WO2009138416A1 - Oxime carbamoyl derivatives as modulators of fatty acid amides hydrolase - Google Patents
Oxime carbamoyl derivatives as modulators of fatty acid amides hydrolase Download PDFInfo
- Publication number
- WO2009138416A1 WO2009138416A1 PCT/EP2009/055748 EP2009055748W WO2009138416A1 WO 2009138416 A1 WO2009138416 A1 WO 2009138416A1 EP 2009055748 W EP2009055748 W EP 2009055748W WO 2009138416 A1 WO2009138416 A1 WO 2009138416A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ethane
- phenyl
- biphenyl
- phenylaminocarbonyloxyimino
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- RMLMNEUQZXHYOC-KGENOOAVSA-N C/C(/c(cc1)ccc1-c1ccc[s]1)=N\OC(Nc1ccccc1)=O Chemical compound C/C(/c(cc1)ccc1-c1ccc[s]1)=N\OC(Nc1ccccc1)=O RMLMNEUQZXHYOC-KGENOOAVSA-N 0.000 description 1
- 0 CC*N(*)C(C)=O Chemical compound CC*N(*)C(C)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/60—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups having oxygen atoms of carbamate groups bound to nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/32—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D207/323—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms with only hydrogen atoms or radicals containing only hydrogen and carbon atoms directly attached to the ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/22—Radicals substituted by doubly bound hetero atoms, or by two hetero atoms other than halogen singly bound to the same carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/36—Nitrogen atoms
Definitions
- the present invention relates to new oxime carbamoyl derivatives, processes for their preparation, and to pharmaceutical compositions containing them for the treatment of neurological disorders, such as neuropathic pain and anxiety.
- Anandamide and other fatty acid amides are known to be chemical messengers that modulate a number of physiological processes (Hanus L.O., Chem. Biodivers., 2007, 4, 1828). Anandamide activates through binding both the central-type (CBl) and peripheral type (CB2) cannabinoid receptors (Devane W. A., et al, Science, 1992, 258, 1946-1949). Anandamide has been reported to be implicated in the modulation of nociception, feeding, emesis, anxiety, cell proliferation, inflammation, and memory (Labar G., et al, Chem. Biodivers., 2007, 4, 1882).
- FAAH fatty-acid-amide- hydrolase
- FAAH is also responsible of the catabolism of many other lipid signaling fatty acid amides (i.e. oleamide, iV-oleoylethanolamine, arachidonylglycerol and palmitoylethanolamide).
- oleamide i.e. oleamide, iV-oleoylethanolamine, arachidonylglycerol and palmitoylethanolamide.
- Modulating the activity of the endocannabinoid system by restoring the levels of endogenous signaling lipids turned out to hold therapeutic promise in a wide range of disparate diseases and pathological conditions such as diseases of energy metabolism (cachexia and anorexia), pain and inflammation, central nervous system disorders (stroke, multiple sclerosis, Parkinson's disease, Huntington disease, Alzheimer disease, epilepsy, schizophrenia, anxiety, depression and insomnia), cardiovascular and respiratory disorders (hypertension, circulatory shock, myocardial reperfusion injury, atherosclerosis and asthma), retinopathy, cancer, gastrointestinal
- FAAH A KO mice cannot metabolize anandamide and, though fertile and generally normal, show signs of enhanced anandamide and related fatty acid amides activity at cannabinoid receptors, such as reduced pain sensation (Cravatt B.F., et al, Proc. Natl. Acad. ScL, 2001, 98, 9371). This suggests the possibility that drugs targeting FAAH may heighten the tonic action of anandamide, while possibly avoiding the multiple, often unwanted effects produced by ⁇ 9 -THC and other direct-acting cannabinoid agonists (Hall W., et al., Lancet, 1998, 352, 1611; Chaperon, F., et al., Crit. Rev. Neurobiol, 1999, 13, 243).
- Eight oxime derivatives are specifically described in this patent application. The same eight oximes derivatives are also present in WO2003051841. Such oximes are not part of the present invention.
- the invention provides novel compounds for inhibiting Fatty Acid Amide Hydrolase (FAAH), compositions that include such compounds as well as methods of treating diseases of energy metabolism, pain and inflammation, central nervous system disorders, cardiovascular and respiratory disorders, retinopathy, cancer, gastrointestinal and liver disorders and musculoskeletal disorders by administering FAAH inhibitors to a patient.
- Fatty Acid Amide Hydrolase FAAH
- compositions that include such compounds as well as methods of treating diseases of energy metabolism, pain and inflammation, central nervous system disorders, cardiovascular and respiratory disorders, retinopathy, cancer, gastrointestinal and liver disorders and musculoskeletal disorders by administering FAAH inhibitors to a patient.
- the invention comprises compounds of general formula I
- R 1 and R 2 are independently H, alkyl, aryl, arylkyl, alkoxyaryl or haloaryl; or R 1 and R 2 taken together with the nitrogen atom to which they are attached form a heterocycle; G is phenylene or thienylene;
- A is phenyl, thienyl, cyclohexyl, pyrrolyl or pyridyl, each of them being optionally substituted once or twice with aminocarbonyl or alkyl; its tautomers, its geometrical isomers, its optically active forms such as enantiomers, diastereomers and its racemate forms, as well as pharmaceutically acceptable salts thereof; with the proviso that when A is cyclohexyl, R 1 cannot be 4-Cl phenyl and 3,4- dichlorophenyl if R 2 is H.
- An embodiment of this invention is that of compounds of formula I, for use as medicaments.
- said medicament is used for treating a neurological disorder, diseases of energy metabolism, cardiovascular and respiratory disorders, gastrointestinal and liver disorders, retinopathy, cancer and musculoskeletal disorders.
- said medicament is used for treating a neurological disorder.
- said medicament is used for treating anxiety and pain.
- alkyl refers to linear or branched alkyl groups having preferably from 1 to about 12 carbon atoms.
- Lower alkyl group is exemplified by groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl, iso-pentyl, neo-pentyl, n-hexyl and the like.
- alkoxy refers to a group -OR where R includes lower alkyl, "C3-C10 cycloalkyl” and “heterocycloalkyl”.
- heterocycloalkyl and/or heterocycle refer to a saturated five- or six-membered ring containing one or two nitrogen, oxygen or sulfur atoms.
- Preferred heterocycloalkyl include pyrrolidine, piperidine, piperazine, morpholine, thiomorpholine and the like.
- aryl refers to an aromatic carbocyclic group of 6 to 14 carbon atoms having a single ring (e. g., phenyl) or multiple rings, which may be attached in a pendent manner or may be fused.
- Preferred aryl include phenyl, naphthyl, biphenyl, indane and the like.
- arylkyl refers to alkyl groups as defined above, having one or more aryl substituent, including benzyl, phenethyl, diphenyl methyl and the like.
- heteroaryl refers to a monocyclic hetero aromatic, or a bicyclic fused-ring heteroaromatic group. Particular examples of heteroaromatic groups include optionally substituted pyridyl, pyrrolyl, furyl, thienyl, or benzothiophene .
- aminocarbonyl refers to the group -C(O)NRR' where each R, R' includes independently H, "alkyl”, “aryl” or “arylaminocarbonyl”.
- “Pharmaceutically acceptable salts” refers to salts of the below identified compounds of formula (I), that retain the desired biological activity.
- examples of such salts include, but are not restricted to acid addition salts formed with inorganic acids (e.g. hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and the like), and salts formed with organic acids such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, fumaric acid, maleic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalene sulfonic acid, toluene sulfonic acid, naphthalene disulfonic acid, methanesulfonic acid and poly-galacturonic acid.
- inorganic acids e.g. hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid,
- the salt is of a mono acid (for example, the hydrochloride, the hydrobromide, the p-toluenesulphonate, or the acetate)
- the hydrogen form of a di-acid for example, the hydrogen sulphate, or the succinate
- the dihydrogen form of a tri-acid for example, the dihydrogen phosphate, or the citrate
- at least one molar equivalent and usually a molar excess of the acid is employed.
- the appropriate and exact chemical equivalents of acid are generally used.
- Suitable pharmaceutically acceptable base addition salts for the compound of the present invention include metallic salts made from aluminium, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from lysine, N, N'- dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine ( ⁇ f-methylglucamine) and procaine.
- Sodium salts are particularly preferred.
- “Enantiomers” refers to the products that are obtained by an asymmetric synthesis, i.e.
- the compounds of the present invention can be prepared by conventional synthetic methods and are described underneath. It will be appreciated that where typical or preferred experimental conditions (i.e. reaction temperatures, time, moles of reagents, solvents, etc.) are given, other experimental conditions can also be used, unless otherwise stated.
- the invention furthermore provides a process for the preparation of compounds of formula I, which can be obtained by reacting compounds of formula II,
- R 1 and R 2 are as described above, in an aprotic solvent such as toluene or benzene at a temperature ranging from 20 0 C to the reflux of the solvent.
- any interfering reactive group can be protected and then deprotected according to well-established procedures described in organic chemistry (see for example: Greene T. W., Wuts P.G.M., "Protective Groups in Organic Synthesis", J. Wiley & Sons, Inc., 3 rd Ed., 1999) and well known to those skilled in the art.
- Another object of the present invention is a method of treating a mammal suffering from disease states, disorders and pathological conditions mediated by fatty acid amide hydrolase; in particular of anxiety and pain, comprising administering a therapeutically effective amount of a compound of Formula (I) as described above.
- therapeutically effective amount refers to an amount of a therapeutic agent needed to treat, ameliorate a targeted disease or condition, or to exhibit a detectable therapeutic effect.
- the therapeutically effective dose can be estimated initially either in cell culture assays or in animal models, usually mice, rats, guinea pigs, rabbits, dogs, or pigs.
- the animal model may also be used to determine the appropriate concentration range and route of administration. Such information can then be used to determine useful doses and routes for administration in humans.
- HED Human Equivalent Dose
- compositions will contain at least one compound of Formula (I) as an active ingredient, in an amount such as to produce a significant therapeutic effect.
- the compositions covered by the present invention are entirely conventional and are obtained with methods which are common practice in the pharmaceutical industry, such as, for example, those illustrated in Remington 's Pharmaceutical Science Handbook, Mack Pub. N. Y. — last edition. According to the administration route chosen, the compositions will be in solid or liquid form, suitable for oral, parenteral or topical administration.
- the compositions according to the present invention contain, along with the active ingredient, at least one pharmaceutically acceptable vehicle or excipient. These may be particularly useful formulation coadjuvants, e.g. solubilising agents, dispersing agents, suspension agents, and emulsifying agents.
- an effective dose will be from 0.01 mg/kg to 100 mg/kg, preferably 0.05 mg/kg to 50 mg/kg.
- the therapeutically effective dose can be estimated initially either in cell culture assays or in animal models, usually mice, rats, guinea pigs, rabbits, dogs, or pigs.
- the precise effective dose for a human subject will depend upon the severity of the disease state, general health of the subject, age, weight, and gender of the subject, diet, time and frequency of administration, drug combination(s), reaction sensitivities, and tolerance/response to therapy. This amount can be determined by routine experimentation and is within the judgement of the clinician.
- compositions may be administered individually to a patient or may be administered in combination with other agents, drugs or hormones.
- the medicament may also contain a pharmaceutically acceptable carrier, for administration of a therapeutic agent.
- Such carriers include antibodies and other polypeptides, genes and other therapeutic agents such as liposomes, provided that the carrier does not itself induce the production of antibodies harmful to the individual receiving the composition, and which may be administered without undue toxicity.
- Suitable carriers may be large, slowly metabolised macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers and inactive virus particles.
- a thorough discussion of pharmaceutically acceptable carriers is available in Remington's Pharmaceutical Sciences (Mack Pub. Co. , N. J. 1991).
- Pharmaceutically acceptable carriers in therapeutic compositions may additionally contain liquids such as water, saline, glycerol and ethanol. Additionally, auxiliary substances, such as wetting or emulsifying agents, pH buffering substances, and the like, may be present in such compositions. Such carriers enable the pharmaceutical compositions to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for ingestion by the patient.
- compositions of the invention can be administered directly to the subject.
- the subjects to be treated can be animals; in particular, human subjects can be treated.
- the medicament of this invention may be administered by any number of routes including, but not limited to, oral, intravenous, intramuscular, intraarterial, intramedullary, intrathecal, intraventricular, transdermal or transcutaneous applications, subcutaneous, intraperitoneal, intranasal, enteral, topical, sublingual, intravaginal or rectal means.
- compositions for oral administration may take the form of bulk liquid solutions or suspensions, or bulk powders. More commonly, however, the compositions are presented in unit dosage forms to facilitate accurate dosing.
- unit dosage forms refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient.
- Typical unit dosage forms include refilled, pre-measured ampoules or syringes of the liquid compositions or pills, tablets, capsules or the like in the case of solid compositions.
- the compound of the invention is usually a minor component (from about 0.1 to about 50% by weight or preferably from about 1 to about 40% by weight) with the remainder being various vehicles or carriers and processing aids helpful for forming the desired dosing form.
- Dosage treatment may be a single dose schedule or a multiple dose schedule.
- An object of the present invention are pharmaceutical compositions containing one or more of the compounds of formula (I) described earlier, in combination with excipients and/or pharmacologically acceptable diluents.
- the compositions in question may, together with the compounds of formula (I), contain known active principles.
- a further object of the invention is a process for the preparation of pharmaceutical compositions characterised by mixing one or more compounds of formula (I) with suitable excipients, stabilizers and/or pharmaceutically acceptable diluents.
- An embodiment of this invention is that of compounds of formula (I) described earlier, wherein R 1 is aryl, alkoxyaryl or haloaryl and R 2 is H.
- a more preferred embodiment of this invention is that of compounds of formula (I) described earlier, wherein R 1 is an optionally substituted aromatic group, R 2 is H and G is phenylene and A is a heteroaryl group.
- An even more preferred embodiment of this invention is that of compounds of formula (I) described earlier, wherein wherein R 1 is an optionally substituted aromatic group, R 2 is H and G is phenylene and A is 2-thienyl.
- Particularly preferred compounds are: l-phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST3715); l-phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3986);
- Figure 1 describes the analgesic effect of the selective FAAH inhibitor ST4020 and URB597 at two different doses and at three time points in a rat model of neuropathic pain induced by intraperitoneal administration vincristine.
- the animals were subjected to the paw withdrawal test 60, 120 and 240 min after oral administration of the tested compounds.
- Na 2 SOzI sodium sulfate
- NaH sodium hydride
- Example 7 1 - ( 1 -piperidinocarbonyloxyimino) - 1 - (3-biphenyl)ethane (ST4017)
- the compounds of the present invention show affinity and inhibit the enzymatic activity of the fatty acid amide hydrolase enzyme.
- the assay of FAAH (EC 3.5.1.4) was performed by measuring the release of [1- 14 C]AA from [1- 14 C]AnNH (52 mCi/mmol), using RP-HPLC. Also [ 3 H]AnNH (205 Ci/mmol) could be used as substrate, measuring the release of [ 3 H]AA under the same experimental conditions described below for [1- 14 C]AnNH.
- the compounds of the present invention were also evaluated with regard to their selectivity profile against the following targets: AMT, NAPE-PLD, MAGL, DAGL, CB1/CB2 and TRPVl according to the procedures described in Maccarrone M., et al, J. Biol. Chem., 2000, 275, 13484; Fezza F., et al, Anal Biochem., 2005, 339, 113; Dinh T.P., et al., 2002, Proc. Natl. Acad. Sci. 99, 10819; Bisogno T., et al, 2003, J. Cell Biol, 163, 463; Maccarrone M., et al, J. Biol Chem., 2000, 275, 31938; Ross R.A., et al, Br. J. Pharmacol, 2001, 132, 631. The results are shown in table 2. Table 2
- the maximum concentration tested corresponds to 10 times those of the IC50 on FAAH
- the maximum concentration tested corresponds to 100 times those of the IC50 on FAAH
- EPM elevated plus maze
- the purpose of the present study was to set up an animal model of anxiety EPM using the anxiolytic effects of benzodiazepine Diazepam in mice and evaluate the effect of FAAH inhibitors URB597 and ST4020. Twelve male CDl mice (Charles River) of about 30 g (2 months old) per group were used.
- the Elevated Plus Maze apparatus was of grey Plexiglas and consisted of two open and two closed arms linked by a common central platform. The maze was elevated 40 cm above floor level and dimly lit. Animals were individually placed on the central platform of the maze facing an open arm. A standard five-min test was employed. The amount of time spent by each animal in either open or closed arm was recorded, as well as the number of entries by each animal into either arm.
- ST4020 reduced anxiety and did not affect the locomotor activity evaluated in elevated plus maze.
- Analgesia animal model The analgesic effect of the selective FAAH inhibitor ST4020 was evaluated in a rat model of neuropathic pain induced by intraperitoneal administration vincristine.
- FAAH inhibitors or vehicle (control group) were given orally.
- the former were administered at 10 and 50 mg/kg doses, and the analgesic effect was measured using the paw withdrawal test, as a model of mechanical hyperalgesia.
- Rats undergoing treatment were divided into two groups: vehicle (9 animals) and vincristine (54 animals). Neuropathy was induced by intraperitoneal injections of 0.15 mg/kg vincristine three times a week. After two weeks, the rats were distributed into six groups of nine animals: vehicle, vincristine + vehicle, vincristine + URB597 (10 mg/kg), vincristine + URB597 (50 mg/kg), vincristine + ST4020 (10 mg/kg) and Vincristine + ST4020 (50 mg/kg). The animals were subjected to the paw withdrawal test 60, 120 and 240 min after oral administration of the tested compounds.
- the acute systemic administration of the selective FAAH inhibitor ST4020 is able to significantly reduce mechanical hyperalgesia associated with a chemotherapeutic model of neuropathic pain.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
The present invention relates to new oxime carbamoyl derivatives of formula (I), processes for their preparation, and to pharmaceutical compositions containing them for the treatment of neurological disorders, such as neuropathic pain and anxiety.
Description
FIELD OF THE INVENTION
The present invention relates to new oxime carbamoyl derivatives, processes for their preparation, and to pharmaceutical compositions containing them for the treatment of neurological disorders, such as neuropathic pain and anxiety. BACKGROUND OF THE INVENTION
Anandamide and other fatty acid amides are known to be chemical messengers that modulate a number of physiological processes (Hanus L.O., Chem. Biodivers., 2007, 4, 1828). Anandamide activates through binding both the central-type (CBl) and peripheral type (CB2) cannabinoid receptors (Devane W. A., et al, Science, 1992, 258, 1946-1949). Anandamide has been reported to be implicated in the modulation of nociception, feeding, emesis, anxiety, cell proliferation, inflammation, and memory (Labar G., et al, Chem. Biodivers., 2007, 4, 1882). The pharmacological action of anandamide is terminated by fatty-acid-amide- hydrolase (FAAH), an enzyme distributed in the central nervous system that degrades fatty acid amides at their site of action (Cravatt B. F., et al., Nature, 1996, 384, 83; Patricelli M.P., et al, Biochemistry, 1999, 38, 9804; WO 98/20119 and U.S. Pat. No. 6,271,015). The crystal structure of a complex of FAAH with a ligand has been solved, confirming that it exerts its catalytic action via the triad Ser-Ser-Lys (Bracey M.H., et al, Science, 2002, 298, 29, 1793).
FAAH is also responsible of the catabolism of many other lipid signaling fatty acid amides (i.e. oleamide, iV-oleoylethanolamine, arachidonylglycerol and palmitoylethanolamide). Modulating the activity of the endocannabinoid system by restoring the levels of endogenous signaling lipids turned out to hold
therapeutic promise in a wide range of disparate diseases and pathological conditions such as diseases of energy metabolism (cachexia and anorexia), pain and inflammation, central nervous system disorders (stroke, multiple sclerosis, Parkinson's disease, Huntington disease, Alzheimer disease, epilepsy, schizophrenia, anxiety, depression and insomnia), cardiovascular and respiratory disorders (hypertension, circulatory shock, myocardial reperfusion injury, atherosclerosis and asthma), retinopathy, cancer, gastrointestinal and liver disorders (inflammatory bowel disease and hepatitis), musculoskeletal disorders (arthritis and osteoporosis) as nicely reviewed lately (Pasher P., et al., Pharmacol. Rev., 2006, 58, 389 and references therein).
FAAH A KO mice cannot metabolize anandamide and, though fertile and generally normal, show signs of enhanced anandamide and related fatty acid amides activity at cannabinoid receptors, such as reduced pain sensation (Cravatt B.F., et al, Proc. Natl. Acad. ScL, 2001, 98, 9371). This suggests the possibility that drugs targeting FAAH may heighten the tonic action of anandamide, while possibly avoiding the multiple, often unwanted effects produced by Δ9-THC and other direct-acting cannabinoid agonists (Hall W., et al., Lancet, 1998, 352, 1611; Chaperon, F., et al., Crit. Rev. Neurobiol, 1999, 13, 243). In particular URB-597, a carbamate-based inhitor, was reported to be efficacious in the zero plus maze animal model of anxiety as well as to have analgesic efficacy in the rat hot plate and formalin tests (Kathuria S., et al, Nat. Med., 2003, 9, 1, 76). WO03007955 describes endonuclease inhibitors as useful agents for treatment of cancer. In this application, the only aminocarbonyloxime derivative to be
described, l-propanone-l-(2'-fluoro[l,l'-biphenyl]-4-yl)-O-[[(3,5- dichlorophenyl)amino]carbonyl]oxime is reported to have an IC50 against Hapl of 2.4 μM. l-propanone-l-(2'-fluoro[l,l'-biphenyl]-4-yl)-O-[(phenylamino)carbonyl]oxime and l-propanonel-(2'-fluoro[l,l'-biphenyl]-4-yl)-O-[[[4-
(trifluoroniethoxy)phenyl] amino] carbonyl]oxime are known compounds but no activity or references could be retrieved among literature.
During a virtual screening aiming at discovering new non-nucleoside inhibitors of HIV-I reverse transcriptase, two commercially available compounds, ethanone-l-(6-benzofuranyl)-O-[[(2- methylphenyl)amino]carbonyl]oxime and ethanone-l-(6-benzofuranyl)-O-[[(3- methylphenyl)amino]carbonyl]oxime were identified but when tested were reported to be deprived of activity in a HIV replication assay in the MT-2 cells at concentrations up to 100 μM (Barreiro G., et al., J Chem. Inf. Model, 2007, 47, 6, 2416).
US5438056 reports oxime carbamates of formula 1, wherein R1 and R2 cannot be alkyl, and R3 and R4 can be H, C4-C20 hydrocarbon chain, as useful agents for reducing excess of cholesterol in humans.

WO2003051842 describes compounds of formula R1R2NCC=X)L wherein X can be O, and L can be of the formula -ON=CRalRa2, as useful agents to modulate the plasma level of free fatty acids by inhibiting the hormone- sensitive lipase
enzyme. Eight oxime derivatives are specifically described in this patent application. The same eight oximes derivatives are also present in WO2003051841. Such oximes are not part of the present invention. US6949574 reports (oxime)carbamoyl fatty acid amide hydrolase inhibitors of formula 2 wherein A can be dibenzofuryl, dibenzothienyl, naphtyl, indolyl, fluorenyl, carbazolyl or a substituted alkoxyphenylalkyl and B can be, among others, a group -N=CR2R3 where R2 is methyl or H, and R3 can be mono- phenyl optionally substituted by halo, Ci-4-haloalkyl and nitro.

Formula 2
Forty years ago, carbamoyl oxime derivatives of general formula 3 wherein Y can be alkyl, R1, R2, and R3 can be alkyl or aryl and X is a lower dialkylamino residue, doted of analgesic properties were described (GB1214077).

Almost thirty years ago, the comparative analgesic, behavioural, and dependence properties of morphine and O-(4-methoxyphenylcarbamoyl)-3- diethylaminopropiophenone oxime hydrochloride were reported. Such a compound showed similar analgesic potency to morphine and suggested a less dependency pattern than the latter (Watzman N., et al., Journal of Pharmaceutical Sciences, 1980, 69, 2, 225).
Recently, the results of a virtual screening utilizing a comparative model of the human monoglyceride lipase enzyme (MGL) were published (Saario S. M., et al., J. Med. Chem. 2006, 49, 4650). None of the hit compounds when tested against MGL enzyme resulted active. When tested against FAAH enzyme, one oxime derivative (MWP00348) resulted to inhibit the enzyme with a sub- micromolar IC50.
The potential therapeutic relevance of inhibiting FAAH has stimulated interest in developing selective and potent inhibitors. Such a strategy potentially represents a safer alternative to the use of exogenous cannabinoid agonists, which have been found to give variable effects. Inhibiting FAAH seems an ideal way of elevating the levels of the endogenous amidated lipids that activate CBl receptors. Therefore, the desire of potent and selective FAAH inhibitors remains an interesting and promising goal. DESCRIPTION OF THE INVENTION The invention provides novel compounds for inhibiting Fatty Acid Amide Hydrolase (FAAH), compositions that include such compounds as well as methods of treating diseases of energy metabolism, pain and inflammation, central nervous system disorders, cardiovascular and respiratory disorders, retinopathy, cancer, gastrointestinal and liver disorders and musculoskeletal disorders by administering FAAH inhibitors to a patient. The invention comprises compounds of general formula I

R1 and R2 are independently H, alkyl, aryl, arylkyl, alkoxyaryl or haloaryl; or R1 and R2 taken together with the nitrogen atom to which they are attached form a heterocycle; G is phenylene or thienylene;
A is phenyl, thienyl, cyclohexyl, pyrrolyl or pyridyl, each of them being optionally substituted once or twice with aminocarbonyl or alkyl; its tautomers, its geometrical isomers, its optically active forms such as enantiomers, diastereomers and its racemate forms, as well as pharmaceutically acceptable salts thereof; with the proviso that when A is cyclohexyl, R1 cannot be 4-Cl phenyl and 3,4- dichlorophenyl if R2 is H.
An embodiment of this invention is that of compounds of formula I, for use as medicaments. In a further embodiment, said medicament is used for treating a neurological disorder, diseases of energy metabolism, cardiovascular and respiratory disorders, gastrointestinal and liver disorders, retinopathy, cancer and musculoskeletal disorders.
In a preferred embodiment, said medicament is used for treating a neurological disorder.
In a more preferred embodiment, said medicament is used for treating anxiety and pain.
The term "alkyl" refers to linear or branched alkyl groups having preferably from 1 to about 12 carbon atoms. Lower alkyl group is exemplified by groups
such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl, iso-pentyl, neo-pentyl, n-hexyl and the like.
The term "alkoxy" refers to a group -OR where R includes lower alkyl, "C3-C10 cycloalkyl" and "heterocycloalkyl". The terms "heterocycloalkyl" and/or heterocycle refer to a saturated five- or six-membered ring containing one or two nitrogen, oxygen or sulfur atoms.
Preferred heterocycloalkyl include pyrrolidine, piperidine, piperazine, morpholine, thiomorpholine and the like.
The term "aryl" refers to an aromatic carbocyclic group of 6 to 14 carbon atoms having a single ring (e. g., phenyl) or multiple rings, which may be attached in a pendent manner or may be fused. Preferred aryl include phenyl, naphthyl, biphenyl, indane and the like.
The term "arylkyl" refers to alkyl groups as defined above, having one or more aryl substituent, including benzyl, phenethyl, diphenyl methyl and the like. The term "heteroaryl" refers to a monocyclic hetero aromatic, or a bicyclic fused-ring heteroaromatic group. Particular examples of heteroaromatic groups include optionally substituted pyridyl, pyrrolyl, furyl, thienyl, or benzothiophene .
The term "aminocarbonyl" refers to the group -C(O)NRR' where each R, R' includes independently H, "alkyl", "aryl" or "arylaminocarbonyl".
"Pharmaceutically acceptable salts" refers to salts of the below identified compounds of formula (I), that retain the desired biological activity. Examples of such salts include, but are not restricted to acid addition salts formed with inorganic acids (e.g. hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid, and the like), and salts formed with organic acids
such as acetic acid, oxalic acid, tartaric acid, succinic acid, malic acid, fumaric acid, maleic acid, ascorbic acid, benzoic acid, tannic acid, pamoic acid, alginic acid, polyglutamic acid, naphthalene sulfonic acid, toluene sulfonic acid, naphthalene disulfonic acid, methanesulfonic acid and poly-galacturonic acid. When the salt is of a mono acid (for example, the hydrochloride, the hydrobromide, the p-toluenesulphonate, or the acetate), the hydrogen form of a di-acid (for example, the hydrogen sulphate, or the succinate), or the dihydrogen form of a tri-acid (for example, the dihydrogen phosphate, or the citrate), at least one molar equivalent and usually a molar excess of the acid is employed. However, when such salts as the sulphate, the hemisuccinate, the hydrogen phosphate, or the phosphate are desired, the appropriate and exact chemical equivalents of acid are generally used. Suitable pharmaceutically acceptable base addition salts for the compound of the present invention include metallic salts made from aluminium, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from lysine, N, N'- dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine (Λf-methylglucamine) and procaine. Sodium salts are particularly preferred. "Enantiomers" refers to the products that are obtained by an asymmetric synthesis, i.e. a synthesis involving non-racemic starting materials and/or reagents or a synthesis comprising at least one enantioselective step, whereby a surplus of one enantiomer in the order of at least about 52% is yielded. The compounds of the present invention can be prepared by conventional synthetic methods and are described underneath. It will be appreciated that where typical or preferred experimental conditions (i.e. reaction temperatures,
time, moles of reagents, solvents, etc.) are given, other experimental conditions can also be used, unless otherwise stated.
The invention furthermore provides a process for the preparation of compounds of formula I, which can be obtained by reacting compounds of formula II,

Formula Il wherein A and G are as described above, with compounds of formula III

wherein R1 and R2 are as described above, in an aprotic solvent such as toluene or benzene at a temperature ranging from 200C to the reflux of the solvent. Alternatively, compounds of formula I wherein R2 is hydrogen can be obtained by reacting compounds of formula II with compounds of formula IV (O=C=NR1) in an aprotic solvent such as toluene or benzene at a temperature ranging from 200C to the reflux of the solvent.
In all said transformations, any interfering reactive group can be protected and then deprotected according to well-established procedures described in organic chemistry (see for example: Greene T. W., Wuts P.G.M., "Protective Groups in Organic Synthesis", J. Wiley & Sons, Inc., 3rd Ed., 1999) and well known to those skilled in the art.
All said transformations are only examples of well-established procedures described in organic chemistry (see for example: March J., "Advanced Organic
Chemistry", J. Wiley & Sons, Inc., 4th Ed., 1992) and well known to those skilled in the art.
We have found that the derivatives (I) and their pharmaceutically acceptable salts, prepared according to the invention, are useful agents for the treatment of disease states, disorders and pathological conditions mediated by fatty acid amide hydrolase; in particular for the treatment of anxiety and pain. Therefore another object of the present invention is a method of treating a mammal suffering from disease states, disorders and pathological conditions mediated by fatty acid amide hydrolase; in particular of anxiety and pain, comprising administering a therapeutically effective amount of a compound of Formula (I) as described above.
The term "therapeutically effective amount" as used herein refers to an amount of a therapeutic agent needed to treat, ameliorate a targeted disease or condition, or to exhibit a detectable therapeutic effect. For any compound, the therapeutically effective dose can be estimated initially either in cell culture assays or in animal models, usually mice, rats, guinea pigs, rabbits, dogs, or pigs. The animal model may also be used to determine the appropriate concentration range and route of administration. Such information can then be used to determine useful doses and routes for administration in humans. In calculating the Human Equivalent Dose (HED) it is recommended to use the conversion table provided in Guidance for Industry and Reviewers document (2002, U.S. Food and Drug Administration, Rockville, Maryland, USA). The pharmaceutical compositions will contain at least one compound of Formula (I) as an active ingredient, in an amount such as to produce a
significant therapeutic effect. The compositions covered by the present invention are entirely conventional and are obtained with methods which are common practice in the pharmaceutical industry, such as, for example, those illustrated in Remington 's Pharmaceutical Science Handbook, Mack Pub. N. Y. — last edition. According to the administration route chosen, the compositions will be in solid or liquid form, suitable for oral, parenteral or topical administration. The compositions according to the present invention contain, along with the active ingredient, at least one pharmaceutically acceptable vehicle or excipient. These may be particularly useful formulation coadjuvants, e.g. solubilising agents, dispersing agents, suspension agents, and emulsifying agents.
The amount of the compound actually administered will typically be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered, drug combination, the age, body weight, and response of the individual patient, the severity of the patient's symptoms, and the like. Generally, an effective dose will be from 0.01 mg/kg to 100 mg/kg, preferably 0.05 mg/kg to 50 mg/kg. For any compound, the therapeutically effective dose can be estimated initially either in cell culture assays or in animal models, usually mice, rats, guinea pigs, rabbits, dogs, or pigs. The precise effective dose for a human subject will depend upon the severity of the disease state, general health of the subject, age, weight, and gender of the subject, diet, time and frequency of administration, drug combination(s), reaction sensitivities, and tolerance/response to therapy. This amount can be
determined by routine experimentation and is within the judgement of the clinician.
Compositions may be administered individually to a patient or may be administered in combination with other agents, drugs or hormones. The medicament may also contain a pharmaceutically acceptable carrier, for administration of a therapeutic agent. Such carriers include antibodies and other polypeptides, genes and other therapeutic agents such as liposomes, provided that the carrier does not itself induce the production of antibodies harmful to the individual receiving the composition, and which may be administered without undue toxicity.
Suitable carriers may be large, slowly metabolised macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers and inactive virus particles. A thorough discussion of pharmaceutically acceptable carriers is available in Remington's Pharmaceutical Sciences (Mack Pub. Co. , N. J. 1991).
Pharmaceutically acceptable carriers in therapeutic compositions may additionally contain liquids such as water, saline, glycerol and ethanol. Additionally, auxiliary substances, such as wetting or emulsifying agents, pH buffering substances, and the like, may be present in such compositions. Such carriers enable the pharmaceutical compositions to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for ingestion by the patient.
Once formulated, the compositions of the invention can be administered directly to the subject. The subjects to be treated can be animals; in particular, human subjects can be treated.
The medicament of this invention may be administered by any number of routes including, but not limited to, oral, intravenous, intramuscular, intraarterial, intramedullary, intrathecal, intraventricular, transdermal or transcutaneous applications, subcutaneous, intraperitoneal, intranasal, enteral, topical, sublingual, intravaginal or rectal means.
The compositions for oral administration may take the form of bulk liquid solutions or suspensions, or bulk powders. More commonly, however, the compositions are presented in unit dosage forms to facilitate accurate dosing. The term "unit dosage forms" refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient. Typical unit dosage forms include refilled, pre-measured ampoules or syringes of the liquid compositions or pills, tablets, capsules or the like in the case of solid compositions. In such compositions, the compound of the invention is usually a minor component (from about 0.1 to about 50% by weight or preferably from about 1 to about 40% by weight) with the remainder being various vehicles or carriers and processing aids helpful for forming the desired dosing form. Dosage treatment may be a single dose schedule or a multiple dose schedule.
An object of the present invention are pharmaceutical compositions containing one or more of the compounds of formula (I) described earlier, in combination with excipients and/or pharmacologically acceptable diluents. The compositions in question may, together with the compounds of formula (I), contain known active principles.
A further object of the invention is a process for the preparation of pharmaceutical compositions characterised by mixing one or more compounds of formula (I) with suitable excipients, stabilizers and/or pharmaceutically acceptable diluents. An embodiment of this invention is that of compounds of formula (I) described earlier, wherein R1 is aryl, alkoxyaryl or haloaryl and R2 is H.
A more preferred embodiment of this invention is that of compounds of formula (I) described earlier, wherein R1 is an optionally substituted aromatic group, R2 is H and G is phenylene and A is a heteroaryl group. An even more preferred embodiment of this invention is that of compounds of formula (I) described earlier, wherein wherein R1 is an optionally substituted aromatic group, R2 is H and G is phenylene and A is 2-thienyl.
Specific embodiments of the present invention are represented by the following compounds: l-phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST3715);
1- dimethylaminocarbonyloxyimino- 1 - (4-biphenyl)ethane (ST3741) ; l-phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3986);
1 - ( 1 - naphthylaminocarbonyloxyimino) - 1 - (3-biphenyl)ethane (ST3987) ; l-(4-fluorophenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3988); l-(4-methoxyphenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3989); l-(l-piperidinocarbonyloxyimino)-l-(3-biphenyl)ethane (ST4017);
1 - (diphenylaminocarbonyloxyimino)- 1 - (3-biphenyl)ethane (ST4018) ; l-(phenylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4020); l-(phenylaminocarbonyloxyimino)-l-(3-(3'-aminocarbonyl)biphenyl)ethane (ST4055);
l-[3'-(l-(phenylaminocarbonyloxyimino)-ethyl)-biphenyl-3-carbonyl]-3-phenyl- urea (ST4056); l-(l-piperidinocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4163); l-(l-dodecylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4164); l-(phenylaminocarbonyloxyimino)-l-(4-(2-thienyl)phenyl)ethane (ST4165); l-(phenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(4-methoxyphenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(l-piperidinocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; 1 - (phenylaminocarbonyloxyimino) - 1 - [4- (2 , 5 - dimethylpyrrol- 1 -yl)- phenyl] ethane; l-(4-fluorophenylaniinocarbonyloxyimino)-l-[4-(2,5-dimethylpyrrol-l-yl)- phenyl] ethane;
1 - naphthylaminocarbonyloxyimino- 1 - [4- (2, 5 - dimethylpyrrol- 1 -yl) - phenyl]ethane (ST7262);
1 - (4- methoxyphenylaminocarbonyloxyimino)- 1 - [4- (2 , 5 - dimethylpyrrol- 1 -yl) - phenyl] ethane; l-(l-piperidinocarbonyloxyimino)-l-[4-(2,5-dimethylpyrrol-l-yl)-phenyl] ethane; l-(phenylaminocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane;
1 - ( 1 - naphthylaminocarbonyloxyimino) - 1 - ( [2, 2 '] bithiophenyl- 5 -yl] )ethane ;
1 - (4- methoxyphenylaminocarbonyloxyimino) - 1 - ( [2 ,2 '] bithiophenyl- 5 -yl] )ethane ; l-(l-piperidinocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane; l-(phenylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane;
l-(l-naphthylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane;
1 - (4- methoxyphenylaminocarbonyloxyimino)- 1 - (5 -phenyl-thiophen- 2 -yl)ethane ;
1 - ( 1 -piperidinocarbonyloxyimino)- 1 - (5 -phenyl- thiophen- 2 -yl)ethane ; l-(phenylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(l-naphthylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(4-methoxyphenylaminocarbonyloxyiniino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(l-piperidinocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(phenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(l-naphthylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(4-niethoxyphenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(l-piperidinocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-^enzothienyl-δ-yl-aminocarbonyloxyimino-l-biphenyl-S-yl-ethane (ST5742); l-(l-naphthylaminocarbonyloxyimino)-l-(4-biphenyl)ethane (ST7380); δ-phenylpentylaminocarbonyloxyimino-l-biphenyl-S-yl-ethane (ST7378);
1 - ( 1 - naphthylaminocarbonyloxyimi.no) - 1 - (4- cy clohexyl-phenyl) - ethane
(ST7261).
Particularly preferred compounds are: l-phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST3715); l-phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3986);
1 - ( 1 - naphthylaminocarbonyloxyimino) - 1 - (4-biphenyl)ethane (ST3987) ; l-(4-fluorophenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3988); l-(4-methoxyphenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3989); l-(l-piperidinocarbonyloxyimino)-l-(3-biphenyl)ethane (ST4017);
l-(phenylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4020); l-(phenylaminocarbonyloxyimino)-l-(3-(3'-aminocarbonyl)-biphenyl)ethane
(ST4055); l-(l-piperidinocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4163); l-(l-dodecylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4164); l-(phenylaminocarbonyloxyimino)-l-(4-(2-thienyl)phenyl)ethane (ST4165); l-(l-naphthylaminocarbonyloxyimino)-l-[4-(2,5-dimethylpyrrol-l-yl)- phenyl] ethane (ST7262); l-φenzothienyl-δ-yl-aminocarbonyloxyimino-l-biphenyl-S-yl-ethane (ST5742); l-(l-naphthylaminocarbonyloxyimino)-l-(4-biphenyl)ethane (ST7380); δ-phenylpentylaminocarbonyloxyimino-l-biphenyl-S-yl-ethane (ST7378) and l-(l-naphthylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)-ethane (ST7261).
DESCRIPTION OF THE DRAWING
Figure 1 describes the analgesic effect of the selective FAAH inhibitor ST4020 and URB597 at two different doses and at three time points in a rat model of neuropathic pain induced by intraperitoneal administration vincristine. The animals were subjected to the paw withdrawal test 60, 120 and 240 min after oral administration of the tested compounds.
EXAMPLES
Abbreviations:
AA: arachidonic acid
AcOEt: ethyl acetate
AnNH: arachidonoylethanolamide (anandamide) DCM: dichloromethane;
DMSO: dimethylsulfoxide
Et2O: diethyl ether
MeOH: methanol
Na2SOzI: sodium sulfate NaH: sodium hydride
RP-HPLC: reversed phase-high-performance liquid chromatography RT: room temperature
General Remarks: 1H spectra were recorded in CDCI3 solution as indicated, at 300 MHz with a Bruker instrument. The chemical shift values are given in ppm. Flash column chromatography was carried out using silica gel (Merck 230-400 mesh).
All reactions were performed with anhydrous solvents under a nitrogen atmosphere unless otherwise specified.
Example 1 l-phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST 3715)

A suspension of oxime of 4-acetylbiphenyl (200 mg, 0.95 mmol) and phenyl isocyanate (340 mg, 2.85 mmol) in 8.5 ml of toluene, was stirred for 2 h at 50°C. Then, the reaction mixture was washed with aqueous ammonia, extracted with AcOEt. The combined organic phases were washed with water and dried over Na2SO4. Solvent was removed under vacuo to afford after purification by silica gel chromatography (hexane/ AcOEt = 4:1) 1- phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane; (280 mg, 89%). 1H NMR (CDCl3) δ: 2.53 (s, 3H), 7.12 (t, IH), 7.35-7.68 (m, 9H), 7.62 (d, 2H), 7.79 (d, 2H), 8.40 (br.s, IH)
Example 2 l-dimethylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST 3741)

A suspension of the oxime of 4-acetylbiphenyl (200 mg, 0.95 mmole) and N, N- dimethylcarbamoyl chloride (306 mg, 2.85 mmol) in 8.5 ml of dichloromethane was stirred for 2 h at 50° C. Then ice was added to the reaction mixture after cooling to 0°C. The organic phase was separated, washed with water and dried over Na2SO4. Solvent was removed under vacuo to afford, after purification by
silica gel chromatography (DCM), l-dimethylaminocarbonyloxyimino-l-(4- biphenyl)ethane; (180 mg, 67%).
1H NMR (CDCl3) δ: 1.80 (br. s, 6 H), 2.52 (s, 3H), 7.3-7.5 (m, 3 H), 7.6-7.78 (m, 4H), 7.82 (m, 2H). Example 3 l-phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3986)

A solution of l-biphenyl-3-yl-ethanone oxime (100 mg, 0.47 mmol) and phenylisocyanate (85 mg, 0.71 mmol) in toluene (45 ml), was refluxed for 1.5 hour. After cooling down to RT, the reaction mixture was washed with aqueous ammonia, extracted with AcOEt. The combined organic phases were washed with water and dried over Na2SO4. Solvent was removed under vacuo to afford, after purification by silica gel chromatography (DCM/Et2θ = 98/2), 1- phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane; as a white solid (140 mg, 90%). 1H NMR (CDCl3) δ: 2.51 (s, 3H), 7.15 (t, IH), 7.2-7.9 (m, 13H), 8.38 (s, IH).
Example 4 l-(l-naphthylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3987)

Example 5 l-(4-fluorophenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3988)

A solution of l-biphenyl-3-yl-ethanone oxime (80 mg, 0.38 mmol) and 4- fluorophenylisocyanate (66 μl, 0.57 mmol) in toluene (7 ml), was refluxed for 1 hour. After removal of the solvent under reduced pressure, the crude reaction mixture was rinsed with Et2θ and MeOH to afford l-(4- fluorophenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane; as a white solid (60 mg, 45%).
1H NMR (DMSO-de) δ: 2.49 (s, 3H), 7.21 (t, 2H), 7.30-7.65 (m, 6H), 7.65-7.90 (m, 4H), 8.03 (s, IH), 9.90 (s, IH).
Example 6 l-(4-methoxyphenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3989)
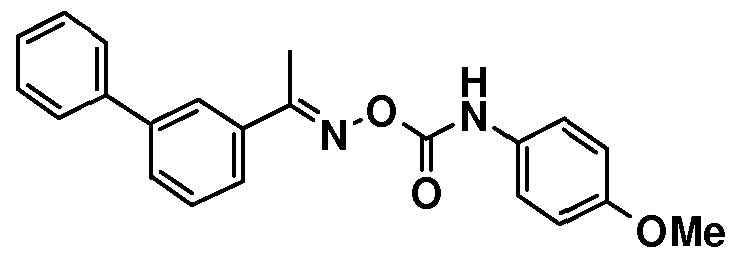
1H NMR (DMSO-de) δ: 2.51 (s, 3H), 3.76 (s, 3H), 6.92 (d, 2H), 7.35-7.65 (m, 6H), 7.70-7.85 (m, 4H), 8.03 (s, IH), 9.63 (s, IH).
Example 7 1 - ( 1 -piperidinocarbonyloxyimino) - 1 - (3-biphenyl)ethane (ST4017)

To a solution of l-biphenyl-3-yl-ethanone oxime (70 mg, 0.33 mmol) in 5 ml of THF were added 26 mg (0.66 mmol) of NaH and the reaction mixture was stirred for 15 minutes. Piperidine-1-carbonyl chloride (130 μl, 1.04 mmol) was added and the mixture was refluxed for 30 minutes. After cooling to RT, the reaction mixture was poured into ice. The mixture was acidified by means of HCl 2N (until pH = 3) and then extracted with AcOEt. The combined organic phases were washed with brine and dried over Na2SO4. Solvent was removed under vacuo to afford after purification by silica gel chromatography (hexane/AcOEt = 9/1) l-(l-piperidinocarbonyloxyimino)-l-(3-biphenyl)ethane; as a white solid (100 mg, 94%).
1H NMR (CDCl3) δ: 1.60 (m, 6H), 2.41 (s, 3H), 3.55 (m, 4H), 7.28-7.50 (m, 4H), 7.60 (m, 3H), 7.72 (d, IH); 7.93 (s, IH).
Example 8 1 - (diphenylaminocarbonyloxyimino) - 1 - (3-biphenvDethane (ST4018)

To a solution of l-biphenyl-3-yl-ethanone oxime (70 mg, 0.33 nimol) in 5 ml of THF were added 26 mg (0.66 mmol) of NaH and the reaction mixture was stirred for 15 minutes. Diphenylamino carbamoyl chloride (230 mg, 1 mmol) was added and the mixture was refluxed for 30 minutes. After cooling to RT, the reaction mixture was poured into ice. The mixture was acidified by means of HCl 2N (until pH = 3) and then extracted with AcOEt. The combined organic phases were washed with brine and dried over Na2SO4. Solvent was removed under vacuo to afford after purification by silica gel chromatography DCM/hexane = 8/2) l-(diphenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane; as a white solid (90 mg, 67%). 1H NMR (CDCl3) δ: 2.02 (s, 3H), 7.21 (m, 2H), 7.30-7.50 (m, HH), 7.55-7.75 (m, 5H), 8.90 (s, IH)
Example 9 l-(phenylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4020)

1H NMR (CDCl3) δ: 2.52 (s, 3H), 7.13 (m, 2H), 7.35 (m, 4H), 7.45 (m, 3H), 7.60 (d, IH), 7.72 (d, IH), 8.91 (s, IH), 8.40 (s, IH).
Example 10 l-(phenylaminocarbonyloxyimino)-l-(3-(3'-aminocarbonyl)biphenyl)ethane (ST4055)

A mixture of 3'-(hydroxyiminoethyl)-biphenyl-3-carboxylic acid amide (100 mg,
0.39 mmol) and phenylisocyanate (64 μl, 0.59 mmol) in toluene (5.7 ml), was refluxed for 1 h. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by silica gel chromatography (DCM/MeOH
= 95/5) to afford l-(phenylaminocarbonyloxyimino)-l-(3-(3'-aminocarbonyl)- biphenyl)ethane; as a white solid (65 mg, 44%).
1H NMR (DMSO-de) δ: 2.50 (s, 3H), 7.02 (t, IH), 7.30 (t, 2H), 7.40-7.70 (m, 5H), 7.86 (m, 4H), 8.04 (s, IH), 7.12 (s, IH), 8.20 (s, IH), 9.81 (s, IH).
Example 11
1 - [3'- ( 1 - (Phenylaminocarboxyimino- ethvDbiphenyl- 3- carbonyll - 3-phenyl-urea
(ST4056)

The above compound was obtained following the procedure described for example 10. After purification by silica gel chromatography (DCM/MeOH = 95/5), the title compound was obtained as a white solid (30 mg, 15%). 1H NMR (DMSOd6) δ: 2.50 (s, 3H), 7.00-7.13 (m, 2H), 7.33 (m, 4H), 7.50-7.70 (m, 6H), 7.83 (d, IH), 7.93 (d, IH), 8.01 (d, 2H), 8.12 (s, IH), 8.36 (s, IH), 9.83 (s, IH), 10.83 (s, IH), 11.18 (s, IH).
Example 12
1 - ( 1 -piperidinocarbonyloxyimino) - 1 - (3- (2 -thienvDphenvDethane (ST4163)

To a solution of l-(3-thiophen-2-yl-phenyl)-ethanone oxime (80 mg, 0.37 mmol) in 3.5 ml of THF was added NaH (13 mg, 0.33 mmol) and the reaction mixture was stirred for 10 minutes. Piperidine-1-carbonyl chloride (140 μl, 1.12 mmol) was added and the mixture was refluxed for 2 h. After cooling to RT, the reaction mixture was poured into ice. The mixture was acidified by means of HCl IN (until pH = 3) and then extracted with AcOEt. The combined organic phases were washed with brine and dried over Na2SO4. Solvent was removed under vacuo and the resulting solid was triturated with Et2θ to afford 1-(1- piperidinocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane; as a white solid (100 mg, 83%).
1H NMR (CDCl3) δ: 1.62 (m, 6H), 2.40 (s, 3H), 3.55 (m, 4H), 7.20 (t, IH), 7.30 (d, IH), 7.39 (m, IH), 7.41 (d, IH), 7.66 (m, 2H), 8.96 (s, IH).
Example 13 1- (phenylaminocarbonyloxyimino)- 1 - (4- (2-thienyl)phenvDethane (ST4165)

A mixture of l-(4-thiophen-2-yl-phenyl)-ethanone oxime (80 mg, 0.37 mmol) and phenylisocyanate (61 μl, 0.56 mmol) in toluene (3.5 ml), was refluxed for
30 minutes. After removal of the solvent under reduced pressure, the crude reaction mixture was triturated with Et2θ to afford 1-
(phenylaminocarbonyloxyimino)-l-(4-(2-thienyl)phenyl)ethane; as a white solid
(40 mg, 32%).
1H NMR (CDCl3) δ: 2.52 (s, 3H), 7.11 (m, 2H), 7.38 (m, 4H), 7.53 (d, 2H), 7.85
(m, 4H), 8.39 (s, NH). Example 14
1 - ( 1 - dodec ylaminocarbonyloxyimino) - 1 - (3- (2 -thienvDphenvDethane (ST4164)

A mixture of l-(3-thiophen-2-yl-phenyl)-ethanone oxime (80 mg, 0.37 mmol) and dodecylisocyanate (130 μl, 0.56 mmol) in toluene (3.5 ml), was refluxed for 2 h. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by silica gel chromatography through gradient elution (DCM/hexane = 9/1 then DCM/Et2O = 9/1) to afford 1-(1-
dodecylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane; as a yellowish oil (60 mg, 38%).
1H NMR (CDCl3) δ: 0.85 (m, 3H), 1.29 (m, 18H), 1.58 (q, 2H), 2.48 (s, 3H), 3.35 (q, 2H), 6.43 (s, NH), 7.12 (t, IH), 7.36 (m, 2H), 7.46 (t, IH), 7.58 (d, IH), 7.71 (d, IH), 7.88 (s, IH).
Example 15 l-(l-naphtylaminocarbonyloxyimino)-142,21bithiophenyl-5-yl-ethane (ST5585)

A solution of 5-acetyl-2,2'-bithiophenyl (200 mg, 0.93 mmol) and NH2OH.HC1 (133 mg, 1.96 mol) in 13 ml of ethanol was refluxed for 10 h. Removal of the solvent under reduced pressure afforded the desired oxime intermediate that was used without further purification in the second step. To a solution of the latter (60 mg) in warm toluene (2.6 ml), was added 1-naphthylisocyanate (59 μl) and the resulting mixture was refluxed for 1 hr. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by silica gel chromatography through gradient elution (DCM/hexane = 9/1 then DCM/Et2θ = 9/1) to afford l-(l-naphtylaminocarbonyloxyimino)-l- [2,2']bithienyl-5-ylethane as a white solid (30 mg). 1H NMR: δ 2.52 (s, 3H), 7.05 (t, IH), 7.18 (d, IH), 7.31 (m, 2H), 7.40 (d, IH), 7.58 (m, 3H), 7.75 (d, IH), 7.93 (d, IH), 8.08 (dd, 2H), 9.08 (s, IH).
Example 16 1 - (benzothien- 5 - yl- aminocarbonyloxyimino) - 1 -biphenyl- 3- yl- ethane (ST5742)
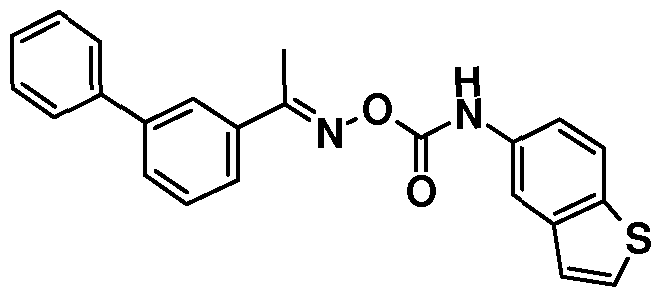
IH NMR: δ 2.59 (s, 3H), 7.33 (d, IH), 7.42 (t, 2H), 7.46-7.61 (m, 4H), 7.62 (m, 2H), 7.73 (m, 2H), 7.84 (d, IH), 7.93 (s, IH), 8.15 (s, IH), 8.51 (d, IH).
Example 17 I-(I- naphth ylaminocarbonyloxyimino) - 1 - (4- c yclohexyl-phenyl) ~ ethane (ST7261)

A mixture of l-(4-cyclohexyl-phenyl)-ethanone oxime (80 mg, 0.37 mmol) and
1-naphthylisocyanate (84 μl, 0.59 mmol) in toluene (3.7 ml) was refluxed for 1 h under a nitrogen atmosphere. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by silica gel chromatography
through gradient elution (DCM/hexane = 9/1 then DCM/Et2O = 9/1) to afford 61 mg of the title compound.
IH NMR: δ 1.17-1.50 (m, 5H), 1.72-1.97 (m, 5H), 2.57 (s, 3H and m, IH), 7.32 (d, 2H), 7.52 (m, 3H), 7.70 (d, 3H), 7.90 (t, 2H), 8.00 (d, IH), 9.00 (d, IH).
Example 18
I-(I- naphth ylaminocarbonyloxyimino) - 1 - (4- [2.5 - dimethylp yrrol- 1 - yll phenyl) ethane (ST7262^

A mixture of l-[4-(2,5-dimethylpyrrol-l-yl)-phenyl]-ethanone oxime (39 mg, 0.17 mmol) and 1-naphthylisocyanate (41 μl, 0.29 mmol) in toluene (18 ml) was refluxed 1 h in under a nitrogen atmosphere. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by silica gel chromatography through gradient elution (DCM/hexane = 9/1 then DCM/Et2θ = 9/1) to afford 30 mg of the title compound,
IH NMR: δ 2.25 (s, 6H), 2.60 (s, 3H), 5.92 (s, 2H), 7.31 (d, 2H), 7.52 (m, 3H), 7.72 (d, IH), 7.80-8.00 (m, 5H), 8.82 (s, IH).
Example 19 l-(5-phenylpentylaminocarbonyloxyimino)-l-biphenyl-3-yl-ethane (ST7378)

1H NMR (CDCl3): δ 7.84 (s, IH), 7.58-7.72 (m, 4H), 7.35-7.52 (m, 4H), 7.10-7.30 (m, 5H), 6.43 (m, IH), 3.30 (q, 2H), 2.60 (t, 2H), 2.47 (m, 3H), 1.62 (m, 4H), 1.40 (m, 2H).
Example 20 l-(l-naphthylaminocarbonyloxyimino)-l-(4-biphenyl)ethane (ST7380)

A mixture of l-biphenyl-4-yl-ethanone oxime (80 mg 0.38 mmol)) and 1- naphthylisocyanate (83 μl, 0.58 mol) in toluene (4 ml) was refluxed for 4 h under a nitrogen atmosphere. After removal of the solvent under reduced pressure, the crude reaction mixture was purified by flash chromatography (DCM / hexane from 9:1 to 95:5) to afford 65 mg (45%) of the title compound.
1H NMR (CDCl3): δ 8.98 (s, IH), 7.80-8.03 (m, 5H), 7.33-7.80 (m, HH), 2.55 (s, 3H).
Biological results FAAH assay
The compounds of the present invention show affinity and inhibit the enzymatic activity of the fatty acid amide hydrolase enzyme. The assay of FAAH (EC 3.5.1.4) was performed by measuring the release of [1- 14C]AA from [1-14C]AnNH (52 mCi/mmol), using RP-HPLC. Also [3H]AnNH (205 Ci/mmol) could be used as substrate, measuring the release of [3H]AA under the same experimental conditions described below for [1-14C]AnNH. Compounds of the invention, at various concentrations, were added in 200 μl hydrolase assay buffer (50 mM Tris-HCl, pH 9.0), in 2-ml Eppendorf tubes, 20 min before adding [l-14C]AnNH2, up to a concentration of 10 μM. The reaction was initiated by the addition of mouse brain homogenate (40 μg), and after incubation at 37°C for 15 min it was stopped by the addition of 800 μl ice-cold methanol/chloroform (2:1, v/v) with vortexing. This mixture was allowed to stand at room temperature for 30 min, then 240 μl chloroform and 240 μl water were added with vortexing. After 10 min at room temperature, the mixture was centrifuged at 300Og for 5 min, the upper aqueous layer was removed by suction and the lower organic phase was dried by spinning the samples in a DNA MINI speedvac (Heto-Holten, Denmark), at 100 mbar and 30°C for 30 min. The residue was dissolved into 50 μl methanol and subjected to RP-HPLC analysis for AA quantitation, as detailed below. FAAH specific activity was expressed as pmol AA released/min/mg protein. Kinetic studies were performed by Lineweaver— Burk analysis, using [1-14C]AnNH, [1- 14C]ODNHEtOH, or [1-14C]ODNH2 in the concentration range 0-12 μM.
Fitting of the experimental points by a linear regression programme (Kaleidagraph 3.0) yielded straight lines with r values>0.97.
Table 1

[++++] [IC50] < 10 nM and/or [Ki] < 10 nM [+++] 10 nM <[ICso] < 100 nM and/or 10 nM < [Ki] < 100 nM
[++] 100 < [IC50] < 500 nM and/or 100 < [Ki] < 500 nM [+] 500 < [IC50] < 5000 nM and/or 500 < [Ki] < 5000 nM NA: not active ND: not determined
Selectivity profile
The compounds of the present invention were also evaluated with regard to their selectivity profile against the following targets: AMT, NAPE-PLD, MAGL, DAGL, CB1/CB2 and TRPVl according to the procedures described in Maccarrone M., et al, J. Biol. Chem., 2000, 275, 13484; Fezza F., et al, Anal Biochem., 2005, 339, 113; Dinh T.P., et al., 2002, Proc. Natl. Acad. Sci. 99, 10819; Bisogno T., et al, 2003, J. Cell Biol, 163, 463; Maccarrone M., et al, J. Biol Chem., 2000, 275, 31938; Ross R.A., et al, Br. J. Pharmacol, 2001, 132, 631. The results are shown in table 2.
Table 2

a: the maximum concentration tested corresponds to 10 times those of the IC50 on FAAH b: the maximum concentration tested corresponds to 100 times those of the IC50 on FAAH
[----] 1000 times [IC50] with an inhibitory activity on the target < 60% [---] 100 times [IC50] with an inhibitory activity on the target < 60% [--] 10 times [IC50] with an inhibitory activity on the target < 60% [-] 5 times [IC50] with an inhibitory activity on the target < 60%
ST4020 was shown to be selective against the above targets. Reversibility
Reversibility was ascertained by incubating FAAH with an excess (i.e., concentrations well above the IC50 values) of the compounds of the present invention for 20 min (as in the enzymatic assay conditions). Subsequently, the FAAH/compound mixtures (in 1 ml volume) were dialyzed overnight against 2 litres of 10 mM Tris-HCl buffer (pH 7.4). The FAAH/compound mixtures were subjected to activity assays as described above, both before and after dialysis.
All compounds from the present invention demonstrated to be reversible, contrarily to URB597, which was found to be irreversible. Anxiety animal model
Many animal models of anxiety are based on the principle of innate general avoidance behaviors. Among them is the elevated plus maze (EPM) (Hogg S., Pharmacol. Biochem. Behav., 1996, 54, 21; Masse F., et al., Behav. Brain Res., 2007, 177, 2, 214) which is based on the natural aversion of rodents for open spaces that uses the conflict between exploration and aversion of elevated open space; the provoked behavior profiles in the EPM appear to include elements of neophobia, exploration and approach/avoidance conflict. EPM is able to demonstrate the anxiolytic effects of drugs. The purpose of the present study was to set up an animal model of anxiety EPM using the anxiolytic effects of benzodiazepine Diazepam in mice and evaluate the effect of FAAH inhibitors URB597 and ST4020. Twelve male CDl mice (Charles River) of about 30 g (2 months old) per group were used. The Elevated Plus Maze apparatus was of grey Plexiglas and consisted of two open and two closed arms linked by a common central platform. The maze was elevated 40 cm above floor level and dimly lit. Animals were individually placed on the central platform of the maze facing an open arm. A standard five-min test was employed. The amount of time spent by each animal in either open or closed arm was recorded, as well as the number of entries by each animal into either arm.
Experiment: URB597 and ST4020 were tested at a dose of 10 mg/10 mL/kg and Diazepam at a dose of 0.5 mg/5 mL/kg. URB597 and ST4020, dispersed in a solution of 5% Tween 80 and 0.5% carboxymethylcellulose, were given orally
60 min before test; Diazepam, dispersed in a solution of 3% Tween 80, was given intraperitoneal^ 30 min before test. Table 3

* = P < 0.05 vs control
One-way analysis of variance: number of total entrances
Source of variation DF F P Between groups 4.55 4.048 0.006
Dunnett's test: § = P<0.05 vs control
ST4020 reduced anxiety and did not affect the locomotor activity evaluated in elevated plus maze.
Analgesia animal model The analgesic effect of the selective FAAH inhibitor ST4020 was evaluated in a rat model of neuropathic pain induced by intraperitoneal administration vincristine. FAAH inhibitors or vehicle (control group) were given orally. The former were administered at 10 and 50 mg/kg doses, and the analgesic effect
was measured using the paw withdrawal test, as a model of mechanical hyperalgesia.
Rats undergoing treatment were divided into two groups: vehicle (9 animals) and vincristine (54 animals). Neuropathy was induced by intraperitoneal injections of 0.15 mg/kg vincristine three times a week. After two weeks, the rats were distributed into six groups of nine animals: vehicle, vincristine + vehicle, vincristine + URB597 (10 mg/kg), vincristine + URB597 (50 mg/kg), vincristine + ST4020 (10 mg/kg) and Vincristine + ST4020 (50 mg/kg). The animals were subjected to the paw withdrawal test 60, 120 and 240 min after oral administration of the tested compounds.
The acute systemic administration of the selective FAAH inhibitor ST4020 is able to significantly reduce mechanical hyperalgesia associated with a chemotherapeutic model of neuropathic pain.
Claims
1. A compound having the general formula I

Formula I wherein:
R1 and R2 are independently H, alkyl, aryl, arylkyl, alkoxyaryl or haloaryl; or R1 and R2 taken together with the nitrogen atom to which they are attached form a heterocycle;
G is phenylene or thienylene; A is phenyl, thienyl, cyclohexyl, pyrrolyl or pyridyl, each of them being optionally substituted once or twice with aminocarbonyl or alkyl; its tautomers, its geometrical isomers, its optically active forms such as enantiomers, diastereomers and its racemate forms, as well as pharmaceutically acceptable salts thereof; with the proviso that when A is cyclohexyl, R1 cannot be 4-Cl phenyl and 3,4- dichlorophenyl if R2 is H.
2. Compounds according to claim 1, wherein R1 is aryl, alkoxyaryl or haloaryl and R2 is H.
3. Compounds according to any of claims 1-2, wherein G is phenylene and A is phenyl, thienyl, cyclohexyl, pyrrolyl or pyridyl.
4. Compounds according to claim 3, wherein A is 2-thienyl.
5. Compounds according to any of claims 1-3 selected from the group consisting of: l-phenylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST3715);
1- dimethylaminocarbonyloxyimino- 1 - (4-biphenyl)ethane (ST3741) ; l-phenylaminocarbonyloxyimino-l-(3-biphenyl)ethane (ST3986);
1 - (1 - naphthylaminocarbonyloxyimino) - 1 - (4-biphenyl)ethane (ST3987) ; l-(4-fluorophenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3988); l-(4-methoxyphenylaminocarbonyloxyimino)-l-(3-biphenyl)ethane (ST3989); l-(l-piperidinocarbonyloxyimino)-l-(3-biphenyl)ethane (ST4017);
1 - (diphenylaminocarbonyloxyimino)- 1 - (3-biphenyl)ethane (ST4018) ; l-(phenylaminocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4020); l-(phenylaminocarbonyloxyimino)-l-(3-(3'-aminocarbonyl)-biphenyl)ethane
(ST4055); l-[3'-(l-(phenylaminocarbonyloxyimino)-ethyl)-biphenyl-3-carbonyl]-3-phenyl- urea (ST4056); l-(l-piperidinocarbonyloxyimino)-l-(3-(2-thienyl)phenyl)ethane (ST4163); l-(l-dodecylaminocarbonyloxyimino)- l-(3-(2-thienyl)phenyl)ethane (ST4164); l-(phenylaminocarbonyloxyimino)-l-(4-(2-thienyl)phenyl)ethane (ST4165); l-(phenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(4-methoxyphenylaminocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(l-piperidinocarbonyloxyimino)-l-(4-cyclohexylphenyl)ethane; l-(phenylaminocarbonyloxyimino)-l-[4-(2,5-dimethyl-pyrrol-l-yl)- phenyl] ethane;
1 - (4-fluorophenylaminocarbonyloxyimino)- 1 - [4- (2 , 5 - dimethyl-pyrrol- 1 -yl) - phenyl] ethane; 1 - ( 1 - naphthylaminocarbonyloxyimi.no) - 1 - [4- (2 , 5 - dimethyl-pyrrol- 1 -yl)- phenyl] ethane (ST7262);
1 - (4- methoxyphenylaminocarbonyloxyimino) - 1 - [4- (2 , 5 - dimethyl-pyrrol- 1 -yl)- phenyl] ethane; l-(l-piperidinocarbonyloxyimino)-l-[4-(2,5-dimethyl-pyrrol-l-yl)- phenyl] ethane; l-(phenylaminocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane;
1 - ( 1 - naphthylaminocarbonyloxyimino) - 1 - ( [2, 2 '] bithiophenyl- 5 -yl] )ethane ; 1 - (4- methoxyphenylaminocarbonyloxyimino) - 1 - ( [2 ,2 '] bithiophenyl- 5 -yl] )ethane ; l-(l-piperidinocarbonyloxyimino)-l-([2,2']bithiophenyl-5-yl])ethane; l-(phenylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane;
1 - (4-fluorophenylaminocarbonyloxyimino)- 1 - (5 -phenyl-thiophen- 2 -yl)ethane ; l-(l-naphthylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane; l-(4-methoxyphenylaminocarbonyloxyimino)-l-(5-phenyl-thiophen-2-yl)ethane;
1 - ( 1 -piperidinocarbonyloxyimino)- 1 - (5 -phenyl- thiophen- 2 -yl)ethane ; l-(phenylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(l-naphthylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(4-methoxyphenylaminocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(l-piperidinocarbonyloxyimino)-l-(4-pyridin-4-yl-phenyl)ethane; l-(phenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(4-fluorophenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(l-naphthylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-(4-methoxyphenylaminocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane; l-naphthylaminocarbonyloxyimino-l-(4-biphenyl)ethane (ST7380); δ-phenylpentylaminocarbonyloxyimino-l-biphenyl-S-yl-ethane (ST7378) and l-(l-piperidinocarbonyloxyimino)-l-(4-pyridin-2-yl-phenyl)ethane.
6. A pharmaceutical composition containing at least one compound according to claims 1-5 as the active ingredient in mixtures with at least one pharmaceutically acceptable vehicle and/or excipient.
7. A process for preparing the pharmaceutical composition according to claim 6, comprising mixing at least one of the compounds according to claims 1-5 with at least one pharmaceutically acceptable vehicle and/or excipient.
8. Use of compounds according to any one of claims 1-5 as medicaments.
9. Use according to claim 8 for the preparation of a medicament for treating a pathological state for which the modulation of FAAH activity would result at improving the health of the patient.
10. Use according to claim 9, wherein said pathological state is a neurological disorder, disease of energy metabolism, cardiovascular and respiratory disorder, gastrointestinal and liver disorders, retinopathy, cancer and musculoskeletal disorder.
11. Use according to claim 10 where the disorder is a neurological disorder.
12. Use according to claim 11 where the disorder is anxiety.
13. Use according to claim 11 where the disorder is neuropathic pain.
14. Process for synthesizing compounds of claim 1, comprising the step of reacting compounds of formula II, 
Formula Il wherein A and G are as described above, with compounds of formula III 
Formula III wherein R1 and R2 are as described above.
15. Process for synthesizing compounds of claim 2, comprising the step of reacting compounds of formula II with compounds of formula IV  wherein R1 is as described above.
wherein R1 is as described above.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08156228 | 2008-05-15 | ||
| EP08156228.2 | 2008-05-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009138416A1 true WO2009138416A1 (en) | 2009-11-19 |
Family
ID=39713717
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/055748 Ceased WO2009138416A1 (en) | 2008-05-15 | 2009-05-13 | Oxime carbamoyl derivatives as modulators of fatty acid amides hydrolase |
Country Status (3)
| Country | Link |
|---|---|
| AR (1) | AR071800A1 (en) |
| TW (1) | TW201006791A (en) |
| WO (1) | WO2009138416A1 (en) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6949574B2 (en) * | 2002-02-08 | 2005-09-27 | Bristol-Myers Squibb Company | (Oxime)carbamoyl fatty acid amide hydrolase inhibitors |
-
2009
- 2009-05-12 TW TW098115695A patent/TW201006791A/en unknown
- 2009-05-13 WO PCT/EP2009/055748 patent/WO2009138416A1/en not_active Ceased
- 2009-05-15 AR ARP090101751A patent/AR071800A1/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6949574B2 (en) * | 2002-02-08 | 2005-09-27 | Bristol-Myers Squibb Company | (Oxime)carbamoyl fatty acid amide hydrolase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| SAARIO, SUSANNA M. ET AL: "Fatty acid amide hydrolase inhibitors from virtual screening of the endocannabinoid system", JOURNAL OF MEDICINAL CHEMISTRY , 49(15), 4650-4656 CODEN: JMCMAR; ISSN: 0022-2623, 2006, XP002494326 * |
Also Published As
| Publication number | Publication date |
|---|---|
| AR071800A1 (en) | 2010-07-14 |
| TW201006791A (en) | 2010-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU645752B2 (en) | N-phenylalkyl substituted alpha-amino carboxamide derivatives and process for their preparation | |
| EP1884513A1 (en) | Pyrazole compound and therapeutic agent for diabetes comprising the same | |
| US8906946B2 (en) | TRPM8 receptor antagonists | |
| US10105363B2 (en) | Histone deacetylase inhibitors and compositions and methods of use thereof | |
| CA2581730C (en) | New histone deacetylases inhibitors | |
| RS50336B (en) | ARILLED FURAN-1 THIOPHENE-AMIDI-CARBONIC ACIDS WITH POTASSIUM CHANNEL BLOCKING | |
| KR20070047763A (en) | Modulators of Alpha7 Nicotinic Acetylcholine Receptor and Their Therapeutic Uses | |
| HRP20020264A2 (en) | 2'-substituted 1,1'-biphenyl-2-carbonamides, method for the production thereof, use thereof as a medicament and pharmaceutical preparations containing said compounds | |
| CS273197B2 (en) | Method of phenoxyacetamidoderivative production | |
| JP4176805B2 (en) | Pyrrole-3-carboxamide derivatives for the treatment of obesity | |
| Choubdar et al. | New classes of carbazoles as potential multi-functional anti-Alzheimer’s agents | |
| Jaiswal et al. | Scaffold hopping-guided design of some isatin based rigid analogs as fatty acid amide hydrolase inhibitors: Synthesis and evaluation | |
| JP2014144947A (en) | Novel histone deacetylase inhibitor | |
| KR100806603B1 (en) | Aminoalkylamide substituted cyclohexyl derivatives | |
| WO2009109504A1 (en) | Enol carbamate derivatives as modulators of fatty acid amide hydrolase | |
| JP2010516787A (en) | Soluble epoxide hydrolase inhibitors for the treatment of metabolic syndrome and related disorders | |
| WO2009138416A1 (en) | Oxime carbamoyl derivatives as modulators of fatty acid amides hydrolase | |
| RU2402543C2 (en) | Anetholdithiolthiones and other dithiolthiones for treating conditions associated with monoamine neurotransmission dysfunction | |
| KR940011151B1 (en) | Cycloalkylurea compounds | |
| US20120252865A1 (en) | Carbamate derivatives in particular for the treatment of neurological disorders | |
| ES2389754T3 (en) | New thiophenediamine derivatives with urea structure | |
| TAKE et al. | Agents for the Treatment of Overactive Detrusor. IV. Synthesis and Structure-Activity Relationships of Cyclic Analogues of Terodiline | |
| WO2004108094A2 (en) | Sulfonamide-substituted chalcone derivatives and their use to treat diseases | |
| CN101208306A (en) | Pyrazole compounds and diabetes therapeutic agent containing the same | |
| Kumar et al. | Novel substituted naphthalen-1-yl-methanone derivatives as anti-hyperglycemic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09745748 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09745748 Country of ref document: EP Kind code of ref document: A1 |