WO2009109998A1 - Novel protein tyrosine phosphatase - ib inhibitors - Google Patents
Novel protein tyrosine phosphatase - ib inhibitors Download PDFInfo
- Publication number
- WO2009109998A1 WO2009109998A1 PCT/IN2009/000135 IN2009000135W WO2009109998A1 WO 2009109998 A1 WO2009109998 A1 WO 2009109998A1 IN 2009000135 W IN2009000135 W IN 2009000135W WO 2009109998 A1 WO2009109998 A1 WO 2009109998A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- methyl
- oxo
- thiazolidin
- ylideneamino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- WUISDMALWQTMCX-UHFFFAOYSA-N CN(CC(N1)=O)S1(=O)=O Chemical compound CN(CC(N1)=O)S1(=O)=O WUISDMALWQTMCX-UHFFFAOYSA-N 0.000 description 1
- 0 N*(C(NC1=Cc(cc2N)ccc2N)=*c(cc2)cc(N)c2N)C1=O Chemical compound N*(C(NC1=Cc(cc2N)ccc2N)=*c(cc2)cc(N)c2N)C1=O 0.000 description 1
- QGULWBQOCMQNFD-UHFFFAOYSA-N O=Cc(cc1)ccc1OCc(cc1)ccc1F Chemical compound O=Cc(cc1)ccc1OCc(cc1)ccc1F QGULWBQOCMQNFD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/54—Nitrogen and either oxygen or sulfur atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention is related to novel compounds of the general formula and their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods of making the above compounds, and their use as Protein tyrosine phosphatase IB (PTP-IB) inhibitors, which are useful in the treatment or prevention of diseases in which PTP-IB enzyme is known to be involved in the pathogenesis.
- PTP-IB Protein tyrosine phosphatase IB
- the PTPlB inhibitors would also find use in the treatment of diseases such as cancer, inflammatory disorders, autoimmune diseases and osteoporosis
- Type 2 diabetes mellitus (hereafter referred as type 2 diabetes, also known as non-insulin-dependent diabetes mellitus, NIDDM) is a heterogeneous disorder, with both genetic and environmental factors contributing to its development.
- the pathogenesis of type 2 diabetes involves multiple mechanisms leading to hyperglycemia, most notably increased hepatic glucose production, impaired insulin secretion by pancreatic ⁇ cells and reduced glucose uptake by skeletal muscle and adipose tissue (peripheral insulin resistance).
- Type 2 diabetic patients are at substantially increased risks of macrovascular disease including coronary heart disease and stroke and microvascular disease including retinopathy, nephropathy and neuropathy.
- Type 2 diabetes is a therapeutic area with huge market potential.
- the number of diabetic patients is projected to increase from 170-175 million in 2000 to over 350 million by 2030 (Wild, S., et al. Diab.Care 27, 1047-1053, 2004; Yach, D., et al. Nat. Med. 12, 62-66, 2006).
- the major part of this numerical increase is expected to occur in developing countries and India will have the distinction of having the largest number of diabetic patients in the world by 2030.
- the treatment approaches for type 2 diabetes include diet, exercise, and a variety of pharmacological agents.
- Clinically established therapies for type 2 diabetes include insulin and its analogs and various oral hypoglycemic agents: sulfonylureas, metformin, ⁇ - glucosidase inhibitors (acarbose, miglitol), non-sulfonylurea insulin secretagogues (repaglinide, nateglinide) and thiazolidinedione (TZD) derivatives (rosiglitazone, pioglitazone) acting via PPARy agonism (Matthaei, S., et al. Endocrine Rev. 21, 585-618, 2000; Skyler, J.S.
- PTPs Protein tyrosine phosphatases
- Unregulated operation of PTPs is responsible to many human diseases including cancer (BIume-Jenscn P., Nature 41 1 , 355-365, 2001), diabetes (Montalibet J., Drug Discov Today: Therap. Strateg. 2, 129-135, 2005), obesity (Cook W.S., Developmental Cell 2, 385-387, 2002), and osteoporosis (Schiller K.R.. J. Cell Biochem. 96, 262-277, 2005).
- PTPlB protein tyrosine phosphatase IB activates c-Src inhuman breast cancer (Bjorge J.D., J Biol Chew 275, 41439-41446, 2000), and also influences lhe down regulation of insulin signaling by dephosphorylating the insulin receptor including insulin receptor substrate- 1 (IRS-I ) and insulin receptor substrate-2 (IRS-2) (Walchli S.. J Biol Chem 275, 9792-9796, 2000). Therefore, PTPl B can be a useful target for diabetes and cancer, and inhibitors of PTPl B may be promising drugs to treat these diseases. In addition, considering that PTPlB knockout mice arc resistant to obesity.
- PTPlB plays critical role in development of obesity (Klanian L.D., MoI Cell Biol 20, 5479-5489. 2000). In spite of the therapeutic potential of PTPlB inhibitor against diabetes, obesity, and cancer, it is difficult to develop selective PTPlB inhibitor over other PTPs including SHP, VHR, LAR, CD45, and cdc25C, because of structural homologies in PTPs (Cheng A., Eur J Biochem 269, 1050-1059, 2002; Penninger J.M., J Nat Immunol 2, 389-396, 2001 ; Qu C.K., Biochim Biophys Acta 1592, 297-301. 2002; Hoffman B.T.. Curr Pharm Dcs 10, 1 161- 1 181 , 2004).
- T-cell PTP ( ' J 1 CPTP) has an 80% homology to PTPl B in the catalytic domains, non-selective inhibition gives rise to severe side effects (Tiganis T., J Biol Chem 274, 27768-27775, 1999; You-Ten K.E., J Exp Med 186, 683-693, 1997) and although, recently, there are different opinions that PTPlB and TCPTP coordinately regulate an insulin signaling process (Galic S., MoI Cell Biol 25, 819-829, 2005).
- the therapeutic potentials of PTP (especially PTPlB) inhibitors in treating human diseases have been extensively reviewed (Lee K..
- Thiazolidine moiety had been screened by various inventors for diversified biological activities (WO2004047760, WO2005082901, WO2006002829, WO2006040050, WO2006040052, WO2006047269).
- Some thiazolidine derivatives were described in WO2007032028 as PTPlB inhibitors.
- PTP inhibitors including inhibition mechanistic study of inhibitors against PTP, structure-activity relationship study, and synthetic and pharmacological study, have been performed in many research groups recently, it is still very challenging to discover specific inhibitors of PTPlB and utilize them in clinical trials.
- the structural homogeneity of active and secondary-binding sites in PTPs family highlights the importance in developing drugs specifically antagonizing PTPl B.
- the main objective of the present invention is therefore to provide novel compounds of the general formula I 5 their pharmaceutically acceptable salts, pharmaceutical compositions containing them, process and intermediates for the preparation of the compounds given in Formula I which have inhibitory activity against PTPlB.
- Another objective of the present invention to develop novel compounds which are effective and useful to lower increased levels of glucose, lipids, to improve insulin resistance, to decrease body weight, for the treatment and/ or prophylaxis of metabolic disorders such as type II diabetis, obesity, hyperlipidemia, with better efficacy and lower toxicity.
- the present invention provides a process for the preparation of novel organic compounds of the general formula (I), their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them.
- a further aspect of the present invention is to provide novel intermediates, a process for their preparation and their use in methods of making compounds of the general formula (I).
- novel organic compounds of present invention represented by the general formula (I) is useful for reducing blood glucose, lowering lipid levels, cholesterol and reducing body weight and also have some excellent effects in the treatment arid/or prophylaxis of diseases caused by insulin resistance such as type II diabetes, hyperlipidemia, obesity, impaired glucose tolerance, diabetic complications with better efficacy, potency, without or reduced toxicity.
- the present invention is related to the compounds of the general formula T
- G 2 is selected from hydrogen, fluoro and methoxy
- G 3 is selected from Ci -4 alkyl:
- G 4 is selected from -CH 2 COOH, -NH-C(O)-COOH, -C(O)-COOH, -CH(F)-COOH, -
- G 5 is selected from hydrogen or Fluoro
- G 6 is selected from -0-, or -N(CH 3 )-
- G 7 is selected from hydrogen or Ci -4 alkyl
- G 8 is unsubstituted phenyl; or phenyl substituted at para position with nitro, fluoro, cyano, Piperidyl, branched or straightchain Ci -6 alkoxy, C 5-6 cycloalkoxy, branched or straightchain Ci -6 alkyl, C 5-6 cycloalkyl; 'a' is O or 1.
- G 2 is selected from hydrogen, fluoro and methoxy
- G 3 is selected from Ci -4 alkyl:
- G 4 is selected from -CH2COOCH 3 , -CH 2 -COOC 2 H 5 , -NO 2 , -C(O)-CH 3 , -CH(F)-
- G 5 is selected from hydrogen or Fluoro
- G 6 is selected from -0-, or -N(CH 3 )-;
- G 7 is selected from hydrogen or Ci -4 alkyl
- G 8 is unsubstituted phenyl; or phenyl substituted dXpara position with nitro, fluoro, cyano, piperidinyl, branched or straightchain Ci -6 alkoxy, C 5-6 cycloalkoxy, branched or straightchain Ci -6 alkyl, or C 5-6 cycloalkyl; 'a' is O or 1.
- the compounds of formula I were synthesized as per 'Scheme F.
- a mixture of aniline derivative of formula (i) and isothiocyanate of compound of formula (ii) in ethanol was heated under reflux for 5-8 hours (h.) to produce thiourea derivative of formula (iii).
- the thiourea derivative (iii) was heated under reflux with ethyl bromoacetate and triethyl amine (TEA) in ethanol for 4-7 h.
- nitro group is converted to -NH-C(O)-COOH as follows: i. Nitro group was reduced to amino function by using suitable reducing agent such as iron in acetic acid. ii. Amino function was acylated with ethyl oxalyl chloride in presence of a base such as triethyl amine to obtain oxalamic acid ethyl ester function [-NH-C(O)-COOC 2 H 5 ], which was further hydrolyzed using base such as lithium hydroxide to obtain oxalamic acid function [-NH- C(O)-COOH].
- the intermediates and the compounds of the present invention are obtained in pure form in a manner known per se, for example by distilling off the solvent in vacuum and recrystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations or subjecting it to one of the purification methods, such as column chromatography on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their combinations.
- a suitable solvent such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations
- a suitable solvent such as pentane, diethyl ether, isopropyl ether, chloroform, dich
- Salts are obtained by dissolving the free compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as dichloromethane or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which contains the desired acid or base or two which the desired acid or base is then added as described in, Berge S. M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1-19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth , wiley- VCH (2002).
- a suitable solvent for example in a chlorinated hydrocarbon, such as dichloromethane or chloroform or a low molecular weight aliphatic alcohol, for example, ethanol or isopropanol, which contains the desired acid or base or two which the desired acid or base is then added as described in, Berge S. M. et
- the present invention also provides pharmaceutical compositions containing compounds of general formula I as defined above and their pharmaceutically acceptable salts in combination with the usual pharmaceutically employed carrier, diluents and the like.
- reaction mixture was cooled to room temperature and solvent was removed under vacuo.
- Step-2 Preparation of Hydroxy-(4-nitro-phenyl)-acetic acid ethyl ester Hydroxy-(4-nitro-phenyl)-acetonitrile (5.0 g, 28.0 mmol) was added to a mixture of acetic acid (20 ml) and ION aqueous hydrochloric acid (20 ml) and heated to 100 0 C for 4 h. Reaction was monitored by thin layer chromatography. Reaction mixture was cooled to room temperature and evaporated under vacuum to dryness to give solid residue and washed with diethyl ether (20 ml).
- Step 3 Preparation of (4-Amino-phenyl)-hydroxy-acetic acid ethyl ester
- a mixture of Hydroxy-(4-nitro-phenyl)-acetic acid ethyl ester (4.7 g, 20.8 mmol) and 10% Pd/C (900 mg) in methanol (50 ml) was stirred under hydrogen atmosphere for 4 h. Catalyst was removed by filtration and filtrate was evaporated under vacuum to give solid residue. Residue was washed with hexane (30 ml) and dried to give off white solid (3.4 g, 85.0 %).
- MS m/z l96 (M+l).
- Step-4 Preparation of Hydroxy-[4-(3-methyl-thioureido)-phenyl]-acetic acid ethyl ester
- 4-Amino-phenyl)-riydroxy-acetic acid ethyl ester 3.3 g, 16.9 mmol
- methylisothiocyanate 1.35 g, 18.6 mmol
- ethanol 50 ml
- Reaction mixture was cooled to room temperature and solvent was removed under reduced pressure.
- To the residue water (40 ml) was added and stirred for 15 min.
- the solid was filtered and washed with 50 % ethanol in water (25 ml) and dried under high vacuum to give a pale yellow solid (4.1 g, 91.0 %).
- MS m/z 269 (M+l).
- Step 5 Preparation of Hydroxy- ⁇ 4 ⁇ [3-memyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl ⁇ - acetic acid ethyl ester
- Step 6 prepration of. ⁇ 4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl ⁇ -hydroxy-acetic acid ethyl ester
- a mixture of Hydroxy- ⁇ 4-[3-methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl ⁇ -acetic acid ethyl ester 300 rag, 0.974 mmol
- 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde 314.9 mg, 1.10 mmol
- piperidine 0.288 ml, 2.92 mmol
- Step 7 Preparation of ⁇ 4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl ⁇ -hydroxy-acetic acid
- Step-1 Preparation of Fluoro- ⁇ 4-[3-methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl ⁇ - acetic acid ethyl ester*.
- Ste ⁇ -2 preparation of ⁇ 4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl] ⁇ meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl ⁇ -fluoro-acetic acid ethyl ester
- a mixture of Fluoro- ⁇ 4-[3-methyl-4-oxo-thiazolidin-2-yHdeneamino]-phenyl ⁇ -acetic acid ethyl ester 300 mg, 0.967 mmol
- 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde 312.9 mg, 1.06 mmol
- piperidine 0.286 ml, 2.90 mmol
- Step-3 Preparation of ⁇ 4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl ⁇ -fluoro-acetic acid
- ⁇ 4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl ⁇ -fluoro-acetic acid ethyl ester 200 mg, 0.341 mmol
- methanol (10 ml) was added a solution of lithium hydroxide monohydrate (28.6 mg, 0.682 mmol) in water (0.5 ml) and stirred at
- Step-1 Preparation of 1 -(4- Acetyl -phenyl)-3-methyl-thiourea
- Step-2 Preparation of 2-(4-Acetyl-phenylimino)-3-methyl-thiazolidin-4-one
- a solution of l-(4-Acetyl-phenyl)-3-methyl-thiourea (2 g, 9.61 mmol), triethylamine (3.4 ml, 23.56 mmol) and ethylbromo acetate (1.2 ml, 11.39 mmol) in ethanol (25 ml) was heated under reflux for 5 h.
- the reaction mixture was cooled to room temperature, water (50 ml) was added and precipitated solid was filtered. Solid was suspended in 50 % ethanol (10 ml), stirred for 15 min, filtered and dried under vacuo to give a yellow solid (1.9 gm, 80 %).
- Step-3 Preparation of 2-(4-Acetyl-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene] -3 -methyl-thiazolidin-4-one
- Step-4 Preparation of (4- ⁇ 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino ⁇ -phenyl)-oxo-acetic acid
- Step-1 Preparation of l-Methyl-3-(4-nitro-phenyl)-thiourea
- Step-2 Preparation of 3-Methyl-2-(4-nitro-phenylimino)-thiazolidin-4-one
- a mixture of l-Methyl-3-(4-nitro-phenyl)-thiourea (4 g, 18.95 mmol), triethylamine (6.8 ml, 47.11 mmol) and ethylbromo acetate (2.2 ml, 20.84 mmol) in ethanol (50 ml) was heated under reflux for 5 h. The reaction mixture was cooled to room temperature and solvent was removed under reduced pressure.
- Step-3 Preparation of 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-2-(4-nitro- phenylimino)-thiazolidin-4-one
- Step-4 Preparation of 2-(4-Amino-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene] -3 -methyl -thiazolidin-4-one
- Step-5 Preparation of N-(4- ⁇ 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino ⁇ -phenyl)-oxalamic acid ethyl ester
- Step-6 Preparation of N-(4- ⁇ 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino ⁇ -phenyl)-oxalamic acid
- Step-1 Preparation of 2-(4-Amino-phenylimino)-3-methyl-thiazolidin-4-one
- 3-Methyl-2-(4-nitro-phenylimino)-thiazolidin-4-one(prepared as per step 2 of example 14; 3.2 gm, 12.74 mmol) in acetic acid (30 ml) was added iron powder (2.1 gm, 37.5 mmol) at room temperature and stirred for 5 h. Inorganics were removed by filtration and filtrate was concentrated under vacuo. Residue was quenched with saturated sodium bicarbonate solution (50 ml) and extracted with dichloromethane (3x 25 ml).
- Step-3 [(4- ⁇ [3-methyl-4-oxo-l,3-thiazolidin-2-ylidene]amino ⁇ phenyl)(tert-butoxycarbonyl- sulfamoyl)amino] aceticacid ethylester
- Step-4 [(4- ⁇ [3 -methyl-4-oxo- 1 ,3 -thiazolidin-2-ylidene] aminophenyl)(sulfamoyl) amino]aceticacid ethylester
- Step 5 Preparation of 5-[4-(3-Methyl-4-oxo-thiazolidin-2-ylideneamino)-phenyl]-l,l-dioxo- 1 ⁇ 6 - [ 1 ,2,5]thiadiazolidin-3 -one
- Step 6 Preparation of 5-(4- ⁇ 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidm-2-ylideneamino ⁇ -phenyl)- 1 ,1 -dioxo-1 ⁇ 6 -[l ,2,5]thiadiazolidin-3-one
- 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde 69 mg, 0.23 mmol
- piperidine 0.056 ml, 0.46 mmol
- Step-1 4-cyclohehyl benzoic acid ethyl ester:
- Step-3 1 -Chloromethyl-4-cyclohexyl -benzene
- Step-4 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde
- reaction mixture was cooled to room temperature and then poured in to cold water (40 ml).
- the precipitated solid was filtered and washed with water (20 ml) to afford off white solid product (2.30 g, 98 %).
- Step-1 4-(l-Ethyl-propoxy)-benzaldehyde :
- reaction mixture was poured into cold water (40 ml).
- the organic material was extracted in ethyl acetate (2x 25 ml).
- the combined organic layer was washed with water (30 ml) and brine (25 ml), dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (1.5 g, 96 %).
- Step-2 4-( 1 -Ethyl-propoxy)-phenyl] -methanol
- Step-3 1 -Chloromethyl-4-( 1 -ethyl-propoxy)-benzene
- Step-4 4- [4-( 1 -Ethyl-propoxy)-benzyloxy] -benzaldehyde
- Step-3 methyl-(4-methyl-benzyl)-amine
- Step-4 4-[Methyl-(4-methyl-benzyl)-amino]-benzaldehyde
- Step 1 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzoic acid ethyl ester
- 1,2,3,4-tetrahydroisoquinoline (1 g, 7.5 mmol) in dry DMF (30 ml)
- potassium carbonate (1.55g, 11.27 mmol
- 4-bromomethyl ethyl benzoate (1.8 g, 8.2 mmol) were added and reaction mixture was heated to 80 °C for 7 h.
- the reaction mixture was cooled to room temperature and then water (40 ml) was added and extracted with ethyl acetate (2x25 ml).
- Step 2 [4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-phenyl]-methanol
- lithium aluminium hydride (0.25 g, 6.7 mmol)
- dry THF 30 ml
- 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzoic acid ethyl ester Ig, 3.5 mmol
- the reaction was quenched by slow addition of cold water (15 ml).
- Triethyl amine (1.95 g, 19.3 mmol) was added to the above reaction mixture and slowly warmed to room temperature. The reaction mixture was then poured into water (25 ml) and diluted with dichloromethane (25 ml). The organic layer was separated, washed with brine (15 ml), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The product was purified by silica gel column chromatography using 10 % solution of ethyl acetate in hexane as the eluent to afford the product as colorless oil (0.54 g, 77 %).
- Step-2 4-[Methyl-(4-isopropyl-benzyl)-amino]-benzaldehyde
- Step 3 Synthesis of 4-(3-Fluoro-4-piperidin-l-yl-benzyloxy)-benzaldehyde
- a solution of (3-Fluoro-4-pi ⁇ eridin-l-yl-phenyl)-methanol 3.5 g, 16.7 mmol
- dimethylformamide 10 ml
- Step-1 Preparation of l-(4-Isobutyl-phenyl)-ethanone
- Step-2 Preparation of l-(4-Isobutyl-phenyl)-ethanol
- Step-3 Prepration Methanesulfonic acid l-(4-isobutyl-phenyl)-ethyl ester
- DCM dimethyl methanesulfonic acid
- l-(4-Isobutyl-phenyl)-ethanol 2.4 g, 13.48 mmol
- triethylamine 3.8, 26.46 mmol
- Reaction mixture was stirred at room temperature for 4 h.
- Reaction mixture was diluted by DCM (50 ml) and poured in to ice water (40 ml). DCM layer was separated, washed with water (30 ml) and dried over anhydrous sodium sulfate. DCM was evaporated under vacuum to afford pale yellow oil (3.2 gm, 90.0 %).
- Step-4 Preparation of 4- [ 1 -(4-Isobutyl-phenyl)-ethoxy] -benzaldehyde
- 4-hydroxy benzaldehyde (1.47 g, 12.10 mmol) and potassium carbonate (2.5 g, 18.11 mmol) in N,N-dimethylformamide ( 20 ml) was added methanesulfonic acid l-(4-isobutyl-phenyl)-ethyl ester (3.1 g, 12.10 mmol) and reaction mixture was stirred at 60 0 C for 5 h.
- the phosphatase activity of human recombinant PTPlB was determined by following a previously described procedure ⁇ Methods 35, 2-8, 2005), but with certain modifications.
- the principle of the assay is based on the hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) and the fluorometric quantitation of the liberated difluoromethylumbelliferone (DiFMU) .
- the reaction mixture (100 ⁇ l) contained 15 ng/well of human recombinant PTPlB enzyme (produced in- house or procured from R&D Systems, USA) in the assay buffer (50 mM Hepes, pH 7.2, 50 mM NaCl, 1 mM EDTA, 1 mM DDT and 0.01 % Triton X-100) and 25 ⁇ M DiFMUP.
- human recombinant PTPlB enzyme produced in- house or procured from R&D Systems, USA
- the assay buffer 50 mM Hepes, pH 7.2, 50 mM NaCl, 1 mM EDTA, 1 mM DDT and 0.01 % Triton X-100
- test compounds The inhibition of PTPlB activity by test compounds was routinely assessed by preincubating the enzyme with test compound (0.1 and 1 ⁇ M for primary screening and 7 concentrations from 0.01 to 10 ⁇ M for the dose-response study) or vehicle (1 % DMSO) for 10 min at 30 0 C, in a total volume of 90 ⁇ l.
- Test compounds were dissolved in DMSO at a concentration of 10 mM and suitably diluted further in assay buffer.
- the enzyme reaction was initiated by the addition of DiFMUP, followed by incubation of assay plates for 5 min at 30 0 C and the liberated product was measured as described above.
- a known inhibitor of PTPlB positive control was always included in the assay. Test compounds at various concentrations were always evaluated in duplicate, along with substrate blanks, vehicle controls and positive controls.
- the results are expressed as percent inhibition of the enzyme activity relative to vehicle controls. Dose-response studies were conducted for those compounds exerting > 50% inhibition of activity at 1 ⁇ M in primary screening.
- the inhibition data expressed as IC 5O, the inhibitor concentration that caused 50% decrease of the activity under assay conditions.
- the IC50 was computed using GraphPad Prism software, version 5.0.
- the PTPlB inhibition data (expressed as IC 50 ) is presented in Table 1.
- T-cell protein tyrosine phosphatase T-cell protein tyrosine phosphatase
- the reaction mixture 100 ⁇ l in 96-well flat- bottom black-well plates
- assay buffer 50 mM Hepes, pH 7.2, 50 mM NaCl, 1 mM EDTA, 1 mM DDT and 0.01 % Triton X-100
- 25 ⁇ M substrate DiFMUP
- 125 mU/well of human recombinant enzyme procured from New England Biolabs, UK
- TCPTP was preincubated with test compound (ranging from 0.01 ⁇ M to 10 ⁇ M) or vehicle (1 % DMSO) for 10 min at 30 0 C, in a total volume of 90 ⁇ l.
- test compound ranging from 0.01 ⁇ M to 10 ⁇ M
- vehicle (1 % DMSO vehicle
- the reaction was started by the addition of 10 ⁇ l substrate (250 ⁇ M running stock in assay buffer) and further incubated for 5 min at 30 0 C.
- the liberated DIFMU was monitored in a fluorescence microplate reader (SpectraMax M5, Molecular Devices, USA), with excitation and emission wavelengths set at 358 nm and 450 run, respectively.
- IC50 was computed using GraphPad Prism software, version 5.0.
- Table 2 Selectivity of PTP-IB inhibitors against TCPTP
- mice Male healthy C57BL/6J mice were randomized into two groups at 4 weeks of age and fed either Lard Control diet (D12450B, 10% kcal from Lard) or High-fat diet (D12451, 45% kcal from Lard) from Research Diets Inc., USA, for 16-20 weeks. Animals were selected on the basis of glucose intolerance to a glucose challenge of 2 g/kg, done at 20-24 weeks of age. Glucose intolerant animals from the high-fat diet group were randomized and grouped on the basis of body weight and plasma triglycerides. Animals were dosed once daily by oral route for 23 days.
- Lard Control diet D12450B, 10% kcal from Lard
- High-fat diet D12451, 45% kcal from Lard
- Test compounds were evaluated in genetically insulin resistant male db/db mice (8-10 weeks of age) at the dose of 10 or 30 mg/kg. Test compounds were administered once daily by oral route for desired duration of treatment. Non-fasted animals were bled under light anesthesia two days before OGTT. OGTT was performed in overnight fasted animals using a glucose load of 1 g/kg/po. Effect of test compounds on body weight gain was also evaluated relative to the vehicle treated animals.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to the novel compounds of the general formula (I), wherein the symbols are same as described in specification, their pharmaceutically acceptable salts, their tautomeric forms, their stereoisomers, pharmaceutical compositions containing them, to process and intermediates for the preparation of the above said compounds, having the utility of these compounds in medicine and to methods for their therapeutic use, and their use in the treatment of diabetes and related diseases.
Description
NOVEL PROTEIN TYROSINE PHOSPHATASE - IB INHIBITORS
Field of the Invention
The present invention is related to novel compounds of the general formula and their pharmaceutically acceptable salts, pharmaceutical compositions containing them, methods of making the above compounds, and their use as Protein tyrosine phosphatase IB (PTP-IB) inhibitors, which are useful in the treatment or prevention of diseases in which PTP-IB enzyme is known to be involved in the pathogenesis. In addition to its therapeutic benefits against type 2 diabetes and obesity, the PTPlB inhibitors would also find use in the treatment of diseases such as cancer, inflammatory disorders, autoimmune diseases and osteoporosis

Background of the invention
Diabetes mellitus is a major, growing health problem worldwide (Yach, D., et al. Nat. Med. 12, 62-66, 2006). Type 2 diabetes mellitus (hereafter referred as type 2 diabetes, also known as non-insulin-dependent diabetes mellitus, NIDDM) is a heterogeneous disorder, with both genetic and environmental factors contributing to its development. The pathogenesis of type 2 diabetes involves multiple mechanisms leading to hyperglycemia, most notably increased hepatic glucose production, impaired insulin secretion by pancreatic β cells and reduced glucose uptake by skeletal muscle and adipose tissue (peripheral insulin resistance). Type 2 diabetic patients are at substantially increased risks of macrovascular disease including coronary heart disease and stroke and microvascular disease including retinopathy, nephropathy and neuropathy.
Type 2 diabetes is a therapeutic area with huge market potential. The number of diabetic patients is projected to increase from 170-175 million in 2000 to over 350 million by 2030 (Wild, S., et al. Diab.Care 27, 1047-1053, 2004; Yach, D., et al. Nat. Med. 12, 62-66, 2006). The major part of this numerical increase is expected to occur in developing countries and India will have the distinction of having the largest number of diabetic patients in the world by 2030.
The treatment approaches for type 2 diabetes include diet, exercise, and a variety of pharmacological agents. Clinically established therapies for type 2 diabetes include insulin and its analogs and various oral hypoglycemic agents: sulfonylureas, metformin, α- glucosidase inhibitors (acarbose, miglitol), non-sulfonylurea insulin secretagogues (repaglinide, nateglinide) and thiazolidinedione (TZD) derivatives (rosiglitazone, pioglitazone) acting via PPARy agonism (Matthaei, S., et al. Endocrine Rev. 21, 585-618, 2000; Skyler, J.S. J.Med.Chem. 47, 4113-4117, 2004). These agents act by different mechanisms to normalize blood glucose levels, but are limited in their abilities, either alone or in combination, to prevent the onset of diabetic complications. Further, each of the above oral agents suffers either from generally inadequate efficacy or number of adverse effects. For example, sulfonylureas, which have been the mainstay of oral treatment for over 5 decades, are known to be associated with a high rate of secondary failure and hypoglycemia. The TZD class of antidiabetic agents (glitazones) improves glucose utilization without stimulating insulin release, but their use is associated with undesirable effects (e.g. risk of myocardial infarction, cardiac hypertrophy, liver toxicity, weight gain).
Considering together the facts that about 90% of all diabetic cases account for NIDDM and the inadequacy of the currently available treatment, the clinical need and market potential for new oral antidiabetic drugs, which maintain tight glycemic control and prevent diabetic complications are very high.
Protein tyrosine phosphatases (PTPs), a large family of signaling enzymes (Alonso A, et a]., Cell; 117. 699-711, 2004). play essential roles in intracellular signal transduction by regulating the cellular level of tyrosine phosphorylation to control cell growth and differentiation, metabolism, cell migration, gene transcription, ion-channel activity, immune response, cell apoptosis, and bone development (Hunter T., Cell 100, 1 13-127, 2000). Unregulated operation of PTPs is responsible to many human diseases including cancer (BIume-Jenscn P., Nature 41 1 , 355-365, 2001), diabetes (Montalibet J., Drug Discov Today: Therap. Strateg. 2, 129-135, 2005), obesity (Cook W.S., Developmental Cell 2, 385-387, 2002), and osteoporosis (Schiller K.R.. J. Cell Biochem. 96, 262-277, 2005).
Among the various PTP family, protein tyrosine phosphatase IB (PTPlB) activates c-Src inhuman breast cancer (Bjorge J.D., J Biol Chew 275, 41439-41446, 2000), and also influences lhe down regulation of insulin signaling by dephosphorylating the insulin receptor including insulin receptor substrate- 1 (IRS-I ) and insulin receptor substrate-2 (IRS-2) (Walchli S.. J Biol Chem 275, 9792-9796, 2000). Therefore, PTPl B can be a useful target for diabetes and cancer, and inhibitors of PTPl B may be promising drugs to treat these diseases. In addition, considering that PTPlB knockout mice arc resistant to obesity. PTPlB plays critical role in development of obesity (Klanian L.D., MoI Cell Biol 20, 5479-5489. 2000). In spite of the therapeutic potential of PTPlB inhibitor against diabetes, obesity, and cancer, it is difficult to develop selective PTPlB inhibitor over other PTPs including SHP, VHR, LAR, CD45, and cdc25C, because of structural homologies in PTPs (Cheng A., Eur J Biochem 269, 1050-1059, 2002; Penninger J.M., J Nat Immunol 2, 389-396, 2001 ; Qu C.K., Biochim Biophys Acta 1592, 297-301. 2002; Hoffman B.T.. Curr Pharm Dcs 10, 1 161- 1 181 , 2004). In particular, because T-cell PTP ('J1CPTP) has an 80% homology to PTPl B in the catalytic domains, non-selective inhibition gives rise to severe side effects (Tiganis T., J Biol Chem 274, 27768-27775, 1999; You-Ten K.E., J Exp Med 186, 683-693, 1997) and although, recently, there are different opinions that PTPlB and TCPTP coordinately regulate an insulin signaling process (Galic S., MoI Cell Biol 25, 819-829, 2005).
The therapeutic potentials of PTP (especially PTPlB) inhibitors in treating human diseases have been extensively reviewed (Lee K.. Ciirr Top Med Chem 3, 797-807, 2003; Zhang Z.Y., Ace Chem Res 36, 385-392. 2003; Hooft van Huijsduijnen R.H., J Med Chem 47, 4142-4146. 2004; Dewang P.M., Curr Med Chem 12, 1-22, 2005; Bialy L.. Angew Chem Int Ed 44, 3814-3839, 2005; Zhang Z. Y., Curr Opin Chem Biol 5, 416-423. 2001; Burke T.R., Biopotymers (Peptide Science) 47, 225-241 , 1998). Especially, structural biology, mechanism, and inhibitors of PTPlB were reviewed by Taylor in Curr Top Med Chem 3, 759-782, 2003 and the small molecule approach to study the function of PTPl B was reviewed by Zhang in Methods 35, 9-21. 2005 which introduced the initial strategies for finding potent and specific PTPlB inhibitors, the synthesis of cell permeable analogs for cellular studies, and the application of those inhibitors to dissect the role of PTPlB in the insulin signaling pathway. Structural and biological features of small molecular PTPlB- specific inhibitors were discussed in depth by Seokjoon Lee and Qian Wang in Medicinal Research Reviews 27, 553-573, 2007, with an emphasis on specificity of small molecular inhibitors against PTPlB over other PTPs. Various classes of Investigational small molecules active against PTPlB enzyme as discussed by Seokjoon Lee et al. arc listed below:
1. Substituted acetophenone, cinnamic acid and analogs.
2. N-oxalylarylaminobenzoic acid derivatives.
3. Isoxazolecarboxylic acid analogs.
4. peptidoniimeli.es.
5. Tetrasubslituted methine derivatives.
6. 3-formylchiOmone.
7. Pyridazinc Derivatives.
8. Abietane-Type Diterpene Pigments.
9. 1 ,2-Naphthoquinone.
Thiazolidine moiety had been screened by various inventors for diversified biological activities (WO2004047760, WO2005082901, WO2006002829, WO2006040050, WO2006040052, WO2006047269). Some thiazolidine derivatives were described in WO2007032028 as PTPlB inhibitors.
Although various kinds of researches about PTP inhibitors including inhibition mechanistic study of inhibitors against PTP, structure-activity relationship study, and synthetic and pharmacological study, have been performed in many research groups recently, it is still very challenging to discover specific inhibitors of PTPlB and utilize them in clinical trials. The structural homogeneity of active and secondary-binding sites in PTPs family highlights the importance in developing drugs specifically antagonizing PTPl B.
Objective of the Invention
The main objective of the present invention is therefore to provide novel compounds of the general formula I5 their pharmaceutically acceptable salts, pharmaceutical compositions containing them, process and intermediates for the preparation of the compounds given in Formula I which have inhibitory activity against PTPlB.
Another objective of the present invention to develop novel compounds which are effective and useful to lower increased levels of glucose, lipids, to improve insulin resistance, to decrease body weight, for the treatment and/ or prophylaxis of metabolic disorders such as type II diabetis, obesity, hyperlipidemia, with better efficacy and lower toxicity.
Summary of the Invention
According to one aspect of the present invention there is provided novel organic compounds represented by the general formula (I), their pharmaceutically acceptable salts, and pharmaceutical compositions containing them or mixture thereof.
In yet another aspect, the present invention provides a process for the preparation of novel organic compounds of the general formula (I), their stereoisomers, their pharmaceutically acceptable salts, pharmaceutical compositions containing them.
A further aspect of the present invention is to provide novel intermediates, a process for their preparation and their use in methods of making compounds of the general formula (I).
Detailed Description of the Invention
The novel organic compounds of present invention represented by the general formula (I) is useful for reducing blood glucose, lowering lipid levels, cholesterol and reducing body weight and also have some excellent effects in the treatment arid/or prophylaxis of diseases caused by insulin resistance such as type II diabetes, hyperlipidemia, obesity, impaired glucose tolerance, diabetic complications with better efficacy, potency, without or reduced toxicity.
The present invention is related to the compounds of the general formula T


(b)
 G2 is selected from hydrogen, fluoro and methoxy;
G2 is selected from hydrogen, fluoro and methoxy;
G3 is selected from Ci-4 alkyl:
G4 is selected from -CH2COOH, -NH-C(O)-COOH, -C(O)-COOH, -CH(F)-COOH, -

G5 is selected from hydrogen or Fluoro; G6 is selected from -0-, or -N(CH3)-; G7 is selected from hydrogen or Ci-4 alkyl;
G8 is unsubstituted phenyl; or phenyl substituted at para position with nitro, fluoro, cyano, Piperidyl, branched or straightchain Ci-6 alkoxy, C5-6 cycloalkoxy, branched or straightchain Ci-6 alkyl, C5-6 cycloalkyl; 'a' is O or 1.
A compound and its pharmaceutically acceptable salts as described herein above wherein the compound of the general formula (I) is selected from:
[4-(3 -Methyl- 5- {4-[4-(l-methyl-butoxy)-benzyloxy]-benzylidene}-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 1)
4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin~2- ylideneamino}-phenyl)-acetic acid (Compound 2)
(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 3)
(4-{5-[4-(4-Isobutyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-ylideneamino}- phenyl)-acetic acid (Compound 4)
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid.(Compound 5)
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-2-fluoro-phenyl] -acetic acid, (Compound 6)
4-{5-[4-(N-Benzyl-methylarnino)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}~phenyl)-acetic acid. (Compound 7)
[4-(5-{4-[l-(4-Isobutyl-phenyl)-ethoxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 8)
(4- { 3 -Methyl-5 - [4-(4-nitro-benzyloxy)-benzylidene] -4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid (Compound 9)
(4-{5-[4-(3,4-Dihydro-lH-isoquinolin-2-yl)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 10)
{4- [5- [ 1 - [4-(4-Fluoro-benzyloxy)-phenyl] -meth-ylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino]-phenyl} -acetic acid (Compound 11)
(4-{5-[4-(4-Ethyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-ylideneamino}- phenyl)-acetic acid (Compound 12)
(4-{5-[4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzylidene]-3-methyl-4-oxo-thiazolidin- 2-ylideneamino}-phenyl)-acetic acid. (Compound 13)
(4-{3-Ethyl-5- [4-(4-ethyl-benzyloxy)-benzylidene]-4-oxo-tliiazolidin-2-ylideneamino}- phenyl)-acetic acid. (Compound 14)
(4- { 3 -Ethyl-5- [4-(4-isopropyl-benzyloxy)-benzylidene] -4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid (Compound 15)
(4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-3 -methoxy-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 16)
5-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)- 1 , 1 -dioxo- 1 λ6-[l ,2,5]thiadiazolidin-3-one (Compound 17)
[4-(3-Methyl-5- {4-[methyl- (4-methyl-benzyl)-amino]-benzylidene} -4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 18)
(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-2-fluoro-phenyl)-acetic acid. (Compound 19)
(2-Fluoro-4-{5-[4-(4-isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidm-2- ylideneamino}-phenyl)-acetic acid. (Compound 20)
(2-Fluoro-4- { 5 - [4-(4-isobutyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 21)
N-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-oxalamic acid (Compound 22).
(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-oxo-acetic acid. (Compound 23)
{4- [5- [ 1 - [4-(4~Cyclohexyl-benzyloxy)-phenyl] -meth-)-ylidene] -3 -methyl-4-oxo-thiazolidin- 2-ylideneamino] -phenyl }-fluoro-acetic acid (Compound 24)
4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-oxo-acetic acid. (Compound 25)
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-fluoro-benzylidene}-3-methyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl] -acetic acid. (Compound 26)
(4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-3 -fluoro-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino) -phenyl)-acetic acid. (Compound 27)
[4-(5-{4-[4-(l-Ethyl-propoxy)-benzyloxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl]-acetic acid. (Compound 28)
(4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -ethyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 29)
{4- [5 - [ 1 - [4-(4-Cyclohexyl-benzyloxy)-phenyl] ~meth-)-ylidene] -3 -methyl-4-oxo-thiazolidin- 2-ylideneamino]-phenyl}-hydroxy-acetic acid (Compound 30)
[4-(5-{3-Fluoro-4-[(4-isopropyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid (Compound 31)
Hydroxy- (4-{3-methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene]-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 32)
(4-{3-Methyl-4-oxo-5-[4-(4-pentyl-benzyloxy)-benzylidene]-thiazolidin-2-ylideneamino}- phenyl)-acetic acid (Compound 33)
[4-(5-{4-[l-(4-Cyclohexyl-phenyl)-ethoxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl]-acetic acid (Compound 34)
(4-{5-[4-(4-Cyclohexyloxy-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 35)
(4- { 5- [4-(4-Cyclohexyloxy-benzyloxy)-benzylidene] -3 -ethyl -4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 36)
5-(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamiηo} -phenyl)- 1,1-dioxo -lλ6- [1,2,5] thiadiazolidin-3- one (Compound 37)
[4-(3-Ethyl-5- {3-fluoro-4-[(4-isopropyl-benzyl)-methyl-amino]-benzylidene}-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid. (Compound 38)
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-fluoro-benzylidene}-3-ethyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid. (Compound 39)
(4- { 5 - [4-(4-Cyano-benzy loxy)-benzylidene]-3 -methyl-4-oxo-thiazolidin-2-lideneamino } - phenyl)-acetic acid (Compound 40)
(4-{5-[4-(3-Fluoro-4-piperidin-l-yl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 41)
{4- [5 - [ 1 - {4- [ 1 -(4-Isobutyl-phenyl)-butoxy]-phenyl } -methylidene]-3 -methyl-4-oxo- thiazolidin-2-ylideneamino] -phenyl} -acetic acid (Compound 42)
The compounds of the invention were prepared as outlined below (Scheme I) according to the methods described herein. However, the invention is not limited to these methods, the compounds may also be prepared as described for structurally related compounds in the literature.
Scheme I

wherein, Gi = (a)


G2 is selected from hydrogen, fluoro and methoxy;
G3 is selected from Ci-4 alkyl:
G4 is selected from -CH2COOCH3, -CH2-COOC2H5, -NO2, -C(O)-CH3, -CH(F)-
COOC2H5, -CH(OH)-COOC2H5, -CH2COOH, -NH-C(O)-COOH, -C(O)-COOH, -
CH(F)-COOH, -CH(OH)-COOH, or
 ;
;
G5 is selected from hydrogen or Fluoro;
G6 is selected from -0-, or -N(CH3)-;
G7 is selected from hydrogen or Ci-4 alkyl;
G8 is unsubstituted phenyl; or phenyl substituted dXpara position with nitro, fluoro, cyano, piperidinyl, branched or straightchain Ci-6 alkoxy, C5-6 cycloalkoxy, branched or straightchain Ci-6 alkyl, or C5-6 cycloalkyl; 'a' is O or 1.
The compounds of formula I were synthesized as per 'Scheme F. A mixture of aniline derivative of formula (i) and isothiocyanate of compound of formula (ii) in ethanol was heated under reflux for 5-8 hours (h.) to produce thiourea derivative of formula (iii). The thiourea derivative (iii) was heated under reflux with ethyl bromoacetate and triethyl amine (TEA) in ethanol for 4-7 h. to get compound of formula (iv); wherein, if G4 is -CH(OH)- COOC2H5, it was further converted to -CHF-COOC2H5 by treatment with diethyl aminosulfur trifluoride (DAST) in dichloromethane, if G4 is nitro, it was converted to 1,1- Dioxo-lλ6-[l,2,5]thiadiazolidin-3-one group as follows (Scheme 2):
utanoi

Scheme 2
(1) compound of formula 'iv', wherein G4 is nitro, is reduced to amino compound 'a' using a suitable reducing agent such as iron / acetic acid.
(2) Compound of formula 'a' is then reacted with ethyl bromo acetate in presence of base such as potassium carbonate or triethyl amine in a polar solvent like N,N- dimethylformamide or ethanol to get compound of formula 'b'.
(3) Reaction of compound of formula 'b' with chlorosulfonylisocyanate and rert-butanol in a suitable solvent gives compound of formula 'c'.
(4) Compound of formula 'c' on deprotection using trifluoroacetic acid and cyclization in presence of a base such as sodium hydroxide provides 1,1-Dioxo-lλ6- [l,2,5]thiadiazolidin-3-one group as in compound of formula 'e'.
Further, mixture of compound of formula (iv), an aromatic aldehyde of formula (v) and piperidine in ethanol were heated under reflux for 10-18 h. to afford compound of formula T.
Wherein,
(a) if G4 is -CH2-COOCH3 or -CH2-COOC2H5, then -CH2-COOCH3 or -CH2- COOC2H5 groups are hydrolyzed to -CH2-COOH using base like sodium hydroxide, lithium hydroxide etc.;
(b) if G4 is -CH(OH)-COOC2H5, then -CH(OH)-COOC2H5 is hydrolyzed to — CH (OH)-COOH using base like sodium hydroxide, lithium hydroxide etc.;
(c) if G4 is -CHF-COOC2H5, then -CHF-COOC2H5 is hydrolyzed to -CHF- COOH using base like sodium hydroxide, lithium hydroxide etc.;
(d) if G4 is -C(O)-CH3, then -C(O)-CH3 is converted to -C(O)-COOH on treatment with oxidizing agent such as selenium dioxide in pyridine;
(e) if G4 is nitro, then nitro group is converted to -NH-C(O)-COOH as follows: i. Nitro group was reduced to amino function by using suitable reducing agent such as iron in acetic acid. ii. Amino function was acylated with ethyl oxalyl chloride in presence of a base such as triethyl amine to obtain oxalamic acid ethyl ester function [-NH-C(O)-COOC2H5], which was further hydrolyzed using base such as lithium hydroxide to obtain oxalamic acid function [-NH- C(O)-COOH].
The intermediates and the compounds of the present invention are obtained in pure form in a manner known per se, for example by distilling off the solvent in vacuum and recrystallizing the residue obtained from a suitable solvent, such as pentane, diethyl ether, isopropyl ether, chloroform, dichloromethane, ethyl acetate, acetone or their combinations or subjecting it to one of the purification methods, such as column chromatography on a suitable support material such as alumina or silica gel using eluent such as dichloromethane, ethyl acetate, hexane, methanol, acetone and their combinations.
Salts are obtained by dissolving the free compound in a suitable solvent, for example in a chlorinated hydrocarbon, such as dichloromethane or chloroform or a low molecular weight
aliphatic alcohol, for example, ethanol or isopropanol, which contains the desired acid or base or two which the desired acid or base is then added as described in, Berge S. M. et al. "Pharmaceutical Salts, a review article in Journal of Pharmaceutical sciences volume 66, page 1-19 (1977)" and in handbook of pharmaceutical salts properties, selection, and use by P.H.Einrich Stahland Camille G.wermuth , wiley- VCH (2002).
The present invention also provides pharmaceutical compositions containing compounds of general formula I as defined above and their pharmaceutically acceptable salts in combination with the usual pharmaceutically employed carrier, diluents and the like.
The following examples are provided to further illustrate the present invention and therefore should not be construed to limit the scope of the invention.' Proton NMR spectra were obtained at 200 and 400 MHz Bruker instruments, with Deuterated chloroform (CDCl3) or deuterated dimethylsulfoxide (DMSO-d6) as solvent.. Chemical shifts (δ) are given in ppm relative to tetramethylsilane [(δ) 0 ppm] or to residual protons in the solvent as internal standard. Melting points correspond to capillary melting points at a heating rate of l°C/min, Molecular ion peaks of all the compounds were determined on Applied Biosystems API- 3000 mass spectrometer.
Example 1
Preparation of {4- [5- [1- [4-(4-Fluoro-benzyIoxy)-phenyl]-meth-ylidene] -3-methyl-4-oxo- thiazolidin-2-ylideneamino]-phenyl}-acetic acid (Compound 11)

To a solution of {4-[5-[l-[4-(4-Fluoro-benzyloxy)-phenyl]-methylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino]-phenyl}-acetic acid methyl ester (140 mg, 0.28 mmol) in THF (5 ml) and methanol (5 ml) was added aqueous lithium hydroxide mono hydrate (18 mg, 0.42 mmol in 0.5 ml water) and stirred at room temperature for 4 h. The solvent was removed under vacuo and residue was suspended in 90 % ethanol (5 ml) and acidified with 2N hydrochloric acid (pH 4). The precipitated solid was filtered, washed with water (10 ml) and triturated with ethanol (5 ml). Solid product was filtered and dried under reduced pressure to give a yellow solid (125 mg, 91 %). MS - m/z 475 (M-I). Melting point = 160-1620C. 1H NMR (DMSO-d6) δ; 3.29(s, 3H)5 3.50(s, 2H), 5.08(s, 2H)5 6.93-6.95 (d, 2H), 7.12-7.14(d, 2H), 7.18-7.22(t, 2H), 7.27-7.29(d, 2H)5 7.47-7.50(m, 4H), 7.68 (s, IH).
Example 2 Preparation of compound of formula I
Analogously, by practicing the chemistry as described in example 1 with appropriate change in the reactants and reaction conditions, following compounds were prepared.
[4-(3 -Methyl-5 - {4- [4-( 1 -methyl-butoxy)-benzyloxy] -benzylidene } -4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 1)
Melting Point: 158-160 0C'
MS: m/z 543 (M-I)
1H NMR (DMSOd6) δ: 0.80-0.88 (t, 3H), 1.16-1.19 (d, 3H), 1.29-1.50 (m, 4H), 3.28(s, 3H),
3.57 (s, 2H), 4.35-4.41 (m, IH), 4.99 (s, 2H), 6.84-6.88 (d, 2H), 6.92-6.96 (d, 2H), 7.05-7.09 (d, 2H), 7.25-7.32 (m, 4H), 7.40-7.44 (d, 2H), 7.64 (s, IH).
4-{5-[4~(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 2)
Melting Point: 220-221°C
MS: m/z 539 (M-I)
1H NMR (DMSOd6) δ: 1.26-1.43 (m, 5H), 1.68-1.76 (m, 5H),' 2.50 (m, IH), 3.32 (s, 3H),
3.58 (s, 2H), 5.08 (s, 2H), 6.97-6.99 (d, 2H), 7.12-7.14 (d, 2H), 7.21-7.23 (d, 2H), 7.28-7.30 (d, 2H), 7.32-7.34 (d, 2H), 7.48-7.50 (d, 2H), 7.70 (s, IH), 12.40 (br s, IH)
(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-tm'azolidin-2- ylideneamino}-plienyl)-acetic acid (Compound 3)
Melting Point: 208-210 °C
MS: m/z 499 (M-I)
1H NMR (DMSOd6) δ: 1.17-1.19 (d, 6H), 2.84-2.91 (m, IH), 3.27 (s, 3H), 3.55 (s, 2H), 5.03
(s, 2H), 6.92-6.94 (d, 2H), 7.25-7.27 (d, 2H), 7.19-7.21 (d, 2H), 7.25-7.27 (d, 2H), 7.28-7.30
(d, 2H), 7.38-7.40 (d, 2H), 7.61 (s, IH).
(4-{5-[4-(4-Isobutyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-ylideneamino}- phenyl)-acetic acid (Compound 4) Melting Point: 301-303 °C MS: m/z 513 (M-I)
1H NMR (DMSOd6) δ: 0.83-0.85 (d, 6H), 1.77-1.84 (m, IH), 2.42-2.44 (d, 2H), 3.29 (s, 3H), 3.35 (s, 2H), 5.07 (s, 2H), 6.89-6.91 (d, 2H), 7.12-7.16 (m, 4H), 7.24-7.26 (d, 2H), 7.32- 7.34 (d, 2H), 7.47-7.49 (d, 2H), 7.68 (s, IH).
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl]-acetic acid.(Compound 5)
Melting Point: 198-199 °C
MS: m/z 552 (M-I)
1H NMR (DMSO-de) δ: 1.18-1.36 (m, 5H), 1.66-1.74 (m, 5H), 2.40-2.48 (m, IH), 3.06 (s,
3H), 3.29 (s, 3H), 3.56 (s, 2H), 4.58 (s, 2H), 6.80-6.82 (d, 2H), 6.95-6.97 (d, 2H), 7.06-7.08
(d, 2H), 7.13-7.15 (d, 2H), 7.27-7.29 (d, 2H), 7.32-7.34 (d, 2H), 7.50 (s, IH)
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-2-fluoro-phenyl]-acetic acid. (Compound 6)
Melting Point: 195-197 0C
MS: m/z 570 (M-I)
1H NMR (DMSOd6) δ: 1.15-1.33 (m, 5H), 1.65-1.74 (m, 5H), 2.35-2.50 (m, IH), 3.06 (s,
3H), 3.38 (s, 3H), 3.62 (s, 2H), 4.59 (s, 2H), 6.80-6.88 (m, 4H), 7.06-7.14 (m, 4H), 7.33-7.35
(m, 3H), 7.62 (s, IH).
4-{5-[4-(N-Benzyl-methylamino)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 7)
Melting Point: 202-204 0C
MS: m/z 470 (M-I)
1H NMR (DMSOd6) δ: 3.08 (s, 3H), 3.27 (s, 3H), 3.55 (s, 2H), 4.64 (br s, 2H), 6.79-6.83 (d,
2H), 6.94-6.98 (d, 2H), 7.15-7.35 (m, 9H), 7.60 (s, IH), 12.33 (br s, IH).
[4-(5-{4-[l-(4-Isobutyl-phenyl)-ethoxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 8)
Melting Point: 161-163 0C
MS: m/z 527 (M-I)
1H NMR (DMSO-d6) δ: 0.81-0.83 (d, 6H), 1.52-1.54 (d, 3H), 1.73-1.83 (m, IH)5 2.37-2.39
(d, 2H), 3.30(s, 3H), 3.59 (s, 2H), 5.45-5.48 (m, IH), 6.92-6.98 (m, 4H), 7.06-7.08 (d, 2H),
7.24 -7.27 (m, 4H), 7.32-7.34 (d, 2H), 7.58 (s, IH), 12.38 (br s, IH).
(4- { 3 -Methyl-5 - [4-(4-nitro-benzyloxy)-benzylidene] -4-oxo-thiazolidin-2-ylideneamino } phenyl) -acetic acid (Compound 9)
Melting Point: 181-183 0C
MS: m/z 502 (M-I)
1H NMR (DMSOd6) δ: 3.32 (s, 3H), 3.57 (s, 2H), 5.32 (s, 2H), 6.96-6.98 (d, 2H), 7.15-7.17
(d, 2H), 7.28-7.30 (d, 2H), 7.50-7.52 (d, 2H), 7.69-7.71 (d, 2H), 7.69-2.71 (m, 3H), 8.24-8.26
(d, 2H).
(4-{5-[4-(3,4-Dihydro-lH-isoquinolin-2-yl)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 10)
Melting Point: 161-163 0C
MS: m/z 482 (M-I)
1H NMR (DMSOd6) δ: 2.88-2.91 (d, 2H), 3.31-3.34 (m, 6H), 3.59-3.63 (m, 4H), 4.50 (s,
2H), 6.97-6.99 (d, 2H), 7.05-7.07 (d, 2H), 7.17-7.20 (d, 4H), 7.28 -7.30 (d, 2H), 7.39-7.41
(d, 2H), 7.64 (s, IH), 12.52 (br s, IH).
(4-{5-[4-(4-Ethyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-ylideneamino} - phenyl) -acetic acid (Compound 12)
Melting Point: 224 - 226 °C
MS: m/z 485 (M-I)
1H NMR (DMSO-d6) δ: 1.09 - 1.25 (t, 3H), 2.51 - 2.65 (m, 2H), 3.29 (s, 3H), 3.35 - 3.55 (s,
2H), 5.08 (s, 2H), 6.98 - 7.03 (d, 2H), 7.14 - 7.46 (m, 10H), 7.69 (s, IH).
(4- { 5-[4-(3 ,4-Dihydro- 1 H-isoquinolin-2-ylmethyl)-benzylidene] -3 -methyl-4-oxo-thiazolidin-
2-ylideneamino} -phenyl)-acetic acid. (Compound 13)
Melting Point: 158-160 0C
MS: m/z 496 (M-I)
1H NMR (DMSOd6) δ: 2.80-3.25 (m, 4H), 3.31 (s, 3H), 3.56(s, 2H), 3.91 (s, 2H), 4.06 (s,
2H), 6.94-6.98 (d, 2H), 7.06-7.15 (m, 4H), 7.25-7.29 (d, 2H), 7.40-7.60 (m, 4H), 7.72 (s,
IH), 12.37 (s, IH)
(4- { 3 -Ethyl-5 - [4-(4-ethyl-benzyloxy)-benzylidene] -4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid. (Compound 14)
Melting Point: 326-329 0C
MS: m/z 499 (M-I)
1H NMR (DMSOd6) δ: 1.13-1.22 (m, 6H), 2.49-2.69 (m, 2H), 3.29-3.39 (m, 2H), 5.06 (s,
2H), 6.88-6.90 (d, 2H)5 7.09-7.10 (d, 2H), 7.13-7.36 (m, 6H), 7.38-7.47 (d, 2H), 7.63 (s, IH).
(4-{3-Ethyl-5-[4-(4-isopropyl-benzyloxy)-benzylidene]-4-oxo-thiazolidin-2-ylideneamino}- phenyl)-acetic acid (Compound 15)
Melting Point: 326-329 0C
MS: m/z 513 (M-I) .
1H NMR (DMSOd6) δ: 1.05-1.28 (m, 9H), 2.80-2.90 (m, IH), 3.59 (s, 2H). 3.90-3.94 (d,
2H), 5.08 (d, 2H), 6.96-7.00 (d, 2H), 7.10-7.14 (d, 2H), 7.22-7.36 (m, 4H), 7.46-7.50 (d, 2H),
7.70 (s, lH), 12.40 (br s, IH).
(4- { 5- [4-(4-Cyclohexyl-benzyloxy)-3 -methoxy-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino) -phenyl)-acetic acid (Compound 16) Melting Point: 185-186 °C MS: m/z 569 (M-I)
1H NMR (DMSO-d6) δ: 1.25-1.42 (m, 5H), 1.71-1.83 (m, 5H), 2.42-2.50 (m,lH), 3.42 (s,3H), 3.68 (s,2H), 3.85 (s,3H), 5.11 (s, 2H), 6.88-6.91 (d, 2H), 6.94 (s,lH), 6.97-6.99 (d, 2H), 7.17-7.19 (d, 2H), 7.29-7.32 (m, 4H), 7.67 (s, IH).
[4-(3-Methyl-5- {4- [methyl- (4-methyl-benzyl)-amino]-benzylidene}-4-oxo-thiazolidin-2- ylideneamino)-phenyl]-acetic acid. (Compound 18)
Melting Point: 248-250 0C
MS: m/z 484 (M-I)
1H NMR (DMSOd6) δ : 2.20 (s, 2H5), 3.03 (s, 3H), 3.26 (s, 3H), 3.54 (s, 2H), 4.54 (s, 2H),
6.73 - 6.75 (d, 2H), 6.92 - 6.94 (m, 3H), 7.02 - 7.05 (m, 3H), 7.24-7.26 (m, 4H), 7.54 (s, IH)
(4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino} -2-fluoro-phenyl)-acetic acid. (Compound 19)
Melting Point: 196-198 0C
MS: m/z 557 (M-I)
1H NMR (DMSOd6) δ: 1.25 - 1.40 (m, 6H), 1.61 - 1.95 (m, 4H), 2.45 - 2.55 (m, IH), 3.42
(s, 3H), 3.75 (s, 2H), 5.03 (s, 2H), 6.76 - 6.82 (m, IH), 6.98 - 7.02 (d, 2H), 7.21 - 7.43 (m,
8H), 7.73 (s, IH)
(2-Fluoro-4-{5-[4-(4-isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino} -phenyl)-acetic acid.(Compound 20)
Melting Point: 273-277 0C
MS: m/z 517 (M-I)
1H NMR (DMSOd6) δ : 1.17 - 1.22 (d, 6H), 2.86 - 2.89 (m, IH), 3.30 (s, 3H), 3.43 (s, 2H),
5.08 (s, 2H), 6.78 - 6.82 (m, 2H), 7.13 - 7.15 (d, 2H), 7.23 - 7.25 (d, 2H), 7.31 - 7.36 (m, 3H),
7.49 - 7.51 (d, 2H), 7.71 (s, IH).
(2-Fluoro-4- { 5 - [4-(4-isobutyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 21)
Melting Point: 189-193 °C
MS: m/z 531 (M-I)
1H NMR (DMSO-d6) δ: - 0.79 - 0.85 (d, 6H), 1.77 - 1.85 (m, IH)3 2.43 - 2.44 (d, 2H), 3.27
(s, 3H), 3.62 (s, 2H), 5.09 (s, 2H), 6.83 - 6.89 (m, 2H), 9.13 - 7.17 (m, 4H), 7.33 - 7.38 (m,
3H), 7.50 - 7.52 (d, 2H), 7.74 (s, IH)
(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-oxo-acetic acid. (Compound 23)
Melting Point: 165-168 0C
MS: m/z 513 (M-I) "
1H NMR (DMSOd6) δ: 1.17 - 1.18 (d, 6H), 2.83 - 2.90 (m, IH), 3.32 (s, 3H), 5.07 (s, 2H),
7.11 - 7.13 (d, 2H), 7.23 - 7.25 (d, 2H), 7.33 - 7.35(d, 2H), 7.47 - 7.49(d, 2H), 7.72(s, IH),
7.88 - 7.90(d, 2H)
4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneammo}-phenyl)-oxo-acetic acid. (Compound 25)
Melting Point: 192-194 0C
MS: m/z 553(M-I)
1E NMR (DMSOd6) δ: - 1.24 - 1.45 (m, 5H), 1.73 - 1.84 (m, 5H), 2.45 - 2.55 (m, IH), 3.45
(s, 3H), 5.03 (s, 2H),.6.99 - 7.01 (d, 2H), 7.14 - 7.16 (d, 2H), 7.21 - 7.23 (d, 2H), 7.31 - 7.33
(d, 2H), 7.38 - 7.40 (d, 2H), 7.77 (s, IH), 8.45 - 8.47 (d, 2H).
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-£luoro-benzylidene}-3-methyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid. (Compound 26) Melting Point: 168-170 0C MS: m/z 570 (M-I)
1H NMR (DMSOd6) δ: - 1.14 - 1.34 (m, 5H), 1.63 - 1.72 (m, 5H), 2.35 - 2.45 (m, IH), 2.83 (s, 3H), 3.29 (s, 3H), 3.56 (s, 2H), 4.39 (s, 2H), 6.91 - 6.97 (m, 3H), 7.08 - 7.16 (m, 5H), 7.23 - 7.28 (m, 3H), 7.59 (s, IH).
(4- { 5- [4-(4-Cyclohexyl-benzyloxy)-3 -fluoro-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 27)
Melting Point: 205-207 °C
MS: m/z 557 (M-I)
1H NMR (DMSOd6) δ: - 1.16 - 1.39 (m, 5H), 1.67 - 1.76 (m, 5H), 2.40 - 2.50 (m, IH)5 3.28
(s, 3H), 3.49 (s, 2H), 5.10 (s, 2H), 6.96 - 6.98 (d, 2H), 7.21 - 7.46 (m, 9H), 7.68 (s, IH).
[4-(5-{4-[4-(l -Ethyl -propoxy)-benzyloxy]-benzylidene} -3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid. (Compound 28)
Melting Point: 162-164 °C
MS: m/z 543 (M-I)
1U NMR (DMSOd6) δ: - 0.87 - 0.90 (t, 6H), 1.50 - 1.60 (m, 4H), 3.32 (s, 3H), 3.59 (d, 2H),
4.19 - 4.22 (t, IH), 5.02 (s, 2H), 6.91 - 6.93 (d, 2H), 6.97 - 6.99 (d, 2H), 7.11 - 7.13 (d, 2H),
7.29 - 7.34 (m, 4H), 7.48 - 7.50 (d, 2H) 7.71 (s, IH), 12.38 (br, IH),
(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-ethyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 29)
Melting Point: 194-196 °C
MS: m/z 553 (M-I)
1H NMR (DMSOd6) δ: 1.23 - 1.39 (m, 8H), 1.67 - 177(m, 5H), 2.40 - 2.55 (m, IH),
3.59 (s, 2H), 3.90 - 3.95 (q, 2H), 5.08 (s, 2H), 6.97 - 6.99 (d, 2H), 7.1 1 - 7.13 (d, 2H), 7.21 -
7.23 (d, 2H), 7.28 - 7.38 (d, 2H), 7.32 - 7.34 (d, 2H), 7.47 - 7.49 (d, 2H), 7.70 (s, IH).
[4-(5 - { 3 -Fluoro-4- [(4-isopropyl-benzyl)-methyl-amino] -benzylidene } -3 -methyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid (Compound 31)
Melting Point: 173-174 0C
MS: m/z 530 (M-I)
1H NMR (DMSOd6) δ: 1.14 - 1.17 (d, 6H), 2.65 - 2.83 (m, 4H), 3.52 (s, 2H), 4.40 (s, 2H)5
7.09 - 7.19 (m, 5H), 7.26 - 7.34(m, 3H), 7.61 (s, IH).
Hydroxy- (4-{3-methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene]-thiazolidin-2- ylideneamino}-phenyl)-acetic acid. (Compound 32)
Melting Point: 202 - 204 0C
MS: m/z 543 (M-I)
1H NMR (DMSOd6) δ: 0.83-0.84 (t, 3H), 1.19 - 1.26 (m, 6H), 1.51 - 1.54 (t, 2H), 3.31 (s,
3H), 3.49 (s, IH), 4.98 (s, IH), 5.08 (s, 2H), 7.01 - 7.31 (m, 8H), 7.40 - 7.46(m, 4H), 7.70
(IH, s), 12.54(br s, IH).
(4- { 3 -Methyl-4-oxo-5 - [4-(4-pentyl-benzyloxy)-benzylidene] -thiazolidin-2-ylideneamino } - phenyl)-acetic acid (Compound 33)
Melting Point: 128-130 0C '
MS:^- m/z 527 (M-I)
1H NMR (DMSOd6) δ : 0.842 (t, 3H), 1.19 - 1.25 (m, 6H), 1.50 - 1.54(t, 2H), 3.26 (s, 3H),
3.47 (m, 2H), 5.02 (s, 2H), 6.95 - 6.96 (d, 2H), 7.14 - 7.30 (m, 8H), 7.43 - 7.45 (d, 2H), 7.68
(s, IH), 12.39 (s, IH)
[4-(5- {4-[ 1 -(4-Cyclohexyl-phenyl)-ethoxy] -benzylidene} -3 -methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid (Compound 34) Melting Point: 165 - 167 0C MS: - m/z 553(M-I)
1U NMR (DMSO-d6) δ: 1.21 - 1.24 (m, 6H), 1.28 - 1.32 (m, 3H), 1.50 - 171 (m, 5H), 2.36- 2.40 (m, IH), 3.23 (s, 3H), 3.60 (s, 2H), 5.50-5.53 (m, IH), 6.94 - 6.98 (m, 4H), 7.14 - 7.18 (d, 2H), 7.27 - 7.31 (m, 4H), 7.37 - 7.41 (d, 2H), 7.64 (s, IH).
(4-{5-[4-(4-Cyclohexyloxy-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino} -phenyl)- acetic acid (Compound 35)
Melting Point: 158 - 159 °C
MS: - m/z 555 (M-I)
1H NMR (DMSOd6) δ: 1.13 - 1.47 (m, 7H), 1.65 - 1.67 (m, 2H), 1.83 - 1.85 (m, 2H), 3.20
(s, 3H), 3.56 (s, 2H), 4.27 - 4.28 (d, IH), 4.9 (s, 2H), 6.87 - 6.96 (m, 4H), 7.06 - 7.09 (d, 2H),
7.26 - 7.31 (m, 4H), 7.42 - 7.44 (d, 2H), 7.65 (s, IH).
(4-{5-[4-(4-Cyclohexyloxy-benzyloxy)-benzylidene]-3-ethyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid (Compound 36)
Melting Point: 128 - 129 °C
MS: - m/z 569 (M-I)
1H NMR (DMSOd6) δ: 1.22 - 1.37 (m, HH), 3.90 - 3.94 (d, 2H), 1.85 - 1.83 (m, 3H), 3.59
(s, 2H), 4.30 - 4.32 (m, IH), 5.02 - 5.12 (s, 2H), 6.8 - 7.0 (m, 4H), 7.10 - 7.14 (d, 2H), 7.27 -
7.35 (m, 4H), 7.46 - 7.50 (d, 2H), 7.7 (s, IH).
[4-(3 -Ethyl-5- { 3 -fluoro-4- [(4-isopropyl-benzyl)-methyl-amino] -benzylidene } -4-oxo- thiazolidin-2-ylideneamino)-phenyl] -acetic acid. (Compound 38)
Melting Point: 159-161 °C
MS: - m/z 544 (M-I)
1H NMR (DMSOd6) δ: 1.15 - 1.16 (d, 6H), 1.25 - 1.26 (t, 3H), 2.79 - 2.85 (m, 4H)5 3.58 (s,
2H), 3.89 - 3.92 (m, 2H), 4.41 (s, 2H), 6.9 - 7.0 (m, 3H), 7.11 - 7.19 (m, 5H), 7.28 - 7.33 (m,
3H), 7.6 (s, IH), 12.38 (s, IH).
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-fluoro-benzylidene}-3-ethyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl] -acetic acid. (Compound 39)
Melting Point: 174 - 175 0C
MS: m/z 584 (M-I)
1H NMR (DMSOd6) δ: 1.18 - 1.40 (m, 8H), 1.66 - 1.74 (m, 5H)5 2.43 - 2.50 (m, IH), 2.84
(s, 3H), 3.58 (s, 2H), 3.89 - 3.94 (m, 2H), 4.41 (s, 2H), 6.97 - 7.01 (m, 3H)5 7.10 - 7.19 (m,
5H), 7.27 - 7.33 (m, 3H), 7.64 (s, IH), 12.37 (s, IH).
(4-{5-[4-(4-Cyano-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-lideneamino}- phenyl)-acetic acid (Compound 40)
Melting Point: 178-180 0C
MS: m/z 482 (M-I)
1H NMR (DMSOd6) δ: 3.30 (s, 3H), 3.55 (s, 2H), 5.25 (s, 2H), 6.95 - 6.97 (d, 2H)5 7.13 -
7.15 (d, 2H), 7.28 - 7.30 (d, 2H), 7.48 - 7.50 (d, 2H), 7.61 - 7.63 (d, 2H)5 7.70 (s, 1H),'7.85 -
7.87 (d, 2H)
(4- { 5- [4-(3 -Fluoro-4-piperidin- 1 -yl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin -2- ylideneamino}-phenyl)-acetic acid (Compound 41)
Melting Point: 289-91 °C
MS: m/z 560 (M + 1)
1H NMR (DMSOd6) δ : 1.50 - 1.51 (m, 2H)5 1.62 - 1.64 (m, 4H), 2.92 - 2.95 (m, 4H), 3.23
(s, 2H), 3.30 (s, 3H)5 5.03 - 5.10 (d, 2H), 6.86 - 6.88 (d, 2H)5 7.00 - 7.25 (m, 6H)5 7.46 - 7.48
(m, 3H), 7.67 (s, IH), 12.38 (s, IH).
{4- [5 - [ 1 - {4- [ 1 -(4-Isobutyl-phenyl)-butoxy] -phenyl } .-methylidene] -3 -methyl-4-oxo- thiazolidin-2-ylideneamino3 -phenyl} -acetic acid (Compound 42) Melting Point: 93-95°C MS: m/z 557 (M+ 1),
'H NMR (DMSO-d6) δ: 0.79 - 0.91 (m, 9H), 1.22-1.35 (m, 2H), 1.73 - 1.76 (m, 2H), 2.35 - 2.39 (m, 3H), 3.29 (s, 3H), 3.58 (s, 2H), 5.31 - 5.32 (m, IH), 6.93 - 7.10 (m, 6H), 7.25 - 7.40 (m, 6H), 12.25 (s, IH).
Example 3
Preparation of {4-[3-Methyl-4-oxo-thiazolidin-2~ylideneamino]-phenyl}-acetic acid methyl ester

A solution of [4-(3-Methyl-thioureido)-phenyl] -acetic acid methyl ester (1.2 g, 5.0 mmol), methylbromo acetate (0.67 ml, 7.0 mmol) and triethylamine (1.5 ml, 10 mmol) in ethanol (20 ml) was heated under reflux for 5 h. The solvent was removed under reduced pressure and residue was dissolved in ethyl acetate (50 ml). Organic solution was washed with water (20 ml) followed by brine (20 ml), and dried over anhydrous sodium sulfate. Solution was evaporated under vacuo and residue was purified by silica gel column chromatography using ethyl acetate: hexane (20:80) as eluent to afford a yellow solid (1.3 g, 93 %). MS = m/z 279 (M+l).
1H NMR (CDCl3) δ; 3.30 (s, 3H), 3.61 (s, 2H)5 3.70 (s, 3H), 3.81 (s, 2H), 6.92 (d, 2H), 25 (d, 2H).
Example 4
Preparation of [4-(3-Ethyl-4-oxo-thiazoIidin-2-yIideneamino)-phenyl]-acetic acid ethyl ester
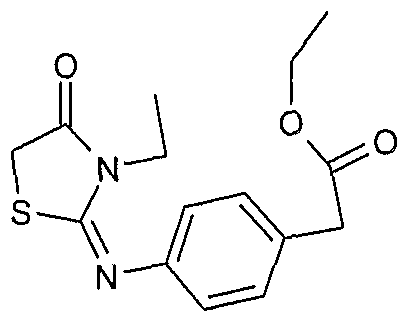
1H NMR (DMSOd6) δ: 1.20 - 1.38 (m, 6H), 3.58 (s, 2H), 3.78 (s, 2H), 3.84 - 3.95 (q, 2H), 4.09 - 4.20 (q, 2H), 6.89 - 6.93 (d, 2H), 7.23 - 7.27 (d, 2H).
Example 5
Preparation of {2-Fluoro-4- [3-methyl-4-oxo-thiazolidinylideneamino] -phenyl}-acetic acid ethyl ester

Analogously, by practicing the chemistry as described in example 3 by substituting [4-(3- methyl-thioureido)-phenyl]-acetic acid methyl ester with [2-Fluoro-4-(3-methyl-thioureido)- phenyl] -acetic acid ethyl ester with appropriate change in the reactants and reaction conditions, { 2-Fluoro-4- [3 -methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl } -acetic acid ethyl ester was prepared.
MS = m/z 311 (M+l).
1H NMR (DMSO-d6) δ: 1.24 - 1.28 (t, 3H), 3.29 (s, 3H), 3.64 (s, 2H), 3.83 (s, 2H), 4.14 -
4.20 (m, 2H), 6.69 - 6.74 (m, 2H), 7.20 - 7.26 (m, IH).
Example 6 Preparation of [4-(3-methyl-thioureido)-phenyI]-acetic acid methyl ester

A solution of p-aminophenylacetic acid methyl ester (1 g, 6.0 mmol) and methylisothiocyanate (480 mg, 6.6 mmol) in ethanol (20 ml) was heated under reflux for 6 h.
The reaction mixture was cooled to room temperature and solvent was removed under vacuo.
Water (30 ml) was added to the residue and stirred for 15 min. The solid was filtered and triturated with 50 % ethanol in water (20 ml) and filtered to give off-white solid (1.3 g, 90
%).
MS = m/z 237 (M-I).
1H NMR (CDCl3) δ:. 3.11 (d, 3H)1 3.63 (s, 2H)1 3.70 (s, 3H), 6.04 (bs, IH), 7.16 (d, 2H),
7.32 (d, 2H).
Example 7 Preparation of [4-(3-ethyl-thioureido)-phenyl]-acetic acid ethyl ester

Analogously, by practicing the chemistry as described in example 6 by substituting methylisothiocyanate with ethylisothiocyanate and p-aminophenylacetic acid methyl ester with p-aminophenylacetic acid ethyl ester with appropriate change in the reactants and reaction conditions, [4-(3-ethyl-thioureido)-phenyl] -acetic acid ethyl ester was prepared. MS = m/z 265 (M-I).
1H NMR (DMSOd6) δ: 1.15 - 1.18 (t, 3H), 1.23 - 1.27 (t, 3H), 3.60 - 3.67 (m, 4H), 4.12 - 4.12 (m, 2H), 6.02 (br s, IH), 7.14 - 7.16 (d, 2H), 7.30 - 7.32 (d, 2H), 8.09 (br s, IH).
Example 8 Preparation of [4-(3-methyl-thioureido)-2-fluoro-phenyl]-acetic acid ethyl ester

Analogously, by practicing the chemistry as described in example 6 by substituting p- aminophenylacetic acid methyl ester with (4-Amino-2-fluoro-phenyl)-acetic acid ethyl ester with appropriate change in the reactants and reaction conditions, [4-(3-Methyl-thioureido)-2- fluoro-phenyl] -acetic acid ethyl ester was prepared.
MS = m/z 269 (M-I)
1H NMR (CDCl3) δ: 1.26 - 1.30 (t, 3H), 3.16 (s, 3H), 3.66 (s, 2H), 4.16 - 4.22 (m, 2H), 6.1
(s, IH), 6.94 - 6.98 (m, 2H), 7.30 - 7.34 (t, IH), 7.68 (s, IH).
Example 9
Preparation of {4- [5- [ 1- [4-(4-Cyclohexyl-benzy Ioxy)-pheny 1] -meth-)-y lidene] -3-methyl- 4-oxo-thiazoIidin-2-ylideneamino]-phenyl}-hydroxy-acetic acid (Compound 30)

Step-1: Preparation of Hydroxy-(4-nitro-phenyl)-acetonitrile
To a stirred solution of p-Nitrobenzaldehyde (5 g, 33.1 mmol) and Trimethyl silyl cyanide (5.2 ml, 39.7 mmol) in dichloromethane (50 ml), Zinc iodide (1.0 g, 3.3 mmol) was added under nitrogen atmosphere and stirred for 2 h at 50-55 0C. 10 % aqueous hydrochloric acid
(20 ml) was added to reaction mixture and stirred at 50-55 0C for 30 min. Reaction mixture was cooled to room temperature and water (100 ml) was added to reaction mixture.
Dichloromethane layer was separated and aqueous layer was extracted with dichloromethane
(2 x 25 ml) and combined dichloromethane layer was washed with water (2 x 25 ml), dried over anhydrous sodium sulphate and evaporated under vacuum to give solid residue. Residue was triturated with hexane (20 ml) and dried under vacuum to give a pale yellow solid (5.0 g,
84.8 %).
MS = m/z l79 (M+l).
1H NMR (DMSO-d6) δ: 5.97 - 6.00 (d, IH), 7.39 - 7.42 (d, IH), 7.74 - 7.85 (d, 2H), 8.29 -
8.33 (d, 2H).
Step-2: Preparation of Hydroxy-(4-nitro-phenyl)-acetic acid ethyl ester Hydroxy-(4-nitro-phenyl)-acetonitrile (5.0 g, 28.0 mmol) was added to a mixture of acetic acid (20 ml) and ION aqueous hydrochloric acid (20 ml) and heated to 100 0C for 4 h. Reaction was monitored by thin layer chromatography. Reaction mixture was cooled to room temperature and evaporated under vacuum to dryness to give solid residue and washed with diethyl ether (20 ml). To the residue, ethanol (50 ml) and con sulfuric acid (1.5 ml, 28.0 mmol) was added and heated to 80-85 0C for 4 h. Reaction mixture was cooled to room temperature and solvent was evaporated under vacuum to give solid residue, which was recrystallized from isopropanol (20 ml) to give pale yellow solid (4.8 g, 76.1 %). MS = m/z 224 (M-I).
1H NMR (DMSO-d6) δ: 1.21 - 1.25 (t, 3H), 3.70 (bs, IH), 4.20 - 4.25 (q, 2H), 5.27 (s, IH), 7.63 - 7.69 (d, 2H), 8.20 - 8.22 (d, 2H).
Step 3: Preparation of (4-Amino-phenyl)-hydroxy-acetic acid ethyl ester A mixture of Hydroxy-(4-nitro-phenyl)-acetic acid ethyl ester (4.7 g, 20.8 mmol) and 10% Pd/C (900 mg) in methanol (50 ml) was stirred under hydrogen atmosphere for 4 h. Catalyst was removed by filtration and filtrate was evaporated under vacuum to give solid residue. Residue was washed with hexane (30 ml) and dried to give off white solid (3.4 g, 85.0 %).
MS = m/z l96 (M+l).
1H NMR (DMSO-d6) δ: 1.09 - 1.13 (t, 3H), 3.38 (br s, IH), 4.02 - 4.09 (q, 2H), 4.90 (s, IH)5
5.74 (bs, 2H), 6.58 - 6.62 (d, 2H), 7.05 - 7.10 (d, 2H).
Step-4: Preparation of Hydroxy-[4-(3-methyl-thioureido)-phenyl]-acetic acid ethyl ester A mixture of (4-Amino-phenyl)-riydroxy-acetic acid ethyl ester (3.3 g, 16.9 mmol) and methylisothiocyanate (1.35 g, 18.6 mmol) in ethanol (50 ml) was heated under reflux for 6 h. Reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. To the residue water (40 ml) was added and stirred for 15 min. The solid was filtered and washed with 50 % ethanol in water (25 ml) and dried under high vacuum to give a pale yellow solid (4.1 g, 91.0 %). MS = m/z 269 (M+l).
Step 5: Preparation of Hydroxy-{4~[3-memyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl}- acetic acid ethyl ester
A mixture of Hydroxy-[4-(3-methyl-thioureido)-phenyl]-acetic acid ethyl ester (4.0 g, 14.9 mmol), triethylamine (4.1 ml, 29.8 mmol) and ethylbromo acetate (1.82 ml, 16.4 mmol) in ethanol (50 ml) was heated under reflux for 5 h. Reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (80 ml) and washed with water (2 x 50 ml) followed by brine (50 ml) and dried over anhydrous sodium sulphate. The solvent was removed under reduced pressure to give residue, which was purified by silica gel column using 20 % Ethyl acetate in hexane as a eluent to afford off-white solid (3.2 g, 69.7 %). MS = m/z 307 (M-I).
Step 6: prepration of.{4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl }-hydroxy-acetic acid ethyl ester A mixture of Hydroxy-{4-[3-methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl}-acetic acid ethyl ester (300 rag, 0.974 mmol), 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde (314.9 mg,
1.10 mmol) and piperidine (0.288 ml, 2.92 mmol) in ethanol(15 ml) was heated under reflux for 16 h. Reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. Water (25 ml) was added to the residue and stirred for 15 min. Solid was filtered and purified by silica gel column using 20 % ethyl acetate in hexane as eluent to afford a yellow solid (240 mg, 42.2 %). MS = m/z 585 (M+l).
1H NMR (CDCl3) 5: 1.09 - 1.13 (t, 3H), 1.22 - 1.36 (m, 5H), 1.76 - 1.98 (m, 5H), 2.50 - 2.53 (m, IH), 3.32 (s, 3H), 3.34 (s, IH), 4.06 - 4.13 (q, 2H), 5.08 (s, 2H), 5.12 - 5.15 (d, IH), 6.08 - 6.11 (d, IH), 6.99 - 7.03 (d, 4H), 7.09 - 7.35 (m, 4H), 7.41 - 7.50 (m, 4H), 7.71 (s, IH).
Step 7: Preparation of {4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl } -hydroxy-acetic acid
To a stirred solution of {4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl}-hydroxy-acetic acid ethyl ester (200 mg,
0.342 mmol) in 1 :1 tetrahydrofuran: methanol (10 ml) was added a solution of lithium hydroxide monohydrate (28.7 mg, 0.684 mmol) in water (0.5 ml) and stirred at room temperature for 2 h. The reaction mixture was acidified with 10 % hydrochloric acid (pH ~ 4) and precipitated solid was filtered, washed with water (10 ml) and hexane (10 ml). It was dried under high vacuum to give a yellow solid (160 mg, 84.2 %).
MS = m/z 555 (M-I).
Melting Point: 206-208 0C.
1H NMR (DMSO-d6) δ: 1.22 - 1.36 (m, 5H), 1.76 - 1.98 (m, 5H), 2.50 (m, IH), 3.30 (s, 3H),
3.59 (s, IH), 4.97 (s, IH), 5.06 (s, 2H), 6.98 - 6.99 (d, 2H), 7.11 - 7.13 (d, 2H), 7.20 - 7.22 (d,
2H), 7.30 - 7.32 (d, 2H), 7.42 - 7.46 (m, 4H), 7.69 (s, IH).
Example 10
Preparation of Hydroxy- (4-{3-methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene] - thiazolidin-2-yIideneamino}-phenyl)-acetic acid. (Compound 32)

Analogously, by practicing the chemistry as described in example 9 for compound 30, by substitution of 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde with 4-(4-Pentyl-benzyloxy)- benzaldehyde with appropriate change in the reactants and reaction conditions, Hydroxy- (4- {3-methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene] -thiazolidin-2-ylideneamino}- phenyl)-acetic acid was prepared. Melting Point: 202-204 °C MS: m/z 543 (M-I)
1H NMR (DMSOd6) δ: 0.83 - 0.84 (t, 3H), 1.19 - 1.26 (m, 6H), 1.51 - 1.54 (t, 2H), 3.31 (s, 3H), 3.49 (s, IH), 4.98 (s, IH), 5.08 (s, 2H), 7.01 - 7.31 (m, 8H), 7.40 - 7.46 (m, 4H), 7.70 (IH, s), 12.54 (s, IH),
Example 11
Preparation of {4-[5-[l-[4-(4-Cyclohexyl-benzyIoxy)-phenyl]-meth-)-yIidene]-3-methyI- 4-oxo-thiazolidin-2-ylideneamino]-phenyl}-fluoro-acetic acid (Compound 24)

Step-1: Preparation of Fluoro-{4-[3-methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl}- acetic acid ethyl ester*.
To a stirred suspension of Hydroxy-{4-[3-methyl-4-oxo-thiazolidin-2-ylideneamino]- phenyl} -acetic acid ethyl ester (prepared as per step 5 of Example 9; 2.0 g, 6.4 mmol) in dry
dichloromethane (50 ml), diethyl aminosulfur trifluoride (0.884 ml, 7.1 mmol) was added under nitrogen atmosphere at 10 °C and stirred for 1 h. Reaction mixture was carefully poured into cold water (50 ml) and stirred for 15 min. dichloromethane layer was separated and aqueous layer was extracted with dichloromethane (50 ml). Combined dichloromethane layer was washed with saturated sodium bicarbonate solution (2 x 25 ml), dried over sodium sulphate and evaporated under vacuum to give yellow liquid (1.8 g, 89.5 %). MS = m/z 311 (M+l).
Steρ-2: preparation of {4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]~meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl }-fluoro-acetic acid ethyl ester A mixture of Fluoro-{4-[3-methyl-4-oxo-thiazolidin-2-yHdeneamino]-phenyl}-acetic acid ethyl ester (300 mg, 0.967 mmol), 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde (312.9 mg, 1.06 mmol) and piperidine (0.286 ml, 2.90 mmol) in ethanol(15 ml) was heated under reflux for 20 h. Reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. Water (25 ml) was added to the residue and stirred for 15 min. Solid was filtered and purified by silica gel column using 15 % ethyl acetate in hexane as eluent to afford a yellow solid (210 mg, 37.0 %). MS = m/z 587 (M+l).
Step-3: Preparation of {4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino] -phenyl }-fluoro-acetic acid To a stirred solution of {4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-ylidene]-3- methyl-4-oxo-thiazolidin-2-ylideneamino]-phenyl}-fluoro-acetic acid ethyl ester (200 mg, 0.341 mmol) in 1 :1 tetrahydrofuran: methanol (10 ml) was added a solution of lithium hydroxide monohydrate (28.6 mg, 0.682 mmol) in water (0.5 ml) and stirred at room temperature for 2 h. The reaction mixture was acidified with 10 % hydrochloric acid (pH ~ 4) and solid was filtered, washed with water (10 ml) and hexane (10 ml). It was dried under high vacuum to give yellow solid (170 mg, 89.4 %). MS = m/z 557 (M-I),
Melting Point: 159 - 160 °C.
1H NMR (DMSO-d6) δ: 1.22 - 1.36 (m, 5H), 1.76 - 1.98 (m, 5H), 2.5 (m, IH), 3.37 (s, 3H), 5.06 (s, 2H), 5.55 - 5.68 (d, IH), 6.99 - 7.01 (d, 2H), 7.11 - 716 (d, 2H), 7.20 - 7.22 (d, 2H), 7.32 - 734 (d, 2H), 7.43 - 7.48 (m, 4H), 7.68 (s, IH), 12.52 (s, IH).
Example 12
Preparation of (4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino}-phenyl)-oxo-acetic acid (compound 25)

Step-1: Preparation of 1 -(4- Acetyl -phenyl)-3-methyl-thiourea
A mixture of 4-amino acetophenone (3 g, 22.22 mmol) and methylisothiocyanate (1.7 g, 23.28 mmol) in ethanol (40 ml) was heated under reflux for 6 h. The reaction mixture was cooled to room temperature and solvent was evaporated under reduced pressure. The residue was treated with 2N hydrochloric acid (25 ml) and stirred for 15 min. Solid was filtered, washed with 50 % ethanol (50 ml) and dried under vacuo to afford an off white solid (2.8 gm, 61 %). MS = m/z 207 (M-I),
Step-2: Preparation of 2-(4-Acetyl-phenylimino)-3-methyl-thiazolidin-4-one A solution of l-(4-Acetyl-phenyl)-3-methyl-thiourea (2 g, 9.61 mmol), triethylamine (3.4 ml, 23.56 mmol) and ethylbromo acetate (1.2 ml, 11.39 mmol) in ethanol (25 ml) was heated under reflux for 5 h. The reaction mixture was cooled to room temperature, water (50 ml) was added and precipitated solid was filtered. Solid was suspended in 50 % ethanol (10 ml), stirred for 15 min, filtered and dried under vacuo to give a yellow solid (1.9 gm, 80 %).
MS: m/z 247 (M-I).
Step-3: Preparation of 2-(4-Acetyl-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene] -3 -methyl-thiazolidin-4-one
A mixture of 2-(4-Acetyl-phenylimino)-3 -methyl-thiazolidin-4-one (210 mg, 0.85 mmol), 4- (4-Cyclohexyl-benzyloxy)-benzaldehyde (250 mg, 0.85 mmol) and piperidine (0.309 ml, 2.55 mmol) in ethanol (20 ml) was heated under reflux for 14 h. The reaction was monitored by thin layer chromatography. The reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. To the residue, Ethyl acetate (40 ml) was added and washed with water (2 x 10 ml). The solvent was removed under vacuo and crude product was purified by silica gel column chromatography using ethyl acetate: hexane (15:85) as eluent to get yellow solid (370 mg, 83 %). MS: m/z 525 (M+l).
Step-4: Preparation of (4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino}-phenyl)-oxo-acetic acid
To a stirred solution of 2-(4-Acetyl-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene] -3 -methyl-thiazolidin-4-one (350 mg, 0.67 mmol) in pyridine (15 ml) was added selenium dioxide (111 mg, 1.0 mmol) and reaction mixture was heated to 110 0C for 3 h. The reaction mixture was cooled to room temperature and inorganics were removed by filtration.
The filtrate was evaporated under vacuo, residue was treated with 2N hydrochloric acid (30 ml) and filtered. It was purified by silica gel column chromatography using ethyl acetate:
Hexane (6:4) as eluent to afford yellow solid (250 mg, 68 %).
MS = m/z 553 (M-I).
Melting Point: 192-194 °C
1H NMR (DMSOd6) δ: 1.24 - 1.45 (m, 5H), 1.73 - 1.84 (m, 5H), 2.45 - 2.55 (m, IH), 3.45
(s, 3H)5 5.03 (s, 2H)5 6.99 - 7.01 (d, 2H)5 7.14 - 7.16 (d5 2H), 7.21 - 7.23 (d, 2H)5 7.31 - 7.33
(d, 2H)5 7.38 - 7.40 (d, 2H), 7.77 (s, IH)5 8.45 - 8.47 (d, 2H), 12.56 (s, IH).
Example 13
Preparation of (4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]~3-methyl-4-oxo- thiazolidin -2-ylideneamino}-phenyl)-oxo-acetic acid. (Compound 23)

Analogously, by practicing the chemistry as described in example 12 for compound 25, by substitution of 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde with 4-(4-Isopropyl-benzyloxy)- benzaldehyde with appropriate change in the reactants and reaction conditions, (4-{5-[4-(4-
Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-ylideneamino}-phenyl)- oxo-acetic acid was prepared.
Melting Point: 165-168 0C
MS: m/z 513 (M-I)
1H NMR (DMSOd6) δ: 1.17 - 1.18 (d, 6H), 2.83 - 2.90 (m, IH), 3.32 (s, 3H), 5.07 (s, 2H),
7.11 - 7.13 (d, 2H), 7.23 - 7.25 (d, 2H), 7.33 - 7.35 (d, 2H), 7.47 - 7.49 (d, 2H), 7.72 (s, IH),
7.88 - 7.90 (d, 2H).
Example 14
Preparation of N-(4- {5- [4-(4-Cyclohexyl-benzyloxy)-benzyIideneJ -3-methyl-4-oxo- thiazolidin-2-ylideneamino}-phenyl)-oxalamic acid (Compound 22)

A mixture of 4-nitroaniline (5 g, 36.75 mmol) and methylisothiocyanate (2.95 g, 40.44 mmol) in ethanol (50 ml) was heated under reflux for 8 h. The reaction mixture was cooled to room temperature and solvent was evaporated under reduced pressure. Residue was dissolved in ethyl acetate (100 ml), washed with 2N hydrochloric acid (40 ml) and water (40 ml). The organic layer was dried over anhydrous sodium sulfate and evaporated under vacuo to afford a yellow solid (4.1 gm, 54 %). MS = m/z 212 (M+l).
Step-2: Preparation of 3-Methyl-2-(4-nitro-phenylimino)-thiazolidin-4-one A mixture of l-Methyl-3-(4-nitro-phenyl)-thiourea (4 g, 18.95 mmol), triethylamine (6.8 ml, 47.11 mmol) and ethylbromo acetate (2.2 ml, 20.84 mmol) in ethanol (50 ml) was heated under reflux for 5 h. The reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. Residue was dissolved in ethyl acetate (60 ml), washed with water (2 X 25 ml) and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure and residue was triturated with 70 % ethanol (20 ml). Solid was filtered and dried under vacuo to afford yellow solid (3.1 gm, 66 %). MS = m/z 252 (M+l).
Step-3: Preparation of 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-2-(4-nitro- phenylimino)-thiazolidin-4-one
A solution of 3-Methyl-2-(4-nitro-phenylimino)-thiazolidin-4-one (500 mg, 1.99 mmol), A- (4-Cyclohexyl-benzyloxy)-benzaldehyde (590 mg, 2.0 mmol) and piperidine (0.725 ml, 5.97 mmol) in EtOH (20 ml) was heated under reflux for 14 h. The reaction was monitored by thin layer chromatography. The reaction mixture was cooled to room temperature and solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (50 ml) and washed with water (2 x 10 ml). Organic layer was dried over anhydrous sodium sulfate and evaporated under vacuum. The residue was purified by silica gel column chromatography using ethyl acetate: hexane (15:85) as eluent to give a yellow solid product (1 gm, 95 %).
MS = m/z 528 (M+l).
Step-4: Preparation of 2-(4-Amino-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene] -3 -methyl -thiazolidin-4-one
To a stirred suspension of 5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-2-(4-nitro- phenylimino)-thiazolidin-4-one (1 gm, 1.9 mmol) in acetic acid (20 ml) was added iron powder (318 mg, 5.7 mmol) at room temperature and stirred for -6 h. Solids were removed by filtration and filtrate was concentrated under vacuum. The residue was quenched with saturated sodium bicarbonate solution (50 ml) and extracted with dichloromethane (3x 30 ml). The combined organic layer was washed with water (20 ml), brine (20 ml) and dried over anhydrous sodium sulphate. Solvent was evaporated under vacuo to give red solid product (700 mg, 74 %). MS = m/z 498 (M+l).
Step-5: Preparation of N-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino}-phenyl)-oxalamic acid ethyl ester
To a stirred solution of 2-(4-Amino-phenylimino)-5-[4-(4-cyclohexyl-benzyloxy)- benzylidene]-3-methyl-thiazolidin-4-one (100 mg, 0.20 mmol) and triethyl amine (0.060 ml, 0.40 mmol) in dichloromethane (10 ml) at 0 °C was added and ethyl oxalyl chloride (0.025 ml, 0.22 mmol). The reaction mixture was stirred at room temperature for 1 h and quenched with water (10 ml). Dichloromethane layer was separated, washed with brine (10 ml) and dried over anhydrous sodium sulfate. The solvent was removed under vacuo to afford a yellow solid product (120 mg, 99 %). MS - m/z 596 (M-I).
Step-6: Preparation of N-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino } -phenyl)-oxalamic acid
To a stirred solution of N-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino}-phenyl)-oxalamic acid ethyl ester (120 mg, 0.20 mmol) in tetrahydrofuran (5 ml) and MeOH (5 ml) was added aqueous solution of lithium hydroxide
monohydrate (17 mg, 0.40 mmol) and stirred at room temperature for 3 h. The solvent was removed under reduced pressure and residue was acidified with 2N HCl (pH 4). Solid was filtered, washed with water (10 ml) and dried under reduced pressure. The solid was triturated with ethanol (5 ml) and filtered to give a yellow solid product (100 mg, 88 %). MS = m/z 568 (M-I). Melting Point: 320-323 0C
1H NMR (DMSOd6) δ: 1.16 - 1.43 (m, 5H), 1.66 - 1.74 (m, 5H), 2.40 - 2.49 (m, IH), 3.3 (s, 3H), 5.07 (s, 2H), 6.96 - 6.98 (d, 2H), 7.10 - 7.13 (d, 2H), 7.20 - 7.22 (d, 2H), 7.32 - 7.34 (d, 2H), 7.47 - 7.50 (d, 2H), 7.69 (s, IH), 7.81 - 7.83 (d, 2H), 10.40 (s, IH).
Example 15
Preparation of 5-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzyIidene]-3-methyl-4-oxo- thiazolidm-2-ylideneamino}-phenyl)-l,l-dioxo-iDD-[l,2,5]thiadiazoIidin-3-one (Compound 17)

Step-1 : Preparation of 2-(4-Amino-phenylimino)-3-methyl-thiazolidin-4-one To a stirred suspension of 3-Methyl-2-(4-nitro-phenylimino)-thiazolidin-4-one(prepared as per step 2 of example 14; 3.2 gm, 12.74 mmol) in acetic acid (30 ml) was added iron powder (2.1 gm, 37.5 mmol) at room temperature and stirred for 5 h. Inorganics were removed by filtration and filtrate was concentrated under vacuo. Residue was quenched with saturated sodium bicarbonate solution (50 ml) and extracted with dichloromethane (3x 25 ml). The combined organic layer was washed with brine (25 ml), dried over anhydrous sodium sulphate and evaporated under vacuo to give red solid (2.7 gm, 96 %). MS = m/z 222 (M+ 1).
Step-2: Preparation of [4-(3 -Methyl -4-oxo-thiazolidin-2-ylideneamino)-phenylamino] -acetic acid ethyl ester
To a stirred solution of 2-(4-Amino-phenylimino)-3 -methyl -thiazolidin-4-one (2.5 gm, 11.31 mmol) and potassium carbonate (3.1 g, 22.46 mmol) in N, N-dimethylformamide (20 ml) at room temperature was added ethylbromo acetate (1.3 ml, 12.30 mmol) and was heated to 60 0C for 1 hour. The reaction mixture was quenched with water (50 ml) and extracted in ethyl acetate (2 x 25 ml). The combined organic layer was washed with brine (2 x 25 ml) and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure and residue was purified by silica gel column chromatography using ethyl acetate: Hexane (15:85) as eluent to afford a brown solid (2.0 gm, 59 %). MS = m/z 308 (M+l).
Step-3: [(4-{[3-methyl-4-oxo-l,3-thiazolidin-2-ylidene]amino}phenyl)(tert-butoxycarbonyl- sulfamoyl)amino] aceticacid ethylester
To a stirred solution of t-butanol (0.150 ml, 2.11 mmol) in dichloromethane (15 ml) at 0 0C was added chlorosulfonylisocyanate (0.172 ml, 1.96 mmol) and stirred for 15 minute. This mixture was added to the previously stirred solution of [4-(3-Methyl-4-oxo-thiazolidin-2- ylideneamino)-phenylamino]-acetic acid ethyl ester (500 mg, 1.63 mmol)) and triethylamine (0.704 ml, 4.88 mmol) in dichloromethane (15 ml) at 0 0C. The reaction mixture was stirred for additional 1 h. Reaction mixture was quenched with water (30 ml), organic layer was separated and washed with brine (2 x 15 ml). Organic layer was dried over anhydrous sodium sulfate and evaporated under reduced pressure to give a yellow solid product (750 mg, 95 %). MS = m/z 485 (M-I).
Step-4 : [(4- { [3 -methyl-4-oxo- 1 ,3 -thiazolidin-2-ylidene] aminophenyl)(sulfamoyl) amino]aceticacid ethylester
To a stirred solution of [(4-{[3-methyl-4-oxo-l,3-thiazolidin-2-ylidene]amino} phenyl)(tert- butoxycarbonyl-sulfamoyl)amino]aceticacid ethylester (750 mg, 1.54 mmol) in dichloromethane (15 ml) at 0 0C was added trifluoroacetic acid (2.2 ml) and stirred at room
temperature for 5 h. The reaction was monitored by thin layer chromatography. The reaction mixture was diluted with DCM (50 ml) and quenched with saturated sodium bicarbonate solution (50 ml). Organic layer was separated, washed with brine (2 x 15 ml) and dried over anhydrous sodium sulfate. The solvent was removed under vacuo and residue was triturated with diethyl ether (15 ml) to give yellow solid product (550 mg, 93 %). MS = m/z 385 (M-I):
Step 5: Preparation of 5-[4-(3-Methyl-4-oxo-thiazolidin-2-ylideneamino)-phenyl]-l,l-dioxo- 1 λ6- [ 1 ,2,5]thiadiazolidin-3 -one
To a stirred solution of ethyl [(4-{[(2)-3-methyl-4-oxo-l,3-thiazolidin-2-ylidene]amino} phenyl)sulfamoyl)amino] acetate (160 mg, 0.41 mmol) in ethanol (10 ml) was added sodium hydroxide (33 mg, 0.83 mmol) dissolved in water (0.5 ml) and stirred for 1 h. The reaction mixture was evaporated and the residue was dissolved in ethyl acetate (50 ml). Ethyl acetate layer was washed with 2N HCl (10 ml), water (10 ml) and dried over sodium sulphate. Solvent was evaporated under vacuo and residue was purified by silica gel column chromatography using Ethyl acetate: Hexane (75:25) as eluent to afford off white solid (80 mg, 57 %). MS = m/z 339 (M-I)
Step 6: Preparation of 5-(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo- thiazolidm-2-ylideneamino} -phenyl)- 1 ,1 -dioxo-1 λ6-[l ,2,5]thiadiazolidin-3-one A mixture of 5-[4-(3-Methyl-4-oxo-thiazolidin-2-ylideneamino)-phenyl]-l,l-dioxo-lλ6- [l,2,5]thiadiazolidin-3-one (80 mg, 0.24 mmol), 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde (69 mg, 0.23 mmol) and piperidine (0.056 ml, 0.46 mmol) in EtOH (15 ml) was heated under reflux for 10 h. The reaction was monitored by thin layer chromatography. The reaction mixture was cool to room temperature and solvent was removed under reduced pressure. The residue was dissolved in ethyl acetate (30 ml) and washed with water (2 x 15 ml). Organic layer was dried over anhydrous sodium sulfate and evaporated under vacuo. The residue was purified by silica gel column chromatography using Ethyl acetate: Hexane (65:35) as eluent to afford yellow solid product (45 mg, 31 %).
MS = m/z 615 (M-I).
Melting Point: 245-248 0C.
1H NMR (DMSO-d6) δ: 1.16 - 1.35 (m, 5H)3 1.62 - 1.71 (m, 5H), 2.35 - 2.48 (m, IH), 3.28
(s, 3H), 4.10 (s, 2H), 5.04 (s, 2H), 7.00 - 7.98 (d, 2H)3 7.12 - 7.14 (d, 4H)3 7.20 - 7.22 (d, 2H),
7.32 - 7.34 (d, 2H), 7.48 - 7.50 (d, 2H), 7.69 (s, IH).
Example 16
Preparation of 5-(4- {5- [4-(4-Isopropyl-benzyloxy)-benzylidene] -3-methyl-4-oxo- thiazolidin-2-ylideneamino} -phenyl)-l,l-dioxo -lλ6- [1,2,5] thiadiazolidin-3- one (Compound 37)

Analogously, by practicing the chemistry as described in example 15 for compound 17, by substitution of 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde with 4-(4-Isopropyl-benzyloxy)- benzaldehyde with appropriate change in the reactants and reaction conditions, 5-(4-{5-[4-(4-
Isopropyl-benzyloxy)-benzylidene]- 3-methyl-4- oxo- thiazolidin-2- ylideneamino} -phenyl)-
1,1-dioxo -lλ6- [1,2,5] thiadiazolidin-3- one was prepared.
Melting Point: 222-224 0C
MS: m/z 575 (M-I)
1H NMR (DMSO-d6) δ: 1.17 - 1.19 (d, 6H), 2.85 - 2.87 (m, IH), 3.32 (s, 3H), 4.46
(s, 2H), 5.09 (s, 2H), 7.03 - 7.05 (d, 2H), 7.12 - 7.14 (d, 2H), 7.18 - 7.20 (d, 2H)3
7.24 - 7.26 (d, 2H), 7.34 - 7.36 (d, 2H), 748 - 7.50 (d, 2H), 7.70 (s, IH).
Example 17 Preparation of 4-(4-Fluoro-benzyloxy)-benzaldehyde

To a stirred solution of 4-hydroxy benzaldehyde (2g, 16.39 mmol) in N, N- dimethylformamide (20 ml) at room temperature was added potassium carbonate (3.4 g, 24.59 mmol) and p-fluorobenzyl bromide (3.1 g, 16.40 mmol). The reaction mixture was heated to 60 °C for 2 h. The reaction mixture was cooled to room temperature, poured in to cold water (50 ml) and precipitated solid was filtered, washed with water (100 ml) and dried under vacuum to afford off white solid (3.70 g, 98 %).
1H NMR (CDCl3) δ: 5.11 (s, 2H), 7.06 - 7.11 (m, 4H), 7.39 - 7.43 (dd, 2H), 7.83 - 7.85 (d, 2H), 9.89 (s, IH).
Example 18 Preparation of 4-(4-cycIohexyl-benzyloxy)-benzaldehyde:

Step-1: 4-cyclohehyl benzoic acid ethyl ester:
To a stirred solution of 4-cyclohehyl benzoic acid (10 g, 49.00 mmol) in ethanol (100 ml) was added sulfuric acid (2 ml) and heated under reflux temperature for 5 h. The reaction mixture was concentrated under vacuo and quenched with saturated sodium bicarbonate solution (50ml). The precipitated solid was filtered, washed with water (25 ml) and dried under reduced pressure to afford white solid (11. 30 g, 99 %).
Step-2: (4-Cyclohexyl-phenyl)-methanol
To a stirred suspension of lithium aluminium hydride (980 mg, 25.86 mmol) in dry tetrahydrofuran (25 ml) at 0 °C was added 4-cyclohehyl benzoic acid ethyl ester (2 g, 8.62 mmol) in tetrahydrofuran (10 ml) via addition funnel. The reaction was continued for another 2 h. The reaction mixture was quenched at 0 °C by addition of ethyl acetate (5 ml) and then acidified with 2N HCl (30 ml). It was extracted in ethyl acetate (3x25 ml). The combined organic layer was washed with water (25 ml) and brine (25 ml) and evaporated under vacuo to afford color less oil (1.60 g, 98 %).
Step-3: 1 -Chloromethyl-4-cyclohexyl -benzene
To a stirred solution of (4-Cyclohexyl-phenyl)-methanol (1.6 g, 8.25 mmol) in dichloromethane (25 ml) at 0 0C was added thionyl chloride (0.800 ml, 10.72 mmol). The reaction was continued for 30 min. The reaction mixture was neutralized with saturated sodium bicarbonate solution (50 ml). The organic layer was separated, washed with water (20 ml) and brine (15 ml). It was dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (1.65 g, 97 %).
Step-4 : 4-(4-Cyclohexyl-benzyloxy)-benzaldehyde
To a stirred solution of 4-hydroxybenzaldehyde (970 mg, 7.95 mmol) in N5N- dimethylformamide (20 ml) at room temperature was added potassium carbonate (2.20 g,
15.90 mmol) and l-Chloromethyl-4-cyclohexyl-benzene(1.65 g, 7.93 mmol). The reaction mixture was heated to 60 °C for 2 h . Reaction was monitored by thin layer chromatography.
The reaction mixture was cooled to room temperature and then poured in to cold water (40 ml). The precipitated solid was filtered and washed with water (20 ml) to afford off white solid product (2.30 g, 98 %).
1H NMR (CDCl3) δ: 1.36 - 1.45 (m, 5H), 1.60 - 1.95 (m, 5H), 2.51 (m, IH), 5.10 (s, 2H),
7.06 - 7.10 (d, 2H), 7.22 - 7.26 (d, 2H), 7.34 - 7.38 (d, 2H), 7.82 - 7.86 (d, 2H), 9.88 (s, IH).
Example 19 Preparation of 4-[4-(l-Ethyl-propoxy)-benzyloxy]-benzaldehyde :

Step-1: 4-(l-Ethyl-propoxy)-benzaldehyde :
To a stiiTed solution of 4-hydroxybenzaldehyde (1 g, 8.19 mmol) in N, N- dimethylformamide (15 ml) at room temperature was added potassium carbonate (2.26 g,
16.39 mmol) and 3-bromopentane (1.2 gm, 9.00 mmol). The reaction mixture was heated to
60 0C for 8 h . The reaction mixture was cooled to room temperature.
The reaction mixture was poured into cold water (40 ml). The organic material was extracted in ethyl acetate (2x 25 ml). The combined organic layer was washed with water (30 ml) and brine (25 ml), dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (1.5 g, 96 %).
Step-2 : 4-( 1 -Ethyl-propoxy)-phenyl] -methanol
To a stirred solution of 4-(l-Ethyl-propoxy)-benzaldehyde (1.4 g, 7.29 mmol) in EtOH (25 ml) at 0 0C was added sodium borohydride (360 mg, 9.47 mmol). The reaction was continued for 30 min. and monitored by thin layer chromatography. The reaction mixture was concentrated, acidified by addition of 2N HCl to pH 2. It was extracted with dichloromethane (2x 25 ml). The combined organic layer was washed with brine (25 ml), dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (1.4 g, 99 %).
Step-3 : 1 -Chloromethyl-4-( 1 -ethyl-propoxy)-benzene
To a stirred solution of 4-(l-Ethyl-propoxy)-phenyl] -methanol (1.35 g, 6.95 mmol) in dichloromethane (30 ml) at 0 0C was added thionyl chloride (0.670 ml, 9.40 mmol). The reaction was continued for 30 min. The reaction mixture was neutralized with saturated sodium bicarbonate solution (20 ml). The organic layer was separated, washed with water
(30 ml) and brine (20 ml). It was dried over anhydrous sodium sulfate and concentrated under vacuo to afford product as colorless oil (1.45 ,g, 98 %).
Step-4 : 4- [4-( 1 -Ethyl-propoxy)-benzyloxy] -benzaldehyde
To a stirred solution of 4-hydroxybenzaldehyde (805 mg, 6.60 mmol) in N, N- dimethylformamide (25 ml) at room temperature was added potassium carbonate (1.36 gm,
9.90 mmol) and chloromethyl-4-(l-ethyl-propoxy)-benzene(1.40 g, 6.60 mmol). It was heated to 60 °C for 2 h . The reaction mixture was allowed to come at room temperature and then poured in to ice water (50 ml). It was extracted in ethyl acetate(2x 25 ml). The combined organic layer was washed with water (30 ml) and brine (15 ml), dried over anhydrous sodium sulfate and concentrated under vacuo to afford pale yellow oil (1.90 g, 97
%).
1H NMR (CDCl3) δ: 0.92 - 0.99 (t, 6H), 1.62 - 1.75 (m, 4H), 4.07 - 4.18 (m, IH), 5.06 (s,
2H), 6.90-6.94 (d, 2H), 7.05 - 7.09 (d, 2H), 7.31 - 7.35 (d, 2H),7.82 - 7.86 (d, 2H) 9.88 (s, 1).
Example 20 Preparation of 4- [Methyl- (4-methyl-benzyl)-amino]-benzaldehyde :

Step-1: 4-methylbenzyl alcohol
To a stirred solution of p-tolualdehyde (3 g, 25 mmol) in EtOH (20 ml) at 0 °C was added sodium borohydride (1.33 g, 35 mmol). The reaction was continued for 30 min. The reaction mixture was concentrated, acidified by careful addition of 2N HCl to pH 2. It was extracted with dichloromethane (2x 20 ml). The combined organic layer was washed with brine (25 ml), dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (3 g, 98 %).
Step-2: 4-methylbenzyl chloride
To a stirred solution, of 4-methylbenzyl alcohol (3 g, 24.59 mmol) in dichloromethane (30 ml) at 0 °C was added thionyl chloride (2.0 ml, 27.05 mmol). The reaction was continued for 30 min. It was monitored by thin layer chromatography. The reaction mixture was quenched with saturated sodium bicarbonate solution (20 ml). The organic layer was separated, washed with water (20 ml) and brine (20 ml). It was dried over anhydrous sodium sulfate and concentrated under vacuo to afford colorless oil (3.3 g, 97%).
Step-3: methyl-(4-methyl-benzyl)-amine
To a stirred solution of 4-methylbenzyl chloride (2.5 g, 17.85 mmol) in EtOH (15 ml) was added 40 % methylamine solution (5 ml) and stirred for 4 h . It was monitored by thin layer chromatography. The reaction mixture was evaporated, quenched with water (30 ml) and extracted in dichloromethane (2x 25 ml). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuo to afford yellow oil (2.1 g, 87%).
Step-4: 4-[Methyl-(4-methyl-benzyl)-amino]-benzaldehyde
To a stirred solution of methyl-(4-methyl-benzyl)-amine (2 g, 14.81 mmol) in N5N- dimethylformamide (20 ml) at room temperature was added potassium carbonate (6.7 g, 48.55 mmol) and 4-Fluorobenzaldehyde (1.83 g, 14.81 mmol). The reaction mixture was heated to 70 0C for 1O h. The reaction was monitored by thin layer chromatography. The reaction mixture was poured in to cold water and extracted in ethyl acetate (2x 25 ml). The combined organic layer was washed with water (25 ml) and brine (25 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum. The crude material was purified by silica gel column EtOAc: Hexane (5: 95) to afford yellow oil (1.8 gm, 51 %). 1HNMR (CDCl3) δ: 2.33 (s, 3H), 3.12 (s, 3H), 4.62 (s, 2H), 6.75 - 6.77 (d, 2H), 7.06 - 7.08 (d, 2H), 7.24 - 7.26 (d, 2H), 7.71 - 7.73 (d, 2H), 9.73 (s, IH).
Example 21 Preparation of 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzaldehyde:

Step 1: 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzoic acid ethyl ester To a stirred solution of 1,2,3,4-tetrahydroisoquinoline (1 g, 7.5 mmol) in dry DMF (30 ml), potassium carbonate (1.55g, 11.27 mmol) and 4-bromomethyl ethyl benzoate (1.8 g, 8.2 mmol) were added and reaction mixture was heated to 80 °C for 7 h. The reaction mixture was cooled to room temperature and then water (40 ml) was added and extracted with ethyl acetate (2x25 ml). The combined extracts was dried over anhydrous sodium sulfate and concentrated under vacuo. Product was purified by silica gel column using 2% ethyl acetate in hexane as a eluent to afford brown liquid (1.6 g, 72.72 %)
Step 2: [4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-phenyl]-methanol To a stirred suspension of lithium aluminium hydride (0.25 g, 6.7 mmol) in dry THF (30 ml) was added 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzoic acid ethyl ester (Ig, 3.5 mmol) dissolved in tetrahydrofuran (15 ml) at O 0C under nitrogen atmosphere and stirred for 30 min. The reaction was quenched by slow addition of cold water (15 ml). The solid precipitated was removed by filtration and the product was extracted by ethyl acetate (3x20 ml). The combined organic layer was washed with brine (10 ml), dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain the product as a brown oil (0.76 g, 89 %).
Step 3: 4-(3,4-Dihydro-lH-isoquinolin-2-ylmethyl)-benzaldehyde
To a solution of oxalyl chloride (0.52g, 4.1 mmol) in dry dichloromethane (15 ml) at -78 °C was added dry dimethylsulfoxide (0.53g, 6.9 mmol) dissolved in dry dichloromethane (10 ml) under nitrogen atmosphere. After stirring for 20 min, [4-(3,4-Dihydro-lH-isoquinolin-2- ylmethyl)-phenyl] -methanol (0.7 g, 2.7 mmol) dissolved in dry dichloromethane (15 ml) was
added. The reaction was continued for Ih at -78 °C and then warmed to -60 to -65 °C. Triethyl amine (1.95 g, 19.3 mmol) was added to the above reaction mixture and slowly warmed to room temperature. The reaction mixture was then poured into water (25 ml) and diluted with dichloromethane (25 ml). The organic layer was separated, washed with brine (15 ml), dried over anhydrous sodium sulfate and concentrated under reduced pressure. The product was purified by silica gel column chromatography using 10 % solution of ethyl acetate in hexane as the eluent to afford the product as colorless oil (0.54 g, 77 %). 1H NMR (CDCl3) δ: 2.74 - 2.80 (t, 2H), 2.89 - 2.95 (t, 2H), 3.56 (s, 2H), 3.77 (s, 2H), 6.97 - 6.99 (m, IH), 7.12 - 7.13 (d, 3H), 7.56 - 7.60 (d, 2H), 7.88 - 7.98 (d, 2H)5 10.08 (s, IH).
Example 22 Preparation of 3-FIuoro-4-[(4-isopropyl-benzyl)-methyl-amino]-benzaldehyde

Step-1: Preparation of (4-Isopropyl-benzyl)-methyl-amine
To a stirred solution of 4-isopropylbenzyl chloride (2.0 gm, 1 1.90 mmol) in ethanol (15 ml) was added 40 % methylamine solution (4 ml) and stirred for 4 h. It was monitored by thin layer chromatography. The reaction mixture was evaporated and the residue was dissolved in dichloromethane (30 ml). The solution was washed with water(25 ml), dried over anhydrous sodium sulfate and concentrated under vacuo. The crude was treated with hexane (20 ml) and filtered to afford white solid (1.80 gm, 92 %).
Step-2: 4-[Methyl-(4-isopropyl-benzyl)-amino]-benzaldehyde
To a stirred solution of (4-Isopropyl-benzyl)-methyl-amine (1.5 g, 9.2 mmol) in N3N- dimethylformamide (20 ml) at room temperature was added potassium carbonate (2.5 g, 18.11 mmol) and 4-Fluorobenzaldehyde (1.43 gm, 10.10 mmol). The reaction mixture was heated at 70 0C for 12 h. The reaction mixture was poured in to cold water(25 ml) and
extracted in ethyl acetate (2x 25 ml). The combined organic layer was washed with brine (25 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under vacuo. The residue was purified by silica gel column chromatography using ethyl acetate and Hexane (5:95) as eluent to afford yellow oil (2.1 g, 79 %).
1H NMR (CDCl3) δ: 1.22 - 1.26 (d, 2H), 2.83 - 2.93 (m, IH), 2.98 (s, 3 H), 4.53 (s, 2H), 6.81 - 6.95 (m, IH), 7.18 - 7.28 (m, 4H), 7.49 - 7.56 (m, 2H), 9.75 (s, IH).
Example 23 Preparation of 4-(3-FIuoro-4-piperidin-l-yl-benzyloxy)-benzaIdehyde

Step 1: Synthesis of 3-Fiuoro-4-piperidin-l-yl-benzaldehyde
A solution of 3,4-Difluoro benzaldehyde (5 g, 35.2 mmol), pieridine (4.1 ml, 42.2 mmol) and potassium carbonate (14.5 g, 105.0 mmol) in dimethylsulfoxide (50 ml) was stirred at 60 °C for 8 h. Reaction mixture was cooled to room temperature, diluted with ethyl acetate (200 ml) and washed with water (2 x 100 ml). Ethyl acetate layer was dried on sodium sulphate and evaporated under vacuum to give brown oil (6.0 g, 83.3 %).
MS = m/z 208 (M+l).
1H NMR (DMSO-d6) δ:1.61 - 1.64 (m, 2H), 1.0 - 1.76 (m, 4H), 3.20 - 3.23 (m, 4H), 6.94 -
6.98 (d, IH), 7.40 - 7.50 (m, 2H), 9.79 (s, IH).
Step 2: Synthesis of (3-Fluoro-4-piperidin-l-yl-phenyl)-methanol
To a stirred solution of 3-Fluoro-4-piperidin-l-yl-benzaldehyde (5.5 g, 24.1 mmol) in methanol (60 ml), sodium borohydride (1.1 g, 28.9 mmol) was added portion wise at 10 °C. Reaction mixture was stirred at room temperature for 1 h. and was quenched by addition of 10 % hydrochloric acid (10 ml) and extracted with ethyl acetate(100 ml). Ethyl acetate layer was washed with water (20 ml.), dried on sodium sulphate and evaporated to give colorless liquid (4.0 g, 90.0 %).
MS = m/z 210 (M+l).
Step 3: Synthesis of 4-(3-Fluoro-4-piperidin-l-yl-benzyloxy)-benzaldehyde To a stirred mixture of 60% suspension of sodium hydride (1.0 g, 25.1 mmol) in N5N- dimethylformamide (30 ml), a solution of (3-Fluoro-4-piρeridin-l-yl-phenyl)-methanol (3.5 g, 16.7 mmol) in dimethylformamide (10 ml) was added at room temperature and stirred at 60 0C for 30 min. 4-Fluoro benzaldehyde (2.2 g, 18.4 mmol) in dimethylformamide (10 ml) was added to above stirred suspension at 60 0C and stirred for 5 h. Reaction mixture was diluted with ethyl acetate (100 ml) and washed with water (2x 50 ml). Ethyl acetate layer was dried on sodium sulphate and evaporated under vacuum to give residue as oil. Residue was purified by silica gel column using 10 % ethyl acetate in hexane as eluent to give colorless oil (2.6 g, 50.6 %). MS = m/z 314 (M+l).
1H NMR (DMSOdO) δ: 1.57 - 1.59 (m, 2H), 1.63 - 1.76 (m, 4H), 3.00 - 3.08 (m, 4H), 5.04 - 5.10 (d, 2H), 6.95 - 7.04 (m, 5H), 7.80 - 7.86 (m, 2H), 9.88 (s, IH).
Example 24 Preparation of 4-(3,4-Dihydro-lH-isoquinoIin-2-yl)-benzaldehyde

To a stirred solution of 4-fluorobenzaldehyde (1 gm, 8 mmol) in dimethylsulfoxide (10 ml) at room temperature was added potassium carbonate (1.78 gm, 13 mmole) and tetrahydroisoquinoline (1.18 gm, 8.8 mmole). The reaction mixture was heated at 80 0C for two hours. The reaction mixture was poured into cold water after completion of reaction. The mixture was extracted in ethyl acetate (2 x 25 ml). The combined organic layer was washed with water (25 ml) and brine (25 ml.). The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The crude material was purified by flash chromatography using ethyl acetate (0.2): Hexane (9.8) as an eluant to afford white solid (1.5 gm. 78%). MS = m/z 238 (M+l).
1H NMR (CDCl3) δ; 2.98 - 3.04 (t, 2H), 3.66 - 3.72 (t, 2H), 4.56 (s, 2H), 6.90 - 6.94 (d, 2H), 7.11 - 7.26 (m, 4H), 7.76 - 7.91 (d, 2H), 9.77 (s, IH).
Example 25 Preparation of 4-[l-(4-IsobutyI-phenyl)-ethoxy]-benzaldehyde

Step-1: Preparation of l-(4-Isobutyl-phenyl)-ethanone
To a stirred solution of Isobutyl benzene (5 g, 37.31 mmol) and acetyl chloride (2.9 ml, 41.04 mmol) in carbon disulphide (40 ml) at room temperature was added anhydrous AlCl3 (5.95 g, 44.77 mmol) and stirred at 45 0C for 3 h. Reaction mixture was cooled to room temperature, poured into ice water (100 ml) under stirring and extracted in DCM (2x 50 ml). Combined DCM layer was washed with 10 % HCl (50' ml), water (50 ml) and dried under anhydrous sodium sulphate. Solvent was evaporated under vacuum to give reddish oil. (6.0 g, 93.0 %)
Step-2: Preparation of l-(4-Isobutyl-phenyl)-ethanol
To a stirred solution bf l-(4-Isobutyl-phenyl)-ethanone (2.7 g, 15.3 mmol) in MeOH (30 ml) at 0 0C was added sodium borohydride (700 mg, 18.40 mmol) and stirred for 30 min at room temperature. The reaction mixture was concentrated under vacuum and residue was acidified by addition of 2N HCl to pH 2. Reaction mixture was extracted with ethyl acetate (2x40 ml). The combined organic layer was washed with brine (25 ml), dried over anhydrous sodium sulfate and concentrated under vacuum to afford colorless oil (2.45 g, 88.0 %).
Step-3: Prepration Methanesulfonic acid l-(4-isobutyl-phenyl)-ethyl ester To a stirred solution of l-(4-Isobutyl-phenyl)-ethanol (2.4 g, 13.48 mmol) and triethylamine (3.8, 26.46 mmol) in DCM (30 ml) at room temperature was added methansulphonyl chloride (1.15 ml, 14.83 mmol). Reaction mixture was stirred at room temperature for 4 h. Reaction mixture was diluted by DCM (50 ml) and poured in to ice water (40 ml). DCM layer was
separated, washed with water (30 ml) and dried over anhydrous sodium sulfate. DCM was evaporated under vacuum to afford pale yellow oil (3.2 gm, 90.0 %).
Step-4 : Preparation of 4- [ 1 -(4-Isobutyl-phenyl)-ethoxy] -benzaldehyde To a stirred solution of 4-hydroxy benzaldehyde (1.47 g, 12.10 mmol) and potassium carbonate (2.5 g, 18.11 mmol) in N,N-dimethylformamide ( 20 ml) was added methanesulfonic acid l-(4-isobutyl-phenyl)-ethyl ester (3.1 g, 12.10 mmol) and reaction mixture was stirred at 60 0C for 5 h. Reaction mixture was poured in to ice water ( 170 ml) and extracted in ethyl acetate (2x 40 ml). The combined organic layer was washed with water (40 ml), dried over anhydrous sodium sulfate and concentrated under vacuum to residue. Residue was purified by silica gel column chromatography using 10 % ethyl acetate in hexane as eluent to give off-white solid. (2.2 g, 66.0 %). MS = m/z 283 (M+l)
1H NMR (CDCl3) δ: 0.88 - 0.89 (d, 6H)5 1.70 - 1.72 (d, 3H), 1.80 - 1.87 (m, IH), 2.43 - 2.46 (d, 3H), 5.36 - 5.41 (m, IH), 6.95 - 6.97 (d, 2H), 7.10 - 7.12 (d, 2H), 7.24 - 7.26 (, 2H), 7.72 - 7.74 (d, 2H), 9.82 (s, IH)
Example 26 Preparation of 4-[l-(4-Isobutyl-phenyI)-butoxy]-benzaIdehyde

Analogously, by practicing the chemistry described in example 25 for 4-[l-(4-Isobutyl- phenyl)-ethoxy] -benzaldehyde, by substitution of acetyl chloride in step 1 with butyryl
chloride with appropriate change in the reactants and reaction conditions, 4-[l-(4-Isobutyl- phenyl)-butoxy]-benzaldehyde was prepared.
MS = m/z 311 (MH-I)
1H NMR (CDCl3) δ: 0.85 - 0.91 (m, 9H), 0.94 - 0.98 (m, 2H), 1.41 - 1.46 (m, 2H), 1.74 -
1.79 (m, IH), 2.41 - 2.44 (d, 2H), 5.13 - 5.19 (m, IH), 6.91 - 6.95 (d, 2H), 7.07 - 7.11 (d,
2H), 7.24 - 7.25 (d, 2H), 7.69 - 7.74 (d, 2H), 9.80 (s, IH).
ExampIe-27: Preparation of compound of formula 'v'
Analogously, by practicing the chemistry described in Examples 17 to 26 with appropriate change in the reactants and reaction conditions, following aldehyde compounds of formula 'v' were prepared.
4-[4-(l-Methyl-butoxy)-benzyloxy]-benzaldehyde: MS = m/z 299 [M+l]. 4-(4-Formyl-phenoxymethyl)-benzonitrile. MS = m/z 238 [M+l]. 4-(4-Cyclohexyl-benzyloxy)-3-methoxy-benzaldehyde. MS = m/z 325 [M+l]. 4-(4-isopropyl-benzyloxy)-benzaldehyde. MS = m/z 255 [M+l]. 4-(4-isobutyl-benzyloxy)-benzaldehyde. MS = m/z 269 [M+l]. 4-(4-Cyclohexyl-phenyl)-ethoxy]-benzaldehyde. MS = m/z 309 [M+l]. 4-(4-Ethyl-benzyloxy)-benzaldehyde. MS = m/z 241 [M+l]. 4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzaldehyde. MS = m/z 308 [M+l]. 4-(Benzyl-methyl-amino)-benzaldehyde. MS = m/z 226 [M+l]. 4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-fluoro-benzaldehyde. MS = m/z 326 [M+l]. 4-(4-nitro-benzyloxy)-benzaldehyde. MS = m/z 258 [M+l]. 4-(4-Cyclohexyl-benzyloxy)-3-flouro-benzaldehyde. MS = m/z 313 [M+l]. 4-(4-Cyclohexyloxy-benzyloxy)-benzaldehyde. MS = m/z 311 [M+l]. 4-(4-Pentyl-benzyloxy)-benzaldehyde. MS = m/z 283 [M+l].
Demonstration of In Vitro Efficacy of Test Compounds A. Inhibition of human recombinant PTPlB
The phosphatase activity of human recombinant PTPlB was determined by following a previously described procedure {Methods 35, 2-8, 2005), but with certain modifications. The principle of the assay is based on the hydrolysis of 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) and the fluorometric quantitation of the liberated difluoromethylumbelliferone (DiFMU) .
Assays were routinely earned out in 96-well flat-bottom black microwell plates. The reaction mixture (100 μl) contained 15 ng/well of human recombinant PTPlB enzyme (produced in- house or procured from R&D Systems, USA) in the assay buffer (50 mM Hepes, pH 7.2, 50 mM NaCl, 1 mM EDTA, 1 mM DDT and 0.01 % Triton X-100) and 25 μM DiFMUP. After incubation of assay plates at 300C for 5 min, the hydrolysis of DiFMUP was monitored in a fluorescence microplate reader (SpectraMax M5 Molecular Devices, USA), with excitation and emission wavelengths set at 358 nm and 450 nm, respectively.
The inhibition of PTPlB activity by test compounds was routinely assessed by preincubating the enzyme with test compound (0.1 and 1 μM for primary screening and 7 concentrations from 0.01 to 10 μM for the dose-response study) or vehicle (1 % DMSO) for 10 min at 300C, in a total volume of 90 μl. Test compounds were dissolved in DMSO at a concentration of 10 mM and suitably diluted further in assay buffer. The enzyme reaction was initiated by the addition of DiFMUP, followed by incubation of assay plates for 5 min at 300C and the liberated product was measured as described above. A known inhibitor of PTPlB (positive control) was always included in the assay. Test compounds at various concentrations were always evaluated in duplicate, along with substrate blanks, vehicle controls and positive controls.
The results are expressed as percent inhibition of the enzyme activity relative to vehicle controls. Dose-response studies were conducted for those compounds exerting > 50%
inhibition of activity at 1 μM in primary screening. The inhibition data, expressed as IC5O, the inhibitor concentration that caused 50% decrease of the activity under assay conditions. The IC50 was computed using GraphPad Prism software, version 5.0. The PTPlB inhibition data (expressed as IC50) is presented in Table 1.
Table 1 : Compounds exerting inhibition of human recombinant PTPlB

B. Determination of Selectivity of PTPlB Inhibitors
PTPlB inhibitors with desired potencies were evaluated for their selectivity against the closely related, T-cell protein tyrosine phosphatase (TCPTP), employing assay conditions similar to that used for PTPlB (see above). The reaction mixture (100 μl in 96-well flat- bottom black-well plates) contained assay buffer (50 mM Hepes, pH 7.2, 50 mM NaCl, 1 mM EDTA, 1 mM DDT and 0.01 % Triton X-100), 25 μM substrate (DiFMUP) and 125 mU/well of human recombinant enzyme (procured from New England Biolabs, UK), with or without test compound. Routinely, TCPTP was preincubated with test compound (ranging from 0.01 μM to 10 μM) or vehicle (1 % DMSO) for 10 min at 300C, in a total volume of 90 μl. The reaction was started by the addition of 10 μl substrate (250 μM running stock in assay buffer) and further incubated for 5 min at 300C. The liberated DIFMU was monitored in a fluorescence microplate reader (SpectraMax M5, Molecular Devices, USA), with excitation and emission wavelengths set at 358 nm and 450 run, respectively. IC50 was computed using GraphPad Prism software, version 5.0. The specificity of inhibition of selected PTPlB inhibitors against TCPTP is shown in Table 2.
Table 2: Selectivity of PTP-IB inhibitors against TCPTP

Demonstration of In Vivo Efficacy of Test Compounds A. Evaluation in Diet-Induced Obese (DIO) Mice
Male healthy C57BL/6J mice were randomized into two groups at 4 weeks of age and fed either Lard Control diet (D12450B, 10% kcal from Lard) or High-fat diet (D12451, 45% kcal from Lard) from Research Diets Inc., USA, for 16-20 weeks. Animals were selected on the basis of glucose intolerance to a glucose challenge of 2 g/kg, done at 20-24 weeks of age. Glucose intolerant animals from the high-fat diet group were randomized and grouped on the basis of body weight and plasma triglycerides. Animals were dosed once daily by oral route for 23 days. On day 21st of treatment non-fasted animals were bled from retro-bulbar venous plexus under light anesthesia to evaluate the effect of test compounds on different metabolic parameters in plasma. Oral glucose tolerance test (OGTT) was performed on day 23 in overnight fasted animals using 2 g/kg/po of glucose load. Effect of test compounds on body weight gain was also evaluated against the vehicle treated animals.
Table 3 : Effect of compound 2 in DIO C57EL/6J Mice

Test compounds were evaluated in genetically insulin resistant male db/db mice (8-10 weeks of age) at the dose of 10 or 30 mg/kg. Test compounds were administered once daily by oral route for desired duration of treatment. Non-fasted animals were bled under light anesthesia two days before OGTT. OGTT was performed in overnight fasted animals using a glucose load of 1 g/kg/po. Effect of test compounds on body weight gain was also evaluated relative to the vehicle treated animals.
Table 4: Effect of 2 in db/db mice

ND= Not determined
Claims
1. A compound of the general formula I

(I)
wherein, G1 = (a)

(b) 
G2 is selected from hydrogen, fluoro and methoxy;
G3 is selected from Ci-4 alkyl:
G4 is selected from -CH2COOH5 -NH-C(O)-COOH, -C(O)-COOH, -CH(F)-COOH,

G5 is selected from hydrogen or Fluoro; G6 is selected from -0-, or -N(CH3)-; G7 is selected from hydrogen or Ci-4 alkyl; G8 is unsubstituted phenyl; or phenyl substituted at para position with nitro, fluoro, cyano, piperidinyl, branched or straight chain C1-6 alkoxy, C5-6 cycloalkoxy, branched or straightchain Ci-6 alkyl, or C5-6 cycloalkyl; 'a' is 0 or 1.
2. A compound and its pharmaceutically acceptable salts as claimed in claim 1 wherein the compound of the general formula (I) is selected from
[4-(3-Methyl-5- {4-[4-(l-methyl-butoxy)-benzyloxy]-benzylidene}-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid;
4- { 5- [4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid;
(4- { 5- [4-(4-Isopropyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
(4- { 5- [4-(4-Isobutyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid;
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid;
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-2-fluoro-phenyl] -acetic acid;
4-{5-[4-(N-Benzyl-methylamino)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid; [4-(5-{4-[l-(4-Isobutyl-phenyl)-ethoxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl]-acetic acid;
(4- { 3 -Methyl-5 - [4-(4-nitro-benzyloxy)-benzylidene]-4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid;
(4-{5-[4-(3,4-Dib.ydro-lH-isoquinolin-2-yl)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
{4- [5 - [ 1 - [4-(4-Fluoro-benzyloxy)-phenyl] -meth-ylidene] -3 -methyl-4~oxo-thiazolidin-2- ylideneamino] -phenyl} -acetic acid;
(4- { 5- [4-(4-Ethyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid;
(4- { 5 - [4-(3 ,4-Dihydro- lH-isoquinolin-2-ylmethyl)-benzylidene] -3 -methyl-4-oxo-thiazolidin- 2-ylideneamino } -phenyl)-acetic acid;
(4- { 3 -Ethyl-5- [4-(4-ethyl-benzyloxy)-benzylidene] -4-oxo-thiazolidin-2-ylideneamino } - phenyl)-acetic acid;
(4-{3-Ethyl-5-[4-(4-isopropyl-benzyloxy)-benzylidene]-4-oxo-thiazolidin-2-ylideneamino}- phenyl)-acetic acid;
(4- { 5- [4-(4-Cyclohexyl-benzyloxy)-3 -methoxy-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
5.(4-{5.[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)- 1 , 1 -dioxo- 1 λ6- [ 1 ,2,5]thiadiazolidin-3 -one; [4-(3-Methyl-5- {4-[methyl- (4-methyl-benzyl)-amino]-benzylidene} -4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid;
(4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino j -2-fluoro-phenyl)-acetic acid;
(2-Fluoro-4-{5-[4-(4-isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
(2-Fluoro-4- { 5- [4-(4-isobutyl-benzyloxy)-benzylidene] -3 -methyl -4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
N-(4- { 5 -[4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -methyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-oxalamic acid;
(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-oxo-acetic acid;
{4-[5-[l-[4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene]-3-methyl-4-oxo-thiazolidin- 2-ylideneamino] -phenyl } -fluoro-acetic acid;
4-{5-[4-(4-Cyclohexyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-oxo-acetic acid;
[4-(5-{4-[(4-Cyclohexyl-benzyl)-methyl-amino]-3-fluoro-benzylidene}-3-methyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl] -acetic acid; (4-{5-[4-(4-Cyclohexyl-benzyloxy)-3-fluoro-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
[4-(5-{4-[4-(l-Ethyl-propoxy)-benzyloxy]-benzylidene}-3-methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid;
(4- { 5 - [4-(4-Cyclohexyl-benzyloxy)-benzylidene] -3 -ethyl-4-oxo-thiazolidin-2- ylideneamino}-phenyl)-acetic acid;
{4- [5 - [ 1 - [4-(4-Cyclohexyl-benzyloxy)-phenyl]-meth-)-ylidene] -3 -methyl-4-oxo-thiazolidin- 2-ylideneamino]-phenyl} -hydroxy-acetic acid;
[4-(5-{3-Fluoro-4-[(4-isopropyl-benzyl)-methyl-amino]-benzylidene}-3-methyl-4-oxo- . thiazolidin-2-ylideneamino)-phenyl] -acetic acid;
Hydroxy- (4-{3-methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene]-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
(4- { 3 -Methyl-4-oxo-5- [4-(4-pentyl-benzyloxy)-benzylidene] -thiazolidin-2-ylideneamino } - phenyl)-acetic acid;
[4-(5- { 4- [ 1 -(4-Cyclohexyl-phenyl)-ethoxy] -benzylidene } -3 -methyl-4-oxo-thiazolidin-2- ylideneamino)-phenyl] -acetic acid;
(4-{5-[4-(4-Cyclohexyloxy-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
(4- { 5 - [4-(4-Cyclohexyloxy-benzyloxy)-benzylidene] -3 -ethyl-4-oxo-thiazolidin-2- ylideneamino } -phenyl)-acetic acid; 5-(4-{5-[4-(4-Isopropyl-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2- ylideneamino] -phenyl)-l,l-dioxo -lλ6- [1,2,5] thiadiazolidin-3- one;
[4-(3-Ethyl-5- {3-fluoro-4-[(4-isoproρyl-benzyl)-methyl-amino]-benzylidene}-4-oxo- thiazolidin-2-ylideneamino)-phenyl] -acetic acid;
[4-(5- { 4- [(4-Cy clohexy l-benzyl)-methyl-amino] -3 -fluoro-benzylidene } -3 -ethyl-4-oxo- thiazolidin-2-ylideneamino)-phenyl]-acetic acid;
(4-{5-[4-(4-Cyano-benzyloxy)-benzylidene]-3-methyl-4-oxo-thiazolidin-2-lideneamino}- phenyl)-acetic acid;
(4- { 5 -[4-(3 -Fluoro-4-piperidin- 1 -yl-benzyloxy)-benzylidene] -3 -methyl-4-oxo~thiazolidin-2- ylideneamino } -phenyl)-acetic acid;
{4-[5-[l-{4-[l-(4-Isobutyl-phenyl)-butoxy]-phenyl}-methylidene]-3-methyl-4-oxo- thiazolidin-2-ylideneamino] -phenyl} -acetic acid.
3. A process for preparation of a compound of formula (I),

wherein, G1 = (a)

(b) 
G2 is selected from hydrogen, fluoro and methoxy;
G3 is selected from Ci-4 alkyl:
G4 is selected from -CH2COOH, -NH-C(O)-COOH, -C(O)-COOH, -CH(F)-COOH, -
CH(OH)-COOH, -CH2COOCH3, -CH2-COOC2H5, -NO2, -C(O)-CH3, -CH(F)-
COOC2H5, -CH(OH)-COOC2H5, or  ;
;
G5 is selected from hydrogen or Fluoro;
G6 is selected from -0-, or -N(CH3)-;
G7 is selected from hydrogen or Ci-4 alkyl;
G8 is unsubstituted phenyl; or phenyl substituted at para position with nitro, fluoro, cyano, piperidinyl, branched or straight chain Ci-6 alkoxy, C5-6 cycloalkoxy, branched or straightchain Ci-6 alkyl, or C5-6 cycloalkyl; 'a' is O or 1; which comprises steps of [A] reaction of compound of formula (i)

G4 = -CH2COOCH3, -CH2-COOC2H55 -NO2, -C(O)-CH3, -CH(F)-COOC2H5, -CH(OH)-

G5 is selected from hydrogen or Fluoro; with compound of formula (ii)
N=C
(») , wherein G3 is selected from Ci-4 alkyl; in a suitable solvent such as ethanol at an elevated temperature to produce thiourea derivative of formula (iii);

[B] reaction of compound of formula (iii) as obtained in [a] with ethyl bromoacetate in presence of a base such as triethyl amine in a suitable solvent such as ethanol to obtain compound of formula (iv);

G4 = -CH2COOCH3, -CH2-COOC2H5, -NO2, -C(O)-CH3, -CH(F)-COOC2H5, -CH(OH)-

G3 is selected from Cj-4 alkyl;
G5 is selected from hydrogen or Fluoro; if G4 is -CH(OH)-COOC2H5, then -CH(OH)-COOC2H5 group is converted to -CHF-
COOC2H5 by treatment with diethyl aminosulfur trifluoride in a suitable solvent such as dichloromethane;
if G4 is nitro, then nitro group is converted to l,l-Dioxo-lλ6-[l,2,5]thiadiazolidin-3-one
group (i.e.  ) by first reducing it to amino group, alkylating amino group with ethyl bromo acetate, treating the compound so obtained with chlorosulfonylisocyanate and tert-butanol in a suitable solvent, deprotecting the compound so obtained in presence of trifluoroacetic acid and further cyclizing the deprotected compound in presence of aqueous sodium hydroxide;
) by first reducing it to amino group, alkylating amino group with ethyl bromo acetate, treating the compound so obtained with chlorosulfonylisocyanate and tert-butanol in a suitable solvent, deprotecting the compound so obtained in presence of trifluoroacetic acid and further cyclizing the deprotected compound in presence of aqueous sodium hydroxide;
[C] condensation of compound of formula (iv) as obtained in [b] with aldehyde of formula I,  wherein, G1 = (a)
wherein, G1 = (a)

(b) 
G2 is selected from hydrogen, fluoro and methoxy;
G6 is selected from -O-, or -N(CH3)-;
G7 is selected from hydrogen or C 1-4 alkyl;
G8 is imsubstituted phenyl; or phenyl substituted at para position with nitro, fluoro, cyano, piperidinyl, branched or straight chain Ci-6 alkoxy, C5-6 cycloalkoxy, branched or straight chain Cj-6 alkyl, or C5-6 cycloalkyl; and 'a' is 0 or 1; in presence of a base such as piperidine in a suitable solvent such as ethanol to obtain compound of formula T;

(g) if G4 is -CH(OH)-COOC2H5, then -CH(OH)-COOC2H5 is hydrolyzed to — CH (OH)-COOH using base like sodium hydroxide or lithium hydroxide;
(h) if G4 is -CHF-COOC2H5, then -CHF-COOC2H5 is hydrolyzed to -CHF- COOH using base like sodium hydroxide or lithium hydroxide;
(i) if G4 is -C(O)-CH3, then -C(O)-CH3 is converted to -C(O)-COOH on treatment with oxidizing agent such as selenium dioxide in pyridine;
(j) if G4 is nitro, then nitro group is converted to -NH-C(O)-COOH, by first reduction to amino group by using suitable reducing agent such as iron in acetic acid, further acylation with ethyl oxalylchloride in presence of a base such as triethyl amine followed by hydrolysis using lithium hydroxide; such that, at each step the product is optionally isolated and purified by standard techniques.
4. A pharmaceutical composition, which comprises a compound of formula I as defined in claim 1 and a pharmaceutically acceptable carrier, diluent, excipient.

5. A pharmaceutical composition as claimed in claim 4, in the form of a tablet, capsule, suspension, powder, syrup, and solution.
6. A method of preventing or treating hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, hyperglycemia, impaired glucose tolerance, obesity, atherosclerosis, insulin resistance or diseases in which the underlying cause is insulin resistance or related diseases, which comprises administering of compound of formula (I) or a pharmaceutical composition as claimed in claims 4 or 5.
7. A method according to claim 6, wherein the disease is type-2 diabetes and insulin resistance, impaired glucose tolerance, dyslipidemia and disorders related to Syndrome X such as hypertension, obesity, eating disorders, hyperlipidemia, atherosclerosis, coronary artery disease, cardiovascular disorders, diseases related to endothelial dysfunction, nephropathy, neuropathy, retinopathy, osteoporosis, polycystic ovary syndrome, pancreatitis, inflammatory bowel diseases, xanthoma or cancer.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN398/KOL/2008 | 2008-03-03 | ||
| IN398KO2008 | 2008-03-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009109998A1 true WO2009109998A1 (en) | 2009-09-11 |
Family
ID=40627391
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2009/000135 Ceased WO2009109998A1 (en) | 2008-03-03 | 2009-03-02 | Novel protein tyrosine phosphatase - ib inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009109998A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012156919A1 (en) | 2011-05-16 | 2012-11-22 | Koninklijke Philips Electronics N.V. | Bio-orthogonal drug activation |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015181336A1 (en) * | 2014-05-28 | 2015-12-03 | Universität Bern | Thiazolidinones as cellular anandamide uptake inhibitors and their use in the treatment of psychiatric or neurological disorders and inflammation, in particular neuroinflammation |
| EP2952188A1 (en) * | 2014-06-03 | 2015-12-09 | Universität Bern | Thiazolidinones as cellular anandamide uptake inhibitors and their use in the treatment of psychiatric or neurological disorders and inflammation, in particular neuroinflammation |
| CN115448892A (en) * | 2022-09-19 | 2022-12-09 | 郑州铁路职业技术学院 | Synthetic method of benzothiadiazole heterocyclic compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005082901A1 (en) * | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Novel chemical compounds |
| WO2008119238A1 (en) * | 2007-03-30 | 2008-10-09 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Substituted five membered heterocycle compounds, preparation method and medical use thereof |
-
2009
- 2009-03-02 WO PCT/IN2009/000135 patent/WO2009109998A1/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005082901A1 (en) * | 2004-02-25 | 2005-09-09 | Smithkline Beecham Corporation | Novel chemical compounds |
| WO2008119238A1 (en) * | 2007-03-30 | 2008-10-09 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Substituted five membered heterocycle compounds, preparation method and medical use thereof |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012156918A1 (en) | 2011-05-16 | 2012-11-22 | Koninklijke Philips Electronics N.V. | Bio-orthogonal drug activation |
| WO2012156919A1 (en) | 2011-05-16 | 2012-11-22 | Koninklijke Philips Electronics N.V. | Bio-orthogonal drug activation |
| EP3973993A1 (en) | 2011-05-16 | 2022-03-30 | Tagworks Pharmaceuticals B.V. | Bio-orthogonal drug activation |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015181336A1 (en) * | 2014-05-28 | 2015-12-03 | Universität Bern | Thiazolidinones as cellular anandamide uptake inhibitors and their use in the treatment of psychiatric or neurological disorders and inflammation, in particular neuroinflammation |
| US10300048B2 (en) | 2014-05-28 | 2019-05-28 | Universitat Bern | Thiazolidinones as cellular anandamide uptake inhibitors and their use in the treatment of psychiatric or neurological disorders and inflammation, in particular neuroinflammation |
| EP2952188A1 (en) * | 2014-06-03 | 2015-12-09 | Universität Bern | Thiazolidinones as cellular anandamide uptake inhibitors and their use in the treatment of psychiatric or neurological disorders and inflammation, in particular neuroinflammation |
| CN115448892A (en) * | 2022-09-19 | 2022-12-09 | 郑州铁路职业技术学院 | Synthetic method of benzothiadiazole heterocyclic compound |
| CN115448892B (en) * | 2022-09-19 | 2023-07-07 | 郑州铁路职业技术学院 | A kind of synthetic method of benzothiadiazole heterocyclic compound |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009109998A1 (en) | Novel protein tyrosine phosphatase - ib inhibitors | |
| JP5632612B2 (en) | Lactam compound or salt thereof and PPAR activator | |
| EP0524781B1 (en) | Therapeutic amides | |
| WO2009109999A1 (en) | Novel protein tyrosine phosphatase - ib inhibitors | |
| JP2025131575A (en) | Modulators of mas-related g-protein receptor x4, and related products and methods | |
| EP1482935B1 (en) | Thiazole and oxazole derivatives which modulate ppar activity | |
| EP1996567A2 (en) | Novel acetyl-coa carboxylase (acc) inhibitors and their use in diabetes, obesity and metabolic syndrome | |
| ZA200109804B (en) | Thiazole and oxazole derivatives and their pharmaceutical use. | |
| SK100099A3 (en) | Thiazole benzenesulfonamides as 'beta'3 agonists for the treatment of diabetes and obesity | |
| HK1003570B (en) | Therapeutic amides | |
| EP2177510A1 (en) | Allosteric protein kinase modulators | |
| NZ554477A (en) | Compounds that modulate PPAR activity and methods for their preparation | |
| WO2008104994A2 (en) | 2,2,2-tri-substituted acetamide derivatives as glucokinase activators, their process and pharmaceutical application | |
| US20090176837A1 (en) | Compounds with activity at retinoic acid receptors | |
| JPWO2002046176A1 (en) | Activator of peroxisome proliferator-activated receptor | |
| EP0439265B1 (en) | Novel 1,3-dicarbonyl compounds and their use | |
| US5116855A (en) | Rhodanine derivatives and pharmaceutical compositions | |
| CN111393372A (en) | A kind of benzimidazole derivative and its preparation method and use | |
| US20110136792A1 (en) | Novel carboxylic acid analogs as glycogen synthase activators | |
| JP3140494B2 (en) | Substituted phenylacetylenes, drugs containing the same, compounds and methods for producing the drugs | |
| JP2005529975A (en) | Amide linker peroxisome proliferator activated receptor modulator | |
| WO2008154023A1 (en) | Novel uses of ppar delta agonists | |
| CZ20033252A3 (en) | Oxazol/ thiazol-derivatives activators of the hppar-alpha receptor | |
| EP2536693A1 (en) | Substituted 2-imidazolidones and analogs | |
| CN109232426A (en) | A kind of N- hydroxyl -5- substitution -1H- pyrazole-3-formamide compound and its preparation method and application |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09716709 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09716709 Country of ref document: EP Kind code of ref document: A1 |