WO2011056806A1 - Process for the preparation of pyrazinone thrombin inhibitor and its intermediates - Google Patents
Process for the preparation of pyrazinone thrombin inhibitor and its intermediates Download PDFInfo
- Publication number
- WO2011056806A1 WO2011056806A1 PCT/US2010/055176 US2010055176W WO2011056806A1 WO 2011056806 A1 WO2011056806 A1 WO 2011056806A1 US 2010055176 W US2010055176 W US 2010055176W WO 2011056806 A1 WO2011056806 A1 WO 2011056806A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- difluoro
- fluoropyridine
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- TYKNQWWXUIYRSZ-UHFFFAOYSA-N NCC(c1ncccc1)(F)F Chemical compound NCC(c1ncccc1)(F)F TYKNQWWXUIYRSZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
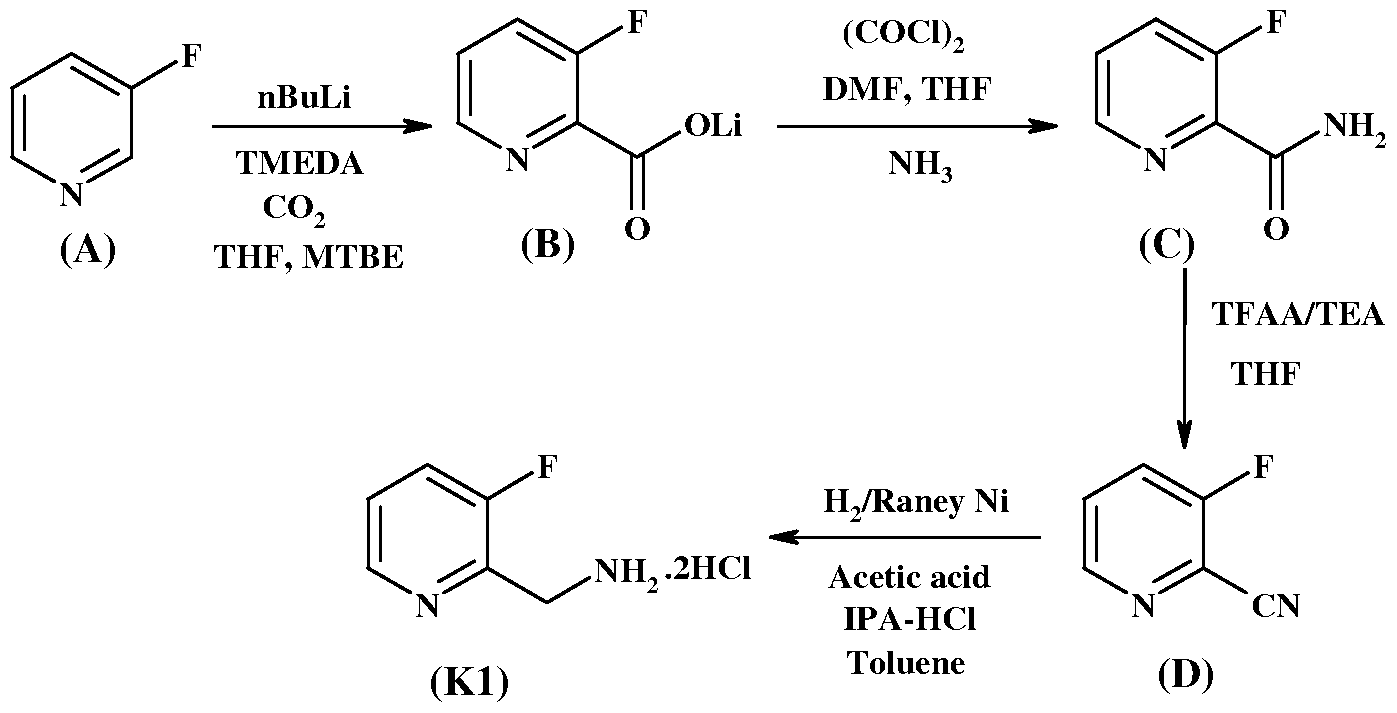
- Described herein is an improved process for the preparation of 3-fluoro-2-pyridyl methyl-3-(2,2-difluoro-2-(2-pyridyl)ethylamino)-6-chloropyrazin-2-one-l-acetamide of compound of formula (I).
- the improvement comprises the preparation of intermediates namely, 2-aminomethyl-3-fluoropyridine or its salt (Kl) and 2,2-difluoro-2-(2-pyridyl) ethylamine or its salt (K2).
- the present invention relates to a simple, commercially viable and industrially scalable process for the preparation of compound of the formula (I) and its intermediates, which involves the use of cost effective raw materials, simple isolation of the product, purification and work-up.
- the present invention also relates to a simple and industrially scalable process for the preparation of compound of formula (I) and its intermediates, which avoids chromatographic purification techniques.
- the present invention provides an improved process for the preparation of com ound of the formula (I).
- the invention comprises processes for making key intermediates namely, 2- aminomethyl-3-fluoropyridine as a hydrochloride salt (Kl) and 2,2-difluoro-2-(2- pyridyl)ethylamine as a benzenesulfonic acid salt (K2).
- the intermediates (Kl) and (K2) are used in the preparation of compound of formula (I), thrombin inhibitor.
- a process for the preparation of 2-aminomethyl-3- fluoropyridine as a hydrochloride salt (Kl) is provided.
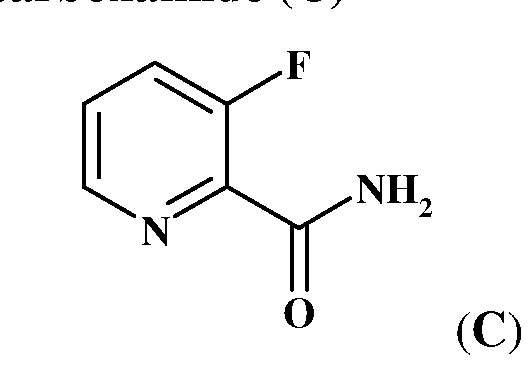
- a process for preparing 2-aminomethyl-3-fluoropyridine.2HCl comprises reduction of 3-fluoropyridine-2-carbonitrile of a compound of formula (D) with the reducing agent H 2 /Raney Ni in the presence of acetic acid or trifluoroacetic acid.
- the reported processes use expensive H 2 /Pd/C that is replaced with cost-effective Raney Nickel.
- the synthetic scheme for the preparation of 2-aminomethyl-3-fluoropyridine.2HCl is depicted in Scheme 1 as follows.
- a process for the preparation of 2,2-difluoro-2-(2- pyridyl)ethylamine as a benzenesulfonic acid (BSA) salt (K2) is provided (Scheme 2).
- the improved process utilizes p-toluenesulfonyl chloride (pTsCl or Tosyl chloride) in the preparation of a tosylate or mesylate (J) from the corresponding difluoro alcohol (H).
- pTsCl or Tosyl chloride p-toluenesulfonyl chloride
- Triflic anhydride was used previously in place of low cost pTsCl or MsCl.
- a process for purifying 3-fluoro-2-pyridylmethyl-3-(2,2- difluoro-2-(2-pyridyl)ethylamino)-6-chloropyrazin-2-one-l-acetamide, a compound of formula (I), using organic solvents including but not limited to ketones, alcohols, esters, ethers, hydrocarbons, halogenated solvents, and the like or a mixture thereof is provided.
- Ketones include, but are not limited to, acetone, butanone and the like.
- Alcohols include, but are not limited to, methanol, ethanol, n-propanol, isopropanol (IPA), n-butanol, t- butanol, hexanol, isoamyl alcohol and the like.
- Esters include, but are not limited to, ethyl acetate, methyl acetate, isopropyl acetate and the like.
- Ethers include, but are not limited to, diisopropyl ether (IPE), methyl t-butyl ether (MTBE) and the like.
- Hydrocarbons include, but are not limited to, hexane, heptane, cyclohexane, decalin, pentane and the like.
- Halogenated solvents include, but not limited to, dichloromethane (MDC), 1,2-dichloroethane and the like or mixture thereof, preferably acetone, isopropyl alcohol or mixtures thereof.
- salts used herein includes inorganic acids such as hydrochloride (hydrochloric), hydrobromide (hydrobromic), sulfuric, sulfamic, phosphoric, nitric and the like, or quaternary ammonium salts, which are formed, e.g., from inorganic or organic acids or bases.
- inorganic acids such as hydrochloride (hydrochloric), hydrobromide (hydrobromic), sulfuric, sulfamic, phosphoric, nitric and the like, or quaternary ammonium salts, which are formed, e.g., from inorganic or organic acids or bases.
- acid addition salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulphonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate, glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2-hydroxyethanesulfonate, lactate, maleate, methanesulfonate, toluenesulfonate, 2-naphthalenesulfonate, nicotinate, nitrate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate,
- Solvates are addition complexes in which a compound is combined with a solvent in some fixed proportion.
- Solvents include, but are not limited to water, methanol, ethanol, 1-propanol, isopropanol, 1-butanol, isobutanol, i-butanol, acetone, methyl ethyl ketone, acetonitrile, ethyl acetate, benzene, toluene, xylene, ethylene glycol, dichloromethane, 1,2-dichloroethane, N-methylformamide, ⁇ , ⁇ -dimethylformamide, N- methylacetamide, pyridine, dioxane, diethyl ether and the like. Hydrates are solvates in which the solvent is water. It is to be understood that the definition of the compounds or intermediates of the present invention encompasses all possible hydrates and solvates, in any proportion.
- thromboin inhibition is used for anticoagulant therapy in individuals having thrombotic conditions, and whenever inhibition of blood coagulation is required such as to prevent coagulation of stored whole blood and to prevent coagulation in other biological samples for testing or storage. Therefore, thrombin inhibitors are added to any medium containing or suspected of containing thrombin and in which it is desired that blood coagulation be inhibited.
- the present invention provides an improved process for the preparation of compound of formula (I); an improved process for the preparation of key intermediates of the compound of formula (I); and/or a pharmaceutical composition comprising the compound; and/or a compound for inhibiting thrombin; and/or an improved method for inhibiting thrombin in blood.
- Step-1 Lithium 3-fluoropyridin -2-carboxylate (B)
- the RM was cooled to -75 °C, then 10 g of 3-fluoropyridine (in 10 mL of MTBE) was added dropwise while maintaining the temperature in the range of - 75 °C to - 70 °C.
- the RM was stirred at the same temperature for 4 hours.
- 100 mL of tetrahydrofuran (THF) was taken and cooled to - 70 °C followed by bubbling of C0 2 gas for 1 hour.
- lithiated 3-fluoropyridine solution (mixture of 3- fluoropyridine + n-BuLi + TMEDA in MTBE) was added dropwise using cannula maintaining the reaction temperature below -70 °C (- 70 °C to - 60 °C). Bubbling of C0 2 gas was continued for another hour and then the reaction temperature was slowly brought to +30 °C. The resulting solid was filtered, dried and washed with 100 mL of 1:1 THF: MTBE, then vacuum dried at 45 °C, resulting in 14.5 g of the titled compound.
- the RM temperature was brought to 10 to 15 °C and maintained for 30 minutes.
- the RM was filtered, and the solid was washed with 5 mL of cold IPA, and dried under vacuum at 40 °C, resulting in 3.8 g of l-(3-fluoropyridin-2-yl)methanamine dihydrochloride salt (Kl).
- the RM was quenched by adding saturated ammonium chloride solution at 0-5 °C and the volume of reaction mixture was reduced to half by distilling under vacuum.
- the solution was basified with 50% NaOH solution and extracted with MTBE.
- Step-4 Preparation of 2,2-Difluoro-2-pyridin-2-ylethanamine benzenesulfonate salt
- Example 3 Process for purifying the compound of formula (I).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Pyridine Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description




















Claims

Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2010800499062A CN102711481A (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates |
| EP10828989.3A EP2496087A4 (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates |
| JP2012537955A JP2013510159A (en) | 2009-11-03 | 2010-11-02 | Pyrazinone thrombin inhibitor and method for producing the same |
| MX2012005165A MX2012005165A (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates. |
| CA2778922A CA2778922A1 (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates |
| RU2012118499/04A RU2012118499A (en) | 2009-11-03 | 2010-11-02 | METHOD FOR PRODUCING THROMBIN PYRAZININE INHIBITOR AND INTERMEDIATE COMPOUNDS FOR ITS PRODUCTION |
| AU2010315338A AU2010315338A1 (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2663CH2009 | 2009-11-03 | ||
| IN2663/CHE/2009 | 2009-11-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011056806A1 true WO2011056806A1 (en) | 2011-05-12 |
Family
ID=43926105
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2010/055176 Ceased WO2011056806A1 (en) | 2009-11-03 | 2010-11-02 | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US8541580B2 (en) |
| EP (1) | EP2496087A4 (en) |
| JP (1) | JP2013510159A (en) |
| KR (1) | KR20120106727A (en) |
| CN (1) | CN102711481A (en) |
| AU (1) | AU2010315338A1 (en) |
| CA (1) | CA2778922A1 (en) |
| MX (1) | MX2012005165A (en) |
| RU (1) | RU2012118499A (en) |
| WO (1) | WO2011056806A1 (en) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140090663A (en) | 2011-11-07 | 2014-07-17 | 디아크론 파마슈티칼스, 인코포레이티드 | An extended release formulation of a direct thrombin inhibitor |
| MX392191B (en) | 2015-10-26 | 2025-03-21 | Bayer Cropscience Ag | DERIVATIVES OF CONDENSED BICYCLIC HETEROCYCLES AS PEST CONTROL AGENTS. |
| JP7535532B2 (en) | 2019-02-26 | 2024-08-16 | バイエル・アクチエンゲゼルシヤフト | Fused bicyclic heterocyclic derivatives as pest control agents. |
| EP3931193A1 (en) | 2019-02-26 | 2022-01-05 | Bayer Aktiengesellschaft | Fused bicyclic heterocycle derivatives as pesticides |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6147078A (en) * | 1998-05-19 | 2000-11-14 | Merck & Co., Inc. | Pyrazinone thrombin inhibitors |
| WO2002046160A2 (en) * | 2000-12-06 | 2002-06-13 | Merck & Co., Inc. | Process for making a thrombin inhibitor |
| US7456290B2 (en) * | 2002-11-20 | 2008-11-25 | Bayer Cropscience S.A. | Process for the preparation of 2-aminomethylpyridine derivative |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA58636C2 (en) * | 1999-06-04 | 2003-08-15 | Мерк Енд Ко., Інк. | Pyrazinone thrombin inhibitors, pharmaceutical composition, method for treatment conditions caused by thrombus formation |
-
2010
- 2010-11-02 CA CA2778922A patent/CA2778922A1/en not_active Abandoned
- 2010-11-02 AU AU2010315338A patent/AU2010315338A1/en not_active Abandoned
- 2010-11-02 EP EP10828989.3A patent/EP2496087A4/en not_active Withdrawn
- 2010-11-02 CN CN2010800499062A patent/CN102711481A/en active Pending
- 2010-11-02 MX MX2012005165A patent/MX2012005165A/en active IP Right Grant
- 2010-11-02 JP JP2012537955A patent/JP2013510159A/en active Pending
- 2010-11-02 US US12/938,297 patent/US8541580B2/en not_active Expired - Fee Related
- 2010-11-02 WO PCT/US2010/055176 patent/WO2011056806A1/en not_active Ceased
- 2010-11-02 RU RU2012118499/04A patent/RU2012118499A/en not_active Application Discontinuation
- 2010-11-02 KR KR1020127010789A patent/KR20120106727A/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6147078A (en) * | 1998-05-19 | 2000-11-14 | Merck & Co., Inc. | Pyrazinone thrombin inhibitors |
| WO2002046160A2 (en) * | 2000-12-06 | 2002-06-13 | Merck & Co., Inc. | Process for making a thrombin inhibitor |
| US7456290B2 (en) * | 2002-11-20 | 2008-11-25 | Bayer Cropscience S.A. | Process for the preparation of 2-aminomethylpyridine derivative |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP2496087A4 * |
| WADE JR.: "Reactions of Alcohols, PowerPoint Presentation", ORGANIC CHEMISTRY, 2003, BLACKBURN, DALLAS COUNTY COMMUNITY COLLEGE DISTRICT, pages 1 - 37 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2778922A1 (en) | 2011-05-12 |
| EP2496087A4 (en) | 2013-06-19 |
| CN102711481A (en) | 2012-10-03 |
| RU2012118499A (en) | 2013-12-10 |
| KR20120106727A (en) | 2012-09-26 |
| AU2010315338A1 (en) | 2012-05-24 |
| US20110105753A1 (en) | 2011-05-05 |
| MX2012005165A (en) | 2012-08-08 |
| EP2496087A1 (en) | 2012-09-12 |
| JP2013510159A (en) | 2013-03-21 |
| US8541580B2 (en) | 2013-09-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101084451B1 (en) | 2-pyrazinecarboxamide derivatives | |
| JP5604424B2 (en) | Method for the preparation of proapoptotic agent ABT-263 | |
| WO2012025944A2 (en) | Sitagliptin, salts and polymorphs thereof | |
| JP2023548666A (en) | 3-Fluoro-5-(((1S,2aR)-1,3,3,4,4-pentafluoro-2a-hydroxy-2,2a,3,4-tetrahydro-1H-cyclopenta[cd]indene-7 Method of preparing -yl)oxy)-benzonitrile | |
| WO2011056806A1 (en) | Process for the preparation of pyrazinone thrombin inhibitor and its intermediates | |
| CA3216002A1 (en) | Preparation method for pyrrole amide compound | |
| Dallanoce et al. | Synthesis and functional characterization of novel derivatives related to oxotremorine and oxotremorine-M | |
| AU751015B2 (en) | Novel sulfonamide substituted chroman derivatives useful as beta-3 adrenoreceptor agonists | |
| US20250145623A1 (en) | Methods of preparation of heterocyclic compounds | |
| Gavhane et al. | Short and stereodivergent syntheses of (−)-5-epi-tashiromine and (−)-tashiromine and the formal synthesis of (−)-isoretronecanol and (−)-trachelanthamidine | |
| US6469172B2 (en) | Process for the preparation of chemical compounds | |
| US8815870B2 (en) | 4-(2-(6-substituted-hexylidene) hydrazinyl)benzonitrile and preparation thereof | |
| CA2437954C (en) | Process for producing quinolinecarboxyaldehyde derivative and intermediate | |
| DE60006125T2 (en) | PREPARATION OF 2- (2-ARYLMORPHOLIN-2-YL) ETHANOL DERIVATIVES AND INTERMEDIATE PRODUCTS | |
| CA2562578C (en) | Process for production of pyrazole-fused ring derivatives | |
| KR20260002957A (en) | Method for producing sulfamoylbenzoic acid derivatives | |
| JP4769464B2 (en) | Method for producing alcohol compound | |
| Zeng et al. | Synthesis of 2, 2, 6, 6-tetrafluoro-4-phenylmethylmorpholin-3-ones: A simple approach from fluorinated triethylene glycol | |
| KR20240052932A (en) | Method for preparing synthetic intermediates of monocyclic pyridine derivatives | |
| AU700600B2 (en) | (S)-5,7-difluoro-1,2,3,4-tetrahydronaphthalen-2-ylamine and its preparation | |
| JP4310511B2 (en) | Method for producing aminomethylpyridine | |
| JP2003221394A (en) | Method for producing quinuclidine derivative | |
| JP2008247889A (en) | Process for producing ε-caprolactone compound | |
| Su et al. | A novel synthesis of N-substituted 2-(benzotriazol-1-yl) acetamides promoted by samarium diiodide | |
| WO2007035003A1 (en) | Process for producing optically active piperazine compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080049906.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10828989 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3577/DELNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2778922 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20127010789 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010315338 Country of ref document: AU |
|
| REEP | Request for entry into the european phase |
Ref document number: 2010828989 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010828989 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012537955 Country of ref document: JP Ref document number: MX/A/2012/005165 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2010315338 Country of ref document: AU Date of ref document: 20101102 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012118499 Country of ref document: RU |