WO2009119692A1 - 新規糖脂質及びその用途 - Google Patents
新規糖脂質及びその用途 Download PDFInfo
- Publication number
- WO2009119692A1 WO2009119692A1 PCT/JP2009/056010 JP2009056010W WO2009119692A1 WO 2009119692 A1 WO2009119692 A1 WO 2009119692A1 JP 2009056010 W JP2009056010 W JP 2009056010W WO 2009119692 A1 WO2009119692 A1 WO 2009119692A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbon atoms
- compound
- group
- hydrogen atom
- salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *CC([C@@](C([C@@]1O)O)O)OC1OCC(C(**)O)*(*)=O Chemical compound *CC([C@@](C([C@@]1O)O)O)OC1OCC(C(**)O)*(*)=O 0.000 description 4
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to a novel glycolipid and its use.
- the immune system has a skillful monitoring function for distinguishing between normal cells and abnormal cells in the living body and eliminating only abnormal cells. However, if the monitoring function breaks down, abnormal cells born due to mutation or the like cannot be eliminated, and growth in vivo is allowed. The mass of abnormal cells thus proliferated is a tumor, that is, a cancer.
- the main cancer treatment methods are surgical removal of cancer or the use of anticancer drugs.
- these treatment methods place a physical burden due to the surgical operation or scars due to side effects of anticancer agents.
- a therapy using immunotherapy is attracting attention.
- cancer cells are attacked by increasing the number of immune cells of the patient and activating them. If the tumor formed by the cancer cells can be reduced, the burden on the body due to the removal operation is small. In addition, since only a few surgical marks are required, the mental burden is greatly reduced.
- Natural killer (NK) T cells are immune cells belonging to a novel lymphocyte lineage that show different characteristics from other lymphocyte lineages (T, B, NK cells). Since NKT cells have cytotoxic perforin granules, they are related to NK cells (Non-patent Document 1). However, since NKT cells express not only NK cell markers but also T cell receptors (TCR), it has been clarified that they are a definitive new cell group (Non-patent Document 2). . NKT cells are composed of Th-1 type cytokines (mainly interferon (IFN) - ⁇ ) produced by helper T (Th) -1 cells that enhance immunostimulatory action and Th-2 cells that enhance immunosuppressive action.
- IFN interferon
- Th-2 helper T
- Th-2 type cytokines mainly interleukin (IL) -4
- IL interleukin
- Non-Patent Document 4 Therefore, by controlling the function of NKT cells, it is possible to adjust the balance of the immune system that has collapsed and enhance the monitoring function to treat cancer.
- NKT cells The most noticeable characteristic of NKT cells is that the ⁇ chain of TCR expressed in NKT cells is the same in all individuals among a single species. This means that all the NKT cells of the same species are activated by the same substance. This ⁇ chain is V ⁇ 24 in humans and V ⁇ 14 in mice, but has very high homology between both species. Also, only a very limited number of ⁇ chains paired with the ⁇ chain are known. For this reason, this TCR is also called an “non-variable TCR”.
- glycosphingolipids The existence of various types of glycosphingolipids is known in vivo. In vivo glycosphingolipids generally have various sugars ⁇ -bonded to ceramide, and their abundance varies depending on the organ, but are present in the cell membranes of various organs (Non-patent Document 5). On the other hand, it has recently been reported that glycosphingolipids in which the sugar is ⁇ -bonded to ceramide have a strong immunostimulatory action and antitumor activity.
- ⁇ -Galactosylceramide typified by Agerasfins is a glycolipid isolated from an extract of Agelas mauritianus, a kind of sponge, and is known to strongly activate NKT cells (Non-patent Document 6). ).
- ⁇ -Galactosylceramide is taken up by antigen-presenting cells (APC) such as dendritic cells (DC) and then on the cell membrane by CD1d protein similar to major histocompatibility complex (MHC) class I molecules.
- APC antigen-presenting cells
- DC dendritic cells
- MHC major histocompatibility complex
- NKT cells are activated by recognizing the thus-presented complex of CD1d protein and ⁇ -galactosylceramide using TCR, and various immune responses are initiated.
- ⁇ -Galactosylceramide is a glycosphingolipid in which galactose is bound in ⁇ -configuration to ceramide formed by acylating a sphingosine base with a long-chain fatty acid, but various analogs have been synthesized so far.
- the correlation between structure and activity has been investigated.
- ⁇ -GalCer represented by the following formula (a) (hereinafter referred to as “ ⁇ -GalCer”) exhibits the strongest activity, and further, the corresponding ⁇ -form ( ⁇ -GalCer) has been shown to have no immunostimulatory activity (Non-patent Document 7).
- NKT cells activated by administration of ⁇ -GalCer have the effect of enhancing IFN- ⁇ production by IFN- ⁇ and NKT cells, cytokines that induce immunostimulatory activity, useful for cancer treatment.
- cytokines that induce immunostimulatory activity useful for cancer treatment.
- IL-12 which is a cytokine produced by dendritic cells
- IL-4 which is a cytokine that induces an immunosuppressive action
- IL-10 which is a cytokine that induces an immunomodulatory action
- Non-Patent Document 8 ⁇ -C-GalCer
- ⁇ -C-GalCer is an analog in which the oxygen atom forming the glucoside bond of ⁇ -GalCer is replaced with a methylene group.
- ⁇ -C-GalCer it has been reported that the bond between sugar and ceramide is converted from a glycosidic bond to a carbon-carbon bond, which increases the in vivo stability and maintains the efficacy for a long time.
- Non-Patent Document 9 Non-Patent Document 9
- ⁇ -C-GalCer shows very weak activity in vitro against human NKT cells, clinical application is difficult.
- Carbaglycolipid A a novel glycolipid having a carbasugar represented by (hereinafter referred to as “carbaglycolipid A”) strongly induces the production of IFN- ⁇ on NKT cells (Non-patent Document 10). ).
- the compound is expected to be clinically applied because it exhibits strong activity in human (in vitro) systems.
- a simpler synthesis method or a simpler Development of novel analogs that can be prepared but have equivalent or higher activity is desired.
- the present invention has been made in view of such circumstances, and a problem to be solved is to provide a novel compound effective for cancer treatment and an intermediate useful for the synthesis of the compound. Moreover, it aims at providing pharmaceuticals, such as an anticancer agent containing this novel compound.
- the present inventors have obtained the knowledge that the compound represented by the following formula (1) selectively produces a specific cytokine. Furthermore, the present inventors have studied in detail, and have found that a specific immunostimulatory ability is expressed by selective production of a specific cytokine and is extremely effective for cancer treatment, and the present invention has been completed.
- compound (1) A compound represented by the following general formula (1) (hereinafter referred to as “compound (1)”) or a salt thereof.
- R 1 represents a hydrogen atom, an alkyl group having 1 to 7 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, or a halogen atom
- R 2 and R 3 each independently represents a substituent having 1 to 28 carbon atoms.
- Y represents —CH 2 —, —CH (OH) — or —CH ⁇ CH—.
- R 1 is a hydrogen atom
- R 2 represents a substituted or unsubstituted hydrocarbon group having 24 to 28 carbon atoms.
- R 1 is an alkyl group having 1 to 7 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, or a halogen atom
- R 2 is a substituted or unsubstituted alkyl group having 1 to 28 carbon atoms, or
- R 1 is a hydrogen atom
- R 2 is a substituted or unsubstituted alkyl group having 24 to 28 carbon atoms, or a salt or a salt thereof according to [1] or [2] above.
- [4] The compound or salt thereof according to any one of [1] to [3] above, wherein R 3 is a substituted or unsubstituted alkyl group having 1 to 28 carbon atoms.
- R 1 represents a hydrogen atom, an alkyl group having 1 to 7 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, or a halogen atom
- R 2 and R 3 each independently represents a substituent having 1 to 28 carbon atoms.
- Y 1 represents —CH 2 —, —CH (OA 1 ) — or —CH ⁇ CH—
- a 1 represents a hydrogen atom or a hydroxyl protecting group
- a 2 represents A protecting group for a hydroxyl group is shown.
- R 1 is a hydrogen atom
- R 2 represents a substituted or unsubstituted hydrocarbon group having 24 to 28 carbon atoms.
- a pharmaceutical comprising compound (1) or a salt thereof.
- a selective IFN- ⁇ production inducer comprising compound (1) or a salt thereof.
- An anticancer agent comprising compound (1) or a salt thereof.
- An immunostimulation method comprising administering an effective amount of compound (1) or a salt thereof to a subject.
- a method for selectively inducing IFN- ⁇ production comprising administering an effective amount of compound (1) or a salt thereof to a subject.
- a method for treating cancer comprising administering an effective amount of compound (1) or a salt thereof to a subject.
- Use of compound (1) or a salt thereof for the production of an anticancer agent Use of compound (1) or a salt thereof for the production of an anticancer agent.
- the present inventors have determined that the hydroxyl group at the 6-position of sugar, which is a part of the general skeleton of galactosylceramide, which is a kind of glycolipid, has other functional groups (one carbon atom). It has been found that a compound converted to an alkyl group of ⁇ 7, an alkoxy group of 1 to 6 carbon atoms, a halogen atom) or a hydrogen atom has a specific immunomodulatory ability and is extremely effective for cancer treatment. The present invention has been completed.
- the compound (1) of the present invention forms a complex with the CD1d protein possessed by the antigen-presenting cell (APC) and is presented to NKT cells.
- APC antigen-presenting cell
- NKT cells recognize this complex via TCR and preferentially produce a large amount of IFN- ⁇ , which is a kind of cytokine that activates the function of immune cells, among its immunoregulatory ability.
- the compound (1) of the present invention strongly activates NKT cells even in a trace amount, and produces a larger amount of IFN- ⁇ than the compounds reported so far. Therefore, sufficient medicinal effects can be obtained with a small amount of administration.
- the compound (1) and salt thereof of the present invention are effective for cancer treatment and the like.
- the compound (2) and a salt thereof of the present invention are useful as a synthetic intermediate for the compound (1) and a salt thereof.
- FIG. 1 is a graph showing the concentration of IFN- ⁇ in serum after administration of a synthetic glycolipid to mice in vivo and after the lapse of the indicated time.
- FIG. 2 is a diagram showing the concentration of IL-4 in serum after administration of a synthetic glycolipid to mice in vivo and after the lapse of the indicated time.
- FIG. 3 is a diagram showing the concentration of IL-12 in serum after the indicated time has elapsed after administration of a synthetic glycolipid to mice in vivo.
- FIG. 4 is a graph showing the concentration of IFN- ⁇ in serum after the indicated time has elapsed after the synthetic glycolipid was administered intravenously to the mouse tail.
- FIG. 5 is a graph showing the concentration of IFN- ⁇ in serum after the indicated time has elapsed after a synthetic glycolipid was administered intravenously to the mouse tail.
- FIG. 6 is a graph showing the concentration of IL-4 in serum after administration of a synthetic glycolipid to the tail of a mouse by intravenous injection and after the indicated time.
- FIG. 7 is a graph showing the concentration of IL-4 in serum after administration of synthetic glycolipids to the tail of mice by intravenous injection and after the lapse of the indicated time.
- FIG. 8 is a diagram showing the concentration of IL-12 in serum after the indicated time has elapsed after intravenous administration of a synthetic glycolipid to the mouse tail.
- FIG. 9 is a diagram showing the concentration of IL-12 in serum after the indicated time has elapsed after intravenous administration of a synthetic glycolipid to the mouse tail.
- R 1 represents a hydrogen atom, an alkyl group having 1 to 7 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, or a halogen atom.
- the alkyl group having 1 to 7 carbon atoms represented by R 1 represents a substituted or unsubstituted alkyl group, which may form a ring.
- the alkoxy group having 1 to 6 carbon atoms represented by R 1 is a group in which a substituted or unsubstituted alkyl group is bonded to an oxygen atom, and the alkyl portion may form a ring.
- Examples include methoxy, ethoxy, n-propyloxy, isopropyloxy, cyclopropyloxy, cyclopropylmethyloxy, n-butoxy, isobutyloxy, sec-butyloxy, tert-butyloxy, pentyloxy, hexyloxy, cyclohexyloxy and the like. , Methoxy, ethoxy and n-propyloxy are preferred.
- Examples of the halogen atom represented by R 1 include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom, and a fluorine atom and a chlorine atom are preferable.
- R 2 and R 3 each independently represents a substituted or unsubstituted hydrocarbon group having 1 to 28 carbon atoms.
- the “hydrocarbon group” means a substituted or unsubstituted alkyl group having 1 to 28 carbon atoms, an alkenyl group having 2 to 28 carbon atoms, an alkynyl group having 2 to 28 carbon atoms, or 3 to 28 carbon atoms.
- a cycloalkyl group, a C3-C28 cycloalkenyl group, and a C6-C14 aryl group which may be linear, branched or cyclic, It may be a saturated hydrocarbon group or an unsaturated hydrocarbon group, and may have an unsaturated bond either in the molecule or at the end.
- R 2 and R 3 a substituted or unsubstituted alkyl group having 1 to 28 carbon atoms is preferable.
- R 2 represents a substituted or unsubstituted hydrocarbon group having 24 to 28 carbon atoms, preferably a substituted or unsubstituted alkyl group having 24 to 28 carbon atoms.
- Examples of the substituent of the hydrocarbon group represented by R 2 and R 3 include a halogen atom (preferably a chlorine atom and a fluorine atom); a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group, a butoxy group, a tert-butoxy group, and the like Alkoxy groups (preferably having 1 to 24 carbon atoms, more preferably 1 to 16 carbon atoms, still more preferably 1 to 10 carbon atoms, particularly preferably 1 to 4 carbon atoms); aryloxy groups such as phenoxy groups (preferably C 6-14); hydroxyl group; amino group; alkylamino group such as methylamino group, dimethylamino group, ethylamino group, diethylamino group; cycloalkylamino group; alkylcarbonylamino group such as acetamide group; cycloalkylcarbonylamino An arylcarbonylamino group such as a benzo
- an alkyl moiety is examples thereof include an electron withdrawing group such as an alkyl-carbonyl group which is a linear or branched alkyl group having 1 to 24 carbon atoms; a carbamoyl group; a trifluoromethyl group.
- an electron withdrawing group such as an alkyl-carbonyl group which is a linear or branched alkyl group having 1 to 24 carbon atoms; a carbamoyl group; a trifluoromethyl group.
- acyl group refers to, for example, a formyl group; an alkyl-carbonyl group (for example, a linear or branched alkyl group having an alkyl moiety of 1 to 24 carbon atoms (preferably 1 to 12 carbon atoms).
- alkyl-carbonyl group for example, acetyl group, propionyl group, butyryl group, isobutyryl group, valeryl group, pivaloyl group, hexanoyl group
- a cycloalkyl-carbonyl group for example, a cycloalkyl moiety having 3 to 3 carbon atoms
- An alkenyl-carbonyl group eg, acryloyl
- alkenyl moiety is a linear or branched alkenyl group having 2 to 12 carbon atoms; Group, methacryloyl group
- aryl-carbonyl group eg ant Le moiety is an aryl group having 6 to 14 carbon atoms, aryl - carbonyl group (e.g., benzoyl group, naphthoyl group) refers to a), or the like.
- the aryl group in the aryl-carbonyl group is, for example, a monocyclic to tricyclic aromatic hydrocarbon group, and specific examples thereof include a phenyl group, a naphthyl group, an anthryl group, and a phenanthryl group.
- a formyl group, an acetyl group, a propionyl group, a butyryl group, an isobutyryl group, a benzoyl group, a naphthoyl group, and the like are preferable, and an acetyl group and a benzoyl group are more preferable.
- alkyl moiety of the alkylamino group and alkylcarbonylamino group examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, Linear or branched alkyl group such as hexyl group, heptyl group, octyl group, nonyl group, decyl group, undecyl group, dodecyl group, tridecyl group, tetradecyl group, pentadecyl group, hexadecyl group, heptadecyl group, octadecyl group (preferably Is exemplified by 1 to 24 carbon atoms, more preferably 1 to 16 carbon atoms, still more preferably 1 to 10 carbon atoms, and particularly preferably 1 to 4 carbon atoms
- Examples of the cycloalkyl portion of the cycloalkylamino group and cycloalkylcarbonylamino group include cycloalkyl groups such as cyclopentyl group and cyclohexyl group (preferably having 3 to 24 carbon atoms, more preferably 3 to 16 carbon atoms, and still more preferably carbon atoms). Examples thereof include 3 to 10, particularly preferably 3 to 6 carbon atoms. Examples of the alkoxy moiety of the alkoxycarbonyl group include those similar to the alkoxy group.
- the above-described substituent is further substituted at a substitutable position among halogen, alkyl group, cycloalkyl group, alkenyl group, alkynyl group, phenyl group, alkoxy group, hydroxyl group, amino group, alkylamino group and cycloalkylamino group. It may be substituted with at least one of the above.
- Examples of the halogen, alkoxy group, alkylamino group and cycloalkylamino group are the same as those described above.
- alkyl group examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group, heptyl group, octyl group, An alkyl group such as a nonyl group, a decyl group, an undecyl group, a dodecyl group, a tridecyl group, a tetradecyl group, a pentadecyl group, a hexadecyl group, a heptadecyl group, an octadecyl group (preferably having 1 to 24 carbon atoms, more preferably 1 to 16 carbon atoms More preferred are those having 1 to 10 carbon atoms, particularly preferably 1 to 4 carbon atoms.
- Examples of the cycloalkyl group include cycloalkyl groups such as a cyclopentyl group and a cyclohexyl group (preferably having 3 to 24 carbon atoms, more preferably 3 to 16 carbon atoms, still more preferably 3 to 10 carbon atoms, and particularly preferably 3 carbon atoms).
- Examples of the alkenyl group include alkenyl groups such as vinyl group, propenyl group, and butenyl group (preferably having 2 to 24 carbon atoms, more preferably 2 to 16 carbon atoms, still more preferably 2 to 10 carbon atoms, and particularly preferably carbon number).
- Examples of the alkenyl group include alkenyl groups such as vinyl group, propenyl group, and butenyl group (preferably having 2 to 24 carbon atoms, more preferably 2 to 16 carbon atoms, still more preferably 2 to 10 carbon atoms, and particularly preferably carbon number).
- alkynyl group examples include alkynyl groups such as ethynyl group, propargyl group, butynyl group, pentynyl group (preferably having 2 to 24 carbon atoms, more preferably 2 to 16 carbon atoms, still more preferably 2 to 10 carbon atoms, and particularly preferably Is exemplified by 2 to 4 carbon atoms.
- R 2 a substituted or unsubstituted alkyl group is preferable, and a linear alkyl group is preferable.
- R 1 is an alkyl group having 1 to 7 carbon atoms, an alkoxy group having 1 to 6 carbon atoms, or a halogen atom
- the carbon number of R 2 is preferably 18 to 26, more preferably 24 to 26.
- R 1 is a hydrogen atom
- the carbon number of R 2 is preferably 24-26.
- Specific examples of R 2 include — (CH 2 ) 23 —CH 3 , — (CH 2 ) 24 —CH 3 , — (CH 2 ) 25 —CH 3 and the like.
- R ⁇ 3 > a substituted or unsubstituted alkyl group is preferable and a linear alkyl group is preferable.
- R 3 preferably has 9 to 20 carbon atoms, more preferably 12 to 18 carbon atoms. Specific examples of R 3 include — (CH 2 ) 11 —CH 3 , — (CH 2 ) 12 —CH 3 , — (CH 2 ) 13 —CH 3 , — (CH 2 ) 14 —CH 3. , — (CH 2 ) 15 —CH 3 , — (CH 2 ) 16 —CH 3 , — (CH 2 ) 17 —CH 3 and the like.
- Y represents —CH 2 —, —CH (OH) — or —CH ⁇ CH—, among which —CH (OH) — is preferred.
- Y 1 represents —CH 2 —, —CH (OA 1 ) — or —CH ⁇ CH—, and —CH (OA 1 ) — is particularly preferred.
- a 1 is as described below.
- a 1 represents a hydrogen atom or a hydroxyl-protecting group, and examples of the hydroxyl-protecting group include an acyl group, a t-butyldimethylsilyl (TBS) group, a benzyl (Bn) group, and a p-methoxybenzyl (PMB) group.
- TBS t-butyldimethylsilyl
- Bn benzyl
- PMB p-methoxybenzyl
- TBS groups and Bn groups are preferred.
- the two A 1 groups may be the same or different, but are preferably the same.
- Y 1 is —CH (OA 1 ) —
- two A 1 may be taken together to form a diol protecting group. Examples of the protecting group for the diol include:
- Examples of the hydroxyl protecting group represented by A 2 include an acyl group, a TBS group, a trimethylsilyl (TMS) group, a Bn group, and a PMB group.
- the acyl group is as described above. Among these, Bn group and PMB group are preferable.
- the ⁇ isomer is adopted among the stereoisomers derived from the cyclic structure of the sugar, but the present inventors have obtained the knowledge that the cytokine production ability is extremely reduced in the ⁇ isomer.
- any isomer is included in the present invention, and is a mixture (including a racemate) of any ratio of two or more isomers. May be.
- compound (1) has an optical isomer derived from the asymmetric carbon of the lipid moiety, but in the present invention, two or more optically active isomers can be used even if they are a single optically active isomer. It may be a mixture in any proportion (including a racemate).
- the asymmetric carbon to which —NHCOR 2 is bonded is preferably S configuration.
- Asymmetric carbon having adjacent asymmetric carbon -NHCOR 2 binds -OH, it is preferred arrangement of anti against asymmetric carbon -NHCOR 2 is attached.
- Y is —CH (OH) —
- the asymmetric carbon in —CH (OH) — represented by Y is preferably R configuration.
- Compound (2) has an optical isomer derived from the asymmetric carbon of the lipid moiety, but in the present invention, two or more optically active isomers may be used, even if they are a single optically active isomer. It may be a mixture in any proportion (including a racemate).
- the asymmetric carbon to which —NHCOR 2 is bonded is preferably S configuration.
- Asymmetric carbon -NHCOR 2 has a -OA 1 adjacent to the asymmetric carbon bonded, it is preferred arrangement of anti against asymmetric carbon -NHCOR 2 is attached.
- Y 1 is —CH (OA 1 ) —
- the asymmetric carbon in —CH (OA 1 ) — represented by Y 1 is preferably in the R configuration.
- the salt of compound (1) and compound (2) is preferably a pharmacologically acceptable salt, such as hydrochloride, hydrobromide, hydroiodide, sulfate, nitrate, phosphate, etc.
- a pharmacologically acceptable salt such as hydrochloride, hydrobromide, hydroiodide, sulfate, nitrate, phosphate, etc.
- Inorganic acid salts organic acid salts such as succinate, fumarate, acetate, methanesulfonate and toluenesulfonate
- alkali metal salts such as sodium salt and potassium salt
- alkali such as magnesium salt and calcium salt Earth metal salts
- ammonium salts such as ammonium salts and alkylammonium salts.
- preferred compound (1) in the present invention include compound 2-9, compound 3-4, compound 3-7, compound 3-10, compound 3-13 and compound 3-16 described in the Examples. Although it is mentioned, it is not limited to these.
- suitable compound (2) in the present invention include, but are not limited to, compounds 2-7 and 2-8 described in the Examples.
- the compound of the present invention can be produced by various methods known per se to those skilled in the art.
- compounds (1) and (2) can be produced according to the method described in the following scheme or a method analogous thereto. Is possible.
- Z represents a halogen atom (for example, a fluorine atom)
- Y 2 represents —CH 2 —, —CH (OA) — or —CH ⁇ CH—
- A represents a hydroxyl-protecting group
- Each symbol has the same meaning as described above.
- the protective group for the hydroxyl group represented by A the same as the protecting group for the hydroxyl group represented by A 1 described above is illustrated.
- a compound (2 ') and a compound (2'') are included by the compound (2) of this invention.
- the raw material compound (A) is obtained, for example, from the compound 2-2 ′ produced by the method described in T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45 or a method analogous thereto. Can be prepared as follows.
- R represents alkyl having 1 to 6 carbon atoms (eg, methyl, ethyl, n-propyl), and other symbols are as defined above.
- Compound 2-3 ′ can be obtained by reacting compound 2-2 ′ with an alkyl halide in the presence of a base.
- the base include sodium hydride and n-butyllithium.
- the amount of the base to be used is generally 1 to 3 equivalents relative to compound 2-2 ′.
- the alkyl halide include methyl iodide, ethyl iodide, propyl bromide and the like.
- the amount of the alkyl halide to be used is generally 1 to 3 equivalents relative to compound 2-2 ′.
- the solvent examples include aprotic solvents such as N, N-dimethylformamide, ethers (eg, diethyl ether, tetrahydrofuran), and mixed solvents thereof.
- the amount of the solvent to be used is generally 10 to 20 times the volume of Compound 2-2 ′.
- the reaction temperature is usually 0 to 80 ° C., and the reaction time is usually 1 to 24 hours.
- Compound 2-3 ′ can be isolated by a conventional method, for example, water is added to the reaction solution, and the mixture is extracted with ethyl acetate. The organic layer is washed with water and saturated brine, and then dried over anhydrous magnesium sulfate.
- Compound 2-3 ′ can be isolated by filtration and concentration.
- Compound 2-3 ′ can be directly reacted with an acid to give compound 2-4 ′.
- compound 2-3 ′ can be obtained by subjecting compound 2-3 ′ to the corresponding O-acetyl form and subjecting it to alcoholysis.
- an O-acetyl form is prepared by treating with a catalytic amount of acid in acetic anhydride, and this is subjected to alcoholysis.
- the acid include concentrated sulfuric acid, concentrated hydrochloric acid, p-toluenesulfonic acid and the like.
- the amount of acetic anhydride used is usually 5 to 20 times the volume of compound 2-3 ′.
- the reaction temperature is usually from 0 ° C.
- O-acetyl compound can be obtained by concentration under reduced pressure. Alcohololysis of the obtained O-acetyl compound is treated with a base such as sodium methoxide or sodium hydroxide in an alcohol solvent such as methanol or ethanol.
- Compound 2-4 ′ can be isolated by a conventional method. For example, it may be acidified with a cation exchange resin, filtered, concentrated and purified.
- Compound 2-5 ′ can be obtained by reacting compound 2-4 ′ with a halogenating agent.
- the halogenating agent include diethylaminosulfur trifluoride, tris (dimethylamino) sulfonium difluorotrimethylsilicate, and the like.
- the amount of the halogenating agent to be used is generally 1 to 3 equivalents relative to compound 2-4 ′.
- the solvent include dichloromethane and the like.
- the amount of the solvent to be used is generally 10 to 30 times the volume of the compound 2-4 ′.
- the reaction temperature is usually ⁇ 78 ° C. to room temperature, and the reaction time is usually 30 minutes to 1 hour.
- Compound 2-5 ′ can be isolated by a conventional method.
- the compound 2-5 ′ can be isolated by drying over anhydrous magnesium sulfate, filtration and concentration.
- each symbol has the same meaning as described above.
- Compound 3-1 ′ produced by the method described in K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203 or a method analogous thereto, and hydrazine monohydrate, hydrogen peroxide solution and The compound 3-2 ′ can be obtained by reaction.
- the amount of hydrazine monohydrate to be used is generally 5 to 20 equivalents relative to compound 3-1 ′.
- the amount of hydrogen peroxide used is usually 5 to 20 equivalents relative to compound 3-1 ′.
- Examples of the solvent include ethanol.
- the amount of the solvent to be used is generally 10 to 50 times the volume of Compound 3-1 ′.
- the reaction temperature is usually from room temperature to 80 ° C., and the reaction time is usually from 1 to 20 hours.
- Compound 3-2 ′ can be isolated by a conventional method. For example, saturated sodium thiosulfate is added to the reaction solution, diluted with ethyl acetate, and the organic layer is washed with water, saturated aqueous sodium thiosulfate solution, and saturated brine. After that, compound 3-2 ′ can be isolated by drying over anhydrous magnesium sulfate, filtering, and concentrating. The conversion from compound 3-2 ′ to compound 3-3 ′ can be performed by the same method as the conversion from compound 2-3 ′ to compound 2-5 ′.
- compound 3-1 ′ can be expressed as follows.
- B 1 and B 2 represent a substituted or unsubstituted alkyl group, and B 1 and B 2 may form a ring.
- other symbols have the same meaning as described above.
- W represents a halogen atom (eg, fluorine atom), and other symbols have the same meanings as described above.
- Compound 3-5 ′ can be obtained by reacting compound 2-2 ′ with a halogenating agent in the presence of a base.
- the base include triethylamine, pyridine and the like.
- the amount of the base to be used is generally 1 to 5 equivalents relative to compound 2-2 ′.
- the halogenating agent include diethylaminosulfur trifluoride, tris (dimethylamino) sulfonium difluorotrimethylsilicate, and the like.
- the amount of the halogenating agent to be used is generally 1 to 3 equivalents relative to compound 2-2 ′.
- Examples of the solvent include dichloromethane and the like.
- the amount of the solvent to be used is generally 10 to 30 times the volume of Compound 2-2 ′.
- the reaction temperature is usually ⁇ 78 ° C. to room temperature, and the reaction time is usually 30 minutes to 2 hours.
- Compound 3-5 ′ can be isolated by a conventional method, for example, methanol is added to the reaction solution, diluted with ethyl acetate, and the organic layer is washed with water, saturated aqueous sodium hydrogen carbonate solution, saturated brine, Compound 3-5 ′ can be isolated by drying over anhydrous magnesium sulfate, filtration and concentration. Conversion from compound 3-5 ′ to compound 3-6 ′ can be carried out in the same manner as the conversion from compound 2-3 ′ to compound 2-5 ′.
- each symbol has the same meaning as described above.
- Compound 3-9 ′ is produced from compound 2-2 ′ by the method described in S. Koto et al. Bull. Chem. Soc. Jpn., 2000, 73, 967-976 or a method analogous thereto.
- Compound 3-8 ′ can be produced by a method similar to the conversion from compound 2-3 ′ to compound 2-5 ′.
- the starting compound (B) can be prepared, for example, according to the method described in H. Takikawa et al. Tetrahedron, 1998, 54, 3141-3150 or S. Kim et al. Synthesis, 2004, 847-850 or a method analogous thereto.
- Step1 In step 1, compound (A) is reacted with compound (B) in the presence of molecular sieves, tin (II) chloride and silver perchlorate to obtain compound (2 ′).
- the amount of compound (A) to be used is generally 1 to 3 equivalents relative to compound (B).
- the amount of molecular sieve used is usually 2 to 10 times the weight of the compound (B).
- the amount of tin (II) chloride used is usually 1.5 to 3 equivalents relative to compound (A).
- the amount of silver perchlorate used is usually 1.5 to 3 equivalents relative to compound (A).
- the solvent include aprotic solvents such as acetonitrile and ethers (eg, diethyl ether and tetrahydrofuran).
- the amount of the solvent used is usually 5 to 20 times the volume of the compound (A).
- the reaction temperature is usually ⁇ 18 to 20 ° C., and the reaction time is usually 1 to 2 hours.
- Compound (2 ′) can be isolated by a conventional method. For example, diethyl ether is added to the reaction solution and filtered, and the filtrate is washed with water and saturated brine, dried over anhydrous magnesium sulfate, and filtered. The compound (2 ′) can be isolated by concentration.
- Step 2 is a step of obtaining the compound (2 ′′) by removing the protecting group A from —OA of the compound (2 ′).
- the removal method is selected depending on the type of protecting group.
- the compound (2 ′) is reacted with quaternary ammonium fluoride (eg, tetra-n-butylammonium fluoride) in a solvent.
- the amount of quaternary ammonium fluoride to be used is generally 1 to 3 equivalents relative to compound (2 ′).
- the reaction temperature is usually from 0 ° C. to room temperature, and the reaction time is usually from 1 to 20 hours.
- the solvent examples include aprotic solvents such as N, N-dimethylformamide and ethers (eg, diethyl ether, tetrahydrofuran).
- the amount of the solvent to be used is generally 10 to 50 times the volume of the compound (2 ′).
- Compound (2 ′′) can be isolated by a conventional method. For example, water is added to the reaction solution, and the mixture is extracted with ethyl acetate. The organic layer is washed with water and saturated brine, and then dried over anhydrous magnesium sulfate. The compound (2 ′′) can be isolated by filtration, concentration, and the like.
- Step 3 is a step of obtaining the compound (1) by removing the protecting group for the hydroxyl group of the sugar moiety in the compound (2 ′′).
- the compound (2 ′′) is reacted in a solvent in a hydrogen atmosphere and in the presence of a reduction catalyst.
- the reduction catalyst include palladium-C, palladium hydroxide, palladium hydroxide-activated carbon, platinum oxide, Raney nickel and the like.
- the amount of the reduction catalyst used may be a catalytic amount with respect to the compound (2 ′′).
- the solvent examples include lower alcohols (eg, methanol, ethanol), halogenated hydrocarbons (eg, dichloromethane, chloroform) and ethers (eg, diethyl ether, tetrahydrofuran). May be.
- the amount of the solvent to be used is generally 20 to 200 times the volume of the compound (2 ′′).
- the reaction temperature is usually from room temperature to 50 ° C., and the reaction time is usually from 5 to 20 hours. After completion of the reaction, the reaction solution is filtered and concentrated to give compound (1).
- the compound (1) and compound (2) of the present invention obtained as described above can be converted into a target salt by a method known per se or a method analogous thereto.
- the pharmaceutical use of the present invention will be described.
- a complex with the CD1d protein possessed by APC is formed and presented to NKT cells.
- NKT cells recognize this complex via TCR, and selectively produce IFN- ⁇ , a kind of cytokine that activates the function of immune cells, among its own immunoregulatory capacity.
- IFN- ⁇ a kind of cytokine that activates the function of immune cells, among its own immunoregulatory capacity.
- the compound (1) of the present invention or a salt thereof induces the production of IL-12 having an action of enhancing IFN- ⁇ production by NKT cells.
- the IFN- ⁇ / IL-4 ratio was 10 or more with respect to 2 of ⁇ -GalCer, and extremely high selective IFN- ⁇ production was confirmed as compared with conventionally known glycolipids (Test Example). 1). Therefore, the compound (1) or a salt thereof of the present invention is useful for an anticancer agent for inhibiting tumor growth, an immunostimulant, and a treatment for correcting cell proliferation disorder and Th1 / Th2 immune balance.
- cancer treatment targets include esophagus, stomach, liver, pancreas, breast, colon, kidney, lung (including small cell lung cancer and non-small cell lung cancer), gallbladder, ovary, testis, bladder, cervix, thyroid, Tumors of prostate and skin (including squamous cell carcinoma); hematopoietic tumors of lymphoid lineage (leukemia, acute lymphocytic leukemia, acute lymphoblastic leukemia, B cell lymphoma, T cell lymphoma, Hodgkin lymphoma, non-Hodgkin lymphoma, hairy Hematopoietic tumors of the myeloid lineage (including acute and chronic myelogenous leukemia, myelodysplastic syndrome and promyelocytic acute leukemia); tumors of mesenchymal origin (fibrosarcoma and rhabdomyosarcoma) Central nervous system and peripheral nervous system tumors (including astrocytoma, neuroblastom
- Cell proliferation disorders include familial adenomatous polyposis, psoriasis, benign prostatic hyperplasia, neurofibromatosis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary fibrosis, arthritis, glomerulonephritis, surgery It is a concept that includes later stenosis and restenosis.
- Examples of the administration target of the compound (1) of the present invention or a salt thereof include mammals such as humans.
- the compound (1) or a salt thereof of the present invention When the compound (1) or a salt thereof of the present invention is administered to humans, it is mixed with itself or a pharmacologically acceptable carrier (for example, excipient, diluent), etc. , Powders, granules, tablets, capsules), parenteral administration agents (for example, injections, suppositories (for example, rectal suppositories, vaginal suppositories)) and the like orally or parenterally safe Can be administered.
- a pharmacologically acceptable carrier for example, excipient, diluent), etc.
- parenteral administration agents for example, injections, suppositories (for example, rectal suppositories, vaginal suppositories)) and the like orally or parenterally safe Can be administered.
- suppositories for example, rectal suppositories, vaginal
- the injection examples include subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection, and infusion.
- the injection comprises compound (1) or a salt thereof as a solubilizer (for example, ⁇ -cyclodextrins), a dispersant (for example, carboxymethylcellulose, sodium alginate), a preservative (for example, methylparaben, propylparaben, benzyl alcohol, Chlorobutanol), isotonic agents (for example, sodium chloride, glycerin, sorbitol, glucose) and the like, and an aqueous injection can be obtained according to a conventional method.
- a solubilizer for example, ⁇ -cyclodextrins
- a dispersant for example, carboxymethylcellulose, sodium alginate
- a preservative for example, methylparaben, propylparaben, benzyl alcohol, Chlorobutanol
- isotonic agents for example, sodium chloride
- the oral administration agent is prepared by adding the compound (1) or a salt thereof to, for example, an excipient (eg, lactose, sucrose, starch), a disintegrant (eg, starch, calcium carbonate), a binder (eg, starch, gum arabic, Carboxymethylcellulose, polyvinylpyrrolidone, hydroxypropylcellulose) or lubricants (eg, talc, magnesium stearate, polyethylene glycol), etc. are added as appropriate and compression molded, followed by coating with hydroxypropylmethylcellulose as necessary. Can also be manufactured. Suppositories can be produced by mixing compound (1) or a salt thereof and a non-irritating excipient (for example, polyethylene glycol, glyceride of higher fatty acid).
- an excipient eg, lactose, sucrose, starch
- a disintegrant eg, starch, calcium carbonate
- a binder eg, starch, gum arabic, Carboxymethyl
- the dose of compound (1) or a salt thereof varies depending on age, body weight, symptom, dosage form, administration method, administration period, etc.
- per patient adult, body weight of about 60 kg
- Step b Synthesis of Compound 2-4
- a solution of Compound 2-3 (733 mg, 1.53 mmol) in acetic anhydride (20 mL) was added to a solution of concentrated sulfuric acid (0.03 mL) in acetic anhydride (10 mL) under ice cooling. Added and stirred for 20 minutes.
- Saturated aqueous sodium hydrogen carbonate solution was added for neutralization, and the mixture was diluted with ethyl acetate.
- the organic layer was washed successively with a saturated aqueous sodium hydrogen carbonate solution, water and saturated brine, and then dried over anhydrous magnesium sulfate. After filtration, the solvent was removed by concentration under reduced pressure.
- Step c Synthesis of Compound 2-5 Diethylaminosulfur trifluoride (0.35 mL, 2.65 mmol) was added at ⁇ 40 ° C. to a solution of Compound 2-4 (602 mg, 1.30 mmol) in dichloromethane (20 mL). After stirring at room temperature for 1 hour, the mixture was cooled again to ⁇ 40 ° C., and methanol (1 mL) was added. The solvent was removed by concentration under reduced pressure, and the residue was diluted with ethyl acetate. The organic layer was washed successively with a saturated aqueous sodium hydrogen carbonate solution, water and saturated brine, and dried over anhydrous magnesium sulfate.
- Step d Synthesis of Compound 2-7
- Compound 2-6 (438 mg, 0.474 mmol) in dry tetrahydrofuran (15 mL) solution, Dry molecular sieves (4.03 g), tin (II) chloride (270 mg, 1.42 mmol) and silver perchlorate (300 mg, 1.45 mmol) were added, and the mixture was stirred in a light-shielded flask for 2 hours.
- n D 24 1.4952.
- Step e Synthesis of Compound 2-8
- tetra-n-butylammonium fluoride in tetrahydrofuran 1.0 M, 370 ⁇ L, 0.37 mmol
- Step f Synthesis of Compound 2-9
- tetrahydrofuran-ethanol-chloroform 5: 8: 2,15 mL
- palladium hydroxide-activated carbon 20%, wet , 33 mg
- the mixture was diluted with chloroform-methanol (5: 1).
- Step g Synthesis of Compound 3-2 Literature known (K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203) Compound 3-1 (1.12 g, 2.43 mmol) and hydrazine monohydrate ( To a solution of 10 mL, 20.6 mmol) in ethanol (50 mL) was slowly added hydrogen peroxide (30%, 4 mL) at room temperature over 3 hours. After the reaction solution was stirred at room temperature for 18 hours, a saturated aqueous sodium thiosulfate solution (20 mL) was added under ice cooling, and the mixture was stirred for 30 minutes.
- IR (film): ⁇ max 1605 (w, arom.), 1500 (s, arom.), 1100 (br.s, CO), 1050 (br.s, CO), 740 (s), 700 (s ) cm -1 .
- Step h Synthesis of Compound 3-5 Literature known (TJ Lucas et al., Carbohydr. Res., 1975, 39, 39-45)
- Compound 2-2 (2.03 g, 4.37 mmol) and triethylamine (1.85 mL, 13.3 mmol) in dichloromethane (30 mL) was slowly added diethylaminosulfur trifluoride (1.20 mL, 9.08 mmol) at ⁇ 40 ° C.
- the reaction mixture was stirred for 5 hours under reflux with heating, methanol (2 mL) was added under ice cooling, and the mixture was stirred for 30 minutes.
- Step i Synthesis of Compound 3-11 Literature known (TJ Lucas et al., Carbohydr. Res., 1975, 39, 39-45) N, N-dimethylformamide of Compound 2-2 (587 mg, 1.26 mmol) -Sodium hydride (60% mineral oil suspension, 158 mg, 3.95 mmol) was added to a solution of tetrahydrofuran (1: 1, 20 mL) under ice cooling. After stirring for 10 minutes under ice cooling, ethyl bromide (295 ⁇ L, 3.95 mmol) and a catalytic amount of tetra-n-butylammonium iodide were added, and the mixture was stirred at room temperature for 12 hours.
- IR (film): ⁇ max 1605 (w, arom.), 1495 (m, arom.), 1115 (br.s, CO), 1050 (br.s, CO), 735 (br.s), 700 (s) cm -1 .
- Step j Synthesis of Compound 3-14 Literature known (TJ Lucas et al., Carbohydr. Res., 1975, 39, 39-45) N, N-dimethylformamide of Compound 2-2 (630 mg, 1.36 mmol) -Sodium hydride (60% mineral oil suspension, 170 mg, 4.25 mmol) was added to a solution of tetrahydrofuran (1: 1, 20 mL) under ice cooling. After stirring for 10 minutes under ice cooling, 1-propyl bromide (390 ⁇ L, 4.28 mmol) and a catalytic amount of tetra-n-butylammonium iodide were added, and the mixture was stirred at room temperature for 12 hours.
- IR (film): ⁇ max 1605 (w, arom.), 1495 (m, arom.), 1110 (br.s, CO), 1050 (br.s, CO), 740 (br.s), 700 (s) cm -1 .
- Test Example 1 Biological Activity Test of Compound 2-9, Compound 3-4, Compound 3-7 and Compound 3-10 ⁇ -GalCer, Carbaglycolipid A, Compound 2-9, Compound 3-4, Compound 3-7 and A DMSO solution with a concentration of 1 mg / mL was prepared for each of compounds 3-10.
- the DMSO solution was added to physiological saline containing 0.5% tween20 (Bio-Rad) so that the dose would be 100 ⁇ g / kg body weight. It was diluted using Otsuka Pharmaceutical Co., Ltd.
- mice One group of 5 C57BL / 6 mice were each injected with 200 ⁇ L of the prepared carbaglycolipid A, compound 2-9, compound 3-4, compound 3-7 and compound 3-10 solution into the mouse tail vein.
- ⁇ -GalCer As a control substance, 200 ⁇ L of ⁇ -GalCer solution prepared to give a dose of 100 ⁇ g / kg body weight by the same method was injected into the tail vein of mice.
- a group to which 200 ⁇ L of physiological saline containing 0.5% tween 20 as a medium was administered was used as a negative control.
- 80 ⁇ L of blood was collected from the inferior venous plexus after 6, 12, 24, 36, 48, and 60 hours after administration, and serum was prepared.
- the content of IFN- ⁇ in serum after 6, 12, 24, 36, 48, and 60 hours after administration was measured by sandwich ELISA (ENDOGEN).
- the measurement results (average value) of IFN- ⁇ production and the standard deviation (STDEV) are shown in FIG.
- the content of IL-4 in serum after 3, 6 and 12 hours after the administration was measured by Cytometric bead array system (BD Biosciences), which is one of ELISA methods.
- FIG. 2 shows the measurement results (average value) of IL-4 production and its standard deviation (STDEV).
- the content of IL-12 in the serum after 3, 6 and 12 hours after administration was measured with a Cytometric bead array system (BD Biosciences), which is one of the ELISA methods.
- the measurement result (average value) of IL-12 production and its standard deviation (STDEV) are shown in FIG.
- Test Example 2 Biological Activity Test of Compound 2-9, Compound 3-4, Compound 3-7, Compound 3-10, Compound 3-13 and Compound 3-16
- ⁇ -GalCer compound 2-9, Compound 3-4, Compound 3-7, Compound 3-10, Compound 3-13 and Compound 3-16
- IFN in serum after 6, 12, 24, 36, 48, 60 hours after administration The content of - ⁇ was measured.
- the measurement results (average value) of IFN- ⁇ production and its standard deviation (STDEV) are shown in FIG.
- FIG. 6 shows the measurement results (average value) of IL-4 production and its standard deviation (STDEV). 3 and 6 hours after administration of ⁇ -GalCer, Compound 2-9, Compound 3-4, Compound 3-7, Compound 3-10, Compound 3-13 and Compound 3-16 in the same manner as in Test Example 1 Later, the content of IL-4 in the serum was measured.
- FIG. 6 shows the measurement results (average value) of IL-4 production and its standard deviation (STDEV).
- Test Example 3 Biological Activity Test of Compound 2-9
- the dose of ⁇ -GalCer was 2 ⁇ g / mouse
- the dose of Compound 2-9 was 2 ⁇ g / mouse, 0.2 ⁇ g / mouse, 0.02 ⁇ g / mouse, 0.002 ⁇ g / mouse, 0.0002 IFN- in serum after 6, 12, 24, 36, 48, and 60 hours after administration of ⁇ -GalCer and Compound 2-9 in the same manner as in Test Example 1 except that the concentration was ⁇ g / mouse.
- the content of ⁇ was measured.
- the measurement result (average value) of IFN- ⁇ production and its standard deviation (STDEV) are shown in FIG.
- ⁇ -GalCer and Compound 2 were administered in the same manner as in Test Example 1 except that the dose of ⁇ -GalCer and the dose of Compound 2-9 were 2 ⁇ g / mouse, 0.2 ⁇ g / mouse, and 0.02 ⁇ g / mouse.
- the content of IL-4 in the serum after 3 to 6 hours after administration of -9 was measured.
- FIG. 7 shows the measurement results (average value) of IL-4 production and its standard deviation (STDEV).
- ⁇ -GalCer and Compound 2 were administered in the same manner as in Test Example 1 except that the dose of ⁇ -GalCer and the dose of Compound 2-9 were 2 ⁇ g / mouse, 0.2 ⁇ g / mouse, and 0.02 ⁇ g / mouse.
- the content of IL-12 (p70) in the serum was measured 3 to 6 hours after administration of -9.
- FIG. 9 shows the measurement results (average value) of IL-12 production and its standard deviation (STDEV).
- Compound 2-9 induced production of IFN- ⁇ in a larger amount than ⁇ -GalCer even when administered at a low concentration.
- Compound 2-9 induced the production of IL-12 in the same amount to 4 times that of ⁇ -GalCer.
- the production amount of IL-4 decreased with decreasing dose of compound 2-9. This indicates that Compound 2-9 selectively induces production of a large amount of IFN- ⁇ compared to ⁇ -GalCer even when administered at a low concentration.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Dermatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本発明は、癌治療等に有効な新規糖脂質及びその新規合成中間体、並びに該新規糖脂質を含有する医薬等を提供する。 下記一般式(1)で表される化合物又はその塩。 (式中、各記号の定義は明細書に記載の通りである。)
Description
本発明は、新規糖脂質及びその用途に関する。
免疫系には、生体において自己の正常細胞と異常細胞とを区別し、異常細胞のみを排除するための巧みな監視機能が存在する。しかしその監視機能が破綻すると、突然変異等によって生まれる異常細胞を排除することができず、生体内での増殖を許してしまう。こうして増殖した異常細胞の塊が腫瘍、即ち癌である。
癌の治療法は、外科手術による癌の摘出、あるいは抗癌剤の使用が主である。しかしながら、これらの治療法は、摘出手術や抗癌剤の副作用による身体的な、あるいは手術痕による精神的な負担をかける。
その様な背景の中、免疫療法を併用した治療法が注目を集めている。免疫療法では、患者自身の免疫細胞数を増やし、さらに活性化することで癌細胞を攻撃する。癌細胞によって形成された腫瘍を小さくすることが出来れば、その摘出手術による身体への負担は小さい。また手術痕もわずかですむため、精神的な負担も大幅に軽減される。
その様な背景の中、免疫療法を併用した治療法が注目を集めている。免疫療法では、患者自身の免疫細胞数を増やし、さらに活性化することで癌細胞を攻撃する。癌細胞によって形成された腫瘍を小さくすることが出来れば、その摘出手術による身体への負担は小さい。また手術痕もわずかですむため、精神的な負担も大幅に軽減される。
ナチュラルキラー(NK)T細胞は、他のリンパ球系列(T, B, NK細胞)と異なる特徴を示す、新規リンパ球系列に属する免疫細胞である。NKT細胞内には細胞障害性パーフォリン顆粒が存在することからNK細胞と類縁である(非特許文献1)。しかしNKT細胞は、NK細胞マーカーのみならずT細胞受容体(TCR)をも発現していることから、決定的に異なる新たな細胞群であることが明らかとなっている(非特許文献2)。NKT細胞は、免疫賦活作用を亢進させるヘルパーT(Th)-1細胞によって産生されるTh-1型サイトカイン(主にインターフェロン(IFN)-γ)と、免疫抑制作用を亢進させるTh-2細胞によって産生されるTh-2型サイトカイン(主にインターロイキン(IL)-4)の両方を産生することができ(非特許文献3)、これによって免疫系のバランスを調節している可能性が示唆されている(非特許文献4)。したがって、NKT細胞の働きを制御することによって、崩れた免疫系のバランスを調整し、監視機能を強化させて癌を治療することが可能となる。
NKT細胞の特性として最も着目されているのは、NKT細胞に発現しているTCRのα鎖が、ある1つの種の間では全個体で同一であるという点である。これは即ち、同種間の生物が持つNKT細胞は全て、同一の物質によって活性化されるということを示している。このα鎖は、ヒトではVα24、ネズミではVα14であるが、両種間でも非常に高い相同性を持っている。また、そのα鎖と対を成すβ鎖も、ごく限られた種類しか知られていない。このため、このTCRは「不可変型TCR」とも呼ばれている。
生体内には、様々な種類のスフィンゴ糖脂質の存在が知られている。生体内のスフィンゴ糖脂質は一般的に様々な糖がセラミドとβ-結合しており、器官によってその存在量は異なるが、様々な器官の細胞膜中に存在している(非特許文献5)。
一方、糖がセラミドにα-結合しているスフィンゴ糖脂質が、強力な免疫賦活作用及び抗腫瘍活性を有することが近年報告された。アゲラスフィン類に代表されるα-ガラクトシルセラミドは、海綿の一種であるAgelas mauritianusの抽出液より単離された糖脂質であり、NKT細胞を強く活性化することが知られている(非特許文献6)。
α-ガラクトシルセラミドは、樹状細胞(DC)などに代表される抗原提示細胞(APC)に取り込まれた後、主要組織適合遺伝子複合体(MHC)クラスI分子に類似したCD1dタンパク質によって細胞膜上に提示される。NKT細胞は、こうして提示されたCD1dタンパク質とα-ガラクトシルセラミドとの複合体を、TCRを用いて認識することにより活性化され、様々な免疫反応が開始される。
一方、糖がセラミドにα-結合しているスフィンゴ糖脂質が、強力な免疫賦活作用及び抗腫瘍活性を有することが近年報告された。アゲラスフィン類に代表されるα-ガラクトシルセラミドは、海綿の一種であるAgelas mauritianusの抽出液より単離された糖脂質であり、NKT細胞を強く活性化することが知られている(非特許文献6)。
α-ガラクトシルセラミドは、樹状細胞(DC)などに代表される抗原提示細胞(APC)に取り込まれた後、主要組織適合遺伝子複合体(MHC)クラスI分子に類似したCD1dタンパク質によって細胞膜上に提示される。NKT細胞は、こうして提示されたCD1dタンパク質とα-ガラクトシルセラミドとの複合体を、TCRを用いて認識することにより活性化され、様々な免疫反応が開始される。
α-ガラクトシルセラミドは、スフィンゴシン塩基が長鎖脂肪酸によりアシル化されて形成されたセラミドに、ガラクトースがα-配置で結合したスフィンゴ糖脂質であるが、これまでに様々な類縁体が合成され、その構造と活性との相関関係が調査されている。一連の合成類縁体の中で、例えば、下記式(a)で表されるα-ガラクトシルセラミド(以下、「α-GalCer」という)が最も強い活性を示すこと、更には対応するβ-体(β-GalCer)には免疫賦活活性は見られないことが明らかとなっている(非特許文献7)。

近年、このようなNKT細胞の機能に着目し、α-GalCerを有効成分として含有する治療薬が提案・開発されている。しかしながら、α-GalCerの投与によって活性化されたNKT細胞は、癌治療のために有用な、免疫賦活活性を誘導するサイトカインであるIFN-γ及びNKT細胞によるIFN-γ産生を増強する作用があり、樹状細胞が産生するサイトカインであるIL-12を産生するとともに、免疫抑制作用を誘導するサイトカインであるIL-4や免疫調節作用を誘導するサイトカインであるIL-10も同時に産生してしまう。その結果、免疫賦活活性の働きが抑制されてしまい、癌治療に対する効果が十分に得難くなるという問題がある。
近年、NKT細胞に対して免疫賦活作用を誘導するサイトカインであるIFN-γを優先的に産生させる糖脂質、α-C-GalCerが開発された(特許文献1~3、非特許文献8)。α-C-GalCerは、α-GalCerのグルコシド結合を形成する酸素原子をメチレン基で置き換えた類縁体である。α-C-GalCerでは、糖とセラミドとの結合がグリコシド結合から炭素-炭素結合へと変換されているため、生体内での安定性が増大し、薬効が長時間持続することが報告されている(非特許文献9)。しかしながら、α-C-GalCerはヒトのNKT細胞に対してはin vitroで非常に弱い活性しか示さないため、臨床応用は難しい。
一方、本発明者のうち、田代らは、独自に式:

で表わされるカルバ糖を有する新規糖脂質(本明細書において、「カルバ糖脂質A」という)が、NKT細胞に対して強力にIFN-γの産生を誘導することを見出した(非特許文献10)。
該化合物はヒト(in vitro)の系に於いても強い活性を示すことから臨床応用が期待されているが、該化合物の合成には多段階を要するため、より簡便な合成方法、あるいは容易に調製が可能でありながら同等あるいはそれ以上の活性を有する新規類縁体の開発が望まれている。
該化合物はヒト(in vitro)の系に於いても強い活性を示すことから臨床応用が期待されているが、該化合物の合成には多段階を要するため、より簡便な合成方法、あるいは容易に調製が可能でありながら同等あるいはそれ以上の活性を有する新規類縁体の開発が望まれている。
糖部分がフコシルである糖脂質としては、(1)下式(b)で表わされる(2S,3R)-1-O-(6’-デオキシ-α-D-ガラクトピラノシル)-2-(N-テトラデカノイルアミノ)-1,3-オクタデカンジオール(特許文献4、化合物12;非特許文献12、13、AGL-571)、式:
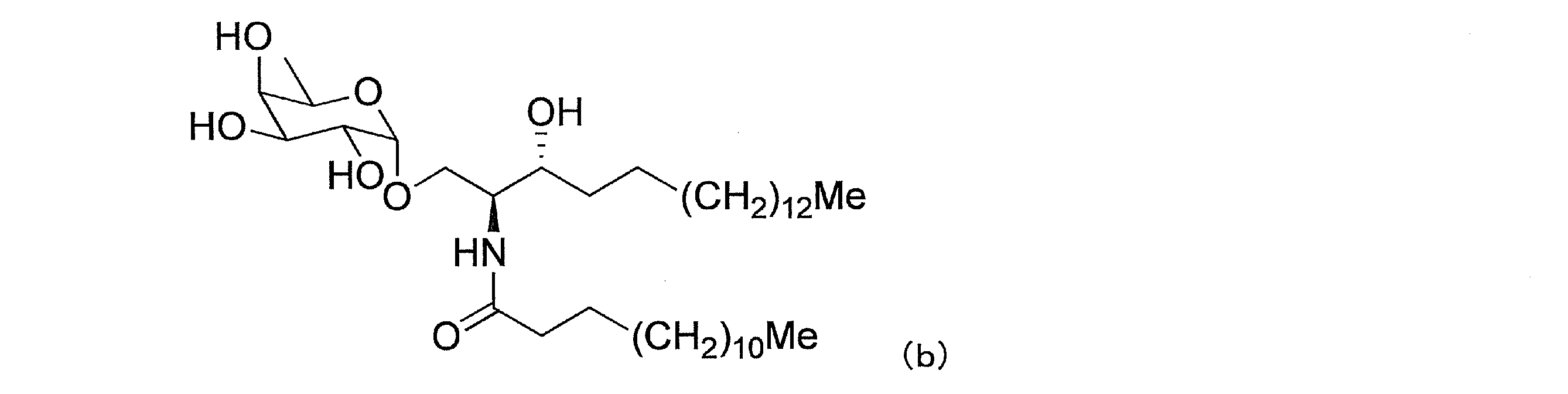
(2)下式(c)で表わされる(2S,3S,4R)-1-O-(6’-デオキシ-α-D-ガラクトピラノシル)-2-(N-テトラコサノイルアミノ)-1,3,4-オクタデカントリオール(特許文献5、DB03-8)、式:
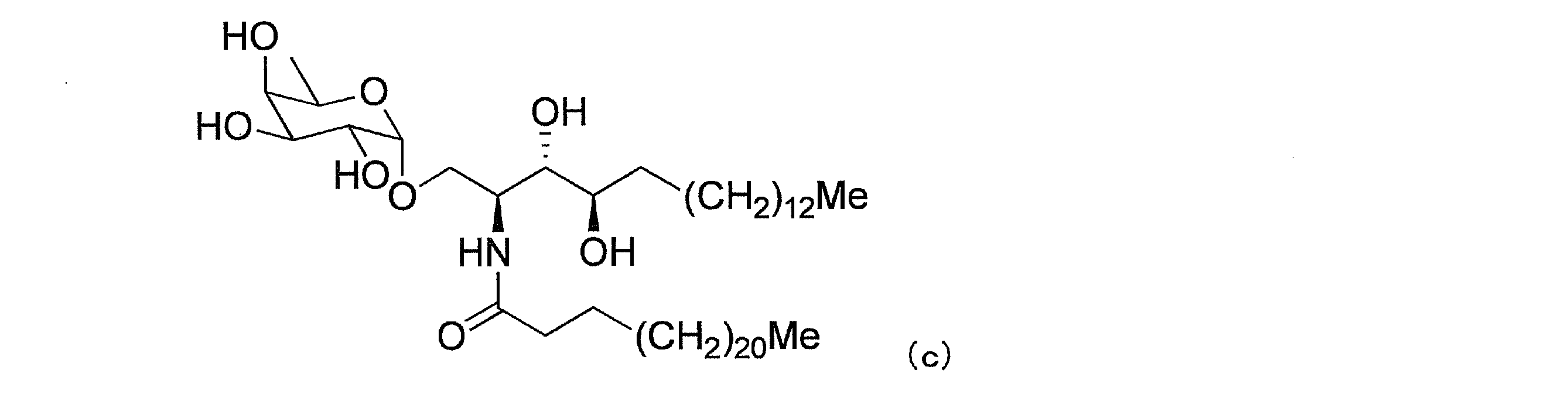
(3)下式(d)で表わされる(2S,3S,4R)-1-O-(α-L-フコピラノシル)-2-(N-ヘキサコサノイルアミノ)-1,3,4-オクタデカントリオール(非特許文献11、化合物27)、式:
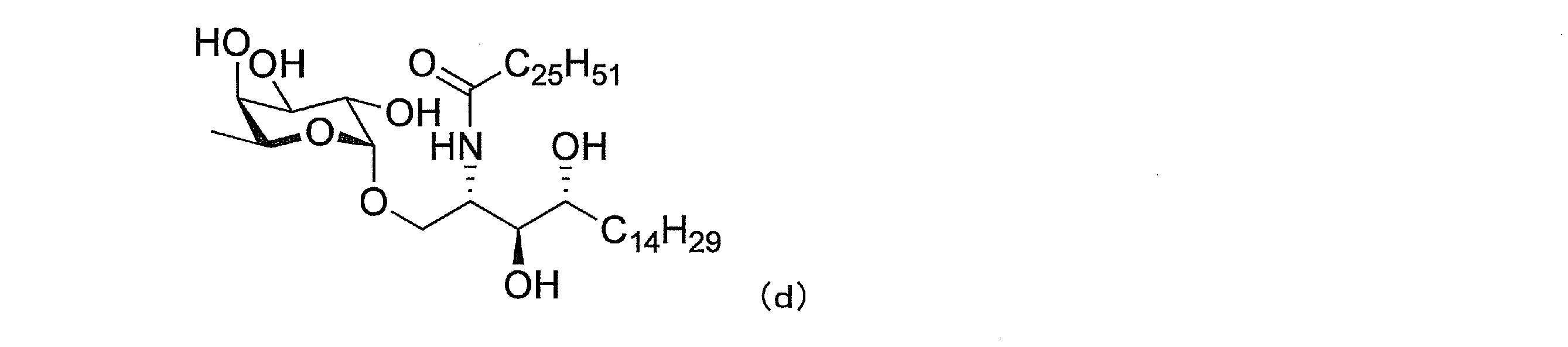
(4)下式(e)で表わされる(2S,3S,4R)-1-O-(β-L-フコピラノシル)-2-(N-ヘキサコサノイルアミノ)-1,3,4-オクタデカントリオール(非特許文献11、化合物30)、式:
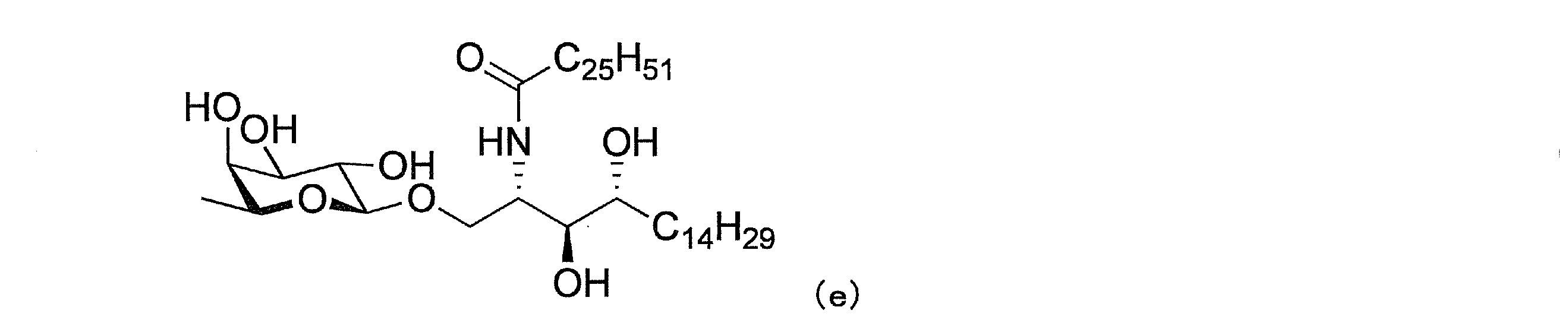
が開示されている。
米国特許出願公開第2005/0222048号明細書
国際公開第2003/105769号パンフレット
独国特許出願公開第10128250号明細書
国際公開第1994/09020号パンフレット
米国特許出願公開第2007/0238673号明細書
Proc. Natl. Acad. Sci. USA 1998, 95, 5690-5693
J. Immunol. 1995, 155, 2972-2983
J. Immunol. 1998, 161, 3271-3281
Nat. Immunol. 2003, 4, 1164-1165
Biochim. Biophys. Acta 1973, 315-335
Science 1997, 278, 1626-1629
J. Med. Chem. 1995, 38, 2176-2187
Angew. Chem. Int. Ed. Engl. 2004, 43, 3818-3822
J. Exp. Med. 2003, 198, 1631-1641
Tetrahedron Lett. 2007, 48, 3343-3347
Tetrahedron 2005, 61, 1855-1862
Biol. Pharm. Bull. 1995, 18, 1487-1491
Bioorg. Med. Chem. 1998, 6, 1905-1910
本発明はこのような実情に鑑みてなされたものであり、その解決しようとする課題は癌治療に有効な新規化合物及び該化合物の合成に有用な中間体を提供することにある。また、かかる新規化合物を含有する抗癌剤等の医薬を提供することを目的とする。
本発明者らは上記課題を解決するため鋭意研究を重ねた結果、下記の式(1)で表わされる化合物が特定のサイトカインを選択的に産生するとの知見を得た。更に本発明者らは詳細に検討したところ、特定サイトカインの選択的産生により特異的な免疫賦活能が発現され、癌治療に極めて有効であることを見出し、本発明を完成するに至った。
すなわち、本発明は以下のとおりである。
[1]下記一般式(1)で表される化合物(以下、「化合物(1)」という)又はその塩。
[1]下記一般式(1)で表される化合物(以下、「化合物(1)」という)又はその塩。
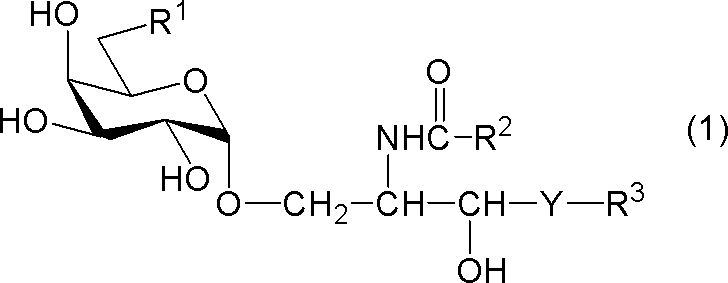
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。]
[2]R1が水素原子、メチル基、メトキシ基、エトキシ基、n-プロピルオキシ基又はフッ素原子である、上記[1]記載の化合物又はその塩。
[3]R1が炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子であり、R2が炭素数1~28の置換又は非置換のアルキル基であるか、又は、R1が水素原子であり、R2が炭素数24~28の置換又は非置換のアルキル基である、上記[1]又は[2]に記載の化合物又はその塩。
[4]R3が炭素数1~28の置換又は非置換のアルキル基である、上記[1]~[3]のいずれかに記載の化合物又はその塩。
[5]Yが-CH(OH)-である、上記[1]~[4]のいずれかに記載の化合物又はその塩。
[6]下記一般式(2)で表される化合物(以下、「化合物(2)」という)又はその塩。
[2]R1が水素原子、メチル基、メトキシ基、エトキシ基、n-プロピルオキシ基又はフッ素原子である、上記[1]記載の化合物又はその塩。
[3]R1が炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子であり、R2が炭素数1~28の置換又は非置換のアルキル基であるか、又は、R1が水素原子であり、R2が炭素数24~28の置換又は非置換のアルキル基である、上記[1]又は[2]に記載の化合物又はその塩。
[4]R3が炭素数1~28の置換又は非置換のアルキル基である、上記[1]~[3]のいずれかに記載の化合物又はその塩。
[5]Yが-CH(OH)-である、上記[1]~[4]のいずれかに記載の化合物又はその塩。
[6]下記一般式(2)で表される化合物(以下、「化合物(2)」という)又はその塩。
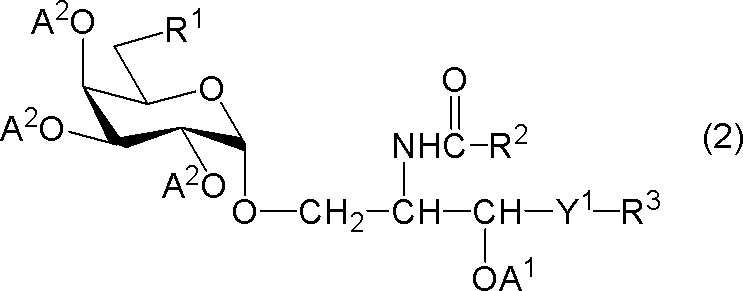
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Y1は-CH2-、-CH(OA1)-又は-CH=CH-を示し、A1は水素原子又は水酸基の保護基を示し、A2は水酸基の保護基を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。]
[7]化合物(1)又はその塩を含有する、医薬。
[8]化合物(1)又はその塩を含有する、免疫賦活剤。
[9]化合物(1)又はその塩を含有する、選択的IFN-γ産生誘導剤。
[10]化合物(1)又はその塩を含有する、抗癌剤。
[11]化合物(1)又はその塩の有効量を対象に投与することを含む、免疫賦活方法。
[12]化合物(1)又はその塩の有効量を対象に投与することを含む、選択的IFN-γ産生誘導方法。
[13]化合物(1)又はその塩の有効量を対象に投与することを含む、癌の治療方法。
[14]免疫賦活剤の製造のための、化合物(1)又はその塩の使用。
[15]選択的IFN-γ産生誘導剤の製造のための、化合物(1)又はその塩の使用。
[16]抗癌剤の製造のための、化合物(1)又はその塩の使用。
[7]化合物(1)又はその塩を含有する、医薬。
[8]化合物(1)又はその塩を含有する、免疫賦活剤。
[9]化合物(1)又はその塩を含有する、選択的IFN-γ産生誘導剤。
[10]化合物(1)又はその塩を含有する、抗癌剤。
[11]化合物(1)又はその塩の有効量を対象に投与することを含む、免疫賦活方法。
[12]化合物(1)又はその塩の有効量を対象に投与することを含む、選択的IFN-γ産生誘導方法。
[13]化合物(1)又はその塩の有効量を対象に投与することを含む、癌の治療方法。
[14]免疫賦活剤の製造のための、化合物(1)又はその塩の使用。
[15]選択的IFN-γ産生誘導剤の製造のための、化合物(1)又はその塩の使用。
[16]抗癌剤の製造のための、化合物(1)又はその塩の使用。
本発明者らは上記課題を解決するため研究を重ねた結果、糖脂質の一種であるガラクトシルセラミドの一般的骨格の一部である糖の6-位の水酸基を他の官能基(炭素数1~7のアルキル基、炭素数1~6のアルコキシ基、ハロゲン原子)又は水素原子に変換した化合物が、特異的な免疫調節能を有していること、癌治療に極めて有効であることを見出し、本発明を完成するに到った。
本発明の化合物(1)は、抗原提示細胞(APC)の持つCD1dタンパク質と複合体を形成し、NKT細胞に提示される。NKT細胞は、この複合体をTCRを介して認識し、それ自身の有する免疫調節能のうち、免疫細胞の働きを活性化するサイトカインの一種であるIFN-γを優先的且つ大量に産生する。
本発明の化合物(1)は微量でもNKT細胞を強力に活性化して、これまで報告されている化合物よりも大量のIFN-γを産生させる。このことから、少量の投与で充分な薬効を得ることが出来る。
本発明の化合物(1)及びその塩は、癌治療等に有効である。
本発明の化合物(2)及びその塩は、化合物(1)及びその塩の合成中間体として有用である。
本発明の化合物(1)は、抗原提示細胞(APC)の持つCD1dタンパク質と複合体を形成し、NKT細胞に提示される。NKT細胞は、この複合体をTCRを介して認識し、それ自身の有する免疫調節能のうち、免疫細胞の働きを活性化するサイトカインの一種であるIFN-γを優先的且つ大量に産生する。
本発明の化合物(1)は微量でもNKT細胞を強力に活性化して、これまで報告されている化合物よりも大量のIFN-γを産生させる。このことから、少量の投与で充分な薬効を得ることが出来る。
本発明の化合物(1)及びその塩は、癌治療等に有効である。
本発明の化合物(2)及びその塩は、化合物(1)及びその塩の合成中間体として有用である。
以下、本発明をその好適な実施形態に即して詳細に説明する。
先ず、本明細書において使用する式中の記号の定義を説明する。
R1は水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示す。
R1で示される炭素数1~7のアルキル基としては、置換、又は非置換のアルキル基を示し、環を形成していても良い。例えば、メチル、エチル、n-プロピル、イソプロピル、シクロプロピル、シクロプロピルメチル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル、へプチル、シクロヘキシルメチル等が挙げられ、メチル、エチルが好ましい。
R1で示される炭素数1~6のアルコキシ基としては、酸素原子に置換、又は非置換のアルキル基が結合したものを示し、そのアルキル部分は環を形成していても良い。例えば、メトキシ、エトキシ、n-プロピルオキシ、イソプロピルオキシ、シクロプロピルオキシ、シクロプロピルメチルオキシ、n-ブトキシ、イソブチルオキシ、sec-ブチルオキシ、tert-ブチルオキシ、ペンチルオキシ、ヘキシルオキシ、シクロヘキシルオキシ等が挙げられ、メトキシ、エトキシ、n-プロピルオキシが好ましい。
R1で示されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられ、フッ素原子、塩素原子が好ましい。
先ず、本明細書において使用する式中の記号の定義を説明する。
R1は水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示す。
R1で示される炭素数1~7のアルキル基としては、置換、又は非置換のアルキル基を示し、環を形成していても良い。例えば、メチル、エチル、n-プロピル、イソプロピル、シクロプロピル、シクロプロピルメチル、n-ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル、へプチル、シクロヘキシルメチル等が挙げられ、メチル、エチルが好ましい。
R1で示される炭素数1~6のアルコキシ基としては、酸素原子に置換、又は非置換のアルキル基が結合したものを示し、そのアルキル部分は環を形成していても良い。例えば、メトキシ、エトキシ、n-プロピルオキシ、イソプロピルオキシ、シクロプロピルオキシ、シクロプロピルメチルオキシ、n-ブトキシ、イソブチルオキシ、sec-ブチルオキシ、tert-ブチルオキシ、ペンチルオキシ、ヘキシルオキシ、シクロヘキシルオキシ等が挙げられ、メトキシ、エトキシ、n-プロピルオキシが好ましい。
R1で示されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられ、フッ素原子、塩素原子が好ましい。
R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換基の炭化水素基を示す。本明細書において「炭化水素基」とは、置換又は非置換の、炭素数1~28のアルキル基、炭素数2~28のアルケニル基、炭素数2~28のアルキニル基、炭素数3~28のシクロアルキル基、炭素数3~28のシクロアルケニル基、炭素数6~14のアリール基をも包含する概念であり、直鎖状、分岐状及び環状のいずれの形態であってもよく、また飽和炭化水素基でも不飽和炭化水素基でもよく、不飽和結合を分子内及び末端のいずれに有していてもよい。中でも、R2及びR3としては、炭素数1~28の置換又は非置換のアルキル基が好ましい。
但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示し、炭素数24~28の置換又は非置換のアルキル基が好ましい。
但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示し、炭素数24~28の置換又は非置換のアルキル基が好ましい。
R2及びR3で示される炭化水素基の置換基としては、ハロゲン原子(好ましくは塩素原子、フッ素原子);メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ基、ブトキシ基、tert-ブトキシ基等のアルコキシ基(好ましくは炭素数1~24、より好ましくは炭素数1~16、更に好ましくは炭素数1~10、特に好ましくは炭素数1~4);フェノキシ基等のアリールオキシ基(好ましくは炭素数6~14);水酸基;アミノ基;メチルアミノ基、ジメチルアミノ基、エチルアミノ基、ジエチルアミノ基等のアルキルアミノ基;シクロアルキルアミノ基;アセトアミド基等のアルキルカルボニルアミノ基;シクロアルキルカルボニルアミノ基;ベンゾイルアミノ基等のアリールカルボニルアミノ基(好ましくは、アリール部分の炭素数が6~14のアリール基である、アリールカルボニルアミノ基)等の電子供与性基、更にはカルボキシル基;アルコキシカルボニル基;アシル基(アシル基としては後述の通りである。好ましくはアルキル部分が炭素数1~24の直鎖又は分岐状のアルキル基である、アルキル-カルボニル基);カルバモイル基;トリフルオロメチル基等の電子求引性基が例示される。
本明細書において「アシル基」とは、例えば、ホルミル基;アルキル-カルボニル基(例えば、アルキル部分が、炭素数1~24(好ましくは炭素数1~12)の直鎖若しくは分岐状のアルキル基である、アルキル-カルボニル基(例えば、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、バレリル基、ピバロイル基、ヘキサノイル基));シクロアルキル-カルボニル基(例えば、シクロアルキル部分が、炭素数3~10のシクロアルキル基である、シクロアルキル-カルボニル基);アルケニル-カルボニル基(例えば、アルケニル部分が炭素数2~12の直鎖若しくは分岐状のアルケニル基である、アルケニル-カルボニル基(例えば、アクリロイル基、メタクリロイル基));アリール-カルボニル基(例えば、アリール部分が、炭素数6~14のアリール基である、アリール-カルボニル基(例えば、ベンゾイル基、ナフトイル基))等をいう。アリール-カルボニル基におけるアリール基とは、例えば、単環~3環式芳香族炭化水素基を示し、具体的に例えば、フェニル基、ナフチル基、アントリル基、フェナントリル基が例示される。中でも、アシル基としては、ホルミル基、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、ベンゾイル基、ナフトイル基等が好ましく、アセチル基、ベンゾイル基がより好ましい。
上記アルキルアミノ基、アルキルカルボニルアミノ基のアルキル部分としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等の直鎖又は分岐状のアルキル基(好ましくは炭素数1~24、より好ましくは炭素数1~16、更に好ましくは炭素数1~10、特に好ましくは炭素数1~4)が例示される。
上記シクロアルキルアミノ基、シクロアルキルカルボニルアミノ基のシクロアルキル部分としては、シクロペンチル基、シクロヘキシル基等のシクロアルキル基(好ましくは炭素数3~24、より好ましくは炭素数3~16、更に好ましくは炭素数3~10、特に好ましくは炭素数3~6)が例示される。
上記アルコキシカルボニル基のアルコキシ部分としては上記アルコキシ基と同様のものが例示される。
上記シクロアルキルアミノ基、シクロアルキルカルボニルアミノ基のシクロアルキル部分としては、シクロペンチル基、シクロヘキシル基等のシクロアルキル基(好ましくは炭素数3~24、より好ましくは炭素数3~16、更に好ましくは炭素数3~10、特に好ましくは炭素数3~6)が例示される。
上記アルコキシカルボニル基のアルコキシ部分としては上記アルコキシ基と同様のものが例示される。
上記した置換基は、置換可能な位置に、さらに、ハロゲン、アルキル基、シクロアルキル基、アルケニル基、アルキニル基、フェニル基、アルコキシ基、水酸基、アミノ基、アルキルアミノ基及びシクロアルキルアミノ基のうちの少なくとも1種で置換されていてもよい。
該ハロゲン、アルコキシ基、アルキルアミノ基、シクロアルキルアミノ基としては上記と同様のものが例示される。
該アルキル基としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基(好ましくは炭素数1~24、より好ましくは炭素数1~16、更に好ましくは炭素数1~10、特に好ましくは炭素数1~4)が例示される。
該シクロアルキル基としては、シクロペンチル基、シクロヘキシル基等のシクロアルキル基(好ましくは炭素数3~24、より好ましくは炭素数3~16、更に好ましくは炭素数3~10、特に好ましくは炭素数3~6)が例示される。
該アルケニル基としては、ビニル基、プロペニル基、ブテニル基等のアルケニル基(好ましくは炭素数2~24、より好ましくは炭素数2~16、更に好ましくは炭素数2~10、特に好ましくは炭素数2~4)が例示される。
該アルキニル基としては、エチニル基、プロパルギル基、ブチニル基、ペンチニル基等のアルキニル基(好ましくは炭素数2~24、より好ましくは炭素数2~16、更に好ましくは炭素数2~10、特に好ましくは炭素数2~4)が例示される。
該ハロゲン、アルコキシ基、アルキルアミノ基、シクロアルキルアミノ基としては上記と同様のものが例示される。
該アルキル基としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等のアルキル基(好ましくは炭素数1~24、より好ましくは炭素数1~16、更に好ましくは炭素数1~10、特に好ましくは炭素数1~4)が例示される。
該シクロアルキル基としては、シクロペンチル基、シクロヘキシル基等のシクロアルキル基(好ましくは炭素数3~24、より好ましくは炭素数3~16、更に好ましくは炭素数3~10、特に好ましくは炭素数3~6)が例示される。
該アルケニル基としては、ビニル基、プロペニル基、ブテニル基等のアルケニル基(好ましくは炭素数2~24、より好ましくは炭素数2~16、更に好ましくは炭素数2~10、特に好ましくは炭素数2~4)が例示される。
該アルキニル基としては、エチニル基、プロパルギル基、ブチニル基、ペンチニル基等のアルキニル基(好ましくは炭素数2~24、より好ましくは炭素数2~16、更に好ましくは炭素数2~10、特に好ましくは炭素数2~4)が例示される。
中でも、R2としては、置換又は非置換のアルキル基が好ましく、また、直鎖状のアルキル基が好ましい。R1が炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子であるときは、R2の炭素数は、好ましくは18~26、より好ましくは24~26である。R1が水素原子であるときは、R2の炭素数は、好ましくは24~26である。R2としては、具体的には例えば、-(CH2)23-CH3、-(CH2)24-CH3、-(CH2)25-CH3等が挙げられる。
また、R3としては、置換又は非置換のアルキル基が好ましく、また、直鎖状のアルキル基が好ましい。R3の炭素数は好ましくは9~20、より好ましくは12~18である。R3としては、具体的には例えば、-(CH2)11-CH3、-(CH2)12-CH3、-(CH2)13-CH3、-(CH2)14-CH3、-(CH2)15-CH3、-(CH2)16-CH3、-(CH2)17-CH3等が挙げられる。
また、R3としては、置換又は非置換のアルキル基が好ましく、また、直鎖状のアルキル基が好ましい。R3の炭素数は好ましくは9~20、より好ましくは12~18である。R3としては、具体的には例えば、-(CH2)11-CH3、-(CH2)12-CH3、-(CH2)13-CH3、-(CH2)14-CH3、-(CH2)15-CH3、-(CH2)16-CH3、-(CH2)17-CH3等が挙げられる。
Yは-CH2-、-CH(OH)-又は-CH=CH-を示し、中でも-CH(OH)-が好適である。
Y1は-CH2-、-CH(OA1)-又は-CH=CH-を示し、中でも-CH(OA1)-が好適である。なお、A1は後述の通りである。
A1は水素原子又は水酸基の保護基を示し、水酸基の保護基としてはアシル基、t-ブチルジメチルシリル(TBS)基、ベンジル(Bn)基、p-メトキシベンジル(PMB)基等が例示される。アシル基としては、前述の通りである。中でも、TBS基、Bn基が好適である。
Y1が-CH(OA1)-である場合、2つのA1は同一であっても異なっていてもよいが同一であることが好ましい。
Y1が-CH(OA1)-である場合、2つのA1が一緒になってジオールの保護基を形成していてもよい。該ジオールの保護基としては、例えば、
Y1は-CH2-、-CH(OA1)-又は-CH=CH-を示し、中でも-CH(OA1)-が好適である。なお、A1は後述の通りである。
A1は水素原子又は水酸基の保護基を示し、水酸基の保護基としてはアシル基、t-ブチルジメチルシリル(TBS)基、ベンジル(Bn)基、p-メトキシベンジル(PMB)基等が例示される。アシル基としては、前述の通りである。中でも、TBS基、Bn基が好適である。
Y1が-CH(OA1)-である場合、2つのA1は同一であっても異なっていてもよいが同一であることが好ましい。
Y1が-CH(OA1)-である場合、2つのA1が一緒になってジオールの保護基を形成していてもよい。該ジオールの保護基としては、例えば、

で示される基(すなわち、ジオールを保護してアセトナイドを形成する基)等が挙げられる。
A2で示される水酸基の保護基としては、例えば、アシル基、TBS基、トリメチルシリル(TMS)基、Bn基、PMB基等が例示される。アシル基としては、前述の通りである。中でも、Bn基、PMB基が好適である。
本発明においては、糖の環状構造に由来する立体異性体の中でα体を採用するが、β体ではサイトカイン産生能が極めて低下するとの知見を本発明者らは得ている。
化合物(1)及び化合物(2)が立体異性体を有する場合には、いずれの異性体も本発明に包含され、2種以上の異性体の任意の割合の混合物(ラセミ体を含む)であってもよい。
特に、化合物(1)には、脂質部分の不斉炭素に由来する光学異性体が存在するが、本発明においては、単一の光学活性体であっても、2種以上の光学活性体の任意の割合の混合物(ラセミ体を含む)であってもよい。-NHCOR2が結合する不斉炭素はS配置が好適である。-NHCOR2が結合する不斉炭素に隣接し-OHを有する不斉炭素は、-NHCOR2が結合する不斉炭素に対してantiの配置が好適である。Yが-CH(OH)-の場合、Yで示される-CH(OH)-中の不斉炭素はR配置が好ましい。
また、化合物(2)には、脂質部分の不斉炭素に由来する光学異性体が存在するが、本発明においては、単一の光学活性体であっても、2種以上の光学活性体の任意の割合の混合物(ラセミ体を含む)であってもよい。-NHCOR2が結合する不斉炭素はS配置が好適である。-NHCOR2が結合する不斉炭素に隣接し-OA1を有する不斉炭素は、-NHCOR2が結合する不斉炭素に対してantiの配置が好適である。Y1が-CH(OA1)-の場合、Y1で示される-CH(OA1)-中の不斉炭素はR配置が好ましい。
化合物(1)及び化合物(2)が立体異性体を有する場合には、いずれの異性体も本発明に包含され、2種以上の異性体の任意の割合の混合物(ラセミ体を含む)であってもよい。
特に、化合物(1)には、脂質部分の不斉炭素に由来する光学異性体が存在するが、本発明においては、単一の光学活性体であっても、2種以上の光学活性体の任意の割合の混合物(ラセミ体を含む)であってもよい。-NHCOR2が結合する不斉炭素はS配置が好適である。-NHCOR2が結合する不斉炭素に隣接し-OHを有する不斉炭素は、-NHCOR2が結合する不斉炭素に対してantiの配置が好適である。Yが-CH(OH)-の場合、Yで示される-CH(OH)-中の不斉炭素はR配置が好ましい。
また、化合物(2)には、脂質部分の不斉炭素に由来する光学異性体が存在するが、本発明においては、単一の光学活性体であっても、2種以上の光学活性体の任意の割合の混合物(ラセミ体を含む)であってもよい。-NHCOR2が結合する不斉炭素はS配置が好適である。-NHCOR2が結合する不斉炭素に隣接し-OA1を有する不斉炭素は、-NHCOR2が結合する不斉炭素に対してantiの配置が好適である。Y1が-CH(OA1)-の場合、Y1で示される-CH(OA1)-中の不斉炭素はR配置が好ましい。
化合物(1)の脂質部分としては、

(式中、各記号は、前述と同義を示す。)等が挙げられる。
化合物(2)の脂質部分としては、
化合物(2)の脂質部分としては、
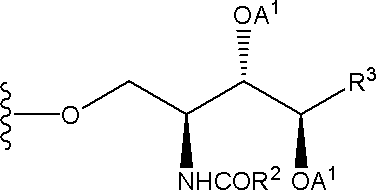
(式中、各記号は、前述と同義を示す。)等が挙げられる。
化合物(1)及び化合物(2)の塩としては、薬理的に許容される塩が好ましく、例えば、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩、リン酸塩等の無機酸塩;コハク酸塩、フマル酸塩、酢酸塩、メタンスルホン酸塩、トルエンスルホン酸塩等の有機酸塩;ナトリウム塩、カリウム塩等のアルカリ金属塩;マグネシウム塩、カルシウム塩等のアルカリ土類金属塩;アンモニウム塩、アルキルアンモニウム塩等のアンモニウム塩等を挙げることができる。
本発明における好適な化合物(1)の具体例を表1に示すが、これらに限定されるものではない。
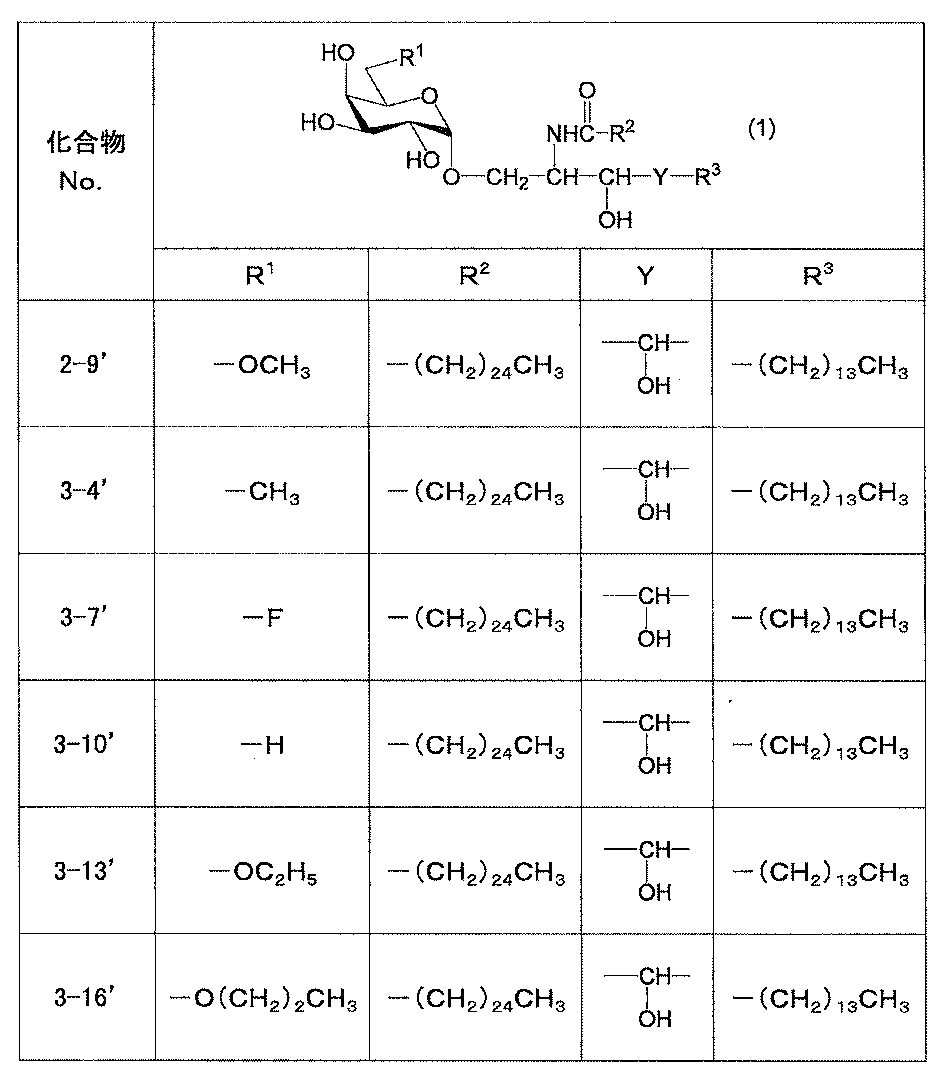
本発明における好適な化合物(1)の具体例としては、実施例に記載の化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13、化合物3-16が挙げられるが、これらに限定されるものではない。
本発明における好適な化合物(2)の具体例としては、実施例に記載の化合物2-7、化合物2-8が挙げられるが、これらに限定されるものではない。
次に、本発明の化合物(1)及び(2)の製造方法について好適な実施形態について説明する。本発明の化合物は当業者にとって自体公知の種々の方法で製造することができるが、例えば、化合物(1)及び(2)は下記スキームに記載の方法又はこれに準ずる方法にしたがって製造することが可能である。

スキーム中、Zはハロゲン原子(例えば、フッ素原子)を示し、Y2は-CH2-、-CH(OA)-又は-CH=CH-を示し、Aは水酸基の保護基を示し、その他の各記号は前述と同義を示す。Aで示される水酸基の保護基としては、前述のA1で示される水酸基の保護基と同様のものが例示される。なお、化合物(2’)及び化合物(2’’)は、本発明の化合物(2)に包含される。
原料化合物(A)は、例えば、T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45に記載の方法又はこれに準ずる方法で製造される化合物2-2’を原料として次のように調製できる。
(i)R1が炭素数1~6のアルコキシ基である化合物(A)(化合物2-5’)
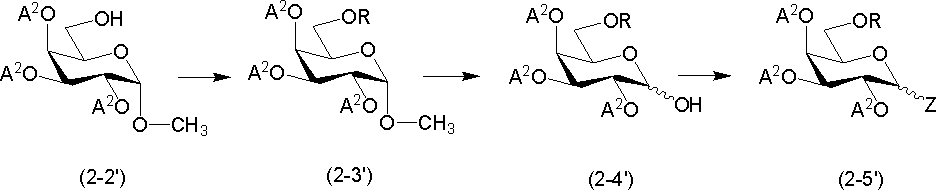
式中、Rは炭素数1~6のアルキル(例、メチル、エチル、n-プロピル)を示し、その他の各記号は前述と同義を示す。
化合物2-2’を塩基存在下、ハロゲン化アルキルと反応させて化合物2-3’を得ることができる。塩基としては、例えば、水素化ナトリウム、n-ブチルリチウム等が挙げられる。塩基の使用量は、化合物2-2’に対して、通常1~3当量である。ハロゲン化アルキルとしては、例えば、ヨウ化メチル、ヨウ化エチル、臭化プロピル等が挙げられる。ハロゲン化アルキルの使用量は、化合物2-2’に対して、通常1~3当量である。
溶媒としては、例えば、N,N-ジメチルホルムアミド、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒、これらの混合溶媒が挙げられる。溶媒の使用量は、化合物2-2’に対して、通常10~20倍容量である。
反応温度は、通常0~80℃、反応時間は、通常1~24時間である。
化合物2-3’は、常法によって単離することができ、例えば反応液に水を加え酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物2-3’を単離することができる。
化合物2-2’を塩基存在下、ハロゲン化アルキルと反応させて化合物2-3’を得ることができる。塩基としては、例えば、水素化ナトリウム、n-ブチルリチウム等が挙げられる。塩基の使用量は、化合物2-2’に対して、通常1~3当量である。ハロゲン化アルキルとしては、例えば、ヨウ化メチル、ヨウ化エチル、臭化プロピル等が挙げられる。ハロゲン化アルキルの使用量は、化合物2-2’に対して、通常1~3当量である。
溶媒としては、例えば、N,N-ジメチルホルムアミド、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒、これらの混合溶媒が挙げられる。溶媒の使用量は、化合物2-2’に対して、通常10~20倍容量である。
反応温度は、通常0~80℃、反応時間は、通常1~24時間である。
化合物2-3’は、常法によって単離することができ、例えば反応液に水を加え酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物2-3’を単離することができる。
化合物2-3’を酸と反応させて直接化合物2-4’を得ることができる。あるいは化合物2-3’を対応するO-アセチル体へと導き、これを加アルコール分解することにより化合物2-4’を得ることができる。
O-アセチル体を経由する場合は、例えば無水酢酸中、触媒量の酸で処理することによりO-アセチル体を調製し、これを加アルコール分解する。酸としては、例えば、濃硫酸、濃塩酸、p-トルエンスルホン酸等が挙げられる。無水酢酸の使用量は、化合物2-3’に対して、通常5~20倍容量である。反応温度は、通常0℃~室温、反応時間は、通常5分~1時間である。中和後、減圧濃縮することによりO-アセチル体を得ることができる。
得られたO-アセチル体の加アルコール分解は、アルコール溶媒、例えばメタノール、エタノール等の溶媒中、ナトリウムメトキシド、水酸化ナトリウム等の塩基で処理する。
化合物2-4’は、常法によって単離することができ、例えば陽イオン交換樹脂で酸性にした後、濾過し、濃縮して精製してもよい。
O-アセチル体を経由する場合は、例えば無水酢酸中、触媒量の酸で処理することによりO-アセチル体を調製し、これを加アルコール分解する。酸としては、例えば、濃硫酸、濃塩酸、p-トルエンスルホン酸等が挙げられる。無水酢酸の使用量は、化合物2-3’に対して、通常5~20倍容量である。反応温度は、通常0℃~室温、反応時間は、通常5分~1時間である。中和後、減圧濃縮することによりO-アセチル体を得ることができる。
得られたO-アセチル体の加アルコール分解は、アルコール溶媒、例えばメタノール、エタノール等の溶媒中、ナトリウムメトキシド、水酸化ナトリウム等の塩基で処理する。
化合物2-4’は、常法によって単離することができ、例えば陽イオン交換樹脂で酸性にした後、濾過し、濃縮して精製してもよい。
化合物2-4’をハロゲン化剤と反応させて化合物2-5’を得ることができる。
ハロゲン化剤としては、例えば、ジエチルアミノサルファートリフルオリド、トリス(ジメチルアミノ)スルホニウムジフルオロトリメチルシリケート等が挙げられる。ハロゲン化剤の使用量は、化合物2-4’に対して、通常1~3当量である。
溶媒としては、ジクロロメタン等が挙げられる。溶媒の使用量は、化合物2-4’に対して、通常10~30倍容量である。
反応温度は、通常-78℃~室温、反応時間は、通常30分~1時間である。
化合物2-5’は、常法によって単離することができ、例えば反応液にメタノールを加え、濃縮し、残渣を酢酸エチルで希釈し、有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物2-5’を単離することができる。
ハロゲン化剤としては、例えば、ジエチルアミノサルファートリフルオリド、トリス(ジメチルアミノ)スルホニウムジフルオロトリメチルシリケート等が挙げられる。ハロゲン化剤の使用量は、化合物2-4’に対して、通常1~3当量である。
溶媒としては、ジクロロメタン等が挙げられる。溶媒の使用量は、化合物2-4’に対して、通常10~30倍容量である。
反応温度は、通常-78℃~室温、反応時間は、通常30分~1時間である。
化合物2-5’は、常法によって単離することができ、例えば反応液にメタノールを加え、濃縮し、残渣を酢酸エチルで希釈し、有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物2-5’を単離することができる。
(ii)R1が炭素数1~7のアルキル基である化合物(A)(化合物3-3’等)

式中、各記号は前述と同義を示す。
K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203に記載された方法、又はこれに準ずる方法により製造される化合物3-1’をヒドラジン1水和物、過酸化水素水と反応させて化合物3-2’を得ることができる。ヒドラジン1水和物の使用量は、化合物3-1’に対して、通常5~20当量である。過酸化水素水の使用量は、化合物3-1’に対して、通常5~20当量である。
溶媒としては、エタノール等が挙げられる。溶媒の使用量は、化合物3-1’に対して、通常10~50倍容量である。
反応温度は、通常室温~80℃、反応時間は、通常1~20時間である。
化合物3-2’は、常法によって単離することができ、例えば反応液に飽和チオ硫酸ナトリウムを加え、酢酸エチルで希釈し、有機層を水、飽和チオ硫酸ナトリウム水溶液、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物3-2’を単離することができる。
化合物3-2’から化合物3-3’への変換は、化合物2-3’から化合物2-5’への変換と同様な手法により行うことができる。
K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203に記載された方法、又はこれに準ずる方法により製造される化合物3-1’をヒドラジン1水和物、過酸化水素水と反応させて化合物3-2’を得ることができる。ヒドラジン1水和物の使用量は、化合物3-1’に対して、通常5~20当量である。過酸化水素水の使用量は、化合物3-1’に対して、通常5~20当量である。
溶媒としては、エタノール等が挙げられる。溶媒の使用量は、化合物3-1’に対して、通常10~50倍容量である。
反応温度は、通常室温~80℃、反応時間は、通常1~20時間である。
化合物3-2’は、常法によって単離することができ、例えば反応液に飽和チオ硫酸ナトリウムを加え、酢酸エチルで希釈し、有機層を水、飽和チオ硫酸ナトリウム水溶液、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物3-2’を単離することができる。
化合物3-2’から化合物3-3’への変換は、化合物2-3’から化合物2-5’への変換と同様な手法により行うことができる。
R1がメチル以外の化合物については、上述のTatsutaらの報告における化合物2-2'から化合物3-1'への変換において、Swern酸化の後に行うWittig反応の際、用いるWittig試薬を選択することで合成することが可能である。この場合化合物3-1'は以下のように表すことができる。B1、B2は置換又は非置換のアルキル基を表し、B1とB2とで環を形成していても良い。式中、その他の記号は前述と同義を示す。
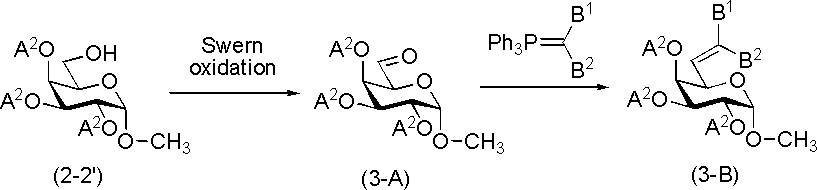
(iii)R1がハロゲン原子である化合物(A)(化合物3-6’)

式中、Wはハロゲン原子(例、フッ素原子)を示し、その他の各記号は前述と同義を示す。
化合物2-2’を塩基存在下、ハロゲン化剤と反応させて化合物3-5’を得ることができる。
塩基としては、トリエチルアミン、ピリジン等が挙げられる。塩基の使用量は、化合物2-2’に対して、通常1~5当量である。ハロゲン化剤としては、例えば、ジエチルアミノサルファートリフルオリド、トリス(ジメチルアミノ)スルホニウムジフルオロトリメチルシリケート等が挙げられる。ハロゲン化剤の使用量は、化合物2-2’に対して、通常1~3当量である。
溶媒としては、ジクロロメタン等が挙げられる。溶媒の使用量は、化合物2-2’に対して、通常10~30倍容量である。
反応温度は、通常-78℃~室温、反応時間は、通常30分~2時間である。
化合物3-5’は、常法によって単離することができ、例えば反応液にメタノールを加え、酢酸エチルで希釈し、有機層を水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物3-5’を単離することができる。
化合物3-5’から化合物3-6’への変換は、化合物2-3’から化合物2-5’への変換と同様な手法により行うことができる。
化合物2-2’を塩基存在下、ハロゲン化剤と反応させて化合物3-5’を得ることができる。
塩基としては、トリエチルアミン、ピリジン等が挙げられる。塩基の使用量は、化合物2-2’に対して、通常1~5当量である。ハロゲン化剤としては、例えば、ジエチルアミノサルファートリフルオリド、トリス(ジメチルアミノ)スルホニウムジフルオロトリメチルシリケート等が挙げられる。ハロゲン化剤の使用量は、化合物2-2’に対して、通常1~3当量である。
溶媒としては、ジクロロメタン等が挙げられる。溶媒の使用量は、化合物2-2’に対して、通常10~30倍容量である。
反応温度は、通常-78℃~室温、反応時間は、通常30分~2時間である。
化合物3-5’は、常法によって単離することができ、例えば反応液にメタノールを加え、酢酸エチルで希釈し、有機層を水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物3-5’を単離することができる。
化合物3-5’から化合物3-6’への変換は、化合物2-3’から化合物2-5’への変換と同様な手法により行うことができる。
(iv)R1が水素原子である化合物(A)(化合物3-9’)

式中、各記号は前述と同義を示す。
化合物3-9'は、S. Koto et al. Bull. Chem. Soc. Jpn., 2000, 73, 967-976に記載の方法、又はこれに準ずる方法により、化合物2-2'から製造される化合物3-8'に対して、化合物2-3'から化合物2-5'への変換と同様な手法により製造することができる。
化合物3-9'は、S. Koto et al. Bull. Chem. Soc. Jpn., 2000, 73, 967-976に記載の方法、又はこれに準ずる方法により、化合物2-2'から製造される化合物3-8'に対して、化合物2-3'から化合物2-5'への変換と同様な手法により製造することができる。
原料化合物(B)は、例えばH. Takikawa et al. Tetrahedron, 1998, 54, 3141-3150あるいはS. Kim et al. Synthesis, 2004, 847-850に記載の方法又はこれに準ずる方法に従って調製できる。
(step1)
step1は、化合物(A)をモレキュラーシーブス、塩化スズ(II)、過塩素酸銀存在下に化合物(B)と反応させて化合物(2’)を得る工程である。
化合物(A)の使用量は、化合物(B)に対して通常1~3当量である。
モレキュラーシーブスの使用量は、化合物(B)に対して通常2~10倍重量である。塩化スズ(II)の使用量は、化合物(A)に対して通常1.5~3当量である。過塩素酸銀の使用量は、化合物(A)に対して通常1.5~3当量である。
溶媒としては、例えば、アセトニトリル、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒が挙げられる。溶媒の使用量は、化合物(A)に対して、通常5~20倍容量である。
反応温度は、通常-18~20℃、反応時間は、通常1~2時間である。
化合物(2’)は、常法によって単離することができ、例えば反応液にジエチルエーテルを加え、濾過し、濾液を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物(2’)を単離することができる。
step1は、化合物(A)をモレキュラーシーブス、塩化スズ(II)、過塩素酸銀存在下に化合物(B)と反応させて化合物(2’)を得る工程である。
化合物(A)の使用量は、化合物(B)に対して通常1~3当量である。
モレキュラーシーブスの使用量は、化合物(B)に対して通常2~10倍重量である。塩化スズ(II)の使用量は、化合物(A)に対して通常1.5~3当量である。過塩素酸銀の使用量は、化合物(A)に対して通常1.5~3当量である。
溶媒としては、例えば、アセトニトリル、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒が挙げられる。溶媒の使用量は、化合物(A)に対して、通常5~20倍容量である。
反応温度は、通常-18~20℃、反応時間は、通常1~2時間である。
化合物(2’)は、常法によって単離することができ、例えば反応液にジエチルエーテルを加え、濾過し、濾液を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物(2’)を単離することができる。
(step2)
step2は、化合物(2’)の-OAにおける保護基Aを除去して化合物(2’’)を得る工程である。除去方法は保護基の種類により選択されるが、例えば、溶媒中で化合物(2’)と第4級アンモニウムフルオリド(例、テトラ-n-ブチルアンモニウムフルオリド)とを反応させる。
第4級アンモニウムフルオリドの使用量は、化合物(2’)に対して、通常1~3当量である。
反応温度は通常0℃~室温であり、反応時間は通常1~20時間である。
溶媒としては、例えば、N,N-ジメチルホルムアミド、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒が挙げられる。溶媒の使用量は、化合物(2’)に対して、通常10~50倍容量である。
化合物(2’’)は、常法によって単離することができ、例えば反応液に水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物(2’’)を単離することができる。
step2は、化合物(2’)の-OAにおける保護基Aを除去して化合物(2’’)を得る工程である。除去方法は保護基の種類により選択されるが、例えば、溶媒中で化合物(2’)と第4級アンモニウムフルオリド(例、テトラ-n-ブチルアンモニウムフルオリド)とを反応させる。
第4級アンモニウムフルオリドの使用量は、化合物(2’)に対して、通常1~3当量である。
反応温度は通常0℃~室温であり、反応時間は通常1~20時間である。
溶媒としては、例えば、N,N-ジメチルホルムアミド、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)等の非プロトン性溶媒が挙げられる。溶媒の使用量は、化合物(2’)に対して、通常10~50倍容量である。
化合物(2’’)は、常法によって単離することができ、例えば反応液に水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥し、濾過し、濃縮することにより化合物(2’’)を単離することができる。
(step3)
step3は、化合物(2’’)における糖部分の水酸基の保護基を除去して化合物(1)を得る工程である。この工程においては、例えば、化合物(2’’)を溶媒中で水素雰囲気下及び還元触媒の存在下に反応させる。
還元触媒としては、パラジウム-C、水酸化パラジウム、水酸化パラジウム-活性炭、酸化白金、ラネーニッケル等が挙げられる。還元触媒の使用量は、化合物(2’’)に対して触媒量であればよい。
溶媒としては、例えば、低級アルコール(例えば、メタノール、エタノール)、ハロゲン化炭化水素類(例えば、ジクロロメタン、クロロホルム)、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)が例示され、これらを混合して使用してもよい。溶媒の使用量は、化合物(2’’)に対して通常20~200倍容量である。
反応温度は通常室温~50℃、反応時間は通常5~20時間である。
反応終了後、反応液を濾過し、濃縮することにより化合物(1)を得ることができる。
step3は、化合物(2’’)における糖部分の水酸基の保護基を除去して化合物(1)を得る工程である。この工程においては、例えば、化合物(2’’)を溶媒中で水素雰囲気下及び還元触媒の存在下に反応させる。
還元触媒としては、パラジウム-C、水酸化パラジウム、水酸化パラジウム-活性炭、酸化白金、ラネーニッケル等が挙げられる。還元触媒の使用量は、化合物(2’’)に対して触媒量であればよい。
溶媒としては、例えば、低級アルコール(例えば、メタノール、エタノール)、ハロゲン化炭化水素類(例えば、ジクロロメタン、クロロホルム)、エーテル類(例、ジエチルエーテル、テトラヒドロフラン)が例示され、これらを混合して使用してもよい。溶媒の使用量は、化合物(2’’)に対して通常20~200倍容量である。
反応温度は通常室温~50℃、反応時間は通常5~20時間である。
反応終了後、反応液を濾過し、濃縮することにより化合物(1)を得ることができる。
上記のようにして得られた本発明の化合物(1)及び化合物(2)は、自体公知の方法あるいはそれに準ずる方法によって、目的とする塩に変換することができる。
次に、本発明の医薬用途について説明する。
本発明の化合物(1)又はその塩を投与することにより、APCの持つCD1dタンパク質と複合体を形成し、NKT細胞に提示される。NKT細胞は、この複合体をTCRを介して認識し、それ自身の有する免疫調節能のうち、免疫細胞の働きを活性化するサイトカインの一種であるIFN-γを選択的かつ大量に産生する一方で、IL-4の産生を抑制することが可能である。また、本発明の化合物(1)又はその塩は、NKT細胞によるIFN-γ産生を増強する作用があるIL-12の産生を誘導する。具体的には、IFN-γ/IL-4比がα-GalCerの2に対して10以上であり、従来公知の糖脂質に比べて極めて高い選択的IFN-γ産生が確認された(試験例1参照)。したがって、本発明の化合物(1)又はその塩は、腫瘍増殖の阻害のための抗癌剤、免疫賦活剤、更には細胞増殖障害やTh1/Th2免疫バランスの是正のための治療に有用である。
本発明の化合物(1)又はその塩を投与することにより、APCの持つCD1dタンパク質と複合体を形成し、NKT細胞に提示される。NKT細胞は、この複合体をTCRを介して認識し、それ自身の有する免疫調節能のうち、免疫細胞の働きを活性化するサイトカインの一種であるIFN-γを選択的かつ大量に産生する一方で、IL-4の産生を抑制することが可能である。また、本発明の化合物(1)又はその塩は、NKT細胞によるIFN-γ産生を増強する作用があるIL-12の産生を誘導する。具体的には、IFN-γ/IL-4比がα-GalCerの2に対して10以上であり、従来公知の糖脂質に比べて極めて高い選択的IFN-γ産生が確認された(試験例1参照)。したがって、本発明の化合物(1)又はその塩は、腫瘍増殖の阻害のための抗癌剤、免疫賦活剤、更には細胞増殖障害やTh1/Th2免疫バランスの是正のための治療に有用である。
癌治療の対象としては、例えば、食道、胃、肝臓、膵臓、乳房、結腸、腎臓、肺(小細胞肺癌、非小細胞肺癌を含む)、胆嚢、卵巣、精巣、膀胱、頸部、甲状腺、前立腺及び皮膚(扁平上皮細胞癌を含む)の腫瘍;リンパ系統の造血腫瘍(白血病、急性リンパ性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリー細胞リンパ腫、バーキットリンパ腫を含む);骨髄系統の造血腫瘍(急性及び慢性の骨髄性白血病、骨髄異形成症候群及び前骨髄急性白血病を含む);間葉起源の腫瘍(線維肉腫及び横紋筋肉腫を含む);中枢神経系及び末梢神経系の腫瘍(星状膠細胞腫、神経芽細胞腫、神経膠腫及び神経鞘腫を含む);他の腫瘍(黒色腫、精上皮腫、奇形癌、骨肉腫、色素性乾皮症、ケラトアカントーマ(keratoacanthoma)、甲状腺濾胞癌、カポージ肉腫を含む)が例示され、これらに限定されない。
また、細胞増殖障害とは、家族性腺腫性ポリポーシス、乾癬、良性前立腺過形成、神経線維腫症、アテローム性動脈硬化症に関連する血管平滑細胞増殖、肺繊維症、関節炎、糸球体腎炎、術後の狭窄、再狭窄を含む概念である。
また、細胞増殖障害とは、家族性腺腫性ポリポーシス、乾癬、良性前立腺過形成、神経線維腫症、アテローム性動脈硬化症に関連する血管平滑細胞増殖、肺繊維症、関節炎、糸球体腎炎、術後の狭窄、再狭窄を含む概念である。
本発明の化合物(1)又はその塩の投与対象は、ヒト等の哺乳動物等が挙げられる。
本発明の化合物(1)又はその塩をヒトに投与する場合、それ自体又はそれを薬理学的に許容される担体(例えば、賦形剤、希釈剤)等と混合し、経口投与剤(例えば、散剤、顆粒剤、錠剤、カプセル剤)、非経口投与剤(例えば、注射剤、坐剤(例えば、直腸坐剤、膣坐剤))等の医薬組成物として経口的又は非経口的に安全に投与することができる。これらの製剤は、従来公知の方法により製造することができる。
注射剤としては、皮下注射、静脈内注射、筋肉内注射、腹腔内注射又は点滴剤等が挙げられる。注射剤は、化合物(1)又はその塩を可溶化剤(例えば、β-シクロデキストリン類)、分散剤(例えば、カルボキシメチルセルロース、アルギン酸ナトリウム)、保存剤(例えば、メチルパラベン、プロピルパラベン、ベンジルアルコール、クロロブタノール)、等張化剤(例えば、塩化ナトリウム、グリセリン、ソルビトール、ブドウ糖)等とともに常法にしたがって水性注射剤にすることもできる。また、植物油(例えば、オリーブ油、ゴマ油、ラッカセイ油、綿実油、コーン油)、プロピレングリコール等に溶解、懸濁又は乳化して油性注射剤にすることもできる。
経口投与剤は、化合物(1)又はその塩に、例えば、賦形剤(例えば、乳糖、白糖、デンプン)、崩壊剤(例えば、デンプン、炭酸カルシウム)、結合剤(例えば、デンプン、アラビアゴム、カルボキシメチルセルロース、ポリビニルピロリドン、ヒドロキシプロピルセルロース)又は滑沢剤(例えば、タルク、ステアリン酸マグネシウム、ポリエチレングリコール)等を適宜添加して圧縮成形し、次いで必要に応じてヒドロキシプロピルメチルセルロース等のコーティングを施すことにより製造することもできる。坐剤は、化合物(1)又はその塩と、非刺激性の賦形剤(例えば、ポリエチレングリコール、高級脂肪酸のグリセライド)とを混合して製造することができる。
化合物(1)又はその塩の投与量は、年齢、体重、症状、剤形、投与方法、投与期間などにより異なるが、例えば、患者(成人、体重約60kg)一人あたり、通常、1日0.1~1mg/kg体重、好ましくは0.5~1mg/kg体重、より好ましくは0.8~1mg/kg体重であり、これを1回から数回に分けて経口又は非経口投与することができる。
以下、本発明を実施例によって更に具体的に説明するが、本発明は以下の実施例に限定されるものではない。
実施例1
化合物2-9の合成
下記のスキームに従って化合物2-9を合成した。
化合物2-9の合成
下記のスキームに従って化合物2-9を合成した。
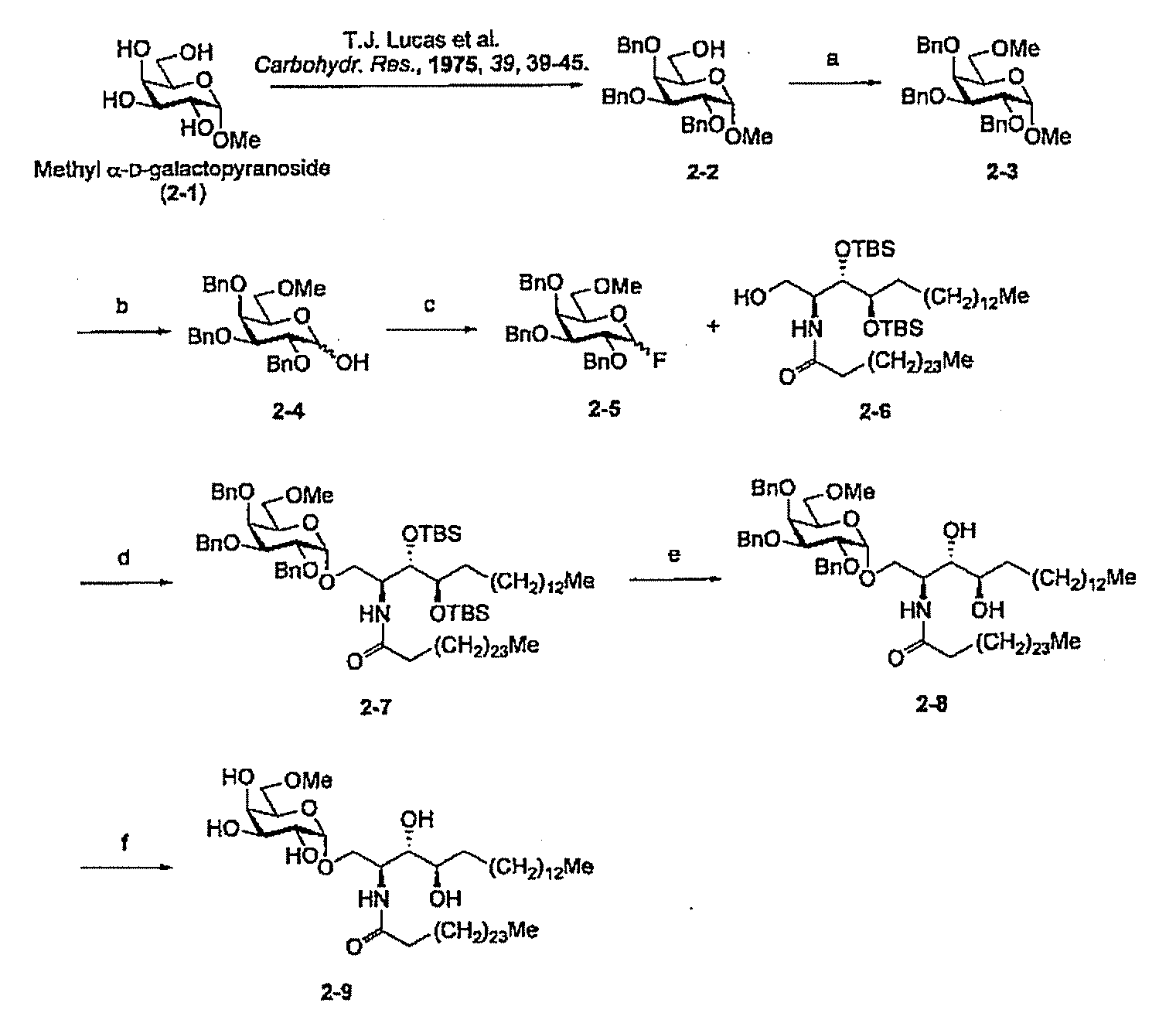
(工程a)化合物2-3の合成
文献既知(T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(1.04 g, 2.24 mmol)のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60%ミネラルオイル懸濁物, 187 mg, 4.68 mmol)を加えた。氷冷下で15分間撹拌した後、ヨウ化メチル(280 μL, 4.50 mmol)を加え、室温下で16時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(30 g, ヘキサン-酢酸エチル = 8 : 1)により精製し、化合物2-3(839 mg, 78%)を無色油状として得た。
nD 22 = 1.5172.
IR (film): νmax = 1600 (w, arom.), 1500 (m, arom.), 1100 (br.s, C-O), 1050 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.26 (15H, m), 4.96 (1H, d, J = 12 Hz), 4.86 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.692 (1H, d, J = 12 Hz), 4.687 (1H, d, J = 3.2 Hz), 4.62 (1H, d, J = 12 Hz), 4.04 (1H, dd, J = 9.6, 3.2 Hz), 3.94 (1H, dd, J = 10, 3.2 Hz), 3.91-3.89 (1H, m), 3.84 (1H, br.t, J = 6.4 Hz), 3.44 (1H, dd, J = 10, 6.4 Hz), 3.37 (3H, s), 3.34 (1H, dd, J = 10, 6.4 Hz), 3.27 (3H, s) ppm.
文献既知(T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(1.04 g, 2.24 mmol)のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60%ミネラルオイル懸濁物, 187 mg, 4.68 mmol)を加えた。氷冷下で15分間撹拌した後、ヨウ化メチル(280 μL, 4.50 mmol)を加え、室温下で16時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(30 g, ヘキサン-酢酸エチル = 8 : 1)により精製し、化合物2-3(839 mg, 78%)を無色油状として得た。
nD 22 = 1.5172.
IR (film): νmax = 1600 (w, arom.), 1500 (m, arom.), 1100 (br.s, C-O), 1050 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.26 (15H, m), 4.96 (1H, d, J = 12 Hz), 4.86 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.692 (1H, d, J = 12 Hz), 4.687 (1H, d, J = 3.2 Hz), 4.62 (1H, d, J = 12 Hz), 4.04 (1H, dd, J = 9.6, 3.2 Hz), 3.94 (1H, dd, J = 10, 3.2 Hz), 3.91-3.89 (1H, m), 3.84 (1H, br.t, J = 6.4 Hz), 3.44 (1H, dd, J = 10, 6.4 Hz), 3.37 (3H, s), 3.34 (1H, dd, J = 10, 6.4 Hz), 3.27 (3H, s) ppm.
(工程b)化合物2-4の合成
化合物2-3(733 mg, 1.53 mmol)の無水酢酸(20 mL)溶液に、濃硫酸(0.03 mL)の無水酢酸(10 mL)溶液を氷冷下で加え、20分間撹拌した。飽和炭酸水素ナトリウム水溶液を加えて中和した後、酢酸エチルで希釈した。有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で順に洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去した。
残渣のメタノール(10 mL) 溶液に、ナトリウムメトキシド(90 mg, 1.7 mmol)を室温下で加え、30分間撹拌した。陽イオン交換樹脂(Dowex 50W-X8)で酸性にした後に濾過し、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 3 : 1)により精製し、化合物2-4(652 mg, 92%)を白色粉末として得た。
IR (KBr): νmax = 3420 (br.s, OH), 1605 (w, arom.), 1495 (m, arom.), 1100 (br.s, C-O), 735 (s), 695 (s) cm-1.
化合物2-3(733 mg, 1.53 mmol)の無水酢酸(20 mL)溶液に、濃硫酸(0.03 mL)の無水酢酸(10 mL)溶液を氷冷下で加え、20分間撹拌した。飽和炭酸水素ナトリウム水溶液を加えて中和した後、酢酸エチルで希釈した。有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で順に洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去した。
残渣のメタノール(10 mL) 溶液に、ナトリウムメトキシド(90 mg, 1.7 mmol)を室温下で加え、30分間撹拌した。陽イオン交換樹脂(Dowex 50W-X8)で酸性にした後に濾過し、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 3 : 1)により精製し、化合物2-4(652 mg, 92%)を白色粉末として得た。
IR (KBr): νmax = 3420 (br.s, OH), 1605 (w, arom.), 1495 (m, arom.), 1100 (br.s, C-O), 735 (s), 695 (s) cm-1.
(工程c)化合物2-5の合成
化合物2-4(602 mg, 1.30 mmol)のジクロロメタン(20 mL)溶液に、ジエチルアミノサルファートリフルオリド (0.35 mL, 2.65 mmol) を-40℃で加えた。室温下で1時間撹拌した後、再び-40℃に冷却し、メタノール(1 mL)を加えた。減圧濃縮により溶媒を留去し、残渣を酢酸エチルで希釈した。有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(15 g, ヘキサン-酢酸エチル = 40 : 3)により精製し、化合物2-5(554 mg, α:β = ca. 1:1, 91%) を無色油状として得た。
1H NMR (400 MHz, CDCl3): δ = 5.59 (1H, dd, J = 54, 3.2 Hz, α-isomer), 5.18 (1H, dd, J = 53, 6.8 Hz, β-isomer) ppm
化合物2-4(602 mg, 1.30 mmol)のジクロロメタン(20 mL)溶液に、ジエチルアミノサルファートリフルオリド (0.35 mL, 2.65 mmol) を-40℃で加えた。室温下で1時間撹拌した後、再び-40℃に冷却し、メタノール(1 mL)を加えた。減圧濃縮により溶媒を留去し、残渣を酢酸エチルで希釈した。有機層を飽和炭酸水素ナトリウム水溶液、水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(15 g, ヘキサン-酢酸エチル = 40 : 3)により精製し、化合物2-5(554 mg, α:β = ca. 1:1, 91%) を無色油状として得た。
1H NMR (400 MHz, CDCl3): δ = 5.59 (1H, dd, J = 54, 3.2 Hz, α-isomer), 5.18 (1H, dd, J = 53, 6.8 Hz, β-isomer) ppm
(工程d)化合物2-7の合成
文献既知(H. Takikawa et al. Tetrahedron, 1998, 54, 3141-3150)化合物2-6(438 mg, 0.474 mmol)の乾燥テトラヒドロフラン(15 mL)溶液に、乾燥させたモレキュラーシーブス(4.03 g)と塩化スズ(II)(270 mg, 1.42 mmol)、過塩素酸銀(300 mg, 1.45 mmol)を加え、遮光したフラスコ内で2時間撹拌した。反応液を-18℃に冷却し、ここへ化合物2-5(252 mg, 0.540 mmol)の乾燥テトラヒドロフラン(10 mL)溶液を加えた。撹拌しながら2時間かけて10℃まで昇温した後、ジエチルエーテル(25 mL)を加え、濾過をした。濾液を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 10 : 1)により精製し、化合物2-7(172 mg, 26%)を無色油状として得た。
nD 24 = 1.4952.
IR (film): νmax = 3360 (m, NH), 1680 (br.s, C=O), 1610 (w, arom.), 1520 (m), 1500 (m, arom.), 1250 (s, t-Bu, Si-Me), 1105 (br.s, C-O), 1060 (br.s, C-O), 835 (s), 780 (s), 735 (br.m), 695 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.38-7.26 (15H, m), 6.26 (1H, d, J = 8.0 Hz), 4.96 (1H, d, J = 12 Hz), 4.800 (1H, d, J = 4.0 Hz), 4.798 (2H, d, J = 12 Hz), 4.72 (1H, d, J = 12 Hz), 4.64 (1H, d, J = 12 Hz), 4.62 (1H, d, J = 12 Hz), 4.10-3.98 (3H, m), 3.93-3.85 (3H, m), 3.79 (1H, br.d, J = 7.6 Hz), 3.69 (1H, dd, J = 11, 2.8 Hz), 3.64-3.60 (1H, m), 3.44 (1H, dd, J = 9.2, 6.0 Hz), 3.29 (1H, dd, J = 9.2, 5.2 Hz), 3.26 (3H, s), 2.05-2.00 (2H, m), 1.59-1.18 (72H, m), 0.90 (9H, s), 0.884 (9H, s), 0.879 (3H, t, J = 6.8 Hz), 0.07 (3H, s), 0.03 (3H, s), 0.024 (3H, s), 0.016 (3H, s) ppm.
文献既知(H. Takikawa et al. Tetrahedron, 1998, 54, 3141-3150)化合物2-6(438 mg, 0.474 mmol)の乾燥テトラヒドロフラン(15 mL)溶液に、乾燥させたモレキュラーシーブス(4.03 g)と塩化スズ(II)(270 mg, 1.42 mmol)、過塩素酸銀(300 mg, 1.45 mmol)を加え、遮光したフラスコ内で2時間撹拌した。反応液を-18℃に冷却し、ここへ化合物2-5(252 mg, 0.540 mmol)の乾燥テトラヒドロフラン(10 mL)溶液を加えた。撹拌しながら2時間かけて10℃まで昇温した後、ジエチルエーテル(25 mL)を加え、濾過をした。濾液を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 10 : 1)により精製し、化合物2-7(172 mg, 26%)を無色油状として得た。
nD 24 = 1.4952.
IR (film): νmax = 3360 (m, NH), 1680 (br.s, C=O), 1610 (w, arom.), 1520 (m), 1500 (m, arom.), 1250 (s, t-Bu, Si-Me), 1105 (br.s, C-O), 1060 (br.s, C-O), 835 (s), 780 (s), 735 (br.m), 695 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.38-7.26 (15H, m), 6.26 (1H, d, J = 8.0 Hz), 4.96 (1H, d, J = 12 Hz), 4.800 (1H, d, J = 4.0 Hz), 4.798 (2H, d, J = 12 Hz), 4.72 (1H, d, J = 12 Hz), 4.64 (1H, d, J = 12 Hz), 4.62 (1H, d, J = 12 Hz), 4.10-3.98 (3H, m), 3.93-3.85 (3H, m), 3.79 (1H, br.d, J = 7.6 Hz), 3.69 (1H, dd, J = 11, 2.8 Hz), 3.64-3.60 (1H, m), 3.44 (1H, dd, J = 9.2, 6.0 Hz), 3.29 (1H, dd, J = 9.2, 5.2 Hz), 3.26 (3H, s), 2.05-2.00 (2H, m), 1.59-1.18 (72H, m), 0.90 (9H, s), 0.884 (9H, s), 0.879 (3H, t, J = 6.8 Hz), 0.07 (3H, s), 0.03 (3H, s), 0.024 (3H, s), 0.016 (3H, s) ppm.
(工程e)化合物2-8の合成
化合物2-7(126 mg, 0.0919 mmol)のテトラヒドロフラン(5 mL)溶液に、テトラ-n-ブチルアンモニウムフルオリドのテトラヒドロフラン溶液(1.0 M, 370 μL, 0.37 mmol)を室温下で加え、18時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(15 g, ヘキサン-酢酸エチル = 7 : 3)により精製し、化合物2-8(98 mg, 93%)を白色固体として得た。
IR (KBr): νmax = 3420 (br.s, OH), 3320 (br.m, NH), 1645 (s, C=O), 1620 (s), 1540 (br.s), 1500 (w, arom.), 1100 (br.s, C-O), 1060 (br.s, C-O), 735 (br.s), 695 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.28 (15H, m), 6.40 (1H, d, J = 8.8 Hz), 4.94 (1H, d, J = 12 Hz), 4.90 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 4.0 Hz), 4.77 (2H, s), 4.68 (1H, d, J = 12 Hz), 4.61 (1H, d, J = 12 Hz), 4.23-4.17 (1H, m), 4.05 (1H, dd, J = 10, 3.6 Hz), 3.94 (1H, br.s), 3.90-3.84 (2H, m), 3.82 (1H, br.t, J = 6.8 Hz), 3.79-3.77 (1H, m), 3.52-3.44 (2H, m), 3.41 (1H, dd, J = 10, 6.4 Hz), 3.34 (1H, dd, J = 8.4, 6.4 Hz), 3.26 (3H, s), 2.18-2.12 (3H, m), 1.64-1.15 (73H, m), 0.88 (6H, t, J = 6.8 Hz) ppm.
化合物2-7(126 mg, 0.0919 mmol)のテトラヒドロフラン(5 mL)溶液に、テトラ-n-ブチルアンモニウムフルオリドのテトラヒドロフラン溶液(1.0 M, 370 μL, 0.37 mmol)を室温下で加え、18時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(15 g, ヘキサン-酢酸エチル = 7 : 3)により精製し、化合物2-8(98 mg, 93%)を白色固体として得た。
IR (KBr): νmax = 3420 (br.s, OH), 3320 (br.m, NH), 1645 (s, C=O), 1620 (s), 1540 (br.s), 1500 (w, arom.), 1100 (br.s, C-O), 1060 (br.s, C-O), 735 (br.s), 695 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.28 (15H, m), 6.40 (1H, d, J = 8.8 Hz), 4.94 (1H, d, J = 12 Hz), 4.90 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 4.0 Hz), 4.77 (2H, s), 4.68 (1H, d, J = 12 Hz), 4.61 (1H, d, J = 12 Hz), 4.23-4.17 (1H, m), 4.05 (1H, dd, J = 10, 3.6 Hz), 3.94 (1H, br.s), 3.90-3.84 (2H, m), 3.82 (1H, br.t, J = 6.8 Hz), 3.79-3.77 (1H, m), 3.52-3.44 (2H, m), 3.41 (1H, dd, J = 10, 6.4 Hz), 3.34 (1H, dd, J = 8.4, 6.4 Hz), 3.26 (3H, s), 2.18-2.12 (3H, m), 1.64-1.15 (73H, m), 0.88 (6H, t, J = 6.8 Hz) ppm.
(工程f)化合物2-9の合成
化合物2-8(73 mg, 0.064 mmol)のテトラヒドロフラン-エタノール-クロロホルム(5 : 8 : 2, 15 mL)溶液に、水酸化パラジウム-活性炭(20%, wet, 33 mg)を室温下で加えた。水素雰囲気下で17時間撹拌した後、クロロホルム-メタノール(5 : 1)で希釈した。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(6 g, ヘキサン-酢酸エチル = 25 : 2)により精製し、化合物2-9(43 mg, 77%)を白色粉末として得た。
IR (KBr): νmax = 3440 (br.s, OH), 3280 (w, NH), 1640 (br.s, C=O), 1540 (br.m), 1080 (br.s, C-O), 720 (w) cm-1.
1H NMR (400 MHz, C5D5N): δ = 8.44 (1H, d, J = 8.8 Hz), 7.05 (1H, br.s), 6.74 (1H, br.s), 6.45 (1H, d, J = 6.4 Hz), 6.39 (1H, br.s), 6.08 (1H, br.s), 5.52 (1H, d, J = 4.0 Hz), 5.28-5.22 (1H, m), 4.64 (1H, dd, J = 10, 5.6 Hz), 4.65-4.58 (1H, m), 4.46 (1H, t, J = 6.4 Hz), 4.40-4.28 (4H, m), 4.14-4.08 (1H, m), 3.97 (1H, dd, J = 9.6, 5.6 Hz), 3.94 (1H, dd, J = 9.6, 6.4 Hz), 3.33 (3H, s), 2.42 (2H, t, J = 7.2 Hz), 2.33-2.20 (1H, m), 1.95-1.60 (5H, m), 1.48-1.16 (68H, m), 0.84 (3H, t, J = 6.8 Hz) ppm.
HRFABMS: calcd for C51H102O9N ([M+H]+) 872.7555; found 872.7553.
化合物2-8(73 mg, 0.064 mmol)のテトラヒドロフラン-エタノール-クロロホルム(5 : 8 : 2, 15 mL)溶液に、水酸化パラジウム-活性炭(20%, wet, 33 mg)を室温下で加えた。水素雰囲気下で17時間撹拌した後、クロロホルム-メタノール(5 : 1)で希釈した。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(6 g, ヘキサン-酢酸エチル = 25 : 2)により精製し、化合物2-9(43 mg, 77%)を白色粉末として得た。
IR (KBr): νmax = 3440 (br.s, OH), 3280 (w, NH), 1640 (br.s, C=O), 1540 (br.m), 1080 (br.s, C-O), 720 (w) cm-1.
1H NMR (400 MHz, C5D5N): δ = 8.44 (1H, d, J = 8.8 Hz), 7.05 (1H, br.s), 6.74 (1H, br.s), 6.45 (1H, d, J = 6.4 Hz), 6.39 (1H, br.s), 6.08 (1H, br.s), 5.52 (1H, d, J = 4.0 Hz), 5.28-5.22 (1H, m), 4.64 (1H, dd, J = 10, 5.6 Hz), 4.65-4.58 (1H, m), 4.46 (1H, t, J = 6.4 Hz), 4.40-4.28 (4H, m), 4.14-4.08 (1H, m), 3.97 (1H, dd, J = 9.6, 5.6 Hz), 3.94 (1H, dd, J = 9.6, 6.4 Hz), 3.33 (3H, s), 2.42 (2H, t, J = 7.2 Hz), 2.33-2.20 (1H, m), 1.95-1.60 (5H, m), 1.48-1.16 (68H, m), 0.84 (3H, t, J = 6.8 Hz) ppm.
HRFABMS: calcd for C51H102O9N ([M+H]+) 872.7555; found 872.7553.
実施例2
化合物3-4の合成
下記のスキームに従って化合物3-4を合成した。
化合物3-4の合成
下記のスキームに従って化合物3-4を合成した。

(工程g)化合物3-2の合成
文献既知(K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203)化合物3-1(1.12 g, 2.43 mmol)とヒドラジン1水和物(10 mL, 20.6 mmol)のエタノール(50 mL)溶液に室温下で過酸化水素水(30%, 4 mL)を3時間かけてゆっくりと加えた。反応液を室温下で18時間撹拌した後、氷冷下で飽和チオ硫酸ナトリウム水溶液(20 mL)を加え、30分間撹拌した。酢酸エチルで希釈し、有機層を水、飽和チオ硫酸ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 20 : 1)により精製し、化合物3-2(981 mg, 87%)を無色油状として得た。
nD 22= 1.5163.
IR (film): νmax = 1605 (w, arom.), 1500 (s, arom.), 1100 (br.s, C-O), 1050 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.25 (15H, m), 4.99 (1H, d, J = 11 Hz), 4.89 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 12 Hz), 4.75 (1H, d, J = 12 Hz), 4.70 (1H, d, J = 12 Hz), 4.66 (1H, d, J = 4.0 Hz), 4.65 (1H, d, J = 11 Hz), 4.05 (1H, dd, J = 10, 4.0 Hz), 3.92 (1H, dd, J = 10, 2.8 Hz), 3.72 (1H, d, J = 2.8 Hz), 3.50 (1H, dd, J = 8.4, 6.0 Hz), 3.35 (3H, s), 1.66 (1H, ddq, J = 14, 7.2, 6.0 Hz), 1.37 (1H, m), 0.81 (3H, t, J = 7.2 Hz) ppm.
文献既知(K. Tatsuta et al. Carbohydr. Res., 1991, 222, 189-203)化合物3-1(1.12 g, 2.43 mmol)とヒドラジン1水和物(10 mL, 20.6 mmol)のエタノール(50 mL)溶液に室温下で過酸化水素水(30%, 4 mL)を3時間かけてゆっくりと加えた。反応液を室温下で18時間撹拌した後、氷冷下で飽和チオ硫酸ナトリウム水溶液(20 mL)を加え、30分間撹拌した。酢酸エチルで希釈し、有機層を水、飽和チオ硫酸ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 20 : 1)により精製し、化合物3-2(981 mg, 87%)を無色油状として得た。
nD 22= 1.5163.
IR (film): νmax = 1605 (w, arom.), 1500 (s, arom.), 1100 (br.s, C-O), 1050 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.25 (15H, m), 4.99 (1H, d, J = 11 Hz), 4.89 (1H, d, J = 12 Hz), 4.84 (1H, d, J = 12 Hz), 4.75 (1H, d, J = 12 Hz), 4.70 (1H, d, J = 12 Hz), 4.66 (1H, d, J = 4.0 Hz), 4.65 (1H, d, J = 11 Hz), 4.05 (1H, dd, J = 10, 4.0 Hz), 3.92 (1H, dd, J = 10, 2.8 Hz), 3.72 (1H, d, J = 2.8 Hz), 3.50 (1H, dd, J = 8.4, 6.0 Hz), 3.35 (3H, s), 1.66 (1H, ddq, J = 14, 7.2, 6.0 Hz), 1.37 (1H, m), 0.81 (3H, t, J = 7.2 Hz) ppm.
化合物3-4の合成
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-2から化合物3-3を経て5段階で化合物3-4を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.49 (1H, d, J = 8.8 Hz), 6.98 (1H, br.s), 6.59 (1H, br.s), 6.44 (1H, br.d, J = 7.2 Hz), 6.13 (2H, br.s), 5.48 (1H, d, J = 3.6 Hz), 5.33-5.26 (1H, m), 4.64 (1H, dd, J = 10, 5.6 Hz), 4.60-4.54 (1H, m), 4.38-4.27 (3H, m), 4.27 (1H, dd, J = 10, 4.8 Hz), 4.17 (1H, br.s), 3.99 (1H, t, J = 6.8 Hz), 2.44 (2H, t, J = 6.8 Hz), 2.35-2.25 (1H, m), 2.15-2.03 (1H, m), 1.98-1.77 (5H, m), 1.74-1.61 (1H, m), 1.47-1.16 (66H, m), 1.05 (3H, t, J = 7.2 Hz), 0.84 (6H, t, J = 7.2 Hz) ppm.
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-2から化合物3-3を経て5段階で化合物3-4を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.49 (1H, d, J = 8.8 Hz), 6.98 (1H, br.s), 6.59 (1H, br.s), 6.44 (1H, br.d, J = 7.2 Hz), 6.13 (2H, br.s), 5.48 (1H, d, J = 3.6 Hz), 5.33-5.26 (1H, m), 4.64 (1H, dd, J = 10, 5.6 Hz), 4.60-4.54 (1H, m), 4.38-4.27 (3H, m), 4.27 (1H, dd, J = 10, 4.8 Hz), 4.17 (1H, br.s), 3.99 (1H, t, J = 6.8 Hz), 2.44 (2H, t, J = 6.8 Hz), 2.35-2.25 (1H, m), 2.15-2.03 (1H, m), 1.98-1.77 (5H, m), 1.74-1.61 (1H, m), 1.47-1.16 (66H, m), 1.05 (3H, t, J = 7.2 Hz), 0.84 (6H, t, J = 7.2 Hz) ppm.
実施例3
化合物3-7の合成
下記のスキームに従って化合物3-7を合成した。
化合物3-7の合成
下記のスキームに従って化合物3-7を合成した。
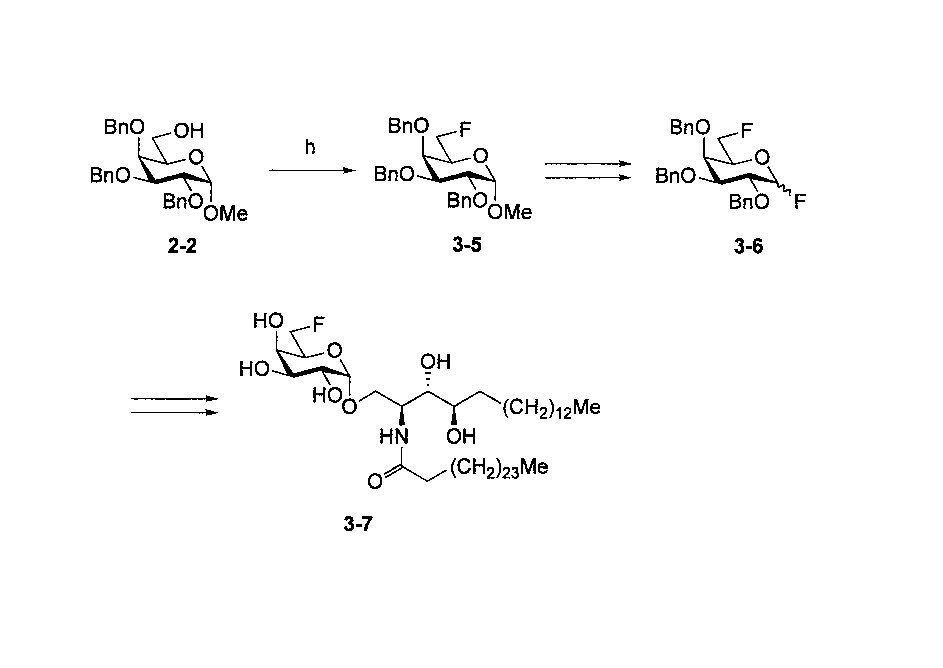
(工程h)化合物3-5の合成
文献既知(T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(2.03 g, 4.37 mmol)とトリエチルアミン(1.85 mL, 13.3 mmol)のジクロロメタン(30 mL)溶液に-40℃下でジエチルアミノサルファートリフルオリド(1.20 mL, 9.08 mmol)をゆっくりと加えた。反応液を加熱還流下で5時間撹拌した後、氷冷下でメタノール(2 mL)を加え、30分間撹拌した。酢酸エチルで希釈し、有機層を水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(30 g, ヘキサン-酢酸エチル= 8 : 1) により精製し、化合物3-5(688 mg, 34%)を無色油状として得た。
nD 22 = 1.5169.
IR (film): νmax = 1600 (w, arom.), 1500 (s, arom.), 1100 (br.s, C-O), 1040 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.24 (15H, m), 4.97 (1H, d, J = 12 Hz), 4.89 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.75 (1H, d, J = 12 Hz), 4.70 (1H, d, J = 12 Hz), 4.69 (1H, d, J = 4.0 Hz), 4.60 (1H, d, J = 12 Hz), 4.44 (1H, ddd, J = 48, 9.2, 6.0 Hz), 4.27 (1H, ddd, J = 46, 9.2, 5.2 Hz), 4.04 (1H, dd, J = 10, 4.0 Hz), 4.00-3.91 (2H, m), 3.89 (1H, br.s), 3.37 (3H, s) ppm.
文献既知(T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(2.03 g, 4.37 mmol)とトリエチルアミン(1.85 mL, 13.3 mmol)のジクロロメタン(30 mL)溶液に-40℃下でジエチルアミノサルファートリフルオリド(1.20 mL, 9.08 mmol)をゆっくりと加えた。反応液を加熱還流下で5時間撹拌した後、氷冷下でメタノール(2 mL)を加え、30分間撹拌した。酢酸エチルで希釈し、有機層を水、飽和炭酸水素ナトリウム水溶液、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(30 g, ヘキサン-酢酸エチル= 8 : 1) により精製し、化合物3-5(688 mg, 34%)を無色油状として得た。
nD 22 = 1.5169.
IR (film): νmax = 1600 (w, arom.), 1500 (s, arom.), 1100 (br.s, C-O), 1040 (br.s, C-O), 740 (s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.42-7.24 (15H, m), 4.97 (1H, d, J = 12 Hz), 4.89 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.75 (1H, d, J = 12 Hz), 4.70 (1H, d, J = 12 Hz), 4.69 (1H, d, J = 4.0 Hz), 4.60 (1H, d, J = 12 Hz), 4.44 (1H, ddd, J = 48, 9.2, 6.0 Hz), 4.27 (1H, ddd, J = 46, 9.2, 5.2 Hz), 4.04 (1H, dd, J = 10, 4.0 Hz), 4.00-3.91 (2H, m), 3.89 (1H, br.s), 3.37 (3H, s) ppm.
化合物3-7の合成
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-5から化合物3-6を経て5段階で化合物3-7を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.47 (1H, d, J = 8.8 Hz), 7.15 (1H, br.s), 6.95 (1H, br.s), 6.67 (1H, br.s), 6.48 (1H, br.s), 6.13 (1H, br.s), 5.56 (1H, d, J = 4.0 Hz), 5.32-5.26 (1H, m), 5.02 (1H, ddd, J = 49, 10, 6.8 Hz), 4.95 (1H, ddd, J = 46, 10, 4.4 Hz), 4.68 (1H, dd, J = 10, 5.6 Hz), 4.61 (1H, dd, J = 10, 4.4 Hz), 4.54-4.47 (1H, m), 4.39 (1H, dd, J = 9.6, 3.2 Hz), 4.31-4.27 (3H, m), 4.12 (1H, t, J = 6.8 Hz), 2.42 (2H, br.t, J = 7.2 Hz), 2.34-2.25 (1H, m), 1.97-1.84 (2H, m), 1.80 (2H, quint., J = 7.2 Hz), 1.73-1.60 (1H, m), 1.48-1.15 (68H, m), 0.84 (6H, t, J = 6.8 Hz) ppm.
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-5から化合物3-6を経て5段階で化合物3-7を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.47 (1H, d, J = 8.8 Hz), 7.15 (1H, br.s), 6.95 (1H, br.s), 6.67 (1H, br.s), 6.48 (1H, br.s), 6.13 (1H, br.s), 5.56 (1H, d, J = 4.0 Hz), 5.32-5.26 (1H, m), 5.02 (1H, ddd, J = 49, 10, 6.8 Hz), 4.95 (1H, ddd, J = 46, 10, 4.4 Hz), 4.68 (1H, dd, J = 10, 5.6 Hz), 4.61 (1H, dd, J = 10, 4.4 Hz), 4.54-4.47 (1H, m), 4.39 (1H, dd, J = 9.6, 3.2 Hz), 4.31-4.27 (3H, m), 4.12 (1H, t, J = 6.8 Hz), 2.42 (2H, br.t, J = 7.2 Hz), 2.34-2.25 (1H, m), 1.97-1.84 (2H, m), 1.80 (2H, quint., J = 7.2 Hz), 1.73-1.60 (1H, m), 1.48-1.15 (68H, m), 0.84 (6H, t, J = 6.8 Hz) ppm.
実施例4
化合物3-10の合成
下記のスキームに従って化合物3-10を合成した。
化合物3-10の合成
下記のスキームに従って化合物3-10を合成した。

化合物3-10の合成
文献既知(S. Koto et al. Bull. Chem. Soc. Jpn., 2000, 73, 967-976)化合物3-8から、化合物2-3から化合物2-9への変換と同様な手法により、化合物3-9を経て5段階で化合物3-10を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.46 (1H, d, J = 8.8 Hz), 6.99 (1H, br.s), 6.44 (1H, br.s), 6.23 (1H, br.s), 6.14 (2H, br.s), 5.48 (1H, d, J = 4.0 Hz), 5.32-5.27 (1H, m), 4.65 (1H, dd, J = 10, 5.6 Hz), 4.57 (1H, dd, J = 10, 4.4 Hz), 4.38 (1H, dd, J = 10, 4.4 Hz), 4.34-4.27 (4H, m), 4.08 (1H, br.d, J = 2.4 Hz), 2.44 (2H, br.t, J = 6.8 Hz), 2.35-2.26 (1H, m), 1.97-1.83 (2H, m), 1.82 (2H, quint., J = 6.8 Hz), 1.74-1.63 (1H, m), 1.50 (3H, d, J = 6.4 Hz), 1.47-1.17 (66H, m), 0.85 (6H, t, J = 6.8 Hz) ppm.
文献既知(S. Koto et al. Bull. Chem. Soc. Jpn., 2000, 73, 967-976)化合物3-8から、化合物2-3から化合物2-9への変換と同様な手法により、化合物3-9を経て5段階で化合物3-10を白色粉末として得た。
1H NMR (400 MHz, C5D5N): δ = 8.46 (1H, d, J = 8.8 Hz), 6.99 (1H, br.s), 6.44 (1H, br.s), 6.23 (1H, br.s), 6.14 (2H, br.s), 5.48 (1H, d, J = 4.0 Hz), 5.32-5.27 (1H, m), 4.65 (1H, dd, J = 10, 5.6 Hz), 4.57 (1H, dd, J = 10, 4.4 Hz), 4.38 (1H, dd, J = 10, 4.4 Hz), 4.34-4.27 (4H, m), 4.08 (1H, br.d, J = 2.4 Hz), 2.44 (2H, br.t, J = 6.8 Hz), 2.35-2.26 (1H, m), 1.97-1.83 (2H, m), 1.82 (2H, quint., J = 6.8 Hz), 1.74-1.63 (1H, m), 1.50 (3H, d, J = 6.4 Hz), 1.47-1.17 (66H, m), 0.85 (6H, t, J = 6.8 Hz) ppm.
実施例5
化合物3-13の合成
下記のスキームに従って化合物3-13を合成した。
化合物3-13の合成
下記のスキームに従って化合物3-13を合成した。
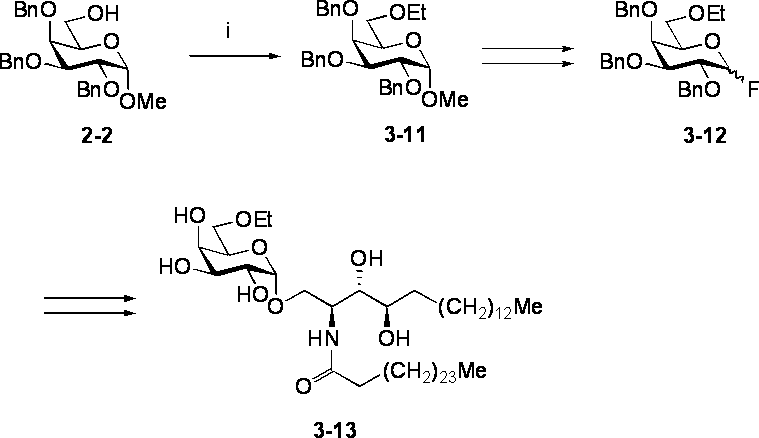
(工程i)化合物3-11の合成
文献既知 (T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(587 mg, 1.26 mmol) のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60% ミネラルオイル懸濁物, 158 mg, 3.95 mmol)を加えた。氷冷下で10分間撹拌した後、臭化エチル(295 μL, 3.95 mmol)と触媒量のヨウ化テトラ-n-ブチルアンモニウムを加え、室温下で12時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 8 : 1)により精製し、化合物3-11(606 mg, 98%)を無色油状として得た。
nD 23 = 1.5170.
IR (film): νmax = 1605 (w, arom.), 1495 (m, arom.), 1115 (br.s, C-O), 1050 (br.s, C-O), 735 (br.s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.24 (15H, m), 4.95 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.83 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.685 (1H, d, J = 12 Hz), 4.682 (1H, d, J = 3.2 Hz), 4.62 (1H, d, J = 12 Hz), 4.06-4.02 (1H, m), 3.96-3.92 (2H, m), 3.85 (1H, br t, J = 6.6 Hz), 3.49-3.42 (3H, m), 3.38-3.34 (1H, m), 3.37 (3H, s), 1.14 (3H, t, J = 7.2 Hz) ppm
文献既知 (T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(587 mg, 1.26 mmol) のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60% ミネラルオイル懸濁物, 158 mg, 3.95 mmol)を加えた。氷冷下で10分間撹拌した後、臭化エチル(295 μL, 3.95 mmol)と触媒量のヨウ化テトラ-n-ブチルアンモニウムを加え、室温下で12時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 8 : 1)により精製し、化合物3-11(606 mg, 98%)を無色油状として得た。
nD 23 = 1.5170.
IR (film): νmax = 1605 (w, arom.), 1495 (m, arom.), 1115 (br.s, C-O), 1050 (br.s, C-O), 735 (br.s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.24 (15H, m), 4.95 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.83 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.685 (1H, d, J = 12 Hz), 4.682 (1H, d, J = 3.2 Hz), 4.62 (1H, d, J = 12 Hz), 4.06-4.02 (1H, m), 3.96-3.92 (2H, m), 3.85 (1H, br t, J = 6.6 Hz), 3.49-3.42 (3H, m), 3.38-3.34 (1H, m), 3.37 (3H, s), 1.14 (3H, t, J = 7.2 Hz) ppm
化合物3-13の合成
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-11から化合物3-12を経て5段階で化合物3-13を白色粉末として得た。
1H NMR (500 MHz, CDCl3): δ = 8.43 (1H, d, J = 9.0 Hz), 7.02 (1H, br s), 6.69 (1H, br s), 6.43 (1H, d, J = 6.5 Hz), 6.36 (1H, br s), 6.07 (1H, d, J = 5.5 Hz), 5.51 (1H, d, J = 3.5 Hz), 5.23 (1H, dq, J = 8.0, 4.0 Hz), 4.63 (1H, dd, J = 10, 5.0 Hz), 4.63-4.58 (1H, m), 4.45 (1H, t, J = 6.5 Hz), 4.40-4.28 (4H, m), 4.36 (1H, dd, J = 10, 5.0 Hz), 4.04 (1H, dd, J = 10, 6.5 Hz), 3.96 (1H, dd, J = 10, 6.5 Hz), 3.55-3.45 (2H, m), 2.42 (2H, dt, J = 7.0, 2.0 Hz), 2.30-2.23 (1H, m), 1.96-1.84 (2H, m), 1.80 (2H, quint., J = 7.0 Hz), 1.72-1.61 (1H, m), 1.48-1.17 (66H, m), 1.14 (3H, t, J = 7.0 Hz), 0.84 (6H, t, J = 7.0 Hz) ppm.
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-11から化合物3-12を経て5段階で化合物3-13を白色粉末として得た。
1H NMR (500 MHz, CDCl3): δ = 8.43 (1H, d, J = 9.0 Hz), 7.02 (1H, br s), 6.69 (1H, br s), 6.43 (1H, d, J = 6.5 Hz), 6.36 (1H, br s), 6.07 (1H, d, J = 5.5 Hz), 5.51 (1H, d, J = 3.5 Hz), 5.23 (1H, dq, J = 8.0, 4.0 Hz), 4.63 (1H, dd, J = 10, 5.0 Hz), 4.63-4.58 (1H, m), 4.45 (1H, t, J = 6.5 Hz), 4.40-4.28 (4H, m), 4.36 (1H, dd, J = 10, 5.0 Hz), 4.04 (1H, dd, J = 10, 6.5 Hz), 3.96 (1H, dd, J = 10, 6.5 Hz), 3.55-3.45 (2H, m), 2.42 (2H, dt, J = 7.0, 2.0 Hz), 2.30-2.23 (1H, m), 1.96-1.84 (2H, m), 1.80 (2H, quint., J = 7.0 Hz), 1.72-1.61 (1H, m), 1.48-1.17 (66H, m), 1.14 (3H, t, J = 7.0 Hz), 0.84 (6H, t, J = 7.0 Hz) ppm.
実施例6
化合物3-16の合成
下記のスキームに従って化合物3-16を合成した。
化合物3-16の合成
下記のスキームに従って化合物3-16を合成した。
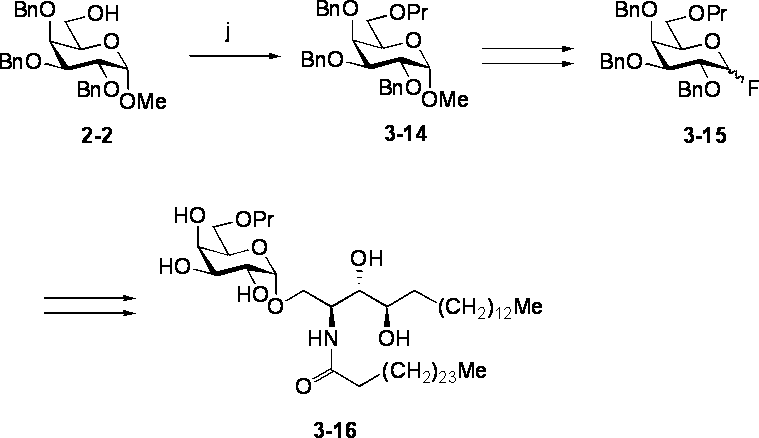
(工程j)化合物3-14の合成
文献既知 (T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(630 mg, 1.36 mmol) のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60% ミネラルオイル懸濁物, 170 mg, 4.25 mmol)を加えた。氷冷下で10分間撹拌した後、1-臭化プロピル(390 μL, 4.28 mmol)と触媒量のヨウ化テトラ-n-ブチルアンモニウムを加え、室温下で12時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 40 : 3)により精製し、化合物3-14(647 mg, 94%)を無色油状として得た。
nD 23 = 1.5177.
IR (film): νmax = 1605 (w, arom.), 1495 (m, arom.), 1110 (br.s, C-O), 1050 (br.s, C-O), 740 (br.s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.24 (15H, m), 4.96 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.83 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.685 (1H, d, J = 12 Hz), 4.680 (1H, d, J = 4.0 Hz), 4.61 (1H, d, J = 12 Hz), 4.06-4.01 (1H, m), 3.96-3.92 (2H, m), 3.86 (1H, t, J = 6.4 Hz), 3.45 (2H, d, J = 6.4 Hz), 3.40-3.34 (1H, m), 3.37 (3H, s), 3.26 (1H, dt, J = 9.6, 7.2 Hz), 1.53 (1H, sext., J = 7.2 Hz), 0.89 (3H, t, J = 7.2 Hz) ppm.
文献既知 (T. J. Lucas et al., Carbohydr. Res., 1975, 39, 39-45)化合物2-2(630 mg, 1.36 mmol) のN,N-ジメチルホルムアミド-テトラヒドロフラン(1 : 1, 20 mL)溶液に氷冷下で水素化ナトリウム(60% ミネラルオイル懸濁物, 170 mg, 4.25 mmol)を加えた。氷冷下で10分間撹拌した後、1-臭化プロピル(390 μL, 4.28 mmol)と触媒量のヨウ化テトラ-n-ブチルアンモニウムを加え、室温下で12時間撹拌した。水を加え、酢酸エチルで抽出し、有機層を水、飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥させた。濾過後、減圧濃縮により溶媒を留去し、残渣をシリカゲルカラムクロマトグラフィー(20 g, ヘキサン-酢酸エチル = 40 : 3)により精製し、化合物3-14(647 mg, 94%)を無色油状として得た。
nD 23 = 1.5177.
IR (film): νmax = 1605 (w, arom.), 1495 (m, arom.), 1110 (br.s, C-O), 1050 (br.s, C-O), 740 (br.s), 700 (s) cm-1.
1H NMR (400 MHz, CDCl3): δ = 7.40-7.24 (15H, m), 4.96 (1H, d, J = 12 Hz), 4.85 (1H, d, J = 12 Hz), 4.83 (1H, d, J = 12 Hz), 4.74 (1H, d, J = 12 Hz), 4.685 (1H, d, J = 12 Hz), 4.680 (1H, d, J = 4.0 Hz), 4.61 (1H, d, J = 12 Hz), 4.06-4.01 (1H, m), 3.96-3.92 (2H, m), 3.86 (1H, t, J = 6.4 Hz), 3.45 (2H, d, J = 6.4 Hz), 3.40-3.34 (1H, m), 3.37 (3H, s), 3.26 (1H, dt, J = 9.6, 7.2 Hz), 1.53 (1H, sext., J = 7.2 Hz), 0.89 (3H, t, J = 7.2 Hz) ppm.
化合物3-16の合成
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-14から化合物3-15を経て5段階で化合物3-16を白色粉末として得た。
1H NMR (500 MHz, CDCl3): δ = 8.43 (1H, d, J = 8.5 Hz), 7.02 (1H, br s), 6.69 (1H, br s), 6.42 (1H, d, J = 6.5 Hz), 6.35 (1H, br s), 6.08 (1H, d, J = 5.5 Hz), 5.52 (1H, d, J = 4.0 Hz), 5.25 (1H, dq, J = 8.0, 4.0 Hz), 4.65 (1H, dd, J = 11, 5.5 Hz), 4.66-4.58 (1H, m), 4.46 (1H, t, J = 5.5 Hz), 4.42-4.28 (4H, m), 4.36 (1H, dd, J = 11, 5.5 Hz), 4.07 (1H, dd, J = 10, 6.0 Hz), 3.97 (1H, dd, J = 10, 6.0 Hz), 3.58-3.48 (2H, m), 2.43 (2H, dt, J = 7.0, 2.0 Hz), 2.31-2.24 (1H, m), 1.96-1.84 (2H, m), 1.81 (2H, quint., J = 7.0 Hz), 1.73-1.64 (1H, m), 1.56 (2H, sext., J = 7.0 Hz), 1.46-1.16 (66H, m), 0.87 (3H, t, J = 7.0 Hz), 0.847 (3H, t, J = 7.0 Hz), 0.845 (3H, t, J = 7.0 Hz) ppm.
化合物2-3から化合物2-9への変換と同様な手法により、化合物3-14から化合物3-15を経て5段階で化合物3-16を白色粉末として得た。
1H NMR (500 MHz, CDCl3): δ = 8.43 (1H, d, J = 8.5 Hz), 7.02 (1H, br s), 6.69 (1H, br s), 6.42 (1H, d, J = 6.5 Hz), 6.35 (1H, br s), 6.08 (1H, d, J = 5.5 Hz), 5.52 (1H, d, J = 4.0 Hz), 5.25 (1H, dq, J = 8.0, 4.0 Hz), 4.65 (1H, dd, J = 11, 5.5 Hz), 4.66-4.58 (1H, m), 4.46 (1H, t, J = 5.5 Hz), 4.42-4.28 (4H, m), 4.36 (1H, dd, J = 11, 5.5 Hz), 4.07 (1H, dd, J = 10, 6.0 Hz), 3.97 (1H, dd, J = 10, 6.0 Hz), 3.58-3.48 (2H, m), 2.43 (2H, dt, J = 7.0, 2.0 Hz), 2.31-2.24 (1H, m), 1.96-1.84 (2H, m), 1.81 (2H, quint., J = 7.0 Hz), 1.73-1.64 (1H, m), 1.56 (2H, sext., J = 7.0 Hz), 1.46-1.16 (66H, m), 0.87 (3H, t, J = 7.0 Hz), 0.847 (3H, t, J = 7.0 Hz), 0.845 (3H, t, J = 7.0 Hz) ppm.
試験例1 化合物2-9、化合物3-4、化合物3-7及び化合物3-10の生物活性試験
α-GalCer、カルバ糖脂質A、化合物2-9、化合物3-4、化合物3-7及び化合物3-10のそれぞれについて、1 mg/mLの濃度のDMSO溶液を調製した。1匹のマウスに200 μLをマウス尾静脈に投与した際、投与量が100 μg/kg体重になるように、上記のDMSO溶液を0.5%のtween20(Bio-Rad)を含有する生理食塩水(大塚製薬株式会社製)を用いて希釈した。
1群5匹のC57BL/6マウスに、調製したカルバ糖脂質A、化合物2-9、化合物3-4、化合物3-7及び化合物3-10の溶液200 μLをそれぞれマウス尾静脈に注射した。対照物質としてα-GalCerを用い、同様の方法により投与量が100 μg/kg体重となるように調製したα-GalCerの溶液200 μLをマウス尾静脈に注射した。媒体である0.5%のtween20を含有する生理食塩水200 μLを投与した群をネガティブコントロールとした。投与後6、12、24、36、48、60時間経過後の血液を眼下静脈叢より80 μL採取し、血清を調製した。
投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を、サンドウィッチELISA(ENDOGEN)により測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図1に示す。
投与後3、6、12時間経過後の血清中のIL-4の含有量を、ELISA法の1つであるCytometric bead arrayシステム(BD Biosciences)で測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図2に示す。
投与後3、6、12時間経過後の血清中のIL-12の含有量を、ELISA法の1つであるCytometric bead arrayシステム(BD Biosciences)で測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図3に示す。
α-GalCer、カルバ糖脂質A、化合物2-9、化合物3-4、化合物3-7及び化合物3-10のそれぞれについて、1 mg/mLの濃度のDMSO溶液を調製した。1匹のマウスに200 μLをマウス尾静脈に投与した際、投与量が100 μg/kg体重になるように、上記のDMSO溶液を0.5%のtween20(Bio-Rad)を含有する生理食塩水(大塚製薬株式会社製)を用いて希釈した。
1群5匹のC57BL/6マウスに、調製したカルバ糖脂質A、化合物2-9、化合物3-4、化合物3-7及び化合物3-10の溶液200 μLをそれぞれマウス尾静脈に注射した。対照物質としてα-GalCerを用い、同様の方法により投与量が100 μg/kg体重となるように調製したα-GalCerの溶液200 μLをマウス尾静脈に注射した。媒体である0.5%のtween20を含有する生理食塩水200 μLを投与した群をネガティブコントロールとした。投与後6、12、24、36、48、60時間経過後の血液を眼下静脈叢より80 μL採取し、血清を調製した。
投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を、サンドウィッチELISA(ENDOGEN)により測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図1に示す。
投与後3、6、12時間経過後の血清中のIL-4の含有量を、ELISA法の1つであるCytometric bead arrayシステム(BD Biosciences)で測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図2に示す。
投与後3、6、12時間経過後の血清中のIL-12の含有量を、ELISA法の1つであるCytometric bead arrayシステム(BD Biosciences)で測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図3に示す。
上記結果より、化合物2-9、化合物3-4、化合物3-7及び化合物3-10のいずれもがα-GalCer及びカルバ糖脂質Aよりも大量のIFN-γの産生を誘導した。特に、化合物2-9が最も強力に産生を誘導した。また、これら何れの化合物もα-GalCerと同程度~3倍量のIL-12の産生を誘導した。一方、IL-4の産生量には大きな差がなかった。このことから化合物2-9、化合物3-4、化合物3-7及び化合物3-10は、α-GalCer及びカルバ糖脂質Aに比べて多量のIFN-γを選択的に産生誘導することが示された。
試験例2 化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の生物活性試験
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図4に示す。
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後3、6時間経過後の血清中のIL-4の含有量を測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図6に示す。
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後3、6時間経過後の血清中のIL-12(p70)の含有量を測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図8に示す。
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図4に示す。
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後3、6時間経過後の血清中のIL-4の含有量を測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図6に示す。
試験例1と同様の方法により、α-GalCer、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16の投与後3、6時間経過後の血清中のIL-12(p70)の含有量を測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図8に示す。
上記結果より、化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16のいずれもがα-GalCerよりも大量のIFN-γの産生を誘導した。特に、化合物2-9が最も強力に産生を誘導した。また、これら何れの化合物もα-GalCerと同程度~2倍量のIL-12の産生を誘導した。一方、IL-4の産生量には大きな差がなかった。このことから化合物2-9、化合物3-4、化合物3-7、化合物3-10、化合物3-13及び化合物3-16のは、α-GalCerに比べて多量のIFN-γを選択的に産生誘導することが示された。
試験例3 化合物2-9の生物活性試験
α-GalCerの投与量が2μg/mouse、化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouse、0.002μg/mouse、0.0002μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図5に示す。
α-GalCerの投与量及び化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後3、6時間経過後の血清中のIL-4の含有量を測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図7に示す。
α-GalCerの投与量及び化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後3、6時間経過後の血清中のIL-12(p70)の含有量を測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図9に示す。
α-GalCerの投与量が2μg/mouse、化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouse、0.002μg/mouse、0.0002μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後6、12、24、36、48、60時間経過後の血清中のIFN-γの含有量を測定した。IFN-γ産生量の測定結果(平均値)及びその標準偏差(STDEV)を図5に示す。
α-GalCerの投与量及び化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後3、6時間経過後の血清中のIL-4の含有量を測定した。IL-4産生量の測定結果(平均値)及びその標準偏差(STDEV)を図7に示す。
α-GalCerの投与量及び化合物2-9の投与量が2μg/mouse、0.2μg/mouse、0.02μg/mouseとなるようにした以外は試験例1と同様の方法により、α-GalCer及び化合物2-9の投与後3、6時間経過後の血清中のIL-12(p70)の含有量を測定した。IL-12産生量の測定結果(平均値)及びその標準偏差(STDEV)を図9に示す。
上記結果より、化合物2-9は低濃度での投与においても、α-GalCerよりも大量のIFN-γの産生を誘導した。また化合物2-9は、α-GalCerと同程度~4倍量のIL-12の産生を誘導した。一方IL-4の産生量は、化合物2-9では投与量を少なくすると、それに伴い減少した。このことから化合物2-9は低濃度での投与においても、α-GalCerに比べて多量のIFN-γを選択的に産生誘導することが示された。
本出願は、日本で出願された特願2008-079265を基礎としており、その内容は本明細書に全て包含されるものである。
Claims (16)
- 下記一般式(1)で表される化合物又はその塩。

[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - R1が水素原子、メチル基、メトキシ基、エトキシ基、n-プロピルオキシ基又はフッ素原子である、請求項1記載の化合物又はその塩。
- R1が炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子であり、R2が炭素数1~28の置換又は非置換のアルキル基であるか、又は、R1が水素原子であり、R2が炭素数24~28の置換又は非置換のアルキル基である、請求項1又は2に記載の化合物又はその塩。
- R3が炭素数1~28の置換又は非置換のアルキル基である、請求項1~3のいずれか一項に記載の化合物又はその塩。
- Yが-CH(OH)-である、請求項1~4のいずれか一項に記載の化合物又はその塩。
- 下記一般式(2)で表される化合物又はその塩。
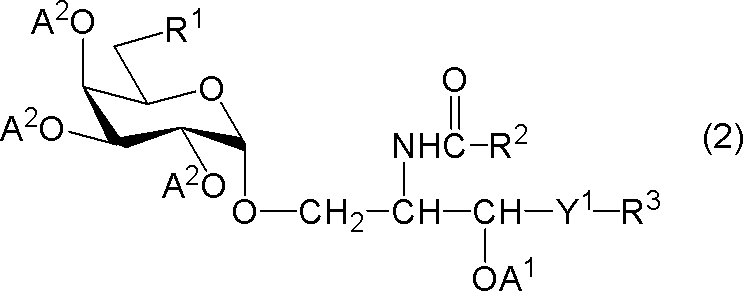
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Y1は-CH2-、-CH(OA1)-又は-CH=CH-を示し、A1は水素原子又は水酸基の保護基を示し、A2は水酸基の保護基を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩を含有する、医薬。
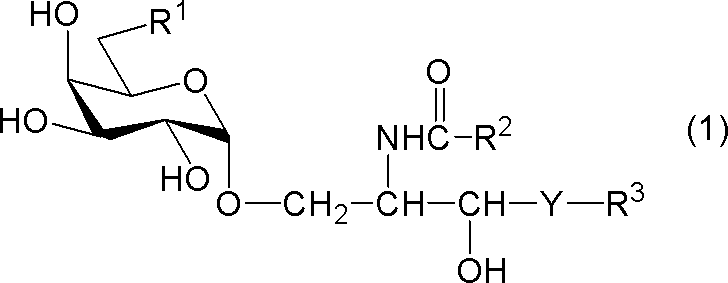
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩を含有する、免疫賦活剤。
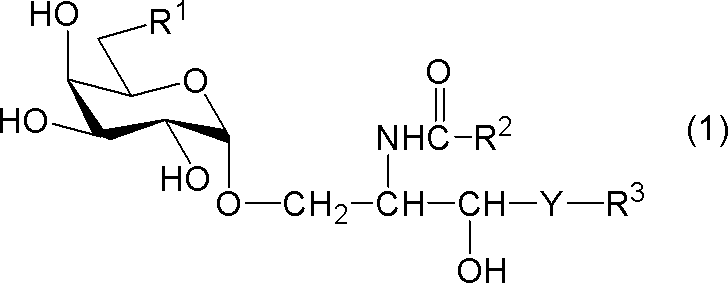
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩を含有する、選択的IFN-γ産生誘導剤。

[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩を含有する、抗癌剤。

[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩の有効量を対象に投与することを含む、免疫賦活方法。
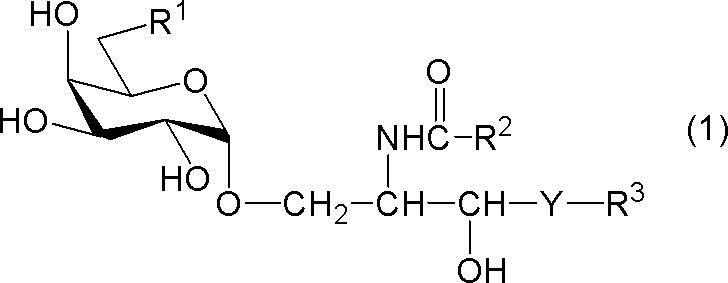
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩の有効量を対象に投与することを含む、選択的IFN-γ産生誘導方法。
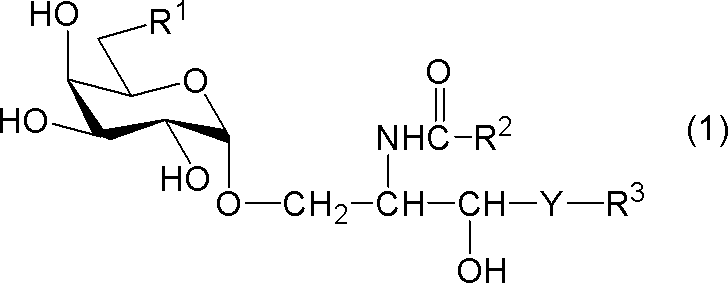
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 下記一般式(1)で表される化合物又はその塩の有効量を対象に投与することを含む、癌の治療方法。
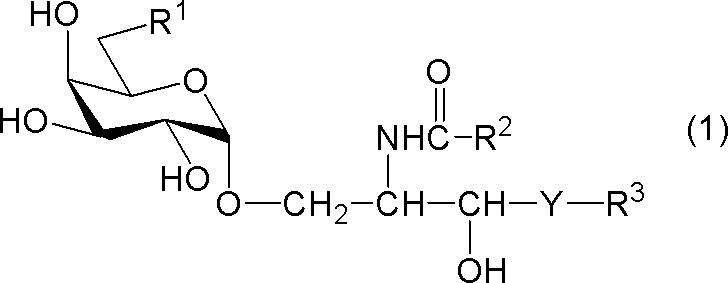
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 免疫賦活剤の製造のための、下記一般式(1)で表される化合物又はその塩の使用。
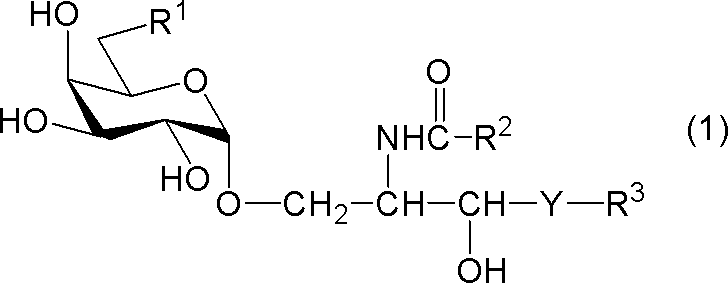
[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 選択的IFN-γ産生誘導剤の製造のための、下記一般式(1)で表される化合物又はその塩の使用。

[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。] - 抗癌剤の製造のための、下記一般式(1)で表される化合物又はその塩の使用。

[式中、R1は、水素原子、炭素数1~7のアルキル基、炭素数1~6のアルコキシ基又はハロゲン原子を示し、R2及びR3はそれぞれ独立に炭素数1~28の置換又は非置換の炭化水素基を示し、Yは-CH2-、-CH(OH)-又は-CH=CH-を示す。但し、R1が水素原子であるときは、R2は炭素数24~28の置換又は非置換の炭化水素基を示す。]
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010505744A JP5561700B2 (ja) | 2008-03-25 | 2009-03-25 | 新規糖脂質及びその用途 |
| US12/934,515 US8551959B2 (en) | 2008-03-25 | 2009-03-25 | Glycolipid and use thereof |
| EP09725339.7A EP2272854B1 (en) | 2008-03-25 | 2009-03-25 | Novel glycolipid and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008079265 | 2008-03-25 | ||
| JP2008-079265 | 2008-03-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009119692A1 true WO2009119692A1 (ja) | 2009-10-01 |
Family
ID=41113882
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/056010 Ceased WO2009119692A1 (ja) | 2008-03-25 | 2009-03-25 | 新規糖脂質及びその用途 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8551959B2 (ja) |
| EP (1) | EP2272854B1 (ja) |
| JP (1) | JP5561700B2 (ja) |
| WO (1) | WO2009119692A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011096536A1 (ja) * | 2010-02-05 | 2011-08-11 | 独立行政法人理化学研究所 | 新規合成糖脂質およびその用途 |
| JP2016529321A (ja) * | 2013-09-06 | 2016-09-23 | アカデミア シニカAcademia Sinica | 改変されたグリコシル基を含む糖脂質を用いたヒトiNKT細胞の活性化 |
| WO2022102557A1 (ja) * | 2020-11-12 | 2022-05-19 | 国立研究開発法人理化学研究所 | 擬似糖脂質誘導体並びにその合成中間体、製造方法及び用途 |
| WO2023153527A1 (ja) | 2022-02-14 | 2023-08-17 | 国立研究開発法人理化学研究所 | Nkt細胞リガンド含有リポソーム組成物 |
| US11744860B2 (en) | 2016-04-28 | 2023-09-05 | Riken | Technology for efficient activation of NKT cells |
| WO2025037627A1 (ja) | 2023-08-15 | 2025-02-20 | 国立研究開発法人理化学研究所 | Nkt細胞リガンド含有リポソーム組成物を含む抗がん剤 |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7960139B2 (en) | 2007-03-23 | 2011-06-14 | Academia Sinica | Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells |
| ES2442024T3 (es) | 2008-07-15 | 2014-02-07 | Academia Sinica | Matrices de glucano sobre portaobjetos de vidrio revestidos con aluminio de tipo PTFE y métodos relacionados |
| US10087236B2 (en) | 2009-12-02 | 2018-10-02 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11377485B2 (en) | 2009-12-02 | 2022-07-05 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US10338069B2 (en) | 2010-04-12 | 2019-07-02 | Academia Sinica | Glycan arrays for high throughput screening of viruses |
| US10130714B2 (en) | 2012-04-14 | 2018-11-20 | Academia Sinica | Enhanced anti-influenza agents conjugated with anti-inflammatory activity |
| WO2014031498A1 (en) | 2012-08-18 | 2014-02-27 | Academia Sinica | Cell-permeable probes for identification and imaging of sialidases |
| EP3013365B1 (en) | 2013-06-26 | 2019-06-05 | Academia Sinica | Rm2 antigens and use thereof |
| EP3013347B1 (en) | 2013-06-27 | 2019-12-11 | Academia Sinica | Glycan conjugates and use thereof |
| AU2015206370A1 (en) | 2014-01-16 | 2016-07-07 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10150818B2 (en) | 2014-01-16 | 2018-12-11 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| CN106415244B (zh) | 2014-03-27 | 2020-04-24 | 中央研究院 | 反应性标记化合物及其用途 |
| CN106661099A (zh) | 2014-05-27 | 2017-05-10 | 中央研究院 | 抗her2醣抗体及其用途 |
| US10118969B2 (en) | 2014-05-27 | 2018-11-06 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| CA2950415A1 (en) | 2014-05-27 | 2015-12-03 | Academia Sinica | Anti-cd20 glycoantibodies and uses thereof |
| JP7093612B2 (ja) | 2014-05-27 | 2022-06-30 | アカデミア シニカ | Bacteroides由来のフコシダーゼおよびそれを使用する方法 |
| WO2015184001A1 (en) | 2014-05-28 | 2015-12-03 | Academia Sinica | Anti-tnf-alpha glycoantibodies and uses thereof |
| TWI745275B (zh) | 2014-09-08 | 2021-11-11 | 中央研究院 | 使用醣脂激活人類iNKT細胞 |
| US10495645B2 (en) | 2015-01-16 | 2019-12-03 | Academia Sinica | Cancer markers and methods of use thereof |
| US9975965B2 (en) | 2015-01-16 | 2018-05-22 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| JP6779887B2 (ja) | 2015-01-24 | 2020-11-04 | アカデミア シニカAcademia Sinica | 新規なグリカンコンジュゲートおよびその使用方法 |
| EP3426693A4 (en) | 2016-03-08 | 2019-11-13 | Academia Sinica | PROCESS FOR MODULAR SYNTHESIS OF N-GLYCANES AND ARRANGEMENTS THEREOF |
| CA3034057A1 (en) | 2016-08-22 | 2018-03-01 | CHO Pharma Inc. | Antibodies, binding fragments, and methods of use |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994009020A1 (fr) | 1992-10-22 | 1994-04-28 | Kirin Beer Kabushiki Kaisha | Nouveau shingoglycolipide et son utilisation |
| WO1998044928A1 (fr) * | 1997-04-10 | 1998-10-15 | Kirin Beer Kabushiki Kaisha | ACTIVATEURS DE CELLULES NKT, CONTENANT DES α-GLYCOSYLCERAMIDES |
| WO1999015627A1 (en) * | 1997-09-22 | 1999-04-01 | Kirin Beer Kabushiki Kaisha | CELLULAR IMMUNOGENICITY POTENTIATING COMPOSITION CONTAINING α-GLYCOSYLCERAMIDE |
| DE10128250A1 (de) | 2000-06-12 | 2001-12-20 | Kotobuki Pharmaceutical Co | Neue Glykolipidderivate |
| WO2003105769A2 (en) | 2002-06-13 | 2003-12-24 | New York University | Synthetic c-glycolipid and its use for treating cancer infectious diseases and autoimmune diseases |
| US20050222048A1 (en) | 2004-03-31 | 2005-10-06 | The Research Foundation Of The City University Of New York | Novel synthetic C-glycolipids, their synthesis and use to treat infections, cancer and autoimmune diseases |
| US20070238673A1 (en) | 2004-08-27 | 2007-10-11 | Porcelli Steven A | Ceramide derivatives as modulators of immunity and autoimmunity |
| US20070238871A1 (en) * | 2004-12-28 | 2007-10-11 | The Rockefeller University | Glycolipids And Analogues Thereof As Antigens For NKT Cells |
-
2009
- 2009-03-25 WO PCT/JP2009/056010 patent/WO2009119692A1/ja not_active Ceased
- 2009-03-25 EP EP09725339.7A patent/EP2272854B1/en active Active
- 2009-03-25 JP JP2010505744A patent/JP5561700B2/ja active Active
- 2009-03-25 US US12/934,515 patent/US8551959B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994009020A1 (fr) | 1992-10-22 | 1994-04-28 | Kirin Beer Kabushiki Kaisha | Nouveau shingoglycolipide et son utilisation |
| WO1998044928A1 (fr) * | 1997-04-10 | 1998-10-15 | Kirin Beer Kabushiki Kaisha | ACTIVATEURS DE CELLULES NKT, CONTENANT DES α-GLYCOSYLCERAMIDES |
| WO1999015627A1 (en) * | 1997-09-22 | 1999-04-01 | Kirin Beer Kabushiki Kaisha | CELLULAR IMMUNOGENICITY POTENTIATING COMPOSITION CONTAINING α-GLYCOSYLCERAMIDE |
| DE10128250A1 (de) | 2000-06-12 | 2001-12-20 | Kotobuki Pharmaceutical Co | Neue Glykolipidderivate |
| WO2003105769A2 (en) | 2002-06-13 | 2003-12-24 | New York University | Synthetic c-glycolipid and its use for treating cancer infectious diseases and autoimmune diseases |
| US20050222048A1 (en) | 2004-03-31 | 2005-10-06 | The Research Foundation Of The City University Of New York | Novel synthetic C-glycolipids, their synthesis and use to treat infections, cancer and autoimmune diseases |
| US20070238673A1 (en) | 2004-08-27 | 2007-10-11 | Porcelli Steven A | Ceramide derivatives as modulators of immunity and autoimmunity |
| US20070238871A1 (en) * | 2004-12-28 | 2007-10-11 | The Rockefeller University | Glycolipids And Analogues Thereof As Antigens For NKT Cells |
Non-Patent Citations (25)
| Title |
|---|
| ANGEW. CHEM. INT. ED. ENGL., vol. 43, 2004, pages 3818 - 3822 |
| BIOCHIM. BIOPHYS. ACTA, 1973, pages 315 - 335 |
| BIOL. PHARM. BULL., vol. 18, 1995, pages 1487 - 1491 |
| BIOORG. MED. CHEM., vol. 6, 1998, pages 1905 - 1910 |
| FAN, GANG-TING ET AL.: "Synthesis of alpha-galactosyl ceramide and the related glycolipids for evaluation of their activities on mouse splenocytes", TETRAHEDRON, vol. 61, 2005, pages 1855 - 1862, XP025383002 * |
| H. TAKIKAWA ET AL., TETRAHEDRON, vol. 54, 1998, pages 3141 - 3150 |
| IIJIMA, HIROSHI ET AL.: "Structure-Activity Relationship and Conformational Analysis of Monoglycosylceramides on the Syngeneic Mixed Leukocyte Reaction", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 6, 1998, pages 1905 - 1910, XP027393083 * |
| J. EXP. MED., vol. 198, 2003, pages 1631 - 1641 |
| J. IMMUNOL., vol. 155, 1995, pages 2972 - 2983 |
| J. IMMUNOL., vol. 161, 1998, pages 3271 - 3281 |
| J. MED. CHEM., vol. 38, 1995, pages 2176 - 2187 |
| K. TATSUTA ET AL., CARBOHYDR. RES., vol. 222, 1991, pages 189 - 203 |
| KIMIKO ANNO ET AL.: "Kabushiki Kaisha Kodansha", TOKAGAKU NO KISO, 11TH PRINT, 10 August 1995 (1995-08-10), pages 54 - 57, XP008145941 * |
| MOTOKI, KAZUHIRO ET AL.: "Immunostimulatory and Antitumor Activities of Monoglycosylceramides Having Various Sugar Moieties", BIOL. PHARM. BULL., vol. 18, 1995, pages 1487 - 1491, XP008143368 * |
| NAT. IMMUNOL., vol. 4, 2003, pages 1164 - 1165 |
| PROC. NATL. ACAD. SCI. USA, vol. 95, 1998, pages 5690 - 5693 |
| S. KIM ET AL., SYNTHESIS, 2004, pages 847 - 850 |
| S. KOTO ET AL., BULL. CHEM. SOC. JPN., vol. 73, 2000, pages 967 - 976 |
| SCIENCE, vol. 278, 1997, pages 1626 - 1629 |
| See also references of EP2272854A4 * |
| T. J. LUCAS ET AL., CARBOHYDR. RES., vol. 39, 1975, pages 39 - 45 |
| T.J. LUCAS ET AL., CARBOHYDR. RES., vol. 39, 1975, pages 39 - 45 |
| TASHIRO, TAKUYA ET AL.: "RCAI-56, a carbocyclic analogue of KRN7000: its synthesis and potent activity for natural killer (NK) T cells to preferentially produce interferon-gamma", TETRAHEDRON LETTERS, vol. 48, 2007, pages 3343 - 3347, XP022031952 * |
| TETRAHEDRON LETT., vol. 48, 2007, pages 3343 - 3347 |
| TETRAHEDRON, vol. 61, 2005, pages 1855 - 1862 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011096536A1 (ja) * | 2010-02-05 | 2011-08-11 | 独立行政法人理化学研究所 | 新規合成糖脂質およびその用途 |
| US8853173B2 (en) | 2010-02-05 | 2014-10-07 | Riken | Synthetic glycolipid and use thereof |
| JP5669215B2 (ja) * | 2010-02-05 | 2015-02-12 | 独立行政法人理化学研究所 | 新規合成糖脂質およびその用途 |
| JP2016529321A (ja) * | 2013-09-06 | 2016-09-23 | アカデミア シニカAcademia Sinica | 改変されたグリコシル基を含む糖脂質を用いたヒトiNKT細胞の活性化 |
| US11744860B2 (en) | 2016-04-28 | 2023-09-05 | Riken | Technology for efficient activation of NKT cells |
| WO2022102557A1 (ja) * | 2020-11-12 | 2022-05-19 | 国立研究開発法人理化学研究所 | 擬似糖脂質誘導体並びにその合成中間体、製造方法及び用途 |
| JPWO2022102557A1 (ja) * | 2020-11-12 | 2022-05-19 | ||
| JP7714237B2 (ja) | 2020-11-12 | 2025-07-29 | 国立研究開発法人理化学研究所 | 擬似糖脂質誘導体並びにその合成中間体、製造方法及び用途 |
| WO2023153527A1 (ja) | 2022-02-14 | 2023-08-17 | 国立研究開発法人理化学研究所 | Nkt細胞リガンド含有リポソーム組成物 |
| WO2025037627A1 (ja) | 2023-08-15 | 2025-02-20 | 国立研究開発法人理化学研究所 | Nkt細胞リガンド含有リポソーム組成物を含む抗がん剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2272854B1 (en) | 2015-08-05 |
| EP2272854A1 (en) | 2011-01-12 |
| US8551959B2 (en) | 2013-10-08 |
| JPWO2009119692A1 (ja) | 2011-07-28 |
| JP5561700B2 (ja) | 2014-07-30 |
| US20110104188A1 (en) | 2011-05-05 |
| EP2272854A4 (en) | 2014-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5561700B2 (ja) | 新規糖脂質及びその用途 | |
| JP3717512B2 (ja) | 新規スフィンゴ糖脂質およびその使用 | |
| JP6192120B2 (ja) | 新規カルバメート糖脂質およびその用途 | |
| CA2854725A1 (en) | Methods for preparation of glycosphingolipids and uses thereof | |
| CA2880191C (en) | Sphingoglycolipid analogues as therapeutic agents | |
| JP5669215B2 (ja) | 新規合成糖脂質およびその用途 | |
| JP5574432B2 (ja) | エステル化α−ガラクトシルセラミド類 | |
| JP5126684B2 (ja) | 新規糖脂質及びその用途 | |
| JP5504891B2 (ja) | 新規擬似糖脂質及びその用途 | |
| US20090215710A1 (en) | Carbohydrate based toll-like receptor (tlr) antagonists | |
| JPWO2004026318A1 (ja) | α−グリコシルセラミドを有効成分とするC型肝炎ウイルス抑制剤 | |
| Pauwels et al. | Divergent synthetic approach to 6′′-modified α-GalCer analogues | |
| HK1207648B (en) | New carbamate glycolipid and use thereof | |
| AU6748598A (en) | Nkt cell activators containing alpha-glycosylceramides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09725339 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010505744 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009725339 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12934515 Country of ref document: US |