WO2009116882A1 - Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol - Google Patents
Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol Download PDFInfo
- Publication number
- WO2009116882A1 WO2009116882A1 PCT/PT2009/000013 PT2009000013W WO2009116882A1 WO 2009116882 A1 WO2009116882 A1 WO 2009116882A1 PT 2009000013 W PT2009000013 W PT 2009000013W WO 2009116882 A1 WO2009116882 A1 WO 2009116882A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- exhibits
- crystal modification
- polymorph
- following
- peaks
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- This invention relates to novel polymorphs of 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy- pyridin-3-yl)-[l,2,4]oxadiazol-5-yl]-3-nitrobenzene-l,2-diol, an inhibitor of catechol-0- methyltransferase (COMT), to processes for their preparation, and to pharmaceutical compositions containing said novel polymorphs as active pharmaceutical ingredient.
- CCT catechol-0- methyltransferase
- L-DOPA levodopa
- L-DOPA induced clinical improvement declines at the end of each dose cycle, giving rise to the so-called 'wearing-off pattern of motor fluctuations.
- a close relationship between the 'wearing-off phenomenon and accumulation of 3-0MD has been described (Tohgi, H., et al., Neurosci. Letters, 132:19-22, 1992). It has been speculated that this may result from impaired brain penetration of L-DOPA due to competition for the transport system across the BBB with 3-OMD (Reches, A.
- COMT inhibition protects L-DOPA from metabolic breakdown in the periphery through Omethylation, such that with repeated doses of L-DOPA, the mean plasma L-DOPA concentration is raised.
- a significantly greater percentage of the orally administered dose of L-DOPA is able to reach the site of action.
- COMT inhibition serves to increase the bioavailability of L-DOPA and therefore the duration of antiparkinsonian action is prolonged with single doses of L-DOPA (Nutt, J.G., Lancet, 351:1221-1222, 1998).
- 5-[3-(2,5-dichloro-4,6-dimethyl- 1 -oxy-pyridin-3-yl)-[ 1 ,2,4]oxadiazol-5-yl]-3-nitrobenzene- 1,2-diol is a COMT inhibitor exhibiting an exceptionally long duration of action as well as balanced properties of bioactivity, bioavailability and safety. It markedly enhances the bioavailability of L-DOPA, increases the delivery of L-DOPA to the brain and significantly augments the levels of dopamine in the brain over extended periods of time.
- 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol-5-yl]-3- nitrobenzene- 1,2-diol is a promising candidate for treating a subject afflicted by a central or peripheral nervous system disorder, in particular for treating mood disorders, movement disorders such as Parkinson's disease and parkinsonian disorders and restless leg syndrome, gastrointestinal disturbances, oedema formation states and hypertension.
- polymorphism The ability of a substance, for example 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3- yl)-[l,2,4]oxadiazol-5-yl]-3-nitrobenzene-l,2-diol, to exist in more than one crystalline form is defined as polymorphism and these different crystalline forms may be referred to as "polymorphic modifications" or "polymorphs".
- polymorphs polymorphs
- the term 'polymorph' may also encompass pseudo-polymorphs.
- polymorphism is caused by the ability of the molecule of a substance to change its conformation or to form different intermolecular and intramolecular interactions, particularly hydrogen bonds, resulting in different atomic arrangements in the crystal lattices of the different polymorphs.
- the polymorphs of a substance possess different crystal lattice energies and, thus, also exhibit different solid state physical properties such as morphology, density, melting point, colour, stability, dissolution rate, milling facility, granulation properties, compacting properties etc.
- the use of different polymorphs often influences factors such as the preparation of pharmaceutical compositions, their stability, dissolution properties, bioavailability and, consequently, their action.
- the use of polymorphs allows modulation of the performance of an active pharmaceutical ingredient (API) such as 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxa-diazol-5-yl]- 3 -nitrobenzene- 1,2-diol as well as affecting the formulation of the API.
- API active pharmaceutical ingredient
- the present invention not only relates to the provision of novel polymorphs of 5-[3-(2,5- dichloro-4,6-dimethyl-l -oxy-pyridin-3-yl)-[ 1 ,2,4]oxadiazol-5-yl]-3-nitrobenzene- 1 ,2-diol (henceforth referred to as "compound of the invention"), but also to processes for their preparation, and to pharmaceutical compositions containing one or more of said novel polymorphs as active ingredient.
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph A.
- a process for the preparation of polymorph A is given in the experimental section.
- Polymorph A is a crystalline polymorph and, thus, characterizable by its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- the diffraction pattern may either be experimentally recorded or calculated from the results of the measurement of the unit cell parameters of the polymorph.
- characteristic peaks of the XRPDs of the polymorphs of the invention are given in degrees 2 ⁇ (Cu-Ka radiation).
- Polymorph A is characterizable by one or more of the peaks given in the following table.
- polymorph A is characterized by one or more of the above peaks in the range of from about 5 to about 25 72 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph A is characterized by 2 to 10, preferably 3 to 5, peaks within the range of from about 5 to about 25 °/2 ⁇ . Even more preferably, the polymorph A is characterized by the signals at 6.6, 13.2, 17.9, 23.2, 23.8 and 24.3 720. Most preferably, polymorph A is characterized by the signals at 6.6, 13.2, 17.9 and 23.8 720.
- polymorph A may also be characterizable by having an exotherm at 251 °C in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- polymorph A may also be characterised by exhibiting a melt onset at 238 °C. Furthermore, polymorph A may also be characterized by being non-hygroscopic over the range of about 5 % to about 95 %, more preferably from about 25 % to about 80 %, and even more preferably from about 40 % to about 60%, relative humidity at 25 0 C over a period of 3 months.
- polymorph A is preferably an anhydrate as is evidenced by a lack of solvent desorption in combined Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) experiments prior to energetic decomposition.
- DSC Differential Scanning Calorimetry
- TGA Thermogravimetric Analysis
- polymorph A is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. More preferably, polymorph A is characterized by one or more of the peaks at 145, 505, 810, 1159, 1228, 1325, 1537, 1589, and 1628 cm “1 . Most preferably, polymorph A is characterized by the peaks at 810, 1325, 1537, 1589, and 1628 cm "1 .
- FT-Raman Peak Positions will generally be reproducible within a range from about +0 cm '1 to +5 cm “1 , preferably from +1 cm “1 to +3 cm “1 , most preferably ⁇ 2 cm “1 . This also applies to the other Raman data presented in this specification.
- Polymorph A is also characterizable by one or more of the following peak positions in Solid State 13 C-NMR: table 3 - Solid State 13C-NMR of polymorph A
- polymorph A is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. Most preferably, polymorph A is characterized by the peaks at 15.7, 114.6, 148.8 and 174.0 ppm.
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph B.
- a process for the preparation of polymorph B is given in the experimental section.
- Polymorph B is a crystalline polymorph and, thus, characterizable by its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- polymorph B is characterized by one or more of the above peaks in the range of from about 5 to about 25 °/2 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph B is characterized by 2 to 10, preferably 3 to 5, peaks within the range of from about 5 to about 25 °/2 ⁇ . Even more preferably, the polymorph B is characterized by the signals at 5.7, 6.9, 11.9, 13.8, 16.9 and 19.6 72 ⁇ . Most preferably, polymorph B is characterized by the signals at 5.7, 6.9, 13.8 and 19.6 °/2 ⁇ .
- polymorph B may also be characterizable by having an exotherm at 237 0 C in a Differential Scanning Calorimetry (DSC) thermogram. Such analyses may also indicate a shoulder peak at 231 °C.
- DSC Differential Scanning Calorimetry
- polymorph B is preferably an anhydrate as is evidenced by a lack of solvent desorption in combined Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) experiments prior to energetic decomposition.
- DSC Differential Scanning Calorimetry
- TGA Thermogravimetric Analysis
- molecular packaging of polymorph B allows the accommodation of crystal water without substantially altering the crystal lattice.
- Polymorph B is also characterizable by one or more of the following FT-Raman Peak Positions: table 5 — Raman spectra of polymorph B
- polymorph B is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. More preferably, polymorph B is characterized by one or more of the peaks at 505, 1292, 1317, 1385, 1537, 1583, and 1630 cm “1 . Most preferably, polymorph B is characterized by the peaks at 505, 1292, 1385, 1583, and 1630 cm- 1 .
- polymorph B is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. Most preferably, polymorph B is characterized by the peaks at 150.3, 133.9, 112.6 and 19.8 ppm.
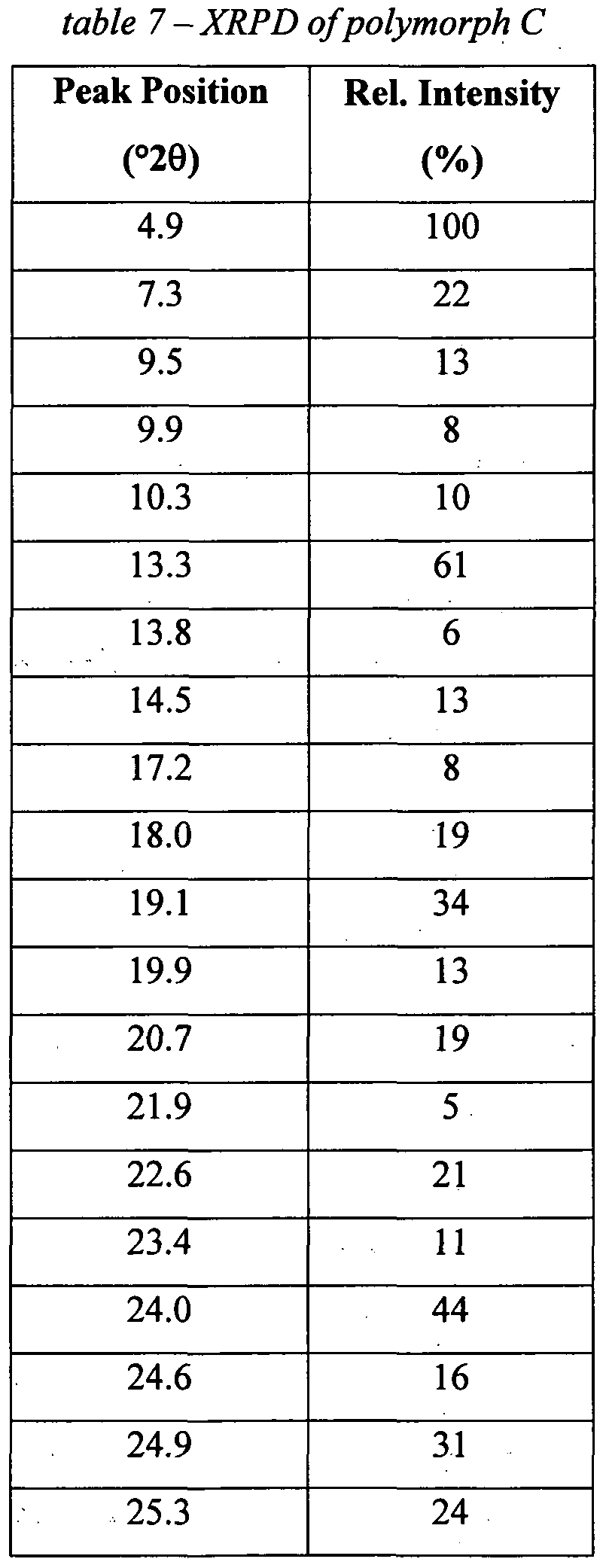
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph C. A process for the preparation of polymorph C is given in the experimental section.
- Polymorph C is a crystalline polymorph and, thus, characterizable by one or more of the peaks of its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- polymorph C is characterized by one or more of the above peaks in the range of from about 5 to about 25 °/2 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph C is characterized by 2 to 10, preferably 3 to 5, peaks within the , range of from about 5 to about 25 °/2 ⁇ . Even more preferably, the polymorph C is characterized by the signals at 4.9, 7.3, 13.3, 19.1, 21.9, 23.4 and 24.0 °/2 ⁇ . Most preferably, polymorph C is characterized by the signals at 4.9, 13.3, 19.1 and 24.0 °/2 ⁇ .
- Polymorph C may also be characterised by showing an exotherm at 203 0 C in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- Polymorph C is also characterizable by one or more of the following FT-Raman Peak Positions: table 8 - Raman spectra of polymorph C
- polymorph C is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. More preferably, polymorph C is characterized by one or more of the peaks at 143, 507, 810, 1244, 1281, 1317, 1352, 1387, 1406, 1537, 1585, and 1630 cm “1 . Most preferably, polymorph C is characterized by the peaks at 1387, 1406, 1537, 1585, and 1630 cm '1 .
- Polymorph C is also characterizable by one or more of the following peak positions in Solid State 13 C-SsNMR: table 9 - Solid State 13C-NMR of polymorph C
- polymorph C is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. Most preferably, polymorph C is characterized by the peaks at 150.7, 133.5, 114.0 and 20.2 ppm.
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph D.
- a process for the preparation of polymorph D is given in the experimental section.
- Polymorph D is a crystalline polymorph and, thus, characterizable by one or more of the peaks of its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- polymorph D is characterized by one or more of the above peaks in the range of from about 5 to about 25 °/2 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph D is characterized by 2 to 10, preferably 3 to 5, peaks within the range of from about 5 to about 25 72 ⁇ . Even more preferably, the polymorph D is characterized by the signals at 11.6, 12.3, 20.0, 20.7, 21.4 and 24.8 720. Most preferably, polymorph D is characterized by the signals at 12.3, 20.0, 20.7 and 21.4 720.
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph E.
- a process for the preparation of polymorph E is given in the experimental section.
- Polymorph E is a crystalline polymorph and, thus, characterizable by one or more of the peaks of its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- polymorph E is characterized by one or more of the above peaks in the range of from about 5 to about 25 °/2 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph E is characterized by 2 to 10, preferably 3 to 5, peaks within the range of from about 5 to about 25 °/2 ⁇ . Even more preferably, the polymorph E is characterized by the signals at 7.8, 9.8, 12.2, 15.6, 16.2, 17.6, 19.8, 21.7, 22.9, and 24.5 72 ⁇ . Most preferably, polymorph E is characterized by the signals at 9.8, 11.2, 19.8 and 24.5 72 ⁇ .
- Polymorph E may be further characterised by showing an endotherm at 159 0 C in a Differential Scanning Calorimetry (DSC) thermogram. Polymorph E may be even further characterised by showing an exotherm at 243 0 C in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- polymorph E is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table. More preferably, polymorph E is characterized by one or more of the peaks at 127, 1022, 1269, 1288, 1327, 1356, 1412, 1533, 1556, 1583, and 1635 cm “1 . Most preferably, polymorph E is characterized by the peaks at 1356, 1533, 1556, 1583, and 1635 c ⁇ f 1 .
- Polymorph E is also characterizable by one or more of the following peak positions in Solid State 13 C-NMR: table 13 - Solid State 1 "3C-NMR of polymorph E
- polymorph E is characterizable by one or more, preferably 2 to 6, and more preferably 3 to 5, peak positions in the above table.
- polymorph C is characterized by the peaks at 151.8, 133.6, 105.1 and 19.4 ppm.
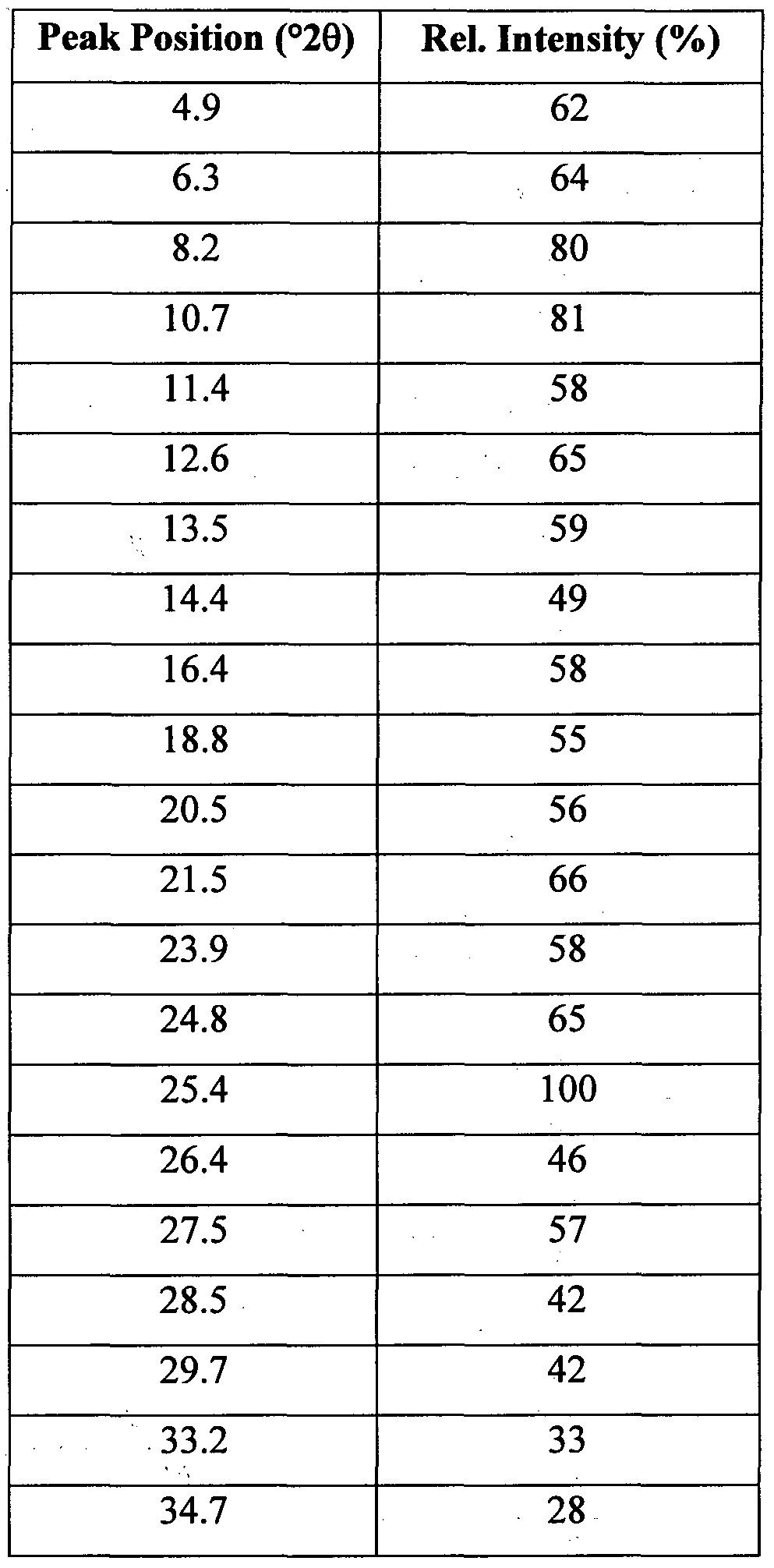
- the present invention relates to a polymorph of the compound of the invention which is, in the following specification, referred to as polymorph F.
- a process for the preparation of polymorph F is given in the experimental section.
- Polymorph F is a crystalline polymorph and, thus, characterizable by one or more of the peaks of its powder X-ray diffraction pattern (XRPD).
- XRPD powder X-ray diffraction pattern
- polymorph F is characterized by one or more of the above peaks in the range of from about 5 to about 25 °/2 ⁇ which is a highly characteristic region of XRPDs. More preferably, polymorph F is characterized by 2 to 10, preferably 3 to 5, peaks within the range of from about 5 to about 25 720. Even more preferably, the polymorph F is characterized by the signals at 8.2, 10.7, 12.6, 13.5, 16.4 and 25.4 720. Most preferably, polymorph F is characterized by the signals at 10.7, 12.6, 16.4 and 25.4 720. In interconversion experiments, it was found that the polymorphs C, D, E, and F convert into polymorph A or B, i.e. modifications A and B are kinetically stable polymorphs. Furthermore, it was found that polymorph B converts into polymorph A when slurried in water containing crystal seeds of modification A over prolonged periods of time. Thus, polymorph A is the thermodynamically stable polymorph.
- modifications A and B are thermodynamically stable and kinetically stable, respectively, they will in particular be characterized by good storage stability.
- Storage stability of the polymorph is defined herein as a lack of rearrangement of one polymorphic modification into another modification under storage conditions of 60 % relative humidity and 25 0 C over a period of 3 months. Accordingly, physical parameters related to the polymorphic modification such as the XRPD, Raman spectra and the Differential Scanning Calorimetry (DSC) thermogram will not change upon storage under the above-specified conditions.
- these polymorphs will also exhibit stable dissolution properties as these properties are dependent on the above defined storage stability of the polymorph. Accordingly, the modifications A and B are also characterized in that they will not be subject to a change in their dissolution profile upon storage.
- a lack of change in the dissolution profile is defined herein as a variation of the time period until 80 % of the polymorph has dissolved under test conditions according to the USP Paddle Test Method, USP, 30th Edition, The National Formulary 25th Edition, 2007, The United States Pharmacopeial Convention, Rockville, volume 1, chapter 711, of less than 10 %, preferably less than 5 % and most preferably less than 1 %, after storage under conditions of 60 % relative humidity at 25 °C over a period of at least 3 months.
- polymorphs of the present invention may be used as active pharmaceutical ingredients in pharmaceutical formulations such as tablets, capsules or injections, without or in combination with one or more pharmaceutically acceptable additives such as sugar, starch, starch derivatives, cellulose, cellulose derivatives, release agents, anti-adhesive agents and agents for regulating flowability.
- pharmaceutically acceptable additives such as sugar, starch, starch derivatives, cellulose, cellulose derivatives, release agents, anti-adhesive agents and agents for regulating flowability.
- the present invention also relates to a pharmaceutical composition, characterized in that it comprises 5-[3-(2,5- dichloro-4,6-dimethyl- 1 -oxy-pyridin-3-yl)- [1,2,4] oxadiazol-5 -yl] -3 -nitrobenzene- 1 ,2-diol and that the X-ray powder diffractogram of the composition exhibits one or more, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, 2 ⁇ angles from the following list of 2 ⁇ angles: 6.6, 6.9, 11.8, 13.2, 17.2, 17.9, 19.8, 22.6, 23.2, 23.8, 24.3, and 25.3.
- said formulation is characterized by an XRPD having one or more of the following characteristic peaks for polymorph A: 6.6, 23.2 and 24.3.
- said pharmaceutical composition may further be characterized in that its Raman spectrum exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [cm '1 ]: 145, 170, 216, 237, 256, 285, 339, 370, 420, 442, 465, 505, 526, 710, 810, 974, 1007, 1059, 1159, 1228, 1254, 1277, 1325, 1387, 1414, 1448, 1498, 1537, 1589, 1628, 2927.
- said pharmaceutical composition may also be further characterized in that its Solid State 13C-NMR spectrum (100 MHz) exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [ppm]: 174.0, 163.9, 150.4, 148.8, 144.5, 140.2, 134.8, 133.2, 129.8, 124.9, 124.0, 122.1, 114.6, 22.5, and 15.7.
- the pharmaceutical formulations comprising polymorph A may also exhibit an exotherm at 251 0 C in a Differential Scanning Calorimetry (DSC) thermogram.
- the present invention relates to a pharmaceutical composition, characterized in that it comprises 5-['3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol- 5-yl]-3-nitrobenzene-l,2-diol and that its X-ray powder diffractogram exhibits one or more, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, 2 ⁇ angles from the following list of 2 ⁇ angles: 5.7, 6.9, 8.0, 9.8, 11.3, 11.9, 13.8, 14.4, 16.9, 19.6, and 20.4.
- said formulation is characterized by an XRPD having one or more of the following characteristic peaks for polymorph B: 5.7, 16.9 and 19.6.
- said pharmaceutical composition may further be characterized in that its Raman spectrum exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [cm '1 ]: 141, 214, 235, 253, 280, 339, 361, 372, 399, 415, 440, 463, 505, 526, 710, 812, 887, 926, 970, 1001, 1059, 1157, 1227, 1292, 1317, 1385, 1406, 1444, 1504, 1537, 1583, 1630, and 2933.
- said pharmaceutical composition may also be further characterized in that its Solid State 13C-NMR spectrum (100 MHz) exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [ppm]: 174.4, 164.7, 150.3, 139.1, 133.9, 132.8, 123.5, 120.3, 114.0, 112.6, 19.8, and 16.6.
- the pharmaceutical formulations comprising polymorph B may also exhibit an exotherm at 237 °C, in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- the present invention relates to a pharmaceutical composition, characterized in that it comprises 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol-
- said formulation is characterized by an XRPD having one or more of the following characteristic peaks for polymorph C: 7.3,
- said pharmaceutical composition may further be characterized in that its Raman spectrum exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [cm "1 ]: 143, 214, 237, 253, 282, 299, 339, 370, 401, 418, 444, 465, 507, 530, 667, 710, 739, 810, 972, 1003, 1061, 1147, 1244, 1281, 1317, 1352, 1387, 1406, 1450, 1483, 1506, 1537, 1585, 1630, and 2918.
- said pharmaceutical composition may also be further characterized in that its Solid State 13C-NMR spectrum (100 MHz) exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [ppm]: 173.9, 165.0, 150.7, 141.0, 138.6, 133.5, 122.8, 120.3, 114.0, 112.4, 41.1, 20.2, and 17.9.
- the pharmaceutical formulations comprising polymorph C may also exhibit an exotherm at 203 °C in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- the present invention relates to a pharmaceutical composition, characterized in that it comprises 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol- 5-yl]-3-nitrobenzene-l,2-diol and that its X-ray powder diffractogram exhibits one or more, preferably 2 to 10, more preferably 3 to 5, and most preferably 4, 20 angles from the following list of 2 ⁇ angles: 11.6, 12.3, 20.0, 20.7, 21.4, 23.8, 24.8, 26.6, 27.4, 28.0, 29.1, 31.1, 32.0, 33.3, 35.0, and 36.0.
- the present invention relates to a pharmaceutical composition, characterized in that it comprises 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol- 5-yl]-3-nitrobenzene-l,2-diol and that its X-ray powder diffractogram exhibits one or more, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, 2 ⁇ angles from the following list of2 ⁇ angles: 4.5, 5.4, 6.2, 6.9, 7.8, 8.4, 9.8, 10.8, 11.2, 11.5, 12.2, 12.4, 13.8, 14.6, 15.6, 16.2, 17.1, 17.6, 18.7, 19.8, 20.1, 21.7, 22.6, 22.9, 24.0, 24.5, 25.1, 26.0, 27.3, 28.8, 30.6, 31.5, 32.7, 33.5, 34.1, 35.8, 36.9, and 39.2.
- said formulation is characterized by an XRPD having one or more of the following characteristic peaks for polymorph E: 7.8, 15.6, 16.2, 17.6, 21.7 and 22.9.
- said pharmaceutical composition may further be characterized in that its Raman spectrum exhibits at least one, preferably 1 to 6, more preferably 2 to 4, and most preferably 3, peaks from the following peak list [cm "1 ]: 127, 216, 231, 260, 341, 384, 461, 498, 526, 712, 739, 769, 816, 893, 922, 972, 1005, 1022, 1057, 1174, 1209, 1234, 1269, 1288, 1327, 1356, 1412, 1460, 1506, 1533, 1556, 1583, 1635, and 2931.
- said pharmaceutical composition may also be further characterized in that its Solid State 13C-NMR spectrum (100 MHz) exhibits at least one, preferably 2 to 10, more preferably 3 to 5, and most preferably 4, peaks from the following peak list [ppm]: 175.2, 164.2, 158.8, 151.8, 147.1, 145.5, 143.4, 139.4, 136.4, 133.6, 130.7, 127.3, 124.8, 123.3, 118.1, 108.3, 105.1, 19.4, and 17.6.
- the pharmaceutical formulations comprising polymorph E may also exhibit an exotherm at 243 0 C in a Differential Scanning Calorimetry (DSC) thermogram.
- DSC Differential Scanning Calorimetry
- the present invention relates to a pharmaceutical composition, characterized in that it comprises 5-[3-(2,5-dichloro-4,6-dimethyl-l-oxy-pyridin-3-yl)-[l,2,4]oxadiazol- 5 -yl] -3 -nitrobenzene- 1,2-diol and that its X-ray powder diffractogram exhibits one or more, preferably 2 to 10, more preferably 3 to 5, and most preferably 4, 20 angles from the following list of 2 ⁇ angles: 4.9, 6.3, 8.2, 10.7, 11.4, 12.6, 13.5, 14.4, 16.4, 18.8, 20.5, 21.5, 23.9, 24.8, 25.4, 26.4, 27.5, 28.5, 29.7, 33.2, and 34.7.
- said formulation is characterized by an XRPD having one or more of the following characteristic peaks for polymorph F: 8.2, 12.6, 13.5, and 16.4.
- pharmaceutical formulations comprising the stable polymorphs A and/or B will also be storage stable.
- pharmaceutical formulations containing modifications A and/or B are further characterized in that they will not be subject to a change in their dissolution profile upon storage.
- a lack of change in the dissolution profile is defined herein as a lack of variation of the time period until 80 % of the active pharmaceutical ingredient has been released under test conditions according to the USP Paddle Test Method, USP, 30th Edition, The National Formulary 25th Edition, 2007, The United States Pharmacopeial Convention, Rockville, volume 1, chapter 711, of less than 10 %, preferably less than 5 % and most preferably less than 1 %, after storage under conditions of 60 % relative humidity at 25 0 C over a period of at least 3 months.
- modifications A and B can be used to convert metastable modifications such as modifications C to F into the more stable modifications A or B.
- mixtures of metastable modifications can be converted to polymorphically pure modifications A and B e.g. by seeding slurries of such mixtures with modification A or B.
- Modification B can also be converted to modification A e.g. by seeding a slurry of modification B with seeding crystals of modification A.
- modifications less stable than modification A such as amorphic, metastable and polymorphically impure modifications to the thermodynamically most stable modification A.
- the most preferred process for doing so is seeding such modifications or mixtures of modifications with seeding crystals of modification A.
- Another preferred process for achieving a conversion is the dissolution of said modifications, followed by seeding with crystals of modification A.
- a 1600L reactor was charged with 245kg formic acid and 10 kg 5-[3-(2,5-dichloro-4,6- dimethyl- 1 -oxy-pyridin-3-yl)-[ 1 ,2,4]oxadiazol-5-yl]-3-nitrobenzene-l ,2-diol.
- the mixture was heated to 70-75 °C until the solid dissolved.
- the hot solution was then filtered to a 250L reactor, and then cooled to 25-35°C. Vacuum was applied and the mixture was distilled at 5O 0 C until 50-70 litres remained.
- To this mixture 160kg isopropanol was introduced. The mixture was cooled to 5-10°C and was allowed to stir for 10 hours.
- the suspension was then centrifuged and washed with 13kg isopropanol.
- the wet material was removed from the centrifuge and dried in a vacuum tray drier.
- the dried material weight was 8.995kg after sampling.
- the yield of polymorph A was 9.395kg, 93.9%.
- Samples of polymorphs A, C and E were analyzed using a PANalytical X'Pert Pro diffractometer.
- the specimen was analyzed using Cu radiation produced using an Optix long fine-focus source.
- An elliptically graded multilayer mirror was used to focus the Cu Ka X-rays of the source through the specimen and onto the detector.
- the specimen was sandwiched between 3-micron thick films, analyzed in transmission geometry, and rotated to optimize orientation statistics.
- a beam-stop and helium were used to minimize the background generated by air scattering.
- Soller slits were used for the incident and diffracted beams to minimize axial divergence.
- Diffraction patterns were collected using a scanning position-sensitive detector (X'Celerator) located 240 mm from the specimen. The data-acquisition parameters of each diffraction pattern are displayed above the image of each pattern in appendix data section.
- a silicon specimen NIST standard reference material 640c
- Samples of polymorph B were analyzed using a Shimadzu XRD-6000 X-ray powder diffractometer using Cu Ka radiation.
- the instrument is equipped with a long fine focus X-ray tube.
- the tube voltage and amperage were set at 40 kV and 40 mA, respectively.
- the divergence and scattering slits were set at 1° and the receiving slit was set at 0.15 mm.
- Diffracted radiation was detected by a NaI scintillation detector.
- a theta-two theta continuous scan at 3 °/min (0.4 sec/0.02° step) from 2.5 to 40 °2 ⁇ was used.
- a silicon standard was analyzed to check the instrument alignment. Samples were prepared for analysis by placing them in an aluminum/silicon sample holder.
- Samples of polymorph D were analyzed using a Bruker D-8 Discover diffractometer and Bruker's General Area Diffraction Detection System (GADDS, v. 4.1.20).
- An incident beam of CuKa radiation was produced using a fine-focus tube (40 kV, 40 mA), a Gobel mirror, and a 0.5 mm double-pinhole collimator. The sample was packed between
- the integrated patterns display diffraction intensity as a function of 20. Prior to the analysis a silicon standard was analyzed to verify the Si 111 peak position. 4. Inel XRG-3000 Diffractometer
- XRG-3000 diffractometer equipped with a CPS (Curved Position Sensitive) detector with a 2q range of 120°.
- Real time data were collected using Cu-Ka radiation.
- the tube voltage and amperage were set to 40 kV and 30 mA, respectively.
- the monochromator slit was set at 1-5 mm by 160 ⁇ m.
- the patterns are displayed from 2.5-40 °2q.
- Samples were prepared for analysis by packing them into thin-walled glass capillaries. Each capillary was mounted onto a goniometer head that is motorized to permit spinning of the capillary during data acquisition. The samples were analyzed for 300 seconds. Instrument calibration was performed using a silicon reference standard.
- DSC for polymorph A was performed using TA Instruments model Q2000 calorimeter. The samples were placed in an aluminium DSC pan, the weight accurately recorded and the pan crimped. The sample cell was equilibrated at 25 0 C and heated under nitrogen purge at a rate of 10 °C/min up to a final temperature of 250 or 300 °C. Indium metal was used as calibration standard. For all other polymorphs analyses were performed using TA Instruments model 2920 calorimeter. The sample cell was equilibrated at 25 °C and heated under nitrogen purge at a rate of 10 °C/min up to a final temperature of 250 or 300 °C. Indium metal was used as calibration standard.
- TGA analyses were performed on a TA Instruments model 2950 thermogravimetric analyzer. The furnace was equilibrated at 25 °C and heated under nitrogen purge at a rate of 10 °C/min up to a final temperature of 300 or 350 °C. Nickel and Alume were used as calibration standards.
- Hotstage microscopy was performed using a Linkam hotstage (model FTIR 600) mounted to a Leica DMLP microscope. Samples were observed using crossed polarized light. Samples were sandwitched between coverslips and visually observed as the stage was heated. The hotstage was calibrated using USP melting point standards. FT-Raman spectra were acquired on an FT-Raman 960 spectrometer (Thermo Nicolet) using an excitation wavelength of 1064 run. Approximately 0.2-0.3 W of Nd: YVO 4 laser power was used to irradiate the samples. The Raman spectra were measured with a germanium detector.
- the samples were prepared for analysis by placing the material in a glass tube and positioning the tube in a gold-coated tube holder in the accessory. A total of 256 sample scans were collected from 3600 - 100 cm “1 at a spectral resolution of 4 cm " , using Happ-Genzel apodization. Wavelength calibration was performed using sulfur and cyclohexane.
- Pulse width 2.2 usec (90.0 deg.) or 2.2usec (76.2 deg) for polymorph E
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Psychology (AREA)
- Anesthesiology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
















Claims
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2009801099261A CN102015696A (en) | 2008-03-17 | 2009-03-16 | 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxopyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitro Crystalline form of phenyl-1,2-diol |
| US12/933,044 US8975410B2 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4] oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
| CA2718772A CA2718772C (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nit robenzene-1,2-diol |
| AU2009226221A AU2009226221A1 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol |
| BRPI0908731A BRPI0908731A2 (en) | 2008-03-17 | 2009-03-16 | 5- [3- (2,5-Dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl) [1,2,4] oxadiazol-5-yl] -3-nitrobenzene-1 crystalline forms 2-diol |
| JP2011500722A JP2011514380A (en) | 2008-03-17 | 2009-03-16 | 5- [3- (2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl) [1,2,4] oxadiazol-5-yl] -3-nitrobenzene-1, 2-Diol crystal form |
| MX2010009610A MX2010009610A (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridi ne-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol. |
| EP09722681.5A EP2276758B1 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,4] oxadiazol-5-yl]-3-nit robenzene-1, 2-diol |
| ES09722681.5T ES2565080T3 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5- [3- (2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl) - [1,2,4] oxadiazol-5-yl] -3-nitrobenzene- 1,2-diol |
| IL207854A IL207854A0 (en) | 2008-03-17 | 2010-08-29 | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nit robenzene-1,2-diol |
| US14/628,630 US9845316B2 (en) | 2008-03-17 | 2015-02-23 | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6972108P | 2008-03-17 | 2008-03-17 | |
| US61/069,721 | 2008-03-17 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/933,044 A-371-Of-International US8975410B2 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4] oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
| US14/628,630 Continuation US9845316B2 (en) | 2008-03-17 | 2015-02-23 | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009116882A1 true WO2009116882A1 (en) | 2009-09-24 |
Family
ID=40585552
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/PT2009/000013 Ceased WO2009116882A1 (en) | 2008-03-17 | 2009-03-16 | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8975410B2 (en) |
| EP (1) | EP2276758B1 (en) |
| JP (2) | JP2011514380A (en) |
| KR (1) | KR20110002462A (en) |
| CN (1) | CN102015696A (en) |
| AR (1) | AR070907A1 (en) |
| AU (1) | AU2009226221A1 (en) |
| BR (1) | BRPI0908731A2 (en) |
| CA (1) | CA2718772C (en) |
| CL (1) | CL2009000628A1 (en) |
| ES (1) | ES2565080T3 (en) |
| IL (1) | IL207854A0 (en) |
| MX (1) | MX2010009610A (en) |
| RU (1) | RU2010139315A (en) |
| TW (1) | TW200942531A (en) |
| WO (1) | WO2009116882A1 (en) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2639131C2 (en) * | 2011-02-11 | 2017-12-19 | Биал- Портела и КА, С.А. | Introduction mode for nitrocatechols |
| US9845316B2 (en) | 2008-03-17 | 2017-12-19 | BIAL—Portela & CA., S.A. | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
| RU2659987C2 (en) * | 2016-12-07 | 2018-07-04 | Федеральное государственное бюджетное образовательное учреждение высшего образования "Московский государственный университет имени М.В. Ломоносова" (МГУ) | Planar solidphase optical sensor for determination of protein compounds by the method of spectroscopy of giant raman scattering and its application for protein compounds detection |
| US10071085B2 (en) | 2009-04-01 | 2018-09-11 | Bial—Portela & Ca, S.A. | Pharmaceutical formulations comprising nitrocatechol derivatives and methods of making thereof |
| WO2021182981A1 (en) | 2020-03-13 | 2021-09-16 | BIAL - PORTELA & Cª, S.A. | Micronised opicapone |
| WO2022025781A1 (en) | 2020-07-28 | 2022-02-03 | BIAL - PORTELA & Cª, S.A. | Solid dispersion of opicapone |
| WO2022081033A1 (en) | 2020-10-16 | 2022-04-21 | BIAL - PORTELA & Cª, S.A. | Opicapone and levodopa for the treatment of parkinson's disease |
| WO2022131944A1 (en) | 2020-12-17 | 2022-06-23 | Bial-Portela & Ca., S.A. | Treatment regimens for early idiopathic parkinson's disease |
| WO2024136689A1 (en) | 2022-12-23 | 2024-06-27 | Bial - Portela & Ca, S.A. | Processes and intermediates for synthesising opicapone |
| WO2024242579A1 (en) | 2023-05-25 | 2024-11-28 | Bial-Portela & Ca., S.A. | Treatment regimens for early idiopathic parkinson's disease |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL1907382T3 (en) | 2005-07-26 | 2016-01-29 | Bial Portela & Ca Sa | Nitrocatechol derivatives as comt inhibitors |
| EP1845097A1 (en) | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| BRPI0721213B1 (en) | 2007-01-31 | 2021-11-09 | Bial - Portela & Ca, S.A. | USE OF REPLACED NITROCATECOLS, AND PACKAGING |
| ES2723729T3 (en) * | 2008-07-29 | 2019-08-30 | Bial Portela & Ca Sa | Nitrocatechol administration regimen |
| BRPI1016132B8 (en) * | 2009-04-01 | 2021-05-25 | Bial Portela & Ca Sa | composition, pharmaceutical formulation, process of obtaining a stable pharmaceutical formulation |
| US9848134B2 (en) | 2010-04-23 | 2017-12-19 | Flir Systems, Inc. | Infrared imager with integrated metal layers |
| JP6456143B2 (en) * | 2011-12-13 | 2019-01-23 | ノヴィファーマ,エス.アー. | Chemical compounds useful as intermediates for preparing catechol-O-methyltransferase inhibitors |
| RU2017120184A (en) | 2014-11-28 | 2018-12-28 | БИАЛ - ПОРТЕЛА ЭНД Ка, С.А. | DRUGS TO DELAY THE COURSE OF PARKINSON'S DISEASE |
| PH12017502097B1 (en) * | 2015-05-20 | 2023-05-05 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (s)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007013830A1 (en) | 2005-07-26 | 2007-02-01 | Portela & Ca. S.A. | Nitrocatechol derivatives as comt inhibitors |
| WO2008094053A1 (en) * | 2007-01-31 | 2008-08-07 | Bial-Portela & Ca, S.A. | Dosage regimen for comt inhibitors |
Family Cites Families (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1532178A (en) | 1921-07-25 | 1925-04-07 | Louis A Godbold | Lubricator |
| FR1260080A (en) | 1960-03-22 | 1961-05-05 | Materiel De Forage Soc De Fab | Sealed roller drill bit |
| US3647809A (en) | 1968-04-26 | 1972-03-07 | Chinoin Gyogyszer Es Vegyeszet | Certain pyridyl-1 2 4-oxadiazole derivatives |
| US4022901A (en) | 1975-03-05 | 1977-05-10 | E. R. Squibb & Sons, Inc. | 3-Pyridinyl-5-isothiocyanophenyl oxadiazoles |
| US4264573A (en) | 1979-05-21 | 1981-04-28 | Rowell Laboratories, Inc. | Pharmaceutical formulation for slow release via controlled surface erosion |
| US4386668A (en) | 1980-09-19 | 1983-06-07 | Hughes Tool Company | Sealed lubricated and air cooled rock bit bearing |
| DK175069B1 (en) | 1986-03-11 | 2004-05-24 | Hoffmann La Roche | Pyrocatechol derivatives |
| US5236952A (en) | 1986-03-11 | 1993-08-17 | Hoffmann-La Roche Inc. | Catechol derivatives |
| YU213587A (en) | 1986-11-28 | 1989-06-30 | Orion Yhtymae Oy | Process for obtaining new pharmacologic active cateholic derivatives |
| DE3840954A1 (en) | 1988-12-05 | 1990-06-07 | Shell Int Research | PREPARATION OF 2-CHLORNICOTINIC ACID ESTERS |
| US5206372A (en) | 1990-06-05 | 1993-04-27 | Shell Research Limited | Preparation of 2-chloropyridine derivatives |
| ES2066092T3 (en) | 1990-11-29 | 1995-03-01 | Wei Ming Pharmaceutical Mfg Co | COADJUVANTE FOR THE DIRECT TRAINING OF TABLETS. |
| EP0619814A1 (en) | 1991-12-31 | 1994-10-19 | Fujisawa Pharmaceutical Co., Ltd. | Oxadiazole derivatives having acetylcholinesterase-inhibitory and muscarinic agonist activity |
| FR2730322B1 (en) | 1995-02-02 | 1997-04-30 | Imago | METALLIC GLASSES |
| DE19628617A1 (en) | 1996-07-16 | 1998-01-22 | Basf Ag | Direct tabletting aid |
| US6206110B1 (en) | 1996-09-09 | 2001-03-27 | Smith International, Inc. | Protected lubricant reservoir with pressure control for sealed bearing earth boring drill bit |
| JP2002526482A (en) | 1998-09-18 | 2002-08-20 | バーテックス ファーマシューティカルズ インコーポレイテッド | inhibitors of p38 |
| GB2344819A (en) | 1998-12-18 | 2000-06-21 | Portela & Ca Sa | 2-Phenyl-1-(3,4-dihydroxy-5-nitrophenyl)-1-ethanones |
| FI109453B (en) | 1999-06-30 | 2002-08-15 | Orion Yhtymae Oyj | Pharmaceutical composition |
| IL131037A (en) | 1999-07-22 | 2004-06-20 | Israel Atomic Energy Comm | Method for making threedimensional photonic band-gap crystals |
| KR100875222B1 (en) | 1999-08-19 | 2008-12-19 | 아스트라제네카 아베 | Heteropolycyclic Compounds and Their Uses as Indirect Glutamate Receptor Antagonists |
| US6660753B2 (en) | 1999-08-19 | 2003-12-09 | Nps Pharmaceuticals, Inc. | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| EP1248869A2 (en) * | 2000-01-07 | 2002-10-16 | Transform Pharmaceuticals, Inc. | High-throughput formation, identification, and analysis of diverse solid-forms |
| FI20000635A0 (en) | 2000-03-17 | 2000-03-17 | Orion Yhtymae Oyj | Use of COMT inhibitors as an analgesic |
| SE0001438D0 (en) | 2000-04-18 | 2000-04-18 | Axon Chemicals Bv | New chemical compounds and their use in therapy |
| DE10029201A1 (en) | 2000-06-19 | 2001-12-20 | Basf Ag | Retarded release oral dosage form, obtained by granulating mixture containing active agent and polyvinyl acetate-polyvinyl pyrrolidone mixture below the melting temperature |
| GB2363792A (en) | 2000-06-21 | 2002-01-09 | Portela & Ca Sa | Nitrocatechols |
| EP1329825A4 (en) | 2000-08-24 | 2006-03-22 | Sagawa Express Co Ltd | Card payment method for service charge concerning to physical distribution or transportation |
| CN1166626C (en) | 2000-08-30 | 2004-09-15 | 李凌松 | Position 3 or 4 substituted phenyl compound, its preparing process and its application |
| WO2002051442A1 (en) | 2000-12-26 | 2002-07-04 | Takeda Chemical Industries, Ltd. | Concomitant drugs |
| US20040097555A1 (en) | 2000-12-26 | 2004-05-20 | Shinegori Ohkawa | Concomitant drugs |
| MXPA03007513A (en) | 2001-02-21 | 2004-07-30 | Nps Pharma Inc | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists. |
| WO2002096867A2 (en) | 2001-05-30 | 2002-12-05 | Lg Biomedical Institute | Inhibitors of protein kinase for the treatment of disease |
| JP2005504014A (en) | 2001-06-08 | 2005-02-10 | サイトビア インコーポレイテッド | Substituted 3-aryl-5-aryl- [1,2,4] -oxadiazoles and analogs as caspase activators and inducers of apoptosis, and methods of use thereof |
| US6627646B2 (en) * | 2001-07-17 | 2003-09-30 | Sepracor Inc. | Norastemizole polymorphs |
| PT1408964E (en) | 2001-07-26 | 2007-05-31 | Merck Patent Gmbh | Use of 2- 5-(4-fluorophenyl)-3-pyridylmethylaminomethyl |
| JP4379853B2 (en) | 2001-10-05 | 2009-12-09 | 惠民製藥股▲分▼有限公司 | Direct tableting formulations and methods of formulating adjuvants |
| CN100364531C (en) | 2002-12-18 | 2008-01-30 | 西托维亚公司 | 3,5-disubstituted-[1,2,4]-oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| WO2005006945A2 (en) | 2003-07-03 | 2005-01-27 | The Salk Institute For Biological Studies | Methods for treating neural disorders and compounds useful therefor |
| WO2005013982A1 (en) | 2003-08-06 | 2005-02-17 | Vertex Pharmaceuticals Incorporated | Aminotriazole compounds useful as inhibitors of protein kinases |
| US7300406B2 (en) | 2003-09-30 | 2007-11-27 | Carter Vandette B | Medical examination apparatus |
| GB0325956D0 (en) | 2003-11-06 | 2003-12-10 | Addex Pharmaceuticals Sa | Novel compounds |
| WO2005105780A2 (en) | 2004-04-28 | 2005-11-10 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of rock and other protein kinases |
| GB0510143D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds A1 |
| US20060173074A1 (en) | 2004-11-10 | 2006-08-03 | Juha Ellmen | Treatment of restless legs syndrome |
| WO2006061697A1 (en) | 2004-12-06 | 2006-06-15 | Themis Laboratories Private Limited | Sulfonylurea compositions and a process for its preparation |
| WO2006071184A1 (en) | 2004-12-28 | 2006-07-06 | Astrazeneca Ab | Aryl sulphonamide modulators |
| US20080051441A1 (en) | 2004-12-28 | 2008-02-28 | Astrazeneca Ab | Aryl Sulphonamide Modulators |
| US8017631B2 (en) | 2005-04-26 | 2011-09-13 | Neurosearch A/S | Oxadiazole derivatives and their medical use |
| US20060257473A1 (en) | 2005-05-11 | 2006-11-16 | Porranee Puranajoti | Extended release tablet |
| GB0510139D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds B1 |
| US7553964B2 (en) | 2005-06-03 | 2009-06-30 | Abbott Laboratories | Cyclobutyl amine derivatives |
| US20090000437A1 (en) | 2005-07-14 | 2009-01-01 | Provo Craft And Novelty, Inc. | Methods for Cutting |
| FR2889525A1 (en) | 2005-08-04 | 2007-02-09 | Palumed Sa | NOVEL POLYQUINOLINE DERIVATIVES AND THEIR THERAPEUTIC USE. |
| US20070048384A1 (en) | 2005-08-26 | 2007-03-01 | Joerg Rosenberg | Pharmaceutical compositions |
| EP1954137A4 (en) | 2005-11-18 | 2008-12-17 | Janssen Pharmaceutica Nv | 2-keto-oxazoles as modulators of fatty acid amide hydrolase |
| WO2007063946A1 (en) | 2005-11-30 | 2007-06-07 | Fujifilm Ri Pharma Co., Ltd. | Diagnostic and remedy for disease caused by amyloid aggregation and/or deposition |
| GB0606774D0 (en) | 2006-04-03 | 2006-05-10 | Novartis Ag | Organic compounds |
| EP1845097A1 (en) | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| PE20080906A1 (en) | 2006-08-17 | 2008-07-05 | Kemia Inc | HETEROARYL DERIVATIVES AS CYTOKINE INHIBITORS |
| US20080167286A1 (en) | 2006-12-12 | 2008-07-10 | Abbott Laboratories | Pharmaceutical compositions and their methods of use |
| US8486979B2 (en) | 2006-12-12 | 2013-07-16 | Abbvie Inc. | 1,2,4 oxadiazole compounds and methods of use thereof |
| JP2008162955A (en) | 2006-12-28 | 2008-07-17 | Chugai Pharmaceut Co Ltd | High-density granules containing valine |
| AR065802A1 (en) | 2007-03-22 | 2009-07-01 | Schering Corp | FORMULATIONS OF TABLETS CONTAINING SALTS OF 8- [(1- (3,5- BIS- (TRIFLUOROMETIL) FENIL) -ETOXI) - METHYL) -8- PHENYL -1, 7- DIAZA- SPIRO [4,5] DECAN - 2- ONA AND TABLETS PREPARED FROM THESE |
| KR101669432B1 (en) | 2007-08-27 | 2016-10-26 | 다트 뉴로사이언스 (케이만) 엘티디. | Therapeutic isoxazole compounds |
| MX2010009043A (en) * | 2008-02-28 | 2010-10-25 | Bial Portela & Ca Sa | Pharmaceutical composition for poorly soluble drugs. |
| WO2009116882A1 (en) | 2008-03-17 | 2009-09-24 | Portela & Ca., S.A. | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,3] oxadiazol-5-yl] -3-nit robenzene-1, 2-diol |
| ES2723729T3 (en) | 2008-07-29 | 2019-08-30 | Bial Portela & Ca Sa | Nitrocatechol administration regimen |
| US8827197B2 (en) | 2008-11-04 | 2014-09-09 | Microgreen Polymers Inc | Apparatus and method for interleaving polymeric roll for gas impregnation and solid-state foam processing |
| PT2413912T (en) | 2009-04-01 | 2019-06-11 | Bial Portela & Ca Sa | Pharmaceutical formulations comprising nitrocatechol derivatives and methods of making thereof |
| BRPI1016132B8 (en) | 2009-04-01 | 2021-05-25 | Bial Portela & Ca Sa | composition, pharmaceutical formulation, process of obtaining a stable pharmaceutical formulation |
| EA026419B1 (en) | 2010-03-04 | 2017-04-28 | Орион Корпорейшн | APPLICATION OF LEVODOPA, CARBIDOPE AND ENTACAPON TO TREAT PARKINSON'S DISEASE |
| US20140045900A1 (en) | 2011-02-11 | 2014-02-13 | Bial-Portela & Ca, S.A. | Administration regime for nitrocatechols |
| JP6456143B2 (en) | 2011-12-13 | 2019-01-23 | ノヴィファーマ,エス.アー. | Chemical compounds useful as intermediates for preparing catechol-O-methyltransferase inhibitors |
-
2009
- 2009-03-16 WO PCT/PT2009/000013 patent/WO2009116882A1/en not_active Ceased
- 2009-03-16 AR ARP090100933A patent/AR070907A1/en unknown
- 2009-03-16 ES ES09722681.5T patent/ES2565080T3/en active Active
- 2009-03-16 US US12/933,044 patent/US8975410B2/en active Active
- 2009-03-16 EP EP09722681.5A patent/EP2276758B1/en active Active
- 2009-03-16 KR KR1020107022717A patent/KR20110002462A/en not_active Withdrawn
- 2009-03-16 TW TW098108415A patent/TW200942531A/en unknown
- 2009-03-16 CN CN2009801099261A patent/CN102015696A/en active Pending
- 2009-03-16 RU RU2010139315/04A patent/RU2010139315A/en unknown
- 2009-03-16 CL CL2009000628A patent/CL2009000628A1/en unknown
- 2009-03-16 AU AU2009226221A patent/AU2009226221A1/en not_active Abandoned
- 2009-03-16 BR BRPI0908731A patent/BRPI0908731A2/en not_active IP Right Cessation
- 2009-03-16 JP JP2011500722A patent/JP2011514380A/en active Pending
- 2009-03-16 MX MX2010009610A patent/MX2010009610A/en not_active Application Discontinuation
- 2009-03-16 CA CA2718772A patent/CA2718772C/en active Active
-
2010
- 2010-08-29 IL IL207854A patent/IL207854A0/en unknown
-
2014
- 2014-10-31 JP JP2014222625A patent/JP2015044837A/en active Pending
-
2015
- 2015-02-23 US US14/628,630 patent/US9845316B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007013830A1 (en) | 2005-07-26 | 2007-02-01 | Portela & Ca. S.A. | Nitrocatechol derivatives as comt inhibitors |
| WO2008094053A1 (en) * | 2007-01-31 | 2008-08-07 | Bial-Portela & Ca, S.A. | Dosage regimen for comt inhibitors |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9845316B2 (en) | 2008-03-17 | 2017-12-19 | BIAL—Portela & CA., S.A. | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol |
| US10071085B2 (en) | 2009-04-01 | 2018-09-11 | Bial—Portela & Ca, S.A. | Pharmaceutical formulations comprising nitrocatechol derivatives and methods of making thereof |
| US10583130B2 (en) | 2009-04-01 | 2020-03-10 | Bial-Portela & Ca, S.A. | Pharmaceutical formulations compromising nitrocatechol derivatives and methods of making thereof |
| RU2639131C2 (en) * | 2011-02-11 | 2017-12-19 | Биал- Портела и КА, С.А. | Introduction mode for nitrocatechols |
| US12129247B2 (en) | 2011-02-11 | 2024-10-29 | Bial-Portela & Ca, S.A. | Administration regime for nitrocatechols |
| RU2659987C2 (en) * | 2016-12-07 | 2018-07-04 | Федеральное государственное бюджетное образовательное учреждение высшего образования "Московский государственный университет имени М.В. Ломоносова" (МГУ) | Planar solidphase optical sensor for determination of protein compounds by the method of spectroscopy of giant raman scattering and its application for protein compounds detection |
| CN115335036A (en) * | 2020-03-13 | 2022-11-11 | 巴尔-波特拉及康邦亚股份有限公司 | Micronized oppicapone |
| WO2021182981A1 (en) | 2020-03-13 | 2021-09-16 | BIAL - PORTELA & Cª, S.A. | Micronised opicapone |
| WO2022025781A1 (en) | 2020-07-28 | 2022-02-03 | BIAL - PORTELA & Cª, S.A. | Solid dispersion of opicapone |
| WO2022081033A1 (en) | 2020-10-16 | 2022-04-21 | BIAL - PORTELA & Cª, S.A. | Opicapone and levodopa for the treatment of parkinson's disease |
| WO2022131944A1 (en) | 2020-12-17 | 2022-06-23 | Bial-Portela & Ca., S.A. | Treatment regimens for early idiopathic parkinson's disease |
| WO2024136689A1 (en) | 2022-12-23 | 2024-06-27 | Bial - Portela & Ca, S.A. | Processes and intermediates for synthesising opicapone |
| WO2024242579A1 (en) | 2023-05-25 | 2024-11-28 | Bial-Portela & Ca., S.A. | Treatment regimens for early idiopathic parkinson's disease |
Also Published As
| Publication number | Publication date |
|---|---|
| AR070907A1 (en) | 2010-05-12 |
| US20150166519A1 (en) | 2015-06-18 |
| CA2718772A1 (en) | 2009-09-24 |
| JP2015044837A (en) | 2015-03-12 |
| JP2011514380A (en) | 2011-05-06 |
| BRPI0908731A2 (en) | 2017-05-16 |
| RU2010139315A (en) | 2012-04-27 |
| AU2009226221A1 (en) | 2009-09-24 |
| US9845316B2 (en) | 2017-12-19 |
| EP2276758A1 (en) | 2011-01-26 |
| IL207854A0 (en) | 2010-12-30 |
| KR20110002462A (en) | 2011-01-07 |
| ES2565080T3 (en) | 2016-03-31 |
| CL2009000628A1 (en) | 2010-04-09 |
| TW200942531A (en) | 2009-10-16 |
| MX2010009610A (en) | 2010-09-30 |
| CN102015696A (en) | 2011-04-13 |
| US8975410B2 (en) | 2015-03-10 |
| US20110112301A1 (en) | 2011-05-12 |
| CA2718772C (en) | 2017-05-02 |
| EP2276758B1 (en) | 2016-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9845316B2 (en) | Crystal forms of 5-[3-(2,5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl)[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol | |
| JP5611846B2 (en) | Substituted heterocyclic fused gamma-carbolines solids | |
| EP3045175B1 (en) | Polymorphic forms of 3-(4-amino-1-oxo-1,3 dihydroisoindol-2-yl)-piperidine-2,6-dione | |
| EP2753603B1 (en) | Polymorphic form of pridopidine hydrochloride | |
| US20120101277A1 (en) | Crystalline form of posaconazole | |
| KR20090052327A (en) | Uses of Azabicyclo Hexane Derivatives | |
| US20250313564A1 (en) | Acidic salt or crystal form of nitrogen-containing fused ring derivative inhibitor, and preparation method therefor and use thereof | |
| KR20130136544A (en) | New crystal form vii of agomelatine, preparation method and use thereof and pharmaceutical composition containing same | |
| JP7152122B2 (en) | edaravone salt | |
| CN115974863B (en) | Malic acid salt of Xanomeline derivative, crystal form A, and preparation method and use thereof | |
| EP4177257B1 (en) | Succinate of octahydrothienoquinoline compound, and crystals thereof | |
| TW202334120A (en) | Crystals of substituted piperidine compounds and salts of substituted piperidine compounds and crystals thereof | |
| JP5077232B2 (en) | Crystals of benzooxadiazole derivatives | |
| US20250304635A1 (en) | Birinapant polymorph h | |
| TW202333694A (en) | Crystal form of fused ring derivative and preparation method and application thereof | |
| WO2017032705A1 (en) | Crystalline form of omarigliptin | |
| WO2015092638A1 (en) | N-carbamoylmethyl-4(r)-phenyl-2-pyrrolidone polymorphic forms | |
| HK1167646B (en) | Methods of making polymorphic forms of 3-(4-amino-1-oxo-1,3 dihydro-isoindol-2-yl)-piperidine-2,6-dione |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980109926.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09722681 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009226221 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 207854 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1854/MUMNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/009610 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2009226221 Country of ref document: AU Date of ref document: 20090316 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011500722 Country of ref document: JP Ref document number: 2718772 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20107022717 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010139315 Country of ref document: RU Ref document number: 2009722681 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12933044 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0908731 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100917 |