WO2010082634A1 - 腫瘍滞留性を有する化合物 - Google Patents
腫瘍滞留性を有する化合物 Download PDFInfo
- Publication number
- WO2010082634A1 WO2010082634A1 PCT/JP2010/050432 JP2010050432W WO2010082634A1 WO 2010082634 A1 WO2010082634 A1 WO 2010082634A1 JP 2010050432 W JP2010050432 W JP 2010050432W WO 2010082634 A1 WO2010082634 A1 WO 2010082634A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mmol
- reaction
- functional group
- tumor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *CC(*)COC([C@@](*)[C@]1*)O[C@](C*)C1=C Chemical compound *CC(*)COC([C@@](*)[C@]1*)O[C@](C*)C1=C 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
- C07H15/10—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical containing unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/26—Acyclic or carbocyclic radicals, substituted by hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
- A61K47/551—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug one of the codrug's components being a vitamin, e.g. niacinamide, vitamin B3, cobalamin, vitamin B12, folate, vitamin A or retinoic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
- C07H15/06—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical being a hydroxyalkyl group esterified by a fatty acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/04—Acyclic radicals, not substituted by cyclic structures attached to an oxygen atom of the saccharide radical
- C07H15/08—Polyoxyalkylene derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/18—Acyclic radicals, substituted by carbocyclic rings
Definitions
- the present invention relates to a novel compound having tumor retention.
- MRI magnetic resonance
- PET PET
- X-ray X-ray
- CT positron emission computed tomography
- cancer detection and diagnosis methods using these have been developed and used.
- a substance that produces a difference in X-ray absorption as compared with surrounding tissues or a substance that affects nuclear magnetic resonance is used as a contrast agent.
- contrast agents When these contrast agents are distributed in the tissue, they give contrast to X-rays and MRI images, and image the shape of the tissue.
- contrast agents used in such a technique include, for example, a molecule such as glucose that is easily taken up by cancer tissues whose metabolism is higher than that of normal cells labeled with a radioactive substance, a fluorescent substance, or a magnetic substance. And those labeled in the same manner with molecules such as sugar chains specifically present on the surface of cancer cells and proteins that gather with antigen as a target.
- These contrast agents are used for detection and / or imaging of cancer tissue by a detector such as PET, MRI, and CT.
- a drug delivery system technology has been developed in which a drug such as an anticancer drug is encapsulated or bound to fine particles and transported to the vicinity of the cancer tissue.
- a drug delivery system include a molecule having affinity for tumor tissue and a molecule in which a molecule for preventing it from being excluded as a foreign substance by the immune system in the body is incorporated into the particle surface.
- An object of the present invention is to provide a novel compound that specifically stays in a tumor.
- a further object of the present invention is to provide a method of retaining the compound in the tumor.
- a further object of the present invention is to provide a method for detecting a tumor using the compound.
- a further object of the present invention is to provide a method for diagnosing a tumor using the compound.
- a further object of the present invention is to provide a method for treating a tumor using the compound.
- R is an anionic group bonded to hydrogen;
- R 1 is OH, OCOH, OCO (CH 2 ) h CH 3 or a functional group, h is an integer of 0 or more,
- R 2 is H, OH, OCOH, OCO (CH 2 ) i CH 3 or a functional group, i is an integer of 0 or more,
- R 3 is OH, SO 3 H or a functional group,
- R 4 is OH, SO 3 H or a functional group, and
- R 5 is OH, SO 3 H or a functional group, At least one of R 1 , R 2 , R 3 , R 4 and R 5 contains a functional group.
- R is an anionic group bonded to hydrogen;
- R 1 is OH, OCOH, OCO (CH 2 ) h CH 3 or a functional group, h is an integer of 0 or more,
- R 2 is H, OH, OCOH or OCO (CH 2 ) i CH 3 or a functional group, i is an integer of 0 or more,
- R 3 is OH, SO 3 H or a functional group,
- R 4 is OH, SO 3 H or a functional group,
- R 5 is OH, SO 3 H or a functional group, j is an integer of 1 or more, and
- X is a functional group or a cationic group,
- k is an integer of 1 or more, At least one of R 1 , R 2 , R 3 , R 4 , R 5 and X is a functional group.
- a tumor comprising detecting the labeling substance at a time when the compound or salt is present at a higher concentration in the tumor than in a tissue other than the tumor, and detecting the tumor in the subject based on the detection result Method for detecting.
- a method for diagnosing a tumor comprising:
- a method for treating a tumor comprising administering the compound or salt according to any one of (1) or (2) above, wherein the functional group is an antitumor substance.
- a novel compound that specifically stays in a tumor is provided.
- a method for retaining the compound in a tumor is provided.
- a method for detecting a tumor using the compound is provided.
- a method for diagnosing a tumor using the compound is provided.
- a method for treating a tumor using the compound is provided.
- FIG. 1 is a graph showing retention of biotinylated ⁇ SQMGMC18: 0 in a tumor.
- FIG. 2 is a graph showing retention of biotinylated ⁇ SQAP C18: 0 in tumors.
- FIG. 3 is a concentration-time transition curve in plasma and organs of nude mice intraperitoneally administered with ⁇ SQMG C18: 0.
- FIG. 4 is a concentration-time course curve in plasma and organs of nude mice intraperitoneally administered with ⁇ SQAP C18: 0.
- FIG. 5 is a concentration-time course curve in plasma and organs of nude mice intravenously administered with ⁇ SQAP C18: 0.
- the novel compound is a compound of the following formula (I) or a pharmaceutically acceptable salt thereof:
- R is an anionic group bonded to hydrogen;
- R 1 is OH, OCOH, OCO (CH 2 ) h CH 3 or a functional group, and h is an integer of 0 or more, preferably 0 or more and 30 or less, more preferably 0 or more and 26 or less, and even more preferably 1 or more and 22 or less.
- R 2 is H, OH, OCOH, OCO (CH 2 ) i CH 3 or a functional group, and i is an integer of 0 or more, preferably 0 or more and 30 or less, more preferably 0 or more and 26 or less, and even more preferably 1 or more.
- R 3 is OH, SO 3 H or a functional group
- R 4 is OH, SO 3 H or a functional group
- R 5 is OH, SO 3 H or a functional group
- At least one of R 1 , R 2 , R 3 , R 4 and R 5 contains a functional group, and preferably any one of R 1 , R 2 , R 3 , R 4 and R 5 contains a functional group.
- the compound according to the invention is a compound of formula (I) or a pharmaceutically acceptable salt thereof,
- R is SO 3 H
- R 3 is OH
- R 4 is OH
- R 5 is OH
- R 1 is OH, OCOH, OCO (CH 2 ) h CH 3 , O— (active group), OCO— (active group) or OCO (CH 2 ) h CH 2 — (active group)
- h is 0 or more
- R 2 is H, OH, OCOH, OCO (CH 2 ) i CH 3 , a functional group, O— (functional group), OCO— (active group) or OCO (CH 2 ) i CH 2 — (functional group).
- I is an integer of 0 or more, preferably 0 or more and 30 or less, more preferably 0 or more and 26 or less, and still more preferably 1 or more and 22 or less, It may be a compound or a pharmaceutically acceptable salt thereof in which at least one of R 1 and R 2 contains a functional group.
- the compound according to the invention is a compound of formula (I) or a pharmaceutically acceptable salt thereof,
- R is SO 3 H;
- R 1 is OH, OCOH, OCO (CH 2 ) h CH 3 or a functional group, and h is an integer of 0 or more, preferably 0 or more and 30 or less, more preferably 0 or more and 26 or less, and still more preferably 1 or more.
- R 2 is H, OH, OCOH, OCO (CH 2 ) i CH 3 or a functional group
- i is an integer of 0 or more, preferably 0 or more and 30 or less, more preferably 0 or more and 26 or less, and still more preferably 1 to 22,
- R 1 and / or R 2 are OCO (CH 2 ) h CH 3 or OCO (CH 2 ) i CH 3 , these are linear or branched saturated Alternatively, it may be an unsaturated fatty acid.
- the compound of the present invention is a sulfopyranosyl (acyl) glycerol, sulfopyranosyl (acyl) propanediol or sulfoquinovosylacylpropanediol having at least one side chain bonded to a functional group.
- a pharmaceutically acceptable salt thereof may be used.
- the pyranose when pyranose which is a sugar skeleton constituting pyranoside is present, the pyranose is ⁇ -D-glucose, ⁇ -D-glucose, ⁇ -D-galactose, ⁇ -D-galactose. , ⁇ -D-mannose, ⁇ -D-mannose, and the like.
- the sugar skeletons of these pyranosides may be either boat-shaped or chair-shaped, or a mixture thereof.
- the chair type is preferable from the viewpoint of stability.
- the absolute configuration when an asymmetric carbon exists, the absolute configuration may be either S or R.
- the compound when the compound is composed of sulfopyranosyl (acyl) glycerol and a functional group, the carbon at the 2-position of the glycerol moiety is an asymmetric carbon, and even in this case, the absolute configuration is S or R.
- the mixture may be sufficient.
- the quinobose ring contained therein may be boat-shaped, chair-shaped, or a mixture thereof. May be. However, the chair shape is generally preferable because it is more stable. Further, the configuration of the propanediol moiety on the quinobose ring may be an ⁇ anomer, a ⁇ anomer, or a mixture thereof.
- anionic group bonded to hydrogen in the compound of the formula (I) include, but are not limited to, SO 3 H, OSO 3 H, PO 3 H 2 and CO 2 H. SO 3 H, OSO 3 H, and PO 3 H 2 are preferable, and SO 3 H is more preferable.
- “functional group” refers to a group exhibiting a desired function that is desired to be transported to a tumor.
- the compound of the formula (I) may contain an active substance such as a labeling substance and an antitumor substance as an active group.
- the agent is maintained by a covalent bond at any position of the compound of formula (I).
- one or more functional groups may be contained in one molecule of the compound of formula (I). Further, when two or more functional groups are present in one molecule of the compound of formula (I), these functional groups may be the same or different from each other.
- the functional group may be one of a binding pair that can specifically bind to each other.
- binding pairs include, but are not limited to, biotin and avidin, an antigen and a specific antibody against the antigen, an antigen and a specific receptor for the antigen, and the like.
- the compound according to the present invention containing one of the binding pairs as an active group is administered to a subject, and after a desired interval, the binding pair having a drug or a labeling substance having a desired effect bound thereto is bound. By administering the other to the subject, the drug or labeling substance can be specifically delivered to the tumor.
- the functional group may or may not contain a linker for binding the active substance and the glycolipid moiety.
- labeling substance used here may be a marker used in a tumor diagnosis technique using an imaging technique generally used in noninvasive diagnosis.
- the labeling substance may be any labeling substance known per se that can be stained with a living body.
- labeling substances include, but are not limited to, radioactive substances, paramagnetic metals, radiopaque metals and dyes.
- radioactive substance examples include, but are not limited to, 11 C, 13 N, 15 O, 18 F, 58 Co, 59 Fe, 62 Cu, 64 Cu, 67 Cu, 67 Ga, 68 Ga, 75 Br, 76 Br, 77 Br , 82 Br, 89 Sr, 90 Y, 111 I, 113 Sn, 117m Sn, 153 Sm, 165 Dy, 166 Ho, 169 Er, 186 Re, 201 Tl, 212 Bi, 213 Bi Etc.
- paramagnetic metal examples include, but are not limited to, Cr (III), Mn (III), Fe (II), Fe (III), Pr (III), Nd (III), Sm (III), Yb (III), Gd (III), Tb (III), Dy (III), Ho (III), Er (III) and the like are included.
- radiopaque metal examples include, but are not limited to, I, Bi, W, Ta, Hf, La, Ln, Ba, Mo, Nb, Zr, and Sr.
- the dyes are not limited to these, but may be any dyes for living body staining known per se, including, for example, dyes such as fluorescent dyes.
- fluorescent dyes include those having the following skeleton in a fluorescent chromophore; naphthalene, anthracene, quinoline, acridine, coumarin, salicylic acid, anthranilic acid, NBD (7-nitrobenz-2-oxa-1,3- Diazole), stilbene, resorufin, BODIPY (4,4-difluoro-4-bora-3a, 4a-diaza-s-indene, carbocyanine, thiacarbocyanine, tetramethylindocarbocyanine, etc.
- the fluorescent dyes Includes substances disclosed in the Molecular Probes Handbook by RP Hoglund (RPHaugland, Molecular Probes Handbook, 6th edition).
- anti-tumor substance used here may be any substance that attacks the tumor due to cytotoxicity. Therefore, the anti-tumor substance may be any substance known per se used for treating tumors.
- antitumor substances include alkylating agents, antimetabolites, anticancer antibiotics, metal complexes, plant alkaloids, topoisomerase inhibitors, microtubule polymerization inhibitors, microtubule depolymerization. Inhibitors, molecular targeted drugs and hormonal agents may be used.
- antitumor substance examples include, but are not limited to, Aceglatone, Aclarubicin hydrochloride, Actinomycin D (also referred to as Dactinomycin)), Amrubicin hydrochloride ( Amrubicin hydrochloride, Anastrozole, Arsenic trioxide, Asparaginase (also called L-Asparaginase)), Bicalutamide, Bleomycin hydrochloride ), Bortezomib, buslufan, capecitabine, carboplatin, carboquone, carmofur, celmoleukin, cisplatin, ribine C, ribine C Cyclophosphamide, Cytarabine, dacarbazine, Daunorubicin hydrochloride (also called Daunomycin), Docetaxcel, Doxifluridine, Doxorubicin hydrochloride (oxorubicin) Also called Enocitabine), Erubicin hydrochloride, Erlotinib hydro
- the agent may be any substance known per se used for treating a tumor.
- it may be a thermal neutron capture material, examples of which include compounds containing elements such as B and Gd.
- the compound according to the invention may be a pharmaceutically acceptable salt of the compound of formula (I).
- a salt comprises an ionic group formed by ionizing at least one of the side chains represented by R, R 1 , R 2 , R 3 , R 4 and / or R 5 of the compound of formula (I) Any salt may be used as long as it is obtained by binding of this ionic group to an ion capable of ion binding.
- the ionized compound of formula (I) and the ionic substance may be bound by one molecule per molecule, that is, by 1: 1, or may be bound by m: n.
- m and n are one or more integers.
- the salt may consist of a plurality of molecules of an ionized compound of formula (I) and one molecule of the ionic substance.
- the salt may be composed of one molecule of the ionized compound of formula (I) and a plurality of molecules of the ionic substance.
- the paired ionic substance may be a cation having a positive charge.
- the cation may be a metal ion including a monovalent metal ion such as Na + and K + , a divalent metal ion such as Ca 2+, and the like.
- the cation is not limited to lithium, sodium, potassium, calcium, magnesium, manganese, iron, zinc, copper, strontium, lead, silver, barium, aluminum, chromium, cobalt, nickel, It may be ammonium, monoalkylammonium, dialkylammonium, trialkylammonium, tetraalkylammonium, monohydroxyalkylammonium, dihydroxyalkylammonium, trihydroxyalkylammonium, tetrahydroxyalkylammonium and the like.
- the compound according to the invention may also be a complex.
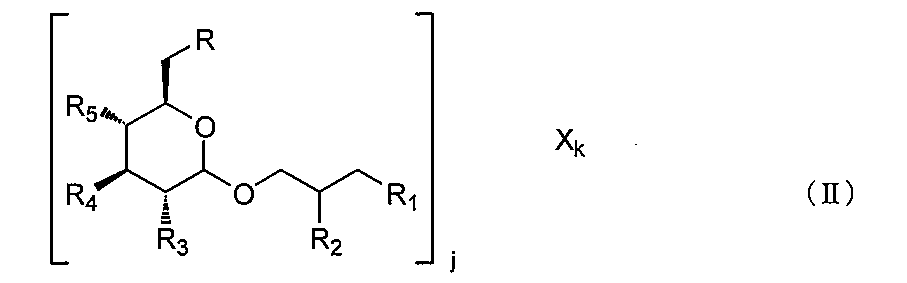
- a complex is a compound represented by the following formula (II) or a pharmaceutically acceptable salt thereof.
- R, R 1 , R 2 , R 3 , R 4 and R 5 are as defined in formula (I); j is an integer of 1 or more, and X is a functional group or a cationic group, and k is an integer of 1 or more, preferably R 1 , R 2 , R 3 , R 4 , R 5 and X At least one is a functional group.
- the cationic group may be a cation having a positive charge.
- the cationic group may be a metal ion including a monovalent metal ion such as Na + and K + , a divalent metal ion such as Ca 2+, and the like.
- the cationic group includes, but is not limited to, lithium, sodium, potassium, calcium, magnesium, manganese, iron, zinc, copper, strontium, lead, silver, barium, aluminum, chromium, cobalt, It may be nickel, ammonium, monoalkylammonium, dialkylammonium, trialkylammonium, tetraalkylammonium, monohydroxyalkylammonium, dihydroxyalkylammonium, trihydroxyalkylammonium and tetrahydroxyalkylammonium.
- X in formula (II) contains an active group
- the active group may be the same active substance as defined in formula (I).
- the active substance and glycolipid May be linked by a coordination bond.
- the compound of the formula (I), the compound of the formula (II) and pharmaceutically acceptable salts thereof can be synthesized using a reaction route known per se.
- the compound according to the present invention exhibits a long-term retention in tumor tissues as compared with other tissues. That is, there is a great difference in distribution concentration between other tissues and tumor tissues. Accordingly, by administering to a subject a compound according to the invention, ie a compound of formula (I) or a pharmaceutically acceptable salt thereof or a compound of formula (II), the tumor is treated with said formula (I) or formula (II). ) Compounds or salts can be retained.
- a method of retaining a compound of formula (I) or formula (II) or a pharmaceutically acceptable salt thereof in a tumor there is also provided.
- the disappearance time in the subject of the compound according to the present invention is different between the tumor tissue and the other tissue, and the compound stays longer in the tumor than in the other tissue. That is, the disappearance time of the compound in the tumor is longer than the disappearance time in the tissue other than the tumor.
- the compound has a functional group in its structure. Therefore, it is possible to detect a tumor, diagnose a tumor, and treat a tumor by using the compound by utilizing the effect of the compound on tumor retention and the functional group.
- the compounds according to the present invention can be used as tumor diagnostic agents, therapeutic agents and contrast agents.
- a labeling substance When detecting a tumor, a labeling substance may be selected as the functional group.
- the compound containing the labeling substance or a pharmaceutically acceptable salt thereof is administered to the subject, and the labeling substance is detected when the compound or salt is present in a higher concentration in the tumor than in the tissue other than the tumor, From the detection result, it is possible to detect whether the subject has a tumor and / or which part of which tissue has a tumor.
- the compound containing the labeling substance or a pharmaceutically acceptable salt thereof is administered to the subject, and the compound or salt is higher in the tumor than in the tissue other than the tumor.
- the labeling substance is detected, and from the detection result, it is possible to diagnose whether the subject has a tumor and / or in which tissue the tumor is present.
- the detection of the labeling substance may be performed by any method known per se according to the type of the labeling substance contained as the functional group.
- the labeling substance when the labeling substance is a radioactive substance, the labeling substance may be detected by a known imaging diagnostic method such as PET and SPECT and / or a detection method using a gamma probe.
- the compounds according to the invention may be interpreted as radioactive diagnostic agents.
- the labeling substance When the labeling substance is a paramagnetic metal, the labeling substance may be detected by a method using nuclear magnetic resonance such as MRI known per se. In this case, the compound according to the invention may be interpreted as a nuclear magnetic resonance diagnostic agent.
- the labeling substance when the labeling substance is a radiopaque substance, the labeling substance may be detected by any known CT and X-ray photography.
- the compound according to the invention may be understood as an X-ray contrast agent.
- the labeling substance is a fluorescent dye
- the labeling substance may be detected by a fluorescent detection method known per se.
- the compound according to the invention may be interpreted as a fluorescent diagnostic agent.
- the detection is not limited to the above detection means, and may be performed by any known noninvasive means.
- the non-invasive means may be any means that can be implemented without subjecting the subject to surgical treatment. It is desirable from the viewpoint of the target QOL to detect the labeling substance by such non-invasive means.
- Such a method may comprise administering to the subject a compound of formula (I) or (II) or a pharmaceutically acceptable salt thereof, wherein the functional group is an antitumor substance.
- a compound according to the invention for use in such a method may be construed as an anticancer or antitumor agent.
- tumor treatment can be performed when the substance contained as the functional group is a thermal neutron capture substance.
- a method comprises administering to a subject a compound of formula (I) or (II) or a pharmaceutically acceptable salt thereof, wherein said functional group is a thermal neutron capture substance, in a tumor rather than in a tissue other than tumor. Irradiating with neutrons when present at a high concentration. Irradiation with neutrons can be performed by any means known per se.
- a compound according to the invention for use in such a method may be understood as a cancer therapeutic agent or a tumor therapeutic agent.
- the irradiation conditions of the aforementioned radiation and the administration conditions of the compound according to the present invention include, as is well known in the field of radiotherapy, the type of radiation source, irradiation method, irradiation site and irradiation period; The type of disease to be treated and the severity of the disease; depending on the age, weight, health status, medical history, etc. of the subject to be irradiated, it can be appropriately selected by a healthcare professional or other specialists.
- subject refers to humans, animals other than humans, for example, mammals such as cats, dogs, horses, cows, sheep, mice, rats and rabbits, reptiles, amphibians, fish and birds.
- the compounds according to the present invention can be used as active ingredients such as diagnostic agents, therapeutic agents and contrast agents.
- the compound can be administered, for example, orally or parenterally.
- the said compound can be made into a pharmaceutical formulation by combining with a suitable pharmaceutically acceptable excipient
- Dosage forms suitable for oral administration include those in a solid, semi-solid, liquid or gas state. Specifically, tablets, capsules, powders, granules, solutions, suspensions, Examples include syrups and elixirs, but are not limited thereto.
- the compound is bound to a binder, tablet disintegrant, lubricant using a method known per se. And further, if necessary, mixed with a diluent, a buffer, a wetting agent, a preservative, a flavoring agent and the like.
- the binder includes crystalline cellulose, cellulose derivatives, corn starch, and gelatin
- the tablet disintegrating agent includes corn starch, potato starch, sodium carboxymethylcellulose
- the lubricant includes talc and magnesium stearate.
- conventionally used additives such as lactose and mannitol can be used.
- the compound is in the form of a liquid, fine powder, together with a gas or liquid propellant, or optionally with a known auxiliary agent such as an infiltrating agent, and a non-residue such as an aerosol container or a nebulizer. It can also be filled in a pressurized container and administered in the form of an aerosol or inhalant.
- a pressurized gas such as dichlorofluoromethane, propane or nitrogen can be used.
- a pharmaceutical preparation containing the compound as an active ingredient When administered parenterally, it can be administered, for example, by rectal administration or injection.
- suppositories are molded by mixing the pharmaceutically active substance of the present invention with excipients such as cocoa butter, carbon wax, polyethylene glycol, which melts at body temperature but solidifies at room temperature, in a manner known per se. Can be formulated.
- injectable preparations are prepared according to methods known per se by dissolving the compounds according to the invention in aqueous or non-aqueous solvents such as vegetable oils, synthetic resin acid glycerides, esters of higher fatty acids, propylene glycol, It can be suspended or emulsified and optionally formulated with conventionally used additives such as solubilizers, osmotic pressure regulators, emulsifiers, stabilizers and preservatives.
- aqueous or non-aqueous solvents such as vegetable oils, synthetic resin acid glycerides, esters of higher fatty acids, propylene glycol, It can be suspended or emulsified and optionally formulated with conventionally used additives such as solubilizers, osmotic pressure regulators, emulsifiers, stabilizers and preservatives.
- a pharmaceutically acceptable solvent such as sterile water for injection or normal physiological saline can be used.
- the compound according to the present invention can be combined with a compound having other pharmaceutically acceptable activity to prepare a pharmaceutical preparation.
- the pharmaceutical preparation according to the present invention can be appropriately set and adjusted according to the administration form, administration route, the type and location of the tumor to be detected, the degree and stage of the disease to be treated, and the like.
- the effective amount of the compound according to the present invention is 0.01 to 1000 mg / kg body weight / day, and when administered as an injection, the effective amount is 0.01 to 500 mg / day.
- the same effective amount can be set to 0.01 to 500 mg / kg body weight / day, but is not limited thereto.
- Tumors include, for example, neurogenic tumors including brain tumors, and the following cancers classified as carcinomas such as squamous cell carcinoma and adenocarcinoma (head and neck cancer, skin cancer, esophageal cancer, thyroid cancer, stomach cancer, lung cancer, gallbladder) Cancer, biliary tract cancer, pancreatic cancer, liver cancer, prostate cancer, uterine cancer, ovarian cancer, breast cancer, kidney cancer, bladder cancer, colon cancer, etc.), melanoma, bone / soft tissue tumor, lymphoma, leukemia, myeloma, etc. Including, but not limited to. “Treatment” as used herein refers to inhibiting the shrinking, disappearing and / or increasing of a tumor as described above.
- carcinomas such as squamous cell carcinoma and adenocarcinoma (head and neck cancer, skin cancer, esophageal cancer, thyroid cancer, stomach cancer, lung cancer, gallbladder) Cancer, biliary tract cancer
- the pharmaceutical preparation according to the present invention comprises, as an active ingredient, one or more effective amounts selected from the group consisting of compounds represented by formulas (I) and (II) and pharmaceutically acceptable salts thereof. You may contain.
- the pharmaceutical preparation can also contain a plurality of different compounds among the compounds according to the present invention. Further, the compound according to the present invention may be combined with other radiosensitizers, antitumor agents or other substances having pharmacological activity and / or substances having pharmacological activity, as long as the activity is not adversely affected. May be used.
- sulfoquinovosylacylpropanediol which is an example of a glycolipid moiety contained in the compound according to the present invention, is also referred to as “SQAP”.
- SQAP Cp: q sulfoquinovosylacylpropanediol
- ⁇ represents an ⁇ anomer
- Cp: q represents the number of carbons contained in the R 1 group of SQAP is “p”
- the double bond is Indicates “q”.
- p is an integer of 1 or more
- q is an integer of 0 or more.
- ⁇ SQAP C18: 0 when “ ⁇ SQAP C18: 0” is described, it is an ⁇ -anomer, a sulfoquinovosylacylpropanediol having 18 carbon atoms in R 1 and 0 double bonds. Indicates that there is.
- sulfoquinovosyl acylglycerol which is an example of a glycolipid moiety contained in the compound according to the present invention
- SQMG sulfoquinovosyl acylglycerol
- ⁇ SQMG C18: 0 is an ⁇ -anomer, a sulfoquinovosylacylglycerol having 18 carbon atoms in R 1 and 0 double bonds. It shows that.
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL).
- the organic layers were combined and washed with 1N HCl aqueous solution to pH 4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. did.
- the concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (1-5). ⁇ 15.3 g (27.6 mmol), 91.4% ⁇ .
- reaction solution was poured into a cold 7.5 M sodium hydroxide (500 mL) solution and extracted with ethyl acetate (4 ⁇ 100 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12: 1) to give the title compound (10) as a colorless wax-like substance. ⁇ 5.2 g (5.24 mmol), 71.2% ⁇ .
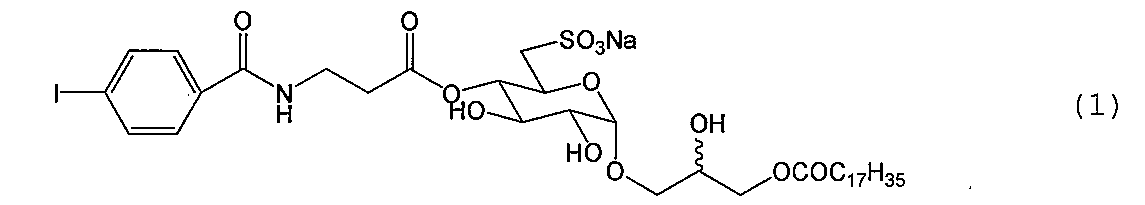
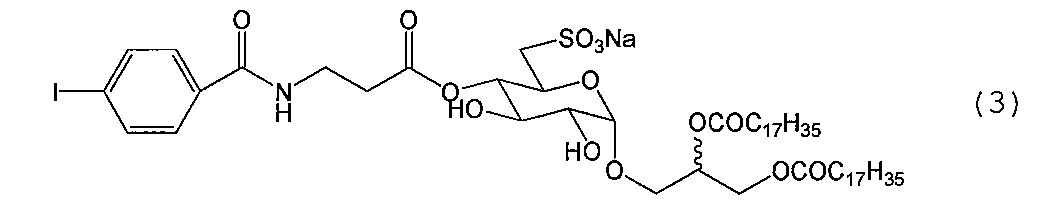
- Compound (1-11) (273.2 mg, 403 ⁇ mol) is dissolved in a mixed solution of anhydrous N, N-dimethylformamide (20 mL) and triethylamine (1 mL), and biotin p-nitrophenyl ester (162 mg, 443 ⁇ mol) The mixture was reacted for 24 hours at room temperature while stirring with a stirrer.
- Synthesis example 2 The production process of the sulfopyranosylacylpropanediol derivative of the present invention is shown in the following scheme 2 by taking as an example an ⁇ -sulfoquinovosylacylpropanediol biotin derivative (also referred to as “biotinylated ⁇ SQAP”).
- ⁇ -sulfoquinovosylacylpropanediol biotin derivative also referred to as “biotinylated ⁇ SQAP”.
- reaction solution was cooled again to 0 ° C., water (20 mL) was first added, then 3M sodium hydroxide solution (70 mL) and 35% hydrogen peroxide solution (70 mL) were sequentially added, and after 1 hour, the temperature was returned to room temperature. Stir for hours. After confirming that the reaction had progressed sufficiently, this solution was extracted with ethyl acetate (3 x 100 mL), and the organic layers were combined, washed with saturated brine (2 x 100 mL), dried over sodium sulfate, and filtered. And concentrated under reduced pressure.
- the concentrate was purified by silica gel chromatography (chloroform-methanol, 100: 0 ⁇ 3: 1) to give the title compound (2-8) as a colorless wax-like substance ⁇ 498 mg (0.87 mmol), 98.7% ⁇ .
- Synthesis example 3 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 3 by taking an ⁇ -sulfoquinovosyl monoacylglycerol monoiodo derivative as an example.
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL). The organic layers were combined and washed with 1N HCl aqueous solution until pH4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure did. The concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (3-5). ⁇ 15.3 g (27.6 mmol), 91.4% ⁇ .
- Pathway F3 1-O-allyl-2,3-di-O-benzyl-4-O- (carbobenzoxy- ⁇ -alanyl) -6-thioacetyl- ⁇ -D-glucopyranoside (3-7)
- compound (3-6) (17.1 g, 22.5 mmol) in anhydrous N, N-dimethylformamide (300 mL) was added potassium thioacetate (5.1 g, 45.0 mmol), and the mixture was stirred at 90 ° C. for 3 hours. After confirming that the reaction had progressed sufficiently, the reaction solution was poured into cold water (400 mL) and extracted with ethyl acetate (3 ⁇ 150 mL).
- reaction solution was poured into a cold 7.5 M sodium hydroxide (500 mL) solution and extracted with ethyl acetate (4 ⁇ 100 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12: 1) to give the title compound (3-10) as a colorless wax-like substance. Obtained ⁇ 5.2 g (5.24 mmol), 71.2% ⁇ .
- Synthesis example 4 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 4 by taking an ⁇ sulfoquinovosyl monoacylglycerol triiodo derivative as an example.
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL). The organic layers were combined and washed with 1N HCl aqueous solution until pH4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure did. The concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (4-5). ⁇ 15.3 g (27.6 mmol), 91.4% ⁇ .
- Pathway F4 1-O-allyl-2,3-di-O-benzyl-4-O- (carbobenzoxy- ⁇ -alanyl) -6-thioacetyl- ⁇ -D-glucopyranoside (4-7)
- compound (4-6) (17.1 g, 22.5 mmol) in anhydrous N, N-dimethylformamide (300 mL) was added potassium thioacetate (5.1 g, 45.0 mmol), and the mixture was stirred at 90 ° C. for 3 hours. After confirming that the reaction had progressed sufficiently, the reaction solution was poured into cold water (400 mL) and extracted with ethyl acetate (3 ⁇ 150 mL).
- reaction solution was poured into a cold 7.5 M sodium hydroxide (500 mL) solution and extracted with ethyl acetate (4 ⁇ 100 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12: 1) to give the title compound (4-10) as a colorless wax-like substance. Obtained ⁇ 5.2 g (5.24 mmol), 71.2% ⁇ .
- Synthesis example 5 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 5 by taking an ⁇ sulfoquinovosyl diacylglycerol monoiodo derivative as an example.
- Pathway D5 1-O-allyl-2,3,4-di-O-benzyl-6-O-tosyl- ⁇ -D-glucopyranoside (5-5)
- compound (5-4) (12.1 g, 30.2 mmol) in anhydrous pyridine (120 mL)
- p-toluenesulfonyl chloride 7.5 g, 39.3 mmol
- 4-dimethylaminopyridine 369 mg, 3.0 mmol
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL). The organic layers were combined and washed with 1N HCl aqueous solution until pH4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure did. The concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (5-5). ⁇ 15.3 g (27.6 mmol), 91.4% ⁇ .
- reaction solution was poured into a cold 7.5 M sodium hydroxide (500 mL) solution and extracted with ethyl acetate (4 ⁇ 100 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the obtained residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12: 1) to give the title compound (5-10) as a colorless wax-like substance. Obtained ⁇ 8.6 g (6.97 mmol), 70.3% ⁇ .
- Synthesis Example 6 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 6 by taking an ⁇ sulfoquinosyl acylglycerol monoiodo derivative as an example.
- Pathway E6 2,3,4-Tri-O- (t-butyldimethylsilyl) -1-O- (2-propenyl) -6-deoxy-6-acetylthio- ⁇ -D-glucose (6-6) 7.9 g (11.0 mmol) of compound (6-5) was dissolved in 20 mL of dry ethanol, 1.8 g of potassium thioacetate was added, and the mixture was reacted for 3 hours with stirring under reflux conditions.
- Pathway F6 3-O- [2,3,4-tri-O- (t-butyldimethylsilyl) -6-deoxy-6-acetylthio- ⁇ -D-glucopyranosyl] -glycerol (6-7)
- Synthesis example 7 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 7 by taking an ⁇ sulfoquinosyl acylglycerol triiodo derivative as an example.
- Synthesis example 8 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 8 by taking a ⁇ sulfoquinobosyl monoacylglycerol monoiodo derivative as an example.
- reaction solution was returned to room temperature, washed with saturated aqueous sodium bicarbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the concentrate was crystallized twice from hot ethanol to give the title compound (8-2) as colorless needle crystals ⁇ 24.8 g (64 mmol), 50.0% ⁇ .
- the concentrate was dissolved in a small amount of ethyl acetate, 200 mL of cold water was added, and the mixture was extracted with ethyl acetate (3 ⁇ 100 mL). The organic layers were combined, washed with saturated brine (2 ⁇ 50 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The obtained residue was crystallized twice from hot ethanol to give the title compound 8-4 as colorless needle crystals.
- the filtrate was concentrated, purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 25: 1 ⁇ 20: 1), and then crystallized from hot ethanol to obtain the same compound (8-4). Obtained. ⁇ 11.6 g (37.6 mmol), 69.6% ⁇ LRMS m / z 311 [M + Na] + .
- Route E8; 1-O-allyl-2,3-di-O-benzyl- ⁇ -D-glucopyranoside (8-6) The compound (8-5) obtained by route D8 was dissolved in acetic acid (72 mL), distilled water (40 mL) was further added, and the mixture was stirred for 1 hour while heating under reflux. After confirming that the reaction had progressed sufficiently, it was cooled to room temperature, the solvent was distilled off with an evaporator, distilled water (15 mL) was added, and the mixture was concentrated again under reduced pressure four times.
- Pathway F8 1-O-allyl-2,3,4-di-O-benzyl-6-O-tosyl- ⁇ -D-glucopyranoside (8-7)
- compound (8-6) (9.3 g, 23.2 mmol) in anhydrous pyridine (120 mL)
- p-toluenesulfonyl chloride (5.8 g, 30.2 mmol)
- 4-dimethylaminopyridine (283 mg, 2.3 mmol
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL). The organic layers were combined and washed with 1N HCl aqueous solution until pH4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure did. The concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (8-7). ⁇ 12.3 g (22.2 mmol), 95.7% ⁇ .
- Route G8 1-O-allyl-2,3-di-O-benzyl-4-O- (carbobenzoxy- ⁇ -alanyl) -6-O-tosyl- ⁇ -D-glucopyranoside (8-8)
- Compound (8-7) (12.3 g, 22.2 mmol), N-carbobenzoxy- ⁇ -alanine (12.1 g, 44.4 mmol), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI ⁇ HCl) (17.0 g, 88.8 mmol) and 4-dimethylaminopyridine (5.4 g, 44.4 mmol) were dissolved in a mixed solution of anhydrous dichloromethane (200 mL) and anhydrous pyridine (50 mL) and reacted at room temperature for 18 hours.
- EDCI ⁇ HCl 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydro
- reaction solution was poured into a cold 7.5 M sodium hydroxide (500 mL) solution and extracted with ethyl acetate (4 ⁇ 100 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12.5: 1 ⁇ 10: 1 ⁇ 8: 1) as a colorless waxy substance The title compound (8-12) was obtained.
- the crude product of compound (8-13) obtained by route L8 was dissolved in a mixed solution of anhydrous dichloromethane (15 mL), pyridine (5 mL) and triethylamine (2 mL), and (4-iodobenzoyl) -p-nitro was dissolved.
- Phenyl ester (563 mg, 1.5 mmol) was added, and the mixture was reacted at room temperature for 24 hours while stirring with a stirrer.
- Synthesis Example 9 The production process of the sulfopyranosyl acylglycerol derivative of the present invention is shown in the following scheme 9 by taking a ⁇ sulfoquinovosyl diacylglycerol monoiodo derivative as an example.
- reaction solution was returned to room temperature, washed with saturated aqueous sodium bicarbonate (2 ⁇ 100 mL) and saturated brine (2 ⁇ 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure.
- the concentrate was crystallized twice from hot ethanol to give the title compound (9-2) as colorless needle crystals ⁇ 24.8 g (64 mmol), 50.0% ⁇ .
- the concentrate was dissolved in a small amount of ethyl acetate and the cold water was extracted with ethyl acetate (3 ⁇ 100 mL). The organic layers were combined, washed with saturated brine (2 ⁇ 50 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The obtained residue was crystallized twice from hot ethanol to give the title compound (9-3) as colorless needle crystals (33.5 g). The filtrate was concentrated, purified by silica gel chromatography (hexane-ethyl acetate, 15: 1 ⁇ 10: 1 ⁇ 8: 1), and crystallized from hot ethanol to give the title compound (9-3) ( 6.63 g). ⁇ Total 40.1 g (82.1 mmol), 84.4% ⁇ LRMS m / z 511 [M + Na] + .
- Pathway D9 1-O-allyl-2,3-di-O-benzyl-4,6-O-benzylidene- ⁇ -D-glucopyranoside (9-5)
- compound (9-4) (11.6 g, 37.6 mmol) in anhydrous N, N-dimethylformamide (DMF, 100 mL)
- benzyl chloride (4.76 g, 4 equivalents)
- sodium hydroxide powder (4.5 g, 3.0 equivalents)
- the reaction solution was poured into cold water (100 mL) and extracted with ethyl acetate (3 ⁇ 50 mL).
- Route E9 1-O-allyl-2,3-di-O-benzyl- ⁇ -D-glucopyranoside (9-6)
- Compound 5 obtained by route D9 was dissolved in acetic acid (72 mL), distilled water (40 mL) was further added, and the mixture was stirred for 1 hour with heating under reflux. After confirming that the reaction had progressed sufficiently, it was cooled to room temperature, the solvent was distilled off with an evaporator, distilled water (15 mL) was added, and the mixture was concentrated again under reduced pressure four times.
- reaction solution was then slowly poured into ice water (100 mL), and the aqueous layer was extracted with ethyl acetate (3 ⁇ 200 mL). The organic layers were combined and washed with 1N HCl aqueous solution until pH4, washed with saturated aqueous sodium bicarbonate (2 x 100 mL) and saturated brine (2 x 100 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure did. The concentrate was purified by silica gel flash chromatography (hexane-ethyl acetate, 6: 1 ⁇ 4: 1 ⁇ 2: 1) to give the title compound (9-7). ⁇ 12.3 g (22.2 mmol), 95.7% ⁇ .
- reaction solution was poured into a cold 7.5 M sodium hydroxide (100 mL) solution and extracted with ethyl acetate (4 ⁇ 50 mL). The organic layers were combined, washed with saturated aqueous sodium hydrogen carbonate (2 ⁇ 50 mL) and saturated brine (2 ⁇ 50 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by silica gel chromatography (chloroform-methanol, 100: 1 ⁇ 50: 1 ⁇ 20: 1 ⁇ 15: 1 ⁇ 12.5: 1 ⁇ 10: 1 ⁇ 8: 1) as a colorless waxy substance The title compound (9-12) was obtained.
- Pathway M9 3-O- [4-O- (4-Iodobenzoyl- ⁇ -alanyl) -6-sulfo- ⁇ -D-quinovopyranosyl] -1,2-di-O-stearoyl-glycerol (9-14)
- the crude product of compound (9-13) obtained by route L9 was dissolved in anhydrous dichloromethane (15 mL), pyridine (5 mL) and triethylamine (2 mL) mixed solution, and (4-iodobenzoyl) -p-nitro Phenyl ester (202 mg, 548 ⁇ mol) was added, and the mixture was reacted at room temperature for 24 hours while stirring with a stirrer.
- Biodistribution 1 Biotinylated ⁇ SQMG 1 ⁇ 10 6 human esophageal squamous cell carcinoma cells TE-8 were transplanted into the right thigh of 11 male KSN nude mice (Japan SLC) and raised until the tumor volume reached 300-900 cm 3 did.
- biotinylated ⁇ SQMG C18: 0 (compound of formula (1-12)) was dissolved in physiological saline, and 3 tumor-bearing nudes were obtained in each group to give 40 mg / kg, 20 mg / kg, and 2 mg / kg.
- Mice were injected intravenously with 0.1 mL at a time.
- Two control tumor-bearing mice were injected with physiological saline.
- each mouse was sacrificed, and the tumor was removed and immediately frozen.
- a frozen section having a thickness of 5 ⁇ m was prepared from the frozen tumor, and each section was stained with avidin labeled with a fluorescent dye TRITC (Tetramethylrhodamine-5- (and 6) -isothiocyanate). Each stained section was observed with a fluorescence microscope, digitized, and then subjected to image processing using Photoshop (Adobe).
- TRITC Tetramethylrhodamine-5- (and 6) -isothiocyanate
- the result is shown in FIG.
- the tumor tissue was strongly stained according to the dose of biotinylated ⁇ SQMG C18: 0, indicating that the amount of biotinylated ⁇ SQMG C18: 0 retained in the tumor was large.
- Biotinylated ⁇ SQAP 1 ⁇ 10 6 human large cell lung cancer cells Lu65 / mouse were transplanted into 6-week-old male KSN nude mice, reared for 14 days, and 100-200 mm 3 tumor masses were formed in each nude mouse.
- biotinylated ⁇ SQAP used is a compound of formula (2-11).
- Control group physiological saline administration
- High dose group biotinylated ⁇ 50 mg / kg administration
- Medium dose group 5 mg / kg administration
- Low dose group Administer 1 mg / kg.
- biotinylated ⁇ SQAP dissolved in physiological saline or physiological saline was administered from the tail vein of each nude mouse to the above amount, and the tumor was excised 1 hour after administration, and the tumor was excised with OCT compound. was immersed and frozen in liquid nitrogen.
- the frozen tumor of each test animal was cut to a thickness of 10 ⁇ m, the section was fixed on a microplate, and the site where biotin was distributed was stained with Alexa488-labeled avidin.
- the image slice was observed with a fluorescence microscope, imaged and captured, and the positive signal of each image was quantified with image analysis software.
- Example 3 Pharmacokinetics 1 Animal test 2 ⁇ 10 6 human colon cancer-derived cells (SW480 strain) were subcutaneously transplanted into 8-week-old male KSN nude mice (Japan SLC), and after 2 weeks (body weight about 25 g, tumor volume about 200 In mm 3 ), ⁇ SQMG C18: 0 or ⁇ SQAP C18: 0 was dissolved in physiological saline, and a dose of 10 mg / kg was intraperitoneally administered (0.1 mL). Blood, liver, kidney, lung, spleen and tumor tissue were collected after death from asphyxiation with carbon dioxide at 30 minutes, 1, 2, 4, 12 and 24 hours after administration. Each tissue used 5 animals for each collection time.
- Plasma Blood was collected from the posterior vena cava using an injection needle moistened with sodium heparin injection (10,000 units / 10 mL), and centrifuged at 6,000 g for 20 minutes to obtain plasma.
- Protein denaturation was performed by adding 5 ⁇ L of internal standard substance ( ⁇ SQMG C16: 0), 5 ⁇ L of distilled water and 4 ⁇ L of phosphoric acid to 90 ⁇ L of plasma, and adding 96 ⁇ L of acetonitrile. To 0.2 mL of this solution, 600 ⁇ L of a 10 mM ammonium acetate aqueous solution was added, stirred well, and centrifuged at 16,000 g for 15 minutes to obtain a supernatant.
- a solid phase extraction operation was performed on 600 ⁇ L of the supernatant as follows.
- Empore (registered trademark) disk cartridge (3M, product number 4115SD) was used, and after adding the measurement sample, it was washed with 1,000 ⁇ L of 10 mM ammonium acetate aqueous solution and 200 ⁇ L of 10 mM ammonium acetate aqueous solution containing 10% acetonitrile.
- the test substance was eluted with 200 ⁇ L of 10 mM aqueous ammonium acetate solution containing 70% acetonitrile.
- LC / MS analysis sample was used.
- An internal standard method was used as a quantitative analysis method. That is, by analyzing the sample to which the test substance was added, a standard line was
- FIG. 3 is a concentration-time course curve in plasma and organs of nude mice administered intraperitoneally with ⁇ SQMG C18: 0 at 10 mg / kg.
- the maximum tissue concentration immediately after administration of ⁇ SQMG ⁇ ⁇ ⁇ C18: 0 was shown in plasma, liver, kidney, spleen, and lung, and ⁇ SQMG C18: 0 decreased rapidly thereafter.
- the value was below the detection limit at 4 hours after administration and at 12 hours in the spleen.
- the tissue concentration immediately after administration was lower than that in other organs, but ⁇ SQMG C18: 0 was detected even 24 hours after administration. This revealed that ⁇ SQMG C18: 0 stays in the tumor tissue for a long time.
- FIG. 4 is a graph showing concentration-time transition curves in plasma and organs of nude mice intraperitoneally administered with ⁇ SQAP C18: 0 at 10 mg / kg.
- the maximum tissue concentration immediately after administration of ⁇ SQAP C18: 0 was shown in plasma, liver, kidney, spleen and lung, and ⁇ SQAP C18: 0 decreased rapidly thereafter.
- the value was below the detection limit at 4 hours after administration and at 12 hours in the liver.
- the tissue concentration immediately after administration of ⁇ SQAPC 18: 0 was lower than that in other organs, but it was also detected 24 hours after the administration. Therefore, it was revealed that ⁇ SQAP C18: 0 stays in the tumor tissue for a long time.
- Example 4 Pharmacokinetics 2 ⁇ SQAP C18: 0 1 mg / s in the same manner as in Example 3 except that human esophageal squamous cell carcinoma cell TE-8 was used as the tumor of the transplanted tumor model and that it was administered by intravenous administration instead of intraperitoneal administration. kg was administered to the animals and tested for pharmacokinetics of ⁇ SQAP C18: 0.
- FIG. 5 As can be seen from FIG. 5, in the plasma, liver, spleen and lung, the highest tissue concentration was shown immediately after administration of ⁇ SQAP C18: 0, and ⁇ SQAP C18: 0 decreased rapidly thereafter. In plasma, liver and lung, it was below the detection limit at 2 hours after administration and at 24 hours in spleen. On the other hand, in the tumor tissue, compared with other organs, the tissue concentration immediately after administration of ⁇ SQAP C18: 0 was low, but no decrease tendency was observed at 2 hours, and it was detected at 24 hours. Therefore, it was revealed that ⁇ SQAP C18: 0 stays in the tumor tissue for a long time.
- the novel compound of the present invention is a substance that specifically stays in the tumor and contains a functional group in its structure. Therefore, depending on the type of the functional group, it is usefully used for drug transport to the tumor, detection and diagnosis of the tumor, and treatment of the tumor.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Saccharide Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description

Rは水素と結合した陰イオン性基であり、
R1はOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数であり、
R3はOH、SO3Hまたは作用基であり、
R4はOH、SO3Hまたは作用基であり、および
R5はOH、SO3Hまたは作用基であり、
R1、R2、R3、R4およびR5の少なくとも1つが作用基を含む。
Rは水素と結合した陰イオン性基であり、
R1はOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、
R2はH、OH、OCOHまたはOCO(CH2)iCH3または作用基であり、iは0以上の整数であり、
R3はOH、SO3Hまたは作用基であり、
R4はOH、SO3Hまたは作用基であり、
R5はOH、SO3Hまたは作用基であり、
jは1以上の整数であり、および
Xは作用基または陽イオン性基であり、kは1以上の整数であり、
R1、R2、R3、R4、R5およびXの少なくとも1つは作用基である。
当該化合物または塩が、腫瘍以外の組織よりも腫瘍において高い濃度で存在する時点で、当該標識物質を検出すること、および
当該検出の結果を基に当該対象において腫瘍を検出すること
を具備する腫瘍を検出するための方法。
当該化合物または塩が、腫瘍以外の組織よりも腫瘍において高い濃度で存在する時点で、当該標識物質を検出すること、および
当該検出の結果を基に当該対象に腫瘍があるか否かを診断すること、
を具備する腫瘍について診断する方法。

Rは水素と結合した陰イオン性基であり、
R1はOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R3はOH、SO3Hまたは作用基であり、
R4はOH、SO3Hまたは作用基であり、および
R5はOH、SO3Hまたは作用基であり、
R1、R2、R3、R4およびR5の少なくとも1つが作用基を含み、好ましくはR1、R2、R3、R4およびR5の何れか1つが作用基を含む。
RがSO3Hであり、R3がOHであり、R4がOHであり、R5がOHであり、
R1がOH、OCOH、OCO(CH2)hCH3、O-(作用基)、OCO-(作用基)またはOCO(CH2)hCH2-(作用基)であり、hは0以上の整数であり、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R2がH、OH、OCOH、OCO(CH2)iCH3、作用基、O-(作用基)、OCO-(作用基)またはOCO(CH2)iCH2-(作用基)であり、iは0以上の整数、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R1およびR2の少なくとも一方が作用基を含む
化合物またはその薬学的に許容される塩であってもよい。
RがSO3Hであり、
R1がOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数であり、好ましくは0以上30以下、より好ましくは0以上26以下、更に好ましくは1以上22以下であり、
R3=R4=R5で且つOHである、またはR3、R4およびR5の少なくとも1つが作用基であり、残りの基がOHであり、
R1、R2、R3、R4およびR5の少なくとも1つが作用基を含む
化合物またはその薬学的に許容される塩であってもよい。
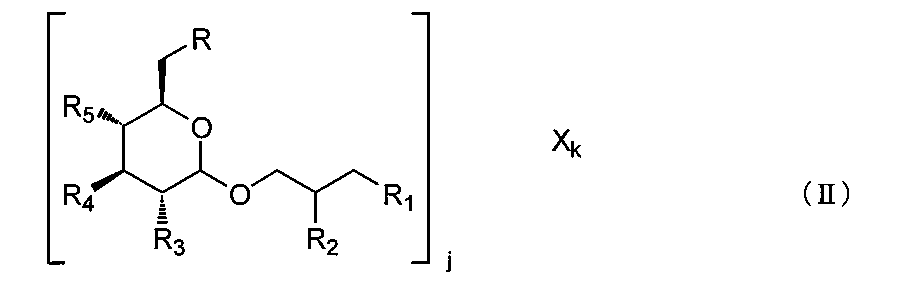
R、R1、R2、R3、R4およびR5は、式(I)において定義した通りであり、
jは1以上の整数であり、および
Xは作用基または陽イオン性基であり、kは1以上の整数であり、好ましくは
R1、R2、R3、R4、R5およびXの少なくとも1つは作用基である。

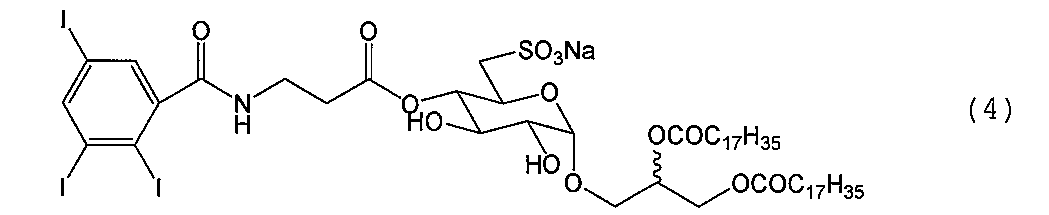
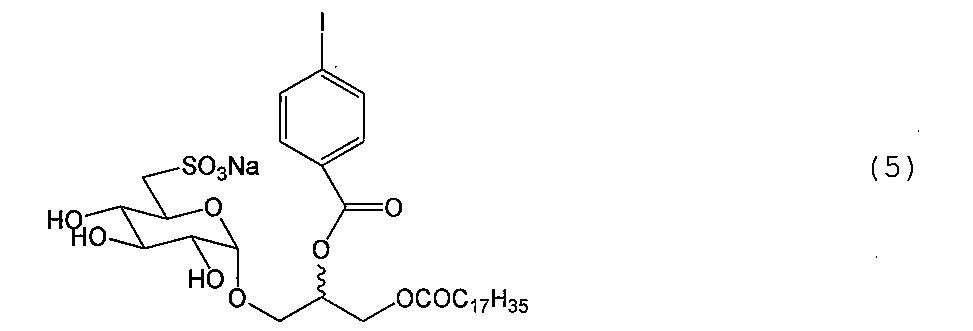

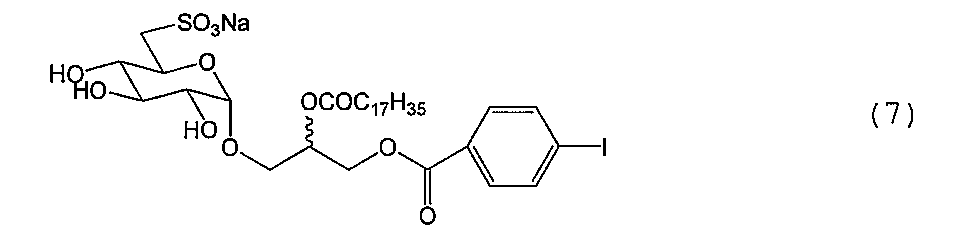
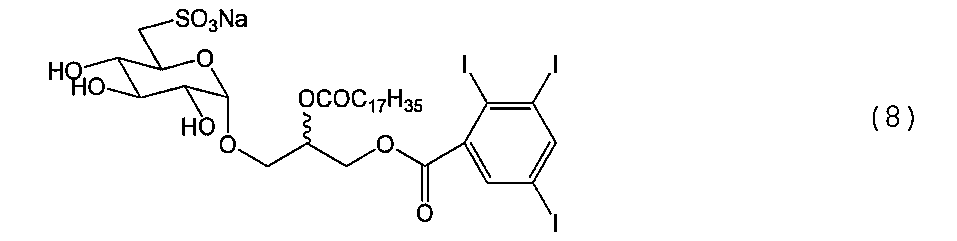



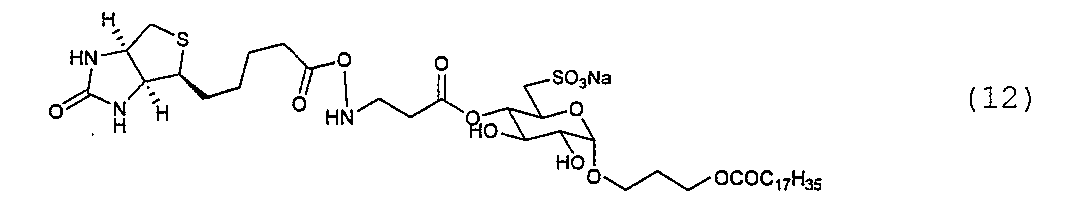
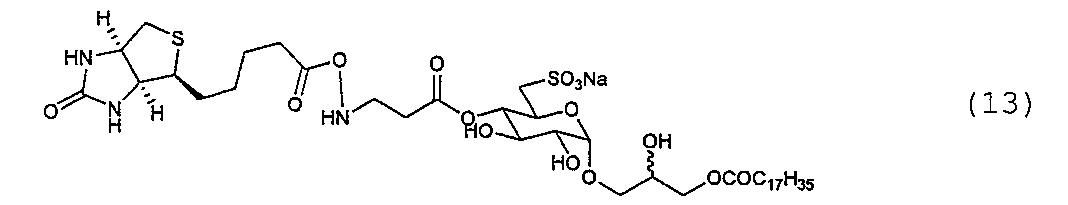
例1.合成
合成例1
本発明のスルホピラノシルグリセロール誘導体の製造工程を、α-スルホキノボシルアシルグリセロールのビオチン誘導体(「ビオチン化αSQMG」とも記す)を例に挙げて次のスキーム1に示す。

化合物(1-1)(100 g、555 mmol)のアリルアルコール(500 mL)懸濁液に、トリフルオロメタンスルホン酸(1.00 mL)を0℃で加え、反応液を80℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3 mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127 g、1.5当量)およびp-トルエンスルホン酸一水和物(5.28 g、0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10 mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000 mL)および水(500 mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(1-2)を得た{34.5 g(112 mmol)、20.2%}。
化合物(1-2)(30.0 g、97.3 mmol)の無水N,N-ジメチルホルムアミド(DMF、300 mL)溶液に、ベンジルブロミド(41.6 g、2.5当量)および水酸化ナトリウム(11.7 g、3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(900 mL)に注ぎ、酢酸エチル(3×300 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(1-3)を得た(33.5 g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同化合物(1-3)を得た(6.63 g)。{計40.1 g (82.1 mmol)、84.4 %}
LRMS 511 m/Z (M+Na)+。
化合物(1-3)(15.6 g、32.0 mmol)を酢酸(90 mL)に溶解し、さらに蒸留水(50 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→2:1→1:1→1:2)で精製し、標題化合物(1-4)を得た{12.1g (30.2 mmol), 94.5%}。
化合物(1-4)(12.1 g、30.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(7.5 g、39.3 mmol)および4-ジメチルアミノピリジン(369 mg、3.0 mmol)を加え、反応液を0℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル、6:1→4:1→2:1)で精製し、標題化合物(1-5)を得た。{15.3 g (27.6 mmol)、91.4 %}。
化合物(1-5)(15.3 g, 27.6 mmol)、N-カルボベンゾキシ-β-アラニン(12.32 g, 55.2 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(15.8 mg, 82.8 mmol)および4-ジメチルアミノピリジン (6.7 g, 55.2 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(1-6)を得た[17.1 g (22.5 mmol ), 81.5 %]。
化合物(1-6)(17.1 g、22.5 mmol)の無水N,N-ジメチルホルムアミド(300 mL)溶液に、チオ酢酸カリウム(5.1 g、45.0 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(1-7)を得た{14.2 g (20.3 mmol ), 90.0 %}。
化合物(1-7)(14.2 g、20.4 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04M四酸化オスミウム-t-ブチルアルコール(3 mL)、トリメチルアミンN-オキシド(3.4 g、30.5 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 2:1→1:1→2:3→1:2→1:4→1:8)で精製し、標題化合物(1-8)を得た{13.2 g (18.9 mmol ), 93.0 %}。
化合物(1-8)(13.1 g、18.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(8.5 g、28.2 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル、6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(1-9)を得た{7.1 g(7.4 mmol)、39.4 %}。
化合物(1-9)(7.1 g、7.4 mmol)の酢酸(160 g、4 mol )溶液に、オキソン(18.1 g、29.5 mmol)および酢酸カリウム(4.0 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(500 mL)溶液に注ぎ、酢酸エチル(4×100 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール、100:1→50:1→20:1→15:1→12:1)で精製し、無色ワックス状物質として標題化合物(10)を得た{5.2 g(5.24 mmol )、71.2 %}。
化合物(1-10)(490 mg、494 μmol)のメタノール(20 mL)および酢酸(0.6 mL)溶液に、10%パラジウム活性炭素(300 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水、100:25:3→65:25:4→65:35:5→65:45:6)で精製して標題化合物(1-11)を得た{221 mg (327 μmol )、66.1 %}。
化合物(1-11)(273.2 mg、403 μmol)を無水N,N-ジメチルホルムアミド(20 mL)及びトリエチルアミン(1 mL)混合溶液に溶解し、ビオチンp-ニトロフェニルエステル(162 mg、443 μmol)を加えて、スターラーで攪拌しながら室温で24時間反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水、40:10:1.2→70:30:2.6→30:10:1.5→65:25:4)で精製して標題化合物(1-12)を得た{320.8 mg(355 μmol)、88.0 %}。
本発明のスルホピラノシルアシルプロパンジオール誘導体の製造工程を、α-スルホキノボシルアシルプロパンジオールビオチン誘導体(「ビオチン化αSQAP」とも記す)を例に挙げて次のスキーム2に示す。

経路A2;1-O-アリル-4,6-O-ベンジリデン-α-D-グルコピラノシド (2-2)
D-グルコース(2-1)(100g, 555mmol)のアリルアルコール(500mL)懸濁液に、トリフルオロメタンスルホン酸(1.00mL)を0℃で加え、反応液を80℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127g, 1.5当量)およびp-トルエンスルホン酸一水和物(5.28g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000mL)および水(500mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(2-2)を得た{34.5g (112mmol), 20.2%}。[α]23 D +97.5° (c1.00 CH3OH), LRMS m/z 331 [M + Na]+, mp 139-141℃
1H NMR (400MHz, CD3OD); δ 7.51-7.47 (m, 2H, ArH), 7.37-7.32 (m, 3H, ArH), 5.99 (dddd, 1H, J=17.2, 10.5, 6.08, 5.32Hz, H2), 5.56 (s, 1H, PhCH), 5.36 (dq, 1H, J=17.3, 1.68Hz, H3a), 5.20 (ddt, 1H, J=10.4, 1.80, 1.28Hz, H3b), 4.88 (d, 1H, J=3.86Hz, H1’), 4.25-4.18 (m, 2H, H1a & H6’a), 4.07 (ddt, 1H, J=13.0, 6.10, 1.36Hz, H1b), 3.85 (t, 1H, J=9.38Hz, H3’), 3.81-3.71 (m, 2H, H5’ & H6’b), 3.52 (dd, 1H, J=9.38, 3.86Hz, H2’), 3.45 (t, 1H, J=9.24Hz, H4’)
13C NMR (100MHz, CD3OD); δ 139.1 (Ar-ipso), 135.4 (C2), 129.9 (Ar), 129.0 (Ar), 127.5 (Ar), 117.8 (C3), 103.0 (PhCH), 100.0 (C1’), 82.9 (C4’), 74.0 (C2’), 72.0 (C3’), 69.9 (C6’), 69.7 (C1), 64.1 (C5)。
化合物(2-2)(30.0g, 97.3mmol)の無水N,N-ジメチルホルムアミド(DMF, 300mL)溶液に、ベンジルブロミド(41.6g, 2.5当量)および水酸化ナトリウム(11.7g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(900mL)に注ぎ、酢酸エチル(3×300mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物3を得た(33.5g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同化合物(2-3)を得た(6.63g)。{計40.1g (82.1mmol), 84.4%}[α]26 D -1.46°(c1.03 CHCl3), LRMS m/z 511 [M + Na]+, mp 86-87℃
1H NMR (400MHz, CDCl3); δ 7.50-7.47 (m, 2H, ArH), 7.40-7.24 (m, 13H, ArH), 5.94 (dddd, 1H, J=17.0, 10.4, 6.70, 5.24Hz, H2), 5.56 (s, 1H, PhCH), 5.33 (dq, 1H, J=17.2, 1.56Hz, H3a), 5.24 (ddt, 1H, J=10.3, 1.56, 1.12Hz, H3b), 4.92 (d, 1H, J=11.2Hz, ArCH2), 4.84 (d, 1H, J=11.2Hz, ArCH2), 4.83 (d, 1H, J=12.1Hz, ArCH2), 4.80 (d, 1H, J=3.76Hz, H1'), 4.68 (d, 1H, J=12.1Hz, ArCH2), 4.26 (dd, 1H, J=10.2, 4.84Hz, H6'a), 4.18 (ddt, 1H, J=12.9, 5.18, 1.40Hz, H1a), 4.79 (t, 1H, J=9.30Hz, H3'), 4.03 (ddt, 1H, J=12.9, 6.68, 1.20Hz, H1b), 3.89 (dt, 1H, J=9.96, 4.80Hz, H5'), 3.70 (t, 1H, J=10.3Hz, H6'b), 3.61 (t, 1H, J=9.44Hz, H4'), 3.57 (dd, 1H, J=8.72, 3.80Hz, H2’)
13C NMR (100MHz, CDCl3); δ 138.7 (Ar-ipso), 138.1 (Ar-ipso), 137.3 (Ar-ipso), 133.5 (C2), 128.9-127.5 (m, Ar), 126.0 (Ar), 118.4 (C3), 101.2 (PhCH), 96.7 (C1’), 82.1 (C3'), 79.1 (C2’), 78.6 (C4'), 75.3 (ArCH2), 73.6 (ArCH2), 69.0 (C6’), 68.4 (C1), 62.5 (C5’)。
水素化リチウムアルミニウム(2.02g, 1.3当量)が懸濁している無水ジクロロメタン(100mL)-無水ジエチルエーテル(100mL)混合溶液に化合物(2-3)(20.0g, 40.9mmol)を加えた。次いで反応液に塩化アルミニウム(7.09g, 1.3当量)の無水ジエチルエーテル溶液200mLを加えて、加熱還流下、4時間攪拌した。反応が充分進行していることを確認した後、水(10mL)をゆっくりと滴下し、一晩後沈殿物を濾別し、ジエチルエーテルで沈殿をリンスした。濾液を水(2 x 100mL)で洗浄し、水層を合わせてジエチルエーテル(2 x 100mL)で抽出した。有機層を合わせて飽和食塩水(2 x 200mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 5:1→4:1→3:1→2:1)で精製し、無色油状物として標題化合物(2-4)を得た{18.1g (36.9mmol), 90.2%}。[α]22 D +45.0° (c1.21 CHCl3), LRMS m/z 513 [M + Na]+
1H NMR (400MHz, CDCl3); δ 7.37-7.26 (m, 15H, ArH), 5.92 (dddd, 1H, J=17.1, 10.4, 6.66, 5.24Hz, H2), 5.31 (dq, 1H, J=17.2, 1.52Hz, H3a), 5.22 (ddt, 1H, J=10.3, 1.46, 1.10Hz, H3b), 5.00 (d, 1H, J=10.9Hz, ArCH2), 4.89 (d, 1H, J=11.0Hz, ArCH2), 4.84 (d, 1H, J=10.9Hz, ArCH2), 4.77 (d, 1H, J=12.0Hz, ArCH2), 4.77 (d, 1H, J=3.60Hz, H1'), 4.65 (d, 1H, J=12.1Hz, ArCH2), 4.64 (d, 1H, J=11.0Hz, ArCH2), 4.14 (ddt, 1H, J=12.9, 5.22, 1.34Hz, H1a), 4.04 (t, 1H, J=9.36Hz, H3'), 3.99 (ddt, 1H, J=12.9, 6.64, 1.08Hz, H1b), 3.79-3.66 (m, 3H, H5' & H6'a & H6'b), 3.54 (t, 1H, J=9.28Hz, H4'), 3.51 (dd, 1H, J=9.60, 3.64Hz, H2'), 1.69 (t, 1H, J=12.0Hz, 6'-OH)
13C NMR (100MHz, CDCl3); δ 138.7 (Ar-ipso), 138.1 (Ar-ipso), 138.1 (Ar-ipso), 133.6 (C2), 128.4-127.6 (m, Ar), 118.3 (C3), 95.6 (C1’), 81.9 (C3’), 79.9 (C2’), 77.3 (C4’), 75.7 (ArCH2), 75.0 (ArCH2), 73.2 (ArCH2), 70.8 (C5’), 68.2 (C1), 61.7 (C6’)。
化合物(2-4)(25.1g, 51.2mmol)の無水ピリジン(250mL)溶液に、p-トルエンスルホニルクロリド(14.6g, 1.5当量)および4-ジメチルアミノピリジン(626mg, 0.1当量)を加え、反応液を室温にて16時間撹拌した。反応が充分進行していることを確認した後、水(10mL)を加えて反応を止め、反応液を減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を0.5M塩酸(200mL)に注ぎ、酢酸エチル(3×200mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100mL)および飽和食塩水(2 x 100mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(2-5)を得た(25.0g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 5:1→4:1→3:1)で精製し、同化合物(2-5)を得た(4.00g)。{計29.0g (45.0mmol), 87.9%} [α]25 D +32.1°(c1.02 CHCl3), LRMS m/z 667 [M+Na]+, mp 86-87℃
1H NMR (400MHz, CDCl3); δ 7.76 (ddd, 2H, J=8.32, 1.96, 1.76Hz, ArH), 7.35-7.26 (m, 15H, ArH), 7.17-7.12 (m, 2H, ArH), 5.88 (dddd, 1H, J=17.2, 10.3, 6.62, 5.24Hz, H2), 5.28 (dq, 1H, J=17.2, 1.56Hz, H3a), 5.20 (ddt, 1H, J=10.3, 1.60, 1.12Hz, H3b), 4.99 (d, 1H, J=10.9Hz, ArCH2), 4.82 (d, 1H, J=10.6Hz, ArCH2), 4.78 (d, 1H, J=10.8Hz, ArCH2), 4.74 (d, 1H, J=12.1Hz, ArCH2), 4.72 (d, 1H, J=3.58Hz, H1'), 4.62 (d, 1H, J=12.1Hz, ArCH2), 4.42 (d, 1H, J=10.6Hz, ArCH2), 4.22 (dd, 1H, J=10.5, 4.20Hz, H6'a), 4.16 (dd, 1H, J=10.5, 2.12Hz, H6'b), 4.07 (ddt, 1H, J=12.9, 5.24, 1.40Hz, H1a), 3.98 (t, 1H, J=9.24Hz, H3'), 3.93 (ddt, 1H, J=12.9, 6.64, 1.16Hz, H1b), 3.81 (ddd, 1H, J=10.1, 4.12, 2.04Hz, H5'), 3.48 (dd, 1H, J=9.62, 3.58Hz, H2'), 3.45 (dd, 1H, J=10.0, 8.90Hz, H4’), 2.39 (s, 3H, Ts-Me)
13C NMR (100MHz, CDCl3); δ 144.8 (Ar-ipso), 138.5 (Ar-ipso), 137.9 (Ar-ipso), 137.7 (Ar-ipso), 133.4 (C2), 132.8 (Ar-ipso), 129.8 Ar(), 128.4-127.6 (m, Ar), 118.4 (C3), 95.4 (C1’), 81.8 (C3’), 79.6 (C2’), 76.9 (C4’), 75.7 (ArCH2), 75.0 (ArCH2), 73.2 (ArCH2), 68.6 (C5’), 68.5 (C6’), 68.3 (C1), 21.6 (Ts-Me)。
化合物(2-5)(29.0g, 45.0mmol)の無水テトラヒドロフラン(THF, 150mL)溶液に、アルゴン雰囲気下、0℃で、0.5M 9-ボラビシクロ[3,3,1]ノナン(9-BBN)のテトラヒドロフラン溶液(180mL, 90.0mmol)を加えた。1時間後、反応液を室温に戻し、引き続き10時間撹拌した。反応液を再び0℃に冷却し、まず水(20mL)を加え、次に3M水酸化ナトリウム溶液(70mL)および35%過酸化水素水(70mL)を順次加え、1時間後室温に戻し、12時間攪拌した。反応が充分進行していることを確認した後、この溶液を酢酸エチル(3 x 100mL)で抽出し、有機層を合わせて飽和食塩水(2 x 100mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 3:2→1:1→2:3)で精製し、無色油状物として標題化合物(2-6)を得た{28.2g (42.5mmol), 94.4%}。 [α]24 D +26.6° (c1.02 CHCl3), LRMS m/z 685 [M + Na]+
1H NMR (400MHz, CDCl3); δ 7.76-7.74 (m, 2H, ArH), 7.35-7.26 (m, 15H, ArH), 7.16-7.12 (m, 2H, ArH), 4.94 (d, 1H, J=10.9Hz, ArCH2), 4.82 (d, 1H, J=10.7Hz, ArCH2), 4.77 (d, 1H, J=10.9Hz, ArCH2), 4.75 (d, 1H, J=12.0Hz, ArCH2), 4.61 (d, 1H, J=12.0Hz, ArCH2), 4.61 (d, 1H, J=3.64Hz, H1'), 4.43 (d, 1H, J=10.7Hz, ArCH2), 4.20-4.13 (m, 2H, H6'a & H6'b), 3.92 (t, 1H, J=9.24Hz, H3'), 3.84-3.74 (m, 4H, H1a & H3a & H3b & H5'), 3.48-3.40 (m, 3H, H1b & H2' & H4'), 2.52 (t, 1H, J=4.74Hz, 3-OH), 2.39 (s, 3H, Ts-Me), 1.88-1.75 (m, 2H, H2a & H2b)
13C NMR (100MHz, CDCl3); δ 144.8 (Ar-ipso), 138.4 (Ar-ipso), 137.9 (Ar-ipso), 137.6 (Ar-ipso), 132.7 (Ar-ipso), 129.8 (Ar), 128.5-127.6 (m, Ar), 97.1 (C1’), 81.8 (C3’), 79.5 (C2’), 76.8 (C4’), 75.6 (ArCH2), 75.0 (ArCH2), 73.4 (ArCH2), 68.7 (C5’), 68.6 (C6’), 67.5 (C1), 61.5 (C3), 31.5 (C2), 21.6 (Ts-Me)。
化合物(2-6)(28.2g, 42.5mmol)の無水DMF (300mL)溶液に、チオ酢酸カリウム(7.28g, 1.5当量)を加え、90℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(900mL)に注ぎ、酢酸エチル(3 x 300mL)で抽出した。有機層を合わせて飽和食塩水(2 x 200mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 2:1→3:2→1:1→2:3)で精製し、淡褐色油状物質として標題化合物(2-7)を得た{21.9g (38.6mmol ), 90.8%}。[α]23 D +33.0°(c1.02 CHCl3), LRMS m/z 584 [M + Na]+
1H NMR (400MHz, CDCl3); δ 7.37-7.24 (m, 15H, ArH), 4.95 (d, 1H, J=10.8Hz, ArCH2), 4.89 (d, 1H, J=10.6Hz, ArCH2), 4.80 (d, 1H, J=10.8Hz, ArCH2), 4.77 (d, 1H, J=12.1Hz, ArCH2), 4.63 (d, 1H, J=12.0Hz, ArCH2), 4.63 (d, 1H, J=3.52Hz, H1'), 4.61 (d, 1H, J=10.7Hz, ArCH2), 3.94 (t, 1H, J=9.22Hz, H3'), 3.88 (ddd, 1H, J=9.86, 6.10, 4.88Hz, H1a), 3.83-3.73 (m, 3H, H3a & H3b & H5'), 3.50 (dd, 1H, J=9.60, 3.64Hz, H2'), 3.45 (ddd, 1H, J=9.92, 5.24, 2.28Hz, H1b), 3.41 (dd, 1H, J=13.6, 3.00Hz, H6'a), 3.30 (dd, 1H, J=9.54, 9.06Hz, H4'), 3.02 (dd, 1H, J=13.7, 7.64Hz, H6'b), 2.67 (br, 1H, 3-OH), 2.32 (s, 3H, SAc-Me), 1.92-1.78 (m, 2H, H2a & H2b)
13C NMR (100MHz, CDCl3); δ 195.0 (SAC-C=O), 138.5 (Ar-ipso), 137.9 (Ar-ipso), 137.8 (Ar-ipso), 128.5-127.6 (m, Ar), 96.9 (C1’), 81.8 (C3’), 80.4 (C4’), 79.8 (C2’), 75.7 (ArCH2), 75.2 (ArCH2), 73.4 (ArCH2), 69.5 (C5’), 67.2 (C1), 61.5 (C3), 31.5 (C2), 30.8 (C6’), 30.5 (SAc-Me)。
式(2-7)の化合物(500 mg, 0.88 mmol)の酢酸(5 mL)溶液に、オキソン(1.63 g, 2.65 mmol)および酢酸カリウム(25 mg)を加え、室温で2日間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5M水酸化ナトリウム(15 mL)溶液に注ぎ、クロロホルム(4×30 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 20mL)および飽和食塩水(2 x 20mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をさらにメタノール(20 mL)に溶解しナトリウムメトキシド(NaOMe)を適量加え室温にて3時間反応した。反応液に氷冷下冷水を加え反応を停止し減圧濃縮した。濃縮物をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:0→3:1)で精製し、無色ワックス状物質として標題化合物(2-8)を得た{498 mg (0.87 mmol ), 98.7 %}。 LRMS m/z 571 [M - Na]- 。
経路H2;3-O-(2,3,4-トリ-O-ベンジル-6-スルホ-α-D-キノボピラノシル) -1-O-(カルボベンゾキシ-β-アラニル)-プロパン-1,3-ジオール ナトリウム塩 (2-9)
化合物(2-8)(900 mg, 1.51 mmol)、N-カルボベンゾキシ-β-アラニン(440 mg, 1.97 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(871 mg, 4.55 mmol)および4-ジメチルアミノピリジン (55 mg, 0.15 mmol)を無水DMF (15 mL)の混合溶液に溶解し、室温にて一晩反応した。反応が充分進行していることを確認し、反応液に水(1 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:0→12:1)で精製し、標題化合物(2-9)を得た{517 mg (0.65 mmol ), 42.8 %}。
化合物(2-9)(517 mg, 0.65 mmol)のメタノール(3.0 mL)、ジクロロメタン(3.0 mL) および酢酸(0.2 mL)溶液に、20%水酸化パラジウム活性炭素(50 mg)を加え水素ガス雰囲気下、室温で一晩反応させた。反応が十分に進まなかったので更にメタノール(3.0 mL)、10%パラジウム活性炭素(50 mg)を加え、水素ガス雰囲気下、室温で2日間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素をセライトで濾別し、濾液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 4:1→2:1→1:1→2:3)で精製し、標題化合物(2-10)を得た{250.8 mg (0.63 mmol ), 98.2%}。LRMS m/z 372 [M - Na]- 。
化合物(2-10)(226 mg, 0.57 mmol)を無水DMF(5 mL)及びトリエチルアミン(2 mL)混合溶液に溶解し、ビオチンp-ニトロフェニルエステル(313 mg, 0.86 mmol)を溶解した無水DMF(2mL)溶液を加えて、スターラーで攪拌しながら室温で一晩反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 10:1→1:4)で精製してナトリウム塩体を得た。塩化カルシウム水溶液にてカルシウム体に交換したイオン交換樹脂(アンバーライト(登録商標)IR-120、15.7 mL)をカラムに充填した。ナトリウム塩体水溶液を反復してカラムを通過させカルシウム塩体へ交換する操作を行い標題化合物(2-11)を得た{210 mg (340 μmol ), 59.5 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、α-スルホキノボシルモノアシルグリセロールモノヨード誘導体を例に挙げて次のスキーム3に示す。

経路A3;1-O-アリル-4,6-O-ベンジリデン-α-D-グルコピラノシド (3-2)
化合物(3-1)(100 g, 555 mmol)のアリルアルコール(500 mL)懸濁液に、トリフルオロメタンスルホン酸(1.00 mL)を0℃で加え、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3 mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127 g, 1.5当量)およびp-トルエンスルホン酸一水和物(5.28 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10 mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000 mL)および水(500 mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(3-2)を得た{34.5 g (112 mmol), 20.2%}。
化合物(3-2)(30.0 g, 97.3 mmol)の無水N,N-ジメチルホルムアミド(DMF, 300 mL)溶液に、ベンジルブロミド(41.6 g, 2.5当量)および水酸化ナトリウム(11.7 g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(900 mL)に注ぎ、酢酸エチル(3×300 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(3-3)を得た(33.5 g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同表題化合物(3-3)を得た(6.63 g)。{計40.1 g (82.1 mmol), 84.4 %}
LRMS m/z 511 [M+Na]+。
化合物(3-3)(15.6 g, 32.0 mmol)を酢酸(90 mL)に溶解し、さらに蒸留水(50 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→2:1→1:1→1:2)で精製し、標題化合物(3-4)を得た{12.1g (30.2 mmol), 94.5%}。
化合物(3-4)(12.1 g, 30.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(7.5 g, 39.3 mmol)および4-ジメチルアミノピリジン(369 mg, 3.0 mmol)を加え、反応液を0 ℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 6:1→4:1→2:1)で精製し、標題化合物(3-5)を得た。{15.3 g (27.6 mmol), 91.4 %}。
化合物(3-5)(15.3 g, 27.6 mmol)、N-カルボベンゾキシ-β-アラニン(12.32 g, 55.2 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(15.8 mg, 82.8 mmol)および4-ジメチルアミノピリジン (6.7 g, 55.2 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(3-6)を得た[17.1 g (22.5 mmol ), 81.5 %]。
化合物(3-6)(17.1 g, 22.5 mmol)の無水N,N-ジメチルホルムアミド(300 mL)溶液に、チオ酢酸カリウム(5.1 g, 45.0 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(3-7)を得た{14.2 g (20.3 mmol ), 90.0 %}。
化合物(3-7)(14.2 g, 20.4 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04 M四酸化オスミウム-t-ブチルアルコール(3 mL)、トリメチルアミンN-オキシド(3.4 g, 30.5 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3 g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 2:1→1:1→2:3→1:2→1:4→1:8)で精製し、標題化合物(3-8)を得た{13.2 g (18.9 mmol ), 93.0 %}。
化合物(3-8)(13.1 g, 18.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(8.5 g, 28.2 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(3-9)を得た{7.1 g (7.4 mmol ), 39.4 %}。
化合物(3-9)(7.1 g, 7.4 mmol)の酢酸(160 g, 4 mol )溶液に、オキソン(18.1 g, 29.5 mmol)および酢酸カリウム(4.0 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(500 mL)溶液に注ぎ、酢酸エチル(4×100 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→20:1→15:1→12:1)で精製し、無色ワックス状物質として標題化合物(3-10)を得た{5.2 g (5.24 mmol ), 71.2 %}。
化合物(3-10)(490 mg, 494 μmol)のメタノール(20 mL)および酢酸(0.6 mL)溶液に、10 %パラジウム活性炭素(300 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 100:25:3→65:25:4→65:35:5→65:45:6)で精製して標題化合物(3-11)を得た{221 mg (327 μmol ), 66.1 %}。
化合物(3-11)(221 mg, 327 μmol)を無水ジクロロメタン(15 mL)、ピリジン(5 mL)及びトリエチルアミン(2 mL)混合溶液に溶解し、(4-ヨードベンゾイル)- p-ニトロフェニルエステル(241 mg, 653 μmol)を加えて、スターラーで攪拌しながら室温で24時間反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 30:5:0.5→30:6:0.7→30:8:0.9→30:10:1.1)で精製して標題化合物(3-12)を得た{262 mg (238 μmol ), 72.8 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、αスルホキノボシルモノアシルグリセロールトリヨード誘導体を例に挙げて次のスキーム4に示す。
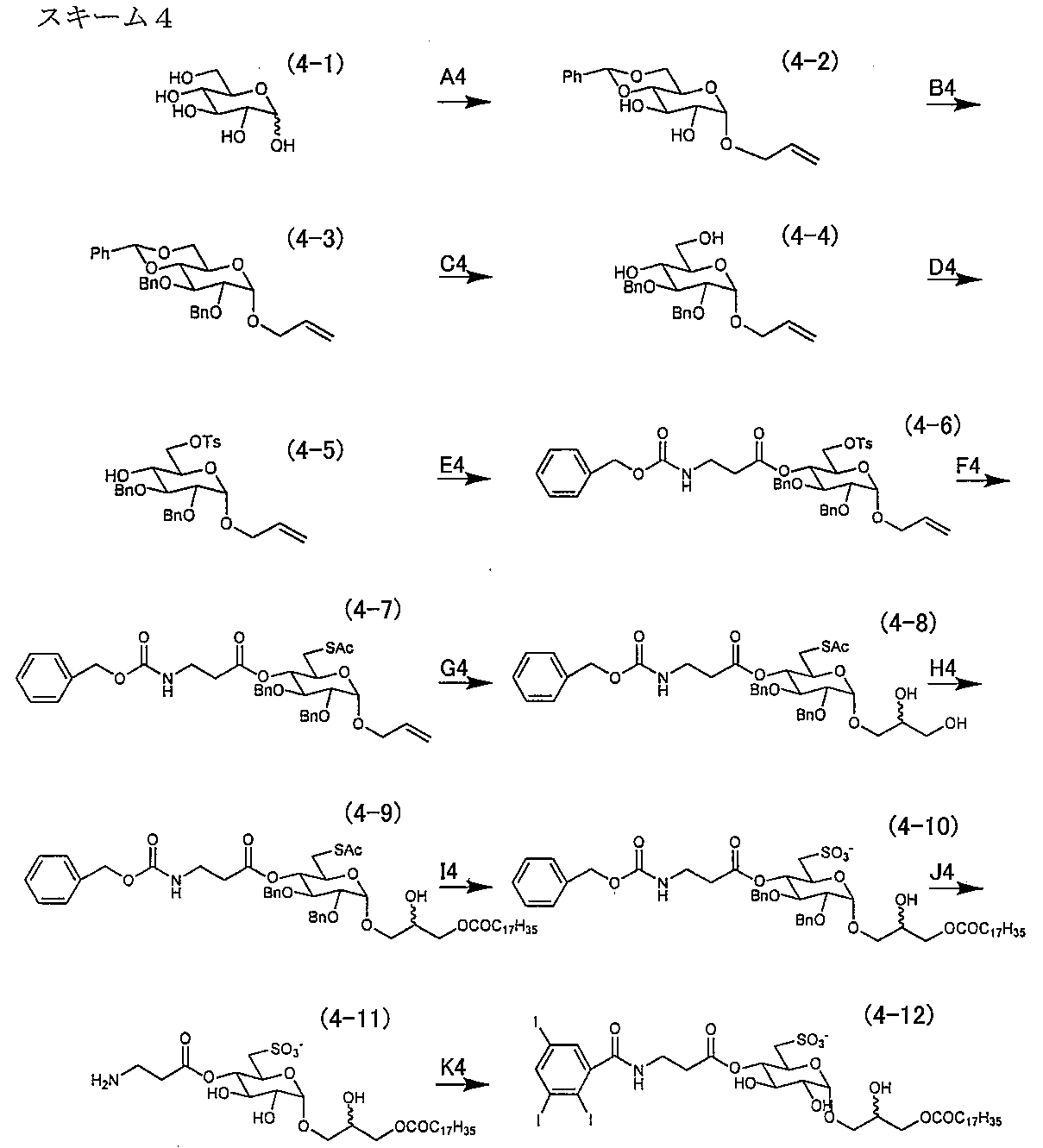
経路A4;1-O-アリル-4,6-O-ベンジリデン-α-D-グルコピラノシド (4-2)
化合物(4-1)(100 g, 555 mmol)のアリルアルコール(500 mL)懸濁液に、トリフルオロメタンスルホン酸(1.00 mL)を0℃で加え、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3 mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127 g, 1.5当量)およびp-トルエンスルホン酸一水和物(5.28 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10 mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000 mL)および水(500 mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(4-2)を得た{34.5 g (112 mmol), 20.2%}。
化合物(4-2)(30.0 g, 97.3 mmol)の無水N,N-ジメチルホルムアミド(DMF, 300 mL)溶液に、ベンジルブロミド(41.6 g, 2.5当量)および水酸化ナトリウム(11.7 g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(900 mL)に注ぎ、酢酸エチル(3×300 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物3を得た(33.5 g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同表題化合物(4-3)を得た(6.63 g)。{計40.1 g (82.1 mmol), 84.4 %}
LRMS m/z 511 [M+Na]+。
化合物(4-3)(15.6 g, 32.0 mmol)を酢酸(90 mL)に溶解し、さらに蒸留水(50 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→2:1→1:1→1:2)で精製し、標題化合物(4-4)を得た{12.1g (30.2 mmol), 94.5%}。
化合物(4-4)(12.1 g, 30.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(7.5 g, 39.3 mmol)および4-ジメチルアミノピリジン(369 mg, 3.0 mmol)を加え、反応液を0 ℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 6:1→4:1→2:1)で精製し、標題化合物(4-5)を得た。{15.3 g (27.6 mmol), 91.4 %}。
化合物(4-5)(15.3 g, 27.6 mmol)、N-カルボベンゾキシ-β-アラニン(12.32 g, 55.2 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(15.8 mg, 82.8 mmol)および4-ジメチルアミノピリジン (6.7 g, 55.2 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(4-6)を得た[17.1 g (22.5 mmol ), 81.5 %]。
化合物(4-6)(17.1 g, 22.5 mmol)の無水N,N-ジメチルホルムアミド(300 mL)溶液に、チオ酢酸カリウム(5.1 g, 45.0 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(4-7)を得た{14.2 g (20.3 mmol ), 90.0 %}。
化合物(4-7)(14.2 g, 20.4 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04 M四酸化オスミウム-t-ブチルアルコール(3 mL)、トリメチルアミンN-オキシド(3.4 g, 30.5 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3 g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 2:1→1:1→2:3→1:2→1:4→1:8)で精製し、標題化合物(4-8)を得た{13.2 g (18.9 mmol ), 93.0 %}。
化合物(4-8)(13.1 g, 18.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(8.5 g, 28.2 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(4-9)を得た{7.1 g (7.4 mmol ), 39.4 %}。
化合物(4-9)(7.1 g, 7.4 mmol)の酢酸(160 g, 4 mol )溶液に、オキソン(18.1 g, 29.5 mmol)および酢酸カリウム(4.0 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(500 mL)溶液に注ぎ、酢酸エチル(4×100 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→20:1→15:1→12:1)で精製し、無色ワックス状物質として標題化合物(4-10)を得た{5.2 g (5.24 mmol ), 71.2 %}。
化合物(4-10)(191 mg, 193 μmol)のメタノール(10 mL)、ジクロロメタン (10mL)および酢酸(0.2 mL)溶液に、10 %水酸化パラジウム活性炭素(160 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、水酸化パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 100:25:3→65:25:4→65:35:5→65:45:6)で精製して標題化合物(4-11)を得た。
化合物(4-11)、2,3,5-トリヨウ素安息香酸(192 mg, 385 μmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(73.8 mg, 385 μmol)および4-ジメチルアミノピリジン (2 mg, 19 μmol)を無水ジクロロメタン(10 mL)及び無水ピリジン(5 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 50:10:1→40:10:1.2→30:10:1.5→20:10:2)で精製して標題化合物(4-12)を得た{81 mg (70 μmol ), (経路J4およびK4二経路での収率)36.3 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、αスルホキノボシルジアシルグリセロールモノヨード誘導体を例に挙げて次のスキーム5に示す。
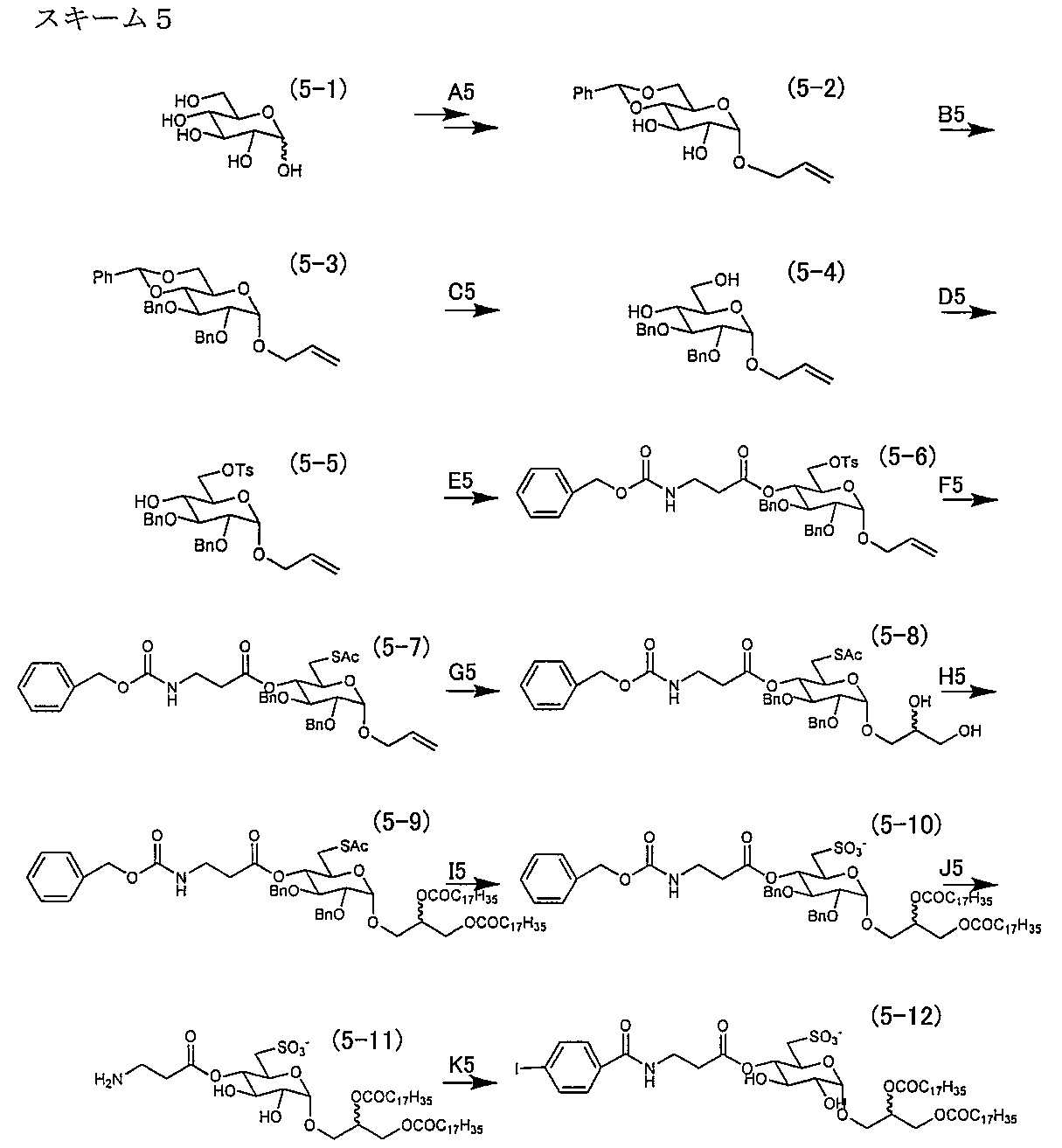
経路A5;1-O-アリル-4,6-O-ベンジリデン-α-D-グルコピラノシド (5-2)
化合物(5-1)(100 g, 555 mmol)のアリルアルコール(500 mL)懸濁液に、トリフルオロメタンスルホン酸(1.00 mL)を0℃で加え、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3 mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127 g, 1.5当量)およびp-トルエンスルホン酸一水和物(5.28 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10 mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000 mL)および水(500 mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(5-2)を得た{34.5 g (112 mmol), 20.2%}。
化合物(5-2)(30.0 g, 97.3 mmol)の無水N,N-ジメチルホルムアミド(DMF, 300 mL)溶液に、ベンジルブロミド(41.6 g, 2.5当量)および水酸化ナトリウム(11.7 g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(900 mL)に注ぎ、酢酸エチル(3×300 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物3を得た(33.5 g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同表題化合物(5-3)を得た(6.63 g)。{計40.1 g (82.1 mmol), 84.4 %}
LRMS m/z 511 [M+Na]+。
化合物(5-3)(15.6 g, 32.0 mmol)を酢酸(90 mL)に溶解し、さらに蒸留水(50 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→2:1→1:1→1:2)で精製し、標題化合物(5-4)を得た{12.1g (30.2 mmol), 94.5%}。
化合物(5-4)(12.1 g, 30.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(7.5 g, 39.3 mmol)および4-ジメチルアミノピリジン(369 mg, 3.0 mmol)を加え、反応液を0 ℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 6:1→4:1→2:1)で精製し、標題化合物(5-5)を得た。{15.3 g (27.6 mmol), 91.4 %}。
化合物(5-5)(15.3 g, 27.6 mmol)、N-カルボベンゾキシ-β-アラニン(12.32 g, 55.2 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(15.8 mg, 82.8 mmol)および4-ジメチルアミノピリジン (6.7 g, 55.2 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(5-6)を得た[17.1 g (22.5 mmol ), 81.5 %]。
化合物(5-6)(17.1 g, 22.5 mmol)の無水N,N-ジメチルホルムアミド(300 mL)溶液に、チオ酢酸カリウム(5.1 g, 45.0 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(5-7)を得た{14.2 g (20.3 mmol ), 90.0 %}。
化合物(5-7)(14.2 g, 20.4 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04 M四酸化オスミウム-t-ブチルアルコール(3 mL)、トリメチルアミンN-オキシド(3.4 g, 30.5 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3 g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 2:1→1:1→2:3→1:2→1:4→1:8)で精製し、標題化合物(5-8)を得た{13.2 g (18.9 mmol ), 93.0 %}。
化合物5-8(13.1 g, 18.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(8.5 g, 28.2 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(5-9)を得た{12.2 g (9.9 mmol ), 52.8 %}。
化合物(5-9)(12.2 g, 9.9 mmol)の酢酸(150 g, 3.8 mol )溶液に、オキソン(24.4 g, 39.7 mmol)および酢酸カリウム(3.8 g)を加え、45℃で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(500 mL)溶液に注ぎ、酢酸エチル(4×100 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→20:1→15:1→12:1)で精製し、無色ワックス状物質として標題化合物(5-10)を得た{8.6 g (6.97 mmol ), 70.3 %}。
化合物(5-10)(480.8 mg, 382μmol)のメタノール(20 mL)、クロロホルム(10 mL)および酢酸(0.5 mL)溶液に、10 %パラジウム活性炭素(300 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 100:25:3→65:25:4→65:35:5→65:45:6)で精製し(5-11)を得た。
化合物(5-11)を無水ジクロロメタン(15 mL)、ピリジン(15 mL)及びトリエチルアミン(2 mL)混合溶液に溶解し、(4-ヨードベンゾイル)- p-ニトロフェニルエステル(635 mg, 1.72 mmol)を加えて、スターラーで攪拌しながら室温で24時間反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 80:10:0.8→70:10:1.0→65:10:1.0→60:10:1.0)で精製して標題化合物(5-12)を得た{279 mg (238μmol ), (経路 J5, K5の二反応で収率55.2 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、αスルホキノボシルアシルグリセロールモノヨード誘導体を例に挙げて次のスキーム6に示す。

経路A6;1-O-アリル-4,6-O-ベンジリデン-α-D-グルコピラノシド (6-2)
化合物(6-1)(100 g, 555 mmol)のアリルアルコール(500 mL)懸濁液に、トリフルオロメタンスルホン酸(1.00 mL)を0℃で加え、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、トリエチルアミン(3 mL)を添加して反応を停止し減圧濃縮した。次いで、残渣を無水アセトニトリル(500 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(127 g, 1.5当量)およびp-トルエンスルホン酸一水和物(5.28 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(10 mL)を添加して反応を停止し減圧濃縮した。残渣をヘキサン(2000 mL)および水(500 mL)中に注ぎ、混合液を激しく撹拌した。生じた沈殿物を濾別し、水およびヘキサンでリンスした。沈殿物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(6-2)を得た{34.5 g (112 mmol), 20.2%}。
化合物(6-2) 10.7g(34.7mmol)を酢酸:水=8:5の溶液260mLに溶解し、100℃、1時間反応後、減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ジクロロメタン:メタノール=6:1)で精製して化合物(6-3)を得た。。(収量6.3g 28.6mmol、収率82.4%)
経路C6; 1-O-(2-プロペニル)-6-O-(4-トリルスルホニル)-α-D-グルコース(6-4)
化合物(6-3) 6.3g(28.6mmol)を乾燥ピリジン200mLに溶解し、p-ジメチルアミノピリジン(DMAP)195mg、p-トルエンスルホニルクロリド7.0gを添加し、撹拌しながら室温で16時間反応した。その後、冷蒸留水20mLを加えて反応を停止し、酢酸エチルで抽出(200mL×3回)し、有機層を合わせて1.0Mおよび0.1M塩酸でpH4まで中和し、飽和食塩水で洗浄(200mL×2回)後、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ジクロロメタン:メタノール=20:1)で精製して表題化合物(6-4)を得た。(収量8.6mg 23.0mmol、収率83.8%)。
化合物(6-4)11.2g(29.9mmol)を乾燥ジクロロメタン25mLに溶解し、t-ブチルジメチルシリルトリフルオロメタンスルホネート23.8g、2,6-ルチジン14.4gを添加し、窒素気流下で撹拌しながら16時間反応した。その後、ジクロロメタン150mLを加えて反応を停止し、飽和食塩水で洗浄(100mL×2回)後、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=30:1)で精製して表題化合物(6-6)を得た。(収量19.6g 27.4mmol、収率91.6%)。
化合物(6-5) 7.9g(11.0mmol)を乾燥エタノール20mLに溶解し、チオ酢酸カリウム1.8gを添加し、還流条件下で撹拌しながら3時間反応した。その後、冷蒸留水100mLを加えて反応を停止し、酢酸エチルで抽出(200mL×3回)し、有機層を合わせて飽和食塩水で洗浄(200mL×2回)後、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=50:1)で精製して表題化合物(6-6)を得た。(収量5.6g 9.02mmol、収率82.0%)。
化合物(6-6) 5.6g(9.02mmol)をt-ブタノール:水=4:1溶液に溶解し、トリメチルアミンN-オキシド二水和物1.5g、四酸化オスミウム-t-ブタノール溶液(0.04M)15mLを添加し、撹拌しながら室温で22時間反応した。その後活性炭15gを加え、撹拌しながら室温で1.5時間放置し、四酸化オスミウムを吸着させた後、吸引濾過した。次に冷蒸留水200mLを加えて反応を停止し、酢酸エチルで抽出(200mL×3回)し、有機層を合わせて飽和食塩水で洗浄(300mL×2回)後、無水硫酸ナトリウムで乾燥、濾過、減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=3:1→2:1)で精製して表題化合物(6-7)を得た。(収量5.2g 7.94mmol、収率88.0%)
経路G6;3-O-[2,3,4-O-tert-ブチルジメチルシリル-6-チオアセチル-α-D-キノボピラノシル]-1-O-ステアロイル-グリセロール(6-8)
化合物(6-7)(1.4 g, 6.1 mmol)の無水ジクロロメタン(20 mL)および無水ピリジン(5 mL)混合溶液に、ステアロイルクロリド(692 mg, 2.3 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(1 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(20 mL)に注ぎ、酢酸エチル(3×20 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 20 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,10:1→8:1→6:1→4:1)で精製し、無色油状物質として標題化合物(6-8)を得た{1.6 g (1.7 mmol ), 83.6 %}。
化合物(6-8)(1.6 g, 1.7 mmol)の無水ジクロロメタン(20 mL)および無水ピリジン(5 mL)混合溶液に、4-ヨード塩化ベンゾイル(555 mg, 2.1 mmol)を加え、室温で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(1 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(20 mL)に注ぎ、酢酸エチル(3×20 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 20 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,12:1→10:1→8:1→6:1→5:1→4:1)で精製し、無色油状物質として標題化合物(6-9)を得た{1.7 g (1.5 mmol ), 85.0 %}。
化合物(6-9)(1.7 g, 1.5 mmol)の酢酸(50 mL)溶液に、オキソン(2.7 g, 4.4 mmol)および酢酸カリウム(1.25 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液に酢酸エチル(200 mL)を注いだ。飽和食塩水(2 x 50 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、酢酸エチルを減圧除去した。酢酸を含む得られた残渣を表題化合物(6-10)として得て、そのまま次の反応に用いることとした。
化合物(6-10)と酢酸を含む混合液にテトラヒドロフラン(1.5 mL)および水(1.5mL)を加え、更にトリフルオロ酢酸(0.3 mL)を加え、室温で48時間攪拌した。反応が充分進行していることを確認した後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 100:10:0.5→80:10:0.5→60:10:1→40:10:1→60:25:2)で精製して標題化合物(6-11)を得た{(経路I6およびJ6の二反応で752 mg (899 μmol ), 60.9 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、αスルホキノボシルアシルグリセロールトリヨード誘導体を例に挙げて次のスキーム7に示す。
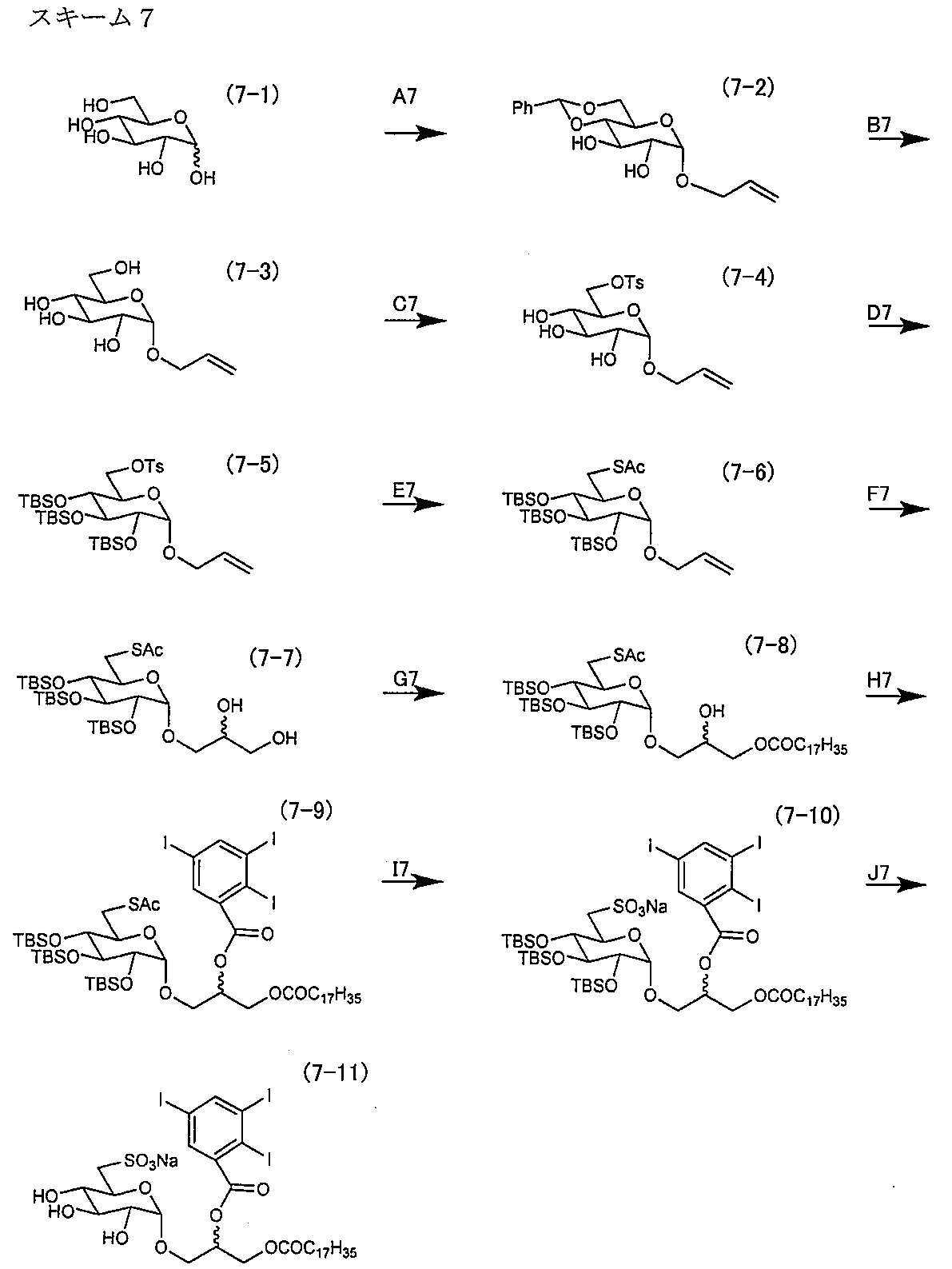
上記の合成例6の経路A6~経路G6と同様の方法により化合物(7-8)を得た。
化合物(7-8)(467 mg, 507 μmol)、2,3,5-トリヨード安息香酸(303.8 mg, 608 μmol)、炭酸ジ-2-ピリジル(DPC)(219 mg, 1013 μmol)および4-ジメチルアミノピリジン (6.2 mg, 51 μmol)を無水ジクロロメタン(10 mL)に溶解し、室温にて48時間反応した。反応が充分進行していることを確認し、反応液に水(30 mL)を注ぎ、反応を停止した後、ジクロロメタン(3×20 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 20 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,12:1→10:1→8:1→6:1→5:1→4:1)で精製し、無色油状物質として標題化合物(7-9)を得た{245 mg (174μmol ), 34.4 %}。
化合物(7-9)(245 mg, 174μmol)の酢酸(10 mL)溶液に、オキソン(322 mg, 523μmol)および酢酸カリウム(250 mg)を加え、室温で24時間激しく攪拌した。反応が充分進行していることを確認した後、反応液に酢酸エチル(50 mL)を注いだ。飽和食塩水(2 x 20 mL)で洗浄、有機層を硫酸ナトリウムで乾燥、濾過後、減圧下濃縮した。酢酸を含む得られた残渣を表題化合物(7-10)としてそのまま次の反応に用いることとした。
経路 I7で得られた化合物(7-10)と酢酸を含む混合液にテトラヒドロフラン(5 mL)、酢酸(10 mL)および水(5 mL)を加え、更にトリフルオロ酢酸(1 mL)を加え、室温で48時間攪拌した。反応が充分進行していることを確認した後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 100:10:0.5→80:10:0.5→60:10:1→40:10:1→60:25:2)で精製して標題化合物(7-11)を得た{(経路I7およびJ7の二反応で102 mg (94μmol ), 53.7 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、βスルホキノボシルモノアシルグリセロールモノヨード誘導体を例に挙げて次のスキーム8に示す。

経路A8;2,3,4,6-テトラ-O-アセチル-1-O-アリル-β-D-グルコピラノシド (8-2)
化合物(8-1)(50.0 g, 128 mmol)、アリルアルコール(22.3 g, 384 mmol)および塩化亜鉛(17.4 g, 128 mmol)のトルエン溶液(500 mL)を、窒素雰囲気下、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、室温に戻し飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(8-2)を得た{24.8 g (64 mmol), 50.0%}。
化合物(8-2)(21.0 g, 54 mmol)をメタノール(200 mL)に溶解し、窒素雰囲気下、室温にて攪拌しているところへ、28%-ナトリウムメチラートメタノール溶液(1 mL)を加え、室温にて2時間攪拌した。反応が十分に進行していることを確認し、反応を終了した。反応液をそのまま減圧濃縮し得られた化合物(8-3)の粗製物を次反応に用いた。
化合物(8-3)(11.9 g, 54 mmol)を無水アセトニトリル(60 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(16.4 g, 2.0当量)およびp-トルエンスルホン酸一水和物(1.0 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(0.5 mL)を添加して反応を停止し減圧濃縮した。濃縮物を少量の酢酸エチルに溶解し、冷水を200 mL加え、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 50 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物8-4を得た。更に濾液を濃縮し、シリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→25:1→20:1)で精製後、熱エタノールから結晶化させることにより同化合物(8-4)を得た。{11.6 g (37.6 mmol), 69.6 %}
LRMS m/z 311 [M+Na]+。
化合物8-4 (11.6 g, 37.6 mmol)の無水N,N-ジメチルホルムアミド(DMF, 100 mL)溶液に、ベンジルクロリド(4.76 g, 4当量)および水酸化ナトリウム粉末(4.5 g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(100 mL)に注ぎ、酢酸エチル(3×50 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 30 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(8-5)を得た。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 12:1→10:1→8:1→6:1→4:1)で精製後、熱エタノールから結晶化させることにより同化合物(8-5)を得た。得られた化合物をフラスコに集めそのまま次反応に用いた。
経路D8で得られた化合物(8-5)を酢酸(72 mL)に溶解し、さらに蒸留水(40 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、得られた残渣をメタノール(150 mL)に溶解し、窒素雰囲気下、室温にて攪拌しているところへ、28%-ナトリウムメチラートメタノール溶液(0.7 mL)を加え、室温にて2時間攪拌した。反応が十分に進行していることを確認し、反応を終了した。反応液をそのまま減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2→1:1→2:3→1:2)で精製し、標題化合物(8-6)を得た{9.3 g (23.2 mmol), 経路 D8, E8の二反応で収率61.7%}。
化合物(8-6)(9.3 g, 23.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(5.8 g, 30.2 mmol)および4-ジメチルアミノピリジン(283 mg, 2.3 mmol)を加え、反応液を0 ℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 6:1→4:1→2:1)で精製し、標題化合物(8-7)を得た。{12.3 g (22.2 mmol), 95.7 %}。
化合物(8-7)(12.3 g, 22.2 mmol)、N-カルボベンゾキシ-β-アラニン(12.1 g, 44.4 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(17.0 g, 88.8 mmol)および4-ジメチルアミノピリジン (5.4 g, 44.4 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(8-8)を得た[15.7 g (20.7 mmol ), 93.0%]。
化合物(8-8)(15.7 g, 20.7 mmol)の無水N,N-ジメチルホルムアミド(200 mL)溶液に、チオ酢酸カリウム(4.7 g, 41.3 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(8-9)を得た{13.6 g (20.5 mmol ), 99.2 %}。
化合物(8-9)(13.6 g, 20.5 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04 M四酸化オスミウム-t-ブチルアルコール(5 mL)、トリメチルアミンN-オキシド(3.4 g, 30.7 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3 g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(トルエン-酢酸エチル, 2:1→3:2→1:1→2:3→1:2→1:4)で精製し、標題化合物(8-10)を得た{11.7 g (16.8 mmol ), 81.8 %}。
化合物(8-10)(11.7 g, 16.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(5.4 g, 26.8 mmol)を加え、0℃で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(8-11)を得た{10.7 g (11.1 mmol ), 66.2%}。
化合物(8-11)(10.7 g, 11.1 mmol)の酢酸(147 g, 2.5 mol )溶液に、オキソン(27.3 g, 44.4 mmol)および酢酸カリウム(3.7 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(500 mL)溶液に注ぎ、酢酸エチル(4×100 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→20:1→15:1→12.5:1→10:1→8:1)で精製し、無色ワックス状物質として標題化合物(8-12)を得た。
化合物(8-12)(756 mg, 762μmol)のメタノール(10 mL)、ジクロロメタン(10 mL)および酢酸(0.5 mL)溶液に、10 %水酸化パラジウム活性炭素(835 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣(化合物(8-13)の粗製物)をそのまま次反応に用いた。
経路L8で得られた化合物(8-13)の粗製物を無水ジクロロメタン(15 mL)、ピリジン(5 mL)及びトリエチルアミン(2 mL)混合溶液に溶解し、(4-ヨードベンゾイル)-p-ニトロフェニルエステル(563 mg, 1.5 mmol)を加えて、スターラーで攪拌しながら室温で24時間反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 70:10:1→60:10:1.1→50:10:1.2→40:10:2→30:10:1.5→65:25:4)で精製して標題化合物(8-14)を得た{442.5mg (487.4μmol ),64.0 %}。
本発明のスルホピラノシルアシルグリセロール誘導体の製造工程を、βスルホキノボシルジアシルグリセロールモノヨード誘導体を例に挙げて次のスキーム9に示す。
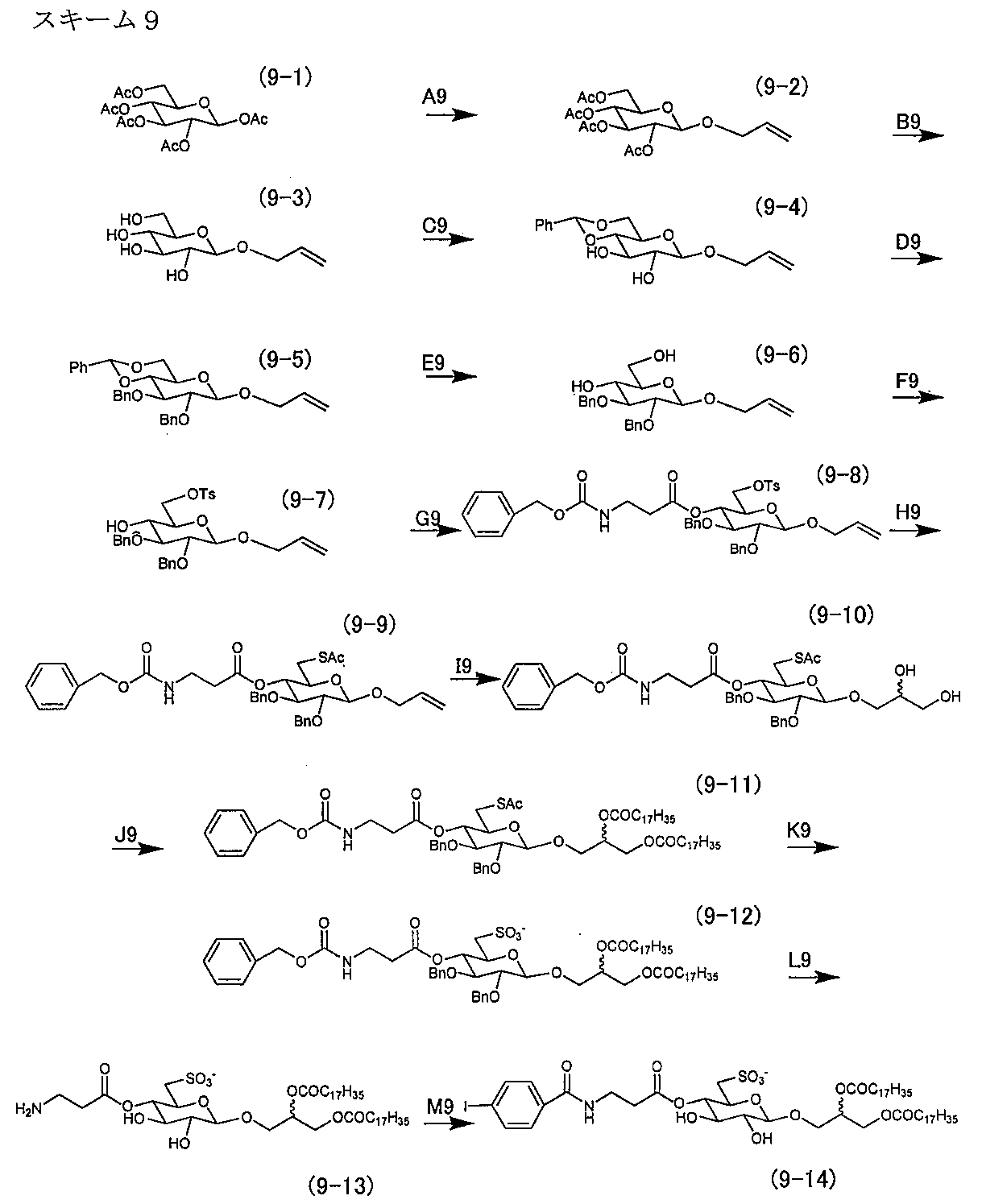
経路A9;2,3,4,6-テトラ-O-アセチル-1-O-アリル-β-D-グルコピラノシド (9-2)
化合物(9-1)(50.0 g, 128 mmol)、アリルアルコール(22.3 g, 384 mmol)および塩化亜鉛(17.4 g, 128 mmol)のトルエン溶液(500 mL)を、窒素雰囲気下、反応液を80 ℃にて48時間激しく撹拌した。反応が充分進行したことを確認した後、室温に戻し飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(9-2)を得た{24.8 g (64 mmol), 50.0%}。
化合物(9-2)(21.0 g, 54 mmol)をメタノール(200 mL)に溶解し、窒素雰囲気下、室温にて攪拌しているところへ、28%-ナトリウムメチラートメタノール溶液(1 mL)を加え、室温にて2時間攪拌した。反応が十分に進行していることを確認し、反応を終了した。反応液をそのまま減圧濃縮し得られた化合物の粗製物を表題化合物(9-3)として次反応に用いた。
化合物(9-3)(11.9 g, 54 mmol)を無水アセトニトリル(60 mL)に懸濁し、ベンズアルデヒドジメチルアセタール(16.4 g, 2.0当量)およびp-トルエンスルホン酸一水和物(1.0 g, 0.05当量)を加えた。反応液を40℃で4時間攪拌後、トリエチルアミン(0.5 mL)を添加して反応を停止し減圧濃縮した。濃縮物を少量の酢酸エチルに溶解し、冷水を酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 50 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(9-3)を得た(33.5 g)。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 15:1→10:1→8:1)で精製後、熱エタノールから結晶化させることにより同表題化合物(9-3)を得た(6.63 g)。{計40.1 g (82.1 mmol), 84.4 %}
LRMS m/z 511 [M+Na]+。
化合物(9-4) (11.6 g, 37.6 mmol)の無水N,N-ジメチルホルムアミド(DMF, 100 mL)溶液に、ベンジルクロリド(4.76 g, 4当量)および水酸化ナトリウム粉末(4.5 g, 3.0当量)を加え、反応液を室温にて24時間激しく撹拌した。反応が充分進行していることを確認した後、反応液を冷水(100 mL)に注ぎ、酢酸エチル(3×50 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 30 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣を熱エタノールから2回結晶化させることにより、無色針状結晶として標題化合物(9-5)を得た。濾液を濃縮し、シリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 12:1→10:1→8:1→6:1→4:1)で精製後、熱エタノールから結晶化させることにより同表題化合物(9-5)を得た。得られた化合物をフラスコに集めそのまま次反応に用いた。
経路D9で得られた化合物5を酢酸(72 mL)に溶解し、さらに蒸留水(40 mL)を加え、加熱還流下、1時間攪拌した。反応が充分進行していることを確認した後、室温まで冷却し、エバポレーターで溶媒を留去し、蒸留水(15 mL)を加えて再び減圧濃縮することを4回繰り返した後、得られた残渣をメタノール(150 mL)に溶解し、窒素雰囲気下、室温にて攪拌しているところへ、28%-ナトリウムメチラートメタノール溶液(0.7 mL)を加え、室温にて2時間攪拌した。反応が十分に進行していることを確認し、反応を終了した。反応液をそのまま減圧濃縮し、シリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2→1:1→2:3→1:2)で精製し、標題化合物(9-6)を得た{9.3 g (23.2 mmol), 経路D9およびE9二反応で収率61.7%}。
化合物(9-6)(9.3 g, 23.2 mmol)の無水ピリジン(120 mL)溶液に、p-トルエンスルホニルクロリド(5.8 g, 30.2 mmol)および4-ジメチルアミノピリジン(283 mg, 2.3 mmol)を加え、反応液を0 ℃で16時間撹拌した。反応が充分進行していることを確認した後、次に反応液を氷水(100 mL)にゆっくり注ぎ、水層を酢酸エチル(3×200 mL)で抽出した。有機層を合わせて1N HCl水溶液でpH4になるまで洗浄し、飽和炭酸水素ナトリウム水(2 x 100 mL)および飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。濃縮物をシリカゲルフラッシュクロマトグラフィー(ヘキサン-酢酸エチル, 6:1→4:1→2:1)で精製し、標題化合物(9-7)を得た。{12.3 g (22.2 mmol), 95.7 %}。
経路G9;1-O-アリル-2,3-ジ-O-ベンジル-4-O-(カルボベンゾキシ-β-アラニル)-6-O-トシル-β-D-グルコピラノシド (9-8)
化合物(9-8)(12.3 g, 22.2 mmol)、N-カルボベンゾキシ-β-アラニン(12.1 g, 44.4 mmol)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI・HCl)(17.0 g, 88.8 mmol)および4-ジメチルアミノピリジン (5.4 g, 44.4 mmol)を無水ジクロロメタン(200 mL)及び無水ピリジン(50 mL)の混合溶液に溶解し、室温にて18時間反応した。反応が充分進行していることを確認し、反応液に水(10 mL)を注ぎ、反応を停止した後、溶液を減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(9-8)を得た[15.7 g (20.7 mmol ), 93.0%]。
化合物(9-8)(15.7 g, 20.7 mmol)の無水N,N-ジメチルホルムアミド(200 mL)溶液に、チオ酢酸カリウム(4.7 g, 41.3 mmol)を加え、90 ℃で3時間攪拌した。反応が充分進行していることを確認した後、反応液を冷水(400 mL)に注ぎ、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル, 4:1→3:1→2:1→3:2)で精製し、標題化合物(9-9)を得た{13.6 g (20.5 mmol ), 99.2 %}。
経路I9;3-O-[2,3-ジ-O-ベンジル-4-O-(カルボベンゾキシ-β-アラニル)-6-チオアセチル-β-D-キノボピラノシル]-グリセロール(9-10)
化合物(9-9)(13.6 g, 20.5 mmol)をt-ブチルアルコール:蒸留水=4:1溶液(200 mL)に溶解し、0.04 M四酸化オスミウム-t-ブチルアルコール(5 mL)、トリメチルアミンN-オキシド(3.4 g, 30.7 mmol)を加え、室温にてスターラーで24時間攪拌した。反応が充分進行していることを確認した後、活性炭を3 g加え、30分間攪拌し触媒を吸着させ、セライトをひいた桐山ロートで吸引濾過し触媒を除き、セライトに残っている反応生成物を酢酸エチルで3回洗浄し回収した。回収した濾液に蒸留水(200 mL)を加え、酢酸エチル(3 x 150 mL)で抽出した。有機層を合わせて飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(トルエン-酢酸エチル, 2:1→3:2→1:1→2:3→1:2→1:4)で精製し、標題化合物(9-10)を得た{11.7 g (16.8 mmol ), 81.8 %}。
経路J9;3-O-[2,3-ジ-O-ベンジル-4-O-(カルボベンゾキシ-β-アラニル)-6-チオアセチル-β-D-キノボピラノシル]-1,2-ジ-O-ステアロイル-グリセロール(9-11)
化合物(9-10)(11.7 g, 16.8 mmol)の無水ジクロロメタン(200 mL)および無水ピリジン(50 mL)混合溶液に、ステアロイルクロリド(5.4 g, 26.8 mmol)を加え、0℃で2時間攪拌した。反応が充分進行していることを確認した後、メタノール(5 mL)を添加して反応を停止し減圧濃縮した。少量の酢酸エチルで懸濁させた残渣を水(200 mL)に注ぎ、酢酸エチル(3×100 mL)で抽出した。有機層を合わせ飽和食塩水(2 x 100 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(ヘキサン-酢酸エチル,6:1→4:1→2:1→3:2→1:1)で精製し、無色油状物質として標題化合物(9-11)を得た{3.3 g (2.7 mmol ), 16.0%}。
化合物(9-11)(3.3 g, 2.7 mmol)の酢酸(100 g, 2.5 mol )溶液に、オキソン(6.6 g, 10.7 mmol)および酢酸カリウム(2.5 g)を加え、室温で48時間激しく攪拌した。反応が充分進行していることを確認した後、反応液を冷7.5 M水酸化ナトリウム(100 mL)溶液に注ぎ、酢酸エチル(4×50 mL)で抽出した。有機層を合わせ飽和炭酸水素ナトリウム水(2 x 50 mL)および飽和食塩水(2 x 50 mL)で洗浄、硫酸ナトリウムで乾燥、濾過後、減圧濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール, 100:1→50:1→20:1→15:1→12.5:1→10:1→8:1)で精製し、無色ワックス状物質として標題化合物(9-12)を得た。
化合物(9-12)(345 mg, 274μmol)のメタノール(10 mL)、ジクロロメタン(15 mL)および酢酸(0.6 mL)溶液に、10 %水酸化パラジウム活性炭素(300 mg)を加え、水素ガス雰囲気下、室温で48時間攪拌した。反応が充分進行していることを確認した後、パラジウム活性炭素を濾別し、濾液を減圧濃縮した。得られた残渣(化合物9-13の粗製物)を表題化合物(9-13)を含む物質としてそのまま次反応に用いた。
経路L9で得られた化合物(9-13)の粗製物を無水ジクロロメタン(15 mL)、ピリジン(5 mL)及びトリエチルアミン(2 mL)混合溶液に溶解し、(4-ヨードベンゾイル)-p-ニトロフェニルエステル(202 mg, 548μmol)を加えて、スターラーで攪拌しながら室温で24時間反応した。反応が充分進行していることを確認した後、トルエン、メタノールを加え共沸させながら減圧下溶媒を留去し濃縮した。得られた残渣をシリカゲルクロマトグラフィー(クロロホルム-メタノール-蒸留水, 70:10:1→60:10:1→60:14:1.6→60:18:2→60:22:2.4→60:25:3)で精製して標題化合物(9-14)を得た{77.6 mg (82.1μmol ),30.0 %}。
1)ビオチン化αSQMG
ヒト食道扁平上皮癌細胞TE-8の1×106個を11匹の雄のKSNヌードマウス(日本エスエルシー社)右大腿部に移植し、腫瘍の体積が300~900cm3になるまで飼育した。

ヒト大細胞肺がん細胞Lu65の1×106個/匹を6週齢の雄のKSNヌードマウスに移植し、14日飼育し、各ヌードマウスに100~200mm3の腫瘍塊を形成させた。
(2)高用量群・・・ビオチン化α 50mg/kg 投与
(3)中用量群・・・同5mg/kg 投与
(4)低用量群・・・同1mg/kg 投与。


(1)動物試験
8週齢の雄性KSNヌードマウス(日本エスエルシー社)にヒト結腸癌由来細胞(SW480株)2×106個を皮下移植し、2週間後(体重約25g腫瘍体積約200 mm3)にαSQMG C18:0またはαSQAP C18:0を生理食塩液に溶解し投与量10mg/kgを腹腔内投与した(0.1mL)。投与後経時的に30分、1、2、4、12および24時間後において二酸化炭素により窒息死後、血液、肝臓、腎臓、肺、脾臓および腫瘍組織を採取した。なお、各組織は採取時間毎に5匹の動物を用いた。
血漿
ヘパリンナトリウム注射液(10,000単位/10mL)で湿らせた注射針を用いて後大静脈より全採血し、6,000gで20分間遠心分離し血漿を得た。血漿90μLに内部標準物質(αSQMG C16:0)5μL、蒸留水5μLおよびリン酸4μLをそれぞれ加え、96μLのアセトニトリルを添加することによりタンパク変性を行った。この溶液0.2mLに10mMの酢酸アンモニウム水溶液600μLを加え、よく攪拌し、16,000gで15分間遠心分離を行って上清を得た。その上清600μLについて以下の通り、固相抽出操作を行った。固相抽出はEmpore(登録商標)ディスクカートリッジ(3M社、製品番号4115SD)を用い、測定試料添加後、10mMの酢酸アンモニウム水溶液1,000μL、10%アセトニトリルを含む10mMの酢酸アンモニウム水溶液200μLで洗浄し、70%アセトニトリルを含む10mMの酢酸アンモニウム水溶液200μLにより被検物質を溶出した。LC/MS分析試料とした。
採血後に各臓器を摘出し、直ちにドライアイス上で凍結した。この試料を凍結粉砕し、重量測定後、1/2倍量(重量比)の内部標準物質(αSQMG C16:0)および蒸留水を添加し、さらに8倍量(重量比)の抽出溶液(2.5% NP-40、2% リン酸、2mM Eserine水溶液)を添加後、ホモジナイズした。上記のホモジナイズ液を遠心分離した上清100μLについて上記血漿の前処理同様、アセトニトリルによるタンパク変性および固相抽出を行うことにより分析試料を得た。
当該分析試料10μLをCapcell Pak MGカラム、(粒子径;3μm、カラムサイズ;2.1×50mm、資生堂)を装着した逆相HPLCシステムで分離した。HPLCは68%アセトニトリル、10mM酢酸アンモニウム水溶液の単一溶媒、1分当たり0.2mLの流速で溶出を行った。質量スペクトルの測定は陰イオンモードで行い、αSQMG C18:0は分子量関連イオン;m/z=583.1、フラグメントイオンm/z=224.7、およびm/z=298.8を測定し、αSQAP C18:0では分子量関連イオン;m/z=567.1、フラグメントイオンm/z=224.7、およびm/z=283.1、また内部標準物質であるαSQMG C16:0は分子量関連イオン;m/z=555.1、フラグメントイオンm/z=224.7、およびm/z=298.8を測定して各イオンに対応したクロマトグラムからピーク面積を積算して内部標準物質のピーク面積比をもとめた。定量分析方法としては内部標準法を用いた。即ち、被検物質を添加した試料を分析することにより内部標準物質のピーク面積比から標準直線を作成し、被検物質の濃度を定量した。
移植腫瘍モデルの腫瘍としてヒト食道扁平上皮癌細胞TE-8を用いたこと、および腹腔内投与に代えて静脈内投与により投与したこと以外は例3と同様の方法によってαSQAP C18:0の1mg/kgを動物に投与して、αSQAP C18:0の薬物動態について試験した。
Claims (11)
- 式(I)の化合物またはその薬学的に許容される塩

Rは水素と結合した陰イオン性基であり、
R1はOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数であり、
R3はOH、SO3Hまたは作用基であり、
R4はOH、SO3Hまたは作用基であり、および
R5はOH、SO3Hまたは作用基であり、
R1、R2、R3、R4およびR5の少なくとも1つが作用基を含む。 - 式(II)の化合物またはその薬学的に許容される塩

Rは水素と結合した陰イオン性基であり、
R1はOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数であり、
R3はOH、SO3Hまたは作用基であり、
R4はOH、SO3Hまたは作用基であり、
R5はOH、SO3Hまたは作用基であり、
jは1以上の整数であり、および
Xは作用基または陽イオン性基であり、kは1以上の整数であり、
R1、R2、R3、R4、R5およびXの少なくとも1つは作用基である。 - RがSO3Hであり、R3がOHであり、R4がOHであり、R5がOHであり、並びに
R1がOH、OCOH、OCO(CH2)hCH3、O-(作用基)、OCO-(作用基)またはOCO(CH2)hCH2-(作用基)であり、hは0以上の整数であり、
R2がH、OH、OCOH、OCO(CH2)iCH3、作用基、O-(作用基)、OCO-(作用基)またはOCO(CH2)iCH2-(作用基)であり、iは0以上の整数であり、
ここで、R1およびR2の少なくとも一方が作用基を含む
請求項1または2の何れか1項に記載の化合物または塩。 - RがSO3Hであり、
R1がOH、OCOH、OCO(CH2)hCH3または作用基であり、hは0以上の整数であり、
R2はH、OH、OCOH、OCO(CH2)iCH3または作用基であり、iは0以上の整数であり、
R3=R4=R5で且つOHである、またはR3、R4およびR5の少なくとも1つが作用基であり、残りの基がOHであり、
R1、R2、R3、R4およびR5の少なくとも1が作用基を含む
請求項1または2の何れか1項に記載の化合物または塩。 - その何れかの側鎖が作用基と結合しているスルホピラノシル(アシル)グリセロール、スルホピラノシル(アシル)プロパンジオール若しくはスルホキノボシルアシルプロパンジオールまたはその薬学的に許容される塩。
- 前記作用基が、標識物質または抗腫瘍物質である請求項1から5の何れかに1項に記載の化合物または塩。
- 請求項1から6の何れか1項に記載の化合物または塩を対象に投与することを特徴とする腫瘍に式(I)若しくは式(II)の化合物または塩を滞留させる方法。
- 前記作用基が標識物質である請求項1から5の何れか1項に記載の化合物または塩を対象に投与すること、
当該化合物または塩が、腫瘍以外の組織よりも腫瘍において高い濃度で存在する時点で、当該標識物質を検出すること、および
当該検出の結果を基に当該対象において腫瘍を検出すること
を具備する腫瘍を検出するための方法。 - 前記作用基が標識物質である請求項1から5の何れか1項に記載の化合物または塩を対象に投与すること、
当該化合物または塩が、腫瘍以外の組織よりも腫瘍において高い濃度で存在する時点で、当該標識物質を検出すること、および
当該検出の結果を基に当該対象に腫瘍の有無を決定すること、
を具備する腫瘍について診断する方法 - 前記検出することが非侵襲的手段により行われる請求項8または9の何れか1項に記載の方法。
- 前記作用基が抗腫瘍物質である請求項1から5の何れか1項に記載の化合物または塩を対象に投与することを具備する腫瘍の治療方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2010800048481A CN102282153A (zh) | 2009-01-16 | 2010-01-15 | 具有肿瘤滞留性的化合物 |
| AU2010205223A AU2010205223B2 (en) | 2009-01-16 | 2010-01-15 | Compound retained in tumor |
| EA201170819A EA201170819A1 (ru) | 2009-01-16 | 2010-01-15 | Соединение, обладающее свойством удерживания в опухоли |
| BRPI1006808A BRPI1006808A2 (pt) | 2009-01-16 | 2010-01-15 | composto tendo propriedade residente de tumor |
| EP10731303A EP2388264A1 (en) | 2009-01-16 | 2010-01-15 | Compound retained in tumor |
| CA2749797A CA2749797A1 (en) | 2009-01-16 | 2010-01-15 | Compound having tumor-resident property |
| SG2011050929A SG172979A1 (en) | 2009-01-16 | 2010-01-15 | Compound having tumor-resident property |
| JP2010546659A JPWO2010082634A1 (ja) | 2009-01-16 | 2010-01-15 | 腫瘍滞留性を有する化合物 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14526709P | 2009-01-16 | 2009-01-16 | |
| US61/145,267 | 2009-01-16 | ||
| JP2009-010158 | 2009-01-20 | ||
| JP2009010158 | 2009-01-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010082634A1 true WO2010082634A1 (ja) | 2010-07-22 |
Family
ID=42339887
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/050432 Ceased WO2010082634A1 (ja) | 2009-01-16 | 2010-01-15 | 腫瘍滞留性を有する化合物 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20100202971A1 (ja) |
| EP (1) | EP2388264A1 (ja) |
| JP (1) | JPWO2010082634A1 (ja) |
| KR (1) | KR20110101200A (ja) |
| CN (1) | CN102282153A (ja) |
| AU (1) | AU2010205223B2 (ja) |
| CA (1) | CA2749797A1 (ja) |
| EA (1) | EA201170819A1 (ja) |
| SG (1) | SG172979A1 (ja) |
| WO (1) | WO2010082634A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020179125A1 (ja) * | 2019-03-05 | 2020-09-10 | 株式会社エム・ティー・スリー | 放射線増感剤 |
| US11267840B1 (en) | 2020-09-25 | 2022-03-08 | M.T.3, Inc. | Compound or salt thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9142025B2 (en) | 2011-10-05 | 2015-09-22 | Electronics And Telecommunications Research Institute | Method and apparatus for obtaining depth information using optical pattern |
| KR101355705B1 (ko) | 2013-01-10 | 2014-01-27 | 제주대학교 산학협력단 | 부챗말 추출물로부터 분리된 화합물 3-O-(6-디옥시-6-설포-α-D-글루코피래노실)-2-O-팔미토일-글리세롤을 유효성분으로 포함하는 탈모방지 또는 발모개선용 조성물 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2365359C (en) * | 1999-02-26 | 2007-09-18 | Toyo Suisan Kaisha, Ltd. | Medicaments containing a sulfopyranosylglycerol derivative |
| US6770729B2 (en) * | 2002-09-30 | 2004-08-03 | Medtronic Minimed, Inc. | Polymer compositions containing bioactive agents and methods for their use |
| CA2567306C (en) * | 2004-06-24 | 2011-06-07 | Toyo Suisan Kaisha, Ltd. | Radiosensitizer |
-
2010
- 2010-01-15 WO PCT/JP2010/050432 patent/WO2010082634A1/ja not_active Ceased
- 2010-01-15 JP JP2010546659A patent/JPWO2010082634A1/ja not_active Ceased
- 2010-01-15 EA EA201170819A patent/EA201170819A1/ru unknown
- 2010-01-15 CA CA2749797A patent/CA2749797A1/en not_active Abandoned
- 2010-01-15 KR KR1020117016195A patent/KR20110101200A/ko not_active Ceased
- 2010-01-15 EP EP10731303A patent/EP2388264A1/en not_active Withdrawn
- 2010-01-15 AU AU2010205223A patent/AU2010205223B2/en not_active Expired - Fee Related
- 2010-01-15 SG SG2011050929A patent/SG172979A1/en unknown
- 2010-01-15 CN CN2010800048481A patent/CN102282153A/zh active Pending
- 2010-01-15 US US12/688,153 patent/US20100202971A1/en not_active Abandoned
Non-Patent Citations (6)
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020179125A1 (ja) * | 2019-03-05 | 2020-09-10 | 株式会社エム・ティー・スリー | 放射線増感剤 |
| JP2020143006A (ja) * | 2019-03-05 | 2020-09-10 | 株式会社エム・ティー・スリー | 放射線増感剤 |
| US12440505B2 (en) | 2019-03-05 | 2025-10-14 | M.T.3, Inc. | Method of treating cancer |
| US11267840B1 (en) | 2020-09-25 | 2022-03-08 | M.T.3, Inc. | Compound or salt thereof |
| JP2022054217A (ja) * | 2020-09-25 | 2022-04-06 | 株式会社エム・ティー・スリー | 化合物又はその塩、及び放射性増感剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2749797A1 (en) | 2010-07-22 |
| JPWO2010082634A1 (ja) | 2012-07-05 |
| EA201170819A1 (ru) | 2012-04-30 |
| AU2010205223B2 (en) | 2013-03-14 |
| KR20110101200A (ko) | 2011-09-15 |
| AU2010205223A1 (en) | 2011-08-11 |
| SG172979A1 (en) | 2011-08-29 |
| US20100202971A1 (en) | 2010-08-12 |
| CN102282153A (zh) | 2011-12-14 |
| EP2388264A1 (en) | 2011-11-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994024142A1 (fr) | Nouveau sphingoglycolipide et son utilisation | |
| WO2014027203A1 (en) | Inflammation imaging and therapy | |
| CN102596220A (zh) | 单磷酸化的脂质a衍生物 | |
| WO2010082634A1 (ja) | 腫瘍滞留性を有する化合物 | |
| Matin et al. | Methyl 4-O-(2-chlorobenzoyl)-α-L-rhamnopyranosides: Synthesis, characterization, and thermodynamic studies | |
| US20100322858A1 (en) | Radiohaloimatinibs and Methods of Their Synthesis and Use in PET Imaging of Cancers | |
| AU2019232652B2 (en) | Releasable antibody conjugates | |
| US8501154B2 (en) | Fluorinated fructose derivatives for PET imaging | |
| CN101641365B (zh) | 新型磺化糖化合物及其医药应用 | |
| US20140088036A1 (en) | Polyphosphate and pyrophosphate derivative of saccharides | |
| EP1881000A1 (en) | Conjugates of 2-fluoro-2-deoxy-glucose and their uses as anti cancer agents | |
| US20210188774A1 (en) | Targeting Nanoparticles | |
| JP7003057B2 (ja) | グリコシル化クロリンe6誘導体、または、その薬学的に許容される塩、医薬組成物、標的を破壊する方法、および、グリコシル化クロリンe6誘導体、またはその薬学的に許容される塩の製造方法 | |
| WO2012153253A2 (en) | Aromatic compounds and metal complexes thereof | |
| EP4130019A1 (en) | Metal-carbohydrate complex | |
| WO2011160216A2 (en) | Compounds useful in imaging and therapy | |
| US20240327450A1 (en) | Glucose derivatives and anticancer agent using same | |
| WO2011147254A1 (zh) | 苯丁酰基姜黄素衍生物及其在制备抗肿瘤药物中的应用 | |
| US6245316B1 (en) | Enhancement of delivery of radioimaging and radioprotective agents | |
| CA2666069C (en) | Fluorinated fructose derivatives for pet imaging | |
| Lees | The synthesis of galectin-3-targeted cancer imaging agents | |
| Maity et al. | Synthesis of alkyne-modified bleomycin disaccharide precursor, its conversion to a 18 F-labeled radiotracer and preliminary in vivo-PET imaging studies |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080004848.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10731303 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2010546659 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20117016195 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2749797 Country of ref document: CA Ref document number: 201170819 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010205223 Country of ref document: AU Ref document number: 5465/DELNP/2011 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010731303 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2010205223 Country of ref document: AU Date of ref document: 20100115 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1006808 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1006808 Country of ref document: BR Kind code of ref document: A2 Effective date: 20110715 |