WO2010121486A1 - 基于吉西他滨结构的前药及其合成方法及应用 - Google Patents
基于吉西他滨结构的前药及其合成方法及应用 Download PDFInfo
- Publication number
- WO2010121486A1 WO2010121486A1 PCT/CN2010/000372 CN2010000372W WO2010121486A1 WO 2010121486 A1 WO2010121486 A1 WO 2010121486A1 CN 2010000372 W CN2010000372 W CN 2010000372W WO 2010121486 A1 WO2010121486 A1 WO 2010121486A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- gemcitabine
- group
- prodrug
- alcohol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention belongs to the field of nucleoside drugs and relates to prodrugs based on the structure of gemcitabine, synthetic methods thereof, and applications related to the treatment of various cancers and related diseases.
- Nucleoside drugs such as cytarabine, gemcitabine, decitabine, azacytidine, cladribine, fludarabine, clofibrate, nerapabine, 6-azide uridine and thiazofurin It is widely used to treat various cancers.
- many of the nucleoside drugs are in clinical research, such as 4, -thio-f luoro-ara-C, 2'-deoxy-2, -f luoromethylcytidine, 4, -thio-ara-C, and 3, -ethynylcytidine (ECYD).
- Gemcitabine Hydrochloride chemically known as 2-deoxy-2,2-difluorocytidine hydrochloride, is a pyrimidine nucleoside analogue developed by Eli Lilly, USA. It was listed in Australia, Finland and other countries in 1995. Named Gemzar, Chinese name is Jianze. Gemcitabine hydrochloride is a cell cycle-specific anti-metabolite drug, which mainly acts on tumor cells in the DNA synthesis phase, that is, S phase cells, under certain conditions, can prevent the progression from G1 phase to S phase.
- Gemcitabine as a prodrug is a good substrate for deoxythymidine kinase phosphorylation in cells and is converted to the following metabolites by enzymes: gemcitabine monophosphate (dFdCMP), gemcitabine diphosphate (dFdCDP) and gemcitabine III Phosphate (dFdCTP), wherein dFdCDP and dFdCTP are active products.
- dFdCDP inhibits ribonucleotide reductase, thereby reducing the amount of deoxynucleotide required for DNA synthesis repair.
- Gemcitabine hydrochloride is suitable for the treatment of non-small cell lung cancer, pancreatic cancer, bladder cancer, breast cancer and other solid tumors. In view of the intestinal toxicity caused by gemcitabine and its low bioavailability, gemcitabine can only be used for intravenous injection, but not for oral administration.
- 5-FU (5-fluorouracil) is the basic drug for the treatment of advanced colorectal cancer.
- Gemcitabine promotes the binding of 5-FU to its target (thymidine synthase) and enhances the latter's inhibition of DNA synthesis.
- Colon cancer is a malignant epithelial tumor of the colon. It is one of the most common malignancies in Europe, North America and Australia. Colon cancer accounts for the second leading cause of cancer death; incidence is low in Africa, Asia and South America, but with the westernization of lifestyle, its incidence is also Increasing.
- gemcitabine hydrochloride cannot be used to treat liver cancer in view of its rapid inactivation in the liver.
- Liver cancer is the third leading cause of cancer death in the world. Risk factors for hepatocellular carcinoma include HBV or HCV. Worldwide, HBV is thought to cause 60%-80% of hepatocellular carcinoma. There has been a lack of effective treatments for a long time, and there is no standard treatment for advanced liver cancer. Doxorubicin is reported to be the most widely used drug, but only one randomized controlled trial involving 60 patients supports its use. Doxorubicin can cause as much as 25% of fatal complications. Mitoxantrone is approved for the treatment of hepatocellular carcinoma, but it is not considered to be a specific drug for the treatment of hepatocellular carcinoma.
- nucleoside drugs are rapidly metabolized into inactive components in the liver and inactivated; so far, no nucleoside drugs have been used to treat liver cancer. Metazasis, Inc.
- liver specific can be targeted to treat liver cancer; b) accelerate the pace of phosphorylation in vivo, enhance the effect; c) improve the physical and chemical properties of absorption, penetration, stability, etc., improve PK performance; d) reduce nucleosides The side effects of the drug itself do not produce additional toxicity.
- Organospecific nucleoside prodrugs are among the most promising new methods.
- Nucleoside prodrugs are the most promising new method to reduce the side effects of anticancer drugs. After the prodrug of the anticancer drug reaches the working organ, it is converted into an active compound by the action of the enzyme to exert a therapeutic effect. Prodrugs such as capecitabine and enoxabine have been studied to overcome the deficiencies of nucleoside drugs. Many pharmaceutical companies are still actively investigating ways to treat cancer with other prodrugs (G. Xu, HL McLeod, Clin. Cancer Res., 2001, 7, 3314 - 3324; M. Rooseboora, JNM Commandeur, NPE Vermeulen, Pharmacol Rev., 2004, 56, 53-102; WD Wu, J. Sigmond, GJ Peters, RF Borch, J. Med. Chem. 2007, 50, 3743-3746).
- V is the end of the protective chain, and V can be a group or atom that can be broken during metabolism, and has a wide range.
- Song et al. reported the protection of gemcitabine glycoside 5'-OH with an amino acid ester group (Song, X. et al. Mol Pharmaceutics 2004, 2, 157-167).
- Wu et al. reported the protection of gemcitabine glycoside 5'-0H with an amine phosphate group (WD Wu, J. Sigmond, GJ Peters, RF Borch, J. Med. Chem. 2007, 50, 3743-3746); more recently, patent W02009/ In 053654, the gemcitabine sugar ring 5'-0H is protected with a phosphate group.
- the patents W098/32762, W004/041203, and W02006098628 and US2007225248 simultaneously protected 0 3 , 0 5 and N 4 with saturated and unsaturated carboxylic acid esters and were subjected to biological tests.
- the gemcitabine of the present invention is the structural basis of a prodrug of a nucleoside anticancer drug; on the one hand, it can overcome the problem of drug resistance existing in lung cancer cells or pancreatic cancer cells, and on the other hand, further increases the solubility by N 4 modification, Bioavailability and organ specificity, the resulting prodrug compounds can overcome the problem of rapid metabolism, reduce gutcitabine-induced intestinal toxicity and increase its bioavailability, making it clinically available for oral administration.
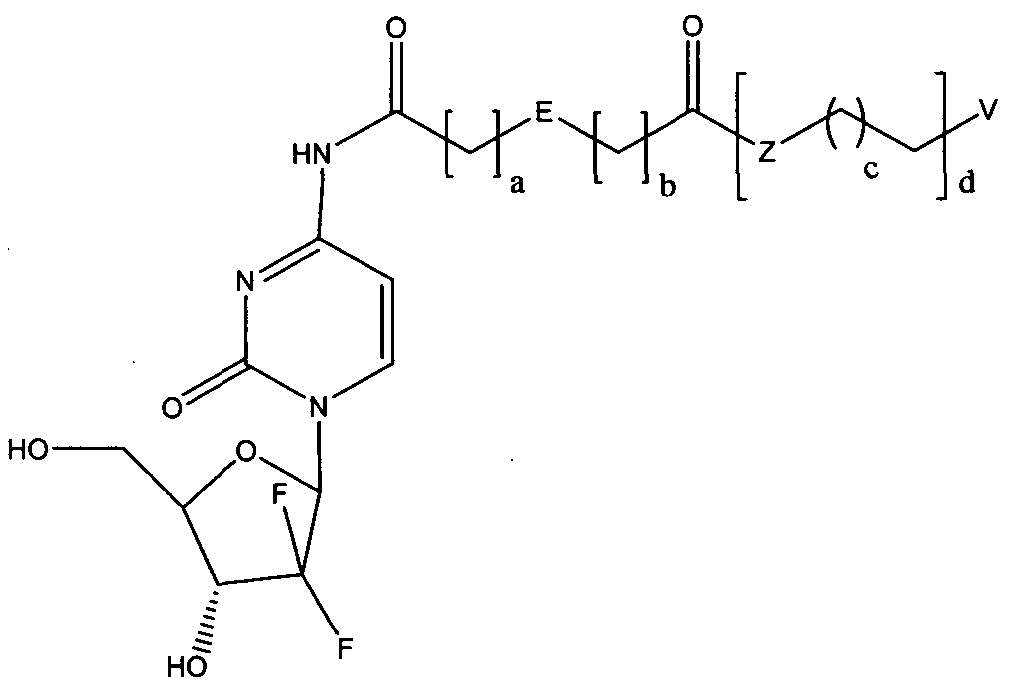
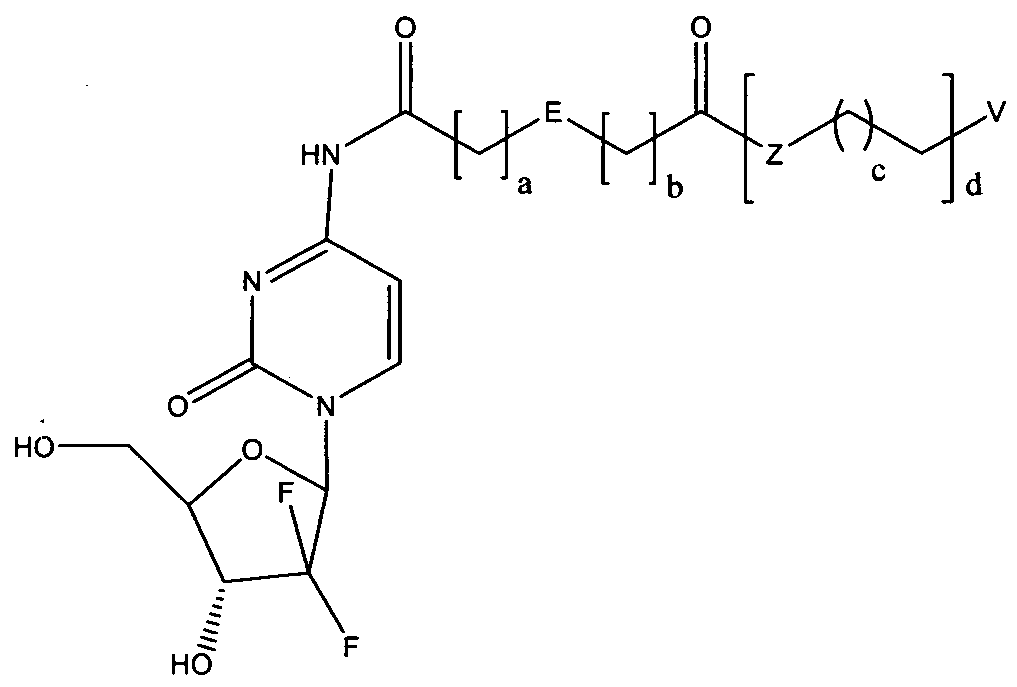
- the invention provides a prodrug based on gemcitabine structure, and the structural formula is as shown in formula (I):
- E is a hydrocarbon group of a 5- to 6-membered ring, containing 1 to 4 impurities A 5- to 5-membered cyclic fluorenyl, aryl or heteroaryl group of an atom.
- the hetero atom is selected from 0, 1 or 5.
- the position of attachment on the ring can be any relative position that is chemically permissible.
- the ring may have no substituent or one or more substituents. Substituents can be anywhere in the ring that can be chemically present.
- Z is selected from 0, ! ⁇ or 5.
- V is selected from the group consisting of hydrogen, a hydrocarbyl group, an alkoxy group, an ester group, a halogen, an amide group, an amino group or a substituted amino group.
- E is selected from one of the following structures:
- a 0 or 1
- b 0 or 1
- Z is 0 or N
- c 10-16
- d 1 or 2; preferably, V is selected from hydrogen or a hydrocarbyl group.
- the invention also provides a method for synthesizing the prodrug based on gemcitabine structure, comprising the following steps:
- the anhydride of step 1) is selected from the group consisting of: phthalic anhydride, phthalic anhydride, 1,2-cyclohexane dianhydride, 1,
- 2-cyclopentane dianhydride 2-cyclopentane dianhydride, pyrazine dianhydride, bismuth dipicolinate, thiophene dianhydride, furan dianhydride, pyrrolic anhydride, 1,2,3,6-tetrahydrophthalic anhydride.
- the alcohol of step 1) is selected from the group consisting of methanol, n-octanol, n-nonanol, n-nonanol, n-undecyl alcohol, lauryl alcohol, n-tetradecyl alcohol, n-hexadecanol or n-octadecyl alcohol.
- the amine of step 1) is selected from the group consisting of: n-octylamine, n-decylamine, n-decylamine, n-undecylamine, n-dodecylamine, n-tetradecylamine, n-hexadecylamine or n-octadecylamine.
- the organic solvent described in steps 1) and 2) is selected from the group consisting of: N,N-dimethylformamide, N,N-dimethylacetamide, tetrahydrofuran, dioxane, dimethyl sulfoxide, sulfolane Or pyridine.
- the acid chloride of step 1) is obtained by reacting a diacid with S0C1 2 , PC1 5 , PC1 3 or P0C1 3 ;
- the diacid is selected from the group consisting of: pyrazine-2,3-dicarboxylic acid, pyridazine -3,6-dicarboxylic acid, isophthalic acid, 4,6-pyrimidine dicarboxylic acid, 2,5-pyridinedicarboxylic acid, 2,3-pyridinedicarboxylic acid, 2,2-diphenyl phthalate, 2, 6 -pyridinedicarboxylic acid, 2,5-thiophenedicarboxylic acid, 3,4-pyridinedicarboxylic acid, 2,4-pyridinedicarboxylic acid, 3,5-pyrazoledicarboxylic acid, phthalic acid, terephthalic acid, 2, 3-thiophene dicarboxylic acid, 3, 4-thiophene dicarboxylic acid, 2, 4-thiophenedicarboxylic
- reaction time of step 1) is 3 to 4 hours;
- step 2) gemcitabine hydrochloride, the substituted acid obtained in step 1), benzotriazole-1-oxytripyrrolephosphonium hexafluorophosphate and 4-dimethylaminopyridine are as follows: 1.1 to 1.3): (1.1 to 1.3): (1.1 to 1.3) molar ratio reaction.
- the invention also provides the use of the prodrug based on gemcitabine structure, characterized in that it is used for the preparation of a medicament for treating lung cancer, pancreatic cancer, gastric cancer, breast cancer, colon cancer and liver cancer.
- Advantageous Effects of the Invention -
- the organ-specific prodrug study of the present invention is based on the above-mentioned cytarabine 0 5 cyclic phosphorylation prodrug, and uses gemcitabine having better antitumor activity as a nucleoside anticancer drug.
- the structural basis of the prodrug on the one hand, it can overcome the problem of drug resistance existing in lung cancer cells or pancreatic cancer cells, on the other hand, by further substituting different groups at the N 4 position, the substituent group at the N 4 position includes The aromatic ring and the heterocyclic fatty chain, by increasing the solubility, bioavailability and organ specificity of the N 4 position, the prodrug compound can overcome the problem of rapid metabolism, reduce the intestinal toxicity caused by gemcitabine and improve Its bioavailability makes it clinically available for oral administration; at the same time, it can better improve anti-tumor, anti-cancer, anti-infective and anti-proliferative capabilities, and can specifically act on the liver or colon.
- the synthesis method of the invention has the advantages of simple steps, easy availability of raw materials, high yield and low cost, and is suitable for industrial production.
- the gemcitabine-based prodrugs of the present invention and preparation methods thereof, including their synthetic preparation methods and screening methods, especially for the treatment of components and methods related to various cancers and related diseases, are also applicable to other cytosine-bearing or It is a substitute or a prodrug of a nucleoside antitumor drug such as 5-azacytosine.
- the present invention encompasses direct manipulation of specific organs, such as the lung, liver or colon, etc., i.e., design synthesis, pharmacy compositions, and methods of use of organ-specific and related nucleoside prodrugs.
- nucleoside anticancer drugs such as azacytidine, decitabine, aza-cytosine, thiazolidine, naribine, 6-nitrogen Glycosides and their cytarabine are also suitable for nucleoside anticancer drugs in clinical research, such as 2 '-deoxy-2 '-fluoro-methylc, 4'-thio-aracitidine, 4, -thiof luoro-aracitidine , 3'-ethynylcytidine, etc.
- nucleoside anticancer drugs such as 2 '-deoxy-2 '-fluoro-methylc, 4'-thio-aracitidine, 4, -thiof luoro-aracitidine , 3'-ethynylcytidine, etc.
- nucleoside prodrug By rationally designing a nucleoside prodrug with a specific function to overcome the intestinal toxicity caused by gemcitabine and improve its bioavailability, it can be used clinically for oral administration; at the same time, to overcome nucleoside drugs in the liver Problems such as excessive metabolism and loss of activity may open up another treatment for advanced liver cancer and colon cancer. detailed description
- Benzotriazole hexafluorophosphate- 1-oxytripyrrolidinylphosphine purchased from Jill Biochemical (Shanghai) Co., Ltd.;
- Example 1 ⁇ 4 - (2-n-dodecyloxyformylbenzoyl) - 2 '-deoxy-2', 2 '-difluorocytidine (Compound NO. 1 ) 5. 0 g Phthalic anhydride (34 mmol), 7. 5 g of lauryl alcohol (40 leg ol) and 240 mg of 4-dimethylaminopyridine (2 mmol) were mixed, heated and melted, reacted for 4 hours, cooled to room temperature, and quantified. 2-n-dodecyloxyformylbenzoic acid.
- Example 2 N 4 -(2-n-undecyloxyformylbenzoyl)-2'-deoxy-2, 2'-difluorocytidine (compound NO .2)
- the lauryl alcohol in Example 1 was replaced with n-undecyl alcohol, and the same as in Example 1 to give the title compound.
- LC UV254 purity 95%.
- LC-MS m/z 564 [M + H] + (M. C 28 H 37 F 0 7 , molecular weight 565).
- Example 3 N 4 -(2-n-hexadecanoyloxybenzoyl)-2'-deoxy-2,2'-difluorocytidine (Compound NO.3) replaced with n-hexadecanol
- the lauryl alcohol, hydrazine, hydrazine-dimethylacetamide in Example 1 was replaced with N,N-dimethylformamide, and the reaction was carried out for 6 hr.
- LC UV254) purity 96%.
- Example 6 N 4 -(3-n-decyloxyformylbenzoyl)-2'-deoxy-2,2'-difluorocytidine (compound N0.6) was replaced with isophthalic acid
- pyrazine-2,3-dicarboxylic acid, sulfolane was used in place of the dioxane in Example 4
- n-decyl alcohol was replaced with n-nonanol in the same manner as in Example 4 to give the title compound.
- LC UV254) purity 96%.
- LC-MS m/z 552 [M + H] + (Molecular formula C 27 H 35 F 2 N 3 0 7 , molecular weight 551).
- Example 7 N 4 -(3-n-undecyloxyformylbenzoyl)-2'-deoxy-2',2'-difluorocytidine (Compound NO. 7) Substituting isophthalic acid for the pyrazine-2,3-dicarboxylic acid in Example 4, dimethyl sulfoxide in place of the dioxane in Example 4, and replacing the lauryl alcohol in Example 4 with n-undecyl alcohol. Others were the same as in Example 4 to give the title compound. LC (UV254) purity 88%. LC-MS m/z 564 [M + H] + (M. C 28 H 37 F 2 N : i 0 7 , molecular weight 565).
- Example 8 N 4 -(5-n-dodecyloxyformylpyridine-2-formyl)-2'-deoxy-2',2'-difluorocytidine (Compound No. 8)
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced by 2,5-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-monohydric alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 99%.
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,5-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 97%.
- LC-MS m/z 564 [M + H] + (M. C 27 H 36 F 0 7 , molecular weight 566).
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,3-pyridinedicarboxylic acid, and the title compound was obtained.
- LC (UV254) purity was 97%.
- LC-MS z 581 [M + H] + (Molecular formula C 28 H 38 F 2 N 4 0 7 , molecular weight 580).
- Example 13 N 4 -(3-n-dodecyloxyformylpyridine-2-formyl)-2'-deoxy-2,2'-difluorocytidine (Compound N0.13)
- Example 14 N 4 -(3-n-undecyloxyformylpyridine-2-formyl)-2'-deoxy-2',2'-difluorocytidine (Compound N0.14)
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced by 2,3-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 98%.
- LC-MS m/z 567 [M + ⁇ (molecular formula C 27 H 36 F 2 N 4 0 7 , molecular weight 566).
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,3-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 97%.
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 4 The pyrazine- 2,3-dicarboxylic acid of Example 4 was replaced with 2,2-diphenyl phthalic acid, and the lauric alcohol of Example 4 was replaced with methanol, and the same as Example 4 to give the title compound.
- Example 18 N 4 -(6-n-dodecyloxyformylpyridine-2-formyl)-2'-deoxy-2,2'-difluorocytidine (Compound NO. 18)
- Example 19 N 4 -(6-n-undecyloxyformylpyridine-2-formyl)-2'-deoxy-2',2'-difluorocytidine (compound N0.19)
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,6-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 95%.
- LC-MS 567 [ ⁇ + ⁇ ] + (molecular formula C 27 H 36 F 0 7 , molecular weight 566).
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,5-thiophenedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 90%.
- LC-MS 572 [M + H] + (M.p. C 26 H 35 F 2 N 3 0 7 S, molecular weight 571).
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced by 3,4-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 97%.
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 3,4-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with a normal ""-alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 97%.
- LC-MS m/z 564 [M + H] + (M. C 27 H 36 F 2 N 4 0 7 , molecular weight 566).
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 25 N 4 -(3-n-dodecyloxyformylpyridine-4-formyl)-2,-deoxy-2,2'-difluorocytidine (Compound No. 25)
- Example 26 N 4 -(4-n-dodecyloxyformylpyridine-2-ylformyl)-2'-deoxy-2,2'-difluorocytidine (after the compound NO.26)
- the code name is: SL-02)
- Example 28 N 4 -(4-n-undecyloxyformylpyridine-2-formyl)-2'-deoxy-2,2'-difluorocytidine (Compound N0.28)
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,4-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 90%.
- LC-MS 567 [ ⁇ + ⁇ ] + (Molecular formula C 27 H 36 F 2 N 4 ( 3 ⁇ 4 , molecular weight 566).
- the structural formula and characterization data of the compound are shown in Table 1.
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 2,4-pyridinedicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-undecyl alcohol, and the same as in Example 4 to give the title compound.
- Example 30 N 4 -(5-n-nonanodecyloxyformylpyrazole-3-formyl)-2'-deoxy-2',2'-difluorocytidine (Compound No. 30)
- Example 4 The pyrazine-2,3-dicarboxylic acid of Example 4 was replaced with 3,5-pyrazoledicarboxylic acid, and the lauryl alcohol of Example 4 was replaced with n-alcohol, and the same as in Example 4 to give the title compound.
- LC UV254 purity 99%.
- the structural formula and characterization data of the compounds are shown in Table 1.
- Example 1 The phthalic anhydride in Example 1 was replaced with phthalic anhydride, and the lauryl alcohol in Example 1 was replaced with n-undecyl alcohol, and the same as in Example 1 to give the title compound.
- LC UV254 purity 90%.
- LC-MS »A580 [M + H]+ M. C 29 H 39 F 2 N 3 0 7 , molecular weight 579).
- Example 1 The phthalic anhydride in Example 1 was replaced with phthalic anhydride, and the lauryl alcohol in Example 1 was replaced with n-undecyl alcohol, and the same as in Example 1 to give the title compound.
- LC UV254 purity 90%.
- LC-MS m/z 580 [M + H] + (M. C 29 H 39 F 2 N 3 0 7 , molecular weight 579).
- Example 1 The phthalic anhydride in Example 1 was replaced with phthalic anhydride, and the same as in Example 1 to give the title compound.
- LC-MS m / z 594 [ M + H] + molecular formula C 3 .H 41 F 2 N 3 0 7, molecular weight 593).
- Example 1 The phthalic anhydride in Example 1 was replaced with phthalic anhydride, and the same as in Example 1 to give the title compound.
- LC- MS m / z 594 [M + H] + molecular formula C 3 .H 41 F 2 N 3 0 7, molecular weight 593).
- Example 37 N 4 -(3-n-dodecylaminoformylpyrazine-2-formyl)-2,-deoxy-2',2,-difluorocytidine (compound N0.37)
- Prodrug compounds were tested for different tumor cells by MTT assay.
- Cell line NCI- ⁇ 43 ⁇ 40, ⁇ 549, LOVO, HT-29, MDA-MB-231, SGC7901, HepG2 or BEL-7402 cell line.
- Culture Adherent growth, cultured in D-MEM cell culture medium of 10% fetal bovine serum (Hyclone), the initial cell concentration of conventional culture was about 3*10 5 /ml, and passaged once every day for 2-3 days. On the day before the experiment, 1: 2 was passaged, and the cell concentration during the experiment was between 5-10*107 ml.
- MTT solution MTT dry powder (Sigma), fully dissolved in PBS to prepare 5 rag/ml, 0.22 ⁇ microporous membrane filtration, and then stored at - 20 °C.
- Cell seeding The cells were cultured 24 hours after passage and grew well. The cells were routinely harvested, and the cell concentration was adjusted to 2 x 10 5 /ml (adherent cells) - 3 x 10 5 /ml (suspended cells) with fresh medium. The adherent cells were inoculated with 100 ⁇ /well, cultured in a 37 ° C, 5% CO 2 incubator for 24 hours, and the old culture solution was discarded, and fresh culture solution was added to 95 ⁇ l/well. Suspension cells were directly inoculated into 95 ⁇ l/well.
- Drug treatment Set 9 concentration gradients for each drug, set 3 replicate wells for each concentration, and set 5 replicate wells for the drug blank control group. Each test was done at the same time as a control. The final concentration of the added drug was 0.25, 0.225, 0.026, 0. 03125, 0. 016, 0. 008, 0. 004, 0. 002 mM, and 5 ⁇ of physiological saline was added to the control group.
- Gem is gemcitabine, as a control
- A549 is a human lung cancer cell line
- NCI-H460 is a human lung cancer cell line
- HT29 is a human colon cancer cell line
- L0V0 is a human colon cancer cell line
- Bel7402 is a human liver cancer cell line
- HepG2 is a human liver cancer cell line
- SGC7901 is a human gastric cancer cell line
- MDA-MB-231 is a human breast cancer cell line
- Gemcitabine SL-01 and SL-02 standard solutions were quantitatively diluted in plasma and sampled at 0, 1, 2, 4, 6, 8, 12, 24 h, respectively, according to standard methods, 20 ⁇ M injections were recorded. The peak area of the chromatogram, the drug concentration was determined, and the stability of gemcitabine, SL-01 and SL-02 in plasma was examined. The results are shown in Table 5.
- a total of 100 healthy mice from Kunming were randomly divided into 2 groups. The first group was the SL- 01 administration group, and the second group was the SL- 02 administration group. Five rats were set in parallel at each time point. SL-01 and SL-02 samples were administered by intragastric administration at a dose of 100 mg/kg (equivalent to 0.2 ml per mouse). The rats were fasted for 12 hours before the administration, and were given water for 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 2, 4, 8 hours, 12 hours, and the blood was taken from the fundus venous plexus, and added with heparin sodium and washed with 2. 5 ⁇ 1 tetrahydrouridine.
- SL-02 is relative to gemcitabine
- Absolute bioavailability of SL-02 relative to gemcitabine AUC SL - 2 and formation of ⁇ surface and XD SL V (AUC JI surface XD iv JI)
- SL-02 relative gemcitabine relative bioavailability AUC SL 2 degradation and generation of gemcitabine And XD SL — 2 / (AUC ⁇ XD oral ⁇ )
- Dosage AUC SL - 2 and formation of ⁇ surface and XD SL V (AUC JI surface XD iv JI)
- the homogenization process is always carried out in an ice bath to avoid a decrease in enzyme activity. Transfer the homogenate to a centrifuge tube, and transfer the homogenate to a centrifuge tube for centrifugation.
- the supernatant was centrifuged for 20 min (12000 r/min), and the obtained centrifugation supernatant was the S9 component.
- the liver microsome S9 fraction was taken and mixed with the drug solution.
- the mixture was centrifuged at 1800 ⁇ l in a test tube, pre-incubated for 5 min in a 37 V water bath, and the reaction was initiated by adding 200 ⁇ l of a 10 mmol/L NADPH solution.
- the final volume of the reaction system was 2 ml.
- lh, 2 h, 4 h, 6 h, 12 h 200 ⁇ 1 incubation system solution was added, and immediately added 1 ml of ice-cold isopropanol to vortex for 30 s, and allowed to stand for 5 min.
- Cell line and culture NCI-H460 lung cancer cell line, HepG2 liver cancer cell line, or HT29 colon cancer cell line, adherent growth, D-MEM medium (Gibco) using 10% fetal bovine serum (Hyclone) Routine culture, 2-3 days 1: 3 pass once. Cells were harvested by subculture to 25 10 cm culture dishes.
- the growth curve was plotted with time as the horizontal axis and tumor volume as the vertical axis.
- the tumor volume of the control group was about 3000 mm 3 , the experiment was stopped. After the eyeballs were taken, the nude mice of each group were sacrificed by cervical dislocation. The tumors were weighed and the growth inhibition rate was calculated.
- Tumor growth inhibition rate (%) (1 - mean tumor weight of the treatment group / mean tumor weight of the control group) X 100%o
- SL-01 dose of 10, 20 and 40 mg/Kg in the low, medium and high dose groups of HepG2 liver cancer cell line transplanted in nude mice From the tumor growth curve or the tumor weight, SL-01 and The inhibitory effect of gemcitabine on the growth of HepG2 nude mice was not obvious.
- the inhibition rate of the SL-01 high-dose group (40 mg/Kg) group was similar to that of the GEM group. From the perspective of assessing drug toxicity by weight loss, the positive control GEM had a weight loss rate of 9% at the end of the trial, while SL-01 was low (10 mg/Kg), medium (20 mg/Kg), and high dose group (40). The weight loss rates of the mg/Kg group were 11.6%, 14%, and 3.5%, respectively.
- the inhibitory effect of SL-01 is slightly better than the equivalent dose of GEM. From the evaluation of drug toxicity by weight loss, the positive control GEM and SL-01 showed no significant toxicity, and the weight loss rate of GEM (20 mg/Kg) and equal dose SL-01 (20 mg/Kg) group (%) ) at dl7 are 4% and 83 ⁇ 4 respectively.
- SL-01 white powder product, insoluble in water, prepared by suspending with 2.5% starch, administered by gavage; gemcitabine, white powder, soluble in water, dissolved in physiological saline during test It was filtered through a microporous membrane and administered to the tail vein.
- Positive control drug furan fluorouracil (FT-207), orally administered preparation, produced by Qilu Pharmaceutical Co., Ltd.
- mice Female 50, 4-5 weeks old, purchased from the Laboratory Animal Science Department of Peking University Medical School, animal production license number: SCXK (Beijing) 2006-0008.
- H460 cells human lung cancer cells, were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences.
- RPMI-1640 medium purchased from Invitrogen, USA.
- Fetal bovine serum purchased from Sino-US cooperation Lanzhou Minhai Biological Engineering Co., Ltd.
- Laboratory equipment cell culture incubator, ultra-clean workbench, centrifuge, large/mouse laminar flow frame, vernier caliper, surgical instruments, syringe, gavage needle, autoclave, etc.
- Tumor cell culture H460 cell line was cultured in RPMI-1640 medium containing 10% fetal bovine serum and grown in a 37 ° C 5% CO 2 incubator, and cultured for passage.
- Method of inoculation of tumor cells in nude mice After the cells are grown to about 80 bottles in a culture flask, the cells are collected. Before digestion, the cells were washed twice with PBS, and 1.5 ml of digestive solution was added to each vial. Under the microscope, the cells were detached and the digestion was terminated. The cell suspension was collected in a 50 ml centrifuge tube, centrifuged and washed twice with PBS. The cells were counted and then suspended in about 10 ml of physiological saline to obtain a tumor cell suspension inoculated into nude mice. Each nude mouse was inoculated with 1 X 1070. 2 ml of cells in the left forelimb. Tumor growth was observed after inoculation.
- nude mice After inoculation of tumor cells in nude mice for 5 days, nude mice were randomly divided into 6 negative control groups, 6 positive control groups, and 6 SL-01 medium dose groups. There were 6 in the SL-01 low-dose group and 6 in the gemcitabine control group.
- Drug efficacy evaluation protocol Weigh the body before and after the administration, and measure the long diameter (a) and short diameter (b) of the tumor.
- W The weight of the tumor in the negative control group, and W is the weight of the tumor in the administration group.
- Test period is 30 days, in which the administration time is 22 days and the administration is 7 times.
- the results are as follows - 1. Effect of drug on tumor volume: Tumor volume of nude mice in each group was measured on days 7, 14, 18 and 22 after administration, as shown in Table 12.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Epidemiology (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Urology & Nephrology (AREA)
- Gastroenterology & Hepatology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Description



















Claims



Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2759474A CA2759474A1 (en) | 2009-04-21 | 2010-03-25 | Prodrugs based on gemcitabine structure and synthetic methods and applications thereof |
| EP10766577.0A EP2423215A4 (en) | 2009-04-21 | 2010-03-25 | COMMITICAL-BASED PRODRUGS AND SYNTHETIC METHOD AND APPLICATION |
| CN2010800179647A CN102428093A (zh) | 2009-04-21 | 2010-03-25 | 基于吉西他滨结构的前药及其合成方法及应用 |
| US13/265,335 US8653048B2 (en) | 2009-04-21 | 2010-03-25 | Prodrugs based on gemcitabine structure and synthetic methods and applications thereof |
| JP2012506308A JP2012524113A (ja) | 2009-04-21 | 2010-03-25 | ゲムシタビン構造に基づくプロドラッグおよびその合成方法と応用 |
| IL215792A IL215792A0 (en) | 2009-04-21 | 2011-10-23 | Prodrugs based on gemcitabine structure as well as synthetic methods and applications thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2009100207167A CN101525361B (zh) | 2009-04-21 | 2009-04-21 | 基于吉西他滨结构的前药及其合成方法及应用 |
| CN200910020716.7 | 2009-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010121486A1 true WO2010121486A1 (zh) | 2010-10-28 |
Family
ID=41093454
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2010/000372 Ceased WO2010121486A1 (zh) | 2009-04-21 | 2010-03-25 | 基于吉西他滨结构的前药及其合成方法及应用 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8653048B2 (zh) |
| EP (1) | EP2423215A4 (zh) |
| JP (1) | JP2012524113A (zh) |
| CN (2) | CN101525361B (zh) |
| CA (1) | CA2759474A1 (zh) |
| IL (1) | IL215792A0 (zh) |
| WO (1) | WO2010121486A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8956613B2 (en) | 2012-11-13 | 2015-02-17 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| CN111298132A (zh) * | 2020-02-22 | 2020-06-19 | 新乡医学院 | 一种树状分子吉西他滨自组装纳米前药及其制备方法和应用 |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101525361B (zh) * | 2009-04-21 | 2010-11-17 | 济南圣鲁金药物技术开发有限公司 | 基于吉西他滨结构的前药及其合成方法及应用 |
| CN102115485A (zh) * | 2009-12-30 | 2011-07-06 | 济南圣鲁金药物技术开发有限公司 | 基于阿糖胞苷结构的前药及其合成方法及应用 |
| CN102432654A (zh) * | 2011-09-26 | 2012-05-02 | 宋云龙 | 吉西他滨酰胺衍生物及其制备方法和用途 |
| CN104955458A (zh) * | 2012-11-07 | 2015-09-30 | Z·索 | 取代的吉西他滨芳基酰胺类似物和使用所述类似物的治疗方法 |
| WO2014145207A1 (en) * | 2013-03-15 | 2014-09-18 | Ohio State Innovation Foundation | Substituted gemcitabine bicyclic amide analogs and treatment methods using same |
| JP6212831B2 (ja) * | 2013-12-04 | 2017-10-18 | 杭州源昶医薬科技有限公司 | ゲムシタビン誘導体、該誘導体を含む組成物及び該誘導体の製薬用途 |
| WO2015116782A1 (en) | 2014-01-29 | 2015-08-06 | Board Of Regents, The University Of Texas System | Nucleobase analogue derivatives and their applications |
| CN106459130A (zh) * | 2014-03-03 | 2017-02-22 | 纽科利制药公司 | 吉西他滨类似物 |
| CN105001291B (zh) * | 2014-04-15 | 2018-12-04 | 上海知萌生物医药科技有限公司 | 吉西他滨化学传递前药及其制备方法和应用 |
| CN104910024B (zh) * | 2015-05-12 | 2017-02-22 | 四川理工学院 | 一种驱油用表面活性剂 |
| CN109153639B (zh) * | 2016-03-14 | 2022-07-29 | 斯法尔制药私人有限公司 | 葫芦巴碱类化合物 |
| US10435429B2 (en) | 2017-10-03 | 2019-10-08 | Nucorion Pharmaceuticals, Inc. | 5-fluorouridine monophosphate cyclic triester compounds |
| KR102708995B1 (ko) | 2018-01-10 | 2024-09-23 | 누코리온 파마슈티컬스, 인코포레이티드. | 포스포르(포스포론)아미다타세탈 및 포스프(온)아탈세탈 화합물 |
| US11427550B2 (en) | 2018-01-19 | 2022-08-30 | Nucorion Pharmaceuticals, Inc. | 5-fluorouracil compounds |
| CN108329371B (zh) * | 2018-03-06 | 2021-04-09 | 沈阳药科大学 | 白蛋白结合型吉西他滨前药及其合成和应用 |
| AU2019258590B2 (en) * | 2018-04-26 | 2022-09-08 | NanoMed Holdings Pty Ltd | Gemcitabine amphiphile prodrugs |
| MX2022000573A (es) | 2019-07-17 | 2022-02-10 | Nucorion Pharmaceuticals Inc | Compuestos ciclicos de desoxirribonucleotido. |
| CN110845560B (zh) * | 2019-11-21 | 2021-08-24 | 广东中科药物研究有限公司 | 苯丙氨酸酰胺化的核苷酸衍生物及其制备方法与应用 |
| JP2023523415A (ja) | 2020-04-21 | 2023-06-05 | リガンド・ファーマシューティカルズ・インコーポレイテッド | ヌクレオチドプロドラッグ化合物 |
| WO2021216431A1 (en) | 2020-04-21 | 2021-10-28 | Ligand Pharmaceuticals, Inc. | Benzyloxy phosph(on)ate compounds |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998032762A1 (en) | 1997-01-24 | 1998-07-30 | Norsk Hydro Asa | Gemcitabine derivatives |
| WO2004041203A2 (en) | 2002-11-04 | 2004-05-21 | Xenoport, Inc. | Gemcitabine prodrugs, pharmaceutical compositions and uses thereof |
| WO2005025552A2 (en) | 2003-09-15 | 2005-03-24 | Drug Discovery Laboratory As | Protein binding compounds |
| US20050101775A1 (en) | 1999-09-08 | 2005-05-12 | Erion Mark D. | Prodrugs for liver specific drug delivery |
| WO2006030217A2 (en) | 2004-09-15 | 2006-03-23 | Drug Discovery Laboratory As | Drug conjugates of long chain fatty acid or ester moieties as protein binding prodrugs |
| WO2006065525A1 (en) | 2004-12-17 | 2006-06-22 | Eli Lilly And Company | Amide prodrug of gemcitabine, compositions and use thereof |
| WO2006098628A1 (en) | 2005-03-18 | 2006-09-21 | Clavis Pharma As | Oral dosage forms of gemcitabine derivatives |
| US20070225248A1 (en) | 2006-03-21 | 2007-09-27 | Clavis Pharma As | Oral dosage forms of gemcitabine derivatives |
| WO2007149891A2 (en) | 2006-06-21 | 2007-12-27 | Eli Lilly And Company | Crystalline forms of gemcitabine amide prodrug, compositions and use thereof |
| WO2009053654A2 (fr) | 2007-10-17 | 2009-04-30 | Hospices Civils De Lyon | Prodrogues phosphoesters de la gemcitabine comme agents anticancereux |
| CN101525361A (zh) * | 2009-04-21 | 2009-09-09 | 济南圣鲁金药物技术开发有限公司 | 基于吉西他滨结构的前药及其合成方法及应用 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5637688A (en) * | 1994-12-13 | 1997-06-10 | Eli Lilly And Company | Process for preparing 1-(2'-deoxy-2'-difluoro-d-ribofuranosyl)-4-aminopyrimidin-2-one hydrochloride |
| US20050137141A1 (en) * | 2003-10-24 | 2005-06-23 | John Hilfinger | Prodrug composition |
| US20090068286A1 (en) * | 2007-09-11 | 2009-03-12 | Resprotect, Gmbh | Method of treating cancer by administration of 5-substituted nucleosides |
-
2009
- 2009-04-21 CN CN2009100207167A patent/CN101525361B/zh not_active Expired - Fee Related
-
2010
- 2010-03-25 CN CN2010800179647A patent/CN102428093A/zh active Pending
- 2010-03-25 CA CA2759474A patent/CA2759474A1/en not_active Abandoned
- 2010-03-25 US US13/265,335 patent/US8653048B2/en not_active Expired - Fee Related
- 2010-03-25 WO PCT/CN2010/000372 patent/WO2010121486A1/zh not_active Ceased
- 2010-03-25 JP JP2012506308A patent/JP2012524113A/ja active Pending
- 2010-03-25 EP EP10766577.0A patent/EP2423215A4/en not_active Withdrawn
-
2011
- 2011-10-23 IL IL215792A patent/IL215792A0/en unknown
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998032762A1 (en) | 1997-01-24 | 1998-07-30 | Norsk Hydro Asa | Gemcitabine derivatives |
| US20050101775A1 (en) | 1999-09-08 | 2005-05-12 | Erion Mark D. | Prodrugs for liver specific drug delivery |
| WO2004041203A2 (en) | 2002-11-04 | 2004-05-21 | Xenoport, Inc. | Gemcitabine prodrugs, pharmaceutical compositions and uses thereof |
| WO2005025552A2 (en) | 2003-09-15 | 2005-03-24 | Drug Discovery Laboratory As | Protein binding compounds |
| WO2006030217A2 (en) | 2004-09-15 | 2006-03-23 | Drug Discovery Laboratory As | Drug conjugates of long chain fatty acid or ester moieties as protein binding prodrugs |
| WO2006065525A1 (en) | 2004-12-17 | 2006-06-22 | Eli Lilly And Company | Amide prodrug of gemcitabine, compositions and use thereof |
| WO2006098628A1 (en) | 2005-03-18 | 2006-09-21 | Clavis Pharma As | Oral dosage forms of gemcitabine derivatives |
| US20070225248A1 (en) | 2006-03-21 | 2007-09-27 | Clavis Pharma As | Oral dosage forms of gemcitabine derivatives |
| WO2007149891A2 (en) | 2006-06-21 | 2007-12-27 | Eli Lilly And Company | Crystalline forms of gemcitabine amide prodrug, compositions and use thereof |
| WO2009053654A2 (fr) | 2007-10-17 | 2009-04-30 | Hospices Civils De Lyon | Prodrogues phosphoesters de la gemcitabine comme agents anticancereux |
| CN101525361A (zh) * | 2009-04-21 | 2009-09-09 | 济南圣鲁金药物技术开发有限公司 | 基于吉西他滨结构的前药及其合成方法及应用 |
Non-Patent Citations (7)
| Title |
|---|
| BENDER ET AL., J. MED. CHEM., vol. 52, 2009, pages 6958 - 6961 |
| G XU; H. L. MCLEOD, CLIN. CANCER RES., vol. 7, 2001, pages 3314 - 3324 |
| M. ROOSEBOOM; J. N. M. COMMANDEUR; N. P. E. VERMEULEN, PHARMACOL. REV., vol. 56, 2004, pages 53 - 102 |
| MARK ERON ET AL., J AM. CHEM. SOC., vol. 126, 2004, pages 5154 - 5163 |
| See also references of EP2423215A4 |
| SONG, X. ET AL., MOL PHARMACEUTICS, vol. 2, 2004, pages 157 - 167 |
| W. D. WU; J. SIGMOND; G J. PETERS; R. F. BORCH, J. MED. CHEM., vol. 50, 2007, pages 3743 - 3746 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8956613B2 (en) | 2012-11-13 | 2015-02-17 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US9540410B2 (en) | 2012-11-13 | 2017-01-10 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| US9890189B2 (en) | 2012-11-13 | 2018-02-13 | BoYen Therapeutics, Inc. | Gemcitabine prodrugs and uses thereof |
| CN111298132A (zh) * | 2020-02-22 | 2020-06-19 | 新乡医学院 | 一种树状分子吉西他滨自组装纳米前药及其制备方法和应用 |
| CN111298132B (zh) * | 2020-02-22 | 2022-06-24 | 新乡医学院 | 一种树状分子吉西他滨自组装纳米前药及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2423215A4 (en) | 2013-05-22 |
| EP2423215A1 (en) | 2012-02-29 |
| CN101525361A (zh) | 2009-09-09 |
| CA2759474A1 (en) | 2010-10-28 |
| CN101525361B (zh) | 2010-11-17 |
| US20120088908A1 (en) | 2012-04-12 |
| CN102428093A (zh) | 2012-04-25 |
| US8653048B2 (en) | 2014-02-18 |
| JP2012524113A (ja) | 2012-10-11 |
| IL215792A0 (en) | 2012-01-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2010121486A1 (zh) | 基于吉西他滨结构的前药及其合成方法及应用 | |
| US10793543B2 (en) | Selective C-KIT kinase inhibitor | |
| CN104530061B (zh) | 埃克替尼盐酸盐晶型、药物组合物和用途 | |
| EP2947070B1 (en) | Multi-targeted ubenimex prodrug derivative and preparation method and use thereof | |
| KR20180041748A (ko) | Parp 억제제, 결정형의 제조방법 및 이의 용도 | |
| CN102432663A (zh) | 雷公藤红素衍生物及其制备和在治备抗肿瘤药物中的应用 | |
| WO2013044811A1 (zh) | 吉西他滨酰胺衍生物及其制备方法和用途 | |
| CN104829596B (zh) | 吡咯取代吲哚酮类衍生物、其制备方法、包含该衍生物的组合物、及其用途 | |
| CN101787064A (zh) | 阿糖胞苷衍生物及其在抗癌抗肿瘤中的用途 | |
| CN102066362A (zh) | 二氢吲哚酮衍生物 | |
| CN104961786A (zh) | 基于吉西他滨结构的前体药物及其应用 | |
| WO2008000743A2 (en) | Novel 2',3'-methylidene acetyl adenosine prodrugs for use as prodrugs for adenosine receptor agonists | |
| TW201734028A (zh) | 抑制癌症及病毒之化合物 | |
| CN113896721B (zh) | 具有肿瘤靶向的烟酰胺磷酸核糖转移酶抑制剂 | |
| WO2017148290A1 (zh) | 一种取代的腺嘌呤化合物及其药物组合物 | |
| JP2015523349A (ja) | ポリフィリンiのアシル化誘導体、その調製方法及び使用 | |
| WO2011079501A1 (zh) | 基于阿糖胞苷结构的前药及其合成方法及应用 | |
| WO2013123745A1 (zh) | 齐多夫定喹啉共轭化合物及其制备方法和抗肝癌之应用 | |
| CN113135909A (zh) | Dpd抑制剂及其制备方法、药物组合物和用途 | |
| CN112279863A (zh) | Hsp90抑制剂与喜树碱衍生物的偶联物及其制备方法与应用 | |
| CN105272975A (zh) | 一类具有1,2,4-恶二唑片段结构的吲哚生物碱及其制备方法和用途 | |
| CN102688250A (zh) | 偶氮类衍生物做为rsk2抑制剂的合成及应用 | |
| WO2011113173A1 (zh) | 阿糖胞苷衍生物及其在抗癌抗肿瘤中的用途 | |
| WO2023138317A1 (zh) | 具有ripk1抑制活性的化合物、其制备方法及其用途 | |
| CN115279761A (zh) | 咪唑并吡啶酮化合物或其盐的晶体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080017964.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10766577 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2759474 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012506308 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 215792 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010766577 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13265335 Country of ref document: US |