WO2011045629A1 - Lipid acyltransferase proteins and methods of making them - Google Patents
Lipid acyltransferase proteins and methods of making them Download PDFInfo
- Publication number
- WO2011045629A1 WO2011045629A1 PCT/IB2009/054535 IB2009054535W WO2011045629A1 WO 2011045629 A1 WO2011045629 A1 WO 2011045629A1 IB 2009054535 W IB2009054535 W IB 2009054535W WO 2011045629 A1 WO2011045629 A1 WO 2011045629A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- lipid acyltransferase
- enzyme
- amino acid
- polypeptide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1025—Acyltransferases (2.3)
- C12N9/1029—Acyltransferases (2.3) transferring groups other than amino-acyl groups (2.3.1)
-
- A—HUMAN NECESSITIES
- A21—BAKING; EDIBLE DOUGHS
- A21D—TREATMENT OF FLOUR OR DOUGH FOR BAKING, e.g. BY ADDITION OF MATERIALS; BAKING; BAKERY PRODUCTS
- A21D8/00—Methods for preparing or baking dough
- A21D8/02—Methods for preparing dough; Treating dough prior to baking
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
-
- A—HUMAN NECESSITIES
- A23—FOODS OR FOODSTUFFS; TREATMENT THEREOF, NOT COVERED BY OTHER CLASSES
- A23V—INDEXING SCHEME RELATING TO FOODS, FOODSTUFFS OR NON-ALCOHOLIC BEVERAGES AND LACTIC OR PROPIONIC ACID BACTERIA USED IN FOODSTUFFS OR FOOD PREPARATION
- A23V2002/00—Food compositions, function of food ingredients or processes for food or foodstuffs
Definitions
- the present invention relates to methods of engineering and producing variant enzymes.
- the present invention further relates to novel variant enzymes and to the use of these novel variant enzymes.
- Lipid cholesterol acyltransferase enzymes have been known for some time (see for example Buckley - Biochemistry 1983, 22, 5490-5493).
- glycerophospholipid cholesterol acyl transferases (GCATs) have been found, which like the plant and/or mammalian lecithinxholesterol acyltransferases (LCATs), will catalyse fatty acid transfer between phosphatidylcholine and cholesterol.
- GCATs cholesterol acyl transferases
- Upton and Buckley (TIBS 20, May 1995, pl78-179) and Brumlik and Buckley (J. of Bacteriology Apr. 1996, p2060-2064) teach a lipase/acyltransferase from Aeromonas hydrophila which has the ability to carry out acyl transfer to alcohol receptors in aqueous media.
- a putative substrate binding domain and active site of the A. hydrophila acyltransferase have been identified (see for example Thornton et al 1988 Biochem. et Biophys. Acta. 959, 153- 159 and Hilton & Buckley 1991 J. Biol. Chem. 266, 997-1000) for this enzyme.
- Buckley et al J. Bacteriol 1996, 178(7) 2060-4) taught that Serl6, Aspl l6 and His291 are essential amino acids which must be retained for enzyme activity to be maintained.
- Robertson et al J. Biol. Chem. 1994, 269, 2146-50
- Y226F, Y230F, Y30F, F13S, S18G, and S18V of the A. hydrophila acyltransferase none of which is encompassed by the present invention.
- WO 2005/066347 relates to variant lipid acyltransferases and methods of making same. These variant enzymes are not encompassed by the present invention.
- the development of variant lipid acyltransferases with modified (enhanced) activity has not been without difficulties, particularly in finding variant lipid acyltransferases which have high specific activity in a number of applications.
- the present invention is predicated upon the finding by the inventors of particular sites of interest for modification.
- the present inventors for the first time have modelled the lipid acyltransferase enzyme and using the tertiary structure of the enzyme identified specific regions for modification.
- the present inventors have prepared and tested enzymes modified in accordance with their findings above and identified advantageous variant lipid acyltransferases for use in a number of applications.
- a method for preparing a variant lipid acyltransferase enzyme comprising expressing in a host organism a nucleotide sequence which has at least 90% identity with a nucleotide sequence encoding a parent lipid acyltransferase and comprises at least one modification (suitably at least two modifications) at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located in a) the canyon region of the enzyme and/or b) insertion site 1 and/or c) insertion site 2, wherein the canyon region, insertion site 1 and/or insertion site 2 enzyme is defined as that region which when aligned based on primary or tertiary structure corresponds to the canyon region, insertion site 1 or msertion site 2 of the enzyme shown herein as SEQ ID No. 6 or SEQ ID No. 16.
- the modification(s) at a position located in the canyon and/or insertion site 1 and/or insertion site 2 is combined with at least one modification at a position which corresponds in the encoded amino acid sequence to an amino acid located outside of the canyon region and/or insertion site 1 and/or insertion site 2.
- a method for preparing a variant lipid acyltransferase enzyme comprising expressing in a host organism a nucleotide sequence which has at least 90% identity with a nucleotide sequence encoding a parent lipid acyltransferase and comprises at least one modification (suitably at least two modifications) at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located at position 27, 31, 85, 86, 122, 119, 120, 201, 245, 232, 235 and/or 236 (preferably at position 27, 31, 85, 86, 119 and/or 120, more preferably at position 27 and/or 31), wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- a method for preparing a variant lipid acyltransferase enzyme comprising expressing in a host organism a nucleotide sequence which has at least 90% identity with a nucleotide sequence encoding a parent lipid acyltransferase and comprises at least one modification at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located at position 27 and/or 31 in combination with at least one further modification, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 119, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207, 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the present invention provides a method of producing a variant lipid acyltransferase, said method comprising modifying a lipid acyltransferase amino acid sequence backbone such that at least one modification (suitably at least two modifications) is made at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located in a) the canyon region of the enzyme and/or b) insertion site 1 and/or c) insertion site 2, wherein the canyon region, insertion site 1 and/or insertion site 2 enzyme is defined as that region which when aligned based on primary or tertiary structure corresponds to the canyon region, insertion site 1 or insertion site 2, respectively, of the enzyme shown herein as SEQ ID No.
- the modification(s) at a position located in the canyon and/or insertion site 1 and/or insertion site 2 is combined with at least one modification at a position which corresponds in the encoded amino acid sequence to an amino acid located outside of the canyon region and/or insertion site 1 and/or insertion site 2.
- a method of producing a variant lipid acyltransferase comprising modifying a lipid acyltransferase amino acid sequence backbone such that at least one modification (suitably at least two modifications) is made at a posiiion(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located in position 27, 31, 85, 86, 122, 119, 120, 201, 245, 232, 235 and/or 236 (preferably at position 27, 31, 85, 86 119 and/or 120, more preferably at position 27 and/or 31), wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No.
- a method of producing a variant lipid acyltransferase comprising modifying a lipid acyltransferase amino acid sequence backbone such that at least one modification (suitably at least two modifications) is made at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located in position 27, 31 in combination with at least one further modification, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the en2yme shown herein as SEQ ID No. 6.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 119, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207, 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- a method comprising: altering the length of a substrate chain length specificity determining segment that lies immediately N- terminal to the catalytic triad (preferably the Asp residue of the catalytic triad) of a parent enzyme (e.g. a parent lipid acyltransferase) that has an amino acid sequence that is at least 70% identical to the lipid acyltransferase from A. salmonicida shown herein as SEQ ID No. 6 or 16, to produce an altered lipid acyltransferase enzyme that has an altered substrate specificity relative to said parent enzyme.
- a parent enzyme e.g. a parent lipid acyltransferase
- the altering comprises making an amino acid insertion or deletion in said substrate chain length specificity determining segment, such as substituting said substrate chain length specificity determining segment of said parent enzyme with the substrate chain length specificity determining segment of a different lipid acyltransferase to produce said altered lipid acyltransferase.
- said altering increases the length of acyl chain that can be transferred by said lipid acyltransferase.
- the method comprises testing said altered lipid acyltransferase enzyme for an altered substrate specificity.
- said testing may include evaluating the ability of said altered or variant lipid acyltransferase enzyme to transfer an acyl group from a substrate to a recipient molecule in an aqueous environment.
- said testing includes evaluating the ability of said variant lipid acyltransferase and said parent enzyme to transfer acyl chains of different lengths.
- the present invention yet further provides an altered or variant lipid acyltransferase comprising an amino acid sequence that is at least 70% identical to the lipid acyltransferase from Aeromonas salmonicida shown herein as SEQ ID No. 6 or 16, wherein a substrate chain length specificity determining segment that lies immediately N-terminal of the Asp residue of the catalytic triad of said altered lipid acyltransferase has an altered length relative to said lipid acyltransferase from Aeromonas salmonicida shown herein as SEQ ID No. 6 or 16.
- the altered lipid acyltransferase comprises an amino acid sequence that is at least 90% identical to the lipid acyltransferase from Aeromonas salmonicida shown herein as SEQ ID No. 6 or 16.
- the present invention provides a (variant) lipid acyltransferase polypeptide obtained by the method(s) according to the present invention.
- nucleic acid preferably an isolated or recombinant nucleic acid
- a vector comprising a nucleotide sequence encoding a lipid acyltransferase enzyme and which nucleotide sequence comprises at least one modification at a position which corresponds in the encoded amino acid sequence to one or more of the following positions: 27, 31, 85, 86, 122, 119, 120, 201, 245, 232, 235 and/or 236 (preferably at position 27, 31, 85, 86 119 and/or 120, more preferably at position 27 and/or 31), wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- nucleic acid preferably an isolated or recombinant nucleic acid
- a vector comprising a nucleotide sequence encoding a lipid acyltransferase enzyme and which nucleotide sequence comprises at least one modification at a position which corresponds in the encoded amino acid sequence to one or more of the following positions: 27 and/or 31, in combination with at least one further modification, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 1 19, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207, 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the nucleotide sequence encoding the lipid acyltransferase enzyme in accordance with the present invention and before modification is a nucleotide sequence shown herein as SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14 or SEQ ID No. 15; or is a nucleotide sequence which has at least 70% identity (preferably at least 80%, more preferably at least 90%, even more preferably at least 95% identity) with a nucleotide sequence shown herein as SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14 or SEQ ID No.
- nucleotide sequence which is related to SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14, SEQ ID No. 15 by the degeneration of the genetic code; or is a nucleotide sequence which hybridises under medium stringency or high stringency conditions to a nucleotide sequence shown herein as SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14 or SEQ ID No. 15.
- the present invention provides in one aspect an isolated or recombinant nucleic acid encoding a polypeptide having lipid acyltransferase activity and comprising a sequence having at least 94% (preferably at least 98%) amino acid sequence identity to the mature region of SEQ ID No.
- the present invention provides in one aspect an isolated or recombinant nucleic acid encoding a polypeptide having lipid acyltransferase activity and comprising a sequence having at least 94% (preferably at least 98%) amino acid sequence identity to the mature region of SEQ ID No. 6 or 16 and which comprises at least one modification (suitably at least two modifications) at a position(s) which corresponds in the encoded amino acid sequence to an amino acid(s) located in position 27 and/or 31 in combination with at least one further modification, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 119, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207, 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- nucleic acid preferably an isolated or recombinant nucleic acid
- a vector comprising a nucleotide sequence encoding a polypeptide having lipid acyltransferase activity, wherein the nucleotide sequence hybridises under medium or high stringency conditions over substantially the entire length of SEQ ID No. 10 or SEQ ID No. 25 or a compliment of SEQ ID No. 10 or SEQ ID No.
- the encoded polypeptide comprising one or more amino acid residues selected from Q, H, N, T, F, Y or C at position 31; R, Y, S, V, I, A, T, M, F, C or L at position 86; R, G, H, K, Y, D, N, V, C, Q, L, E, S or F at position 27; H, R, D, E 85; T or I at position 119; K or E at position 120; S, L, A, F, W, Y, R, H, or C at position 122; R at position 201; S as position 245; A or V at position 235; G or S at position 232; G or E at position 236, wherein the positions are equivalent amino acid positions with respect of SEQ ID No. 6.
- the present invention provides a variant lipid acyltransferase polypeptide encoded by the nucleic acid or nucleotide sequence according to the present invention.
- the present invention provides a variant lipid acyltransferase polypeptide encoded by a nucleic acid or nucleotide sequence according to the present invention when expressed in a Bacillus expression host, in particular in a B. Uc eniformis expression host.
- the present invention provides a method of producing a polypeptide comprising introducing the nucleic acid or a vector into a host cell (preferably a Bacillus expression host, in particular in a B. Ucheniformis expression host), wherein said nucleic acid or vector comprising said nucleotide sequence encoding said polypeptide operably linked to a regulatory sequence capable of directing expression of a polypeptide encoded by the nucleic acid, culturing the host cell under conditions in which the regulatory sequence directs expression of the polypeptide encoded by the nucleic acid or vector.
- a host cell preferably a Bacillus expression host, in particular in a B. Ucheniformis expression host
- the present invention provides a pro-peptide or a polypeptide which has lipid acyltransferase activity and comprises an amino acid sequence which is at least 90% (preferably at least 95%, more preferably at least 98%, more preferably at least 99%) identical with the amino acid sequence shown as SEQ ID No. 6 or 16 and comprises one or more modifications at one or more of the following positions: 27, 31, 85, 86, 122, 1 19, 120, 201, 245, 232, 235 and/or 236 (preferably at position 27, 31, 85, 86, 119 and/or 120 more preferably at position 27 and/or 31).
- the present invention provides a pro-peptide or a polypeptide which has lipid acyltransferase activity and comprises an amino acid sequence shown as SEQ ID No. 6 or 16 except for one or more modifications at one or more of the following positions: 27, 31, 85, 86, 122, 119, 120, 201, 245, 232, 235 and/or 236 (preferably at position 27, 31, 85, 86, 11 and/or 120 more preferably at position 27 and/or 31 ).
- the present invention provides a pro-peptide or a polypeptide which has lipid acyltransferase activity and comprises an amino acid sequence which is at least 90% (preferably at least 95%», more preferably at least 98%, more preferably at least 99%) identical with the amino acid sequence shown as SEQ ID No. 6 or 16 and comprises one or more modifications at positions 27 and/or 31 in combination with at least one further modification, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 119, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207, 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the present invention provides a pro-peptide or a polypeptide which has lipid acyltransferase activity and comprises an amino acid sequence shown as SEQ ID No. 6 or 16 except for one or more modifications at one or more of the following positions: 27 and/or 31 in combination with at least one further modification.
- the at least one further modification may be at one or more of the following positions 85, 86, 122, 119, 120, 201, 245, 23, 81, 82, 289, 227, 229, 233, 33, 207 and/or 130, wherein the position numbering is defined as that position which when aligned based on primary or tertiary structure corresponds to the same position of the enzyme shown herein as SEQ ID No. 6.
- the polypeptide of the present invention may be a pro-peptide which undergoes further post- translational modification to a mature peptide, i.e. a polypeptide which has lipid acyltransferase activity.
- SEQ ID No. 16 is the same as SEQ ID No. 6 except that SEQ ID No. 16 has undergone post-translational and/or post-transcriptional modification to remove some amino acids, more specifically 38 amino acids. Therefore the polypeptide shown herein as SEQ ID No. 6 could be considered in some circumstances (i.e. in some host cells) as a pro-peptide - which is further processed to a mature peptide by post- translational and/or post-transcriptional modification.
- cleavage site(s), in respect of the post-translational and/or post-transcriptional modification may vary slightly depending on host species. In some host species there may be no post translational and/or post-transcriptional modification, hence the pro-peptide would then be equivalent to the mature peptide (i.e. a polypeptide which has lipid acyltransferase activity).
- the cleavage site(s) may be shifted by a few residues (e.g. 1, 2 or 3 residues) in either direction compared with the cleavage site shown by reference to SEQ ID No. 16 compared with SEQ ID No. 6.
- the cleavage may commence at residue 232, 233, 234, 235, 236, 237 or 238 for example.
- the cleavage may end at residue 270, 271, 272, 273, 274, 275 or 276 for example.
- the cleavage may result in the removal of about 38 amino acids, in some embodiments the cleavage may result in the removal of between 30-45 residues, such as 34- 42 residues, such as 36-40 residues, preferably 38 residues.
- the present invention further provides a method of making a foodstuff comprising adding a polypeptide according to the present invention to one or more ingredients of the foodstuff.
- the present invention provides a method of making a baked product comprising adding a polypeptide according to the present invention to a dough and baking the dough to make the baked product.
- the present invention provides the use of a variant lipid acyltransferase enzyme of the present invention or obtainable (preferably, obtained) by the method according to the present invention in a process of treating egg or egg-based products to produce lysophospholipids .
- the present invention further provides a method of preparing a lyso-phospholipid comprising treating a phospholipid with a polypeptide according to the present invention to produce the lyso-phospholipid.
- the present invention provides a method of preparing a lyso- glycolipid comprising treating a glycolipid with a polypeptide according to the present invention to produce a lyso-glycolipid.
- the present invention further provides a process of enzymatic degumming of vegetable or edible oils, comprising treating the edible or vegetable oil with a polypeptide according to the present invention so as to hydrolyse a major part of the polar lipids present therein.
- a variant lipid acyltransferase enzyme according to the present invention or obtainable (preferably, obtained) by the method according to the present invention in a process for reducing the content of a phospholipid in an edible oil, comprising treating the oil with said variant lipolytic enzyme so as to hydrolyse a major part of the phospholipid, and separating an aqueous phase containing the hydrolysed phospholipid from the oil.
- the present invention provides a foodstuff or a baked product obtained by the method of the present invention.
- the present invention provides a cleaning composition or a detergent composition comprising a variant lipid acy!transferase according to the present invention.
- a bread and/or dough improving composition comprising a variant lipid acyltransferase according to the present invention.
- the present invention also encompasses methods of expressing the nucleotide sequence for use in the present invention using the same, such as expression in a host cell; including methods for transferring same.
- the present invention further encompasses methods of isolating the nucleotide sequence, such as isolating from a host cell.
- Other aspects concerning the amino acid sequence for use in the present invention include: a construct encoding the amino acid sequences for use in the present invention; a vector encoding the amino acid sequences for use in the present invention; a plasmid encoding the amino acid sequences for use in the present invention; a transformed cell expressing the amino acid sequences for use in the present invention; a transformed tissue expressing the amino acid sequences for use in the present invention; a transformed organ expressing the amino acid sequences for use in the present invention; a transformed host expressing the amino acid sequences for use in the present invention; a transformed organism expressing the amino acid sequences for use in the present invention.
- the present invention also encompasses methods of purifying the amino acid sequence for use in the present invention using the same, such as expression in a host cell; including methods of transferring same, and then purifying said sequence. DETAILED ASPECTS
- amino acid position numbering as used herein unless stated otherwise relates to the corresponding position when aligned based on primary and/or tertiary structure (preferably primary structure) to the enzyme shown herein as SEQ ID No. 6. In some embodiments however the amino acid position numbering as used herein may relate to the corresponding position when aligned based on tertiary structure to the enzyme shown herein as SEQ ID No. 16.
- amino acid position numbering as used herein may relate to the corresponding position when aligned based on tertiary structure to the enzyme shown herein as SEQ ID No. 16.
- the modification(s) result in improved properties which may be include a) altering the substrate specificity of the lipid acyltransferase, for instance and by way of example only: i) altering the enzymes ability to use certain compounds as acceptors, for example improving the enzymes ability to utilise a carbohydrate (e.g.
- maltose as an acceptor molecule thus improving the enzymes ability to produce a carbohydrate ester): or ii) altering the enzymes ability to use saturated or unsaturated fatty acids as a substrate: or iii) changing the enzymes specificity such that it preferentially utilises the fatty acid from the Snl or Sn2 position of the lipid: or iv) altering the substrate chain length specificity of the enzyme; b) altering the kinetics of the enzyme; and/or c) lowering the enzymes ability to carry out a hydrolysis reaction whilst maintaining or enhancing the enzymes ability to carry out an acyl transferase reaction.
- the tertiary structure of the lipid acyltransferases has revealed an unusual and interesting structure which allows lipid acyltransferases to be engineered more successfully.
- the lipid acyltransferase tertiary structure has revealed a cave and canyon structure.
- Alterations in the cave region may (for example) alter the enzyme's substrate chain length specificity for example. Alterations in the canyon (particularly some preferred key modifications) have been found to be important in for example enhancing or changing the enzyme's substrate specificity.
- positions 31, 27, 85, 86, 119 and 120 can be found at positions 31, 27, 85, 86, 119 and 120.
- positions 31 and/or 27 are highly preferred.
- lipid acyltransferase enzymes in the cave and/or canyon region preferably the canyon region thereof to provide modified enzymes having altered activity/properties.
- At least one modification in the canyon region in combination with at least one modification either elsewhere in the canyon or outside of the canyon.
- alternations are not within the cave region.
- the present invention may provide a method of modifying a lipid acyltransferase (either by modifying the nucleotide sequence or the amino acid sequence) thus to result in one or modifications in the following residues: 27, 31, 85, 86, 122, 119, 120, 201, 245, 232, 235, 236, and nucleic acids and lipid acyltransferases having said modification(s).
- the present invention may provide a method for modifying a lipid acyltransferase (either by modifying the nucleotide sequence or the amino acid sequence) thus to result in one or modifications in the following residues: 27, 31, 85, 86, 122, 119, 120, 201, 245, this/these can in some instances be in combination with one or more further modifications in one or more of the following residues 23, 81, 82, 289, 227, 229, 233, 33, 207 or 130.
- the present invention also provides nucleic acids and lipid acyltransferases having these modification(s).
- the variant enzyme must comprise at least one amino acid modification compared with the parent enzyme.
- the variant enzyme may comprise at least 2, preferably at least 3, preferably at least 4 amino acid modifications compared with the parent enzyme.
- the DNA sequence encoding the lipid acyltransferase shown herein as SEQ ID No. 6 or 16 is modified.
- the methods according to the present invention may comprise a further step of formulating the variant enzyme into an enzyme composition and/or a foodstuff composition, such as a bread improving composition.
- the present invention further provides for a bread improving composition comprising said variant lipid acyltransferase according to the present invention.
- the method of producing a variant lipid acyltransferase enzyme further comprises one or more of the following steps:
- An amino acid residue of a lipid acyltransferase may be equivalent to a residue of the lipid acyltransferase shown herein as SEQ ID No. 16 or 6 if it is either homologous (i.e. having a corresponding position in either the primary and/or tertiary structure) or analogous to a specific residue or portion of that residue in the lipid acyltransferase shown in SEQ ID no. 6 or 16 (i.e. having the same or similar functional capacity to combine, react, and/or chemically interact).
- the amino acid sequence of a lipid acyltransferase is directly compared to the lipid acyltransferase enzyme shown herein as SEQ ID No. 6 or 16 primary sequence and particularly to a set of residues known to be invariant in all or most lipid acyltransferases for which sequences are known.
- the residues equivalent to particular amino acids in the primary sequence of SEQ ID No. 6 or 16 are defined.
- alignment of conserved residues conserves 100% of such residues.
- alignment of greater than 75% or as little as 50% of conserved residues are also adequate to define equivalent residues.
- conservation of the catalytic serine and histidine residues are maintained. conserveed residues are used to define the corresponding equivalent amino acid residues of the lipid acyitransferase shown in SEQ ID No. 6 or 16 in other lipid
- acyltransferases such as from other Aeromonas species, as well as any other organisms.
- sequence alignment such as pairwise alignment can be used fhttp://www.ebi.ac.ul ⁇ emboss/aligji/index.html).
- equivalent amino acids in alternative parental lipid acyitransferase polypeptides which correspond to one or more of the amino acids defined with reference to SEQ ID No. 16 or SEQ ID No. 6 can be determined and modified.
- emboss pairwise alignment standard settings usually suffice.
- Corresponding residues can be identified using "needle" in order to make an alignment that covers the whole length of both sequences.
- equivalent residues may be defined by determining homology at the level of tertiary structure for a lipid acyitransferase whose tertiary structure has been determined by x-ray crystallography.
- "equivalent residues” are defined as those for which the atomic coordinates of two or more of the main chain atoms of a particular amino acid residue of the lipid acyitransferase shown herein as SEQ ID No. 6 or 16 (N on N, CA on CA, C on C, and O on O) are within 0.13nm and preferably 0.1 nm after alignment.
- Alignment is achieved after the best model has been oriented and positioned to give the maximum overlap of atomic coordinates of non-hydrogen protein atoms of the lipid acyitransferase in question to the lipid acyitransferase shown herein as SEQ ID No. 6 or 16.
- the best model is the crystallographic model giving the lowest R factor for experimental diffraction data at the highest resolution available.
- Equivalent residues which are functionally and/or structurally analogous to a specific residue of the lipid acyitransferase as shown herein as SEQ ID No.
- 6 or 16 are defined as those amino acids of the lipid acyltransferase that preferentially adopt a conformation such that they either alter, modify or modulate the protein structure, to effect changes in substrate specification, e.g. substrate binding and/or catalysis in a manner defined and attributed to a specific residue of the lipid acyltransferase shown herein as SEQ ID No. 6 or 16.
- residues of the lipid acyltransferase in cases where a tertiary structure has been obtained by x-ray crystallography), which occupy an analogous position to the extent that although the main chain atoms of the given residue may not satisfy the criteria of equivalence on the basis of occupying a homologous position, the atomic coordinates of at least two of the side chain atoms of the residue lie with 0.13 nm of the corresponding side chain atoms of the lipid acyltransferase shown herein as SEQ ID No. 6 or 16.
- SEQ ID No. 16 which is a Aeromonas salmonicida lipid acyltransferase comprising an N80D mutation
- Crystallization Crystals of the enzyme were obtained using hanging drop diffusion whereby a drop (5 ⁇ 1 protein solution ( mg/ml in ) was mixed with 5 ⁇ 1 of a reservoir solution 1.4 M ammonium phosphate, 0.10 M potassium phosphate, 44 mM sodium chloride, 94 mM ammonium chloride, 2 mM magnesium sulphate, 2 mM calcium chloride) was placed on a plastic coverslip and inverted over a well of a 6x4 Linbro plate containing 1 ml of a reservoir solution and allowed to reach equilibrium for a period lasting several days to 2 weeks. Crystals of the (NH4)2PtC14 were obtained by soaking pregrown crystals in the reservoir solution above containing lOmM (NH4)2PtC14.
- Multiwavelength anomalous diffraction data were collected for the derivative at the Stanford Synchrotron Radiation Laboratory (SSRL, Menlo Park, USA) on beamline 11.1 at wavelengths corresponding to the inflection ( ⁇ ), low energy remote (XT), and the peak ( ⁇ 3) of a platinum MAD experiment. Later, a data set ( ⁇ ) was collected to 1.5 A resolution. The data sets were collected at ⁇ 00 ⁇ using Quantum 315 CCD. Data were integrated using Mosflm (Leslie, A.G.W. (1999) Acta Crystallogr., D55, 1696- 1702) and scaled with the SCALA program from the CCP4 suite (Collaborate computer project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Chrystallogr., D50, 760-763). Data statistics are summarized in Table A.
- Multiwavelength anomalous diffraction data were collected for the (NH 4 ) 2 PtCl 4 derivative at the Stanford Synchrotron Radiation Laboratory (SSRL, Menlo Park, USA) on beamline 11.1 at wavelengths corresponding to the peak ( ⁇ ) of a platinum SAD experiment. Later, a data set ( ⁇ ) was collected to 1.5 A resolution. The data sets were collected at 100K using Quantum 315 CCD. Data were integrated using Mosflm [supra] and scaled with the SCALA program from the CCP4 suite [supra]. Data statistics are summarized in the Table below (mod).
- the initial structure was determined using the 2.8 A platinum SAD data ( ⁇ 3 or ⁇
- the final model includes a protein dimer and 1 198 water molecules in the asymmetric unit. No electron density was observed for the first methionine residue in any of the molecules.
- PROCHECK11 (Laskowski, G.N. et al. (1993) J. Appl. Chrystallogr., 26, 91- 07.) indicates that 94% of the residues in all of the monomers are located in the core regions of the Ramachandran plot (Ramachandran, G. N. et al. (1 68) Advan. Protein Chem., 23, 283- 437) with no residues in the disallowed or generously allowed regions.
- ESU Estimated overall coordinate error (Tickle et al., 1998).
- Rcryst ⁇
- F ca i c and F 0 b s are the calculated and observed structure factor amplitudes, respectively.
- Rfree as for Rcryst, but for 5.0% of the total reflections chosen at random and omitted from refinement.
- the crystal structure shows that the enzyme is an catalytic triad is composed of Ser 16, Asp 288 and His 291.
- a structural-homoiogy search may be performed using the program DALI (Holm and Sander, Trends Biochem. Sci., 478-480 [1 95]), which is based on a distance criterion and does not use sequence information for the comparison to obtain closely related proteins.
- DALI Holm and Sander, Trends Biochem. Sci., 478-480 [1 95]
- IIVN E. coli thioesterase I
- the thioesterase I is an enzyme which has a very different activity and specificity compared with lipid acyltransferases and it has a completely different amino acid sequence when viewed on a primary level.
- the tertiary structure of thioesterase I can be compared with lipid acyltransferases. The fact that these enzymes have very different activities despite the structural similarities are useful when engineering lipid acyltransferases and selecting modifications as the small differences in the tertiary structure (e.g. the cave, canyon regions and the insertion regions must be contributing to the different activity of the lipid acyltransferase enzyme compared with the thioesterase I (IIVN).
- lipid acyltransferase was found to share a common structural motif, having the five-stranded parallel ⁇ -sheet structure sandwiched by a-helices on either side.
- the three-dimensional structure of the Aeromonas salmonicida lipid acyltransferase which has undergone post-translational modification (i.e. clipping) and which comprises the N80D mutation (SEQ ID No. 16) showed that lipid acyltransferases, in particular the lipid acyltransferase enzyme, contains large insertions between common elements of secondary structure.
- Segments 3 and 4 precede insertions 3 and 4 respectively, and segment 5 immediately follows insertion 4. Insertions 4 and 5 also contribute to the over enclosure resulting in the cave, thus the cave is different to the canyon in that insertions 1 and 2 form the lining of the canyon while insertions 3 and 4 form the overlaying structure. Insertions 3 and insertion 4 cover the cave.
- a lipid acyltransferase may be altered by modifying the amino acid residues in one or more of the canyon, the cave, the insertion 1, the insertion 2, the insertion 3 or the insertion 4. In one embodiment of the present invention a lipid acyltransferase may be altered by modifying the amino acid residues in one or more of the canyon, insertion 1 or insertion 2.
- the dimensions of the acyl chain binding cavity of a lipid acyltransferase may be altered by making changes to the amino acid residues that form the larger cave. This may be done by modulating the size the regions that link the common features of secondary structure as discussed above.
- the size of the cave may be altered by changing the amino acids in the region between the last (fifth) beta strand of the enzyme and the Asp-X-X- His motif that forms part of the catalytic triad.
- the substrate chain length specificity determining segment of a lipid acyltransferase is a region of contiguous amino acids that lies between the ⁇ 5 ⁇ -strand of the enzyme and the Asp residue of the catalytic triad of that enzyme (the Asp residue being part of the Asp-Xaa-Xaa- His motif).
- GID:33357066 each showing a signature three-layer alpha/beta/alpha structure, where the beta-sheets are composed of five parallel strands allow the substrate chain length specificity determining segments of each of the lipid acyltransferase enzymes to be determined.
- the substrate chain length specificity determining segment of the Aeromonas salmonicida lipid acyltransferase lies immediately N-terminal to the Asp residue of the catalytic triad of the enzyme.
- the length of the substrate chain length specificity determining segment may vary according to the distance between the Asp residue and the ⁇ 5 ⁇ -strand of the enzyme.
- the substrate chain length specificity determining segments of the lipid acyltransferase are about 13 amino, 19 amino acids and about 70 amino acids in length, respectively.
- a substrate chain length specificity determining segment may be in the range of 10 to 70 amino acids in length, e.g., in the range of 10 to 30 amino acids in length, 30 to 50 amino acids in length, or 50 to 70 amino acids.
- the Table below provides an exemplary sequence for the substrate chain length specificity determining segment of the lipid acyltransferase enzyme
- the amino acid sequence of a substrate chain length specificity determining segment may or may not be the amino acid sequence of a wild-type enzyme.
- the substrate chain length specificity determining segment may have an amino acid sequence that is at least 70%, e.g., at least 80%, at least 90% or at least 95% identical to the substrate chain length specificity determining segment of a wild type lipid acyltransferase.
- the variant enzyme may be prepared using site directed mutagenesis.
- the mutations may be prepared randomly for instance using a commercial kit such as the GeneMorph PCR mutagenesis kit from Stratagene, or the Diversify PCR random mutagenesis kit from Clontech.
- EP 0 583 265 refers to methods of optimising PCR based mutagenesis, which can also be combined with the use of mutagenic DNA analogues such as those described in EP 0 866 796.
- Error prone PCR technologies are suitable for the production of variants of lipid acyl transferases with preferred characteristics.
- WO0206457 refers to molecular evolution of lipases.
- a third method to obtain novel sequences is to fragment non-identical nucleotide sequences, either by using any number of restriction enzymes or an enzyme such as Dnase I, and reassembling full nucleotide sequences coding for functional proteins (hereinafter referred to as "shuffling").
- shuffling Alternatively one can use one or multiple non-identical nucleotide sequences and introduce mutations during the reassembly of the full nucleotide sequence.
- DNA shuffling and family shuffling technologies are suitable for the production of variants of lipid acyl transferases with preferred characteristics. Suitable methods for performing 'shuffling' can be found in EPO 752 008, EP1 138 763, EP1 103 606. Shuffling can also be combined with other forms of DNA mutagenesis as described in US 6,180,406 and WO 01/34835.
- key modifications include one or more of the following modifications: L31Q, H, N, T, F, Y or C (preferably L31 Q); M27R, G, H, K, Y, D, N, V, C, Q, L, E, S or F (preferably M27V); V85H, R, D or E; I86R,Y, S, V, I, A, T, M, F, C or L (preferably I86S or A); Al 19T or I; Y120K or E; W122S, L or A (preferably W122L); E201R; Q245S; F235A or V; W232G or S; and/or A236G or E.
- the modification(s) are made at one or more of the following positions: 31 , 27, 85, 86, 1 19, 120.
- key modifications in the canyon include one or more of the following modifications: L31Q, H, N, T, F, Y or C (preferably L31 Q); 27R, G, H, , Y, D, N, V, C, Q, L, E, S or F (preferably M27V); V85H, R, D or E; I86R,Y, S, V, I, A, T, M, F, C or L (preferably I86S or A); Al 19T or I; Y120K or E, which may be in combination with one another and/or in combination with a further modification.
- the modifications are made at one or more positions 31 and/or 27.
- the modifications may be L31Q, H, N, T, F, Y or C (preferably L31 Q) and/or M27R, G, H, K, Y, D, N, V, C, Q, L, E, S or F (preferably M27V).
- the modifications are made at positions are 085, 086.
- the modifications may be V85H, R, D or E and/or I86R, Y, S, V, I, A, T, M, F, C or L.
- the modification may be Q245S.
- the modification is made in at least insertion site 1.
- a modification is made in at least insertion site 1 in combination with a further modification in insertion site 2 and/or 4 and/or at one or more of the following positions 119, 120, 122, 201, 77, 130, 82, 120, 207, 167, 227, 215, 230, 289.
- a modification is made in at least the canyon region in combination with a further modification in insertion site 4 and/or at one or more of the following positions 122, 201, 77, 130, 82, 120, 207, 167, 227, 215, 230, 289.
- Preferred modifications are given for particular site:
- the modification may be one or more of the following: L31Q, H, N, T, F, Y or C (preferably L31 Q); M27R, G, H, , Y, D, N, V, C, Q, L, E, S or F (preferably M27V); V85H, R, D or E; I86R,Y, S, V, I, A, T, M, F, C or L (preferably I86S or A); Al 19T or I; Y120K or E; W122S, L or A (preferably W122L); E201R; Q245S; F235A or V; W232G or S; and/or A236G or E) suitably the variant lipid acyltransferase may be additionally modified at one or more of the following positions 130, 82, 121, 74, 83, 77, 207
- the lipid acyltransferase backbone when aligned (on a primary or tertiary basis) with the lipid acyltransferase enzyme shown herein as SEQ ID No. 6 preferably has D in position 80.
- N80D we have therefore shown in many of the combinations taught herein N80D as a modification. If N80D is not mentioned as a suitable modification and the parent backbone does not comprise D in position 80, then an additional modification of N80D should be incorporated into the variant lipid acyltransferase to ensure that the variant comprises D in position 80.
- N80D modification is a preferred modification and a backbone enzyme or parent enzyme is preferably used which already possesses amino acid D in position 80. If, however, a backbone is used which does not contain amino acid D in position (such as one more of the lipid acyltransferases shown here as SEQ ID No. 1, 3, 4, 5, 8, or 9 for instance) then preferably an additional modification of N80D is included.
- the present invention relates to a method of producing a variant lipid acyltransferase, said method comprising:
- substitution at position 27 or 31 is identified by alignment of the parent sequence with SEQ ID No. 16 or SEQ ID No. 6.
- the substitution at position 31 may be a substitution to an amino acid residue selected from the group consisting of: Q, H, Y, N, T, F, Y and C, preferably Q.
- the substitution at position 27 may be a substitution to an amino acid residue selected from the group consisting of: , G, H, , Y, D, N, V, C, Q and L preferably V.
- the present invention also relates to a method of preparing a variant lipid acyltransferase said method comprising:
- lipid acyltransferase a) substituting at least one amino acid residue at at least position 31 of a parent lipid acyltransferase to glutamine, which position is identified by alignment of the parent sequence with SEQ ID No. 16 or SEQ ID No. 6; b) wherein the variant lipid acyltransferase has a maltose transferase activity of at least 0.0282% when using the maltose transferase assay provided in protocol 1.
- the present invention further relates to a variant lipid acyltransferase obtainable (or obtained) by any of the methods of the present invention.
- the substitution at position 31 identified by alignment of the parent sequence with SEQ ID No. 1 or SEQ ID No. 6 may be a substitution to an amino acid residue selected from the group consisting of: Q, H, Y and F, preferably Q.
- the variant polypeptide comprises one or more further modification(s) at any one or more of amino acid residue positions: 27, 77, 80, 82, 85, 85, 86, 121, 122, 130, 167, 207, 227, 230 and 289, which position is identified by alignment of the parent sequence with SEQ ID No. 16,
- at least one of the one or more further modification(s) may be at amino acid residue position: 86, 122 or 130, which position is identified by alignment of the parent sequence with SEQ ID No, 16,
- the variant lipid acyltransferase comprises one or more of the following further substitutions: 186 (A, C, F, L, M, S, T, V, R, I or Y); W 122 (S, A, F, W, C, H, L, M, R or Y); R130 A, C, D, G, H, I, K, L, M, N, Q, T, V, R, F or Y); or any combination thereof.
- the variant lipid acyltransferase may comprises at least one mutation as taught in Tables 1 and/or 2 of the Examples presented herewith or a combination of modifications as taught in Tables 3 and/or 4 of the Examples presented herewith.
- the variant lipid acyltransferase may comprise one of the following combinations of modifications (where the parent back bone already comprises amino acid D in position 80, the modification can be expressed without reference to N80D):
- G67A, N80D, V85H wherein said positions are identified by alignment of the parent sequence with SEQ ID No. 16 or SEQ ID No. 6.
- the variant lipid acyltransferase may be identical to the parent lipid acyltransferase except for a modification at position 31 and, optionally, one or more further modification(s) at any one or more of amino acid residue positions: 27, 77, 80, 82, 85, 85, 86, 121, 122, 130, 167, 207, 227, 230 and 289, which position is identified by alignment of the parent sequence with SEQ ID No. 16 or SEQ ID No. 6.
- the variant lipid acyltransferase may be identical to the parent lipid acyltransferase except for a modification at position 31 and, optionally, one or more further modification(s) at any one or more of amino acid residue positions: 86, 122 or 130, which position is identified by alignment of the parent sequence with SEQ ID No. 16 or SEQ ID No. 6.
- the variant polypeptide has any one of the modifications as detailed above, except for a modification at position 80.
- SEQ ID No. 6, SEQ ID No. 16 or a polypeptide encoded by SEQ ID No. 10 or SEQ ID No. 25 will already have aspartic acid at position 80, when said positions are identified by alignment of the parent sequence with SEQ ID No. 6.
- the variant lipid acyltransferase or the variant lipid acyltransferase obtainable by a method according to the present invention may have at least 75% identity to the parent lipid acyltransferase, suitably the variant lipid acyltransferase may have at least 75% or at least 80% or at least 85% or at least 90% or at least 95% or at least 98% identity to the parent lipid acyltransferase.
- the present invention also relates to a variant polypeptide having lipid acyltransferase activity, wherein the variant comprises a modification at at least position 31 compared to a parent lipid acyltransferase, wherein position 31 is identified by alignment with SEQ ID No. 16 or SEQ ID No. 6 and wherein the variant lipid acyltransferase has a maltose transferase activity of at least 0.0282% when using the maltose transferase assay provided in protocol 1.
- the variant lipid acyltransferase has the following modifications and/or the following modifications are made in the methods of the present invention:
- the variant lipid acyltransferase has the following modifications and/or the following modifications are made in the methods of the present invention:
- Al 19T, N80D (which can be expressed as Al 19T where the backbone enzyme already has D in position 80);
- A119A, N80D (which can be expressed as A119A where the backbone enzyme already has D in position 80);
- G67A, V85H, N80D which can be expressed as G67A, V85H where the backbone enzyme already has D in position 80).
- the present invention provides a variant polypeptide having lipid acyltransferase activity, wherein the variant comprises a modification at at least position 31 compared to a parent lipid acyltransferase, wherein position 31 is identified by alignment with SEQ ID No. 16 and wherein the variant lipid acyltransferase has a carbohydrate (preferably maltose) transferase activity of at least 0.0282% when using the carbohydrate transferase assay provided in protocol 1 herein after disclosed.
- the improved property relates to an improved carbohydrate transferase activity (e.g.
- the variant lipid acyltransferase has a carbohydrate transferase activity (e.g. maltose transferase activity) of at least 0.0282% when using the maltose transferase assay provided in protocol 1 herein defined below.
- modifying means adding, substituting and/or deleting.
- modifying means "substituting”.
- lipid acyltransferase as used herein means an enzyme which has acyltransferase activity (for example an enzyme classified as E.C. 2.3.1.x, in particular 2.3.1.43 in accordance with the Enzyme Nomenclature Recommendations (1992) of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology), whereby the enzyme is capable of transferring an acyl group from a lipid to one or more acceptor substrates, such as one or more of the following: a sterol; a stanol; a carbohydrate (e.g. maltose and/or glucose); a protein; a protein subunit; cholesterol, an alcohol, glycerol.
- acceptor substrates such as one or more of the following: a sterol; a stanol; a carbohydrate (e.g. maltose and/or glucose); a protein; a protein subunit; cholesterol, an alcohol, glycerol.
- galactolipid as
- phospholipid as used herein means lecithin, including phosphatidylcholine.
- lecithin as used herein encompasses phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine and phosphatidylglycerol .
- polar lipid as used herein means a phospholipid and/or a galactolipid, preferably a phospholipid and a galactolipid.
- transferase as used herein is interchangeable with the term “lipid acyltransferase”.
- galactolipid transferase activity means the ability of the enzyme to catalyse the transfer of an acyl group from a galactolipid donor to an acceptor molecule (other than water).
- phospholipids transferase activity means the ability of the enzyme to catalyse the transfer of an acyl group from a phospholipids donor to an acceptor molecule (other than water).
- the lipid acyltransferase as defined herein catalyses one or more of the following reactions: interesterification, transesterification, alcoholysis, hydrolysis.
- the lipid acyltransferase as defined herein catalyses one or more of the following reactions: transesterification, alcoholysis.
- the term “interesterification” refers to the enzymatic catalysed transfer of acyl groups between a lipid donor and lipid acceptor, wherein the lipid donor is not a free acyl group.
- transesterification as used herein means the enzymatic catalysed transfer of an acyl group from a lipid donor (other than a free fatty acid) to an acyl acceptor (other than water).
- alcoholysis refers to the enzymatic cleavage of a covalent bond of an acid derivative by reaction with an alcohol ROH so that one of the products combines with the H of the alcohol and the other product combines with the OR group of the alcohol.
- alcohol refers to an alkyl compound containing a hydroxyl group.
- hydrolysis refers to the enzymatic catalysed transfer of an acyl group from a lipid to the OH group of a water molecule. Acyl transfer which results from hydrolysis requires the separation of the water molecule.
- improved properties or “improved activity” as used herein means that the variant enzyme has a property or activity which is more desired (i.e. it could be higher or lower) when compared with that property or activity of the parent (or backbone) enzyme.
- an increased ratio of transferase activity compared with hydrolysis activity means the variant enzyme when compared with the parent enzyme is able to catalyse lipid transferase at a higher rate compared with lipid hydrolysis. This may mean that both lipid transferase activity and lipid hydrolysis activity are increased compared with the parent enzyme or that lipid transferase activity is increased whilst lipid hydrolysis activity is decreased compared with the parent enzyme. It is the final relation between the two activities which is important.
- the lipid substrate upon which the parent lipid acyltransferase and/or the variant lipid acyltransferase according to the present invention acts is one or more of the following lipids: a phospholipid, such as a lecithin, e.g. phosphatidylcholine, a triacylglyceride, a cardiolipin, a diglyceride, or a glycolipid, such as digalactosyldiglyceride (DGDG) or monogalactosyldiglyceride (MGDG) for example.
- a phospholipid such as a lecithin, e.g. phosphatidylcholine, a triacylglyceride, a cardiolipin, a diglyceride, or a glycolipid, such as digalactosyldiglyceride (DGDG) or monogalactosyldiglyceride (MGDG) for example.
- DGDG digalactosyldiglycer
- This lipid substrate may be referred to herein as the "lipid acyl donor”.
- the lipid substrate upon which the parent lipid acyltransferase and/or the variant lipid acyltransferase acts is a phospholipid, such as lecithin, for example phosphatidylcholine .
- the lipid substrate may be a food lipid, that is to say a lipid component of a foodstuff.
- the lipid substrate or lipid acyl donor may be one or more lipids present in one or more of the following substrates: fats, including lard, tallow and butter fat; oils including oils extracted from or derived from palm oil, sunflower oil, soya bean oil, safflower oil, cotton seed oil, ground nut oil, corn oil, olive oil, peanut oil, coconut oil, and rape seed oil.
- Lecithin from soya, rape seed or egg yolk is also a suitable lipid substrate.
- the lipid substrate may be an oat lipid or other plant based material containing galactolipids.
- the lipid acyl donor is preferably lecithin (such as phosphatidylcholine) in egg yolk.
- the lipid may be selected from lipids having a fatty acid chain length of from 8 to 22 carbons.
- the lipid may be selected from lipids having a fatty acid chain length of from 16 to 22 carbons, more preferably of from 16 to 20 carbons.
- the lipid may be selected from lipids having a fatty acid chain length of no greater than 14 carbons, suitably from lipids having a fatty acid chain length of from 4 to 14 carbons, suitably 4 to 10 carbons, suitably 4 to 8 carbons.
- the variant lipid acyltransferase according to the present invention may utilize a protein as the acyl acceptor.
- the protein may be one or more of the proteins found in a food product, for example in a dairy product and/or a meat product.
- suitable proteins may be those found in curd or whey, such as lactoglobulin.
- Other suitable proteins include ovalbumin from egg, gliadin, glutenin, puroindoline, lipid transfer proteins from grains, and myosin from meat.
- the parent enzymes may include any enzyme with esterase or lipase activity - and therefore an esterase or a lipase could be used as a parent enzyme in the methods of the present invention in some instances.
- the parent enzyme is a lipid acyltransferase enzyme.
- the parent enzyme aligns to the pfam00657 consensus sequence.
- parent lipid acyltransferase and “parent sequence” is used interchangeably herein.
- the "parent lipid acyltransferase” may refer to the sequence that is modified (i.e. the sequence to which a modification is made).
- the parent lipid acyl transferase for use in any one of the methods of the present invention may be a natural lipid acyl transferase or a variant lipid acyl transferase.
- nucleotide sequence encoding a parent lipid acyl transferase for use in the present invention may be one as described in WO2004/064537, WO2004/064987, WO2005/066347, or WO2006/008508. These documents are incorporated herein by reference.
- lipid acyltransferase as used herein means an enzyme which has acyltransferase activity (for example an enzyme classified as E.C. 2.3.1.x, in particular 2.3.1.43 in accordance with the Enzyme Nomenclature Recommendations (1992) of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology), whereby the enzyme is capable of transferring an acyl group from a lipid to one or more acceptor substrates, such as one or more of the following: a sterol; a stanol; a carbohydrate (e.g. maltose and/or glucose); a protein; a protein subunit; cholesterol, an alcohol, glycerol.
- acceptor substrates such as one or more of the following: a sterol; a stanol; a carbohydrate (e.g. maltose and/or glucose); a protein; a protein subunit; cholesterol, an alcohol, glycerol.
- the parent and/or variant lipid acyltransferase is one classified under the Enzyme Nomenclature classification (E.C. 2.3.1.43).
- Suitable lipid acyltransferases for use as parent enzymes in accordance with the present invention may be identified by use of the following assay entitled “Lipid acyltransferase assay”.
- variant enzymes in accordance with the present invention also preferably are lipid acyltransferases when classified using the "Lipid acyltransferase assay” and/or the assay detailed in Protocol 1 and/or the assay detailed in Protocol 2 shown below.
- Substrate 50 mg Cholesterol (Sigma C8503) and 450 mg Soya phosphatidylcholine(PC), Avanti #441601 is dissolved in chloroform, and chloroform is evaporated at 40°C under vacuum.
- 250 ⁇ substrate is added in a glass with lid at 40°C.
- the enzyme added should esterify 2-5% of the cholesterol in the assay.
- the amount of cholesterol ester is analysed by HPTLC using Cholesteryl stearate
- Transferase activiiy is calculated as the amount of cholesterol ester formation per minute under assay conditions.
- Transferase Unit is defined as ⁇ cholesterol ester produced per minute at 40°C and pH 7 in accordance with the transferase assay given above. If the enzyme exhibits a specific transferase unit (TrU) per mg enzyme of at least 25 TrU/mg enzyme protein then it would be considered a lipid acyitransferase suitable for use as a parent enzyme in the methods of the present invention. In addition preferably the variant lipid acyltransferases of the present invention and for use in the present invention have a specific transferase unit (TrU) per mg enzyme of at least 25 TrU/mg enzyme protein when determined using the lipid acyitransferase assay given above.
- the variant lipid acyitransferase for use in the present invention may be dosed in amount of 0.05 to 50 TrU per g oil, suitably in an amount of 0.5 to 5 TrU per g oil.
- the parent and/or variant lipid acyitransferase for use in the methods or uses of the invention may have at least one, preferably more than one, preferably more than two, of the following, a GDSx block, a GANDY block, a HPT block.
- the lipid acyitransferase may have a GDSx block and a GANDY block.
- the enzyme may have a GDSx block and a HPT block.
- the enzyme comprises at least a GDSx block. See WO2004/064537 or WO2004/064987 for further details.
- residues of the GANDY motif are selected from GANDY, GGNDA, GGNDL, most preferably GANDY.
- the parent and/or variant lipid acyitransferase may comprise a GDSx motif and/ or a GANDY motif.
- the parent lipid acyitransferase enzyme and/or the variant lipid acyitransferase may be characterised as an enzyme which possesses lipid acyitransferase activity and which comprises the amino acid sequence motif GDSX, wherein X is one or more of the following amino acid residues L, A, V, I, F, Y, H, Q, T, N, M or S.
- the parent and/or the variant lipid acyitransferase comprises a GDSX motif.
- the GDSX motif is comprised of four conserved amino acids.
- the serine within the motif is a catalytic serine of the lipid acyltransferase enzyme.
- the serine of the GDSX motif may be in a position corresponding to Ser-16 in Aeromonas kydrophila lipolytic enzyme taught in Brumlik & Buckley (Journal of Bacteriology Apr. 1996, Vol. 178, No. 7, p 2060-2064).
- the sequence is preferably compared with the hidden markov model profiles (HMM profiles) of the pfam database.
- Pfam is a database of protein domain families. Pfam contains curated multiple sequence alignments for each family as well as profile hidden Markov models (profile HMMs) for identifying these domains in new sequences.
- profile HMMs profile hidden Markov models
- An introduction to Pfam can be found in Bateman A et al. (2002) Nucleic Acids Res. 30; 276-280.
- the GDSX motif can be identified using the Hammer software package, the instructions are provided in Durbin R, Eddy S, and Krogh A (1998) Biological sequence analysis; probabilistic models of proteins and nucleic acids. Cambridge University Press, ISBN 0-521-62041-4 and the references therein, and the HMMER2 profile provided within this specification.
- the PFAM database can be accessed, for example, through several servers which are currently located at the following websites.
- the database offers a search facility where one can enter a protein sequence. Using the default parameters of the database the protein sequence will then be analysed for the presence of Pfam domains.
- the GDSX domain is an established domain in the database and as such its presence in any query sequence will be recognised.
- the database will return the alignment of the Pfam00657 consensus sequence to the query sequence.
- the parent lipid acyltransferase may be obtainable, preferably obtained, from an organism from one of the following genera: Aeromonas, Streptomyces, Saccharomyces, Lactococcus, Mycobacterium, Streptococcus, Lactobacillus, Desulfitobacterium, Bacillus, Campylobacter, Vibrionaceae, Xylella, Sulfolobus, Aspergillus, Schizosaccharomyces, Listeria, Neisseria, Mesorhizobium, Ralstonia, Xanthomonas and Candida.
- the lipid acyltransferase may be obtainable, suitably may be obtained, from an organism from the genus Aeromonas.
- the parent lipid acyltransferase enzyme may be encoded by one of the following nucleotide sequences:
- nucleotide sequence which has 70% or more (preferably 75% or more, 85% or more, 95% or more or 98% or more) identity with any one of the sequences shown as SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14 or SEQ ID No. 15;
- nucleotide sequence which is related to SEQ ID No. 25, SEQ ID No. 10, SEQ ID No. 11, SEQ ID No. 12, SEQ ID No. 13, SEQ ID No. 14, SEQ ID No. 15 by the degeneration of the genetic code; or
- nucleotide sequence encoding a parent lipid acyltransferase enzyme in accordance with the present invention may be a nucleotide sequence which has 70% or more, 75% or more, 80% or more, preferably 85% or more, more preferably 90% or more, even more preferably 95% or more, such as 98 or 99% ore more, identity the sequence shown as SEQ ID No. 10 or SEQ ID No. 25.
- the parent lipid acyl transferase enzyme may comprise (or consist of) one of the following amino acid sequences:
- SEQ ID No. 1 an amino acid sequence which has 75%, 80%, 85%, 90%, 95%, 98% or more identity with any one of the sequences shown as SEQ ID No. 1 , SEQ ID No. 1, SEQ ID No. 3, SEQ ID No. 4, or SEQ ID No. 5, SEQ ID No. 6, SEQ ID No. 8 or SEQ ID No. 9.
- the parent lipid acyl transferase enzyme of the present invention may be a lipid acyltransferase that comprises (or consists of) either the amino acid sequence shown as SEQ ID No. 6 or 16, or comprises (or consists of) an amino acid sequence which has 75% or more, preferably 80% or more, preferably 85% or more, preferably 90% or more, preferably 95% or more, identity with the amino acid sequence shown as SEQ ID No. 16 or the amino acid sequence shown as SEQ ID No. 6.
- Suitable parent lipid acyltransferases are known in the art.
- the lipid acyltransferases taught in WO2009/081094 are suitable as parent lipid acyltransferases in accordance with the present invention.
- the parent lipid acyltransferase enzyme may be a phospholipid glycerol acyl transferase.
- Phospholipid glycerol acyl transferases include those isolated from Aeromonas spp., preferably Aeromonas hydrophila or A. salmonicida, most preferably A. salmonicida or variants thereof.
- the signal peptides of the acyl transferase has been cleaved during expression of the transferase.
- the signal peptide of SEQ ID No.s 1, 3, 4 and 5 are amino acids 1-18. Therefore the most preferred regions are amino acids 19-335 for SEQ ID No. 1 and SEQ ID No. 3 ⁇ A. hydrophila) and amino acids 19- 336 for SEQ ID No. 4 and SEQ ID No. 5 ⁇ A. salmonicida).
- the alignments as herein described use the mature sequence, i.e. without the signal peptide for instance.
- the lipid acyl transferase for use in the present invention comprises (or consists of) the amino acid sequence shown in SEQ ID No. 16 or comprises (or consists of) an amino acid sequence which has at least 70%, at least 75%, at least 85%, at least 90%, at least 95%, at least 98% identity to SEQ ID No. 16.
- SEQ ID No.s 8 and 9 are mature protein sequences of a lipid acyl transferase from A. hydrophila and A. salmonicida respectively which may or may not undergo further post-translational modification.
- Suitable parent lipid acyltransferases for use in accordance with the present invention and/or in the methods of the present invention may comprise any one of the following amino acid sequences and/or be encoded by the following nucleotide sequences:
- nucleic acid which encodes a polypeptide exhibiting lipid acyltransferase activity and is at least 70% identical (preferably at least 80%, more preferably at least 90%, even more preferably at least 95, such as at least 98%, identical) with the polypeptide sequence shown in SEQ ID No. 6 or with the polypeptide shown in SEQ ID No. 16;
- a (isolated) polypeptide comprising (or consisting of) an amino acid sequence as shown in SEQ ID No. 1 or SEQ ID No. 6 or an amino acid sequence which is at least 70% identical (preferably at least 80% identical, more preferably at least 90% identical, even more preferably at least 95, such as at least 98%,) with SEQ ID No. 6 or SEQ ID No. 16;
- nucleic acid encoding a lipid acyltransferase, which nucleic acid comprises (or consists of) a nucleotide sequence shown as SEQ ID No. 10 or SEQ ID No. 25 or a nucleotide sequence which is at least 70% identical (preferably at least 80%, more preferably at least 90% identical) with the nucleotide sequence shown as SEQ ID No. 10 or SEQ ID No. 25; d) a nucleic acid which hybridises under medium or high stringency conditions to a nucleic acid probe comprising the nucleotide sequence shown as SEQ ID No. 10 or SEQ ID No. 25 and encodes for a polypeptide exhibiting lipid acyltransferase activity;
- nucleic acid which is a fragment of the nucleic acid sequences specified in a), c) or d); or f) a polypeptide which is a fragment of the polypeptide specified in b).
- nucleotide sequences encoding a parent lipid acyltransferase for use in the present invention may be a polynucleotide encoding a lipid acyltransferase (such as SEQ ID No. 6 or SEQ ID No. 16).
- the nucleotide sequence is expressed in Bacillus licheniformis by transforming said B. licheniformis with a nucleotide sequence shown in SEQ ID No. 10 or SEQ ID No. 25 or a nucleotide sequence having at least 75% with SEQ ID No. 10 or SEQ ID No. 25 (more preferably at least 80%, more preferably at least 85%, more preferably at least 95%, more preferably at least 98% identity therewith) or with a modified nucleotide sequence in accordance with the present invention; culturing said B. licheniformis and isolating the lipid acyltransferase(s) produced thereby.
- the parent lipid acyltransferase enzyme according to the present invention comprises the following catalytic triad: Ser 16, Asp 288 and His 291 (these residue numbers refer to the mature sequences shown in SEQ ID No. 6 and 16 for example).
- Ser 16, Asp 288 and His 291 comprises a serine residue, an aspartic acid residue and a histidine residue, respectively, at positions corresponding to Ser-34, Asp-306 and His-309 in the Aeromonas hydrophila lipid acyl transferase enzyme shown in SEQ ID No. 3 or SEQ ID No. 1, i.e. the immature sequences which comprise a signal peptide - which forms the first 18 amino acid residues of these sequences.
- the active site residues correspond to Ser-7, Asp-345 and His-348.
- the variant lipid acyltransferase is one classified under the Enzyme Nomenclature classification (E.C. 2.3.1.43).
- the variant lipid acyltransferase according to the present invention may exhibit one or more of the following lipase activities: glycolipase activity (E.C. 3.1.1.26), triacylglycerol lipase activity (E.C. 3.1.1.3), phospholipase A2 activity (E.C. 3.1.1.4) or phospholipase Al activity (E.C. 3.1.1.32).
- glycolipase activity E.C. 3.1.1.26
- triacylglycerol lipase activity E.C. 3.1.1.3
- phospholipase A2 activity E.C. 3.1.1.4
- phospholipase Al activity E.C. 3.1.1.32
- phospholipase Al activity E.C. 3.1.1.32
- the variant lipid acyltransferase according to the present invention may have at least one or more of the following activities: glycolipase activity (E.C. 3.1.1.26) and/or phospholipase Al activity (E.C. 3.1.1.32) and/or phospholipase A2 activity (E.C. 3.1.1.4).
- the variant lipid acyltransferase according to the present invention may have at least glycolipase activity (E.C. 3.1.1.26).
- the variant lipid acyltransferase according to the present invention may be capable of transferring an acyl group from a glycolipid and/or a phospholipid to one or more of the following acceptor substrates: a sterol, a stanol, a carbohydrate (e.g. maltose, and/or glucose for example), a protein, glycerol.
- acceptor substrates e.g. a sterol, a stanol, a carbohydrate (e.g. maltose, and/or glucose for example), a protein, glycerol.
- the variant lipid acyltransferase according to the present invention does not exhibit triacylglycerol lipase activity (E.C. 3.1.1.3).
- the variant lipid acyltransferase may have one or more modifications as discussed in detail above and/or may be prepared by the methods of the present invention.
- the variant lipid acyltransferase according to the present invention retains at least 70%, preferably at least 80%, preferably at least 90%, preferably at least 95%, preferably at least 97%, preferably at least 99% homology with the parent enzyme.
- the variant lipid acyltransferase enzyme may comprise the amino acid sequence motif GDSXj wherein X is one or more of the following amino acid residues L, A, V, I, F, Y, H, Q, T, N, M or S.
- X of the GDSX motif is L.
- substitution is not intended to cover the replacement of an amino acid with the same amino acid.
- substitution of one or more amino acid residues at a position other than position 31 may cover the replacement of an amino acid with the same amino acid provided that this results in a change of codon at the nucleotide sequence level.
- WO 2005/066347 discloses particular positions of a lipid acyltransferase that can be modified to enhance the ratio of transferase activity from galactolipids compared with phospholipids.
- the variant lipid acyltransferase of the present application may in addition to the modifications taught herein comprise one or more further modifications as disclosed in WO 2005/066347 (which is incorporated herein by reference).
- a variant lipid acyltransferase enzyme retains or incorporates at least one or more of the pfam00657 consensus sequence amino acid residues found in the GDSx, GA DY and HPT blocks.
- Enzymes such as lipases with no or low lipid acyltransferase activity in an aqueous environment may be mutated using molecular evolution tools to introduce or enhance the transferase activity, thereby producing a variant lipid acyltransferase enzyme with significant transferase activity suitable for use in the compositions and methods of the present invention.
- the variant lipid acyltransferase according to the present invention and produced by the methods of the present invention has at least one improved property compared with a parent (i.e. backbone) or unmodified lipid acyltransferase.
- improved property may include a) an altered substrate specificity of the lipid acyltransferase, for instance and by way of example only i) an altered ability of the enzymes to use certain compounds as acceptors, for example an improved ability to utilise a carbohydrate (e.g.
- maltose as an acceptor molecule thus improving the enzymes ability to produce a carbohydrate ester) or ii) an altering ability to use saturated or unsaturated fatty acids as a substrate or iii) a changed specificity such that the variant lipid acyltransferase preferentially utilises the fatty acid from the Snl or Sn2 position of a lipid substrate or iv) an altered substrate chain length specificity of in the variant enzyme; b) altered kinetics of the enzyme; and/or c) lowered ability of the variant lipid acyltransferase to carry out a hydrolysis reaction whilst maintaining or enhancing the enzymes ability to carry out an acyl transferase reaction.
- Other improved properties may be for example related to improvements and/or changes in pH and/or temperature stability, and/or detergent and or oxidative stability. Indeed, it is contemplated that enzymes having various degrees of stability in one or more of these characteristics (pH, temperature, proteolytic stability, detergent stability, and/or oxidative stability) can be prepared in accordance with the present invention. Characterization of wild-type (e.g. parent lipid acyltransferase) and mutant (e.g. variant lipid acyltransferase) proteins is accomplished via any means suitable and is preferably based on the assessment of properties of interest. Suitably, the variant lipid acyltransferase may have a carbohydrate (e.g.
- the variant lipid acyltransferase may have a carbohydrate (e.g. maltose) transferase activity of at least 0.0059% when using the assay provided in protocol 2 detailed hereinbelow.
- the variant lipid acyltransferase may have a maltose transferase activity of at least 0.009, 0.01, 0.02, 0.03, 0.035, 0.04, 0.045, 0.05, 0.055 or 0.06% when using the assay provided in Protocol 2.
- the variant lipid acyltransferase may have a carbohydrate (e.g. maltose) transferase activity of at least 0.0282% + 10-20%.
- carbohydrate e.g. maltose
- the methods according to the present invention may comprise the steps of testing the variant lipid acyltransferase for one or more of the improved properties and/or selecting a variant enzyme, which when compared with the parent enzyme, which has the improved property.
- the methods according to the present invention may comprise the steps of measuring the ability of the variant lipid acyltransferase to transfer an acyl group to a carbohydrate (e.g. maltose and/or glucose) compared with the parent lipid acyltransferase; and selecting a variant lipid acyltransferase having an improved ability to transfer an acyl group to maltose as compared with the parent lipid acyltransferase.
- the variant enzyme of the present invention when compared with the parent enzyme, may have an increased transferase activity and either the same or less hydrolytic activity. In other words, suitably the variant enzyme may have a higher transferase activity to hydrolytic activity (e.g.
- the variant enzyme may preferentially transfer an acyl group from a lipid (including phospholipid, galactolipid or tnacylglycerol) to an acyl acceptor rather than simply hydrolysing the lipid.
- a lipid including phospholipid, galactolipid or tnacylglycerol
- the lipid acyltransferase according to the present invention and for use in the invention may be a variant with enhanced enzyme activity on polar lipids, preferably phospholipids and/or glycolipids, when compared to the parent enzyme.
- such variants also have low or no activity on lyso-polar lipids.
- the enhanced activity on polar lipids, preferably phospholipids and/or glycolipids may be the result of hydrolysis and/or transferase activity or a combination of both.
- the enhanced activity on polar lipids in the result of transferase activity.
- Variant lipid acyl transferases according to the present invention and for use in the invention may have decreased activity on triglycerides, and/or monoglycerides and/or diglycerides compared with the parent enzyme.
- the variant enzyme may have no activity on triglycerides and/or monoglycerides and/or diglycerides. Low activity on triglycerides is preferred in variant enzymes which are to be used for bakery applications, for treatment of egg or egg-based products and/or for degumming oils.
- variant lipid acyltransferase of the present invention or composition comprising said variant lipid acyltransferase of the present invention may be used in combination with other components.
- Examples of other components include one or more of: thickeners, gelling agents, emulsifiers, binders, crystal modifiers, sweeteners (including artificial sweeteners), rheology modifiers, stabilisers, antioxidants, dyes, enzymes, carriers, vehicles, excipients, diluents, lubricating agents, flavouring agents, colouring matter, suspending agents, disintegrants, granulation binders etc.
- These other components may be natural.
- These other components may be prepared by use of chemical and or enzymatic techniques.
- thickener or gelling agent refers to a product that prevents separation by slowing or preventing the movement of particles, either droplets of immiscible liquids, air or insoluble solids. Thickening occurs when individual hydrated molecules cause an increase in viscosity, slowing the separation. Gelation occurs when the hydrated molecules link to form a three-dimensional network that traps the particles, thereby immobilizing them.
- stabiliser as used here is defined as an ingredient or combination of ingredients that keeps a product (e.g. a food product) from changing over time.
- Emulsifier refers to an ingredient (e.g. a food product ingredient) that prevents the separation of emulsions.
- Emulsions are two immiscible substances, one present in droplet form, contained within the other.
- Emulsions can consist of oil-in-water, where the droplet or dispersed phase is oil and the continuous phase is water; or water-in-oil, where the water becomes the dispersed phase and the continuous phase is oil.
- Foams, which are gas-in-liquid, and suspensions, which are solid-in-liquid, can also be stabilised through the use of emulsifiers.
- Emulsifiers have a polar group with an affinity for water (hydrophilic) and a non-polar group which is attracted to oil (lipophilic). They are absorbed at the interfaces of the two substances, providing an interfacial film acting to stabilise the emulsion.
- the hydrophilic/lipophilic properties of emulsifiers are affected by the structure of the molecule. These properties are identified by the hydrophilic/lipophilic balance (HLB) value. Low HLB values indicate greater lipophilic tendencies which are used to stabilise water-in-oil emulsions.
- High HLB values are assigned to hydrophilic emulsifiers, typically used in oil-in- water emulsions. These values are derived from simple systems. Because foods often contain other ingredients that affect the emulsification properties, the HLB values may not always be a reliable guide for emulsifier selection.
- the term "binder” refers to an ingredient (e.g. a food ingredient) that binds the product together through a physical or chemical reaction. During “elation” for instance, water is absorbed, providing a binding effect. However, binders can absorb other liquids, such as oils, holding them within the product. In the context of the present invention binders would typically be used in solid or low-moisture products for instance baking products: pastries, doughnuts, bread and others.
- crystal modifier refers to an ingredient (e.g. a food ingredient) that affects the crystallisation of either fat or water. Stabilisation of ice crystals is important for two reasons. The first is directly related to the product stability from a separation standpoint. The more freeze/thaw cycles a product encounters, the larger the ice crystals become. These large crystals can break down product structure, either naturally occurring, as in the case of cell walls, or that which is created by "elation". Because the water is no longer held in place, the product may exhibit syneresis, or weeping, after thawing. Secondly, in the case of a product which is consumed frozen, these large crystals result in an undesirable, gritty mouth feel.
- Carriers or “vehicles” mean materials suitable for compound administration and include any such material known in the art such as, for example, any liquid, gel, solvent, liquid diluent, solubilizer, or the like, which is non-toxic and which does not interact with any components of the composition in a deleterious manner.
- nutritionally acceptable carriers include, for example, water, salt solutions, alcohol, silicone, waxes, petroleum jelly, vegetable oils, polyethylene glycols, propylene glycol, liposomes, sugars, gelatin, lactose, amylose, magnesium stearate, talc, surfactants, silicic acid, viscous paraffin, perfume oil, fatty acid monoglycerides and diglycerides, petroethral fatty acid esters, hydroxymethyl-cellulose, polyvinylpyrrolidone, and the like.
- excipients include one or more of: microcrystalline cellulose and other celluloses, lactose, sodium citrate, calcium carbonate, dibasic calcium phosphate, glycine, starch, milk sugar and high molecular weight polyethylene glycols.
- disintegrants include one or more of: starch (preferably corn, potato or tapioca starch), sodium starch glycollate, croscarmellose sodium and certain complex silicates.
- granulation binders include one or more of: polyvinylpyrrolidone, hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose (HPC), sucrose, maltose, gelatin and acacia.
- lubricating agents include one or more of: magnesium stearate, stearic acid, glyceryl behenate and talc.
- diluents include one or more of: water, ethanol, propylene glycol and glycerin, and combinations thereof.
- the other components may be used simultaneously (e.g. when they are in admixture together or even when they are delivered by different routes) or sequentially (e.g. they may be delivered by different routes).
- the term "component suitable for animal or human consumption” means a compound which is or can be added to the composition of the present invention as a supplement which may be of nutritional benefit, a fibre substitute or have a generally beneficial effect to the consumer.
- the ingredients can be used in a wide variety of products that require gelling, texturising, stabilising, suspending, film-forming and structuring, retention of juiciness, without adding unnecessary viscosity.
- the ingredients will be able to improve the shelf live and stability of the viable culture.
- the components may be prebiotics such as alginate, xanthan, pectin, locust bean gum (LBG), inulin, guar gum, galacto-oligosaccharide (GOS), fructo- oligosaccharide (FOS), lactosucrose, soybean oligosaccharides, palatinose, isomalto- oligosaccharides, gluco-oligosaccharides and xylo-oligosaccharides.
- prebiotics such as alginate, xanthan, pectin, locust bean gum (LBG), inulin, guar gum, galacto-oligosaccharide (GOS), fructo- oligosaccharide (FOS), lactosucrose, soybean oligosaccharides, palatinose, isomalto- oligosaccharides, gluco-oligosaccharides and xylo-oligosaccharides.
- the variant lipid acyltransferase according to the present invention may be used in combination with the fungal lipolytic enzyme taught in WO2005/087918 (the teachings of which are incorporated herein by reference). This may be a particularly useful combination for use in baking.
- variant lipid acyltransferases according to the present invention in some applications may be used in combination with one or more other enzymes, such as lipases, phospholipases, cellulases, hemicellulases, xylanases, starch degrading enzymes (including amylases, particularly maltogenic amylases), oxidoreductases and proteases.
- other enzymes such as lipases, phospholipases, cellulases, hemicellulases, xylanases, starch degrading enzymes (including amylases, particularly maltogenic amylases), oxidoreductases and proteases.
- lipid acyltransferase of the present invention When used in degumming applications it may be used in combination with a phospholipase A enzyme and/or a phospholipase C enzyme.
- ASSAYS ASSAYS
- the acyltransferase activity accounts for at least 5%, more preferably at least 10%, more preferably at least 20%, more preferably at least 30%, more preferably at least 40%, more preferably 50%, more preferably at least 60%, more preferably at least 70%, more preferably at least 80%, more preferably at least 90% and more preferably at least 98% of the total enzyme activity.
- the % transferase activity i.e. the transferase activity as a percentage of flie total enzymatic activity
- a foodstuff to which a lipid acyltransferase according to the present invention has been added may be extracted following the enzymatic reaction with CHC1 3 :CH 3 0H 2:1 and the organic phase containing the lipid material is isolated and analysed by GLC according to the procedure detailed hereinbelow. From the GLC analysis (and if necessary HPLC analysis) the amount of free fatty acids and one or more of sterol/stanol esters; carbohydrate esters, protein esters; diglycerides; or monoglycerides are determined.
- a control foodstuff to which no enzyme according to the present invention has been added is analysed in the same way. Calculation:
- % sterol/stanol ester(control) and Mv sterol ester average molecular weight of the sterol/stanol esters) ⁇ applicable where the acyl acceptor is a sterol and/or stanol;
- Carrier gas Helium.
- PSSI cold split injection (initial temp 50°C heated to 385°C), volume ⁇ . ⁇
- Sample preparation 30 mg of sample was dissolved in 9 ml Heptane:Pyridin, 2:1 containing internal standard heptadecane, 0.5 mg ml. 300 ⁇ 1 sample solution was transferred to a crimp vial, 300 ⁇ MSTFA (N-Methyl-N-trimethylsilyl-trifluoraceamid) was added and reacted for 20 minutes at 60°C.
- an assay may contain the following components: a test acyl substrate (e.g., an acyl ester) having a carbon chain of a particular length, a recipient molecule, e.g., a carbohydrate, sterol/stanol or an alcohol, and a lipid acyltransferase enzyme, where the assay detects transfer of an acyl group from the acyl substrate to a recipient molecule in an aqueous environment.
- a library of variant lipid acyltransferases may be made, and the members of the library tested for altered substrate specificity. Altered lipid acyltransferases having altered substrate specificity may be identified using such methods.
- Protocol 1 Determination of maltose transferase activity f 20h protocol
- 14g egg yolk and 7g carbohydate monohydrate e.g. maltose monohydrate, glucose monohydrate, lactose monohydrate and/or galactose monohydrate
- lg egg yolk/carbohydrate substrate e.g. maltose substrate, glucose substrate, lactose substrate and/or galactose substrate
- ⁇ enzyme solution or control solution
- 7.5ml CHCl 3 :MeOH 2;1 is added and mixed for 30 minutes before centrifuging at ISOOrpm (700g) for 10 minutes.
- the organic phase is then transferred to a TLC vial.
- a TLC plate is activated by incubation at 160°C for 10 minutes.
- each sample is then applied to a HPTLC Si-plate.
- carbohydrate-monooleate e.g. maltose- monooleate, glucose-monooleate, lactose- monooleate, and/or galactose-monooleate
- the TLC-plate is then eluted in solvent 4-1 (CHCl 3 :Methanol:Water 64:26:4) for 20 minutes, before being dried in a fume cupboard for approximately 10 minutes and incubated at 160°C for 10 minutes.
- the plate is then developed by submerging in 6% Cu-acetate in 16% H3PO4 for 10 seconds, then drying (with dry filter paper) and incubating at 160°C for 8 minutes.
- the % carbohydrate transferase activity (e.g. maltose, glucose, lactose and/or galactose transferase activity) is defined as the % carbohydate-ester (e.g. maltose-ester, glucose-ester, lactose-ester and/or galactose ester) determined using this protocol.
- % carbohydate-ester e.g. maltose-ester, glucose-ester, lactose-ester and/or galactose ester
- 14g egg yolk and 7g carbohydate monohydrate e.g. maltose monohydrate, glucose monohydrate, lactose monohydrate and/or galactose monohydrate
- lg egg yolk/ carbohydrate substrate e.g. maltose substrate, glucose substrate, lactose substrate and/or galactose substrate
- ⁇ enzyme solution or control solution
- 7.5ml CHCl 3 :MeOH 2;1 is added and mixed for 30 minutes before centrifuging at lSOOrpm (700g) for 10 minutes.
- the organic phase is then transferred to a TLC vial.
- a TLC plate is activated by incubation at 160°C for 10 minutes.
- the % carbohydrate transferase activity (e.g. maltose, glucose, lactose and/or galactose transferase activity) is defined as the % carbohydate-ester (e.g. maltose-ester, glucose-ester, lactose-ester and/or galactose ester) determined using this protocol.
- % carbohydate-ester e.g. maltose-ester, glucose-ester, lactose-ester and/or galactose ester
- compositions comprising an altered or variant lipid acyltransferase are also provided.
- the altered lipid acyltransferase comprises an amino acid sequence that is at least 70%» identical with a wild-type lipid acyltransferase or a backbone lipid acyltransferase.
- the composition may be a food product, e.g., an edible product for human or animal consumption, or an intermediate in the manufacture of an edible food product (e.g. a food supplement), or a cleaning and/or detergent composition, for example.
- a subject variant lipid acyltransferase as described herein
- Rj comprises a substituted or unsubstituted carbon chain of at least 5 carbon atoms and R 2 is any organic moiety
- a source of hydrogen peroxide may be present in a subject cleaning composition.
- a subject cleaning composition may be employed for example, in laundry applications, hard surface cleaning, automatic dishwashing applications, as well as cosmetic applications such as dentures, teeth, hair and skin.
- the subject enzymes are ideally suited for laundry applications such as the bleaching of fabrics.
- the enzymes of the present invention find use in both granular and liquid compositions.
- the subject enzyme also finds use in cleaning additive products.
- the subject cleaning additive products are ideally suited for inclusion in wash processes where additional bleaching effectiveness is desired. Such instances include, but are not limited to, low temperature solution cleaning applications.
- the additive product may be, in its simplest form, one or more of the enzymes of the present invention.
- Such additive may be packaged in dosage form for addition to a cleaning process where a source of peroxygen is employed and increased bleaching effectiveness is desired.
- Such single dosage form may comprise a pill, tablet, gelcap or other single dosage unit such as pre-measured powders or liquids.
- a filler or carrier material may be included to increase the volume of such composition.
- Suitable filler or carrier materials include, but are not limited to, various salts of sulfate, carbonate and silicate as well as talc, clay and the like.
- Filler or carrier materials for liquid compositions may be water or low molecular weight primary and secondary alcohols including polyols and diols. Examples of such alcohols include, but are not limited to, methanol, ethanol, propanol and isopropanol. The compositions may contain from about 5% to about 90% of such materials. Acidic fillers can be used to reduce pH.
- the cleaning additive may include activated peroxygen source such as esters of alcohols, esters of diols, or esters of polyols or as defined below or the adjunct ingredients as also defined below.
- the cleaning compositions and cleaning additives of the present invention require an effective amount of the enzyme provided by the present invention.
- a cleaning composition of the present invention comprise at least 0.0001 weight percent, from about 0.0001 to about 1 , from about 0.001 to about 0.5, or even from about 0.01 to about 0.1 weight percent of at least one enzyme of the present invention.
- cleaning compositions and “cleaning formulations” refer to compositions that find use in the removal of undesired compounds from items to be cleaned, such as fabric, dishes, contact lenses, other solid substrates, hair (shampoos), skin (soaps and creams), teeth (mouthwashes, toothpastes), etc.
- the term encompasses any materials/compounds selected for the particular type of cleaning composition desired and the form of the product ⁇ e.g., liquid, gel, granule, or spray composition), as long as the composition is compatible with the perhydrolase and other enzyme(s) used in the composition.
- the specific selection of cleaning composition materials are readily made by considering the surface, item or fabric to be cleaned, and the desired form of the composition for the cleaning conditions during use.
- the terms further refer to any composition that is suited for cleaning, bleaching, disinfecting, and/or sterilizing any object and/or surface. It is intended that the terms include, but are not limited to detergent compositions ⁇ e.g., liquid and/or solid laundry detergents and fine fabric detergents; hard surface cleaning formulations, such as for glass, wood, ceramic and metal counter tops and windows; carpet cleaners; oven cleaners; fabric fresheners; fabric softeners; and textile and laundry pre-spotters, as well as dish detergents).
- detergent compositions e.g., liquid and/or solid laundry detergents and fine fabric detergents
- hard surface cleaning formulations such as for glass, wood, ceramic and metal counter tops and windows
- carpet cleaners oven cleaners
- fabric fresheners fabric softeners
- textile and laundry pre-spotters as well as dish detergents
- cleaning composition includes unless otherwise indicated, granular or powder-form all-purpose or heavy-duty washing agents, especially cleaning detergents; liquid, gel or paste-form all-purpose washing agents, especially the so-called heavy-duty liquid (HDL) types; liquid fine-fabric detergents; hand dishwashing agents or light duty dishwashing agents, especially those of the high-foaming type; machine dishwashing agents, including the various tablet, granular, liquid and rinse-aid types for household and institutional use; liquid cleaning and disinfecting agents, including antibacterial hand-wash types, cleaning bars, mouthwashes, denture cleaners, car or carpet shampoos, bathroom cleaners; hair shampoos and hair-rinses; shower gels and foam baths and metal cleaners; as well as cleaning auxiliaries such as bleach additives and "stain-stick" or pre-treat types.
- HDL heavy-duty liquid
- cleaning and disinfecting agents including antibacterial hand-wash types, cleaning bars, mouthwashes, denture cleaners, car or carpet shampoos, bathroom cleaners; hair shampoos and
- detergent composition and “detergent formulation” are used in reference to mixtures which are intended for use in a wash medium for the cleaning of soiled objects.
- the term is used in reference to laundering fabrics and/or garments (e.g., “laundry detergents”).
- laundry detergents e.g., "laundry detergents”
- the term refers to other detergents, such as those used to clean dishes, cutlery, etc. (e.g., "dishwashing detergents”). It is not intended that the present invention be limited to any particular detergent formulation or composition.
- the term encompasses detergents that contain surfactants, transferase(s), hydrolytic enzymes, oxido reductases, builders, bleaching agents, bleach activators, bluing agents and fluorescent dyes, caking inhibitors, masking agents, enzyme activators, antioxidants, and solubilizers.
- hard surface cleaning composition refers to detergent compositions for cleaning hard surfaces such as floors, walls, tile, bath and kitchen fixtures, and the like. Such compositions are provided in any form, including but not limited to solids, liquids, emulsions, etc.
- washing composition refers to all forms for compositions for cleaning dishes, including but not limited to granular and liquid forms.
- fabric cleaning composition refers to all forms of detergent compositions for cleaning fabrics, including but not limited to, granular, liquid and bar forms.
- textile refers to woven fabrics, as well as staple fibers and filaments suitable for conversion to or use as yarns, woven, knit, and non-woven fabrics.
- the term encompasses yarns made from natural, as well as synthetic (e.g., manufactured) fibers.
- textile materials is a general term for fibers, yarn intermediates, yarn, fabrics, and products made from fabrics (e.g., garments and other articles).
- fabric encompasses any textile material. Thus, it is intended that the term encompass garments, as well as fabrics, yarns, fibers, non-woven materials, natural materials, synthetic materials, and any other textile material.
- the term “compatible,” means that the cleaning composition materials do not reduce the enzymatic activity of the perhydrolase to such an extent that the perhydrolase is not effective as desired during normal use situations. Specific cleaning composition materials are exemplified in detail hereinafter.
- composition may be employed in a variety of methods or uses.
- the method or use may include contacting a subject altered lipid acyltransferase with a substrate under conditions suitable for the lipid acyltransferase to transfer an acyl group from said substrate onto an acceptor molecule.
- the contacting may be done in aqueous conditions.
- esters produced when the variant enzymes are used as biocatalysts can be used in cosmetics, in foodstuffs (including in baked products, in meat products, in egg based products and in dairy products), in pharmaceuticals, in soap, in candles, in oils, in lubricants, in varnishes, in linoleum, in inks, in prints, in textile dyes, and in or as surfactants.
- the above-described enzyme may be employed in a variety of food applications.
- the enzyme may be present in, or may be used to make, a foodstuff, where a foodstuff is any substance which is suitable for human and/or animal consumption.
- a foodstuff encompasses both baked goods produced from dough as well as the dough used in the preparation of said baked goods.
- a foodstuff may be a water-containing foodstuff.
- An exemplary water- containing foodstuff comprise 10-98% water, e.g. 14-98%, 18-98%, 20-98%, 40-98%, 50- 98%, 70-98%, 75-98% water, excluding solid components.
- a foodstuff may be selected from one or more of the following: eggs, egg-based products, including but not limited to mayonnaise, salad dressings, sauces, ice creams, egg powder, modified egg yolk and products made therefrom; baked goods, including breads, cakes, sweet dough products, laminated doughs, liquid batters, muffins, doughnuts, biscuits, crackers and cookies; confectionery, includmg chocolate, candies, caramels, halawa, gums, including sugar free and sugar sweetened gums, bubble gum, soft bubble gum, chewing gum and puddings; frozen products including sorbets, preferably frozen dairy products, including ice cream and ice milk; dairy products, including cheese, cream, butter, milk, coffee cream, whipped cream, custard cream, milk drinks and yoghurts; mousses, whipped vegetable creams, meat products, including processed meat products; edible oils and fats, aerated and non-aerated whipped products, oil-in-water emulsions, water-in-oil
- the foodstuff in accordance with the present invention may be a dough product or a baked product, such as a bread, a fried product, a snack, cakes, pies, brownies, cookies, noodles, snack items such as crackers, graham crackers, pretzels, and potato chips, and pasta.
- a foodstuff may be a flour, pre-mix, oil, fat, cocoa butter, coffee whitener, salad dressing, margarine, spread, peanut butter, shortenings, ice cream or cooking oil.
- the foodstuff in accordance with the present invention may be a dairy product, including butter, milk, cream, cheese such as natural, processed, and imitation cheeses in a variety of forms (including shredded, block, slices or grated), cream cheese, ice cream, frozen desserts, yoghurt, yoghurt drinks, butter fat, anhydrous milk fat, other dairy products.
- the enzyme according to the present invention may improve fat stability in dairy products.
- a method of preparing a foodstuff generally comprises adding an above-described enzyme to the foodstuff or an ingredient thereof.
- a foodstuff comprising a variant enzyme is provided.
- enzyme may be employed in the following methods: in situ production of an emulsifier without an increase in free fatty acids; a reduction in the accumulation of free fatty acids in the foodstuff; a reduction in free cholesterol levels in the foodstuff; an increase in sterol esters and/or stanol esters; a reduction in blood serum cholesterol and/or low density lipoproteins; an increase in carbohydrate esters; a reduction in unwanted free carbohydrates.
- the acyl acceptor molecule in the foodstuff may be any compound containing a hydroxy group (-OH), such as for example, polyvalent alcohols, including glycerol; sterol; stanols; carbohydrates; hydroxy acids including fruit acids, citric acid, tartaric acid, lactic acid and ascorbic acid; proteins or a sub-unit thereof, such as amino acids, protein hydrolysates and peptides (partly hydrolysed protein) for example; and mixtures and derivatives thereof.
- the acyl acceptor is not water.
- a sterol and/or stanol may comprise one or more of the following structural features: i) a 3- beta hydroxy group or a 3-alpha hydroxy group; and/or ii) A:B rings in the cis position or A:B rings in the trans position or C5-C6 is unsaturated.
- Suitable sterol acyl acceptors include cholesterol and phytosterols, for example alpha-sitosterol, beta-sitosterol, stigmasterol, ergosterol, campesterol, 5,6-dihydrosterol, brassicasterol, alpha-spinasterol, beta-spinasterol, gamma-spinasterol, deltaspinasterol, fucosterol, dimosterol, ascosterol, serebisterol, episterol, anasterol, hyposterol, chondrillasterol, desmosterol, chalinosterol, poriferasterol, clionasterol, sterol glycosides, and other natural or synthetic isomeric forms and derivatives.
- phytosterols for example alpha-sitosterol, beta-sitosterol, stigmasterol, ergosterol, campesterol, 5,6-dihydrosterol, brassicasterol, alpha-spinasterol, beta-spinasterol, gam
- suitably more than one sterol and/or stanol may act as the acyl acceptor, suitably more than two sterols and or stanols may act as the acyl acceptor.
- suitably more than one sterol ester and/or stanol ester may be produced.
- the present invention provides a method for the in situ production of both a cholesterol ester and at least one sterol or stanol ester in combination.
- the lipid acyltransferase for some aspects of the present invention may transfer an acyl group from a lipid to both cholesterol and at least one further sterol and/or at least one stanol.
- the sterol acyl acceptor may be cholesterol.
- the amount of free cholesterol in the foodstuff may be reduced as compared with the foodstuff prior to exposure to the enzyme and/or as compared with an equivalent foodstuff which has not been treated with the enzyme.
- a subject enzyme may be used in the production of an egg-based product.
- a method that includes contacting a subject enzyme with an egg or egg- based product is provided.
- An egg-based product comprising a subject enzyme is also provided.
- Egg yolk may comprise up to 1% glucose and, egg or egg based products may be treated with glucose oxidase to remove some or all of this glucose.
- this unwanted sugar can be readily removed by "esterifying" the sugar to form a sugar ester.
- a carbohydrate ester can function as an emulsifier in foodstuffs.
- the enzyme can be employed to transfer an acyl group to a sugar, the invention encompasses the production of an emulsifier, in situ, in the foodstuff.
- a subject enzyme may utilize a sterol and/or stanol and a carbohydrate as an acyl acceptor, which method is particularly useful for the production of foodstuffs containing eggs or egg products.
- the ester produced e.g., stanol ester or the sterol ester
- a subject enzyme may be added to dough, for example, as part of a baking method.
- the method may also include baking dough containing the enzyme to make a baked product from the dough.
- a subject enzyme may result in one or more of the following technical effects in dough and/or baked products: an improved specific volume of either the dough or the baked products (for example of bread and/or of cake); an improved dough stability; an improved crust score (for example a thinner and/or crispier bread crust), an improved crumb score (for example a more homogenous crumb distribution and/or a finer crumb structure and/or a softer crumb); an improved appearance (for example a smooth surface without blisters or holes or substantially without blisters or holes); a reduced staling; an enhanced softness; an improved odour; an improved taste.
- Flour used in dough and baked products typically contains about 2% maltose. Maltose unlike other sugars is not used by yeast. Therefore in one embodiment of the present invention it may be pertinent to increase the maltose utilization of the enzyme, i.e. to increase the ability of the enzyme to transfer an acyl group from a lipid to maltose to form a maltose ester. This can be particularly important and lead to substantial advantages in baked products, such as a bread, a fried product, a snack, cakes, pies, brownies, cookies, noodles, snack items such as crackers, graham crackers, pretzels, and potato chips, and pasta.
- variant lipid acyltransferases of the present invention it is possible to replace the commonly used emulsifier "DATEMTM" in baked products.
- DATEMTM emulsifier
- carbohydrate ester e.g. maltose ester
- a subject enzyme may be employed to degum (i.e., reduce the amount of polar lipid, e.g., phospholipids and/or glycolipid such as lecithin, i.e., phosphatidylcholine and cephalin) in vegetable or edible oils.
- a subject variant lipid acyltransferase may be contacted with the oil so as to hydrolyse a polar lipid(s) in the oil
- a variant lipid acyltransferase in accordance with the present invention may be employed to reduce phospholipid in an edible oil, comprising treating the oil with a subject enzyme so as to hydrolyse a major part of the phospholipid, and separating an aqueous phase containing the hydrolysed phospholipid from the oil.
- a subject enzyme may be employed to convert polar lipids (e.g.
- glycolipids and/or phospholipids into a higher value product, such as carbohydrate esters, protein esters (e.g., via reaction with a serine, threonine, tyrosine, or cysteine residue), and a hydroxy acid ester.
- a subject enzyme may be employed to transfer any acyl chain onto a sterol, a stanol, a carbohydrate, a protein, or glycerol, for example.
- an emulsifier may be prepared in situ in the foodstuff without an increase in the free fatty acid content of the foodstuff.
- the production of free fatty acids can be detrimental to foodstuffs.
- free fatty acids have been linked with off-odours and/or off-flavours in foodstuffs, as well other detrimental effects, including a soapy taste in cheese for instance.
- this method results in the in situ preparation of an emulsifier(s) (preferably two emulsifiers) wherein the accumulation of free fatty acids is reduced and/or eliminated.
- the fatty acid that is removed from the lipid is transferred by the enzyme to an acyl acceptor, for example a sterol and/or a stanol.
- an acyl acceptor for example a sterol and/or a stanol.
- the instant method may result in no significant increase in the level of free fatty acids in the foodstuff. Such methods may be particularly employed on foodstuffs containing eggs.
- the variant lipid acyltransferase according to the present invention may utilize a protein as the acyl acceptor.
- the protein may be one or more of the proteins found in a food product, for example in a dairy product and/or a meat product.
- suitable proteins may be those found in curd or whey, such as lactoglobulin.
- suitable proteins include ovalbumin from egg, gliadin, glutenin, puroindoline, lipid transfer proteins from grains, and myosin from meat.
- the present invention provides methods and compositions for the use of peracids in textile bleaching and in various other applications.
- the present invention provides one-step methods for textile processing applications, including but not limited to one-step desizing, scouring and bleaching processes ⁇ See e.g., EP WO 03002810, EP 1255888, WO 0164993, and US 20020007516, all of which are hereby incorporated by reference).
- bleaching involves processing textile material before it is dyed and/or after it is incorporated into textile goods.
- the lipid acyltransferase in accordance with the present invention may be encoded by any one of the nucleotide sequences taught herein. Depending upon the host cell used post-transcriptional and/or post-translational modifications may be made. It is envisaged that the lipid acyltransferase for use in the present methods and/or uses encompasses lipid acyltransferases which have undergone post-transcriptional and/or post-translational modification.
- SEQ ID No. 16 is the same as SEQ ID No. 6 except that SEQ ID No. 16 has undergone post- translational and/or post-transcriptional modification to remove some amino acids, more specifically 38 amino acids. Notably the N-terminal and C-terminal part of the molecule are covalently linked by an S-S bridge between two cysteines. Amino residues 236 and 236 of SEQ ID No. 16 are not covalently linked following post-translational modification. The two peptides formed are held together by one or more S-S bridges.
- the precise cleavage site(s) in respect of the post-translational and/or post-transcriptional modification may vary slightly such that by way of example only the 38 amino acids removed (as shown in SEQ ID No. 16 compared with SEQ ID No. 6) may vary slightly.
- the cleavage site may be shifted by a few residues (e.g. 1 , 2 or 3 residues) in either direction compared with the cleavage site shown by reference to SEQ ID No. 16 compared with SEQ ID No. 6.
- the cleavage may commence at residue 232, 233, 234, 235, 236, 237 or 238 for example.
- the cleavage may result in the removal of about 38 amino acids, in some embodiments the cleavage may result in the removal of between 30-45 residues, such as 34-42 residues, such as 36-40 residues, preferably 38 residues.
- the lipid acyltransferase is a recovered/isolated lipid acyltransferase.
- the parent or variant lipid acyltransferase produced may be in an isolated form.
- the nucleotide sequence encoding a parent or variant lipid acyltransferase for use in the present invention may be isolated form.
- isolated means that the sequence or protein is at least substantially free from at least one other component with which the sequence or protein is naturally associated in nature and as found in nature.
- the lipid acyltransferase may be in a purified form.
- nucleotide sequence encoding a lipid acyltransferase for use in the present invention may be in a purified form.
- purified means that the sequence is in a relatively pure state - e.g. at least about 51% pure, or at least about 75%, or at least about 80%, or at least about 90% pure, or at least about 95% pure or at least about 98% pure.
- a nucleotide sequence encoding either a polypeptide which has the specific properties as defined herein or a polypeptide which is suitable for modification may be isolated from any cell or organism producing said polypeptide. Various methods are well known within the art for the isolation of nucleotide sequences.
- a genomic DNA and/or cDNA library may be constructed using chromosomal DNA or messenger RNA from the organism producing the polypeptide. If the amino acid sequence of the polypeptide is known, labelled oligonucleotide probes may be synthesised and used to identify polypeptide-encoding clones from the genomic library prepared from the organism. Alternatively, a labelled oligonucleotide probe containing sequences homologous to another known polypeptide gene could be used to identify polypeptide-encoding clones. In the latter case, hybridisation and washing conditions of lower stringency are used.
- polypeptide-encoding clones could be identified by inserting fragments of genomic DNA into an expression vector, such as a plasmid, transforming enzyme-negative bacteria with the resulting genomic DNA library, and then plating the transformed bacteria onto agar containing an enzyme inhibited by the polypeptide, thereby allowing clones expressing the polypeptide to be identified.
- an expression vector such as a plasmid, transforming enzyme-negative bacteria with the resulting genomic DNA library
- the nucleotide sequence encoding the polypeptide may be prepared synthetically by established standard methods, e.g. the phosphoroamidite method described by Beucage S.L. et al (1981) Tetrahedron Letters 22, p 1859-1869, or the method described by Matthes et al (1984) EMBO J. 3, p 801-805.
- the phosphoroamidite method oligonucleotides are synthesised, e.g. in an automatic DNA synthesiser, purified, annealed, ligated and cloned in appropriate vectors.
- the nucleotide sequence may be of mixed genomic and synthetic origin, mixed synthetic and cDNA origin, or mixed genomic and cDNA origin, prepared by ligating fragments of synthetic, genomic or cDNA origin (as appropriate) in accordance with standard techniques. Each ligated fragment corresponds to various parts of the entire nucleotide sequence.
- the DNA sequence may also be prepared by polymerase chain reaction (PCR) using specific primers, for instance as described in US 4,683,202 or in Saiki R et al (Science (1988) 239, pp 487-491).
- PCR polymerase chain reaction
- nucleotide sequence refers to an oligonucleotide sequence or polynucleotide sequence, and variant, homologues, fragments and derivatives thereof (such as portions thereof).
- the nucleotide sequence may be of genomic or synthetic or recombinant origin, which may be double-stranded or single-stranded whether representing the sense or antisense strand.
- nucleotide sequence in relation to the present invention includes genomic DNA, cDNA, synthetic DNA, and RNA. Preferably it means DNA, more preferably cDNA for the coding sequence.
- the nucleotide sequence per se encoding a polypeptide having the specific properties as defined herein does not cover the native nucleotide sequence in its natural environment when it is linked to its naturally associated sequence(s) that is/are also in its/their natural environment.
- the term "non- native nucleotide sequence" means an entire nucleotide sequence that is in its native environment and when operatively linked to an entire promoter with which it is naturally associated, which promoter is also in its native environment.
- the polypeptide of the present invention can be expressed by a nucleotide sequence in its native organism but wherein the nucleotide sequence is not under the control of the promoter with which it is naturally associated within that organism.
- the polypeptide is not a native polypeptide.
- the term "native polypeptide” means an entire polypeptide that is in its native environment and when it has been expressed by its native nucleotide sequence.
- the nucleotide sequence encoding polypeptides having the specific properties as defined herein is prepared using recombinant DNA techniques (i.e. recombinant DNA).
- the nucleotide sequence could be synthesised, in whole or in part, using chemical methods well known in the art (see Caruthers MH et al (1980) Nuc Acids Res Symp Ser 215-23 and Horn T et al (1980) Nuc Acids Res Symp Ser 225-232).
- an enzyme-encoding nucleotide sequence has been isolated, or a putative enzyme- encoding nucleotide sequence has been identified, it may be desirable to modify the selected nucleotide sequence, for example it may be desirable to mutate the sequence in order to prepare an enzyme in accordance with the present invention. Mutations may be introduced using synthetic oligonucleotides. These oligonucleotides contain nucleotide sequences flanking the desired mutation sites.
- EP 0 583 265 refers to methods of optimising PCR based mutagenesis, which can also be combined with the use of mutagenic DNA analogues such as those described in EP 0 866 796.
- Error prone PCR technologies are suitable for the production of variants of lipid acyl transferases with preferred characteristics.
- WO0206457 refers to molecular evolution of lipases.
- a third method to obtain novel sequences is to fragment non-identical nucleotide sequences, either by using any number of restriction enzymes or an enzyme such as Dnase I, and reassembling full nucleotide sequences coding for functional proteins. Alternatively one can use one or multiple non-identical nucleotide sequences and introduce mutations during the reassembly of the full nucleotide sequence.
- DNA shuffling and family shuffling technologies are suitable for the production of variants of lipid acyl transferases with preferred characteristics. Suitable methods for performing 'shuffling' can be found in EP0 752 008, EP1 138 763, EP1 103 606. Shuffling can also be combined with other forms of DNA mutagenesis as described in US 6,180,406 and WO 01/34835.
- mutations or natural variants of a polynucleotide sequence can be recombined with either the wild type or other mutations or natural variants to produce new variants.
- Such new variants can also be screened for improved functionality of the encoded polypeptide.
- the nucleotide sequence encoding a lipid acyltransferase used in the invention may encode a variant lipid acyltransferase, i.e. the lipid acyltransferase may contain at least one amino acid substitution, deletion or addition, when compared to a parental enzyme.
- the variant enzymes retain at least 40%, 50 %, 60%, 70%, 80%, 90%, 95%, 97%, 99% homology with the parent enzyme.
- Suitable parent enzymes may include any enzyme with esterase or lipase activity.
- the parent enzyme aligns to the pfam00657 consensus sequence.
- the present invention also encompasses the use of amino acid sequences encoded by a nucleotide sequence which encodes a lipid acyltransferase for use in any one of the methods and/or uses of the present invention.
- amino acid sequence is synonymous with the term “polypeptide” and/or the term “protein”.
- amino acid sequence is synonymous with the term "peptide”.
- the amino acid sequence may be prepared/isolated from a suitable source, or it may be made synthetically or it may be prepared by use of recombinant DNA techniques.
- amino acid sequences may be obtained from the isolated polypeptides taught herein by standard techniques.
- Purified polypeptide may be freeze-dried and 100 ⁇ g of the freeze-dried material may be dissolved in 50 ⁇ of a mixture of 8 M urea and 0.4 M ammonium hydrogen carbonate, pH 8.4. The dissolved protein may be denatured and reduced for 15 minutes at 50°C following overlay with nitrogen and addition of 5 ⁇ of 45 mM dithiothreitol. After cooling to room temperature, 5 ⁇ of 100 mM iodoacetamide may be added for the cysteine residues to be derivatized for 15 minutes at room temperature in the dark under nitrogen.
- the resulting peptides may be separated by reverse phase HPLC on a VYDAC C18 column (0.46x15cm; ⁇ ; The Separation Group, California, USA) using solvent A: 0.1% TFA in water and solvent B: 0.1% TFA in acetonitrile.
- Selected peptides may be re-chromatographed on a Develosil C18 column using the same solvent system, prior to N-terminal sequencing. Sequencing may be done using an Applied Biosystems 476A sequencer using pulsed liquid fast cycles according to the manufacturer's instructions (Applied Biosystems, California, USA).
- homologue means an entity having a certain homology with the subject amino acid sequences and the subject nucleotide sequences.
- homology can be equated with “identity”.
- the homologous amino acid sequence and/or nucleotide sequence should provide and/or encode a polypeptide which retains the functional activity and/or enhances the activity of the enzyme.
- a homologous sequence is taken to include an amino acid sequence which may be at least 75, 85 or 90% identical, preferably at least 95 or 98% identical to the subject sequence.
- the homologues will comprise the same active sites etc. as the subject amino acid sequence.
- homology can also be considered in terms of similarity (i.e. amino acid residues having similar chemical properties/functions), in the context of the present invention it is preferred to express homology in terms of sequence identity.
- a homologous sequence is taken to include a nucleotide sequence which may be at least 75, 85 or 90% identical, preferably at least 95 or 98% identical to a nucleotide sequence encoding a polypeptide of the present invention (the subject sequence).
- the homologues will comprise the same sequences that code for the active sites etc. as the subject sequence.
- homology can also be considered in terms of similarity (i.e. amino acid residues having similar chemical properties/functions), in the context of the present invention it is preferred to express homology in terms of sequence identity.
- Homology comparisons can be conducted by eye, or more usually, with the aid of readily available sequence comparison programs. These commercially available computer programs can calculate % homology between two or more sequences. % homology may be calculated over contiguous sequences, i.e. one sequence is aligned with the other sequence and each amino acid in one sequence is directly compared with the corresponding amino acid in the other sequence, one residue at a time. This is called an "ungapped" alignment. Typically, such ungapped alignments are performed only over a relatively short number of residues.
- Calculation of maximum % homology therefore firstly requires the production of an optimal alignment, taking into consideration gap penalties.
- a suitable computer program for carrying out such an alignment is the Vector NTI (Invitrogen Corp.).
- Other software that can perform sequence comparisons include, but are not limited to, the BLAST package (see Ausubel et al 1999 Short Protocols in Molecular Biology, 4 th Ed ⁇ Chapter 18), and FASTA (Altschul et al 1990 J. Mol. Biol. 403-410). Both BLAST and FASTA are available for offline and online searching (see Ausubel et al 1999, pages 7-58 to 7-60). However, for some applications, it is preferred to use the Vector NTI program.
- BLAST 2 Sequences is also available for comparing protein and nucleotide sequence (see FEMS Microbiol Lett 1999 174(2): 247-50; FEMS Microbiol Lett 1999 177(1): 187-8 and tatiana@ncbi.nlm.nih.gov).
- a scaled similarity score matrix is generally used that assigns scores to each pairwise comparison based on chemical similarity or evolutionary distance.
- An example of such a matrix commonly used is the BLOSUM62 matrix - the default matrix for the BLAST suite of programs.
- Vector ⁇ programs generally use either the public default values or a custom symbol comparison table if supplied (see user manual for further details). For some applications, it is preferred to use the default values for the Vector NTI package.
- percentage homologies may be calculated using the multiple alignment feature in Vector NTI (Invitrogen Corp.), based on an algorithm, analogous to CLUSTAL (Higgins DG & Sharp PM (1988), Gene 73(1), 237-244).
- % homology preferably % sequence identity.
- the software typically does this as part of the sequence comparison and generates a numerical result.
- sequence identity for the nucleotide sequences is determined using CLUSTAL with the gap penalty and gap extension set as defined above.
- the degree of identity with regard to a nucleotide sequence is determined over at least 20 contiguous nucleotides, preferably over at least 30 contiguous nucleotides, preferably over at least 40 contiguous nucleotides, preferably over at least 50 contiguous nucleotides, preferably over at least 60 contiguous nucleotides, preferably over at least 100 contiguous nucleotides.
- the degree of identity with regard to a nucleotide sequence may be determined over the whole sequence.
- the degree of amino acid sequence identity in accordance with the present invention may be suitably determined by means of computer programs known in the art, such as Vector NTI 10 (Invitrogen Corp.).
- the matrix used is preferably BLOSUM62 with Gap opening penalty of 10.0 and Gap extension penalty of 0.1.
- the degree of identity with regard to an amino acid sequence is determined over at least 20 contiguous amino acids, preferably over at least 30 contiguous amino acids, preferably over at least 40 contiguous amino acids, preferably over at least 50 contiguous amino acids, preferably over at least 60 contiguous amino acids.
- the degree of identity with regard to an amino acid sequence may be determined over the whole sequence.
- sequences may also have deletions, insertions or substitutions of amino acid residues which produce a silent change and result in a functionally equivalent substance.
- Deliberate amino acid substitutions may be made on the basis of similarity in polarity, charge, solubility, hydrophobicity, hydrophilicity, and/or the amphipathic nature of the residues as long as the secondary binding activity of the substance is retained.
- negatively charged amino acids include aspartic acid and glutamic acid; positively charged amino acids include lysine and arginine; and amino acids with uncharged polar head groups having similar hydrophilicity values include leucine, isoleucine, valine, glycine, alanine, asparagine, glutamine, serine, threonine, phenylalanine, and tyrosine.
- the present invention also encompasses homologous substitution (substitution and replacement are both used herein to mean the interchange of an existing amino acid residue, with an alternative residue) that may occur i.e. like-for-like substitution such as basic for basic, acidic for acidic, polar for polar etc.
- Non-homologous substitution may also occur i.e.
- Z ornithine
- B diaminobutyric acid ornithine
- O norleucine ornithine
- pyriylalanine thienylalanine
- naphthylalanine phenylglycine
- Variant amino acid sequences may include suitable spacer groups that may be inserted between any two amino acid residues of the sequence including alkyl groups such as methyl, ethyl or propyl groups in addition to amino acid spacers such as glycine or ⁇ -alanine residues.
- alkyl groups such as methyl, ethyl or propyl groups
- amino acid spacers such as glycine or ⁇ -alanine residues.
- a further form of variation involves the presence of one or more amino acid residues in peptoid form, will be well understood by those skilled in the art.
- the peptoid form is used to refer to variant amino acid residues wherein the ex-carbon substituent group is on the residue's nitrogen atom rather than the -carbon.
- Nucleotide sequences for use in the present invention or encoding a polypeptide having the specific properties defined herein may include within them synthetic or modified nucleotides.
- a number of different types of modification to oligonucleotides are known in the art. These include methylphosphonate and phosphorothioate backbones and/or the addition of acridine or polylysine chains at the 3' and/or 5' ends of the molecule.
- nucleotide sequences described herein may be modified by any method available in the art. Such modifications may be carried out in order to enhance the in vivo activity or life span of nucleotide sequences.
- the present invention also encompasses the use of nucleotide sequences that are complementary to the sequences discussed herein, or any derivative, fragment or derivative thereof. If the sequence is complementary to a fragment thereof then that sequence can be used as a probe to identify similar coding sequences in other organisms etc.
- Polynucleotides which are not 100% homologous to the sequences of the present invention but fall within the scope of the invention can be obtained in a number of ways.
- Other variants of the sequences described herein may be obtained for example by probing DNA libraries made from a range of individuals, for example individuals from different populations.
- other viral/bacterial, or cellular homologues particularly cellular homologues found in mammalian cells e.g. rat, mouse, bovine and primate cells
- such homologues and fragments thereof in general will be capable of selectively hybridising to the sequences shown in the sequence listing herein.
- sequences may be obtained by probing cDNA libraries made from or genomic DNA libraries from other animal species, and probing such libraries with probes comprising all or part of any one of the sequences in the attached sequence listings under conditions of medium to high stringency. Similar considerations apply to obtaining species homologues and allelic variants of the polypeptide or nucleotide sequences of the invention.
- Variants and strain/species homologues may also be obtained using degenerate PC which will use primers designed to target sequences within the variants and homologues encoding conserved amino acid sequences within the sequences of the present invention.
- conserved sequences can be predicted, for example, by aligning the amino acid sequences from several variants/homologues. Sequence alignments can be performed using computer software known in the art. For example the GCG Wisconsin PileUp program is widely used.
- the primers used in degenerate PC will contain one or more degenerate positions and will be used at stringency conditions lower than those used for cloning sequences with single sequence primers against known sequences.
- polynucleotides may be obtained by site directed mutagenesis of characterised sequences. This may be useful where for example silent codon sequence changes are required to optimise codon preferences for a particular host cell in which the polynucleotide sequences are being expressed. Other sequence changes may be desired in order to introduce restriction polypeptide recognition sites, or to alter the property or function of the polypeptides encoded by the polynucleotides.
- Polynucleotides (nucleotide sequences) of the invention may be used to produce a primer, e.g. a PCR primer, a primer for an alternative amplification reaction, a probe e.g. labelled with a revealing label by conventional means using radioactive or non-radioactive labels, or the polynucleotides may be cloned into vectors.
- a primer e.g. a PCR primer, a primer for an alternative amplification reaction, a probe e.g. labelled with a revealing label by conventional means using radioactive or non-radioactive labels, or the polynucleotides may be cloned into vectors.
- primers, probes and other fragments will be at least 15, preferably at least 20, for example at least 25, 30 or 40 nucleotides in length, and are also encompassed by the term polynucleotides of the invention as used herein.
- Polynucleotides such as DNA polynucleotides and probes according to the invention may be produced recombinantly, synthetically, or by any means available to those of skill in the art. They may also be cloned by standard techniques.
- primers will be produced by synthetic means, involving a stepwise manufacture of the desired nucleic acid sequence one nucleotide at a time. Techniques for accomplishing this using automated techniques are readily available in the art.
- Longer polynucleotides will generally be produced using recombinant means, for example using a PCR (polymerase chain reaction) cloning techniques. This will involve making a pair of primers (e.g. of about 15 to 30 nucleotides) flanking a region of the lipid targeting sequence which it is desired to clone, bringing the primers into contact with mRNA or cDNA obtained from an animal or human cell, performing a polymerase chain reaction under conditions which bring about amplification of the desired region, isolating the amplified fragment (e.g. by purifying the reaction mixture on an agarose gel) and recovering the amplified DNA.
- the primers may be designed to contain suitable restriction errzyme recognition sites so that the amplified DNA can be cloned into a suitable cloning vector.
- the present invention also encompasses the use of sequences that are complementary to the sequences of the present invention or sequences that are capable of hybridising either to the sequences of the present invention or to sequences that are complementary thereto.
- hybridisation shall include “the process by which a strand of nucleic acid joins with a complementary strand through base pairing” as well as the process of amplification as carried out in polymerase chain reaction (PCR) technologies.
- the present invention also encompasses the use of nucleotide sequences that are capable of hybridising to the sequences that are complementary to the subject sequences discussed herein, or any derivative, fragment or derivative thereof.
- the present invention also encompasses sequences that are complementary to sequences that are capable of hybridising to the nucleotide sequences discussed herein.
- Hybridisation conditions are based on the melting temperature (Tm) of the nucleotide binding complex, as taught in Berger and immel (1987, Guide to Molecular Cloning Techniques, Methods in Enzymology, Vol. 152, Academic Press, San Diego CA), and confer a defined "stringency” as explained below.
- Maximum stringency typically occurs at about Tm-5°C (5°C below the Tm of the probe); high stringency at about 5°C to 10°C below Tm; intermediate stringency at about 10°C to 20°C below Tm; and low stringency at about 20°C to 25°C below Tm.
- a maximum stringency hybridisation can be used to identify or detect identical nucleotide sequences while an intermediate (or low) stringency hybridisation can be used to identify or detect similar or related polynucleotide sequences.
- the present invention encompasses the use of sequences that are complementary to sequences that are capable of hybridising under high stringency conditions or intermediate stringency conditions to nucleotide sequences encoding polypeptides having the specific properties as defined herein.
- the present invention encompasses the use of sequences that are complementary to sequences that are capable of hybridising under high stringency conditions (e.g. 65°C and O.lxSSC ⁇ lxSSC - 0.15 M NaCl, 0.015 M Na-citrate pH 7.0 ⁇ ) to nucleotide sequences encoding polypeptides having the specific properties as defined herein.
- high stringency conditions e.g. 65°C and O.lxSSC ⁇ lxSSC - 0.15 M NaCl, 0.015 M Na-citrate pH 7.0 ⁇
- the present invention also relates to the use of nucleotide sequences that can hybridise to the nucleotide sequences discussed herein (including complementary sequences of those discussed herein).
- the present invention also relates to the use of nucleotide sequences that are complementary to sequences that can hybridise to the nucleotide sequences discussed herein (including complementary sequences of those discussed herein).
- polynucleotide sequences that are capable of hybridising to the nucleotide sequences discussed herein under conditions of intermediate to maximal stringency.
- the present invention covers the use of nucleotide sequences that can hybridise to the nucleotide sequences discussed herein, or the complement thereof, under stringent conditions (e.g. 50°C and 0.2xSSC). In a more preferred aspect, the present invention covers the use of nucleotide sequences that can hybridise to the nucleotide sequences discussed herein, or the complement thereof, under high stringency conditions (e.g. 65°C and O.lxSSC).
- stringent conditions e.g. 50°C and 0.2xSSC
- high stringency conditions e.g. 65°C and O.lxSSC.
- microbial eukaryotic or prokaryotic expression hosts may be used.
- low copy number, preferably single event chromosomal expression systems may be preferred.
- Expression systems with high transformation frequencies are also preferred, particularly for the expression of large variant libraries (>1000 colonies), such as those prepared using random mutagenesis and/or shuffling technologies.
- a eukaryotic expression host namely yeast
- Microbial eukaryotic expression hosts such as yeast, may be preferred for the expression of variant libraries produced using a eukaryotic acyltransferase parent gene.
- Bacillus i.e. Bacillus subtilis t as an expression host in the production of enzymes are described in WO02/ 14490.
- Microbial prokaryotic expression hosts such as Bacillus, may be preferred for the expression of variant libraries produced using a prokaryotic acyltransferase parent gene.
- the expression host is Bacillus licheniformis.
- Expression of lipid acyltransferases in Bacillus licheniformis is taught in WO2008/090395 (the teachings of which are incorporated herein by reference).
- a nucleotide sequence for use in the present invention or for encoding a polypeptide having the specific properties as defined herein can be incorporated into a recombinant replicable vector.
- the vector may be used to replicate and express the nucleotide sequence, in polypeptide form, in and/or from a compatible host cell. Expression may be controlled using control sequences which include promoters/enhancers and other expression regulation signals. Prokaryotic promoters and promoters functional in eukaryotic cells may be used. Tissue specific or stimuli specific promoters may be used. Chimeric promoters may also be used comprising sequence elements from two or more different promoters described above.
- the polypeptide produced by a host recombinant cell by expression of the nucleotide sequence may be secreted or may be contained intracellularly depending on the sequence and/or the vector used.
- the coding sequences can be designed with signal sequences which direct secretion of the substance coding sequences through a particular prokaryotic or eukaryotic cell membrane.
- construct which is synonymous with terms such as “conjugate”, “cassette” and “hybrid” - includes a nucleotide sequence encoding a polypeptide having the specific properties as defined herein for use according to the present invention directly or indirectly attached to a promoter.
- An example of an indirect attachment is the provision of a suitable spacer group such as an intron sequence, such as the Shl-intron or the ADH intron, intermediate the promoter and the nucleotide sequence of the present invention.
- fused in relation to the present invention which includes direct or indirect attachment. In some cases, the terms do not cover the natural combination of the nucleotide sequence coding for the protein ordinarily associated with the wild type gene promoter and when they are both in their natural environment.
- the construct may even contain or express a marker which allows for the selection of the genetic construct.
- the construct comprises at least a nucleotide sequence of the present invention or a nucleotide sequence encoding a polypeptide having the specific properties as defined herein operably linked to a promoter.
- organism in relation to the present invention includes any organism that could comprise a nucleotide sequence according to the present invention or a nucleotide sequence encoding for a polypeptide having the specific properties as defined herein and/or products obtained therefrom.
- transgenic organism in relation to the present invention includes any organism that comprises a nucleotide sequence coding for a polypeptide having the specific properties as defined herein and/or the products obtained therefrom, and/or wherein a promoter can allow expression of the nucleotide sequence coding for a polypeptide having the specific properties as defined herein within the organism.
- the nucleotide sequence is incorporated in the genome of the organism.
- transgenic organism does not cover native nucleotide coding sequences in their natural environment when they are under the control of their native promoter which is also in its natural environment.
- the transgenic organism of the present invention includes an organism comprising any one of, or combinations of, a nucleotide sequence coding for a polypeptide having the specific properties as defined herein, constructs as defined herein, vectors as defined herein, piasmids as defined herein, cells as defined herein, or the products thereof.
- the transgenic organism can also comprise a nucleotide sequence coding for a polypeptide having the specific properties as defined herein under the control of a promoter not associated with a sequence encoding a lipid acyltransf erase in nature.
- the host organism can be a prokaryotic or a eukaryotic organism.
- Suitable prokaryotic hosts include bacteria such as E. coli and Bacillus Hcheniformis, preferably B. licheniformis.
- prokaryotic hosts are well documented in the art, for example see Sambrook et al (Molecular Cloning: A Laboratory Manual, 2nd edition, 1989, Cold Spring Harbor Laboratory Press). If a prokaryotic host is used then the nucleotide sequence may need to be suitably modified before transformation - such as by removal of introns. In another embodiment the transgenic organism can be a yeast.
- Filamentous fungi cells may be transformed using various methods known in the art - such as a process involving protoplast formation and transformation of the protoplasts followed by regeneration of the cell wall in a manner known.
- Aspergillus as a host microorganism is described in EP 0 238 023.
- Another host organism can be a plant.
- a review of the general techniques used for transforming plants may be found in articles by Potrykus (Annu Rev Plant Physiol Plant Mol Biol [1991] 42:205-225) and Christou (Agro-Food-Industry Hi-Tech March/April 1994 17- 27). Further teachings on plant transformation may be found in EP-A-0449375.
- the polypeptide may be secreted from the expression host into the culture medium from where the enzyme may be more easily recovered.
- the secretion leader sequence may be selected on the basis of the desired expression host.
- Hybrid signal sequences may also be used with the context of the present invention.
- Typical examples of secretion leader sequences not associated with a nucleotide sequence encoding a lipid acyltransferase in nature are those originating from the fungal amyloglucosidase (AG) gene (glaK - both 18 and 24 amino acid versions e.g. from Aspergillus), the a-factor gene (yeasts e.g. Saccharomyces, Kluyveromyces and Hansenula) or the a-amylase gene ⁇ Bacillus).
- AG fungal amyloglucosidase
- a-factor gene e.g. Saccharomyces, Kluyveromyces and Hansenula
- a-amylase gene ⁇ Bacillus a-amylase gene ⁇ Bacillus
- the lipid acyltransferase for use in the present invention may be produced as a fusion protein, for example to aid in extraction and purification thereof.
- fusion protein partners include glutathione-S-transferase (GST), 6xHis, GAL4 (DNA binding and/or transcriptional activation domains) and ⁇ -galactosidase. It may also be convenient to include a proteolytic cleavage site between the fusion protein partner and the protein sequence of interest to allow removal of fusion protein sequences. Preferably the fusion protein will not hinder the activity of the protein sequence.
- amino acid sequence of a polypeptide having the specific properties as defined herein may be ligated to a non-native sequence to encode a fusion protein.
- a non-native sequence For example, for screening of peptide libraries for agents capable of affecting the substance activity, it may be useful to encode a chimeric substance expressing a non-native epitope that is recognised by a commercially available antibody.
- Figure 1 shows the amino acid sequence of a mutant Aeromonas salmonicida mature lipid acyltransferase (GCAT) with a mutation of Asn80Asp (notably, amino acid 80 is in the mature sequence) (SEQ ID No. 6);
- Figure 2 shows an amino acid sequence (SEQ ID No. 1) a lipid acyl transferase from Aeromonas hydrophila (ATCC #7965);
- Figure 3 shows a pfam00657 consensus sequence from database version 6 (SEQ ID No. 2);
- Figure 4 shows an amino acid sequence (SEQ ID No. 3) obtained from the organism Aeromonas hydrophila (P1048O; GL121051);
- Figure 5 shows an amino acid sequence (SEQ ID No. 4) obtained from the organism Aeromonas salmonicida (AAG098404; GE9964017);
- Figure 6 shows an amino acid sequence (SEQ ID No. 5) of a lipid acyltransferase from Aeromonas salmonicida subsp. Salmonicida (ATCC#14174);
- Figure 7 shows an amino acid sequence (SEQ ID No. 7) of the fusion construct used for mutagenesis of the Aeromonas hydrophila lipid acyltransferase gene.
- the underlined amino acids is a xylanase signal peptide;
- Figure 8 shows an amino acid sequence (SEQ ID No. 8) obtained from the organism Aeromonas hydrophila (P10480; GI: 121051);
- Figure 9 shows the amino acid sequence (SEQ ID No. 9) of a mutant Aeromonas salmonicida mature lipid acyltransferase (GCAT);
- Figure 10 shows a ribbon representation of the HVN.PDB crystal structure which has glycerol in the active site. The Figure was made using the Deep View Swiss-PDB viewer;
- Figure 11 shows IIYN.PDB Crystal Structure - Side View using Deep View Swiss-PDB viewer, with glycerol in active site - residues within IOA of active site glycerol are coloured black;
- Figure 12 shows 1IVN.PDB Crystal Structure - Top View using Deep View Swiss-PDB viewer, with glycerol in active site - residues within ioA of active site glycerol are coloured black;
- Figure 13 shows alignment 1
- Figure 14 shows alignment 2; Figures 15 and 16 show an alignment of 1IVN to PI 0480 (PI 0480 is the database sequence for A. hydrophila enzyme), this alignment was obtained from the PFAM database and used in the model building process; Figure 17 shows an alignment where PI 0480 is the database sequence for Aeromonas hydrophila. This sequence is used for the model construction and the site selection. Note that the full protein (SEQ ID No. 3) is depicted, the mature protein (equivalent to SEQ ID No. 8) starts at residue 19.
- A. sal Aeromonas salmonicida (SEQ ID No. 9) GDSX lipase, A. hyd is Aeromonas hydrophila (SEQ ID No. 8) GDSX lipase.
- the consensus sequence contains a * at the position of a difference between the listed sequences;
- Figure 18 shows a gene construct
- Figure 19 shows a codon optimised gene construct (no. 052907)
- Figure 20 shows the sequence of the Xhol insert containing the a lipid acyltransferase precursor gene, the -35 and -10 boxes are underlined;
- Figure 21 shows BML780-KLM3'CAP50 (comprising SEQ ID No. 16 - upper colony) and BML780 (the empty host strain - lower colony) after 48h growth at 7°C on 1% tributyrin agar;
- Figure 22 shows a nucleotide sequence from Aeromonas salmonicida (SEQ ID No. 10) including the signal sequence (preLAT - positions 1 to 87);
- Figure 23 shows a nucleotide sequence (SEQ ID No. I I) encoding a lipid acyl transferase according to the present invention obtained from the organism Aeromonas hydrophila;
- Figure 24 shows a nucleotide sequence (SEQ ID No. 12) encoding a lipid acyl transferase according to the present invention obtained from the organism Aeromonas salmonicida;
- Figure 25 shows a nucleotide sequence (SEQ ID No. 13) encoding a lipid acyltransferase from Aeromonas hydrophila (ATCC #7965);
- Figure 26 shows a nucleotide sequence (SEQ ID No. 14) encoding a lipid acyltransferase from Aeromonas salmonicida subsp. Salmonicida (ATCC#14174);
- Figure 27 shows a nucleotide sequence (SEQ ID No. 15) encoding an enzyme from Aeromonas hydrophila including a xylanase signal peptide;
- Figure 28 shows the amino acid sequence of a mutant Aeromonas salmonicida mature lipid acyltransferase (GCAT) with a mutation of Asn80Asp (notably, amino acid 80 is in the mature sequence) ⁇ shown herein as SEQ ID No. 6 - and after undergoing post-translational modification as SEQ ID No.
- GCAT Aeromonas salmonicida mature lipid acyltransferase
- SEQ ID No. 16 amino acid residues 235 and 236 of SEQ ID No. 1 are not covalently linked following post-translational modification. The two peptides formed are held together by one or more S-S bridges.
- Amino acid 236 in SEQ ID No. 16 corresponds with the amino acid residue number 274 in SEQ ID No. 6 shown herein [SEQ ID No. 16 is the same as SEQ ID No. 6 except SEQ ID No. 15 shows the sequence after post-translational clipping or cleavage to remove 38 amino acids];
- Figure 29 shows the plasmid pLA52, containing the nucleotide sequence (SEQ ID No. 25) encoding lipid acyltransferase enzyme, used for generation of site evaluation libraries;
- Figure 30 shows the "canyon" and “cave” structure of lipid acyltransferase s, in particular of the Aeromonas salmonicida lipid acyltransferase enzyme taught herein as SEQ ID No. 16;
- Figure 31 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry IIVN);
- Figure 32 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry IIVN) and identifies the residues of "insertion 1" (residues 22-36) as sticks;
- Figure 33 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E.
- FIG. 34 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry 1IVN) and identifies the residues of "insertion 3" (residues 162-168) as sticks;
- Figure 35 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry UVN) and identifies the residues of "insertion 4" (residues 213-281) as sticks;
- Figure 36 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry 1IVN) and shows insertion 4 (residues 213-381) in cartoon and the catalytic triad of the lipid acyltransferase (S16, D288, H291) is shown in space filling;
- Figure 37 shows the structural alignment between the lipid acyltransferase of Aeromonas salmonicida lipid acyltransferase comprising the N80D mutation and which is the mature sequence having undergone post-translational clipping [shown herein as SEQ ID No. 16] (white) and E. coli thioesterase (black) (PDB entry UVN) and shows the catalytic triad (SI , D288, H291) is shown in dark grey space filling and the residues forming the canyon (including Y117, A119 and Y120) shown in white space filling;
- Figure 39 shows a graph of the screening results in vitro which evaluated maltose-ester generation g methyl ester (ME)/mg enzyme protein with the backbone lipid acyltransferase enzyme from Aeromonas salmonicida (comprising the N80D variation); and 4 variant enzymes;
- ME methyl ester
- Figure 40 shows a graph from 3 rd round screening in vitro which evaluates the hydrolysis activity of the enzyme (LATU/mg enzyme protein) with the backbone lipid acyltransferase enzyme from Aeromonas salmonicida (comprising the N80D variation); and 4 variant enzymes;
- Figure 41 shows a graph in a dough slurry showing % maltose ester generation using the backbone lipid acyltransferase enzyme from Aeromonas salmonicida (comprising the N80D variation), and two variants M27K and L 1Q, W122L when dosed at lOmg/kg flour;
- Figure 42 shows SEQ ID No. 25 a nucleotide sequence encoding a backbone lipid acyltransferase enzyme from Aeromonas salmonicida which was used in the preparation of the variant lipid acyltransferase enzyme in the examples hereinbelow;
- Figure 43 shows a Standard curve based on attachment (0.1, 0.3, 0.5, 0.8 and 1.5 ⁇ ) of 0.5% refined rapeseed oil (standard) as a function of measured triglyceride area on the TLC plate;
- Figure 44 shows oil yield (%) calculated from the amount of gum (control) subtracted amount of gum (enzymatic sample).
- Sample 5 is not shown due to analysis error.
- Sample 2 EDS 226 (0.1 LATU-K g), 3: K710 (0.2 LATU-K/g), 4: K710 (0.4 LATU-K/g), 6: K916 (0.04 LATU- K/g), 7: K916 (0.1 LATU-K/g) and 8: K91 (0.2 LATU-K/g);
- Figure 45 shows GC-resu!ts. Contents (%) of FFA's, phytosterols and phytosierol esters in oils degummed with the lipid acyltransferase mutants.
- Sample 1 control
- 2 EDS 226 (0.1 LATU-K g)
- 3 K710 (0.2 LATU-K g)
- 4 K710 (0.4 LATU-K/g)
- 6 K916 (0.04 LATU-K/g)
- 8 K916 (0.2 LATU-K/g);
- Figure 46 shows lyso-activity shown as degradation of 2-lyso-phosphatidylcholine (2-LPC) to a free fatty acid (FFA) and glycerophosphatidylcholine (GPC);
- Figure 47 shows results from TLC analysis of the gum phase. Relative degradation of phospholipids (PE and PA) in gums. Results are based on oils, degummed with different lipid acyltransferase mutants.
- Sample 1 control, 2: EDS 226 (0.1 LATU-K/g), 3: 710 (0.2 LATU-K/g), 4: 710 (0.4 LATU-K/g), 5: K710 (0.9 LATU-K g) 6: K916 (0.04 LATU-K g), 7: K916 (0.1 LATU-K/g) and 8: K916 (0.2 LATU-K g); and
- Figure 48 shows results from TLC analysis of the gum phase. Triglyceride content (%) in gums, obtained from degumming with lipid acyltransferase mutants.
- Sample 1 control
- 2 EDS 226 (0.1 LATU-K/g)
- 3 K710 (0.2 LATU-K g)
- 4 K710 (0.4 LATU-K/g)
- 5 K710 (0.9 LATU-K/g)
- 6 K916 (0.04 LATU-K g)
- 8 K916 (0.2 LATU- K/g).
- lipid acyltransferase with improved properties compared with the backbone enzyme (i.e. a enzyme expressed from the nucleotide sequence SEQ ID No. 25), for example the lipid acyltransferase may have an altered substrate specificity, e.g. an improved ability to generate carbohydrate esters (e.g. a maltose-ester) and/or may have a reduced hydrolysis activity.
- the backbone enzyme i.e. a enzyme expressed from the nucleotide sequence SEQ ID No. 25
- the lipid acyltransferase may have an altered substrate specificity, e.g. an improved ability to generate carbohydrate esters (e.g. a maltose-ester) and/or may have a reduced hydrolysis activity.
- the vector used for the site evaluation libraries was pCS32new N80D, also named pLA52, containing the Aeromonas salmonicida GCAT gene with an Asn to Asp substitution at position 80 (the nucleotide sequence for this gene is provided herein as SEQ ID No. 25).
- the N80D mutation enhances the expression of the enzyme in Bacillus subtilis.
- the gene is under control of the p32 promoter and expressed with a bacillus Cyclodextrin glucanotransferase (CGTase) signal sequence. All variants were screened in the Bacillus subtilis expression host OS21DAprE.
- Figure 29 shows the plasmid pLA52, containing the Aeromonas salmonicida GCAT gene with an Asn to Asp substitution at position 80 (the nucleotide sequence for this gene is provided herein as SEQ ID No. 25) lipid acyltransferase enzyme, used for generation of site evaluation libraries.
- Site evaluation library construction containing the Aeromonas salmonicida GCAT gene with an Asn to Asp substitution at position 80 (the nucleotide sequence for this gene is provided herein as SEQ ID No. 25) lipid acyltransferase enzyme, used for generation of site evaluation libraries.
- Combination libraries were generated in the same way as site evaluation libraries except that up to four different primers containing specific mutations were used in the same MSDM reaction and the combination variants were identified by sequencing of E. coli colonies and subsequently transformed into Bacillus subtilis OS21DAprE.
- the bacillus libraries were plated on LB plates and 96 colonies were picked at random from each library into 96- well plates. A replica of each 96-well plate was sent to AGOWA Gmbh, Berlin, Germany, for nucleic acid sequencing. The sequences of the 96 colonies were analysed at the mutated site and all available individual mutants representing each codon were selected for screening. Selection of combination variants
- This method was developed for analysis of maltose ester formation by transferase of fatty acids from lecithin to maltose.
- Substrate Pasteurized egg yolk
- Enzymation Weigh 1 g egg yolk/maltose substrate into a 12 mL glass vial with screw lid.
- TLC Sample application: Apply 8 ⁇ of each sample to a HPTLC Si-plate. As a standard,
- a 500 ml measuring flask is half filled with water. Add powder and acid, and adjust with water. Place on magnetic stirrer for 2-3 hours.
- Table 1 shows the maltose transferase activity as % maltose-ester after 20 hours, for each of the mutant variant lipid acyltransferases (with single point mutations as taught herein).
- the backbone is the Aeromonas salmonicida lipid acyltransferase comprising the N80D variation and encoded by the nucleotide sequence SEQ ID No. 25 (sometimes referred to herein as the N80D backbone) produced in Bacillus licheniformis production strain.
- ⁇ - Backbone is the Aeromonas salmonicida lipid acyltransferase comprising the N80D variation and encoded by the nucleotide sequence SEQ ID No. 25 (sometimes referred to herein as the N80D backbone) produced in Bacillus licheniformis production strain.
- Table 2 shows the maltose transferase activity as % maltose-ester after 1 hour, for each of the mutant variant lipid acyltransferases (with single point mutations as taught herein).
- the backbone is the Aeromonas salmonicida lipid acyltransferase comprising the N80D variation and encoded by the nucleotide sequence SEQ ID No. 25) - produced in Bacillus licheniformis production strain:
- key modifications include L031Q, F 5 H, T, Y, C; W122A, L, S; V85R, D, E; I86R, Y; M27N, H, Y, K, R; F235A, V; W232G, S; A236G, E; E201R, Al 19T, Y120E.
- Tables 3 and 4 show the combination winners when analysing maltose transferase activity as % maltose-ester after 20 hours and 1 hour respectively, for each of the mutant variant lipid acyltransferases.
- the lipid acyltransferase backbone when aligned (on a primary or tertiary basis) with the lipid acyltransferase enzyme shown herein as SEQ ID No. 16 or 6 preferably has D in position 80.
- N80D we have therefore shown in the above tables N80D as a modification.
- the backbone used in the above experiments already contained the N80D modification and therefore the other modifications could have been expressed without referencing the N80D modification, i.e. L31Q, N80D, W122L could have been expressed as L31Q, W122L.
- the N80D modification is a preferred modification and a backbone enzyme is preferably used which already possesses amino acid D in position 80.
- a backbone which does not contain amino acid D in position (such as one more of the lipid acyltransferases shown here as SEQ ID No. 1, 3, 4, 5, 8, or 9 for instance) then preferably an additional modification of N80D is included.
- Butano Ethanol (85:15 (v/v)) was immediately added to the 2.3 g sample withdrawn and then mixed for 15 sec. on a vortex before being placed on a rotormixer at 35 rpm for 5 min. Afterwards the sample was placed in a water bath at 97°C for 10 minutes, followed by mixing on a rotormixer at 35 rpm for one hour. The sample was then cenirifuged at 1370g for ten minutes. The supernatant being the organic phase containing the extracted lipid was then transferred to new glass tube.
- HPTLC plates (20 x 10 cm, Merck no. 1.05641) were activated by drying (160°C, 20-30 minutes) and standard and samples were applied using an Automatic HPTLC Applicator (ATS4, CAMAG). Plate eiution was performed using an Automatic Developing Chamber (ADC2, CAMAG) (7 cm). After eiution, plates were dried (160°C, 10 minutes), cooled, and immersed (10 seconds) in developing fluid (6 % cupric acetate in 16 % H 3 P0 4 ). After drying (160°C, 6 minutes) plates were evaluated visually using a TLC scanner (TLC Scanner 3, CAMAG).
- the variant enzymes has a significantly reduced hydrolysis activity (see the data shown in Figure 40). In fact, the variant enzymes had very little hydrolysis activity left..
- M27K, N80D otherwise known as a single point mutation M27 - where the backbone enzyme has amino acid D at position 80
- L31Q, N80D, W122L otherwise known as L31Q, W122L where the backbone enzyme has amino acid D at position 80. The results are shown in Figure 41. The variants showed a significantly changed acceptor preference.
- the variant enzymes were able use carbohydrate (particularly maltose) as an acyl acceptor more effectively than the backbone lipid acyltransferase (N80D) - referred to therein as KLM3'.
- EXAMPLE 3 Lipid acyltransferase variants in degumming
- the aim of the present experiment was to analyse the performance of three lipid acyltransferase variants according to the present invention (designated EDS 226 [having N80D, AI M mutation], 710 [having N80D, A119T mutation] and K916 [having N80D, G67A, V85H mutations]) in water degumming of crude soy bean oil.
- EDS 226 having N80D, AI M mutation
- 710 having N80D, A119T mutation
- K916 having N80D, G67A, V85H mutations
- the results are compared with previous water degumming experiments with the backbone lipid acyltransferase (sometimes referred to herein as LM3'), namely the lipid acyltransferase from Aeromonas salmonicida with an N80D mutation and which is post-translationally clipped when expressed in Bacillus licheniformis (the sequence for which is shown herein as SEQ ID No. 16).
- the evaluated mutants are variants of the Aeromonas Salmonicida glycerophospholipid cholesterol acyltransferase (lipid acyltransferase), being expressed in Bacillus lichiniformis.
- K710 proved to be highly hydrolytic, as observed from the total phosphor content of the oil, being reduced to approximately 32 ppm compared to 42 and 56 ppm in oils degummed with EDS 226 and 916. Furthermore 710 attributed to a higher content of phytosterol esters and free fatty acids (FFA's) in the oil than EDS 226 and K916.
- FFA's phytosterol esters and free fatty acids
- lipid acyltransferase mutants originating from Aeromon s salmonicida were tested.
- the enzymes are glycerophospholipid cholesterol acyltransferases (GCAT) with mutations, other than NSOD, as indicated in the Table below:.
- GCAT glycerophospholipid cholesterol acyltransferases
- LATU Lipid Acyl Transferase Unit
- 1 LATU is defined according to a standard enzyme. The assay is based on the enzyme's ability to hydrolyse lecithin and liberate free fatty acids ( ⁇ FFA/min*ml). Assays conditions: (substrate; phosphatidylcholine, temperature 30°C, pH 7.0). LATU-K assays denote that the enzyme is diluted in 3% NaCI and is carried out to avoid precipitation of the enzyme. Oil
- Samples for water degumming trials Sample 1: control, 2: EDS 226 (0.1 LATU- K/g), 3: 710 (0.2 LATU-K/g), 4: 710 (0.4 LATU-K/g), 5: K710 (0.9 LATU-K/g) 6:
- LATU-K/g oil 0.00 0.1 0.2 0.4 0.9 0.04 0.1 0.2
- HPTLC High-performance-Thin-Layer-Chromatography
- Solution B was applied (0.3 ⁇ 1) to the HPTLC plate by an automatic TLC applicator.
- Phospholipid standard no. 16 (0.5% phospholipid (Spectra Lipid, Germany) diluted in CHCI 3 ) was applied (0.1, 0.3, 0.5, 0.8 and 1.5 ⁇ 1) to the HPTLC plate by an automatic
- Figure 43 shows the standard curve is calculated on the basis of applied amount (0.1, 0.3, 0.5, 0.8 and 1.5 ⁇ ) of 0.5% refined rapeseed oil as function of the area of triglycerides (measured on the TLC plate).
- sample 3 the area of triglyceride is measured to 9647 (Figure 43) on the TLC plate, corresponding to applying 0.231 ⁇ of the standard on the TLC plate.
- a standard curve was made, based on gum from the control (no enzyme) in order to calculate the phospholipid content in enzymatic samples.
- Control gum was applied in 0.1, 0.3, 0.5, 0.8 and ⁇ . ⁇ to the TLC plate.
- PA the detected area (1779) of PA in sample 3 corresponds to 0.983 ⁇ of the standard (attached on the TLC plate, graph not shown).
- the standard contains 0.5% phospholipid, whereof PA makes out 5.13%.
- Carrier gas Helium
- sample was dissolved in 12ml Heptane:Pyridin, 2:1 containing internal standard heptadecane, 0.5mg/ml. 500 ⁇ 1 sample solution was transferred to a crimp vial, ⁇ MSTFA (TST-Methyl-N-trimethylsilyl-trifluoraceamid) was added and reacted for 15 minutes at 60°C.
- ⁇ MSTFA TST-Methyl-N-trimethylsilyl-trifluoraceamid
- the Table below shows the content (ppm) of phospholipids (phosphatidylcholine, phosphatidyl-ethanolamine and phosphatidic acid) in water degummed oils (control and enzymatic samples).
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- Enzymes And Modification Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description






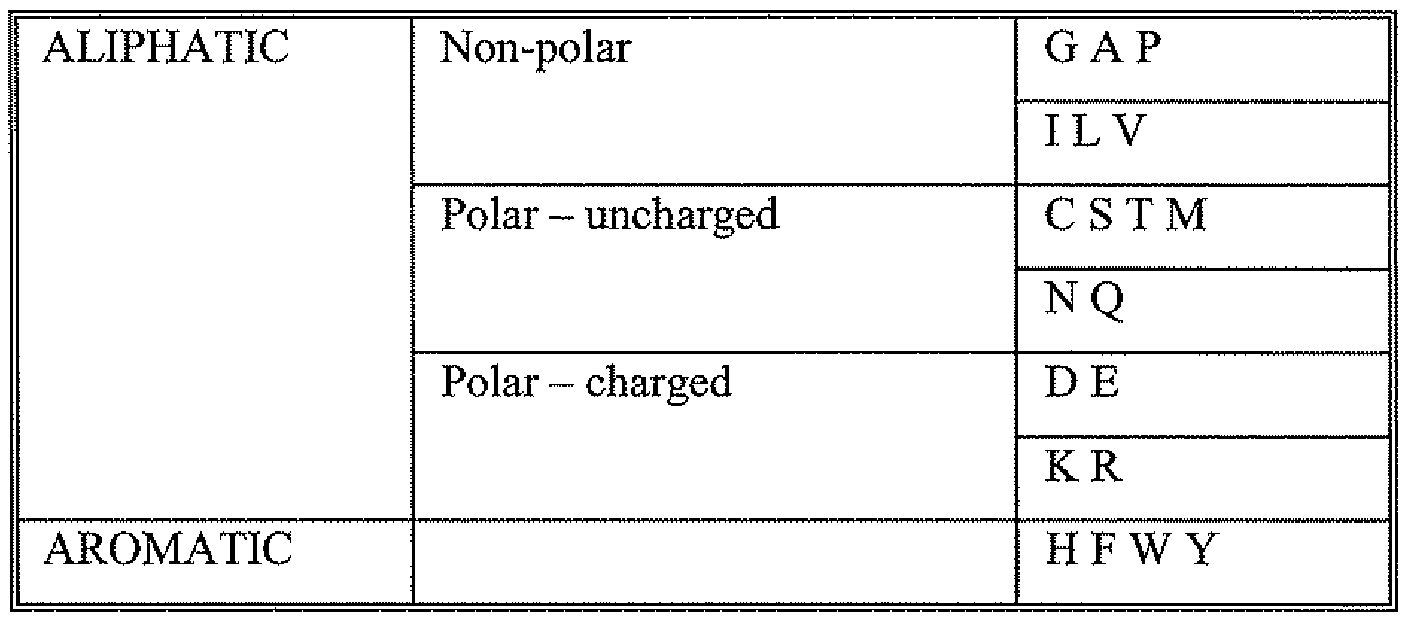







Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2009801629450A CN102656264A (en) | 2009-10-15 | 2009-10-15 | Lipid acyltransferase proteins and methods of making them |
| PCT/IB2009/054535 WO2011045629A1 (en) | 2009-10-15 | 2009-10-15 | Lipid acyltransferase proteins and methods of making them |
| EP09748481A EP2488638A1 (en) | 2009-10-15 | 2009-10-15 | Lipid acyltransferase proteins and methods of making them |
| US13/444,302 US9175271B2 (en) | 2009-10-15 | 2012-04-11 | Lipid acyltransferase proteins and methods of making them |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2009/054535 WO2011045629A1 (en) | 2009-10-15 | 2009-10-15 | Lipid acyltransferase proteins and methods of making them |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/444,302 Continuation-In-Part US9175271B2 (en) | 2009-10-15 | 2012-04-11 | Lipid acyltransferase proteins and methods of making them |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011045629A1 true WO2011045629A1 (en) | 2011-04-21 |
Family
ID=41615823
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2009/054535 Ceased WO2011045629A1 (en) | 2009-10-15 | 2009-10-15 | Lipid acyltransferase proteins and methods of making them |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9175271B2 (en) |
| EP (1) | EP2488638A1 (en) |
| CN (1) | CN102656264A (en) |
| WO (1) | WO2011045629A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015150372A1 (en) | 2014-04-01 | 2015-10-08 | Dupont Nutrition Biosciences Aps | Method for increasing crude palm oil yields |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009081094A2 (en) * | 2007-12-21 | 2009-07-02 | Danisco A/S | Process |
| CN104513838A (en) * | 2013-09-29 | 2015-04-15 | 丰益(上海)生物技术研发中心有限公司 | Lipositol, composition and product comprising same and preparation method of the product |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004064537A2 (en) * | 2003-01-17 | 2004-08-05 | Danisco A/S | Method for the in situ production of an emulsifier in foodstuff |
| WO2005066351A2 (en) * | 2003-12-24 | 2005-07-21 | Danisco A/S | Enzymatic treatment of oils |
| WO2008090395A1 (en) * | 2007-01-25 | 2008-07-31 | Danisco A/S | Production of a lipid acyltransferase from transformed bacillus licheniformis cells |
| WO2009081094A2 (en) * | 2007-12-21 | 2009-07-02 | Danisco A/S | Process |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6344328B1 (en) | 1995-12-07 | 2002-02-05 | Diversa Corporation | Method for screening for enzyme activity |
| US6361974B1 (en) | 1995-12-07 | 2002-03-26 | Diversa Corporation | Exonuclease-mediated nucleic acid reassembly in directed evolution |
| ATE231186T1 (en) | 1998-07-21 | 2003-02-15 | Danisco | GROCERIES |
| EP1131416B1 (en) | 1998-11-27 | 2009-09-02 | Novozymes A/S | Lipolytic enzyme variants |
| EP1307548A2 (en) | 2000-07-13 | 2003-05-07 | Maxygen, Inc. | Novel lipase genes |
| CA2418317A1 (en) | 2000-08-11 | 2002-02-21 | Genencor International, Inc. | Bacillus transformation, transformants and mutant libraries |
| EP1425462A4 (en) | 2001-06-29 | 2008-01-02 | Novozymes North America Inc | Single-bath preparation of cellulosic materials |
| GB0301117D0 (en) * | 2003-01-17 | 2003-02-19 | Danisco | Method |
| MXPA05007653A (en) | 2003-01-17 | 2005-09-30 | Danisco | Method. |
| US7955814B2 (en) | 2003-01-17 | 2011-06-07 | Danisco A/S | Method |
| US20050196766A1 (en) | 2003-12-24 | 2005-09-08 | Soe Jorn B. | Proteins |
| DK2119771T3 (en) | 2003-12-24 | 2018-12-10 | Dupont Nutrition Biosci Aps | PROTEINS |
| CN102533440B (en) | 2004-07-16 | 2017-09-19 | 杜邦营养生物科学有限公司 | Method for degumming oil with enzymes |
-
2009
- 2009-10-15 EP EP09748481A patent/EP2488638A1/en not_active Withdrawn
- 2009-10-15 CN CN2009801629450A patent/CN102656264A/en active Pending
- 2009-10-15 WO PCT/IB2009/054535 patent/WO2011045629A1/en not_active Ceased
-
2012
- 2012-04-11 US US13/444,302 patent/US9175271B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004064537A2 (en) * | 2003-01-17 | 2004-08-05 | Danisco A/S | Method for the in situ production of an emulsifier in foodstuff |
| WO2005066351A2 (en) * | 2003-12-24 | 2005-07-21 | Danisco A/S | Enzymatic treatment of oils |
| WO2008090395A1 (en) * | 2007-01-25 | 2008-07-31 | Danisco A/S | Production of a lipid acyltransferase from transformed bacillus licheniformis cells |
| WO2009081094A2 (en) * | 2007-12-21 | 2009-07-02 | Danisco A/S | Process |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015150372A1 (en) | 2014-04-01 | 2015-10-08 | Dupont Nutrition Biosciences Aps | Method for increasing crude palm oil yields |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2488638A1 (en) | 2012-08-22 |
| CN102656264A (en) | 2012-09-05 |
| US9175271B2 (en) | 2015-11-03 |
| US20130034627A1 (en) | 2013-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5697294B2 (en) | Glycolipid acyltransferase mutant and method for producing the same | |
| JP4804283B2 (en) | Use of lipid acyltransferases | |
| JP4804340B2 (en) | Use of lipid acyltransferases | |
| JP2007516717A6 (en) | protein | |
| RU2518345C2 (en) | Proteins | |
| CN101917862A (en) | method | |
| US20150359806A1 (en) | Method for producing phytosterol/phytostanol phospholipid esters | |
| US8030044B2 (en) | Lipid acyltransferases | |
| US9175271B2 (en) | Lipid acyltransferase proteins and methods of making them | |
| DK1704236T3 (en) | PROTEINS | |
| HK1137484A (en) | Proteins | |
| MXPA06007423A (en) | Proteins | |
| HK1095362B (en) | Method for the in situ production of an emulsifier in a foodstuff |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980162945.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09748481 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3285/CHENP/2012 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2009748481 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009748481 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012008781 Country of ref document: BR |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: 112012008781 Country of ref document: BR |
|
| ENPW | Started to enter national phase and was withdrawn or failed for other reasons |
Ref document number: 112012008781 Country of ref document: BR |