WO2009143684A1 - 培美曲塞二钠及其中间体4-(4-甲酯基苯基)丁醛的制备方法 - Google Patents
培美曲塞二钠及其中间体4-(4-甲酯基苯基)丁醛的制备方法 Download PDFInfo
- Publication number
- WO2009143684A1 WO2009143684A1 PCT/CN2008/073182 CN2008073182W WO2009143684A1 WO 2009143684 A1 WO2009143684 A1 WO 2009143684A1 CN 2008073182 W CN2008073182 W CN 2008073182W WO 2009143684 A1 WO2009143684 A1 WO 2009143684A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- butanal
- carbomethoxyphenyl
- organic solvent
- ethyl
- reaction mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
Definitions
- This invention relates to the field of chemistry, and more particularly to a process for the preparation of 4-(4-methylphenyl)butanal of pemetrexed disodium and its intermediates. Background technique
- pemetrexed disodium N- ⁇ 4-[2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl) Ethyl]-L-glutamate disodium, the structural formula is as follows:
- Pemetrexed disodium is an anti-tumor drug against folate metabolism that acts by interfering with folate-dependent metabolic processes during cell replication. In vitro tests have shown that it inhibits folate-dependent enzymes such as thymidylate synthase, dihydrofolate reductase, glycine ribonucleoside formyltransferase, which are involved in the biosynthesis of thymidine and purine nucleosides.
- An object of the present invention is to provide a process for synthesizing 4-(4-carbomethoxyphenyl)butanal to overcome the deficiencies of the above techniques for industrial production.
- a process for the preparation of 4-(4-carbomethoxyphenyl)butanal comprising the steps of:
- the organic solvent is selected from one or more of (i) a C5-C8 alkane, a petroleum ether or a C2-C4 ether; (ii) a solvent in the (i) group a mixed solvent with ethyl acetate; (iii) ethyl acetate.
- the C5-C8 alkane is selected from n-hexane or cyclohexane
- the C2-C4 ether is selected from the group consisting of isopropyl ether.
- the organic solvent is selected from the group consisting of n-hexane or a mixed solvent of cyclohexane and ethyl acetate in a volume ratio of 4 to 8:1.
- the step (a) includes adding water to terminate the reaction to obtain a reaction mixture; and in the step (c), the pH of the organic phase extract is adjusted to 7 to 8 with a sodium hydrogencarbonate solution, and then Wash with water and then remove the color with silica gel.
- the organic solvent is removed by evaporating under reduced pressure or vacuum drying in step (c).
- the sodium hydrogencarbonate solution has a weight concentration of 1% to a saturated solution.
- the sodium bicarbonate solution was a saturated sodium bicarbonate solution.
- the volume ratio of the 4-(4-carbomethoxyphenyl)butanal-containing reaction mixture to the organic solvent in the step (b) is 1:0. For 1: 0. 15 ⁇ 0. 50.
- a method for preparing pemetrexed disodium comprises the steps of: (a) condensing methyl p-bromobenzoate and 3-buten-1-ol to form a reaction mixture containing 4-(4-carbomethoxyphenyl)butanal;
- a third aspect of the invention there is provided an improved process for the preparation of pemetrexed disodium, wherein the improvement is that the intermediate 4-(4-methyl ester) is prepared by the process of the first aspect of the invention Phenyl phenyl) butanal.
- the inventors have extensively and intensively studied and found for the first time that the reaction mixture after condensation of methyl bromobenzoate with 3-buten-1-ol can be very efficiently obtained by extraction with an organic solvent, decolorization of silica gel, and removal of an organic solvent. High purity 4-(4-carbomethoxyphenyl)butanal is obtained, and can be directly used for the next bromination reaction of the synthesis of pemetrexed disodium without purification.
- the method of the invention comprises the following steps:
- Methyl p-bromobenzoate and 3-buten-1-ol are condensed, then extracted with an organic solvent, and the extract is added to silica gel for decolorization, and the organic solvent is distilled off to obtain 4-(4-carbomethoxyphenyl).
- the method of the invention comprises the following steps:
- the organic solvent is selected from the group consisting of:
- a mixed solvent of a solvent and ethyl acetate in the (i) group is preferably n-hexane or cyclohexane, and the C2-C4 ether is preferably isopropyl ether;
- a mixed solvent of n-hexane (or cyclohexane) and ethyl acetate is preferable, and the volume ratio of the mixing is not particularly limited, and is usually 4 to 8:1.
- the sodium hydrogencarbonate solution has a weight concentration of 1% to a saturated solution, preferably a saturated sodium hydrogencarbonate solution.
- the volume ratio of the reaction mixture (aqueous solution) to the organic solvent in step (b) is 1: 1 ⁇ 1, preferably 0. 15 ⁇ 0 ⁇ 50;
- the yield of the product obtained by the method of the invention can reach above 80%, and the purity of the GC detection can reach over 95%. It can be directly used to synthesize the next bromination reaction of pemetrexed disodium without purification.
- the present invention provides an improved process for the preparation of pemetrexed disodium, which is improved by the preparation of the intermediate 4-(4-carbophenylphenyl)butanal by the above process of the present invention.
- the intermediate 4-(4-carbomethoxyphenyl)butanal prepared by the method of the present invention can be used to prepare pemetrexed disodium according to a conventional conventional method.
- the intermediate 4-(4-carbophenyl)butanal prepared by the present invention can be directly used in the bromination reaction in the above synthesis method without purification.
- the main advantages of the preparation method of the intermediate of the invention include: simple process operation, low cost reagents and easy availability, and suitable for industrial production. detailed description
- the reaction mixture was cooled to 25 ° C, 200 ml of water was added, and extracted with 100 ml of a mixture of petroleum ether (60-90 ° C) and ethyl acetate (7:1), and the organic layer was combined. After the sodium hydrogen solution was adjusted to pH 7, it was washed with water (5 X 50 ml). 1%, The purity of the GC is 96.1%. The purity of the GC is 96.1%. The purity of the GC is 96.1%. . Example 3
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
培美曲塞二钠及其中间体 4-(4-甲酯基苯基)丁醛的制备方法
技术领域
本发明涉及化学领域, 更具体地涉及培美曲塞二钠及其中间体的 4-(4-甲 酯基苯基)丁醛的制备方法。 背景技术
培美曲塞二钠的化学名是 N-{4-[2-(2-氨基 -4-氧 -4,7-二氢 -1H-吡咯并 [2, 3-d]嘧啶 -5-基)乙基] -L-谷氨酸二钠, 结构式如下:

目前,常用的培美曲塞二钠的合成流程是 EP 0905128中所公开的合成流程。 在该合成流程中, 4-(4-甲酯基苯基)丁醛是合成抗肿瘤药培美曲塞二钠的关键 中间体。
关于 4-(4-甲酯基苯基)丁醛合成方法报道的主要有三种:
一^ J. Org. Chem., 1990, 55: 3222-7报道采用 3-丁炔 -1-醇与对溴苯甲酸 甲酯缩合, 然后再经钯炭氢化还原, PCC 氧化醇得到产品, 此法步骤繁琐, 且 试剂昂贵。
二是 Tetrahedron Letters, 1989, 30:6629-32报道采用 3-丁烯- 1-醇与 对溴苯甲酸甲酯缩合, 经柱层析分离得到纯品, 此法需要柱层析, 不适合工业 化生产。
三是化工时刊, 2006, 20:54-55.报道将 3-丁烯 -1-醇与对溴苯甲酸甲酯缩 合得到的 4-(4-甲酯基苯基)丁醛粗品与亚硫酸氢钠反应形成磺酸盐, 再用盐酸 处理得到纯品, 此法得到的产品纯度高, 但总收率只有 38%。
因此, 本领域迫切需要改进中间体 4- (4-甲酯基苯基)丁醛的制备工艺, 以 便能够工业化、 更高效地制备培美曲塞二钠。 发明内容
本发明的目的是提供一种合成 4- (4-甲酯基苯基)丁醛的方法, 以克服上述 技术的不足, 以便于工业化生产。 在本发明的第一方面, 提供了一种 4- (4-甲酯基苯基)丁醛的制备方法, 它 包括如下步骤:
(a)将对溴苯甲酸甲酯和 3-丁烯 -1-醇进行缩合,形成含 4- (4-甲酯基苯基) 丁醛的反应混合物;
(b)对所述的反应混合物用有机溶剂萃取, 获得有机相萃取物;
(c)对所述的有机相萃取物用硅胶进行脱色,然后去除有机溶剂, 从而得到 4_ (4-甲酯基苯基)丁醛。
在另一优选例中, 所述的有机溶剂选自(i) C5-C8的烷烃、 石油醚或 C2-C4 的醚中的一种或一种以上; (i i)第(i)组中溶剂与乙酸乙酯的混合溶剂; (i i i) 乙酸乙酯。
在另一优选例中,所述的 C5-C8的烷烃选自正己烷或环己烷,所述的 C2-C4 的醚选自异丙醚。
在另一优选例中, 所述的有机溶剂选自正己烷或环己烷和乙酸乙酯的混合 溶剂, 混合的体积比例为: 4〜8: 1。
在另一优选例中, 在步骤(a)中包括加入水终止反应, 获得反应混合物; 并 且在步骤(c)中包括用碳酸氢钠溶液调节有机相萃取物的 pH至 7〜8后, 再用 水洗, 然后硅胶脱色。
在另一优选例中, 在步骤(c)中通过减压蒸干或真空干燥去除有机溶剂。 在另一优选例中, 所述的碳酸氢钠溶液的重量浓度为 1%〜饱和溶液。 碳酸 氢钠溶液为饱和碳酸氢钠溶液。
在另一优选例中, 步骤 (b)中所述的含 4- (4-甲酯基苯基)丁醛的反应混合 物与有机溶剂的体积比为 1: 0. 1〜1, 更佳地为 1 : 0. 15〜0. 50。
在本发明的第二方面, 提供了一种培美曲塞二钠的制备方法, 它包括如下 步骤:
(a)将对溴苯甲酸甲酯和 3-丁烯 -1-醇进行缩合,形成含 4- (4-甲酯基苯基) 丁醛的反应混合物;
(b)对所述的反应混合物用有机溶剂萃取, 获得有机相萃取物;
(c)对所述的有机相萃取物用硅胶进行脱色,然后去除有机溶剂, 从而得到 4- (4-甲酯基苯基)丁醛;
(d)将 4- (4-甲酯基苯基)丁醛与 Br2进行溴代反应, 形成 2-溴 -4- (4-甲酯 基苯基)丁醛;
(e)将 2-溴 -4- (4-甲酯基苯基)丁醛与 2, 4-二氨基 -6-羟基嘧啶反应, 形成 4- [2- (2-氨基 -4, 7-二氢 -4-氧 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酸甲 酯;
(f)将 4- [2- (2-氨基- 4, 7-二氢- 4-氧- 1H-吡咯 [2, 3-d]嘧啶- 5-烃基)乙基] 苯甲酸甲酯进行水解, 从而形成 4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯
[2, 3-d]嘧啶 -5-烃基)乙基]苯甲酸;
(g)将 4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙 基]苯甲酸与 L-谷氨酸二乙酯盐酸盐进行缩合反应, 形成 N- [4- [2- (2-氨基
-4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酰 -L-谷氨酸二乙 酯, 然后再形成对甲苯磺酸盐; 和
(h)将 N- [4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基) 乙基]苯甲酰 -L-谷氨酸二乙酯对甲苯磺酸盐与氢氧化钠反应, 形成培美曲塞二 钠。
在本发明的第三方面, 提供了一种改进的培美曲塞二钠的制备方法, 其中 改进之处在于用本发明第一方面中所述的方法制备中间体 4- (4-甲酯基苯基) 丁醛。 发明详述
本发明人经过广泛而深入的研究, 首次发现, 对于溴苯甲酸甲酯与 3-丁烯 -1-醇缩合后的反应混合物, 通过有机溶剂萃取、 硅胶脱色, 再去除有机溶剂, 可以非常高效地获得高纯度的 4- (4-甲酯基苯基)丁醛, 并且可以不需精制直接 用于合成培美曲塞二钠的下一步溴代反应。 在此基础上, 本发明人完成了本发 明。
本发明的方法包括如下步骤:
将对溴苯甲酸甲酯和 3-丁烯 -1-醇进行缩合, 然后采用有机溶剂萃取, 萃 取物加入硅胶脱色, 蒸去有机溶剂后即可得到 4- (4-甲酯基苯基)丁醛;
在一优选例中, 本发明方法包括如下步骤:
(a)将对溴苯甲酸甲酯溶于合适的溶剂(DMF、 DMA或 DMS0等极性较大的溶 剂)中, 加入二水合乙酸锂, 氯化锂, 四丁基溴化铵, 鼓氮气, 再加入 3-丁烯 -1-醇, 乙酸钯进行缩合反应, 然后加入水终止反应, 获得反应混合物;
(b)向反应混合物中加入有机溶剂萃取, 将得到的有机相(萃取物);
(c)用碳酸氢钠溶液调 pH至 7〜8后再用水洗, 然后硅胶脱色, 减压蒸去溶 剂得到产品;
在本发明方法中, 所述的有机溶剂选自:
(i) C5-C8的烷烃、 石油醚或 C2-C4的醚中的一种或一种以上; 或
(i i)第(i)组中溶剂与乙酸乙酯的混合溶剂。所述的 C5-C8的烷烃优选正己 烷或环己烷, C2-C4的醚优选异丙醚;
优选正己烷(或环己烷)和乙酸乙酯的混合溶剂, 混合的体积比例没有特别 限制, 通常为 4〜8: 1。
所述的碳酸氢钠溶液的重量浓度为 1%〜饱和溶液, 优选饱和碳酸氢钠溶 液。
优选地, 步骤(b)中反应混合物(水溶液)与有机溶剂的体积比为 1 : 0. 1〜 1, 优选 0. 15〜0· 50;
用本发明方法得到的产品收率可达 80%以上, GC检测纯度可达 95%以上。 可以不需精制直接用于合成培美曲塞二钠的下一步溴代反应。
另一方面, 本发明还提供了一种改进的培美曲塞二钠的制备方法, 其改进 之处在于用本发明上述的方法制备中间体 4- (4-甲酯基苯基)丁醛。 换言之, 用 本发明方法制备的中间体 4- (4-甲酯基苯基)丁醛,可以按现有的常规方法用于 制备培美曲塞二钠。
一种优选的制备培美曲塞二钠的方法可参见 ΕΡ 0905128,其反应路线如下:
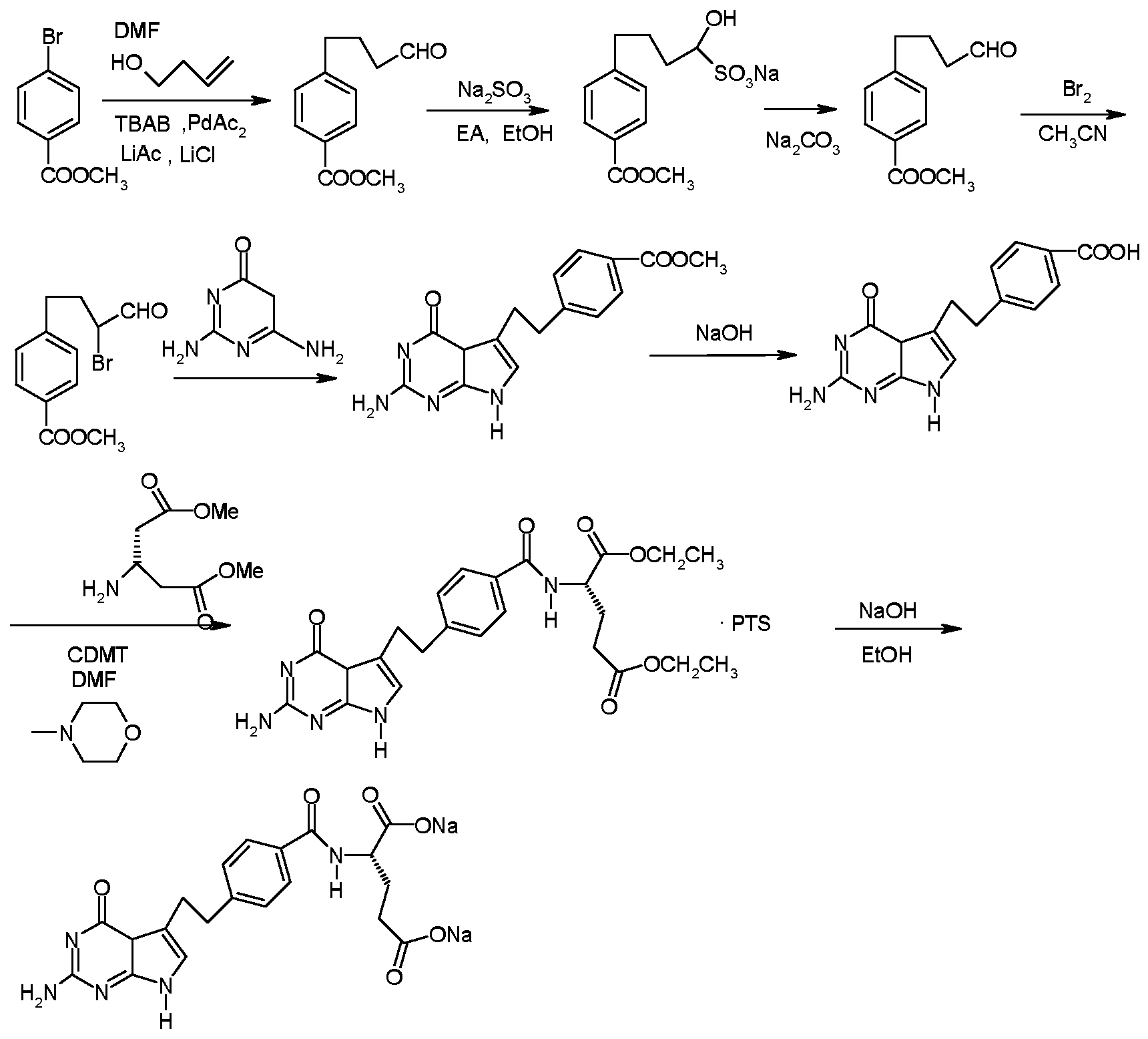
本发明制备的中间体 4- (4-甲酯基苯基)丁醛可以不需精制而直接用于上 述合成方法中的溴代反应。 本发明的中间体制备方法的主要优点包括: 工艺操作简单, 所用试剂价廉 且来源易得, 适合工业化生产。 具体实施方式
下面结合具体实施例, 进一步阐述本发明。 应理解, 这些实施例仅用于说 明本发明而不用于限制本发明的范围。 下列实施例中未注明具体条件的实验方 法, 通常按照常规条件, 或按照制造厂商所建议的条件。 除非另外说明, 否则 份数和百分比为重量份和重量百分比。 实施例 1
4- (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(85. 0g, 0. 395mol)溶于 1L DMF中, 加入二 水合乙酸锂 45. 0g, 氯化锂 50. 0g, 四丁基溴化铵 60. 0g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(34. 0g, 0. 472mol), 乙酸钯 2. 3g,于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 1L, 用环己烷和乙酸乙酯(6 : 1) 混合溶液 300ml萃取 3次, 合并有机层, 有机层用饱和碳酸氢钠溶液调 pH至 8 后,再用水(4 X 500ml)洗涤。加硅胶 80g搅拌半小时脱色,所得淡黄色溶液 40°C 减压蒸干得 4- (4-甲酯基苯基)丁醛 70. 0g, 收率 86. 0%, GC检测纯度为 96. 3%。 实施例 2
4- (4-甲酯基苯基)丁醛的制备缩合反应: 将对溴苯甲酸甲酯(10. 8g,
0. 050mol)溶于 150ml DMF中, 加入二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基 溴化铵 7. 6g, 鼓氮气 5分钟, 再加入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 200ml, 用石油醚(60〜90°C )和乙 酸乙酯(7 : 1)混合溶液 100ml分 3次萃取, 合并有机层, 有机层用饱和碳酸氢 钠溶液调 pH至 7后, 再用水(5 X 50ml)洗涤。 加硅胶 10g搅拌半小时脱色, 所 得淡黄色溶液 4CTC减压蒸干得 4- (4-甲酯基苯基)丁醛 8. 4g, 收率 81. 6%, GC 检测纯度为 96. 1%。 实施例 3
4- (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(10. 8g, 0. 050mol)溶于 150ml DMF中, 加入 二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基溴化铵 7. 6g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 200ml, 用异丙醚和乙酸乙酯(9 :
1)混合溶液 100ml分 3次萃取, 合并有机层, 有机层用饱和碳酸氢钠溶液调 pH 至 7. 5后, 再用水(5 X 50ml)洗涤。 加硅胶 10g搅拌半小时脱色, 所得淡黄色 溶液 4CTC减压蒸干得 4- (4-甲酯基苯基)丁醛 8. 0g, 收率 77. 7%, GC检测纯度 为 95· 2%。 实施例 4
4- (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(10. 8g, 0. 050mol)溶于 150ml DMF中, 加入 二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基溴化铵 7. 6g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 200ml, 用正己烷和乙酸乙酯(4:
1)混合溶液 100ml分 3次萃取, 合并有机层, 有机层用 5%碳酸氢钠溶液调 pH 至 7. 6后, 再用水(5 X 50ml)洗涤。 加硅胶 10g搅拌半小时脱色, 所得淡黄色 溶液 4CTC减压蒸干得 4- (4-甲酯基苯基)丁醛 8. 2g, 收率 79. 6%, GC检测纯度 为 95· 0%。 实施例 5
4_ (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(10. 8g, 0. 050mol)溶于 150ml DMF中, 加入 二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基溴化铵 7. 6g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 200ml, 用正己烷和乙酸乙酯(8 : 1)混合溶液 120ml分 3次萃取, 合并有机层, 有机层用 2%碳酸氢钠溶液调 pH 至 7后, 再用水(5 X 50ml)洗涤。 加硅胶 10g搅拌半小时脱色, 所得淡黄色溶 液 40°C减压蒸干得 4- (4-甲酯基苯基)丁醛 7. 6§, 收率 73. 8%, GC检测纯度为 96. 5%。 实施例 6
4- (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(10. 8g, 0. 050mol)溶于 150ml DMF中, 加入 二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基溴化铵 7. 6g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 20°C, 加水 200ml, 用环己烷 100ml分 3次萃 取,合并有机层,有机层用饱和碳酸氢钠溶液调 pH至 7〜8后,再用水(5 X 50ml) 洗涤。 加硅胶 10g搅拌半小时脱色, 所得淡黄色溶液 4CTC减压蒸干得 4- (4-甲 酯基苯基)丁醛 5. 4g, 收率 52. 4%, GC检测纯度为 96. 8%。
实施例 7
4- (4-甲酯基苯基)丁醛的制备
缩合反应: 将对溴苯甲酸甲酯(10. 8g, 0. 050mol)溶于 150ml DMF中, 加入 二水合乙酸锂 5. 7g, 氯化锂 6. 4g, 四丁基溴化铵 7. 6g, 鼓氮气 5分钟, 再加 入 3-丁烯 -1-醇(4. 3g, 0. 060mol), 乙酸钯 0. 3g, 于 60°C下搅拌反应 10小时。
分离: 将反应混合物冷却至 25 °C, 加水 200ml, 用乙酸乙酯 120ml分 3次 萃取,合并有机层,有机层用饱和碳酸氢钠溶液调 pH至 8后,再用水(5 X 50ml) 洗涤。 加硅胶 10g搅拌半小时脱色, 所得淡黄色溶液 4CTC减压蒸干得 4- (4-甲 酯基苯基)丁醛 9. 0g, 收率 87. 4%, GC检测纯度为 81. 6%。 实施例 8
下一步反应实施例:
4_ [2- (2-氨基 -4, 7-二氢 -4-氧 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲 酸甲酯
将实施例 1中制备的 4- (4-甲酯基苯基)丁醛 18. 9g加入至 100ml乙腈中, 冰浴冷却至 0°C, 保持 0°C缓慢滴加 4. 72ml Br2, 滴毕, 撤去冰浴, 室温下搅 拌反应 2 小时, 在室温下减压旋蒸 20分钟后, 残留物中加入水 100ml, 加入 12. 3g 2, 4-二氨基 -6-羟基嘧啶, 30g 无水乙酸钠, 鼓氮气 5 分钟, 氮气保护 下于 45 °C反应过夜, 反应结束后冷却至 20°C结晶。 过滤, 得土黄色固体湿重 约 26. 2g。
4_ [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯 甲酸
26. 2g上一步的产物加入到 310ml 2N NaOH溶液中, 40°C下搅拌反应 2小时, 加入乙醇 460ml, 冰水浴下以 6N盐酸调节 pH值至 3〜4, 有大量固体析出, 过 滤得淡黄色固体, 用 1 : 1乙醇水溶液洗涤, 干燥后得淡黄色固体 18. 8g, 收率 75. 3%。
N- [4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3_d]嘧啶 -5-烃基)乙基] 苯甲酰 -L-谷氨酸二乙酯对甲苯磺酸盐
18. 8g上一步的产物悬浮于 150ml DMF中,混合物搅拌 15分钟后加入 19. 0g
N-甲基吗啉, 冰水浴下加入 14. 3g 2-氯 -4,6-二甲氧-1,3,5-三嗪(00 了)。 撤去 冰浴混合物室温下搅拌反应 40分钟, 加入 L-谷氨酸二乙酯盐酸盐 19. 5g, 维 持于 35 °C下继续反应 2小时。 反应结束后混合物加入 350ml二氯甲烷和 350ml 去离子水, 搅拌 15 分钟, 静止分层, 分出有机相。 45 °C减压蒸去二氯甲烷, 最后加乙醇使体积至 600ml, 加热溶液至 70〜75 °C, 于 1小时内滴加 30. 8g对 甲苯磺酸(溶于 500ml乙醇的溶液), 有大量固体析出, 继续回流 2小时。 冷却 至室温。 过滤, 滤饼用 750ml无水乙醇洗涤。 干燥得 15. 2g产物, 颜色为灰白 色。 N- [4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3_d]嘧啶 -5-烃基)乙基] 苯甲酰 -L-谷氨酸二钠(培美曲塞二钠)
15. 2g上一步的产物在氮气保护下, 冰浴下溶于 IN 90ml氢氧化钠溶液, 搅拌直至固体完全溶解。 冰浴下以 2N盐酸调节 pH至 3,有大量白色固体析出, 抽干固体。 所得固体搅拌下缓慢滴加 1N氢氧化钠溶液, 控制滴加量使固体恰 好全溶(此时 pH约为 7. 5-8. 5)。 加入 150ml乙醇, 过滤, 滤液中再加入 400ml 乙醇后放冰箱过夜,有大量白色晶体析出。过滤,干燥得 N- [4- [2- (2-氨基 -4, 7- 二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酰 -L-谷氨酸二钠 9. 9g。 产品经 ¾ NMR和 13C NMR检测, 结果与 EP0905128报道的一致。 应理解, 在阅读了本发明的上述讲授内容之后, 本领域技术人员可以对本 发明作各种改动或修改, 这些等价形式同样落于本申请所附权利要求书所限定 的范围。
Claims
1 . 一种 4- (4-甲酯基苯基)丁醛的制备方法, 其特征在于, 包括如下步骤:
(a)将对溴苯甲酸甲酯和 3-丁烯 -1-醇进行缩合,形成含 4- (4-甲酯基苯基) 丁醛的反应混合物;
(b)对所述的反应混合物用有机溶剂萃取, 获得有机相萃取物;
(c)对所述的有机相萃取物用硅胶进行脱色,然后去除有机溶剂, 从而得到 4_ (4-甲酯基苯基)丁醛。
2.根据权利要求 1所述的方法,其特征在于,所述的有机溶剂选自(i) C5-C8 的烷烃、 石油醚或 C2-C4 的醚中的一种或一种以上; (i i)第(i)组中溶剂与乙 酸乙酯的混合溶剂;和(i i i)乙酸乙酯。
3. 根据权利要求 2所述的方法, 其特征在于, 所述的 C5-C8的烷烃选自正 己烷或环己烷, 所述的 C2-C4的醚选自异丙醚。
4. 根据权利要求 2所述的方法, 其特征在于, 所述的有机溶剂选自正己烷 或环己烷和乙酸乙酯的混合溶剂, 混合的体积比例为: 4〜8: 1。
5. 根据权利要求 1所述的方法, 其特征在于, 在步骤(a)中包括加入水终 止反应, 获得反应混合物; 并且在步骤(c)中包括用碳酸氢钠溶液调节有机相 萃取物的 pH至 7〜8后, 再用水洗, 然后硅胶脱色。
6. 根据权利要求 1所述的方法, 其特征在于, 在步骤(c)中通过减压蒸干 或真空干燥去除有机溶剂。
7. 根据权利要求 5所述的方法, 其特征在于, 所述的碳酸氢钠溶液的重量 浓度为 1%〜饱和溶液。 碳酸氢钠溶液为饱和碳酸氢钠溶液。
8. 根据权利要求 1所述的方法, 其特征在于, 步骤(b)中所述的含 4- (4- 甲酯基苯基)丁醛的反应混合物与有机溶剂的体积比为 1 : 0. 1〜1,更佳地为 1 : 0· 15〜0· 50。
9. 一种培美曲塞二钠的制备方法, 其特征在于, 包括如下步骤:
(a)将对溴苯甲酸甲酯和 3-丁烯 -1-醇进行缩合,形成含 4- (4-甲酯基苯基) 丁醛的反应混合物;
(b)对所述的反应混合物用有机溶剂萃取, 获得有机相萃取物;
(c)对所述的有机相萃取物用硅胶进行脱色,然后去除有机溶剂, 从而得到
4_ (4-甲酯基苯基)丁醛;
(d)将 4- (4-甲酯基苯基)丁醛与 Br2进行溴代反应, 形成 2-溴 -4- (4-甲酯 基苯基)丁醛;
(e)将 2-溴 -4- (4-甲酯基苯基)丁醛与 2, 4-二氨基 -6-羟基嘧啶反应, 形成 4- [2- (2-氨基 -4, 7-二氢 -4-氧 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酸甲 酯;
(f)将 4- [2- (2-氨基- 4, 7-二氢- 4-氧- 1H-吡咯 [2, 3-d]嘧啶- 5-烃基)乙基] 苯甲酸甲酯进行水解, 从而形成 4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酸;
(g)将 4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙 基]苯甲酸与 L-谷氨酸二乙酯盐酸盐进行缩合反应, 形成 N- [4- [2- (2-氨基
-4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基)乙基]苯甲酰 -L-谷氨酸二乙 酯, 然后再形成对甲苯磺酸盐; 和
(h)将 N- [4- [2- (2-氨基 -4, 7-二氢 -4-氧代 -1H-吡咯 [2, 3-d]嘧啶 -5-烃基) 乙基]苯甲酰 -L-谷氨酸二乙酯对甲苯磺酸盐与氢氧化钠反应, 形成培美曲塞二 钠。
10.一种改进的培美曲塞二钠的制备方法, 其特征在于, 其改进之处在于用 权利要求 1所述的方法制备中间体 4- (4-甲酯基苯基)丁醛。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/995,257 US8507716B2 (en) | 2008-05-30 | 2008-11-25 | Process for preparing pemetrexed disodium and its intermediate, 4-(4-carbomethoxyphenyl) butanal |
| EP08874449A EP2301909A4 (en) | 2008-05-30 | 2008-11-25 | PROCESS FOR THE PREPARATION OF PEMETREXED DISODIUM AND ITS INTERMEDIATE 4- (4-CARBOMETHYXYPHENYL) BUTANAL |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200810038375.1 | 2008-05-30 | ||
| CN200810038375A CN101591247B (zh) | 2008-05-30 | 2008-05-30 | 合成4-(4-甲酯基苯基)丁醛的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009143684A1 true WO2009143684A1 (zh) | 2009-12-03 |
Family
ID=41376560
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2008/073182 Ceased WO2009143684A1 (zh) | 2008-05-30 | 2008-11-25 | 培美曲塞二钠及其中间体4-(4-甲酯基苯基)丁醛的制备方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8507716B2 (zh) |
| EP (1) | EP2301909A4 (zh) |
| CN (1) | CN101591247B (zh) |
| AR (1) | AR076821A1 (zh) |
| WO (1) | WO2009143684A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010028105A3 (en) * | 2008-09-08 | 2010-06-10 | Dr. Reddy's Laboratories Ltd. | Amorphous pemetrexed disodium |
| US20120245349A1 (en) * | 2011-03-23 | 2012-09-27 | Scinopharm Taiwan, Ltd. | Process for the production of a pemetrexed salt |
| CN105531276A (zh) * | 2013-09-05 | 2016-04-27 | 株式会社三养生物制药 | 用于制备高纯度培美曲塞的得到提高的中间体的制备方法及利用其来制备高纯度培美曲塞的方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012111027A2 (en) * | 2011-02-15 | 2012-08-23 | Hetero Research Foundation | Process for pemetrexed disodium |
| ITRM20120398A1 (it) * | 2012-08-08 | 2014-02-09 | Berlin Chemie Ag | Procedimento di sintesi pemetrexed e suo sale di lisina. |
| KR101372788B1 (ko) * | 2013-08-12 | 2014-03-10 | 제일약품주식회사 | 고순도의 페메트렉시드 이나트륨 염의 제조방법 |
| CN107628947B (zh) * | 2017-09-14 | 2020-07-28 | 浙江工业大学 | 一种培美曲塞二钠关键中间体的制备方法 |
| CN115093415A (zh) * | 2019-05-13 | 2022-09-23 | 南京制药厂有限公司 | 培美曲塞二钠原料药中残留金属钯的去除方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0905128A1 (en) | 1997-09-26 | 1999-03-31 | Eli Lilly And Company | Processes and intermediates useful to make antifolates |
| CN1800169A (zh) * | 2004-12-30 | 2006-07-12 | 上海金色医药科技发展有限公司 | 培美曲塞二钠盐关键中间体及其合成方法,以及由该中间体合成培美曲塞二钠盐的方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3210325A (en) * | 1962-10-15 | 1965-10-05 | Goodrich Co B F | Copolymers of polyesters derived from hydroxymethyl stearic acid |
| BR9812524A (pt) * | 1997-09-26 | 2000-07-25 | Lilly Co Eli | Processos e intermediários utilizáveis para produzir antifolatos |
-
2008
- 2008-05-30 CN CN200810038375A patent/CN101591247B/zh active Active
- 2008-11-25 US US12/995,257 patent/US8507716B2/en not_active Expired - Fee Related
- 2008-11-25 EP EP08874449A patent/EP2301909A4/en not_active Withdrawn
- 2008-11-25 WO PCT/CN2008/073182 patent/WO2009143684A1/zh not_active Ceased
-
2009
- 2009-01-08 AR ARP090100049A patent/AR076821A1/es unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0905128A1 (en) | 1997-09-26 | 1999-03-31 | Eli Lilly And Company | Processes and intermediates useful to make antifolates |
| CN1800169A (zh) * | 2004-12-30 | 2006-07-12 | 上海金色医药科技发展有限公司 | 培美曲塞二钠盐关键中间体及其合成方法,以及由该中间体合成培美曲塞二钠盐的方法 |
Non-Patent Citations (4)
| Title |
|---|
| CHEMICAL INDUSTRY TIMES, vol. 20, 2006, pages 54 - 55 |
| J. ORG. CHEM., vol. 55, 1990, pages 3222 - 7 |
| See also references of EP2301909A4 |
| TETRAHEDRON LETTERS, vol. 30, 1989, pages 6629 - 32 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010028105A3 (en) * | 2008-09-08 | 2010-06-10 | Dr. Reddy's Laboratories Ltd. | Amorphous pemetrexed disodium |
| US20120245349A1 (en) * | 2011-03-23 | 2012-09-27 | Scinopharm Taiwan, Ltd. | Process for the production of a pemetrexed salt |
| US9051322B2 (en) * | 2011-03-23 | 2015-06-09 | Scinopharm Taiwan, Ltd. | Process for the production of a pemetrexed salt |
| CN105531276A (zh) * | 2013-09-05 | 2016-04-27 | 株式会社三养生物制药 | 用于制备高纯度培美曲塞的得到提高的中间体的制备方法及利用其来制备高纯度培美曲塞的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2301909A4 (en) | 2011-11-30 |
| US8507716B2 (en) | 2013-08-13 |
| CN101591247A (zh) | 2009-12-02 |
| US20110124861A1 (en) | 2011-05-26 |
| CN101591247B (zh) | 2012-09-05 |
| AR076821A1 (es) | 2011-07-13 |
| EP2301909A1 (en) | 2011-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009143684A1 (zh) | 培美曲塞二钠及其中间体4-(4-甲酯基苯基)丁醛的制备方法 | |
| CN112062767B (zh) | 一种卢美哌隆的制备方法及其中间体 | |
| AU2018102141A4 (en) | Method for preparing Baricitinib | |
| JP6114881B2 (ja) | 「3−(5−置換オキシ−2,4−ジニトロ−フェニル)−2−オキソ−プロピオン酸エステル」の化合物、そのプロセスおよび用途 | |
| CN102093245B (zh) | 一种西他列汀中间体、西他列汀或其盐的制备方法 | |
| TWI900761B (zh) | 吡咯醯胺化合物的製備方法 | |
| CN101085775A (zh) | 培美曲塞中间体及制备方法 | |
| CN103373989B (zh) | 盐酸帕唑帕尼的中间体的制备方法 | |
| CN102675135B (zh) | 一种合成α-氨基酸酯的方法 | |
| EP2279186A1 (en) | CONVERSION OF TRYPTOPHAN INTO ß-CARBOLINE DERIVATIVES | |
| JP2016531925A (ja) | ペメトレキセド製造のための中間体製造方法及びこれを用いて高純度ペメトレキセドを製造する方法 | |
| CN101607971A (zh) | 9-[2-(二乙氧基膦酰甲氧基)乙基]腺嘌呤的合成方法 | |
| CN102532109B (zh) | 一种拉帕替尼及其盐的合成方法 | |
| CN113214197B (zh) | 一种维生素c乙基醚的制备方法 | |
| TW202302605A (zh) | 新穎方法 | |
| CN103373963B (zh) | 盐酸帕唑帕尼的中间体及其制备方法 | |
| CN107365312B (zh) | 一种制备Oclacitinib的新方法 | |
| CN100379729C (zh) | 硝基化合物及其在培美曲塞制备中的应用 | |
| CN101293854B (zh) | 培美曲塞的新中间体及制备方法与应用 | |
| CN100408577C (zh) | 咪唑并(1,2-a)吡啶-3-乙酰胺类的制备方法 | |
| TWI330642B (en) | Method for producing triterpene | |
| CN118047762B (zh) | 一种利扎曲普坦ep杂质c的制备方法 | |
| CN105377826A (zh) | 杂环化合物的制造方法 | |
| CN118930551B (zh) | 制备β-氨基吡嗪乙酸酯的方法 | |
| JP5507579B2 (ja) | N−[5−(3−ジメチルアミノ−アクリロイル)−2−フルオロ−フェニル]−n−メチル−アセトアミドの調製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08874449 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008874449 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12995257 Country of ref document: US |