WO2010113832A1 - コリネ型細菌形質転換体及びそれを用いるイソブタノールの製造方法 - Google Patents
コリネ型細菌形質転換体及びそれを用いるイソブタノールの製造方法 Download PDFInfo
- Publication number
- WO2010113832A1 WO2010113832A1 PCT/JP2010/055504 JP2010055504W WO2010113832A1 WO 2010113832 A1 WO2010113832 A1 WO 2010113832A1 JP 2010055504 W JP2010055504 W JP 2010055504W WO 2010113832 A1 WO2010113832 A1 WO 2010113832A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- dna
- seq
- corynebacterium glutamicum
- activity
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/02—Preparation of oxygen-containing organic compounds containing a hydroxy group
- C12P7/04—Preparation of oxygen-containing organic compounds containing a hydroxy group acyclic
- C12P7/16—Butanols
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0006—Oxidoreductases (1.) acting on CH-OH groups as donors (1.1)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/88—Lyases (4.)
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E50/00—Technologies for the production of fuel of non-fossil origin
- Y02E50/10—Biofuels, e.g. bio-diesel
Definitions
- the present invention relates to isobutanol production technology. More specifically, the present invention relates to a transformant of Corynebacterium glutamicum that has been subjected to a specific genetic manipulation to impart an isobutanol production function, and an efficient method for producing isobutanol using the transformant.
- Biofuels using renewable resources as a raw material are attracting attention against the background of global warming and fossil resource depletion.
- Isobutanol and n-butanol are the next generation biofuels following bioethanol, and have many advantages such as high heat content, low corrosiveness, low water content, high mixing with gasoline, and easy mixing with diesel fuel. Have.
- Isobutanol is found as one by-product called fusel alcohol during alcoholic fermentation by yeast.
- the fusel alcohol is a name for higher alcohols such as isoamyl alcohol and active amyl alcohol in addition to isobutanol, and is named from “fusel” which means “bad alcohol” in German.
- Isobutanol is derived from 2-ketoisovalerate, a biosynthetic metabolic intermediate to valine (Non-patent Document 1).
- Patent Document 2 it is shown that the amount of isobutanol (fusel alcohol) produced as a by-product in alcohol fermentation by yeasts depends on the growth of yeast.
- the following techniques are known as techniques for producing isobutanol using genetically modified bacteria.
- Patent Document 1 discloses an acetohydroxyacid synthase gene derived from Klebsiella pneumoniae or Bacillus subtilis, Bacillus subtilis, Escherichia coli, or Saccharomyces cerevisiae acehydroxyacid cerevisiae.
- Meloloreductase gene Escherichia coli or Saccharomyces cerevisiae-derived dihydroxy acid dehydratase gene, Lactococcus lactis-derived 2-keto acid decarboxylase gene, Escherichia coli or Clostridium acetobutylicum-derived alcohol dehydrogenase As Escherichia coli, Saccharomyces cerevisiae, Bacillus subtilis, A technique for producing isobutanol by expressing using Lactococcus plantarum or Enterococcus faecalis is disclosed.
- the technology disclosed in this document does not disclose a butanol production technology using Corynebacterium glutamicum, which is a specific species belonging to a coryneform bacterium, as a host.
- genes derived from Corynebacterium glutamicum, which is the host of the present invention do not contain genes, and when these endogenous genes are highly expressed, transformants excellent in isobutanol productivity can be obtained. It is not shown to come out.
- the above disclosed technologies are technologies for producing isobutanol accompanied by the growth of microorganisms to be used, whereas in the present invention, a production method that does not involve substantial growth under reducing conditions allows growth. It has been proposed to be a better method than the accompanying production method (see comparative example below).
- Patent Document 2 and Non-Patent Document 3 include an acetohydroxyacid synthase gene derived from Escherichia coli or Bacillus subtilis, an acetohydroxyacid isomeroreductase gene derived from Escherichia coli, a dihydroxy acid dehydratase gene derived from Escherichia coli, Lactococcus lactis, A technique for producing isobutanol by expressing a 2-keto acid decarboxylase gene derived from Saccharomyces cerevisiae or Clostridium acetobutylicum and an alcohol dehydrogenase gene derived from Saccharomyces cerevisiae in Escherichia coli as a host is disclosed.
- Patent Document 3 a technology for producing lactic acid, succinic acid, or ethanol with high efficiency in a state that does not substantially proliferate by reacting genetically modified Corynebacterium glutenmicum in a reaction solution under reducing conditions.
- Patent Document 3 has been disclosed (Patent Document 3), but no mention is made of isobutanol having a different metabolic pathway of formation from the organic compound described in Patent Document 3, and isobutanol is produced with high efficiency even in the state without growth.
- the production method is superior to the method involving growth.
- An object of the present invention is to provide a microorganism capable of efficiently producing isobutanol and a method capable of efficiently producing isobutanol using the microorganism.
- Corynebacterium which functions in glutamicum (1) a gene encoding an enzyme having acetohydroxy acid synthase activity, (2) a gene encoding an enzyme having acetohydroxy acid isomeroreductase activity, (3) dihydroxy acid dehydratase activity A gene encoding an enzyme having (4) a gene encoding an enzyme having 2-keto acid decarboxylase activity, and (5) Corynebacterium glutamicum having a gene encoding an enzyme having alcohol dehydrogenase activity.
- One or more of (1) to (5) is an endogenous gene of Corynebacterium glutamicum, and one or more of (1) to (5) is a foreign gene. To produce efficiently.
- the present invention has been completed by further research based on the above findings, and provides the following microorganisms and a method for producing isobutanol.
- Item 1 Corynebacterium glutamicum having the following genes (1) to (5), wherein one or more of (1) to (5) is an endogenous gene of Corynebacterium glutamicum, (1 A transformant having the ability to produce isobutanol, wherein one or more of () to (5) is a foreign gene.
- the endogenous gene is (1) a gene encoding an enzyme having acetohydroxy acid synthase activity, (2) a gene encoding an enzyme having acetohydroxy acid isomeroreductase activity, and (3) an enzyme having dihydroxy acid dehydratase activity Item 3.
- the transformant according to Item 1 or 2 which comprises a gene selected from genes encoding.
- Item 4. The gene according to any one of Items 1 to 3, wherein the gene encoding the enzyme having alcohol dehydrogenase activity is a gene derived from Escherichia coli or a gene derived from Pseudomonas putida. Transformant.
- a gene encoding an enzyme having acetohydroxy acid synthase activity is complementary to DNA consisting of the base sequence of SEQ ID NO: 34, DNA consisting of the base sequence of SEQ ID NO: 58, or DNA consisting of the base sequence of SEQ ID NO: 34 or 58
- a gene selected from any gene of a DNA that hybridizes under stringent conditions with DNA consisting of a simple nucleotide sequence and encodes a polypeptide having acetohydroxyacid synthase activity and Stringent conditions for a gene encoding an enzyme having acetohydroxy acid isomeroreductase activity to be a DNA comprising the nucleotide sequence of SEQ ID NO: 61 or a DNA comprising a nucleotide sequence complementary to the DNA comprising the nucleotide sequence of SEQ ID NO: 61
- a gene selected from any gene of DNA encoding a polypeptide that hybridizes with and has acetohydroxy acid isomeroreductase activity and A
- Item 6. The transformant according to any one of Items 1 to 5, wherein the Corynebacterium glutamicum used as a host is Corynebacterium glutamicum R (FERM BP-18976), ATCC 13032, or ATCC 13869.
- Item 7. Corynebacterium glutamicum IBU1 (Accession No. NITE BP-718), Corynebacterium glutamicum IBU2 (Accession No. NITE BP-719), Corynebacterium glutamicum IBU3 (Accession No. NITE BP-720), or Corynebacterium glutamicum IBU4 (Accession No. P72, Transit P-NTE) .
- Item 8 The method comprises reacting the transformant according to any one of Items 1 to 7 or a processed product thereof in a reaction medium containing a saccharide under reducing conditions, and recovering the produced isobutanol.
- a process for producing isobutanol. Item 9.
- Item 9. The method for producing isobutanol according to Item 8, wherein the transformant does not substantially grow in the reaction step.
- Item 10. Item 10.
- the transformant of the present invention can produce isobutanol from a carbohydrate or the like without substantially growing under reduced conditions. For this reason, the carbonaceous material used for substance production is exclusively consumed for the production of isobutanol, which is the target product, and is not consumed for growth, so that isobutanol can be produced with high efficiency.
- isobutanol which is the target substance, can be easily recovered and purified. According to the present invention, efficient isobutanol production using renewable resources as raw materials becomes possible, and a rational process in industrial production can be realized.
- FIG. 1 shows pCRB1-ilvB-ilvN-ilvC / CG, pCRB207-alsS / BS, pCRB207-ilvD / CG, pCRB207-kivD / LL, pCRB207-ipd / SE, pCRB207- produced in Example 1 (4) It is a schematic diagram which shows adh2 / SC, pCRB207-adhP / EC, and pCRB207-adh / PP.
- FIG. 1 shows pCRB1-ilvB-ilvN-ilvC / CG, pCRB207-alsS / BS, pCRB207-ilvD / CG, pCRB207-kivD / LL, pCRB207-ipd / SE, pCRB207- produced in Example 1 (4) It is a schematic diagram which shows adh2 / SC, pCRB207-adhP
- FIG. 3 shows the aerobic growth curve of Corynebacterium glutamicum IBU2 described in Comparative Example 3.
- the transformant having isobutanol-producing ability of the present invention is a corynebacteria having the genes (1) to (5) described above, which functions in Corynebacterium glutamicum.
- An um glutamicum having at least one of (1) to (5) is an endogenous gene and at least one of (1) to (5) is an exogenous gene and has an isobutanol-producing ability It is a transformant.
- the endogenous gene is preferably highly expressed. That is, the endogenous gene originally possessed by the host, Corynebacterium glutamicum, is preferably highly expressed by being introduced as a plasmid or the like. “Function in Corynebacterium glutamicum” means that the enzyme encoded by DNA is expressed in Corynebacterium glutamicum and exhibits a catalytic action.
- Hosts to be transformed in the present invention are transformed with recombinant vectors containing isobutanol production-related genes, and the enzymes involved in isobutanol production encoded by these genes are expressed, resulting in isobutanol.
- isobutanol There is no particular limitation as long as it is Corynebacterium glutamicum.
- Corynebacterium glutamicum used as a host is a group of microorganisms defined in Bergey's Manual of Determinative Bacteriology, Vol. 8, 599 (1974).
- Corynebacterium glutamicum R (FERM P-18976), ATCC13032, ATCC13869, ATCC13058, ATCC13059, ATCC13060, ATCC13232, ATCC13286, ATCC13287, ATCC13655, ATCC13745, ATCC13746, ATCC13761, 318 -233 (FERM BP-1497) or MJ-233AB-41 (FERM BP-1498).
- corynetypes such as Brevibacterium flavum, Brevibacterium lactofermentum, Brevibacterium divaricatum, Corynebacterium lilium, etc.
- the name of the bacterium is the same as Corynebacterium glutamicum (Liebl, W. et al., Trans Brevibacterium divaricatum DSM 20297T, "Brevibacterium flavum” DSM 20411, “Brevibacterium lactofermentum DSM DSM DSM DSM DSM , And Corynebacterium glutamicum and their distinction by rRNA gene restriction patterns.
- Komagata Kazuo et al. Coryneform bacteria classification, fermentation and industry, 45: 944-963 (1987) Therefore, it is included in the present invention.
- Particularly preferred hosts include Corynebacterium glutamicum R (FERM P-18976), ATCC13032, ATCC13869, and the like. These Corynebacterium glutamicum may be a mutant strain or an artificial genetic recombinant in addition to the wild strain.
- a gene disruption strain in which one or more of genes such as lactate dehydrogenase, phosphoenolpyrvate carboxylase, and malate dehydrogenase are disrupted can be mentioned.
- a gene disruption strain in which one or more of genes such as lactate dehydrogenase, phosphoenolpyrvate carboxylase, and malate dehydrogenase are disrupted can be mentioned.
- a disrupted strain of Corynebacterium glutamicum R (FERM P-18976) or its lactate (lactate dehydrogenase: LDH) gene.
- Lactate dehydrogenase-disrupted strain of Corynebacterium glutamicum R (FERM P-18976) was recombined to disrupt the metabolic pathway from pyruvate to lactic acid by disrupting the lactate dehydrogenase gene using genetic engineering techniques.
- Corynebacterium glutamicum R Such a lactate dehydrogenase-disrupted strain and its production method are described in WO2005 / 010182A1.
- Isobutanol production-related enzyme gene In the present specification, the following genes (1) to (5) are also referred to as isobutanol production-related genes.
- a gene encoding an enzyme having acetohydroxy acid synthase (AHAS) activity As the main gene, DNA comprising the base sequence represented by SEQ ID NO: 34 and DNA comprising the base sequence represented by SEQ ID NO: 58 are preferred. And a DNA that hybridizes under stringent conditions with a DNA consisting of a base sequence complementary to the DNA consisting of the base sequence represented by SEQ ID NO: 34 or 58 and that encodes an enzyme having acetohydroxyacid synthase activity At least one DNA selected from the group.
- DNA comprising the nucleotide sequence represented by SEQ ID NO: 61 and the nucleotide sequence represented by SEQ ID NO: 61 are preferred. At least one DNA selected from the group consisting of DNAs that hybridize under stringent conditions with DNA consisting of a complementary nucleotide sequence and DNA encoding an enzyme having acetohydroxy acid isomeroreductase activity It is.
- DNA comprising the base sequence represented by SEQ ID NO: 62 and DNA comprising the base sequence represented by SEQ ID NO: 62 are preferred. And at least one DNA selected from the group consisting of DNAs encoding an enzyme having a dihydroxy acid dehydratase activity.
- a gene encoding an enzyme having 2-keto acid decarboxylase (KDC) activity As this gene, a DNA comprising the base sequence represented by SEQ ID NO: 43 and a base sequence represented by SEQ ID NO: 52 are preferred. It encodes an enzyme that hybridizes under stringent conditions with DNA and DNA consisting of a base sequence complementary to DNA consisting of the base sequence represented by SEQ ID NO: 43 or 52 and has 2-keto acid decarboxylase activity At least one kind of DNA selected from the group consisting of DNA.
- DNA comprising the nucleotide sequence represented by SEQ ID NO: 40, DNA comprising the nucleotide sequence represented by SEQ ID NO: 46, and sequence Hybridizes under stringent conditions with DNA consisting of the base sequence represented by No. 49, and DNA comprising a base sequence complementary to the DNA consisting of the base sequence represented by SEQ ID No. 40, 46 or 49, and alcohol It is at least one DNA selected from the group consisting of DNAs encoding enzymes having dehydrogenase activity.
- the gene (1) in the present invention is a gene encoding an enzyme having acetohydroxyacid synthase activity.
- the gene (2) is a gene encoding an enzyme having acetohydroxyacid isomer reductase activity.
- the gene of (3) is a gene encoding an enzyme having dihydroxy acid dehydratase activity.
- the koji gene is a gene encoding an enzyme having 2-keto acid koji decarboxylase activity.
- the gene (5) is a gene encoding an enzyme having alcohol-dehydrogenase activity.
- DNA fragments obtained by synthesizing DNA fragments containing these genes according to their sequences can be used. Moreover, it is possible to obtain fragments by a hybridization method or a PCR method based on an amino acid sequence conserved among isobutanol production-related enzyme proteins. Furthermore, it is possible to obtain fragments by degenerate PCR using mixed primers designed based on other known isobutanol production-related gene sequences.
- a part of the natural nucleotide sequence is the other. It may be substituted with a base (nucleotide), may be deleted, a base (nucleotide) may be newly inserted, and a part of the base sequence may be rearranged. Any of these gene derivatives can be used in the present invention.
- the part may be, for example, 1 to several amino acid residues (1 to 5, preferably 1 to 3, more preferably 1 to 2).
- the origin of the genes (1) to (5) may be a bacterium that produces isobutanol or a bacterium that does not have the ability to produce isobutanol.
- Isobutanol is found as one of the by-products called fusel alcohol during alcohol fermentation by yeast (Hazelwood, LA et al., The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism.Appl. Environ Microbiol. 74: 2259-2266 (2008)].
- This fusel alcohol is a name for higher alcohols such as isoamyl alcohol and active amyl alcohol in addition to isobutanol. It was first discovered by Enrlich a century ago that it originated from an amino acid metabolic system.
- This metabolic system is called the Enrlich pathway. It is converted from 2-keto acid, which is a biosynthetic metabolic intermediate such as valine, leucine, isoleucine, methionine, and phenylalanine, to aldehyde by 2-keto acid decarboxylase with broad substrate specificity. Converted to alcohol by alcohol dehydrogenase. Isobutanol is derived from 2-ketoisovalerate, an intermediate of biosynthetic metabolism to valine (Dickinson, JR et al., An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae. J. Biol. Chem. 273: 25751-25756 (1998)].
- the isobutanol production pathway that is, the 5-step metabolic pathway from pyruvate to isobutanol (the first 3 steps are the same as the valine biosynthesis pathway) is specifically: (1) The reaction from pyruvate to acetolactate Acetohydroxy acid synthase catalyzed (hereinafter referred to as “AHAS”), (2) acetohydroxy acid isomeroreductase catalyzing the reaction from acetolactate to 2,3-dihydroxyisovalerate ( Acetohydroxy acid isomeroreductase (hereinafter referred to as “AHAIR”), (3) Dihydroxy acid dehydratase (hereinafter referred to as “dihydroxy acid dehydratase”) that catalyzes the reaction from 2,3-dihydroxyisovalerate to 2-ketoisovalerate The enzyme is referred to as “DHAD”), (4) 2-catalyzing the reaction from 2-ketoisovalerate to isobutyrylaldehyde 2-Keto
- the first three steps (1) to (3) are identical to the essential amino acid valine biosynthesis pathway, they are widely encoded on the genomes of bacteria, archaea and eukaryotes. Therefore, any gene that can be obtained from any organism and expressed and function in Corynebacterium glutamicum bacteria can be used.
- bacteria such as Bacillus subtilis function in order to prevent acidification by discharging excessive pyruvic acid accumulated in the microbial cell rather than the valine biosynthesis pathway.
- Possible acetohydroxyacid synthase (gene name is “alsS”) [Cruz Ramos, H. et al., Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. J.
- Bacteriol. 182, 3072-3080 (2000 )] Exists, but this gene may be used. In particular, it may be preferable to use a gene derived from the host Corynebacterium glutamicum as the gene for the steps (1) to (3). Specific examples of other metabolic steps (4) and (5) include the following.
- the Lactococcus lactis has a kivD gene [de la Plaza, M. et al., Biochemical and molecular characterization of alpha- ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis. FEMS Microbiol. Lett. 238, 367-374 (2004)], Staphylococcus epidermidis Zhang, YQ et al., Genome-based analysis ofulvirulence genes in a-non-biofilm-forming Staphylococcusiderepidermidis strain (ATCC 12228). Mol. Microbiol.
- the aforementioned kivD gene derived from Lactococcus lactis or ipd gene derived from Staphylococcus epidermidis may be used.
- the present invention it has been clarified that, in the host Corynebacterium ⁇ ⁇ ⁇ ⁇ glutamicum, when a kivD gene derived from Lactococcus ip lactis or an ipd gene derived from Staphylococcus epidermidis is introduced, it shows high KDC activity (Examples described later) reference).
- ADH in metabolic step (5) is generally mild substrate specificity and is known to react with aldehydes such as acetaldehyde and butyraldehyde in addition to isobutyryl aldehyde.
- aldehydes such as acetaldehyde and butyraldehyde in addition to isobutyryl aldehyde.
- a gene (adh) [Accession number; YP_001267259] encoding ADH derived from Pseudomonas putida can be used.
- the strain introduced with the adhP gene derived from Escherichia coli or the strain introduced with the adh gene derived from Pseudomonas putida is more than the strain introduced with the adh2 gene derived from Saccharomyces cerevisiae.
- isobutyrylaldehyde as a substrate, high ADH activity was detected (see Examples described later). Therefore, regarding the types, combinations, and order of introduction of microorganisms derived from the genes (1) to (5) above, those derived from Corynebacterium glutamicum, at least one of which is the host. There is no particular limitation, except for including.
- one endogenous gene and four foreign genes two endogenous genes, three foreign genes, three endogenous genes and two foreign genes
- the number may be either 4 or 4 endogenous genes and 1 foreign gene.
- the endogenous gene is preferably a gene containing (1) to (3), and the exogenous gene is preferably a gene containing (4) and (5).
- Preferred examples of the isobutanol production-related genes (1) to (5) in the present invention include the following.
- the DNA consisting of the base sequence represented by SEQ ID NO: 34 is a gene derived from Corynebacterium glutamicum (ilvBN), and the DNA consisting of the base sequence represented by SEQ ID NO: 58 is a gene derived from Bacillus subtilis. (AlsS) is preferred.
- the DNA consisting of the base sequence represented by SEQ ID NO: 61 is preferably a gene derived from Corynebacterium glutamicum (ilvC).
- DNA which consists of a base sequence represented by sequence number 62 is a gene (ilvD) derived from Corynebacterium glutamicum.
- the DNA consisting of the base sequence represented by SEQ ID NO: 43 is a gene derived from Lactococcus lactis (kivD)
- the DNA consisting of the base sequence represented by SEQ ID NO: 52 is derived from Staphylococcus epidermidis
- the gene (ipd) is derived from Lactococcus lactis
- the DNA consisting of the base sequence represented by SEQ ID NO: 40 is a gene derived from Escherichia coli (adhP), and the DNA consisting of the base sequence represented by SEQ ID NO: 46 is a gene derived from Pseudomonas putida ( adh), and the DNA comprising the nucleotide sequence represented by SEQ ID NO: 49 is preferably a gene (adh2) derived from Saccharomyces cerevisiae.
- the genes (1) to (5) described above may be hybridized under stringent conditions with DNA each consisting of a complementary base sequence.
- (1) is DNA consisting of the base sequence represented by SEQ ID NO: 34 derived from Corynebacterium glutamicum or DNA consisting of the base sequence represented by SEQ ID NO: 58 derived from Bacillus subtilis
- (2) is coryne DNA consisting of the base sequence represented by SEQ ID NO: 61 derived from bacterial glutamicum
- (3) is the DNA consisting of the base sequence represented by SEQ ID NO: 62 derived from Corynebacterium glutamicum
- (4) is the lacto A DNA comprising the nucleotide sequence represented by SEQ ID NO: 43 derived from Coccus aeruginosa or a DNA comprising the nucleotide sequence represented by SEQ ID NO: 52 derived from the Staphylococcus epidermidis
- (5) is SEQ ID NO: 40 derived from Escherichia coli, Pseudomonas It is represented by SEQ ID NO: 46 derived from putida or SEQ ID NO: 49 derived from Saccharo
- a DNA sequence that hybridizes under stringent conditions with a DNA comprising a base sequence complementary to the DNA comprising the base sequence represented by SEQ ID NO: 34 refers to the base sequence represented by SEQ ID NO: 34 DNA obtained by colony hybridization method or plaque hybridization method using DNA consisting of a complementary nucleotide sequence to DNA as a probe.
- stringent conditions refers to general conditions such as those described in Molecular Cloning, “A Laboratory Manual”, “Second Edition”, 1989, Vol 2, p11.45, and the like. Specifically, it refers to the case where hybridization occurs at a temperature 5 to 10 ° C. lower than the melting temperature (Tm) of the complete hybrid.
- the DNA has a sequence homology of about 90% or more with the DNA comprising the sequence. More preferably, the sequence has a sequence homology of about 92% or more, more preferably about 95% or more, and particularly preferably about 98% or more.
- the homology is a value calculated using GENETYX® (registered trademark) Ver. 8 (manufactured by Genetics).
- the transformant having the ability to produce isobutanol of the present invention introduces at least one foreign gene from the DNAs of (1) to (5) described above which functions in Corynebacterium glutamicum into Corynebacterium glutamicum. And can be obtained by imparting productivity.
- the endogenous gene derived from Corynebacterium glutamicum is introduced.
- Oligonucleotide primers for amplifying the sequences of genes encoding AHAS, AHAIR, DHAD, KDC, and ADH derived from various organisms by the PCR (polymerase chain reaction) method include the following. Examples of such primers include primers represented by the nucleotide sequences of SEQ ID NOs: 36 and 37 for amplifying the gene encoding AHAS; and nucleotides of SEQ ID NOs: 63 and 64 for amplifying the gene encoding AHAIR.
- a known PCR device such as a thermal cycler can be used.
- the PCR cycle may be determined according to a known technique. For example, denaturation, annealing, and extension are defined as one cycle, and usually 10 to 100 cycles, preferably about 20 to 50 cycles.
- cDNA isolated from a microorganism exhibiting the above-mentioned butanol production pathway enzyme activity can be used to amplify cDNAs of genes encoding AHAS, AHAIR, DHAD, KDC, and ADH by PCR. .
- the gene obtained by the PCR method can be introduced into an appropriate cloning vector.
- Cloning methods include commercially available PCR cloning systems such as pGEM-T easy vector system (Promega), TOPO TA-cloning system (Invitrogen), Mighty Cloning Kit (Takara). You can also In addition, one example of the method will be described in detail in Examples, and a DNA fragment containing the region was appropriately designed based on the base sequences of genes encoding known AHAS, AHAIR, DHAD, KDC, and ADH. It can also be obtained by a hybridization method using a synthetic primer as a template.
- a cloning vector containing the gene obtained by PCR is introduced into a microorganism such as Escherichia coli JM109 strain, and the strain is transformed.
- the transformed strain is cultured in a medium containing an appropriate antibiotic (for example, ampicillin, chloramphenicol, etc.) corresponding to the marker gene in the vector, and the cells are recovered from the culture.
- Plasmid DNA is extracted from the collected cells. Plasmid DNA can be extracted by a known technique, or can be easily extracted using a commercially available plasmid extraction kit. Examples of commercially available plasmid extraction kits include Qiaquick plasmid purification kit (trade name: Qiaquick plasmid purification kit, manufactured by Qiagen).
- the base sequence of DNA can be determined by a known method such as a dioxynucleotide enzyme method.
- the base sequence can be determined using a capillary electrophoresis system using a multi-fluorescence technique for detection. It can also be determined using a DNA sequencer such as ABI PRISM 3730xl DNA Analyzer (Applied Biosystems).
- Said method can be performed based on the conventional method of genetic engineering experiment.
- Introduction method and expression method of the vector and the foreign gene of various microorganisms because they are described in many experimental books [e.g., Sambrook, J. & Russel, D. W. Molecular Cloning: A Laboratory Manual (3 rd Edition) CSHL Press (2001) or Ausubel, F. et al. Current protocols in molecular biology. Green Publishing and Wiley Interscience, New York (1987), etc.], vector selection, gene introduction, and expression can be performed accordingly.
- promoters are suitable for use in the present invention.
- Such a promoter may be any nucleotide sequence obtained from many known sources including yeast, bacteria and other cell sources, and having a function of initiating transcription of a target gene in coryneform bacteria. May be.
- lac, trc, tac promoters, etc. can be used in coryneform bacteria.
- the promoter used in the present invention can be modified as necessary to change its regulatory mechanism.
- the terminator under the control sequence arranged downstream of the target gene may be any nucleotide sequence as long as it has a function of terminating transcription of the gene in coryneform bacteria.
- Each gene encoding AHAS, AHAIR, DHAD, KDC, and ADH is expressed on a plasmid or chromosome in Corynebacterium glutamicum as a host.
- these genes are introduced under expressible regulatory sequences.
- “under the control sequence” means that these genes can be transcribed and translated by joint work with a promoter, an inducer, an operator, a ribosome binding site, a transcription terminator, and the like.
- any plasmid vector may be used as long as it contains a gene that controls the autonomous replication function in coryneform bacteria.
- pAM330 derived from Brevibacterium lactofermentum 2256 [JP 58-67699], [Miwa, K. et al., Cryptic plasmids in glutamic acid-producing bacteria. Agric . Biol. Chem. 48: 2901-2903 (1984)] and [Yamaguchi, R. et al., Determination of the complete nucleotide sequence of the Brevibacterium lactofermentum plasmid pAM330 and the analysis of its genetic information. Sucericympid 16: 265-267 (1985)], pHM1519 derived from Corynebacterium glutamicum ATCC13058 [Miwa, K.
- phage DNA etc. are mentioned, Any other vector can be used as long as it can replicate in a host. Moreover, it is preferable that the vector contains a multiple cloning site having various restriction enzyme sites therein or a single restriction enzyme site.
- the nucleotide sequence is confirmed.
- the gene can be constructed by connecting a control sequence such as an appropriate promoter and terminator to the gene and inserting it into an appropriate restriction enzyme site of any of the plasmid vectors exemplified above. Details are described in the Examples.
- a known method can be used without limitation.
- a known method such as electroporation or conjugation can be used.
- the electric pulse method is a known method [Kurusu, Y. et al., Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation. Agric. Biol. Chem. 54: 443-447 (1990)] and [ Vertes AA et al., Presence of mrr- and mcr-like restriction systems in Coryneform bacteria. Res. Microbiol. 144: 181-185 (1993)].
- the above method can be performed based on a conventional method of genetic engineering experiment.
- Information on vectors of various microorganisms such as E. coli and actinomycetes, and methods for introducing and expressing foreign genes are described in many experiments (for example, Sambrook, J., Russel, DW, Molecular Cloning A Laboratory).
- transformants of Corynebacterium glutamicum created by the above method include Corynebacterium glutamicum IBU1 (Accession number: NITE BP-718), Corynebacterium glutamicum (Corynebacterium glutamicum).
- IBU2 Accession number: NITE BP-719
- Corynebacterium glutamicum IBU3 Accession number: NITE BP-720
- Corynebacterium glutamicum IBU4 accesion number: NITE BP-721 All have been deposited with the Patent Microorganism Deposit Center (National Institute of Technology and Evaluation, 2-5-8, Kazusa Kamashi, Kisarazu City, Chiba, Japan (Postal code 292-0818). Date of deposit: March 17, 2009 Date of notification of commissioning: March 2, 2009 6th).
- the transformant of the present invention has an increased glycolytic flow rate, increased resistance to isobutanol, osmotic pressure or organic acids, and by-products (other than the desired product). Further included can be a genetic modification that results in one or more of the characteristics selected from the group consisting of reduced production (meaning carbon-containing molecules). Such genetic modification can be specifically introduced by overexpression of a foreign gene and / or inactivation of an endogenous gene, classical mutagenesis, screening and / or selection of a target mutant.
- the transformant can be mutated by an artificial mutagenesis method using ultraviolet rays, X-rays, chemicals, etc., and any mutant strain obtained in this way can produce isobutanol as the object of the present invention. As long as it has the ability, it can be used as the transformed microorganism of the present invention.
- the transformant of Corynebacterium glutamicum of the present invention thus created (hereinafter simply referred to as “transformant”) may be cultured using a medium usually used for culturing microorganisms.
- a natural medium or a synthetic medium containing a carbon source, a nitrogen source, inorganic salts, and other nutrient substances can be used.
- Examples of the carbon source include sugars or sugar alcohols such as glucose, fructose, sucrose, mannose, maltose, mannitol, xylose, galactose, starch, molasses, sorbitol or glycerin, acetic acid, citric fermentation, lactic acid, fumaric acid, maleic acid or Examples include organic acids such as gluconic acid and alcohols such as ethanol or propanol. Moreover, hydrocarbons, such as normal paraffin, etc. can be used if desired. A carbon source may be used individually by 1 type, and may mix and use 2 or more types. The concentration of these carbon sources in the medium is usually about 0.1 to 10% (wt).
- the nitrogen source examples include, but are not limited to, nitrogen compounds such as inorganic or organic ammonium compounds such as ammonium chloride, ammonium sulfate, ammonium nitrate, and ammonium acetate, urea, aqueous ammonia, sodium nitrate, or potassium nitrate.
- nitrogen compounds such as inorganic or organic ammonium compounds such as ammonium chloride, ammonium sulfate, ammonium nitrate, and ammonium acetate, urea, aqueous ammonia, sodium nitrate, or potassium nitrate.
- corn steep liquor, meat extract, bepton, NZ-amine, protein hydrolyzate, or nitrogen-containing organic compounds such as amino acids can be used.
- a nitrogen source may be used individually by 1 type, and may mix and use 2 or more types.
- the medium concentration of the nitrogen source varies depending on the nitrogen compound used, but is usually about 0.1 to 10% (wt).
- inorganic salts examples include monopotassium phosphate, dipotassium phosphate, magnesium sulfate, sodium chloride, ferrous nitrate, manganese sulfate, zinc sulfate, cobalt sulfate, and calcium carbonate. These inorganic salts may be used alone or in a combination of two or more.
- the medium concentration of inorganic salts varies depending on the inorganic salt used, but is usually about 0.01 to 1.0% (wt).
- the nutrient substance examples include meat extract, peptone, polypeptone, yeast extract, dry yeast, corn steep liquor, defatted milk powder, defatted soy hydrochloride hydrolyzate, extracts of animals and plants or microbial cells, and degradation products thereof.
- the medium concentration of the nutrient substance varies depending on the nutrient substance used, but is usually about 0.1 to 10% (wt).
- vitamins can be added as necessary. Examples of vitamins include biotin, thiamine (vitamin B1), pyridoxine (vitamin B6), pantothenic acid, inositol, nicotinic acid and the like.
- the pH of the medium is preferably about 5-8.
- a preferred microorganism culture medium is A medium (Inui, M. et al., Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions.J. Mol. Microbiol. Biotechnol. 7: 182-196 (2004)).
- BT medium [Omumasaba, CA et al., Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J. Mol. Microbiol. Biotechnol. 8: 91-103 (2004)].
- the culture temperature may be about 15 to 45 ° C.
- the culture time may be about 1 to 7 days.
- the method for recovering and separating the cultured cells from the culture obtained as described above is not particularly limited, and for example, a known method such as centrifugation or membrane separation can be used.
- the collected cultured microbial cells may be treated, and the resulting microbial cell processed product may be used in the next step.
- the cell-treated product may be any product obtained by applying some treatment to cultured cells, and examples thereof include immobilized cells obtained by immobilizing cells with acrylamide or carrageenan.
- the cultured cells of the transformant recovered or separated from the culture obtained as described above or the treated cells thereof are subjected to isobutanol production reaction in a reaction medium under reducing conditions.
- the treated microbial cell include an immobilized microbial cell in which the microbial cell is immobilized with, for example, acrylamide, carrageenan or the like.
- Isobutanol comprising a step of causing isobutanol to be produced in a medium containing saccharide (reaction medium) under reducing conditions by the transformant or the treated product of the microbial cell, and a step of recovering the generated isobutanol
- the isobutanol production method can be any of batch production method, fed-batch production method, and continuous production method.
- the reaction medium only needs to contain an organic carbon source (for example, saccharides) as a raw material for isobutanol.
- an organic carbon source for example, saccharides
- Any organic carbon source may be used as long as the transformant of the present invention can be used for biochemical reactions.
- the saccharide include monosaccharides such as glucose, xylose, arabinose, galactose, fructose or mannose, disaccharides such as cellobiose, sucrose or lactose, maltose, and polysaccharides such as dextrin or soluble starch. It is done.
- monosaccharides and disaccharides are preferable, monosaccharides are more preferable, and glucose is even more preferable.
- a mixed sugar of two or more kinds of sugars can also be used.
- the concentration of the carbon source is preferably about 0.1 to 10% by weight, more preferably about 0.5 to 10% by weight.
- the reaction medium includes other components necessary for the transformant or its processed product to maintain its metabolic function, that is, carbon sources such as various sugars; nitrogen sources necessary for protein synthesis; phosphorus, potassium, sodium, etc.
- trace metal salts such as iron, manganese or calcium can be included. These addition amounts can be appropriately determined depending on the required reaction time, the type of target organic compound product or the type of transformant used. Depending on the transformant used, the addition of specific vitamins may be preferred.
- the carbon source nitrogen source, inorganic salts, vitamins and trace metal salts, known ones, for example, those exemplified for the growth culture step of the transformant can be used.
- the pH of the reaction medium is preferably about 6-8.
- the reaction of the transformant or its cell-treated product with a saccharide is preferably carried out under temperature conditions where the transformant of the present invention or its cell-treated product can act, and the transformant or its cell-treated product. It can be selected as appropriate depending on the type of the above. Usually, the temperature may be about 25 to 35 ° C.
- the production of isobutanol according to the present invention is carried out under reducing conditions.
- the reducing conditions in the present invention are conditions defined by the redox potential of the reaction medium.
- the oxidation-reduction potential of the reaction medium is preferably about ⁇ 100 mV (millivolt) to ⁇ 500 mV, more preferably about ⁇ 200 mV to ⁇ 500 mV. These can be measured using an oxidation-reduction potentiometer (for example, ORP Electrodes manufactured by BROADLEY JAMES).
- the above reducing conditions can be achieved by appropriately setting the oxygen concentration, temperature, and pH of the reaction medium.
- the reaction medium may be sealed, or anaerobic conditions may be set by a method such as nitrogen gas sealing.
- One of the features of the isobutanol production method of the present invention is that it is substantially not accompanied by growth of transformants used in a reaction medium containing a saccharide under reducing conditions. The fact that isobutanol is produced without the growth of transformants in the reaction medium, the reaction solution is collected over time, and the isobutanol concentration and cell concentration (for example, the optical concentration of the cell) are determined. You can know by measuring.
- the culture time may be about 2 to 72 hours, for example.
- Isobutanol can be recovered by a known method used in a bioprocess. As such known methods, there are a distillation method, a membrane permeation method, an organic solvent extraction method, and the like, and the separation, purification, and collection method can be appropriately determined according to the reaction solution composition, the type and amount of by-products.
- Example 1 Cloning and expression of isobutanol production gene (1) Chromosomal DNA extraction from the extraction Bacillus subtilis 168 NBRC14144 chromosomal DNA from microorganisms, NBRC Medium No.802 medium [Polypepton 10g, Yeast extract 2g, MnSO 4 .7H 2 dissolved O 1 g of distilled water 1L] to After inoculation with platinum ears, shake culture at 37 ° C until logarithmic growth, collect cells, and use DNA genome extraction kit (trade name: GenomicPrep Cells and Tissue DNA Isolation Kit, manufactured by Amersham) Then, according to the instruction manual, chromosomal DNA was recovered from the collected cells.
- DNA genome extraction kit trade name: GenomicPrep Cells and Tissue DNA Isolation Kit, manufactured by Amersham
- Chromosomal DNA extraction from Escherichia coli K12-MG1655 was performed by inoculating with LB medium (tryptone 10 g, yeast extract 5 g, NaCl 5 g in 1 L of distilled water) using platinum ears and until the logarithmic growth phase. Cultivate culture at °C, collect the cells, and then use the DNA genome extraction kit (trade name: GenomicPrep Cells and Tissue DNA Isolation ⁇ Kit, manufactured by Amersham) to collect chromosomal DNA from the collected cells according to the instruction manual. It was collected.
- LB medium tryptone 10 g, yeast extract 5 g, NaCl 5 g in 1 L of distilled water
- Chromosomal DNA extraction from Lactococcus lactis NBRC100933 is inoculated into MRS medium (manufactured by DIFCO) using platinum ears and permeabilized at 30 ° C until the logarithmic growth phase.
- MRS medium manufactured by DIFCO
- chromosomal DNA was collected from the collected cells according to the instruction manual.
- Chromosomal DNA extraction from Pseudomonas putida F1 ATCC 700007 is inoculated using a platinum loop in Nutrient Broth medium (manufactured by DIFCO), followed by osmotic culture at 30 ° C until the logarithmic growth phase.
- a kit (trade name: GenomicPrep ⁇ Cells ⁇ ⁇ and Tissue DNA Isolation Kit, manufactured by Amersham), chromosomal DNA was collected from the collected cells according to the instruction manual.
- Chromosomal DNA extraction from Saccharomyces cerevisiae NBRC2376 was performed by inoculating YM medium [Glucose 10g, Peptone 5g, Yeast extract 3g, Malt extract 3g in 1L of distilled water], inoculating with platinum ears, and at 28 ° C until the logarithmic growth phase. After microbial culture, collect microbial cells and collect chromosomal DNA from the collected bacterial cells using the DNA genome extraction kit (trade name: GenomicPrep Cells and Tissue DNA Isolation Kit, manufactured by Amersham) according to the instruction manual did.
- YM medium Glucose 10g, Peptone 5g, Yeast extract 3g, Malt extract 3g in 1L of distilled water
- Chromosomal DNA extraction from Staphylococcus epidermidis NBRC12993 is, NBRC Medium No.802 medium [Polypepton 10g, Yeast extract 2g, MnSO 4 .7H dissolved in distilled water 1L of 2 O 1 g], after inoculation with platinum loop After osmotic culture at 30 ° C. until the logarithmic growth phase, the cells were collected and collected according to the instruction manual using a DNA genome extraction kit (trade name: GenomicPrep Cells and Tissue DNA Isolation Kit, manufactured by Amersham). Chromosomal DNA was recovered from the cells.
- cloning vector pCRB22 DNA fragment containing the DNA replication origin (hereinafter referred to as pCASE1-ori) of plasmid pCASE1 derived from Corynebacterium casei JCM12072 and the DNA fragment containing cloning vector pHSG298 (manufactured by Takara Bio Inc.) as follows Amplified by During PCR, in order to clone the pCASE1-ori sequence and the cloning vector pHSG298, respectively, the following pairs of primers were synthesized based on SEQ ID NO: 1 (pCASE1-ori sequence) and SEQ ID NO: 2 (cloning vector-pHSG298). ,used.
- pCASE1-ori sequence amplification primer (a-1); 5'- AT AGATCT AGAACGTCCGTAGGAGC -3 '(SEQ ID NO: 3) (B-1); 5'- AT AGATCT GACTTGGTTACGATGGAC -3 '(SEQ ID NO: 4)
- the primers (a-1) and (b-1) have a BglII restriction enzyme site added.
- Cloning vector pHSG298 amplification primer (a-2); 5'- AT AGATCT AGGTTTCCCGACTGGAAAG -3 (SEQ ID NO: 5) (B-2); 5'- AT AGATCT CGTGCCAGCTGCATTAATGA -3 '(SEQ ID NO: 6)
- the BglII restriction enzyme site is added to the primers (a-2) and (b-2).
- template DNA total DNA extracted from Corynebacterium casei JCM12072 obtained from Japan. Collection of Microorganisms (JCM) and cloning vector pHSG298 (manufactured by Takara Bio Inc.) were used. Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the pCASE1-ori sequence, use the combination of primers (a-1) and (b-1). When amplifying the cloning vector pHSG298, use the combination of primers (a-2) and (b-2). It was.
- PCR cycle Denaturation process: 94 °C, 60 seconds Annealing process: 52 °C, 60 seconds Extension process: 72 °C pCASE1-ori sequence: 150 seconds Cloning vector pHSG298: 180 seconds One cycle was performed for 30 cycles.
- the resulting ligation A solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing 50 ⁇ g / ml kanamycin [1% polypeptone, 0.5% yeast Extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme BglII to confirm the inserted fragment.
- BglII restriction enzyme
- the cloning vector containing the pCASE1-ori sequence was named pCRB22.
- pCG1-ori DNA replication origin derived from plasmid pCG1 [(JP-A-57-134500)] capable of replicating in Corynebacterium glutamicum
- cloning vector pHSG398 Tikara Bio Inc.
- pCG1-ori sequence amplification primer (a-3); 5'- AT AGATCT AGCATGGTCGTCACAGAG -3 '(SEQ ID NO: 9) (B-3); 5'- AT AGATCT GGAACCGTTATCTGCCTATG -3 '(SEQ ID NO: 10)
- B-3 5'- AT AGATCT GGAACCGTTATCTGCCTATG -3 '(SEQ ID NO: 10)
- BglII restriction enzyme site is added to the primers (a-3) and (b-3).
- Cloning vector pHSG398 amplification primer (a-4); 5'- AT AGATCT GTCGAACGGAAGATCACTTC -3 '(SEQ ID NO: 11) (B-4); 5'- AT AGATCT AGTTCCACTGAGCGTCAG -3 '(SEQ ID NO: 12)
- a BglII restriction enzyme site is added to primers (a-4) and (b-4).
- pCG1 (JP-A-57-134500)] and cloning vector pHSG398 (manufactured by Takara Bio Inc.) were used.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the pCG1-ori sequence, use a combination of primers (a-3) and (b-3). To amplify the cloning vector pHSG398, use a combination of primers (a-4) and (b-4). It was.
- PCR cycle Denaturation process: 94 °C, 60 seconds Annealing process: 52 °C, 60 seconds Extension process: 72 °C pCG1-ori sequence: 120 seconds Cloning vector pHSG398: 150 seconds One cycle was performed for 30 cycles.
- the resulting ligation solution B was transformed into Escherichia coli JM109 by the calcium chloride method (Journal of Molecular Biology, 53, 159 (1970)), and an LB agar medium (1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme BglII to confirm the inserted fragment.
- the restriction enzyme BglII the restriction enzyme to confirm the inserted fragment.
- an about 1.9-kb DNA fragment of the pCG1-ori sequence was observed in addition to the about 2.2-kb DNA fragment of the cloning vector pHSG398.
- the cloning vector containing the pCG1-ori sequence was named pCRB11.
- Cloning Vector pCRB15 A DNA fragment containing the cloning vector pCRB11 and a DNA fragment containing a zeocin resistance gene derived from pSELECT-zeo-mcs (manufactured by Invitrogen Corporation) were amplified by the following PCR method. During PCR, the following pair of primers were synthesized and used based on SEQ ID NO: 13 (pCRB11) and SEQ ID NO: 14 (zeocin resistance gene) in order to clone the cloning vector pCRB11 and the zeocin resistance gene, respectively.
- Cloning vector pCRB11 sequence amplification primer (a-5); 5'- AT GATATC CGAAGTGATCTTCCGTTCGA -3 '(SEQ ID NO: 15) (B-5); 5'- AT GATATC AAGGCAGTTATTGGTGCCCT -3 '(SEQ ID NO: 16)
- EcoRV restriction enzyme sites are added to primers (a-5) and (b-5).
- Primer for amplification of zeocin resistance gene (a-6); 5'- AT GATATC TAGCTTATCCTCAGTCCTGC -3 '(SEQ ID NO: 17) (B-6); 5'- AT GATATC CCATCCACGCTGTTTTGACA -3 '(SEQ ID NO: 18)
- EcoRV restriction enzyme sites are added to primers (a-6) and (b-6).
- cloning vectors pCRB11 and pSELECT-zeo-mcs (manufactured by Invitrogen Corporation) were used. Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the cloning vector pCRB11 sequence, use a combination of primers (a-5) and (b-5); to amplify a zeocin resistance gene, use a combination of primers (a-6) and (b-6). It was.
- PCR cycle Denaturation process: 94 °C, 60 seconds Annealing process: 52 °C, 60 seconds Extension process: 72 °C pCRB11 sequence: 200 seconds Zeocin resistance gene: 45 seconds More than 30 cycles were performed for 30 cycles.
- the resulting ligation C solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing 1 ⁇ g / ml of zeocin (1% polypeptone, 0.5% yeast). Extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme EcoRV to confirm the inserted fragment.
- EcoRV restriction enzyme
- an approximately 0.5-kb DNA fragment was observed in the case of the zeocin resistance gene.
- the cloning vector containing the zeocin resistance gene was named pCRB15.
- cloning vector pCRB205 DNA containing cloning vector pCRC200 [Yasuda, K. et al., Analyzes of the acetate-producing pathways in Corynebacterium glutamicum under oxygen-deprived conditions. Applied Microbiology and Biotechnology. 77: 853-860 (2007)] The fragment was amplified by the following PCR method. During PCR, the following pair of primers were synthesized and used based on SEQ ID NO: 19 (pCRC200) in order to clone pCRC200.
- pCRC200 amplification primer (a-7); 5'- CTCT ACTAGT GTCGAC GGATCC TTGTGTGGAATTGTGAGCGG -3 ' (SEQ ID NO: 20) (B-7); 5'- CTCT ACTAGT CATACGAGCCGGAAGCATAA -3 ' (SEQ ID NO: 21)
- SpeI, SalI, and BamHI restriction enzyme sites are added to the primer (a-7), and a SpeI restriction enzyme site is added to the primer (b-7).
- the cloning vector pCRC200 containing the tac promoter was used as the template DNA.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When the pCRC200 gene was amplified, the primer (a-7) and (b-7) were combined.
- PCR cycle Denaturation process: 94 ° C., 60 seconds Annealing process: 52 ° C., 60 seconds Extension process: 72 ° C., 300 seconds One cycle was performed for 30 cycles.
- Ligation D solution This was designated as Ligation D solution.
- the resulting ligation D solution was transformed into Escherichia coli JM109 by the calcium chloride method (Journal of Molecular Biology, 53, 159 (1970)), and an LB agar medium (1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with the restriction enzyme SpeI to confirm the insertion restriction enzyme site.
- a cloning vector obtained by adding restriction enzyme sites SpeI, SalI and BamHI sites to the cloning vector pCRC200 was named pCRB205.
- cloning vector pCRB207 DNA fragment containing the promoter sequence of the gapA gene (hereinafter referred to as PgapA) encoding glyceraldehyde-3-phosphate dehydrogenase derived from Corynebacterium glutamicum R, and cloning vector
- PgapA promoter sequence of the gapA gene
- a terminator sequence derived from pKK223-3 (Pharmacia) was amplified by the following method.
- Primer for PgapA sequence amplification In addition, a SalI restriction enzyme site is added to the primer (a-8), and SalI, BamHI and NcoI restriction enzyme sites are added to the primer (b-8).
- Terminator sequence amplification primer (a-9); 5'- CTCT GCATGC CCATGG CTGTTTTGGCGGATGAGAGA -3 ' (SEQ ID NO: 26) (B-9); 5'- CTCT GCATGC TCATGA AAGAGTTTGTAGAAACGCAAAAAGG -3 (SEQ ID NO: 27)
- the primer (a-9) has SphI and NcoI restriction enzyme sites
- the primer (b-9) has SphI and BspHI restriction enzyme sites.
- chromosomal DNA extracted from Corynebacterium glutamicum R (FERM P-18976) and pKK223-3 plasmid (Pharmacia) were used.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the PgapA sequence, a combination of primers (a-8) and (b-8) was used, and when a terminator sequence was amplified, a combination of primers (a-9) and (b-9) was used.
- PCR cycle Denaturation process: 94 °C, 60 seconds Annealing process: 52 °C, 60 seconds Extension process: 72 °C PgapA sequence: 45 seconds Terminator sequence: 30 seconds A cycle of 30 seconds or more was performed for 30 cycles.
- the resulting ligation E solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium [1% polypeptone, 0.5% yeast containing 50 ⁇ g / ml of kanamycin. Extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with restriction enzyme SalI to confirm the inserted fragment.
- restriction enzyme SalI restriction enzyme to confirm the inserted fragment.
- about 0.6-kb DNA fragment of PgapA sequence was recognized in addition to about 4.1-kb DNA fragment of cloning vector pCRB22.
- the cloning vector containing the PgapA sequence was named pCRB206.
- the resulting ligation F solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing 50 ⁇ g / ml kanamycin [1% polypeptone, 0.5% yeast Extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- an about 0.4-kb DNA fragment of the terminator sequence was observed.
- the cloning vector containing the rrnBT1T2 terminator sequence was named pCRB207.
- PldhA sequence amplification primer (a-10); 5'- CTCT GTCGAC CGGAACTAGCTCTGCAATGA -3 ' (SEQ ID NO: 29) (B-10); 5'- CTCT GTCGAC GGATCC CATATG CGATCCCACTTCCTGATTTC -3 ' (SEQ ID NO: 30)
- a SalI restriction enzyme site is added to the primer (a-10)
- SalI, BamHI and NcoI restriction enzyme sites are added to the primer (b-10).
- chromosomal DNA extracted from Corynebacterium glutamicum R (FERM P-18976) was used.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the PldhA sequence, the primers (a-10) and (b-10) were combined.
- PCR cycle Denaturation process: 94 ° C., 60 seconds Annealing process: 52 ° C., 60 seconds Extension process: 72 ° C., 30 seconds More than 30 cycles were performed for 30 cycles.
- the resulting ligation G solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing 50 ⁇ g / ml kanamycin [1% polypeptone, 0.5% yeast Extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with restriction enzyme SalI to confirm the inserted fragment.
- restriction enzyme SalI restriction enzyme to confirm the inserted fragment.
- pCRB208 The cloning vector containing the PldhA sequence was named pCRB208.
- alsS gene amplification primer (a-11); 5'- CTCT CCATGG TGACAAAAGCAACAAAAGAACAAAAATCC -3 ' (SEQ ID NO: 32) (B-11); 5'- CTCT CCATGG AGATCT CTAGAGAGCTTTCGTTTTCATGAG -3 ' (SEQ ID NO: 33)
- the primer (a-11) has an NcoI restriction enzyme site
- the primer (b-11) has an NcoI and BglII restriction enzyme site.
- ilvB-ilvN-ilvC gene amplification primer (a-12); 5'- GA CCCGGG AGTAAAGGAGCCAGAAAGTCGTGAA -3 ' (SEQ ID NO: 36) (B-12); 5'- GA CCCGGG CCTGCAGG TGCCTTATGTACAAAGTGCACAGCA -3 ' (SEQ ID NO: 37)
- the SmaI restriction enzyme site is added to the primer (a-12), and the SmaI and Sse8387I restriction enzyme sites are added to the primer (b-12).
- ilvD gene amplification primer (a-13); 5'- CTCT TCATGA TCCCACTTCGTTCAAAAGTC -3 ' (SEQ ID NO: 38) (B-13); 5'- CTCT TCATGA TTAGTCGACCTGACGGAC -3 ' (SEQ ID NO: 39)
- a BspHI restriction enzyme site is added to primers (a-13) and (b-13).
- adhP gene amplification primer (a-14); 5'- CTCT CCATGG AGGCTGCAGTTGTTACGAAG -3 ' (SEQ ID NO: 41) (B-14); 5'- CTCT CCATGG AGATCT TTAGTGACGGAAATCAATCACCAT -3 ' (SEQ ID NO: 42)
- the primer (a-14) has an NcoI restriction enzyme site
- the primer (b-14) has an NcoI and BglII restriction enzyme site.
- Lactococcus lactis-derived isobutanol production gene group A DNA fragment containing the kivD gene encoding 2-keto acid decarboxylase derived from Lactococcus lactis was amplified by the following PCR method. In order to clone the kivD gene during PCR, the following pair of primers were used based on SEQ ID NO: 43 (Lactococcus lactis kivD gene), respectively, and “394 DNA / RNA synthesizer (Applied Biosystems)” synthesizer) ”and used.
- the primer (a-15) has an NcoI restriction enzyme site, and the primer (b-15) has an NcoI and BglII restriction enzyme site.
- adh gene amplification primer (a-16); 5'- CTCT TCATGA AAGCTGCTGTCGTTGC -3 '(SEQ ID NO: 47) (B-16); 5'- CTCT TCATGA CTCGAG TCAGCCTTCGAACTGTATCAC -3 ' (SEQ ID NO: 48)
- the primer (a-16) has a BspHI restriction enzyme site
- the primer (b-16) has a BspHI and XhoI restriction enzyme site.
- Primer for adh2 gene amplification The primer (a-17) has an NcoI restriction enzyme site, and the primer (b-17) has an NcoI and BglII restriction enzyme site.
- the primer (a-18) has an NcoI restriction enzyme site, and the primer (b-18) has an NcoI and BglII restriction enzyme site.
- chromosomal DNA extracted from Bacillus subtilis 168 NBRC14144 obtained from NITE Biological Resource Center (NBRC) was used.
- the chromosomal DNA extracted from Corynebacterium glutamicum R was used as Corynebacterium glutamicum.
- Escherichia coli chromosomal DNA extracted from Escherichia coli K12-MG1655 was used.
- Lactococcus lactis chromosomal DNA extracted from Lactococcus lactis NBRC100933 obtained from NITE Biological Resource Center (NBRC) was used.
- chromosomal DNA extracted from Saccharomyces cerevisiae NBRC-2376 obtained from NITE Biological Resource Center (NBRC) was used.
- Staphylococcus epidermidis used chromosomal DNA extracted from Staphylococcus epidermidis NBRC12993 obtained from NITE Biological Resource Center (NBRC).
- NBRC NITE Biological Resource Center
- chromosomal DNA extracted from Pseudomonas putida F1 ATCC700007 obtained from American Type Culture Collection (ATCC) was used.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR.
- Bacillus Subtilis alsS gene is amplified by combining primer (a-11) and (b-11) ⁇
- Corynebacterium glutamicum ilvB-ilvN-ilvC gene is amplified by primer (a-12) and (b -12), when amplifying the ilvD gene, a combination of primer (a-13) and (b-13) ⁇ , when amplifying the Escherichia coli adhP gene, a combination of primer (a-14) and (b-14) ⁇
- primer (a-15) and (b-15) ⁇ When amplifying the Lactococcus lactis kivD gene, a combination of primer (a-15) and (b-15) ⁇ , when amplifying Pseudomonas putida adh gene, a combination of primer (a-16) and (b-16) ⁇
- Saccharomyces When amplifying the cerevisiae adh2 gene, a combination of primer (a-17) and (b-17) ⁇ , and when
- PCR cycle The above was regarded as one cycle, and 30 cycles were performed.
- the resulting ligation H solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)] and LB agar medium containing 1 ⁇ polypeptone, 0.5% yeast containing 50 ⁇ g / ml of ampicillin. Extract, 0.5% sodium chloride, and 1.5% straw agar).
- plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme SmaI to confirm the inserted fragment.
- SmaI restriction enzyme
- Ptac-ilvB-ilvN-ilvC sequence amplification primer (a-19); 5'- AT GCAAGC TTCGGCTGTGCAGGTCGTAAAT -3 '(SEQ ID NO: 56) (B-19); 5'-AC GCAAGC TTCGCTTATGTACAAAGTGCAC -3 '(SEQ ID NO: 57)
- a HindIII restriction enzyme site is added to the primers (a-19) and (b-19).
- the aforementioned plasmid pKK223-3-ilvB-ilvN-ilvC / CG was used as the template DNA.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) Amplification of the Ptac-ilvB-ilvN-ilvC sequence was performed with a combination of primers (a-19) and (b-19).
- PCR cycle Denaturation process: 94 ° C., 60 seconds Annealing process: 52 ° C., 60 seconds Extension process: 72 ° C., 240 seconds More than one cycle, 30 cycles were performed.
- the cloning vector pCRB1 (Nakata, K. et al., Vectors) of about 3.9-kb DNA fragment containing the tac promoter sequence amplified by PCR and the ilvB-ilvN-ilvC sequence derived from Corynebacterium glutamicum R and about 4.1-kb for the genetics engineering of corynebacteria; in Saha, BC (ed.): Fermentation Biotechnology, ACS Symposium Series 862.Washington, American Chemical Society: 175-191 (2003)) 2 ⁇ l each was cleaved with the restriction enzyme HindIII at 70 ° C.
- T4 DNA ligase 10 ⁇ buffer 1 ⁇ l and T4 DNA ligase (Takara Bio Inc.) 1 unit was added to this, 10 ⁇ l with sterile distilled water, reacted at 15 ° C. for 3 hours and bound. This was designated as Ligation I solution.
- the resulting ligation I solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium (1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with the restriction enzyme HindIII to confirm the inserted fragment.
- HindIII restriction enzyme
- Cloning of isobutanol production gene into pCRB207 About 1.8-kb DNA fragment containing alsS gene from Bacillus subtilis strain amplified by PCR shown in (3) above, about 1.0-kb DNA fragment containing adhP gene from Escherichia coli strain About 1.7-kb DNA fragment containing the kivD gene from Lactococcus lactis strain, about 1.1-kb DNA fragment containing the adh2 gene from Saccharomyces cerevisiae strain and about 10 ⁇ l of about 1.7-kb DNA fragment containing the ipd gene from Staphylococcus epidermidis strain and Cloning vector pCRB207 containing PgapA promoter 2 ⁇ l each with restriction enzyme NcoI and inactivating the restriction enzyme by treating at 70 ° C.
- T4 DNA ligase 10 ⁇ buffer 1 ⁇ l of T4 DNA ligase (Takara Bio Inc.) Add 1 unit of each component, make 10 ⁇ l with sterile distilled water, and react at 15 ° C for 3 hours. And combined. This was designated as Ligation J solution, K solution, L solution, M solution and N solution.
- the seven types of ligation J solution, K solution, L solution, M solution, N solution, O solution and P solution thus obtained were each converted into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)].
- LB agar medium 1% polypeptone, 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar
- 50 ⁇ g / ml kanamycin was applied to an LB agar medium (1% polypeptone, 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar) containing 50 ⁇ g / ml kanamycin.
- plasmid DNA was extracted from the culture solution, the plasmid was cut with a restriction enzyme, and the inserted fragment was confirmed.
- the inserted fragment in addition to the plasmid pCRB207 about 5.1-kb DNA fragment, in the case of the alsS gene derived from the Bacillus subtilis strain (Ligation J solution), the inserted fragment of about 1.8-kb in length is transformed into the adhP gene derived from the Escherichia coli strain (Ligation K solution).
- the inserted fragment of about 1.0-kb in length is the Lactococcus lactis-derived kivD gene (Ligation L solution), and the inserted fragment of about 1.7-kb in length is the Saccharomyces cerevisiae-derived adh2 gene (ligation M).
- the insert fragment of about 1.1-kb in length is the ipd gene derived from Staphylococcus epidermidis strain (Ligation N solution), and the insert fragment of about 1.7-kb in length is derived from Corynebacterium glutamicum strain ilvD
- the inserted fragment of about 2.0-kb in length, the adh gene from the Pseudomonas ⁇ ⁇ ⁇ putida strain (ligation P solution) Insert a length of about 1.0-kb was observed.
- Plasmid containing alsS gene from Bacillus subtilis strain is pCRB207-alsS / BS
- plasmid containing adhP gene from Escherichia coli strain is pCRB207-adhP / EC
- plasmid containing kivD gene from Lactococcus pylori strain is pCRB207-kivD / LL
- plasmids containing the adh gene derived from Pseudomonas sputida strain were named pCRB207-adh / PP, respectively (FIG. 1).
- Electropulse method [Agric. Biol. Chem., Vol. 54, 443-447 (1990) and Res. Microbiol., Vol. 144, 181-185 (1993)], Corynebacterium glutamicum R ldhA mutant [J Mol. Microbiol. Biotechnol., Vol. 8, 243-254 (2004)] were transformed and applied to A agar medium containing 50 ⁇ g / ml kanamycin.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- the plasmids pCRB1-ilvB-ilvN-ilvC / CG, pCRB207-alsS / BS, pCRB207-adhP / EC, pCRB207-kivD / LL, pCRB207-adh2 / SC, pCRB207-ipd / SE, pCRB207- Introduction of ilvD / CG and pCRB207-adh / PP was observed.
- the resulting strains are Corynebacterium glutamicum R ldhA mutant / pCRB1-ilvB-ilvN-ilvC / CG, ldhA mutant / pCRB207-alsS / BS, ldhA mutant / pCRB207-adhP / EC, ldhA mutant / pCRB207-kivD / LL They were named ldhA mutant / pCRB207-adh2 / SC, ldhA mutant / pCRB207-ipd / SE, ldhA mutant / pCRB207-ilvD / CG and ldhA mutant / pCRB207-adh / PP.
- Example 2 Measurement of Enzyme Activity in Corynebacterium glutamicum Introduced with Isoisobutanol Production Gene
- Corynebacterium glutamicum R ldhA mutant into which isoisobutanol production gene constructed in Example 1 was independently introduced (hereinafter, Corynebacterium glutamicum single) (Referred to as a transgenic strain) (with the exception that ilvB-ilvN-ilvC is linked on the same plasmid), isoisobutanol production-related enzymes, ie, acetohydroxy acid synthase (AHAS), acetohydroxy acid isomeroreductase ( AHAIR), dihydroxy acid dehydratase (DHAD), 2-keto acid decarboxylase (KDC), and alcohol dehydrogenase (ADH) were measured by the following methods.
- Corynebacterium glutamicum R ldhA mutant used as a host was also measured in the same manner.
- Corynebacterium glutamicum single gene-introduced strain grown on the above plate was mixed with A liquid medium containing 50 ⁇ g / ml kanamycin [(NH 2 ) 2 CO 2 g, (NH 4 ) 2 SO 4 7 g, KH 2 PO 4 0.5 g , K 2 HPO 4 0.5 g, MgSO 4 .7H 2 O 0.5 g, 0.06% (w / v) Fe 2 SO 4 .7H 2 O + 0.042% (w / v) MnSO 4 .2H 2 O 1 ml, 0.02 % (w / v) biotin solution 1 ml, 0.01% (w / v) thiamin solution 2 ml, yeast extract 2 g, vitamin assay casamino acid 7 g, glucose 40 g dissolved in 1 L of distilled water] Test with 10 ml One platinum ear was inoculated into the tube and aerobically shaken at 28 ° C. for 15 hours.
- Corynebacterium glutamicum single gene-introduced strain grown under the above conditions was inoculated into 100 ml of A liquid medium containing 50 ⁇ g / ml of kanamycin, and aerobically shaken and cultured at 28 ° C. for 15 hours.
- Corynebacterium glutamicum R ldhA mutant was cultured under the same conditions except that kanamycin was not added to medium A.
- Each microbial cell thus grown in culture was collected by a centrifugal separator (4 ° C., 8,000 ⁇ g, 10 minutes). The obtained cells were washed with disruption solution 1 (100 mM Tris-HCl buffer solution pH 7.5, 2 mM dithiothreitol). Centrifugation was performed again and the collected cells were suspended in 1 ml of crushing liquid 1, 1 g of glass beads were added, and homogenized with an ultrasonic crusher (Biorupter, manufactured by Cosmo Bio) for 30 minutes. Thereafter, insoluble substances were removed by centrifugation (4 ° C., 15,000 ⁇ g, 10 minutes) to obtain enzyme solution 1. The amount of protein was measured by Bio-Rad protein assay (manufactured by Bio-Rad). Using the enzyme solution thus obtained, AHAS, AHAIR, DHAD and ADH activities were measured by the method described below.
- disruption solution 1 100 mM Tris-HCl buffer solution pH 7.5, 2 mM dithiothrei
- AHAS activity was determined using pyruvate as a substrate and a decrease in absorbance at 333 nm as pyruvate decreased (Craig M. et.al., Mutagenesis of Escherichia coli acetohydroxyacid synthase isoenzyme II and characterization of three herbicide-insensitive forms. Biochem. J., 335: 653-661 (1998)).
- the reaction solution used for the activity measurement is 50 mM potassium phosphate pH 8.0, 10 mM magnesium chloride, 0.1 mM PPP, 0.1 mM FAD, 50 mM sodium pyruvate, and the spectrophotometer DU-800 (manufactured by BECKMAN) is used for the absorbance measurement. It was.
- the results of AHAS activity measurement of Corynebacterium glutamicum R ldhA mutant used as 1 are shown in the following table (1U indicates the amount of enzyme that consumes 1 ⁇ mol of substrate per minute).
- AHAIR activity was measured using the decrease in absorbance at 340 nm accompanying the decrease in NADPH when 2,3-dihydroxyisovaleric acid was formed from acetolactate and NADPH (Ruyex., Et.al., Characterization of enzymes of the branched-chain amino acid biosynthetic pathway in Methanococcus spp. J. Bacteriol., 173: 2086-2092 (1991)).
- the reaction solution used for the activity measurement was 100 mM Tris-HCl buffer pH 7.6, 10 mM magnesium chloride, 0.15 mM NADPH, 10 mM acetolactic acid, and a spectrophotometer DU-800 (manufactured by BECKMAN) was used for the absorbance measurement.
- the reaction was started by adding enzyme solution 1 to the reaction solution at 30 ° C., and the molar extinction coefficient was 6.22 M ⁇ 1 from the difference between the slope of the absorbance decrease of the reaction solution and the absorbance decrease of the reaction solution not containing acetolactate. Activity values were calculated using cm ⁇ 1 .
- DHAD activity was measured using the increase in absorbance at 340 nm accompanying the increase in 2-ketoisovaleric acid when 2-ketoisovaleric acid was formed from 2,3-dihydroxyisovaleric acid (Dennis HF, et.al., The role and properties of the iron-sulfur cluster in Escherichia coli dihydroxy-acid dehydratase. J. Biol. Chem., 268: 14732-14742 (1993)).
- the reaction solution used for the activity measurement was 50 mM Tris-HCl buffer pH 8.0, 10 mM magnesium chloride, 6 mM 2,3-dihydroxyisovaleric acid, and the spectrophotometer DU-800 (manufactured by BECKMAN) was used for the absorbance measurement. Using. The reaction was started by adding enzyme solution 1 to the reaction solution at 30 ° C. From the difference between the slope of absorbance decrease of the reaction solution and the slope of absorbance decrease of the reaction solution not containing 2,3-dihydroxyisovaleric acid, Activity values were calculated using an extinction coefficient of 190 M ⁇ 1 cm ⁇ 1 .
- ADH activity was measured using the decrease in absorbance at 340 nm as an index of decrease in NADH when isobutanol was formed from isobutyraldehyde (Durre, P. et al., Enzymaticommegtations on butanol dehydrogenase and butyraldehyde dehydrogenase in extracts of Clostridium acetobutylicum. Appl. Microbiol. Biotechnol. 26: 268-272 (1987)).
- the composition of the reaction solution is 50 mM MES buffer (pH 6.0), 0.15 mM NADH, 40 mM isobutyraldehyde, and the absorbance measurement is 95% N 2 using a spectrophotometer Ultraspec 2100 pro (manufactured by GE Healthcare Biosciences). And in an anaerobic chamber (COY) substituted with 5% H 2 . Add enzyme solution 2 to the reaction solution at 30 ° C and let stand for 10 minutes, then add isobutyraldehyde or the same volume of pure water as a control sample to 40 mM, and the molar extinction coefficient of 6,220 M from the difference in each absorbance decrease The activity value was calculated using -1 cm- 1 .
- the results of ADH activity measurement of Corynebacterium glutamicum R ldhA mutant used as the host are shown in the following table (1U indicates the amount of enzyme that consumes 1 ⁇ mol of substrate per minute).
- Corynebacterium glutamicum single gene-introduced strain grown on the above plate was mixed with A liquid medium containing 50 ⁇ g / ml kanamycin [(NH 2 ) 2 CO 2 g, (NH 4 ) 2 SO 4 7 g, KH 2 PO 4 0.5 g , K 2 HPO 4 0.5 g, MgSO 4 .7H 2 O 0.5 g, 0.06% (w / v) Fe 2 SO 4 .7H 2 O + 0.042% (w / v) MnSO 4 .2H 2 O 1 ml, 0.02 % (w / v) biotin solution 1 ml, 0.01% (w / v) thiamin solution 2 ml, yeast extract 2 g, vitamin assay casamino acid 7 g, glucose 40 g dissolved in 1 L of distilled water] Test with 10 ml One platinum ear was inoculated into the tube and aerobically shaken at 28 ° C. for 15 hours.
- Corynebacterium glutamicum single gene-introduced strain grown under the above conditions was inoculated into 100 ml of A liquid medium containing 50 ⁇ g / ml kanamycin, and aerobically shaken at 28 ° C. for 15 hours.
- Corynebacterium glutamicum R ldhA mutant was cultured under the same conditions except that kanamycin was not added to the A medium.
- the respective cells grown in this way were collected by centrifugation (4 ° C., 8,000 ⁇ g, 10 minutes).
- the obtained cells BT (- glucose) broth [(NH 2) 2 CO 2g , (NH 4) 2 SO 4 7g, KH 2 PO 4 0.5 g, K 2 HPO 4 0.5 g, MgSO 4 .7H 2 O 0.5 g, 0.06% ( w / v) Fe 2 SO 4 .7H 2 O + 0.042% (w / v) MnSO 4 .2H 2 O 1 ml, 0.02% (w / v) biotin solution 1 ml, 0.01 Dissolve 2 ml of% (w / v) thiamin solution in 1 L of distilled water] It was suspended in 40 ml and transferred to a 50 ml medium bottle.
- the subsequent operation was performed in an anaerobic chamber (COY) substituted with 95% N 2 and 5% H 2 .
- the medium bottle and the cells were anaerobically replaced by opening the medium bottle in the anaerobic chamber and stirring for 2 hours.
- the cells were collected by centrifugation (4 ° C., 5,000 ⁇ g, 10 minutes), and washed with disruption solution 2 (50 mM MOPS buffer pH 7.0). Centrifuge again and suspend the collected cells in 1 ml of disruption solution 2. Then, add 2 g of glass beads, homogenize with a vortex mixer for 1 minute and leave on ice for 10 minutes to disrupt the cells. did. Thereafter, the insoluble material was removed by centrifugation (4 ° C., 5,000 ⁇ g, 10 minutes) to obtain enzyme solution 2. Using the obtained enzyme solution 2, the KDC activity was measured by the method described below.
- KDC activity was measured using a two-step reaction in combination with a yeast-derived alcohol dehydrogenase (Roche). That is, simultaneously with conversion of 2-ketoisovaleric acid to isobutyraldehyde by KDC, conversion of isobutyraldehyde to isobutanol by yeast-derived alcohol dehydrogenase is performed. In this reaction, when isobutanol is formed from 1 molecule of 2-ketoisovaleric acid, 1 molecule of NADH is consumed. Therefore, the KDC activity was determined by the decrease in absorbance at 340 nm accompanying the decrease of NADH.
- the composition of the reaction solution is 50 mM MES buffer pH 6.0, 5 mM magnesium chloride, 1.5 mM TPP, 0.2 mM 2-ketoisovaleric acid, 0.15 mM NADH, 20 U alcohol-derived alcohol dehydrogenase, and the spectrophotometer Ultraspec
- the measurement was performed in an anaerobic chamber (COY) substituted with 95% N 2 and 5% H 2 using 2100 pro (GE Healthcare Bioscience).
- COY anaerobic chamber
- 2100 pro GE Healthcare Bioscience
- Corynebacterium glutamicum R ldhA mutant / pCRB207-kivD / LL and ldhA mutant / pCRB207-ipd / SE which are introduced strains of the gene encoding KDC constructed in Example 1, and coryne used as a host as a control
- the results of HBD activity measurement of bacteria Glutamicum R ldhA mutant are shown in the following table (1U indicates the amount of enzyme that consumes 1 ⁇ mol of substrate per minute).
- alsS gene amplification primer (a-20); 5'-CTCT CCCGGG AAACTTTTTAGAAAGGTGTGTTTCACCCGTGTTGACAAAAGCAACAAAAGAAC -3 ' (SEQ ID NO: 59) (B-20); 5'-CTCT CCCGGG AGATCT CTAGAGAGCTTTCGTTTTCATGA -3 ' (SEQ ID NO: 60)
- the SmaI restriction enzyme site is added to the primer (a-20), and the SmaI and BglII restriction enzyme sites are added to the primer (b-20).
- ilvB-ilvN-ilvC gene amplification primer (a-12); 5'- GA CCCGGG AGTAAAGGAGCCAGAAAGTCGTGAA -3 ' (SEQ ID NO: 36) (B-12); 5'- GA CCCGGG CCTGCAGG TGCCTTATGTACAAAGTGCACAGCA -3 ' (SEQ ID NO: 37)
- the SmaI restriction enzyme site is added to the primer (a-12), and the SmaI and Sse8387I restriction enzyme sites are added to the primer (b-12).
- ilvC gene amplification primer (a-21); 5'- CTCT CCATGG CTATTGAACTGCTTTATGATG -3 ' (SEQ ID NO: 63) (B-21); 5'- CTCT CCATGG AGATCT TTAAGCGGTTTCTGCGCGA -3 ' (SEQ ID NO: 64)
- the primer (a-21) has an NcoI restriction enzyme site
- the primer (b-21) has an NcoI and BglII restriction enzyme site.
- ilvD gene amplification primer (a-22); 5'- GA CCCGGG GAGCAGATTTGAAAAGCGCATCATG -3 ' (SEQ ID NO: 65) (B-22); 5'- GA CCCGGG GGTACC GTATTTGCAACGGGGAGCTCCACCA -3 ' (SEQ ID NO: 66)
- the SmaI restriction enzyme site is added to the primer (a-22), and the SmaI and KpnI restriction enzyme sites are added to the primer (b-22).
- the Escherichia coli sequence (SEQ ID NO: 67; ilvB-ilvN gene), (SEQ ID NO: 68; ilvC gene), (SEQ ID NO: 69) to clone the ilvB-ilvN gene, ilvC gene, ilvD gene and adhP gene.
- SEQ ID NO: 67; ilvB-ilvN gene SEQ ID NO: 68; ilvC gene
- SEQ ID NO: 69 to clone the ilvB-ilvN gene, ilvC gene, ilvD gene and adhP gene.
- the following pair of primers was synthesized using “394 DNA / RNA synthesizer” (Applied Biosystems), respectively. ,used.
- ilvB-ilvN gene amplification primer (a-23); 5'- CTCT CCCGGG ATGGCAAGTTCGGGCACAA -3 ' (SEQ ID NO: 70) (B-23); 5'- CTCT CCCGGG AGATCT TTACTGAAAAAACACCGCGATCTT-3 ' (SEQ ID NO: 71)
- the SmaI restriction enzyme site is added to the primer (a-23), and the SmaI and BglII restriction enzyme sites are added to the primer (b-23).
- ilvC gene amplification primer (a-24); 5'- CTCT CCCGGG ATGGCTAACTACTTCAATACACTG -3 ' (SEQ ID NO: 72) (B-24); 5'- CTCT CCCGGG AGATCT TTAACCCGCAACAGCAATACG-3 ' (SEQ ID NO: 73)
- SmaI restriction enzyme site is added to the primer (a-24) and the SmaI and BglII restriction enzyme sites are added to the primer (b-24).
- ilvD gene amplification primer (a-25); 5'- CTCT TTTAAA ATGCCTAAGTACCGTTCCG -3 ' (SEQ ID NO: 74) (B-25); 5'- CTCT TTTAAA AGATCT TTAACCCCCCAGTTTCGATTTAT-3 ' (SEQ ID NO: 75)
- the DraI restriction enzyme site is added to the primer (a-25), and the DraI and BglII restriction enzyme sites are added to the primer (b-25).
- adhP gene amplification primer (a-14); 5'- CTCT CCATGG AGGCTGCAGTTGTTACGAAG -3 ' (SEQ ID NO: 41) (B-14); 5'- CTCT CCATGG AGATCT TTAGTGACGGAAATCAATCACCAT -3 ' (SEQ ID NO: 42)
- the primer (a-14) has an NcoI restriction enzyme site
- the primer (b-14) has an NcoI and BglII restriction enzyme site.
- the primer (a-15) has an NcoI restriction enzyme site, and the primer (b-15) has an NcoI and BglII restriction enzyme site.
- adh gene amplification primer (a-16); 5'- CTCT TCATGA AAGCTGCTGTCGTTGC -3 '(SEQ ID NO: 47) (B-16); 5'- CTCT TCATGA CTCGAG TCAGCCTTCGAACTGTATCAC -3 ' (SEQ ID NO: 48)
- the primer (a-16) has a BspHI restriction enzyme site
- the primer (b-16) has a BspHI and XhoI restriction enzyme site.
- Primer for adh2 gene amplification The primer (a-17) has an NcoI restriction enzyme site, and the primer (b-17) has an NcoI and BglII restriction enzyme site.
- Bacillus subtilis used was chromosomal DNA extracted from Bacillus subtilis 168 NBRC14144 obtained from NITE Biological Resource Center (NBRC).
- the chromosomal DNA extracted from Corynebacterium glutamicum R was used as Corynebacterium glutamicum.
- Escherichia coli chromosomal DNA extracted from Escherichia coli K12-MG1655 was used.
- Lactococcus lactis chromosomal DNA extracted from Lactococcus lactis NBRC100933 obtained from NITE Biological Resource Center (NBRC) was used.
- Pseudomonas putida is Saccharomyces cerevisiae using chromosomal DNA extracted from Pseudomonas putida F1 ATCC 700007 obtained from American Type Collection AT Collection (ATCC), Saccharomyces cerevisiae NBRC 2376 obtained from NITE Biological Resource Center (NBRC) Using.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR.
- Bacillus Subtilis alsS gene is amplified by combining primer (a-20) and (b-20) ⁇
- Corynebacterium glutamicum ilvB-ilvN-ilvC gene is amplified by primer (a-12) and (b -12), primer (a-21) and (b-21) for amplifying the ilvC gene, primer (a-22) and (b-22) ⁇ ⁇ ⁇ ⁇ ⁇ for amplifying the ilvD gene, Escherichia coli ilvB- Primers (a-23) and (b-23) for amplifying the ilvN gene, primers (a-24) and (b-24) for amplifying the ilvC gene, and primers (a -25) and (b-25), when adhP gene is amplified, primer (a-14) and ⁇ (b-14) ⁇ ⁇ ⁇ ⁇ ⁇ , and when a Lactococcus lactis kivD gene is amplified, primer (a-15) and ( b-15
- PCR cycle The above was regarded as one cycle, and 30 cycles were performed.
- T4 DNA ligase 10 ⁇ buffer 1 ⁇ l T4 DNA ligase (manufactured by Takara Bio Inc.) 1 unit of each component was added, made up to 10 ⁇ l with sterile distilled water, reacted at 15 ° C. for 3 hours, and bound. This was designated as Ligation Q solution, R solution, S solution and T solution.
- T4 DNA ligase 10 ⁇ buffer and 1 unit of T4 DNA ligase Takara Bio Inc.
- Each component was added, made up to 10 ⁇ l with sterile distilled water, reacted at 15 ° C. for 3 hours, and bound. This was designated as Ligation U solution.
- Each of the obtained 5 types of ligation Q solution, R solution, S solution, T solution and U solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)] to give ampicillin.
- LB agar medium 1% polypeptone, 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar
- each of the growing strains on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, the plasmid was cut with a restriction enzyme, and the inserted fragment was confirmed.
- an insertion fragment of about 3.7-kb in length is In the case of the ilvD gene (ligation R solution) derived from the um glutamicum strain, the inserted fragment of about 2.0-kb in length is approximately 2.0-kb in the case of the ilvB-ilvN gene derived from the Escherichia coli strain (ligation S solution).
- the insert fragment of about 1.5-kb in length is in the case of the Escherichia coli strain-derived ilvD gene (ligation U solution), in the case of the insert fragment of about 1.9-kb in length.
- PKK223-3-ilvB-ilvN-ilvC / CG is a plasmid containing the ilvB-ilvN-ilvC gene derived from Corynebacterium glutamicum
- pKK223-3-ilvD / CG is a plasmid containing the ilvD gene derived from Corynebacterium glutamicum
- Escherichia coli PKK223-3-ilvB-ilvN / EC containing the ilvB-ilvN gene derived from the strain
- pKK223-3-ilvC / EC containing the ilvC gene derived from the Escherichia coli strain
- pKK223- containing the ilvD gene derived from the Escherichia coli strain were named 3-ilvD / EC, respectively.
- Ptac-ilvB-ilvN-ilvC sequence amplification primer (a-19); 5'- AT GCAAGC TTCGGCTGTGCAGGTCGTAAAT -3 '(SEQ ID NO: 56) (B-19); 5'-AC GCAAGC TTCGCTTATGTACAAAGTGCAC-3 '(SEQ ID NO: 57)
- a HindIII restriction enzyme site is added to the primers (a-19) and (b-19).
- Primer for amplification of Ptac-ilvD sequence (a-26); 5'- ATAT CCTGCAGG CTAGCGCTGTGCAGGTCGTAAATCAACT -3 ' (SEQ ID NO: 77) (B-26); 5'- ATATGCTAGCT CCTGCAGG TATTTGCAACGGGGAGCTC -3 ' (SEQ ID NO: 78)
- Sse8387I restriction enzyme site is added to the primers (a-26) and (b-26).
- the aforementioned plasmids pKK223-3-ilvB-ilbN-ilvC / CG and pKK223-3-ilvD / CG were used.
- Actual PCR was performed using the thermal cycler GeneAmp PCR System 9700 (manufactured by Applied Biosystems) and TaKaRa LA Taq (manufactured by Takara Bio Inc.) as a reaction reagent under the following conditions.
- Reaction solution The above was mixed, and 50 ⁇ l of the reaction solution was subjected to PCR. *) When amplifying the Ptac-ilvB-ilvN-ilvC sequence, use a combination of primers (a-19) and (b-19) .To amplify the Ptac-ilvD sequence, primers (a-26) and (b-26 ).
- PCR cycle Denaturation process: 94 °C, 60 seconds Annealing process: 52 °C, 60 seconds Extension process: 72 °C Ptac-ilvB-ilvN-ilvC sequence: 240 sec Ptac-ilvD sequence: 150 sec One cycle was performed for 30 cycles.
- the cloning vector pCRB1 (Nakata, K. et al., Vectors) of about 3.9-kb DNA fragment containing tac promoter sequence amplified by PCR and ilvB-ilvN-ilvC sequence derived from Corynebacterium glutamicum R and about 4.1-kb for the genetics engineering of corynebacteria; in Saha, BC (ed.): Fermentation Biotechnology, ACS Symposium Series 862.Washington, American Chemical Society: 175-191 (2003)] 2 ⁇ l each was cleaved with the restriction enzyme HindIII at 70 ° C.
- T4 DNA ligase 10 ⁇ buffer 1 ⁇ l and T4 DNA ligase (Takara Bio Inc.) 1 unit was added to this, 10 ⁇ l with sterile distilled water, reacted at 15 ° C. for 3 hours and bound. This was designated as Ligation V solution.
- the obtained ligation V solution was transformed into Escherichia coli JM109 by the calcium chloride method (Journal of Molecular Biology, 53, 159 (1970)), and an LB agar medium (1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with the restriction enzyme HindIII to confirm the inserted fragment.
- HindIII restriction enzyme
- the resulting ligation W solution was transformed into Escherichia coli JM109 by the calcium chloride method (Journal of Molecular Biology, 53, 159 (1970)), and an LB agar medium (1% polypeptone, 1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% koji yeast extract, 0.5% koji sodium chloride, and 1.5% koji agar].
- the growing strain on the medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme Sse8387I to confirm the inserted fragment.
- Sse8387I the restriction enzyme
- the plasmid containing the ilvB-ilvN-ilvC gene derived from Corynebacterium glutamicum and ilvD was named pCRB1-ilvB-ilvN-ilvC / CG-ilvD / CG (FIG. 2).
- the above plasmid pKK223-3-ilvB-ilvN / EC was cleaved with restriction enzymes BamHI and BglII, and after agarose electrophoresis, the tac promoter and Escherichia recovered from the agarose gel by QIAquick Gel Extraction Kit (manufactured by Qiagen). About 2.3-kb DNA fragment ligated with E.
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and LB agar medium containing 50 ⁇ g / ml of chloramphenicol (1% polypeptone) , 0.5% koji yeast extract, 0.5% koji sodium chloride, and 1.5% koji agar].
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- an insert fragment of about 2.3-kb in length was observed in the case of the Escherichia coli strain-derived ilvB-ilvN gene (ligation X solution).
- the plasmid containing the ilvB-ilvN gene derived from the Escherichia coli strain was named pCRB1-ilvB-ilvN / EC. This plasmid has only one restriction enzyme site BamHI.
- the above plasmid pKK223-3-ilvC / EC was cleaved with the restriction enzymes BamHI and BglII, and after agarose electrophoresis, the tac promoter and Escherichia coli recovered from the agarose gel by QIAquick Gel Extraction Kit (manufactured by Qiagen).
- the resulting ligation Y solution was used to transform Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium [1% polypeptone containing 50 ⁇ g / ml of chloramphenicol. , 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- a plasmid containing the ilvB-ilvN gene derived from the Escherichia coli strain and the ilvC gene derived from the Escherichia coli strain was named pCRB1-ilvB-ilvN / EC-ilvC / EC. This plasmid has only one restriction enzyme site BamHI.
- the above-described plasmid pKK223-3-ilvD / EC was cleaved with restriction enzymes BamHI and BglII, and after agarose electrophoresis, the tac promoter and Escherichia coli strain recovered from the agarose gel by QIAquick Gel Extraction Kit (manufactured by Qiagen).
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium [1% polypeptone containing 50 ⁇ g / ml of chloramphenicol. , 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- an Escherichia coli strain-derived ilvD gene (ligation Z solution) has an insertion fragment of about 2.1-kb in length. It was.
- the plasmid containing the Escherichia coli strain-derived ilvB-ilvN gene, the Escherichia coli strain-derived ilvC gene and the Escherichia coli strain-derived ilvD gene was designated as pCRB1-ilvB-ilvN / EC-ilvC / EC-ilvD / EC.
- This plasmid has only one restriction enzyme site BamHI.
- ligation AE solution and AF solution Each component of 1 ⁇ l of solution, 1 ⁇ l of T4 DNA ligase (manufactured by Takara Bio Inc.) was added to 10 ⁇ l of sterile distilled water, reacted at 15 ° C. for 3 hours, and bound. This was designated as ligation AE solution and AF solution.
- the inserted fragment of about 1.0-kb is a kivD gene derived from Lactococcus ⁇ ⁇ lactis strain (ligation AC solution)
- the inserted fragment of about 1.7-kb is adh2 gene derived from Saccharomyces cerevisiae strain (ligation AD solution)
- the insertion fragment of about 1.1-kb in length is the ilvD gene derived from Corynebacterium glutamicum strain (Ligation AE solution)
- the insertion fragment of about 2.0-kb in length is the adh gene derived from Pseudomonas putida strain (Ligation AF)
- an inserted fragment having a length of about 1.0-kb was observed.
- the plasmid containing the ilvC gene from Corynebacterium glutamicum strain is pCRB207-ilvC / CG
- the plasmid containing the adhP gene from Escherichia coli strain is pCRB207-adhP / EC
- the plasmid containing the kivD gene from Lactococcus lactis strain is pCRB207-kivD / LL
- PCRB207-adh2 / SC is a plasmid containing the adh2 gene from Saccharomyces cerevisiae strain
- pCRB207-ilvD / CG is a plasmid containing the ilvD gene from Corynebacterium glutamicum strain
- pCRB207-adh / PP is a plasmid containing the adh gene from Pseudomonas coconut Respectively.
- the resulting ligation AG solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium (1% polypeptone, containing 50 ⁇ g / ml of chloramphenicol). 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the above-described plasmid pCRB207-ilvC / CG was cleaved with the restriction enzyme BamHI, and after agarose electrophoresis, the gapA gene recovered from the agarose gel by the QIAquick Gel Extraction Kit (manufactured by Qiagen) and the ilvC gene derived from Corynebacterium glutamicum strain and After a terminal blunting reaction was performed using the DNA Blunting Kit (manufactured by TAKARA BIO INC.) With the approximately 2.0-kb DNA fragment ligated with the terminator sequence and the plasmid pCRB205-alsS / BS cleaved with SphI.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- a plasmid containing the alsS gene derived from the Bacillus subtilis strain and the ilvC gene derived from the Corynebacterium glutamicum strain was named pCRB205-alsS / BS-ilvC / CG (FIG. 2).
- Cloning of isobutanol-producing gene into pCRB208 Cloning vector pCRB208 containing about 10 ⁇ l of a 1.7-kb DNA fragment containing the Lactococcus lactis kivD gene amplified by PCR shown in (1) above and the ldhA promoter Cleave 2 ⁇ l each with the restriction enzyme NcoI and inactivate the restriction enzyme by treating at 70 ° C. for 10 minutes. Then, mix both, and add 1 ⁇ l of T4 DNA ligase 10 ⁇ buffer, T4 DNA ligase (Takara Bio) 1 unit of each component was added, made up to 10 ⁇ l with sterile distilled water, reacted at 15 ° C.
- the resulting ligation AI solution was transformed into Escherichia coli JM109 by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing 50 ⁇ g / ml kanamycin [1% polypeptone, 0.5% yeast Extract, 0.5% sodium chloride, and 1.5% agar).
- plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme NcoI to confirm the inserted fragment.
- NcoI restriction enzyme
- an inserted fragment of about 1.7-kb in length was observed in the case of the Lactococcus lactis kivD gene (ligation AI solution).
- the plasmid containing the Lactococcus lactis kivD gene was named pCRB208-kivD / LL (FIG. 2).
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and LB containing zeocin 25 ⁇ g / ml It was applied to an agar medium (1% polypeptone, 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with the restriction enzyme BamHI to confirm the inserted fragment.
- the inserted fragment of about 2.0-kb in length is converted to the Pseudomonas putida strain-derived adh gene (ligation AK solution).
- Saccharomyces cerevisiae strain adh2 gene ligation AL solution
- an insertion fragment of about 2.1-kb in length was observed.
- the plasmid containing the adhP gene derived from the Escherichia coli strain was named pCRB15-adhP / EC
- the plasmid containing the Pseudomonas putida strain adh gene was named pCRB15-adh / PP
- the plasmid containing the adh2 gene derived from the Saccharomyces cerevisiae strain was named pCRB15-adh2 / SC. ( Figure 2).
- the above-described plasmid pCRB207-ilvD / CG was digested with restriction enzymes KpnI and PstI, and after agarose electrophoresis, the gapA promoter and the Corynebacterium glutamicum strain recovered from the agarose gel by the QIAquick Gel Extraction Kit (manufactured by Qiagen).
- Escherichia coli JM109 was transformed by the calcium chloride method [Journal of Molecular Biology, 53, 159 (1970)], and an LB agar medium containing zeocin 25 ⁇ g / ml [1% polypeptone, 0.5% Yeast extract, 0.5% sodium chloride, and 1.5% agar).
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- a restriction enzyme to confirm the inserted fragment.
- a plasmid containing the adhP gene derived from the Escherichia coli strain and the ilvD gene derived from the Corynebacterium glutamicum strain was named pCRB15-adhP / EC-ilvD / CG (FIG. 2).
- strain was transformed and applied to an A agar medium containing chloramphenicol 5 ⁇ g / ml, kanamycin 50 ⁇ g / ml, and zeocin 25 ⁇ g / ml.
- chloramphenicol 5 ⁇ g / ml kanamycin 50 ⁇ g / ml
- zeocin 25 ⁇ g / ml plasmids that can coexist in Corynebacterium glutamicum.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- introduction of the plasmids pCRB1-ilvB-ilvN-ilvC / CG-ilvD / CG, pCRB208-kivD / LL and pCRB15-adh2 / SC prepared above was confirmed.
- the resulting strain was named Corynebacterium glutamicum IBU1.
- the outline of gene recombination of this strain is summarized in Table 6.
- Corynebacterium glutamicum IBU1 has been deposited at the Patent Biological Depository Center of the National Institute of Technology and Evaluation, 2-5-8 Kazusa Kamashi, Kisarazu, Chiba, Japan (zip code 292-0818) Date: March 17, 2009, accession number: BP-718).
- the strain was transformed and applied to an A agar medium containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin, and 25 ⁇ g / ml zeocin.
- These three types of plasmids are plasmids that can coexist in Corynebacterium glutamicum.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- introduction of the plasmids pCRB1-ilvB-ilvN-ilvC / CG-ilvD / CG, pCRB208-kivD / LL and pCRB15-adhP / EC prepared above was confirmed.
- the resulting strain was named Corynebacterium glutamicum IBU2.
- the outline of gene recombination of this strain is summarized in Table 6.
- Corynebacterium glutamicum IBU2 has been deposited with the Patent Biological Depository Center of the National Institute of Technology and Evaluation, 2-5-8 Kazusa Kamashi, Kisarazu, Chiba, Japan (zip code 292-0818) Date: March 17, 2009, accession number: BP-719).
- the strain was transformed and applied to an A agar medium containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin, and 25 ⁇ g / ml zeocin.
- These three types of plasmids are plasmids that can coexist in Corynebacterium glutamicum.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- introduction of the plasmids pCRB1-ilvB-ilvN-ilvC / CG-ilvD / CG, pCRB208-kivD / LL and pCRB15-adh / PP prepared above was confirmed.
- the resulting strain was named Corynebacterium glutamicum IBU3. The outline of gene recombination of this strain is summarized in Table 6.
- Corynebacterium glutamicum IBU3 has been deposited with the Patent Biological Depository Center of the National Institute of Technology and Evaluation, 2-5-8 Kazusa Kamashi, Kisarazu City, Chiba, Japan (zip code 292-0818) Date: March 17, 2009, accession number: BP-720).
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- introduction of the plasmids pCRB205-alsS / BS-ilvC / CG, pCRB207-kivD / LL and pCRB15-adhP / EC-ilvD / CG prepared above was confirmed.
- the obtained strain was named Corynebacterium glutamicum IBU4. The outline of gene recombination of this strain is summarized in Table 6.
- Corynebacterium glutamicum IBU4 has been deposited with the Patent Biological Depository Center of the National Institute of Technology and Evaluation, 2-5-8 Kazusa Kamashi, Kisarazu, Chiba, Japan (zip code 292-0818) Date: March 17, 2009, accession number: BP-721).
- strain was transformed and applied to A agar medium containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin, and 25 ⁇ g / ml zeocin.
- plasmids that can coexist in Corynebacterium glutamicum.
- the growing strain on this medium was subjected to liquid culture by a conventional method, plasmid DNA was extracted from the culture solution, and the plasmid was cleaved with a restriction enzyme to confirm the inserted fragment.
- introduction of the plasmids pCRB1-ilvB-ilvN / EC-ilvC / EC-ilvD / EC, pCRB208-kivD / LL and pCRB15-adh2 / SC prepared above was confirmed.
- the resulting strain was named Corynebacterium glutamicum IBU5.
- Example 4 Corynebacterium glutamicum Isobutanol production experiment under reducing conditions using IBU1, IBU2 and IBU3 Corynebacterium glutamicum IBU1, IBU2 and IBU3, which were created in Example 3, were treated with chloramphenicol 5 ⁇ g / It was applied to an A agar medium containing ml, kanamycin 50 ⁇ g / ml and zeocin 25 ⁇ g / ml, and allowed to stand in the dark at 28 ° C. for 20 hours.
- Corynebacterium glutamicum IBU1, IBU2 and IBU3 grown on the above plate was placed in a test tube containing 10 ml of A liquid medium containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin and 25 ⁇ g / ml zeocin.
- the cells were aerobically shaken at 28 ° C. for 15 hours.
- Quantification of isobutanol was performed by centrifuging the sampled reaction solution (4 ° C., 15,000 ⁇ g, 10 minutes) and analyzing the resulting supernatant by gas chromatography.
- the gas chromatography analysis was performed using Shimadzu Gas Chromatography (GC-2014) in which a capillary column (Stabilwax; 60m ⁇ 0.53mmID ⁇ 1 ⁇ mID; RESTEK) was set. Analysis conditions are set at a carrier gas flow rate of 318 ml / min and a split ratio of 1:20.
- GC oven conditions are maintained at 40 ° C. for 5 minutes, then 40 to 120 ° C. at 20 ° C. / min, 120 ° C. to 220 ° C. The temperature was raised to 50 ° C. at 50 ° C./min and held at 220 ° C. for 4 minutes.
- Corynebacterium glutamicum IBU1 was 184 mM 22 hours after the start of the isobutanol production reaction under reducing conditions, 251 ⁇ ⁇ mM isobutanol 36 hours later, and Corynebacterium glutamicum IBU2 was reduced under reducing conditions. 22 hours after the start of the isobutanol formation reaction, 252 mM and 36 hours after the addition of 365 ⁇ ⁇ mM isobutanol, Corynebacterium glutamicum IBU3 is 255 MmM, 36 hours after the start of the isobutanol formation reaction under reducing conditions. Later, 370 mM isobutanol was produced in each reaction solution.
- Corynebacterium glutamicum IBU1, IBU2, and IBU3 share the same gene encoding AHAS, AHAIR, DHAD, and KDC. The only difference is that IBU1 is a Saccharomyces cerevisiae gene that encodes ADH.
- IBU2 Derived from adh2 (adh2 / SC) gene, IBU2 introduced an adhP (adhP / EC) gene derived from Escherichia coli, and IBU3 introduced an adh (adh / PP) gene derived from Pseudomonas sputida (see Table 6),
- AdhP / EC adhP
- IBU3 introduced an adh (adh / PP) gene derived from Pseudomonas sputida
- the ADH activity when using isobutyraldehyde as a substrate in Corynebacterium glutamicum showed higher values in the order of adh2 / SC2 / ⁇ adhP / EC ⁇ adh / PP (see Table 4). This difference in ADH activity is thought to reflect the difference in isobutanol productivity.
- Example 5 Isobutanol production experiment under reducing conditions using Corynebacterium glutamicum IBU4 Isobutanol production was evaluated in the same manner as in Example 4 except that Corynebacterium glutamicum IBU4 was used. As a result, Corynebacterium glutamicum IBU4 produced 97 mM isobutanol in the reaction solution 22 hours after the start of the isobutanol production reaction under reducing conditions, and 146 mM after 36 hours.
- Corynebacterium glutamicum IBU4 is common in that the same genes encoding AHAIR, DHAD, KDC, and ADH are introduced compared to IBU2. The only difference is that IBU4 is a gene encoding AHAS. Whereas the alsS gene derived from Bacillus subtilis has been introduced, IBU2 introduces the ilvBN (ilvB-ilvN) gene derived from Corynebacterium glutamicum (see Table 6). In this example, with regard to the gene encoding AHAS, when the ilvBN gene endogenous to Corynebacterium glutamicum was introduced, the productivity was higher than when the alsS gene derived from exogenous (from Bacillus subtilis) was introduced. It is an experimental example which shows giving.
- IBU2 introduces the ilvBN, ilvC, and ilvD genes derived from Corynebacterium glutamicum. That is, exogenous genes are all introduced into IBU5, whereas endogenous ilvBN, ilvC, and ilvD genes are introduced into IBU2. Therefore, IBU2 introduced with the ilvBN, ilvC, and ilvD genes endogenous to Corynebacterium glutamicum showed higher productivity than IBU5 introduced with the exogenous Escherichia coli ilvBN, ilvC, and ilvD genes.
- Corynebacterium glutamicum IBU2 grown on the above plate was inoculated into a test tube containing 10 ml of A liquid medium containing chloramphenicol 5 ⁇ g / ml, kanamycin 50 ⁇ g / ml and zeocin 25 ⁇ g / ml, and 28 The culture was aerobically shaken at 15 ° C. for 15 hours.
- Corynebacterium glutamicum IBU2 grown under the above conditions was prepared using A (-urea) liquid medium (350 mM (5.22%) glucose, 0.7% ammonium sulfate sulfate containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin and 25 ⁇ g / ml zeocin.
- a (-urea) liquid medium 350 mM (5.22%) glucose, 0.7% ammonium sulfate sulfate containing 5 ⁇ g / ml chloramphenicol, 50 ⁇ g / ml kanamycin and 25 ⁇ g / ml zeocin.
- the introduced plasmids used were pCRB1-ilvB-ilvN-ilvC / CG-ilvD / CG, pCRB208-kivD / LL and pCRB15-adhP / EC similar to Corynebacterium glutamicum IBU2.
- isobutanol can be efficiently produced from saccharides.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
遺伝子組換え菌によるイソブタノールの生産技術としては以下のような技術が知られている。例えば、特許文献1は、クレブジーラ ニューモニエ(Klebsiella pneumoniae)又はバチルス サブチリス(Bacillus subtilis)由来のアセトヒドロキシ酸 シンターゼ遺伝子、バチルス サブチリス、エシェリヒア コリ(Escherichia coli)又はサッカロマイセス セレビシエ(Saccharomyces cerevisiae)由来のアセトヒドロキシ酸 イソメロレダクターゼ遺伝子、エシェリヒア コリ又はサッカロマイセス セレビシエ由来のジヒドロキシ酸 デヒドラターゼ遺伝子、ラクトコッカス ラクティス(Lactococcus lactis)由来の2-ケト酸 デカルボキシラーゼ遺伝子、エシェリヒア コリ又はクロストリジウム アセトブチリカム(Clostridium acetobutylicum)由来のアルコール デヒドロゲナーゼ遺伝子を、宿主としてエシェリヒア コリ、サッカロマイセス セレビシエ、バチルス サブチリス、ラクトコッカス プランタラム(Lactococcus plantarum)又はエンテロコッカス フェカリス(Enterococcus faecalis)等を使用して発現させイソブタノールを生産する技術を開示している。
コリネバクテリウム グルタミカム中で機能する(1)アセトヒドロキシ酸 シンターゼ活性を有する酵素をコードする遺伝子、(2)アセトヒドロキシ酸 イソメロレダクターゼ活性を有する酵素をコードする遺伝子、(3) ジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードする遺伝子、(4) 2-ケト酸 デカルボキシラーゼ活性を有する酵素をコードする遺伝子、及び(5)アルコール デヒドロゲナーゼ活性を有する酵素をコードする遺伝子を有するコリネバクテリウム グルタミカム(Corynebacterium glutamicum)であって、(1)~(5)の1以上がコリネバクテリウム グルタミカムの内在性の遺伝子であり、(1)~(5)の1以上が外来性の遺伝子である形質転換体は、イソブタノールを効率よく生産する。
項1. 下記の(1)~(5)の遺伝子を有するコリネバクテリウム グルタミカム(Corynebacterium glutamicum)であって、(1)~(5)の1以上がコリネバクテリウム グルタミカムの内在性の遺伝子であり、(1)~(5)の1以上が外来性の遺伝子であることを特徴とする、イソブタノール生産能を有する形質転換体。
(1)アセトヒドロキシ酸 シンターゼ(acetohydroxy acid synthase)活性を有する酵素をコードする遺伝子
(2)アセトヒドロキシ酸 イソメロレダクターゼ(acetohydroxy acid isomeroreductase)活性を有する酵素をコードする遺伝子
(3)ジヒドロキシ酸 デヒドラターゼ(dihydroxy acid dehydratase)活性を有する酵素をコードする遺伝子
(4)2-ケト酸 デカルボキシラーゼ(2-keto acid decarboxylase)活性を有する酵素をコードする遺伝子
(5)アルコール デヒドロゲナーゼ(alcohol dehydrogenase)活性を有する酵素をコードする遺伝子
項2. 内在性の遺伝子が高発現されていることを特徴とする項1に記載の形質転換体。
項3. 内在性の遺伝子が(1)アセトヒドロキシ酸シンターゼ活性を有する酵素をコードする遺伝子、(2)アセトヒドロキシ酸イソメロレダクターゼ活性を有する酵素をコードする遺伝子、および(3)ジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードする遺伝子から選ばれる遺伝子を含むことを特徴とする項1又は2に記載の形質転換体。
アセトヒドロキシ酸 イソメロレダクターゼ活性を有する酵素をコードする遺伝子が、配列番号61の塩基配列からなるDNA、又は配列番号61の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつアセトヒドロキシ酸 イソメロレダクターゼ活性を有するポリペプチドをコードするDNAのいずれかかの遺伝子から選ばれる遺伝子であり、そして、
ジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードする遺伝子が、配列番号62の塩基配列からなるDNA、又は配列番号62の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつジヒドロキシ酸 デヒドラターゼ活性を有するポリペプチドをコードするDNAのいずれかの遺伝子から選ばれる遺伝子であり、そして、
2-ケト酸 デカルボキシラーゼ活性を有する酵素をコードする遺伝子が、配列番号43の塩基配列からなるDNA、配列番号52の塩基配列からなるDNA、あるいは、配列番号43又は52の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ2-ケト酸 デカルボキシラーゼ活性を有するポリペプチドをコードするDNAのいずれかの遺伝子から選ばれる遺伝子であり、そして、
アルコール デヒドロゲナーゼ活性を有する酵素をコードする遺伝子が、配列番号40の塩基配列からなるDNA、配列番号46の塩基配列からなるDNA、あるいは、配列番号40、又は46の塩基配列からなるDNAと相補的な塩基配列からなるDNAとスコリネバクテリウム グルタミカムトリンジェントな条件でハイブリダイズし、かつアルコール デヒドロゲナーゼ活性を有するポリペプチドをコードするDNAから選ばれる遺伝子であることを特徴とする項1~4のいずれかに記載の形質転換体。
項7. Corynebacterium glutamicumIBU1(受託番号 NITE BP-718)、Corynebacterium glutamicumIBU2(受託番号 NITE BP-719)、Corynebacterium glutamicumIBU3(受託番号 NITE BP-720)、又は、Corynebacterium glutamicumIBU4(受託番号 NITE BP-721)である形質転換体。
項9. 反応工程において、形質転換体が実質的に増殖しない項8に記載のイソブタノールの製造方法。
項10. 還元条件下の反応培地の酸化還元電位が-100~-500ミリボルトであることを特徴とする項8又は9に記載のイソブタノールの製造方法。
本発明により、再生可能資源を原料とした効率的なイソブタノール生産が可能となり、工業生産における合理的プロセスを実現することが出来る。
(I)イソブタノール生産能を有する形質転換体
本発明のイソブタノール生産能を有する形質転換体は、コリネバクテリウム グルタミカム中で機能する、前述した(1)~(5)の遺伝子を有するコリネバクテリウム グルタミカムであって、(1)~(5)の少なくとも1が内在性の遺伝子であり、かつ、(1)~(5)の少なくとも1が外来性の遺伝子である、イソブタノール生産能を有する形質転換体である。内在性の遺伝子は高発現していることが好ましい。即ち、宿主であるコリネバクテリウム グルタミカムが本来有する内在性の遺伝子は、さらにプラスミド等として導入されていることにより高発現していることが好ましい。
「コリネバクテリウム グルタミカム中で機能する」とは、DNAがコードする酵素がコリネバクテリウム グルタミカム中で発現し、触媒作用を呈することを意味する。
本発明において形質転換の対象となる宿主は、イソブタノール生産関連遺伝子群を含む組換えベクターにより形質転換され、これらの遺伝子がコードするイソブタノール生産に関与する酵素が発現し、結果としてイソブタノールを生産することができるコリネバクテリウム グルタミカムであれば特に限定されない。
宿主として用いられるコリネバクテリウム グルタミカムとは、バージーズ・マニュアル・デターミネイティブ・バクテリオロジー〔Bergey’s Manual of Determinative Bacteriology、Vol. 8、599(1974)〕に定義されている一群の微生物である。
具体的には、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R(FERM P-18976)、ATCC13032、ATCC13869、ATCC13058、ATCC13059、ATCC13060、ATCC13232、ATCC13286、ATCC13287、ATCC13655、ATCC13745、ATCC13746、ATCC13761、ATCC14020、ATCC31831、 MJ-233(FERM BP-1497)又はMJ-233AB-41(FERM BP-1498)等が挙げられる。
また、これらコリネバクテリウム グルタミカムは、野生株の他に、その変異株や人為的な遺伝子組換え体であってもよい。例えば、ラクテートデヒドロゲナーゼ(lactate dehydrogenase)、フォスフォエノールピルベートカルボキシラーゼ(phosphoenolpyrvate carboxylase)、マレートデヒドロゲナーゼ(malate dehydrogenase)などの遺伝子の1又は2以上を破壊した遺伝子破壊株が挙げられる。このような遺伝子破壊株を宿主として用いることにより、イソブタノールの生産性を向上させたり、副生成物の生成を抑制したりすることができる。
本明細書中、下記の(1)~(5)の遺伝子をイソブタノール生産関連遺伝子ともいう。
本遺伝子として、好ましいのは、配列番号34で表わされる塩基配列からなるDNA及び配列番号58で表わされる塩基配列からなるDNA、並びに、配列番号34又は58で表わされる塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつアセトヒドロキシ酸 シンターゼ活性を有する酵素をコードするDNAからなる群より選択される少なくとも1種のDNAである。
本遺伝子として、好ましいのは、配列番号61で表わされる塩基配列からなるDNA、並びに、配列番号61で表わされる塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつアセトヒドロキシ酸 イソメロレダクターゼ活性を有する酵素をコードするDNAからなる群より選択される少なくとも1種のDNAである。
本遺伝子として、好ましいのは、配列番号62で表わされる塩基配列からなるDNA、並びに、配列番号62で表わされる塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードするDNAからなる群より選択される少なくとも1種のDNAである。
本遺伝子として、好ましいのは、配列番号43で表わされる塩基配列からなるDNA及び配列番号52で表わされる塩基配列からなるDNA、並びに、配列番号43又は52で表わされる塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ2-ケト酸 デカルボキシラーゼ活性を有する酵素をコードするDNAからなる群より選択される少なくとも1種のDNAである。
本遺伝子として、好ましいのは、配列番号40で表わされる塩基配列からなるDNA、配列番号46で表わされる塩基配列からなるDNA、及び配列番号49で表わされる塩基配列からなるDNA、並びに、配列番号40、46又は49で表わされる塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつアルコール デヒドロゲナーゼ活性を有する酵素をコードするDNAからなる群より選択される少なくとも1種のDNAである。
イソブタノールは、酵母によるアルコール発酵時のfusel alcoholと呼ばれる副生物の一つとして見出される〔Hazelwood, L.A. et al., The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 74:2259-2266 (2008)〕。このfusel alcoholとは、イソブタノールの他、イソアミルアルコール、活性アミルアルコール等の高級アルコールの呼称であり、アミノ酸代謝系に由来することが1世紀前Enrlichにより初めて見出された。この代謝系はEnrlich経路と呼ばれ、バリン、ロイシン、イソロイシン、メチオニン、フェニールアラニンなどの生合成代謝中間体である2-ケト酸から、広い基質特異性を有する2-ケト酸 デカルボキシラーゼによりアルデヒドに変換し、さらにアルコール デヒドロゲナーゼによりアルコールに変換される。イソブタノールは、バリンへの生合成代謝中間体である2-ketoisovalerateに由来する〔Dickinson, J.R. et al., An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae. J. Biol. Chem. 273:25751-25756 (1998)〕。
さらに、これらの遺伝子は、イソブタノール生産能を有さない菌から取得してもよい。
中でも、(1)~(3)のステップに関する遺伝子としては、宿主であるコリネバクテリウム グルタミカム由来の遺伝子を用いると好ましい場合もある。
その他の代謝ステップ(4)、 (5)に関する具体的例として、以下があげられる。
従って、上記(1)~(5)の遺伝子の由来微生物の種類、組み合わせ、及び導入の順番等に関しては、これらの各種の遺伝子の内、少なくとも一つが宿主であるコリネバクテリウム グルタミカム由来のものを含む点を除いては、特段の限定があるものではない。
上記(4)においては、配列番号43で表わされる塩基配列からなるDNAが、ラクトコッカス ラクティス由来の遺伝子(kivD)であり、配列番号52で表わされる塩基配列からなるDNAが、スタフィロコッカス エピデルミディス由来の遺伝子(ipd)であることが好ましい。
本発明ではまた、上記記載の遺伝子(1)~(5)について、それぞれが相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズしたものであっても良い。
次いで、PCR法で得られた遺伝子を含むクローニングベクターを、微生物、例えばエシェリヒア コリ(Escherichia coli)JM109菌株などに導入し、該菌株を形質転換する。この形質転換された菌株を、ベクター中のマーカー遺伝子に対応した適当な抗生物質(例えばアンピシリン、クロラムフェニコールなど)を含む培地で培養し、培養物から菌体を回収する。回収された菌体からプラスミドDNAを抽出する。プラスミドDNAの抽出は、公知の技術によって行なうことができ、また市販のプラスミド抽出キットを用いて簡便に抽出することもできる。市販のプラスミド抽出キットとしては、キアクイックプラスミド精製キット(商品名:Qiaquick plasmid purification kit、キアゲン社製)などが挙げられる。この抽出されたプラスミドDNAの塩基配列を決定することにより、AHAS、AHAIR、DHAD、KDC、ADHをコードする遺伝子配列を確認することができる。DNAの塩基配列の決定は、公知の方法、例えばジオキシヌクレオチド酵素法などにより決定することができる。また、キャピラリー電気泳動システムを用いて、検出には多蛍光技術を使用して塩基配列を決定することもできる。また、DNAシーケンサー、例えばABI PRISM 3730xl DNA Analyzer(アプライドバイオシステム社製)などを使用して決定することもできる。
目的遺伝子を含むプラスミドベクターのコリネバクテリウム グルタミカムへの導入方法としては、公知の方法を制限無く使用できる。このような公知の方法として、例えば電気穿孔法、コンジュゲーション法などの公知の方法で行うことができる。具体的には、電気パルス法を、公知の方法 〔Kurusu, Y. et al., Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation. Agric. Biol. Chem. 54:443-447 (1990)〕 及び 〔Vertes A.A. et al., Presence of mrr- and mcr-like restriction systems in Coryneform bacteria. Res. Microbiol. 144:181-185 (1993)〕により行うことができる。
また、形質転換体は、紫外線、エックス線又は薬品等を用いる人工的な変異導入方法により変異しうるが、このように得られるどのような変異株であっても本発明の目的とするイソブタノール生産能を有するかぎり、本発明の形質転換された微生物として使用することができる。
かくして創製された本発明のコリネバクテリウム グルタミカムの形質転換体(以下、単に「形質転換体」という)は、微生物の培養に通常使用される培地を用いて培養すればよい。この培地としては、通常、炭素源、窒素源、無機塩類及びその他の栄養物質等を含有する天然培地又は合成培地等を用いることができる。
培地のpHは約5~8が好ましい。
培養温度は約15~45℃とすればよく、培養時間は約1~7日間とすればよい。
回収された培養菌体に対して処理を加え、得られる菌体処理物を次工程に用いてもよい。前記菌体処理物としては、培養菌体に何らかの処理が加えられたものであればよく、例えば、菌体をアクリルアミド又はカラギーナン等で固定化した固定化菌体等が挙げられる。
上記の如くして得られる培養物から回収分離された形質転換体の培養菌体又はその菌体処理物は、還元条件下の反応培地でのイソブタノール生成反応に供せられる。菌体処理物としては、菌体を例えばアクリルアミド、カラギーナン等で固定化した固定化菌体などが挙げられる。上記形質転換体、又は菌体処理物により、還元条件下で、糖類を含有する培地(反応培地)中でイソブタノール生成を行なわせる工程と、生成したイソブタノールを回収する工程とを含むイソブタノールの製造方法も本発明の1つである。
イソブタノール生成方式は、回分式、流加式、連続式のいずれの生成方式も可能である。
形質転換体又はその菌体処理物と糖類との反応は、本発明の形質転換体又はその菌体処理物が活動できる温度条件下で行なわれることが好ましく、形質転換体又はその菌体処理物の種類などにより適宜選択することができる。通常、約25~35℃とすればよい。
本発明のイソブタノール製造方法の特徴の一つは、還元条件下の糖類を含有する反応培地で使用される形質転換体の増殖を実質的に伴わないことである。反応培地中の形質転換体の増殖を伴わないでイソブタノールが生成されることは、経時的に適宜反応液を採取し、そのイソブタノール濃度と菌体濃度(例えば、菌体の光学濃度)を測定することにより知ることができる。
また、培養時間は、例えば約2~72時間とすればよい。
(1) 微生物からの染色体DNAの抽出
バチルス サブチリス168 NBRC14144からの染色体DNA抽出は、NBRC Medium No.802培地 [Polypepton 10g、Yeast extract 2g、MnSO4.7H2O 1gを蒸留水1Lに溶解] に、白金耳を用いて植菌後、対数増殖期まで37℃で振盪培養し、菌体を集菌後、DNAゲノム抽出キット(商品名:GenomicPrep Cells and Tissue DNA Isolation Kit、アマシャム社製)を用いて、取扱説明書に従い、集めた菌体から染色体DNAを回収した。
クローニングベクターpCRB22の構築
コリネバクテリウム カゼイ JCM12072由来のプラスミドpCASE1のDNA複製起点(以降、pCASE1-oriと記す)配列、及びクローニングベクターpHSG298(タカラバイオ株式会社製)をそれぞれ含むDNA断片を以下のPCR法により増幅した。
PCRに際して、pCASE1-ori配列、クローニングベクターpHSG298をそれぞれクローン化するべく、配列番号1(pCASE1-ori配列)、配列番号2(クローニングベクター-pHSG298)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-1); 5’- AT AGATCT AGAACGTCCGTAGGAGC -3’ (配列番号3)
(b-1); 5’- AT AGATCT GACTTGGTTACGATGGAC -3’ (配列番号4)
尚、プライマー(a-1)及び(b-1)には、BglII制限酵素部位が付加されている。
(a-2); 5’- AT AGATCT AGGTTTCCCGACTGGAAAG -3 (配列番号5)
(b-2); 5’- AT AGATCT CGTGCCAGCTGCATTAATGA -3’ (配列番号6)
尚、プライマー(a-2)及び(b-2)には、BglII制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) pCASE1-ori配列を増幅する場合はプライマー(a-1)と(b-1)の組み合わせ、クローニングベクターpHSG298を増幅する場合はプライマー(a-2)と(b-2)の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃
pCASE1-ori配列:150秒
クローニングベクターpHSG298:180秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションA液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、カナマイシン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
pCASE1-ori配列を含むクローニングベクターをpCRB22と命名した。
コリネバクテリウム グルタミカム内で複製可能なプラスミドpCG1 [(特開昭57-134500)] 由来のDNA複製起点(以降、pCG1-oriと記す)配列、及びクローニングベクターpHSG398(タカラバイオ株式会社製)をそれぞれ含むDNA断片を以下のPCR法により増幅した。
PCRに際して、pCG1-ori配列、クローニングベクターpHSG398をそれぞれクローン化するべく、配列番号7(pCG1-ori配列)、配列番号8(クローニングベクターpHSG398)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-3); 5’- AT AGATCT AGCATGGTCGTCACAGAG -3’ (配列番号9)
(b-3); 5’- AT AGATCT GGAACCGTTATCTGCCTATG -3’ (配列番号10)
尚、プライマー(a-3)及び(b-3)には、BglII制限酵素部位が付加されている。
(a-4); 5’- AT AGATCT GTCGAACGGAAGATCACTTC -3’ (配列番号11)
(b-4); 5’- AT AGATCT AGTTCCACTGAGCGTCAG -3’ (配列番号12)
尚、プライマー(a-4)及び(b-4)には、BglII制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) pCG1-ori配列を増幅する場合はプライマー(a-3)と(b-3)の組み合わせ、クローニングベクターpHSG398を増幅する場合はプライマー(a-4) と (b-4) の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃
pCG1-ori配列:120秒
クローニングベクターpHSG398:150秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションB液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
pCG1-ori配列を含むクローニングベクターをpCRB11と命名した。
クローニングベクターpCRB11を含むDNA断片及びpSELECT-zeo-mcs(インビトロジェン株式会社製)由来のゼオシン耐性遺伝子をそれぞれ含むDNA断片を以下のPCR法により増幅した。
PCRに際して、クローニングベクターpCRB11及びゼオシン耐性遺伝子をそれぞれクローン化するべく、配列番号13(pCRB11)及び配列番号14(ゼオシン耐性遺伝子)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-5); 5’- AT GATATC CGAAGTGATCTTCCGTTCGA -3’ (配列番号15)
(b-5); 5’- AT GATATC AAGGCAGTTATTGGTGCCCT -3’ (配列番号16)
尚、プライマー(a-5)及び(b-5)には、EcoRV制限酵素部位が付加されている。
(a-6); 5’- AT GATATC TAGCTTATCCTCAGTCCTGC -3’ (配列番号17)
(b-6); 5’- AT GATATC CCATCCACGCTGTTTTGACA -3’ (配列番号18)
尚、プライマー(a-6)及び(b-6)には、EcoRV制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) クローニングベクターpCRB11配列を増幅する場合はプライマー(a-5)と(b-5)の組み合わせ、ゼオシン耐性遺伝子を増幅する場合はプライマー(a-6) と (b-6) の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃
pCRB11配列:200秒
ゼオシン耐性遺伝子:45秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションC液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、ゼオシン25μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
ゼオシン耐性遺伝子を含むクローニングベクターをpCRB15と命名した。
クローニングベクターpCRC200〔Yasuda, K. et al., Analyses of the acetate-producing pathways in Corynebacterium glutamicum under oxygen-deprived conditions. Applied Microbiology and Biotechnology. 77:853-860 (2007)〕を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、pCRC200をクローン化するべく、配列番号19(pCRC200)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-7); 5’- CTCT ACTAGT GTCGAC GGATCC TTGTGTGGAATTGTGAGCGG -3’
(配列番号20)
(b-7); 5’- CTCT ACTAGT CATACGAGCCGGAAGCATAA -3’
(配列番号21)
尚、プライマー(a-7)には、SpeI、SalI及びBamHI制限酵素部位が、プライマー(b-7)には、SpeI制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) pCRC200遺伝子を増幅する場合はプライマー(a-7)と(b-7)の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃、300秒
以上を1サイクルとし、30サイクル行った。
上記のPCRにより増幅したpCRC200由来遺伝子を含む約5.0-kb DNA断片10μlを制限酵素SpeIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ(タカラバイオ株式会社製)1unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションD液とした。
得られたライゲーションD液を、塩化カルシウム法 〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
クローニングベクターpCRC200に制限酵素部位SpeI、SalI及びBamHIサイトを付加したクローニングベクターをpCRB205と命名した。
コリネバクテリウム グルタミカムR由来のグリセルアルデヒド3リン酸デヒドロゲナーゼ(glyceraldehyde-3-phosphate dehydrogenase)をコードするgapA遺伝子のプロモーター配列(以降、PgapAと記す)を含むDNA断片、及びクローニングベクターpKK223-3(ファルマシア社製)由来rrnBT1T2双方向ターミネーター配列(以降、ターミネーター配列と記す)を含むDNA断片をを以下の方法により増幅した。
PCRに際して、PgapA配列及びターミネーター配列をそれぞれクローン化するべく、配列番号22(PgapA配列)、配列番号23(ターミネーター配列)を基に、それぞれ下記の一対のプライマーを合成し、使用した。

(a-9); 5’- CTCT GCATGC CCATGG CTGTTTTGGCGGATGAGAGA -3’
(配列番号26)
(b-9); 5’- CTCT GCATGC TCATGA AAGAGTTTGTAGAAACGCAAAAAGG -3
(配列番号27)
尚、プライマー(a-9)には、SphI及びNcoI制限酵素部位が、プライマー(b-9)には、SphI及びBspHI制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) PgapA配列を増幅する場合はプライマー(a-8)と(b-8)の組み合わせ、ターミネーター配列を増幅する場合はプライマー(a-9) と (b-9) の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃
PgapA配列:45秒
ターミネーター配列:30秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションE液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、カナマイシン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
PgapA配列を含むクローニングベクターをpCRB206と命名した。
得られたライゲーションF液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、カナマイシン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
rrnBT1T2ターミネーター配列を含むクローニングベクターをpCRB207と命名した。
コリネバクテリウム グルタミカムR由来のL-乳酸デヒドロゲナーゼ(L-lactate dehydrogenase)をコードするldhA遺伝子のプロモーター配列(以降、PldhAと記す)を含むDNA断片を以下の方法により増幅した。
PCRに際して、PldhA配列をクローン化するべく、配列番号28(PldhA配列)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-10); 5’- CTCT GTCGAC CGGAACTAGCTCTGCAATGA -3’
(配列番号29)
(b-10); 5’- CTCT GTCGAC GGATCC CATATG CGATCCCACTTCCTGATTTC -3’
(配列番号30)
尚、プライマー(a-10)には、SalI制限酵素部位が、プライマー(b-10)には、SalI、BamHI及びNcoI制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) PldhA配列を増幅する場合はプライマー(a-10)と(b-10)の組み合わせ行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃、30秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションG液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、カナマイシン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
PldhA配列を含むクローニングベクターをpCRB208と命名した。
バチルス サブチリス由来のイソブタノール生産遺伝子のクローニング
バチルス サブチリス由来のアセトヒドロキシ酸 シンターゼをコードするalsS遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、alsS遺伝子をクローン化するべく、配列番号31(バチルス サブチリスalsS遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。
(a-11); 5’- CTCT CCATGG TGACAAAAGCAACAAAAGAACAAAAATCC -3’
(配列番号32)
(b-11); 5’- CTCT CCATGG AGATCT CTAGAGAGCTTTCGTTTTCATGAG -3’
(配列番号33)
尚、プライマー(a-11)には、NcoI制限酵素部位が、プライマー(b-11)には、NcoI及びBglII制限酵素部位が付加されている。
コリネバクテリウム グルタミカム由来のアセトヒドロキシ酸 シンターゼをコードするilvB-ilvN遺伝子、アセトヒドロキシ酸 イソメロレダクターゼをコードするilvC遺伝子、ジヒドロキシ酸 デヒドラターゼをコードするilvD遺伝子をそれぞれ含むDNA断片を以下のPCR法により増幅した。
(a-12); 5’- GA CCCGGG AGTAAAGGAGCCAGAAAGTCGTGAA -3’
(配列番号36)
(b-12); 5’- GA CCCGGG CCTGCAGG TGCCTTATGTACAAAGTGCACAGCA -3’
(配列番号37)
尚、プライマー(a-12)には、SmaI制限酵素部位が、プライマー(b-12)には、SmaI及びSse8387I制限酵素部位が付加されている。
(a-13); 5’- CTCT TCATGA TCCCACTTCGTTCAAAAGTC -3’
(配列番号38)
(b-13); 5’- CTCT TCATGA TTAGTCGACCTGACGGAC -3’
(配列番号39)
尚、プライマー(a-13)及び(b-13)には、BspHI制限酵素部位が付加されている。
エシェリヒア コリ由来のアルコール デヒドロゲナーゼをコードするadhP遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、adhP遺伝子をクローン化するべく、配列番号40(エシェリヒア コリadhP遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。
(a-14); 5’- CTCT CCATGG AGGCTGCAGTTGTTACGAAG -3’
(配列番号41)
(b-14); 5’- CTCT CCATGG AGATCT TTAGTGACGGAAATCAATCACCAT -3’
(配列番号42)
尚、プライマー(a-14)には、NcoI制限酵素部位が、プライマー(b-14)には、NcoI及びBglII制限酵素部位が付加されている。
ラクトコッカス ラクティス由来の2-ケト酸 デカルボキシラーゼをコードするkivD遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、kivD遺伝子をクローン化するべく、配列番号43(ラクトコッカス ラクティスkivD遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。

シュードモナス プチダ由来のアルコール デヒドロゲナーゼをコードするadh遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際してadh遺伝子をクローン化するべく、配列番号46(シュードモナス プチダadh遺伝子)を基に、それぞれ下記の一対のプライマーをアプライド・バイオシステムズ(Applied Biosystems) 社製「394 DNA/RNA シンセサイザー (synthesizer)」を用いて合成し、使用した。
(a-16); 5’- CTCT TCATGA AAGCTGCTGTCGTTGC -3’ (配列番号47)
(b-16); 5’- CTCT TCATGA CTCGAG TCAGCCTTCGAACTGTATCAC -3’
(配列番号48)
尚、プライマー(a-16)には、BspHI制限酵素部位が、プライマー(b-16)には、BspHI及びXhoI制限酵素部位が付加されている。
サッカロマイセス セレビシエ由来のアルコール デヒドロゲナーゼをコードするadh2遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際してadh2遺伝子をクローン化するべく、配列番号49(サッカロマイセス セレビシエadh2遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。

スタフィロコッカス エピデルミディス由来の2-ケト酸 デカルボキシラーゼ(別名;インドール-3-ピルベート デカルボキシラーゼ)をコードするipd遺伝子(locus tag; NP_765765)を含むDNA断片を以下のPCR法により増幅した。
PCRに際してipd遺伝子をクローン化するべく、配列番号52(スタフィロコッカス エピデルミディスipd遺伝子)を基に、それぞれ下記の一対のプライマーを合成し、使用した。



イソブタノール生産遺伝子のpKK223-3へのクローニング
上記(3)のPCRにより増幅したコリネバクテリウム グルタミカム株由来ilvB-ilvN-ilvC遺伝子を含む約3.7-kb DNA断片10μl及びtacプロモーターを含有するクローニングベクターpKK223-3(ファルマシア社製)2μlを各々制限酵素SmaIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションH液とした。
コリネバクテリウム グルタミカム株由来ilvB-ilvN-ilvC遺伝子を含むプラスミドをpKK223-3-ilvB-ilvN-ilvC/CGと命名した。
上述のプラスミドpKK223-3-ilvB-ilvN-ilvC/CGから、tacプロモーター及びコリネバクテリウム グルタミカムR由来ilvB-ilvN-ilvC遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、tacプロモーター及びコリネバクテリウム グルタミカムR由来ilvB-ilvN-ilvC遺伝子(配列番号55;Ptac-ilvB-ilvN-ilvC配列)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-19); 5’- AT GCAAGC TTCGGCTGTGCAGGTCGTAAAT -3’ (配列番号56)
(b-19); 5’- AC GCAAGC TTCGCTTATGTACAAAGTGCAC -3’ (配列番号57)
尚、プライマー(a-19)及び(b-19)には、HindIII制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。

*) Ptac-ilvB-ilvN-ilvC配列を増幅する場合はプライマー(a-19)と(b-19)の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃、240秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションI液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
コリネバクテリウム グルタミカム株由来ilvB-ilvN-ilvC遺伝子を含むプラスミドをpCRB1-ilvB-ilvN-ilvC/CG(図1)と命名した。
上記項(3)に示したPCRにより増幅したバチルス サブチリス株由来alsS遺伝子を含む約1.8-kb DNA断片、エシェリヒア コリ株由来adhP遺伝子を含む約1.0-kb DNA断片、ラクトコッカス ラクティス株由来kivD遺伝子を含む約1.7-kb DNA断片、サッカロマイセス セレビシエ株由来adh2遺伝子を含む約1.1-kb DNA断片及びスタフィロコッカス エピデルミディス株由来ipd遺伝子を含む約1.7-kb DNA断片 10μl及びPgapAプロモーターを含有するクローニングベクターpCRB207 2μlを各々制限酵素NcoIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションJ液、K液、L液、M液及びN液とした。
上述のプラスミド1種類pCRB1-ilvB-ilvN-ilvC/CGを用いて、電気パルス法 [Agric. Biol. Chem.、Vol.54、443-447(1990) 及び Res. Microbiol.、Vol.144、181-185(1993)] により、コリネバクテリウム グルタミカムR ldhA mutant [J. Mol. Microbiol. Biotechnol.、Vol. 8、243-254(2004)] を形質転換し、クロラムフェニコール5μg/mlを含むA寒天培地に塗布した。
実施例1において構築したイソイソブタノール生産遺伝子をそれぞれ単独で導入したコリネバクテリウム グルタミカムR ldhA mutant(以降、コリネバクテリウム グルタミカム単一遺伝子導入株と記す)(ただし、例外としてilvB-ilvN-ilvCは同一のプラスミド上に連結されている)におけるイソイソブタノール生産関連酵素、すなわちアセトヒドロキシ酸シンターゼ(AHAS)、アセトヒドロキシ酸イソメロレダクターゼ(AHAIR)、ジヒドロキシ酸デヒドラターゼ(DHAD)、2-ケト酸デカルボキシラーゼ(KDC)、アルコールデヒドロゲナーゼ(ADH)の活性を以下の方法により測定した。尚、コントロールとして、宿主として用いたコリネバクテリウム グルタミカムR ldhA mutantも同様の方法にて測定した。
コリネバクテリウム グルタミカム単一遺伝子導入株を、カナマイシン50μg/mlを含むA寒天培地[(NH2)2CO 2g、(NH4)2SO4 7g、KH2PO4 0.5 g、K2HPO4 0.5 g、MgSO4.7H2O 0.5 g、0.06% (w/v) Fe2 SO4.7H2O + 0.042% (w/v) MnSO4.2H2O 1 ml、0.02% (w/v) biotin solution 1 ml、0.01% (w/v) thiamin solution 2 ml、yeast extract 2 g、vitamin assay casamino acid 7 g、glucose 40 g、寒天 15 gを蒸留水1Lに懸濁] に塗布し、28℃、20時間暗所に静置した。




コリネバクテリウム グルタミカム単一遺伝子導入株を、カナマイシン 50μg/ml含むA寒天培地 [(NH2)2CO 2g、(NH4)2SO4 7g、KH2PO4 0.5 g、K2HPO4 0.5 g、MgSO4.7H2O 0.5 g、0.06% (w/v) Fe2 SO4.7H2O + 0.042% (w/v) MnSO4.2H2O 1 ml、0.02% (w/v) biotin solution 1 ml、0.01% (w/v) thiamin solution 2 ml、yeast extract 2 g、vitamin assay casamino acid 7 g、glucose 40 g、寒天 15 gを蒸留水1Lに懸濁] に塗布し、28℃、20時間暗所に静置した。

(1) ブタノール生産遺伝子のクローニング
バチルス サブチリス由来のイソブタノール生産遺伝子のクローニング
バチルス サブチリス由来のアセトヒドロキシ酸 シンターゼをコードするalsS遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、alsS遺伝子をクローン化するべく、バチルス サブチリスの配列(配列番号58;alsS遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。
(a-20);5’-CTCT CCCGGG AAACTTTTTAGAAAGGTGTGTTTCACCCGTGTTGACAAAAGCAACAAAAGAAC -3’
(配列番号59)
(b-20);5’-CTCT CCCGGG AGATCT CTAGAGAGCTTTCGTTTTCATGA -3’
(配列番号60)
コリネバクテリウム グルタミカム由来のアセトヒドロキシ酸 シンターゼをコードするilvB-ilvN遺伝子、アセトヒドロキシ酸 イソメロレダクターゼをコードするilvC遺伝子、ジヒドロキシ酸 デヒドラターゼをコードするilvD遺伝子をそれぞれ含むDNA断片を以下のPCR法により増幅した。
(a-12); 5’- GA CCCGGG AGTAAAGGAGCCAGAAAGTCGTGAA -3’
(配列番号36)
(b-12); 5’- GA CCCGGG CCTGCAGG TGCCTTATGTACAAAGTGCACAGCA -3’
(配列番号37)
尚、プライマー(a-12)には、SmaI制限酵素部位が、プライマー(b-12)には、SmaI及びSse8387I制限酵素部位が付加されている。
(a-21); 5’- CTCT CCATGG CTATTGAACTGCTTTATGATG -3’
(配列番号63)
(b-21); 5’- CTCT CCATGG AGATCT TTAAGCGGTTTCTGCGCGA -3’
(配列番号64)
尚、プライマー(a-21)には、NcoI制限酵素部位が、プライマー(b-21)には、NcoI及びBglII制限酵素部位が付加されている。
(a-22); 5’- GA CCCGGG GAGCAGATTTGAAAAGCGCATCATG -3’
(配列番号65)
(b-22); 5’- GA CCCGGG GGTACC GTATTTGCAACGGGGAGCTCCACCA -3’
(配列番号66)
尚、プライマー(a-22)には、SmaI制限酵素部位が、プライマー(b-22)には、SmaI及びKpnI制限酵素部位が付加されている。
エシェリヒア コリ由来のアセトヒドロキシ酸 シンターゼをコードするilvB-ilvN遺伝子、アセトヒドロキシ酸 イソメロレダクターゼをコードするilvC遺伝子、ジヒドロキシ酸 デヒドラターゼをコードするilvD遺伝子及びアルコールデヒドロゲナーゼをコードするadhP遺伝子を含むDNA断片を以下のPCR法により増幅した。
(a-23); 5’- CTCT CCCGGG ATGGCAAGTTCGGGCACAA -3’
(配列番号70)
(b-23); 5’- CTCT CCCGGG AGATCT TTACTGAAAAAACACCGCGATCTT-3’
(配列番号71)
尚、プライマー(a-23)には、SmaI制限酵素部位が、プライマー(b-23)には、SmaI及びBglII制限酵素部位が付加されている。
(a-24); 5’- CTCT CCCGGG ATGGCTAACTACTTCAATACACTG -3’
(配列番号72)
(b-24); 5’- CTCT CCCGGG AGATCT TTAACCCGCAACAGCAATACG-3’
(配列番号73)
尚、プライマー(a-24)には、SmaI制限酵素部位が、プライマー(b-24)には、SmaI及びBglII制限酵素部位が付加されている。
(a-25); 5’- CTCT TTTAAA ATGCCTAAGTACCGTTCCG -3’
(配列番号74)
(b-25); 5’- CTCT TTTAAA AGATCT TTAACCCCCCAGTTTCGATTTAT-3’
(配列番号75)
尚、プライマー(a-25)には、DraI制限酵素部位が、プライマー(b-25)には、DraI及びBglII制限酵素部位が付加されている。
(a-14); 5’- CTCT CCATGG AGGCTGCAGTTGTTACGAAG -3’
(配列番号41)
(b-14); 5’- CTCT CCATGG AGATCT TTAGTGACGGAAATCAATCACCAT -3’
(配列番号42)
尚、プライマー(a-14)には、NcoI制限酵素部位が、プライマー(b-14)には、NcoI及びBglII制限酵素部位が付加されている。
ラクトコッカス ラクティス由来の2-ケト酸 デカルボキシラーゼをコードするkivD遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際して、kivD遺伝子をクローン化するべく、ラクトコッカス ラクティスの配列(配列番号43;kivD遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。

シュードモナス プチダ由来のアルコール デヒドロゲナーゼをコードするadh遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際してadh遺伝子をクローン化するべく、シュードモナス プチダの配列(配列番号46;adh遺伝子)を基に、それぞれ下記の一対のプライマーをアプライド・バイオシステムズ(Applied Biosystems) 社製「394 DNA/RNA シンセサイザー (synthesizer)」を用いて合成し、使用した。
(a-16); 5’- CTCT TCATGA AAGCTGCTGTCGTTGC -3’ (配列番号47)
(b-16); 5’- CTCT TCATGA CTCGAG TCAGCCTTCGAACTGTATCAC -3’
(配列番号48)
尚、プライマー(a-16)には、BspHI制限酵素部位が、プライマー(b-16)には、BspHI及びXhoI制限酵素部位が付加されている。
サッカロマイセス セレビシエ由来のアルコール デヒドロゲナーゼをコードするadh2遺伝子を含むDNA断片を以下のPCR法により増幅した。
PCRに際してadh2遺伝子をクローン化するべく、サッカロマイセス セレビシエの配列(配列番号49;adh2遺伝子)を基に、それぞれ下記の一対のプライマーを、アプライド・バイオシステムズ(Applied Biosystems)社製「394 DNA/RNAシンセサイザー(synthesizer)」を用いて合成し、使用した。



イソブタノール生産遺伝子のpKK223-3へのクローニング
上記(1)のPCRにより増幅したコリネバクテリウム グルタミカム株由来ilvB-ilvN-ilvC遺伝子を含む約3.7-kb DNA断片、コリネバクテリウム グルタミカム株由来ilvD遺伝子を含む約2.0-kb DNA断片、エシェリヒア コリ株由来ilvB-ilvN遺伝子を含む約2.0-kb DNA断片及びエシェリヒア コリ株由来ilvC遺伝子を含む約1.5-kb DNA断片10μl及びtacプロモーターを含有するクローニングベクターpKK223-3(ファルマシア社製)2μlを各々制限酵素SmaIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションQ液、R液、S液及びT液とした。
得られた5種のライゲーションQ液、R液、S液、T液及びU液それぞれを、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、アンピシリン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
上述のプラスミドpKK223-3-ilvB-ilbN-ilvC/CG及びpKK223-3-ilvD/CGから、tacプロモーターを含むコリネバクテリウム グルタミカムR由来ilvB-ilvN-ilvC遺伝子及びilvD遺伝子のDNA断片を以下のPCR法により増幅した。
PCRに際して、tacプロモーター及びコリネバクテリウム グルタミカムR由来ilvB-ilvN-ilvC遺伝子(配列番号55;Ptac-ilvB-ilvN-ilvC配列)、ilvD遺伝子(配列番号76;Ptac-ilvD配列)を基に、それぞれ下記の一対のプライマーを合成し、使用した。
(a-19); 5’- AT GCAAGC TTCGGCTGTGCAGGTCGTAAAT -3’(配列番号56)
(b-19); 5’- AC GCAAGC TTCGCTTATGTACAAAGTGCAC -3’(配列番号57)
尚、プライマー(a-19)及び(b-19)には、HindIII制限酵素部位が付加されている。
(a-26); 5’- ATAT CCTGCAGG CTAGCGCTGTGCAGGTCGTAAATCAACT -3’
(配列番号77)
(b-26); 5’- ATATGCTAGCT CCTGCAGG TATTTGCAACGGGGAGCTC -3’
(配列番号78)
尚、プライマー(a-26)及び(b-26)には、Sse8387I制限酵素部位が付加されている。
実際のPCRは、サーマルサイクラー GeneAmp PCR System 9700(アプライド・バイオシステムズ社製)を用い、反応試薬としてTaKaRa LA Taq(タカラバイオ株式会社製)を用いて下記の条件で行った。
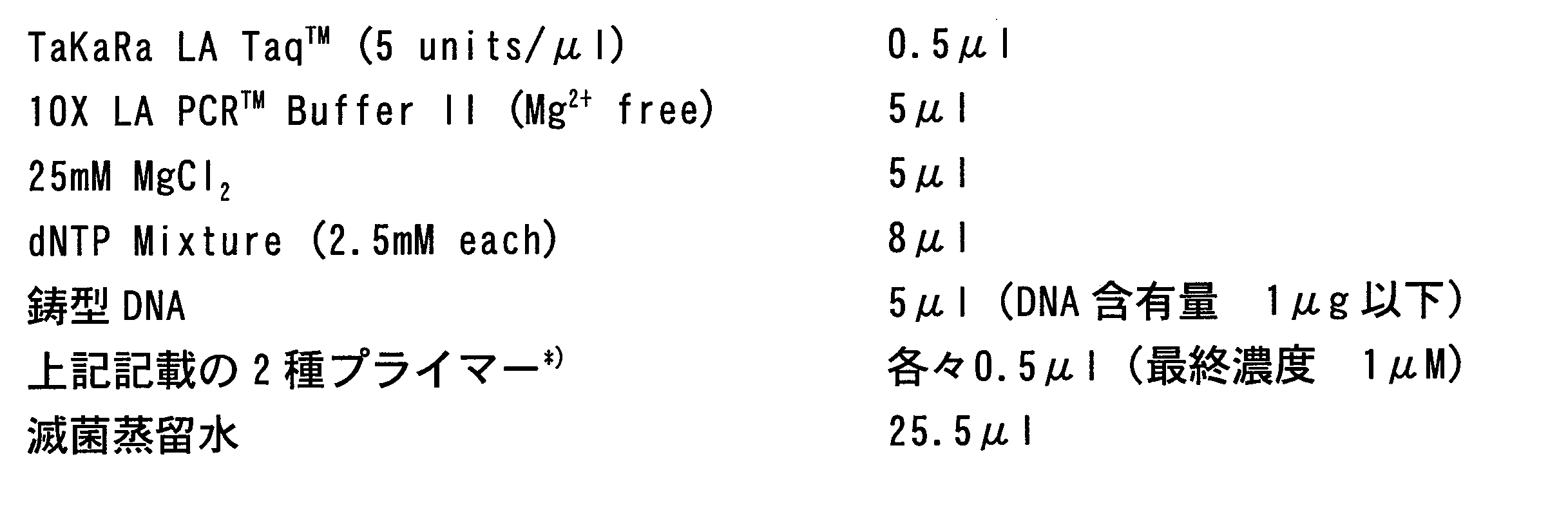
*) Ptac-ilvB-ilvN-ilvC配列を増幅する場合はプライマー(a-19)と(b-19)の組み合わせ、Ptac-ilvD配列を増幅する場合はプライマー(a-26)と(b-26)の組み合わせで行った。
デナチュレーション過程 :94℃、60秒
アニーリング過程 :52℃、60秒
エクステンション過程 :72℃
Ptac-ilvB-ilvN-ilvC配列:240秒
Ptac-ilvD配列:150秒
以上を1サイクルとし、30サイクル行った。
得られたライゲーションV液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
コリネバクテリウム グルタミカム株由来ilvB-ilvN-ilvC遺伝子を含むプラスミドをpCRB1-ilvB-ilvN-ilvC/CGと命名した。
エシェリヒア コリ株由来ilvB-ilvN遺伝子を含むプラスミドをpCRB1-ilvB-ilvN/ECと命名した。
尚、このプラスミドは、制限酵素部位BamHIが1箇所のみ存在する。
得られたライゲーションY液を用い、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール 50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
エシェリヒア コリ株由来ilvB-ilvN遺伝子及びエシェリヒア コリ株由来ilvC遺伝子を含むプラスミドをpCRB1-ilvB-ilvN/EC-ilvC/ECと命名した。
尚、このプラスミドは、制限酵素部位BamHIが1箇所のみ存在する。
得られたライゲーションZ液を用い、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール 50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
エシェリヒア コリ株由来ilvB-ilvN遺伝子、エシェリヒア コリ株由来ilvC遺伝子及びエシェリヒア コリ株由来ilvD遺伝子を含むプラスミドをpCRB1-ilvB-ilvN/EC-ilvC/EC-ilvD/ECと命名した。
尚、このプラスミドは、制限酵素部位BamHIが1箇所のみ存在する。
上記項(1)に示したPCRにより増幅したコリネバクテリウム グルタミカム株由来ilvC遺伝子を含む約1.0-kb DNA断片、エシェリヒア コリ株由来adhP遺伝子を含む約1.0-kb DNA断片、ラクトコッカス ラクティス株由来kivD遺伝子を含む約1.7-kb DNA断片、サッカロマイセス セレビシエ株由来adh2遺伝子を含む約1.1-kb DNA断片10μl及びPgapAプロモーターを含有するクローニングベクターpCRB207 2μlを各々制限酵素NcoIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションAA液、AB液、AC液及びAD液とした。
上記項(1)に示したPCRにより増幅したバチルス サブチリス株由来alsS遺伝子を含む約1.8-kb DNA断片10μl及びtacプロモーターを含有するクローニングベクターpCRB205 2μlを各々制限酵素SmaIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションAG液とした。
得られたライゲーションAG液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
バチルス サブチリス株由来alsS遺伝子を含むプラスミドをpCRB205-alsS/BSと命名した。
得られたライゲーションAH液を用い、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、クロラムフェニコール 50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
バチルス サブチリス株由来alsS遺伝子及びコリネバクテリウム グルタミカム株由来ilvC遺伝子を含むプラスミドをpCRB205-alsS/BS-ilvC/CGと命名した(図2)。
上記項(1)に示したPCRにより増幅したラクトコッカス ラクティスkivD遺伝子を含む約1.7-kb DNA断片10μl及びldhAプロモーターを含有するクローニングベクターpCRB208 2μlを各々制限酵素NcoIで切断し、70℃で10分処理させることにより制限酵素を失活させた後、両者を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ (タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションAI液とした。
得られたライゲーションAI液を、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、カナマイシン50μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
ラクトコッカス ラクティスkivD遺伝子を含むプラスミドをpCRB208-kivD/LLと命名した(図2)。
上述のプラスミドpCRB207-adhP/EC、pCRB207-adh/PP及びpCRB207-adh2/SCを制限酵素BamHIで切断し、アガロース電気泳動後、アガロースゲルからQIAquick Gel Extraction Kit(株式会社キアゲン社製)によって回収したgapAプロモーターとエシェリヒア コリ株由来adhP遺伝子及びターミネーター配列を連結した約2.0-kbのDNA断片と、gapAプロモーターとシュードモナス プチダ株由来adh遺伝子及びターミネーター配列を連結した約2.0-kbのDNA断片と、gapAプロモーターとサッカロマイセス セレビシエ株由来adh2遺伝子及びターミネーター配列を連結した約2.1-kbのDNA断片とBamHIで切断した上述のプラスミドpCRB15約3.8-kbを70℃で10分処理させることにより制限酵素を失活させたDNA断片を混合し、これにT4 DNAリガーゼ10×緩衝液 1μl 、T4 DNAリガーゼ(タカラバイオ株式会社製) 1 unitの各成分を添加し、滅菌蒸留水で10μl にして、15℃で3時間反応させ、結合させた。これをライゲーションAJ液、AK液及びAL液とした。
得られたライゲーションAM液を用い、塩化カルシウム法〔Journal of Molecular Biology, 53, 159 (1970)〕によりエシェリヒア コリJM109を形質転換し、ゼオシン 25μg/mlを含むLB寒天培地〔1% ポリペプトン、0.5% 酵母エキス、0.5% 塩化ナトリウム、および1.5% 寒天〕に塗布した。
エシェリヒア コリ株由来adhP遺伝子及びコリネバクテリウム グルタミカム株由来ilvD遺伝子を含むプラスミドをpCRB15-adhP/EC-ilvD/CGと命名した(図2)。
上述のプラスミドpCRB1-ilvB-ilvN-ilvC/CG-ilvD/CG、pCRB208-kivD/LL及びpCRB15-adh2/SCを用いて、電気パルス法 [Agric. Biol. Chem.、Vol. 54、443-447(1990) 及びRes. Microbiol.、Vol. 144、181-185(1993)] により、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R ldhA mutant [J. Mol. Microbiol. Biotechnol.、Vol. 8、243-254(2004)] 株を形質転換し、クロラムフェニコール 5μg/ml、カナマイシン 50μg/ml、ゼオシン25μg/mlを含むA寒天培地に塗布した。尚、これら3種類のプラスミドは、コリネバクテリウム グルタミカム内で共存可能なプラスミドである。

<遺伝子起源略語>
BS; バチルス サブチリス(Bacillus subtilis)
CG; コリネバクテリウム グルタミカム R(Corynebacterium glutamicum)
EC; エシェリヒア コリ(Escherichia coli)
LL; ラクトコッカス ラクティス(Lactococcus lactis)
PP; シュードモナス プチダ(Pseudomonas putida)
SC; サッカロマイセス セレビシエ(Saccharomyces cerevisiae)
上述のプラスミドpCRB1-ilvB-ilvN/EC-ilvC/EC-ilvD/EC、pCRB208-kivD/LL及びpCRB15-adh2/SCを用いて、電気パルス法 [Agric. Biol. Chem.、Vol. 54、443-447(1990) 及びRes. Microbiol.、Vol. 144、181-185(1993)] により、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R ldhA mutant [J. Mol. Microbiol. Biotechnol.、Vol. 8、243-254(2004)] 株を形質転換し、クロラムフェニコール 5μg/ml、カナマイシン 50μg/ml、ゼオシン25μg/mlを含むA寒天培地に塗布した。尚、これら3種類のプラスミドは、コリネバクテリウム グルタミカム内で共存可能なプラスミドである。
実施例3で創製したコリネバクテリウム グルタミカムIBU1、IBU2及びIBU3を、クロラムフェニコール 5μg/ml、カナマイシン 50μg/mlおよびゼオシン25μg/mlを含むA寒天培地に塗布し、28℃、20時間暗所に静置した。
上記のプレートで生育したコリネバクテリウム グルタミカム IBU1、IBU2及びIBU3を、クロラムフェニコール 5μg/ml、カナマイシン 50μg/mlおよびゼオシン25μg/mlを含むA液体培地10mlの入った試験管に一白金耳植菌し、28℃にて15時間、好気的に振盪培養を行った。
上記条件で生育したコリネバクテリウム グルタミカムIBU1、IBU2及びIBU3を、クロラムフェニコール 5μg/ml、カナマイシン 50μg/mlおよびゼオシン25μg/mlを含むA液体培地500mlの入った容量2Lの三角フラスコに植菌し、28℃にて15時間、好気的に振盪培養を行った。
コリネバクテリウム グルタミカム (Corynebacterium glutamicum) IBU4を用いた還元条件下におけるイソブタノール生産実験
コリネバクテリウム グルタミカム IBU4を用いた以外は、実施例4と同様の方法でイソブタノール生産を評価した。
この結果、コリネバクテリウム グルタミカムIBU4は、還元条件下でのイソブタノール生成反応の開始から22時間後に97 mM、36時間後に146 mMのイソブタノールを反応液中にそれぞれ生産していた。
本実施例は、AHASをコードする遺伝子に関して、コリネバクテリウム グルタミカム内在性のilvBN遺伝子を導入した場合の方が、外来性(バチルス サブチリス由来)のalsS遺伝子を導入した場合よりも、高い生産性を与えることを示す実験例である。
コリネバクテリウム グルタミカム IBU5を用いた以外は、実施例4と同様の方法でイソブタノール生産を評価した。
この結果、コリネバクテリウム グルタミカムIBU5は、還元条件下でのイソブタノール生成反応の開始から22時間後に72 mM、36時間後に97 mMのイソブタノールを反応液中にそれぞれ生産していた。
コリネバクテリウム グルタミカムIBU5は、IBU2と比較して、KDC、ADHをコードする同じ遺伝子が導入されていることは共通であり、異なる点は、AHAS、AHAIR、DHADをコードする遺伝子として、IBU5がエシェリヒア コリ由来のilvBN、ilvC、ilvD遺伝子が導入されているのに対して、IBU2はコリネバクテリウム グルタミカム由来のilvBN、ilvC、ilvD遺伝子を導入させている点である。すなわち、IBU5はすべて外来性遺伝子が導入されているのに対して、IBU2は、内在性のilvBN、ilvC、ilvD遺伝子が導入されている。従って、コリネバクテリウム グルタミカム内在性のilvBN、ilvC、ilvD遺伝子を導入したIBU2の方が、外来性エシェリヒア コリ由来のilvBN、ilvC、ilvD遺伝子を導入したIBU5に比べて高い生産性を示した。
実施例3で創製したコリネバクテリウム グルタミカムIBU2を、クロラムフェニコール 5μg/ml、カナマイシン 50μg/mlおよびゼオシン25μg/mlを含むA寒天培地に塗布し、28℃、20時間暗所に静置した。
上記のプレートで生育したコリネバクテリウム グルタミカムIBU2を、クロラムフェニコール 5μg/ml、カナマイシン 50μg/mlおよびゼオシン25μg/mlを含むA液体培地10mlの入った試験管に一白金耳植菌し、28℃にて15時間、好気的に振盪培養を行った。
この結果、コリネバクテリウム グルタミカムIBU2は、図3に示すように増殖した。この時、好気培養開始から4時間後に0 mM、10時間後に0 mM、16時間後に0.3 mMのイソブタノールを生産した。そして、16時間以降では、イソブタノールの生成濃度の増大は殆ど認められなっかた。コリネバクテリウム グルタミカムIBU2は、増殖を伴う好気条件下ではほとんどイソブタノールを生産しなかったのに対して、増殖を伴わない還元条件下(実施例4参照)では、著量のイソブタノールを生産することが分かった。
イソブタノールを生産する宿主としてコリネバクテリウム グルタミカム(Corynebacterium glutamicum)に代わりに、コリネバクテリウム キャピトビス(Corynebacterium capitovis)、コリネバクテリウム カゼイ(Corynebacterium casei)、コリネバクテリウム ハロトレランス(Corynebacterium halotolerans)及びコリネバクテリウム テルペノタビディウム(Corynebacterium terpenotabidum)を用いる以外は、実施例4と同様の方法でイソブタノール生産を評価した。尚、導入プラスミドは、コリネバクテリウム グルタミカム IBU2と同様のpCRB1-ilvB-ilvN-ilvC/CG-ilvD/CG、pCRB208-kivD/LL及びpCRB15-adhP/ECを用いた。
このイソブタノールを生産する場合の宿主として、コリネバクテリウム属の中で、コリネバクテリウム グルタミカムがもっとも優れていることが明らかになった。
Corynebacterium glutamicum IBU2 NITE BP-719
Corynebacterium glutamicum IBU3 NITE BP-720
Corynebacterium glutamicum IBU4 NITE BP-721
Claims (10)
- 下記の(1)~(5)の遺伝子を有するコリネバクテリウム グルタミカム(Corynebacterium glutamicum)であって、(1)~(5)の1以上がコリネバクテリウム グルタミカムの内在性の遺伝子であり、(1)~(5)の1以上が外来性の遺伝子であることを特徴とする、イソブタノール生産能を有する形質転換体。
(1)アセトヒドロキシ酸 シンターゼ(acetohydroxy acid synthase)活性を有する酵素をコードする遺伝子
(2)アセトヒドロキシ酸 イソメロレダクターゼ(acetohydroxy acid isomeroreductase)活性を有する酵素をコードする遺伝子
(3)ジヒドロキシ酸 デヒドラターゼ(dihydroxy acid dehydratase)活性を有する酵素をコードする遺伝子
(4)2-ケト酸 デカルボキシラーゼ(2-keto acid decarboxylase)活性を有する酵素をコードする遺伝子
(5)アルコール デヒドロゲナーゼ(alcohol dehydrogenase)活性を有する酵素をコードする遺伝子 - 内在性の遺伝子が高発現されていることを特徴とする請求項1に記載の形質転換体。
- 内在性の遺伝子が(1)アセトヒドロキシ酸シンターゼ活性を有する酵素をコードする遺伝子、(2)アセトヒドロキシ酸イソメロレダクターゼ活性を有する酵素をコードする遺伝子、および(3)ジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードする遺伝子から選ばれる遺伝子を含むことを特徴とする請求項1に記載の形質転換体。
- アルコール デヒドロゲナーゼ活性を有する酵素をコードする遺伝子が、エシェリヒア コリ(Escherichia coli)由来の遺伝子、または、シュードモナス プチダ(Pseudomonas putida) 由来の遺伝子であることを特徴とする請求項1に記載の形質転換体。
- アセトヒドロキシ酸 シンターゼ活性を有する酵素をコードする遺伝子が、配列番号34の塩基配列からなるDNA、配列番号58の塩基配列からなるDNA、あるいは、配列番号34又は58の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつアセトヒドロキシ酸 シンターゼ活性を有するポリペプチドをコードするDNAのいずれかの遺伝子から選ばれる遺伝子であり、そして、
アセトヒドロキシ酸 イソメロレダクターゼ活性を有する酵素をコードする遺伝子が、配列番号61の塩基配列からなるDNA、又は配列番号61の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつアセトヒドロキシ酸 イソメロレダクターゼ活性を有するポリペプチドをコードするDNAのいずれかかの遺伝子から選ばれる遺伝子であり、そして、
ジヒドロキシ酸 デヒドラターゼ活性を有する酵素をコードする遺伝子が、配列番号62の塩基配列からなるDNA、又は配列番号62の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつジヒドロキシ酸 デヒドラターゼ活性を有するポリペプチドをコードするDNAのいずれかの遺伝子から選ばれる遺伝子であり、そして、
2-ケト酸 デカルボキシラーゼ活性を有する酵素をコードする遺伝子が、配列番号43の塩基配列からなるDNA、配列番号52の塩基配列からなるDNA、あるいは、配列番号43又は52の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつ2-ケト酸 デカルボキシラーゼ活性を有するポリペプチドをコードするDNAのいずれかの遺伝子から選ばれる遺伝子であり、そして、
アルコール デヒドロゲナーゼ活性を有する酵素をコードする遺伝子が、配列番号40の塩基配列からなるDNA、配列番号46の塩基配列からなるDNA、あるいは、配列番号40、又は46の塩基配列からなるDNAと相補的な塩基配列からなるDNAとスコリネバクテリウム グルタミカムトリンジェントな条件でハイブリダイズし、かつアルコール デヒドロゲナーゼ活性を有するポリペプチドをコードするDNAから選ばれる遺伝子であることを特徴とする請求項1に記載の形質転換体。 - 宿主として用いられるコリネバクテリウム グルタミカムが、コリネバクテリウム グルタミカムR(FERM P-18976)、ATCC13032、又はATCC13869である請求項1に記載の形質転換体。
- Corynebacterium glutamicumIBU1(受託番号 NITE BP-718)、Corynebacterium glutamicumIBU2(受託番号 NITE BP-719)、Corynebacterium glutamicumIBU3(受託番号 NITE BP-720)、又は、Corynebacterium glutamicumIBU4(受託番号 NITE BP-721)である形質転換体。
- 請求項1又は7に記載の形質転換体、又はその処理物を、還元条件下の、糖類を含有する反応培地で反応させる工程と、生産されたイソブタノールを回収する工程とを含むことを特徴とするイソブタノールの製造方法。
- 反応工程において、形質転換体が実質的に増殖しない請求項8に記載のイソブタノールの製造方法。
- 還元条件下の反応培地の酸化還元電位が-100~-500ミリボルトであることを特徴とする請求項8に記載のイソブタノールの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011507169A JP5698655B2 (ja) | 2009-03-30 | 2010-03-29 | コリネ型細菌形質転換体及びそれを用いるイソブタノールの製造方法 |
| CA2755310A CA2755310A1 (en) | 2009-03-30 | 2010-03-29 | Coryneform bacterium transformant and process for producing isobutanol using the same |
| US13/258,670 US8871478B2 (en) | 2009-03-30 | 2010-03-29 | Coryneform bacterium transformant and process for producing isobutanol using the same |
| EP10758603A EP2415860A4 (en) | 2009-03-30 | 2010-03-29 | TRANSFORMANT CORYNEFORMER BACTERIA AND PROCESS FOR ISOBUTANOL PRODUCTION THEREWITH |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-083668 | 2009-03-30 | ||
| JP2009083668 | 2009-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010113832A1 true WO2010113832A1 (ja) | 2010-10-07 |
Family
ID=42828122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/055504 Ceased WO2010113832A1 (ja) | 2009-03-30 | 2010-03-29 | コリネ型細菌形質転換体及びそれを用いるイソブタノールの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8871478B2 (ja) |
| EP (1) | EP2415860A4 (ja) |
| JP (1) | JP5698655B2 (ja) |
| CA (1) | CA2755310A1 (ja) |
| WO (1) | WO2010113832A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013005764A (ja) * | 2011-06-24 | 2013-01-10 | National Institute Of Advanced Industrial Science & Technology | 大腸菌において混合糖から有機物質を生産する方法 |
| KR20130139258A (ko) * | 2010-11-10 | 2013-12-20 | 그린 페놀 고키노 페놀 주시 세이조 기쥬쓰 겐큐 구미아이 | 코리네형 세균 형질 전환체 및 그것을 사용하는 페놀의 제조 방법 |
| EP2749638A4 (en) * | 2011-08-22 | 2015-04-22 | Res Inst Innovative Tech Earth | MEANS FOR TRANSFORMING CORYNEFORMER BACTERIA AND PROCESS FOR VALIN PRODUCTION THEREWITH |
| JP6450912B1 (ja) * | 2018-04-27 | 2019-01-16 | 株式会社Co2資源化研究所 | ヒドロゲノフィラス属細菌形質転換体 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11541105B2 (en) | 2018-06-01 | 2023-01-03 | The Research Foundation For The State University Of New York | Compositions and methods for disrupting biofilm formation and maintenance |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57134500A (en) | 1981-02-12 | 1982-08-19 | Kyowa Hakko Kogyo Co Ltd | Plasmid pcg1 |
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5867699A (ja) | 1981-10-16 | 1983-04-22 | Ajinomoto Co Inc | プラスミド |
| JPS62166890A (ja) | 1986-01-20 | 1987-07-23 | Asahi Chem Ind Co Ltd | イソクエン酸デヒドロゲナ−ゼ産生遺伝子を含むdna断片 |
| WO2005010182A1 (ja) | 2003-07-29 | 2005-02-03 | Research Institute Of Innovative Technology For The Earth | コリネ型細菌形質転換体及びそれを用いるジカルボン酸の製造方法 |
| JP3869788B2 (ja) | 2002-12-18 | 2007-01-17 | 財団法人地球環境産業技術研究機構 | コリネ型細菌を用いる有機化合物の製造方法 |
| WO2007050671A2 (en) | 2005-10-26 | 2007-05-03 | E. I. Du Pont De Nemours And Company | Fermentive production of four carbon alcohols |
| WO2008098227A2 (en) | 2007-02-09 | 2008-08-14 | The Regents Of The University Of California | Biofuel production by recombinant microorganisms |
| JP2009039031A (ja) * | 2007-08-08 | 2009-02-26 | Research Institute Of Innovative Technology For The Earth | ブタノール生産能を有する形質転換体 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3003925A (en) * | 1956-05-17 | 1961-10-10 | Kyowa Hakko Kogyo Kk | Method of producing l-glutamic acid by fermentation |
| US3136702A (en) * | 1961-07-19 | 1964-06-09 | Ajinomoto Kk | Process for producing l-glutamic acid |
| DE102004055508A1 (de) * | 2004-11-17 | 2006-06-01 | Basf Ag | Verfahren zur Herstellung optisch aktiver Alkohole |
| US20080274526A1 (en) | 2007-05-02 | 2008-11-06 | Bramucci Michael G | Method for the production of isobutanol |
| US20090111154A1 (en) | 2007-04-04 | 2009-04-30 | The Regents Of The University Of California | Butanol production by recombinant microorganisms |
| WO2010068921A2 (en) * | 2008-12-11 | 2010-06-17 | Bio Architecture Lab, Inc. | Biosynthesis of commodity chemicals |
-
2010
- 2010-03-29 EP EP10758603A patent/EP2415860A4/en not_active Withdrawn
- 2010-03-29 CA CA2755310A patent/CA2755310A1/en not_active Abandoned
- 2010-03-29 US US13/258,670 patent/US8871478B2/en active Active
- 2010-03-29 WO PCT/JP2010/055504 patent/WO2010113832A1/ja not_active Ceased
- 2010-03-29 JP JP2011507169A patent/JP5698655B2/ja active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57134500A (en) | 1981-02-12 | 1982-08-19 | Kyowa Hakko Kogyo Co Ltd | Plasmid pcg1 |
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5867699A (ja) | 1981-10-16 | 1983-04-22 | Ajinomoto Co Inc | プラスミド |
| JPS62166890A (ja) | 1986-01-20 | 1987-07-23 | Asahi Chem Ind Co Ltd | イソクエン酸デヒドロゲナ−ゼ産生遺伝子を含むdna断片 |
| JP3869788B2 (ja) | 2002-12-18 | 2007-01-17 | 財団法人地球環境産業技術研究機構 | コリネ型細菌を用いる有機化合物の製造方法 |
| WO2005010182A1 (ja) | 2003-07-29 | 2005-02-03 | Research Institute Of Innovative Technology For The Earth | コリネ型細菌形質転換体及びそれを用いるジカルボン酸の製造方法 |
| WO2007050671A2 (en) | 2005-10-26 | 2007-05-03 | E. I. Du Pont De Nemours And Company | Fermentive production of four carbon alcohols |
| WO2008098227A2 (en) | 2007-02-09 | 2008-08-14 | The Regents Of The University Of California | Biofuel production by recombinant microorganisms |
| JP2009039031A (ja) * | 2007-08-08 | 2009-02-26 | Research Institute Of Innovative Technology For The Earth | ブタノール生産能を有する形質転換体 |
Non-Patent Citations (41)
| Title |
|---|
| "Bergeys Manual of Determinative Bacteriology", vol. 8, 1974, pages: 599 |
| "Molecular Cloning, A Laboratory Manual", vol. 2, 1989, pages: 11.45 |
| AGRIC. BIOL. CHEM., vol. 54, 1990, pages 443 - 447 |
| ATSUMI, S. ET AL.: "Acetolactate Synthase from Bacillus subtilis Serves as a 2-Ketoisovalerate Decarboxylase for Isobutanol Biosynthesis in Escherichia coli", APPLIED AND ENVIRONMENTAL MICROBIOLOGY, vol. 75, no. 19, 14 August 2009 (2009-08-14), pages 6306 - 6311, XP008153201 * |
| ATSUMI, S. ET AL.: "Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde", NATURE BIOTECHNOLOGY, vol. 27, no. 12, December 2009 (2009-12-01), pages 1177 - 1180, XP008153202 * |
| ATSUMI, S. ET AL.: "Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels", NATURE, vol. 451, 3 January 2008 (2008-01-03), pages 86 - 89, XP002491157 * |
| AUSUBEL, F. ET AL.: "Current protocols in molecular biology", 1987, GREEN PUBLISHING AND WILEY INTERSCIENCE |
| BIOCHEM. J., vol. 335, 1998, pages 653 - 661 |
| BLATTNER, F. R. ET AL.: "The complete genome sequence of Escherichia coli K-12", SCIENCE, vol. 277, 1977, pages 1453 - 1474 |
| CRUZ RAMOS, H. ET AL.: "Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression", J. BACTERIOL., vol. 182, 2000, pages 3072 - 3080 |
| DE LA PLAZA, M. ET AL.: "Biochemical and molecular characterization of a-ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis", FEMS MICROBIOL., LETT., vol. 238, 2004, pages 367 - 374 |
| DENNIS H. F.: "The role and properties of the iron-sulfur cluster in Escherichia coli dihydroxy-acid dehydratase", J. BIOL. CHEM., vol. 268, 1993, pages 14732 - 14742 |
| DICKINSON, J.R. ET AL.: "An investigation of the metabolism of valine to isobutyl alcohol in Saccharomyces cerevisiae", J., BIOL. CHEM., vol. 273, 1998, pages 25751 - 25756 |
| DURRE, P. ET AL.: "Enzymatic investigations on butanol dehydrogenase and butyraldehyde dehydrogenase in extracts of Clostridium acetobutylicum", APPL. MICROBIOL. BIOTECHNOL., vol. 26, 1987, pages 268 - 272 |
| EIKMANNS, B.J. ET AL.: "A family of Corynebacterium glutamicum/Escherichia coli shuttle vectors for cloning, controlled gene expression, and promoter probing", GENE, vol. 102, 1991, pages 93 - 98 |
| HAZELWOOD, L.A. ET AL.: "The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism", APPL. ENVIRON. MICROBIOL., vol. 74, 2008, pages 2259 - 2266 |
| HOPWOOD, D.A., BIBB, M.J., CHARTER, K.F., BRUTON, C.J., KIESER, H.M., LYDIATE, D.J., SMITH, C.P., WARD, J.M., SCHREMPF, H.: "Genetic manipulation of Streptomyces: A Laboratory manual", 1985, THE JOHN INNES INSTITUTE |
| INUI, M. ET AL.: "Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions", J. MOL. MICROBIOL. BIOTECHNOL., vol. 7, 2004, pages 182 - 196 |
| J. MOL. MICROBIOL. BIOTECHNOL., vol. 8, 2004, pages 243 - 254 |
| JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 273, 1998, pages 25751 - 25756 |
| JOURNAL OF MOLECULAR BIOLOGY, vol. 53, 1970, pages 159 |
| KATSUMATA, R. ET AL.: "Protoplast transformation of glutamate-producing bacteria with plasmid DNA", J. BACTERIOL., vol. 159, 1984, pages 306 - 311 |
| KAZUO KOMAGATA ET AL.: "Classification of the cryneform group of bacteria", FERMENTATION AND INDUSTRY, vol. 45, 1987, pages 944 - 963 |
| KURUSU, Y. ET AL.: "Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation", AGRIC., BIOL. CHEM., vol. 54, 1990, pages 443 - 447 |
| KURUSU, Y. ET AL.: "Identification of plasmid partition function in coryneform bacteria", APPL. ENVIRON. MICROBIOL., vol. 57, 1991, pages 759 - 764 |
| LIEBL, W. ET AL.: "Transfer of Brevibacterium divaricatum DSM 20297T, ''Brevibacterium flavums'' DSM 20411, ''Brevibacterium lactofermentum'' DSM 20412 and DSM 1412, and Corynebacterium glutamicum and their distinction by rRNA, gene restriction patterns", INT J SYST BACTERIOL., vol. 41, 1991, pages 255 - 260 |
| MIWA, K. ET AL.: "Cryptic plasmids in glutamic acid-producing bacteria", AGRIC. BIOL. CHEM., vol. 48, 1984, pages 2901 - 2903 |
| MIWA, K. ET AL.: "Cryptic plasmids in glutamic acid-producing bacteria", AGRIC., BIOL., CHEM., vol. 48, 1984, pages 2901 - 2903 |
| NAKATA, K. ET AL.: "Fermentation Biotechnology", 2003, AMERICAN CHEMICAL SOCIETY, article "Vectors for the genetics engineering of corynebacteria", pages: 175 - 191 |
| NATURE, vol. 451, 2008, pages 86 - 90 |
| OMUMASABA, C. A. ET AL.: "Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation", J. MOL. MICROBIOL. BIOTECHNOL., vol. 8, 2004, pages 91 - 103 |
| PROCESS BIOCHEMISTRY, vol. 29, 1994, pages 303 - 309 |
| RES. MICROBIOL., vol. 144, 1993, pages 181 - 185 |
| RUYEX: "Characterization of enzymes of the branched-chain amino acid biosynthetic pathway in Methanococcus spp", J. BACTERIOL., vol. 173, 1991, pages 2086 - 2092 |
| SAMBROOK, J., RUSSELL, D. W.: "Molecular Cloning: A Laboratory Manual", 2001, CSHL PRESS |
| See also references of EP2415860A4 * |
| VERTES A.A. ET AL.: "Presence of mrr- and mcr-like restriction systems in Coryneform bacteria", RES. MICROBIOL., vol. 144, 1993, pages 181 - 185 |
| YAMAGUCHI, R. ET AL.: "Determination of the complete nucleotide sequence of the Brevibacterium lactofermentum plasmid pAM330 and the analysis of its genetic information", NUCLEIC ACIDS SYMP. SER., vol. 16, 1985, pages 265 - 267 |
| YASUDA, K. ET AL.: "Analyses of the acetate-producing pathways in Corynebacterium glutamicum under oxygen-deprived conditions", APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, vol. 77, 2007, pages 853 - 860 |
| YOSHIMOTO, H. ET AL.: "Genetic and physiological analysis of branched-chain alcohols and isoamyl acetate production in Saccharomyces cerevisiae", APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, vol. 59, no. 4-5, 1 January 2002 (2002-01-01), pages 501 - 508, XP002425678 * |
| ZHANG, Y. Q. ET AL.: "Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228", MOL. MICROBIOL., vol. 49, 2003, pages 1577 - 1593 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20130139258A (ko) * | 2010-11-10 | 2013-12-20 | 그린 페놀 고키노 페놀 주시 세이조 기쥬쓰 겐큐 구미아이 | 코리네형 세균 형질 전환체 및 그것을 사용하는 페놀의 제조 방법 |
| JP2013005764A (ja) * | 2011-06-24 | 2013-01-10 | National Institute Of Advanced Industrial Science & Technology | 大腸菌において混合糖から有機物質を生産する方法 |
| EP2749638A4 (en) * | 2011-08-22 | 2015-04-22 | Res Inst Innovative Tech Earth | MEANS FOR TRANSFORMING CORYNEFORMER BACTERIA AND PROCESS FOR VALIN PRODUCTION THEREWITH |
| US9290770B2 (en) | 2011-08-22 | 2016-03-22 | Research Institute Of Innovative Technology For The Earth | Coryneform bacterium transformant and process for producing valine using the same |
| JP6450912B1 (ja) * | 2018-04-27 | 2019-01-16 | 株式会社Co2資源化研究所 | ヒドロゲノフィラス属細菌形質転換体 |
| WO2019207812A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社Co2資源化研究所 | ヒドロゲノフィラス属細菌形質転換体 |
| US11697817B2 (en) | 2018-04-27 | 2023-07-11 | Utilization Of Carbon Dioxide Institute Co., Ltd. | Genus Hydrogenophilus bacterium transformant |
| US12188025B2 (en) | 2018-04-27 | 2025-01-07 | Utilization Of Carbon Dioxide Institute Co., Ltd. | Genus hydrogenophilus bacterium transformant |
| US12188024B2 (en) | 2018-04-27 | 2025-01-07 | Utilization Of Carbon Dioxide Institute Co., Ltd. | Genus Hydrogenophilus bacterium transformant |
Also Published As
| Publication number | Publication date |
|---|---|
| US8871478B2 (en) | 2014-10-28 |
| EP2415860A1 (en) | 2012-02-08 |
| JPWO2010113832A1 (ja) | 2012-10-11 |
| EP2415860A4 (en) | 2012-09-05 |
| CA2755310A1 (en) | 2010-10-07 |
| US20120115196A1 (en) | 2012-05-10 |
| JP5698655B2 (ja) | 2015-04-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7139478B2 (ja) | 発酵経路を経由するフラックスの増大を示す組み換え微生物体 | |
| JP6528295B1 (ja) | ヒドロゲノフィラス属細菌形質転換体 | |
| US8216820B2 (en) | Transformant of coryneform bacteria capable of producing isopropanol | |
| JP6199747B2 (ja) | 組換え微生物およびそれらの使用 | |
| JP5960701B2 (ja) | コリネ型細菌形質転換体及びそれを用いるバリンの製造方法 | |
| JP5395667B2 (ja) | イソプロパノール生産能を有する形質転換体 | |
| CN106459958B (zh) | 使高活性突变型酶高表达的棒状细菌转化体及使用它的4-羟基苯甲酸或其盐的制造方法 | |
| JP5252940B2 (ja) | コリネ型細菌形質転換体及びそれを用いるブタノールの製造方法 | |
| JP5243748B2 (ja) | ブタノール生産能を有する形質転換体 | |
| JP5698655B2 (ja) | コリネ型細菌形質転換体及びそれを用いるイソブタノールの製造方法 | |
| JP6668577B1 (ja) | 1,3−プロパンジオールの製造方法 | |
| JP4927297B2 (ja) | 組換え型コリネ型細菌を用いるエタノールの製造方法 | |
| JPWO2001096573A1 (ja) | 組換え型コリネ型細菌を用いるエタノールの製造方法 | |
| NZ614459B2 (en) | Recombinant microorganisms and uses therefor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10758603 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011507169 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3731/KOLNP/2011 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2755310 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010758603 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13258670 Country of ref document: US |