WO2009090320A1 - Procede de preparation d'une piperidine disubstituee et intermediaires - Google Patents
Procede de preparation d'une piperidine disubstituee et intermediaires Download PDFInfo
- Publication number
- WO2009090320A1 WO2009090320A1 PCT/FR2008/001280 FR2008001280W WO2009090320A1 WO 2009090320 A1 WO2009090320 A1 WO 2009090320A1 FR 2008001280 W FR2008001280 W FR 2008001280W WO 2009090320 A1 WO2009090320 A1 WO 2009090320A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- acid
- action
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C1C(*)(*)CNC(*)C1 Chemical compound *C1C(*)(*)CNC(*)C1 0.000 description 1
- ATSPEBLEIQXAOO-LURJTMIESA-N CCC(CC[C@@H](C(O)=O)N)=N Chemical compound CCC(CC[C@@H](C(O)=O)N)=N ATSPEBLEIQXAOO-LURJTMIESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/72—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D211/78—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/32—Oximes
- C07C251/34—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C251/36—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with the carbon atoms of the oxyimino groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C251/38—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with the carbon atoms of the oxyimino groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the subject of the invention is a process for preparing a disubstituted 2,5-piperidine and new intermediates.
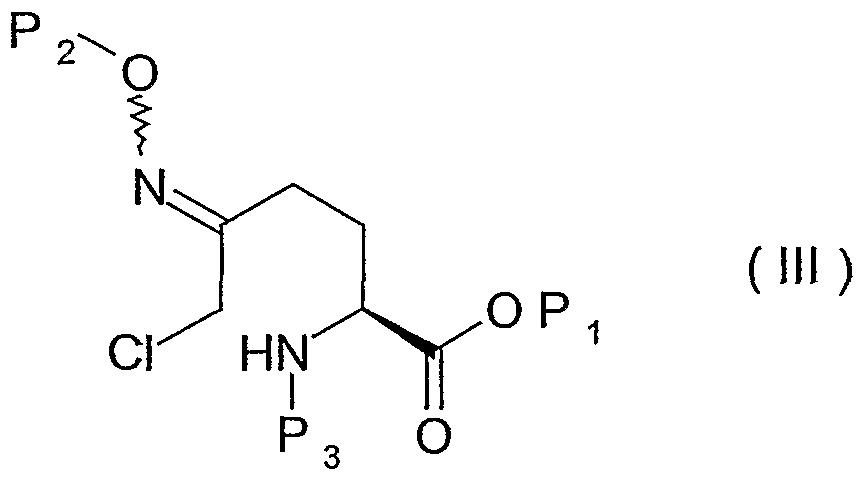
- P 1 and P 2 represent protective groups of the carboxylic acid and oxyamine functions known to those skilled in the art and in particular those cited in application WO 02/10172.
- the compound of formula (I) is in the form of a mixture of isomers (2S, 5R) and (2S, 5S).
- the compound of formula (I) can be obtained as described in application WO 02/10172, in particular in example 32, starting from the protected cis-5-hydroxy-piperidme-2-carboxylic acid.
- the subject of the present invention is a new process for preparing the compound of formula (I), characterized in that the compound of formula (b) is treated:
- the beta-acetosulfoxonium compound of formula (b) can be obtained starting from the protected (S) pyrroglutamic acid of formula (a):
- the protective group of the carboxylic acid function Pi is in particular a residue of alkyl, allyl, benzyl or p-nitrobenzyl ester, equivalent residues known to those skilled in the art could of course also be suitable.
- P 1 is preferably a benzyl group.
- the nitrogen protection group P 3 forms in particular a carbamate and is preferably a tert-butoxycarbonyl or benzyloxycarbonyl, it can also be an electron-withdrawing group such as those known to those skilled in the art referenced in the "Greene” (Protective Groups in Organic Synthesis, 3rd edition).
- P 3 is preferably a tert-butoxycarbonyl group.
- the protective group of hydroxylamine P 2 is in particular a benzyl or allyl radical.
- P 2 is preferably a benzyl group.
- the conditions generating HCl and making it possible to prepare the compound of formula (II) preferably consist in using lithium chloride in the presence of a strong acid.
- hydrochloric acid is, for example, hydrochloric acid, sulfuric acid, a sulfonic acid such as methanesulfonic acid or ethanesulphonic acid.
- lithium chloride is used in the presence of methanesulfonic acid.
- the deprotection of the amine function is carried out by the action of an acid, for example hydrochloric acid, sulfonic acid, trifluoroacetic acid or an alkanesulfonic acid.
- an acid for example hydrochloric acid, sulfonic acid, trifluoroacetic acid or an alkanesulfonic acid.
- these conditions are known to those skilled in the art.
- Advantageous conditions are to use a protecting group tert-butoxycarbonyl and cleaving it by the action of methanesulphonic acid. It can be carried out for example in ethyl acetate.
- the protected ⁇ -chlorooxime of formula (III) is preferably used without being isolated, that is to say in solution in the reaction solvent. It is the same for the protected ⁇ -chlorooxime of formula (IV).
- the base used to cyclize the compound of formula (IV) is for example an alkali metal hydroxide, carbonate or bicarbonate, preferably sodium bicarbonate, or an amine base, for example triethylamine.
- the reducing agent used to reduce the oxime function is, for example, an alkaline borohydride, diborane or borane-pyridine type reactant in the presence of an acid, for example hydrochloric acid. It can be carried out in an alcohol such as methanol or ethanol, or in another organic solvent such as dichloromethane.
- the salification of the compound of formula (I) is optionally carried out by adding an acid in the soluble phase to the compound.
- an acid in the soluble phase to the compound.
- acid salts of the products of formula (I) mention may be made, inter alia, of those formed with mineral acids, such as hydrochloric, hydrobromic, hydroiodic, sulfuric or phosphoric acids, or with organic acids such as formic acid.
- acetic, trifluoroacetic, propionic benzoic, maleic, fumaric, succinic, tartaric, citric, oxalic, glyoxylic, aspartic, alkanesulfonic, such as methane and ethanesulphonic, arylsulfonic acids such as benzene and para-toluenesulphonic acids.
- the salts are preferably those allowing crystallization under easy conditions.
- the oxalic acid salt is particularly preferred.
- the present invention provides a process for preparing the intermediate of formula (I) under particularly attractive conditions, with an overall yield of the order of 70% and which thus makes it possible to overcome the failure previously encountered.
- the compounds of formula (III), (IV) and (V) obtained during the implementation of the process are new and also constitute one of the objects of the invention, as new industrial compounds and in particular intermediates. necessary in the preparation of the compounds of formula (I).
- the following example illustrates the invention.
- the reaction mixture is warmed to 0 ° C., stirred for 45 minutes and then added to a mixture of ammonium chloride (450 g), water (1.5 l) and ethyl acetate (0.5 l). ) at 20 ° C.
- the organic phase is isolated and washed with a solution of ammonium chloride (180 g) in water (0.6 L) and then with a solution of sodium chloride (200 g) in water (0.6 L). ).
- the aqueous phases are extracted with ethyl acetate.
- the combined organic phases are dried and the product is precipitated by concentrating the solution under reduced pressure at 20 ° C to a volume of 0.4 l and then adding methyl tert-butyl ether (0.25 l).
- the suspension is cooled to -10 ° C, stirred for 2 h, filtered and washed with ethyl acetate-methyl-tert-butyl ether (7: 3, 2x50 ml).
- the crystals are dried at 40 ° C under reduced pressure to give the expected ⁇ -keto-sulfoxonium (114.7 g, 279 moles, 89% yield).
- Step A (S) -5- (benzyloxyimino) -2-tert-butyloxycarbonylamino-6-chloro-hexanoic acid, benzyl ester (E + Z) Methanesulfonic acid (29.3 g, 0.305 mol) is added slowly to a mixture of ⁇ -ketosulfoxonium (114 g, 0.277 mol) and lithium chloride (13.3 g, 0.314 mol) in THF (1.71 ml) at room temperature. The reaction mixture is heated at 50 ° C for 5 hours and then cooled to room temperature.
- Benzylhydroxylamine hydrochloride (46.4 g, 0.291 mol) and sodium acetate (29.6 g, 0.361 mol) are added.
- the reaction medium is diluted with ethyl acetate (0.5 l) and water (0.5 l) and then stirred at room temperature for 18 hours.
- the organic phase is isolated, concentrated to a volume of 0.4 l and then washed with a solution of sodium chloride (25 g) in water (0.25 l).
- the aqueous phase is separated and then extracted with ethyl acetate (0.2 l).
- the organic phases are combined and stirred for 1 hour with sodium sulfate (100 g).
- the mixture is filtered and rinsed with ethyl acetate (2 x 0.1 L).
- the ⁇ -chlorooxime solution is stored in the refrigerator for the next step where it is used as it is. (130.8 g, 0.275 mole, 99.3% yield).
- the ⁇ -chlorooxime solution (0.275 mol) is dried by azeotropic distillation to a volume of 0.5 l and then diluted with ethyl acetate (0.5 l). The solution is cooled to 0 ° C. The methanesulfonic acid (136 g, 1.42 mol) is added over 15 minutes. The reaction mixture is heated at 40 ° C for 1 hour and then cooled to room temperature before being added to a solution of sodium bicarbonate (279 g, 3.40 moles) in water (1 L). The reaction mixture is heated at 50 ° C for 3 hours and then cooled to room temperature. The organic phase is isolated and then washed with a solution of sodium chloride (50 g) in water (0.5 l).
- the aqueous phases are extracted with ethyl acetate (0.5 l).
- the organic phases are united.
- the solution is mixed with silica (100 g) and stirred for 20 minutes.
- the solution is filtered and then washed with ethyl acetate.
- the piperidine oxime solution is concentrated to a volume of 0.2 l and then stored in the refrigerator for the next step. (88.8 g, 0.262 moles, 95.3% yield).
- the E and Z isomers of the oxime are separated by chromatography and then analyzed by NMR;
- Stage C (2S) -5-benzyloxyamino-piperidine-2-carboxylic acid, benzyl ester and its oxalate (mixture (2S.5R) and (2S, 5S) ⁇ 50/50).
- the ethyl acetate, in which the piperidinoxime obtained in stage B is in solution, is replaced by methanol by distillation and then diluted to a volume of 0.2.
- the piperidinoxime solution (0.261 mole) is added in 30 ml. minutes to a solution of HCl (1.32 mol) in methanol (0.31 L) at 0 ° C.
- borane pyridine (45.4 g, 0.49 mol) is added in 4 hours to the reaction medium at 0 ° C. The medium is warmed to room temperature and then stirred overnight. The solution is concentrated to a volume of 0.18 l and then diluted with dichloromethane (0.36 L) and water (0.36 L). An aqueous solution of 50% sodium hydroxide is added until pH 7. The aqueous phase is separated and then extracted with dichloromethane (0.27 l). The organic phase is washed with water.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description








Claims








Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT08870986T ATE539057T1 (de) | 2007-09-14 | 2008-09-12 | Verfahren zur herstellung von disubstituiertem piperidin und zwischenprodukten davon |
| ES08870986T ES2380098T3 (es) | 2007-09-14 | 2008-09-12 | Procedimiento de preparación de una piperidina disustituida y productos intermedios |
| EP08870986A EP2200983B1 (fr) | 2007-09-14 | 2008-09-12 | Procede de preparation d'une piperidine disubstituee et intermediaires |
| HK10111791.6A HK1145324B (en) | 2007-09-14 | 2008-09-12 | Method for preparing disubstituted piperidine and intermediates |
| CN200880106715.8A CN102056901B (zh) | 2007-09-14 | 2008-09-12 | 二取代哌啶及其中间体的制备方法 |
| US12/677,741 US8288553B2 (en) | 2007-09-14 | 2008-09-12 | Method for preparing disubstituted piperidine and intermediates |
| CA2699438A CA2699438C (fr) | 2007-09-14 | 2008-09-12 | Procede de preparation d'une piperidine disubstituee et intermediaires |
| JP2010524543A JP5421265B2 (ja) | 2007-09-14 | 2008-09-12 | 二置換ピペリジン及び中間体の製法 |
| US13/616,995 US20130012712A1 (en) | 2007-09-14 | 2012-09-14 | Method for preparing disubstituted piperidine and intermediates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0706449A FR2921060B1 (fr) | 2007-09-14 | 2007-09-14 | Nouveau procede de preparation d'une piperidine disubsituee et nouveaux intermediaires |
| FR0706449 | 2007-09-14 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/616,995 Division US20130012712A1 (en) | 2007-09-14 | 2012-09-14 | Method for preparing disubstituted piperidine and intermediates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009090320A1 true WO2009090320A1 (fr) | 2009-07-23 |
Family
ID=39428023
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2008/001280 Ceased WO2009090320A1 (fr) | 2007-09-14 | 2008-09-12 | Procede de preparation d'une piperidine disubstituee et intermediaires |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US8288553B2 (fr) |
| EP (1) | EP2200983B1 (fr) |
| JP (1) | JP5421265B2 (fr) |
| CN (1) | CN102056901B (fr) |
| AT (1) | ATE539057T1 (fr) |
| CA (1) | CA2699438C (fr) |
| ES (1) | ES2380098T3 (fr) |
| FR (1) | FR2921060B1 (fr) |
| WO (1) | WO2009090320A1 (fr) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012086241A1 (fr) * | 2010-12-22 | 2012-06-28 | Meiji Seikaファルマ株式会社 | Dérivé de diazabicyclooctane optiquement actif, et procédé de fabrication de celui-ci |
| WO2012133486A1 (fr) | 2011-03-31 | 2012-10-04 | 株式会社カネカ | Procédé de préparation d'un composé amine cyclique |
| WO2014091268A1 (fr) | 2012-12-11 | 2014-06-19 | Naeja Pharmaceutical Inc. | Nouveaux composés bicycliques et leur utilisation en tant qu'agents antibactériens et inhibiteurs de β-lactamase |
| US8796257B2 (en) | 2011-12-02 | 2014-08-05 | Naeja Pharmaceutical Inc. | Bicyclic compounds and their use as antibacterial agents and β-lactamase inhibitors |
| US20150141401A1 (en) * | 2012-05-30 | 2015-05-21 | Meiji Seika Pharma Co., Ltd. | Novel beta-lactamase inhibitor and process for preparing the same |
| US9505761B2 (en) | 2011-12-02 | 2016-11-29 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and beta-lactamase inhibitors |
| CN106699756A (zh) * | 2016-12-30 | 2017-05-24 | 淄博鑫泉医药技术服务有限公司 | β内酰胺酶抑制剂阿维巴坦的合成方法 |
| US9676777B2 (en) | 2010-12-22 | 2017-06-13 | Meiji Seika Pharma Co., Ltd. | Compounds useful for producing an optically active diazabicyclooctane compound |
| US10000491B2 (en) | 2013-09-24 | 2018-06-19 | Meiji Seika Pharma Co., Ltd. | Process for producing diazabicyclooctane derivative and intermediate thereof |
| US10131665B2 (en) | 2013-10-08 | 2018-11-20 | Meiji Seika Pharma Co., Ltd. | Processes for producing diazabicyclooctane compounds |
| US10294224B2 (en) | 2014-12-05 | 2019-05-21 | Meiji Seika Pharma Co., Ltd. | Lyophilized composition of a diazabicyclooctane compound and process of producing the same |
| RU2713400C1 (ru) * | 2017-08-18 | 2020-02-05 | Синьфа Фармасьютикал Ко., Лтд | Способ получения 5r-[(бензилокси)амино]пиперидин-2s-карбоксилата и его оксалатов |
| WO2022047603A1 (fr) | 2020-09-01 | 2022-03-10 | Ningxia Academy Of Agriculture And Forestry Sciences | Inhibiteurs de bêta-lactamase et leur préparation |
| WO2022233181A1 (fr) | 2021-05-07 | 2022-11-10 | Ningxia Academy Of Agriculture And Forestry Sciences | Composés substitués de sulfonylamidine et leur utilisation en tant qu'inhibiteurs de bêta-lactamase |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2951171A1 (fr) | 2009-10-09 | 2011-04-15 | Novexel | Nouveau sel de sodium d'un compose azabicyclique sous forme enantiomere cristallisee et nouvelles formes polymorphes et pseudopolymorphes ainsi que leur preparation |
| RU2610091C2 (ru) | 2011-06-17 | 2017-02-07 | Астразенека Аб | (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид |
| US8940897B2 (en) | 2012-03-30 | 2015-01-27 | Cubist Pharmaceuticals, Inc. | 1,3,4-oxadiazole and 1,3,4-thiadiazole β-lactamase inhibitors |
| US8916709B2 (en) | 2012-03-30 | 2014-12-23 | Cubist Pharmaceuticals, Inc. | 1,2,4-oxadiazole and 1,2,4-thiadiazole β-lactamase inhibitors |
| WO2013149136A1 (fr) | 2012-03-30 | 2013-10-03 | Cubist Pharmaceuticals, Inc. | Inhibiteurs des β-lactamases dérivés d'isoxazole |
| US8969570B2 (en) | 2012-03-30 | 2015-03-03 | Cubist Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| BR112015021332B1 (pt) | 2013-03-08 | 2020-06-30 | Wockhardt Limited | sal de sódio de ácido (2s, 5r)-6-benzilóxi-7-oxo-1,6-diaza-biciclo [3.2.1] octano-2-carboxílico e sua preparação |
| WO2014152996A1 (fr) | 2013-03-14 | 2014-09-25 | Cubist Pharmaceuticals, Inc. | Forme cristalline d'un inhibiteur de bêta-lactamase |
| US9120796B2 (en) | 2013-10-02 | 2015-09-01 | Cubist Pharmaceuticals, Inc. | B-lactamase inhibitor picoline salt |
| WO2017136254A1 (fr) * | 2016-02-04 | 2017-08-10 | Merck Sharp & Dohme Corp. | Procédés de préparation de dérivés d'hydroxylamine utiles dans la préparation d'agents anti-infectieux |
| CN107540601B (zh) * | 2016-06-28 | 2019-07-19 | 新发药业有限公司 | 5r-苄氧氨基哌啶-2s-甲酸酯及其草酸盐的便捷制备方法 |
| CN107540600B (zh) * | 2016-06-28 | 2019-07-09 | 新发药业有限公司 | 一种阿维巴坦中间体生产废液的回收利用方法 |
| CN109956941B (zh) | 2017-12-25 | 2020-08-04 | 新发药业有限公司 | 一种阿维巴坦的简便制备方法 |
| CN109970625B (zh) * | 2017-12-28 | 2021-02-26 | 新发药业有限公司 | 一种5r-苄氧氨基哌啶-2s-甲酸或其衍生物的制备方法 |
| CN114206851B (zh) * | 2019-04-26 | 2025-02-11 | 默沙东有限责任公司 | 可用于制备(2s,5r)-7-氧代-n-哌啶-4-基-6-(硫酸基)-1,6-二氮杂双环[3.2.1]辛烷-2-甲酰胺的中间体的制备方法 |
| CN110498762B (zh) * | 2019-08-28 | 2020-10-27 | 山东安信制药有限公司 | 一种(2s,5r)-5-[(苄氧基)氨基]-哌啶-2-甲酸乙酯的合成方法 |
| CN114989169A (zh) * | 2022-07-04 | 2022-09-02 | 哈药集团技术中心 | 一种阿维巴坦钠的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002010172A1 (fr) * | 2000-08-01 | 2002-02-07 | Aventis Pharma Sa | Composes azabicycliques, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens |
| WO2002100860A2 (fr) * | 2001-06-08 | 2002-12-19 | Aventis Pharma S.A. | Composes heterocycliques, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens |
| WO2003063864A2 (fr) * | 2002-01-28 | 2003-08-07 | Aventis Pharma S.A. | Composes heterocycliques, actifs comme inhibiteurs de beta-lactamases |
| WO2004052891A1 (fr) * | 2002-12-06 | 2004-06-24 | Aventis Pharma S.A. | Composes heterocyclique, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens et inhibiteurs de beta-lactamases |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5059377A (fr) * | 1973-10-04 | 1975-05-22 | ||
| JPS63162666A (ja) * | 1986-12-26 | 1988-07-06 | Asahi Chem Ind Co Ltd | アミノオキシ酢酸誘導体およびその製造方法 |
| JPH0393762A (ja) * | 1989-09-04 | 1991-04-18 | Wako Pure Chem Ind Ltd | 4‐ハロゲノ‐2‐アルコキシイミノ‐3‐オキソ脂肪酸の製造方法 |
| US6399793B1 (en) * | 2000-08-16 | 2002-06-04 | Bristol-Myers Squibb Company | Process for the preparation of α' chloroketones |
| FR2833596B1 (fr) | 2001-12-14 | 2005-02-18 | Aventis Pharma Sa | Procede de preparation de derives d'echinocandine |
| US7439253B2 (en) | 2002-12-06 | 2008-10-21 | Novexel | Heterocyclic compounds, their preparation and their use as medicaments, in particular as antibacterials and beta-lactamase inhibitors |
| JP3935126B2 (ja) * | 2003-09-24 | 2007-06-20 | 雅裕 亀田 | 装飾用灯具 |
| US7396934B2 (en) | 2003-11-17 | 2008-07-08 | Youssef El-Ahmad | Process for preparing 3-fluoroquinolines |
| JP2007126358A (ja) * | 2004-01-20 | 2007-05-24 | Fujiyakuhin Co Ltd | フェニルアラニン誘導体 |
-
2007
- 2007-09-14 FR FR0706449A patent/FR2921060B1/fr active Active
-
2008
- 2008-09-12 CA CA2699438A patent/CA2699438C/fr active Active
- 2008-09-12 AT AT08870986T patent/ATE539057T1/de active
- 2008-09-12 US US12/677,741 patent/US8288553B2/en active Active
- 2008-09-12 ES ES08870986T patent/ES2380098T3/es active Active
- 2008-09-12 EP EP08870986A patent/EP2200983B1/fr active Active
- 2008-09-12 WO PCT/FR2008/001280 patent/WO2009090320A1/fr not_active Ceased
- 2008-09-12 JP JP2010524543A patent/JP5421265B2/ja active Active
- 2008-09-12 CN CN200880106715.8A patent/CN102056901B/zh active Active
-
2012
- 2012-09-14 US US13/616,995 patent/US20130012712A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002010172A1 (fr) * | 2000-08-01 | 2002-02-07 | Aventis Pharma Sa | Composes azabicycliques, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens |
| WO2002100860A2 (fr) * | 2001-06-08 | 2002-12-19 | Aventis Pharma S.A. | Composes heterocycliques, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens |
| WO2003063864A2 (fr) * | 2002-01-28 | 2003-08-07 | Aventis Pharma S.A. | Composes heterocycliques, actifs comme inhibiteurs de beta-lactamases |
| WO2004052891A1 (fr) * | 2002-12-06 | 2004-06-24 | Aventis Pharma S.A. | Composes heterocyclique, leur preparation et leur utilisation comme medicaments, notamment comme anti-bacteriens et inhibiteurs de beta-lactamases |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016117742A (ja) * | 2010-12-22 | 2016-06-30 | Meiji Seikaファルマ株式会社 | 光学活性なジアザビシクロオクタン誘導体およびその製造法 |
| JPWO2012086241A1 (ja) * | 2010-12-22 | 2014-05-22 | Meiji Seikaファルマ株式会社 | 光学活性なジアザビシクロオクタン誘導体およびその製造法 |
| WO2012086241A1 (fr) * | 2010-12-22 | 2012-06-28 | Meiji Seikaファルマ株式会社 | Dérivé de diazabicyclooctane optiquement actif, et procédé de fabrication de celui-ci |
| JP2018009008A (ja) * | 2010-12-22 | 2018-01-18 | Meiji Seikaファルマ株式会社 | 光学活性なジアザビシクロオクタン誘導体およびその製造法 |
| US9676777B2 (en) | 2010-12-22 | 2017-06-13 | Meiji Seika Pharma Co., Ltd. | Compounds useful for producing an optically active diazabicyclooctane compound |
| WO2012133486A1 (fr) | 2011-03-31 | 2012-10-04 | 株式会社カネカ | Procédé de préparation d'un composé amine cyclique |
| JP5952806B2 (ja) * | 2011-03-31 | 2016-07-13 | 株式会社カネカ | 環状アミン化合物の製造法 |
| US9096523B2 (en) | 2011-03-31 | 2015-08-04 | Kaneka Corporation | Process for preparing cyclic amine compounds |
| US8877743B2 (en) | 2011-12-02 | 2014-11-04 | Naeja Pharmaceutical Inc. | Bicyclic compounds and their use as antibacterial agents and β-lactamase inhibitors |
| US10544146B2 (en) | 2011-12-02 | 2020-01-28 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and β-lactamase inhibitors |
| US10030019B2 (en) | 2011-12-02 | 2018-07-24 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and β-lactamase inhibitors |
| US9393239B2 (en) | 2011-12-02 | 2016-07-19 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and betalactamase inhibitors |
| US9505761B2 (en) | 2011-12-02 | 2016-11-29 | Fedora Pharmaceuticals Inc. | Bicyclic compounds and their use as antibacterial agents and beta-lactamase inhibitors |
| US8796257B2 (en) | 2011-12-02 | 2014-08-05 | Naeja Pharmaceutical Inc. | Bicyclic compounds and their use as antibacterial agents and β-lactamase inhibitors |
| US9708320B2 (en) | 2012-05-30 | 2017-07-18 | Meiji Seika Pharma Co., Ltd. | β-lactamase inhibitor and process for preparing the same |
| US9181250B2 (en) | 2012-05-30 | 2015-11-10 | Meiji Seika Pharma Co., Ltd. | Beta-lactamase inhibitor and process for preparing the same |
| US12103928B2 (en) | 2012-05-30 | 2024-10-01 | Meiji Seika Pharma Co., Ltd. | Processes for preparing a diazabicyclooctane compound |
| US11731971B2 (en) | 2012-05-30 | 2023-08-22 | Meiji Seika Pharma Co., Ltd. | Processes for preparing a diazabicyclooctane compound |
| US10023573B2 (en) | 2012-05-30 | 2018-07-17 | Meiji Seika Pharma Co., Ltd. | Beta-lactamase inhibitor and process for preparing the same |
| US20150141401A1 (en) * | 2012-05-30 | 2015-05-21 | Meiji Seika Pharma Co., Ltd. | Novel beta-lactamase inhibitor and process for preparing the same |
| US11117896B2 (en) | 2012-05-30 | 2021-09-14 | Meiji Seika Pharma Co., Ltd. | Processes for preparing a diazabicyclooctane compound |
| US10556905B2 (en) * | 2012-05-30 | 2020-02-11 | Meiji Seika Pharma Co., Ltd. | Processes for preparing a diazabicyclooctane compound |
| WO2014091268A1 (fr) | 2012-12-11 | 2014-06-19 | Naeja Pharmaceutical Inc. | Nouveaux composés bicycliques et leur utilisation en tant qu'agents antibactériens et inhibiteurs de β-lactamase |
| US10000492B2 (en) | 2013-09-24 | 2018-06-19 | Meiji Seika Pharma Co., Ltd. | Process for producing diazabicyclooctane derivative |
| US10000491B2 (en) | 2013-09-24 | 2018-06-19 | Meiji Seika Pharma Co., Ltd. | Process for producing diazabicyclooctane derivative and intermediate thereof |
| US10604522B2 (en) | 2013-10-08 | 2020-03-31 | Meiji Seika Pharma Co., Ltd. | Processes for producing diazabicyclooctane compounds |
| US10131665B2 (en) | 2013-10-08 | 2018-11-20 | Meiji Seika Pharma Co., Ltd. | Processes for producing diazabicyclooctane compounds |
| US11414417B2 (en) | 2013-10-08 | 2022-08-16 | Meiji Seika Pharma Co., Ltd. | Crystalline forms of diazabicyclooctane derivative and production process thereof |
| US12221443B2 (en) | 2013-10-08 | 2025-02-11 | Meiji Seika Pharma Co., Ltd. | Crystalline forms of diazabicyclooctane derivative and production process thereof |
| US10294224B2 (en) | 2014-12-05 | 2019-05-21 | Meiji Seika Pharma Co., Ltd. | Lyophilized composition of a diazabicyclooctane compound and process of producing the same |
| US11117895B2 (en) | 2014-12-05 | 2021-09-14 | Meiji Seika Pharma Co., Ltd. | Process for producing crystals of a diazabicyclooctane derivative |
| CN106699756A (zh) * | 2016-12-30 | 2017-05-24 | 淄博鑫泉医药技术服务有限公司 | β内酰胺酶抑制剂阿维巴坦的合成方法 |
| RU2713400C1 (ru) * | 2017-08-18 | 2020-02-05 | Синьфа Фармасьютикал Ко., Лтд | Способ получения 5r-[(бензилокси)амино]пиперидин-2s-карбоксилата и его оксалатов |
| RU2713400C9 (ru) * | 2017-08-18 | 2020-06-16 | Синьфа Фармасьютикал Ко., Лтд | Способ получения 5r-[(бензилокси)амино]пиперидин-2s-карбоксилата и его оксалатов |
| WO2022047603A1 (fr) | 2020-09-01 | 2022-03-10 | Ningxia Academy Of Agriculture And Forestry Sciences | Inhibiteurs de bêta-lactamase et leur préparation |
| WO2022233181A1 (fr) | 2021-05-07 | 2022-11-10 | Ningxia Academy Of Agriculture And Forestry Sciences | Composés substitués de sulfonylamidine et leur utilisation en tant qu'inhibiteurs de bêta-lactamase |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2200983A1 (fr) | 2010-06-30 |
| CA2699438C (fr) | 2016-11-22 |
| US20100197928A1 (en) | 2010-08-05 |
| US8288553B2 (en) | 2012-10-16 |
| CA2699438A1 (fr) | 2009-07-23 |
| US20130012712A1 (en) | 2013-01-10 |
| CN102056901A (zh) | 2011-05-11 |
| ES2380098T3 (es) | 2012-05-08 |
| JP5421265B2 (ja) | 2014-02-19 |
| FR2921060A1 (fr) | 2009-03-20 |
| EP2200983B1 (fr) | 2011-12-28 |
| JP2010539147A (ja) | 2010-12-16 |
| ATE539057T1 (de) | 2012-01-15 |
| HK1145324A1 (en) | 2011-04-15 |
| CN102056901B (zh) | 2014-09-03 |
| FR2921060B1 (fr) | 2012-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2200983B1 (fr) | Procede de preparation d'une piperidine disubstituee et intermediaires | |
| FR2963006A1 (fr) | Procede de preparation de derives de nitro-benzofurane | |
| JP4299677B2 (ja) | コンブレタスタチンの製造方法 | |
| US20100113778A1 (en) | Process for preparing o-chloromethylphenylglyoxylic esters, improved process for preparing (e)-2-(2-chloromethylphenyl)-2-alkoximinoacetic esters, and novel intermediates for their preparation | |
| CZ257593A3 (en) | Derivatives of phenylacetic acid esters and process for preparing thereof | |
| KR102748251B1 (ko) | 리나글립틴 및 이의 염의 제조를 위한 중간체 및 방법 | |
| WO1996038447A1 (fr) | Derives du 2-azabicyclo[2.2.1]heptane, leur preparation et leur application | |
| EP2938595B1 (fr) | Procede de synthese d'une hydrazine utile dans le traitement du virus du papillome | |
| FR2689890A1 (fr) | Procédé de fabrication d'un dérivé à groupe benzo[b]thiophényle-5 et intermédiaire de fabrication. | |
| FR2792635A1 (fr) | Derives de cyclobutene-3,4-dione leur preparation et leur application en therapeutique | |
| HU223138B1 (hu) | Új eljárás racém alkil-4,6,7,8,9,9a-hexahidro-2H, 3H-pirido-[1,2-a] pirazin-1-on-7-karbonsavészterek előállítására | |
| JP2003171354A (ja) | 光学活性アミン化合物又はその塩の製造方法 | |
| WO2010020719A1 (fr) | Procede de preparation de l'ester ethylique de l'acide 4- [trans-4-[(phenylmethyl)-amino]cyclohexyl] benzoïque et de son sel hemifumarate | |
| WO2000020412A1 (fr) | Derives de quinazolinedione, leurs preparations et leurs applications en therapeutique | |
| BE891687A (fr) | Composes heterocycliques contenant un groupe c-acetyle et leur procede de preparation | |
| EP1347956A1 (fr) | Procede de cyclisation pour la synthese de composes derives de pyrrolidine | |
| HK1145324B (en) | Method for preparing disubstituted piperidine and intermediates | |
| FR2519339A1 (fr) | Composes heterocycliques contenant un groupe c-acetyle et leur procede de preparation | |
| JPH07247275A (ja) | オキサゾリジノン誘導体の単一のジアステレオマーの製造方法及びウレタン化合物の単一のジアステレオマーのフマル酸塩 | |
| EP0828740A1 (fr) | Derives du 2-azabicyclo(2.2.1)heptane, leur preparation et leur application | |
| JP2003026672A (ja) | 3−アミノオキセタンの新規製造法並びに中間体 | |
| BE566396A (fr) | ||
| WO1999006392A1 (fr) | Procede de preparation de 2h-1 benzopyranes et intermediaires de synthese | |
| JPS5811420B2 (ja) | ベンゾビシクロアルカンアミン類の製法 | |
| WO2000073305A1 (fr) | Preparation de derives de la camptothecine et de la nothapodytine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880106715.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08870986 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2699438 Country of ref document: CA Ref document number: 12677741 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2010524543 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1724/DELNP/2010 Country of ref document: IN Ref document number: 2008870986 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |