WO2009120655A1 - Indole derivatives - Google Patents
Indole derivatives Download PDFInfo
- Publication number
- WO2009120655A1 WO2009120655A1 PCT/US2009/038022 US2009038022W WO2009120655A1 WO 2009120655 A1 WO2009120655 A1 WO 2009120655A1 US 2009038022 W US2009038022 W US 2009038022W WO 2009120655 A1 WO2009120655 A1 WO 2009120655A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- indol
- methyl
- azetidin
- pyridin
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 COC1(C*C1)C1C2=C**C(*)=C2**1 Chemical compound COC1(C*C1)C1C2=C**C(*)=C2**1 0.000 description 4
- ZIWIZSMFIDQUKT-UHFFFAOYSA-N CC(C)N(C1)CC1c1c[n](C)c2c1ccc(N(C=CC(c(cc1)ncc1Cl)=C1)C1=O)c2 Chemical compound CC(C)N(C1)CC1c1c[n](C)c2c1ccc(N(C=CC(c(cc1)ncc1Cl)=C1)C1=O)c2 ZIWIZSMFIDQUKT-UHFFFAOYSA-N 0.000 description 1
- KNUNCONSRJCMTQ-UHFFFAOYSA-O CN(C1)CC1c1c[nH]c2c1ccc([NH+](CCC(CSc(cc1)ccc1F)=C1)C1=O)c2 Chemical compound CN(C1)CC1c1c[nH]c2c1ccc([NH+](CCC(CSc(cc1)ccc1F)=C1)C1=O)c2 KNUNCONSRJCMTQ-UHFFFAOYSA-O 0.000 description 1
- OXGFSMPNOGNFBU-UHFFFAOYSA-N C[n]1c(cc(cc2)N(C=CC(c(nc3)ccc3Cl)=C3)C3=O)c2c(C2CN(CC3CC3)C2)c1 Chemical compound C[n]1c(cc(cc2)N(C=CC(c(nc3)ccc3Cl)=C3)C3=O)c2c(C2CN(CC3CC3)C2)c1 OXGFSMPNOGNFBU-UHFFFAOYSA-N 0.000 description 1
- WDEXSZIHJSHWQK-UHFFFAOYSA-N O=C(CC(F)(F)F)N(C1)CC1c1c[nH]c2cc(N(C=CC(c(cc3)ccc3Cl)=C3)C3=O)ccc12 Chemical compound O=C(CC(F)(F)F)N(C1)CC1c1c[nH]c2cc(N(C=CC(c(cc3)ccc3Cl)=C3)C3=O)ccc12 WDEXSZIHJSHWQK-UHFFFAOYSA-N 0.000 description 1
- AXFABVAPHSWFMD-UHFFFAOYSA-N Oc1ncnc(Cl)c1 Chemical compound Oc1ncnc(Cl)c1 AXFABVAPHSWFMD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention is directed to indole derivatives which bind to the MCHl receptor.
- the subject invention relates to uses of said compounds in the preparation of a pharmaceutical composition for the treatment of obesity and CNS related disorders and to methods of treating said disorders comprising administering a therapeutically effective amount of a compound of the invention.
- MCH Melanin-concentrating hormone
- MCH exerts several physiological effects through interaction with its receptors. For example, an icv injection of MCH in rats stimulates food intake (Levens, et al. Int. J. Obesity 2002, 26, 1289-1295), and chronic administration leads to increased body weight (Kanatani, et al. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E583-E588).
- the link between MCHl-R and the effects of MCH on feeding is demonstrated by reports on the phenotype of MCHl-R knockout mice. Independent groups generated knock-out mice with the targeted deletion of MCHl-R.
- mice The phenotype of these mice was lean, hyperphagic and hypermetabolic, with an increased resistance to diet-induced obesity (Marsh, et al. Proc. Natl. Acad. ScL 2002, 99, 3240-3245). These observations suggest that MCHl-R antagonists could be useful for the treatment of obesity related disorders.
- MCHl -R binding sites in the CNS such as the amygdala, accumbens nucleus, dorsal raphe and locus cocruleus is suggestive of a role for MCH in the regulation of mood and stress
- several groups have identified selective, high affinity MCHl-R antagonists and evaluated their effects in in-vivo behavioral paradigms predictive of antidepressant and/or anxiolytic activity.
- mice or rats spend immobile in the forced-swim test decrease the amount of time mice or rats spend immobile in the forced-swim test (Porsolt, et al. Arch bit Phannacodyn Ther. 1977, 229, 327- 336 and Luki, ct al. Psychopharmacology 2001 , 155, 315-322).
- Pretreatmenl of rats with a single oral dose of SNAP-7941, a selective MCHl-R antagonist, or fluoxetine decreased the duration of immobility compared with vehicle-treated controls, and increased the time these animals spent swimming (Borowsky, et al. Nature Medicine 2002, 8, 825-830).
- the profile of SNAP-7941 in the rat forced-swim test is similar to that of clinically used antidepressants, indicating that MCH l-R receptor blockage may be a therapeutic modality for the treatment of mood-disorders such as depression.
- the rat social interaction test has been used as a model of anxiety (File and Hyde Br. J. Pharmacol. 1987, 62, 19-24).
- Acute treatment with 3, 10 and 30 mg/kg SNAP-7941 or 5 mg/kg chlordiazepoxide increased social interaction time compared with vehicle- treated controls (Borowsky, et al. Nature Medicine 2002, 8, 825-830) without an overall increase in locomotor activity.
- the response to the two lower doses of SNAP-7941 was as robust as the response to 5 mg/kg chlordiazepoxide.
- the profile of this potent MCHl-R antagonist in the rat social interaction test suggests that MCHl-R antagonists may have potential as anxiolytic agents.
- the compounds of the subject invention can be used to treat obesity, mood and anxiety related disorders as well as the additional indications which are disclosed herein in the detailed description section.
- the objective of the subject invention is to provide compounds which are ligands at the MCHl receptor. Accordingly, the present invention relates to compounds of Formula Ia and Ib:
- each B 1 , B 2 and B 3 is independently CH or N;
- R 1 is -(CHR 7 ) m N(R 8 )(R 9 ) or -(CHR 7 J n R 10 , and where the R 1 containing moiety of formula Ib is connected to one A but not when A is N;
- R 2 is H or straight chained or branched C 1 -C 7 alkyl optionally substituted with one or more fluorine and where the carbons of the Ci-C 7 alkyl are optionally replaced with one to three N, O or S atoms;
- R ⁇ is -X(CH 2 ) p YR l ' or X(CHCH)YR 1 ', and where B 1 of the R 3 containing moiety of formula Ia is connected to one A but not when A is N;
- each X and Y is independently CH 2 , O, S, NH or a bond, provided that an O, S or NH is separated from another O, S or NH by at least two carbon atoms;
- each R 4 , R ⁇ and R 6 is independently H, halogen or straight chained or branched Ci-C 6 alkyl;
- each R 7 is independently H, OH, or wherein one R 7 can combine with another R 7 on an adjacent carbon atom to form C 3 -C f1 cycloalkyl and wherein one R 7 can combine with a H on the shared carbon atom to form CrQs cycloalkyl; wherein each R 8 and R 9 is independently H or straight chained or branched Ci-C 7 alkyl;
- R 10 is a nitrogen containing heterocyclic moiety optionally substituted with one or
- halogen -COR ", a S or O containing heterocyclic ring, or straight chained or branched CpC 7 alkyl optionally substituted with halogen or -OCH- ? ;
- R 1 1 is CrC 6 cycloalkyl, straight chained or branched alkoxy-Cs-Q, cycloaikyl, straight chained or branched Ci-C 4 alkyl/alkoxy, phenyl, napthyl, 5 to 6-membered heteroaryl, benzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C]-C 4 alkyl optionally substituted with F;
- n is an integer from I to 4 inclusive;
- n is an integer from 0 to 4 inclusive
- p is independently an integer from 0 to 3 inclusive, or a pharmaceutically acceptable salt thereof.
- the compound is selected from one of the exemplified compounds which are disclosed in the Experimental Section.
- the subject invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula Ia or Ib, or a pharmaceutically acceptable salt and a pharmaceutically acceptable carrier.
- the present invention provides a method of treating a subject suffering from mood disorders, anxiety or obesity comprising administering to the subject a therapeutically effective amount of a compound of Formula Ia or Ib.
- the present invention further provides uses of a compound of Formula Ia or Ib in the manufacture of a pharmaceutical composition for the treatment of mood disorders, anxiety or obesity.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disorder, stabilized (i.e., not worsening) state of disorder, delay or slowing of disorder progression, amelioration or palliation of the disorder stale, and remission (whether partial or total), whether detectable or undetectable.
- terapéuticaally effective amount is an amount sufficient to effect beneficial or desired clinical or biochemical results.
- a “therapeutically effective amount” can be administered one or more times.
- a therapeutically effective amount” of a compound is an amount that is sufficient to palliate, ameliorate, stabilize, reverse, slow or delay the progression of the disorder state.
- the term "antagonist” refers to a compound which binds to, and decreases the activity of, a receptor in the presence of an agonist.
- activation may be measured using any appropriate second messenger system which is coupled to the receptor in a cell or tissue in which the receptor is expressed.
- second messenger systems are adenylate cyclase, intracellular calcium mobilization, ion channel activation, guanylate cyclase and inositol phospholipid hydrolysis.
- agonist refers to a compound which binds to, and increases activity of, a receptor as compared with the activity of the receptor in the absence of any agonist.
- the term "straight chained or branched Ci-C 7 alkyl” refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive.
- substiluents include, but are not limited to, methyl, ethyl, 1 -propyl, 2-propyl, 1 -butyl, 2 -butyl, 2-melhyl-2-propyl, 2-mcthyl-l -propyl and n-heptyl.
- the term “straight chained or branched C]-C 4 alkyl” refers to a saturated hydrocarbon having from one to four carbon atoms inclusive.
- C ⁇ -C 6 cycloalkyl refers to the group consisting of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- a nitrogen containing heterocyclic moiety refers Io the following groups:
- a S or O containing heterocyclic moiety refers to the following groups:
- R % containing moiety of formula Ia refers to the following moiety:
- heteroaryl is used to include five and six membered unsaturated rings that contain one or more oxygen, sulfur, or nitrogen atoms.
- heteroaryl groups include, but are not limited to, furanyl, thienyl, pyrrolyl, oxazolyl, thiazoiyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, and tria/inyl.
- halogen refers to F, Cl, Br of I.
- the compounds of the invention may have an advantage over other compounds in the art in that they possess reduced p-glycoproiein (pGP) activity and accordingly, the compounds may have unexpectedly enhanced brain-blood penetration. Additionally, the present invention further provides certain embodiments which are immediately described below.
- pGP p-glycoproiein
- each A is independently CR 6 or N provided that when one A is N, the other A is CR 6 ;
- each B 1 , B 2 and B 3 is independently CH or N;
- R 1 is -(CHR 7 ), n N(R 8 )(R 9 ) or -(CHR 7 ) n R 10 , and where the R 1 moiety of formula Ib is connected to one A but not when A is N;
- R 2 is H or straight chained or branched Ci-C 7 alkyi optionally substituted with one or more fluorines and where the carbons of the C 1 -C 7 alkyl arc optionally replaced with one to three N, O or S atoms;
- R 1 is -X(CH 2 ) P YR' ' or X(CHCH)YR 1 1 , and where B 1 of the R 1 containing moiety of formula
- Ia is connected to one A but not when A is N;
- each X and Y is independently CH 2 , O, S, NH or a bond, provided that an O, S or NH is separated from another O, S or NH by at least two carbon atoms;
- each R 4 , R 5 and R 6 is independently H, halogen or straight chained or branched CpC 7 alkyl
- each R 7 is independently H, OH, or wherein one R 7 can combine with another R 7 on an adjacent carbon atom to form C 3 -C U cycloalkyl and wherein one R 7 can combine with a H on the shared carbon atom to form C 3 -C 6 cycloalkyl;
- each R and R is independently H or straight chained or branched Cj -C 7 alkyl
- R 1( is a nitrogen containing heterocyclic moiety optionally substituted with one or more halogen or straight chained or branched Cj-C 7 alkyl;
- R 11 is C 3 -Q, cycloalkyl, phenyl or a 5 to 6-membered heteroaryl, where the phenyl or 5 to 6- membered heteroaryl can be optionally substituted with F, Cl, Br, or straight chained or branched Cj-C 4 alkyl optionally substituted with F;
- n is an integer from 1 to 4 inclusive
- n is an integer from 0 to 4 inclusive; and p is independently an integer from 0 to 3 inclusive, or a pharmaceutically acceptable salt thereof.
- the compound has the following structure:
- B 1 is N and B 2 and B 3 are CH;
- R 1 is -(CHR 7 ) m N(R 8 )(R 9 ) or -(CHR 7 ) n R 10 ;
- R 2 is H or straight chained or branched C 1 -C 7 alkyl optionally substituted with one or more fluorine or -OH and where the carbons of the Ci-C 7 alkyl are optionally replaced with one to three N, O or S atoms;
- each R and R " is independently H, halogen or straight chained or branched Ci-C 7 alkyl
- each R 7 is independently H, OH, or wherein one R 7 can combine with another R 7 on an adjacent carbon atom to form CrQ cycloalkyl and wherein one R 7 can combine with a H on the shared carbon atom to form C 3 -C 6 cycloalkyl;
- each R 8 and R 9 is independently H or straight chained or branched Ci-C 7 alkyl optionally substituted with one or more fluorine; wherein R J is a nitrogen containing heterocyclic moiety optionally substituted with one or more halogen, -COR 12 , a S or O containing heterocyclic ring, or .straight chained or branched Ci -C 7 alkyl optionally substituted with halogen or -OCH- ? ;
- R 1 1 is C 3 -C 6 cycloalkyl, straight chained or branched alkoxy-CrC ⁇ cycloalkyl, straight chained or branched C 1 -C 4 aikyl/alkoxy, phenyl, napthyl, 5 to 6-membcred heteroaryl, benzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC 4 alkyl optionally substituted with F;
- R " is straight chained or branched C]-C 7 alkyl or Ci-C 6 cycloalkyl, where each of which is optionally substituted with one or more halogen or methoxy;
- n is an integer from 0 to 4 inclusive, or a pharmaceutically acceptable salt thereof.
- R 1 is-(CHR 7 ) m N(R 8 )(R 9 ).
- R 1 is-(CHR 7 ) n R 10 .
- each R 4 and R 5 is independently H or straight chained or branched C 1 -C 4 alkyl.
- R 10 is piperazinyl or morpholinyl, where each of which is optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C 4 alkyl optionally substituted with halogen.
- R 10 is piperidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C 4 alkyl optionally substituted with halogen.
- R 10 is pyrrolidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C 4 alkyl optionally substituted with halogen. In one embodiment, R 10 is azetidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C 4 alkyl optionally substituted with halogen.
- R 10 is piperazinyl or morpholinyl, where each of which is optionally substituted with straight chained or branched C 1 -C 4 alkyl optionally substituted with halogen.
- R 10 is piperidinyl optionally straight chained or branched C 1 -C 4 alkyl optionally substituted with fluorine.
- R K) is pyrrolidinyl optionally straight chained or branched C 1 -C 4 alky] optionally substituted with fluorine,
- R 10 is azetidinyl optionally straight chained or branched Ci-C 4 alkyl optionally substituted with fluorine.
- R 7 is H; and R 4 and R 5 is independently H, or straight chained or branched C 1 -C 4 alkyl.
- n is 0 and R 2 is H, methyl or ethyl.
- n 1 or 2.
- R 1 ' is C 3 -C 6 cycloalkyl or alkoxy-C 3 -Cc, cycloalkyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C 1 - C 4 alkyl optionally substituted with F.
- R 1 ' is phenyl or napthyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C 1 -C 4 alkyl optionally substituted with F.
- R ⁇ is a 5 to 6-membered heteroaryl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC 4 alkyl optionally substituted with F.
- R.” is bcnzothiophcnyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C 1 -C 4 alkyl optionally substituted with F.
- R s ' is phenyl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Cj-C 4 alkyl optionally substituted with F.
- R 1 1 is napthy! optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Ci-C 4 alkyl optionally substituted with F.
- the compound falls under formula Ia.
- the compound falls under formula Ib.
- B 1 is CH. In a separate embodiment, B 1 is N.
- B " is CH. In a separate embodiment, B" is N,
- B 1 is CH. In a separate embodiment, B 3 is N.
- B 1 is CH and B 3 is CH. In a separate embodiment, B 1 is CH, B 1 is N and
- B 3 is CH.
- each R 4 , R 5 and R 6 is independently H, halogen or straight chained or branched Ci-C 4 alkyl; and wherein each R 8 and R 9 is independently H or straight chained or branched Cj-C 4 alkyl.
- R 10 is azetidinyl, cyclopropyl, cyclobutyl, cyclopenyl or cyciohcxyl, wherein the azetidinyl, cyclopropyl, cyclobutyl, cyclopenyl or cyclohexyl is optionally substituted with with one or more halogen or straight chained or branched Ci-C 4 alkyl.
- R 3 is -X(CH 2 ) P YR' '
- R 3 is -X(CHCH)YR 1 '.
- R 2 is H or straight chained or branched Ci-C 4 alkyl.
- X is CH 2 or O; and p is 1 or 2.
- X is S; and p is 1 or 2.
- R 1 1 is is C 3 -C 6 cycloalkyl or phenyl, where the phenyl can be optionally substituted with F, Cl, Br, or straight chained or branched Cj-C 4 alkyl optionally substituted with F.
- R 1 ' is is C 3 -C 6 cycloalkyl
- R 1 ' is phenyl optionally substituted with F, CI, Br, or straight chained or branched CpC 4 alkyl optionally substituted with F.
- R l ' is heteroaryl optionally substituted with F, Cl, Br, or straight chained or branched Ci -C 4 alkyl optionally substituted with F.
- the subject invention is also directed to a compound selected from the group consisting of A-
- the subject invention is also directed to a compound selected from the group consisting of 4- (4-chIoro-phenyl)- 1 -[3 ⁇ ( 1 -methyl-azetidin-3-yI)- 1 h-indol-6-yl]- lh-pyridin-2-one; 4-(4-chloro- phenyl)- 1 - ⁇ 3-[ 1 -(2-fluoro-ethyl)-azctidin-3-yl]- 1 h-indol-6-yl ⁇ - 1 h-pyridin-2-one; 4 ⁇ (4 ⁇ chlo ⁇ > phenyl)- 1 - ⁇ 3-[ 1 -(2-methoxy-cthyl)-azetidin-3-yl]- 1 h-indol- ⁇ -yl ⁇ - lh-pyridin-2-one; 4-(4- chloro-phenyl)- 1 -[ 1 -methyl-3-(
- the subject invention is also directed to a compound selected from the group consisting of3- [3-(l -methyl-azetidin-3-yl)- lh-indol-6-yl]-6-(4-trifluoromethyl-phenyl)-3h-pyrimidin-4-one; 1 - ⁇ 3-[ 1 -(2-fluoro-cthyl) ⁇ azetidin-3 ⁇ yI]- 1 -methyl- 1 h ⁇ indol ⁇ 6 ⁇ yl ⁇ -4-(4-fluoro-phenyI)- 1 h- pyridin-2-one; 5-fluoro- 1 '- ⁇ 3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yl]- 1 -methyl- 1 h-indol-6-yI ⁇ - l'h- [2,4'lbipyridinyl-2'-onc; 1'- ⁇ 3-[ 1 -(2-fluoro
- the present invention is also directed to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention and an acceptable pharmaceutical carrier,
- the present invention is also directed to a method of treating mood disorders in a subject comprising administering a therapeutically effective amount of a compound of the invention.
- the present invention is also directed to method of treating anxiety in a subject comprising administering a therapeutically effective amount of a compound of the invention,
- the present invention is also directed to a method of treating obesity in a subject comprising administering a therapeutically effective amount of a compound of the invention.
- the present invention is also directed to method of treating urinary disorders in a subject comprising administering a therapeutically effective amount of a compound of the invention,
- the invention is directed to uses of a compound of the invention for the manufacture of a pharmaceutical composition for treating a disorder selected from the group consisting of mood, anxiety and obesity related disorders.
- Pharmaceutically Acceptable Salts are provided.
- the present invention also comprises salts of the present compounds, typically, pharmaceutically acceptable salts.
- Such salts include pharmaceutically acceptable acid addition salts.
- Acid addition salts include salts of inorganic acids as well as organic acids.
- suitable inorganic acids include hydrochloric, hydrobromic, hydroiodic, phosphoric, sulfuric, sulfamic, nitric acids and the like.
- suitable organic acids include formic, acetic, trichloroacetic, trifluoroacetic, propionic, benzoic, cinnamic, citric, fumaric, glycolic, itaconic, lactic, methanesulfonic, malcic, malic, malonic, mandelic, oxalic, pyruvic, salicylic, succinic, methane sulfonic, elhanesulfonic, tartaric, ascorbic, pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic, aspartic, stearic, palmitic, EDTA, glycoiic, p-aminobenzoic, glutamic, bcnzenesulfonic, p- to
- compositions include the pharmaceutically acceptable salts listed in S. M. Berge, et al., J. Pharw, Set,, 1977, 66, 2 and Paulekuhn, el al. J. Med. Chem. 2007 (December online publication), the contents of all which are hereby incorporated by reference.
- the compounds of this invention may exist in unsolvated as well as in solvatcd forms with pharmaceutically acceptable solvents such as water, ethanol and the like.
- Racemic forms may be resolved into the optical antipodes by known methods, for example, by separation of diastcreomeric salts thereof with an optically active acid, and liberating the optically active amine compound by treatment with a base. Separation of such diastereomeric salts can be achieved, e.g. by fractional crystallization.
- the optically active acids suitable for this purpose may include, but are not limited to d- or 1- tartaric, mandelic or camphorsulfonic acids.
- Another method for resolving racematcs into the optical antipodes is based upon chromatography on an optically active matrix.
- the compounds of the present invention may also be resolved by the formation and chromatographic separation of diastereomeric derivatives from chiral derivatizing reagents, such as, e.g., chiral alkylating or acylating reagents, followed by cleavage of the chiral auxiliary. Any of the above methods may be applied either to resolve the optical antipodes of the compounds of the invention per se or to resolve the optical antipodes of synthetic intermediates, which can then be converted by methods described herein into the optically resolved final products which are the compound of the invention.
- chiral derivatizing reagents such as, e.g., chiral alkylating or acylating reagents
- Optically active compounds may also be prepared from optically active starting materials.
- the present invention further provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention and a pharmaceutically acceptable carrier.
- the present invention also provides a pharmaceutical composition comprising a therapeutically effective amount of one of the specific compounds disclosed in the Experimental Section and a pharmaceutically acceptable carrier.
- the compounds of the invention may be administered alone or in combination with pharmaceutically acceptable carriers or excipients, in either single or multiple doses.
- the pharmaceutical compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19 lh Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
- compositions may be specifically formulated for administration by any suitable route such as oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) routes, ⁇ t will be appreciated that the route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient.
- Pharmaceutical compositions for oral administration include solid dosage forms such as capsules, tablets, dragees, pills, lozenges, powders and granules.
- compositions may be prepared with coatings such as enteric coatings or they may be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art.
- Liquid dosage forms for oral administration include solutions, emulsions, suspensions, syrups and elixirs.
- compositions for parenteral administration include sterile aqueous and nonaqueous injectable solutions, dispersions, suspensions or emulsions as well as sterile powders to be reconstituted in sterile injectable solutions or dispersions prior to use.
- Suitable administration forms include, but arc not limited to, suppositories, sprays, ointments, creams, gels, inhalants, dermal patches and implants.
- Typical oral dosages range from about 0.001 to about 100 mg/kg body weight per day. Typical oral dosages also range from about 0.01 to about 50 mg/kg body weight per day. Typical oral dosages further range from about 0.05 to about 10 mg/kg body weight per day. Oral dosages are usually administered in one or more dosages, typically, one Io three dosages per day. The exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art.
- a typical unit dosage form for oral administration may contain from about 0.01 to about 1000 mg, from about 0.05 to about 500 mg, or from about 0.5 mg to about 200 mg.
- the present invention also provides a process for making a pharmaceutical composition comprising admixing a therapeutically effective amount of a compound of Formula the invention and a pharmaceutically acceptable carrier,
- the compound utilized in the aforementioned process is one of the specific compounds disclosed in the Experimental Section.
- the compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof.
- One example is an acid addition salt of a compound having the utility of a free base.
- a compound of the invention contains a free base such salts are prepared in a conventional manner by treating a solution or suspension of a free base of Formula Ia or Ib with a molar equivalent of a pharmaceutically acceptable acid.
- suitable organic and inorganic acids arc described above.
- solutions of the compounds of the invention in sterile aqueous solution aqueous propylene glycol, aqueous vitamin E or sesame or peanut oil may be employed.
- aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose.
- the aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration.
- the compounds of the invention may be readily incorporated into known sterile aqueous media using standard techniques known to those skilled in the art.
- Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solutions and various organic solvents.
- solid carriers include lactose, terra alba, sucrose, cyclodextrin, talc, gelatin, agar, pectin, acacia, magnesium slearatc, stearic acid and lower alkyl ethers of cellulose.
- liquid carriers include, but are not limited to, syrup, peanut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water.
- the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostcarate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical compositions formed by combining the compounds of the invention and a pharmaceutically acceptable carrier arc then readily administered in a variety of dosage forms suitable for the disclosed routes of administration.
- the formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and optionally a suitable excipient.
- the orally available formulations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or waier-in-oil liquid emulsion.
- the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it may be in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but will range from about 25 mg to about 1 g per dosage unit.
- the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
- the compounds of Formula Ia and Ib bind to the MCHl receptor. Accordingly, the present invention provides a method of treating depression in a subject which comprises administering to the subject a therapeutically effective amount of a compound of this invention. This invention further provides a method of treating a subject suffering from anxiety which comprises administering to the subject a therapeutically effective amount of a compound of this invention,
- This invention also provides a method of treating a subject suffering from obesity which comprises administering to the subject a therapeutically effective amount of a compound of this invention.
- the obesity indication includes in particular exogenic obesity, hypei ⁇ nsulinaemic obesity, hyperplasmic obesity, hyperphyseal adiposity, hypoplasmic obesity, hypothyroid obesity, hypothalamic obesity, symptomatic obesity, infantile obesity, upper body obesity, alimentary obesity, hypogonadal obesity, central obesity.
- This range of indications also includes cachexia, anorexia and hyperphagia.
- the treatment of obesity is expected to weight loss in a subject.
- This invention also provides a method of treating a subject suffering from urinary disorders which comprises administering to the subject a therapeutically effective amount of a compound of this invention.
- Another range of indications for which the compounds according to the invention are advantageously suitable is the prevention and/or treatment of micturition disorders, such as for example urinary incontinence, hyperactive bladder, urgency, nycturia, enuresis, while the hyperactive bladder and urgency may or may not be connected with benign prostatic hyperplasia.
- the subject is a human being.
- TLC Thin-layer chromatography
- the compounds of Formula Ia may be synthesized according to the procedures described in Scheme 1.
- the compounds of Formula II and III are commercially available or may be synthesized by those skilled in the art.
- the compounds of Formula II may be synthesized according to the procedures described in Scheme 14, 15, and 16.
- the compounds of Formula III may be synthesized according to the procedures described in Scheme 6, 7, 8, 9, 10, 1 1, 12, and 13. Copper-catalyzed coupling reaction of compounds II with compounds III in the presence of copper iodide, potassium carbonate and trans-N,N'-DimethyI- cyclohexane- 1 ,2-di amine in DMF at about 100 0 C affords the compounds of Formula Ia.
- the compounds of Formula Ia may be prepared according to the procedures described in Scheme 2.
- the compounds of Formula V may be synthesized via dehydroxylation of alcohols of Formula IV with TFA and lriethyl silane in DCM at about 0 0 C for about 10 min.
- Pyridinoncs of Formula III arc coupled with compound V in the presence of copper iodide, potassium carbonate and trans-N,N'-dmiethyl-cyclohexane-l,2-diamine in DMF at about 100 0 C for about 18 h, followed by dcprotection of N-Boc in the presence of TFA, to afford the compound VI.
- the reductive amination of compound VI with aldehydes (or ketones) or the alkylation with alkyl halides affords the compounds of Formula Ia.
- the compounds of Formula Ia may be prepared according to the procedures described in Scheme 3.
- Scheme 4
- the compounds of Formula Ia may be prepared according to the procedures described in Scheme 4.
- the compounds of Formula VIII are prepared via Suzuki reactions of pyridinones of Formula III and indole VII in the presence of palladium chloride, base and a suitable ligand in DMF at about 80 0 C for about 12 h.
- the compounds of Formula Ia are prepared via iridium-catalyzed reactions of indoles of Formula VIII and alcohols IX in the presence of [Cp*IrCl 2 ]2 and potassium hydroxide in DCM in sealed tube at about 110 0 C for about 24 h.
- reaction conditions in connection with the iridium-catalyzed reactions sec R. Grigg et al., Org. Letters 2007, 9 (17), 3299-3302 and references cited therein.
- the compounds of Formula Ia may be prepared according to the procedures described in Scheme 5. Mitsunobu reaction of compounds of Formula X and phenols of Formula XI in the presence of triphenylphosphine and DBAD in THF affords ethers or thio ethers of Formula XII.
- Pyridine-N-oxidcs of Formula XIII can be prepared via oxidation of XII with mCPBA in DCM at room temperature.
- the compounds of Formula XIV are prepared via reaction of pyridine-N-oxidc of Formula XIII with POCl 3 in toluene at about 90 0 C for about 12 h.
- the pyridinoncs of Formula XV are synthesized from hydrolysis of XIV in acetic acid and water at about 90 0 C for about 24 h.
- the compounds of Formula Ia are prepared via Suzuki coupling reactions of XV and XVI in the presence of palladium, base and a suitable ligand in DMF at about 80 0 C for about 12 h.
- the compounds of Formula III may be prepared according to the procedures described in Scheme 6.
- the compounds of Formula XIX are synthesized via nucleophilic substitution of 4-chloropyridine N-oxide of Formula XVIII with alcohols or thio alcohols of Formula XVII in the presence of tris[2-(2-melhoxycthoxy)- ⁇ thyl]amine (TDA-I), potassium hydroxide and potassium carbonate in toluene under reflux for about 3 h.
- TDA-I tris[2-(2-melhoxycthoxy)- ⁇ thyl]amine
- Pyridinones of Formula III are prepared via reaction of compounds XIX in acetic anhydride at about 140 0 C overnight.
- the compounds of Formula III may be synthesized according to the procedures described in Scheme 7.
- the compounds of Formula XX which are commercially available or synthesized by those skilled in the art, are treated with alcohols or thio alcohols XI in the presence of triphenylphosphine and DBAD in THF at room temperature for about 2 h to afford ethers or thio ethers of Formula XXI.
- the compounds of Formula XXI are treated with acetic acid in water at about 120 0 C for about 12 h to afford pyridinones of Formula HI.
- the compounds of Formula III may be prepared according to the procedures described in Scheme 8.
- the mesylates of Formula XXII are synthesized from reaction of alcohols of Formula XX and mesyl chloride in the presence of base.
- the compounds of Formula III are prepared via substitution reaction of mesylates of Formula XXII with alcohols or thio alcohols XI in the presence of base such as cesium carbonate.
- R 3 aryl, heteroaryl
- the compounds of Formula III may be prepared according to the procedures described in Scheme 9.
- the compounds of Formula XXIV arc synthesized via Suzuki reaction of 4-chloropyridinc N-oxide XVIII with aryl boronic acid or aryl boronic ester of Formula XXIII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPh- ⁇ ) 4 and Na 2 CO 3 in 1 ,2-dimelhoxy ethane/water under reflux for about 16 h.
- Pyridinones of Formula III are prepared via reaction of compounds XXIV in acetic anhydride at about 140 0 C overnight.
- the compounds of Formula IH may be prepared according to the procedures described in Scheme 10.
- the compounds of Formula XXV can be treated with aryl boronic acid or aryl boronic ester XXIII, which are commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPlIs) 4 and Na 2 CO- ? in 1,2-dimethoxy ethane under reflux for about 16 h to afford the compounds of Formula XXVI.
- the compounds of Formula XXVI can be treated with acetic acid at about 110 0 C for overnight to afford pyridinones of Formula III.
- the compounds of Formula III may be prepared according to the procedures described in Scheme 11.
- the compounds of Formula XXVIII which are commercially available or synthesized by those skilled in the art, are treated with pyridinyl boronic acid or pyridinyl boronic ester XXVII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPh 3 ) 4 and Na 2 CO 3 in 1,2-dimethoxy ethane under reflux for about 16 h to afford Formula XXVI.
- the compounds of Formula XXVI are treated with acetic acid at about 1 10 0 C for overnight to afford pyridinones of Formula HI.
- the compounds of Formula III may be prepared according to the procedures described in Scheme 12.
- the compounds of Formula XXIX which are commercially available or may be synthesized by those skilled in the art, can be treated with aryl boronic acid or aryi boronic ester XXIII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPhO 4 and Na 2 CO 3 in 1 ,2-dimethoxy ethane under reflux for 16 h to afford pyridinones of Formula III.
- the compounds of pyridinone XXXII may be prepared according to the procedures described in Scheme 13.
- the compounds of Formula XXXII are prepared via Wittig reaction of aldehydes XXX and phosphonates XXXI in the presence of potasium f ⁇ r/ ⁇ butoxide in THF at room temperature for about 4 h.
- Scheme 14
- the compounds of Formula XXXV may be synthesized according to the procedures described in Scheme 14.
- the compounds of Formula XXXIII and VII are commercially available or may be synthesized by those skilled in the art.
- the azetidinone of Formula XXXIV is prepared by reaction of XXXIII with sulphur trioxide pyridine complex in the presence of triethylamine in DMSO and DCM at about 10 0 C for about 30 min and at about 20 0 C for about 1 h.
- l-(3 ⁇ Indolyl)azetidin-3 ⁇ ol IV is prepared via condensation of azetidone XXXIV with indoles of Formula VII in the presence of potassium hydroxide in methanol at about 45-50 0 C for about 6 h.
- the compound XXXV is prepared via reduction of l-(3-indolyl)azetidin»3-ol IV with LAH in THF under reflux for about 6 h.
- the substituted indoles VII may be synthesized according to the procedures described in
- Boc-azetidin-3-one (68.0 g, 397 mmol) was added in one portion. The resulting solution was stirred at 45-50 0 C for 6 h and concentrated in vacuo. The residue was diluted with EtOAc
- Example Ib I -[ I -Ethyl-3-((S)- 1 -mcthyl- ⁇ yrroIidin-2-yImethyl)- 1 H-indol-6-yl]-4-(pyridin-3- ylmethoxy)- 1 H-pyridin-2-one
- the compound was prepared from 4-(4-fluoro-phenoxyniethyl)-i H-pyrid in-2-one and 6- bromo-3-(l-melhyI-azelidin-3-yl)-lH-indolc VI.
- Example Id 4-(4-Chloro ⁇ benzyloxy)- 1 -[3-( 1 -melhyl-azetidin-3-yl)- 1 H-indol-6-yl j- 1 H- pyridin-2-one
- the compound was prepared from 4-(4-chloro-benzyloxy)-lH-pyridin-2-onc and bromo-3-(l - methyl-azetidin-3-yl)-lH-indole.
- ESMS m/e: 420 (M+H) + .
- the compound was prepared from 4-(4-fluoro-phenylsulfanylmethyl)-lH-pyridin-2-one and 6-bromo-3-( 1 -methyl-azetidin-3-yl)- I H-indole.
- the compound was prepared from 4-[(E)-2 ⁇ (4»fluoro ⁇ phenyl) ⁇ vinyl] ⁇ l-[3 ⁇ (l-melhyl-azelidin- 3-yl)- lH-indol-6-yl] - 1 H-pyridin-2-one and 6-bromo-3-( 1 -methyl-azetidin-3-yl)- 1 H-indoic.
- Example Ig 4-( 1 -Cyclopropyl-ethoxy)- 1 -[3-( 1 -melhyl-azclidin-3-yl)- 1 H-indol-6-yll- 1 H- pyridin ⁇ 2-onc
- the compound was prepared from 4-(l-cyclopropyI-cthoxy)-l H-pyridin-2-one and 6-bromo- 3-( 1 -methyl-azetidin-3-yl)" 1 H ⁇ indole ESMS m/e: 364.0 (M+H) + .
- Example 2a 4-(4-Chloro-phenyl)-l -[3-(I -methyl-azetidin-3-yl)- lH-indol-6-yl]-lH-pyridin-2-one
- Example 2b 4-(4-Chloro-phenyl)- l- ⁇ 3-[ l-(2-fluoro-ethyl)-azetidin-3-yl]- lH-indol-6-yl ⁇ - lH- pyi ⁇ din-2-one
- the compound was prepared from 4-(4-chloro-phenyI)- 1 H-pyridin-2-one and 6-bromo-3-[ 1 - (2-fluoro-cthyl)-azelidin-3-yl]- lH-indole.
- Example 2c 4-(4-Chloro-phenyl)- 1 - ⁇ 3-[ 1 -(2-mcthoxy-ethyl)-azelidin-3-yI]- 1 H-indol-6-yl ⁇ - 1 H-pyridin-2-one
- the compound was prepared from 6-bromo-3-[l-(2-mcthoxy-ethyI)-azetidin-3-yl]-lH-indole and 4-(4-chlor ⁇ - ⁇ hcnyI)-l H- ⁇ yridin-2-one.
- ESMS m/e: 433.9 (M+H) + .
- Example 2d 4-(4-ChIoro-phenyl)- 1 -[ 1 -methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one
- the compound was prepared from 6-bromo- 1 -methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indole and 4-(4-chloro-phenyl)-l H-pyridin-2-one.
- the compound was prepared from 6-biOmo-3-(l-isopropyl ⁇ azetidin-3-yl)- lH-indole and 5- chloro-rH-[2,4']bipyridinyl-2'-one.
- ESMS m/e:419.0 (M+H) + .
- the compound was prepared from 6-bromo-3-[l-(2-fluoiO-ethyl)-azetidin-3-yl]-lH-indolc and 6-trifluoromethyl" rH-[3,4']bipyridinyl ⁇ 2'-one.
- ESMS m/e: 456.9 (M+H) + .
- Example 2g 4-(2-FI uoro-4-trifluoiOtnethyl -phenyl)- 1 -[3-( 1 -methyl ⁇ aze ⁇ din ⁇ 3 ⁇ yl) ⁇ 1 H ⁇ indo! ⁇ yll-1 H-pyridin-2-one
- the compound was prepared from 6-biOmo-3-( l-methyl-azctidin-3-yl)-lH-indolc and 4-(2- fluoro-4-trifluor ⁇ methyl- ⁇ henyl)- 1 H-pyridin-2-one.
- Example 2h 4-[4-( 1 -Hydroxy- 1 -methyl-ethyl) ⁇ phenyl] ⁇ 1 ⁇ [3-( I -methyl-azetidin ⁇ 3-yl) ⁇ 1 H- indoI-6-yl]-l H-pyridin-2-one
- the compound was prepared from 6-bromo-3-( l-methyl-azetidin-3-yl)-lH-indole and 6-(4- chloro- ⁇ henyI)-3H-pyrimidin-4-one.
- the compound was prepared from 6 ⁇ bromo-2-methyl-3-(l-methyI-azetidin-3-y!)-l H-indole and ⁇ -trifluoiOmethyl-l ⁇ -tS ⁇ 'Jbipyridinyl ⁇ '-one.
- the compound was prepared from 6-bromo-3-(l-mcthyl-azetidin-3-yl)-l H-indole and 4-(5- methyl-bcnzo[b]thiophen-2-yl)-I H-pyridin-2-one.
- ESMS m/e:425.9 (M+H) + .
- Example 21 1 -[3-( l-Methyl-azetidin-3-yl)- 1 H-indol-6-yI]-4-( 1 ⁇ methyl- 1 H-indol-5-yl)- 1 H- pyridin-2-one
- the compound was prepared from 6-bromo-3-(l-mcthyl-azetidin-3-yl)-lH-indole and 4- na ⁇ hthalen-2-yl-l H- ⁇ yridin-2-onc.
- ESMS m/e: 406.1 (M+H) + .
- the compound was prepared from 3-(l-methyl-azelidin-3-yl)-l H-indolc and 4-(5-fluoro- benzooxazol-2-yl)-l H-pyridin-2-one ESMS m/e: 415.0 (M+H) + .
- the compound was prepared from 6-bromo-3-[l-(2-fluoro-ethyl)-azetidin-3-yl]-lH-indole and 4-(5-ethyl-pyrimidin-2-yl)-l H-pyridin-2-one.
- ESMS m/e: 417.9 (M+H) + .
- the compound was prepared from 6-bromo-3-[ l-(2-fluoiO-ethyt)-azetidin-3-yl]- l -methyl- IH- indole and S-chloro- l ⁇ - ⁇ 'Jbipyridinyl-a'-one.
- the compound was prepared from 4-(4-tTrifluoroniethoxy-phenyl)-lH-pyridin-2-one and 6- bromo-3-(l-methyl-azetidin-3-yl)-lH-indole ESMS m/e: 439.9 (M+H) ⁇
- the compound was prepared from 6-(4-trifluoromethyl-phenyl)-3H-pyrimidin-4-one and 6- bromo-3-(l-methyl-azetidin-3-yl)-lH-indole.
- ESMS m/e: 424.9 (M+H) + .
- the compound was prepared from 4-(4-fluoro-phenyl)-l H-pyridin-2-one and 6-bromo-3-[l- (2-fluoro-ethyI)-azetidin-3 -yl]-l -methyl- IH-indole.
- the compound was prepared from 5-trifluoromethyI-l 'H-[2,4']bipyridinyl-2'-one and 6- bromo-3-[ 1 -(2-fluoro-elhyl)-azetidin-3-yIJ- 1 -methyl- 1 H-indole.
- the compound was prepared from 5-trifluorornethyi-l ⁇ -[2,4']bipyridinyI ⁇ 2' ⁇ onc and 6- bromo- 1 -methyl-3-( 1 -methyI-aze ⁇ din-3-yl)- 1 H-indole.
- the compound was prepared from 6-trifluoiOmethyI-rH-[3,4']bipyridinyi-2'-one and 6- bromo-3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yI J-I -methyl- 1 H-indole.
- ESMS m/e: 470.9 (M+H) + .
- the compound was prepared from 6-(4-chloro-phenyl)-3H-pyrimidin-4-one and 6-bromo-2- methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indole.
- Example 3a 4-(4-Chloro-phenyl)- 1 - ⁇ 3 ⁇ [ 1 -(tetrahydro-thiophcn-3-yl)-azetidin-3-yl]- 1 H-indol- 6-yl ⁇ - 1 H-pyridin-2-one
- Example 3b 4-(4-Chloro-phenyl)- 1-[3-Cl -oxetan-3-yl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one
- the compound was prepared from l-(3-azetidin-3-yl- l H-indoI-6-yl)-4-(4-chloro-phenyl)- lH- pyridin-2-one and oxclan-3-one.
- ESMS m/e: 432.0 (M+H) + .
- the compound was prepared from l-[3-azctidin-3-yl-l-(2-hydroxy-cthy])-l H-indol-6-yi]-4- (4-chloro-phenyI)-l H-pyridin-2-onc and acetaldehyde.
- ESMS m/e: 447.9 (M+H) + .
- Example 3d 4-(4-Chloro-phenyl)- 1 - ⁇ 3-f l-(3,3,3 ⁇ trifluoro-piOpionyl)-azctidin-3-yl]-lH-indol-
- the compound of example 3d were prepared in an analogous manner to the procedure described for example 3e and was prepared from l-(3-azetidin-3-yl-l H-indol-6-yl)-4-(4- chloro-phenyl)- l H-pyridin-2-one and 3,3,3-t ⁇ fluoro-propionyl chloride.
- Example 3e 4-(4-Chloro-phcnyl)- 1 -[3-( 3 -cyclopropanecarbonyl-azetidin-S-yl)- 1 -methyl- 1 H- indol-6-yl] ⁇ l H-pyridin-2-one
- Example 3f 4-(4-Chloro-phenyl)-l ⁇ l-mcthyl-3-[ l-(tetrahydro-furan-3 ⁇ yl) ⁇ azetidin-3-yl]- 1 H-indol-6-yl ⁇ - 1 H-pyridin-2-one
- the compound was prepared from l-(3-azetidin-3-yl- l -methyl- 1 H-indol-6-yl)-4-(4-chloro- phcnyl)- lH-pyridin-2-onc and dihydro-furan-3-one.
- ESMS m/e: 459.9 (M+H) + .
- the compound was prepared from l' ⁇ (3-azetidin ⁇ 3-yl-l -methyl- lH-indoI-6-yl)-5-chloro- l 'H-
- the compound was prepared from l'-(3-azetidin-3-yl-l -methyl- lH ⁇ indol ⁇ 6 ⁇ yl)-5-chloro-l 1 H- [2,4']bipy ⁇ dinylTM2'-onc and acetone.
- ESMS m/e: 432.9 (M+H) + .
- Example 3i 1 -[ 1 -Methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indoI-6-yl]-4-(4-trifluoromethyl- phenyl)- lH-pyridin-2-one
- the compound was prepared from l-(3-azetidin-3-yl- l -methyl- lH-indol-6- yl)-4-(4- trifluoiOmethyl-phenyl)- lH-pyridin-2-onc and formaldehyde.
- ESMS m/e: 437.9 (M+H) + .
- the compound of example 3j were prepared in an analogous manner to the procedure described for example 3e and was prepared from I -(3-azetidin-3-yl- 1 -methyl- lH-indol-6-yl)- 4-(4-trifluoromethyl-phenyl)-l H-pyridin-2-one and acetyl chloride.
- the compound was prepared from 3-(3-azetidin-3-yl-lH-indoI-6-yl) ⁇ 6-(4-chloro-phenyl)-3H- pyrimidin-4-one and tetrahydro-furan-3-carbaldehyde.
- Example 31 4-(4-Fluoro-phenyl)- 1 -[3-( 1 -isopropyl-azetidin-3-yl)- 1 -methyl- 1 H-indol-6-yl] - 1 H-pyridin-2-one
- the compound was prepared from I -(3-azetidin-3-yl ⁇ l -methyl- l H-indol-6-yl)-4-(4-fluoro- phcnyl)-lH- ⁇ yridin-2-one and acetone.
- ESMS m/e: 41 ⁇ .0(M+H) + .
- Example 4b 1 -[ 1 -Methyl-3-( 1 -methyl-azetidin-3-y ⁇ )- 1 H-indol-6-yl]-4-(2,2,2-tri ⁇ uoiO- ethoxy)- 1 H-pyridin-2-one
- the compound was prepared from l-[3-(l-methyl-azetidin-3-yl)-l H-indof-6-yll-4-(2,2,2- trifluoro-elhoxy)-lH-pyridin-2-onc and iodomethane.
- ESMS m/e: 392.0 (M+H) + .
- the compound was prepared from 5-chloro-l '-[3-(l -ethyl-azetidin-3-yl)- l H-indol-6-yl]-l TI- [2,4']bipyridinyl-2'-one and 1 -bromo-2-fluor ⁇ -elhane.
- Thc compound was prepared 5-Chloro- 1 '-[3-(I -ethyl-azetidin-3-yl)- lH-indol-6-yll- l 1 H- [2,4'JbipyridinyI-2'-one and iodomethane.
- ESMS m/e: 419.2 (M+H) + .
- the affinity of the compounds of the invention at the MCHl receptor was determined by measuring the inhibition of binding of a radioactive ligand ant the MCHl receptor.
- the procedure for determining specific binding of a test compound may be used as described by Audinot, et al. British Journal of Pharmacology, 2001, 133, 371-378.
- the specific binding of test compounds can be measured at the rat MCHl receptor (GenBank Accession No. NM_031758) using [ 125 I]-S3 ⁇ 057 (NEX396; PcrkinElmcr Life Sciences, Inc.) as the radioligand.
- the compounds of the invention were tested for their affinity at the MCH IR receptor. All the compounds disclosed herein show Ki values of less than about 5.0 ⁇ M. Nearly all the compounds show Ki values of less than about l .O ⁇ M. Most of the compounds have Ki values of less than about 20OnM with many of compounds having Ki values of less than about 5OnM.
- Functional activity of the compounds of the invention can be measured by receptor assays which determine the degree of intracellular second messenger response.
- receptor assays which determine the degree of intracellular second messenger response.
- Cos-7 ceils are transfected with the MCHl receptor using the DEAE-dcxtran method (Gerald, ct al. J. Biol. Chcm. 1995, 270, 26758-26761).
- Other cell transfcction methods employing various host cells, are well-known in the art. Certain compounds were tested to determine their functional activity and were determined to be potent antagonists at the MCHl receptor.
- the in-vivo effects of the compounds of the present invention may be evaluated by using the following in-vivo behavioral animal models.
- the behavioral models described below are not intended to be the only models used to determine the efficacy of a compound of the invention to treat the corresponding disorder.
- the marble burying experiment can also be used to screen for compounds for potential as anxiolytics. The skilled artisan would recognize the changes in certain parameters of the experiments to acquire the most exact data.
- Rat Forced-swim Test Male Long-Evans rats are used and housed individually, maintained on about 12 h reverse light/dark cycle with lights off at about 09:00, and given free access to either a high-fat diet (#D 12451; fat percentage, about 45% kcal; Research Diets, New Brunswick, New Jersey) or a control diet (#D124508, fat percentage, about 10% kcal; Research Diets, New Brunswick, New Jersey) and water. After about 1 1 weeks, rats on the high fat diet began receiving a compound of the invention or vehicle by i.p. injection twice daily, about 1 h before lights off and about 10 h later, for about 4 weeks. Rat Forced-swim Test:
- Rat Social-interaction Test The procedure is performed for about 15 min as previously described (File and Hyde Br. J, Pharmacol. 1987, 62, 19-24) under low-light conditions using pairs of unfamiliar male Sprague-Dawley rats previously housed singly and exposed to the test arena for about 15 min on the previous day.
- a compound of the invention, chlordiazepoxide or vehicle is injected i.p. as ⁇ 1.0 ml/kg solution. All test sessions are videotaped and recorded for later scoring.
- Active social interaction defined as sniffing, grooming, biting, boxing and crawling over and under, as well as locomotor activity (defined as squares crossed), is scored by a single rater, who is blinded to the treatment of each pair.
- Rat Models of Micturition Compounds useful in the treatment of urinary disorders are assessed in various animal models of the micturition reflex as described in the art. (See, e.g., Maggi, CA, et al. J Pharmacol Exp Ther, 1987, 240, 998-1005; Morikawa, K, et al. Eur. J Pharmacol., 1992 213:409-415; Yoshiyama, M, et al. Eur J Pharmacol 287:73-78; and Yoshiyama, Urology, 1999, 54(5), 929- 33.)
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to indole derivatives which bind to the MCHl receptor. In separate aspects, the subject invention is directed to uses of said compounds in the preparation of a pharmaceutical composition for the treatment of obesity and CNS related disorders and to methods of treating said disorders comprising administering a therapeutically effective amount of a compound of the invention.
Description
INDOLE DERIVATIVES
FIELD OF THE INVENTION
The present invention is directed to indole derivatives which bind to the MCHl receptor. In separate aspects, the subject invention relates to uses of said compounds in the preparation of a pharmaceutical composition for the treatment of obesity and CNS related disorders and to methods of treating said disorders comprising administering a therapeutically effective amount of a compound of the invention.
BACKGROUND OF THE INVENTION Throughout this application, various publications are referenced to in full citations. The disclosures of these publications are hereby incorporated by reference into the subject application to describe more fully the state of the art to which this invention pertains.
Melanin-concentrating hormone (MCH) is a cyclic 19-amino acid peptide produced predominantly by neurons in the lateral hypothalamus and zona inperta of the brain which project broadly throughout the brain. Mammalian MCH is conserved between rat, mouse, and human, exhibiting 100 % amino acid homology, and the effects of MCH are mediated through its interaction with receptors that belong in the rhodopsin superfamily of G protein-coupled receptors. Presently, two receptor subtypes for MCH have been identified in humans, MCHl- R and MCH2-R.
MCH exerts several physiological effects through interaction with its receptors. For example, an icv injection of MCH in rats stimulates food intake (Levens, et al. Int. J. Obesity 2002, 26, 1289-1295), and chronic administration leads to increased body weight (Kanatani, et al. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E583-E588). The link between MCHl-R and the effects of MCH on feeding is demonstrated by reports on the phenotype of MCHl-R knockout mice. Independent groups generated knock-out mice with the targeted deletion of MCHl-R. The phenotype of these mice was lean, hyperphagic and hypermetabolic, with an increased resistance to diet-induced obesity (Marsh, et al. Proc. Natl. Acad. ScL 2002, 99, 3240-3245). These observations suggest that MCHl-R antagonists could be useful for the treatment of obesity related disorders.
As the distribution of MCHl -R binding sites in the CNS such as the amygdala, accumbens nucleus, dorsal raphe and locus cocruleus is suggestive of a role for MCH in the regulation of mood and stress, several groups have identified selective, high affinity MCHl-R antagonists and evaluated their effects in in-vivo behavioral paradigms predictive of antidepressant and/or anxiolytic activity.
For example, clinically used antidepressants decrease the amount of time mice or rats spend immobile in the forced-swim test (Porsolt, et al. Arch bit Phannacodyn Ther. 1977, 229, 327- 336 and Luki, ct al. Psychopharmacology 2001 , 155, 315-322). Pretreatmenl of rats with a single oral dose of SNAP-7941, a selective MCHl-R antagonist, or fluoxetine decreased the duration of immobility compared with vehicle-treated controls, and increased the time these animals spent swimming (Borowsky, et al. Nature Medicine 2002, 8, 825-830). The profile of SNAP-7941 in the rat forced-swim test is similar to that of clinically used antidepressants, indicating that MCH l-R receptor blockage may be a therapeutic modality for the treatment of mood-disorders such as depression.
The rat social interaction test has been used as a model of anxiety (File and Hyde Br. J. Pharmacol. 1987, 62, 19-24). Acute treatment with 3, 10 and 30 mg/kg SNAP-7941 or 5 mg/kg chlordiazepoxide increased social interaction time compared with vehicle- treated controls (Borowsky, et al. Nature Medicine 2002, 8, 825-830) without an overall increase in locomotor activity. The response to the two lower doses of SNAP-7941 was as robust as the response to 5 mg/kg chlordiazepoxide. The profile of this potent MCHl-R antagonist in the rat social interaction test suggests that MCHl-R antagonists may have potential as anxiolytic agents.
Accordingly, it is expected that the compounds of the subject invention can be used to treat obesity, mood and anxiety related disorders as well as the additional indications which are disclosed herein in the detailed description section.
SUMMARY QF THE INVENTION
The objective of the subject invention is to provide compounds which are ligands at the MCHl receptor. Accordingly, the present invention relates to compounds of Formula Ia and Ib:

wherein each B1 , B2 and B3 is independently CH or N;
wherein R1 is -(CHR7)mN(R8)(R9) or -(CHR7JnR10, and where the R1 containing moiety of formula Ib is connected to one A but not when A is N;
wherein R2 is H or straight chained or branched C1-C7 alkyl optionally substituted with one or more fluorine and where the carbons of the Ci-C7 alkyl are optionally replaced with one to three N, O or S atoms;
wherein R^ is -X(CH2)pYRl ' or X(CHCH)YR1 ', and where B1 of the R3 containing moiety of formula Ia is connected to one A but not when A is N;
wherein each X and Y is independently CH2, O, S, NH or a bond, provided that an O, S or NH is separated from another O, S or NH by at least two carbon atoms;
wherein each R4, R^ and R6 is independently H, halogen or straight chained or branched Ci-C6 alkyl;
wherein each R7 is independently H, OH, or wherein one R7 can combine with another R7 on an adjacent carbon atom to form C3-Cf1 cycloalkyl and wherein one R7 can combine with a H on the shared carbon atom to form CrQs cycloalkyl;
wherein each R8 and R9 is independently H or straight chained or branched Ci-C7 alkyl;
wherein R10 is a nitrogen containing heterocyclic moiety optionally substituted with one or
I 0 more halogen, -COR ", a S or O containing heterocyclic ring, or straight chained or branched CpC7 alkyl optionally substituted with halogen or -OCH-?;
wherein R1 1 is CrC6 cycloalkyl, straight chained or branched alkoxy-Cs-Q, cycloaikyl, straight chained or branched Ci-C4 alkyl/alkoxy, phenyl, napthyl, 5 to 6-membered heteroaryl, benzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C]-C4 alkyl optionally substituted with F;
wherein m is an integer from I to 4 inclusive;
wherein n is an integer from 0 to 4 inclusive; and
wherein p is independently an integer from 0 to 3 inclusive, or a pharmaceutically acceptable salt thereof.
In separate aspects of the invention, the compound is selected from one of the exemplified compounds which are disclosed in the Experimental Section.
Furthermore, the subject invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula Ia or Ib, or a pharmaceutically acceptable salt and a pharmaceutically acceptable carrier.
Moreover, the present invention provides a method of treating a subject suffering from mood disorders, anxiety or obesity comprising administering to the subject a therapeutically effective amount of a compound of Formula Ia or Ib. The present invention further provides uses of a compound of Formula Ia or Ib in the manufacture of a pharmaceutical composition for the treatment of mood disorders, anxiety or obesity.
DETAILED DESCRIPTION QF THE INVENTION Definitions
As used herein, "treating" and "treatment" refer to an approach for obtaining beneficial or desired clinical results. For purposes of this invention, beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disorder, stabilized (i.e., not worsening) state of disorder, delay or slowing of disorder progression, amelioration or palliation of the disorder stale, and remission (whether partial or total), whether detectable or undetectable.
As used herein, "therapeutically effective amount" is an amount sufficient to effect beneficial or desired clinical or biochemical results. A "therapeutically effective amount" can be administered one or more times. For purposes of this invention, "a therapeutically effective amount" of a compound is an amount that is sufficient to palliate, ameliorate, stabilize, reverse, slow or delay the progression of the disorder state.
As used in this invention, the term "antagonist" refers to a compound which binds to, and decreases the activity of, a receptor in the presence of an agonist. In the case of a G-protein coupled receptor, activation may be measured using any appropriate second messenger system which is coupled to the receptor in a cell or tissue in which the receptor is expressed. Some specific, but by no means limiting, examples of well-known second messenger systems are adenylate cyclase, intracellular calcium mobilization, ion channel activation, guanylate cyclase and inositol phospholipid hydrolysis. Conversely, the term "agonist" refers to a compound which binds to, and increases activity of, a receptor as compared with the activity of the receptor in the absence of any agonist.
In the present invention, the term "straight chained or branched Ci-C7 alkyl" refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive. Examples of such substiluents include, but are not limited to, methyl, ethyl, 1 -propyl, 2-propyl, 1 -butyl, 2 -butyl, 2-melhyl-2-propyl, 2-mcthyl-l -propyl and n-heptyl. Similarly, the term "straight chained or branched C]-C4 alkyl" refers to a saturated hydrocarbon having from one to four carbon atoms inclusive.
The term "C^-C6 cycloalkyl" refers to the group consisting of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
The term "a nitrogen containing heterocyclic moiety", as used herein, refers Io the following groups:

The term "a S or O containing heterocyclic moiety", as used herein, refers to the following groups:

As used in herein, "the R% containing moiety of formula Ia" refers to the following moiety:

As used in the present invention, the term "heteroaryl" is used to include five and six membered unsaturated rings that contain one or more oxygen, sulfur, or nitrogen atoms. Examples of heteroaryl groups include, but are not limited to, furanyl, thienyl, pyrrolyl, oxazolyl, thiazoiyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, pyridyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, and tria/inyl.
As used herein, halogen refers to F, Cl, Br of I.
Additionally, the compounds of the invention may have an advantage over other compounds in the art in that they possess reduced p-glycoproiein (pGP) activity and accordingly, the compounds may have unexpectedly enhanced brain-blood penetration.
Additionally, the present invention further provides certain embodiments which are immediately described below.
In one embodiment, each A is independently CR6or N provided that when one A is N, the other A is CR6;
each B1, B2 and B3 is independently CH or N;
R1 is -(CHR7),nN(R8)(R9) or -(CHR7)nR10, and where the R1 moiety of formula Ib is connected to one A but not when A is N;
R2 is H or straight chained or branched Ci-C7 alkyi optionally substituted with one or more fluorines and where the carbons of the C1-C7 alkyl arc optionally replaced with one to three N, O or S atoms;
R1 is -X(CH2)PYR' ' or X(CHCH)YR1 1 , and where B1 of the R1 containing moiety of formula
Ia is connected to one A but not when A is N;
each X and Y is independently CH2, O, S, NH or a bond, provided that an O, S or NH is separated from another O, S or NH by at least two carbon atoms;
each R4, R5 and R6 is independently H, halogen or straight chained or branched CpC7 alkyl;
each R7 is independently H, OH, or wherein one R7 can combine with another R7 on an adjacent carbon atom to form C3-CU cycloalkyl and wherein one R7 can combine with a H on the shared carbon atom to form C3-C6 cycloalkyl;
each R and R is independently H or straight chained or branched Cj -C7 alkyl;
R1( is a nitrogen containing heterocyclic moiety optionally substituted with one or more halogen or straight chained or branched Cj-C7 alkyl;
R11 is C3-Q, cycloalkyl, phenyl or a 5 to 6-membered heteroaryl, where the phenyl or 5 to 6- membered heteroaryl can be optionally substituted with F, Cl, Br, or straight chained or branched Cj-C4 alkyl optionally substituted with F;
m is an integer from 1 to 4 inclusive;
n is an integer from 0 to 4 inclusive; and p is independently an integer from 0 to 3 inclusive, or a pharmaceutically acceptable salt thereof.
In another embodiment of the present invention, and as a subgenus of formula Ia, the compound has the following structure:
R'

B1 is N and B2 and B3 are CH;
R1 is -(CHR7)mN(R8)(R9) or -(CHR7)nR10;
R2 is H or straight chained or branched C1-C7 alkyl optionally substituted with one or more fluorine or -OH and where the carbons of the Ci-C7 alkyl are optionally replaced with one to three N, O or S atoms;
each R and R" is independently H, halogen or straight chained or branched Ci-C7 alkyl;
each R7 is independently H, OH, or wherein one R7 can combine with another R7 on an adjacent carbon atom to form CrQ cycloalkyl and wherein one R7 can combine with a H on the shared carbon atom to form C3-C6 cycloalkyl;
each R8 and R9 is independently H or straight chained or branched Ci-C7 alkyl optionally substituted with one or more fluorine;
wherein R J is a nitrogen containing heterocyclic moiety optionally substituted with one or more halogen, -COR12, a S or O containing heterocyclic ring, or .straight chained or branched Ci -C7 alkyl optionally substituted with halogen or -OCH-?;
R1 1 is C3-C6 cycloalkyl, straight chained or branched alkoxy-CrCβ cycloalkyl, straight chained or branched C1-C4 aikyl/alkoxy, phenyl, napthyl, 5 to 6-membcred heteroaryl, benzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC4 alkyl optionally substituted with F;
R " is straight chained or branched C]-C7 alkyl or Ci-C6 cycloalkyl, where each of which is optionally substituted with one or more halogen or methoxy;
m is an integer from 1 to 4 inclusive; and n is independently an integer from 0 to 4 inclusive, or a pharmaceutically acceptable salt thereof.
In another embodiment, R1 is-(CHR7)mN(R8)(R9).
In yet another embodiment, R1 is-(CHR7)nR10.
In one embodiment, each R4 and R5 is independently H or straight chained or branched C1-C4 alkyl.
ϊn one embodiment, R10 is piperazinyl or morpholinyl, where each of which is optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C4 alkyl optionally substituted with halogen.
In one embodiment, R10 is piperidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C4 alkyl optionally substituted with halogen.
In one embodiment, R10 is pyrrolidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C4 alkyl optionally substituted with halogen.
In one embodiment, R10 is azetidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C4 alkyl optionally substituted with halogen.
In one embodiment, R10 is piperazinyl or morpholinyl, where each of which is optionally substituted with straight chained or branched C1-C4 alkyl optionally substituted with halogen.
In one embodiment, R10 is piperidinyl optionally straight chained or branched C1-C4 alkyl optionally substituted with fluorine.
In one embodiment, RK) is pyrrolidinyl optionally straight chained or branched C1-C4 alky] optionally substituted with fluorine,
In one embodiment, R10 is azetidinyl optionally straight chained or branched Ci-C4 alkyl optionally substituted with fluorine.
In one embodiment, R7 is H; and R4 and R5 is independently H, or straight chained or branched C1-C4 alkyl.
In one embodiment, n is 0 and R2 is H, methyl or ethyl.
In one embodiment, n is 1 or 2.
In one embodiment, R1 ' is C3-C6 cycloalkyl or alkoxy-C3-Cc, cycloalkyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C1- C4 alkyl optionally substituted with F.
In one embodiment, R1 ' is phenyl or napthyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C1-C4 alkyl optionally substituted with F.
In one embodiment, Rπ is a 5 to 6-membered heteroaryl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC4 alkyl optionally substituted with F.
In one embodiment, R." is bcnzothiophcnyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched C1-C4 alkyl optionally substituted with F.
In one embodiment, Rs ' is phenyl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Cj-C4 alkyl optionally substituted with F.
In one embodiment, R1 1 is napthy! optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Ci-C4 alkyl optionally substituted with F.
In one embodiment, the compound falls under formula Ia.
In another embodiment, the compound falls under formula Ib.
In one embodiment, B1 is CH. In a separate embodiment, B1 is N.
In one embodiment, B" is CH. In a separate embodiment, B" is N,
In one embodiment, B1 is CH. In a separate embodiment, B3 is N.
In one embodiment, B1 is CH and B3 is CH. In a separate embodiment, B1 is CH, B1 is N and
B3 is CH.
In a separate embodiment, each R4, R5and R6 is independently H, halogen or straight chained or branched Ci-C4 alkyl; and wherein each R8 and R9 is independently H or straight chained or branched Cj-C4 alkyl.
In yet another embodiment, R10 is azetidinyl, cyclopropyl, cyclobutyl, cyclopenyl or cyciohcxyl, wherein the azetidinyl, cyclopropyl, cyclobutyl, cyclopenyl or cyclohexyl is
optionally substituted with with one or more halogen or straight chained or branched Ci-C4 alkyl.
In one embodiment, R3 is -X(CH2)PYR' '
In one embodiment, R3 is -X(CHCH)YR1 '.
In one embodiment, R2 is H or straight chained or branched Ci-C4 alkyl.
In one embodiment, X is CH2 or O; and p is 1 or 2.
In one embodiment, X is S; and p is 1 or 2.
In one embodiment, R1 1 is is C3-C6 cycloalkyl or phenyl, where the phenyl can be optionally substituted with F, Cl, Br, or straight chained or branched Cj-C4 alkyl optionally substituted with F.
In one embodiment, R1 ' is is C3-C6 cycloalkyl
In one embodiment, R1 ' is phenyl optionally substituted with F, CI, Br, or straight chained or branched CpC4 alkyl optionally substituted with F.
In one embodiment, R l ' is heteroaryl optionally substituted with F, Cl, Br, or straight chained or branched Ci -C4 alkyl optionally substituted with F.
The subject invention is also directed to a compound selected from the group consisting of A-
Benzyloxy- 1 -[3-( 1 -methyl-azetidin-3-yI)- 1 H-indol-6-yl]- lH-pyridin-2-one, 4-(4-Fluoro- phenoxymethyl)- 1 -[3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H-pyridin-2-one, 4-(4-
Chloro-benzyloxy)- 1 -[3-( 1 -methyl-azetidin-3-yl)- 1 H~indol~6~yl]~ 1 H-pyridin-2-one, A- Benzyloxy- 1 -[ 1 -methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H-pyridin-2-one, 4-(4- Fluoro-phenylsυlfanyimethyl)- 1 -[3-( 1 -methyl -azetidin-3-yl)- lH-indol-6-yl] - 1 H-pyridin-2-one and 4-[(E)-2-(4-fluoro-phcnyl)-vinyI]- 1 -[3-(I -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one.
The subject invention is also directed to a compound selected from the group consisting of 4- (4-chIoro-phenyl)- 1 -[3~( 1 -methyl-azetidin-3-yI)- 1 h-indol-6-yl]- lh-pyridin-2-one; 4-(4-chloro- phenyl)- 1 - { 3-[ 1 -(2-fluoro-ethyl)-azctidin-3-yl]- 1 h-indol-6-yl } - 1 h-pyridin-2-one; 4~(4~chloπ> phenyl)- 1 - { 3-[ 1 -(2-methoxy-cthyl)-azetidin-3-yl]- 1 h-indol-ό-yl } - lh-pyridin-2-one; 4-(4- chloro-phenyl)- 1 -[ 1 -methyl-3-( l-methyl-azetidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 5- chloro- 1 '-[3-(I -isopropyl-azetidin-3-yl)- 1 h-indol-6-yl]- lih-[2)4']bipyridinyl-21-one; I 1- { 3-[ 1 -(2- fluoro-ethyl)-azetidin-3-yl]- l h-indol-6-yl }-6-tπfluoromethyl-rh-[3,4']bipyridinyi-2'-one; A- (2-fluoro-4-trifIuoromethyl-phenyl)- 1 -[3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin- 2-one; 4-(2-fluoro-4-trifluoromcthyl-phenyl)- 1 h-pyridin-2-one; 4-[4-( 1 -hydroxy- 1 -melhyl- ethyl)-phenylj- 1 - [3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 6-(4-chloro- phenyl)-3-l3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-yl] ~3h-pyrimidin-4-one; I i-[2-methyl-3-( 1 - methyl-azetidin-3»yl)»lh»indol-6-yl]-6-trifluoiOmethyl-rh-[3,4']bipyridinyl-2'-one; 4- bcnzo[b]thiophen-2-yl-l-[3-(l-methyI-azetidin-3-yl)- l h-indoI-6-yIJ- lh-pyridin-2-one; 1 ~[3~ ( 1 -methyl-azetidin-3-yl)- lh-indol-6-yl]-4-( 1 -methyl- 1 h-indol-5-yl)- 1 h-pyridin-2-one; 1 -13-( 1 - methyl-azetidin-3-yl)- l h-indoI-6-yI]-4-naphthalcn-2-yl-I h-pyridin-2-one; 4-(5-fIuoro- benzooxazol-2-yl)- 1 -[3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 4~(5~ethyl- pyrimidin-2-yl)- 1 - { 3-f 1 -(2-fluoro-ethyl)-azetidin-3-yl] - 1 h-indol-6-yl } - 1 h~pyridin-2-one; 5- chloro- 1 '-{ 3-[ 1 -(2-fluoiO-ethyl)-azetidin-3-ylj- 1 -methyl- 1 h-indol-6-yl } - 1 'h-[ 2,4']bipyridinyi- 2'-one; 1 -[3-(I -methyl-azctidin-3-yl)- 1 h-indol-6-yl]-4-(4-trifluoromethoxy-ρhcnyl)- 1 h- pyridin-2-one; 4-(4-chloro-phenyl)- 1 - { 3-[ 1 -(telrahydro-thiophen-3-yl)~azetidin-3-yl]- 1 h- indol-6-yl } - 1 h-pyridin-2-one; 4-(4-chIoro-phenyl)~ 1 ~[3~( 1 -oxetan-3-yl-azetidin-3-yl)- 1 h- indol-6-yl]- 1 h-pyridin-2-one; 4~(4~chloro-phenyl)- 1 -[3-( 1 -ethyl-azetidin-3-yl)- 1 -(2-hydroxy- ethyl)- 1 h~indol~6~yl]» 1 h-pyridin-2-one; 4-(4-chloro-phenyl)- 1 - { 3-[ 1 -(3 ,3,3-lrifIuoro- propionyl)-azetidin-3-yl]- 1 h-indol-6-yl } - 1 h-pyridin-2-one; 1 ~[ 1 ~(2-fluoiO-ethyi)-3-( 1 -methyl- azetidin-3-yl)- 1 h-indol-6-yl]-4-(2,2,2-trifluoro-ethoxy)- 1 h-pyridin-2-onc; 1 -f 1 -methyl-3~( 1 - melhyl-a/eiidin~3-yl)-l h-indol-6-yl]-4-(2,212-trifluoiO-ethoxy)- lh-pyridin-2-one; 5-chforø-l1- [3-( 1 ~cthyl-azctidin-3-yl)- 1 -(2-fluoro-ethyl)- 1 h-indol-6-yl]- 1 'h-[2,4']bipyridinyl-2'-one; and 5- Chloro- 1 '-[3-( 1 -ethyl-azetidin-3-yl)- 1 -methyl- 1 H-indol-6-yl]- 1 Η~[2,4']bipyridinyl-2'-one.
The subject invention is also directed to a compound selected from the group consisting of3- [3-(l -methyl-azetidin-3-yl)- lh-indol-6-yl]-6-(4-trifluoromethyl-phenyl)-3h-pyrimidin-4-one;
1 -{ 3-[ 1 -(2-fluoro-cthyl)~azetidin-3~yI]- 1 -methyl- 1 h~indol~6~yl } -4-(4-fluoro-phenyI)- 1 h- pyridin-2-one; 5-fluoro- 1 '- { 3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yl]- 1 -methyl- 1 h-indol-6-yI } - l'h- [2,4'lbipyridinyl-2'-onc; 1'- { 3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yl]- 1 -methyl- 1 h-indol-6-yl } -5- trifluoromethyl- 1 'h~[2,4']bipyridinyl~2'~one; 1 '-[ 1 -mcthyl-3-( 1 ~methyl-azetidin~3-yl)- lh-indol- 6-yl]-5-trifluoromethyl- 1 'h-[2,4']bipyπdinyl-2'-one; T- { 3-[ 1 -(2-Ωuoro-ethy])-azetidin-3-yl]- 1 - methyl- 1 h-indol-6-yl } -6-trifluoromethyI- 1 'h-[3,4']bipyridinyl-21-one; 6-(4-chloro-phenyl)-3- { 3-[ 1 -(2-fluoro-ethyl)-azetidin-3-ylj- 1 -methyl- 1 h-indol-6-yl } -3h-pyrimidin-4-one; 6-(4- chloiO-phcnyl)-3-[2-methyl-3-(l-methyl-azctidin-3-yl)- lh-indoi-6-yi]-3h-pyrimidin-4-one; 4- (4-chloro-phenyl)- 1 - { 1 -methyl-3-f 1 -(tetrahydro-furan-3-yl)-azctidin-3-yl]- 1 h-indol-6-yl } - 1 h- pyridin-2-one; 5-chloro- 1 '-[3-( 1 -cyclopropyhnethyl-azelidin-S-yl)- 1 -methyl- 1 h-indol-6-yl]- ] 'h~[2,4']bipyridinyl~2r~one; 5-chloro~ 1 '-[3-( 1 -isopropyl-azctidin-3-yl)- 1 -methyl- 1 h-indol-6- yl j- 1 'h-[2,4'lbipyridinyl-2'-one; 1 -L I -methyl-3-( 1 -methyl-azctidin-3-yl)- 1 h-indol-6-yl]-4-(4- trifluoromethyl-phenyl)-lh-pyridin-2-one; ] -[3-(l-acetyl-azetidin-3-yl)-l-methyl- lh-indol-6- yl]-4-(4-tπfluoi'omethyl~phenyl)- 1 h-pyridin-2-one; 6-(4~chloπ>phenyl)-3- { 3-[ 1 -(tetrahydro- furan-3-ylmethyl)-azetidin-3-ylJ-lh-indoi-6-yl }-3h-pyrimidin-4-onc; and 4-(4-fluoro- phenyl)- 1 -[3-(I -isopropyl-azetidin-3-yl)- 1 -methyl- 1 h-indol-6-yl]- 1 h-pyridin-2-one.
The present invention is also directed to a pharmaceutical composition comprising a compound of the invention and an acceptable pharmaceutical carrier,
Additionally, the present invention is also directed to a method of treating mood disorders in a subject comprising administering a therapeutically effective amount of a compound of the invention. The present invention is also directed to method of treating anxiety in a subject comprising administering a therapeutically effective amount of a compound of the invention, Moreover, the present invention is also directed to a method of treating obesity in a subject comprising administering a therapeutically effective amount of a compound of the invention. The present invention is also directed to method of treating urinary disorders in a subject comprising administering a therapeutically effective amount of a compound of the invention,
In separate embodiment, the invention is directed to uses of a compound of the invention for the manufacture of a pharmaceutical composition for treating a disorder selected from the group consisting of mood, anxiety and obesity related disorders.
Pharmaceutically Acceptable Salts
The present invention also comprises salts of the present compounds, typically, pharmaceutically acceptable salts. Such salts include pharmaceutically acceptable acid addition salts. Acid addition salts include salts of inorganic acids as well as organic acids.
Representative examples of suitable inorganic acids include hydrochloric, hydrobromic, hydroiodic, phosphoric, sulfuric, sulfamic, nitric acids and the like. Representative examples of suitable organic acids include formic, acetic, trichloroacetic, trifluoroacetic, propionic, benzoic, cinnamic, citric, fumaric, glycolic, itaconic, lactic, methanesulfonic, malcic, malic, malonic, mandelic, oxalic, pyruvic, salicylic, succinic, methane sulfonic, elhanesulfonic, tartaric, ascorbic, pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic, aspartic, stearic, palmitic, EDTA, glycoiic, p-aminobenzoic, glutamic, bcnzenesulfonic, p- toluenesulfonic acids, theophylline acetic acids, as well as the 8-halotheophyllines, for example 8-bromolheophyllinc and the like. Further examples of pharmaceutically acceptable inorganic or organic acid addition salts include the pharmaceutically acceptable salts listed in S. M. Berge, et al., J. Pharw, Set,, 1977, 66, 2 and Paulekuhn, el al. J. Med. Chem. 2007 (December online publication), the contents of all which are hereby incorporated by reference.
Furthermore, the compounds of this invention may exist in unsolvated as well as in solvatcd forms with pharmaceutically acceptable solvents such as water, ethanol and the like.
Racemic forms may be resolved into the optical antipodes by known methods, for example, by separation of diastcreomeric salts thereof with an optically active acid, and liberating the optically active amine compound by treatment with a base. Separation of such diastereomeric salts can be achieved, e.g. by fractional crystallization. The optically active acids suitable for this purpose may include, but are not limited to d- or 1- tartaric, mandelic or camphorsulfonic acids. Another method for resolving racematcs into the optical antipodes is based upon chromatography on an optically active matrix. The compounds of the present invention may also be resolved by the formation and chromatographic separation of diastereomeric derivatives from chiral derivatizing reagents, such as, e.g., chiral alkylating or acylating
reagents, followed by cleavage of the chiral auxiliary. Any of the above methods may be applied either to resolve the optical antipodes of the compounds of the invention per se or to resolve the optical antipodes of synthetic intermediates, which can then be converted by methods described herein into the optically resolved final products which are the compound of the invention.
Additional methods for the resolution of optical isomers, known to those skilled in the art, may be used. Such methods include those discussed by J. Jaques, A. Collet and S. Wilcn in Enantiomers, Racemates, and Resolutions, John Wiley and Sons, New York 1981. Optically active compounds may also be prepared from optically active starting materials.
Pharmaceutical compositions
The present invention further provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention and a pharmaceutically acceptable carrier. The present invention also provides a pharmaceutical composition comprising a therapeutically effective amount of one of the specific compounds disclosed in the Experimental Section and a pharmaceutically acceptable carrier.
The compounds of the invention may be administered alone or in combination with pharmaceutically acceptable carriers or excipients, in either single or multiple doses. The pharmaceutical compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19lh Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
The pharmaceutical compositions may be specifically formulated for administration by any suitable route such as oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) routes, ϊt will be appreciated that the route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient.
Pharmaceutical compositions for oral administration include solid dosage forms such as capsules, tablets, dragees, pills, lozenges, powders and granules. Where appropriate, the compositions may be prepared with coatings such as enteric coatings or they may be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art. Liquid dosage forms for oral administration include solutions, emulsions, suspensions, syrups and elixirs.
Pharmaceutical compositions for parenteral administration include sterile aqueous and nonaqueous injectable solutions, dispersions, suspensions or emulsions as well as sterile powders to be reconstituted in sterile injectable solutions or dispersions prior to use.
Other suitable administration forms include, but arc not limited to, suppositories, sprays, ointments, creams, gels, inhalants, dermal patches and implants.
Typical oral dosages range from about 0.001 to about 100 mg/kg body weight per day. Typical oral dosages also range from about 0.01 to about 50 mg/kg body weight per day. Typical oral dosages further range from about 0.05 to about 10 mg/kg body weight per day. Oral dosages are usually administered in one or more dosages, typically, one Io three dosages per day. The exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art.
The formulations may also be presented in a unit dosage form by methods known to those skilled in the art. For illustrative purposes, a typical unit dosage form for oral administration may contain from about 0.01 to about 1000 mg, from about 0.05 to about 500 mg, or from about 0.5 mg to about 200 mg.
For parenteral routes such as intravenous, intrathecal, intramuscular and similar administration, typical doses arc in the order of half the dose employed for oral administration.
The present invention also provides a process for making a pharmaceutical composition comprising admixing a therapeutically effective amount of a compound of Formula the invention and a pharmaceutically acceptable carrier, In an embodiment of the present invention, the compound utilized in the aforementioned process is one of the specific compounds disclosed in the Experimental Section.
The compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof. One example is an acid addition salt of a compound having the utility of a free base. When a compound of the invention contains a free base such salts are prepared in a conventional manner by treating a solution or suspension of a free base of Formula Ia or Ib with a molar equivalent of a pharmaceutically acceptable acid. Representative examples of suitable organic and inorganic acids arc described above.
For parenteral administration, solutions of the compounds of the invention in sterile aqueous solution, aqueous propylene glycol, aqueous vitamin E or sesame or peanut oil may be employed. Such aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose. The aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration. The compounds of the invention may be readily incorporated into known sterile aqueous media using standard techniques known to those skilled in the art.
Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solutions and various organic solvents. Examples of solid carriers include lactose, terra alba, sucrose, cyclodextrin, talc, gelatin, agar, pectin, acacia, magnesium slearatc, stearic acid and lower alkyl ethers of cellulose. Examples of liquid carriers include, but are not limited to, syrup, peanut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water. Similarly, the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostcarate or glyceryl distearate, alone or mixed with a wax. The pharmaceutical compositions formed by combining the compounds of the invention and a pharmaceutically acceptable carrier arc then readily administered in a variety of dosage forms suitable for the disclosed routes of administration. The formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and optionally a suitable excipient. Furthermore, the orally available formulations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or waier-in-oil liquid emulsion.
If a solid carrier is used for oral administration, the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it may be in the form of a troche or lozenge. The amount of solid carrier will vary widely but will range from about 25 mg to about 1 g per dosage unit.
If a liquid carrier is used, the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
Treatment of Disorders
As mentioned above, the compounds of Formula Ia and Ib bind to the MCHl receptor. Accordingly, the present invention provides a method of treating depression in a subject which comprises administering to the subject a therapeutically effective amount of a compound of this invention. This invention further provides a method of treating a subject suffering from anxiety which comprises administering to the subject a therapeutically effective amount of a compound of this invention,
This invention also provides a method of treating a subject suffering from obesity which comprises administering to the subject a therapeutically effective amount of a compound of this invention. The obesity indication includes in particular exogenic obesity, hypeiϊnsulinaemic obesity, hyperplasmic obesity, hyperphyseal adiposity, hypoplasmic obesity, hypothyroid obesity, hypothalamic obesity, symptomatic obesity, infantile obesity, upper body obesity, alimentary obesity, hypogonadal obesity, central obesity. This range of indications also includes cachexia, anorexia and hyperphagia. The treatment of obesity is expected to weight loss in a subject.
This invention also provides a method of treating a subject suffering from urinary disorders which comprises administering to the subject a therapeutically effective amount of a compound of this invention. Another range of indications for which the compounds according to the invention are advantageously suitable is the prevention and/or treatment of micturition disorders, such as for example urinary incontinence, hyperactive bladder, urgency, nycturia, enuresis, while the hyperactive bladder and urgency may or may not be connected with benign prostatic hyperplasia. In an embodiment of this invention, the subject is a human being.
EXPERIMENTAL SECTION
General Methods: AU reactions were performed under a nitrogen atmosphere and the reagents, neat or in appropriate solvents, were transferred to the reaction vessel via syringe and cannula techniques. Anhydrous solvents were purchased from Aldrich Chemical Company and used as received. The NMR spectra were recorded at Bruker Avance (400 MHz) or GE QEPIus300 in CDCi3, McOH-d4 or DMSO-dό as solvent with iclramethylsilane as the internal standard unless otherwise noted. Chemical shifts (5) are expressed in ppm, coupling constants (J) are expressed in Hz, and splitting patterns are described as follows: s = singlet; d = doublet; t = triplet; q = quartet; quintet; sextet; septet; br = broad; m = multiplet; dd = doublet of doublets; dt = doublet of triplets; td = triplet of doublets; dm = doublet of multiplets; ddd = doublet of doublet of doublets. Unless otherwise noted, mass spectra were obtained using clectrospray ionization (ESMS, Micromass Platform II or Quattro Micro) and (M+H)+ is reported. Thin-layer chromatography (TLC) was carried out on glass plates pre- coated with silica gel 60 F254 (0.25 mm, EM Separations Tech.), Preparative TLC was carried out on glass sheets pre-coated with silica gel GF (2 mm, Analtech). Flash column chromatography was performed on Merck silica gel 60 (230-400 mesh).
Source of chemicals Most of the reagents used in the experimental section were purchased from Aldrich-Sigma. 1- Boc-3~(hydroxy)azetidine was purchased from CHN Technologies Inc. (2-Bromo-pyridin-4- yl)-methanol was purchased from Alfa Aesar.
Scheme 1

Ia
(a) CuI/ K2CO3/ trans-N,N'-Dimethyl-cyclohexane-l,2-diamine/ DMF/ about 100 0C, about 18 h
The compounds of Formula Ia may be synthesized according to the procedures described in Scheme 1. The compounds of Formula II and III are commercially available or may be synthesized by those skilled in the art. For example, the compounds of Formula II may be synthesized according to the procedures described in Scheme 14, 15, and 16. The compounds of Formula III may be synthesized according to the procedures described in Scheme 6, 7, 8, 9, 10, 1 1, 12, and 13. Copper-catalyzed coupling reaction of compounds II with compounds III in the presence of copper iodide, potassium carbonate and trans-N,N'-DimethyI- cyclohexane- 1 ,2-di amine in DMF at about 1000C affords the compounds of Formula Ia.
For representative reaction conditions in connection with copper-catalyzed coupling see S. L. Buchwald et al, Org. Letters 2007, 9 (4), 643-646 and references cited therein.
Scheme 2

The compounds of Formula Ia may be prepared according to the procedures described in Scheme 2. The compounds of Formula V may be synthesized via dehydroxylation of alcohols of Formula IV with TFA and lriethyl silane in DCM at about 0 0C for about 10 min. Pyridinoncs of Formula III arc coupled with compound V in the presence of copper iodide, potassium carbonate and trans-N,N'-dmiethyl-cyclohexane-l,2-diamine in DMF at about 100 0C for about 18 h, followed by dcprotection of N-Boc in the presence of TFA, to afford the compound VI. The reductive amination of compound VI with aldehydes (or ketones) or the alkylation with alkyl halides affords the compounds of Formula Ia.
Scheme 3

(a) alkyl halides/ 18-Crown-6/ KO'Bu/ THF.
The compounds of Formula Ia may be prepared according to the procedures described in Scheme 3. The compounds of Formula Ia (R2≠H) are prepared from Ia (R2=H) via reaction of Ia and alkyl halides in the presence of potasium fert-butoxidc and 18-crown~6-cther in THF.
Scheme 4

Ia
(a) K2CO3/ PdCl2(dppf)2/ DMF/ about 80 0C, about 12 h (b) [Cp+IrCl2I2/ KOH/ DCM/ 1 10
0C, about 24 h The compounds of Formula Ia may be prepared according to the procedures described in Scheme 4. The compounds of Formula VIII are prepared via Suzuki reactions of pyridinones of Formula III and indole VII in the presence of palladium chloride, base and a suitable ligand in DMF at about 80 0C for about 12 h. The compounds of Formula Ia are prepared via iridium-catalyzed reactions of indoles of Formula VIII and alcohols IX in the presence of [Cp*IrCl2]2 and potassium hydroxide in DCM in sealed tube at about 110 0C for about 24 h. For reaction conditions in connection with the iridium-catalyzed reactions, sec R. Grigg et al., Org. Letters 2007, 9 (17), 3299-3302 and references cited therein.
Scheme 5

X X! XiI XIII
X = O1 S XIV
X = O, S

(a) PPh3/ DBAD/ THF/ rt, about 24 h (b) mCPBA/ DCM/ rl, about Ih (c) POCIV toluene/ about 90 0C, about 12 h (d) AcOH/ H2O/ about 90 0C, about 24 h (e) K2CO3/ Pd(PPh^)4/
DMF/ about 60-80 0C, about 12 h
The compounds of Formula Ia may be prepared according to the procedures described in Scheme 5. Mitsunobu reaction of compounds of Formula X and phenols of Formula XI in the presence of triphenylphosphine and DBAD in THF affords ethers or thio ethers of Formula XII. Pyridine-N-oxidcs of Formula XIII can be prepared via oxidation of XII with mCPBA in DCM at room temperature. The compounds of Formula XIV are prepared via reaction of pyridine-N-oxidc of Formula XIII with POCl3 in toluene at about 90 0C for about 12 h. The pyridinoncs of Formula XV are synthesized from hydrolysis of XIV in acetic acid and water at about 90 0C for about 24 h. The compounds of Formula Ia are prepared via Suzuki coupling reactions of XV and XVI in the presence of palladium, base and a suitable ligand in DMF at about 80 0C for about 12 h.
Scheme 6

XViI XVIl! XiX ill
X = O1 S
(a) TDA-I/ KOH/ K2CO3/ toluene/ reflux, about 3 h (b) Ac2O/ about 140 0C, overnight
The compounds of Formula III may be prepared according to the procedures described in Scheme 6. The compounds of Formula XIX are synthesized via nucleophilic substitution of 4-chloropyridine N-oxide of Formula XVIII with alcohols or thio alcohols of Formula XVII in the presence of tris[2-(2-melhoxycthoxy)-ιthyl]amine (TDA-I), potassium hydroxide and potassium carbonate in toluene under reflux for about 3 h. Pyridinones of Formula III are prepared via reaction of compounds XIX in acetic anhydride at about 140 0C overnight. For representative general reactions in connection with synthesis of pyridinones see Parra, M. et ul, Current Org. Chem. 2005, 9, 1757- 1779 and references cited therein.
Scheme 7

XX Xl XXI III W = CL Br O1 S
(a) PPh3/ DBAD/ THF/ it, about 2 h (b) AcOH/ H2O/ about 120 0C, about 12 h
Alternatively, the compounds of Formula III may be synthesized according to the procedures described in Scheme 7. The compounds of Formula XX, which are commercially available or synthesized by those skilled in the art, are treated with alcohols or thio alcohols XI in the presence of triphenylphosphine and DBAD in THF at room temperature for about 2 h to afford ethers or thio ethers of Formula XXI. The compounds of Formula XXI are treated with acetic acid in water at about 120 0C for about 12 h to afford pyridinones of Formula HI.
Scheme 8

XX XXII Xl
W = CI1 Br X =^ O1 S
(a) MsCl/ TEA/ DCM/ rt, about 12 h (b) Cs2CO3/ MeCN/ rt, about 16 h
Alternatively, the compounds of Formula III may be prepared according to the procedures described in Scheme 8. The mesylates of Formula XXII are synthesized from reaction of alcohols of Formula XX and mesyl chloride in the presence of base. The compounds of Formula III are prepared via substitution reaction of mesylates of Formula XXII with alcohols or thio alcohols XI in the presence of base such as cesium carbonate.
Scheme 9

XXIiI XVIiI XXIV IN
R3 = aryl, heteroaryl
(a) Pd(PPh3V Na2CO3/ 1 ,2-dimclhoxy ethane-water/ reflux, about 16 h (b) Ac2O/ about 140 0C, overnight
Alternatively, the compounds of Formula III may be prepared according to the procedures described in Scheme 9. The compounds of Formula XXIV arc synthesized via Suzuki reaction of 4-chloropyridinc N-oxide XVIII with aryl boronic acid or aryl boronic ester of Formula XXIII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPh-^)4 and Na2CO3 in 1 ,2-dimelhoxy ethane/water under reflux for about 16 h. Pyridinones of Formula III are prepared via reaction of compounds XXIV in acetic anhydride at about 1400C overnight.
Scheme 10

XXV XXIII XXVI Ii!
W = F1 Cl R3 = aryS, heteroaryl
(a) (PPh-,)4/ Na2COV 1,2-dimethoxy ethane-water/ reflux, about 16 h. (b) AcOH/ about 1 10
C, overnight
The compounds of Formula IH may be prepared according to the procedures described in Scheme 10. The compounds of Formula XXV can be treated with aryl boronic acid or aryl boronic ester XXIII, which are commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPlIs)4 and Na2CO-? in 1,2-dimethoxy ethane under reflux for about 16 h to afford the compounds of Formula XXVI. The compounds of Formula XXVI can be treated with acetic acid at about 110 0C for overnight to afford pyridinones of Formula III.
Scheme 11

XXVl! XXVIIf XXVI
W = F, Cl V = CI1 Br , 1 R3 = aryl, heteroaryl
(a) (PPh3)4/ Na2CO3/ 1 ,2-dimethoxy ethane-water/ reflux, about 16 h (b) AcOH/ about 1 10 0C, overnight
The compounds of Formula III may be prepared according to the procedures described in Scheme 11. The compounds of Formula XXVIII, which are commercially available or synthesized by those skilled in the art, are treated with pyridinyl boronic acid or pyridinyl boronic ester XXVII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPh3)4 and Na2CO3 in 1,2-dimethoxy ethane under reflux for
about 16 h to afford Formula XXVI. The compounds of Formula XXVI are treated with acetic acid at about 1 10 0C for overnight to afford pyridinones of Formula HI.
Scheme 12

R3 = aryi, heteroaryl B2 = N (a) (PPh^)4/ Na2CO3/ 3 ,2-dimethoxy ethane-water/ reflux, about 16 h
The compounds of Formula III may be prepared according to the procedures described in Scheme 12. The compounds of Formula XXIX, which are commercially available or may be synthesized by those skilled in the art, can be treated with aryl boronic acid or aryi boronic ester XXIII, which is commercially available or may be synthesized by those skilled in the art, in the presence of Pd(PPhO4 and Na2CO3 in 1 ,2-dimethoxy ethane under reflux for 16 h to afford pyridinones of Formula III.
Scheme 13

The compounds of pyridinone XXXII may be prepared according to the procedures described in Scheme 13. The compounds of Formula XXXII are prepared via Wittig reaction of aldehydes XXX and phosphonates XXXI in the presence of potasium før/~butoxide in THF at room temperature for about 4 h.
Scheme 14

XXXIIi XXXIV VII

(a) SO3 Py/ DMSO/ TEA/ DCM/ about 10-20 0C, about 1.5 h (b) KOH/ MeOH/ about 45-50 0C, about 6 h (c) LAH/ THF/ reflux, about 6 h
The compounds of Formula XXXV may be synthesized according to the procedures described in Scheme 14. The compounds of Formula XXXIII and VII are commercially available or may be synthesized by those skilled in the art. In summary, the azetidinone of Formula XXXIV is prepared by reaction of
 XXXIII with sulphur trioxide pyridine complex in the presence of triethylamine in DMSO and DCM at about 10 0C for about 30 min and at about 20 0C for about 1 h. l-(3~Indolyl)azetidin-3~ol IV is prepared via condensation of azetidone XXXIV with indoles of Formula VII in the presence of potassium hydroxide in methanol at about 45-500C for about 6 h. The compound XXXV is prepared via reduction of l-(3-indolyl)azetidin»3-ol IV with LAH in THF under reflux for about 6 h.
XXXIII with sulphur trioxide pyridine complex in the presence of triethylamine in DMSO and DCM at about 10 0C for about 30 min and at about 20 0C for about 1 h. l-(3~Indolyl)azetidin-3~ol IV is prepared via condensation of azetidone XXXIV with indoles of Formula VII in the presence of potassium hydroxide in methanol at about 45-500C for about 6 h. The compound XXXV is prepared via reduction of l-(3-indolyl)azetidin»3-ol IV with LAH in THF under reflux for about 6 h.
Scheme 15

VII VII VII VII Ra=RS=H R^=PhSO2, R5=H R2=PhSO2, R5=/=H R2=H,R5=/=H
(a) NaH/ sulfonyl chloride/ DMF/ about 20 0C, about 18 h (b) LDA/ halides/ THF/ about -78 0C to about 20 0C, about 18 h (c) KOH/ MeOH/ reflux, about 18 h
The substituted indoles VII (R"=H, R' ≠H) may be synthesized according to the procedures described in Scheme 15. The protected indoles of Formula VII (R2=PhSO2, R5=H) are prepared via reaction of VII (R"= R =H) with benzeiiesulfonyl chloride in the presence of sodium hydride in DMF at room temperature for about 38 h. The substituted indoles VII
(R2= PhSO2, R5≠H) are prepared via reaction of VII (R2-PhSO2, R5 ^H) with halides in the presence of LDA in THF at about -78 0C for about 1 h and at about 20 0C for about 18 h. The unprotected indoles VII (R2=H, R5≠H) is prepared via reaction of VII (R2= PhSO2, R5≠H) in the presence of potassium hydroxide in methanol at reflux for 18 h.
Scheme 16

The substituted indoles VII may be synthesized according to the procedures described in
Scheme 16. The compounds VII are prepared via reaction of XXXVI with vinyl magnesium bromide in THF at about -40 0C for about 30 min.
Preparation of intermediates Representative intermediate compounds were synthesized as follows:
3-(6-Bromo-l H-indol-3-yl)-azetidine-l-carboxylic acid tert-buiyl ester: To a suspension of 3-(6-bromo-lH-indol-3-yl)-3-hydroxy-azetidirie- l-carboxylic acid /erf- butyl ester (1Og, 27.2 mmol) in DCM (50 mL) was added triethylsilane (40 mL, 200 mmol) at O0C, followed by addition of trifluoroacetic acid (7 mL, 90 mmol) dropwise until the suspension disappeared. The reaction was maintained at O0C for 10 min and poured into sat. NaHCO^. The mixture was extracted with DCM three times. The combined organic phase was washed with brine, dried with MgSO4, and concentrated in vacuo to afford 3~(6~bromo-l H- indol-3-yl)-azelidine- l -carboxylic acid tcrl-bulyl ester (9.78g, 100%). ESMS m/e: 352.9 (M+H)+.
3- { 6-[4-(4-Chloro-ρhcnyl)-2-oxo-2H-pyridin- 1 -yll- 1 H-indol-3-yl }-azetidine- 1-carboxylic acid tert-butyl ester:
To a stirred mixture of 3-(6-bromo-l H~indol~3~yl)~azetidinc- 1-carboxylic acid tert-butyl ester (1.70 g, 4.84 mmol), 4-(4-chloro-phenyl)- lH-pyridin-2-one (1.0 g, 4.86 mmol), K2CO3 (1.50 g, 10.85 mmol) and trans N,N'-dimethyiaminocyclohexane-l ,2-dimanie (0.38 g, 2.67 mmol) in DMF (25.0 niL) was added copper (I) iodide (0.51 g, 2.67 mmol) at room temperature. The resulting green color mixture was heated at 90 0C for overnight. After heating at 9O0C overnight, the reaction was cool to room temperature and quenched with drops of water. Solvent DMF was removed using high vacuum to yield the crude product. The residue was dissolved in DCM (300 mL) and washed successively with a saturated solution of EDTA, water, and brine. Solvents were concentrated to yield brown color solid. The brown color solid was redissolved in DCM (50 mL) and triturated with diethyl ether (- 100 mL) to yield the desired compound as a brown color solid (1.80 g, 78%). ESMS m/e: 475.9 (M+H)+.
1 -(3-Azelidin-3-yl- 1 H-indol-6-yl)-4-(4-chloro-phenyl)- 1 H-ρyridin-2-one-2HCl:
To a stirred solution of 3-{ 6-[4-(4-chloro-phcnyl)-2-oxo-2H-pyridin-l-ylJ-l H-indol-3-yl }- azetidine- 1-carboxylic acid tert-butyl ester (1.70 g, 3.57 mmol) in DCM (25.0 mL) was added 2M solution of HCl in diethyl ether (0.52 g, 14.28 mmol) at room temperature under nitrogen atmosphere. After stirring Ih at room temperature, the solid was filtered and washed with DCM. The crude product was redissolved in methanol ( 15 mL) and triturated with diethyl ether to afford the desired product as its hydrochloride salt (1.50 g, 94%), ESMS m/e 375.9 (M+H)+.
4-(4-Chloro-benzyloxy)-pyπdine-N-oxide: To a suspension of powdered potassium hydroxide (7.60 g, 136 mmol) and potassium carbonate (4.70 g, 34.1 mmol) in dry toluene were added (4-chloro-phenyl)-methanol (7.26 g, 50.9 mmol), 4-chloropyridinc-N-oxide (4.39 g, 34.0 mmol) and TDA-I (1.10 mL, 3.44 mmol). After the mixture was heated at reflux for 3 hours, the organic layer was separated and dried over anhydrous sodium sulfate. The solvent was removed in vacuo and the residue was purified through a silica gel column to give 4-(4-chloro-benzyloxy)-pyridine-N-oxide in
40% yield as light yellowish solid. 1H NMR (CDCl1) 5: 8.13 (m, 2H), 7.37 (m, 4H), 6.85 (m, 2H), 5.06 (s, 2H); ESMS m/e: 235.9 (M+H)+.
4-(4-Chloro-benzyloxy)- 1 H-pyridin-2-one: A solution of 4-(4-chloro-benzyIoxy)-pyridine-N-oxidc (0.950 g, 4.04 mmol) in acetic anhydride (40 mL) was heated at 140 0C for overnight. The volatiles were removed in vacuo and the residue was purified through a silica gel column to give 4-(4-chloro-benzyloxy)- lH- pyridin-2-one (0.674 g, 71 %). 1H NMR (CDCl3) δ: 12.4 (br s, IH), 7.38 (m, 4H), 7.22 (m, IH), 6.03 (m, IH), 5.93 (m, I H), 4.99 (s, 2H); ESMS m/e: 236.0 (M+H)+.
2-Chloro-4-(4-fluoro-phenoxymelhyl)-pyridinc:
To a mixture of (2~chIoro-pyridin-4-yI)-mcthaiiol (0.500 g, 3.5 mmol), 4-fluoro-phcnol (0.430 g, 3.83 mmol) and triphenylphosphine (1.00 g, 3.83 mmol) in THF (10 mL) was added DBAD (0.890 g, 3.83 mmol) in a portion. The resulting mixture was stirred at room temperature for 2 h. The resulting mixture was concentrated in vacuo. The crude material was purified over silica gel eluting with 0 to 50% EtOAc/hexanes to give 2-chloro-4-(4- fluoro-phenoxymethyl)-pyridine (1.60 g, di-Boc-hydrizine co-eluted). The material was carried on to the next step without further purification. 1H NMR (300 MHz, CDCl3) δ 8.42 (m, IH), 7.45 (s, IH), 7.29 (m, IH), 7.05 (m, 2H), 6.90 (m, 2H), 5.05 (s, 2H); ESMS m/e: 238.0 (M+H)+.
4-(4-Fluoro-phcnoxymethyl)- 1 H-pyridin-2-onc:
To a mixture of 2-chloro-4-(4-fluoro-phenoxymethyl)-pyπdine ( 1.6 g) in acetic acid (50 mL) was added water (15 mL). The resulting mixture was rcfluxed at 120 0C for overnight. After removal of all the solvent in vacuo, the residue was quenched with sat. NaHCO3 and extracted with EtOAc (3x50 mL). The combined organic phases were washed with brine (50 mL), dried over MgSO4, and concentrated in vacuo. The crude material was purified over silica gel eluting with 0 to 10% MeOH/DCM to give 4-(4-fluoro-phenoxymethyi)- lH-pyridin-2-one ( 143 mg, 19% for 2 step) and 4-(4-Fluoro-phenoxymethyl)-l H-pyridin-2-one (0.53 g) was recovered. 1H NMR (300 MHz, MeOD) δ 7.42 (m, IH), 7.00 (m, 4H), 6.60 (s, IH), 6.42 (m, IH), 5.00 (s. 2H); ESMS m/e: 220.0 (M+H)+.
4-(2-Fluoro-4-trifluoiOmethyi-phcnyl)-pyridine 1 -oxide:
Into a 20 mL vial with septum cap was added 2-fluoiO-4-(trifluoromethyl)phenylboronic acid
(578 mg, 2.78mmol), 4-chloro-pyridine 3 -oxide (300 mg, 2.32 mmol), sodium carbonate (638 mg, 6.02 mmol, dissolved in minimum water) and 1,2-dimethoxycthanc (5 mL). The reaction was degassed with nitrogen for 5 minutes and then tetrakis(lriphenylphosphine)palladium(0)
(134 mg, 0.1 16 mmol) was added. The reaction was heated and stirred overnight at 80 0C. The reaction was cooled to room temperature and then was extracted with water and ethyl acetate.
The organic layer was collected and concentrated in vacuo. The residue was purified over silica gel eluting with 5% 2M ammonia/methanol in DCM to give 4-(2-fluoro-4- trifluoromelhyl-phenyl)-pyridinc 1 -oxide in 54.7% yield. ESMS m/e: 257.9 (M+H)+.
4-(2"Fluorθ"4-trifluoromethyl-phenyl)~ 1 H-pyridin-2-one
Into a Radley reaction tube (24x 150mm) was added 4-(2-fluoro-4-trifluoromethyl-phenyl)- pyridine 1 -oxide (326 mg, 1.27 mmol) and acetic anhydride (10 mL), The reaction was heated to reflux and stirred overnight. The reaction mixture was then concentrated and used in next step without further purification. ESMS m/e: 257.9 (M+H)+.
4-(4-Chlorophenyl)- 1 H-pyridin-2-one:
A mixture of 4-bromo-2-fluoropyridine (1.50 g, 8.52 mmol), 4-chloro-phenylboronic acid (1.33 g, 8.52 mmol), tetrakis(lriphenylphosphine)palladium (0.985 g, 0.852 mmol), and Na2CO-) (1.81 g, 17.0 mmol) in a 80-mL scaled tube was purged with nitrogen and 1,2- dimcthoxyethane (30 mL) and water (5 mL) were added into the sealed tube. The reaction mixture was heated at 1 10 0C overnight. The mixture was cooled to room temperature, diluted with EtOAc (100 mL) and washed with water. The organic layer was concentrated in vacuo and the crude product was used for the next step without further purification. The crude 4-(4-chlorophenyl)-2-fluoropyridine was placed into a sealed tube and acetic acid (15 mL) was added. The reaction mixture was heated at 1 10 0C for 16 h and concentrated. The crude residue was dissolved in IN NaOH and titrated up to pH7 with 1 N HCl. The aqueous layer was extracted with EtOAc (30 mL x 3) and the combined organic layer was concentrated in vacuo. The crude residue was purified by column chromatography (DCM to 10% methanol (2N NH3) in DCM) to provide the desired product (535 mg, 30 %) as pale brown solid. ESMS m/e: 206.0 (M+H)+
6-(4-Chloro-phenyl)-3H~pyrimidin-4~one:
To a mixture of 4-chlorophenylboronic acid ( 1 .20 g, 7.66 mmol), 6-chloro-4- hydroxypyrimidine (1.00 g, 7,66 mmol), sodium carbonate (1.6 g, 15 mmol), water (7.0 mL) and 1 ,2-Dimethoxycthanc (16 mL) was added tetrakis(triphenylphosphinc)paIladium(0) (0.443 g, 0.383 mmol). Nitrogen was bubbled through the mixture for five minutes. The mixture was then refluxed for 16 h. The reaction mixture was cooled to room temperature and water (50 mL) was added and extracted with EtOAc. The organic layers were combined, washed with brine, dried over sodium sulfate, filtered, and evaporated to provide the crude product. The crude product was purified by silica gel chromatography using 90: 10: 1 DCM: methanol: ammonium hydroxide. The fractions were collected and rotovapped to provide 6-(4-ChIoro-ρhenyl)-3H-ρyrimidin-4-one. 1H NMR (400 MHz, CD3OD) δ: 9.04 (s, IH), 7.84 (m, 2H), 7.58 (m, 2H), and 6.94 (s, IH); ESMS m/c: 207.0 (M+H)+.
4-[(E)-2-(4-Fluoro-phenyl)-vinyl]-pyridin-2-ol:
To a solution of dieihyl-4-fIιiorobenzyl-phosphonatc (515 mg, 2.09 mmol) in THF ( 10.0 mL) was added potassium tert-buloxide (246 mg, 2.09 mmol). After stirring at 20 0C for 1 h, the mixture was treated with 2-hydroxypyridoaIdehyde (246 mg, 2.00 mmol), and allowed to stir for 3 h at 20 0C. The reaction mixture was quenched with water, and the aqueous phase was extracted with DCM, the combined organic phases were dried over MgSO4 and concentrated in vacuo. The crude oil was purified by column chromatography (DCM to 20%methanol in DCM) gave the desired product 4-[(£)-2-(4-fluoro-phenyO-vinyl]-pyridin-2-ol (32.0 mg, 7.5%, 5: 1 transxis) as white crystal. 1H NMR (400 MHz, CDCh) δ: 7.56 (m, 2H), 7.33 (m, I H), 7.26 (m, IH), 7.08 (m, 2H), 6.87 (m, IH), 6.64 (m, IH), 6.54 (m, IH); ESMS m/e: 216 (M+H)+.
3-Oxo-azetidine- l-carboxylic acid tert-butyl ester:
To a solution of l-Z?<xs-3-(hydroxy)azetidine (54.4 g, 314 mmol) and triethylamine (109 mL, 785 mmol) in DMSO (100 mL) and DCM (8.00 mL) in ice-water bath was added sulfur trioxide pyridine complex (100 g, 628 mmol) portionwisc to keep the temperature below 10 0C. After the addition of sulfur trioxide pyridine complex, the reaction mixture was stirred at below 10 0C for about 30 min and at about 20 0C for about 1 hr. The reaction mixture was
quenched with cold waster (400 mL) and the aqueous layer was extracted with EtOAc (3x300 mL). The combined organic layers were washed with brine (2x100 mL), concentrated up to 100 mL in vacuo, dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The residue was kept at temperature below 0 0C and the resulting solid was suspended in cold hexanes, filtered and washed with cold hexanes to give 3-oxo-azetidine-l-carboxylic acid tert-butyl ester (47.0 g, 87.1%) as a white solid. 1H NMR (400 MHz, CDCl3) δ: 4.71 (s, 4H), 1.49 (s, 9H).
3-(6-Bromo-lH-indol-3-yl)-3-hydroxy-azetidine- l-carboxylic acid tert-bulyl ester: To a solution of 6-bromoindole (70.8 g, 361 mmoi) in methanol (450 mL) was added potassium hydroxide (22.3 g, 397 mmol). After all potassium hydroxide was dissolved, 1-
Boc-azetidin-3-one (68.0 g, 397 mmol) was added in one portion. The resulting solution was stirred at 45-50 0C for 6 h and concentrated in vacuo. The residue was diluted with EtOAc
(600 mL), washed with water (2x 100 mL) and brine (100 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. The crude brown solid was suspended with 20%
EtOAc in hexanes (80 mL), filtered, and washed with 20% EtOAc in hexanes (180 mL) to give 3-(6-bromo~ lH™indol-3-yl)-3-hydroxy-azetidine-l-carboxylic acid tert-butyl ester (53.8 g, 41.1%) as a white solid. 1H NMR (400 MHz, CDCh) δ: 8.15 (s, IH), 7.60-7.54 (m, 2H),
7.30-7.21 (m, 2H), 4.42-4.35 (m, 2H), 4.29-4.24 (m, 2H), 2.39 (s, I H), 1.49 (s, 9H); ESMS m/e: 367 (M-S-H)+.
6~Bronio~3-( 1 -methyl-azelidin-3-yl)- 1 H-indole:
To a suspension of lithium aluminum hydride (1 1.4 g, 300 mmol) in THF (230 mL) was added a solution of l-Z?oc-3-(6-bromoindol-3-yl)azetidin-3-ol (20.0 g, 54.6 mmol) in THF (70 mL) dropwise at 0 0C. The reaction mixture was refluxed for 6 h, cooled to the room temperature, and quenched with water ( 1 1.4 mL), sodium hydroxide (3M solution, 11.4 mL), and water (34.2 mL) in ice-water bath. The resulting suspension was filtered and washed with THF, EtOAc and DCM subsequently. The combined organic filtrates were concentrated in vacuo, suspended in 10% methanol in DCM, and filtered to give 6-bromo-3-( l-mcthyl- azetidin-3-yl)-lH-indolc (8.00 g, two-time filiations) as a white solid and the filtrate was further purified by column chromatography on SiO2 (2M ammonia in methanol:DCM = 0: 1 to
1 :9) to give an additional desired product (3.00 g) as a brown solid. The total desired product (1 1.0 g, 76.1%) was obtained. 1H NMR (400 MHz, CDCl3) δ: 8.08 (s, IH), 7.53-7.45 (m, 2H), 7.25-7.20 (m, IH)5 7.06 (s, I H), 3.92-3.78 (m, 3H), 3.28-3.22 (m, 2H), 2.43 (s, 3H); ESMS m/e: 265 (M+H)+.
1 -Benzenesulfonyl-6-benzyloxy- 1 H-iiidole:
To a solution of 6-benzyloxy-l H~indole (5.00 g, 22.4 mmol) in DMF (50 mL) was added sodium hydride (897 mg, 22.4 mmol). After stirring at 23 0C for 0.5 hour, the mixture was treated with benzenesulfonyl chloride (3.15 mL, 24,7 mmol), and allowed to stir for 18 h at 23 0C. The reaction mixture was quenched with water, and the aqueous phase was extracted with EtOAc. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The crude oil was purified by column chromatography (hexanes to 20% EtOAc in hexanes gradient) to give the desired product l-benzenesuIfonyl-6-benzyloxy- l H-indole (5.65 g, 69%) as white solid. 1H NMR (400 MHz, CDCl3) δ: 7.75 (m, IH), 7.73 (m, IH), 7.64 (m, I H), 7.60-7.49 (m, 3H), 1 Al -135 (m, 7H), 6.98 (m, IH), 6.59 (m, I H), 5.20 (s, 2H); ESMS m/e: 364 (M+H)+
l-Benzenesulfonyl-6-benzyIoxy-2-methyl- lH-indole:
To the LDA solution [15.3 mmol, generated in situ from di-isopropylamine (2.17 mL, 15.3 mmol) and n-butyl lithium (8.20 mL, 16.3 mmol)] in THF (50 mL) was added 1- benzenesulfonyl-6-bcnzyloxy-l H-indole (5.27 g, 14.6 mmol) at -78 0C. The mixture was stirred at -78 0C for I h, then 23 0C for 1 h. The mixture was cooled to -78 0C again, then treated with methyl iodide (2.90 g, 20.4 mmol), and stirred at 23 0C for 18 h. The reaction was quenched with water, the aqueous phase was extracted with EtOAc. The combined organic phase were dried over MgSO4 and concentrated in vacuo. The crude oil was purified by column chromatography (hexanes to 20% EtOAc in hexanes gradient) to give 1- benzenesulfonyl-6-benzyloxy-2-methyl-l H-indoIe (3.97 g, 72%) as pale yellow solid. 1H NMR (400 MHz, CDCI3) δ: 7.85 (m, IH), 7.68 (m, IH), 7.66 (m, I H), 7.56-7.49 (m, 3H), 7,46-7.34 (m, 5H), 7.28 (m, IH), 6.95 (m, IH), 6.28 (m, I H), 5.20 (s, 2H), 2.57 (s, 3H); ESMS m/e: 378 (MH-H)+
6-Benzyloxy-2-methyl- 1 H-indole:
The mixture of l~benzenesulfonyl~6-benzyloxy-2-methyl-lH-indole (3.97 g, 10.5 mmol) and potassium hydroxide (4.09 g, 73.0 mmol) in methanol (100 mL) was heated to reflux for 18 h.
The reaction mixture was cooled to room temperature. The volaliles were removed in vacuo. The residue was redissolved in water and extracted with EtOAc. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The crude oil was purified by column chromatography (hexanes to 20% EtOAc in hexanes gradient) to give ό-benzyloxy-2- methyl- IH-indolc (2.15 g, 86%) as brown solid. 1H NMR (400 MHz, CDCl3) δ: 7.71 (br,
IH), 7.51-7.45 (m, 2H), 7.43-7.37 (m, 3H), 7.33 (m, IH), 6.88 (m, IH), 6.84 (m, IH), 6.16 (m, IH), 5.12 (s, 2H), 2.42 (m, 3H); ESMS m/e: 238 (M+H)+
6-Methoxy-7-methyl- 1 H-indolc:
To a solution of l-methoxy-2-methyl~3~nitro-benzene (5.00 g, 29.9 mmol) in THF ( 100 mL) was added vinyl magnesium bromide (89.9 mL, 89.9 mmol) in one portion at -40 0C. The mixture was stirred at -40 0C for 30 min, was quenched by pouring into aqueous ammonium chloride saturated solution. The aqueous solution was extracted with EtOAc. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The crude oil was purified by column chromatography (hexanes to 20% EtOAc in hexanes gradient) to give 6-methoxy- 7-methyl- l H-indoie (1.32 g, 27%) as brown solid. 1H NMR (400 MHz, CDCh) δ: 7.94 (br, IH), 7.46 (m, IH), 7.16 (m, IH), 6.89 (m, IH), 6.54 (m, IH), 3.94 (s, 3H), 2.40 (s, 3H); ESMS mA- 162 (M+H)+
Compounds of the Invention
Thc following compounds were synthesized according to the schemes described in the experimental section:
Example Ia
4-Benzyloxy- 1 -[3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H-pyridin-2-onc

To a solution of 4-benzyloxy-lH-pyridin-2-one (2.01 g, 10.0 mmol), 6-bromo-3-(l-melhyl- azetidin-3-yl)- lH-indolc (2.64 g, 10.0 mmol), CuI (1.89 g, 10 mmol), K2CO3 (2.76 g, 20.0 mmol) in DMF (20 niL) was added irans-N,N'-dimcthyl-cyclohcxanc-l,2-diamine ( 1.42 g, 10.0 mmol) at room temperature. The resulting mixture was heated at 100 0C overnight. The resulting mixture was concentrated in vacuo. The residue was quenched with Ammonium hydroxide (15% solution) and extracted with 3: 1 Chloroform: 2-propanol solution (3x100 mL). The combined organic phases were concentrated /Vi vacuo. The crude material was purified over silica gel eluting with 0 to 10% McOH (2M NH^)/DCM to give 4-benzyloxy-l- [3-( l-melhyl-azelidin-3-yl)-l H-indol-6-yl] -lH-pyridin-2-one (2.20 g. 57.1%). 1H NMR (300 MHz, CDCl3) δ 9.06 (s, IH), 7.58 (m, IH), 7.42 (m, 5H), 7.31 (m, I H), 7.29 (m, IH), 6.96 (m, 2H), 6.08 (m, 2H), 5.07 (s, 2H), 3.87 (m, 3H), 3.28 (m, 2H), 2.46 (s, 3H). ESMS m/e: 386.1 (M+H)+.
The compounds of examples Ib-I h were prepared in an analogous manner to the procedure described for example 1 a.
Example Ib I -[ I -Ethyl-3-((S)- 1 -mcthyl-ρyrroIidin-2-yImethyl)- 1 H-indol-6-yl]-4-(pyridin-3- ylmethoxy)- 1 H-pyridin-2-one

The compound was prepared from 6-biOmo-l-ethyl-3-((5)-l-methyl-pyrroiidin-2-ylmcthyl)- I H-indolc and 4-(ρyridin-3-ylmethoxy)-lH-ρyridin-2-one. ESMS m/e: 443.1 (M+H)+.
Example Ic 4-(4-Fluoro-phenoxymethyl)- 1 - [3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one

Example Id 4-(4-Chloro~benzyloxy)- 1 -[3-( 1 -melhyl-azetidin-3-yl)- 1 H-indol-6-yl j- 1 H- pyridin-2-one

The compound was prepared from 4-(4-chloro-benzyloxy)-lH-pyridin-2-onc and bromo-3-(l - methyl-azetidin-3-yl)-lH-indole. ESMS m/e: 420 (M+H)+.
Example Ie 4-(4-FIuoro-phenyIsul fan ylmethyl)- l -[3-(I -methyl -azelidin-3-yl)- l H-indol-6- yl]- 1 H-pyridin-2-onc

The compound was prepared from 4-(4-fluoro-phenylsulfanylmethyl)-lH-pyridin-2-one and 6-bromo-3-( 1 -methyl-azetidin-3-yl)- I H-indole. ESMS m/e: 420 (M+H)+.
Example If 4-(4-FluoiO-phenylsulfanylmethyl)- 1 -[3-( 1 -meLhyl-azetidin~3~yl)~ 1 H-indol-6- ylj-l H-pyridin-2-one

Example Ig 4-( 1 -Cyclopropyl-ethoxy)- 1 -[3-( 1 -melhyl-azclidin-3-yl)- 1 H-indol-6-yll- 1 H- pyridin~2-onc

The compound was prepared from 4-(l-cyclopropyI-cthoxy)-l H-pyridin-2-one and 6-bromo- 3-( 1 -methyl-azetidin-3-yl)" 1 H~indole ESMS m/e: 364.0 (M+H)+.
Example Ih 4-Benzyloxy-l-[ l-methyl-3-(l -mcthyl-azctidin-3-yl)-lH-indol-6-yl |-IH- pyridin-2-one

The compound was prepared from 4-(4-Chloro-benzyloxy)- l -[3-(I -melhyl-azclidin-3-yl)- IH- indol-6-yl]-] H-pyridin-2-one and iodomethane. ESMS m/c: 400 (M+H)+
Example 2a 4-(4-Chloro-phenyl)-l -[3-(I -methyl-azetidin-3-yl)- lH-indol-6-yl]-lH-pyridin-2-one

Into a vial was added 4-(4-ChIoro-phenyl)- l H-pyridin-2-one (78 mg, 0.38 mmol), ό-Bromo- 3-(l -methyl-azetidin-3-yl)- lH-indole (100 mg, 0.38 mmol), K2CO3 (79 mg, 0.57 mmol), CuI (72 mg, 0.38 mmol), rra/Ly-MN'-Dimcthyl-cyclohexane-l ^-diaminc (0.054g, 0.38 mmol) and DMF (5 niL), The resulting mixture was heated at 100 0C overnight. The resulting mixture was quenched with 30% NH4OH and extracted with EtOAc three times. The combined organic phase was washed with water two times and brine once, dried with MgSO4 and concentrated in vacuo. The crude material was purified over silica gel eluting with 0 to 10% MeOH (2M NH,) : DCM to afford 4-(4-Chloro-phenyl)-l -[3-(I -methyl-azetidin-3-yl)- ] H-
4]
indol-6-yl]-l H-pyridin-2-one (30 mg). 1H NMR (400 mHz, CDCl3) δ: 9.7 (s, IH), 7.5-6.4 (m, 1 IH), 3.75 (m, 3H), 3.15 (m, 2H), 2.34 (s, 3H). ESMS m/e: 390 (M+H)+
The compounds of examples 2b-2y were prepared in an analogous manner to the procedure for example 2a.
Example 2b 4-(4-Chloro-phenyl)- l-{ 3-[ l-(2-fluoro-ethyl)-azetidin-3-yl]- lH-indol-6-yl}- lH- pyiϊdin-2-one

Example 2c 4-(4-Chloro-phenyl)- 1 - { 3-[ 1 -(2-mcthoxy-ethyl)-azelidin-3-yI]- 1 H-indol-6-yl } - 1 H-pyridin-2-one

Example 2d 4-(4-ChIoro-phenyl)- 1 -[ 1 -methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one

Example 2e 5-Chloro- 1 '-[3-( 1 -isopropyI-azelidin-3-yl)- 1 H-indol-6-yl]- 11H-[2,4'JbiρyridinyI-21- one

The compound was prepared from 6-biOmo-3-(l-isopropyl~azetidin-3-yl)- lH-indole and 5- chloro-rH-[2,4']bipyridinyl-2'-one. ESMS m/e:419.0 (M+H)+.
Example 2f 1 '- { 3-[ 1 -(2-Fluoro-ethyl)-azetidin-3-yl]- 1 H-indoI-6-yI } -6-trifluoromethyl- 11H- [3,4']bipyridinyI-2'-one

The compound was prepared from 6-bromo-3-[l-(2-fluoiO-ethyl)-azetidin-3-yl]-lH-indolc and 6-trifluoromethyl" rH-[3,4']bipyridinyl~2'-one. ESMS m/e: 456.9 (M+H)+.
Example 2g 4-(2-FI uoro-4-trifluoiOtnethyl -phenyl)- 1 -[3-( 1 -methyl ~azeϋdin~3~yl)~ 1 H~indo!~ό~ yll-1 H-pyridin-2-one

The compound was prepared from 6-biOmo-3-( l-methyl-azctidin-3-yl)-lH-indolc and 4-(2- fluoro-4-trifluorømethyl-ρhenyl)- 1 H-pyridin-2-one. ESMS m/e: 441.9 (M+H)+.
Example 2h 4-[4-( 1 -Hydroxy- 1 -methyl-ethyl)~phenyl]~ 1 ~[3-( I -methyl-azetidin~3-yl)~ 1 H- indoI-6-yl]-l H-pyridin-2-one

The compound was prepared from 6-biOmo-3-( l-methyl-azctidin-3-yl)-lH-indole and 4-[4~ ( 1 -hydroxy- l-methyl-ethyl)-phenyl]- l H-pyridin-2-one. ESMS m/e: 414.0 <M+H)+.
Example 2i 6-(4-Chloro-phenyl)-3-[3-( 1 -methyl-azelidin~3~yl)- 1 H-indol-6-yl]-3H-pyrirnidin-
4-one

The compound was prepared from 6-bromo-3-( l-methyl-azetidin-3-yl)-lH-indole and 6-(4- chloro-ρhenyI)-3H-pyrimidin-4-one. ESMS m/e: 390.9 (M+H)+.
Example 2j 1 '-[2-Methyl-3-( 1 -methyl-a/.elidin-3-yl)- 1 H-indol-6-yl]-6-trifluoromethyl- 11H- f3,4']bipyridinyl-2'-one
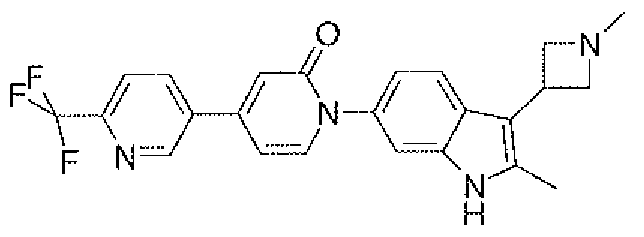
Example 2k 4-Benzo[b]thiophen-2-yl- 1-[3-( 1 -mcthyl-azeiidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one

The compound was prepared from 6-bromo-3-(l-mcthyl-azetidin-3-yl)-l H-indole and 4-(5- methyl-bcnzo[b]thiophen-2-yl)-I H-pyridin-2-one. ESMS m/e:425.9 (M+H)+.
Example 21 1 -[3-( l-Methyl-azetidin-3-yl)- 1 H-indol-6-yI]-4-( 1 ^methyl- 1 H-indol-5-yl)- 1 H- pyridin-2-one

The compound was prepared from 6-bromo-3-( l -mcthyl-azetidin-3-yl)-lH-indole and 4-( l - methyl- I H-indoI-5-yl)-lH-ρyridin-2-one. ESMS m/c: 409.0 (M+H)+.
Example 2m 1 ~[3-( 1 -Meihyl-azetidin-3-yI)- 1 H-indol-6-yi]-4-naphthalen-2-yl- 1 H-pyridin-2-one

The compound was prepared from 6-bromo-3-(l-mcthyl-azetidin-3-yl)-lH-indole and 4- naρhthalen-2-yl-l H-ρyridin-2-onc. ESMS m/e: 406.1 (M+H)+.
Example 2n 4~(5-Fiuoro-benzooxazoi~2~yl)- 1 -[3-( 1 -mcthyl-azctidin-3-yl)- 1 H-indoI-6-yl]- 1 H-pyridin-2-one

The compound was prepared from 3-(l-methyl-azelidin-3-yl)-l H-indolc and 4-(5-fluoro- benzooxazol-2-yl)-l H-pyridin-2-one ESMS m/e: 415.0 (M+H)+.
Example 2o 4-(5-Ethyl-pyrimidin-2-yl)- 1 - { 3-[ 1 -(2-fluoiO-clhyl)-azclidin-3-yl]- i H-indol-6- yl }-l H-pyridin-2-one

Example 2p 5-Chloro- 1 '- { 3-[ 1 -(2-fluoro-ethyl)-azetidm-3-yll - 1 -methyl- 1 H-indol-6-yl }- 11H-
[2,4']bipyridinyl-2'-one

The compound was prepared from 6-bromo-3-[ l-(2-fluoiO-ethyt)-azetidin-3-yl]- l -methyl- IH- indole and S-chloro- lΗ-^^'Jbipyridinyl-a'-one. ESMS m/e: 436.9 (M+H)+.
Example 2q 1 -[3-(l-Methyl-azetidin-3-yl)-lH-indol-6-yll-4-(4-trifluoiOmethoxy-phenyl)- 1 H-pyridin-2-one

The compound was prepared from 4-(4-tTrifluoroniethoxy-phenyl)-lH-pyridin-2-one and 6- bromo-3-(l-methyl-azetidin-3-yl)-lH-indole ESMS m/e: 439.9 (M+H)\
Example 2r 3-[3-(l-MethyI-azetidin-3-yl)-lH-indol-6-yl]-6-(4-triπuoromethyl-phenyl)-3H- pyrimidin-4-one

The compound was prepared from 6-(4-trifluoromethyl-phenyl)-3H-pyrimidin-4-one and 6- bromo-3-(l-methyl-azetidin-3-yl)-lH-indole. ESMS m/e: 424.9 (M+H)+.
Example 2s I -{3-[I -(2-Fluoro-ethyl)-azetidin-3-yI]-l-methyl-l H-indol-6-yI}-4-(4-fluoro- phenyl)- 1 H-pyridin-2-one

The compound was prepared from 4-(4-fluoro-phenyl)-l H-pyridin-2-one and 6-bromo-3-[l- (2-fluoro-ethyI)-azetidin-3 -yl]-l -methyl- IH-indole. 1H NMR (400 MHz, CDCl3) δ: 7.61-7.66 (m, IH), 7.52-7.58 (m, 2H), 7.40-7.44 (m, IH), 7.29-7.31 (m, IH), 7.07-7.14 (m, 2H), 6.99- 7.04 (m, IH), 6.96 (s, IH), 6.78-6.81 (m, IH), 6.38-6.44 (m, IH), 4.48-4.56 (m, I H), 4.36- 4.45 (m, IH), 3.84-3.99 (m, 3H), 3.71 (s, 3H), 3.24-3.36 (m, 2H), 2.81-2.89 (m, IH), 2.73- 2.80 (m, 1 H). ESMS m/e: 420.0 (M+H)4.
Example 2t 5-Fluoro- 1 '- {3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yl]- 1 -methyl- 1 H-indol-6-yl} - 11H-
[2,4']bipyridinyl-2'-one

The compound was prepared from 5-fiuoro-lΗ-[2541]bipyridinyI-2'-one and 6-bromo-3-[l-(2- fluoro-ethyl)-azetidin-3-yI]-l -methyl- IH-indole. ESMS m/e: 421.0 (M+H)'.
Example 2u V- { 3-[ 1 -(2-Fluoro-cthyl)-azctidin-3-yl]- 1 -methyl- 1 H~indol-6-yl ) -5- trifluoromethyl- 1 Η-[2,4']bipyridinyl-2'-one

The compound was prepared from 5-trifluoromethyI-l 'H-[2,4']bipyridinyl-2'-one and 6- bromo-3-[ 1 -(2-fluoro-elhyl)-azetidin-3-yIJ- 1 -methyl- 1 H-indole. ESMS m/e: 471.0 (M+H)+.
Example 2v 1 '-[ 1 -Melhyl-3-( 1 -methyI-azelidin-3-yl)- 1 H-indol-6-yl]-5-trifluoromethyI- 11H-
[2,4']bipyridinyl-2'-one

The compound was prepared from 5-trifluorornethyi-lΗ-[2,4']bipyridinyI~2'~onc and 6- bromo- 1 -methyl-3-( 1 -methyI-azeϋdin-3-yl)- 1 H-indole. ESMS m/c: 438.9 (M+H)+.
Example 2w 1 '- { 3-[ 1 -(2-Fluoro-ethyl)-azctidin-3-yI] - 1 -methyl- 1 H-indol-6-yl }-6- trifluoromcthyl- 1 Η-[3,4']bipyridinyl-2'-one

The compound was prepared from 6-trifluoiOmethyI-rH-[3,4']bipyridinyi-2'-one and 6- bromo-3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yI J-I -methyl- 1 H-indole. ESMS m/e: 470.9 (M+H)+.
Example 2x 6-(4-Chloro-phcnyl)-3- { 3-[ 1 -(2-fluoro-ethyl)-a/etidin-3-ylJ- 1 -methyl- 1 H-indol- 6-yl }-3H-pyrimidin-4-one

The compound was prepared from 6-(4-chloro-phenyl)-3H-pyiiimidin-4-onc and 6-bromo-3- [ 1 -(2-fluoro-ethyl)-azetidin-3-yl J- 1 -methyl- 1 H-indole. ESMS m/e: 436.9 (M+H)+.
Example 2y 6-(4-Chloro-phenyl)-3-[2-methyl-3-( l-methyl~azetidin-3-yl)- lH-indol-6-yl]-3H- pyrimidin-4-one

The compound was prepared from 6-(4-chloro-phenyl)-3H-pyrimidin-4-one and 6-bromo-2- methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indole. ESMS m/e: 404.9 (Mn-H)+.
Example 3a 4-(4-Chloro-phenyl)- 1 - { 3~[ 1 -(tetrahydro-thiophcn-3-yl)-azetidin-3-yl]- 1 H-indol- 6-yl } - 1 H-pyridin-2-one

The compounds of examples 3b, 3c, 3f, 3g, 3h, 3i, and 31 were prepared in an analogous manner to the procedure described for example 3a.
Example 3b 4-(4-Chloro-phenyl)- 1-[3-Cl -oxetan-3-yl-azetidin-3-yl)- 1 H-indol-6-yl]- 1 H- pyridin-2-one

The compound was prepared from l-(3-azetidin-3-yl- l H-indoI-6-yl)-4-(4-chloro-phenyl)- lH- pyridin-2-one and oxclan-3-one. ESMS m/e: 432.0 (M+H)+.
Example 3c 4-(4-Chloro-phenyl)-l-[3-(l-cthyl-azctidin-3-yl)-l-(2-hydroxy-ethyl)-lH-indol- 6-yl] - 1 H-pyridin-2-onc

The compound was prepared from l-[3-azctidin-3-yl-l-(2-hydroxy-cthy])-l H-indol-6-yi]-4- (4-chloro-phenyI)-l H-pyridin-2-onc and acetaldehyde. ESMS m/e: 447.9 (M+H)+.
Example 3d 4-(4-Chloro-phenyl)- 1 -{ 3-f l-(3,3,3~trifluoro-piOpionyl)-azctidin-3-yl]-lH-indol-
6-yl } - 1 H-pyridin-2-onc

The compound of example 3d were prepared in an analogous manner to the procedure described for example 3e and was prepared from l-(3-azetidin-3-yl-l H-indol-6-yl)-4-(4- chloro-phenyl)- l H-pyridin-2-one and 3,3,3-tπfluoro-propionyl chloride. ESMS m/e: 485.8 (M+H)+.
Example 3e 4-(4-Chloro-phcnyl)- 1 -[3-( 3 -cyclopropanecarbonyl-azetidin-S-yl)- 1 -methyl- 1 H- indol-6-yl]~l H-pyridin-2-one

To a solution of l -(3-azetidin-3-yl-l -methyl- IH- indol-6-yl)-4-(4-chloro-phenyl)- lH-pyridin- 2-one-2TFA (0.130g, 0.258 mrnol) and irielhylamine (3.0 mL, 20 mmol) in DCM (10 itiL)
was added cyclopropanccarbonyl chloride (3.0 mL, 30 mmol) dropwise. The reaction mixture was stirred at room temperature for 2 h and quenched with saturated NaHCO3. The organic layer was separated and the aqueous layer was extracted with EtOAc (x3). The combined organic phases were washed with brine, dried over MaSO4, and concentrated in vacuo. The crude residue was purified over silica gel eluting with 0 to 10% MeOH (2M NH3):DCM to afford 4-(4-chloro-phenyl)- 1 -[3-( 1 -cyclopropanccarbonyl-azctidin-3-yl)- 1 -methyl- 1 H-indol- 6-yll-lH-pyridin-2-one (85 mg, 72%). ESMS ra/e: 457.9 (M+H)+.
The compounds of examples 3d and 3j were prepared in an analogous manner to the procedure described for example Ia.
Example 3f 4-(4-Chloro-phenyl)-l~{ l-mcthyl-3-[ l-(tetrahydro-furan-3~yl)~azetidin-3-yl]- 1 H-indol-6-yl } - 1 H-pyridin-2-one

Example 3g 5-Chloro- 1 '-[3-( 1 »cyclopiOpylmethyl-azetidin-3-yl)~ 1 -methyl- 1 H-indol-6-yl]- 1 Η-[2,4']bipyridinyl-21-one

The compound was prepared from l'~(3-azetidin~3-yl-l -methyl- lH-indoI-6-yl)-5-chloro- l 'H-
[2,4']bipyridinyl-2'-one and cyclopropanccarbaldehydc. ESMS m/e: 444.9 (M+H)+.
Example 3h 5-Chloro- 1 '-[3-( 1 -isopropyl-azetidin-3-yl)- 1 -methyl- 1 H-indol-6-yl J- 1 'H- [2,41]bipyridinyl-21-one

Example 3i 1 -[ 1 -Methyl-3-( 1 -methyl-azetidin-3-yl)- 1 H-indoI-6-yl]-4-(4-trifluoromethyl- phenyl)- lH-pyridin-2-one

The compound was prepared from l-(3-azetidin-3-yl- l -methyl- lH-indol-6- yl)-4-(4- trifluoiOmethyl-phenyl)- lH-pyridin-2-onc and formaldehyde. ESMS m/e: 437.9 (M+H)+.
Example 3j 1 -f 3-( 1 - Acetyl-azetidin-3-yl)- 1 -methyl- 1 H-indol~6-yl]-4-(4-trifluoiOmethyl- phenyl)- 1 H-pyridin-2-one
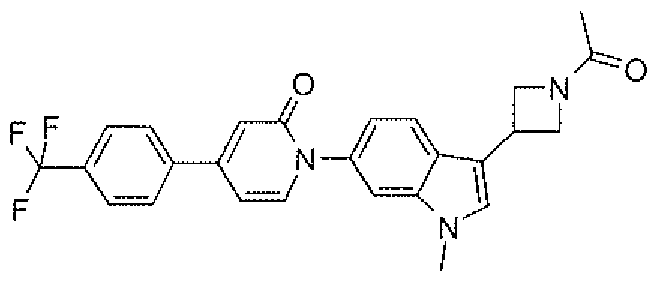
The compound of example 3j were prepared in an analogous manner to the procedure described for example 3e and was prepared from I -(3-azetidin-3-yl- 1 -methyl- lH-indol-6-yl)- 4-(4-trifluoromethyl-phenyl)-l H-pyridin-2-one and acetyl chloride. ESMS m/e: 465.9 (M+H)+.
Example 3k 6-(4-Chloro-phenyl)-3- { 3-[ 1 -(tetrahydiO-furan-3-ylmethyl)-azetidin-3-yl]- 1 H- indol-6-yl } -3H-pyrimidin-4-one

The compound was prepared from 3-(3-azetidin-3-yl-lH-indoI-6-yl)~6-(4-chloro-phenyl)-3H- pyrimidin-4-one and tetrahydro-furan-3-carbaldehyde. ESMS m/e: 460.9 (M+H)+.
Example 31 4-(4-Fluoro-phenyl)- 1 -[3-( 1 -isopropyl-azetidin-3-yl)- 1 -methyl- 1 H-indol-6-yl] - 1 H-pyridin-2-one

The compound was prepared from I -(3-azetidin-3-yl~l -methyl- l H-indol-6-yl)-4-(4-fluoro- phcnyl)-lH-ρyridin-2-one and acetone. ESMS m/e: 41ό.0(M+H)+.
Example 4a l -[ l-(2-Fluoro-ethyl)-3-(l-methyl-azetidin-3-yl)-lH-indol-6-yl]-4-(2,2,2- Irifluoro-ethoxy)- 1 H-pyridin-2-one
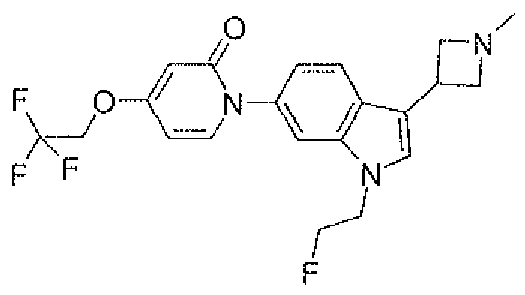
To a mixture of , 4-(2,2-difluoro-propoxy)-l-[3-( l-methyl-azelidin-3-yl)-IH-indol-6-yl]- lH- pyridin-2-one (57.0 mg, 0.153 mmol), potassium tert-butoxiάe (25.7 mg, 0.229 mmol) and 1 ,4,7, 10, 13, 16-hexaoxacyclooctadecane (60.5 mg, 0.229 mmol) , in tetrahydrofuran (5 iiiL, 60 mmol) was added 1 -bromo-2-fluoroethane (38.8 mg, 0.305 mmol) . The resulting mixture was stirred at room temperature for 3 hours. The resulting mixture was quenched with sat NaHCO3 (20 ml) and was extracted with 3: 1 (chloroform: 2-propanol) solution (2 x 20 ml). The combined organic phase was concentrated in vacuo. The crude material was purified over silica gel, eluting with 0 to 10% NH3 (2M solution in MeOH)/DCM, to give l-[l-(2-fluoro- ethyl)-3-(I -methyl-azetidin-3-yI)~ lH-indol--6-yl]-4-(2,2,2-trifluoiO-ethoxy)-lH-pyridiπ-2-one (14 mg, 22%). ESMS m/e: 423.9 (M+H)\
The compounds of examples 4b-4d were prepared in an analogous manner to the procedure of example 4a.
Example 4b 1 -[ 1 -Methyl-3-( 1 -methyl-azetidin-3-yϊ)- 1 H-indol-6-yl]-4-(2,2,2-triΩuoiO- ethoxy)- 1 H-pyridin-2-one
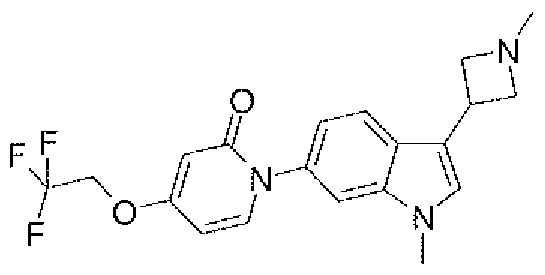
Example 4c 5-Chloro- 1 '-[3-( 1 -clhyl-azetidin-3-yl)- 1 -(2-fluoro-cthyl)- 1 H-indol-6-yl]- 11H- [2,4']bipyridinyl-2'-one

The compound was prepared from 5-chloro-l '-[3-(l -ethyl-azetidin-3-yl)- l H-indol-6-yl]-l TI- [2,4']bipyridinyl-2'-one and 1 -bromo-2-fluorø-elhane. ESMS m/e: 450.9 (M+H)+.
Example 4d 5-Chloro- 11- [3-( 1 -ethyl-azetidin-3-yl)- 1 -methyl- lH-indol-6-yIl-l 1H-
[2,4']bipyridinyl-2'-one

Thc compound was prepared 5-Chloro- 1 '-[3-(I -ethyl-azetidin-3-yl)- lH-indol-6-yll- l 1H- [2,4'JbipyridinyI-2'-one and iodomethane. ESMS m/e: 419.2 (M+H)+.
Pharmacological Evaluation of Compounds
In» Vitro Assays
The affinity of the compounds of the invention at the MCHl receptor was determined by measuring the inhibition of binding of a radioactive ligand ant the MCHl receptor. The procedure for determining specific binding of a test compound may be used as described by Audinot, et al. British Journal of Pharmacology, 2001, 133, 371-378. Similarly, the specific binding of test compounds can be measured at the rat MCHl receptor (GenBank Accession No. NM_031758) using [125I]-S3ό057 (NEX396; PcrkinElmcr Life Sciences, Inc.) as the radioligand.
The compounds of the invention were tested for their affinity at the MCH IR receptor. All the compounds disclosed herein show Ki values of less than about 5.0μM. Nearly all the compounds show Ki values of less than about l .OμM. Most of the compounds have Ki values
of less than about 20OnM with many of compounds having Ki values of less than about 5OnM.
Table I: Binding affinity of selected compounds at the MCHl receptor

Functional Activity
Functional activity of the compounds of the invention can be measured by receptor assays which determine the degree of intracellular second messenger response. For example, Cos-7 ceils are transfected with the MCHl receptor using the DEAE-dcxtran method (Gerald, ct al. J. Biol. Chcm. 1995, 270, 26758-26761). Other cell transfcction methods, employing various host cells, are well-known in the art. Certain compounds were tested to determine their functional activity and were determined to be potent antagonists at the MCHl receptor.
In-Vivo Assays
The in-vivo effects of the compounds of the present invention may be evaluated by using the following in-vivo behavioral animal models. The behavioral models described below are not intended to be the only models used to determine the efficacy of a compound of the invention to treat the corresponding disorder. For example, in addition to the Rat Social-interaction test, the marble burying experiment can also be used to screen for compounds for potential as anxiolytics. The skilled artisan would recognize the changes in certain parameters of the experiments to acquire the most exact data.
Diet Induced Obesity (DIO) Model:
Male Long-Evans rats are used and housed individually, maintained on about 12 h reverse light/dark cycle with lights off at about 09:00, and given free access to either a high-fat diet (#D 12451; fat percentage, about 45% kcal; Research Diets, New Brunswick, New Jersey) or a control diet (#D124508, fat percentage, about 10% kcal; Research Diets, New Brunswick, New Jersey) and water. After about 1 1 weeks, rats on the high fat diet began receiving a compound of the invention or vehicle by i.p. injection twice daily, about 1 h before lights off and about 10 h later, for about 4 weeks.
Rat Forced-swim Test:
The procedure which may be used here is similar to that previously described (Luki, et al. Psychopharmacohgy 2001 , 155, 315-322) with the following modifications. Male Sprague- Dawley rats may be used. Swim sessions are conducted for about 5 min, by placing rats in a plexiglass cylinder (about 46 cm tall x 20 cm in diameter) filled about 30 cm deep with water at about 230C, A compound of the invention or vehicle (about 0.01% lactic acid, about pH 6) is administered orally as a 1 ml/kg solution. Test sessions arc videotaped and recorded for later scoring by a single rater, who is blinded to the treatment condition. Immobility is scored as the time a rat remained floating in the water making only movements necessary to keep its head above the water. Swimming is scored as the time a rat made active swimming motions, more than necessary to maintain its head above water.
Rat Social-interaction Test: The procedure is performed for about 15 min as previously described (File and Hyde Br. J, Pharmacol. 1987, 62, 19-24) under low-light conditions using pairs of unfamiliar male Sprague-Dawley rats previously housed singly and exposed to the test arena for about 15 min on the previous day. A compound of the invention, chlordiazepoxide or vehicle is injected i.p. as ~ 1.0 ml/kg solution. All test sessions are videotaped and recorded for later scoring. Active social interaction, defined as sniffing, grooming, biting, boxing and crawling over and under, as well as locomotor activity (defined as squares crossed), is scored by a single rater, who is blinded to the treatment of each pair.
Rat Models of Micturition Compounds useful in the treatment of urinary disorders are assessed in various animal models of the micturition reflex as described in the art. (See, e.g., Maggi, CA, et al. J Pharmacol Exp Ther, 1987, 240, 998-1005; Morikawa, K, et al. Eur. J Pharmacol., 1992 213:409-415; Yoshiyama, M, et al. Eur J Pharmacol 287:73-78; and Yoshiyama, Urology, 1999, 54(5), 929- 33.)
Claims
Claim 1. A compound having the structure:
R1

wherein R1 is -(CHR7)mN(R8)(R9) or -(CHR7)nR10;
wherein R" is H or straight chained or branched Ci-C7 alkyl optionally substituted with one or more fluorine or -OH and where the carbons of the CrC7 alkyl are optionally replaced with one to three N, O or S atoms;
wherein each R4 and R5 is independently H, halogen or straight chained or branched Ci-C7 alkyl;
wherein each R7 is independently H, OH, or wherein one R7 can combine with another R7 on an adjacent carbon atom to form C-; -CO cycloalkyl and wherein one R7 can combine with a H on the shared carbon atom to form CrC6 cycloalkyl;
wherein each R8 and R9 is independently H or straight chained or branched Ci-C7 alkyl optionally substituted with one or more fluorine;
wherein R10 is a nitrogen containing heterocyclic moiety optionally substituted with one or more halogen, -COR12, a S or O containing heterocyclic ring, or straight chained or branched Cs-C7 alkyl optionally substituted with halogen or -OCH3;
wherein R1 1 is C3-Q cycloalkyl, straight chained or branched alkoxy-C3-C6 cycloalkyl, straight chained or branched Ci-C4 alkyl/alkoxy, phenyl, napthyl, 5 to 6-membered heteroaryl, benzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC4 alkyl optionally substituted with F;
wherein R is straight chained or branched Cs-Cy alkyl or C3-C6 cycloalkyl, where each of which is optionally substituted with one or more halogen or methoxy;
wherein m is an integer from 1 to 4 inclusive; and wherein n is independently an integer from 0 to 4 inclusive, or a pharmaceutically acceptable salt thereof.
Claim 2. The compound of claim 1 , wherein R1 is-(CHR7)mN(R8)(R9).
Claim 3. The compound of claim 1, wherein R1 is-(CHR7)nR10.
Claim 4, The compound of claim 1 , wherein each R4 and Rs is independently H or straight chained or branched Ci-C4 alkyl;
Claim 5. The compound of claim 1 , wherein R10 is pipera/inyl or morpholinyl, where each of which is optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched C]-C4 alky] optionally substituted with halogen.
Claim 6. The compound of claim 1, wherein R1 is piperidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched C]-C4 alkyl optionally substituted with halogen.
Claim 7. The compound of claim 1 , wherein R1 is pyrrolidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched Ci-C4 alkyl optionally substituted with halogen.
Claim 8. The compound of claim 1 , wherein Ri0 is azetidinyl optionally substituted with a S or O containing heterocyclic ring or one or more straight chained or branched C1-C4 alkyl optionally substituted with halogen.
Claim 9. The compound of claim 1 , wherein R7 is H; and R4 and R5 are each independently H, or straight chained or branched Ci -C4 alkyl.
Claim 10. The compound of claim 1 , wherein n is 0 and R~ is H, methyl or ethyl.
Claim 1 1. The compound of claim 1 , wherein n is 1 or 2.
Claim 12. The compound of claim 1, wherein R1 1 is C3-C6 cycloalkyl or alkoxy-QrCό cycloalkyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Ci-C4 alkyl optionally substituted with F.
Claim 13. The compound of claim I , wherein Rπ is phenyl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC4 alkyl optionally substituted with F.
Claim 14. The compound of claim ! , wherein R1 1 is a 5 to 6-mcmbered heteroaryl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Ci-C4 alkyl optionally substituted with F.
Claim 15. The compound of claim 1 , wherein R1 1 is bcnzothiophenyl, indolyl or benzoxazolyl, where each of which is optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched Ci-C4 alky! optionally substituted with F.
Claim 16. The compound of claim 1, wherein R1 1 is napthyl optionally substituted with one or more F, Cl, Br, -OH, or straight chained or branched CpC4 alkyl optionally substituted with F,
Claim 17. A compound selected from the group consisting of 4-(4-chloro-phenyl)-l-[3-(l- meihyl-azetidin-3-yl)- 1 h-indoI-6-yl j~ I h-pyridin-2-one; 4-(4-chloro-phenyl)- 1 ~ { 3-[ 1 ~(2~fluoro- ethyl)~azetidin-3-yI] - 1 h-indol-6-yl } - ih-pyridin-2-one; 4-(4-chloro-phenyI)- 1 - { 3-[ 1 -(2- methoxy-ethyl)-azetidtn-3-yl]- 1 h-indol-6-yl } - lh-pyridin-2-one; 4-(4-chloro-phenyl)- 1 -f 1 - methyl -3-( 1 -methyl-azclidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 5-chloro- 1 '-[3-( 1 - isoprøpyl-azetidin-3-yl)-l h-indol-6-yll- 1 'h-[2,4']bipyridinyl-21-one; 1 '- { 3-[ I -(2-fluoro-ethyl)- azetidin-3-yl]-l h-indol-6-yl }-6-trifluoromethyl-rh-[3,4iJbipyridinyl-2'-one; 4-(2-fluoro-4- trifluoromethyl-phenyl)- 1 -f 3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-ylJ - 1 h-pyridin-2-one; 4-(2- fIuoiO-4-trifluoromethyl-phenyl)- lh-pyridin-2-one; 4-[4-(l -hydroxy- l -methyl-ethyl)-phenyl]- 1 -[3-( 1 -methyl-azctidin-3-yl)" 1 h-indol-6-yl]- 1 h-pyridin-2-one; 6-(4-chloro-phenyl)-3- [3-( 1 - methyl-azetidin-3-yI)-lh-indoI-6-yI]-3h-pyrimidin-4-one; r-[2-methyl-3-( l-methyl-azetidin- 3-yl)-lh-indol-6-ylJ-6-trifluoiOmethyl-rh-[3,4'jbipyridinyl-2'-one; 4-benzo[b]thiophen~2~yl~ 1 -[3-( 1 -methyl-azelidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 1 -[3-( 1 -mcthyl-azetidin-3-yl)- 1 h-indoI-6-yI]-4-( 1 -methyl- 1 h-indol-5-yl)- 1 h-pyridin-2-one; 1 -[3-(I -methyl-azetidin-3-yl)- Ih- indol~6~yl]-4~naphthalen-2-yl-l h-pyridin-2-one; 4-(5-fluoro-benzooxazol-2-yl)-l -[3-(l-methyl- azetidin-3-yI)- 1 h-indol-6-yl] -1 h-pyridin-2-one; 4-(5-ethyl-pyrimidin-2-yI)-l-{3-[l-(2-fluoiO- ethyl)-azetidin-3-yl]- 1 h-indoi-6-yl } - 1 h-pyridin-2-one; 5-chloro- T- { 3-[ 1 -(2-fluoro-ethyl)- azelidin-3-yl]- 1 -methyl- 1 h-indol-6-yl }- 1 'h-[2,4']bipyridinyl-2'-one; 1 -[3-( 1 -methyl-azetidin- 3-yl)- 1 h-indol-6-yl]-4-(4-trifluoiOmcthoxy-phenyl)- 1 h-pyridin-2-one; 4-(4-chloro-phenyI)- 1 - { 3-f 1 -(tetrahydiO-thiophen-3-yl)-azetidin-3-yll- 1 h-indol-6-yl } - 1 h-pyridin-2-one; 4-(4-chloro- phenyl)- 1 -[3-( 1 -oxetan-3-yl-azetidin-3-yl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 4-(4-chloro- phcnyl)- 1 -[3-( 1 -cthyl~azetidin~3-yl)- 1 -(2-hydroxy-ethyl)- 1 h-indol-6-yl]- 1 h-pyridin-2-one; 4- (4-chloro-phenyl)- 1 - { 3-[ 1 -(3,3 ,3-U'ifluoiO-piOpionyl)-azetidin-3-yl]- 1 h-indol-6-yl } - 1 h- pyridin-2-one; I-[ l -(2-fluorø-ethyl)-3-(l-methyl-azetidin-3-yl)-lh-indol-6-yl]-4-(2,2,2- trifluoro-ethoxy)-! h-pyridin-2-one; 1 -[ I -methyl-3-(l-methyl-azetidin-3-y!)-l h-indol-6-yl J-4- (2,2,2-trifluoro-ethoxy)- 1 h-pyridin-2-one; 5-chloro- 1 '-[3-( I-ethyl-azetidin-3-yl)- 1 -(2-fluoro- ethyl)- i h-indol-6-yl]- 11h-[2,4t]bipyridinyl-2'-one; and 5-Chloro- 1 '-[3-( 1 -ethyl-azetidin-3-yI)- 1 -methyl- 1 H-indol-6-yl j- 1 Η-[2,4'jbiρyridinyl-2'-one.
Claim 18. A compound selected from the group consisting of3-[3-(l -methyl -azelidin-3-yl)- l h-indoi-6-yi]-6-(4-trifiuoromcthyl-phcnyl)-3h-pyrimidin-4-one; l -{3-[ l-(2-fluoro-ethyl)- azctidin-3-yl]- 1 -methyl- 1 h-indol-6-yl } -4-(4-fluoro-phenyl)- 1 h-pyridin-2-one; 5-fluoro- 1 '- { 3- [ 1 -(2-fluoro-ethyl)-azetidin-3-ylj- 1 -methyl- 1 h-indol-6-yl } - 1 'h-^'Jbipyridinyl-T-one; 1'- { 3- [ 1 ~(2-fluoro-ethyl)-azctidin-3-yl]- 1 -methyl- 1 h-indol-6~yl }-5-trifluoromethyl- 1 'h- [2,4']bipyridinyl-2'-one; 1 '-[ 1 -methyl-3-( 1 -methyl-azetidin-3-yl)- 1 h-indol-6-yl]-5- trifluoromethyl- 1 'h-f 2,4']bipyridinyl-2'-one; I1- { 3-[ 1 -(2-fluoro-ethyl)-azetidin-3-yl]- 1 -methyl - 1 h-indol-6-yl } -6-trifluoromethyl- 3 'h-[3 ,4']bipyridinyl-2'-one; 6-(4-chloro-phenyl)-3- { 3-[ 1 -(2- fluoro-cthyl)-azctidin-3-yl]- 1 -methyl- 1 h-indol-6-yl }-3h~pyrimidin-4-one; 6-(4-chloro- phenyl)-3-f2-methyl-3-( I -methyl-azctidin-3-yl)- 1 h-indol-6-yl]-3h-pyrimidin-4-one; 4-(4- chloro-phenyl)- 1 - { 1 -methyl-3-f 1 -(tctrahydro-furan-3-yl)-azetidin-3-yl]- 1 h-indol-6-yl } - 1 h- pyridin-2-one; 5-chloro- 1 '-[3-( 1 -cyclopiOpyImethyl-azetidin-3-yl)- 1 -methyl- 1 h-indol-6-yl]- 1 'h-[2,4'Jbipyridinyl-2'-one; 5-chloro- 1 '-[3-( l-isopropyl-azetidin~3-yl)- 1 -methyl- 1 h-indol-6- yl]- 1 'h-[2,4'3bipyπdinyl-2'-one; 1 -[ 1 -methyl-3-< 1 -mcthyl-azetidin-3-yl)- 1 h-indol-ό-yl"|-4-(4- trifluoromethyl-phenyl)- 1 h-pyridin-2-one; 1-[3-(I -acetyI-azetidin-3-yl)- 1 -methyl- 1 h-indol-6- yl]-4-(4-trifluoromethyl-phenyl)-I h-pyridin-2-one; 6-(4-chloro-phenyl)-3-{3-[l-(tetrahydro- furan-S-ylmcthylJ-azctidin-S-y^-lh-indol-ό-yl J-Sh-pyrimidin^-onc; and 4-(4-fluoro- phenyl)-l-[3-( l-isopiOpyl-azctidin-3-yl)-l -methyl- lh-indol-6-ylj-l h-pyridin-2-one.
Claim 19. A pharmaceutical composition comprising a compound of claim 1 and an acceptable pharmaceutical carrier.
Claim 20. A method of treating mood disorders in a subject comprising administering a therapeutically effective amount of a compound of claim I .
Claim 23. A method of treating anxiety in a subject comprising administering a therapeutically effective amount of a compound of claim 1.
Claim 22. A method of treating obesity in a subject comprising administering a therapeutically effective amount of a compound of claim 1.
Claim 23. A method of treating urinary disorders in a subject comprising administering a therapeutically effective amount of a compound of claim 1 .
Claim 24. The method of claim 1 , wherein the mood disorder is depression.
Claim 25. Use of a compound of claim 1 in the manufacture of a pharmaceutical composition for treating a disorder selected from the group consisting of mood, anxiety and obesity related disorders.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US4055908P | 2008-03-28 | 2008-03-28 | |
| US61/040,559 | 2008-03-28 | ||
| US5698208P | 2008-05-29 | 2008-05-29 | |
| US61/056,982 | 2008-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009120655A1 true WO2009120655A1 (en) | 2009-10-01 |
Family
ID=41114297
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/038022 Ceased WO2009120655A1 (en) | 2008-03-28 | 2009-03-24 | Indole derivatives |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009120655A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103214408A (en) * | 2013-05-07 | 2013-07-24 | 南京理工大学 | Method for synthesizing bisindole methane derivative |
| EP2558094A4 (en) * | 2010-04-12 | 2013-09-25 | Merck Sharp & Dohme | PYRIDONE DERIVATIVES |
| JP2015503502A (en) * | 2012-01-12 | 2015-02-02 | 武田薬品工業株式会社 | Benzimidazole derivatives as MCH receptor antagonists |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000043393A1 (en) * | 1999-01-20 | 2000-07-27 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| US6358992B1 (en) * | 1998-11-25 | 2002-03-19 | Cell Pathways, Inc. | Method of inhibiting neoplastic cells with indole derivatives |
| WO2003074047A1 (en) * | 2002-03-04 | 2003-09-12 | 4Sc Ag | Indole derivatives as modulators of potassium channels |
| US20070299068A1 (en) * | 2004-07-14 | 2007-12-27 | Karp Gary M | Methods for treating hepatitis C |
-
2009
- 2009-03-24 WO PCT/US2009/038022 patent/WO2009120655A1/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6358992B1 (en) * | 1998-11-25 | 2002-03-19 | Cell Pathways, Inc. | Method of inhibiting neoplastic cells with indole derivatives |
| WO2000043393A1 (en) * | 1999-01-20 | 2000-07-27 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| WO2003074047A1 (en) * | 2002-03-04 | 2003-09-12 | 4Sc Ag | Indole derivatives as modulators of potassium channels |
| US20070299068A1 (en) * | 2004-07-14 | 2007-12-27 | Karp Gary M | Methods for treating hepatitis C |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2558094A4 (en) * | 2010-04-12 | 2013-09-25 | Merck Sharp & Dohme | PYRIDONE DERIVATIVES |
| JP2015503502A (en) * | 2012-01-12 | 2015-02-02 | 武田薬品工業株式会社 | Benzimidazole derivatives as MCH receptor antagonists |
| CN103214408A (en) * | 2013-05-07 | 2013-07-24 | 南京理工大学 | Method for synthesizing bisindole methane derivative |
| CN103214408B (en) * | 2013-05-07 | 2015-09-30 | 南京理工大学 | A kind of method of synthesizing bisindole methane derivative |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW201103930A (en) | Azetidinyl diamides as monoacylglycerol lipase inhibitors | |
| EP0464558B1 (en) | Antimigraine alkoxypyrimidine derivatives | |
| HU208132B (en) | Process for producing 3-arylcarbonyl-1h-indole derivatives and pharmaceutical compositions comprising same | |
| TW200306186A (en) | Pyridinoylpiperidines as 5-HT1F agonists | |
| KR20040017325A (en) | Phenylsulfonyl-1,3-dihydro-2h-indole-2-one derivatives, their preparation and their therapeutic use | |
| NO327378B1 (en) | Arylmethylamine derivatives, the use of the compounds as well as pharmaceutical compositions. | |
| CA2159772A1 (en) | 5-ht1f agonists for the treatment of migraine | |
| TW200401641A (en) | 1-Heterocyclylalkyl-3-sulfonylindole or-indazole derivatives as 5-hydroxytryptamine-6 ligands | |
| EP0687267B1 (en) | Pyrrolo-pyridine derivatives as ligands of dopamine receptor | |
| US5576336A (en) | Indole derivatives as dopamine D4 antagonists | |
| AU773164B2 (en) | Indolylpiperidine derivatives as antihistaminic and antiallergic agents | |
| HUT74096A (en) | 3-indolylpiperidine derivatives | |
| AU2006298852A1 (en) | Indane derivatives as MCH receptor antagonists | |
| WO2009120655A1 (en) | Indole derivatives | |
| WO2015152368A1 (en) | Oxazolidinone and oxazinanone derivatives | |
| US4870087A (en) | Indole containing, 2-thienyl-1,2,3,6-tetrahydropyridyls and their pharmaceutical use | |
| US4547576A (en) | Tetrahydrocarbazole derivatives | |
| AU673098B2 (en) | Antimigraine cyclobutenedione derivatives of indolylalkyl-pyridinyl and pyrimidinylpiperazines | |
| JP4177461B2 (en) | (4-Piperidinyl) -1H-2-benzopyran derivatives useful as antipsychotic agents | |
| JP2005104896A (en) | 2-alkoxy-6-amino-5-halogenopyridine-3-carboxamide derivatives and pharmaceutical compositions containing the same | |
| WO2009137270A2 (en) | Azetidine derivatives | |
| US5418242A (en) | Piperidinylthioindole derivatives, their methods of preparation and pharmaceutical compositions in which they are present, useful especially as analgesics | |
| US5521188A (en) | Antimigraine cyclobutenedione derivatives of indolylalkyl-pyridinyl and pyrimidinylpiperazines | |
| WO1999055695A1 (en) | Indolyl derivatives as serotonergic agents | |
| CN101460460A (en) | Piperidine derivatives useful as serotonin transporter inhibitors and neurokinin-1 receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09725157 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09725157 Country of ref document: EP Kind code of ref document: A1 |