WO2011040559A1 - 水銀酸化触媒及びその製造方法 - Google Patents
水銀酸化触媒及びその製造方法 Download PDFInfo
- Publication number
- WO2011040559A1 WO2011040559A1 PCT/JP2010/067132 JP2010067132W WO2011040559A1 WO 2011040559 A1 WO2011040559 A1 WO 2011040559A1 JP 2010067132 W JP2010067132 W JP 2010067132W WO 2011040559 A1 WO2011040559 A1 WO 2011040559A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mercury
- moo
- catalyst
- raw material
- carrier
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images




Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/28—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/30—Tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/74—General processes for purification of waste gases; Apparatus or devices specially adapted therefor
- B01D53/86—Catalytic processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/74—General processes for purification of waste gases; Apparatus or devices specially adapted therefor
- B01D53/86—Catalytic processes
- B01D53/8665—Removing heavy metals or compounds thereof, e.g. mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/063—Titanium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/882—Molybdenum and cobalt
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/883—Molybdenum and nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/885—Molybdenum and copper
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8873—Zinc, cadmium or mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8877—Vanadium, tantalum, niobium or polonium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/50—Catalysts, in general, characterised by their form or physical properties characterised by their shape or configuration
- B01J35/56—Foraminous structures having flow-through passages or channels, e.g. grids or three-dimensional [3D] monoliths
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0201—Impregnation
- B01J37/0203—Impregnation the impregnation liquid containing organic compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0234—Impregnation and coating simultaneously
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2251/00—Reactants
- B01D2251/20—Reductants
- B01D2251/206—Ammonium compounds
- B01D2251/2062—Ammonia
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2251/00—Reactants
- B01D2251/50—Inorganic acids
- B01D2251/502—Hydrochloric acid
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/207—Transition metals
- B01D2255/20723—Vanadium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2255/00—Catalysts
- B01D2255/20—Metals or compounds thereof
- B01D2255/207—Transition metals
- B01D2255/20769—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2257/00—Components to be removed
- B01D2257/60—Heavy metals or heavy metal compounds
- B01D2257/602—Mercury or mercury compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/46—Removing components of defined structure
- B01D53/48—Sulfur compounds
- B01D53/50—Sulfur oxides
- B01D53/501—Sulfur oxides by treating the gases with a solution or a suspension of an alkali or earth-alkali or ammonium compound
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/46—Removing components of defined structure
- B01D53/64—Heavy metals or compounds thereof, e.g. mercury
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/46—Removing components of defined structure
- B01D53/68—Halogens or halogen compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/74—General processes for purification of waste gases; Apparatus or devices specially adapted therefor
- B01D53/86—Catalytic processes
- B01D53/8621—Removing nitrogen compounds
- B01D53/8625—Nitrogen oxides
Definitions
- the present invention relates to a mercury oxidation catalyst and a method for producing the same.
- mercury present in flue gas is thought to contain water-insoluble metallic mercury and water-soluble mercury compounds, and if metallic mercury can be converted into mercury compounds in the presence of a catalyst such as a denitration catalyst, Mercury compounds can be removed by a downstream desulfurization apparatus (see, for example, Patent Document 1).
- the inventors of the present invention have made extensive studies on a mercury oxidation catalyst that functions as a denitration catalyst and can convert metallic mercury into a water-soluble mercury compound.
- the present invention relates to a novel mercury oxidation catalyst developed by such a process and a method for producing the same.
- An object of the present invention is to provide a mercury oxidation catalyst containing V 2 O 5 and MoO 3 as active components, suppressing the volatilization of MoO 3 , and providing a durable and stable novel mercury oxidation catalyst and a method for producing the same. It is in.
- the present invention is a mercury oxidation catalyst that oxidizes metallic mercury in exhaust gas to mercury oxide, using TiO 2 as a carrier and activating V 2 O 5 and MoO 3 on the carrier. It is supported as a component, and at least one atom or compound selected from the group consisting of W, Cu, Co, Ni, and Zn and their compounds is supported as a MoO 3 volatilization suppressing component.
- the present invention is a method for producing a novel catalyst having excellent durability with a mercury oxidation catalyst that oxidizes metallic mercury in exhaust gas into a mercury compound, A step of preparing a catalyst raw material solution A containing MoO 3 serving as an active component and a MoO 3 volatilization suppressing component; A step of supporting the components in the catalyst raw material solution A on a TiO 2 carrier; A step of drying and calcining the carrier that has undergone the supporting step of the catalyst raw material solution A; Preparing a catalyst raw material solution B containing V 2 O 5 as an active component; And a step of supporting the components in the catalyst raw material solution B on the previously calcined carrier and a step of drying and calcining the carrier after the catalyst raw material solution B carrying step.
- the method for producing a mercury oxidation catalyst according to the present invention is, in one form thereof, the Mo volatilization suppressing component is at least one atom selected from the group consisting of W, Cu, Co, Ni, Zn, and compounds thereof. Or a compound.
- the mercury oxidation catalyst for the V 2 O 5 and MoO 3 as an active ingredient to suppress volatilization of MoO 3 ingredients, durability new high mercury oxidation catalyst and a method for producing the same.
- 10 is a graph showing XRD analysis results after pulverization and mixing of honeycomb catalysts according to Test Examples 7 to 9. It is a graph which expands and shows FIG. 2 partially. 10 is a graph showing the activity of a honeycomb catalyst according to Test Example 9.
- the mercury oxidation catalyst according to the present invention is a mercury oxidation catalyst that oxidizes metallic mercury in exhaust gas to a mercury compound.
- the mercury oxidation catalyst according to the present invention uses TiO 2 as a carrier, V 2 O 5 and MoO 3 as active components on the carrier, W, Cu, Co, Ni, Zn, and compounds thereof. At least one kind of atom or compound selected from the group consisting of the above is supported as a MoO 3 volatilization suppressing component.
- the fact that the compounds of W, Cu, Co, Ni, and Zn are suitable as the MoO 3 volatilization suppressing component was found as a result of intensive studies by the present inventors, and the effects thereof are further shown in the following examples. It is.
- components that can be dissolved on the phase diagram are also listed as volatilization suppressing components.
- a catalyst raw material solution containing an active component and a MoO 3 volatilization suppressing component is prepared.
- the base material containing the TiO 2 carrier is immersed in the catalyst raw material solution, impregnated with the catalyst raw material solution, and dried and fired.
- the base material containing TiO 2 carriers for example, produced as extrusion.
- the substrate is again immersed in the catalyst raw material solution, and the base material impregnated with the catalyst raw material solution is dried and fired.
- the target mercury oxidation catalyst can be obtained.
- a MoO 3 as the active ingredient, to prepare a catalyst raw material solution A containing the MoO 3 volatilization suppressing component. Then, by immersing the substrate containing TiO 2 carrier catalyst stock solution A, it is impregnated with a catalyst raw material solution A. Next, the base material immersed in the catalyst raw material solution A is dried and fired. On the other hand, a catalyst raw material solution B containing V 2 O 5 serving as another active ingredient is prepared. Further, the previously fired base material is immersed in the catalyst raw material solution B and impregnated with the catalyst raw material solution B. Finally, the base material that has been impregnated with the catalyst raw material solution B is dried and fired.
- the composition ratio of the mercury oxidation catalyst according to the present invention is not particularly limited.
- the active components V 2 O 5 and MoO 3
- the active components are used as oxides with respect to 100 parts by weight of the TiO 2 carrier.
- a composition of 0.1 to 20 parts by weight is preferred.
- the composition of the MoO 3 volatilization inhibiting component is preferably 0.06 to 50 parts by weight with respect to 1 part by weight of MoO 3 .
- the mercury oxidation catalyst according to the present invention can arbitrarily select the shape of the substrate in accordance with the system configuration. For example, it is integrally formed in a pellet shape, a plate shape, a cylindrical shape, a corrugated shape, a honeycomb shape, or the like. It can be set to any shape.
- the honeycomb shape may be an extrusion type or a coat type.
- the mercury oxidation catalyst of the present invention can be coated, and the catalyst regenerated by the regeneration method disclosed in JP-A-2009-226388 can be coated. It is also possible to produce this catalyst by a kneading method.
- a molybdenum raw material, a tungsten raw material, a vanadium raw material and a titanium dioxide or titanium dioxide raw material may be kneaded and extruded.
- a molybdenum raw material, tungsten raw material and titanium dioxide or titanium dioxide raw material, etc. after appropriately performing treatments such as drying, firing, pulverization, etc., kneading and extruding them with vanadium raw material etc. It is mentioned as a form.
- the vanadium raw material or the like can be kneaded and then dried, filled in the denitration apparatus without firing, and fired at the operating temperature of the denitration apparatus.
- V (vanadium) raw material of the active ingredient vanadium dioxide, vanadyl oxalate, vanadyl sulfate, ammonium metavanadate and the like can be used.
- Mo (molybdenum) raw material of the active ingredient salts such as ammonium molybdate and sodium molybdate can be used in addition to molybdenum trioxide.
- salts such as ammonium paratungstate and ammonium metatungstate can be used as the W (tungsten) raw material.
- tungsten tungsten
- Cu (copper) raw material copper nitrate, copper acetate, copper hydroxide, etc.
- Co (cobalt) raw material cobalt nitrate, cobalt acetate, basic cobalt carbonate, etc.
- Ni (nickel) raw material nitric acid
- nickel, nickel acetate, basic nickel carbonate, etc., and Zn (zinc) raw material, zinc nitrate, basic zinc carbonate, etc. can be used.
- Solution solution and MoO 3 volatilization inhibiting component of the active ingredient may be each prepared by dissolving the known solvents.
- Exhaust gas to be treated in the present invention is, for example, boiler exhaust gas of a thermal power plant or factory that burns fuel containing sulfur, mercury, etc. of coal, heavy oil, etc., metal factory, oil refinery factory, petrochemical factory, etc. Exhaust gas from a heating furnace with a low NO x concentration, carbon dioxide, oxygen, SO x , dust, or moisture, and a large amount of exhaust gas emission.
- an ammonia injection device 2 and an HCl injection device 4 for injecting NH 3 supplied from an ammonia tank 3 into exhaust gas are installed in a flow path from the boiler 1 to the reduction denitration device 5.
- the exhaust gas from the boiler 1 is introduced into the reduction denitration device 5.
- the exhaust gas into which NH 3 and HCl have been injected undergoes a reaction between NH 3 and NO x in a reductive denitration apparatus, and at the same time, metal Hg is oxidized to HgCl 2 in the presence of HCl.
- the mercury oxidation catalyst according to the present invention When the mercury oxidation catalyst according to the present invention is employed in the treatment in the reduction denitration apparatus 5, MoO 3 that is an active component is combined with the MoO 3 volatilization suppressing component, thereby suppressing the volatilization of MoO 3. , Durability is improved.
- the mercury oxidation reaction by the denitration catalyst is inhibited by unburned coal (CO, HC) (5 and 6), but the HC oxidation function is added to the denitration catalyst with mercury oxidation function. Further, the effect of inhibiting the mercury oxidation reaction can be reduced.
- oxidizers used for oxidizing metal mercury include HCl, HBr, and the like, and the addition amount may be a very small amount, and in some cases, a new oxidizer may not be added. . That is, for example, when a metal mercury oxidant such as HCl derived from coal contains several tens of ppm in the exhaust gas, it is not necessary to install an apparatus for spraying a metal mercury oxidant such as HCl. In that case, it is possible to greatly reduce the equipment cost for safety management measures for metal mercury oxidizers such as HCl which require attention in handling.
- any mercury oxidizing agent may be used as long as it reacts with mercury in exhaust gas to produce a water-soluble mercury compound.
- any mercury oxidizing agent may be used as long as it reacts with mercury in exhaust gas to produce a water-soluble mercury compound.
- Halogen compounds, other amine salts of the above acids, other salts, and the like can be used.
- the amount of mercury oxidant added to the exhaust gas should be a stoichiometric amount or more than the water-insoluble mercury such as metallic mercury. When coal or heavy oil is used as the fuel, it is added to the exhaust gas.
- the concentration of the mercury oxidant is 1000 ppm or less with respect to the exhaust gas, and is actually about 1 to 300 ppm.
- hydrogen chloride may be used as a drug, or hydrochloric acid may be used.
- the concentration of hydrochloric acid is not particularly limited, and examples thereof include concentrated hydrochloric acid to dilute hydrochloric acid of about 5%.
- a conventional metering pump for chemicals and a spray nozzle can be used as a device for adding HCl to exhaust gas.
- the addition of salts such as ammonium chloride is preferably carried out using an aqueous salt solution.
- Mercury oxidant may be added before or after addition of ammonia to the exhaust gas.
- the exhaust gas passes through the air preheater 6 and the heat exchanger 7, and then the dust is removed by the electric dust collector 8. Then, the SO 2 in the exhaust gas is removed by the wet desulfurization device 9. At the same time, mercury compounds are removed.
- Exhaust gas exiting the reductive denitration device contains an excess of a metal mercury oxidizer such as HCl, but is absorbed by the alkaline aqueous solution of the desulfurization device, so that it is not discharged from the chimney.
- a mercury oxidant is provided upstream of the denitration apparatus. Is going to be added.
- NH 3 is only necessary for denitration, and even if NH 3 is not added upstream of the reduction denitration device, mercury is converted to chloride by the mercury oxidizing agent in the presence of the catalyst of the reduction denitration device. The effect of removing mercury oxide by wet desulfurization equipment remains the same.
- Example 1 (Test on Mo volatilization suppression) A TiO 2 carrier was formed into a honeycomb and fired to prepare a base material containing the TiO 2 carrier. The substrate was immersed in the following mixed solutions (Test Examples 1 to 6), dried, and then fired at 500 ° C. for 3 hours to obtain a honeycomb catalyst.
- Test Example 1 24.94 g of ammonium molybdate was dissolved in 100 g of water.
- Test Example 2 A mixture was prepared so that 28.477 g of ammonium molybdate and 26.191 g of ammonium paratungstate were contained in 100 g of a 40 wt% methylamine solution.
- Test Example 3 A mixture was prepared so that 27.95 g of ammonium molybdate and 15.95 g of copper hydroxide were contained in 100 g of a 40 wt% methylamine solution.
- Test Example 4 A mixture was prepared so that 24.821 g of ammonium molybdate and 41.607 g of cobalt nitrate were contained in 100 g of water.
- Test Example 5 A mixture was prepared so that 25.639 g of ammonium molybdate and 79.223 g of nickel nitrate were contained in 100 g of water.
- Test Example 6 A mixture was prepared so that 24.828 g of ammonium molybdate and 42.347 g of zinc nitrate were contained in 100 g of water.
- the honeycomb catalysts of Test Examples 1 to 6 were exposed for 8 hours at 550 ° C. under a simulated gas flow, and the MoO 3 concentrations before and after the treatment were compared to determine the volatility of MoO 3 .
- the results are shown in Table 1 below. Simulated gas composition: O 2 3% vol (dry), CO 2 10% vol (dry), H 2 O 10% (wet), SO 2 500 ppm (dry), remaining N 2
- Test Examples 2 to 6 that include any one of W, Cu, Co, Ni, and Zn are all MoO. The volatilization of 3 was suppressed. That is, it is understood that these components function as MoO 3 volatilization suppressing components.
- Example 2 (Verification of manufacturing method) Test Example 7 A TiO 2 carrier was formed into a honeycomb and fired to prepare a base material containing the TiO 2 carrier. Then, a solution was prepared by dissolving 19.636 g of ammonium molybdate and 1.478 g of ammonium metavanadate in 80 g of water. The honeycomb substrate was immersed in the catalyst raw material solution, dried, and then fired at 500 ° C. for 3 hours. The obtained honeycomb catalyst was exposed for 8 hours at 550 ° C. under a simulated gas flow, and the concentration of MoO 3 before and after the treatment was compared to determine the volatility of MoO 3 , which was 18.4 wt%.
- Test Example 8 a solution in which 22.55 g of ammonium molybdate, 20.0.697 g of ammonium paratungstate, and 1.485 g of ammonium metavanadate were dissolved in 80 g of a 40 wt% methylamine solution was prepared.
- the honeycomb substrate was immersed in the catalyst raw material solution, dried, and then fired at 500 ° C. for 3 hours.
- the obtained honeycomb catalyst was exposed at 550 ° C. for 8 hours under the flow of simulated gas, and the concentration of MoO 3 before and after the treatment was compared to determine the sublimation rate of MoO 3 , which was 16.7 wt%. Thus, the volatilization rate slightly decreased.
- V (vanadium) is a main active component of the denitration reaction, and it is preferable that a small amount of adjustment can be made independently in the catalyst design. Therefore, it is different from Japanese Patent Application No. 2006-256639 in that the influence of V (vanadium) addition is reduced.
- Test Example 9 A solution in which 21.777 g of ammonium molybdate and 19.903 g of ammonium paratungstate were dissolved in 80 g of 40 wt% methylamine solution was prepared, and a solution in which 3.347 g of vanadyl sulfate was dissolved in 80 g of water was prepared. A mixed solution of an ammonium molybdate solution and an ammonium paratungstate solution was used as a catalyst raw material solution A. The honeycomb substrate was immersed in the catalyst raw material solution A and fired at 500 ° C. for 3 hours.
- this base material was immersed in the vanadyl sulfate solution (catalyst raw material solution B), and baked at 500 ° C. for 3 hours.
- the obtained honeycomb catalyst was exposed at 550 ° C. for 8 hours under the flow of simulated gas, and the concentrations of MoO 3 before and after the treatment were compared to determine the volatility of MoO 3 , which was 12.1 wt%. This result is in line with the result of the previous Example 1, and eliminates the inconvenience associated with the addition of V (vanadium).
- the composition of the simulated gas was the same as that of Example 1 in Test Examples 7 to 9.
- Example 3 The results of XRD analysis of the honeycomb catalysts obtained in Test Examples 7 to 9 are shown in FIGS. Region theta A is, WO 3, or a region where the characteristic appears in a composite oxide of WO 3 and MoO 3.
- the region ⁇ B is a region where MoO 3 appears.
- a peak related to WoO 3 appears (sub-region ⁇ C ).
- a peak appears in a region that seems to be a composite oxide of WO 3 and MoO 3 , whereas this does not appear in Test Example 9 (sub-region ⁇ D ).
- Test Example 7 a peak of MoO 3 appears, but in Test Examples 8 and 9, it does not appear (region ⁇ B ). From this result, it is understood that Test Example 9 has a small amount of composite oxide of WO 3 and MoO 3 and MoO 3 is suitably dissolved in the crystallized WO 3 . That is, it is understood that the production method in which the immersion in the catalyst raw material solution A and the immersion in the catalyst raw material solution B are performed in two stages shows the grounds for superiority. In Test Example 9, a peak attributed to MoO3 was not observed in the range of 27 to 28 deg (2 ⁇ ) (region ⁇ B ) in the XRD analysis, and in the range of 22 to 25 deg (2 ⁇ ) (region ⁇ A ).
- a peak attributed to the volatilization suppressing component or the composite oxide of MoO3 and the volatilization suppressing component appears.
- the XRD peak in the region ⁇ A can change depending on the drying and firing conditions, but no peak attributed to MoO 3 appears in the region ⁇ B.
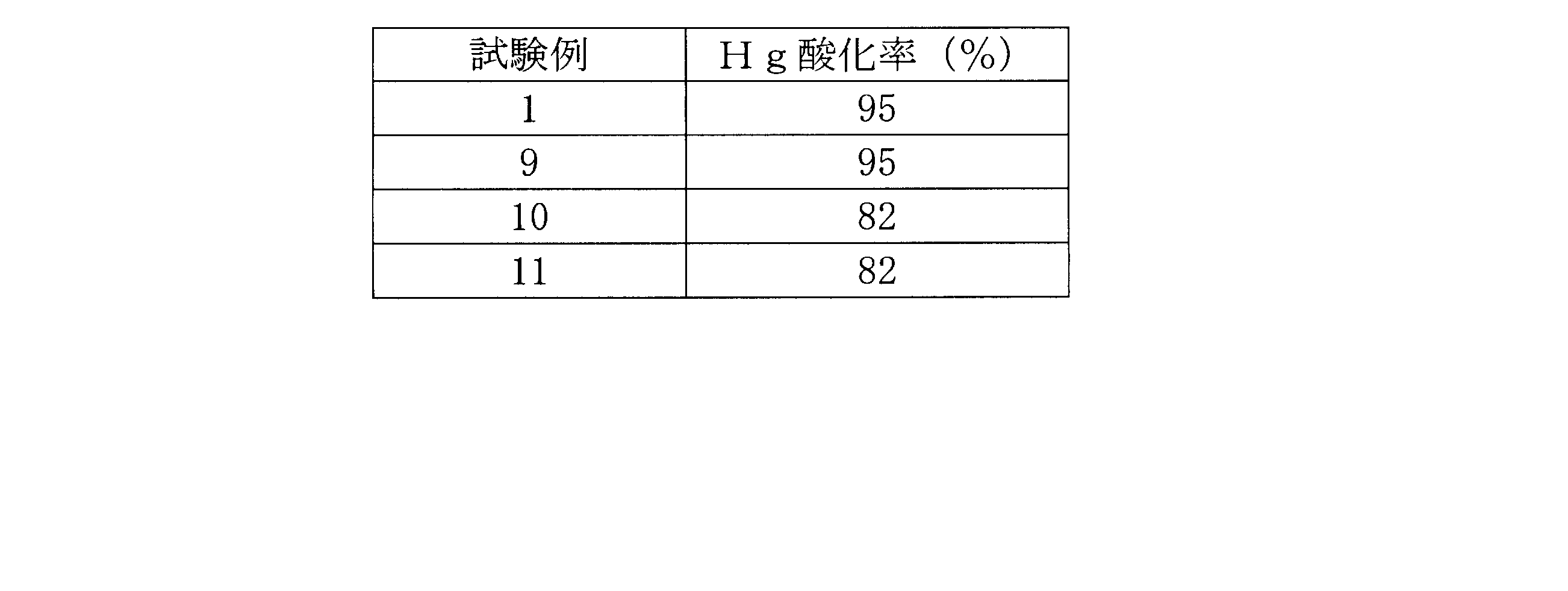
- Example 4 The Hg oxidation rate of the honeycomb catalyst according to Test Example 9 is shown in FIG.
- the horizontal axis represents the HCl concentration in the simulated exhaust gas on a dry basis. It is understood that the oxidation ability itself is comparable with that of the test example 7 honeycomb catalyst. In addition, it has been verified by the present inventors that the oxidation ability of the honeycomb catalyst according to Test Example 7 is superior to the conventional one.
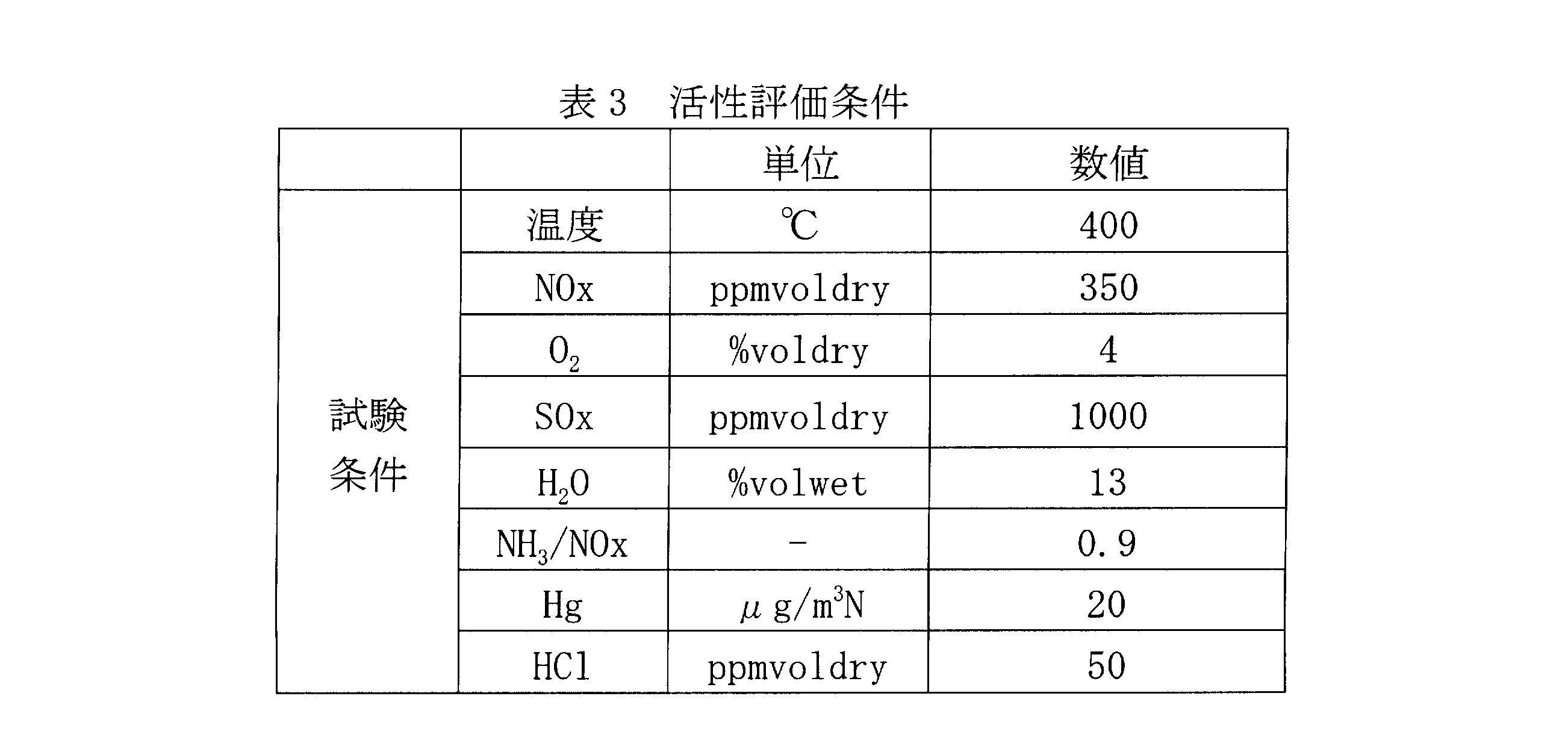
- the activity evaluation conditions are as shown in Table 2.
- Test Example 10 A catalyst containing vanadium and tungsten was prepared as follows. To the titanium dioxide powder, an aqueous solution of ammonium metavanadate was added in an amount of 0.5% by mass in terms of V 2 O 5 , and an aqueous solution of ammonium tungstate in an amount of 3% by mass in terms of WO 3. After baking for 4 hours, a powder (A) composed of titanium oxide [TiO 2 ] -vanadium oxide [V 2 O 5 ] -molybdenum oxide [WO 3 ] was obtained.
- Powder (A) 1000 g was placed carboxymethylcellulose 25 g, polyethylene oxide 12.5g into a kneader, and 30 minutes kneaded to moderate water, extruded into a honeycomb shape, and fired for five hours at 500 ° C. After drying, WO 3 (3) A -V 2 O 5 (0.5) / TiO 2 catalyst was prepared.
- Test Example 11 The catalyst prepared in Test Example 10 was immersed in an aqueous solution of ammonium molybdate, dried and calcined, and a MoO 3 (7) -WO 3 (3) -V 2 O 5 (0.5) / TiO 2 catalyst was prepared. .
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Environmental & Geological Engineering (AREA)
- Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Catalysts (AREA)
- Exhaust Gas Treatment By Means Of Catalyst (AREA)
Abstract
Description
活性成分となるMoO3と、MoO3揮発抑制成分とを含有する触媒原料溶液Aを調製する工程と、
TiO2担体に上記触媒原料溶液A中の成分を担持する工程と、
上記触媒原料溶液Aの担持工程を経た上記担体を乾燥、焼成する工程と、
活性成分となるV2O5を含有する触媒原料溶液Bを調製する工程と、
先に焼成した上記担体に上記触媒原料溶液B中の成分を担持する工程と、上記触媒原料溶液Bの担持工程を経た上記担体を乾燥、焼成する工程と、を備える。
本発明に係る水銀酸化触媒は、TiO2を担体とし、該担体上に、V2O5及びMoO3を活性成分として担持し、W,Cu,Co,Ni,及びZn、並びにそれらの化合物からなる群より選ばれる少なくとも1種類の原子、又は化合物をMoO3揮発抑制成分として担持している。
W,Cu,Co,Ni,及びZnの化合物がMoO3揮発抑制成分として好適であることは、本発明者らが鋭意検討した結果見出したものであり、後の実施例でその効果がさらに示される。その他、相図の上で固溶し得る成分も揮発抑制成分として挙げられる。
そして、TiO2担体を含む基材をこの触媒原料溶液に浸漬し、触媒原料溶液を含浸させ、乾燥、焼成を行う。
なお、TiO2担体を含む基材は、例えば、押出し成型のようにして製造する。
次いで、再び触媒原料溶液に浸漬し、触媒原料溶液を含浸させた基材を乾燥、焼成する。
以上によって、目的とする水銀酸化触媒を得ることができる。
そして、TiO2担体を含む基材をこの触媒原料溶液Aに浸漬し、触媒原料溶液Aを含浸させる。
次いで、触媒原料溶液Aに浸漬した基材を乾燥、焼成する。
一方、別の活性成分となるV2O5を含有する触媒原料溶液Bを調製する。
さらに、先に焼成した基材を上記触媒原料溶液Bに浸漬し、触媒原料溶液Bを含浸させる。
最後に、触媒原料溶液Bの含浸工程を経た基材を乾燥、焼成する。
以上のように、V2O5を先の焼成工程の後に添加することよって、MoO3の揮発をより抑制した目的とする水銀酸化触媒を得ることができる。このことは、本発明者らが鋭意検討した結果見出したものであり、後の実施例でその効果がさらに示される。なお、ここで、バナジウム原料等を含浸後、乾燥を行い、焼成せずに脱硝装置内に充填し、脱硝装置の運転温度で焼成することもできる。
押出し成型したハニカム基材の上に、本発明の水銀酸化触媒をコーティングすることができ特開2009-226388にある再生方法で再生された触媒にコートすることが可能である。
また、混錬法で本触媒を製造することも可能である。
例えば、二酸化チタンのハニカムに活性成分を含浸するのではなく、モリブデン原料、タングステン原料及びバナジウム原料と二酸化チタンあるいは二酸化チタン原料等を混錬して押出すようにした方法として実施できる。
また、モリブデン原料、タングステン原料と二酸化チタンあるいは二酸化チタン原料等を押出したあと、適宜乾燥、焼成、粉砕等の処理を施した後、それらとバナジウム原料等を混錬して押出す方法も、他の形態として挙げられる。なお、ここで、バナジウム原料等を混錬後、乾燥を行い、焼成せずに脱硝装置内に充填し、脱硝装置の運転温度で焼成することもできる。
活性成分の他方のMo(モリブデン)原料としては、三酸化モリブデンの他、モリブデン酸アンモニウム、モリブデン酸ナトリウム等の塩を用いることができる。
MoO3揮発抑制成分のうち、W(タングステン)原料としては、パラタングステン酸アンモニウム、メタタングステン酸アンモニウム等の塩を用いることができる。また、Cu(銅)原料としては、硝酸銅、酢酸銅、水酸化銅 等、Co(コバルト)原料としては、硝酸コバルト、酢酸コバルト、塩基性炭酸コバルト 等、Ni(ニッケル)原料としては、硝酸ニッケル、酢酸ニッケル、塩基性炭酸ニッケル等、及びZn(亜鉛)原料としては、硝酸亜鉛、塩基性炭酸亜鉛等を用いることができる。
本発明で処理対象とする排ガスは、例えば、石炭、重質油等の硫黄、水銀等を含む燃料を燃焼する火力発電所、工場等のボイラ排ガス、金属工場、石油精製工場、石油化学工場等の加熱炉排ガスであり、NOx濃度が低く、二酸化炭素、酸素、SOx、ばいじん又は水分を含むものであり、排ガスの排出量が多いものである。
ボイラ1からの排ガスは還元脱硝装置5へ導入される。NH3とHClが注入された排ガスは還元脱硝装置においてNH3とNOxとの反応が行われると同時にHCl存在下で金属HgがHgCl2に酸化される。
(1)Hg+2HCl+1/2O2→HgCl2+H2O
(2)HgCl2+NH3+1/4O2
→Hg+1/2N2+2HCl+1/2H2O
(3)4NO+4NH3+O2→4N2+6H2O
(4)HgCl2+SO2+H2O→Hg+2HCl+SO3
(5)HgCl2+CO+H2O→Hg+2HCl+CO2
(6)HgCl2+HC(ex.HCHO,C2H4,C6H6)
+H2O+O2→Hg+2HCl+CO2
(7)HC+O2→CO,CO2+H2O
(8)2NH3+3/2O2→N2+3H2O
なお、上記一連の反応において、脱硝触媒による水銀酸化反応は、石炭の未燃分(CO,HC)により阻害されるが(5式,6式)、水銀酸化機能付きの脱硝触媒にHC酸化機能も付与することで更に水銀酸化反応への阻害効果を低減できる。
すなわち、例えば、石炭由来のHCl等の金属水銀酸化剤が排ガス中に数十ppm含まれる場合は、HCl等の金属水銀酸化剤噴霧装置の設置は不要である。その場合、取扱い上注意を要するHCl等の金属水銀酸化剤の安全管理対策にかかる設備コストを大幅に低減できる。
例えば、HClの他、塩化アンモニウム、塩素、次亜塩素酸、次亜塩素酸アンモニウム、亜塩素酸、亜塩素酸アンモニウム、塩素酸、塩素酸アンモニウム、過塩素酸、過塩素酸アンモニウム、臭素等のハロゲン化合物、その他上記酸のアミン塩類、その他の塩類等を用いることができる。
TiO2担体をハニカム状に成形し、これを焼成してTiO2担体を含む基材を準備した。そして、この基材を以下の各混合溶液(試験例1~6)に浸漬し、乾燥後、その後基材を500℃で3時間焼成し、ハニカム触媒を得た。
試験例2: モリブデン酸アンモニウム28.477gとパラタングステン酸アンモニウム26.191gが100gの40wt%メチルアミン溶液に含まれるように調合した。
試験例3: モリブデン酸アンモニウム27.95gと水酸化銅15.95gが100gの40wt%メチルアミン溶液に含まれるように調合した。
試験例4: モリブデン酸アンモニウム24.821gと硝酸コバルト41.607gが100gの水に含まれるように調合した。
試験例5: モリブデン酸アンモニウム25.639gと硝酸ニッケル79.223gが100gの水に含まれるように調合した。
試験例6: モリブデン酸アンモニウム24.828gと硝酸亜鉛42.347gが100gの水に含まれるように調合した。
結果を、以下の表1に示す。
模擬ガスの組成:
O2 3%vol(dry), CO2 10%vol(dry), H2O 10%(wet), SO2 500ppm(dry), 残N2

試験例7
TiO2担体をハニカム状に成形し、これを焼成してTiO2担体を含む基材を準備した。
そして、モリブデン酸アンモニウム19.636gとメタバナジン酸アンモニウム1.478gを80gの水に溶解した溶液を調製した。
ハニカム基材を触媒原料溶液に浸漬し、乾燥後、500℃で3時間焼成した。
得られたハニカム触媒を模擬ガスの流通下550℃で8時間曝露し、処理前後のMoO3の濃度を比較し、MoO3の揮発率を求めたところ18.4wt%であった。
次に、モリブデン酸アンモニウム22.569gとパラタングステン酸アンモニウム20.697gとメタバナジン酸アンモニウム1.685gを80gの40wt%メチルアミン溶液に溶解した溶液を調製した。
ハニカム基材を触媒原料溶液に浸漬し、乾燥後、500℃で3時間焼成した。
得られたハニカム触媒を模擬ガスの流通下550℃で8時間曝露し、処理前後のMoO3の濃度を比較し、MoO3の昇華率を求めたところ16.7wt%であった。
このように、揮発率は若干低下した。
その結果、V(バナジウム)を先の焼成工程の後に添加することよって、MoとV(バナジウム)の複合酸化物よりMoO3の揮発をより抑制した目的とする水銀酸化触媒を得ることができることに想到した。以下の手順で行った試験例9でもそのことが実証された。また、V(バナジウム)は脱硝反応の主活性成分であり、触媒設計上も単独で微量の調整が可能な方が良い。したがって、V(バナジウム)添加による影響を低減している点で特願2006-256639とは異なる。
モリブデン酸アンモニウム21.677gとパラタングステン酸アンモニウム19.903gを80gの40wt%メチルアミン溶液に溶解した溶液を調製し、さらに硫酸バナジル3.347gを80gの水に溶解した溶液を調製した。
モリブデン酸アンモニウムの溶液と、パラタングステン酸アンモニウムの溶液との混合溶液を触媒原料溶液Aとした。
ハニカム基材を触媒原料溶液Aに浸漬し、500℃で3時間焼成した。
得られたハニカム触媒を模擬ガスの流通下550℃で8時間曝露し、処理前後のMoO3の濃度を比較し、MoO3の揮発率を求めたところ12.1wt%であった。
この結果は、先の実施例1の結果に沿うものであり、V(バナジウム)の添加に伴う不都合を解消している。
なお、模擬ガスの組成は、試験例7~9とも実施例1と同様であった。
上記試験例7~9で得られたハニカム触媒についてXRD解析を行った結果を図2、図3に示す。
領域θAは、WO3、又はWO3とMoO3との複合酸化物の特徴が現れる領域である。
領域θBは、MoO3の現われる領域である。
試験例8、9ともWoO3に関連するピークが現われている(サブ領域θC)。
しかし、試験例8では、WO3とMoO3との複合酸化物と思われる領域でピークが現れるのに対し、試験例9ではこれが現われていない(サブ領域θD)。
この結果から、試験例9は、WO3とMoO3との複合酸化物の生成が少なく、MoO3が,結晶化したWO3中に好適に固溶しているものと理解される。すなわち、触媒原料溶液Aでの浸漬と触媒原料溶液Bでの浸漬とを二段階で行う製造方法が優れている根拠を示しているものと理解される。
なお、試験例9では、XRD分析にて27~28 deg(2θ)の範囲(領域θB)にMoO3に帰属するピークが見られず、22~25deg(2θ)の範囲(領域θA)に揮発抑制成分あるいはMoO3と揮発抑制成分との複合酸化物に帰属するピークが現れる。また、領域θAのXRDピークは乾燥、焼成条件により変化し得るが、領域θBにMoO3に帰属されるピークは現れない。
試験例9によるハニカム触媒のHg酸化率を図4に示す。
横軸は、模擬排ガス中のHCl濃度をドライベースで示した。基準とした試験例7のハニカム触媒と比較し、酸化能自体についても、比肩するものであることが了解される。なお、試験例7に係るハニカム触媒の酸化能は、従来のものに比して優れていることは、本発明者らによって検証されている。活性評価条件は表2の通りである。

ただし、例えば試験例10で製造した触媒を試験例11のハニカム基材とする場合、あるいは試験例9のハニカム基材とする場合、試験例10で製造したV2O5とWO3の量がごく少量であれば、酸化能が発揮されることは有り得る。
バナジウムとタングステンを含む触媒は以下のように調製した。
二酸化チタン粉末にメタバナジン酸アンモンの水溶液をV2O5換算で0.5質量%、タングステン酸アンモニウムの水溶液をWO3換算で3質量%となる様添加し、充分混合した後乾燥し450℃で4時間焼成して、酸化チタン[TiO2]―酸化バナジウム[V2O5]―酸化モリブデン[WO3]からなる粉末(A)を得た。粉末(A)1000gにカルボキシメチルセルロース25g、ポリエチレンオキサイド12.5gをニーダーに入れ、適度の水を加えて30分混練し、ハニカム形状に押出し成型し、乾燥後500℃で5時間焼成し、WO3(3)-V2O5(0.5)/TiO2触媒を調製した。
試験例10で調製した触媒をモリブデン酸アンモニウムの水溶液に浸漬し、乾燥、焼成後、MoO3(7)-WO3(3)-V2O5(0.5)/TiO2触媒を調製した。
2 アンモニア注入装置
3 アンモニア・タンク
4 HCl注入装置
5 還元脱硝装置
6 空気予熱器
7 熱交換器
8 電気集塵器
9 湿式脱硫装置
Claims (5)
- 排ガス中の水銀を水溶性の水銀化合物に酸化する水銀酸化触媒であって、TiO2を担体とし、該担体上に、V2O5及びMoO3を活性成分として担持し、W,Cu,Co,Ni,及びZn、並びにこれらの原子の化合物からなる群より選ばれる少なくとも1種類の原子、又は化合物をMoO3揮発抑制成分として担持した水銀酸化触媒。
- 排ガス中の水銀を水溶性の水銀化合物に酸化する水銀酸化触媒であって、TiO2を担体とし、該担体上に、V2O5及びMoO3を活性成分として担持し、W,Cu,Co,Ni,及びZn、並びにこれらの原子の化合物からなる群より選ばれる少なくとも1種類の原子、又は化合物をMoO3揮発抑制成分として担持しており、XRD分析にて、MoO3に帰属するピークが現れないことを特徴とする水銀酸化触媒。
- 排ガス中の水銀を水溶性の水銀化合物に酸化する水銀酸化触媒の製造方法であって、
活性成分となるMoO3と、MoO3揮発抑制成分とを含有する触媒原料溶液Aを調製する工程と、
TiO2担体に上記触媒原料溶液A中の成分を担持する工程と、
上記触媒原料溶液Aの担持工程を経た上記担体を乾燥、焼成する工程とを備える水銀酸化触媒の製造方法。 - 排ガス中の水銀を水溶性の水銀化合物に酸化する水銀酸化触媒の製造方法であって、
活性成分となるMoO3と、MoO3揮発抑制成分とを含有する触媒原料溶液Aを調製する工程と、
TiO2担体に上記触媒原料溶液A中の成分を担持する工程と、
上記触媒原料溶液Aの担持工程を経た上記担体を乾燥、焼成する工程と、
活性成分となるV2O5を含有する触媒原料溶液Bを調製する工程と、
先に焼成した上記担体に上記触媒原料溶液B中の成分を担持する工程と、
上記触媒原料溶液Bの担持工程を経た上記担体を乾燥、焼成する工程と、
を備える水銀酸化触媒の製造方法。 - 上記MoO3揮発抑制成分が、W,Cu,Co,Ni,及びZn、並びにこれらの原子の化合物からなる群より選ばれる少なくとも1種類の原子、化合物である請求項3又は4に記載の水銀酸化触媒の製造方法。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI1015063A BRPI1015063A2 (pt) | 2009-10-01 | 2010-09-30 | "catalisador de oxidação de mercúrio, e, método para produzir um catalisador de oxidação de mercúrio." |
| CA2764132A CA2764132C (en) | 2009-10-01 | 2010-09-30 | Mercury oxidation catalyst and method for producing the same |
| CN201080029023.5A CN102470345B (zh) | 2009-10-01 | 2010-09-30 | 汞氧化催化剂及其制造方法 |
| EP10820674.9A EP2484441B1 (en) | 2009-10-01 | 2010-09-30 | Mercury oxidation catalyst and method for producing the same |
| JP2011534325A JP5357974B2 (ja) | 2009-10-01 | 2010-09-30 | 水銀酸化触媒及びその製造方法 |
| KR1020117030998A KR101373372B1 (ko) | 2009-10-01 | 2010-09-30 | 수은 산화 촉매 및 이의 제조방법 |
| RU2011153281/04A RU2493908C2 (ru) | 2009-10-01 | 2010-09-30 | Катализатор окисления ртути и способ его приготовления |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24771609P | 2009-10-01 | 2009-10-01 | |
| US61/247,716 | 2009-10-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011040559A1 true WO2011040559A1 (ja) | 2011-04-07 |
Family
ID=43823634
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/067132 Ceased WO2011040559A1 (ja) | 2009-10-01 | 2010-09-30 | 水銀酸化触媒及びその製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US8288309B2 (ja) |
| EP (1) | EP2484441B1 (ja) |
| JP (1) | JP5357974B2 (ja) |
| KR (1) | KR101373372B1 (ja) |
| CN (1) | CN102470345B (ja) |
| BR (1) | BRPI1015063A2 (ja) |
| CA (1) | CA2764132C (ja) |
| RU (1) | RU2493908C2 (ja) |
| WO (1) | WO2011040559A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106975331A (zh) * | 2017-04-27 | 2017-07-25 | 北京清新环境技术股份有限公司 | 一种烟气催化吸附协同脱汞装置及其方法 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104415766B (zh) * | 2013-08-22 | 2017-02-01 | 上海郎特电力环保科技有限公司 | 一种燃煤电站烟气脱汞脱硝复合催化剂及其制备方法 |
| JP6257775B2 (ja) * | 2013-12-11 | 2018-01-10 | ゼァージァン ユニバーシティ | 脱硝性能と水銀酸化性能を有する触媒とその製法 |
| WO2017153236A1 (en) * | 2016-03-07 | 2017-09-14 | Haldor Topsøe A/S | Preparation of a catalytic fabric filter with lower pressure drop |
| CN108525514B (zh) * | 2018-04-19 | 2021-03-30 | 南京信息工程大学 | 一种抗硫抗水除尘脱硝脱汞多功能滤料及其制备方法 |
| CN110215768B (zh) * | 2019-05-31 | 2021-08-17 | 南京杰科丰环保技术装备研究院有限公司 | 一种除尘脱硝脱汞一体化滤料及其制备方法 |
| CN113655170A (zh) * | 2021-08-04 | 2021-11-16 | 安徽迪诺环保新材料科技有限公司 | 烟气脱硝催化剂用纳米二氧化钛粉体脱硝率的测定方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0999239A (ja) * | 1995-10-06 | 1997-04-15 | Babcock Hitachi Kk | 排ガス脱硝触媒の製造方法 |
| JPH10230137A (ja) | 1997-02-19 | 1998-09-02 | Mitsubishi Heavy Ind Ltd | 排ガス処理方法及び排ガス処理装置 |
| JP2003053142A (ja) * | 2001-08-09 | 2003-02-25 | Mitsubishi Heavy Ind Ltd | 排ガスの水銀除去方法及び装置 |
| JP2005125213A (ja) * | 2003-10-22 | 2005-05-19 | Nippon Shokubai Co Ltd | 排ガス処理方法 |
| JP2006256639A (ja) | 2005-03-15 | 2006-09-28 | Fuji Photo Film Co Ltd | 平版印刷版の梱包構造 |
| WO2008035773A1 (fr) * | 2006-09-22 | 2008-03-27 | Babcock-Hitachi Kabushiki Kaisha | Catalyseur pour l'oxydation du mercure métallique |
| JP2009226388A (ja) | 2008-02-29 | 2009-10-08 | Mitsubishi Heavy Ind Ltd | 排ガス処理触媒の再生方法及びこの方法を使用した排ガス処理触媒 |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU490489A1 (ru) * | 1972-10-30 | 1975-11-05 | Предприятие П/Я Р-6991 | Способ очистки газов от ртути |
| EP0214085B1 (en) * | 1985-08-19 | 1992-04-08 | Mitsubishi Jukogyo Kabushiki Kaisha | Process for preparing a catalyst for removing nitrogen oxides |
| JP2991431B2 (ja) | 1987-06-05 | 1999-12-20 | バブコツク日立株式会社 | アンモニア接触還元脱硝用触媒およびその製法 |
| JP2583911B2 (ja) | 1987-10-26 | 1997-02-19 | バブコツク日立株式会社 | 窒素酸化物除去用触媒 |
| US5221484A (en) * | 1991-01-10 | 1993-06-22 | Ceramem Separations Limited Partnership | Catalytic filtration device and method |
| JP3270084B2 (ja) | 1991-12-11 | 2002-04-02 | バブコック日立株式会社 | 窒素酸化物除去用触媒およびその製造法 |
| FR2684899B1 (fr) * | 1991-12-16 | 1994-03-25 | Rhone Poulenc Chimie | Catalyseur de reduction selective des oxydes d'azote contenus dans un flux gazeux et application desdits catalyseurs. |
| JP3310926B2 (ja) | 1997-09-30 | 2002-08-05 | 三菱重工業株式会社 | 脱硝触媒及びその製造方法 |
| JP3924418B2 (ja) | 1999-08-12 | 2007-06-06 | 株式会社日本触媒 | 排ガス処理用触媒および排ガス処理方法 |
| JP4175465B2 (ja) | 2003-02-07 | 2008-11-05 | 三菱重工業株式会社 | 排ガス中の水銀除去方法およびそのシステム |
| ATE458543T1 (de) * | 2003-10-15 | 2010-03-15 | Haldor Topsoe As | Katalysator-träger, daraus hergestellter katalysator und prozess für die reinigung von abgasen |
| US7264784B2 (en) | 2003-10-22 | 2007-09-04 | Nippon Shokubai Co., Ltd. | Method for treating exhaust gas |
| JP4113090B2 (ja) | 2003-10-22 | 2008-07-02 | 株式会社日本触媒 | 排ガス処理方法 |
| JP5051977B2 (ja) * | 2005-01-31 | 2012-10-17 | バブコック日立株式会社 | 排ガス中微量有害物質の除去装置及びその運転方法 |
| JP4981318B2 (ja) * | 2005-12-19 | 2012-07-18 | 三菱重工業株式会社 | 排ガス処理装置および排ガス処理方法 |
-
2010
- 2010-09-30 BR BRPI1015063A patent/BRPI1015063A2/pt not_active Application Discontinuation
- 2010-09-30 CN CN201080029023.5A patent/CN102470345B/zh active Active
- 2010-09-30 RU RU2011153281/04A patent/RU2493908C2/ru active
- 2010-09-30 EP EP10820674.9A patent/EP2484441B1/en active Active
- 2010-09-30 CA CA2764132A patent/CA2764132C/en active Active
- 2010-09-30 WO PCT/JP2010/067132 patent/WO2011040559A1/ja not_active Ceased
- 2010-09-30 JP JP2011534325A patent/JP5357974B2/ja active Active
- 2010-09-30 US US12/895,194 patent/US8288309B2/en active Active
- 2010-09-30 KR KR1020117030998A patent/KR101373372B1/ko active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0999239A (ja) * | 1995-10-06 | 1997-04-15 | Babcock Hitachi Kk | 排ガス脱硝触媒の製造方法 |
| JPH10230137A (ja) | 1997-02-19 | 1998-09-02 | Mitsubishi Heavy Ind Ltd | 排ガス処理方法及び排ガス処理装置 |
| JP2003053142A (ja) * | 2001-08-09 | 2003-02-25 | Mitsubishi Heavy Ind Ltd | 排ガスの水銀除去方法及び装置 |
| JP2005125213A (ja) * | 2003-10-22 | 2005-05-19 | Nippon Shokubai Co Ltd | 排ガス処理方法 |
| JP2006256639A (ja) | 2005-03-15 | 2006-09-28 | Fuji Photo Film Co Ltd | 平版印刷版の梱包構造 |
| WO2008035773A1 (fr) * | 2006-09-22 | 2008-03-27 | Babcock-Hitachi Kabushiki Kaisha | Catalyseur pour l'oxydation du mercure métallique |
| JP2009226388A (ja) | 2008-02-29 | 2009-10-08 | Mitsubishi Heavy Ind Ltd | 排ガス処理触媒の再生方法及びこの方法を使用した排ガス処理触媒 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2484441A4 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106975331A (zh) * | 2017-04-27 | 2017-07-25 | 北京清新环境技术股份有限公司 | 一种烟气催化吸附协同脱汞装置及其方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20120012993A (ko) | 2012-02-13 |
| JPWO2011040559A1 (ja) | 2013-02-28 |
| RU2493908C2 (ru) | 2013-09-27 |
| CA2764132A1 (en) | 2011-04-07 |
| CA2764132C (en) | 2013-10-29 |
| EP2484441A1 (en) | 2012-08-08 |
| RU2011153281A (ru) | 2013-07-10 |
| EP2484441B1 (en) | 2017-09-06 |
| US20110082028A1 (en) | 2011-04-07 |
| EP2484441A4 (en) | 2013-07-24 |
| CN102470345B (zh) | 2014-02-26 |
| CN102470345A (zh) | 2012-05-23 |
| US8288309B2 (en) | 2012-10-16 |
| BRPI1015063A2 (pt) | 2016-04-19 |
| KR101373372B1 (ko) | 2014-03-13 |
| JP5357974B2 (ja) | 2013-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5357974B2 (ja) | 水銀酸化触媒及びその製造方法 | |
| US10746073B2 (en) | Denitration catalyst and method for producing the same | |
| CN114870833A (zh) | 一种低温低钒scr脱硝催化剂及其制备方法 | |
| WO2015079721A1 (ja) | 排ガス処理触媒 | |
| TWI555569B (zh) | A method for purifying exhaust gas containing metallic mercury, an oxidation catalyst for metallic mercury in exhaust gas, and a method of manufacturing the same | |
| JP4508584B2 (ja) | 高温排ガス用脱硝触媒 | |
| KR20160058639A (ko) | 선택적 촉매환원반응용 탈질촉매의 제조방법 | |
| JP5386096B2 (ja) | 排ガス処理触媒 | |
| US20160375404A1 (en) | Exhaust gas treatment system | |
| JP2005087815A (ja) | 排ガス処理方法 | |
| CN108236943A (zh) | 一种钒基氧化物催化剂的制备方法 | |
| KR102558168B1 (ko) | 암모니아 산화용 촉매 및 이의 제조 방법 | |
| KR20030078258A (ko) | 넓은 활성온도 대역을 갖는 질소산화물 및/또는 다이옥신제거용 삼산화 바나듐을 포함한 바나듐/티타니아계 촉매 | |
| JP5638982B2 (ja) | 脱硝装置 | |
| KR20020058179A (ko) | 이산화황을 포함하고 있는 배가스 중의 질소산화물을저온에서 제거하기 위한 선택적 환원촉매 | |
| JP2005342710A (ja) | 耐熱性脱硝触媒 | |
| KR102579933B1 (ko) | 실란 열처리를 통한 작용기층을 포함하는 선택적 환원 촉매 및 그의 제조방법 | |
| JP7278555B1 (ja) | 排ガスの脱硝方法 | |
| JP6640357B2 (ja) | 排ガス脱硝触媒、排ガス処理システム、及び排ガス処理方法 | |
| KR102634719B1 (ko) | 표면처리층을 포함하는 선택적 환원 촉매 및 그의 제조방법 | |
| KR102847399B1 (ko) | 실리카를 이용한 촉매 성형체의 제조방법 | |
| JP2012035150A (ja) | 排ガス浄化用触媒およびその製造方法 | |
| JP2006326578A (ja) | アンモニアおよび窒素酸化物の分解用触媒 | |
| KR20030061379A (ko) | 배가스 정화촉매 화합물, 이 화합물을 함유한 촉매 및 그제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080029023.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10820674 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2011534325 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2764132 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9430/DELNP/2011 Country of ref document: IN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010820674 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010820674 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2011153281 Country of ref document: RU Kind code of ref document: A Ref document number: 20117030998 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: PI1015063 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: PI1015063 Country of ref document: BR Kind code of ref document: A2 Effective date: 20111226 |