WO2011030786A1 - リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池 - Google Patents
リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池 Download PDFInfo
- Publication number
- WO2011030786A1 WO2011030786A1 PCT/JP2010/065404 JP2010065404W WO2011030786A1 WO 2011030786 A1 WO2011030786 A1 WO 2011030786A1 JP 2010065404 W JP2010065404 W JP 2010065404W WO 2011030786 A1 WO2011030786 A1 WO 2011030786A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- powder
- particles
- ferric phosphate
- particle powder
- olivine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images
















Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/5825—Oxygenated metallic salts or polyanionic structures, e.g. borates, phosphates, silicates, olivines
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/37—Phosphates of heavy metals
- C01B25/375—Phosphates of heavy metals of iron
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/45—Phosphates containing plural metal, or metal and ammonium
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/60—Particles characterised by their size
- C01P2004/61—Micrometer sized, i.e. from 1-100 micrometer
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/11—Powder tap density
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/80—Compositional purity
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
Definitions
- Crystalline ferric phosphate hydrate particle powder suitable as a precursor of olivine type lithium iron phosphate particle powder, its production method, and olivine type iron phosphate for non-aqueous electrolyte secondary battery positive electrode using the precursor A method for producing a lithium particle powder is provided.
- lithium-ion secondary batteries having a small size, light weight, and high energy density are attracting attention.
- lithium ion secondary batteries use rare metal-containing compounds such as Co and Ni as positive electrode materials, and in order for lithium ion secondary batteries to become widespread, there are major issues in terms of cost and supply stability. is there.
- ignition and explosion accidents of lithium ion secondary batteries caused by the positive electrode material have been reported, and safety issues have been pointed out. From such a background, development of a safe positive electrode material for lithium ion secondary electric fields that does not contain a rare metal is expected.
- olivine-type lithium iron phosphate is attracting attention as a positive electrode active material for 3.5 V-class lithium ion secondary batteries.
- Olivine-type lithium iron phosphate is a positive electrode material that has a very stable crystal structure and a very low risk of ignition and explosion because the P atom and the O atom are strongly bonded by a covalent bond in the crystal structure.
- the transition metal atom contributing to the oxidation-reduction reaction is Fe, not Co or Ni, it is a material that can be expected to greatly improve in terms of cost and supply stability.
- the solid-phase method is a method by which olivine-type lithium iron phosphate can be synthesized relatively easily.
- solid phase methods a method using a divalent iron compound as an iron raw material and a method using a trivalent iron compound are disclosed.
- Patent Document 1 discloses a method using iron oxalate as a divalent iron compound raw material.
- iron oxalate is used as a raw material, there is a problem in terms of cost that it is too expensive as an Fe raw material for olivine-type lithium iron phosphate aimed at an inexpensive positive electrode material.
- Patent Document 2 discloses a method using ferrous phosphate octahydrate (Fe 3 (PO 4 ) 2 .8H 2 O) as a divalent iron compound raw material. Here, it reports that it contains 0.1% or more and 1% or less of Na ions although it discloses an invention related to fine ferrous phosphate octahydrate with a low Na content. In such a solid phase reaction using ferrous phosphate octahydrate as a raw material, only olivine-type lithium iron phosphate particle powder containing a large amount of Na can be obtained. Since Na shows only a function of lowering the battery characteristics, further reduction of the Na content is desired as a positive electrode material for a lithium ion secondary battery.
- ferrous phosphate octahydrate Fe 3 (PO 4 ) 2 .8H 2 O
- Patent Document 3 Patent Document 4 and Patent Document 5 disclose a method for producing olivine-type lithium iron phosphate particles using ferric phosphate dihydrate as a trivalent iron compound raw material.
- Fe and P are uniformly present in the raw material in the raw material, and if the composition ratio also manages the P / Fe molar ratio in the ferric phosphate dihydrate, there is a risk of causing a composition shift.
- ferric phosphate hydrate which is the main raw material, in an industrially inexpensive manner in a short time has not yet been established. Further, the amount of impurities in the ferric phosphate hydrate is an important factor that affects the amount of impurities in the olivine-type lithium iron phosphate particles, but a method for reducing it is not yet known.
- the crystal phase was an amorphous phase, and Na exceeding 10,000 ppm was detected.
- Non-Patent Document 1 ferric phosphate dihydrate whose crystal structure is a strenite structure is synthesized by a hydrothermal reaction under high temperature and high pressure. Since this manufacturing method is carried out under high temperature and high pressure, the ferric phosphate dihydrate particles produced are large crystalline particles having primary particles of micron order to submillimeter order.
- the olivine-type lithium iron phosphate synthesized from crystalline powder of ferric phosphate hydrate particles with large primary particles becomes a powder with primary particles in the micron order, and fine olivine-type lithium iron phosphate particles in the submicron order. It is not suitable as a raw material for synthesizing powder.
- Non-Patent Document 2 ferric phosphate dihydrate is synthesized by a wet reaction under normal pressure.
- the amount of impurities is not mentioned, and the reduction method is not described.
- this method since this method has an extremely low reaction concentration of 0.01 M or less, ferric phosphate hydrate cannot be produced industrially at low cost.
- the bulky olivine type lithium iron phosphate particle powder is disadvantageous in terms of volume energy density because it is difficult to increase the positive electrode density in the coating film when applied to the electrode. Therefore, the olivine-type lithium iron phosphate particle powder for a nonaqueous electrolyte lithium ion secondary battery positive electrode is preferably an aggregated particle in which fine primary particles are aggregated.
- the present invention provides a fine, extremely low-impurity ferric phosphate hydrate particle powder suitable as a precursor of an olivine-type lithium iron phosphate particle powder for a positive electrode active material for a nonaqueous electrolyte secondary battery, and its production Establishing a law is a technical issue. Further, it is a technical problem to establish a method for producing olivine-type lithium iron phosphate particle powder that is fine and contains very few impurities, using the ferric phosphate hydrate particles synthesized by the production method of the present invention. To do.
- the present invention is a precursor of olivine type lithium iron phosphate particles, ferric phosphate having a Na content of 100 ppm or less and a phosphorus / iron molar ratio of 0.9 to 1.1. It is a hydrate particle powder (Invention 1).
- the present invention is the ferric phosphate hydrate particles powder according to the present invention 1, wherein the crystal structure is at least one of a strengite structure and a metastrength structure. (Invention 2).
- the present invention is the ferric phosphate hydrate particles powder according to the present invention 1 or 2, wherein the secondary particles are formed by aggregation of the plate-like primary particles, and the average secondary particles Ferric phosphate hydrate particles powder having a diameter of 5 to 20 ⁇ m (Invention 3).
- the present invention also provides a ferric phosphate hydrated powder according to any one of the present inventions 1 to 3, wherein the hydrated ferric phosphate has a tap density of 0.7 to 1.5 g / cc. It is a product particle powder (Invention 4).
- the present invention also provides a method for producing ferric phosphate hydrate particles by reacting iron oxide particles or hydrated iron oxide particles with a phosphorus compound in a solution, wherein the BET specific surface area is 50 m 2 / g.
- the reaction concentration on the basis of the iron concentration in the solution is in the range of 0.1 to 3.0 mol / L, and the addition amount of the phosphorus compound used for charging is such that the phosphorus ion is compared to the iron ion.
- This is the method for producing ferric phosphate hydrate particles according to the present invention 5, wherein the molar percentage is 100 to 1000% and the pH of the reaction solution is 3 or less (the present invention 6).
- the present invention also includes mixing the ferric phosphate hydrate particles powder according to any one of the present inventions 1 to 4, a lithium compound, and an organic compound at 300 to 800 ° C. in an inert atmosphere or a reducing atmosphere. It is a manufacturing method of the olivine type lithium iron phosphate particle powder to heat-process (this invention 7).
- the present invention is an olivine-type lithium iron phosphate particle powder for a non-aqueous electrolyte secondary battery obtained by the method for producing an olivine-type lithium iron phosphate particle powder according to the present invention 7 (Invention 8).
- the present invention is a non-aqueous electrolyte secondary battery using the olivine type lithium iron phosphate particle powder described in the present invention 8 (Invention 9).
- the method for producing ferric phosphate hydrate particles according to the present invention does not require a high-pressure vessel such as an autoclave, and can produce ferric phosphate hydrate particles at a low cost.
- the obtained ferric phosphate hydrate particles powder is a high purity crystalline ferric phosphate hydrate particles powder that is fine, has very few impurities, and has a small P / Fe ratio deviation.
- the ferric phosphate hydrate particles powder obtained in the present invention is an aggregate of ferric phosphate hydrate particles in which fine primary particles are aggregated, and is a high purity crystalline phosphoric acid having a high tap density.
- Ferric hydrate particles powder By using such a powder as a precursor, olivine-type lithium iron phosphate particle powder in which fine primary particles are aggregated can be produced inexpensively and easily.
- the olivine-type lithium iron phosphate particles according to the present invention are fine and have few impurities by using the precursor, and a high discharge capacity is obtained when a secondary battery is obtained, the non-aqueous electrolyte secondary battery Suitable as a positive electrode active material.
- ferric phosphate hydrate particles according to the present invention will be described.
- the composition of the ferric phosphate hydrate particles according to the present invention is represented by FePO 4 .nH 2 O (0 ⁇ n ⁇ 2) (n represents the amount of hydrated water), and dihydrate. Is the most stable. However, the amount of hydration water varies depending on the drying temperature, and the amount of hydration water is not a problem when used as a precursor of olivine type lithium iron phosphate.
- the ferric phosphate hydrate particles according to the present invention have a P / Fe molar ratio of 0.9 to 1.1. Since the deviation in the P / Fe molar ratio is directly reflected in the deviation in the P / Fe molar ratio of the olivine-type lithium iron phosphate particle powder, it is required to be controlled around 1.0 which is the theoretical composition. A more preferable P / Fe molar ratio is 0.95 to 1.05.
- the crystal structure of the ferric phosphate hydrate particles according to the present invention is one or more selected from a strength stone structure (JCPDS card No. 33-0667) and a metastrength stone structure (JCPDS card No. 33-0666). It is. These crystal structures have a P / Fe theoretical molar ratio of 1.0.
- the ferric phosphate hydrate particles according to the present invention preferably have a BET specific surface area in the range of 10 m 2 to 50 m 2 / g.
- a powder having a BET specific surface area of more than 50 m 2 / g is difficult to synthesize, and a powder having a BET specific surface area of less than 10 m 2 / g is too large as a precursor of olivine type lithium iron phosphate particles.
- a larger BET specific surface area is more preferable.
- a more preferable BET specific surface area is 10 to 40 m 2 / g.
- the average primary particle diameter of the ferric phosphate hydrate particles according to the present invention is preferably in the range of 50 to 1000 nm.
- a powder having an average primary particle diameter exceeding 1000 nm is too large as a precursor of olivine-type lithium iron phosphate particle powder, and a powder having an average primary particle diameter of less than 50 nm is difficult to synthesize.
- a more preferable average primary particle size is 150 to 500 nm.
- the average secondary particle size of the ferric phosphate hydrate particles according to the present invention is preferably in the range of 3 to 20 ⁇ m.
- ferric phosphate hydrate particles having an average secondary particle size of less than 3 ⁇ m are used as precursors, olivine-type lithium iron phosphate particles having a high tap density cannot be synthesized.
- ferric phosphate hydrate particles having an average secondary particle size of more than 20 ⁇ m are used as a precursor, the resulting olivine-type lithium iron phosphate particles are too large.
- the ferric phosphate hydrate particles according to the present invention preferably have a tap density of 0.4 to 1.5 g / cc.
- the tap density is less than 0.4 g / cc, the powder becomes bulky, and when applied to the electrode, the density of the positive electrode material in the coating film is difficult to increase.
- the tap density is preferably larger, more preferably 0.7 to 1.5 g / cc, and even more preferably 0.75 to 1.45 g / cc.
- the ferric phosphate hydrate particles according to the present invention have an extremely low Na content of 100 ppm or less.
- the Na content exceeds 100 ppm, the Na content of the olivine-type lithium iron phosphate obtained using the ferric phosphate hydrate particles is undesirably high.
- the residual of the unreacted iron oxide particle powder or hydrous iron oxide particle powder derived from the raw material is extremely small.
- the Na content is more preferably 70 ppm or less, and still more preferably 1 to 50 ppm.
- ferric phosphate hydrate particles according to the present invention use a raw material that does not substantially contain sulfur compound ions and nitrogen compound ions, they do not adsorb or contain such ions. In addition, no harmful gases such as SO X and NO X are generated during firing.
- the ferric phosphate hydrate particles according to the present invention comprise fine iron oxide particles having a BET specific surface area of 50 m 2 / g or hydrated iron oxide particles and a phosphorus compound at 60 to 100 ° C. in a solution. It can be obtained by reacting with stirring in the temperature range.
- iron oxide particle powder or a hydrous iron oxide particle powder having a BET specific surface area of less than 50 m 2 / g When an iron oxide particle powder or a hydrous iron oxide particle powder having a BET specific surface area of less than 50 m 2 / g is used, only a large ferric phosphate hydrate particle powder of the order of several ⁇ m is obtained. In addition, unreacted iron oxide particle powder or hydrous iron oxide particle powder tends to remain, causing the P / Fe molar ratio to deviate greatly from 1.0, which is the theoretical composition.
- an iron oxide particle powder or a hydrous iron oxide particle powder having a BET specific surface area of 50 m 2 / g or more a fine ferric phosphate hydrate particle powder having no P / Fe molar ratio deviation can be obtained. it can. More preferably, iron oxide particle powder or hydrous iron oxide particle powder having a BET specific surface area of 80 to 150 m 2 / g may be used.
- iron oxide particle powder or the hydrous iron oxide particle powder used in the present invention it is more preferable to use a fine goethite powder ( ⁇ -FeOOH) having a large BET specific surface area.
- ⁇ -FeOOH fine goethite powder
- Hydrous iron oxide powder powder generally has a spindle shape, needle shape, or rod shape.
- the primary particle diameter of the hydrous iron oxide powder used in the present invention is preferably an average major axis diameter of 50 to 200 nm.
- the Na content contained in the iron oxide particle powder or the hydrous iron oxide particle powder varies greatly depending on the production method and reaction conditions. Of course, when the amount of Na in the raw material is small, the possibility of Na being mixed into the ferric phosphate hydrate particles powder is reduced. The product particle powder cannot be manufactured. In the present invention, since the iron raw material is dissolved in the ferric phosphate hydrate particles after being dissolved in the acidic solution, Na can be eluted and removed in the filtrate. Alternatively, the Na content in the hydrous iron oxide particle powder is not limited, but is preferably about 1000 to 3000 ppm.
- the addition amount of the phosphorus compound is preferably in the range of 100 to 1000%, more preferably in the range of 100 to 600% in terms of mole percent with respect to the iron ions contained in the iron oxide particle powder or the hydrous iron oxide particle powder. It is.
- the phosphorus compound orthophosphoric acid, metaphosphoric acid, phosphorus pentoxide and the like can be preferably used.
- the reaction temperature in the solution is preferably 60 to 100 ° C.
- the temperature is lower than 60 ° C., an amorphous substance is by-produced as an impurity phase, and the P / Fe molar ratio becomes smaller than 1.0.
- it exceeds 100 ° C. it is difficult to say that it is industrial because a pressure vessel is required. More preferably, fine particles can be obtained by reacting in a temperature range of 60 to 80 ° C.
- the reaction pH in the solution is preferably pH 3 or less. When the pH exceeds 3, the iron oxide particle powder or the hydrous iron oxide particle powder does not sufficiently react, and the P / Fe molar ratio of the product becomes smaller than 1.0.
- an acidic solution such as sulfuric acid, hydrochloric acid or nitric acid can be used.
- the pH during the reaction affects the Na content and the content of impurity anions.
- Na can be eluted and removed in the filtrate, so that the Na content of the ferric phosphate hydrate particles can be reduced to 100 ppm or less, but the pH of the reaction solution is further controlled. As a result, the Na content can be reduced.
- the pH range of 1.5 to 2.5 is most preferable as a pH range with a low Na content and a small amount of impurity anions.
- the reaction concentration on the basis of the iron concentration of the solution is preferably in the range of 0.1 to 3.0 mol / L.
- the reaction concentration is lower than 0.1 mol / L, it is not preferable for industrially obtaining ferric phosphate hydrate particles, and when the reaction concentration exceeds 3.0 mol / L, the viscosity of the reaction slurry is It is high and uniform stirring is difficult.
- a more preferable reaction concentration is 0.1 to 1.0 mol / L on the basis of iron concentration.
- the inside of the reactor must be uniformly stirred.
- the stirring speed can be appropriately adjusted in order to control the aggregate particle diameter.
- the stirring speed varies depending on the shape and size of the stirring blade, but in the present invention, it is preferable to stir at a peripheral speed of 1.0 to 4.0 m / s and a stirring speed of 1-1000 rpm.
- the raw material iron oxide particle powder or hydrous iron oxide particle powder can be obtained relatively easily by a coprecipitation reaction from iron sulfate.
- the iron oxide particle powder or the hydrous iron oxide particle powder Prior to reaction in the solution, the iron oxide particle powder or the hydrous iron oxide particle powder is pulverized or crushed using a dry and wet mixer such as a Henschel mixer, rake machine, high speed mixer, universal stirrer, or ball mill. And mix with an aqueous solution containing a phosphorus compound.
- a dry and wet mixer such as a Henschel mixer, rake machine, high speed mixer, universal stirrer, or ball mill.
- excess water can be removed using a ventilation dryer, freeze vacuum dryer, spray dryer, filter press, vacuum filter, filter thickener or the like.
- the crystal structure of the ferric phosphate hydrate particles according to the present invention is at least one selected from a strength stone structure (JCPDS card No. 33-0667) and a metastrength stone structure (JCPDS card No. 33-0666).
- the ratio between the strengite structure and the metastrength structure varies and is influenced by a plurality of factors such as the raw material mixing ratio, reaction temperature, reaction pH, and reaction concentration.
- the ratio is evaluated by comparing the plane diffraction peak intensity with the (110) plane diffraction peak intensity derived from the metastrengthite-structured ferric phosphate hydrate particles.
- the (122) plane diffraction peak intensity derived from the strengthite structure / (110) plane diffraction peak intensity derived from the metastrength structure is defined as “(122) / (110) peak intensity ratio”
- the (122) / (110) peak intensity ratio of the ferric phosphate hydrate particles according to the present invention is preferably in the range of 0.01 to 10, more preferably 0.01 to 3.
- the strength stone structure is assumed to be the first phase.
- Particles having a strenite structure as the first phase tend to have a primary particle shape.
- the phosphorus compound is added in an amount of 100 to 100 in terms of mole percent with respect to the iron ions contained in the iron oxide particle powder or the hydrous iron oxide particle powder. A range of 120% is preferred.
- ferric phosphate hydrate particles according to the present invention when the strength stone structure is the first phase, ferric phosphate hydrate particles powder is applied by applying a strong shear force during the reaction. Is preferably made into fine particles.
- ferric phosphate hydrate particles according to the present invention In order to make the ferric phosphate hydrate particles according to the present invention into fine particles, it is preferable to stir at a peripheral speed of 2.5 to 3.2 m / s and a stirring speed of 800 to 1000 rpm.
- the average secondary particle size of the secondary particles (aggregates) of fine ferric phosphate hydrate particles having the first strength strengite structure according to the present invention is preferably in the range of 3 to 10 ⁇ m.
- a more preferable average secondary particle diameter is 5 to 8 ⁇ m.
- the metastrengthite structure is assumed to be the first phase. Particles having a metastrengite structure as the first phase tend to have a thin primary particle shape.
- the amount of phosphorus compound added is 120 to 120 in terms of mole percent with respect to the iron ions in which the phosphorus ions are contained in the iron oxide particle powder or the hydrous iron oxide particle powder. A range of 1000% is preferable, and a range of 200 to 600% is more preferable.
- ferric phosphate hydrate particles when the metastrengite structure is the first phase, the shear force applied during the reaction is suppressed, and the plate-like primary particles agglomerate densely. It is preferable to form an aggregate of the ferric phosphate hydrate particles constituting the secondary particles.
- stirring may be performed at a peripheral speed of 1.0 to 1.5 m / s and a stirring rotational speed of 150 to 350 rpm. preferable.
- the tap density is high by making the ferric phosphate hydrate particles according to the present invention an aggregate of ferric phosphate hydrate particles in which plate-like primary particles aggregate to form secondary particles. Ferric phosphate hydrate particles can be obtained.
- the average secondary particle diameter of secondary particles (aggregates) of ferric phosphate hydrate particles in which the metastrength structure according to the present invention is the first phase and the plate-like primary particles are aggregated is 5 to 20 ⁇ m. It is preferable that it exists in the range. A more preferable average secondary particle size is 8 to 18 ⁇ m.
- the aggregate powder of ferric phosphate hydrate particles in which the thin plate-like primary particles whose metastrengthite structure is the first phase according to the present invention is densely aggregated to form secondary particles has a tap density of 0. It is preferably 7 to 1.5 g / cc.
- the tap density is preferably larger, more preferably 0.7 to 1.45 g / cc, and even more preferably 0.75 to 1.45 g / cc.
- the method for producing olivine-type lithium iron phosphate particles according to the present invention comprises ferric phosphate hydrate particles according to the present invention, a lithium compound and an organic compound, uniformly pulverized and mixed dry or wet, and reduced. Heat treatment is performed at 300 to 800 ° C. in an atmosphere or an inert atmosphere.
- the apparatus there is no limitation on the apparatus as long as it can be uniformly pulverized and mixed, such as a ball mill, a vibration mill, a planetary ball mill, a paint shaker, a high-speed rotating blade type mill, a jet mill, etc.
- the amount of lithium compound added is preferably in the range of 100 to 120% in terms of mole percent with respect to the iron ions contained in the ferric phosphate hydrate particles.
- the lithium compound lithium hydroxide, lithium carbonate and the like are preferable.
- an organic compound is added for the purpose of reducing the organic compound during firing and the conductive carbonaceous material generated after firing is olivine-type lithium iron phosphate. It is to adhere to the particle powder surface.
- Organic compounds used are organic compounds that exhibit reducing properties during firing, such as saccharides such as monosaccharides, disaccharides, trisaccharides, and polysaccharides, fatty acids such as saturated fatty acids and unsaturated fatty acids, and resins such as polyethylene and polyvinyl alcohol. If it is a compound, there will be no restriction
- olivine-type lithium iron phosphate particle powder which is known to have poor conductivity, is used as a positive electrode material for a non-aqueous electrolyte secondary battery, attaching a conductive carbonaceous material to the surface of the powder can improve battery characteristics. It is effective to improve.
- the conductive carbonaceous material such as carbon black, ketjen black, or carbon fiber
- the conductive carbonaceous material is mixed in advance as an organic compound, and then an inert atmosphere or You may bake in a reducing atmosphere.
- Calcination may be performed after mixing an organic compound as a reducing agent and a conductive carbonaceous material as a conductive agent.
- the organic compound is preferably added in an amount of 5 wt% to 20 wt% with respect to the total weight of the ferric phosphate hydrate particles and the lithium compound according to the present invention.
- Calcination can be heat-treated in a gas flow type box muffle furnace, a gas flow type rotary furnace, a fluidized heat treatment furnace or the like.
- the inert atmosphere include nitrogen, helium, and argon
- examples of the reducing atmosphere include hydrogen and carbon monoxide.
- the heating and baking temperature is preferably 300 ° C to 800 ° C. If it is less than 300 ° C., the reduction reaction of iron ions does not proceed sufficiently, and a crystal phase other than olivine-type lithium iron phosphate remains, and if it exceeds 800 ° C., another crystal phase appears, which is not preferable.
- the olivine-type lithium iron phosphate particles according to the present invention preferably have a BET specific surface area of 10 m 2 to 100 m 2 / g.
- the olivine type lithium iron phosphate particle powder according to the present invention preferably contains 1 wt% to 10 wt% of carbon.
- the olivine-type lithium iron phosphate particle powder according to the present invention preferably has a Na content of 100 ppm or less.
- the average primary particle diameter of the olivine type lithium iron phosphate particles according to the present invention is preferably 50 to 300 nm.
- the olivine type lithium iron phosphate particles according to the present invention preferably have an average secondary particle size of 5 to 20 ⁇ m. A more preferable average secondary particle size is 8 to 18 ⁇ m.
- the olivine type lithium iron phosphate particle powder according to the present invention preferably has a tap density of 0.7 to 1.5 g / cc.
- a more preferable tap density is 0.75 to 1.45 g / cc, and still more preferably 0.8 to 1.45 g / cc.
- a conductive agent and a binder are added and mixed according to a conventional method.
- the conductive agent acetylene black, carbon black, graphite and the like are preferable
- the binder polytetrafluoroethylene, polyvinylidene fluoride and the like are preferable.
- a secondary battery manufactured using the olivine-type composite oxide particle powder according to the present invention includes the positive electrode, the negative electrode, and an electrolyte.
- lithium metal lithium metal, lithium / aluminum alloy, lithium / tin alloy, graphite, graphite or the like can be used.
- an organic solvent containing at least one of carbonates such as propylene carbonate and dimethyl carbonate and ethers such as dimethoxyethane can be used as the solvent for the electrolytic solution.
- At least one lithium salt such as lithium perchlorate and lithium tetrafluoroborate can be dissolved in the above solvent and used.
- the nonaqueous electrolyte secondary battery manufactured using the olivine-type lithium iron phosphate particles according to the present invention exhibits excellent charge / discharge characteristics with an initial discharge capacity of 150 to 165 mAh / g at a charge / discharge rate of C / 10. can do.
- the specific surface area is a specific surface area determined by a BET one-point continuous method using MONOSORB (manufactured by Yuasa Ionics Co., Ltd.) after drying and deaerating the sample under nitrogen gas at 110 ° C. for 45 minutes.
- MONOSORB manufactured by Yuasa Ionics Co., Ltd.
- Carbon amount was measured using a carbon sulfur analyzer (EMIA-820 manufactured by Horiba, Ltd.).
- the Na content and the P / Fe molar ratio were measured by dissolving the sample and using an emission plasma analyzer ICAP-6500 (manufactured by Thermo Fisher Scientific).
- the crystal structure of the particles was measured using an X-ray diffractometer RINT 2500 (manufactured by Rigaku Corporation) with Cu-K ⁇ , 40 kV, 300 mA.
- the crystallite size was calculated from the measured X-ray diffraction pattern by Rietveld analysis using an analysis program “Rietan-2000”.
- the average primary particle diameter was measured by Hitachi ultra-high resolution field emission scanning electron microscope S-4800 (average value of 50 points on the photograph).
- the tap density was determined by measuring the powder density after tapping 500 times using a tap denser (KYT-3000, manufactured by Seishin Enterprise Co., Ltd.).
- the initial charge / discharge characteristics and rate characteristics of the coin cell were evaluated using olivine type lithium iron phosphate particles.
- olivine type lithium iron phosphate particle powder as a positive electrode active material
- acetylene black as a conductive material
- polyvinylidene fluoride dissolved in N-methylpyrrolidone as a binder
- a CR2032-type coin cell was prepared by using metallic lithium punched to 16 mm ⁇ as a negative electrode and a solution obtained by mixing EC and DMC in which 1 mol / L LiPF 6 was dissolved in a volume ratio of 1: 2 as an electrolyte.
- the initial charge / discharge characteristics were measured at a constant temperature of 25 ° C., and charging was performed up to 4.3V and discharging was performed up to 2.0V.
- the rate characteristics were measured at 0.1 C and 5.0 C with a theoretical capacity of 170 mAh / g.
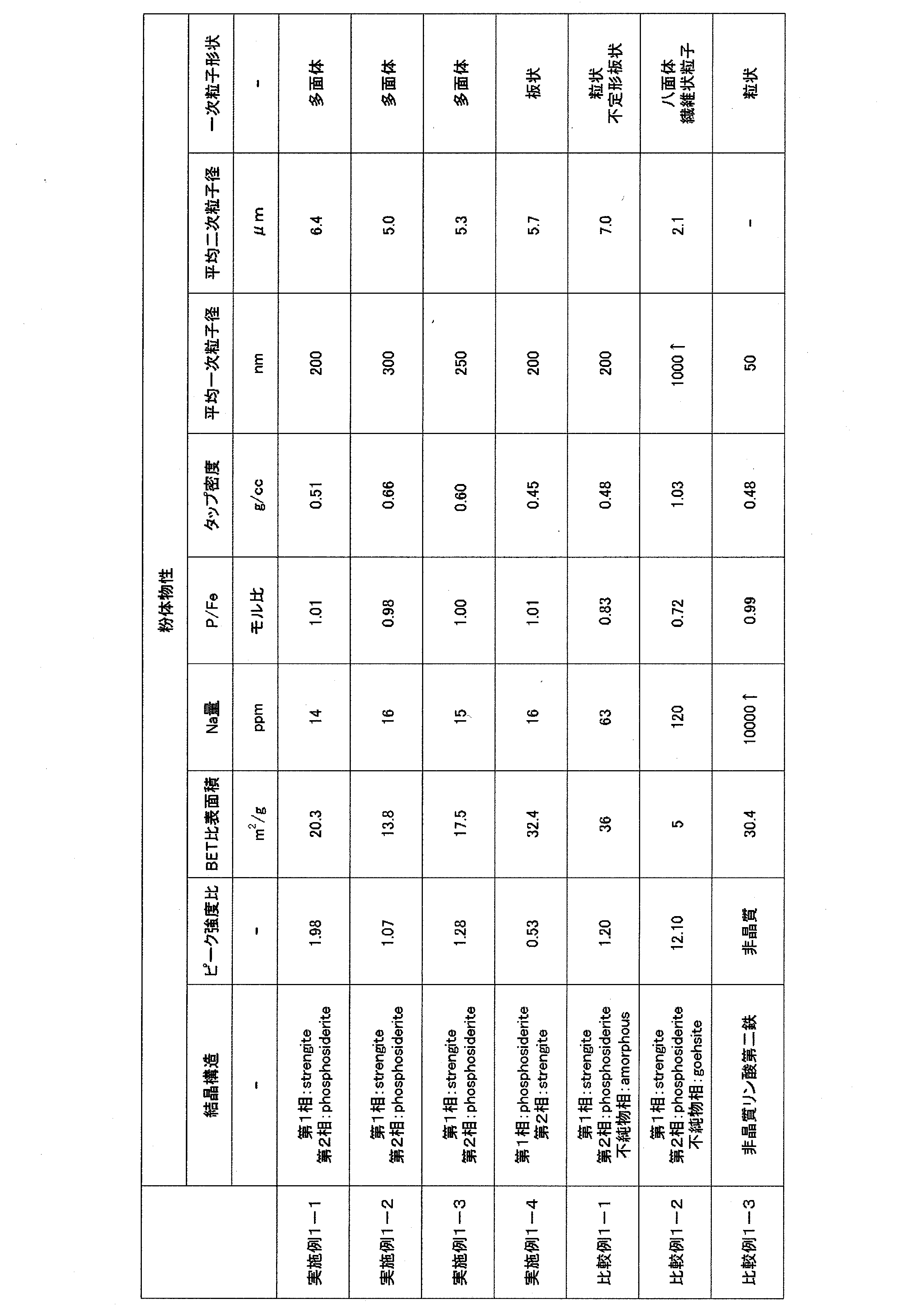
- Example 1-1 1147 g of a 25% slurry of hydrous iron oxide particles having a BET specific surface area of 98.5 m 2 / g was put into a heating type mixing stirrer, and the orthophosphoric acid solution was added while stirring so that the molar ratio of P / Fe was 1.1. Then, the amount of the mixed slurry was adjusted with ion-exchanged water so that the volume became 10 L, and the reaction concentration on the iron concentration basis was set to 0.3 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 2.8 m / s and a rotation speed of 900 rpm and heated to 60 ° C.
- the reaction was continued for 16 hours while maintaining the temperature in the mixing stirrer at 60 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a press filter, and then dried at 110 ° C. for 12 hours with a ventilator to obtain 555 g of dry powder.
- the BET specific surface area of the obtained powder was 20.3 m 2 / g, and the Na content was 14 ppm. Moreover, the P / Fe molar ratio of the obtained powder was 1.01, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the obtained powder was photographed with a scanning electron microscope, it was a polyhedral fine particle having an average primary particle diameter of 200 nm, and the secondary particle shape was indefinite.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 6.4 ⁇ m, and the tap density was 0.51 g / cc.
- a scanning electron micrograph of the obtained powder is shown in FIG. 1, and a powder X-ray diffraction diagram is shown in FIG.
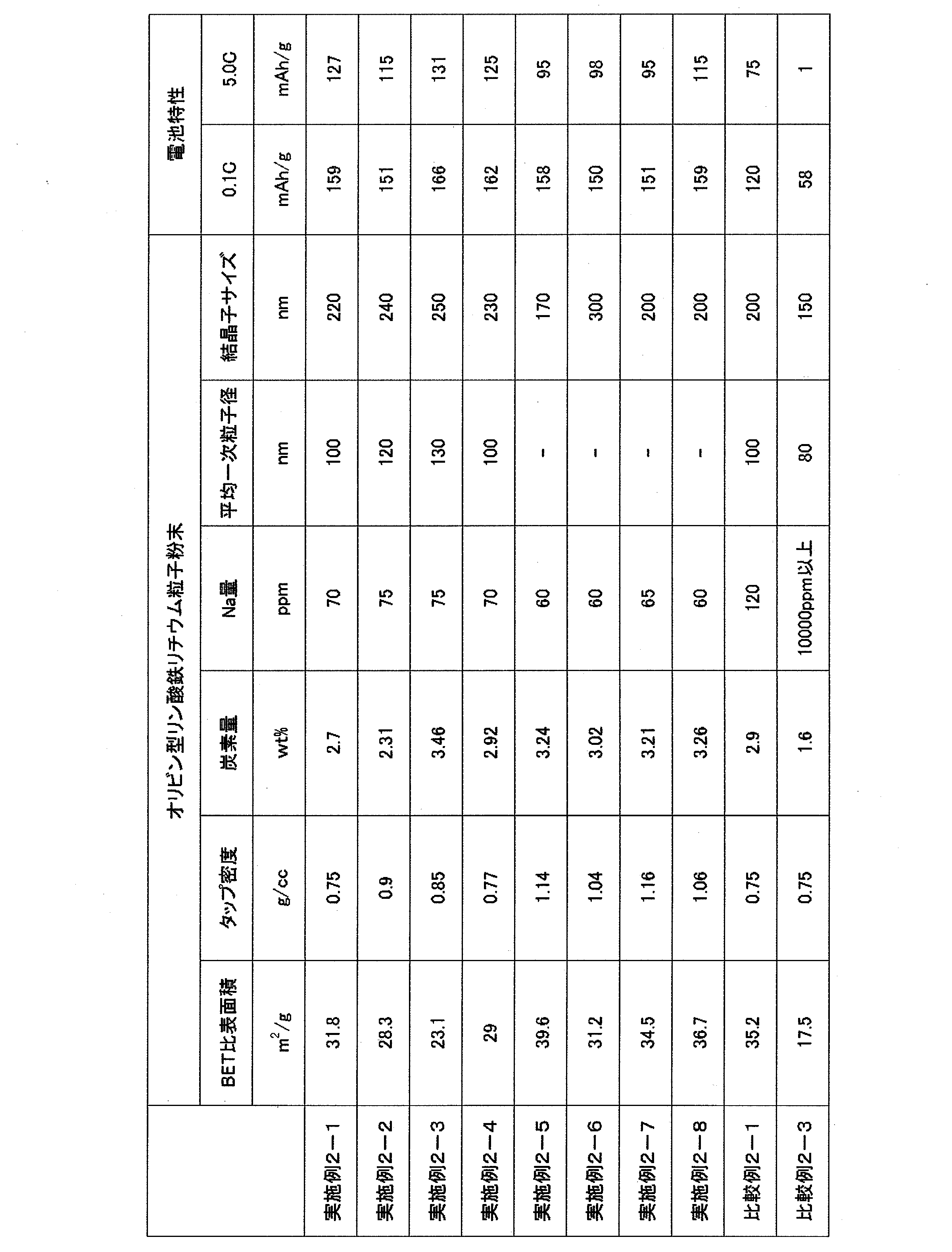
- Example 2-1> After putting 35 g of the ferric phosphate hydrate particles powder obtained in Example 1-1, 8.15 g of lithium hydroxide monohydrate, 4.32 g of sucrose and 120 mL of ethanol into a zirconia planetary ball mill pot, ⁇ 3 mm 450 g of zirconia beads were added, and wet mixing and pulverization were performed at 300 rpm for 2 hours. The slurry after the pulverization was solid-liquid separated with a Nutsche, and then dried with a dryer at 80 ° C. for 6 hours.
- the obtained dried product was loosened in a mortar, then calcined at 725 ° C. for 3 hours in a nitrogen atmosphere, and passed through a 75 ⁇ m sieve to obtain olivine-type lithium iron phosphate particle powder.
- the obtained fired product had a BET specific surface area of 31.8 m 2 / g, a tap density of 0.75 g / cc, a carbon content of 2.70 wt%, and an Na content of 70 ppm.
- the average primary particle size was as fine as 100 nm.
- the crystallite size determined by Rietveld analysis was around 220 nm.
- the secondary particle shape was indefinite.
- a scanning electron micrograph of the obtained lithium iron phosphate particle powder is shown in FIG.
- Example 1-2 A product was obtained in the same manner as in Example 1-1 except that the wet reaction temperature was changed from 60 ° C. to 80 ° C.
- the powder was ferric phosphate hydrate particles with the main phase being a compound corresponding to the strengite structure and the second phase being a compound corresponding to the metastrength structure. .
- the (122) / (110) peak intensity ratio of the obtained powder was 1.07. Moreover, the impurity phase was not confirmed.
- the BET specific surface area of the obtained powder was 13.8 m 2 / g, and the Na content was 16 ppm. Moreover, the P / Fe molar ratio of the obtained powder was 0.98, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- Scanning electron micrographs of the obtained powder were found to be polyhedral fine particles having an average primary particle size of 300 nm, and the secondary particle shape was irregular.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 5.0 ⁇ m, and the tap density was 0.66 g / cc.
- Example 2-2> After putting 35 g of the ferric phosphate hydrate particles powder obtained in Example 1-2, 8.15 g of lithium hydroxide monohydrate, 4.32 g of sucrose, and 120 mL of ethanol into a zirconia planetary ball mill pot, ⁇ 5 mm 300 g of zirconia beads were added, and wet mixing and pulverization were performed at 300 rpm for 4 hours. The slurry after the pulverization was solid-liquid separated with a Nutsche, and then dried with a dryer at 80 ° C. for 6 hours.
- the obtained dried product was loosened in a mortar, then calcined at 725 ° C. for 3 hours in a nitrogen atmosphere, and passed through a 75 ⁇ m sieve to obtain olivine-type lithium iron phosphate particle powder.
- the fired product obtained had a BET specific surface area of 28.3 m 2 / g, a tap density of 0.9 g / cc, a carbon content of 2.31 wt%, and an Na content of 75 ppm.
- the average primary particle size was as fine as 120 nm.
- the crystallite size determined by Rietveld analysis was around 240 nm.
- the secondary particle shape was indefinite.
- Example 1-3 A 25% slurry of hydrous iron oxide particles having a BET specific surface area of 120.0 m 2 / g was peptized using zirconia balls having a diameter of 5 mm for 12 hours. Next, 1147 g of this slurry was put into a heating type mixing stirrer, and while stirring, the orthophosphoric acid solution was added so that the molar ratio of P / Fe was 1.02, and the slurry mixed with ion-exchanged water so that the volume became 10 L The reaction concentration on the basis of iron concentration was adjusted to 0.3 mol / L.
- the stirring blade was rotated at a peripheral speed of 2.8 m / s and a rotation speed of 900 rpm and heated to 70 ° C. while stirring at high speed, and the reaction was continued for 18 hours while maintaining the temperature in the mixing stirrer at 70 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a press filter, and then dried at 110 ° C. for 12 hours with a ventilator to obtain 555 g of dry powder.
- the powder was ferric phosphate hydrate particles with the main phase being a compound corresponding to the strengite structure and the second phase being a compound corresponding to the metastrength structure. .
- the (122) / (110) peak intensity ratio of the obtained powder was 1.28. Moreover, the impurity phase was not confirmed.
- the BET specific surface area of the obtained powder was 17.5 m 2 / g, and the Na content was 15 ppm. Moreover, P / Fe molar ratio of the obtained powder was 1.00, and it was confirmed that the ferric phosphate hydrate particles powder according to the theoretical composition was obtained.
- Scanning electron micrographs of the obtained powder were found to be polyhedral fine particles having an average primary particle size of 250 nm, and the secondary particle shape was irregular.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 5.3 ⁇ m, and the tap density was 0.60 g / cc.
- Example 2-3 After putting 10 g of the ferric phosphate hydrate particles powder obtained in Example 1-3, 2.30 g of lithium hydroxide monohydrate, 1.23 g of sucrose, and 120 mL of ethanol into a polypropylene pot, zirconia with a diameter of 5 mm was obtained. 850 g of beads were added, and wet mixing / pulverization was performed at 200 rpm for 24 hours. The slurry after the pulverization was solid-liquid separated with a Nutsche, and then dried with a dryer at 80 ° C. for 6 hours.
- the obtained dried product was loosened in a mortar and then calcined at 725 ° C. for 3 hours in a nitrogen atmosphere to obtain olivine-type lithium iron phosphate particle powder.
- the obtained fired product had a BET specific surface area of 23.1 m 2 / g, a tap density of 0.85 g / cc, a carbon content of 3.46 wt%, and an Na content of 75 ppm.
- the average primary particle size was as small as 130 nm.
- the crystallite size determined by Rietveld analysis was around 250 nm.
- the secondary particle shape was indefinite.
- Example 1-4 3057 g of a 25% slurry of hydrous iron oxide particles having a BET specific surface area of 120.0 m 2 / g was put into a heating type mixing stirrer, and the orthophosphoric acid solution was added while stirring so that the molar ratio of P / Fe was 1.1. Then, the amount of the mixed slurry was adjusted with ion-exchanged water so that the volume became 10 L, and the reaction concentration on the iron concentration basis was set to 0.8 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 2.8 m / s and a rotation speed of 900 rpm and heated to 70 ° C.
- the reaction was carried out for 19 hours while maintaining the temperature in the mixing stirrer at 70 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a press filter, and then dried at 110 ° C. for 12 hours with a ventilating dryer to obtain 1490 g of dry powder.
- the powder was ferric phosphate hydrate particles with the main phase being a compound corresponding to the metastrengthite structure and the second phase being a compound corresponding to the strehlenite structure. .
- the (122) / (110) peak intensity ratio of the obtained powder was 0.53. Moreover, the impurity phase was not confirmed.
- the BET specific surface area of the obtained powder was 32.4 m 2 / g, and the Na content was 16 ppm. Moreover, the P / Fe molar ratio of the obtained powder was 1.01, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 5.7 ⁇ m, and the tap density was 0.45 g / cc.
- a scanning electron micrograph of the obtained powder is shown in FIG.
- Example 2-4 After putting 10 g of the ferric phosphate hydrate particles powder obtained in Example 1-4, 2.30 g of lithium hydroxide monohydrate, 1.23 g of sucrose, and 120 mL of ethanol into a polypropylene pot, 850 g of zirconia beads were added, and wet mixing and pulverization were performed at 200 rpm for 24 hours. The slurry after the pulverization was solid-liquid separated with a Nutsche, and then dried with a dryer at 80 ° C. for 6 hours.
- the obtained dried product was loosened in a mortar and then calcined at 725 ° C. for 3 hours in a nitrogen atmosphere to obtain olivine-type lithium iron phosphate particle powder.
- the obtained fired product had a BET specific surface area of 29.0 m 2 / g, a tap density of 0.77 g / cc, a carbon content of 2.92 wt%, and an Na content of 70 ppm.
- the average primary particle size was as fine as 100 nm.
- the crystallite size determined by Rietveld analysis was around 230 nm.
- the secondary particle shape was indefinite.
- ferric phosphate containing an amorphous phase as an impurity phase in addition to a compound that matches the strengite structure as the main phase and a compound that matches the metastrength structure as the second phase. It was a hydrate particle powder.
- the (122) / (110) peak intensity ratio of the obtained powder was 1.20.
- the BET specific surface area of the obtained powder was 36.0 m 2 / g, and the Na content was 63 ppm.
- the P / Fe molar ratio of the obtained powder was 0.83, which was found to deviate from the theoretical composition by about 17%.
- the obtained dried product was loosened in a mortar and then calcined at 725 ° C. for 3 hours in a nitrogen atmosphere to obtain olivine-type lithium iron phosphate particle powder.
- the fired product obtained had a BET specific surface area of 35.2 m 2 / g, a tap density of 0.75 g / cc, a carbon content of 2.9 wt%, and an Na content of 120 ppm.
- the average primary particle size was as fine as 100 nm.
- the crystallite size determined by Rietveld analysis was around 200 nm.
- ⁇ Comparative Example 1-2> Place 1056 g of 5.8% slurry of hydrous iron oxide particles having a BET specific surface area of 32.0 m 2 / g in a heating type mixing stirrer, and stir the orthophosphoric acid solution so that the P / Fe molar ratio becomes 1.1.
- the amount of the mixed slurry was adjusted with ion-exchanged water so that the volume was 10 L, and the reaction concentration based on the iron concentration was set to 0.3 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 2.8 m / s and a rotation speed of 900 rpm and heated to 80 ° C.
- the reaction was continued for 16 hours while maintaining the temperature in the mixing stirrer at 80 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a press filter, and then dried at 110 ° C. for 12 hours with a ventilator to obtain 550 g of dry powder.
- the obtained powder had a BET specific surface area of 5.0 m 2 / g and an Na content of 120 ppm. Moreover, it was found that the P / Fe molar ratio of the obtained powder was 0.72, which was shifted by about 28% from the theoretical composition.
- the obtained powder is deviated from P / Fe, when used as it is as a raw material for olivine-type lithium iron phosphate particles, a heterogeneous phase is generated after firing and the capacity is reduced. Moreover, since it was a large particle with an average primary particle diameter of 1 ⁇ m or more and was too large as a precursor of submicron order olivine type lithium iron phosphate particles, no firing was performed.
- Aldrich ferric phosphate dihydrate powder had a BET specific surface area of 30.4 m 2 / g and an Na content of over 10,000 ppm.
- the P / Fe molar ratio was 0.99, confirming that there was almost no deviation from the theoretical composition.
- FIG. 7 shows a scanning electron micrograph of Aldrich ferric phosphate dihydrate powder.
- the obtained fired product had a BET specific surface area of 17.5 m 2 / g, a tap density of 0.75 g / cc, a carbon content of 1.6 wt%, and a Na content of more than 10,000 ppm.
- the average primary particle size was as fine as 80 nm.
- the crystallite size determined by Rietveld analysis was around 150 nm.
- Table 1 shows the synthesis conditions of ferric phosphate hydrate particles obtained in Examples 1-1 to 1-4 and Comparative Examples 1-1 to 1-3, and Table 2 shows various properties.
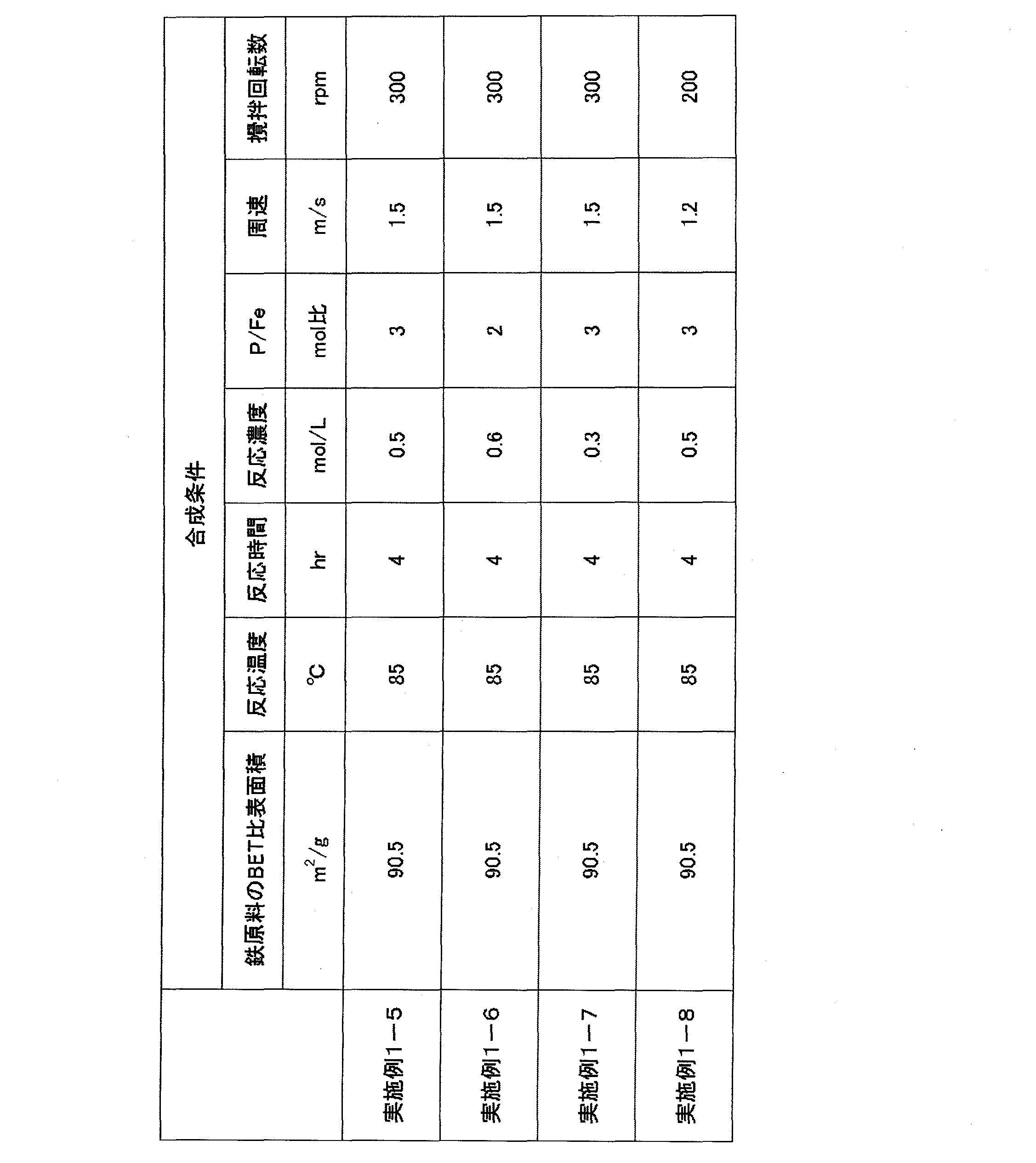
- Example 1-5 1176 g of a 6% slurry of hydrous iron oxide particles having a BET specific surface area of 90.5 m 2 / g was put into a heating type mixing stirrer, and the orthophosphoric acid solution was added while stirring so that the molar ratio of P / Fe was 3.0. Then, the liquid volume of the mixed slurry was adjusted with ion-exchanged water so that the volume became 1.5 L, and the reaction concentration based on the iron concentration was set to 0.5 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 1.5 m / s and a rotational speed of 300 rpm and heated to 85 ° C.
- the reaction was carried out for 4 hours while maintaining the temperature in the mixing stirrer at 85 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a Nutsche, and then subjected to a drying treatment at 110 ° C. for 12 hours with a ventilator to obtain 140 g of a dry powder.
- the obtained powder had a BET specific surface area of 31.5 m 2 / g and an Na content of 10 ppm or less, which was not detectable. Moreover, the P / Fe molar ratio of the obtained powder was 0.98, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the obtained powder When the obtained powder was photographed with a scanning electron microscope, it was a rounded square columnar secondary particle in which the thin plate-like primary particles were densely aggregated. Measurement of primary particle size was attempted from a scanning electron micrograph, but measurement of the plate surface direction was difficult because the plate surface of the primary particles grew in the center direction of the aggregated particles. Although the thickness direction could be measured, it grew to very thin plate-like particles of 50 nm or less.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 15.7 ⁇ m, and the tap density was 0.93 g / cc.
- a scanning electron micrograph of the obtained powder is shown in FIG. 8, and a powder X-ray diffraction diagram is shown in FIG.
- Example 2-5 35 g of the ferric phosphate hydrate particles obtained in Example 1-5, 8.15 g of lithium hydroxide monohydrate and 4.32 g of sucrose were mixed in a mortar, and then mixed at 725 ° C. under a nitrogen atmosphere.
- the olivine type lithium iron phosphate particle powder was obtained by baking for a period of time and passing through a 75 ⁇ m sieve.
- the fired product obtained had a BET specific surface area of 39.6 m 2 / g, a tap density of 1.14 g / cc, a carbon content of 3.24%, and an Na content of 60 ppm.
- the crystallite size determined by Rietveld analysis was 170 nm, and the secondary particle shape substantially maintained the aggregate shape of ferric phosphate hydrate particles as a precursor.
- a scanning electron micrograph of the obtained powder is shown in FIG. 10, and a powder X-ray diffraction diagram is shown in FIG.
- Example 1-6 1412 g of a 6% slurry of hydrous iron oxide particles having a BET specific surface area of 90.5 m 2 / g was put into a heating type mixing stirrer, and the orthophosphoric acid solution was added while stirring so that the molar ratio of P / Fe was 2.0. Then, the amount of the mixed slurry was adjusted with ion-exchanged water so that the volume was 1.5 L, and the reaction concentration on the iron concentration basis was 0.6 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 1.5 m / s and a rotational speed of 300 rpm and heated to 85 ° C.
- the reaction was carried out for 4 hours while maintaining the temperature in the mixing stirrer at 85 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a press filter, and then dried at 110 ° C. for 12 hours with a ventilating dryer to obtain 168 g of dry powder.
- the obtained powder had a BET specific surface area of 23.7 m 2 / g and an Na content of 10 ppm or less, which was not detectable. Moreover, the P / Fe molar ratio of the obtained powder was 0.96, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the obtained powder When the obtained powder was photographed with a scanning electron microscope, it was a rounded square columnar secondary particle in which the thin plate-like primary particles were densely aggregated. An attempt was made to measure the primary particle size from a scanning electron micrograph, but the measurement was difficult because the plate surface of the primary particles grew in the center direction of the aggregated particles. Although the thickness direction could be measured, it grew to very thin plate-like particles of 50 nm or less.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 9.8 ⁇ m, and the tap density was 0.80 g / cc.
- a scanning electron micrograph of the obtained powder is shown in FIG. 12, and a powder X-ray diffraction diagram is shown in FIG.
- Example 2-6 35 g of the ferric phosphate hydrate particles obtained in Example 1-6, 8.15 g of lithium hydroxide monohydrate and 4.32 g of sucrose were mixed in a mortar, and then mixed at 725 ° C. under a nitrogen atmosphere.
- the olivine type lithium iron phosphate particle powder was obtained by baking for a period of time and passing through a 75 ⁇ m sieve.
- the obtained fired product had a BET specific surface area of 31.2 m 2 / g, a tap density of 1.04 g / cc, a carbon content of 3.02%, and an Na content of 60 ppm.
- the crystallite size determined by Rietveld analysis was 300 nm, and the secondary particle shape substantially maintained the aggregate shape of ferric phosphate hydrate particles as a precursor.
- a scanning electron micrograph of the obtained powder is shown in FIG.
- Example 1-7 Put 1059 g of 4% slurry of hydrous iron oxide particles with a BET specific surface area of 90.5 m 2 / g in a heating type mixing stirrer and add the orthophosphoric acid solution with stirring so that the molar ratio of P / Fe is 3.0. Then, the amount of the mixed slurry was adjusted with ion-exchanged water so that the volume became 1.5 L, and the reaction concentration on the iron concentration basis was set to 0.3 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 1.5 m / s and a rotational speed of 300 rpm and heated to 85 ° C.
- the reaction was carried out for 4 hours while maintaining the temperature in the mixing stirrer at 85 ° C. .
- the slurry taken out was washed with 3 times the amount of water using a Nutsche, and then dried at 110 ° C. for 12 hours with a ventilated dryer to obtain 84 g of dry powder.
- the obtained powder had a BET specific surface area of 21.1 m 2 / g and an Na content of 10 ppm or less, which was not detectable. Moreover, the P / Fe molar ratio of the obtained powder was 0.97, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the obtained powder When the obtained powder was photographed with a scanning electron microscope, it was a rounded square columnar secondary particle in which the thin plate-like primary particles were densely aggregated. An attempt was made to measure the primary particle size from a scanning electron micrograph, but the measurement was difficult because the plate surface of the primary particles grew in the center direction of the aggregated particles. Although the thickness direction could be measured, it grew to very thin plate-like particles of 50 nm or less.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 13.3 ⁇ m, and the tap density was 0.95 g / cc.
- a scanning electron micrograph of the obtained powder is shown in FIG.
- Example 2-7 35 g of the ferric phosphate hydrate particles obtained in Example 1-7, 8.15 g of lithium hydroxide monohydrate, and 4.32 g of sucrose were mixed in a mortar, and then mixed at 725 ° C. under a nitrogen atmosphere.
- the olivine type lithium iron phosphate particle powder was obtained by baking for a period of time and passing through a 75 ⁇ m sieve.
- the obtained fired product had a BET specific surface area of 34.5 m 2 / g, a tap density of 1.16 g / cc, a carbon content of 3.21%, and an Na content of 65 ppm.
- the crystallite size determined by Rietveld analysis was 200 nm, and the secondary particle shape almost maintained the aggregate shape of ferric phosphate hydrate particles as a precursor.
- a scanning electron micrograph of the obtained powder is shown in FIG.
- Example 1-8 7842 g of 6% slurry of hydrous iron oxide particles having a BET specific surface area of 90.5 m 2 / g was put into a heating type mixing stirrer, and the orthophosphoric acid solution was added while stirring so that the molar ratio of P / Fe was 3.0. Then, the liquid volume of the mixed slurry was adjusted with ion-exchanged water so that the volume became 10 L, and the reaction concentration on the basis of iron concentration was set to 0.5 mol / L. Thereafter, the stirring blade was rotated at a peripheral speed of 1.2 m / s and a rotational speed of 200 rpm and heated to 85 ° C.
- the reaction was carried out for 4 hours while maintaining the temperature in the mixing stirrer at 85 ° C. .
- the taken-out slurry was washed with 3 times the amount of water using a press filter, and then subjected to a drying treatment at 110 ° C. for 12 hours with a ventilating dryer to obtain 934 g of a dry powder.
- the obtained powder had a BET specific surface area of 28.1 m 2 / g and an Na content of 10 ppm or less, which was not detectable. Moreover, the P / Fe molar ratio of the obtained powder was 1.01, and it was confirmed that a ferric phosphate hydrate particle powder having a P / Fe molar ratio very close to the theoretical composition was obtained.
- the obtained powder When the obtained powder was photographed with a scanning electron microscope, it was a rounded square columnar secondary particle in which the thin plate-like primary particles were densely aggregated. Measurement of primary particle size was attempted from a scanning electron micrograph, but measurement of the plate surface direction was difficult because the plate surface of the primary particles grew in the center direction of the aggregated particles. Although the thickness direction could be measured, it grew to very thin plate-like particles of 50 nm or less.
- the result of particle diameter measurement by laser diffraction method was an average secondary particle diameter of 10.6 ⁇ m, and the tap density was 0.83 g / cc.
- Example 2-8 35 g of the ferric phosphate hydrate particles obtained in Example 1-8, 8.15 g of lithium hydroxide monohydrate and 4.32 g of sucrose were mixed in a mortar, and then mixed at 725 ° C. under a nitrogen atmosphere.
- the olivine type lithium iron phosphate particle powder was obtained by baking for a period of time and passing through a 75 ⁇ m sieve.
- the fired product obtained had a BET specific surface area of 36.7 m 2 / g, a tap density of 1.06, a carbon content of 3.26%, and an Na content of 60 ppm.
- the crystallite size determined by Rietveld analysis was 200 nm, and the secondary particle shape almost maintained the aggregate shape of ferric phosphate hydrate particles as a precursor.
- Table 3 shows the synthesis conditions of ferric phosphate hydrate particles obtained in Examples 1-5 to 1-8, and Table 4 shows the various properties.
- the olivine-type lithium iron phosphate particle powder according to the present invention has a large charge / discharge capacity, excellent rate characteristics during charge / discharge, and is effective as an active material for non-aqueous electrolyte secondary batteries. It was.
- the ferric phosphate hydrate particles according to the present invention can be easily produced from inexpensive raw materials, and the resulting powder is fine and contains very few impurities. Therefore, the olivine-type lithium iron phosphate for non-aqueous electrolyte secondary batteries Suitable as a precursor of particle powder. Furthermore, since the ferric phosphate hydrate particles according to the present invention are secondary particles in which fine primary particles are aggregated, as a precursor of olivine-type lithium iron phosphate particles for non-aqueous electrolyte secondary batteries. Is preferred. Moreover, the positive electrode active material for nonaqueous electrolyte secondary batteries which exhibits the outstanding battery performance can be manufactured by using the ferric phosphate hydrate particle powder obtained by this invention as a raw material.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Crystallography & Structural Chemistry (AREA)
- Battery Electrode And Active Subsutance (AREA)
Abstract
Description
BET比表面積が98.5m2/gの含水酸化鉄粒子の25%スラリー1147gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が1.1になるように投入し、容積が10Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.3mol/Lとした。その後、攪拌羽根を周速2.8m/s、900rpmの回転数で回転させて高速攪拌しながら60℃に加熱し、混合攪拌機内の温度を60℃に保持させた状態で16時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末555gを得た。
実施例1-1で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gおよびエタノール120mLをジルコニア製遊星ボールミルポットに入れた後、φ3mmのジルコニアビーズ450gを加えて、300rpmで2時間湿式混合・粉砕処理を行った。粉砕処理後のスラリーをヌッチェで固液分離した後、80℃で6時間乾燥機で乾燥を行った。
湿式反応温度を60℃から80℃へ変更した以外は実施例1-1と同様に反応を行い、生成物を得た。
実施例1-2で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15gとスクロース4.32gおよびエタノール120mLをジルコニア製遊星ボールミルポットに入れた後、φ5mmのジルコニアビーズ300gを加えて、300rpmで4時間湿式混合・粉砕処理を行った。粉砕処理後のスラリーをヌッチェで固液分離した後、80℃で6時間乾燥機で乾燥を行った。
BET比表面積が120.0m2/gの含水酸化鉄粒子の25%スラリーを、φ5mmのジルコニアボールを用いて12時間解こうを行った。次に、このスラリー1147gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が1.02になるように投入し、容積が10Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.3mol/Lとした。その後、攪拌羽根を周速2.8m/s、900rpmの回転数で回転させて高速攪拌しながら70℃に加熱し、混合攪拌機内の温度を70℃に保持させた状態で18時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末555gを得た。
実施例1-3で得られたリン酸第二鉄含水物粒子粉末10gと水酸化リチウム一水和物2.30g、スクロース1.23gおよびエタノール120mLをポリプロピレン製ポットに入れた後、φ5mmのジルコニアビーズ850gを加えて、200rpmで24時間湿式混合・粉砕処理を行った。粉砕処理後のスラリーをヌッチェで固液分離した後、80℃で6時間乾燥機で乾燥を行った。
BET比表面積が120.0m2/gの含水酸化鉄粒子の25%スラリー3057gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が1.1になるように投入し、容積が10Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.8mol/Lとした。その後、攪拌羽根を周速2.8m/s、900rpmの回転数で回転させて高速攪拌しながら70℃に加熱し、混合攪拌機内の温度を70℃に保持させた状態で19時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末1490gを得た。
実施例1-4で得られたリン酸第二鉄含水物粒子粉末10gと水酸化リチウム一水和物2.30g、スクロース1.23g、およびエタノール120mLをポリプロピレン製ポットに入れた後、φ5mmのジルコニアビーズ850gを加えて、200rpmで24時間湿式混合・粉砕処理を行った。粉砕処理後のスラリーをヌッチェで固液分離した後、80℃で6時間乾燥機で乾燥を行った。
湿式反応温度を60℃から50℃へ変更した以外は実施例1-1と同様に反応を行い、生成物を得た。
比較例1-1で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gおよびエタノール120mLをジルコニア製遊星ボールミルポットに入れた後、3mmのジルコニアビーズ450gを加えて、300rpmで6時間湿式混合・粉砕処理を行った。粉砕処理後のスラリーをヌッチェで固液分離した後、80℃で6時間乾燥機で乾燥を行った。
BET比表面積が32.0m2/gの含水酸化鉄粒子の5.8%スラリー1056gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が1.1になるように投入し、容積が10Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.3mol/Lとした。その後、攪拌羽根を周速2.8m/s、900rpmの回転数で回転させて高速攪拌しながら80℃に加熱し、混合攪拌機内の温度を80℃に保持させた状態で16時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末550gを得た。
リン酸第二鉄含水物粒子粉末として、市販されているAldrich製リン酸第二鉄二水和物粉末を用いた。
このAldrich製リン酸第二鉄二水和物粉末を用いた以外は実施例2-1と同様の条件で焼成を行い、焼成物を得た。

BET比表面積が90.5m2/gの含水酸化鉄粒子の6%スラリー1176gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が3.0になるように投入し、容積が1.5Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.5mol/Lとした。その後、攪拌羽根を周速1.5m/s、300rpmの回転数で回転させて低速攪拌しながら85℃に加熱し、混合攪拌機内の温度を85℃に保持させた状態で4時間反応させた。反応後、取り出したスラリーを、ヌッチェを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末140gを得た。
実施例1-5で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gを乳鉢で混合した後、窒素雰囲気下、725℃で5時間焼成し、75μmの篩を通すことによってオリビン型リン酸鉄リチウム粒子粉末を得た。
BET比表面積が90.5m2/gの含水酸化鉄粒子の6%スラリー1412gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が2.0になるように投入し、容積が1.5Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.6mol/Lとした。その後、攪拌羽根を周速1.5m/s、300rpmの回転数で回転させて低速攪拌しながら85℃に加熱し、混合攪拌機内の温度を85℃に保持させた状態で4時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末168gを得た。
実施例1-6で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gを乳鉢で混合した後、窒素雰囲気下、725℃で5時間焼成し、75μmの篩を通すことによってオリビン型リン酸鉄リチウム粒子粉末を得た。
BET比表面積が90.5m2/gの含水酸化鉄粒子の4%スラリー1059gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が3.0になるように投入し、容積が1.5Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.3mol/Lとした。その後、攪拌羽根を周速1.5m/s、300rpmの回転数で回転させて低速攪拌しながら85℃に加熱し、混合攪拌機内の温度を85℃に保持させた状態で4時間反応させた。反応後、取り出したスラリーを、ヌッチェを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末84gを得た。
実施例1-7で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gを乳鉢で混合した後、窒素雰囲気下、725℃で5時間焼成し、75μmの篩を通すことによってオリビン型リン酸鉄リチウム粒子粉末を得た。
BET比表面積が90.5m2/gの含水酸化鉄粒子の6%スラリー7842gを加熱式混合攪拌機に入れ、攪拌しながらオルトリン酸溶液をP/Feのモル比が3.0になるように投入し、容積が10Lとなるようイオン交換水で混合スラリーの液量を調整して鉄濃度基準での反応濃度を0.5mol/Lとした。その後、攪拌羽根を周速1.2m/s、200rpmの回転数で回転させて低速攪拌しながら85℃に加熱し、混合攪拌機内の温度を85℃に保持させた状態で4時間反応させた。反応後、取り出したスラリーを、プレスフィルターを用いて3倍量の水で水洗を行った後、110℃で12時間通風式乾燥機で乾燥処理を行い、乾燥粉末934gを得た。
実施例1-8で得られたリン酸第二鉄含水物粒子粉末35gと水酸化リチウム一水和物8.15g、スクロース4.32gを乳鉢で混合した後、窒素雰囲気下、725℃で5時間焼成し、75μmの篩を通すことによってオリビン型リン酸鉄リチウム粒子粉末を得た。

Claims (9)
- オリビン型リン酸鉄リチウム粒子粉末の前駆体であり、Na含有量が100ppm以下であり、リン/鉄のモル比が0.9~1.1であるリン酸第二鉄含水物粒子粉末。
- リン酸第二鉄含水物粒子粉末の結晶構造がストレング石構造およびメタストレング石構造のうち少なくとも一種である請求項1に記載のリン酸第二鉄含水物粒子粉末。
- リン酸第二鉄含水物粒子粉末が板状の一次粒子が凝集することによって形成される二次粒子であり、平均二次粒子径が5~20μmである請求項1または2に記載のリン酸第二鉄含水物粒子粉末。
- タップ密度が0.7~1.5g/ccである請求項1~3のいずれかに記載のリン酸第二鉄含水物粒子粉末。
- 酸化鉄粒子粉末又は含水酸化鉄粒子粉末とリン化合物とを溶液中で反応して請求項1~4のいずれかに記載のリン酸第二鉄含水物粒子粉末を製造する方法において、BET比表面積が50m2/g以上の酸化鉄粒子粉末又は含水酸化鉄粒子粉末を鉄原料として用い、かつ、60~100℃の温度領域で反応を行うことを特徴とするリン酸第二鉄含水物粒子粉末の製造法。
- 溶液中の鉄濃度基準での反応濃度が0.1~3.0mol/Lの範囲であり、かつ、仕込みに使用するリン化合物の添加量が、リンイオンが鉄イオンに対してモルパーセント換算で100~1000%であり、反応溶液のpHが3以下である請求項5に記載のリン酸第二鉄含水物粒子粉末の製造法。
- 請求項1~4のいずれかに記載のリン酸第二鉄含水物粒子粉末とリチウム化合物と有機化合物とを混合し、不活性雰囲気下または還元雰囲気下300~800℃で熱処理するオリビン型リン酸鉄リチウム粒子粉末の製造法。
- 請求項7に記載のオリビン型リン酸鉄リチウム粒子粉末の製造法によって得られる非水電解質二次電池用オリビン型リン酸鉄リチウム粒子粉末。
- 請求項8に記載のオリビン型リン酸鉄リチウム粒子粉末を使用した非水電解質二次電池。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10815381.8A EP2476647B1 (en) | 2009-09-09 | 2010-09-08 | Process for production of olivine-type lithium iron phosphate particle powder |
| US13/394,990 US20120237425A1 (en) | 2009-09-09 | 2010-09-08 | Ferric phosphate hydrate particles and process for producing the same, olivine type lithium iron phosphate particles and process for producing the same, and non-aqueous electrolyte secondary battery |
| CN2010800398095A CN102612487A (zh) | 2009-09-09 | 2010-09-08 | 磷酸铁水合物颗粒粉末及其制造方法、橄榄石型磷酸铁锂颗粒粉末及其制造方法以及非水电解质二次电池 |
| KR1020127005889A KR101718918B1 (ko) | 2009-09-09 | 2010-09-08 | 인산제2철 함수물 입자 분말 및 그의 제조법, 올리빈형 인산철리튬 입자 분말 및 그의 제조법 및 비수전해질 이차 전지 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009208404 | 2009-09-09 | ||
| JP2009-208404 | 2009-09-09 | ||
| JP2010-125084 | 2010-05-31 | ||
| JP2010125084 | 2010-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011030786A1 true WO2011030786A1 (ja) | 2011-03-17 |
Family
ID=43732455
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/065404 Ceased WO2011030786A1 (ja) | 2009-09-09 | 2010-09-08 | リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20120237425A1 (ja) |
| EP (1) | EP2476647B1 (ja) |
| JP (1) | JP5835540B2 (ja) |
| KR (1) | KR101718918B1 (ja) |
| CN (1) | CN102612487A (ja) |
| TW (1) | TW201124337A (ja) |
| WO (1) | WO2011030786A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012012279A (ja) * | 2009-09-09 | 2012-01-19 | Toda Kogyo Corp | リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池。 |
| JP2013505193A (ja) * | 2009-09-18 | 2013-02-14 | エー123 システムズ, インコーポレイテッド | リン酸第二鉄およびその調製の方法 |
| JP2013032257A (ja) * | 2011-06-28 | 2013-02-14 | Nichia Corp | オリビン型リチウム遷移金属酸化物及びその製造方法 |
| JP2014088283A (ja) * | 2012-10-30 | 2014-05-15 | Rin Kagaku Kogyo Kk | リン酸第二鉄含水和物粒子粉末及びその製造方法 |
| JP2014088282A (ja) * | 2012-10-30 | 2014-05-15 | Rin Kagaku Kogyo Kk | リン酸第二鉄含水和物粒子粉末およびその製造方法 |
| US20140147586A1 (en) * | 2012-11-27 | 2014-05-29 | Universite De Montreal | Process for making an alkali metal oxyanion comprising iron |
| CN103864044A (zh) * | 2014-03-10 | 2014-06-18 | 瓮福(集团)有限责任公司 | 利用微波法将磷铁转化为电池级磷酸铁的方法 |
| US9660267B2 (en) | 2009-09-18 | 2017-05-23 | A123 Systems, LLC | High power electrode materials |
| JP2018018828A (ja) * | 2010-04-28 | 2018-02-01 | 株式会社半導体エネルギー研究所 | 二次電池用電極 |
| JP2019061917A (ja) * | 2017-09-28 | 2019-04-18 | 住友大阪セメント株式会社 | リチウムイオン二次電池用正極材料およびその製造方法、リチウムイオン二次電池用正極、リチウムイオン二次電池 |
| CN109850862A (zh) * | 2019-04-23 | 2019-06-07 | 王柯娜 | 一种电池级无水磷酸铁的制备方法 |
| CN116216681A (zh) * | 2022-12-28 | 2023-06-06 | 湖北虹润高科新材料有限公司 | 一种碱式磷酸铁铵的制备方法 |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5589338B2 (ja) * | 2009-10-09 | 2014-09-17 | 東洋インキScホールディングス株式会社 | リチウム二次電池用正極活物質材料の製造方法、及びそれを用いたリチウム二次電池 |
| JP5776573B2 (ja) * | 2012-02-06 | 2015-09-09 | 住友金属鉱山株式会社 | リチウム二次電池用正極活物質とその製造方法および該正極活物質の前駆体とその製造方法、並びに該正極活物質を用いたリチウム二次電池 |
| KR20140021843A (ko) * | 2012-08-10 | 2014-02-21 | 삼성정밀화학 주식회사 | 나노사이즈 철인산염 입자의 제조 방법 |
| KR101973052B1 (ko) * | 2012-08-10 | 2019-04-26 | 삼성에스디아이 주식회사 | 리튬 금속인산화물의 제조방법 |
| CN102842717B (zh) * | 2012-09-26 | 2014-09-17 | 河北工业大学 | 自组装纺锤体形纳米结构磷酸铁锂的制备方法 |
| US9660266B2 (en) | 2012-11-21 | 2017-05-23 | Lg Chem, Ltd. | Lithium secondary battery |
| WO2014081221A1 (ko) * | 2012-11-21 | 2014-05-30 | 주식회사 엘지화학 | 리튬 이차전지 |
| EP2778126A1 (en) * | 2013-03-15 | 2014-09-17 | Clariant International Ltd. | Lithium transition metal phosphate secondary agglomerates and process for its manufacture |
| WO2014180334A1 (zh) | 2013-05-08 | 2014-11-13 | 台湾立凯电能科技股份有限公司 | 电池复合材料及其前驱物的制备方法 |
| KR102062705B1 (ko) * | 2013-06-14 | 2020-01-06 | 삼성에스디아이 주식회사 | 리튬 금속인산화물의 제조방법 |
| CN107601447A (zh) * | 2016-07-12 | 2018-01-19 | 南通亨利锂电新材料有限公司 | 一种复合晶相正磷酸铁及其制备方法 |
| CN106450295B (zh) * | 2016-09-14 | 2019-10-18 | 上海电力学院 | 一种钠离子电池正极材料Na3Fe2(PO4)3及其制备方法 |
| JP7035054B2 (ja) * | 2016-12-15 | 2022-03-14 | ハイドロ-ケベック | 炭素の添加による炭素非含有カンラン石の脱リチウム化 |
| CN112384477A (zh) * | 2018-06-21 | 2021-02-19 | 学校法人工学院大学 | 氧化铁粉末、组合物、陶瓷器、氧化铁粉末前体、氧化铁粉末前体的制造方法及氧化铁粉末的制造方法 |
| CN109305663A (zh) * | 2018-08-15 | 2019-02-05 | 湖南鸿跃电池材料有限公司 | 电池级无水磷酸铁及其制备方法 |
| KR102713210B1 (ko) | 2019-02-13 | 2024-10-02 | 주식회사 엘지에너지솔루션 | 괴타이트를 포함하는 리튬 이차전지용 양극 및 이를 구비한 리튬 이차전지 |
| GB201905177D0 (en) * | 2019-04-11 | 2019-05-29 | Johnson Matthey Plc | Lithium metal phosphate, its preparation and use |
| KR102737304B1 (ko) * | 2019-12-16 | 2024-12-02 | 주식회사 엘지에너지솔루션 | 양극재 수명 예측방법, 수명 특성이 우수한 양극재를 포함하는 양극 합제, 양극 및 이를 포함하는 이차전지 |
| CN112838206B (zh) * | 2020-12-31 | 2022-06-03 | 福建师范大学 | 一类空气稳定性优异的层状氧化物正极材料以及通过调节钠含量改善空气稳定性的方法 |
| CN113479861B (zh) * | 2021-07-01 | 2023-02-14 | 广东邦普循环科技有限公司 | 低硫含量纳米磷酸铁的制备方法 |
| CN113979417B (zh) * | 2021-11-30 | 2023-06-16 | 中钢天源股份有限公司 | 一种硫酸体系中低硫高纯磷酸铁的制备方法 |
| CN115974031A (zh) * | 2022-10-11 | 2023-04-18 | 贝特瑞(天津)纳米材料制造有限公司 | 高压实磷酸铁锂正极材料及其制备方法和锂离子电池 |
| KR20260032587A (ko) * | 2023-07-12 | 2026-03-09 | 테슬라, 인크. | 활성 물질 입자, 이의 제조 공정, 및 이의 제조에 유용한 장치 |
| CN119517946B (zh) * | 2023-08-23 | 2026-01-27 | 宁德时代新能源科技股份有限公司 | 一种正极活性材料、正极极片、二次电池和用电装置 |
| CN121666360A (zh) * | 2023-09-07 | 2026-03-13 | 太阳化学公司 | 包含磷酸铁的材料 |
| CN117623255A (zh) * | 2023-10-19 | 2024-03-01 | 贵州雅友新材料有限公司 | 一种片状的磷酸铁的制备方法 |
| CN117602602A (zh) * | 2023-11-23 | 2024-02-27 | 贵州中伟兴阳储能科技有限公司 | 一种无水磷酸铁及其制备方法 |
| CN118545688B (zh) * | 2024-07-24 | 2024-11-12 | 广东邦普循环科技有限公司 | 一种磷酸铁材料及其制备方法与应用 |
| CN118867196B (zh) * | 2024-08-16 | 2026-04-03 | 内蒙古大学 | 用于钠离子电池的铁基磷酸盐复合正极材料及其制备方法 |
| CN119954118A (zh) * | 2025-01-26 | 2025-05-09 | 湖北虹润高科新材料有限公司 | 一种磷酸铁、其制备方法和生产装置及磷酸铁锂正极材料 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001053198A1 (en) | 2000-01-18 | 2001-07-26 | Valence Technology, Inc. | Preparation of lithium-containing materials |
| JP2003292307A (ja) | 2002-01-31 | 2003-10-15 | Nippon Chem Ind Co Ltd | リン酸第一鉄含水塩結晶、その製造方法及びリチウム鉄リン系複合酸化物の製造方法 |
| JP2004509447A (ja) | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | 制御されたサイズを持つ炭素によって被覆された、酸化還元物質の合成方法 |
| JP2004095386A (ja) | 2002-08-30 | 2004-03-25 | Sumitomo Osaka Cement Co Ltd | リチウムイオン電池用正極材料の製造方法およびリチウムイオン電池 |
| WO2005041327A1 (ja) | 2003-10-27 | 2005-05-06 | Mitsui Engineering & Shipbuilding Co.,Ltd. | 二次電池用正極材料、二次電池用正極材料の製造方法、および二次電池 |
| JP2006032241A (ja) | 2004-07-21 | 2006-02-02 | Mitsui Mining Co Ltd | リチウムイオン二次電池用正極材料、その製造方法、及びリチウムイオン二次電池 |
| WO2007000251A1 (en) | 2005-06-29 | 2007-01-04 | Umicore | Crystalline nanometric lifepo4 |
| JP2007511458A (ja) | 2003-11-14 | 2007-05-10 | ジュート−ヒェミー アクチェンゲゼルシャフト | リン酸鉄リチウム、その製造方法及び電極剤としてのそれの使用 |
| JP2009266813A (ja) * | 2008-03-31 | 2009-11-12 | Toda Kogyo Corp | 非水電解質二次電池用オリビン型複合酸化物粒子粉末及びその製造方法、並びに二次電池 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6140001A (en) * | 1999-05-04 | 2000-10-31 | Mitsui Mining & Smelting Co., Ltd. | Iron oxide microparticles and a process for producing them |
| US6777132B2 (en) * | 2000-04-27 | 2004-08-17 | Valence Technology, Inc. | Alkali/transition metal halo—and hydroxy-phosphates and related electrode active materials |
| KR101061702B1 (ko) * | 2002-10-18 | 2011-09-01 | 미쯔이 죠센 가부시키가이샤 | 리튬 전지용 양극 재료의 제조방법 및 리튬 전지 |
| KR100790280B1 (ko) * | 2004-03-30 | 2008-01-02 | 마쯔시다덴기산교 가부시키가이샤 | 비수전해액 2차 전지 |
| CA2622675A1 (en) * | 2007-02-28 | 2008-08-28 | Sanyo Electric Co., Ltd. | Method of producing active material for lithium secondary battery, method of producing electrode for lithium secondary battery, method of producing lithium secondary battery, and method of monitoring quality of active material for lithium secondary battery |
| WO2008145034A1 (en) * | 2007-05-28 | 2008-12-04 | Byd Company Limited | Method for preparing lithium iron phosphate as a positive electrode active material for a lithium ion secondary battery |
| DE102007049757A1 (de) * | 2007-10-16 | 2009-04-23 | Chemische Fabrik Budenheim Kg | Eisen(III)orthophosphat für Li-Ionen-Akkumulatoren |
| CN100595949C (zh) * | 2008-02-26 | 2010-03-24 | 郑州瑞普生物工程有限公司 | 用于锂电池正极材料正磷酸铁的制备方法 |
| DE102009001204A1 (de) * | 2009-02-26 | 2010-09-02 | Chemische Fabrik Budenheim Kg | Herstellung von Eisenorthophosphat |
| US9682861B2 (en) * | 2009-05-04 | 2017-06-20 | Meecotech, Inc. | Electrode active composite materials and methods of making thereof |
| WO2011030786A1 (ja) * | 2009-09-09 | 2011-03-17 | 戸田工業株式会社 | リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池 |
-
2010
- 2010-09-08 WO PCT/JP2010/065404 patent/WO2011030786A1/ja not_active Ceased
- 2010-09-08 KR KR1020127005889A patent/KR101718918B1/ko active Active
- 2010-09-08 JP JP2010201001A patent/JP5835540B2/ja active Active
- 2010-09-08 EP EP10815381.8A patent/EP2476647B1/en active Active
- 2010-09-08 TW TW099130317A patent/TW201124337A/zh unknown
- 2010-09-08 CN CN2010800398095A patent/CN102612487A/zh active Pending
- 2010-09-08 US US13/394,990 patent/US20120237425A1/en not_active Abandoned
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001053198A1 (en) | 2000-01-18 | 2001-07-26 | Valence Technology, Inc. | Preparation of lithium-containing materials |
| JP2004509447A (ja) | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | 制御されたサイズを持つ炭素によって被覆された、酸化還元物質の合成方法 |
| JP2004509058A (ja) | 2000-09-26 | 2004-03-25 | ハイドロ−ケベック | Lixm1−ym’y(xo4)nを主成分とする物質の合成法 |
| JP2003292307A (ja) | 2002-01-31 | 2003-10-15 | Nippon Chem Ind Co Ltd | リン酸第一鉄含水塩結晶、その製造方法及びリチウム鉄リン系複合酸化物の製造方法 |
| JP2004095386A (ja) | 2002-08-30 | 2004-03-25 | Sumitomo Osaka Cement Co Ltd | リチウムイオン電池用正極材料の製造方法およびリチウムイオン電池 |
| WO2005041327A1 (ja) | 2003-10-27 | 2005-05-06 | Mitsui Engineering & Shipbuilding Co.,Ltd. | 二次電池用正極材料、二次電池用正極材料の製造方法、および二次電池 |
| JP2007511458A (ja) | 2003-11-14 | 2007-05-10 | ジュート−ヒェミー アクチェンゲゼルシャフト | リン酸鉄リチウム、その製造方法及び電極剤としてのそれの使用 |
| JP2006032241A (ja) | 2004-07-21 | 2006-02-02 | Mitsui Mining Co Ltd | リチウムイオン二次電池用正極材料、その製造方法、及びリチウムイオン二次電池 |
| WO2007000251A1 (en) | 2005-06-29 | 2007-01-04 | Umicore | Crystalline nanometric lifepo4 |
| JP2009266813A (ja) * | 2008-03-31 | 2009-11-12 | Toda Kogyo Corp | 非水電解質二次電池用オリビン型複合酸化物粒子粉末及びその製造方法、並びに二次電池 |
Non-Patent Citations (5)
| Title |
|---|
| CHARLES DELACOURT ET AL.: "Chemistry of Materials", vol. 15, 2003, AMERICAN CHEMICAL SOCIETY, pages: 5051 - 5058 |
| REALE, P. ET AL.: "Synthesis and Thermal Behavior of Crystalline Hydrated Iron(III) Phosphates of Interest as Positive Electrodes", LI BATTERIES, CHEM. MATER., vol. 15, no. 26, 2003, pages 5051 - 5058, XP002523335 * |
| See also references of EP2476647A4 |
| SONG, Y. ET AL.: "New Iron(III) Phosphate Phases: Crystal Structure and Electrochemical and Magnetic Properties", INORG. CHEM., vol. 41, no. 22, 2002, pages 5778 - 5786, XP008152430 * |
| YANNING SONG ET AL.: "Inorganic Chemistry", vol. 41, 2002, AMERICAN CHEMICAL SOCIETY, pages: 5778 - 5786 |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012012279A (ja) * | 2009-09-09 | 2012-01-19 | Toda Kogyo Corp | リン酸第二鉄含水物粒子粉末及びその製造法、オリビン型リン酸鉄リチウム粒子粉末及びその製造法、並びに非水電解質二次電池。 |
| US10522833B2 (en) | 2009-09-18 | 2019-12-31 | A123 Systems, LLC | High power electrode materials |
| JP2013505193A (ja) * | 2009-09-18 | 2013-02-14 | エー123 システムズ, インコーポレイテッド | リン酸第二鉄およびその調製の方法 |
| US11652207B2 (en) | 2009-09-18 | 2023-05-16 | A123 Systems Llc | High power electrode materials |
| US9660267B2 (en) | 2009-09-18 | 2017-05-23 | A123 Systems, LLC | High power electrode materials |
| US9954228B2 (en) | 2009-09-18 | 2018-04-24 | A123 Systems, LLC | High power electrode materials |
| US12027702B2 (en) | 2010-04-28 | 2024-07-02 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material of power storage device, power storage device, electrically propelled vehicle, and method for manufacturing power storage |
| US10916774B2 (en) | 2010-04-28 | 2021-02-09 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material of power storage device, power storage device, electrically propelled vehicle, and method for manufacturing power storage |
| JP2018018828A (ja) * | 2010-04-28 | 2018-02-01 | 株式会社半導体エネルギー研究所 | 二次電池用電極 |
| US10224548B2 (en) | 2010-04-28 | 2019-03-05 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material of power storage device, power storage device, electrically propelled vehicle, and method for manufacturing power storage |
| JP2013032257A (ja) * | 2011-06-28 | 2013-02-14 | Nichia Corp | オリビン型リチウム遷移金属酸化物及びその製造方法 |
| JP2014088282A (ja) * | 2012-10-30 | 2014-05-15 | Rin Kagaku Kogyo Kk | リン酸第二鉄含水和物粒子粉末およびその製造方法 |
| JP2014088283A (ja) * | 2012-10-30 | 2014-05-15 | Rin Kagaku Kogyo Kk | リン酸第二鉄含水和物粒子粉末及びその製造方法 |
| US20140147586A1 (en) * | 2012-11-27 | 2014-05-29 | Universite De Montreal | Process for making an alkali metal oxyanion comprising iron |
| CN103864044A (zh) * | 2014-03-10 | 2014-06-18 | 瓮福(集团)有限责任公司 | 利用微波法将磷铁转化为电池级磷酸铁的方法 |
| JP2019061917A (ja) * | 2017-09-28 | 2019-04-18 | 住友大阪セメント株式会社 | リチウムイオン二次電池用正極材料およびその製造方法、リチウムイオン二次電池用正極、リチウムイオン二次電池 |
| US10566622B2 (en) | 2017-09-28 | 2020-02-18 | Sumitomo Osaka Cement Co., Ltd. | Cathode material for lithium-ion secondary battery and method for manufacturing the same, cathode for lithium-ion secondary battery, and lithium-ion secondary battery |
| CN109850862A (zh) * | 2019-04-23 | 2019-06-07 | 王柯娜 | 一种电池级无水磷酸铁的制备方法 |
| CN116216681A (zh) * | 2022-12-28 | 2023-06-06 | 湖北虹润高科新材料有限公司 | 一种碱式磷酸铁铵的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR101718918B1 (ko) | 2017-03-22 |
| TW201124337A (en) | 2011-07-16 |
| JP2012012279A (ja) | 2012-01-19 |
| EP2476647A1 (en) | 2012-07-18 |
| EP2476647B1 (en) | 2023-07-05 |
| CN102612487A (zh) | 2012-07-25 |
| EP2476647A4 (en) | 2016-03-09 |
| JP5835540B2 (ja) | 2015-12-24 |
| KR20120079058A (ko) | 2012-07-11 |
| US20120237425A1 (en) | 2012-09-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5835540B2 (ja) | リン酸第二鉄含水物粒子粉末の製造法、オリビン型リン酸鉄リチウム粒子粉末の製造法、並びに非水電解質二次電池の製造法。 | |
| JP5293936B2 (ja) | 非水電解質二次電池用オリビン型複合酸化物及びその製造方法、並びに二次電池 | |
| JP5464322B2 (ja) | リン酸鉄リチウム粒子粉末の製造方法、オリビン型構造のリン酸鉄リチウム粒子粉末、該リン酸鉄リチウム粒子粉末を用いた正極材シート及び非水溶媒系二次電池 | |
| JP5517032B2 (ja) | 非水電解質二次電池用オリビン型複合酸化物粒子粉末及びその製造方法、並びに二次電池 | |
| KR101365578B1 (ko) | 리튬-망간 스피넬을 함유하는 혼합 산화물 및 그의 제조 방법 | |
| JP6097306B2 (ja) | マンガン含有金属リン酸塩及びその製造方法 | |
| US9082525B2 (en) | Lithium silicate-based compound and production process for the same, positive-electrode active material and positive electrode for use in lithium-ion secondary battery as well as secondary battery | |
| JP5678685B2 (ja) | リチウム二次電池用正極活物質の前駆体とその製造方法およびリチウム二次電池用正極活物質の製造方法 | |
| WO2012176904A1 (ja) | リチウムイオン二次電池用正極活物質の製造方法 | |
| WO2011115211A1 (ja) | リン酸マンガン鉄リチウム粒子粉末の製造方法、リン酸マンガン鉄リチウム粒子粉末、及び該粒子粉末を用いた非水電解質二次電池 | |
| JP4829557B2 (ja) | リチウム鉄複合酸化物の製造方法 | |
| US20130183584A1 (en) | Lithium-silicate-based compound and production process for the same | |
| JP7792023B2 (ja) | リン酸マンガン鉄リチウム正極材料及びその調製方法、リン酸マンガン鉄前駆体及びその調製方法、リチウムイオン電池 | |
| JP7125302B2 (ja) | ナトリウムイオン二次電池用nasicon型負極活物質粒子の製造方法 | |
| US12142761B2 (en) | Transition metal oxide particles encapsulated in nanostructured lithium titanate or lithium aluminate, and the use thereof in lithium ion batteries | |
| JP2010232091A (ja) | リチウムイオン電池用正極活物質の製造方法とリチウムイオン電池用正極活物質及びリチウムイオン電池用電極並びにリチウムイオン電池 | |
| TW201803803A (zh) | 磷酸釩鋰的製造方法 | |
| JP7217514B2 (ja) | チタン酸化物、チタン酸化物の製造方法、およびチタン酸化物を含む電極活物質を用いたリチウム二次電池 | |
| JP7089982B2 (ja) | ナトリウムイオン二次電池用負極活物質粒子及びその製造方法 | |
| JP7617182B2 (ja) | 電池複合材料及びその前駆体の製造方法 | |
| JP2025126780A (ja) | リン酸バナジウムリチウムの製造方法 | |
| JP2025125626A (ja) | リン酸バナジウムリチウムの製造方法 | |
| JP2025125625A (ja) | リン酸バナジウムリチウムの製造方法 | |
| HK1152412A (en) | Mixed oxide containing a lithium-manganese spinel and process for producing it |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080039809.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10815381 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20127005889 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010815381 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13394990 Country of ref document: US |