WO2011029001A1 - Compounds as tyrosine kinase modulators - Google Patents
Compounds as tyrosine kinase modulators Download PDFInfo
- Publication number
- WO2011029001A1 WO2011029001A1 PCT/US2010/047816 US2010047816W WO2011029001A1 WO 2011029001 A1 WO2011029001 A1 WO 2011029001A1 US 2010047816 W US2010047816 W US 2010047816W WO 2011029001 A1 WO2011029001 A1 WO 2011029001A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- independently selected
- hydrogen
- alkoxycarbonyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *1c2cccnc2C=C1 Chemical compound *1c2cccnc2C=C1 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention is directed to novel compounds with multiple aromatic components capable of modulating, regulating and/or inhibiting tyrosine kinase signal transduction.
- the present invention is also directed to methods of prevention and/or treatment of disorders related to unregulated tyrosine kinase signal transduction, including but not limited to, cell growth disorders, metabolic disorders, blood vessel proliferative disorders, inflammatory disorders, neurodegenerative diseases and immune disorders.
- PTKs Protein tyrosine kinases
- RTKs receptor tyrosine kinases
- Binding sites are thereby created for intracellular signal transduction molecules and lead to the formation of complexes with a spectrum of cytoplasmic signaling molecules that facilitate the appropriate cellular response (e.g., cell division, metabolic homeostasis, and responses to the extracellular microenvironment).
- a specific growth factor i.e., a ligand
- receptor dimerization i.e., a ligand
- Binding sites are thereby created for intracellular signal transduction molecules and lead to the formation of complexes with a spectrum of cytoplasmic signaling molecules that facilitate the appropriate cellular response (e.g., cell division, metabolic homeostasis, and responses to the extracellular microenvironment).
- cytoplasmic signaling molecules e.g., cell division, metabolic homeostasis, and responses to the extracellular microenvironment.
- tyrosine phosphorylation sites function as high-affinity binding sites for SH2 (src homology) domains of signaling molecules.
- substrates which have a catalytic domain and (2) substrates which lack a catalytic domain but serve as adapters and associate with catalytically active molecules.
- substrates which have a catalytic domain and (2) substrates which lack a catalytic domain but serve as adapters and associate with catalytically active molecules.
- the specificity of the interactions between receptors or proteins and SH2 domains of their substrates is determined by the amino acid residues immediately surrounding the phosphorylated tyrosine residue. Differences in binding affinities between SH2 domains and the amino acid sequences surrounding the
- phospho tyrosine residues on particular receptors are consistent with the observed differences in their substrate phosphorylation profiles. These observations suggest that the function of each RTK is determined not only by its pattern of expression and ligand availability, but also by the array of downstream signal transduction pathways that are activated by a particular receptor. Thus, phosphorylation provides an important regulatory step which determines the selectivity of signaling pathways recruited by specific growth factor receptors, as well as differentiation factor receptors.
- the RTKs comprise a large family of transmembrane receptors with diverse biological activities.
- the intrinsic function of RTKs is activated upon ligand binding, which results in phophorylation of the receptor and multiple cellular substrates, and subsequently in a variety of cellular responses.
- At present, at least nineteen distinct RTK subfamilies have been identified.
- One RTK subfamily, designated the HER subfamily is believed to be comprised of EGFR, HER2, HER3 and HER4.
- Ligands to the HER subfamily of receptors include epithelial growth factor (EGF), TGF-a, amphiregulin, HB-EGF, betacellulin and heregulin.
- the second subfamily of RTKs designated the insulin subfamily, is comprised of the INS-R, the IGF-IR and the IR-R.
- the third RTK subfamily, the "PDGF" family includes the PDGF a and ⁇ receptors, CSFIR, c-kit and FLK-II.
- Another subfamily of RTKs, identified as the FLK family is believed to be comprised of the kinase insert domain-receptor fetal liver kinase- 1 (KDR/FLK-1), the fetal liver kinase 4 (FLK-4) and the fms-like tyrosine kinase 1 (fit- 1 ) .
- KDR/FLK-1 fetal liver kinase 4
- FLK-4 fetal liver kinase 4
- fit- 1 fms-like tyrosine kinase 1
- RTKs Two other subfamilies of RTKs have been designated as the FGF receptor family (FGFR1, FGFR2, FGFR3 and FGFR4) and the Met subfamily (c-met and Ron). Because of the similarities between the PDGF and FLK subfamilies, the two subfamilies are often considered together.
- the known RTK subfamilies are identified in Plowman et al, 1994, DN&P 7(6): 334-339, which is incorporated herein by reference.
- the non-receptor tyrosine kinases represent a collection of cellular enzymes which lack extracellular and transmembrane sequences. At present, over twenty- four individual non- receptor tyrosine kinases, comprising eleven subfamilies (Src, Frk, Btk, Csk, Abl, Zap70, Fes/Fps, Fak, Jak, Ack and LIMK) have been identified. At present, the Src subfamily of non-receptor tyrosine kinases is comprised of the largest number of PTKs, and include Src, Yes, Fyn, Lyn, Lck, Blk, Hck, Fgr and Yrk. The Src subfamily of enzymes has been linked to oncogenesis. A more detailed discussion of non-receptor tyrosine kinases is provided in Bolen, 1993, Oncogen 8: 2025-2031, which is incorporated herein by reference.
- PTKs protein tyrosine kinases
- RTKs protein tyrosine kinases
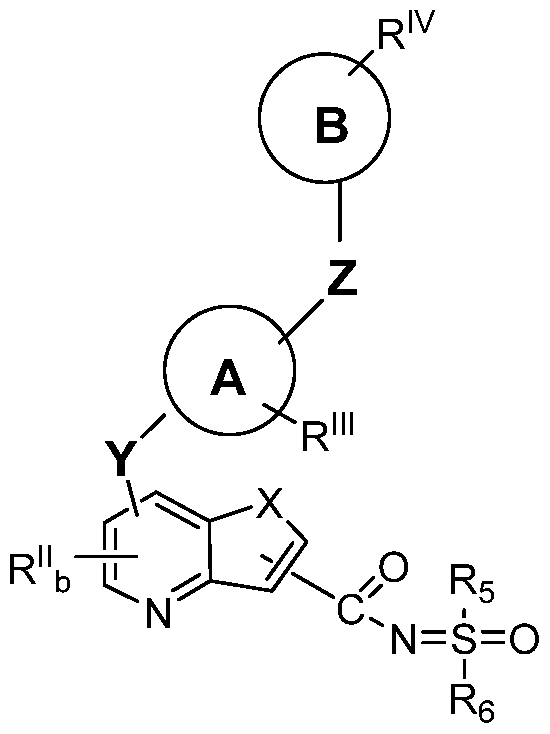
- the present invention is directed to compounds represented by Formula I capable of modulating, regulating and/or inhibiting tyrosine kinase signal transduction, and uses of the compounds and compositions incorporating the compounds for disease treatment and prevention.
- X is selected from the group consisting of NR , O, S(0) n ;
- n is 0 or an integer of from 1 to 2;
- R 1 is independently selected from the group consisting of hydrogen, alkenyl, alkoxyalkyl, CF 3 , alkyl, alkylcarbonyl, alkoxycarbonyl, aryl, heterocycloalkyl, hydroxyalkyl, and alkyl(N R 2 R 3 ), wherein R 2 and R 3 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 and R 3 and may be taken together to form a 5-7 membered heterocyclic ring with N;
- R 1 is selected from the group consisting of hydrogen, halogen, Ci to C 8 alkyl, S(0) f R 4 , (CR 5 R 6 )dC(0)OR 4 , S(0) f (CR 5 R 6 )dC(0)OR 4 , (CR 5 R 6 ) d Ar, NR 4 (CR 5 R 6 ) d Ar,
- hydroxycarbonyl, hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl, sulfonate and CR 5 R 6 may represent a carbocyclic or heterocyclic ring of from 5 to 6 carbons or alternatively, (CR 5 R 6 ) d and (CR 5 R 6 ) e may form a 3-7 membered carbocyclic or heterocyclic ring, wherein the ring may be optionally substituted with up to three of hydroxyl, halo, Ci-Cg alkyl, Ci-Cg hydroxyalkyl, Ci-Cg alkoxyalkyl,
- alkoxycarbonylalkyl alkoxycarbonyl, alkoxycarbonyl, hydroxycarbonyl, hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl and sulfonate; a is 0 or an integer of from 1 to 3;
- d is 0 or an integer of from 1 to 5;
- e is an integer of from 1 to 4.
- R is independently selected from the group consisting of hydrogen, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkyl, aryloxy, aryloxyalkyl, halo, haloalkoxy, haloalkyl, hydroxy, hydroxyalkoxy, hydroxyalkyl, (NR 2 R 3 )alkoxy, (NR 2 R 3 )alkenyl, (NR 2 R 3 )alkyl,
- R 2 andR 3 independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 andR 3 and may be taken together to form a 5-7 membered heterocyclic ring with N; b is 0 or an integer of from 1 to 2; Y is selected from the group consisting of
- g is 0 or an integer of from 1 to 3;
- h is 0 or an integer of from 1 to 3;
- R 1 is independently selected from the group consisting of hydrogen, alkenyl, alkoxyalkyl, CF 3 , alkyl, alkylcarbonyl, alkoxycarbonyl, aryl, heterocycloalkyl, hydroxyalkyl, and alkyl(N R 2 R 3 ), wherein R 2 and R 3 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 and R 3 and may be taken together to form a 5-7 membered heterocyclic ring with N;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkylcarbonyl,
- alkoxycarbonyl alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and
- Ring A is selected from the group consisting of:
- An 8 to 10 membered bicyclic heteroaryl group which has 1-6 heteroatoms independently selected from the group consisting of O, N and S;
- R nI represents optionally 1-3 substituents independently selected from the group consisting of C1-C5 linear or branched alkyl, C 1-C5 linear or branched haloalkyl, C1-C5 alkoxy, hydroxy, amino, C1-C5 alkylamino, C1-C6 dialkylamino, halogen, cyano, and nitro;
- Z is selected from the group consisting of
- i 0 or 1 ;
- j is 0 or 1 ;
- R 7 and R 8 are independently selected from the group consisting of hydrogen and alkyl.
- Ring B is selected from the group consisting of:
- R IV represents optionally 1-3 substituents, independently selected from the group consisting of alkoxy, alkoxyalkyl, alkoxycarbonyl, alkyl, aryloxy, arylalkyl, carboxy, cyano, halo, haloalkoxy, haloalkyl, hydroxy, hydroxyalkyl, nitro, and— NR 9 R 10 ; wherein R 9 and R 10 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl.
- a compound according to Formula I including any tautomer, stereoisomer, diastereoisomeric form, polymorphic form, crystal form, a solvate, a hydrate, a metabolite, a pharmaceutically acceptable salt or prodrug, mixture of different stereoisomers, mixture of different crystal forms.
- Formula II including any tautomer, stereoisomer, diastereoisomeric form, crystal form, polymorphic form, mixture of stereoisomers, mixture of polymorphic forms, mixture of crystal forms, a solvate, a hydrate, a metabolite, a pharmaceutically acceptable salt or a prodrug.
- R 1 is selected from the group consisting of hydrogen, halogen, Ci to Cg alkyl, (CR 5 R 6 )dC(0)OR 4 , (CR 5 R 6 ) d Ar, NR 4 (CR 5 R 6 ) d Ar, (CR 5 R 6 ) d C(0)N(R 4 ) 2 , NR 4 (CR 5 R 6 ) d C(0)N(R 4 ) 2 , 0(CR 5 R 6 ) d C(0)N(R 4 ) 2 , (CR 5 R 6 ) d OR 4 , OC(0)(CR 5 R 6 ) d N(R 4 ) 2 ,
- each R 4 is independently selected from the group consisting of hydrogen, hydroxyl, Ci-Cg alkyl, aryl, Ci-Cg hydroxyalkyl, Ci- Cg alkoxyalkyl, (CR 5 R 6 )d and N(R 4 ) 2 may form a 3-7 membered heterocyclic ring, comprising of aziridine, azetidine, pyrrolidine, 5-fluoropyrrolidine, piperidine, 6- fluoropiperidine, N-methylpiperazine, morpholine, 2,6-dimethylmorpholine, thiomorpholine, and wherein said heterocyclic ring may be optionally substituted with up to three of R 5 ; wherein R 5 and R 6 are independently selected from the group consisting of hydrogen, halo, hydroxyl, Ci-Cg alkyl, Ci-Cg hydroxyalkyl, Ci-Cg alkoxyal
- hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl, sulfonate and CR 5 R 6 may represent a carbocyclic or heterocyclic ring of from 5 to 6 carbons or alternatively, (CR 5 R 6 )d and (CR 5 R 6 ) e may form a 3-7 membered carbocyclic or heterocyclic ring, wherein the ring may be optionally substituted with up to three of hydroxyl, halo, Ci-Cg alkyl, Ci-Cg hydroxyalkyl, Ci-Cg alkoxyalkyl, alkoxycarbonylalkyl, alkoxycarbonyl, hydroxycarbonyl, hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl and sulfonate.
- the diseases or conditions are selected from the group consisting of colorectal cancer, lung cancer, hematological cancer, renal cancer, liver cancer, breast cancer, diabetic retinopathy, macular degeneration, age-related macular degeneration, retinopathy of prematurity, ocular angiogenesis, retinal edema, retinal ischemia, diabetic macular edema, cystoid macular edema, retinal vein occlusion, branch vein occlusion, preretinal neovascularization, laser-induced choroidal neovascularization, neovascularization associated with keratoplasty, glaucoma and ocular tumors, arthritis, restenosis, hepatic cirrhosis, atherosclerosis, psoriasis, diabetes mellitus, wound healing, inflammation, neurodegenerative diseases and immune disorders.
- the diseases or conditions are selected from the group consisting of colorectal cancer, lung cancer, hematological cancer, renal cancer, liver cancer, breast
- a pharmaceutical composition comprising a therapeutic effective amount of a compound according to paragraphs 1 - 10 together with a pharmaceutically acceptable carrier which is suitable for systematic, parenteral, local or topical delivery.
- compositions of paragraph 15 which are in the form selected from the group consisting of tablets, capsules, intravenous injections, intramuscular injections, local injections, topical creams, gels and ointments, eye drops, ophthalmic solutions, ophthalmic suspensions, ophthalmic emulsions, intravitreal injections, subtenon injections, ophthalmic biodrodible implant, and non-bioeordible ophthalmic inserts or depots.
- the present invention is directed to a series of compounds with multiple aromatic components useful as protein tyrosine kinase inhibitors.
- the compounds of the present invention are useful for treating diseases related to unregulated tyrosine kinase signal transduction, for example, cancer, blood vessel proliferative disorders, fibrotic disorders, and neurodegenerative diseases.
- compounds of the present invention are useful for the treatment of colorectal cancer, lung cancer, hematological cancer, renal cancer, liver cancer, breast cancer, diabetic retinopathy, macular degeneration, age-related macular degeneration, retinopathy of prematurity, ocular angiogenesis, retinal edema, retinal ischemia, diabetic macular edema, cystoid macular edema, retinal vein occlusion, branch vein occlusion, preretinal neovascularization, laser-induced choroidal neovascularization, neovascularization associated with keratoplasty, glaucoma and ocular tumors, arthritis, restenosis, hepatic cirrhosis, atherosclerosis, psoriasis, diabetes mellitus, wound healing, inflammation, neurodegenerative diseases and immune disorders.
- the present invention is directed to a compound of Formula I:
- X is selected from the group consisting of NR 1 , O, S(0) n ; n is 0 or an integer of from 1 to 2;
- R 1 is independently selected from the group consisting of hydrogen, alkenyl, alkoxyalkyl, CF 3 , alkyl, alkylcarbonyl, alkoxycarbonyl, aryl, heterocycloalkyl, hydroxyalkyl, and alkyl(N R 2 R 3 ), wherein R 2 and R 3 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 and R 3 and may be taken together to form a 5-7 membered heterocyclic ring with N; R 1 is selected from the group consisting of hydrogen, halogen, Ci to Cg alkyl, S(0) f R 4 ,
- hydroxycarbonyl, hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl, sulfonate and CR 5 R 6 may represent a carbocyclic or heterocyclic ring of from 5 to 6 carbons or alternatively, (CR 5 R 6 ) d and (CR 5 R 6 ) e may form a 3-7 membered carbocyclic or heterocyclic ring, wherein the ring may be optionally substituted with up to three of hydroxyl, halo, Ci-Cg alkyl, Ci-Cg hydroxyalkyl, Ci-Cg alkoxyalkyl, alkoxycarbonylalkyl, alkoxycarbonyl, hydroxycarbonyl, hydroxycarbonylalkyl, amide, alkylamide, amidoalkyl and sulfonate; a is 0 or an integer of from 1 to 3;
- d is 0 or an integer of from 1 to 5;
- e is an integer of from 1 to 4.
- f is 0 or an integer of from 1 to 2;
- R n is independently selected from the group consisting of hydrogen, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkyl, aryloxy, aryloxyalkyl, halo, haloalkoxy, haloalkyl, hydroxy, hydroxyalkoxy, hydroxyalkyl, (NR 2 R 3 )alkoxy, (NR 2 R 3 )alkenyl, (NR 2 R 3 )alkyl,
- R 2 andR 3 independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 andR 3 and may be taken together to form a 5-7 membered heterocyclic ring with N; b is 0 or an integer of from 1 to 2;
- Y is selected from the group consisting of
- g is 0 or an integer of from 1 to 3;
- h is 0 or an integer of from 1 to 3;
- R 1 is independently selected from the group consisting of hydrogen, alkenyl, alkoxyalkyl, CF 3 , alkyl, alkylcarbonyl, alkoxycarbonyl, aryl, heterocycloalkyl, hydroxyalkyl, and alkyl(N R 2 R 3 ), wherein R 2 and R 3 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and heterocyclylsulfonyl; alternatively R 2 and R 3 and may be taken together to form a 5-7 membered heterocyclic ring with N;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, arylsulfonyl, haloalkylsulfonyl, and
- Ring A is selected from the group consisting of:
- An 8 to 10 membered bicyclic heteroaryl group which has 1-6 heteroatoms independently selected from the group consisting of O, N and S;
- R in represents optionally 1-3 substituents independently selected from the group consisting of C1-C5 linear or branched alkyl, C1-C5 linear or branched haloalkyl, C1-C5 alkoxy, hydroxy, amino, C1-C5 alkylamino, C1-C6 dialkylamino, halogen, cyano, and nitro;
- Z is selected from the group consisting of
- i 0 or 1 ;
- j is 0 or 1 ;
- R 7 and R 8 are independently selected from the group consisting of hydrogen and alkyl; Rin B is selected from the group consisting
- R IV represents optionally 1-3 substituents, independently selected from the group consisting of alkoxy, alkoxyalkyl, alkoxycarbonyl, alkyl, aryloxy, arylalkyl, carboxy, cyano, halo, haloalkoxy, haloalkyl, hydroxy, hydroxyalkyl, nitro, and— NR 9 R 10 ; wherein R 9 and R 10 are independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, aryl, arylalkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl.
- reference to a compound should be construed broadly to include compounds, pharmaceutically acceptable salts, prodrugs, tautomers, stereoisomers, diastereoisomers, alternate solid forms, crystal forms, polymorphic forms, hydrates, solvates, metabolites, mixtures of stereoisomers, mixtures of crystal forms, non-covalent complexes, and combinations thereof, of a chemical entity of a depicted structure or a chemical name.
- a pharmaceutically acceptable salt is any salt of the parent compound that is suitable for administration to an animal or human.
- a pharmaceutically acceptable salt also refers to any salt which may form in vivo as a result of administration of an acid, another salt, or a prodrug which is converted into an acid or salt.
- a salt comprises one or more ionic forms of the compound, such as a conjugate acid or base, associated with one or more corresponding counter-ions. Salts can form from or incorporate one or more deprotonated acidic groups (e.g. carboxylic acids), one or more protonated basic groups (e.g. amines), or both (e.g. zwitterions).
- a “prodrug” is a compound, which when administered to the body of a subject (such as a mammal), breaks down in the subject's metabolic pathway to provide an active compound of Formula I. More specifically, a prodrug is an active or inactive "masked” compound that is modified chemically through in vivo physiological action, such as hydrolysis, metabolism and the like, into a compound of this invention following
- a prodrug is a masked carboxylic acid group.
- a masked carboxylate anion include a variety of esters, such as alkyl (for example, methyl, ethyl), cycloalkyl (for example, cyclohexyl), aralkyl (for example, benzyl, p-methoxybenzyl), and alkylcarbonyloxyalkyl (for example, pivaloyloxymethyl).
- esters such as alkyl (for example, methyl, ethyl), cycloalkyl (for example, cyclohexyl), aralkyl (for example, benzyl, p-methoxybenzyl), and alkylcarbonyloxyalkyl (for example, pivaloyloxymethyl).
- Amines have been masked as arylcarbonyloxymethyl substituted derivatives which are cleaved by esterases in vivo releasing the free drug and formaldehy
- Tautomers are isomers that are in rapid equilibrium with one another.
- tautomers may be related by transfer of a proton, hydrogen atom, or hydride ion.
- Alternate solid forms are different solid forms than those that may result from practicing the procedures described herein.
- alternate solid forms may be amorphous forms, crystal forms, polymorphs, and the mixtures thereof.
- Non-covalent complexes are complexes that may form between the compound and one or more additional chemical species that do not involve a covalent bonding interaction between the compound and the additional chemical species. They may or may not have a specific ratio between the compound and the additional chemical species. Examples might include solvates, hydrates, charge transfer complexes, and the like.
- the present invention is also directed to the use of the compounds as protein tyrosine kinase modulators and inhibitors. These compounds can be used to treat diseases related to unregulated tyrosine kinase signal transduction, for example, various cancers, blood vessel proliferative disorders, fibrotic disorders, and neurodegenerative diseases.
- compounds of the present invention are useful for the treatment and/or prevention of colorectal cancer, lung cancer, hematological cancer, renal cancer, liver cancer, breast cancer, diabetic retinopathy, macular degeneration, age-related macular degeneration, retinopathy of prematurity, ocular angiogenesis, retinal edema, retinal ischemia, diabetic macular edema, cystoid macular edema, retinal vein occlusion, branch vein occlusion, preretinal
- neovascularization laser-induced choroidal neovascularization, neovascularization associated with keratoplasty, glaucoma and ocular tumors, arthritis, restenosis, hepatic cirrhosis, atherosclerosis, psoriasis, diabetes mellitus, wound healing, inflammation, neurodegenerative diseases and immune disorders in the human being.
- the present invention is also directed to the preparation of a medicament for the treatment and prevention of diseases and conditions related with abnormal activities of tyrosine kinase receptors.
- the medicament contains a pharmaceutical acceptable composition, which comprises the therapeutic effective amount of the compounds of present invention, together with a pharmaceutical acceptable carrier.
- treat refers to the diagnosis, cure, mitigation, treatment, or prevention of disease or other undesirable conditions.
- compositions contain therapeutic effective amount of the compounds of the present invention. These compositions can be used as a medicament and administered to a mammal, such as a person, in need thereof.
- suitable dosage forms and medicaments are well known in the art, and can be readily adapted for delivery of the compounds of the present invention, such as, but not limited to, systematic, parenteral, local and topical delivery.
- the dosage forms can be tablets, capsules, intravenous injections, intramuscular injections, local injections, topical creams, gels and ointments, eye drops, ophthalmic solutions, ophthalmic suspensions, ophthalmic emulsions, intravitreal injections, subtenon injections, ophthalmic biodrodible implant, and non-bioeordible ophthalmic inserts or depots, nasal sprays and ointment, various rectal or vaginal preparations.
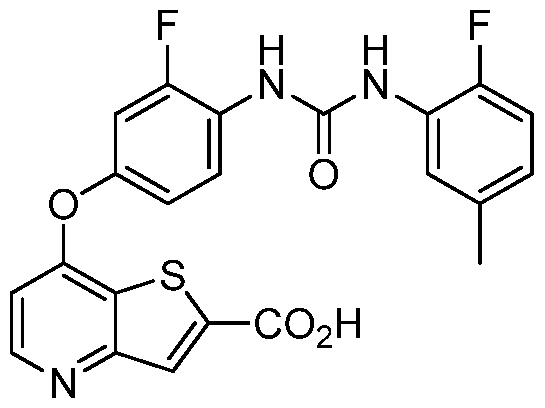
- Methyl 7-chlorothieno[3,2-b]pyridine-2-carboxylate (5 g, 0.022 mol) and the 4-amino-3- fluorophenol (3.3 g, 0.026 mol) were added to a round bottom flask containing cesium carbonate (14.8 g, 0.045 mol), ethyl-2-cyclohexanone carboxylate (0.73 g, 0.004 mol), and copper (I) chloride (0.22 g, 0.002 mol).
- the mixture was diluted with DMSO (250 mL) and stirred at 70 C under an atmosphere of nitrogen for 2 hours.
- Methyl 7-(4-amino-3-fluorophenoxy)thieno[3,2-b]pyridine-2-carboxylate (1.62 g, 5.1 mmol) was taken up in 55 mL of ethyl acetate followed by the dropwise addition of 2-fluoro-5- methylphenyl isocyanate (0.85 g, 5.6 mmol) in 5 mL ethyl acetate. The solution afforded a lavender solid after stirring at room temperature for overnight.
- Methyl 7-(3-fluoro-4-(3-(2-fluoro-5-methylphenyl)ureido)phenoxy)thieno[3,2-b]pyridine-2- carboxylate (1.84 g, 3.92 mmol) was taken up in 100 mL THF followed by the dropwise addition of IN sodium hydroxide (4.8 mL, 4.8 mmol). The solution was stirred at room temperature for 3 hours, at which time an additional 2.4 mL of IN sodium hydroxide was added. The solution was stirred at room temperature for overnight and the resulting mixture was diluted with 75 mL of water and acidified using IN HC1.
- the insoluble material was separated by filtration and the filter cake was suspended in ethyl acetate and stirred for several minutes before filtering.
- the filter cake was washed several times with ethyl acetate and dried under high vacuum to give 7-[3-fluoro-4-( ⁇ [(2-fluoro-5- methylphenyl)amino]carbonyl ⁇ amino)phenoxy]thieno[3,2-b]pyridine-2-carboxylic acid as an off white solid.
- Biological data for the compounds of the present invention was generated by the use of one or more of the following assays.
- DMSO concentration 0.1%.
- test agents for screening, cells were pre-incubated with test agents for 30 minutes, at a single concentration (10 .mu.M) or at concentrations ranging from 0.01 to 10.0 .mu.M followed by VEGF stimulation (5 ng/mL). Changes in fluorescence at 516 nm were measured simultaneously in all 96 wells using a cooled CCD camera. Data were generated by determining max-min fluorescence levels for unstimulated, stimulated, and drug treated samples. IC.sub.50 values for test compounds were calculated from % inhibition of VEGF stimulated responses in the absence of inhibitor. VEGFR2 Kinase Assay
- the cytoplasmic domain of the human VEGF receptor (VEGFR-2) was expressed as a Histidine-tagged fusion protein following infection of insect cells using an His engineered baculovirus. His-VEGFR-2 was purified to homogeneity, as determined by SDS-PAGE, using nickel resin chromatography. Kinase assays were performed in 96 well microtiter plates that were coated overnight with 30 .mu.g of poly-Glu-Tyr (4: 1) in 10 mM Phosphate
- mice Male Hartley guinea pigs (300-600 g) were anesthetized with isof uorane, sheared, and given a single dose of drug or the respective vehicle. The guinea pigs were dosed orally unless indicated otherwise in Table 3. Ten minutes prior to the end of drug treatment, guinea pigs were anesthetized with isofluorane, and 0.5%> Evans blue dye (EBD) in PBS (13-15 mg/kg dose of EBD) was injected intravenously. After 5 minutes, triplicate intradermal injections of 100 ng rhVEGF.sub.165 in 100 .mu.l PBS and of 100 .mu.l PBS alone were administered on the flank. After 20 minutes, each animal was euthanized with Pentosol, and the skin containing the intradermal injection sites was removed for image analysis.
- EBD Evans blue dye
- the percent inhibition data was plotted as a function of oral dose, using the " best-fi analysis within MicroSoft Excel software.
- the ID. sub.50 value was verified visually by using the plotted data (horizontal line from 50% y value, at intersection with best- fit line drop vertical line to x axis (dose).
- FITC-dextran MW 2.times.10. sup.6
- images were obtained from the flat mounts of the RPE-choroid-sclera from each eye, and the area occupied by hyperfluorescent neovessels within each laser lesion was measured using ImagePro 4 software.
- the percent inhibition data was plotted as a function of oral dose, using the " best-fi analysis within MicroSoft Excel software. The ID. sub.50 value was verified visually by using the plotted data
- test compounds were reconstituted in 100% DMSO and added to the cells to give a final DMSO concentration of 0.1%).
- test agents for screening, cells were pre-incubated with test agents for 30 minutes, at a single concentration (10 ⁇ ) or at concentrations ranging from 0.00 InM to 10 ⁇ followed by
- IC 50 values for test compounds were calculated from % inhibition of PDGF stimulated responses in the absence of inhibitor.
- TaMe II Biological Activities of Compounds of the Present Invention
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Ophthalmology & Optometry (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Dermatology (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Hospice & Palliative Care (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
- Oncology (AREA)
- Emergency Medicine (AREA)
- Psychiatry (AREA)
- Obesity (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Gastroenterology & Hepatology (AREA)
- Pulmonology (AREA)
Abstract
Description




























Claims








Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2012109233/04A RU2012109233A (en) | 2009-09-03 | 2010-09-03 | COMPOUNDS AS TYROSINKINASE MODULATORS |
| SG2012015038A SG178963A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| BR112012004718A BR112012004718A2 (en) | 2009-09-03 | 2010-09-03 | compounds as tyrosine kinase modulators |
| AU2010289359A AU2010289359A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| MX2012002591A MX2012002591A (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators. |
| CN2010800389522A CN102498114A (en) | 2009-09-03 | 2010-09-03 | Compounds as Modulators of Tyrosine Kinases |
| JP2012528089A JP2013503903A (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| NZ598455A NZ598455A (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| CA2772625A CA2772625A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| EP10752975.2A EP2473513B1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| IL218332A IL218332A0 (en) | 2009-09-03 | 2012-02-27 | Compounds as tyrosine kinase modulators |
| ZA2012/01592A ZA201201592B (en) | 2009-09-03 | 2012-03-02 | Compounds as tyrosine kinase modulators |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US23960309P | 2009-09-03 | 2009-09-03 | |
| US61/239,603 | 2009-09-03 | ||
| US30661610P | 2010-02-22 | 2010-02-22 | |
| US61/306,616 | 2010-02-22 | ||
| US35669910P | 2010-06-21 | 2010-06-21 | |
| US61/356,699 | 2010-06-21 | ||
| US36053110P | 2010-07-01 | 2010-07-01 | |
| US61/360,531 | 2010-07-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011029001A1 true WO2011029001A1 (en) | 2011-03-10 |
Family
ID=43086186
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/047816 Ceased WO2011029001A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
| PCT/US2010/047800 Ceased WO2011028995A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/047800 Ceased WO2011028995A1 (en) | 2009-09-03 | 2010-09-03 | Compounds as tyrosine kinase modulators |
Country Status (19)
| Country | Link |
|---|---|
| US (5) | US8809534B2 (en) |
| EP (2) | EP2473501B1 (en) |
| JP (3) | JP5868855B2 (en) |
| KR (2) | KR20120082890A (en) |
| CN (2) | CN102498114A (en) |
| AU (2) | AU2010289353B2 (en) |
| BR (2) | BR112012004843A2 (en) |
| CA (2) | CA2772625A1 (en) |
| CL (2) | CL2012000587A1 (en) |
| ES (1) | ES2730086T3 (en) |
| IL (2) | IL218332A0 (en) |
| IN (1) | IN2012DN02493A (en) |
| MX (2) | MX2012002591A (en) |
| NZ (2) | NZ598455A (en) |
| PH (1) | PH12014500281A1 (en) |
| RU (2) | RU2012112151A (en) |
| SG (2) | SG178965A1 (en) |
| WO (2) | WO2011029001A1 (en) |
| ZA (2) | ZA201201592B (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8232294B2 (en) | 2009-03-21 | 2012-07-31 | Ning Xi | Amino ester derivatives, sailts thereof and methods of use |
| US8293897B2 (en) | 2008-10-14 | 2012-10-23 | Ning Xi | Compounds comprising a spiro-ring and methods of use |
| US10829496B2 (en) | 2017-05-11 | 2020-11-10 | Bristol-Myers Squibb Company | Thienopyridines and benzothiophenes useful as IRAK4 inhibitors |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7431710B2 (en) | 2002-04-08 | 2008-10-07 | Glaukos Corporation | Ocular implants with anchors and methods thereof |
| BRPI0914772A2 (en) | 2008-06-25 | 2015-10-20 | Envivo Pharmaceuticals Inc | 1,2-disubstituted heterocyclic compounds |
| CN105125547A (en) | 2009-05-07 | 2015-12-09 | 费瑞姆医药有限公司 | Forum pharmaceuticals inc |
| US12478503B2 (en) | 2009-05-18 | 2025-11-25 | Glaukos Corporation | Implants with controlled drug delivery features and methods of using same |
| US10206813B2 (en) | 2009-05-18 | 2019-02-19 | Dose Medical Corporation | Implants with controlled drug delivery features and methods of using same |
| EP2432420A4 (en) | 2009-05-18 | 2018-01-10 | Dose Medical Corporation | Drug eluting ocular implant |
| US9340555B2 (en) | 2009-09-03 | 2016-05-17 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| US8906944B2 (en) * | 2009-09-03 | 2014-12-09 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| AU2010289353B2 (en) * | 2009-09-03 | 2016-12-08 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| EP2654715B1 (en) | 2010-11-24 | 2017-01-25 | Dose Medical Corporation | Drug eluting ocular implant |
| US10245178B1 (en) | 2011-06-07 | 2019-04-02 | Glaukos Corporation | Anterior chamber drug-eluting ocular implant |
| US9045493B2 (en) * | 2012-02-09 | 2015-06-02 | Merck Patent Gmbh | Furo[3,2-b]- and thieno[3,2-b]pyridin derivatives |
| WO2015069287A1 (en) * | 2013-11-08 | 2015-05-14 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| CA2932353A1 (en) | 2013-12-13 | 2015-06-18 | Steven P. Treon | Methods to treat lymphoplasmacytic lymphoma |
| EP3079683A4 (en) * | 2013-12-13 | 2017-12-20 | Dana-Farber Cancer Institute, Inc. | Methods to treat lymphoplasmacytic lymphoma |
| CN106866623A (en) * | 2014-04-08 | 2017-06-20 | 北大方正集团有限公司 | Polysubstituted pyridine compounds, preparation method, purposes and pharmaceutical composition |
| AU2015266850B2 (en) | 2014-05-29 | 2019-12-05 | Glaukos Corporation | Implants with controlled drug delivery features and methods of using same |
| KR102431436B1 (en) | 2014-08-29 | 2022-08-10 | 테스 파마 에스.알.엘. | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| CN104326985A (en) * | 2014-09-24 | 2015-02-04 | 安润医药科技(苏州)有限公司 | Preparation method of linifanib |
| EP3715346B1 (en) | 2014-10-22 | 2024-01-03 | Dana-Farber Cancer Institute, Inc. | Thiazolyl-containing compounds for treating proliferative diseases |
| WO2017040853A1 (en) | 2015-09-02 | 2017-03-09 | Glaukos Corporation | Drug delivery implants with bi-directional delivery capacity |
| WO2017053885A1 (en) | 2015-09-25 | 2017-03-30 | Glaukos Corporation | Punctal implants with controlled drug delivery features and methods of using same |
| CN107531679B (en) * | 2016-03-18 | 2021-07-02 | 江苏恒瑞医药股份有限公司 | Aromatic amide derivatives, their preparation method and their application in medicine |
| AU2017252294B2 (en) | 2016-04-20 | 2021-12-02 | Glaukos Corporation | Bioresorbable ocular drug delivery device |
| CN107663202B (en) * | 2016-07-29 | 2020-09-04 | 西华大学 | 3-(ureido-methyl)-4-aryl-pyridine derivative and preparation method thereof and application as anti-cancer drug |
| EP3600333A4 (en) | 2017-03-21 | 2020-11-25 | The Scripps Research Institute | CU- AND NI-CATALYZED DECARBOXYLATIVE BORYLATION REACTIONS |
| WO2018211442A1 (en) * | 2017-05-18 | 2018-11-22 | Pi Industries Ltd. | Formimidamidine compounds useful against phytopathogenic microorganisms |
| MX2021003904A (en) | 2018-10-05 | 2021-10-26 | Annapurna Bio Inc | COMPOUNDS AND COMPOSITIONS FOR THE TREATMENT OF CONDITIONS ASSOCIATED WITH APJ RECEPTOR ACTIVITY. |
| CA3121202A1 (en) | 2018-11-30 | 2020-06-04 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0039051A2 (en) | 1980-04-24 | 1981-11-04 | Merck & Co. Inc. | Mannich-base hydroxamic acid prodrugs for the improved bioavailability of non-steroidal anti-inflammatory agents, a process for preparing and a pharmaceutical composition containing them |
| US4966849A (en) | 1985-09-20 | 1990-10-30 | President And Fellows Of Harvard College | CDNA and genes for human angiogenin (angiogenesis factor) and methods of expression |
| WO1991015495A1 (en) | 1990-04-02 | 1991-10-17 | Pfizer Inc. | Benzylphosphonic acid tyrosine kinase inhibitors |
| WO1992020642A1 (en) | 1991-05-10 | 1992-11-26 | Rhone-Poulenc Rorer International (Holdings) Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| WO1992021660A1 (en) | 1991-05-29 | 1992-12-10 | Pfizer, Inc. | Tricyclic polyhydroxylic tyrosine kinase inhibitors |
| US5217999A (en) | 1987-12-24 | 1993-06-08 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Styryl compounds which inhibit EGF receptor protein tyrosine kinase |
| WO1994003427A1 (en) | 1992-08-06 | 1994-02-17 | Warner-Lambert Company | 2-thioindoles (selenoindoles) and related disulfides (selenides) which inhibit protein tyrosine kinases and which have antitumor properties |
| US5302606A (en) | 1990-04-16 | 1994-04-12 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Styryl-substituted pyridyl compounds which inhibit EGF receptor tyrosine kinase |
| WO1994014808A1 (en) | 1992-12-23 | 1994-07-07 | Farmitalia Carlo Erba Srl | Vinylene-azaindole derivatives and process for their preparation |
| US5330992A (en) | 1992-10-23 | 1994-07-19 | Sterling Winthrop Inc. | 1-cyclopropyl-4-pyridyl-quinolinones |
| US5792783A (en) | 1995-06-07 | 1998-08-11 | Sugen, Inc. | 3-heteroaryl-2-indolinone compounds for the treatment of disease |
| US6541504B1 (en) | 2002-04-03 | 2003-04-01 | Allergan Sales, Llc | (3Z)-3-(2,3-dihydro-1H-inden-1-ylidene)-1,3-dihydro-2H-indol-2-ones as kinase inhibitors |
| WO2003106462A1 (en) * | 2002-06-14 | 2003-12-24 | Pfizer Inc. | Benzofused heteroaryl amide derivatives of thienopyridines useful as therapeutic agents, pharmaceutical compositions including the same, and methods for their use |
| US6747025B1 (en) | 2002-11-27 | 2004-06-08 | Allergan, Inc. | Kinase inhibitors for the treatment of disease |
| US6765012B2 (en) | 2001-09-27 | 2004-07-20 | Allergan, Inc. | 3-(Arylamino)methylene-1,3-dihydro-2H-indol-2-ones as kinase inhibitors |
| US20070197537A1 (en) * | 2006-01-30 | 2007-08-23 | Blake James F | Heterobicyclic thiophene compounds and methods of use |
| WO2009026717A1 (en) * | 2007-08-29 | 2009-03-05 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US20090118276A1 (en) * | 2007-11-02 | 2009-05-07 | Wyeth | Thienopyrimidines, thienopyridines, and pyrrolopyrimidines as b-raf inhibitors |
| WO2009109035A1 (en) * | 2008-03-05 | 2009-09-11 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US20100081675A1 (en) * | 2008-09-26 | 2010-04-01 | National Health Research Institutes | Fused multicyclic compounds as protein kinase inhibitors |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU9802198A (en) * | 1997-10-21 | 1999-05-10 | Pharmacia & Upjohn Company | Antiinflammatory thiadiazolyl ureas which act as lfa-1 and mac-1 inhibitors |
| AU9598601A (en) * | 2000-10-20 | 2002-04-29 | Eisai Co Ltd | Nitrogenous aromatic ring compounds |
| WO2002090352A2 (en) * | 2001-05-08 | 2002-11-14 | Schering Aktiengesellschaft | Selective anthranilamide pyridine amides as inhibitors of vegfr-2 and vegfr-3 |
| TWI319387B (en) * | 2002-04-05 | 2010-01-11 | Astrazeneca Ab | Benzamide derivatives |
| US20050058689A1 (en) | 2003-07-03 | 2005-03-17 | Reactive Surfaces, Ltd. | Antifungal paints and coatings |
| GB0229022D0 (en) * | 2002-12-12 | 2003-01-15 | Novartis Ag | Organic Compounds |
| MXPA05009102A (en) * | 2003-02-28 | 2006-05-31 | Bayer Pharmaceuticals Corp | Substituted pyridine derivatives useful in the treatment of cancer and other disorders. |
| EP1636585B2 (en) * | 2003-05-20 | 2012-06-13 | Bayer HealthCare LLC | Diaryl ureas with kinase inhibiting activity |
| ES2314444T3 (en) * | 2003-08-29 | 2009-03-16 | Pfizer Inc. | TIENOPIRIDINE-PHENYLACETAMIN AND ITS USEFUL DERIVATIVES AS NEW ANTIANGIOGEN AGENTS. |
| MXPA06002256A (en) * | 2003-08-29 | 2006-05-17 | Pfizer | Naphthalene carboxamides and their derivatives useful as new anti-angiogenic agents. |
| WO2005110410A2 (en) * | 2004-05-14 | 2005-11-24 | Abbott Laboratories | Kinase inhibitors as therapeutic agents |
| CN101128454A (en) * | 2004-12-22 | 2008-02-20 | 阿斯利康(瑞典)有限公司 | Pyridine carboxamide derivatives used as anticancer drugs |
| EP1846388A4 (en) * | 2005-01-25 | 2011-12-07 | Synta Pharmaceuticals Corp | Thiophene compounds for inflammation and immune-related uses |
| PL1848435T3 (en) | 2005-01-25 | 2016-08-31 | Synta Pharmaceuticals Corp | Compounds against inflammations and immune-related uses |
| US20060223849A1 (en) | 2005-03-14 | 2006-10-05 | Mjalli Adnan M | Benzazole derivatives, compositions, and methods of use as beta-secretase inhibitors |
| JO2787B1 (en) * | 2005-04-27 | 2014-03-15 | امجين إنك, | Substituted Amid derivatives & methods of use |
| AU2006313456B2 (en) | 2005-05-20 | 2011-06-23 | Methylgene Inc. | Inhibitors of VEGF receptor and HGF receptor signaling |
| CA2610509A1 (en) * | 2005-06-03 | 2006-12-14 | Bayer Healthcare Ag | 1-methyl-1h-pyrazole-4-carboxamides useful as cancer chemotherapeutic agents |
| DE102005062742A1 (en) * | 2005-12-22 | 2007-06-28 | Bayer Schering Pharma Ag | New sulfoximine-substituted pyrimidines useful for treating e.g. diseases caused by inflammatory, allergic or proliferative processes, oncological diseases, cancer, eye, autoimmune and neurodegerative diseases |
| US20070155746A1 (en) * | 2005-12-23 | 2007-07-05 | Kalypsys, Inc. | Novel substituted pyridinyloxy and pyrimidinyloxy amides useful as inhibitors of protein kinases |
| JP2009533327A (en) * | 2006-03-22 | 2009-09-17 | バーテックス ファーマシューティカルズ インコーポレイテッド | C-MET Kinase Inhibitors for Treating Proliferative Diseases |
| AR060061A1 (en) * | 2006-03-22 | 2008-05-21 | Methylgene Inc | INHIBITORS OF THE ACTIVITY OF PROTEIN TIROSINA QUINASA |
| US7790756B2 (en) * | 2006-10-11 | 2010-09-07 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of myleoproliferative diseases and other proliferative diseases |
| WO2008131276A1 (en) * | 2007-04-20 | 2008-10-30 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of myleoproliferative diseases and other proliferative diseases |
| NZ580660A (en) * | 2007-04-30 | 2012-02-24 | Abbott Lab | Inhibitors of diacylglycerol o-acyltransferase type 1 enzyme |
| ES2593279T3 (en) | 2007-08-29 | 2016-12-07 | Methylgene Inc. | Processes and intermediates for preparing condensed heterocyclic kinase inhibitors |
| WO2009070328A1 (en) | 2007-11-26 | 2009-06-04 | The Regents Of The University Of California | Modulators of the epidermal growth factor receptor (egfr) pathway for use in the treatment or prevention of substance abuse |
| WO2009103778A1 (en) * | 2008-02-19 | 2009-08-27 | Novasaid Ab | Compounds and methods |
| US8906944B2 (en) * | 2009-09-03 | 2014-12-09 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
| AU2010289353B2 (en) * | 2009-09-03 | 2016-12-08 | Allergan, Inc. | Compounds as tyrosine kinase modulators |
-
2010
- 2010-09-03 AU AU2010289353A patent/AU2010289353B2/en not_active Ceased
- 2010-09-03 CA CA2772625A patent/CA2772625A1/en not_active Abandoned
- 2010-09-03 CN CN2010800389522A patent/CN102498114A/en active Pending
- 2010-09-03 WO PCT/US2010/047816 patent/WO2011029001A1/en not_active Ceased
- 2010-09-03 US US12/875,223 patent/US8809534B2/en active Active
- 2010-09-03 SG SG2012015053A patent/SG178965A1/en unknown
- 2010-09-03 US US12/875,218 patent/US8614234B2/en active Active
- 2010-09-03 RU RU2012112151/04A patent/RU2012112151A/en not_active Application Discontinuation
- 2010-09-03 KR KR1020127008525A patent/KR20120082890A/en not_active Withdrawn
- 2010-09-03 EP EP10760159.3A patent/EP2473501B1/en not_active Not-in-force
- 2010-09-03 WO PCT/US2010/047800 patent/WO2011028995A1/en not_active Ceased
- 2010-09-03 AU AU2010289359A patent/AU2010289359A1/en not_active Abandoned
- 2010-09-03 MX MX2012002591A patent/MX2012002591A/en not_active Application Discontinuation
- 2010-09-03 NZ NZ598455A patent/NZ598455A/en not_active IP Right Cessation
- 2010-09-03 MX MX2012002596A patent/MX2012002596A/en not_active Application Discontinuation
- 2010-09-03 IN IN2493DEN2012 patent/IN2012DN02493A/en unknown
- 2010-09-03 ES ES10760159T patent/ES2730086T3/en active Active
- 2010-09-03 JP JP2012528084A patent/JP5868855B2/en not_active Expired - Fee Related
- 2010-09-03 RU RU2012109233/04A patent/RU2012109233A/en not_active Application Discontinuation
- 2010-09-03 JP JP2012528089A patent/JP2013503903A/en active Pending
- 2010-09-03 EP EP10752975.2A patent/EP2473513B1/en not_active Not-in-force
- 2010-09-03 CA CA2772718A patent/CA2772718C/en active Active
- 2010-09-03 BR BR112012004843A patent/BR112012004843A2/en not_active IP Right Cessation
- 2010-09-03 KR KR1020127008527A patent/KR20120047313A/en not_active Withdrawn
- 2010-09-03 BR BR112012004718A patent/BR112012004718A2/en not_active IP Right Cessation
- 2010-09-03 SG SG2012015038A patent/SG178963A1/en unknown
- 2010-09-03 CN CN2010800482682A patent/CN102686577A/en active Pending
- 2010-09-03 NZ NZ598781A patent/NZ598781A/en not_active IP Right Cessation
-
2012
- 2012-02-27 IL IL218332A patent/IL218332A0/en unknown
- 2012-02-28 IL IL218374A patent/IL218374A0/en unknown
- 2012-03-02 CL CL2012000587A patent/CL2012000587A1/en unknown
- 2012-03-02 ZA ZA2012/01592A patent/ZA201201592B/en unknown
- 2012-03-02 CL CL2012000592A patent/CL2012000592A1/en unknown
- 2012-03-05 ZA ZA2012/01627A patent/ZA201201627B/en unknown
-
2013
- 2013-10-15 US US14/054,444 patent/US9328103B2/en active Active
-
2014
- 2014-02-03 PH PH12014500281A patent/PH12014500281A1/en unknown
-
2015
- 2015-07-08 US US14/793,913 patent/US9475801B2/en active Active
-
2016
- 2016-01-06 JP JP2016001387A patent/JP6109974B2/en not_active Expired - Fee Related
- 2016-09-19 US US15/269,612 patent/US9725433B2/en not_active Expired - Fee Related
Patent Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0039051A2 (en) | 1980-04-24 | 1981-11-04 | Merck & Co. Inc. | Mannich-base hydroxamic acid prodrugs for the improved bioavailability of non-steroidal anti-inflammatory agents, a process for preparing and a pharmaceutical composition containing them |
| US4966849A (en) | 1985-09-20 | 1990-10-30 | President And Fellows Of Harvard College | CDNA and genes for human angiogenin (angiogenesis factor) and methods of expression |
| US5217999A (en) | 1987-12-24 | 1993-06-08 | Yissum Research Development Company Of The Hebrew University Of Jerusalem | Styryl compounds which inhibit EGF receptor protein tyrosine kinase |
| WO1991015495A1 (en) | 1990-04-02 | 1991-10-17 | Pfizer Inc. | Benzylphosphonic acid tyrosine kinase inhibitors |
| US5302606A (en) | 1990-04-16 | 1994-04-12 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Styryl-substituted pyridyl compounds which inhibit EGF receptor tyrosine kinase |
| WO1992020642A1 (en) | 1991-05-10 | 1992-11-26 | Rhone-Poulenc Rorer International (Holdings) Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| WO1992021660A1 (en) | 1991-05-29 | 1992-12-10 | Pfizer, Inc. | Tricyclic polyhydroxylic tyrosine kinase inhibitors |
| WO1994003427A1 (en) | 1992-08-06 | 1994-02-17 | Warner-Lambert Company | 2-thioindoles (selenoindoles) and related disulfides (selenides) which inhibit protein tyrosine kinases and which have antitumor properties |
| US5330992A (en) | 1992-10-23 | 1994-07-19 | Sterling Winthrop Inc. | 1-cyclopropyl-4-pyridyl-quinolinones |
| WO1994014808A1 (en) | 1992-12-23 | 1994-07-07 | Farmitalia Carlo Erba Srl | Vinylene-azaindole derivatives and process for their preparation |
| US5883116A (en) | 1995-06-07 | 1999-03-16 | Sugen, Inc. | 3-(2'-alkoxybenzylidenyl)-2-indolinone and analogues thereof for the treatment of disease |
| US5792783A (en) | 1995-06-07 | 1998-08-11 | Sugen, Inc. | 3-heteroaryl-2-indolinone compounds for the treatment of disease |
| US5883113A (en) | 1995-06-07 | 1999-03-16 | Sugen, Inc. | 3-(4'-Bromobenzylindenyl)-2-indolinone and analogues thereof for the treatment of disease |
| US5886020A (en) | 1995-06-07 | 1999-03-23 | Sugen, Inc. | 3-(4'-dimethylaminobenzylidenyl)-2-indolinone and analogues thereof for the treatment of disease |
| US5834504A (en) | 1995-06-07 | 1998-11-10 | Sugen, Inc. | 3-(2'-halobenzylidenyl)-2-indolinone compounds for the treatment of disease |
| US6765012B2 (en) | 2001-09-27 | 2004-07-20 | Allergan, Inc. | 3-(Arylamino)methylene-1,3-dihydro-2H-indol-2-ones as kinase inhibitors |
| US6541504B1 (en) | 2002-04-03 | 2003-04-01 | Allergan Sales, Llc | (3Z)-3-(2,3-dihydro-1H-inden-1-ylidene)-1,3-dihydro-2H-indol-2-ones as kinase inhibitors |
| WO2003106462A1 (en) * | 2002-06-14 | 2003-12-24 | Pfizer Inc. | Benzofused heteroaryl amide derivatives of thienopyridines useful as therapeutic agents, pharmaceutical compositions including the same, and methods for their use |
| US6747025B1 (en) | 2002-11-27 | 2004-06-08 | Allergan, Inc. | Kinase inhibitors for the treatment of disease |
| US20070197537A1 (en) * | 2006-01-30 | 2007-08-23 | Blake James F | Heterobicyclic thiophene compounds and methods of use |
| WO2009026717A1 (en) * | 2007-08-29 | 2009-03-05 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US20090118276A1 (en) * | 2007-11-02 | 2009-05-07 | Wyeth | Thienopyrimidines, thienopyridines, and pyrrolopyrimidines as b-raf inhibitors |
| WO2009109035A1 (en) * | 2008-03-05 | 2009-09-11 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US20100081675A1 (en) * | 2008-09-26 | 2010-04-01 | National Health Research Institutes | Fused multicyclic compounds as protein kinase inhibitors |
Non-Patent Citations (15)
| Title |
|---|
| "Bundgaard Design of Prodrugs", 1985, ELSEVIER |
| BOLEN, ONCOGEN, vol. 8, 1993, pages 2025 - 2031 |
| BUNDGAARD, J. MED. CHEM., 1989, pages 2503 |
| EDELMAN; CASTRO, EXP. EYE RES., vol. 71, 2000, pages 523 - 533 |
| JEFFREY EDELMAN, EXP. EYE. RES., vol. 80, 2005, pages 249 - 258 |
| JELLINEK ET AL., BIOCHEMISTRY, vol. 33, pages 10450 - 56 |
| KENDALL; THOMAS, PROC. NAT'L ACAD. SCI, vol. 90, 1994, pages 10705 - 09 |
| KIM ET AL., NATURE, vol. 362, 1993, pages 841 - 844 |
| KINSELLA ET AL., EXP. CELL RES., vol. 199, 1992, pages 56 - 62 |
| MARIANI ET AL., PROC. AM. ASSOC. CANCER RES., vol. 35, 1994, pages 2268 |
| PLOWMAN ET AL., DN&P, vol. 7, no. 6, 1994, pages 334 - 339 |
| RAEPPEL S ET AL: "Identification of a novel series of potent RON receptor tyrosine kinase inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 20, no. 9, 1 May 2010 (2010-05-01), pages 2745 - 2749, XP027012825, ISSN: 0960-894X, [retrieved on 20100319] * |
| RICHARD B. SILVERMAN: "Organic Chemistry ofDrug Design and Drug Action, 2d Ed.,", 2004, ELSEVIER ACADEMIC PRESS, article "Prodrugs and Drug Delivery Systems", pages: 496 - 557 |
| TAKANO ET AL., MOL. BIO. CELL, vol. 4, 1993, pages 358A |
| WRIGHT ET AL., J. CELLULAR PHYS., vol. 152, 1992, pages 448 - 57 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8293897B2 (en) | 2008-10-14 | 2012-10-23 | Ning Xi | Compounds comprising a spiro-ring and methods of use |
| US8426585B2 (en) | 2008-10-14 | 2013-04-23 | Ning Xi | Compounds comprising a spiro-ring |
| US8232294B2 (en) | 2009-03-21 | 2012-07-31 | Ning Xi | Amino ester derivatives, sailts thereof and methods of use |
| US10829496B2 (en) | 2017-05-11 | 2020-11-10 | Bristol-Myers Squibb Company | Thienopyridines and benzothiophenes useful as IRAK4 inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8809534B2 (en) | Compounds as tyrosine kinase modulators | |
| US10221192B2 (en) | Compounds as tyrosine kinase modulators | |
| US6747025B1 (en) | Kinase inhibitors for the treatment of disease | |
| EP3749646B1 (en) | Heteroaryl compounds as kinase inhibitor | |
| JP6239149B2 (en) | Heteroaromatic compounds and their use as dopamine D1 ligands | |
| US6699863B1 (en) | Kinase inhibitors for the treatment of disease | |
| CN120265619A (en) | Isoquinolones as PI3K inhibitors | |
| US20130065880A1 (en) | Compounds as tyrosine kinase modulators | |
| JP6564394B2 (en) | Heterocyclic compounds and their use as dopamine D1 ligands | |
| TWI565698B (en) | Quinoxaline compound, its production method and use | |
| WO2004050621A2 (en) | Indol derivatives and their use as kinase inhibitors | |
| JP2006512400A (en) | Kinase inhibitors for the treatment of diseases | |
| HK40026650A (en) | Aminopyrimidine derivatives, preparation method therefor and use thereof | |
| HK40026650B (en) | Aminopyrimidine derivatives, preparation method therefor and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080038952.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10752975 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 218332 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2772625 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/002591 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010289359 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012000592 Country of ref document: CL Ref document number: 2012528089 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1201000929 Country of ref document: TH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1990/DELNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010752975 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201203714 Country of ref document: UA |
|
| ENP | Entry into the national phase |
Ref document number: 2010289359 Country of ref document: AU Date of ref document: 20100903 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20127008525 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012109233 Country of ref document: RU |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012004718 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012004718 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120301 |