WO2010122506A2 - Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof - Google Patents
Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof Download PDFInfo
- Publication number
- WO2010122506A2 WO2010122506A2 PCT/IB2010/051746 IB2010051746W WO2010122506A2 WO 2010122506 A2 WO2010122506 A2 WO 2010122506A2 IB 2010051746 W IB2010051746 W IB 2010051746W WO 2010122506 A2 WO2010122506 A2 WO 2010122506A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- variants
- target
- hiv1
- positions
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases [RNase]; Deoxyribonucleases [DNase]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16022—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Definitions
- the invention relates to the use of meganuclease variants which cleave at least one target in the provirus of a retrovirus and in particular cleave the genomic insertion of an integrating Virus genome and in particular to meganuclease variants which cleave the Human Immunodeficiency Virus genome following genomic insertion, for the treatment of an infection of one or more of these viruses.
- the present Invention also relates to such variants and to vectors encoding such variants, as well as to a cell or multi-cellular organism modified by such a vector and to the use of said meganuclease variant and derived products for genome engineering and for in vivo and ex vivo (gene cell therapy) genome therapy.
- viruses present specific treatment and control problems as they always comprise an intracellular stage to their life cycle, in which the nucleic acid genome of the virus is inserted into a host cell and normally transported to the nucleus. During this stage of the virus life cycle, the virus genome can enter into a dormant state whilst inside a host cell, during which time the production of new virus particles/proteins/copies of the viral genome ceases.
- medicaments and treatments for viral infection consist of compounds which affect aspects of virus biology involved in the active stages of the virus life cycle, such as compounds which target/inactivate a viral enzyme or structural protein. Therefore whilst in a dormant state the viral genome resident in the cytoplasm or nucleus of a host cell can not be affected by most conventional anti-virus medicaments and therefore persists.
- Retroviruses which are contained with the family Retroviridae which comprises in turn seven genera. Alpharetrovirus, Betaretrovirus, Gammaretrovirus, Deltaretrovirus, Epsilonretro- virus, Lentivirus and Spumavirus. These groups of viruses are responsible for several important diseases such as Human T-lymphotrophic virus ⁇ Gammaretrovirus), Rous Sarcoma ⁇ Alpharetrovirus) and Human Immunodeficiency Virus ⁇ Lentivirus).
- HIV Human Immunodeficiency Virus
- Figure 1 is an example of a Retrovirus which is responsible for a significant and ongoing global medical crisis. HIV viruses persist and continue to replicate for many years in the infected individual before causing overt signs of disease. HIV is the causative agent of the Acquired Immune Deficiency Syndrome (AIDS), which is characterized by a susceptibility to infection with opportunistic pathogens, mainly as a result of a profound decrease in the number of CD4+ T cells,
- AIDS Acquired Immune Deficiency Syndrome
- a characteristic feature of the Retroviridae family of viruses is that viral particles contain two copies of an RNA genome. After infection, the genomic RNA is reverse transcribed by a viral enzyme into DNA, which is then permanently integrated into the host genome.
- the retroviral genome harbors the sequences coding for the viral enzymatic, structural and regulatory proteins.
- the genomic RNA molecule contains a series of non-coding sequences that have important functions in different steps of the viral life cycle ( Figure 2).
- the "2007 AIDS epidemic update” report issued by the UNAIDS (Joint United Nations Programme on HIV/AIDS), indicates that 33.2 million [30.6 - 36.1 million] people were estimated to be living with HIV, 2.5 million [1.8 - 4.1 million] people became newly infected with HIV and 2.1 million [1.9 - 2.4 million] people died of AIDS in 2007.
- HIV is characterized by a high genetic variability, due to the rapid viral turnover (10 10 - 10 12 viral particles produced per day) in an HIV-infected individual, combined with the high mutation rate arising during reverse transcription (10 "4 per nucleotide).
- Two types of HIV, HIV-I and HIV-2, which are closely related to each other, have been identified to date (Sharp et al., Philos Trans R Soc Lond B Biol Sci, 2001, 356, 867-76). Most AIDS worldwide is caused by the more virulent HIV-I, while HIV-2 is endemic in West Africa.
- HIV is transmitted by direct sexual contact, by blood or blood products, and from an infected mother to infant, either intrapartum, perinatally, or via breast milk. Infection of humans with HIV- 1 causes a dramatic decline in the number of CD4+ T lymphocytes. When the number of CD4+ cells is very reduced, opportunistic infections and neoplasms occur (Simon et al., Lancet, 2006, 368 , 489-504).
- Anti retro viral treatment for HIV infection consists of drugs which work by slowing down the replication of HIV in the body.
- nucleoside or nucleotide reverse transcriptase inhibitors There are several classes of anti-HIV drugs that attack the virus in different ways and the most common classes of antiretrovirals are nucleoside or nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors and entry inhibitors (Flexncr C, Nature Reviews Drug Discovery, 2007, 6, 959-966).
- HAART Highly Active Antiretroviral Therapy
- HAART typically combines drugs from at least two different classes of antiretroviral drugs and has been shown to effectively suppress the virus when used properly.
- Highly active antiretroviral therapy has revolutionalized how people infected with HIV arc treated, and reduces the rate at which resistance develops.
- HIV vaccines will not be able to completely eliminate HIV infection, because the virus "hides" in certain cells of the body, where it can last silent for decades meaning that any effect of the vaccine will have been lost.
- a new field for the treatment of HIV infection is the development of genetic therapies against HIV. Gene therapy could allow the prevention of progressive HIV infection by persistently blocking viral replication.
- RNA-based agents such as ribozymes, aptamers and small interfering RNAs and protein-based agents.
- zinc- finger nucleases against the CCR5 receptor a protein present on the surface of immune cells that is required to mediate viral entry
- the disruption of the CCR5 receptor from the immune cells by the nucleases is proposed to render the patient's cells permanently resistant to CCR5- specific strains of HIV. This approach is based on the fact that people with natural mutations on this receptor are resistant to HIV infection.
- An interesting target that has not been pursued in the fight against the AIDS pandemic and more generally retroviruses is the genomically integrated provirus and/or the reverse transcribed DNA version of the retrovirus genome prior to its integration, since targeting the proviral DNA could lead to the elimination or inactivation of the structure that allows the virus to multiply and the infection to propagate.
- One novel way to inactivate the provirus which the inventors have decided to investigate is by the use of nucleases that could cleave the integrated form of the virus and generate mutations and/or deletions in the provirus following the action of the cellular DNA repair machinery.
- the target sequences should be located in the coding sequences of essential genes, since the inactivation of an accessory gene may not lead to viral eradication.
- the viral genome also contains essential regulatory sequences that are located in the long terminal repeats (LTRs) that flank the viral genome in the provirus. Even if mutations in these regions would be expected to have a less drastic effect than a mutation in an essential gene, the fact that they are duplicated sequences could be useful in an approach of "virus clipping", meaning the excision of long regions of the proviral DNA by the action of a nuclease cleaving twice in the viral sequence.
- LTRs long terminal repeats
- HIV is characterized by a high degree of sequence variability due to the nature of the viral reverse transcriptase. It is therefore essential to check the sequence conservation of the target among the different isolates.
- the inventors have developed a new molecular medicine approach based on the inactivation of the retrovirus provirus through the use of tailored meganucleases specifically targeting the proviral DNA, using the HIV-I provirus in the genome of the infected cell as a model.
- the principle of this new therapeutic strategy is that the tailored meganucleases against targets in the provirus will generate a double strand break (DSB) at their target sequences, chosen to be located in genes/regulatory sequences/structural sequences that are essential for the virus to replicate or alternatively target sequences which are present in multiple copies in the provirus, for instance in the two flanking LTR regions, so allowing the provirus or a portion thereof to be excised.
- DLB double strand break
- the epidemiology of HIV makes research into the HIV virus a major and extremely active area of research.
- the manipulation of the HIV provirus is one area of research in which to date reagents have not been readily available as workers have instead concentrated on attempting to manipulate the HIV virion per se. Therefore the means to easily engineer the HIV provirus in situ in the genome of an infected cell/organism would likely provide valuable insights into this aspect of HIV biology and potentially open new avenues of attack in combating HIV.
- HIV shows a very high level of genetic change, not all of the components of the HIV genome are as capable of supporting change as others. Generally speaking it is those portions of the virus which are immunogenic, that is present upon the exterior of the virus particle where they can interact with the components of the hosts immune system, which are most able to support high levels of variability. Whereas the essential internal structural or packaging components of HIV are less able to continue to function following changes in their coding sequences.
- HEs Homing Endonucleases
- proteins families Cholier, B. S. and B.L. Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774.
- proteins are encoded by mobile genetic elements which propagate by a process called "homing”: the endonuclease cleaves a cognate allele from which the mobile element is absent, thereby stimulating a homologous recombination event that duplicates the mobile DNA into the recipient locus.
- homologous recombination event that duplicates the mobile DNA into the recipient locus.
- LAGLIDADG The LAGLIDADG family, named after a conserved peptide motif involved in the catalytic center, is the most widespread and the best characterized group. Seven structures are now available. Whereas most proteins from this family are monomelic and display two LAGLIDADG motifs, a few have only one motif, and thus dimerize to cleave palindromic or pseudo-palindromic target sequences.
- LAGLIDADG peptide is the only conserved region among members of the family, these proteins share a very similar architecture ( Figure 3).
- the catalytic core is flanked by two DNA-binding domains with a perfect two-fold symmetry for homodimers such as l-Crel (Chevalier, et ⁇ /., Nat. Struct. Biol., 2001, 8, 312-316) , ⁇ -Mso ⁇ (Chevalier et al., J. MoI.
- the two LAGLIDADG peptides also play an essential role in the dimerization interface. DNA binding depends on two typical saddle-shaped ⁇ folds, sitting on the DNA major groove. Other domains can be found, for example in inteins such as ⁇ l-PfuX (Ichiyanagi et al, J. MoI. Biol., 2000, 300 ; 889-901) and PI- Seel (Moure et al , Nat. Struct. Biol, 2002, 9, 764-770), whose protein splicing domain is also involved in DNA binding.
- inteins such as ⁇ l-PfuX (Ichiyanagi et al, J. MoI. Biol., 2000, 300 ; 889-901)
- PI- Seel PI- Seel
- residues 28 to 40 and 44 to 77 of I-Crel were shown to form two separable functional subdomains, able to bind distinct parts of a homing endonuclease half-site target sequence (Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/049095 and WO 2007/057781).
- the combination of the two former steps allows a larger combinatorial approach, involving four different subdomains.
- the different subdomains can be modified separately and combined to obtain an entirely redesigned meganuclease variant (heterodimer or single-chain molecule) with chosen specificity, as illustrated in Figure 5.
- couples of novel meganucleases are combined in new molecules ("half-meganuclcascs") cleaving palindromic targets derived from the target one wants to cleave.
- the combination of such "half-meganucleases” can result in a heterodimeric species cleaving the target of interest.
- XPC gene (WO2007093918), RAG gene (WO2008010093), HPRT gene (WO2008059382), beta-2 microglobulin gene (WO2008102274), Rosa26 gene (WO2008152523) and Human hemoglobin beta gene (WO200913622).
- base-pairs ⁇ 1 and ⁇ 2 do not display any contact with the protein, it has been shown that these positions are not devoid of content information (Chevalier et al, J. MoL Biol., 2003, 329, 253-269), especially for the base-pair ⁇ 1 and could be a source of additional substrate specificity (Argast et al., J. MoI. Biol., 1998, 280, 345-353; Jurica et al., MoL Cell., 1998, 2, 469-476; Chevalier, B.S. and B.L. Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774).
- an l-Crel variant which cleaves a target in the provirus of a pathogenic virus, for use in treating an infection of said virus.
- the inventors therefore provide a set of l-Crel variants which can recognise and cut targets in a genomically integrated provirus (GIP).
- I-Crel variants provide a new therapeutic route to retrovirus and in particular HIV treatment by HIV provirus inactivation or alteration.
- This new class of enzymes is also potentially useful in studies into the transcriptional and regulatory behaviour of the provirus.
- This new class of anti-HIV medicament can act in a number of ways including by non-homologous end joining, the replacement/removal by homologous recombination with an introduced DNA targeting construct of a portion of the provirus or the removal of the provirus following recombination between chromosome arms. Each of these different mechanisms is discussed in detail below.
- the genomically integrated provirus refers to the DNA sequence present in one or several places in the host cell genome which was inserted following reverse transcription of the RNA virus genome and its integration into the host genome.
- meganuclease (s) and variant (s) and variant meganuclease (s) will be used interchangeably herein.
- the inventors have therefore created a new class of meganuclease based reagents which are useful for the treatment of a retrovirus infection and the most important and potentially useful feature of these enzymes is that instead of acting upon the virion or any component thereof they act upon the genomic insertion of the virus.
- Targeting the integrated provirus would allow a clinician to eliminate the structure which leads to the generation of further viral particles, acting at a level that no other anti-viral therapeutic approaches have yet been developed.
- prior art therapies which act upon the different steps of the viral life cycle allow to a clinician to inhibit viral replication, but do not eliminate the source of the virions, which therefore allows for the amplification of the viral infection when the treatment is withdrawn or resistance develops.
- These variants also allow the targeting of the DNA version of the virus genome before it has integrated into the host cell genome. By inactivating the virus genome before it can integrate into the host cell genome, the claimed variants can act during the early step of cell infection in a way which no current antiretroviral medicament can.
- the Inventors have validated this new class of anti-retrovirus reagents by generating meganuclease variants to a series of DNA targets in the genome of the HIV provirus ( Figures 7, 24, 35 and 48). Seven targets in the HIV pro virus were chosen [one in U3 LTR (target HIVlJ), one in U5 LTR (target HIVlJ), two in the ⁇ 24 gene (target HIV 1_4) and (target HIV1_7), two in the protease gene (target HIV 3 _5) and (target HIV 1_9) and one in the p7 gene (target HIV 1_8)] and the inventors set out to determine whether it was possible to generate meganucleases capable of cleaving these.
- targets in the HIV pro virus were chosen [one in U3 LTR (target HIVlJ), one in U5 LTR (target HIVlJ), two in the ⁇ 24 gene (target HIV 1_4) and (target HIV1_7), two in the protease gene (target HIV 3 _5) and (target HIV 1_
- target sequences are present in the U3 and U5 LTR regions, the coding sequence of the structural gene gag and more specifically in the p7 and p24 proteins therein and in the structural gene pol, specifically in the protease gene. These seven targets were selected based on their therapeutic potential.
- the p24 protein is a structural component of the viral capsid and is essential for the virus to replicate.
- the inventors have shown that it is possible to generate l-Crel variants which can cleave targets in the p24 gene (target HIV 1_4) and
- the HIV protease is also an essential protein that is needed for viral particle maturation, without which viral particles remain in an immature state and are not infectious.
- the inventors have shown that it is possible to generate l-Crel variants which can cleave targets in the protease gene (target HIV 1_5) and (target HIV 1_9) in the present Patent Application. These two targets do not overlap and hence these two enzymes could be used simultaneously so further reducing the chances of resistance developing and/or causing an excision of the portion of protease situated between the two cleavage sites.
- the HIV nucleocapsid protein (p7, ou NC) is bound to the single- stranded RNA geneome. This protein plays a key role in the HIV life cycle since, being an RNA chaperone, its activity is required for efficient reverse transcription, making it an interesting target for antiviral therapy.
- the inventors have shown that it is possible to generate ⁇ -Crel variants which can cleave targets in the p7 gene (target HIV1_8).
- the inventors have therefore established that meganuclease variants can be generated in both the sequences of essential genes as well as in regulatory non- coding sequences essential for viral replication.
- essential genes of the GIP provirus are those genes which must remain active in order for the GIP provirus to be converted into further virions which are able to exit the host cell and infect further cells.
- essential genes other types of essential genetic elements can exist such as the regulatory elements of essential genes and/or structural sequence elements of the HIV provirus that are necessary for its packaging and/or insertion into the genome.
- the pathogenic virus is from a genus selected from the group consisiting of: Alpharetrovirus, Beiaretrovirus, Gammaretrovirus, Dellaretroviru$ > Epsilonretrovirus, Lentivirus and Spumavirus.
- Biotechnology Information http://www.ncbi.nlm.nih.gov/) or the virus genomics and bioinformatics resources centre at University College London (http://www.biochem.ucl.ac.uk/bsm/virus_database/VIDA.html).
- the virus is selected from the group consisting of: Human T-lymphotrophic virus, Rous Sarcoma and Human Immunodeficiency Virus.
- the virus is either Human Immunodeficiency Virus Type 1 (HIVl) or Human Immunodeficiency Virus Type 2 (HIV2).
- HIVl Human Immunodeficiency Virus Type 1
- HIV 2 Human Immunodeficiency Virus Type 2
- the DNA target is within a DNA sequence essential for
- I HV replication I HV replication, viability, packaging or virulence.
- the DNA target is within an essential gene or regulatory element or structural clement of the HIV provirus.
- the DNA target is within the open reading frame of the HIV provirus encoding a gene or regulatory element of a gene selected from the group: GAG, POL, ENV, TAT and REV.
- the target in the HIVl provirus is selected from the group consisting of the sequences SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368.
- the variant is selected from one of the sequences SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368.
- the variant is characterized in that at least one of the two l-Crel monomers has at least two substitutions, one in each of the two functional subdomains of the LAGLIDADG core domain situated from positions; in particular said substitution(s) in the first functional subdomain comprise a substitution in at least one of positions 26, 28, 30, 32, 33, 38 and/or 40 and said substitution(s) in the second functional subdomain comprise a substitution in at least one of positions positions 44, 68, 70, 75 and/or 77 and being obtainable by a method comprising at least the steps of: (a) constructing a first series of l-Crel variants having at least one substitution in a first functional subdomain of the LAGL ⁇ DADG core domain comprising at least one substitution at a position selected from the group: 26, 28, 30, 32, 33, 38 and/or 40 of l-Crel, (b) constructing a second series of l-Crel variants having at least one substitution in a second functional subdomain of the LAGLIDADG core domain comprising at least
- step (c) selecting and/or screening the variants from the first series of step (a) which are able to cleave a DNA target sequence selected from the group SEQ
- step (d) selecting and/or screening the variants from the second series of step (b) which are able to cleave a mutant 1-OeI site wherein at least one of (i) the nucleotide triplet in positions -5 to -3 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions -5 to -3 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions +3 to +5 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in position -5 to -3 of said DNA target sequence from said provirus,
- step (e) selecting and/or screening the variants from the first series of step (a) which are able to cleave a mutant l-Crel site wherein at least one of (i) the nucleotide triplet in positions +8 to +10 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus,
- step (f) selecting and/or screening the variants from the second series of step (b) which are able to cleave a mutant l-Crel site wherein at least one of (i) the nucleotide triplet in positions +3 to +5 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -5 to -3 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus,
- step (g) combining in a single variant, the mutation(s) in positions 26, 28, 30, 32, 33, 38 and/or 40 and 44, 68, 70, 75 and/or 77 of two variants from step (c) and step (d), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions -10 to -8 is identical to the nucleotide triplet which is present in positions -10 to -8 of said DNA target sequence from said provirus , (ii) the nucleotide triplet in positions +8 to +10 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions -10 to - 8 of said DNA target sequence from said provirus, (iii) the nucleotide triplet in positions -5 to -3 is identical to the nucleotide triplet which is present in positions -5 to -3 of said DNA target sequence from said provirus and (iv) the nucleotide triplet in
- step (h) combining in a single variant, the mutation(s) in positions 26, 28, 30, 32, 33, 38 and/or 40, and 44, 68, 70, 75 and/or 77 of of two variants from step (e) and step (f), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions +8 to +10 of the 1-OeI site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 is identical to the reverse complementary sequence of the nucleotide triplet in positions +8 to +10 of said DNA target sequence from said provirus, (iii) the nucleotide triplet in positions +3 to +5 is identical to the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus, (iv) the nucleo
- step (j) selecting and/or screening the heterodimers from step (i) which are able to cleave said DNA target sequence from said pro virus.
- a combinatorial approach, as illustrated schematically in Figure 6 was used to entirely redesign the DNA binding domain of the l-Crel protein and thereby engineer novel meganucl eases with fully engineered specificity.
- heterodimer of step (i) may comprise monomers obtained in steps (g) and (h), with the same DNA target recognition and cleavage activity properties.
- the heterodimer of step (i) may comprise monomers obtained in steps (g) and (h), with different DNA target recognition and cleavage activity properties.
- first series of l-Crel valiants of step (a) are derived from a first parent meganuclease.
- step (b) are derived from a second parent meganuclease.
- first and second parent meganucleases are identical.
- first and second parent meganucleases are different.
- the variant may be obtained by a method comprising the additional steps of:
- step (k) selecting heterodimers from step (j) and constructing a third series of variants having at least one substitution in at least one of the monomers of said selected heterodimers,
- step (k) (1) combining said third series variants of step (k) and screening the resulting heterodimers for enhanced cleavage activity against said DNA target from the GIP.
- the inventors have found that although specific meganucleases can be generated to a particular target in the GIP using the above method, that such meganucleases can be improved further by the additional rounds of substitution and selection against the intended target. Meganuclease generated to targets in the GIP using other methods are also comprised within the present Patent Application.
- the substitutions in the third series of variants arc introduced by site ditected mutagenesis in a DNA molecule encoding said third series of variants, and/or by random mutageneis in a DNA molecule encoding said third series of variants.
- the substitution of residues in the meganucleases can be performed randomly, that is wherein the chances of a substitution event occurring are equal chance across all the residues of the meganuclease. Or on a site directed basis wherein the chances of certain residues being subject to a substitution is higher than other residues.
- steps (k) and (1) are repeated at least two times and wherein the heterodimers selected in step (k) of each further iteration are selected from heterodimers screened in step (1) of the previous iteration which showed increased cleavage activity against said DNA target from the GIP.
- the inventors have found that the meganucleases can be further improved by using multiple iterations of the additional steps (k) and (1).
- the variant comprises one or more substitutions in posi- tions 137 to 143 of l-Crel that modify the specificity of the variant towards the nucleotide in positions ⁇ 1 to 2, ⁇ 6 to 7 and/or ⁇ 1 1 to 12 of the target site in the GIP.
- the variant comprises one or more substitutions on the entire l-Crel sequence that improve the binding and/or the cleavage properties of the variant towards said DNA target sequence from the GIP.
- the present invention also encompasses the substitution of any of the residues present in the l-Crel enzyme.
- the variant is a heterodimer, resulting from the association of a first and a second monomer having different mutations in positions 26, 28, 30, 32, 33, 38 and/or 40, and 44, 68, 70, 75 and/or 77 of l-Crel, said heterodimer being able Io cleave a non-palindromic DNA target sequence from the HIV provirus.
- the l-Crel enzyme acts as a dimer, by ensuring that the variant is a heterodimer this allows a specific combination of two different I- OeI monomers which increases the possible targets cleaved by the variant,
- the heterodimeric variant is an obligate heterodimer variant having at least one pair of mutations in corresponding residues of the first and the second monomers which mediate an intermolecular interaction between the two I- Crel monomers, wherein the first mutation of said pair(s) is in the first monomer and the second mutation of said pair(s) is in the second monomer and said pair(s) of mutations impairs the formation of functional homodimers from each monomer without preventing the formation of a functional heterodimer, able to cleave the genomic DNA target from the HIV provirus.
- the monomers have at least one of the following pairs of mutations, respectively for the first and the second monomer: a) the substitution of the glutamic acid in position 8 with a basic amino acid, preferably an arginine (first monomer) and the substitution of the lysine in position 7 with an acidic amino acid, preferably a glutamic acid (second monomer); the first monomer may further comprise the substitution of at least one of the lysine residues in positions 7 and 96, by an arginine, b) the substitution of the glutamic acid in position 61 with a basic amino acid, preferably an arginine (first monomer) and the substitution of the lysine in position 96 with an acidic amino acid, preferably a glutamic acid (second monomer); the first monomer may further comprise the substitution of at least one of the lysine residues in positions 7 and 96, by an arginine, c) the substitution of the leucine in position 97 with an aromatic amino acid, preferably a phen
- the variant which is an obligate heterodimer
- the first monomer further comprises the K7R, E8R, E61R, K96R and L97F or K7R, E8R, F54W, E61R, K96R and L97F mutations
- the second monomer further comprises the K7E, F54G, L58M and K96E or K7E, F54G, K57M and K96E mutations.
- a single-chain chimeric meganuclease which comprises two monomers or core domains of one or two variant(s) according to the first aspect of the present invention, or a combination of both.
- the single-chain meganuclease comprises a first and a second monomer according to the first aspect of the present invention, connected by a peptidic linker.
- the l-Crel variant is combined with other antiretro viral drugs.
- Most antiretroviral drugs have at least three names. Sometimes a drug is referred to by its research or chemical name, such as AZT, The second name is the generic name for all drugs with the same chemical structure; for example AZT is also known as zidovudine. The third name is the brand name given by the pharmaceutical company; one of the brand names for zidovudine is Retrovir. Lastly, an abbreviation of the common name might sometimes also be used, such as ZDV, which is the fourth name given to zidovudine.
- the I- OeI variant is combined with other antiretroviral agents such as those listed above or with other meganucleases directed against different targets in the HIV provirus.
- l-Crel variants according to the present invention are used only once the viral load of an individual has been reduced significantly using antiretroviral drugs.
- the 1-OeI variants are then used to elimate as many proviruses as possible whilst the HIV virus population is in its enforced dormant state.
- kits of parts comprising at least one 1-OeI according to the present invention either in the form of a peptide or a nucleotide encoding the variant(s) and one or more other anti-HIV medicaments, together with instructions for the administration of the variant and other anti-HIV medicaments to a patient.
- the meganuclease when used as a polypeptide is associated with:
- the sequence of the variant/single-chain meganuclease is fused with the sequence of a membrane translocating peptide (fusion protein).
- the meganuclease in the form of a polynucleotide encoding said meganuclease in a vector can be introduced into a cell by a variety of methods (e.g., injection, direct uptake, projectile bombardment, liposomes, electroporation). Meganucleases can be stably or transiently expressed into cells using expression vectors. Techniques of expression in eukaryotic cells are well known to those in the art. (See Current Protocols in Human Genetics: Chapter 12 "Vectors For Gene Therapy” & Chapter 13 "Delivery Systems for Gene Therapy”).
- the meganuclease may also comprise a nuclear localization signal (NLS) which is an amino acid sequence which acts like a 'tag' on the exposed surface of a protein.
- NLS nuclear localization signal
- the NLS is used to target the protein to the cell nucleus through the Nuclear Pore Complex and to direct a newly synthesized protein into the nucleus via its recognition by cytosolic nuclear transport receptors.
- this signal consists of one or more short sequences of positively charged lysines or arginines.
- a polynucleotide fragment encoding the variant according to the first aspect of the present invention or the single-chain chimeric meganuclease according to a second aspect of the present invention.
- an expression vector comprising at least one polynucleotide fragment according to the second aspect of the present invention.
- the expression vector includes a targeting construct comprising a sequence to be introduced flanked by sequences sharing homologies with the regions surrounding said DNA target sequence from the provirus.
- the present invention therefore also relates to a unified genetic construct which encodes the variant under the control of suitable regulatory sequences as well as sequences homologous to portions of the provirus surrounding the variant DNA target site. Following cleavage of the target site by the variant these homologous portions can act as complementary sequences in a homologous recombination reaction with the provirus replacing the existing provirus sequence with a new sequence engineered between the two homologous portions in the unified genetic construct.
- homologous sequences of at least 50 bp preferably more than 100 bp and more preferably more than 200 bp are used.
- Shared DNA homologies are located in regions flanking upstream and downstream the site of the break and the DNA sequence to be introduced should be located between the two arms. Therefore, the targeting construct is preferably from 200 bp to 6000 bp, more preferably from 1000 bp to 2000 bp; it comprises: a sequence which has at least 200 bp of homologous sequence flanking the target site, for repairing the cleavage and a sequence for inactivating the provirus and/or a sequence of an exogenous gene of interest which it is intended to insert at the site of the DN ⁇ repair event by homologous recombination.
- DNA homologies are generally located in regions directly upstream and downstream to the site of the break (sequences immediately adjacent to the break; minimal repair matrix). However, when the insertion is associated with a deletion of ORF sequences flanking the cleavage site, shared DNA homologies are located in regions upstream and downstream the region of the deletion.
- a vector which can be used in the present invention includes, but is not limited to, a viral vector, a plasmid, a RNA vector or a linear or circular DNA or RNA molecule which may consists of a chromosomal, non chromosomal, semisynthetic or synthetic nucleic acids.
- Preferred vectors are those capable of autonomous replication (episomal vector) and/or expression of nucleic acids to which they are linked (expression vectors). Large numbers of suitable vectors are known to those of skill in the art and commercially available.
- Viral vectors include retrovirus, adenovirus, parvovirus (e.g. adeno associated viruses), coronavirus, negative strand RNA viruses such as orthomyxovirus (e.
- influenza virus rhabdovirus
- paramyxovirus e. g. measles and Sendai
- positive strand RNA viruses such as picor- navirus and alphavirus
- double-stranded DNA viruses including adenovirus, herpesvirus (e. g., Herpes Simplex virus types 1 and 2, Epstein-Barr virus, cytomegalovirus), and poxvirus (e. g,, vaccinia, fowlpox and canarypox).
- viruses include Norwalk virus, togavirus, flavivirus, reoviruses, papovavirus, hepadnavirus, and hepatitis virus, for example.
- retroviruses include: avian leukosis-sarcoma, mammalian C-type, B-type viruses, D type viruses, HTLV- BLV group, lentivirus, spumavirus (Coffin, J. M., Retroviridae: The viruses and their replication, In Fundamental Virology, Third Edition, B. N. Fields, et al., Eds., Lippincott-Raven Publishers, Philadelphia, 1996).
- Vectors can comprise selectable markers, for example: neomycin phosphotransferase, histidinoi dehydrogenase, dihydro folate reductase, hygromycin phosphotransferase, herpes simplex virus thymidine kinase, adenosine deaminase, glutamine synthetase, and hypoxanthine-guanine phosphoribosyl transferase (HRPT) for eukaryotic cell culture; TRPl for S. cerevisiae; tetracycline, rifampicin or ampicillin resistance in E. coli.
- selectable markers for example: neomycin phosphotransferase, histidinoi dehydrogenase, dihydro folate reductase, hygromycin phosphotransferase, herpes simplex virus thymidine kinase, adenosine deamina
- the viral vector is selected from the group comprising lentiviruses, Adeno-associated viruses (AAV) and Adenoviruses.
- AAV Adeno-associated viruses
- the variant and targeting construct may be on different nucleic acid constructs.
- the variant in the form of a peptide and the targeting construct as a nucleic acid molecule may be used in combination.
- said sequence to be introduced is a sequence which inactivates the HIV provirus.
- sequence which inactivates the HIV provirus comprises in the 5' to 3' orientation: a first transcription termination sequence and a marker cassette including a promoter, the marker open reading frame and a second transcription termination sequence, and said sequence interrupts the transcription of the coding sequence.
- sequence sharing homologies with the regions surrounding DNA target sequence is from the HIV provirus or a fragment of the HIV provirus comprising sequences upstream and downstream of the cleavage site, so as to allow the deletion of coding sequences flanking the cleavage site.
- a cell according to the present invention may be made according to a method, comprising at least the step of: (a) introducing into a cell, a meganuclcase, as defined above, so as to induce a double stranded cleavage at a site of interest of the GIP comprising a DNA recognition and cleavage site of said meganuclease, and thereby generate a genomically modified cell having repaired the double-strands break, by non- homologous end joining, and
- step (b) isolating the genomically modified cell of step (a), by any appropriate mean.
- the cell which is modified may be any cell of interest.
- the cells are pluripotent precursor cells such as embryo- derived stem (ES) cells, which are well-known in the art.
- the cells may advantageously be human cells, for example PerC6 (Fallaux et al., Hum. Gene Ther. 9, 1909-1917, 1998) or HEK293 (ATCC # CRL- 1573) cells or an immortal T lymphocyte line such as Jurkat (Schneider et al (1977). Int J Cancer 19 (5): 621-6.).
- the meganuclease can be provided directly to the cell or through an expression vector comprising the polynucleotide sequence encoding said meganuclease linked to regulatory sequences suitable for directing its expression in the cell used.
- modified cell line would have a number of potential uses including the elucidation of aspects of the biology of the modified GIP as well as a model for screening compounds and other substances for therapeutic effects against ceils comprising the modified GIP.
- a non-human transgenic animal which is modified by a polynucleotide according to a second aspect of the present invention or a vector according to a third aspect of the present invention.
- the subject-matter of the present invention is also a method for making an animal which comprises a modified GIP, comprising at least the step of:
- step (a) introducing into a pluripotent precursor cell or an embryo of an animal, a meganuclease, as defined above, so as to induce a double stranded cleavage at a site of interest of the GIP comprising a DNA recognition and cleavage site of said meganuclease, and thereby generate a genomically modified precursor cell or embryo having repaired the double-strands break by non-homologous end joining, (b) developing the genomically modified animal precursor cell or embryo of step (a) into a chimeric animal, and
- step (c) deriving a transgenic animal from a chimeric animal of step (b).
- the GIP may be inactivated by insertion of a sequence of interest by homologous recombination between the genome of the animal and a targeting DNA construct according to the present invention.
- targeting DNA is introduced into the cell under conditions appropriate for introduction of the targeting DNA into the site of interest.
- step (b) comprises the introduction of the genoraically modified precursor cell obtained in step (a), into blastocysts, so as to generate chimeric animals.
- transgenic animal could be used as a multicellular animal model to elucidate aspects of the biology of the GIP, by means of engineering the provirus present in the progenitor cell line. Such transgenic animals also could be used to screen and characterise the effects of for instance novel anti-HIV medicaments.
- the targeting DNA construct is inserted in a vector.
- the targeting DNA comprises the sequence of the exogenous gene encoding the protein of interest, and eventually a marker gene, flanked by sequences upstream and downstream of and essential gene in the HIV provirus, as defined above, so as to generate genomically modified cells (animal precursor cell or embryo/animal or human cell) having replaced the HIV gene by the exogenous gene of interest, by homologous recombination.
- the exogenous gene and the marker gene are inserted in an appropriate expression cassette, as defined above, in order to allow expression of the heterologous protein/marker in the transgenic animal/recombinant cell line.
- the meganuclease can be used either as a polypeptide or as a polynucleotide construct encoding said polypeptide. It is introduced into somatic cells of an individual, by any convenient means well-known to those in the art, which are appropriate for the particular cell type, alone or in association with either at least an appropriate vehicle or carrier and/or with the targeting DNA. Once in a cell, the meganucleasc and if present, the vector comprising targeting DNA and/or nucleic acid encoding a meganuclcase are imported or translocated by the cell from the cytoplasm to the site of action in the nucleus.
- transgenic plant which is modified by a polynucleotide according to a second aspect of the present invention or a vector according to a third aspect of the present invention.
- variant or single-chain chimeric meganuclease or vector is associated with a targeting DNA construct.
- the use of the variant is for inducing a double-strand break in a site of interest within the GIP, thereby inducing a DNA recombination event, a DNA loss or cell death.
- said double-strand break is for: modifying a specific sequence in the GIP, so as to induce restoration of a GIP function such as replication in studies upon the biology of the virus, or to attenuate or activate the GIP or a gene therein, introducing a mutation into a site of interest of a GIP gene, introducing an exogenous gene or a part thereof, inactivating or deleting the GIP or a part thereof or leaving the DNA unrepaired and degraded.
- this present aspect of the present invention relates to the use of a meganuclease variant to treat HIV infection, by inactivating the HIV provirus by therapeutic genome engineering.
- the use of the meganuclease according to the present invention comprises at least the following steps:
- the meganuclease is provided directly to the cell or through an expression vector comprising the polynucleotide sequence encoding of the meganuclease and is suitable for its expression in the host cell.
- This strategy is used to introduce a DN ⁇ sequence at the target site, for example to generate a HIV provirus knock-in or knock-out animal model or cell lines that can be used for drug testing or in the case of a cell line, which can be used for administration into a patient from whom it was derived.
- the use of the meganuclease comprises at least the following steps:
- inter chromosome arm recombination events are also expected to occur following cleavage of the provirus target.
- the recombination of chromosome arms occurs most frequently during mitosis, but can also occur as part of the repair mechanism for DNA strand breaks.
- Such an inter chromosome arm recombination event would either lead to the elimination of the non homologous portions on either side of the break (e.g. the provirus) or more likely cause portions of the provirus to be recombined onto different chromosome arms. In either event this would lead to the inactivation of the provirus.
- the use of the meganuclease comprises at least the following steps:
- the variant is used for genome therapy to knock-out in animals/cells the GIP, in particular a sequence is introduced which inactivates the HIV provirus.
- the introduced sequence may also delete the HIV provirus or part thereof, and introduce an exogenous gene or part thereof (knock-in/gene replacement).
- the DNA which repairs the site of interest may comprise the sequence of an exogenous gene of interest, and a selection marker, such as the G418 resistance gene.
- the sequence to be introduced can be any other sequence used to alter the chromosomal DNA in some specific way including a sequence used to modify a specific sequence, to attenuate or activate the endogenous gene of interest in the H ⁇ V provirus or to introduce a mutation into a site of interest in the HIV provirus.
- Such chromosomal DNA alterations may be used for genome engineering (animal models and recombinant cell lines including human cell lines).
- the sequence to be introduced comprises, in the 5' to 3' orientation: at least a transcription termination sequence (polyAl), preferably said sequence fuithei comprises a marker cassette including a promoter and the marker open reading frame (ORF) and a second transcription termination sequence for the marker gene ORF (polyA2).
- polyAl transcription termination sequence
- said sequence fuithei comprises a marker cassette including a promoter and the marker open reading frame (ORF) and a second transcription termination sequence for the marker gene ORF (polyA2).
- Inactivation of the HIV provirus may also occur by insertion of a marker gene within an essential gene of HIV, which would disrupt the coding sequence.
- the insertion can in addition be associated with deletions of ORF sequences flanking the cleavage site and eventually, the insertion of an exogenous gene of interest (gene replacement).
- inactivation of the HIV provirus may also occur by insertion of a sequence that would destabilize the mRNA transcript of an essential gene.
- the present invention also provides a composition characterized in that it comprises at least one variant as defined above (variant or single-chain derived chimeric meganucleasc) and/or at least one expression vector encoding the variant, as defined above.
- provirus targeting variant in as both a peptide and nucleotide form allows for the immeadiate action of the variant as as its persistence in the target cell.
- composition comprises more than one variant, wherein each of the variants is directed towards a different target sequence in the provirus.
- composition comprises a targeting DNA construct comprising a sequence which inactivates the HlV provirus, flanked by sequences sharing homologies with the genomic DNA cleavage site of said variant, as defined above.
- said targeting DNA construct is either included in a recombinant vector or it is included in an expression vector comprising the polynucleotide(s) encoding the variant according to the invention.
- the subject-matter of the present invention is also the use of at least one meganuclease and/or one expression vector, as defined above, for the preparation of a medicament for preventing, improving or curing HIV infection in an individual in need thereof.
- the subject-matter of the present invention is also the use of at least one variant and/or one expression vector, as defined above, for the preparation of a medicament for preventing, improving or curing a pathological condition associated with HIV infection in an individual in need thereof.
- the variants according to the present invention provide a possible means to prevent chromosomal integration of a target cell with the retrovirus genome.
- the first step of the viral infection following viral entry into the target cell is the reverse transcription (RT) of the viral genomic RNA.
- RT reverse transcription
- a linear double stranded DNA molecule is formed which then enters the nucleus so that it can be integrated in the cellular genome.
- Meganuclease variants of the present invention are also able to cleave the pre-integration complex (PlC), which is an episomal double stranded DNA molecule, conferring a protective effect during the earliest steps of viral infection, of a cell population.
- PlC pre-integration complex
- the use of the meganuclease may comprise at least the step of (a) inducing in somatic tissue(s) of the donor/ individual a double stranded cleavage at a site of interest of the HIV provirus comprising at least one recognition and cleavage site of said meganuclease by contacting said cleavage site with said meganuclease, and (b) introducing into said somatic tissue(s) a targeting DNA, wherein said targeting DNA comprises (1) DNA sharing homologies to the region surrounding the cleavage site and (2) DNA which inactivates the HIV provirus upon recombination between the targeting DNA and the chromosomal DNA, as defined above.
- the targeting DNA is introduced into the somatic tissues(s) under conditions appropriate for introduction of the targeting DNA into the site of interest.
- the targeting construct may comprise sequences for deleting the HIV provirus or a portion thereof and introducing the sequence of an exogenous gene of interest (gene replacement).
- the use of the meganuclease comprises at least the step of: inducing in somatic tissue(s) of the donor/individual a double stranded cleavage at a site of interest of the HIV provirus comprising at least one recognition and cleavage site of the meganuclease by contacting the cleavage site with the meganuclease, and thereby inducing mutagenesis of an open reading frame in the HIV provirus by repair of the double-strands break by non-homologous end joining.
- said double-stranded cleavage may be induced, ex vivo by introduction of said meganuclease into infected cells isolated for instance from the circulatory system of the donor/individual and then transplantation of the modified cells back into the diseased individual.
- the subject-matter of the present invention is also a method for preventing, improving or curing HIV infection, in an individual in need thereof, said method comprising at least the step of administering to said individual a composition as defined above, by any means.
- the meganucleases and a pharmaceutically acceptable excipient are administered in a therapeutically effective amount.
- Such a combination is said to be administered in a "therapeutically effective amount” if the amount administered is physiologically significant.
- An agent is physiologically significant if its presence results in a detectable change in the physiology of the recipient.
- an agent is physiologically significant if its presence results in a decrease in the severity of one or more symptoms of the targeted HIV infection.
- the meganuclease comprising compo- sitions should be non-immunogenic, i.e., engender little or no adverse immunological response.
- a variety of methods for ameliorating or eliminating deleterious immunological reactions of this sort can be used in accordance with the invention.
- One means of achieving this is to ensure that the meganuclease is substantially free of N-formyl methionine.
- Another way to avoid unwanted immunological reactions is to conjugate meganucleases to polyethylene glycol (“PEG”) or polypropylene glycol (“PPG”) (preferably of 500 to 20,000 daltons average molecular weight (MW)). Conjugation with PEG or PPG 5 as described by Davis et al.
- the invention also relates to meganuclease variants, related materials and uses thereof which recognize non-virus retroelements and/or the integrated genomes of viruses which do not have mechanisms to integrate into the host cell genome.
- Non-virus retroelements are endogenous genomic DNA elements that include the gene for reverse transcriptase and are also known as class I transposable elements. These retrotransposons, include the long terminal repeat (LTR) retrotransposons, non-LTR retroposons and group II mitochondrial introns. They are though to be derived from partially inactivated retroviruses which have lost the ability to form infective virus particles. These genetic elements however are increasingly becoming associated with various diseases, in particular cancers and immune disorders which result form the integration of the element into a site close to a gene (s) whose misregulation leads to the observed disease phenotype.
- LTR long terminal repeat
- the present invention therefore also relates to meganuclease variants which can be used to cleave a genomic retrotransposon either in a specific tissue or cell type or more generally so as to treat the disease phenotype using one or more of the mechanisms described above.
- the present invention also relates to meganuclease variants which can recognise and cleave targets in genomic insertions of viruses which do not normally insert into the host cell genome.
- the non-specific insertion of viral genetic material into the host cell genome is a disease causing mechanism which is currently being investigated. For example in the important virus hepatitis B, chronic infection with this virus is associated with a greatly elevated risk of hepatocellular carcinoma.
- Hepatocellular carcinoma is one of the most common cancers in the world and hence a treatment for this condition, using a meganuclease variant which can cleave the randomly integrated hepatitis B genome and have a therapeutic affect upon hepatocytes via one or more of mechanisms detailed above is therefore also within the scope of the present invention as are more generally meganuclease variants to genomically integrated copies of virus genetic material which cause a disease phenotype.
- - Amino acid residues in a polypeptide sequence are designated herein according to the one-letter code, in which, for example, Q means GIn or Glutamine residue, R means Arg or Arginine residue and D means Asp or Aspartic acid residue.
- - Amino acid substitution means the replacement of one amino acid residue with another, for instance the replacement of an Arginine residue with a Glutamine residue in a peptide sequence is an amino acid substitution.
- - Altered/enhanced/increased cleavage activity refers to an increase in the detected level of meganuclease cleavage activity (see below) against a target DNA sequence by a first meganuclease in comparison to the activity of a second meganuclease against the target DNA sequence.
- the first meganuclease will be a variant of the second and comprise one or moie substituted amino acid residues in comparison to the second meganuclease.
- beta-hairpin it is intended two consecutive beta-strands of the antiparallel beta-sheet of a LAGLIDADG homing endonuclease core domain ( ⁇ ip2 or ⁇ 3 ⁇ 4 ) which are connected by a loop or a turn,
- chimeric DNA target or “hybrid DNA target” it is intended the fusion of a different half of two parent meganuclease target sequences.
- at least one half of said target may comprise the combination of nucleotides which are bound by at least two separate subdomains (combined DNA target).
- the cleavage activity of the variant according to the invention may be measured by any well-known, in vitro or in vivo cleavage assay, such as those described in the International PCT Application WO 2004/067736; Epinat et at, Nucleic Acids Res., 2003, 31, 2952-2962; Chames el at, Nucleic Acids Res., 2005, 33, el78; Arnould et al., J. MoI. Biol, 2006, 355, 443-458, and ⁇ rnould et al, J. MoI. Biol., 2007, 371, 49-65.
- the cleavage activity of the variant of the invention may be measured by a direct repeat recombination assay, in yeast or mammalian cells, using a reporter vector.
- the reporter vector comprises two truncated, non-functional copies of a reporter gene (direct repeats) and the genomic (non-palindromic) DNA target sequence within the intervening sequence, cloned in a yeast or a mammalian expression vector.
- the genomic DNA target sequence comprises one different half of each (palindromic or pseudo-palindromic) parent homodimcric meganuclease target sequence.
- hcterodi merle variant results in a functional endonuclease which is able to cleave the genomic DNA target sequence.
- This cleavage induces homologous recombination between the direct repeats, resulting in a functional reporter gene (LacZ, for example), whose expression can be monitored by an appropriate assay.
- the specificity of the cleavage by the variant may be assessed by comparing the cleavage of the (non-palindromic) DNA target sequence with that of the two palindromic sequences cleaved by the parent homodimeric meganucleases or compared with wild type meganuclease.
- selection or selecting it is intended to mean the isolation of one or more meganuclease variants based upon an observed specified phenotype, for instance altered cleavage activity.
- This selection can be of the variant in a peptide form upon which the observation is made or alternatively the selection can be of a nucleotide coding for selected meganuclease variant.
- screening it is intended to mean the sequential or simulta- neous selection of one or more meganuclease variant (s) which exhibits a specified phenotype such as altered cleavage activity.
- derived from it is intended to mean a meganuclease variant which is created from a parent meganuclease and hence the peptide sequence of the meganuclease variant is related to (primary sequence level) but derived from (muta- tions) the sequence peptide sequence of the parent meganuclease.
- domain or “core domain” it is intended the "LAGLIDADG homing endonuclease core domain” which is the characteristic ⁇ i ⁇ i ⁇ 2 ⁇ 2 ⁇ 3 ⁇ 4 ⁇ 3 fold of the homing endonucleases of the LAGLIDADG family, corresponding to a sequence of about one hundred amino acid residues.
- Said domain comprises four beta-strands ( ⁇ i ⁇ 2 ⁇ 3 ⁇ 4 ) folded in an antiparallel beta-sheet which interacts with one half of the DNA target.
- This domain is able to associate with another LAGLIDADG homing endonuclease core domain which interacts with the other half of the DNA target to form a functional endonuclease able to cleave said DNA target.
- the LAGLIDADG homing endonuclease core domain corresponds to the residues 6 to 94.
- the DNA target is defined by the 5' to 3' sequence of one strand of the double-stranded polynucleotide, as indicated for C1221 (see figure 1). Cleavage of the DNA target occurs at the nucleotides at positions +2 and -2, respectively for the sense and the antisense strand. Unless otherwise indicated, the position at which cleavage of the DNA target by an I-Cre I meganuclease variant occurs, corresponds to the cleavage site on the sense strand of the DNA target.
- DNA target half-site by "DNA target half-site", "half cleavage site” or half-site” it is intended the portion of the DNA target which is bound by each LAGLIDADG homing endonuclease core domain.
- DNA target sequence from the HIV provirus it is intended a 20 to 24 bp sequence of the HIV provirus which is recognized and cleaved by a meganuclease variant.
- the DNA target sequence from then HIV provirus is in an essential gene sequence and/or within an essential regulatory sequence and/or within an essential structural sequence of the HIV provirus.
- first/second/third/n f!l series of variants it is intended a collection of variant meganucleases, each of which comprises one or more amino acid substitution in comparison to a parent meganuclease from which all the variants in the series are derived.
- functional variant it is intended a variant which is able to cleave a DNA target sequence, preferably said target is a new target which is not cleaved by the parent meganuclease.
- such variants have amino acid variation at positions contacting the DNA target sequence or interacting directly or indirectly with said DNA target.
- heterodimer it is intended to mean a meganuclease comprising two non-identical monomers. In particular the monomers may differ from each other in their peptide sequence and/or in the DNA target half-site which they recognise and cleave.
- homologous is intended a sequence with enough identity to another one to lead to a homologous recombination between sequences, more particularly having at least 95 % identity, preferably 97 % identity and more preferably 99 %.
- all the l-Crel variants described comprise an additional Alanine after the first Methionine of the wild type 1-OeI sequence (SEQ ID NO: 344). These variants also comprise two additional Alanine residues and an Aspartic Acid residue after the final Proline of the wild type l-Crel sequence.
- l-Crel sites include the wild-type (natural) non- palindromic l-Crel homing site and the derived palindromic sequences such as the sequence 5'- Li2C- ⁇ a-ioa.9a ⁇ a-7C -6 g -5 t.4C-3g -2 tia + ic+2gi3a + 4C + sg+6tf7tH 8t+9t) iogi i ia» ⁇ 2 (SEQ ID NO: 343), also called C 1221 .
- identity refers to sequence identity between two nucleic acid molecules or polypeptides. Identity can be determined by comparing a position in each sequence which may be aligned for purposes of comparison.
- nucleic acid or amino acid sequences When a position in the compared sequence is occupied by the same base, then the molecules are identical at that position.
- a degree of similarity or identity between nucleic acid or amino acid sequences is a function of the number of identical or matching nucleotides at positions shared by the nucleic acid sequences.
- Various alignment algorithms and/or programs may be used to calculate the identity between two sequences, including FASTA, or BLAST which are available as a part of the GCG sequence analysis package (University of Wisconsin, Madison, Wis.), and can be used with, e.g., default settings.
- meganuclease an cndonuclease having a double-stranded DNA target sequence of 12 to 45 bp.
- the meganuclease is either a dimeric enzyme, wherein each domain is on a monomer or a monomelic enzyme comprising the two domains on a single polypeptide.
- meganuclease domain the region which interacts with one half of the DNA target of a meganuclease and is able to associate with the other domain of the same meganuclease which interacts with the other half of the DNA target to form a functional meganuclease able to cleave said DNA target.
- meganuclease variant or “variant” it is intended a meganuclease obtained by replacement of at least one residue in the amino acid sequence of the parent meganuclease (natuial oi variant meganuclease) with a different amino acid.
- monomer it is intended to mean a peptide encoded by the open reading frame of the 1-Crel gene or a variant thereof, which when allowed to dimcrise forms a functional l-Cre ⁇ enzyme. In particular the monomers dimerise via interactions mediated by the L ⁇ GLIDADG motif.
- mutant is intended the substitution, deletion, insertion of one or more nucleotides/amino acids in a polynucleotide (cDNA, gene) or a polypeptide sequence.
- Said mutation can affect the coding sequence of a gene or its regulatory sequence. It may also affect the structure of the genomic sequence or the structure/stability of the encoded mRNA.
- onc-lctter code is used for designating the base of a nucleoside: a is adenine, t is thymine, c is cytosine, and g is guanine.
- r represents g or a (purine nucleotides)
- k represents g or t
- s represents g or c
- w represents a or t
- m represents a or c
- y repre- sents t or c pyrimidine nucleotides
- d represents g, a or t
- v represents g, a or c
- b represents g, t or c
- h represents a, t or c
- n represents g, a, t or c.
- parent meganuclease it is intended to mean a wild type meganuclease or a variant of such a wild type meganuclease with identical properties or alternatively a meganuclease with some altered characteristic in comparison to a wild type version of the same meganuclease.
- the parent meganuclease can refer to the initial meganuclease from which the first series of variants are derived in step a. or the meganuclease from which the second series of variants are derived in step b., or the meganuclease from which the third series of variants are derived in step k.
- peptide linker it is intended to mean a peptide sequence of at least 10 and preferably at least 17 amino acids which links the C terminal amino acid residue of the first monomer to the N terminal residue of the second monomer and which allows the two variant monomers to adopt the correct conformation for activity and which does not alter the specificity of either of the monomers for their targets.
- provirus it is intended to mean a DNA version of a retrovirus genome.
- the provirus may be the DNA molecule directly resulting from the reverse transcription of the RNA genome of a virus or alternatively it may be the chromosomally integrated version of the virus genome present at one or more sites in one or more chromosomes of the target cell.
- subdomain it is intended the region of a LAGLIDADG homing endonuclease core domain which interacts with a distinct part of a homing endonuclease DNA target half-site.
- single-chain meganuclease a meganuclease comprising two LAGLIDADG homing endonuclease domains or core domains linked by a peptidic spacer.
- the single-chain meganuclease is able to cleave a chimeric DNA target sequence comprising one different half of each parent meganuclease target sequence.
- targeting DNA construct/minimal repair matrix/repair matrix it is intended to mean a DNA construct comprising a first and second portions which are homologous to regions 5' and 3 5 of the DNA target in situ.
- the DNA construct also comprises a third portion positioned between the first and second portion which comprise some homology with the corresponding DNA sequence in situ or alternatively comprise no homology with the regions 5' and 3 ' of the DNA target in situ.
- a homologous recombination event is stimulated between the genome containing the HIV provirus and the repair matrix, wherein the genomic sequence containing the DNA target is replaced by the third portion of the repair matrix and a variable part of the first and second portions of the repair matrix.
- vector a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked into a host cell in vitro, in vivo or ex vivo.
- Figure 1 Schematic representation of an HIV-I viral particle. The two molecules of genomic RNA are represented, together with the RT, inside the viral capsid.
- the envelope derived from the membrane of the infected cells, contains the envelope glycoproteins gp41and g ⁇ l20.
- Figure 2 A: Organization of the HIV-I genomic RNA molecules, Different genes are represented with different shades of grey, and the proteins encoded by these genes are represented in the lower part of the panel.
- B Genetic organization of the integrated HIV-I provirus, showing the structure of the LTRs after duplication of the U3 and U5 sequences during reverse transcription.
- Figure 3 Tridimensional structure of the I-Oel homing endonuclease bound to its DNA target.
- the catalytic core is surrounded by two ⁇ oc ⁇ folds forming a saddle-shaped interaction interface above the DNA major groove.
- Figure 4 Different 1-OeI variants binding different sequences derived from the l-Crel target sequence (top right and bottom left) to obtain heterodimers or single chain fusion molecules cleaving non palindromic chimeric targets (bottom right).
- Figure 5 Shows a schematic representation of the smaller independent subunits of the l-Crel meganuclease, i.e., s ⁇ bunit within a single monomer or ⁇ fold (top right and bottom left). These independent subunits allow for the design of novel chimeric molecules (bottom right), by combination of mutations within a same monomer. Such molecules would cleave palindromic chimeric targets (bottom right).
- Figure 6 Shows a schematic representation of a method to combine four different subdomains so as to generate a custom meganuclease which cleaves a selected target.
- Figure 7 The IIIV1_1 target sequence (SEQ ID NO:319) and its derivatives. In the HIV1_1.2 target (SEQ ID NO:320), the AC ⁇ C sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343).
- HIV1JL3 (SEQ ID NO:321) is the palindromic sequence derived from the left part of HIVlJ .2, (SEQ ID NO:320) and HIVlJ .4 (SEQ ID NO:322) is the palindromic sequence derived from the right part of HIV IJ .2 (SEQ ID NO:320).
- HIVlJ .5 (SEQ ID NO:323) and HIVlJ .6 (SEQ ID NO:324) are pseudo- palindromic targets derived, respectively, from HIV IJ .3 (SEQ ID NO:321) and HIVl J .4 (SEQ ID NO:322), containing the natural ACAC sequence in the middle of the target.
- the boxed motives from 1 OAGAJ 3 , 1 OTGGJP, 5TACJP and 5CTG_P are found in the HIVlJ series of targets.
- Figure 8 pCLS1055 plasmid map.
- Figure 9 pCLS0542 plasmid map.
- Figure 10 Cleavage of HIVlJ .3 (SEQ ID NO:321) target by combinatorial variants.
- the figure displays an example of screening of I-Crel combi- natorial valiants with the HIVl J.3 target (SEQ ID NO:321).
- the positive variants correspond to: BlO, SEQ ID NO: 1; Cl, SEQ ID NO:2; C7, SEQ ID NO:3; ClO, SEQ ID NO:4; C3 : SEQ ID NO:5; all described in Table II.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIVl J .3 target (SEQ ID NO:321) has been mated with another yeast strain containing the meganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 11 pCLS 1 107 plasmid map.
- Figure 12 Cleavage of HIVlJ .4 (SEQ ID NO:322) and H ⁇ V1J .6 (SEQ ID NO:324) targets by combinatorial variants.
- the figure displays an example of screening of l-Crel combinatorial variants with the HIVl J .4 (SEQ ID NO:322) and HIVlJ .6 (SEQ ID NO:324) targets.
- the positive variants correspond to: C8, SEQ ID NO:7; A5, SEQ ID NO:8; Al, SEQ ID NO:9; A12, SEQ ID NO: 10; C3, SEQ ID NO:1 1; all described in Table IV.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIVl J.4 or the HIVlJ .6 targets have been mated with another yeast strain containing the meganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3)
- Figure 13 Cleavage of the HIVlJ .2 (SEQ ID NO:320) and HIVlJ (SEQ ID NO:319) target sequences by heterodimeric combinatorial variants.
- Left panel Example of screening of combinations of l-Crel variants against the HIV IJ.2 target.
- Right panel Screening of the same combinations of 1-OeI variants against the HIVlJ target.
- Some heterodimers resulted in cleavage of the HIV IJ .2 target (SEQ ID NO:320).
- the heterodimer displaying a signal with HIVlJ target (SEQ ID NO:319) is observed at positions D3.
- each cluster contains 6 spots.
- a yeast strain harboring the HIVlJ target SEQ ID NO:319
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3)
- Figure 14 Cleavage of HIVlJ .3 (SEQ ID NO:321) and HIVl J.5 (SEQ ID NO: 323) targets by meganuclease variants improved by random mutagenesis in example 5.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_1.3 and HIV1_L5 targets.
- the positive variants presented correspond to: F3, SEQ ID NO:27; CI l , SEQ ID NO:26; 118, SEQ ID NO:28; El 2, SEQ ID NO:29; all described in Table VIII. Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_1.3 (SEQ ID NO:321) or the HIVlJ .5 (SEQ ID NO:323) targets have been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIVl J.3 target (SEQ ID NO:321).
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 15 Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by random mutagenesis in example 5.
- the figure displays an example of screening of I-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:46) cleaving the HIV IJ.4 target.
- the positive variants presented correspond to: F3, SEQ ID NO:27; Cl 1 , SEQ ID NO:26; I ⁇ 8, SEQ ID NO:28; E12, SEQ ID NO:29; all described in Table VIIL Each cluster contains 6 spots.
- a yeast strain harboring the HIVlJ .4 mutant and the HIVlJ target have been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant.
- the two spots on the right contain the same negative or positive controls, These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 16 Cleavage of HIVlJ .3 (SEQ ID NO:321) and HIVl J.5 (SEQ ID NO:323) targets by meganuclease variants improved by a second round of random mutagenesis in example 5bis.
- the figure displays an example of screening of I-CVel meganuclease variants with the HIVl J .3 and HIVl J .5 targets.
- the positive variants presented correspond to: A12 5 SEQ ID NO:42; D8, SEQ ID NO:38; G8, SEQ ID NO:36; G3, SEQ ID NO:40; all described in Table IX. Each cluster contains 4 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 17 Cleavage of HIVlJ (SEQ ID NO:319) target by meganuciease variants improved by a second round of random mutagenesis in example 5bis.
- the figure displays an example of screening of 1-OeI meganuciease variants with the HIV IJ target, when mated with a meganuciease (SEQ ID NO:46) cleaving the HIVl J .4 target.
- the positive variants presented correspond to: A12, SEQ ID NO:42; D8, SEQ ID NO:38; G8, SEQ ID NO:36; G3, SEQ ID NO:40; all described in Table IX. Each cluster contains 6 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 18 Cleavage of HIVl J.3 (SEQ ID NO:321) and HIV1J .5 (SEQ ID NO:323) targets by meganuciease variants improved by site-directed mutagenesis in example 6.
- the figure displays an example of screening of l-Cre ⁇ meganuciease variants with the HIVl J .3 and HIVl J .5 targets.
- the positive variants presented correspond to: FlO, SEQ ID NO:63; H2, SEQ ID NO:60; H3, SEQ ID NO:59; A3, SEQ ID NO:64; F4, SEQ ID NO:65; some of them described in Table XI.
- Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIVlJ target (see Figure 19). This is due to the presence of the Gl 9S mutation in these variants.
- Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIVl J.3 or the HIVl J .5 targets have been mated with another yeast strain containing the meganuciease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 19 Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by site-directed mutagenesis in example 6.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:46) cleaving the HIV IJ .4 target.
- the positive variants presented correspond to: Fl O, SEQ ID NO:63; H2, SEQ ID NO:60; H3, SEQ ID NO:59; A3, SEQ ID NO:64; F4, SEQ ID NO:65; some of them described in Table XI.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV IJ .4 mutant and the HIVlJ target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 20 Cleavage of HIVlJ .4 (SEQ ID NO:322) and HIVLl .6 (SEQ ID NO:324) targets by meganuclease variants improved by random mutagenesis in example 7.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets.
- the positive variants presented correspond to: B7, SEQ ID NO:46; B9, SEQ ID NO:68; B 12, SEQ ID NO:69; A9, SEQ ID NO:70; E5, SEQ ID NO:71 ; all described in Table XIII. Each cluster contains 6 spots.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:26) cleaving the HIVlJ .3 target.
- the positive variants presented correspond to: B7, SEQ ID NO:46; B9, SEQ ID NO:68; B12, SEQ ID NO:69; A9, SEQ ID NO:70; E5, SEQ ID NO:71; all described in Table XIII.
- Each cluster contains 6 spots.
- a yeast strain harboring the HIVl J.3 mutant (SEQ ID NO:26) and the IHVlJ target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster AI ), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 22 Cleavage of HIVlJ .4 (SEQ ID NO:322) and HIVlJ .6
- SEQ ID NO:324 targets by meganuclease variants improved by a second round of random mutagenesis in example 7bis.
- the figure displays an example of screening of I-Crel meganuclease variants with the HIVl J .4 and HIVl J .6 targets.
- the positive variants presented correspond to: A3, SEQ ID NO:76; Bl, SEQ ID NO:77; Cl, SEQ ID NO:78; D3, SEQ ID NO:79; D5, SEQ ID NO:80; all described in Table XIV. Each cluster contains 4 spots.
- a yeast strain harboring the 1IIV1 J .4 or the HIVl J.6 targets have been mated with another yeast strain containing the rneganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 23 Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by a second round of random mutagenesis in example 7bis.
- the figure displays an example of screening of 1-OeI meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:26) cleaving the HIVl J .3 target.
- the positive variants presented correspond to: A3, SEQ ID NO:76; Bl , SEQ ID NO:77; Cl , SEQ ID NO:78; D3, SEQ ID NO:79; D5, SEQ ID NO: 80; all described in Table XIV.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV 1_1 .3 mutant (SEQ ID NO:26) and the HIV 1_1 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2) ; and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 24 The HIVlJ target sequence (SEQ ID NO:325) and its derivatives.
- HIV1_3.2 target (SEQ ID NO:32 ⁇ )
- the TTTA sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343).
- HIV1_3.3 (SEQ ID NO: 327) is the palindromic sequence derived from the left part of HIV IJ, .2, and H ⁇ V1_3.4 (SEQ ID NO:328) is the palindromic sequence derived from the right part of HIVl J.2.
- HIVl J.5 (SEQ ID NO:329) and HIVl _3.6 (SEQ ID NO:330) are pseudo-palindromic targets derived, respectively, from HIV1_3.3 and HIV1_3,4, containing the natural TTTA sequence in the middle of the target.
- FIG. 25 Cleavage of HIV1_3.3 target (SEQ ID NO:327) by combinatorial variants.
- the figure displays an example of screening of l-Crel combinatorial variants with the HIV1_3.3 target.
- the positive variants correspond to: A6, SEQ ID NO:89; Al, SEQ ID NO:91 ; A8, SEQ ID NO.90; ⁇ 4, SEQ ID NO:88; all described in Table XVI. Each cluster contains 4 spots.
- SEQ ID NO:330 targets by combinatorial variants.
- the figure displays an example of screening of l-Crel combinatorial variants with the HIV1_3.4 and HIV1_3.6 targets.
- the positive variants correspond to: C12, SEQ ID NO:98; C8, SEQ ID NO:99; E4, SEQ ID NO: 100; G4, SEQ ID NO:97; E9, SEQ ID NO: 101; all described in Table XVIII.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_3.4 or the HIV1_3.6 targets has been mated with another yeast strain containing the mcganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 27 Cleavage of HIV1_3.3 target (SEQ ID NO:327) by meganuclease variants improved by random mutagenesis in example 12.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_3.3 target.
- the positive variants presented correspond to: El, SEQ ID NO: 105; C8, SEQ ID NO: 106; A2, SEQ ID NO:107; A7, SEQ ⁇ D NO:108; BlO, SEQ ID NO: 109; all described in Table XIX. Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_3.3 target has been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant cleaving the IIIV1_3.3 target.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 28 Cleavage of HIV1_3.3 target (SEQ ID NO:327) by mcganuclease variants improved by a second round of random mutagenesis in example 12bis.
- the figure displays an example of screening of 1-OeI meganuclease variants with the HIV1_3.3 target.
- the positive variants presented correspond to: Al l, SEQ ID NO:115; B7, SEQ ID NO:1 16; F12, SEQ ID NO:117; G2, SEQ ID NO: 118; H9, SEQ ID NO:1 19; all described in Table XX. Each cluster contains 4 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
- Figure 29 Cleavage of H ⁇ V1J.3 (SEQ ID NO:327) and HIV1_3.5 (SEQ ID NO: 329) targets by meganuclease variants improved by site-directed mutagenesis in example 13.
- the figure displays an example of screening of 1-OeI meganuclease variants with the HIV1_3.3 and HIV1J3.5 targets.
- the positive variants presented correspond to: Al, SEQ ID NO: 126; G3, SEQ ID NO: 127; Cl, SEQ ID NO:128; H6, SEQ ID NO: 129; E5, SEQ ID NO: 130; described in Table XXI.
- Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIV1_3 target (see Figure 30). This is due to the presence of the Gl 9S mutation in these variants.
- Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_3.3 or the HIV1_3.5 targets have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a previously improved variant.
- Figure 30 Cleavage of HIV1_3 (SEQ ID NO:325) target by meganuclease variants improved by site-directed mutagenesis in example 13.
- the figure displays an example of screening of ⁇ -Cre ⁇ meganuclease variants with the HIV 1_3 target, when mated with a meganuclease (SEQ ID NO: 125) cleaving the HIV1_3.4 target.
- the positive variants presented correspond to: Al, SEQ ID NO:12 ⁇ ; G3, SEQ ID NO:127; Cl, SEQ ID NO: 128; H ⁇ , SEQ ID NO:129; E5, SEQ ID NO: 130; described in Table XXI.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV 1_3.4 mutant (SEQ ID NO: 125) and the HIV1_3 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a previously improved variant.
- Figure 31 Cleavage of H1V1_3.4 (SEQ ID NO:328) and HIVlJ.6 (SEQ ID NO: 330) targets by meganuclease variants improved by random mutagenesis in example 14.
- the figure displays an example of screening of 1-CVeI meganuclease variants with the HIVl J.4 and HIVl J, 6 targets.
- the positive variants presented correspond to: E8, SEQ ID NO: 136; B 12, SEQ ID NO : 137; B l , SEQ ID NO: 138; B8, SEQ ID NO: 139; D6, SEQ ID NO: 140; all described in Table XXII. Each cluster contains 6 spots.
- a yeast strain harboring the HIVl J.4 or the HIVlJ.6 targets has been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV IJ.4 target.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3),
- Figure 32 Cleavage of HIVl J.4 (SEQ ID NO:328) and HIVl J.6 (SEQ ID NO:330) targets by meganuclease variants improved by a second round of random mutagenesis in example 14bis.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets.
- the positive variants presented correspond to: F7, SEQ ID NO:146; B12, SEQ ID NO: 147; G7, SEQ ID NO:148; D2, SEQ ID NO:149; A5, SEQ ID NO: 150; all described in Table XXIII. Each cluster contains 4 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a previously improved variant.
- Figure 33 Cleavage of HIVl J.4 (SEQ ID NO:328) and HIVl J.6 (SEQ ID NO: 330) targets by meganuclease variants improved by site-directed mutagenesis in example 15.
- the figure displays an example of screening of I-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets.
- the positive variants presented correspond to: Dl 5 SEQ ID NO:156; C2, SEQ ID NO:157; F2, SEQ ID NO:158; A4, SEQ ID NO:159; G7, SEQ ID NO: 160; described in Table XXIV. Each cluster contains 6 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a non-improved variant.
- Figure 34 Cleavage of HIV1_3 target (SKQ ID NO:325) by meganuclease variants improved by site-directed mutagenesis in example 15.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_3 target, when mated with a meganuclease (SEQ ID NO: 109) cleaving the HIV1_3,3 target.
- the positive variants presented correspond to: Dl , SEQ ID NO:156; C2 5 SEQ ID NO: 157; F2, SEQ ID NO: 158; A4, SEQ ID NO: 159; G7, SEQ ID NO: 160; described in Table XXIV.
- Each cluster contains 6 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a previously improved variant.
- Figure 35 The H ⁇ V1_4 (SEQ ID NO:331) target sequence and its derivatives.
- HIV1__4.2 target SEQ ID NO:332
- the GGAC sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343).
- HIV1_4.3 SEQ ID NO:333
- HIV1_4.4 SEQ ID NO:3344
- HIV1_4.5 (SEQ ID NO:335) and HIV1_4.6 (SEQ ID NO:336) are pseudo-palindromic targets derived, respectively, from HIV1_4.3 and HIV1_4.4, containing the natural GGAC sequence in the middle of the target. As shown in the Figure, the boxed motives from 10AGC_P, 10TGT_P, 5TCT_P and 5TATJP are found in the HIV 1_4 series of targets.
- Figure 36 Cleavage of HIV1_4.3 (SEQ ID NO:333) target by combinatorial variants.
- the figure displays an example of screening of ⁇ -Crel combi- natorial variants with the HIV1_4.3 target.
- the positive variants correspond to: Al l , SEQ ID NO:168; A5, SEQ ID NO: 170; A2, SEQ ID NO: 171 ; A4 ; SEQ ID NO: 173; A3, SEQ ID NO: 174; all described in Table XXVl.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_4.3 target has been mated with another yeast strain containing the meganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 37 Cleavage of HIV1_4.4 (SEQ ID NO:334) and HIV1_4.6 (SEQ ID NO:336) targets by combinatorial variants.
- the figure displays an example of screening of l-Crel combinatorial variants with the HIV1_4.4 and HIV1__4.6 targets.
- the positive variants correspond to: A7, SEQ ID NO: 177; A5, SEQ ID NO:178; B8, SEQ ⁇ D NO: 179; E6, SEQ ID NO:180; F2, SEQ ID NO: 181 ; all described in Table XXVIII. Each cluster contains 4 spots.
- HIV1_4 (SEQ ID NO:331) target sequences by heterodimeric combinatorial variants.
- Some heterodimers resulted in cleavage of the HIV1__4.2 target, while no cleavage activity was detected on the HIV 1_4 target.
- the position of mutants in certain positions as an example is: line A, SEQ ID NO: 170; line B, SEQ ID NO:171 ; column 1, SEQ ID NO: 177; column 2, SEQ ID NO: 178; column 3; SEQ ID NO: 179.
- These mutants have been described in Tables XXVI and XXVIII. Each cluster contains 6 spots.
- a yeast strain harboring the HIV 1_4 or HIV1_4.2 target have been mated with another yeast strain containing the meganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 39 Cleavage of HIV1_4,3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by random mutagenesis in example 20.
- the figure displays an example of screening of I-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets.
- the positive variants presented correspond to: F8, SEQ ID NO:189; C6 5 SEQ ID NO: 190; E12, SEQ ID NO: 191; G12, SEQ ID NO:192; G6, SEQ ID NO:193; GI l, SEQ ID NO:194; all described in Table XXX.
- Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_4.3 or the H ⁇ V1_4.5 targets have been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV1_4.3 target.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 40 Cleavage of HIV1_4.3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by a second round of random mutagenesis in example 20bis.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets.
- the positive variants presented correspond to: E7, SEQ ID NO:200; Al , SEQ ID NO:201 ; E9, SEQ ID NO:202; A4, SEQ ID NO:203; Al l, SEQ ID NO:204; all described in Table XXXI. Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_4.3 or the HIV1_4.5 targets has been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al) 5 positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 41 Cleavage of HIV1_4 (SEQ ID NO:331) target by meganuclease variants improved by a second round of random mutagenesis in example 20bis.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4 target, when mated with a meganuclease (SEQ ID NO: 199) cleaving the HIV1_4.4 target.
- the positive variants presented correspond to: E7, SEQ ID NO:200; Al, SEQ ID NO:201 ; E9, SEQ ID NO:202; ⁇ 4, SEQ ID NO:203; Al 1 , SEQ ID NO:204; all described in Table XXXI.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_4.4 mutant (SEQ ID NO: 199) and the HIV1__4 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 42 Cleavage of HIV1_4 (SEQ ID NO:331) target by meganuclease variants improved by site-directed mutagenesis in example 21.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO:210) cleaving the HIV1_4.4 target.
- the positive variants presented correspond to: Al, SEQ ID NO:211 ; A2, SEQ ID NO:212; A5 ; SEQ ID NO:213; A7 ; SEQ ID NO:214; A8, SEQ ID NO:215; G2, SEQ ID NO:216; described in Table XXXII.
- Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.4 mutant (SEQ ID NO:210) and the HIV 1_4 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 43 Cleavage of HIV1_4.3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by site-directed mutagenesis in example 21.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets.
- the variants presented correspond to: Al, SEQ ID NO:211 ; A2, SEQ ID NO:212; A5, SEQ ID NO:213; A7, SEQ ID NO:214; A8, SEQ ID NO:215; G2, SEQ ID NO:21 ⁇ ; described in Table XXX ⁇ .
- SEQ ID NO:336 targets by meganuclease variants improved by random mutagenesis in example 22.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.4 and H ⁇ V1__4.6 targets.
- the positive variants presented correspond to: D4, SEQ ID NO: 199; D5, SEQ ID NO:2 ⁇ O; C8, SEQ ID NO:221 ; ClO, SEQ ID NO:222; E8, SEQ ID NO:223; all described in Table XXXIlI. Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_4.4 or the HIV1_4.6 targets have been mated with another yeast strain containing the meganuclease variants.
- the two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV1_4.4 target.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 45 Cleavage of HIV 1_4 (SEQ ID NO:33 I) target by meganuclease variants improved by random mutagenesis in example 22.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO: 190) cleaving the HIV1_4.3 target.
- the positive variants presented correspond to: D4 ; SEQ ID NO:199; D5, SEQ ID NO:210; C8, SEQ ID NO:221; ClO, SEQ ID NO:222; E8, SEQ ID NO:223; all described in Table XXXIII.
- Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_4.3 mutant (SEQ ID NO: 190) and the HIV 1_4 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al) 5 positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a non- improved variant.
- Figure 46 Cleavage of HIV 1_4 (SEQ ID NO:331) target by meganuclease variants improved by site-directed mutagenesis in example 23.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO: 190) cleaving the HIV1_4.3 target.
- the positive variants presented correspond to: B5, SEQ ID NO:229; B4 : SEQ ID NO:231 ; A5 > SEQ ID NO:235; A8, SEQ ID NO:236; Al l , SEQ ID NO:237; described in Table XXXIV.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV 1_4 target and the HIVl j4.3 mutant (SEQ ID NO: 190) has been mated with another yeast strain containing different meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 47 Cleavage of HIV1_4.4 (SEQ ID NO:334) and H1V1_4.6 (SEQ ID NO:336) targets by meganuclease variants improved by site-directed mutagenesis in example 23.
- the figure displays an example of screening of I-Crel meganuclease variants with the HIV1_4.4 and HIV1__4.6 targets.
- the positive variants presented correspond to: B5, SEQ ID NO:229; B4, SEQ ID NO:231 ; A5. SEQ ID NO:235; A8, SEQ ID NO:236; Al l, SEQ ID NO:237; described in Table XXXIV. Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_4.4 or the HIV1_4.6 targets have been mated with another yeast strain containing different meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 48 The HIV 1_5 target sequence (SEQ ID NO:337) and its derivatives.
- HIV1_5.2 target (SEQ ID NO:338) 5 the ATAC sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343).
- HIV1_5.3 (SEQ ID NO:339) is the palindromic sequence derived from the left pail of HIV1_5.2
- HIV1_5.4 (SEQ ID NO:340) is the palindromic sequence derived from the right part of I-IIVl_5.2.
- HIV1_5.5 (SEQ ID NO:341) and HIV1_5.6 (SEQ ID NO:342) are pseudo-palindromic targets derived, respectively, from HIV1_5,3 and HIVl J5.4, containing the natural ATAC sequence in the middle of the target.
- the boxed motives from 1 OTCTJP, 10CTG_P, 5TAG_P and 5CCTJP are found in the HIV1_5 series of targets.
- Figure 49 Cleavage of HIV1_5.3 (SEQ ID NO:339) target by combinatorial variants.
- the figure displays an example of screening of I-Od combinatorial variants with the HIV1_5.3 target.
- the two positive variants correspond to: Al , SEQ ID NO:242; A2, SEQ ID NO:241; described in Table XXXVI.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIVlJj.3 target has been mated with another yeast strain containing the meganucleasc variants.
- the two spots on the right contain the same negative or positive controls. These controls are: negative control (cluster Al), positive control (cluster A2), and strong positive control (cluster A3).
- Figure 50 Cleavage of HIV1_5.4 (SEQ ID NO:340) target by combinatorial variants.
- the figure displays an example of screening of ⁇ -Crel combinatorial variants with the HIV1_5.4 target.
- the positive variants correspond to: Al, SEQ ID NO:249; A3, SEQ ID NO:245; ⁇ 4, SEQ ID NO:252; A7, SEQ ID NO:250; AlO, SEQ ID NO:246; all described in Table XXXVIII.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_5.4 target has been mated with another yeast strain containing the meganuclease variants.
- the two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- Figure 51 Cleavage of the HIV1_5.2 target sequence (SEQ ID NO:338) by heterodimeric combinatorial variants.
- One hcterodimer resulted in cleavage of the HIV1_5,2 target.
- the heterodimer displaying a signal with HIV1_5.2 target is observed at position B4.
- the position of certain mutants as an example is: line A, SEQ ID NO:242; line B, SEQ ID NO:241; column 3, SEQ ID NO:245; column 4, SEQ ID NO:252; column 5; SEQ ID NO:251.
- Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV1_5.2 target has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- negative control i.e. cluster Al
- positive control i.e. cluster A2
- strong positive control i.e. cluster A3
- Figure 52 Cleavage of HIV1_5.3 target (SEQ ID NO:339) by meganuclease variants improved by random mutagenesis in example28.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.3 target.
- the positive variants presented correspond to: A6, SEQ ID NO:256; A12, SEQ ID NO:257; Al l, SEQ ID NO:258; AlO, SEQ ID NO:259; A2, SEQ ID NO:260 ;all described in Table XXXIX. Each cluster contains 6 spots.
- the spot on the low- right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a non-improved variant.
- Figure 53 Cleavage of HIV1_5.3 (SEQ ID NO:339) and HIV1_5.5 (SEQ ID NO:341) targets by meganuclease variants improved by a second round of random mutagenesis in example 28bis.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.3 and HIV1_5.5 targets.
- the positive variants presented correspond to: G2, SEQ ID NO:266; E4, SEQ ID NO:267; C2, SEQ ID NO:268; A12, SEQ ID NO:269; CI l, SEQ ID NO:270; all described in Table XL. Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_5.3 or the HIV1_5.5 targets have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 54 Cleavage of HIV 1_5 target (SEQ ID NO:337) by meganuclease variants improved by a second round of random mutagenesis in example 28bis.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5 target, when mated with a meganuclease (SEQ ID NO:276) cleaving the HIV1_5.4 target.
- the positive variants presented correspond to; G2, SEQ ID NO:266; E4, SEQ ID NO:267; C2, SEQ ID NO:268; A12, SEQ ID NO:269; CI l, SEQ ID NO:270; all described in Table XL.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_5.4 mutant and the HIV 1_5 target have been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- negative control i.e. cluster Al
- positive control i.e. cluster A2
- strong positive control i.e. cluster A3
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 55 Cleavage of HIV1_5.3 (SEQ ID NO:339) and HIV1_5.5 (SEQ ID NO:341) targets by meganuclease variants improved by site-directed mutagenesis in example 29.
- the figure displays an example of screening of 1-CVeI meganuclease variants with the HIV1_5.3 and HIV1_5.5 targets.
- the positive variants presented correspond to: C6, SEQ ID NO:278; F8, SEQ ID NO:279; H7, SEQ ID NO:280; Fl, SEQ ID NO:281 ; G 12, SEQ ID NO:282; described in Table XLI.
- Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIV1__5 target (see Figure 56). This is due to the presence of the G19S mutation in these variants.
- Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_5.3 or the HIV1_5.5 targets has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right is a negative control. The spot in the upper-right contains, as an internal control, an improved variant.
- Figure 56 Cleavage of HIV1_5 target (SEQ ID NO:337) by meganuclease variants improved by site-directed mutagenesis in example 29.
- the figure displays an example of screening of 1-Crel meganuclease variants with the HIV 1_5 target, when mated with a meganuclease (SEQ ID NO:276) cleaving the HIV1_5.4 target.
- the positive variants presented correspond to: C ⁇ , SEQ ID NO:278; F8, SEQ ID NO:279; H7, SEQ ID NO:280; Fl 5 SEQ ID NO:281 ; G12, SEQ ID NO:282; described in Table XLI.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV1_5.4 mutant (SEQ ID NO:276) and the HIV 1_5 target has been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 57 Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIV1J.6 (SEQ ID NO:342) targets by meganuclease variants improved by random mutagenesis in example 30.
- the figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.4 and HIV1_5.6 targets.
- the positive variants presented correspond to: D6, SEQ ID NO:276; A4, SEQ ID NO:288; ClO, SEQ ID NO:289; A9, SEQ ID NO:290; Al , SEQ ID NO:291 ; all described in Table XLII. Each cluster contains 6 spots.
- a yeast strain harboring the HIV1_5.4 or the H ⁇ V1_5.6 targets has been mated with another yeast strain containing the meganuclease variants.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, a non- improved variant cleaving the HIV1_5.4 target.
- Figure 58 Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIVl_5, ⁇ (SEQ ID NO: 342) targets by meganuclease variants improved by a second round of random mutagenesis in example 30bis.
- the figure displays an example of screening of 1-Crel meganuclease variants with the H ⁇ V1_5.4 and IIIV1 5.6 targets.
- the positive variants presented correspond to: A12, SEQ ID NO:297; Al, SEQ ID NO:298; Al l , SEQ ID NO:299; A8, SEQ ID NO:300; B4, SEQ ID NO:301; all described in Table XLIIL Each cluster contains 4 spots.
- the spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 59 Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIV1_5.6 (SEQ ID NO:342) targets by meganuciease variants improved by site-directed mutagenesis in example 31.
- the figure displays an example of screening of l-Crel meganuciease variants with the HIV1_5.4 and HIVlJ).6 targets.
- the positive variants presented correspond to: Hl , SEQ ID NO:307; H2, SEQ ID NO:308; H9, SEQ ID NO:309; B3, SEQ ID NO:310; H3, SEQ ID NO:31 1; described in Table XLIV.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV 1__5.4 or the HIV1_5.6 targets has been mated with another yeast strain containing different meganuciease variants.
- the spot on the low-right contains negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 60 Cleavage of HIV1__5 (SEQ ID NO:337) target by meganuciease variants improved by site-directed mutagenesis in example 31.
- the figure displays an example of screening of 1-OeI meganuciease variants with the H ⁇ V1_5 target, when mated with a meganuciease (SEQ ID NO:256) cleaving the HIV1_5.3 target.
- the positive variants presented correspond to: Hl , SEQ ID NO:307; H2, SEQ ID NO:308; H9, SEQ ID NO:309; B3, SEQ ID NO:310; H3 ; SEQ ID NO:311 ; described in Table XLIV.
- Each cluster contains 4 spots.
- a yeast strain harboring the HIV 1_5 target and the HI V 1 5.3 mutant (SEQ ID NO:256) has been mated with another yeast strain containing different meganuciease variants.
- the spot on the iow-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
- the spot in the upper-right contains, as an internal control, an improved variant.
- Figure 61 pCLS1853 plasmid map.
- Figure 62 Schematic representation of the pseudo-H ⁇ V provirus integrated in the HEK293-VLP-CL40 cell line used for validation of the activity of HlV meganucleases.
- the LTRs encompassing the U3, R and U5 regulatory sequences are duplicated and flanking the viral genes gag and pol.
- the env gene has been partially deleted and a pEFla-PuroR-IRES-BGFP cassette has been introduced between the 5' portion of env and the 3' LTR.
- the ORF of the TAT and REV genes have been introduced in the cellular genome using different retroviral vectors.
- Figure 63 Levels of p24 produced by the HEK293-VLP-CL40 cell line 48 hours after transfection with 1 ⁇ g of meganuclease expression plasmid.
- the amount of p24 present in cell culture supernatants was determined by ELISA.
- a sample transfected by a non related meganuclease (NRM, see text) is used for normalization.
- NRM non related meganuclease
- the amount of p24 produced by HIV meganuclease transfected cells is represented as the percentage of VLP production respect to the amount produced by the NRM transfected cells.
- the values represent the data from at least 3 independent transfections.
- Figure 64 represents a scheme of the mechanism leading to the generation of small deletions and insertions (InDeI) during repair of double-strand break by non homologous end-joining (NHEJ).
- Example 1 Strategy for engineering meganucleases cleaving the HIVI l target from the HIVl virus
- the HIV1__1 target is a 22 bp (non-palindromic) target located in U3 region of the proviral LTRs ( Figures 2 and 7). Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, the HIV 1_1 target is present twice in the HIV_1 provirus. This target is precisely located at positions 84- 105 and 8159-9180 of the HIV-I pNL4-3 vector (accession number AF324493, ⁇ dachi et al., J.
- the HIV 1_1 sequence is partly a patchwork of the 1 OAGAJP, 10TGG_P, 5TAC JP and 5_CTG_P targets (these designations describe the 3b ⁇ starting at the indicated nucleotide of the l-Crel target, for instance 10AG ⁇ _P indicates that nucleotides -10, -9 and -8 are A(-10) G(-9) A(-8) ( Figure 7)) which are cleaved by previously identified meganucleases. These meganucleases were obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould et al., J. MoI, Biol., 2006, 355, 443-458; Smith et al., Nucleic Acids Res., 2006.
- the 10AGA_P, 10TGG_P, 5TACJP and 5_CTG_P target sequences are 24 bp derivatives of C 1221, a palindromic sequence cleaved by 1-OeI (Arnould et al., precited).
- 1-OeI bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- HIV1__1 series of targets were defined as 22 bp sequences instead of 24 bp.
- HIVlJ differs from C1221 (SEQ ID NO: 343) in the 4 bp central region. According to the structure of the I-Oel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-OeI protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- Two other pseudo- palindromic targets were derived from these two containing the ACAC sequence in -2 to 2 (targets HIVlJ .5 and HIVlJ .6, Figure 7).
- proteins able to cleave HIVl J.3 and HIVlJ .4 targets or, preferentially, the pseudo-palindromic targets as homodimers were first designed (examples 2 and 3) and then co-expressed to obtain heterodimers cleaving HIVlJ (example 4).
- Heterodimcrs cleaving the HIVl J .2 and HIVlJ targets could be identified.
- Example 2 Identification of meganucleases cleaving HIVl 1.3 This example shows that l-Crel variants can cut the HIVl J .3 DNA target sequence derived from the left part of the HIVlJ .2 target in a palindromic form ( Figure 7).
- HIVlJ .3 is similar to 1 OAGAJ 1 at positions ⁇ 1 , ⁇ 2, ⁇ 6, ⁇ 8, ⁇ 9, and ⁇ 10 and to 5TACJP at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 6. It was hypothesized that positions ⁇ 7 and ⁇ 11 would have little effect on the binding and cleavage activity. Variants able to cleave the 1 OAGAJ 1 target were obtained by mutagenesis of I-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- Variants able to cleave 5TAC_P were obtained by mutagenesis on 1-OeI N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al., J. MoL Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
- the target was cloned as follows: an oligonucleotide corresponding to the HIV1_1.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTGCAGAACTACGTACGTAGTTCTGCCAATCGTCTGTCA 3' (SEQ ID NO: 14). The same procedure was followed for cloning the HIV 1_1.5 target, using the oligonucleotide:
- Double-stranded target DNA generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8).
- Yeast reporter vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B ⁇ MAT a, ura3 ⁇ 851, trpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202), resulting in a reporter strain, b) Construction of combinatorial mutants l-Cre ⁇ variants cleaving 10AGA_P or 5TAC_P were previously identified, as described in Smith el al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al., L MoI.
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgatlggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542, Figure 9) and primers (assF 5'-ctannnttgacctttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgatlggagacttgacc-3'(SEQ ID
- PCR fragments resulting from the amplification reaction using the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an cquimolar ratio.
- filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidasc activity. Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants
- yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA. Alternatively, ORFs were amplified from yeast DNA by PCR (Akada et al, Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MlLLEGEN SA. B) Results
- I-Crel combinatorial variants were constructed by associating muta- tions at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TAC_P with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10AGA_P on the 1-OeI scaffold, resulting in a library of complexity 1600. Examples of combinatorial variants are displayed in Table I. In Table I the peptide sequence of these two subdomains are provided in the first column and second row respectively. This library was transformed into yeast and 3348 clones (2 times the diversity) were screened for cleavage against the HIVlJL 3 and HIV1_1.5 DNA targets.
- variants 36 positive clones were found to cleave the HIV1__1.3 target, which after sequencing turned out to correspond to 31 different novel endonuclease variants (Table II). Those variants showed no cleavage activity of the HIV1_1.5 DNA target. Examples of positives are shown in Figure 10. Some of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77. Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be I-O ⁇ l combined variants resulting from micro- recombination between two original variants during in vivo homologous recombina- tion in yeast.
- Example 3 Making of meganucleases cleaving HIVl 1.4 This example shows that 1-OeI variants can cleave the HIV1_1.4 DNA target sequence derived from the right part of the HIV1_1.2 target in a palindromic form ( Figure 7).
- HIVlJ .4 is similar to 5CTG P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 8 and to 10TGGJP at positions ⁇ 1 , ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 8, ⁇ 9 and ⁇ 10. It was hypothesized that positions ⁇ 6, ⁇ 7 and ⁇ 11 would have little effect on the binding and cleavage activity. Variants able to cleave 5CTG_P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould el al., J. MoL Biol., 2006, 355, 443-458; Smith et al.
- Variants able to cleave the 10TGG_P target were obtained by mutagenesis of l-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith el al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- I-Oel variants cleaving 10TGG_P or 5CTG_P were previously identified, as described in Smith el al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arno ⁇ ld et al, J. MoI. Biol, 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10TGG_P and 5CTGJ? targets.
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1 107, Figure 1 1) and primers (assF 5'-ctannnttgacctttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and Gail OR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.O 5 0.1 % SDS, 6% dimethyl formamide (DMF), 7 raM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
- variants showed cleavage activity on the HIV1_1.6 DNA target. Examples of positives are shown in Figure 12.
- the sequence of several of the variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 as well as additional mutations (see examples Table IV). Such variants likely result from PCR artifacts during the combinatorial process.
- the variants may be ⁇ -Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.
- Example 4 Making of ineganucleases cleaving HIV1 1.2 and HIVI l l-Crel variants able to cleave each of the palindromic HIV1_1.2 derived targets (HIV1_1,3 and HIV1_1.4) were identified in example 2 and example 3. Pairs of such variants (one cutting HIV1_1.3 and one cutting HIV1_1.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_1.2 and the non palindromic HIV 1_1 targets.
- A) Materials and Methods a) Construction of target vector The experimental procedure is as described in example 2, with the exception that an oligonucleotide corresponding to the HIV 1_1.2 target sequence:
- Yeast DNA was extracted from variants cleaving the HIV1_1.4 taiget in the pCLS 1107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_1.3 target in the pCLS0542 expression vector. Transformanls were selected on synthetic medium lacking leucine and containing
- Mating was performed using a colony gridder (QpixII, Genetix). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm 2 ). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0 C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors.
- Example 5 Improvement of meganucleases cleaving HIVI l by random mutagenesis of proteins cleaving HIVl 1.3 and assembly with proteins cleaving HIV1_1.4
- I-Crel variants able Io cleave the H ⁇ V3_1.2 and HIV 1_1 target by assembly of variants cleaving the palindromic HIVlJ .3 and HIV1_1.4 target have been previously identified in example 4. However, these variants display stronger activity with the H1V1_1.2 target compared to the HIV1__1 target.
- Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn 2+ .
- PCR reactions were carried out that amplify the 1-OeI coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRcv (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25), which are common to the pCLS0542 ( Figure 9) and pCLSl 107 ( Figure 11) vectors.
- the yeast strain FYBL2-7B (MATa, ura3 ⁇ 851, (rpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202) containing the HIV1__1 target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with one variant, in the kanamycin vector (pCLS1 107), cutting the HIV1_1.4 target, using a high efficiency LiAc transformation protocol.
- Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
- the 93 clones showing the highest cleavage activity on target HIV1_1.3 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (I- Crel 33T,40K,44R,68Y,70S,77N +132V or KNSTQK/RYSDN +132V, according to the nomenclature of Table I). After mating with this yeast strain, 41 clones were found to cleave the HIV 1_1 target more efficiently than the original variant.
- 41 positives contained proteins able to form hetcrodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46), that showed cleavage activity on the HIV 1_J target.
- An example of positive clones is shown in Figure 15. Sequencing of these 41 positive clones indicates that 31 distinct variants were identified. Ten of these 31 variants are presented as an example in Table VIII.
- Example 5bis Improvement of meganucleases cleaving HIVI l by a second round of random mutagenesis of proteins cleaving HIV1 1.3 and assembly with proteins cleaving HIV1_1.4
- a second round of random mutagenesis was carried out following the same rationale of example 5.
- four variants cleaving HIV1_1,3 were mutagenized, and variants were screened for cleavage activity of HIV1_1.3 and HIV1_1.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIV1_1 when co-expressed with a variant cleaving HIV 1_J .4.
- the 79 clones showing cleaving target HIV1_1.3 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (1-OeI 33T,40K,44R,68Y,70S ; 77N,132V or KNSTQK/RYSDN +132V, according to the nomenclature of Table I). After mating with this yeast strain, 76 clones were found to cleave the HIV 1_1 target.
- 76 positives contained proteins able to form heterodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46) showing cleavage activity on the HIV 1_1 target.
- SEQ ID NO: 46 An example of positives is shown in Figure 17. Sequencing of these 76 positive clones indicates that 44 distinct variants were identified. Ten of these 44 variants are presented as an example in Table IX.
- Example 6 Improvement of meganucleases cleaving HIVl ⁇ by site-directed mutagenesis of proteins cleaving HIVl 1.3 and assembly with proteins cleaving HIVl 1.4
- the 1-OeI variants cleaving HIV1_1.3 described in Table IX issued from random mutagenesis in examples 5 and 5bis were also mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV 1_1 in combination with a variant cleaving HIVl J.4.
- Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or Gl 9SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
- the same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V 105 A and 1132V substitutions in the coding
- F54LF 5'-acccagcgccgttggctgctggacaactaglg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
- E80KF 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
- V 105AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
- I132VF 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA ( ⁇ CLS0542, Figure 9), linearized by digestion with Ncol and Eagl.
- This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trpl ⁇ 63, Ieu2 ⁇ l, his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods EnzymoL, 2002, 350, 87-96).
- Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast.
- a library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and
- Isoleucinc 132 with Valine was constructed on a pool of five variants cleaving
- HIV1_1.3 (described in Tabic X). 558 transformed clones were screened for cleavage against the HIV1_1.3 and H ⁇ V1_1,5 DNA targets. A total of 395 positive clones were found to cleave H1V1_J .3, while 349 of those cleaved also the HIV1_1.5 target. An example of positive variants is shown in figure 18
- the 558 transformed clones were also mated with a yeast strain that contains (i) the HIV1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (l-Crel 33T,40K,44R,68Y,70S,77N +132V or KNSTQK/RYSDN + 132V, according to the nomenclature of Table I).
- 458 clones were found to cleave the HIV 1_1.
- 458 positives contained proteins able to form heterodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46) showing cleavage activity on the IHVl 1 target.
- An example of positives is shown in Figure 19.
- Example 7 Improvement of meganucleases cleaving HIV1_1 by random mutagenesis of proteins cleaving HIV1 1.4 and assembly with proteins cleaving HIV1JL3
- Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn 2+ .
- PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagltatcagtcggccgc ⁇ '; SEQ ID NO: 25).
- yeast strain FYBL2-7B (MAT a, ura3 ⁇ 851, trpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202) containing the HIVM target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_1.3 target, using a high efficiency LiAc transformation protocol.
- Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
- the 89 clones showing the highest cleavage activity on target HIV3_1.4 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid
- Example 7bis Improvement of meganucleases cleaving HIVI l by a second round of random mutagenesis of proteins cleaving HIV1 I.4 and assembly with proteins cleaving HIV1_1.3
- a second round of random mutagenesis was carried out following the same rationale of example 7.
- four variants cleaving HIV1_1.4 were mutagenized, and variants were screened for cleavage activity of HIV1J .4 and HIV1_1.6 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HiVl ⁇ l when co-expressed with a variant cleaving HIV 1_1.3.
- the 59 clones showing cleaving target HIV1_1.4 were then mated with a yeast strain that contains (i) the HIV1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_J .3 target (1-OeI 30G,38R,44N,68Y,70S,75R,77Y +79N or KGSYRS/NYSRY +79N, according to the nomenclature of Table I). After mating with this yeast strain, 42 clones were found to cleave the HIV 1_1.
- 42 positives contained proteins able to form heterodimers with KGSYRS/NYSRY +79N (SEQ ID NO: 28) showing cleavage activity on the HIV 1_1 target.
- An example of positives is shown in Figure 23. Sequencing of these 42 positive clones indicates that 35 distinct variants were identified. Ten of these 35 variants are presented as an example in Table XIV.
- Example 8 Strategy for engineering meganuc ⁇ eases cleaving the HIV1 3 target from the HIVl virus
- the HIV 1_3 target is a 22 bp (non-palindromic) target located in U5 region of the proviral LTRs. Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, the HIV1_3 target is present twice in the HIVl provirus. This target is precisely located at positions 599-620 and 9674-9695 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et ai., J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
- the HIV 1_3 sequence (SEQ ID NO: 325) is partly a patchwork of the 10C ⁇ GJP, 10ACA_P ; 5CCT_P and 5_GAC_P targets ( Figure 24) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould et al, J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006.
- HIVlJ could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
- the 1 OCAGJ 3 , 10ACA_P, 5CCT_P and 5_GAC_P target sequences are 24 bp derivatives of C 1221, a palindromic sequence cleaved by ⁇ -Oel (Arnould et al., precited).
- the structure of l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al., Nat. Struct. Biol, 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- HIV 1_3 differs from C 1221 in the 4 bp central region. According to the structure of the I- Oe I protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-OeI protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region.
- the TTTA sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_3.2 (SEQ ID NO: 326, Figure 24). Then, two palindromic targets, HIV1_3.3 (SEQ ID NO: 327) and HIV1_3.4 (SEQ ID NO: 328), were derived from HIV1_3.2 ( Figure 24). Since HIV1J.3 and HIV1_3.4 arc palindromic, they should be cleaved by homodimeric proteins.
- Two other pseudo-paiindromic targets were derived from these two, containing the TTTA sequence in -2 to 2 (targets HIV1J.5 (SEQ ID NO: 329) and HIV1_3.6 (SEQ ID NO: 330), figure 24).
- proteins able to cleave HIV1_3.3 and HIV1_3.4 targets or, preferentially, the pseudo- palindromic targets as homodimers were first designed (examples 9 and 10) and then co-expressed to obtain heterodimers cleaving HIV 1_3 (example 11). Hetcrodimers cleaving the HIV1_3.2 or HIV 1_3 targets could not be identified.
- HIV1 ⁇ 3.3 is similar to 10CAG_P at positions ⁇ 1 , ⁇ 2, ⁇ 6, ⁇ 8, ⁇ 9, and ⁇ 10 and to 5CCT_P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 6. It was hypothesized that positions ⁇ 7 and ⁇ 1 1 would have little effect on the binding and cleavage activity.
- Variants able to cleave the 1OCAG_P target were obtained by mutagenesis of I-Crel N75 or D75, at positions 28, 30, 32 ; 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- Variants able to cleave 5CCTJP were obtained by mutagenesis on 1-OeI N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
- A) Material and Methods a) Construction of target vector
- the target was cloned as follows: an oligonucleotide corresponding to the HIV1_3.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTCTCAGACCCTGTACAGGGTCTGAGCAATCGTCTGTCA 3' (SEQ ID NO: 86). The same procedure was followed for cloning the HIV3_1.5 target, using the oligonucleotide: 5' TGGCATACAAGTTTCTCAGACCCTTTTAAGGGTCTGAGCAATCGTCTGTCA 3' (SEQ ID NO: 87).
- Double-stranded target DNA generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8).
- yeast reporter vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B ⁇ MAT a, ura3 ⁇ 851, trpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202), resulting in a reporter strain.
- I-Crel variants cleaving 10CAG_P or 5CCT_P were previously identified, as described in Smith et at. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et aL, J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10AGA_P and 5TACJP targets.
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7,0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37 0 C, to monitor ⁇ -galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants
- yeast DN ⁇ was extracted using standard protocols and used to transform E, coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA.
- ORFs were amplified from yeast DNA by PCR (Akada et al, 5 Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MILLEGEN SA.
- variants showed no cleavage activity of the HIV1_3.5 DNA target. Examples of positives are shown in Figure 25. Some of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 (SEQ ID NO: 92 to 94, Table XVI). Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.
- Example 10 Making of meganucleases cleaving HIV1 3.4
- HIV1_3,4 is similar to 5GAC_P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 8 and to 1 OACAJ 3 at positions ⁇ 1 , ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 8, ⁇ 9 and ⁇ 10. It was hypothesized that positions ⁇ 6, ⁇ 7 and ⁇ 11 would have little effect on the binding and cleavage activity. Variants able to cleave 5GAC P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould et al., J. MoI.
- mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5 GACJP were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10ACA_P.
- A) Material and Methods a) Construction of target vector
- the experimental procedure is as described in example 2, with the exception that different oligonucleotides corresponding to the H ⁇ V1_3.4 and H ⁇ Vl_3. ⁇ targets.
- the oligonucleotide used for the HIV1_3.4 target was: 5'
- PCR amplification is carried out using primers (GaIl OF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1107, Figure 1 1) and primers (assF 5'-ctannnttgacctttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the I-Oel coding sequence for amino acids 39-43.
- primers GaIl OF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLSH07, Figure 1 1) linearized by digestion with Dralll and NgoMYV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6 ⁇ (MAT ⁇ , trpl ⁇ 63, Ieu2 ⁇ l , his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96).
- An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast, c) Mating of meganuclease expressing clones and screening in yeast
- filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
- DNA Sequencing of these 93 strongest clones allowed the identification of 64 novel cndonuclease variants. Examples of positives are shown in Figure 26. Some variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44 > 68, 70, 75, 77 as well as additional mutations (see examples Table XVIII 5 SEQ ID NO: 102 to 104), Such variants likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.
- Example 11 Making of meganucleases cleaving HIV1 3.2 and HIV1_3 ⁇ -Cre ⁇ variants able to cleave each of the palindromic HIV1_3.2 derived targets (H ⁇ V1_3.3 and HIV1_3.4) were identified in example 9 and example 10. Pairs of such variants (one cutting HIV1_3.3 and one cutting HIV1_3.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_3.2 and the non palindromic HIVlJ targets.
- A) Materials and Methods a) Construction of target vector The experimental procedure is as described in example 9, with the exception that an oligonucleotide corresponding to the HlVl J3.2 target sequence:
- Yeast DNA was extracted from variants cleaving the HIV1J3.4 target in the pCLS 1 107 expression vector using standard protocols and was used to transform E. coll The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_3.3 target in the pCLS0542 expression vector. Transformants were selected on synthetic medium lacking leucine and containing G418. c) Mating of meganucleases coexpressing clones and screening in yeast Mating was performed using a colony gridder (QpixII, Genetix).
- Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm 2 ). ⁇ second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target.
- Membranes were placed on solid agar YPD rich medium, and incubated at 3O 0 C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors.
- I-Crel variants able to cleave the HIV1_3.3 target have been previously identified in example 9.
- variants display, however, weak cleavage activity and where therefore mutagenized in order to improve their activity.
- Four mutants were selected for random mutagenesis and the variants obtained were screened for cleavage activity of HIV1_3.3 and HIV1_3.5 targets.
- the 1-OeI protein bound to its target there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-Crel protein (Chevalier et al, Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al , J. MoI. Biol., 2003, 329, 253-269).
- HIV1_3.3 and HIV1_3.5 DNA targets A total of 51 positive clones were found to cleave HIV1__3.3, while none of those cleaved the HIV1_3.5 target. Sequencing of the
- Example 12bis Improvement of meganucleases cleaving HIV1 3.3 by a second round of random mutagenesis of proteins cleaving HIV1 3.3
- Example 13 Improvement of meganucleases cleaving HIV1 3 by site-directed mutagenesis of proteins cleaving HIV1 3.3 and assembly with proteins cleaving HIVl 3.4
- Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the G19S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- Crel coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
- G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' SEQ ID NO: 47
- F54LF 5'-acccagcgccgttggctgctggacaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
- E80KF 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3 ' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
- V 105AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattUcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
- 1132VF 5'-acctgggtggatcaggttgcagctctgaacgat-3' and 1132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en cquimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Nco I and Eagl.
- This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trpl ⁇ 63, ku2 ⁇ l, his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96).
- Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast.
- HIV1_3,3 (described in Table XX, SEQ ID 1 15 to 119). 558 transformed clones were screened for cleavage against the HIV1_3.3 and HIV1_3.5 DNA targets. A total of 376 positive clones were found to cleave H ⁇ V1_3.3, while 54 of those cleaved also the
- HIV1__3.5 target An example of positive variants is shown in figure 29.
- the 558 transformed clones were also mated with a yeast strain that contains (i) the HIV 1J3 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_3.4 target (38Y,44Y,68S,70S,75R,77V,43L,81V,105A,107R or KNSYYS/YSSRV
- ten 1-OeI variants cleaving the HIV 1_3 target when forming a heterodimer with the KNSYYS/YSSRV variant are listed in Table XXI.
- Example 14 Improvement of meganucleases cleaving HIV1_3.4 by random mutagenesis of initial proteins cleaving HIV1 3.4
- Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn 2+ .
- PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25).
- Example 14bis Improvement of meganucleases cleaving HIV1 3.4 by a second round of random mutagenesis of proteins cleaving HIV1 3.3
- HIV1_3.4 and HIVl m 3.6 targets A total of 178 positive clones were found to cleave HIV1_3.4, while 63 of those cleaved also the HIV1_3.6 target. Sequencing of the 93 clones showing the strongest cleavage activity in the HIV1_3.4 target allowed the identification of 62 novel endonuclease variants. An example of the identified variants is presented in table XXIII and figure 32.
- Example 15 Improvement of meganucleases cleaving HIV1 3 by site-directed mutagenesis of proteins cleaving HIV1JS.4 and assembly with proteins cleaving HIV1_3.3
- Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the l-Crel coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G 19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5' ⁇ gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
- G 19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' SEQ ID NO: 47
- F54LF 5'-acccagcgccgttggctgctggacaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50); * E80KF: 5'-ttaagcaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattitgcttaa-3' SEQ ID NO: 51 and 52);
- F87LF 5'-aagccgctgcacaacctgctgactcaactgcag-3'
- F87LR 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3 ! SEQ ID NO: 53 and 54);
- V105AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3'
- V105AR 5'- ttcgataattttcagagccaggttlgcctgttt-3' SEQ ID NO: 55 and 56);
- I132VF 5'-acctgggtggatcaggltgcagctctgaacgat-3 ' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA ( ⁇ CLS0542, Figure 9), linearized by digestion with Ncol and Eagl.
- This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trpl ⁇ 63, leu2 ⁇ l, his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96).
- Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast.
- the 317 transformed clones were also mated with a yeast strain that contains (i) the HIV 1_3 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_3.3 target (I-Crel 32K,33A,44K,68E,70S,75N,77R, +132N or KNKAQS/KESNR +132N, according to the nomenclature of Table I). After mating with this yeast strain, 264 clones were found to cleave the HIV 1_3.
- 264 positives contained proteins able to form heterodimers with KNKAQS/KESNR +132N (SEQ ID NO: 109, Table XIX) showing cleavage activity on the HIV1_3 target.
- KNKAQS/KESNR +132N SEQ ID NO: 109, Table XIX
- An example of positive clones is shown in Figure 34.
- Example 16 Strategy for engineering meganucleases cleaving the HIV1 4 target from the HIVl virus
- the HIV 1_4 target is a 22 bp (non-palindromic) target located in the gag gene of the HIVl provims. This target is precisely located at positions 1629-1650 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al., J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
- the HIV 1_4 sequence (SEQ ID NO: 331) is partly a patchwork of the 1 OAGCJP, 1 OTGTJP, 5TCTJP and 5 JTATJP targets ( Figure 35) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; ⁇ rnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith et al., Nucleic Acids Res., 2006.
- HIV1_4 could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
- the 10AGC_P, 10TGT_P, 5TCT_P and 5 JTATJP target sequences are 24 bp derivatives of C1221, a palindromic sequence cleaved by 1-OeI (Arnould et al., precited).
- the structure of l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al., Nat. Struct. Biol., 2001 , 8, 312-316; Chevalier and Sloddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- HIV1_4 differs from C1221 in the 4 bp central region. According to the structure of the l-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the l-Crel protein (Chevalier et al., Nat. Struct. Biol, 2001 , 8, 312-316; Chevalier and Stoddard, Nucleic ⁇ cids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region.
- the GGAC sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_4.2 (SEQ ID NO: 332, Figure 35). Then, two palindromic targets, HIV1_4.3 (SEQ ID NO: 333) and HIV1_4.4 (SEQ ID NO: 334), were derived from HIV1_4.2 ( Figure 35). Since HIV1__4.3 and HIV 1 4.4 are palindromic, they should be cleaved by homodimeric proteins.
- Two other pseudo-palindromic targets were derived from these two, containing the GGAC sequence in -2 to 2 (targets HIV1_4.5 (SEQ ID NO: 335) and HIV1_4.6 (SEQ ID NO: 336), figure 35).
- proteins able to cleave HIV1_4.3 and HIV1_4.4 targets or, preferentially, the pseudo- palindromic targets as homodimers were first designed (examplesl7 and 18) and then co-expressed to obtain heterodimers cleaving HIV 1_4 (example 19).
- Heterodimers cleaving the HIV1_4.2 and I ⁇ IVl_j4 targets could be identified.
- HIV1_4.3 is similar to 10AGC_P at positions ⁇ 1, ⁇ 2, ⁇ 6, ⁇ 8, ⁇ 9, and ⁇ 10 and to 5TCT_P at positions ⁇ 1 , ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 6. It was hypothesized that positions ⁇ 7 and ⁇ 1 1 would have little effect on the binding and cleavage activity.
- Variants able to cleave the 1 OAGCJP target were obtained by mutagenesis of l-Oel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- Variants able to cleave 5TCT_P were obtained by mutagenesis on l-Crel N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould el al., J. MoI. Biol, 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
- the target was cloned as follows: an oligonucleotide corresponding to the HIV1_4,3 target sequence flanked by gateway cloning sequences was ordered from PROL ⁇ GO: 5' TGGCATACAAGTTTCCAGCATTCTGTACAGAATGCTGGCAATCGTCTGTCA 3' (SEQ ID NO: 166). The same procedure was followed for cloning the HIV1_4.5 target, using the oligonucleotide:
- Double-stranded target DNA generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8).
- yeast reporter vector was transformed into S ⁇ cch ⁇ romyces cerevisi ⁇ e strain FYBL2-7B ⁇ MAT ⁇ , ur ⁇ 3 ⁇ 851, trpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202), resulting in a reporter strain.
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542, Figure 9) and primers (assF 5'-ctannnttgacctlt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C 3 to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS 3 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37 0 C, to monitor ⁇ -galactosidase activity.
- DMF dimethyl formamide
- Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants
- yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA.
- ORFs were amplified from yeast DNA by PCR (Akada et al., Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by M ⁇ LLEGEN SA.
- variants showed no cleavage activity of the HIV1_4.5 DNA target. Examples of positives are shown in Figure 36. Two of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 (SEQ ID 168 and 174, Table XXVI). Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro- recombination between two original variants during in vivo homologous recombination in yeast. Example 18: Making of meganucleases cleaving HIV1 4.4
- H1V1_4.4 DNA target sequence derived from the right part of the HIV1_4.2 target in a palindromic form ( Figure 35).
- H1V1_4.4 is similar to 5TAT_P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1 107, Figure 1 1) and primers (assF 5'-ctannnttgacctttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS 5 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2. B) Results
- I-C>el combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TATJP with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OTGTJP on the 1-Crel scaffold, resulting in a library of complexity 1406. Examples of combinatorial variants are displayed in Table XXVII. This library was transformed into yeast and 3348 clones (2.3 times the diversity) were screened for cleavage against the UWl_4A and HIV1_4.6 DNA targets. A total of 210 positive clones were found to cleave HIV1_4.4. 40 of these clones were also able to cleave the HIV1_4.6 DNA target.
- variants Sequencing of these 93 clones with the strongest activity allowed the identification of 45 novel endonuclease variants. Examples of positives are shown in Figure 37.
- the sequence of several of the variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 as well as additional mutations (see examples in Table XXVIII, SEQ ID 178 and 184). Such variants likely result from PCR artifacts during the combinatorial process.
- the variants may be l-Crel combined variants resulting from micro -recombination between two original variants during in vivo homologous recombination in yeast.
- Example 19 Making of meganucleases cleaving HIV1 4.2 and HIV1 4 l-Crel variants able to cleave each of the palindromic HIV1_4.2 derived targets (HIV 1_4.3 and HIV1_4.4) were identified in example 2 and example 3. Pairs of such variants (one cutting HIV1_4.3 and one cutting HIV1_4.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_4.2 and the non palindromic HI V 1_4 targets.
- Yeast DNA was extracted from variants cleaving the HIV1_4.4 target in the pCLS1 107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_4 ' 3 target in the pCLS0542 expression vector. Transformants were selected on synthetic medium lacking leucine and containing G418. c) Mating of meganucleases coexpressing clones and screening in yeast
- Mating was performed using a colony gridder (QpixII, Genetix). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm 2 ). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at 3O 0 C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors.
- proteins cleaving HIV1_4.3 were mutagenized and their homodimeric cleavage activity was determined, and in a second step, it was assessed whether they could cleave HIV 1_4 when co-expressed with a protein cleaving HIV 1_4.4.
- yeast strain FYBL2-7B (MAT a, ura3 ⁇ 851, tr ⁇ ! ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202) containing the HIV 1_4 target in the yeast reporter vector (pCLS 1055, Figure 8) was transformed with one variant, in the kanamycin vector (pCLS1107), cutting the HIVl m 4.4 target, using a high efficiency LiAc transformation protocol.
- Variant-target yeast strains were used as target strains for mating assays as described in example 19. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 17.
- the 93 clones showing the highest cleavage activity on target HIV1__4.3 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (I- OeI 30H,33M,38A,44N,68Y,70S ⁇ 75Y,77R or KHSMAS/NYSYR, according to the nomenclature of Table I). After mating with this yeast strain, no clones were found to cleave the HIV 1_4 when forming heterodimers with KHSMAS/NYSYR (SEQ ID NO: 177, TaWe XXIX).
- Example 20bis Improvement of meganucleases cleaving HIV1_4.3 by a second round of random mutagenesis of proteins and assembly with proteins cleaving HIV1_4.4
- a second round of random mutagenesis was carried out following the same rationale of example 20.
- four variants cleaving HIVl_4-3 were mutagenized, and variants were screened for cleavage activity of H ⁇ V1__4.3 and HIV1_4.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIVl _4 when co-expressed with a variant cleaving HIV1_4.4.
- the materials and methods have previously been described in example 20.
- HIV1_4.3 and HIV1_4.5 DNA targets A total of 377 positive clones were found to cleave HIV1_4.3, while 208 of those cleaved also the HIV1_4.5 target. Sequencing of the 93 clones with the highest activity allowed the identification of 53 novel endonuclease variants. An example of the identified variants is presented in table XXXI and figure 40.
- the 93 clones showing cleaving target HIV1_4,3 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (l-Crel 30H,33M,38A,44A,68Y,70S,75Y,77R,155R or KIISMAS/AYSYR +155R, according to the nomenclature of Table I). After mating with this yeast strain, all the 93 clones were found to cleave the HIV 1_4.
- 93 positives contained proteins able to form heterodimers with KHSMAS/AYSYR +155R (SEQ ID NO: 199) showing cleavage activity on the HIV1_4 target.
- An example of positives is shown in Figure 41. Sequencing of these 93 positive clones indicates, as mentioned before, that 53 distinct variants were identified. Ten of these 53 variants are presented as an example in Table XXXI.
- Example 21 Improvement of meganucleases cleaving HIV1 4 by site-directed mutagenesis of proteins cleaving HIV1 4.3 and assembly with proteins cleaving HIVI 4.4
- I-CVel variants cleaving HIV1_4.3 were also mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving H ⁇ V1_4 in combination with a variant cleaving HIVl _4.4.
- Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIl OF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5 5 -acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G 19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
- F54LF 5'-acccagcgccgttggctgctggacaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
- E80KF 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3 ' SEQ ID NO: 51 and 52);
- F87LF 5'-aagccgctgcacaacctgctgactcaactgcag-3'
- F87LR 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54
- V105AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3'
- V105AR 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
- I132VF 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Ncol and Eagl.
- This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa 1 irpl ⁇ 63, leu2 ⁇ l, his3 ⁇ 200) using a high effi- ciency LiAc transformation protocol (Gietz and Woods, Methods EnzymoL, 2002, 350, 87-96).
- Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast, c) Mating of meganuclease expressing clones and screening in yeast
- HIV1_4.3 (described in Table XXXI, SEQ ID NO:200 to 205).
- 558 transformed clones were mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (30H ⁇ 33M,38A,44N,68Y,70S,75Y,77R or KHSMAS/NYS YR, according to the nomenclature of Table I).
- 486 clones were found to cleave the HlV 1_4.
- 486 positives contained proteins able to form heterodimers with KHSMAS/NYSYR (SEQ ID NO: 177) showing cleavage activity on the HIV 1_4 target.
- An example of positive variants is shown in figure 42. Sequencing of the 93clones with the highest cleavage activity on the
- HIV1_4 target allowed the identification of 34 different endonuclease variants.
- These 93 clones were also tested for their ability to cleave the HIV1_4.3 and HIV1_4.5 targets. In this case, 71 clones were able to cleave the HIV1_4.3 target, and 69 the HIV1_4.5 target (see Figure 43 for an example). Sequence analysis of these clones showed the presence of 25 different endonuclease variants. Comparison of sequences of the positive clones in all the targets indicated the presence of a total of 40 novel endonuclease variants.
- Example 22 Improvement of meganuc ⁇ eases cleaving HIV1 4.4 by random mutagenesis and assembly with proteins cleaving HIV1 4.3
- the assembly of l-Crel variants cleaving the palindromic HIV1_4.3 and HIV1_4.4 target to cleave the HIV1_4.2 and HIV1__4 have been previously described in example 19. However, these variants display activity with the HIV1_4.2 target and not with the HIV 1_4 target.
- Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn " .
- PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5 s - gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3 '; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc- ⁇ '; SEQ ID NO: 25).
- yeast strain FYBL2-7B (MATa, ura3 ⁇ 851, (rpl ⁇ 63, leu2 ⁇ l, lys2 ⁇ 202) containing the HIV Ij4 target in the yeast reporter vector (pCLS 1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_4.3 target, using a high efficiency LiAc transformation protocol.
- Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 17.
- the 93 clones showing the highest cleavage activity on target HIV1_4.4 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.3 target (I- Cre ⁇ 28Q,38R,40K ! 44K ! 68T ; 70G,75N +132V or QNSYRK/KTGN ⁇ +132V, according to the nomenclature of Table I). After mating with this yeast strain, 90 clones were found to cleave the HIV 1_4 target.
- 90 positives contained proteins able to form heterodimers with QNSYRK/KTGNI +132V (SEQ ID NO: 190, Table XXX), that showed cleavage activity on the HIV 1_4 target.
- An example of positives is shown in Figure 45. Sequencing of these 90 positive clones indicates that 65 distinct variants were identified. Ten of these 65 variants are presented as an example in Table XXXIII.
- Example 23 Improvement of meganucleases cleaving HIV1 4 by site-directed mutagenesis of proteins cleaving HIV1 4.4 and assembly with proteins cleaving HIV1 4.3
- Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' or GaIlOR 5'-acaaccttgattggagacttgacc-3') and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'- gatgatgctaccgtcagagtccacaaagccggc-3' (SEQ ID NO: 48)).
- G19SF 5'-gccggcttttgtggactctgacggtagcatcatc-3' SEQ ID NO: 47
- G19SR 5'- gatgatgctaccgtcagagtccacaaagccggc-3' SEQ ID NO: 48
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified. Approximately 25ng of each of the two overlapping PCR fragments and 75ng of vector DNA (pCLSl 107, Figure 1 1) linearized by digestion with DraIII and NgoMlY were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, (rpl ⁇ 63, lei ⁇ l, his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombi- nation in yeast.
- F54LF 5'-acccagcgccgttggctgctggacaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
- E80KF 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
- Vl 05AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and Vl 05AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56); * I132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgtlcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- a library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and Isoleucine 132 with Valine) was constructed on a pool of four variants cleaving HIV1_4.4 (see Table XXXIII, SEQ ID NO: 199, 177, 221 and 228).
- 558 transformed clones were mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.3 target (28Q,38R,40K,44K,68T,70G,75N or QNSYRK/KTGNI, according to the nomenclature of Table I).
- 16 clones were found to cleave the HIV 1_4.
- 16 positives contained proteins able to form heterodimers with QNSYRK7KTGN1 +132V (SEQ ID NO: 190, Table XXX) showing cleavage activity on the HIV 1_4 target.
- FIG 46 An example of positive variants is shown in figure 46. Sequencing of these positive clones allowed the identification of 10 different endonuclease variants.
- the clones cleaving the HIV 1_4 target were also tested for their ability to cleave the HIV1_4.4 and HIV1_4.6 targets (see Figure 47 for an example). In this case, 15 of the clones were able to cleave the HIV1_4.3 and the HIV1_4.5 targets. Sequence analysis of these clones showed the presence of 10 different endonuclease variants. Comparison of sequences of the positive clones in all the targets indicated the presence of a total of 1 1 novel endonuclease variants.
- the sequence of ten l-Crel variants cleaving the HIV 1_4 target when forming a heterodimer with the KHSMAS/NYSYR variant arc listed in Table XXXIV.
- Example 24 Strategy for engineering meganucleases cleaving the HIV1_5 target from the HIVl virus
- the HIV 1_5 target is a 22 bp (non-palindromic) target located in the pol gene of the HIVl provirus. This target is precisely located at positions 2317-2338 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al., J. Virol, 1986, 59, 284-291), a subtype B infectious molecular clone.
- the HIV1_5 sequence (SEQ ID NO: 337) is partly a patchwork of the 1 OTCTJP, 10CTG_P, 5TAG_P and 5 CCTJ targets ( Figure 48) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould ct al., J. MoL Biol, 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006.
- HIV1_5 could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
- the 10TCT_P, 10CTG_P, 5TAG_P and 5_CCTJP target sequences are 24 bp derivatives of C 1221 , a palindromic sequence cleaved by l-Crel (Arnould et al, precited).
- l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al, Nat. Struct. Biol, 2001, 8, 312-316; Chevalier and Stoddard, Nucleic ⁇ cids Res., 2001 , 29, 3757-3774; Chevalier el a!., J.
- HlV 1__5 series of targets were defined as 22 bp sequences instead of 24 bp.
- H ⁇ V1_5 differs from C1221 in the 4 bp central region. According to the structure of the I-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the l-Crel protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI.
- the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region.
- the ATAC sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_5.2 (SEQ ID NO: 338, Figure 48).
- two palindromic targets, HIV1_5.3 (SEQ ID NO: 339) and HIV1_5.4 (SEQ ID NO: 340) were derived from HIV1 ⁇ 5.2 ( Figure 48). Since HIV1_5.3 and HIV1_5.4 are palindromic, they should be cleaved by homodimeric proteins.
- Two other quasi-palindromic targets were derived from these two, containing the ATAC sequence in -2 to 2 (targets H ⁇ V1_5.5 (SEQ ID NO: 341) and HIV1_5.6 (SEQ ID NO: 342), figure 48).
- proteins able to cleave HIVl m 5.3 and HIV1_5.4 targets or, preferentially, the quasi- palindromic targets as homodimers were first designed (examples 25 and 26) and then co-expressed to obtain heterodimers cleaving HIV 1_5 (example 27).
- Heterodimers cleaving the HIV1_5.2 and HIV 1_5 targets could be identified.
- Example 25 Identification of meganucleases cleaving HIV1 5.3 This example shows that l-Crel variants can cut the HIV 1__5.3 DNA target sequence derived from the left part of the HIV1_5.2 target in a palindromic form ( Figure 48).
- HIV1__5.3 is similar to 10TCT_P at positions ⁇ 1, ⁇ 2, ⁇ 6, ⁇ 8, ⁇ 9, and ⁇ 10 and to 5TAG_P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 6. It was hypothesized that positions ⁇ 7 and ⁇ 11 would have little effect on the binding and cleavage activity. Variants able to cleave the 1 OTCTJP target were obtained by mutagenesis of l-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al Nucleic ⁇ cids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- Variants able to cleave 5TAG_JP were obtained by mutagenesis on l-Crel N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al, J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
- the target was cloned as follows: an oligonucleotide corresponding to the HIV1JJ.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTGCTCTATTAGGTACCTAATAGAGCCAATCGTCTGTCA 3' (SEQ ID NO: 52). The same procedure was followed for cloning the HIV1_5.5 target, using the oligonucleotide: 5'
- Double-stranded target DNA generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8).
- yeast reporter vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B (MAT a, ura3 ⁇ 851, trpl ⁇ 63, leu2 ⁇ l, tys2 ⁇ 202), resulting in a reporter strain.
- I-Crel variants cleaving 10TCT_P or 5TAG_P were previously identified, as described in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al, J. MoL Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10TCT_P and 5TAGJ > targets.
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3 J (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542 5 Figure 9) and primers (assF 5'-ctannnttgaccltt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3 J (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidase activity.
- DMF dimethyl formamide
- results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants
- yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA.
- ORFs were amplified from yeast DNA by PCR (Akada et al., Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MILLEGEN SA.
- variants Two positive clones were found (though having weak cleavage activity), which after sequencing turned out to correspond to 2 different novel endonuclease variants (Table XXXVI). These two positives are shown in Figure 49. These two variants display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77. Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.
- Example 26 Making of meganucleases cleaving HIV1 5.4
- HIV1_5.4 DNA target sequence derived from the right part of the HIV1_5.2 target in a palindromic form ( Figure 4).
- HIV1_5.4 is similar to 5CCTJP at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 5 and ⁇ 8 and to 10CTG_P at positions ⁇ 1, ⁇ 2, ⁇ 3, ⁇ 4, ⁇ 8, ⁇ 9 and ⁇ 10. It was hypothesized that positions ⁇ 6, ⁇ 7 and ⁇ 1 1 would have little effect on the binding and cleavage activity.
- Variants able to cleave 5CCT_P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould el al, J. MoI. Biol., 2006, 355, 443-458; Smith et al Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156).
- Variants able to cleave the 1 OTGGJ* target were obtained by mutagenesis of 1-OeI N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
- HIV1_5.4 target mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5CCT JP were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10CTG_P.
- A) Material and Methods a) Construction of target vector
- the experimental procedure is as described in example 2, with the exception that different oligonucleotides corresponding to the HIV 1_5.4 and H1V1_5.6 targets.
- the oligonucleotide used for the HIV1_5.4 target was: 5' TGGCATACAAGTTTATCTGCTCCTGTACAGGAGCAGATCAATCGTCTGTCA 3' (SEQ ID NO: 243), and 5'
- I-CVel variants cleaving 1 OCTGJ 5 or 5CCTJP were previously identified, as described in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al, J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10CTG_P and 5CCT _P targets.
- PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1107, Figure 11) and primers (assF 5'-ctannnttgacctttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43.
- primers GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO
- PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio.
- filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0 C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF) 3 7 mM ⁇ -mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor ⁇ -galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software.
- Example 27 Making of meganucleases cleaving HIV1 S.2 and HIV1 5
- A3' (SEQ ID NO: 254) or the HIV1_5 target sequence: 5TGGCATACAAGTTTGCTCTATTAGATACAGGAGCAGATCAATCGTCTGTC
- Yeast DNA was extracted from variants cleaving the HIV1_5.4 target in the pCLS 1107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_5.3 target in the pCLS0542 expression vector.
- Transformants were selected on synthetic medium lacking leucine and containing
- Mating was performed using a colony gridder (Qpix ⁇ , Genetix).
- Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm 2 ). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at
- the functional combination cleaving the HIV1_5.2 target correspond to mutants KNSCYS/AYQNI (SEQ ID 241, cleaving HIV1_5.3) and KTSGQS/KYSDR +151 A (SEQ ID 252, cleaving HIVlJ .4)
- Example 28 Improvement of meganucleases cleaving HIVl 5.3 by random mutagenesis and assembly with proteins cleaving HIVl 5.4 l-Crel variants able to cleave the HIVl J.3 have been identified in example 25. Since these two variants show a weak activity, and only one of them is able to cleave the HIVl J.2 target when assembled with a meganuclease cleaving the HIV IJ.4, these two variants were mutagenized, and the clones generated were screened for cleavage activity of HIVl J.3 and HIVl J.5 targets.
- mutants with the strongest activity were screened for cleavage activity of HIVlJ when co-expressed with a variant cleaving HIVl J.4.
- the structure of the l-Crel protein bound to its target there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-CVeI protein (Chevalier el ai, Nat. Struct. Biol, 2001 , 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al , J. MoI. Biol., 2003, 329, 253-269).
- Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn 2+ .
- PCR reactions were carried out that amplify the 1-OeI coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-B'; SEQ ID NO: 25) ⁇ which are common to the pCLS0542 ( Figure 9) and pCLSl 107 ( Figure 1 1) vectors.
- HIV1_5.3 and HIV1_5.5 DNA targets A total of 20 positive clones were found to cleave HIV1_5.3, while none of those cleaved the HIV1_5.5 target. Sequencing of the 20 clones allowed the identification of 13 novel cndonuclcasc variants. An example of these variants is presented in table XXXIX and in figure 52.
- the 20 clones showing cleavage activity on target HIV1_5.3 were also mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid ( ⁇ ) an expression plasmid containing a variant that cleaves the HIV1_5.4 target (SEQ ID 252; 1-OeI 30T,33G,44K,68Y,70S,77R +151A or KTSGQS/KYSDR +15IA, according to the nomenclature of Table I). After mating with this yeast strain, no clones were found to cleave the HIV 1_5 target.
- Example 28bis Improvement of meganucleases cleaving HTV1 5.3 by a second round of random mutagenesis and assembly with proteins cleaving HIV1_5.4
- a second round of random mutagenesis was carried out following the same rationale of example 28.
- ten variants cleaving HIV1_5.3 were mutagenized, and variants were screened for cleavage activity of HIV1_5.3 and H ⁇ V1_5.5 targets. Additionally, the mutants with the strongest activity were screened for cleavage activity of HIV 1_5 when co-expressed with a variant cleaving HIV1_5.4.
- the 80 clones showing cleavage activity on target HIV1_5.3 were then mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.4 target (I- Crel 30S 5 33N,44K,68Y ; 70S,77R +103T or KSSNQS/KYSDR +103T, according to the nomenclature of Table I). After mating with this yeast strain, 4 clones were found to cleave the HIV 1_5.
- 4 positives contained proteins able to form heterodimers with KSSNQS/KYSDR +103T (SEQ ID NO: 276) showing cleavage activity on the HIV 1_5 target.
- An example of positives is shown in Figure 54.
- These 4 variants are presented as an example in Table XXXX (SEQ ID 266 to 269).
- Example 29 Improvement of meganucleases cleaving HIV1 5 by site-directed mutagenesis of proteins cleaving HIV1 5.3 and assembly with proteins cleaving HIV1_5.4
- Site- directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the G19S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- Crel coding sequence.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIl OR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3 '(SEQ ID NO: 48)).
- E80KF 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcUgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagtlgagtcagcaggttgtgcagcggctt-3 ' SEQ ID NO: 53 and 54);
- Vl 05AF 5'-aaacaggcaaacctggctctgaaaattatcgaa ⁇ 3'
- V 105AR 5'- tlcgataattttcagagccaggtttgcctgltl-3' SEQ ID NO: 55 and 56);
- I132VF 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Nc ⁇ i and Eagl.
- This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATcc, trpl ⁇ 63, leu2 ⁇ l, his3 ⁇ 200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymoi., 2002, 350, 87-96).
- Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast.
- the 558 transformed clones were also mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.4 target ( ⁇ -Crel
- Example 30 Improvement of meganucleases cleaving HIV1 5.4 by random mutagenesis and assembly with proteins cleaving HIV1 5.3
- yeast strain FYBL2-7B (MAT a, ura3 ⁇ 851, trpl ⁇ 63, lei ⁇ l, lys2 ⁇ 202) containing the HIV IJ target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_5.3 target, using a high efficiency LiAc transformation protocol.
- Variant-target yeast strains were used as target strains for mating assays as described in example 27. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 25.
- the 53 positive clones showing the highest cleavage activity on target HIV1_5.4 were then mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.3 target (l-Cre ⁇ 33C,38Y,44A,68Y ; 70Q,75N +89A or KNSCYS/AYQNI +89A, according to the nomenclature of Table I; SEQ ID 256). After mating with this yeast strain, no clones were found to cleave the HIV 1_5 target.
- Example 30bis Improvement of meganucleases cleaving HIV1_5 by a second round of random mutagenesis of proteins cleaving H1V1 5.4 and assembly with proteins cleaving HIV1_5.3
- a second round of random mutagenesis was carried out following the same rationale of example 30.
- six variants cleaving HIV1_5.4 were mutagenized, and variants were screened for cleavage activity of HIV1_5.4 and HIV1_5.6 targets. Additionally the mutants were screened for cleavage activity of HIV 1_5 when co- expressed with a variant cleaving HIV1_5.3.
- H ⁇ V1_5.4 and HIV1_5.6 DNA targets A total of 21 positive clones were found to cleave HIV1_5.4, while 9 of those cleaved also the HIV1_5.6 target. Sequencing of the 21 clones allowed the identification of 16 novel endonuclease variants. An example of the identified variants is presented in Table XLIII and figure 58.
- the 21 positive clones showing cleavage activity on target H ⁇ V1_5.4 were then mated with a yeast strain that contains (i) the HIV IJ target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the H ⁇ V1J5.3 target (l-Crel 33C,38Y,44A,68Y,70Q,75N +89A or KNSCYS/AYQNI +89A 3 according to the nomenclature of Table I; SEQ ID 256). After mating with this yeast strain, no clones were found to cleave the HIV 1_5 target.
- Example 31 Improvement of meganucleases cleaving HIV1 5 by site-directed mutagenesis of proteins cleaving HIV1_5.4 and assembly with proteins cleaving HIV1_5.3
- Two of the ⁇ -Crel variants cleaving HIVI_5.4 described in Table XL ⁇ II were mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV1_5.4 and HIV1_5,6, as well as for cleavage of the IIIV1_5 target when in combination with a variant cleaving HIV 1 5.3.
- PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3 ' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctltgtggactclgacggtagcatcatc-3' (SEQ ID NO: 47) or Gl 9SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
- the same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V 105 A and Il 32V substitutions in
- F54LF 5'-acccagcgccgttggctgctggacaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50); * E80KF: 5'-ttaagcaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52);
- F 87LF 5 ! -aagccgctgcacaacctgctgactcaactgcag-3'
- F 87LR 5'- ctgcagltgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
- V 105AF 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
- Il 32VF 5'-acctgggtggatcaggttgcagctctgaacgat-3' and Il 32VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
- the resulting PCR products contain 33bp of homology with each other.
- the PCR fragments were purified.
- the ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Nco ⁇ and Eagl.
- the 558 transformed clones were also mated with a yeast strain that contains (i) the HIV1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.3 target (l-Crel 33C ) 38Y,44A,68Y,70Q,75N +89A or KNSCYS/AYQNI +89A, according to the nomenclature of Table I).
- 137 clones were found to cleave the HIV 1_5.
- 137 positives contained proteins able to form heterodimers with KNSCYS/AYQNI +89A (SEQ ID NO: 256) showing cleavage activity on the HIV1_5 target.
- An example of positives is shown in figure 60. Sequencing of the 93 clones with the highest cleavage activity on the HIV 1_5 target allowed the identification of 48 different endonuclease variants.
- Example 32 Covalent assembly as single chain and improvement of meganucleases cleaving different HIVl targets by site-directed mutagenesis
- Example 33 Determination of antiviral effect of HIVl meganuclease variants derived from I-Crel
- the efficacy of HIV meganucleases to cleave the corresponding proviral DNA target was assessed in a cellular system containing a defective integrated provirus.
- This cellular model produces viral-like particles (VLPs) containing all the essential HIVl proteins with the exception of the viral envelope glycoproteins. Nevertheless, the produced VLPs are not able to infect the cells due to the absence of entry-mediating proteins in the viral envelope. Production of VLPs can be measured in the supernatants of cultured cells using an HIVl-p24 ELISA kit.
- the VLP-producing cells were transfected with the plasmids coding for the different versions of the SCOH-HIVl meganucleases and the antiviral effect was measured by the reduction in the litres of p24 present in the supernatants of transfected cells respect to a "control" sample in which the cells were transfected by a non-related meganuclease (NRM), which has no cleavage activity on the HIVl proviral DNA.
- NEM non-related meganuclease
- two retroviral vectors were generated harbouring either the tat or the rev coding sequences. These two vectors were used to sequentially transduce HEK-293 cells, leading to the generation of a cell line able to produce the tat and rev proteins after integration of the retroviral vectors in the cellular genome.
- the generated cell line was then transduced by a lentiviral expression vector that, after integration of the dsDNA resulting from reverse transcription, would generate the pseudo-Ill Vl provirus containing the meganuclease target hits.
- the cells were tested for their ability to produce VLPs by determining the presence of the HIVI p24 protein in the culture supernatants using the Alliance® HIVl-p24 ELISA Kit (Perkin Elmer Inc, Waltham, MA, USA).
- the VLP producing cells were subjected to clonal dilutions in order to characterize the number of pseudo HIVl integrated provirus in different clones.
- a cellular clone (HEK293-VLP-CL40) containing between 1 and 2 copies of the pseudo HIVl provirus (as determined by qPCR) was used for assessing the antiviral activity of meganucleascs.
- HEK293-VLP-CL40 cells were cultured in DMEM media supplemented with 2mM L-glutamine, penicillin (100 IU/ml), streptomycin (100 mg/ml), amphotericin B (Fongizone: 0.25 mg/ml, Invitrogen-Life Science) and 10% of foetal bovine serum (FBS).
- FBS foetal bovine serum
- HEK293-VLP-CL40 cells were seeded in 12-well culture plates (Falcon, Becton Dickinson, Le Pont De Claix, France) at 10 5 cells per well and incubated overnight at 37 0 C in 1 ml of complete growth medium. The cultures were about 70% confluent on the day of transfection. Transfection with 1 ⁇ g of plasmid expressing I-Crel variants cleaving different HIVl target sequences was done using FuGENE ⁇ HD Transfection Reagent (Roche Diagnostics, Indianapolis, IN 5 USA) according to manufacturer's instruction, Transfection media was replaced 24h after transfection and cells were kept at 37°C in complete growth medium for other 24 hours. c) Cell harversting and p24 determination
- HEK-293-CL40 transfected cells were then recovered and counted, prior to centrifugation at 1500 rpm for 5 minutes and storage of the dry cellular pellet at -20 0 C for ulterior extraction of the genomic DNA.
- the amount of p24 present in cellular supernatants was determined using the Alliance® HlVl-p24 ELISA Kit (Pcrkin Elmer Inc, Waltham, MA, USA) according to the manufacturer's instructions. Results were expressed as p24 in pg/ml (or as pg/well, according to the cell culture conditions). The production of p24 was normalized by the number of cells present in the well at the moment of media harvesting, and expressed as p24 levels in fg/cell. 2) Results
- the single chain molecules described in Table XLV were tested for their ability to target the HIVl provirus and reduce the amount of VLPs produced in the HEK293-VLP-CL40 cellular model.
- Cells were transfected with 1 ⁇ g of plasmid expressing the meganuclease variants and the level of p24 present in the culture supernatants was determined 48h after transfection, as previously described.
- a non related meganuclease was transfected. This NRM is not active against the HlVl provirus and should have no effect on the level of p24 produced by NRM transfected cells.
- the meganuclease target sites have already been described except for the HIV 1_7, HIVlJ and HIV1_9 targets.
- the HIV 1_1 target, described in example 1, is located in the U3 region of the proviral LTRs; while the HIV 1_3 target, described in example 8, is located in the U5 region of the proviral LTRs. Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, each of these two targets are present twice in the HIVl provirus.
- the HIV 1_4 target has been described in example 16, and is located in the gag gene of the HIVl provirus, more precisely in the coding sequence of the p24 (CApsid) protein.
- the HIV IJ target (G GAG CC ACC CCAC AAG AT TTA A,
- SEQ ID NO: 366) also cleaves the coding sequence of the ⁇ 24 protein, though at a different position.
- the HIV 1_7 target is also a 22 bp (non-palindromic) target precisely located at positions 1321-1342 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al, J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
- the HIV 1_5 target has been described in example 24, and is located in the pol gene of the HIVl provirus, more precisely in the sequence coding for the PRotease protein.
- the HIV 1_9 target also cleaves the coding sequence of the protease, though at a different position.
- the HIV1_9 target (A GAA AT CTG TTGA CTC AG ATT G, SEQ ID NO: 368) is also a 22 bp (non-palindromic) target located at positions 251 1-2532 of the HIV-I pNL4-3 vector.
- the HIV 1_8 target (G GGC CC CTA GGAA AAA GG GCT G, SEQ ID NO: 367) is a 22 bp (non-palindromic) target located in the gag gene of the HIVl provirus. This target is precisely located at positions 2006-2027 of the HIV-I pNL4-3 vector, on the coding sequence of the p7 (NC, NucleoCapsid) protein.
- HEK293-VLP-CL40 cells was confirmed by sequencing and their position is represented in figure 62.
- I-Crcl variants targeting the HIVl genome induce a decrease in p24 litres in a cellular model harbouring an HIVl provirus p24 titres were determined 48 hours after transfection with the HIVl meganucleascs as previously described.
- Figure 63 shows the levels of p24 (in %) produced by the cells transfected with the different meganuclease plasmids. A reduction of p24 production is observed in samples transfected with HIV meganucleases. The meganucleases showing a higher reduction in p24 titers correspond to variants SCOH-HIV1_3-B and
- SCOH-HIVlJ-D (SEQ ID NO: 350 and 352), leading to nearly a 50% reduction of p24 levels compared to cells transfected with the NRM.
- HIV1_8-D > SEQ ID NO: 362; and SCOH-HIV 1_9-B, SEQ ID NO: 364).
- Example 34 Detection of cleavage activity at the HIV1 8 locus in a human cell line harbouring an integrated HIVl provirus.
- HIV provirus HEK293-VLP-CL40 cells. Repair of double-strand break by non homologous end-joining
- Example 34.1 Detection of induced mutagenesis at the endogenous site Two Single Chain l-Crel variants targeting the HIV 1_8 target cloned in the pCLS1853 plasmid were used for this experiment. The day previous to the experiment, cells derived from the human embryonic kidney cell line, 293 -H (HEK293-VLP-CL40) were seeded in a 10 cm dish at density of 10 6 ceUs/dish.
- cells were transfccted with 3 ⁇ g of an empty plasmid or a meganuclease-expressing plasmid using FuGene® HD Transfcction Reagent (Roche Diagnostics, Indianapolis, IN, USA) according to manufacturer's instruction. 72 hours after transfection, cells were collected and diluted (dilution 1/20) in fresh culture medium. After 7 days of culture, cells were collected and genomic DNA extracted. 200 ng of genomic DNA were used to amplify the endogenous locus surrounding the meganuclease cleavage site by PCR amplification.
- a 325 bp fragment corresponding to the HIV 1_8 locus was amplified using specific PCR primers HI8f (SEQ ID NO 369; 5 '-GACCCGGCCATAAAGCAAGAGTTTTGGCTG-S ') and IIISr (SEQ ID NO 370; 5'-AAGCTCTCTTCTGGTGGGGCTGTTGGCTCT-S ').
- PCR amplification was performed to obtain a fragment flanked by specific adaptator sequences (SEQ ID NO 371 ; 5'-CCATCTCATCCCTGCGTGTCTCCGACTCAG-S ' and 25 SEQ ID NO: 372 5'-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-S') provided by the company offering sequencing service (GATC Biotech AG, Germany) on the 454 sequencing system (454 Life Sciences).
- specific adaptator sequences SEQ ID NO 371 ; 5'-CCATCTCATCCCTGCGTGTCTCCGACTCAG-S ' and 25 SEQ ID NO: 372 5'-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-S'
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Medicinal Chemistry (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Communicable Diseases (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Oncology (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Enzymes And Modification Thereof (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Meganuclease variants which cleave at least one target in the provirus of a retrovirus and in particular which cleave the genomic insertion of the provirus. The present invention in particular relates to meganuclease variants which cleave the provirus of the Human Immunodeficiency Virus genome following genomic insertion. Vector encoding such variants, as well as to a cell or multi-cellular organism modified by such a vector and use of said meganuclease variants and derived products for genome engineering and for in vivo and ex vivo (gene cell therapy) genome therapy.
Description
MEGANUCLEASE VARIANTS CLEAVING AT LEAST ONE TARGET IN THE GENOME OF A RETROVIRUS AND USES THEREOF
The invention relates to the use of meganuclease variants which cleave at least one target in the provirus of a retrovirus and in particular cleave the genomic insertion of an integrating Virus genome and in particular to meganuclease variants which cleave the Human Immunodeficiency Virus genome following genomic insertion, for the treatment of an infection of one or more of these viruses.
The present Invention also relates to such variants and to vectors encoding such variants, as well as to a cell or multi-cellular organism modified by such a vector and to the use of said meganuclease variant and derived products for genome engineering and for in vivo and ex vivo (gene cell therapy) genome therapy.
Viral infections of various sorts are a serious and continuing health, agricultural and economic problem worldwide. In particular viruses present specific treatment and control problems as they always comprise an intracellular stage to their life cycle, in which the nucleic acid genome of the virus is inserted into a host cell and normally transported to the nucleus. During this stage of the virus life cycle, the virus genome can enter into a dormant state whilst inside a host cell, during which time the production of new virus particles/proteins/copies of the viral genome ceases. These characteristics present a significant problem as most medicaments and treatments for viral infection consist of compounds which affect aspects of virus biology involved in the active stages of the virus life cycle, such as compounds which target/inactivate a viral enzyme or structural protein. Therefore whilst in a dormant state the viral genome resident in the cytoplasm or nucleus of a host cell can not be affected by most conventional anti-virus medicaments and therefore persists.
One group of viruses presents additional problems as they integrate into the host cell genome. This group, called retroviruses, like other viruses are transmitted via the infection of new host cells by virus particles and can also cause the endemic infection of the progeny cells of a host cell in which they are genomically integrated. This second mode of transmission, particularly when the retrovirus genome is dormant can result in the clonal expansion of the retrovirus containing cells, which in turn can cause significant problems once the retrovirus genomes activate.
The present invention therefore relates to Retroviruses which are contained with the family Retroviridae which comprises in turn seven genera. Alpharetrovirus, Betaretrovirus, Gammaretrovirus, Deltaretrovirus, Epsilonretro- virus, Lentivirus and Spumavirus. These groups of viruses are responsible for several important diseases such as Human T-lymphotrophic virus {Gammaretrovirus), Rous Sarcoma {Alpharetrovirus) and Human Immunodeficiency Virus {Lentivirus).
The Human Immunodeficiency Virus (HIV) (Figure 1) is an example of a Retrovirus which is responsible for a significant and ongoing global medical crisis. HIV viruses persist and continue to replicate for many years in the infected individual before causing overt signs of disease. HIV is the causative agent of the Acquired Immune Deficiency Syndrome (AIDS), which is characterized by a susceptibility to infection with opportunistic pathogens, mainly as a result of a profound decrease in the number of CD4+ T cells, A characteristic feature of the Retroviridae family of viruses is that viral particles contain two copies of an RNA genome. After infection, the genomic RNA is reverse transcribed by a viral enzyme into DNA, which is then permanently integrated into the host genome.
The retroviral genome harbors the sequences coding for the viral enzymatic, structural and regulatory proteins. In addition, the genomic RNA molecule contains a series of non-coding sequences that have important functions in different steps of the viral life cycle (Figure 2).
The "2007 AIDS epidemic update" report, issued by the UNAIDS (Joint United Nations Programme on HIV/AIDS), indicates that 33.2 million [30.6 - 36.1 million] people were estimated to be living with HIV, 2.5 million [1.8 - 4.1 million] people became newly infected with HIV and 2.1 million [1.9 - 2.4 million] people died of AIDS in 2007.
HIV is characterized by a high genetic variability, due to the rapid viral turnover (1010 - 1012 viral particles produced per day) in an HIV-infected individual, combined with the high mutation rate arising during reverse transcription (10"4 per nucleotide). Two types of HIV, HIV-I and HIV-2, which are closely related to each other, have been identified to date (Sharp et al., Philos Trans R Soc Lond B Biol Sci, 2001, 356, 867-76). Most AIDS worldwide is caused by the more virulent HIV-I, while HIV-2 is endemic in West Africa. Both viruses appear to have spread to
humans from other primate species and the best evidence from sequence relationships suggests that HIV-I has passed to humans on at least three independent occasions from the chimpanzee, Pan troglodytes and HIV-2 from the sooty mangabey, Cercocebus atys. The three zoonotic transmissions that generated the HIV-I type viruses gave rise to three different viral groups: M, O and N. The M group (for main), represents the substantial majority of worldwide infections. The O (for outlier) and N (for non-M/non-0) groups remain essentially restricted to Central Africa (Sharp et al., Philos Trans R Soc Lond B Biol Sci, 2001 , 356 , 867-76). HIV is transmitted by direct sexual contact, by blood or blood products, and from an infected mother to infant, either intrapartum, perinatally, or via breast milk. Infection of humans with HIV- 1 causes a dramatic decline in the number of CD4+ T lymphocytes. When the number of CD4+ cells is very reduced, opportunistic infections and neoplasms occur (Simon et al., Lancet, 2006, 368 , 489-504). Anti retro viral treatment for HIV infection consists of drugs which work by slowing down the replication of HIV in the body. Currently, there are around 30 anti retroviral drugs approved to treat people infected with HΪV in various countries around the world. There are several classes of anti-HIV drugs that attack the virus in different ways and the most common classes of antiretrovirals are nucleoside or nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors and entry inhibitors (Flexncr C, Nature Reviews Drug Discovery, 2007, 6, 959-966).
People with HIV need to continuously take antiretroviral drugs. Furthermore for antiretroviral treatment to be effective for a long time, it has been found that more than one antiretroviral drug must be taken at a time as single drug treatment regimes invariably lead to HIV resistance to the single drug negating its therapeutic effects.
Combination Therapy, wherein at least two and normally three different medicaments are taken simultaneously prolongs the period of time before resistance develops for one or more of the medicaments. The term Highly Active Antiretroviral Therapy (HAART) is used to describe a combination of three or more anti-HIV drugs. HAART typically combines drugs from at least two different classes
of antiretroviral drugs and has been shown to effectively suppress the virus when used properly. Highly active antiretroviral therapy has revolutionalized how people infected with HIV arc treated, and reduces the rate at which resistance develops.
Normally when anti-HIV treatment is started, the viral load drops to an undetectable level. When drug resistance develops, the amount of HIV in the blood rises and the risk of the person becoming ill increases and this usually means that the drug regimen needs to be changed (Martinez- Caj as and Weinberg, Drugs, 2008, 68,
43-72).
Currently available HIV treatments have converted HIV infection into a chronic disease, increasing the lifespan of infected individuals. Λnti-HIV drugs can reduce the rate of viral replication, retarding therefore the onset of AIDS. Nevertheless, the emergence of strains resistant to these existing treatments, require the continual development of new therapeutic strategies (Rossi et al, Nat. Biotechnol., 2007, 25, 1444-54). Although there are currently no vaccines to prevent or treat HIV, researchers are developing and testing several potential HIV vaccines, either for preventive and/or therapeutic purposes. However, vaccine development encounters the same problem as anti-HIV drugs concerning the rapid viral evolution and the subsequent development of resistance or in the case of a vaccine an evolved IHV strain which no longer comprises the epitope used in the vaccine and hence is not affected by the immune response elicited by the vaccine. At the present time the general consensus in the scientific and medical community is that therapeutic HIV vaccines will not be able to completely eliminate HIV infection, because the virus "hides" in certain cells of the body, where it can last silent for decades meaning that any effect of the vaccine will have been lost. A new field for the treatment of HIV infection is the development of genetic therapies against HIV. Gene therapy could allow the prevention of progressive HIV infection by persistently blocking viral replication. Gene-targeting strategies are being developed with RNA-based agents such as ribozymes, aptamers and small interfering RNAs and protein-based agents. Among the last group, the use of zinc- finger nucleases against the CCR5 receptor, a protein present on the surface of immune cells that is required to mediate viral entry, is currently in Phase I clinical trials. In this case, the disruption of the CCR5 receptor from the immune cells by the
nucleases is proposed to render the patient's cells permanently resistant to CCR5- specific strains of HIV. This approach is based on the fact that people with natural mutations on this receptor are resistant to HIV infection.
To date however the number of effective anti-HIV/retro virus therapies is very small, due in part to the limited number of target genes/proteins/pathways present in the relatively simple retrovirus genome/life cycle as well as to the rapid creation of 'escape' mutants by the retrovirus during replication which allow members of the virus population to evade therapeutic compounds that more slowly evolving pathogens such as bacteria or protozoa would not be able to develop resistance to with the same speed.
In addition due to the existence of dormant intragenomic copies of the provirus which are not affected by any current therapy, the curing of HIV infection (AIDS) is currently simply not possible.
An interesting target that has not been pursued in the fight against the AIDS pandemic and more generally retroviruses is the genomically integrated provirus and/or the reverse transcribed DNA version of the retrovirus genome prior to its integration, since targeting the proviral DNA could lead to the elimination or inactivation of the structure that allows the virus to multiply and the infection to propagate. One novel way to inactivate the provirus which the inventors have decided to investigate is by the use of nucleases that could cleave the integrated form of the virus and generate mutations and/or deletions in the provirus following the action of the cellular DNA repair machinery.
An important point to be considered in this kind of approach is the choice of the target sequences. In a first instance, the target sequences should be located in the coding sequences of essential genes, since the inactivation of an accessory gene may not lead to viral eradication. The viral genome also contains essential regulatory sequences that are located in the long terminal repeats (LTRs) that flank the viral genome in the provirus. Even if mutations in these regions would be expected to have a less drastic effect than a mutation in an essential gene, the fact that they are duplicated sequences could be useful in an approach of "virus clipping", meaning the excision of long regions of the proviral DNA by the action of a nuclease cleaving twice in the viral sequence. Another important point that should be
considered is the degree of sequence variation that is observed in the target sequences among different circulating viral isolates. As discussed above HIV is characterized by a high degree of sequence variability due to the nature of the viral reverse transcriptase. It is therefore essential to check the sequence conservation of the target among the different isolates.
The inventors have developed a new molecular medicine approach based on the inactivation of the retrovirus provirus through the use of tailored meganucleases specifically targeting the proviral DNA, using the HIV-I provirus in the genome of the infected cell as a model. The principle of this new therapeutic strategy is that the tailored meganucleases against targets in the provirus will generate a double strand break (DSB) at their target sequences, chosen to be located in genes/regulatory sequences/structural sequences that are essential for the virus to replicate or alternatively target sequences which are present in multiple copies in the provirus, for instance in the two flanking LTR regions, so allowing the provirus or a portion thereof to be excised.
The epidemiology of HIV, particularly in sub Saharan Africa, makes research into the HIV virus a major and extremely active area of research. The manipulation of the HIV provirus is one area of research in which to date reagents have not been readily available as workers have instead concentrated on attempting to manipulate the HIV virion per se. Therefore the means to easily engineer the HIV provirus in situ in the genome of an infected cell/organism would likely provide valuable insights into this aspect of HIV biology and potentially open new avenues of attack in combating HIV.
Even if the meganuclease targets have been selected following the criteria mentioned above, namely in essential genes and particularly in sequences showing the highest degree of conservation, the capacity of the virus to generate escape mutants under the selective pressure of a drug/therapy must be considered.
To minimize the effect of drug resistance(s), "Combination Therapy" has already been shown to counter act this feature of HIV biology. In the same way, the possibility of using a combination of meganucleases could help to prevent any resistance that could be generated during viral replication. In addition although HIV shows a very high level of genetic change, not all of the components of
the HIV genome are as capable of supporting change as others. Generally speaking it is those portions of the virus which are immunogenic, that is present upon the exterior of the virus particle where they can interact with the components of the hosts immune system, which are most able to support high levels of variability. Whereas the essential internal structural or packaging components of HIV are less able to continue to function following changes in their coding sequences. These differences do not affect the ability of HIV to evolve so as to elude the host immune response, but have proven useful in specifically engineering drugs for which it is more difficult for HIV to develop resistance. The increased levels of conservation of some provirus sequences can also be used to further hone the meganuclease(s) according to the present invention.
In vivo meganucleases are essentially represented by homing endonuclcases. Homing Endonucleases (HEs) are a widespread family of natural meganucleases including hundreds of proteins families (Chevalier, B. S. and B.L. Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774). These proteins are encoded by mobile genetic elements which propagate by a process called "homing": the endonuclease cleaves a cognate allele from which the mobile element is absent, thereby stimulating a homologous recombination event that duplicates the mobile DNA into the recipient locus. Given their exceptional cleavage properties in terms of efficacy and specificity, they could represent ideal scaffolds to derive novel, highly specific endonucleases.
HEs belong to four major families. The LAGLIDADG family, named after a conserved peptide motif involved in the catalytic center, is the most widespread and the best characterized group. Seven structures are now available. Whereas most proteins from this family are monomelic and display two LAGLIDADG motifs, a few have only one motif, and thus dimerize to cleave palindromic or pseudo-palindromic target sequences.
Although the LAGLIDADG peptide is the only conserved region among members of the family, these proteins share a very similar architecture (Figure 3). The catalytic core is flanked by two DNA-binding domains with a perfect two-fold symmetry for homodimers such as l-Crel (Chevalier, et α/., Nat. Struct. Biol., 2001, 8, 312-316) , ϊ-Msoϊ (Chevalier et al., J. MoI. Biol., 2003, 329, 253-269) and Ϊ-Ceul
(Spiegel et al., Structure, 2006, 14, 869-880) and with a pseudo symmetry for monomers such as l-Scel (Moure el al , J. MoI. Biol., 2003, 334, 685-69, l-Dmol (Suva et al, J. MoI. Biol., 1999, 286, 1 123-1 136) or 1-Anil (Bolduc et al , Genes Dev., 2003, 17, 2875-2888). Both monomers and both domains (for monomelic proteins) contribute to the catalytic core, organized around divalent cations. Just above the catalytic core, the two LAGLIDADG peptides also play an essential role in the dimerization interface. DNA binding depends on two typical saddle-shaped αββαββα folds, sitting on the DNA major groove. Other domains can be found, for example in inteins such as Ϋl-PfuX (Ichiyanagi et al, J. MoI. Biol., 2000, 300; 889-901) and PI- Seel (Moure et al , Nat. Struct. Biol, 2002, 9, 764-770), whose protein splicing domain is also involved in DNA binding.
The making of functional chimeric meganucleases, by fusing the N- terminal l-Dmol domain with an I-Crel monomer (Chevalier et al , MoI. Cell., 2002, 10, 895-905 ; Epinat et al, Nucleic Acids Res; 2003, 31, 2952-62; International PCT Applications WO 03/078619 and WO 2004/031346) have demonstrated the plasticity of LAGLID ADG proteins.
Different groups have also used a semi -rational approach to locally alter the specificity of the Ϊ-Creϊ (Seligman et al., Genetics, 1997, 147, 1653-1664; Sussman et al., J. MoI. Biol., 2004, 342, 31-41 ; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156; Arnould et al., J. MoI. Biol., 2006, 355, 443-458; Rosen et al, Nucleic Acids Res., 2006, 34, 4791-4800 ; Smith et al , Nucleic Acids Res., 2006, 34, el 49), l-Scel (Doyon et al., J. Am. Chem. Soc, 2006, 128, 2477-2484), Pl-Scel (Gimble et al, J. MoI. Biol., 2003, 334, 993-1008 ) and l-Msol (Ashworth et al, Nature, 2006, 441 , 656-659). In addition, hundreds of l-Crel derivatives with locally altered specificity were engineered by combining the semi-rational approach and High Throughput Screening:
- Residues Q44, R68 and R70 or Q44, R68, D75 and 177 of l-Crel were mutagenized and a collection of variants with altered specificity at positions ± 3 to 5 of the DNA target (5NNN DNA target) were identified by screening (International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith et al, Nucleic Acids Res., 2006, 34, el49).
- Residues K28, N30 and Q38 or N30; Y33 and Q38 or K28, Y335
Q38 and S40 of l-Crel were mutagenized and a collection of variants with altered specificity at positions ± 8 to 10 of the DNA target (10NNN DNA target) were identified by screening (Smith et al, Nucleic Acids Res., 2006, 34, e!49; International PCT Applications WO 2007/060495 and WO 2007/049156).
Two different variants were combined and assembled in a functional heterodimeric endonuclease able to cleave a chimeric target resulting from the fusion of two different halves of each variant DNA target sequence (Λrnould et al, precited; International PCT Applications WO 2006/097854 and WO 2007/034262), as illustrates in Figure 4.
Furthermore, residues 28 to 40 and 44 to 77 of I-Crel were shown to form two separable functional subdomains, able to bind distinct parts of a homing endonuclease half-site target sequence (Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/049095 and WO 2007/057781). The combination of mutations from the two subdomains of l-Crel within the same monomer allowed the design of novel chimeric molecules (homodimers) able to cleave a palindromic combined DNA target sequence comprising the nucleotides at positions ± 3 to 5 and + 8 to 10 which are bound by each subdomain (Smith et al, Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/049095 and WO 2007/057781 ).
The combination of the two former steps allows a larger combinatorial approach, involving four different subdomains. The different subdomains can be modified separately and combined to obtain an entirely redesigned meganuclease variant (heterodimer or single-chain molecule) with chosen specificity, as illustrated in Figure 5. In a first step, couples of novel meganucleases are combined in new molecules ("half-meganuclcascs") cleaving palindromic targets derived from the target one wants to cleave. Then, the combination of such "half-meganucleases" can result in a heterodimeric species cleaving the target of interest. The assembly of four sets of mutations into heterodimeric endonucleases cleaving a model target sequence or a sequence from different genes has been for instance described in the following patent applications: XPC gene (WO2007093918), RAG gene (WO2008010093),
HPRT gene (WO2008059382), beta-2 microglobulin gene (WO2008102274), Rosa26 gene (WO2008152523) and Human hemoglobin beta gene (WO200913622).
The method for producing meganuclease variants and the assays based on cleavage-induced recombination in mammal or yeast cells, which are used for screening variants with altered specificity are described in the International PCT Application WO 2004/067736; Epinat et al, Nucleic Acids Res., 2003, 31, 2952- 2962; Chames et al., Nucleic Acids Res., 2005, 33, el78, and Arnould et al, J. MoI. Biol., 2006, 355, 443-458. These assays result in a functional LacZ reporter gene which can be monitored by standard methods. These variants can be used to cleave genuine chromosomal sequences and have paved the way for novel perspectives in several fields, including gene therapy.
Even though the base-pairs ±1 and ±2 do not display any contact with the protein, it has been shown that these positions are not devoid of content information (Chevalier et al, J. MoL Biol., 2003, 329, 253-269), especially for the base-pair ±1 and could be a source of additional substrate specificity (Argast et al., J. MoI. Biol., 1998, 280, 345-353; Jurica et al., MoL Cell., 1998, 2, 469-476; Chevalier, B.S. and B.L. Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774). In vitro selection of cleavable \-Cre\ targets (Argast et ah, precited) randomly mutagenized, revealed the importance of these four base-pairs on protein binding and cleavage activity. It has been suggested that the network of ordered water molecules found in the active site was important for positioning the DNA target (Chevalier et al , Biochemistry, 2004, 43, 14015-14026). In addition, the extensive conformational changes that appear in this region upon l-Crel binding suggest that the four central nucleotides could contribute to the substrate specificity, possibly by sequence dependent conformational preferences (Chevalier et al., 2003, precited).
Therefore the inventors seeing the problems associated with retroviruses and in particular HIV, have generated a new class of reagents which can be used to specifically target and manipulate the retroviral provirus. This new class of anti -retroviral molecules can recognize and cleave the integrated provirus either in vitro or in vivo, these reagents can be used for a variety of purposes for instance in research as well as in novel treatment regimes.
According to a first aspect of the present invention there is provided an l-Crel variant which cleaves a target in the provirus of a pathogenic virus, for use in treating an infection of said virus.
The inventors therefore provide a set of l-Crel variants which can recognise and cut targets in a genomically integrated provirus (GIP). Such I-Crel variants provide a new therapeutic route to retrovirus and in particular HIV treatment by HIV provirus inactivation or alteration. This new class of enzymes is also potentially useful in studies into the transcriptional and regulatory behaviour of the provirus. This new class of anti-HIV medicament can act in a number of ways including by non-homologous end joining, the replacement/removal by homologous recombination with an introduced DNA targeting construct of a portion of the provirus or the removal of the provirus following recombination between chromosome arms. Each of these different mechanisms is discussed in detail below. In the present Patent Application the genomically integrated provirus (GIP) refers to the DNA sequence present in one or several places in the host cell genome which was inserted following reverse transcription of the RNA virus genome and its integration into the host genome.
In the present Patent Application the terms meganuclease (s) and variant (s) and variant meganuclease (s) will be used interchangeably herein.
The inventors have therefore created a new class of meganuclease based reagents which are useful for the treatment of a retrovirus infection and the most important and potentially useful feature of these enzymes is that instead of acting upon the virion or any component thereof they act upon the genomic insertion of the virus.
Targeting the integrated provirus would allow a clinician to eliminate the structure which leads to the generation of further viral particles, acting at a level that no other anti-viral therapeutic approaches have yet been developed. Conversely, prior art therapies which act upon the different steps of the viral life cycle allow to a clinician to inhibit viral replication, but do not eliminate the source of the virions, which therefore allows for the amplification of the viral infection when the treatment is withdrawn or resistance develops.
These variants also allow the targeting of the DNA version of the virus genome before it has integrated into the host cell genome. By inactivating the virus genome before it can integrate into the host cell genome, the claimed variants can act during the early step of cell infection in a way which no current antiretroviral medicament can.
The Inventors have validated this new class of anti-retrovirus reagents by generating meganuclease variants to a series of DNA targets in the genome of the HIV provirus (Figures 7, 24, 35 and 48). Seven targets in the HIV pro virus were chosen [one in U3 LTR (target HIVlJ), one in U5 LTR (target HIVlJ), two in the ρ24 gene (target HIV 1_4) and (target HIV1_7), two in the protease gene (target HIV 3 _5) and (target HIV 1_9) and one in the p7 gene (target HIV 1_8)] and the inventors set out to determine whether it was possible to generate meganucleases capable of cleaving these.
These target sequences are present in the U3 and U5 LTR regions, the coding sequence of the structural gene gag and more specifically in the p7 and p24 proteins therein and in the structural gene pol, specifically in the protease gene. These seven targets were selected based on their therapeutic potential.
As mentioned before, one potential therapeutic approach would be to cleave both LTRs of the integrated provirus which would in turn lead to excision of the viral genome from the infected cells. The inventors have shown that it is possible to generate l-Crel variants which can cleave targets in the U3 (target HIVlJ) and U5
(target HIVlJ) LTRs in the present Patent Application.
An alternative therapeutic approach would be to targeting one or more essential genes, the p24 protein is a structural component of the viral capsid and is essential for the virus to replicate. The inventors have shown that it is possible to generate l-Crel variants which can cleave targets in the p24 gene (target HIV 1_4) and
(target HIV IJ) in the present Patent Application. These two targets do not overlap and hence these two enzymes could be used simultaneously so further reducing the chances of resistance developing and/or causing an excision of the portion of p24 situated between the two cleavage sites.
The HIV protease is also an essential protein that is needed for viral particle maturation, without which viral particles remain in an immature state and are
not infectious. The inventors have shown that it is possible to generate l-Crel variants which can cleave targets in the protease gene (target HIV 1_5) and (target HIV 1_9) in the present Patent Application. These two targets do not overlap and hence these two enzymes could be used simultaneously so further reducing the chances of resistance developing and/or causing an excision of the portion of protease situated between the two cleavage sites.
The HIV nucleocapsid protein (p7, ou NC) is bound to the single- stranded RNA geneome. This protein plays a key role in the HIV life cycle since, being an RNA chaperone, its activity is required for efficient reverse transcription, making it an interesting target for antiviral therapy. The inventors have shown that it is possible to generate Ϊ-Crel variants which can cleave targets in the p7 gene (target HIV1_8).
The inventors have therefore established that meganuclease variants can be generated in both the sequences of essential genes as well as in regulatory non- coding sequences essential for viral replication.
These targets were also selected based on a screen on the "Los
Alamos National Laboratory" Sequence database (www.hiv.lanl.gov) to determine their degree of conservation among circulating isolates, which showed a high degree of sequence conservation among the different viral strains for which the complete sequence of their genome is available.
In the present Patent Application essential genes of the GIP provirus are those genes which must remain active in order for the GIP provirus to be converted into further virions which are able to exit the host cell and infect further cells. In addition to essential genes, other types of essential genetic elements can exist such as the regulatory elements of essential genes and/or structural sequence elements of the HIV provirus that are necessary for its packaging and/or insertion into the genome.
According to a further aspect of the present invention the pathogenic virus is from a genus selected from the group consisiting of: Alpharetrovirus, Beiaretrovirus, Gammaretrovirus, Dellaretroviru$> Epsilonretrovirus, Lentivirus and Spumavirus.
Multiple examples of genomic sequences for viruses of the specified types are available from public databases such as the National Center for
Biotechnology Information (http://www.ncbi.nlm.nih.gov/) or the virus genomics and bioinformatics resources centre at University College London (http://www.biochem.ucl.ac.uk/bsm/virus_database/VIDA.html).
In particular the virus is selected from the group consisting of: Human T-lymphotrophic virus, Rous Sarcoma and Human Immunodeficiency Virus.
Most particularly the virus is either Human Immunodeficiency Virus Type 1 (HIVl) or Human Immunodeficiency Virus Type 2 (HIV2). In particular the DNA target is within a DNA sequence essential for
I HV replication, viability, packaging or virulence.
In particular the DNA target is within an essential gene or regulatory element or structural clement of the HIV provirus.
In particular the DNA target is within the open reading frame of the HIV provirus encoding a gene or regulatory element of a gene selected from the group: GAG, POL, ENV, TAT and REV.
In particular the target in the HIVl provirus is selected from the group consisting of the sequences SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368. In particular the variant is selected from one of the sequences SEQ
ID NO: 1-13; SEQ ID NO: 26-46; SEQ ID NO: 59-85; SEQ ID NO: 88-94; SEQ ID NO: 97-165; SEQ ID NO: 168-174; SEQ ID NO: 177-186; SEQ ID NO: 189-238; SEQ ID NO: 241 -242; SEQ ID NO: 245-253; SEQ ID NO: 256-316; SEQ ID NO: 346-365. In particular the variant is characterized in that at least one of the two l-Crel monomers has at least two substitutions, one in each of the two functional subdomains of the LAGLIDADG core domain situated from positions; in particular said substitution(s) in the first functional subdomain comprise a substitution in at least one of positions 26, 28, 30, 32, 33, 38 and/or 40 and said substitution(s) in the second functional subdomain comprise a substitution in at least one of positions positions 44, 68, 70, 75 and/or 77 and being obtainable by a method comprising at least the steps of:
(a) constructing a first series of l-Crel variants having at least one substitution in a first functional subdomain of the LAGLΪDADG core domain comprising at least one substitution at a position selected from the group: 26, 28, 30, 32, 33, 38 and/or 40 of l-Crel, (b) constructing a second series of l-Crel variants having at least one substitution in a second functional subdomain of the LAGLIDADG core domain comprising at least one substitution at a position selected from the group: 44, 68, 70, 75 and/or 77of l-Crel,
(c) selecting and/or screening the variants from the first series of step (a) which are able to cleave a DNA target sequence selected from the group SEQ
ID NO: 319 to 342 and SEQ ID NO: 366 to 368, wherein at least one of (i) the nucleotide triplet in positions -10 to -8 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions -10 to -8 of the selected DNA target sequence from said provirus and (U) the nucleotide triplet in positions +8 to +10 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in position -10 to -8 of said DNA target sequence from said provirus,
(d) selecting and/or screening the variants from the second series of step (b) which are able to cleave a mutant 1-OeI site wherein at least one of (i) the nucleotide triplet in positions -5 to -3 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions -5 to -3 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions +3 to +5 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in position -5 to -3 of said DNA target sequence from said provirus,
(e) selecting and/or screening the variants from the first series of step (a) which are able to cleave a mutant l-Crel site wherein at least one of (i) the nucleotide triplet in positions +8 to +10 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus,
(f) selecting and/or screening the variants from the second series of step (b) which are able to cleave a mutant l-Crel site wherein at least one of (i) the
nucleotide triplet in positions +3 to +5 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -5 to -3 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus,
(g) combining in a single variant, the mutation(s) in positions 26, 28, 30, 32, 33, 38 and/or 40 and 44, 68, 70, 75 and/or 77 of two variants from step (c) and step (d), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions -10 to -8 is identical to the nucleotide triplet which is present in positions -10 to -8 of said DNA target sequence from said provirus , (ii) the nucleotide triplet in positions +8 to +10 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions -10 to - 8 of said DNA target sequence from said provirus, (iii) the nucleotide triplet in positions -5 to -3 is identical to the nucleotide triplet which is present in positions -5 to -3 of said DNA target sequence from said provirus and (iv) the nucleotide triplet in positions +3 to +5 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions -5 to -3 of said DNA target sequence from said provirus , and/or
(h) combining in a single variant, the mutation(s) in positions 26, 28, 30, 32, 33, 38 and/or 40, and 44, 68, 70, 75 and/or 77 of of two variants from step (e) and step (f), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions +8 to +10 of the 1-OeI site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of said DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 is identical to the reverse complementary sequence of the nucleotide triplet in positions +8 to +10 of said DNA target sequence from said provirus, (iii) the nucleotide triplet in positions +3 to +5 is identical to the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus, (iv) the nucleotide triplet in positions -5 to -3 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions +3 to +5 of said DNA target sequence from said provirus,
(i) combining the variants obtained in steps (g) and (h) to form heterodimers, and
(j) selecting and/or screening the heterodimers from step (i) which are able to cleave said DNA target sequence from said pro virus. A combinatorial approach, as illustrated schematically in Figure 6 was used to entirely redesign the DNA binding domain of the l-Crel protein and thereby engineer novel meganucl eases with fully engineered specificity.
In particular the heterodimer of step (i) may comprise monomers obtained in steps (g) and (h), with the same DNA target recognition and cleavage activity properties.
Alternatively the heterodimer of step (i) may comprise monomers obtained in steps (g) and (h), with different DNA target recognition and cleavage activity properties.
In particular the first series of l-Crel valiants of step (a) are derived from a first parent meganuclease.
In particular the second series of variants of step (b) are derived from a second parent meganuclease.
In particular the first and second parent meganucleases are identical. Alternatively the first and second parent meganucleases are different.
In particular the variant may be obtained by a method comprising the additional steps of:
(k) selecting heterodimers from step (j) and constructing a third series of variants having at least one substitution in at least one of the monomers of said selected heterodimers,
(1) combining said third series variants of step (k) and screening the resulting heterodimers for enhanced cleavage activity against said DNA target from the GIP.
The inventors have found that although specific meganucleases can be generated to a particular target in the GIP using the above method, that such meganucleases can be improved further by the additional rounds of substitution and
selection against the intended target. Meganuclease generated to targets in the GIP using other methods are also comprised within the present Patent Application.
In particular in said step (k) the substitutions in the third series of variants arc introduced by site ditected mutagenesis in a DNA molecule encoding said third series of variants, and/or by random mutageneis in a DNA molecule encoding said third series of variants.
In the additional rounds of substitution and selection, the substitution of residues in the meganucleases can be performed randomly, that is wherein the chances of a substitution event occurring are equal chance across all the residues of the meganuclease. Or on a site directed basis wherein the chances of certain residues being subject to a substitution is higher than other residues.
In particular steps (k) and (1) are repeated at least two times and wherein the heterodimers selected in step (k) of each further iteration are selected from heterodimers screened in step (1) of the previous iteration which showed increased cleavage activity against said DNA target from the GIP.
The inventors have found that the meganucleases can be further improved by using multiple iterations of the additional steps (k) and (1).
Through the inventors work they have identified the residues in the first subdomain which when altered have most effect upon altering the l-Crel enzymes specificity.
Through the inventors work they have identified the residues in the second subdomain which when altered have most effect upon altering the Ϊ-Crel enzymes specificity.
In particular the variant comprises one or more substitutions in posi- tions 137 to 143 of l-Crel that modify the specificity of the variant towards the nucleotide in positions ± 1 to 2, ± 6 to 7 and/or ± 1 1 to 12 of the target site in the GIP.
In particular the variant comprises one or more substitutions on the entire l-Crel sequence that improve the binding and/or the cleavage properties of the variant towards said DNA target sequence from the GIP. As well as specific mutations at the residue identified above, the present invention also encompasses the substitution of any of the residues present in the l-Crel enzyme.
In particular the variant is a heterodimer, resulting from the association of a first and a second monomer having different mutations in positions 26, 28, 30, 32, 33, 38 and/or 40, and 44, 68, 70, 75 and/or 77 of l-Crel, said heterodimer being able Io cleave a non-palindromic DNA target sequence from the HIV provirus. As explained above the l-Crel enzyme acts as a dimer, by ensuring that the variant is a heterodimer this allows a specific combination of two different I- OeI monomers which increases the possible targets cleaved by the variant,
In particular the heterodimeric variant is an obligate heterodimer variant having at least one pair of mutations in corresponding residues of the first and the second monomers which mediate an intermolecular interaction between the two I- Crel monomers, wherein the first mutation of said pair(s) is in the first monomer and the second mutation of said pair(s) is in the second monomer and said pair(s) of mutations impairs the formation of functional homodimers from each monomer without preventing the formation of a functional heterodimer, able to cleave the genomic DNA target from the HIV provirus.
The inventors have previously established a number of residue changes which can ensure an l-Crel monomer is an obligate heterodimer (WO2008/093249).
In particular the monomers have at least one of the following pairs of mutations, respectively for the first and the second monomer: a) the substitution of the glutamic acid in position 8 with a basic amino acid, preferably an arginine (first monomer) and the substitution of the lysine in position 7 with an acidic amino acid, preferably a glutamic acid (second monomer); the first monomer may further comprise the substitution of at least one of the lysine residues in positions 7 and 96, by an arginine, b) the substitution of the glutamic acid in position 61 with a basic amino acid, preferably an arginine (first monomer) and the substitution of the lysine in position 96 with an acidic amino acid, preferably a glutamic acid (second monomer); the first monomer may further comprise the substitution of at least one of the lysine residues in positions 7 and 96, by an arginine, c) the substitution of the leucine in position 97 with an aromatic amino acid, preferably a phenylalanine (first monomer) and the substitution of the
phenylalanine in position 54 with a small amino acid, preferably a glycine (second monomer); the first monomer may further comprise the substitution of the phenylalanine in position 54 by a tryptophane and the second monomer may further comprise the substitution of the leucine in position 58 or lysine in position 57, by a methionine, and d) the substitution of the aspartic acid in position 137 with a basic amino acid, preferably an arginine (first monomer) and the substitution of the arginine in position 51 with an acidic amino acid, preferably a glutamic acid (second monomer). In particular the variant, which is an obligate heterodimer, wherein the first and the second monomer, respectively, further comprises the D137R mutation and the R51D mutation.
In particular the variant, which is an obligate heterodimer, wherein the first monomer further comprises the K7R, E8R, E61R, K96R and L97F or K7R, E8R, F54W, E61R, K96R and L97F mutations and the second monomer further comprises the K7E, F54G, L58M and K96E or K7E, F54G, K57M and K96E mutations.
According to a further aspect of the present invention there is provided a single-chain chimeric meganuclease which comprises two monomers or core domains of one or two variant(s) according to the first aspect of the present invention, or a combination of both.
An alternative approach to ensuring that the variant consists of a specific combination o[ monomers is to link the selected monomers for instance using a peptide linker. In particular the single-chain meganuclease comprises a first and a second monomer according to the first aspect of the present invention, connected by a peptidic linker.
According to a further aspect of the present invention the l-Crel variant is combined with other antiretro viral drugs. Most antiretroviral drugs have at least three names. Sometimes a drug is referred to by its research or chemical name, such as AZT, The second name is the generic name for all drugs with the same chemical structure; for example AZT is
also known as zidovudine. The third name is the brand name given by the pharmaceutical company; one of the brand names for zidovudine is Retrovir. Lastly, an abbreviation of the common name might sometimes also be used, such as ZDV, which is the fourth name given to zidovudine.
Lists of drugs approved for use in the USA are provided below:


Due to the constant evolution of resistance to existing HIV medicaments additional antiretroviral drugs continue to be developed and approved for the treatlent of HIV infections.
In accordance with this further aspect of the present invention the I- OeI variant is combined with other antiretroviral agents such as those listed above or with other meganucleases directed against different targets in the HIV provirus.
According to a preferred embodiment of the present invention l-Crel variants according to the present invention are used only once the viral load of an individual has been reduced significantly using antiretroviral drugs. The 1-OeI
variants are then used to elimate as many proviruses as possible whilst the HIV virus population is in its enforced dormant state.
Using this strategy it is conceivable that an existant HIV infection could be cured. Perhaps more likely the reduction in the number of active proviruses will lead to a decrease in the number of new virus particles being produced which in turn will reduce the chances of resistant virus particles being generated against any of the medicaments being used to suppress HIV replication. Allowing the use for longer periods of time of the medicaments, so reducing the chances that an individual will ever be infected with HIV particles which are resistant to all anti-HIV medicaments. In accordance with a further aspect of the present invention there is also provided a kit of parts comprising at least one 1-OeI according to the present invention either in the form of a peptide or a nucleotide encoding the variant(s) and one or more other anti-HIV medicaments, together with instructions for the administration of the variant and other anti-HIV medicaments to a patient. According to the present invention, the meganuclease when used as a polypeptide is associated with:
- liposomes, polyethyleneimine (PEI); in such a case said association is administered and therefore introduced into somatic target cells.
- membrane translocating peptides (Bonetta, The Scientist, 2002, 16, 38; Ford et al., Gene Ther., 2001, 8, 1-4 ; Wadia and Dowdy, Curr. Opin. Biotechnol.,
2002, 13, 52-56); in such a case, the sequence of the variant/single-chain meganuclease is fused with the sequence of a membrane translocating peptide (fusion protein).
Alternatively, the meganuclease in the form of a polynucleotide encoding said meganuclease in a vector. Vectors comprising targeting DNA and/or nucleic acid encoding a meganuclease can be introduced into a cell by a variety of methods (e.g., injection, direct uptake, projectile bombardment, liposomes, electroporation). Meganucleases can be stably or transiently expressed into cells using expression vectors. Techniques of expression in eukaryotic cells are well known to those in the art. (See Current Protocols in Human Genetics: Chapter 12 "Vectors For Gene Therapy" & Chapter 13 "Delivery Systems for Gene Therapy"). Optionally, it
may be preferable to incorporate a nuclear localization signal into the recombinant protein to be sure that it is expressed within the nucleus.
The meganuclease may also comprise a nuclear localization signal (NLS) which is an amino acid sequence which acts like a 'tag' on the exposed surface of a protein. The NLS is used to target the protein to the cell nucleus through the Nuclear Pore Complex and to direct a newly synthesized protein into the nucleus via its recognition by cytosolic nuclear transport receptors. Typically, this signal consists of one or more short sequences of positively charged lysines or arginines.
According to a second aspect of the present invention there is provided a polynucleotide fragment encoding the variant according to the first aspect of the present invention or the single-chain chimeric meganuclease according to a second aspect of the present invention.
According to a third aspect of the present invention there is provided an expression vector comprising at least one polynucleotide fragment according to the second aspect of the present invention.
In particular the expression vector, includes a targeting construct comprising a sequence to be introduced flanked by sequences sharing homologies with the regions surrounding said DNA target sequence from the provirus.
One important use of a variant according to the present invention is in increasing the incidence of homologous recombination events at or around the site where the variant cleaves its target. The present invention therefore also relates to a unified genetic construct which encodes the variant under the control of suitable regulatory sequences as well as sequences homologous to portions of the provirus surrounding the variant DNA target site. Following cleavage of the target site by the variant these homologous portions can act as complementary sequences in a homologous recombination reaction with the provirus replacing the existing provirus sequence with a new sequence engineered between the two homologous portions in the unified genetic construct.
Preferably, homologous sequences of at least 50 bp, preferably more than 100 bp and more preferably more than 200 bp are used. Shared DNA homologies are located in regions flanking upstream and downstream the site of the break and the DNA sequence to be introduced should be located between the two arms.
Therefore, the targeting construct is preferably from 200 bp to 6000 bp, more preferably from 1000 bp to 2000 bp; it comprises: a sequence which has at least 200 bp of homologous sequence flanking the target site, for repairing the cleavage and a sequence for inactivating the provirus and/or a sequence of an exogenous gene of interest which it is intended to insert at the site of the DNΛ repair event by homologous recombination.
For the insertion of a sequence, DNA homologies are generally located in regions directly upstream and downstream to the site of the break (sequences immediately adjacent to the break; minimal repair matrix). However, when the insertion is associated with a deletion of ORF sequences flanking the cleavage site, shared DNA homologies are located in regions upstream and downstream the region of the deletion.
A vector which can be used in the present invention includes, but is not limited to, a viral vector, a plasmid, a RNA vector or a linear or circular DNA or RNA molecule which may consists of a chromosomal, non chromosomal, semisynthetic or synthetic nucleic acids. Preferred vectors are those capable of autonomous replication (episomal vector) and/or expression of nucleic acids to which they are linked (expression vectors). Large numbers of suitable vectors are known to those of skill in the art and commercially available. Viral vectors include retrovirus, adenovirus, parvovirus (e.g. adeno associated viruses), coronavirus, negative strand RNA viruses such as orthomyxovirus (e. g., influenza virus), rhabdovirus (e. g., rabies and vesicular stomatitis virus), paramyxovirus (e. g. measles and Sendai), positive strand RNA viruses such as picor- navirus and alphavirus, and double-stranded DNA viruses including adenovirus, herpesvirus (e. g., Herpes Simplex virus types 1 and 2, Epstein-Barr virus, cytomegalovirus), and poxvirus (e. g,, vaccinia, fowlpox and canarypox). Other viruses include Norwalk virus, togavirus, flavivirus, reoviruses, papovavirus, hepadnavirus, and hepatitis virus, for example. Examples of retroviruses include: avian leukosis-sarcoma, mammalian C-type, B-type viruses, D type viruses, HTLV- BLV group, lentivirus, spumavirus (Coffin, J. M., Retroviridae: The viruses and their replication, In Fundamental Virology, Third Edition, B. N. Fields, et al., Eds., Lippincott-Raven Publishers, Philadelphia, 1996).
Vectors can comprise selectable markers, for example: neomycin phosphotransferase, histidinoi dehydrogenase, dihydro folate reductase, hygromycin phosphotransferase, herpes simplex virus thymidine kinase, adenosine deaminase, glutamine synthetase, and hypoxanthine-guanine phosphoribosyl transferase (HRPT) for eukaryotic cell culture; TRPl for S. cerevisiae; tetracycline, rifampicin or ampicillin resistance in E. coli.
In particular for the purposes of gene therapy and in accordance with a preferred embodiment of the present invention, the viral vector is selected from the group comprising lentiviruses, Adeno-associated viruses (AAV) and Adenoviruses. In accordance with another aspect of the present invention the variant and targeting construct may be on different nucleic acid constructs.
In accordance with another aspect of the present invention the variant in the form of a peptide and the targeting construct as a nucleic acid molecule may be used in combination. In particular, wherein said sequence to be introduced is a sequence which inactivates the HIV provirus.
In particular, wherein the sequence which inactivates the HIV provirus comprises in the 5' to 3' orientation: a first transcription termination sequence and a marker cassette including a promoter, the marker open reading frame and a second transcription termination sequence, and said sequence interrupts the transcription of the coding sequence.
In particular, wherein said sequence sharing homologies with the regions surrounding DNA target sequence is from the HIV provirus or a fragment of the HIV provirus comprising sequences upstream and downstream of the cleavage site, so as to allow the deletion of coding sequences flanking the cleavage site.
According to a fourth aspect of the present invention there is provided a host cell which is modified by a polynucleotide according to a second aspect of the present invention or a vector according to a third aspect of the present invention. A cell according to the present invention may be made according to a method, comprising at least the step of:
(a) introducing into a cell, a meganuclcase, as defined above, so as to induce a double stranded cleavage at a site of interest of the GIP comprising a DNA recognition and cleavage site of said meganuclease, and thereby generate a genomically modified cell having repaired the double-strands break, by non- homologous end joining, and
(b) isolating the genomically modified cell of step (a), by any appropriate mean.
The cell which is modified may be any cell of interest. For making transgenic/knock-out animals, the cells are pluripotent precursor cells such as embryo- derived stem (ES) cells, which are well-known in the art. For making recombinant cell lines, the cells may advantageously be human cells, for example PerC6 (Fallaux et al., Hum. Gene Ther. 9, 1909-1917, 1998) or HEK293 (ATCC # CRL- 1573) cells or an immortal T lymphocyte line such as Jurkat (Schneider et al (1977). Int J Cancer 19 (5): 621-6.). The meganuclease can be provided directly to the cell or through an expression vector comprising the polynucleotide sequence encoding said meganuclease linked to regulatory sequences suitable for directing its expression in the cell used.
Such a modified cell line would have a number of potential uses including the elucidation of aspects of the biology of the modified GIP as well as a model for screening compounds and other substances for therapeutic effects against ceils comprising the modified GIP.
According to a fifth aspect of the present invention there is provided a non-human transgenic animal which is modified by a polynucleotide according to a second aspect of the present invention or a vector according to a third aspect of the present invention.
The subject-matter of the present invention is also a method for making an animal which comprises a modified GIP, comprising at least the step of:
(a) introducing into a pluripotent precursor cell or an embryo of an animal, a meganuclease, as defined above, so as to induce a double stranded cleavage at a site of interest of the GIP comprising a DNA recognition and cleavage site of said meganuclease, and thereby generate a genomically modified precursor cell or embryo having repaired the double-strands break by non-homologous end joining,
(b) developing the genomically modified animal precursor cell or embryo of step (a) into a chimeric animal, and
(c) deriving a transgenic animal from a chimeric animal of step (b). Alternatively, the GIP may be inactivated by insertion of a sequence of interest by homologous recombination between the genome of the animal and a targeting DNA construct according to the present invention.
In particular the targeting DNA is introduced into the cell under conditions appropriate for introduction of the targeting DNA into the site of interest.
In particular, step (b) comprises the introduction of the genoraically modified precursor cell obtained in step (a), into blastocysts, so as to generate chimeric animals.
Such a transgenic animal could be used as a multicellular animal model to elucidate aspects of the biology of the GIP, by means of engineering the provirus present in the progenitor cell line. Such transgenic animals also could be used to screen and characterise the effects of for instance novel anti-HIV medicaments.
In particular the targeting DNA construct is inserted in a vector. For making transgenic animals/recombinant cell lines, including human cell lines expressing an heterologous protein of interest, the targeting DNA comprises the sequence of the exogenous gene encoding the protein of interest, and eventually a marker gene, flanked by sequences upstream and downstream of and essential gene in the HIV provirus, as defined above, so as to generate genomically modified cells (animal precursor cell or embryo/animal or human cell) having replaced the HIV gene by the exogenous gene of interest, by homologous recombination. The exogenous gene and the marker gene are inserted in an appropriate expression cassette, as defined above, in order to allow expression of the heterologous protein/marker in the transgenic animal/recombinant cell line.
The meganuclease can be used either as a polypeptide or as a polynucleotide construct encoding said polypeptide. It is introduced into somatic cells of an individual, by any convenient means well-known to those in the art, which are appropriate for the particular cell type, alone or in association with either at least an appropriate vehicle or carrier and/or with the targeting DNA.
Once in a cell, the meganucleasc and if present, the vector comprising targeting DNA and/or nucleic acid encoding a meganuclcase are imported or translocated by the cell from the cytoplasm to the site of action in the nucleus.
According to a sixth aspect of the present invention there is provided a transgenic plant which is modified by a polynucleotide according to a second aspect of the present invention or a vector according to a third aspect of the present invention.
According to a further aspect of the present invention there is provided the use of at least one variant or at least one single-chain chimeric meganuclease as defined above, or at least one vector according to the third aspect of the present invention, for genome engineering for non-therapeutic purposes.
In particular the variant or single-chain chimeric meganuclease or vector is associated with a targeting DNA construct.
In particular the use of the variant is for inducing a double-strand break in a site of interest within the GIP, thereby inducing a DNA recombination event, a DNA loss or cell death.
According to the invention, said double-strand break is for: modifying a specific sequence in the GIP, so as to induce restoration of a GIP function such as replication in studies upon the biology of the virus, or to attenuate or activate the GIP or a gene therein, introducing a mutation into a site of interest of a GIP gene, introducing an exogenous gene or a part thereof, inactivating or deleting the GIP or a part thereof or leaving the DNA unrepaired and degraded.
In particular this present aspect of the present invention relates to the use of a meganuclease variant to treat HIV infection, by inactivating the HIV provirus by therapeutic genome engineering.
According to one aspect of the present invention the use of the meganuclease according to the present invention, comprises at least the following steps:
1) introducing a double-strand break at at least one site of interest in the HIV provirus comprising at least one recognition and cleavage site of said meganuclease, by contacting said cleavage site with said meganuclease;
2) providing a targeting DNΛ construct comprising the sequence to be introduced flanked by sequences sharing homologies to the targeted locus.
Wherein the meganuclease is provided directly to the cell or through an expression vector comprising the polynucleotide sequence encoding of the meganuclease and is suitable for its expression in the host cell.
This strategy is used to introduce a DNΛ sequence at the target site, for example to generate a HIV provirus knock-in or knock-out animal model or cell lines that can be used for drug testing or in the case of a cell line, which can be used for administration into a patient from whom it was derived. According to a further aspect of the present invention the use of the meganuclease, comprises at least the following steps:
1) introducing a double-strand break at a site of interest of the IHV provirus comprising at least one recognition and cleavage site of said meganuclease, by contacting said cleavage site with said meganuclease; 2) maintaining said broken genomic locus under conditions appropriate for homologous recombination with chromosomal DNA sharing homologies to regions surrounding the cleavage site.
As well as inactivating the provirus using a targeting construct, a significant number of inter chromosome arm recombination events are also expected to occur following cleavage of the provirus target. The recombination of chromosome arms occurs most frequently during mitosis, but can also occur as part of the repair mechanism for DNA strand breaks. Such an inter chromosome arm recombination event would either lead to the elimination of the non homologous portions on either side of the break (e.g. the provirus) or more likely cause portions of the provirus to be recombined onto different chromosome arms. In either event this would lead to the inactivation of the provirus.
According to still further aspect of the present invention the use of the meganuclease, comprises at least the following steps:
1) introducing a double-strand break at a site of interest of the HIV provirus comprising at least one recognition and cleavage site of said meganuclease, by contacting said cleavage site with said meganuclease ;
2) maintaining said broken genomic locus under conditions appropriate for repair of the double-strands break by non-homologous end joining.
According to a further aspect of the present invention the variant is used for genome therapy to knock-out in animals/cells the GIP, in particular a sequence is introduced which inactivates the HIV provirus.
All HIV proviruses present in the cell have to be targeted in order to totally inactivate the pathogenicity of the virus. In addition, the introduced sequence may also delete the HIV provirus or part thereof, and introduce an exogenous gene or part thereof (knock-in/gene replacement). For making knock-in animals/cells the DNA which repairs the site of interest may comprise the sequence of an exogenous gene of interest, and a selection marker, such as the G418 resistance gene. Alternatively, the sequence to be introduced can be any other sequence used to alter the chromosomal DNA in some specific way including a sequence used to modify a specific sequence, to attenuate or activate the endogenous gene of interest in the HΪV provirus or to introduce a mutation into a site of interest in the HIV provirus. Such chromosomal DNA alterations may be used for genome engineering (animal models and recombinant cell lines including human cell lines).
Inactivation of the HIV provirus may occur by insertion of a transcription termination signal that will interrupt the transcription of an essential gene such as GAG, POL and ENV and result in a truncated protein. In this case, the sequence to be introduced comprises, in the 5' to 3' orientation: at least a transcription termination sequence (polyAl), preferably said sequence fuithei comprises a marker cassette including a promoter and the marker open reading frame (ORF) and a second transcription termination sequence for the marker gene ORF (polyA2). This strategy can be used with any variant cleaving a target downstream of the relevant gene promoter and upstream of the stop codon.
Inactivation of the HIV provirus may also occur by insertion of a marker gene within an essential gene of HIV, which would disrupt the coding sequence. The insertion can in addition be associated with deletions of ORF sequences flanking the cleavage site and eventually, the insertion of an exogenous gene of interest (gene replacement).
In addition, inactivation of the HIV provirus may also occur by insertion of a sequence that would destabilize the mRNA transcript of an essential gene.
The present invention also provides a composition characterized in that it comprises at least one variant as defined above (variant or single-chain derived chimeric meganucleasc) and/or at least one expression vector encoding the variant, as defined above.
The administration of the provirus targeting variant in as both a peptide and nucleotide form allows for the immeadiate action of the variant as as its persistence in the target cell.
In particular the composition comprises more than one variant, wherein each of the variants is directed towards a different target sequence in the provirus.
In particular the composition comprises a targeting DNA construct comprising a sequence which inactivates the HlV provirus, flanked by sequences sharing homologies with the genomic DNA cleavage site of said variant, as defined above.
Preferably, said targeting DNA construct is either included in a recombinant vector or it is included in an expression vector comprising the polynucleotide(s) encoding the variant according to the invention.
The subject-matter of the present invention is also the use of at least one meganuclease and/or one expression vector, as defined above, for the preparation of a medicament for preventing, improving or curing HIV infection in an individual in need thereof. The subject-matter of the present invention is also the use of at least one variant and/or one expression vector, as defined above, for the preparation of a medicament for preventing, improving or curing a pathological condition associated with HIV infection in an individual in need thereof.
As discussed above the variants according to the present invention provide a possible means to prevent chromosomal integration of a target cell with the retrovirus genome. The first step of the viral infection following viral entry into the target cell is the reverse transcription (RT) of the viral genomic RNA. During this RT
process, a linear double stranded DNA molecule is formed which then enters the nucleus so that it can be integrated in the cellular genome. Meganuclease variants of the present invention are also able to cleave the pre-integration complex (PlC), which is an episomal double stranded DNA molecule, conferring a protective effect during the earliest steps of viral infection, of a cell population.
The use of the meganuclease may comprise at least the step of (a) inducing in somatic tissue(s) of the donor/ individual a double stranded cleavage at a site of interest of the HIV provirus comprising at least one recognition and cleavage site of said meganuclease by contacting said cleavage site with said meganuclease, and (b) introducing into said somatic tissue(s) a targeting DNA, wherein said targeting DNA comprises (1) DNA sharing homologies to the region surrounding the cleavage site and (2) DNA which inactivates the HIV provirus upon recombination between the targeting DNA and the chromosomal DNA, as defined above. The targeting DNA is introduced into the somatic tissues(s) under conditions appropriate for introduction of the targeting DNA into the site of interest. The targeting construct may comprise sequences for deleting the HIV provirus or a portion thereof and introducing the sequence of an exogenous gene of interest (gene replacement).
In this case the use of the meganuclease comprises at least the step of: inducing in somatic tissue(s) of the donor/individual a double stranded cleavage at a site of interest of the HIV provirus comprising at least one recognition and cleavage site of the meganuclease by contacting the cleavage site with the meganuclease, and thereby inducing mutagenesis of an open reading frame in the HIV provirus by repair of the double-strands break by non-homologous end joining.
According to the present invention, said double-stranded cleavage may be induced, ex vivo by introduction of said meganuclease into infected cells isolated for instance from the circulatory system of the donor/individual and then transplantation of the modified cells back into the diseased individual.
The subject-matter of the present invention is also a method for preventing, improving or curing HIV infection, in an individual in need thereof, said method comprising at least the step of administering to said individual a composition as defined above, by any means.
For purposes of therapy, the meganucleases and a pharmaceutically acceptable excipient are administered in a therapeutically effective amount. Such a combination is said to be administered in a "therapeutically effective amount" if the amount administered is physiologically significant. An agent is physiologically significant if its presence results in a detectable change in the physiology of the recipient. In the present context, an agent is physiologically significant if its presence results in a decrease in the severity of one or more symptoms of the targeted HIV infection.
In particular as far as possible the meganuclease comprising compo- sitions should be non-immunogenic, i.e., engender little or no adverse immunological response. A variety of methods for ameliorating or eliminating deleterious immunological reactions of this sort can be used in accordance with the invention. One means of achieving this is to ensure that the meganuclease is substantially free of N-formyl methionine. Another way to avoid unwanted immunological reactions is to conjugate meganucleases to polyethylene glycol ("PEG") or polypropylene glycol ("PPG") (preferably of 500 to 20,000 daltons average molecular weight (MW)). Conjugation with PEG or PPG5 as described by Davis et al. (US 4,179,337) for example, can provide non-immunogenic, physiologically active, water soluble endonuclease conjugates with anti-viral activity. Similar methods also using a poly- ethylene—polypropylene glycol copolymer are described in Saifer et al. (US 5,006,333).
In accordance with a further aspect of the present invention, the invention also relates to meganuclease variants, related materials and uses thereof which recognize non-virus retroelements and/or the integrated genomes of viruses which do not have mechanisms to integrate into the host cell genome.
Non-virus retroelements are endogenous genomic DNA elements that include the gene for reverse transcriptase and are also known as class I transposable elements. These retrotransposons, include the long terminal repeat (LTR) retrotransposons, non-LTR retroposons and group II mitochondrial introns. They are though to be derived from partially inactivated retroviruses which have lost the ability to form infective virus particles. These genetic elements however are increasingly becoming associated with various diseases, in particular cancers and immune
disorders which result form the integration of the element into a site close to a gene (s) whose misregulation leads to the observed disease phenotype.
The present invention therefore also relates to meganuclease variants which can be used to cleave a genomic retrotransposon either in a specific tissue or cell type or more generally so as to treat the disease phenotype using one or more of the mechanisms described above.
The present invention also relates to meganuclease variants which can recognise and cleave targets in genomic insertions of viruses which do not normally insert into the host cell genome. The non-specific insertion of viral genetic material into the host cell genome is a disease causing mechanism which is currently being investigated. For example in the important virus hepatitis B, chronic infection with this virus is associated with a greatly elevated risk of hepatocellular carcinoma.
In the past this association has been explained as a side effect of the episomal hepatitis
B genome upon the hepatocyte host cells. Although this is doubtless true, recently the random genomic insertion of copies of the hepatitis B genome into the host cell genome has also been shown to be a causative factor in hepatocyte carcinoma
(Goodarzi et al., 2008, Hep. Mon; 8 (2): 129-133).
Hepatocellular carcinoma is one of the most common cancers in the world and hence a treatment for this condition, using a meganuclease variant which can cleave the randomly integrated hepatitis B genome and have a therapeutic affect upon hepatocytes via one or more of mechanisms detailed above is therefore also within the scope of the present invention as are more generally meganuclease variants to genomically integrated copies of virus genetic material which cause a disease phenotype. Definitions
Throughout the present Patent Application a number of terms and features are used to present and describe the present invention, to clarify the meaning of these terms a number of definitions are set out below and wherein a feature or term is not otherwise specifically defined or obvious from its context the following definitions apply.
- Amino acid residues in a polypeptide sequence are designated herein according to the one-letter code, in which, for example, Q means GIn or Glutamine residue, R means Arg or Arginine residue and D means Asp or Aspartic acid residue. - Amino acid substitution means the replacement of one amino acid residue with another, for instance the replacement of an Arginine residue with a Glutamine residue in a peptide sequence is an amino acid substitution.
- Altered/enhanced/increased cleavage activity, refers to an increase in the detected level of meganuclease cleavage activity (see below) against a target DNA sequence by a first meganuclease in comparison to the activity of a second meganuclease against the target DNA sequence. Normally the first meganuclease will be a variant of the second and comprise one or moie substituted amino acid residues in comparison to the second meganuclease.
- by "beta-hairpin" it is intended two consecutive beta-strands of the antiparallel beta-sheet of a LAGLIDADG homing endonuclease core domain (βip2 or β3β4) which are connected by a loop or a turn,
- by "chimeric DNA target" or "hybrid DNA target" it is intended the fusion of a different half of two parent meganuclease target sequences. In addition at least one half of said target may comprise the combination of nucleotides which are bound by at least two separate subdomains (combined DNA target).
- Cleavage activity: the cleavage activity of the variant according to the invention may be measured by any well-known, in vitro or in vivo cleavage assay, such as those described in the International PCT Application WO 2004/067736; Epinat et at, Nucleic Acids Res., 2003, 31, 2952-2962; Chames el at, Nucleic Acids Res., 2005, 33, el78; Arnould et al., J. MoI. Biol, 2006, 355, 443-458, and Λrnould et al, J. MoI. Biol., 2007, 371, 49-65. For example, the cleavage activity of the variant of the invention may be measured by a direct repeat recombination assay, in yeast or mammalian cells, using a reporter vector. The reporter vector comprises two truncated, non-functional copies of a reporter gene (direct repeats) and the genomic (non-palindromic) DNA target sequence within the intervening sequence, cloned in a yeast or a mammalian expression vector. Usually, the genomic DNA target sequence comprises one different half of each (palindromic or pseudo-palindromic) parent
homodimcric meganuclease target sequence. Expression of the hcterodi merle variant results in a functional endonuclease which is able to cleave the genomic DNA target sequence. This cleavage induces homologous recombination between the direct repeats, resulting in a functional reporter gene (LacZ, for example), whose expression can be monitored by an appropriate assay. The specificity of the cleavage by the variant may be assessed by comparing the cleavage of the (non-palindromic) DNA target sequence with that of the two palindromic sequences cleaved by the parent homodimeric meganucleases or compared with wild type meganuclease.
- by "selection or selecting" it is intended to mean the isolation of one or more meganuclease variants based upon an observed specified phenotype, for instance altered cleavage activity. This selection can be of the variant in a peptide form upon which the observation is made or alternatively the selection can be of a nucleotide coding for selected meganuclease variant.
- by "screening" it is intended to mean the sequential or simulta- neous selection of one or more meganuclease variant (s) which exhibits a specified phenotype such as altered cleavage activity.
- by "derived from" it is intended to mean a meganuclease variant which is created from a parent meganuclease and hence the peptide sequence of the meganuclease variant is related to (primary sequence level) but derived from (muta- tions) the sequence peptide sequence of the parent meganuclease.
- by "domain" or "core domain" it is intended the "LAGLIDADG homing endonuclease core domain" which is the characteristic αiβiβ2α2β3β4α3 fold of the homing endonucleases of the LAGLIDADG family, corresponding to a sequence of about one hundred amino acid residues. Said domain comprises four beta-strands (βiβ2β3β4) folded in an antiparallel beta-sheet which interacts with one half of the DNA target. This domain is able to associate with another LAGLIDADG homing endonuclease core domain which interacts with the other half of the DNA target to form a functional endonuclease able to cleave said DNA target. For example, in the case of the dimeric homing endonuclease l-Crel (163 amino acids), the LAGLIDADG homing endonuclease core domain corresponds to the residues 6 to 94.
- by "DNA target", "DNA target sequence", "target sequence" , "target-site", "target" , "site"; "site of interest"; "recognition site", "recognition
sequence", "homing recognition site", "homing site", "cleavage site" it is intended a 20 to 24 bp double-stranded palindromic, partially palindromic (pseudo-palindromic) or non-palindromic polynucleotide sequence that is recognized and cleaved by a LAGLIDADG homing endonuclease such as l-Crel, or a variant, or a single-chain chimeric meganuclease derived from l-Crel. These terms refer to a distinct DNA location, preferably a genomic location, at which a double stranded break (cleavage) is to be induced by the meganuclease. The DNA target is defined by the 5' to 3' sequence of one strand of the double-stranded polynucleotide, as indicated for C1221 (see figure 1). Cleavage of the DNA target occurs at the nucleotides at positions +2 and -2, respectively for the sense and the antisense strand. Unless otherwise indicated, the position at which cleavage of the DNA target by an I-Cre I meganuclease variant occurs, corresponds to the cleavage site on the sense strand of the DNA target.
- by "DNA target half-site", "half cleavage site" or half-site" it is intended the portion of the DNA target which is bound by each LAGLIDADG homing endonuclease core domain.
- by "DNA target sequence from the HIV provirus" it is intended a 20 to 24 bp sequence of the HIV provirus which is recognized and cleaved by a meganuclease variant. In particular the DNA target sequence from then HIV provirus is in an essential gene sequence and/or within an essential regulatory sequence and/or within an essential structural sequence of the HIV provirus.
- by "first/second/third/nf!l series of variants" it is intended a collection of variant meganucleases, each of which comprises one or more amino acid substitution in comparison to a parent meganuclease from which all the variants in the series are derived. - by "functional variant" it is intended a variant which is able to cleave a DNA target sequence, preferably said target is a new target which is not cleaved by the parent meganuclease. For example, such variants have amino acid variation at positions contacting the DNA target sequence or interacting directly or indirectly with said DNA target. - by "heterodimer" it is intended to mean a meganuclease comprising two non-identical monomers. In particular the monomers may differ from each other
in their peptide sequence and/or in the DNA target half-site which they recognise and cleave.
- by "homologous" is intended a sequence with enough identity to another one to lead to a homologous recombination between sequences, more particularly having at least 95 % identity, preferably 97 % identity and more preferably 99 %.
- by "I-Grel" it is intended the wild-type l-Crel having the sequence of pdb accession code Ig9y, corresponding to the sequence SEQ ID NO: 344 in the sequence listing, - by "l-Crel variant with novel specificity" it is intended a variant having a pattern of cleaved targets different from that of the parent mcganuclease. The terms "novel specificity", "modified specificity", "novel cleavage specificity", "novel substrate specificity" which are equivalent and used indifferently, refer to the specificity of the variant towards the nucleotides of the DNA target sequence. In the present Patent Application all the l-Crel variants described comprise an additional Alanine after the first Methionine of the wild type 1-OeI sequence (SEQ ID NO: 344). These variants also comprise two additional Alanine residues and an Aspartic Acid residue after the final Proline of the wild type l-Crel sequence. These additional residues do not affect the properties of the enzyme and to avoid confusion these additional residues do not affect the numeiation of the residues in l-Crel or a variant referred in the present Patent Application, as these references exclusively refer to residues of the wild type l-Crel enzyme (SEQ ID NO: 344) as present in the variant, so for instance residue 2 of l-Crel is in fact residue 3 of a variant which comprises an additional Alanine after the first Methionine. - by "l-Crel site" it is intended a 22 to 24 bp double-stranded DNA sequence which is cleaved by l-Crel. l-Crel sites include the wild-type (natural) non- palindromic l-Crel homing site and the derived palindromic sequences such as the sequence 5'- Li2C-πa-ioa.9a^a-7C-6g-5t.4C-3g-2tia+ic+2gi3a+4C+sg+6tf7tH 8t+9t) iogi i ia» \2 (SEQ ID NO: 343), also called C 1221 . - "identity" refers to sequence identity between two nucleic acid molecules or polypeptides. Identity can be determined by comparing a position in each sequence which may be aligned for purposes of comparison. When a position in
the compared sequence is occupied by the same base, then the molecules are identical at that position. A degree of similarity or identity between nucleic acid or amino acid sequences is a function of the number of identical or matching nucleotides at positions shared by the nucleic acid sequences. Various alignment algorithms and/or programs may be used to calculate the identity between two sequences, including FASTA, or BLAST which are available as a part of the GCG sequence analysis package (University of Wisconsin, Madison, Wis.), and can be used with, e.g., default settings.
- by "meganuclease", it is intended an cndonuclease having a double-stranded DNA target sequence of 12 to 45 bp. The meganuclease is either a dimeric enzyme, wherein each domain is on a monomer or a monomelic enzyme comprising the two domains on a single polypeptide.
- by "meganuclease domain", it is intended the region which interacts with one half of the DNA target of a meganuclease and is able to associate with the other domain of the same meganuclease which interacts with the other half of the DNA target to form a functional meganuclease able to cleave said DNA target.
- by "meganuclease variant" or "variant" it is intended a meganuclease obtained by replacement of at least one residue in the amino acid sequence of the parent meganuclease (natuial oi variant meganuclease) with a different amino acid. - by "monomer" it is intended to mean a peptide encoded by the open reading frame of the 1-Crel gene or a variant thereof, which when allowed to dimcrise forms a functional l-Creϊ enzyme. In particular the monomers dimerise via interactions mediated by the LΛGLIDADG motif.
- by "mutation" is intended the substitution, deletion, insertion of one or more nucleotides/amino acids in a polynucleotide (cDNA, gene) or a polypeptide sequence. Said mutation can affect the coding sequence of a gene or its regulatory sequence. It may also affect the structure of the genomic sequence or the structure/stability of the encoded mRNA.
- Nucleotides are designated as follows: onc-lctter code is used for designating the base of a nucleoside: a is adenine, t is thymine, c is cytosine, and g is guanine. For the degenerated nucleotides, r represents g or a (purine nucleotides), k represents g or t, s represents g or c, w represents a or t, m represents a or c, y repre-
sents t or c (pyrimidine nucleotides), d represents g, a or t, v represents g, a or c, b represents g, t or c, h represents a, t or c, and n represents g, a, t or c.
- by "parent meganuclease" it is intended to mean a wild type meganuclease or a variant of such a wild type meganuclease with identical properties or alternatively a meganuclease with some altered characteristic in comparison to a wild type version of the same meganuclease. In the present invention the parent meganuclease can refer to the initial meganuclease from which the first series of variants are derived in step a. or the meganuclease from which the second series of variants are derived in step b., or the meganuclease from which the third series of variants are derived in step k.
- by "peptide linker" it is intended to mean a peptide sequence of at least 10 and preferably at least 17 amino acids which links the C terminal amino acid residue of the first monomer to the N terminal residue of the second monomer and which allows the two variant monomers to adopt the correct conformation for activity and which does not alter the specificity of either of the monomers for their targets.
- by "provirus" it is intended to mean a DNA version of a retrovirus genome. In particular the provirus may be the DNA molecule directly resulting from the reverse transcription of the RNA genome of a virus or alternatively it may be the chromosomally integrated version of the virus genome present at one or more sites in one or more chromosomes of the target cell.
- by "subdomain" it is intended the region of a LAGLIDADG homing endonuclease core domain which interacts with a distinct part of a homing endonuclease DNA target half-site.
- by "single-chain meganuclease", "single-chain chimeric meganu- clease", "single-chain meganuclease derivative", "single-chain chimeric meganuclease derivative" or "single-chain derivative" it is intended a meganuclease comprising two LAGLIDADG homing endonuclease domains or core domains linked by a peptidic spacer. The single-chain meganuclease is able to cleave a chimeric DNA target sequence comprising one different half of each parent meganuclease target sequence. - by "targeting DNA construct/minimal repair matrix/repair matrix" it is intended to mean a DNA construct comprising a first and second portions which are homologous to regions 5' and 35 of the DNA target in situ. The DNA construct
also comprises a third portion positioned between the first and second portion which comprise some homology with the corresponding DNA sequence in situ or alternatively comprise no homology with the regions 5' and 3 ' of the DNA target in situ. Following cleavage of the DNA target, a homologous recombination event is stimulated between the genome containing the HIV provirus and the repair matrix, wherein the genomic sequence containing the DNA target is replaced by the third portion of the repair matrix and a variable part of the first and second portions of the repair matrix.
- by "vector" is intended a nucleic acid molecule capable of transporting another nucleic acid to which it has been linked into a host cell in vitro, in vivo or ex vivo. For a better understanding of the invention and to show how the same may be carried into effect, there will now be shown by way of example only, specific embodiments, methods and processes according to the present invention with reference to the accompanying drawings in which: Figure 1: Schematic representation of an HIV-I viral particle. The two molecules of genomic RNA are represented, together with the RT, inside the viral capsid. The envelope, derived from the membrane of the infected cells, contains the envelope glycoproteins gp41and gρl20.
Figure 2: A: Organization of the HIV-I genomic RNA molecules, Different genes are represented with different shades of grey, and the proteins encoded by these genes are represented in the lower part of the panel. B; Genetic organization of the integrated HIV-I provirus, showing the structure of the LTRs after duplication of the U3 and U5 sequences during reverse transcription.
Figure 3: Tridimensional structure of the I-Oel homing endonuclease bound to its DNA target. The catalytic core is surrounded by two αββocββα folds forming a saddle-shaped interaction interface above the DNA major groove.
Figure 4: Different 1-OeI variants binding different sequences derived from the l-Crel target sequence (top right and bottom left) to obtain heterodimers or single chain fusion molecules cleaving non palindromic chimeric targets (bottom right).
Figure 5: Shows a schematic representation of the smaller independent subunits of the l-Crel meganuclease, i.e., sυbunit within a single monomer or αββαββα fold (top right and bottom left). These independent subunits allow for the design of novel chimeric molecules (bottom right), by combination of mutations within a same monomer. Such molecules would cleave palindromic chimeric targets (bottom right).
Figure 6: Shows a schematic representation of a method to combine four different subdomains so as to generate a custom meganuclease which cleaves a selected target. Figure 7: The IIIV1_1 target sequence (SEQ ID NO:319) and its derivatives. In the HIV1_1.2 target (SEQ ID NO:320), the ACΛC sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343). HIV1JL3 (SEQ ID NO:321) is the palindromic sequence derived from the left part of HIVlJ .2, (SEQ ID NO:320) and HIVlJ .4 (SEQ ID NO:322) is the palindromic sequence derived from the right part of HIV IJ .2 (SEQ ID NO:320). HIVlJ .5 (SEQ ID NO:323) and HIVlJ .6 (SEQ ID NO:324) are pseudo- palindromic targets derived, respectively, from HIV IJ .3 (SEQ ID NO:321) and HIVl J .4 (SEQ ID NO:322), containing the natural ACAC sequence in the middle of the target. As shown in the Figure, the boxed motives from 1 OAGAJ3, 1 OTGGJP, 5TACJP and 5CTG_P are found in the HIVlJ series of targets.
Figure 8: pCLS1055 plasmid map. Figure 9: pCLS0542 plasmid map.
Figure 10: Cleavage of HIVlJ .3 (SEQ ID NO:321) target by combinatorial variants. The figure displays an example of screening of I-Crel combi- natorial valiants with the HIVl J.3 target (SEQ ID NO:321). On the filter, the positive variants correspond to: BlO, SEQ ID NO: 1; Cl, SEQ ID NO:2; C7, SEQ ID NO:3; ClO, SEQ ID NO:4; C3: SEQ ID NO:5; all described in Table II. Each cluster contains 4 spots. On the two spots on the left, a yeast strain harboring the HIVl J .3 target (SEQ ID NO:321) has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows:
negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 11 : pCLS 1 107 plasmid map.
Figure 12: Cleavage of HIVlJ .4 (SEQ ID NO:322) and HΪV1J .6 (SEQ ID NO:324) targets by combinatorial variants. The figure displays an example of screening of l-Crel combinatorial variants with the HIVl J .4 (SEQ ID NO:322) and HIVlJ .6 (SEQ ID NO:324) targets. On the filter, the positive variants correspond to: C8, SEQ ID NO:7; A5, SEQ ID NO:8; Al, SEQ ID NO:9; A12, SEQ ID NO: 10; C3, SEQ ID NO:1 1; all described in Table IV. Each cluster contains 4 spots. On the two spots on the left, a yeast strain harboring the HIVl J.4 or the HIVlJ .6 targets have been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3)
Figure 13: Cleavage of the HIVlJ .2 (SEQ ID NO:320) and HIVlJ (SEQ ID NO:319) target sequences by heterodimeric combinatorial variants. Left panel: Example of screening of combinations of l-Crel variants against the HIV IJ.2 target. Right panel: Screening of the same combinations of 1-OeI variants against the HIVlJ target. Some heterodimers resulted in cleavage of the HIV IJ .2 target (SEQ ID NO:320). The heterodimer displaying a signal with HIVlJ target (SEQ ID NO:319) is observed at positions D3. On the filter, the position of mutants in certain positions as an example is: line C, SEQ ID NO: 10; line D, SEQ ID NO: 11 ; column 2, SEQ ID NO:1 ; column 3, SEQ ID NO:2; column 4; SEQ ID NO:5. These mutants have been described in Tables II and IV. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIVlJ target (SEQ ID NO:319) has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3)
Figure 14: Cleavage of HIVlJ .3 (SEQ ID NO:321) and HIVl J.5 (SEQ ID NO: 323) targets by meganuclease variants improved by random mutagenesis
in example 5. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_1.3 and HIV1_L5 targets. On the filter, the positive variants presented correspond to: F3, SEQ ID NO:27; CI l , SEQ ID NO:26; 118, SEQ ID NO:28; El 2, SEQ ID NO:29; all described in Table VIII. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_1.3 (SEQ ID NO:321) or the HIVlJ .5 (SEQ ID NO:323) targets have been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIVl J.3 target (SEQ ID NO:321). The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 15: Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by random mutagenesis in example 5. The figure displays an example of screening of I-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:46) cleaving the HIV IJ.4 target. On the filter, the positive variants presented correspond to: F3, SEQ ID NO:27; Cl 1 , SEQ ID NO:26; IΪ8, SEQ ID NO:28; E12, SEQ ID NO:29; all described in Table VIIL Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIVlJ .4 mutant and the HIVlJ target have been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant. The two spots on the right contain the same negative or positive controls, These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 16: Cleavage of HIVlJ .3 (SEQ ID NO:321) and HIVl J.5 (SEQ ID NO:323) targets by meganuclease variants improved by a second round of random mutagenesis in example 5bis. The figure displays an example of screening of I-CVel meganuclease variants with the HIVl J .3 and HIVl J .5 targets. On the filter, the positive variants presented correspond to: A125 SEQ ID NO:42; D8, SEQ ID NO:38; G8, SEQ ID NO:36; G3, SEQ ID NO:40; all described in Table IX. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the
HIV1_1.3 or the HIV1_1.5 targets have been mated with another yeast strain containing the meganucleasc variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
Figure 17: Cleavage of HIVlJ (SEQ ID NO:319) target by meganuciease variants improved by a second round of random mutagenesis in example 5bis. The figure displays an example of screening of 1-OeI meganuciease variants with the HIV IJ target, when mated with a meganuciease (SEQ ID NO:46) cleaving the HIVl J .4 target. On the filter, the positive variants presented correspond to: A12, SEQ ID NO:42; D8, SEQ ID NO:38; G8, SEQ ID NO:36; G3, SEQ ID NO:40; all described in Table IX. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIVl J .4 mutant (SEQ ID NO:46) and the HIVlJ target (SEQ ID NO:319) have been mated with another yeast strain containing the meganuciease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
Figure 18: Cleavage of HIVl J.3 (SEQ ID NO:321) and HIV1J .5 (SEQ ID NO:323) targets by meganuciease variants improved by site-directed mutagenesis in example 6. The figure displays an example of screening of l-Creϊ meganuciease variants with the HIVl J .3 and HIVl J .5 targets. On the filter, the positive variants presented correspond to: FlO, SEQ ID NO:63; H2, SEQ ID NO:60; H3, SEQ ID NO:59; A3, SEQ ID NO:64; F4, SEQ ID NO:65; some of them described in Table XI. Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIVlJ target (see Figure 19). This is due to the presence of the Gl 9S mutation in these variants. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIVl J.3 or the HIVl J .5 targets have been mated with another yeast strain containing the meganuciease variants. The spot on the low-right contain negative or positive controls. These controls are serially
repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement. Figure 19: Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by site-directed mutagenesis in example 6. The figure displays an example of screening of l-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:46) cleaving the HIV IJ .4 target. On the Filter, the positive variants presented correspond to: Fl O, SEQ ID NO:63; H2, SEQ ID NO:60; H3, SEQ ID NO:59; A3, SEQ ID NO:64; F4, SEQ ID NO:65; some of them described in Table XI. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV IJ .4 mutant and the HIVlJ target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
Figure 20: Cleavage of HIVlJ .4 (SEQ ID NO:322) and HIVLl .6 (SEQ ID NO:324) targets by meganuclease variants improved by random mutagenesis in example 7. The figure displays an example of screening of l-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets. On the filter, the positive variants presented correspond to: B7, SEQ ID NO:46; B9, SEQ ID NO:68; B 12, SEQ ID NO:69; A9, SEQ ID NO:70; E5, SEQ ID NO:71 ; all described in Table XIII. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIVl J.4 or the HIVI J .6 targets have been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIVl J, 4 target. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 21 : Cleavage of IHVlJ target (SEQ ID NO:319) by mcganuclease variants improved by random mutagenesis in example 7. The figure displays an example of screening of l-Crel meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:26) cleaving the HIVlJ .3 target. On the filter, the positive variants presented correspond to: B7, SEQ ID NO:46; B9, SEQ ID NO:68; B12, SEQ ID NO:69; A9, SEQ ID NO:70; E5, SEQ ID NO:71; all described in Table XIII. Each cluster contains 6 spots. On the 2 spots on the left, as well as those in the middle, a yeast strain harboring the HIVl J.3 mutant (SEQ ID NO:26) and the IHVlJ target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster AI ), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement. Figure 22: Cleavage of HIVlJ .4 (SEQ ID NO:322) and HIVlJ .6
(SEQ ID NO:324) targets by meganuclease variants improved by a second round of random mutagenesis in example 7bis. The figure displays an example of screening of I-Crel meganuclease variants with the HIVl J .4 and HIVl J .6 targets. On the filter, the positive variants presented correspond to: A3, SEQ ID NO:76; Bl, SEQ ID NO:77; Cl, SEQ ID NO:78; D3, SEQ ID NO:79; D5, SEQ ID NO:80; all described in Table XIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the 1IIV1 J .4 or the HIVl J.6 targets have been mated with another yeast strain containing the rneganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
Figure 23: Cleavage of HIVlJ target (SEQ ID NO:319) by meganuclease variants improved by a second round of random mutagenesis in example 7bis. The figure displays an example of screening of 1-OeI meganuclease variants with the HIVlJ target, when mated with a meganuclease (SEQ ID NO:26) cleaving the HIVl J .3 target. On the filter, the positive variants presented correspond
to: A3, SEQ ID NO:76; Bl , SEQ ID NO:77; Cl , SEQ ID NO:78; D3, SEQ ID NO:79; D5, SEQ ID NO: 80; all described in Table XIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV 1_1 .3 mutant (SEQ ID NO:26) and the HIV 1_1 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2); and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a variant issued from the first round of improvement. Figure 24: The HIVlJ target sequence (SEQ ID NO:325) and its derivatives. In the HIV1_3.2 target (SEQ ID NO:32ό), the TTTA sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343). HIV1_3.3 (SEQ ID NO: 327) is the palindromic sequence derived from the left part of HIV IJ, .2, and HΪV1_3.4 (SEQ ID NO:328) is the palindromic sequence derived from the right part of HIVl J.2. HIVl J.5 (SEQ ID NO:329) and HIVl _3.6 (SEQ ID NO:330) are pseudo-palindromic targets derived, respectively, from HIV1_3.3 and HIV1_3,4, containing the natural TTTA sequence in the middle of the target. As shown in the Figure, the boxed motives from 10CΛG_P, 10ACA_P} 5CCT_P and 5GAC_P are found in the HIV 1_3 series of targets. Figure 25: Cleavage of HIV1_3.3 target (SEQ ID NO:327) by combinatorial variants. The figure displays an example of screening of l-Crel combinatorial variants with the HIV1_3.3 target. On the filter, the positive variants correspond to: A6, SEQ ID NO:89; Al, SEQ ID NO:91 ; A8, SEQ ID NO.90; Λ4, SEQ ID NO:88; all described in Table XVI. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIV1_3.3 target has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive contiol (i.e. cluster A3). Figure 26: Cleavage of HIV1_3.4 (SEQ ID NO:328) and HIVl_3.ό
(SEQ ID NO:330) targets by combinatorial variants. The figure displays an example of screening of l-Crel combinatorial variants with the HIV1_3.4 and HIV1_3.6
targets. On the filter, the positive variants correspond to: C12, SEQ ID NO:98; C8, SEQ ID NO:99; E4, SEQ ID NO: 100; G4, SEQ ID NO:97; E9, SEQ ID NO: 101; all described in Table XVIII. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIV1_3.4 or the HIV1_3.6 targets has been mated with another yeast strain containing the mcganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 27: Cleavage of HIV1_3.3 target (SEQ ID NO:327) by meganuclease variants improved by random mutagenesis in example 12. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_3.3 target. On the filter, the positive variants presented correspond to: El, SEQ ID NO: 105; C8, SEQ ID NO: 106; A2, SEQ ID NO:107; A7, SEQ ΪD NO:108; BlO, SEQ ID NO: 109; all described in Table XIX. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_3.3 target has been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the IIIV1_3.3 target. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 28: Cleavage of HIV1_3.3 target (SEQ ID NO:327) by mcganuclease variants improved by a second round of random mutagenesis in example 12bis. The figure displays an example of screening of 1-OeI meganuclease variants with the HIV1_3.3 target. On the filter, the positive variants presented correspond to: Al l, SEQ ID NO:115; B7, SEQ ID NO:1 16; F12, SEQ ID NO:117; G2, SEQ ID NO: 118; H9, SEQ ID NO:1 19; all described in Table XX. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_3.3 target has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot
in the upper-right contains, as an internal control, a variant issued from the first round of improvement.
Figure 29: Cleavage of HΪV1J.3 (SEQ ID NO:327) and HIV1_3.5 (SEQ ID NO: 329) targets by meganuclease variants improved by site-directed mutagenesis in example 13. The figure displays an example of screening of 1-OeI meganuclease variants with the HIV1_3.3 and HIV1J3.5 targets. On the filter, the positive variants presented correspond to: Al, SEQ ID NO: 126; G3, SEQ ID NO: 127; Cl, SEQ ID NO:128; H6, SEQ ID NO: 129; E5, SEQ ID NO: 130; described in Table XXI. Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIV1_3 target (see Figure 30). This is due to the presence of the Gl 9S mutation in these variants. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_3.3 or the HIV1_3.5 targets have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a previously improved variant.
Figure 30: Cleavage of HIV1_3 (SEQ ID NO:325) target by meganuclease variants improved by site-directed mutagenesis in example 13. The figure displays an example of screening of \-Cre\ meganuclease variants with the HIV 1_3 target, when mated with a meganuclease (SEQ ID NO: 125) cleaving the HIV1_3.4 target. On the filter, the positive variants presented correspond to: Al, SEQ ID NO:12ό; G3, SEQ ID NO:127; Cl, SEQ ID NO: 128; Hό, SEQ ID NO:129; E5, SEQ ID NO: 130; described in Table XXI. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV 1_3.4 mutant (SEQ ID NO: 125) and the HIV1_3 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a previously improved variant.
Figure 31 : Cleavage of H1V1_3.4 (SEQ ID NO:328) and HIVlJ.6 (SEQ ID NO: 330) targets by meganuclease variants improved by random mutagenesis in example 14. The figure displays an example of screening of 1-CVeI meganuclease variants with the HIVl J.4 and HIVl J, 6 targets. On the filter, the positive variants presented correspond to: E8, SEQ ID NO: 136; B 12, SEQ ID NO : 137; B l , SEQ ID NO: 138; B8, SEQ ID NO: 139; D6, SEQ ID NO: 140; all described in Table XXII. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIVl J.4 or the HIVlJ.6 targets has been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV IJ.4 target. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3),
Figure 32: Cleavage of HIVl J.4 (SEQ ID NO:328) and HIVl J.6 (SEQ ID NO:330) targets by meganuclease variants improved by a second round of random mutagenesis in example 14bis. The figure displays an example of screening of l-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets. On the filter, the positive variants presented correspond to: F7, SEQ ID NO:146; B12, SEQ ID NO: 147; G7, SEQ ID NO:148; D2, SEQ ID NO:149; A5, SEQ ID NO: 150; all described in Table XXIII. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIVl J.4 or the HIVl J.6 targets have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a previously improved variant.
Figure 33: Cleavage of HIVl J.4 (SEQ ID NO:328) and HIVl J.6 (SEQ ID NO: 330) targets by meganuclease variants improved by site-directed mutagenesis in example 15. The figure displays an example of screening of I-Crel meganuclease variants with the HIVl J.4 and HIVl J.6 targets. On the filter, the positive variants presented correspond to: Dl5 SEQ ID NO:156; C2, SEQ ID NO:157; F2, SEQ ID NO:158; A4, SEQ ID NO:159; G7, SEQ ID NO: 160; described in Table
XXIV. Each cluster contains 6 spots. On the 4 spots on the left, a yeast stiain harboring the HIV1_3.4 or the HIV1_3.6 targets have been mated with another yeast strain containing 4 different meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a non-improved variant.
Figure 34: Cleavage of HIV1_3 target (SKQ ID NO:325) by meganuclease variants improved by site-directed mutagenesis in example 15. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_3 target, when mated with a meganuclease (SEQ ID NO: 109) cleaving the HIV1_3,3 target. On the filter, the positive variants presented correspond to: Dl , SEQ ID NO:156; C25 SEQ ID NO: 157; F2, SEQ ID NO: 158; A4, SEQ ID NO: 159; G7, SEQ ID NO: 160; described in Table XXIV. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV 1_3 target and the HIV1_3.3 mutant has been mated with another yeast strain containing different meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a previously improved variant.
Figure 35: The HΪV1_4 (SEQ ID NO:331) target sequence and its derivatives. In the HIV1__4.2 target (SEQ ID NO:332), the GGAC sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343). HIV1_4.3 (SEQ ID NO:333) is the palindromic sequence derived from the left part of HIV1_4.2, and HIV1_4.4 (SEQ ID NO:334) is the palindromic sequence derived from the right part of HIV1_4.2. HIV1_4.5 (SEQ ID NO:335) and HIV1_4.6 (SEQ ID NO:336) are pseudo-palindromic targets derived, respectively, from HIV1_4.3 and HIV1_4.4, containing the natural GGAC sequence in the middle of the target. As shown in the Figure, the boxed motives from 10AGC_P, 10TGT_P, 5TCT_P and 5TATJP are found in the HIV 1_4 series of targets.
Figure 36: Cleavage of HIV1_4.3 (SEQ ID NO:333) target by combinatorial variants. The figure displays an example of screening of Ϊ-Crel combi-
natorial variants with the HIV1_4.3 target. On the filter, the positive variants correspond to: Al l , SEQ ID NO:168; A5, SEQ ID NO: 170; A2, SEQ ID NO: 171 ; A4; SEQ ID NO: 173; A3, SEQ ID NO: 174; all described in Table XXVl. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIV1_4.3 target has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 37: Cleavage of HIV1_4.4 (SEQ ID NO:334) and HIV1_4.6 (SEQ ID NO:336) targets by combinatorial variants. The figure displays an example of screening of l-Crel combinatorial variants with the HIV1_4.4 and HIV1__4.6 targets. On the filter, the positive variants correspond to: A7, SEQ ID NO: 177; A5, SEQ ID NO:178; B8, SEQ ΪD NO: 179; E6, SEQ ID NO:180; F2, SEQ ID NO: 181 ; all described in Table XXVIII. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIV1_4.4 or the HIV1_4.6 targets has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al)5 positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). Figure 38: Cleavage of the HIV1_4.2 (SEQ ID NO:332) and
HIV1_4 (SEQ ID NO:331) target sequences by heterodimeric combinatorial variants. Example of screening of combinations of l-Creϊ variants against the I-HV1_4.2 target. Some heterodimers resulted in cleavage of the HIV1__4.2 target, while no cleavage activity was detected on the HIV 1_4 target. On the filter, the position of mutants in certain positions as an example is: line A, SEQ ID NO: 170; line B, SEQ ID NO:171 ; column 1, SEQ ID NO: 177; column 2, SEQ ID NO: 178; column 3; SEQ ID NO: 179. These mutants have been described in Tables XXVI and XXVIII. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV 1_4 or HIV1_4.2 target have been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows:
negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 39: Cleavage of HIV1_4,3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by random mutagenesis in example 20. The figure displays an example of screening of I-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets. On the filter, the positive variants presented correspond to: F8, SEQ ID NO:189; C65 SEQ ID NO: 190; E12, SEQ ID NO: 191; G12, SEQ ID NO:192; G6, SEQ ID NO:193; GI l, SEQ ID NO:194; all described in Table XXX. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.3 or the HΪV1_4.5 targets have been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV1_4.3 target. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 40: Cleavage of HIV1_4.3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by a second round of random mutagenesis in example 20bis. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets. On the filter, the positive variants presented correspond to: E7, SEQ ID NO:200; Al , SEQ ID NO:201 ; E9, SEQ ID NO:202; A4, SEQ ID NO:203; Al l, SEQ ID NO:204; all described in Table XXXI. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.3 or the HIV1_4.5 targets has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al)5 positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant. Figure 41: Cleavage of HIV1_4 (SEQ ID NO:331) target by meganuclease variants improved by a second round of random mutagenesis in example 20bis. The figure displays an example of screening of l-Crel meganuclease
variants with the HIV1_4 target, when mated with a meganuclease (SEQ ID NO: 199) cleaving the HIV1_4.4 target. On the filter, the positive variants presented correspond to: E7, SEQ ID NO:200; Al, SEQ ID NO:201 ; E9, SEQ ID NO:202; Λ4, SEQ ID NO:203; Al 1 , SEQ ID NO:204; all described in Table XXXI. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.4 mutant (SEQ ID NO: 199) and the HIV1__4 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 42: Cleavage of HIV1_4 (SEQ ID NO:331) target by meganuclease variants improved by site-directed mutagenesis in example 21. The figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO:210) cleaving the HIV1_4.4 target. On the filter, the positive variants presented correspond to: Al, SEQ ID NO:211 ; A2, SEQ ID NO:212; A5; SEQ ID NO:213; A7; SEQ ID NO:214; A8, SEQ ID NO:215; G2, SEQ ID NO:216; described in Table XXXII. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.4 mutant (SEQ ID NO:210) and the HIV 1_4 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 43: Cleavage of HIV1_4.3 (SEQ ID NO:333) and HIV1_4.5 (SEQ ID NO:335) targets by meganuclease variants improved by site-directed mutagenesis in example 21. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.3 and HIV1_4.5 targets. On the filter, the variants presented correspond to: Al, SEQ ID NO:211 ; A2, SEQ ID NO:212; A5, SEQ ID NO:213; A7, SEQ ID NO:214; A8, SEQ ID NO:215; G2, SEQ ID NO:21ό; described in Table XXXΪΪ. Some of these variants show no cleavage activity as
homodimers while they are active as heterodimcrs on the HIV \ A target (see Figure 42). This is due to the presence of the Gl 9S mutation in these variants. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.3 or the HIV1_4.5 targets have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant. Figure 44: Cleavage of HIV1_4.4 (SEQ ID NO:334) and HIV1_ 4.6
(SEQ ID NO:336) targets by meganuclease variants improved by random mutagenesis in example 22. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_4.4 and HΪV1__4.6 targets. On the filter, the positive variants presented correspond to: D4, SEQ ID NO: 199; D5, SEQ ID NO:2ΪO; C8, SEQ ID NO:221 ; ClO, SEQ ID NO:222; E8, SEQ ID NO:223; all described in Table XXXIlI. Each cluster contains 6 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.4 or the HIV1_4.6 targets have been mated with another yeast strain containing the meganuclease variants. The two spots in the middle contain, as an internal control, a non-improved variant cleaving the HIV1_4.4 target. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 45: Cleavage of HIV 1_4 (SEQ ID NO:33 I) target by meganuclease variants improved by random mutagenesis in example 22. The figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO: 190) cleaving the HIV1_4.3 target. On the filter, the positive variants presented correspond to: D4; SEQ ID NO:199; D5, SEQ ID NO:210; C8, SEQ ID NO:221; ClO, SEQ ID NO:222; E8, SEQ ID NO:223; all described in Table XXXIII. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV1_4.3 mutant (SEQ ID NO: 190) and the HIV 1_4 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive
controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al)5 positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a non- improved variant. Figure 46: Cleavage of HIV 1_4 (SEQ ID NO:331) target by meganuclease variants improved by site-directed mutagenesis in example 23. The figure displays an example of screening of l-Crel meganuclease variants with the HIV 1_4 target, when mated with a meganuclease (SEQ ID NO: 190) cleaving the HIV1_4.3 target. On the filter, the positive variants presented correspond to: B5, SEQ ID NO:229; B4: SEQ ID NO:231 ; A5> SEQ ID NO:235; A8, SEQ ID NO:236; Al l , SEQ ID NO:237; described in Table XXXIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV 1_4 target and the HIVl j4.3 mutant (SEQ ID NO: 190) has been mated with another yeast strain containing different meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 47: Cleavage of HIV1_4.4 (SEQ ID NO:334) and H1V1_4.6 (SEQ ID NO:336) targets by meganuclease variants improved by site-directed mutagenesis in example 23. The figure displays an example of screening of I-Crel meganuclease variants with the HIV1_4.4 and HIV1__4.6 targets. On the filter, the positive variants presented correspond to: B5, SEQ ID NO:229; B4, SEQ ID NO:231 ; A5. SEQ ID NO:235; A8, SEQ ID NO:236; Al l, SEQ ID NO:237; described in Table XXXIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_4.4 or the HIV1_4.6 targets have been mated with another yeast strain containing different meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 48: The HIV 1_5 target sequence (SEQ ID NO:337) and its derivatives. In the HIV1_5.2 target (SEQ ID NO:338)5 the ATAC sequence in the middle of the target is replaced with GTAC, the bases found in C 1221 (SEQ ID NO:343). HIV1_5.3 (SEQ ID NO:339) is the palindromic sequence derived from the left pail of HIV1_5.2, and HIV1_5.4 (SEQ ID NO:340) is the palindromic sequence derived from the right part of I-IIVl_5.2. HIV1_5.5 (SEQ ID NO:341) and HIV1_5.6 (SEQ ID NO:342) are pseudo-palindromic targets derived, respectively, from HIV1_5,3 and HIVl J5.4, containing the natural ATAC sequence in the middle of the target. As shown in the Figure, the boxed motives from 1 OTCTJP, 10CTG_P, 5TAG_P and 5CCTJP are found in the HIV1_5 series of targets.
Figure 49: Cleavage of HIV1_5.3 (SEQ ID NO:339) target by combinatorial variants. The figure displays an example of screening of I-Od combinatorial variants with the HIV1_5.3 target. On the filter, the two positive variants correspond to: Al , SEQ ID NO:242; A2, SEQ ID NO:241; described in Table XXXVI. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIVlJj.3 target has been mated with another yeast strain containing the meganucleasc variants. The two spots on the right contain the same negative or positive controls. These controls are: negative control (cluster Al), positive control (cluster A2), and strong positive control (cluster A3). Figure 50: Cleavage of HIV1_5.4 (SEQ ID NO:340) target by combinatorial variants. The figure displays an example of screening of Ϊ-Crel combinatorial variants with the HIV1_5.4 target. On the filter, the positive variants correspond to: Al, SEQ ID NO:249; A3, SEQ ID NO:245; Λ4, SEQ ID NO:252; A7, SEQ ID NO:250; AlO, SEQ ID NO:246; all described in Table XXXVIII. Each cluster contains 4 spots. On the spots on the left, a yeast strain harboring the HIV1_5.4 target has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 51 : Cleavage of the HIV1_5.2 target sequence (SEQ ID NO:338) by heterodimeric combinatorial variants. Example of screening of combina-
tions of I-Crel variants against the HIV1_5.2 target. One hcterodimer resulted in cleavage of the HIV1_5,2 target. The heterodimer displaying a signal with HIV1_5.2 target is observed at position B4. On the filter, the position of certain mutants as an example is: line A, SEQ ID NO:242; line B, SEQ ID NO:241; column 3, SEQ ID NO:245; column 4, SEQ ID NO:252; column 5; SEQ ID NO:251. These mutants have been described in Tables XXXVI and XXXVΪII. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV1_5.2 target has been mated with another yeast strain containing the meganuclease variants. The two spots on the right contain the same negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3).
Figure 52: Cleavage of HIV1_5.3 target (SEQ ID NO:339) by meganuclease variants improved by random mutagenesis in example28. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.3 target. On the filter, the positive variants presented correspond to: A6, SEQ ID NO:256; A12, SEQ ID NO:257; Al l, SEQ ID NO:258; AlO, SEQ ID NO:259; A2, SEQ ID NO:260 ;all described in Table XXXIX. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV1_5.3 or target has been mated with another yeast strain containing the meganuclease variants. The spot on the low- right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a non-improved variant.
Figure 53: Cleavage of HIV1_5.3 (SEQ ID NO:339) and HIV1_5.5 (SEQ ID NO:341) targets by meganuclease variants improved by a second round of random mutagenesis in example 28bis. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.3 and HIV1_5.5 targets. On the filter, the positive variants presented correspond to: G2, SEQ ID NO:266; E4, SEQ ID NO:267; C2, SEQ ID NO:268; A12, SEQ ID NO:269; CI l, SEQ ID NO:270; all described in Table XL. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_5.3 or the HIV1_5.5 targets have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right
contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant. Figure 54: Cleavage of HIV 1_5 target (SEQ ID NO:337) by meganuclease variants improved by a second round of random mutagenesis in example 28bis. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5 target, when mated with a meganuclease (SEQ ID NO:276) cleaving the HIV1_5.4 target. On the filter, the positive variants presented correspond to; G2, SEQ ID NO:266; E4, SEQ ID NO:267; C2, SEQ ID NO:268; A12, SEQ ID NO:269; CI l, SEQ ID NO:270; all described in Table XL. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_5.4 mutant and the HIV 1_5 target have been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 55: Cleavage of HIV1_5.3 (SEQ ID NO:339) and HIV1_5.5 (SEQ ID NO:341) targets by meganuclease variants improved by site-directed mutagenesis in example 29. The figure displays an example of screening of 1-CVeI meganuclease variants with the HIV1_5.3 and HIV1_5.5 targets. On the filter, the positive variants presented correspond to: C6, SEQ ID NO:278; F8, SEQ ID NO:279; H7, SEQ ID NO:280; Fl, SEQ ID NO:281 ; G 12, SEQ ID NO:282; described in Table XLI. Some of these variants show no cleavage activity as homodimers while they are active as heterodimers on the HIV1__5 target (see Figure 56). This is due to the presence of the G19S mutation in these variants. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_5.3 or the HIV1_5.5 targets has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right is a negative control. The spot in the upper-right contains, as an internal control, an improved variant.
Figure 56: Cleavage of HIV1_5 target (SEQ ID NO:337) by meganuclease variants improved by site-directed mutagenesis in example 29. The
figure displays an example of screening of 1-Crel meganuclease variants with the HIV 1_5 target, when mated with a meganuclease (SEQ ID NO:276) cleaving the HIV1_5.4 target. On the filter, the positive variants presented correspond to: Cβ, SEQ ID NO:278; F8, SEQ ID NO:279; H7, SEQ ID NO:280; Fl 5 SEQ ID NO:281 ; G12, SEQ ID NO:282; described in Table XLI. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV1_5.4 mutant (SEQ ID NO:276) and the HIV 1_5 target has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 57: Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIV1J.6 (SEQ ID NO:342) targets by meganuclease variants improved by random mutagenesis in example 30. The figure displays an example of screening of l-Crel meganuclease variants with the HIV1_5.4 and HIV1_5.6 targets. On the filter, the positive variants presented correspond to: D6, SEQ ID NO:276; A4, SEQ ID NO:288; ClO, SEQ ID NO:289; A9, SEQ ID NO:290; Al , SEQ ID NO:291 ; all described in Table XLII. Each cluster contains 6 spots. On the 4 spots on the left, a yeast strain harboring the HIV1_5.4 or the HΪV1_5.6 targets has been mated with another yeast strain containing the meganuclease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, a non- improved variant cleaving the HIV1_5.4 target.
Figure 58: Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIVl_5,ό (SEQ ID NO: 342) targets by meganuclease variants improved by a second round of random mutagenesis in example 30bis. The figure displays an example of screening of 1-Crel meganuclease variants with the HΪV1_5.4 and IIIV1 5.6 targets. On the filter, the positive variants presented correspond to: A12, SEQ ID NO:297; Al, SEQ ID NO:298; Al l , SEQ ID NO:299; A8, SEQ ID NO:300; B4, SEQ ID NO:301; all described in Table XLIIL Each cluster contains 4 spots. On the 2 spots on the left, a
yeast strain harboring the HIV1J5.4 or the HIVl_5.ό targets have been mated with another yeast strain containing the meganuciease variants. The spot on the low-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 59: Cleavage of HIV1_5.4 (SEQ ID NO:340) and HIV1_5.6 (SEQ ID NO:342) targets by meganuciease variants improved by site-directed mutagenesis in example 31. The figure displays an example of screening of l-Crel meganuciease variants with the HIV1_5.4 and HIVlJ).6 targets. On the filter, the positive variants presented correspond to: Hl , SEQ ID NO:307; H2, SEQ ID NO:308; H9, SEQ ID NO:309; B3, SEQ ID NO:310; H3, SEQ ID NO:31 1; described in Table XLIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV 1__5.4 or the HIV1_5.6 targets has been mated with another yeast strain containing different meganuciease variants. The spot on the low-right contains negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control (i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant. Figure 60: Cleavage of HIV1__5 (SEQ ID NO:337) target by meganuciease variants improved by site-directed mutagenesis in example 31. The figure displays an example of screening of 1-OeI meganuciease variants with the HΪV1_5 target, when mated with a meganuciease (SEQ ID NO:256) cleaving the HIV1_5.3 target. On the filter, the positive variants presented correspond to: Hl , SEQ ID NO:307; H2, SEQ ID NO:308; H9, SEQ ID NO:309; B3, SEQ ID NO:310; H3; SEQ ID NO:311 ; described in Table XLIV. Each cluster contains 4 spots. On the 2 spots on the left, a yeast strain harboring the HIV 1_5 target and the HI V 1 5.3 mutant (SEQ ID NO:256) has been mated with another yeast strain containing different meganuciease variants. The spot on the iow-right contain negative or positive controls. These controls are serially repeated every three clusters as follows: negative control (i.e. cluster Al), positive control (i.e. cluster A2), and strong positive control
(i.e. cluster A3). The spot in the upper-right contains, as an internal control, an improved variant.
Figure 61 : pCLS1853 plasmid map.
Figure 62: Schematic representation of the pseudo-HΪV provirus integrated in the HEK293-VLP-CL40 cell line used for validation of the activity of HlV meganucleases. The LTRs, encompassing the U3, R and U5 regulatory sequences are duplicated and flanking the viral genes gag and pol. The env gene has been partially deleted and a pEFla-PuroR-IRES-BGFP cassette has been introduced between the 5' portion of env and the 3' LTR. The location of the meganuclcasc targets HIVlJ (SEQ ID NO:319); HΪV1_3 (SEQ ID NO:325), HIV 1_4 (SEQ ID NO:331), HIVlJ (SEQ ID NO:337)5 HIV 1J7 (SEQ ID NO:366), HIV1_8 (SEQ ID NO:367) and HIV1^9 (SEQ ID NO:368) are represented. The ORF of the TAT and REV genes have been introduced in the cellular genome using different retroviral vectors. Figure 63: Levels of p24 produced by the HEK293-VLP-CL40 cell line 48 hours after transfection with 1 μg of meganuclease expression plasmid.
The amount of p24 present in cell culture supernatants was determined by ELISA. A sample transfected by a non related meganuclease (NRM, see text) is used for normalization. In this way, the amount of p24 produced by these cells, expressed in fg/cell is considered as 100% of VLP production. The amount of p24 produced by HIV meganuclease transfected cells is represented as the percentage of VLP production respect to the amount produced by the NRM transfected cells. The values represent the data from at least 3 independent transfections.
Figure 64: represents a scheme of the mechanism leading to the generation of small deletions and insertions (InDeI) during repair of double-strand break by non homologous end-joining (NHEJ).
There will now be described by way of example a specific mode contemplated by the Inventors. In the following description numerous specific details are set forth in order to provide a thorough understanding. It will be apparent however, to one skilled in the art, that the present invention may be practiced without limitation to these specific details. In other instances, well known methods and structures have not been described so as not to unnecessarily obscure the description.
Example 1: Strategy for engineering meganucleases cleaving the HIVI l target from the HIVl virus
The HIV1__1 target is a 22 bp (non-palindromic) target located in U3 region of the proviral LTRs (Figures 2 and 7). Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, the HIV 1_1 target is present twice in the HIV_1 provirus. This target is precisely located at positions 84- 105 and 8159-9180 of the HIV-I pNL4-3 vector (accession number AF324493, Λdachi et al., J. Virol, 1986, 59, 284-291), this infective molecular clone was generated from the NY5 strain (Barre-Sinoussi et al., Science, 1983, 220, 868-871 and Benn et al., Science , 1985, 230, 949-951) a subtype B infectious molecular clone.
The HIV 1_1 sequence is partly a patchwork of the 1 OAGAJP, 10TGG_P, 5TAC JP and 5_CTG_P targets (these designations describe the 3bρ starting at the indicated nucleotide of the l-Crel target, for instance 10AGΛ_P indicates that nucleotides -10, -9 and -8 are A(-10) G(-9) A(-8) (Figure 7)) which are cleaved by previously identified meganucleases. These meganucleases were obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould et al., J. MoI, Biol., 2006, 355, 443-458; Smith et al., Nucleic Acids Res., 2006.
The 10AGA_P, 10TGG_P, 5TACJP and 5_CTG_P target sequences are 24 bp derivatives of C 1221, a palindromic sequence cleaved by 1-OeI (Arnould et al., precited). However, the structure of 1-OeI bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269), and in this study, only positions -11 to 11 were considered. Consequently, the HIV1__1 series of targets were defined as 22 bp sequences instead of 24 bp. HIVlJ differs from C1221 (SEQ ID NO: 343) in the 4 bp central region. According to the structure of the I-Oel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-OeI protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269). Thus, the bases at these positions should not impact the binding efficiency. However, they could
affect cleavage, which results from two nicks at the edge of this region. Thus, the ACAC sequence in -2 to 2 was first substituted with the GTΛC sequence from Cl 221 , resulting in target IIIV1_1.2 (Figure 7). Then, two palindromic targets, HIV1_1.3 and HIVLl .4, were derived from HIVlJ .2 (Figure 7). Since HIVl J .3 and HIVl J .4 are palindromic, they should be cleaved by homodimeric proteins. Two other pseudo- palindromic targets were derived from these two containing the ACAC sequence in -2 to 2 (targets HIVlJ .5 and HIVlJ .6, Figure 7). Thus, proteins able to cleave HIVl J.3 and HIVlJ .4 targets or, preferentially, the pseudo-palindromic targets as homodimers were first designed (examples 2 and 3) and then co-expressed to obtain heterodimers cleaving HIVlJ (example 4). Heterodimcrs cleaving the HIVl J .2 and HIVlJ targets could be identified. In order to improve cleavage activity for the HIVlJ target, a series of variants cleaving HIVl J.3 and HIVl J.4 was chosen, and then refined. The chosen variants were subjected to random or site-directed mutagenesis, and used to form novel heterodimers that were screened against the HIVlJ target (examples 5, 6, 7 and 8). Heterodimers could be identified with an improved cleavage activity for the HIVlJ target. Example 2: Identification of meganucleases cleaving HIVl 1.3 This example shows that l-Crel variants can cut the HIVl J .3 DNA target sequence derived from the left part of the HIVlJ .2 target in a palindromic form (Figure 7). HIVlJ .3 is similar to 1 OAGAJ1 at positions ±1 , ±2, ±6, ±8, ±9, and ±10 and to 5TACJP at positions ±1, ±2, ±3, ±4, ±5 and ±6. It was hypothesized that positions ±7 and ±11 would have little effect on the binding and cleavage activity. Variants able to cleave the 1 OAGAJ1 target were obtained by mutagenesis of I-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156. Variants able to cleave 5TAC_P were obtained by mutagenesis on 1-OeI N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al., J. MoL Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that
this position has little impact on the specificity at bases 10 to 8 of the target. Mutations at positions 24 found in variants cleaving the 5TAC_P target will be lost during the combinatorial process. But it was hypothesized that this will have little impact on the capacity of the combined variants to cleave the HIV 1_1.3 target. Therefore, to check whether combined variants could cleave the
HIV1_1.3 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TACJ? were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10AGA_P. A) Material and Methods a) Construction of target vector
The target was cloned as follows: an oligonucleotide corresponding to the HIV1_1.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTGCAGAACTACGTACGTAGTTCTGCCAATCGTCTGTCA 3' (SEQ ID NO: 14). The same procedure was followed for cloning the HIV 1_1.5 target, using the oligonucleotide:
5' TGGCATACAAGTTTGCAGAACTACACACGTAGTTCTGCCAATCGTCTGTCA 3' (SEQ ID NO: 15). Double-stranded target DNA, generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8). Yeast reporter vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B {MAT a, ura3Δ851, trplΔ63, leu2Δl, lys2Δ202), resulting in a reporter strain, b) Construction of combinatorial mutants l-Creϊ variants cleaving 10AGA_P or 5TAC_P were previously identified, as described in Smith el al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al., L MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10AGA_P and 5TAC_P targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the \-Crel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-
acaaccttgatlggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542, Figure 9) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction using the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an cquimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS0542, Figure 9) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et aL, J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETIX). Variants were giϊdded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidasc activity. Results were analyzed by scanning and quantification was performed using appropriate software,
d) Sequencing of variants
To recover the variant expression plasmids, yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA. Alternatively, ORFs were amplified from yeast DNA by PCR (Akada et al, Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MlLLEGEN SA. B) Results
I-Crel combinatorial variants were constructed by associating muta- tions at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TAC_P with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10AGA_P on the 1-OeI scaffold, resulting in a library of complexity 1600. Examples of combinatorial variants are displayed in Table I. In Table I the peptide sequence of these two subdomains are provided in the first column and second row respectively. This library was transformed into yeast and 3348 clones (2 times the diversity) were screened for cleavage against the HIVlJL 3 and HIV1_1.5 DNA targets. 36 positive clones were found to cleave the HIV1__1.3 target, which after sequencing turned out to correspond to 31 different novel endonuclease variants (Table II). Those variants showed no cleavage activity of the HIV1_1.5 DNA target. Examples of positives are shown in Figure 10. Some of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77. Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be I-Oøl combined variants resulting from micro- recombination between two original variants during in vivo homologous recombina- tion in yeast.
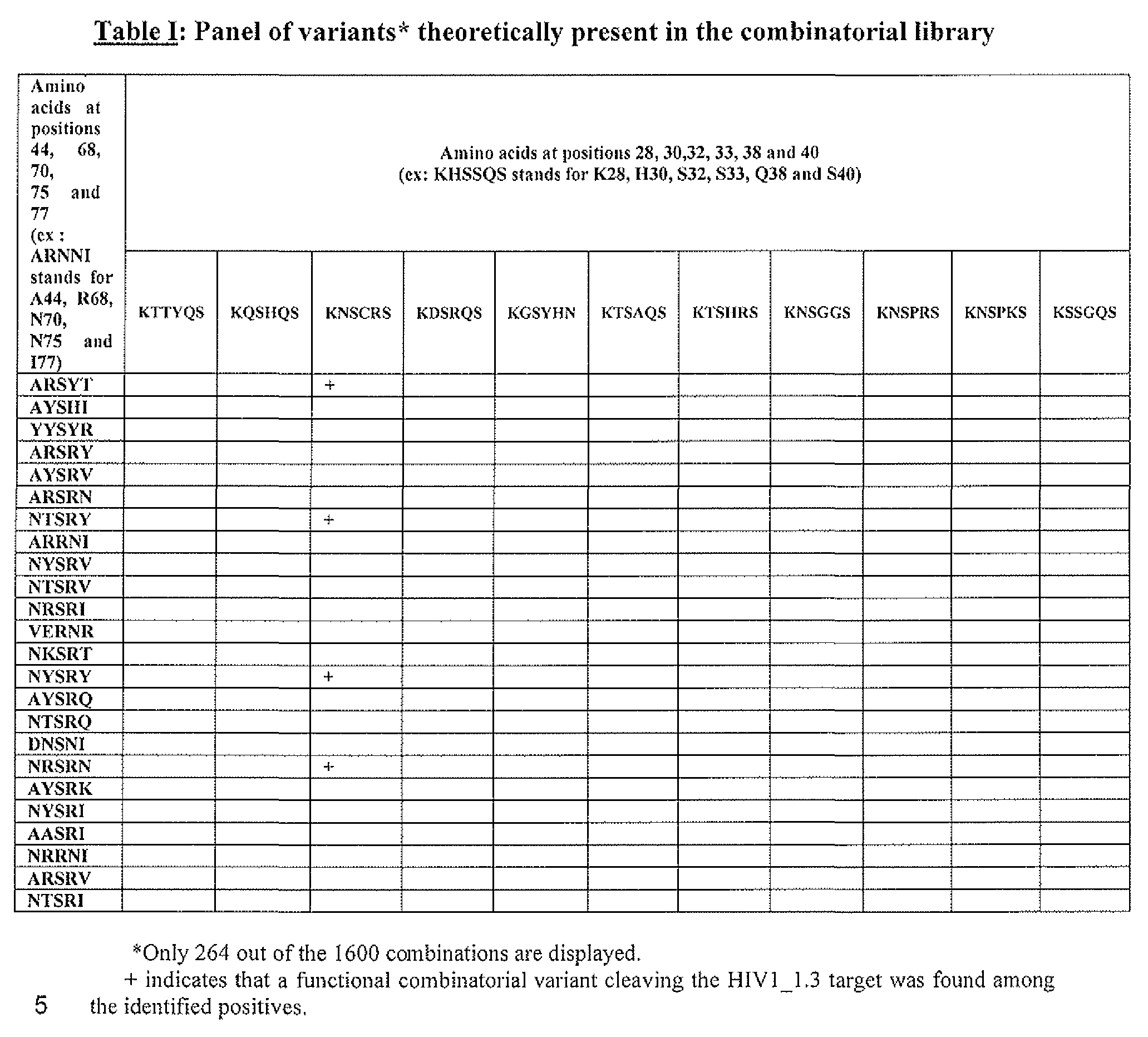
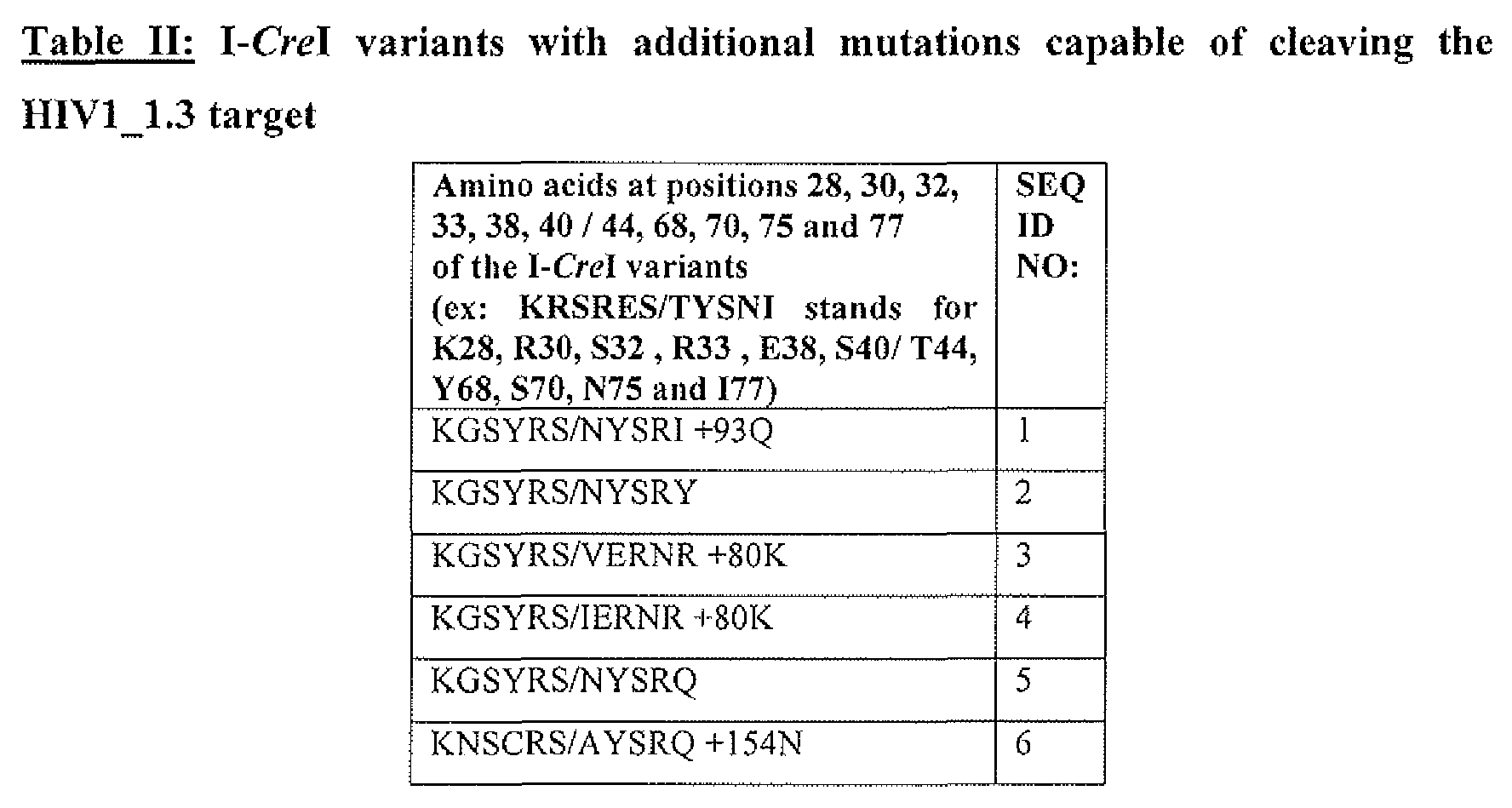
Example 3: Making of meganucleases cleaving HIVl 1.4 This example shows that 1-OeI variants can cleave the HIV1_1.4 DNA target sequence derived from the right part of the HIV1_1.2 target in a palindromic form (Figure 7).
HIVlJ .4 is similar to 5CTG P at positions ±1, ±2, ±3, ±4, ±5 and ±8 and to 10TGGJP at positions ±1 , ±2, ±3, ±4, ±8, ±9 and ±10. It was hypothesized that positions ±6, ±7 and ±11 would have little effect on the binding and cleavage activity. Variants able to cleave 5CTG_P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould el al., J. MoL Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156). Variants able to cleave the 10TGG_P target were obtained by mutagenesis of l-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith el al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target.
Therefore, to check whether combined variants could cleave the HIVlJ .4 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving
5TTCJP were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10GGA_P.
A) Material and Methods a) Construction of target vector The experimental procedure is as described in example 2, with the exception that different oligonucleotides corresponding to the HΪV1__1.4 and HIVl_l .ό targets. The oligonucleotide used for the HIV 1__1 A target was:
S'TGGCATACAAGTTTCCTGGCCCTGGTACCAGGGCCAGGCAATCGTCTGTCA 3' (SEQ ID NO: 20), and
5 'TGGCATACAAGTTTCCTGGCCCTGΛCACCAGGGCCAGGC AATCGTCTGTCA 3 ' (SEQ ID NO: 21 ) for HIV IJ.6 target. b) Construction of combinatorial variants
I-Oel variants cleaving 10TGG_P or 5CTG_P were previously identified, as described in Smith el al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnoυld et al, J. MoI. Biol, 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10TGG_P and 5CTGJ? targets. In order to generate I-O<?I derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the I-CVel coding sequence. For both the 5! and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1 107, Figure 1 1) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and Gail OR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS1 107, Figure 11)
linearized by digestion with DraIII and NgoMiV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, leu2Δ l , his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods EnzymoL, 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast, c) Mating of meganuclease expressing, clones and screening in yeast
Screening was performed as described previously (Arnould et al.} J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETΪX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.O5 0.1 % SDS, 6% dimethyl formamide (DMF), 7 raM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
B) Results l-Crel combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5CTG_P with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10TGG_P on the 1-CVeI scaffold, resulting in a library of complexity 1600. Examples of combinatorial variants are displayed in Table III. This library was transformed into yeast and 3348 clones (2 times the diversity) were screened for cleavage against the HIV1_1.4 and HΪV1_1.6 DNA targets. A total of 32 positive clones were found to cleave HIVl-IA Sequencing of these 32 clones allowed the identification of 25 novel endonuclease variants. One of those variants showed cleavage activity on the HIV1_1.6 DNA target. Examples of positives are shown in Figure 12. The sequence of several of the variants
identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 as well as additional mutations (see examples Table IV). Such variants likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be \-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.


Example 4: Making of ineganucleases cleaving HIV1 1.2 and HIVI l l-Crel variants able to cleave each of the palindromic HIV1_1.2 derived targets (HIV1_1,3 and HIV1_1.4) were identified in example 2 and example 3. Pairs of such variants (one cutting HIV1_1.3 and one cutting HIV1_1.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_1.2 and the non palindromic HIV 1_1 targets. A) Materials and Methods a) Construction of target vector The experimental procedure is as described in example 2, with the exception that an oligonucleotide corresponding to the HIV 1_1.2 target sequence:
5 'TGGCATACAAGTTTGCAGAACTACGTACCAGGGCCAGGCAATCGTCTGTCA 3 ' (SEQ ID NO: 22) or the HIV 1_1 target sequence:
5'TGGCATACAAGTTTGCAGAACTACACACCAGGGCCAGGCAATCGTCTGTCA 3 ' (SEQ ID NO: 23) was used. b) Co-expression of variants
Yeast DNA was extracted from variants cleaving the HIV1_1.4 taiget in the pCLS 1107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_1.3 target in the pCLS0542 expression vector.
Transformanls were selected on synthetic medium lacking leucine and containing
G418. c) Mating of meganucleases coexpressing clones and screening in yeast
Mating was performed using a colony gridder (QpixII, Genetix). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at 300C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS5 6% dimethyl formamide (DMF), 7mM β-mercaptoethanol, 1% agarose, and incubated at 370C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. B) Results
Co-expression of variants cleaving the HIV1_J,4 target (6 variants chosen among those described in Table III and Table IV) and six variants cleaving the HIVlJ .3 target (described in Tables I and II) resulted in cleavage of the HIV1_1.2 target in most of the cases (Figure 13). Nevertheless, only one of these combinations was able to weakly cut the HIV 1_1 natural target that differs from the HIV1_1.2 sequence by 2 bp at positions 1 and 2 (Figure 13). Examples of functional combinations are summarized in Table V and Table VL

I-Crel variants able Io cleave the HΪV3_1.2 and HIV 1_1 target by assembly of variants cleaving the palindromic HIVlJ .3 and HIV1_1.4 target have been previously identified in example 4. However, these variants display stronger activity with the H1V1_1.2 target compared to the HIV1__1 target.
Therefore six variants cleaving HlV 1_ 1.3 were mutagenized, and variants were screened for cleavage activity of HIVlJ.3 and HIV1_1.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIVl J when co-expressed with a variant cleaving HIV1__1.4. According to the structure of the I-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the l-Crel protein (Chevalier et al, Nat. Struct. Biol, 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001 , 29, 3757-3774; Chevalier et al. , J. MoI. Biol., 2003, 329, 253-269). Thus, it is difficult to rationally choose a set of positions to mutagenize, and mutagenesis was performed on the whole protein. Random mutagenesis results in high complexity libraries. Therefore, to limit the complexity of the variant libraries to be tested, only one of the two components of the heterodimcrs cleaving HIVlJ was mutagenized. Thus, in a first step, proteins cleaving HIV1_1.3 were mutagenized and their homodimeric cleavage activity was determined, and in a second step, it was assessed whether they could cleave HIV 1_1 when co-expressed with a protein cleaving HIVlJ .4. A) Material and Methods a) Construction of libraries by random mutagenesis
Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the 1-OeI coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRcv (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25), which are common to the pCLS0542 (Figure 9) and pCLSl 107 (Figure 11) vectors. Approximately 25 ng of the PCR product and 75 ng of vector DNA
(pCLS0542) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, hιs3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the l-Crel variant were generated by in vivo homologous recombination in yeast. b) Mating of meganuclease expressing clones and screening in yeast
Mating was performed as previously described in example 2. Positive resulting clones were verified by sequencing (MlLLEGEN) as described in example 2. c) Variant-target yeast strains, screening and sequencing
The yeast strain FYBL2-7B (MATa, ura3Δ851, (rplΔ63, leu2Δl, lys2Δ202) containing the HIV1__1 target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with one variant, in the kanamycin vector (pCLS1 107), cutting the HIV1_1.4 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2. B) Results Six variants cleaving HIV1__1.3, were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table VIL
2232 transformed clones were screened for cleavage against the HIV1_1.3 and HIV1_1.5 DNA targets. A total of 297 positive clones were found to cleave HIV1_1.3, while only 6 of those cleaved the HIV1_1.5 target. Sequencing of the 93 clones showing the strongest activity allowed the identification of 51 novel endonuclease variants. An example of the identified variants is presented in table VIII and in figure 14.

The 93 clones showing the highest cleavage activity on target HIV1_1.3 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (I- Crel 33T,40K,44R,68Y,70S,77N +132V or KNSTQK/RYSDN +132V, according to the nomenclature of Table I). After mating with this yeast strain, 41 clones were found to cleave the HIV 1_1 target more efficiently than the original variant. Thus, 41 positives contained proteins able to form hetcrodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46), that showed cleavage activity on the HIV 1_J target. An
example of positive clones is shown in Figure 15. Sequencing of these 41 positive clones indicates that 31 distinct variants were identified. Ten of these 31 variants are presented as an example in Table VIII.
Example 5bis: Improvement of meganucleases cleaving HIVI l by a second round of random mutagenesis of proteins cleaving HIV1 1.3 and assembly with proteins cleaving HIV1_1.4
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 5. For this purpose, four variants cleaving HIV1_1,3 were mutagenized, and variants were screened for cleavage activity of HIV1_1.3 and HIV1_1.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIV1_1 when co-expressed with a variant cleaving HIV 1_J .4.
The materials and methods have previously been described in example 5, A) Results Four variants cleaving HIV1_1.3, were pooled, randomly mutagenized and transformed into yeast. The four variants submitted to random mutagenesis correspond to variants described in Table VIII (SEQ ID NO: 26, 27, 28 and 29).
2232 transformed clones were screened for cleavage against the Ϊ-HV1_ 1.3 and HIVI l .5 DNA targets. A total of 79 positive clones were found to cleave HIV1_1.3, while 60 of those cleaved also the HIV1_1.5 target. Sequencing of the 79 clones allowed the identification of 47 novel endonuclease variants. An example of the identified variants is presented in table IX and figure 16.
The 79 clones showing cleaving target HIV1_1.3 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (1-OeI 33T,40K,44R,68Y,70S;77N,132V or KNSTQK/RYSDN +132V, according to the nomenclature of Table I). After mating with this yeast strain, 76 clones were found to cleave the HIV 1_1 target. Thus, 76 positives contained proteins able to form heterodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46) showing cleavage activity on the HIV 1_1 target. An example of positives is shown in Figure 17. Sequencing of these 76 positive clones indicates that 44 distinct variants were identified. Ten of these 44 variants are presented as an example in Table IX.

Example 6: Improvement of meganucleases cleaving HIVl ϊ by site-directed mutagenesis of proteins cleaving HIVl 1.3 and assembly with proteins cleaving HIVl 1.4
The 1-OeI variants cleaving HIV1_1.3 described in Table IX issued from random mutagenesis in examples 5 and 5bis were also mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV 1_1 in combination with a variant cleaving HIVl J.4.
Six amino-acid substitutions have been found in previous studies to enhance the activity of 1-OeI derivatives: these mutations correspond to the replace- ment of Glycine 19 with Serine (Gl 9S), Phenylalanine 54 with Leucine (F54L),
Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine
105 with Alanine (V 105A) and Isoleucine 132 with Valine (1132V). These mutations were introduced into the coding sequence of proteins cleaving HIV1_1.3, and the resulting proteins were tested for their ability to induce cleavage of the HIV 1_1 target, upon co-expression with a variant cleaving HIV IJ .4, as well as for the ability to cleave targets HIV1_1.3 and HIVlJ .5.
A) Materia! and Methods a) Site-directed mutagenesis
Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or Gl 9SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)). The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V 105 A and 1132V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactaglg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
* E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
* V 105AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
* I132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (ρCLS0542, Figure 9), linearized by digestion with Ncol and Eagl. This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, Ieu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods EnzymoL, 2002,
350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 5, d) Sequencing of variants
The experimental procedure is as described in example 2. B) Results
A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and
Isoleucinc 132 with Valine) was constructed on a pool of five variants cleaving
HIV1_1.3 (described in Tabic X). 558 transformed clones were screened for cleavage against the HIV1_1.3 and HΪV1_1,5 DNA targets. A total of 395 positive clones were found to cleave H1V1_J .3, while 349 of those cleaved also the HIV1_1.5 target. An example of positive variants is shown in figure 18
The 558 transformed clones were also mated with a yeast strain that contains (i) the HIV1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_1.4 target (l-Crel 33T,40K,44R,68Y,70S,77N +132V or KNSTQK/RYSDN + 132V, according to the nomenclature of Table I). After mating with this yeast strain, 458 clones were found to cleave the HIV 1_1. Thus, 458 positives contained proteins able to form heterodimers with KNSTQK/RYSDN +132V (SEQ ID NO: 46) showing cleavage activity on the IHVl 1 target. An example of positives is shown in Figure 19.
Sequencing of the 186 clones with the highest cleavage activity on the IHVl- I target allowed the identification of 138 different endonuclease variants.
The sequence of the five best 1-OeI variants cleaving the HIV1_1 target when forming a heterodimer with the KNSTQK/RYSDN +132V variant are listed in Table XI.
 Example 7: Improvement of meganucleases cleaving HIV1_1 by random mutagenesis of proteins cleaving HIV1 1.4 and assembly with proteins cleaving HIV1JL3
Example 7: Improvement of meganucleases cleaving HIV1_1 by random mutagenesis of proteins cleaving HIV1 1.4 and assembly with proteins cleaving HIV1JL3
As a complement to example 4 we also decided to perform random mutagenesis with variants that cleave HIV1_1.4. The mutagenized proteins cleaving HIV 1_ 1.4 were then tested to determine if they could efficiently cleave HIV 1_1 when co-expressed with a protein cleaving HIV1_1.3.
A) Material and Methods a) Construction of libraries by random mutagenesis
Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagltatcagtcggccgc^'; SEQ ID NO: 25). Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLSl 107, Figure 1 1) linearized by digestion with DralW and NgoMW were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the I-Crel variant were generated by in vivo homologous recombination in yeast. b) Variant-target yeast strains, screening and sequencing
The yeast strain FYBL2-7B (MAT a, ura3Δ851, trplΔ63, leu2Δl, lys2Δ202) containing the HIVM target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_1.3 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
B) Results
Six variants cleaving HIV1_1 ,4 were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table XII.
2232 transformed clones were screened for cleavage against the HIVM .4 and HIVM.6 DNA targets. A total of 388 positive clones were found to cleave HIVM .4, while 88 of those also cleaved the HIVM .6 target. Sequencing of the 89 clones showing the strongest activity allowed the identification of 50 novel endonuclease variants. An example of the identified variants is presented in table XIII and in figure 20.

The 89 clones showing the highest cleavage activity on target HIV3_1.4 were then mated with a yeast strain that contains (i) the HIV 1_1 target in a reporter plasmid
(ii) an expression plasmid containing a variant that cleaves the HΪV1_1.3 target (I-
CVeI 30G,38R,44V,ό8E,75N,77R,54L,80K,81T,132V,163R or KGSYRS/VERNR
+54L+80K+81T+132V+163R, according to the nomenclature of Table I). After mating with this yeast strain, 88 clones were found to cleave the HIV 1_1 target. Thus, 46 positives contained proteins able to form heterodimers with KGSYRS/VERNR
+54L+80K+81T+132V+163R (SEQ ID NO: 26), that showed cleavage activity on the
HIV1_1 target. An example of positives is shown in Figure 21. Sequencing of these 88 positive clones indicates that 46 distinct variants were identified. Ten of these 46 variants are presented as an example in Table XIII.
Example 7bis: Improvement of meganucleases cleaving HIVI l by a second round of random mutagenesis of proteins cleaving HIV1 I.4 and assembly with proteins cleaving HIV1_1.3
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 7. For this purpose, four variants cleaving HIV1_1.4 were mutagenized, and variants were screened for cleavage activity of HIV1J .4 and HIV1_1.6 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HiVl^l when co-expressed with a variant cleaving HIV 1_1.3.
The materials and methods have previously been described in example 7. A) Results Four variants cleaving HIV1_1.4, were pooled, randomly mutagenized and transformed into yeast. The four variants submitted to random mutagenesis correspond to variants described in Table XIII (SEQ ID NO: 46, 68, 69 and 71).
2232 transformed clones were screened for cleavage against the HIV1_1.4 and HIV1__1.6 DNA targets. A total of 59 positive clones were found to cleave HIV1_1.4, while 16 of those cleaved also the HIV1_1.6 target. Sequencing of the 49 clones allowed the identification of 35 novel endonuclease variants. An example of the identified variants is presented in table XIV and figure 22.
The 59 clones showing cleaving target HIV1_1.4 were then mated with a yeast strain that contains (i) the HIV1_1 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_J .3 target (1-OeI 30G,38R,44N,68Y,70S,75R,77Y +79N or KGSYRS/NYSRY +79N, according to the nomenclature of Table I). After mating with this yeast strain, 42 clones were found to cleave the HIV 1_1. Thus, 42 positives contained proteins able to form heterodimers with KGSYRS/NYSRY +79N (SEQ ID NO: 28) showing cleavage activity on the HIV 1_1 target. An example of positives is shown in Figure 23. Sequencing of these
42 positive clones indicates that 35 distinct variants were identified. Ten of these 35 variants are presented as an example in Table XIV.

Example 8: Strategy for engineering meganucϊeases cleaving the HIV1 3 target from the HIVl virus The HIV 1_3 target is a 22 bp (non-palindromic) target located in U5 region of the proviral LTRs. Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, the HIV1_3 target is present twice in the HIVl provirus. This target is precisely located at positions 599-620 and 9674-9695 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et ai., J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
The HIV 1_3 sequence (SEQ ID NO: 325) is partly a patchwork of the 10CΛGJP, 10ACA_P; 5CCT_P and 5_GAC_P targets (Figure 24) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould et al, J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006. Thus, HIVlJ could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
The 1 OCAGJ3, 10ACA_P, 5CCT_P and 5_GAC_P target sequences are 24 bp derivatives of C 1221, a palindromic sequence cleaved by Ϊ-Oel (Arnould et al., precited). However, the structure of l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on
binding and cleavage (Chevalier et al., Nat. Struct. Biol, 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269), and in this study, only positions -1 1 to 11 were considered. Consequently, the HIV1__3 series of targets were defined as 22 bp sequences instead of 24 bp. HIV 1_3 differs from C 1221 in the 4 bp central region. According to the structure of the I- Oe I protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-OeI protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269). Thus, the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region. Thus, the TTTA sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_3.2 (SEQ ID NO: 326, Figure 24). Then, two palindromic targets, HIV1_3.3 (SEQ ID NO: 327) and HIV1_3.4 (SEQ ID NO: 328), were derived from HIV1_3.2 (Figure 24). Since HIV1J.3 and HIV1_3.4 arc palindromic, they should be cleaved by homodimeric proteins. Two other pseudo-paiindromic targets were derived from these two, containing the TTTA sequence in -2 to 2 (targets HIV1J.5 (SEQ ID NO: 329) and HIV1_3.6 (SEQ ID NO: 330), figure 24). Thus, proteins able to cleave HIV1_3.3 and HIV1_3.4 targets or, preferentially, the pseudo- palindromic targets as homodimers were first designed (examples 9 and 10) and then co-expressed to obtain heterodimers cleaving HIV 1_3 (example 11). Hetcrodimers cleaving the HIV1_3.2 or HIV 1_3 targets could not be identified. In order to obtain cleavage activity for the HIV1_3 target, a series of variants cleaving HIV1_3.3 and HIV1_3.4 was chosen, and then refined. The chosen variants were subjected to random or site-directed mutagenesis, and used to form novel heterodimers that were screened against the HIV1__3 target (examples 12, 13, 14 and 15). Heterodimers could be identified with an improved cleavage activity for the HIV 1 3 target, Example 9: Identification of meganucleases cleaving HIV1_3.3
This example shows that 1-OeI variants can cut the HIV1_3.3 target sequence derived from the left part of the HΪV1_3.2 target in a palindromic form (Figure 24).
HIV1^3.3 is similar to 10CAG_P at positions ±1 , ±2, ±6, ±8, ±9, and ±10 and to 5CCT_P at positions ±1, ±2, ±3, ±4, ±5 and ±6. It was hypothesized that positions ±7 and ±1 1 would have little effect on the binding and cleavage activity. Variants able to cleave the 1OCAG_P target were obtained by mutagenesis of I-Crel N75 or D75, at positions 28, 30, 32; 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156. Variants able to cleave 5CCTJP were obtained by mutagenesis on 1-OeI N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target. Mutations at positions 24 found in variants cleaving the 5CCT_P target will be lost during the combinatorial process. But it was hypothesized that this will have little impact on the capacity of the combined variants to cleave the HIV1_3.3 target.
Therefore, to check whether combined variants could cleave the HIV1_3.3 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5CCT_P were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OCAGJP.
A) Material and Methods a) Construction of target vector The target was cloned as follows: an oligonucleotide corresponding to the HIV1_3.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTCTCAGACCCTGTACAGGGTCTGAGCAATCGTCTGTCA 3' (SEQ ID NO: 86). The same procedure was followed for cloning the HIV3_1.5 target, using the oligonucleotide: 5' TGGCATACAAGTTTCTCAGACCCTTTTAAGGGTCTGAGCAATCGTCTGTCA 3' (SEQ ID NO: 87). Double-stranded target DNA, generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8). Yeast reporter
vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B {MAT a, ura3Δ851, trplΔ63, leu2Δl, lys2Δ202), resulting in a reporter strain. b) Construction of combinatorial mutants
I-Crel variants cleaving 10CAG_P or 5CCT_P were previously identified, as described in Smith et at. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et aL, J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10AGA_P and 5TACJP targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the I-CVel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5!-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542, Figure 9) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS0542, Figure 9) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, Ieu2Δ l, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et al., J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETIX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was
performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7,0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 370C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants
To recover the variant expression plasmids, yeast DNΛ was extracted using standard protocols and used to transform E, coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA. Alternatively, ORFs were amplified from yeast DNA by PCR (Akada et al,5 Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MILLEGEN SA. B) Results l-Crel combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5CCT JP with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10CAG_P on the l-Crel scaffold, resulting in a library of complexity 1600. Examples of combinatorial variants are displayed in Table XV. This library was transformed into yeast and 3348 clones (2 times the diversity) were screened for cleavage against the HIV1_3.3 and HIV1_3,5 DNA targets. 10 positive clones were found to cleave the HIV1_3.3 target, which after sequencing turned out to correspond to 7 different novel endonuclease variants (Table XVI). These variants showed no cleavage activity of the HIV1_3.5 DNA target. Examples of positives are shown in Figure 25. Some of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 (SEQ ID NO: 92 to 94, Table XVI). Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants
resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.


Example 10; Making of meganucleases cleaving HIV1 3.4
This example shows that l-Crel variants can cleave the HIV1_3.4 DNA target sequence derived from the right part of the HIV1_3.2 target in a palindromic form (Figure 24).
HIV1_3,4 is similar to 5GAC_P at positions ±1, ±2, ±3, ±4, ±5 and ±8 and to 1 OACAJ3 at positions ±1 , ±2, ±3, ±4, ±8, ±9 and ±10. It was hypothesized that positions ±6, ±7 and ±11 would have little effect on the binding and cleavage activity. Variants able to cleave 5GAC P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith el al Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156). Variants able to cleave the 10ACA_P target were obtained by mutagenesis of 1-OeI N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target.
Therefore, to check whether combined variants could cleave the HIV1__3,4 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving
5 GACJP were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10ACA_P.
A) Material and Methods a) Construction of target vector The experimental procedure is as described in example 2, with the exception that different oligonucleotides corresponding to the HΪV1_3.4 and HΪVl_3.ό targets. The oligonucleotide used for the HIV1_3.4 target was: 5'
TGGCATACAAGTTTCCACACTGACGTACGTCAGTGTGGCAATCGTCTGTCA 3' (SEQ ID NO: 95), and 5'
TGGCΛTACAAGTTTCCACACTGACTTTAGTCAGTGTGGCAATCGTCTGTCA 3' (SEQ ID NO: 96) for HIV1_ 3.6 target. b) Construction of combinatorial variants l-Crel variants cleaving 10ACA_P or 5GAC J3 were previously identified, as described in Smith et al Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al., J. MoI. Biol, 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10ACA_P and 5GAC_P targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the 1-OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIl OF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1107, Figure 1 1) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the I-Oel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two
overlapping PCR fragments and 75 ng of vector DNA (pCLSH07, Figure 1 1) linearized by digestion with Dralll and NgoMYV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6Λ (MATα, trpl Δ63, Ieu2Δ l , his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast, c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et a!., J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (Qpixϊl, GENETIX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2), A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2.
B) Results l-Crel combinatorial variants were constructed by associating muta- tions at positions 44, 68, 70, 75 and 77 from proteins cleaving 5GAC_P with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10ACA_P on the l-Crel scaffold, resulting in a library of complexity 2280. Examples of combinatorial variants are displayed in Table XVII. This library was transformed into yeast and 3348 clones (1.5 times the diversity) were screened for cleavage against the HIV1_3.4 and HIV1_3.6 DNA targets. A total of 305 positive clones were found to cleave HIV1_3.4, and two of those variants showed cleavage activity on the HIV1__3.6 target. DNA Sequencing of these 93 strongest clones allowed the identification of 64 novel
cndonuclease variants. Examples of positives are shown in Figure 26. Some variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44> 68, 70, 75, 77 as well as additional mutations (see examples Table XVIII5 SEQ ID NO: 102 to 104), Such variants likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.


Example 11: Making of meganucleases cleaving HIV1 3.2 and HIV1_3 Ϊ-Creϊ variants able to cleave each of the palindromic HIV1_3.2 derived targets (HΪV1_3.3 and HIV1_3.4) were identified in example 9 and example 10. Pairs of such variants (one cutting HIV1_3.3 and one cutting HIV1_3.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_3.2 and the non palindromic HIVlJ targets. A) Materials and Methods a) Construction of target vector The experimental procedure is as described in example 9, with the exception that an oligonucleotide corresponding to the HlVl J3.2 target sequence:
5 ' TGGCATACAAGTTTCTCAGACCCTGTACGTCAGTGTGGCA ATCGTCTGTCA
3'(SEQ ID NO: 317) or the HIV 1_3 target sequence:
5' TGGCATACAAGTTTCTCAGACCCTTTTAGTCAGTGTGGCAATCGTCTGTCA 3' (SEQ ID NO: 318) was used. b) Co-expression of variants
Yeast DNA was extracted from variants cleaving the HIV1J3.4 target in the pCLS 1 107 expression vector using standard protocols and was used to transform E. coll The resulting plasmid DNA was then used to transform yeast strains
expressing a variant cutting the HIV1_3.3 target in the pCLS0542 expression vector. Transformants were selected on synthetic medium lacking leucine and containing G418. c) Mating of meganucleases coexpressing clones and screening in yeast Mating was performed using a colony gridder (QpixII, Genetix).
Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). Λ second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at 3O0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pll 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. B) Results
Co-expression of variants cleaving the HIV1_3.4 target (4 variants) and five variants cleaving the HIV1_3.3 target didn't result in cleavage of the HIV1_3 target, though most of the couples were able to cleave the HIV1__3.2 target. Example 12: Improvement of meganucleases cleaving HIV1 3.3 by random mutagenesis of initial proteins cleaving HIV1 3.3
I-Crel variants able to cleave the HIV1_3.3 target have been previously identified in example 9.
These variants display, however, weak cleavage activity and where therefore mutagenized in order to improve their activity. Four mutants were selected for random mutagenesis and the variants obtained were screened for cleavage activity of HIV1_3.3 and HIV1_3.5 targets. According to the structure of the 1-OeI protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-Crel protein (Chevalier et al, Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al , J.
MoI. Biol., 2003, 329, 253-269). Thus, it is difficult to rationally choose a set of positions to mutagenize, and mutagenesis was performed on the whole protein.
A) Material and Methods a) Construction of libraries by random mutagenesis Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the I-Crel coding sequence using the primers preATGCrcFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRcv (S'-ggctcgaggagctcgtclagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25), which are common to the pCLS0542 (Figure 9) and pCLS1107 (Figure 11) vectors. Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLS0542) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplAόS, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the l-Crel variant were generated by in vivo homologous recombination in yeast. b) Mating of meganuclease expressing clones and screening in yeast
Experiments were performed as previously described in example 9. Positive resulting clones were verified by sequencing (MILLEGEN) as described in example 9.
B) Results
Four variants cleaving HΪV1_3.3, were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in tabic XVI (SEQ ID 88 to 91).
2232 transformed clones were screened for cleavage against the
HIV1_3.3 and HIV1_3.5 DNA targets. A total of 51 positive clones were found to cleave HIV1__3.3, while none of those cleaved the HIV1_3.5 target. Sequencing of the
51 clones allowed the identification of 35 novel endonuclease variants. An example of the identified variants is presented in table XIX and in figure 27.

Example 12bis: Improvement of meganucleases cleaving HIV1 3.3 by a second round of random mutagenesis of proteins cleaving HIV1 3.3
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 7bis. For this purpose, ten variants cleaving HIV1_3.3 were mutagenized, and variants were screened for cleavage activity of HIVl J3.3 and HIV1_3.5 targets. The materials and methods have previously been described in example 1 1. A) Results
Ten variants cleaving HIV1_3.3, were pooled, randomly mutagenized and transformed into yeast. The variants submitted to random mutagenesis correspond to variants described in Table XIX (SEQ ID NO: 105 to 114).
2232 transformed clones were screened for cleavage against the HIV1_3.3 and HIV1_3.5 DNA targets. A total of 262 positive clones were found to cleave HIV1_3.3, while 24 of those cleaved also, though weakly, the HIV1_3.5 target. Sequencing of the 93 clones showing the strongest cleavage activity in the HIV1_3.3 target allowed the identification of 69 novel endonuclease variants. An example of the identified variants is presented in table XX and figure 28.

Example 13: Improvement of meganucleases cleaving HIV1 3 by site-directed mutagenesis of proteins cleaving HIV1 3.3 and assembly with proteins cleaving HIVl 3.4
Five l-Crel variants cleaving HIV1_3.3 after two cycles of random mutagenesis (examples 12 and 12bis) were mutagenized by introducing selected amiπo-acid substitutions in the proteins and screening for more efficient variants cleaving HIV 1_3 in combination with a variant cleaving HIV1J3.4.
Six amino-acid substitutions have been found in previous studies to enhance the activity of I-Oøl derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (G 19 S), Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K); Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (Vl 05A) and Isoleucinc 132 with Valine (Il 32V). These mutations were introduced into the coding sequence of proteins cleaving HIVl 3.3, and the resulting proteins were tested for their ability to induce cleavage of the HIV1__3 target, upon co-expression with a variant cleaving HIV1_3.4, as well as for the ability to cleave targets HIV 1 J3.3 and HIV1_3.5. A) Material and Methods a) Site-directed mutagenesis
Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the G19S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried
out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- Crel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)). The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V105A and I132V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
* E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3 ' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
* V 105AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattUcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
* 1132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and 1132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en cquimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Nco I and Eagl. This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trpl Δ63, ku2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 11.
d) Sequencing of variants
The experimental procedure is as described in example 9. B) Results
A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid
80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and lsoleucine 132 with Valine) was constructed on a pool of five variants cleaving
HIV1_3,3 (described in Table XX, SEQ ID 1 15 to 119). 558 transformed clones were screened for cleavage against the HIV1_3.3 and HIV1_3.5 DNA targets. A total of 376 positive clones were found to cleave HΪV1_3.3, while 54 of those cleaved also the
HIV1__3.5 target. An example of positive variants is shown in figure 29.
The 558 transformed clones were also mated with a yeast strain that contains (i) the HIV 1J3 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_3.4 target (38Y,44Y,68S,70S,75R,77V,43L,81V,105A,107R or KNSYYS/YSSRV
+43L+81V+105A+107R, according to the nomenclature of Table I). After mating with this yeast strain, 386 clones were found to cleave the HIV1_3, Thus, 386 positives contained proteins able to form heterodimers with KNSYYS/YSSRV
+43L+81V+105A+107R (SEQ ID NO: 125) showing cleavage activity on the HIVlJ target. An example of positives is shown in Figure 30.
Sequencing of 93 clones with the high cleavage activity on the HIVl _3 and/or HIV1_3.3 target allowed the identification of 62 different endonuclease variants.
As an example, ten 1-OeI variants cleaving the HIV 1_3 target when forming a heterodimer with the KNSYYS/YSSRV variant are listed in Table XXI.

As a complement to example 5 we also decided to perform random mutagenesis with variants that cleave HIV1__3.4. The mutagenized proteins cleaving HIV1_3.4 were then tested to determine the efficiency of cleavage of the H1V1__3.4 and HI V 1_3.6 targets. A) Material and Methods a) Construction of libraries by random mutagenesis
Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25). Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLS l 107, Figure 11) linearized by digestion with Drάlϊt and NgoMlV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplA63, leu2Al, hi$3A200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the l-Crel variant were generated by in vivo homologous recombination in yeast.
b) Mating of meganuclease expressing clones and screening in yeast
Mating was performed as previously described in example 9. Positive resulting clones were verified by sequencing (MILLEGEN) as described in example 9. B) Results
Five variants cleaving HIV1_3.4 were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table XVIII (SEQ ID NO:97 to 101).
2232 transformed clones were screened for cleavage against the HIV I JJ .4 and HlVl J3.6 DNA targets. A total of 645 positive clones were found to cleave HIV1J3.4, while 156 of those also cleaved the HIV1_3.6 target. Sequencing of the 93 clones showing the strongest activity allowed the identification of 52 novel endonuclease variants. An example of the identified variants is presented in table XXII and in figure 31

Example 14bis: Improvement of meganucleases cleaving HIV1 3.4 by a second round of random mutagenesis of proteins cleaving HIV1 3.3
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 6. For this purpose, ten variants cleaving HIV1_3,4 were mutagenized, and variants were screened for cleavage activity of HIVl _3.4 and HIV1_3.6 targets. The materials and methods have previously been described in example 1 1.
A) Results
Ten variants cleaving HIV1_3.4, were pooled, randomly mutagenized and transformed into yeast. The variants submitted to random mutagenesis correspond to variants described in Table XXII (SEQ ID NO: 136 to 145). 2232 transformed clones were screened for cleavage against the
HIV1_3.4 and HIVlm3.6 targets. A total of 178 positive clones were found to cleave HIV1_3.4, while 63 of those cleaved also the HIV1_3.6 target. Sequencing of the 93 clones showing the strongest cleavage activity in the HIV1_3.4 target allowed the identification of 62 novel endonuclease variants. An example of the identified variants is presented in table XXIII and figure 32.

Example 15: Improvement of meganucleases cleaving HIV1 3 by site-directed mutagenesis of proteins cleaving HIV1JS.4 and assembly with proteins cleaving HIV1_3.3
Four of the improved I-Oel variants cleaving HIV1_3.4 described in Table XXIII and used for a second round of random mutagenesis in example 14bis were also mutagenized by introducing selected amino-acid substitutions in the proteins and screening for variants cleaving HIV1_3.1 in combination with a variant cleaving HIVl J.3.
Six amino-acid substitutions have been found in previous studies to enhance the activity of l-Crel derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (Gl 9S), Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (V105A) and Isoleucine 132 with Valine (Il 32V). These mutations
were introduced into the coding sequence of proteins cleaving HIV1_3.3, and the resulting proteins were tested for their ability to induce cleavage of the HIV IJ target, upon co-expression with a variant cleaving HIV1_3.4. A) Material and Methods Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the l-Crel coding sequence. For both the 5' and 3 ' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G 19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'~gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)). The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V 105 A and 1132V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50); * E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattitgcttaa-3' SEQ ID NO: 51 and 52);
* F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3! SEQ ID NO: 53 and 54);
* V105AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V105AR: 5'- ttcgataattttcagagccaggttlgcctgttt-3' SEQ ID NO: 55 and 56);
* I132VF: 5'-acctgggtggatcaggltgcagctctgaacgat-3 ' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (ρCLS0542, Figure 9), linearized by digestion with Ncol and Eagl. This mix was used to transform the yeast Saccharomyces
cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 1 1. d) Sequencing of variants
The experimental procedure is as described in example 9. B) Results A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and Isoleucine 132 with Valine) was constructed on a pool of four variants cleaving HIV1_3.4 (SEQ ID 136 to 139, Table XXII ). 317 transformed clones were screened for cleavage against the HIV1_3.4 and HIV1_3.6 DNA targets. A total of 311 positive clones were found to cleave HIVI__3.4, while 262 of those cleaved also the HIV1_3.6 target. An example of positive variants is shown in figure 33.
The 317 transformed clones were also mated with a yeast strain that contains (i) the HIV 1_3 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_3.3 target (I-Crel 32K,33A,44K,68E,70S,75N,77R, +132N or KNKAQS/KESNR +132N, according to the nomenclature of Table I). After mating with this yeast strain, 264 clones were found to cleave the HIV 1_3. Thus, 264 positives contained proteins able to form heterodimers with KNKAQS/KESNR +132N (SEQ ID NO: 109, Table XIX) showing cleavage activity on the HIV1_3 target. An example of positive clones is shown in Figure 34.
Sequencing of the 317 clones allowed the identification of 69 different endonuclease variants.
As an example, ten l-Crel variants cleaving the HIV1__3 target when forming a heterodimer with the KNKAQS/KESNR +132N variant are listed in Table XXIV.
 Example 16: Strategy for engineering meganucleases cleaving the HIV1 4 target from the HIVl virus
Example 16: Strategy for engineering meganucleases cleaving the HIV1 4 target from the HIVl virus
The HIV 1_4 target is a 22 bp (non-palindromic) target located in the gag gene of the HIVl provims. This target is precisely located at positions 1629-1650 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al., J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
The HIV 1_4 sequence (SEQ ID NO: 331) is partly a patchwork of the 1 OAGCJP, 1 OTGTJP, 5TCTJP and 5 JTATJP targets (Figure 35) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Λrnould et al., J. MoI. Biol., 2006, 355, 443-458; Smith et al., Nucleic Acids Res., 2006. Thus, HIV1_4 could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
The 10AGC_P, 10TGT_P, 5TCT_P and 5 JTATJP target sequences are 24 bp derivatives of C1221, a palindromic sequence cleaved by 1-OeI (Arnould et al., precited). However, the structure of l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al., Nat. Struct. Biol., 2001 , 8, 312-316; Chevalier and Sloddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol.,
2003, 329, 253-269), and in this study, only positions -11 to 1 1 were considered. Consequently, the HIV 1_4 scries of targets were defined as 22 bp sequences instead of 24 bp. HIV1_4 differs from C1221 in the 4 bp central region. According to the structure of the l-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the l-Crel protein (Chevalier et al., Nat. Struct. Biol, 2001 , 8, 312-316; Chevalier and Stoddard, Nucleic Λcids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269). Thus, the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region. Thus, the GGAC sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_4.2 (SEQ ID NO: 332, Figure 35). Then, two palindromic targets, HIV1_4.3 (SEQ ID NO: 333) and HIV1_4.4 (SEQ ID NO: 334), were derived from HIV1_4.2 (Figure 35). Since HIV1__4.3 and HIV 1 4.4 are palindromic, they should be cleaved by homodimeric proteins. Two other pseudo-palindromic targets were derived from these two, containing the GGAC sequence in -2 to 2 (targets HIV1_4.5 (SEQ ID NO: 335) and HIV1_4.6 (SEQ ID NO: 336), figure 35). Thus, proteins able to cleave HIV1_4.3 and HIV1_4.4 targets or, preferentially, the pseudo- palindromic targets as homodimers were first designed (examplesl7 and 18) and then co-expressed to obtain heterodimers cleaving HIV 1_4 (example 19). Heterodimers cleaving the HIV1_4.2 and IΪIVl_j4 targets could be identified. In order to improve cleavage activity for the HIV1_4 target, a series of variants cleaving HIV1_4'3 and HIV1_4.4 was chosen, and then refined. The chosen variants were subjected to random or site-directed mutagenesis, and used to form novel heterodimers that were screened against the HIV1_4 target (examples 20, 21, 22 and 23). Heterodimers could be identified with an improved cleavage activity for the HIV 1_4 target. Example 17: Identification of meganucleases cleaving HIV1 4.3
This example shows that l-Crel variants can cut the HIV1_4.3 DNA target sequence derived from the left part of the HIV1_4.2 target in a palindromic form (Figure 35). HIV1_4.3 is similar to 10AGC_P at positions ±1, ±2, ±6, ±8, ±9, and ±10 and to 5TCT_P at positions ±1 , ±2, ±3, ±4, ±5 and ±6. It was hypothesized that positions ±7 and ±1 1 would have little effect on the binding and cleavage activity.
Variants able to cleave the 1 OAGCJP target were obtained by mutagenesis of l-Oel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156. Variants able to cleave 5TCT_P were obtained by mutagenesis on l-Crel N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould el al., J. MoI. Biol, 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70, However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target. Mutations at positions 24 found in variants cleaving the 5TCT_P target will be lost during the combinatorial process. But it was hypothesized that this will have little impact on the capacity of the combined variants to cleave the HIV1_4.3 target. Therefore, to check whether combined variants could cleave the HIVl_4-3 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TCT_P were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10AGC_P. A) Material and Methods a) Construction of target vector
The target was cloned as follows: an oligonucleotide corresponding to the HIV1_4,3 target sequence flanked by gateway cloning sequences was ordered from PROLΪGO: 5' TGGCATACAAGTTTCCAGCATTCTGTACAGAATGCTGGCAATCGTCTGTCA 3' (SEQ ID NO: 166). The same procedure was followed for cloning the HIV1_4.5 target, using the oligonucleotide:
5 'TGGCATACAAGTTTCCAGCATTCTGGACAGAATGCTGGCAATCGTCTGTCA 3 ' (SEQ ID NO: 167). Double-stranded target DNA, generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8). Yeast reporter vector was transformed into Sαcchαromyces cerevisiαe strain FYBL2-7B {MAT α, urα3Δ851, trplΔ63, leu2Δl, lys2Δ202), resulting in a reporter strain.
b) Construction of combinatorial mutants l-Crel variants cleaving 10AGC_P or 5TCT_P were previously identified, as described in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al.t J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10AGC_P and 5TCT_P targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the l-Crel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS0542, Figure 9) and primers (assF 5'-ctannnttgacctlt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS0542, Figure 9) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, leu2Δ l , his3Δ200) using a high efficiency LiAc transformation protocol (Gictz and Woods, Methods EnzymoL, 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et al., J. MoI. Biol, 2006, 355, 443-458). Mating was performed using a colony gridder (Qpixlϊ, GENETΪX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and
incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C3 to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS3 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 370C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants To recover the variant expression plasmids, yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA. Alternatively, ORFs were amplified from yeast DNA by PCR (Akada et al., Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MΪLLEGEN SA. B) Results l-Crel combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TCTJP with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OAGCJP on the Ϊ-Crel scaffold, resulting in a library of complexity 3800. Examples of combinatorial variants are displayed in Table XXV. This library was transformed into yeast and 3348 clones were screened for cleavage against the HIV1_4.3 and HIV1_4.5 DNA targets. 7 positive clones were found to cleave the HΪV1_4.3 target, which after sequencing turned out to correspond to 7 different novel endonuclease variants (Table XXVI). Those variants showed no cleavage activity of the HIV1_4.5 DNA target. Examples of positives are shown in Figure 36. Two of the variants obtained display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 (SEQ ID 168 and 174, Table XXVI). Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro- recombination between two original variants during in vivo homologous recombination in yeast.
 Example 18: Making of meganucleases cleaving HIV1 4.4
Example 18: Making of meganucleases cleaving HIV1 4.4
This example shows that 1-OeI variants can cleave the H1V1_4.4 DNA target sequence derived from the right part of the HIV1_4.2 target in a palindromic form (Figure 35). H1V1_4.4 is similar to 5TAT_P at positions ±1, ±2, ±3, ±4, ±5 and
±8 and to 10TGTJP at positions ±1, ±2, ±3, ±4, ±8, ±9 and ±10. It was hypothesized that positions ±6, ±7 and ±11 would have little effect on the binding and cleavage activity. Variants able to cleave 5TAT_P were obtained by mutagenesis of I -Od N75 at positions 44, 68, 70, 75 and 77, as described previously (Amould el al., J. MoL Biol., 2006, 355, 443-458; Smith et al Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156). Variants able to cleave the 10TGT_P target were obtained by mutagenesis of l-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target.
Therefore, to check whether combined variants could cleave the HIV1_4.4 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TAT_P were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OTGTJP. A) Material and Methods a) Construction of target vector The experimental procedure is as described in example 17, with the exception that different oligonucleotides corresponding to the HIV1_4.4 and HIV 1 4.6 targets. The oligonucleotide used for the HIV1_4.4 target was:
5 TGGCATACAAGTTTCTTGTCTT ATGTACATA AGACAAGCAATCGTCTGTCA3 ' (SEQ ID NO: 175), and
5 'TGGCATACAAGTTTCTTGTCTT ATGGACATAΛGACAAGCAATCGTCTGTCA3 ' (SEQ ID NO: 176) for HIV1_4.6 target.
b) Construction of combinatorial variants l-Crel variants cleaving 10TGT_P or 5TAT_P were previously identified, as described in Smith et ah Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould ei a!., J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 1 OTGTJP and 5TΛT_P targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the l-Crel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1 107, Figure 1 1) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS1107, Figure 11) linearized by digestion with DraIII and NgoMlY were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, leu2Δ l , his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et al.> J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETIX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and
incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS5 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2. B) Results
I-C>el combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TATJP with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OTGTJP on the 1-Crel scaffold, resulting in a library of complexity 1406. Examples of combinatorial variants are displayed in Table XXVII. This library was transformed into yeast and 3348 clones (2.3 times the diversity) were screened for cleavage against the UWl_4A and HIV1_4.6 DNA targets. A total of 210 positive clones were found to cleave HIV1_4.4. 40 of these clones were also able to cleave the HIV1_4.6 DNA target. Sequencing of these 93 clones with the strongest activity allowed the identification of 45 novel endonuclease variants. Examples of positives are shown in Figure 37. The sequence of several of the variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 as well as additional mutations (see examples in Table XXVIII, SEQ ID 178 and 184). Such variants likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro -recombination between two original variants during in vivo homologous recombination in yeast.


Example 19: Making of meganucleases cleaving HIV1 4.2 and HIV1 4 l-Crel variants able to cleave each of the palindromic HIV1_4.2 derived targets (HIV 1_4.3 and HIV1_4.4) were identified in example 2 and example 3. Pairs of such variants (one cutting HIV1_4.3 and one cutting HIV1_4.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_4.2 and the non palindromic HI V 1_4 targets. A) Materials and Methods a) Construction of target vector The experimental procedure is as described in example 2, with the exception that an oligonucleotide corresponding to the HIV1_4.2 target sequence: S'TGGCATACAAGTTTCCAGCATTCTGTACATAAGACAAGCAATCGTCTGTC A
3'(SEQ ID NO: 187) or the HIV1^4 target sequence: 5' TGGCATACAAGTTTCCAGCATTCTGGACATAAGACAAGCAATCGTCTGTC A3'
(SEQ ID NO: 188) was used.
b) Co-expression of variants
Yeast DNA was extracted from variants cleaving the HIV1_4.4 target in the pCLS1 107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_4'3 target in the pCLS0542 expression vector. Transformants were selected on synthetic medium lacking leucine and containing G418. c) Mating of meganucleases coexpressing clones and screening in yeast
Mating was performed using a colony gridder (QpixII, Genetix). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at 3O0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. B) Results
Co-expression of variants cleaving the HIV1__4.4 target (10 variants corresponding to those described in Table XXVIII, SEQ ID 177 to 186) and six variants cleaving the HIVl „4.3 target (Table XXVI, SEQ ID 168 and 170 to 174) resulted in cleavage of the HIV 1 4.2 target in most of the cases (Figure 38). Nevertheless, none of these combinations was able to cut the HIV 1_4 natural target that differs from the HIV1_4.2 sequence by 2 bp at positions 1 and 2 (Figure 35). Examples of functional combinations are summarized in Table XXIX.
 Example 20: Improvement of meganucleases cleaving HIV1_4.3 by random mutagenesis of proteins and assembly with proteins cleaving HIV1 4.4
Example 20: Improvement of meganucleases cleaving HIV1_4.3 by random mutagenesis of proteins and assembly with proteins cleaving HIV1 4.4
The assembly of Ϊ-Creϊ variants cleaving the palindromic HIV1_4.3 and HIV1_4.4 target to cleave the HIV1_4.2 and HIV 1_4 have been previously identified in example 4. However, these variants display activity with the HIV1_4.2 target and not with the HIV 1 _4 target.
Therefore seven variants cleaving HIV1_4.3 were mutagenized, and variants were screened for cleavage activity of HTV1_4.3 and HIV1__4.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HTVl _4 when co-expressed with a variant cleaving HΪV1_4.4. According to the structure of the 1-OeI protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the I-Crel protein (Chevalier et at, , Nat. Struct. Biol., 2001 , 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al , J. MoL Biol, 2003, 329, 253-269). Thus, it is difficult to rationally choose a set of positions to mutagenize, and mutagenesis was performed on the whole protein. Random mutagenesis results in high complexity libraries. Therefore, to limit the complexity of the variant libraries to be tested, only one of the two components of the heterodimers cleaving HIV1_4 was mutagenized.
Thus, in a first step, proteins cleaving HIV1_4.3 were mutagenized and their homodimeric cleavage activity was determined, and in a second step, it was
assessed whether they could cleave HIV 1_4 when co-expressed with a protein cleaving HIV 1_4.4.
A) Material and Methods a) Construction of libraries by random mutagenesis Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-S'; SEQ ID NO: 25), which are common to the ρCLS0542 (Figure 9) and pCLSl 107 (Figure 11) vectors. Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLS0542) linearized by digestion with Ncol and Eagϊ were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the 1-OeI variant were generated by in vivo homologous recombination in yeast. b) Mating of meganuclease expressing clones and screening in yeast
Experiments were performed as previously described in example 17. Positive resulting clones were verified by sequencing (MILLEGEN) as described in example 17. c) Variant-target yeast strains, screening and sequencing
The yeast strain FYBL2-7B (MAT a, ura3Δ851, trρ!Δ63, leu2Δl, lys2Δ202) containing the HIV 1_4 target in the yeast reporter vector (pCLS 1055, Figure 8) was transformed with one variant, in the kanamycin vector (pCLS1107), cutting the HIVlm4.4 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 19. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 17.
B) Results
Seven variants cleaving HIV1_4.3, were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table XXVL
2232 transformed clones were screened for cleavage against the HIV1_4.3 and HIVl j4.5 DNA targets. A total of 249 positive clones were found to cleave HIV1_4.3, while 12 of them cleaved also the HIV1_4.5 target. Sequencing of the 93 clones showing the strongest activity allowed the identification of 60 novel endonuciease variants. An example of the identified variants is presented in table XXX and in figure 39.

The 93 clones showing the highest cleavage activity on target HIV1__4.3 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (I- OeI 30H,33M,38A,44N,68Y,70S}75Y,77R or KHSMAS/NYSYR, according to the nomenclature of Table I). After mating with this yeast strain, no clones were found to cleave the HIV 1_4 when forming heterodimers with KHSMAS/NYSYR (SEQ ID NO: 177, TaWe XXIX).
Example 20bis: Improvement of meganucleases cleaving HIV1_4.3 by a second round of random mutagenesis of proteins and assembly with proteins cleaving HIV1_4.4
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 20. For this purpose, four variants cleaving HIVl_4-3 were mutagenized, and variants were screened for cleavage activity of HΪV1__4.3 and HIV1_4.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIVl _4 when co-expressed with a variant cleaving HIV1_4.4. The materials and methods have previously been described in example 20.
A) Results
Six variants cleaving HIV1_4.3, were pooled, randomly mutagenized and transformed into yeast. The six variants submitted to random mutagenesis correspond to variants described in Table XXX (SEQ ID NO: 189 to 194). 2232 transformed clones were screened for cleavage against the
HIV1_4.3 and HIV1_4.5 DNA targets. A total of 377 positive clones were found to cleave HIV1_4.3, while 208 of those cleaved also the HIV1_4.5 target. Sequencing of the 93 clones with the highest activity allowed the identification of 53 novel endonuclease variants. An example of the identified variants is presented in table XXXI and figure 40.
The 93 clones showing cleaving target HIV1_4,3 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (l-Crel 30H,33M,38A,44A,68Y,70S,75Y,77R,155R or KIISMAS/AYSYR +155R, according to the nomenclature of Table I). After mating with this yeast strain, all the 93 clones were found to cleave the HIV 1_4. Thus, 93 positives contained proteins able to form heterodimers with KHSMAS/AYSYR +155R (SEQ ID NO: 199) showing cleavage activity on the HIV1_4 target. An example of positives is shown in Figure 41. Sequencing of these 93 positive clones indicates, as mentioned before, that 53 distinct variants were identified. Ten of these 53 variants are presented as an example in Table XXXI.

I-CVel variants cleaving HIV1_4.3 were also mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HΪV1_4 in combination with a variant cleaving HIVl _4.4.
Six amino-acid substitutions have been found in previous studies to enhance the activity of I-CVel derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (G 19S), Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (V 105A) and Isoleucine 132 with Valine (1132V). These mutations were introduced into the coding sequence of proteins cleaving HIVl _4.3, and the resulting proteins were tested for their ability to induce cleavage of the HIV ϊ_4 target, upon co-expression with a variant cleaving HIV1_4.4, as well as for the ability to cleave targets HIV1_4.3 and HIV1_4.5. A) Material and Methods a) Site-directed mutagenesis
Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out
using a primer with homology to the vector (GaIl OF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 55-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G 19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)).
The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V105Λ and Il 32V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
* E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3 ' SEQ ID NO: 51 and 52);
* F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54); * V105AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V105AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
* I132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Ncol and Eagl. This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa1 irplΔ63, leu2Δl, his3Δ200) using a high effi- ciency LiAc transformation protocol (Gietz and Woods, Methods EnzymoL, 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast, c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 20. d) Sequencing of variants
The experimental procedure is as described in example 17.
B) Results
A library containing a population harboring the six aminoacid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid
80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and Isoleucine 132 with Valine) was constructed on a pool of six variants cleaving
HIV1_4.3 (described in Table XXXI, SEQ ID NO:200 to 205).
558 transformed clones were mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.4 target (30H}33M,38A,44N,68Y,70S,75Y,77R or KHSMAS/NYS YR, according to the nomenclature of Table I). After mating with this yeast strain, 486 clones were found to cleave the HlV 1_4. Thus, 486 positives contained proteins able to form heterodimers with KHSMAS/NYSYR (SEQ ID NO: 177) showing cleavage activity on the HIV 1_4 target. An example of positive variants is shown in figure 42. Sequencing of the 93clones with the highest cleavage activity on the
HIV1_4 target allowed the identification of 34 different endonuclease variants. These 93 clones were also tested for their ability to cleave the HIV1_4.3 and HIV1_4.5 targets. In this case, 71 clones were able to cleave the HIV1_4.3 target, and 69 the HIV1_4.5 target (see Figure 43 for an example). Sequence analysis of these clones showed the presence of 25 different endonuclease variants. Comparison of sequences of the positive clones in all the targets indicated the presence of a total of 40 novel endonuclease variants.
The sequence of ten l-Crel variants cleaving the HIV 1_4 target when forming a heterodimer with the KHSMAS/NYSYR variant are listed in Table XXXII.

Example 22: Improvement of meganucϊeases cleaving HIV1 4.4 by random mutagenesis and assembly with proteins cleaving HIV1 4.3 The assembly of l-Crel variants cleaving the palindromic HIV1_4.3 and HIV1_4.4 target to cleave the HIV1_4.2 and HIV1__4 have been previously described in example 19. However, these variants display activity with the HIV1_4.2 target and not with the HIV 1_4 target.
As a complement to example 4 we also decided to perform random mutagenesis with variants that cleave HIV1_4.4. Therefore ten variants cleaving HIV1_4.3 were mutagenizcd, and variants were screened for cleavage activity of HIV1_4.4 and HIV1_4.6 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIV 1_4 when co-expressed with a variant cleaving HIV1_4.3. A) Material and Methods a) Construction of libraries by random mutagenesis
Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn ". PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5 s- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3 '; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-β'; SEQ ID NO: 25). Approximately 25 ng of the PCR product and 75 ng of vector DNA
(pCLSl 107, Figure 1 1) linearized by digestion with Dralll and NgoMlV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, UψlΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the I-Crel variant were generated by in vivo homologous recombination in yeast, b) Variant-target yeast strains, screening and sequencing
The yeast strain FYBL2-7B (MATa, ura3Δ851, (rplΔ63, leu2Δl, lys2Δ202) containing the HIV Ij4 target in the yeast reporter vector (pCLS 1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_4.3 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 4. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 17. B) Results
Ten variants cleaving HIV1_4.4 were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table XXXII.
2232 transformed clones were screened for cleavage against the HIV1_4.4 and HIV1_4.6 DNA targets. A total of 210 positive clones were found to cleave HIV1_4.4, while 32 of those also cleaved the HIVl_4.ό target. Sequencing of the 93 clones showing the strongest activity allowed the identification of 65 novel endonuclease variants. An example of the identified variants is presented in table XXXIII and in figure 44.

The 93 clones showing the highest cleavage activity on target HIV1_4.4 were then mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.3 target (I- Creϊ 28Q,38R,40K!44K!68T;70G,75N +132V or QNSYRK/KTGNΪ +132V, according to the nomenclature of Table I). After mating with this yeast strain, 90 clones were found to cleave the HIV 1_4 target. Thus, 90 positives contained proteins able to form heterodimers with QNSYRK/KTGNI +132V (SEQ ID NO: 190, Table XXX), that showed cleavage activity on the HIV 1_4 target. An example of positives is shown in Figure 45. Sequencing of these 90 positive clones indicates that 65 distinct variants were identified. Ten of these 65 variants are presented as an example in Table XXXIII.
Example 23: Improvement of meganucleases cleaving HIV1 4 by site-directed mutagenesis of proteins cleaving HIV1 4.4 and assembly with proteins cleaving HIV1 4.3
Four of the 1-OeI variants cleaving HIV1_4.4 described in Table XXXVII were mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV 1_4 in combination with a variant cleaving HIV1_4.3. Six amino-acid substitutions have been found in previous studies to enhance the activity of 1-OeI derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (G 19S)3 Phenylalanine 54 with Leucine (F54L),
Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (V105A) and Isoleucine 132 with Valine (1132V). These mutations were introduced into the coding sequence of proteins cleaving HIV1_4.4, and the resulting proteins were tested for their ability to induce cleavage of the HIV1__4 target, upon co-expression with a variant cleaving HIV 1_4.3 A) Material and Methods a) Site-directed mutagenesis
Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the Gl 9S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' or GaIlOR 5'-acaaccttgattggagacttgacc-3') and a primer specific to the 1-OeI coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'- gatgatgctaccgtcagagtccacaaagccggc-3' (SEQ ID NO: 48)). The resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. Approximately 25ng of each of the two overlapping PCR fragments and 75ng of vector DNA (pCLSl 107, Figure 1 1) linearized by digestion with DraIII and NgoMlY were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, (rplΔ63, leiύΔl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombi- nation in yeast.
The same strategy is used with the following pair of oligonucleotides to create other libraries containing the F54L, E80K, F87L, V105A and Il 32V substitutions, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50);
* E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52);
* F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagttgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
* Vl 05AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and Vl 05AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56); * I132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgtlcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58). c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 22. d) Sequencing of variants The experimental procedure is as described in example 17.
B) Results
A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and Isoleucine 132 with Valine) was constructed on a pool of four variants cleaving HIV1_4.4 (see Table XXXIII, SEQ ID NO: 199, 177, 221 and 228).
558 transformed clones were mated with a yeast strain that contains (i) the HIV 1_4 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_4.3 target (28Q,38R,40K,44K,68T,70G,75N or QNSYRK/KTGNI, according to the nomenclature of Table I). After mating with this yeast strain, 16 clones were found to cleave the HIV 1_4. Thus, 16 positives contained proteins able to form heterodimers with QNSYRK7KTGN1 +132V (SEQ ID NO: 190, Table XXX) showing cleavage activity on the HIV 1_4 target. An example of positive variants is shown in figure 46. Sequencing of these positive clones allowed the identification of 10 different endonuclease variants. The clones cleaving the HIV 1_4 target were also tested for their ability to cleave the HIV1_4.4 and HIV1_4.6 targets (see Figure 47 for an example). In this case, 15 of the clones were able to cleave the HIV1_4.3 and the HIV1_4.5 targets. Sequence analysis of these clones showed the presence of 10 different endonuclease variants. Comparison of sequences of the positive clones in all the targets indicated the presence of a total of 1 1 novel endonuclease variants.
The sequence of ten l-Crel variants cleaving the HIV 1_4 target when forming a heterodimer with the KHSMAS/NYSYR variant arc listed in Table XXXIV.

Example 24: Strategy for engineering meganucleases cleaving the HIV1_5 target from the HIVl virus
The HIV 1_5 target is a 22 bp (non-palindromic) target located in the pol gene of the HIVl provirus. This target is precisely located at positions 2317-2338 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al., J. Virol, 1986, 59, 284-291), a subtype B infectious molecular clone.
The HIV1_5 sequence (SEQ ID NO: 337) is partly a patchwork of the 1 OTCTJP, 10CTG_P, 5TAG_P and 5 CCTJ targets (Figure 48) which are cleaved by previously identified meganucleases, obtained as described in International PCT Applications WO 2006/097784 and WO 2006/097853; Arnould ct al., J. MoL Biol, 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006. Thus, HIV1_5 could be cleaved by combinatorial variants resulting from these previously identified meganucleases.
The 10TCT_P, 10CTG_P, 5TAG_P and 5_CCTJP target sequences are 24 bp derivatives of C 1221 , a palindromic sequence cleaved by l-Crel (Arnould et al, precited). However, the structure of l-Crel bound to its DNA target suggests that the two external base pairs of these targets (positions -12 and 12) have no impact on binding and cleavage (Chevalier et al, Nat. Struct. Biol, 2001, 8, 312-316; Chevalier
and Stoddard, Nucleic Λcids Res., 2001 , 29, 3757-3774; Chevalier el a!., J. MoL Biol., 2003, 329, 253-269), and in this study, only positions -11 to 1 1 were considered. Consequently, the HlV 1__5 series of targets were defined as 22 bp sequences instead of 24 bp. HΪV1_5 differs from C1221 in the 4 bp central region. According to the structure of the I-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the l-Crel protein (Chevalier et al., Nat. Struct. Biol., 2001, 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al., J. MoI. Biol., 2003, 329, 253-269). Thus, the bases at these positions should not impact the binding efficiency. However, they could affect cleavage, which results from two nicks at the edge of this region. Thus, the ATAC sequence in -2 to 2 was first substituted with the GTAC sequence from C 1221, resulting in target HIV1_5.2 (SEQ ID NO: 338, Figure 48). Then, two palindromic targets, HIV1_5.3 (SEQ ID NO: 339) and HIV1_5.4 (SEQ ID NO: 340), were derived from HIV1^5.2 (Figure 48). Since HIV1_5.3 and HIV1_5.4 are palindromic, they should be cleaved by homodimeric proteins. Two other quasi-palindromic targets were derived from these two, containing the ATAC sequence in -2 to 2 (targets HΪV1_5.5 (SEQ ID NO: 341) and HIV1_5.6 (SEQ ID NO: 342), figure 48). Thus, proteins able to cleave HIVlm5.3 and HIV1_5.4 targets or, preferentially, the quasi- palindromic targets as homodimers were first designed (examples 25 and 26) and then co-expressed to obtain heterodimers cleaving HIV 1_5 (example 27). Heterodimers cleaving the HIV1_5.2 and HIV 1_5 targets could be identified. In order to improve cleavage activity for the HIV1_5 target, a series of variants cleaving HIV1_5.3 and HIV1_5.4 was chosen, and then refined. The chosen variants were subjected to random or site-directed mutagenesis, and used to form novel heterodimers that were screened against the HIV1__5 target (examples 28, 29, 30 and 31). Heterodimers could be identified with an improved cleavage activity for the HIV 1_5 target. Example 25: Identification of meganucleases cleaving HIV1 5.3 This example shows that l-Crel variants can cut the HIV 1__5.3 DNA target sequence derived from the left part of the HIV1_5.2 target in a palindromic form (Figure 48). HIV1__5.3 is similar to 10TCT_P at positions ±1, ±2, ±6, ±8, ±9, and ±10 and to 5TAG_P at positions ±1, ±2, ±3, ±4, ±5 and ±6. It was hypothesized that positions ±7 and ±11 would have little effect on the binding and cleavage activity. Variants able to
cleave the 1 OTCTJP target were obtained by mutagenesis of l-Crel N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al Nucleic Λcids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156. Variants able to cleave 5TAG_JP were obtained by mutagenesis on l-Crel N75 at positions 24, 44, 68, 70, 75 and 77 as described in Arnould et al, J. MoI. Biol., 2006, 355, 443-458; Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional subdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target. Mutations at positions 24 found in variants cleaving the 5TAG_P target will be lost during the combinatorial process. But it was hypothesized that this will have little impact on the capacity of the combined variants to cleave the HΪV1_5.3 target. Therefore, to check whether combined variants could cleave the IIΪV1_5.3 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5TAG_P were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10TCT_P. A) Material and Methods a) Construction of target vector
The target was cloned as follows: an oligonucleotide corresponding to the HIV1JJ.3 target sequence flanked by gateway cloning sequences was ordered from PROLIGO: 5' TGGCATACAAGTTTGCTCTATTAGGTACCTAATAGAGCCAATCGTCTGTCA 3' (SEQ ID NO: 52). The same procedure was followed for cloning the HIV1_5.5 target, using the oligonucleotide: 5'
TGGCATACAAGTTTGCTCTATTAGATACCTAATAGAGCCAATCGTCTGTCA 3' (SEQ ID NO: 53). Double-stranded target DNA, generated by PCR amplification of the single stranded oligonucleotide, was cloned using the Gateway protocol (INVITROGEN) into the yeast reporter vector (pCLS1055, Figure 8). Yeast reporter vector was transformed into Saccharomyces cerevisiae strain FYBL2-7B (MAT a, ura3Δ851, trplΔ63, leu2Δl, tys2Δ202), resulting in a reporter strain.
b) Construction of combinatorial mutants
I-Crel variants cleaving 10TCT_P or 5TAG_P were previously identified, as described in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al, J. MoL Biol., 2006, 355, 443-458; International PCT Applications WO 2006/097784 and WO 2006/097853, respectively for the 10TCT_P and 5TAGJ> targets. In order to generate l-Creϊ derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the 1-OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3J (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) specific to the vector (pCLS05425 Figure 9) and primers (assF 5'-ctannnttgaccltt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19)), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS0542, Figure 9) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trpl Δ63, leu2Δ l , his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et al., J. MoI. Biol, 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETIX). Variants were gridded on nylon fillers covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and
incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software, d) Sequencing of variants To recover the variant expression plasmids, yeast DNA was extracted using standard protocols and used to transform E. coli. Sequencing of variant ORFs was then performed on the plasmids by MILLEGEN SA. Alternatively, ORFs were amplified from yeast DNA by PCR (Akada et al., Biotechniques, 2000, 28, 668-670), and sequencing was performed directly on the PCR product by MILLEGEN SA. B) Results l-Crel combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5 TAGJP with the 28, 30, 32, 33} 38 and 40 mutations from proteins cleaving 1 OTCTJP on the l-Crel scaffold, resulting in a library of complexity 3920. Examples of combinatorial variants are displayed in Table XXXV, none of the variants tested from the combinatorial library produced a positive result. This library was transformed into yeast and 3348 clones (1.7 times the diversity) were screened for cleavage against the HIV1_5,3 and HIV1J5.5 DNA targets. Two positive clones were found (though having weak cleavage activity), which after sequencing turned out to correspond to 2 different novel endonuclease variants (Table XXXVI). These two positives are shown in Figure 49. These two variants display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77. Such combinations likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be l-Crel combined variants resulting from micro-recombination between two original variants during in vivo homologous recombination in yeast.

Example 26: Making of meganucleases cleaving HIV1 5.4
This example shows thai I-O<?I variants can cleave the HIV1_5.4 DNA target sequence derived from the right part of the HIV1_5.2 target in a palindromic form (Figure 4).
HIV1_5.4 is similar to 5CCTJP at positions ±1, ±2, ±3, ±4, ±5 and ±8 and to 10CTG_P at positions ±1, ±2, ±3, ±4, ±8, ±9 and ±10. It was hypothesized that positions ±6, ±7 and ±1 1 would have little effect on the binding and cleavage activity. Variants able to cleave 5CCT_P were obtained by mutagenesis of l-Crel N75 at positions 44, 68, 70, 75 and 77, as described previously (Arnould el al, J. MoI. Biol., 2006, 355, 443-458; Smith et al Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2006/097784, WO 2006/097853, WO 2007/060495 and WO 2007/049156). Variants able to cleave the 1 OTGGJ* target were obtained by mutagenesis of 1-OeI N75 or D75, at positions 28, 30, 32, 33, 38, 40 and 70, as described previously in Smith et al. Nucleic Acids Res., 2006, 34, el49; International PCT Applications WO 2007/060495 and WO 2007/049156.
Both sets of proteins are mutated at position 70. However, the existence of two separable functional sυbdomains was hypothesized. This implies that this position has little impact on the specificity at bases 10 to 8 of the target. Therefore, to check whether combined variants could cleave the
HIV1_5.4 target, mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5CCT JP were combined with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 10CTG_P. A) Material and Methods a) Construction of target vector
The experimental procedure is as described in example 2, with the exception that different oligonucleotides corresponding to the HIV 1_5.4 and H1V1_5.6 targets. The oligonucleotide used for the HIV1_5.4 target was: 5' TGGCATACAAGTTTATCTGCTCCTGTACAGGAGCAGATCAATCGTCTGTCA 3' (SEQ ID NO: 243), and 5'
TGGCATACAAGTTTATCTGCTCCTATACAGGAGCAGATCAATCGTCTGTCA 3' (SEQ ID NO: 244) for HIV1_5.6 target, b) Construction of combinatorial variants
I-CVel variants cleaving 1 OCTGJ5 or 5CCTJP were previously identified, as described in Smith et al Nucleic Acids Res., 2006, 34, el 49; International PCT Applications WO 2007/060495 and WO 2007/049156, and Arnould et al, J. MoI. Biol., 2006, 355, 443-458; International PCT Applications WO
2006/097784 and WO 2006/097853, respectively for the 10CTG_P and 5CCT _P targets. In order to generate l-Crel derived coding sequences containing mutations from both series, separate overlapping PCR reactions were carried out that amplify the 5' end (aa positions 1-43) or the 3' end (positions 39-167) of the 1-OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using primers (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIlOR 5'- acaaccttgattggagacttgacc-3' (SEQ ID NO: 17)) specific to the vector (pCLS1107, Figure 11) and primers (assF 5'-ctannnttgaccttt-3' (SEQ ID NO: 18) or assR 5'- aaaggtcaannntag-3'(SEQ ID NO: 19), where nnn codes for residue 40, specific to the l-Crel coding sequence for amino acids 39-43. The PCR fragments resulting from the amplification reaction realized with the same primers and with the same coding sequence for residue 40 were pooled. Then, each pool of PCR fragments resulting from the reaction with primers GaIlOF and assR or assF and GaIlOR was mixed in an equimolar ratio. Finally, approximately 25 ng of each final pool of the two overlapping PCR fragments and 75 ng of vector DNA (pCLS1107, Figure U) linearized by digestion with DraIII and NgoMV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATα, trplΔ63, leu2Δ l, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). An intact coding sequence containing both groups of mutations is generated by in vivo homologous recombination in yeast, c) Mating of meganuclease expressing clones and screening in yeast
Screening was performed as described previously (Arnould et aL, J. MoI. Biol., 2006, 355, 443-458). Mating was performed using a colony gridder (QpixII, GENETIX). Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of the reporter- harboring yeast strain. Membranes were placed on solid agar YPD rich medium, and incubated at 30 0C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking tryptophan, adding G418, with galactose (2 %) as a carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pH 7.0, 0.1 % SDS,
6% dimethyl formamide (DMF)3 7 mM β-mercaptoethanol, 1% agarose, and incubated at 37°C, to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 2. B) Results l-Crel combinatorial variants were constructed by associating mutations at positions 44, 68, 70, 75 and 77 from proteins cleaving 5 CCTJP with the 28, 30, 32, 33, 38 and 40 mutations from proteins cleaving 1 OCTGJ? on the l-Crel scaffold, resulting in a library of complexity 1600. Examples of combinatorial variants are displayed in Table XXXXI. This library was transformed into yeast and 3348 clones (2 times the diversity) were screened for cleavage against the HIV1__5.4 and HIV1_5,6 DNA targets. A total of 10 positive clones were found to cleave HIV1_5.4. Sequencing of these 10 clones allowed the identification of 9 novel endonuclease variants, which are represented in Table XXXVII. Examples of positives arc shown in Figure 50. The sequence of several of the variants identified display non parental combinations at positions 28, 30, 32, 33, 38, 40 or 44, 68, 70, 75, 77 as well as additional mutations (Table XXXVIII, SEQ ID 246, 247, 251, 252 and 253). Such variants likely result from PCR artifacts during the combinatorial process. Alternatively, the variants may be 1-OeI combined variants resulting from micro- recombination between two original variants during in vivo homologous recombination in yeast.

1-OeI variants able to cleave each of the palindromic HΪV1_5.2 derived targets (HIV 1_5.3 and HIV1_5.4) were identified in example 25 and example 26. Pairs of such variants (one cutting HIV1_5.3 and one cutting HIV1_5.4) were co- expressed in yeast. Upon co-expression, there should be three active molecular species, two homodimers, and one heterodimer. It was assayed whether the heterodimers that should be formed, cut the HIV1_5.2 and the non palindromic HIV 1_5 targets. A) Materials and Methods a) Construction of target vector
The experimental procedure is as described in example 2, with the exception that an oligonucleotide corresponding to the HΪV1_5.2 target sequence:
STGGCATΛCΛΛGTTTGCTCTATTAGGTACAGGAGCΛGΛTCAATCGTCTGTC
A3' (SEQ ID NO: 254) or the HIV1_5 target sequence: 5TGGCATACAAGTTTGCTCTATTAGATACAGGAGCAGATCAATCGTCTGTC
A 3'
(SEQ ID NO: 255) was used. b) Co-expression of variants
Yeast DNA was extracted from variants cleaving the HIV1_5.4 target in the pCLS 1107 expression vector using standard protocols and was used to transform E. coli. The resulting plasmid DNA was then used to transform yeast strains expressing a variant cutting the HIV1_5.3 target in the pCLS0542 expression vector.
Transformants were selected on synthetic medium lacking leucine and containing
G418. c) Mating of meganucleases coexpressing clones and screening in yeast
Mating was performed using a colony gridder (Qpixϊϊ, Genetix).
Variants were gridded on nylon filters covering YPD plates, using a low gridding density (4-6 spots/cm2). A second gridding process was performed on the same filters to spot a second layer consisting of different reporter-harboring yeast strains for each target. Membranes were placed on solid agar YPD rich medium, and incubated at
300C for one night, to allow mating. Next, filters were transferred to synthetic medium, lacking leucine and tryptophan, adding G418, with galactose (2 %) as a
carbon source, and incubated for five days at 37 0C, to select for diploids carrying the expression and target vectors. After 5 days, filters were placed on solid agarose medium with 0.02 % X-GaI in 0.5 M sodium phosphate buffer, pϊl 7.0, 0.1 % SDS, 6% dimethyl formamide (DMF), 7mM β-mercaptoethanol, 1% agarose, and incubated at 370C5 to monitor β-galactosidase activity. Results were analyzed by scanning and quantification was performed using appropriate software. B) Results
Co-expression of variants cleaving the IIIV1J.4 target (9 variants chosen among those described in Table XXXVIII) and the two variants cleaving the HIV1_5.3 target (described in Table XXXVI) resulted in cleavage of the HIV1_5.2 target in one of the cases (Figure 51). Nevertheless, this combination was not able to cut the HIV1_5 natural target, that differs from the HIV1_5.2 sequence by 2 bp at positions 1 and 2 (Figure 48). The functional combination cleaving the HIV1_5.2 target correspond to mutants KNSCYS/AYQNI (SEQ ID 241, cleaving HIV1_5.3) and KTSGQS/KYSDR +151 A (SEQ ID 252, cleaving HIVlJ .4)
Example 28: Improvement of meganucleases cleaving HIVl 5.3 by random mutagenesis and assembly with proteins cleaving HIVl 5.4 l-Crel variants able to cleave the HIVl J.3 have been identified in example 25. Since these two variants show a weak activity, and only one of them is able to cleave the HIVl J.2 target when assembled with a meganuclease cleaving the HIV IJ.4, these two variants were mutagenized, and the clones generated were screened for cleavage activity of HIVl J.3 and HIVl J.5 targets. Additionally the mutants with the strongest activity were screened for cleavage activity of HIVlJ when co-expressed with a variant cleaving HIVl J.4. According to the structure of the l-Crel protein bound to its target, there is no contact between the 4 central base pairs (positions -2 to 2) and the 1-CVeI protein (Chevalier el ai, Nat. Struct. Biol, 2001 , 8, 312-316; Chevalier and Stoddard, Nucleic Acids Res., 2001, 29, 3757-3774; Chevalier et al , J. MoI. Biol., 2003, 329, 253-269). Thus, it is difficult to rationally choose a set of positions to mutagenize, and mutagenesis was performed on the whole protein. Random mutagenesis results in high complexity libraries. Therefore, to limit the complexity of the variant libraries to be tested, only one of the two components of the heterodimers cleaving HIVlJ was mutagenized.
Thus, in a first step, proteins cleaving HIV1_5.3 were mutagenized and their homodimeric cleavage activity was determined, and in a second step, it was assessed whether they could cleave HIV 1_5 when co-expressed with a protein cleaving HIV 1_5.4. A) Material and Methods a) Construction of libraries by random mutagenesis
Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn2+. PCR reactions were carried out that amplify the 1-OeI coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (S'-ggctcgaggagctcgtctagaggatcgctcgagttatcagtcggccgc-B'; SEQ ID NO: 25)} which are common to the pCLS0542 (Figure 9) and pCLSl 107 (Figure 1 1) vectors. Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLS0542) linearized by digestion with Ncol and Eagl were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the l-Crel variant were generated by in vivo homologous recombination in yeast. b) Mating of meaanuclease expressing clones and screening in yeast
Experiment were performed as previously described in example 25. Positive resulting clones were verified by sequencing (MlLLEGEN) as described in example 25. c) Variant-target yeast strains, screening and sequencing The yeast strain FYBL2-7B (MATa, ura3Δ851, trp!Δ63, leu2Δl, lys2Δ202) containing the HIV 1_5 target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with one variant, in the kanamycin vector (pCLS1 107), cutting the HIV1_5.4 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 27. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 25.
B) Results
Two variants cleaving HIV1_5.3, were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in table XXXVI. 2232 transformed clones were screened for cleavage against the
HIV1_5.3 and HIV1_5.5 DNA targets. A total of 20 positive clones were found to cleave HIV1_5.3, while none of those cleaved the HIV1_5.5 target. Sequencing of the 20 clones allowed the identification of 13 novel cndonuclcasc variants. An example of these variants is presented in table XXXIX and in figure 52.

Example 28bis: Improvement of meganucleases cleaving HTV1 5.3 by a second round of random mutagenesis and assembly with proteins cleaving HIV1_5.4
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 28. For this purpose, ten variants cleaving HIV1_5.3 were mutagenized, and variants were screened for cleavage activity of HIV1_5.3 and HΪV1_5.5 targets.
Additionally, the mutants with the strongest activity were screened for cleavage activity of HIV 1_5 when co-expressed with a variant cleaving HIV1_5.4.
The materials and methods have previously been described in example 28. A) Results Ten variants cleaving HIV1_5.3, were pooled, randomly mutagcnized and transformed into yeast. The variants submitted to random mutagenesis correspond to variants described in Table XXXIX (SEQ ID NO: 256 to 265).
2232 transformed clones were screened for cleavage against the HIV1__5.3 and HIV1_5.5 DNA targets. A total of 80 positive clones were found to cleave HIV1_5.3; while 25 of those cleaved also the HIV1_5.5 target. Sequencing of the 80 clones allowed the identification of 39 novel endonuclease variants. An example of the identified variants is presented in table XXXX and figure 53.
The 80 clones showing cleavage activity on target HIV1_5.3 were then mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.4 target (I- Crel 30S533N,44K,68Y;70S,77R +103T or KSSNQS/KYSDR +103T, according to the nomenclature of Table I). After mating with this yeast strain, 4 clones were found to cleave the HIV 1_5. Thus, 4 positives contained proteins able to form heterodimers with KSSNQS/KYSDR +103T (SEQ ID NO: 276) showing cleavage activity on the HIV 1_5 target. An example of positives is shown in Figure 54. These 4 variants are presented as an example in Table XXXX (SEQ ID 266 to 269).

Three of the I-Crel variants cleaving HIV1_5.3 described in Table XLwcre mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV 1_5 in combination with a variant cleaving HIV1_5.4.
Six amino-acid substitutions have been found in previous studies to enhance the activity of l-Crel derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (Gl 9S), Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (V 105A) and Isoleucine 132 with Valine (1132V). These mutations were introduced into the coding sequence of proteins cleaving HIV1_5.3, and the resulting proteins were tested for their ability to induce cleavage of the HIV 1_5 target, upon co-expression with a variant cleaving HΪV1_5.4, as well as for the ability to cleave targets HIV1_5.3 and HIV1_5.5. A) Material and Methods a) Site-directed mutagenesis
Site- directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the G19S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- Crel coding sequence. For both the 5' and 3' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3' (SEQ ID NO: 16) or GaIl OR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctttgtggactctgacggtagcatcatc-3' (SEQ ID NO: 47) or G19SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3 '(SEQ ID NO: 48)). The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V105A and I132V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgclgggt-3' (SEQ ID NO: 49 and 50);
* E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcUgattttgcttaa-3' SEQ ID NO: 51 and 52); * F87LF: 5'-aagccgctgcacaacctgctgactcaactgcag-3' and F87LR: 5'- ctgcagtlgagtcagcaggttgtgcagcggctt-3 ' SEQ ID NO: 53 and 54);
* Vl 05AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa~3' and V 105AR: 5'- tlcgataattttcagagccaggtttgcctgltl-3' SEQ ID NO: 55 and 56);
* I132VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and I132VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Ncόi and Eagl. This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATcc, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymoi., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast. c) Mating of meganuclease expressing clones and screening in yeast
The experimental procedure is as described in example 28. d) Sequencing of variants
The experimental procedure is as described in example 25. B) Results A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and Isoleucine 132 with Valine) was constructed on a pool of three variants cleaving HIV1J.3 (SEQ ID NO: 266, 269 and 270; described in Table XL). 558 transformed clones were screened for cleavage against the HIV1_5.3 and HIV1__5.5 DNA targets. A total of 450 positive clones were found to cleave HIV1__5.3, while 435 of those
cleaved also the HIV1_5.5 target. An example of positive variants is shown in figure 55.
The 558 transformed clones were also mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.4 target (Ϊ-Crel
30S,33N,44K,68Y,70S,77R +103T or KSSNQS/KYSDR +103T, according to the nomenclature of Table I). After mating with this yeast strain, 444 clones were found to cleave the HΪV1__5. Thus, 444 positives contained proteins able to form heterodimers with KSSNQS/KYSDR +103T (SEQ ID NO: 276) showing cleavage activity on the HIVl _5 target. An example of positive clones is shown in Figure 56.
Sequencing of the 93 clones with the highest cleavage activity on the HIV 1_5 target allowed the identification of 50 different endonuclease variants.
The sequence of ten l-Crel variants cleaving the HIV 1_5 target when forming a heterodimer with the KSSNQS/KYSDR +103T variant are listed in Table XLI.

Example 30: Improvement of meganucleases cleaving HIV1 5.4 by random mutagenesis and assembly with proteins cleaving HIV1 5.3
As a complement to example 29 we also decided to perform random mutagenesis with variants that cleave HIV1_5.4, The variants generated were screened for their cleavage activity on targets HIV1_5.4 and IIIV1_5.6; and the
mutagenized proteins cleaving 11IV1_5.4 were then tested to determine if they could efficiently cleave HIV1 5 when co-expressed with a protein cleaving IIIV1 5.3.
A) Material and Methods a) Construction of libraries by random mutagenesis Random mutagenesis was performed on a pool of chosen variants, by PCR using Mn +. PCR reactions were carried out that amplify the l-Crel coding sequence using the primers preATGCreFor (5'- gcataaattactatacttctatagacacgcaaacacaaatacacagcggccttgccacc-3'; SEQ ID NO: 24) and ICrelpostRev (5'-ggctcgaggagctcgtclagaggatcgctcgagtlalcagtcggccgc-35; SEQ ID NO: 25). Approximately 25 ng of the PCR product and 75 ng of vector DNA (pCLS U07, Figure 1 1) linearized by digestion with Dralll and NgoMlV were used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa1 trplΔόS, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Expression plasmids containing an intact coding sequence for the I-Crel variant were generated by in vivo homologous recombination in yeast. b) Variant-target yeast strains, screening and sequencing
The yeast strain FYBL2-7B (MAT a, ura3Δ851, trplΔ63, leiύΔl, lys2Δ202) containing the HIV IJ target in the yeast reporter vector (pCLS1055, Figure 8) was transformed with variants, in the leucine vector (pCLS0542), cutting the HIV1_5.3 target, using a high efficiency LiAc transformation protocol. Variant-target yeast strains were used as target strains for mating assays as described in example 27. Positives resulting clones were verified by sequencing (MILLEGEN) as described in example 25. B) Results
Nine variants cleaving HIV1_5.4 were pooled, randomly mutagenized and transformed into yeast. The sequences of the variants subjected to random mutagenesis are described in Table XXXVIII.
2232 transformed clones were screened for cleavage against the HIV1_5.4 and IΪIV1__5.6 DNA targets. A total of 53 positive clones were found to cleave HIV1_5.4, while 6 of those also cleaved the HIV1_5,6 target. Sequencing of the 53 clones showing the strongest activity allowed the identification of 42 novel
endonuclease variants. An example of the identified variants is presented in Table XLIIand in figure 57.

The 53 positive clones showing the highest cleavage activity on target HIV1_5.4 were then mated with a yeast strain that contains (i) the HIV 1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.3 target (l-Creϊ 33C,38Y,44A,68Y;70Q,75N +89A or KNSCYS/AYQNI +89A, according to the nomenclature of Table I; SEQ ID 256). After mating with this yeast strain, no clones were found to cleave the HIV 1_5 target. Example 30bis: Improvement of meganucleases cleaving HIV1_5 by a second round of random mutagenesis of proteins cleaving H1V1 5.4 and assembly with proteins cleaving HIV1_5.3
In order to further improve the activity of the obtained meganucleases, a second round of random mutagenesis was carried out following the same rationale of example 30. For this purpose, six variants cleaving HIV1_5.4 were mutagenized, and variants were screened for cleavage activity of HIV1_5.4 and HIV1_5.6 targets. Additionally the mutants were screened for cleavage activity of HIV 1_5 when co- expressed with a variant cleaving HIV1_5.3.
The materials and methods have previously been described in example 30.
A) Results
Six variants cleaving HIV1_5.4, were pooled, randomly raυtagenizcd and transformed into yeast. The six variants submitted to random mutagenesis correspond to variants described in Table XLII (SEQ ID NO: 276 and 288 to 292). 2232 transformed clones were screened for cleavage against the
HΪV1_5.4 and HIV1_5.6 DNA targets. A total of 21 positive clones were found to cleave HIV1_5.4, while 9 of those cleaved also the HIV1_5.6 target. Sequencing of the 21 clones allowed the identification of 16 novel endonuclease variants. An example of the identified variants is presented in Table XLIII and figure 58. The 21 positive clones showing cleavage activity on target HΪV1_5.4 were then mated with a yeast strain that contains (i) the HIV IJ target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HΪV1J5.3 target (l-Crel 33C,38Y,44A,68Y,70Q,75N +89A or KNSCYS/AYQNI +89A3 according to the nomenclature of Table I; SEQ ID 256). After mating with this yeast strain, no clones were found to cleave the HIV 1_5 target.

Example 31: Improvement of meganucleases cleaving HIV1 5 by site-directed mutagenesis of proteins cleaving HIV1_5.4 and assembly with proteins cleaving HIV1_5.3
Two of the \-Crel variants cleaving HIVI_5.4 described in Table XLΪII were mutagenized by introducing selected amino-acid substitutions in the proteins and screening for more efficient variants cleaving HIV1_5.4 and HIV1_5,6,
as well as for cleavage of the IIIV1_5 target when in combination with a variant cleaving HIV 1 5.3.
Six amino-acid substitutions have been found in previous studies to enhance the activity of l-Oel derivatives: these mutations correspond to the replacement of Glycine 19 with Serine (G 19S), Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K), Phenylalanine 87 with Leucine (F87L), Valine 105 with Alanine (V105A) and Isoleucine 132 with Valine (Il 32V). A) Material and Methods a) Site-directed mutagenesis Site-directed mutagenesis libraries were created by PCR on a pool of chosen variants. For example, to introduce the G19S substitution into the coding sequence of the variants, two separate overlapping PCR reactions were carried out that amplify the 5' end (residues 1-24) or the 3' end (residues 14-167) of the I- OeI coding sequence. For both the 5' and 3' end, PCR amplification is carried out using a primer with homology to the vector (GaIlOF 5'-gcaactttagtgctgacacatacagg-3 ' (SEQ ID NO: 16) or GaIlOR 5'-acaaccttgattggagacttgacc-3'(SEQ ID NO: 17)) and a primer specific to the l-Crel coding sequence for amino acids 14-24 that contains the substitution mutation G19S (G19SF 5'-gccggctltgtggactclgacggtagcatcatc-3' (SEQ ID NO: 47) or Gl 9SR 5'-gatgatgctaccgtcagagtccacaaagccggc-3'(SEQ ID NO: 48)). The same strategy is used with the following pair of oligonucleotides to introduce the mutations leading to the F54L, E80K, F87L, V 105 A and Il 32V substitutions in the coding sequences of the variants, respectively:
* F54LF: 5'-acccagcgccgttggctgctggacaaactagtg-3' and F54LR: 5'- cactagtttgtccagcagccaacggcgctgggt-3' (SEQ ID NO: 49 and 50); * E80KF: 5'-ttaagcaaaatcaagccgctgcacaacttcctg-3' and E80KR: 5'- caggaagttgtgcagcggcttgattttgcttaa-3' SEQ ID NO: 51 and 52);
* F 87LF: 5!-aagccgctgcacaacctgctgactcaactgcag-3' and F 87LR: 5'- ctgcagltgagtcagcaggttgtgcagcggctt-3' SEQ ID NO: 53 and 54);
* V 105AF: 5'-aaacaggcaaacctggctctgaaaattatcgaa-3' and V 105AR: 5'- ttcgataattttcagagccaggtttgcctgttt-3' SEQ ID NO: 55 and 56);
* Il 32VF: 5'-acctgggtggatcaggttgcagctctgaacgat-3' and Il 32VR: 5'- atcgttcagagctgcaacctgatccacccaggt-3' SEQ ID NO: 57 and 58).
For each substitution to be introduced, the resulting PCR products contain 33bp of homology with each other. The PCR fragments were purified. The ten PCR fragments were pooled en equimolar amounts to generate a mix containing 50ng of PCR DNA and 75ng of vector DNA (pCLS0542, Figure 9), linearized by digestion with Ncoϊ and Eagl. This mix was used to transform the yeast Saccharomyces cerevisiae strain FYC2-6A (MATa, trplΔ63, leu2Δl, his3Δ200) using a high efficiency LiAc transformation protocol (Gietz and Woods, Methods Enzymol., 2002, 350, 87-96). Intact coding sequences containing the substitutions are generated in vivo by homologous recombination in yeast. c) Mating of meganucleasc expressing clones and screening in yeast
The experimental procedure is as described in example 28. d) Sequencing of variants
The experimental procedure is as described in example 25. B) Results A library containing a population harboring the six amino-acid substitutions (Glycine 19 with Serine, Phenylalanine 54 with Leucine, Glutamic acid 80 with Lysine, Phenylalanine 87 with Leucine, Valine 105 with Alanine and lsoleucine 132 with Valine) was constructed on a pool of two variants cleaving HIV1_5.4 (SEQ ID NO: 297 and 299; described in Table XLIII). 558 transformed clones were screened for cleavage against the HIV1_5.4 and HIVl_5.ό DNA targets. A total of 378 positive clones were found to cleave HIV1_5.4, while 321 of those cleaved also the HIVl_5.ό target. An example of positive variants is shown in figure 59.
The 558 transformed clones were also mated with a yeast strain that contains (i) the HIV1_5 target in a reporter plasmid (ii) an expression plasmid containing a variant that cleaves the HIV1_5.3 target (l-Crel 33C)38Y,44A,68Y,70Q,75N +89A or KNSCYS/AYQNI +89A, according to the nomenclature of Table I). After mating with this yeast strain, 137 clones were found to cleave the HIV 1_5. Thus, 137 positives contained proteins able to form heterodimers with KNSCYS/AYQNI +89A (SEQ ID NO: 256) showing cleavage activity on the HIV1_5 target. An example of positives is shown in figure 60.
Sequencing of the 93 clones with the highest cleavage activity on the HIV 1_5 target allowed the identification of 48 different endonuclease variants.
The sequence of ten 1-OeI variants cleaving the HIV 1_J5 target when forming a heterodimer with the KNSCYS/AYQNI +89A variant arc listed in Table XXXXIV.

Example 32: Covalent assembly as single chain and improvement of meganucleases cleaving different HIVl targets by site-directed mutagenesis
Coexpression of the variants cleaving the non-palindromic targets used during the custom meganuclease development process described in previous examples leads to cleavage of the corresponding DNA target in yeast. Different mutants were selected, either showing a high cleavage activity as heterodimers in the corresponding non-palindromic targets, or a high cleavage activity as homodimers in the HIV IJSf.5 and in the HIVl JSI.6 pseudo-palindromic targets (N standing for any of the targets described in the present patent application: 1 , 3, 4, 5, 7, 8 and 9). In all cases the mutant cleaving the HIV1_N,5 target and the mutant cleaving the HIVl-N.6 target will be called Ma and Mb. This nomenclature is not related to the identity of the HIV IJSf.5 or HIVlJN'.ό cutter, but to the position in the single chain molecule (Ma being the N-terminal mutant and Mb being the C- terminal mutant). Single chain constructs were engineered using the linker RM2
(AAGGSDKYNQALSKYNQALSKYNQALSGGGGS) (SEQ ID NO: 345) resulting in the production of the canonical single chain molecule: Ma-RM2-Mb. During this
design step, the Gl 9S mutation was introduced in the C-terminaf (Mb) mutant. In addition, mutations K7E and K96E were introduced into the Ma mutant, while mutations E8K and E61R were introduced into the Mb mutant. This leads to the generation of the single chain molecule: Ma(K7E K96E)-RM2-Mb(E8K E61R) that is called SCOH-HIVl-MaMb.
Four additional amino-acid substitutions have been found in previous studies to enhance the activity of I-Crel derivatives: these mutations correspond to the replacement of Phenylalanine 54 with Leucine (F54L), Glutamic acid 80 with Lysine (E80K)> Valine 105 with Alanine (V 105A) and Isoleucine 132 with Valine (1132V). Certain combinations of these mutations were introduced into the coding sequence of N-terminal and C-terminal protein fragment (if these mutations were not present in the original mutants). The coding sequences of the single chain proteins were cloned into a mammalian expression vector, and their activity on the corresponding target in the HIVl genome was tested in a cellular model developed for this purpose. Table XLV shows an example of the single chain molecules that have been generated for the different HIVl targets.



Λ series of synthetic gene assemblies were ordered to MWG- EUROFINS. Synthetic genes coding for the different single chain variants targeting the HIVl pro virus were cloned in pCLS 1853 (figure 61) using Ascl and Xhol restriction sites.
Example 33: Determination of antiviral effect of HIVl meganuclease variants derived from I-Crel The efficacy of HIV meganucleases to cleave the corresponding proviral DNA target was assessed in a cellular system containing a defective integrated provirus. This cellular model produces viral-like particles (VLPs) containing all the essential HIVl proteins with the exception of the viral envelope glycoproteins. Nevertheless, the produced VLPs are not able to infect the cells due to the absence of entry-mediating proteins in the viral envelope. Production of VLPs can be measured in the supernatants of cultured cells using an HIVl-p24 ELISA kit. The VLP-producing cells were transfected with the plasmids coding for the different versions of the SCOH-HIVl meganucleases and the antiviral effect was measured by the reduction in the litres of p24 present in the supernatants of transfected cells respect to a "control" sample in which the cells were transfected by a non-related meganuclease (NRM), which has no cleavage activity on the HIVl proviral DNA. 1) Material and Methods a) Generation of a cellular system allowing to test the antiviral activity of HIVl meganucleases A cell line capable of producing non-replicative VLPs was generated in order Io dispose of a model allowing to determine the efficacy of antiviral meganucleases. With the aim of introducing an HIV provirus in the cells, a lenti viral vector pseudotyped by the VSV envelope protein was used to transduce the HEK-293 human cell line. In order to avoid viral replication on the cellular model, the integrated provirus harbours deletion of the HIVl accessory proteins (Vif, Vpr, Vpu and NeJ) as well as of the viral envelope glycoprotein (env). A cassette conferring puromycin
resistance to the cell line was introduced, as well as the EGFP coding sequence (EFlalfa.p-PuroR-IRES-EGFP) to replace the env coding sequence.
For safety reasons, two other HIVl essential proteins have been deleted from the proviral sequence, those of the Tat and the Rev proteins, which are essential for the production of viral progeny.
To produce the cellular system, two retroviral vectors were generated harbouring either the tat or the rev coding sequences. These two vectors were used to sequentially transduce HEK-293 cells, leading to the generation of a cell line able to produce the tat and rev proteins after integration of the retroviral vectors in the cellular genome. The generated cell line was then transduced by a lentiviral expression vector that, after integration of the dsDNA resulting from reverse transcription, would generate the pseudo-Ill Vl provirus containing the meganuclease target hits. The structure of the integrated provirus correspond to the sequence elements USRUS(HIV)-PSiGAGPOL(HIV)-EFIa=PUrOIIRESiGFP-USRUS(HIV) and is schematically represented in figure 62.
The cells were tested for their ability to produce VLPs by determining the presence of the HIVI p24 protein in the culture supernatants using the Alliance® HIVl-p24 ELISA Kit (Perkin Elmer Inc, Waltham, MA, USA). In a next step, the VLP producing cells were subjected to clonal dilutions in order to characterize the number of pseudo HIVl integrated provirus in different clones. A cellular clone (HEK293-VLP-CL40) containing between 1 and 2 copies of the pseudo HIVl provirus (as determined by qPCR) was used for assessing the antiviral activity of meganucleascs.
HEK293-VLP-CL40 cells were cultured in DMEM media supplemented with 2mM L-glutamine, penicillin (100 IU/ml), streptomycin (100 mg/ml), amphotericin B (Fongizone: 0.25 mg/ml, Invitrogen-Life Science) and 10% of foetal bovine serum (FBS). b) Transfection of HEK293-VLP-CL40 cells
The day before transfection, HEK293-VLP-CL40 cells were seeded in 12-well culture plates (Falcon, Becton Dickinson, Le Pont De Claix, France) at 105 cells per well and incubated overnight at 370C in 1 ml of complete growth medium. The cultures were about 70% confluent on the day of transfection. Transfection with
1 μg of plasmid expressing I-Crel variants cleaving different HIVl target sequences was done using FuGENE© HD Transfection Reagent (Roche Diagnostics, Indianapolis, IN5 USA) according to manufacturer's instruction, Transfection media was replaced 24h after transfection and cells were kept at 37°C in complete growth medium for other 24 hours. c) Cell harversting and p24 determination
Cell supcrnatants were harvested 48h post-transfection and p24 titres were either measured immediately or the supernatants were kept at -200C for ulterior quantification of the p24. HEK-293-CL40 transfected cells were then recovered and counted, prior to centrifugation at 1500 rpm for 5 minutes and storage of the dry cellular pellet at -200C for ulterior extraction of the genomic DNA.
The amount of p24 present in cellular supernatants was determined using the Alliance® HlVl-p24 ELISA Kit (Pcrkin Elmer Inc, Waltham, MA, USA) according to the manufacturer's instructions. Results were expressed as p24 in pg/ml (or as pg/well, according to the cell culture conditions). The production of p24 was normalized by the number of cells present in the well at the moment of media harvesting, and expressed as p24 levels in fg/cell. 2) Results
The single chain molecules described in Table XLV (SEQ ID NO: 346 to 365) were tested for their ability to target the HIVl provirus and reduce the amount of VLPs produced in the HEK293-VLP-CL40 cellular model. Cells were transfected with 1 μg of plasmid expressing the meganuclease variants and the level of p24 present in the culture supernatants was determined 48h after transfection, as previously described. As a control, a non related meganuclease (NRM) was transfected. This NRM is not active against the HlVl provirus and should have no effect on the level of p24 produced by NRM transfected cells. The p24 levels of NRM transfected cells, expressed in fg/ccll, was considered as 100% of VLP production, and the p24 levels present in samples transfected with HIV meganucleases were compared to the NRM value, in order to determine the percentage of VLP production in these samples.
a) Sequences targeted in the HlVl pro virus by the HIVl meganucleases
The meganuclease target sites have already been described except for the HIV 1_7, HIVlJ and HIV1_9 targets.
The HIV 1_1 target, described in example 1, is located in the U3 region of the proviral LTRs; while the HIV 1_3 target, described in example 8, is located in the U5 region of the proviral LTRs. Since the LTRs are duplicated sequences flanking the viral ORFs in the integrated provirus, each of these two targets are present twice in the HIVl provirus.
The HIV 1_4 target has been described in example 16, and is located in the gag gene of the HIVl provirus, more precisely in the coding sequence of the p24 (CApsid) protein. The HIV IJ target (G GAG CC ACC CCAC AAG AT TTA A,
SEQ ID NO: 366) also cleaves the coding sequence of the ρ24 protein, though at a different position. The HIV 1_7 target is also a 22 bp (non-palindromic) target precisely located at positions 1321-1342 of the HIV-I pNL4-3 vector (accession number AF324493, Adachi et al, J. Virol., 1986, 59, 284-291), a subtype B infectious molecular clone.
The HIV 1_5 target has been described in example 24, and is located in the pol gene of the HIVl provirus, more precisely in the sequence coding for the PRotease protein. The HIV 1_9 target also cleaves the coding sequence of the protease, though at a different position. The HIV1_9 target (A GAA AT CTG TTGA CTC AG ATT G, SEQ ID NO: 368) is also a 22 bp (non-palindromic) target located at positions 251 1-2532 of the HIV-I pNL4-3 vector.
The HIV 1_8 target (G GGC CC CTA GGAA AAA GG GCT G, SEQ ID NO: 367) is a 22 bp (non-palindromic) target located in the gag gene of the HIVl provirus. This target is precisely located at positions 2006-2027 of the HIV-I pNL4-3 vector, on the coding sequence of the p7 (NC, NucleoCapsid) protein.
Over again, it should be noted that two cleavage sites are present in the HIVl proviral DNA for targets HIV 1_1 and HIV1_3, while the remaining targets present only one cleavage site in the integrated provirus. The presence of the HIVl meganuclease cleavage sites in the
HEK293-VLP-CL40 cells was confirmed by sequencing and their position is represented in figure 62.
b) I-Crcl variants targeting the HIVl genome induce a decrease in p24 litres in a cellular model harbouring an HIVl provirus p24 titres were determined 48 hours after transfection with the HIVl meganucleascs as previously described. The values, expressed as p24 in fg/cell, were normalized respect to the amount of p24 released in a well transfected by a NRM, which was considered to be 100% for VLP production.
Figure 63 shows the levels of p24 (in %) produced by the cells transfected with the different meganuclease plasmids. A reduction of p24 production is observed in samples transfected with HIV meganucleases. The meganucleases showing a higher reduction in p24 titers correspond to variants SCOH-HIV1_3-B and
SCOH-HIVlJ-D (SEQ ID NO: 350 and 352), leading to nearly a 50% reduction of p24 levels compared to cells transfected with the NRM.
A significant reduction of p24 titers, ranging from 35-40%, is observed also for other I-Crel variants cleaving different targets in the HIVl provirus (SCOH-HIV1 J-B, SEQ ID NO: 347; SCOH-HIVlJ-A, SEQ ID NO: 357; SCOH-
HIV1_8-D> SEQ ID NO: 362; and SCOH-HIV 1_9-B, SEQ ID NO: 364).
Example 34: Detection of cleavage activity at the HIV1 8 locus in a human cell line harbouring an integrated HIVl provirus.
VCr el variants targeting the HIV 1_8 target, as well as their activity have been described in Examples 32 and 33. The efficiency of two of the HIV 1_8 meganucleases to cleave their endogenous DNA target sequence was next tested. This example will demonstrate that meganucleases engineered to cleave the HIV ]_8 target sequence cleave their cognate endogenous site in human cells harboring an integrated
HIVl provirus (HEK293-VLP-CL40 cells). Repair of double-strand break by non homologous end-joining
(NHEJ) can generate small deletions and insertions (InDeI) (Figure 64). In nature, this error-prone mechanism can be deleterious for the cells survival but provides a rapid indicator of meganucleases activity at endogenous loci.
Example 34.1 : Detection of induced mutagenesis at the endogenous site Two Single Chain l-Crel variants targeting the HIV 1_8 target cloned in the pCLS1853 plasmid were used for this experiment. The day previous to
the experiment, cells derived from the human embryonic kidney cell line, 293 -H (HEK293-VLP-CL40) were seeded in a 10 cm dish at density of 106ceUs/dish.
The following day, cells were transfccted with 3 μg of an empty plasmid or a meganuclease-expressing plasmid using FuGene® HD Transfcction Reagent (Roche Diagnostics, Indianapolis, IN, USA) according to manufacturer's instruction. 72 hours after transfection, cells were collected and diluted (dilution 1/20) in fresh culture medium. After 7 days of culture, cells were collected and genomic DNA extracted. 200 ng of genomic DNA were used to amplify the endogenous locus surrounding the meganuclease cleavage site by PCR amplification. A 325 bp fragment corresponding to the HIV 1_8 locus was amplified using specific PCR primers HI8f (SEQ ID NO 369; 5 '-GACCCGGCCATAAAGCAAGAGTTTTGGCTG-S ') and IIISr (SEQ ID NO 370; 5'-AAGCTCTCTTCTGGTGGGGCTGTTGGCTCT-S '). PCR amplification was performed to obtain a fragment flanked by specific adaptator sequences (SEQ ID NO 371 ; 5'-CCATCTCATCCCTGCGTGTCTCCGACTCAG-S ' and 25 SEQ ID NO: 372 5'-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-S') provided by the company offering sequencing service (GATC Biotech AG, Germany) on the 454 sequencing system (454 Life Sciences).
An average of 3,000 sequences was obtained from pools of the amplicons (500 ng). After sequencing, different samples were identified based on barcode sequences introduced in the first of the above adaptators. 15 sequences showed the presence of insertions or deletions in the cleavage site of HIV 1_8 meganucleases.

The analysis of the genomic DNA extracted from cells transfected with the meganucleases targeting the HIV1__8 locus showed that around 1% of the
analyzed sequences contained InDeI events within the recognition site of HIV 1_8 meganucleases (Table XLVI). Since small deletions or insertions could be related to PCR or sequencing artefacts, the same locus was analyzed after transfection with a plasmid that does not express the meganuclease. The analysis of the HIV IJ locus revealed that no InDeI events could be detected. These data demonstrate that meganucleases engineered to target the HIV1_8 locus are active in human cells and can cleave their cognate endogenous sequence. Moreover, it shows that meganucleases have the ability to generate small InDeI events within a sequence which would disrupt a gene ORF and thus inactivate the corresponding gene expression product.
Claims
1. An l-Crel variant which cleaves a DNA target in the provirus of a pathogenic retrovirus, for use in treating an infection of said retrovirus.
2. The variant of claim 1, wherein said pathogenic retrovirus is from a genus selected from the group consisiting of: Alpharelrovirus, Betaretrovirus,
Gammaretrovirus, Deltaretrovirus, Epsilonretrovirus, Lentivirus and Spumavirus.
3. The variant of claim 1 or 2, wherein said retrovirus is selected from the group consisting of: Human T-lymphotrophic virus, Rous Sarcoma and Human Immunodeficiency Virus.
4. The variant of claim 3, wherein said Human Immunodeficiency
Virus is selected from the group consisting of Human Immunodeficiency Virus Type 1 (HIVl) and Human Immunodeficiency Virus Type 2 (HIV2).
5. The variant of claim 1 wherein said DNA target is selected from the group consisting of the sequences SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368.
6. The variant of claim 1 consisting of the sequences SEQ ID NO: 350; SEQ ID NO: 352; SEQ ID NO: 1-13; SEQ ID NO: 26-46; SEQ ID NO: 59-85; SEQ ID NO: 88-94; SEQ ID NO: 97-165; SEQ ID NO: 168-174; SEQ ID NO: 177- 186; SEQ ID NO: 189-238; SEQ ID NO: 241-242; SEQ ID NO: 245-253; SEQ ID NO: 256-316; SEQ ID NO: 346-349; SEQ ID NO: 351 ; SEQ ID NO: 353-365.
7. The variant of claim 1, characterized in that at least one of the two 1-OeI monomers has at least two substitutions, one in each of the two functional subdomains of the LAGLIDADG core domain, in particular the first functional subdomain comprises a substitution(s) in at least one of positions 26, 28, 30, 32, 33, 38 and/or 40 and in the second functional subdomain comprises a substitution in at least one of positions positions 44, 68, 70, 75 and/or 77; said variant being obtainable by a method comprising at least the steps of:
(a) constructing a first series of l-Crel variants having at least one substitution in a first functional subdomain of the LAGLIDADG core domain comprising at least one substitution at a position selected from the group: 26, 28, 30,
32, 33, 38 and/or 40 of I-C«?I, (b) constructing a second series of l-Crel variants having at least one substitution in a second functional subdomain of the LAGLIDADG core domain comprising at least one substitution at a position selected from the group: 44, 68, 70, 75 and/or 77 of I-CreI, (c) selecting and/or screening the variants from the first series of step (a) which are able to cleave a DNA target sequence selected from the group SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368, wherein at least one of (i) the nucleotide triplet in positions -10 to -8 of the I-Od site has been replaced with the nucleotide triplet which is present in positions -10 to -8 of the selected DNA target sequence from said provirus and (ii) the nucleotide triplet in positions +8 to +10 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in position -10 to -8 of said selected DNA target sequence from said provirus,
(d) selecting and/or screening the variants from the second series of step (b) which are able to cleave a DNA target sequence selected from the group SEQ
ID NO: 319 to 342 and SEQ ID NO: 366 to 368, wherein at least one of (i) the nucleotide triplet in positions -5 to -3 of the I-CVeϊ site has been replaced with the nucleotide triplet which is present in positions -5 to -3 of the selected DNA target sequence from said provirus and (ii) the nucleotide triplet in positions +3 to +5 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in position -5 to -3 of said selected DNA target sequence from said provirus,
(e) selecting and/or screening the variants from the first series of step (a) which are able to cleave a DNA target sequence selected from the group SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368, wherein at least one of (i) the nucleotide triplet in positions +8 to +10 of the I-Oβl site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of the selected DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +8 to +10 of said selected DNA target sequence from said provirus, (f) selecting and/or screening the variants from the second series of step (b) which are able to cleave a DNΛ target sequence selected from the group SEQ ID NO: 319 to 342 and SEQ ID NO: 366 to 368, wherein at least one of (i) the nucleotide triplet in positions +3 to +5 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +3 to +5 of the selected DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -5 to -3 has been replaced with the reverse complementary sequence of the nucleotide triplet which is present in positions +3 to +5 of said selected DNA target sequence from said provirus, (g) combining in a single variant, the mutation(s) in positions 26,
28, 30, 32, 33, 38 and/or 40 and 44, 68, 70, 75 and/or 77 of two variants from step (c) and step (d), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions -10 to -8 is identical to the nucleotide triplet which is present in positions -10 to -8 of said selected DNA target sequence from said provirus , (ii) the nucleotide triplet in positions +8 to +10 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions -10 to -8 of said selected DNA target sequence from said provirus, (iii) the nucleotide triplet in positions -5 to -3 is identical to the nucleotide triplet which is present in positions -5 to -3 of said selected DNA target sequence from said provirus and (iv) the nucleotide triplet in positions +3 to +5 is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions -5 to -3 of said selected DNA target sequence from said provirus, and/or
(h) combining in a single variant, the mutation(s) in positions 26, 28, 30, 32, 33, 38 and/or 40, and 44, 68, 70, 75 and/or 77 of two variants from step (c) and step (f), to obtain a novel homodimeric l-Crel variant which cleaves a sequence wherein (i) the nucleotide triplet in positions +8 to +10 of the l-Crel site has been replaced with the nucleotide triplet which is present in positions +8 to +10 of said selected DNA target sequence from said provirus and (ii) the nucleotide triplet in positions -10 to -8 is identical to the reverse complementary sequence of the nucleo- tide triplet in positions +8 to +10 of said selected DNA target sequence from said provirus, (iii) the nucleotide triplet in positions +3 to +5 is identical to the nucleotide triplet which is present in positions +3 to +5 of said selected DNA target sequence from said provirus, (iv) the nucleotide triplet in positions -5 to -3 Is identical to the reverse complementary sequence of the nucleotide triplet which is present in positions +3 to +5 of said selected DNA target sequence from said provirus,
(i) combining the variants obtained in steps (g) and (h) to form heterodimers, and
(i) selecting and/or screening the heterodimers from step (i) which are able to cleave said DNA target sequence from said provirus.
8. The variant of any one of claims 1 to 7, combined with another anti-retro viral medicament.
9. A polynucleotide fragment encoding the variant of any one of claims 1 to 7.
10. An expression vector comprising at least one polynucleotide fragment of claim 9.
1 1. A host cell which is modified by a polynucleotide of claim 9 or a vector of claim 10,
12. A non-human transgenic animal which is modified by a polynucleotide of claim 9 or a vector of claim 10.
13. Use of at least one variant of any one of claims 1 to 7, or at least one vector according to claim 9, for genome engineering, for non-therapeutic purposes.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10719387A EP2421964A2 (en) | 2009-04-21 | 2010-04-21 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
| US13/265,575 US20120260356A1 (en) | 2009-04-21 | 2010-04-21 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
| US14/064,775 US20140178942A1 (en) | 2009-04-21 | 2013-10-28 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2009/005582 WO2010122367A2 (en) | 2009-04-21 | 2009-04-21 | Meganuclease variants cleaving the genomic insertion of a virus and uses thereof |
| IBPCT/IB2009/005582 | 2009-04-21 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/265,575 A-371-Of-International US20120260356A1 (en) | 2009-04-21 | 2010-04-21 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
| US14/064,775 Division US20140178942A1 (en) | 2009-04-21 | 2013-10-28 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010122506A2 true WO2010122506A2 (en) | 2010-10-28 |
| WO2010122506A3 WO2010122506A3 (en) | 2011-01-27 |
Family
ID=41225988
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/005582 Ceased WO2010122367A2 (en) | 2009-04-21 | 2009-04-21 | Meganuclease variants cleaving the genomic insertion of a virus and uses thereof |
| PCT/IB2010/051746 Ceased WO2010122506A2 (en) | 2009-04-21 | 2010-04-21 | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/005582 Ceased WO2010122367A2 (en) | 2009-04-21 | 2009-04-21 | Meganuclease variants cleaving the genomic insertion of a virus and uses thereof |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US20120260356A1 (en) |
| EP (1) | EP2421964A2 (en) |
| WO (2) | WO2010122367A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013010062A3 (en) * | 2011-07-14 | 2013-03-07 | Life Technologies Corporation | Nucleic acid complexity reduction |
| US10619205B2 (en) | 2016-05-06 | 2020-04-14 | Life Technologies Corporation | Combinatorial barcode sequences, and related systems and methods |
| US10704164B2 (en) | 2011-08-31 | 2020-07-07 | Life Technologies Corporation | Methods, systems, computer readable media, and kits for sample identification |
| US10978174B2 (en) | 2015-05-14 | 2021-04-13 | Life Technologies Corporation | Barcode sequences, and related systems and methods |
| US12146189B2 (en) | 2011-08-31 | 2024-11-19 | Life Technologies Corporation | Methods, systems, computer readable media, and kits for sample identification |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1863909B2 (en) | 2005-03-15 | 2014-09-10 | Cellectis | I-crei meganuclease variants with modified specificity, method of preparation and uses thereof |
| WO2009019528A1 (en) * | 2007-08-03 | 2009-02-12 | Cellectis | Meganuclease variants cleaving a dna target sequence from the human interleukin-2 receptor gamma chain gene and uses thereof |
| WO2009074842A1 (en) * | 2007-12-13 | 2009-06-18 | Cellectis | Improved chimeric meganuclease enzymes and uses thereof |
| CN102177235A (en) * | 2008-09-08 | 2011-09-07 | 赛莱克蒂斯公司 | Meganuclease variants that cleave a DNA target sequence from the glutamine synthetase gene and uses thereof |
| EP2180058A1 (en) | 2008-10-23 | 2010-04-28 | Cellectis | Meganuclease recombination system |
| EP2480659A2 (en) | 2009-09-24 | 2012-08-01 | Cellectis | Meganuclease reagents of uses thereof for treating genetic diseases caused by frame shift/non sense mutations |
| US9044492B2 (en) | 2011-02-04 | 2015-06-02 | Cellectis Sa | Method for modulating the efficiency of double-strand break-induced mutagenesis |
| CA3237482A1 (en) | 2021-11-03 | 2023-05-11 | The J. David Gladstone Institutes, A Testamentary Trust Established Under The Will Of J. David Gladstone | Precise genome editing using retrons |
| WO2023141602A2 (en) | 2022-01-21 | 2023-07-27 | Renagade Therapeutics Management Inc. | Engineered retrons and methods of use |
| WO2024044723A1 (en) | 2022-08-25 | 2024-02-29 | Renagade Therapeutics Management Inc. | Engineered retrons and methods of use |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003215869B2 (en) * | 2002-03-15 | 2008-04-24 | Cellectis | Hybrid and single chain meganucleases and use thereof |
| EP3202899B1 (en) * | 2003-01-28 | 2020-10-21 | Cellectis | Custom-made meganuclease and use thereof |
| ES2582091T3 (en) * | 2005-10-18 | 2016-09-09 | Precision Biosciences | Rationally designed meganucleases with sequence specificity and altered DNA binding affinity |
| WO2007049095A1 (en) * | 2005-10-25 | 2007-05-03 | Cellectis | Laglidadg homing endonuclease variants having mutations in two functional subdomains and use thereof |
| US20100146651A1 (en) * | 2006-11-14 | 2010-06-10 | Cellectis | Meganuclease variants cleaving a dna target sequence from the hprt gene and uses thereof |
| WO2008102199A1 (en) * | 2007-02-20 | 2008-08-28 | Cellectis | Meganuclease variants cleaving a dna target sequence from the beta-2-microglobulin gene and uses thereof |
| WO2009013559A1 (en) * | 2007-07-23 | 2009-01-29 | Cellectis | Meganuclease variants cleaving a dna target sequence from the human hemoglobin beta gene and uses thereof |
-
2009
- 2009-04-21 WO PCT/IB2009/005582 patent/WO2010122367A2/en not_active Ceased
-
2010
- 2010-04-21 WO PCT/IB2010/051746 patent/WO2010122506A2/en not_active Ceased
- 2010-04-21 US US13/265,575 patent/US20120260356A1/en not_active Abandoned
- 2010-04-21 EP EP10719387A patent/EP2421964A2/en not_active Withdrawn
-
2013
- 2013-10-28 US US14/064,775 patent/US20140178942A1/en not_active Abandoned
Non-Patent Citations (52)
| Title |
|---|
| ADACHI ET AL., J. VIROI., vol. 59, 1986, pages 284 - 291 |
| ADACHI ET AL., J. VIROL., vol. 59, 1986, pages 284 - 291 |
| AKADA ET AL., BIOTECHNIQUES, vol. 28, 2000, pages 668 - 670 |
| AMOULD ET AL., J. MOL. BIOL., vol. 355, 2006, pages 443 - 458 |
| ARGAST ET AL., J. MOL. BIOL., vol. 280, 1998, pages 345 - 353 |
| ARNOULD ET AL., J. MOL. BIOL., vol. 355, 2006, pages 443 - 458 |
| ARNOULD ET AL., J. MOL. BIOL., vol. 371, 2007, pages 49 - 65 |
| ARNOULD, J. MOL. BIOL., vol. 355, 2006, pages 443 - 458 |
| ASHWORTH ET AL., NATURE, vol. 441, 2006, pages 656 - 659 |
| BARRE-SINOUSSI ET AL., SCIENCE, vol. 220, 1983, pages 868 - 871 |
| BENN ET AL., SCIENCE, vol. 230, 1985, pages 949 - 951 |
| BOLDUC ET AL., GENES DEV., vol. 17, 2003, pages 2875 - 2888 |
| BONETTA, THE SCIENTIST, vol. 16, 2002, pages 38 |
| CHAMES ET AL., NUCLEIC ACIDS RES., vol. 33, 2005, pages E178 |
| CHAMES ET AL., NUCLEIC ACIDS RES., vol. 33, 2005, pages EL78 |
| CHEVALIER ET AL., BIOCHEMISTRY, vol. 43, 2004, pages 14015 - 14026 |
| CHEVALIER ET AL., J. MOL. BIOL., vol. 329, 2003, pages 253 - 269 |
| CHEVALIER ET AL., MOL. CELL., vol. 10, 2002, pages 895 - 905 |
| CHEVALIER ET AL., NAT. STRUCT. BIOL, vol. 8, 2001, pages 312 - 316 |
| CHEVALIER ET AL., NAT. STRUCT. BIOL., vol. 8, 2001, pages 312 - 316 |
| CHEVALIER, B.S.; B.L. STODDARD, NUCLEIC ACIDS RES., vol. 29, 2001, pages 3757 - 3774 |
| CHEVALIER; STODDARD, NUCLEIC ACIDS RES., vol. 29, 2001, pages 3757 - 3774 |
| DOYON ET AL., J. AM. CHEM. SOC., vol. 128, 2006, pages 2477 - 2484 |
| EPINAT ET AL., NUCLEIC ACIDS RES, vol. 31, 2003, pages 2952 - 62 |
| EPINAT ET AL., NUCLEIC ACIDS RES., vol. 31, 2003, pages 2952 - 2962 |
| FALLAUX ET AL., HUM. GENE THER., vol. 9, 1998, pages 1909 - 1917 |
| FLEXNER C, NATURE REVIEWS DRUG DISCOVERY, vol. 6, 2007, pages 959 - 966 |
| FORD ET AL., GENE THER., vol. 8, 2001, pages 1 - 4 |
| GIETZ; WOODS, METHODS ENZYMOJ., vol. 350, 2002, pages 87 - 96 |
| GIETZ; WOODS, METHODS ENZYMOL., vol. 350, 2002, pages 87 - 96 |
| GIMBLE ET AL., J. MOL. BIOL., vol. 334, 2003, pages 993 - 1008 |
| GOODARZI ET AL., HEP. MON, vol. 8, no. 2, 2008, pages 129 - 133 |
| ICHIYANAGI ET AL., J. MOL. BIOL., vol. 300, 2000, pages 889 - 901 |
| JURICA ET AL., MOL. CELL, vol. 2, 1998, pages 469 - 476 |
| MARTINEZ-CAJAS; WAINBERG, DRUGS, vol. 68, 2008, pages 43 - 72 |
| MOURE ET AL., NAT. STRUCT. BIOL., vol. 9, 2002, pages 764 - 770 |
| MOURE, J. MOL. BIOL., vol. 334, 2003, pages 685 - 69 |
| ROSEN ET AL., NUCLEIC ACIDS RES., vol. 34, 2006, pages 4791 - 4800 |
| ROSSI ET AL., NAT. BIOTECHNOL., vol. 25, 2007, pages 1444 - 54 |
| SCHNEIDER ET AL., IN1 J CANCER, vol. 19, no. 5, 1977, pages 621 - 6 |
| SELIGMAN ET AL., GENETICS, vol. 147, 1997, pages 1653 - 1664 |
| SHARP ET AL., PHILOS TRANS R SOC LOND B BIOL SCI, vol. 356, 2001, pages 867 - 76 |
| SILVA ET AL., J. MOL. BIOL., vol. 286, 1999, pages 1123 - 1136 |
| SIMON ET AL., LANCET, vol. 368, 2006, pages 489 - 504 |
| SMITH ET AL., NUCLEIC ACIDS RES., 2006 |
| SMITH ET AL., NUCLEIC ACIDS RES., vol. 34, 2006, pages E149 |
| SMITH ET AL., NUCLEIC ACIDS RES., vol. 34, 2006, pages EL49 |
| SMITH, NUCLEIC ACIDS RES., vol. 34, 2006, pages 149 |
| SMITH, NUCLEIC ACIDS RES., vol. 34, 2006, pages E149 |
| SPIEGEL ET AL., STRUCTURE, vol. 14, 2006, pages 869 - 880 |
| SUSSMAN ET AL., J. MOL. BIOL., vol. 342, 2004, pages 31 - 41 |
| WADIA; DOWDY, CURR. OPIN. BIOTECHNOL., vol. 13, 2002, pages 52 - 56 |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013010062A3 (en) * | 2011-07-14 | 2013-03-07 | Life Technologies Corporation | Nucleic acid complexity reduction |
| US10704164B2 (en) | 2011-08-31 | 2020-07-07 | Life Technologies Corporation | Methods, systems, computer readable media, and kits for sample identification |
| US12146189B2 (en) | 2011-08-31 | 2024-11-19 | Life Technologies Corporation | Methods, systems, computer readable media, and kits for sample identification |
| US10978174B2 (en) | 2015-05-14 | 2021-04-13 | Life Technologies Corporation | Barcode sequences, and related systems and methods |
| US12315598B2 (en) | 2015-05-14 | 2025-05-27 | Life Technologies Corporation | Barcode sequences, and related systems and methods |
| US10619205B2 (en) | 2016-05-06 | 2020-04-14 | Life Technologies Corporation | Combinatorial barcode sequences, and related systems and methods |
| US11208692B2 (en) | 2016-05-06 | 2021-12-28 | Life Technologies Corporation | Combinatorial barcode sequences, and related systems and methods |
| US12264363B2 (en) | 2016-05-06 | 2025-04-01 | Life Technologies Corporation | Combinatorial barcode sequences, and related systems and methods |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010122367A2 (en) | 2010-10-28 |
| US20140178942A1 (en) | 2014-06-26 |
| EP2421964A2 (en) | 2012-02-29 |
| WO2010122367A3 (en) | 2011-03-17 |
| WO2010122506A3 (en) | 2011-01-27 |
| US20120260356A1 (en) | 2012-10-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2421964A2 (en) | Meganuclease variants cleaving at least one target in the genome of a retrovirus and uses thereof | |
| EP2046950B1 (en) | Meganuclease variants cleaving a DNA target sequence from a RAG1 gene and uses thereof | |
| Lee et al. | Nonrandom distribution of gp120 N-linked glycosylation sites important for infectivity of human immunodeficiency virus type 1. | |
| US20150315557A1 (en) | Meganuclease variants cleaving the genome of a pathogenic non-integrating virus and uses thereof | |
| Hizi et al. | dUTPase: the frequently overlooked enzyme encoded by many retroviruses | |
| WO2011007193A1 (en) | Viral vectors encoding a dna repair matrix and containing a virion-associated site specific meganuclease for gene targeting | |
| Prokofjeva et al. | Therapy of HIV infection: current approaches and prospects | |
| WO2008102199A1 (en) | Meganuclease variants cleaving a dna target sequence from the beta-2-microglobulin gene and uses thereof | |
| AU2008263502A1 (en) | Method for enhancing the cleavage activity of I-Crei derived meganucleases | |
| WO2011036510A1 (en) | Meganuclease variants cleaving the genome of the herpes simplex virus and uses thereof | |
| Jubier-Maurin et al. | Genetic characterization of the nef gene from human immunodeficiency virus type 1 group M strains representing genetic subtypes A, B, C, E, F, G, and H | |
| WO2010136841A2 (en) | Meganuclease variants cleaving the genome of a non-genomically integrating virus and uses thereof | |
| US7803582B2 (en) | Recombinant vector and use in gene therapy | |
| Zheng et al. | Intracellular immunity to HIV-1: newly defined retroviral battles inside infected cells | |
| CA2415187C (en) | Dna vaccines encoding hiv accessory proteins | |
| Schaller et al. | The Early Bird Catches the Worm–Can Evolution Teach us Lessons in Fighting HIV? | |
| Kim | HIV-1 Evasion of Human TRIM5α via Cyclophilin A | |
| Valeri | Studio dei meccanismi di immunità innata che ostacolano la manipolazione genetica delle cellule ematopoietiche staminali | |
| Bovolenta | Blocking HIV-1 Vif restores a natural mechanism of intracellular antiviral defense | |
| DE LYON | Evolutionary and functional characterization of SAMD9/SAMD9L innate immunity proteins and their involvement in HIV infection | |
| Schüssler | SAMHD1: HIV-1 restriction, post-translational regulation and function | |
| Prelli Bozzo | Replication competent HIV-guided CRISPR screen identifies novel antiviral factors | |
| Bozzo | Replication Competent HIV-guided CRISPR Screen Identifies Novel Antiviral Factors | |
| de Sousa Pereira | Is the European Rabbit (Oryctolagus Cuniculus) a Good Animal Model to Study HIV-1 Pathogenesis and Virus-Host Interactions? | |
| Warner | Investigating SAMHD1 restriction of HIV-1 in stem cell-derived macrophages |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10719387 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010719387 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13265575 Country of ref document: US |