WO2010138577A1 - Radiolabeled pde10 inhibitors - Google Patents
Radiolabeled pde10 inhibitors Download PDFInfo
- Publication number
- WO2010138577A1 WO2010138577A1 PCT/US2010/036186 US2010036186W WO2010138577A1 WO 2010138577 A1 WO2010138577 A1 WO 2010138577A1 US 2010036186 W US2010036186 W US 2010036186W WO 2010138577 A1 WO2010138577 A1 WO 2010138577A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pdelo
- compound
- aryl
- heterocyclyl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 **(C=C1)C=CC(C(O2)=O)=C1C2=O Chemical compound **(C=C1)C=CC(C(O2)=O)=C1C2=O 0.000 description 1
- FKEZXKKALAXIGI-OVCLIPMQSA-N CC(C)Oc1cccc(C(N2CCC(N3/C=C/CC(C=C)O)=Nc4ccccc4C3=O)=O)c1C2=O Chemical compound CC(C)Oc1cccc(C(N2CCC(N3/C=C/CC(C=C)O)=Nc4ccccc4C3=O)=O)c1C2=O FKEZXKKALAXIGI-OVCLIPMQSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0459—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having six-membered rings with two nitrogen atoms as the only ring hetero atoms, e.g. piperazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/74—Quinazolines; Hydrogenated quinazolines with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached to ring carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the invention relates generally to novel PDElO inhibitors and to their use as radiotracers for quantitative imaging of PDElO in mammals.
- Noninvasive, nuclear imaging techniques can be used to obtain basic and diagnostic information about the physiology and biochemistry of a variety of living subjects including experimental animals, normal humans and patients. These techniques rely on the use of sophisticated imaging instrumentation that is capable of detecting radiation emitted from radiotracers administered to such living subjects. The information obtained can be reconstructed to provide planar and tomographic images that reveal distribution of the radiotracer as a function of time. Use of appropriately designed radiotracers can result in images which contain information on the structure, function and most importantly, the physiology and biochemistry of the subject Much of this information cannot be obtained by other means.
- radiotracers used in these studies are designed to have defined behaviors in vivo which permit the determination of specific information concerning the physiology or biochemistry of the subject or the effects that various diseases or drugs have on the physiology or biochemistry of the subject.
- radiotracers are available for obtaining useful information concerning such things as cardiac function, myocardial blood flow, lung perfusion, liver function, brain blood flow, regional brain glucose and oxygen metabolism.
- Compounds can be labeled with either positron or gamma emitting radionuclides.
- PET positron emitting
- the most commonly used positron emitting (PET) radionuclides are ⁇ C, ⁇ F, ⁇ O and 13N_, all of which are accelerator produced, and have half lives of 20, 1 10, 2 and 10 minutes, respectively. Since the half-lives of these radionuclides are so short, it is only feasible to use them at institutions that have an accelerator on site or very close by for their production., thus limiting their use.
- gamma emitting radiotracers are available which can be used by essentially any hospital in the U.S. and in most hospitals worldwide. The most widely used of these are 99m TCj 201 ⁇ j m & 123 ⁇ .
- Radiotracers bind with high affinity and specificity to selective receptors and neuroreceptors.
- Successful examples include radiotracers for imaging the following receptor systems: estrogen, muscarinic, dopamine Dl and D2, opiate, neuropeptide- Y, cannabinoid-1 and neurokinin- 1.
- Schizophrenia is debilitating disorder affecting the psychic and motor functions of the brain. It is typically diagnosed in individuals in their early to mid-twenties and symptoms include hallucinations and delusions or at the other extreme, anhedonia or social withdrawal. Across the spectrum, the symptoms are indicative of cognitive impairment and functional disabilities. Notwithstanding improvements in antipsychotic treatments, current therapies, including typical (haloperidol) and atypical (clozapine or olanzapine) antipsychotics, have been less than acceptable and result in an extremely high rate of noncompliance or discontinuation of medication. Dissatisfaction with therapy is attributed to lack of efficacy or intolerable and unacceptable side affects.
- Cyclic AMP is thought to regulate the activity of cAMP-dependent protein kinase (PKA), which in turns phosphorylates and regulates many types of proteins including ion channels, enzymes and transcription factors.
- PKA cAMP-dependent protein kinase
- cGMP is also responsible for downstream regulation of kinases and ion channels.
- PDEs 3', 5 '-cyclic nucleotide specific phosphodiesterases
- families are further subdivided based on catalytic domain homology and substrate specificity and include the 1) cAMP specific, PDE4A- D, 7A and 7B, and 8A and 8B, 2) cGMP specific, PDE 5A, 6A-C, and 9A, and 3) those that are dual substrate, PDE IA-C, 2A, 3 A and 3B, 1OA, and 1 IA.
- the homology between the families ranging from 20% to 45% suggests that it may be possible to develop selective inhibitors for each of these subtypes.
- the PDElO subtype at present consists of a sole member, PDElOA, having alternative splice variants at both the N-term ⁇ nus (three variants) and C-terminus (two variants), but that does not affect the GAF domain in the N-terminus or the catalytic site in C-terminus.
- the N-terminus splice variants, PDElOAl and PDE10A2 differ in that the A2 variant has a PKA phosphorylation site that upon activation, i.e. PICA phosphorylation in response to elevated cAMP levels, results in intracellular changes to the localization of the enzyme.
- PDElOA is unique relative to other PDE families also having the conserved GAF domain in that its ligand is cAMP, while for the other GAF-domain PDEs the ligand is cGMP (Kehler et al., Expert Opin. Ther. Patents (2007) 17(2): 147-158).
- PDElOA has limited but high expression in the brain and testes. The high expression in the brain and, in particular, the neurons of the striatum, unique to PDElO, suggests that inhibitors thereto may be well suited from treating neurological and psychiatric disorders and conditions.
- PET Pierositron Emission Tomography
- radiotracers and imaging technology may provide a powerful method for clinical evaluation and dose selection of PDElO inhibitors.
- the invention herein is directed to radiolabeled PDElO inhibitors that would be useful for exploratory and diagnostic imaging applications, both in vitro and in vivo, and for competition studies using radiolabeled and unlabeled PDElO inhibitors.
- the present invention is directed to pyrimidinone compounds of general structural and radiolabeled derivatives that are useful as therapeutic agents for the treatment of central nervous system disorders associated with phosphodiesterase 10 (PDElO).
- the present invention also relates to PDElO inhibitors that are useful in treating neurological and psychiatric disorders and, in particular, schizophrenia, Huntington's disease, or psychosis associated with striatal hypofunction or basal ganglia dysfunction.
- the compounds of the invention are also directed to the use of radiolabeled PDElO inhibitors via PET tracer technology for the in vivo quantitative imaging of PDElO in mammals.
- the present invention is directed to PDEl 0 inhibitors and derivatives of structural formula I:
- A is selected from the group consisting of
- R is selected from the group consisting of
- R2 and R3 are each independently selected from the group consisting of
- (f) -C6-10 aryU n is independently 0 to 4; and p is independently 0 or 1 ; or a radiolabeled derivative and pharmaceutically acceptable salts thereof.
- Ra is selected from the group consisting of (1) halogen, (2) hydroxyl,
- R a is selected from the group consisting of
- An embodiment of the present invention includes compounds where A is -(CH2)nC6-10 aryl optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Another embodiment of this invention is when said aryl is phenyl.
- An embodiment of the present includes compounds where A is -(CH2)nC5-10 heteroaryl optionally substituted with 1 to 3 groups of Ra and all other variables are as previously described. Another embodiment of this invention is when said heteroaryl is pyridyl.
- An embodiment of the present invention includes compounds where Ar is
- (CH2)nC6-10 sryl optionally substituted with 1 to 3 groups of R ⁇ and all other variables are as previously described.
- Another embodiment of this invention is when said aryl is phenyl.
- An embodiment of the present invention includes compounds where Ar is -(CH2)nC5-10 heterocyclyl optionally substituted with 1 to 3 groups of R a and all other variables are as previously described.
- Another embodiment of this invention is when said heterocyclyl is selected from the group consisting of indole and indazole.
- An embodiment of the present invention includes compounds where R is -(CH2)nC5-10 heterocyclyl optionally substituted with 1 to 3 groups of R a and all other variables are as previously described.
- Another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of (a) indole, and
- An embodiment of the present invention includes compounds where R is -(CH2)nNR2C(O)NR2R3 optionally substituted with 1 to 3 groups of Ra and all other variables are as previously described. Another embodiment of this invention is when R2 is hydrogen and R3 is -Cg-CiO aryl- Another embodiment of this invention is when said -C ⁇ -C ⁇ Q aryl is biphenyl.
- An embodiment of the present invention is where A and Ar is -(CH2)nC6-10 a ryl > each optionally substituted with 1 to 3 groups of R a and all other variables are as previously described.
- An embodiment of this invention is where A and Ar is phenyl and R is -
- (CH2)nC5-l ⁇ heterocyclyl each optionally substituted with 1 to 3 groups of R a and all other variables are as previously described.
- Another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- Ar is selected from the group consisting of
- R is -(CH2)nC5-lO heterocyclyl or -(CH2) n NR 2 C(O)NR2R3 optionally substituted with 1 to 3 groups of Ra and all other variables are as previously described.
- n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- phthalimidyl Another embodiment of the compound of formula Ha is where Ar is phenyl and R is -(CH2) n C5-10 heterocyclyl or -(CH2)nNR 2 C(O)NR 2 R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of (1) indole, and
- Another embodiment of the compound of formula Ha is where Ar is indole and R is -(CH2) n C5- 10 heterocyclyl or -(CH2)nNR 2 C(O)NR 2 R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- Another embodiment of the compound of formula Ha is where Ar is indazole and R is -(CH2) n C5-10 heterocyclyl or -(CH2)nNR 2 C(O)NR 2 R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- R is -(CB2)nC5-10 heterocyclyl or -(CH2)nNR2C(O)NR2R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described.
- Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of (1) indole,
- Ra is selected from the group consisting of (1) halogen
- Ra is selected from the group consisting of
- Ar is selected from the group consisting of (1) phenyl,
- Another embodiment of the compound of formula lib is where Ar is phenyl and R is -(CH2)nC5-10 heterocyclyl or -(CH2)nNR2C(O)NR2R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- phthalimidyl Another embodiment of the compound of formula Hb is where Ar is indole and R is -(CH2) n C5-10 heterocyclyl or -(CH2) n NR 2 C(O)NR2R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of (1) indole, and
- Another embodiment of the compound of formula Hb is where Ar is indazole and R is -(CH2)nC5-10 heterocyclyl or -(CH2) n NR 2 C(O)NR2R3 optionally substituted with 1 to 3 groups of Ra and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- Another embodiment of the compound of formula lib is where Ar is biphenyl and R is -(CH2)nC5-l 0 heterocyclyl or -(CH2) n NR 2 C(O)NR2R3 optionally substituted with 1 to 3 groups of R a and all other variables are as previously described. Still another embodiment of this invention is when n is 1 to 3, preferably 2, and said heterocyclyl is selected from the group consisting of
- R a is selected from the group consisting of
- R a is selected from the group consisting of
- R a is selected from the group consisting of
- R a is selected from the group consisting of (1) fluorine,
- the invention is directed to radiolabeled compounds of formula I, Ha, lib, HIa or IHb, for example ⁇ C or 18F labeled compounds.
- the present invention is also directed to a method for quantitative imaging of PDElO in a mammal which comprises administering to a mammal in need of such imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is also directed to a method for quantitative imaging of tissues bearing PDElO in a mammal which comprises administering to a mammal in need of such imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is also directed to a method for the quantitative imaging of
- PDElO in tissues of a mammalian species which comprises administering to the mammalian species in need of such imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is also directed to a method for quantitative imaging of PDElO in the brain in a mammal which comprises administering to a mammal in need of such imaging an effective amount of the radiolabeled compound of the present invention.
- radiolabeled compounds and methods are for use in a human.
- Specific embodiments of the present invention include a compound which is selected from the group consisting of the subject compounds of the Examples herein and pharmaceutically acceptable salts thereof and individual enantiomers and diastereomers thereof.
- any variable e.g. aryl, heterocycle, Ra etc.
- its definition on each occurrence is independent at every other occurrence.
- combinations of substituents or variables are permissible only if such combinations result in stable compounds.
- R a When R a is -O- and attached to a carbon it is referred to as a carbonyl group and when it is attached to a nitrogen (e.g., nitrogen atom on a pyridyl group) or sulfur atom it is referred to a N-oxide and sulfoxide group, respectively.
- alkyl encompasses groups having the prefix "alk” such as, for example, alkoxy, alkanoyl, alkenyl, and alkynyl and means carbon chains which may be linear or branched or combinations thereof.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, and hepty ⁇ .
- Alkenyl refers to a hydrocarbon radical straight, branched or cyclic containing from 2 to 10 carbon atoms and at least one carbon to carbon double bond.
- Preferred alkenyl groups include ethenyl, propenyl, butenyl and cyclohexenyl.
- alkenyl is C2-Cg alkenyl.
- Preferred alkynyls are C2-C6 alkynyl.
- alkenyl alkynyl and other like terms include carbon chains containing at least one unsaturated C-C bond.
- fluoroalkyl refers to an alkyl substituent as described herein containing at least one fluorine substituent.
- cycloalkyl refers to a saturated hydrocarbon containing one ring having a specified number of carbon atoms. Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- Ci-g includes alkyls containing 6, 5, 4, 3, 2, or 1 carbon atoms.
- alkoxy as used herein, alone or in combination, includes an alkyl group connected to the oxy connecting atom.
- alkoxy also includes alkyl ether groups, where the term ' alkyl' is defined above, and 'ether' means two alkyl groups with an oxygen atom between them.
- suitable alkoxy groups include methoxy, ethoxy, n- propoxy, i-propoxy, n-butoxy, s-butoxy, t-butoxy, methoxymethane (also referred to as 'dimethyl ether'), and methoxyethane (also referred to as 'ethyl methyl ether').
- aryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic. Examples of such aryl elements include phenyl, napthyl, tetrahydronapthyl, indanyl, or biphenyl.
- heterocycle, heterocyclyl, or heterocyclic represents a stable 5- to 7-membered monocyclic or stable 8- to 11-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heterocycle or heterocyclic includes heteroaryl moieties.
- heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, be ⁇ zofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, 1,3-dioxolanyl, furyl, im ⁇ dazolidinyl, imidazolinyl, imidazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isotbiazoKdinyl, morpholinyl, nap
- heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl, benzothiazolyl, benzothienyl, benzoxazolyl, chromanyl, cinnolinyl, dihydrobenzofuryl, dihydrobenzothienyl, dihydrobenzothiopyranyl, dihydrobenzothiopyranyl sulfone, furyl, imldazolidinyl, imidazolinyl, im ⁇ dazolyl, indolinyl, indolyl, isochromanyl, isoindolinyl, isoquinolinyl, isothiazolidinyl, isothiazolyl, isothiazolidinyl, morpholinyl, naphthyridinyl, o
- the heterocyclic group is a heteroaryl group.
- heteroaryl refers to groups having 5 to 14 ring atoms, preferably 5, 6, 9, or 10 ring atoms; having 6, 10, or 14 ⁇ electrons shared in a cyclic array; and having, in addition to carbon atoms, between one and about three hetero atoms selected from the group consisting of N, 0, and S heteroaryl groups include, without limitation, thienyl, benzothienyl, furyl, benzofuryl, dibenzofuryl, pyrrolyl, imidazolyl, pyrazoiyl, pyridyl, pyrazinyl, pyrimidinyl, indolyl, quinolyl, ⁇ soquinolyl, quinoxalinyl, tetrazolyl, oxazolyl, thiazolyl, and isoxazolyl.
- the heterocyclic group is fused to an aryl or heteroaryl group.
- fused heterocycles include, without limitation, tetrahydroquinolinyl and dihydrobenzofuranyl.
- heteroaryl represents a stable 5- to 7-membered monocyclic- or stable 9- to 10-membered fused bicyclic heterocyclic ring system which contains an aromatic ring, any ring of which may be saturated, such as piperidinyl, partially saturated, or unsaturated, such as pyridinyl, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O and S 5 and wherein the nitrogen and sulfur heteroatoms may optionally be oxidized, and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heteroaryl groups include, but are not limited to, benzimidazole, benzisothiazole, benzisoxazole, benzofuran, benzothiazole, benzothiophene, benzotriazole, benzoxazole, carboline, cinnoline, furan, furazan, imidazole, indazole, indole, indolizine, isoquinoline, isothiazole, isoxazole, naphthyridine, oxadiazole, oxazole, phthalazine, pteridme, purine, pyran, pyrazine, pyrazole, pyridazine, pyridine, pyrimidine, pyrrole, quinazoline, quinoline, quinoxaline, tetrazole, thiadiazole, thiazo
- heterocycloalkyls examples include azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, tetrahydrofuranyl, imidazolinyl, pyrolidin-2-one, piperidin-2-one, and thiomo ⁇ holinyl.
- heteroatom means O, S or N, selected on an independent basis.
- a moiety that is substituted is one in which one or more hydrogens have been independently replaced with another chemical substituent.
- substituted phenyls include 2-flurophenyl, 3,4-dichlorophenyl, 3-chloro-4-fluoro-phenyl, 2,4- fluor-3-propylphenyl.
- substituted n-octyls include 2,4 dimethyl- 5 -ethyl-octyl and 3-cyclopentyloctyl. Included within this definition are methylenes (-CH 2 -) substituted with oxygen to form carbonyl (-CO-).
- Suitable substituents include, without limitation, halo, hydroxy, oxo (e.g., an annular -CH- substituted with oxo is -C(O)-), nitro, halohydrocarbyl, hydrocarbyl, aryl, aralkyl, a ⁇ koxy, aryloxy, amino, acylamino, alkylcarbamoyl, arylcarbamoyl, aminoalkyl, acyl, carboxy, hydroxyalkyl, , alkanesulfonyl, arenesulfonyl, alkanesulfonamido, arenesulfonamido, aralkylsulfonamido, alkylcarbonyl, acyloxy, cyano, and ureido groups.
- Preferred substituents, which are themselves not further substituted are:
- Halogen refers to fluorine, chlorine, bromine and iodine. Compounds described herein may contain one or more double bonds and may thus give rise to cis/trans isomers as well as other conformational isomers. The present invention includes all such possible isomers as well as mixtures of such isomers unless specifically stated otherwise.
- the compounds of the present invention may contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereoraers. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the ambit of this invention. Any formulas, structures or names of compounds described in this specification that do not specify a particular stereochemistry are meant to encompass any and all existing isomers as described above and mixtures thereof in any proportion.
- stereochemistry is meant to encompass that particular isomer in pure form or as part of a mixture with other isomers in any proportion.
- the independent syntheses of these diastereomers or their chromatographic separations may be achieved as known in the art by appropriate modification of the methodology disclosed herein.
- Their absolute stereochemistry may be determined by the x-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration. If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- the compounds of the present invention may be labeled as radiotracers for in vitro imaging.
- the compounds of the invention may be prepared as Positron Emission Tomograph (PET) tracers for in vivo imaging and quantification of PDElO.
- PET Positron Emission Tomograph
- Suitable radionuclides that may be incorporated in the instant compounds include, but not limited, 3 H (also written as T), 1 1 C, 18 F 3 35s, 125 I, 82 Br, 123 1, 131 1, 75 Br, 15 O 5 13 N 5
- radionuclide that is incorporated in the instant radiolabeled compounds will depend on the specific analytical or pharmaceutical application of that radiolabeled compound.
- compounds that incorporate 3 H, 35s, 125j or 82 ⁇ r W UJ generally be most useful.
- compounds that incorporate a radionuclide selected from ⁇ C, ⁇ 8 F, ⁇ 23 I, * 3 ⁇ I 5 7 %r, 7 ⁇ Br or 77 ⁇ r are preferred.
- incorporation of a chelating radionuclide such as Tc"" m may also be useful.
- * 8 F may be preferable over 1 * C because with the longer half-life of 1 8 F, imaging can be carried out long enough to allow a more specific signal to develop and improved conditions for receptor quantification studies.
- Compounds can be radiolabeled with either positron or gamma emitting radionuclides.
- Radiolabeled PDElO inhibitors when labeled with the appropriate radionuclide, are potentially useful for a variety of in vitro and/or in vivo imaging applications.
- Specific examples of possible imaging applications include, but are not limited to, determining the location of, the relative activity of and/or quantifying PDElO, radioimmunoassays of PDElO inhibitors j and autoradiography to determine the distribution of PDElO in a mammal or an organ or tissue sample thereof.
- the dose required to effectively inhibit the PDElO enzyme can be determined by the blockade of the PET radiotracer image in humans.
- the instant radiolabeled PDElO inhibitors when labeled with the positron emitting radionuclide, such as 11 C, 1 %V, 15 ⁇ and 13N, are useful for positron emission tomographic (PET) imaging of PDElO in the brain of living humans and experimental animals.
- PET positron emission tomographic
- These radiolabeled PDElO inhibitors may be used as research tools to study the interaction of unlabeled PDElO inhibitors with PDElO in vivo via competition between the unlabeled drug and the radiolabeled compound for binding to the receptor.
- the radiolabeled PDElO inhibitors may be used to help define a clinically efficacious dose of a PDElO inhibitor.
- the radiolabeled PDElO inhibitors can be used to provide information that is useful for choosing between potential drug candidates for selection for clinical development.
- the radiolabeled PDElO inhibitors may also be used to study the regional distribution and concentration of PDElO in the living human brain, as well as the brain of living experimental animals and in tissue samples.
- the radiolabeled PDElO inhibitors may also be used to study disease or pharmacologically related changes in PDElO concentrations.
- PET tracers such as the present radiolabeled PDElO inhibitors and currently available PET technology can be used, but is not limited to, to obtain the following information: relationship between level of receptor occupancy by candidate PDElO inhibitors and clinical efficacy in patients; dose selection for clinical trials of PDElO inhibitors prior to initiation of long term clinical studies; comparative potencies of structurally novel PDElO inhibitors; investigating the influence of PDElO inhibitors on in vivo transporter affinity and density during the treatment of clinical targets with PDElO inhibitors and other agents; changes in the density and distribution of PDElO, for example, 1) during the active stage of a psychiatric disease or condition, 2) for the evaluation of efficacy during treatment, or 3) during remission; changes in PDElO expression and distribution in CNS disorders; imaging neurodegenerative disease when PDElO is upregulated; imaging neurodegenerative disease when PDElO is involved; and the like.
- Isotopically-labeled compounds of formula I, Ha, lib, Ilia or HIb can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed to produce radiolabeled derivatives.
- a compound of Formula I, Ha, or Ilia 2- ⁇ 2-[3-(4-hydroxyphenyl)-4- oxo-3,4-dihydroquinazolin-2-yl] ethyl ⁇ -4-isopropoxy-l/f-isoindole-l,3(2/f)-dione (D-6), can be labeled with * ⁇ C, to produce 4-isopropoxy-2- ⁇ 2-[3-(4-l lC-methoxyphenyl)-4-oxo-3,4- dihydroquinazolin-2-yl] ethyl ⁇ -lH-isoindole-1 ,3(2H)-dione (E-I ), which in turn can be used in
- a compound of Formula I, Ua, or IHa, 2- ⁇ 2-[3-(4- methoxyphenyl)-4-oxo-3 ,4-dihydroquinazolin-2-y 1] ethyl ⁇ -4 -pyr ⁇ din-4-yl- 1 H-isoindole- 1 , 3 (2H)- dione, can be labeled and used in PET studies.
- radiolabeled PDElO inhibitors of the present invention have utility in imaging PDElO or for diagnostic imaging with respect to any of the mentioned neurological and psychiatric disorders associated with PDElO dysfunction.
- the present invention is also directed to a method for quantitative imaging of
- the present invention is also directed to a method for quantitative imaging of tissues bearing PDElO in a mammal which comprises administering to a mammal in need of such quantitative imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is also directed to a method for quantitative imaging of PDElO in tissues of a mammalian species which comprises administering to the mammalian species in need of such quantitative imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is also directed to a method for quantitative imaging of PDEl 0 in the brain in a mammal which comprises administering to a mammal in need of such quantitative imaging an effective amount of the radiolabeled compound of the present invention.
- the present invention is further directed to a method for the detection or quantification of PDEl 0 in mammalian tissue which comprises administering to a mammal in which such quantification is desired an effective amount of the radiolabeled compound of the present invention.
- the mammal is a human. It will be understood that, as used herein, references to the compounds of structural formulas I, Ha, Hb, IHa, and HIb are meant to also include the pharmaceutically acceptable salts, and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or in other synthetic manipulations.
- the compounds of the present invention may be administered in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids.
- inorganic bases and organic bases include aluminum, ammonium, calcium, copper (cupris and cuprous), ferric, ferrous, lithium, magnesium, manganese (manganic and manganous), potassium, sodium, zinc and the like salts.
- Particular embodiments include the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'- dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylamino-ethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidme, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, raandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid and the like.
- composition as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- This term in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- compositions of the present invention encompass any composition made by combining a compound of the present invention and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- the pharmaceutical compositions of the present invention comprise compounds of the invention (or pharmaceutically acceptable salts thereof) as an active ingredient, a pharmaceutically acceptable carrier, and optionally one or more additional therapeutic agents or adjuvants.
- Such additional therapeutic agents can include, for example, i) opiate agonists or antagonists, ii) calcium channel antagonists, in) 5HT receptor agonists or antagonists, iv) sodium channel antagonists, v) NMDA receptor agonists or antagonists, vi) COX-2 selective inhibitors, vii) NKl antagonists, viii) non-steroidal anti-inflammatory drugs ("NSAID”), ix) selective serotonin reuptake inhibitors ("SSRI”) and/or selective serotonin and norepinephrine reuptake inhibitors (“SSNRI”), x) tricyclic antidepressant drugs, xi) norepinephrine modulators, xii) lithium, xiii) valproate, xiv) neurontin (gabapentin), xv) pregabalin, and xvi) sodium channel blockers.
- NSAID non-steroidal anti-inflammatory drugs
- SSNRI selective serotonin reuptake inhibitor
- compositions suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
- Treating or “treatment of a disease state includes: 1) preventing the disease state, i.e. causing the clinical symptoms of the disease state not to develop in a subject that may be exposed to or predisposed to the disease state, but does not yet experience or display symptoms of the disease state; 2) inhibiting the disease state, i.e., arresting the development of the disease state or its clinical symptoms; 3) or relieving the disease state, i.e., causing temporary or permanent regression of the disease state or its clinical symptoms.
- the subject treated in the present methods is generally a mammal, in particular, a human being, male or female, in whom therapy is desired.
- the term "therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician. It is recognized that one skilled in the art may affect the neurological and psychiatric disorders by treating a patient presently afflicted with the disorders or by prophylactically treating a patient afflicted with such disorders with an effective amount of the compound of the present invention.
- treatment refers to all processes wherein there may be a slowing, interrupting, arresting, controlling,, or stopping of the progression of the neurological and psychiatric disorders described herein, but does not necessarily indicate a total elimination of all disorder symptoms, as well as the prophylactic therapy to retard the progression or reduce the risk of the noted conditions, particularly in a patient who is predisposed to such disease or disorder.
- the compounds of the invention are useful in methods of treating a neurological or psychiatric disorder associated with PDElO dysfunction in a patient such as a mammal in need of such inhibition comprising the administration of an effective amount of the compound.
- a variety of other mammals can be treated according to the method of the present invention.
- the subject compounds are useful in a method of inhibiting PDElO activity in a patient such as a mammal in need of such inhibition comprising the administration of an effective amount of the compound.
- primates, especially humans a variety of other mammals can be treated according to the method of the present invention.
- inhibitors of PDElO and, in particular inhibitors of PDElOA will provide therapeutic benefit to those individuals suffering from psychiatric and cognitive disorders.
- the unique and exclusive distribution of PDElOA in the medium spiny projection neurons of the striatum, which form the principle site for cortical and dopaminergic input within basal ganglia suggests that it may be possible and desirable to identify inhibitors of PDElO to ameliorate or eliminate unwanted cellular signaling within this site.
- Applicants believe that inhibition of PDElOA in the striatum will result in increased cAMP/cGMP signaling and striatal output, which has the potential to restore behavioral inhibition that is impaired in cognitive disease such as schizophrenia.
- compounds of the invention provide a method for treating or ameliorating diseases or conditions in which striatal hypofanction is a prominent feature or ones in which basal ganglia dysfunction plays a role, such as, Parkinson's disease, Huntington's disease, schizophrenia, obsessive-compulsive disorders, addiction and psychosis.
- Other conditions for which the inhibitors described herein may have a desirable and useful effect include those requiring a reduction in activity and reduced response to psychomotor stimulants or where it would be desirable to reduce conditional avoidance responses, which, is often predictive of clinical antipsychotic activity.
- a selective PDElO inhibitor refers to an organic molecule that effectively inhibits an enzyme from the PDElO family to a greater extent than enzymes from the PDE 1-9 or PDEl 1 families
- a selective PDElO inhibitor Is an organic molecule having a Ki for inhibition of PDElO that is less than or about one-tenth that for a substance that is an inhibitor for another PDE enzyme.
- the organic molecule inhibits PDElO activity to the same degree at a concentration of about one-tenth or less than the concentration required for any other PDE enzyme.
- a selective PDElO inhibitor is an organic molecule, having a Ki for inhibition of PDEl 0 that is less than or about one-hundredth that for a substance that is an inhibitor for another PDE enzyme.
- the organic molecule inhibits PDElO activity to the same degree at a concentration of about one- hundredth or less than the concentration required for any other PDE enzyme.
- a "selective PDElO inhibitor” can be identified, for example, by comparing the ability of an organic molecule to inhibit PDElO activity to its ability to inhibit PDE enzymes from the other PDE families.
- an organic molecule may be assayed for its ability to inhibit PDElO activity, as well as PDElA, PDElB 5 PDElC, PDE2A, PDE3A, PDE3B, PDE4A, PDE4B, PDE4C, PDE4D, PDE5A, PDE6A, PDE6B, PDE6C, PDE7A, PDE7B, PDE8A, PDE8B, PDE9 A, and/or PDEI l A.
- compounds of the present invention provide a method for treating schizophrenia or psychosis comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- DSM-IV-TR The Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) (2000, American Psychiatric Association, Washington DC) provides a diagnostic tool that includes paranoid, disorganized, catatonic or undifferentiated schizophrenia and substance-induced psychotic disorders.
- DSM-IV-TR The Diagnostic and Statistical Manual of Mental Disorders
- Schizophrenia or psychosis includes the diagnosis and classification of these mental disorders as described in DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- Disorders and conditions encompassed herein include, but are not limited to, conditions or diseases such as schizophrenia or psychosis, including schizophrenia (paranoid, disorganized, catatonic, undifferentiated, or residual type), schizophreniform disorder, schizoaffective disorder, for example of the delusional type or the depressive type, delusional disorder, psychotic disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder due to a general medical condition and substance-induced or drug-induced (for example psychosis induced by alcohol, amphetamine, cannabis, cocaine, hallucinogens, inhalants, opioids, phencyclidine, ketamine and other dissociative anaesthetics, and other psychostimulants), psychosispsychotic disorder, psychosis associated with affective disorders, brief reactive psychosis, schizoaffective psychosis,
- the compounds of the present invention provide a method for treating cognitive disorders comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- the DSM-IV-TR also provides a diagnostic tool that includes cognitive disorders including dementia, delirium, amnestic disorders and age-related cognitive decline.
- cognitive disorders includes the diagnosis and classification of these disorders as described in DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- disorders and conditions encompassed herein include, but are not limited to, disorders that comprise as a symptom a deficiency in attention and/or cognition, such as dementia (associated with Alzheimer's disease, ischemia, multi-infarct dementia, trauma, intracranial tumors, cerebral trauma, vascular problems or stroke, alcoholic dementia or other drag-related dementia, AIDS, HIV disease, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt Jacob disease, perinatal hypoxia, other general medical conditions or substance abuse), Alzheimer's disease, multi-infarct dementia, AIDS-related dementia, and Fronto temperal dementia, delirium, amnestic disorders or age related cognitive decline.
- dementia associated with Alzheimer's disease, ischemia, multi-infarct dementia, trauma, intracranial tumors, cerebral trauma, vascular problems or stroke, alcoholic dementia or other drag-related dementia, AIDS, HIV disease, Parkinson's disease, Huntington's disease, Pick's disease, Creutzfeldt Jacob disease, perinatal hypoxia
- compounds of the present invention provide a method for treating anxiety disorders comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- the DSM-IV-TR also provides a diagnostic tool that includes anxiety disorders as generalized anxiety disorder, obsessive- compulsive disorder and panic attack.
- anxiety disorders includes the diagnosis and classification of these mental disorders as described in DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- disorders and conditions encompassed herein include, but are not limited to, anxiety disorders such as, acute stress disorder, agoraphobia, generalized anxiety disorder, obsessive-compulsive disorder, panic attack, panic disorder, post-traumatic stress disorder, separation anxiety disorder, social phobia, specific phobia, substance-induced anxiety disorder and anxiety due to a general medical condition.
- compounds of the present invention provide a method for treating substance-related disorders and addictive behaviors comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- the DSM-IV-TR also provides a diagnostic tool that includes persisting dementia, persisting amnestic disorder, psychotic disorder or anxiety disorder induced by substance abuse, and tolerance of, dependence on or withdrawal from substances of abuse.
- disorders and addictive behaviors includes the diagnosis and classification of these mental disorders as described in DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- Disorders and conditions encompassed herein include, but are not limited to, substance-related disorders and addictive behaviors, such as substance-induced delirium, persisting dementia, persisting amnestic disorder, psychotic disorder or anxiety disorder, drug addiction, tolerance, and dependence or withdrawal from substances including alcohol, amphetamines, cannabis, cocaine, hallucinogens, inhalants, nicotine, opioids, phencyclidine, sedatives, hypnotics or anxiolytics.
- compounds of the present invention provide a method for treating obesity or eating disorders associated with excessive food intake, and complications associated therewith, comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- obesity is included in the tenth edition of the International Classification of Diseases and Related Health Problems (ICD- 10) (1992 World Health Organization) as a general medical condition.
- the DSM-IV-TR also provides a diagnostic tool that includes obesity in the presence of psychological factors affecting medical condition.
- the term "obesity or eating disorders associated with excessive food intake” includes the diagnosis and classification of these medical conditions and disorders described in ICD-10 and DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- Disorders and conditions encompassed herein include, but are not limited to, obesity, bulimia nervosa and compulsive eating disorders.
- compounds of the present invention provide a method for treating mood and depressive disorders comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- mood and depressive disorders includes the diagnosis and classification of these medical conditions and disorders described in the DSM-IV-TR and the term is intended to include similar disorders described in other sources.
- disorders and conditions encompassed herein include, but are not limited to, bipolar disorders, mood disorders including depressive disorders, major depressive episode of the mild, moderate or severe type, a manic or mixed mood episode, a hypomanic mood episode, a depressive episode with atypical features, a depressive episode with melancholic features, a depressive episode with catatonic features, a mood episode with postpartum onset, post-stroke depression; major depressive disorder, dysthymic disorder, minor depressive disorder, premenstrual dysphoric disorder, post-psychotic depressive disorder of schizophrenia, a major depressive disorder superimposed on a psychotic disorder such as delusional disorder or schizophrenia, a bipolar disorder, for example, bipolar I disorder, bipolar II disorder, cyclothymic disorder, depression including unipolar depression, seasonal depression and post-partum depression, premenstrual syndrome (PMS) and premenstrual dysphoric disorder (PDD), mood disorders due to a general medical condition, and substance-induced mood disorders.
- a bipolar disorder
- compounds of the invention provide methods for treating other types of cognitive, learning and mental related disorders including, but not limited to, learning disorders, such as a reading disorder, a mathematics disorder, or a disorder of written expression, attention-deficit/hyperactivity disorder, age-related cognitive decline, pervasive developmental disorder including autistic disorder, attention disorders such as attention-deficit hyperactivity disorder (ADHD) and conduct disorder; an NMDA receptor- related disorder, such as autism, depression, benign forgetfulness, childhood learning disorders and closed head injury; a neurodegenerative disorder or condition, such as neurodegeneration associated with cerebral trauma, stroke, cerebral infarct, epileptic seizure, neurotoxin poisoning, or hypoglycemia-induced neurodegeneration; multi-system atrophy; movement disorders, such as akinesias and akinetic-rigid syndromes (including, Parkinson's disease, drug-induced parkinsonism, postencephalitic parkinsonism, progressive supranuclear palsy, multiple system atrophy, corticobas
- compounds of the present invention provide a method for treating pain comprising administering to a patient in need thereof an effective amount of a compound of the present invention.
- pain embodiments are bone and j oint pain (osteoarthritis), repetitive motion pain, dental pain, cancer pain, myofascial pain (muscular injury, fibromyalgia), perioperative pain (general surgery, gynecological), chronic pain and neuropathic pain.
- schizophrenia bipolar disorder
- depression including unipolar depression, seasonal depression and post-partum depression
- premenstrual syndrome PMS
- premenstrual dysphoric disorder PDD
- learning disorders pervasive developmental disorders, including autistic disorder, attention disorders including Attention-Deficit/Hyperactivity Disorder, autism, tic disorders including Tourette's disorder, anxiety disorders including phobia and post traumatic stress disorder, cognitive disorders associated with dementia, AIDS dementia, Alzheimer's, Parkinson's, Huntington's disease, spasticity, myoclonus, muscle spasm, tinnitus and hearing impairment and loss are of particular importance.
- the present invention is further directed to a method for the manufacture of a medicament for treating neurological or psychiatric disorders associated with PDElO dysfunction, including those disorders and conditions listed above, in humans and animals comprising combining a compound of the present invention with one or more additional therapeutic agents, carriers, or diluents.
- the present invention is also directed to compounds of the invention for use in the treatment of neurological or psychiatric disorders associated with PDElO dysfunction, including those disorders and conditions listed above, in humans and animals comprising combining a compound of the present invention with one or more additional therapeutic agents, carriers, or diluents.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reduction of risk of the diseases, disorders and conditions noted herein.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reduction of risk of the aforementioned diseases, disorders and conditions in combination with other agents.
- the compounds of the present invention may be used in combination with one or more other drags in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of the present invention or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- the terms "administration of and or "administering a" compound should be understood to mean providing a compound of the invention or a prodrug of a compound of the invention to the individual in need of treatment.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention may be desirable.
- the combination therapy may also includes therapies in which the compound of the present invention and one or more other drugs are administered on different overlapping schedules.
- the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used.
- the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1:1000, such as about 200:1 to about 1:200.
- Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
- the compound of the present invention and other active agents may be administered separately or in conjunction.
- the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- the subject compounds may be used alone or in combination with other agents which are known to be beneficial in the subject indications or other drugs that affect receptors or enzymes that either increase the efficacy, safety, convenience, or reduce unwanted side effects or toxicity of the compounds of the present invention.
- the subject compound and the other agent may be co-administered, either in concomitant therapy or in a fixed combination.
- the subject compound may be employed in combination with anti- Alzheimer' s agents, beta-secretase inhibitors, gamma-secretase inhibitors, HMG-CoA reductase inhibitors, NSAID 's including ibuprofen, vitamin E, and anti-amyloid antibodies.
- the subject compound may be employed in combination with sedatives, hypnotics, anxiolytics, antipsychotics, antianxiety agents, cyclopyrrolones, imidazopyridines, pyrazolopyrimidines, minor tranquilizers, melatonin agonists and antagonists, melatonergic agents, benzodiazepines, barbiturates, 5HT-2 antagonists, and the like, such as: adinazolam, allobarbital, alonimid, alprazolam, amisulpride, amitriptyline, amobarbital, amoxapine, aripiprazole, atypical antipsychotics, bentazepam, benzoctamine, brotizolam, bupropion, busprione, butabarbital, butalbital, capuride, carbocloral, chloral betaine, chloral hydrate, clomipramine, clonazepam, cloperidone,
- the subject compound may be employed in combination with levodopa (with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide), anticholinergics such as biperiden (optionally as its hydrochloride or lactate salt) and trihexyphenidyl (benzhexol) hydrochloride, COMT inhibitors such as entacapone, MOA-B inhibitors, antioxidants, A2a adenosine receptor antagonists, cholinergic agonists, NMDA receptor antagonists, serotonin receptor antagonists and dopamine receptor agonists such as alentemol, bromocriptine, fenoldopam, lisuride, naxagolide, pergolide and pramipexole.
- levodopa with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide
- anticholinergics such as biperi
- the dopamine agonist may be in the form of a pharmaceutically acceptable salt, for example, alentemol hydrobromide, bromocriptine mesylate, fenoldopam mesylate, naxagolide hydrochloride and pergolide mesylate. Lisuride and pramipexol are commonly used in a non-salt form.
- the subject compound may be employed in combination with a compound from the phenothiazine, thioxanthene, heterocyclic dibenzazepine, butyrophenone, diphenylbutylpiperidine and indolone classes of neuroleptic agent.
- Suitable examples of phenothiazines include chlorpromazine, mesoridazine, thioridazine, acetophenazine, fluphenazine, perphenazine and trifluoperazine.
- Suitable examples of thioxanthenes include chlorprothixene and thiothixene.
- An example of a dibenzazepine is clozapine.
- An example of a butyrophenone is haloperidol.
- An example of a diphenylbutylpiperidine is pimozide.
- An example of an indolone is molindolone.
- Other neuroleptic agents include loxapine, sulpiride and risperidone.
- the neuroleptic agents when used in combination with thesubject compound may be in the form of a pharmaceutically acceptable salt, for example, chlorpromazine hydrochloride, mesoridazine besylate, thioridazine hydrochloride, acetophenazine maleate, fluphenazine hydrochloride, flurphenazine enathate, fluphenazine decanoate, trifluoperazine hydrochloride, thiothixene hydrochloride, haloperidol decanoate, loxapine succinate and molindone hydrochloride.
- a pharmaceutically acceptable salt for example, chlorpromazine hydrochloride, mesoridazine besylate, thioridazine hydrochloride, acetophenazine maleate, fluphenazine hydrochloride, flurphenazine enathate, fluphenazine decanoate, trifluoperazine hydrochloride, thiothix
- Perphenazine, chlorprothixene, clozapine, haloperidol, pimozide and risperidone are commonly used in a non-salt form.
- the subject compound may be employed in combination with acetophenazine, alenternol, aripiprazole, amisulpride, benzhexol, bromocriptine, biperiden, chlorpromazine, chlorprothixene, clozapine, diazepam, fenoldopam, fluphenazine, haloperidol, levodopa, levodopa with benserazide, levodopa with carbidopa, lisuride, loxapine, mesoridazine, molindolone, naxagolide, olanzapine, pergolide, pe ⁇ henazine, pimozide, pramipexole, quetiapine,
- the subject compound may be employed in combination with an anti-depressant or anti-anxiety agent, including norepinephrine reuptake inhibitors
- SSRIs selective serotonin reuptake inhibitors
- MAOIs monoamine oxidase inhibitors
- RIMAs reversible inhibitors of monoamine oxidase
- SNRIs noradrenaline reuptake inhibitors
- corticotropin releasing factor (CRF) antagonists ⁇ -adrenoreceptor antagonists
- neurokinin- 1 receptor antagonists atypical anti-depressants
- benzodiazepines 5-HT IA agonists or antagonists, especially 5-HT JA partial agonists, and corticotropin releasing factor (CRP) antagonists.
- Specific agents include: amitriptyline, clomipramine, doxepin, imipramine and trimipramine; amoxapine, desipramine, maprotiline, nortriptyline and protriptyline; fluoxetine, fluvoxamine, paroxetine and sertraline; isocarboxazid, phenelzine, tranylcypromine and selegiline; moclobemide: venlafaxine; duloxetine; aprepitant; bupropion, lithium, nefazodone, trazodone and viloxazine; alprazolam, chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam, oxazepam and prazepam; buspirone, flesinoxan, gepirone and ipsapirone, and pharmaceutically acceptable salts thereof.
- the compounds of the present invention may be administered by oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant), by inhalation spray, nasal, vaginal, rectal, sublingual, or topical routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- parenteral e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant
- inhalation spray nasal, vaginal, rectal, sublingual, or topical routes of administration
- nasal, vaginal, rectal, sublingual, or topical routes of administration may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- the compounds of the invention are effective for
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients that are suitable for the manufacture of tablets.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredients are mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions, oily suspensions, dispersible powders or granules, oil-in- water emulsions, and sterile injectable aqueous or oleagenous suspension may be prepared by standard methods known in the art.
- the compounds of the invention are radiolabeled and/or are used as PET tracers, it is preferable that administration be done intravenously.
- Radiotracers labeled with positron emitting radionuclides are generally administered via intravenous injection within one hour of their synthesis due to the short half-life of the radionuclides involved, which is typically 20 and 110 minutes for C-1 1 and F-18, respectively.
- the amount required for imaging will normally be determined by the prescribing physician with the dosage generally varying according to the quantity of emission from the radionuclide used.
- an effective amount will be the amount of compound sufficient to produce emissions in the range of from about l-5mCi.
- the mass associated with a PET tracer is in the form of the natural isotope, for example, 1 ⁇ C for an 11 C PET tracer and 19F for an 1 ⁇ F PET tracer, respectively.
- This mass comprises from about 1 ⁇ g to about 50 ⁇ g of a radiolabeled PDElO inhibitor in order to avoid significant inhibition of PDElO.
- the following illustrative procedure may be utilized when performing PET imaging studies on patients in a clinical setting.
- the human subject is either unmedicated or pre- medicated with unlabeled PDElO inhibitor or other pharmacological intervention some time prior to the day of the experiment and is fasted for at least 12 hours allowing water intake ad libitum.
- a 20 G two inch venous catheter is inserted into the contralateral ulnar vein for radiotracer administration.
- Administration of the PET tracer is often timed to coincide with time of maximum (Tmax) or minimum (Tmin) of PDElO inhibitor (or other compound of intervention) concentration in the blood.
- the human subject is positioned in the PET camera and a tracer dose of [llCJExample 5 ( ⁇ 20 mCi) is administered via i.v. catheter.
- Either arterial or venous blood samples are taken at appropriate time intervals throughout the PET scan in order to analyze and quantitate the fraction of umetabolized [ ⁇ C] (Example 5) in plasma.
- Images are acquired for up to 120 minutes. Within ten minutes of the injection of radiotracer and at the end of the imaging session, 1 ml blood samples are obtained for determining the plasma concentration of any unlabeled PDElO inhibitor (or other compound of intervention) which may have been administered before the PET tracer.
- TAC time activity curves
- Inhibition of PDElO is then calculated based on the change of BP in the presence of PDElO inhibitors at the various dosing paradigms as compared to the BP in the unmedicated state.
- Inhibition curves are generated by plotting the above data vs the dose (concentration) of PDElO inhibitors.
- the ID50 values are obtained by curve fitting the dose- rate/inhibition curves with the following equation:
- B Ao ⁇ Ao*I/( ID 5O + I ) + NS
- B is the %-Dose/g of radiotracer in tissues for each dose of clinical candidate
- Ao is the specifically bound radiotracer in a tissue in the absence of PDElO inhibitors
- I is the injected dose of antagonist
- ID50 is the dose of compound which inhibits 50% of specific radiotracer binding to PDElO
- NS is the amount of non-specifically bond radiotracer.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reduction of risk of the diseases, disorders and conditions noted herein.
- the dose of the active ingredient in the composition may be varied, however, it is necessary that the amount of the active ingredient be such that a suitable dosage form is obtained.
- the active ingredient may be administered to patients (animals and human) in need of such treatment in dosages that will provide optimal pharmaceutical efficacy.
- the selected dosage depends upon the desired therapeutic effect, on the route of administration, and on the duration of the treatment.
- the dose will vary from patient to patient depending upon the nature and severity of disease, the patient's weight, special diets being adhered to by the patient, concurrent medication, and other factors which those skilled in the art will recognize.
- dosage levels between 0.01 to 10 mg/kg of body weight daily are administered to the patient, e.g., humans and elderly humans.
- the dosage range will generally be about 0.5 mg to 1.0 g per patient per day which may be administered in single or multiple doses.
- the dosage range will be about 0.5 mg to 500 mg per patient per day; in another embodiment about 0.5 mg to 200 mg per patient per day; and in yet another embodiment about 5 mg to 50 mg per patient per day.
- Pharmaceutical compositions of the present invention may be provided in a solid dosage formulation such as comprising about 0.5 mg to 500 mg active ingredient, or comprising about 1 mg to 250 mg active ingredient.
- the pharmaceutical composition may be provided in a solid dosage formulation comprising about 1 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg active ingredient.
- compositions may be provided in the form of tablets containing 1.0 to 1000 mg of the active ingredient, such as, 1, 5, 10, 15, 20, 25, 50, 75, 100, 15O 5 200, 250, 300, 400, 500, 60O 5 750, 800, 900, and 1000 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably in a regimen of once or twice per day.
- the compounds of the following examples had activity in inhibiting the human
- PDElO enzyme as described in the biological assay that follows, generally with a Ki of less than about 1 ⁇ M.
- Many of the compounds within the present invention had activity in inhibiting the human PDElO enzyme in the aforementioned assay, generally with a Ki of less than about 0.1 ⁇ M.
- Such a result is indicative of the intrinsic activity of the compounds in use as inhibitors of the PDElO enzyme.
- a substance is considered to effectively inhibit PDElO activity if it has a Ki of less than or about 1 ⁇ M, preferably less than or about 0.1 ⁇ M.
- the present invention also includes compounds within the generic scope of the invention which possess activity as inhibitors of other phosphodiesterase enzymes.
- the PDElO Ki is a measure of the ability of the test compound to inhibit the action of the PDElO enzyme.
- the Ki of the compound was determined for PDEs 1-5, 7-9, and 11.
- the selectivity is defined as the Ki of the test compound for the most potently inhibited PDE other than PDElO, divided by the Ki for PDElO.
- the PDE enzyme most potently inhibited other than PDElO is listed.
- Reactions used to generate the compounds of this invention are prepared by employing reactions as shown in the schemes and examples herein, in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures. Starting materials are made according to procedures known in the art or as illustrated herein. In some cases the final product may be further modified, for example, by manipulation of substituents. These manipulations may include, but are not limited to, reduction, oxidation, alkylation, acylation, and hydrolysis reactions which are commonly known to those skilled in the art. In some cases the order of carrying out the foregoing reaction schemes may be varied to facilitate the reaction or to avoid unwanted reaction products. The following examples are provided so that the invention might be more fully understood. These examples are illustrative only and should not be construed as limiting the invention in any way.
- Me methyl; Et: ethyl; t-Bu: tert- butyl; Ar: aryl; Ph: phenyl; Bn: benzyl; Ac: acetyl; THF: tetrahydrofuran; DEAD: diethylazodicarboxylate; DIPEA: N,N-diisopropylethylamine; DMSO: dimethylsulfoxide; EDC: N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide; HOAT: 1- hydroxy-7-aza-benzotriazole; HOBT: hydroxybenzotriazole hydrate; Boc: tert-butyloxy carbonyl; Et 3 N: triethylamine; DCM: dichloromethane; DCE: dichloroethane; BSA: bovine serum albumin; TFA: trifluoracetic acid; DMF: N,N
- anthranilic acid (or alternatively a l-amino-2- carboxy-heterocycle) can be coupled in one pot to an aliphatic carboxylic acid with triphenylphosphite, followed by addition of aniline (or an amino-substituted heterocycle) to provide compounds A-I of the current invention.
- D-3 (835 g, 2.50 mmol) was dissolved in DCM (5 mL) and TFA (5 mL) and stirred at room temperature for 2 hours. Solvents were removed and the residue azeotroped with toluene. This provided D-4 (690 g, 100%) as an off-white solid. Data for D-4: LRMS: calculated M + H for C14H15NO5: 278.27; Found: 278.62.
- Anthranilic acid (1.0 g, 7.29 mmol), D-4 (2.02 g, 7.29 mmol), and triphenylphosphite (2.01 mL, 7.66 mmol) were dissolved in pyridine (20 mL) and heated in a sealed tube at 100 0 C for 2 hours. After cooling to room temperature, the tube was opened, 4- benzyloxyaniline hydrochloride (1.89 g, 8.02 mmol) was added and heating at 100 0 C was resumed for 4 hours. The pyridine was removed by azeolroping with toluene, and the residue was suspended in CHCI3 and toluene.
- [1 *C]CO2 was provided by Siemens, North Wales, PA.
- the [1 lC]CO2 was converted to [1 lCJmethyl iodide using a GE Medical Systems TRACERlab FXc system.
- Radiochemical procedures were carried out using a Gilson 233XL liquid handler.
- Radiotracers were purified by reverse-phase HPLC using a Waters 600E controller and the preparative HPLC runs were monitored at 254nm using a Pharmacia-Biotech UV-MII UV detector and a Bioscan FlowCount photodiode detector.
- the radiochemical purities and identities were determined by co-injection with authentic standards on an analytical Waters 600E HPLC system equipped with a Waters 996 UV detector and a photodiode radiodetector (Bioscan FlowCount).
- reaction mixture was heated at 45°C for 3 minutes, diluted with H 2 O (0.5 mL) and purified by HPLC (Phenomenex Gemini C18 column (1O x 150 mm, 5 ⁇ m), acetoiiitrile (solvent A) and 10 mMNa2HPO4 (solvent B) under 15-min linear gradient condition consisting of 50%A 50%B to 70%A 30%B at 5 ml/min.
- HPLC Henomenex Gemini C18 column (1O x 150 mm, 5 ⁇ m), acetoiiitrile (solvent A) and 10 mMNa2HPO4 (solvent B) under 15-min linear gradient condition consisting of 50%A 50%B to 70%A 30%B at 5 ml/min.
- the radiochemical and chemical purity was determined at 270 nm using a Waters XTerra Cl 8 column (4.6 x 150 mm, 5 ⁇ m), isocratic 60% acetonitrile, 40% H2O (0.1% TFA) @ 1 niL/rnin, providing a retention time of 6 minutes for E- 1.
- the specific activity was determined by counting an aliquot of E-I in a dose calibrator and determining the mass by analytical HPLC against a mass calibration curve for D-7.
- 2-Amino-4-methylbenzoic acid 150 mg, 0.99 mmol
- D-4 275 mg, 0.99 mmol
- triphenylphosphite 286 ⁇ L, 1.1 mmol
- 6- aminoindazole 132 mg, 0.99 mmol
- heating at 100 0 C was resumed for 4 hours.
- the reaction was partitioned between EtOAc and saturated NaHCO3. After separation, the organic layer was washed with brine, dried over Na 2 SO 4 , and concentrated by rotary evaporation.
- [1 lcjMethyl iodide was converted to [1 lC]methyl triflate by distillation through a column ( ⁇ 5 x 30 mm) of silver triflate heated to 200 0 C.
- the [1 lC]methyl triflate was bubbled into mixture of G-I (0.2-0.5 mg) in DMF (0.25 mL) containing IM sodium hydroxide (3 uL) at room temperature. When the amount of radioactivity trapped in solution peaked, the mixture was allowed to set at room temperature for 3 min.
- reaction mixture was then diluted with H 2 O (0.5 mL) and purified by HPLC (Phenomenex Synergi Polar-RP 10 x 150 mm, 5 ⁇ m), using acetonitrile (solvent A) and 0.1% trifluoracetic acid in water (solvent B) under isocratic conditions of 55% A 45%B at 5 ml/min.
- HPLC Henomenex Synergi Polar-RP 10 x 150 mm, 5 ⁇ m
- the activity of the compounds in accordance with the present invention as PDElO inhibitors may be readily determined without undue experimentation using a fluorescence polarization (FP) methodology well known in the art (Huang, W., et al., J. Biomol Screen, 2002, 7: 215).
- FP fluorescence polarization
- the compounds of the Examples had activity in reference assays by exhibiting their ability to inhibit the hydrolysis of the phosphate ester bond of a cyclic nucleotide. Any compound exhibiting a Ki (inhibitory constant) below 1 ⁇ M would be considered a PDElO inhibitor as defined herein.
- Ki inhibitory constant
- PDEl 0A2 was amplified from human fetal brain cDNA (Clontech, Mountain View, CA) using a forward primer corresponding to nucleotides 56-77 of human PDE10A2 (Accession No. AF 127480, Genbank Identifier 4894716), containing a Kozak consensus sequence, and a reverse primer Corresponding to nucleotides 2406-2413 of human PDE10A2 (Accession No. AF127480, Genbank Identifier 4894716).
- Amplification with Easy-A polymerase (Stratagene, La Jolla, CA) was 95 0 C for 2 minutes followed by thirty three cycles of 95 0 C for 40 seconds, 55 0 C for 30 seconds, and 72 0 C for 2 minutes 48 seconds. Final extension was 72 0 C for 7 minutes.
- the PCR product was TA cloned into pcDNA3.2-TOPO (Invitrogen, Carlsbad, CA) according to standard protocol.
- AD293 cells with 70-80% confiuency were transiently transfected with human PDB10A2/pcDNA3.2-TOPO using Lipofectamine 2000 according to manufacturer specifications (Invitrogen, Carlsbad, CA). Cells were harvested 48 hours post-transfection and lysed by sonication (setting 3, 10 X 5 sec pulses) in a buffer containing 20 mM HEPES, 1 mM EDTA and protease inhibitor cocktail (Roche). Lysate was collected by centrifugation at 75,000 xg for 20 minutes. Supernatant containing the cytoplasmic fraction was used for evaluation of PDE10A2 activity.
- the fluorescence polarization assay for cyclic nucleotide phosphodiesterases was performed using an IMAP® FP kit supplied by Molecular Devices, Sunnyvale, CA (product # R8139). IMAP® technology has been applied previously to phosphodiesterase assays (Huang, W., et al., J. Biomol Screen, 2002, 7: 215). Assays were performed at room temperature in 384- well microliter plates with an incubation volume of 20.2 ⁇ L. Solutions of test compounds were prepared in DMSO and serially diluted with DMSO to yield 8 ⁇ L of each of 10 solutions differing by 3-fold in concentration, at 32 serial dilutions per plate.
- PDElO inhibitor can be any compound that is present at 5,000 times its Ki value in the assay described as follows, such as papaverine (see Siuciak, et al. Neuropharmacology (2006) 51 :386-396; Becker, et al.
- 0% of inhibition is determined by using DMSO (1% final concentrations).
- a Labcyte Echo 555 (Labcyte, Sunnyvale, CA) is used to dispense 200 nL from each well of the titration plate to the 384 well assay plate.
- a solution of enzyme (1/1600 dilution from aliquots; sufficient to produce 20% substrate conversion) and a separate solution of FAM- labeled cAMP PDE from Molecular Devices (product # R7506), at a final concentration of 50 nM are made in the assay buffer (10 mM Tris HCl, pH 7.2, 10 mM MgCl2, 0.05% NaN3 0.01% Tween-20, and 1 mM DTT).
- the enzyme and the substrate are then added to the assay plates in two consecutive additions of 10 ⁇ L, and then shaken to mix.
- the reaction is allowed to proceed at room temperature for 30 minutes.
- a binding solution is then made from the kit components, comprised of 80% Solution A, 20% Solution B and binding reagent at a volume of 1/600 the total binding solution.
- the enzymatic reaction is stopped by addition of 60 ⁇ L of the binding solution to each well of the assay plates and the plates are sealed and shaken for 10 seconds.
- the plate was incubated at room temperature for at least one hour prior to determining the fluorescence polarization (FP).
- the parallel and perpendicular fluorescence of each well of the plate was measured using a Perkin Elmer EnVisionTM plate reader (Waltham, MA). Fluorescence polarization (mP) was calculated from the parallel (S) and perpendicular (P) fluorescence of each sample well and the analogous values for the median control well, containing only substrate (So and Po), using the following
- Polarization (mP) 1000*(S/So-P/Po)/(S/So+P/Po).
- Dose-inhibition profiles for each compound were characterized by fitting the mP data to a four-parameter equation given below.
- n (0%r ⁇ P -100%mP)(Imax- Imm) 1 ⁇ ft/ n / ⁇ n / admir , ⁇ n / n w, r ⁇ , mP — — ⁇ + 100% mP + (0%mP - 100%mP)( 1 - Imax)
- the median signal of the "0% inhibition controls” (0%mP) and the median signal of the "100% inhibition controls” (100%mP) are constants determined from the controls located in columns 1-2 and 23-24 of each assay plate.
- An apparent (K 1n ) for FAM-labeled cAMP of 150 nM was determined in separate experiments through simultaneous variation of substrate and selected drug concentrations.
- PDElA (Cat# 60010), PDE3A (Cat# 60030), PDE4A1A (Cat# 60040), PDE5A1 (Cat# 6005O) 5 PDE6C (Cat# 60060), PDE7A (Cat# 6007O) 5 PDE8A1 (Cat# 6008O) 5 PDE9A2 (Cat# 60090), PDEl 1 A4 (Cat# 60110).
- Assays for PDE 1 through 11 were performed in parallel at room temperature in 384-well microliter plates with an incubation volume of 20.2 ⁇ L. Solutions of test compounds were prepared in DMSO and serially diluted with DMSO to yield 30 ⁇ L of each often solutions differing by 3-fold in concentration, at 32 serial dilutions per plate. 100% inhibition was determined by adding buffer in place of the enzyme and 0% inhibition is determined by using DMSO (1% final concentrations). A Labcyte POD 810 (Labcyte, Sunnyvale, CA) was used to dispense 200 nL from each well of the titration plate to make eleven copies of the assay plate for each titration, one copy for each PDE enzyme.
- a solution of each enzyme (dilution from aliquots, sufficient to produce 20% substrate conversion) and a separate solution of FAM- labeled cAMP or FAM-labeled cGMP from Molecular Devices ( Sunnyvale, CA, product # R7506 or cGMP#R7508), at a final concentration of 50 nM were made in the assay buffer (10 mM Tris HCl, pH 7.2, 10 rnM MgCl 2/ 0.05% NaN 3 0.01% Tween-20, and 1 mM DTT). Note that the substrate for PDE2 is 50 nM FAM c AMP containing 1000 nM of cGMP.
- the enzyme and the substrate were then added to the assay plates in two consecutive additions of 10 ⁇ L and then shaken to mix.
- the reaction was allowed to proceed at room temperature for 60 minutes.
- a binding solution was then made from the kit components, comprised of 80% Solution A, 20% Solution B and binding reagent at a volume of 1/600 the total binding solution.
- the enzymatic reaction was stopped by addition of 60 ⁇ L of the binding solution to each well of the assay plate.
- the plates were sealed and shaken for 10 seconds.
- the plates were incubated at room temperature for one hour, then the parallel and perpendicular fluorescence was measured using a Tecan Genios Pro plate reader (Tecan, Switzerland).
- the apparent inhibition constants for the compounds against all 11 PDE's was determined from the parallel and perpendicular fluorescent readings as described for PDElO FP assay using the following apparent KM values for each enzyme and substrate combination: PDElA (FAM cGMP) 70 nM, rhesus PD2A3 (FAM cAMP) 10,000 nM, PDE3A (FAM cAMP) 50 nM, PDE4A1A (FAM cAMP) 1500 nM 5 PDE5A1 (FAM cGMP) 400 nM, PDE6C (FAM cGMP) 700 nM, PDE7A (FAM cAMP) 150 nM, PDE8A1 (FAM cAMP) 50 nM, PDE9A2 (FAM cGMP) 60 nM, PDE10A2 (FAM cAMP) 15OnM, PDEl 1 A4 (FAM cAMP) 1000 nM.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Psychiatry (AREA)
- Addiction (AREA)
- Optics & Photonics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pain & Pain Management (AREA)
- Psychology (AREA)
- Epidemiology (AREA)
- Physics & Mathematics (AREA)
- Cardiology (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Hospice & Palliative Care (AREA)
- Ophthalmology & Optometry (AREA)
- Child & Adolescent Psychology (AREA)
- Heart & Thoracic Surgery (AREA)
- Vascular Medicine (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
Description














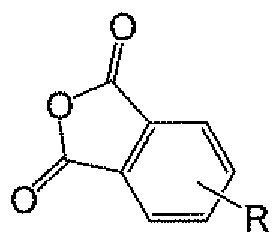




















Claims

Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012513203A JP5718323B2 (en) | 2009-05-29 | 2010-05-26 | Radiolabeled PDE10 inhibitor |
| ES10781130T ES2739973T3 (en) | 2009-05-29 | 2010-05-26 | Radiolabelled PDE10 inhibitors |
| EP10781130.9A EP2435048B1 (en) | 2009-05-29 | 2010-05-26 | Radiolabeled pde10 inhibitors |
| CA2763130A CA2763130C (en) | 2009-05-29 | 2010-05-26 | Radiolabeled pde10 inhibitors |
| US13/320,816 US8846000B2 (en) | 2009-05-29 | 2010-05-26 | Radiolabeled PDE10 inhibitors |
| AU2010254149A AU2010254149B2 (en) | 2009-05-29 | 2010-05-26 | Radiolabeled PDE10 inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18214909P | 2009-05-29 | 2009-05-29 | |
| US61/182,149 | 2009-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010138577A1 true WO2010138577A1 (en) | 2010-12-02 |
Family
ID=43223044
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/036186 Ceased WO2010138577A1 (en) | 2009-05-29 | 2010-05-26 | Radiolabeled pde10 inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8846000B2 (en) |
| EP (1) | EP2435048B1 (en) |
| JP (1) | JP5718323B2 (en) |
| AU (1) | AU2010254149B2 (en) |
| CA (1) | CA2763130C (en) |
| ES (1) | ES2739973T3 (en) |
| WO (1) | WO2010138577A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012112946A1 (en) | 2011-02-18 | 2012-08-23 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2013027845A1 (en) * | 2011-08-22 | 2013-02-28 | Takeda Pharmaceutical Company Limited | Radiolabeled compounds and their use as radiotracers for quantitative imaging of phosphodiesterase (pde10a) in mammals |
| US8691827B2 (en) | 2009-08-17 | 2014-04-08 | Merck Sharp & Dohme Corp. | Amino tetrahydro-pyridopyrimidine PDE10 inhibitors |
| WO2014071044A1 (en) | 2012-11-01 | 2014-05-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014118039A1 (en) * | 2013-01-31 | 2014-08-07 | F. Hoffmann-La Roche Ag | Radiolabeled compounds |
| US9200016B2 (en) | 2013-12-05 | 2015-12-01 | Allergan, Inc. | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| US10214519B2 (en) | 2016-09-23 | 2019-02-26 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10227350B2 (en) | 2016-09-23 | 2019-03-12 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10479770B2 (en) | 2016-09-23 | 2019-11-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USD977386S1 (en) | 2020-09-22 | 2023-02-07 | Positec Power Tools (Suzhou) Co., Ltd. | Battery pack |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001046183A1 (en) * | 1999-12-21 | 2001-06-28 | Celgene Corporation | SUBSTITUTED 1,3,4-OXADIAZOLES AND A METHOD OF REDUCING TNF-α LEVELS |
| WO2002102315A2 (en) * | 2001-06-19 | 2002-12-27 | Bristol-Myers Squibb Company | QUINAZOLINE AND PYRIDO[2,3-d]PYRIMIDINE INHIBITORS OF PHOSPHODIESTERASE (PDE) 7 |
| WO2005087919A1 (en) * | 2004-03-12 | 2005-09-22 | Pfizer Products Inc. | Crystal structure of 3',5'-cyclic nucleotide phosphodiesterase (pde10a) and uses thereof |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TR199902094T2 (en) * | 1997-02-28 | 1999-12-21 | Prizer Products Inc. | 3-Heteroareyl-4(3H)-quinazolinonal�n atropisomerleri. |
| WO2008020302A2 (en) * | 2006-08-17 | 2008-02-21 | Pfizer Products Inc. | Heteroaromatic quinoline-based compounds as phosphodiesterase (pde) inhibitors |
| KR100933559B1 (en) * | 2007-10-01 | 2009-12-23 | 재단법인서울대학교산학협력재단 | HsK0 inhibitors containing 3H-quinazolin-4-one derivatives and anticancer agents using the same |
| BRPI1012333A2 (en) * | 2009-03-24 | 2016-03-29 | Gilead Calistoga Llc | atropisomers of 2-purinyl-3-tolyl-quinazolinones derivatives and methods of use |
-
2010
- 2010-05-26 AU AU2010254149A patent/AU2010254149B2/en not_active Ceased
- 2010-05-26 CA CA2763130A patent/CA2763130C/en active Active
- 2010-05-26 ES ES10781130T patent/ES2739973T3/en active Active
- 2010-05-26 JP JP2012513203A patent/JP5718323B2/en not_active Expired - Fee Related
- 2010-05-26 US US13/320,816 patent/US8846000B2/en active Active
- 2010-05-26 EP EP10781130.9A patent/EP2435048B1/en active Active
- 2010-05-26 WO PCT/US2010/036186 patent/WO2010138577A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001046183A1 (en) * | 1999-12-21 | 2001-06-28 | Celgene Corporation | SUBSTITUTED 1,3,4-OXADIAZOLES AND A METHOD OF REDUCING TNF-α LEVELS |
| WO2002102315A2 (en) * | 2001-06-19 | 2002-12-27 | Bristol-Myers Squibb Company | QUINAZOLINE AND PYRIDO[2,3-d]PYRIMIDINE INHIBITORS OF PHOSPHODIESTERASE (PDE) 7 |
| WO2005087919A1 (en) * | 2004-03-12 | 2005-09-22 | Pfizer Products Inc. | Crystal structure of 3',5'-cyclic nucleotide phosphodiesterase (pde10a) and uses thereof |
Non-Patent Citations (14)
| Title |
|---|
| "Genbank", Database accession no. 4894716 |
| BECKER ET AL., BEHAV BRAIN RES, vol. 186, no. 2, 2008, pages 155 - 60 |
| FUJISHIGE ET AL., J. BIOL. CHEM., vol. 274, 1999, pages 18438 - 18445 |
| FUJITA ET AL.: "Quantification of brain phosphodiesterase 4 in rat with (R)-[11C]Rolipram-PET", NEUROLMAGE, vol. 26, 2005, pages 1201 - 1210, XP004929572 * |
| HUANG, W. ET AL., J. BIOMOL SCREEN, vol. 7, 2002, pages 215 |
| KEHLER ET AL., EXPERT OPIN. THER. PATENTS, vol. 17, no. 2, 2007, pages 147 - 158 |
| LIEBERMAN ET AL., N. ENGL. J. MED., vol. 353, 2005, pages 1209 - 1223 |
| LOUGHNEY ET AL., GENE, vol. 234, 1999, pages 109 - 117 |
| MOSSER ET AL., JALA, vol. 8, 2003, pages 54 - 63 |
| SCHMIDT ET AL., J PHARMACOL EXP THER, vol. 325, 2008, pages 681 - 690 |
| See also references of EP2435048A4 |
| SIUCIAK ET AL., NEUROPHARMACOLOGY, vol. 51, 2006, pages 386 - 396 |
| SODERLING ET AL., PNAS, USA, vol. 96, 1999, pages 7071 - 7076 |
| THRELFELL ET AL., J PHARMACOL EXP THER, vol. 328, no. 3, 2009, pages 785 - 795 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8691827B2 (en) | 2009-08-17 | 2014-04-08 | Merck Sharp & Dohme Corp. | Amino tetrahydro-pyridopyrimidine PDE10 inhibitors |
| US8772316B2 (en) | 2011-02-18 | 2014-07-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (PDE10A) |
| WO2012112946A1 (en) | 2011-02-18 | 2012-08-23 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| US9670181B2 (en) | 2011-02-18 | 2017-06-06 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| CN103889974B (en) * | 2011-08-22 | 2017-10-03 | 武田药品工业株式会社 | Radiolabeled compound and they in mammal as phosphodiesterase (PDE10A) quantitative imaging radiotracer purposes |
| WO2013027845A1 (en) * | 2011-08-22 | 2013-02-28 | Takeda Pharmaceutical Company Limited | Radiolabeled compounds and their use as radiotracers for quantitative imaging of phosphodiesterase (pde10a) in mammals |
| CN103889974A (en) * | 2011-08-22 | 2014-06-25 | 武田药品工业株式会社 | Radiolabeled compounds and their use as radiotracers for quantitative imaging of phosphodiesterase (pde10a) in mammals |
| JP2014524407A (en) * | 2011-08-22 | 2014-09-22 | 武田薬品工業株式会社 | Radiolabeled compounds as radiotracers for quantitative imaging of phosphodiesterase (PDE10A) in mammals and uses thereof |
| US9579407B2 (en) | 2011-08-22 | 2017-02-28 | Takeda Pharmaceutical Company Limited | Radiolabeled compounds and their use as radiotracers for quantitative imaging of phosphodiesterase (PDE10A) in mammals |
| WO2014071044A1 (en) | 2012-11-01 | 2014-05-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014118039A1 (en) * | 2013-01-31 | 2014-08-07 | F. Hoffmann-La Roche Ag | Radiolabeled compounds |
| US9200016B2 (en) | 2013-12-05 | 2015-12-01 | Allergan, Inc. | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| US9902710B2 (en) | 2013-12-05 | 2018-02-27 | Exonhit Therapeutics, Sa | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| US10214519B2 (en) | 2016-09-23 | 2019-02-26 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10227350B2 (en) | 2016-09-23 | 2019-03-12 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| US10479770B2 (en) | 2016-09-23 | 2019-11-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5718323B2 (en) | 2015-05-13 |
| ES2739973T3 (en) | 2020-02-05 |
| CA2763130C (en) | 2017-01-17 |
| EP2435048A4 (en) | 2013-07-10 |
| JP2012528176A (en) | 2012-11-12 |
| US20120064005A1 (en) | 2012-03-15 |
| US8846000B2 (en) | 2014-09-30 |
| CA2763130A1 (en) | 2010-12-02 |
| EP2435048A1 (en) | 2012-04-04 |
| AU2010254149B2 (en) | 2014-08-21 |
| AU2010254149A1 (en) | 2011-12-22 |
| EP2435048B1 (en) | 2019-07-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2763130C (en) | Radiolabeled pde10 inhibitors | |
| US9266881B2 (en) | Triazolopyridinone PDE10 inhibitors | |
| US10376504B2 (en) | Substituted quinolinones as PDE9 inhibitors | |
| US8691827B2 (en) | Amino tetrahydro-pyridopyrimidine PDE10 inhibitors | |
| US10370336B2 (en) | Phenyl-cyanoquinolinone PDE9 inhibitors | |
| EP2621276B1 (en) | 2-alkoxy pyrimidine pde10 inhibitors | |
| US20130203756A1 (en) | Isoindoline pde10 inhibitors | |
| US10370337B2 (en) | Oxy-cyanoquinolinone PDE9 inhibitors | |
| WO2010138585A1 (en) | Pyrimidinones as pde10 inhibitors | |
| US9284302B2 (en) | Cyclobutyl benzimidazoles as PDE 10 inhibitors | |
| WO2014078214A1 (en) | Azetidine benzimidazoles as pde10 inhibitors | |
| US20110319409A1 (en) | 7-aza-quinazoline pde10 inhibitors | |
| EP2919782B1 (en) | Secondary alcohol subsituted triazoles as pde10 inhibitors | |
| EP2968324B1 (en) | Substituted pyridazinone derivatives as pde10 inhibitors | |
| US9359348B2 (en) | Cyclopropyl imidazopyridine PDE10 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10781130 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13320816 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2763130 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012513203 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010254149 Country of ref document: AU Ref document number: 2010781130 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2010254149 Country of ref document: AU Date of ref document: 20100526 Kind code of ref document: A |