WO2010144522A1 - Ureidophenyl substituted triazine derivatives and their therapeutical applications - Google Patents
Ureidophenyl substituted triazine derivatives and their therapeutical applications Download PDFInfo
- Publication number
- WO2010144522A1 WO2010144522A1 PCT/US2010/037890 US2010037890W WO2010144522A1 WO 2010144522 A1 WO2010144522 A1 WO 2010144522A1 US 2010037890 W US2010037890 W US 2010037890W WO 2010144522 A1 WO2010144522 A1 WO 2010144522A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- disease
- pharmaceutically acceptable
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCCC(C)C*C(N)NC Chemical compound CCCC(C)C*C(N)NC 0.000 description 3
- QFCKYNDJEALQAV-UHFFFAOYSA-N CC(C)NC(Nc(cc1)ccc1N)=O Chemical compound CC(C)NC(Nc(cc1)ccc1N)=O QFCKYNDJEALQAV-UHFFFAOYSA-N 0.000 description 1
- ZAWKATPGKOLINO-UHFFFAOYSA-N CC1=C(Nc2nc(Cl)nc(Cl)n2)SC(C)=CCC1 Chemical compound CC1=C(Nc2nc(Cl)nc(Cl)n2)SC(C)=CCC1 ZAWKATPGKOLINO-UHFFFAOYSA-N 0.000 description 1
- HESHRJKEOOGGBU-UHFFFAOYSA-O CCCCNCNCC(C)(C)[NH3+] Chemical compound CCCCNCNCC(C)(C)[NH3+] HESHRJKEOOGGBU-UHFFFAOYSA-O 0.000 description 1
- VASXKOXECMYATF-UHFFFAOYSA-N CCNC(Nc(cc1)ccc1Sc1nc(Nc2ncc(C(C)C)[s]2)nc(N2CCN(C)CC2)n1)=O Chemical compound CCNC(Nc(cc1)ccc1Sc1nc(Nc2ncc(C(C)C)[s]2)nc(N2CCN(C)CC2)n1)=O VASXKOXECMYATF-UHFFFAOYSA-N 0.000 description 1
- ONHVBXJIPXRBEN-UHFFFAOYSA-N CCNC(Nc(cc1)ccc1Sc1nc(Nc2ncc(C)[s]2)nc(N2CCN(C)CC2)n1)=O Chemical compound CCNC(Nc(cc1)ccc1Sc1nc(Nc2ncc(C)[s]2)nc(N2CCN(C)CC2)n1)=O ONHVBXJIPXRBEN-UHFFFAOYSA-N 0.000 description 1
- ADVXAMGGIRUOKU-UHFFFAOYSA-N CCc1nc(Cl)nc(Cl)n1 Chemical compound CCc1nc(Cl)nc(Cl)n1 ADVXAMGGIRUOKU-UHFFFAOYSA-N 0.000 description 1
- VROIXQLRAWCCAO-UHFFFAOYSA-N CCc1nc(Nc2n[nH]c(C)c2)nc(Nc(cc2)ccc2NC(NCc2ccccc2)=O)n1 Chemical compound CCc1nc(Nc2n[nH]c(C)c2)nc(Nc(cc2)ccc2NC(NCc2ccccc2)=O)n1 VROIXQLRAWCCAO-UHFFFAOYSA-N 0.000 description 1
- FSOUAFCWYPVFBO-UHFFFAOYSA-N CCc1nc(Nc2n[n](C(NC(C)C)=O)c(C)c2)nc(Nc(cc2)ccc2NC(NC(C)C)=O)n1 Chemical compound CCc1nc(Nc2n[n](C(NC(C)C)=O)c(C)c2)nc(Nc(cc2)ccc2NC(NC(C)C)=O)n1 FSOUAFCWYPVFBO-UHFFFAOYSA-N 0.000 description 1
- HKTFJSZRSYFYQS-UHFFFAOYSA-N Cc1cc(Nc2nc(C3CC3)nc(Nc(cc3)ccc3N)n2)n[nH]1 Chemical compound Cc1cc(Nc2nc(C3CC3)nc(Nc(cc3)ccc3N)n2)n[nH]1 HKTFJSZRSYFYQS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates generally to the use of compounds to treat a variety of disorders, diseases and pathologic conditions and more specifically to the use of triazine compounds to modulate protein kinases and for treating protein kinase-mediated diseases.
- Protein kinases constitute a large family of structurally related enzymes that are responsible for the control of a variety of signal transduction processes within the cell. Protein kinases, containing a similar 250-300 amino acid catalytic domain, catalyze the phosphorylation of target protein substrates.
- the kinases may be categorized into families by the substrates in the phosphorylate (e.g., protein-tyrosine, protein-serine/threonine, lipids, etc.). Tyrosine phosphorylation is a central event in the regulation of a variety of biological processes such as cell proliferation, migration, differentiation and survival. Several families of receptor and non-receptor tyrosine kinases control these events by catalyzing the transfer of phosphate from ATP to a tyrosine residue of specific cell protein targets.
- families of receptor and non-receptor tyrosine kinases control these events by catalyzing the transfer of phosphate from ATP to a tyrosine residue of specific cell protein targets.
- kinases in the protein kinase family include, without limitation, abl, Akt, bcr-abl, BIk, Brk, Btk, c- kit, c-Met, c-src, c-fms, CDK1 , CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, cRafl , CSF1 R, CSK, EGFR, ErbB2, ErbB3, ErbB4, Erk, Fak, fes, FGFR1 , FGFR2, FGFR3, FGFR4, FGFR5, Fgr, flt-1 , Fps, Frk, Fyn, Hck, IGF-1 R, INS-R, Jak, KDR, Lck, Lyn, MEK, p38, PDGFR, PIK, PKC, PYK2, ros, Tie, Tie-2,
- kinase activity acts as molecular switches regulating cell proliferation, activation, and/or differentiation. Uncontrolled or excessive kinase activity has been observed in many disease states including benign and malignant proliferation disorders as well as diseases resulting from inappropriate activation of the immune system (autoimmune disorders), allograft rejection, and graft vs host disease.
- Src kinase is involved in proliferation and migration responses in many cell types, cell activation, adhesion, motility, and survival, growth factor receptor signaling, and osteoclast activation (Biscardi et al., Adv. Cancer Res. (1999), 76, 61-119; Yeatman et al., Nat. Rev. Cancer (2004), 4, 470-480; Owens, D. W.; McLean et al., MoI. Biol. Cell (2000), 11, 51-64).
- Src Src
- Fyn Yes, Fgr, Lyn, Hck, Lck
- BIk BIk
- SH4 domain contains the myristylation signals that guide the Src molecule to the cell membrane. This unique domain of Src proteins is responsible for their specific interaction with particular receptors and protein targets (Thomas et al., Annu Rev Cell Dev Biol (1997), 13, 513-609).
- the modulating regions, SH3 and SH2 control intra- as well as intermolecular interactions with protein substrates which affect Src catalytic activity, localization and association with protein targets (Pawson T., Nature (1995), 373, 573-580).
- the kinase domain, SH1 found in all proteins of the Src family, is responsible for the tyrosine kinase activity and has a central role in binding of substrates.
- the N-terminal half of Src kinase contains the site(s) for its tyrosine phosphorylation and regulates the catalytic activity of Src (Thomas et al., Annu Rev Cell Dev Biol (1997), 13: 513-609).
- v-Src differs from cellular Src (c-Src) on the basis of the structural differences in C-terminal region responsible for regulation of kinase activity.
- the prototype member of the Src family protein tyrosine kinases was originally identified as the transforming protein (v-Src) of the oncogenic retrovirus, Rous sarcoma virus, RSV (Brugge et al., Nature (1977), 269, 346- 348); Hamaguchi et al. (1995), Oncogene 10: 1037-1043).
- Viral v-Src is a mutated and activated version of a normal cellular protein (c-Src) with intrinsic tyrosine kinase activity (Collett et al., Proc Natl Acad Sci U S A (1978), 75, 2021- 2024). This kinase phosphorylates its protein substrates exclusively on tyrosyl residues (Hunter et al., Proc Natl Acad Sci U S A (1980), 77, 1311 -1315).
- Src is a cytoplasmic protein tyrosine kinase, whose activation and recruitment to perimembranal signaling complexes has important implications for cellular fate. It has well-documented that Src protein levels and Src kinase activity are significantly elevated in human breast cancers (Muthuswamy et al., Oncogene, (1995), 11 , 1801-1810); Wang et al., Oncogene (1999), 18, 1227-1237; Warmuth et al., Curr. Pharm. Des.
- NSCLCs non-small cell lung cancers
- bladder cancer bladder cancer
- oesophageal cancer Jankowski et al., Gut, (1992), 33, 1033-8
- prostate and ovarian cancer Wiener et al., Clin.
- Src kinase modulates signal transduction through multiple oncogenic pathways, including EGFR, Her2/neu, PDGFR, FGFR, and VEGFR (Frame et al., Biochim. Biophys. Acta (2002), 1602, 114-130; Sakamoto et al., Jpn J Cancer Res, (2001 ), 92: 941-946).
- Src kinase inhibitors may be useful anti-cancer agents (Abram et al., Exp. Cell Res., (2000), 254, 1 ). It is reported that inhibitors of src kinase had significant antiproliferative activity against cancer cell lines (M. M. Moasser et al., Cancer Res., (1999), 59, 6145; Tatosyan et al., Biochemistry (Moscow) (2000), 65, 49-58).) and inhibited the transformation of cells to an oncogenic phenotype (R.
- Src-family kinases are also important for signaling downstream of other immune cell receptors.
- Fyn like Lck, is involved in TCR signaling in T cells (Appleby et al., Cell, (1992), 70, 751 ).
- Hck and Fgr are involved in Fey receptor signaling leading to neutrophil activation (Vicentini et al., J. Immunol. (2002), 168, 6446). Lyn and Src also participate in Fey receptor signaling leading to release of histamine and other allergic mediators (Turner, H. and Kinet, J-P Nature (1999), 402, B24).
- Lck plays a role in T-cell signaling. Mice that lack the Lck gene have a poor ability to develop thymocytes. The function of Lck as a positive activator of T-cell signaling suggests that Lck inhibitors may be useful for treating autoimmune disease such as rheumatoid arthritis (Molina et al., Nature, (1992), 357, 161 ).
- Hck is a member of the Src protein-tyrosine kinase family and is expressed strongly in macrophages, an important HIV target cell and its inhibition in HIV-infected macrophages might slow disease progression (Ye et al., Biochemistry, (2004), 43 (50), 15775 -15784).
- Hck, Fgr and Lyn have been identified as important mediators of integrin signaling in myeloid leukocytes (Lowell et al., J. Leukoc. Biol., (1999), 65, 313). Inhibition of these kinase mediators may therefore be useful for treating inflammation (Boschelli et al., Drugs of the Future (2000), 25(7), 717). It is reported that Syk is a tyrosine kinase that plays a critical role in the cell degranulation and eosinophil activation and Syk kinase is implicated in various allergic disorders, in particular asthma (Taylor et al., MoI. Cell. Biol. (1995), 15, 4149).
- BCR-ABL encodes the BCR-AEL protein, a constitutively active cytoplasmic tyrosine kinase present in 90% of all patients with chronic myelogenous leukemia (CML) and in 15-30% of adult patients with acute lymphoblastic leukemia (ALL). Numerous studies have demonstrated that the activity of BCR-ABL is required for the cancer causing ability of this chimeric protein.
- Src kinases play a role in the replication of hepatitis B virus.
- the virally encoded transcription factor HBx activates Src in a step required for propagation of the virus (Klein et al., EMBO J. (1999), 18, 5019; Klein et al., MoI. Cell. Biol. (1997), 17, 6427).
- Some genetic and biochemical data clearly demonstrate that Src-family tyrosine kinases serve as a critical signal relay, via phosphorylation of c-Cbl, for fat accumulation, and provide potential new strategies for treating obesity (Sun et al., Biochemistry, (2005), 44 (44), 14455 -14462).
- Src inhibitors are also being pursued for the treatment of other diseases including osteoporosis and stroke (Susva et al., Trends Pharmacol. Sci. (2000), 21, 489-495; Paul et al., Nat. Med. (2001 ), 7, 222-227). It is also possible that inhibitors of the Src kinase activity are useful in the treatment of osteoporosis (Soriano et al., Cell (1991), 64, 693; Boyce et al. J Clin. Invest (1992), 90, 1622; Owens et al., MoI. Biol. Cell (2000), 11, 51-64), T cell mediated inflammation (Anderson et al., Adv. Immunol. (1994), 56, 151 ; Goldman, F D et al. J. Clin. Invest. (1998), 102, 421 ), and cerebral ischemia (Paul et al. Nature Medicine (2001 ), 7, 222).
- src family kinases participate in signal transduction in several cell types.
- fyn like lck, is involved in T-cell activation.
- Hck and fgr are involved in Fe gamma receptor mediated oxidative burst of neutrophils.
- Src and lyn are believed to be important in Fc epsilon induced degranulation of mast cells, and so may play a role in asthma and other allergic diseases.
- the kinase lyn is known to be involved in the cellular response to DNA damage induced by UV light (Hiwasa et al., FEBS Lett.
- T cells play a pivotal role in the regulation of immune responses and are important for establishing immunity to pathogens.
- T cells are often activated during inflammatory autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, type I diabetes, multiple sclerosis, Sjogren's disease, myasthenia gravis, psoriasis, and lupus.
- T cell activation is also an important component of transplant rejection, allergic reactions, and asthma.
- T cells are activated by specific antigens through the T cell receptor, which is expressed on the cell surface. This activation triggers a series of intracellular signaling cascades mediated by enzymes expressed within the cell (Kane et al. Current Opinion in Immunol. (2000), 12, 242). These cascades lead to gene regulation events that result in the production of cytokines, like interleukin-2 (IL- 2). IL-2 is a necessary cytokine in T cell activation, leading to proliferation and amplification of specific immune responses.
- IL-2 interleukin-2
- 6,440,965 disclosed substituted pyrimidine derivatives in the treatment of neurodegenerative or neurological disorders
- PCT WO 02/08205 reported the pyrimidine derivatives having neurotrophic activity
- PCT WO 03/014111 disclosed arylpiperazines and arylpiperidines and their use as metalloproteinase inhibiting agents
- PCT WO 03/024448 described compounds as inhibitors of histone deacetylase enzymatic activity
- PCT WO 04/058776 disclosed compounds which possess anti-angiogenic activity.
- PCT WO 01/94341 and WO 02/16352 disclosed Src kinase inhibitors of quinazoline derivatives.
- PCT WO03/026666A1and WO03/018021A1 disclosed pyrimidinyl derivatives as kinase inhibitors.
- U.S. Pat. No 6498165 reported Src kinase inhibitor compounds of pyrimidine compounds.
- Peptides as Src Tyrosine Kinase Inhibitors is reported recently (Kumar et al., J. Med. Chem., (2006), 49 (11 ), 3395 -3401).
- the quinolinecarbonitriles derivatives was reported to be potent dual Inhibitors of Src and AbI Kinases (Diane et al., J. Med. Chem., (2004), 47 (7), 1599 -1601 ).
- Another kinase family of particular interest is the aurora kinases.
- the aurora kinases is reported to be potent dual Inhibitors of Src and AbI Kinases.
- Aurora kinase family is a collection of highly related serine/threonine kinase that are key regulators of mitosis, essential for accurate and equal segtion of genomic material from parent to daught cells.
- Members of the Aurora kinase family include three related kinases kown as Aurora-A, Aurora-B, and Aurora-C. Despite significant sequence homology, the localization and functions of these kinases are largely distinct from one another (Richard D.Carvajal, et al. Clin Cancer Res 2006; 12(23): 6869-6875; Daruka Mahadevan, et al. Expert Opin. Drug Discov. 2007 2(7): 1011-1026) .
- Aurora-A is ubiquitously expressed and regulates cell cycle events occurring from late S phase through M phase, including centrosome maturation (Berdnik D, et al. Curr Biol 2002; 12:640-7), mitotic entry (Hirota T, et al. Cell 2003;114:585- 98; Dutertre S, et al. J Cell Sci 2004;117:2523-31 ), centrosome separation (Marumoto T, et al. J Biol Chem 2003;278:51786-95), bipolar-spindle assembly (Kufer TA, et al. J CeII Biol 2002;158:617-23; Eyers PA, et al.
- Aurora-B is a chromosomal passenger protein critical for accurate chromosomal segregation, cytokinesis (Hauf S, et al. J Cell Biol 2003;161 :281- 94; Ditchfield C, et al. J Cell Biol 2003; 161 :267-80; Giet R, et al. J Cell Biol 2001 ;152:669-82; Goto H, et al. J Biol Chem 2003;278:8526-30), protein localization to the centromere and kinetochore, correct microtubule-kinetochore attachments (Murata-Hori M, et al. Curr Biol 2002; 12:894-9), and regulation of the mitotic checkpoint.
- Aurora-B localizes first to the chromosomes during prophase and then to the inner centromere region between sister chromatids during prometaphase and metaphase (Zeitlin SG, et al. J Cell Biol
- Aurora-B participates in the establishment of chromosomal biohentation, a condition where sister kinetochores are linked to opposite poles of the bipolar spindle via amphitelic attachments. Errors in this process, manifesting as a merotelic attachment state (one kinetochore attached to microtubules from both poles) or a syntelic attachment state (both sister kinetochores attached to microtubules from the same pole), lead to chromosomal instability and aneuploidy if not corrected before the onset of anaphase.
- Aurora-B The primary role of Aurora-B at this point of mitosis is to repair incorrect microtubule- kinetochore attachments (Hauf S, et al. J Cell Biol 2003;161 :281-94; Ditchfield C, et al. J Cell Biol 2003;161 :267-80; Lan W, et al. Curr Biol 2004; 14:273-86.).
- Aurora-A overexpression is a necessary feature of Aurora-A-induced tumorigenesis.
- mitosis is characterized by the presence of multiple centrosomes and multipolar spindles (Meraldi P et al. EMBO J 2002;21 :483-92.).
- cells abrogate the mitotic checkpoint and progress from metaphase to anaphase, resulting in numerous chromosomal separation defects.
- These cells fail to undergo cytokinesis, and, with additional cell cycles, polyploidy and progressive chromosomal instability develop (Anand S, et al. Cancer Cell 2003;3:51-62).
- Aurora-A inhibition results in delayed, but not blocked, mitotic entry, centrosome separation defects resulting in unipolar mitotic spindles, and failure of cytokinesis (Marumoto T, et al. J Biol Chem 2003;278:51786-95).
- Encouraging antitumor effects with Aurora-A inhibition were shown in three human pancreatic cancer cell lines (Panc-1 , MIA PaCa-2, and SU.86.86), with growth suppression in cell culture and near-total abrogation of tumorigenicity in mouse xenografts (Hata T, et al. Cancer Res 2005;65:2899-905.).
- Aurora-B inhibition results in abnormal kinetochore-microtubule attachments, failure to achieve chromosomal biorientation, and failure of cytokinesis (Goto H, et al. J Biol Chem 2003;278:8526-30; Severson AF, et al. Curr Biol 2000;10:1162-71 ). Recurrent cycles of aberrant mitosis without cytokinesis result in massive polyploidy and, ultimately, to apoptosis (Hauf S, et al. J Cell Biol
- the first three small-molecule inhibitors of Aurora described include ZM447439 (Ditchfield C, et al. J Cell Biol 2003;161 :267-80), Hesperadin (Hauf S, et al. J Cell Biol 2003;161 :281-94), and
- MK0457 (VX680) (Harrington EA, et al. Nat Med 2004; 10:262-7).
- the following agents are nonspecific inhibitors: ZM447439 inhibits Aurora-A and Aurora-B; Herperadin inhibits primarily Aurora-B; MK0457 inhibits all three Aurora kinases.
- ZM447439 inhibits Aurora-A and Aurora-B
- Herperadin inhibits primarily Aurora-B
- MK0457 inhibits all three Aurora kinases.
- Selective inhibitors of Aurora have also been developed.
- a selective Aurora-A inhibitor is MLN8054 (Hoar HM, et al. [abstract C40].
- Aurora-B inhibitor A expmple of selective Aurora-B inhibitor is AZD1152 (Schellens J, et al. [abstract 3008]. Proc Am Soc Clin Oncol 2006;24:122s).
- the next generation of Aurora inhibitors is currently being developed, including agents by Nerviano Medical Sciences (PHA-680632 and PHA-739358), Rigel (R763), Sunesis (SNS-314), NCE Discovery Ltd. (NCED#17), Astex Therapeutics (AT9283), and Montigen Pharmaceuticals (MP- 235 and MP-529).
- PHA-680632 and PHA-739358 The next generation of Aurora inhibitors is currently being developed, including agents by Nerviano Medical Sciences (PHA-680632 and PHA-739358), Rigel (R763), Sunesis (SNS-314), NCE Discovery Ltd. (NCED#17), Astex Therapeutics (AT9283), and Montigen Pharmaceuticals (MP- 235 and MP-529).
- RTKs Receptor tyrosine kinases
- cytoplasm and/or nucleus of a cell are transmembrane proteins that generally include an extracellular ligand-binding domain, a membrane-spanning domain and a catalytic cytoplasmic tyrosine kinase domain. The binding of ligand to the extracellular potion is believed to promote dimerization, resulting in trans-phosphorylation and activation of the intracellular tyrosine kinase domain (Schlessinger et al. Neuron 1992;9:383-391 ).
- FLT3 FMS-related tyrosine kinase 3
- FLK-2 fetal liver kinase 2
- STK-1 human stem cell kinase 1
- FLT3 FMS-related tyrosine kinase 3
- FLKIII class III receptor tyrosine kinase family that include KIT, PDGFR, FMS and FLT1
- FLT3 is a membrane-spanning protein and composed of four domains; an extracellular ligand-binding domains consisting of five immunoglobin-like structures, a transmembrane (TM) domain, a juxtamembrane (JM) domain and a cytoplasmic C-Terminal tyrosine kinase (TK) domain.
- TM transmembrane
- JM juxtamembrane
- TK cytoplasmic C-Terminal tyrosine kinase
- the ligand for FLT3 was cloned in 1993 and shown to be a Type I transmembrane protein expressed in cells of the hematopoietic bone marrow microenvironment, including bone marrow fibroblasts and other cells (Lyman SD, et al. Cell 1993;75:1157-1167). Both the membrane-bound and soluable forms can activate the tyrosine kinase activity of the receptor and stimulate growth of progenitor cells in the marrow and blood.
- Binding of ligand to receptor induces dimerisation of the receptor and activation of the kinase domains; which then autophosphorylate and catalyse phosphorylation of substrate proteins of various signal transduction pathways such as signal transducer and activator of transcription 5 (STAT5), RAS/mitogen-activated protein kinase (RAS/MAPK), phosphoinositide 3-kinase (PI3K), src homologous and collagen gene (SHC), SH2-containing inositol-5-phosphatase (SHIP), and cytoplasmic tyrosine phosphatase with 2 Src-homology 2 (SH2) domains (SHP2), which play important roles in cellular proliferation, differentiation, and survival (Dosil M, et al.
- RAS/MAPK RAS/mitogen-activated protein kinase
- PI3K phosphoinositide 3-kinase
- SHC src homologous and collagen gene
- FLT3 gene is also expressed in placenta, gonads and brain (Maroc N, et al. Oncogene 1993;8:909-918) and also plays an importand role in the immune response (deLapeyriere O, et al. Leukemia 1995;9:1212-1218).
- FLT3 is overexpressed at the levels in 70-100% of cases of acute myeloid leukemias (AML), and in a high percentage of T-acute lymphocytic leukemia (ALL) cases (Griffin JD, et al. Haematol J. 2004;5: 188-190). It is also overexpressed in a smaller subset of chronic myeloid leukemia (CML) in blast crisis. Studies have shown that the leukemic cells of B lineage ALL and AML frequently co-express FL, setting up autocrine or paracrine signaling loops that result in the constitutive activation of FLT3 (Zheng R, et. al. Blood. 2004; 103:267-274).
- FLT3 mutations are one of the most frequent somatic alterations in AML, occurring in approximately 1/3 of patients.
- FLT3-ITD has also been detected at low frequency in myelodysplastic syndrome (MDS) (Yokota S, et al. Leukemia 1997;11 :1605-1609; Horiike S, et al. Leukemia 1997; 11 :1442-1446).
- MDS myelodysplastic syndrome
- the ITDs are always in-frame, and are limited to the JM domain. However, they vary in length and position from patient to patient. These repeat sequences may serve to disrupt the autoinhibitory activity of the JM domain resulting in the constitutive activation of FLT3.
- FLT3-ITD and FLT3- Asp835 mutations are associated with FLT3 autophosphorylation and phosphorylation of downstream targets (Mizuki M, et al. Blood 2000;96:3907- 3914; Mizuki M, et al. Blood 2003;101 :3164-3173; Hayakawa F, et al. Oncogene
- FLT3 inhibitors such as PKC412 (N-benzoyl staurosporine) (Fabbro D, et al. Anticancer Drug Des 2000;15:17-28; Weisberg E, et al. Cancer Cell 2002; 1 :433-443), CT53518 (also known as MLN518) (Kelly LM, et al. Cancer Cell 2002;1 :421-432), SU11248 (O'Farrell AM, et al.
- PKC412 N-benzoyl staurosporine
- CT53518 also known as MLN53518
- SU11248 O'Farrell AM, et al.
- Aurora kinase inhibors are of special intrest in treating certain disorders, including cancer.
- an antitumor agent comprising a triazine derivative as described in formula (I), pharmaceutically-acceptable formulations thereof, methods for making novel compounds and compositions for using the compounds.
- the compounds and compositions comprising the compounds in formula (I) have utility in treatment of a variety of diseases.
- the combination therapy described herein may be provided by the preparation of the triazine derivative of formula (I) and the other therapeutic agent as separate pharmaceutical formulations followed by the administration thereof to a patient simultaneously, semi-simultaneously, separately or over regular intervals.
- the present invention provides methods of use for certain chemical compounds such as kinase inhibitors for treatment of various diseases, disorders, and pathologies, for example, cancer, and vascular disorders, such as myocardial infarction (Ml), stroke, or ischemia.
- kinase inhibitors for treatment of various diseases, disorders, and pathologies, for example, cancer, and vascular disorders, such as myocardial infarction (Ml), stroke, or ischemia.
- Ml myocardial infarction
- the triazine compounds described in this invention may block the enzymatic activity of some or many of the members of the Aurora kinase family, in addition to blocking the activity of other receptor and non-receptor kinase.
- Such compounds may be beneficial for treatment of the diseases where disorders affect cell motility, adhesion, and cell cycle progression, and in addition, diseases with related hypoxic conditions, osteoporosis and conditions, which result from or are related to increases in vascular permeability, inflammation or respiratory distress, tumor growth, invasion, angiogenesis, metastases and apoptosis.
- the present invention is related to compounds showed as in Formula (I)
- W and Y are independently selected from S, O, NR4, or CR4
- R4 is independently selected from hydrogen or an optionally substituted C1-4 aliphatic group.
- K is selected from -NR4, O, or S
- R1 represents hydrogen, halogen, hydroxy, amino, cyano, alkyl, cycloalkyl, alkenyl, alkynyl, alkylthio, aryi, arylalkyl, heterocyclic, heteroaryl, heterocycloalkyl, alkylsulfonyl, alkoxycarbonyl and alkylcarbonyl.
- R2 is selected from:
- X is CH, when R 6 is hydrogen; or X-R 6 is O; or X is N, R 6 represents groups of hydrogen, C 1 -C 6 alkyl, C 2 -Ce alkenyl, C2-C 6 alkynyl, C3-C- 1 0 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, d- Ce haloalkyl, C 1 -C ⁇ alkoxy, C-i- C 6 alkylthio, C 2 -C 6 alkanoyl, d- C 6 alkoxycarbonyl, C 2 - Ce alkanoyloxy, mono- and di-(C 3 -Cs cycloalkyl)aminoCo-C 4 alkyl, (4- to 7- membered heterocycle)Co-C 4 alkyl, C 1 -C 6 alkylsulfonyl, mono- and di-(C 1 - C 6 alkyl)
- halo refers to fluorine, chlorine, bromine or iodine.
- alkyl herein alone or as part of another group refers to a monovalent alkane (hydrocarbon) derived radical containing from 1 to 12 carbon atoms unless otherwise defined. Alkyl groups may be substituted at any available point of attachment. An alkyl group substituted with another alkyl group is also referred to as a "branched alkyl group”.
- Exemplary alkyl groups include methyl, ethyl, propyl, isopropyl, n-butyl, t-butyl, isobutyl, pentyl, hexyl, isohexyl, heptyl, dimethylpentyl, octyl, 2,2,4-trimethylpentyl, nonyl, decyl, undecyl, dodecyl, and the like.
- substituents include but are not limited to one or more of the following groups: alkyl, aryl, halo (such as F, Cl, Br, I), haloalkyl (such as CCb or CF 3 ), alkoxy, alkylthio, hydroxy, carboxy (-COOH), alkyloxycarbonyl (-C(O)R), alkylcarbonyloxy (- OCOR), amino (-NH2), carbamoyl (-NHCOOR- or -OCONHR- ), urea (-NHCONHR-) or thiol (-SH).
- alkyl groups are substituted with, for example, amino, heterocycloalkyl, such as morpholine, piperazine, piperidine, azetidine, hydroxyl, methoxy, or heteroaryl groups such as pyrrolidine,
- the term 'cycloalkyl" herein alone or as part of another group refers to fully saturated and partially unsaturated hydrocarbon rings of 3 to 9, preferably 3 to 7 carbon atoms.
- the examples include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, and like. Further, a cycloalkyl may be substituted.
- alkenyl herein alone or as part of another group refers to a hydrocarbon radical straight, branched or cyclic containing from 2 to 12 carbon atoms and at least one carbon to carbon double bond.
- groups include the vinyl, allyl, 1-propenyl, isopropenyl, 2-methyl-1-propenyl, 1-butenyl, 2- butenyl, 3-butenyl, 1-pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 1-hexenyl, 2- hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 1-heptenyl, and like.
- Alkenyl groups may also be substituted at any available point of attachment.
- exemplary substituents for alkenyl groups include those listed above for alkyl groups, and especially include C3 to C7 cycloalkyl groups such as cyclopropyl, cyclopentyl and cyclohexyl, which may be further substituted with, for example, amino, oxo, hydroxyl, etc.
- alkynyl refers to straight or branched chain alkyne. groups, which have one or more unsaturated carbon-carbon bonds, at least one of which is a triple bond.
- Alkynyl groups include C2-C8 alkynyl, C2-C6 alkynyl and C2-C4 alkynyl groups, which have from 2 to 8, 2 to 6 or 2 to 4 carbon atoms, respectively.
- Illustrative of the alkynyl group include ethenyl, propenyl, isopropenyl, butenyl, isobutenyl, pentenyl, and hexenyl.
- Alkynyl groups may also be substituted at any available point of attachment.
- Exemplary substituents for alkynyl groups include those listed above for alkyl groups such as amino, alkylamino, etc.
- the numbers in the subscript after the symbol "C" define the number of carbon atoms a particular group can contain.
- alkoxy alone or as part of another group denotes an alkyl group as described above bonded through an oxygen linkage (-O-).
- Preferred alkoxy groups have from 1 to 8 carbon atoms. Examples of such groups include the methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, tert-butoxy, n-pentyloxy, isopentyloxy, n-hexyloxy, cyclohexyloxy, n-heptyloxy, n- octyloxy and 2-ethylhexyloxy.
- alkylthio refers to an alkyl group as described above attached via a sulfur bridge.
- Preferred alkoxy and alkylthio groups are those in which an alkyl group is attached via the heteroatom bridge.
- Preferred alkylthio groups have from 1 to 8 carbon atoms. Examples of such groups include the methylthio, ethylthio, n-propythiol, n-butylthiol, and like.
- alkoxycarbonyl herein alone or as part of another group denotes an alkoxy group bonded through a carbonyl group.
- An alkoxycarbonyl radical is represented by the formula: -C(O)OR, where the R group is a straight or branched C1-C6 alkyl group, cycloalkyl, aryl, or heteroaryl.
- alkylcarbonyl herein alone or as part of another group refers to an alkyl group bonded through a carbonyl group or -C(O)R.
- arylalkyl herein alone or as part of another group denotes an aromatic ring bonded through an alkyl group (such as benzyl) as described above.
- aryl herein alone or as part of another group refers to monocyclic or bicyclic aromatic rings, e.g. phenyl, substituted phenyl and the like, as well as groups which are fused, e.g., napthyl, phenanthrenyl and the like.
- An aryl group thus contains at least one ring having at least 6 atoms, with up to five such rings being present, containing up to 20 atoms therein, with alternating (resonating) double bonds between adjacent carbon atoms or suitable heteroatoms.
- halogen such as I, Br, F, or Cl
- alkyl such as methyl, ethyl, propyl
- alkoxy such as methoxy or ethoxy
- hydroxy carboxy
- carbamoyl alkyloxycarbonyl
- nitro alkenyloxy
- amino, cycloalkyl, aryl, heteroaryl cyano, alkyl S(0)m (m
- aromatic refers to a cyclically conjugated molecular entity with a stability, due to derealization, significantly greater than that of a hypothetical localized structure, such as the Kekule structure.
- amino herein alone or as part of another group refers to -NH2.
- An “amino” may optionally be substituted with one or two substituents, which may be the same or different, such as alkyl, aryl, arylalkyl, alkenyl, alkynyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl, cycloheteroalkylalkyl, cycloalkyl, cycloalkylalkyl, haloalkyl, hydroxyalkyl, alkoxyalkyl, thioalkyl, carbonyl or carboxyl.
- substituents may be further substituted with a carboxylic acid, any of the alkyl or aryl substituents set out herein.
- the amino groups are substituted with carboxyl or carbonyl to form N-acyl or N- carbamoyl derivatives.
- alkylsulfonyl refers to groups of the formula (SO2)-alkyl, in which the sulfur atom is the point of attachment.
- alkylsulfonyl groups include C1- C6 alkylsulfonyl groups, which have from 1 to 6 carbon atoms.
- Methylsulfonyl is one representative alkylsulfonyl group.
- heteroatom refers to any atom other than carbon, for example, N, O, or S.
- heteroaryl herein alone or as part of another group refers to substituted and unsubstituted aromatic 5 or 6 membered monocyclic groups, 9 or 10 membered bicyclic groups, and 11 to 14 membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings.
- Each ring of the heteroaryl group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom.
- the fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated.
- the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- Heteroaryl groups which are bicyclic or tricyclic must include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non- aromatic.
- the heteroaryl group may be attached at any available nitrogen or carbon atom of any ring.
- monocyclic heteroaryl groups include pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, diazolyl, isoxazolyl, thiazolyl, thiadiazolyl, S isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like.
- bicyclic heteroaryl groups include indolyl, benzothiazolyl, benzodioxolyl, benzoxaxolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, dihydroisoindolyl, tetrahydroquinolinyl and the like.
- tricyclic heteroaryl groups include carbazolyl, benzidolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like.
- heterocycle or “heterocycloalkyl” herein alone or as part of another group refers to a cycloalkyl group (nonaromatic) in which one of the carbon atoms in the ring is replaced by a heteroatom selected from O, S or N.
- the "heterocycle” has from 1 to 3 fused, pendant or spiro rings, at least one of which is a heterocyclic ring (i.e. , one or more ring atoms is a heteroatom, with the remaining ring atoms being carbon).
- the heterocyclic ring may be optionally substituted which means that the heterocyclic ring may be substituted at one or more substitutable ring positions by one or more groups independently selected from alkyl (preferably lower alkyl), heterocycloalkyl, heteroaryl, alkoxy (preferably lower alkoxy), nitro, monoalkylamino (preferably a lower alkylamino), dialkylamino (preferably a alkylamino), cyano, halo, haloalkyl (preferably trifluoromethyl), alkanoyl, aminocarbonyl, monoalkylaminocarbonyl, dialkylaminocarbonyl, alkyl amido (preferably lower alkyl amido), alkoxyalkyl (preferably a lower alkoxy; lower alkyl), alkoxycarbonyl (preferably a lower alkoxycarbonyl), alkylcarbonyloxy (preferably a lower alkylcarbonyloxy) and aryl (preferably phenyl), said aryl being optionally substituted by
- a heterocyclic ring comprises 1-4 heteroatoms; within certain embodiments each heterocyclic ring has 1 or 2 heteroatoms per ring.
- Each heterocyclic ring generally contains from 3 to 8 ring members (rings having from to 7 ring members are recited in certain embodiments), and heterocycles comprising fused, pendant or spiro rings typically contain from 9 to 14 ring members which consists of carbon atoms and contains one, two, or three heteroatoms selected from nitrogen, oxygen and/or sulfur.
- heterocycle or “heterocycloalkyl groups include piperazine, piperidine, morpholine, thiomorpholine, pyrrolidine, imidazolidine and thiazolide.
- substituted refers to a molecular moiety that is covalently bonded to an atom within a molecule of interest.
- a “ring substituent” may be a moiety such as a halogen, alkyl group, haloalkyl group or other group discussed herein that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member.
- aryl or heterocyclyl or other group may be substituted at one or more substitutable positions by one or more groups independently selected from alkyl (preferably lower alkyl), alkoxy (preferably lower alkoxy), nitro, monoalkylamino (preferably with one to six carbons), dialkylamino (preferably with one to six carbons), cyano, halo, haloalkyl (preferably trifluoromethyl), alkanoyl, aminocarbonyl, monoalkylaminocarbonyl, dialkylaminocarbonyl, alkyl amido (preferably lower alkyl amido), alkoxyalkyl (preferably a lower alkoxy and lower alkyl), alkoxycarbonyl (preferably a lower alkoxycarbonyl), alkylcarbonyloxy (preferably a lower alkylcarbonyloxy) and aryl (preferably phenyl), said aryl being optionally substituted by halo, lower alkyl and lower alkoxy groups
- Optional substitution is also indicated by the phrase "substituted with from 0 to X substituents," where X is the maximum number of possible substituents. Certain optionally substituted groups are substituted with from 0 to 2, 3 or 4 independently selected substituents. A dash (“-") that is not between two letters or symbols is used to indicate a point of t attachment for a substituent. For example, -CONH2 is attached through the carbon atom.
- a dashed cycle that locates inside of a heterocyle ring is used to indicate a conjugated system.
- the bonds between two atomes may be single bond or double bond.
- anticancer agent includes any known agent that is useful for the treatment of cancer including, but is not limited, Acivicin; Aclarubicin;
- Acodazole Hydrochloride AcrQnine; Adozelesin; Aldesleukin; Altretamine; Ambomycin; Ametantrone Acetate; Aminoglutethimide; Amsacrine; Anastrozole;
- Anthramycin Asparaginase; Asperlin; Azacitidine; Azetepa; Azotomycin;
- Busulfan Cactinomycin; Calusterone; Caracemide; Carbetimer; Carboplatin; Carmustine; Carubicin Hydrochloride; Carzelesin; Cedefingol; Chlorambucil;
- Cirolemycin Cisplatin; Cladribine; Crisnatol Mesylate; Cyclophosphamide;
- Cytarabine dacarbazine; Dactinomycin; Daunorubicin Hydrochloride;
- Docetaxel Docetaxel; Doxorubicin; Doxorubicin Hydrochloride; Droloxifene; Droloxifene Citrate; Dromostanolone Propionate; Duazomycin; Edatrexate; Eflomithine
- Interferon Alfa-2b Interferon Alfa-n1 ; Interferon Alfa-n3; Interferon Beta- 1 a;
- Interferon Gamma- 1 b Interferon Gamma- 1 b; Iproplatin; lrinotecan Hydrochloride; Lanreotide Acetate; Letrozole; Leuprolide Acetate; Liarozole Hydrochloride; Lometrexol Sodium;
- Metoprine Meturedepa; Mitindomide; Mitocarcin; Mitocromin; Mitogillin;
- Mitomalcin Mitomycin; Mitosper; Mitotane; Mitoxantrone Hydrochloride; Mycophenolic Acid; Nocodazole; Nogalamycin; Ormaplatin; Oxisuran; Paclitaxel;
- Pipobroman Piposulfan; Piroxantrone Hydrochloride; Plicamycin; Plomestane;
- Porfimer Sodium Porfiromycin; Prednimustine; Procarbazine Hydrochloride;
- Puromycin Puromycin Hydrochloride; Pyrazofurin; Riboprine; Rogletimide; Safmgol; Safingol Hydrochloride; Semustine; Simtrazene; Sparfosate Sodium;
- Taxane Taxoid; Tecogalan Sodium; Tegafur; Teloxantrone Hydrochloride;
- Temoporfin Teniposide; Teroxirone; Testolactone; Thiamiprine; Thioguanine; Thiotepa; Tiazofurin; Tirapazamine; Topotecan Hydrochloride; Toremifene
- Vapreotide Verteporfin; Vinblastine Sulfate; Vincristine Sulfate; Vindesine;
- kinase refers to any enzyme that catalyzes the addition of phosphate groups to a protein residue; for example, serine and threonine kineses catalyze the addition of phosphate groups to serine and threonine residues.
- the terms "Src kinase,” “Src kinase family,” and “Src family” refer to the related homologs or analogs belonging to the mammalian family of Src kineses, including, for example, c-Src, Fyn, Yes and Lyn kineses and the hematopoietic- restricted kineses Hck, Fgr, Lck and BIk.
- terapéuticaally effective amount refers to the amount of the compound or pharmaceutical composition that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician, e.g., restoration or maintenance of vasculostasis or prevention of the compromise or loss or vasculostasis; reduction of tumor burden; reduction of morbidity and/or mortality.
- pharmaceutically acceptable refers to the fact that the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a compound refers to the act of providing a compound of the invention or pharmaceutical composition to the subject in need of treatment.
- protected refers that the group is in modified form to preclude undesired side reactions at the protected site. Suitable protecting groups for the compounds of the present invention will be recognized from the present application taking into account the level of skill in the art, and with reference to standard textbooks, such as Greene, T. W. et al., Protective Groups in Organic Synthesis, John Wiley & Sons, New York (1999).
- pharmaceutically acceptable salt of a compound recited herein is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals without excessive toxicity or carcinogenicity, and preferably without irritation, allergic response, or other problem or complication.
- Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids.
- Specific pharmaceutical salts include, but are not limited to, salts of acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric, sulfamic, sulfanilic, formic, toluenesulfonic, methanesulfonic, benzene sulfonic, ethane disulfonic, 2- hydroxyethylsulfonic, nitric, benzoic, 2-acetoxybenzoic, citric, tartaric, lactic, stearic, salicylic, glutamic, ascorbic, pamoic, succinic, fumaric, maleic, propionic, hydroxymaleic, hydroiodic, phenylacetic, alkanoic such as acetic, HOOC- (CH2)n-COOH where n is 0-4, and the like.
- acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric, s
- pharmaceutically acceptable cations include, but are not limited to sodium, potassium, calcium, aluminum, lithium and ammonium.
- a pharmaceutically acceptable acid or base salt can be synthesized from a parent compound that contains a basic or acidic moiety by any conventional chemical method. Briefly, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanol, isopropanol or acetonitrile, is preferred.
- each compound of Formula I may, but need not, be formulated as a hydrate, solvate or non- covalent complex.
- the various crystal forms and polymorphs are within the scope of the present invention.
- prodrugs of the compounds of Formula I are also provided herein.
- prodrug refers a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce a compound of Formula I, or other formula provided herein.
- a prodrug may be an acylated derivative of a compound as provided herein.
- Prodrugs include compounds wherein hydroxy, amine or thiol groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxy, amino, or thiol group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups within the compounds provided herein.
- Prodrugs of the compounds provided herein may be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved in vivo to yield the parent compounds.
- Groups that are "optionally substituted” are unsubstituted or are substituted by other than hydrogen at one or more available positions.
- Such optional substituents include, for example, hydroxy, halogen, cyano, nitro, C1-C6 alkyl, C2-C6 alkenyl, C2- C6 alkynyl, C1-C6 alkoxy, C2-C6 alkyl ether, C3-C6 alkanone, C2-C6 alkylthio, amino, mono- or di-(C1-C6 alkyl)amino, C1-C6 haloalkyl, -COOH, -CONH2, mono- or di-(C1-C6 alkyl)aminocarbonyl, -SO2NH2, and/or mono or di(C1-C6 alkyl) sulfonamido, as well as carbocyclic and heterocyclic groups.
- -H, -CH3, -CH2CH3, -CH CHCH3, -CH2CH2CH3, -CH2CH2CH2CH3, iso-propyl, cyclopropyl, cyclobutyl, tert-butyl, phenyl (-Ph), -CH2OH, - COOCH2CH3, -Cl, -F, -Br.
- R3 groups of formula (I) are list below:
- the compounds of the invention may be compounds of formula (I) wherein
- R1 groups of formula (I) are listed below:
- -H, -CH3, -CH2CH3, -CH CHCH3, -CH2CH2CH3, -CH2CH2CH2CH3, iso-propyl, cyclopropyl, cyclobutyl, tert-butyl, phenyl (-Ph), -CH2OH, - COOCH2CH3, -Cl, -F, -Br.
- W and Y are independently selected from S, O, NR4, or CR4;
- R4 is independently selected from hydrogen or an optionally substituted C1-4 aliphatic group.
- K is selected from -NR4, O, or S R2 is selected from:
- R 5 represents hydrogen, C 1 -C 4 alkyl, oxo
- X is CH, when R 6 is hydrogen; or X-R 6 is O; or X is N, R 6 represents groups of hydrogen, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C6 alkynyl, C3-C 1 0 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, d- C 6 haloalkyl, C 1 -C 6 alkoxy, Cr C 6 alkylthio, C2-C6 alkanoyl, Cr Ce alkoxycarbonyl, C2- Ce alkanoyloxy, mono- and di-(C 3 -Cs cycloalkyl)aminoCo-C 4 alkyl, (4- to 7- membered heterocycle)Co-C 4 alkyl, C 1 -Ce alkylsulfonyl, mono- and di-(Cr Ce alkyl) sulfonamido, and mono
- R3 is Hydrogene, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 1 0 aryl or heteroaryl, (C 3 -C7cycloalkyl)C 1 -C 4 alkyl, d- C 6 haloalkyl, each of which is substituted with from O to 4 substituents independently chosen from halogen, hydroxy, cyano, amino, -COOH and oxo;
- W and Y are independently selected from S, O, NR4, or CR4; R4 is independently selected from hydrogen or an optionally substituted
- K is selected from -NR4, O, or S
- R2 is selected from:
- R 5 represents hydrogen, C 1 -C 4 alkyl, oxo
- X is CH, when R 6 is hydrogen; or X-R 6 is O; or X is N, R 6 represents groups of hydrogen, C 1 -C 6 alkyl, C 2 -C6 alkenyl, C 2 -C 6 alkynyl, C3-C10 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, d- C 6 haloalkyl, C 1 -C 6 alkoxy, C r C 6 alkylthio, C 2 -C 6 alkanoyl, C 1 - C 6 alkoxycarbonyl, C 2 - C 6 alkanoyloxy, mono- and di-(C 3 -C 8 cycloalkyl)aminoCo-C 4 alkyl, (4- to 7- membered heterocycle)C 0 -C 4 alkyl, C 1 -C 6 alkylsulfonyl, mono- and di-(C- ⁇ - C 6
- R3 is Hydrogene, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 10 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, d- C 6 haioalkyl, each of which is substituted with from O to 4 substituents independently chosen from halogen, hydroxy, cyano, amino, -COOH and oxo;
- W and Y are independently selected from S, O, NH, or CH
- K is selected from -NH, O, or S
- R2 is selected from: (i) amino, alkyl amino, aryl amino, heteroaryl amino;
- R 5 represents hydrogen, C 1 -C 4 alkyl, oxo
- X is CH, when R 6 is hydrogen; or X-R 6 is O; or X is N, R 6 represents groups of hydrogen, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C3-C10 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, Cr C 6 haloalkyl, C 1 -C 6 alkoxy, Cr C 6 alkylthio, C 2 -C 6 alkanoyl, Cr C 6 alkoxycarbonyl, C 2 - C 6 alkanoyloxy, mono- and di-(C 3 -Cs cycloalkyl)aminoCo-C 4 alkyl, (4- to 7- membered heterocycle)Co-C 4 alkyl, C 1 -C 6 alkylsulfonyl, mono- and di-(Cr C 6 alkyl) sulfonamido
- R3 is Hydrogene, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 3 -C 10 aryl or heteroaryl, (C 3 -C 7 cycloalkyl)C 1 -C 4 alkyl, C r C 6 haloalkyl, each of which is substituted with from O to 4 substituents independently chosen from halogen, hydroxy, cyano, amino, -COOH and oxo;
- Preferred heterocyclic groups in compounds of Formula (I) include
- the present invention relates to a compound of formula (I) wherein R1 is hydrogen. According to another embodiment, the present invention relates to a compound of formula (I) wherein R1 is chloro.
- the present invention relates to a compound of formula (I) wherein R1 is methyl.
- the present invention relates to a compound of formula I wherein R1 is ethyl.
- the present invention relates to a compound of formula I wherein R1 is propyl.
- the present invention relates to a compound of formula I wherein R1 is isopropyl. According to another embodiment, the present invention relates to a compound of formula I wherein R1 is isobutyl.
- the present invention relates to a compound of formula I wherein R1 is tert-butyl.
- the present invention relates to a compound of formula I wherein R1 is cyclopropyl. According to another embodiment, the present invention relates to a compound of formula I wherein R1 is cyclobutyl.
- the present invention relates to a compound of formula I wherein R2 is methyl-piperazinyl. According to another embodiment, the present invention relates to a compound of formula I wherein R2 is (2-hydroxylethyl)-piperazinyl.
- the present invention relates to a compound of formula I wherein R2 is (4-pyridinyl)-piperazinyl.
- the present invention relates to a compound of formula I wherein R2 is methyl.
- the present invention relates to a compound of formula I wherein R2 is ethyl.
- the present invention relates to a compound of formula I wherein R2 is cyclopropyl.
- Examples of specific compounds of the present invention are those compounds defined in the following:
- a method of preparing the inventive compounds is provided.
- the compounds of the present invention can be generally prepared using cyanuric chloride as a starting material.
- Compound (I) may contain various stereoisomers, geometric isomers, tautomeric isomers, and the like. All of possible isomers and their mixtures are included in the present invention, and the mixing ratio is not particularly limited.
- the triazine derivative compounds of Formula (I) in this invention can be prepared by known procedure in the prior art. The examples could be found in US patent No. 2005250945A1 ; US patent No. 20050227983A1 ; PCT WO
- reduction refers to the process of reducing a nitro functionality to an amino functionality, or the process of transforming an ester functionality to an alcohol.
- the reduction of a nitro group can be carried out in a number of ways well known to those skilled in the art of organic synthesis including, but not limited to, catalytic hydrogenation, reduction with SnCI2 and reduction with titanium bichloride.
- the reduction of an ester group is typically performed using metal hydride reagents including, but not limited to, diisobutyl-aluminum hydride (DIBAL), lithium aluminum hydride (LAH), and sodium borohydride.
- DIBAL diisobutyl-aluminum hydride
- LAH lithium aluminum hydride
- sodium borohydride sodium borohydride
- hydrolyze refers to the reaction of a substrate or reactant with water. More specifically, “hydrolyze” refers to the conversion of an ester or nitrite functionality into a carboxylic acid. This process can be catalyzed by a variety of acids or bases well known to those skilled in the art of organic synthesis.
- the compounds of Formula (I) may be prepared by use of known chemical reactions and procedures. The following general preparative methods are presented to aid one of skill in the art in synthesizing the inhibitors, with more detailed examples being presented in the experimental section describing the working examples.
- Heterocyclic amines are defined in formula (II). Some of heterocyclic amines are commercially available, others may be prepared by known procedure in the prior art (Katritzky, et al. Comprehensive Heterocyclic Chemistry; Permagon Press: Oxford, UK, 1984, March. Advanced Organic Chemistry, 3" Ed.; John Wiley: New York, 1985), or by using common knowledge of organic chemistry.
- substituted heterocyclic amines can be generated using standard methods (March, J. Advanced Organic Chemistry, 4th Ed.; John Wiley, New York (1992); Larock, R. C. Comprehensive Organic Transformations, 2 nd Ed., John Wiley, New York (1999); World patent No. WO 99/32106).
- heterocyclic amines can be commonly synthesized by reduction of nitroheteros using a metal catalyst, such as Ni, Pd, or Pt, and H 2 ⁇ r a hydride transfer agent, such as formate, cyclohexadiene, or a borohydride (Rylander. Hydrogenation Methods; Academic Press: London, UK (1985)).
- Nitroheteros may also be directly reduced using a strong hydride source, such as LAH 1 (Seyden- Penne. Reductions by the Alumino- and Borohydrides in Organic Synthesis; VCH Publishers: New York (1991 )), or using a zero valent metal, such as Fe, Sn or Ca, often in acidic media.
- LAH 1 Sud- Penne. Reductions by the Alumino- and Borohydrides in Organic Synthesis; VCH Publishers: New York (1991 )
- a zero valent metal such as Fe, Sn or Ca
- thiazole amine with a substituent (lib) can be prepared from commercial compounds as illustrated in Scheme 2.
- a substituted aldehyde which may be commercially available or prepared by oxidizing an alcohole, can be brominated by broming or NBS (N- Bromosuccinimide); after bromination, the aldehyde can be converted to the corresponding thiazole amine (lib) by reacting with thiourea.
- oxidizing reagent for the oxidation step, a variety of oxidizing reagent can be used, such as pyridinium chlorochromate (PCC) activated dimethyl sulfoxide (DMSO), hypervalent iodide compounds, Tetrapropylammonium perruthenate (TPAP) or 2,2,6,6- Tetramethylpiperidine-1-oxyl (TEMPO).
- PCC pyridinium chlorochromate
- DMSO dimethyl sulfoxide
- TPAP Tetrapropylammonium perruthenate
- TEMPO 2,2,6,6- Tetramethylpiperidine-1-oxyl
- pyrazole amines with a substituent can be prepared by known procedure in the prior art, such as US Patent 6407238; F. Gabrera Escribano, et al. Tetrahedron Letters, Vol. 29, No. 46, pp. 6001-6004, 1988; Org. Biomol. Chem., 2006, 4, 4158 - 4164; WO/2003/026666.
- Precursors R 2 H can be purchased from suppliers such as Alderich.
- Intermediate of urea compound (III) can be prepared by general knowledge on urea synthesis know in the arts. As illustrated in scheme 3, substituted isocyanate can be be used to react with a substituted aniline under appropriate conditions to produce intermediate (III).
- Scheme 4 illustrated the synthesis method for compounds with alkyl or aryl as R 2 .
- the 6-alkyl or aryl substituted dichloro-triazine (b) may be synthesized by the methods known in the art (e.g., J. Med. Chem. 1999, 42, 805-818 and J. Med. Chem. 2004, 47, 600-611 ) from cyanuric chloride (a) and Grignard reagents.
- the monochloro-triazine (c) can be formed from the reaction of a 6- alkyl or aryl substituted dichloro-triazine (b) with heterocyclic amine, which can be converted to triazine derivatives (I) by reaction with intermiate (111).
- dichloro-triazine (b) can be converted to monochloro-triazine (d) by reacting with intermediate (III), which also can be converted to triazine derivative (I) by reacting with a heterocyclic amine.
- the triazine derivative can also be synthesized by the reaction of cyanuric chloride with a sequence of heterocyclic amines and HR 2 to give2,4-disubstituted-6-chloro-1 ,3,5-triazines.
- the displacement of the last chlorine by intermediate (III) can be achieved by increasing the temperature, affording the trisubstituted-1 ,3,5-triazines (I).
- the reaction can be completed in one pot or step by step.
- reaction sequence can also be used to make triazine derivatives such as illustrated in Scheme 6.
- other reaction sequence can also be utilized.
- the triazine derivative can be obtained by synthesizinf intermediate (IV), which react with an appropriate regent such as substituted isocyanate to produce compound (I).
- Othe methods can also be used for the prepreation of urea compound from an amine such as thos kown in the arts (E. Artuso, I. Degani, R. Fochi, C. Magistris, Synthesis, 2007, 3497-3506; C. Han, J. A. Porco, Jr, Org. Lett, 2007, 9, 1517-1520; H. Lebel, O. Leogane, Org. Lett, 2006, 8, 5717-5720; M. B. Bertrand, J. P.
- the reaction is preferably conducted in the presence of an inert solvent.
- an inert solvent there is no particular restriction on the nature of the solvent to be employed, provided that it has no adverse effect on the reaction or on the reagents involved and that it can dissolve the reagents, at least to some extent.
- suitable solvents include: aliphatic hydrocarbons, such as hexane, heptane, ligroin and petroleum ether; aromatic hydrocarbons, such as benzene, toluene and xylene; halogenated hydrocarbons, especially aromatic and aliphatic hydrocarbons, such as methylene chloride, chloroform, carbon tetrachloride, dichloroethane, chlorobenzene and the dichlorobenzenes; esters, such as ethyl formate, ethyl acetate, propyl acetate, butyl acetate and diethyl carbonate; ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran, dioxane.
- aliphatic hydrocarbons such as hexane, heptane, ligroin and petroleum ether
- aromatic hydrocarbons such as benzene, toluene and
- ketones such as acetone, methyl ethyl ketone, methyl isobutyl ketone, isophorone and cyclohexanone
- nitro compounds which may be nitroalkanes or nitroaranes, such as nitroethane and nitrobenzene
- nitriles such as acetonitrile and isobutyronitrile
- amides which may be fatty acid amides, such as formamide, dimethylformamide, dimethylacetamide and hexamethylphosphoric triamide
- sulphoxides such as dimethyl sulphoxide and sulpholane.
- the reaction can take place over a wide range of temperatures, and the precise reaction temperature is not critical to the invention. In general, we find it convenient to carry out the reaction at a temperature of from -50°C to 100°C.
- the present invention provides compositions of matter that are formulations of one or more active drugs and a pharmaceutically-acceptable carrier.
- the invention provides a composition for administration to a mammalian subject, which may include a compound of formula I, or its pharmaceutically acceptable salts.
- Pharmaceutically acceptable salts of the compounds of this invention include those derived from pharmaceutically acceptable inorganic and organic acids and bases.
- suitable acid salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, formate, fumarate, glucoheptanoate, glycerophosphate, glycolate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, malonate, methanesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oxalate, palmoate, pec
- Salts derived from appropriate bases include alkali metal (e.g., sodium and potassium), alkaline earth metal (e.g., magnesium), ammonium and N+(C1-4 alkyl)4 salts.
- alkali metal e.g., sodium and potassium
- alkaline earth metal e.g., magnesium
- ammonium and N+(C1-4 alkyl)4 salts This invention also envisions the quaternization of any basic nitrogen-containing groups of the compounds disclosed herein. Water or oil- soluble or dispersible products may be obtained by such quaternization.
- the compositions of the present invention may be administered orally, parenterally, by inhalation spray, topically, rectally, nasally, buccally, vaginally or via an implanted reservoir.
- parenteral includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- the compositions are administered orally, intrapehtoneally or intravenously.
- compositions of this invention may be orally administered in any orally acceptable dosage form including, but not limited to, capsules, tablets, troches, elixirs, suspensions, syrups, wafers, chewing gums, aqueous suspensions or solutions.
- the oral compositions may contain additional ingredients such as: a binder such as microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as starch or lactose, a disintegrating agent such as alginic acid, corn starch and the like; a lubricant such as magnesium stearate; a glidant such as colloidal silicon dioxide; and a sweetening agent such as sucrose or saccharin or flavoring agent such as peppermint, methyl salicylate, or orange flavoring.
- a binder such as microcrystalline cellulose, gum tragacanth or gelatin
- an excipient such as starch or lactose, a disintegrating agent such as alginic acid, corn starch and the like
- a lubricant such as magnesium stearate
- a glidant such as colloidal silicon dioxide
- a sweetening agent such as sucrose or saccharin or flavoring agent such as peppermint, methyl salicylate, or orange flavoring.
- tablets or pills may be coated with sugar, shellac, or other enteric coating agents.
- a syrup may contain, in addition to the active ingredients, sucrose as a sweetening agent and certain preservatives, dyes and colorings and flavors. Materials used in preparing these various compositions should be pharmaceutically or veterinarally pure and non-toxic in the amounts used.
- the active ingredient may be incorporated into a solution or suspension.
- the solutions or suspensions may also include the following components: a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose.
- the parenteral preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of glass or plastic.
- the pharmaceutical forms suitable for injectable use include sterile solutions, dispersions, emulsions, and sterile powders.
- the final form should be stable under conditions of manufacture and storage. Furthermore, the final pharmaceutical form should be protected against contamination and should, therefore, be able to inhibit the growth of microorganisms such as bacteria or fungi.
- a single intravenous or intraperitoneal dose can be administered. Alternatively, a slow long-term infusion or multiple short-term daily infusions may be utilized, typically lasting from 1 to 8 days. Alternate day dosing or dosing once every several days may also be utilized.
- Sterile, injectable solutions may be prepared by incorporating a compound in the required amount into one or more appropriate solvents to which other ingredients, listed above or known to those skilled in the art, may be added as required.
- Sterile injectable solutions may be prepared by incorporating the compound in the required amount in the appropriate solvent with various other ingredients as required. Sterilizing procedures, such as filtration, may then follow.
- dispersions are made by incorporating the compound into a sterile vehicle which also contains the dispersion medium and the required other ingredients as indicated above. In the case of a sterile powder, the preferred methods include vacuum drying or freeze drying to which any required ingredients are added.
- Suitable pharmaceutical carriers include sterile water; saline, dextrose; dextrose in water or saline; condensation products of castor oil and ethylene oxide combining about 30 to about 35 moles of ethylene oxide per mole of castor oil; liquid acid; lower alkanols; oils such as corn oil; peanut oil, sesame oil and the like, with emulsifiers such as mono- or di-glyceride of a fatty acid, or a phosphatide, e.g., lecithin, and the like; glycols; polyalkylene glycols; aqueous media in the presence of a suspending agent, for example, sodium carboxymethylcellulose; sodium alginate; poly(vinylpyrolidone) ; and the like, alone, or with suitable dispensing agents such as lecithin; polyoxyethylene stearate; and the like.
- a suspending agent for example, sodium carboxymethylcellulose; sodium alginate; poly(vinylpyrolidone)
- the carrier may also contain adjuvants such as preserving stabilizing, wetting, emulsifying agents and the like together with the penetration enhancer.
- adjuvants such as preserving stabilizing, wetting, emulsifying agents and the like together with the penetration enhancer.
- the final form must be sterile and should also be able to pass readily through an injection device such as a hollow needle.
- the proper viscosity may be achieved and maintained by the proper choice of solvents or excipients.
- the use of molecular or particulate coatings such as lecithin, the proper selection of particle size in dispersions, or the use of materials with surfactant properties may be utilized.
- compositions containing triazine derivatives and methods useful for the in vivo delivery of triazine derivatives in the form of nanoparticles which are suitable for any of the aforesaid routes of administration.
- United States Patent Nos. 5,916,596, 6,506,405 and 6,537,579 teach the preparation of nanoparticles from the biocompatible polymers, such as albumin.
- methods for the formation of nanoparticles of the present invention by a solvent evaporation technique from an oil-in-water emulsion prepared under conditions of high shear forces (e.g., sonication, high pressure homogenization, or the like).
- compositions of this invention may be administered in the form of suppositories for rectal administration.
- suppositories for rectal administration.
- suppositories can be prepared by mixing the agent with a suitable non- irritating excipient that is solid at room temperature but liquid at rectal temperature and therefore will melt in the rectum to release the drug.
- suitable non- irritating excipient include cocoa butter, beeswax and polyethylene glycols.
- compositions of this invention may also be administered topically, especially when the target of treatment includes areas or organs readily accessible by topical application, including diseases of the eye, the skin, or the lower intestinal tract. Suitable topical formulations are readily prepared for each of these areas or organs.
- Topical application for the lower intestinal tract can be effected in a rectal suppository formulation (see above) or in a suitable enema formulation. Topically-transdermal patches may also be used.
- the pharmaceutically acceptable compositions may be formulated in a suitable ointment containing the active component suspended or dissolved in one or more carriers.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petrolatum, white petrolatum, propylene glycol, polyoxyethylene, polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutically acceptable compositions can be formulated in a suitable lotion or cream containing the active components suspended or dissolved in one or more pharmaceutically acceptable carriers.
- suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutically acceptable compositions may be formulated as micronized suspensions in isotonic, pH adjusted sterile saline, or, preferably, as solutions in isotonic, pH adjusted sterile saline, either with or without a preservative such as benzylalkonium chloride.
- the pharmaceutically acceptable compositions may be formulated in an ointment such as petrolatum.
- the pharmaceutically acceptable compositions of this invention may also be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- compositions of this invention are formulated for oral administration.
- the compounds of the invention may be used to treat diseases associated with cellular proliferation or hyperproliferation, such as cancers which include but are not limited to tumors of the nasal cavity, paranasal sinuses, nasopharynx, oral cavity, oropharynx, larynx, hypopharynx, salivary glands, and paragangliomas.
- cancers which include but are not limited to tumors of the nasal cavity, paranasal sinuses, nasopharynx, oral cavity, oropharynx, larynx, hypopharynx, salivary glands, and paragangliomas.
- the compounds of the invention may also be used to treat cancers of the liver and biliary tree (particularly hepatocellular carcinoma), intestinal cancers, particularly colorectal cancer, ovarian cancer, small cell and non-small cell lung cancer, breast cancer, sarcomas (including fibrosarcoma, malignant fibrous histiocytoma, embryonal rhabdomysocarcoma, leiomysosarcoma, neuro-fibrosarcoma, osteosarcoma, synovial sarcoma, liposarcoma, and alveolar soft part sarcoma), neoplasms of the central nervous systems (particularly brain cancer), and lymphomas (including Hodgkin's lymphoma, lymphoplasmacytoid lymphoma, follicular lymphoma, mucosa- associated lymphoid tissue lymphoma, mantle cell lymphoma, B-lineage large cell lymphoma, Burkitt's lymphoma, and
- the compounds and methods of the present invention are also useful in treating a variety of disorders, including but not limited to, for example: stroke, cardiovascular disease, myocardial infarction, congestive heart failure, cardiomyopathy, myocarditis, ischemic heart disease, coronary artery disease, cardiogenic shock, vascular shock, pulmonary hypertension, pulmonary edema (including cardiogenic pulmonary edema), pleural effusions, rheumatoid arthritis, diabetic retinopathy, retinitis pigmentosa, and retinopathies, including diabetic retinopathy and retinopathy of prematurity, inflammatory diseases, restenosis, asthma, acute or adult respiratory distress syndrome (ARDS), lupus, vascular leakage, protection from ischemic or reperfusion injury such as ischemic or reperfusion injury incurred during organ transplantation, transplantation tolerance induction; ischemic or
- T- cell mediated hypersensitivity diseases including contact hypersensitivity, delayed- type hypersensitivity, and gluten-sensitive enteropathy (Celiac disease); Type 1 diabetes; psoriasis; contact dermatitis (including that due to poison ivy); Hashimoto's thyroiditis; Sjogren's syndrome; Autoimmune Hyperthyroidism, such as Graves' disease; Addison's disease (autoimmune disease of the adrenal glands); autoimmune polyglandular disease (also known as autoimmune polyglandular syndrome); autoimmune alopecia; pernicious anemia; vitiligo; autoimmune hypopituatarism; Guillain-Barre syndrome; other autoimmune diseases; cancers, including those where kineses such as Src-family kineses are activated or overexpressed, such as colon carcinoma and thymoma, or cancers where kinase activity facilitates tumor growth or survival; glomerulonephritis, serum sickness; uticaria; allergic diseases such as respiratory allergies
- the compounds of the invention may be used to treat diseases associated with undesired cellular proliferation or hyperproliferation comprising identifying the mammal afflicted with said disease or condition and administering to said afflicted mammal a composition comprising the compound of formula 1 , wherein the disease or condition is associated with a kinase.
- the compounds of the invention may be used to treat diseases associated with undesired cellular proliferation or hyperproliferation comprising identifying the mammal afflicted with said disease or condition and administering to said afflicted mammal a composition comprising the compound of formula 1 , wherein the disease or condition is associated with a tyrosine kinase.
- the compounds of the invention may be used to treat diseases associated with undesired cellular proliferation or hyperproliferation comprising identifying the mammal afflicted with said disease or condition and administering to said afflicted mammal a composition comprising the compound of formula 1 , wherein the disease or condition is associated with the kinase that is a serine kinase or a threonine kinase.
- the compounds of the invention may be used to treat diseases associated with undesired cellular proliferation or hyperproiiferation comprising identifying the mammal afflicted with said disease or condition and administering to said afflicted mammal a composition comprising the compound of formula 1 , wherein the disease or condition is associated with the kinase that is a Src family kinase.
- the invention also provides methods of treating a mammal afflicted with the above diseases and conditions.
- the amount of the compounds of the present invention that may be combined with the carrier materials to produce a composition in a single dosage form will vary depending upon the host treated, the particular mode of administration.
- the compositions should be formulated so that a dosage of between 0.01-100 mg/kg body weight/day of the inhibitor can be administered to a patient receiving these compositions.
- the invention compounds are administered in combination with chemotherapeutic agent, an anti-inflammatory agent, antihistamines, chemotherapeutic agent, immunomodulator, therapeutic antibody or a protein kinase inhibitor, e.g., a tyrosine kinase inhibitor, to a subject in need of such treatment.
- chemotherapeutic agent an anti-inflammatory agent, antihistamines, chemotherapeutic agent, immunomodulator, therapeutic antibody or a protein kinase inhibitor, e.g., a tyrosine kinase inhibitor
- the method includes administering one or more of the inventive compounds to the afflicted mammal.
- the method may further include the administration of a second active agent, such as a cytotoxic agent, including alkylating agents, tumor necrosis factors, intercalators, microtubulin inhibitors, and topoisomerase inhibitors.
- a second active agent such as a cytotoxic agent, including alkylating agents, tumor necrosis factors, intercalators, microtubulin inhibitors, and topoisomerase inhibitors.
- the second active agent may be co-administered in the same composition or in a second composition.
- suitable second active agents include, but are not limited to, a cytotoxic drug such as Acivicin; Aclarubicin; Acodazole Hydrochloride; AcrQnine; Adozelesin; Aldesleukin; Altretamine; Ambomycin; Ametantrone Acetate; Aminoglutethimide; Amsacrine; Anastrozole; Anthramycin; Asparaginase; Asperlin; Azacitidine;
- a cytotoxic drug such as Acivicin; Aclarubicin; Acodazole Hydrochloride; AcrQnine; Adozelesin; Aldesleukin; Altretamine; Ambomycin; Ametantrone Acetate; Aminoglutethimide; Amsacrine; Anastrozole; Anthramycin; Asparaginase; Asperlin; Azacitidine;
- Cyclophosphamide Cytarabine; dacarbazine; Dactinomycin; Daunorubicin Hydrochloride; Decitabine; Dexormaplatin; Dezaguanine; Dezaguanine Mesylate; Diaziquone; Docetaxel; Doxorubicin; Doxorubicin Hydrochloride; Droloxifene; Droloxifene Citrate; Dromostanolone Propionate; Duazomycin; Edatrexate; Eflomithine Hydrochloride; Elsamitrucin; Enloplatin; Enpromate; Epipropidine;
- Fenretinide Floxuridine; Fludarabine Phosphate; Fluorouracil; Flurocitabine;
- Fosquidone Fostriecin Sodium; Gemcitabine; Gemcitabine Hydrochloride; Gold Au 198; Hydroxyurea; ldarubicin Hydrochloride; Ifosfamide; llmofosine; Interferon
- Alfa-2a Interferon Alfa-2b; Interferon Alfa-n1; Interferon Alfa-n3; Interferon Beta- La; Interferon Gamma- Ib; Iproplatin; lrinotecan Hydrochloride; Lanreotide
- Metoprine Meturedepa; Mitindomide; Mitocarcin; Mitocromin; Mitogillin;
- Mitomalcin Mitomycin; Mitosper; Mitotane; Mitoxantrone Hydrochloride;
- Pipobroman Piposulfan; Piroxantrone Hydrochloride; Plicamycin; Plomestane;
- Porfimer Sodium Porfiromycin; Prednimustine; Procarbazine Hydrochloride;
- Puromycin Puromycin Hydrochloride; Pyrazofurin; Riboprine; Rogletimide;
- Taxane Taxoid; Tecogalan Sodium; Tegafur; Teloxantrone Hydrochloride;
- Temoporfin Teniposide; Teroxirone; Testolactone; Thiamiprine; Thioguanine;
- Vapreotide Verteporfin; Vinblastine Sulfate; Vincristine Sulfate; Vindesine;
- Vindesine Sulfate Vinepidine Sulfate; Vinglycinate Sulfate; Vinleurosine Sulfate;
- the present invention also relates to compounds as shown in Formula (A):
- Y is selected from C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, -NR 4 R 5 , and -Q-R 3 ;
- Q is heterocycloalkyl, which is optionally substituted with C 1 -C 4 alkyl or oxo;
- R 3 is selected from H, C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, aryl, and heteroaryl;
- R 4 and R 5 are each independently selected from H, and C 1 -C 6 alkyl
- X is -K- Ar '-R 1 ;
- K is selected from NR 4 , S, and O;
- Ar 1 is phenyl
- R 1 is -NHC(O)NH-R 7 ;
- R 7 is selected from H, C)-C 6 alkyl, aryl, aryl(C 1 -C 6 )alkyl, each of which is optionally substituted with halo, hydroxy, C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, and C 1 -C 6 alkoxy;
- Z is -NH-Ar 2 -R 2 ;
- Ar 2 is heteroaryl including at least one nitrogen
- R 2 is one or more substituents independently selected from halo, hydroxy, C 1 -C 6 alkyl, -C(O)NH-W, C 2 -C 6 alkenyl, and C 2 -C 6 alkynyl; W is C 1 -C 6 alkyl.
- the present invention also relates to compounds as shown in Formula (A):
- Y is selected from C-C 6 alkyl and -Q-R 3 ;
- Q is piperazinyl;
- R 3 is C 1 -C 6 alkyl;
- X is -K- Ar '-R 1 ; K is selected from NH and S;
- Ar 1 is phenyl
- R 1 is -NHC(O)NH-R 7 ;
- R 7 is selected from C 1 -C 6 alkyl, phenyl, benzyl, which phenyl and benzyl are optionally substituted with C 1 -C 6 alkyl, Cj-C 6 haloalkyl, and C 1 -C 6 alkoxy;
- Z is -NH-Ar 2 -R 2 ;
- Ar 2 is selected from thiazolyl and pyrazolyl;
- R 2 is one or more substituents independently selected from C 1 -C 6 alkyl and -C(O)NH-W;
- W is C 1 -C 6 alkyl.
- the compounds and compositions may be used at sub-cytotoxic levels in combination with other agents in order to achieve highly selective activity in the treatment of non-neoplastic disorders, such as heart disease, stroke and neurodegenerative diseases (Whitesell et al., Curr Cancer Drug Targets (2003), 3(5), 349-58).
- the exemplary therapeutical agents that may be administered in combination with invention compounds include EGFR inhibitors, such as gefitinib, erlotinib, and cetuximab.
- Her2 inhibitors include canertinib, EKB-569, and GW- 572016.
- Src inhibitors include Src inhibitors, dasatinib, as well as Casodex (bicalutamide), Tamoxifen, MEK-1 kinase inhibitors, MARK kinase inhibitors, PI3 inhibitors, and PDGF inhibitors, such as imatinib, Hsp90 inhibitors, such as 17- AAG and 17-DMAG.
- anti-angiogenic and antivascular agents which, by interrupting blood flow to solid tumors, render cancer cells quiescent by depriving them of nutrition. Castration, which also renders androgen dependent carcinomas non-proliferative, may also be utilized. Also included are IGF1 R inhibitors, inhibitors of non- receptor and receptor tyrosine kineses, and inhibitors of integrin.
- the pharmaceutical composition and method of the present invention may further combine other protein therapeutic agents such as cytokines, immunomodulatory agents and antibodies.
- cytokine encompasses chemokines, interleukins, lymphokines, monokines, colony stimulating factors, and receptor associated proteins, and functional fragments thereof.
- functional fragment refers to a polypeptide or peptide which possesses biological function or activity that is identified through a defined functional assay.
- the cytokines include endothelial monocyte activating polypeptide Il (EMAP- II), granulocyte-macrophage-CSF (GM-CSF), granulocyte- CSF (G- CSF), macrophage- CSF (M-CSF), IL-1 , lL-2, IL-3, IL- 4, IL-5, IL-6, IL- 12, and IL-13, interferons, and the like and which is associated with a particular biologic, morphologic, or phenotypic alteration in a cell or cell mechanism.
- Other therapeutic agents for the combinatory therapy include cyclosporins
- CD40 and gp39 agents blocking the interaction between CD40 and gp39, such as antibodies specific for CD40 and for gpn39 (i.e., CD154), fusion proteins constructed from CD40 and gp39 (CD40lg and CD8gp39), inhibitors, such as nuclear translocation inhibitors, of NF-kappa B function, such as deoxyspergualin (DSG), cholesterol biosynthesis inhibitors such as HM:G CoA reductase inhibitors (lovastatin and simvastatin), non-steroidal antiinflammatory drugs (NSAIDs) such as ibuprofen and cyclooxygenase inhibitors such as rofecoxib, steroids such as prednisone or dexamethasone, gold
- NSAIDs non-steroidal antiinflammatory drugs
- Electrospray (ES+) ionization was used.
- Analytical gradient consisted of 10% ACN in water ramping up to 100% ACN over 5 minutes unless otherwise stated.
- HPLC High performance liquid chromatography
- Example 7 To thesulution of compound 6 (40mg, 0.12mMol) in 2mL of CH 2 CI 2 was added phenylisocyanate (15 ⁇ L, 17mg, 0.14mMol). Reaction mixture was stirred overnight at room temperature. Solvent was evaporated. Flash column chromatography (silica, CH 2 Cl 2 MeOH 100/0 to 95/5to 90/10) yielded 7mg (13%) of desired product of compound 7.
- Example 9 To the solution of compound 8 (3g, 16.9mMol) in 1OmL of THF was added 3-amino-5- methylpyrazole (1.64g, 16.9mMol) and DIPEA (3.24mL, 2.41g, 18.6mMol) in 5mL of THF. Reaction mixture was stirred for 2 hours at room temperature. 1,4- phenylenediamine (1.83g, 16.9mMol) and DIPEA (3.24mL, 2.41g, 18.6mMol) was added and reaction was microwaved at 100°C for 60 minutes.
- Example 11 To the solution of compound 9 (150mg, 0.48mMol) in 15mL of CH 2 CI 2 was added phenylisocyanate (57 ⁇ L, 63mg, 0.53mMol). Reaction mixture was stirred overnight at room temperature. Solvent was evaporated. Flash column chromatography (silica, CH 2 Cl 2 /Me0H 98/2 to 95/5 to 90/10) yielded 45mg (22%) of desired product of compound 11.
- This example illustrated c-Src kinase, Aurora-A kinase, Flt3 kinase, Ret kinase and TrkA Kinase Assays of selected Compounds from this invention (referred to
- kinaseProfilerTM Service Assay Protocols (Millipore) were used to test the kinase inhibiting activity of novel compounds from this invention.
- the buffer composition was as: 20 mM MOPS, 1 mM EDTA, 0.01 % Brij-35, 5% Glycerol, 0.1 % ⁇ -mercaptoethanol, 1 mg/mL BSA.
- Test compounds were initially dissolved in DMSO at the desired concentration, then serially diluted to the kinase assay buffer.
- Aurora-A(h) (5-10 mU) is incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 200 ⁇ M LRRASLG (Kemptide), 10 mM MgAcetate and [ ⁇ 33 P-ATP].
- the reaction was initiated by the addition of the MgATP mix. After incubation for 40 minute at room temperature, the reaction was stopped by addition of 5 ⁇ L of a 3% phosphoric acid solution. 10 ⁇ L of the reaction was then spotted onto a P30 filtermat and washed three times for 5 minutes in 50 mM phosphoric acid and once in methanol prior to drying and scintillation counting. Wells containing substrate but no kinase and wells containing a phosphopeptide control were used to set 0% and 100% phosphorylation value, repectively.
- Kinase Hotspot SM kinase assay was used to test the compounds for IC50 or % inhibitions (Reaction Biology Corp.). Inhibitor IC50 values were determined by titration of compound at the optimal kinase concentration (Kinase EC50).
- Table 1 shows representative data for the inhibition of c-Src kinase, Aurora-A kinase, Flt3 kinase, Ret kinase and TrkA Kinase by the compounds of this invention at a concentration of 1 ⁇ M.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Ophthalmology & Optometry (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
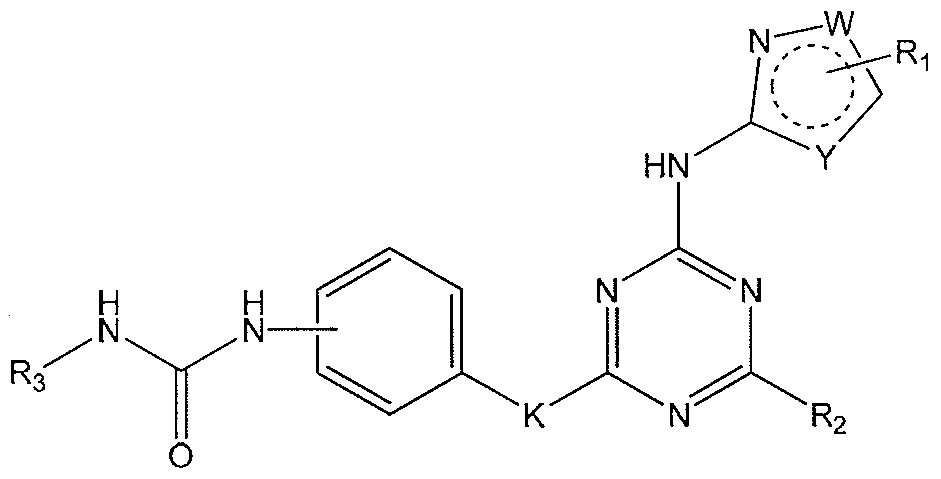












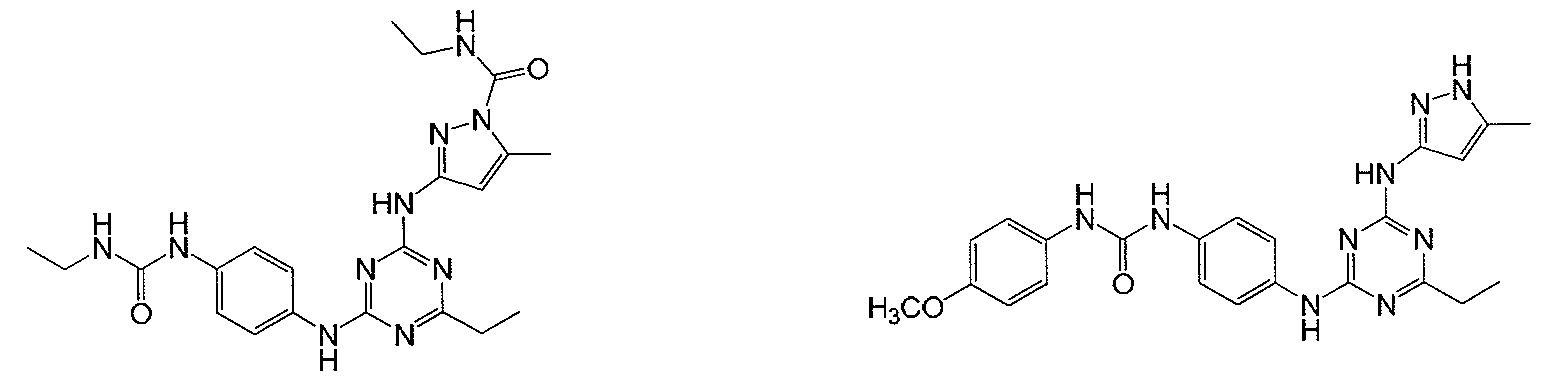




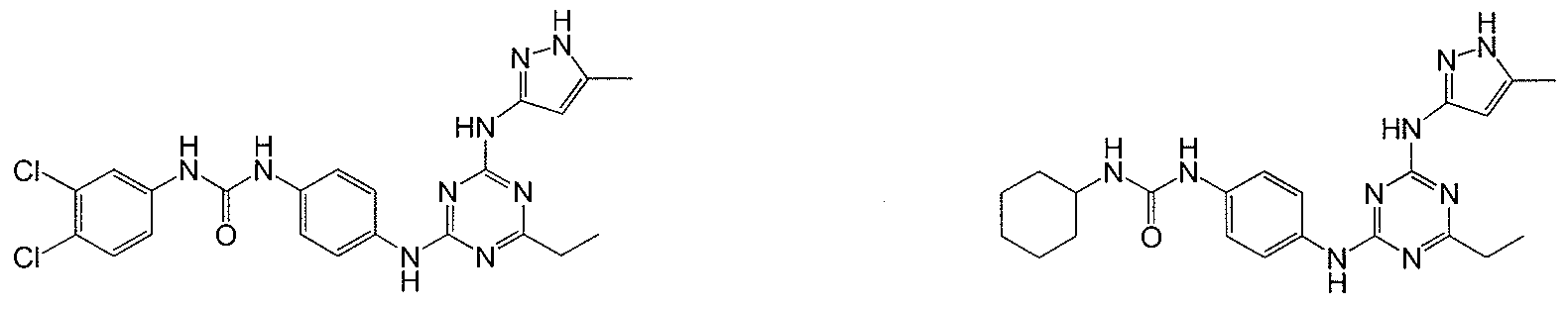























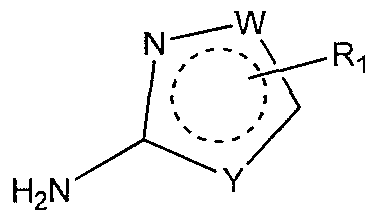















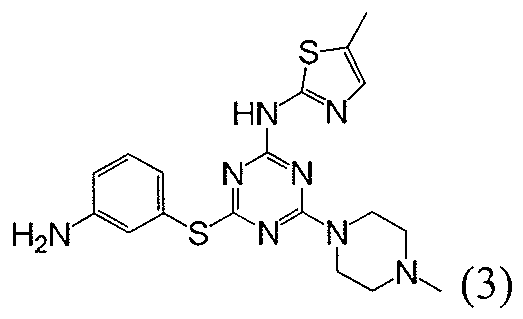
























Claims








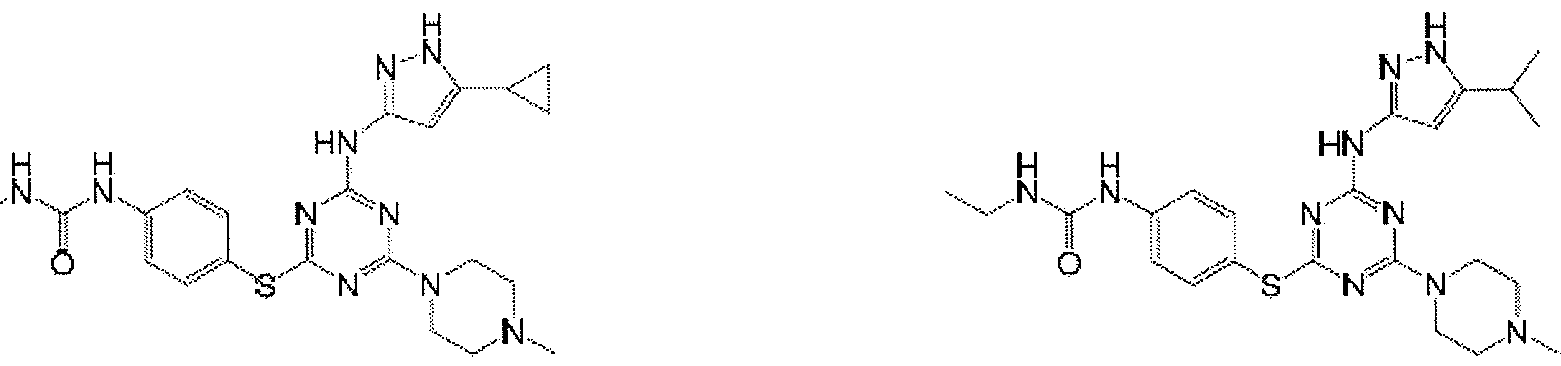












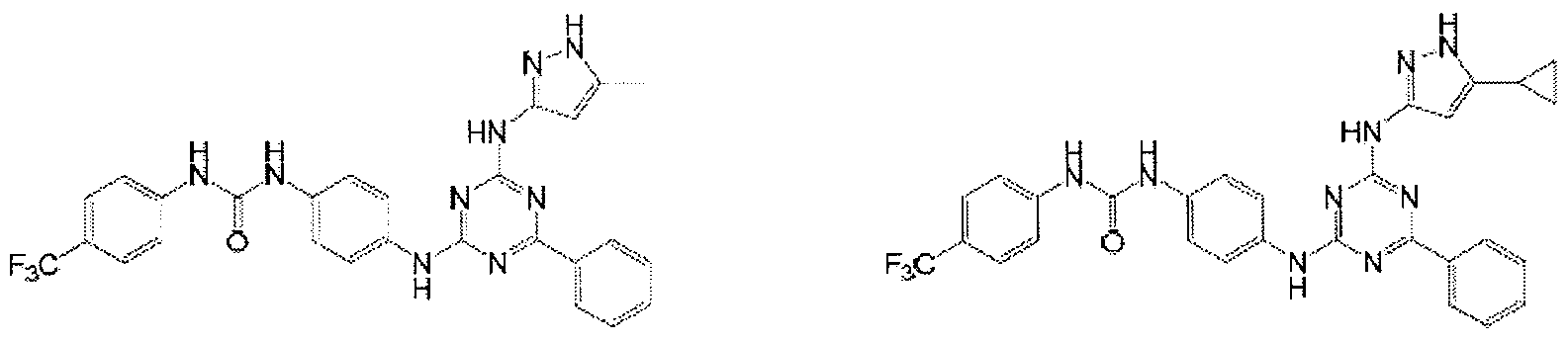




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2765050A CA2765050A1 (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
| JP2012515092A JP2012529527A (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutic applications |
| BRPI1011527A BRPI1011527A2 (en) | 2009-06-09 | 2010-06-09 | ureidophenyl substituted triazine derivatives and their therapeutic applications. |
| EP10786737A EP2440056A4 (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
| US13/376,818 US20120202818A1 (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
| CN201080032850XA CN102573481A (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
| AU2010258825A AU2010258825B2 (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
| IL216829A IL216829A0 (en) | 2009-06-09 | 2011-12-07 | Ureidophenyl substituted triazine derivatives, compositions comprising the same and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18542709P | 2009-06-09 | 2009-06-09 | |
| US61/185,427 | 2009-06-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010144522A1 true WO2010144522A1 (en) | 2010-12-16 |
Family
ID=43309205
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/037890 Ceased WO2010144522A1 (en) | 2009-06-09 | 2010-06-09 | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20120202818A1 (en) |
| EP (1) | EP2440056A4 (en) |
| JP (1) | JP2012529527A (en) |
| KR (1) | KR20120016676A (en) |
| CN (1) | CN102573481A (en) |
| AU (1) | AU2010258825B2 (en) |
| BR (1) | BRPI1011527A2 (en) |
| CA (1) | CA2765050A1 (en) |
| IL (1) | IL216829A0 (en) |
| WO (1) | WO2010144522A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102250065A (en) * | 2011-05-20 | 2011-11-23 | 浙江海正药业股份有限公司 | Substituted triazine phenyl urea derivatives and application thereof |
| EP2440050A4 (en) * | 2009-06-08 | 2013-04-03 | California Capital Equity Llc | Triazine derivatives and their therapeutical applications |
| WO2013161919A1 (en) * | 2012-04-26 | 2013-10-31 | 小野薬品工業株式会社 | Trk-INHIBITING COMPOUND |
| JP2014525418A (en) * | 2011-09-05 | 2014-09-29 | 浙江海正薬業股▲ふん▼有限公司 | 4-Substituted- (3-substituted-1H-pyrazole-5-amino) -pyrimidine derivatives having protein kinase inhibitory activity and uses thereof |
| US9463192B2 (en) | 2013-02-19 | 2016-10-11 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US10017495B2 (en) | 2013-07-11 | 2018-07-10 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10028961B2 (en) | 2013-07-11 | 2018-07-24 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10376510B2 (en) | 2013-07-11 | 2019-08-13 | Agios Pharmaceuticals, Inc. | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| WO2022046861A1 (en) * | 2020-08-26 | 2022-03-03 | Nalo Therapeutics | Modulators of myc family proto-oncogene protein |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120121515A1 (en) | 2009-03-13 | 2012-05-17 | Lenny Dang | Methods and compositions for cell-proliferation-related disorders |
| TWI598337B (en) | 2009-06-29 | 2017-09-11 | 阿吉歐斯製藥公司 | Therapeutic compounds and compositions |
| WO2011050210A1 (en) | 2009-10-21 | 2011-04-28 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| SMT202400081T1 (en) | 2011-05-03 | 2024-03-13 | Agios Pharmaceuticals Inc | Pyruvate kinase activators for use in therapy |
| CN102827170A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Active treatment compositions and use method thereof |
| CN102827073A (en) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | Therapeutically active compositions and application methods thereof |
| PE20142098A1 (en) | 2012-01-06 | 2015-01-08 | Agios Pharmaceuticals Inc | THERAPEUTICALLY ACTIVE COMPOUNDS AND THEIR METHODS OF USE |
| US9474779B2 (en) | 2012-01-19 | 2016-10-25 | Agios Pharmaceuticals, Inc. | Therapeutically active compositions and their methods of use |
| EP2906212A4 (en) | 2012-10-15 | 2016-06-08 | Agios Pharmaceuticals Inc | COMPOUNDS AND THERAPEUTIC COMPOSITIONS |
| CA2949048A1 (en) | 2013-05-22 | 2014-11-27 | The Regents Of The University Of California | Aurora kinase inhibitors |
| CN105473560B (en) * | 2013-07-11 | 2020-01-17 | 安吉奥斯医药品有限公司 | Therapeutic active compounds and methods of use |
| AU2014287122B9 (en) | 2013-07-11 | 2018-11-01 | Les Laboratoires Servier | N,6-bis(aryl or heteroaryl)-1,3,5-triazine-2,4-diamine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| WO2015010297A1 (en) | 2013-07-25 | 2015-01-29 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| NZ723859A (en) | 2014-03-14 | 2023-01-27 | Servier Lab | Pharmaceutical compositions of therapeutically active compounds and their uses |
| CA2989111C (en) | 2015-06-11 | 2023-10-03 | Agios Pharmaceuticals, Inc. | Methods of using pyruvate kinase activators |
| FI3362065T3 (en) | 2015-10-15 | 2024-06-19 | Servier Lab | Combination therapy comprising ivosidenib, cytarabine and daunorubicin or idarubicin for treating acute myelogenous leukemia |
| KR102699521B1 (en) | 2015-10-15 | 2024-08-26 | 르 라보레또레 쎄르비에르 | Combination therapy for the treatment of malignant tumors |
| CA3023854A1 (en) * | 2016-05-12 | 2017-11-16 | Nanjing Shiqi Pharmaceutical Co. Ltd. | Novel 2,4,6-trisubstituted s-triazine compounds, preparation method therefor, and use thereof |
| US10980788B2 (en) | 2018-06-08 | 2021-04-20 | Agios Pharmaceuticals, Inc. | Therapy for treating malignancies |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008076883A2 (en) * | 2006-12-15 | 2008-06-26 | Abraxis Bioscience, Inc. | Triazine derivatives and their therapeutical applications |
| US7473691B2 (en) * | 2000-09-15 | 2009-01-06 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002022605A1 (en) * | 2000-09-15 | 2002-03-21 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| US6989385B2 (en) * | 2000-12-21 | 2006-01-24 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| EP1485381B8 (en) * | 2002-03-15 | 2010-05-12 | Vertex Pharmaceuticals Incorporated | Azolylaminoazine as inhibitors of protein kinases |
| PE20040522A1 (en) * | 2002-05-29 | 2004-09-28 | Novartis Ag | DIARYLUREA DERIVATIVES DEPENDENT ON PROTEIN KINASE |
| US20050014753A1 (en) * | 2003-04-04 | 2005-01-20 | Irm Llc | Novel compounds and compositions as protein kinase inhibitors |
| MX2008005814A (en) * | 2005-11-03 | 2008-10-15 | Vertex Pharma | Aminopyrimidines useful as kinase inhibitors. |
-
2010
- 2010-06-09 WO PCT/US2010/037890 patent/WO2010144522A1/en not_active Ceased
- 2010-06-09 AU AU2010258825A patent/AU2010258825B2/en active Active
- 2010-06-09 US US13/376,818 patent/US20120202818A1/en not_active Abandoned
- 2010-06-09 CN CN201080032850XA patent/CN102573481A/en active Pending
- 2010-06-09 JP JP2012515092A patent/JP2012529527A/en active Pending
- 2010-06-09 BR BRPI1011527A patent/BRPI1011527A2/en not_active IP Right Cessation
- 2010-06-09 KR KR1020127000625A patent/KR20120016676A/en not_active Ceased
- 2010-06-09 EP EP10786737A patent/EP2440056A4/en not_active Withdrawn
- 2010-06-09 CA CA2765050A patent/CA2765050A1/en not_active Abandoned
-
2011
- 2011-12-07 IL IL216829A patent/IL216829A0/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7473691B2 (en) * | 2000-09-15 | 2009-01-06 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| WO2008076883A2 (en) * | 2006-12-15 | 2008-06-26 | Abraxis Bioscience, Inc. | Triazine derivatives and their therapeutical applications |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP2440056A4 * |
| WILLIAMS ET AL.: "Crystal Structures of the Lyn Protein Tyrosine Kinase Domain in Its Apo- and Inhibitor-bound State.", J BIOL CHEM, vol. 284, no. 1, 2 January 2009 (2009-01-02), pages 284 - 291, XP008124733 * |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2440050A4 (en) * | 2009-06-08 | 2013-04-03 | California Capital Equity Llc | Triazine derivatives and their therapeutical applications |
| CN102250065A (en) * | 2011-05-20 | 2011-11-23 | 浙江海正药业股份有限公司 | Substituted triazine phenyl urea derivatives and application thereof |
| WO2012159557A1 (en) * | 2011-05-20 | 2012-11-29 | 浙江海正药业股份有限公司 | Substituted triazine phenylurea derivative and use thereof against tumors |
| JP2014525418A (en) * | 2011-09-05 | 2014-09-29 | 浙江海正薬業股▲ふん▼有限公司 | 4-Substituted- (3-substituted-1H-pyrazole-5-amino) -pyrimidine derivatives having protein kinase inhibitory activity and uses thereof |
| WO2013161919A1 (en) * | 2012-04-26 | 2013-10-31 | 小野薬品工業株式会社 | Trk-INHIBITING COMPOUND |
| US9242977B2 (en) | 2012-04-26 | 2016-01-26 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US10765676B2 (en) | 2013-02-19 | 2020-09-08 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US10300060B2 (en) | 2013-02-19 | 2019-05-28 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US9763943B2 (en) | 2013-02-19 | 2017-09-19 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US9993479B2 (en) | 2013-02-19 | 2018-06-12 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US9498453B2 (en) | 2013-02-19 | 2016-11-22 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US9463192B2 (en) | 2013-02-19 | 2016-10-11 | Ono Pharmaceutical Co., Ltd. | Trk-inhibiting compound |
| US10017495B2 (en) | 2013-07-11 | 2018-07-10 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10172864B2 (en) | 2013-07-11 | 2019-01-08 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10376510B2 (en) | 2013-07-11 | 2019-08-13 | Agios Pharmaceuticals, Inc. | 2,4- or 4,6-diaminopyrimidine compounds as IDH2 mutants inhibitors for the treatment of cancer |
| US10028961B2 (en) | 2013-07-11 | 2018-07-24 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US10946023B2 (en) | 2013-07-11 | 2021-03-16 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| US11844758B2 (en) | 2013-07-11 | 2023-12-19 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| US12433895B2 (en) | 2013-07-11 | 2025-10-07 | Servier Pharmaceuticals Llc | Therapeutically active compounds and their methods of use |
| WO2022046861A1 (en) * | 2020-08-26 | 2022-03-03 | Nalo Therapeutics | Modulators of myc family proto-oncogene protein |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2010258825A1 (en) | 2012-01-12 |
| KR20120016676A (en) | 2012-02-24 |
| EP2440056A1 (en) | 2012-04-18 |
| JP2012529527A (en) | 2012-11-22 |
| EP2440056A4 (en) | 2012-12-05 |
| CN102573481A (en) | 2012-07-11 |
| BRPI1011527A2 (en) | 2016-07-26 |
| AU2010258825B2 (en) | 2014-08-21 |
| US20120202818A1 (en) | 2012-08-09 |
| IL216829A0 (en) | 2012-02-29 |
| CA2765050A1 (en) | 2010-12-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2010258825B2 (en) | Ureidophenyl substituted triazine derivatives and their therapeutical applications | |
| AU2010258964B2 (en) | Benzyl substituted triazine derivatives and their therapeutical applications | |
| WO2010144338A1 (en) | Triazine derivatives and their therapeutical applications | |
| WO2010144345A1 (en) | Triazine derivatives and their therapeutical applications | |
| EP2440055B1 (en) | Styryl-triazine derivatives and their therapeutical applications | |
| US9078902B2 (en) | Triazine derivatives and their therapeutical applications | |
| AU2014227487A1 (en) | Triazine derivatives and their therapeutical applications | |
| HK1172509A (en) | Benzyl substituted triazine derivatives and their therapeutical applications | |
| HK1172514A (en) | Styryl-triazine derivatives and their therapeutical applications | |
| HK1172506A (en) | Ureidophenyl substituted triazine derivatives and their therapeutical applications |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080032850.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10786737 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010258825 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2765050 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012515092 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9966/DELNP/2011 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 20127000625 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010786737 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010786737 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2010258825 Country of ref document: AU Date of ref document: 20100609 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13376818 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1011527 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1011527 Country of ref document: BR Kind code of ref document: A2 Effective date: 20111209 |