WO2011030356A2 - Process for the preparation of indoline derivatives and their intermediates thereof - Google Patents
Process for the preparation of indoline derivatives and their intermediates thereof Download PDFInfo
- Publication number
- WO2011030356A2 WO2011030356A2 PCT/IN2010/000607 IN2010000607W WO2011030356A2 WO 2011030356 A2 WO2011030356 A2 WO 2011030356A2 IN 2010000607 W IN2010000607 W IN 2010000607W WO 2011030356 A2 WO2011030356 A2 WO 2011030356A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- reaction mixture
- solvent
- residue
- stirring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/18—Radicals substituted by singly bound oxygen or sulfur atoms
- C07D317/22—Radicals substituted by singly bound oxygen or sulfur atoms etherified
Definitions
- the present invention relates to novel compounds, which can be used as an intermediates in the process for the preparation of indoline derivative. More specifically the present invention relates to an intermediate compounds, their preparation and use in the process for preparation of Silodosin and its related compounds. Background of the Invention
- Silodosin is an indoline compound, chemically known as l-(3-Hydroxypropyl)-5-[(2R)-2- ( ⁇ 2- [2-(2,2,2-trifluoro-ethoxy)phenoxy] ethyl ⁇ amino)propyl] -2 ,3 -dihydro- 1 H-indole-7- carboxamide and represented by Formula (I).
- Silodosin was disclosed in U.S. Patent No. 5,387,603 as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia.
- EP 1806340 patent application relates to a process for the preparation of silodosin comprising mixing 3- ⁇ 7-cyano-5-[(2R)-2-( ⁇ 2-[2-(2,2,2-trifluoroethoxy)phenoxy]- ethyl ⁇ amino)propyl]-2,3-dihydro-lH-indol-l-yl ⁇ propyl benzoate of Formula (1) with oxalic acid to produce corresponding monooxalate salt compound.
- step (d) Extracting the reaction mixture of step (c) with an aromatic hydrocarbon solvent followed by separation of aqueous and organic layers;
- step (f) Dissolving the oil (compound X) obtained in step (e) in halogenated hydrocarbon solvent followed by addition of boron tribromide at about -10°C to -25°C;
- step (g) Quenching the reaction mixture of step (f) with aqueous solution of an inorganic base followed by separation of aqueous and organic layers;
- step (h) Concentrating the organic layer of step (g) to get a compound (XI) as an oil;
- step (i) Dissolving the oily compound (XI) of step (k) in an aprotic polar organic solvent followed by addition of an inorganic base and bromo acetaldehyde ethylene glycol under stirring;
- step (j) Stirring the reaction mixture of step (i) at 110-135°C for 4-6 hours;
- step (k) Adding water to the reaction mixture of step (j) at 25-40°C;
- step (p) Dissolving the compound (XII) obtained step (o) in solvent 1,4-dioxane and water followed by the addition of concentrated sulphuric acid;
- step (3) Extracting the residue of step (2) in to an organic solvent followed by concentration of the organic layer under reduced pressure to get residue;
- step (3) Purifying the residue of step (3) by conventional methods to get a solid compound
- step (4) Dissolving the solid of step (4) in a halogenated hydrocarbon solvent
- step (6) Adding to a solution of step (5), oxalyl chloride and halogenated hydrocarbon solvent, followed by triethyl amine at a temperature below -70°C;
- step (6) Quenching the reaction mixture of step (6) with water and extracting the reaction solution with a halogenated solvent
- step (8) Purifying the residue of step (8) by conventional methods to get the solid compound (VIII).
- step b Adding a reducing agent to the reaction mixture of step a and stirring for about 1-2 hours;
- step (b) Stirring the reaction mixture of step (b) at a temperature between 35 and 45°C for about 2-3 hours;
- step (c ) Concentrating the reaction mixture of step (c ) by evaporating the solvent under reduced pressure;
- step (d) Acidifying the residue of step (d) with mineral acid;
- step (c) Adding a weak organic acid , an alcohol solvent and a compound, [2- (2,2,2-Trifluoro-ethoxy)-phenoxy]-acetaldehyde (VIII) to the residue of step (c) at (25-40°) and continue stirring for about 1-2 hours;
- step (e) Adding a reducing agent to the reaction mixture of step (d) and stirring for about 1-2 hours;
- step (e) Stirring the reaction mixture of step (e) at a temperature between 35 and 45°C for about 2-3 hours;
- step (f) Concentrating the reaction mixture of step (f) by evaporating the solvent under reduced pressure;
- step (h) Adding aqueous mineral acid to the reaction mixture of step (g); i) Extracting the reaction mixture of step (h) into aprotic polar organic solvent;
- step (i) Concentrating the extraction of step (i) under reduced pressure to get the residue;
- step (j) Purifying the residue of step (j) by conventional methods;
- step (k) 1) Dissolving the solid obtained in step (k) in an aprotic polar organic solvent and adding an oxidizing agent followed by inorganic aqueous basic solution;
- step (1) Stirring the contents of step (1) for about 2-3 hours at about 25-35°C;
- step (m) Adding water to the reaction mixture of step (m) and extracting the reaction solution in to an organic solvent;
- step (o) Purifying the residue of step (o) by conventional methods to get the compound (I).
- step (b) Stirring the reaction mixture of step (a) at a temperature 90-130°C for about 4-8 hours;
- step (c) Adding water to the reaction mixture of step (b) at about 30-45°C;
- step (d) Extracting the reaction mixture of step (c) in to an aromatic hydrocarbon solvent followed by separation of aqueous and organic layers;
- step (f) Dissolving the oily compound (XI) of step (e) in an aprotic polar organic solvent followed by addition of an inorganic base and bromo acetaldehyde ethylene glycol under stirring;
- step (g) Stirring the reaction mixture of step (f) at about 110-135°C for about 4-6 hours; (h) Adding water to the reaction mixture of step (g) at about 25 - 40 C;
- step (j) Extracting the reaction mixture of step (i) in to a organic solvent followed by separation of aqueous and organic layers;
- step (k) Purifying the residue obtained in step (k) to get the solid compound (XII); (m) Dissolving the compound (XII) obtained step (1) in solvent 1,4-dioxane and water followed by the addition of concentrated sulphuric acid or oxalic acid;
- the present invention provides a process for the preparation of Silodosin comprising condensation of compound of formula (VII) and (VIII) via reductive amination using sodiumcyanoborohydride or reduction of in situ formed imine with reducing agents such as sodiumborohydride, sodium triacetoxyborohydride , Raney Ni, H 2 /Pd-C.
- An embodiment of the present invention provides a process for the preparation of key intermediate compound (VII) from 7-cyano indoline.
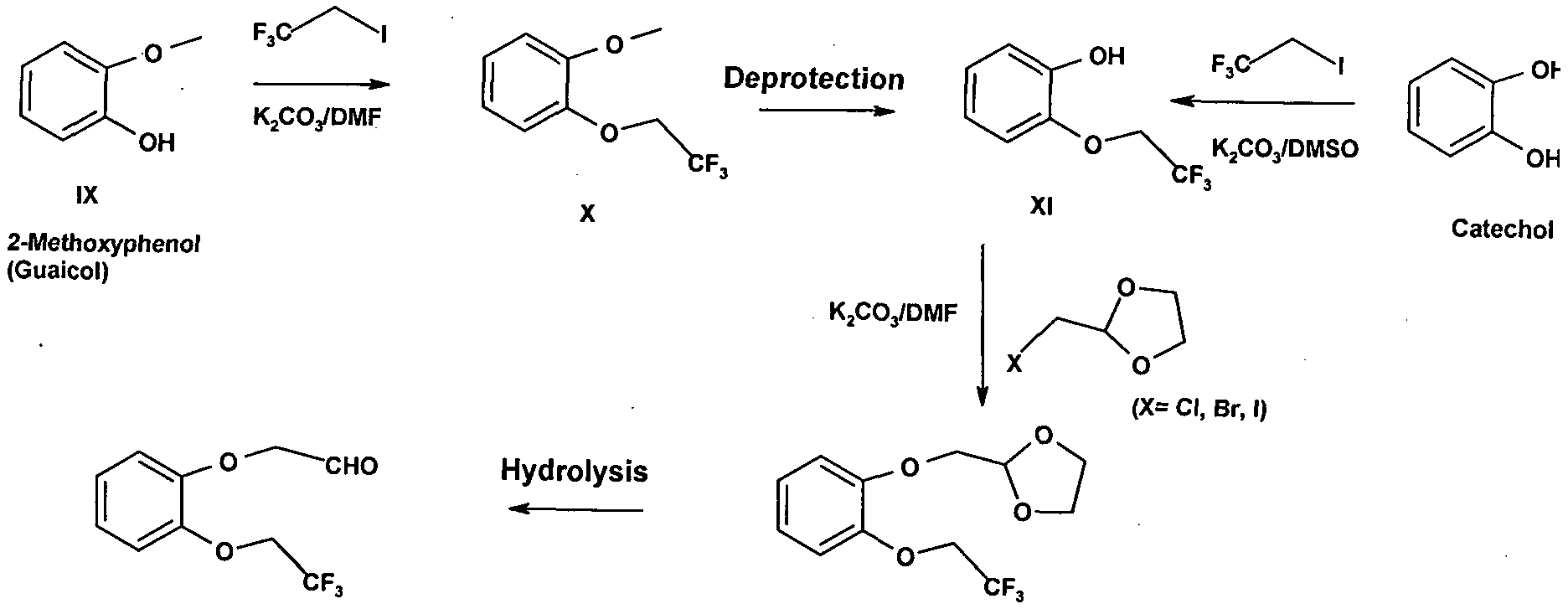
- Another embodiment of the present invention provides a process for the preparation of key intermediate compound of Formula (VIII) from 2-methoxy phenol comprising.
- step (e) extracting the reaction solution of step (d) in to an aromatic hydrocarbon solvent
- step (i) dissolving the oil (compound X) obtained in step (h) in halogenated hydrocarbon solvent;
- step (j) addition of boron tribromide to the solution of step (i) at about -10°C;
- step (k) quenching the reaction mixture of step (k) with aqueous solution of an inorganic base
- step (n) concentrating the organic solvent of step (m) to get a compound (XI) as an oil;
- step (p) addition of an inorganic base and bromo acetaldehyde ethylene glycol to the solution of step (o) under stirring;
- step (q) stirring the reaction mixture of step (p) at about 110-135°C for about 4-6 hours;
- step (r) addition of water to the reaction mixture of step (q) at a temperature between about 25 and 40°C;
- step (s) adjusting the pH of the reaction solution of step (r) to 3-4 with mineral acid;
- step (t) extracting the reaction solution of step (s) in to a organic solvent
- step (u) separating the organic layer from the extraction of step (t) and washing with water;
- step (v) concentrating the organic solvent of step (u) under reduced pressure toget the residue;
- step (w) purifying the residue obtained in step (v) to get the solid compound (XII);
- step (x) dissolving the compound (XII) obtained step (w) in solvent 1,4-dioxane and water;
- step (y) addition of concentrated sulphuric acid or oxalic acid to the reaction solution of step (x) and stirring the contents for about 3-6 hours at about 85-110°C;
- step (bb) concentrating the organic solvent from step (aa) under reduced pressure
- step (cc) purifying the residue of step (bb) to get the compound (VIII).
- step (e) extracting the reaction solution of step (d) in to an aromatic hydrocarbon solvent
- step (i) dissolving the oily compound (XI) of step (h) in an aprotic polar organic solvent;
- step (j) addition of an inorganic base and bromo acetaldehyde ethylene glycol to the solution of step (i) under stirring;
- step (k) stirring the reaction mixture of step (j) at about 110-135°C for about 4-6 hours; (1) addition of water to the reaction mixture of step (k) at a temperature between about 25 and 40°C;
- step (n) extracting the reaction solution of step (m) in to a organic solvent
- step (o) separating the organic layer from the extraction of step (n) and washing with water;
- step (p) concentrating the organic solvent of step (o) under reduced pressure to get the residue;
- step (q) purifying the residue obtained in step (p) to get the solid compound (XII); (r) dissolving the compound (XII) obtained step (q) in solvent 1,4-dioxane and water;
- step (s) addition of concentrated sulphuric acid or oxalic acid to the reaction solution of step (r) and stirring the contents for about 3-6 hours at about 85-110°C; (t) diluting the reaction mixture of step(s) with water and extracting the reaction solution with an organic solvent;
- step (u) separating the organic layer form the extractions of step (t) and washing with water;
- step (v) concentrating the organic solvent from step (u) under reduced pressure (w) purifying the residue of step (v) to get the compound (VIII).
- An inorganic base used in the reaction selected form the group but not limited to alkali metal hydroxide, alkali metal cabonate and alkali metal bicarbonate.
- the inorganic base is alkali metal cabonate and potassium carbonate is especially preferable.
- the halogenated hydrocarbon solvent used in the above reaction is selected from the group consisting methylene chloride, chloroform, carbon tetrachloride, ethylene dichloride and the like.
- the compound (VIII) can also be obtained from a compound 2-(2, 2, 2- trifluoroethoxy) phenol (XI) by the process comprising,
- step (4) purifying the residue of step (4) by conventional methods to get a solid compound
- step (6) dissolving the solid of step (5) in a halogenated hydrocarbon solvent and adding to a solution of oxalyl chloride and halogenated hydrocarbon solvent, followed by triethyl amine at a temperature below -70°C;
- step (4) quenching the reaction mixture of step (4) with water and extracting the reaction solution with a halogenated solvent
- step (8) washing the organic extractions of step (5) with water and concentrating the solvent under reduced pressure to get a residue
- step (6) purifying the residue of step (6) by conventional methods to get the solid compound (VIII).
- An inorganic base used in the above reaction selected form the group but not limited to alkali metal hydroxide, alkali metal cabonate and alkali metal bicarbonate.
- the inorganic base is alkali metal cabonate and potassium carbonate is especially preferable.
- the aprotic polar solvent used in the above reaction is selected from the group consisting dimethyl formamide, N-methyl pyrrolidone, acetonitrile, dimethyl sulfoxide and the like.
- the halogenated hydrocarbon solvent used in the above reaction is selected from the group containing methylene chloride, chloroform, carbon tetrachloride, ethylene dichloride and the like.
- Purification technique used for isolation of the solid from the residue obtained in above reactions comprises column chromatography, recrystallization and the like.
- step d add a weak organic acid , an alcohol solvent and a compound, [2-(2,2,2- Trifluoro-ethoxy)-phenoxy]-acetaldehyde (VIII) to the residue of step c at ambient temperature (25-40°) and continue stirring for about 1-2 hours; e. addition of a reducing agent to the reaction mixture of step d and stirring for about 1-2 hours;
- step f stirring the reaction mixture of step e at a temperature between 35 and 45°C for about 2-3 hours;
- step f concentrating the reaction mixture of step f by evaporating the solvent under reduced pressure
- step h addition of aqueous mineral acid to the reaction mixture of step g; i. extracting the reaction mixture of step h into an organic solvent; j. concentrating the extractions of step i under reduced pressure to get the residue;
- step j purifying the residue of step j by conventional methods
- step k 1. dissolving the solid obtained in step k in an aprotic polar organic solvent and adding an oxidizing agent followed by inorganic aqueous basic solution;
- step 1 stirring the contents of step 1 for about 2-3 hours at about 25-35°C; n. addition of water to the reaction mixture of step m and extracting the reaction solution in to an organic solvent;
- step o purifying the residue of step o by conventional methods to get the compound (I).
- the weak organic base used in the above reaction selected form the group but not limited to acetic acid, trifluoro acetic acid, formic acid.
- An alcohol solvent used in the above reaction is selected from the group consisting lower alcohol solvent like methanol, ethanol, propanol, isopropanol, and tertiary butanol.
- the reducing agent used in the above reaction is selected from the group consisting alkaliborohydride, alkali cyanoborohydride, ranynickel, and Palladium on carbon, preferably alkali cyanoborohydride most preferably sodiumcyanoboro hydride.
- the aprotic polar solvent used in the above reaction is selected from the group consisting dimethyl formamide, N-methyl pyrrolidone, acetonitrile, dimethly sulfoxide and the like.
- the oxidizing agent used in the above reaction is hydrogen peroxide.
- Purification technique used for isolation of the solid from the residue obtained in above reactions comprises column chromatography, recrystallization and the like.
- in yet another embodiement of the present invention provides a process for the preparation of compound (VII) comprising the steps for the preparation of compounds (II), (IV), (V), (VI) and conversion to compound (VIII).
- the first embodiment of the present invention which provides a process for preparing compound (VII) from 7-Cyano indoline comprising steps, which can be shown by Scheme- 1.
- Scheme 1 In an embodiment of the present invention provide a process for the preparation of compound (VIII) comprises reaction of compound of formula XI with bromo acetaldehyde ethylene glycol to give compound of formula XII; followed by the hydrolysis of compound of formula XII to yield compound of Formula VIII, wherein compound XI is prepared from catechol
- Another embodiment of the invention is process for the preparation of Silodosin (compound of Formula I) comprising condensation of novel intermediate compound (VIII) and compound (XV) followed by deprotection and oxidation.
- step a addition of a reducing agent to the reaction mixture of step a and stirring for about 1-2 hours;
- step b stirring the reaction mixture of step b at a temperature between 35 and 45°C for about 2-3 hours;
- step f (d) concentrating the reaction mixture of step f by evaporating the solvent under reduced pressure;
- step d (e) acidify the residue of step d with mineral acid and extract product in ethyl acetate
- reaction mass was then heated to 40-45°C and stirred for 2 hours. After the completion of reaction solvent, was evaporated under reduced pressure and water was added to the residue. Reaction mass was acidified with aqueous mineral acid and extracted the mixture in ethyl acetate. Organic layer was washed with water and dried over sodium sulphate. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane (5/95) as eluent to give 9.6 g of (XVI) as oil.
- the reaction mixture was extracted with ethyl acetate.
- the combined ethyl acetate layer was extracted 2N hydrochloric acid.
- the aqueous layer was neutralized with sodium bicarbonate and extracted the product in ethyl acetate.
- the organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate.
- the solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate.
- the resulting solution was cooled to 5°C and filtered to get 4.51 g of (I) as solid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Indole Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description

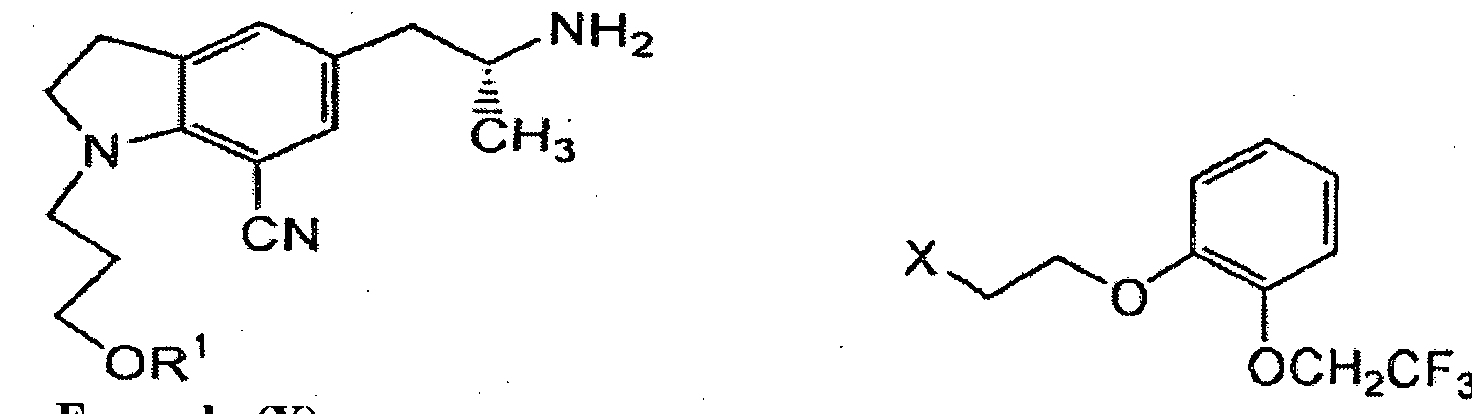










Claims




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012528507A JP5661773B2 (en) | 2009-09-12 | 2010-09-13 | Process for the preparation of indoline derivatives and intermediates thereof |
| CA2772997A CA2772997A1 (en) | 2009-09-12 | 2010-09-13 | Process for the preparation of indoline derivatives and their intermediates thereof |
| EP10803270.7A EP2475634B1 (en) | 2009-09-12 | 2010-09-13 | Process for the preparation of indoline derivatives and their intermediates thereof |
| US13/392,564 US8471039B2 (en) | 2009-09-12 | 2010-09-13 | Process for the preparation of indoline derivatives and their intermediates thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1420/MUM/2009 | 2009-09-12 | ||
| IN1420MU2009 | 2009-09-12 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011030356A2 true WO2011030356A2 (en) | 2011-03-17 |
| WO2011030356A3 WO2011030356A3 (en) | 2011-05-05 |
Family
ID=43569457
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IN2010/000607 Ceased WO2011030356A2 (en) | 2009-09-12 | 2010-09-13 | Process for the preparation of indoline derivatives and their intermediates thereof |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8471039B2 (en) |
| EP (1) | EP2475634B1 (en) |
| JP (1) | JP5661773B2 (en) |
| CA (1) | CA2772997A1 (en) |
| WO (1) | WO2011030356A2 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011101864A1 (en) * | 2010-02-17 | 2011-08-25 | Panacea Biotec Ltd | Novel process for the synthesis of phenoxyethyl derivatives |
| CN102320996A (en) * | 2011-07-14 | 2012-01-18 | 四川大学 | A kind of preparation of silodosin midbody and purification process |
| WO2012014186A1 (en) | 2010-07-30 | 2012-02-02 | Ranbaxy Laboratories Limited | Process for the preparation of silodosin and its novel intermediates |
| CN102382029A (en) * | 2011-07-26 | 2012-03-21 | 浙江华海药业股份有限公司 | Preparation method of salt formation of silodosin intermediate |
| WO2013056842A1 (en) * | 2011-10-21 | 2013-04-25 | Sandoz Ag | Method for preparing silodosin |
| WO2013072935A2 (en) | 2011-10-10 | 2013-05-23 | Cadila Healthcare Limited | Process for the preparation of silodosin |
| CN103396352A (en) * | 2013-08-07 | 2013-11-20 | 苏州明锐医药科技有限公司 | Preparation method of Silodosin |
| CN104230782A (en) * | 2013-06-09 | 2014-12-24 | 昆明积大制药股份有限公司 | Synthetic method of silodosin |
| WO2015010594A1 (en) * | 2013-07-22 | 2015-01-29 | 中国科学院上海药物研究所 | Indoline compound, preparation method therefor, pharmaceutical composition, and application thereof |
| CN104557662A (en) * | 2014-12-26 | 2015-04-29 | 华润赛科药业有限责任公司 | Synthesis method of silodosin dialkyl compound |
| KR20150066782A (en) * | 2013-12-09 | 2015-06-17 | 동우신테크 주식회사 | Method of preparing 1-(indolin-5-yl)propan-2-ol derivatives |
| CN106083689A (en) * | 2016-06-14 | 2016-11-09 | 齐鲁制药有限公司 | A kind of new preparation process of Silodosin compound |
| WO2017080414A1 (en) * | 2015-11-09 | 2017-05-18 | 黄欢 | Method for preparing silodosin intermediate |
| US10421719B2 (en) | 2015-09-30 | 2019-09-24 | Urquima S.A. | Maleic acid salt of a silodosin intermediate |
| CN120758094A (en) * | 2025-09-03 | 2025-10-10 | 山东正诺化工设备有限公司 | Anti-stress corrosion material based on slurry steam generator tube sheet and preparation method thereof |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104211631A (en) * | 2013-05-30 | 2014-12-17 | 中国科学院上海药物研究所 | Indoles compound, preparation method thereof, pharmaceutical composition and application thereof |
| KR20160110517A (en) * | 2014-02-06 | 2016-09-21 | 우베 고산 가부시키가이샤 | Method for producing indoline compound |
| JP2016088847A (en) * | 2014-10-30 | 2016-05-23 | 株式会社トクヤマ | (-)-1- (3-hydroxypropyl) -5-[[(2R) -2-({2- [2- (2,2,2-trifluoroethoxy) phenoxy] ethyl} amino) propyl]- 2,3-Dihydro-1H-indole-7-carboxamide]) |
| WO2016139773A1 (en) * | 2015-03-04 | 2016-09-09 | 株式会社三洋化学研究所 | Novel synthesis method for silodosin synthetic intermediate |
| CN114262291B (en) * | 2022-01-04 | 2023-05-19 | 重庆医科大学 | A kind of synthetic method of alvacopam |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387603A (en) | 1992-12-02 | 1995-02-07 | Kissei Pharmaceutical Co., Ltd. | 1,5,7-trisubstituted indoline compounds and salts thereof |
| EP1806340A1 (en) | 2004-10-27 | 2007-07-11 | Kissei Pharmaceutical Co., Ltd. | Indoline compound and process for producing the same |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62114952A (en) * | 1985-11-13 | 1987-05-26 | Yamanouchi Pharmaceut Co Ltd | Production of substituted phenethylamine derivative |
| JP4324266B2 (en) * | 1999-02-26 | 2009-09-02 | キッセイ薬品工業株式会社 | α1A adrenergic receptor mutant, measurement method using the mutant, and therapeutic agent for dysuria associated with prostatic hypertrophy |
| JP4634560B2 (en) * | 2000-01-14 | 2011-02-16 | キッセイ薬品工業株式会社 | Process for producing optically active indoline derivative and production intermediate thereof |
| JP4634580B2 (en) | 2000-07-03 | 2011-02-16 | 富士通株式会社 | Electrode structure for oxide dielectric film, capacitor element using the same, and manufacturing method thereof |
| JP4921646B2 (en) * | 2001-03-08 | 2012-04-25 | キッセイ薬品工業株式会社 | 1- (3-Benzyloxypropyl) -5- (2-substituted propyl) indoline derivatives and methods of use thereof |
-
2010
- 2010-09-13 JP JP2012528507A patent/JP5661773B2/en not_active Expired - Fee Related
- 2010-09-13 CA CA2772997A patent/CA2772997A1/en not_active Abandoned
- 2010-09-13 EP EP10803270.7A patent/EP2475634B1/en active Active
- 2010-09-13 WO PCT/IN2010/000607 patent/WO2011030356A2/en not_active Ceased
- 2010-09-13 US US13/392,564 patent/US8471039B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5387603A (en) | 1992-12-02 | 1995-02-07 | Kissei Pharmaceutical Co., Ltd. | 1,5,7-trisubstituted indoline compounds and salts thereof |
| EP1806340A1 (en) | 2004-10-27 | 2007-07-11 | Kissei Pharmaceutical Co., Ltd. | Indoline compound and process for producing the same |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011101864A1 (en) * | 2010-02-17 | 2011-08-25 | Panacea Biotec Ltd | Novel process for the synthesis of phenoxyethyl derivatives |
| WO2012014186A1 (en) | 2010-07-30 | 2012-02-02 | Ranbaxy Laboratories Limited | Process for the preparation of silodosin and its novel intermediates |
| CN102320996A (en) * | 2011-07-14 | 2012-01-18 | 四川大学 | A kind of preparation of silodosin midbody and purification process |
| CN102382029B (en) * | 2011-07-26 | 2016-06-29 | 浙江华海药业股份有限公司 | A kind of salt formation of silodosin intermediate preparation method |
| CN102382029A (en) * | 2011-07-26 | 2012-03-21 | 浙江华海药业股份有限公司 | Preparation method of salt formation of silodosin intermediate |
| WO2013072935A2 (en) | 2011-10-10 | 2013-05-23 | Cadila Healthcare Limited | Process for the preparation of silodosin |
| WO2013056842A1 (en) * | 2011-10-21 | 2013-04-25 | Sandoz Ag | Method for preparing silodosin |
| CN104302621A (en) * | 2011-10-21 | 2015-01-21 | 桑多斯股份公司 | Method for preparing silodosin |
| US9938239B2 (en) | 2011-10-21 | 2018-04-10 | Sandoz Ag | Method for preparing silodosin |
| CN104230782A (en) * | 2013-06-09 | 2014-12-24 | 昆明积大制药股份有限公司 | Synthetic method of silodosin |
| WO2015010594A1 (en) * | 2013-07-22 | 2015-01-29 | 中国科学院上海药物研究所 | Indoline compound, preparation method therefor, pharmaceutical composition, and application thereof |
| CN103396352A (en) * | 2013-08-07 | 2013-11-20 | 苏州明锐医药科技有限公司 | Preparation method of Silodosin |
| KR20150066782A (en) * | 2013-12-09 | 2015-06-17 | 동우신테크 주식회사 | Method of preparing 1-(indolin-5-yl)propan-2-ol derivatives |
| CN104557662A (en) * | 2014-12-26 | 2015-04-29 | 华润赛科药业有限责任公司 | Synthesis method of silodosin dialkyl compound |
| US10421719B2 (en) | 2015-09-30 | 2019-09-24 | Urquima S.A. | Maleic acid salt of a silodosin intermediate |
| WO2017080414A1 (en) * | 2015-11-09 | 2017-05-18 | 黄欢 | Method for preparing silodosin intermediate |
| CN106083689A (en) * | 2016-06-14 | 2016-11-09 | 齐鲁制药有限公司 | A kind of new preparation process of Silodosin compound |
| CN106083689B (en) * | 2016-06-14 | 2020-07-31 | 齐鲁制药有限公司 | Preparation method of silodosin compound |
| CN120758094A (en) * | 2025-09-03 | 2025-10-10 | 山东正诺化工设备有限公司 | Anti-stress corrosion material based on slurry steam generator tube sheet and preparation method thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2772997A1 (en) | 2011-03-17 |
| JP2013504563A (en) | 2013-02-07 |
| US20120165548A1 (en) | 2012-06-28 |
| JP5661773B2 (en) | 2015-01-28 |
| EP2475634A2 (en) | 2012-07-18 |
| US8471039B2 (en) | 2013-06-25 |
| EP2475634B1 (en) | 2017-05-24 |
| WO2011030356A3 (en) | 2011-05-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2475634B1 (en) | Process for the preparation of indoline derivatives and their intermediates thereof | |
| US20080221338A1 (en) | Processes for preparing darifenacin hydrobromide | |
| KR20090080516A (en) | Method for preparing trityl olmesartan medoxomill and olmesartan medoxomill | |
| KR101540435B1 (en) | Stereoselective synthesis of valiolamine | |
| KR20110036055A (en) | Improved process for the preparation of high purity sunitinib and its pharmaceutically acceptable salts | |
| EP3230280A1 (en) | Process for preparation of luliconazole | |
| FI93835C (en) | Process for the preparation of 3- (benzothiazol-2-ylmethyl) -4-oxo-3-H-phthalazin-1-ylacetic acid derivative | |
| EP0011059B1 (en) | Process for the preparation of (+,-) vincadifformine and other related pentacyclic derivatives | |
| FR2950343A1 (en) | NOVEL PROCESS FOR THE SYNTHESIS OF IVABRADINE AND ITS SALTS OF ADDITION TO A PHARMACEUTICALLY ACCEPTABLE ACID | |
| US20240336603A1 (en) | Process for the preparation of a cyp11a1 inhibitor and intermediates thereof | |
| US8569483B2 (en) | Process for the preparation of bazedoxifene acetate and intermediates thereof | |
| JP4828863B2 (en) | Process for producing (Z) -1-phenyl-1- (N, N-diethylaminocarbonyl) -2-phthalimidomethylcyclopropane | |
| KR20180116371A (en) | Process for producing 4-alkoxy-3-hydroxypicolic acid | |
| US20060287535A1 (en) | Process for the manufacture of 1,2-benzisoxazole-3-methanesulphonamide | |
| US7321055B2 (en) | Production method of optically active dephenylalanine compounds | |
| AU2003269331A1 (en) | Acyl derivatives of 5-(2-(4-(1,2 benzisothiazole-3-yl)-1-piperazinyl)ethyl)-6-chloro-1,3-dihydro-2h-indol-2-one having neuroleptic activity | |
| KR20170123132A (en) | Process for Preparing Treprostinil | |
| JPS62228058A (en) | Novel isoindoline derivative and production thereof | |
| AASAASAAAS | 3 CONH | |
| JP2022529000A (en) | Method for Producing Substituted 2- [2- (Phenyl) Ethylamino] Alkaneamide Derivative | |
| US11459291B2 (en) | Method of preparation of (1R,3S)-3-amino-1-cyclopentanol and salt thereof | |
| KR20150044559A (en) | A new process for the preparation of 3-amino-9,13b-dihydro-1H-dibenz [c,f] imidazo[1,5-a]azepine bromic acid salt | |
| KR100377578B1 (en) | Process for the preparation of ondansetron and pharmaceutically acceptable salts thereof | |
| KR100665794B1 (en) | Novel Production Method of 4- (4-Fluorophenyl) -2-isobutyryl-3-phenyl-4-oxo-N-phenyl-butylamide | |
| US20060035906A1 (en) | Process for production of 1-[2-(benzimidazol-2-yl- thio)ethy]piperazine or salts thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10803270 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13392564 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2772997 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012528507 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010803270 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010803270 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 833/KOLNP/2012 Country of ref document: IN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10803270 Country of ref document: EP Kind code of ref document: A2 |