WO2009144288A1 - Process to prepare paliperidone and intermediates thereof - Google Patents
Process to prepare paliperidone and intermediates thereof Download PDFInfo
- Publication number
- WO2009144288A1 WO2009144288A1 PCT/EP2009/056578 EP2009056578W WO2009144288A1 WO 2009144288 A1 WO2009144288 A1 WO 2009144288A1 EP 2009056578 W EP2009056578 W EP 2009056578W WO 2009144288 A1 WO2009144288 A1 WO 2009144288A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- process according
- compound
- solvent
- salt
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- NMALKTKBJPTUDK-UHFFFAOYSA-N CC(N=C1N2C=CC=C1O)=C(CCCl)C2=O Chemical compound CC(N=C1N2C=CC=C1O)=C(CCCl)C2=O NMALKTKBJPTUDK-UHFFFAOYSA-N 0.000 description 2
- OMQHDIHZSDEIFH-UHFFFAOYSA-N CC(C(CCO1)C1=O)=O Chemical compound CC(C(CCO1)C1=O)=O OMQHDIHZSDEIFH-UHFFFAOYSA-N 0.000 description 1
- GNJWAVGJDQQQSS-UHFFFAOYSA-N CC(N=C1N2C=CC=C1O)=C(CCO)C2=O Chemical compound CC(N=C1N2C=CC=C1O)=C(CCO)C2=O GNJWAVGJDQQQSS-UHFFFAOYSA-N 0.000 description 1
- JKVUGXRJSYRXFN-UHFFFAOYSA-N CC(N=C1N2CCCC1O)=C(CCCl)C2=O Chemical compound CC(N=C1N2CCCC1O)=C(CCCl)C2=O JKVUGXRJSYRXFN-UHFFFAOYSA-N 0.000 description 1
- PMXMIIMHBWHSKN-UHFFFAOYSA-N CC(N=C1N2CCCC1O)=C(CCN(CC1)CCC1c1n[o]c3c1ccc(F)c3)C2=O Chemical compound CC(N=C1N2CCCC1O)=C(CCN(CC1)CCC1c1n[o]c3c1ccc(F)c3)C2=O PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 description 1
- BMTSZVZQNMNPCT-UHFFFAOYSA-N Nc(nccc1)c1O Chemical compound Nc(nccc1)c1O BMTSZVZQNMNPCT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the invention relates to the synthesis of Paliperidone, a drug useful to treat schizophrenia, and processes to prepare intermediates thereof with improved yield and atomic economy.
- Paliperidone, I is an orally active drug useful for the treating of schizophrenia. Its chemical name is ( ⁇ )-3-[2-[4-(6-fluoro-l,2-benzisoxazol-3-yl)-l- piperidinyl]ethyl]-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4/f-pyrido[l,2- a]pyrimidin-4-one and its chemical structure is depicted below:
- Paliperidone was first disclosed in European patent application EP0368388. In said application Paliperidone is prepared as depicted in Scheme 1.
- Said compound V is coupled with compound VI in the presence of diisopropylamine in methanol to yield the desired compound I in a 21% yield.
- protecting groups are widely present in the field of organic synthesis, and they are useful to differentiate two or more moieties with similar reactivity. Even that, its use reduces the atomic economy of the reaction and increases the waste by-products. All this increases the costs and the environmental problems of the reaction.
- the use of the benzyl protecting group increases the amount of hydrogen gas, which is a toxic and flammable gas, consumed during the synthesis process.
- the invention relates to a process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with compound V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent (Scheme 4).
- the invention also relates to a process for obtaining 3-(2-chloroethyl)-2- methyl-9-hydroxy-4H-pyrido[l,2-a]pyrimidin-4-one, IX, or a salt thereof, comprising two steps (Scheme 6): a) reaction of 2-amino-3-hydroxypyridine, VII, and 2-acetylbutyrolactone,
- diol VIII in a solvent and a cosolvent to obtain the diol VIII, b) diol VIII is reacted with a suitable chlorinating agent, [Cl ⁇ ], to selectively obtain compound IX, and c) optionally, compound IX is converted into a salt thereof.
- FIG.l illustrates the X-ray powder diffraction pattern of IX. ⁇ C1;
- FIG.2 illustrates the X-ray powder diffraction pattern of an embodiment of purified IX.HC1.
- FIG.3 illustrates the Differential scanning calorimetry of a second embodiment of purified IX.HC1.
- FIG.4 illustrates the X-ray powder diffraction pattern of a second embodiment of purified IX.HC1. DETAILED DESCRIPTION OF THE INVENTION
- one -pot reaction means two or more reactions that take place without isolating intermediate compounds, wherein all the reactants are added at the beginning of the first reaction or adding the reactants sequentially during the course of the reaction.
- polar aprotic solvent relates to a polar solvent that is not capable of exchanging protons with the reagents and that has no polarizable proton.
- polar aprotic solvents are dimethylformamide (DMF), dimethylsulphoxide (DMSO), N-methylpyrrolidone and dimethylacetamide (DMAc).
- the FT-IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrophotometer in an ATR accessory, from 650 to 4000 cm "1 .
- the present invention relates to a process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with compound V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent.
- the solvent used in the process for obtaining Paliperidone of the present invention is selected from an alcohol such as methanol, ethanol or isopropanol or water or a mixture of said solvents.
- an alcohol such as methanol, ethanol or isopropanol or water or a mixture of said solvents.
- the base triethylamine or diisopropylethylamine, is used as solvent.
- the process of the present invention preferably takes place from room temperature to the reflux temperature of the solvent.
- the preferred temperature is from 50 0 C to the reflux temperature of the solvent.
- Compound V, or a salt thereof, used in the process of the present invention can be prepared by hydrogenation of compound IX, or a salt thereof.
- the hydrogenation of compound IX can be performed using a palladium hydrogenation catalyst in an alcoholic solution.
- the alcohol is preferably methanol, ethanol or isopropanol.
- the alcoholic solution consists of an alcohol of commercial purity (see Example 3).
- the alcoholic solution consists of a mixture of the alcohol with water, from 1 :0.01 to a ratio of 1 :1 (v/v) (i.e. from 99% alcohol up to 50% by volume of alcohol in the mixture), preferably from 99% to 80% of alcohol to water (see Example 4).
- the preparation of intermediate 3-(2-chloroethyl)-2-methyl-9-hydroxy-4H- pyrido[l,2-a]pyrimidin-4-one, IX, or a salt thereof can comprise the following steps: a) reaction of 2-amino-3-hydroxypyridine, VII, and 2-acetylbutyrolactone, III, in a solvent and a cosolvent to obtain the diol VIII, b) diol VIII is reacted with a suitable chlorinating agent, [Cl ], to selectively obtain compound IX, and c) optionally, compound IX is converted into a salt thereof.
- the solvent used in the aforementioned step a) is an aromatic hydrocarbon or a mixture thereof such as benzene, toluene or xylenes.
- the cosolvent used in step a) is a polar aprotic solvent such as JV-methylpyrrolidone, DMSO, DMAc or DMF.
- an acidic catalyst such as methanesulphonic acid, /?-toluensulphonic acid, sulphuric acid or acetic acid is added.
- the catalyst is methanesulphonic acid.
- Step a) of the process of the present invention to prepare compound IX can be performed from 80 0 C to the reflux temperature of the solvent or mixture of solvents, preferably it is performed at reflux temperature.
- the suitable chlorinating agent, [CF], of the aforementioned step b) is selected from POCI3, PCI3, PCI5, ⁇ f-chlorosuccinimide, cyanuric chloride or the like.
- Step b) of the process of the present invention to prepare compound IX can be performed from room temperature to the reflux temperature of the solvent or mixture of solvents.
- the temperature is 80-120 0 C, more preferably 90-100
- the process of the present invention to prepare compound IX is performed with means to remove the evolved water, such as a Dean-Stark receiver or molecular sieves.
- the process of the present invention to prepare compound IX can be performed as a one pot reaction, or the intermediate alcohol VIII can be isolated.
- compound IX is prepared as a one pot reaction without isolation of VIII. Since a chlorinating agent is used in step b), compound IX is likely to be obtained in the form of the hydrochloride salt.
- Other salts such as sulphate, hydrogensulphate, oxalate or acetate can also be formed from the free base and used for the purposes of the present invention.
- IR 2650, 2603, 2523, 1701, 1636, 1616, 1580, 1508, 1441, 1403, 1358, 1334, 1302, 1229, 1189, 1172, 1160, 1138, 1101, 1074, 1039, 1014, 998, 938, 918, 888, 874, 825, 800, 789, 755, 708, 678;
- XRD Peak list 7.85, 10.51, 13.25, 14.60, 16.65, 17.20, 19.50, 21.04, 22.63, 23.13, 23.29, 24.00, 24.97, 25.91, 26.30, 26.68, 27.09, 29.21, 29.72, 30.51, 32.74,
- the obtained XRD pattern was the same as that obtained in the previous embodiment.
- the product obtained as described above can be further dried in the vacuum oven at 60 0 C for 24 hours.
- the product was characterised by IR and DSC:
- the mixture is allowed to cooled down to 70-75 0 C and POCl 3 (12.5 ml, 136.55 mmol) is added, then the mixture is heated to 90-95 0 C and maintained until disappearance of the reactants. Then, the reaction mixture is allowed to cool to room temperature and water is added. The mixture is neutralised with NH 3 aq and the product is extracted with DCM (3x 100ml). The solvent is evaporated under reduced pressure and an oily residue is obtained. Next, IPA is added and HClaq is added until pH 2-3. An off- white solid precipitates during the addition. The suspension is allowed to cool and the solid is filtered and washed with IPA.
- the solution is placed in a hydrogenation reactor and 3.75 mg of Pd/C are added to the mixture.
- the reactor is then purged with nitrogen once and three times with H 2 .
- the reaction mixture was heated to 50 0 C and the reaction was controlled until disappearance of the reactants at atmospheric pressure.
- IR 1651.6, 1597.0, 153.9, 1486.4, 1447.2, 1326.0, 1270.4, 1 182.1, 1118.8, 1074.2, 1020.7, 981.0, 951.1, 907.6, 802.1, 736.0, 706.5, 659.7.
- IR 1691.5, 1661.8, 1591.8, 1548.8, 1454.4, 1432.4, 1388.5, 1329.8, 1188.3, 1113.0, 1058.0, 1025.6, 906.1, 832.3, 789.5, 728.8.
- reaction vessel is purged an extra five times with hydrogen (50-60 psi) and heated up to 55°C under hydrogen (at atmospheric pressure) over 20 minutes. After that, the vessel is pressurized with hydrogen (15 psi) and mechanical stirring supplied (1500 rpm) at 55°C for 3 hours. The reaction vessel is then cooled to room temperature and carefully vented.
- reaction solution is filtered over celite, the filtered cake washed with MeOH and the combined filtrates evaporated to dryness to give V.HC1, as an off-white solid.
- a solution of potassium acetate in water is added drop wise, until complete precipitation of compound V is achieved.
- the obtained precipitate is filtered and washed with water, yielding 2.23g (92% yield) of compound V.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
The present invention relates to an improved process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with compound V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent. Moreover, the invention relies on the preparation of intermediates used in such process.
Description
PROCESS TO PREPARE PALIPERIDONE AND INTERMEDIATES
THEREOF
FIELD OF THE INVENTION
The invention relates to the synthesis of Paliperidone, a drug useful to treat schizophrenia, and processes to prepare intermediates thereof with improved yield and atomic economy.
BACKGROUND
Paliperidone, I, is an orally active drug useful for the treating of schizophrenia. Its chemical name is (±)-3-[2-[4-(6-fluoro-l,2-benzisoxazol-3-yl)-l- piperidinyl]ethyl]-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4/f-pyrido[l,2- a]pyrimidin-4-one and its chemical structure is depicted below:

Paliperidone was first disclosed in European patent application EP0368388. In said application Paliperidone is prepared as depicted in Scheme 1.

Scheme 1
In the synthesis exemplified in said document it is used a benzyl protecting group in the hydroxyl of pyridine II. Protected pyridine II is reacted with the lactone III where the protecting group in the hydroxyl avoids selectivity problems during the coupling step. Then, the phosphoryl chloride substitutes the resulting hydroxyl group with chlorine and compound IV is obtained in a 62.3% yield, again the benzyl group avoids selectivity problems. Compound IV is then hydrogenated in the presence of a palladium catalyst to remove the benzyl protecting group and to reduce the pyridine ring. Compound V is obtained quantitatively.
Said compound V is coupled with compound VI in the presence of diisopropylamine in methanol to yield the desired compound I in a 21% yield.
International patent applications WO2008021345A and WO2008024415A also relate to the synthesis of Paliperidone and use a similar approach, also using, a benzyl protecting group with low to moderate yields.
The use of protecting groups is widely present in the field of organic synthesis, and they are useful to differentiate two or more moieties with similar reactivity. Even that, its use reduces the atomic economy of the reaction and increases the waste by-products. All this increases the costs and the environmental problems of the reaction.
Not only that, but in this case the use of the benzyl protecting group increases the amount of hydrogen gas, which is a toxic and flammable gas, consumed during the synthesis process.
There are documents describing the synthesis of suitable intermediates, such as EP0730594 and EP0808313 that prepare compound VIII in xylene and /?-toluenesulphonic acid (Scheme 2). The resulting hydroxy ethyl moiety is activated with a mesyl group.

In WO2008021345 it is described the preparation of Paliperidone, I, via the coupling of compounds V and VI using an inorganic base (Scheme 3).



Scheme 3
Therefore, there is a need in the art to develop a new process to obtain Paliperidone with higher yield, increase atomic economy and less environmental drawbacks.
All the documents cited therein are enclosed in its entirety by reference.
BRIEF DESCRIPTION OF THE INVENTION
The invention relates to a process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with compound V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent (Scheme 4).

Scheme 4
Moreover, the invention relies on to a process for preparing compound V or a salt thereof by hydrogenation of compound IX or a salt thereof (Scheme 5).

Scheme 5
The invention also relates to a process for obtaining 3-(2-chloroethyl)-2- methyl-9-hydroxy-4H-pyrido[l,2-a]pyrimidin-4-one, IX, or a salt thereof, comprising two steps (Scheme 6): a) reaction of 2-amino-3-hydroxypyridine, VII, and 2-acetylbutyrolactone,
III, in a solvent and a cosolvent to obtain the diol VIII, b) diol VIII is reacted with a suitable chlorinating agent, [Cl~], to selectively obtain compound IX, and c) optionally, compound IX is converted into a salt thereof.

Scheme 6
FIGURES
FIG.l illustrates the X-ray powder diffraction pattern of IX.ΗC1;
FIG.2 illustrates the X-ray powder diffraction pattern of an embodiment of purified IX.HC1.
FIG.3 illustrates the Differential scanning calorimetry of a second embodiment of purified IX.HC1.
FIG.4 illustrates the X-ray powder diffraction pattern of a second embodiment of purified IX.HC1.
DETAILED DESCRIPTION OF THE INVENTION
In the context of the present invention, the following terms have the meaning detailed below:
The term "one -pot reaction" means two or more reactions that take place without isolating intermediate compounds, wherein all the reactants are added at the beginning of the first reaction or adding the reactants sequentially during the course of the reaction.
The term "polar aprotic solvent" relates to a polar solvent that is not capable of exchanging protons with the reagents and that has no polarizable proton. Examples of polar aprotic solvents are dimethylformamide (DMF), dimethylsulphoxide (DMSO), N-methylpyrrolidone and dimethylacetamide (DMAc).
The FT-IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrophotometer in an ATR accessory, from 650 to 4000 cm"1.
The present invention relates to a process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with compound V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent.

It has been surprisingly found that the change from the diisopropylamine base used in the prior art (EP0368388) to triethylamine or diisopropylethylamine allows to obtain the desired product Paliperidone, I, not only with a great increase on the yield (93%) but also without requiring column chromatography. In fact, only one crystallization is needed. Therefore, the use of triethylamine or diisopropylethylamine allows increasing the yield of the process and avoids the use of column chromatographic solvents and other related materials. Moreover, another
advantage of avoiding purification through column chromatography is the use of no special equipment which means that the process can be undertaken with common reactors.
The increase on the yield and the use of common reactors to perform the reaction reduces the costs of the overall process, and the reduction of the solvents and other materials reduces the environmental drawbacks of the reaction.
The solvent used in the process for obtaining Paliperidone of the present invention is selected from an alcohol such as methanol, ethanol or isopropanol or water or a mixture of said solvents. When the process is performed in the absence of a solvent, the base, triethylamine or diisopropylethylamine, is used as solvent.
The process of the present invention preferably takes place from room temperature to the reflux temperature of the solvent. The preferred temperature is from 50 0C to the reflux temperature of the solvent.
Compound V, or a salt thereof, used in the process of the present invention can be prepared by hydrogenation of compound IX, or a salt thereof.

The hydrogenation of compound IX can be performed using a palladium hydrogenation catalyst in an alcoholic solution. The alcohol is preferably methanol, ethanol or isopropanol. According to one embodiment of the present invention, the alcoholic solution consists of an alcohol of commercial purity (see Example 3).
According to another embodiment of the present invention, the alcoholic solution consists of a mixture of the alcohol with water, from 1 :0.01 to a ratio of 1 :1 (v/v) (i.e. from 99% alcohol up to 50% by volume of alcohol in the mixture), preferably from 99% to 80% of alcohol to water (see Example 4).
The preparation of intermediate 3-(2-chloroethyl)-2-methyl-9-hydroxy-4H- pyrido[l,2-a]pyrimidin-4-one, IX, or a salt thereof, can comprise the following steps: a) reaction of 2-amino-3-hydroxypyridine, VII, and 2-acetylbutyrolactone, III, in a solvent and a cosolvent to obtain the diol VIII,
b) diol VIII is reacted with a suitable chlorinating agent, [Cl ], to selectively obtain compound IX, and c) optionally, compound IX is converted into a salt thereof.

The use of a cosolvent facilitates the solubility of the starting and final products resulting in an easier and faster reaction. To facilitate this step a) of the process, an acidic catalyst such as methanesulphonic acid, /?-toluensulphonic acid, sulphuric acid or acetic acid is added. Preferably, the catalyst is methanesulphonic acid.
Step a) of the process of the present invention to prepare compound IX, can be performed from 80 0C to the reflux temperature of the solvent or mixture of solvents, preferably it is performed at reflux temperature.
The suitable chlorinating agent, [CF], of the aforementioned step b) is selected from POCI3, PCI3, PCI5, Λf-chlorosuccinimide, cyanuric chloride or the like.
Step b) of the process of the present invention to prepare compound IX, can be performed from room temperature to the reflux temperature of the solvent or mixture of solvents. Preferably the temperature is 80-120 0C, more preferably 90-100
0C.
The process of the present invention to prepare compound IX is performed with means to remove the evolved water, such as a Dean-Stark receiver or molecular sieves. The process of the present invention to prepare compound IX can be performed as a one pot reaction, or the intermediate alcohol VIII can be isolated. Preferably, compound IX is prepared as a one pot reaction without isolation of VIII.
Since a chlorinating agent is used in step b), compound IX is likely to be obtained in the form of the hydrochloride salt. Other salts such as sulphate, hydrogensulphate, oxalate or acetate can also be formed from the free base and used for the purposes of the present invention.
The following non-limiting examples will further illustrate specific embodiments of the invention. They are, however, not intended to be limiting the scope of the present invention in any way.
EXAMPLES
Example 1 : 3-(2-chloroethyl)-2-methyl-9-hydroxy-4H-pyrido [ 1 ,2-a] pyrimidin-4- one hydrochloride, IX-HCl
Solvent

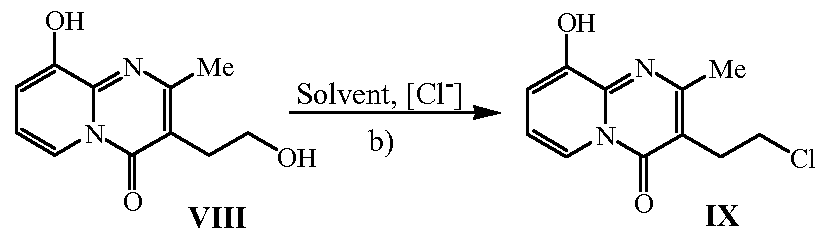
5.O g (45.41 mmol) of 2-amino-3-hydroxypyridine, VII, are suspended in xylene (35 ml) and JV-methylpyrrolidone (5 ml). Next, methanesulphonic acid (0.021 ml; 0.31 mmol) and 2-acetylbutyrolactone, III, (2.9 ml, 26.93 mmol) were added. The resulting suspension is refluxed collecting the water evolved in a Dean-Stark receiver during two hours. Then a second portion of the 2-acetylbutyrolactone, III, (2.4 ml, 22.29 mmol) is added to the reaction mixture and the reflux is maintained until disappearance of the reactants.
The mixture is allowed to cooled down to 70-75 0C and POCI3 (12.5 ml, 136.55 mmol) is added, then the mixture is heated to 90-95 0C and maintained until disappearance of the reactants. Then the mixture is allowed to cool to room temperature and water is added and the mixture cooled to 10 0C and a off- white solid precipitates that is filtered and washed with water. 9.4 g (34.4 mmol, 75% yield) of IXΗC1 were obtained.
Analytical data:
RMN 1H (D2O) δ (ppm): 2.70 (s, CH3); 3.21 0,CH2); 3.87 0,CH2Cl); 7.58 (t, αrCH); 7.75 (d, αrCH); 8.71(J, αrCH). m.p.: 213 - 218 °C
DSC: m.p. = 212.9 0C
K.F. = 5.6 % Of H2O
IR: 2844.8, 1703.5, 1641.4, 1584.10, 1515,9, 1406.8, 1297.7, 1227.0, 1162.2, 1142.3, 999.8, 877.2, 815.6, 778; 756.6; 722; 686 cm"1 XRD Peak list: 8.68, 12.25, 13.71, 16.01, 16.58, 19.39, 20.67, 23.67, 25.58,
25.99, 28.02, 28.21, 31.02,
Purification of IX.HC1
The product IX-HCl (3-(2-chloroethyl)-2-methyl-9-hydroxy-4H-pyrido[l,2- a]pyrimidin-4-one monohydro chloride) thus obtained (9.4 g; 34.15 mmol), is placed in a round bottom flask and isopropyl alcohol 47ml is added. The suspension is taken to 60 0C and water is added until a clear solution is obtained. The solution is then allowed to cool and taken to 0 0C. The off-white precipitate was filtered and washed with isopropyl alcohol and dried.7.01 g (75% yield of a 99% HPLC purity pure product) of compound IX are obtained. Analytical data of IX-HCl:
RMN 1H (D2O) δ (ppm): 2.70 (s, CH3); 3.21 0,CH2); 3.87 0,CH2Cl); 7.57 (t, αrCH); 7.74 (d, αrCH); 8.70(J, αrCH). DSC: m.p.= 228 0C
K.F: 4.7 % OfH2O
IR: 3493; 3290; 1692.3; 1644.5; 1619.1; 1589.4; 1511.6, 1411,7; 1304,02; 1230.6; 1174.4; 1146.7; 1075.1; 914.1, 874.7, 811.1, 781.7, 760.9; 739.7; 721; 686.
XRD Peak list: 10.49, 11.08, 11.91, 16.54, 22.47, 23.23, 23.94, 26.08, 26.77, 27.00, 28.01, 35.07.
The product obtained as described above can be further dried in the vacuum oven at 60 0C for 24 hours. Analytical data:
K.F: 0.1 % Of H2O DSC: m.p.= 227 0C
IR: 2650, 2603, 2523, 1701, 1636, 1616, 1580, 1508, 1441, 1403, 1358, 1334, 1302, 1229, 1189, 1172, 1160, 1138, 1101, 1074, 1039, 1014, 998, 938, 918, 888, 874, 825, 800, 789, 755, 708, 678;
XRD Peak list: 7.85, 10.51, 13.25, 14.60, 16.65, 17.20, 19.50, 21.04, 22.63, 23.13, 23.29, 24.00, 24.97, 25.91, 26.30, 26.68, 27.09, 29.21, 29.72, 30.51, 32.74,
33.39.
Alternative purification method of IX.HC1
3-(2-chloroethyl)-2-methyl-9-hydroxy-4H-pyrido[ 1 ,2-a]pyrimidin-4-one monohydrochloride, IX.HC1, (15.Og; 54.5mmol) was placed in a round bottom flask and isopropyl alcohol was added. The suspension was taken to reflux and it was kept for 30 minutes. The reaction mixture was then allowed to cool and taken to 0 0C. The off-white precipitate was filtered and washed with isopropyl alcohol and dried. 13.3g (88.7% yield of a 98% HPLC purity pure product) of compound IX.HC1 were obtained.
Analytical data:
K.F= 0.1% Of H2O DSC: m.p.= 227.6 0C.
RMN 1H (D2O) δ (ppm): 2.70 (s, CH3); 3.22 (f,CH2); 3.88 (^CH2Cl); 7.59 (t, αrCH); 7.75 (d, αrCH); 8.71(J, αrCH).
IR: 1709.7; 1642.6; 1620.4; 1583.2; 1514.2; 1410.73; 1303.02; 1228.6; 1171.1; 1144.7; 1076.7; 919.1; 876.6; 815.2 ; 781.65; 736.7; 719.9; 679.9 cm"1. XRD Peak list: 7.94, 10.57, 13.37, 14.64, 16.45, 17.28, 19.55, 21.11, 22.26,
22.70, 23.54, 24.08, 25.03, 26.00, 26.20, 26.39, 26.83, 27.21, 28.85, 29.43, 30.59,
32.67. The obtained XRD pattern was the same as that obtained in the previous embodiment.
The product obtained as described above can be further dried in the vacuum oven at 60 0C for 24 hours. The product was characterised by IR and DSC:
DSC: m.p.= 222.7-230.74 0C
IR: 2439.4; 1712.5; 1640.6; 1618.3; 1578.9; 1522.4; 1442.6; 1410.4; 1356; 1333.3; 1300.9; 1270.1; 1228.33; 1189.4; 1169; 1140.8; 1076.1; 1039.5; 1020.9; 921.47; 876.6; 849.9; 789.9; 755.9; 736.0; 705.73; 676.8 cm"1.
Example 2 : 3-(2-chloroethyl)-2-methyl-9-hydroxy-4H-pyrido [ 1 ,2-a] pyrimidin-4- one monohydrochloride, IX.
5.O g (45.41 mmol) of 2-amino-3-hydroxypyridine, VII, are suspended in xylene (35 ml) and JV-methylpyrrolidone (5 ml). Next, methanesulphonic acid
(0.021ml; 0.31mmol) and 2-acetylbutyrolactone, III, (2.9 ml, 26.93 mmol) are added to the suspension. The resulting suspension is refluxed collecting the water evolved in a Dean-Stark receiver during two hours. Then a second portion of the 2- acetylbutyrolactone, III, (2.4 ml, 22.29 mmol) is added to the reaction mixture and the reflux is maintained until disappearance of the reactants.
The mixture is allowed to cooled down to 70-75 0C and POCl3 (12.5 ml, 136.55 mmol) is added, then the mixture is heated to 90-95 0C and maintained until disappearance of the reactants. Then, the reaction mixture is allowed to cool to room temperature and water is added. The mixture is neutralised with NH3aq and the product is extracted with DCM (3x 100ml). The solvent is evaporated under reduced pressure and an oily residue is obtained. Next, IPA is added and HClaq is added until pH 2-3. An off- white solid precipitates during the addition. The suspension is allowed to cool and the solid is filtered and washed with IPA. 9.4 g (34.4 mmol, 75% yield of a 98% HPLC pure product) of IX are obtained.
Example 3 : 3-(2-chloroethyl)-6,7,8,9-tetrahydro-2-methyl-9-hydroxy-4H- pyrido[l,2-a]pyrimidin-4-one, V
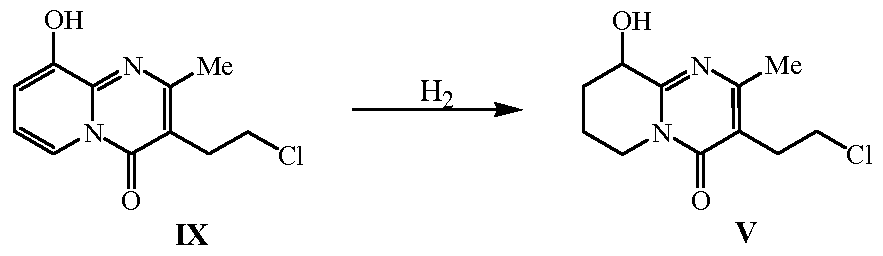
25.0 g (90.86 mmol) of compound IX hydrochloride are dissolved in methanol (225ml) and the solution is then treated with carbon and Celite during Ih at
40 0C. The mixture was then filtered and the cake was washed with hot methanol.
The solution is placed in a hydrogenation reactor and 3.75 mg of Pd/C are added to the mixture. The reactor is then purged with nitrogen once and three times with H2.
The reaction mixture was heated to 50 0C and the reaction was controlled until disappearance of the reactants at atmospheric pressure.
When the reaction is over the catalyst is filtered and the solvent is evaporated until an oily residue is obtained. Then, water is added and a solution of potassium acetate in water is added dropwise to the above solution. The compound precipitates and is filtered and washed with water. 19.7 g. (81.17 mmol; 89.5% yield) of the desired product V are obtained.
Analytical data of V: NMR 1H (CDCl3) δ (ppm): 1.72-2.33 (m, CH2-CH2); 2.37 (s, CH3); 3.01
(f,CH2); 3.77 (t, CH2Cl); 3.94 (at, CH2); 4.52 (t, CH-OH). m.p.: 102-106 0C
IR: 1651.6, 1597.0, 153.9, 1486.4, 1447.2, 1326.0, 1270.4, 1 182.1, 1118.8, 1074.2, 1020.7, 981.0, 951.1, 907.6, 802.1, 736.0, 706.5, 659.7.
Preparation of VΗCl
0.500 g (2.06mmol) of compound V, obtained according to the process described above, are dissolved in 2-butanone (3.5 ml) and concentrated hydrochloric acid is added to the solution until pH 2 is obtained. The solvent was then removed
under vacuum till an oily residue is obtained. 2-Butanone is added to the residue and evaporated under vacuum. This procedure is repeated three times.
2-Butanone is added to the oily residue and the product is allowed to precipitate. Then, the suspension is cooled to 0 0C. The precipitated product is filtered and washed with 2-butanone. 0.263 g. (0.942 mmol; 46% yield) of the desired product V.HC1 are obtained.
Analytical data of Y UCl:
NMR 1H (D2O) δ (ppm): 1.91-2.37 (m, CH2-CH2); 2.53 (s, CH3); 3.07 (/,CH2); 3.80 (t, CH2Cl); 4.01 (at, CH2); 4.97 (t, CH-OH). m.p.: 118-121 0C
IR: 1691.5, 1661.8, 1591.8, 1548.8, 1454.4, 1432.4, 1388.5, 1329.8, 1188.3, 1113.0, 1058.0, 1025.6, 906.1, 832.3, 789.5, 728.8.
Example 4: 3-(2-chloroethyl)6,7,8,9-tetrhydro-2-methyl-9-hydroxy-4H- pyrido[l,2-a]pyrimidin-4-one, V
2.75 g (10 mmol) of IX.HC1, 40 ml of IPA/H2O (19: 1 v/v, 0.25 M) and 404 mg of Pd/C catalyst (5% w/w (dry), containing 66.20% water) are placed in a reaction vessel and purged with nitrogen (~40 psi) five times at room temperature.
Afterwards, the reaction vessel is purged an extra five times with hydrogen (50-60 psi) and heated up to 55°C under hydrogen (at atmospheric pressure) over 20 minutes. After that, the vessel is pressurized with hydrogen (15 psi) and mechanical stirring supplied (1500 rpm) at 55°C for 3 hours. The reaction vessel is then cooled to room temperature and carefully vented.
Analysis of the supernatant by HPLC showed that high conversions are achieved, over 98 %, and less than 1% of the de-chlorinated by-product is produced.
The reaction solution is filtered over celite, the filtered cake washed with MeOH and the combined filtrates evaporated to dryness to give V.HC1, as an off-white solid. A solution of potassium acetate in water is added drop wise, until complete
precipitation of compound V is achieved. The obtained precipitate is filtered and washed with water, yielding 2.23g (92% yield) of compound V.
Analytical data of V. HCl:
RMN 1H (D2O) δ (ppm): 1.91-2.37 (m, CH2-CH2), 2.53 (s, CH3); 3.07 (t, CH2); 3.80
(t, CH2Cl); 4.01 (at, CH2); 4.97 (t, CH-OH).
Analytical data of V:
RMN 1U (CDCl3) δ (ppm): 1.72-2.33 (m, CH2-CH2), 2.37 (s, CH3); 3.01 (t, CH2);
3.77 (t, CH2Cl); 3.94 (at, CH2); 4.52 (t, CH-OH).
Example 5: Preparation of Paliperidone, I
5.7 g (23.49 mmol) of compound V are placed in a reaction vessel and methanol is added (29 ml). To this suspension 6.0 g (23.41 mmol) of compound VI are added and then Et3N (9.8 ml; 70.41 mmol) is added. The resulting reaction mixture is then taken to reflux and maintained at this temperature overnight. The reaction mixture is then cooled down to 0 0C and the solid precipitated is filtered and washed with MeOH. 7.4 g (17.35 mmol, 74%) of Paliperidone I are obtained.
Example 6: Preparation of Paliperidone, I.

10.0 g (41.20 mmol) of compound V and 50 ml of methanol are placed in a reaction vessel. Afterwards, 10.6 g (41.33 mmol) of compound VI and 17.6 ml of DIPEA (102.81 mmol) are added. The resulting suspension is heated under reflux overnight. The reaction mixture is then cooled down to 0 0C and the solid precipitate
filtered and washed firstly with MeOH and then with water. 16.3 g (38.22 mmol, 93% yield) of Paliperidone I are obtained.
Claims
1. A process for obtaining Paliperidone, I, comprising alkylating compound VI, or a salt thereof, with V, or a salt thereof, using a base selected from triethylamine or diisopropylethylamine and, optionally, a solvent.

2. The process according to claim 1 wherein the solvent is an alcohol such as methanol, ethanol, isopropanol, water or a mixture thereof.
3. The process according to claim 1 which takes place from room temperature to the reflux temperature of the solvent, preferably from 50 0C to the reflux temperature of the solvent.
4. The process according to claim 1 wherein compound V, or a salt thereof, is prepared by hydrogenation of compound IX, or a salt thereof.

5. The process according to claim 4 wherein compound IX is hydrogenated using a palladium hydrogenation catalyst.
6. The process according to claim 4 wherein compound IX is hydrogenated in an alcoholic solution.
7. The process according to claim 6 wherein the alcohol in the alcoholic solution is selected from methanol, ethanol or isopropanol.
8. The process according to claim 6 wherein the alcoholic solution consist of a mixture of an alcohol with water from 1 :0.01 to a ratio of 1 : 1 (v/v).
9. The process according to claim 4 wherein the preparation of compound IX or a salt thereof comprises the following steps: a) reaction of 2-amino-3-hydroxypyridine, VII, and 2-acetylbutyrolactone, III, in a solvent and a cosolvent to obtain the diol VIII, b) diol VIII is reacted with a chlorinating agent, [Cl~], to selectively obtain compound IX, and c) optionally, compound IX is converted into a salt thereof.

10. The process according to claim 9 wherein the solvent is an aromatic hydrocarbon or a mixture thereof such as benzene, toluene or xylenes.
11. The process according to claim 9 wherein the cosolvent is a polar aprotic solvent such as JV-methylpyrrolidone, DMSO or DMF.
12. The process according to claim 9 wherein an acidic catalysis is used in step a), being the preferred acid methanesulphonic acid.
13. The process according to claim 9 wherein step a) is performed at reflux temperature.
14. The process according to claim 9 wherein the chlorinating agent, [Cl~], in step b) is POCl3.
15. The process according to claim 9 wherein step b) is performed at 80-120 0C, preferably at 90-100 0C.
16. The process according to claim 9 which is performed with means to remove the evolved water, such as a Dean-Stark receiver or molecular sieves.
17. The process according to claim 9 which is performed as a one pot reaction.
18. The process according to claim 9 wherein compound IX is obtained in the form of a hydrochloride salt.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES09753943.1T ES2625323T3 (en) | 2008-05-29 | 2009-05-28 | Process to prepare paliperidone and intermediates thereof |
| US12/994,693 US8309717B2 (en) | 2008-05-29 | 2009-05-28 | Process to prepare paliperidone and intermediates thereof |
| EP09753943.1A EP2303877B1 (en) | 2008-05-29 | 2009-05-28 | Process to prepare paliperidone and intermediates thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08157224.0 | 2008-05-29 | ||
| EP08157224 | 2008-05-29 | ||
| US5869208P | 2008-06-04 | 2008-06-04 | |
| US61/058,692 | 2008-06-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009144288A1 true WO2009144288A1 (en) | 2009-12-03 |
Family
ID=40934150
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/056578 Ceased WO2009144288A1 (en) | 2008-05-29 | 2009-05-28 | Process to prepare paliperidone and intermediates thereof |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8309717B2 (en) |
| EP (1) | EP2303877B1 (en) |
| ES (1) | ES2625323T3 (en) |
| WO (1) | WO2009144288A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2303877A1 (en) | 2008-05-29 | 2011-04-06 | Inke, S.A. | Process to prepare paliperidone and intermediates thereof |
| WO2011073997A3 (en) * | 2009-12-14 | 2011-12-22 | Cadila Healthcare Limited | Process for preparing paliperidone and pharmaceutically acceptable salts thereof |
| WO2012042368A1 (en) * | 2010-09-30 | 2012-04-05 | Aurobindo Pharma Limited | Process for preparation of paliperidone |
| US20140200228A1 (en) * | 2011-05-30 | 2014-07-17 | Cipla Limited | Process for the preparation of paliperidone |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0368388A2 (en) * | 1988-11-07 | 1990-05-16 | Janssen Pharmaceutica N.V. | 3-Piperidinyl-1,2-benzisoxazoles |
| US5158952A (en) * | 1988-11-07 | 1992-10-27 | Janssen Pharmaceutica N.V. | 3-[2-[4-(6-fluoro-1,2-benzisoxozol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9 tetrahydro-9-hydroxy-2-methyl-4H-pyrido [1,2-a]pyrimidin-4-one, compositions and method of use |
| US5254556A (en) * | 1988-11-07 | 1993-10-19 | Janssen Pharmaceutica N.V. | 3-piperidinyl-1,2-benzisoxazoles |
| US20060004199A1 (en) * | 2004-07-01 | 2006-01-05 | Dr. Reddy's Laboratories Limited | Process for the preparation of pure 3-[2-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)-1-piperidinyl]-ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido [1,2-a] pyrimidin-4-one |
| WO2009010988A1 (en) * | 2007-07-19 | 2009-01-22 | Natco Pharma Limited | An improved, industrially viable process for the preparation of high purity paliperidone |
| WO2009015828A1 (en) * | 2007-07-27 | 2009-02-05 | Synthon B.V. | Paliperidone derivatives |
| US20090048272A1 (en) * | 2007-08-16 | 2009-02-19 | Pratap Reddy Padi | Preparation of paliperidone |
| WO2009044413A2 (en) * | 2007-10-05 | 2009-04-09 | Matrix Laboratories Limited | Improved process for preparing paliperidone, novel polymorphic forms of the same and process thereof |
| WO2009047499A2 (en) * | 2007-10-09 | 2009-04-16 | Cipla Limited | Processes for the preparation of paliperidone and pharmaceutically acceptable salts thereof and intermediates for use in the processes |
| WO2009060297A2 (en) * | 2007-11-07 | 2009-05-14 | Orchid Chemicals & Pharmaceuticals Limited | An improved process for the preparation of paliperidone and its intermediates |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2137486T3 (en) | 1993-11-23 | 1999-12-16 | Janssen Pharmaceutica Nv | DERIVATIVES OF 9-HYDROXY-PIRIDO (1,2-A) PIRIMIDIN-4-ONA-ETER. |
| EP1791839B1 (en) * | 2004-09-09 | 2015-06-10 | Janssen Pharmaceutica NV | Preparation of 9-hydroxy-3-(2-hydroxyethyl)-2-methyl-4h-pyrido[1,2-a]pyrimidin-4-one and crystalls thereof |
| JP2009524574A (en) * | 2006-08-23 | 2009-07-02 | テバ ファーマシューティカル インダストリーズ リミティド | Method for synthesizing CMHTP and its intermediate |
| WO2009045489A2 (en) | 2007-10-03 | 2009-04-09 | Teva Pharmaceutical Industries Ltd. | Process for the synthesis of cmhtp, a paliperidone intermediate |
| US8309717B2 (en) | 2008-05-29 | 2012-11-13 | Inke, S.A. | Process to prepare paliperidone and intermediates thereof |
-
2009
- 2009-05-28 US US12/994,693 patent/US8309717B2/en not_active Expired - Fee Related
- 2009-05-28 ES ES09753943.1T patent/ES2625323T3/en active Active
- 2009-05-28 EP EP09753943.1A patent/EP2303877B1/en not_active Not-in-force
- 2009-05-28 WO PCT/EP2009/056578 patent/WO2009144288A1/en not_active Ceased
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0368388A2 (en) * | 1988-11-07 | 1990-05-16 | Janssen Pharmaceutica N.V. | 3-Piperidinyl-1,2-benzisoxazoles |
| US5158952A (en) * | 1988-11-07 | 1992-10-27 | Janssen Pharmaceutica N.V. | 3-[2-[4-(6-fluoro-1,2-benzisoxozol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9 tetrahydro-9-hydroxy-2-methyl-4H-pyrido [1,2-a]pyrimidin-4-one, compositions and method of use |
| US5254556A (en) * | 1988-11-07 | 1993-10-19 | Janssen Pharmaceutica N.V. | 3-piperidinyl-1,2-benzisoxazoles |
| US20060004199A1 (en) * | 2004-07-01 | 2006-01-05 | Dr. Reddy's Laboratories Limited | Process for the preparation of pure 3-[2-[4-(6-fluoro-1, 2-benzisoxazol-3-yl)-1-piperidinyl]-ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido [1,2-a] pyrimidin-4-one |
| WO2009010988A1 (en) * | 2007-07-19 | 2009-01-22 | Natco Pharma Limited | An improved, industrially viable process for the preparation of high purity paliperidone |
| WO2009015828A1 (en) * | 2007-07-27 | 2009-02-05 | Synthon B.V. | Paliperidone derivatives |
| US20090048272A1 (en) * | 2007-08-16 | 2009-02-19 | Pratap Reddy Padi | Preparation of paliperidone |
| WO2009044413A2 (en) * | 2007-10-05 | 2009-04-09 | Matrix Laboratories Limited | Improved process for preparing paliperidone, novel polymorphic forms of the same and process thereof |
| WO2009047499A2 (en) * | 2007-10-09 | 2009-04-16 | Cipla Limited | Processes for the preparation of paliperidone and pharmaceutically acceptable salts thereof and intermediates for use in the processes |
| WO2009060297A2 (en) * | 2007-11-07 | 2009-05-14 | Orchid Chemicals & Pharmaceuticals Limited | An improved process for the preparation of paliperidone and its intermediates |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2303877A1 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2303877A1 (en) | 2008-05-29 | 2011-04-06 | Inke, S.A. | Process to prepare paliperidone and intermediates thereof |
| WO2011073997A3 (en) * | 2009-12-14 | 2011-12-22 | Cadila Healthcare Limited | Process for preparing paliperidone and pharmaceutically acceptable salts thereof |
| WO2012042368A1 (en) * | 2010-09-30 | 2012-04-05 | Aurobindo Pharma Limited | Process for preparation of paliperidone |
| US20140200228A1 (en) * | 2011-05-30 | 2014-07-17 | Cipla Limited | Process for the preparation of paliperidone |
| US9062049B2 (en) * | 2011-05-30 | 2015-06-23 | Cipla Limited | Process for the preparation of paliperidone |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2303877A1 (en) | 2011-04-06 |
| US20110124863A1 (en) | 2011-05-26 |
| ES2625323T3 (en) | 2017-07-19 |
| US8309717B2 (en) | 2012-11-13 |
| EP2303877B1 (en) | 2017-02-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK2641897T3 (en) | A process for the preparation of 6- (7 - ((1-aminocyclopropyl) methoxy) -6-methoxy-quinolin-4-yloxy) -N-methyl-1-naphthamide and synthetic intermediates thereof | |
| CN113227045B (en) | Synthesis of Substituted Heterocyclic Fused γ-Carbolines | |
| CN109641891B (en) | Novel compounds and methods | |
| US9133188B2 (en) | Methods for preparing naphthyridines | |
| CN106928275A (en) | The Preparation Method And Their Intermediate and crystal formation of volution amine aryl phosphoric-oxygenic compound | |
| EP2303877B1 (en) | Process to prepare paliperidone and intermediates thereof | |
| CN101600716A (en) | Improved process for the preparation of 9-hydroxy-3-(2-chloroethyl)-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one hydrochloride | |
| CN116745261A (en) | A new method for the synthesis of 8-[(2-hydroxybenzoyl)amino)octanoic acid (salcaprozic acid) via amide formation | |
| CN111285847A (en) | Method for preparing alectinib | |
| JP5635418B2 (en) | Process for the preparation of enantiomerically pure indolopyridine. | |
| WO2009060297A2 (en) | An improved process for the preparation of paliperidone and its intermediates | |
| US20100267954A1 (en) | Process for the purification of paliperidone | |
| WO2006134491A2 (en) | New crystalline form of moxifloxacin hydrochloride and process for its preparation | |
| US20090221828A1 (en) | Process for Preparing 1-Halo-2,7-Naphthyridinyl Derivatives | |
| EP1845091A1 (en) | Process for producing muscarine receptor antagonist and intermediate therefor | |
| JP2008525521A (en) | Process for the preparation of substantially pure 4-amino-1-isobutyl-1H-imidazo [4,5-c] -quinoline (imiquimod) | |
| US8093384B2 (en) | Processes for the preparation of alfuzosin | |
| KR100346619B1 (en) | Process for preparing 8-cyclopentyl-6-ethyl-3-[substituted]-5,8-dihydro-4h-1,2,3a,7,8-pentaaza-as-indacenes and intermediates useful therein | |
| HK1231871A1 (en) | A process for the preparation of compound and synthetic intermediates thereof | |
| HK1167136B (en) | A process for the preparation of 6-(7-((1-aminocyclopropyl)methoxy)-6-methoxyquinolin-4-yloxy)-n-methyl-1-naphthamide and synthetic intermediates thereof | |
| WO2008053324A1 (en) | An improved process for the preparation of gemifloxacin mesylate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09753943 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2009753943 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009753943 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12994693 Country of ref document: US |