WO2009133278A1 - Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique - Google Patents
Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique Download PDFInfo
- Publication number
- WO2009133278A1 WO2009133278A1 PCT/FR2009/000381 FR2009000381W WO2009133278A1 WO 2009133278 A1 WO2009133278 A1 WO 2009133278A1 FR 2009000381 W FR2009000381 W FR 2009000381W WO 2009133278 A1 WO2009133278 A1 WO 2009133278A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- solid
- mil
- mmol
- curve
- gas
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/02—Iron compounds
- C07F15/025—Iron compounds without a metal-carbon linkage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/02—Cosmetics or similar toiletry preparations characterised by special physical form
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/19—Cosmetics or similar toiletry preparations characterised by the composition containing inorganic ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/19—Cosmetics or similar toiletry preparations characterised by the composition containing inorganic ingredients
- A61K8/28—Zirconium; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/19—Cosmetics or similar toiletry preparations characterised by the composition containing inorganic ingredients
- A61K8/29—Titanium; Compounds thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/49—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds containing heterocyclic compounds
- A61K8/494—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds containing heterocyclic compounds with more than one nitrogen as the only hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/58—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds containing atoms other than carbon, hydrogen, halogen, oxygen, nitrogen, sulfur or phosphorus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/223—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material containing metals, e.g. organo-metallic compounds, coordination complexes
- B01J20/226—Coordination polymers, e.g. metal-organic frameworks [MOF], zeolitic imidazolate frameworks [ZIF]
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28014—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their form
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28054—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their surface properties or porosity
- B01J20/28069—Pore volume, e.g. total pore volume, mesopore volume, micropore volume
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28054—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their surface properties or porosity
- B01J20/28078—Pore diameter
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F17—STORING OR DISTRIBUTING GASES OR LIQUIDS
- F17C—VESSELS FOR CONTAINING OR STORING COMPRESSED, LIQUEFIED OR SOLIDIFIED GASES; FIXED-CAPACITY GAS-HOLDERS; FILLING VESSELS WITH, OR DISCHARGING FROM VESSELS, COMPRESSED, LIQUEFIED, OR SOLIDIFIED GASES
- F17C11/00—Use of gas-solvents or gas-sorbents in vessels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2800/00—Properties of cosmetic compositions or active ingredients thereof or formulation aids used therein and process related aspects
- A61K2800/40—Chemical, physico-chemical or functional or structural properties of particular ingredients
- A61K2800/58—Metal complex; Coordination compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/80—Complexes comprising metals of Group VIII as the central metal
- B01J2531/84—Metals of the iron group
- B01J2531/842—Iron
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/1691—Coordination polymers, e.g. metal-organic frameworks [MOF]
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2226—Anionic ligands, i.e. the overall ligand carries at least one formal negative charge
- B01J31/223—At least two oxygen atoms present in one at least bidentate or bridging ligand
- B01J31/2239—Bridging ligands, e.g. OAc in Cr2(OAc)4, Pt4(OAc)8 or dicarboxylate ligands
Definitions
- the present invention relates to solids consisting of metal-organic network or "Metal-Organic
- the MOF solids of the present invention are capable of adsorbing and releasing controlled gases of biological interest in a controlled manner. They can be used in the pharmaceutical field and / or for applications in the cosmetics field. They can also be used in the food field.
- Metal-Organic Framework are inorganic-organic hybrid framework coordination polymers comprising metal ions and organic ligands coordinated to metal ions. These materials are organized into mono-, bi- or three-dimensional networks where the clusters Metals are interconnected by spacer ligands periodically. These materials have a crystalline structure, are most often porous and are used in many industrial applications such as gas storage, adsorption of liquids, separation of liquids or gases, catalysis, etc.
- US patent application 10 / 039,733 [1] describes a reaction process involving a catalyst system comprising a MOF material based on Zinc. This same material is also used for storing gas in US patent application 10 / 061,147 [2].
- MOF materials based on networks of the same topology are called "isoreactive". These networks organized in space have made it possible to obtain a more homogeneous porosity.
- patent application US 10 / 137,043 [3] describes several zinc-based IRMOF (Isoreticular Metal-Organic Framework) materials used for gas storage.
- gases of biological interest such as NO, CO and H 2 S are of extreme importance for the biological functioning of mammals. They are involved in a large number of processes such as vasodilatation, prevention of platelet aggregation and thrombosis formation, neurotransmission and wound healing.
- NO has been extensively studied in biology [32].
- the biological activity of NO includes [33]: - anti-inflammatory activity,
- NO is involved in the activity of many drugs (calcium channel blocker, ACE inhibitors and ANGII type 1 receptor antagonists, ⁇ -blocker and hydroxymethylglutaryl-CoA reductase inhibitors).
- CO is involved in biological activities such as:
- pancreatic beta cells The protection of pancreatic beta cells from apoptosis [40];
- US Patent 2002155166 describes for example the use of CO as a biological marker and therapeutic agent for various types of diseases and in the field of transplantation.
- WO 0278684 relates to methods and compositions for treating vascular, inflammatory and immune system diseases using compounds capable of generating CO in vivo.
- CH 2 Cl 2 is described as the preferred compound capable of generating CO in vivo. Indeed, CH 2 Cl 2 is metabolized to CO and thus provides a source of gas.
- WO 02092075 uses carbonylated metals as the source of CO gas emissions.
- H 2 S as a signaling molecule is increasingly important [49]. Since the pKa of H 2 S is 6.8, at physiological pH it is mainly present as H 2 S and [HS] 2 . The concentration of H 2 S is 50-160 mM in the brain tissue and 10-100 mM in the blood.
- H 2 S is active in the cardiovascular system and in the central nervous system. For example, it can cause vasodilation [50]. Moreover, it is able to easily coordinate hemo groups and certain cytochromes. More research is needed to demonstrate the importance of H 2 S as a signaling molecule.
- NO can cause harmful side effects: this is the case in septicemia, where excessive production of NO by macrophages leads to massive vasodilation, the main cause of hypotension in septic shock, neurotransmitter, nerve cells, relaxant smooth muscle, digestive tract, regulator of apoptosis, antiapoptotic or apoptotic depending on the presence or absence of cell reducers).
- NONOate or metal-component polymers have low storage capacity, require high pressure to charge NO, are relatively expensive and potentially toxic [7,8].
- nitrate-based acidic creams which are potential NO donors, are pro-inflammatory and are not suitable for sensitive skin [9].
- antithrombotic apparatuses such as, for example, stents, catheters and cannulas which are inserted into the flow.
- the present invention is specifically intended to meet these needs and disadvantages of art prior art by providing porous crystalline MOF solids charged with at least one Lewis-based gas selected from the group consisting of NO, CO and H 2 S, at least a portion of which is coordinated with M, said solid comprising a three-dimensional succession of patterns corresponding to the following formula (I):
- Each occurrence of M independently represents an ion of a transition metal selected from the group consisting of Fe, Ti, Zr, Mn and wherein z is 2 to 4 or a mixture thereof;
- M is 1 to 12
- P is 1 to 6; • X is an anion selected from the group consisting of OH " is as defined below, o ù R 1 is hydrogen, C 1 -C 12 linear or branched, optionally substituted, aryl, wherein n is an integer from 1 to 4;
- L is a spacer ligand comprising a radical R
- an organic radical comprising a metal element selected from the group consisting of ferrocene, porphyrin, phthalocyanine; the radical R being optionally substituted by one or more groups R 2 independently selected from the group comprising
- aryl, heteroaryl or heterocyclic radical is optionally substituted; or, when G represents R G1 and R G2 together with the nitrogen atom to which they are bonded form an optionally substituted heterocycle or heteroaryl.
- MOFs according to the invention have, among others, the advantage: - of being based on non-toxic metals and therefore adapted to an application in the pharmaceutical, medical and / or cosmetic fields,
- 20 - Have a greater stability than that described for metals such as copper or aluminum, 20 - have a modular stability depending on the choice of the structure and the organic ligand, thus making it possible to adapt the MOF to different desired applications.
- the terms “salting out / releasing”, “releasing / releasing” and “delivering / delivering” will be; indifferently- used to signify the partial or total release of the gases present in the MOF solids.
- partial is meant a release of less than 100% of the initially adsorbed amount.
- substituted for example means the replacement of a hydrogen radical in a given structure by a radical R 2 as defined above. When more than one position may be substituted, the substituents may be the same or different at each position.
- the term "spacer ligand” means a ligand (including, for example, neutral species and ions) coordinated with at least two metals, participating in the distance between these metals and in the formation of empty spaces. or pores.
- the spacer ligand can comprise 1 to 6 carboxylate groups, as defined above, which can be monodentates or bidentates, that is to say include one or two points of attachment to the metal.
- the points of attachment to the metal are represented by the symbol # in the formulas. When the structure of a function A has two points of attachment #, it means that the coordination to the metal can be done by one or the other or the two points of attachment.
- alkyl means a linear, branched or cyclic, saturated or unsaturated, optionally substituted carbon radical comprising 1 to 12 carbon atoms, for example 1 to 10 carbon atoms, for example 1 to 10 carbon atoms. 8 carbon atoms, for example 1 to 6 carbon atoms
- alkene means an alkyl radical, as defined above, having at least one carbon-carbon double bond.
- alkyne in the sense of the present invention, an alkyl radical, as defined above, having at least one carbon-carbon triple bond.
- aryl means an aromatic system comprising at least one ring which satisfies Hekel's aromaticity rule. Said aryl is optionally substituted and may comprise from 6 to 50 carbon atoms, for example 6 to 20 carbon atoms, for example 6 to 10 carbon atoms.
- heteroaryl means a system comprising at least one aromatic ring of 5 to 50 members, among which at least one member of the aromatic ring is a heteroatom, chosen in particular from the group comprising sulfur, oxygen, nitrogen, boron.
- Said heteroaryl is optionally substituted and may comprise from 1 to 50 carbon atoms, preferably 1 to 20 carbon atoms, preferably 3 to 10 carbon atoms.
- cycloalkyl in the sense of the present invention, an optionally substituted cyclic, saturated or unsaturated carbon radical, which may comprise 3 to 20 carbon atoms, preferably 3 to
- haloalkyl in the sense of the present invention, an alkyl radical as defined above, said alkyl system comprising at least one halogen.
- heteroalkyl in the sense of the present invention, an alkyl radical as defined previously, said alkyl system comprising at least one heteroatom, in particular selected from the group consisting of sulfur, oxygen, nitrogen, boron.
- heterocycle in the sense of the present invention, a cyclic carbon radical comprising at least one heteroatom, saturated or unsaturated, optionally substituted, and which may comprise 2 to 20 carbon atoms, preferably 5 to 20 carbon atoms, preferably 5 to 10 carbon atoms.
- the heteroatom may for example be selected from the group consisting of sulfur, oxygen, nitrogen and boron.
- Heteroalkoxy and “heteroaryloxy” in the sense of the present invention respectively an alkyl, aryl, heteroalkyl and heteroaryl radical bonded to an oxygen atom.
- Alkylthio means "arylthio"
- Heteroalkylthio and “heteroarylthio” in the sense of the present invention respectively an alkyl, aryl, heteroalkyl and heteroaryl linked to a sulfur atom.
- the Lewis gases according to the invention are gases of biological interest chosen from the group comprising NO, CO and H 2 S.
- said gas is NO.
- M is selected from the group consisting of Fe, Ti, Mn, Zr. M can also be a mixture of these metals. M is advantageously Fe. As previously indicated, M is a transition metal ion M z + in which z is 2 to 4.
- the solids of the invention comprise a three-dimensional succession of units of formula (I) in which M may represent a single type of ion M z + , for example Fe, in which z may be identical or different, for example 2, 3 or a mixture of 2 and 3.
- the solids of the invention may comprise a three-dimensional succession of units of formula (I) in which M may represent a mixture of different ions M z + , for example Fe and
- each metal ion z may be the same or different, for example 2, 3, 4 or a mixture of 2, 3 and 4.
- M z + represents the trivalent octahedral Fe with z equal to 3.
- the Fe has a coordination number of 6.
- the coordination number is meant the number of bonds for which the two electrons shared in the bond originate from the same atom.
- the electron donor atom acquires a positive charge while the electron acceptor atom acquires a negative charge.
- the metal ions can be isolated or grouped into metal "clusters".
- the MOF solids according to the invention can for example be constructed from chains of octahedra or trimers of octahedron.
- metal cluster means a set of atoms containing at least two metal ions bonded by ionocovalent bonds, either directly by anions, for example O, OH, Cl, etc., or by the organic ligand.
- MOF solids according to the invention can be in different forms or "phases" taking into account the various possibilities of organization and connections of the ligands to the metal ion or metal group.
- phase means a hybrid composition comprising at least one metal and at least one organic ligand having a defined crystalline structure.
- the crystalline spatial organization of the solids of the present invention is the basis of the particular characteristics and properties of these materials. In particular, it governs the size of the pores, which has an influence on the specific surface of the materials and on the adsorption characteristics.
- the pore size can be adjusted by choosing appropriate ligands L.
- the ligand L of the unit of formula (I) of the present invention may be a di-, tri-, tetra- or hexa-carboxylate ligand chosen from the group comprising: (succinate), C 3 H 6 (CO 2 -) 2 (glutarate), C 4 H 4 (CO 2 -) 2 (muconate), C 4 H 8 (CO 2 -) 2 (adipate), C 5 H 3 S (CO2 -) 2 (2,5 thiophènedicarboxylate), C 6 H 4 (CO2 -) 2 (terephthalate), C 6 H 2 N 2 (CO 2 ") 2 (2, 5-pyrazine dicarboxylate), C 10 H 6 (CO 2 -) 2 (naphthalene-2,6-dicarboxylate), C 12 H ⁇ (CO 2 " ) 2 (biphenyl-4,4'-dicarboxylate), C 12 H 8 N 2 (CO 2 -) 2
- the MOF solid according to the invention may comprise a mass percentage of M in the dry phase of 5 to 50%, preferably 18 to 31%.
- the mass percentage (% m) is a unit of measurement used in chemistry and metallurgy to designate the composition of a mixture or an alloy, that is to say the proportions of each component in the mixture.
- 1% m of a component component Ig per 100 g of mixture or 1 kg of said component per 100 kg of mixture.
- the MOF solids of the present invention have the particular advantage of having a thermal stability up to a temperature of 350oC. More particularly, these solids have a thermal stability of 120oC and 350oC).
- the MOF solid according to the invention may have a pore size of 0.4 to 6 nm, preferably 0.5 to 5.2 nm, and more preferably 0.5 to 3.4 nm. .
- the MOF solid according to the invention may have a specific surface area (BET) of 5 to 6000 m 2 / g, preferably 5 to 4500 m 2 / g.
- BET specific surface area
- the MOF solid according to the invention may have a pore volume of 0 to 4 cm 2 / g, preferably of 0.05 to 2 cm 3 / g.
- the pore volume means the accessible volume for the gas molecules.
- the MOF solid of the invention can have a gas loading capacity of 0.5 to 50 mmol of gas per gram of dry solid.
- the load capacity means the gas storage capacity or the amount of adsorbed gas in the material.
- the loading capacity can be expressed in mass capacity (gram / gram) or in molar capacity (mol / mol) or in others (mol / gram, gram / mol, etc.)
- the Lewis base coordinates with M.
- at least 1 to 5 mmol of gas per gram of dry solid is coordinated with M.
- the part of the gas or gases which does not coordinate with M can advantageously fill the free space in the pores.
- the MOF solid of the present invention can be in the form of a robust structure, which has a rigid framework and contracts only very little when the pores are empty of their contents which can
- solvent non-coordinative carboxylic acid, etc. It can also be in the form of a flexible structure, which can swell and deflate varying the pore opening depending on the nature of the adsorbed molecules which
- the term "rigid structure” means structures which swell or contract only very slightly, that is to say with an amplitude of up to 10%.
- the MOF solid according to the invention can have a rigid structure which is swells or contracts with an amplitude ranging from 0 to 10%.
- the rigid structures may for example be built based on chains or trimers of octahedra.
- the solid MOF solid may have a mass percentage of M dry phase of 5 to 50%, preferably 18 to 31%.
- M will represent iron here.
- the MOF solid of rigid structure according to the invention may have a pore size of 0.4 to 6 nm, in particular from 0.5 to 5.2 nm, more particularly from 0.5 to 3.4 nm.
- the MOF solid of rigid structure according to the invention may have a pore volume of 0.5 to 4 cm 3 / g, in particular of 0.05 to 2 cm 3 / g.
- the term "flexible structure” means structures that swell or contract with a large amplitude, in particular with an amplitude greater than 10%, preferably greater than 50%.
- the flexible structures may for example be constructed based on chains or trimers of octahedra.
- the MOF material according to the invention may have a flexible structure which swells or contracts with an amplitude of from 10% to 300%, preferably from 50% to 300%.
- the MOF solid of flexible structure may have a mass percentage of M in the dry phase of 40%, preferably 18 to 31%.
- M will represent iron here.
- the MOF solid of flexible structure may have a pore size of 0.4 to 6 nm, in particular 0.5 to 5.2 nm, and more particularly 0.5 at 1.6 nm.
- the MOF solid of flexible structure according to the invention may have a pore volume of 0 to 3 cm 3 / g, in particular from 0 to 2 cm 3 / g.
- the inventors have experimentally demonstrated that the amplitude of the flexibility depends on the nature of the ligand and the solvent used, as described in the "Examples" section below.
- MOF solids may comprise a three-dimensional succession of units corresponding to formula (I).
- the MOF solids may comprise a three-dimensional succession of iron (III) carboxylates of formula (I).
- iron (III) carboxylates can be chosen from the group comprising MIL-88, MIL-89, MIL-96, MIL-100, MIL-101 and MIL-102, and more particularly in the group comprising MIL-88A. , MIL-88B, MIL-88Bt, MIL-88C, MIL-88D, MIL-88E, MIL-89, MIL-96, MIL-100, MIL-101, MIL-102. These reasons are presented in the "Examples" section.
- the MOF solids may comprise a three-dimensional succession of units corresponding to formula (I), chosen from the group comprising: flexible structure, for example MIL-88A of flexible structure, for example MIL-88B of flexible structure, for example MIL-88C of flexible structure, for example MIL-88D flexible structure, for example MIL-89 of rigid structure, for example MIL-96 of rigid structure, for example MIL- 100 rigid structure, for example MIL-101 of rigid structure, for example MIL-102 in which X is as defined above.
- flexible structure for example MIL-88A of flexible structure, for example MIL-88B of flexible structure, for example MIL-88C of flexible structure, for example MIL-88D flexible structure, for example MIL-89 of rigid structure, for example MIL-96 of rigid structure, for example MIL- 100 rigid structure, for example MIL-101 of rigid structure, for example MIL-102 in which X is as defined above.
- MOF materials of the same general formula (I) but of different structures This is the case, for example, of MIL-88B and MIL-101 solids. Indeed, the difference between MIL-88B and MIL-101 solids lies in the way ligands are connected.
- the octahedron trimers in the MIL-101 solid, the ligands L assemble in the form of rigid tetrahedra, while in the solid MIL-88B, they form trigonal bipyramids, making possible the separation between the trimers.
- the mode of assembly of these ligands can be controlled during the synthesis for example by adjusting the pH.
- the MIL-88 solid is obtained in a less acidic medium than the MIL-101 solid as described in the "Examples” section below.
- the MOF solids of the invention can make possible the grafting of molecules on their surface in order to meet the needs related to the vectorization of compounds towards specific biological targets and / or the stealthiness of the particles. This thus makes it possible to improve the biodistribution of the active ingredients in a more targeted manner.
- the MOF solids according to the invention may optionally comprise at their surface at least one organic surface agent. This can be grafted or deposited on the surface of solids, for example adsorbed on the surface or covalently bound, by hydrogen bonding, by Van der Waals bond, by electrostatic interaction.
- the surfactant may also be incorporated by entanglement in the manufacture of MOF solids [10, 28].
- surfactant means a molecule covering in part or in part all the surface of the solid allowing to modulate the surface properties of the material, for example: to modify its biodistribution, for example to avoid its recognition by the reticuloendothelial system ("stealth"), and / or to give it bioadhesion properties interesting when administered orally, ocularly, nasally, and / or
- surfactants can be used to combine the above properties.
- the organic surface agent can be chosen for example from the group comprising:
- an oligosaccharide such as cyclodextrins
- a polysaccharide such as chitosan, dextran, fucoidan, alginate, pectin, amylose, starch, cellulose, xylan, a glycosaminoglycan such as hyaluronic acid, heparin,
- polyethylene glycol PEG
- polyvinyl alcohol polyvinyl alcohol
- polyethylene imine PEG
- a surfactant such as pluronic, lecithin.
- - vitamins such as biotin
- coenzymes such as lipoic acid
- the surfactant can also be a targeting molecule, i.e. a molecule recognizing or being specifically recognized by a biological target.
- a targeting molecule i.e. a molecule recognizing or being specifically recognized by a biological target.
- the organic surfactant may be a targeting molecule selected from the group consisting of biotin, chitosan, lipoic acid, an antibody or antibody fragment, a peptide.
- a targeting molecule selected from the group consisting of biotin, chitosan, lipoic acid, an antibody or antibody fragment, a peptide.
- biotin at the surface can be used to couple ligands easily, for example by simple incubation.
- This method of surface modification has the advantage of not disturbing the core of the MOF solids, even when they contain gas, and can be done after the synthesis of MOF solids, and thus to offer a variety of possible overlaps.
- the subject of the invention is also a method for preparing MOF solids as defined in the present invention, comprising at least one reaction step consisting of
- At least one ligand L 'comprising a radical R having q groups * -C ( O) -R 3 , where
- step (ii) activating the MOF material obtained in (i); and (iii) contacting the material.
- M is an ion of a transition metal as defined above.
- the preparation of MOF materials may preferably be carried out in the presence of energy which may be provided, for example, by heating, such as, for example, hydrothermal or solvothermal conditions, but also by microwaves, ultrasonics, grinding, a process involving a supercritical fluid, etc.
- energy which may be provided, for example, by heating, such as, for example, hydrothermal or solvothermal conditions, but also by microwaves, ultrasonics, grinding, a process involving a supercritical fluid, etc.
- the corresponding protocols are those known to those skilled in the art. Non-limiting examples of protocols that can be used for hydrothermal or solvothermal conditions are described for example in [20]. For the microwave synthesis, non-limiting examples of usable protocols are described, for example [21, 22, 23, 24].
- the conditions in the presence of a roller mill may for example refer to the publications [25, 26]. , 27].
- hydrothermal or solvothermal conditions whose reaction temperatures may vary between 0 and 220oC, are generally carried out in glass (or plastic) containers when the temperature is below the boiling point of the solvent.
- Teflon bodies inserted into metal bombs are used [20]
- the solvents used are generally polar.
- the following solvents may be used: water, alcohols, dimethylformamide, dimethylsulfoxide, acetonitrile, tetrahydrofuran, diethylformamide, chloroform, cyclohexane, acetone, cyanobenzene, dichloromethane, nitrobenzene, ethylene glycol, dimethylacetamide or mixtures of these solvents.
- One or more co-solvents may also be added at any stage of the synthesis for better solubilization of the compounds of the mixture. It may in particular be composed of monocarboxylic acids, such as acetic acid or formic acid. benzoic acid, etc.
- One or more additives may also be added during synthesis to modulate the pH of the mixture. These additives are chosen from the acids organic or inorganic or organic bases. By way of example, the additive may be chosen from the group comprising: HF, HCl, HNO 3 , H 2 SO 4 , NaOH, KOH, lutidine, ethylamine, methylamine, ammonia, urea , EDTA, tripropylamine, pyridine.
- the reaction step (i) can be carried out according to at least one of the following reaction conditions: with a reaction temperature of 0 ° C. to 220 ° C., preferably of 50 ° to 150 ° C.;
- co-solvent chosen from the group comprising acetic acid, formic acid, benzoic acid;
- a solvent in the presence of a solvent is chosen from the group comprising water, the alcohols R s -OH where R s is a linear or branched C 1 -C 6 alkyl radical, dimethylformamide, dimethylsulfoxide, acetonitrile and tetrahydrofuran; diethylformamide, chloroform, cyclohexane, acetone, cyanobenzene, dichloromethane, nitrobenzene, ethylene glycol, dimethylacetamide or mixtures of these solvents, miscible or not; in a supercritical medium, for example in supercritical CO 2 ;
- step (iii) As indicated, prior to contacting the MOF material with the Lewis base gas in step (iii), it is necessary for the material from step (i) to be activated in step (ii).
- This activation step (ii) makes it possible to empty the pores of the MOF material and to make them accessible for the coordination of the gas or gases.
- the emptying can take place, for example, by the departure of water molecules and / or solvents present in the reaction medium either by activation under primary or secondary vacuum or under a gas flow (helium, nitrogen, air, etc.), with or without the heating of the solid at a temperature of 25 to 300oC, in particular of 50 to 250oC, and more particularly of 100 to 250oC.
- the heating can be carried out for a period of time between 1 hour and 96 hours, typically between 3 and 15 hours.
- Step (ii) may also be a step of reducing the metal centers M of said MOF material to M z + ions in which z is 2 to 4.
- the metal centers may be partially or even totally reduced.
- the metal centers may consist of identical or different metals, for example only iron, or a mixture of one or more metals such as iron in the presence of titanium, manganese or zirconium.
- the partial reduction of the metal centers has an influence on the duration of gas release by the MOF solid, which increases significantly.
- the activation step (ii) may, in addition, be carried out at a high temperature and under reduced pressure.
- Reduced pressure means a pressure ranging from 1 to 10 -2 Pa, preferably 10 -3 to 10 -5 Pa.
- the activation may be at 50-250 ° C under a pressure of 1 to 10 -2 Pa, or 10 -3 to 10 -5 Pa.
- step (iii) of the process of the invention the activated MOF material in step (ii) is contacted with at least one of Lewis base gas.
- the gas may be in pure form or in admixture with an inert gas.
- the contacting of the solid with the gas in step (iii) can be carried out at a temperature ranging from -150 to 100 oC.
- Step (iii) can also be carried out at a pressure ranging from 10 4 to 10 7 Pa.
- the Lewis base gas is preferably NO.
- Step (iii) can then be carried out at a temperature ranging from
- the NO contact with the solid can be carried out at a pressure ranging from 10 5 to 10 6 Pa.
- a Lewis base gas mixture may be used in step (iii) of the process.
- the MOF solid according to the invention can have a gas loading capacity of 0.5 to 50 mmol of gas per gram of dry solid.
- the solid according to the invention can have at least 1 to 5 mmol of gas per gram of dry solid coordinated with M.
- the process for preparing the MOF solids according to the invention may further comprise a step
- This fixing step (iv) can be carried out during or after the reaction step (i) or after the activation step (ii) and before step (iii) contacting the MOF material with the gas. Examples are provided below.
- the preparation method of the invention has the advantage of allowing the obtaining of pure crystalline MOF solids, homogeneous, in a reduced number of stages and with high yields. This reduces the synthesis time and manufacturing costs.
- the inventors have also demonstrated that the particular structural characteristics of the solids of the present invention, especially in terms of flexibility or pore size, make them adsorbents with a high loading capacity, high selectivity and high purity. . They thus make possible the selective adsorption of Lewis-based gas molecules of biological interest, such as molecules of NO, CO or H 2 S, with a favorable energy cost and a higher release time.
- Lewis-based gas molecules of biological interest such as molecules of NO, CO or H 2 S
- the research work carried out by the inventors has allowed them to highlight the interest of the MOF materials according to the invention for the adsorption and the controlled release, in terms of quantity of gas and release time, of gas Biological interest.
- the MOF solids of the invention make it possible to control the duration of release of the gases.
- the gas release time can range from 1 to 100 hours whereas with the zeolites of the state of the art, the duration of this release does not exceed 10 hours under a water vapor pressure [10, 11].
- the MOF solids of the invention have a Lewis base gas adsorption and storage capacity substantially greater than that of known zeolites.
- the MOFs of the invention may have a NO adsorption capacity ranging from 2.5 to 4.5 mmol / g whereas with known zeolites, this capacity is less than 1.5 mmol / g. This reduces the amount of material to be used for a given application.
- this greater adsorption capacity makes it possible to have materials in which the release of the gases can be done continuously.
- the release of the gases can also be targeted when the materials comprise on their surface at least one targeting agent as defined above.
- the invention also relates to the use of MOF solids according to the invention, charged with at least one Lewis gas of biological interest, at least a part of which is coordinated with M as a medicament.
- Said gas or the gas mixture can be contained in the pores and at least partially coordinated with the invention.
- the MOF solids according to the invention have the advantage of having high adsorption capacities.
- they make it possible to effectively adsorb gas molecules which may present particular difficulties, for example because of their instability, their high reactivity, their low solubility, and so on.
- the Lewis base gas may be any electron donor gas of biological interest, such as, for example, NO, CO, H 2 S. [10, 28].
- MOF solids of the invention can be used, for example, as a coating for the aforementioned medical articles. As a coating, they can be used alone or in combination with other therapeutic agents. MOFs being charged with gas, are capable of releasing the gas (or the gas mixture). They can then be an effective means to prevent the formation of thromboses in contact with the foreign body.
- the MOF solids of the invention can therefore be used as an antithrombotic drug. It can also be considered to use them alone or in combination with other known antithrombotic agents such as clopidogrel.
- the invention also extends to medical articles, such as, for example, stents, catheters, prosthetic conduits and other medical implants in the body following a surgical procedure, comprising an MOE solid according to the Keefer L. invention.
- the MOF solid according to the invention can be loaded with at least one Lewis gas of biological interest for use in the field of cosmetics or dermatology.
- the activity For example, antibacterial NO may be of great interest for applications particularly in the field of cosmetic creams.
- a modulable release of NO will allow to disinfect the skin for a prolonged period.
- the inorganic-organic character of MOFS solids will facilitate their dispersion in cosmetic creams.
- the beneficial cosmetic or dermatological effects that may result from the use of an MOF solid according to the invention loaded with NO are, for example: the anti-wrinkle effects, the reconstitution of the fullness of the lips and the exaltation of their natural red color, the stimulation of the natural rosy color of the skin and the homogenization of the complexion, by a vasodilatation action and / or an increase in the blood circulation of the skin; - reduction of skin damage caused by UV light; - the anti-bacterial effect in the treatment of acne.
- the MOF solids of the invention have the advantage of presenting unexpected load capacities, never before attained in the materials of the prior art.
- MOF solids with a flexible structure can in particular allow a release with a flexible duration.
- MOF solids of flexible structure comprising hydrophobic ligands, will allow to modulate the duration of the release of the gas.
- MOFs of flexible structures as defined above can thus constitute a credible alternative for a release of gas of biological interest such as NO over a longer period.
- these solids whose first toxicity tests show that they are not a priori toxic, can also be of great interest in the food industry, particularly as antibacterial agent, for example for the preservation of food and industrial preparations.
- the introduction of a small amount of MOF solid according to the invention combined with the presence of water naturally present in the food can lead to a slow and continuous release of NO to inhibit bacterial growth and destroy microorganisms.
- MOF solids according to the invention are pharmaceutical, cosmetic or dermatological compositions comprising an MOF solid according to the invention and a pharmaceutically or cosmetically acceptable vehicle.
- MOF solids according to the invention have been the subject of very positive toxicity studies, described in the "Examples" section. They also appear to be biodegradable and degradability studies are still in progress.
- the MOF solids of the present invention used for the release of gas of biological interest make it possible to overcome the problems related to the toxicity in the prior art cited above.
- MOFs based on low toxicity metals make it possible to envisage applications in biological conditions.
- the MOF solids can be both loaded with gas and in principle pharmaceutically active.
- the pharmaceutically active principle may be contained in the pores. This provides a combined therapeutic effect.
- the MOF solids according to the invention can be loaded in principle pharmaceutically active with a loading capacity of 1 to 200% by weight of dry solid, for example from 1 to 70% by weight of dry solid, or about 10% by weight. at 700 mg per gram of dry solid.
- the invention extends to the use of such solids as a medicament.
- their preparation process may comprise, in addition, a step (v) of introduction into said solid of at least one molecule. of interest, which may be a pharmaceutically active ingredient.
- Said introduction step can be carried out during the reaction step (i) or after it so as to obtain a solid loaded with a molecule of interest.
- the molecule of interest may for example be introduced into the MOF material of the present invention:
- the gas is adsorbed by the material
- the MOF solids of the invention loaded in one or a mixture of gases, comprises an L ligand which itself has a therapeutic activity.
- a combined therapeutic effect can then be obtained by the release of the (or) gas and the activity of L after dissolution of the material.
- FIG. 1 represents an X-ray diffractogram of the solid MIL-100 (Fe).
- Figure 3 shows a thermogravimetric analysis (under air, with a heating rate of 2 ° C / minute) of the compound MIL-100 (Fe).
- FIG. 4 represents the X-ray diffractogram of the solid MIL-101 (Fe)
- Figure 5 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the compound MIL-101 (Fe).
- Figure 6 shows RX diffractograms of raw MIL-88A solids (top curve) and suspended in water (bottom curve).
- FIG. 7 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the compound MIL-88A (Fe) hydrated.
- FIG. 8 X-ray diffractograms of solids MIL-88B dry (bottom curve (b)) and hydrated (curve (a) from the top).
- FIG. 9 represents a thermogravimetric analysis (under air, with a heating rate of 2 ° C./minute) of the hydrated MIL-88B compound.
- Figure 10 shows X-ray diffractograms of dry MIL-89 solids (curve a), DMF (curve b) and hydrated solids (curve c).
- Figure 11 shows an X-ray diffractogram of the MIL-88C solid.
- FIG. 12 represents a thermogravimetric analysis (under air, with a heating rate of 2 ° C./minute) of the crude synthetic compound MIL-88C.
- FIG. 13 represents X-ray diffractograms of the raw MIL-88D (bottom curve (b)) and hydrated (top curve (a)) solids.
- FIG. 14 represents a thermogravimetric analysis (under air, with a heating rate of 2 ° C./minute) of the hydrated compound MIL-88D (Fe).
- Figure 15 shows X-ray diffractograms of crude MIL-88B-NO 2 solid (curve (a) at the top) and hydrated (curve (b) at bottom).
- Figure 16 shows a thermogravimetric analysis (under air, with a heating rate of 2 ° C / minute) of the compound MIL-88B-NO 2 (Fe) after washing and drying.
- FIG. 17 represents X-ray diffractograms of the solid MIL-88B-2OH crude (curve (c) below), hydrated (curve (b) of the medium) and dried under vacuum (curve (a) at the top).
- Figure 18 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the hydrous MIL-88B-2OH (Fe) compound.
- FIG. 19 represents X-ray diffractograms of the crude MIL-88B-NH 2 solid (curve (b) at the bottom) and of the dry MIL-88B-NH 2 solid (curve (a) at the top).
- Figure 20 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the hydrous MIL-88B-NH 2 (Fe) compound.
- FIG. 21 shows RX diffractograms of the raw MIL-88B-CH 3 solids (curve (a) at the top), hydrated (curve (b) of the medium) and solvated with DMF (curve (c) at the bottom).
- FIG. 22 represents X-ray diffractograms of the solid MIL-88B-C1 solid (curve (b) below) and hydrated (curve (a) at the top).
- FIG. 23 represents a thermogravimetric analysis (under air, with a speed of heating 2 ° C / minute) of the compound MIL-88B-Cl (Fe) hydrated.
- FIG. 25 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the hydrated compound MIL-88B-4CH 3 (Fe).
- FIG. 26 represents X-ray diffractograms of the raw MIL-88B-4F solids (curve (c) at the bottom), hydrated (curve (b)) and solvated with EtOH (curve (a) at the top).
- Figure 27 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the hydrous MIL-88B-4F (Fe) compound.
- FIG. 28 represents X-ray diffractograms of the raw MIL-88B-Br solids (curve (b) at the bottom) and hydrated solids (curve (a) at the top).
- FIG. 29 shows a thermogravimetric analysis (in air, with a heating rate of 2oC / minute) of the compound MIL-88B-Br (Fe) hydrated.
- FIG. 30 represents an X-ray diffractogram of the solid MIL-88E (Fe).
- Figure 31 shows RX diffractograms of the raw MIL-88F (curve (b) bottom) and hydrated solids (curve (a) at the top).
- FIG. 32 represents a thermogravimetric analysis (under air, with a heating rate of 2 ° C./minute) of the hydrated compound MIL-88F (Fe).
- Figure 33 shows X-ray diffractograms of MIL-88D- crude solid 4CH 3 (curve (b) below) and hydrated (curve (a) above).
- FIG. 34 represents a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the compound MIL-88D-4CH 3 (Fe) hydrated.
- Figure 35 shows X-ray diffractograms of the raw MIL-88D-2CH 3 (curve (c) bottom), hydrated (curve (b)) and wet (curve (a) top).
- FIG. 36 represents a thermogravimetric analysis (in air, with a heating rate of 2 ° C./minute) of the crude synthetic compound MIL-88D-2CH 3 (Fe).
- FIG. 37 represents an X-ray diffractogram of the crude MIL-88G solid (curve (c) below), solvated with DMF (curve (b) of the medium) and solvated with pyridine (curve (a) at the top).
- Figure 38 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / min) of the crude synthetic MIL-88G (Fe) compound.
- Figure 39 shows a diffractogram RX of the solid MIL-88G-2C1 crude (curve (b) down) and dry (curve (a) at the top).
- FIG. 40 shows a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the crude synthetic MIL-88G-2C1 (Fe) compound.
- FIG. 41 represents X-ray diffractograms of the solid MIL-102 (Fe) solids (curve (a)) and reference MIL-102 (Cr) (curve (b)).
- FIG. 42 represents a the ⁇ ogravimetric analysis (under air) of the crude synthetic MIL-102 (Fe) compound.
- Figure 43 shows a reaction scheme for obtaining 3,5,3 ', 5'-tetramethylbiphenyl 4,4'-dicarboxylic acid.
- FIG. 44 shows a reaction scheme for obtaining 3,3'-dimethylbiphenyl 4,4'-dicarboxylic acid.
- FIG. 45 represents a photograph obtained by SEM (Scanning Electron Microscopy) of the solid MIL-89nano.
- FIG. 46 represents a photograph obtained by SEM (Scanning Electron Microscopy) of the MIL-88Anano solid.
- Figure 47 shows a picture obtained by SEM (Scanning Electron Microscopy) of the solid MIL-100nano.
- Figure 48 shows a SEM image obtained from the solid MIL-88Btnano.
- FIG. 50 represents a quantity of unsaturated iron sites present in the MIL-100 Fe activated under vacuum at different temperatures.
- Figure 51 shows an NO adsorption isotherm at 298 K for iron carboxylates
- MIL-IOO (Fe) activated at 120oC overnight.
- FIG. 52 represents a NO release profile (NO rel in mmol / g) under vapor pressure of the solid MIL-100 (Fe) as a function of time t (in hours).
- Figure 53 shows (left): an NO adsorption isotherm at 298 K for MIL-100 (Fe) activated at 250 ° C under vacuum overnight; (on the right): a profile of release of NO under steam pressure of the solid MIL-IOO (Fe) activated at 250oC under vacuum overnight.
- Figure 54 is a schematic view of the breathing phenomenon (swelling and contraction) in MIL-88A, MIL-88B, MIL-88C, MIL-88D and MIL-89 solids.
- the swelling amplitude between dry forms (top) and open forms (bottom) is shown in% at the bottom of the figure.
- Figure 55 shows NO adsorption isotherms at 298 K for iron carboxylates MIL-88A (Fe) and MIL-88B (Fe) activated at 150 ° C overnight.
- Figure 56 shows NO steam release profiles under water vapor pressure of MIL-88A (Fe) (top) and MIL-88B (Fe) (bottom) solids activated at 150oC overnight.
- FIG. 57 represents, at the top, a study of the reversibility of the swelling of the MIL-88A solid by X-ray diffraction ( ⁇ ⁇ 1.79 ⁇ ), at the bottom, of the X-ray diffractograms of the solid MIL-88A in the presence of solvents ( ⁇ - 1.5406 ⁇ ).
- FIG. 58 represents an explanatory diagram of 1-a-flexibility-in the hybrid "phases " ⁇ MIL-53 ⁇ " ( ⁇ a ⁇ ) .r and
- FIG 59 shows the crystallographic structure of MIL-126 (Fe).
- the FeO 6 polyhedra are represented with or without a star, indicating the two MIL-88D networks.
- the carbon atoms are in black.
- Figure 60 shows an X-ray diffractograirane of the solid MIL-126 (Fe) -
- Figure 61 represents a thermogravimetric analysis (in air, with a heating rate of 2 ° C / minute) of the compound MIL-126 (Fe).
- Fig. 62 shows nitrogen adsorption isotherms for MIL-126 (Fe) atmosphere).
- Figure 63 shows an X-ray diffractogram of iron 3, 3 ', 5, 5'-azobenzenetetracarboxylate
- FIG. 64 represents the X-ray diffractogram of the crude iron 2, 5-dihydroxterephthalate solid.
- the phase, of trigonal symmetry, is an isotype of that published by Dietzel et al. cobalt and nickel (space group R3) [61].
- FIG. 65 represents a thermogravimetric analysis (in air, with a heating rate of 2 ° C./minute) of iron 3,3 ', 5,5'-azobenzenetetracarboxylate compound.
- Figure 67 shows the XRD diagrams of unmodified MIL-88A material before and after the addition of a drop of water (MIL88A + H 2 O); MIL-88A modified with chitosan 7% before (MIL88AQ100) and after adding a drop of water (MIL88A Q100 +
- MIL-88A modified with chitosan 2% before (MIL88AQ25) and after the addition of a drop of water (MIL88A Q125 + H 2 O).
- Figure 68 shows thermogravimetric analysis of unmodified MIL-88A material (MIL88A, green), modified with 2% chitosan (MIL-88A-
- Figure 69 shows the confocal microscopy images of the MIL-100 (Fe) material superficially modified with Dextran-Fluorescein-Biotin.
- Figure 70 shows the evolution of the particle size (P in nm) of MIL-88A surface-modified with polyethylene glycol, as a function of time (t in min).
- FIG. 71 represents the profiles of release of NO under water vapor pressure (curve (a)) and in the phosphate buffer (curve (b)) of solid MIL-88A (Fe).
- the amount of NO released (N0 rel in mmol. G -1 on the left and ppm NO on the right) is expressed as a function of time t (in hours).
- FIG. 72 represents the profiles of release of NO under pressure of water vapor (curve
- Figure 73 shows the X-ray diffractogram of the MIC-88A-nano solid obtained by microwave synthesis.
- Figure 74 shows NO adsorption isotherms at 298 K for iron carboxylates MIL-88A (Fe) -nano activated at 150oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol.g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is represented as a function of the pressure P (in mmHg).
- FIG. 75 represents the NO release profiles under water vapor pressure of MIL-88A (Fe) solids (5 microns, curve (a)) and MIL-88A (Fe) -nano (120 nm, curve (b) )).
- the amount of NO released (N0 rel in mmol.g -1 on the left and ppm NO on the right) is expressed as a function of time t (in hours).
- Figure 76 shows the NO adsorption isotherms at 298 K for iron carboxylates MIL-88B (Fe) -NO2 activated at 150oC under vacuum overnight.
- the amount of NO (N0 abg in mmol.g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is represented as a function of the pressure P (in mmHg).
- FIG. 77 represents the NO adsorption isotherms at 298 K for iron carboxylates MIL-88B (Fe) -20H activated at 80oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol.g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is represented as a function of the pressure P (in mmHg).
- FIG. 78 represents the profiles of release of NO under pressure of water vapor (curve
- FIG. 79 represents the profiles of release of NO under pressure of water vapor (curve (a)) and in the phosphate buffer (curve (b)) of solid MIL-88B (Fe) -2OH.
- the amount of NO released (NO rel in mmol.g -1 , on the left, and in ppm NO, on the right) is expressed as a function of the time t (in hours).
- FIG. 80 shows the release profiles of NO under vapor pressure of water of MIL-IOOFe solids (curve (a)), MIL-88A (curve (b)) f MIL-88B (curve (C)), MIL-88-2OH (curve (d)) and MIL-88B-NO2 (curve (e)).
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- Figure 81 shows the NO adsorption isotherms at 298 K for the activated MIL-22 solid at 350oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol., O1 ) adsorbed (curve (a)) and desorbed (curve (b)) is plotted as a function of pressure P (in mm Hg).
- FIG. 82 shows the profiles of NO release of solid MIL-22 under steam pressure.
- the amount of NO released (NO rel in mmol.g -1 on the left and ppm NO on the right) is expressed as a function of the time t (in hours).
- Figure 83 shows the CO adsorption isotherm ( ODg in mmol / g) for the solid MIL-
- Figure 84 shows the histological sections of the liver are observed by staining Proust (iron blue). They show an accumulation of iron in the liver.
- FIG. 86 represents the profiles of release of NO under water vapor pressure of the solid CPO-27 (Co dihydroxoterephthalate) (curve (a)), CPO-27 (2,5-dihydroxoteraphthalate of Ni; M 2 (dhtp) ) (H 2 O).
- MIL-53 Al terephthalate
- Curve (d) and MIL-53 (Cr terephthalate)
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- Figure 87 shows the profiles of release of NO under steam pressure of solid MIL-88A (3 samples under the same conditions) in the form of cream.
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- Figure 88 shows the profiles of release of NO under water vapor pressure of the solid MIL-88A-nano in the form of cream (curve (b)) and in powder form (curve (a)) in comparison with the release in a PBS solution (curve (c)).
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- This example describes the synthesis of various iron carboxylates.
- the solids obtained were then characterized according to the methods described below.
- the crystalline structure of iron carboxylate solids was analyzed by X-ray diffraction (RX) using a Siemens D5000 diffractometer (radiation). ⁇ , ⁇ -2 ⁇ mode), at room temperature in air. The diagrams are represented either in angular distances (2 ⁇ , degrees °) or inter-reticular distances (d, in ⁇ or Angstrom). Characterization of the porosity (Langmuir surface area and pore volume) of the solids was measured by nitrogen adsorption at 77 K with a Micromeretics ASAP-2010. The solids were previously dehydrated at 150oC under primary vacuum overnight.
- Thermogravimetric analysis was performed under an air atmosphere using a Model TA instrument 2050 instrument. The heating speed was 2 ° C / minute.
- the curve resulting from the thermogravimetric analysis of the solids represents the loss of mass Pm (in%) as a function of the temperature T (in oC). Elemental analysis of solids was carried out by the CNRS Central Analysis Service of
- ICP-AES Inductive Coupled Plasma - Atomic Emission
- ICP-MS Inductively Coupled Plasma-Mass Spectrometry or Inductively Coupled Plasma Mass Spectrometry
- the iron carboxylate MIL-100 (Fe) was synthesized according to two conditions: with and without hydrofluoric acid.
- the space group is Fd-3m (no. 227).
- the surface (Langmuir) specific for this solid is close to 2900 m 2 . g -1 .
- the curve resulting from the thermogravimetric analysis of the compound MIL-100 (Fe) is given in FIG. 3. This diagram represents the loss of mass Pm (in%) as a function of the temperature T (in oC).
- the characteristics of the crystalline structure are as follows: - the space group is Fd-3m (no. 227).
- the solid (200 mg) is suspended in 100 ml of distilled water with stirring for 12 hours to remove the solvent remaining in the pores. The solid is then recovered by filtration.
- the diffractogram RX is given in FIG.
- thermogravimetric analysis of the hydrated compound MIL-88A under air, at a heating rate of 2 ° C./minute are shown in FIG. 7.
- the mass loss Pm (in%) is represented as a function of the temperature T (in oC).
- MIL-88A does not have an accessible surface (greater than 20 m 2 / g) at 77 K nitrogen, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- Table 4 mesh parameters of solid MIL-88B, dry and hydrated.
- Figure 8 shows the digits . RX grams of dry solid and hydrated solid.
- MIL-88B does not have an accessible surface (greater than 20 m 2 / g) at 77 K nitrogen, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- Figure 10L shows the RX diffractograms a), b) and c) respectively of the dry MIL-89 (Fe) solid, the MIL-89 (Fe) solid solvated with DMF and the hydrated MIL-89 (Fe) solid.
- the compound MIL-89 (Fe) does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen. .
- Figure 11 shows the X-ray diffractogram of the MIL-88C solid.
- the solid is then dried at 150oC in air for 15 hours.
- Figure 13 shows the X-ray diffractogram of the solid MIL-88D crude (bottom curve (b)) and hydrated (top curve (a)).
- thermogravimetric analysis of the hydrated compound MIL-88D (Fe) (in air, at a heating rate of 2 ° C./minute) are represented in FIG. 14 (loss of mass Pm as a function of the temperature T).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- FIG. 15 represents the X-ray diffractogram of the crude MIL-88B-NO 2 solid (curve (a) from above) and hydrated (curve (b) from the bottom).
- thermogravimetric analysis under air, at a heating rate of 2 ° C / minute
- Pm is represented as a function of the temperature T (in ° C).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- the product is calcined at 150oC under vacuum for 15 hours.
- Figure 17 shows the RX diffractogram of the crude MIL-88B-2OH solid (curve (c) below), hydrated (middle curve (b)) and dried under vacuum (top (a)).
- MIL-88B-2OH compound (Fe), after washing and drying, are represented in FIG. 18.
- the mass loss Pm (in%) is represented as a function of the temperature T (in oC).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- the solid is calcined at 200oC for 2 days.
- FIG. 19 represents the X-ray diffractogram of the crude MIL-88B-NH 2 solid (curve (b) below), and dried under vacuum (curve (a) at the top).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- FIG. 21 represents the X-ray diffractogram of the crude MIL-88B-CH 3 solid (curve (a) at the top), hydrated (curve (b) of the medium) and solvated with DMF (curve (c) at the bottom).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- FIG. 21 represents the X-ray diffractogram of the solid MIL-88B-C1 solid (curve (a) at the top), hydrated (curve (b) of the medium) and solvated with DMF (curve (C) at the bottom).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C / minute) of the solid MIL-88B-Cl (Fe) hydrate is shown in FIG.
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- This compound has an accessible surface on the order of 1200 m 2 / g (Langmuir) with nitrogen at 77 K, since the dry structure has a sufficient pore size (6-7 A) to incorporate nitrogen N 2 .
- FIG. 26 shows the X-ray diffractogram of the crude solid (bottom curve (c)), the hydrated solid (curve (b)) and the solvated solid with ethanol (curve (a) at the top).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C / minute) of the solid MIL-88B-4F (Fe) hydrate is shown in FIG. 27.
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- the solid is calcined at 150oC under vacuum for 15 hours.
- Figure 28 shows the X-ray diffractogram of the crude solid (curve (b) below) and the hydrated solid (curve (a) at the top).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C / minute) of the solid MIL-88B-Br (Fe) hydrate is shown in FIG.
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- Figure 30 shows the X-ray diffractogram of the crude synthetic MIL-88E (Fe) solid. This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- the solid is recovered by filtration.
- Figure 31 shows the X-ray diffractograms of the crude solid (curve (b) below) and the hydrated solid (curve (a) at the top).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C / minute) of the hydrated solid MIL-88F (Fe) is shown in FIG.
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- Figure 33 shows the X-ray diffractograms of the crude solid (curve (b) below) and the hydrated solid (curve (a) at the top). Thermogravimetric analysis (under air, at a heating rate of 2 ° C./minute) of the solid MIL-88D-4CH 3 (Fe) hydrate is shown in FIG.
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- the solid is calcined at 150oC under vacuum for 15 hours.
- FIG. 35 represents the X-ray diffractograms of the crude solid (curve (c) below), of the hydrated solid
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- FIG. 37 shows the X-ray diffractograirans of the crude MIL-88G solid (bottom curve (c)), of the solid solvated with DMF (curve (b) of the medium) and of the solid solvated with pyridine (curve (a) at the top ).
- This compound does not have an accessible surface (greater than 20 m 2 / g) with nitrogen at 77 K, since the dry structure has a pore size too small to incorporate N 2 nitrogen.
- Figure 39 shows the X-ray diffractograms of the crude MIL-88G-2C1 solid (curve (b) at the bottom) and the dry MIL-88G-2C1 solid (curve (a) at the top).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C./minute) of the crude solid MIL-88G-2Cl (Fe) is shown in FIG.
- Figure 41 shows the X-ray diffractograirans of the solid MIL-102 (Fe) crude (curve (a)) and the solid MIL-102 (Cr) (curve (b)).
- Thermogravimetric analysis (under air, at a heating rate of 2 ° C./min) of the crude solid MIL-102 (Fe) is shown in FIG. 42.
- This compound has a low surface area (Langmuir surface: 101 m 2 / g) with nitrogen at 77 K.
- the solid is then dried at 150 ° C. under primary vacuum for 15 hours.
- the crystallographic structure of the MIL-126 (Fe) solid is an interpenetrating form of the MIL-88D (Fe) structure, that is, it has two entangled MIL-88D-type crystalline subarrays (FIG. ).
- Figure 60 shows the X-ray diffractogram of the crude MIL-126 solid.
- thermogravimetric analysis of the crude synthetic compound MIL-126 (Fe) (under air, at a heating rate of 2 ° C./minute) are represented in FIG. 61 (loss of mass Pm as a function of the temperature T ).
- This compound has a large accessible surface (Langmuir) (greater than 2100 m 2 / g) with nitrogen at 77 K ( Figure 62).
- Teflon 23 ml put in a PAAR metal bomb for 3 days at 150oC with a rise of 1 hour. The solid is recovered by filtration.
- the solid is then dried at 200 ° C. under primary vacuum for 15 hours.
- Figure 63 shows the X-ray diffractograirane of the crude iron 3,3 ', 5,5'-azobenzenetetracarboxylate solid.
- the phase, of cubic symmetry, is an isotype of that published by the group of Professor Eddaoudi with indium (space group Pa3) [51].
- the results of the thermogravimetric analysis of the crude synthetic 3,3 ', 5,5'-azobenzenetetracarboxylate compound (under air, at a heating rate of 2 ° C./minute) are shown in FIG. Pm as a function of temperature T).
- This compound has a large accessible surface (Langmuir) (greater than 1200 m 2 / g) with nitrogen at 77 K ( Figure 66).
- the solid is then dried at 150 ° C. under primary vacuum for 15 hours.
- Figure 64 shows the RX diffractogram of the crude iron 2, 5-dihydroxterephthalate solid.
- the phase, of trigonal symmetry, is an isotype of that published by Dietzel et al. cobalt and nickel (space group R3) [61].
- the iron (III) acetate used in the examples below for the synthesis of the MOF materials according to the invention, is synthesized according to the following protocol. This synthesis may refer to the publication by Dziobkowski et al., Inorg. Chem. , 1982, 21, 671 [ref. 14].
- Aesar are suspended in 39 ml of concentrated hydrochloric acid (37%, marketed by Aldrich) at 0 ° C.
- Diazotization was performed by adding a solution of sodium nitrite (6 g in 50 ml of water). After stirring for 15 minutes at 0 ° C., a solution of potassium iodide (70 g in 200 ml of water) was added slowly to the resulting violet solution. After the addition is complete, the mixture is stirred for 2 hours at room temperature. The resulting black slurry is filtered to recover a black precipitate, washed with water.
- the solid is suspended in dichloromethane (DCM, 98%, sold by the company SDS) and a saturated solution of sodium thiosulfate is added, causing discoloration. After stirring for 1 hour, the organic phase is decanted and the aqueous phase is extracted with DCM. The organic phase is dried over sodium sulphate and then evaporated to give the diiodinated intermediate in the form of a greyish solid. Elution with pure pentane on a column of silica (marketed by the company SDS) makes it possible to obtain the mixture of the compounds monoiodé-and-diiodév-the-mixture-of-these "was directly used in the stage next step 2:.
- DCM dichloromethane
- SDS silica
- the diester is saponified with 9.7 g of potassium hydroxide (marketed by the company VWR) in 100 ml of 95% ethanol (marketed by the company SDS), at reflux for 5 days.
- the solution was concentrated in vacuo and the product is solubilized in eauv ⁇ " ⁇ concentrated hydrochloric acid is added until pH 1, and a white precipitate is formed.
- MIL-89nano The synthesis of MIL-89nano is carried out from iron acetate (1 mmol, synthesized according to the synthesis described in example 2) and muconic acid (1 mmol, Fluka, 97%) in 5 ml of ethanol (Riedel-de Ha ⁇ n, 99.8%) with the addition of 0.25 ml of 2M sodium hydroxide (Alfa Aesar, 98%) in an autoclave (Paar bomb) at 100 ° C. for 12 hours. After cooling the container, the product is recovered by centrifugation at 5000 rpm (rotation per minute) for 10 minutes. 200 mg of the solid are suspended in
- Figure 45 is a scanning electron microscopy (SEM) image of the MIL-89nano solid.
- the nanoparticles show a rounded and slightly elongated morphology, with a very homogeneous particle size of 50-100 nm ( Figure 45).
- FeCl 3 .6H 2 O (1 mmol, Alfa Aesar, 98%) and fumaric acid (1 mmol, Acros, 99%) are dispersed in 15 ml of ethanol. (Riedel-de Ha ⁇ n, 99.8%). Then, to this solution is added 1 mL of acetic acid (Aldrich, 99.7%). The solution is placed in a glass bottle and heated at 65oC for 2 hours. The solid is recovered by centrifugation at 5000 rpm for 10 minutes.
- the particle size measured by light scattering is 250 nm.
- the scanning electron microscopy (SEM) of the MIL-88Anano solid is shown in FIG.
- the SEM images show elongated particles with edges. There are two particle sizes: about 500 nm and 150 nm.
- MIL-100nano The synthesis of MIL-100nano is carried out by mixing FeCl 3 .6H 2 O (1 mmol, Alfa Aesar, 98%) and 1,3,5-benzenetricarboxylic acid (1,3,5-BTC; 1 mmol). Aldrich, 95%) in 3 mL of distilled water. The mixture is placed in a PAAR bomb at 100oC for 12 hours. The product is recovered by centrifugation at 5000 rpm (10 minutes). 200 mg of the solid are suspended in 100 ml of distilled water with stirring and reflux for 3 hours to remove the acid-remaining in the pores. Then, the solid is recovered by centrifugation at 5000rpm (10 minutes). The particle size measured by light scattering is 536 nm.
- SEM Scanning electron microscopy
- the product is heated at 200oC under vacuum for 1 day.
- the product is stored under vacuum or inert atmosphere given its low air stability.
- the particle size is measured by light scattering is 310 nm.
- the solid MIL-88Btnano is synthesized from a solution of FeCl 3 .6H 2 O (1 mmol, Alfa Aesar, 98%) and 1,4-benzenetetramethyldicarboxylic acid (1 mmol, Chem Service) in 10 mL of dimethylformamide (Fluka, 98%) with 0.4 mL of 2M NaOH. This solution is placed in a Paar bomb and heated at 100oC for 2 hours. Then, the container is cooled with water cold, the product is recovered by centrifugation at 5000rpm (10 minutes).
- Measurement of particle size by light scattering shows two populations of nanoparticles of 50 and 140 nm.
- the nanoparticles of the solid MIL-88Btnano have a spherical morphology with a size of about
- the scanning electron microscopy (SEM) of the solid MIL-88Btnano is shown in FIG. 48.
- the solid MIL-88Bnano is synthesized from a solution of iron acetate (1 mmol, synthesized according to the synthesis described in Example 2) and 1,4-benzenedicarboxylic acid (1 mmol; BDC Aldrich, 98%) in 5 mL of methanol (Aldrich, 99%).
- This solution is placed in a Paar bomb and heated at 100 ° C for 2 hours.
- the container is then cooled with cold water, the product is recovered by centrifugation at 5000 rpm (10 minutes).
- 200 mg of the solid are suspended in 100 ml of distilled water with stirring under reflux for 15 hours to exchange the solvent which remains in the pores. Then, the solid is recovered by centrifugation at 5000rpm (10 minutes).
- Measurement of particle size by light scattering shows a bimodal distribution of nanoparticles of 156 and 498 nm.
- the scanning electron microscopy (SEM) of the solid MIL-88Bnano is shown in FIG. 49.
- the morphology of the particles is very irregular, with a size of 300 nm. Determination of particle size by light scattering was performed on a Malvern Zetasizer Nano Series - Nano-ZS (Zen 3600 Series, Serial No. 500180, UK).
- the surface modification of the nanoparticles with chitosan makes it possible to envisage different routes of administration of the nanoparticles thanks to the bio-adhesion properties specific to this polymer.
- the solution is stirred for 45 minutes.
- the Teflon bomb is placed in a hermetically sealed metallic body and heated in an oven at 80 Q C for 12 hours.
- the solid obtained is recovered by centrifugation at 5000 rpm for 10 minutes and washed with distilled water and acetone.
- the particle size obtained is measured with a potential device Z Malvern Zetasizer Nano series - Nano-ZS; model Zen 3600; Serial No. 500180;
- X-ray diffraction patterns are collected with a diffractometer Siemens D5000 X ' Pert CDM from 3 to 20o (2 ⁇ ) with a step size of 0.04o and 2s per step.
- the XRD diagrams presented in FIG. 67 made it possible to verify that the phase obtained is indeed the
- MIL-88A The flexibility of the material is also verified by DRX by adding a drop of water on the solid.
- the amount of chitosan incorporated in the material is estimated by thermogravimetric analysis (TGA) shown in FIG. 68.
- TGA thermogravimetric analysis
- the apparatus used is a TGA2050 TA apparatus of 25 to 500oC with a heating ramp of 2 ° C./min under oxygen flow ( lOOmL / min).
- the amount of fumaric acid is around 45% (relative to the dehydrated product).
- the MIL88A-Q25 and MIL-88A-Q100 materials contain an amount of chitosan of the order of 16% and 22% (weight) based on the dehydrated product, respectively.
- the dextran used is grafted on the one hand with fluorescein and on the other hand with biotin (Dex B FITC 10000 g / mol, anionic, fixable lysine, Molecular Probes, catalog D7178).
- biotin Dex B FITC 10000 g / mol, anionic, fixable lysine, Molecular Probes, catalog D7178.
- the characteristics of dextran are as follows: Dextran fluorescein and biotin, molecular weight 10,000 g / mol, anionic, capable of binding lysine ("mini-emerald") lot 3603IA, D7178, "Molecular probes", In vitro detection technology 1 mole fluorescein / mole, 2 moles biotin / mole. a) Preparation of surface-modified nanoparticles
- the iron 1,3,5-benzenetricarboxylate MIL-100 particles (particle diameter 1.79 microns) were washed with MiIIiQ water.
- Fluorescein allows the detection of particles using a confocal scanning laser microscope, while hydrophobic biotin allows:
- Figure 69 shows the optical sections thus obtained. There is a halo around the particles, indicating the presence of dextran - (only fluorescent compound) only on the surface. Indeed, the long polymer chains could not penetrate to the heart of the particles.
- This method of surface modification has the advantage of not disturbing the heart of particles (containing active ingredients) and to be post-synthesis, so to offer a variety of possible overlays.
- the nanoparticles must be prevented from being directed to the liver; the best method is to graft on the surface of the hybrid nanoparticles hydrophilic chains of the polyethylene glycol (PEG) type, in order to reduce their accumulation in this organ.
- PEG polyethylene glycol
- the PEG chains may have different terminal groups to graft onto the surface of the materials.
- the interaction of PEG with the particle surface can be modified using different types of PEG.
- PEG-NH 2 alpha-t-butyloxycarbonylamino-omega-amino poly (ethylenglycol) (PEG, Boc-NH-PEG-NH 2, 5000 MW, Iris Biotech) - PEG-COOH ( ⁇ oly (ethylenglycol) carboxylic acid, Iris Biotech)
- PEG-PO 4 synthesized in the laboratory according to the following method:
- the phosphonate group is attached to PEG-NH 2 by an amide condensation with an ester bound to a phosphonate group.
- the sodium salt of phosphonate was used.
- the coupling was carried out from trimethylphosphonoformate [CAS 31142-23-1] according to the procedure described by Robert A. Moss, Hugo Morales-Rojas, Saketh Vijayaraghavan and Jingzhi Tian, J. Am. Chem. Soc., 2004, 126, (35), 10923-10936 [52].
- the polyethylene glycol modification may be carried out during or after the synthesis as indicated below.
- the iron acetate (1 mmol, synthesized according to the synthesis A described in Example 2) and the muconic acid (1 mmol, Fluka 97%) are mixed in 10 ml of methanol (Aldrich, 99.9%). The whole is introduced into a Teflon body of 23 ml. The monoacid PEG is then introduced at a level of 3; 8.5 or 13% by weight relative to the total weight of solid. 0.35 mL of 2 M sodium hydroxide is optionally added. The solution is stirred for 20 minutes.
- the Teflon bomb is placed in a metal body hermetically closed and heated in an oven at 100oC for 12 hours.
- the solid obtained is recovered by centrifugation at 5000 rpm for 10 minutes and washed with distilled water and acetone.
- the PEG assay was carried out by UV spectrophotometry (at the wavelength of 500 nm), according to the method described in B Baleux et al. CR.
- the weight% of PEG in the nanoparticles is greater than the mass% of PEG introduced at the beginning of synthesis
- the nanoparticles of MIL-100 are synthesized by microwave (CEM Microwaves) starting from a solution of 9.7 g iron nitrate hexahydrate (Aldrich, 97%), 3.38 g of 1, 3,5-benzenetricarboxylic acid (1.3.5-BTC, Aldrich, 99%) and 40 g of distilled water at 180oC for 30min (600W power).
- the particle size measured by light scattering is 400 nm.
- PEGylated nanoparticles of MIL-100 modified with polyethylene glycol are obtained by the surface modification of the previously mentioned particles.
- 30 mg of MIL-100 are suspended in 3 ml of an aqueous solution of 10 mg of polyethylene glycol aminoteminal (PEG-NH 2 5000 g / mol, Aldrich, 97%) at 30 ° C. for 3 hours with stirring.
- These nanoparticles are recovered by centrifugation (10000 rpm / 10 min) and washed with distilled water.
- the amount of PEG at the surface is determined by the method of Baleux and Champertier, based on the formation of a complex colored by iodine-iodide on PEG, which is selectively measured by spectrophotometry at 500 nm).
- the amount of PEG is 19% by weight and the particle size after the modification by polyethylene glycol increases to 800 nm.
- SEM scanning electron microscopy
- aqueous solutions of iron chloride III (2.7 mg / ml, FeCl 3 , 6H 2 O marketed by ACROS 97%) and fumaric acid (1.16 mg / ml; 4 H 4 O 4 marketed by ACROS 99%) are prepared.
- the two solid reactants are weighed and dissolved separately in water in the proportions given in the examples below.
- the fumaric acid solution is brought to 70 ° C. with stirring for 120 min to solubilize the product.
- the iron chloride is stirred with a magnetic stirrer for 30 minutes.
- a total of 8 vials are prepared.
- 5 ml of iron III chloride solution (2.7 mg / ml) and 5 ml of fumaric acid solution (1.16 mg / ml): - 4 vials serve as a control where the reactions are carried out for the 4 synthesis times: 30, 60, 90 and 120 min,
- the 8 vials are simultaneously placed in a sonic bath at 0 ° C for the corresponding times t (30, 60, 90 and 120 min).
- a total of 8 vials are prepared.
- 5 ml of iron III chloride solution (2.7 mg / ml) and 5 ml of fumaric acid solution (1.16 mg / ml): - 4 vials serve as a control where the reactions are carried out for the 4 synthesis times: 30, 60, 90 and 120 min,
- the 8 vials are simultaneously placed in a sonic bath at 0 ° C for the corresponding times t (30, 60, 90 and 120 min). After synthesis, a volume of 0.1 ml of solution is taken from each vial to determine the particle size by light scattering using a Dynamic Light Scattering (DLS, Nanosizer) apparatus. The rest of the solution was then centrifuged at 10,000 rpm at 0 ° C for 15 minutes to separate the supernatant from the solid formed. The supernatant is removed using a pasteur pipette and the pellet recovered is placed under the hood at room temperature (about 20oC). Equipment used:
- 100 mg of nanoparticles of MIL100, MIL88, MIL53 or MIL101 are dispersed under sonication in 100 mL of 17.9 mM solution of 2- (methoxy (polyethyleneoxy) propyl) trimethoxysilane in the anhydrous toluene.
- the mixture is subjected to ultrasound at 60oC for 4 h, under flow of inert gas (nitrogen).
- the resulting colloidal suspension, containing PEG-surface-modified nanoparticles is washed twice with ethanol and twice in 20 mM sodium phosphate solution (pH 8.0) and finally resuspended in water. .
- FA has been attached to nanoparticles thanks to a bifunctional spacer, silane-PEG- trifluoroethyl ester (TFEE) synthesized according to a method described in the literature by Kohler N. et al., J. Am Chem Soc 2004; 126; 7206-7211 [55].
- TFEE silane-PEG- trifluoroethyl ester
- nanoparticles 100 mg are coated with PEG-TFEE according to the same method as described above, using silane-PEG-TFEE in place of 2-methoxy (polyethyleneoxy) -propyltrimethoxysilane.
- the resulting nanoparticles, coated with PEG-TFEE, are washed twice and then resuspended in 100 mL of dry toluene.
- a primary amine was grafted to the end groups of the PEG chains by adding 1 mL of ethylenediamine (Sigma) to the nanoparticles maintained under nitrogen flow. The mixture was sonicated (4h, 60oC).
- the resulting nanoparticles, coated with amine were washed three times with ethanol, and three times with dimethyl sulfoxide (DMSO). The nanoparticles were finally resuspended in 50 mL of anhydrous DMSO.
- FA was coupled to the terminal amino groups of the PEG chains by adding 50 mL of FA solution (23 mM FA in DMSO) with equimolar amounts of dicyclohexylcarbodiimide (DCC) (Sigma) and 10 ⁇ L of pyridine. The mixture was protected from light and allowed to react overnight with two-dimensional agitation (180 rpm). Nanoparticles conjugated with PEGH and FA (NP-PEG-FA) were washed twice with ethanol, and twice with 20 mM sodium citrate solution (pH 8.0) and resuspended finally in this same solution of sodium citrate. b) Synthesis n ° 2: surface modification during nanoparticle synthesis
- the modification of the surface of the MOF solids can also be done during the synthesis.
- the surface modification is carried out with chitosan previously grafted with folic acid (FA).
- chitosan (Mn molecular weight of 255 kDa, viscosity: 200-800 cps in 1% acetic acid, marketed by Sigma-Aldrich)
- the chitosan is previously deacetylated to obtain a degree of acetylation of 82% (determined by 1 H-NMR) according to the method described by Wang LS (Thesis, National University of Singapore, Singapore, 2001).
- reaction product was dried under vacuum and then dissolved in 1.5 mL of 2: 1 (v: v) DMSO and TEA.
- An equimolar amount of 2-aminoethanethiol (Wako) was added and the reaction allowed to continue overnight under anhydrous conditions.
- a thiol group could be introduced to folic acid and the product results is called FA-SH.
- chitosan 100 mg are dissolved in 50 mL of acetic acid solution (20%).
- the pH of the solution is adjusted to 6 by addition of sodium hydroxide and 100 mg of NHS-PEG-MaI are introduced into the reaction mixture.
- the mixture is left to react for 3 hours to room temperature (about 20oC) with stirring, then the pH is adjusted to 7.
- the mixture is allowed to react overnight, under anhydrous conditions.
- the FA-SH synthesized as before was added in a graduated manner with stirring and the pH was adjusted to 6.5-7.5 with sodium hydroxide.
- the conjugate obtained called FA-PEG chi carries FA coupled to chitosan via a PEG spacer arm, which is an advantage to reach the receptor of
- the degree of substitution can be adjusted by varying the weight ratio PEG: chitosan used in the reaction. This polymer was dialyzed for 48 hours against de-ionized water using a membrane
- Hybrid solids can be surface modified by adsorption of polysaccharides such as dextran grafted with biotin.
- polysaccharides such as dextran grafted with biotin.
- We can therefore consider adsorbing, in place of dextran grafted with biotin, chitosan grafted with folic acid (synthesized as described in the publication cited above), and optionally, if necessary, also grafted with d other hydrophobic compounds such as cholesterol or aliphatic links, to ensure better adhesion at the surface of the nanoparticles.
- Hybrid solids can be surface modified with PEG during synthesis.
- the monomethoxy PEG monoacid used during this synthesis is substituted by PEG monoacid having a reactive function blocked at the chain end as the commercial product:
- Boc-PEG-carbonate NHS MW 5000
- Boc tert-butoxycarbonyl (Reference SUNBRIGHT * BO-050TS, NOF Corporation).
- MeO-PEG-COOH and Boc-PEG-carbonateNHS mixtures (1: 0.05 to 1: 0.5 mass ratios) are used in place of MeO-PEG-COOH.
- Deprotection will be by addition of trifluoroacetic acid (TFA).
- TFA 0.6 ml of TFA is added to a suspension of 300 mg of nanoparticles in 2 ml of water. It is allowed to react for 1 h at room temperature (approximately 20 ° C.) with magnetic stirring. The particles are isolated by centrifugation and washed three times with distilled water.
- Reactive groups at the surface are functionalized with ligands such as FA, for example as in synthesis No. 1 indicated in paragraph a).
- ligands such as FA
- the quantity of folic acid effectively coupled to the nanoparticles can be determined after degradation thereof in an acidic medium, neutralization at pH 7, and then redissolution in a suitable solvent, such as dichloromethane, DMSO, or a mixture of these two solvents. . Folic acid can then be quantified by measuring the UV absorbance
- BIAcore plasmon surface resonance
- EXAMPLE 10 Synthesis of MOF Materials Based on Bioactive Ligands
- the use of ligands with a biological activity is of interest for: the release of active compound by degradation of the MOF material encapsulation of other active molecules for combined therapies
- Antimicrobial activity assays, as well as degradation in physiological media and activity on cells will be performed on flexible porous iron carboxylates of type MIL-88 using 4, 4 'azobenzene dicarboxylic acids and 3 3'-4,4'-dichlorobenzene dicarboxylic acid, among others.
- Azobenzene, azelaic acid and nicotinic acid Azobenzene, azelaic acid and nicotinic acid.
- the rigid structure of the azo molecules allows them to behave as liquid crystal mesogens in many materials.
- azobenzene can be photoisomerized (cis or trans isomer) hence its use for photomodulating the affinity of a ligand (for example a drug) for a protein.
- azobenzene can act as a photointerruptor between a ligand and a protein by allowing or preventing the protein-drug binding according to the cis or trans isomer of azobenzene (one end of the azobenzene can for example be substituted by a group that binds to the target protein while the other end is linked to a ligand (drug) of the protein).
- Azelaic acid (HO 2 C- (CH 2 ) 7 -CO 2 H) is a saturated dicarboxylic acid which has antribacterial, keratolytic and comedolytic properties. he is used especially in the treatment of acne and rosacea.
- Nicotinic acid (C 5 H 4 N-CO 2 H) is one of two forms of vitamin B3 with nicotinamide. Vitamin B3 is particularly necessary for the metabolism of carbohydrates, lipids and proteins.
- 200 mg of the solid are suspended in 10 mL of DMF with stirring at room temperature for 2 hours to exchange the acid remaining in the pores.
- the solid is then recovered by filtration and then calcined at 150 ° C. under vacuum for 15 hours to remove the DMF remaining in the pores.
- the particle size measured by light scattering is> 1 micron.
- the particle size measured by light scattering is> 1 micron.
- the particle size measured by light scattering is 498 nm, with a second minority population of 1100 nm.
- the particle size measured by light scattering is> 1 micron (1500 nm).
- the synthesis conditions in DMF are as follows: 135 mg of FeCl 3 .6H 2 O (1 mmol, Aldrich, 99%) and
- nicotinic acid (Aldrich, 99%) are dispersed in 5 ml of DMF (Fluka, 98%). Everything is left in a 23 ml Teflon body placed in a PAAR metal bomb for 16 hours at 100oC. The solid is recovered by filtration and washed with acetone.
- Iron Nicotinate 2 The synthesis conditions of iron nicotinate 2 are as follows:
- the activation of the material MIL-100 was carried out by heating at 150 ° C. under a primary vacuum for 15 hours.
- the resulting solid has only iron at the oxidation state + III.
- the partial reduction of the MIL-100 (Fe) material was carried out by heating at 250 ° C. under primary vacuum for 15 hours. Infrared spectroscopy quantified the relative iron (II) / iron (III) content around 20/80% ( Figure 50).
- Figure 50 shows the amount of coordinately unsaturated iron sites present in the activated MIL-100 (Fe) solid as a function of the heat treatment performed.
- the solid MIL-100 (Fe) is activated under residual vacuum (about 10 -5 Torr) at different temperatures and for different durations.
- T (Fe) represents the content of coordinately initiated iron sites and T (Fe 2+ ) represents the content of co-ordinated Fe 2+ sites (in ⁇ mol of unsaturated sites per gram of activated solid or in% of unsaturated iron sites).
- the amounts of unsaturated iron sites are determined by adsorption of CO at 100K followed by infrared spectroscopy. The uncertainty in the values is estimated at +/- 10%.
- MIL-88 The category of flexible solid hybrids based on trimers of trivalent transition metals is called MIL-88. These compounds are typically constructed from iron octahedra trimers, i.e. three iron atoms connected by a central oxygen and six carboxylate functions connecting two to two iron atoms; a molecule of terminal water, coordinated with each iron atom, then completes the octahedral coordination of the metal.
- trimers are then linked together by aliphatic or aromatic dicarboxylic acids to form the MIL-88A, B, C, D and MIL-89 (-A for fumaric acid, -B for terephthalic acid, -C for acid 2, 6-naphthalenedicarboxylic acid, -D for 4,4'-biphenyldicarboxylic acid and MIL-89 for trans, trans muconic acid), as described in Serre et al., Angew. Chem. Int. Ed. 2004, 43, 6286 [17].
- Other analogues with other dicarboxylic acids have also been synthesized and are named MIL-88E, F, G ...
- the distance between trimers in the swollen form goes from 13.8 ⁇ with fumaric acid (MIL-88A) to 20.5 ⁇ with the biphenyl ligand (MIL-88D).
- the size of the pores of the inflated forms thus varies between 7A (MIL-88A) to 16A (MIL-88D).
- the swelling is reversible, as shown by the example of solid MIL-88A in the presence of water in Figure 57 and also depends on the nature of the solvent used as described in Serre et al. J. Am. Chem. Soc., 2005, 127, 16273-16278 [19]. "Breathing" is continuous, with no apparent breaks in the connections during breathing. Moreover, returning to room temperature, the solid swells again by resolvation, confirming the reversibility of respiration.
- each trimer is connected to six other trimers, three below and three above, by the. dicarboxylates which leads to the formation of bipyramidal cages of trimers. Within these cages, the connection between trimers occurs only along the axis c and the absence of any link in the plane (ab) is at the origin of the flexibility ( Figure 58).
- MIL100-Fe exhibiting flexible (MIL-100-Fe) redox activity (MIL88) (MIL88) and substituted (on the ligand) (MIL88-FeNO 2 ).
- porous hybrid solids of the MIL-n type based on high oxidation (+3) metals seem to have the ideal profile for prolonged release of NO.
- MIL-100 consisting of trimers of chromium or iron octahedra connected by trimesic acids
- the latter is easily evacuated by heating under vacuum and gives way to unsaturated and accessible metal centers (metal in coordination five).
- Most iron-based MIL solids possess this type of trimers and thus can all potentially adsorb on their metal centers organic molecules having a character of electron donor (Lewis base), such as NO via the free doublet located in the orbitals 5 ⁇ .
- Nitrogen Oxide MOF materials previously activated are exposed to about 2 bar of NO (99.5%, marketed by Air Liquide) for 30 minutes. They are then evacuated under vacuum (to avoid the release of physisorbed NO and thus the so-called "initial burst" phenomenon corresponding to a very large amount of NO released from the first minutes of release) and placed under a dry argon atmosphere. This last operation (evacuation / argon) is repeated 3 times to ensure that all physisorbed NO has been removed.
- NO Nitrogen Oxide
- the NO adsorption / desorption measurements are carried out using a thermostated gravimetric type apparatus to eliminate any effect of the external environment.
- a CI microbalance marketed by the manufacturer
- the pressure is measured by two BOC Edwards Active probes (measuring range: 1x10 "8- lxl0 " 2 and 1x10 "4- lxl0 3 mbar) .
- the MOF sample (-50mg) is
- Air Liquide, 99.5% Air Liquide, 99.5%
- the NO measurements are carried out using a "Sievers NOA 28Oi Chemiluminescence Nitric Oxide Analyzer” analyzer.
- the instrument is calibrated by passing air through a zero filter (Sievers, ⁇ 1 ppb NO) and 89.48 ppm NO gaseous (Air Products, nitrogen balance).
- the gas flow is set at 180 mL / min with a pressure in the cell of 8.5 torr and an oxygen pressure of 6.1 psi.
- the solid For applications where the solid is in contact with the blood (tubes, catheters, etc.), the presence of an aqueous medium is necessary. For such applications, the release of NO by a sample in powder form has been studied in a physiological fluid simulated.
- the NO-loaded solid 50mg + NO
- the NO-loaded solid 50mg + NO
- 4 mL of phosphate buffered saline pH -5.5, PBS
- the amount of NO released at different times is analyzed by the Sievers NOA 28O Chemiluminescence Nitric Oxide Analyzer.
- the solid MIL100 (Fe) therefore adsorbs large amounts of NO (2-4 mmol.g -1 ) at ambient temperature (approximately 20 ° C.) (FIG. 51) and releases it slowly and very partially (under a moisture flow of 11%). ) as shown by the preliminary results obtained with MIL-100 (Fe) solids ( Figure 52).
- the release profiles of NO under water vapor pressure are, moreover, very interesting.
- Figure 51 shows the NO adsorption isotherm (N0 ada in mmol / g) at the temperature of 298 K for iron carboxylate MIL-100 (Fe) activated at 120oC overnight.
- This figure represents the amount of NO adsorbed (curve (a)) and desorbed (curve (b)) as a function of the pressure P (in mmHg).
- Figure 52 shows the release profile of NO (N0 rel in mmol / g) under vapor pressure of the solid MIL-100 (Fe).
- the NO is represented in ppm (or “parts per million”) or in ppb (or “parts per billion”) as a function of time t in hours.
- the inventors partially reduced the iron (III) of the MIL-100 compound (Fe) by activation under a primary vacuum (12 hours at 250 ° C.). Infrared spectroscopy showed that under these conditions this led to the reduction of about 15-20% iron (III) to iron (II).
- Low-oxidation transition metals, such as iron (II) have additional electrons in orbitals d. It is known that the electronic transfer of orbital d from metal to 2 ⁇ ⁇ orbital molecules such as NO and CO reinforces the adsorbate metal bond (phenomenon of retro-donation) and thus stabilizes the species coordinated on the metal center.
- Figure 53 shows, on the left: the NO adsorption isotherm at 298 K for the MIL-100 (Fe) activated at
- Figure 54 is a schematic view of the respiration phenomenon in MIL-88 solids (-A, -B, -C and -D).
- Figure 55 shows NO adsorption isotherms at 298 K for iron carboxylates MIL-88A (Fe) and MIL-88B (Fe) activated at 150oC under vacuum overnight.
- the amount of NO (N0 abg in mmol / g) adsorbed (curve (a)) and desorbed (curve (b)) is represented as a function of the pressure P (in mmHg). The release of NO is then tested under water vapor pressure (11% humidity in a neutral gas).
- Figure 56 shows the NO release profiles under water vapor pressure of MIL-88A (Fe) (top) and MIL-88B (Fe) (bottom) solids activated at 150oC overnight.
- the amount of NO released (NO rel in mol / g) is expressed as a function of time t (in seconds).
- FIG. 71 represents the profiles - of release of NO under water vapor pressure (curve (a)) and in the phosphate buffer (curve (b)) of solid MIL-88A (Fe).
- the quantity of NO released (N0 rel in mmol.g -1 , on the left, and ppm NO, on the right) is expressed as a function of the time t (in hours).
- the amount released is lower than that released for MIL-88A (Fe).
- the amount of NO released is very low (0.002 mmol.g -1 ), either in the gas stream or in the PBS (FIG. 58).
- the release at active concentrations is very rapid (1 hour in PBS and 4 hours in the gas stream).
- FIG. 72 represents the profiles of release of NO under pressure of water vapor (curve
- the microwave synthesis conditions are as follows: 270 mg (1 mmol) of FeCl 3 .6H 2 O, 116 mg of fumaric acid (1.0 mmol, 99% Acros) dispersed in 30 ml of distilled water, the everything left in a teflon body 2 min at 100oC with a heating ramp of 1 min (power 1600W). The solid is recovered by centrifugation at 10000 rpm for 10 min.
- 200 mg of the product are suspended in 100 ml of distilled water to exchange the fumaric acid which remains free.
- the hydrated solid is recovered by centrifugation at 10,000 rpm for 10 min.
- Figure 73 shows the X-ray diffractogram of the MIC-88A-nano solid obtained by microwave synthesis.
- the monodisperse particle size measured by light scattering is 120 nm.
- Figure 74 shows NO adsorption isotherms at 298 K for iron carboxylates MIL-88A (Fe) -nano activated at 150oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol ⁇ g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is plotted as a function of pressure P (in mmHg).
- FIG. 75 represents the NO release profiles under water vapor pressure of the MIL-88A (Fe) -nano (120 nm, curve (b)) and MIL-88A (Fe) solids (5 micron, curve (a) )).
- the amount of NO released (N0 rel in mmol.g "1 , on the left, and ppm NO, on the right) is expressed as a function of time t (in hours)
- Micrometric MIL-88A (Fe) appears to have a slower release than MIL-88A (Fe) -nano in the first 10 hours. This effect is probably due to a smaller characteristic NO diffusion distance in the MIL88A nanoparticles compared to the micrometric MIL88A.
- the MIL-88B flexible crystalline structure exhibits isotypes by modification of the spacer organic with different functional groups.
- these functional groups will substitute one or more hydrogens of the organic spacer (terephthalic acid), thereby modulating the hydrophobicity and stability of these flexible phases, consequently the adsorption and release of biological gases.
- electron acceptor groups may be able to create new interactions with biological gauzes (Lewis bases).
- the inventors have tested the adsorption and the release of NO from flexible phases of the MIL-88B type, based on the organic ligands: nitroterephthalate (MIL88B-NO2) and 2,5-dihydroxoteraphthalate (MIL88B-2OH).
- Figure 76 shows NO adsorption isotherms at 298 K for iron carboxylates MIL-88B (Fe) -NO2 activated at 150 ° C under vacuum overnight.
- the amount of NO (N0 aba in mmol.g -1) adsorbed (curve (a)) and desorbed (curve (b)) is represented as "pressure function P (in mmHg).
- Figure 77 shows NO adsorption isotherms at 298 K for activated iron carboxylates MIL-88B (Fe) -2OH at 80oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol.g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is plotted against pressure P (in mmHg).
- FIG. 78 represents the profiles of release of NO under pressure of water vapor (curve (a)) and in the phosphate buffer (curve (b)) of solid
- the amount of NO released (N0 rel in mmol.g -1 , on the left, and in ppm NO, on the right) is expressed as a function of time t (in hours).
- FIG. 79 represents the profiles of release of NO under pressure of water vapor (curve (a)) and in the phosphate buffer (curve (b)) of solid MIL-88B (Fe) -2OH.
- the amount of NO released (N0 rel in ⁇ unol.g -1 on the left and in ppm NO on the right) is expressed as a function of the time t (in hours).
- Table 15 Capacity and kinetics of adsorption and release of iron carboxylates in PBS solution and under gas flow conditions.
- FIG. 80 shows the NO release profiles under water vapor pressure of MIL-IOOFe solids (curve (a)), MIL-88A (curve (b)), MIL-88B (curve (C)), MIL -88-2OH (curve (d)) and MIL-88B-NO2 (curve (e)).
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- Example 14 adsorption and release of NO from the titanium diphosphonate MIL-22 (Ti 3 O 2 (H 2 O) 2 ( ⁇ 3 P- (CH 2) -PO 3) 2 (H 2 O) 2)
- the titanium diphosphonate MIL-22 was obtained according to the method reported by C. Serre, G. Ferey, Inorg. Chem. 1999, 38, 5370-5373 [60].
- the amount of theoretical adsorbed NO is -4 mmol.g -1
- the experimental NO adsorption capacity is only 0.18 mmol.g -1 . This difference can be explained because the activation conditions are insufficient to eliminate all the coordinated water. Thus, larger capacities can be obtained with solids activation conditions such as 500oC under vacuum for 16 hours.
- Figure 81 shows the NO adsorption isotherms at 298 K for the activated MIL-22 solid at 350oC under vacuum overnight.
- the amount of NO (N0 ab3 in mmol.g -1 ) adsorbed (curve (a)) and desorbed (curve (b)) is represented as a function of the pressure P (in mm Hg).
- Figure 82 shows NO release profiles by solid MIL-22 under water vapor pressure.
- the amount of NO released (N0 rel in mmol.g -1 on the left and ppm NO on the right) is expressed as a function of the time t (in hours).
- the CO adsorption measurements are carried out at 303K up to 2 bar in a laboratory-developed gauze metering system connected to a thermostatically gravimetric type apparatus (Rubotherm Prâzisionsme ⁇ technik GmbH).
- the MIL-100 (Fe) sample (500mg) is activated beforehand at the required temperature (100oC or 250oC) under vacuum (at 2 ⁇ 10 -3 mbar) for 12 or 20 hours.
- the dry CO gas Air Liquide, 99, 9%
- the measured weight gain as a function of time until stabilization.
- FIG. 83 represents the CO adsorption isotherm (Co ada in mmol / g) at the temperature of 303 K as a function of the pressure P (in bar) for the iron carboxylate MIL-100 (Fe) activated at 100 ° C. during a 12h (100oC curve), 250oC during 12h (curve 205oC (I)) and 250oC during 2Oh (curve 250 ° C (2)).
- the solid MIL-IOO (Fe) adsorb considerable amounts of CO to "room temperature (0.4-1.3 mmol ⁇ g -1) and at low pressure (up to 2 bars) ( Figure 83).
- the ability The adsorption of CO in the MIL-100 (Fe) increases drastically when the iron (III) of the compound MIL-100 (Fe) is partially reduced by activation at 250oC under primary vacuum (12 and 20 hours at 250oC). Infra-Red spectroscopy showed that under these conditions, this led to the reduction of about 15-20% iron (III) iron (II).
- Low-oxidation transition metals, such as iron (II) have additional electrons in orbitals d.
- MIL-88A Fe of composition Fe 3 O [O 2 CC 2 H 2 -
- the nanoparticles are stable in this medium.
- Table 16 Serum parameters measured 7 days after intravenous introduction of iron carboxylates MIL-88A (Fe) and MIL-88Bt (Fe).
- the animals used for the experiment are four week old female Wistar rats with a weight of 161.36 ⁇ 16.1 g.
- MIL-88A 150 and 500 nm
- MIL-88Bt 50 and 140 nm
- glucose 5% control group
- Blood samples taken in the jugular under isoflurane anesthesia were also performed at different times: 1 and 3 days, 1 and 2 weeks, 1, 2 and 3 months.
- the serum was isolated to measure serum parameters such as IL-6 (interleukin 6), albumin, serum Fe, PAS, GGT, bilirubin, cholesterol, and transaminases.
- each group of animals was sacrificed after 1 day, 1 week, 1 and 3 days, respectively. month.
- the animals were anesthetized with isoflurane and then the spleen, kidneys, liver and heart were removed and preserved for histological studies. Four times were also used for microsomal extraction to measure activation of cytochrome P450.
- intrajugular injection per day is performed for 4 consecutive days in 26 randomly assigned rats in different groups where the animals are sacrificed after 5 or 10 days.
- the blood undergoes the same treatment as for the acute toxicity test and the serum obtained is intended for the same analyzes.
- the animals On days of sacrifice, at 5 and 10 days, the animals are anesthetized with isoflurane and then the spleen, kidneys, liver, heart and lungs are removed and treated as well as for the acute toxicity test.
- Evolution of the weight of the animals The animals were weighed every day in order to compare the evolution of the weight of the different groups. An average was done for each day and in each of these groups.
- Cytochrome P450 is an enzyme associated with the smooth inner endoplasmic reticulum that is heavily involved in the degradation of exogenous molecules. This enzyme has a very low substrate specificity and is able to catalyze the transformation of newly synthesized compounds such as drugs. Most P450 cytochromes can be induced or repressed at the transcriptional level by different xenobiotics; this is often the cause of medication side effects. The determination of this enzyme makes it possible to determine whether the MOF material used is metabolized by cytochrome P450 in which case it would activate or inhibit its activity. The amount of cytochrome can only be interpreted if it has been related to the total amount of protein contained in each sample.
- the assay of the proteins contained in the sample was carried out thanks to the use of a BCA kit supplied by PIERCE (lot # HI106096).
- This method combines the reduction of Cu 2+ to Cu + by proteins in alkaline medium with colorimetric detection, very sensitive and selectively, Cu + cation with a reagent containing Bicinchoninic acid (BCA).
- BCA Bicinchoninic acid
- MIL-88A (having received glucose) and the group "MIL-88A" whose material is not metabolized by Cyp450.
- the MIL-88Bt material does not appear to be metabolized by Cyp450 either.
- Interleukin 6 is a multifunctional cytokine that plays an important role in host defense, immune responses, nerve cell function, and hematopoiesis.
- a high level of IL-6 in serum has been observed, for example, in viral and bacteriological infections, trauma, autoimmune diseases, inflammation, or cancer.
- the assay was performed using a "Quantikine, Rat IL-6" kit provided by R & D Systems laboratories.
- Subacute toxicity results the variations are not significant.
- An increase in the rate Plasma observed appears to be due to a puncture phenomenon during injections that produce local inflammation, if the individual groups are compared in isolation with the control group (glucose).
- Acute toxicity results The variations are not significant and lead to the same conclusions as in the case of subacute toxicity.
- liver nanoparticle injections transaminase levels (Alanine amino transferase or ALAT and aspartate amino transferase or ASAT), alkaline phosphatase (PAS), ⁇ -glutamate transferase (GGT) , bilirubin, cholesterol, albumin and serum iron.
- transaminase levels Alkaline phosphatase (PAS)
- PAS alkaline phosphatase
- GTT ⁇ -glutamate transferase
- bilirubin cholesterol
- albumin serum iron
- Serum albumin levels decreased slightly after the first day of injection for both materials, consistent with a local inflammatory process at injection and with the increase in IL-6 observed previously. After 3 days, levels return to normal. Serum levels of ASAT are increased one day after injection, which may indicate a process of cytolysis. However, 3 days later the administration of the nanoparticles, the values return to normal. Similarly, alkaline phosphatase is increased after 1 day, indicating a process of cytolysis, but the situation returns to normal after 3 days. The return to normal after 3 days indicates that it is a transient and non-permanent cytolysis process. There is no loss of cellular function. Cholesterol levels are normal. Serum iron levels are decreased in comparison with the control group, and this is more pronounced in the MIL-88A group. This could be explained by a complexation of serum iron by nanoparticles. The situation returns to normal 3 days after administration.
- the serum parameters were also measured at 1 week and according to these results, there is no longer any difference between the 3 groups in serum iron; rats treated with MIL-88A and MIL-88Bt recovered a serum iron concentration comparable to that of the control group. Moreover, regarding the levels of the other serum parameters, there is not a significant difference in comparison with the control group.
- Histologic sections Histological sections, with a thickness of
- Acute Toxicity Results Histologic liver sections show iron accumulation in the liver after injection of materials that is more important for MIL-88A solid. The material appears to be in the form of undegraded nanoparticles. Accumulation is less important for MIL-88Bt material, which may mean lower uptake for the liver or faster reuse of stored iron. After 1 and 3 months the iron content in the spleen and liver returns to normal.
- the determination of the iron contained in the suspensions of MIL-88A and MIL-88Bt injected into the animals is carried out by UV-Vis spectrophotometry at the wavelength of 520 nm, by specific colorimetry of the ferrous ions with the bipyridine (formation of a red complex), after solubilization of iron oxide in concentrated sulfuric acid, and reduction of ferric ions to ferrous ions by ascorbic acid.
- the determination of iron in the organs is carried out as well as for the iron determination of the suspensions explained above, after grinding . .. of the organ to be tested. This assay makes it possible to determine the route of elimination of the compounds of the material or their storage in certain organs: liver, kidneys, spleen and lungs, the heart being used as a control. d) Conclusion
- a cytochrome P-450 assay observed the state of cytochrome P-450 activity over a long period. This cytochrome is known for its ability to metabolize certain xenobiotics. The study shows that the activity rate, although fluctuating, remains below the values observed in control rats that received an injection of phenobarbital, a cytochrome P-450 activator, indicating that the materials are not metabolized. Cyp450, consistent with the high polarity of the dicarboxylic ligands. The results are very promising and already indicate that MIL-88A and MIL-88Bt 'materials induce no sign of severe toxicity, even if additional toxicity studies are to be performed.
- MOFs have many advantages over inorganic porous solids, zeolites.
- the high stability of zeolites in aqueous media does not allow its elimination by the body, producing the storage of the product in the body.
- most zeolites have aluminum in its structure, element with a very important toxicity.
- the composition of other MOFs based on metals well known for their toxicity (cobalt, nickel, chromium or copper) does not allow to consider applications in the medical or cosmetic field of these MOFs.
- the solids of the invention have a priori low toxicity composition (Fe, Ti). Indeed, in Example 16, we demonstrate the absence of toxicity of two iron carboxylates (iron fumarate and iron tetramethylterephthalate) by toxicity tests in vivo on rats.
- the solids of the invention it is possible to control their degradation rate according to the desired applications.
- the solid MIL-100 (Fe) is stable under hydrothermal conditions, whereas the solid MIL-101 (Fe) is degraded rapidly in the presence of water at room temperature.
- the amount of NO released (N0 rel in immol g -1 ) is expressed as a function of time t (in hours).
- the cream used is composed of 50% by weight of paraffin and 50% by weight of polyethylene glycol (PEG), both are blended with the aid of an automatic pipette for 30 seconds.
- PEG polyethylene glycol
- the amount of NO released by the cream is 6-8 times smaller than the powder form ( Figures 87 and 88) because the paraffin which is hydrophobic, does not allow the contact of the solid with the flow of water vapor.
- the release at biologically active concentrations is carried out for 10 hours in the case of the MIL-88A micrometric size solid
- FIG. 85 represents the profiles of release of NO under water vapor pressure of the solid MIL-88A (3 samples under the same conditions) in the form of cream.
- the amount of NO released (NO rel in mmol.g -1 ) is expressed as a function of time t (in hours).
- FIG. 86 represents the profiles of release of NO under water vapor pressure of the solid
- Topically applied nitric oxide induces T-lymphocyte infiltration in human skin, minimal inflammation goal, Mowbray M, Tan XJ, Wheatley
- Flavahan A. M. Choi, C. Weiner M., J. Vase. Res. 1999, 36, 114 - 119; T. Morita, S. At Mitsialis,
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Birds (AREA)
- Analytical Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Vascular Medicine (AREA)
- Oncology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Cosmetics (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Compounds Of Iron (AREA)
Abstract
La présente invention se rapporte à des solides constitués de réseau métal-organique ou « Metal-Organic Framework » (MOF), cristallin poreux chargé en au moins un gaz d'intérêt biologique, ainsi, qu'à leur procédé de préparation. Les solides MOF de la présente invention sont capables d'adsorber et de relarguer de manière contrôlée des gaz à intérêt biologique. Ils peuvent être utilisés dans le domaine pharmaceutique et/ou pour des applications dans le domaine cosmétique. Ils peuvent également être utilisés dans le domaine alimentaire.
Description
SOLIDE HYBRIDE CRISTALLIN POREUX POUR L'ADSORPTION ET LA LIBERATION DE GAZ A INTERET BIOLOGIQUE
DESCRIPTION
Domaine technique
La présente invention se rapporte à des solides constitués de réseau métal-organique ou « Metal-Organic
Framework » (MOF), cristallin poreux chargé en au moins un gaz d'intérêt biologique, ainsi, qu'à leur procédé de préparation.
Les solides MOF de la présente invention sont capables d'adsorber et de relarguer de manière contrôlée des gaz à intérêt biologique. Ils peuvent être utilisés dans le domaine pharmaceutique et/ou pour des applications dans le domaine cosmétique. Ils peuvent également être utilisés dans le domaine alimentaire.
Les références entre crochets [X] renvoient à la liste des références à la fin des exemples.
État de la technique
Les réseaux métal-organique ou « Metal-Organic Framework » (MOF) sont des polymères de coordination de charpente hybride inorganique-organique comprenant des ions métalliques et des ligands organiques coordinés aux ions métalliques. Ces matériaux sont organisés en réseau mono-, bi- ou tridimensionnels où les clusters
métalliques sont reliés entre eux par des ligands espaceurs de façon périodique. Ces matériaux ont une structure cristalline, sont le plus souvent poreux et sont utilisés dans de nombreuses applications industrielles telles que le stockage de gaz, l'adsorption de liquides, la séparation de liquides ou de gaz, la catalyse, etc.
On peut citer par exemple la demande de brevet US 10/039,733 [1] qui décrit un procédé réactionnel faisant intervenir un système de catalyse comprenant un matériau MOF à base de Zinc. Ce même matériau est également utilisé pour le stockage de gaz dans la demande de brevet US 10/061,147 [2].
En outre, les matériaux MOF basés sur des réseaux de même topologie sont qualifiés de « isoréticulaires » . Ces réseaux organisés dans l'espace ont permis d'obtenir une porosité plus homogène. Ainsi, la demande de brevet US 10/137,043 [3] décrit plusieurs matériaux IRMOF (Isoreticular Metal- Organic Framework) à base de zinc utilisés pour le stockage de gaz.
Par ailleurs, les gaz d'intérêt biologique tels que le NO, CO et H2S sont d'une importance extrême pour le fonctionnement biologique des mammifères. Ils interviennent dans un grand nombre de processus comme par exemple la vasodilatation, la prévention de l'agrégation des plaquettes- et la formation de thrombose, la neurotransmission et la guérison des plaies.
II est connu que tant le monoxyde de carbone (CO) comme l'oxyde nitrique (NO) à très faibles
concentrations ont une activité importante comme molécules de signalisation dans le corps.
NO a été amplement étudié dans la biologie [32]. L'activité biologique du NO comprend [33] : - activité anti-inflammatoire,
- régulation de la dysfonction sexuelle,
- indications cardiovasculaires (traitement de l'angine de poitrine.
De plus, NO est impliqué dans l'activité de nombreux médicaments (bloqueur de chanel de calcium, inhibiteurs de l'ACE et antagonistes du récepteur ANGII type 1, β- bloqueur et inhibiteurs de la réductase hydroxymethylglutaryl-CoA) .
Concernant le CO, même si le mécanisme d'action doit encore être élucidé pour le CO, son implication dans nombreux effets physiologiques est connue et décrite [34], Ainsi, le CO est impliqué dans des activités biologiques comme par exemple :
1. l'activité anti-inflammatoire - diminue le endotoxique shock [35],
- diminue l'inflammation allergique [36] ;
2. la suppresion du rejet des organes greffés [37] ;
3. la protection contre l'hyperoxia [38] ; 4. la protection contre l'ischémie [39] ;
5. la protection des cellules bêta pancréatiques de 1 ' apoptose [40] ;
6. la modulation de la spermatogenèse dans des conditions de stress à travers des cellules de Leydig [41] ;
7. la diminution de la pression de perfusion dans les régions isolées de la placenta humaine [42] ;
8. la protection contre le choc septique et des lésions pulmonaires chez les modèles animaux, 9. la modulation du tonus des muscles lisses vasculaires [44] ;
10. la régulation de la pression artérielle en situation de stress [45] ;
11. la suppression des lésions artériosclérotiques associés aux maladies chroniques et le rejet de greffe [46].
Les recherches scientifiques sur le rôle des émissions de CO dans l'organisme sont encore à un stade précoce. Il y a des recherches qui laissent entendre qu'il peut aussi intervenir significativement dans d'autres domaines de la médecine y compris la chirurgie de transplantation, la neuroprotection dans les accidents vasculaires cérébraux, la maladie d'Alzheimer [47]. ϋ Le CO est aussi impliqué dans le contrôle de la fonction vasculaire placentaire [48].
Le. rôle bénéfique de CO dans l'organisme a également été décrit dans trois demandes de brevet : US Patent 2002155166, WO 0278684, WO 02092075. Ces demandes décrivent l ' application du gaz CO dans le domaine médicale. US 2002155166 décrit par exemple l'utilisation de CO en tant que marqueur biologique et agent thérapeutique pour différentes types de maladies et dans le domaine de la transplantation. WO 0278684 porte sur les méthodes et les compositions pour traiter les maladies vasculaire, inflammatoire et du système immunitaire utilisant des composés capables de générer
du CO in vivo. CH2Cl2 est décrit comme étant le composé préféré capable de générer du CO in vivo. En effet, le CH2Cl2 est métabolisé en CO et fournit ainsi une source de gaz. WO 02092075 utilise les métaux carbonylés comme la source des émissions de CO gaz.
L ' intérêt du H2S comme une molécule de signalisation est de plus en plus important [49]. Le pKa de H2S étant de 6,8, à pH physiologique il est surtout présent en tant que H2S et [HS]2. La concentration de H2S est de 50-160 mM dans le tissu cérébral et de 10-100 mM dans le sang.
H2S est actif dans le système cardio-vasculaire et dans le système nerveux central. Par exemple, il peut causer une vasodilatation [50]. Par ailleurs, il est capable de coordonner facilement les groupes hemo et certains cytochromes. Plus de recherche est nécessaire pour démontrer l'importance de H2S en tant que molécule de signalisation.
L'utilisation de ces gaz exogènes ouvre de très nombreuses possibilités et est devenu un enjeu important pour le développement notamment de nouveaux médicaments ou des procédés prophylactiques et thérapeutiques incluant des applications potentielles dans les procédés anti-thrombogéniques , l'amélioration de l'efficacité du traitement de plaies et des ulcères, le traitement des infections fongiques et bactériennes etc rXa-—libération—contrôlée-de- ce type de gaz,, en particulier NO, de par ses propriétés antibactériennes, pourrait également s'avérer utile pour des applications en cosmétiques, en particulier pour les crèmes cosmétiques [4], mais également pour la
conservation des aliments (effet antibactérien et antioxydant ) [ 5 ] .
Dans le cas particulier de NO, la délivrance homogène de ce gaz à partir d'une solution est déjà connue pour certaines pathologies (i.e. à partir de trinitrate de glycéryl pour traiter l ' angine ) . Toutefois, cette approche est restreinte à cause d'effets secondaires indésirables comme conséquence de la grande variété d'effets qu'il a en fonction de la localisation (vasodilatateur et inhibiteur de l'agrégation plaquettaire, endothélium ; microbicide, macrophages dans certaines circonstances le NO peut provoquer des effets secondaires nuisibles : c'est le cas dans les septicémies, où la production excessive de NO par les macrophages conduit à une vasodilatation massive, cause principale de l'hypotension rencontrée dans le choc septique ; neurotransmetteur, cellules nerveuses ; relaxante des muscles lisses, tube digestif ; régulateur de l'apoptose, antiapoptotique ou apoptotique en fonction de la présence ou non de réducteurs cellulaires ) .
L'inhalation de NO gaz a également été utilisée pour traiter certaines pathologies des poumons. Cependant, la délivrance de NO sous forme gazeuse à partir de bouteilles de gaz n'est pas pratique et limite l'intérêt d'une telle méthode. par ailleurs, une. part significative des thérapies liées au gaz d'intérêt biologique nécessite un relargage ou libération contrôlé et ciblé dudit gaz dans des zones spécifiques du corps humain évitant ainsi des effets systémiques [6]. Dans le cas
particulier de NO, ceci est très important car NO a une durée de vie biologique courte.
Les technologies actuelles possèdent de nombreux inconvénients. Par exemple, les polymères de type NONOate ou à composante métallique ont une faible capacité de stockage, nécessitent une pression élevée pour charger le NO, sont relativement coûteux et potentiellement toxiques [7,8].
Dans des applications dermatologiques, les crèmes acides à base de nitrates, qui sont des donneurs potentiels de NO, sont pro-inflammatoires et ne sont pas adaptées aux peaux sensibles [9].
Récemment, l'utilisation de solides poreux de types zéolites ou de solides hybrides pour le stockage de NO a été décrite [10, 11, 12]. Ces solides possèdent une capacité de stockage qui surpasse celle des autres matériaux, une durée de vie longue (la capacité de relargage de NO après 2 ans de stockage reste intacte), un faible coût et ne semblent pas présenter de toxicité les rendant ainsi très attractifs.
De plus, les tests dermatologiques ont montré la compatibilité des zéolites chargées de NO avec la peau humaine y compris avec les peaux sensibles [13].
Malgré les avantages précités, la délivrance de NO avec les zéolithes ne peut se faire que sur une courte durée, les rendant ainsi inadaptés pour une application pour laquelle une libiération de longue durée est souhaitée
L'adsorption, le stockage et la libération du NO par des réseaux métal-organique poreux à base de cuivre ou aluminium pour inhiber l'agrégation
plaquettaire ont également été décrites. Malgré la grande capacité d'adsorption et de stockage de NO par ces solides (adsorption significativement améliorée par rapport aux autres solides comme les polymères 5 organiques ou les zéolithes ) , une fois en contact avec une solution biologique/physiologique (plasma riche en plaquette), ces solides montre une faible stabilité.
Comme mentionné, l'une des applications
10 particulièrement importante dans le domaine du relargage des gaz à intérêt biologique et en particulier du NO, concerne les appareillages antithrombotiques comme par exemple les stents, les cathéters et les canules qui sont insérés dans le flux
15 sanguin avec des durées variables pour des motifs thérapeutiques ou diagnostiques ainsi que des circuits extracorporels utilisés pour la dialyse rénale et en chirurgie.
En effet, la prévention des thromboses est
20 d'une importance vitale après l'insertion des stents, cathéters, conduits prosthétiques et autres implants médicaux dans le corps pendant l'opération chirurgicale, ce qui peut conduire souvent à des complications dangereuses notamment pour cause
25 d'occlusion des vaisseaux sanguins.
Parmi les inconvénients des systèmes déjà connus on peut citer par exemple: : - manque de libération ciblée des gaz, qui conduit à de nombreux effets secondaires indésirables, 30 - quantité libérée mal contrôlée, et donc potentiellement non adaptée à l'application requise,
durée de libération des gaz en milieu physiologique courte ou accompagné de libération de substances indésirables, et/ou une faible stabilité en milieu biologique/physiologique limitant ainsi leur utilisation dans de tels milieux.
Il existe donc un besoin réel de développer des systèmes permettant l'adsorption et le relargage de gaz d'intérêt biologique, dans lesquels lesdits systèmes possèdent une forte capacité d'adsorption et le relargage des gaz peut se faire de manière continue et ciblée.
De plus, il existe un besoin réel de disposer de systèmes permettant de délivrer de manière contrôlée la quantité optimale de gaz nécessaire pour une application donnée.
En outre, il existe un réel besoin de disposer de systèmes permettant de contrôler la durée de libération des gaz en milieu physiologique, notamment pour pouvoir avoir des libérations de longue durée pouvant aller au-delà de 6 heures tout en préservant leur stabilité en milieu biologique/physiologique pendant toute la durée de cette libération.
De plus, il existe un réel besoin de disposer de systèmes ayant des capacités de charge suffisantes surtout si l' on envisage des administrations - répétées de gaz.
Exposé de l'invention
La présente invention a précisément pour but de répondre à ces besoins et inconvénients de l'art
antérieur en fournissant des solides MOF cristallins poreux chargés en au moins un gaz base de Lewis choisi dans le groupe comprenant NO, CO et H2S dont au moins une partie se coordine avec M, ledit solide comprenant une succession tridimensionnelle de motifs répondant à la formule ( I ) suivante :

• chaque occurrence de M, identique ou différent, représente indépendamment un ion d'un métal de transition
 choisi dans le groupe comprenant Fe, Ti, Zr, Mn et dans lequel z est de 2 à 4 ou un mélange de ceux-ci;
choisi dans le groupe comprenant Fe, Ti, Zr, Mn et dans lequel z est de 2 à 4 ou un mélange de ceux-ci;
• m est 1 à 12 ;
• k est 0 à 4 ;
• 1 est 0 à 18 ;
• p est 1 à 6 ; • X est un anion choisi dans le groupe comprenant OH"

 est tel que défini ci-dessous,
est tel que défini ci-dessous,
 où R1 est un hydrogène, un alkyle en C1 à C12, linéaire ou ramifié, éventuellement substitué, un aryle, où n est un nombre entier de 1 à 4 ;
où R1 est un hydrogène, un alkyle en C1 à C12, linéaire ou ramifié, éventuellement substitué, un aryle, où n est un nombre entier de 1 à 4 ;
• L est un ligand espaceur comprenant un radical R
comportant q groupements carboxylates
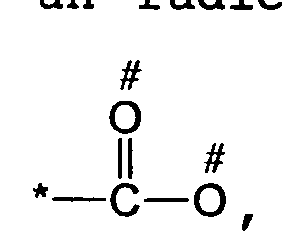 , où
, où
 q désigne le point d'attachement du carboxylate avec le radical R ;
- # désigne les points d'attachement possibles du carboxylate à l'ion métallique ;
q désigne le point d'attachement du carboxylate avec le radical R ;
- # désigne les points d'attachement possibles du carboxylate à l'ion métallique ;
- R représente :
(i) un radical C2_12alcène ou
 C2_12alcyne ;
C2_12alcyne ;
(ii) un radical aryle mono- ou poly-cyclique, fusionné ou non, comprenant 6 à 50 atomes de carbone ;
(iii) un hétéroaryle mono- ou poly-cyclique, fusionné ou non, comprenant 1 à 50 atomes de carbone ;
(iv) un radical organique comprenant un élément métallique choisi dans le groupe comprenant le ferrocène, la porphyrine, la phthalocyanine; le radical R étant éventuellement substitué par un ou plusieurs groupes R2, indépendamment choisis dans le groupe comprenant

C2.10alcène; C3_10cycloalkyle;

 hétéroalkyle;
hétéroalkyle;
 C6_10aryle;
C6_10aryle;
 3 20hétérσcyclique;
3 20hétérσcyclique;

 hétéroaryle ; F ; Cl ; Br; I;
hétéroaryle ; F ; Cl ; Br; I;


5
 ou
ou
 dans lequel le radical aryle, hétéroaryle ou hétérocyclique est éventuellement substitué ; ou bien, lorsque G représente
dans lequel le radical aryle, hétéroaryle ou hétérocyclique est éventuellement substitué ; ou bien, lorsque G représente
 RG1 et RG2 conjointement avec l'atome d'azote auquel ils 10 sont liés forment un hétérocycle ou un hétéroaryle éventuellement substitué.
RG1 et RG2 conjointement avec l'atome d'azote auquel ils 10 sont liés forment un hétérocycle ou un hétéroaryle éventuellement substitué.
Les MOF de selon invention, présente, entre autres, l'avantage : 15 - d'être à base de métaux non toxiques et donc adaptés à une application dans les domains pharmaceutique, médicales et/ou cosmétiques,
- présenter une stabilité supérieure à celle décrites pour les métaux tels que le cuivre ou l'aluminium, 20 - présenter une stabilité modulable en fonction du choix de la structure et du ligand organique, permettant ainsi d'adapter les MOF aux différentes applications souhaitées.
25 Dans le cadre de la présente invention, les termes « relarguer/relargage », « libérer/libération » et «délivrer/délivrance» » seront; indifféremment- utilisés pour signifier le dégagement partiel ou total des gaz présents dans les solides MOF. 30 Par « partiel », on entend une libération de moins de 100% de la quantité initialement adsorbée.
Le terme « substitué » désigne par exemple le remplacement d'un radical hydrogène dans une structure donnée par un radical R2 tel que défini précédemment. Lorsque plus d'une position peut être substituée, les substituants peuvent être les mêmes ou différents à chaque position.
On entend par « ligand espaceur » au sens de la présente invention, un ligand (incluant par exemple les espèces neutres et les ions) coordiné à au moins deux métaux, participant à l'éloignement entre ces métaux et à la formation d'espaces vides ou pores. Le ligand espaceur peut comprendre 1 à 6 groupements carboxylates, tels que définis précédemment, qui peuvent être monodentates ou bidentates, c'est-à-dire comprendre un ou deux points d'attachement au métal. Les points d'attachement au métal sont représentés par le sigle # dans les formules. Lorsque la structure d'une fonction A comporte deux points d'attachement #, cela signifie que la coordination au métal peut se faire par l'un, l'autre ou les deux points d ' attachement .
On entend par « alkyle » au sens de la présente invention, un radical carboné linéaire, ramifié ou cyclique, saturé ou insaturé, éventuellement substitué, comprenant 1 à 12 atomes de carbone, par exemple 1 à 10 atomes de carbone, par exemple 1 à 8 atomes de carbone, par exemple 1 à 6 atomes de-carboneτ
On entend par « alcène » au sens de la présente invention, un radical alkyle, tel que défini précédemment, présentant au moins une double liaison carbone-carbone .
On entend par « alcyne » au sens de la présente invention, un radical alkyle, tel que défini précédemment, présentant au moins une triple liaison carbone-carbone . On entend par « aryle » au sens de la présente invention, un système aromatique comprenant au moins un cycle satisfaisant la règle d'aromaticité de Hϋckel. Ledit aryle est éventuellement substitué et peut comprendre de 6 à 50 atomes de carbone, par exemple 6 à 20 atomes de carbone, par exemple 6 à 10 atomes de carbone.
On entend par « hétéroaryle » au sens de la présente invention, un système comprenant au moins un cycle aromatique de 5 à 50 chaînons parmi lesquels au moins un chaînon du cycle aromatique est un hétéroatome, notamment choisi dans le groupe comprenant le soufre, l'oxygène, l'azote, le bore. Ledit hétéroaryle est éventuellement substitué et peut comprendre de 1 à 50 atomes de carbone, de préférence 1 à 20 atomes de carbone, de préférence 3 à 10 atomes de carbone.
On entend par « cycloalkyle » au sens de la présente invention, un radical carboné cyclique, saturé ou insaturé, éventuellement substitué, qui peut comprendre 3 à 20 atomes de carbone, de préférence 3 à
10 atomes de carbone. On entend par «-haloalkyle » au sens de la présente invention, un radical alkyle tel que défini précédemment, ledit système alkyle comprenant au moins un halogène.
On entend par « hétéroalkyle » au sens de la présente invention, un radical alkyle tel que défini
précédemment, ledit système alkyle comprenant au moins un hétéroatome, notamment choisi dans le groupe comprenant le soufre, l'oxygène, l'azote, le bore.
On entend par « hétérocycle » au sens de la présente invention, un radical carboné cyclique comprenant au moins un hétéroatome, saturé ou insaturé, éventuellement substitué, et qui peut comprendre 2 à 20 atomes de carbone, de préférence 5 à 20 atomes de carbone, de préférence 5 à 10 atomes de carbone. L 'hétéroatome peut être par exemple choisi dans le groupe comprenant le soufre, l'oxygène, l'azote, le bore.
On entend par « alkoxy » , « aryloxy » ,
« hétéroalkoxy » et « hétéroaryloxy » au sens de la présente invention, respectivement un radical alkyle, aryle, hétéroalkyle et hétéroaryle liés à un atome d ' oxygène .
On entend par « alkylthio » , « arylthio » ,
« hétéroalkylthio » et « hétéroarylthio » au sens de la présente invention, respectivement un radical alkyle, aryle, hétéroalkyle et hétéroaryle liés à un atome de soufre.
Les gaz de Lewis selon l'invention sont des gaz à intérêt biologique choisis dans le groupe comprenant NO, CO et H2S. De préférence, ledit gaz est du NO.
La structure cristalline particulière des solides MOF selon l'invention procure à ces matériaux des propriétés spécifiques.
Dans les solides MOF de l'invention, M est choisi dans le groupe comprenant Fe, Ti, Mn, Zr. M peut également être un mélange de ces métaux. M est avantageusement le Fe.
Comme indiqué précédemment, M est un ion de métal de transition Mz+ dans lequel z est de 2 à 4.
Lorsque M est un mélange de métaux, pour chaque métal, z peut être avoir une valeur identique ou différente. Dans un mode de réalisation de l'invention, les solides de l'invention comprennent une succession tridimensionnelle de motifs de formule (I) dans laquelle M peut représenter un seul type d'ion Mz+, par exemple le Fe, dans lequel z peut être identique ou différent, par exemple 2, 3 ou un mélange de 2 et de 3.
Dans un autre mode de réalisation de l'invention, les solides de l'invention peuvent comprendre une succession tridimensionnelle de motifs de formule (I) dans laquelle M peut représenter un mélange de différents ions Mz+, par exemple le Fe et le
Ti, dans lequel pour chaque ion métallique z peut être identique ou différent, par exemple 2, 3, 4 ou un mélange de 2 , 3 et 4.
Dans un mode de réalisation particulier, Mz+ représente le Fe trivalent octaédrique avec z égal à 3. Dans ce mode de réalisation, le Fe présente un nombre de coordination de 6.
Par le nombre coordination, on entend le nombre de liaison pour laquelle les deux électrons partagés dans la liaison proviennent du même atome. L'atome donneur d'électrons acquiert une charge positive alors que l'atome accepteur d'électrons acquiert une charge négative.
Les ions métalliques peuvent être isolés ou regroupés en « clusters » métalliques. Les solides MOF selon l'invention peuvent par exemple êtres construits
à partir de chaînes d'octaèdres ou de trimères d'octaèdre.
On entend par « cluster métallique » au sens de la présente invention un ensemble d'atomes contenant au moins deux ions métalliques liés par des liaisons ionocovalentes , soit directement par des anions, par exemple O, OH, Cl, etc., soit par le ligand organique.
De plus, les solides MOF selon l'invention peuvent se présenter sous différentes formes ou « phases » compte tenu des diverses possibilités d'organisation et de connections des ligands à l'ion métallique ou au groupement métallique.
On entend par « phase » au sens de la présente invention une composition hybride comprenant au moins un métal et au moins un ligand organique possédant une structure cristalline définie.
L'organisation spatiale cristalline des solides de la présente invention est à la base des caractéristiques et propriétés particulières de ces matériaux. Elle régit notamment la taille des pores, qui a une influence sur la surface spécifique des matériaux et sur les caractéristiques d'adsorption.
Elle régit également la densité des matériaux, celle-ci étant relativement faible, la proportion de métal dans ces matériaux, la stabilité des matériaux, la rigidité et la flexibilité des structures, etc. En outre, la taille -des- pores peut être ajustée par le choix de ligands L appropriés.
En particulier, le ligand L du motif de formule (I) de la présente invention peut être un ligand di-, tri-, tétra- ou hexa-carboxylate choisi dans le groupe comprenant :
 (succinate), C3H6 ( CO2- )2 (glutarate), C4H4 ( CO2- )2 (muconate), C4H8 (CO2-)2 (adipate), C5H3S (CO2-)2 (2,5- thiophènedicarboxylate ) , C6H4 (CO2-) 2 ( téréphtalate ) , C6H2N2 (CO2 ") 2 (2, 5-pyrazine dicarboxylate), C10H6 ( CO2- )2 (naphtalène-2, 6-dicarboxylate) , C12Hβ(CO2 ")2 (biphényle- 4,4' -dicarboxylate ) , C12H8N2 (CO2-) 2
(succinate), C3H6 ( CO2- )2 (glutarate), C4H4 ( CO2- )2 (muconate), C4H8 (CO2-)2 (adipate), C5H3S (CO2-)2 (2,5- thiophènedicarboxylate ) , C6H4 (CO2-) 2 ( téréphtalate ) , C6H2N2 (CO2 ") 2 (2, 5-pyrazine dicarboxylate), C10H6 ( CO2- )2 (naphtalène-2, 6-dicarboxylate) , C12Hβ(CO2 ")2 (biphényle- 4,4' -dicarboxylate ) , C12H8N2 (CO2-) 2
( azobenzènedicarboxylate ) , C12H6Cl2N2 (CO2-) 2
( dichloroazobenzènedicarboxylate ) , C12H6N2 (CO2-) 4 ( azobenzènetetracarboxylate ) , C12H6N2 (OH ) 2 (CO2 " ) 2 (dihydroxoazobenzènedicarboxylate) , C6H3(CO2 ")3 (benzène- 1,2,4-tricarboxylate) , C6H3 ( CO2- )3 (benzène-1,3, 5- tricarboxylate) , C24H15(CO2 ")3 (benzène-1,3,5- tribenzoate) , C42H27 (CO2-)3 ( 1,3,5-tris[4'-carboxy( 1, 1 '- biphenyl-4-yl )benzène ), C6H2(CO2-)4 (benzène-1, 2,4,5- tétracarboxylate, C10H4(CO2 ")4 (naphtalène-2,3,6, 7- tétracarboxylate) , C10H4(CO2-)4 (naphtalène-1, 4,5,8- tétracarboxylate), C12H6 (CO2-)4 (biphényl-3,5,3 ' ,5'- tétracarboxylate, et les analogues modifiés choisis dans le groupe comprenant le 2-aminotéréphtalate, le 2- nitrotéréphtalate, le 2-méthyltéréphtalate, le 2- chlorotéréphtalate, le 2-bromotéréphtalate, le 2,5- dihydroxotéréphtalate, le tétrafluorotéréphtalate, le 2 , 5-dicarboxytéréphtalate le diméthyl-4 , 4 ' - biphénydicarboxylate, le tétraméthyl-4,4 '- biphénydicarboxylate le dicarboxy-4 , 4 ' - biphénydicarboxylate . : En particulier, anion X du motif de formule
(I) de la présente invention peut être choisi dans le groupe comprenant OH-, Cl-, F-, R-(COO)n-, PF6-, ClO4-, avec R et n tel que défini précédemment.
En particulier, le solide MOF selon l'invention, peut comprendre un pourcentage massique de M en phase sèche de 5 à 50%, de préférence de 18 A 31%. Le pourcentage massique (%m) est une unité de mesure utilisée en chimie et en métallurgie pour désigner la composition d'un mélange ou d'un alliage, c'est-à-dire les proportions de chaque composant dans le mélange.
1 %m d'un composant = Ig du composant pour 100 g de mélange ou encore 1 kg dudit composant pour 100 kg de mélange.
Les solides MOF de la présente invention présentent notamment l'avantage d'avoir une stabilité thermique jusqu'à une température de 350ºC. Plus particulièrement, ces solides présentent une stabilité thermique de 120ºC et 350ºC).
En particulier, le solide MOF selon l'invention peut avoir une taille de pores de 0,4 à 6 nm, de préférence de 0,5 à 5,2 nm, et de manière plus préférée de 0,5 à 3,4 nm.
En particulier, le solide MOF selon l'invention peut avoir une surface spécifique (BET) de 5 à 6000 m2/g, de préférence de 5 à 4500 m2/g.
En particulier, le solide MOF selon l'invention peut avoir un volume poreux de 0 à 4 cmVg, de préférence de 0,05 à 2 cmVg. Dans le-cadre de l' invention, le volume poreux-signifie le volume accessible pour les molécules de gaz.
Le solide MOF de l'invention peut avoir une capacité de charge en gaz de 0,5 à 50 mmol de gaz par gramme de solide sec.
Dans le cadre de la présente invention, la capacité de charge signifie la capacité de stockage de gaz ou la quantité de gaz adsorbé dans le matériau. La capacité de charge peut être exprimée en capacité 5 massique (gramme/gramme) ou en capacité molaire (mol/mol) ou en d'autres ( mol/gramme, gramme/mol, etc. )
Comme indiqué précédemment, dans les solides
MOF de l'invention, au moins une partie du ou des gaz
10 base de Lewis se coordine avec M. Avantageusement, au moins 1 à 5 mmol de gaz par gramme de solide sec se coordine avec M.
La partie du ou des gaz qui ne se coordine pas avec M peut remplir avantageusement l'espace libre dans 15 les pores.
Le solide MOF de la présente invention peut se présenter sous la forme d'une structure robuste, qui a une charpente rigide et ne se contracte que très peu lorsque les pores se vident de leur contenu qui peut
20 être, par exemple, du solvant, de l'acide carboxylique non coordine, etc. Il peut également se présenter sous la forme d'une structure flexible, qui peut se gonfler et se dégonfler faisant varier l'ouverture des pores en fonction de la nature des molécules adsorbées qui
25 peuvent être, par exemple, des (solvants et/ou des gaz) . On entend par :« structure rigide », au sens de la présente invention des structures qui se gonflent ou se contractent que très faiblement, c'est-à-dire avec 30 une amplitude jusqu'à 10%.
En particulier, le solide MOF selon l'invention, peut avoir une structure rigide qui se
gonfle ou se contracte avec une amplitude allant de 0 à 10%.
Les structures rigides peuvent par exemple être construites à base de chaînes ou de trimères d'octaèdres.
Selon un mode de réalisation de l'invention, le solide MOF de structure rigide peut avoir un pourcentage massique de M en phase sèche de 5 à 50%, de préférence de 18 à 31%. Avantageusement, M représentera ici le fer.
Le solide MOF de structure rigide selon l'invention peut avoir une taille de pores de 0,4 à 6 nm, en particulier de 0,5 à 5,2 nm, plus particulièrement de 0,5 à 3,4 nm. Le solide MOF de structure rigide selon l'invention peut avoir un volume poreux de 0,5 à 4 cmVg, en particulier de 0,05 à 2 cmVg.
On entend par « structure flexible », au sens de la présente invention des structures qui se gonflent ou se contractent avec une grande amplitude, notamment avec une amplitude supérieure à 10%, de préférence supérieure à 50%.
Les structures flexibles peuvent par exemple être construites à base de chaînes ou de trimères d'octaèdres.
En particulier, le matériau MOF selon l-'invention, peut- avoir une structure- flexible- qui se gonfle ou se contracte avec une amplitude de 10% à 300%, de préférence de 50 à 300%. Dans un mode de réalisation particulier de l'invention, le solide MOF de structure flexible peut avoir un pourcentage massique de M en phase sèche de 5
à 40%, de préférence de 18 à 31%. Avantageusement, M représentera ici le fer.
Par exemple, dans le cadre de l'invention, le solide MOF de structure flexible peut avoir une taille de pores de 0,4 à 6 nm, en particulier de 0,5 à 5,2 nm, et plus particulièrement de 0,5 à 1,6 nm.
Par exemple, le solide MOF de structure flexible selon l'invention peut avoir un volume poreux de 0 à 3 cm3/g, en particulier de 0 à 2 cmVg. En outre, les inventeurs ont mis en évidence expérimentalement que l'amplitude de la flexibilité dépend de la nature du ligand et du solvant utilisé, comme décrit dans la partie « Exemples » ci-dessous.
Différents matériaux MOF ont été élaborés par les inventeurs à l'Institut Lavoisier de Versailles nommées « MIL » (pour « Matériau Institut Lavoisier »). L'appellation « MIL » de ces structures est suivie d'un nombre arbitraire n donnée par les inventeurs pour identifier les différents solides. Dans le cadre de la présente invention, les inventeurs ont mis en évidence que des solides MOF peuvent comprendre une succession tridimensionnelle de motifs répondant à la formule (I).
Dans un mode de réalisation de l'invention, les solides MOF peuvent comprendre une succession tridimensionnelle de carboxylates de fer (III) répondant à la formule (I). Ces carboxylates de fer (III )- peuvent- être choisis dans le groupe comprenant MIL-88, MIL-89, MIL-96, MIL-100, MIL-101 et MIL-102, et plus particulièrement dans le groupe comprenant MIL-88A, MIL-88B, MIL-88Bt, MIL-88C, MIL-88D, MIL-88E, MIL-89,
MIL-96, MIL-100, MIL-101, MIL-102. Ces motifs sont présentés dans la partie « Exemples » .
En particulier, les solides MOF peuvent comprendre une succession tridimensionnelle de motifs répondant à la formule (I) , choisis dans le groupe comprenant :
 de structure flexible, par exemple MIL-88A
de structure flexible, par exemple MIL-88A
 de structure flexible, par exemple MIL-88B de structure flexible, par exemple
de structure flexible, par exemple MIL-88B de structure flexible, par exemple
 MIL-88C de structure flexible, par exemple
MIL-88C de structure flexible, par exemple
 MIL-88D
MIL-88D
 de structure flexible, par exemple MIL-89
de structure flexible, par exemple MIL-89
 de structure rigide, par exemple MIL-96 de structure rigide, par exemple MIL-
de structure rigide, par exemple MIL-96 de structure rigide, par exemple MIL-
 100
100
 de structure rigide, par exemple MIL-101
de structure rigide, par exemple MIL-101
 de structure rigide, par exemple MIL-102 dans laquelle X est tel que défini précédemment.
de structure rigide, par exemple MIL-102 dans laquelle X est tel que défini précédemment.
En outre, à partir d'un même ligand acide carboxylique L et des mêmes bases de fer (trimères), les inventeurs ont pu obtenir des matériaux MOF de même formule générale (I) mais de structures différentes. C'est par exemple le cas des solides MIL-88B et MIL- 101. En effet, la différence des solides MIL-88B et MIL-101 réside dans le mode de connexions des ligands
aux trimères d'octaèdre : dans le solide MIL-101, les ligands L s'assemblent sous forme de tétraèdres rigides, tandis que dans le solide MIL-88B, ils forment des bipyramides trigonales, rendant possible l'écartement entre les trimères.
Ces différents matériaux sont présentés dans la partie « Exemples » ci-dessous. Le mode d'assemblage de ces ligands peut être contrôlé lors de la synthèse par exemple par l'ajustement du pH. Par exemple, le solide MIL-88 est obtenu en milieu moins acide que le solide MIL-101 comme décrit dans la partie « Exemples » ci- dessous.
En outre, les solides MOF de l'invention peuvent rendre possible le greffage de molécules à leur surface afin de répondre aux besoins liés à la vectorisation de composés vers des cibles biologiques spécifiques et/ou à la furtivité des particules. Ceci permet ainsi d'améliorer la biodistribution des principes actifs de manière plus ciblée. Ainsi, selon un mode de réalisation particulier, les solides MOFs selon l'invention, peuvent éventuellement comprendre à leur surface au moins un agent de surface organique. Celui-ci peut être greffé ou déposé à la surface des solides, par exemple adsorbé en surface ou lié par liaison covalente, par liaison hydrogène, par liaison de Van der Waals, parinteraction électrostatique. L'agent de- surface- peut- également être incorporé par enchevêtrement lors de la fabrication des solides MOF [10, 28]. On entend par « agent de surface » selon l ' invention une molécule recouvrant en partie ou en
totalité la surface du solide permettant de moduler les propriétés de surface du matériau, par exemple : de modifier sa biodistribution, par exemple pour éviter sa reconnaissance par le système réticulo endothélial ( « furtivité » ) , et/ou de lui conférer des propriétés de bioadhésion intéressantes lors de l'administration par voies orale, oculaire, nasale, et/ou
- de lui permettre un ciblage spécifique de certains organes/tissus malades, etc.
Selon l'invention, plusieurs agents de surface peuvent être utilisés pour combiner les propriétés précitées.
Selon l'invention, l'agent de surface organique peut être choisi par exemple dans le groupe comprenant:
- un oligosaccharide comme les cyclodextrines,
- un polysaccharide comme le chitosan, le dextran, le fucoïdane, l'alginate, la pectine, l'amylose, l'amidon, la cellulose, le xylane, - un glycosaminoglycanne comme l'acide hyaluronique, l'héparine,
- un polymère comme le polyéthylène glycol (PEG), l'alcool polyvinylique, le polyéthylène imine,
- un tensioactif comme le pluronic, la lécithine. - des vitamines cpmme la biotine
- des coenzymes comme l'acide lipoique
- des anticorps ou fragments d'anticorps
- des aminoacides ou peptides.
L'agent de surface peut également être une molécule de ciblage, c'est-à-dire une molécule
reconnaissant ou étant reconnue spécifiquement par une cible biologique. L'association des solides MOF de l'invention avec une molécule de ciblage permet ainsi de vectoriser les produits vers cette cible biologique, cellule, tissus, organe.
Ainsi, l'agent de surface organique peut être une molécule de ciblage choisie dans le groupe comprenant la biotine, le chitosan, l'acide lipoique, un anticorps ou fragment d'anticorps, un peptide. Par exemple, la présence de la biotine en surface peut être mise à profit afin de coupler des ligands de manière aisée, par exemple par simple incubation. Pour cela, il est possible d'utiliser des protocoles décrits dans les publications [29, 30]. Cette méthode de modification de surface présente l ' avantage de ne pas perturber le cœur des solides MOF, même lorsqu'ils contiennent du gaz, et de pouvoir se faire après la synthèse des solides MOF, et donc d'offrir une variété de recouvrements possibles. II est également possible d'utiliser un mélange de polymères portant des fonctions susceptibles d' interagir avec la particule (MOF) en tant qu'agent de surface afin de répondre à un cahier de charge précis, par exemple la bioadhésion, la reconnaissance spécifique, etc.
L'invention a également pour objet un procédé de préparation de solides MOF tels que définis dans la présente invention, comprenant au moins une étape réactionnelle consistant
(i) à mélanger dans un solvant polaire :
- au moins une solution comprenant au moins un précurseur inorganique métallique se présentant sous la forme de métal M, d'un sel métallique de M ou d'un complexe de coordination comprenant un ion métallique de M
- au moins un ligand L' comprenant un radical R comportant q groupements *-C(=O)-R3, où
• q et R sont tels que définis précédemment,
• * désigne le point d'attachement du groupement avec le radical R,
• R3 est choisi dans le groupe comprenant un OH, un OY, avec Y est un cation alcalin, un halogène, un radical —OR4, -O-C(=O)R4, -NR4R4', où R4 et R4' sont des radicaux alkyles en C1-12, pour obtenir un matériau MOF ;
( ii ) à activer le matériau MOF obtenu en ( i ) ; et ( iii ) à mettre en contact le matériau. MOF obtenu à l'étape (ii) avec un gaz base de Lewis dont au moins une partie se coordine avec M, de façon à obtenir ledit solide.
M est un ion d'un métal de transition comme défini précédemment.
La préparation de matériaux MOF peut être de préférence réalisée en présence d'énergie qui peut être apportée par exemple par le chauffage, comme par exemple des conditions hydrothermales ou solvothermales, mais égalementr par micro-ondes , par ultrasons, par broyage, par un procédé faisant intervenir un fluide supercritique, etc. Les protocoles correspondants sont ceux connus de l'homme du métier. Des exemples non limitatifs de protocoles utilisables pour les conditions hydrothermales ou solvothermales
sont décrits par exemple dans [20]. Pour la synthèse par voie micro-ondes, des exemples non limitatifs de protocoles utilisables sont décrits par exemple [21, 22, 23, 24] Les conditions en présence d'un broyeur à cylindre peuvent par exemple se référer aux publications [25, 26, 27].
Les conditions hydrothermales ou solvothermales, dont les températures de réactions peuvent varier entre 0 et 220ºC, sont généralement effectuées dans des récipients en verre (ou en plastique) lorsque la température est inférieure à la température d'ébullition du solvant. Lorsque la température est supérieure ou lorsque la réaction s'effectue en présence de fluor, des corps en téflon insérés dans des bombes métalliques sont employés [20],
Les solvants utilisés sont généralement polaires . Notamment les solvants suivants peuvent être utilisés : l'eau, les alcools, le diméthylformamide, le diméthylsulfoxide, l'acétonitrile, le tétrahydrofurane, le diéthylformamide, le chloroforme, le cyclohexane, l'acétone, le cyanobenzène, le dichlorométhane , le nitrobenzène, l'éthylèneglycol, le diméthylacétamide ou des mélanges de ces solvants.
Un ou plusieurs co-solvants peuvent également être ajoutés à n'importe quelle étape de la synthèse pour une meilleure solubilisation des composés du mélange II peut s-agir notamment d'acides monocarboxyliques, tels que l'acide acétique, l'acide formique, l'acide benzoïque, etc. Un ou plusieurs additifs peuvent également être ajoutés au cours de la synthèse afin de moduler le pH du mélange. Ces additifs sont choisis parmi les acides
inéraux ou organiques ou les bases minérales ou organiques. À titre d'exemple, l'additif peut être choisi dans le groupe comprenant : HF, HCl, HNO3, H2SO4, NaOH, KOH, la lutidine, l'éthylamine, la méthylamine, l'ammoniac, l'urée, l'EDTA, la tripropylamine, pyridine .
De préférence, l'étape réactionnelle (i) peut être réalisée suivant au moins une des conditions réactionnelles suivantes : - avec une température de réaction de 0ºC à 220ºC, de préférence de 50 à 150ºC ;
- avec une vitesse d'agitation de 0 à 1000 rpm (ou rotation par minute), de préférence de 0 à 500 rpm ; - avec un temps de réaction de 1 minute à 144 heures, de préférence de 1 minute à 15 heures ;
- avec un pH de 0 à 7, de préférence de 1 à 5 ;
- avec l'addition d'au moins un co-solvant au solvant, au précurseur, au ligand ou au mélange de ceux-ci, ledit co-solvant étant choisi dans le groupe comprenant l'acide acétique, l'acide formique, l'acide benzoïque ;
- en présence d'un solvant est choisi dans le groupe comprenant l'eau, les alcools Rs—OH où Rs est un radical alkyle en C1 à C6 linéaire ou ramifié, le diméthylformamide, diméthylsulfoxide l ' acétonitrile, le tétrahydrofurane le diéthylformamide, le chloroforme, le cyclohexane, l'acétone, le cyanobenzène, le dichlorométhane, le nitrobenzène, l'éthylèneglycol, le diméthylacétamide ou des mélanges de ces solvants, miscibles ou non ;
- dans un milieu supercritique, par exemple dans le CO2 supercritique ;
- sous micro-ondes et/ou sous ultrasons ;
- dans des conditions d'électrolyse électrochimique ; dans des conditions utilisant un broyeur à cylindres ;
- dans un flux gazeux.
Comme indiqué, préalablement à la mise en contact du matériau MOF avec le gaz base de Lewis dans l'étape (iii), il est nécessaire que le matériau issu de l'étape (i) soit activé dans l'étape (ii).
Cette étape d'activation (ii) permet de vider les pores du matériau MOF et de les rendre accessibles pour la coordination du ou des gaz. Le vidage peut se faire, par exemple, par le départ des molécules d'eau et/ou des solvants présents dans le milieu réactionnel soit par activation sous vide primaire ou secondaire ou sous flux gazeux (hélium, azote, air...), avec ou non le chauffage du solide à une température de 25 à 300ºC, en particulier de 50 à 250ºC, et plus particulièrement de 100 à 250ºC. Le chauffage peut être effectué pendant une période de temps entre 1 heure et 96 heures, typiquement entre 3 et 15 heures. L'étape (ii) peut également être une étape de réduction des centres métalliques M dudit matériau MOF en ions Mz+ dans lequel- z est de 2 à 4. Selon les conditions d'activation, les centres métalliques peuvent être partiellement voire totalement réduits. Les centres métalliques peuvent être constitués de métaux identiques ou différents, par exemple uniquement le fer, ou un mélange d'un ou plusieurs
métaux tels que le fer en présence de titane, de manganèse ou de zirconium.
Lorsque les centres métalliques sont en partie réduits, en particulier si une partie du fer est au degré d'oxydation + I I ( z=2 ) ou une partie du manganèse au degré d'oxydation +III (z=3), une partie des molécules de gaz peuvent alors se coordiner plus fortement avec le métal par effet de rétrodonation. Par rétrodonation, on entend le transfert de la densité électronique du métal M dans une orbitale antiliante du gaz . Cela conduit à une augmentation du nombre de molécules de gaz coordinées par centre métallique. Le solide MOF final comportera alors une plus grande quantité de molécules de gaz coordinées aux ions métalliques réduits. Les solides MOF résultants posséderont alors une plus forte capacité de stockage de gaz.
Par ailleurs, la réduction partielle des centres métalliques a une influence sur la durée du relargage des gaz par le solide MOF, celle-ci augmentant de manière conséquente.
L'étape d'activation (ii) peut, en outre, être réalisée à une température élevée et sous pression réduite. Par pression réduite, on entend une pression allant de 1 à 10-2 Pa, avantageusement de 10-3 à 10-5 Pa.
Par exemple, l'activation peut se faire à 50- 250ºC sous_une_ pression de 1 à 10-2 Pa, ou de 10-3 à 10-5 Pa.
Dans l'étape (iii) du procédé de l'invention, le matériau MOF activé dans l'étape (ii) est mis en contact avec au moins un de gaz base de Lewis. Le gaz
peut être sous forme pure ou en mélange avec un gaz inerte .
La mise en contact du solide avec le gaz dans l'étape (iii) peut être réalisée à une température allant de -150 à 100 ºC.
L'étape (iii) peut également être réalisée à une pression allant de 104 à 107 Pa.
Dans un mode de réalisation particulier, le gaz base de Lewis est de préférence le NO. L'étape (iii) peut alors être réalisée à une température allant de
-100ºC à +50ºC. La mise en contact du NO avec le solide peut se faire à une pression allant de 105 à 106 Pa.
Selon l'application envisagée, on peut utiliser un mélange de gaz base de Lewis dans l'étape (iii) du procédé.
Le solide MOF selon l'invention peut avoir une capacité de charge en gaz de 0,5 à 50 mmol de gaz par gramme de solide sec .
Comme indiqué précédemment, au moins une partie du gaz peut se coordiner avec M. Avantageusement, le solide selon l ' invention peut avoir au moins 1 à 5 mmol de gaz par gramme de solide sec coordiné avec M.
Selon un mode particulier de réalisation de l'invention, lorsque les solides MOFs de l'invention, comprennent à leur surface au moins un agent de surface, le procédé de préparation des solides MOF selon l' invention, peut comprendre en outre une étape-
(iv) de fixation sur ledit solide d'au moins un agent de surface organique. Cette étape de fixation (iv) peut être réalisée pendant ou après l'étape réactionnelle (i) ou encore après l'étape d'activation (ii) et avant l'étape (iii)
de mise en contact du matériau MOF avec le gaz. Des exemples sont fournis ci-dessous.
Le procédé de préparation de l ' invention a l'avantage de permettre l'obtention des solides MOF cristallins purs, et homogènes, en un nombre réduit d'étapes et avec des rendements élevés. Ceci réduit la durée de synthèse et les coûts de fabrication.
Par ailleurs, les inventeurs ont également mis en évidence que les caractéristiques structurales particulières des solides de la présente invention, notamment en termes de flexibilité ou de taille des pores, en font des adsorbants de grande capacité de charge, de grande sélectivité et de grande pureté. Ils rendent donc possible l'adsorption sélective de molécules de gaz base de Lewis à intérêt biologique, comme par exemple des molécules de NO, CO ou H2S, avec un coût énergétique favorable et un temps de relargage plus élevé. Ainsi, les travaux de recherche menés par les inventeurs leur ont permis de mettre en évidence l'intérêt les matériaux MOF selon l'invention pour l'adsorption et la libération contrôlée, en termes de quantité de gaz et de durée de libération, de gaz à intérêt biologique.
De par leur structure, les solides MOF de l'invention permettent de contrôler la durée de libération des gaz. Ainsi, avec les solides MOF de l'iinvention la durée de libération des gaz peuvent aller de 1 à 100 heures alors qu'avec les zéolithes de l'état de la technique, la durée de cette libération n'excède pas 10 heures sous une pression de vapeur d'eau [10, 11].
Par ailleurs, les solides MOF de l'invention ont une capacité d'adsorption et de stockage de gaz base de Lewis sensiblement supérieure à celle des zéolithes connus. Par exemple, les MOF de l'invention peuvent avoir une capacité d'adsorption du NO allant de 2,5 à 4,5 mmol/g alors qu'avec les zéolithes connus, cette capacité est inférieure à 1,5 mmol/g. Ceci permet de diminuer la quantité de matériau à utiliser pour une application donnée. De plus, cette plus grande capacité d'adsorption permet de disposer de matériaux dans lesquels la libération des gaz peut se faire de manière continue. La libération des gaz peut également être ciblée lorsque les matériaux comprennent à leur surface au moins un agent de ciblage tel que défini précédemment .
L'invention concerne également l'utilisation de solides MOF selon l'invention, chargés en au moins un gaz base de Lewis à intérêt biologique dont au moins une partie se coordine avec M comme médicament. Ledit gaz ou le mélange de gaz peut être contenu dans les pores et au moins en partie coordine avec M 1 'invention.
En effet, les solides MOF selon l'invention ont l'avantage de présenter de grandes capacités d'adsorption. En outre, ils permettent d'adsorber efficacement des molécules de gaz qui peuvent présenter- des difficultés particulières, par exemple en raison de leur instabilité, leur grande réactivité, leur faible solubilité, etc.
En outre, le gaz base de Lewis peut être tout gaz donneur d'électrons ayant un intérêt biologique, comme par exemple, le NO, CO, H2S. [10, 28].
Une utilisation particulièrement intéressante des MOF selon l'invention est dans la prévention des thromboses, notamment après l'insertion des stents, cathéters, conduits prosthétiques et autres implants médicaux dans le corps suite à une intervention chirurgicale. Dans ce type d'utilisation, les solides MOF de l'invention peuvent servir, par exemple, de revêtement pour les articles médicaux précités. En tant que revêtement, ils peuvent être utilisés seuls ou en association avec d'autres agents thérapeutiques. Les MOF étant chargés en gaz, sont capables de relarguer le gaz (ou le mélange de gaz). Ils peuvent alors constituer un moyen efficace pour prévenir la formation des thromboses au contact du corps étranger. Les solides MOF de l'invention peuvent donc être utilisés comme médicament antithrombotique . On peut également envisager de les utiliser seuls ou en association avec d'autres agents antithrombotiques connues comme par exemple le clopidogrel.
L'invention s'étend également aux articles médicaux, comme par exemple les stents, cathéters, conduits prosthétiques et autres implants médicaux dans le corps suite à une intervention chirurgicale, comprenant un solide MOE selon l'invention Keefer L.
K.., Nat. Mater. 2003, VOL 2, 357 [31].
En outre, le solide MOF selon l'invention peut être chargé en au moins un gaz base de Lewis à intérêt biologique pour une utilisation dans le domaine de la cosmétique ou de la dermatologie. L'activité
antibactérienne du NO peut, par exemple, être très intéressante pour des applications notamment dans le domaine des crèmes cosmétiques. Selon la teneur en eau de l'émulsion dans laquelle les particules de solides MOF de l'invention peuvent être dispersées, un relargage modulable du NO va permettre de désinfecter la peau de manière prolongée. De plus, le caractère inorganique-organique des solides MOFS va faciliter leur dispersion dans les crèmes cosmétiques. Les effets cosmétiques ou dermatologiques bénéfiques pouvant résulter de l'utilisation d'un solide MOF selon l'invention chargé en NO sont, par exemple : les effets antirides, la reconstitution de la plénitude des lèvres et l'exaltation de leur couleur rouge naturelle, la stimulation de la couleur rosé naturelle de la peau et l'homogénéisation du teint, par une action vasodilatation et/ou augmentation de la circulation sanguine de la peau ; - la réduction des dommages de la peau causés par la lumière UV ; - l'effet anti-bactérien dans le traitement de l'acné.
Comme déjà indiqué, les solides MOF de l'invention ont l'avantage de présenter des capacités de charge inattendues, encore jamais atteintes dans les matériau de l'art antérieur.
Ils présentent, en outre, l'avantage de permettre des temps de relargage modulables grâce à leur structure. En effet, la nature rigide ou flexible des structures des solides MOFs de l'invention a une influence sur la cinétique de relargage de gaz et la teneur en gaz libéré. Les solides MOF avec une
structure flexible peuvent notamment permettre une libération avec une durée modulable. Ainsi, des solides MOF de structure flexible comprenant des ligands hydrophobes, vont permettre de moduler la durée de la libération du gaz.
L'utilisation des MOFs de structures flexibles tels que définis précédemment peut ainsi constituer une alternative crédible pour un relargage de gaz d'intérêt biologique comme le NO sur une plus longue durée. Chargés en un gaz (ou un mélange de gaz) approprié, ces solides, dont les premiers tests de toxicité montrent qu'ils ne sont a priori pas toxiques, peuvent également être d'un grand intérêt dans l'industrie alimentaire, notamment en tant qu'agent antibactérien, par exemple pour la conservation des aliments et des préparations industrielles. En particulier, lors de la mise sous vide de préparations alimentaires, l'introduction d'une faible quantité de solide MOF selon l'invention combinée à la présence d'eau naturellement présente dans les aliments peut conduire à un relargage lent et continu de NO permettant d'inhiber la croissance bactérienne et de détruire les microorganismes .
Un autre objet de l'invention est un composition pharmaceutique , cosmétique ou- dermatologique comprenant un solide MOF selon 1 ' invention et un véhicule pharmaceutiquement ou cosmétiquement acceptable. De plus, les solides MOF selon l'invention ont fait l'objet d'études de toxicité très positives,
décrites dans la partie « Exemples » . Ils semblent également biodégradables et les études de dégradabilité sont encore en cours .
Ainsi, les solides MOF de la présente invention utilisés pour le relargage de gaz à intérêt biologique permettent de pallier les problèmes liés à la toxicité dans l'art antérieur cités précédemment.
En effet, l'utilisation de MOFs à base de métaux de basse toxicité permettent d'envisager des applications en conditions biologiques.
Dans un mode de réalisation particulier de l'invention, les solides MOF peuvent être à la fois chargés en gaz et en principe pharmaceutiquement actif. Le principe pharmaceutiquement actif peut être contenu dans les pores. Cela permet d'obtenir un effet thérapeutique combiné.
En particulier, les solides MOF selon l'invention peut être chargée en principe pharmaceutiquement actif avec une capacité de charge de l à 200% en poids de solide sec, par exemple de 1 à 70% en poids de solide sec, soit près de 10 à 700 mg par gramme de solide sec.
Ainsi, l'invention s'étend à l'utilisation de tels solides comme médicament. Lorsque les solides MOF de l'invention sont chargés à la fois en gaz et en en principe pharmaceutiquement actif, leur procédé de préparation peut comprendre, en outre, une étape (v) d'introduction dans ledit solide, d'au moins une molécule d'intérêt, qui peut être un principe pharmaceutiquement actif.
Ladite étape d'introduction peut être réalisée au cours de l'étape réactionnelle (i) ou après celle-ci
de façon à obtenir un solide chargé en molécule d'intérêt.
Toute méthode connue de l'homme du métier peut être utilisée au cours de l'étape d'introduction (v). La molécule d'intérêt peut être par exemple introduite dans le matériau MOF de la présente invention :
• par imprégnation, en immergeant le matériau dans une solution de la molécule d'intérêt ;
• par sublimation de la molécule d'intérêt, puis le gaz est adsorbé par le matériau ; ou
Dans un autre mode de réalisation, les solides MOF de l'invention, chargés en un ou en un mélange de gaz, comprend un ligand L qui possède lui-même une activité thérapeutique. Un effet thérapeutique combiné peut alors être obtenu par le relargage du (ou des) gaz et l'activité de L après dissolution du matériau.
D'autres avantages pourront apparaître à l'homme du métier à la lecture des exemples ci-dessous, illustrés par les figures annexées, donnés à titre illustratifs .
Brève description des figures - La figure 1 représente un diffractogramme RX du solide MIL-IOO(Fe) . La figure 2 représente un isotherme d'adsorption d'azote à 77 K du solide MIL-100 (Po=1 atm) . La figure 3 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-IOO(Fe).
La figure 4 représente le diffractogramme RX du solide MIL-IOl(Fe)

La figure 5 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-IOl(Fe).
La figure 6 représente des diffractogrammes RX des solides MIL-88A brut (courbe du haut) et suspendu dans l'eau (courbe du bas).
- La figure 7 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88A(Fe) hydraté.
La figure 8 des diffractogrammes RX des solides MIL-88B sec (courbe (b) du bas) et hydraté (courbe (a) du haut ) . - La figure 9 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B hydraté.
La figure 10 représente des diffractogrammes RX des solides MIL-89 sec (courbe a), DMF (courbe b) et hydraté (courbe c).
La figure 11 représente un diffractogramme RX du solide MIL-88C.
- La figure 12 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88C brut de synthèse. - La figure 13 représente des diffractogrammes- RX des solides MIL-88D brut (courbe (b) du bas) et hydraté (courbe (a) du haut). - La figure 14 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88D(Fe) hydraté.
La figure 15 représente des diffractogrammes RX du solide MIL-88B-NO2 brut (courbe (a) en haut) et hydraté (courbe (b) en bas).
La figure 16 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B-NO2(Fe) après lavage et séchage.
- La figure 17 représente des diffractogrammes RX du solide MIL-88B-2OH brut (courbe (c) en bas), hydraté (courbe (b) du milieu) et séché sous vide (courbe (a) en haut ) .
La figure 18 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B-2OH(Fe) hydraté .
La figure 19 représente des diffractogrammes RX du solide MIL-88B-NH2 brut (courbe (b) en bas) et du solide MIL-88B-NH2 sec (courbe (a) en haut).
La figure 20 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B-NH2(Fe) hydraté .
La figure 21 représente des diffractogrammes RX des solides MIL-88B-CH3 brut (courbe (a) en haut), hydraté (courbe (b) du milieu) et solvaté avec le DMF (courbe (c) en bas). La figure 22 représente des diffractogrammes RX du solide MIL-88B-C1 brut (courbe (b) en bas) et hydraté (courbe (a) en haut). La figure 23 représente une analyse thermogravimétrique (sous air, avec une vitesse de
chauffe de 2°C/minute) du composé MIL-88B-Cl(Fe) hydraté .
La figure 24 représente des diffractogrammes RX du solide MIL-88B-4CH3 brut (courbe (b) en bas) et hydraté ( courbe ( a ) en haut ) .
La figure 25 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B-4CH3(Fe) hydraté . - La figure 26 représente des diffractogrammes RX des solides MIL-88B-4F brut (courbe (c) en bas), hydraté (courbe (b)) et solvaté avec EtOH (courbe (a) en haut) .
La figure 27 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88B-4F (Fe) hydraté .
- La figure 28 représente des diffractogrammes RX des solides MIL-88B-Br brut (courbe (b) en bas) et hydraté ( courbe ( a) en haut ) .
- La figure 29 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2ºC/minute) du composé MIL-88B-Br(Fe) hydraté . - La figure 30 représente un diffractogramme RX du solide MIL-88E(Fe). La figure 31 représente des diffractogrammes RX des solides MIL-88F brut (courbe (b) en bas) et hydraté ( courbe ( a ) en haut ) . - La figure 32 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88F (Fe) hydraté.
La figure 33 représente des diffractogrammes RX des solides MIL-88D- 4CH3 brut (courbe (b) en bas) et hydraté ( courbe ( a ) en haut ) .
- La figure 34 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88D-4CH3 (Fe) hydraté .
La figure 35 représente des diffractogrammes RX des solides MIL-88D-2CH3 brute (courbe (c) en bas), hydraté (courbe (b)) et mouillé (courbe (a) en haut).
La figure 36 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88D-2CH3 (Fe) brut de synthèse. - La figure 37 représente une diffractogramme RX du solide MIL-88G brut (courbe (c) en bas), solvaté avec le DMF (courbe (b) du milieu) et solvaté avec la pyridine (courbe (a) en haut).
La figure 38 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88G (Fe) brut de synthèse.
La figure 39 représente une diffractogramme RX du solide MIL-88G-2C1 brut (courbe (b) en bas) et sec (courbe (a) en haut).
La figure 40 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-88G-2C1 (Fe) brut de synthèse. - La figure 41 représente des diffractogrammes RX des solides MIL-102(Fe) brut (courbe (a)) et référence MIL-102(Cr) (courbe (b)).
La figure 42 représente une analyse theππogravimétrique (sous air) du composé MIL-102 (Fe) brut de synthèse.
La figure 43 représente un schéma réactionnel pour l'obtention de l'acide 3,5, 3 ',5'- tétraméthylbiphényl 4,4' -dicarboxylique .
La figure 44 représente un schéma de réaction pour l'obtention de l'acide 3 , 3 ' -dimethylbiphenyl 4,4'- dicarboxylique . - La figure 45 représente un cliché obtenu par MEB (Microscopie Electronique à Balayage) du solide MIL-89nano.
- La figure 46 représente un cliché obtenu par MEB (Microscopie Electronique à Balayage) du solide MIL-88Anano.
La figure 47 représente un cliché obtenu par MEB (Microscopie Electronique à Balayage) du solide MIL-lOOnano.
La figure 48 représente un cliché obtenu par MEB du solide MIL-88Btnano.
- La figure 49 représente un cliché obtenu par MEB du solide MIL-88Bnano.
- La figure 50 représente une quantité de sites Fer insaturés présents dans le MIL-100 Fe activé sous vide à différentes températures.
La figure 51 représente un isotherme d'adsorption de NO à 298 K pour le carboxylates de fer
MIL-IOO(Fe) activé à 120ºC pendant une nuit.
- La figure 52 représente un profil de relargage de NO (NOrel en mmol/g) sous pression de vapeur du solide MIL-IOO(Fe) en fonction du temps t (en heure).
La figure 53 représente (à gauche) : un isotherme d'adsorption de NO à 298 K pour le MIL- 100(Fe) activé à 250ºC sous vide pendant une nuit ; (à droite) : un profil de relargage de NO sous pression de vapeur du solide MIL-IOO(Fe) activé à 250ºC sous vide pendant une nuit.
La figure 54 représente une vue schématique du phénomène de respiration (gonflement et contraction) dans les solides MIL-88A, MIL-88B, MIL-88C, MIL-88D et MIL-89. L'amplitude de gonflement entre formes sèches (en haut) et formes ouvertes (en bas) est représentée en % en bas de la figure.
La figure 55 représente des isothermes d'adsorption de NO à 298 K pour les carboxylates de fer MIL-88A(Fe) et MIL-88B(Fe) activés à 150ºC pendant une nuit.
La figure 56 représente des profils de relargage de NO sous pression de vapeur d'eau des solides MIL-88A(Fe) (en haut) et MIL-88B(Fe) (en bas) activés à 150ºC pendant une nuit.
- La figure 57 représente, en haut, une étude de la réversibilité du gonflement du solide MIL-88A par diffraction des RX (λ~1.79Â), en bas, des diffractogrammes RX du solide MIL-88A en présence de solvants (λ-1.5406Â).
La figure 58 représente un schéma explicatif de 1-a—flexibilité—dans—les" phases"hybrides ~MIL-53~"(~a~).r et
MIL-88 (b et c) .
La figure 59 représente la structure cristallographique du MIL-126(Fe). Les polyèdres FeO6 sont représentés avec ou sans étoile, en indiquant les
deux réseaux MIL-88D. Les atomes de carbone sont en noir.
La figure 60 représente une diffractograirane des rayons X du solide MIL-126(Fe)
 - La figure 61 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-126(Fe).
- La figure 61 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé MIL-126(Fe).
- La figure 62 représente des isothermes d'adsorption d'azote pour le MIL-126(Fe)
 atmosphère) .
atmosphère) .
La figure 63 représente une diffractogramme des rayons X du 3, 3' ,5, 5'-azobenzènetétracarboxylate de fer

- La figure 64 représente le diffractogramme RX du solide 2 , 5-dihydroxotéréphthalate de fer brut. La phase, de symétrie trigonal, est un isotype de celle publiée par Dietzel et al. au cobalt et nickel (groupe de espace R3) [61] .
La figure 65 représente une analyse thermogravimétrique (sous air, avec une vitesse de chauffe de 2°C/minute) du composé 3,3 ',5,5'- azobenzènetetracarboxylate de fer.
La figure 66 des isothermes d'adsorption d'azote pour le 3,3' ,5,5 '-azobenzènetetracarboxylate de fer (P0=I atmosphère).
La figure 67 représente les diagrammes de DRX du matériau MIL-88A non modifié- avant
 et après l'addition d'une goutte d'eau (MIL88A + H2O) ; MIL-88A modifié avec le chitosane 7% avant (MIL88AQ100) et après l'addition d'une goutte d'eau (MIL88A Q100 +
et après l'addition d'une goutte d'eau (MIL88A + H2O) ; MIL-88A modifié avec le chitosane 7% avant (MIL88AQ100) et après l'addition d'une goutte d'eau (MIL88A Q100 +
H2O) ; MIL-88A modifié avec le chitosane 2% avant
(MIL88AQ25) et après l'adition d'une goutte d'eau (MIL88A Q125 + H2O) .
La figure 68 représente l'analyse thermogravimétrique du matériau MIL-88A non modifié (MIL88A ; vert), modifié avec le chitosane 2% (MIL-88A-
Q25, noir) et modifié avec le chitosane 7% (MIL-88A-
QlOO, rouge).
La figure 69 représente les images de microscopie confocale du matériau MIL-IOO(Fe) modifié superficiellement avec du Dextrane-Fluoresceine- Biotine.
La figure 70 représente l'évolution de la taille des particules (P en nm) de MIL-88A à surface modifiée par le polyéthylène glycol, en fonction du temps (t en min).
- La figure 71 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe (a)) et dans le tampon phosphate (courbe (b)) de solide MIL-88A(Fe). La quantité de NO relarguée (N0rel en mmol. g"1, à gauche, et ppm NO, à droite) est exprimée en fonction du temps t (en heures).
La figure 72 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe
(a)) et dans le tampon phosphate (courbe (b) ) de solide MIL-88B(Fe). La quantité de NO relargué (NOrel en mmol. g"1, a gauche, et ppm NO, a droite) est exprimée en fonction du temps t ( en heures).
La figure 73 représente le diffractogramme des rayons X du solide MIL-88A-nano obtenu par synthèse microondes.
La figure 74 représente les isothermes d'adsorption de NO à 298 K pour les carboxylates de fer
MIL-88A(Fe)-nano activé à 150ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol.g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg ) . La figure 75 représente les profils de relargage de NO sous pression de vapeur d'eau des solides MIL-88A(Fe) (5 microns, courbe (a)) et MIL- 88A(Fe)-nano (120 nm, courbe (b) ) . La quantité de NO relargué (N0rel en mmol.g"1, à gauche, et ppm NO, à droite) est exprimée en fonction du temps t (en heures) .
La figure 76 représente les isothermes d'adsorption de NO à 298 K pour les carboxylates de fer MIL-88B(Fe)-NO2 activé à 150ºC sous vide pendant une nuit. La quantité de NO (N0abg en mmol.g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg) .
- La figure 77 représente les isothermes d'adsorption de NO à 298 K pour les carboxylates de fer MIL-88B(Fe) -20H activé à 80ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol.g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg).
- La figure 78 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe
(a)) et dans le tampon phosphate (courbe (b)) de solide MIL-88B-(Fe)-NΘ2. La quantité de NO relargué - (N0rel en mmol.g-1, à gauche, et en ppm NO, a droite) est exprimée en fonction du temps t (en heures). - La figure 79 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe (a)) et dans le tampon phosphate (courbe (b)) de solide
MIL-88B(Fe)-2OH. La quantité de NO relargué (NOrel en mmol.g-1, à gauche, et en ppm NO, à droite) est exprimée en fonction du temps t (en heures).
- La figure 80 représente les profils de relargage de NO sous pression de vapeur d'eau des solides MIL-IOOFe (courbe (a)), MIL-88A (courbe (b))f MIL-88B (courbe (C)), MIL-88-2OH (courbe (d)) et MIL- 88B-NO2 (courbe (e)). La quantité de NO relargué (NOrel en mmol.g-1) est exprimée en fonction du temps t (en heures) .
La figure 81 représente les isothermes d'adsorption de NO à 298 K pour le solide MIL-22 activé à 350ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol.,O1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mm Hg) .
- La figure 82 représente les profils de relargage de NO par solide MIL-22 sous pression de vapeur d'eau. La quantité de NO relarguée (NOrel en mmol.g"1 à gauche et en ppm NO à droite) est exprimée en fonction du temps t (en heures).
La figure 83 représente l'isotherme d'adsorption de CO (C0adg en mmol/g) pour le solide MIL-
100(Fe) à la température de 303 K en fonction de la pression P (en bar) pour le carboxylate de fer MIL-
100(Fe) activé à 100ºC pendant une 12h (courbe 100ºC), 250ºC pendant 12h (courbe-205oC(1) et 250°C pendant 20h
(courbe 250°C(2) ) .
La figure 84 représente les coupes histologiques du foie sont observées par coloration de Proust (fer en bleu). Elles montrent une accumulation de fer dans le foie.
La figure 85 représente représente les isothermes d'adsorption de NO à 298 K pour les solides CPO-27(dihydroxotéréphthalate de Co) (courbes (a) et (b)), CPO-27(2,5-dihydroxotéréphthalate de Ni ; M2 (dhtp) (H2O) .XH2O (M = Ni or Co dhtp = acide 2,5- dihydroxytéréphthalique, x-8)) (courbes (c) et (d) ) , MIL-100 (trimésate de Fe) (courbes (e) et (f)), HKUST(trimésate de Cu) (courbes (g) et (h)), MIL- 53(téréphthalate de Al) (courbes (i) et (J)) et MIL- 53(téréphthalate de Cr) (courbes (k) et (I)).
La figure 86 représente les profils de relargage de NO sous pression de vapeur d'eau du solide CPO-27(dihydroxotéréphthalate de Co) (courbe (a)), CPO- 27(2,5-dihydroxotéréphthalate de Ni ; M2 ( dhtp ) ( H2O ) . xH20 (M = Ni or Co dhtp = acide 2 ,5-dihydroxytéréphthalique acid, x-8)) (courbe (b)), HKUST-I (trimésate de Cu) (courbe (c)), MIL-53(téréphthalate de Al) (courbe (d)) et MIL-53(téréphthalate de Cr) (courbe (e)). La quantité de NO relarguée (NOrel en mmol.g-1) est exprimée en fonction du temps t (en heures).
La figure 87 représente les profils de relargage de NO sous pression de vapeur d'eau du solide MIL-88A (3 échantillons sous les mêmes conditions) sous forme de crème. La quantité de NO relargué (NOrel en mmol.g-1) est exprimée en fonction du temps t (en heures ) . La figure 88 représente les profils de relargage de NO sous pression de vapeur d'eau du solide MIL-88A-nano sous forme de crème (courbe (b)) et sous forme de poudre (courbe (a)) en comparaison avec la libération dans une solution PBS (courbe (c)). La
quantité de NO relarguée (NOrel en mmol.g -1) est exprimée en fonction du temps t (en heures).
La figure 89 représente une diffractogramme des rayons X de nicotinate de fer 2 : Groupe de space P 21/n : a = 16.422899, b = 21.423401, c = 11.048300 beta = 91.806999.
EXEMPLES
Exemple 1 : Synthèse et données sur les carboxylates de fer de la présente invention
Cet exemple décrit la synthèse de divers carboxylates de fer. Les solides obtenus ont ensuite été caractérisés selon les méthodes décrites ci- dessous.
L'analyse de la structure cristalline des solides carboxylates de fer a été effectuée par diffraction des rayons X (RX) à l'aide d'un diffractomètre Siemens D5000 (radiation
 Â, mode Θ-2Θ) , à température ambiante à l'air. Les diagrammes sont représentés soit en distances angulaires (2Θ, en degrés °) soit en distances inter- réticulaire (d, en  ou Angstrôm) . La caractérisation de la porosité (surface spécifique Langmuir et volume de pore) des solides a été mesurée par l'adsorption d'azote à 77 K avec un appareil Micromeretics ASAP-2010. Les solides ont été préalablement déshydratés à 150ºC sous vide primaire pendant une nuit. L'isotherme d'adsorption d'azote par les solides est donnée par une courbe représentant le volume d'azote adsorbé V (en cmVg) en fonction du rapport de la pression P sur la pression de référence P0=I atm. L'analyse thermogravimétrique a été effectuée sous atmosphère d'air à l'aide d'un appareil Modèle TA- instrument 2050 . La vitesse de chauffe était de
2°C/minute. La courbe résultant de l'analyse thermogravimétrique des solides représente la perte de masse Pm (en %) en fonction de la température T (en ºC) . L'analyse élémentaire des solides a été effectuée par le Service Central d'Analyse du CNRS de
Â, mode Θ-2Θ) , à température ambiante à l'air. Les diagrammes sont représentés soit en distances angulaires (2Θ, en degrés °) soit en distances inter- réticulaire (d, en  ou Angstrôm) . La caractérisation de la porosité (surface spécifique Langmuir et volume de pore) des solides a été mesurée par l'adsorption d'azote à 77 K avec un appareil Micromeretics ASAP-2010. Les solides ont été préalablement déshydratés à 150ºC sous vide primaire pendant une nuit. L'isotherme d'adsorption d'azote par les solides est donnée par une courbe représentant le volume d'azote adsorbé V (en cmVg) en fonction du rapport de la pression P sur la pression de référence P0=I atm. L'analyse thermogravimétrique a été effectuée sous atmosphère d'air à l'aide d'un appareil Modèle TA- instrument 2050 . La vitesse de chauffe était de
2°C/minute. La courbe résultant de l'analyse thermogravimétrique des solides représente la perte de masse Pm (en %) en fonction de la température T (en ºC) . L'analyse élémentaire des solides a été effectuée par le Service Central d'Analyse du CNRS de
Vernaison :
Analyse organique :
Microanalyses C,H,N,O,S dans les produits pharmaceutiques, les polymères et d'une façon générale les produits de synthèse, par détections coulométrique, catharométrique ou cellule infrarouge.
Analyse inorganique :
Les principales techniques misent en oeuvre : - ICP-AES (« Inductive Coupled Plasma - Atomic Emission
Spectroscopy » ou « Spectrométrie d'émission atomique par plasma à couplage inductif » ) avec différents types de détecteurs
- ICP-MS ( « Inductively Coupled Plasma-Mass Spectrometry » ou « Spectrométrie de masse par Plasma à couplage inductif » ) avec des spectromètres de masse quadripolaires ou à secteur magnétique
- CVAAS ( « Cold-Vapor Atomic Absorption Spectroscopy » ou « Spectroscopie d'absorption atomique à vapeur froide »)
- Couplage ICP / MS / HPLC ( « Inductively Coupled Plasma / Mass Spectrometry / High Performance Liquid
Chromatography » ou « Chromatographie liquide Haute Performance / Spectrométrie de Masse par Plasma à Couplage Inductif ») - Fluorescence X
Traitements d'échantillons par voie humide, par voie sèche ou micro-ondes .

Le carboxylate de fer MIL-IOO(Fe) a été synthétisé selon deux conditions: avec et sans acide fluorhydrique .
Conditions de synthèse avec de l'acide fluorhydrique :
56 mg de fer métallique en poudre (1 mmol, commercialisé par la société Riedel de Haen, 99%), 140 mg d'acide 1,3,5-benzenetricarboxylique (0,6 mmol, 1,3,5-BTC ; commercialisé par la société Aldrich, 99%) sont dispersés dans 5 mL d'eau distillée avec 0,6 mL d'acide nitrique 2M (commercialisé par la société VWR, 50%) et 0,4 mL d'acide fluorhydrique 5M (commercialisé par la société SDS, 50%). Le tout est placé dans un corps en Téflon de 23 ml mis dans une bombe métallique de la société PAAR et laissé pendant 6 jours à 150ºC avec un palier de monté en température de 12 heures et un palier de descente en température de 24 heures. Le solide est récupéré par filtration. Puis, le solide (200 mg) est suspendu dans 100 mL d'eau distillée à reflux sous agitation pendant 3h pour éliminer l'acide trimésique restant dans les pores. Le solide est ensuite récupéré par filtration à chaud.
Conditions de synthèse sans acide fluorhydrique
0,27 g de FeCl3.6H2O (1 mmol , commercialisé par la société Alfa Aesar, 98%), 140 mg (0,6 mmol) d'acide 1,3,5-benzenetricarboxylique (1,3,5-BTC ; commercialisé par la société Aldrich, 99%) sont dispersés dans 5 mL de l'eau distillée. Le tout est laissé dans un corps en Téflon de 23 mL mis dans une Bombe métallique PAAR pendant 3 jours à 130ºC. Le solide est ensuite filtré et lavé avec de l ' acétone
Le solide (200 mg) est ensuite suspendu dans 100 mL d'eau distillée à reflux sous agitation pendant
3h pour éliminer l'acide trimésique restant dans les pores. Le solide est ensuite récupéré par filtration à chaud.
Données caractéristiques du solide carboxylate de fer MIL-IOO(Fe) :
L'analyse de la structure cristalline du solide MIL-IOO(Fe) par diffraction des rayons X donne le diffractogramme RX représenté en figure 1.
Les caractéristiques de la structure cristalline sont les suivantes :
- le groupe d'espace est Fd-3m (n°227).
- les paramètres de maille sont : a=73,l  ; volume de la maille V=393000

L'isotherme d'absorption d'azote à 77 K du solide MIL-IOO(Fe) (à la pression P0=1 atm) est donnée en figure 2. La surface (Langmuir) spécifique de ce solide est proche de 2900 m2. g-1.
La courbe résultant de l'analyse thermogravimétrique du composé MIL-IOO(Fe) est donné en figure 3. Ce diagramme représente la perte de masse Pm (en %) en fonction de la température T (en ºC).
Le tableau ci-dessous donne l'analyse élémentaire du solide MIL-IOO(Fe) ou Fe3O[C6H3- dans le cas où X=F.
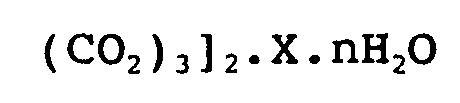


Synthèse du solide MIL-IOl ( Fe ) :
0,27 g (1 mmol) de FeCl3.6H2O, 249 mg (1,5 mmol) d'acide 1,4-benzenedicarboxylique (1,4-BDC, commercialisé par la société Aldrich, 99%) sont dispersés dans 10 mL de diméthylformamide (DMF, commercialisé par la société Fluka, 98%). Le mélange est laissé 12 heures à 100ºC dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR. Le solide est ensuite filtré et lavé avec de l'acétone.
Données caractéristiques du solide MIL-IOl(Fe) :
Le diffractogramme RX du solide MIL-IOl(Fe) est représenté dans la figure 4.
Les caractéristiques de la structure cristalline sont les suivantes :
- le groupe d'espace est Fd-3m (n°227).
- les paramètres de maille du solide MIL-IOl(Fe) à 298 K sont : a=89,0 Â ; volume de la maille V=707000 Â3.
La composition élémentaire théorique du solide sec (avec X=F) est la suivante : Fe 24,2 % ; C 41,4 % ; F 2,7 % ; H l,7 %.
Synthèse du solide MIL-88A(Fe) :
0,27 g (1 mmol) de FeCl3.6H2O (commercialisé par la société Alfa Aesar, 98%) et 116 mg (1 mmol) d'acide fumarique (Aldrich, 99%) sont dispersés dans 5 ml de dimethylformamide (DMF, Fluka, 98%) avec 0,4 mL NaOH 2M (Alfa Aesar, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 100ºC. Le solide est ensuite filtré et lavé avec de l'acétone.
Puis, le solide (200 mg) est mis en suspension dans 100 mL d'eau distillée sous agitation pendant 12h pour éliminer le solvant restant dans les pores. Le solide est ensuite récupéré par filtration.
Données caractéristiques du solide MIL-88A(Fe) :
L'analyse de la structure cristalline du solide donne les caractéristiques répertoriées dans le tableau suivant :
Tableau 2 : paramètres de maille du solide MIL-88A, sec et hydraté.

Le diffractogramme RX est donné en figure 6.
Les résultats de l'analyse thermogravimétrique du composé MIL-88A hydraté (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 7. La perte de masse Pm (en %) est représentée en fonction de la température T (en ºC).
Le composé MIL-88A ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
L'analyse élémentaire est donnée dans le tableau ci-dessous : Tableau 3 :


Synthèse du solide MIL-88B(Fe) :
0,27 g (1 πunol) de FeCl3.6H2O (Alfa Aesar, 98%) et 116 mg (1 mmol ) d'acide 1 , 4-benzenedicarboxylique (Aldrich, 98%) sont dispersés dans 5 inL de diméthylformamide (DMF, Fluka, 98%) avec 0,4 mL de soude 2M (Alfa Aesar, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 100ºC. Le solide est ensuite filtré et lavé avec de l'acétone.
200 mg du solide sont mis en suspension dans 10OmL d'eau distillée sous agitation pendant 12h pour éliminer le solvant restant dans les pores. Puis, le solide est récupéré par filtration.
Données caractéristiques du solide MIL-88B(Fe) :
L'analyse de la structure cristalline du solide donne les caractéristiques répertoriées dans le tableau suivant :
Tableau 4 : paramètres de maille du solide MIL-88B, sec et hydraté.

La figure 8 représente les_diffraçto.grammes RX du solide sec et du solide hydraté.
Les résultats de l'analyse thermogravimétrique du composé MIL-88B hydraté (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 9. La
perte de masse Pm (en %) est représentée en fonction de la température T (en ºC).
Le composé MIL-88B ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-89(Fe) :
172 mg (1 mmol) d'acétate de fer (préparé suivant la synthèse décrite par Dziobkowski et al., Inorg. Chem. 1982, 20, 671 [réf. 14]) et 150 mg (1 mmol) d'acide muconique (Fluka, 97%) sont dispersés dans 10 ml de méthanol (Fluka, 98%) avec 0,35 mL de soude 2M (Alfa Aesar, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 100ºC. Le solide est ensuite filtré et lavé avec de l'acétone.
200 mg du solide sont mis en suspension dans 10OmL d'eau distillée sous agitation pendant 12h pour éliminer le solvant restant dans les pores. Le solide est ensuite récupéré par filtration.
Données caractéristiques du solide MIL 89 (Fe) :
La figure 10L représente les diffractogrammes RX a), b) et c) respectivement du solide MIL-89(Fe) sec, du solide MIL-89(Fe) solvaté avec le DMF et du solide MIL-89(Fe) hydraté .
Le composé MIL-89(Fe) ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
 Synthèse du solide MIL-88C(Fe) :
Synthèse du solide MIL-88C(Fe) :
172 mg (1 mmol) d'acétate de fer (synthétisé selon l'exemple 2) et 140 mg (1 mmol) d'acide 2,6- naphtalenedicarboxylique (Aldrich, 95%) sont dispersés dans 5 ml de diméthylformamide (DMF, Fluka, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 150ºC avec un palier de monté de 12 heures et un palier de descente de 24 heures. Le solide est récupéré par filtration. Le solide est séché à 150ºC sous air pendant 15 heures.
Données caractéristiques du solide MIL-88C(Fe) :
L'analyse de la structure cristalline du solide donne les caractéristiques répertoriées dans le tableau suivant :
Tableau 5 : paramètres de maille du solide MIL-88C, sec et solvaté

Les résultats de l ' analyse thermogravimétrique du composé MIL-88C brut de synthèse (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 12.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
 Synthèse du solide MIL-88D(Fe) :
Synthèse du solide MIL-88D(Fe) :
270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 140 mg (0,6 mmol) d'acide 4,4'- biphenyldicarboxylique (Fluka, 95%) sont dispersés dans
5 ml de Diméthylformamide (DMF, Aldrich, 99%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 100ºC avec un palier de montée d'une heure et un palier de descente d'une heure. Le solide est récupéré par filtration.
Le solide est ensuite séché à 150ºC sous air pendant 15 heures.
Données caractéristiques du solide MIL-88D(Fe) :
L'analyse de la structure cristalline du solide donne les caractéristiques répertoriées dans le tableau suivant :
Tableau 6 : paramètres de maille du solide MIL-88D, sec et solvaté (pyridine).

La figure 13 représente le diffractogramme RX du solide MIL-88D brut (courbe (b) du bas) et hydraté ( courbe ( a ) du haut ) .
Les résultats de l'analyse thermogravimétrique du composé MIL-88D(Fe) hydraté (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 14 (perte de masse Pm en fonction de la température T).
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88B-NO2(Fe)
0,27 g (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 211 mg (1 mmol) d'acide 2-nitrotéréphthalique
(Acros, 99%) sont dispersés dans 5 ml d'eau distillée. Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 12 heures à 100ºC. Le solide est récupéré par filtration. 200 mg du solide sont mis en suspension dans 1OmL d'éthanol absolu dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 12 heures à 100ºC pour éliminer l'acide restant dans les pores. Le solide est ensuite récupéré par filtration et séché à 100ºC.
Données caractéristiques du solide MIL-88B-NO2 (Fe) :
La figure 15 représente le diffractogramme RX du solide MIL-88B-NO2 brut (courbe (a) du haut) et hydraté ( courbe ( b ) du bas ) .
Les résultats de l'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du composé MIL-88B-NO2 ( Fe ), après lavage et séchage, sont représentés en figure 16. La perte de masse Pm (en %) est représentée en fonction de la température T (en °C).
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
L'analyse élémentaire est donnée dans le tableau ci-dessous :
Tableau 7 :

5
 Synthèse du solide MIL-88B-20H(Fe) :
Synthèse du solide MIL-88B-20H(Fe) :
354 mg (1 mmol) de Fe(ClO4)3.xH2O (Aldrich, 99%)
10 et 198 mg (1 mmol) d'acide 2,5-dihydroxotéréphthalique (obtenu par hydrolyse de l'ester diéthylique correspondant, Aldrich 97%) sont dispersés dans 5 ml de DMF (Fluka, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR
15 pendant 12 heures à 85°C. Le solide est récupéré par filtration.
Pour éliminer l'acide restant dans les pores, le produit est calciné à 150ºC sous vide pendant 15 heures .
20
Données caractéristiques du solide MIL-88B-2OH(Fe) :
La figure 17 représente le diffractogramme RX du solide MIL-88B-2OH brut (courbe (c) en bas), hydraté25 (courbe (b) du milieu) et séché sous vide (côùrbë (a) en haut) .
Les résultats de l'analyse thermogravimétrique
(sous air, à une vitesse de chauffe de 2°C/minute) du
30 composé MIL-88B-2OH(Fe) , après lavage et séchage, sont
représentés en figure 18. La perte de masse Pm (en %) est représentée en fonction de la température T (en ºC) .
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
L'analyse élémentaire est donnée dans le tableau ci-dessous : Tableau 8 :


Synthèse du solide MIL-88B-NH2 (Fe) :
0.27 g (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 180 mg (1 mmol) d'acide 2-aminotéréphthalique (Fluka, 98%) sont dispersés dans 5 ml de l'éthanol absolu. Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 100oC. Le solide est récupéré par filtration.
Pour éliminer l'acide restant dans les pores, le solide est calciné à 200ºC pendant 2 jours.
Données caractéristiques du solide MIL-88B-NH2(Fe)
La figure 19 représente le diffractogramme RX du solide MIL-88B-NH2 brut (courbe (b) en bas), et séché sous vide (courbe (a) en haut).
Les résultats de l'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88B-NH2(Fe) hydraté sont représentés en figure 20.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l ' azote N2.

Synthèse du solide MIL-88B-CH3(Fe) :
354 mg (1 mmol) de Fe(ClO4)3.xH20 (Aldrich, 99%) et 180 mg (1 mmol) d'acide 2-méthyltéréphthalique (préparé suivant la synthèse décrite par Anzalone et al., J. Org. Chem. 1985, 50, 2128 [réf. 15]) sont dispersés dans 5 ml de méthanol (Fluka, 99%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 100ºC. Le-solide- est récupéré par filtration..
200 mg du solide sont mis en suspension dans 1OmL de DMF sous agitation à température ambiante pour échanger l'acide présent dans les pores par le DMF, puis le DMF est éliminé par chauffage à 150ºC sous vide pendant 12 heures.
Données caractéristiques du solide MIL-88B-CH3 (Fe) :
La figure 21 représente le diffractogramme RX du solide MIL-88B-CH3 brut (courbe (a) en haut), hydraté (courbe (b) du milieu) et solvaté avec le DMF (courbe (c) en bas).
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88B-Cl(Fe) :
354 mg (1 mmol) de Fe(ClO4)3.xH2O (Aldrich, 99%) et 200 mg (1 mmol) d'acide 2-chlorotéréρhthalique
(synthétisé selon la synthèse A de l'exemple 3) sont dispersés dans 10 ml de DMF avec 0,1 mL de HF 5M (SDS,
50%) et 0,1 mL de HCl IM (Aldrich, 37%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 5 jours à 100ºC. Le solide est récupéré par filtration. Le solide obtenu est calciné à 150ºC sous vide.
Données caractéristiques du solide MIL-88B-C1 (Fe) :
La figure 21 représente le diffractogramme RX du solide MIL-88B-C1 brut (courbe (a) en haut), hydraté
(courbe (b) du milieu) et solvaté avec le DMF (courbe (C) en bas) .
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88B- Cl(Fe) hydraté est représentée en figure 23.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

0,27 g (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%), 222 mg (1 mmol) d'acide 1,4-tétraméthyltéréphthalique (Chem Service, 95%) sont dispersés dans 10 ml de DMF (Fluka, 98%) avec 0,4 mL de soude 2M (Alfa Aesar, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 100ºC. Le solide est récupéré par filtration. 200 mg du solide sont mis en suspension dans
10OmL d'eau sous agitation à température ambiante pendant 12 heures pour éliminer l'acide restant dans les pores. Le solide est ensuite récupéré par filtration.
Données caractéristiques du solide MIL-88B-4CH3 (Fe)
La figure 24 représente le diffractogramme RX du solide brut (courbe (b) en bas) et du solide hydraté (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88B- 4CH3(Fe) hydraté est représentée en figure 25.
Ce composé présente une surface accessible de l'ordre de 1200 m2/g (Langmuir) à l'azote à 77 K, puisque la structure sèche possède une taille de pores suffisante (6-7 Â) pour incorporer de l'azote N2.

Synthèse du solide MIL-88B-4F (Fe) :
270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 230 mg (1 mmol) d'acide tétrafluorotéréphthalique (Aldrich, 98%) sont dispersés dans 10 . ml d'eau distillée. Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 85°C. Le solide est récupéré par filtration. 200 mg du solide sont mis en- suspension dans
20mL de l'eau sous agitation à température ambiante pendant 2 heures pour éliminer l'acide restant dans les pores. Le solide est ensuite récupéré par filtration.
Données caractéristiques du solide MIL-88B-4F (Fe) :
La figure 26 représente le diffractogramme RX du solide brut (courbe (c) en bas), du solide hydraté (courbe (b)) et du solide solvaté avec l'éthanol (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88B- 4F(Fe) hydraté est représentée en figure 27.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
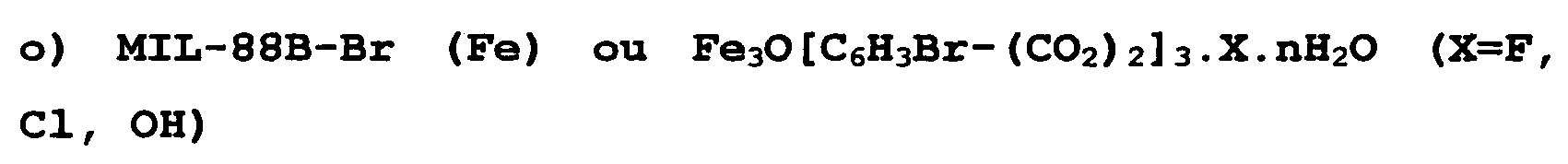
Synthèse du solide MIL-88B-Br (Fe) :
270 mg (1 mmol) de FeC13.6H2O (Alfa Aesar, 98%) et 250 mg (1 mmol) d'acide 2-bromotéréphthalique
(Fluka, 95%) sont dispersés dans 10 ml de DMF (Fluka,
98%) avec 0,2 mL d'acide fluorhydrique 5M (SDS, 50%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 150ºC . Le solide est récupéré par filtrtation.
Pour éliminer l'acide restant dans les pores, le solide est calciné à 150ºC sous vide pendant 15 heures.
Données caractéristiques du solide MIL-88B-Br (Fe) :
La figure 28 représente le diffractogramme RX du solide brut (courbe (b) en bas) et du solide hydraté (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88B- Br(Fe) hydraté est représentée en figure 29.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88E (Fe) :
270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 204 mg (1 mmol) d'acide 2, 5-pyrazinedicarboxylique (Aldrich, 98%) sont dispersés dans 5 ml de DMF (Fluka, 98%) avec 0,05 mL de HF 5M (SDS, 50%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 100ºC. Le solide est récupéré par filtration.
Données caractéristiques du solide MIL-88E (Fe) :
La figure 30 représente le diffractogramme RX du solide MIL-88E(Fe) brut de synthèse.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

354 mg (1 mmol) de Fe(ClO4)3.xH20 (Aldrich, 99%) et 258 mg (1 mmol) d'acide 2 , 5-tiophenedicarboxylique
(Aldrich, 99%) sont dispersés dans 2,5 ml de DMF
(Fluka, 98%) avec 0,1 mL de HF 5M (SDS, 50%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 100ºC.
Le solide est récupéré par filtration.
200 mg du solide sont mis en suspension dans
10OmL d'eau sous agitation à température ambiante pendant 12 heures pour éliminer l'acide restant dans les pores. Le solide est ensuite récupéré par filtration.
Données caractéristiques du solide MIL-88F(Fe) :
La figure 31 représente les diffractogrammes RX du solide brut (courbe (b) en bas) et du solide hydraté (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88F(Fe) hydraté est représentée en figure 32.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

354 mg (1 mmol) de Fe(ClO4)3.xH2O (Aldrich, 99%) et 298 mg (1 mmol) d'acide tétraméthylbiphényl-4,4 '- dicarboxylique (synthétisé selon la synthèse B décrite dans l'exemple 3) sont dispersés dans 5 ml de DMF (Fluka, 98%) avec 0,2 mL de soude 2M (Alfa Aesar, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 100ºC. Le solide est récupéré par filtration. 200 mg du solide sont mis en suspension dans 1OmL de DMF sous agitation à température ambiante pendant 2 heures pour échanger l'acide restant dans les pores. Le solide est ensuite récupéré par filtration, et le DMF présent dans les pores éliminé par calcination à 150ºC sous vide pendant 15 heures.
Données earaetéristiques du solide MIL-88D-4CH3( Fe) :
La figure 33 représente les diffractogrammes RX du solide brut (courbe (b) en bas) et du solide hydraté (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88D- 4CH3(Fe) hydraté est représentée en figure 34.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88D-2CH3(Fe) :
270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 268 mg (1 mmol) d'acide diméthylbiphén'yl - 4,4'- dicarboxylique (synthétisé selon la synthèse C décrite dans l'exemple 3) sont dispersés dans 5 ml de DMF (Fluka, 98%) avec 0,25 mL de HF 5M (SDS, 50%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 150ºC. Le solide est récupéré par filtration.
Pour éliminer l'acide restant dans les pores, le solide est calciné à 150ºC sous vide pendant 15 heures.
Données caraetéristiques du solide-MïL-.88D-2CH3 ( Fe ) :
La figure 35 représente les diffractogrammes RX du solide brut (courbe (c) en bas), du solide hydraté
MIL-88D-2CH3 (H2O) (courbe (b) du milieu) et du solide en
suspension dans l'eau MIL-88D-2CH3( goutte H2O) (courbe (a) en haut) .
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88D- 2CH3(Fe) brut est représentée en figure 36.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88G(Fe) :
118 mg (0,33 mmol) de Fe(C104)3.xH20 (Aldrich, 99%) et 90 mg (0,33 mmol) d'acide 4,4'- azobenzènedicarboxylique (synthétisé selon la méthode décrite par Ameerunisha et a1., J. Chem. Soc. Perkin Trans.2 1995, 1679 [réf. 16]) sont dispersés dans.15 ml de DMF (Fluka, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 15OºC. Le solide est récupéré par filtration. 200 mg du solide sont mis en suspension dans
10 mL de DMF sous agitation à température ambiante pendant 2 heures pour échanger l'acide restant dans les pores. Le solide est ensuite récupéré par filtration et le DMF restant dans les pores éliminé par calcination à 150ºC sous vide pendant 15 heures.
onnées caractéristiques du solide MIL-88G(Fe) :
La figure 37 représente les diffractograiranes RX du solide MIL-88G brut (courbe (c) en bas), du solide solvaté avec le DMF (courbe (b) du milieu) et du solide solvaté avec la pyridine (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88G(Fe) brut est représentée en figure 38.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.

Synthèse du solide MIL-88G-2Cl(Fe) :
177 mg (0,5 mmol) de Fe(C104)3.xH20 (Aldrich, 99%) et 169 mg (0,5 mmol) d'acide dichloro-4,4' - azobenzenedicarboxylique (synthétisé selon la synthèse D décrite dans l'exemple 3) sont dispersés dans 15 ml de DMF (Fiuka, 98%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 12 heures à 15OºC. Le solide est récupéré par filtration.
200 mg du solide sont mis en suspension dans 1OmL de DMF sous agitation à température ambiante
pendant 2 heures pour échanger l'acide restant dans les pores. Le solide est ensuite récupéré par filtration, et le DMF restant dans les pores est éliminé par calcination à 150ºC sous vide pendant 15 heures.
Données caractéristiques du solide MIL-88G-2Cl(Fe) :
La figure 39 représente les diffractogrammes RX du solide MIL-88G-2C1 brut (courbe (b) en bas) et du solide MIL-88G-2C1 sec (courbe (a) en haut).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/minute) du solide MIL-88G- 2Cl(Fe) brut est représentée en figure 40.
Ce composé ne présente pas de surface accessible (supérieure à 20 m2/g) à l'azote à 77 K, puisque la structure sèche possède une taille de pores trop faible pour incorporer de l'azote N2.
 Synthèse du solide MIL-102(Fe) :
Synthèse du solide MIL-102(Fe) :
270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 268 mg (1 mmol) d'acide 1,4,5,8- naphthaTenetétracarbσxyrique sont dispersés- dans- 5 ml d'eau distillée. Le mélange est laissé dans un corps en
Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 15 heures à 100ºC. Le solide est récupéré par filtration.
Données caractéristiques du solide MIL-102(Fe) :
La figure 41 représente les diffractograiranes RX du solide MIL-102(Fe) brut (courbe (a)) et du solide MIL-102(Cr) (courbe (b)).
L'analyse thermogravimétrique (sous air, à une vitesse de chauffe de 2°C/min) du solide MIL-102(Fe) brut est représentée en figure 42.
Ce composé présente une surface spécifique faible (surface de Langmuir : 101 m2/g) à l'azote à 77 K.

270 mg (1 mmol) de FeCl3.6H2O (Alfa Aesar, 98%) et 140 mg (0,6 mmol) d'acide 4,4'- biphenyldicarboxylique (Fluka, 95%) sont dispersés dans 5 ml de diméthylformamide (DMF, Aldrich, 99%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 12 heures à 150ºC avec un palier de montée de 1 heure et un palier de descende de 1 heure. Le solide est récupéré par filtration.
Le solide est ensuite séché à 15OºC sous vide primaire pendant 15 heures.
Données caractéristiques du solide MIL-126(Fe)
La structure cristallographique du solide MIL- 126(Fe) est une forme interpénétrée de la structure MIL-88D(Fe), c'est-à-dire qu'elle possède deux sous- réseaux cristallins de type MIL-88D enchevêtrés (figure 59).
L'analyse de la structure cristalline du solide donne les caractéristiques répertoriées dans le tableau suivant.
Tableau 9 : paramètres de maille du solide MIL-126, sec et solvaté (diméthylformamide) .

La figure 60 représente le diffractogramme RX du solide MIL-126 brut.
Les résultats de l'analyse thermogravimétrique du composé MIL-126(Fe) brut de synthèse (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 61 (perte de masse Pm en fonction de la température T) .
Ce composé présente une surface accessible importante ( Langmuir ) (supérieure à 2100 m2/g) à l'azote à 77 K (figure 62) .
y ) 3 , 3 ' , 5 , 5 ' -azobenzènetétracarboxylate de fer ou
 Synthèse du solide 3,3 ' ,5,5 ' -azobenzènetétracarboxylate de fer:
Synthèse du solide 3,3 ' ,5,5 ' -azobenzènetétracarboxylate de fer:
118 mg (0.3 πunol) de Fe(ClO4)3.nH2O (Aldrich, 98%) et 119 mg (0,6 iranol) d'acide 3, 3 ',5,5'- azobenzènetetracarboxylique (préparé selon le mode opératoire indiqué ci-dessous dans « synthèse I » ) sont dispersés dans 5 ml de diméthylformamide (DMF, Aldrich,
99%) avec l'addition de 0,1 mL d'acide fluorhydrique 5M (HF, SDS 50%). Le mélange est laissé dans un corps en
Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 150ºC avec un palier de montée de 1 heure. Le solide est récupéré par filtration.
Le solide est ensuite séché à 200ºC sous vide primaire pendant 15 heures.
Synthèse I: acide 3,3 ' ,5,5'-azobenzènetétracarboxylique
15g d'acide 2-nitroisophthalique (commercialisé par la société Aldrich, 98%) et 50g de soude sont placés dans 225 mL d'eau distillée, et chauffés à 50ºC sous agitation. 100g de glucose (Aldrich, 96%) dissout dans 150 mL d'eau sont ajoutés. Le mélange est agité 15 minutes, puis un bullage d'air est effectué pendant 3 heures, à température ambiante (20ºC ). Le sel de disodium est récupéré par filtration, lavé à l'éthanol, puis dissout à nouveau- dans 120 mL d'eau De l'acide chlorhydrique (commercialisé par la société Aldrich VWR, 37%) est ajouté jusqu'à obtenir un pH égal à 1. Le solide est récupéré par filtration et séché sous vide à 90ºC.
Données caractéristiques du solide 3,3 ',5,5'- azobenzènetétracarboxylate de fer :
La figure 63 représente le diffractograirane RX du solide 3,3 ' ,5,5'-azobenzènetétracarboxylate de fer brut. La phase, de symétrie cubique, est un isotype de celle publie par le groupe du Professeur Eddaoudi à l'indium (groupe de espace Pa3) [51]. Les résultats de l'analyse thermogravimétrique du composé 3,3' ,5,5'-azobenzènetétracarboxylate de fer brut de synthèse (sous air, à une vitesse de chauffe de 2°C/minute) sont représentés en figure 65 (perte de masse Pm en fonction de la température T). Ce composé présente une surface accessible importante (Langmuir) (supérieure à 1200 m2/g) à l'azote à 77 K (figure 66).
z ) 2, 5-dihydroxoterephthalate de fer ou Fe2 (2OC- C6H2(OH)2-CO2) (H2O) -XH2O
Synthèse du solide 2,5-dihydroxotéréphthalate de fer:
270 mg (1 mmol) de FeClO3.6H2O (Alfa Aesar, 98%) et 200 mg (1 mmol) d'acide 2,5-dihydroxotéréphthalique
(obtenu par hydrolyse de l'ester diéthylique rcorrespondant, Aldrich 97%) sont- dispersés dans 5—ml de diméthylformamide (DMF, Aldrich, 99%). Le mélange est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 150ºC avec un palier de montée de 12 heures et un palier de
refroidissement de 24 heures. Le solide est récupéré par filtration.
Le solide est ensuite séché à 150ºC sous vide primaire pendant 15 heures.
Données caractéristiques du solide 2,5- dihydroxoterephthalate de fer :
La figure 64 représente le diffractogramme RX du solide 2, 5-dihydroxotéréphthalate de fer brut. La phase, de symétrie trigonal, est un isotype de celle publiée par Dietzel et al. au cobalt et nickel (groupe de espace R3) [61].
Exemple 2 : Synthèse de l'acétate de fer(III)
L'acétate de fer (III), utilisé dans les exemples ci-dessous pour la synthèse des matériaux MOF selon l'invention, est synthétisé selon le protocole suivant. Cette synthèse peut se référer à la publication de Dziobkowski et al., Inorg. Chem. , 1982, 21, 671 [réf. 14].
6,72 g de poudre de fer métallique (Riedel-de Haën, 99%), 64 mL d'eau déionisée et 33,6 mL d'acide perchlorique à 70 % dans l'eau (Riedel-de Haën) sont mélangés sous agitation magnétique et chauffés à 50ºC pendant 3 heures. Après arrêt du chauffage, la solution est agitée pendant 12 heures. Le fer métallique résiduel est éliminé par décantation suivie d'un changement de récipient. 20,6 mL de solution de peroxyde d'hydrogène dans l'eau (commercialisé par la
société Alfa Aesar, 35%) sont ajoutés goutte à goutte sous agitation, le tout étant maintenu dans un bain de glace à 0ºC. On ajoute 19,7 g d'acétate de sodium (Aldrich, 99%) à la solution bleue sous agitation en maintenant la solution à 0-5ºC. On laisse la solution s'évaporer pendant 3 jours sous la hotte dans un cristallisoir (Volume=0,5 1) en verre. Finalement, les cristaux rouges d'acétate de fer sont récupérés par filtration et lavés très rapidement avec de l'eau déionisée glacée. Les cristaux sont ensuite séchés sous atmosphère d'air.
Exemple 3 : Synthèse des ligands
a) Synthèse A : synthèse de l'acide chlorotéréphtalique
6 g (0,043 mol) de chloroxylene (commercialisé par la société Aldrich, > 99%), 16 mL d'acide nitrique (commercialisé par la société VWR, 70%) et 60 mL d'eau distillée sont introduits dans un corps en téflon de 120 mL. Celui-ci est placé dans une bombe métallique PAAR, chauffé à 170ºC pendant 12h. Le produit est récupéré par filtration, puis lavé abondamment à l'eau distillée. Un rendement de 75% est obtenu.
RMN 1H (300 MHz, d6-DMSO) : δ (ppm) : 7,86 (d, J = 7,8 Hz), 7,93 (dd, J = 7,8; 1,2 Hz), 7,96 (d, J = 1,2 Hz)
b) Synthèse B : synthèse de l'acide 3,5,3' ,5' - tétraméthylbiphényl-4 , 4' -dicarboxylique
Le schéma réactionnel de cette synthèse est représenté en figure 43.
lèrβ étape : 10,2 g de tétraméthylbenzidine (98%, Alfa
Aesar) sont mis en suspension dans 39 mL d'acide chlorhydrique concentré (37%, commercialisé par la société Aldrich) à 0ºC. La diazotation a été effectuée par addition d'une solution du nitrite de sodium (6 g dans 50 mL d'eau). Après 15 min d'agitation à 0ºC, une solution d'iodure de potassium (70 g dans 200 mL d'eau) a été ajoutée lentement à la solution violette résultante. Une fois l'addition terminée, le mélange est agité pendant 2 heures à température ambiante. La suspension noire résultante est filtrée pour récupérer un précipité noir, lavé à l'eau. Le solide est mis en suspension dans le dichlorométhane (DCM, 98%, commercialisé par la société SDS) et une solution saturée de thiosulfate de sodium est ajoutée, causant la décoloration. Après 1 heure d'agitation, la phase organique est décantée et la phase aqueuse est extraite au DCM. La phase organique est séchée sur sulfate de sodium, puis évaporée pour donner l'intermédiaire diiodé sous forme d'un solide grisâtre. L'élution avec du pentane pur sur colonne de silice (commercialisé par la société SDS) permet d'obtenir le mélangé des composés- monoiodé- et- diiodév- Le-mélange -de-ces derniers" a été directement employé dans l'étape suivante. 2e étape : 7,2 g du composé iodé brut est solubilisé dans 100 mL de tétrahydrofurane (THF, distillé sur sodium) . Après refroidissement à -78°C, 35 mL de n-butyllithium
dans le cyclohexane (2,5 M, commercialisé par la société Aldrich) sont ajoutés. La solution est laissée revenir à température ambiante, une suspension blanche apparait après 2 heures. Elle est à nouveau refroidie à -78°C et 12 mL d'éthylchloroformate sont ajoutés. Le mélange est laissé à température ambiante, une solution jaune limpide est obtenu après 1 heure. Une partition entre l'eau et le dichlorométhane, suivie d'une extraction au dichlorométhane donne le diester brut. Celui-ci est purifié par chromatographie sur gel de silice, avec pour éluant un mélange Et2O/Pentane : 1/9, (rapport frontal : Rf = 0,3). 6,3 g de diester sont obtenus sous la forme d'un solide incolore (rendement de 42 % à partir de la benzidine). Caractérisation du diester obtenu : RMN 1H (300 MHz, CDCl3): δ (ppm) : 1,29 (t, J = 7,2 Hz, 6H), 2,29 (s, 13H); 4,31 (q, J = 7,2 Hz, 4H); 7,12 (s, 4H). RMN 13C (75 MHz, CDCl3): δ (ppm) : 14,3 (CH3), 19,9 (CH3), 61,0 (CH2), 126,5 (CH), 133,2 (Cq), 135,5 (Cq), 141,4 (Cq), 169,8 (Cq). 3e étape :
Finalement, le diester est saponifié avec 9,7 g d'hydroxyde de potassium (commercialisé par la société VWR) dans 100 mL d'éthanol 95% (commercialisé par la société SDS), au reflux pendant 5 jours. La solution est concentrée sous vide et le produit est solubilisé dans l'eauv De ï' acide chlorhydrique" concentré ~ est ajouté jusqu'à pH 1, et un précipité blanc est formé.
Il est récupéré par filtration, lavé à l'eau et séché. 5,3 g de diacide sont ainsi obtenus sous forme de solide blanc (rendement quantitatif).
c) Synthèse C : synthèse de l'acide 3,3'- diméthylbiphényl 4,4' -dicarboxylique
Le schéma réactionnel de cette synthèse est représenté en figure 44.
La même procédure que celle décrite pour la synthèse B a été utilisée en partant de 12,1 g de diméthylbenzidine. A l'issue de la 1ère étape, 18,4 g de 3,3' -diméthy1-4 , 4 ' -diiodo-biphényle sont obtenus
(rendement: 74%).
Caractérisation du composé diiodé obtenu : RMN 1H (300 MHz, CDCl3): δ (ppm) : 2,54 (s, 6H), 7,10 (dd, J = 2,2 et 8,1 Hz, 2H), 7,46 (d, J = 2,2 Hz, 2H), 7,90 (d, J = 8,1 Hz, 2H). RMN 13C (75 MHz, CDCl3): δ (ppm) : 28,3 (CH3), 100,3 (Cq), 126,0 (CH), 128,3 (CH), 139,4 (CH), 140,4 (Cq), 141,9 (Cq).
A l'issue des 2e et 3e étapes, 6,9 g d'acide 3 ,3 ' -dimethyl-biphenyl-4 , 4 ' -dicarboxylique sont obtenus à partir des 18,4 g de composé diiodé.
Caractérisation des composés obtenus : Le diester obtenu à l'issue de la 2e étape et le diacide obtenu à l'issue de la 3Θ étape ont des signatures spectroscopiques identiques à celles décrites dans la littérature (Shiotani Akinori, Z. Naturforsch. 1994, 49, 12, 1731-1736 [réf. 31]).
d) Synthèse D : synthèse de l'acide 3,3' -dichloro4, 4' - azobenzènedicarboxylique
15g d'acide o-chlorobenzoïque (commercialisé par la société Aldrich, 98%) et 50g de soude sont
placés dans 225 mL d'eau distillée, et chauffés à 50ºC sous agitation. 100g de glucose (Aldrich, 96%) dissout dans 150 mL d'eau sont ajoutés. Le mélange est agité 15 minutes, puis un bullage d'air est effectué pendant 3 heures, à température ambiante. Le sel de disodium est récupéré par filtration, lavé à l'éthanol, puis dissout à nouveau dans 120 mL d'eau. De l'acide chlorhydrique (commercialisé par la société Aldrich VWR, 37%) est ajouté jusqu'à obtenir un pH égal à 1. Le solide est récupéré par filtration et séché sous vide à 90ºC.
e) Synthèse E : acide 3,5,3 ' ,5 '-azobenzènetétra- carboxylique
19 g d'acide 5-nitroisophthalique (Aldrich, 98%) et 50 g de soude sont placés dans 250 mL d'eau distillée, et chauffés à 50ºC sous agitation. Une solution de 100g de glucose (Aldrich, 96%) dissous dans
150 mL d'eau est ajoutée. Le mélange est agité 15 minutes, puis un bullage d'air est effectué pendant 3 h à température ambiante. Le sel de disodium résultant est récupéré par filtration, dissout dans 300 mL d'eau à température ambiante. De l'acide chlorhydrique (VWR,
37%) est ajouté jusqu'à obtenir un pH égal à 1. Le solide est récupéré par filtration et séché sous vide à 90ºC.
Exemple 4 : Synthèse dé nanoparticules dé . MOF selon l ' invention
a) MIL-89nano
La synthèse du MIL-89nano est effectuée à partir de l'acétate de fer (1 mmol ; synthétisé selon la synthèse décrite dans l'exemple 2) et l'acide muconique (1 mmol; Fluka, 97%) en 5 mL d'éthanol (Riedel-de Haën, 99,8%) avec l'adition de 0,25 mL de hydroxyde de sodium 2M (Alfa Aesar, 98%) dans un autoclave (Paar bomb) à 100ºC pendant 12h. Après refroidissement du container, le produit est récupéré par centrifugation à 5000 rpm (rotation par minute) pendant 10 minutes. 200 mg du solide sont mis en suspension dans
100 mL d'eau distillée sous agitation pendant 15h pour éliminer le solvant restant dans les pores. Puis, le solide est récupéré par centrifugation à 5000 rpm pendant 10 minutes. La taille de particule mesurée par diffusion de lumière est de 400 nm (nanomètres) .
La figure 45 représente un cliché obtenu par microscopie électronique à balayage (MEB) du solide MIL-89nano. Les nanoparticules montrent une morphologie arrondie et légèrement allongée, avec une taille de particules très homogène de 50-100 nm (Figure 45).
b) MIL-88Anano
Pour l'obtention du matériau MIL-88Anano, du FeCl3.6H2O (1 mmol; AIfa Aesar, 98%) et de l'acide fumarique (1 mmol; Acros, 99%) sont dispersés dans 15 mL d'éthanol (Riedel-de Haën, 99,8%). Puis, à cette solution est ajouté 1 mL d'acide acétique (Aldrich, 99.7%). La solution est placée dans un flacon en verre et chauffée à 65ºC pendant 2 heures. Le solide est
récupéré par centrifugation à 5000rpm pendant 10 minutes .
200 mg du solide sont mis en suspension dans
10OmL d'eau distillée sous agitation pendant 15h pour éliminer le solvant res ant dans les pores. Puis, le solide est récupéré par centrifugation à 5000 rpm pendant 10 minutes.
La taille de particule mesurée par diffusion de lumière est de 250 nm. La microscopie électronique à balayage (MEB) du solide MIL-88Anano est représentée en figure 46.
Les images de MEB (Figure 46) montrent des particules allongées avec des arêtes . Il existe deux tailles de particules : environ 500 nm et 150 nm.
c) MIL-lOOnano
La synthèse du MIL-lOOnano est effectuée en mélangeant du FeCl3.6H2O (1 mmol; Alfa Aesar, 98%) et de l'acide 1, 3,5-benzenetricarboxylique (1,3 ,5-BTC ; 1 mmol; Aldrich, 95%) dans 3 mL d'eau distillée. Le mélange est placé dans une bombe PAAR à 100ºC pendant 12h. Le produit est récupéré par centrifugation à 5000rpm (10 minutes). 200 mg du solide sont mis en suspension dans 100 mL d'eau distillée sous agitation et reflux pendant 3h pour éliminer l'acide—restant dans les pores . Puis, le solide est récupéré par centrifugation à 5000rpm (10 minutes). La taille de particule mesurée par diffusion de lumière est 536 nm.
La microscopie électronique à balayage (MEB) du solide MIL-lOOnano est représentée en figure 47.
On peut observer sur les images de MEB une grande agrégation particulaire (Figure 47). Les nanoparticules sont plutôt sphériques, mais la taille reste difficile à déterminer compte tenu de la grande agrégation. On peut estimer une taille de 40-600 nm.
d) MIL-lOlnano
Pour l'obtention du solide MIL-lOlnano une solution de FeCl3.6H2O (1 mmol; Alfa Aesar, 98%) et d'acide 1,4-benzenedicarboxylique (1,5 mmol; 1,4-BDC
Aldrich, 98%) dans 10 mL de diméthylformamide (Fluka,
98%) est placée dans une bombe Paar et chauffé à 100ºC pendant 15 heures. Le solide est récupéré par centrifugation à 5000rpm (10 minutes).
Pour éliminer l'acide qui reste dans les pores, le produit est chauffé à 200ºC sous vide pendant 1 jour. Le produit est conservé sous vide ou atmosphère inerte compte tenu de sa faible stabilité à l'air. La taille de particule est mesurée par diffusion de lumière est 310 nm.
e) MIL-88Btnano
Le solide MIL-88Btnano est synthétisé à partir d'une solution de FeCl3.6H2O (1 mmol; Alfa Aesar, 98%) et d'aeide 1,4-benzenetetramethyldicarboxylique ( 1 mmol; Chem Service) dans 10 mL de diméthylformamide (Fluka, 98%) avec 0,4 mL de NaOH 2M. Cette solution est placée dans une bombe Paar et chauffée à 100ºC pendant 2 heures. Puis, le container est refroidi avec de l'eau
froide, le produit est récupéré par centrifugation à 5000rpm (10 minutes).
200 mg du solide sont mis suspension dans 10OmL d'eau distillée sous agitation pendant 15h pour éliminer le solvant restant dans les pores. Le solide est ensuite récupéré par centrifugation à 5000rpm (10 minutes ) .
La mesure de la taille de particule par diffusion de lumière montre deux populations de nanoparticules de 50 et 140 nm.
Les nanoparticules du solide MIL-88Btnano ont une morphologie sphérique avec une taille d'environ
50 nm. Seulement une fraction minoritaire possède une taille d'environ 200 nm. On peut y observer aussi des agglomérats de petites particules.
La microscopie électronique à balayage (MEB) du solide MIL-88Btnano est représentée en figure 48.
f) MIL-88Bnano
Le solide MIL-88Bnano est synthétisé à partir d'une solution d'acétate de fer (1 mmol; synthétisé selon la synthèse décrite dans l'exemple 2) et d'acide 1,4-benzenedicarboxylique (1 mmol; 1,4-BDC Aldrich, 98%) dans 5 mL de méthanol (Aldrich, 99%). Cette solution est placée dans une bombe Paar et chauffé à 100°C pendant 2 heures. Le container est- ensuite- refroidit avec de l'eau froide, le produit est récupéré par centrifugation à 5000rpm (10 minutes). 200 mg du solide sont mis en suspension dans 100 mL d'eau distillée sous agitation à reflux pendant 15h pour échanger le solvant qui reste dans les pores.
Puis, le solide est récupéré par centrifugation à 5000rpm (10 minutes).
La mesure de la taille de particule par diffusion de lumière montre une distribution bimodale de nanoparticules de 156 et 498 nm.
La microscopie électronique à balayage (MEB) du solide MIL-88Bnano est représentée en figure 49.
La morphologie des particules est très irrégulière, avec une taille de 300 nm. La détermination de la taille de particule par diffusion de lumière a été effectué sur un appareil Malvern Zetasizer Nano série - Nano-ZS (modèle Zen 3600 ; sériai N° 500180 ; UK).
La microscopie électronique à balayage a été réalisé en utilisant un microscope Topcon (Akashi) EM 002B ultra-haute résolution 20OkV.
Les différences entre les valeurs apportées par les deux techniques sont dues d'une part à la coloration orange des particules de carboxylates de fer, le faisceau laser de l'appareil de diffusion de lumière étant rouge, et d'autre part à des phénomènes d'agrégation particulaire.
Exemple 5 : Synthèse de carboxylates de fer (III) à surface modifiée par le chitosane
La modification de surface des nanoparticules avec le chitosane permet d'envisager différentes voies d'administration des nanoparticules grâce aux propriétés de bio-adhésion spécifiques à ce polymère.
Dans cet exemple la modification superficielle est réalisée pendant la synthèse du matériau MIL-88A.
a) Préparation des nanoparticules à surface modifiée
A une solution de FeCl3.6H2O (1 mmol, 270 mg; Alfa Aesar, 98%) et d'acide fumarique (1 iranol, 116 mg;
Acros , 99%) dans 5 mL d'eau distillée, dans un bombe en téflon de 23 mL, ont été ajoutés 7 mg de l'agent modifiant de surface, le chitosane modifié. Deux types de chitosane modifiés avec des chaînes alkyle (C12, lauryle) ont été utilisés ; l'un avec un modification de 2% de chaînes alkyles (Q25) et l'autre modifie avec
7% (QlOO).
Pour la dissolution complète du chitosane, la solution est placée sous agitation pendant 45 minutes. La bombe en téflon est placée dans un corps métallique fermé hermétiquement et chauffé dans un étuve à 80QC pendant 12 heures.
Le solide obtenu est récupéré par centrifugation à 5000 rpm pendant 10 minutes et lavé avec de l'eau distillée et l'acétone.
b) Analyse et caractérisation
La taille de particules obtenues est mesurée avec un appareil potentiel Z Malvern Zetasizer Nano série - Nano-ZS; model Zen 3600 ; sériai N° 500180 ;
UK, en observant une taille de 2,64 et 0,91 microns pour MIL88A-Q 25 et MIL88A-Q 100, respectivement.
Les diagrammes de diffraction de rayon X (DRX) sont collectés avec un diffractomètre Siemens D5000 X'Pert MDP
 de 3 à 20º (2θ) avec un taille de pas de 0,04º et 2 s par pas.
Les diagrammes de DRX présentés en figure 67 ont permit de vérifier que la phase obtenue est bien le
de 3 à 20º (2θ) avec un taille de pas de 0,04º et 2 s par pas.
Les diagrammes de DRX présentés en figure 67 ont permit de vérifier que la phase obtenue est bien le
MIL-88A. La flexibilité du matériau est également vérifiée par DRX en ajoutant une goutte d'eau sur le solide.
La quantité de chitosane incorporée au matériau est estimée par analyse thermogravimétrique (ATG) présentée en figure 68. L'appareil utilisé est un appareil TGA2050 TA de 25 à 500ºC avec un rampe de chauffage de 2°C/min sous flux d'oxygène ( lOOmL/min) . Dans les matériaux, la quantité d'acide fumarique est bien autour du 45% (par rapport au produit déshydraté). Les matériaux MIL88A-Q25 et MIL-88A-Q100 contiennent une quantité de chitosane de l'ordre de 16% et de 22% (poids) par rapport au produit déshydraté, respectivement .
Exemple 6 : Synthèse de carboxylates de fer (III) à surface modifiée par le dextrane fluoresceine biotin
Dans cet exemple, le dextrane utilisé est greffé d'une part avec la fluoresceine et d'autre part avec de la biotine (Dex B FITC 10000 g/mol, anionique, lysine fixable, Molecular Probes, catalog D7178). Les caractéristiques du dextrane sont les suivantes : Dextrane fluorescein and biotin, poids moléculaire 10 000 g/mole, anionique, capable de fixer la lysine (« mini-emerald ») lot 3603IA, D7178, « Molecular probes » , Technologie de détection in vitro, 1 mole fluoresceine/mole, 2 moles biotine/mole.
a) Préparation des nanoparticules à surface modifiée
Les particules de 1,3,5-benzenetricarboxylate de fer MIL-100 (diamètre de particule 1,79 microns) ont été lavées avec de l'eau MiIIiQ.
Cinq milligrammes de particules ont été dispersées dans 0,5 mL eau MiIIiQ. Nous avons rajouté à cette suspension 0,5 mL de solution aqueuse de Dex B FITC (5 mg/mL). Elles ont été incubées a température ambiante pendant 24 h, puis récupérées par centrifugation (3800 rpm, 10 minutes). Le surnageant a été prélevé, puis le culot (particules) a été resuspendu dans 0,5 ml d'eau MiIIiQ. Après une nouvelle centrifugation, les particules ainsi lavées du Dex B FITC en excès ont été placées sur une lamelle afin d'être observées dans le microscope confocal (excitation 488 nm, émission 515 nm) .
b) Analyse et caractérisation La fluorescéine permet la détection des particules à l'aide d'un microscope confocal à balayage laser, alors que la biotine, hydrophobe, permet :
1. l'ancrage dans le cœur des particules
2. la fonctionnalisation avec des ligands biotinylés. La figure 69 présente les coupes optiques ainsi obtenues. On distingue une auréole autour des particules, indiquant la présence de_ dextrane - (seul composé fluorescent) uniquement à la surface. En effet, les longues chaînes de polymère n'ont pas pu pénétrer au cœur des particules.
Cette méthode de modification de surface présente l'avantage de ne pas perturber le cœur des
particules (contenant des principes actifs) et de se faire post-synthèse, donc d'offrir une variété de recouvrements possibles.
Exemple 7: Synthèse de carboxylates de fer (III) à surface modifiée par le polyéthylène glycol (PEG)
Pour réduire au minimum la toxicité du Busulfan dans le foie, il faut empêcher les nanoparticules d'être dirigées vers le foie ; la meilleure méthode consiste à greffer en surface des nanoparticules hybrides des chaînes hydrophiles de type polyéthylène glycol (PEG), afin de diminuer leur accumulation dans cet organe. Nous envisageons une étude complète de la dégradation in vitro des particules recouvertes ou non de PEG, dans des milieux différents.
Les chaînes du PEG pourront avoir différent groupes terminaux pour se greffer à la surface des matériaux. Ainsi, l'interaction du PEG avec la surface de particule peut être modifié en utilisant différents types de PEG.
PEG-NH2 ( alpha-t-butyloxycarbonylamino-omega-amino poly(ethylenglycol) (PEG ; Boc-NH-PEG-NH2, 5000 MW, Iris Biotech) - PEG-COOH (ρoly(ethylenglycol) carboxylic acid, Iris Biotech)
PEG-PO4 synthétisé au laboratoire selon le procédé suivant :
 Le groupe phosphonate est attaché au PEG-NH2 par une condensation amide avec un ester lié à un groupe phosphonate. Le sel de sodium du phosphonate a été utilisé. Ensuite, le couplage a été réalisé a partir du triméthyl-phosphonoformate [CAS 31142-23-1] selon la procédure décrite par Robert A. Moss, Hugo Morales- Rojas, Saketh Vijayaraghavan and Jingzhi Tian, J. Am. Chem. Soc, 2004, 126,(35), 10923 - 10936 [52].
Le groupe phosphonate est attaché au PEG-NH2 par une condensation amide avec un ester lié à un groupe phosphonate. Le sel de sodium du phosphonate a été utilisé. Ensuite, le couplage a été réalisé a partir du triméthyl-phosphonoformate [CAS 31142-23-1] selon la procédure décrite par Robert A. Moss, Hugo Morales- Rojas, Saketh Vijayaraghavan and Jingzhi Tian, J. Am. Chem. Soc, 2004, 126,(35), 10923 - 10936 [52].
Une dissolution du PEG-NH2 (87,6 mg, M = 5400, Iris Biotech GmbH, PEG1069) dans 2 mL de DMF (Fluka, 97%) avec un excès de disodium méthylphosphonoformate (50 mg, M = 183,99) a été chauffé a 100ºC pendant 15h sous agitation. Après, le solvant est éliminé sous vide et le résidu suspendu dans éthanol absolu. L'excès de phosphonoformate est insoluble, donc il peut être éliminé par filtration. Le filtrat est concentré pour obtenir le produit (85 mg) . NMR 31P, (D2O), d = 1,3 ppm.
La modification par polyéthylène glycol pourra se réaliser pendant ou après la synthèse comme indiqué ci-dessous.
a) Modification superficielle avec du PE6-C00H pendant la synthèse des nanoparticules
Les synthèses des MOFs se font directement en présence _de monpméthoxy. PEG.. monoacide.- (MeO-PEG-COOH) de- formule générale CH3-O-(CH2-CH2-O)n-CH2-CH2-COOH (Sigma, masse molaire 5000 g/mole). Le monométhoxy PEG monoacide est introduit à 3 ; 8,5 ou 13 % en masse par rapport au poids total de solide utilisé en synthèse.
Procédé de préparation :
L'acétate de fer (1 mmol ; synthétisé selon la synthèse A décrite dans l'exemple 2) et l'acide muconique (1 mmol; Fluka 97%) sont mélangés dans 10 mL de méthanol (Aldrich, 99,9%). Le tout est introduit dans un corps en téflon de 23 mL. Le monoacide PEG est ensuite introduit à hauteur de 3 ; 8,5 ou 13 % en masse par rapport au poids total de solide. 0,35 mL de soude 2 M est éventuellement ajoutée. La solution est placée sous agitation pendant 20 minutes.
La bombe en téflon est placée dans un corps métallique fermé hermétiquement et chauffé dans une étuve à 100ºC pendant 12 heures. Le solide obtenu est récupéré par centrifugation à 5000 rpm pendant 10 minutes et lavé avec de l'eau distillée et l'acétone.
Le dosage du PEG dans les carboxylates de fer est effectué comme suit : les particules sont dégradées totalement en milieu acide (HCl 5M) afin de libérer le
PEG associé. Après neutralisation des solutions obtenues (à pH=7) et destruction des nanoparticules avec de la soude, le dosage du PEG a été effectué par spectrophotométrie UV (à la longueur d'onde de 500 nm) , d'après la méthode décrite dans B. Baleux et al. CR.
Acad. Scienœs Paris, série C, 274 ( 1972) pages 1617-
1620 [53]. Les principaux résultats sont répertoriés dans le tableau suivant.
Tableau 10. Modification du matériau MIL88A avec le PEG
5000 g/mole.

Nous pouvons constater que :
- l'ajout de soude permet de diminuer la taille des nanoparticules
- le % massique de PEG dans les nanoparticules est supérieur au % massique de PEG introduit en début de synthèse
- de manière remarquable, il est possible d'obtenir des particules d'environ 230 nm contenant 13 % en poids de PEG, ce qui est intéressant pour des applications médicales ( « furtivité » )
En effet, les nanoparticules « furtives » décrites dans la littérature contiennent généralement moins de 10 % mass de PEG, comme décrit dans R. Gref et al. Colloids and Surfaces B: Biointerfaces, Volume 18, Issues 3-4, October 2000, pages 301-313 [54].
b) Modification superficielle des nanoparticules MIL- 100 avec du PEG après la synthèse desdites nanoparticules
Les nanoparticules du MIL-100 sont synthétisées par voie microondes (Microondes CEM) en partant d'une solution de 9,7 g nitrate de fer hexahydrate (Aldrich, 97%), 3,38g d'acide 1, 3,5-benzenetricarboxylique (1,3,5-BTC, Aldrich, 99%) et 40 g d'eau distillée à 180ºC pendant 30min (puissance 600W). La taille de particule mesurée par diffusion de lumière est de 400nm.
Les nanoparticules pegylées du MIL-100 modifiées par polyéthylène glycol sont obtenues par la modification de surface des particules citées antérieurement. 30 mg du MIL-100 sont suspendus dans 3mL d'une solution aqueuse de lOmg du polyéthylène glycol aminoteminal (PEG-NH2 5000 g/mole, Aldrich, 97%) à 30ºC pendant 3 heures sous agitation. Ces nanoparticules sont récupérées par centrifugation (10000 rpm/10 min) et lavées avec de l'eau distillée.
La quantité du PEG en surface est déterminée par la méthode de Baleux et Champertier, basée dans la formation d'un complexe coloré par l'iodine-iodide sur le PEG, lequel est sélectivement mesuré par spectrophotométrie à 500nm) . La quantité du PEG est de 19% en masse et la taille de particules après la modification par le polyéthylène glycol augmente à 800nm. Par contre, l'observation de nanoparticules modifiées et non modifiées par le PEG par microscopie électronique à balayage. (SEM) montre de nanoparticules de 150nm dans les deux cas. Cette différence peut être due a des phénomènes d'agrégation particulaires.
Exemple 8 : Synthèse par voie ultrasons de carboxylates de fer (III) à surface modifiée par le polyéthylène glycol (PEG)
La synthèse par voie ultrasons de nanoparticules de solide MIL-88A à surface modifiée par le PEG a été réalisée à différents temps de reaction (30, 60, 90 et 120 minutes).
Dans les exemples qui suivent deux modes opératoire ont été effectués : a) dans le 1er mode opératoire, le PEG est ajouté 15 minutes avant la fin de la synthèse, b) dans le second mode opératoire, le PEG est ajouté dès le début de la synthèse (t = 0 min).
Pour chacune des synthèses ci-dessous, des solutions aqueuses de chlorure de fer III (2,7 mg/ml ; FeCl3, 6H2O commercialisé par ACROS 97%) et d'acide fumarique (1,16 mg/ml ; C4H4O4 commercialisé par ACROS 99%) sont préparées. Les deux réactifs solides sont pesés et dissous séparément dans l'eau dans les proportions données dans les exemples ci-dessous. La solution d'acide fumarique est portée à 70ºC sous agitation pendant 120 min pour solubiliser le produit. Le chlorure de fer est agité à l'aide d'un agitateur magnétique pendant 30 min.
a) Synthèse n° 1 :
Au total 8 fioles sont préparées. Dans chacune des 8 fioles de 20 mL, sont ajoutés 5 mL de solution de chlorure de fer III (2,7 mg/ml) et 5 mL de solution d'acide fumarique (1,16 mg/ml) :
- 4 fioles servent de témoin où les réactions sont réalisées pour les 4 temps de synthèse : 30, 60, 90 et 120 min,
- dans les 4 autres fioles, 5 mg de PEG sont ajoutés 15 min avant la fin de chacune des synthèses de durée 30,
60, 90 et 120 min (la fin d'une synthèse correspond à la sortie du bain à ultrasons).
Les 8 fioles sont placés en même temps dans un bain de sonication à 0ºC, pendant les durées t correspondantes (30, 60, 90 et 120 min).
Après la synthèse, un volume de 0,1 ml de solution est prélevé dans chaque fiole afin de déterminer la taille des particules par diffusion de lumière grâce à un appareil de Dynamic Light Scattering (DLS, Nanosizer). Le reste de solution est ensuite passé à la centrifugeuse à 10 000 rpm à 0ºC pendant 15 min afin de séparer le surnageant du solide formé. Le surnageant est éliminé à l'aide d'une pipette pasteur et le culot récupéré est placé sous la hotte à température ambiante (environ 20ºC). Appareillage utilisé :
Bain de sonication : Labo-moderne TK 52H n° de série : 164046192 SONOCLEAN
- Centrifugeuse : JOUAN MR 1812 - Nanosizer : Coulter N4 PLUS USA ; Malvern
b) Synthèse n° 2 : _
Au total 8 fioles sont préparées. Dans chacune des 8 fioles de 20 mL, sont ajoutés 5 mL de solution de chlorure de fer III (2,7 mg/ml) et 5 mL de solution d'acide fumarique (1,16 mg/ml) :
- 4 fioles servent de témoin où les réactions sont réalisées pour les 4 temps de synthèse : 30, 60, 90 et 120 min,
- dans les 4 autres fioles, 5 mg de PEG sont ajoutés dès le début de chacune des synthèses de durée 30, 60,
90 et 120 min.
Les 8 fioles sont placés en même temps dans un bain de sonication à 0ºC, pendant les durées t correspondantes (30, 60, 90 et 120 min). Après la synthèse, un volume de 0,1 ml de solution est prélevé dans chaque fiole afin de déterminer la taille des particules par diffusion de lumière grâce à un appareil de Dynamic Light Scattering (DLS, Nanosizer). Le reste de solution est ensuite passé à la centrifugeuse à 10 000 rpm à 0ºC pendant 15 min afin de séparer le surnageant du solide formé. Le surnageant est éliminé à l'aide d'une pipette pasteur et le culot récupéré est placé sous la hotte à température ambiante (environ 20ºC). Appareillage utilisé :
Bain de sonication : Labo-moderne TK 52H n° de série : 164046192 SONOCLEAN
- Centrifugeuse : JOUAN MR 1812
- Nanosizer : Coulter N4 PLUS USA ; Malvern L'évolution de la taille de particule (P en nm) en fonction du temps (t en min) est représenté dans la figure 7.Q. Celle-ci. montre, qu'il n'y a pas de variation significative après l'ajoute du PEG au temps initial de synthèse. Que ce soit en présence ou non de PEG au temps initial de la synthèse, il est possible d'observer en DRX un epaulement à 11°, caractéristique de la phase
MIL-88A qui semble augmenter en intensité avec le temps de synthèse. c) Conclusion de l'étude:
Le but de cette étude est d'optimiser la taille des particules qui doivent être inférieures à 200 nm afin de pouvoir les rendre compatible avec une administration par voie intraveineuse. Les résultats obtenus sont satisfaisants puisque les diamètres de particules obtenus sont inférieurs à 200 nm (avec vérification des structures cristallines type MIL-88A dans la plupart de solides). De plus, même si les rendements sont inférieurs à ceux obtenus par voie solvothermale ou voie microondes, on peut les considérer comme acceptables (tableau ci-dessous).
Tableau 11. Rendements de synthèse par voie ultrasons

II est possible d'observer que la taille des particules augmente en fonction du temps de synthèse.
De même, la modification par le PEG à t=0 min aboutit à des diamètres de particules plus petits que la modification par PEG à t=f-15min, due probablement au fait que là croissance cristalline soit arrêtée plus tôt.
Exemple 9 : Synthèse de solides MOF à surface modifiée par le polyéthylène glycol (PEG) et l'acide folique (FA)
Acide folique :

a) Synthèse n° 1 : modification de surface après synthèse des nanoparticules
Modification de surface avec du PEG :
100 mg de nanoparticules de MIL100, MIL88, MIL53 ou MILlOl (préalablement deshydraté à 100°C/nuit) sont dispersées sous sonication dans 100 mL de solution à 17,9 mM de 2-(méthoxy(polyéthylèneoxy)- propyl)triméthoxysilane dans le toluène anhydre. Le mélange est soumis à des ultrasons à 60ºC pendant 4 h, sous débit de gaz inerte (azote). La suspension colloïdale résultante, contenant les nanoparticules à surface modifiée avec du PEG, est lavée deux fois avec de l'éthanol et deux fois dans une solution de citrate de sodium 20 mM- (pH 8, 0) et resuspendue finalement dans l'eau.
Modification de surface avec du PEG et du FA :
Le FA a été attaché aux nanoparticules grâce à un espaceur bifonctionnel, le silane-PEG-
trifluoroéthylester (TFEE) synthétisé selon une méthode décrite dans la littérature par Kohler N. et al., J Am Chem Soc 2004; 126 ; 7206-7211 [55].
100 mg de nanoparticules sont recouvertes par du PEG-TFEE selon la même méthode que décrit ci-dessus, en utilisant le silane-PEG-TFEE à la place du 2-méthoxy (polyéthylèneoxy)-propyltriméthoxysilane.
Les nanoparticules résultantes, recouvertes de PEG-TFEE, sont lavées deux fois puis resuspendues dans 100 mL de toluène sec. Une amine primaire a été greffée aux groupements terminaux des chaines de PEG en rajoutant 1 mL de éthylènediamine (Sigma) aux nanoparticules maintenues sous débit d'azote. Le mélange a été soumis aux ultrasons (4h, 60ºC). Les nanoparticules résultantes, recouvertes par l'aminé, ont été lavées trois fois avec de l'éthanol, et trois fois avec du diméthyl sulfoxide (DMSO) . Les nanoparticules ont été finalement resuspendues dans 50 mL de DMSO anhydre. Le FA a été couplé aux groupements aminé terminaux des chaines de PEG en rajoutant 50 mL de solution de FA (23 mM FA dans du DMSO) avec des quantités équimolaires de dicyclohexylcarbodiimide (DCC) (Sigma) et 10 μL de pyridine. Le mélange a été protégé de la lumière et laissé réagir pendant une nuit sous agitation bidimensionnelle (180 rpm) . Les nanoparticules conjuguées avec du PEGH et du FA (NP- PEG-FA) ont été lavées deux fois avec-de l'éthanol, et deux fois avec une solution de citrate de sodium 20 mM (pH 8,0) et resuspendues finalement dans cette même solution de citrate de sodium.
b) Synthèse n°2 : modification de surface pendant la synthèse des nanoparticules
La modification de la surface des solides MOF peut également se faire pendant la synthèse. Dans l'exemple qui suit, la modification de surface est réalisée avec du chitosane préalablement greffé avec de l'acide folique (FA).
Un exemple de synthèse de chitosane greffé avec de l'acide folique via un espaceur PEG est décrite dans la publication de Peggy Chan et al., Biomaterials, Volume 28, Issue 3, 2007, pp 540-549 [56].
Les réactifs suivants ont été utilisés pour la réalisation de cet exemple :
- chitosane (masse molaire Mn de 255 kDa, viscosité: 200—800 cps dans 1% d'acide acétique, commercialisé par la société Sigma-Aldrich)
- le W-hydroxylsuccinimide-PEG-Maleimide (NHS-PEG-MAL, Mn 3400 Da, commercialisé par la société Nektar, NOF Corporation, Tokyo, Japan) - l'ester succinimidyl de monométhoxy-PEG (mPEG-SPA, Mn 5000 Da, commercialisé par la société Nektar, NOF Corporation, Tokyo, Japan) .
Le chitosane est préalablement déacétylé pour obtenir un degré d'acétylation de 82% (déterminé par 1H-NMR) selon le procédé décrit par Wang LS (Thesis, National University of Singapore, Singapore, 2001).
100 mg de chi^sane__pnt__été_diss_ous._dans__50-- mL- de solution d'acide acétique (20%). Le pH de la solution a été ajusté à 6 par addition d'hydroxyde de sodium et le mPEG-SPA a été introduit dans le mélange réactionnel. Le mélange est laissé réagir pendant 24 h à température ambiante sous agitation. Le produit
obtenu est dialyse pendant 24 h contre de l'eau de- ionisée en utilisant une membrane avec un seuil de coupure de 12,000 Da (Spectrum Laboratories, USA) et finalement, lyophilisé. Pour synthétiser le chitosane greffé avec du PEG et du FA, l'ester de N-hydroxysuccinimide de FA a été préparé selon la méthode décrite par J. H. Van Steenis et al., J Control Release 87 (2003), pp. 167- 176 [57]. Brièvement, 1 g de FA a été ajouté à un mélange de DMSO anhydre (40 mL) et triéthylamine (TEA, 0,5 mL). Le mélange a été agité à l'abri de la lumière pendant la nuit, dans des conditions anhydres. Les autres réactifs, dicyclohexylcarbodiimide (DCC, 0,5 g) et N- hydroxysuccinimide (NHS, 0,52 g) ont été ajoutés et le mélange est laissé réagir pendant 18h à l'abri de la lumière et dans des conditions anhydres. Le produit secondaire, la dicyclohexylurée (DCU) ayant précipité, a été enlevé par filtration. Le DMSO et le TFA ont été évaporés sous vide. Le produit de la réaction a été séché sous vide, puis dissout dans 1,5 mL de mélange 2:1 (v:v) de DMSO et TEA. Une quantité équimolaire de 2-aminoéthanethiol (Wako) a été rajoutée et la reaction a été laissée se poursuivre pendant la nuit dans des conditions anhydres. Ainsi, un groupement thiol a pu être introduit à l'acide folique et le produit résultats est dénommé FA-SH .
100 mg de chitosane sont dissous dans 50 mL de solution d'acide acétique (20%). Le pH de la solution est ajusté à 6 par addition d'hydroxyde de sodium et 100 mg de NHS-PEG-MaI sont introduits dans le mélange réactionnel. La mélange est laissé réagir pendant 3 h à
température ambiante (environ 20ºC) sous agitation, puis le pH est ajusté à 7. Le mélange est laissé réagir pendant la nuit, dans des conditions anhydres. Le FA-SH synthétisé comme précédemment a été rajouté de manière 5 graduée sous agitation et le pH a été ajusté à 6,5—7,5 avec de la soude.
Le conjugué obtenu dénommé FA-PEG chi porte du FA couplé au chitosane via un bras espaceur de PEG, ce qui est un avantage pour atteindre le récepteur de
10 l'acide folique (comme il a été décrit dans la littérature, voir par exemple : A. Gabizon, H. Shmeeda, A. T. Horowitz and S. Zalipsky, Tumor cell targeting of liposome-entrapped drugs with phospholipids-anchored folie acid-PEG conjugates, Adv Drug Deliv Rev 56
15 (2004), pp. 1177-1192 [58].
Le degré de substitution peut être ajusté en variant le rapport massique PEG : chitosane utilisé dans la réaction. Ce polymère a été dialyse pendant 48 h contre de l'eau de-ionisée en utilisant une membrane
20 avec un seuil de coupure de 12,000 Da (Spectrum Laboratories, USA) et finalement, lyophilisé.
c) Synthèse n°3 :
Les solides hybrides peuvent être modifiés en 25 surface par adsorption de polysaccharides tels que le dextrane greffé avec la biotine. Nous pouvons donc envisager d'adsorber, à la place du dextrane greffé avec la biotine, du chitosane greffé avec l'acide folique (synthétisé comme décrit 30 dans la publication citée plus haut), et, éventuellement, si nécessaire, greffé aussi avec d'autres composés hydrophobes comme le cholestérol ou
des chaînons aliphatiques, afin d'assurer une meilleure adhésion au niveau de la surface des nanoparticules.
Une fonctionnalisation de surface pourrait également se faire via l'adsorption d'autres polysaccharides greffés avec de le FA.
d) Synthèse n°4 :
Les solides hybrides peuvent être modifiés en surface avec du PEG lors de leur synthèse. Le monométhoxy PEG monoacide utilisé lors de cette synthèse est substitué par du PEG monoacide comportant une fonction réactive bloquée en bout de chaine comme le produit commercial :
Boc-PEG-carbonateNHS, MW 5000, Boc = tert- butoxycarbonyle (Référence SUNBRIGHT* BO-050TS, NOF Corporation) .
Après réaction, comme indiqué dans l'exemple, des mélanges MeO-PEG-COOH et Boc-PEG-carbonateNHS (rapports massiques 1:0.05 à 1:0,5) sont utilisés à la place de MeO-PEG-COOH. La déprotection se fera par ajout d'acide trifluoroacétique (TFA). Protocole opératoire:
On ajoute 0,6 mL de TFA à une suspension de 300 mg de nanoparticules dans 2 mL d'eau. On laisse réagir pendant Ih à température ambiante (environ 20ºC) sous agitation magnétique. On isole les particules par centrifugation et on es lave trois fois avec de-l'eau- distillée.
On fonctionnalise les groupements réactifs en surface avec des ligands tels le FA, par exemple comme dans la synthèse n°l indiqué dans le paragraphe a) .
e) Caractérisation des nanoparticules :
La quantité d'acide folique effectivement couplée aux nanoparticules pourra être déterminée après dégradation de celles-ci dans un milieu acide, neutralisation à pH 7, puis redissolution dans un solvant approprié, tel le dichlorométhane, le DMSO, ou un mélange de ces deux solvants. L'acide folique pourra alors être quantifié par une mesure de l'absorbance UV
(à 358 nm, le coefficient d'extinction molaire ε de l'acide folique est 15,76 M-1 cm-1).
Pour vérifier que l'acide folique se trouve bien en surface des nanoparticules, la technique de la résonance de surface du plasmon (BIAcore) est utilisé. La protéine qui se lie à l'acide folique (« folate binding protein ») est immobilisée au niveau de la surface du détecteur, sur un mince film de dextrane activé (procédure classique recommandée par le constructeur BIAcore). La quantité de nanoparticules effectivement attachée à ce support est évaluée par rapport à celle de nanoparticules non recouvertes d'acide folique.
Exemple 10 : Synthèse de matériaux MOF à base de ligands bioactifs L'utilisation des ligands avec une activité biologique présente un intérêt pour : la libération de composé actif par dégradation du matériau MOF l'encapsulation d'autres molécules actives pour des thérapies combinées
Des tests de l'activité antimicrobienne , ainsi que la dégradation dans des milieux physiologiques et l'activité sur des cellules seront réalisés sur des carboxylates de fer poreux a structure flexible de type MIL-88 utilisant les acides 4, 4 'azobenzene dicarboxylique et 3, 3 '-dichlore 4,4 '-azobenzene dicarboxylique, entre autres.
Dans les synthèses qui suivent, divers molécules bioactives sont utilisées pour préparer les matériaux MOF de la présente invention, et notamment :
1 'azobenzene, l'acide azélaïque et l'acide nicotinique.
L 'azobenzene (AzBz), de formule C6H5-N=N-C6H5, peut être incorporé aux matrices de polymère comme stabilisateur. De plus, la structure rigide des molécules azoïques leur permet de se comporter en tant que mésogènes à cristal liquide dans de nombreux matériaux. Par ailleurs, l' azobenzene peut être photoisomérisé (isomère cis ou trans) d'où son utilisation pour photomoduler l'affinité d'un ligand (par exemple un médicament) pour une protéine. En effet, l' azobenzene peut jouer le rôle de photointerrupteur entre un ligand et une protéine en permettant ou en empêchant la liaison protéine- médicament suivant l'isomère cis ou trans de 1' azobenzene (une extrémité de l' azobenzene peut par exemple être substituée par un groupement se liant à la protéine cible tandis que l ' autre extrémité est reliée- à un ligand (médicament) de la protéine).
L'acide azélaïque ( HO2C- (CH2)7-CO2H) est un acide dicarboxylique saturé qui a des propriétés antribactérienne, kératolytique et comédolytique . Il
est utilisé notamment dans le traitement de l'acné et la rosacea.
L'acide nicotinique (C5H4N-CO2H) est l'une des deux forme de la vitamine B3 avec la nicotinamide. La vitamine B3 est notamment nécessaire au métabolisme des glucides, lipides et protéines.
a) MIL-88G(AzBz) (Fe) ou Fe3O[C12H8N2- (CO2) 2]3. X. nH20 (X=F, Cl, OH) 118 mg de Fe(ClO4)3.xH2O (0.33 mmol, Aldrich,
99%) et 90 mg (0,33 mmol) d'acide 4,4'- azobenzenedicarboxylique ( synthétisé selon la méthode décrite par Ameerunisha et al., J. Chem. Soc. Perkin Trans. 2, 1679, 1995 [59]) sont dispersés dans 15 ml de DMF (Fluka, 98%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 150ºC. Le solide est récupéré par filtration.
200 mg du solide sont suspendus dans 1OmL de DMF sous agitation à température ambiante pendant 2 h pour échanger l'acide restant dans les pores. Le solide est ensuite récupéré par filtration puis calciné à 150ºC sous vide pendant 15 heures pour éliminer le DMF restant dans les pores. La taille de particule mesurée par diffusion de lumière est > 1 micron.
b) MIL-886-2Cl (AzBz-2Cl) (Fe) ou Fe3O[C12H6N2Cl2- (CO2) 2] 3. X . nH2O (X=F , Cl , OH) 177 mg de Fe(ClO4)3.xH2O (0,5 mmol, Aldrich,
99%) et 169 mg (0,5 mmol) d'acide dichloro-4,4'- azobenzenedicarboxylique (préparé selon la synthèse D
décrite dans l'exemple 3) sont dispersés dans 15 ml de DMF (Fluka, 98%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 12 h à 150ºC. Le solide est récupéré par filtration.
200 mg du solide sont suspendus dans 1OmL de DMF sous agitation à température ambiante pendant 2 h pour échanger l'acide restant dans les pores. Le solide est ensuite récupéré par filtration puis calciné à 150ºC sous vide pendant 15 h pour éliminer le DMF restant dans les pores .
La taille de particule mesurée par diffusion de lumière est > 1 micron.
c) Azobenzène 3 , 3 ' , 5 , 5 ' -tétracarboxylate de fer 1
118 mg de Fe(ClO4)3.xH2O (0,3 mmol, Aldrich, 99%) et 119 mg (0,3 mmol) d'acide 3, 3', 5,5'- azobenzènetétracarboxylique (préparé selon la synthèse E décrite dans l'exemple 3) sont dispersés dans 15 ml de DMF (Fluka, 98%) avec 0,1 mL de HF 5M (SDS, 50%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 3 jours à 150ºC. Le solide est récupéré par filtration et lavé avec de 1 ' acétone . Le solide obtenu a une structure cubique rigide.
La taille de particule,mesurée, par diffusion de- lumière est > 1 micron.
d) Azobenzène 3,3 ' ,5,5 '-tétracarboxylate de fer 2
118 mg de Fe(C104)3.xH20 (0,3 mmol, Aldrich, 99%) et 119 mg (0,3 mmol) d'acide 3, 3', 5, 5'-
azobenzènetétracarboxylique (préparé selon la synthèse E décrite dans l'exemple 3) sont dispersés dans 15 ml de l'eau distillée avec 0,1 inL de HF 5M (SDS, 50%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 150ºC. Le solide est récupéré par filtration et lavé avec 1 ' acétone .
La taille de particule mesurée par diffusion de lumière est 498 nm, avec une deuxième population minoritaire de 1100 nm.
e) Azélate de fer 1
270 mg de FeCl3.6H2O (1 mmol, Aldrich, 99%) et 188 mg (1 mmol) d'acide azelaique (Aldrich, 99%) sont dispersés dans 5 ml d'eau distillée. Le tout est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 3 jours à 100ºC. Le solide est récupéré par filtration et lavé à l'acétone.
200 mg de solide sont suspendus dans 50 mL d'éthanol absolu sous agitation pendant 5 h pour l'activer. Le solide est récupéré par filtration.
La taille de particule mesurée par diffusion de lumière est > 1 micron (1500 nm) .
f) Nicotinate de fer 1
Les conditions de synthèse dans l'eau sont les suivantes :
135 mg de FeCl3.6H2O (1 mmol, Aldrich, 99%) et
62 mg (1 mmol) d'acide nicotinique (Aldrich, 99%) sont dispersés dans 5 ml d'eau distillée avec 0,1 mL de NaOH
2M. Le tout est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 16 heures à
100ºC. Le solide est récupéré par filtration et lavée avec l'acétone.
Les conditions de synthèse dans le DMF sont les suivantes : 135 mg de FeCl3.6H2O (1 mmol, Aldrich, 99%) et
62 mg (1 mmol) d'acide nicotinique (Aldrich, 99%) sont dispersés dans 5 ml de DMF (Fluka, 98%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une Bombe métallique PAAR pendant 16 h à 100ºC. Le solide est récupéré par filtration et lavée avec l'acétone.
La taille de particule monodisperse (PDI=O, 241) mesurée par diffusion de lumière est 662 nm.
g) Nicotinate de fer 2 Les conditions de synthèses du nicotinate de fer 2 sont les suivantes :
71 mg d'acétate de fer (III) (0,12 mmol, selon la procède décrit anterieurment) et 73,8 mg (0,6 mmol) d'acide nicotinique (Aldrich, 99%) sont dispersés dans 5 ml de DMF (Fluka, 98%). Le tout est laissé dans un corps en Téflon de 23 ml mis dans une bombe métallique PAAR pendant 24 heures à 140ºC. Le solide est récupéré par filtration et lavée avec l'acétone.
Les donnes cristallographiques de cette phase sont dans le figure 89:
Groupe de space P 21/n a = 16.422899 b = 21.423401 c = 11.048300 beta = 91.806999
Exemple 11 : Détermination de la teneur en fer dans le solide MIL-IOO(Fe)
Activation :
Afin de vider les pores du matériau (solvants, acides résiduels) et de libérer les sites de coordination du métal, l' activation du matériau MIL- 100(Fe) a été effectuée par chauffage à 150ºC sous vide primaire pendant 15 heures. Le solide résultant ne possède que du fer au degré d'oxydation +III.
Réduction

La réduction partielle du matériau MIL-100(Fe) a été effectuée par chauffage a 250ºC sous vide primaire pendant 15 heures. La spectroscopie infrarouge a permis de quantifier la teneur relative en fer(II)/fer(III) autour de 20/80 % (Figure 50). La figure 50 représente la quantité de sites Fer coordinativement insaturés présent dans le solide MIL-IOO(Fe) activé en fonction du traitement thermique effectué. Le solide MIL-IOO(Fe) est activé sous vide résiduel (environ 10-5 Torr) à différentes températures et pendant différentes durées. T(Fe) représente la teneur en sites Fer coordinativement instaurés et T(Fe2+) représente la teneur en sites Fe2+ coordinativement instaurés (en μmole de sites insaturés par gramme de solide activé ou en % de sites Fer insaturés).
Les quantités de sites Fer insaturés sont déterminées par adsorption de CO à 100K suivie par
spectroscopie infrarouge. L'incertitude sur les valeurs est estimée à +/-10 %.
Exemple 12: Mise en évidence de la flexibilité des solides
La catégorie de solides hybrides flexibles à base de trimères de métaux de transition trivalents est dénommée MIL-88. Ces composés sont typiquement construits à partir de trimères d'octaèdres de fer, c'est-à-dire trois atomes de fer connectés par un oxygène central et par six fonctions carboxylates connectant deux à deux les atomes de fer ; une molécule d'eau terminale, coordinée à chaque atome de fer vient ensuite compléter la coordinence octaédrique du métal. Ces trimères sont ensuite reliés entre eux par des acides dicarboxyliques aliphatiques ou aromatiques pour former les solides MIL-88A, B, C, D et MIL-89 (-A pour acide fumarique, -B pour acide téréphthalique, -C pour acide 2,6-naphtalenedicarboxylique, -D pour acide 4,4'- biphényldicarboxylique et MIL-89 pour l'acide trans, trans muconique ) , comme décrit dans le document de Serre et al., Angew. Chem. Int. Ed. 2004, 43, 6286 [17]. D'autres analogues avec d'autres diacides carboxyliques ont également été synthétisés et sont nommés MIL-88E, F, G...
L'étude du comportement de ces solides par diffraction des RX, a permis d'établir que ces composés sont flexibles avec des amplitudes de « respiration » (c'est-à-dire de gonflement ou de contraction) considérables entre leur forme sèche et leur forme solvatée. Il en résulte des variations de volume de
maille entre 85 et 230 % selon la nature de l'espaceur organique (figure 54), comme décrit dans le document Serre et al., Science, 2007, 315, 1828 [18]. Les inventeurs ont noté que les formes sèches ne sont pas poreuses avec une taille de pores (tunnels) à peu près identique quel que soit le ligand carboxylique utilisé. Par contre, le gonflement du solide hybride en phase liquide est fonction de la longueur de l'espaceur organique. Ainsi, la distance entre trimères dans la forme gonflée passe de 13,8 Â avec l'acide fumarique (MIL-88A) à 20,5 Â avec le ligand biphényl (MIL-88D). La taille des pores des formes gonflées varie ainsi entre 7 Â (MIL-88A) à 16 Â (MIL-88D) . Le gonflement est réversible, comme le montre l'exemple du solide MIL-88A en présence d'eau dans la figure 57 et dépend également de la nature du solvant utilisé comme décrit dans le document Serre et al. J. Am. Chem. Soc, 2005, 127, 16273-16278 [19]. La « respiration » s'effectue de manière continue, sans rupture apparente de liaisons durant la respiration. Par ailleurs, de retour à température ambiante, le solide gonfle à nouveau par resolvatation, confirmant le caractère réversible de la respiration.
Si l'on regarde de près l'arrangement entre les trimères constitutifs de la structure, chaque trimère est relié à six autres trimères, trois en dessous et trois au dessus, par les . dicarboxylates ce qui conduit à la formation de cages bipyramidales de trimères. Au sein de ces cages, la connexion entre trimères s'effectue uniquement selon l'axe c et l'absence de tout lien dans le plan (ab) est à l'origine de la flexibilité (figure 58).

En effet, lorsque l'on insère un solvant dans le matériau, la cage se déforme avec un rapprochement des trimères selon l'axe c et un éloignement dans les directions a et b, ce qui provoque une augmentation globale du volume de la cage (figure 58). Finalement, la flexibilité de ces solides hybrides est remarquable, mais cependant comparable à celle de certains polymères. La principale différence concerne la cristallinité des solides hybrides, les polymères étant amorphes. Enfin, contrairement aux polymères, le gonflement se produit dans les solides hybrides de manière anisotrope.

qgpieeauraic Ol yOe MtsF .san
O (Dv W n
D) rt h (Dv ggaaer (D u> σ n a
01 •0 rt ta H
(Dv Φ Φ π
H- (Dv 11 α fer G. 01 ( (D Φ W ft 01 re •dsι• rt (D ni
01 α
01 rt 01
01 O
H α- α ; rt-
(D H-
(D O (D O
3 -
01 a
O
(D (D
CO Φ
O Ct
• • 01
C C (D M φ
CU U)
H rt i
C (D H- 0)
H- O n
U) a (0 £3 0)
(D U)
(D Φ rt
(D Ol α Φ
CO (D a
P) o
(D α 1O
(U M eu
O S
H- α O O rt (D *l
(Dt 01 rt D)

(MILlOO-Fe), présentant une activité redox (MILlOO-Fe), flexibles (MIL88), flexibles et substitués (sur le ligand) (MIL88-FeNO2) .
En, effet, si la délivrance de NO en quantité importante sur une courte période est facile à produire avec les zéolithes, les solides hybrides poreux de type MIL-n à base de métaux à haut degré d'oxydation (+3) semblent avoir le profil idéal pour le relargage prolongé de NO. En effet, les inventeurs ont montré précédemment que le MOF à larges pores dénoté MIL-100 constitué de trimères d'octaèdres de chrome ou de fer relié par des acides trimésiques, est stable, même après le départ de l'eau coordinée sur les centres métalliques . Cette dernière est facilement évacuée par chauffage sous vide et laisse place à des centres métalliques insaturés et accessibles (métal en coordinence cinq). La plupart des solides MIL au fer possèdent ce type de trimères et donc peuvent tous potentiellement adsorber sur leurs centres métalliques des molécules organiques possédant un caractère de donneur d'électrons (base de Lewis), tel que NO via le doublet libre situé dans les orbitales 5σ.
Chargement en oxyde d'azote (NO) : Les matériaux MOFs préalablement activés sont exposés à environ 2 bars de NO (99,5%, commercialisé par la société. Air Liquide) pendant 30 minutes. Ils sont ensuite évacués sous vide (pour éviter la libération du NO physisorbé et donc le phénomène dit de « initial burst » correspondant à une très grande quantité de NO libéré dès les premières minutes de relargage) et placés sous atmosphère d'argon sec. Cette
dernière opération (évacuation/argon) est répétée 3 fois pour s'assurer que tout le NO physisorbé a été éliminé.
5 Mesure des isothermes d ' adsorption/désorption de NO
Les mesures d' adsorption/désorption de NO sont effectuées à l'aide d'un appareil de type gravimétrique thermostaté pour éliminer tout effet de l'environnement extérieur. Une microbalance CI, commercialisée par la
10 société CI Electronics Ltd) est utilisée (sensibilité : 0,1 μg, reproductibilité de la mesure de la masse : 0,01%). La pression est mesurée par deux sondes BOC Edwards Active (domaine de mesure : lxl0"8-lxl0"2 et lxl0"4-lxl03 mbar). L'échantillon de MOF (-50mg) est
15 préalablement activé à la température requise (voir plus haut) à 2xlO-3 mbar jusqu'à ce que plus aucune perte de masse ne soit observée. L'échantillon est ensuite refroidi à la température de mesure et conservé a cette température soit par un bain d'eau thermostaté
20 (température précise à ±0,02 K), soit par trempage dans l'azote liquide. La contrebalance est maintenue à la même température que l'échantillon pour minimiser les effets de différence de température sur la lecture de la température, elle-même mesurée à l'aide d'un
25 thermocouple de type K placé proche de l'échantillon (< 5 mm). La variation de température de l'échantillon durant la mesure est inférieure à 0, 1 K. Le gaz NO sec
(Air Liquide, 99,5 %) est introduit dans le système jusqu'à ce que la pression désirée soit atteinte, et la
30 prise de masse mesurée en fonction du temps jusqu'à stabilisation.
De cette façon, une isotherme d'adsorption est obtenue en incrémentant la pression et en relevant le gain de masse de l'échantillon à l'équilibre. La désorption de NO est effectuée en diminuant graduellement la pression jusqu'à la valeur désirée. (2 x 10'3 mbar) .
Quantification du relargage de NO par chimiluminecsence
Les mesures de NO sont réalisées à l'aide d'un analyseur « Sievers NOA 28Oi chemiluminescence Nitric Oxide Analyzer ». L'instrument est calibré en passant de l'air dans un filtre zéro (Sievers, < 1 ppb NO) et 89,48 ppm de NO gazeux (Air Products, balance azote). Le flux gazeux est fixé à 180 mL/min avec une pression dans la cellule de 8,5 torr et une pression d'oxygène de 6,1 psi.
Pour mesurer le relargage de NO par un échantillon sous forme de poudre, de l'azote gazeux d'humidité connue (11% d'eau par passage du flux gazeux sur une solution aqueuse de LiCl) est passé sur la poudre et le flux gazeux résultant envoyé vers l'analyseur, et la quantité de NO (en ppm et ppb) enregistrée. Cette approche est valable par exemple pour des applications cutanées, où le solide est en contact avec la peau et la libération du NO se réalise en_p_r_és.e.nc_e._de_.lJ-humidité—de_la-peau.
Pour des applications où le solide est en contact avec le sang (tubes, cathéters, etc.), la présence d'un milieu aqueux est nécessaire. Pour de telles applications, le relargage de NO par un échantillon sous forme de poudre a été étudiée dans un
fluide physiologique simulé. Ainsi, le solide chargé en NO (50mg + NO) est suspendu dans 4 mL d'un tampon phosphate saline (pH -5,5 ; PBS) à 22°C sous agitation. La quantité de NO libéré à différents temps est analysée par l'analyseur « Sievers NOA 28Oi chemiluminescence Nitric Oxide Analyzer » .
13.1. Solide MILlOO(Fe)
Le solide MILlOO(Fe) adsorbe donc de fortes quantités de NO (2-4 mmol.g-1) à température ambiante (environ 20ºC) (figure 51) et le relargue lentement et très partiellement (sous flux d'humidité de 11%) comme le montrent les résultats préliminaires obtenus avec les solides MIL-IOO(Fe) (figure 52). Les profils de relargage de NO sous pression de vapeur d'eau sont, en outre, très intéressants.
La figure 51 représente l'isotherme d'adsorption de NO (N0ada en mmol/g) à la température de 298 K pour le carboxylate de fer MIL-IOO(Fe) activé à 120ºC pendant une nuit. Cette figure représente la quantité de NO adsorbé (courbe (a)) et désorbé (courbe (b)) en fonction de la pression P (en mmHg) .
La figure 52 représente le profil de relargage de NO (N0rel en mmol/g) sous pression de vapeur du solide MIL-IOO(Fe). Le NO est représenté en ppm (ou « partie par million ») ou en ppb (ou « partie par milliard ») en fonction du temps t en heure.
On considère que le relargage de NO à un niveau utile sur le plan biologique est fini lorsque le taux de libération est inférieur à quelques ppb/minutes.
Notons aussi que tout le gaz NO n'est que très
partiellement relargué dans ces conditions (11% d'eau dans un flux de gaz neutre), et qu'il reste à peu près 75 % du NO encore adsorbé. Il restera donc à voir avec quelle vitesse ce dernier va se désorber lors de tests réels, i.e. lors de la mise en contact du solide avec le milieu physiologique (sur la peau ou en contact avec le sang) .
Dans un second temps, les inventeurs ont réduit partiellement le fer (III) du composé MIL-IOO(Fe) par activâtion sous vide primaire (12 heures à 250ºC). La spectroscopie infrarouge a montré que dans ces conditions, cela conduisait à la réduction d'environ 15-20 % du fer(III) en fer(II). Les métaux de transition de faibles degrés d'oxydation, tel que le fer(II), possèdent des électrons supplémentaires dans les orbitales d. Il est connu que le transfert électronique des orbitales d du métal vers les orbitales 2π● de molécules telles que NO et CO renforce la liaison métal adsorbat (phénomène de rétro-donation) et stabilise ainsi les espèces coordinées sur le centre métallique. La quantité de NO augmente considérablement avec l'introduction de fer(II) (4,5 mmol.g-1 à 1 bar au lieu de 2,5 mmol.g-1 pour le solide pur fer(III)), car celui-ci est susceptible d' interagir avec plus d'une molécule de NO par centre métallique (figure 53). De plus, le relargage est alors plus lent, la libération de. NO étant toujours présente après 17 heures au lieu de 12 heures avec l'analogue non réduit. Notons ici que la quantité totale de NO relarguée (<1 mmol.g-1) est encore une fois largement inférieure à celle adsorbée (2,5-4,5 mmol.g-1). Cela provient de la présence de sites très forts d'adsorption, que l'eau sous forme de
vapeur (11% d'humidité dans le gaz neutre) ne parvient pas à désorber. Des tests en milieu aqueux permettront de connaître les performances réelles de ces solides.
La figure 53 représente, à gauche : l'isotherme d'adsorption de NO à 298 K pour le MIL-IOO(Fe) activé à
250ºC sous vide pendant une nuit ; à droite : le profil de relargage de NO sous pression de vapeur du solide
MIL-IOO(Fe) activé à 250ºC sous vide pendant une nuit.
13.2 Solides MIL88A et MIL88B
Finalement, les inventeurs ont testé l'adsorption et le relargage de NO à partir des phases flexibles MIL-88A et MIL-88B. Ces solides possèdent les mêmes trimères d'octaèdres que le composé MIL-100 mais avec une structure flexible. On peut noter ici que les inventeurs ont prouvé précédemment que ces deux composés gonflent en présence de liquides, mais ne sont pas poreux sous leur forme sèche (figure 54). Il n'était donc pas du tout évident de penser que ces solides allaient adsorber le gaz NO. Les inventeurs ont observé une adsorption de près de 2,5 mmol.g"1 de NO à 298 K et à des pressions inférieures à 1 atmosphère (figure 55) .
La figure 54 représente la vue schématique du phénomène de respiration dans les solides MIL-88 (-A, -B, -C et -D) .
La figure 55 représente les isothermes d'adsorption de NO à 298 K pour les carboxylates de fer MIL-88A(Fe) et MIL-88B(Fe) activés à 150ºC sous vide pendant une nuit. La quantité de NO (N0abg en mmol/g) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg) .
Le relargage de NO est ensuite testé sous pression de vapeur d'eau (11% d'humidité dans un gaz neutre). Dans les deux cas, la quantité de NO relarguée est encore plus faible qu'avec le solide MIL-100 (<0,055 et 0,002 mmol.g-1) en comparaison avec les quantités adsorbées (2,5 mmol.g-1) (figure 56), ce qui laisserait penser, à première vue, que le gaz est beaucoup plus fortement adsorbé que sur le composé MIL- 100(Fe). C'est la première fois que l'on observe une si faible proportion de NO relargué avec un solide poreux ou un polymère.
La figure 56 représente les profils de relargage de NO sous pression de vapeur d'eau des solides MIL-88A(Fe) (en haut) et MIL-88B(Fe) (en bas) activés à 150ºC pendant une nuit. La quantité de NO relargué (NOrel en mol/g) est exprimée en fonction du temps t (en secondes).
Pour des applications ou le solide est en contact avec le sang (tubes, cathéters, etc.), et donc en présence d'un milieu aqueux, cela va sans doute se traduire par un relargage extrêmement lent (quelques jours), ce qui, associé à la composition a priori biocompatible de ces solides (fer, acides carboxyliques ) . rend ces solides hautement intéressants pour les applications recherchées. Une explication possible à ce comportement singulier pourrait être la suivante : les phases flexibles MIL-88 sont « fermées » après vidage des pores par chauffage et donc sans porosité accessible pour les gaz habituels (H2, CO2,
CH4, N2, etc.). Les mesures d'adsorption de N2 à 77 K ont montré précédemment la quasi-absence d'adsorption dans ces solides. Si le gaz NO est tout de même adsorbé en quantité importante, sans doute sur les centres métallique insaturés, c'est que l'interaction entre ce gaz et le fer est beaucoup forte qu'avec les autres types de molécules gazeuses, en tout cas suffisante pour « ouvrir » un peu le matériau. À ce stade, NO est rentré dans le solide chimisorbé sur le centre métallique mais ces derniers n'ayant que très peu ouverts leurs pores, vont laisser très difficilement l'eau diffuser dans les pores et donc cette dernière connaît les plus grandes difficultés à chasser le NO du solide. En milieu aqueux, lors de tests réels, la libération de NO va sans doute être conditionnée par 1 'hydrophobicité et la stabilité de ces phases flexibles. La possibilité de changer presque à souhait l'espaceur organique des phases MIL-88 va donc en théorie nous permettre de moduler la cinétique de relargage du NO en milieu physiologique.
La libération a été aussi étudiée dans des conditions plus proches de conditions réelles dans le plasma. Ainsi, le solide chargé en NO a été place dans 4mL d'un tampon phosphate (pH ~5.5) a 22ºC sous agitation. La figure 71 représente les profils - de relargage de NO sous pression de vapeur d'eau (courbe (a)) et dans le tampon phosphate (courbe (b)) de solide MIL-88A(Fe). La quantité de NO relarguée (N0rel en mmol. g-1, à gauche, et ppm NO, à droite) est exprimée en fonction du temps t (en heures).
La quantité libérée est beaucoup plus importante quand la libération se produit dans le PBS que sous flux de vapeur d'eau ; ce qui est raisonnable en considérant que le contact avec le milieu liquide est plus important (figure 71). De cette façon, bien l'eau bien les phosphates présents dans le milieu pourront déplacer le gaz adsorbé/coordonné a métal.
Il y a une forte libération de NO dans les premières 2 heures et après la libération est maintenue dans des concentrations biologiquement actives (>10 ppb) jusqu'à 20 heures. Ca peut être intéressant pour avoir un effet de choque dans un premier temps (anticoagulant par exemple) et le maintenir sur quelques heures.
Concernant le solide MIL-88B(Fe) , la quantité libérée est plus faible que celle libérée pour le MIL- 88A(Fe). La quantité de NO libéré est très faible (0,002 mmol.g-1), ca soit dans le flux gazeux ou dans le PBS (figure 58). La libération à des concentrations actives est très rapide ( 1 heure dans le PBS et 4 heures dans le flux gazeux).
La figure 72 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe
(a)) et dans le tampon phosphate (courbe (b) ) de solideMIL-88B(Fe). La quantité de NO relargué (NOrel en mmol . g-1, a gauche, et ppm NO, a droite) est exprimée en fonction du temps t (en heures).

Les conditions de synthèse microondes sont les suivantes : 270 mg (1 mmol ) de FeCl3.6H2O, 116 mg d'acide fumarique (1,0 mmol, Acros 99%) dispersés dans 30 mL de l'eau distillée, le tout laissé dans un corps en téflon 2 min à 100ºC avec une rampe de chauffage de 1 min (puissance 1600W). Le solide est récupéré par centrifugation à 10000 rpm pendant 10 min.
200 mg du produit sont suspendus dans 100 mL de l'eau distille pour échanger l'acide fumarique qui reste libre. Le solide hydraté est récupéré par centrifugation à 10000rpm pendant 10min.
La figure 73 représente le diffractogramme des rayons X du solide MIL-88A-nano obtenu par synthèse microondes .
La taille de particule monodisperse mesurée par diffusion de lumière est 120 nm.
Adsorption NO
50 mg de nanoparticules du MIL-88A-nano préalablement activés, sous vide a température ambiante (environ 20°C)pendant 5h et sous vide a 150ºC pendant la
15 heures, sont exposés à environ 2 bars de NO 99,5%, commercialisé par la société Air Liquide) pendant 30minutes (voir exemple 13).
La quantité de NO adsorbée, 2,5 mmol. g-1, est considérable et tout a fait comparable avec celle obtenu pour la même structure avec une taille de cristallite plus grande (exemple précédente, taille de particule ~ 5 microns).
La figure 74 représente les isothermes d'adsorption de NO à 298 K pour les carboxylates de fer MIL-88A(Fe)-nano activé à 150ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol. g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg).
Libération de NO
Le relargage de NO est ensuite testé sous pression de vapeur d'eau (11% d'humidité dans un gaz neutre ) .
La figure 75 représente les profils de relargage de NO sous pression de vapeur d'eau des solides MIL-88A(Fe)-nano (120 nm, courbe (b)) et MIL- 88A(Fe) (5 microns, courbe (a)) . La quantité de NO relargué (N0rel en mmol.g"1, à gauche, et ppm NO, à droite) est exprimée en fonction du temps t (en heures)
Dans les deux cas, matériaux nano et micrométrique, la quantité de NO relarguée est comparable (0,055 mmol.g-1, Figure 75). MIL-88A(Fe) micrométrique semble avoir une libération plus lente que MIL-88A( Fe) -nano dans les premières 10 heures. Cet effet est probablement dû à une distance caractéristique de diffusion du NO plus petite dans les nanoparticules de MIL88A par rapport au MIL88A micrométrique .
13.4. Solides MIL88B modifiés avec différents groupes fonctionnels
La structure cristalline flexible MIL-88B présente des isotypes par la modification de l'espaceur
organique avec différents groupes fonctionnels. Ainsi, ces groupes fonctionnels vont substituer un ou plusieurs hydrogènes de l'espaceur organique (acide téréphtalique) , en modulant ainsi l'hydrophobicité et la stabilité de ces phases flexibles, par conséquence l'adsorption et libération de gazes biologiques. De plus, les groupes accepteurs d'électrons pourront peut être de créer des nouvelles interactions avec les gazes biologiques (bases de Lewis). Les inventeurs ont testé l'adsorption et le relargage de NO à partir des phases flexibles du type MIL-88B, à base des ligands organiques : nitrotéréphthalate (MIL88B-NO2) et 2,5- dihydroxotéréphthalate (MIL88B-2OH) .
Adsorption de NO
Les deux matériaux ont montré une capacité d' adsorption similaire (1 mmol.g-1), diminuée en comparaison avec le solide MIL-88B non modifié. Cet effet peut être expliqué par l'élimination incomplète de l'eau coordonnée.
La figure 76 représente les isothermes d' adsorption de NO à 298 K pour les carboxylates de fer MIL-88B(Fe)-N02 activé à 150ºC sous vide pendant une nuit. La quantité de NO (N0aba en mmol.g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en" fonction de la pression P (en mmHg).
La figure 77 représente les isothermes d' adsorption de NO à 298 K pour les carboxylates de fer MIL-88B(Fe)-2OH activé à 80ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol.g-1) adsorbé
(courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mmHg) .
Libération de NO La fonctionnalisation avec de groupes nitro ou dihydroxo dans le solide du type MIL-88B, permet un important ralentissement de la libération de NO dans le milieu PBS.
Ainsi, on observe la libération du NO à des concentrations biologiquement actives (>10 ppm) jusqu'à 11 et 24 heures avec MIL-88B-2OH et MIL-88B-NO2 respectivement (figures 78 et 79), en comparaison avec le solide non modifié MIL-88B qui libère sa charge en 1 heure. De même, la quantité de NO libérée est aussi plus importante dans le MIL-88B-2OH et MIL-88B-NO2 (0,13 et 0,25 mmol.g"1, respectivement) par rapport avec le MIL-88B non modifié(0,01 mmol.g-1).
On observe que quand la libération du NO est menée à bien dans la solution PBS, la quantité libérée est beaucoup plus importante que sous conditions de flux gazeux (Tableau 15).
La figure 78 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe (a)) et dans le tampon phosphate.. ( courbe (b) ) de solide
MIL-88B(Fe)-NO2. La quantité de NO relargué (N0rel en mmol.g-1, à gauche, et en ppm NO, a droite) est exprimée en fonction du temps t (en heures).
La figure 79 représente les profils de relargage de NO sous pression de vapeur d'eau (courbe
(a)) et dans le tampon phosphate (courbe (b)) de solide MIL-88B(Fe)-2OH. La quantité de NO relargué (N0rel en πunol.g"1, à gauche, et en ppm NO, à droite) est exprimée en fonction du temps t (en heures).
Comparaison des MILs
On peut conclure au vu des résultats (Tableau 15 et Figure 80), que l'on peut moduler la capacité et la cinétique d'adsorption et de libération des carboxylates de fer en fonction de leur caractère rigide, flexible ou fonctionnalisé. Si le solide mésoporeux adsorbe et libère les plus grandes quantités de NO, les phases flexibles adsorbent moins mais libèrent avec des cinétiques très différentes en fonction du ligand choisi. Ainsi, les solides non modifiés libèrent très rapidement (<lh) une très faible quantité tandis que l'introduction d'une fonctionnalité permet non seulement d'augmenter la quantité libérée mais sur des temps caractéristiques beaucoup plus grands (11 et 24h). Cela vient sans doute de la plus grande facilité qu'a l'eau à diffuser dans ces phases à cause d'une ouverture de pores plus importantes et d'un caractère hydrophile des substituants (OH, NO2).
Tableau 15: Capacité et cinétique d'adsorption et de libération des carboxylates de fer en solution PBS et sous conditions de flux gazeux.

La figure 80 représente les profils de relargage de NO sous pression de vapeur d'eau des solides MIL-IOOFe (courbe (a)), MIL-88A (courbe (b) ) , MIL-88B (courbe (C)), MIL-88-2OH (courbe (d)) et MIL- 88B-NO2 (courbe (e)). La quantité de NO relargué (NOrel en mmol.g-1) est exprimée en fonction du temps t (en heures) .
Exemple 14 : adsorption et libération de NO à partir du diphosphonate de titane MIL-22 (Ti3θ2(H2θ)2(θ3P-(CH2)- PO3)2. (H2θ)2)
Le diphosphonate de titane MIL-22 a été obtenu suivant la méthode rapporté par C. Serre, G. Férey, Inorg. Chem. 1999, 38, 5370-5373 [60].
50 mg du MIL-22 préalablement activés, sous vide a 300ºC pendant 15 heures, sont exposés à environ 2 bars de NO 99,5%, commercialisé par la société Air Liquide) pendant 30 minutes (voir exemple 13).
La quantité de NO adsorbé théorique est de -4 mmol.g"1. Par contre, la capacité d'adsorption de NO expérimentale n'est que de 0,18 mmol.g"1. Cette différence peut être expliquée car les conditions d'activation sont insuffisantes pour éliminer la totalité de l'eau coordonnée. Ainsi, capacités plus importantes pourront être obtenues avec des conditionnes d'activation du solides tels que 500ºC sous vide pendant 16 heures.
La figure 81 représente les isothermes d'adsorption de NO à 298 K pour le solide MIL-22 activé à 350ºC sous vide pendant une nuit. La quantité de NO (N0ab3 en mmol.g-1) adsorbé (courbe (a)) et désorbé (courbe (b)) est représentée en fonction de la pression P (en mm Hg) .
La figure 82 représente les profils de relargage de NO par lee solide MIL-22 sous pression de vapeur d'eau. La quantité de NO relargué (N0rel en mmol.g"1 à gauche et en ppm NO à droite) est exprimée en fonction du temps t (en heures). . —
La libération partielle du NO (0.0016 mmol/g) est réalisé dans des concentrations biologiquement actives pendant 0,8 heures.
Exemple 15 : Mesure des isothermes d'adsorption de CO pour le solide MIL-IOO(Fe)
Les mesures d'adsorption de CO sont effectuées à 303K jusqu'à 2 bars dans un système de dosage de gazes mis au point au laboratoire et connecté à un appareil de type gravimétrique thermostaté (Rubotherm Prâzisionsmeβtechnik GmbH). L'échantillon du MIL- 100(Fe) (500mg) est préalablement activé à la température requise (100ºC ou 250ºC) sous vide (à 2xlO"3 mbar) pendant 12 ou 20 heures. Le gaz CO sec (Air Liquide, 99,9 %) est introduit dans le système jusqu'à ce que la pression désirée soit atteinte, et la prise de masse mesurée en fonction du temps jusqu'à stabilisation.
De cette façon, une isotherme d'adsorption est obtenue en incrémentant la pression et en relevant le gain de masse de l'échantillon à l'équilibre.
La figure 83 représente l'isotherme d'adsorption de CO (C0ada en mmol/g) à la température de 303 K en fonction de la pression P (en bar) pour le carboxylate de fer MIL-IOO(Fe) activé à 100ºC pendant une 12h (courbe 100ºC), 250ºC pendant 12h (courbe 205ºC(I)) et 250ºC pendant 2Oh (courbe 250°C(2)).
Le solide MIL-IOO(Fe) adsorbe de quantités considérables de CO à " température ambiante (0,4-1,3 mmol.g-1) et à base pression (jusqu'à 2 bars) (Figure 83). La capacité d'adsorption du CO dans le MIL-IOO(Fe) augmente drastiquement quand le fer(III) du composé MIL-IOO(Fe) est réduit partialement par activation a 250ºC sous vide primaire (12 et 20 heures à 250ºC). La
spectroscopie Infra-Rouge a montré que dans ces conditions, cela conduisait à la réduction d'environ 15-20 % du fer(III) en fer(II). Les métaux de transition de faibles degrés d'oxydation, tel que le fer (II), possèdent des électrons supplémentaires dans les orbitales d. Il est connu que le transfert électronique des orbitales d du métal vers les orbitales 2π* de molécules telles que NO et CO renforce la liaison métal—adsorbat (phénomène de rétro-donation) et stabilise ainsi les espèces coordinnées sur le centre métallique. La quantité de CO augmente considérablement avec l'introduction de fer(II) (1,3 mmol.g"1 à 2 bars au lieu de 0,4 mmol.g"1 pour le solide pur fer (III)), car celui-ci est susceptible d' interagir avec plus d'une molécule de CO par centre métallique.
Exemple 16 : Essais in vivo de toxicité des carboxylates de fer (III)
a) Carboxylates de fer testés
Les deux solides carboxylates de fer suivants (synthétisés selon les modes opératoires de l'exemple 1 ) sont respectivement testés : 3. MIL-88A(Fe) de composition Fe3O[O2C-C2H2-
CO2] 3 •OH • nH2O
4. MIL-88Btnano(Fe) de composition Fe3O[O2C-C6 (CH3) 4- CO2] 3 •OH • nH20
b) Tests de toxicité
L'étude de toxicité aiguë in vivo est effectuée sur des rats Wistar femelles de 4 semaines (125 g) en injectant par voie intraveineuse aux rats des doses croissantes (50, 100 et 200 mg/kg) de nanoparticules MIL-88A (de 210 nm) et MIL-88Bt (de 100 nm) suspendus dans 0,5 mL d'une solution de glucose 5%.
Les nanoparticules sont stables dans ce milieu.
Le temps de stabilité de ces suspensions est réduit à quelques minutes quand la concentration de particules est maximale (200 mg/kg, 25 mg/0,5mL). Pour cette raison, les prélèvements sont réalisés sous agitation douce des suspensions de nanoparticules. Il n'a pas été possible d'administrer des doses supérieures à 200 mg/kg, puisque le volume maximal injectable à des rats est 0,5 ml.
Les résultats sont prometteurs étant donné qu'aucun signe de toxicité majeur n'est observé après 7 jours d'essai. Les valeurs sériques de l'albumine, cholestérol et transaminases (ASAT/ALAT) ne présentent pas de variation significative après 7 jours d'essai et le poids des organes par rapport au poids corporel ne varie pas significativement (Tableau 16).
Tableau 16 : Paramètres sériques mesurés 7 jours après l'introduction intraveineuse des carboxylates de fer MIL-88A(Fe) et MIL-88Bt (Fe) .

Les coupes histologiques du foie sont observées par coloration de Proust (fer en bleu), et présentées en figure 84. Elles montrent une accumulation de fer dans le foie. Bien qu'il soit nécessaire d'effectuer
une étude plus approfondie sur les effets de ces solides dans l'organisme à long terme, ces résultats sont très prometteurs, et permettent d'envisager des applications biomédicales pour ces matériaux.
Des études de toxicité aiguë et subaiguë plus approfondies ont été réalisées.
Les animaux utilisés pour l'expérimentation sont des rats Wistar femelles de quatre semaines avec un poids de 161,36 ± 16,1 g.
Tous les essais ont été réalisés dans l'animalerie de l'Université de Pharmacie dans des conditions de température et humidité, et après 3 jours d'adaptation des animaux a l'animalerie (3 jours).
Pour les tests de toxicité aiguë, une injection intrajugulaire unique des matériaux MIL-88A (150 et 500 nm) , MIL-88Bt (50 et 140 nm) ou de glucose 5% (groupe témoin) est effectuée à 4 groupes (à respectivement 1 jour, 1 semaine, 1 mois et 3 mois) de 8 rats choisies au hasard et anesthésiés sous isoflurane.
L'évolution du poids des animaux ainsi que leur comportement ont été suivis.
Des prélèvements de sang dans la jugulaire sous anesthésie par isoflurane ont également été réalisés à différents temps : 1 et 3 jours, 1 et 2 semaines, 1, 2 et 3 mois . Le sérum a été isolé pour mesurer des paramètres sériques tels que IL-6 (interleukine 6), albumine, Fe sérique, PAS, GGT, bilirubine, cholestérol et transaminases.
Par ailleurs, chaque groupe d'animaux a été sacrifié après respectivement 1 jour, 1 semaine, 1 et 3
mois. Les animaux ont été anesthésiés à l'isoflurane puis la rate, les reins, le foie et le coeur ont été prélevés et conservés pour des études histologiques. Quatre fois ont également été utilisés pour réaliser une extraction microsomale afin de mesurer l'activation du cytochrome P450.
Pour les tests de toxicité subaiguë, une injection intrajugulaire par jour est effectuée pendant 4 jours consécutifs sur 26 rats répartis au hasard dans différents groupes où les animaux sont sacrifiés au bout de 5 ou 10 jours.
L'évolution du poids des animaux isolés ainsi que leur comportement alimentaire (mesure des quantités d'eau et de nourriture consommées) ont été suivis. Les urines et déjections ont également été récupérés.
Des prélèvements de sang dans la jugulaire ont également été effectués sur différents groupes de rat à
3 et 5 jours, 8 et 10 jours. Le sang subit le même traitement que pour l'essai de toxicité aiguë et le sérum obtenu est destiné aux mêmes analyses .
Les jours des sacrifices, aux 5 et 10 jours, les animaux sont anesthésiés à l'isoflurane puis la rate, les reins, le foie, le coeur et les poumons sont prélevés et traités de même que pour l'essai de toxicité aiguë.
c) Résultats
Evolution du poids des animaux : Les animaux ont été pesés tous les jours dans le but de comparer l'évolution du poids des différents
groupes. Une moyenne a été effectuée pour chaque jour et dans chacun de ces groupes.
Pour les tests de toxicité subaiguë, l'augmentation du poids observée avec le groupe du glucose est légèrement diminuée quand on administre le matériau. Cette variation est plus évidente quand la dose administrée est plus élevée
Les études en toxicité aiguë montrent que l'administration des matériaux MIL-88A et MIL-88Bt ne produit pas de variation significative du poids au cours du temps .
Evolution de la consommation d'eau et de nourriture :
En toxicité subaiguë, l'évolution est globalement similaire pour le groupe témoin et le groupe ayant reçu une injection de 25 mg/kg. Une différence plus marquée est observée dans le groupe ayant reçu la plus forte dose et se caractérise par une plus faible consommation de nourriture pendant l'étude. Cette observation est confirmée et en totale concordance avec les résultats obtenus par l'évolution du poids.
Comparaison du poids des organes prélevés : Résultats de toxicité subaiguë : il n'apparait pas de différence significative entre le poids de rate, reins et coeur des différents groupes. Le poids des poumons parait être légèrement augmenté à 5 jours comme à 10 jours. Toxicité aiguë : on observe une augmentation du poids de la rate jusqu'à une semaine après l'administration, et un retour a la normale à 1 et 3
mois pour le MIL-88A et MIL-88Bt, respectivement. Le poids du foie augmente de façon importante quand les matériaux sont injectés, ce qui traduit peut être l'accumulation du fer dans le foie. On observe que la situation revient à la normale pour le MIL-88A après 3 mois, mais pas pour le MIL-88Bt ou le poids reste élevé.
Dosage du cytochrome P450 dans les suspensions microsomales :
Le cytochrome P450 est un enzyme associé à la face interne du réticulum endoplasmique lisse qui est fortement impliqué dans la dégradation des molécules exogènes. Cet enzyme possède une très faible spécificité de substrat et est capable de catalyser la transformation de composés nouvellement synthétisés tels que les médicaments. La plupart des cytochromes P450 peuvent être induits ou réprimés, au niveau transcriptionnel, par différents xénobiotiques ; ceci est souvent la cause d'effets secondaires des médicaments. Le dosage de cet enzyme permet de déterminer si le matériau MOF utilisé est métabolisé par le cytochrome P450 auquel cas il activerait ou inhiberait son activité. La quantité de cytochrome n'est interprétable qu'à condition d'avoir été rapportée à la quantité totale de protéines contenue dans chaqμe échantillon..
Le dosage de protéines contenues dans l'échantillon a été effectué grâce à l'utilisation d'un kit BCA fourni par PIERCE (lot #HI106096). Cette méthode combine la réduction du Cu2+ en Cu+ par les protéines en milieu alcalin avec la détection colorimétrique, très sensible
et sélective, du cation Cu+ grâce à un réactif contenant l'acide Bicinchoninique (BCA).
Le rapport entre la concentration de cytochrome et la quantité de protéines totale donne l'activité du cytochrome exprimée en mol. g-1. Les résultats en toxicité aiguë montrent qu'il n'y a pas de grosse différence d'activité entre le groupe témoin négatif
(ayant reçu le glucose) et le groupe « MIL-88A » dont le matériau n'est pas métabolisé par le Cyp450. Le matériau MIL-88Bt ne semble pas non plus être métabolisé par le Cyp450.
Dosage de 1 'interleukine 6 dans le sérum :
L'interleukine 6 (IL-6) est une cytokine multifonctionnelle qui joue un rôle important dans la défense de l'hôte, les réponses immunitaires, les fonctions des cellules nerveuses et l ' hématopoïèse . Un niveau élevé d'IL-6 dans le sérum a par exemple été observé lors d'infections virales et bactériologiques, de trauma, de maladies auto-immunes, d'inflammation, ou de cancer.
Cette étude a pour de déterminer s ' il existe une réaction inflammatoire après l'administration des nanoparticules de carboxylates de fer. Ainsi, il est possible de voir si le niveau d'IL-6 est augmenté par rapport aux groupes témoins (injection de glucose, donc réaction inflammatoire locale par la piqûre)
Le dosage a été effectué par l'utilisation d'un kit « Quantikine, Rat IL-6 » fourni par les laboratoires R&D Systems.
Résultats de toxicité subaiguë : les variations ne sont pas significatives. Une augmentation du taux
plasmatique observé (activation de la production d'IL- 6) semble être dû phénomène de piqûre lors des injections qui produisent une inflammation locale, si on compare de façon isolée les différents groupes avec le groupe témoin ( glucose ) .
Résultats de toxicité aiguë : les variations ne sont pas significatives et conduisent aux mêmes conclusions que dans le cas de la toxicité subaiguë.
Dosage des paramètres sériques :
Tous les dosages ont été réalisés en utilisant des dispositifs automatiques. Quelques paramètres clés ont été déterminés pour évaluer les conséquences des injections de nanoparticules au niveau du foie, des taux de transaminases (Alanine amino transférase ou ALAT et aspartate amino transférase ou ASAT), phosphatases alcalines (PAS), γ-glutamate transférase (GGT), bilirubine, cholestérol, albumine et fer sérique . Les résultats montrent que les niveaux sériques de l'ALAT sont tout à fait normaux, tout comme les niveaux de bilirubine (< 2 μmol/L) et de γ-glutamique transférase (<2 Ul/L).
Les niveaux d'albumine sérique ont légèrement diminués après le premier jour d'injection pour les deux matériaux, en accord avec un processus inflammatoire local du à la piqûre et avec l'augmentation de la IL-6 observée précédemment. Après 3 jours, les niveaux reviennent à la normale. Les niveaux sériques de l'ASAT sont augmentés un jour après l'injection, ce qui peut indiquer un processus de cytolyse. Toutefois, 3 jours après
l'administration des nanoparticules, les valeurs retournent à la normale. De la même façon, la phosphatase alcaline se trouve augmentés après 1 jour, indiquant un processus de cytolyse, mais la situation revient à la normale après 3 jours. Le retour à la normale après 3 jours indique qu'il s'agit d'un processus de cytolyse transitoire et non permanent. Il n'y a donc pas de perte de fonction cellulaire. Les niveaux de cholestérol sont normaux. Les niveaux de fer sériques sont diminués en comparaison avec le groupe témoin, et cela est plus prononcé dans le groupe MIL-88A. Cela pourrait s'expliquer par une complexation du fer sérique par les nanoparticules. La situation revient à la normale 3 jours après l'administration.
Les paramètres sériques ont aussi été dosés à 1 semaine et d'après ces résultats, il n'y a plus de différence entre les 3 groupes au niveau du fer sérique ; les rats traités au MIL-88A et MIL-88Bt ont récupéré une concentration en fer sérique comparable à celle du groupe témoin. Par ailleurs, concernant les niveaux des autres paramètres sériques, il n'y a pas une différence significative en comparaison avec le groupe témoin.
Coupes histologisque : Les coupes histologiques, d'une épaisseur de
5 μm, sont effectuées dans un cryostat, déshydratées et colorées (coloration hématoxyline/éosine puis coloration au bleu Proust : coloration du fer en bleu) . Par l'observation des coupes histologiques, il est possible de déterminer la voie d'élimination des
composés du matériau ou leur stockage dans certains organes : foie, reins, rate et poumons, le coeur étant utilisé comme témoin.
Résultats de toxicité aiguë : les coupes histologigues de foie montrent une accumulation du fer dans le foie après l'injection des matériaux qui est plus importante pour le solide MIL-88A. Le matériau semble être sous sa forme de nanoparticules non dégradées. L'accumulation est moins importante pour le matériau MIL-88Bt, ce qui peut signifier une plus faible captation pour le foie ou la réutilisation plus rapide du fer stocké. Apres 1 et 3 mois le contenu en fer dans la rate et le foie revient à la normale.
Dosage du fer dans les suspensions injectées et dans les organes :
Le dosage du fer contenu dans les suspensions de MIL-88A et MIL-88Bt injectées aux animaux est effectué par spectrophotométrie UV-Visible à la longueur d'onde de 520 nm, par colorimétrie spécifique des ions ferreux avec la bipyridine (formation d'un complexe rouge), après solubilisation de l'oxyde de fer dans de l'acide sulfurique concentré, et réduction des ions ferriques en ions ferreux par l'acide ascorbique. Le dosage du fer dans les organes est réalisé de même que pour le dosage en fer des suspensions expliqué précédemment, après_ broyage... de l'organe à tester. Ce dosage permet de déterminer la voie d'élimination des composés du matériau ou leur stockage dans certains organes : foie, reins, rate et poumons, le coeur étant utilisé comme témoin.
d) Conclusion
Lors des essais de toxicité, l'observation minutieuse des animaux n'a révélé aucun signe apparent de nocivité du matériau injecté. En effet, les animaux ont gardé un comportement tout à fait normal. Pendant les études, les animaux ont bien grossi en comparaison avec le groupe témoin, même si pour l'étude de toxicité subaiguë l'augmentation du poids est moins importante que pour le groupe témoin, probablement lié a l'administration consécutive des doses importantes. La consommation d'eau reste elle globalement normale dans l'essai de toxicité subaiguë.
Un dosage du cytochrome P-450 a permis d'observer l'état de l'activité du cytochrome P-450 sur une longue période. Ce cytochrome est connu pour sa capacité à métaboliser certains xénobiotiques. L'étude montre que le taux d'activité, bien que fluctueux, reste en dessous des valeurs observées sur les rats témoins qui ont reçu une injection de phénobarbital, activateur de cytochrome P-450, ce qui indique que les matériaux ne sont pas métabolisés par voie Cyp450, en concordance avec la polarité importante des ligands dicarboxyliques . Les résultats sont très prometteurs et indiquent déjà que les matériaux MIL-88A et MIL-88Btn' induisent aucun signe de toxicité sévère, même si des études de toxicité complémentaires doivent être effectuées. Le devenir et les effets des nanoparticules dans l'organisme sont en cours d'étude afin de mettre en relation le bénéfice apporté par ces matériaux par la vectorisation de médicaments délicats à encapsuler
et présentant un grand potentiel thérapeutique. Des études similaires sont également en cours de réalisation avec d'autres nanovecteurs de structure et/ou de composition différentes.
Exemple 17 : Comparaison performances différents solides poreux pour l'adsorption et libération de NO
Les matériaux MOFs présentent nombreuses avantages par rapport aux solides poreux inorganiques, les zéolites. Ainsi, la grande stabilité des zéolites dans des milieux aqueux ne permet pas son élimination par l'organisme, en produisant le stockage du produit dans le corps. De plus, la plus part de zéolites ont de l'aluminium dans sa structure, élément avec une très importante toxicité.
De même, la composition d'autres MOFs, à base de métaux bien connus pour leur toxicité (cobalt, nickel, chrome ou cuivre) ne permet pas envisager des applications dans le domaine médical ou cosmétique de ces MOFs. Contrairement à ces MOFs, les solides de l'invention possèdent une composition a priori peu toxique (Fe, Ti). En effet, dans l'exemple 16, nous démontrons l'absence de toxicité de deux carboxylates de fer (fumarate de fer et tetramethylterephthalate de fer) par des tests de toxicité in vivo sur de rats.
De plus, avec les solides de l'invention il est possible de contrôler leur vitesse de dégradation selon les applications recherchées. Par exemple, le solide MIL-IOO(Fe) est stable sous des conditions hydrothermales, tandis que le solide MIL-IOl(Fe) est
dégradé rapidement en présence d'eau a température ambiante .
Les performances de nos MOFs ont été aussi comparés avec les MOFs rapportés dans la bibliographie (y compris dans la demande WO 2008/020218. Concernant leur capacité d'adsorption, les matériaux reportés dans cette invention ont montré des capacités toute a fait comparables a celles des autres MOF a base de métaux toxiques (4,5 mmol.g-1) (Figure 84) avec des libérations contrôlées dans le temps. De plus, nous avons obtenu le solide CPO-27 a base de fer(II) (2,5- dihydroxotéréphthalate de fer), de composition biocompatible, dont la capacité d'adsorption escomptée de NO sera proche de celle des analogues a base de Ni et/ou Co (6-7 mmol.g-1) avec une libération controllée sur une très longue durée (Figure 86).
La figure 85 représente représente les isothermes d'adsorption de NO à 298 K pour les solides CPO-27(dihydroxotéréphthalate de Co) (courbes (a) et (b)), CPO-27(2,5-dihydroxotéréphthalate de Ni ; M2 (dhtp) (H2O) .xH20 (M = Ni or Co dhtp = acide 2,5- dihydroxytéréphthalique, x-8)) (courbes (c) et (d)), MIL-100(trimésate de Fe) (courbes (e) et (f)), HKUST(trimésate de Cu) (courbes (g) et (h)), MIL- 53(téréphthalate de Al) (courbes (i) et (j)) et MIL- 53(téréphthalate de Cr) (courbes (k) et (I)). La quantité de NO (N0ab3 en mmol.g-1) adsorbé et désorbé est représentée en fonction de la pression P (en mbar ) . La figure 86 représente les profils de relargage de NO sous pression de vapeur d'eau du solide CPO-27(dihydroxotéréphthalate de Co) (courbe (a)), CPO-
27(2,5-dihydroxotéréphthalate de Ni ; M2 (dhtp) (H2O) .XH2O (M = Ni or Co dhtp = acide 2 ,5-dihydroxytéréphthalique acid, x-8)) (courbe (b)), HKUST-I (trimésate de Cu) (courbe (c)), MIL-53(téréphthalate de Al) (courbe (d)) et MIL-53(téréphthalate de Cr) (courbe (e)). La quantité de NO relarguée (N0rel en immol. g-1) est exprimée en fonction du temps t (en heures).
Exemple 18 : Formulation sous forme de crème comprenant un solide MOF selon l'invention
La crème utilisée est composée de 50% en poids de paraffine et 50% en poids de polyéthylène glycol (PEG), le deux sont mélanges à l'aide d'une pipette automatique pendant 30 secondes.
10% en poids du solide MIL-88A ou MIL-88A-nano chargé en NO sont ensuite mélangés de la même façon avec la crème.
Pour mesurer le relargage de NO par un échantillon sous forme de crème, de l'azote gazeux d'humidité connue (11% d'eau par passage du flux gazeux sur une solution aqueuse de LiCl) est passé sur le mélange de crème et de poudre ; le flux gazeux résultant est ensuite envoyé vers l'analyseur, et la quantité de NO (en ppm et ppb) enregistrée. Cette approche est valable pour des applications cutanées, où le solide est en contact avec la peau et la libération, du NO se réalise en présence de l'humidité de la peau.
La quantité de NO libérée par la crème est 6-8 fois plus petite que la forme poudre (Figures 87 et 88) car la paraffine qui est hydrophobe, ne permet pas le contact du solide avec le flux de vapeur d'eau.
La libération à des concentrations biologiquement actives est réalisée pendant 10 heures dans le cas du solide MIL-88A de taille micrométrique
(Figure 87). Par contre, on observe une libération très rapide (10 minutes ; Figure 88) lorsque la crème comprend du MIL-88A-nano. Ces résultats, en total concordance avec ceux obtenus avec la poudre, sont dues à une longueur de diffusion plus petite et aussi à un meilleur contact de la crème grâce à la surface plus importante de ces nanoparticules.
La figure 85 représente les profils de relargage de NO sous pression de vapeur d'eau du solide MIL-88A (3 échantillons sous les mêmes conditions) sous forme de crème. La quantité de NO relargué (NOrel en mmol.g-1) est exprimée en fonction du temps t (en heures ) .
La figure 86 représente les profils de relargage de NO sous pression de vapeur d'eau du solide
MIL-88A-nano sous forme de crème (courbe (b)) et sous forme de poudre (courbe (a)) en comparaison avec la libération dans une solution PBS (courbe (C)). La quantité de NO relarguée (N0rel en mmol.g-1) est exprimée en fonction du temps t (en heures).
LISTE DES REFERENCES
[I] Demande de brevet US 10/039,733 [2] Demande de brevet US 10/061,147 [3] Demande de brevet US 10/137,043
[4] Nitric oxide function in the skin, Cals-Grierson MM, Ormerod AD, NITRIC OXIDE-BIOLOGY AND CHEMISTRY 2004, 10(4), 179-193
Topically applied nitric oxide induces T- Lymphocyte infiltration in human skin, but minimal inflammation, Mowbray M, Tan XJ, Wheatley
PS , et al . , JOURNAL OF INVESTIGATIVE DERMATOLOGY
2008, 128(2), 352-360
[5] Effects of nitric oxide and oxidation in vivo and postmortem on méat tenderness, Warner RD, Dunshea FR, Ponnampalam EN, et al. MEAT SCIENCE Volume: 71 Issue: 1 Pages: 205-217, 2005
[6] L.K. Keefer Nature Materials, 2, 357(2003)
[7] P. G. Parzuchowski et al, J. Am. Chem. Soc. 124, 12182(2002)
[8] J.T. Mitchell-Koch et al, Angew Chemie, 43, 2806 (2004)
[9] A. D. Ormerod et al, J. Invest Dermatol 113, 392- 397(1999) [10] WO 2005/003032
[II] P. S. Wheatley et al, Stud Surf Sci Catal, 158, 2033-2040 (2005) ;
P. S. Wheatleyet al, J. Am. Chem. Soc, 128, 502- 509, (2006)
B. Xiao et al, J. Am. Chem. Soc, 129, 1203-1209 (2007)
[12] Pollution fighter turns clôt buster, New Scientist, 5th February (2005) [13] M. Mowbray et al, J. Invest Dermatol, 126, 102
(2006)
[14] C. T. Dziobkowski, T. J. Wrobleski, D. B. Brown, Inorg. Chem. 1982, 20, 671
[15] L. Anzalone et al., J. Org. Chem. 1985, 50, 2128 [16] S. Ameerunisha et al., J. Chem. Soc. Perkin Trans. 2 1995, 1679
[17] C. Serre et al., Angew. Chem. Int. Ed. 2004, 43, 6286
C. Serre et al., Chem. Comm. 2006, 284-286 [29] [18] Serre et al., Science, 2007, 315, 1828 [30]
[19] C. Serre et al. J. Am. Chem. Soc, 2005, 127, 16273-16278
[20] K. Byrapsa, M. Yoshimura, « Handbook of hydrothermal technology », Noyés Publications, Parkridge, New Jersey USA, William Andrew Publishing, LLC, Norwich NY USA, 2001
[21] G. Tompsett, W. C. Conner, K. S. Yngvesson, ChemPhysChem. 2006, 7, 296
[22] S. -E. Park, J. -S. Chang, Y. K. Hwang, D. S. Kim, S. H. Jhung, J. -S. Hwang, Catal. Survey Asia 2004, 8, 91
[23] C. S. Cundy, Collect. Czech. Chem. Commum. 1998, 63, 1699
[24] S. H. Jhung, J. -H. Lee, J. -S. Chang, Bull. Kor. Chem. Soc. 2005, 26, 880
[25] A. Pichon, Cryst. Eng. Comm. 8, 2006, 211-214 [26] D. Braga, Angew. Chem. Int. Ed. 45, 2006, 142-246 [27] D. Braga, Dalton Trans., 2006, 1249-1263.
[28] R. ALBERTO AND R. MOTTERLINI, DALTON TRANS, 2007, 1650
[29] Sabine Balthasar, Kerstin Michaelis, Norbert Dinauer, Hagen von Briesen, Jôrg Kreuter and Klaus Langer, "Préparation and characterisation of antibody modified gelatin nanoparticles as drug carrier System for uptake in lymphocytes", Biomaterials, 2005, 26, 15, 2723-2732.
[30] Ruxandra Gref, Patrick Couvreur, Gillian Barratt and Evgueni Mysiakine, "Surface-engineered nanoparticles for multiple ligand coupling", Biomaterials, 2003, 24, 24, 4529-4537.
[31] Keefer L. K., Nat. Mater. 2003, vol 2, 357.
[32] A. R. Butler and R. Nicholson, Life, Death and Nitric Oxide, The Royal Society of Chemistry, Cambridge, 2003; F. Murad, N. Engl. J. Med. , 'Nitric Oxide and Cyclic GMP in CeIl Signaling and Drug Development' 2006, 355, 2003.
[33] C. Napoli and L. J. Ignarro, 'NO-Releasing Drugs' Annu. Rev. Pharmacol. Toxicol. , 2003, 43, 97.
[34] T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. Green and R. Motterlini, 'Métal carbonyls : a new class of pharmaceuticâls?'
Angew. Chem., Int. Ed., 2003, 42, 3722; S. W.
Ryter, J. Alam and A. M. K. Choi, 'CO-metal interaction: vital signalling from a lethal gas' Physiol. Rev., 2006, 86, 583 ; J. Boczkowski, J.
J. Poderoso and R. Motterlini, 'CO-metal
interaction: vital signalling from a lethal gas' Trends Biochem. Sci., 2006, 31, 614 ; R. Motterlini, B. E. Mann and R. Foresti, 'Therapeutic applications of carbon-monoxide- releasing molécules' Expert Opin. Invest. Drugs, 2005, 14, 1305.
[35] L. E. Otterbein, F. H. Bach, J. Alam, M. Soares, H. T. Lu, M.Wysk, R. J. Davis, R. A. Flavell, A. M. K. Choi, Nat. Med. 2000,6, 422 - 428. [36] J. T. Chapman, L. E. Otterbein, J. A. Elias, A. M. K. Choi, Am. J.Physiol. Lung CeIl Mol. Physiol. 2001, 281, L209 - L215.
[37] K. Sato, J. Balla, L. Otterbein, R. N. Smith, S. Brouard, Y. Lin, E. Csizmadia, J. Sevigny, S. C. Robson, G. Vercellotti, A. M. Choi, F. H. Bach, M. P. Soares, J. Immunol. 2001, 166, 4185 — 4194K.
[38] M. Stupfel, G. Bouley, Ann. N. Y. Acad. Sci. 1970, 174, 342 - 368.
[39] F. Amersi, X. D. Shen, D. Anselmo, J. Melinek, S. Iyer, D. J. Southard, M. Katori, H. D. VoIk, R.W. Busuttil, R. Buelow,J. W. Kupiec-Weglinski, Hepatology 2002, 35, 815 - 823.
[40] L. Gunther, P. 0. Berberat, M. Haga, S. Brouard, R. N. Smith,M. P. Soares, F. H. Bach, E. Tobiasch, Diabètes 2002, 51, 994 -999. [41] N. Ozawa, N. Goda, N. Makino, T. Yamaguchi , Y.
Yoshimura,M. Suematsu, J. Clin. Invest. 2002, 109, 457 - 467.
[42] S. A. Bainbridge, A. E. Farley, B. E. McLaughlin, C H. Graham, G. S. Marks, K. Nakatsu, J. F. Brien, G. N. Smith, Placenta 2002, 23, 563 - 569.
[43] D. Morse, J. Sethi, A. M. K. Choi, Crit. Care Med. 2002, 30, S12 -S17.
[44] S. P. Gaine, G. Booth, L. Otterbein, N. A.
Flavahan, A. M. Choi, C. M. Weiner, J. Vase. Res. 1999, 36, 114 — 119; T. Morita, S. A Mitsialis,
H. Koike, Y. Liu, S. Kourembanas , J. Biol. Chem.
1997,272, 32 804 - 32809.
[45] R. Motterlini, A. Gonzales, R. Foresti, J. E. Clark, C. J. Green, R. M. Winslow, Cire. Res. 1998, 83, 568 - 577; I. A. Sairanut, R Foresti, J. E. Clark, D. J. Exon, M. J. Vesely, P. Sarathchandra,C. J. Green, R. Motterlini, Br. J. Pharmacol. 1998, 125, 1437 -1444.
[46] L. E. Otterbein, B. S. Zuckerbraun, M. Haga, F. Liu, R. Song, A.Usheva, C. Stachulak, N. Bodyak, R. N. Smith, E. Csizmadia, S.Tyagi, Y. Akamatsu, R. J. Flavell, T. R. Billiar, E. Tzeng, F. H. Bach, A. M. K. Choi, M. G. Soares, Nat. Med. 2003, 9, 183 - 190. [47] D. E. Baranano, S. Dore, C. D. Ferris, S. H. Snyder, Clin.Neurosci. Res. 2001, 1, 46 — 52) et de l'ovulation (W. Tschugguel, F. Stonek, Z. Zhegu, W. Dietrich, C. Schneeberger,T. Stimpfl, T. Waldhoer, W. Vycudilik, J. C. Huber, J. Clin. Endocrinol. Metab. 2001, 86, 3833 - 3839.
[48] M. McLean, M. Bowman, V. Clifton, R. Smith, A. B.
Grossman,J. Clin. Endocrinol. Metab. 2000, 85, 2345 - 2349.
[49] R. Wang, FASEB J., 2002, 16, 1792; R. Wang, Antioxid. Redox Signaling, 2003, 5, 493.
[50] W. M. Zhoa, K. N. Houk and L. P. Oison, EMBO J. , 2001, 20, 6008.
[51] Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. Ch. Kravtsov, R. Luebke, M. Eddaoudi, Angew. Chem. Int. Ed. 2007, 46, 3278-3283.
[52] Robert A. Moss, Hugo Morales-Rojas, Saketh Vijayaraghavan and Jingzhi Tian, J. Am. Chem. Soc, 2004, 126,(35), 10923 - 10936.
[53] B. Baleux et al. CR. Acad. Sciences Paris, série C, 274 (1972) pages 1617-1620.
[54] R. Gref et al. Cσlloids and Surfaces B: Biointerfaces, Volume 18, Issues 3-4, October 2000, pages 301-313.
[55] Kohler N. et al., J Am Chem Soc 2004; 126 ; 7206- 7211.
[56] Peggy Chan et al., Biomaterials, Volume 28, Issue 3, 2007, pp 540-549.
[57] J. H. Van Steenis et al., J Control Release 87 (2003), pp. 167-176. [58] A. Gabizon, H. Shmeeda, A. T. Horowitz and S. Zalipsky, Tumor cell targeting of liposome- entrapped drugs with phospholipids-anchored folie acid-PEG conjugates, Adv Drug Deliv Rev 56 (2004), pp. 1177-1192. [59] Ameerunisha et al., J. Chem. Soc. Perkin Trans. 2 1679-,--1995-.~
[60] C. Serre, G. Férey, Inorg. Chem. 1999, 38, 5370- 5373.
[61] P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom, H. Fjellvag, Chem. Commun., 2006, 956-961 et
P. D. C. Dietzel, R. E. Johnsen, R. Blom, H. Fjellvag, P.Chem. Eur. J., 2008, 14, 2389-2397.
Claims
1. Solide MOF cristallin poreux chargé en au moins un gaz base de Lewis choisi dans le groupe constitué de NO, CO et H2S dont au moins une partie se coordine avec M, ledit solide comprenant une succession tridimensionnelle de motifs répondant à la formule (I) suivante : 
dans laquelle :
• chaque occurrence de M représente indépendamment un ion d'un métal de transition Mz+ choisi dans le groupe constitué de Fe, Ti, Zr, Mn et dans lequel z est de 2 à 4 ou un mélange de ceux-ci;
• m est 1 à 12 ;
• k est 0 à 4 ;
• 1 est 0 à 18 ;
• p est 1 à 6 ; • X est un anion choisi dans le groupe constitué de OH", Cl", F", I", Br", SO4 2', NO3 ", ClO4 ", PF6 ", BF4 ", R-(COO)n " où R est tel que défini ci-dessous, R1-COO)n-, R1-SO3)n-, R1HPO3)n-, où R1 est un hydrogène, un alkyle en C1 à C12, linéaire ou ramifié, éventuellement substitué, un aryle, où n est un nombre entier de 1 à 4 ;
• L est un ligand espaceur constitué d'un radical R
comportant q groupements carboxylates  où
où
- q est 1, 2, 3, 4, 5 ou 6 ; * désigne le point d'attachement du carboxylate avec le radical R ; - # désigne les points d'attachement possibles xylate à l'ion métallique ;
- R représente :
(i) un radical  ,
,  ou C2_12alcyne ;
ou C2_12alcyne ;
(ii) un radical aryle mono- ou poly-cyclique, fusionné ou non, comprenant 6 à 50 atomes de carbone ;
(iii) un hétéroaryle mono- ou poly-cyclique, fusionné ou non, comprenant 1 à 50 atomes de carbone ;
(iv) un radical organique comprenant un élément métallique choisi dans le groupe comprenant le ferrocène, la porphyrine, la phthalocyanine; le radical R étant éventuellement substitué par un ou plusieurs groupes R2, indépendamment choisis dans le groupe comprenant 

 C
C  y
y  6 10 y
6 10 y  hétérocyclique;
hétérocyclique;  ;
;
C1_10alkylC3_10hétéroaryle ; F ; Cl ; Br; I;
-NO2 ; -CN ; -CF3 ; -CH2CF3 ; -OH ; -CH2OH ;
-CH2CH2OH ; -NH2 ; -CH2NH2 ; -NHCHO ; -COOH ; -CONH2 ; -SO3H ; -CH2SO2CH3 ; -PO3H2 ; ou une fonction —GRG1 dans laquelle G est —0-, -S-, -  -C(=0)-, -S(=0)-, -SO2- ,___ -C ( =0)O- ,
-C(=0)-, -S(=0)-, -SO2- ,___ -C ( =0)O- ,  C( O)NRG2 -0C(=0)-,
C( O)NRG2 -0C(=0)-,  , -0C(=0)0-,
, -0C(=0)0-,
-OC(=O)NRG2-, -NRG2C ( =0 ) 0- , 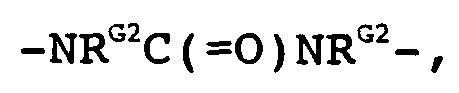 -C(=S)-, où chaque occurrence de
-C(=S)-, où chaque occurrence de  est indépendamment des autres occurrences de
est indépendamment des autres occurrences de  un atome d'hydrogène ; ou une fonction C1_12hétéroalkyle, C2_10alcène ou C2_10alcyne, linéaire, ramifiée ou cyclique, éventuellement substituée ; ou un groupe C6_10aryle, C3.10hétéroaryle, C5_10hétérocyclique,
un atome d'hydrogène ; ou une fonction C1_12hétéroalkyle, C2_10alcène ou C2_10alcyne, linéaire, ramifiée ou cyclique, éventuellement substituée ; ou un groupe C6_10aryle, C3.10hétéroaryle, C5_10hétérocyclique,  10aryle ou C1.10alkyleC3_10hétéroaryle dans lequel le radical aryle, hétéroaryle ou hétérocyclique est éventuellement substitué ; ou bien, lorsque G représente -NRG2-, RG1 et RG2 conjointement avec l'atome d'azote auquel ils sont liés forment un hétérocycle ou un hétéroaryle éventuellement substitué.
10aryle ou C1.10alkyleC3_10hétéroaryle dans lequel le radical aryle, hétéroaryle ou hétérocyclique est éventuellement substitué ; ou bien, lorsque G représente -NRG2-, RG1 et RG2 conjointement avec l'atome d'azote auquel ils sont liés forment un hétérocycle ou un hétéroaryle éventuellement substitué.
2. Solide selon la revendication 1, dans lequel le ligand L est un ligand di-, tri-, tétra- ou hexa- carboxylate choisi dans le groupe constitué de : fumarate, succinate, glutarate, muconate, adipate, 2,5- thiophènedicarboxylate, téréphtalate, 2,5-pyrazine dicarboxylate, naphtalène-2, 6-dicarboxylate, biphényle- 4,4' -dicarboxylate , azobenzènedicarboxylate , dichloroazobenzènedicarboxylate, azobenzènetétra- carboxylate, dihydroxoazobenzènedicarboxylate, benzène- 1 , 2 , 4-tricarboxylate , benzène-1 , 3 , 5-tricarboxylate , benzèήe-l,3,5-tribenzoate, l,3,5-tris[4'- carboxy( 1, 1 ' - biphenyl-4-yl )benzène , benzène-1 , 2,4,5- tétracarboxylate, naphtalène-2,3, 6,7-tétracarboxylate, naphtalène-1 , 4,5, 8-tétracarboxaeylate biphényl- te , 3,5,3',5' , 5 '-tétraca.rbcocylate, et les analogues modifiés choisis dans le groupe comprenant le 2- aminotéréphtalate, le 2-nitrotéréphtalate, le 2- méthyltéréphtalate, le 2-chlorotéréphtalate, le 2- bromotéréphtalate, le 2,5-dihydroxotéréphtalate, le tétrafluorotéréphtalate, le 2,5-dicarboxytéréphtalate, le diméthyl-4,4'-biphénydicarboxylate, le tétraméthyl- 4,4'-biphénydicarboxylate, le dicarboxy-4, 4 '- biphénydicarboxylate .
3. Solide selon l'une des revendications 1 ou 2, dans lequel l'anion X est choisi dans le groupe constitué de OH", Cl", F", R-(COO)n ", PF6 ", ClO4 ", avec R et n tel que défini dans la revendication 1.
4. Solide selon l'une quelconque des revendications 1 à 3, comprenant un pourcentage massique de M en phase sèche de 5 à 50%.
5. Solide selon l'une quelconque des revendications 1 à 4, dans lequel la taille de pores du matériau MOF est de 0,4 à 6 nm.
6. Solide selon l'une quelconque des revendications 1 à 5, dans lequel le solide a un volume poreux de 0 à 4 cmVg.
7. Solide selon l'une quelconque des revendications 1 à 6, dans lequel le solide a une capacité de charge en gaz de 0,5 à 50 mmol de gaz par gramme de solide sec.
8. Solide selon l'une quelconque des revendications_
1 à 7, dans lequel au moins 1 à 5 mmol de gaz par gramme de solide sec se coordine avec M.
9. Solide selon l'une quelconque des revendications 1 à 8, dans lequel ledit solide a une structure flexible qui se gonfle ou se contracte avec une amplitude allant de 10 à 300%.
10. Solide selon l'une quelconque des revendications 1 à 8 , dans lequel ledit solide a une structure rigide qui se gonfle ou se contracte avec une amplitude allant de 0 à 10%.
11. Solide selon la revendication 10, dans lequel le solide a un volume poreux de 0,5 à 4 cm3/g.
12. Solide selon l'une quelconque des revendications 1 à 11, dans lequel ledit solide comprend une succession tridimensionnelle de motifs répondant à la formule (I) choisis dans le groupe constitué de :
- Fe3OX[C2H2 ( CO2 )2]3 de structure flexible
- Fe3OX[C6H4 (CO2)]3 de structure flexible
- Fe3OX[C10H6(CO2)]3 de structure flexible - Fe3OX[C12H8(CO2)]3 de structure flexible
- Fe3OX[C4H4(CO2)J3 de structure flexible
- Fe12O(OH)18(H2O)3[C6H3(CO2)J6 de structure rigide
- Fe3OX[C6H3 ( CO2 )3]2 de structure rigide
- Fe3OX[C6H4 ( CO2 )J3 de structure rigide - Fe6O2X2[C10H2(CO2)J3 de structure rigide dans laquelle, X est tel que défini dans les revendications- 1 ou 2. ,—
13. Solide selon la revendication l'une quelconque des revendications 1 à 12, dans lequel le gaz est NO.
14. Solide selon l'une quelconque des revendications 1 à 13, comprenant à sa surface au moins un agent de surface organique.
15. Solide selon la revendication 14, dans lequel l'agent de surface organique est choisi dans le groupe comprenant
- un oligosaccharide comme les cyclodextrines,
- un polysaccharide comme le chitosan, le dextran, le fucoïdane, l'alginate, la pectine, l'amylose, l'amidon, la cellulose, le xylane, un glycosaminoglycanne comme l'acide hyaluronique, l'héparine,
- un polymère comme le polyéthylène glycol (PEG), l'alcool polyvinylique, le polyéthylène imine,
- un tensioactif comme le pluronic, la lécithine,
- des vitamines comme la biotine,
- des coenzymes comme l'acide lipoique,
- des anticorps ou fragments d'anticorps, - des aminoacides ou peptides.
16. Solide selon l'une des revendications 14 ou 15, dans lequel l ' agent de surface organique est une molécule de ciblage choisie dans le groupe comprenant : la biotine, le chitosan, l'acide lipoique, un anticorps ou fragment d'anticorps, un peptide.
17. Procédé de préparation d'un solide tel que défini dans l'une quelconque des revendications 1 à 16, comprenant au moins une étape réactionnelle consistant (i) à mélanger dans un solvant polaire : au moins une solution comprenant au moins un précurseur inorganique métallique se présentant sous la forme de métal M, d'un sel métallique de M ou d'un complexe de coordination comprenant un ion métallique de M
- au moins un ligand L' comprenant un radical R comportant q groupements  où
où
• q et R sont tels que définis précédemment,
• * désigne le point d'attachement du groupement avec le radical R,
• R3 est choisi dans le groupe comprenant un OH, un OY, avec Y est un cation alcalin, un halogène, un radical  où R4 et R4' sont des radicaux alkyles en
où R4 et R4' sont des radicaux alkyles en  pour obtenir un matériau MOF ;
pour obtenir un matériau MOF ;
(ii) à activer le matériau MOF obtenu en (i) ; et (ϋi) à mettre en contact le matériau MOF obtenu à l'étape (ii) avec un gaz base de Lewis dont au moins une partie se coordine avec M, de façon à obtenir ledit solide.
18. Procédé selon la revendication 17, dans lequel l'étape (ii) est également une étape de réduction des centres métalliques M dudit matériau MOF en ions Mz+ dans lequel z est de 2 à 4.
19. Procédé selon l'une des revendications 17 ou 18, dans lequel l'étape (ii) d'activation est réalisée à une température de 25 à 300ºC . 
20. Procédé selon l'une quelconque des revendications 17 à 19, dans lequel l'étape (ii) d'activation est réalisée à une pression de 1 à 10-2 Pa.
21. Procédé selon l'une quelconque des revendications 17 à 20, dans lequel, dans l'étape (iii) de mise en contact, le gaz est sous forme pure ou en mélange avec un gaz inerte.
5
22. Procédé de préparation selon l'une quelconque des revendications 17 à 21 comprenant, en outre, une étape (iv) de fixation d'au moins un agent de surface organique, laquelle étape intervenant pendant ou après
10 l'étape réactionnelle (i) ou après l'étape d'activation (ii) et avant l'étape (iii) de mise en contact du matériau MOF avec le gaz .
23. Procédé selon l'une quelconque des 15 revendications 17 à 21, dans lequel dans l'étape (iii) le matériau MOF obtenu à l'étape (ii) est mis en contact avec le NO.
24. Procédé selon la revendication 22, dans lequel 20 l'étape (iii) est réalisée à une température de -100ºC à +50ºC .
25. Procédé selon l'une des revendications 22 ou 23, dans lequel l'étape (iii) est réalisée à une
25 pression de 105 à 106 Pa.
26. Composition pharmaceutique, cosmétique ou dermatologique comprenant un solide selon l'une quelconque des revendications 1 à 16 et un véhicule 30 pharmaceutiquement ou cosmétiquement acceptable.
27. Article médical comprenant un solide MOF cristallin comme défini dans l'une quelconque des revendications 1 à 16.
28. Utilisation d'un solide selon l'une quelconque des revendications 1 à 16 pour le relargage d'un gaz base de Lewis dont au moins une partie se coordine avec
M.
29. Médicament comprenant un solide selon l'une quelconque des revendications 1 à 16.
30. Utilisation d'un solide selon l'une quelconque des revendications 1 à 16 comme médicament anti- thrombotique ou anti-microbien.
31. Utilisation d'un solide selon l'une quelconque des revendications 1 à 16 pour la fabrication d'un médicament ou d'une composition cosmétique.
32. Utilisation d'un solide selon l'une quelconque des revendications 1 à 16 dans le domaine alimentaire.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011502414A JP5907727B2 (ja) | 2008-04-01 | 2009-03-31 | 生物学的に関心のあるガスを吸着し、放出するための多孔性結晶ハイブリッド固体 |
| US12/935,642 US20110052650A1 (en) | 2008-04-01 | 2009-03-31 | Porous crystalline hybrid solid for adsorbing and releasing gas of biological interest |
| EP09738312A EP2260048B1 (fr) | 2008-04-01 | 2009-03-31 | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique |
| CA2720008A CA2720008C (fr) | 2008-04-01 | 2009-03-31 | Solide cristallin poreux constitues de reseau metal-organique pour l`adsorption et la liberation de gaz a interet biologique |
| AT09738312T ATE551348T1 (de) | 2008-04-01 | 2009-03-31 | Poröser und kristalliner hybridfestkörper zur aufnahme und freigabe eines gases von biologischem interesse |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0852150 | 2008-04-01 | ||
| FR0852150A FR2929278A1 (fr) | 2008-04-01 | 2008-04-01 | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique. |
| FR0803214A FR2929277B1 (fr) | 2008-04-01 | 2008-06-10 | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique. |
| FR0803214 | 2008-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009133278A1 true WO2009133278A1 (fr) | 2009-11-05 |
Family
ID=39926571
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2009/000381 Ceased WO2009133278A1 (fr) | 2008-04-01 | 2009-03-31 | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20110052650A1 (fr) |
| EP (1) | EP2260048B1 (fr) |
| JP (1) | JP5907727B2 (fr) |
| AT (1) | ATE551348T1 (fr) |
| CA (1) | CA2720008C (fr) |
| FR (2) | FR2929278A1 (fr) |
| WO (1) | WO2009133278A1 (fr) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010136677A1 (fr) * | 2009-05-28 | 2010-12-02 | Centre National De La Recherche Scientifique -Cnrs | Utilisation d'un solide hybride cristallin poreux comme catalyseur de reduction d'oxydes d'azote et dispositifs |
| WO2011057090A2 (fr) | 2009-11-06 | 2011-05-12 | Colgate-Palmolive Company | Composition et procédé d'administration |
| WO2015188941A1 (fr) | 2014-06-13 | 2015-12-17 | Lorenz Meinel | Système de libération de gaz thérapeutique |
| EP3254755A1 (fr) * | 2016-06-10 | 2017-12-13 | Centre National de la Recherche Scientifique - CNRS - | Matériau solide hybride inorganique-organique à base de titane à degré élevé de condensation, son procédé de préparation et ses utilisations |
| CN108893459A (zh) * | 2018-06-07 | 2018-11-27 | 宁夏大学 | 一种MOFs固载酶,其制备方法及应用 |
| CN109865141A (zh) * | 2019-03-27 | 2019-06-11 | 南京工业大学 | 一种通过压力合成抗癌药物/MOFs复合功能材料的方法 |
| US10329243B2 (en) | 2015-01-13 | 2019-06-25 | Xi'an Libang Pharmaceutical Co., Ltd | Biphenyl derivative and uses thereof |
| EP3808351A1 (fr) | 2019-10-14 | 2021-04-21 | Julius-Maximilians-Universität Würzburg | Système de libération de gaz thérapeutique à deux composants basé sur membrane pour administration orale |
| EP3878471A1 (fr) | 2020-03-13 | 2021-09-15 | Julius-Maximilians-Universitaet Wuerzburg | Système thérapeutique pour l'application topique, transdermique et transcutanée de monoxyde de carbone |
| US11206824B2 (en) | 2015-01-08 | 2021-12-28 | Julius-Maximilians-Universitaet Wuerzburg | Gas delivery device comprising a gas releasing molecule and a gas permeable membrane |
| CN120392091A (zh) * | 2025-06-27 | 2025-08-01 | 浙江大学 | 一种非侵入式便携的多模式可穿戴汗液传感器件 |
| WO2026020020A1 (fr) * | 2024-07-17 | 2026-01-22 | Massachusetts Institute Of Technology | Matériaux de stabilisation de micronutriments |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8779175B2 (en) | 2004-10-25 | 2014-07-15 | Synthonics, Inc. | Coordination complexes, pharmaceutical solutions comprising coordination complexes, and methods of treating patients |
| EP1871433B1 (fr) | 2005-03-24 | 2009-04-22 | NOLabs AB | Traitement cosmétique, dispositif pour application du traitment et méthode de production |
| FR2921660B1 (fr) | 2007-10-01 | 2015-09-25 | Centre Nat Rech Scient | Nanoparticules hybrides organiques inorganiques a base de carboxylates de fer. |
| FR2938539B1 (fr) * | 2008-11-18 | 2012-12-21 | Centre Nat Rech Scient | Procede de preparation d'azocarboxylates aromatiques d'aluminium poreux et cristallises de type "metal-organic framework" |
| US8236787B2 (en) * | 2008-12-09 | 2012-08-07 | Synthonics, Inc. | Frequency modulated drug delivery (FMDD) |
| WO2011069541A1 (fr) * | 2009-12-09 | 2011-06-16 | Technische Universität Bergakademie Freiberg | Procédé de production de matériaux structuraux organométalliques à base d'oxyde par intégration d'oxyde avec contrôle de la teneur en eau |
| KR101663184B1 (ko) * | 2009-12-15 | 2016-10-06 | 삼성전자주식회사 | 하이브리드 다공성 물질 및 그의 제조방법 |
| US8771756B2 (en) | 2009-12-28 | 2014-07-08 | Colorado State University Research Foundation | Biocompatible materials for medical devices |
| US10150792B2 (en) | 2010-11-08 | 2018-12-11 | Synthonics, Inc. | Bismuth-containing compounds, coordination polymers, methods for modulating pharmacokinetic properties of biologically active agents, and methods for treating patients |
| ES2804263T3 (es) | 2011-07-05 | 2021-02-05 | Novan Inc | Composiciones tópicas |
| WO2013006613A1 (fr) | 2011-07-05 | 2013-01-10 | Novan, Inc. | Procédés de fabrication de compositions topiques et appareil afférent |
| US9675923B2 (en) * | 2011-08-25 | 2017-06-13 | The Regents Of The University Of California | Gas separations with redox-active metal-organic frameworks |
| WO2013063354A1 (fr) | 2011-10-27 | 2013-05-02 | Novan, Inc. | Compositions pour bain libérant de l'oxyde nitrique et leurs procédés d'utilisation |
| CN102425010A (zh) * | 2011-11-28 | 2012-04-25 | 宁波大学 | 1,2,3,4-丁烷四羧酸锰铁电材料及其制备方法 |
| ES2861439T3 (es) | 2012-03-14 | 2021-10-06 | Novan Inc | Composiciones farmacéuticas que liberan óxido nítrico |
| FR2991325B1 (fr) * | 2012-05-31 | 2015-01-16 | Centre Nat Rech Scient | Solide hybride organique inorganique ameliore a surface externe modifiee |
| US20150306566A1 (en) * | 2012-11-30 | 2015-10-29 | Cedar Advanced Technology Group Ltd. | Sorption material for the sorption of gas molecules, in particular co2, in minimally invasive surgical procedures |
| US9855211B2 (en) | 2013-02-28 | 2018-01-02 | Novan, Inc. | Topical compositions and methods of using the same |
| US11813284B2 (en) | 2013-08-08 | 2023-11-14 | Novan, Inc. | Topical compositions and methods of using the same |
| ES2887110T3 (es) | 2014-01-16 | 2021-12-21 | Grace W R & Co | Medios para cromatografía de afinidad y dispositivos para cromatografía |
| PL3137209T3 (pl) * | 2014-05-02 | 2023-01-02 | W.R. Grace & Co. - Conn. | Funkcjonalizowany materiał nośnikowy i sposoby wytwarzania oraz stosowania funkcjonalizowanego materiału nośnikowego |
| EP3166593B1 (fr) | 2014-07-11 | 2020-05-20 | Novan, Inc. | Compositions antivirales topiques et méthodes d'utilisation de celles-ci |
| US10322082B2 (en) | 2014-07-11 | 2019-06-18 | Novan, Inc. | Topical antiviral compositions and methods of using the same |
| US10925689B2 (en) | 2014-07-14 | 2021-02-23 | Novan, Inc. | Nitric oxide releasing nail coating compositions, nitric oxide releasing nail coatings, and methods of using the same |
| EP3177262A4 (fr) | 2014-08-08 | 2018-04-18 | Novan Inc. | Émulsions topiques |
| CN105524117B (zh) * | 2014-09-28 | 2018-02-23 | 中国科学院大连化学物理研究所 | 一种超声雾化制备纳米有机金属框架物的方法 |
| KR102566292B1 (ko) | 2015-06-05 | 2023-08-10 | 더블유.알. 그레이스 앤드 캄파니-콘. | 흡착성 바이오프로세싱 정화제와 이의 제조 및 사용 방법 |
| WO2017019614A1 (fr) | 2015-07-28 | 2017-02-02 | Novan, Inc. | Associations et méthodes de traitement et/ou de prévention d'infections fongiques |
| US9808788B2 (en) | 2015-07-29 | 2017-11-07 | Panaceanano, Inc. | Method of using cyclodextrin-based metal organic frameworks |
| US9834803B2 (en) | 2015-08-31 | 2017-12-05 | Panaceanano, Inc. | Methods to isolate cyclodextrins |
| US20170079296A1 (en) * | 2015-09-22 | 2017-03-23 | The United States Of America, As Represented By The Secretary Of Agriculture | Methods For Treating Plants Or Fruits |
| WO2017087501A1 (fr) | 2015-11-17 | 2017-05-26 | Panaceanano, Inc. | Réseaux organométalliques à base de cyclodextrine contenant un parfum |
| US10736967B2 (en) | 2016-01-05 | 2020-08-11 | Panaceanano, Inc. | Method of preparing cyclodextrin complexes |
| US10912743B2 (en) | 2016-03-02 | 2021-02-09 | Novan, Inc. | Compositions for treating inflammation and methods of treating the same |
| CN108883194A (zh) * | 2016-03-24 | 2018-11-23 | 帕那刻亚纳诺有限公司 | 含有环糊精类金属有机框架的组合物 |
| US11166980B2 (en) | 2016-04-13 | 2021-11-09 | Novan, Inc. | Compositions, systems, kits, and methods for treating an infection |
| EP3357929B1 (fr) * | 2017-02-02 | 2020-06-17 | Centre National de la Recherche Scientifique | Procédé de préparation de mof-carboxylate-nanoparticules à basse température |
| JP7229654B2 (ja) * | 2017-03-16 | 2023-02-28 | 日本製鉄株式会社 | 多孔性高分子金属錯体の賦形体 |
| US11883807B2 (en) | 2017-04-11 | 2024-01-30 | Colorado State University Research Foundation | Functionalization of metal-organic frameworks |
| JPWO2018225784A1 (ja) * | 2017-06-06 | 2020-04-09 | 株式会社Atomis | ワクチン組成物及びアジュバント |
| WO2018236803A1 (fr) | 2017-06-19 | 2018-12-27 | Novan, Inc. | Compositions topiques et leurs procédés d'utilisation |
| CN107266767A (zh) * | 2017-06-26 | 2017-10-20 | 台山长江塑料制品有限公司 | 一种可降解塑料及其制备方法 |
| WO2019014348A1 (fr) | 2017-07-11 | 2019-01-17 | Colorado State University Research Foundation | Matériau composite chitosane et structure métallo-organique |
| CN107602872B (zh) * | 2017-09-08 | 2020-06-19 | 聊城大学 | 一种发光多孔金属有机框架新材料的制备及其对痕量硝基爆炸物的检测方法 |
| KR102050597B1 (ko) | 2018-01-02 | 2019-12-03 | 재단법인대구경북과학기술원 | 이온성 고분자 내의 확산 조절을 통한 금속-유기 구조체의 인-시츄 제조방법 |
| WO2019135565A1 (fr) * | 2018-01-02 | 2019-07-11 | 재단법인대구경북과학기술원 | Procédé pour la préparation, in situ, d'un réseau métallo-organique à l'aide du réglage de la diffusion au sein d'un polymère ionique |
| WO2019169221A1 (fr) | 2018-03-01 | 2019-09-06 | Novan, Inc. | Suppositoires libérant de l'oxyde nitrique et leurs procédés d'utilisation |
| KR102831159B1 (ko) | 2018-04-16 | 2025-07-07 | 온퀄리티 파마슈티컬스 차이나 리미티드 | 암 치료의 부작용을 예방하거나 치료하기 위한 방법 |
| CN109201009B (zh) * | 2018-11-22 | 2021-10-29 | 天津工业大学 | 负载偶氮的光敏铬金属有机骨架多孔材料的制备和应用 |
| US20220158173A1 (en) * | 2019-03-07 | 2022-05-19 | Cornell University | Mof-sulfur materials and composite materials, methods of making same, and uses thereof |
| US12343419B2 (en) * | 2019-03-29 | 2025-07-01 | Framergy Inc. | Topical composition |
| CN111229171B (zh) * | 2020-01-19 | 2022-05-10 | 武汉工程大学 | 一种秸秆负载mof材料吸附剂及其制备方法和应用 |
| CN114073922B (zh) * | 2020-08-20 | 2024-02-20 | 中国科学院苏州纳米技术与纳米仿生研究所 | 卟啉基金属-有机骨架纳米球及其制备方法与应用 |
| CN112473629A (zh) * | 2020-11-10 | 2021-03-12 | 武汉理工大学 | 一种负载金属有机框架的柔性材料及其制备方法和应用 |
| FR3119104B1 (fr) * | 2021-01-28 | 2024-02-02 | Commissariat Energie Atomique | Procédé de formation d’un réseau métallo-organique |
| CN113004531B (zh) * | 2021-02-18 | 2022-09-16 | 上海健康医学院 | 一种铜金属有机框架材料及其制备与应用 |
| CN113040169B (zh) * | 2021-03-09 | 2021-11-23 | 泉州师范学院 | 碳掺杂MoS2/CoP/C复合抗菌材料及其制备方法与应用 |
| CN113797896B (zh) * | 2021-10-09 | 2023-11-24 | 湖北中烟工业有限责任公司 | 一种金属有机框架吸附材料的制备方法及其所得到的吸附材料 |
| CN114426676B (zh) * | 2021-12-20 | 2023-03-24 | 南京师范大学 | 一种磁性铁基mof微生物载体材料及其制备方法 |
| CN114487083B (zh) * | 2022-01-19 | 2024-05-07 | 中国地质大学(北京) | 一种磁性羟基纳米材料Fe3O4@COFs及其在磺胺质谱检测领域的应用 |
| CN115191479A (zh) * | 2022-07-15 | 2022-10-18 | 山东农业大学 | 一种水果保鲜剂及其制备方法与应用 |
| CN116273188B (zh) * | 2023-03-01 | 2024-06-25 | 岭南师范学院 | 一种负载金属的多孔有机框架材料在催化Pictet-Spengler反应中的应用 |
| CN117264229B (zh) * | 2023-09-19 | 2025-09-12 | 浙江大学杭州国际科创中心 | 金属有机框架材料及其制备方法和在空气集水中的应用 |
| EP4578543A1 (fr) * | 2023-12-29 | 2025-07-02 | Immaterial Ltd | Corps à structure organométallique |
| CN118186454A (zh) * | 2024-03-25 | 2024-06-14 | 河南省科学院激光制造研究所 | 不同尺寸mil-101纳米颗粒的制备方法 |
| CN118955938B (zh) * | 2024-10-16 | 2024-12-31 | 合肥工业大学 | 具有pH和湿度双重响应的硫辛酸@Cu-MOF材料及其制法与应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008020218A1 (fr) * | 2006-08-17 | 2008-02-21 | The University Court Of The University Of St Andrews | Adsorption et libération d'oxyde nitrique dans des milieux organométalliques |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7678390B2 (en) * | 1999-04-01 | 2010-03-16 | Yale University | Carbon monoxide as a biomarker and therapeutic agent |
| ATE334992T1 (de) * | 2001-04-30 | 2006-08-15 | Univ Michigan | Isoretikuläre organometallische grundstrukturen, verfahren zu deren bildung und systematische entwicklung von deren porengrösse und funktionalität, mit anwendung für die gasspeicherung |
| US20030078311A1 (en) * | 2001-10-19 | 2003-04-24 | Ulrich Muller | Process for the alkoxylation of organic compounds in the presence of novel framework materials |
| US6929679B2 (en) * | 2002-02-01 | 2005-08-16 | Basf Aktiengesellschaft | Method of storing, uptaking, releasing of gases by novel framework materials |
| WO2005062110A1 (fr) * | 2003-12-22 | 2005-07-07 | Lg Chem, Ltd. | Matiere electrochromique a duree de vie prolongee |
-
2008
- 2008-04-01 FR FR0852150A patent/FR2929278A1/fr active Pending
- 2008-06-10 FR FR0803214A patent/FR2929277B1/fr active Active
-
2009
- 2009-03-31 WO PCT/FR2009/000381 patent/WO2009133278A1/fr not_active Ceased
- 2009-03-31 EP EP09738312A patent/EP2260048B1/fr active Active
- 2009-03-31 AT AT09738312T patent/ATE551348T1/de active
- 2009-03-31 US US12/935,642 patent/US20110052650A1/en not_active Abandoned
- 2009-03-31 CA CA2720008A patent/CA2720008C/fr active Active
- 2009-03-31 JP JP2011502414A patent/JP5907727B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008020218A1 (fr) * | 2006-08-17 | 2008-02-21 | The University Court Of The University Of St Andrews | Adsorption et libération d'oxyde nitrique dans des milieux organométalliques |
Non-Patent Citations (5)
| Title |
|---|
| HORCAJADA PATRICIA ET AL: "Metal-organic frameworks as efficient materials for drug delivery", ANGEWANDTE CHEMIE. INTERNATIONAL EDITION, vol. 45, no. 36, 1 January 2006 (2006-01-01), pages 5974 - 5978, XP002476583, ISSN: 1433-7851 * |
| HORCAJADA PATRICIA ET AL: "Synthesis and catalytic properties of MIL - 100 ( Fe ), an iron(III) carboxylate with large pores", CHEMICAL COMMUNICATIONS - CHEMCOM, no. 27, 1 January 2007 (2007-01-01), pages 2820 - 2822, XP002476454, ISSN: 1359-7345, [retrieved on 20070515] * |
| LLEWELLYN PHILIP L ET AL: "High Uptakes of CO2 and CH4 in Mesoporous Metal-Organic Frameworks MIL-100 and MIL-101", LANGMUIR, 21 March 2008 (2008-03-21), pages A - F, XP002476457, ISSN: 0743-7463, [retrieved on 20080321] * |
| MELLOT-DRAZNIEKS CAROLINE ET AL: "Very Large Swelling in Hybrid Frameworks: A Combined Computational and Powder Diffraction Study", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 127, no. 46, 1 January 2005 (2005-01-01), pages 16273 - 16278, XP002476453, ISSN: 0002-7863, [retrieved on 20051028] * |
| XIAO BO ET AL: "High-capacity hydrogen and nitric oxide adsorption and storage in a metal-organic framework", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 129, no. 5, 7 February 2007 (2007-02-07), pages 1203 - 1209, XP009093205, ISSN: 0002-7863 * |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2945966A1 (fr) * | 2009-05-28 | 2010-12-03 | Centre Nat Rech Scient | Utilisation d'un solide hybride cristallin poreux comme catalyseur de reduction d'oxydes d'azote et dispositifs |
| WO2010136677A1 (fr) * | 2009-05-28 | 2010-12-02 | Centre National De La Recherche Scientifique -Cnrs | Utilisation d'un solide hybride cristallin poreux comme catalyseur de reduction d'oxydes d'azote et dispositifs |
| WO2011057090A2 (fr) | 2009-11-06 | 2011-05-12 | Colgate-Palmolive Company | Composition et procédé d'administration |
| WO2011057090A3 (fr) * | 2009-11-06 | 2012-09-27 | Colgate-Palmolive Company | Composition et procédé d'administration |
| AU2010315033B2 (en) * | 2009-11-06 | 2013-03-21 | Colgate-Palmolive Company | Composition and method of delivery |
| US8637444B2 (en) | 2009-11-06 | 2014-01-28 | Colgate-Palmolive Company | Compositions and method of delivery |
| US9375394B2 (en) | 2009-11-06 | 2016-06-28 | Colgate-Palmolive Company | Composition and method of delivery |
| WO2015188941A1 (fr) | 2014-06-13 | 2015-12-17 | Lorenz Meinel | Système de libération de gaz thérapeutique |
| DE102014008685A1 (de) | 2014-06-13 | 2015-12-17 | Lorenz Meinel | Freisetzungssystem für therapeutisches Gas |
| US11206824B2 (en) | 2015-01-08 | 2021-12-28 | Julius-Maximilians-Universitaet Wuerzburg | Gas delivery device comprising a gas releasing molecule and a gas permeable membrane |
| US10329243B2 (en) | 2015-01-13 | 2019-06-25 | Xi'an Libang Pharmaceutical Co., Ltd | Biphenyl derivative and uses thereof |
| WO2017211923A1 (fr) * | 2016-06-10 | 2017-12-14 | Centre National De La Recherche Scientifique | Matériau mof solide hybridé inorganique-organique à base de titane, avec un degré élevé de condensation cristalline , son procédé de préparation et ses utilisations |
| US10941166B2 (en) | 2016-06-10 | 2021-03-09 | Centre National De La Recherche Scientifique | Crystalline high degree of condensation titanium-based inorganic-organic hybrid solid MOF material, method for preparing same and uses thereof |
| EP3254755A1 (fr) * | 2016-06-10 | 2017-12-13 | Centre National de la Recherche Scientifique - CNRS - | Matériau solide hybride inorganique-organique à base de titane à degré élevé de condensation, son procédé de préparation et ses utilisations |
| CN108893459A (zh) * | 2018-06-07 | 2018-11-27 | 宁夏大学 | 一种MOFs固载酶,其制备方法及应用 |
| CN109865141A (zh) * | 2019-03-27 | 2019-06-11 | 南京工业大学 | 一种通过压力合成抗癌药物/MOFs复合功能材料的方法 |
| CN109865141B (zh) * | 2019-03-27 | 2022-02-18 | 南京工业大学 | 一种通过压力合成抗癌药物/MOFs复合功能材料的方法 |
| EP3808351A1 (fr) | 2019-10-14 | 2021-04-21 | Julius-Maximilians-Universität Würzburg | Système de libération de gaz thérapeutique à deux composants basé sur membrane pour administration orale |
| EP3878471A1 (fr) | 2020-03-13 | 2021-09-15 | Julius-Maximilians-Universitaet Wuerzburg | Système thérapeutique pour l'application topique, transdermique et transcutanée de monoxyde de carbone |
| WO2021180908A1 (fr) | 2020-03-13 | 2021-09-16 | Julius-Maximilians-Universitaet Wuerzburg | Système thérapeutique pour l'application topique, transdermique et transcutanée de monoxyde de carbone |
| WO2026020020A1 (fr) * | 2024-07-17 | 2026-01-22 | Massachusetts Institute Of Technology | Matériaux de stabilisation de micronutriments |
| CN120392091A (zh) * | 2025-06-27 | 2025-08-01 | 浙江大学 | 一种非侵入式便携的多模式可穿戴汗液传感器件 |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2929277A1 (fr) | 2009-10-02 |
| CA2720008C (fr) | 2017-02-14 |
| FR2929278A1 (fr) | 2009-10-02 |
| ATE551348T1 (de) | 2012-04-15 |
| EP2260048A1 (fr) | 2010-12-15 |
| JP5907727B2 (ja) | 2016-04-26 |
| EP2260048B1 (fr) | 2012-03-28 |
| US20110052650A1 (en) | 2011-03-03 |
| FR2929277B1 (fr) | 2019-12-20 |
| CA2720008A1 (fr) | 2009-11-05 |
| JP2011523624A (ja) | 2011-08-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2260048B1 (fr) | Solide hybride cristallin poreux pour l'adsorption et la liberation de gaz a interet biologique | |
| EP2193137B1 (fr) | Nanoparticules hybrides organiques inorganiques a base de carboxylates de fer | |
| EP2193136B1 (fr) | Solide hybride organique inorganique a surface modifiee | |
| CA2873379C (fr) | Solide de reseau metal-organique (mof) cristallin poreux, a surface externe modifiee et son procede de preparation | |
| Keskin et al. | Biomedical applications of metal organic frameworks | |
| Xing et al. | Recent advances in metal–organic frameworks for stimuli-responsive drug delivery | |
| US20210277042A1 (en) | Low temperature process for the synthesis of mof carboxylate nanoparticles | |
| EP3006474B1 (fr) | Solide poreux ayant une surface externe greffée avec un polymère | |
| Vahed et al. | (Fe) MIL-100-Met@ alginate: a hybrid polymer–MOF for enhancement of metformin's bioavailability and pH-controlled release | |
| CN114939178B (zh) | 氨基酸/多肽配位聚合物及其制备方法和应用 | |
| Liu et al. | Post-therapy via integrated curcumin and doxorubicin modified cerium-based UiO-66 MOFs using an antioxidant and anticancer therapeutic strategy | |
| Christodoulou | A comprehensive study of the erosion mechanism of porous hybrid particles for biomedical applications | |
| Lian | Design and Synthesis of Enzyme-MOF (Metal-Organic Framework) Composites for Long-Persistent Biomedical Applications | |
| Chen | Hemoglobin nanoparticles equipped with antioxidant coatings as a novel type of therapeutic oxygen carriers | |
| CN118903450A (zh) | 一种五肽基超分子组装颗粒及其作为药物递送载体的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09738312 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2720008 Country of ref document: CA Ref document number: 2009738312 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011502414 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |