[M+1]
第二步
4-(3-三氟甲基 咪唑并 [1,5-a]哌嗪 -1-基) -苯胺 将粗品 1-(4-硝基苯基) -3-三氟甲基-咪唑并 [l,5-a]吡嗪 6a(240 mg, 0.779 mmol) 搅拌下溶解于 5 mL无水乙醇中, 加入 72 mg lO%Pd/C, 抽换气三次, 在氢气氛下 室温搅拌过夜, 用硅藻土将反应液过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 4-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯胺 6b(150 mg, 白色固体), 收率: 68.5 %。
MS m/z (ESI): 283[M+1]
第三步
RM3-[l-(4-氨基苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 :l-(2,4,5-三氟苄 基) -丙基 } -氨基甲酸叔丁酯
氮气氛下,将 4-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯胺 6b(150 mg, 0.5 mmol) 搅拌下溶解于 5 mL二氯甲烷中, 依次加入二异丙基乙胺 (0.35 mL, 2 mmol), (R)-3- 叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(167 mg, 0.5 mmol)和双 (2-氧代 -3-噁唑 烷基)次膦酰氯 (191 mg, 0.75 mmol),室温下搅拌反应过夜, 薄层色谱跟踪反应, 原 料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(4-氨基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5-三氟苄 基) -丙基 氨基甲酸叔丁酯 6c(150 mg, 白色固体), 收率: 50.1 %。
MS m/z (ESI): 598[M+1]
第四步
(R)-3-氨基 -l-[l-(4-氨基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮二盐酸盐
将 (RM3-[l-(4-氨基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 6c(250 mg, 0.42 mmol)搅拌下溶解于 5 mL 4.3N 氯化氫甲醇溶液中, 室温下搅拌 2小时后反应完毕, 蒸干溶剂, 用硅胶柱色谱法 纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-(1-苯基 -3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -7-基) -4-(2,4,5-三氟苯基) -丁小酮二盐酸盐 6(150 mg, 白色固体), 收率: 62.5%。 MS m/z (ESI): 498[M+1]
1應 MR (400MHz, CD3OD): 87.80(d, 2Η), 7.48(m, 2H), 7.40(m, 1H), 7.19 (m, 1H), 4.97-5.07(m, 2H), 4.27-4.37 (m, 2H), 3.85-4.09(m, 3H), 2.93-3.10(m, 4H) 实施例 7
(R)- 3-氨基小 [l-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯
, 将 3-三氟甲基-咪唑并 [1,5-a]吡嗪 lh(3.5 g, 18.7 mmol)溶解于 50 mL乙醇中,搅 拌下加入 0.5 g Pd/C, 抽换气三次, 在氢气氛下搅拌过夜, 反应完毕, 用硅藻土将 反应液过滤, 减压蒸干滤液, 得到的残留物用 100 mL乙醇洗涤, 搅拌下向得到的 溶液中逐渐滴加二碳酸二叔丁酯 (6.2 g, 28.1 mmol)的 100 mL乙醇溶液, 滴加完毕 后, 继续搅拌 30分钟后反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残 余物, 得到标题产物 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7a(3.7 g, 白色固 体), 收率: 68%。
第二步
1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯
在 100 mL干燥的烧瓶中加入 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7a(300 mg, 1.04 mmol)和 50 mL乙醇, 搅拌溶解后, 加入 N-溴琥珀酰亚胺 (369 mg, 2.08 mmol), 得到的混合物在室温下搅拌, 1小时后反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -7-羧酸叔丁酯 7b(220 mg, 白色固体), 收率: 57.8%。
第三步
1-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 3-甲氧基苯硼酸 (133.6 mg, 0.89 mmol), 四三苯基膦化钯 (92.8 mg, 0.083 mmol), 碳 酸钾 (220.8 mg, 1.62 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管 中, 在氩气保护下, 120°C, 在微波下反应 15分钟后, 反应完毕, 将反应液在减 压下浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(3-甲氧基苯基) -3- 三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7c(300 mg, 白色固体), 收率: 93.5%。 MS m/z (ESI): 398[M+1]
第四步
1-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 将 1-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7c(300 mg, 0.756 mmol)溶解于 30 mL二氯甲烷中,搅拌下滴加三氟乙酸 (2.6 g, 22.6 mmol),所 得的溶液继续搅拌 5小时后反应完毕, 减压浓缩反应液, 得到粗品标题产物 1-(3- 甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 7d(300 mg,黄色油状物), 不经分离直接进行下一步反应。 .
第五步
(R)-{3-[l-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 苄基) -丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 1-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐
7d(271 mg, 0.81 mmol)搅拌下溶解于 30 mL二氯甲烷中, 依次加入二异丙基乙胺 (0.49 g, 4.9 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(310 mg, 0.81 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (200 mg, 1.21 mmol), 室温下搅拌反应过 夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (R)-{3-[l-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 7e(215 mg, 白色固体),收率: 45%。
MS m/z (ESI): 613[M+1]
第六步
(R)-3-氨基 -1-[1-(3-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l -(3-甲氧基苯基 )-3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 基 ]-3-氧代 小(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 7e(215 mg, 0.35 mmol)溶解于 4 mL 4.3N 氯化氢甲醇溶液中, 搅拌反应 2小时后反应完毕, 减压浓缩反应液, 用硅胶柱色 谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(3-甲氧基苯基) -3-三氟甲基- 咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁- 1-酮盐酸盐 7(140 mg,黄色固体), 收率: 72.6%。
MS m/z (ESI): 513[M+1]
!HNMR (400MHz, CD3OD): δ 6.91-7.37(m, 6H), 4.82-5.04(m, 2H), 4.26-4.34(t, 3H), 3.84-3.98 (m, 6H), 1.28-1.32(m, 3H) 实施例 8
(R)-3-氨基 -l-[l-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基]- 4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐

第一步
1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(200 mg, 0.54 mmol), 4-氟苯硼酸 (83 mg, 0.594 mmol), 四三苯基膦化钯 (62 mg, 0.054 mmol),碳酸钾 (149 mg, 1.08 mmol), 1.5 mL乙二醇二甲醚和 1.5 mL水放入 20 mL微波反应管中, 在 氩气保护下, 120°C, 在微波下反应 10分钟后, 反应完毕, 反应液用乙酸乙酯萃 取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 抽滤, 减压浓縮滤液, 用硅胶柱 色谱法纯化所得残余物,得到标题产物 1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 8a(250 mg, 白色固体), 收率: 60%。
MS m/z (ESI): 386[M+1]
第二步
1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 将 1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 8a(250 mg, 0.649 mmol)溶解于 5 mL二氯甲烷中, 搅拌下加入 11 mL三氟乙酸, 室温下搅拌 1.5小 时后反应完毕, 减压浓缩反应液, 得到粗品标题产物 1-(4-氟苯基 )-3-三氟甲基-咪 唑并 [1,5-a]哌嗪三氟乙酸盐 8b(450 mg,黄色固体),不经分离直接进行下一步反应。
第三步
(R)-{3-[l-(4-氟苯基 )- 3-三氟甲基 -咪唑 [1,5-a]呢嗪 -7-基] -3-氧代 -l-(2,4,5-三氟 基) - 丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 8b(450 mg, 0.649 mmol)搅拌下溶解于 10 mL二氯甲烷中, 依次加入二异丙基乙胺 (0.51 g, 3.9 mmol), (R)- 3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(216 mg, 0.65 mmol) 和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (248 mg, 0.975 mmol), 室温下搅拌反应 1小时 后, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (R)-{3-〖l-(4-氟苯基 )-3-三氟甲基-咪唑 [1,5-a]哌嗪- 7-基〗 -3-氧 代小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 8C(330 nig, 白色固体), 收率: 84%。 MS m/z (ESI): 601 [ +l]
第四步
(R)-3-氨基 -1-[1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(4-氟苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基]- 3-氧代 -1-(2,4,5-三氟 苄基) -丙基 } -氮基甲酸叔丁酯 8c(330 mg, 0.55 mmol)溶解于 5 mL 4.3 氯化氢甲醇 溶液中, 搅拌反应 2小时后反应完毕, 减压浓縮反应液, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(4-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]—4— (2,4,5-三氟苯基) -丁 -1-酮盐酸盐 8(60 mg,黄色固体), 收率: 20.3 % MS m/z (ESI): 501 [M+l]
1HNMR (400MHz, CD3OD): δ 7.63(d, 2H), 7.35(m, 1H), 7.19(m, 3H), 4.94-5.08(m, 2H), 4.27-435 (m, 2H), 3.90-4.10(m, 3H), 2.95-3.08(m, 4H) 实施例 9
(R)-3-氨基 -1 -[1 -(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一歩
1-(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 4-
甲氧基苯硼酸 (167 mg, 1.7 mmol), 四三苯基膦化钯 (116 mg, 0.1 mmol),碳酸钾 (276 mg, 2 mmol), 3 mL 乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 在氩气保 护下, 120°C,微波下反应 12分钟后,反应完毕,反应液用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 1-(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸 叔丁酯 9a(250 mg, 白色固体), 收率: 63 %。
MS m/z (ESI): 398[M+1]
第二步
1-(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 将 1-(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 9a(250 mg,
0.629 mmol)溶解于 5 mL二氯甲烷中, 搅拌下滴加三氟乙酸 (1.5 mL, 18.9 mmol), 所得的溶液继续搅拌 1小时后反应完毕, 减压浓缩反应液, 得到标题产物 1-(4-甲 氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 9b(400 mg,黄色固体), 产物 不经分离直接进行下一步反应。
第三步
(R)-{3-[l-(4-甲氧基苯基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 苄基) -丙基 氨基甲酸叔丁酯
氮气氛下, 将 1-(4-甲氧基苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 9b(400 mg, 0.63 mmol)搅拌下溶解于 10 mL二氯甲烷中, 依次加入二异丙基乙胺 (0.49 g, 3.78 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(210 mg, 0.63 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (240 mg, 0.945 mmol), 室温下搅拌反 应 1 小时, 薄层色谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法 纯化所得残余物, 得到标题产物 (R)-{3-[l-(4-甲氧基苯基) -3-三氟甲基 -咪唑 [1,5-a] 哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 9c(280 mg,黄色固 体), 收率: 72.7%。
MS lz (ESI): 613[M+1]
第四步
(R)-3-氨基 -l-[l-(4-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基]斗(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(4-甲氧基苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基 ]-3-氧代
-1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 9c(280 mg, 0.45 mmol)溶解于 5 mL 4.3N 氯化氢甲醇溶液中, 溶液在搅拌下反应 2小时后反应完毕, 减压浓缩反应液, 用 硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1 -[1 -(4-甲氧基苯基) -3-三 氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 9(120 mg,黄色 固体), 收率: 48 %
MS m/z (ESI): 513[M+1]
lHNMR (400MHz, CD3OD): δ 7.53(d, 2H), 7.10-7.45(m, 2H), 7.03(m, 2H),
4.92-5.01(m, 2H), 4.28-4.37 (m, 2H), 3.83-4.10(m, 6H), 2.90-3.10(m, 4H) 实施例 10
(R)-3-氨基小[1-(2-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
1-(2-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 2- 甲氧基苯硼酸 (167 mg, 1.1 mmol), 四三苯基膦化钯 (116 mg, 0.1 mmol),碳酸钾 (276 mg, 2 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氩气保护 下, 120°C,在微波下反应 50分钟后,反应完毕,反应液用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 1-(2-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸 叔丁酯 10a(300 mg, 白色固体), 收率: 75 %。
MS m/z (ESI): 398[M+1]
第二步
l-(2-甲氧基苯基) -3-三氟甲基 咪唑并 [1,5-a]哌嗪 将 1-(2-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 10a(300 mg, 0.756 mmol)溶解于 5 mL二氯甲烷中, 搅拌下滴加三氟乙酸 (2.6 g, 22.6 mmol), 所 得的溶液继续搅拌 1 小时后反应完毕, 减压浓缩反应液, 得到黄色粘稠状标题产 物 1-(2-甲氧基苯基) -3-三氟甲基 -5咪唑并 [1,5-a]哌嗪三氟乙酸盐 10b, 产物不经分 离直接进行下一步反应。
第三步
(R)-{3-[l-(2-甲氧基苯基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 基) -丙基 氨基甲酸叔丁酯
氮气氛下, 将 1-(2-甲氧基苯基) -3-三氟甲基 咪唑并 [1,5-a]哌嗪三氟乙酸盐 10b(308 mg, 0.75 mmol)搅拌下溶解于 10 mL二氯甲烷中,依次加入三乙胺 (303 mg, 3 mmol), (R 3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(250 mg, 0.75 mmol)和
双 (2-氧代 -3-噁唑烷基)次膦酰氯 (286 mg, 1.12 mrnol), 室温下搅拌 1.5小时后, 薄 层色谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化所得残余 物, 得到标题产物 (R)-{3- [1-(2-甲氧基苯基) -3-三氟甲基 -咪唑 [1,5- a]哌嗪 -7-基] -3-氧 代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 10c(380 mg,黄色固体), 收率: 82.8 %。
MS n /z (ESI): 613[M+1]
第四歩
(R)-3-氨基 -l-[l-(2-甲氧基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2-甲氧基苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基 ]-3-氧代
-1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 10c(380 mg, 0.62 mmol)溶解于 4 mL 4.3N氯化氢甲醇溶液中,溶液在搅拌下反应 2小时后反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基- 1-[1-(2-甲氧基苯基) -3- 三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 10(100 mg,黄 色固体), 收率: 31 %。
MS m/z (ESI): 513[M+1]
^MR (400MHz, DMSO- ): δ 8.33(bs, 2H), 7.57(m, 1H), 7.47(m, 2H), 7.38(m, 1H), 7.11(m, 1H), 7.03(m, 1H), 4.65(m, 2H), 4.18-4.32(m, 2H), 3.84-3.95(m, 5H), 3.73(s, 1H), 2.76-3.17(m, 4H) 实施例 11
(R)-3-氨基小[1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基 KT-1-酮盐酸盐

1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 2,3- 二氯苯硼酸 (210 mg, 1.1 mmol),四三苯基膦化钯 (116 mg, 0.1 mmol),碳酸钾 (276 mg, 2 liimol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氩气保护下, 120 , 在微波下反应 15分钟后, 反应完毕, 反应液用乙酸乙酯萃取 (15 mLx3),
合并有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所 得残余物,得到标题产物 1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔 丁酯 lla(220 mg,黄色固体), 收率: 50%。
MS m/z (ESI): 398[M+1]
第二步
1-(2,3-二氯苯基 )-3-三氟甲基--咪唑并 [1,5-a]哌嗪 将 1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 lla(220 mg, 0.504 mmol)溶解于 5 mL二氯甲烷中,搅拌下滴加三氟乙酸 (1.72 g, 15.1 mmol),所 得的溶液继续搅拌 1 小时后反应完毕, 减压浓缩反应液, 得到黄色粘稠状标题产 物 1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 lib, 产物不经分离 直接进行下一步反应。
第三步
(R)-{3-[l-(2,3-二氯苯基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟苄 基) -丙基 }-氨基甲酸叔丁酯
氮气氛下, 将 1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 llb(225 mg, 0.5 mmol)搅拌下溶解于 8 mL二氯甲烷中, 依次加入三乙胺 (202 mg, 2 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(167 mg, 0.5 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (191 mg, 0.75 mmol), 室温下搅拌 1.5小时, 薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(2,3-二氯苯基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代 -i-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 llc(290 mg,黄色固体), 收率: 89.2%。 MS m/z (ESI): 651[M+1]
第四步
(R)-3-氯基小[1-(2,3-二氯苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2,3-二氯苯基)-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基 ]-3-氧代 -1- (2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 lle(290 mg, 0.45 mmol)溶解于 4 mL 4.3N氯化氢甲醇溶液中,溶液在搅拌下反应 2小时后反应完毕,减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(2,3-二氯苯基) -3- 三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 11(100 mg,黄 色固体), 收率: 38%
MS m/z (ESI): 551[M+1]
HNMR (400MHz, DMSO-c¾): δ 8.21 (bs, 2H), 7.72(d, 2H), 7.43-7.56(m, 3H), 4.68(d, 2H), 4.36(m, 2H), 3.95(s, 2H), 3.62(s, 1H), 2.90-3.10(m, 4H) 实施例 12
(R)-3-氨基 -l-[l-(3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐

第一步
1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(2.6 g, 7.02 mmol)溶解 于 40 mL二氯甲烷中, 搅拌下加入三氟乙酸 (16 mL, 211 mmol), 所得溶液在室温 下搅拌 30分钟后反应完毕, 减压浓缩反应液, 得到黄色粘稠状标题产物 1-溴 -3- 三氟甲基-咪唑并 [l,5-a]哌嗪三氟乙酸盐 12a, 不经分离直接进行下一步反应。 MS m/z (ESI): 272[M+1]
第二步
(R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] - 氨基甲酸叔丁酯
将粗品 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 12a溶解于 70 mL二氯 甲烷中,依次加入三乙胺 (4.9 mL, 35.1 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟 -苯基) -丁酸 le(2,34 g, 7.02 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (2.68 g, 10.5 mmol), 室温下搅拌 1小时反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 12b(3 g, 白色固体), 收率: 70%。
第三步
(R)-{3-[l-(3-氟苯基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) - 丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(100 mg, 0.171 mmol), 3-氟苯硼酸 (26 mg, 0.188 mmol), 四三苯基膦化钯 (20 mg, 0.0171 mmol), 碳酸钾 (47 mg, 0.342 mmol), 2 mL乙二醇
二甲醚和 2 mL水放入 10 mL微波反应管中, 氩气保护下, 140Ό, 在微波下反应 15分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合 并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得 残余物, 得到标题产物 (R)-{3-[l-(3-氟苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧 代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 12c(84 mg, 白色固体), 收率: 81 %。 MS m/z (ESI): 601 [M+l]
第四步
(R)-3-氨基 -l-[l-(3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基:) -丁 -1-酮盐酸盐
将 (R)-{3- [1-(3-氟苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 苄基) -丙基 } -氨基甲酸叔丁酯 12c(84 mg, 0.2 mmol)溶解于 3 mL 3.1N氯化氢甲醇溶 液中, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R 3-氨基 -1-[1-(3-氟苯基 )-3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 12(70 mg, 白色固 体), 收率: 93.3 %。
MS m/z (ESI): 501 [M+l]
1HNMR (400MHz, CD3OD): δ 7.07-7.48(m, 6H), 4.97-5.10(m, 2H), 4.27-4.35(m, 2H), 3.89-4.10(m, 3H), 2.84-3.08(m, 4H) 实施例 13
(R)-3-氨基 -l-[l-(2,4-二氟苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基 丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2,4-二氟苯基) -3-三氟甲基-咪唑 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄 基) -丙基 氨基甲酸叔丁酯
在氩气保护下, 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基)-丙基] -氨基甲酸叔丁酯 12b (100 mg, 0.171 mmol), 2,4-二氟苯硼 酸 (30 mg, 0.188 mmol),四三苯基膦化钯 (20 mg, 0.0171 mmol),碳酸钾 (47 mg, 0.342
mmol), 1 mL乙二醇二甲醚和 1 mL水放入 lO mL微波反应管中, 120 °C, 在微波 下反应 20分钟后,反应完毕,向反应液中加入 5 mL 7j,用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R)-{3-[l-(2,4-二氟苯基) -3-三氟甲基 -咪唑 [1 ,5-a]哌嗪 -7- 基] -3-氧代 -1 -(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 13a(50 mg, 白色固体),收率: 48 %。
MS m/z (ESI): 619[M+1]
第二步
(R)-3-氨基 -1 -[1 -(2,4-二氟苯基)- 3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2,4-二氟苯基) -3-三氟甲基 -咪唑 [1 ,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 13a(50 mg, 0.08 mrnol)溶解于 3 mL二氯甲垸中, 加入 2.5 mL 3.1N氯化氢甲醇溶液中,室温下搅拌反应 1小时,薄层色谱跟踪反应, 原料消失,减压浓缩反应液,用硅胶柱色谱法纯化所得残余物,得到标题产物 (R)-3- 氨基小[1 -(2,4-二氟苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗉 -7-基] -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐 13(40 mg, 白色固体), 收率: 81 %。
MS m/z (ESI): 519[M+1]
'HNMR (400MHZ, CD3OD): δ 7.62(m, 2H), 7.38(m, 2H), 7.05-7.18 (m, 3H),
4.75-4.80(m, 2H), 4.28-4.37(m, 2H), 3.98-4.07(m, 2H), 3.87(s, 1H), 2.75-3.23(m, 4H) 实施例 14
(R)-4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰 ]-3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -1 -基} -苄 腈三氟乙酸盐

第一步
4-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苄腈三氟乙酸盐 将 1-溴 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 4- 氰基苯硼酸 (161.7 mg, 1.1 mmol), 四三苯基膦化钯 (116 mg, 0.1 mmol), 碳酸钾 (276 mg, 2 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氩气保护 下, 120Ό , 在微波下反应 30分钟后, 反应完毕, 减压浓缩反应液, 用硅胶柱色
谱法纯化所得残余物, 将得到的黄色油状物溶解于 30 mL二氯甲烷中, 搅拌下滴 加三氟乙酸 (2.6 g, 22.6 mmol), 所得的溶液继续搅拌半小时后反应完毕, 减压浓缩 反应液, 得到粗品标题产物 4-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)-苄腈三氟乙酸 盐 14a(270 mg,黄色油状物 ), 不经分离直接进行下一步反应。
第二步
(R)-{3-[l-(4-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄 基) -丙基 氨基甲酸叔丁酯
氮气氛下,将 4-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苄腈三氟乙酸盐 14a(251 mg, 0.85 mmol)搅拌下溶解于 20 mL二氯甲烷中,依次加入二异丙基乙胺 (404 mg, 4 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(283 mg, 0.85 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (323 mg, 1.28 mmol), 室温下搅拌反应 3小时, 薄层 色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化, 得到标题产 物 (R)-{3-[l-(4-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 苄基) -丙基 氨基甲酸叔丁酯 14b(400 mg, 白色固体), 收率: 77.5 %。
MS m/z (ESI): 608[M+1]
' 第三步
(R)-4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -苄 腈三氟乙酸盐
将 (R)-{3-[l-(4-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氣基甲酸叔丁酯 14b(400 mg, 0.66 mmol)溶解于 4 mL二氯甲烷 中, 搅拌下加入 5 mL三氟乙酸, 半个小时后反应完毕, 减压浓縮反应液, 用硅胶 柱色谱法纯化所得残余物, 得到标题产物 (R)-4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁 酰] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苄腈三氟乙酸盐 14(200 mg, 白色固体), 收率: 48.9%。
MS m/z (ESI): 509[M+1]
]HNMR (400MHz, CD3OD): δ 7.79-7.85(m, 4H), 7.21-7.32(m, 2H), 5.01-5.10(m, 2H), 4.27-4.35 (m, 2H), 3.98-4.10 (m, 3H), 3.02-3.30(m, 3H), 2.82(m, 1H) 实施例 15
(R)-3-氨基 -l-[l-(4-甲基 -噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -基] -4-(2,4,5-三 氟苯基) -丁 -1-酮盐酸盐


第一步
l-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 5- 甲基噻吩 -2-基硼酸 (170 mg, 1.2 mmoI), 四三苯基膦化钯 (116 mg, 0.1 rnrnol), 碳酸 钾 (276 mg, 2 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氩 气保护下, 120°C, 在微波下反应 20分钟后, 反应完毕, 将反应液在减压下浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(4-甲基噻吩 -3-基) -3-三氟甲基- 咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 15a(250 mg,黄色油状物), 收率: 67.6%。
MS m/z (ESI): 388[M+1]
第二步
1-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 将 1-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 15a(250 mg, 0.65 mmol)溶解于 10 mL二氯甲烷中,搅拌下滴加三氟乙酸 (1.2 mL, 16 mmol), 所得的溶液继续搅拌 3 小时后反应完毕, 减压浓缩反应液, 得到黄色油状液体粗 品标题产物 1-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 15b,不经分离直接 进行下一步反应。
笛二丄!^
一少
(R)-{3-[l-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 1-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 15b(187 mg, 0.65 mmol)搅拌下溶解于 10 mL二氯甲烷中,依次加入二异丙基乙胺 (0.57 mL, 3.25 mmol), (R 3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(217 mg, 0.65 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (254 mg, 1 mmol), 室温下搅拌反应 6小时, 薄层色 谱跟踪反应, 原嵙消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产 物 (R)-{3-[l-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]-3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 15c(220 mg,无色油状液体),收率: 56.2 %。
MS m/z (ESI): 603 [M+1]
第四步
(R)-3-氨基小 [1-(4-甲基 -噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(4-甲基噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-l-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 15c(220 mg, 0.36 mmol)溶解于 5 mL 3.1 氯化氢甲醇溶液中, 室温下搅拌反应过夜, 减压浓缩反应液, 用硅胶柱色谱 法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(4-甲基 -噻吩 -3-基) -3-三氟甲基- 咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 15(100 mg, 白色固体), 收率: 50%。
MS m/z (ESI): 504[M+1]
JHNMR (400MHz, CD3OD): 6 7.37(dd, 1H), 7.17(dd, 1H), 7.10-7.36(m, 2H), 4.80-4.92 (m, 2H), 4.52(t, 1H), 4.29(t, 1H), 4.01-4.10(m, 2H), 3.50-3.64(m, 1H), 2.86(m, 2H), 2.63-2.70(m, 1H), 2.3 l(s, 3H), 2.07(m, 1H) 实施例 16
(R)-3-氨基 -1-[1-(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

笛 _ .At
1-(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 7b(740 mg, 2 mmol), 4- 吡唑硼酸 (246 mg, 2.2 mmol), 四三苯基膦化钯 (232 mg, 0.2 mmol), 碳酸钾 (540 mg, 4 mmol), 5 mL乙二醇二甲醚和 5 mL水放入 20 mL微波反应管中, 氩气保护下, 140 , 在微波下反应 50分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙 酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镜干燥, 过滤, 减压浓缩滤液, 用硅 胶柱色谱法纯化所得残余物, 得到标题产物 1-(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 16a(80 mg, 白色固体), 收率: 11.2%。
MS m/z (ESI): 358[M+1]
第二步
1_(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪盐酸盐 将 1-(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 16a(80 mg, 0.224 mmol)溶解于 5 mL 4. IN氯化氢甲醇溶液中, 室温下搅拌反应过夜, 薄层色 谱跟踪反应,原料消失,减压浓缩反应液,得到黄色油状液体粗品标题产物 1-(1H- 吡唑 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪盐酸盐 16b, 不经分离直接进行下一步反
应。
第三步
(R)-{3-氧代 -3-[1-(1Η-吡唑 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基 ]小(2,4,5-三氟 苄基) -丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 1-(1H-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪盐酸盐 16b(64.6 mg, 0.22 mmol)搅拌下溶解于 10 mL二氯甲垸中,依次加入二异丙基乙胺 (113.7 mg, 0.88 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(73.3 mg, 0.22 mmol) 和双 (2-氧代 -3-噁唑垸基)次膦酰氯 (84 mg, 0.33 mmol), 室温下搅拌反应过夜, 反应 完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3- 氧代 -3-[1-(1Η-吡唑 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -1-(2,4,5-三氟苄基) - 丙基 氨基甲酸叔丁酯 16c(60 mg, 白色固体), 收率: 47.7 %。
MS m/z (ESI): 573[M+1]
第四步
(R)-3-氨基 -1-[1-(1H-吡唑 -4-基)- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将(R)-{3-氧代 -3-[1-(1Η-吡唑 -4-基)-3-三氟甲基-咪唑并 [1,5- a]哌嗪 -7- 基] -1 -(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 16c(60 mg, 0.1 mmol)溶解于 5 mL 3.1N氯化氢甲醇溶液中, 室温下搅拌反应过夜, 减压浓缩反应液, 用硅胶柱色谱 法纯化所得残余物,得到标题产物 (R)-3-氨基 -1-[1-(1H-吡唑 -4-基) -3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 16(30 mg,无色油状液体), 收率: 63.5 %。
MS m/z (ESI): 473 [M+1]
ifiNMR (400MHz, DMSO-^): δ 8.26 (s, 3H), 7.95(s, 1H), 7.86(s, 1H), 7.50-7.60(m, 2H), 4.74-4.9 l(m, 2H), 4.27(m, 1H), 4.12(m, 1H), 3.94(m, 2H), 3.75(m, 1H), 3.09(m, 1H), 2.98(m, 2H), 2.84(m, 1H) 实施例 17
(R)-3-氨基 -1-[1- (吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯
- 基:) -丁 -1-酮盐酸盐

(R)-[3-氧代 -3-(l-吡啶 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
将 (R)_[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪- 7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(204 mg, 0.171 mmol), 3-吡啶硼酸 (47 mg, 0.382 mmol), 四三苯基膦化,巴 (38 mg, 0.035 mmol), 碳酸钾 (96 mg, 0.348 mmol), 2.5 mL乙二醇 二甲醚和 2.5 mL水放入 10 mL微波反应管中, 氩气保护下, 140°C, 在微波下反 应 20分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 m] 3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R)-[3-氧代 -3-(1-吡啶- 3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 17a(120 mg,黄色固体),收率: 60 %。
MS m/z (ESI): 584[M+1]
第二步
(R)-3-氨基 -1-(1-吡啶 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 )-4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐
将 (R)-[3-氧代 -3-(1-吡啶 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 17a(120 mg, 0.2 mmol)溶解于 5 mL 3.1N氯化氢甲 醇溶液中, 室温下搅拌反应过夜, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残 余物, 得到标题产物 (R)-3-氨基小 [1- (吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 17(50 mg,黄色固体), 收率: 48.2 %。
MS m/z (ESI): 484[M+1]
!HNMR (400MHz, CD3OD): δ 9.10(d, 1H), 8.72(m, 2H), 8.05(m, 1H), 7.39(m, 1H), 7.18(m, 1H), 5.14(m, 2H), 4.34(m, 2H), 4.05(m, 2H), 3.91(s, 1H), 2.86-3. l l(m, 4H) 实施例 18
(R)-3-氨基 (吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐

第一步
(R)-[3-氧代 -3-(l-吡啶- 4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -l-(2,4,5-三氟苄 基)-丙基] -氨基甲酸叔丁酯
将 (R)_[3-(l-溴 -3-三氟甲基-咪唑并 [l,5_a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(102 mg, 0.174 mmol) , 4-吡啶硼酸 (23.5 mg, 0.191 mmol), 四三苯基膦化钯 (19 mg, 0.0174 mmol), 碳酸钾 (48 mg, 0.348 mmol), 2 mL 乙二醇二甲醚和 2 mL水放入 10 mL微波反应管中, 氩气保护下, O °C, 在微波 下反应 20分钟后,反应完毕,向反应液中加入 5 mL水,用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R [3-氧代 _3-(1-吡啶 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟苄基)-丙基] -氨基甲酸叔丁酯 18a(90 mg, 白色固体), 收率: 90 %。
MS m/z (ESI): 584[M+1]
第二步
(R)-3-氨基 吡啶 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐
将 (R)-[3-氧代 -3-(1-吡啶- 4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三 氟苄基) -丙基 1]-氨基甲酸叔丁酯 18a(90 mg, 0.15 mmol)溶解于 5 mL 3.1N氯化氢甲 醇溶液中, 室温下搅拌反应过夜, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残 余物, 得到标题产物 (R)-3-氨基 (吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 18(20 mg,无色油状液体), 收率: 25.6%。 MS m/z (ESI): 484[M+1]
^MR (400MHz, CD3OD): δ 8.66(d, 2H), 7.94(d, 2H), 7.34(m, 2H), 5.12-5.19 (m, 2H), 4.29-4.37(m, 2H), 3.91-4.10(m, 3H), 3.65(s, 2H), 3.06(m, 2H), 2.88-2.96(m, 2H) 实施例 19
(R)-3-氨基 -l-[l-(2-氯噻吩 -3-基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁小酮三氟乙酸盐

第一步
1-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, lmmol), 2-氯
噻吩 -3-基硼酸 (195 mg, 1.2 mmol),四三苯基膦化钯 (116 mg, 0.1 mmol),碳酸钾 (276 mg, 2 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氮气保护 下, 搅拌均匀后封口, 140°C, 在微波下反应 20分钟后, 反应完毕, 向反应液中 力口入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过 滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 19a(285 mg,黄色油状液体), 收率: 70%
MS m/z (ESI): 408[M+1]
第二步
1-(2-氯噻吩 -3-基)- 3-三氟甲基-咪唑并 [1,5-a]哌嗪盐酸盐 将 1-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 19a(285 mg, 0.7 mmol)溶解于 5 mL 3.1N氯化氢甲醇溶液, 混合液在室温下搅拌过夜, 反应完 毕, 减压浓缩反应液, 得到标题产物 1-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a] 哌嗪盐酸盐 19b, 不经分离直接进行下一步反应。
第三步
(R)-{3-[l-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 1-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪盐酸盐 19b(240 mg, 0.22 mmol)搅拌下溶解于 10 mL二氯甲垸中, 依次加入三乙胺 (282.8 mg, 2.8 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(233.3 mg, 0.7 mmol)和 双 (2-氧代 -3-噁唑垸基)次膦酰氯 (267 mg, 1.05 mmol), 室温下搅拌反应过夜, 反应 完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟 基) -丙基 } -氨基甲酸叔丁酯 19c(100 mg,黄色固体), 收率: 22.9%。
MS m/z (ESI): 622[M-1]
第四步
(R)-3-氨基小 [1- (2-氯噻吩 -3-基) -3-三氟甲基 -咪唑 [1,5- a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮三氟乙酸盐
将 (R)-[3-[l-(2-氯噻吩 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 19c(100 mg, 0.16 mmol)溶液于 3 mL三 氟乙酸和 15 mL二氯甲烷中, 所得的溶液在室温下搅拌 3小时后反应完毕, 减压 浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(2- 氯噻吩 -3-基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮三氟乙 酸盐 19(20 mg,无色油状液体), 收率: 22.5 %。
MS m/z (ESI): 522[M-1]
iHNMR (400MHz, CD3OD): δ 7.43(m, 1H), 7.29(m, 1H), 7.14(m, 2H), 4.78-4.93(m,
4H), 4.14-4.3 l(m, 2H), 4.01-4.10(m, 2H), 3.53(m, 1H), 2.78-2.84(m, 2H), 2.57- 2.65(m: 2H) 实施例 20
(R)-3-氨基小 {1-[1-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7- 基}-4-(2,4,5-三氟苯基) -丁小酮三氟乙酸盐

第一步
(R)-[3-{l-[l-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基}-3-氧 代小(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 将 (R)- [3-(1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代- 1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(181 mg, 0.31 mmol), 1-异戊基 -1H-吡唑 -4-基硼酸 (62 mg, 0.34 mmol),四三苯基膦化钯 (36 mg, 0.03 mmol),碳酸钾 (85 mg, 0.616 mmol), 2 mL 乙二醇二甲醚和 2 mL水放入 10 mL微波反应管中, 氩气保护下, 140°C , 在微波 下反应 15分钟后,反应完毕,向反应液中加入 5 mL水,用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所 得残余物,得到标題产物 (R)-[3-{l-[l-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基}-3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 20a(160 mg, 淡黄色固体), 收率: 80?^。
MS m/z (ESI): 643[M+1]
(R)-3-氨基 -1-{1-[1-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7- 基 }_4_(2,4,5_三氟苯基) -丁 _1_酮三氟乙酸盐 将 (R)-[3-{l-[l-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪- 7- 基 }-3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 20a(160 mg, 0.25 mmol)溶解 于 5 mL二氯甲烷中, 搅拌下加入三氟乙酸 (0.5 mL, 6.67 mmol), 室温下搅拌 6小 时后反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产 物 (R)-3-氨基 -1-{1-[1-(3-甲基丁基) -1H-吡唑 -4-基] -3-三氟甲基 -咪唑 [1,5-a]哌嗉 -7- 基 }-4-(2,4,5-三氟苯基) -丁小酮三氟乙酸盐 20(100 mg,黄色固体), 收率: 61 %。 MS m/z (ESI): 543 [M+1]
!HNMR (400MHz, CD3OD): δ 7.93(dd, 1H), 7.74(s, 1H), 7.32(m, 2H), 4.89-4.94 (m, 2H), 4.20-4.28(m, 4H), 3.94-4.19(m, 3H), 3.05(t, 2H), 2.94(m, 1H), 2.82(dd, 1H), 1.78(m, 2H), 1.56(m, 1H), 1.29(d, 2H), 0.96(t, 6H) 实施例 21
(R)-3-氨基 -l-[3-三氟甲基小(2-三氟甲基苯基) -咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三
氟苯基) -丁 -1-酮盐酸盐

第一步
3-三氟甲基 -1-(2-三氟甲基苯基) -咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 2- 三氟甲基苯硼酸 (209 mg, 1.1 mmol), 四三苯基膦化钯 (116 mg, 0.1 mmol), 碳酸钾 (276 mg, 2 mmol), 3 mL乙二醇二甲醚和 3 mL水放入 20 mL微波反应管中, 氩气 保护下, 120°C, 在微波下反应 30分钟后, 反应完毕, 用乙酸乙酯萃取反应液 (10 mLx3), 合并有机相, 用无水硫酸樣干燥, 抽滤, 减压浓縮滤液, 用硅胶柱色谱法 纯化所得残余物,得到标题产物 3-三氟甲基 -1-(2-三氟甲基苯基)-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 21a(323 mg,黄色固体), 收率: 74%。
MS m/z (ESI): 436[M+1]
第二步 '
3-三氟甲基 -1-(2-三氟甲基苯基) -咪唑并 [1,5-a]哌嗪盐酸盐 将 3-三氟甲基小(2 -三氟甲基苯基) -咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 21a(323 mg, 0.74 mmol)溶解于 2 mL甲醇中,搅拌下滴加 3 mL 4.3N氯化氢甲醇溶液,所得 的溶液继续搅拌 1小时后反应完毕, 减压浓缩反应液, 得到标题产物 3-三氟甲基 -1-(2 -三氟甲基苯基) -咪唑并 [1,5-a]哌嗪盐酸盐 21b, 不经分离直接进行下一步反 应。
第三步
(R)-{3-氧代 -1-(2,4,5-三氟苄基) -3-[3-三氟甲基 -1-(2-三氟甲基苯基) -咪唑并 [1,5-a]哌
嗪 -7-基] -丙基 } -氨基甲酸叔丁酯
将 3-三氟甲基 -1-(2 -三氟甲基苯基 咪唑并 [1,5-a]哌嗪三氟乙酸盐 21b(276 mg, 0.74 mmol)搅拌下溶解于 30 mL二氯甲烷中,依次加入三乙胺 (300 mg, 2.97 ΙΏΙΊΙΟΙ), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(247 mg, 0.74 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (282 mg, 1.11 mmol), 室温下搅拌反应过夜, 薄层色谱跟踪 反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化, 得到标题产物 (R)-{3- 氧代 -1-(2,4,5-三氟苄基) -3-[3-三氟甲基 -1-(2-三氟甲基苯基) -咪唑并 [1,5-a]哌嗪 -7- 基] -丙基 氨基甲酸叔丁酯 21c(420 mg,黄色固体), 收率: 87 %。
MS m/z (ESI): 651 [M+l]
第四步
(R)-3-氨基 -l-[3-三氟甲基小(2-三氟甲基苯基) -咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-氧代 -1-(2,4,5-三氟苄基) -3-[3-三氟甲基 -1-(2-三氟甲基苯基) -咪唑并 [l,5-a]哌嗪 -7-基] -丙基 氨基甲酸叔丁酯 21c(420 mg, 0.65 mmol)溶解于 5 mL 3. IN 氯化氢甲醇溶液中, 搅拌反应 30分钟后反应完毕, 减压浓缩反应液, 用硅胶柱色 谱法纯化所得残余物,得到标题产物 (R)-3-氨基 -1-[3-三氟甲基 -1-(2-三氟甲基苯基) - 咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 21(200 mg, 白色固体), 收率: 57%。
MS m/z (ESI): 551 [M+l]
1HNMR (400MHz, CD3OD): δ 7.76(m, 1H), 7.69(m, 2H), 7.36(d, 1H), 7.03(m, 1H), 6.87 (m, 1H), 4.65(m, 1H), 4.5 l(s, 1H), 3.88-4.29(m, 5H), 2.30-2.73(m, 4H) 实施例 22
(R)-3-氨基 -1-[1-吗啉 -3-三氟甲基-咪唑并 [1,5- a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1- 酮盐酸盐

1-吗啉 _4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(200 mg, 0.54mmol), 吗啉 (56 mg, 0.65mmol),三 (二亚苄基丙酮)二钯 (2.4 mg, 0.0026 mmol), 2- (二叔丁基 膦)联苯 (3.2 mg, 0.022 mmol), 叔丁醇钠 (78 mg, 0.81 mmol), 2 mL干燥的甲苯放入 20 mL微波反应管中, 100°C, 在微波下反应 70分钟后, 反应完毕, 减压浓缩反应 液, 用硅胶柱色谱法纯化所得残余物,得到标题产物 1-吗啉 -4-基 -3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-羧酸叔丁酯 22a(100 mg, 白色固体), 收率: 49%。
MS m/z (ESI): 377[M+1]
第二步
1_吗啉 _4_基 -3—三氟甲基-咪唑并 [l,5_a]哌嗪三氟乙酸盐 将 1-吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 22a(150 mg, 0.40 mmol)溶解于 10 mL二氯甲垸中, 搅拌下滴加三氟乙酸 (2.76 g, 24 mmol), 所得的 溶液继续搅拌 2小时后反应完毕, 减压浓缩反应液, 得到粗品标题产物 1-吗啉 -4- 基 -3-三氟甲基-咪唑并 [1,5- a]哌嗪三氟乙酸盐 22b, 不经分离直接进行下一步反应。
第三步
(R)-[3-(l-吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基 )-丙基] -氨基甲酸叔丁酯
氮气氛下, 将 1-吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪三氟乙酸盐 22b搅拌 下溶解于 20 mL二氯甲垸中 , 依次加入三乙胺 (0.40 g, 4 mmol)和 (R)-3-叔丁氧羰基 氨基 _4-(2,4,5-三氟-苯基) -丁酸 le(147 mg, 0.44 mmol), 搅拌 10分钟后, 加入双 (2- 氧代 -3-噁唑烷基)次膦酰氯 (152 mg, 0.6 mmol), 室温下搅拌反应 2小时, 薄层色 谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化, 得到标题产物 (R)-[3-(l-吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 22c(200 mg, 浅黄色油状物), 收率: 84%。
MS m/z (ESI): 592[M+1]
第四步
(R)-3-氣基小(1-吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐
将 (R)-[3-(l-吗啉 -4-基 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基) -3-氧代- 1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 22c(200 mg, 0.34 mmol)溶解于 4 mL 4.3N氯化氢甲 醇溶液中, 搅拌反应 2小时后反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化 所得残余物, 得到标题产物 (R)-3-氨基 吗啉 -4-基 -3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -7-基) -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 22(42 mg,黄色固体), 收率: 25 %。 MS m/z (ESI): 492[M+1]
'HNMR (400MHz, CD3OD): δ 7.28(m, 2H), 4.72-4.83(m, 4H), 3.59-4.24(m, 8H),
2.68-3.03(m, 2H) 实施例 23
(R)- 3-氨基 -1- [ (4-二甲氨基-苯基)- 3-三氟甲基-咪唑并 [1, -a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮三氟乙酸盐

第一歩
(R)-{3-[l-(4-二甲氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 4-二甲氨基苯硼酸 (62 mg, 0.376 mmol),四三苯基膦化钯 (39.4 mg, 0.034 mmol),碳酸钾 (94.3 mg, 0.683 mmol), 3 mL 乙二醇二甲醚和 3 mL水放入 10 mL微波反应管中, 氩气保护下, 130°C, 在微波 下反应 30分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 用硅胶柱色 谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(4-二甲氨基-苯基) -3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 23a(150 mg, 浅黄色固体), 收率: 70%。
MS m/z (ESI): 626[M+1]
第二步
(R)-3-氨基 -l-[l-(4-二甲氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮三氟乙酸盐
将 (R)-{3-[l-(4-二甲氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 23a(150 mg, 0.24 mmol)溶解于 5 mL二 氯甲烷中, 搅拌下加入 2 mL三氟乙酸, 室温下搅拌 3小时后反应完毕, 减压浓缩 反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 -1-[1-(4-二甲 基氨苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮三氟乙 酸盐 23(90 mg, 白色固体), 收率: 59%。
MS m/z (ESI): 526[M+1]
JHNMR (400MHz, CD3OD): δ 7.55(m, 2H), 7.10-7.40(m, 4H), 7.34(m, 2H), 4.95-5.01 (m, 2H), 4.27(m, 2H), 3.81-4.10(m, 3H), 3.63(m, 3H), 3.05(m, 7H) 实施例 24
(R)-N-[4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-苯基] -乙酰胺盐酸盐

第一步
(R)-{3-[l-(4-乙酰氨基苯基 )-3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟 苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基 >3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(146 mg, 0.25 mmol), 4-乙酰氨基苯硼酸 (67 mg, 0.375 mmol), 二 (二亚苄基丙酮)钯 (2.3 mg, 0.005 mmol), 2-二环己基膦基-2,,4,,6,-三异丙 基联苯 (4.8 mg, 0.01 mmol),磷酸钾 (106 mg, 0.5 mmol), 1.5 mL叔丁醇加入到 10 mL 微波反应管中, 氩气保护下, ΙΟΟΌ , 在微波下反应 30分钟后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫 酸镁干燥, 过滤, 减压浓縮滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产 物 (R)-{3-[l-(4-乙酰氨基苯基 )-3-三氟甲基-咪唑 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 24a(100 mg, 白色固体), 收率: 62 %。
MS m/z (ESI): 640[M+1]
第二步
(R)— N-[4-{7-[3-氣基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 苯基] -乙酰胺盐酸盐
将 (R)-{3-[l -(4-乙酰氨基苯基 )-3-三氟甲基 -咪唑 [1 ,5-a]哌嗪 -7-基 ]-3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 24a(100 mg, 0.156 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 有白色固体析出, 室温下搅拌 10 分钟后, 补加 2 mL 2N氯化氢乙酸乙酯溶液, 继续反应 1小时, 薄层色谱跟踪反 应, 原料消失, 过滤, 得到标题产物 (R)-N-[4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰 基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}-苯基] -乙酰胺盐酸盐 24(62 mg, 浅黄色固 体), 收率: 69%。
MS m/z (ESI): 540[M+1]
'HNMR (400MHZ, CD3OD): δ 7.69(m, 2H), 7.56(m, 2H), 7.20(m, 1H), 7.13(m, 1H); 5.04(m, 2H), 4.4 l(m, 2H), 3.89(s, 1H), 2.86-3.08(m, 4H), 2.14(s, 3H) 实施例 25
(R)-3-氣基小[1-(2,4-二三氟甲基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] (2,4,5-三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l -(2,4-二三氟甲基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基)-丙基 } -氨基甲酸叔丁酯
在氩气保护下, 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2,4-二三氟甲 基苯硼酸 (97 mg, 0.376 mmol),四三苯基膦化钯 (40 mg, 0.0342 mmol),碳酸钾 (94 mg, 0.684 mmol), 2 mL乙二醇二甲醚和 2 mL水放入 10 mL微波反应管中, 130Ό, 在微波下反应 35分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 反应液 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅 胶柱色谱法纯化所得残余物,得到标题产物 (R)-{3-[l-(2,4-二三氟甲基-苯基) -3-三氟 甲基-咪唑并 [1,5-aJ哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 25a(70 mg,浅黄色固体), 收率: 30%。
MS m/z (ESI): 719[M+1]
第二步
(R)-3-氣基 -1-[1-(2,4-二三氟甲基-苯基) -3-三氟甲基-咪唑并 [1,5-a]'哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁小酮盐酸盐
将 (R)-{3-[l-(2,4-二三氟甲基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 25a(70 mg, 0.097 mmol)溶解于 2 tnL二 氯甲垸中,搅拌下加入 2 mL 3.1N氯化氢甲醇溶液,室温下搅拌 2小时,反应完毕, 减压浓缩反应液,得到标题产物 (R)-3-氨基 -1-[1-(2,4-二三氟甲基-苯基) -3-三氟甲基 -咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 25(60 mg,黄色固体),
收率: 100 %。
MS m z (ESl): 619[M+1]
!HNMR (400MHz, CD3OD): δ 8.28(s, 4H), 8.17(s, 1H), 7.75(dd, 1H), 7.52(m, 2H), 4.70(d, 2H), 4.15-4.40 (m, 2H), 3.95(m, 2H), 3.73(s, 1H), 2.70-3. l l(m, 4H) 实施例 26
(R)-3-氨基小 [l-(6-氨基 -P比啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮二盐酸盐

第一步
(R)-{3-[l-(6-氨基-吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基)-丙基 氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(293 mg, 0.5 mmol), 6-氨基吡啶 -3-基硼酸 (165 mg, 0.75 mmol), 二 (二亚苄基丙酮)钯 (5.8 mg, 0.01 mmol), 2-二环已基膦基 -2,,4,,6'-三异丙 基联苯 (9.5 mg, 0.02 mmol), 磷酸钾 (212 mg, 1 mmol), 2 mL叔丁醇加入到 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 30分钟后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫 酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产 物 (R)-{3-[l-(6-氨基-吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]-3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 26a(130 mg,浅黄色固体),收率: 43 %。 MS m/z (ESI): 599[M+1] .
第二步
(R)-3-氨基小 [l-(6-氨基-吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三 氟苯基) -丁 -1-酮二盐酸盐
将 (R)-{3-[l-(6-氨基-吡啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 26a(130 mg, 0.21 mmol)溶解于 2 mL二 氯甲烷和 2 mL甲醇的混合溶剂中, 搅拌下加入 10 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌 2小时,反应完毕,减压浓缩反应液,得到标题产物 (R)-3-氨基 -1-[1-(6- 氨基-吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮二
盐酸盐 26(120 mg,黄色固体), 收率: 100 %。
MS m/z (ESI): 499[M+1]
!HNMR (400MHz, CD3OD): δ 8.15-8.40(m, 6H), 7.55(m, 2H), 7.18(m, 1H)
4.89-5.05(m, 2H), 3.70-4.40(m, 5H), 2.75-3.20(m, 4H) 实施例 27
(R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯乙腈盐酸盐

第一步
(R)-{3-[l-(3-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟苄 基) -丙基) -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 3-氰基苯硼酸 (75.4 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4,,6'-三异丙 基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入到 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 30分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx4), 合并有机相, 用无 水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标 题产物 (R)-{3-[l-(3-氰基苯基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 27a(187 nig, 白色固体), 收率: 90%。
MS m/z (ESI): 630[M+23]
第二步
(R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯乙腈盐酸盐
将 (R)-{3-[l-(3-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 27a(187 mg, 0.308 mmol)溶解于 2 mL乙酸乙酯 中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 有白色固体析出, 室温下搅拌反应 0分 钟后, 补加 2 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色谱跟踪 反应, 原料消失, 过滤, 得到标题产物 (R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰 基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}-苯乙腈盐酸盐 27(110 mg,黄色固体), 收
率: 66%
MS m z (ESI): 508[M+1]
!HNMR (400MHz, CD3OD): δ 7.97(m, 2H), 7.65(m, 2H), 7.10-7.40(m, 2H), 5.07 (m; 2H), 4.28-4.36(01, 2H), 3.92-4.10(m, 3H), 3.09(m, 4H) 实施例 28
(R)-3-氨基小 [1- (呋喃 -3-基) -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐

第一步
(R)-[3-(l-呋喃 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基〗 -氨基甲酸叔丁酯 12b(293 mg, 0.5 mmol), 呋喃 -3-基硼酸 (84 mg, 0.75 mmol), 二 (二亚苄基丙酮)钯 (5.8 mg, 0.01 mmol), 2-二环己基膦基-2,,4,,6,-三异丙基联苯 (8.2 mg, 0.02 mmol),磷酸钾 (212 mg, 1 mmol), 1.5 mL叔丁醇加入 10 mL微波反应 管中, 氩气保护下, 100Ό , 在微波下反应 30分钟后, 反应完毕, 向反应液中加 入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-(l- 呋喃 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基)-丙基] -氨 基甲酸叔丁酯 28a(200 mg, 浅黄色固体), 收率: 70%。
MS m/z (ESI): 573 [M+l]
第二步
(R)-3-氨基小 (1-呋喃 -3-基 -3-三氟甲基 -咪唑 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) -丁
- 1-酮盐酸盐
将 (R)-[3-(l-呋喃 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 28a(200 mg, 0.349 mmol)溶解于 2 mL乙酸乙酯中, 加入 10 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌 1.5小时, 反应完毕, 减压浓缩 反应液, 得到标题产物 (R)-3-氨基小 [1- (呋喃 -3-基) -3-三氟甲基-咪唑 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁小酮盐酸盐 28(150 mg,黄色固体), 收率: 91 %。
MS m/z (ESI): 473 [M+l]
'HNMR (400MHZ, CD3OD): δ 6.96(m, 1H), 6.73(s, 1H), 6.33(m, 1H), 6.30 (m, 1H): 5.95(m, 1H), 4.01 (m, 2H), 3.45(m, 2H), 3.14(m, 3H), 2.14(m, 4H) 实施例 29
(R)-2-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯乙腈盐酸盐

第一步
(R)-{3-[l-(2-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄 基:) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氰基苯硼酸 (75.4 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2',4',6'-三异丙 基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入到 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无 水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标 题产物 (R)-{3-[l-(2-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 29a(100 mg, 白色固体), 收率: 51 %。
MS m/z (ESI): 630[M+23]
第二步
(R)-2-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 苯乙腈盐酸盐
将 (R)-{3-[l-(2-氰基苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 29a(100 mg, 0.165 mmol)溶解于 2 mL乙酸乙酯 中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色谱跟踪反 应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化,得到标题产物 (R)-2-{7-[3- 氨基 (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -苯乙腈盐酸 盐 29(40 mg,黄色固体), 收率: 48 %。
MS m/z (ESI): 508[M+1]
!HNMR (400MHz, CD3OD): δ 7.86(m, 2H), 7.76(m, 2H), 7.20-7.33(m, 2H), 4.92 (m, 2H), 4.40(m, 2H), 4.08(m, 2H), 3.85(s, 1H), 2.92-3.07(m, 4H) 实施例 30
(R)-3-氨基小 [l-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐

第一步
■ (R)-{3-[l-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1 -(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氯 -3-氟吡啶 -4-基硼酸 (89.8 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入到 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 30分钟后, 反应 完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx4), 合并有机相, 用无水硫酸镁干燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得 到标题产物 (R)-{3-[l-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3- 氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 30a(120 mg, 浅黄色固体), 收率: 55 %。
MS m/z (ESI): 636[M+1]
第二步
(R)-3-氨基 -1-[1-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3- [1-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 30a(120 mg, 0.189 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色谱 跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到 标题产物 (R)-3-氨基 -1-[1-(2-氯 -3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-
] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 30(80 mg,黄色固体), 收率: 80%。
S m/z (ESI): 536[M+1] 实施例 31
(R)-3-氨基- l-[l-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]呃嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 6-二甲氨基吡啶 -3-基硼酸 (85 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mrnol), 2-二环己基膦基-2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 rnrnol), 2 mL叔丁醇加 入到 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应 完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫酸镁千燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得 到标题产物 (R)- {3-[1-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 31a(115 mg,浅黄色固体), 收率: 53 %。
MS m/z (ESI): 627[M+1]
第二步
(R)-3-氨基 -1-[1-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁小酮盐酸盐
将 (R)-{3-[l-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 31a(115 mg, 0.183 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色谱 跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到 标题产物 (R)-3-氨基 -1-[1-(6-二甲氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-
基] -4_(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 31(70 mg,黄色固体), 收率: 73 %。
lH MR (400MHz, CD3OD): 5 8.26(m, 2H), 7.42(m, 2H), 7.23(m, 1H), 5.10(m, 2H), 4.34(m, 2H), 4.05(m, 3H), 3.15(m, 3H), 2.04(s, 1H), 1.24(m, 1H) 实施例 32
(R)-3-氨基 -l-[l-(3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(3-氟吡啶 -4-基 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 3-氟吡啶 -4-基硼酸 (72.3 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),璘酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C , 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mIX3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 减压浓缩滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟节基) -丙基) -氨基甲酸叔丁酯 32a(125 mg,浅黄色固体), 收率: 60%。
MS m/z (ESI): 602[M+1]
第二步
(R)-3-氨基小 [l-(3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(3-氟吡啶 -4-基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基)-丙基}-氨基甲酸叔丁酯 32a(125 mg, 0.21 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色谱 跟踪反应, 原料消失, 减压浓縮反应液, 用硅胶柱色谱法纯化所得残余物, 得到 标题产物 (R)-3-氨基 -1-[1-(3-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-
基] -4-(2,4,5-三氟苯基) -丁小酮盐酸盐 32(80 mg,黄色固体), 收率: 77%。
MS m/z (ESI): 502[M+1] 实施例 33
(R)-3-氨基小[1- (嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基]斗(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐
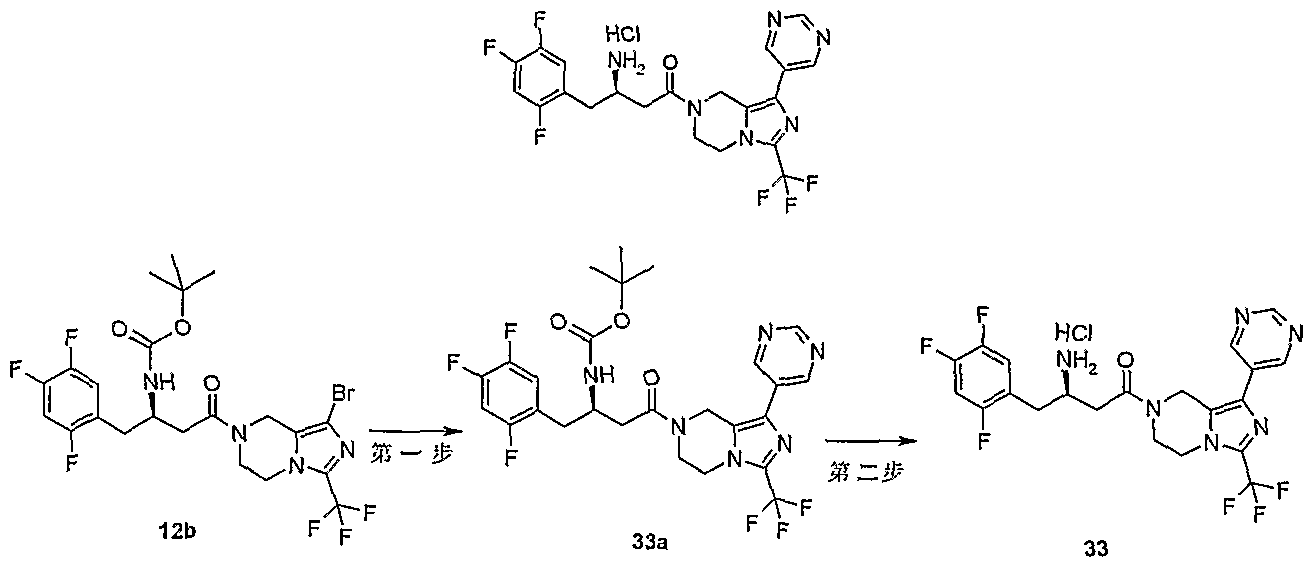
第一步
(R)-[3-(l-嘧啶 -5-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342mmol), 嘧啶 -5-基硼酸 (63.6 mg, 0.513 nrniol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4',6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取反应液 (15 mLx3), 合并有机相, 用无水硫 酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产 物 (R)-[3-(l-嘧啶 -5-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 33a(110 mg,浅黄色固体), 收率: 55 %。
MS m/z (ESI): 585[M+1]
(R)-3-氨基小 (1-嘧啶 -5-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐
将 (R)-[3-(l-嘧啶 -5-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 33a(110 mg, 0.188 mmol)溶解于 2 mL乙酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消 失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨基 -1-[1- (嘧 啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 33(75 mg,黄色固体), 收率: 82%。
MS m/z (ESI): 485 [M+l]
实施例 34
(R)-3-氨基 -ί-[1-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基]- 4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小 (2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基)- 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2,3-二氯吡啶 -4-基硼酸 (98 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4',6,- 三异丙基联苯 (6.52 mg, 0.014 rnmoi), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100Ό , 在微波下反应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 34a(140 mg,浅黄色固体),收率: 63 %。 MS m/z (ESI): 652[M+1]
第二步
(R)-3-氨基 -1-[1-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基)-丙基}-氨基甲酸叔丁酯34 140 mg, 0.214 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2,3-二氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐 34(90 mg,黄色固体), 收率: 78%。
MS m/z (ESI): 552[M+1]
实施例 35
(R)-3-氨基 -l-[l-(3-氯吡啶 -4-基)- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4- (2,4,5- 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(3-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 3-氯吡啶 -4-基硼酸 (80.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2',4',6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(3-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 35a(130 mg, 浅黄色固体), 收率: 63 %。
MS m/z (ESI): 618[M+1]
第二步
(R)-3-氨基小 [1-(3-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)_{3-[1 -(3-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 35a(130 mg, 0.21 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(3-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 小酮盐酸盐 35(80 mg,黄色固体), 收率: 72%。
MS m/z (ESI): 518[M+1] 实施例 36
(R)-3-氨基 -1-[1-(6-氯吡啶 -3-基 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟
苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(6-氯吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟苄基:) -丙基 氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 6-氯吡啶 -3-基硼酸 (80.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氣气保护下, 100Ό , 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(6-氯吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 36a(130 mg, 浅黄色固体), 收率: 61 %。
MS m/z (ESI): 618[M+1]
第二步
(R)-3-氨基小[1-(6-氯吡啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l -(6-氯吡啶 -3-基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基 ]-3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 36a(130 mg, 0.21 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基小 [1-(6-氯吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐36(100 11^, 黄色固体), 收率: 92%。
MS m/z (ESI): 518[M+1] 实施例 37
(R)-3-氨基小 [l-(2-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3- (1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氯吡啶 -4-基硼酸 (80.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2',4,,6'-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol),2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(6-氨基 -吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2-氯吡 啶 -4-基) -丙基 氨基甲酸叔丁酯 37a(145 mg, 浅黄色固体), 收率: 68 %。
MS m/z (ESI): 618[M+1]
第二步
(R)-3-氨基小[1-(2-氯吡啶 -4-基 >3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 37a(145 mg, 0.239 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2-氯吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 37(85 mg,黄色固体), 收率: 71 %。
MS m/z (ESI): 518[M+1] 实施例 38
(R)-3-氨基小 [1-(2-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -l-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R [3-(1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氟吡啶 -3-基硼酸 (72.3 mg, 0.513 mrnol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 无水硫酸镁干燥, 过滤,滤液减压浓缩,用硅胶柱色谱法纯化所得残余物,得到标题产物 (R)-{3-[l-(2- 氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙 基} -氨基甲酸叔丁酯 38a(100 mg, 浅黄色固体), 收率: 48%。
MS m/z (ESI): 602[M+1]
第二步
(R)-3-氨基 -1-[1-(2-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 38a(100 mg, 0.166 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁
-1-酮盐酸盐 38(55 mg,黄色固体), 收率: 66%。
S m/z (ESI): 502[M+1]
1HNMR (400MHz, CD3OD): δ 8.23-8.29(m, 2H), 7.18-7.50(m, 3H), 4.87-4.95(m, 2H),
3.93-4.43(m, 5H), 2.79-3. ll(m, 4H)。 实施例 39
(R)-3-氨基 -l-[l-(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]-3-氧代 -l-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 6-氟吡啶 -3-基硼酸 (72.3 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4,,6'-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 39a(150 mg, 浅黄色固体), 收率: 73 %。
MS m/z (ESI): 602[M+1]
第二步
(R)-3-氨基 -1 -[1 -(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l -(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -3_氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 39a(l 50 mg, 0.249 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基小[1-(6-氟吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 39(90 mg,黄色固体), 收率: 72%。
MS m/z (ESI): 502[M+1] 实施例 40
(R)-3-氨基 -l-[l-(2-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1 -a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -l-(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氟吡啶 -4-基硼酸 (72.3 mg, 0.513 mmoi), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL 微波反应管中, 氩气保护下, 100Ό, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸续干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(2-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 40a(130 mg,浅黄色固体), 收率: 63 %。
MS m/z (ESI): 602[M+1]
第二步
(R)-3-氨基 -1-[1-(2-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2-氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1 -(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 40a(l 30 mg, 0.215 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2-氟吡啶 -4-基) -3-三氟申基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐 40(80 mg,黄色固体), 收率: 76%。
MS m/z (ESl): 502[M+1] 实施例 41
(R)-3-氨基小 [1-(6-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基)-丁小酮盐酸盐

第一步
(R)-{3-[l-(6-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12bC200 mg, 0.342 mmol), 6-甲氧基吡啶 -3-基硼酸 (78.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100Ό , 在微波下反应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (RM3-[l-(6-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 41a(155 mg, 浅黄色固体),收率: 74%。 MS m/z (ESI): 614[M+1]
第二步
(R)-3-氨基 -l-[l-(6-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(6-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 41a(155 mg, 0.252 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基— ^(6-甲氧基吡啶 _3_基 )—3_三氟甲基-咪唑并 [ —a]哌嗪 _7_基] _4_(2,4,5_三氟苯 基) -丁 -1-酮盐酸盐 41(100 mg,黄色固体), 收率: 77%。
MS m/z (ESI): 514[M+1]
iHNMR (400MHz, CD3OD): δ 8.50(m, 1H), 8.38(m, 1H), 7.39(m, 1H), 7.23(m, 1H), 5.12(m, 2H), 4.35(m, 2H), 4.18(m, 4H), 3.95(m, 2H), 3.14(m, 2H), 2.95(m, 2H), 2.04(m; 1H), 2.02(m, 1H) 实施例 42
(R)-3-氨基 -l-[l-(4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 _1-酮盐酸盐

第一歩
(R)-{3-[l-(4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 4-甲氧基吡啶 -3-基硼酸 (78.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4',6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL i敫波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL zK, 用乙酸乙酯萃取 (15 ml 3), 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (RM3-[l-(4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 42a(125 mg, 浅黄色固体),收率: 60%。 MS m/z (ESI): 614[M+1]
第二步
(R)-3-氨基- 1-[1-(4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 42a(125 mg, 0.204 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1- (4-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐 42(70 mg,黄色固体), 收率: 68 %。
MS m/z (ESI): 514[M+1]
^MR (400MHz, CD3OD): δ 8.86-8.77(m, 2H), 7.81-7.79(m, 1H), 7.43-7.42(m, 1H), 7.23-7.19(m, 1H), 4.43-4.32(m, 4H), 4.13-3.95(m, 3H), 3.14-2.97(m, 4H) 实施例 43
(R)- 3-氨基 -l-[l-(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁小酮盐酸盐

第一步
(R)-{3-[l-(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1 ,5- a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基)- 3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-甲氧基吡啶 -3-基硼酸 (78.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 - 2,,4',6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100 , 在微波下反应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL z , 用乙酸乙酯萃取 (15 ml 3) , 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 43a(130 mg, 浅黄色固体),收率: 62%。 MS m/z (ESI): 614[M+1]
第二步
(R)-3-氨基 -1 -[1-(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 _1-酮盐酸盐
将 (R)-{3-[l -(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 43a(130 mg, 0.211 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2-甲氧基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐 43(60 mg,黄色固体), 收率: 56 %。
MS m/z (ESI): 514[M+1] 实施例 44
(R)-3-氨基 -l-[l-(3-甲氧基吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(3-甲氧基吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 3-甲氧基吡啶 -4-基硼酸 (78.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反.应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(3-甲氧基吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 44a(110 mg, 浅黄色固体),收率: 52%。 MS m/z (ESI): 614[M+1]
第二步
(R)-3-氨基 -1-[1-(3-甲氧基吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(3-甲氧基吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 44a(110 mg, 0.179 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)- 3-氨 基 -l- -p—甲氧基吡啶 _4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯 基) -丁 -1-酮盐酸盐 44(80 mg,黄色固体), 收率: 86%。
MS m/z (ESI): 514[M+1] 实施例 45
(R)-3-氨基 -1-(1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯基) -丁 -1- 酮盐酸盐

将 (R)- [3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(038 g, 0.65 mmol)搅拌下溶解于 2 mL 3N氯化氢的乙酸 乙酯溶液中, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反 应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-氨基 溴 -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯基) -丁 -1-酮盐酸盐 45(0.25 g,淡黄色 固体), 收率: 73.5%。
MS m/z (ESI): 485.2[M+1]
1HNMR (400MHz, CD3OD): δ 7.35(m, 1H), 7.18(m, 1H), 4.71(m, 2H), 4.3 l(m, 1H), 4.20(m, 1H), 4.02(m, 2H), 3.74(m, 1H), 3.00(m, 2H), 2.78(m, 2H) 实施例 46
(R)-3-氨基 -1-[1-(2H-吡唑 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2H-吡唑 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟苄基) - 丙基〗 -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2H-吡唑 -3-基硼酸 (65 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2,,4',6'-三异丙 基联苯 (6.52 mg, 0.014 mmol),磷酸钾 (145 mg, 0.684 mmoi),2 mL叔丁醇加入 10 mL
微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反 应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(2H-吡唑 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟 苄基) -丙基 } -氨基甲酸叔丁酯 46a(110 mg, 浅黄色固体), 收率: 56%。
MS m/z (ESI): 573 [M+1]
第二步
(R)-3-氨基 -1-[1-(2H-吡唑 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(lH-吡唑 -4-基)-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]-3-氧代 小 (2,4,5-三氟苄基) -丙基 }-氨基甲酸叔丁酯 46a(110 mg, 0.192 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(2H-吡唑 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) -丁 小酮盐酸盐 46(50 mg,黄色固体), 收率: 56%。
MS m/z (ESI): 473 [M+1] 实施例 47
(R)-3-氨基 -1-[1-(4-三氟甲基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(4-三氟甲基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)- [3-(1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 4-三氟甲基吡啶 -3-基硼酸 (98 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基-2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100Ό , 在微波下反应 1.5小时后, 反应完
毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 niL><3), 合并有机相, 用无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(4-三氟甲基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 47a(145 mg, 浅黄色固体),收率: 65 %。 MS m/z (ESI): 652[M+1]
第二步
(R)-3-氨基 -1 -[1-(4-三氟甲基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基] -4-(2,4,5-三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(4-三氟甲基吡啶 - 3-基) -3-三氟甲基-咪唑并 [1,5- a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 47a(145 mg, 0.222 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨 基 -1-[1-(4-三氟甲基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟 苯基) -丁 -1-酮盐酸盐 47(90 mg,黄色固体), 收率: 74 %。
MS m/z (ESI): 552[M+1] 实施例 48
(R)-3-氨基 -1-[1-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐

第一步
(R)-{3-[l-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1 -(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2,6-二氟吡啶 -4-基硼酸 (81.5 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol) , 2-二环己基膦基 -2,,4,,6,- 三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加 入 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完 毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水
硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,6-二氟吡啶 -4-基) -丙基 } -氮基甲酸叔丁酯 48a(125 mg,浅黄色固体),收率: 60
%。
MS m/z (ESI): 620[M+1]
第二歩
(R)-3-氨基 -1-[1-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 48a(125 mg, 0.2 mnol)溶解于 2 mL乙酸 乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消失., 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-3-氨基 -1-[1-(2,6-二氟吡啶 -4-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐 48(85 mg,黄色固体), 收率: 76%。
MS m/z (ESI): 520[M+1] 实施例 49
(R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 哌啶 -2-酮盐酸盐

第一步
1-(2-氧代哌啶 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 哌啶 -2-酮 (96 mg, 0.97 mmol),碘化亚铜 (46 mg, 0.24 mmol), 反式 -Ν,Ν'-二甲基环己 烷 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL 微波反应管中, 氩气保护下, 135°C, 在微波下反应 2小时后, 反应完毕, 将反应
液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(2-氧代哌啶 -1- 基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 49a(120 mg,黄色油状物), 收率- 38 %。
MS m/z (ESI): 389[M+1] 第二步
1-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -2-酮盐酸盐 将 1-(2-氧代哌啶 -1-基) -3-三氟甲基-咪唑并 [1,5- a]哌嗪 -7-羧酸叔丁酯 49a(120 mg, 0.31 mmol)溶解于 1 mL 2.4 氯化氢乙酸乙酯溶液中, 室温下搅拌 40分钟后, 补加 2 mL 2.4N氯化氢乙酸乙酯溶液, 继续反应 1小时, 薄层色谱跟踪反应, 原料 消失, 减压浓缩反应液, 得到黄色油状液体粗品标题产物 三氟甲基-咪唑并 [1,5-a]哌嗉 -1-基) -哌啶 -2-酮盐酸盐 49b, 不经分离直接进行下一步反应。
第三步
(R)-{3-[l-(2-氧代哌啶小基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟 基) -丙基 } -氨基甲酸叔丁酯
氮气氛下,将 1-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -2-酮盐酸盐 49b(100 mg, 0.31 mmoi)搅拌下溶解于 5 mL二氯甲烷中, 依次加入三乙胺 (0.21 mL, 1.54 mmol),(R)-3-叔丁氧羰基氨基- 4-(2,4,5-三氟-苯基) -丁酸 le(102 mg, 0.31 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (117.6 mg, 0.46 mrnol),室温下搅拌反应过夜,薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标題产 物 (R)-{3-[l-(2-氧代哌啶 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 49c(105 mg,无色油状液体),收率: 56.2 %。
MS m/z (ESI): 603[M+1]
第四步
(R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基]- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 哌啶 -2-酮盐酸盐
将 (R)-{3-[l-(2-氧代哌啶 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 49c(105 mg, 0.17 mmol)溶解于 5 mL 3.1N氯化氢甲醇溶液中, 室温下搅拌过夜, 减压浓缩反应液, 用硅胶柱色谱法纯 化所得残余物, 得到标题产物 (R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -1-基} -哌啶 -2-酮盐酸盐 49(44 mg, 白色固体),收率: 50 %。 MS m/z (ESI): 503[M+1]
ifi MR (400MHz, CD3OD): δ 7.40(m, 1H), 7.26(m, IH), 4.65(m, 2H), 4.33(m, IH), 4.25(d, IH), 4.08(d, 1H), 3.98(m, 2H), 3.81(d, 2H), 3.12(s, 2H), 2.98(m, IH), 2.85(m, IH), 2.60(m, 2H), 2.03(m, 4H)。
实施例 50
(R)-l-{7-[3-氨基 (2,4,5-三氟苯基)-丁酰基 ]_3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 乙酰胺盐酸盐

第一步
1-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 乙酰胺 (57.4 mg, 0.97 mmol), 碘化亚铜 (30.85 mg, 0.162 mmol), 反式 -Ν,Ν'-二甲基 环己烷 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL微波反应管中, 氩气保护下, 130Ό , 在微波下反应 2小时后, 反应完毕, 加 入 10 mL水, 用乙酸乙酯萃取 (15 mLx4), 合并有机相, 用 20 mL饱和氯化钠溶液 洗涤, 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 50a(200 mg, 白色固体:), 收率: 71 %。
MS m/z (ESI): 349[M+1]
1HNMR (400MHz, CDC13): δ 8.08(s, 1H), 4.68(s, 2H), 4.11(m, 2H), 3.86(m, 2H), 2.13(s, 3H), 1.49(s, 9H)
第二步
N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪小基) -乙酰胺三氟乙酸盐 将 1-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 50a(190 mg, 0.545 mmol)溶解于 5 mL二氯甲垸中, 缓慢滴加三氟乙酸 (1.26 mL, 16.36 mmol), 室温搅 拌 2小时, 薄层色谱跟踪反应, 原料消失, 减压浓縮反应液, 得到黄色油状液体 粗品标题产物 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -乙酰胺三氟乙酸盐 50b, 不 经分离直接进行下一步反应。
第三步
(R)-[3-(l-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基)- 3-氧代 -l-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯
氮气氛下, 将 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -乙酰胺三氟乙酸盐 50b(190 mg, 0.545 mmol)搅拌下溶解于 5 mL二氯甲烷中,依次加入三乙胺 (0.38 mL, 2.72 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(181.6 mg, 0.545 mmol)和双 (2-氧代 -3-噁唑垸基)次膦酰氯 (208 mg, 0.818 mmol), 室温下搅拌反应 2 小时, 薄层色谱跟踪反应, 原料消失, 加入 10 mL水, 用二氯甲烷萃取 (15 mLx4), 合并有机相, 用 20 mL饱和氯化钠溶液洗涤, 无水硫酸镁干燥, 过滤, 滤液减压 浓縮, 用硅胶柱色谱法纯化, 得到标题产物 (R)-[3-(l-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 50c(160 mg,无 色油状液体), 收率: 52%。
MS m/z (ESI): 564[M+1]
第四步
(R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 乙酰胺盐酸盐
将 (R)-[3-(l-乙酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 50c(160 mg, 0.28 mmol)溶解于 5 mL 3. IN氯化氢甲 醇溶液中, 室温下搅拌过夜, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}-乙酰胺盐酸盐 50(156 mg,黄色固体), 收率: 95 %。
MS m/z (ESI): 464[M+1]
!HNMR (400MHz, CD3OD): 5 7.37-7.3 l(m, 1H), 7.23-7.12(m, 1H), 4.90-4.66(m, 2H), 4.36-4.23(m, 2H), 4.12-3.97(m, 2H), 3.57-3.56(m, 1H), 2.91-2.81(m, 2H), 2.72-2.57(m, 2H), 2.21-2.19(s, 3H) 实施例 51
(R)-l-{7-[3-氣基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 甲酰胺盐酸盐


第一步
1-甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 甲酰胺 (43 mg, 0.97 mmol), 碘化亚铜 (46 mg, 0.24 inniol), 反式 -Ν,Ν'-二甲基环己烷 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL微 波反应管中, 氩气保护下, 135°C, 在微波下反应 2小时后, 反应完毕, 减压浓缩 反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-甲酰氨基 -3-三氟甲基- 咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 51a(190 mg, 白色固体), 收率: 70%。
MS m z (ESI): 335[M+1]
第二步
N-(3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -1 -基) -甲酰胺盐酸盐 将 1-甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 51a(190 mg, 0.57 mmol)溶解于 4 mL 2.4N氯化氢乙酸乙酯溶液中, 室温下搅拌 2小时, 薄层色谱跟 踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 N-(3-三氟甲基-咪唑并 [l,5-a] 哌嗪 -1-基) -甲酰胺盐酸盐 51b, 不经分离直接进行下一步反应。
第三步
(R)-[3-(l-甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟 基) - 丙基] -氨基甲酸叔丁酯
氮气氛下, 将 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)-甲酰胺盐酸盐 51b(154 mg, 0.57 mmol)搅拌下溶解于 5 mL二氯甲烷中, 依次加入三乙胺 (0.38 mL, 1.54 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(187 mg, 0.58 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (216 mg, 0.85 mmol), 室温下搅拌反应过夜, 薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产 物 (R)-[3-(l-甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 51c(l63 mg,无色油状液体), 收率: 52%。
MS m/z (ESI): 550[M+1]
第四歩
(R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -
甲酰胺盐酸盐
将 (R)-[3-(l-甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1- (2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 51c(163 mg, 0.29 mmol)溶解于 5 mL 3.1 氯化氢甲 醇溶液中, 室温下搅拌过夜, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -1-基}-甲酰胺盐酸盐 51(79 mg, 白色固体), 收率: 55 %。
MS m/z (ESI): 450[M+1] 实施例 52
(R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 吡咯烷 -2-酮盐酸盐

第一步
1-(2-氧代吡咯烷 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 吡咯垸 -2-酮 (82 mg, 0.97 mmol),碘化亚铜 (46 mg, 0.24 mmol), 反式 -Ν,Ν'-二甲基环 己烷 -1,2-二胺 (23 mg, 0.16 mmol),碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL 微波反应管中, 氩气保护下, 135°C, 在微波下反应 2小时后, 反应完毕, 减压浓 缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(2-氧代吡咯烷 -1- 基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 52a(180 mg, 白色固体),收率: 61 %。
MS m/z (ESI): 361 [M+1]
第二歩
1-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -吡咯垸 -2-酮盐酸盐 将 1-(2-氧代吡咯垸 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 52a(l 80 mg, 0.50 mmol)溶解于 4 mL 2.4N氯化氢乙酸乙酯溶液中, 室温下搅拌 2
小时, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 1-(3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -1-基) -吡咯垸 -2-酮盐酸盐 52b,不经分离直接进行下一歩反 应。
第二歩
(RM3-[l-(2-氧代吡咯烷小基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基] -3-氧代
小 (2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 氮气氛下, 将 1-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -吡咯烷 -2-酮盐酸盐 52b(148 mg, 0.5 mmol)搅拌下溶解于 5 mL二氯甲垸中, 依次加入三乙胺 (0.34 mL, 2.48 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(164 mg, 0.5 mmol) 和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (189 mg, 0.74 mmol), 室温下搅拌过夜, 薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产 物 (R {3- [1-(2-氧代吡咯烷 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 52e(144 mg,无色油状液体), 收率: 50 %。
MS m/z (ESI): 576[M+1]
第四步
(R)-l-{7- [3-氨基斗 (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 吡咯垸 -2-酮盐酸盐
将 (R)-{3-[l-(2-氧代吡咯垸 -1-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 52c(144 mg, 0.25 mmol)溶解于 5 mL 3.1 氯化氢甲醇溶液中, 室温下搅拌过夜, 减压浓缩反应液, 用硅胶柱色谱法纯 化所得残余物, 得到标题产物 (R)-l-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -1-基}-吡咯烷 -2-酮盐酸盐 52(69 mg, 白色固体), 收率: 55 %。
MS m/z (ESI): 476[M+1] 实施例 53
(R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 1,1-二甲基脲盐酸盐


第一步
l-(3,3-二甲基 -脲基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯
将 1-溴 -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-羧酸叔丁酯 7b(500 mg, 1.35 mmol), 1,1-二甲基脲 (143 mg, 1.62 mmol), 碘化亚铜 (51.3 mg, 0.27 mmol), 反式 -Ν,Ν'-二甲 基环己烷 -1,2-二胺 (39 mg, 0.27 mmol),碳酸钾 (347 mg, 2.71 mmol) , 5 mL二甲苯力口 入 20 mL微波反应管中,氩气保护下, 135°C,在微波下反应 2小时后,反应完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-(3,3-二甲基- 脲基) -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-羧酸叔丁酯 53a(77 mg,浅黄色固体),收率: 15 %。
MS ni/z (ESI): 378[M+1]
第二步
U-二甲基 -3-(3-三氟甲基-咪唑并 [1,5-a]哌嗪小基) -脲盐酸盐 将 1-(3,3-二甲基 -脲基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 53a(77 mg, 0.2 mmol)溶解于 5 mL乙酸乙酯中, 加入 5 mL 2.4N氯化氢乙酸乙酯溶液, 室温下 搅拌 2小时, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 1,1- 二甲基 -3-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -脲盐酸盐 53b, 不经分离直接进行 下一步反应。
第三步
(R)-{3-[l-(3,3-二甲基 -脲基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯
氮气氛下, 将 U-二甲基 -3-(3-三氟甲基-咪唑并 [l,5-a]哌嗪 -1-基) -脲盐酸盐 53b(70 mg, 0.22 mmol)搅拌下溶解于 5 mL二氯甲垸中, 依次加入三乙胺 (89 mg, 0.88 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(73 mg, 0.22 mmol) 和双 (2-氧代 -3-噁唑垸基)次膦酰氯 (84 mg, 0.33 mmol), 室温下搅拌过夜, 薄层色谱 跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-{3-[l-(3,3-二甲基 -脲基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯 53c(78 mg, 浅黄色油状液体), 收率: 60%。
MS m/z (ESI): 593 [M+1]
第四步
(R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪小 基}-1,1-二甲基脲盐酸盐
将 (R)-{3-[l-(3,3-二甲基 -脲基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 53c(78 mg, 0.13 mmol)溶解于 5 mL 3. IN 氯化氢甲醇溶液中, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压 浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -1-基}-1,1-二甲基脲盐 酸盐 53(41 mg, 白色固体), 收率: 59%。
MS m/z (ESI): 493[M+1]
!HNMR (400MHz, CD3OD): δ 7.39(m, 1H), 7.24(m, 1H), 4.76(m, 2H), 4.32(m, 2H), 4.02(m, 4H), 2.85-3.10(m, 9H), 实施例 54
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪小 基}-^-甲基乙酰胺盐酸盐

第一步
1- (乙酰基甲基氨基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), N-甲基-乙酰胺 (71 mg, 0.97 mmol), 碘化亚铜 (46 mg, 0.24 mmol), 反式 -Ν,Ν'-二甲 基环己垸 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL微波反应管中, 氩气保护下, 135°C , 在微波下反应 2小时后, 反应完毕, 减压浓縮反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1- (乙酰基甲基 氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 54a(206 mg, 白色固体), 收率: 70%。
MS m/z (ESI): 363 [M+l]
+ 第二步
N-甲基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)-乙酰胺盐酸盐 将 1- (乙酰基甲基氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 54a(206 mg, 0.57 mmol)溶解于 4 mL 2.4N氯化氢乙酸乙酯溶液中,室温下搅拌反应 2小时, 薄层色谱跟踪反应, 原料消失,减压浓缩反应液,得到标题产物 N-甲基 -N-(3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -1-基)-乙酰胺盐酸盐 54b, 不经分离直接进行下一步反应。
第三步
(R)-{3-[l- (乙酰基甲基氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟苄基) -丙基 氨基甲酸叔丁酯
氮气氛下, 将 N-甲基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -乙酰胺盐酸盐 54b(171 mg, 0.57 mmol)搅拌下溶解于 5 mL二氯甲垸中,依次加入三乙胺 (0.39 mL, 2.83 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(187 mg, 0.57 mmol) 和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (216 mg, 0.85 mmol), 室温下搅拌反应过夜, 薄 层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化, 得到标题 产物 (R)- {3-[1- (乙酰基甲基氨基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 54c(171 mg,无色油状液体), 收率: 52 %。
MS m/z (ESI): 578[M+1]
第四步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基]- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-N-甲基乙酰胺盐酸盐
将 (R)-{3-[l- (乙酰基甲基氨基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 54c(171 mg, 0.3 mmol)溶解于 5 mL 3.1N 氯化氢甲醇溶液中, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压 浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}- -甲基乙酰胺盐 酸盐 54(84 mg, 白色固体), 收率: 55 %。
MS m/z (ESI): 478[M+1] 实施例 55
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 甲磺酰胺盐酸盐


第一步
1-甲磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol), 甲磺酰胺 (92 mg, 0.97 mmol), 碘化亚铜 (46 mg 0.24 mmol), 反式 -Ν,Ν'-二甲基环己 烷 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL 微波反应管中, 氩气保护下, 135 C, 在微波下反应 2小时后, 反应完毕, 减压浓 缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-甲磺酰氨基 -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 _7-羧酸叔丁酯 55a(158 mg, 白色固体), 收率: 75 %
MS m/z (ESI): 385[M+1]
第二步
N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪小基) -甲磺酰胺盐酸盐 将 1-甲磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 55a(158 mg, 0.41 mmol)溶解于 4 mL 2.4N氯化氢乙酸乙酯溶液中, 室温下搅拌 2小时, 薄层色 谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -甲磺酰胺盐酸盐 55b, 不经分离直接进行下一步反应。
(R)-[3-(l-甲磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
氮气氛下,将 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -甲磺酰胺盐酸盐 55b(131 mg, 0.41 mmol)搅拌下溶解于 5 mL二氯甲烷中, 依次加入三乙胺 (0.28 mL, 2.03 mmol), (R)-3-叔丁氧羰基氨基- 4-(2,4,5-三氟-苯基) -丁酸 le(135 mg, 0.41 ol)和双 (2-氧代 -3-噁唑垸基)次膦酰氯 (155 mg, 0.61 mmol), 室温下搅拌反应过夜, 薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产 物 (R)-[3-(l-甲磺酰氨基 -3-Ξ氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟 苄基) -丙基] -氨基甲酸叔丁酯 55c(133 mg,无色油状液体), 收率: 54%
MS m/z (ESI): 600[M+1]
第四步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 甲磺酰胺盐酸盐
将 (R)- [3-(l-甲磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -l-(2,4,5- 三氟苄基) -丙基] -氨基甲酸叔丁酯 55c(133 mg, 0.22 mmol)溶解于 5 mL 3.1N氯化氢 甲醇溶液中, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反 应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-N-{7-[3-氨基 -4-(2,4,5- 三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -甲磺酰胺盐酸盐 55(71 mg, 白色固体), 收率: 60%。
MS m/z (ESI): 500[M+1] 实施例 56
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯甲磺酰胺盐酸盐

1-苯磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 氮气氛下, 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81 mmol),苯磺酰胺 (152 mg, 0.97 mmol),碘化亚铜 (46 mg, 0.24 mmol),反式 -Ν,Ν'- 二甲基环己垸 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯 加入封管中, 135°C, 反应 16小时后, 反应完毕, 减压浓缩反应液, 用硅胶柱色 .谱法纯化所得残余物, 得到标题产物 1-苯磺酰氨基 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 56a(253 mg, 白色固体), 收率: 70 %。
MS m/z (ESI): 447[M+1]
第二步
N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯磺酰胺盐酸盐 将 1-苯磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 56a(253 mg,
0.57 mmol)溶解于 4 mL 2.4N氯化氢乙酸乙酯溶液中, 室温下搅拌 2小时, 薄层色
谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯磺酰胺盐酸盐 56b, 不经分离直接进行下一步反应。
第三歩
(R)-[3-(l-苯磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基)- 3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
氮气氛下,将 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯磺酰胺盐酸盐 56b(218 mg, 0.57 mmol)搅拌下溶解于 5 mL二氯甲烷中, 依次加入三乙胺 (0.39 mL, 2.83 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(187 mg, 0.57 mmol)和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (216 mg, 0.85 mmol), 室温下搅拌过夜, 薄层色谱跟 踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)- [3-(1-苯磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟苄 基) -丙基]-氨基甲酸叔丁酯 56c(146 mg,无色油状液体), 收率: 52%。
MS m/z (ESI): 662[M+1]
第四步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯磺酰胺盐酸盐
将 (R)-[3-(l-苯磺酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5- 三氟苄基) -丙基] -氨基甲酸叔丁酯 56c(146 mg, 0.22 mmol)溶解于 5 mL 3.1N氯化氢 甲醇溶液中, 室温下搅拌过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯 基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯磺酰胺盐酸盐 56(73 mg, 白色 固体), 收率: 55 %。
MS m/z (ESI): 562[M+1] 实施例 57
(R)-3-氨基 -1-(1-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐


第一步
1-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(200 mg, 0.54 mmol), 二甲胺 (29 mg, 0.65 mmol), 三 (二亚苄基丙酮)二钯 (2.4 mg, 0.0026 mmol), 2- (二叔 丁基膦)联苯 (3.2 mg, 0.022 mmol), 叔丁醇钠 (78 mg, 0.81 mmol), 2 mL干燥的甲苯 放入 20 mL微波反应管中, 氩气保护下, 100T;, 在微波下反应 70分钟后, 反应 完毕, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-二甲 氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 57a(90 mg, 白色固体), 收率: 50%。
MS m/z (ESI): 335[M+1]
第二步
二甲基 -(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -胺三氟乙酸盐 将 1-二甲氨基- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 57a(90 mg, 0.27mmol)溶解于 10 mL二氯甲烷中, 搅拌下滴加三氟乙酸 (1.84 g, 16mmol), 所得 的溶液继续搅拌 2小时后反应完毕, 减压浓缩反应液, 得到标题产物二甲基 -(3-三 氟甲基-咪唑并 [l,5-a]哌嗪- 1-基) -胺三氟乙酸盐 57b, 不经分离直接进行下一步反 应。
第三步
(R)-[3-(l-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯
氮气氛下, 将二甲基 -(3-三氟甲基-咪唑并 [1,5-a]哌嗪小基) -胺三氟乙酸盐 57b 搅拌下溶解于 20 mL二氯甲烷中, 依次加入三乙胺 (0.40 g, 4 mniol)和 (R)-3-叔丁氧 羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(147 mg, 0.44 mmol), 搅拌 10分钟后, 加入 双 (2-氧代 -3-噁唑垸基)次膦酰氯 (152 mg, 0.6 mmol), 室温下搅拌 2小时后, 薄层色 谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产 物 (R)-[3-(l-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯 57c(l l l mg,无色油状液体), 收率: 75 %。
MS m/z (ESI): 550[M+1]
第四步
(R)-3-氨基 -1-(1-二甲氨基- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟苯基) - 丁 -1-酮盐酸盐
将 (R)-[3-(l-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三 氟苄基) -丙基] -氨基甲酸叔丁酯 57c(l 11 mg, 0.2 mmol)溶解于 4 mL 4.3N氯化氢甲醇 溶液中, 溶液在搅拌下反应 2小时后反应完毕, 减压浓缩反应液, 用硅胶柱色谱 法纯化所得残余物, 得到标题产物 (R)-3_氨基 -1-(1-二甲氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4- (2,4,5-三氟苯基) -丁 -1-酮盐酸盐 57(27 mg, 白色固体), 收率: 30%。
(R)- N-(5-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪小 基} -吡啶 -2基) -乙酰胺盐酸盐 '

第一步
(R)-{3-[l-(6-乙酰氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 将 (R)-{3-[l-(6-氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1 -(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 56a(155 mg, 0.252 mmol)溶解于 2 mL 吡啶中, 加入 0.5 mL乙酸酐, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消失, 将 反应液减压浓缩, 得到标题产物 (R)-{3-[l-(6-乙酰氨基吡啶 -3-基) -3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 58a(155 mg, 黄色固体), 收率: 97%。
MS ni/z (ESI): 641 [M+1]
第二步
(R)-N-(5-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基} -吡啶 -2基) -乙酰胺盐酸盐
将 (R)-{3-[l-(6-乙酰氨基吡啶 -3-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 58a(155 mg, 0.242 mmol)溶解于 6 mL二
氯甲烷和 2 mL甲醇中,搅拌下加入 5 mL 2N氯化氢乙酸乙酯溶液, 室温反应 2小 时, 薄层色谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-N-(5-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 吡啶 -2基) -乙酰胺盐酸盐 58(125 mg,黄色固体), 收率: 90%。
MS m/z (ESI): 541 [M+1]
!HNMR (400MHz, CD3OD): δ 8.70(m, 2H), 7.63(m, 1H), 7.43(m, 1H), 7.24(m, 1H), 5.16(m, 2H), 4.31-4.41(d, 2H), 3.96-4.14(d, 2H), 3.96(s, 1H), 3.02-3.14(m, 4H), 2.38(s, 3H) 实施例 59
(R)-N-{7-[3-氨基 -4- (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪小基} - 苯甲酰胺盐酸盐

第一步
1-苯甲酰氨基 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴- 3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(300 mg, 0.81mmol), 苯甲酰胺 (117.8 mg, 0.97mmol), 碘化亚铜 (46.3 mg, 0.24 mmol), 反式 -Ν,Ν'-二甲基 环己烷 -1,2-二胺 (23 mg, 0.16 mmol), 碳酸钾 (224 mg, 1.6 mmol), 5 mL甲苯加入 20 mL微波反应管中, 氩气保护下, 135°C , 在微波下反应 2小时后, 反应完毕, 减 压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-苯甲酰氨基 -3- 三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 59a(170 mg, 白色固体), 收率: 52 %。
MS m/z (ESI): 411[M+1]
一少
N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯甲酰胺盐酸盐 将 1-苯甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 59a(90 mg, 0.27mmol)溶解于 4 mL 2.1N氯化氢乙酸乙酯溶液中, 室温下搅拌过夜, 薄层色谱
跟踪反应,原料消失,减压浓缩反应液,得到标题产物 N-(3-三氟甲基-咪唑并 [1,5-a] 哌嗪 -1-基) -苯甲酰胺盐酸盐 59b, 不经分离直接进行下一步反应。
第三步
(R)-[3-(l-苯甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3 -氧代 -1-(2,4,5-三氟苄 基) -丙基] -氨基甲酸叔丁酯
氮气氛下,将 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯甲酰胺盐酸盐 59b(140 mg, 0.4 mmol)搅拌下溶解于 10 mL二氯甲烷中, 依次加入三乙胺 (204 mg, 2.02 mmol)和 (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(206 mg, 0.81 mmol),搅 拌 10分钟后, 加入双 (2-氧代 -3-噁唑烷基)次膦酰氯 (152 mg, 0.6 mmol), 室温下搅 拌过夜, 薄层色谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯 化,得到标题产物 (R)-[3-(l-苯甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 59c(170 mg, 白色固体), 收率: 67%。 MS m/z (ESI): 626[M+1]
第四步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 苯甲酰胺盐酸盐
将 (R)- [3-(1-苯甲酰氨基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5- 三氟苄基) -丙基] -氨基甲酸叔丁酯 59c(170 mg, 0.27 mmol)溶解于 5 mL乙酸乙酯中, 加入 5 mL 2.1N氯化氢乙酸乙酯溶液,室温下搅拌过夜, 反应完毕, 减压浓缩反应 液, 得到标题产物 (R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -苯甲酰胺盐酸盐 59(150 mg, 白色固体), 收率: 98 %。
MS m/z (ESI): 526[M+1]
'HNMR (400MHz, CD3OD): δ 8.01 (m, 2H), 7.65(m, 1H), 7.55(m, 2H), 7.40(m, 1H), 7.17(m, 1H), 4.87(m, 2H), 4.27-4.36(m, 2H), 3.94-4.12(m, 3H), 2.85-3.24(m, 4H), 实施例 60
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-4-甲基苯甲磺酰胺盐酸盐
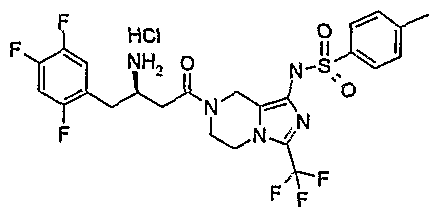

第一步
i - (甲苯 _4-磺酰氨基 3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-羧酸叔丁酯 氮气氛下, 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol), 对甲苯磺酰胺 (206 mg, 1.2 mmol), 碘化亚铜 (38 mg, 0.2 mmol), 反式 -Ν,Ν'- 二甲基环己烷 -1,2-二胺 (28 mg, 0.2 mmol), 碳酸钾 (276 mg, 2 mmol), 5 mL甲苯加 入封管中, 135°C下, 反应 16小时后, 反应完毕, 将反应液减压浓缩, 用硅胶柱 色谱法纯化所得残佘物, 得到标题产物 1- (甲苯 -4-磺酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 60a(280 mg, 白色固体), 收率: 61 %。
MS m/z (ESI): 461 [M+l]
第二步
4-甲基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)-苯磺酰胺盐酸盐 将 1- (甲苯 -4-磺酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 60(280 mg, 0.61 mmol)溶解于 5 mL 2.1N氯化氢乙酸乙酯溶液中, 室温下搅拌过夜, 薄层 色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 4-甲基 -N-(3-三氟甲基 -咪唑并 [1,5-a]哌嗪 -1-基) -苯磺酰胺盐酸盐 60b, 不经分离直接进行下一步反应。
第三步
(R -{3-[l- (甲苯 -4-磺酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5- 三氟苄基) -丙基 } -氨基甲酸叔丁酯 .
氮气氛下, 将 4-甲基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -苯磺酰胺盐酸盐
60b(250 mg, 0.63 mmol)搅拌下溶解于 10 mL二氯甲烷中,依次加入三乙胺 (0.2 mL, 1.26 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(210 mg, 0.63 mmol) 和双 (2-氧代 -3-噁唑烷基)次膦酰氯 (240 mg, 0.95 mmol), 室温下搅拌 3小时, 薄层 色谱跟踪反应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题 产物 (R)-{3-[l - (甲苯 -4-磺酰氨基) -3-三氟甲基-咪唑并 [ 1 ,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 60c(100 mg,无色油状液体),收率:23.5 %。
MS m/z (ESI): 676[M+1]
第四步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-4-甲基苯甲磺酰胺盐酸盐
将 (R)-{3-[l- (甲苯 -4-磺酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氣基甲酸叔丁酯 60c(100 mg, 0.148 mmol)溶解于 5 mL 2.1N氯化氢乙酸乙酯溶液中, 室温下搅拌 5小时, 减压浓缩反应液, 用硅胶柱色 谱法纯化所得残余物, 得到标题产物 (R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰 基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 4-甲基苯甲磺酰胺盐酸盐 60(80 mg, 白色 固体), 收率: 88 %。
MS m/z (ESI): 576[M+1]
^MR (400MHz, CD3OD): δ 7.64(t, 2H), 7.34(d, 2H), 7.18(m, 2H), 4.80(m, 2H), 4.23(d, 2H), 4.03(m, 2H), 3.76(s, 1H), 2.99(s, 2H), 2.80(m, 2H), 2.44(s, 3H) 实施例 61
(R)-N-(4-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-2-氟苯基) -乙酰胺盐酸盐

第一步
2-氟 -4-(4,4,5,5-四甲基 -1,3,2-二氧硼戊环 -2-基)苯胺 氮气氛下, 将 4-溴 -2-氟苯胺 (570 mg, 3 mmol), 二氯二乙腈钯 (23.3 mg, 0.09 mmol), 2-二环己基膦基 -2,,6,-二甲氧基联苯 (73.9 mg, 0.18 mmol)溶解于 15 mL甲 苯中,搅拌下加入三乙胺 (758 mg, 7.5 mmol)和 4,4,5,5-四甲基 -1,3,2-二氧硼戊环 (461 mg, 3.6 mmol), 90°C下搅拌 5小时, 薄层色谱跟踪反应, 原料消失, 将反应液减 压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 2-氟 -4-(4,4,5,5-四甲基 -1,3,2-二氧硼
戊环 -2-基)苯胺 61a(400 mg,无色油状液体), 收率: 56%。
MS m/z (ESI): 238[M+1]
_ .
(R)-{3-[l- (4-氨基 -3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟节基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 2-氟 -4-(4,4,5,5-四甲基 -1,3,2-二氧 硼戊环 -2-基)苯胺 61a(122 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2',4,,6,-三异丙基联苯 (6.52 mg, 0.014 mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 1.5小时后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(4-氨基 -3-氟苯基 )-3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基] -3-氧代 -1- (2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 61b(120 mg, 浅黄色固体), 收率: 57%。
MS m/z (ESI): 616[M+1]
第三步
(RM3-[l-(4-乙酰氣基 -3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-{3-[l-(4-氨基 -3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 61b(120 mg, 0.19 mmol)溶解于 2 mL吡 啶中, 加入 0.5 mL乙酸酐, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消失, 将反 应液减压浓缩, 得到标题产物 (R {3-[l-(4-乙酰氨基 -3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 61c(115 mg,黄 色固体), 收率: 93 %。
MS m/z (ESI): 658[M+1]
第四步
(R)_N-(4-{7-[3-氨基 -4- (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-2-氟苯基) -乙酰胺盐酸盐
将 (R)-{3-[l-(4-乙酰氨基 -3-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 61c(115 mg, 0.174皿 nol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)_N_(4-{7_[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-2-氟苯基) -乙酰胺盐酸盐 61(70 mg,黄色固体), 收率: 72%。
MS m/z (ESI): 558[M+1]
1H MR (400MHz, CD3OD): δ 8.06(m, 1H), 7.40(m, 4H), 5.10(m, 2H), 4.38(m, 2H), 4.05(m, 3H), 3.05(m, 4H), 2.20(s, 3H) 实施例 62
(R)_N— (4_{7_[3-氣基—4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-2-氟苯基) -乙酰胺盐酸盐

第一步
4-氟 -3-(4,4,5,5-四甲基 -1,3,2-二氧硼戊环 -2-基)苯胺 氮气氛下, 将 3-溴 -4-氟苯胺 (570 mg, 3 mmol), 二氯二乙腈钯 (23.3 mg, 0.09 mmol), 2-二环己基膦基 -2,,6,-二甲氧基联苯 (73.9 mg, 0.18 mmol)溶解于 15 mL甲 苯中,搅拌下加入三乙胺 (758 mg, 7.5 mmol)和 4,4,5,5-四甲基 -1,3,2-二氧硼戊环 (461 mg, 3.6 mmol), 90°C下搅拌 5小时, 薄层色谱跟踪反应, 原料消失, 将反应液减 压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 4-氟 -3-(4,4,5,5-四甲基 -1,3,2-二氧硼 戊环 -2-基)苯胺 62a(400 mg,无色油状液体), 收率: 56%。
MS m/z (ESI): 238[M+1]
第二步
(R)-{3-[l-(5-氨基 -2-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟节基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(200 mg, 0.342 mmol), 4-氟 -3-(4,4,5,5-四甲基 -1,3,2-二氧 硼戊环 -2-基)苯胺 62a(122 mg, 0.513 mmol), 二 (二亚苄基丙酮)钯 (4 mg, 0.0068 mmol), 2-二环己基膦基 -2',4,,6'-三异丙基联苯 (6.52 mg, O.O mmol), 磷酸钾 (145 mg, 0.684 mmol), 2 mL叔丁醇加入 10 mL微波反应管中, 氩气保护下, 100°C, 在微波下反应 L5小时后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取
(15 mLx3), 合并有机相, 用无水硫酸镜干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(5-氨基 -2-氟苯基 )-3-三氟甲基 -咪唑 并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 62b(120 mg, 浅黄色固体), 收率: 57%。
MS ra/z (ESI): 616[M+1]
第三步
(R)-{3-[l-(5-乙酰氨基 -2-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-{3-[l-(5-氨基 -2-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 62b(120 mg, 0.19 mmol)溶解于 2 mL吡 啶中, 加入 0.5 mL乙酸酐, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消失, 将反 应液减压浓縮, 得到标题产物 (R)-{3-[l-(5-乙酰氨基 -2-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 62c(115 mg,黄 色固体), 收率: 93 %。
MS m/z (ESI): 658[M+1]
第四步
(R)_N-(4-{7- [3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 2-氟苯基) -乙酰胺盐酸盐
将 (R)-{3-[l-(5-乙酰氨基 -2-氟苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 62c(115 mg, 0.174 mmol)溶解于 2 mL乙 酸乙酯中, 加入 2 mL 2N氯化氢乙酸乙酯溶液, 室温搅拌过夜, 薄层色谱跟踪反 应, 原料消失, 将反应液减压浓缩, 用硅胶柱色谱法纯化, 得到标题产物 (R)-N-(4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -1- 基}-2-氟苯基) -乙酰胺盐酸盐 62(70 mg,黄色固体), 收率: 71%。
MS m/z (ESI): 558[M+1] 实施例 63
(R)-4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 苯甲酰胺盐酸盐

MS m/z (ESI): 525[M+1]
'HNMR (400MHZ, CD3OD): δ 8.34(s, 2H), 7.97-8.05(m, 2H), 7.56-7.74(m, 2H), 4.97-5.05(m, 2H), 4.33-4.35(s, 1H), 3.95-3.98(m, 2H), 3.76(m, 1H), 3.17(s, 1H), 2.99-3.16(m, 3H), 1.24(s, 3H) 实施例 64
(R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 苯甲酰胺盐酸盐

在 25 mL茄形瓶中加入化合物 (R)-3-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰] -3-三 氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -苄腈盐酸盐 27(60 mg, 0.11 mmol),加入 1 mL三氟 乙酸 /浓硫酸 0:v=4:l)的混合溶剂, 室温搅拌过夜, 薄层色谱跟踪反应, 原料消失, 反应液用冰浴冷却, 慢慢滴加氨水调 pH为 8-9, 用乙酸乙酯萃取 (20 mLx3), 合并 有机相, 用无水硫酸镁干燥, 抽滤, 减压浓缩滤液, 向残留物中加入 4 mL 4.3N氯 化氢甲醇溶液,搅拌 10分钟后,减压浓缩,得到标题产物 (R)-3-{7-[3-氨基 -4-(2,4,5- 三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -苯甲酰胺盐酸盐 64(40 mg, 黄色固体), 收率: 65 %。
MS m/z (ESI): 525[M+1]
lWMR (400MHz, CD3OD): δ 8.15(d, 1H), 7.84(m, 2H), 7.57(m, 1H), 7.49(m, 1H), 7.19(m, 1H), 5.09(m, 1H), 4.36(d, 2H), 4.06(m, 2H), 3.60(m, 2H), 3.09(m, 2H), 2.91(ra,
实施例 65
(R)-N-(4-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪小 基 苯基) -甲磺酰胺盐酸盐

第一步
(R)-{3-[l-(4-甲磺酰氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(150 mg, 0.256 mmol), 4- (甲磺酰氨基)苯硼酸 (83.8 mg, 0.282 mmol),四三苯基膦化钯 (30 mg, 0.0256 mmol),碳酸钾 (70.8 mg, 0.512 mmol), 2 mL乙二醇二甲醚和 2 mL水放入 10 mL微波反应管中, 氩气保护下, 130°C, 在 微波下反应 30分钟后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法 纯化所得残余物, 得到标题产物 (R)-{3-[l-(4-甲磺酰氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 65a(120 mg, 白 色固体), 收率: 70 %。
MS m/z (ESI): 676[M+1]
第二步
(R)-N-(4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 苯基) -甲磺酰胺盐酸盐
将 (R)-{3-[l-(4-甲磺酰氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗉 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 氨基甲酸叔丁酯 65a(150 mg, 0.222 mmol)溶解于 5 mL 2N氯化氢乙酸乙酯溶液, 析出白色固体, 室温搅拌 2小时, 薄层色谱跟踪反应, 原料消失,将反应液过滤,得到白色固体,真空干燥,得到标题产物 (R)-N-(4-{7-[3- 氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基 苯基) -甲磺 酰胺盐酸盐 65(100 mg,黄色固体), 收率: 73 %。
MS m/z (ESI): 576[M+1]
JHNMR (400MHz, CD3OD): δ 7.66-7.62(m, 2H), 7.43-7.37(m, 3H), 7.27-7.16(m, 1H), 5.11-4.93(m, 2H), 4.40-4.31(m, 2H), 4.15-3.92(m, 3H), 3.12-2.82(m, 7H) 实施例 66
〗00
(R)-3-氨基 -1-(1-噻吩 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-( 4,5-三氟-苯 基) -丁 -1-酮盐酸盐

第一步
(R)-[3-氧代 -3-(1-噻吩 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟-苯 基) -丙基 ]-氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(293 mg, 0.5 mmol), 噻吩 -3-基硼酸 (96 mg, 0.75 mmol), 四三苯基膦化钯 (58 mg, 0.05 mmol), 碳酸钾 (138 mg, 1 mmol), 3 mL乙二醇二甲醚 和 3 mL 7K放入 20 mL微波反应管中,氩气保护下, 120°C, 在微波下反应 30分钟 后, 反应完毕, 向反应液中加入 5 mL水, 用乙酸乙酯萃取 (15 mLx3), 合并有机 相, 用无水硫酸镁千燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-氧代 -3-(1 -噻吩 -3-基 -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -Ί- 基) -1-(2,4,5-三氟-苯基)-丙基] -氨基甲酸叔丁酯 66a(200 mg, 浅黄色固体), 收率- 68 %。
MS m/z (ESI): 589[M+1]
笛一丄 !=·
(R)-3-氨基 -1-(1-噻吩 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯 基) -丁 -1-酮盐酸盐
将 (R)-[3-氧代 -3-(1-噻吩 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基)小(2,4,5-三 氟-苯基) -丙基] -氨基甲酸叔丁酯 66a(0.2 g, 0.34 mmol)放入反应瓶中, 加入 2 mL乙 酸乙酯,搅拌使其溶解, 再加入 3 mL 2.0N的氯化氢乙酸乙酯溶液, 室温下搅拌反 应 1.5小时, 薄层色谱跟踪反应, 原料消失, 过滤反应液, 收集固体, 真空千燥, 得到标题产物 (R)-3-氨基 噻吩 -3-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基) -4-(2,4,5-三氟-苯基)-丁小酮盐酸盐 66(0.13 g,黄色固体), 收率: 72.9%。
MS m/z (ESI): 489.2[M+1] 实施例 67
(R)-3-氨基小[1-(4-甲磺酰 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 4-(2,4,5-三氟
-苯基) -丁 -1 -酮盐酸盐

第一步
(R)-{3-[l-(4-甲磺酰 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三 氟苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(0.3 g, 0.5 mmol), 4- (甲磺酰)苯硼酸 (123 mg, 0.6 mmol), 2-二环己基膦基-2,,4,,6,-三异丙基联苯(9.53 mg, 0.02 mmol), 二 (二亚苄基丙酮)二 钯 (0.006 g, 0.01 mmol)以及磷酸钾 (0.22 g, 1 mmol)搅拌下溶解于 10 mL正丁醇中, 氩气保护下, 130Ό , 在微波下反应 30分钟后, 反应完毕, 过滤反应液, 固体用 二氯甲烷 /甲醇 (v:V=10:l)洗涤, 收集洗涤液,抽干,得到标题产物 (R)-{3-[l-(4-甲磺 酰-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨 基甲酸叔丁酯 67a(150 mg, 白色固体), 收率: 44%。
MS m/z (ESI): 661.2[M+1]
第二步
将 (R)-{3-[l-(4-甲磺酰 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟苄基) -丙基 } -氨基甲酸叔丁酯 67a(150 mg, 0.227 mmol)搅拌下溶解于 5 mL二氯甲烷 /乙酸乙酯 (v:v=l :l)混合溶剂中, 加入 10 mL 2N氯化氢乙酸乙酯溶 液, 反应 1小时, 薄层色谱跟踪反应, 原料消失, 抽干溶剂, 得到标题产物 (R)-3- 氨基 -1-[1-(4-甲磺酰 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟-苯 基) -丁 -1-酮盐酸盐 67(0.12 g, 白色固体), 收率: 88.6%。
MS m/z (ESI): 561.3[M+1]
iHNMR (400MHz, CD3OD): δ 8.01-7.97(m, 2H), 7.88-7.85(m, 2H), 7.31-7.29(m, 1H), 7.11-7.20(m, 1H), 5.00-5.08(m, 2H), 4.24-4.32(m, 2H), 2.85-3.08(m, 3H), 2.80-3.12(m, 5H),2.64-2.80(m, 2H)。 实施例 68
(R)-N-[4-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-2-氟-苯基] -环丙基甲酰胺盐酸盐

第一步
(R)-[3-{l- [4- (环丙基甲酰氨基) -3-氟-苯基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基}-3- 氧代 -1-(2,4,5-三氟-苄基) -丙基] -氮基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(0.292 g, 0.5 mmol), 环丙基甲酰 [3-氟 -5-(4,4,5-三甲基 -[1,3,2]二氧硼戊环 -2-基)-苯基] -胺 (123 mg, 0.6 mmol), 二 (二亚苄基丙酮)二钯 (5.75 mg, 0.01 mmol), 2-二环己基膦基-2,,4,,6,-三异丙基联苯(9.53 mg, 0.02 mmol)以及磷 酸钾 (0.22 g, 1 mmol)放入微波反应管中,搅拌下溶解于 10 mL正丁醇中,氩气保护 下, 130Ό, 在微波下反应 30分钟后, 反应完毕, 过滤反应液, 固体用二氯甲烷 / 甲醇 (V:V=10:1)洗漆, 收集洗涤液,减压浓缩,得到标题产物 (R)-[3-{l-[4- (环丙基甲 酰氨基 )-3-氟-苯基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 3-氧代 -1-(2,4,5-三氟-苄 基)-丙基] -氨基甲酸叔丁酯 68a(150 mg, 白色固体), 收率: 44%。
MS m/z (ESI): 661.2[M+1]
第二步
(R)-N-[4-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -1 - 基}-2-氟-苯基]-环丙基甲酰胺盐酸盐
将 ( )-[3-{l-[4- (环丙基甲酰氨基) -3-氟-苯基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基}-3-氧代 -1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 68a(0.1 g, 0.146 mmol)与 4 mL 6.5N的氯化氢的乙酸乙酯溶液一同放入反应瓶中,室温下搅拌反应 4小时,薄 层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 (R)-N-[4-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}-2-氟-苯基] -环丙 基甲酰胺盐酸盐 68(0.085 g,淡黄色固体), 收率: 94%。
MS m/z (ESI): 584.1[M+1]。 实施例 69
(R)-N-{7-[3-氮基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} - 环丙基甲酰胺盐酸盐

第一步
1- (环丙基甲酰胺基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯
将 1 -溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(0.555 g, 1.5 mmol)与 碳酸钾以及 5 mL甲苯放入到反应管中,再加入环丙基甲酰胺 (0.255 g, 3 mmol),碘 化亚铜(0.057 g, 0.3 mmol)和( ^I^-N^N2-二甲基环己基 -1,2-二胺(0.042g, 0.3mmol), 氩气保护下, 封管, 于油浴中控制外温 125 °C搅拌反应过夜, 薄层色谱 跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到 标题产物 1- (环丙基甲酰胺基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯 69a(0.25 g,黄色固体), 收率: 44.6%。
MS m/z (ESI): 375.0[M+1〗。
第二步
N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)环丙基甲酰胺盐酸盐
将 1- (环丙基甲酰胺基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 69a(0.25 g, 0.67 mmol)放入反应瓶中, 加入 5 mL 2.3N的氯化氨的乙酸乙酯溶液, 室温下搅 拌反应 4小时, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基)环丙基甲酰胺盐酸盐 69b(0.2 g,黄色固体), 收率: 95.8°/。。
MS m/z (ESI): 275.2[M+1]。
(R)-{3-[l- (环丙基甲酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小 (2,4,5- 三氟-苯基) -丙基 } -氮基甲酸叔丁酯
将 N-(3-三氟甲基-咪唑并 [l,5-a]哌嗪 -1-基)环丙基甲酰胺盐酸盐 69b(0.2 g, 0.67 mmol)放入到反应瓶中, 加入 10 mL二氯甲垸, 三乙胺 (0.30 mL, 1.34 mmol), 室温 下搅拌使其溶解,再加入 (R 3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(0.223 g, 0.67 mmol), 以及双 (2-氧代 -3-噁唑烷基)次磷酰氯 (0.254 g, 1 mmol), 室温下搅拌反
应过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化 所得残余物, 得到标题产物 (R)-{3-[l- (环丙基甲酰氨基) -3-三氟甲基-咪唑并 [1,5-a] 哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟-苯基) -丙基 } -氨基甲酸叔丁酯 69c(0.23 g, 白色固 体), 收率: 58.3 %。
MS m/z (ESI): 589.9[M+1]。
第四步
(R)-N-{7-[3-氨基 -4- (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基卜 环丙基甲酰胺盐酸盐
将 (R)-{3-[l- (环丙基甲酰氨基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟-苯基) -丙基 } -氨基甲酸叔丁酯 69c(0.23 g, 0.39 mmol)放入到反应瓶 中, 加入 5 mL 2.3N的氯化氢的乙酸乙酯溶液, 室温下搅拌反应 4小时, 薄层色谱 跟踪反应, 原料消失, 旋干反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-N-{7-[3-氨基- 4- (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 环丙基甲酰胺盐酸盐 69(0.15 g, 白色固体), 收率: 73.1 %。
MS m/z (ESI): 490.2[M+1]。
1HNMR (400MHz, CD3OD): δ 7.3 l(m, 1H), 7.16(m, 1H), 4.58(m, 2H), 4.3 l(t, 1H), 4.2 l(t, 1H), 3.99(m, 2H), 3.62(m, 1H), 2.88(m, 2H), 2.72(m, 1H), 2.61 (m, 1H), 1.94(s, 1H), 1.83(m, 1H), 1.36(m, 1H), 0.94(m, 4H) 实施例 70
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基 甲磺酰基 -哌啶 -4-甲酰胺盐酸盐

第一步
1-甲磺酰基 -哌啶 -4-甲酰胺
将哌啶 -4-甲酰胺 70a(1.025 g, 8 ramol)溶解于 80mL干燥的乙腈中, 加入碳酸 钾 (4.42 g, 32 mmol),于冰浴下将甲磺酰氯 (2075 g, 24 mmol)滴入反应液中,冰浴下 搅拌反应 2小时后逐渐升至室温反应 2小时, 薄层色谱跟踪反应, 原料消失, 过
滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-甲磺酰基- 哌啶 -4-甲酰胺 70b(1.27 g, 白色固体), 收率: 77.0%。
MS m/z (ESI): 207.1 [M+l]。
第二步
1-[(1-甲磺酰基 -哌啶 -4-羰基) -氨基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(370 mg, 1 mmol)放入 反应管中,依次加入磷酸钾 (424 mg, 2 mmol), 5 mL邻二甲苯, 1-甲磺酰基 -哌啶 -4- 甲酰胺 70b(412.5 mg, 2 mmol), 碘化亚铜 (38 mg, 0.2 mmol)以及 (1R,2R)-N1,N2-二 甲基环己基 -1,2-二胺 (28.4 mg, 0.2 mmol), 氩气保护下封管, 于油浴中控温 125 搅拌反应过夜, 薄层色谱跟踪反应, 原料未反应完, 于 140°C油浴中控温搅拌发应 8小时, 薄层色谱跟踪反应, 原料大部分已反应完, 旋干反应液, 用硅胶柱色谱法 纯化所得残余物,得到标题产物 1-[(1-甲磺酰基 -哌啶 -4-羰基) -氨基 ]-3-三氟甲基-咪 唑并 [l,5-a]¾ 嗪 -7-羧酸叔丁酯 70c(95 mg,黄色固体), 收率: 19.2%。
MS m z (ESI): 496.0[M+1]。
第三步
1-甲磺酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -4-甲酰胺盐酸盐 将 1-[(1-甲磺酰基 -哌啶 -4-羰基) -氨基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔 丁酯 70c(190 mg, 0.38 mmol)放入反应瓶中, 加入 3 mL 2.3N的氯化氢的乙酸乙酯 溶液, 室温下搅拌反应 4小时, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 1-甲磺酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -4-甲酰胺 盐酸盐 70d(150 mg,黄色固体), 收率: 91.5 %。
MS m/z (ESI): 396.3 [M+l]。
第四歩
(R)-[3-{l-[(l-甲磺酰 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基}-3-氧代
- 1 -(2,4,5-三氟-苯基) -丙基] -氨基甲酸叔丁酯 将 1-甲磺酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -4-甲酰胺盐酸盐 70d(165.6 mg, 0.38 mmol)溶解于8 mL二氯甲烷中, 再加入 0.2 mL三乙胺, (R)-3- 叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(127.8 mg, 0.38 mmol)以及双 (2-氧代 -3-噁唑烷基)次磷酰氯 (144.8 mg, 0.57 mmol),室温下搅拌反应 2小时, 薄层色谱跟 踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物,·得到标 题产物 (R)-[3-{ l-[(l-甲磺酰 -呃啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基) -3-氧代 -1-(2,4,5-三氟-苯基) -丙基] -氨基甲酸叔丁酯 70e(100 mg, 白色固体), 收 率: 37.1 %。
MS m/z (ESI): 710.9[M+l] c
第五步
(R)-N-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-
基}-1-甲磺酰基 -哌啶 -4-甲酰胺盐酸盐
将 (R)-[3-{ 甲磺酰 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基}-3-氧代 -1-(2,4,5-三氟-苯基) -丙基] -氨基甲酸叔丁酯 70e(100 mg, 0.14 mmol)放入 反应瓶中, 加入 4 mL 2.3N的氯化氢的乙酸乙酯溶液, 室温下搅拌反应 3小时, 薄 层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 (R)-N-{7-[3-氨基 -4- (2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-眯唑并 [1,5-a]哌嗪 -1-基}-1-甲磺酰基 -哌啶 -4-甲酰胺盐酸盐 70(80 mg, 白色固体), 收率: 88.4%。
MS m/z (ESI): 611.2[M+1]。
'HNMR (400MHZ, CD3OD): δ 7.36(m, IH), 7.20(m, IH), 4.75(m, 2H), 4.23 (m, 2H), 4.01 (m, 2H), 3.83(m, 3H), 3.02(m, 2H), 2.90(m, 5H), 2.62(m, IH), 2.50(m, IH), 2.01(m: 2H), 1.85(m, 2H), 1.30(m, 2H) 实施例 71
(R)-l-乙酰基 -N-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -1-基 哌啶 -4-甲酰胺盐酸盐
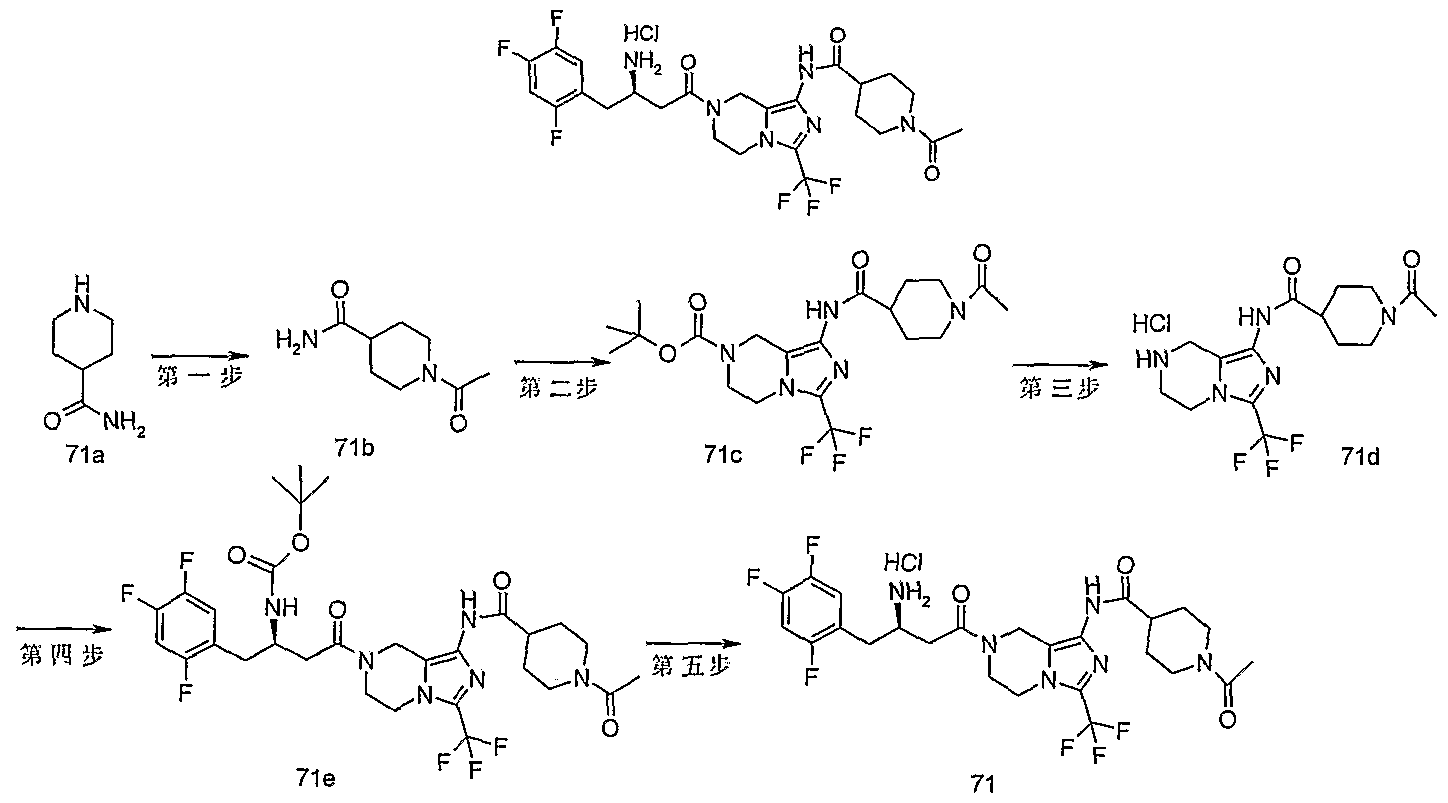
第一步
1-乙酰基 -哌啶 -4-甲酰胺
将哌啶 -4-甲酰胺 71a(1.025 g, 8 mmol)溶解于干燥的乙腈中,加入碳酸钾 (4.4 g,
32 mmol), 于冰浴下向反应液中滴加乙酰氯 (1.9 g, 24 mtnol), 冰浴下搅拌反应 2小 时后慢慢升至室温反应 4小时, 薄层色谱跟踪反应, 原料消失, 过滤, 减压浓缩 滤液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 1-乙酰基 -哌啶 -4-甲酰胺 71b(1.14 g, 白色固体), 收率: 83.8%。
MS m/z (ESI): 171.2[M+1]。
第二步
H(l-乙酰基 -哌啶斗羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 将 1-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 7b(740 mg, 2 mmol)放入
反应管中, 依次加入碳酸钾 (550 mg, 4 mmol), 8 mL邻二甲苯, 1-乙酰基 -哌啶 -4- 甲酰胺 71b(680 mg, 4 mmol), 碘化亚铜 (76 mg, 0.4 mmol)以及 (1R,2R)-N1,N2-二甲 基环己基 -1,2-二胺 (56 mg, 0.4 mmol), 氣气置换后封管, 于油浴中控温 145 搅拌 反应 6小时, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法 纯化所得残余物,得到标题产物 1-[(1-乙酰基 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-羧酸叔丁酯 71c(560 mg,淡黄色油状物), 收率: 60.9%。
MS m/z (ESI): 460.3 [M+l
第三步
1-乙酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -呢啶 -4-甲酰胺盐酸盐 将 1-[(1-乙酰基 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-羧酸叔丁酯
71c(560 mg, 1.21 mmol)溶解于 5 mL 2.3N的氯化氢的乙酸乙酯溶液中,室温下搅拌 反应过夜, 析出大量黄色固体, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 1-乙酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -4-甲酰胺盐 酸盐 71d(480 mg,黄色固体;), 收率: 100%。
MS m/z (ESI): 360·5[Μ+1]。
第四步
(R)-[3-{l-[(l-乙酰基 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 3-氧代
-1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 将 1-乙酰基 -N-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基) -哌啶 -4-甲酰胺盐酸盐 71d(200 mg, 0.56 mmol)溶解于 6 mL二氯甲垸中, 再加入三乙胺 (112 mg, 0.84 mmol), (R)-3-叔丁氧羰基氨基 -4-(2,4,5-三氟-苯基) -丁酸 le(186 mg, 0.56 mmol)以及 双 (2-氧代 -3-噁唑垸基)次磷酰氯 (213 mg, 0.84 mmol), 室温下搅拌反应过夜, 薄层 色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-{l-[(l-乙酰基 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基}-3-氧代 -1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 71e(270 mg,淡黄色固 体), 收率: 71.4%。
MS m/z (ESI): 675.0[M+l
第五步
(R)-l-乙酰基 -N-{7-[3-氨基 -4- (2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -1-基} -哌啶 -4-甲酰胺盐酸盐
将 (R)-[3-{l-[(l-乙酰基 -哌啶 -4-羰基) -胺] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7- 基}-3-氧代 -1-(2,4,5-三氟-节基)-丙基] -氨基甲酸叔丁酯 71e(150 mg, 0.22 mmol)放入 反应瓶中, 加入 5 mL 2.3N氯化氢的乙酸乙酯溶液, 室温下搅拌反应 3小时, 薄层 色谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 (R)-l-乙酰基 -N-{7-[3_ 氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -哌啶 -4-甲酰 胺盐酸盐 71(133 mg,淡黄色固体), 收率: 99.3%。
MS m/z (ESI): 575.1 [M+l]。
'HNMR (400MHZ, CD3OD): δ 7.37(m, IH), 7.22(m, IH), 4.76(m, IH), 4.60(m, IH), 4.52(m, IH), 4.36(m, I H), 4.2 l(m, I H), 4.05(m, 4H), 3.27(m, I H), 3.12(m, 2H), 2.95(m, IH), 2.77(m, 3H), 2.18(s, 3H), 1.94(m, 3H), 1.73(m, 2H) 实施例 72
(R)- 3-氨基 -1-(1-噻吩 -2-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯 基) -丁 -1-酮盐酸盐

第一歩
(R)-[3-氧代 -3-(1-噻吩 -2-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟-苄 基) -丙基] -氨基甲酸叔丁酯
在氩气保护下, 将化合物 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3- 氧代 -1-(2,4,5-三氟苄基) -丙基] -氨基甲酸叔丁酯 12b(0.293 g, 0.5 mmol)与噻吩 -2-基 硼酸 (0.096 g, 0.75 mmol)以及四三苯基膦化钯 (0.058 g, 0.05 mmol),碳酸钾 (0.138 g, 1 mmol), 3 mL乙二醇二甲醚, 3 mL水一同放入 20 mL的微波反应管中, 封管, 于 12CTC下微波反应 30分钟,薄层色谱跟踪反应,原料消失,向反应液中加入 5 mL 水, 用乙酸乙酯萃取 (15 mLx3), 合并有机相, 无水硫酸镁千燥, 过滤, 滤液减压 浓缩, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-氧代 -3-(1-噻吩 -2-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 72a(0.2 g,淡黄色固体), 收率: 68 %。
MS m/z (ESI): 589[M+1]。
第二步
将 (R)-[3-氧代 -3-(1-噻吩 -2-基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 )小(2,4,5-三 氟-苄基) -丙基] -氨基甲酸叔丁酯 72a(0.2 g, 0.34 mmol)与 2 mL乙酸乙酯放入到反应 瓶中, 再加入 3 mL 2.0 N氯化氢的乙酸乙酯溶液, 室温下搅拌反应过夜, 薄层色 谱跟踪反应, 原料消失, 减压浓缩反应液, 得到标题产物 (R 3-氨基 -1-(1-噻吩 -2- 基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯基) -丁 -1-酮盐酸盐 72(0.13 g,黄色固体), 收率: 72.9%。
MS m/z (ESI): 489.2[M+1]。
实施例 73
(R)-N-[3-{7-[3-氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-苯基] -乙酰胺盐酸盐

第一步
(R)-{3-[l-(3-氨基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟- 苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(0.3 g, 0.51 mmol), 3-(4,4,5,5-四甲基 -[1,3,2]二氧硼戊环 -2-基) -苯胺 (0.128 g, 0.76 mmol), 磷酸钾 (0.218 g, 1.03 mmol), 二 (二亚苄基丙酮)钯 (0.006 g, 0.01 mmol)以及 2-二环己基膦基-2,,4,,6,-三异丙基联苯(0.01 g, 0.02 mmol) 放入反应瓶中, 搅拌下溶解于 3 mL正丁醇中, 氩气保护下, 100°C, 微波下反应 30分钟, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化 所得残余物, 得到标题产物 (R)-{3-[l-(3-氣基-苯基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 73a(0.2 g,黄色固体), 收 率: 65.4%。
第二步
(R)-{3-[l-(3-乙酰胺 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三 氟-苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-{3-[l-(3-氨基 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基 ]-3-氧代 -1-(2,4,5-三氟-苄基) -丙基 氨基甲酸叔丁酯 73a(0.2 g, 0.33 mmol)放入反应瓶中,加 入 2 mL乙酸酐, 搅拌下溶解于 4 mL吡啶中, 室温下搅拌反应过夜, 薄层色谱跟 踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标 题产物 (R)-{3-[l-(3-乙酰胺 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 73b(0.14 g, 白色固体), 收率: 65.4%。 MS m/z (ESI): 662.2[M+23]
第三步
(R)-N-[3-{7-[3-氨基 -4-(2,4,5-三氟苯基)-丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-
基 }-苯基] -乙酰胺盐酸盐
将 (R)-{3-[l-(3-乙酰胺 -苯基 )-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟-苄基)-丙基}-氨基甲酸叔丁酯 73b(0.14 g, 0.11 mmol)溶解于 2 mL甲 醇中, 向反应液中加入 7 mL氯化氢的甲醇溶液, 室温下搅拌反应 5小时, 薄层色 谱跟踪反应,原料消失,减压浓缩反应液,真空干燥,得到标题产物 (R)-N-[3-{7-[3- 氨基 -4-(2,4,5-三氟苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基}-苯基] -乙酰 胺盐酸盐 73(0.12 g,黄色固体), 收率: 96.5 %。
MS m/z (ESI): 540.2[M+1] 实施例 74
(R)-3-氨基 -1-[1-(2-甲氧基 -嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟-苯基) -丁 -1-酮盐酸盐

第一歩
(R)-{3-[l-(2-甲氧基 -嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1-(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(0.293 g, 0.5 mmol), 2-甲氧基嘧啶 -5-基硼酸 (118 mg, 0.75 mmol), 二 (二亚苄基丙酮)钯 (5.8 mg, 0.01 mmol), 2-二环己基膦基 -2,,4',6,-三异丙 基联苯 (9.5 mg, 0.02 mmol), 磷酸钾 (0.212 g, 1 mmol)搅拌下溶解于 2.5 mL正丁醇 中, 氩气保护下, 100°C, 在微波下反应 30分钟后, 反应完毕, 有大量固体析出, 冷却后加入 15 mL正己烷, 过滤, 除去滤液, 固体用二氯甲垸 /甲醇 (v:V=10:l)混合 液溶解, 再次过滤, 除去残渣, 滤液减压浓縮, 得到标题产物 (R)-{3-[l-(2-甲氧基- 嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代小(2,4,5-三氟-苄基) -丙基 } - 氨基甲酸叔丁酯 74a(220 mg, 黄褐色固体), 收率: 71.7%。
MS m/z (ESI): 615.1[M+1]
第二步
(R)-3-氨基 -l-[l-(2-甲氧基 -嘧啶 -5-基) -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基] -4-(2,4,5- 三氟-苯基) -丁 -1-酮盐酸盐
将 (R)-{3-[l-(2-甲氧基 -嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1- (2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 74a(220 mg, 0.36 mmol)搅拌下溶解于 3 mL乙酸乙酯中, 加入 5 mL 2.7N氯化氢乙酸乙酯溶液, 室温下反应 1小时, 薄层 色谱跟踪反应, 原料消失, 蒸千溶剂, 用硅胶柱色谱法纯化, 得到标题产物 (R 3- 氨基 -1-[1-(2-甲氧基 -嘧啶 -5-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -4-(2,4,5-三氟- 苯基) -丁 -1-酮盐酸盐 74(80 mg,淡黄色固体), 收率: 41.6 %。
MS m/z (ESI): 515.2[M+1]
iHNMR (400MHz, CD3OD): δ 8.92(m, 2H), 7.40(m, 1H), 7.23(m, 1H), 5.06(m, 2H), 4.35(m, 2H), 4.35(m, 2H), 4.13(m, 3H), 4.00(m, 2H), 3.47(s, 2H), 2.98(m, 3H), 实施例 75
(R)-5-{7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -呋 喃 -2-甲醛盐酸盐

第一步
(R)-{3-[l-(5-甲酰基 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
小(2,4,5-三氟-苄基) -丙基 氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1 -(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(0.123 g, 0.21 mmol), 5-甲酰基呋喃 -2-基硼酸 (35.3 mg, 0.252 mmol) , 四 (三苯基膦)钯 (12.1 mg, 0.01 mmol), 四丁基溴化胺 (13.5 mg, 0.042 mmol),碳酸钾 (87.12 mg, 0.63 mmol) ,搅拌下溶解于 1.5 mLl,4-二氧六环和 0.75 mL 水的混合溶剂中, 氩气氛下, 100°C, 在微波下反应 30分钟后, 反应完毕, 蒸干 溶剂, 用硅胶柱色谱法纯化, 得到标题产物 (R)-{3-[l-(5-甲酰基 -呋喃 -2-基) -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 75a(65 mg,黄色固体), 收率: 51.6 %。
MS m/z (ESI): 623.1 [M+23]
第二步
(R)-5-{7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -呋 喃 -2-甲醛盐酸盐
将 (R)-{3-[l-(5-甲酰基 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-l-(2,4,5-三氟-节基) -丙基 氨基甲酸叔丁酯 75a(65 mg, 0.11 mmol)搅拌下溶解于 1 mL乙酸乙酯中, 加入 l mL 2.7N氯化氢乙酸乙酯溶液, 室温下反应 4小时, 薄层 色谱跟踪反应, 原料消失, 蒸干溶剂, 用硅胶柱色谱法纯化, 得到标题产物 (R)- 5-{7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁基 ]-3-三氟甲基-咪唑并 [1,5- a]哌嗪 -1-基} -呋 喃 -2-甲醛盐酸盐 75(80 mg,淡黄色固体), 收率: 41.6%。
MS m/z (ESI): 515.2[M+1] 实施例 76
(R)-7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-腈盐酸

第一步
(R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 羧酸甲酯
将八羰基二钴 (3.5 g, 10.23 mmol),氯乙酸乙酯 (0.62 g, 5.12 mmol),碳酸钾 (1.41 g, 10.23 mmol)以及 10mL 甲醇放入反应瓶中, 室温下搅拌反应 5 分钟, 加入
(R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟苄基) -丙基] - 氨基甲酸叔丁酯 12b(2.0 g, 3.41 mmol), 于油浴中控温 60°C反应过夜, 薄层色谱跟 踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标 题产物 (R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a] 哌嗪 -1-羧酸甲酯 76a(l.l g, 白色固体), 收率: 57.3 %。
MS m/z (ESI): 565.0[M+1]
第二步
(R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 羧酸
将 (R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]
哌嗪 -1-羧酸甲酯 76a(1.0 g, 1.77 mmol)溶解于 10 mL甲醇中, 加入 4 N的氢氧化钠 溶液 13.25 niL, 室温下搅拌反应 30分钟, 反应结束, 加入 2 N的盐酸溶液调 pH 值为 4-5 , 用乙酸乙酯萃取 (20 mLx3), 收集有机相, 用 20 mL饱和食盐水洗一次, 合并有机相, 用无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法纯化所 得残余物, 得到标题产物 (R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟 甲基-咪唑并 [1,5-a]哌嗪 -1-羧酸 76b(0.78 g,淡黄色固体), 收率: 80.1 %。
MS m/z (ESI): 573.1 [M+23]
第三步
(R)-[3-(l-氨甲酰基 -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -7-基)- 3-氧代 -1-(2,4,5-三氟-苄 基: I-丙基] -氨基甲酸叔丁酯
将 (R)-7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a] 哌嗪 -1-羧酸 76b(0.3 g, 0.54 mmol), 碳酸胺 (0.156 g, 1.62 mmol), 双 (2-氧代 -3-噁唑 烷基)次磷酰氯 (0.412 g, 1.62 mmol)放入反应瓶中,加入 30 mL四氢呋喃以及 0.5 mL 三乙胺, 室温下搅拌反应过夜, 薄层色谱跟踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-[3-(l-氨甲酰基 -3-三氟甲基-咪 唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 76c(284 mg, 白色固体), 收率: 95.0%。
MS m/z (ESI): 549.9[M+1]
第四步
(R)-[3-(l-氰基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小 (2,4,5-三氟-苄基) -丙 基]-氨基甲酸叔丁酯
氮气保护下,将 (RM3-(1-氨甲酰基 -3-三氟甲基-咪唑并 [l,5-a]呃嗪 -7-基) -3-氧代 -1-(2,4,5-三氟-苄基) -丙基] -氨基甲酸叔丁酯 76c(0.35 g, 0.636 mmol), 咪唑 (0.091 g, 1.34 mmol)放入到干燥的反应瓶中, 加入 10 mL吡啶溶解, 用干冰 -丙酮浴冷至 -35°C, 保持 -35 °C向反应液中滴加三氯氧磷 (0.4 g, 2.6 mmol), 加毕, 控温 -35°C反 应 1 小时, 后升至室温反应过夜, 薄层色谱跟踪反应, 原料消失, 旋千反应液中 的吡啶, 加入 5 mL饱和氯化钠溶液溶解, 用乙酸乙酯萃取 (10 mLx4), 合并有机 相, 依次用饱和硫酸铜溶液洗 (5 mLx2), 5 mL饱和碳酸氢钠溶液洗, 5 mL饱和氯 化钠溶液洗, 收集有机相, 用无水硫酸镁千燥, 过滤, 滤液减压浓缩, 用硅胶柱 色谱法纯化所得残余物, 得到标题产物 (R)-[3-(l-氰基 -3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -7-基) -3-氧代 -l-(2,4,5-三氟-苄基) -丙基] -氣基甲酸叔丁酯 76d(0.12 g, 白色固体), 收率: 35.5 %。
MS m/z (ESI): 554.0[M+23]
第五步
(R)-7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [l,5-a]哌嗪 -1-腈盐酸
±
将 (R)-[3-(l-氰基 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代小(2,4,5-三氟-苄 基)-丙基] -氨基甲酸叔丁酯 76d(0.12 g, 0.23 mmol)搅拌下溶解于 4 mL 2.0N氯化氢 乙酸乙酯溶液中, 室温下搅拌反应 4小时, 薄层色谱跟踪反应, 原料消失, 减压 浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-7-[3-氨基 -4-(2,4,5-三氟-苯基) -丁酰基] -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-腈盐酸盐 76(0.06 g, 白色固体), 收率: 56.7%。
MS m/z (ESI): 432.1[M+1]
^MR (400MHz, CD3OD): δ 7.43-7.39(ra, 1H), 7.30-7.26(m, 1H), 5.01(m, 2H), 4.36-4.28(m, 2H), 4.12-4.09(m, 1H), 4.02-3.94(m, 2H), 3.12-3.10(m, 2H), 3.03-2.99(m, 1H), 2.88-2.81(m, 1H) 实施例 77
(R)-5-{7-[3-氨基 -4-(2,4,5-三氟苯基)丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基}-^-甲基呋喃 -2-甲酰胺盐酸盐

第一步
(R)-{3-[l-(5-醛基 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟-苄基) -丙基 } -氨基甲酸叔丁酯
将 (R)-[3-(l-溴 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟苄基) - 丙基] -氨基甲酸叔丁酯 12b(1.308 g, 2.23 mmol), 5-醛基呋喃 -2-基硼酸 (0.469 g, 3.35 mmol), 四三苯基磷化钯 (0.127 g, 0.11 mmol), 四丁基溴化胺 (0.145 g, 0.45 mmol) 以及碳酸钾 (0.925 g, 6.69 mmol)放入微波反应管中, 再加入 7 mL二甲醚和 3.5 mL 水, 氩气置换后封管, 140°C, 微波下反应 70分钟, 薄层色谱跟踪反应, 有少量 原料未反应, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-{3-[l-(5-醛基 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 -1-(2,4,5- 三氟-苄基) -丙基 氨基甲酸叔丁酯 77a(0.8 g,黄色固体), 收率: 59.7%。
MS m/z (ESI): 623.0[M+23]
第二歩
(R)-5-{7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰 ]-3-三氟甲基-咪唑并 [1,5- a]哌嗪
- 1-基} -呋喃 -2-羧酸
将 (R)-{3-[l-(5-醛基 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代
-1- (2,4,5-三氟-苄基) -丙基 氨基甲酸叔丁酯 77a(0.85 g, 1.42 mmol)搅拌下溶解于 30 mL四氢呋喃和 15 mL水的混合溶液中, 然后依次加入二水合磷酸二氢钠 (0.663 g, 4.25 mmol), 亚氯酸钠 (0.384 g, 4.25 mmol)以及 2-甲基 -2-丁烯 (0.298 g, 4.25 mmol), 室温下搅拌反应过夜, 薄层色谱跟踪反应, 有少量原料未反应, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题产物 (R)-5-{7-[3-叔丁氧酰胺 -4-(2,4,5- 三氟-苯基) -丁酰 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1-基} -呋喃 -2-羧酸 77b(0.13 g,黄 色油状固体), 收率: 14.9%。
MS m/z (ESI): 615.2[M-1]
第三步
(R)-{3-[l-(5-甲基氨甲酰 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧代 小(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 将 5-{7-[3-叔丁氧酰胺 -4-(2,4,5-三氟-苯基) -丁酰 ]-3-三氟甲基-咪唑并 [1,5-a]哌 嗪 -1-基 呋喃 -2-羧酸 77b(0.13 g, 0.21 mmol)搅拌下溶解于 5 mL二氯甲垸中, 依次 加入一甲胺盐酸盐 (0.029 g, 0.42 mmol), 双 (2-氧代 -3-噁唑垸基)次磷酰氯 (0.107 g, 0.42 mmol)以及三乙胺 (0.064 g, 0.63 mmol), 室温下搅拌反应过夜, 薄层色谱跟踪 反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标题 产物 (R)-{3-[l-(5-甲基氨甲酰 -呋喃 _2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3-氧 代 -1-(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 77c(0.05 g, 白色固体), 收率: 37.9 %。
MS m/z (ESI): 652.2[M+23]
第四步
(R)-5-{7-[3-氨基 -4-(2,4,5-三氟苯基)丁酰基 ]-3-三氟甲基-咪唑并 [1,5-a]哌嗪 -1- 基} -N-甲基呋喃 -2-甲酰胺盐酸盐
将 (R)-{3-[l-(5-甲基氨甲酰 -呋喃 -2-基) -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基] -3- 氧代 -1-(2,4,5-三氟-苄基) -丙基 } -氨基甲酸叔丁酯 77c(0.05 g, 0.08 mmol)搅拌下溶解 于 3tnL乙酸乙酯中, 再加入 2 mL 3N氯化氢的乙酸乙酯溶液, 室温下撹拌反应过 夜,薄层色谱跟踪反应,原料消失,过滤, 固体用乙酸乙酯洗 (2 mLx3),收集固体, 真空干燥, 得到标题产物 (R)-5-{7-[3-氨基 -4-(2,4,5-三氟苯基)丁酰基 ]-3-三氟甲基- 咪唑并 [1,5-a]哌嗪 -1-基}- -甲基呋喃 -2-甲酰胺盐酸盐 77(0.036 g,黄色固体), 收 率: 80.0%。
MS m/z (ESI): 530.2[M+1]
实施例 78
(R)-3-氨基 -1 -(1 -氯 -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7-基) -4-(2,4,5- 酮盐酸盐

第一步
(R)-[3-(l-氯 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟-苄基)-丙基] - 氨基甲酸叔丁酯
将 (R)-[3-氧代 -1-(2,4,5-三氟苄基) -3-(3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -丙 基] -氨基甲酸叔丁酯 lj(0.11 g, 0.217 mmol)搅拌下溶解于 15 mL无水乙醇中, 加入 1-氯-四氢吡咯 -2,5-二酮 (0.058 g, 0.435 mmol), 室温下搅拌反应过夜, 薄层色谱跟 踪反应, 原料消失, 减压浓缩反应液, 用硅胶柱色谱法纯化所得残余物, 得到标 题产物 (R)-[3-(l-氯 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -3-氧代 -1-(2,4,5-三氟-苄 基) -丙基] -氨基甲酸叔丁酯 78a(0.06 g, 白色固体), 收率: 51.3 %。
MS m/z (ESI): 540.8[M+1]
第二步
(R)-3-氨基- 1-(1-氯 -3-三氟甲基-咪唑并 [1,5-a]哌嗪 -7-基) -4-(2,4,5-三氟-苯基) -丁小 酮盐酸盐
将 (R)-[3-(l-氯 -3-三氟甲基-眯唑并 [1,5-a]哌嗪 -7-基) -3-氧代 4-(2,4,5-三氟-苄 基) -丙基] -氨基甲酸叔丁酯 78a(55 mg, 0.1 mmol)放入反应瓶中, 加入 6 mL乙酸乙 酯, 搅拌使其溶解, 再加入 3 mL 3N氯化氢乙酸乙酯溶液, 室温下搅拌反应 2小 时, 薄层色谱跟踪反应, 原料消失, 减压浓縮反应液, 用硅胶柱层析法纯化所得 残余物, 得到标题产物 (R)-3-氨基 -1-(1 -氯 -3-三氟甲基-咪唑并 [1 ,5-a]哌嗪 -7- 基) -4-(2,4,5-三氟-苯基) -丁 -1-酮盐酸盐 78(40 mg,淡黄色固体), 收率: 85.1 %。
JHNMR (400MHz, CD3OD): δ 7.37(m, 1Η), 7.23(m, 1H), 4.76(m, 2H), 4.30(m, 2H), 3.93(m, 3H), 3.10(m, 2H), 2.85(m, 2H), 2.00(s, 2H)。
测试例:
生物学评价
DPP IV抑制活性的测定
下面的方法是用来测定本发明化合物抑制 DPPIV酶活性的能力。 每个化合物 的抑制率或半抑制浓度 IC5Q(把酶活性抑制至 50%时所测化合物的浓度)是以固定 量的酶混合底物及不同浓度的待测化合物来测定的。
DPP IV抑制活性的测定
材料和方法:
材料:
a. 白色 96孔板 (BMG)
b. Tris缓冲液: 制备 100mL 2mM的 Tris缓冲液, 将 0.0242g Tris溶解于约 90mL dH20中, 用 HC1和 NaOH调节 pH到 8.00, 最后加 d¾0至 100mL。
c. DPPIV酶 (CalBiochem Catalog no.317630), 溶解于 Tris缓冲液中至 2mM。 d. DPPIV— GloTM底物 (Promega Catalog no.G8350), 溶解于 dH20中至 lmM。 e. DPPIV—Glo.缓冲液 (Promega Catalog no.G8350)
f. 荧光素检测试剂 (Promega Catalog no.G8350)
g. DMSO
h. d¾0
操作:
按以下操作顺序进行:
1. 解冻 DPPIV— Glo.使用前缓冲并平衡到室温。
2. 使用前缓冲冻存的荧光素检测试剂。
3. 悬浮 DPPIV— Glo.在底物中加入超纯水轻微混合均勾后, 制成 ImM的底物。
4. 将荧光素检测试剂放入茶色瓶中, 加入 DPPIV— Glo.。 荧光素检测试剂应在 1 分钟内溶解。
5. 用 DMSO溶解所测化合物至最终操作浓度的 50倍。
6. 每个试管中加入 50倍浓度的所测化合物 2μί, 在反面对照合空白对照中加入 2 LDMSO。
7. 在每个试管中加入 46 L Tris缓冲液, 在空白对照中加入 48μί Tris缓冲液。 8. 在反面对照和测试样的每个试管中加入 2μΙ )ΡΡΐν酶。
9. 振动混合并离心试管。 将试管中物质全部转移到 96-well平板上。
10.混合底物和 DPPIV— Glo.比例为 1:49。 振动混合至充分混合。 使用前在室温下 静置 30-60分钟。
11.在每个 96-well平板孔中加入 5(^L DPPIV— Glo.和底物的混合液, 用封膜封住 平板。
12.用平板振荡器在 300-500rpm/30s下慢慢混合%孔中物质。在室温下培养 30分
钟到 3小时。
13.记录发光。
抑制率定义: [1-(S-B)/(N-B)]*100%
S: 样品
B: 空白对照
N: 反面对照
IC50值


S6Z.lOO/800ZN3/X3d 86ZS90/600J OAV
Claims
1. 一种由通式 (I)表示的化合物或其药学上可接受的盐:

其中:
Ar是苯基, 该苯基是未取代的或者进一步被 1〜5个 R7所取代;
R1选自氢原子、烷基、三氟甲基、环烷基、 杂环烷基、 芳基或杂芳基, 其中垸 基、 环烷基、 杂环烷基、 芳基、 杂芳基进一步被一个或多个选自卤素、 氰基、 芳 基、 羟基或氨基的取代基所取代;
R2选自氢原子、 卤素、氰基、氨基、烷基、环烷基、 杂环烷基、 芳基、 杂芳基、 -NR3R4、 -NR3C(0)R4或 -NC(0)NR3R4, 其中环烷基、 杂环烷基、 芳基、 杂芳基进 一步被一个或多个选自卤素、 氨基、 氰基、 硝基、 羟基、 烷基、 环烷基、 烷氧基、 杂芳基、三卤代烷基、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4、 -NC(0)NR3R\ -COR5 或 -S02R6的取代基所取代;
R3和 R4各自独立地选自氢原子、 烷基、 环烷基、 杂环烷基、 芳基、 杂芳基或 -S02R6, 其中烷基、 环烷基、 杂环烷基、 芳基或者杂芳基进一步被一个或多个选 自卤素、羟基、氨基、垸氧基、烷基、芳基、杂环烷基、 -COR5、 -S02R6、 - NR3R4、 羧酸或羧酸酯的取代基所取代;
或者, R3和 R4—起形成一个 4〜8元杂环基, 其中 4〜8元杂环内含有一个或 多个 N、 0、 S原子, 并且 4〜8元杂环上进一步被一个或多个选自卤素、 烷基、 芳基、 杂芳基、 羟基、 氰基、 垸氧基、 羟烷基、 杂环烷基或 -NR3R4的取代基所取 代;
R5选自氢原子或垸基;
R6选自垸基或芳基, 其中芳基进一步被一个或多个垸基取代。
R7选自卤素、氰基、羟基、烷基或者垸氧基, 其中烷基或者烷氧基是未取代的 或者进一步被一个或多个卤素取代。
2.根据权利要求 1所述的化合物或其药学上可接受的盐, 其中 R1为三氟甲基。
3. 根据权利要求 1所述的化合物或其药学上可接受的盐, 其中该化合物选自-

86ZS90/6001 ΟΛλ
S6LlOO/800lND/13d

£6/.lOO/800lN3/13d 86ZS9 /6001 OAV


4. 根据权利要求 1所述的化合物或其药学上可接受的盐, 所述的盐为所述化合物 与选自以下的酸形成的盐: 磷酸盐、 苹果酸、 乳酸、 马来酸、 盐酸、 甲磺酸、 硫 酸、 磷酸、 柠檬酸、 酒石酸、 乙酸或三氟乙酸。
5. 通式化合物 (IA)或通式化合物 (ΙΒ), 所述化合物为合成根据权利要求 1所述的通 式 (I)化合物的中间体:

其中:
X选自卤素;
R1选自氢原子、 烷基、三氟甲基、 环烷基、 杂环烷基、 芳基或杂芳基, 其中垸 基、 环烷基、 杂环烷基、 芳基、 杂芳基进一步被一个或多个选自卤素、 氰基、 芳 基、 羟基或氨基的取代基所取代;
R2选自氢原子、 卤素、 氰基、 氨基、烷基、 环烷基、 杂环烷基、 芳基、 杂芳基、 -NR3R4、 -NR3C(0)R4或 -NC(0)NR3R4, 其中环烷基、 杂环烷基、 芳基、 杂芳基进 一步被一个或多个选自卤素、 氨基、 氰基、 硝基、 羟基、 烷基、 环烷基、 烷氧基、 杂芳基、 三卤代烷基、 -NR3R4、 -NR3C(0)R4、 -C(0)NR3R4、 -NC(0)NR3R4、 -COR5 或 -S02R6的取代基所取代;
R3和 R4各自独立地选自氢原子、 垸基、 环烷基、 杂环烷基、 芳基、 杂芳基或 -S02R6, 其中烷基、 环垸基、 杂环烷基、 芳基或者杂芳基进一步被一个或多个选 自卤素、羟基、氨基、烷氧基、垸基、 芳基、杂环烷基、 -COR5、 -S02R6、 - NR3R4、 羧酸或羧酸酯的取代基所取代;
或者, R3和 R4—起形成一个 4〜 8元杂环基, 其中 4〜8元杂环内含有一个或 多个 N、 0、 S原子, 并且 4〜8元杂环上进一步被一个或多个选自卤素、 烷基、 芳基、 杂芳基、 羟基、 氰基、 烷氧基、 羟垸基、 杂环烷基或 -NR3R4的取代基所取 代;
R5选氢原子或烷基;
R6选自烷基或芳基, 其中芳基进一歩被一个或多个烷基取代。
6.根据权利要求 5所述的通式化合物 (IA)或通式化合物 (IB),其中 R1是三氟甲基。
7. 根据权利要求 5所述的通式化合物 (IA)的制备方法, 该方法包括:
 将原料 R2取代的吡嗪 2-甲胺与酸酐进行反应, 生成的酰胺产物与三氯氧磷在 室温下混合搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催 化下, 氢化还原生成通式化合物 (IA)。
将原料 R2取代的吡嗪 2-甲胺与酸酐进行反应, 生成的酰胺产物与三氯氧磷在 室温下混合搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催 化下, 氢化还原生成通式化合物 (IA)。
8. 根据权利要求 5所述的通式化合物 (IB)的制备方法, 该方法包括:

将原料吡嗪 2-甲胺与酸酐进行反应,生成的酰胺产物与三氯氧磷在室温下混合 搅拌后, 加入五氧化二磷, 生成咪唑并 [1,5-a]吡嗪环, 然后在 Pd/C催化下, 氢气 还原后与二碳酸二叔丁酯反应, 生成叔丁氧基羰基保护的咪唑并 [1,5-a]哌嗪产物, 然后在 N-卤代琥珀酰亚胺条件下反应, 得到通式化合物 (IB)。
9. 根据权利要求 5所述的通式化合物 (IA)的制备方法, 该方法包括:

将通式化合物 (IB)在钯类试剂催化下, 与硼酸或硼酸酯在微波下反应, 进行 Suzuki偶联, 或与取代胺类在催化条件下进行 Buchwald偶联, 得到产物在酸性条 件下脱掉氨基的保护基, 得到通式化合物 (IA)。
10. 根据权利要求 1所述的通式 (I)化合物的制备方法, 该方法包括:

根据权利要求 5所述的通式化合物 (IA)与 3-叔丁氧基羰基氨基 -4-芳基-丁酸在 缩合试剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行缩合,得到的产物在酸性条 件下脱掉氨基保护基, 得到通式化合物 (1)。
11 . 根据权利要求 1所述的通式 (I)化合物的制备方法, 该方法包括:

根据权利要求 5所述的通式化合物 (IB)脱保护基后与 3-叔丁氧基羰基氨基 -4- 芳基 -丁酸在缩合试剂双 (2-氧代 -3-噁唑烷基)次膦酰氯的条件下进行反应, 得到的 缩合产物在钯类试剂催化下, 进一步与硼酸或硼酸酯在微波下反应, 进行 Suzuki 偶联, 得到通式化合物 (I) ; 缩合产物也可以与取代胺类在催化条件下, 进行 Buchwald偶联, 得到通式化合物 (I); 缩合产物也可以在油浴中, 以八羰基二钴和 氯乙酸乙酯作为催化剂, 在一氧化碳气氛下与醇反应, 得到的取代羧酸酯化合物 进一步水解成羧酸, 与碳酸铵反应生成的酰胺, 进一步脱水生成 R2为氯基的通式 化合物 (1)。
12. 根据权利要求 10〜11中任一项所述的制备方法, 其中还包括将得到的通式 (I) 化合物经纯化后在酸的甲醇、 二氯甲烷或乙酸乙酯溶液中反应, 得到其酸加成产 物盐。
13. 根据权利要求 12所述的制备方法,其中所述的酸选自磷酸盐、苹果酸、乳酸、 马来酸、 盐酸、 甲磺酸、 硫酸、 磷酸、 柠檬酸、 酒石酸、 乙酸或三氟乙酸。
14. 一种药用组合物, 其含有治疗有效剂量的根据权利要求 1〜4中任何一项所述 的化合物或其药学上可接受的盐, 及药学上可以接受的载体或赋形剂。
15. 根据权利要求 1所述的化合物或其药学上可接受的盐在制备治疗 II型糖尿病、 高血糖症、 肥胖症或胰岛素抵抗症的药物中的用途。
16. 一种抑制二肽基肽酶 IV催化活性的方法, 其中包括将所述的二肽基肽酶 IV 与权利要求 1〜4中任何一项所述的化合物或盐接触。
17. 根据权利要求 14所述的药物组合物在制备治疗 II型糖尿病、 髙血糖症、肥胖 症或胰岛素抵抗症的药物中的用途。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200880009683XA CN101641360B (zh) | 2007-10-25 | 2008-10-24 | 哌嗪类衍生物,其制备方法及其在医药上的应用 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200710163499.8 | 2007-10-25 | ||
| CNA2007101634998A CN101417999A (zh) | 2007-10-25 | 2007-10-25 | 哌嗪类衍生物,其制备方法及其在医药上的应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009065298A1 true WO2009065298A1 (en) | 2009-05-28 |
Family
ID=40629017
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2008/001795 Ceased WO2009065298A1 (en) | 2007-10-25 | 2008-10-24 | Piperazine derivatives, preparation process and pharmaceutical use thereof |
Country Status (3)
| Country | Link |
|---|---|
| CN (2) | CN101417999A (zh) |
| TW (1) | TW201019937A (zh) |
| WO (1) | WO2009065298A1 (zh) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011508735A (ja) * | 2007-12-26 | 2011-03-17 | ジエンス ヘンルイ メデイシンカンパニー リミテッド | テトラヒドロ・イミダゾ[1,5−α]ピラジン誘導体、その調製プロセスおよび医薬的使用 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| US9273058B2 (en) | 2013-11-14 | 2016-03-01 | Bristol-Myers Squibb Company | Substituted pyrazolo-piperazines as casein kinase 1 δ/ε inhibitors |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| JP2021519308A (ja) * | 2018-03-29 | 2021-08-10 | ボード オブ レジェンツ, ザ ユニバーシティ オブ テキサス システムBoard Of Regents, The University Of Texas System | 転写活性化タンパク質のイミダゾピペラジン阻害剤 |
| US12540139B2 (en) | 2020-10-02 | 2026-02-03 | Board Of Regents, The University Of Texas System | Imidazopiperazine inhibitors of transcription activating proteins |
| US12559497B2 (en) | 2020-10-02 | 2026-02-24 | Board Of Regents, The University Of Texas System | Imidazopiperazine inhibitors of transcription activating proteins |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101899048B (zh) * | 2009-05-27 | 2013-04-17 | 上海恒瑞医药有限公司 | (R)-7-[3-氨基-4-(2,4,5-三氟-苯基)-丁酰]-3-三氟甲基-5,6,7,8-四氢-咪唑并[1,5-a]吡嗪-1-羧酸甲酯的盐 |
| CN102030683B (zh) * | 2009-09-27 | 2013-07-31 | 浙江九洲药业股份有限公司 | 西他列汀中间体及其制备方法和用途 |
| CN102702205B (zh) * | 2012-05-16 | 2014-01-15 | 苏州新凯生物医药技术有限公司 | 西他列汀的制备方法 |
| CN103724352B (zh) * | 2012-10-16 | 2017-11-28 | 江苏盛迪医药有限公司 | 一种dpp‑iv抑制剂的中间体、其制备方法和通过其制备dpp‑iv抑制剂的方法 |
| CN103910634B (zh) * | 2012-12-29 | 2018-04-27 | 广东东阳光药业有限公司 | 西格列汀中间体的制备方法 |
| WO2014127747A1 (zh) * | 2013-02-22 | 2014-08-28 | 成都先导药物开发有限公司 | 一种抑制dpp-iv的化合物及其中间体 |
| CN103664962A (zh) * | 2013-12-20 | 2014-03-26 | 南京华威医药科技开发有限公司 | 哌嗪类衍生物 |
| CN109928888B (zh) * | 2017-12-19 | 2023-04-11 | 浙江瑞博制药有限公司 | 一种西格列汀中间体的制备方法 |
| CN113620957B (zh) * | 2020-05-08 | 2022-04-19 | 盛世泰科生物医药技术(苏州)有限公司 | 一种胺甲基吡嗪类化合物的制备方法 |
| CN114057751B (zh) * | 2022-01-17 | 2022-04-12 | 盛世泰科生物医药技术(苏州)有限公司 | 一种dpp-iv抑制剂及其关键中间体的制备方法 |
| CN115583935A (zh) * | 2022-11-03 | 2023-01-10 | 浙江工业大学 | 一种4-(2,4,5-三氟苯基)-3-氧丁酸酯的制备方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07101959A (ja) * | 1993-09-30 | 1995-04-18 | Sankyo Co Ltd | 1−メチルカルバペネム誘導体 |
| WO2003076427A1 (en) * | 2002-03-14 | 2003-09-18 | Pfizer Limited | Quinazoline compounds useful in therapy |
| CN1484640A (zh) * | 2001-01-02 | 2004-03-24 | - | 用作α1A/B肾上腺素能受体拮抗剂的喹唑酮衍生物 |
| WO2004058266A1 (en) * | 2002-12-20 | 2004-07-15 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| CN1524082A (zh) * | 2001-07-06 | 2004-08-25 | 作为治疗或预防糖尿病的二肽基肽酶抑制剂的β-氨基四氢咪唑并(1,2-A)吡嗪和四氢三唑并(4,3-A)吡嗪 |
-
2007
- 2007-10-25 CN CNA2007101634998A patent/CN101417999A/zh active Pending
-
2008
- 2008-10-24 CN CN200880009683XA patent/CN101641360B/zh not_active Expired - Fee Related
- 2008-10-24 WO PCT/CN2008/001795 patent/WO2009065298A1/zh not_active Ceased
- 2008-11-20 TW TW097144812A patent/TW201019937A/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07101959A (ja) * | 1993-09-30 | 1995-04-18 | Sankyo Co Ltd | 1−メチルカルバペネム誘導体 |
| CN1484640A (zh) * | 2001-01-02 | 2004-03-24 | - | 用作α1A/B肾上腺素能受体拮抗剂的喹唑酮衍生物 |
| CN1524082A (zh) * | 2001-07-06 | 2004-08-25 | 作为治疗或预防糖尿病的二肽基肽酶抑制剂的β-氨基四氢咪唑并(1,2-A)吡嗪和四氢三唑并(4,3-A)吡嗪 | |
| WO2003076427A1 (en) * | 2002-03-14 | 2003-09-18 | Pfizer Limited | Quinazoline compounds useful in therapy |
| WO2004058266A1 (en) * | 2002-12-20 | 2004-07-15 | Merck & Co., Inc. | 3-amino-4-phenylbutanoic acid derivatives as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
Non-Patent Citations (1)
| Title |
|---|
| DHAR, T. G. MURALI ET AL.: "Synthesis and SAR of p38a MAP kinase inhibitors based on heterobicyclic scaffolds", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 17, no. 18, 15 September 2007 (2007-09-15), pages 5019 - 5024 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011508735A (ja) * | 2007-12-26 | 2011-03-17 | ジエンス ヘンルイ メデイシンカンパニー リミテッド | テトラヒドロ・イミダゾ[1,5−α]ピラジン誘導体、その調製プロセスおよび医薬的使用 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| US9273058B2 (en) | 2013-11-14 | 2016-03-01 | Bristol-Myers Squibb Company | Substituted pyrazolo-piperazines as casein kinase 1 δ/ε inhibitors |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| JP2021519308A (ja) * | 2018-03-29 | 2021-08-10 | ボード オブ レジェンツ, ザ ユニバーシティ オブ テキサス システムBoard Of Regents, The University Of Texas System | 転写活性化タンパク質のイミダゾピペラジン阻害剤 |
| JP7387627B2 (ja) | 2018-03-29 | 2023-11-28 | ボード オブ レジェンツ,ザ ユニバーシティ オブ テキサス システム | 転写活性化タンパク質のイミダゾピペラジン阻害剤 |
| US12186324B2 (en) | 2018-03-29 | 2025-01-07 | Board Of Regents, The University Of Texas System | Imidazopiperazine inhibitors of transcription activating proteins |
| US12540139B2 (en) | 2020-10-02 | 2026-02-03 | Board Of Regents, The University Of Texas System | Imidazopiperazine inhibitors of transcription activating proteins |
| US12559497B2 (en) | 2020-10-02 | 2026-02-24 | Board Of Regents, The University Of Texas System | Imidazopiperazine inhibitors of transcription activating proteins |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101641360B (zh) | 2012-07-11 |
| CN101417999A (zh) | 2009-04-29 |
| TW201019937A (en) | 2010-06-01 |
| CN101641360A (zh) | 2010-02-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009065298A1 (en) | Piperazine derivatives, preparation process and pharmaceutical use thereof | |
| CA2947338C (en) | Multi-fluoro-substituted compound as bruton's tyrosine kinase (btk) inhibitor | |
| KR102429419B1 (ko) | Rho-키나아제 억제제로서 티로신 아마이드 유도체 | |
| KR101994381B1 (ko) | 키나아제 억제제 | |
| CN103974948B (zh) | 在治疗过度增殖性疾病中用作bub1激酶抑制剂的取代的苄基吲唑 | |
| JP4572194B2 (ja) | 置換された8−パーフルオロアルキル−6,7,8,9−テトラヒドロピリミド[1,2−a]ピリミジン−4−オン誘導体 | |
| WO2009082881A1 (en) | Tetrahydro-imidazo[1,5-a]pyrazine derivatives, preparation methods and medical uses thereof | |
| JP7686741B2 (ja) | Alk5阻害剤としてのピリダジニルアミノ誘導体 | |
| AU2017382029A1 (en) | Bicyclic dihydropyrimidine-carboxamide derivatives as Rho-kinase inhibitors | |
| AU2010271270A1 (en) | Substituted pyrazolo[1,5-a]pyrimidine compounds as Trk kinase inhibitors | |
| EP3704118A1 (en) | Aminoimidazopyridazines as kinase inhibitors | |
| KR20210113287A (ko) | 할로-알릴아민 화합물 및 그의 용도 | |
| JPWO2005021550A1 (ja) | 二環性ピラゾール誘導体 | |
| WO2018133875A1 (zh) | Jak酶抑制剂及其制备方法和用途 | |
| KR102000382B1 (ko) | 신규 시클로프로파벤조푸라닐 피리도피라진디온 | |
| KR20100050485A (ko) | 1,2,3,4-테트라히드로피롤로[1,2-a]피라진-6-카르복스아미드 및 2,3,4,5-테트라히드로피롤로[1,2-a][1,4]-디아제핀-7-카르복스아미드 유도체, 그의 제조 및 치료적 용도 | |
| WO2006030847A1 (ja) | 新規二環性ピラゾール誘導体 | |
| JP2013527226A (ja) | 3,4−ジヒドロピロロ[1,2−a]ピラジン−2,8(1H)−ジカルボキサミド誘導体、その調製、およびその治療的使用 | |
| KR20220090553A (ko) | Ssao 억제제 및 그의 용도 | |
| WO2023118253A1 (en) | Cyclohexane acid derivatives as lpa receptor inhibitors | |
| EP1942897A1 (en) | Use of pyrimidine derivatives in the manufacture of a medicament for prevention and/or treatment of alzheimer's disease | |
| CN117043144A (zh) | 作为lpa受体2抑制剂的8-环-取代的喹唑啉衍生物 | |
| JPWO2006112331A1 (ja) | 新規縮合ピロール誘導体 | |
| JP2024534028A (ja) | サイクリン依存性キナーゼ阻害剤 | |
| WO2021116257A1 (en) | Thienopyrimidine derivatives as lpa receptor 2 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880009683.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08851197 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08851197 Country of ref document: EP Kind code of ref document: A1 |