WO2011083870A1 - マウス人工染色体ベクター - Google Patents
マウス人工染色体ベクター Download PDFInfo
- Publication number
- WO2011083870A1 WO2011083870A1 PCT/JP2011/050490 JP2011050490W WO2011083870A1 WO 2011083870 A1 WO2011083870 A1 WO 2011083870A1 JP 2011050490 W JP2011050490 W JP 2011050490W WO 2011083870 A1 WO2011083870 A1 WO 2011083870A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mouse
- artificial chromosome
- cell
- gene
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


































































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
- A01K67/0278—Knock-in vertebrates, e.g. humanised vertebrates
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1086—Preparation or screening of expression libraries, e.g. reporter assays
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/8509—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells for producing genetically modified animals, e.g. transgenic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2207/00—Modified animals
- A01K2207/15—Humanized animals
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/05—Animals comprising random inserted nucleic acids (transgenic)
- A01K2217/052—Animals comprising random inserted nucleic acids (transgenic) inducing gain of function
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/20—Animal model comprising regulated expression system
- A01K2217/206—Animal model comprising tissue-specific expression system, e.g. tissue specific expression of transgene, of Cre recombinase
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/105—Murine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/01—Animal expressing industrially exogenous proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/20—Pseudochromosomes, minichrosomosomes
- C12N2800/208—Pseudochromosomes, minichrosomosomes of mammalian origin, e.g. minichromosome
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/30—Vector systems comprising sequences for excision in presence of a recombinase, e.g. loxP or FRT
Definitions
- the present invention relates to a mouse artificial chromosome vector that is stably maintained in rodents and capable of transmitting offspring.
- the present invention also relates to a cell carrying the mouse artificial chromosome vector.
- the present invention further relates to a non-human animal such as a mouse carrying the mouse artificial chromosome vector.
- Transgenic mice are widely used to utilize target genes and expression products by introducing gene-loaded vectors.
- the site of gene introduction is random, and the introduction of the introduced gene may suppress the expression of the introduced gene.
- the conventional gene introduction method does not control the copy number.
- the size is limited to about 200 kb. For these reasons, it has been difficult to clone a gene or gene cluster having a size exceeding 200 kb, which is not uncommon as a mammalian gene, into a vector including a regulatory region. Therefore, the conventional gene transfer method has a limitation that the original function of the transgene cannot be reproduced and verified.
- Non-patent Document 1 a technique for producing a chromosome-introduced mouse using a new chromosome introduction method called gene introduction at the chromosome level.
- ES mouse embryonic stem
- Human chromosome fragments were retained independently, multiple human genes were expressed in a tissue-specific manner, and meiosis was achieved. It has been shown that there are also human chromosomes that can be transmitted in a fractional manner through.
- the present inventors introduced the full length of human chromosome 21 (about 35 Mb) into a mouse, and produced a Down syndrome model mouse having high practical value (Non-patent Document 2).
- Non-patent Document 2 the introduced gene on human chromosome 21 reproduced the physiological expression pattern, indicating the effectiveness of the chromosome vector.
- the present inventors utilized chromosome deletion by telomere truncation technology using artificial telomere sequences, which are chromosome engineering techniques, and chromosome cloning by the Cre / loxP system, to obtain a human artificial chromosome containing only the target region.
- HAC human artificial chromosome
- Mb megabase
- the drug metabolizing enzyme CYP3A gene cluster (1 Mb) on human chromosome 7 and the human DMD gene (2.5 Mb), which is the causative gene of human X-linked muscular dystrophy It was cloned into a HAC vector (CYP3A-HAC, DMD-HAC) and introduced into mouse ES cells to prepare mice (Patent Document 1, Non-Patent Document 5). From the tissue retention rate and expression analysis of mice introduced with CYP3A-HAC, the CYP3A gene cluster on HAC is retained in each tissue of the mouse (FIG.
- Mammalian artificial chromosome vectors including human artificial chromosomes, have advantages not found in conventional vector systems (viruses, YACs, BACs, PACs, cosmids, plasmids). It is expected to be useful as an animal production system.
- Patent Documents 2 and 3 disclose HAC vectors that are relatively stably retained in cells, which are composed of chromosome fragments obtained by modifying human chromosome 14 or human chromosome 21 and reducing the size thereof. .
- human chromosomes including human artificial chromosome vectors have a constant retention rate in mouse cells
- artificial chromosome vectors it has not fully demonstrated its advantages.
- Human artificial chromosomes are unstable in each mouse tissue, and the ratio (retention rate) of human artificial chromosomes held in mouse tissue cells tends to decrease. Retention is not constant. Alternatively, the same holds true between mouse individuals, and the retention rate of human artificial chromosomes between mouse individuals is not constant. Due to this heterogeneous retention between mouse tissues and individuals, detailed, accurate and highly reproducible analysis of transgenes using mouse cells or mouse individuals into which the target gene (s) has been introduced by human artificial chromosomes Is difficult to do. As described above, there are very few reports on rodent-derived artificial chromosomes including mice, and there are no artificial chromosomes that are stably maintained in rodent cells or individuals.
- An object of the present invention is to provide a mouse artificial chromosome in which a target gene (s) to be introduced is stably maintained in a rodent cell or a rodent individual, thereby enabling detailed, accurate and highly reproducible analysis.
- a target gene (s) to be introduced is stably maintained in a rodent cell or a rodent individual, thereby enabling detailed, accurate and highly reproducible analysis.
- the present invention encompasses the following features.
- a mouse-derived natural centromere a mouse chromosome-derived long arm fragment obtained by deleting the distal long arm from the site of the mouse chromosome long arm near the centromere, and a telomere sequence, and mammalian cells and tissues
- a mouse artificial chromosome vector characterized by being stably maintained in
- the long arm fragment derived from mouse chromosome consists of the remainder from which at least 99.5% of the total number of endogenous genes is deleted from any one long arm of mouse chromosomes 1 to 19 (1) Or the mouse
- the DNA sequence insertion site is a loxP sequence, FRT sequence, ⁇ C31attB and ⁇ C31attP sequence, R4attB and R4attP sequence, TP901-1attB and TP901-1attP sequence, or Bxb1attB and Bxb1attP sequence (7) or (8) The mouse artificial chromosome vector described.
- (11) The mouse artificial chromosome vector according to any one of (1) to (10), further comprising a foreign DNA sequence.
- (13) The mouse artificial chromosome vector according to (11) or (12) above, wherein the foreign DNA sequence is a human DNA sequence.
- the enzyme involved in the first-phase reaction is selected from the group consisting of CYP such as CYP1A, CYP1B, CYP2A, CYP2B, CYP2C, CYP2D, CYP2E, CYP2J, CYP3A, CYP4A, CYP4B and their subfamily, and CES.
- the gene encoding the transporter encodes at least one gene selected from the group consisting of MDR1, MDR2, MRP2, OAT, OATP, OCT and BCRP Chromosome vector.
- the mouse artificial chromosome vector according to any one of the above (11) to (21), comprising sequences of at least two genes to be selected.
- the foreign DNA sequence is a polymorphism such as cytokines, hormones, growth factors, nutrition factors, hematopoietic factors, blood coagulation / lysis factors, immunoglobulins, G protein-coupled receptors, enzymes, etc. Sequence of gene or DNA encoding peptides, or therapeutic gene or DNA related to diseases such as tumor, muscular dystrophy, hemophilia, neurodegenerative disease, autoimmune disease, allergic disease, genetic disease.
- the mouse artificial chromosome vector according to any one of (11) to (13) above.
- the cell is a hepatocyte, intestinal cell, kidney cell, spleen cell, lung cell, heart cell, skeletal muscle cell, brain cell, bone marrow cell, lymphocyte cell, megakaryocyte cell, sperm or egg
- the stem cell is an embryonic stem (ES) cell or an induced pluripotent stem (iPS) cell.
- ES embryonic stem
- iPS induced pluripotent stem
- a pharmaceutical composition comprising the cell according to any one of (28) to (32) above, which retains a mouse artificial chromosome vector containing a foreign DNA sequence for disease treatment.
- a non-human animal characterized by retaining the mouse artificial chromosome vector described in any one of (1) to (27) above.
- the non-human animal according to (34) above which is a disease model animal.
- the non-human animal according to (34) above which is an animal that enables expression of a foreign human drug metabolism-related gene.
- the non-human animal according to (34) above which is an animal that enables production of human antibodies.
- a method for screening a substance effective for treating a disease which comprises administering a candidate drug to the non-human animal according to (35) which is a disease model animal and evaluating the therapeutic effect of the drug. .
- a drug or food is administered to a non-human animal or a cell, organ or tissue derived from the animal described in (36) or (38) above, which holds a mouse artificial chromosome vector containing a gene related to human drug metabolism,
- a method for testing a pharmacological action and / or metabolism and / or toxicity of a drug or food comprising measuring the pharmacological action and / or metabolism and / or toxicity of the drug or food.
- a mouse artificial chromosome vector provided with a DNA sequence insertion site can be used to introduce any cell or tissue that has been difficult in the prior art when a target gene (group) is introduced into a rodent cell or a rodent individual. Can stably hold the target gene (s) at the same retention rate, and the reporter gene can be inserted together with the target foreign DNA sequence or gene, so that vector-introduced cells can be visualized. It enables accurate and highly reproducible analysis or efficient recovery of expression products.
- This specification includes the contents described in the specification and / or drawings of Japanese Patent Application No. 2010-1425 which is the basis of the priority of the present application.
- FIG. 1 shows a schematic diagram of the procedures of Examples 1 and 2.
- the cell names etc. in the figure are shown in the following format.
- Cell name Intracellular gene modification; retained chromosome fragment name, transgene-retained chromosome name).
- the symbols in the figure are as follows.
- BSr blasticidin (BS) resistance gene
- puro puromycin resistance gene
- artificial telomere artificial telomere (TTAGGG) repeat sequence
- EGFP green fluorescent protein expression gene
- neo neomycin (G418) resistance gene
- loxP site-specific 3'HPRT: Exon sequence from the 3rd to the 9th of the HPRT gene.
- FIG. 2 shows a schematic diagram of the procedure of the third embodiment.
- telomere artificial telomere (TTAGGG) repeat sequence
- EGFP green fluorescent protein expression gene
- neo neomycin (G418) resistance gene
- 3′HPRT third to ninth exon sequence of HPRT gene
- puro Puromycin resistance gene.
- FIG. 3 shows a schematic diagram of the procedure of the fourth embodiment.
- the cell names etc. in the figure are shown in the following format.
- Cell name Intracellular gene modification; retained chromosome fragment name, transgene-retained chromosome name).
- the symbols in the figure are as follows. puro: puromycin resistance gene, artificial telomere: artificial telomere (TTAGGG) repeat sequence, 5'HPRT: first and second exon sequences of HPRT gene, hyg: hygromycin resistance gene, loxP: site-specific DNA sequence insertion site .
- FIG. 4 shows a schematic diagram of the procedure of the fifth embodiment.
- the cell names etc. in the figure are shown in the following format.
- FIG. 5 shows a schematic diagram of the procedure of the sixth embodiment.
- the cell names etc. in the figure are shown in the following format.
- Cell name (intracellular gene modification; retained chromosome fragment name, transgene-retained chromosome name).
- the symbols in the figure are as follows.
- FIG. 6 shows mouse A9 cells (mouse A9 (neo)) (left) and cell fusion clones of mouse A9 cells and mouse fibroblasts (mouse embryonic fibroblast (mChr11-BSr)) (mouse A9xmouse fibronic; mChr11-BSr)).
- FIG. 6 shows mouse A9 cells (mouse A9 (neo)) (left) and cell fusion clones of mouse A9 cells and mouse fibroblasts (mouse embryonic fibroblast (mChr11-BSr)) (mouse A9xmouse fibronic; mChr11-BSr)).
- FIG. 7 shows the results of FISH analysis of a DT40 (mChr11-Bsr) clone using mouse Cot-1 DNA as a probe.
- FIG. 8 shows the SKY FISH analysis result (left) and the SKY FISH stained image (right) showing that the mouse chromosome (mChr11-BSr) introduced into the chicken DT40 cell is the 11th mouse chromosome.
- FIG. 9 shows the telomere truncation vector in the AL671968 region of mouse chromosome 11 and the partial structure of the mouse chromosome 11 allele subjected to homologous recombination with the vector.
- DT40 (MAC) [DT40 (B6bT-1)] clone monono containing the allele of mouse artificial chromosome MAC telomeric truncation in mouse chromosome 11 region AL671968 using the pBS-TEL / puro_MAC vector.
- -A color FISH analysis result (right) is shown.
- DT40 (mChr11-BSr) in the left figure shows a DT40 (mChr11-BSr) clone before telomere truncation.
- FIG. 11 shows a partial structure of a GFP-neo-loxP-3′HPRT type loxP targeting vector (pMAC1) and an allele of mouse artificial chromosome MAC subjected to homologous recombination with the vector.
- FIG. 12 shows a two-color FISH analysis result of DT40 (MAC1) clone when mouse Cot-1 DNA and GFP-PGKneo-loxP-3′HPRT cassette are used as probes.
- FIG. 13 shows a mono-color FISH analysis result of a CHO (HPRT ⁇ ; MAC1) clone using mouse Cot-1 DNA as a probe.
- FIG. 14 shows the results of a mono-color FISH analysis of a mouse ES (MAC1) clone when mouse minor satellite DNA is used as a probe.
- FIG. 15 shows a targeting vector (pMPloxPHyg) for inserting loxP into the AC004922 region located very close to the CYP3A locus on human chromosome 7 and on the centromere side (about 300 Kb centromere side), and homologous recombination with the vector
- FIG. 15a shows the partial structure of the human chromosome 7 allele subjected to the above (FIG. 15a).
- FIG. 15 shows a targeting vector for inserting loxP into the AC004922 region located very close to the CYP3A locus on human chromosome 7 and on the centromere side (about 300 Kb centromere side), and homologous recombination with the vector
- FIG. 15a shows the partial structure of the human chromosome 7 allele subjected to the above (FIG. 15
- FIG. 15b shows the results of analysis of homologous recombinants by Southern hybridization of hygromycin-resistant strains of the human chromosome 7-containing DT40 cell clone transfected with the linearized vector.
- the upper arrowhead represents a non-homologous recombinant (approximately 10.9 kb) and the lower arrowhead represents a homologous recombinant (approximately 8.9 kb).
- FIG. 16 shows a targeting vector (pTELhisD-PT) for inserting a human telomere sequence into the AC073842 region located very close to the CYP3A locus on human chromosome 7 and on the telomere side (about 150 Kb telomere side), and The partial structure of the human chromosome 7 allele that has undergone homologous recombination is shown.
- FIG. 17 shows a two-color FISH analysis result of a CHO (HPRT-, MAC1 + hChr7-loxP-tel) clone using mouse Cot-1 DNA and human Cot-1 DNA as probes.
- FIG. 18 shows the construction of a mouse artificial chromosome CYP3A-MAC in which about 1 Mb around the human CYP3A gene cluster region (AC004922-human CYP3A gene cluster-AC073842) was translocated and cloned into MAC1.
- FIG. 19 shows a two-color FISH analysis result of a CHO (CYP3A-MAC, hChr7- ⁇ CYP3A) clone using human Cot-1 DNA and mouse Cot-1 DNA as probes.
- FIG. 20 shows a targeting vector (pMAC2) for constructing a mouse artificial chromosome vector MAC2 and a partial structure of an allele of mouse artificial chromosome MAC that has undergone homologous recombination with the vector.
- FIG. 1 shows the construction of a mouse artificial chromosome CYP3A-MAC in which about 1 Mb around the human CYP3A gene cluster region (AC004922-human CYP3A gene cluster-AC07
- FIG. 21 shows a two-color FISH analysis result of DT40 (MAC2) clone when using mouse Cot-1 DNA and 5 ′ HPRT-loxP-PGK hygro cassette as probes.
- FIG. 22 shows a two-color FISH analysis result of a CHO (HPRT ⁇ ; MAC2) clone using mouse Cot-1 DNA and 5′HPRT-loxP-PGK hygro cassette as probes.
- FIG. 23 shows the results of a mono-color FISH analysis of a mouse ES (HPRT-, MAC2) clone when mouse minor satellite DNA is used as a probe.
- FIG. 24 shows a partial structure of a PGKneo-loxP-3′HPRT type loxP targeting vector (pMAC3) and an allele of mouse artificial chromosome MAC3 subjected to homologous recombination with the vector.
- FIG. 25 shows the two-color FISH analysis results of DT40 (MAC3) clone when mouse Cot-1 DNA and mouse minor satellite DNA are used as probes.
- FIG. 26 shows a mono-color FISH analysis result of a CHO (HPRT ⁇ ; MAC3) clone using mouse Cot-1 DNA as a probe.
- Figure 27 is a drug-resistant clones B6 when the mouse minor satellite DNA probe -; indicating the mono-color FISH analysis results of (HPRT MAC3).
- FIG. 28 shows the retention analysis result of long-term culture of B6 (HPRT ⁇ ; MAC3) clone.
- FIG. 29 shows a procedure for inserting a specific gene (for example, GFP) into mouse artificial chromosome vector MAC3 by the Cre-loxP method, and GFP insertion vector and homologous recombination are performed by the vector.
- the partial structure of the broken mouse chromosome 11 allele is shown.
- FIG. 30 shows a two-color FISH analysis result of a CHO (GFP-MAC) clone which is a CHO cell holding mouse artificial chromosome GFP-MAC when mouse Cot-1 DNA and X6.1EGFP are used as probes.
- FIG. 31 shows the results of FISH analysis after long-term culture of mouse artificial chromosome GFP-MAC in mouse ES cells (B6-ES strain) using mouse minor satellite DNA and GFP as probes.
- FIG. 32 shows the retention analysis result of long-term culture of B6 (GFP-MAC) clone. Diamonds indicate the long-term culture with drug selection, and squares indicate the long-term culture with no drug selection.
- FIG. 33 shows a progeny transmission individual born from a chimeric mouse carrying a mouse artificial chromosome vector (GFP-MAC).
- FIG. 34 shows the retention ratio of 21HAC1 or 21HAC2 and MAC1 in CHO cells after long-term culture (25PDL).
- FIG. 35 shows the retention rate of 21HAC2 or MAC1 in ES cells after long-term culture (75PDL).
- FIG. 36 shows a stereoscopic fluorescence microscope image in TC (MAC1) mouse (female) tissue.
- FIG. 37 shows the GFP positive rate in bone marrow-derived cells of TC (MAC1) mice or TC (21 HAC2) mice.
- FIG. 38 shows the GFP positive rate in blood cells of spleen-derived cells of TC (MAC1) mice or TC (21 HAC2) mice.
- FIG. 39 shows the results of a mono-color FISH analysis of tail fibroblasts derived from TC (MAC1) mice using a mouse minor satellite DNA probe.
- FIG. 40 shows the two-color FISH analysis results of CYP3A-BAC (RP11-757A13) and A9 (CYP3A-MAC) using mouse minor satellite DNA probe.
- FIG. 41 shows a mono-color FISH analysis result of TT2F (CYP3A-MAC) using a CYP3A-BAC (RP11-757A13) DNA probe.
- FIG. 42 shows the retention rate of CYP3A-MAC in ES cells after long-term culture (100PDL).
- FIG. 43 shows a stereoscopic fluorescence microscope image in TC (CYP3A-MAC) mouse (male) tissue.
- FIG. 44 shows the GFP positive rate in the blood cell of the bone marrow origin cell of a TC (CYP3A-MAC) mouse or a TC (CYP3A-HAC ⁇ ) mouse.
- FIG. 45 shows the retention of CYP3A-MAC or CYP3A-HAC ⁇ in each tissue of TC (CYP3A-MAC) mice or TC (CYP3A-HAC ⁇ ) mice.
- FIG. 43 shows a stereoscopic fluorescence microscope image in TC (CYP3A-MAC) mouse (male) tissue.
- FIG. 44 shows the GFP positive rate in the blood cell of the bone marrow origin cell of a TC (CYP3A-MAC) mouse or a TC (CYP3A
- FIG. 46 shows a mono-color FISH analysis result in a TC (CYP3A-MAC) hetero mouse or a TC (CYP3A-MAC) homo mouse using a CYP3A-BAC (RP11-757A13) DNA probe.
- FIG. 47 shows the results of tissue-specific gene expression analysis of CYP3A gene cluster in each tissue of TC (CYP3A-MAC) mice.
- GAPDH represents glyceraldehyde 3-phosphate dehydrogenase (glyceraldehyde 3-phosphate dehydrogenase).
- FIG. 48 shows the results of a time-specific gene expression analysis of the CYP3A gene cluster in TC (CYP3A-MAC) mouse liver.
- FIG. 49 shows the two-color FISH analysis results of rat ES (CYP3A-MAC) using CYP3A-BAC (RP11-757A13) and mouse Cot-1 DNA probe.
- Figure 50 is a mouse Cot-1 DNA and human Cot-1 DNA and using the probe CHO -; shows the two-color FISH analysis results of (HPRT MAC1, hChr21-loxP) .
- FIG. 51 shows the construction of a mouse artificial chromosome hChr21q-MAC obtained by translocation cloning about 33 Mb of the hChr21q region to MAC1.
- FIG. 52 shows a two-color FISH analysis result of a CHO (hChr21q-MAC, hChr21-hChr21q) clone using human Cot-1 DNA and mouse Cot-1 DNA as probes.
- FIG. 53 shows a two-color FISH analysis result of a TT2F (hChr21q-MAC) clone using human Cot-1 DNA and mouse minor satellite DNA probe.
- FIG. 54 shows the retention rate of hChr21q-MAC in ES cells after long-term culture (50PDL).
- FIG. 55 shows a fluorescent photograph of a chimeric mouse carrying hChr21q-MAC.
- FIG. 56 shows a targeting vector (pCKloxPHyg), a target sequence and a chromosome generated by homologous recombination for inserting a loxP sequence into AP001721 proximal to DSCR (Down syndrome disease-causing region cluster) of human chromosome 21 (hChr21).
- the allele is shown in Fig. 57 shows the results of Southern blot analysis for DT40 (hChr21q22.12-loxP).
- FIG. 58 shows a two-color FISH analysis result of DT40 (hChr21q22.12-loxP) clone using human Cot-1 DNA and hygromycin DNA as probes.
- FIG. 60 shows the construction of a mouse artificial chromosome hChr21q22.12-MAC obtained by translocation cloning about 12 Mb of the hChr21q22.12-qter region to MAC1.
- FIG. 61 shows a two-color FISH analysis result of a CHO (hChr21q22.12-MAC, hChr21-hChr21q22.12) clone using human Cot-1 DNA and mouse Cot-1 DNA as probes.
- FIG. 60 shows the construction of a mouse artificial chromosome hChr21q22.12-MAC obtained by translocation cloning about 12 Mb of the hChr21q22.12-qter region to MAC1.
- FIG. 61 shows a two-color FISH analysis result of a CHO (hChr21q22.12-MAC, hChr21-hChr21q22.12) clone using human Cot-1 DNA and mouse Cot-1 DNA as probes.
- FIG. 62 shows a two-color FISH analysis result of TT2F (hChr21q22.12-MAC) clone using human Cot-1 DNA and mouse minor satellite DNA probe.
- FIG. 63 shows the retention of hChr21q22.12-MAC in ES cells after long-term culture (50PDL).
- FIG. 64 shows the retention rate of 21HAC1 or 21HAC2 and MAC2 in CHO cells after long-term culture (25PDL).
- FIG. 65 shows the structure of 1 copy FVIII-PAC.
- FIG. 66 shows the construction method of 1 to 16 copies FVIII-PAC.
- FIG. 67 shows the results of Clotting assay (comparison of FVIII activity) after long-term culture in CHO (FVIIIx1-MAC) 1-3 and CHO (FVIIIx1-HAC) 1-2.
- FIG. 68 shows the results of the cloning assay (comparison of FVIII activity) in CHO (FVIIIx1-MAC) and CHO (FVIIIx16-MAC).
- FIG. 69 shows an entry vector construction method for construction of a multi-integration platform cassette.
- FIG. 70 shows a method for constructing a multi-integrate platform cassette.
- FIG. 71 shows a method for constructing the MI-MAC vector.
- FIG. 72 shows a method for inserting a gene into an MI-MAC vector.
- FIG. 73 shows a construction method of PXR-MAC.
- FIG. 74 shows a two-color FISH analysis result of a CHO (PXR-MAC) clone using mouse cot-1 DNA and DNA derived from human PXR-BAC (RP11-169N13) (CHORI) as probes.
- FIG. 75 shows a mono-color FISH analysis result of a TT2F (PXR-MAC) clone using a human PXR-BAC-derived DNA (RP11-169N13) (CHORI) as a probe.
- FIG. TT2F PXR-MAC
- FIG. 76 shows a partial structure of a GFP-5′HPRT-loxP-hyg type loxP targeting vector (pMAC4) and an allele of mouse artificial chromosome MAC4 subjected to homologous recombination with the vector.
- FIG. 77 shows the results of two-color FISH analysis of DT40 (MAC4) clone using mouse cot-1 DNA and GFP-5′HPRT-loxP-hyg cassette as probes.
- FIG. 78 shows a mono-color FISH analysis result of a CHO (HPRT ⁇ ; MAC4) clone using mouse Cot-1 DNA as a probe.
- FIG. 80 shows a two-color FISH analysis result of DT40 (hChr4-tel) using human cot-1 DNA and puromycin DNA as probes. The left figure shows DT40 (hChr4) before modification, and the right figure shows DT40 (hChr4-tel) after modification.
- FIG. 80 shows a targeting vector (pTELpuro-UGT2) for inserting a human telomere sequence into the AC1252392 region located very close to the UGT2 locus on the human 4 chromosome and on the telomere side (about 150 Kb telomere side), and The partial structure of the human chromosome 4 allele in which homologous recombination was performed is shown.
- FIG. 80 shows a two-color FISH analysis result of DT40 (hChr4-tel) using human cot-1 DNA and puromycin DNA as probes. The left figure shows DT40 (hChr4) before modification, and the right figure shows DT40 (hChr4-tel) after modification.
- FIG. 81 shows a targeting vector (pUGT2loxPneo), a target sequence and a chromosomal allele resulting from homologous recombination for inserting the loxP sequence into human chromosome 4 AC0743378.
- Fig. 82 shows the results of two-color FISH analysis of DT40 (hChr4-loxP-tel) using human cot-1 DNA and neomycin DNA as probes.
- FIG. 83 shows a two-color FISH analysis result of a CHO (HPRT ⁇ ; MAC4, hChr4-loxP-tel) clone using human Cot-1 DNA and mouse Cot-1 DNA as probes.
- FIG. 85 shows a two-color FISH analysis result of a CHO (UGT2-MAC, hChr4- ⁇ UGT2) clone using UGT2-BAC (RP11-643N16) (CHORI) DNA and mouse Cot-1 DNA as probes.
- FIG. 86 shows a two-color FISH analysis result of A9 (UGT2-MAC) clone using UGT2-BAC (RP11-643N16) (CHORI) and mouse minor satellite DNA as probes.
- FIG. 85 shows a two-color FISH analysis result of A9 (UGT2-MAC) clone using UGT2-BAC (RP11-643N16) (CHORI) and mouse minor satellite DNA as probes.
- FIG. 87 shows a mono-color FISH analysis result of a TT2F (UGT2-MAC) clone when UGT2-BAC (RP11-643N16) (CHORI) DNA is used as a probe.
- FIG. 88 shows the retention rate of UGT2-MAC in ES cells after long-term culture (75PDL).
- FIG. 89 shows a targeting vector (pTELpuro-CYP2C) for inserting a human telomere sequence into the AL157734 region located in the vicinity of the CYP2C locus on the human chromosome 10 and on the telomere side (about 150 Kb telomere side), and The partial structure of the human chromosome 10 allele in which homologous recombination was performed is shown.
- FIG. 90 shows a two-color FISH analysis result of DT40 (hChr10-tel) using human cot-1 DNA and puromycin DNA as probes.
- the left figure shows DT40 (hChr10) before modification, and the right figure shows DT40 (hChr10-tel) after modification.
- FIG. 91 shows a targeting vector (pCYP2CloxPneo), a target sequence and a chromosome allele resulting from homologous recombination for inserting a loxP sequence into AL138759 of human chromosome 10.
- FIG. 92 shows the construction of a mouse artificial chromosome CYP2C-MAC obtained by translocation cloning 380 kb of the vicinity of the human CYP2C gene cluster region (AL138759-human CYP2C gene cluster-AL158784) to MAC4.
- FIG. 93 shows the construction of a mouse artificial chromosome CYP2C-MAC obtained by translocation cloning 380 kb of the vicinity of the human CYP2C gene cluster region (AL138759-human CYP2C gene cluster-AL158784) to MAC4.
- FIG. 95 shows a two-color FISH analysis result of a CHO (CYP2C-MAC, hChr10- ⁇ CYP2C) clone using CYP2C-BAC (RP11-466J14) (CHORI) DNA and mouse Cot-1 DNA as probes.
- FIG. 95 shows the targeting vector (pMDR1loxPbs), the target sequence and the chromosomal allele resulting from homologous recombination for inserting the loxP sequence into AC005045 of human chromosome 7.
- FIG. 96 shows a targeting vector (pTELpuro-MDR1) for inserting a human telomere sequence into the AC003083 region located very close to the MDR1 locus on human chromosome 7 and on the telomere side (about 50 Kb telomere side), and The partial structure of the human chromosome 7 allele that has undergone homologous recombination is shown.
- FIG. 97 shows the results of two-color FISH analysis of DT40 (hChr7M-loxP-tel) using human cot-1 DNA and puromycin DNA as probes.
- FIG. 98 shows a two-color FISH analysis result of a CHO (HPRT ⁇ ; MAC4, hChr7M-loxP-tel) clone using mouse Cot-1 DNA and human Cot-1 DNA as probes.
- FIG. 99 shows the construction of a mouse artificial chromosome MDR1-MAC obtained by translocation cloning 210 kb around the human MDR1 gene region (AC005045-human MDR1 gene-AC003083).
- FIG. 100 shows a two-color FISH analysis result of a CHO (MDR1-MAC, hChr7- ⁇ MDR1) clone using MDR1-BAC (RP11-784L5) (CHORI) DNA and mouse Cot-1 DNA as probes.
- the natural centromere derived from the mouse chromosome, the long arm fragment derived from the mouse chromosome obtained by deleting the distal long arm from the site of the long arm of the mouse chromosome near the centromere, and the telomere A mouse artificial chromosome vector characterized in that it comprises a sequence and is stably retained in mammalian cells and individual tissues.
- the term “natural centromere derived from a mouse chromosome” refers to the entire centromere (complete centromere) of any one mouse chromosome.
- a centromere does not include a structure having a centromere function that is accidentally or artificially obtained by using a part of a centromere sequence of a mouse chromosome, and a centromere of a chromosome of another animal species.
- a “mouse artificial chromosome” or “mouse artificial chromosome vector” is an artificial chromosome constructed by a top-down approach, not an artificial chromosome constructed by a bottom-up approach.
- the top-down approach is a method of constructing a natural centromere as an artificial chromosome vector by deleting a gene region from a natural chromosome by chromosomal modification.
- the bottom-up approach is a method for constructing an artificial chromosome having a centromere function by obtaining a part of a centromere sequence as cloned DNA and transfecting it into a mammalian cell.
- a long arm fragment derived from a mouse chromosome obtained by deleting the distal long arm from the site of the long arm of the mouse chromosome in the vicinity of the centromere is a vector in which the vector of the present invention is stably maintained in a mouse cell or individual tissue. It is desirable to eliminate the effects of endogenous genes as much as possible so that they do not interfere with mouse ontogeny and offspring transmission, so that the endogenous genes in the long arm of the mouse chromosome are removed.
- DNA in the present specification is used for all types of DNA nucleic acids including genes or loci, cDNAs, and chemically modified DNAs.
- retention ratio refers to the proportion of cells in which artificial chromosomes are present in cultured cells or mouse tissue cells.
- the phrase “stablely maintained” of the chromosomal vector of the present invention means that the chromosomal vector does not easily fall off during cell division, that is, it is stably maintained in the cell even after division. This means that the chromosome vector is efficiently transmitted to daughter cells and offspring mice.
- the mouse chromosome may be any of mouse chromosomes 1 to 19, X and Y, but preferably any of chromosomes 1 to 19.
- chromosome 11 is exemplified, but as long as it has the above-described configuration, it is possible to produce a mouse artificial chromosome vector in the same manner even with other chromosomes.
- the long arm fragment is, for example, without limitation, AL671968 of the long arm of chromosome 11 or BX572640 (located on the centromere side from AL671968).
- CR954170 located on the centromere side from AL671968 and BX572640
- AL713875 located on the centromere side from AL671968
- the long arm fragment may contain, as a basic structure, a mouse artificial chromosome contained in the deposited cell line DT40 B6bT-1 (FERM BP-11128), which is DT40 (MAC) in the present specification (FIG. 1). , 3 and 4).
- the long arm fragment is, for example, a long arm fragment from which a region farther from the position such as AC121307, AC161799 is deleted.
- the long arm fragment is composed of a long arm fragment from which a region farther from the position such as AC127687, AC140982, etc. is deleted.
- These basic structures can further include DNA sequence insertion sites such as loxP for inserting foreign DNA or genes (eg, MAC1, MAC2, MAC3, MAC4, etc .; see FIGS. 1, 3, 4). Since the vector of the present invention can contain a site for inserting foreign DNA or gene sequence, when the vector is introduced into any cell by incorporating the desired foreign DNA or gene into this site. Therefore, it is possible to express the target foreign DNA or gene.
- the vector of the present invention also modifies the mouse chromosome and uses the mouse-derived natural centromere as it is for the production of the vector.
- Conventionally known mouse artificial chromosome vectors include satellite DNA-based mammalian artificial chromosomes (referred to as Aces and SATAC) prepared by using a part of the centromere sequence. A mouse artificial chromosome produced using the above has never been produced.
- the retention rate of the mammalian artificial chromosome is not stable in the tissue of the mouse individual as in the HAC vector (Co Do et al., Chromesome Res. 2000; 8 (3): 83-91).
- Useful and surprising properties of the vectors of the present invention include improved retention in mammalian cells or individual tissues including rodents such as mice, rats, hamsters, etc., thereby being stably retained in the cells and thus the target gene (Group) can be stably maintained for a long period of time, can be expressed for a long period of time without variation in the amount of transgenes between rodent individuals or tissues, and differentiated pluripotent cells (for example, ES cells) , IPS cells, etc.), and can improve the efficiency of rodent ontogeny and offspring transmission.
- rodents such as mice, rats, hamsters, etc.
- HACs human artificial chromosomes
- mouse artificial chromosome or “mouse artificial chromosome vector”
- this term refers to an artificial chromosome having the above-mentioned characteristics prepared from a mouse-derived chromosome fragment as exemplified above, and is top-down. It is an artificial chromosome constructed by an approach and not constructed by a bottom-up approach. To repeat, the top-down approach is a method of constructing a natural centromere as an artificial chromosome vector by deleting a gene region from a natural chromosome by chromosome modification.
- the bottom-up approach is a method of constructing a structure having a centromere function by obtaining a part of a centromere sequence as cloned DNA and transfecting it into a mammalian cell.
- the artificial chromosome can be stably replicated and distributed as a chromosome independent of the original chromosome of the cell to be introduced.
- the mouse-derived chromosomal fragment is a fragment of any chromosome among the 1-19th mouse, X and Y chromosomes (long arm fragment from which at least 99.5% of the total number of endogenous genes in the long arm has been deleted).
- the fragment includes a long arm fragment in which the distal long arm is deleted from the site of the long arm of the mouse chromosome near the centromere, as defined above.
- the production of the artificial chromosome of the present invention is described in Examples described later, particularly Examples 1 to 5 and FIGS. 1 to 4, and a technique for producing an artificial chromosome from mouse chromosome 11 fragment is exemplified. .
- the production of mouse artificial chromosomes from other chromosome fragments can be carried out in exactly the same manner.
- Mouse chromosome sequence information is available from DDBJ / EMBL / GenBank, Santa Cruz Biotechnology, Inc. Available from Chromosome Databases.
- the “long arm” of a chromosome refers to a chromosomal region including a gene region from the centromere side of a mouse chromosome.
- distal means a region far from the centromere (that is, the telomere side).
- proximal means an area located on the centromere side of a specific part of the long arm
- the long arm proximal means an area located on the centromere side of the specific part of the long arm.
- This specific site is at least 99.5%, preferably at least 99.7%, more preferably at least 99.8%, most preferably, of all endogenous genes (number) present in the long arm of one chromosome from mouse. 99.9 to 100% are deleted positions.
- the “retention ratio” in the present specification refers to the proportion of cells in which artificial chromosomes are present in cultured cells or mouse tissue cells.
- the “DNA sequence insertion site” in the present specification means a site in the artificial chromosome where a target DNA (including gene) sequence can be inserted, such as a site-specific recombinase recognition site.
- Such recognition sites include, but are not limited to, for example, loxP (Cre recombinase recognition site), FRT (Flp recombinase recognition site), ⁇ C31attB and ⁇ C31attP ( ⁇ C31 recombinase recognition site), R4attB and R4attP (R4 recombinase recognition site), TP901-1attB and TP901-1attP (TP901-1 recombinase recognition site) or Bxb1attB and Bxb1attP (Bxb1 recombinase recognition site) are included.
- the “site-specific recombination enzyme” in the present specification is an enzyme for causing recombination specifically with a target DNA sequence at a recognition site of these enzymes.
- examples include Cre integrase (also referred to as Cre recombinase), ⁇ C31 integrase, R4 integrase, TP901-1 integrase, and Bxb1 integrase.
- the “telomere sequence” in the present specification is a homologous or heterologous natural telomere sequence or an artificial telomere sequence.
- the same species means an animal of the same species as the mouse from which the chromosome fragment of the artificial chromosome vector is derived, while the heterogeneous means mammals other than the mouse (including humans).
- the artificial telomere sequence refers to a sequence having a telomere function produced artificially, such as a (TTAGGG) n sequence (n means repetition).
- Introduction of a telomere sequence into an artificial chromosome can be performed by telomere truncation (substitution of telomere sequence) as described in, for example, International Publication WO 00/10383. Telomere truncation can be used for chromosome shortening in the production of the artificial chromosome of the present invention.
- foreign gene or “foreign DNA” in the present specification is a gene or DNA of interest that is inserted into a vector and inserted into a vector, and is not inherently present in the target cell. Means a gene or DNA to be expressed in a target cell, or a sequence thereof.
- mamal refers to primates such as humans, monkeys and chimpanzees, rodents such as mice, rats, hamsters and guinea pigs, and ungulates such as cows, pigs, sheep and goats. Including, but not limited to.
- an “embryonic stem cell” or “ES cell” in the present specification is a stem cell having a differentiation pluripotency and a semi-permanent proliferation ability established from an inner cell mass of a blastocyst of a fertilized egg derived from a mammal.
- the artificial chromosome vector of the present invention includes the following steps (a) to (c): (A) obtaining a cell carrying a mouse chromosome; (B) deleting the distal long arm of the mouse chromosome so as not to include the majority (99.5% to 100%) of the endogenous gene (number); and (C) inserting one or more DNA sequence insertion sites proximal to the long arm It can produce by the method containing.
- the order of the steps (b) and (c) may be reversed.
- Mouse A9 hybrid mouse ne; mChr11-BSr
- mouse A9 hybrid cell which carries a mouse fusion with mouse A9 cell (ATCC VA20110-2209) mouse A9 (neo) and retains a mouse chromosome labeled with a drug resistance gene.
- Mouse fibroblasts can be obtained based on methods described in the literature. For example, mouse fibroblasts can be established from C57B6 strain mice available from CLEA Japan. As cells having a high homologous recombination rate, for example, chicken DT40 cells (Dieken et al., Nature Genetics, 12: 174-182, 1996) can be used. Furthermore, the transfer can be performed by a known chromosome transfer method, for example, the micronucleus cell fusion method (Koi et al., Jpn. J. Cancer Res., 80: 413-418, 1973).
- telomere truncation described in WO 00/10383. Specifically, in a cell holding a mouse chromosome, construct a targeting vector holding an artificial telomere sequence, obtain a clone in which the (artificial) telomere sequence is inserted at a desired position on the chromosome by homologous recombination, This results in deletion mutants by telomere truncation.
- the desired position is the cutting position of the distal long arm to be deleted, and the artificial telomere sequence is replaced and inserted at this position by homologous recombination, and the distal long arm is deleted.
- This position can be appropriately set by designing the target sequence when constructing the targeting vector.
- a target sequence is designed based on the DNA sequence of AL671968 (GenBank accession number) of mouse chromosome 11 long arm, and telomere truncation is set to occur on the telomere side of the target sequence. (See FIG. 9). As a result, a mouse chromosome 11 fragment from which most of the endogenous gene has been deleted is obtained.
- telomere truncation can be performed for other chromosomes.
- a site-specific recombination enzyme recognition site can be preferably inserted. That is, a phenomenon in which a certain enzyme recognizes a specific recognition site and specifically causes DNA recombination at the recognition site is known.
- an enzyme and its enzyme By using a system comprising an enzyme recognition site, a target gene or DNA sequence can be inserted and mounted. Examples of such a system include a Cre enzyme derived from bacteriophage P1 and a system of loxP sequence that is a recognition site thereof (Cre / loxP system; B.
- Flp enzyme derived from Saccharomyces cerevisiae and FRT (Flp Recombination Target) sequence system Flp / FRT system
- FRT Flp Recombination Target sequence system
- ⁇ C31 integrase derived from Streptomyces phage and ⁇ C31attB / attP sequence that is the recognition site System
- R4 integrase and its recognition site R4attB / attP sequence system TP901-1 integrase, and its recognition site TP901-1 attB / attP sequence system
- Bxb1 integrase and its recognition unit The Bxb1attB / attP sequence system, which is a position, can be mentioned, but the system is not limited to the above system as long as it can function as a DNA sequence insertion site.
- a known method such as homologous recombination can be used, and the insertion position and number are within the proximal and proximal arm. It can be set appropriately. In the present invention, it is also possible to insert one type of recognition site or one different type of recognition site. By setting the recognition site, the insertion position of the foreign gene or DNA can be specified, so that the insertion position becomes constant, and no unexpected position effect is received.
- the gene inserted in the recognition site loxP sequence of the site-specific recombinase inserted into BX572640 on mouse chromosome 11 is expressed in a tissue-specific manner. (FIGS. 11, 20, and 24).
- a reporter gene may be inserted in advance, preferably leaving the insertion site of the target gene or DNA sequence.
- reporter gene for example, fluorescent protein (For example, green fluorescent protein (GFP or EGFP) gene, yellow fluorescent protein (YFP), etc.), tag protein encoding DNA, (beta) -galactosidase gene, Examples include luciferase gene, and GFP or EGFP is preferable.
- the mouse artificial chromosome vector of the present invention may further contain a selection marker gene.
- the selectable marker is effective in selecting cells transformed with the vector. Examples of the selection marker gene include either a positive selection marker gene and a negative selection marker gene, or both.
- Positive selectable marker genes include drug resistance genes such as neomycin resistance gene, ampicillin resistance gene, blasticidin S (BS) resistance gene, puromycin resistance gene, geneticin (G418) resistance gene, hygromycin resistance gene and the like.
- the negative selection marker gene includes, for example, herpes simplex thymidine kinase (HSV-TK) gene, diphtheria toxin A fragment (DT-A) gene and the like. In general, HSV-TK is used in combination with ganciclovir or cyclovir.
- homologous recombination can be preferably used as a technique for inserting a reporter gene or a desired foreign gene or DNA into the mouse artificial chromosome vector of the present invention.
- Homologous recombination involves both sequences (5′arm and 3 ′) that are homologous to the nucleotide sequences of the 5 ′ region and 3 ′ region of the insertion position on the mouse chromosome (each about 1 to 4 kb, preferably about 2 to 4 kb). arm) can be performed using a targeting vector obtained by ligating the DNA cassette to be inserted.
- a targeting vector obtained by ligating the DNA cassette to be inserted.
- the vector used for this purpose include plasmids, phages, cosmids, viruses and the like, with plasmids being preferred.
- Examples of basic plasmids for constructing the targeting vector include, but are not limited to, V907 or V913 (Lexicon Genetics).
- a basic vector may include one or more sequences or elements that are commonly inserted in vector construction, such as a promoter, enhancer, selectable marker gene, origin of replication, and the like.
- the mouse artificial chromosome produced by the above-described method comprises a mouse-derived chromosome fragment (including a natural centromere, a long arm fragment from which at least 99%, preferably at least 99.5% endogenous genes have been deleted, and Short arms (if present)) and artificial telomere sequences.
- the centromere is the entire centromere structure of the mouse chromosome used for the production of an artificial chromosome.
- mouse artificial chromosome vector of the present invention is a mouse artificial chromosome vector prepared in Examples described later, and this artificial chromosome is a vector obtained by deleting the distal long arm of mouse chromosome 11 at AL671968. (FIGS. 1, 3, 4, 9, and 10).
- This vector contains as a basic structure a mouse artificial chromosome contained in the deposited cell line DT40B6bT-1 (FERM BP-11128), which is DT40 (MAC) herein. Since it is a basic structure, the following DNA sequence insertion site, selection marker gene, foreign gene (or DNA), etc. can be inserted into this DNA structure.
- the mouse artificial chromosome vector preferably includes one or a plurality of DNA sequence insertion sites, for example, a site-specific recombinase recognition site (for example, a loxP sequence which is a Cre enzyme recognition site) (FIGS. 1 and 3). 4, FIG. 11, FIG. 20, FIG. 24).
- the recognition site of the site-specific recombinase is, for example, a GFP-PGKneo-loxP-3′HPRT type loxP sequence, a 5′HPRT-loxP-hyg type, or a PGKneo-loxP.
- GFP is a green fluorescent protein gene
- PGKneo is a phosphoglycerate kinase promoter / neomycin resistance gene cassette
- HPRT is a hypoxanthine-guanine phosphoribosyltransferase gene
- hyg is a hygromycin resistance gene.
- the mouse artificial chromosome vector may further contain a reporter gene and a selection marker gene (eg, a positive selection marker gene and a negative selection marker gene).
- the vector may further contain a target foreign gene or DNA sequence.
- mouse artificial chromosome vector of the present invention are the advantages of the conventional artificial chromosome vector: 1) Since it is not inserted into the host chromosome and is maintained independently, it does not destroy the host gene. 2) Constant copy number (Multiple (multiple) copies are possible) are retained stably and are subject to physiological expression control of the host cell, so there is no overexpression or loss of expression of the inserted gene. 3) There is a restriction on the size of DNA that can be introduced. In addition to being able to introduce genes and multiple genes / isoforms containing expression control regions, the retention rate in rodent cells or rodents is improved compared to conventional artificial chromosomes.
- Tissue after vector introduction which realizes stable expression of the transgene for a long period of time and improves the offspring transmission rate, thereby improving the production efficiency of the transgenic mouse.
- the retention rate is 90% or higher in any tissue, and even in the case of HAC, the retention rate is 90% or higher even for blood-related tissues that are less than 20%.
- the size of the foreign gene or DNA sequence is not particularly limited and may be 20 kb or less, or may exceed 20 kb, for example, 50 kb or more, 100 kb or more, 200 kb or more, 500 kb or more, 700 kb or more, 1 Mb or more, It is 10 Mb or more, 20 Mb or more, 30 Mb or more, 40 Mb or more, or 50 Mb or more.
- the vector of the present invention can be loaded with foreign DNA (chromosomal fragment) having a size that is difficult for artificial chromosome vectors such as BAC, PAC, and YAC, that is, 1 Mb or more.
- the vector of the present invention can be used in mammalian cells, tissues, or a large foreign gene or DNA of 200 kb or more, such as 1 Mb or more, more stably and at a higher retention rate (90% or more) than HAC. It is possible to keep in a non-human animal individual, preferably in a rodent cell, tissue or individual. As one embodiment of the present invention, the present invention can stably maintain a foreign gene or DNA having a huge size of 200 kb or more in a rodent cell or rodent with a retention rate of 90% or more. And a method for producing the vector.
- the foreign gene or DNA is a nucleic acid sequence introduced from the outside into the target cell, and is not particularly limited, and is preferably a gene or DNA derived from any biological species or from any tissue or cell, Is a gene or DNA derived from a mammal, more preferably a gene or DNA derived from a human.
- genes or DNA include genes encoding polypeptides such as cytokines, hormones, growth factors, nutrient factors, hematopoietic factors, immunoglobulins, G protein-coupled receptors, enzymes, etc.
- DNA DNA
- other tumors muscular dystrophy, hemophilia
- neurodegenerative diseases eg Alzheimer's disease, Huntington's disease, Parkinson's disease, etc.
- autoimmune diseases allergic diseases, genetic diseases, etc.
- therapeutic genes or DNA include therapeutic genes or DNA, (human) drug-metabolizing enzyme gene (s) or DNA, (human) drug metabolism-related genes, human chromosome long or short arm DNA, (human) genomic libraries, etc.
- s drug-metabolizing enzyme gene
- human chromosome long or short arm DNA human chromosome long or short arm DNA
- human genomic libraries etc.
- Cytokines include, for example, interferons (eg, IF- ⁇ , IF- ⁇ , IF- ⁇ , etc.), interleukins (eg, IL-1, IL-2, IL-4, IL-6, IL-11, IL-12 etc.), tumor necrosis factor (eg TNF- ⁇ , TNF- ⁇ ), TGF- ⁇ family protein (eg osteogenesis promoting protein (BMP), etc.) and the like.
- interferons eg, IF- ⁇ , IF- ⁇ , IF- ⁇ , etc.
- interleukins eg, IL-1, IL-2, IL-4, IL-6, IL-11, IL-12 etc.
- tumor necrosis factor eg TNF- ⁇ , TNF- ⁇
- TGF- ⁇ family protein eg osteogenesis promoting protein (BMP), etc.
- Hormones include, for example, growth hormone, human chorionic gonadotropin (hCG), human placental lactogen (hPL), human pituitary gonadotropin, thyroid stimulating hormone (TSH), luteinizing hormone releasing factor, insulin, Glucagon, somatostatin, prolactin and the like are included.
- growth factors or nutrient factors include insulin-like growth factor, brain-derived neurotrophic factor (BDNF), albumin-fused villous-like neurotrophic factor, platelet-derived neurotrophic factor (PDNF), transforming growth factor, nerve growth Factor (NGF), TNF growth factor and the like are included.
- blood coagulation / lysis factors include Factor VII, Factor VIII, Factor X, t-PA, and the like.
- Hematopoietic factors include, for example, erythropoietin, (granulocyte) colony-stimulating factor, thrombopoietin and the like.
- Immunoglobulins include, for example, human antibodies against various antigens, humanized antibodies, chimeric antibodies, recombinant antibodies such as synthetic antibodies, and the like.
- G protein-coupled receptors include adrenergic receptors, muscarinic acetylcholine receptors, adenosine receptors, GABA receptors (type B), angiotensin receptors, cholecystokinin receptors, dopamine receptors, glucagon receptors, Histamine receptors, olfactory receptors, opioid receptors, secretin receptors, somatostatin receptors, gastrin receptors, P2Y receptors and the like are included.
- Enzymes include, for example, asparaginase, superoxide dismutase, uricase, streptokinase, dopamine synthase, adenosine deaminase and the like.
- Examples of therapeutic genes related to various diseases including tumors, muscular dystrophy, neurodegenerative diseases (for example, Alzheimer's disease, Huntington's disease, Parkinson's disease, etc.), autoimmune diseases, allergic diseases, genetic diseases, etc. , IL-12 gene, TNF- ⁇ gene, tumor suppressor gene, dopamine synthesizing enzyme gene, hereditary defective enzyme gene, and the like.
- Drug metabolizing enzymes are enzymes involved in metabolic reactions for decomposing and discharging foreign substances such as drugs and poisons, and are used for enzymes involved in first-phase reactions (oxidation, reduction, hydrolysis) and second-phase reactions (conjugation). Contains the enzymes involved.
- Examples of the enzyme involved in the first phase reaction include known enzymes such as cytochrome P450 (“CYP”), specifically CYP1A, CYP1B, CYP2A, CYP2B, CYP2C, CYP2D, CYP2E, CYP2J, CYP3A, CYP4A, and CYP4B.
- CYP cytochrome P450
- CYP3A subfamily includes CYP3A4, CYP3A43, CYP3A5, CYP3A7 and the like
- CYP2C subfamily includes CYP2C8, CYP2C9, CYP2C18, CYP2C19 and the like
- CYP3A-MAC described in the examples described later means a CYP3A cluster, and consists of CYP3A4, CYP3A43, CYP3A5, and CYP3A7.
- enzymes involved in the second phase reaction include, for example, UGT1 and UGT2.
- Examples of the drug metabolism-related gene include a gene encoding a transporter and a gene encoding a nuclear receptor.
- Examples of genes encoding transporters are MDR1, MDR2, MRP2, OAT, OATP, OCT, BCRP and the like, and examples of genes encoding nuclear receptors are PXR, AhR, CAR, PPAR ⁇ and the like.
- the foreign DNA sequence related to drug metabolism that can be introduced into the vector of the present invention includes a gene encoding an enzyme related to the first phase reaction, a gene encoding an enzyme related to the second phase, and a transporter.
- at least one insulator sequence can be present in the vicinity of the foreign gene or foreign DNA insertion site or on both sides of the insertion site.
- the insulator sequence has an enhancer blocking effect (that is, adjacent genes are not affected by each other) or a chromosomal boundary effect (distinguish between a region that guarantees gene expression and a region where gene expression is suppressed).
- Such sequences include, for example, human ⁇ globin HS1 to HS5, chicken ⁇ globin HS4, and the like.
- the introduction of a foreign gene or DNA can be carried out by utilizing the site-specific recombinase system as exemplified above, which is inserted as the above DNA sequence insertion site.
- a targeting vector that holds a loxP sequence that is a recognition site of Cre enzyme and a foreign gene or DNA, or a chromosome fragment that holds a foreign gene or DNA into which a loxP sequence that is a recognition site of Cre enzyme is inserted is constructed.
- Cre enzyme By expressing the Cre enzyme in a cell carrying the mouse artificial chromosome vector, a foreign gene or DNA can be introduced by site-specific recombination with the targeting vector or the chromosome fragment in the loxP sequence.
- a circular DNA having a recognition site for a site-specific recombinase (eg, loxP sequence, FRT sequence, etc.) can be inserted.
- DNA cloned by an existing vector such as circular YAC as a host can also be inserted.
- the preferred loxP sequence is a wild sequence derived from the P1 phage, and the insertion reaction of the circular insert into the loxP sequence on the artificial chromosome vector by Cre enzyme is reversible. Once the circular insert is inserted, two loxP sequences remain on the artificial chromosome vector.
- micronucleus cell fusion method comprises micronucleus fusion between a cell having the ability to form a micronucleus (for example, mouse A9 cell) containing the mouse artificial chromosome vector of the present invention and other desired cells, and then transferring the vector to the other cell. It is a method to transfer to.
- a micronucleus for example, mouse A9 cell
- the cell into which the vector can be introduced is an animal cell, preferably a mammalian cell including a human cell, such as an oocyte, a germ line cell such as a sperm cell, an embryonic stem (ES) cell, a sperm stem (GS) cell, It includes stem cells such as somatic stem cells, somatic cells, fetal cells, adult cells, normal cells, disease cells, primary cultured cells, passaged cells or established cells.
- a polyploid inducer for example, colcemid, colchicine, etc.
- Stem cells include, for example, ES cells, embryonic germ (EG) cells, embryonal carcinoma (EC) cells, mGS cells, pluripotent stem cells such as human mesenchymal stem cells, induced pluripotent stem (iPS) cells, nuclei Examples include transplanted cloned embryo-derived embryonic stem (ntES) cells.
- Preferred cells are selected from the group consisting of somatic cells from mammals (preferably rodents including mice), non-human germline cells, stem cells and progenitor cells.
- the vector When the cell is a cell derived from a mammal such as a rodent, the vector is more stably maintained in the cell or tissue of a mammal (for example, a rodent such as a mouse) into which the vector of the present invention has been introduced.
- the dropout of the vector from is significantly reduced or no dropout occurs.
- the cells are, for example, hepatocytes, intestinal cells, kidney cells, spleen cells, lung cells, heart cells, skeletal muscle cells, brain cells, bone marrow cells, lymphocyte cells, megakaryocyte cells, sperm, eggs and the like.
- the tissue is a tissue such as liver, intestine, kidney, spleen, lung, heart, skeletal muscle, brain, bone marrow, testis, ovary, etc.
- ES cells can be established and maintained by removing the inner cell mass from the blastocyst of the fertilized egg of the subject animal and using mitomycin C-treated mouse fetal fibroblasts as a feeder (MJ Evans and MH). Kaufman (1981) Nature 292: 154-156). iPS cells can be obtained in about 3 to 5 weeks by introducing a specific reprogramming factor (DNA or protein) into somatic cells (including somatic stem cells), culturing them in an appropriate medium, and subculturing them. Generate colonies.
- DNA or protein a specific reprogramming factor
- the reprogramming factor is, for example, a combination consisting of Oct3 / 4, Sox2, Klf4 and c-Myc; a combination consisting of Oct3 / 4, Sox2 and Klf4; a combination consisting of Oct4, Sox2, Nanog and Lin28; or Oct3 / 4,
- a combination comprising Sox2, Klf4, c-Myc, Nanog and Lin28 is known (K. Takahashi and S. Yamanaka, Cell 126: 663-676 (2006); WO 2007/069666; M. Nakagawa et al.
- a mouse embryo fibroblast cell line for example, STO
- a vector-transfected somatic cell about 10 cells
- ES cell medium 4 ⁇ 10 5 Cells / cm 2
- the basic medium is, for example, Dulbecco's modified Eagle medium (DMEM), Ham F-12 medium, a mixed medium thereof, and the ES cell medium is a mouse ES cell medium, a primate ES cell medium (Reprocell), or the like. Can be used. Since ES cells and iPS cells are known to contribute to the germline, these cells into which the mouse artificial chromosome vector of the present invention containing the gene or DNA of interest is introduced are the same type of mammals from which the cells are derived.
- DMEM Dulbecco's modified Eagle medium
- Reprocell primate ES cell medium
- Non-human animals can be created by techniques including injection into blastocysts of animal embryos, transplanting the embryos into the womb of a surrogate parent and giving birth . Furthermore, homozygous animals and their progeny animals can be produced by mating the resulting male and female transgenic animals.
- the pluripotent cells such as ES cells and iPS cells via the mouse artificial chromosome vector of the present invention, and other cells mentioned above, foreign genes or DNA such as human antibody genes, disease treatment genes, drug metabolism related genes, etc.
- Disease model non-human animals such as related diseases can be produced.
- the endogenous gene corresponding to the foreign gene contained in the mouse artificial chromosome vector is destroyed or the expression of the endogenous gene is reduced.
- the gene targeting method can be used as the destruction method.
- the RNAi method can be used as a method for reducing the expression of an endogenous gene.
- foreign genes include drug metabolism-related genes and human antibody genes.
- a cell and a transgenic non-human animal holding a mouse artificial chromosome vector can be produced.
- a specific example of a non-human animal is a rodent such as a mouse or a rat carrying a mouse artificial chromosome vector. That is, the present invention provides a cell and a non-human animal characterized by holding a mouse artificial chromosome vector. Furthermore, the cells, tissues or organs obtained from the non-human animals of the present invention can be used to produce cell lines that produce proteins expressed by foreign genes.
- the present invention further provides a method for producing a protein, comprising culturing a cell holding a mouse artificial chromosome vector containing an exogenous DNA sequence in an expressible manner and recovering the protein encoded by the produced DNA.
- the protein include proteins and polypeptides useful in the above-mentioned medical and agricultural industries. DNA encoding these proteins or polypeptides is inserted into a mouse artificial chromosome vector so that it can be expressed in the presence of a promoter (and enhancer, if necessary), and appropriate cells are transformed or transfected. To do. The resulting cells are cultured and the DNA is expressed to produce the protein or polypeptide, which is recovered from the cells or medium.
- insect cells such as Sf cells, avian cells, yeast cells, and eukaryotic cells such as plant cells can be used as the cells.
- the culture conditions including the medium are selected according to the cell type, and known conditions can be used as the culture conditions.
- the medium for animal cells includes MEM medium, DMEM medium, Ham's F12 medium, Eagle's MEM medium, Iskov EME medium, RPMI 1640 medium, and mixed media thereof.
- the recovery (or isolation) of proteins or polypeptides is gel filtration chromatography, ion exchange chromatography, affinity chromatography, HPLC, chromatography methods such as FPLC, salting-out method, ammonium sulfate precipitation method, organic solvent precipitation Known methods such as a method, an ultrafiltration method, and crystallization may be carried out singly or in combination.
- Manufacturing method of human antibody The present invention further provides a method for producing a human antibody, which comprises producing a human antibody using the above-mentioned non-human animal carrying a mouse artificial chromosome vector containing a human antibody gene and recovering the human antibody.
- the human antibody gene is a gene encoding any class of human IgG, IgM, IgA, IgD, and IgE, or any subclass of human IgG1, IgG2, IgG3, IgG4, IgA1, and IgA2.
- Preferred human antibody genes are the IgG class and its subclasses.
- a human antibody is composed of two identical heavy chains (H) and two identical light chains (L). Both H and L chains are composed of a variable region and a constant region. Yes.
- the human H chain and L chain variable regions are composed of three hypervariable regions (in order of CDR1, CDR2, CDR3 from the N-terminal side to the C-terminal side) and four framework regions (FR1 from the N-terminal side to the C-terminal side). , FR2, FR3, FR4), and the specificity of the antibody is determined by three CDR sequences each of human H chain and L chain.
- FR2, FR3, FR4 the specificity of the antibody is determined by three CDR sequences each of human H chain and L chain.
- a human IgG antibody it is composed of a heavy chain ⁇ chain and a light chain ⁇ chain or ⁇ chain, and these antibody chain genes are human chromosome 14, chromosome 22, chromosome 2, respectively. Exists on.
- human antibody genes used in the present invention human chromosome fragments containing each antibody locus are used, and these are integrated into different or identical mouse artificial chromosomes.
- Antibody gene sequences can be obtained from NCBI (USA) databases and the like. This series of techniques is an improvement of the technique described in, for example, JP-A-2005-230020.
- a non-human animal capable of producing a fully human antibody includes a non-human animal holding a mouse artificial chromosome vector containing a human ⁇ chain locus, and a mouse artificial chromosome vector containing a human ⁇ chain locus and / or a human ⁇ chain locus It is possible to obtain chimeric non-human animals and their progeny animals that possess both the H chain and L chain loci by mating with the same kind of non-human animals carrying A non-human animal (such as a rodent such as a mouse) capable of producing a fully human antibody thus immunized with a specific antigen peptide or antigen polypeptide, and the human antibody is separated from the blood of the animal Human antibodies can be produced by a method comprising Alternatively, a spleen of a non-human animal immunized with a specific antigen can be removed and fused with myeloma cells to obtain a hybridoma that produces a monoclonal antibody.
- the present invention further provides a method for screening a substance effective for treating the disease, comprising administering a candidate drug to the non-human animal, which is a disease model animal, and evaluating the therapeutic effect of the drug.
- a disease model non-human animal is an artificially produced animal having a disease caused by an abnormality such as a biological function abnormality, a drug metabolism abnormality, a chromosomal abnormality or the like due to a defect or mutation of a certain protein.
- model non-human animals having chromosomal abnormalities include, but are not limited to, animals with human chromosome 18 or 21 trisomy.
- Such a non-human animal prepares a gene or chromosome fragment containing the above-mentioned abnormality in the gene or chromosome, incorporates it into the mouse artificial chromosome vector of the present invention, introduces it into an ES cell or iPS cell, and fertilizes an egg.
- a substance effective for treating the disease can be screened by administering a candidate drug to the non-human animal produced as described above and evaluating the therapeutic effect of the drug.
- Candidate agents include, but are not limited to, for example, low molecular weight compounds, high molecular weight compounds, (sugar) proteins, peptides, (phosphorus or sugar) lipids, saccharides, and the like. (7) Methods for testing pharmacological action, metabolism or toxicity of drugs or foods according to an embodiment of the present invention, the present invention also administers a drug or food to the non-human animal or a cell, organ or tissue derived from the above-mentioned non-human animal carrying a mouse artificial chromosome vector containing a human drug metabolism-related gene.
- a method for testing the pharmacological action and / or metabolism and / or toxicity of a drug or food comprising measuring the pharmacological action and / or metabolism and / or toxicity of the drug or food.
- the present invention also provides a method for culturing a microsome or microsomal fraction S9 obtained from the above non-human animal carrying a mouse artificial chromosome vector containing a human drug metabolism-related gene, together with cultured cells or bacteria and a drug or / and food, Alternatively, the present invention provides a method for testing the toxicity of a drug or food, which comprises measuring the (bad) effect (eg, mutation, etc.) that the food has on the cells or bacteria.
- Human drug metabolism related genes are those exemplified above.
- the method for producing a non-human animal is also the same as described above.
- the pharmacology of the drug or food can be determined by, for example, observing the state of the animal or testing the effect on the organ or chromosome. Action, metabolism or toxicity can be determined.
- the microsome or microsomal fraction S9 obtained from the non-human animal (9000 g fraction containing a large number of enzymes that catalyze hydrolysis, reduction, oxidation, binding, etc.) is obtained.
- Detection of the toxicity of a drug or food to cells can be performed by an Ames test or a micronucleus test. In the Ames test, toxicity is determined based on Salmonella mutations. In micronucleus tests, toxicity is determined based on abnormalities in the chromosomes of cell nuclei. These test methods are well known and can be used in the methods of the present invention. EXAMPLES Hereinafter, although an Example is given and this invention is demonstrated in more detail, the scope of the present invention is not limited to these specific examples.
- mouse artificial chromosome vector MAC A mouse artificial chromosome MAC [DT40 (B6bT)] that does not contain an endogenous gene is constructed by performing telomere truncation on the mouse chromosome (FIG. 1).
- a mouse artificial chromosome MAC [DT40 (B6bT)] that does not contain an endogenous gene is constructed by performing telomere truncation on the mouse chromosome (FIG. 1).
- a mouse artificial chromosome MAC [DT40 (B6bT)] that does not contain an endogenous gene is constructed by performing telomere truncation on the mouse chromosome (FIG. 1).
- mChr11-BSr Mouse embryonic fibroblast (mChr11-BSr), which is a mouse fibroblast containing mouse chromosome 11 labeled with a drug resistance gene (Bsr gene), and a mouse in which a neo gene, which is a G418 resistance gene
- a mouse A9 xmouse embryonic hybrid (neo; mChr11-BSr), which is a mouse A9 hybrid cell that carries a cell fusion with A9 (neo) and retains a mouse chromosome labeled with a drug resistance gene, is established.
- drug resistance genes are introduced into mouse A9 cells, which are known to have a high micronucleus formation rate.
- the mouse chromosome labeled with is introduced by cell fusion.
- Mouse embryonic fibroblast (mChr11-BSr), which is a mouse fibroblast in which a drug resistance gene (Bsr gene) is inserted on the mouse chromosome, and a G418 resistance gene.
- Mouse A9 (neo), which is a mouse A9 cell into which the neo gene has been inserted, is washed with PBS ( ⁇ ), and the cells are dispersed by adding trypsin and suspended in a culture solution (10% FBS, DMEM). , Each cell 1x10 6 Individual flasks for culture (25 cm 2 ) At the same time and cultured for one day.
- the PEG solution was aspirated, washed 3 times with serum-free DMEM, and cultured in a normal culture solution (10% FBS, DMEM) for 1 day. After washing the cell surface with PBS ( ⁇ ), the cells were dispersed by adding trypsin and placed in a double selective culture solution (10% FBS, DMEM) containing G418 (800 ⁇ g / ml) and blasticidin S (4 ⁇ g / ml). The suspended cells were implanted in a plastic culture dish and selectively cultured for 2 to 3 weeks.
- PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle conditions were 95 ° C for 10 minutes after heat denaturation and 35 cycles of 94 ° C for 30 seconds, 60 ° C for 30 seconds and 72 ° C for 30 seconds.
- 3 out of 3 clones were positive.
- [A. 2.2] quinacrine and Hoechst double staining For clones that were positive by the PCR analysis, double quinacrine-Hoechst staining was performed.
- a chromosome slide was immersed in 50 ml of a macilben solution [11.18 g of citric acid monohydrate and 13.29 g of disodium hydrogen phosphate were dissolved in 1 L of water and autoclaved] Then, immerse in quinacrine [cat: Q2876, SIGMA] dissolved in 50 ml of makilben solution at 60 ⁇ g / ml for 20 minutes, wash the back of the chromosome slide with tap water, and then soak in makilben solution.
- quinacrine cat: Q2876, SIGMA
- mouse A9xmouse embryonic fibroblast hybrid (neo; mChr11-BSr) retains a labeled mouse chromosome.
- telomere truncation which is chromosome site-specific cleavage by insertion of artificial telomere
- loxP sequence which is DNA sequence insertion site into mouse chromosome by homologous recombination
- DT40 which is a chicken DT40 cell having a high homologous recombination frequency
- mice chromosome was transferred from A9 hybrid embryonic hybrid hybrid (neo; mChr11-BSr) 7 which is an A9 hybrid cell clone to DT40 which is a chicken DT40 cell having high homologous recombination frequency. .
- the flask was inserted into a container for exclusive use of a large high-speed centrifuge (BECKMAN), hot water (34 ° C.) was added to such an extent that the flask was not hidden, and centrifuged (Rotor ID 10.500, 8,000 rpm, 1 h, 34 ° C.). After the centrifugation, cytochalasin B was recovered, and the pellets in each flask were recovered in 15 ml tubes with 2 ml of serum-free medium DMEM.
- BECKMAN high-speed centrifuge
- each tube was centrifuged (1,200 rpm, 5 minutes RT), the supernatant was aspirated, and the pellets in each tube were combined into 5 ml serum-free The medium was collected and suspended in DMEM, and centrifuged (2000 rpm for 5 minutes). Since DT40 cells, which are recipient cells, are floating cells, they need to be once attached. Coated by incubating 1 hole at 37 ° C overnight with 1.5 ml of poly-L-lysine (SIGMA) adjusted to 50 ⁇ g / ml to attach DT40 to 1 hole of 6-well plate (Nunc) did.
- SIGMA poly-L-lysine
- Poly-L-lysine is recovered and the plate is washed with PBS ( ⁇ ), approximately 1 ⁇ 10 7 Individual DT40 cells were gently seeded on a plate with 2 ml of serum-free medium (DMEM). The whole plate was set in a centrifuge (Beckman), and centrifuged at 37 ° C., 1200 rpm for 3 minutes to obtain attached DT40. The purified micronucleated cells were resuspended in 2 ml of serum-free medium containing PHA-P (SIGMA) and gently seeded on the adherent DT40 from which the serum-free medium (DMEM) was removed. The plate was centrifuged at 37 ° C., 1200 rpm, 3 minutes.
- SIGMA PHA-P
- PEG1000 (Wako) [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 1 ml for exactly 1 minute.
- Serum-free culture medium (DMEM) is washed 4 times with 4 ml, pipetted with 3 ml of normal DT40 culture medium, adhering DT40 is returned to a floating state, and seeded on two 24-well plates at 37 ° C. overnight. Incubated with.
- Blasticidin S was added to 1500 ⁇ g / ml and selective culture was performed for 3 to 4 weeks.
- telomere truncation vector Site-specific cleavage by telomere truncation from mouse chromosome 11 region AL671968 in chicken DT40 cells Less endogenous genes other than the target gene to be introduced as a mouse artificial chromosome vector, the effect on the experimental system is reduced, and among the endogenous genes, genes that affect the ontogenesis of mice due to changes in gene expression, such as imprint genes Delete most of the mouse's long arm.
- C. 1 Construction of telomere truncation vector The pBS-TEL / puro construct (Kuroiwa et al. Nature Biotech 2002) was used as the basic vector for short-arm proximal site-specific cleavage.
- a homologous recombination target sequence was designed from the base sequence (AL671968) of the long arm of mouse chromosome 11 obtained from the GenBank database.
- the sequence of a primer for PCR amplification of a homologous recombination target sequence is shown below by extracting genomic DNA from DT40 (mChr11-BSr) as a template.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- DT40 (MAC) -1 which is a chicken DT40 cell carrying the mouse artificial chromosome vector MAC, has an identification name of DT40 B6bT-1 and is a patent biological deposit center of the National Institute of Advanced Industrial Science and Technology (1st East, Tsukuba City, Ibaraki, Japan). No. 1 No. 1 (Chuo No. 6) was deposited internationally based on the provisions of the Budapest Treaty on May 14, 2009 and was given the deposit number FERM BP-11128.
- mouse artificial chromosome vector MAC1 was constructed by inserting a GFP-PGKneo-loxP-3′HPRT type loxP sequence into the mouse artificial chromosome MAC as a DNA insertion sequence, and the stability of MAC1 in mouse ES cells was verified. The stability in an individual tissue is verified by producing a descendant transmission mouse into which MAC1 is introduced.
- Insertion of GFP-PGKneo-loxP-3′HPRT type loxP sequence into mouse artificial chromosome vector MAC [A.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 10 seconds and 68 ° C for 5 minutes.
- Each PCR product was digested with BglII (TAKARA), separated and purified by agarose gel, and then cloned into the V913 BglII or BamHI site (vector name: VH21-12).
- 3′HPRT-loxP the loxP sequence oligo-synthesized at the XbaI site of V820 (Lexicon genetics) was cloned.
- 3′HPRT-loxP which is the third to ninth exons of the HPRT gene, was cloned into EcoRI and AscI of V907 (Lexicon genetics) (vector name: X3.1).
- the PGKneo sequence excised with KpnI and NotI was cloned into the KpnI site and EcoRI site of X3.1 (vector name: X4.1).
- HS4-CAG-EGFP-HS4 (distributed by Dr. Okabe and NIH, Dr. Felsenfeld) smoothed after NotI and SalI digestion was cloned into the EcoRV site of pVNLH (vector name: p VGNLH).
- FIG. 11 shows a targeting vector, a target sequence, and a chromosomal allele resulting from homologous recombination.
- DT40 (MAC) -1 7 Cells were washed once with no-addition RPMI1640 medium, suspended in 0.5 ml of no-addition RPMI1640 medium, and 25 ⁇ g of the targeting vector pMAC1 linearized with restriction enzyme NotI (TAKARA) was added, and a cuvette for electroporation was added. (Bio-Rad) and allowed to stand at room temperature for 10 minutes. The cuvette was set on Gene Pulser (Bio-Rad), and voltage was applied under the conditions of 550 V and 25 ⁇ F. After standing at room temperature for 10 minutes, the cells were cultured for 24 hours.
- TAKARA restriction enzyme NotI
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 10 seconds and 68 ° C for 7 minutes were performed.
- the subsequent analysis was performed on these 2 clones.
- mouse artificial chromosome vector MAC1-containing DT40 cells In order to introduce the mouse artificial chromosome vector MAC1 into mouse ES cells via CHO cells, or the target gene (group) and the like stably via loxP which is the DNA sequence insertion site of mouse artificial chromosome vector MAC1 in CHO cells, for example In order to insert a CYP3A cluster or the like, it is introduced into CHO cells. [B.
- PCR analysis In order to extract the genomic DNA of the G418 resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether the mouse artificial chromosome MAC1 could be introduced into CHO cells.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- mouse artificial chromosome vector MAC1 could be introduced into CHO cells.
- mouse artificial chromosome vector MAC1 is introduced into mouse ES cells to produce mouse artificial chromosome vector MAC1-containing chimeric mice and offspring transmitting mice. .
- the microcell was filled into a centrifuge flask, followed by centrifugation at 34 ° C., 8000 rpm for 1 hour.
- the microcell was suspended in serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes and suspended in 5 ml of serum-free DMEM medium.
- the microcell was suspended in 5 ml of serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes.
- Donor cells include B6-ES, which is an ES cell of a C57B6 strain mouse, and B6 (HPRT, which is an HPRT-deficient strain obtained by 6TG treatment of B6-ES cells. ⁇ ), And C57B6xCBA strain F1 mouse ES cells TT2F, and TT2F cells treated with 6TG, KO56 (HPRT ⁇ ) Was used.
- DMEM Dulbecco's Modified Eagle's Medium-high glucose: SIGMA
- 10% FCS 10% FCS
- LIF Meerin Leukemia Inhibitory Factor
- L-glutamine 3.5 g / ml: GIBCO
- sodium pyruvate solution 3.5 g / ml: GIBCO
- MEM Nonesential amino acid (0.125 mM: GIBCO)
- the culture was performed at 37 ° C.
- Mouse ES cells were washed twice with PBS ( ⁇ ), and the cells were dispersed by trypsin treatment.
- the cells were collected with a culture solution obtained by adding 10% FBS to DMEM medium, centrifuged at 1500 rpm, and the supernatant was removed.
- the suspension was resuspended in 5 ml of serum-free culture, gently added to serum-free medium containing the pellet after microcell centrifugation, and centrifuged at 1200 rpm.
- the supernatant was removed, and PEG1000 (Wako) solution [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 0.5 ml for exactly 1 minute and 30 seconds. .
- MAC1 B6-ES
- B6 HPRT ⁇ MAC1
- KO56 HPRT
- MAC1 a total of 32 resistant colonies obtained by two micronucleus cell fusions were isolated and expanded for subsequent analysis.
- TT2F a total of 30 resistant colonies obtained by four micronucleus cell fusions were isolated and expanded, and analysis after FISH analysis was performed.
- C. 2 Selection of drug resistant clones
- C. 2.1 PCR analysis In order to extract the genomic DNA of the G418 resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether or not the mouse artificial chromosome MAC1 could be introduced into mouse ES cells. The primer sequences are shown below.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation, followed by 30 cycles of 98 ° C for 10 seconds and 68 ° C for 10 minutes.
- 30 clones out of 32 clones were positive in all of the primer sets, and 14 clones were subjected to subsequent analysis. [C.
- mouse artificial chromosome vector MAC1 could be introduced into mouse ES cells (FIG. 14).
- FIG. 14 Construction of mouse artificial chromosome vector CYP3A-MAC
- the CYP3A cluster which is a group of human drug metabolizing enzyme genes, is translocated to the mouse artificial chromosome vector MAC1 using the Cre / loxP system to construct CYP3A-MAC.
- targeting vector pMPloxPHyg for inserting the recognition sequence loxP of Cre recombinase into the AC004922 region located very close to the CYP3A locus on human chromosome 7 and on the centromere side (about 300 Kb centromere side) is prepared as follows. did. First, the AC004922 genomic region was amplified by PCR using the following primers. V901 (Lexicon genetics) was used as a basic plasmid for inserting the loxP sequence.
- PCR Gene Amp 9600 manufactured by Perkin-Elmer is used as a thermal cycler, LATaq (Takara Shuzo) is used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 20 seconds and 68 ° C for 7 minutes. The PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech).
- V901-NP21 was cleaved with restriction enzymes AscI (NEB) and KpnI, and restriction enzymes AscI and KpnI were obtained from the cassette vector 5′HPRT-loxP-Hyg-TK (Kazuki et al., Gene Therapy: PMID: 21085194, 2010).
- the DNA fragment containing loxP was excised and ligated.
- a vector having the loxP sequence in the same direction as the cloned AC004922 genomic fragment was designated as a targeting vector pMPloxPHyg.
- the size of the final loxP insertion construct is 12 kb.
- the targeting vector, the target sequence and the chromosomal allele resulting from homologous recombination are shown in FIG.
- PCR uses Perkin-Elmer's Gene Amp 9600 as thermal cycler, Taq polymerase uses LATaq (TAKARA), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 10 seconds and 68 ° C for 4 minutes. As a result of screening 96 clones, 36 clones were identified as homologous recombinants. [A. 3.2] Southern blot analysis Southern blot analysis was performed on the 6 clones confirmed to be recombined by PCR analysis as follows.
- This genomic DNA was treated with restriction enzyme EcoRI (TAKARA), electrophoresed in a 0.8% agarose gel, and alkaline blotted onto a GeneScreen Plus TM hybridization transfer membrane (NENTM Life Science Products, Inc.).
- EcoRI restriction enzyme
- This filter was subjected to Southern hybridization using an MPp probe obtained by amplifying the gene sequence in AC004922 by PCR to identify a homologous recombinant.
- the MPp probe is prepared by PCR using the following primers and DF141 genomic DNA as a template, and using the PCR product as a template and random priming. 32 A P-labeled DNA probe was made (Amersham, following the attached protocol).
- Primers for MPp probe preparation For PCR, GeneAmp 9600 manufactured by Perkin-Elmer was used as a thermal cycler, EX Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. The temperature and cycle conditions were 35 cycles of heat denaturation at 93 ° C. for 5 minutes, 93 ° C. for 1 minute, 54 ° C. for 1 minute, and 72 ° C. for 1 minute. Southern hybridization predicted that a band of about 10.9 kb was detected for the non-homologous recombinant and about 8.9 kb for the homologous recombinant (FIG.
- targeting vector pTELhisD-PT for inserting a human telomere sequence into the AC073842 region located very close to the CYP3A locus on human chromosome 7 and on the telomere side (about 150 Kb telomere side) was prepared as follows. First, the AC073842 genomic region was amplified by PCR using the following primers. For PCR, Gene Amp 9600 manufactured by Perkin-Elmer was used as a thermal cycler, LATaq (TAKARA) was used as Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 20 seconds and 68 ° C for 8 minutes were performed.
- the PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech). Then, it cleaved with restriction enzymes BamHI (Boehringer) and BglII (Nippon Gene), and gel-filtered with CHROMASPIN-TE 1000 (Clontech). This PCR fragment was cloned into the BamHI site of plasmid pTELhisD (Kuroiwa et al., Nature Biotech., 20:88, 2002).
- the cloned AC073842 genomic fragment having the same direction as the human telomere sequence was designated as the target targeting vector pTELhisD-PT.
- the final long arm proximal site specific cleavage construct size is 14.4 kb.
- the targeting vector, target sequence, and chromosomal allele resulting from homologous recombination are shown in FIG. [B.
- PCR analysis In order to select recombinants using the genomic DNA of a histidinol-resistant strain as a template, PCR was performed as a primary screening using the following primers located on the telomere side of the cleavage site to confirm whether site-specific cleavage occurred. The primer sequences are shown below. PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle conditions were 95 ° C for 10 minutes after heat denaturation, followed by 30 cycles of 95 ° C for 20 seconds, 55 ° C for 30 seconds, and 72 ° C for 30 seconds.
- 2 out of 433 clones were positive.
- the sequence is as follows. PCR was performed using LATaq (Takara Shuzo) with the above primers, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions.
- hChr7-loxP-tel is introduced into the mouse artificial chromosome vector MAC1-containing CHO cell.
- C. 1 Micronucleus cell fusion and isolation of drug resistant clones Using recipient cells DT40 (hChr7-loxP-tel) 608 and 748, MAC1-containing CHO hprt-deficient cells (obtained from Human Science Research Resource Bank, registration number JCRB0218), CHO (HPRT ⁇ MAC1) was subjected to the micronucleus cell fusion method.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, DGTP, dTTP) were used according to the recommended conditions. . Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C. for 10 seconds and 68 ° C. for 7 minutes. As a result of PCR, 19 clones out of 48 clones were positive in all primer sets, and 20 clones including 1 negative clone were analyzed thereafter. [C.
- human CYP3A gene cluster which is a DNA of 1 Mb size, in the mouse individual, it is translocated into the mouse artificial chromosome vector MAC1 (FIG. 18).
- PCR analysis In order to extract the genomic DNA of HAT-resistant strains and select reciprocal translocation clones as templates, PCR is performed using the following primers to confirm whether chromosomal reciprocal translocation occurs on human chromosome 7 fragment and MAC1. did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- mouse artificial chromosome vector MAC2 A mouse artificial chromosome vector MAC2 in which a 5'HPRT-loxP-PGKhyg type loxP sequence is inserted as a DNA insertion sequence into the mouse artificial chromosome vector MAC is constructed (FIG. 3).
- the loxP sequence of 5′HPRT-loxP-PGK hyg type is inserted into a GFP-loaded HAC vector (21HAC2) derived from chromosome 21 described in a report by Kazuki et al. (Gene Therapy: PMID: 21085194, 2010), HAC and MAC gene expression can be compared in the same vector.
- a gene transfer vector for insertion into 21HAC2 can be used as it is without going through a vector preparation step.
- Insertion of 5′HPRT-loxP-PGKhyg type loxP sequence into mouse artificial chromosome MAC [A. 1] Construction of loxP targeting vector of 5'HPRT-loxP-PGKhyg type VH21-12 prepared above was used as the basic plasmid for inserting the loxP sequence. The 5′HPRT-loxP-PGKhygro cassette excised from K6.1I with KpnI and AscI was cloned into the KpnI and AscI sites of V907 (Lexicon genetics) (vector name: pV907-AML).
- FIG. 20 shows a targeting vector, a target sequence, and a chromosomal allele resulting from homologous recombination.
- the targeting vector pMAC2 prepared above was linearized with the restriction enzyme NotI (TAKARA), and then transfected into the clone DT40 (MAC) prepared above, which contains hygromycin (1.5 mg / ml).
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 10 seconds and 68 ° C for 7 minutes were performed.
- 8 clones out of 45 clones were positive in all primer sets, and the subsequent analysis was performed on 6 clones randomly selected from these 8 clones.
- mouse artificial chromosome vector MAC2-containing chicken DT40 cells into MAC2 CHO cells
- a target gene etc. for example, GFP gene etc.
- PCR analysis In order to extract the genomic DNA of a hygromycin resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether the mouse artificial chromosome vector MAC2 could be introduced into CHO cells.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- mouse artificial chromosome vector MAC2 in which the loxP sequence as the gene insertion site was inserted into the mouse artificial chromosome MAC, which is a chromosome fragment derived from mouse chromosome 11, could be introduced into CHO cells.
- the medium When confluent, the medium was replaced with F12 medium supplemented with 20% FBS and 0.1 ⁇ g / ml colcemid. After further incubation for 48 hours, 20% FBS, 0 The medium was changed with F12 medium supplemented with 1 ⁇ g / ml colcemid, and further incubated overnight to form microcells.
- the culture solution was removed, and a cytochalasin B (10 ⁇ g / ml, sigma) solution that had been preliminarily kept at 37 ° C. was filled into a centrifuge flask, followed by centrifugation at 34 ° C., 8000 rpm for 1 hour.
- Micronuclear cells (also referred to as “microcells”) were suspended in serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes and suspended in 5 ml of serum-free DMEM medium. The microcell was suspended in 5 ml of serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes.
- Donor cells include B6-ES, which is an ES cell of a C57B6 strain mouse obtained from CLEA Japan, and B6 (HPRT), an HPRT-deficient strain obtained by performing 6TG treatment on the ES cell. ⁇ ), And KO56 (HPRT), an HPRT-deficient strain of TT2F cells ⁇ ) was used.
- DMEM Dulbecco's Modified Eagle's Medium-high glucose: SIGMA
- 10% FCS 10% FCS
- LIF Meerin Leukemia Inhibitory Factor
- L-glutamine 3.5 g / ml: GIBCO
- sodium pyruvate solution 3.5 g / ml: GIBCO
- MEM non-essential amino acid (0.125 mM: GIBCO)
- CO 2 5% CO 2
- the culture was performed at 37 ° C.
- Mouse ES cells were washed twice with PBS ( ⁇ ), and the cells were dispersed by trypsin treatment.
- the cells were collected with a culture solution obtained by adding 10% FBS to DMEM medium, centrifuged at 1500 rpm, and the supernatant was removed.
- the suspension was resuspended in 5 ml of serum-free culture, gently added to serum-free medium containing the pellet after microcell centrifugation, and centrifuged at 1200 rpm.
- the supernatant was removed, and PEG1000 (Wako) solution [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 0.5 ml for exactly 1 minute and 30 seconds. .
- PCR analysis In order to extract a genomic DNA of a hygromycin resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether the mouse artificial chromosome MAC2 could be introduced into mouse ES cells. The primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- mice artificial chromosome vector MAC2 in which the loxP sequence as the gene insertion site was inserted into the mouse artificial chromosome MAC, which is a chromosome fragment derived from mouse chromosome 11, could be introduced into mouse ES cells (FIG. 23).
- in vitro stability can be verified using mouse ES cells carrying mouse artificial chromosome vector MAC2.
- a chimeric mouse can be prepared from the ES cells, and a mouse strain TC (MAC2) in which MAC2 is transmitted to offspring can be prepared. Moreover, the stability of MAC2 in somatic cells can be verified using the TC (MAC2) mouse strain.
- MAC2 mouse strain in somatic cells
- MAC2 mouse strain in somatic cells
- mice artificial chromosome vector MAC3 in mouse ES cells is verified, and further, the progeny transmission mouse into which MAC3 has been introduced is prepared, thereby verifying the stability in individual tissues.
- Insertion of loxP sequence of PGKneo-loxP-3′HPRT type into mouse artificial chromosome MAC [A. 1] Preparation of PGKneo-loxP-3'HPRT type loxP targeting vector VH21-12 prepared above was used as the basic plasmid for inserting the loxP sequence.
- Chicken DT40 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (Gibco, hereinafter referred to as FBS), 1% chicken serum (Gibco), 10-4M 2-mercaptoethanol (Sigma). .
- DT40 (MAC) -1 7 Cells were washed once with no-addition RPMI1640 medium, suspended in 0.5 ml of no-addition RPMI1640 medium, and 25 ⁇ g of the targeting vector pMAC3 linearized with the restriction enzyme NotI (TAKARA) was added, and a cuvette for electroporation was added. (Bio-Rad) and allowed to stand at room temperature for 10 minutes. The cuvette was set on Gene Pulser (Bio-Rad), and voltage was applied under the conditions of 550 V and 25 ⁇ F. After standing at room temperature for 10 minutes, the cells were cultured for 24 hours.
- TAKARA restriction enzyme NotI
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 10 seconds and 68 ° C for 9 minutes.
- 16 clones out of 17 clones were positive in all the primer sets. Therefore, 2 clones were randomly selected from the 16 clones and the subsequent analysis was performed.
- cytochalasin B (10 ⁇ g / ml, sigma) solution pre-incubated at 37 ° C. was filled in the centrifuge flask, followed by centrifugation at 34 ° C., 8000 rpm for 1 hour.
- the microcell was suspended in serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 1700 rpm for 10 minutes and suspended in 5 ml of serum-free DMEM medium.
- Donor cells include CHO (HPRT), which is a CHO hprt-deficient cell (obtained from Human Science Research Resource Bank, registration number JCRB0218). ⁇ ) was used. The purified micronuclei were resuspended in 2 ml of serum-free culture medium containing PHA-P (SIGMA), and gently seeded on CHO cells from which the culture supernatant (F12 medium supplemented with 10% FBS (invitrogen)) was removed. The plate was incubated at 37 ° C. for 15 minutes.
- SIGMA serum-free culture medium containing PHA-P
- F12 medium supplemented with 10% FBS (invitrogen) was removed. The plate was incubated at 37 ° C. for 15 minutes.
- PEG1000 (Wako) solution [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 1 ml for exactly 1 minute.
- Serum-free culture medium (DMEM) was washed 4 times with 4 ml, and 5 ml of normal CHO cell culture medium was added and incubated overnight. After washing the cell surface twice with PBS ( ⁇ ), the cells were dispersed by trypsin treatment, seeded on 5 cell culture dishes having a diameter of 10 cm, G418 was added to 800 ⁇ g / ml, and selective culture was performed for 3 to 4 weeks.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation, followed by 30 cycles of 98 ° C for 10 seconds and 68 ° C for 10 minutes.
- 6 clones out of 7 clones were positive in all primer sets, and the subsequent analysis was performed on these 6 clones. [B.
- mouse artificial chromosome MAC3 is introduced into mouse ES cells, and mouse artificial chromosome vector MAC3-containing chimeric mice and offspring transfer mice are produced. . [C.
- the microcell was filled into a centrifuge flask, followed by centrifugation at 34 ° C., 8000 rpm for 1 hour.
- the microcell was suspended in serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes and suspended in 5 ml of serum-free DMEM medium.
- the microcell was suspended in 5 ml of serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes.
- Donor cells include B6 (HPRT), an HPRT-deficient strain obtained by 6TG treatment of ES cells of C57B6 strain mice obtained from CLEA Japan. ⁇ ) Was used.
- DMEM Dulbecco's Modified Eagle's Medium-high glucose: SIGMA
- 10% FCS 10% FCS
- LIF Meerin Leukemia Inhibitory Factor
- L-glutamine 3.5 g / ml: GIBCO
- sodium pyruvate solution 3.5 g / ml: GIBCO
- MEM Nonesential amino acid (0.125 mM: GIBCO) And add 5% CO 2
- the culture was performed at 37 ° C.
- Mouse ES cells were washed twice with PBS ( ⁇ ), and the cells were dispersed by trypsin treatment. The cells were collected with a culture solution obtained by adding 10% FBS to DMEM medium, centrifuged at 1500 rpm, and the supernatant was removed. The suspension was resuspended in 5 ml of serum-free culture, gently added to serum-free medium containing the pellet after microcell centrifugation, and centrifuged at 1200 rpm.
- PEG1000 (Wako) solution [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 0.5 ml for exactly 1 minute and 30 seconds. . 13 ml of serum-free medium (DMEM) was gently added and centrifuged at 1200 rpm. Supernatant was removed, normal mouse ES cell culture medium was added, mitomycin-treated G418-resistant mouse embryonic fibroblasts were used as feeder cells, seeded in two 10 cm diameter cell culture dishes, and incubated overnight did.
- DMEM serum-free medium
- G418 was added at 250 ⁇ g / ml and selective culture was performed for 3 to 4 weeks. A total of 28 resistant colonies obtained by two micronucleus cell fusions were isolated and expanded, and the subsequent analysis was performed (clone name: B6 (HPRT ⁇ MAC3)).
- B6 HPRT ⁇ MAC3
- C. 2 Selection of drug resistant clones
- C. 2.1 PCR analysis In order to select the recombinant DNA as a template by extracting the genomic DNA of the G418 resistant strain, PCR was performed using the following primers to confirm whether site-specific cleavage occurred on mouse chromosome 11. The primer sequences are shown below.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation, followed by 30 cycles of 98 ° C for 10 seconds and 68 ° C for 10 minutes.
- 27 clones out of 28 clones were positive in all primer sets, and the subsequent analysis was performed on these 27 clones. [C.
- [D] Stability of mouse artificial chromosome vector MAC3 in mouse ES cells Mouse ES clones obtained above (eg B6 (HPRT ⁇ MAC3) -3, -s6, obtained in the above [C]), the ratio of MAC1-retaining cells was measured by FISH analysis after long-term culture in non-selective culture of 0 to 100 PDL. (FIG. 28). As a result, it was confirmed that the mouse artificial chromosome vector MAC3 is maintained very stably at a ratio of 95% or more in mouse ES cells (in vitro).
- ICR MCH
- a chimeric mouse carrying the mouse artificial chromosome vector MAC3 and a wild-type mouse can be crossed to produce a mouse strain TC (MAC3) in which MAC3 is transmitted to offspring. Moreover, the stability of MAC3 in somatic cells can be verified using the TC (MAC3) mouse strain.
- TC mouse strain
- Example 6 Construction of mouse artificial chromosome vector GFP-MAC As an example of a gene encoding a useful protein in the mouse artificial chromosome vector MAC3, EGFP, which is a fluorescent gene, is inserted using the Cre / loxP system to verify the expression and long-term stability of the functional protein (FIG. 5).
- 5′HPRT-loxP the loxP sequence oligo-synthesized at the XbaI site of V820 (Lexicon genetics) was cloned.
- 5′HPRT-loxP was cloned into C907 and AscI of V907 (Lexicon genetics), and PGKhygro was cloned into the ClaI and KpnI sites (vector name: pX6.1).
- HS4-CAG-EGFP-HS4 distributed by Osaka University, Dr. Okabe and NIH, Dr. Felsenfeld
- FIG. 29 shows the chromosomal site-specific DNA insertion caused by GFP insertion in the HPRT reconstruction system using the Cre / loxP system.
- PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle condition were 94 ° C. for 10 minutes, followed by 35 cycles of 94 ° C. for 30 seconds, 60 ° C. for 30 seconds and 72 ° C. for 30 seconds.
- all 22 clones were positive, and the subsequent analysis was performed on these 22 clones.
- CHO (GFP-MAC) -4, 10, 12 are cultured in a cell culture dish, and when confluent, replaced with F12 medium supplemented with 20% FBS, 0.05 ⁇ g / ml colcemid, After culturing for 48 hours, the medium was changed with F12 medium supplemented with 20% FBS and 0.05 ⁇ g / ml colcemid, and further incubated overnight to form microcells. The culture solution was removed, and a cytochalasin B (10 ⁇ g / ml, sigma) solution that had been preliminarily kept at 37 ° C.
- the microcell was filled into a centrifuge flask, followed by centrifugation at 34 ° C., 8000 rpm for 1 hour.
- the microcell was suspended in serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes and suspended in 5 ml of serum-free DMEM medium.
- the microcell was suspended in 5 ml of serum-free DMEM medium and purified with 8 ⁇ m, 5 ⁇ m, and 3 ⁇ m filters. After purification, it was centrifuged at 2000 rpm for 10 minutes.
- wild type B6 cells established from ES cells of C57B6 strain mice obtained from CLEA Japan and mouse ES cells of wild type TT2F cells were used.
- DMEM Dulbecco's Modified Eagle's Medium-high glucose: SIGMA
- 10% FCS 10% FCS
- LIF Meerin Leukemia Inhibitory Factor
- L-glutamine 3.5 g / ml: GIBCO
- sodium pyruvate solution 3.5 g / ml: GIBCO
- MEM Nonsessential amino acid (0.125 mM: GIBCO)
- Add 5% CO 2 The culture was performed at 37 ° C.
- Mouse ES cells were washed twice with PBS ( ⁇ ), and the cells were dispersed by trypsin treatment. The cells were collected with a culture solution obtained by adding 10% FBS to DMEM medium, centrifuged at 1500 rpm, and the supernatant was removed. The suspension was resuspended in 5 ml of serum-free culture, gently added to serum-free medium containing the pellet after microcell centrifugation, and centrifuged at 1200 rpm.
- PEG1000 (Wako) solution [5 g of PEG1000 was completely dissolved in serum-free DMEM medium and 1 ml of dimethyl sulfoxide was added and sterilized by filtration] was fused with 0.5 ml for exactly 1 minute and 30 seconds. . 13 ml of serum-free medium (DMEM) was gently added and centrifuged at 1200 rpm. Supernatant was removed, normal mouse ES cell culture medium was added, mitomycin-treated G418-resistant mouse embryonic fibroblasts were used as feeder cells, seeded in two 10 cm diameter cell culture dishes, and incubated overnight did.
- DMEM serum-free medium
- G418 was added to 250 ⁇ g / ml and selective culture was performed for 3 to 4 weeks. A total of 36 resistant colonies obtained by two micronucleus cell fusions were isolated and expanded for subsequent analysis (clone names: TT2F (GFP-MAC) and B6-ES (GFP-MAC)).
- TT2F GFP-MAC
- B6-ES GTP-MAC
- B. 2.1 PCR analysis In order to select the recombinant DNA as a template by extracting the genomic DNA of the G418 resistant strain, PCR was performed using the following primers to confirm whether site-specific cleavage occurred on mouse chromosome 11. The primer sequences are shown below.
- PCR GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, LA Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions of TRANS L1 / R1 were heat denaturation at 94 ° C. for 1 minute, followed by 35 cycles of 98 ° C. for 10 seconds and 68 ° C. for 1 minute.
- the temperature and cycle conditions of TRANS L1 / m11 6R were heat denaturation at 94 ° C for 1 minute, followed by 35 cycles of 98 ° C for 10 seconds and 68 ° C for 7 minutes.
- mouse ES cells introduced with mouse artificial chromosome GFP-MAC are usually karyotype and can be used for long-term culture and production of chimeric mice.
- GFP-MAC mouse artificial chromosome
- mouse artificial chromosome vector GFP-MAC could be introduced into mouse ES cells.
- mice derived from male wild-type B6 (GFP-MAC) clone were born, 20 of which were male mice, 1 GFP positive 50% chimeric mouse, 5 40% chimeric mice, 30% chimeric mice 1 mouse, 7 20% chimeric mice, 3 10% chimeric mice and 3 5% chimeric mice.
- 14 chimeric mice derived from the wild type TT2F (GFP-MAC) clone were born, and one of them was a GFP positive individual with a chimera rate of about 100% in which the white portion was hardly observable.
- the ES cell lines (B6 and TT2F) carrying the mouse artificial chromosome vector GFP-MAC have the ability to form chimeras, that is, the ability to differentiate into normal tissues of individual mice. Indicated.
- GFP fluorescence was observed in the whole body of one of the three pups, indicating that the mouse artificial chromosome is stable even in the individual mouse (FIG. 33).
- a mouse strain in which GFP-MAC is transmitted as a descendant is called TC (GFP-MAC). Further, as described in Example 8, the stability of GFP-MAC in somatic cells can be verified using the TC (GFP-MAC) mouse strain.
- TC GFP-MAC
- mice artificial chromosome vector MAC1 Stability of mouse artificial chromosome vector MAC1 in mouse ES cells
- mouse ES clones obtained above for example, KO56 (MAC1) -5 and TT2F (MAC1) -23, obtained in Example 2 above
- MAC1 by FISH analysis after long-term culture in non-selective culture of 0 to 75 PDL As a result of measuring the ratio of the retained cells, the retention rate was 90% or more even in 75PDL.
- mouse ES cells carrying the GFP-loaded HAC vector (21HAC2) derived from chromosome 21 described in the report of Kazuki et al. (Gene Therapy: PMID: 21085194, 2010 the retention rate is 75% or less in 75PDL. there were.
- FIG. [A. 3] Production of chimeric mouse carrying mouse artificial chromosome vector MAC1 Using the ES cell clone obtained in Example 2 above, a chimeric mouse was prepared by the method of Tomizuka et al. (Nature Genet. 16: 133, 1997). As a host, an 8-cell embryo obtained by male-male mating of MCH (ICR) (white, purchased from Nippon Claire) was used. It is possible to determine whether or not a pup mouse born as a result of transplanting an injected embryo into a temporary parent is a chimera based on the hair color.
- ICR MCH
- TC mouse strain in which MAC1 is transmitted as a descendant
- TC mouse strain in which MAC1 is transmitted as a descendant
- a mouse strain in which MAC1 is transmitted as a descendant is referred to as TC (MAC1).
- MAC1 A mouse strain in which MAC1 is transmitted as a descendant
- a mouse strain in which MAC1 is transmitted as a descendant is referred to as TC (MAC1).
- MAC1 A mouse strain in which MAC1 is transmitted as a descendant
- MAC1 A mouse strain in which MAC1 is transmitted as a descendant
- FISH Fluorescence in situ hybridization
- mouse artificial chromosome vector MAC1 was maintained very stably at a rate of 90% or more in mouse ES cells (in vitro) and mouse tissues (in vivo).
- Example 8 Preparation and stability of mouse artificial chromosome vector CYP3A-MAC-carrying mouse [A] Transfer of CYP3A-MAC from CHO cells to mouse A9 cells In order to produce mouse ES cells that retain CYP3A-MAC, CHO cells (CHO (CYP3A-MAC, hChr7- ⁇ CYP3A) 22, 26, 34, 35, etc.) that retain CYP3A-MAC obtained in Example 3 above.
- mice A9 cells were introduced into mouse A9 cells with high microcell-forming ability by the micronucleus cell fusion method.
- a total of 25 resistant colonies obtained by 8 micronucleus cell fusions were isolated and expanded, and the subsequent analysis was performed (clone name: A9 (CYP3A-MAC)).
- CYP3A-MAC clone name: A9 (CYP3A-MAC)
- FISH analysis Tomizuka et al., Nature Genet. 16: 133, 1997) using CYP3A-BAC (RP11757A13) (CHORI) and mouse minor satellite DNA probe, CYP3A specifically detected by the probe was detected.
- Microcells were purified from A9 cells (such as A9 (CYP3A-MAC) 8, 9) that retained a single CYP3A-MAC and suspended in 5 ml of DMEM.
- A9 cells such as A9 (CYP3A-MAC) 8, 9
- CYP3A-MAC CYP3A-MAC 8
- DMEM fetal calf serum
- Each mouse ES cell TT2F was peeled off by trypsin treatment, washed 3 times with DMEM, suspended in 5 ml of DMEM, added to the centrifuged microcell, and centrifuged at 1250 rpm for 10 minutes to completely remove the supernatant.
- the precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds. Thereafter, 10 ml of DMEM was slowly added, centrifuged at 1250 rpm for 10 minutes, suspended in 30 ml of ES medium, dispensed into three 100 mm diameter Petri dishes (Corning) pre-spread with feeder cells, and cultured. After 24 hours, the medium was replaced with a medium containing 300 ⁇ g / ml G418, and selective culture was performed for about 1 week.
- a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30
- ICR MCH
- MAC1-retaining ES clone eg TT2F (CYP3A-MAC) 8-5, 8-16, 8-22, 9-4, 9-7, 9-9, 9-10, etc., acquired in [B] above
- TT2F CYP3A-MAC
- 28 chimeric mice a dark brown part was recognized in the hair color
- Five of them were individuals with a chimera rate of about 100% in which almost no white portion was observable. That is, it was shown that the ES cell line (TT2F) holding the mouse artificial chromosome vector CYP3A-MAC has the ability to form chimeras, that is, has the ability to differentiate into normal tissues of mouse individuals.
- [E] Progeny transmission of CYP3A-MAC from chimeric mice carrying CYP3A-MAC
- Five female chimeric mice (chimera rate: about 100%) prepared in the above [D] were mated with MCH (ICR) (white, purchased from CLEA Japan) male mouse. Of the 60 pups born from the chimeric mice, 50 were dark brown indicating that they possessed dominant inheritance derived from ES cells. That is, it was shown that the ES cell line carrying CYP3A-MAC differentiated into functional egg cells in female chimeric mice.
- TC CYP3A-MAC
- TC CYP3A-MAC
- TC CYP3A-MAC
- FISH analysis probed with CYP3A-BAC (RP11757A13) DNA using the tail fibroblasts of 18 pups using the method described by Shinohara et al. (Human Molecular Genetics, 10: 1163-1175, 2001). As a result, the presence of CYP3A-MAC was visually confirmed.
- CYP3A-MAC is maintained at a rate of 95% or higher for mouse ES cells (in vitro) very stably for a long period of time, and is highly stable at a rate of 90% or higher for mouse tissues (in vivo).
- Primers for detecting human CYP3A gene cluster expression Primer for detecting mouse Cyp3a gene cluster expression: Control gene expression detection primer: For PCR, GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, EX Taq (Takara Shuzo) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. The temperature and cycle conditions were 35 cycles of heat denaturation at 93 ° C for 5 minutes followed by 93 ° C for 1 minute, 56 ° C for 1 minute, and 72 ° C for 1 minute.
- CYP3A4 is only in the liver and small intestine
- CYP3A5 is only in the liver, small intestine and lung
- CYP3A7 is only in the liver, small intestine, kidney and lung
- CYP3A43 is in the liver and small intestine.
- Cyp3a11 was detected only in the liver and small intestine
- Cyp3a13 was detected only in the liver and small intestine.
- Control GAPDH was detected in all tissues.
- a representative result of the female (14) is shown in FIG. Thus, tissue-specific expression was observed as seen in humans, indicating that it became humanized.
- Example 9 Production of TC (CYP3A-MAC) / ⁇ cyp mouse strain
- A Construction of a mouse strain that retains CYP3A-MAC and in which both alleles of the endogenous Cyp3a gene group have been disrupted About GFP-positive mouse individuals obtained by backcrossing the TC (CYP3A-MAC) prepared in Example 8 to the ⁇ cyp strain prepared in Example 7 of WO 2009/063722 (PCT / JP2008 / 068928) The genotype was analyzed using the PCR method described above. A part of the tail of 51 pups obtained by mating was cut out, and genomic DNA was prepared from the sample.
- the obtained DNA was subjected to PCR using the primers for CYP3A-MAC detection and the primers described in Table 1 of WO 2009/063722 (PCT / JP2008 / 068928) in the same manner as described above, and the CYP3A-MAC was retained and the Cyp3a gene cluster was isolated.
- the mice were CLINE3A-MAC in 24 mice and one of the alleles of the endogenous Cyp3a gene group was destroyed (hetero KO).
- the Cyp3a gene cluster was disrupted in this heterozygous and backcrossed to the ⁇ cyp strain with a mouse carrying CYP3A-MAC.
- TC CYP3A-MAC
- ⁇ cyp mouse strain can reproduce human drug metabolism, it can be used as a model mouse for in vivo tests for examining the efficacy and toxicity of the first phase reaction in drug development.
- Example 10 Preparation of mouse artificial chromosome vector CYP3A-MAC-carrying rat
- rESWIv3i-1 Hirabayashi et al., Mol.
- rESWIv3i-1 which is a rat ES cell capable of progeny transmission from A9 cells carrying CYP3A-MAC obtained in Example 8 above.
- the precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds. Thereafter, 10 ml of DMEM was slowly added, centrifuged at 1250 rpm for 10 minutes, suspended in 30 ml of ES medium, dispensed into three 100 mm diameter Petri dishes (Corning) pre-spread with feeder cells, and cultured. After 24 hours, the medium was replaced with a medium containing 300 ⁇ g / ml G418, and selective culture was performed for about 1 week.
- a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30
- liver microsomes derived from rTC (CYP3A-MAC) rat strain can be used as a sample for examining the efficacy and toxicity of the first phase reaction in drug development.
- rTC (CYP3A-MAC) rat strain can reproduce human drug metabolism, it can be used as a model rat for an in vivo test for examining the efficacy and toxicity of the first phase reaction in drug development.
- Example 11 Construction of mouse artificial chromosome vector hChr21q-MAC
- translocation cloning was performed using a Cre / loxP system on a DNA sequence containing a region 33 Mb distal to AP001657 of the long arm of human chromosome 21 in mouse artificial chromosome vector MAC1.
- hChr21q-MAC is constructed.
- hChr21-loxP from DT40 containing hChr21-loxP into MAC1-containing CHO cells
- hChr21-loxP is introduced into mouse artificial chromosome vector MAC1-containing CHO cells.
- PCR analysis In order to extract the genomic DNA of the HAT-resistant strain and select the reciprocal translocation clone as a template, PCR is performed using the following primers to confirm whether a reciprocal chromosome translocation occurs on human chromosome 21 fragment and MAC1. did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Each mouse ES cell TT2F was peeled off by trypsin treatment, washed 3 times with DMEM, suspended in 5 ml of DMEM, added to the centrifuged microcell, and centrifuged at 1250 rpm for 10 minutes to completely remove the supernatant. The precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds.
- a mouse strain TC in which hChr21q-MAC is transmitted to offspring can be produced from a chimeric mouse carrying the mouse artificial chromosome vector hChr21q-MAC.
- the stability of hChr21q-MAC in somatic cells can be verified using the TC (hChr21q-MAC) mouse strain.
- the TC (hChr21q-MAC) strain can be used as a model mouse for Down syndrome, and can be used for elucidating the mechanism of Down syndrome symptom onset and developing therapeutics for symptom improvement.
- Example 12 Construction of mouse artificial chromosome vector hChr21q22.12-MAC
- translocation cloning was performed using a Cre / loxP system on a mouse artificial chromosome vector MAC1 and a DNA sequence containing a region distal to AP00172 of the long arm of human chromosome 21 using the Cre / loxP system.
- hChr21q22.12-MAC is constructed.
- the loxP sequence is inserted into AP001721 proximal to the DSCR (down syndrome disease cause cluster) of human chromosome 21 (hChr21) in DT40 cells.
- the targeting vector pCKloxPHyg for inserting the recognition sequence loxP of Cre recombinase into the Down syndrome causative gene region (DSCR) located in the vicinity of AP001721 on human chromosome 21 and on the centromere side (about 50 Kb centromere side) is as follows: In this way, it was produced. First, the AP001721 genomic region was amplified by PCR using the following primers. V901 (Lexicon genetics) was used as a basic plasmid for inserting the loxP sequence.
- PCR Gene Amp 9600 manufactured by Perkin-Elmer is used as a thermal cycler, LATaq (Takara Shuzo) is used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 20 seconds and 68 ° C for 5 minutes. After the PCR product was treated with proteinase K (Gibco), the PCR fragment (2.9 kb and 2.0 kb) was gel-filtered with CHROMASPIN-TE400 (Clontech).
- 5′-HPRT-loxP-Hyg was excised with KpnI and AscI from the 5′-HPRT-loxP-Hyg-TK vector described in the report of Kazuki et al. (Gene Therapy: PMID: 21085194, 2010), and V901T-1HR2 Cloning into the AscI and KpnI sites (pCKloxPHyg).
- the size of the final loxP insertion construct is 11.2 kb.
- a targeting vector, a target sequence and a chromosomal allele resulting from homologous recombination are shown in FIG. [A.
- This filter was subjected to Southern hybridization using an SP7 probe obtained by amplifying the gene sequence in AP001721 by PCR to identify a homologous recombinant.
- the SP7 probe was prepared by PCR using the following primers and DT40 (21-2-3) genomic DNA as a template, and using the PCR product as a template by random priming. 32 A P-labeled DNA probe was made (Amersham, following the attached protocol).
- SP7 probe preparation primer For PCR, GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler, Ex Taq (TAKARA) was used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. The temperature and cycle conditions were 35 cycles of heat denaturation at 93 ° C. for 5 minutes, 93 ° C. for 1 minute, 54 ° C. for 1 minute, and 72 ° C. for 1 minute. By Southern hybridization, it was predicted that a band of about 7.5 kb was detected in the non-homologous recombinant and about 9.4 kb in the homologous recombinant (FIG.
- FIG. [A. 3.3] two-color FISH analysis FISH analysis was performed according to Matsubara et al. (FISH experimental protocol, Shujunsha, 1994). When FISH analysis was performed on 10 clones among the clones confirmed to be recombination using human cot-1 DNA and hygromycin as probes, human chromosome 21 was not translocated to the host chromosome in all clones. Since a signal derived from hygromycin was detected in the vicinity of 21q22, it was confirmed that recombination occurred in a site-specific manner (FIG. 58).
- PCR analysis In order to extract the genomic DNA of a hygromycin resistant strain and select a recombinant as a template, 20 clones out of the 140 clones were subjected to PCR using the following primers, and the human chromosome 21 fragment was transformed into MAC1-containing CHO. It was confirmed whether it was introduced into the cells.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- PCR analysis In order to extract the genomic DNA of the HAT-resistant strain and select the reciprocal translocation clone as a template, PCR is performed using the following primers to confirm whether a reciprocal chromosome translocation occurs on human chromosome 21 fragment and MAC1. did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Microcells were purified from CHO cells (such as CHO (hChr21q22.12-MAC, hChr21-hChr21q22.12) 1,12) that retain a single hChr21q-MAC and suspended in 5 ml of DMEM.
- CHO cells such as CHO (hChr21q22.12-MAC, hChr21-hChr21q22.12) 1,12
- DMEM fetal
- Each mouse ES cell TT2F was peeled off by trypsin treatment, washed 3 times with DMEM, suspended in 5 ml of DMEM, added to the centrifuged microcell, and centrifuged at 1250 rpm for 10 minutes to completely remove the supernatant.
- the precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds. Thereafter, 10 ml of DMEM was slowly added, centrifuged at 1250 rpm for 10 minutes, suspended in 30 ml of ES medium, dispensed into three 100 mm diameter Petri dishes (Corning) pre-spread with feeder cells, and cultured. After 24 hours, the medium was replaced with a medium containing 300 ⁇ g / ml G418, and selective culture was performed for about 1 week.
- a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30
- a mouse strain TC (hChr21q22.12-MAC) carrying hChr21q22.12-MAC as a descendant can be produced from a chimeric mouse carrying the mouse artificial chromosome vector hChr21q22.12-MAC. .
- the stability of hChr21q22.12-MAC in somatic cells can be verified using the TC (hChr21q22.12-MAC) mouse strain.
- the hChr21q22.12-MAC strain can be used as a model mouse for Down syndrome, and can be used for elucidating the mechanism of Down syndrome symptom onset and developing therapeutic agents for symptom improvement.
- the causative gene region of Down's syndrome can be identified by comparing the phenotypes of the TC (hChr21q-MAC) mouse strain and the TC (hChr21q22.12-MAC) mouse strain.
- Example 13 Stability of mouse artificial chromosome vector MAC2
- A Stability of mouse artificial chromosome vector MAC2 in CHO cells CHO clones obtained above (eg CHO (HPRT ⁇ MAC2) -13,18, obtained in Example 4 above), the proportion of MAC2-retaining cells was measured by FISH analysis after long-term culture in non-selective culture of 0 to 25 PDL. As a result, 90% or more in 25PDL Retention rate.
- FIG. 14 Construction of mouse artificial chromosome vector FVIII-MAC As an example of a gene encoding a useful protein in the mouse artificial chromosome vector MAC2, the eighth factor (FVIII), which is a causative gene of hemophilia A, is inserted using the Cre / loxP system, and the expression and long-term stability of the functional protein To verify.
- the CAG-F8-pA region of pB-CAGF8 was isolated by the SalI and AvrII sites and cloned into the SalI and AvrII sites so as to be sandwiched between the two HS4 insulator sequences on pB3ins2 (pB3-F8ins2).
- pPAC4 Choildren's Hospital Oakland Research Institute (CHORI), BAC / PAC Resources
- CHORI Oakland Research Institute
- BAC / PAC Resources was introduced into a vector incorporating a 3′HPRT-loxP sequence.
- the FVIII expression cassette (HS4-CAG-F8-pA-HS4), which is the AscI and FseI regions on pB3-F8ins2, was cloned into the AscI and FseI sites of pPAC4 to obtain a 1-copy FVIII insertion construct of the HPRT reconstruction system (vector) Name: pPAC4 F8ins2 H3-9 (1 copy FVIII-PAC)) (FIG. 65).
- PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ex Taq (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and the cycle conditions were 94 ° C. for 10 minutes, followed by 35 cycles of 94 ° C. for 30 seconds, 60 ° C. for 30 seconds, and 72 ° C. for 1 minute. As a result of PCR, 4 clones derived from CHO (FVIIIx1-MAC), 17 clones derived from CHO (FVIIIx1-HAC), and 3 clones derived from CHO (FVIIIx16-MAC) were positive.
- the cells cultured to 0 PDL and 25 PDL in non-selective culture were cultured in a 6-well dish, and the medium was changed from 100% confluent to a new medium. After 24 hours, the culture supernatant was collected, and the degree of FVIII activation by FVIII activity was measured.
- 0PDL there was almost no difference in activity between CHO (FVIIIx1-MAC) 1-3 and CHO (FVIIIx1-HAC) 1-2.
- the activity of CHO (FVIIIx1-MAC) 1-3 was increased 1.8 times in 25PDL
- the activity of CHO (FVIIIx1-HAC) 1-2 was 1/6 times in 25PDL. It decreased (FIG.
- mouse artificial chromosome vector MI-MAC capable of introducing multiple genes
- a multi-integrate platform having five site-specific recombination enzyme recognition sites ⁇ C31 attP, R4 attP, TP901-1 attP, Bxb1 attP, FRT
- Cre site-specific recombination enzyme recognition sites
- LoxP system is used to verify introduction / expression of multiple genes.
- a cassette having a plurality of gene-loading sites for introducing a plurality of genes into a mouse artificial chromosome vector was prepared using a Multisite-Gateway kit (Invitrogen) as follows. First, using PGK-hyg (Clontech) as a template, ⁇ C31 attP site, R4 attP site, TP901-1 attP site, Bxb1 attP site, and FRT site, which are gene introduction sites, are used for first PCR (the following primer primer R1) To the PGK promoter sequence.
- PCR uses Perkin-Elmer Gene Amp 9600 as thermal cycler, Taq polymerase uses kodplus (Toyobo), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C. for 2 minutes and 20 cycles of 94 ° C. for 15 seconds, 68 ° C. for 30 seconds, and 72 ° C. for 90 seconds. The PCR product was treated with proteinase K (Gibco) and then purified with CHROMASPIN-TE400 (Clontech).
- Second PCR for adding the gateway attB sequence necessary for the Multisite-Gateway BP reaction was performed. PCR conditions and the like are the same as above except that the number of cycles is 25.
- the sequence of primer F1 is also as described above.
- a donor vector (Invitrogen: pDONR221 P1-P5r, pDONR221 P5-P4, pDONR221 P4r-P3r, pDONR221 P3-P2) having a gateway attP sequence corresponding to these PCR fragments was mixed, and in vitro recombination using BP clonease By reaction (BP reaction), entry vector (pENTR L1-FRT-PGK- ⁇ C31-R5, pENTR L5-PGK-R4-L4, pENTR R4-PGK-TP901-1-R3, pENTR L3-PGK-Bxb1-L2) was made (FIG. 69). BP reaction was performed according to the recommended conditions.
- plasmids incorporating 3′HPRT-loxP sequences necessary for introducing these gene introduction sites into mouse artificial chromosome vectors were prepared by the following procedure. Using the above X3.1 as a template, amplification was performed using the following primers. PCR conditions are the same as those described above (cycle number 25). This PCR fragment and DEST cassette (Invitrogen: R1-ccdB-Cm-R2) were blunt-ligated to obtain pDEST.
- this pDEST and the entry vector prepared above (pENTR L1-FRT-PGK- ⁇ C31-R5, pENTR L5-PGK-R4-L4, pENTR R4-PGK-TP901-1-R3, pENTR L3-PGK-Bxb1 -L2) was mixed, and a multi-integration platform cassette was prepared by in vitro recombination reaction (LR reaction) using LR clone (FIG. 70). LR reaction was performed according to the recommended conditions. [A.
- the multi-integrate platform cassette is the above-mentioned CHO (HPRT ⁇ MAC2) and CHO (HPRT) described below ⁇ MAC4) can be inserted into mouse artificial chromosome vectors MAC2 and MAC4 by obtaining Cre-loxP recombination as described above (referred to as MI-MAC) (FIG. 71).
- MI-MAC Cre-loxP recombination as described above
- the PCR conditions are the same as the above conditions (cycle number 25). Thereafter, this PCR fragment was blunt-cloned into SLR test (Toyobo) cleaved with restriction enzymes EcoRV and SmaI site to prepare pNeo. Next, the recombination sequences ( ⁇ C31 attB, R4 attB, TP901-1 attB, Bxb1 attB, FRT) corresponding to each attP site or FRT site were synthesized de novo ( ⁇ C31, Bxb1, FRT: Integrated DNA technology. R4, TP901-1: Invitrogen).
- pNeo was cleaved with the restriction enzyme SalI, and a DNA fragment containing ⁇ C31 attB or R4 attB was excised from the synthesized vector with the restriction enzyme SalI and ligated (pNeo- ⁇ C31 attB, pNeo-R4 attB).
- pNeo is cleaved with the restriction enzyme ClaI
- a DNA fragment containing TP901-1 attB or FRT is cut out from the above synthesized vector with the restriction enzyme ClaI
- ligated pNeo-TP901-1 attB, pNeo-FRT, pNeo-FRT was cleaved with the restriction enzyme NheI
- a DNA fragment containing Bxb1 attB was cut out from the synthesized vector with the restriction enzyme NheI to prepare a ligated pNeo-Bxb1 attB.
- These vectors are cassette vectors that can insert an arbitrary foreign gene into the BamHI site and can be mounted on a mouse artificial chromosome having a plurality of gene mounting sites (FIG. 72).
- Site-specific recombination enzymes ⁇ C31 integral, R4 integral, TP901-1 integral, Bxb1 integral
- ⁇ C31 Codon device, et al .: Invitrogen
- These integrals are synthesized by optimizing codon usage to mammals so as to achieve high expression in mammalian cells.
- PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle condition were 94 ° C. for 10 minutes, followed by 35 cycles of 94 ° C. for 30 seconds, 60 ° C. for 30 seconds and 72 ° C. for 30 seconds.
- 6 clones out of 11 clones were positive with all primers, and the subsequent analysis was performed with these 6 clones.
- mice carrying the mouse artificial chromosome vector PXR-MAC were obtained by mounting the human PXR gene on the mouse artificial chromosome MAC3.
- the CHO cells retaining PXR-MAC obtained in [A] above were introduced into mouse ES cells (wild type TT2F) by the micronucleus cell fusion method. According to the method of Tomizuka et al. (Nature Genet.
- Microcells were purified from CHO cells (such as CHO (PXR-MAC) 7, 9, 10) that retained one PXR-MAC and suspended in 5 ml of DMEM. About 10 7 Each mouse ES cell TT2F was peeled off by trypsin treatment, washed 3 times with DMEM, suspended in 5 ml of DMEM, added to the centrifuged microcell, and centrifuged at 1250 rpm for 10 minutes to completely remove the supernatant.
- the precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds. Thereafter, 10 ml of DMEM was slowly added, centrifuged at 1250 rpm for 10 minutes, suspended in 30 ml of ES medium, dispensed into three 100 mm diameter Petri dishes (Corning) pre-spread with feeder cells, and cultured. After 24 hours, the medium was replaced with a medium containing 300 ⁇ g / ml G418, and selective culture was performed for about 1 week.
- a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30
- a chimeric mouse can be prepared from mouse ES cells carrying the mouse artificial chromosome vector PXR-MAC, and a mouse strain TC (PXR-MAC) in which PXR-MAC is transmitted to offspring can be prepared. Moreover, the stability of PXR-MAC in somatic cells can be verified using the TC (PXR-MAC) mouse strain. In addition, the TC (PXR-MAC) mouse strain can reproduce CYP gene expression induction by drugs in humans.
- a TC (CYP3A-MAC / PXR-MAC) strain can be produced by crossing with the above TC (CYP3A-MAC) mouse strain, and used as a model mouse for in vivo testing for examining drug efficacy and toxicity in drug development. it can.
- Construction of mouse artificial chromosome vector MAC4 A mouse artificial chromosome vector MAC4 in which a GFP-5′HPRT-loxP-PGKhyg type loxP sequence is inserted into the mouse artificial chromosome MAC as a DNA insertion sequence is constructed.
- the loxP sequence of 5′HPRT-loxP-PGKhyg type has been inserted into a GFP-loaded HAC vector (21HAC2) derived from chromosome 21 described in a report by Kazuki et al. (Gene Therapy: PMID: 21085194, 2010). And MAC gene expression can be compared with the same vector. Further, a gene transfer vector for insertion into 21HAC2 can be used as it is without going through a vector preparation step. [A] Insertion of loxP sequence of GFP-5′HPRT-loxP-hyg type into mouse artificial chromosome MAC [A.
- FIG. 76 shows a targeting vector, a target sequence, and a chromosomal allele resulting from homologous recombination.
- PCR analysis In order to select genomic DNA of hygromycin-resistant strains and select recombinants as templates, PCR was performed using the following primers, and it was confirmed whether recombination was occurring specifically on mouse chromosome vector MAC .
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- PCR analysis In order to extract a genomic DNA of a hygromycin resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether or not the mouse artificial chromosome vector MAC4 could be introduced into CHO cells.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- mouse artificial chromosome vector MAC4 could be introduced into CHO cells.
- mouse ES cells carrying the mouse artificial chromosome vector MAC4 can be produced, and in vitro stability can be verified using the ES cells.
- a chimeric mouse can be produced from the ES cells, and a mouse strain TC (MAC4) in which MAC4 is transmitted to offspring can be produced.
- MAC4 mouse strain in which MAC4 is transmitted to offspring
- the stability of MAC4 in somatic cells can be verified using the TC (MAC4) mouse strain.
- Example 18 Construction of mouse artificial chromosome vector UGT2-MAC UGT2-MAC is constructed in the same manner as in Example 3 by translocating the UGT2 cluster, which is a group of human drug metabolizing enzyme genes, into the mouse artificial chromosome vector MAC4 using the Cre / loxP system. In addition, the stability of UGT2-MAC in mouse ES cells is verified.
- UGT2 cluster which is a group of human drug metabolizing enzyme genes
- MAC4 the mouse artificial chromosome vector MAC4 using the Cre / loxP system.
- Cre / loxP the stability of UGT2-MAC in mouse ES cells is verified.
- targeting vector pTELpuro-UGT2 for inserting a human telomere sequence into the AC125239 region located very close to the UGT2 locus on human chromosome 4 and on the telomere side (about 150 Kb telomere side) was prepared as follows. First, the AC125239 genomic region was amplified by PCR using the following primers. For PCR, Gene Amp 9600 manufactured by Perkin-Elmer was used as a thermal cycler, LATaq (TAKARA) was used as Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- TAKARA LATaq
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 20 seconds and 68 ° C for 8 minutes were performed.
- the PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech). Then, it cleaved with restriction enzymes PstI (Nippon Gene) and BglII (Nippon Gene), and gel-filtered with CHROMASPIN-TE 1000 (Clontech).
- FIG. 79 shows a targeting vector, a target sequence, and a chromosomal allele resulting from homologous recombination.
- PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle conditions were 95 ° C for 10 minutes after heat denaturation, followed by 30 cycles of 95 ° C for 20 seconds, 55 ° C for 30 seconds, and 72 ° C for 30 seconds.
- the sequence is as follows.
- PCR was performed using LATaq (Takara Shuzo) with the above primers, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions.
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 20 seconds and 68 ° C for 8 minutes were performed.
- a band of about 8 kb was detected only in 2 clones in which site-specific recombination occurred. No band was detected in the negative controls DT40 and DT40 (hChr4) 1.
- FISH experimental protocol Shujunsha, 1994.
- FISH analysis was performed using two clones of human cot-1 DNA and puromycin DNA as a probe.
- human chromosome 4 did not translocate to the host chromosome in all clones.
- a puromycin-derived signal was detected at the end of the human chromosome 4 fragment, and it was confirmed that recombination occurred in a site-specific manner because it was cleaved at the target site (FIG. 80).
- targeting vector pUGT2loxPneo for inserting the recognition sequence loxP of the Cre recombinase into the AC074378 region located very close to the UGT2 locus on human chromosome 4 and on the centromere side (about 300 Kb centromere side) is prepared as follows. did. First, the AC0737478 genomic region was amplified by PCR using the following primers. V907 (Lexicon genetics) was used as a basic plasmid for inserting the loxP sequence.
- PCR Gene Amp 9600 manufactured by Perkin-Elmer is used as a thermal cycler, LATaq (Takara Shuzo) is used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 20 seconds and 68 ° C for 7 minutes. The PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech).
- FRT-pGKneo-FRT was excised with EcoRI and BamHI from the loxP-FRT-pGKneo-FRT-loxP cassette pNT1.1 (distributed from Osaka University, Genetic Information Experiment Center), and the above-mentioned X3.1 BglII site was excised.
- Cloned vector name: X3.1-FRT-pGKneo-FRT.
- V907-UGT2HR2 was cleaved with the restriction enzyme EcoRI, and a DNA fragment containing loxP was excised from the X3.1-FRT-pGKneo-FRT with the restriction enzyme EcoRI and ligated.
- a vector having the loxP sequence in the same direction as the cloned AC0743378 genomic fragment was designated as a targeting vector pUGT2loxPneo.
- the size of the final loxP insertion construct is 11.1 kb.
- FIG. 81 shows a targeting vector, a target sequence, and a chromosome allele resulting from homologous recombination.
- TAKARA restriction enzyme NotI
- PCR uses Perkin-Elmer's Gene Amp 9600 as thermal cycler
- Taq polymerase uses LATaq (TAKARA)
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 10 seconds and 68 ° C for 4 minutes.
- 5 clones were identified as homologous recombinants.
- two-color FISH analysis FISH analysis was performed according to Matsubara et al. (FISH experimental protocol, Shujunsha, 1994).
- FISH analysis was performed using human cot-1 DNA and neomycin DNA as probes, and human chromosome 4 was not translocated to the host chromosome in all clones. Since a signal derived from neomycin was detected in the vicinity of 4q13, it was confirmed that recombination occurred in a site-specific manner (FIG. 82). From the above results, it was concluded that the loxP sequence, which is a gene introduction site, was inserted site-specifically into AC0743378 on human chromosome 4.
- PCR analysis In order to extract the genomic DNA of a neomycin resistant strain and select a recombinant as a template, PCR was performed using the following primers to confirm whether the human chromosome 4 fragment was introduced into MAC4-containing CHO cells. The primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- PCR analysis In order to extract the genomic DNA of HAT-resistant strains and select reciprocal translocation clones as templates, PCR is performed using the following primers to confirm whether chromosomal reciprocal translocation occurs on human chromosome 4 fragment and MAC4 did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- Microcells were purified from A9 cells (such as A9 (UGT2-MAC) 13, 15) that retained a single UGT2-MAC and suspended in 5 ml of DMEM. About 10 7 Each mouse ES cell TT2F was peeled off by trypsin treatment, washed 3 times with DMEM, suspended in 5 ml of DMEM, added to the centrifuged microcell, and centrifuged at 1250 rpm for 10 minutes to completely remove the supernatant.
- A9 cells such as A9 (UGT2-MAC) 13, 15
- UGT2-MAC UGT2-MAC
- the precipitate was sufficiently loosened by tapping, 0.5 ml of a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1 minute and 30 seconds. Thereafter, 10 ml of DMEM was slowly added, centrifuged at 1250 rpm for 10 minutes, suspended in 30 ml of ES medium, and dispensed into three 100 mm diameter petri dishes (Corning) in which feeder cells had been pre-spread and cultured. After 24 hours, the medium was replaced with a medium containing 300 ⁇ g / ml G418, and selective culture was performed for about 1 week.
- a 1: 1.4 PEG solution [5 g PEG1000 (Wako Pure Chemical Industries), 1 ml DMSO (Sigma) dissolved in 6 ml of DMEM] was added, and the mixture was sufficiently stirred for about 1
- a chimeric mouse can be prepared from mouse ES cells carrying the mouse artificial chromosome vector UGT2-MAC, and a mouse strain TC (UGT2-MAC) in which UGT2-MAC is transmitted to offspring can be prepared. Moreover, the stability of UGT2-MAC in somatic cells can be verified using the TC (UGT2-MAC) mouse strain. In addition, since the TC (UGT2-MAC) mouse strain can reproduce human drug metabolism, it can be used as a model mouse for in vivo tests for examining the efficacy and toxicity of the second phase reaction in drug development.
- Example 19 Construction of mouse artificial chromosome vector CYP2C-MAC CYP2C-MAC is constructed in the same manner as in Example 3 by translocating the CYP2C cluster, which is a human drug metabolizing enzyme gene group, into the mouse artificial chromosome vector MAC4 using the Cre / loxP system.
- targeting vector pTELpuro-CYP2C for inserting a human telomeric sequence into the AL157834 region located very close to the CYP2C locus on human chromosome 10 and on the telomere side (about 150 Kb telomere side) was prepared as follows. First, the AL157834 genomic region was amplified by PCR using the following primers.
- PCR Gene Amp 9600 manufactured by Perkin-Elmer was used as a thermal cycler, LATaq (TAKARA) was used as Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 20 seconds and 68 ° C for 8 minutes were performed.
- the PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech).
- FIG. 1 A targeting vector, a target sequence, and a chromosomal allele resulting from homologous recombination are shown in FIG. [A. 2] Transfection and isolation of drug resistant clones Kazuki et al. Chicken DT40 cells carrying human chromosome 10 from A9 (KM32-2) and A9 (KM26-3) (Kugoh et al. DNA research 1999) carrying human chromosome 10 were prepared by the method described in BBRC 2004. (Clone name: DT40 (hChr10)).
- the targeting vector pTELpuro-CYP2C prepared above was linearized with the restriction enzyme PstI (Nippon Gene), and then transfected into the clone DT40 (hChr10) 1,42 prepared above, and puromycin (0 .3 ug / ml), and dispensed into 10 96-well culture plates for selective culture for about 2 weeks. A total of 96 resistant colonies obtained by four transfections were isolated and expanded, and the subsequent analysis was performed (clone name: DT40 (hChr10-tel)). [A. 3] Selection of homologous recombinants [A.
- PCR analysis In order to select recombinants using the genomic DNA of a puromycin-resistant strain as a template, PCR was performed as a primary screening using the following primers located on the telomere side from the cleavage site to confirm whether site-specific cleavage occurred. The primer sequences are shown below. PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle conditions were 95 ° C for 10 minutes after heat denaturation, followed by 30 cycles of 95 ° C for 20 seconds, 55 ° C for 30 seconds, and 72 ° C for 30 seconds.
- PCR was confirmed by PCR whether site-specific homologous recombination had occurred in the 3 clones that were not detected by the above primers using the following primers.
- the sequence is as follows. PCR was performed using LATaq (Takara Shuzo) with the above primers, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions.
- targeting vector pCYP2CloxPneo for inserting the recognition sequence loxP of the Cre recombinase into the AL138759 region located very close to the CYP2C locus on human chromosome 10 and on the centromere side (about 300 Kb centromere side) is prepared as follows. did. First, the AL138759 genomic region was amplified by PCR using the following primers. V907 (Lexicon genetics) was used as a basic plasmid for inserting the loxP sequence.
- PCR Gene Amp 9600 manufactured by Perkin-Elmer is used as a thermal cycler, LATaq (Takara Shuzo) is used for Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was. The temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 20 seconds and 68 ° C for 7 minutes. The PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech).
- V907-CYP2CHR2 V907-CYP2CHR2 was cleaved with the restriction enzyme EcoRI, and a DNA fragment containing loxP was cut out from the X3.1-FRT-pGKneo-FRT with the restriction enzyme EcoRI and ligated.
- a vector having the loxP sequence in the same direction as the cloned AL138759 genomic fragment was designated as a targeting vector pCYP2CloxPneo.
- the size of the final loxP insertion construct is 10.3 kb.
- FIG. 91 shows a targeting vector, a target sequence, and a chromosome allele resulting from homologous recombination.
- TAKARA restriction enzyme NotI
- the medium was replaced with a medium containing neomycin (1.5 mg / ml), dispensed into three 96-well culture plates, and subjected to selective culture for about 2 weeks, a total of 15 resistances obtained by two transfections. Colonies were isolated and expanded, and the subsequent analysis was performed (clone name: DT40 (hChr10-tel-loxP)).
- DT40 hChr10-tel-loxP
- PCR uses Perkin-Elmer's Gene Amp 9600 as thermal cycler
- Taq polymerase uses LATaq (TAKARA)
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 10 seconds and 68 ° C for 4 minutes.
- a homologous recombinant was identified as a homologous recombinant.
- FISH analysis was performed according to Matsubara et al. (FISH experimental protocol, Shujunsha, 1994).
- FISH analysis was performed on one clone among the clones confirmed to have recombination using human cot-1 DNA and neomycin as probes, human clone 10 did not translocate to the host chromosome in all clones.
- a neomycin-derived signal was detected nearby, confirming that site-specific recombination had occurred. From the above results, it was concluded that the loxP sequence, which is the gene introduction site, was inserted site-specifically into AL138759 on human chromosome 10.
- PCR analysis In order to extract a neomycin resistant strain genomic DNA and select a recombinant as a template, PCR was performed using the following primers to confirm whether the human chromosome 10 fragment was introduced into MAC4-containing CHO cells. The primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- a human CYP2C gene cluster which is 380 kb size DNA
- it is translocated into the mouse artificial chromosome vector MAC4 (FIG. 93).
- PCR analysis In order to select HAT-resistant strain genomic DNA and select reciprocal translocation clones as templates, PCR is performed using the following primers to confirm whether chromosomal reciprocal translocation occurs on human chromosome 10 fragment and MAC4. did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- mice were obtained from CHO cells (CHO (CYP2C-MAC, hChr10- ⁇ CYP2C) 2, 8, 10) retaining CYP2C-MAC obtained in [D] above.
- the A9 cells were introduced into mouse A9 cells having high microcell-forming ability by the micronucleus cell fusion method.
- a total of 4 resistant colonies obtained by four micronucleus cell fusions were isolated and expanded, and the subsequent analysis was performed (clone name: A9 (CYP2C-MAC)).
- mouse ES cells carrying the mouse artificial chromosome vector CYP2C-MAC can be prepared, and in vitro stability can be verified using the ES cells.
- a chimeric mouse can be prepared from the ES cells, and a mouse strain TC (CYP2C-MAC) in which CYP2C-MAC is transmitted to offspring can be prepared.
- the stability of CYP2C-MAC in somatic cells can be verified using the TC (CYP2C-MAC) mouse strain.
- liver microsomes derived from the TC (CYP2C-MAC) mouse strain can be used as a sample for examining the efficacy and toxicity of the first phase reaction in drug development.
- mouse artificial chromosome vector MDR1-MAC MDR1-MAC is constructed in the same manner as in Example 3 by translocating the MDR1 gene, which is a human drug metabolizing enzyme gene group, into the mouse artificial chromosome vector MAC4 using the Cre / loxP system.
- the loxP sequence is inserted into the AC005045 proximal to the MDR1 gene of human chromosome 7 (hChr7) in DT40 cells.
- Construction of targeting vector pMDR1loxPbs A targeting vector pMDR1loxPbs for inserting the recognition sequence loxP of the Cre recombinase into the AC005045 region located very close to the MDR1 locus on human chromosome 7 and on the centromere side (about 50 Kb centromere side) is prepared as follows. did. First, the AC005045 genomic region was amplified by PCR using the following primers.
- V901 (Lexicon genetics) was used as a basic plasmid for inserting the loxP sequence.
- Gene Amp 9600 manufactured by Perkin-Elmer is used as a thermal cycler
- LATaq (Takara Shuzo) is used for Taq polymerase
- buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was.
- the temperature and cycle conditions were 94 ° C for 1 minute after heat denaturation and 35 cycles of 98 ° C for 20 seconds and 68 ° C for 7 minutes.
- the PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech). Then, it cut
- V901-MDR1HR2 was cleaved with restriction enzymes AscI (NEB) and KpnI, and the restriction vector was obtained from the cassette vector Bs-loxP-3′HPRT (Hoshiya et al., Mol Ther. 2009; 17 (2): 309-17).
- a DNA fragment containing loxP was cut out with AscI and KpnI and ligated.
- the vector having the loxP sequence in the same direction as the cloned AC005045 genomic fragment was designated as the targeting vector pMDR1loxPbs.
- the final loxP insertion construct size is 13.0 kb.
- FIG. 95 shows the targeting vector, the target sequence, and the chromosome allele resulting from homologous recombination.
- A. 3 Selection of homologous recombinants [A. 3.1] PCR analysis Genomic DNA was extracted from a blasticidin S resistant clone using Puregene DNA Isolation Kit (Gentra System), and homologous recombinants were identified by PCR using the following two sets of primers. Homologous recombinants were identified by PCR using the following two sets of primers. PCR uses Perkin-Elmer's Gene Amp 9600 as thermal cycler, Taq polymerase uses LATaq (TAKARA), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) are used according to the recommended conditions. It was.
- targeting vector pTELpuro-MDR1 for inserting a human telomere sequence into the AC003083 region located very close to the MDR1 locus on human chromosome 7 and on the telomere side (about 50 Kb telomere side) was prepared as follows. First, the AC003083 genomic region was amplified by PCR using the following primers. For PCR, Gene Amp 9600 manufactured by Perkin-Elmer was used as a thermal cycler, LATaq (TAKARA) was used as Taq polymerase, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions. .
- TAKARA LATaq
- the temperature and cycle conditions were such that after heat denaturation at 94 ° C for 1 minute, 35 cycles of 98 ° C for 20 seconds and 68 ° C for 8 minutes were performed.
- the PCR product was treated with proteinase K (Gibco) and then gel filtered with CHROMASPIN-TE400 (Clontech). After that, it was cleaved with restriction enzymes EcoRI (Nippon Gene) and PstI (Nippon Gene), and gel filtered with CHROMASPIN-TE 1000 (Clontech). This PCR fragment was cloned into the EcoRI and PstI sites of the plasmid pTELpuro (Kuroiwa et al., Nature Biotech., 20:88, 2002).
- the cloned AC003083 genome fragment was in the same direction as the human telomere sequence as the target targeting vector pTELpuro-MDR1.
- the final long arm proximal site specific cleavage construct size is 13.1 kb.
- the targeting vector, target sequence, and chromosomal allele resulting from homologous recombination are shown in FIG. [B.
- PCR analysis In order to select recombinants using the genomic DNA of a puromycin-resistant strain as a template, PCR was performed as a primary screening using the following primers located on the telomere side from the cleavage site to confirm whether site-specific cleavage occurred. The primer sequences are shown below. PCR uses Perkin-Elmer's GeneAmp9600 as thermal cycler, Taq polymerase uses Ampli Taq Gold (Applied Biosystems), and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) according to the recommended conditions Using.
- the temperature and cycle conditions were 95 ° C for 10 minutes after heat denaturation, followed by 30 cycles of 95 ° C for 20 seconds, 55 ° C for 30 seconds, and 72 ° C for 30 seconds.
- PCR was confirmed by PCR whether site-specific homologous recombination had occurred in the 3 clones that were not detected by the above primers using the following primers.
- the sequence is as follows. PCR was performed using LATaq (Takara Shuzo) with the above primers, and buffers and dNTPs (dATP, dCTP, dGTP, dTTP) were used according to the recommended conditions.
- PCR analysis In order to extract the genomic DNA of the blasticidin S resistant strain and select the recombinant as a template, PCR is performed using the following primers to confirm whether the human chromosome 7 fragment has been introduced into MAC4-containing CHO cells did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- PCR analysis In order to extract the genomic DNA of HAT-resistant strains and select reciprocal translocation clones as templates, PCR is performed using the following primers to confirm whether or not chromosomal reciprocal translocation occurs on human chromosome 7 fragment and MAC4. did.
- the primer sequences are shown below.
- GeneAmp9600 manufactured by Perkin-Elmer was used as a thermal cycler
- LA Taq (TAKARA) was used for Taq polymerase
- buffers and dNTPs dATP, dCTP, dGTP, dTTP
- Temperature and cycle conditions were heat denaturation at 94 ° C. for 1 minute, followed by 30 cycles of 98 ° C.
- mice were obtained from CHO cells (CHO (MDR1-MAC, hChr7- ⁇ MDR1) 1, 2, 4) that retained MDR1-MAC obtained in [D] above.
- the A9 cells were introduced into mouse A9 cells with high microcell forming ability by the micronucleus cell fusion method.
- mouse ES cells carrying the mouse artificial chromosome vector MDR1-MAC can be prepared, and in vitro stability can be verified using the ES cells.
- a chimeric mouse can be produced from the ES cells, and a mouse strain TC (MDR1-MAC) in which MDR1-MAC is transmitted to offspring can be produced.
- TC mouse strain
- the stability of MDR1-MAC in somatic cells can be verified using the TC (MDR1-MAC) mouse strain.
- the TC (MDR1-MAC) mouse strain can reproduce drug transport and the like in humans.
- TC CYP3A-MAC / MDR1-MAC
- TC CYP3A-MAC / MDR1-MAC
- the mouse artificial chromosome vector of the present invention has the same usefulness as the human artificial chromosome described in WO 2009/063722, but further, the retention rate is improved in rodent cells or rodent individuals. Individually maintained in a rodent cell, can stably retain a target gene (s) for a long period of time, and there is no variation in the amount of transgene between rodent individuals and tissues such as mice. Allows more accurate analysis of transgenes between cells or tissues.
- the mouse artificial chromosome vector of the present invention includes, for example, introduction of foreign genes into recipient cells, establishment of iPS cells and use in regenerative medicine, production of cells expressing foreign genes and useful non-human animals, production of proteins, It can be used for various purposes and purposes such as analysis of gene function.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- General Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Environmental Sciences (AREA)
- Immunology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Animal Husbandry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Biodiversity & Conservation Biology (AREA)
- Psychology (AREA)
- Psychiatry (AREA)
- Oncology (AREA)
- Pulmonology (AREA)
- Communicable Diseases (AREA)
Abstract
Description
本発明はまた、上記マウス人工染色体ベクターを保持する細胞に関する。
本発明はさらに、上記マウス人工染色体ベクターを保持する、マウスなどの非ヒト動物に関する。
このような問題が存在するなかで、本発明者らは、染色体レベルでの遺伝子導入という新しい染色体導入法を用いた染色体導入マウスの作製技術を開発した(非特許文献1)。この技術によりヒト染色体又はその断片をマウス胚性幹(ES)細胞に導入しキメラマウスを作製したところ、ヒト染色体断片が独立に保持され、複数のヒト遺伝子が組織特異的に発現し、減数分裂を経て断片的に子孫伝達可能なヒト染色体も存在することを示した。また本発明者らは、ヒト21番染色体全長(約35Mb)をマウスに導入し、実用的にも価値の高いダウン症候群モデルマウスを作製した(非特許文献2)。このマウスを解析したところ、導入したヒト21番染色体上の遺伝子は生理的な発現様式を再現し染色体ベクターの有効性が示された。
さらに、本発明者らは、染色体工学的手法である人工テロメア配列を使用したテロメアトランケーション技術による染色体削除、ならびに、Cre/loxPシステムによる染色体クローニングを利用し、目的の領域のみを含むヒト人工染色体の構築を可能とし、その結果、メガベース(Mb)サイズの特定ヒト染色体領域を含むヒト人工染色体(HAC,Human Artificial Chromosme)ベクターの構築に成功し、該ベクターがマウス個体で機能することも示した(非特許文献3)。さらに上述の技術を応用して既知遺伝子を含まない新規HACベクターを構築した(非特許文献4)。また、本発明者らは上記背景に基づき、目的遺伝子を搭載したHACベクターを任意の細胞へ導入し、目的遺伝子の安定的発現に成功している。さらにHACベクターのヒト型モデルマウスへの適用例として、ヒト7番染色体上の薬物代謝酵素CYP3A遺伝子クラスター(1Mb)とヒトX連鎖型筋ジストロフィーの原因遺伝子であるヒトDMD遺伝子(2.5Mb)をそれぞれHACベクターにクローニングし(CYP3A−HAC、DMD−HAC)、マウスES細胞に導入してマウスを作製した(特許文献1、非特許文献5)。
CYP3A−HACを導入したマウスの組織保持率と発現解析から、HAC上のCYP3A遺伝子クラスターはマウスの各組織において保持され(特許文献1の図8)、その発現様式はヒト組織でのパターンと同様に、肝臓、小腸特異的であった。さらにDMD−HAC導入マウスの組織保持率と発現解析の解析から、マウスのそれぞれの組織でDMD−HACは保持され(非特許文献5のFig4A)、ヒトにおいて組織特異的発現が知られているスプライシングアイソフォームの中で少なくとも3種がヒトと同様に発現することが明らかとなった。これら一連の結果から従来の遺伝子導入マウスに代わる新たな遺伝子(群)導入方法としてHAC導入マウスの有用性が示唆された。
ヒト人工染色体を含む哺乳類人工染色体ベクターは、従来のベクター系(ウイルス、YAC、BAC、PAC、コスミド、プラスミド)にはない利点を有しているために、新たな遺伝子の機能解析やヒト型モデル動物の作製系として有用であると期待されている。例えば、特許文献2及び3では、ヒト14番染色体やヒト21番染色体を改変し、そのサイズを縮小した染色体の断片からなる、細胞中で比較的安定に保持されるHACベクターが開示されている。
しかしながら、従来の遺伝子改変マウスでは不可能なMb単位の遺伝子導入を可能にしたヒト21番染色体導入マウス(ダウン症候群モデルマウス)やHACベクター導入マウスでは、これらのヒト染色体ベクターの保持率の低下、組織間や個体間にばらつきが生じること、子孫伝達の頻度が不安定などの問題が存在する。このため、HACベクターなどの保持率を常に考慮する必要があり、また、特定の遺伝子領域が持つ機能や疾患との関わりについて研究する場合、目的遺伝子の発現動態や発現産物を組織細胞レベルで詳細かつ正確に解析することが難しい場合もあり、再現性の高い均一的な解析の障害となる。
さらにまた、マウス細胞とヒト細胞を細胞融合するとき、マウス細胞内ではヒト染色体は不安定であることが知られている。このように、ヒト人工染色体ベクターを含むヒト染色体はマウス細胞内では保持率が一定しないことから、ヒト人工染色体ベクターをマウス細胞に導入する場合、及び遺伝子導入マウスを作製する場合において、人工染色体ベクターとしての利点を十分に発揮できていない。遺伝子導入マウス細胞、又は遺伝子導入マウスを作製していく上で、今後は導入遺伝子の保持率を向上及び一定にすることによって、より詳細かつ正確で再現性の高い遺伝子機能の解析又は遺伝子発現産物の効率的な回収を可能にする。
天然のヒト染色体断片をマウス細胞に移入すると、前述のとおり、ヒト染色体断片はマウス細胞内で不安定である(例えば、Shinohara et al.(2000)Chromosome Research,8:713−725)。これはマウス個体内でも同様で、マウス各組織においてヒト人工染色体は不安定であり、マウス組織細胞で保持されているヒト人工染色体の割合(保持率)が低下する傾向にあり、マウス組織細胞の保持率が一定ではなくなる。あるいは、マウス個体間においても同様で、マウス個体間でのヒト人工染色体の保持率が一定ではなくなる。このようなマウス組織間や個体間での不均一な保持率によって、ヒト人工染色体によって目的遺伝子(群)を導入したマウス細胞あるいはマウス個体を用いて導入遺伝子の詳細かつ正確で再現性の高い解析を行うことが困難である。
マウスを含めたげっ歯類由来の人工染色体については、上記のとおり、それに関連する報告は非常に少ないうえに、げっ歯類細胞内又は個体内で安定に維持される人工染色体は存在しない。本発明の目的は、導入される目的遺伝子(群)がげっ歯類細胞内又はげっ歯類個体内で安定に維持され、それにより詳細かつ正確で再現性の高い解析を可能にするマウス人工染色体ベクターを提供することにある。
(1)マウス染色体由来の天然型セントロメア、セントロメア近傍のマウス染色体長腕の部位から長腕遠位を削除したマウス染色体由来の長腕断片、及びテロメア配列を含むこと、ならびに、哺乳類の細胞及び組織において安定に保持されることを特徴とする、マウス人工染色体ベクター。
(2)マウス染色体が1~19番染色体のいずれか1つである、上記(1)記載のマウス人工染色体ベクター。
(3)マウス染色体由来の長腕断片が、マウス1~19番染色体のいずれか1つの長腕からその全内在性遺伝子数の少なくとも99.5%が削除された残部からなる、上記(1)又は(2)記載のマウス人工染色体ベクター。
(4)寄託細胞株DT40 B6bT−1(FERM BP−11128)に含まれるマウス人工染色体を基本構造として含む、上記(1)~(3)のいずれか記載のマウス人工染色体ベクター。
(5)哺乳類がげっ歯類である、上記(1)~(4)のいずれか記載のマウス人工染色体ベクター。
(6)げっ歯類がマウス、ラット又はハムスターである、上記(5)記載のマウス人工染色体ベクター。
(7)1つ又は複数のDNA配列挿入部位をさらに含む、上記(1)~(6)のいずれか記載のマウス人工染色体ベクター。
(8)DNA配列挿入部位が、部位特異的組換え酵素の認識部位である、上記(7)記載のマウス人工染色体ベクター。
(9)DNA配列挿入部位が、loxP配列、FRT配列、φC31attB及びφC31attP配列、R4attB及びR4attP配列、TP901−1attB及びTP901−1attP配列、或いはBxb1attB及びBxb1attP配列である、上記(7)又は(8)記載のマウス人工染色体ベクター。
(10)レポーター遺伝子、選択マーカー遺伝子又はその両方をさらに含む、上記(1)~(9)のいずれか記載のマウス人工染色体ベクター。
(11)外来DNA配列をさらに含む、上記(1)~(10)のいずれか記載のマウス人工染色体ベクター。
(12)外来DNA配列のサイズが200kb以上である、上記(1)~(11)のいずれか記載のマウス人工染色体ベクター。
(13)外来DNA配列がヒトDNA配列である、上記(11)又は(12)記載のマウス人工染色体ベクター。
(14)外来DNA配列が、薬物代謝関連遺伝子のDNA配列である、上記(11)~(13)のいずれか記載のマウス人工染色体ベクター。
(15)薬物代謝関連遺伝子が、第一相反応又は第二相反応に関わる酵素をコードする遺伝子である、上記(14)記載のマウス人工染色体ベクター。
(16)第一相反応に関わる酵素が、CYP1A、CYP1B、CYP2A、CYP2B、CYP2C、CYP2D、CYP2E、CYP2J、CYP3A、CYP4A、CYP4B及びそれらのサブファミリーなどのCYP、並びにCES、からなる群より選択される少なくとも1つの酵素をコードするものである、上記(15)記載のマウス人工染色体ベクター。
(17)第二相反応に関わる酵素が、UGT1及びUGT2からなる群より選択される少なくとも1つの酵素をコードするものである、上記(15)記載のマウス人工染色体ベクター。
(18)薬物代謝関連遺伝子が、トランスポーターをコードする遺伝子である、上記(14)記載のマウス人工染色体ベクター。
(19)トランスポーターをコードする遺伝子が、MDR1、MDR2、MRP2、OAT、OATP、OCT及びBCRPからなる群より選択される少なくとも1つの遺伝子をコードするものである、上記(18)記載のマウス人工染色体ベクター。
(20)薬物代謝関連遺伝子が、核内受容体をコードする遺伝子である、上記(14)記載のマウス人工染色体ベクター。
(21)核内受容体をコードする遺伝子が、PXR、AhR、CAR及びPPARαからなる群より選択される少なくとも1つの遺伝子をコードするものである、上記(20)記載のマウス人工染色体ベクター。
(22)外来DNA配列が、ヒト染色体の長腕又は短腕のDNA配列である、上記(11)~(13)のいずれか記載のマウス人工染色体ベクター。
(23)外来DNA配列が、第一相反応に関わる酵素をコードする遺伝子、第二相に関わる酵素をコードする遺伝子、トランスポーターをコードする遺伝子及び核内受容体をコードする遺伝子からなる群より選択される少なくとも2つの遺伝子の配列を含む、上記(11)~(21)のいずれか記載のマウス人工染色体ベクター。
(24)ヒト染色体が、疾患遺伝子の原因領域を含む、ヒト染色体の長腕又は短腕のDNA配列である、上記(22)記載のマウス人工染色体ベクター。
(25)外来DNA配列が、サイトカイン類、ホルモン類、成長因子類、栄養因子類、造血因子類、血液凝固・溶解因子類、イムノグロブリン類、G蛋白質共役型受容体類、酵素類などのポリペプチド類をコードする遺伝子又はDNAの配列、或いは、腫瘍、筋ジストロフィー、血友病、神経変性疾患、自己免疫疾患、アレルギー性疾患、遺伝性疾患などの疾患に関連する治療用遺伝子又はDNAの配列である、上記(11)~(13)のいずれか記載のマウス人工染色体ベクター。
(26)細胞が、肝細胞、腸細胞、腎細胞、脾細胞、肺細胞、心臓細胞、骨格筋細胞、脳細胞、骨髄細胞、リンパ球細胞、巨核球細胞、精子又は卵子である、上記(1)~(25)のいずれか記載のマウス人工染色体ベクター。
(27)組織が、肝臓、腸、腎臓、脾臓、肺、心臓、骨格筋、脳、骨髄、精巣又は卵巣由来の組織である、上記(1)~(25)のいずれか記載のマウス人工染色体ベクター。
(28)上記(1)~(27)のいずれか記載のマウス人工染色体ベクターを保持する細胞。
(29)細胞が、体細胞、非ヒト生殖系列細胞、幹細胞及び前駆細胞からなる群から選択される、上記(28)記載の細胞。
(30)幹細胞が、胚性幹(ES)細胞又は人工多能性幹(iPS)細胞である、上記(29)記載の細胞。
(31)細胞が、初代培養細胞、継代細胞又は株化細胞である、上記(28)~(30)のいずれか記載の細胞。
(32)細胞がヒト抗体産生を可能にする細胞である、上記(28)~(31)のいずれか記載の細胞。
(33)疾患治療用の外来DNA配列を含むマウス人工染色体ベクターを保持する上記(28)~(32)のいずれかに記載の細胞を含む医薬組成物。
(34)上記(1)~(27)のいずれか記載のマウス人工染色体ベクターを保持することを特徴とする、非ヒト動物。
(35)疾患モデル動物である、上記(34)記載の非ヒト動物。
(36)外来のヒト薬物代謝関連遺伝子を発現可能にする動物である、上記(34)記載の非ヒト動物。
(37)ヒト抗体を産生可能にする動物である、上記(34)記載の非ヒト動物。
(38)マウス人工染色体ベクターに含まれる外来DNAに対応する内在遺伝子が破壊されている又は内在遺伝子の発現が低下している、上記(34)~(37)のいずれか記載の非ヒト動物。
(39)外来DNA配列を含むマウス人工染色体ベクターを保持する上記(28)~(32)のいずれか記載の細胞を培養し、産生された該DNAによってコードされるタンパク質を回収することを含む、タンパク質の生産方法。
(40)ヒト抗体遺伝子を含むマウス人工染色体ベクターを保持する上記(37)又は(38)記載の非ヒト動物を用いてヒト抗体を産生し、該ヒト抗体を回収することを含む、ヒト抗体の製造方法。
(41)疾患モデル動物である上記(35)記載の非ヒト動物に候補薬剤を投与し、該薬剤の治療効果を評価することを含む、該疾患を治療するのに有効な物質をスクリーニングする方法。
(42)ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する上記(36)又は(38)記載の非ヒト動物或いは該動物由来の細胞、器官又は組織に、薬物又は食品を投与し、該薬物又は食品の薬理作用及び/又は代謝及び/又は毒性を測定することを含む、薬物又は食品の薬理作用及び/又は代謝及び/又は毒性の試験方法。
(43)ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する上記(36)又は(38)記載の非ヒト動物から得られたミクロソーム又はミクロソーム画分S9を、培養細胞若しくは細菌と、薬物及び/若しくは食品と共に培養し、薬物又は食品が該細胞又は細菌に及ぼす影響を測定することを含む、薬物又は食品の毒性の試験方法。
(44)上記(1)~(27)のいずれか記載のマウス人工染色体ベクターを使用して、200kb以上の大サイズの外来DNAをげっ歯類細胞又はげっ歯類個体内で90%以上の保持率で安定に維持させることを含む、細胞又は個体内での大サイズDNAの安定化方法。
本発明により、DNA配列挿入部位を設けたマウス人工染色体ベクターは、目的遺伝子(群)をげっ歯類細胞内又はげっ歯類個体内に導入する場合に、従来困難であったすべての細胞あるいは組織において目的遺伝子(群)を安定にかつ同一の保持率で保持させることが可能となり、また目的とする外来のDNA配列や遺伝子と共にレポーター遺伝子を挿入することができるのでベクター導入細胞を可視化でき、詳細かつ正確で再現性の高い解析又は発現産物の効率的な回収を可能にする。
本明細書は本願の優先権の基礎である日本国特許出願2010−1425号の明細書及び/又は図面に記載される内容を包含する。
図2は、実施例3の手順の模式図を示す。図中の細胞名等は次の形式で示してある。細胞名(細胞内遺伝子改変;保持染色体断片名,導入遺伝子−保持染色体名)。図中の記号等は次のとおりである。hChr7:ヒト7番染色体、CYP3A cluster:ヒトCYP3A遺伝子クラスター、5’HPRT:HPRT遺伝子の1番目と2番目のエクソン配列、loxP:部位特異的DNA配列挿入部位、hyg:ハイグロマイシン耐性遺伝子、hisD:ヒスチジノール耐性遺伝子、人工テロメア:人工テロメア(TTAGGG)繰り返し配列、EGFP:緑色蛍光タンパク発現遺伝子、neo:ネオマイシン(G418)耐性遺伝子、3’HPRT:HPRT遺伝子の3番目から9番目までのエクソン配列、puro:ピューロマイシン耐性遺伝子。
図3は、実施例4の手順の模式図を示す。図中の細胞名等は次の形式で示してある。細胞名(細胞内遺伝子改変;保持染色体断片名,導入遺伝子−保持染色体名)。図中の記号等は次のとおりである。puro:ピューロマイシン耐性遺伝子、人工テロメア:人工テロメア(TTAGGG)繰り返し配列、5’HPRT:HPRT遺伝子の1番目と2番目のエクソン配列、hyg:ハイグロマイシン耐性遺伝子、loxP:部位特異的DNA配列挿入部位。
図4は、実施例5の手順の模式図を示す。図中の細胞名等は次の形式で示してある。細胞名(細胞内遺伝子改変;保持染色体断片名,導入遺伝子−保持染色体名)。図中の記号等は次のとおりである。puro:ピューロマイシン耐性遺伝子、人工テロメア:人工テロメア(TTAGGG)繰り返し配列、neo:ネオマイシン(G418)耐性遺伝子、loxP:部位特異的DNA配列挿入部位、3’HPRT:HPRT遺伝子の3番目から9番目までのエクソン配列。
図5は、実施例6の手順の模式図を示す。図中の細胞名等は次の形式で示してある。細胞名(細胞内遺伝子改変;保持染色体断片名,導入遺伝子−保持染色体名)。図中の記号等は次のとおりである。neo:ネオマイシン(G418)耐性遺伝子、loxP:部位特異的DNA配列挿入部位、3’HPRT:HPRT遺伝子の3番目から9番目までのエクソン配列、puro:ピューロマイシン耐性遺伝子、人工テロメア:人工テロメア(TTAGGG)繰り返し配列、EGFP:緑色蛍光タンパク発現遺伝子、5’HPRT:HPRT遺伝子の1番目と2番目のエクソン配列。
図6は、マウスA9細胞(mouseA9(neo))(左)及び、マウスA9細胞とマウス線維芽細胞(mouse embryonic fibroblast(mChr11−BSr))との細胞融合クローン(mouse A9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr))を示す。
図7は、マウスCot−1 DNAをプローブとしたときのDT40(mChr11−Bsr)クローンのFISH解析の結果を示す。
図8は、ニワトリDT40細胞内に導入されたマウス染色体(mChr11−BSr)がマウス11番染色体であることを示すSKY FISHの解析結果(左)、並びに、SKY FISH染色像(右)を示す。
図9は、マウス11番染色体のAL671968領域におけるテロメアトランケーション用のベクター、及び、該ベクターにより相同組換えが行われたマウス11番染色体アレルの部分構造を示す。
図10は、pBS−TEL/puro_MACベクターを用いて、マウス11番染色体領域AL671968においてテロメアトランケーションが行われたマウス人工染色体MACのアレルを含むDT40(MAC)[DT40(B6bT−1)]クローンのmono−color FISH解析結果(右)を示す。左図のDT40(mChr11−BSr)は、テロメアトランケーション実施前であるDT40(mChr11−BSr)クローンを示す。
図11は、GFP−neo−loxP−3’HPRTタイプのloxPターゲティングベクター(pMAC1)、及び、該ベクターにより相同組換えが行われたマウス人工染色体MACのアレルの部分構造を示す。
図12は、マウスCot−1 DNA及びGFP−PGKneo−loxP−3’HPRTカセットをプローブとしたときのDT40(MAC1)クローンのtwo−color FISH解析結果を示す。
図13は、マウスCot−1 DNAをプローブとしたときのCHO(HPRT−;MAC1)クローンのmono−color FISH解析結果を示す。
図14は、マウスminor satellite DNAをプローブとしたときのmouseES(MAC1)クローンのmono−color FISH解析結果を示す。
図15は、ヒト7染色体上のCYP3A遺伝子座のごく近傍かつセントロメア側(約300Kbセントロメア側)に位置するAC004922領域にloxPを挿入するためのターゲティングベクター(pMPloxPHyg)、及び、該ベクターにより相同組換えが行われたヒト7番染色体アレルの部分構造を示す(図15a)。また、図15bは、線状化した該ベクターでトランスフェクションしたヒト7番染色体断片含有DT40細胞クローンのハイグロマイシン耐性株について、サザンハイブリダイゼーションによる相同組換え体の分析結果を示す。図15b中の矢頭について、上の矢頭が非相同組換え体(約10.9kb)を表し、下の矢頭が相同組換え体(約8.9kb)を表す。
図16は、ヒト7染色体上のCYP3A遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAC073842領域にヒトテロメア配列を挿入するためのターゲティングベクター(pTELhisD−PT)、及び、該ベクターにより相同組換えが行われたヒト7番染色体アレルの部分構造を示す。
図17は、マウスCot−1 DNA及びヒトCot−1 DNAをプローブとしたときのCHO(HPRT−,MAC1+hChr7−loxP−tel)クローンのtwo−color FISH解析結果を示す。
図18は、ヒトCYP3A遺伝子クラスター領域周辺(AC004922−ヒトCYP3A遺伝子クラスター−AC073842)の約1MbをMAC1へ転座クローニングしたマウス人工染色体CYP3A−MACの構築を示す。
図19は、ヒトCot−1 DNA及びマウスCot−1DNAをプローブとしたときのCHO(CYP3A−MAC,hChr7−ΔCYP3A)クローンのtwo−color FISH解析結果を示す。
図20は、マウス人工染色体ベクターMAC2を構築するためのターゲティングベクター(pMAC2)、及び、該ベクターにより相同組換えが行われたマウス人工染色体MACのアレルの部分構造を示す。
図21は、マウスCot−1DNA及び5’HPRT−loxP−PGK hygroカセットをプローブとしたときのDT40(MAC2)クローンのtwo−color FISH解析結果を示す。
図22は、マウスCot−1DNA及び5’HPRT−loxP−PGK hygroカセットををプローブとしたときのCHO(HPRT−;MAC2)クローンのtwo−color FISH解析結果を示す。
図23は、マウスminor satellite DNAをプローブとしたときのmouseES(HPRT−,MAC2)クローンのmono−color FISH解析結果を示す。
図24は、PGKneo−loxP−3’HPRTタイプのloxPターゲティングベクター(pMAC3)、及び、該ベクターにより相同組換えが行われたマウス人工染色体MAC3のアレルの部分構造を示す。
図25は、マウスCot−1DNA及びmouse minor satellite DNAをプローブとしたときのDT40(MAC3)クローンのtwo−color FISH解析結果を示す。
図26は、マウスCot−1 DNAをプローブとしたときのCHO(HPRT−;MAC3)クローンのmono−color FISH解析結果を示す。
図27は、マウスminor satellite DNAをプローブとしたときの薬剤耐性クローンB6(HPRT−;MAC3)のmono−color FISH解析結果を示す。
図28は、B6(HPRT−;MAC3)クローンの長期培養の保持率解析結果を示す。実線はB6(HPRT−:MAC3)−3、破線はB6(HPRT−:MAC3)−s6のMACベクター保持率をそれぞれ示す。
図29は、マウス人工染色体ベクターMAC3に特定遺伝子(例えばGFP)をCre−loxP法にて部位特異的遺伝子挿入するための手順を示し、GFP挿入用ベクター、及び、該ベクターにより相同組換えが行われたマウス11番染色体アレルの部分構造を示す。
図30は、マウスCot−1DNA及びX6.1EGFPをプローブとしたときのマウス人工染色体GFP−MACを保持するCHO細胞であるCHO(GFP−MAC)クローンのtwo−color FISH解析結果を示す。
図31は、mouse minor satellite DNA及びGFPをプローブとしたときのマウスES細胞(B6−ES株)中のマウス人工染色体GFP−MACの長期培養後のFISH解析結果を示す。
図32は、B6(GFP−MAC)クローンの長期培養の保持率解析結果を示す。菱形は薬剤選択を行った長期培養、四角は薬剤選択を行っていない長期培養の保持率をそれぞれ示す。
図33は、マウス人工染色体ベクター(GFP−MAC)を保持するキメラマウスから生まれた子孫伝達個体を示す。
図34は、長期培養後(25PDL)のCHO細胞における21HAC1または21HAC2とMAC1の保持率を示す。
図35は、長期培養後(75PDL)のES細胞における21HAC2またはMAC1の保持率を示す。
図36は、TC(MAC1)マウス(雌)組織における実体蛍光顕微鏡画像を示す。
図37は、TC(MAC1)マウスまたはTC(21HAC2)マウスの骨髄由来細胞の血液系細胞におけるGFP陽性率を示す。
図38は、TC(MAC1)マウスまたはTC(21HAC2)マウスの脾臓由来細胞の血液系細胞におけるGFP陽性率を示す。
図39は、マウスminor satellite DNAプローブを用いたTC(MAC1)マウス由来の尻尾線維芽細胞のmono−color FISH解析結果を示す。
図40は、CYP3A−BAC(RP11−757A13)ならびにマウスminor satellite DNAプローブを用いたA9(CYP3A−MAC)のtwo−color FISH解析結果を示す。
図41は、CYP3A−BAC(RP11−757A13)DNAプローブを用いたTT2F(CYP3A−MAC)のmono−color FISH解析結果を示す。
図42は、長期培養後(100PDL)のES細胞におけるCYP3A−MACの保持率を示す。
図43は、TC(CYP3A−MAC)マウス(雄)組織における実体蛍光顕微鏡画像を示す。
図44は、TC(CYP3A−MAC)マウスまたはTC(CYP3A−HACΔ)マウスの骨髄由来細胞の血液系細胞におけるGFP陽性率を示す。
図45は、TC(CYP3A−MAC)マウスまたはTC(CYP3A−HACΔ)マウスの各組織におけるCYP3A−MACまたはCYP3A−HACΔの保持率を示す。
図46は、CYP3A−BAC(RP11−757A13)DNAプローブを用いたTC(CYP3A−MAC)ヘテロマウスまたはTC(CYP3A−MAC)ホモマウスにおける、mono−color FISH解析結果を示す。
図47は、TC(CYP3A−MAC)マウスの各組織におけるCYP3A遺伝子クラスターの組織特異的遺伝子発現解析の結果を示した図である。GAPDHはグリセルアルデヒド3−リン酸デヒドロゲナーゼ(glyceraldehyde 3−phosphate dehydrogenase)を表す。
図48は、TC(CYP3A−MAC)マウス肝臓におけるCYP3A遺伝子クラスターの時期特異的遺伝子発現解析の結果を示した図である。
図49は、CYP3A−BAC(RP11−757A13)ならびにマウスCot−1 DNAプローブを用いたラットES(CYP3A−MAC)のtwo−color FISH解析結果を示す。
図50は、マウスCot−1 DNA及びヒトCot−1 DNAをプローブを用いたCHO(HPRT−;MAC1,hChr21−loxP)のtwo−color FISH解析結果を示す。
図51は、hChr21q領域の約33MbをMAC1へ転座クローニングしたマウス人工染色体hChr21q−MACの構築を示す。
図52は、ヒトCot−1 DNAおよびマウスCot−1DNAをプローブとしたときのCHO(hChr21q−MAC,hChr21−hChr21q)クローンのtwo−color FISH解析結果を示す。
図53は、ヒトCot−1 DNAならびにマウスminor satellite DNAプローブを用いたTT2F(hChr21q−MAC)クローンのtwo−color FISH解析結果を示す。
図54は、長期培養後(50PDL)のES細胞におけるhChr21q−MACの保持率を示す。
図55は、hChr21q−MACを保持するキメラマウスの蛍光写真を示す。
図56は、ヒト21番染色体(hChr21)のDSCR(ダウン症候群疾患原因領域クラスター)の近位のAP001721へloxP配列を挿入するための、ターゲティングベクター(pCKloxPHyg)、標的配列および相同組換えにより生じる染色体アレルをに示す。
図57は、DT40(hChr21q22.12−loxP)におけるサザンブロット解析結果を示す。
図58は、ヒトCot−1 DNAおよびハイグロマイシンDNAをプローブとしたときのDT40(hChr21q22.12−loxP)クローンのtwo−color FISH解析結果を示す。
図59は、ヒトCot−1 DNAおよびマウスCot−1DNAをプローブとしたときのCHO(HPRT−;MAC1,hChr21q22.12−loxP)クローンのtwo−color FISH解析結果を示す。
図60は、hChr21q22.12−qter領域の約12MbをMAC1へ転座クローニングしたマウス人工染色体hChr21q22.12−MACの構築を示す。
図61は、ヒトCot−1 DNAおよびマウスCot−1DNAをプローブとしたときのCHO(hChr21q22.12−MAC,hChr21−hChr21q22.12)クローンのtwo−color FISH解析結果を示す。
図62は、ヒトCot−1 DNAならびにマウスminor satellite DNAプローブを用いたTT2F(hChr21q22.12−MAC)クローンのtwo−color FISH解析結果を示す。
図63は、長期培養後(50PDL)のES細胞におけるhChr21q22.12−MACの保持率を示す。
図64は、長期培養後(25PDL)のCHO細胞における21HAC1または21HAC2とMAC2の保持率を示す。
図65は、1コピーFVIII−PACの構造を示す。
図66は、1から16コピーFVIII−PACの構築方法を示す。
図67は、CHO(FVIIIx1−MAC)1−3およびCHO(FVIIIx1−HAC)1−2における、長期培養後のClotting assay(FVIIIの活性比較)の結果を示す。
図68は、CHO(FVIIIx1−MAC)およびCHO(FVIIIx16−MAC)におけるClotting assayの結果(FVIIIの活性比較)を示す。
図69は、複数遺伝子搭載部位(multi−integrase platform)カセットの構築のためのentry vector構築方法を示す。
図70は、複数遺伝子搭載部位(multi−integrase platform)カセットの構築方法を示す。
図71は、MI−MACベクターの構築方法を示す。
図72は、MI−MACベクターへの遺伝子挿入方法を示す。
図73は、PXR−MACの構築方法を示す。
図74は、マウスcot−1 DNAおよびヒトPXR−BAC由来のDNA(RP11−169N13)(CHORI)をプローブとしたときのCHO(PXR−MAC)クローンのtwo−color FISH解析結果を示す。
図75は、ヒトPXR−BAC由来のDNA(RP11−169N13)(CHORI)をプローブとしたときのTT2F(PXR−MAC)クローンのmono−color FISH解析結果を示す。
図76は、GFP−5’HPRT−loxP−hygタイプのloxPターゲティングベクター(pMAC4)、及び、該ベクターにより相同組換えが行われたマウス人工染色体MAC4のアレルの部分構造を示す。
図77は、マウスcot−1 DNAおよびGFP−5’HPRT−loxP−hygカセットをプローブとしたときのDT40(MAC4)クローンのtwo−color FISH解析結果を示す。
図78は、マウスCot−1DNAをプローブとしたときのCHO(HPRT−;MAC4)クローンのmono−color FISH解析結果を示す。
図79は、ヒト4染色体上のUGT2遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAC1252392領域にヒトテロメア配列を挿入するためのターゲティングベクター(pTELpuro−UGT2)、及び、該ベクターにより相同組換えが行われたヒト4番染色体アレルの部分構造を示す。
図80は、ヒトcot−1 DNAおよびピューロマイシンDNAをプローブに用いたDT40(hChr4−tel)のtwo−color FISH解析結果を示す。左図は改変前のDT40(hChr4)を示し、右図は改変後のDT40(hChr4−tel)を示す。
図81は、ヒト4番染色体のAC074378へのloxP配列を挿入するための、ターゲティングベクター(pUGT2loxPneo)、標的配列および相同組換えにより生じる染色体アレルをに示す。
図82は、ヒトcot−1 DNAおよびネオマイシンDNAをプローブに用いたDT40(hChr4−loxP−tel)のtwo−color FISH解析結果を示す。
図83は、ヒトCot−1 DNAおよびマウスCot−1DNAをプローブとしたときのCHO(HPRT−;MAC4,hChr4−loxP−tel)クローンのtwo−color FISH解析結果を示す。
図84は、ヒトUGT2遺伝子クラスター領域周辺(AC074378−ヒトUGT2遺伝子クラスター−AC125239)2MbをMAC4へ転座クローニングしたマウス人工染色体UGT2−MACの構築を示す。
図85は、UGT2−BAC(RP11−643N16)(CHORI)DNA及びマウスCot−1 DNAをプローブとしたときのCHO(UGT2−MAC,hChr4−ΔUGT2)クローンのtwo−color FISH解析結果を示す。
図86は、UGT2−BAC(RP11−643N16)(CHORI)及びマウスminor satellite DNAをプローブとしたときのA9(UGT2−MAC)クローンのtwo−color FISH解析結果を示す。
図87は、UGT2−BAC(RP11−643N16)(CHORI)DNAをプローブとしたときのTT2F(UGT2−MAC)クローンのmono−color FISH解析結果を示す。
図88は、長期培養後(75PDL)のES細胞におけるUGT2−MACの保持率を示す。
図89は、ヒト10染色体上のCYP2C遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAL157834領域にヒトテロメア配列を挿入するためのターゲティングベクター(pTELpuro−CYP2C)、及び、該ベクターにより相同組換えが行われたヒト10番染色体アレルの部分構造を示す。
図90は、ヒトcot−1 DNAおよびピューロマイシンDNAをプローブに用いたDT40(hChr10−tel)のtwo−color FISH解析結果を示す。左図は改変前のDT40(hChr10)を示し、右図は改変後のDT40(hChr10−tel)を示す。
図91は、ヒト10番染色体のAL138759へのloxP配列を挿入するための、ターゲティングベクター(pCYP2CloxPneo)、標的配列および相同組換えにより生じる染色体アレルをに示す。
図92は、マウスCot−1 DNA及びヒトCot−1 DNAをプローブとしたときの(HPRT−;MAC4,hChr10−loxP−tel)クローンのtwo−color FISH解析結果を示す。
図93は、ヒトCYP2C遺伝子クラスター領域周辺(AL138759−ヒトCYP2C遺伝子クラスター−AL157834)380kbをMAC4へ転座クローニングしたマウス人工染色体CYP2C−MACの構築を示す。
図94は、CYP2C−BAC(RP11−466J14)(CHORI)DNA及びマウスCot−1 DNAをプローブとしたときのCHO(CYP2C−MAC,hChr10−ΔCYP2C)クローンのtwo−color FISH解析結果を示す。
図95は、ヒト7番染色体のAC005045へのloxP配列を挿入するための、ターゲティングベクター(pMDR1loxPbs)、標的配列および相同組換えにより生じる染色体アレルをに示す。
図96は、ヒト7染色体上のMDR1遺伝子座のごく近傍かつテロメア側(約50Kbテロメア側)に位置するAC003083領域にヒトテロメア配列を挿入するためのターゲティングベクター(pTELpuro−MDR1)、及び、該ベクターにより相同組換えが行われたヒト7番染色体アレルの部分構造を示す。
図97は、ヒトcot−1 DNAおよびピューロマイシンDNAをプローブに用いたDT40(hChr7M−loxP−tel)のtwo−color FISH解析結果を示す。
図98は、マウスCot−1 DNA及びヒトCot−1 DNAをプローブとしたときのCHO(HPRT−;MAC4,hChr7M−loxP−tel)クローンのtwo−color FISH解析結果を示す。
図99は、ヒトMDR1遺伝子領域周辺(AC005045−ヒトMDR1遺伝子−AC003083)210kbをMAC4ベクター転座クローニングしたマウス人工染色体MDR1−MACの構築を示す。
図100は、MDR1−BAC(RP11−784L5)(CHORI)DNA及びマウスCot−1 DNAをプローブとしたときのCHO(MDR1−MAC,hChr7−ΔMDR1)クローンのtwo−color FISH解析結果を示す。
上記のとおり、本発明は、その第1の態様において、マウス染色体由来の天然型セントロメア、セントロメア近傍のマウス染色体長腕の部位から長腕遠位を削除したマウス染色体由来の長腕断片、及びテロメア配列を含むこと、ならびに、哺乳類の細胞及び個体組織において安定に保持されることを特徴とする、マウス人工染色体ベクターを提供する。
本明細書中で使用する「マウス染色体由来の天然型セントロメア」という用語は、いずれか1つのマウス染色体のセントロメア全体(完全なセントロメア)を指す。したがって、このようなセントロメアには、マウス染色体のセントロメア配列の一部を用いて偶発的又は人工的に得られたセントロメア機能を有する構造体、及び、他の動物種の染色体のセントロメアは含まれない。
本明細書中で使用する「マウス人工染色体」又は「マウス人工染色体ベクター」はトップダウンアプローチで構築された人工染色体であり、ボトムアップアプローチで構築された人工染色体ではない。トップダウンアプローチとは、自然染色体から遺伝子領域を染色体改変により削除し天然セントロメアを人工染色体ベクターとして構築する方法である。ボトムアップアプローチとは、セントロメア配列の一部をクローン化DNAとして取得し、哺乳類細胞にトランスフェクションすることによりセントロメア機能を有する人工染色体を構築する方法である。
本明細書中で使用する「セントロメア近傍のマウス染色体長腕の部位から長腕遠位を削除したマウス染色体由来の長腕断片」は、本発明のベクターがマウスの細胞又は個体組織において安定に保持されるように、かつマウスの個体発生と子孫伝達の妨げにならないように、可能な限り内在遺伝子の影響を排除することが望ましく、そのために、マウス染色体の長腕中の内在遺伝子を除去するようにセントロメアに近い長腕部位で削除して得られる長腕断片を指す。これは全内在遺伝子(数)の少なくとも99.5%、好ましくは少なくとも99.7%、より好ましくは少なくとも99.8%、最も好ましくは99.9~100%が除去されるようにセントロメアに近い長腕部位で削除して得られる長腕断片を指す。
本明細書中の「DNA」は、特に断らない限り、遺伝子若しくは遺伝子座、cDNA、化学修飾DNAを含むすべての種類のDNA核酸に対し使用するものとする。
本明細書中で使用する「保持率」とは、培養細胞、又はマウスの組織細胞の中で人工染色体が存在している細胞の割合を指す。
本発明の染色体ベクターが「安定に保持される」とは、細胞分裂の際に該染色体ベクターの脱落を起こし難く、すなわち、分裂後であっても細胞内で安定に保持されること、それゆえに、該染色体ベクターが娘細胞や子孫マウスに効率よく子孫伝達されることを意味する。
マウス染色体は、マウス染色体1~19、X及びYのいずれでもよいが、好ましくは1番~19番染色体のいずれかである。後述の実施例では、11番染色体を例示しているが、上記の構成を有するかぎり、他の染色体であっても同様にマウス人工染色体ベクターを作製することが可能である。
マウス11番染色体断片由来の人工染色体ベクターの場合には、前記長腕断片は、非限定的に例えば、該11番染色体の長腕のAL671968、或いはBX572640(AL671968よりセントロメア側に位置する。)、CR954170(AL671968及びBX572640よりセントロメア側に位置する。)又はAL713875(AL671968よりセントロメア側に位置する。)、よりも遠位の領域が削除された長腕断片からなる。あるいは、前記長腕断片は、本明細書中でのDT40(MAC)である、寄託細胞株DT40 B6bT−1(FERM BP−11128)に含まれるマウス人工染色体を基本構造として含んでもよい(図1,3,4参照)。その他、例えばマウス15番染色体断片由来の人工染色体ベクターの場合、前記長腕断片は、非限定的に例えば、AC121307、AC161799などの位置よりも遠位の領域が削除された長腕断片からなる。マウス16番染色体断片由来の人工染色体ベクターの場合、前記長腕断片は、非限定的に例えば、AC127687、AC140982などの位置よりも遠位の領域が削除された長腕断片からなる。これらの基本構造には、外来DNA又は遺伝子を挿入するためのloxPなどのDNA配列挿入部位をさらに含むことができる(例えば、MAC1,MAC2,MAC3,MAC4など;図1,3,4参照)。
本発明のベクターは、外来DNA又は遺伝子配列を挿入するための部位を含むことができるため、この部位に、目的の外来DNA又は遺伝子を組み込むことによって、該ベクターが任意の細胞に導入されたときに該目的の外来DNA又は遺伝子を発現することが可能となり、したがって、タンパク質生産、治療用薬剤のスクリーニング、薬物代謝試験、DNAの機能解析、iPS細胞の誘導、遺伝子治療、有用な非ヒト動物の作製などへの応用に使用することが可能である(例えば、CYP3A−MAC,GFP−MACなど;図2,5参照)。
本発明のベクターはまた、マウス染色体を改変し、マウス由来の天然型セントロメアをそのままベクターの作製のために利用している。従来公知のマウス人工染色体ベクターとしては、セントロメア配列の一部を利用して作製されたサテライトDNAベースの哺乳類人工染色体(Aces及びSATACと称される)が知られているが、マウス染色体のセントロメア全体を利用して作製されたマウス人工染色体は、これまで作製されたことがない。また、上記哺乳類人工染色体はHACベクターと同様にマウス個体の組織で保持率がばらつき安定ではない(Co Doら,Chromosome Res.2000;8(3):83−91)。
本発明のベクターの有用なかつ意外な特性として、マウス、ラット、ハムスターなどのげっ歯類を含む哺乳類の細胞又は個体組織において保持率が向上し、これによって細胞内で安定に保持され、したがって目的遺伝子(群)を長期間安定に保持することができ、げっ歯類の個体間又は組織間において導入遺伝子量にバラつきがなく長期間発現させることができ、また、分化多能性細胞(例えばES細胞、iPS細胞、等)を介したげっ歯類の個体発生及び子孫伝達の効率を向上させうる、などの特性を挙げることができる。ヒト人工染色体(HAC)と比較される興味深い特性として、HACの保持率が20%未満と非常に低い血液系組織を含めて、組織間のばらつきが極端に少なく、保持率は、試験したどの組織(例えば、肝臓、腸、腎臓、脾臓、肺、心臓、骨格筋、脳又は骨髄由来の組織)でも90%以上であるし、また、本発明のマウス人工染色体は、HACと比べて効率よく繁殖可能であり、HACでは不可能であった、細胞での複数(多)コピーの保持を可能にする。
定義:
本明細書で使用する用語の定義は、当業界で使用される一般的な意味に加えて、具体的に以下の意味を包含するものとする。
本明細書中で「マウス人工染色体」又は「マウス人工染色体ベクター」と称する場合、この用語は、上記例示のようなマウス由来の染色体断片から作製された上記特徴を有する人工染色体を指し、トップダウンアプローチで構築された人工染色体であり、ボトムアップアプローチで構築されていない。繰り返しになるが、トップダウンアプローチとは、自然染色体から遺伝子領域を染色体改変により削除し天然セントロメアを人工染色体ベクターとして構築する方法である。一方、ボトムアップアプローチとは、セントロメア配列の一部をクローン化DNAとして取得し、哺乳類細胞にトランスフェクションすることによりセントロメア機能を有する構造体を構築する方法である。人工染色体は、導入すべき細胞本来の染色体から独立した染色体として安定に複製及び分配が可能である。マウス由来の染色体断片は、マウスの1~19番、X及びY染色体のうちの任意の染色体の断片(長腕の全内在遺伝子数の少なくとも99.5%が削除された長腕断片)であり、該断片には、上で定義したような、セントロメア近傍のマウス染色体長腕の部位から長腕遠位が削除された長腕断片が含まれる。本発明の人工染色体の作製については、後述の実施例、特に実施例1~5、図1~図4に記載されており、マウス11番染色体断片から人工染色体を作製する手法が例示されている。他の染色体断片からのマウス人工染色体の作製もまったく同様に実施しうる。
マウスの染色体の配列情報は、DDBJ/EMBL/GenBank、Santa Cruz Biotechnology,Inc.などのChromosome Databasesから入手可能である。
本明細書中の染色体の「長腕」とはマウス染色体のセントロメア側から遺伝子領域を含む染色体領域を指す。一方、マウス染色体には、短腕がほとんど存在しない。
本明細書中の「遠位」とは、セントロメアから遠い領域(すなわち、テロメア側)を意味する。反対に、セントロメアに近い領域(すなわち、セントロメア側)は「近位」と称する。長腕遠位は、長腕の特定部位よりもテロメア側に位置する領域を意味し、長腕近位は、長腕の特定部位よりもセントロメア側に位置する領域を意味する。この特定部位は、マウス由来の1つの染色体の長腕に存在する全内在遺伝子(数)の少なくとも99.5%、好ましくは少なくとも99.7%、より好ましくは少なくとも99.8%、最も好ましくは99.9~100%が削除される位置である。
本明細書中の「保持率」とは、培養細胞、又はマウスの組織細胞の中で人工染色体が存在している細胞の割合を指す。
本明細書中の「DNA配列挿入部位」とは、人工染色体における、目的DNA(遺伝子を含む)配列を挿入できる部位、例えば、部位特異的組換え酵素の認識部位等を意味する。このような認識部位には、非限定的に、例えばloxP(Creリコンビナーゼ認識部位)、FRT(Flpリコンビナーゼ認識部位)、φC31attB及びφC31attP(φC31リコンビナーゼ認識部位)、R4attB及びR4attP(R4リコンビナーゼ認識部位)、TP901−1attB及びTP901−1attP(TP901−1リコンビナーゼ認識部位)、或いはBxb1attB及びBxb1attP(Bxb1リコンビナーゼ認識部位)などが含まれる。
本明細書中の「部位特異的組換え酵素」とは、これら酵素の認識部位で特異的に目的のDNA配列と組換えを起こすための酵素である。その例は、Creインテグレース(Creリコンビナーゼとも称する。)、φC31インテグレース、R4インテグレース、TP901−1インテグレース、Bxb1インテグレースなどである。
本明細書中の「テロメア配列」は、同種又は異種の天然テロメア配列、或いは、人工テロメア配列である。ここで、同種とは、人工染色体ベクターの染色体断片が由来するマウスと同種の動物を意味し、一方、異種とは、該マウス以外の哺乳動物(これには、ヒトを含む)を意味する。また、人工テロメア配列は、(TTAGGG)n配列(nは、繰り返しを意味する。)などの人工的に作製されたテロメア機能を有する配列を指す。人工染色体へのテロメア配列の導入は、例えば国際公開WO 00/10383に記載されるようなテロメアトランケーション(テロメア配列の置換)によって行うことができる。テロメアトランケーションは、本発明の人工染色体の作製において染色体の短縮のために使用することができる。
本明細書中の「外来遺伝子」又は「外来DNA」とは、ベクターの遺伝子挿入部位に挿入する、ベクターに搭載する目的の遺伝子又はDNAであり、対象とする細胞内に本来的には存在しない、対象とする細胞内で発現させるべき遺伝子又はDNA、或いはそれらの配列、を意味する。
本明細書中の「哺乳動物」という用語は、ヒト、サル、チンパンジーなどの霊長類、マウス、ラット、ハムスター、モルモットなどのげっ歯類、ウシ、ブタ、ヒツジ、ヤギなどの有蹄類などを含むが、これらに限定されない。
本明細書中の「胚性幹細胞」又は「ES細胞」は、哺乳動物由来の受精卵の胚盤胞の内部細胞塊から樹立された分化多能性と半永久的増殖能とを備えた幹細胞である(M.J.Evans and M.H.Kaufman(1981)Nature 292:154−156;J.A.Thomson et al.(1999)Science 282:1145−1147;J.A.Thomson et al.(1995)Proc.Natl.Acad.Sci.USA 92:7844−7848;J.A.Thomson et al.(1996)Biol.Reprod.55:254−259;J.A.Thomson and V.S.Marshall(1998)Curr.Top.Dev.Biol.38:133−165)。この細胞と同等の性質をもつ、体細胞の再プログラミングによって人工的に誘導された細胞が「人工多能性幹細胞」又は「iPS細胞」である(K.Takahashi and S.Yamanaka(2006)Cell 126:663−676;K.Takahashi et al.(2007)Cell 131:861−872;J.Yu et al.(2007)Science 318:1917−1920)。
マウス人工染色体ベクターの作製及び用途:
以下に、本発明のマウス人工染色体ベクターの作製及びその用途について説明する。具体的には、後述の実施例1~5(図1~4)にその手順が記載されている。
(1)マウス人工染色体ベクターの作製
本発明の人工染色体ベクターは、以下の工程(a)~(c):
(a)マウス染色体を保持する細胞を得る工程、
(b)内在遺伝子(数)の大部分(99.5%以上100%以下)を含まないようにマウス染色体の長腕遠位を削除する工程、及び
(c)長腕近位に1つ以上のDNA配列挿入部位を挿入する工程
を含む方法によって作製することができる。ここで、工程(b)及び(c)の順序は逆であってもよい。
工程(a):
本発明の人工染色体ベクターを作製するには、まず、マウス染色体を保持する細胞を作製する。例えば、薬剤耐性遺伝子(例えば、blasticidin S registance gene(BSr))で標識されたマウス染色体を保持するマウス線維芽細胞であるmouse embryonic fibroblast(mChr11−BSr)とG418耐性遺伝子であるneo遺伝子を導入したマウスA9細胞(ATCC VA20110−2209)であるmouse A9(neo)と細胞融合し、薬剤耐性遺伝子で標識されたマウス染色体を保持するマウスA9雑種細胞であるmouse A9x mouse embryonic fibroblast(neo;mChr11−BSr)から、その染色体を相同組換え率の高い細胞に移入することにより作製することができる。マウス繊維芽細胞は、文献記載の方法に基づいて入手することが可能であり、例えば、マウス繊維芽細胞は日本クレアより入手可能なC57B6系統のマウスより樹立可能である。相同組換え率の高い細胞としては、例えば、ニワトリDT40細胞(Dieken et al.,Nature Genetics,12:174−182,1996)を利用できる。さらにまた、上記移入は、公知の染色体移入法、例えば、微小核細胞融合法(Koi et al.,Jpn.J.Cancer Res.,80:413−418,1973)によって行うことができる。
工程(b):
マウス由来の単一の染色体を保持する細胞において、該マウス染色体の長腕遠位を削除する。このとき、重要なことは、長腕上に存在する内在遺伝子の大部分を削除(又は、除去もしくは欠失)しマウスセントロメアを保持する人工染色体を構築することである。これは長腕上に存在する全内在遺伝子の少なくとも99.5%、好ましくは少なくとも99.7%、より好ましくは少なくとも99.8%、最も好ましくは99.9~100%を削除(又は、除去もしくは欠失)するように切断位置を決定することである。そうすることによって、人工染色体が導入された、げっ歯類などの哺乳動物由来の、好ましくはマウス由来の、細胞、組織又は個体において安定かつ高保持率で保持され、目的遺伝子(群)の正確な解析、物質生産などに用いるすることができる。上記内在遺伝子の削除は、例えば、WO 00/10383号に記載のテロメアトランケーションにより行うことができる。具体的には、マウス染色体を保持する細胞において、人工テロメア配列を保持するターゲティングベクターを構築し、相同組換えにより染色体上の所望の位置に(人工)テロメア配列が挿入されたクローンを取得し、これによってテロメアトランケーションにより欠失変異体が得られる。すなわち、所望の位置(又は、部位)が削除すべき長腕遠位の切断位置であり、この位置に人工テロメア配列が相同組換えにより置換、挿入されて長腕遠位が削除される。この位置は、ターゲティングベクターを構築する際の標的配列の設計により、適宜設定できる。例えば、後述の実施例では、マウス11番染色体長腕のAL671968(GenBank登録番号)のDNA配列に基づいて標的配列を設計し、その標的配列よりもテロメア側でテロメトランケーションが起こるように設定されている(図9参照)。これにより、内在遺伝子の大部分が削除されたマウス11番染色体断片が得られる。他の染色体の場合にも同様にテロメアトランケーションを実施できる。
工程(c):
DNA配列挿入部位として、好ましくは部位特異的組換え酵素の認識部位を挿入することができる。すなわち、ある種の酵素が特定の認識部位を認識して特異的にその認識部位でDNAの組換えを起こす現象が知られており、本発明のマウス人工染色体ベクターでは、このような酵素とその酵素の認識部位からなる系を利用して、目的とする遺伝子又はDNA配列を挿入、搭載できる。このような系として、例えば、バクテリオファージP1由来のCre酵素と、その認識部位であるloxP配列の系(Cre/loxP系;B.Sauer in Methods of Enzymology;1993,225:890−900)や、出芽酵母由来のFlp酵素と、その認識部位であるFRT(Flp Recombination Target)配列の系(Flp/FRT系)や、ストレプトミセスファージ由来のφC31インテグレースと、その認識部位であるφC31attB/attP配列の系、R4インテグレースと、その認識部位であるR4attB/attP配列の系、TP901−1インテグレースと、その認識部位であるTP901−1attB/attP配列の系、Bxb1インテグレースと、その認識部位であるBxb1attB/attP配列の系、などを挙げることができるが、DNA配列挿入部位として機能しうるのであれば、上記の系に限定されないものとする。
このような部位特異的組換え酵素の認識部位の挿入のためには、公知の方法、例えば、相同組換え法が利用でき、挿入位置及び数は、長腕近位及び短腕近位内に適宜設定することができる。
本発明においては、1つの種類の認識部位又は異なる種類の認識部位を1つ挿入することも可能である。認識部位の設定により、外来遺伝子又は外来DNAの挿入位置を特定することができるので、挿入位置が一定となり、想定外の位置効果(position effect)を受けることもなくなる。後述の実施例に例示されるマウス人工染色体の場合には、マウス11番染色体上のBX572640に挿入されている部位特異的組換え酵素の認識部位loxP配列において挿入された遺伝子を組織特異的に発現させることを可能にする(図11、図20、図24)。
本発明の、DNA配列挿入部位を有するマウス人工染色体ベクターには、好ましくは、目的とする遺伝子又はDNA配列の挿入部位を残して、レポーター遺伝子をあらかじめ挿入しておいてもよい。レポーター遺伝子としては、特に限定するものではないが、例えば、蛍光タンパク質(例えば、緑色蛍光タンパク質(GFP又はEGFP)遺伝子、黄色蛍光タンパク質(YFP)、等)、タグタンパク質コードDNA、β−ガラクトシダーゼ遺伝子、ルシフェラーゼ遺伝子などが挙げられるが、GFP又はEGFPが好ましい。
本発明のマウス人工染色体ベクターにはさらに、選択マーカー遺伝子を含んでもよい。選択マーカーは、該ベクターで形質転換された細胞を選別する際に有効である。選択マーカー遺伝子としては、ポジティブ選択マーカー遺伝子及びネガティブ選択マーカー遺伝子のいずれか、又はその両方が例示される。ポジティブ選択マーカー遺伝子には、薬剤耐性遺伝子、例えばネオマイシン耐性遺伝子、アンピシリン耐性遺伝子、ブラストサイジンS(BS)耐性遺伝子、ピューロマイシン耐性遺伝子、ゲネチシン(G418)耐性遺伝子、ハイグロマイシン耐性遺伝子などが含まれる。また、ネガティブ選択マーカー遺伝子には、例えば単純ヘルペスチミジンキナーゼ(HSV−TK)遺伝子、ジフテリアトキシンA断片(DT−A)遺伝子などが包含される。一般に、HSV−TKは、ガンシクロビル又はシクロビルと組み合わせて使用される。
本発明のマウス人工染色体ベクターに、レポーター遺伝子又は目的の外来遺伝子もしくはDNAを挿入する手法としては、相同組換え法を好ましく使用できる。相同組換えは、マウス染色体上の挿入位置の5’側領域及び3’側領域の塩基配列(各々約1~4kb、好ましくは約2~4kb)と相同な両配列(5’arm及び3’arm)の間に、挿入すべきDNAカセットを連結して得られたターゲティングベクターを用いて行うことができる。この目的で使用されるベクターとしては、例えばプラスミド、ファージ、コスミド、ウイルスなどが挙げられ、好ましくはプラスミドである。ターゲティングベクター構築のための基本プラスミドの例は、V907又はV913(Lexicon Genetics)などであるが、これらに限定されない。基本ベクターには、プロモーター、エンハンサー、選択マーカー遺伝子、複製開始点などの、ベクター構築において一般的に挿入される1つ又は2つ以上の配列又はエレメントが含まれていてもよい。
上記の手法で作製されたマウス人工染色体は、マウス由来の染色体断片(これには、天然型セントロメア、少なくとも99%、好ましくは少なくとも99.5%、の内在遺伝子が削除された長腕断片、及び(存在する場合の)短腕が含まれる。)と、人工テロメア配列とを含む。また、上記セントロメアは、人工染色体の作製のために利用されたマウスの染色体のセントロメア構造の全体である。
本発明のマウス人工染色体ベクターの例は、後述の実施例で作製されたマウス人工染色体ベクターであり、この人工染色体は、マウス11番染色体において長腕遠位をAL671968において削除して得られたベクターである(図1、図3、図4、図9、図10)。このベクターは、本明細書中でのDT40(MAC)である、寄託細胞株DT40B6bT−1(FERM BP−11128)に含まれるマウス人工染色体を基本構造として含む。基本構造であることから、このDNA構造中に以下のようなDNA配列挿入部位、選択マーカー遺伝子、外来遺伝子(又は、DNA)などを挿入することができる。
上記マウス人工染色体ベクターには、好ましくは、1つ又は複数のDNA配列挿入部位、例えば部位特異的組換え酵素の認識部位(例えばCre酵素認識部位であるloxP配列)を含む(図1、図3、図4、図11、図20、図24)。ここで、部位特異的組換え酵素の認識部位は、例えばGFP−PGKneo−loxP−3’HPRTタイプのloxP配列であるか、或いは、5’HPRT−loxP−hygタイプであるか、或いはPGKneo−loxP−3’HPRTタイプのloxP配列或いはGFP−5’HPRT−loxP−PGKhygタイプのloxP配列であるが、これらに限定されない。ここで、GFPは、緑色蛍光タンパク質遺伝子であり、PGKneoは、ホスホグリセリン酸キナーゼプロモーター/ネオマイシン耐性遺伝子カセットであり、HPRTは、ヒポキサンチン−グアニンホスホリボシルトランスフェラーゼ遺伝子であり、hygはハイグロマイシン耐性遺伝子である。
上記マウス人工染色体ベクターにはさらに、レポーター遺伝子、選択マーカー遺伝子(ポジティブ選択マーカー遺伝子、ネガティブ選択マーカー遺伝子など)を含むことができる。該ベクターにはさらに、目的の外来遺伝子又はDNA配列を含んでもよい。
本発明のマウス人工染色体ベクターの利点としては従来の人工染色体ベクターの利点である、1)宿主染色体に挿入されず独立して維持されることから、宿主遺伝子を破壊しない、2)一定のコピー数(複数(多)コピー可能)で安定に保持され、宿主細胞の生理的発現制御を受けることから、挿入された遺伝子の過剰発現や発現消失が起きない、3)導入可能なDNAサイズに制約がないことから、発現調節領域を含む遺伝子や複数遺伝子/アイソフォームの導入が可能となることに加え、げっ歯類細胞或いはげっ歯類個体中における保持率が従来の人工染色体に比べて向上すること、導入遺伝子の長期間における安定発現を実現し、かつ子孫伝達率が向上することで遺伝子導入マウスの作製効率が向上する、(4)ベクター導入後の組織間のばらつきが少なく、すなわち保持率はどの組織でも90%以上であり、HACの場合20%未満の保持率である血液系組織でさえも90%以上の保持率である、などの利点が挙げられる。
(2)外来遺伝子又はDNAの導入
本発明のマウス人工染色体ベクターには、外来遺伝子又はDNAを導入することができる。
外来遺伝子又はDNA配列のサイズは、特に制限されず、20kb以下であってもよいし、或いは20kbを超えてもよく、例えば50kb以上、100kb以上、200kb以上、500kb以上、700kb以上、1Mb以上、10Mb以上、20Mb以上、30Mb以上、40Mb以上、又は50Mb以上である。本発明のベクターは、HACベクターと同様に、BAC、PAC、YACなどの人工染色体ベクターでは困難なサイズ、すなわち1Mb以上の外来DNA(染色体断片)を搭載可能である。そのうえ、本発明のベクターは、そのように200kb以上、例えば1Mb以上、の大サイズの外来遺伝子又はDNAをHACよりも安定にかつ高保持率(90%以上)で哺乳類の細胞内、組織内又は非ヒト動物個体内、好ましくはげっ歯類の細胞内、組織内又は個体内、に保持することを可能にする。
本発明の実施形態の一つとして、本発明は、200kb以上の巨大なサイズの外来遺伝子又はDNAをげっ歯類細胞またはげっ歯類個体で90%以上の保持率で安定に維持させることが可能なベクター、並びに、該ベクターを作製する方法を提供する。
外来遺伝子又はDNAは、対象となる細胞にとって外部から導入される核酸配列であり、特に限定されるものではなく、任意の生物種由来の或いは任意の組織又は細胞由来の遺伝子又はDNAであり、好ましくは哺乳動物由来の遺伝子又はDNA、より好ましくはヒト由来の遺伝子又はDNAである。そのような遺伝子又はDNAには、サイトカイン類、ホルモン類、成長因子類、栄養因子類、造血因子類、イムノグロブリン類、G蛋白質共役型受容体類、酵素類などのポリペプチド類をコードする遺伝子又はDNA、その他、腫瘍、筋ジストロフィー、血友病、神経変性疾患(例えばアルツハイマー病、ハンチントン病、パーキンソン病、等)、自己免疫疾患、アレルギー性疾患、遺伝性疾患などを含む種々の疾患に関連する治療用遺伝子又はDNA、(ヒト)薬物代謝酵素の遺伝子(群)又はDNAや(ヒト)薬物代謝関連遺伝子、ヒト染色体の長腕又は短腕のDNA、(ヒト)ゲノムライブラリーなどが含まれるが、これらに限定されない。
サイトカイン類には、例えば、インターフェロン類(例えばIF−α,IF−β,IF−γ等)、インターロイキン類(例えばIL−1,IL−2,IL−4,IL−6,IL−11,IL−12等)、腫瘍壊死因子(例えばTNF−α,TNF−β)、TGF−βファミリータンパク質(例えば骨形成促進タンパク質(BMP)、等)などが含まれる。
ホルモン類には、例えば、成長ホルモン、、ヒト絨毛性ゴナドトロピン(hCG)、ヒト胎盤性ラクトゲン(hPL)、ヒト下垂体性性腺刺激ホルモン、甲状腺刺激ホルモン(TSH)、黄体形成ホルモン放出因子、インスリン、グルカゴン、ソマトスタチン、プロラクチンなどが含まれる。
成長因子類又は栄養因子類には、例えば、インスリン様増殖因子、脳由来神経栄養因子(BDNF)、アルブミン融合絨毛様神経栄養因子、血小板由来神経栄養因子(PDNF)、形質転換成長因子、神経成長因子(NGF)、TNF成長因子などが含まれる。
血液凝固・溶解因子類には、例えば、第VII因子、第VIII因子、第X因子、t−PAなどが含まれる。
造血因子類には、例えば、エリスロポエチン、(顆粒球)コロニー形成刺激因子、トロンボポエチンなどが含まれる。
イムノグロブリン類には、例えば、各種抗原に対するヒト抗体類、ヒト化抗体類、キメラ抗体類、合成抗体などの組換え抗体類などが含まれる。
G蛋白質共役型受容体類には、アドレナリン受容体、ムスカリン性アセチルコリン受容体、アデノシン受容体、GABA受容体(B型)、アンギオテンシン受容体、コレシストキニン受容体、ドパミン受容体、グルカゴン受容体、ヒスタミン受容体、嗅覚受容体、オピオイド受容体、セクレチン受容体、ソマトスタチン受容体、ガストリンの受容体、P2Y受容体などが含まれる。
酵素類には、例えば、アスパラギナーゼ、スーパーオキシドジスムターゼ、ウリカーゼ、ストレプトキナーゼ、ドパミン合成酵素、アデノシン・デアミナーゼなどが含まれる。
腫瘍、筋ジストロフィー、神経変性疾患(例えばアルツハイマー病、ハンチントン病、パーキンソン病、等)、自己免疫疾患、アレルギー性疾患、遺伝性疾患などを含む種々の疾患に関連する治療用遺伝子には、例えばジストロフィン遺伝子、IL−12遺伝子、TNF−α遺伝子、腫瘍抑制遺伝子、ドパミン合成系酵素遺伝子、遺伝性欠損酵素の遺伝子類、などが含まれる。
薬物代謝酵素は、薬や毒物などの異物を分解・排出するための代謝反応に関わる酵素であり、第一相反応(酸化、還元、加水分解)に関わる酵素及び第二相反応(抱合)に関わる酵素を含む。第一相反応に関わる酵素には、例えばチトクロムP450(「CYP」)などの公知の酵素、具体的にはCYP1A、CYP1B、CYP2A、CYP2B、CYP2C、CYP2D、CYP2E、CYP2J、CYP3A、CYP4A、CYP4B及びそれらのサブファミリー、並びにCES、などが含まれる。CYPサブファミリーとしては、例えばCYP3AのサブファミリーにはCYP3A4,CYP3A43,CYP3A5,CYP3A7などが含まれるし、また、CYP2CのサブファミリーにはCYP2C8,CYP2C9,CYP2C18,CYP2C19などが含まれる。ちなみに、後述の実施例に記載されるCYP3A−MACは、CYP3Aクラスターを意味しており、CYP3A4,CYP3A43,CYP3A5及びCYP3A7からなる。一方、第二相反応(抱合)に関わる酵素には、例えばUGT1及びUGT2などが含まれる。
薬物代謝関連遺伝子には、例えばトランスポーターをコードする遺伝子、核内受容体をコードする遺伝子などが含まれる。トランスポーターをコードする遺伝子の例は、MDR1,MDR2,MRP2,OAT,OATP,OCT,BCRPなどであり、核内受容体をコードする遺伝子の例は、PXR,AhR,CAR,PPARαなどである。
このように、本発明のベクターに導入可能な、薬物代謝に関係する外来DNA配列は、第一相反応に関わる酵素をコードする遺伝子、第二相に関わる酵素をコードする遺伝子、トランスポーターをコードする遺伝子及び核内受容体をコードする遺伝子からなる群より選択される少なくとも1つの遺伝子の配列、或いは、少なくとも2つの遺伝子の配列を含むことができる。
本発明のマウス人工染色体ベクターにおける外来遺伝子又は外来DNAの挿入部位の近傍又は挿入部位の両側には、少なくとも1つのインスレーター配列を存在させることができる。インスレーター配列は、エンハンサーブロッキング効果(すなわち、隣り合う遺伝子が互いに影響を受けない)又は染色体バウンダリー効果(遺伝子発現を保証する領域と遺伝子発現が抑制される領域を隔て区別する)を有する。このような配列には、例えばヒトβグロビンHS1~HS5、ニワトリβグロビンHS4などが包含される。
外来遺伝子又はDNAの導入は、上記のDNA配列挿入部位として挿入されている、上で例示されるような部位特異的組換え酵素の系を利用して行うことができる。例えば、Cre酵素の認識部位であるloxP配列と外来遺伝子又はDNAを保持するターゲティングベクター或いは、Cre酵素の認識部位であるloxP配列を挿入した外来遺伝子又はDNAを保持する染色体断片を構築し、本発明のマウス人工染色体ベクターを保持する細胞内でCre酵素を発現させることにより、loxP配列において該ターゲティングベクター或いは該染色体断片との部位特異的組換えにより、外来遺伝子又はDNAを導入できる。
本発明のマウス人工染色体ベクターには、部位特異的組換え酵素の認識部位(例えばloxP配列、FRT配列など)を保持する環状DNAを挿入することもでき、大腸菌を宿主としたプラスミドや、酵母を宿主とした環状YAC等の既存のベクターによりクローン化されたDNAも挿入できる。好適なloxP配列はP1ファージに由来する野生の配列であり、Cre酵素による人工染色体ベクター上のloxP配列への環状インサートの挿入反応は可逆的である。一旦環状インサートが挿入されると、人工染色体ベクター上に2つのloxP配列が残る。このため再度Cre酵素を発現させると環状インサートを切り出す逆反応が起きる可能性もあり、二次的にインサートを挿入するようなさらなる人工染色体ベクターの改変が困難となる。しかし、塩基置換を行った変異loxP配列やφC31インテグレースの認識部位であるattB/attP配列の組み合わせ等によっては、逆反応を起こさず、複数の環状インサートを逐次挿入する系も構築できる。
(3)マウス人工染色体ベクターの細胞への移入及び非ヒト動物の作出
本発明のマウス人工染色体ベクター、或いは、外来遺伝子若しくはDNAを含む本発明のマウス人工染色体ベクターは、任意の細胞に移入又は導入することができる。そのための手法には、例えば、微小核細胞融合法、リポフェクション、リン酸カルシウム法、マイクロインジェクション、エレクトロポレーションなどが含まれるが、好ましい手法は微小核細胞融合法である。
微小核細胞融合法は、本発明のマウス人工染色体ベクターを含有する微小核形成能を有する細胞(例えばマウスA9細胞)と、他の所望の細胞との微小核融合によって該ベクターを該他の細胞に移入する方法である。微小核形成能を有する細胞は、倍数体誘発剤(例えばコルセミド、コルヒチンなど)で処理して微小核多核細胞を形成し、サイトカラシン処理により微小核体を形成する処理を行ったのちに、所望の細胞との細胞融合を行う。
上記のベクター導入可能な細胞は、動物細胞、好ましくはヒト細胞を含む哺乳動物細胞、例えば卵母細胞、精子細胞などの生殖系列細胞、胚性幹(ES)細胞、精子幹(GS)細胞、体性幹細胞などの幹細胞、体細胞、胎児細胞、成体細胞、正常細胞、疾患細胞、初代培養細胞、継代細胞又は株化細胞など、を包含する。幹細胞には、例えばES細胞、胚性生殖(EG)細胞、胚性癌腫(EC)細胞、mGS細胞、ヒト間葉系幹細胞などの多能性幹細胞、人工多能性幹(iPS)細胞、核移植クローン胚由来胚性幹(ntES)細胞などが含まれる。好ましい細胞は、哺乳動物(好ましくは、マウスを含むげっ歯類)由来の体細胞、非ヒト生殖系列細胞、幹細胞及び前駆細胞からなる群から選択される。細胞がげっ歯類などの哺乳類由来の細胞である場合、本発明のベクターが導入された哺乳類(例えばマウスなどのげっ歯類)の細胞又は組織において、ベクターがより安定に保持される、すなわち細胞からのベクターの脱落が有意に低下する、又は脱落が起こらない。
細胞は、例えば、肝細胞、腸細胞、腎細胞、脾細胞、肺細胞、心臓細胞、骨格筋細胞、脳細胞、骨髄細胞、リンパ球細胞、巨核球細胞、精子、卵子などである。
組織は、例えば肝臓、腸、腎臓、脾臓、肺、心臓、骨格筋、脳、骨髄、精巣、卵巣などの組織である、
ES細胞は、対象動物の受精卵の胚盤胞から内部細胞塊を取出し、マイトマイシンC処理マウス胎仔線維芽細胞をフィーダーにして樹立し維持することができる(M.J.EvansとM.H.Kaufman(1981)Nature 292:154−156)。
iPS細胞は、体細胞(体性幹細胞を含む)に、ある特定の再プログラム化因子(DNA又はタンパク質)を導入し、適当な培地にて培養、継代培養することによって約3~5週間でコロニーを生成する。再プログラム化因子は、例えばOct3/4、Sox2、Klf4及びc−Mycからなる組み合わせ;Oct3/4、Sox2及びKlf4からなる組み合わせ;Oct4、Sox2、Nanog及びLin28からなる組み合わせ;あるいは、Oct3/4、Sox2、Klf4、c−Myc、Nanog及びLin28からなる組み合わせなどが知られている(K.Takahashi and S.Yamanaka,Cell 126:663−676(2006);WO 2007/069666;M.Nakagawa et al.,Nat.Biotechnol.26:101−106(2008);K.Takahashi et al.,Cell 131:861−872(2007);J.Yu et al.,Science 318:1917−1920(2007);J.Liao et al.,Cell Res.18,600−603(2008))。培養例は、マイトマイシンC処理したマウス胎仔線維芽細胞株(例えばSTO)をフィーダー細胞とし、このフィーダー細胞層上でES細胞用培地を用いて、ベクター導入体細胞(約104~105細胞/cm2)を約37℃の温度で培養することを含む。フィーダー細胞は必ずしも必要ではない(Takahashi,K.et al.,Cell 131:861−872(2007))。基本培地は、例えばダルベッコ改変イーグル培地(DMEM)、ハムF−12培地、それらの混合培地などであり、ES細胞用培地は、マウスES細胞用培地、霊長類ES細胞用培地(リプロセル社)などを使用することができる。
ES細胞及びiPS細胞は、生殖系列に寄与することが知られているので、目的の遺伝子又はDNAを含む本発明のマウス人工染色体ベクターを導入したこれらの細胞を、該細胞が由来する同種の哺乳動物の胚の胚盤胞に注入し、この胚を仮親の子宮に移植し、出産させることを含む手法によって、非ヒト動物(又は、トランスジェニック動物(ヒトを除く))を作出することができる。さらにまた、得られた雌雄のトランスジェニック動物を交配することによって、ホモ接合性動物、さらにその子孫動物を作出することができる。
本発明のマウス人工染色体ベクターを介してES細胞やiPS細胞などの分化多能性細胞、その他の上記細胞類に、ヒト抗体遺伝子、疾患治療用遺伝子、薬物代謝関連遺伝子などの外来の遺伝子若しくはDNAを導入することによって、ヒト抗体を産生可能にする細胞や非ヒト動物を作製することができるし、また、治療用タンパク質を産生可能にする細胞を作製することができるし、さらにまた、薬物代謝関連疾患などの疾患モデル非ヒト動物を作製することができる。
そのような非ヒト動物では、マウス人工染色体ベクターに含まれる外来遺伝子に対応する内在遺伝子が破壊されている又は内在遺伝子の発現が低下していることが好ましい場合もある。破壊の方法には、ジーンターゲティング法を使用することができる。内在遺伝子の発現の低下法には、RNAi法を用いることができる。例えば、そのような外来遺伝子の例には、薬物代謝関連遺伝子、ヒト抗体遺伝子などが挙げられる。内在遺伝子が破壊された非ヒト動物は、外来遺伝子を含むマウス人工染色体ベクターを保持するキメラ非ヒト動物若しくはその子孫と、対応する内在遺伝子をクラスターごと欠失させたキメラ動物若しくは子孫とを交配させて得られた該内在遺伝子がヘテロに欠失した動物同士をさらに交配させることによって作出することができる。
上記の手法によって、マウス人工染色体ベクターを保持する細胞及びトランスジェニック非ヒト動物を作製することができる。具体的な非ヒト動物の例は、マウス人工染色体ベクターを保持する、マウスやラットなどのげっ歯類である。
すなわち、本発明は、マウス人工染色体ベクターを保持することを特徴とする、細胞及び非ヒト動物を提供する。
さらにまた、本発明の非ヒト動物から得られる細胞、組織又は器官は、それらから、外来遺伝子によって発現されたタンパク質を産生する細胞株を作製するために使用することも可能である。
(4)有用なタンパク質の生産法
本発明はさらに、外来DNA配列を発現可能に含むマウス人工染色体ベクターを保持する細胞を培養し、産生された該DNAによってコードされるタンパク質を回収することを含む、タンパク質の生産方法を提供する。
タンパク質として、例えば上記の医療上、農業上などの産業上有用なタンパク質又はポリペプチド類が挙げられる。これらのタンパク質又はポリペプチド類をコードするDNAを、プロモーター(及び必要であれば、エンハンサー)の存在下で発現可能となるようにマウス人工染色体ベクターに挿入し、適当な細胞を形質転換若しくはトランスフェクションする。得られた細胞を培養し、該DNAを発現させて該タンパク質又はポリペプチドを産生させ、これを、細胞又は培地から回収する。
細胞は、哺乳動物細胞の他に、Sf細胞などの昆虫細胞、鳥類細胞、酵母細胞、植物細胞などの真核細胞の使用も可能である。
培地を含む培養条件は、細胞の種類に応じて選択され、培養条件として公知の条件を使用しうる。例えば、動物細胞の培地としては、MEM培地、DMEM培地、Ham’s F12培地、Eagle’s MEM培地、イスコフEME培地、RPMI1640培地、これらの混合培地などが含まれる。
タンパク質又はポリペプチド類の回収(又は、単離)は、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィー、親和性クロマトグラフィー、HPLC、FPLCなどのクロマトグラフィー法、塩析法、硫安沈殿法、有機溶媒沈殿法、限外ろ過法、結晶化などの公知の手段を単独で又は組み合わせて実施しうる。
(5)ヒト抗体の製造法
本発明はさらに、ヒト抗体遺伝子を含むマウス人工染色体ベクターを保持する上記の非ヒト動物を用いてヒト抗体を産生し、該ヒト抗体を回収することを含む、ヒト抗体の製造方法を提供する。
ヒト抗体遺伝子は、ヒトIgG,IgM,IgA,IgD,IgEのいずれかのクラス、或いは、ヒトIgG1,IgG2,IgG3,IgG4,IgA1,IgA2のいずれかのサブクラスをコードする遺伝子である。好ましいヒト抗体遺伝子は、IgGクラス及びそのサブクラスである。
ヒト抗体は、2本の同一配列の重鎖(H)と2本の同一配列の軽鎖(L)から構成されており、H鎖もL鎖もともに、可変領域と定常領域から構成されている。ヒトH鎖及びL鎖の可変領域はともに、3つの超可変領域(N末端側からC末端側にCDR1,CDR2,CDR3の順番)と4つのフレームワーク領域(N末端側からC末端側にFR1,FR2,FR3,FR4の順番)からなり、ヒトH鎖及びL鎖のそれぞれ3つずつのCDR配列によって抗体の特異性が決まる。
ヒトIgG抗体の場合、重鎖であるμ鎖と、軽鎖であるλ鎖若しくはκ鎖とによって構成されており、これらの抗体鎖遺伝子はそれぞれ、ヒト14番染色体、22番染色体、2番染色体上に存在する。本発明で使用するヒト抗体遺伝子としては、各抗体遺伝子座を含むヒト染色体断片を使用し、これらを異なる又は同一のマウス人工染色体に組み込む。抗体遺伝子配列は、NCBI(米国)のデータベースなどから入手可能である。この一連の手法は、例えば特開2005−230020号公報に記載される技術の改良である。
完全ヒト抗体を生産可能な非ヒト動物は、ヒトμ鎖遺伝子座を含むマウス人工染色体ベクターを保持する非ヒト動物と、ヒトλ鎖遺伝子座及び/又はヒトκ鎖遺伝子座を含むマウス人工染色体ベクターを保持する同種の非ヒト動物とを交配することによって、両方のH鎖及びL鎖遺伝子座を保有するキメラ非ヒト動物及びその子孫動物を得ることが可能である。
このようにして作製された完全ヒト抗体産生可能な非ヒト動物(例えばマウスなどのげっ歯類)に、ある特定の抗原ペプチド若しくは抗原ポリペプチドを免疫し、該動物の血液からヒト抗体を分離することを含む方法により、ヒト抗体を生産することができる。
或いは、ある特定の抗原で免疫した非ヒト動物の脾臓を摘出し、ミエローマ細胞と融合させ、モノクローナル抗体を産生するハイブリドーマを得ることも可能である。
(6)治療物質のスクリーニング法
本発明はさらに、疾患モデル動物である上記非ヒト動物に候補薬剤を投与し、該薬剤の治療効果を評価することを含む、該疾患を治療するのに有効な物質をスクリーニングする方法を提供する。
疾患モデル非ヒト動物は、ある種のタンパク質の欠損、変異などによる生物機能異常、薬物代謝異常、染色体異常などの異常を原因とする疾患を持つ人工的に作製された動物である。染色体異常をもつモデル非ヒト動物の例は、以下に限定されないが、ヒト18番又は21番染色体トリソミーをもつ動物である。
このような非ヒト動物は、遺伝子又は染色体に上記のような異常を含む遺伝子又は染色体断片を作製し、これを本発明のマウス人工染色体ベクターに組み込み、ES細胞又はiPS細胞に導入し、受精卵の胚盤胞に注入し、この細胞を非ヒト動物の仮親の子宮に移植し、出産させることを含む方法によって作出することができる。
上記のようにして作製された非ヒト動物に、候補薬剤を投与し、該薬剤の治療効果を評価することによって、該疾患を治療するのに有効な物質をスクリーニングすることができる。
候補薬剤は、非限定的に、例えば、低分子化合物、高分子化合物、(糖)タンパク質、ペプチド、(リン又は糖)脂質、糖類、などである。
(7)薬物又は食品の薬理作用、代謝又は毒性の試験方法
本発明の実施形態によれば、本発明はまた、ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する上記非ヒト動物或いは該動物由来の細胞、器官又は組織に、薬物又は食品を投与し、該薬物又は食品の薬理作用及び/又は代謝及び/又は毒性を測定することを含む、薬物又は食品の薬理作用及び/又は代謝及び/又は毒性の試験方法を提供する。
本発明はまた、ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する上記非ヒト動物から得られたミクロソーム又はミクロソーム画分S9を、培養細胞若しくは細菌及び薬物若しくは/及び食品と共に培養し、薬物又は食品が該細胞又は細菌に及ぼす(悪)影響(例えば変異、等)を測定することを含む、薬物又は食品の毒性の試験方法を提供する。
ヒト薬物代謝関連遺伝子は、上に例示したものである。また、非ヒト動物の作出方法も上に記載したものと同様である。
ヒト薬物代謝関連遺伝子を有するマウス人工染色体ベクターを保持する上記の非ヒト動物を用いる上記方法では、例えば、該動物の状態の観察、臓器や染色体に及ぼす影響の試験などによって、薬物又は食品の薬理作用、代謝又は毒性を判定することができる。
本発明のもうひとつの方法では、該非ヒト動物から得られたミクロソーム若しくはミクロソーム画分S9(9000g画分であって加水分解、還元、酸化、結合などを触媒する多数の酵素を含む画分)を、培養細胞(特に、動物細胞、好ましくは哺乳類細胞)又は細菌(好ましくは、サルモネラ菌)と一緒に、薬物及び/又は食品の存在下で培養する。薬物又は食品の、細胞に対する毒性の検出は、エイムス試験又は小核試験によって行うことができる。エイムス試験では、サルモネラ菌の変異に基づいて毒性を判定する。小核試験では、細胞核の染色体の異常に基づいて毒性を判定する。これらの試験法はよく知られており、本発明の方法で使用しうる。
以下、実施例を挙げて本発明をさらに詳しく説明するが、本発明の範囲はこれらの具体例に限定されるものではない。
マウス人工染色体ベクターMACの構築
マウス染色体にテロメアトランケーションを行うことで内在遺伝子を含まないマウス人工染色体MAC[DT40(B6bT)]を構築する(図1)。
[A]A9細胞とマウス線維芽細胞(neo;mChr11−BSr)のハイブリッド細胞樹立
薬剤耐性遺伝子(Bsr遺伝子)で標識されたマウス11番染色体を含有するマウス線維芽細胞であるmouse embryonic fibroblast(mChr11−BSr)と公知のマウスA9細胞にG418耐性遺伝子であるneo遺伝子を挿入したmosue A9(neo)と細胞融合し、薬剤耐性遺伝子で標識されたマウス染色体を保持するマウスA9雑種細胞であるmouse A9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr)を樹立する。薬剤耐性遺伝子で標識されたマウス染色体を微小核細胞融合法により相同組換頻度の高いニワトリDT40細胞へ導入するために、微小核形成率が高いことが知られているマウスA9細胞に薬剤耐性遺伝子で標識されたマウス染色体を細胞融合によって導入する。
[A.1]細胞融合及び二重薬剤耐性クローンの単離
日本クレアより入手可能なC57B6系統のマウス胎仔より樹立し、薬剤耐性遺伝子(Bsr遺伝子)をマウス染色体上に挿入したマウス繊維芽細胞であるmouse embryonic fibroblast(mChr11−BSr)と、G418耐性遺伝子であるneo遺伝子が挿入されているマウスA9細胞であるmosue A9(neo)をそれぞれPBS(−)で細胞表面を洗浄後、トリプシン添加によって細胞を分散し、培養液(10%FBS、DMEM)に懸濁し、それぞれの細胞1x106個を培養用フラスコ(25cm2)に同時に植え込み、一日間培養する。PBS(−)で細胞表面を2回洗浄した後に3mlのPEG(1:1.4)溶液[5g,PEG1000,cat:165−09085,wakoを無血清DMEM6mlに溶解させ、ジメチルスルホキシド1mlを加えて濾過滅菌する]で1分間処理し、さらに3mlのPEG(1:3)溶液[5g,PEG1000,cat:165−09085,wakoを無血清DMEM15mlに溶解させて濾過滅菌する]に換えて1分間処理した。PEG溶液を吸引後、無血清DMEMで3回洗浄し、通常の培養液(10%FBS、DMEM)で一日間培養した。PBS(−)で細胞表面を洗浄後、トリプシン添加によって細胞を分散し、G418(800μg/ml)及びブラストサイジンS(4μg/ml)を含む二重選択培養液(10%FBS、DMEM)に懸濁した細胞をプラスチック培養皿に植え込み、2~3週間選択培養した。2回の細胞融合で得た合計3個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:mouse A9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr))。
[A.2]雑種細胞の選択
[A.2.1]PCR
二重薬剤耐性クローンからゲノムDNAを抽出して、以下のプライマーを用いてPCRを行い、薬剤耐性遺伝子(Bsr遺伝子)で標識されたマウス染色体が保持されていることを確認した。

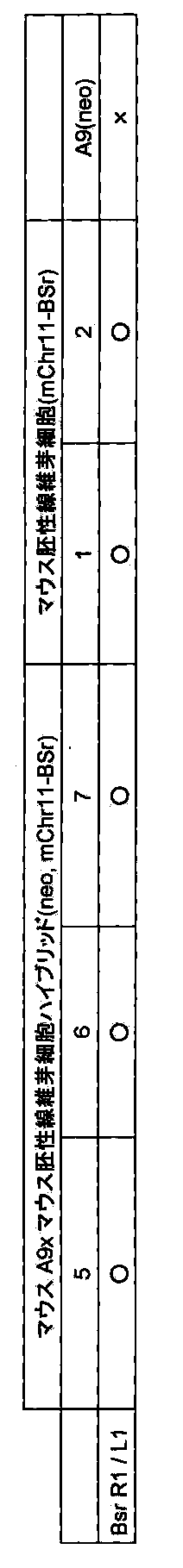
上記PCR解析によって陽性であったクローンについて、キナクリン・ヘキスト二重染色を行った。キナクリン・ヘキスト二重染色は、まず、50mlのマキルベン溶液[クエン酸一水和物11.18gとリン酸水素二ナトリウム13.29gを1Lの水に溶解させ、オートクレーブする]に染色体スライドを浸し、ついで50mlのマキルベン溶液に60μg/mlになるようにキナクリン[cat:Q2876,SIGMA]を溶解したものに20分間浸し、水道水で染色体スライドの裏面を洗浄後、マキルベン溶液に浸し、50mlのマキルベン溶液に0.5μg/mlになるようにヘキスト[cat:B−2883]を溶解したものに15分浸したあとに、カバーガラスで覆った。蛍光顕微鏡により観察したところ、3クローンすべてにおいて通常の核型が2nのところが大半の核型が4n以上になっていた。特に、クローンA9(21−B6b)7において薬剤耐性遺伝子(Bsr遺伝子)で標識されたマウス染色体を保持するマウス線維芽細胞と、G418耐性遺伝子であるneo遺伝子が挿入されているマウスA9細胞が一対一で細胞融合したことがわかった(図6)。

[B]薬剤耐性遺伝子で標識されたマウス染色体のDT40細胞への導入
薬剤耐性遺伝子で標識されたマウス染色体を含有するマウスA9雑種細胞であるmouse A9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr)から薬剤耐性遺伝子で標識されたマウス染色体をニワトリDT40細胞であるDT40へ導入する。マウス染色体番号の同定、及び人工テロメアの挿入による染色体部位特異的切断であるテロメアトランケーション、及び相同組換によりマウス染色体にDNA配列挿入部位であるloxP配列の挿入を効率的に行うために、薬剤耐性遺伝子で標識されたマウス染色体を微小核細胞融合法により相同組換頻度の高いニワトリDT40細胞であるDT40へ導入する。
[B.1]微小核細胞融合及び薬剤耐性クローンの単離
染色体同定と染色体改変を効率的に行うために、マウス染色体をA9雑種細胞クローンであるA9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr)7から相同組換え頻度の高いニワトリDT40細胞であるDT40へ移入した。フラスコ×24で培養していたドナー細胞であるA9xmouse embryonic fibroblast hybrid(neo;mChr11−BSr)7がそれぞれのフラスコにおいて70%コンフルエントになった時点で、コルセミド処理(コルセミド0.05μg/ml、20%FCS、DMEM)を37℃5%CO2の条件下にて48時間行った。コルセミド処理が終了したら、フラスコ内のmediumをアスピレートし、そのフラスコの9分目までをサイトカラシンBで満たした。フラスコを大型高速遠心機(BECKMAN)専用の容器に挿入し、温湯(34℃)をフラスコが隠れない程度に加え、遠心(Rortor ID10.500、8,000rpm、1h、34℃)をした。遠心終了後、サイトカラシンBを回収し、各フラスコ内のペレットを、それぞれ2mlの無血清培地DMEMにて15mlチューブに回収した。8μm→5μm→3μmフィルターの順にゆっくりとフィルトレーションした後、それぞれのチューブを遠心(1,200rpm5分R.T)し、上清をアスピレートした後、各チューブのペレットをまとめて5mlの無血清培地DMEMに回収、懸濁し、遠心(2000rpm5分)をした。
レシピエント細胞であるDT40細胞は浮遊細胞であるため、一度付着状態にする必要がある。DT40を6穴プレート(Nunc)のうちの1穴に付着させるために50μg/mlに調整したポリ−L−リジン(SIGMA)1.5mlで1穴を37℃、オーバーナイトでインキュベートすることでコーティングした。ポリ−L−リジンを回収し、プレートをPBS(‐)で洗浄し、約1x107個のDT40細胞を2mlの無血清培養液(DMEM)でプレートに静かに播種した。プレートごと遠心機(Beckman)にセットし、37℃、1200rpm、3分間遠心して付着DT40にした。
精製した微小核細胞をPHA−P(SIGMA)を含む無血清培養液2mlに再度懸濁し、無血清培養液(DMEM)を除去した付着DT40上に静かに播種した。プレートを37℃、1200rpm、3分間遠心した。上清を除去し、PEG1000(Wako)[5gのPEG1000を無血清DMEM培地に完全に溶解し、ジメチルスルホキシドを1ml添加して濾過滅菌する]溶液を1mlで正確に1分間融合した。無血清培養液(DMEM)を4mlで4回洗浄し、通常のDT40の培養液3mlでピペッティングして、付着DT40を浮遊状態に戻し、37℃、24穴プレート2枚に播種し、オーバーナイトでインキュベートした。ブラストサイジンSを1500μg/mlになるように加え、3~4週間選択培養した。1回の微小核細胞融合で得た合計2個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:DT40(mChr11−BSr))
[B.2]薬剤耐性クローンの選別
[B.2.1]FISH解析
上記で得られたDT40(mChr11−BSr)のクローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところDT40(mChr11−BSr)−1において、95%で正常核型(2n)あたりのマウス染色体数が1コピーであったため、以降の解析を行った(図7)。
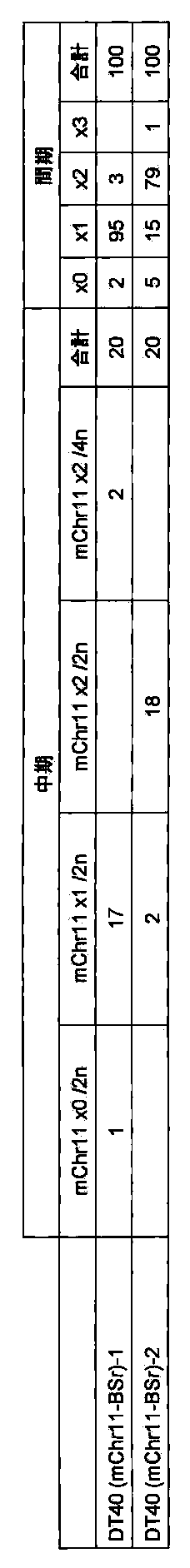
Kaiら(Cell Res,19:247−58,2009)の報告に記された方法でSKY−FISHを行ったところ、ニワトリDT40細胞に導入されたマウス染色体はマウス11番染色体であることがわかった(図8)。
[C]ニワトリDT40細胞内におけるマウス11番染色体領域AL671968から遠位のテロメアトランケーションによる部位特異的切断
マウス人工染色体ベクターとして導入目的遺伝子以外の内在遺伝子は少ない方が実験系に及ぼす影響が軽減され、かつ内在遺伝子のうちインプリント遺伝子のようにマウス個体発生に遺伝子発現量の変化による影響を及ぼす遺伝子を極力残さないことが必要であるので、マウス長腕の大部分を削除する。
[C.1]テロメアトランケーションベクター作製
短腕近位部位特異的切断用の基本ベクターにはpBS−TEL/puroコンストラクト(Kuroiwa et al.Nature Biotech 2002)を用いた。GenBankデータベースより得たマウス11番染色体長腕近位の塩基配列(AL671968)から相同組換え標的配列を設計した。DT40(mChr11−BSr)からゲノムDNAを抽出して鋳型とし、相同組換え標的配列をPCR増幅するためのプライマーの配列を以下に示す。
[C.2]相同組換体の選別
マウス11番染色体領域AL671968から遠位において部位特異的切断を行うベクターは上述のpBS−TEL/puro_MACを用いてトランスフェクションを行い、ピューロマイシン耐性及びブラストサイジンS非耐性クローンの単離及び相同組換え体の選別を行った。その結果、マウス11番染色体領域を切断できた5クローンを確認した(クローン名:DT40(MAC))。ターゲティングベクター、標的配列、及び相同組換えにより生じる染色体アレルを図9に示した。
[C.3]mono−color FISH解析による薬剤耐性クローンの選別
上記で得られたDT40(MAC)の5クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところ5クローンうち2クローンにおいてマウス11番染色体の長腕部分がセントロメア近傍で切断されていることを確認した(図10)。
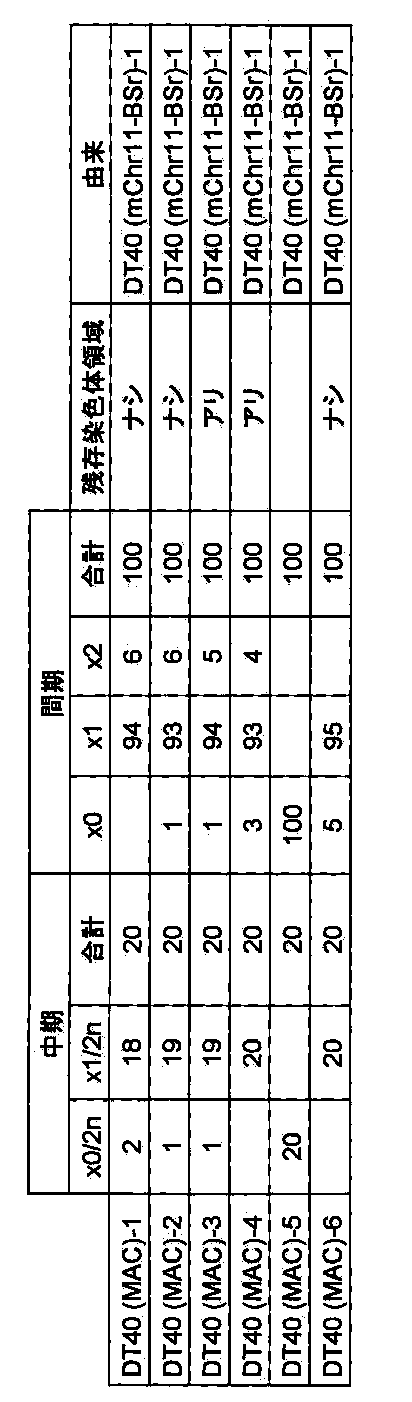
[実施例2]マウス人工染色体ベクターMAC1の構築
マウス人工染色体MACにDNA挿入配列としてGFP−PGKneo−loxP−3’HPRTタイプのloxP配列を挿入することでマウス人工染色体ベクターMAC1を構築し、MAC1のマウスES細胞での安定性を検証し、さらにMAC1を導入した子孫伝達マウスを作製することで、個体組織での安定性を検証する。
[A]マウス人工染色体ベクターMACへのGFP−PGKneo−loxP−3’HPRTタイプのloxP配列の挿入
[A.1]GFP−PGKneo−loxP−3’HPRTタイプのloxPターゲティングベクター作製
DT40(MAC)にloxP配列を挿入するための基本プラスミドにはV913(Lexicon genetics)を用いた。loxP挿入部位であるマウス11番染色体のDNA配列はGenBankデータベースより得た(BX572640.9)。薬剤耐性クローンからゲノムDNAを抽出して鋳型とし、相同組換えの二つの標的配列の増幅に用いたプライマーの配列を以下に示す。
それぞれのPCR産物をBglII(TAKARA)で消化して、アガロースゲルにより分離し精製後、V913のBglIIあるいはBamHIサイトにクローニングした(ベクター名:VH21−12)。3’HPRT−loxPはV820(Lexicon genetics)のXbaIサイトにオリゴ合成したloxP配列をクローニングした。HPRT遺伝子の3番目から9番目のエクソンである3’HPRT−loxPをV907(Lexicon genetics)のEcoRIとAscIにクローニングした(ベクター名:X3.1)。さらにX3.1のKpnIサイトとEcoRIサイトにKpnIとNotIにより切り出したPGKneo配列をクローニングした(ベクター名:X4.1)。X4.1からKpnIとAscIにより切り出したPGKneo−loxP−3’HPRTをV913のKpnIサイトとAscIサイトにクローニングした(ベクター名:p VNLH)。pVNLHのEcoRVサイトにNotIとSalI消化後に平滑化をおこなったHS4−CAG−EGFP−HS4(大阪大学、岡部博士及びNIH、Felsenfeld博士より分与)をクローニングした(ベクター名:p VGNLH)。p VGNLHからSalIとAscIにより切り出したGFP−PGKneo−loxP−3’HPRTカセットをVH21−12のXhoIサイトとAscIサイトにクローニングした(ベクター名:pMAC1)。ターゲティングベクター、標的配列および相同組換えにより生じる染色体アレルを図11に示す。
[A.2]トランスフェクションおよびG418耐性クローンの単離
ニワトリDT40細胞の培養は10%ウシ胎仔血清(ギブコ、以下FBSで記す)、1%ニワトリ血清(ギブコ)、10−4M 2−メルカプトエタノール(シグマ)を添加したRPMI1640培地(ギブコ)中で行った。DT40(MAC)−1の約107個の細胞を無添加RPMI1640培地で一回洗浄し、0.5mlの無添加RPMI1640培地に懸濁し、制限酵素NotI(TAKARA)で線状化したターゲティングベクターpMAC1を25μg加え、エレクトロポレーション用のキュベット(バイオラッド)に移し、室温で10分間静置した。キュベットをジーンパルサー(バイオラッド)にセットし、550V、25μFの条件で電圧印加した。室温で10分間静置後、24時間培養した。G418(1.5mg/ml)を含む培地に交換し、96穴培養プレート2枚に分注して約2週間の選択培養を行った。2回のトランスフェクションで得た合計14個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(MAC1))
[A.3]相同組換体の選別
[A.3.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス11番染色体上で部位特異的に組換え起こっているかを確認した。そのプライマー配列を以下に示す。
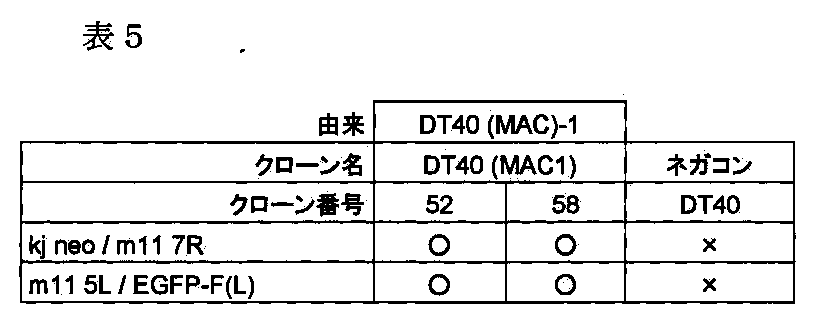
上記から得られたDT40(MAC1)−52とDT40(MAC1)−58において、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNAおよびGFP−PGKneo−loxP−3’HPRTカセットをプローブにしてFISH解析を行ったところ、loxP配列がターゲティングされたマウス11番染色体断片のセントロメア付近にプローブ由来のFITCシグナルが検出され、かつネガティブコントロールのターゲティングする前のマウス11番染色体断片(例えばDT40(MAC)−1)では現れなかったシグナルが検出されたことから、部位特異的に組換えが起こったことが視覚的に確かめられた(図12)。これらの結果から、マウス人工染色体ベクターMAC1を保持するDT40細胞クローンが得られたと結論できた。
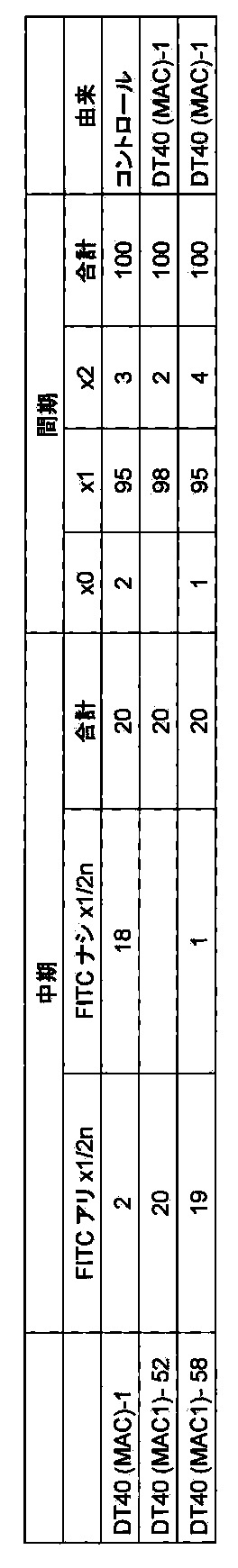
CHO細胞を介してマウス人工染色体ベクターMAC1をマウスES細胞に導入するため、或いはCHO細胞内でマウス人工染色体ベクターMAC1のDNA配列挿入部位であるloxPを介して安定に目的遺伝子(群)等、例えばCYP3Aクラスターなどを挿入するため、CHO細胞に導入する。
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(MAC1)52及び58を用いて、上記と同様にCHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)であるCHO(HPRT−)に微小核細胞融合法を行った。2回の微小核細胞融合で得た合計24個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC1))
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工番染色体MAC1がCHO細胞に導入できているかを確認した。そのプライマー配列を以下に示す。

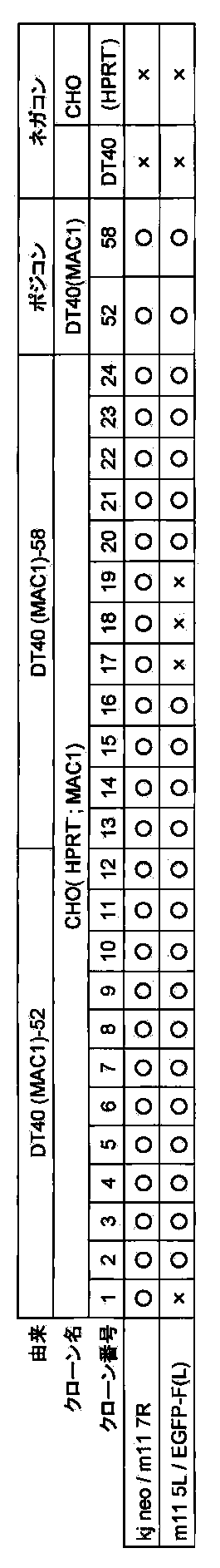
上記で得られたCHO(HPRT−;MAC1)の20クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところ20クローン中、5クローンで95%の割合でマウス人工染色体ベクターMAC1がCHO細胞に導入されていることを確認した(図13)。
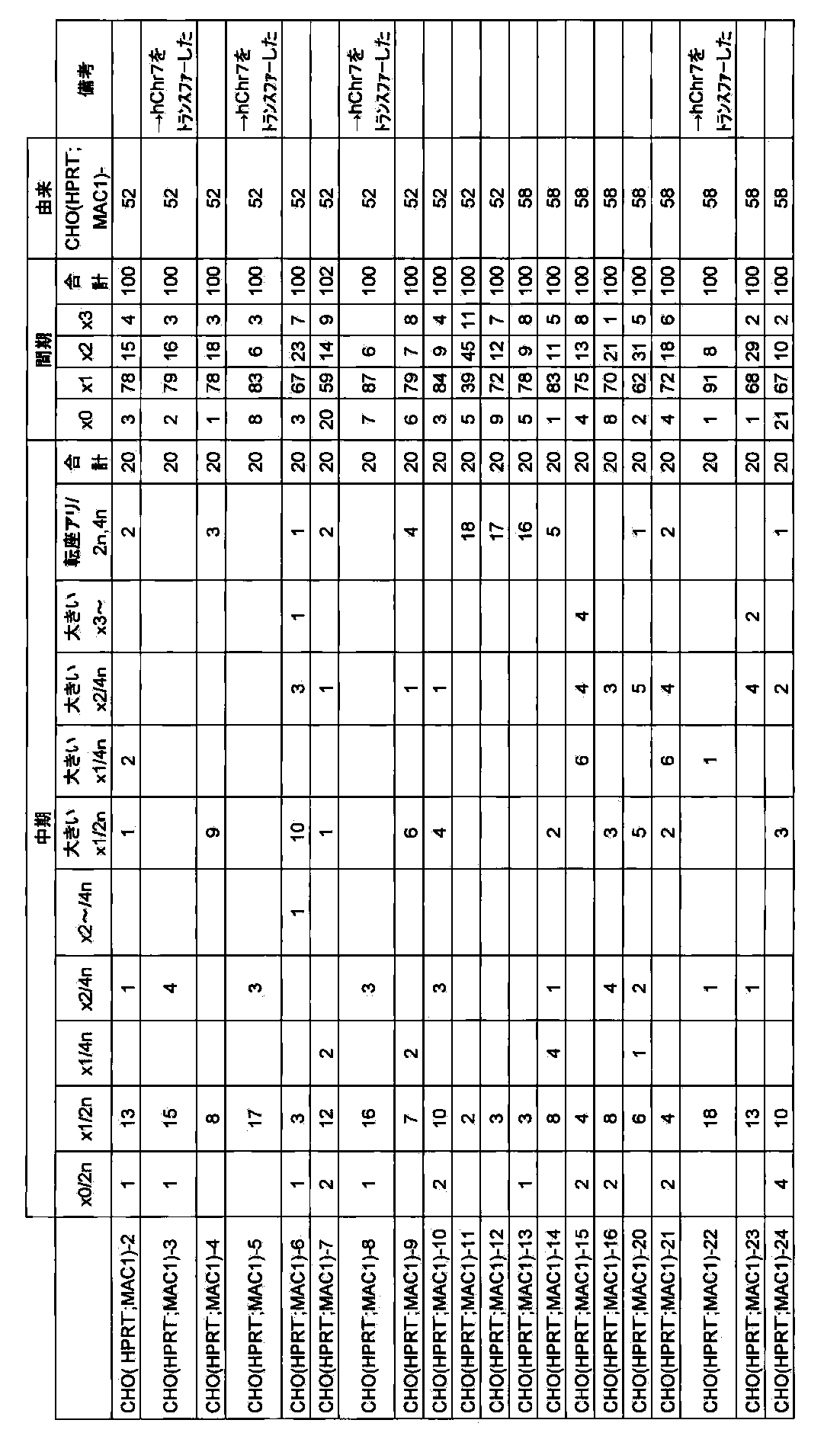
[C]マウス人工染色体ベクターMAC1含有CHO細胞からマウスES細胞へマウス人工染色体ベクターMAC1の導入
マウスES細胞及びマウス個体内でのマウス人工染色体ベクターMAC1の安定性を検証するために、マウス人工染色体MAC1をマウスES細胞に導入し、マウス人工染色体ベクターMAC1含有キメラマウスならびに子孫伝達マウスを作製する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるCHO(HPRT−;MAC1)−3,5,8,22を細胞培養皿で培養し、コンフルエントになった時点で20% FBS、0.1μg/mlコルセミドを添加したF12培地に交換し、さらに48時間培養後に20% FBS、0.1μg/mlコルセミドを添加したF12培地で培地交換し、さらにオーバーナイトでインキュベートしてミクロセルを形成させた。培養液を除去し、予め37℃で保温したサイトカラシンB(10μg/ml,シグマ)溶液を遠心用フラスコに満たし、34℃、8000rpm、1時間の遠心を行った。ミクロセルを無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心し、無血清DMEM培地5mlに懸濁した。ミクロセルを5mlの無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心した。
ドナー細胞にはC57B6系統マウスのES細胞であるB6−ES、及びB6−ES細胞に6TG処理を行ったHPRT欠損株であるB6(HPRT−)、及びC57B6xCBA系統F1マウスのES細胞であるTT2F、及びTT2F細胞に6TG処理を行ったHPRT欠損株であるKO56(HPRT−)を用いた。培養には、DMEM(Dulbecco’s Modified Eagle’s Medium−high glucose:SIGMA)に、10%FCS、LIF(Muerin Leukemia Inhibitory Factor)、1×10−5M 2−ME(2−メルカプトエタノール:SIGMA)、L−グルタミン(3.5g/ml:GIBCO)、Sodium pyruvate solution(3.5g/ml:GIBCO)、MEM Nonessential amino acid(0.125mM:GIBCO)を添加し、5% CO2、37℃にて培養をおこなった。マウスES細胞をPBS(−)で細胞表面を2回洗浄後にトリプシン処理により細胞を分散させ、DMEM培地に10%FBSを添加した培養液で回収し、1500rpmで遠心し、上清を除去し、無血清培養液5mlに再度懸濁し、ミクロセルの遠心後のペレットを含む無血清培地に静かに添加し、さらに1200rpmで遠心した。上清を除去し、PEG1000(Wako)溶液[5gのPEG1000を無血清DMEM培地に完全に溶解し、ジメチルスルホキシドを1ml添加して濾過滅菌する]を0.5mlで正確に1分30秒間融合した。13mlの無血清培養液(DMEM)を静かに添加し、1200rpmで遠心した。上清を除去し、通常のマウスES細胞の培養液を添加し、マイトマイシン処理したG418耐性マウス胎生線維芽細胞をフィーダー細胞として使用し、直径10cm細胞培養皿2枚に播種し、オーバーナイトでインキュベートした。G418を250μg/mlになるように加え、3~4週間選択培養した(クローン名:B6−ES(MAC1)及びB6(HPRT−;MAC1)及びKO56(HPRT−;MAC1))。B6−ES(MAC1)及びB6(HPRT−;MAC1)及びKO56(HPRT−;MAC1)については、それぞれ2回の微小核細胞融合で得た合計32個の耐性コロニーを単離し増殖させ、以降の解析を行った。TT2F(MAC1)については、4回の微小核細胞融合で得た合計30個の耐性コロニーを単離し増殖させ、FISH解析以降の解析を行った
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工番染色体MAC1がマウスES細胞に導入できているかを確認した。そのプライマー配列を以下に示す。

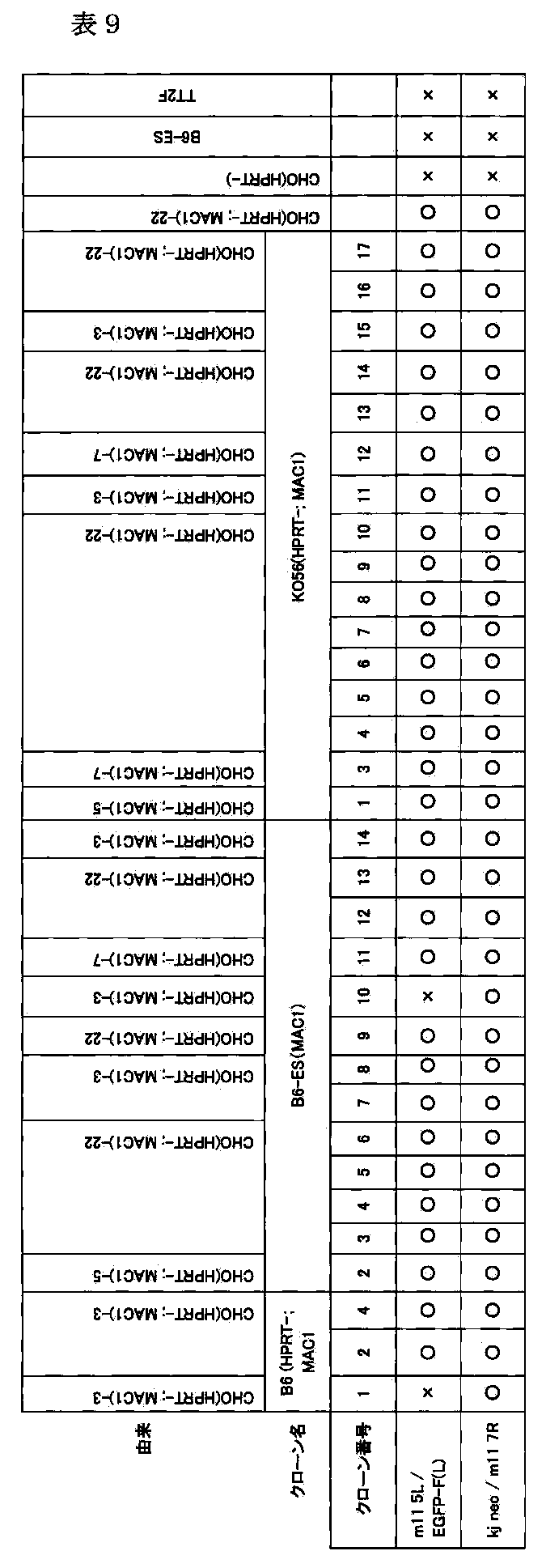
上記で得られたB6−ES(MAC1)及びB6(HPRT−;MAC1)及びKO56(HPRT−;MAC1)のクローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスminor satellite DNAをプローブにしたFISH解析を行ったところ14クローン中、全てのクローンで85%以上の割合でMAC1がマウスES細胞に導入されていることを確認した。また、正常核型である内在マウス染色体の本数がB6−ESの場合40本、またはKO56の場合39本であることを確認した。B6(HPRT−;MAC1)の場合、40本の核型クローンは得られなかった。また、同様にTT2F(MAC1)については、7クローンについて解析し、7クローン中全てのクローンで、90%以上の割合で導入されていることを確認した。また、また、正常核型である内在マウス染色体の本数が39本であることを、7クローン中3クローンについて確認した。
以上の結果から、マウス人工染色体ベクターMAC1がマウスES細胞に導入できたと結論付けた(図14)。

マウス人工染色体ベクターMAC1にヒト薬物代謝酵素遺伝子群であるCYP3AクラスターをCre/loxPシステムを用いて転座クローニングを行い、CYP3A−MACを構築する。また、CYP3A−MACのマウスES細胞での安定性を検証し、さらにCYP3A−MACを導入した子孫伝達マウスを作製することで、個体組織での安定性を検証する。また、子孫伝達マウスにおいて、CYP3A遺伝子の組織特異的遺伝子発現を検証する(図2)。
[A]ヒト7番染色体のAC004922へのloxP配列の部位特異的挿入
マウス人工染色体ベクターMAC1にloxP配列を介して転座挿入するために、DT40細胞内でヒト7番染色体(hChr7)のCYP3A遺伝子クラスターの近位のAC004922へloxP配列を挿入する。
[A.1]ターゲティングベクターpMPloxPHygの作製
ヒト7番染色体上のCYP3A遺伝子座のごく近傍かつセントロメア側(約300Kbセントロメア側)に位置するAC004922領域にCre組換え酵素の認識配列loxPを挿入するためのターゲティングベクターpMPloxPHygを以下のようにして作製した。まず、AC004922ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
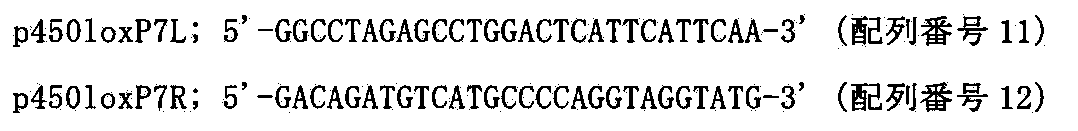
[A.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpMPloxPHygを制限酵素NotI(TAKARA)で線状化後、WO01/011951に記載された方法で作製されたヒト7番染色体断片(AF006752座で部位特異的に切断されている)を保持するニワトリDT40細胞(クローンDF141)にトランスフェクションし、ハイグロマイシンB(1.5mg/ml)を含む培地に交換し、96穴培養プレート3枚に分注して約2週間の選択培養を行った。5回のトランスフェクションで得た合計96個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr7−loxP))。
[A.3]相同組換え体の選別
[A.3.1]PCR解析
ハイグロマイシン耐性クローンからPuregene DNA Isolation Kit(Gentra System社)を用いてゲノムDNAを抽出して、以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。

[A.3.2]サザンブロット解析
上記PCR解析にて組換えを確認した6クローンについて以下のようにサザンブロット解析を行った。このゲノムDNAを制限酵素EcoRI(TAKARA)で処理し、0.8%アガロースゲル中で電気泳動し、GeneScreen PlusTMハイブリダイゼーショントランスファーメンブレン(NENTM Life Science Products,Inc.)へアルカリブロッティングした。このフィルターに対してAC004922中の遺伝子配列をPCRにより増幅したMPpプローブを用いてサザンハイブリダイゼーションを行い相同組換え体の同定を行った。MPpプローブの作製は以下のプライマーを用いDF141のゲノムDNAを鋳型としてPCRを行い、そのPCR産物を鋳型としてランダムプライミングによる32P標識DNAプローブを作製した(アマシャム、添付のプロトコールに従った)。
MPpプローブ作製用プライマー:
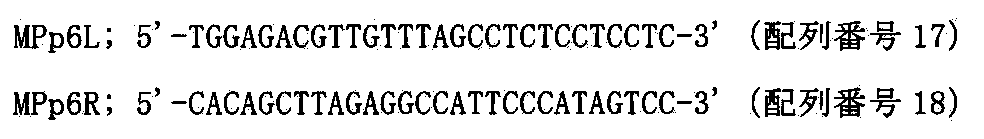
[A.3.3]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち6クローンをヒトcot−1 DNAおよびハイグロマイシンをプローブにしてFISH解析を行ったところ、ヒト7番染色体は全てのクローンで宿主染色体に転座することなく、7q22付近にハイグロマイシン由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた。以上の結果から、ヒト7番染色体断片に遺伝子導入部位であるloxP配列が部位特異的に挿入されたと結論づけた。
[B]hChr7−loxPにおけるヒト7番染色体領域AC073842での部位特異的切断
WO 2009/063722(PCT/JP2008/068928)にあるように、ヒト7番染色体のCYP3A遺伝子クラスターから遠位側のマウス個体の発生に強く関わる遺伝子を削除するために、部位特異的染色体削除であるテロメアトランケーションを行う。
[B.1]ターゲティングベクターpTELhisD−PTの作製
ヒト7染色体上のCYP3A遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAC073842領域にヒトテロメア配列を挿入するためのターゲティングベクターpTELhisD−PTを以下のようにして作製した。まず、AC073842ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
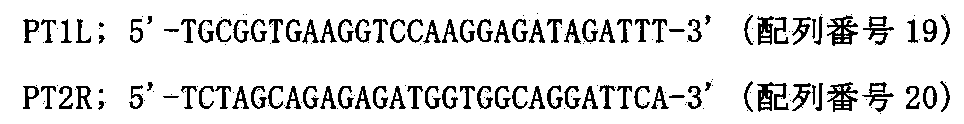
[B.2]トランスフェクション及びヒスチジノール耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpTELhisD−PTを制限酵素SrfI(東洋紡)で線状化後、上記で作製したクローンDT40(hChr7−loxP)122にトランスフェクションし、ヒスチジノール(0.5mg/ml)を含む培地に交換し、96穴培養プレート10枚に分注して約2週間の選択培養を行った。5回のトランスフェクションで得た合計335個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr7−loxP−tel))。
[B.3]相同組換え体の選別
[B.3.1]PCR解析
ヒスチジノール耐性株のゲノムDNAを鋳型として組換え体を選別するため一次スクリーニングとして切断部位よりテロメア側に位置する以下のプライマーを用いてPCRを行い、部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。

次に上記プライマーで検出されなかった433クローンのうちの2クローンについて以下のプライマーを用いて部位特異的相同組換えが起こっているかをPCRにて確認した。配列は以下のとおりである。


FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち2クローンをヒトcot−1 DNA及びヒスチジノールをプローブにしてFISH解析を行ったところ、loxP配列が挿入されたヒト7番染色体は全てのクローンで宿主染色体に転座することなく、ヒト7番染色体断片末端にヒスチジノール由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた。
以上の結果から、クローンDT40(hChr7−loxP−tel)608及び748において、CYP3A遺伝子クラスター領域よりテロメア側のAC073842から遠位において切断できたと結論付けた。
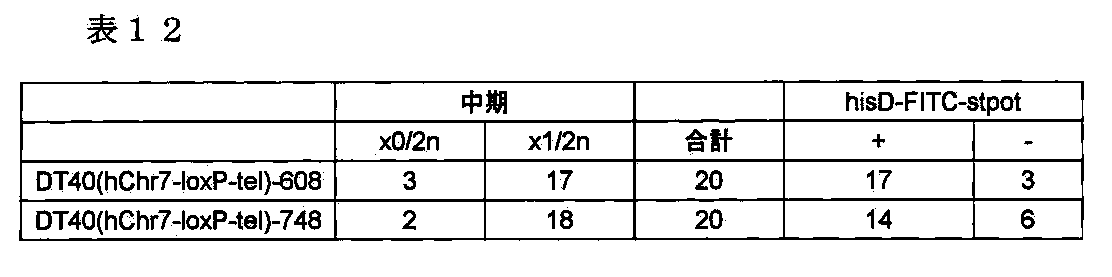
CHO細胞内でマウス人工染色体ベクターMAC1にloxP配列を介してヒトCYP3A遺伝子クラスター領域を転座挿入するために、hChr7−loxP−telをマウス人工染色体ベクターMAC1含有CHO細胞に導入する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(hChr7−loxP−tel)608及び748を用いて、上記と同様にMAC1含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC1)に微小核細胞融合法を行った。5回の微小核細胞融合で得た合計48個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC1,hChr7−loxP−tel))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、ヒト7番染色体断片がMAC1含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。



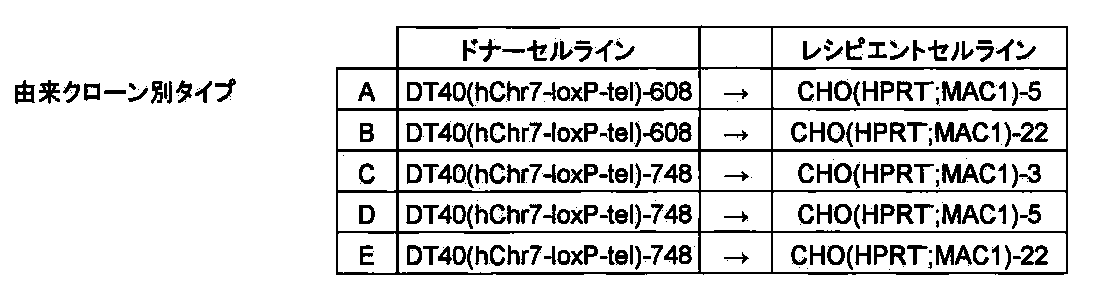
上記で得られたCHO(HPRT−;MAC1,hChr7−loxP−tel)の19クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ、上記陰性の1クローンを除いた18全てにおいてMAC1ならびにhChr7−loxP−telがCHO細胞に1コピーもしくは2コピーで導入されていることを確認した(図17)。


[D]CHO(HPRT−;MAC1,hChr7−loxP−tel)クローンにおけるヒトCYP3A遺伝子クラスター領域周辺(AC004922−ヒトCYP3A遺伝子クラスター−AC073842)1MbのMAC1ベクターへの部位特異的転座
1MbサイズのDNAであるヒトCYP3A遺伝子クラスターをマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC1に転座挿入する(図18)。
[D.1]トランスフェクション及びHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC1,hChr7−loxP−tel)−6、9、12、47にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、4回の導入で得た合計42個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(CYP3A−MAC1,hChr7−ΔCYP3A))。
[D.2]薬剤耐性クローンの選別
[D.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト7番染色体断片とMAC1上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。



上記で得られたCHO(CYP3A−MAC1,hChr7−ΔCYP3A)の27クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ27クローン中、25クローンで50%以上の割合でloxP配列を含むマウス11番染色体断片からなるMAC1上にヒト7番染色体由来によるシグナルが観察されていることを確認した(図19)。
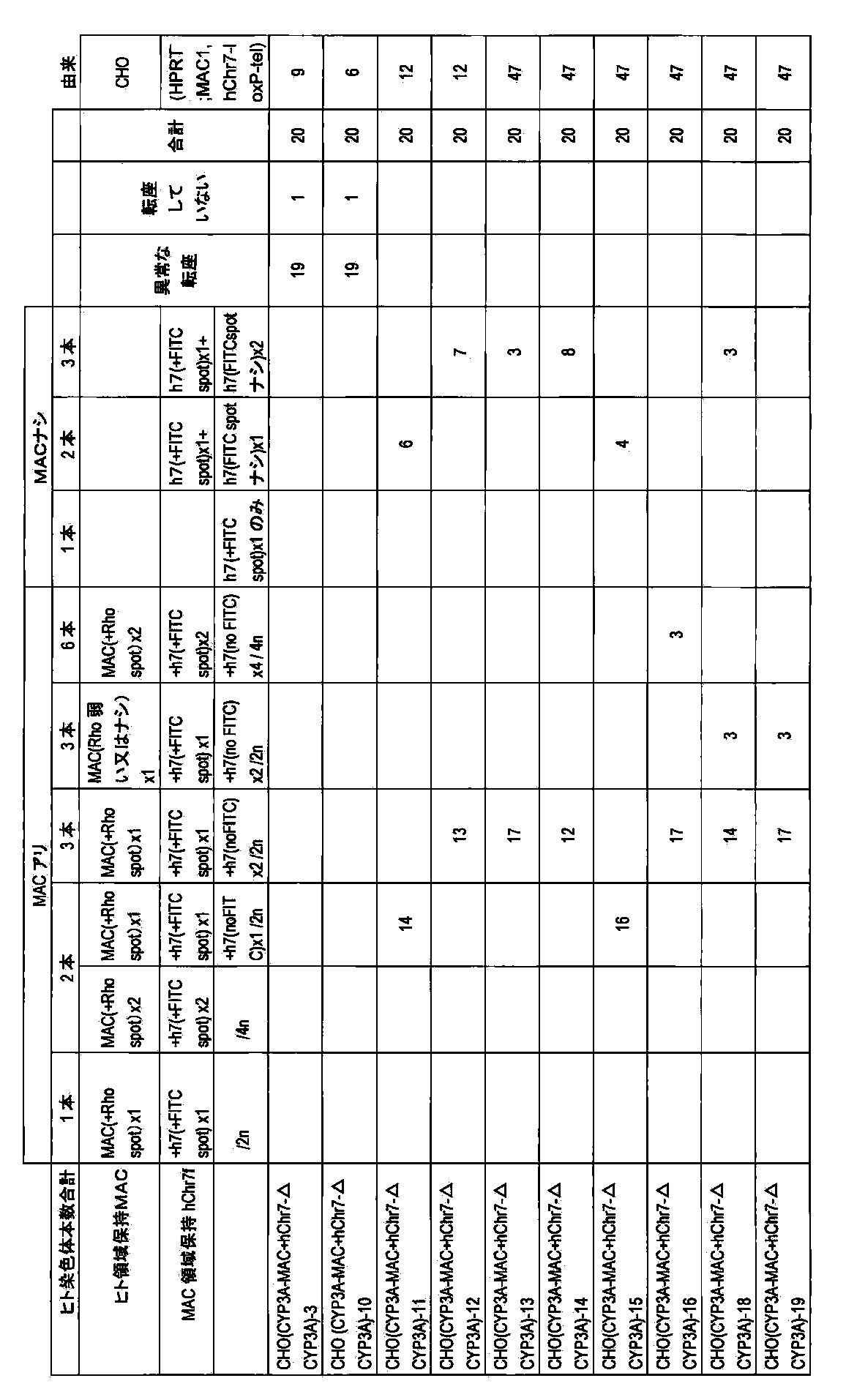
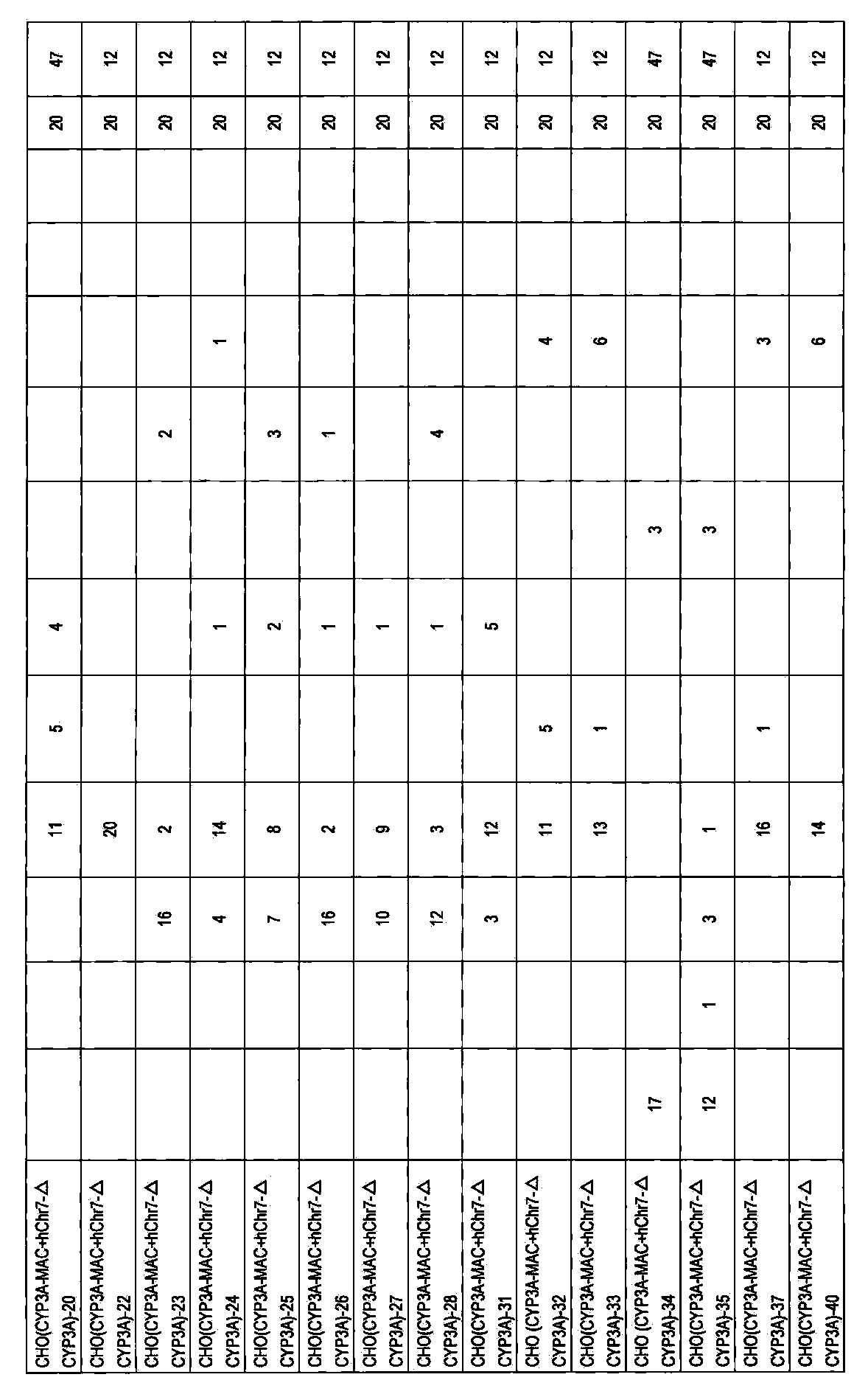
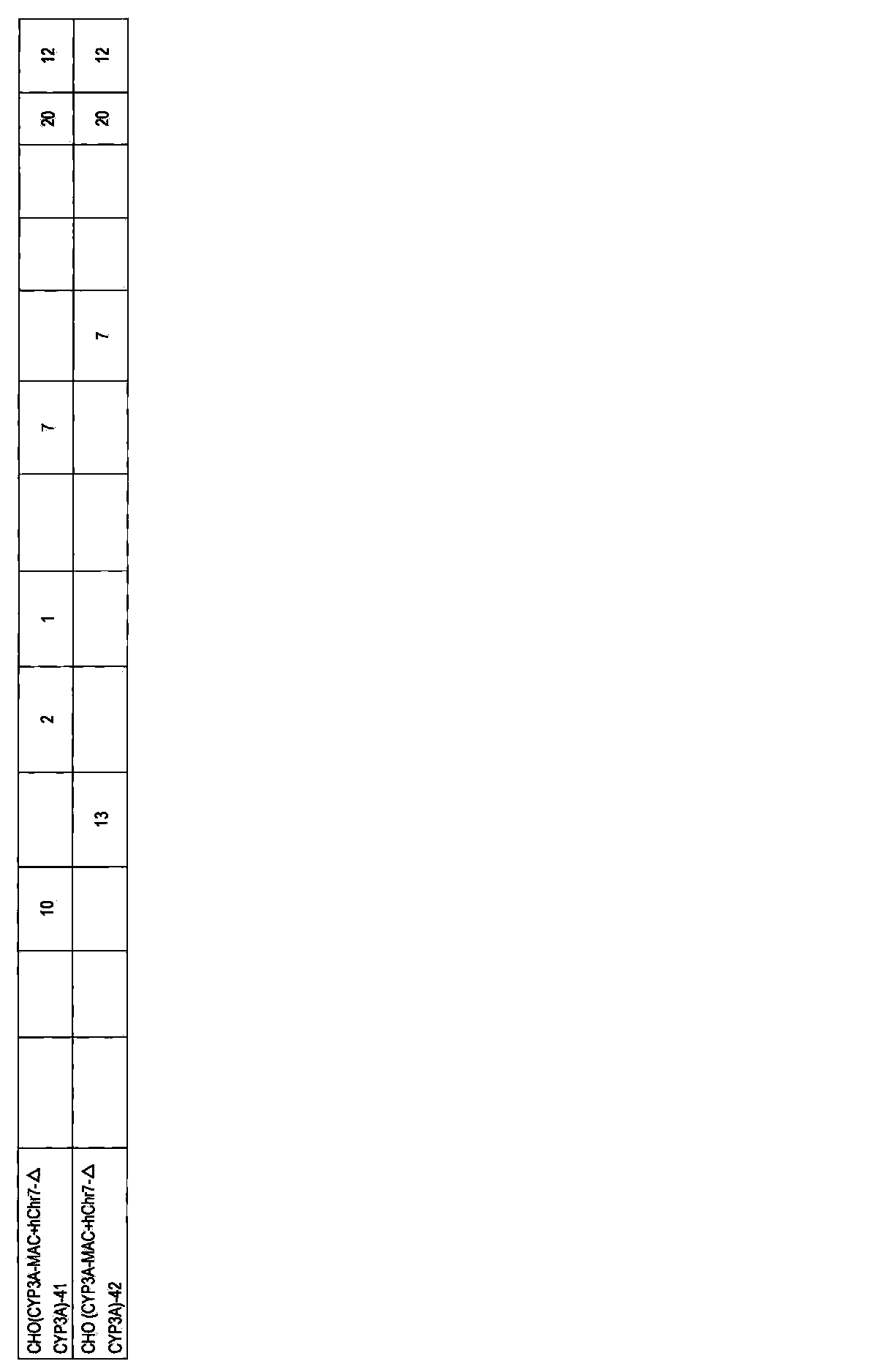
[実施例4]マウス人工染色体ベクターMAC2の構築
マウス人工染色体ベクターMACにDNA挿入配列として5’HPRT−loxP−PGKhygタイプのloxP配列を挿入したマウス人工染色体ベクターMAC2を構築する(図3)。5’HPRT−loxP−PGK hygタイプのloxP配列は、Kazukiらの報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)に挿入されており、HACとMACの遺伝子発現を同じベクターで比較することができる。また、21HAC2への挿入のための遺伝子導入ベクターがベクター作製のステップを経ることなく、そのまま利用可能である。
[A]マウス人工染色体MACへの5’HPRT−loxP−PGKhygタイプのloxP配列の挿入
[A.1]5’HPRT−loxP−PGKhygタイプのloxPターゲティングベクター作製
loxP配列を挿入するための基本プラスミドには上記で作成したVH21−12を用いた。上記X6.1からKpnIとAscIにより切り出した5’HPRT−loxP−PGKhygroカセットをV907(Lexicon genetics)のKpnIとAscIサイトにクローニングした(ベクター名:p V907−AML)。さらにp V907−AMLから5’HPRT−loxP−PGK hygroカセットをXhoIとSalIにより切り出してVH21−12のXhoIサイトにクローニングした(ベクター名:pMAC2)。ターゲティングベクター、標的配列および相同組換えにより生じる染色体アレルを図20に示す。
[A.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpMAC2を制限酵素NotI(TAKARA)で線状化後、上記で作製したクローンDT40(MAC)にトランスフェクションし、ハイグロマイシン(1.5mg/ml)を含む培地に交換し、96穴培養プレート2枚に分注して約2週間の選択培養を行った。1回のトランスフェクションで得た合計45個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(MAC2))。
[A.3]相同組換体の選別
[A.3.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工染色体ベクターMAC上で部位特異的に組換え起こっているかを確認した。そのプライマー配列を以下に示す。



上記から得られたDT40(MAC2)の6クローンにおいて、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNAおよび5’HPRT−loxP−PGK hygroカセットをプローブにしてFISH解析を行ったところ、ネガティブコントロールのターゲティングする前のマウス11番染色体断片であるマウス人工染色体ベクターMACにおいては、明らかにプローブ由来のシグナルが検出された割合が10%であったのに対し、DT40(MAC2)の6クローンでは、50%以上の割合でプローブ由来のシグナルが検出されたことから、上記6クローンにおいて、部位特異的に組換えが起こったことが視覚的に確かめられた(図21)。これらの結果から、マウス人工染色体ベクターMAC2を保持するDT40細胞クローンが得られたと結論できた。
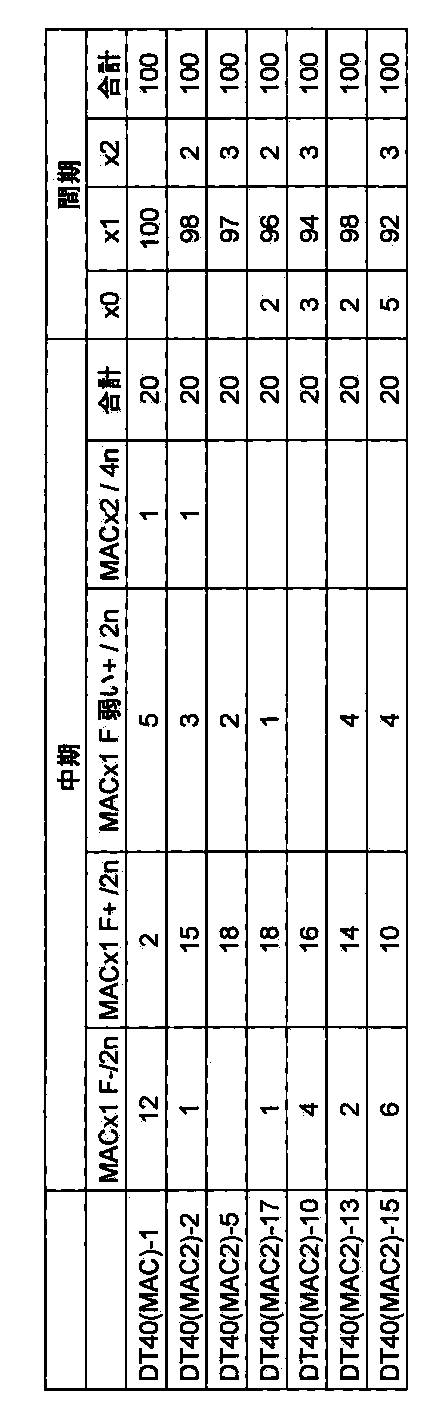
マウス人工染色体ベクターMAC2をマウスES細胞へ安定に導入するために、CHO細胞へ導入する。またマウス人工染色体ベクターMAC2のDNA配列挿入部位であるloxPを介して安定に目的遺伝子等(例えばGFP遺伝子など)を挿入するためにCHO細胞に導入する。
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(MAC2)−5及び17を用いて、上記と同様にCHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)であるCHO(HPRT−)に微小核細胞融合法を行った。2回の微小核細胞融合で得た合計44個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC2))。
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工染色体ベクターMAC2がCHO細胞に導入できているかを確認した。そのプライマー配列を以下に示す。

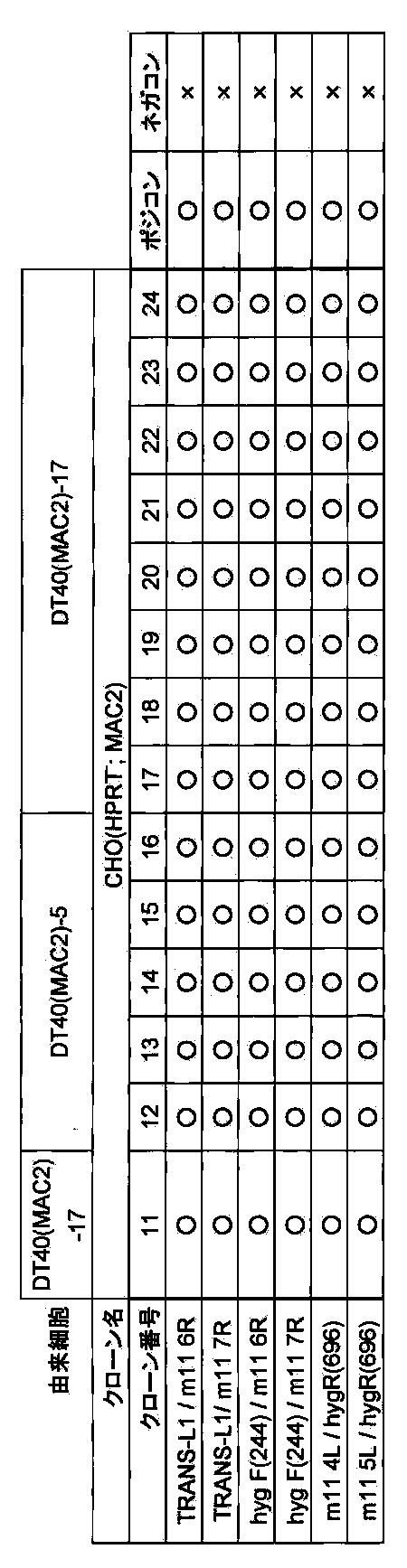
上記で得られたCHO(HPRT−;MAC2)の14クローンからランダムに選んだ9クローンについて松原ら(FISH実験プロトコール、秀潤社、1994)に従った方法でマウスCot−1 DNAと5’HPRT−loxP−PGK hygroカセットをプローブにしたFISH解析を行ったところ9クローン中、8クローンで95%の割合でMAC2がCHO細胞に導入されていることを確認した(図22)。
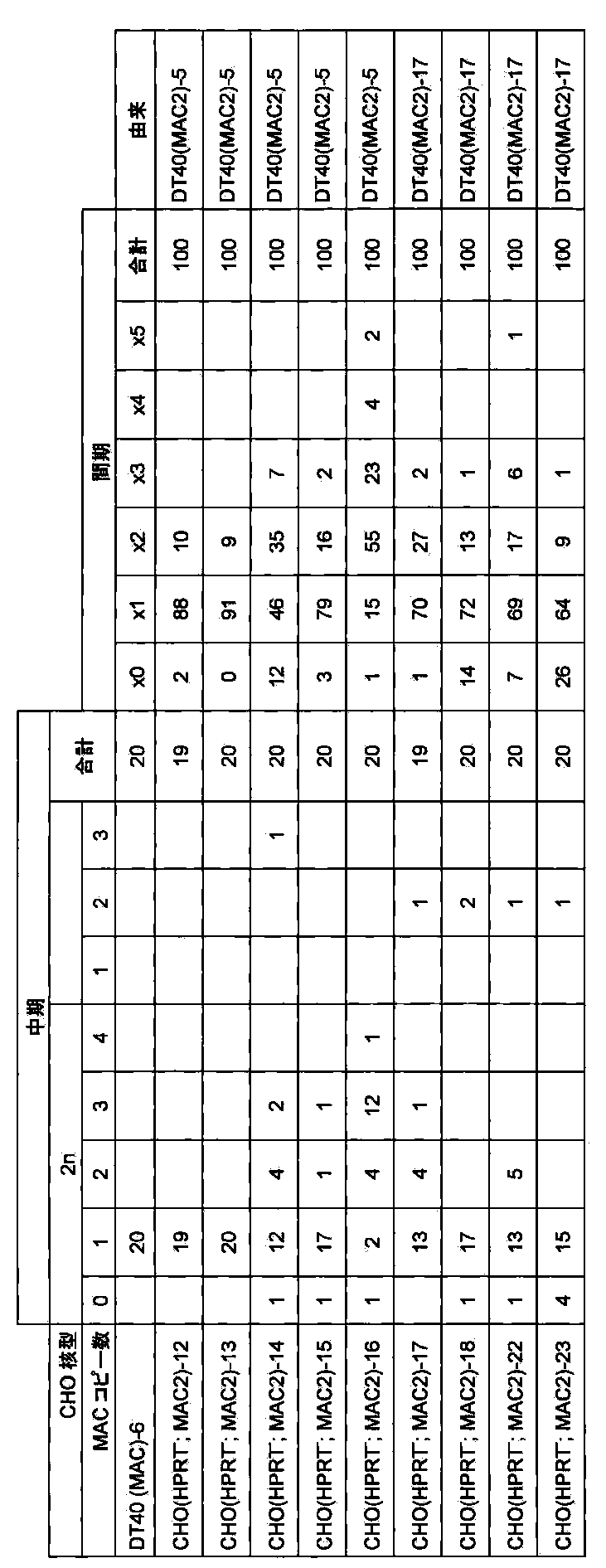
[C]マウス人工染色体ベクターMAC2含有CHO細胞からマウスES細胞へのマウス人工染色体ベクターMAC2の導入
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるCHO(HPRT−;MAC2)−13,18を細胞培養皿で培養し、コンフルエントになった時点で20% FBS、0.1μg/mlコルセミドを添加したF12培地に交換し、さらに48時間培養後に20% FBS、0.1μg/mlコルセミドを添加したF12培地で培地交換し、さらにオーバーナイトでインキュベートしてミクロセルを形成させた。培養液を除去し、予め37℃で保温したサイトカラシンB(10μg/ml,シグマ)溶液を遠心用フラスコに満たし、34℃、8000rpm、1時間の遠心を行った。微小核細胞(「ミクロセル」ともいう)を無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心し、無血清DMEM培地5mlに懸濁した。ミクロセルを5mlの無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心した。
ドナー細胞には日本クレアより入手したC57B6系統マウスのES細胞であるB6−ES、及び該ES細胞に6TG処理を行ったHPRT欠損株であるB6(HPRT−)、及びTT2F細胞のHPRT欠損株であるKO56(HPRT−)を用いた。培養には、DMEM(Dulbecco’s Modified Eagle’s Medium−high glucose:SIGMA)に、10%FCS、LIF(Muerin Leukemia Inhibitory Factor)、1×10−5M 2−ME(2−メルカプトエタノール:SIGMA)、L−グルタミン(3.5g/ml:GIBCO)、Sodium pyruvate溶液(3.5g/ml:GIBCO)、MEM非必須アミノ酸(0.125mM:GIBCO)を添加し、5% CO2、37℃にて培養をおこなった。マウスES細胞をPBS(−)で細胞表面を2回洗浄後にトリプシン処理により細胞を分散させ、DMEM培地に10%FBSを添加した培養液で回収し、1500rpmで遠心し、上清を除去し、無血清培養液5mlに再度懸濁し、ミクロセルの遠心後のペレットを含む無血清培地に静かに添加し、さらに1200rpmで遠心した。上清を除去し、PEG1000(Wako)溶液[5gのPEG1000を無血清DMEM培地に完全に溶解し、ジメチルスルホキシドを1ml添加して濾過滅菌する]を0.5mlで正確に1分30秒間融合した。13mlの無血清培養液(DMEM)を静かに添加し、1200rpmで遠心した。上清を除去し、通常のマウスES細胞の培養液を添加し、マイトマイシン処理したG418耐性マウス胎生線維芽細胞をフィーダー細胞として使用し、直径10cm細胞培養皿2枚に播種し、オーバーナイトでインキュベートした。ハイググロマイシンを250μg/mlになるように加え、3~4週間選択培養した。2回の微小核細胞融合で得た合計28個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:B6−ES(MAC2)及びB6(HPRT−;MAC2)及びKO56(HPRT−;MAC2))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工番染色体MAC2がマウスES細胞に導入できているかを確認した。そのプライマー配列を以下に示す。


上記で得られたmouse ES(MAC2)のクローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスminor sattelite DNAをプローブにしたFISH解析を行ったところ3クローン中、1クローンで80%以上の割合でMAC2がマウスES細胞に1コピーで導入され、かつKO56細胞の通常核型である内在マウス染色体の本数が39本であることを確認した。
以上の結果から、マウス11番染色体由来の染色体断片であるマウス人工染色体MACに遺伝子挿入部位であるloxP配列が挿入されたマウス人工染色体ベクターMAC2がマウスES細胞に導入できたと結論付けた(図23)。
[実施例5]マウス人工染色体ベクターMAC3の構築
マウス人工染色体MACにDNA挿入配列としてPGKneo−loxP−3’HPRTタイプのloxP配列を挿入したマウス人工染色体ベクターMAC3を構築する(図4)。マウス人工染色体ベクターMAC3のマウスES細胞での安定性を検証し、さらにMAC3を導入した子孫伝達マウスを作製することで、個体組織での安定性を検証する。
[A]マウス人工染色体MACへのPGKneo−loxP−3’HPRTタイプのloxP配列の挿入
[A.1]PGKneo−loxP−3’HPRTタイプのloxPターゲティングベクターの作製
loxP配列を挿入するための基本プラスミドには上記で作成したVH21−12を用いた。p VNLHからSalIとAscIにより切り出したPGKneo−loxP−3’HPRTカセットをVH21−12のXhoIサイトとAscIサイトにクローニングした(ベクター名:pMAC3)。ターゲティングベクター、標的配列および相同組換えにより生じる染色体アレルを示す(図24)。
[A.2]トランスフェクションおよびG418耐性クローンの単離
ニワトリDT40細胞の培養は10%ウシ胎仔血清(ギブコ、以下FBSで記す)、1%ニワトリ血清(ギブコ)、10−4M 2−メルカプトエタノール(シグマ)を添加したRPMI1640培地(ギブコ)中で行った。DT40(MAC)−1の約107個の細胞を無添加RPMI1640培地で一回洗浄し、0.5mlの無添加RPMI1640培地に懸濁し、制限酵素NotI(TAKARA)で線状化したターゲティングベクターpMAC3を25μg加え、エレクトロポレーション用のキュベット(バイオラッド)に移し、室温で10分間静置した。キュベットをジーンパルサー(バイオラッド)にセットし、550V、25μFの条件で電圧印加した。室温で10分間静置後、24時間培養した。G418(1.5mg/ml)を含む培地に交換し、96穴培養プレート2枚に分注して約2週間の選択培養を行った。2回のトランスフェクションで得た合計14個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(MAC3))
[A.3]相同組換体の選別
[A.3.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス11番染色体上で部位特異的に組換え起こっているかを確認した。そのプライマー配列を以下に示す。

上記で得られたDT40(MAC3)の2クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところすべてのクローンで染色体転座等が起きずに90%以上の割合で独立に保持されていた。

上記からランダムに選んだDT40(MAC3)−160及び187において、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNA及びマウスminor satellite DNAをプローブにしてFISH解析を行ったところ、マウス人工染色体MAC3が1コピーで独立に存在していることがわかった(図25)。
これらの結果から、DNA挿入配列としてloxP配列をマウスセントロメア付近に挿入したマウス人工染色体ベクターMAC3を保持するDT40細胞クローンが得られたと結論付けた。
[B]マウス人工染色体ベクターMAC3含有ニワトリDT40細胞からMAC3のCHO細胞への導入
マウス人工染色体ベクターMAC3のDNA配列挿入部位であるloxPを介して安定に目的遺伝子等(例えばGFP遺伝子など)を挿入するためにCHO細胞に導入する。
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(MAC3)−160を細胞培養皿で培養し、コンフルエントになった時点で20% FBS、1% ニワトリ血清、10−4M 2−メルカプトエタノール、0.05μg/mlコルセミドを添加したRPMI1640培地に交換し、さらに12時間培養してミクロセルを形成させた。培養液を24mlの無血清DMEM培地に置換し、予め100μ/mlのポリL−リジンでコートした遠心用25cm2フラスコ12本(コーニング)に2mlずつ分注し、37℃で30分培養し、細胞をフラスコの底に付着させた。上清を除去し、予め37℃で保温したサイトカラシンB(10μg/ml,シグマ)溶液を遠心用フラスコに満たし、34℃、8000rpm、1時間の遠心を行った。ミクロセルを無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、1700rpm,10分間遠心し、無血清DMEM培地5mlに懸濁した。
ドナー細胞にはCHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)であるCHO(HPRT−)を用いた。精製した微小核をPHA−P(SIGMA)を含む無血清培養液2mlに再度懸濁し、培養上清液[10% FBS添加F12培地(invitrogen)]を除去したCHO細胞上に静かに播種した。プレートを37℃、15分間インキュベートした。上清を除去し、PEG1000(Wako)溶液[5gのPEG1000を無血清DMEM培地に完全に溶解し、ヂメチルスルホキシドを1ml添加して濾過滅菌する]を1mlで正確に1分間融合した。無血清培養液(DMEM)を4mlで4回洗浄し、通常のCHO細胞の培養液5ml加えてオーバーナイトでインキュベートした。PBS(−)で細胞表面を2回洗浄後にトリプシン処理により細胞を分散させ、直径10cm細胞培養皿5枚に播種し、G418を800μg/mlになるように加え、3~4週間選択培養した。2回の微小核細胞融合で得た合計12個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC3))。
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工染色体ベクターMAC3がCHO細胞に導入できているかをを確認した。そのプライマー配列を以下に示す。


上記で得られたCHO(HPRT−;MAC3)の6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところ6クローン中、3クローンで90%以上の割合でCHO細胞にMAC3が導入されていることを確認した(図26)。

[C]マウス人工染色体ベクターMAC3含有CHO細胞からMAC3のマウスES細胞への導入
マウスES細胞及びマウス個体内でのマウス人工染色体ベクターMAC3の安定性を検証するために、マウス人工染色体MAC3をマウスES細胞に導入し、マウス人工染色体ベクターMAC3含有キメラマウスならびに子孫伝達マウスを作製する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるCHO(HPRT−;MAC3)−1,6を細胞培養皿で培養し、コンフルエントになった時点で20% FBS、0.1μg/mlコルセミドを添加したF12培地に交換し、さらに48時間培養後に20% FBS、0.1μg/mlコルセミドを添加したF12培地で培地交換し、さらにオーバーナイトでインキュベートしてミクロセルを形成させた。培養液を除去し、予め37℃で保温したサイトカラシンB(10μg/ml,シグマ)溶液を遠心用フラスコに満たし、34℃、8000rpm、1時間の遠心を行った。ミクロセルを無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心し、無血清DMEM培地5mlに懸濁した。ミクロセルを5mlの無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心した。
ドナー細胞には日本クレアより入手したC57B6系統マウスのES細胞に6TG処理を行ったHPRT欠損株であるB6(HPRT−)を用いた。培養には、DMEM(Dulbecco’s Modified Eagle’s Medium−high glucose:SIGMA)に、10%FCS、LIF(Muerin Leukemia Inhibitory Factor)、1×10−5M 2−ME(2−メルカプトエタノール:SIGMA)、L−グルタミン(3.5g/ml:GIBCO)、Sodium pyruvate solution(3.5g/ml:GIBCO)、MEM Nonessential amino acid(0.125mM:GIBCO)を添加し、5% CO2、37℃にて培養をおこなった。マウスES細胞をPBS(−)で細胞表面を2回洗浄後にトリプシン処理により細胞を分散させ、DMEM培地に10%FBSを添加した培養液で回収し、1500rpmで遠心し、上清を除去し、無血清培養液5mlに再度懸濁し、ミクロセルの遠心後のペレットを含む無血清培地に静かに添加し、さらに1200rpmで遠心した。上清を除去し、PEG1000(Wako)溶液[5gのPEG1000を無血清DMEM培地に完全に溶解し、ジメチルスルホキシドを1ml添加して濾過滅菌する]を0.5mlで正確に1分30秒間融合した。13mlの無血清培養液(DMEM)を静かに添加し、1200rpmで遠心した。上清を除去し、通常のマウスES細胞の培養液を添加し、マイトマイシン処理したG418耐性マウス胎生線維芽細胞をフィーダー細胞として使用し、直径10cm細胞培養皿2枚に播種し、オーバーナイトでインキュベートした。G418を250μg/mlになるように加え、3~4週間選択培養した。2回の微小核細胞融合で得た合計28個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:B6(HPRT−;MAC3))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス11番染色体上で部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。


上記で得られたB6(HPRT−;MAC3)のうち16クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスminor satellite DNAをプローブにしたFISH解析を行ったところ16クローン中、5クローンで95%以上の割合でMAC3がマウスES細胞に導入されていることを確認した。
以上の結果から、マウス人工染色体ベクターMAC3がマウスES細胞に導入できたと結論付けた(図27)。
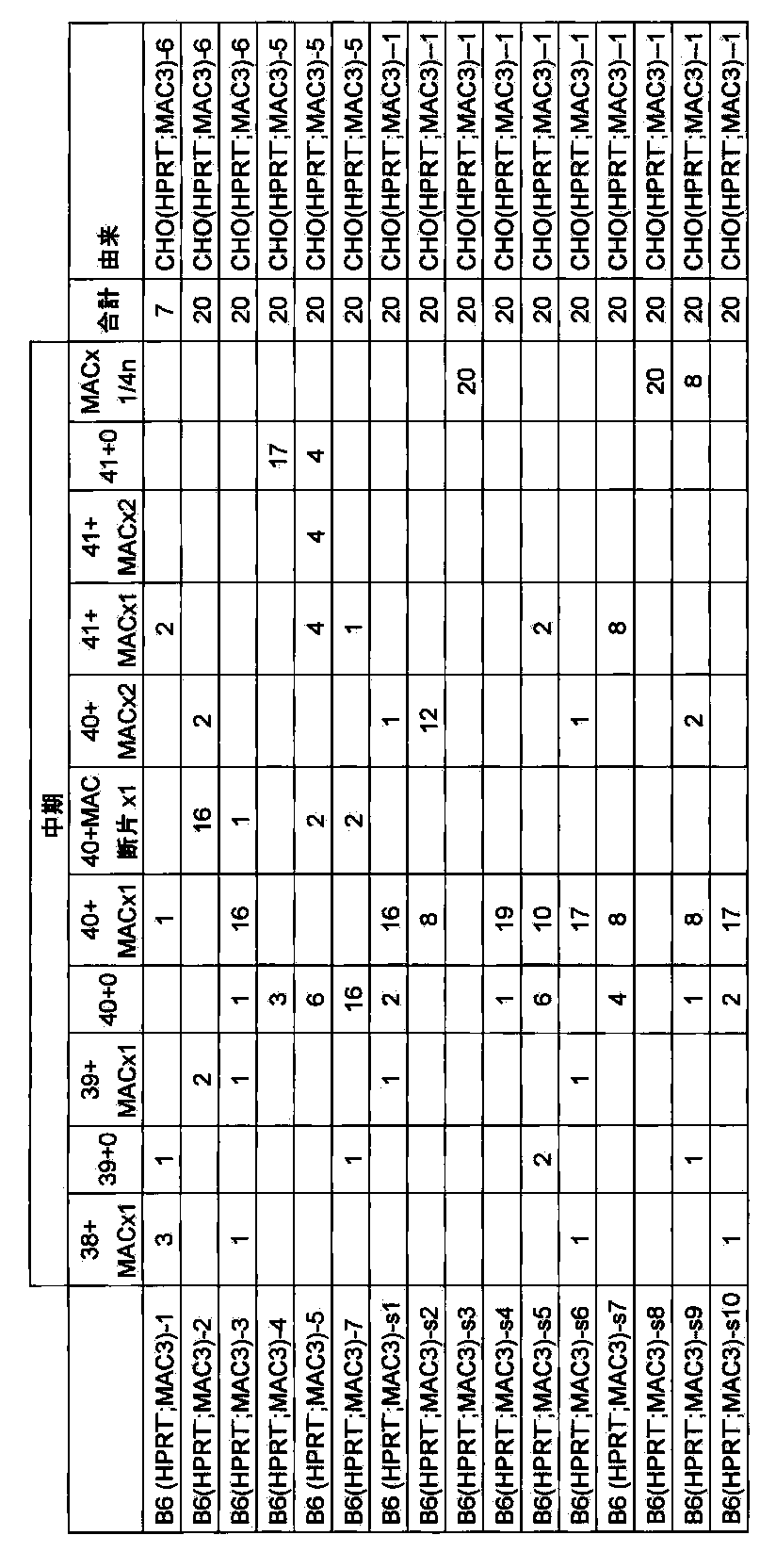
上記で得られたマウスESクローン(例えばB6(HPRT−;MAC3)−3,−s6、上記[C]で取得)について0~100PDLの非選択培養下での長期培養後におけるFISH解析によるMAC1保持細胞の割合を計測した結果、100PDLにおいても95%以上の保持率であった(図28)。
以上の結果、マウス人工染色体ベクターMAC3はマウスES細胞(in vitro)において、95%以上の割合で非常に安定に維持されることが確かめられた。
[E]マウス人工染色体ベクターMAC3を保持するキメラマウスの作製
上記で得られたES細胞クローンを用いて(ジーンターゲティング、実験医学、1995)の手法に従い、キメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる桑実胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうか、またICRの胚細胞に対してES細胞が個体を形成する細胞の中での寄与率としてキメラ率を判定できる。
B6(HPRT;MAC3)クローン(例えばB6(HPRT;MAC3)−ES(MAC3)−s6上記で取得)を注入した60個の胚を仮親に移植した結果、8匹のキメラマウス(毛色に濃茶色の部分の認められる)が誕生した。8匹のうち、2匹は雄で10%キメラマウスが1匹、5%キメラマウスが1匹生まれ、6匹は雌で10%キメラマウスが4匹、5%キメラマウスが2匹生まれた。すなわち、マウス人工染色体MAC3を保持するES細胞株(B6HPRT−/−株)はキメラ形成能を保持している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[F]実施例8記載のように、マウス人工染色体ベクターMAC3を保持するキメラマウスと野生型マウスと交配させてMAC3が子孫伝達されたマウス系統TC(MAC3)をを作製できる。また、上記TC(MAC3)マウス系統を用いて、体細胞におけるMAC3の安定性を検証できる。
[実施例6]マウス人工染色体ベクターGFP−MACの構築
マウス人工染色体ベクターMAC3に有用タンパク質をコードする遺伝子の例として、蛍光遺伝子であるEGFPをCre/loxPシステムを用いて挿入し、機能タンパク質の発現と長期安定性を検証する(図5)。
[A]マウス人工染色体ベクターMAC3ベクター含有CHO細胞における特定の遺伝子(例えばGFP)のCre/loxPシステムによるマウス人工染色体ベクターMAC3への挿入
マウス人工染色体MACにDNA挿入配列としてPGKneo−loxP−3’HPRTタイプのloxP配列を挿入したマウス人工染色体ベクターMAC3においてloxPが稼働しプラスミドDNAが部位特異的に挿入できるかどうか検証する。
[A.1]EGFP挿入ベクターの作製
loxP配列を挿入するための基本プラスミドにはV913(Lexicon genetics)を用いた。5’HPRT−loxPはV820(Lexicon genetics)のXbaIサイトにオリゴ合成したloxP配列をクローニングした。5’HPRT−loxPをV907(Lexicon genetics)のClaIとAscIにクローニングし、PGKhygroをClaIとKpnIサイトにクローニングした(ベクター名:pX6.1)。さらにX6.1のNotIサイトとSalIサイトにNotIとSalIにより切り出したHS4−CAG−EGFP−HS4(大阪大学、岡部博士及びNIH、Felsenfeld博士より分与)をクローニングし、HPRT再構築系のGFP挿入コンストラクトとした(ベクター名:p X6.1 EGFP)。Cre/loxPシステムによるHPRT再構築系のGFP挿入により生じる染色体部位特異的DNA挿入を図29に示す。
[A.2]トランスフェクション及びHAT耐性クローンの単離
遺伝子導入はリポフェクション法を用いて行い、90%コンフルエント状態になった6wellの細胞に対して、Cre1μgとGFP挿入ベクター2μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計22個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(GFP−MAC))。
[A.3]薬剤耐性クローンの選別
[A.3.1]蛍光顕微鏡観察によるGFP挿入体の確認
クローニングした22個のコロニーを蛍光顕微鏡下にて観察したところ、全てのクローンにてGFP陽性細胞が観察され、その陽性率はほぼ100%であった。
[A.3.2]PCR解析
HAT耐性株のゲノムDNAを鋳型として組換え体を選別するため以下のプライマーを用いてPCRを行い、部位特異的にGFP遺伝子の挿入が起こっているかを確認した。そのプライマー配列を以下に示す。

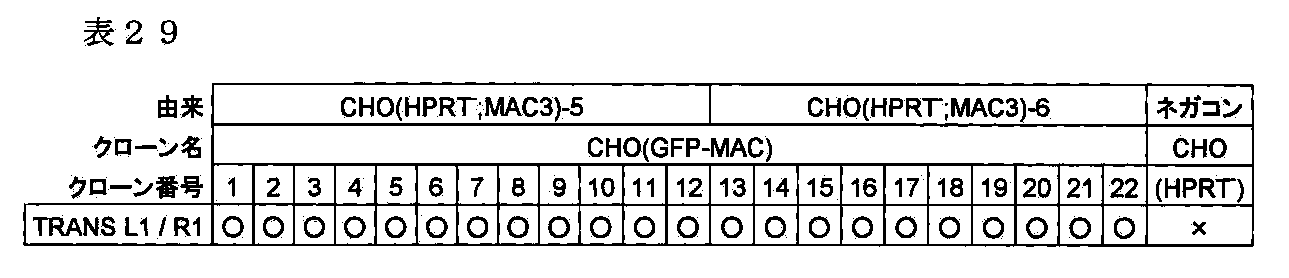
上記から結果からランダムに選んだ6クローンにおいて、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNA及びX6.1EGFPをプローブにしてFISH解析を行ったところ、6クローンのうち、3クローンにおいて50%以上の割合でGFP−MACが1コピー保持され、さらにX6.1EGFP由来のシグナルが現れ、ネガティブコントロールであるEGFPを部位特異的挿入する前のMAC3上にはシグナルが検出されなかったことから、部位特異的にEGFPが挿入されたことが確かめられた(図30)。
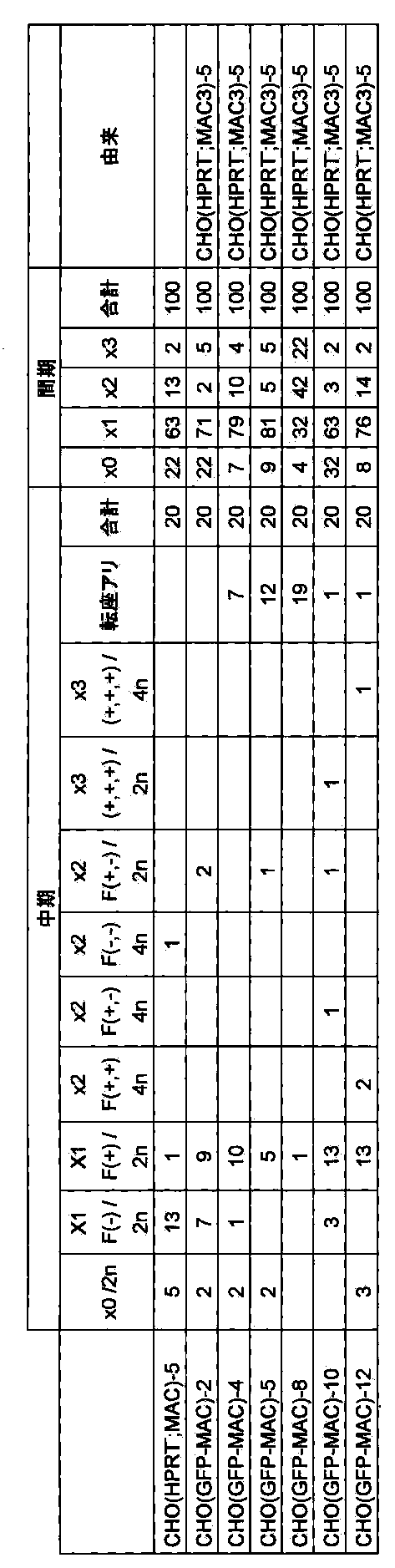
[B]マウス人工染色体ベクターGFP−MAC含有CHO細胞からGFP−MACのマウスES細胞への導入
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるCHO(GFP−MAC)−4、10、12を細胞培養皿で培養し、コンフルエントになった時点で20% FBS、0.05μg/mlコルセミドを添加したF12培地に交換し、さらに48時間培養後に20% FBS、0.05μg/mlコルセミドを添加したF12培地で培地交換し、さらにオーバーナイトでインキュベートしてミクロセルを形成させた。培養液を除去し、予め37℃で保温したサイトカラシンB(10μg/ml,シグマ)溶液を遠心用フラスコに満たし、34℃、8000rpm、1時間の遠心を行った。ミクロセルを無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心し、無血清DMEM培地5mlに懸濁した。
ミクロセルを5mlの無血清DMEM培地に懸濁し、8μm,5μm,3μmフィルターにて精製した。精製後、2000rpm,10分間遠心した。
ドナー細胞には日本クレアより入手したC57B6系統マウスのES細胞から樹立した野生型B6細胞及び、野生型TT2F細胞のマウスES細胞を用いた。培養には、DMEM(Dulbecco’s Modified Eagle’s Medium−high glucose:SIGMA)に、10%FCS、LIF(Muerin Leukemia Inhibitory Factor)、1×10−5M2−ME(2−メルカプトエタノール:SIGMA)、L−グルタミン(3.5g/ml:GIBCO)、Sodium pyruvate solution(3.5g/ml:GIBCO)、MEM Nonessential amino acid(0.125mM:GIBCO)を添加し、5%CO2、37℃にて培養をおこなった。マウスES細胞をPBS(−)で細胞表面を2回洗浄後にトリプシン処理により細胞を分散させ、DMEM培地に10%FBSを添加した培養液で回収し、1500rpmで遠心し、上清を除去し、無血清培養液5mlに再度懸濁し、ミクロセルの遠心後のペレットを含む無血清培地に静かに添加し、さらに1200rpmで遠心した。上清を除去し、PEG1000(Wako)溶液[5gのPEG1000を無血清DMEM培地に完全に溶解し、ジメチルスルホキシドを1ml添加して濾過滅菌する]を0.5mlで正確に1分30秒間融合した。13mlの無血清培養液(DMEM)を静かに添加し、1200rpmで遠心した。上清を除去し、通常のマウスES細胞の培養液を添加し、マイトマイシン処理したG418耐性マウス胎生線維芽細胞をフィーダー細胞として使用し、直径10cm細胞培養皿2枚に播種し、オーバーナイトでインキュベートした。G418を250μg/mlになるように加え、3~4週間選択培養した。2回の微小核細胞融合で得た合計36個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:TT2F(GFP−MAC)及びB6−ES(GFP−MAC))。
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
G418耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス11番染色体上で部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。


上記PCR解析によって陽性であったクローンについて、上述の方法と同じようにキナクリン・ヘキスト二重染色を行った。キナクリン・ヘキスト二重染色したクローンの染色体像を蛍光顕微鏡により観察したところ、24クローン中、18クローンにおいて100%の割合でマウス人工染色体GFP−MACが保持されていることがわかった。
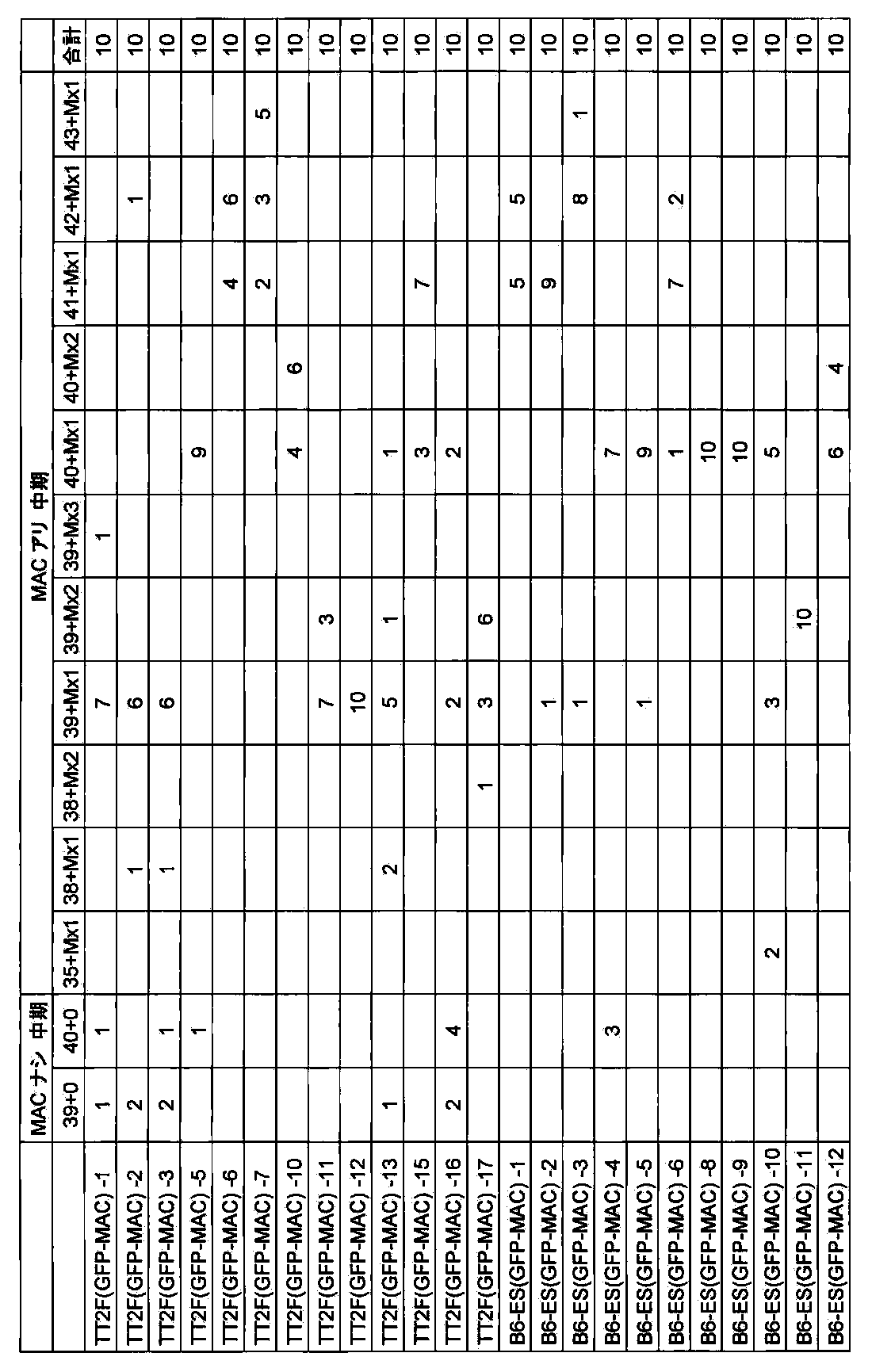
[B.2.3]two−color FISH解析
上記で得られたマウスES(GFP−MAC)のクローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスminor satellite DNA及びp X6.1EをプローブにしたFISH解析を行ったところ12クローン中、6クローンで95%以上の割合でGFP−MACがマウスES細胞に導入されていることを確認した。

[C]マウス人工染色体ベクターGFP−MACのマウスES細胞における安定性
上記で得られたマウスESクローン(例えばB6−ES(MAC3)−9、上記[B]で取得)について0~100PDLの非選択培養下での長期培養後におけるFISH解析によるGFP−MAC3保持細胞の割合を計測した結果、100PDLにおいても95%以上の保持率であった(図31、図32)。また、コロニーを蛍光顕微鏡下にて観察したところ、全てのクローンにてGFP陽性細胞が観察され、その陽性率はほぼ100%であった。
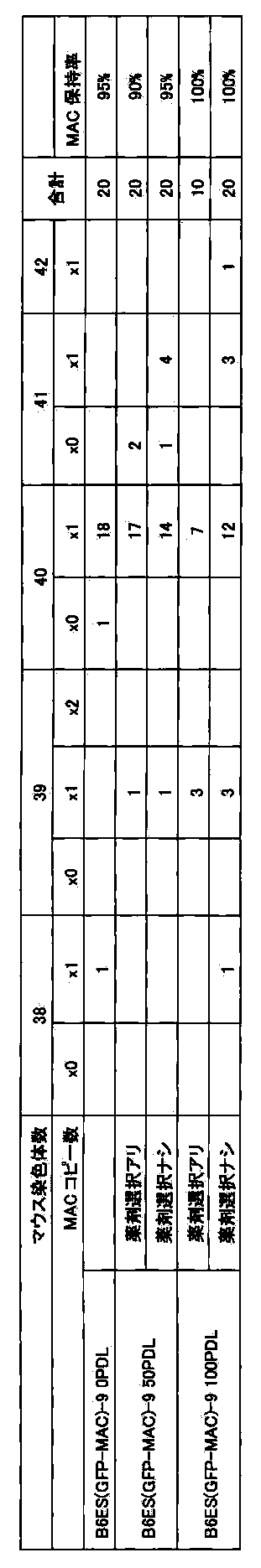
[D]マウス人工染色体ベクターGFP−MACを保持するキメラマウスの作製
上記[B]で得られたES細胞クローンを用いて(ジーンターゲティング、実験医学、1995)の手法に従い、キメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる桑実胚及び8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。
野生型雄B6(GFP−MAC)クローン及び野性型(GFP−MAC)TT2F雌クローン(例えばB6−ES(GFP−MAC)4および18、TT2F(GFP−MAC)−12上記で取得)をそれぞれ注入した胚(野生型雄B6(GFP−MAC)クローン260個及び野性型雌TT2F(GFP−MAC)クローン180個)を仮親に移植した結果、キメラマウス(毛色に濃茶色の部分の認められる)が誕生した。雄野生型B6(GFP−MAC)クローン由来のキメラマウスは42匹生まれ、そのうち20匹は雄マウスであり、GFP陽性50%キメラマウスが1匹、40%キメラマウスが5匹、30%キメラマウスが1匹、20%キメラマウスが7匹、10%キメラマウスが3匹、5%キメラマウスが3匹生まれた。また野性型TT2F(GFP−MAC)クローン由来のキメラマウスは14匹生まれ、そのうち1匹は白色の部分がほとんど観察できないキメラ率約100%かつGFP陽性の個体であった。
以上のことから、マウス人工染色体ベクターGFP−MACを保持するES細胞株(B6及びTT2F)はキメラ形成能を有している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[E]マウス人工染色体ベクターGFP−MACを保持するキメラマウスからのマウス人工染色体の子孫伝達
上記[D]で作製された雌キメラマウス(キメラ率約100%)をC57B6(黒色、日本クレア社より購入)雄マウスと交配したキメラマウスより誕生した4匹の仔マウスのうち、3匹がES細胞由来のGFP−MACの優性遺伝形質である、GFPの蛍光が観察された。また3匹のうち1匹の仔マウスについては、全身でGFPの蛍光が観察され、マウス個体においてもマウス人工染色体が安定であることが示された(図33)。GFP−MACが子孫伝達されたマウス系統をTC(GFP−MAC)と呼ぶ。また、実施例8記載のように、上記TC(GFP−MAC)マウス系統を用いて、体細胞におけるGFP−MACの安定性を検証できる。
[実施例7]マウス人工染色体ベクターMAC1の安定性
[A.1]マウス人工染色体ベクターMAC1のCHO細胞における安定性
上記で得られたCHOクローン(例えばCHO(HPRT−;MAC1)−8,22、上記実施例2で取得)について0~25PDLの非選択培養下での長期培養後におけるFISH解析によるMAC1保持細胞の割合を計測した結果、25PDLにおいても90%以上の保持率であった。一方、Kazukiら報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)を保持するCHO細胞では、25PDLにおいては70%以下の保持率であった。その代表的な結果を図34に示す。
[A.2]マウス人工染色体ベクターMAC1のマウスES細胞における安定性
上記で得られたマウスESクローン(例えばKO56(MAC1)−5およびTT2F(MAC1)−23、上記実施例2で取得)について0~75PDLの非選択培養下での長期培養後におけるFISH解析によるMAC1保持細胞の割合を計測した結果、75PDLにおいても90%以上の保持率であった。一方、Kazukiらの報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)を保持するマウスES細胞では、75PDLにおいては70%以下の保持率であった。その代表的な結果を図35に示す。
[A.3]マウス人工染色体ベクターMAC1を保持するキメラマウスの作製
上記実施例2で得られたES細胞クローンを用いてTomizukaらの方法(Nature Genet.16:133,1997)でキメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。MAC1保持ESクローン(例えばKO56MAC1−5およびTT2FMAC1−4、上記実施例2で取得)を注入した1620個の胚を仮親に移植した結果、56匹のキメラマウス(毛色に濃茶色の部分の認められる)が誕生した。うち13匹は白色の部分がほとんど観察できないキメラ率約100%の個体であった。すなわち、マウス人工染色体ベクターMAC1を保持するES細胞株(KO56およびTT2F)はキメラ形成能を保持している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[A.4]マウス人工染色体ベクターMAC1を保持するキメラマウスからのMAC1の子孫伝達
上記[A.3]で作製された雌キメラマウス(キメラ率約100%)2匹をMCH(ICR)(白色、日本クレア社より購入)雄マウスと交配した。キメラマウスより誕生した18匹の仔マウスのうち、13匹がES細胞由来の優性遺伝形質を保持することを示す濃茶色であった。すなわちMAC1を保持するES細胞株は雌キメラマウスにおいて機能的な卵細胞に分化したことが示された。また、GFP蛍光によりMAC1の保持を検討した結果、13匹中6匹(46%)においてGFP陽性であり、キメラマウス子孫におけるMAC1の保持が確認された。すなわち、メンデル遺伝の法則従い、MAC1形質が約50%の頻度で出現したことが確かめられ、卵子におけるMAC1の保持率が100%に近いことが示された。MAC1が子孫伝達されたマウス系統をTC(MAC1)と呼ぶ。
[A.5]TC(MAC1)マウス系統の体細胞におけるMAC1の安定性
[A.5.1]実体蛍光顕微鏡観察
上記で得られたTC(MAC1)マウスのうち雄(5)および雌(2)1匹ずつについて脳、胸腺、心臓、肺、肝臓、腎臓、脾臓、小腸、筋肉、精巣(または卵巣)を実体蛍光顕微鏡観察下にて観察したところ、全ての組織にてGFP陽性が観察され、その陽性率は100%であった。その雌(5)の代表的な結果を図36に示す。
[A.5.2]血液系細胞のFACS解析
B細胞(CD19)、T細胞(CD4,CD8)、巨核球(CD41)に対する特異的抗体(Becton,Dickinson and Company)を用いて、骨髄ならびに脾臓細胞におけるGFP陽性率を検討した結果、全ての組織で、陽性率は95%以上であった。一方、Kazukiらの報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)を保持するマウスでは、全ての組織で、陽性率は15%以下であった。その代表的な結果を図37、図38に示す。
[A.5.3]蛍光in situハイブリダイゼーション(FISH)解析
また上記と同様の個体から調製した尻尾繊維芽細胞を用いて、Shinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスminor satellite DNAをプローブにしたFISH解析を行ったところ、視覚的にMAC1の存在を確認し、マウス染色体とは独立に95%以上の細胞でMAC1が存在していることが確かめられた(図39)。
以上の結果、マウス人工染色体ベクターMAC1は、マウスES細胞(in vitro)ならびにマウス組織(in vivo)において、90%以上の割合で非常に安定に維持されることが確かめられた。
[実施例8]マウス人工染色体ベクターCYP3A−MAC保持マウスの作製と安定性
[A]CHO細胞からのCYP3A−MACのマウスA9細胞への移入
CYP3A−MACを保持するマウスES細を作製するために、上記実施例3で得られたCYP3A−MACを保持するCHO細胞(CHO(CYP3A−MAC,hChr7−ΔCYP3A)22,26,34,35など)からマウスA9細胞へミクロセル形成能の高いマウスA9細胞へ微小核細胞融合法により導入した。8回の微小核細胞融合で得た合計25個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:A9(CYP3A−MAC))。その結果、CYP3A−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であるクローンが6クローンあった。さらに、CYP3A−BAC(RP11757A13)(CHORI)ならびにマウスminor satellite DNAプローブを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出されるCYP3A−MACの存在が6クローン中、3クローンで確認された(図40)。以上のことから、CYP3A−MAC保持A9細胞が3クローン得られたと結論付けた。
[B]A9細胞からのCYP3A−MACのマウスES細胞への移入
CYP3A−MACを保持するキメラマウスを作製するために、上記[A]で得られたCYP3A−MACを保持するA9細胞からマウスES細胞(野性型TT2F)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のCYP3A−MACを保持するA9細胞(A9(CYP3A−MAC)8,9など)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のマウスES細胞TT2Fをトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4 PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計34個のコロニーを単離し増殖させ、以後の解析を行った。A9(CYP3A−MAC)8からは14クローン、A9(CYP3A−MAC)9からは7クローンがCYP3A−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であった。さらに、上記のうち20クローンについて、CYP3A−BAC(RP11−757A13)(CHORI)由来のDNAを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出され、かつマウスの核型が正常なクローンは8クローンであった(図41)。以上のことから、CYP3A−MAC保持TT2F細胞が8クローン得られたと結論付けた。
[C]CYP3A−MACのマウスES細胞における安定性
上記[B]で得られたマウスESクローン(例えばTT2F(CYP3A−MAC)8−5,8−22,9−4,9−7,9−9,上記[B]で取得)について0~100PDLの非選択培養下での長期培養後におけるFISH解析によるCYP3A−MAC保持細胞の割合を計測した結果、100PDLにおいても95%以上の保持率であった。(図42)。
[D]CYP3A−MACを保持するキメラマウスの作製
上記[B]で得られたCYP3A−MAC保持ES細胞クローンを用いてTomizukaらの方法(Nature Genet.16:133,1997)でキメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。MAC1保持ESクローン(例えばTT2F(CYP3A−MAC)8−5,8−16,8−22,9−4,9−7,9−9,9−10など,上記[B]で取得)を注入した840個の胚を仮親に移植した結果、28匹のキメラマウス(毛色に濃茶色の部分の認められる)が誕生した。うち5匹は白色の部分がほとんど観察できないキメラ率約100%の個体であった。すなわち、マウス人工染色体ベクターCYP3A−MACを保持するES細胞株(TT2F)はキメラ形成能を保持している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[E]CYP3A−MACを保持するキメラマウスからのCYP3A−MACの子孫伝達
上記[D]で作製された雌キメラマウス(キメラ率約100%)5匹をMCH(ICR)(白色、日本クレア社より購入)雄マウスと交配した。キメラマウスより誕生した60匹の仔マウスのうち、50匹がES細胞由来の優性遺伝形質を保持することを示す濃茶色であった。すなわちCYP3A−MACを保持するES細胞株は雌キメラマウスにおいて機能的な卵細胞に分化したことが示された。また、GFP蛍光によりCYP3A−MACの保持を検討した結果、50匹中29匹(58%)においてGFP陽性であり、キメラマウス子孫におけるCYP3A−MACの保持が確認された。すなわち、メンデル遺伝の法則従い、CYP3A−MAC形質が約50%の頻度で出現したことが確かめられ、卵子におけるCYP3A−MACの保持率が100%に近いことが示された。CYP3A−MACが子孫伝達されたマウス系統をTC(CYP3A−MAC)と呼ぶ。
[F]TC(CYP3A−MAC)マウス系統の体細胞におけるCYP3A−MACの安定性
[F.1]実体蛍光顕微鏡観察
上記で得られたTC(CYP3A−MAC)マウスのうち雄(2)1匹、雌(14)1匹について脳、胸腺、心臓、肺、肝臓、腎臓、脾臓、小腸、筋肉、精巣を実体蛍光顕微鏡観察下にて観察したところ、全ての組織にてGFP陽性が観察され、その陽性率は100%であった。その雄(2)の代表的な結果を図43に示す。
[F.2]血液系細胞のFACS解析
B細胞(CD19)、T細胞(CD4,CD8)、巨核球(CD41)に対する特異的抗体(Becton,Dickinson and Company)を用いて、骨髄におけるGFP陽性率を検討した結果、全ての組織で、陽性率は94%以上であった。一方、WO 2009/063722(PCT/JP2008/068928)に記載された14番染色体由来のHACベクター(CYP3A−HACΔ)を保持するマウスでは、全ての組織で、陽性率は20%以下であった。その代表的な結果を図44に示す。
[F.3]蛍光in situハイブリダイゼーション(FISH)解析
また上記と同様の個体や組織において、Shinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でCYP3A−BAC(RP11−757A13)DNAをプローブにしたFISH解析を行ったところ、視覚的にCYP3A−MACの存在を確認し、90−98%の細胞でCYP3A−MACが存在していることが確かめられた。一方、WO 2009/063722(PCT/JP2008/068928)に記載されたヒト14番染色体由来のHACベクター(CYP3A−HACΔ)を保持するマウスでは、全ての組織で、陽性率は56−97%であった。その代表的な結果を図45に示す。
[F.4]TC(CYP3A−MAC)系統の伝達率
TC(CYP3A−MAC)雌マウス8匹とMCH(ICR)(白色、日本クレア社より購入)雄マウス8匹を交配することで伝達率を検討した。81匹の仔マウスが得られ、38匹がGFP陰性、43匹がGFP陽性個体であった(伝達率は53%)。すなわち、伝達率はメンデル遺伝の法則に合致しており、CYP3A−MAC形質が約50%の頻度で出現したことが確かめられ、卵子におけるCYP3A−MACの保持率が100%に近いことが示された。
[F.5]CYP3A−MACが2本保持されたTC(CYP3A−MAC)ホモ系統の作製と伝達率
上記で得られたTC(CYP3A−MAC)雄マウスとTC(CYP3A−MAC)雌マウスを交配し、CYP3A−MACが2本保持されたTC(CYP3A−MAC)ホモ系統の樹立を試みた。18匹の仔マウスの尻尾繊維芽細胞を用いて、Shinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でCYP3A−BAC(RP11757A13)DNAをプローブにしたFISH解析を行ったところ、視覚的にCYP3A−MACの存在を確認した。4x36の系統では、12匹の仔マウスのうち、3匹は2コピー、5匹は1コピー、4匹は0コピーであった。24x37の系統では、6匹の仔マウスのうち、1匹は2コピー、3匹は1コピー、2匹は0コピーであった。合計18匹が得られ、CYP3A−MACの2コピー(4匹)、1コピー(8匹)、0コピー(6匹)の比率は、1:2:1.5であり、メンデル遺伝の法則にほぼ合致していた。その代表的な結果を図46に示す。
以上の結果、CYP3A−MACは、マウスES細胞(in vitro)において、95%以上の割合で非常に安定に長期間維持され、マウス組織(in vivo)において、90%以上の割合で非常に安定にかつホモ系統で維持されることが確かめられた。
[G]TC(CYP3A−MAC)マウス系統における組織特異的CYP3A遺伝子クラスターの遺伝子発現
上記でTC(CYP3A−MAC)マウスのうち雄(2)および雌(14)1匹ずつについて、脳、胸腺、心臓、肺、肝臓、腎臓、脾臓、小腸、筋肉において、トータルRNAを市販のプロトコール(QIAGEN)に従い抽出後、市販のプロトコール(invitrogen)に従い、cDNA合成を行い、それを鋳型としてPCRを行い、ヒトCYP3A遺伝子クラスターおよびマウスCyp3a遺伝子クラスターの発現を検出した。そのプライマー配列を以下に示す。
ヒトCYP3A遺伝子クラスター発現検出用プライマー:


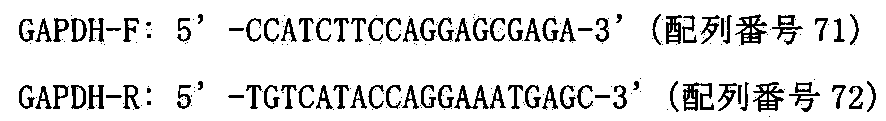
その結果TC(CYP3A−MAC)を保持するマウスにおいて、CYP3A4は肝臓と小腸でのみ、CYP3A5は肝臓と小腸と肺でのみ、CYP3A7は肝臓と小腸と腎臓と肺でのみ、CYP3A43は肝臓と小腸と腎臓でのみ、Cyp3a11は肝臓と小腸でのみ、Cyp3a13は肝臓と小腸でのみ発現を検出できた。また、コントロールのGAPDHは全ての組織で検出された。その雌(14)の代表的な結果を図47に示す。このようにヒトでみられるよう組織特異的発現が認められ、ヒト型化になったことが示された。
[H]TC(CYP3A−MAC)マウス系統における時期特異的CYP3A遺伝子クラスターの遺伝子発現
GFP陽性のTC(CYP3A−MAC)マウスの雄および雌の胎齢14.5日、胎齢16.5日、胎齢18.5日、生後0日、4週齢、6週齢、12週齢、24週齢における肝臓からのトータルRNAを市販のプロトコール(QIAGEN)に従い抽出後、市販のプロトコール(invitrogen)に従い、cDNA合成を行い、それを鋳型としてPCRを行い、上述のヒトCYP3A遺伝子クラスター発現およびマウスCyp3a遺伝子クラスター発現を検出するプライマーを用いて発現を検出した。
その結果、成体型発現のヒトCYP3A4、ヒトCYP3A5、マウスcyp3a11、マウスCyp3a13は成体期において、胎児型のCYP3A7は胎児において強く発現していることが確かめられた。また、コントロールのGAPDHは全ての胎齢、週齡で同程度の発現量が検出された。その代表的な結果を図48に示す。このようにヒトでみられるような時期特異的発現が認められ、ヒト型化になったことが示された。
[実施例9]TC(CYP3A−MAC)/Δcypマウス系統の作製
[A]CYP3A−MACを保持し、かつ内因性Cyp3a遺伝子群の両アレルが共に破壊されたマウス系統の構築
上記実施例8で作製されたTC(CYP3A−MAC)を、WO 2009/063722(PCT/JP2008/068928)の実施例7で作製されたΔcyp系統に戻し交配し、得られたGFP陽性マウス個体について、上記記載のPCR法を用いて遺伝子型の解析を行った。交配して得られた仔マウス51匹中の尻尾を一部切り取り、その試料よりゲノムDNAを調製した。得られたDNAについてCYP3A−MAC検出用プライマーおよびWO 2009/063722(PCT/JP2008/068928)の表1記載のプライマーを用いたPCRを前記と同様に行い、CYP3A−MACの保持およびCyp3a遺伝子クラスターのKOを検討した結果、24匹においてCYP3A−MACを保持し、かつ内因性Cyp3a遺伝子群の片方のアレルが破壊された(ヘテロKO)マウス系統であることが確認された。さらにこのヘテロにCyp3a遺伝子群が破壊され、かつCYP3A−MACを保持するマウスとΔcyp系統に戻し交配し、得られたGFP陽性マウス38匹中の尻尾を一部切り取り、その試料よりゲノムDNAを調製し、上記と同様のPCR法を用いて遺伝子型の解析を行った。その結果18匹においてCYP3A−MACを保持し、かつ内因性Cyp3a遺伝子群の両方のアレルが破壊された(ホモKO)マウス系統であることが確認された。(以下、TC(CYP3A−MAC)/Δcypと記す)。
[B]TC(CYP3A−MAC)/Δcypマウス系統における代謝解析
TC(CYP3A−MAC)/Δcypマウス、Δcypマウス個体の肝臓ミクロソームをOmuraら(J.Biol.Chem.,239,2370,1964)に従い、CYP3A4で代謝されることがわかっているトリアゾラム(Triazolam)(200μM)と混合し、その代謝物であるα−OH−triazolamおよび4−OH−triazolamを測定できる。WO 2009/063722(PCT/JP2008/068928)に記載のように、TC(CYP3A−MACΔ)/Δcypマウスにおいて、同系統のマウスやヒト(HLM:human liver microsome)と活性が同程度であることを確かめられる。以上のことから、TC(CYP3A−MAC)/Δcypマウス系統においてCYP3A−MAC上のヒトCYP3A遺伝子が機能的でかつヒトと同等であることが確かめられる。
[C]従って、TC(CYP3A−MAC)/Δcypマウス系統由来の肝臓ミクロソームは医薬品開発における第一相反応の薬効・毒性を調べるための試料として利用できる。また、TC(CYP3A−MAC)/Δcypマウス系統はヒトの薬物代謝を再現できるので、医薬品開発における第一相反応の薬効・毒性を調べるためのin vivo試験用のモデルマウスとして利用できる。
[実施例10]マウス人工染色体ベクターCYP3A−MAC保持ラットの作製
[A]A9細胞からのCYP3A−MACのラットES細胞への移入
CYP3A−MACを保持するキメララットを作製するために、上記実施例8で得られたCYP3A−MACを保持するA9細胞から、子孫伝達可能なラットES細胞であるrESWIv3i−1(Hirabayashi et al.,Mol Reprod Dev.2010 Feb;77(2):94.)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のCYP3A−MACを保持するA9細胞(A9(CYP3A−MAC)8,9など)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のラットES細胞rESWIv3i−1をトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計10個のコロニーを単離し増殖させ、以後の解析を行った。A9(CYP3A−MAC)8からは2クローン、A9(CYP3A−MAC)8からは3クローンがCYP3A−MAC領域のみを検出する前出のプライーマーを用いたPCRで陽性であった。さらに、上記の5クローンについて、CYP3A−BAC(RP11−757A13)(CHORI)ならびにマウスCot−1 DNAを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出され、かつラットの核型が正常なクローンは3クローンであった(図49)。以上のことから、CYP3A−MAC保持ラットES細胞が3クローン得られたと結論付けた。
[B]実施例8記載のように、マウス人工染色体ベクターCYP3A−MACを保持するラットES細胞を用いて、in vitroの安定性を検証できる。また、上記ES細胞からキメララットを作製し、ラットが子孫伝達されたラット系統rTC(CYP3A−MAC)を作製できる。また、上記rTC(CYP3A−MAC)ラット系統を用いて、体細胞におけるCYP3A−MACの安定性を検証できる。また、rTC(CYP3A−MAC)ラット系統由来の肝臓ミクロソームは医薬品開発における第一相反応の薬効・毒性を調べるための試料として利用できる。また、rTC(CYP3A−MAC)ラット系統はヒトの薬物代謝を再現できるので、医薬品開発における第一相反応の薬効・毒性を調べるためのin vivo試験用のモデルラットとして利用できる。
[実施例11]マウス人工染色体ベクターhChr21q−MACの構築
ダウン症候群モデルマウスを作製するため、マウス人工染色体ベクターMAC1にヒト21番染色体長腕のAP001657よりも遠位の領域33Mbを含むDNA配列をCre/loxPシステムを用いて転座クローニングを行い、実施例3と同様にして、hChr21q−MACを構築する。
[A]hChr21−loxP含有DT40からhChr21−loxPのMAC1含有CHO細胞への導入
CHO細胞内でマウス人工染色体ベクターMAC1にloxP配列を介してヒト21番染色体長腕のAP001657よりも遠位の領域を転座挿入するために、loxP配列をAP001657に挿入したヒト21番染色体であるhChr21−loxPをマウス人工染色体ベクターMAC1含有CHO細胞に導入する。
[A.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるhChr21−loxP含有DT40細胞、DT40(kk139)(特開2007−295860)用いて、上記と同様にMAC1含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC1)に微小核細胞融合法を行った。14回の微小核細胞融合で得た合計114個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC1,hChr21−loxP))。
[A.2]薬剤耐性クローンの選別
[A.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、上記114クローンのうちの60クローンについて、以下のプライマーを用いてPCRを行い、ヒト21番染色体断片がMAC1含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。


[A.2.2]two−color FISH解析
上記で得られたCHO(HPRT−;MAC1,hChr21−loxP)の11クローンのうち6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ6クローン中、1クローンで70%の割合でMAC1ならびにhChr21−loxPがCHO細胞に1コピーで導入されていることを確認した(図50)。
以上の結果から、hChr21−loxPがマウス人工染色体ベクターMAC1含有CHO細胞に導入できたと結論付けた。
[B]CHO(HPRT−;MAC1,hChr21−loxP)クローンにおけるヒト21番染色体長腕のAP001657よりも遠位の領域33MbのMAC1ベクターへの部位特異的転座
33MbサイズのDNAであるヒト21番染色体長腕のAP001657よりも遠位の領域をマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC1に転座挿入する(図51)。
[B.1]トランスフェクションおよびHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC1,hChr21−loxP)−37にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計2個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(hChr21q−MAC,hChr21−hChr21q))。
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト21番染色体断片とMAC1上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。


[B.2.2]two−color FISH解析
上記で得られたCHO(hChr21q−MAC,hChr21−hChr21q)の2クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ2クローン中、2クローンで90%以上の割合でMAC1上にヒト21番染色体由来によるシグナルが観察されていることを確認した(図52)。
以上の結果から、ヒト21番染色体長腕のAP001657よりも遠位の領域33Mbがマウス人工染色体ベクターMAC1に相互転座によるクローニングができたと結論付けた。
[C]CHO細胞からのhChr21q−MACのマウスES細胞への移入
hChr21q−MACを保持するキメラマウスを作製するために、上記[B]で得られたhChr21q−MACを保持するCHO細胞からマウスES細胞(野性型TT2F)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のhChr21q−MACを保持するCHO細胞(CHO(hChr21q−MAC,hChr21−hChr21q)1,2)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のマウスES細胞TT2Fをトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計24個のコロニーを単離し増殖させ、以後の解析を行った。CHO(hChr21q−MAC,hChr21−hChr21q)1からは2クローン、CHO(hChr21q−MAC,hChr21−hChr21q)2からは6クローンがhChr21q−MAC領域のみを検出する前出のプライーマーを用いたPCRで陽性であった。さらに、上記のうち8クローンについて、ヒトCot−1 DNAならびにマウスminor satellite DNAを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出され、かつマウスの核型が正常なクローンは2クローンであった(図53)。以上のことから、hChr21q−MAC保持TT2F細胞が2クローン得られたと結論付けた。
[D]hChr21q−MACのマウスES細胞における安定性
上記で得られたマウスESクローン(例えばTT2F(hChr21q−MAC)22、上記[C]で取得)について0~50PDLの非選択培養下での長期培養後におけるFISH解析によるhChr21q−MAC保持細胞の割合を計測した結果、50PDLにおいても95%以上の保持率であった。(図54)。
[E]hChr21q−MACを保持するキメラマウスの作製
上記[C]で得られたhChr21q−MAC保持ES細胞クローンを用いてTomizukaらの方法(Nature Genet.16:133,1997)でキメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。MAC1保持ESクローン(例えばTT2F(hChr21q−MAC)20,22など,上記[C]で取得)を注入した220個の胚を仮親に移植した結果、18匹のキメラマウス(毛色に濃茶色の部分の認められる)が誕生した。うち2匹は白色の部分がほとんど観察できないキメラ率約100%の個体であった。また、うち1匹はGFPが陽性の個体であった(図55)。すなわち、マウス人工染色体ベクターhChr21q−MACを保持するES細胞株(TT2F)はキメラ形成能を保持している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[F]実施例8記載のように、マウス人工染色体ベクターhChr21q−MACを保持するキメラマウスから、hChr21q−MACが子孫伝達されたマウス系統TC(hChr21q−MAC)をを作製できる。また、上記TC(hChr21q−MAC)マウス系統を用いて、体細胞におけるhChr21q−MACの安定性を検証できる。また、TC(hChr21q−MAC)系統はダウン症候群のモデルマウスとして利用でき、ダウン症候群の症状発症のメカニズム解明や症状改善のための治療薬開発に利用できる。
[実施例12]マウス人工染色体ベクターhChr21q22.12−MACの構築
ダウン症候群も出るマウスを作製するため、マウス人工染色体ベクターMAC1にヒト21番染色体長腕のAP00172よりも遠位の領域を含むDNA配列をCre/loxPシステムを用いて転座クローニングを行い、実施例3と同様にして、hChr21q22.12−MACを構築する。
[A]ヒト21番染色体のAP001721へのloxP配列の部位特異的挿入
マウス人工染色体ベクターMAC1にloxP配列を介して転座挿入するために、DT40細胞内でヒト21番染色体(hChr21)のDSCR(ダウン症候群疾患原因領域クラスター)の近位のAP001721へloxP配列を挿入する。
[A.1]ターゲティングベクターpCKloxPHygの作製
ヒト21番染色体上のAP001721のごく近傍かつセントロメア側(約50Kbセントロメア側)に位置するダウン症候群原因遺伝子領域(DSCR)にCre組換え酵素の認識配列loxPを挿入するためのターゲティングベクターpCKloxPHygを以下のようにして作製した。まず、AP001721ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
[A.2]トランスフェクションおよびハイグロマイシン耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpCKloxPHygを制限酵素NotI(TAKARA)で線状化後、ヒト21番染色体を保持するDT40雑種細胞(Kazuki et al.BBRC 2004,DT40(21−2−3))にトランスフェクションし、ハイグロマイシンB(1.5mg/ml)を含む培地に交換し、96穴培養プレート3枚に分注して約2週間の選択培養を行った。4回のトランスフェクションで得た合計178個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr21q22.12−loxP))。
[A.3]相同組換え体の選別
[A.3.1]PCR解析
ハイグロマイシン耐性クローンからPuregene DNA Isolation Kit(Gentra System社)
を用いてゲノムDNAを抽出して、以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
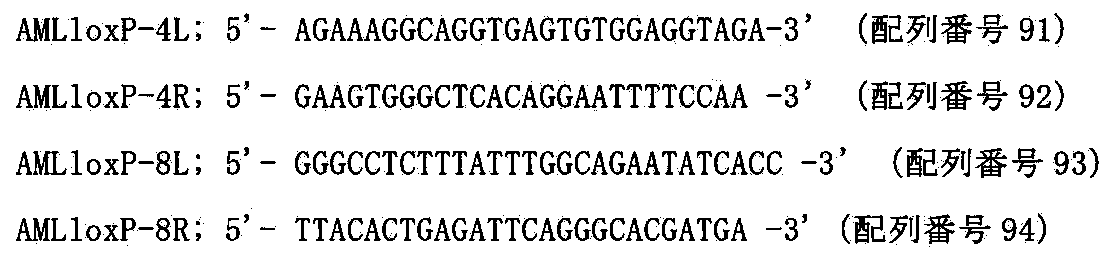
[A.3.2]サザンブロット解析
上記PCR解析にて組換えを確認したうちの10クローンについて以下のようにサザンブロット解析を行った。このゲノムDNAを制限酵素EcoRI(TAKARA)で処理し、0.8%アガロースゲル中で電気泳動し、GeneScreen PlusTM ハイブリダイゼーショントランスファーメンブレン(NENTM Life Science Products,Inc.)へアルカリブロッティングした。このフィルターに対してAP001721中の遺伝子配列をPCRにより増幅したSP7プローブを用いてサザンハイブリダイゼーションを行い相同組換え体の同定を行った。SP7プローブの作製は以下のプライマーを用いDT40(21−2−3)のゲノムDNAを鋳型としてPCRを行い、そのPCR産物を鋳型としてランダムプライミングによる32P標識DNAプローブを作製した(アマシャム、添付のプロトコールに従った)。
SP7プローブ作製用プライマー:
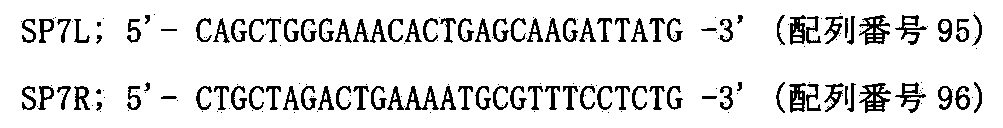
[A.3.3]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち10クローンをヒトcot−1 DNAおよびハイグロマイシンをプローブにしてFISH解析を行ったところ、ヒト21番染色体は全てのクローンで宿主染色体に転座することなく、21q22付近にハイグロマイシン由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた(図58)。以上の結果から、ヒト21番染色体断片に遺伝子導入部位であるloxP配列が部位特異的に挿入されたと結論づけた。
[B]hChr21q22.12−loxP含有DT40からhChr21q22.12−loxPのMAC1含有CHO細胞への導入
CHO細胞内でマウス人工染色体ベクターMAC1にloxP配列を介してヒト21番染色体長腕のAP001721よりも遠位の領域を転座挿入するために、loxP配列をAP001721に挿入したヒト21番染色体であるhChr21q22.12−loxPをマウス人工染色体ベクターMAC1含有CHO細胞に導入する。
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるhChr21q22.12−loxP含有DT40細胞、DT40(hChr21q22.12−loxP)47を用いて、上記と同様にMAC1含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC1)に微小核細胞融合法を行った。15回の微小核細胞融合で得た合計140個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC1,hChr21q22.12−loxP))。
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、上記140クローンのうちの20クローンについて、以下のプライマーを用いてPCRを行い、ヒト21番染色体断片がMAC1含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。


[B.2.2]two−color FISH解析
上記で得られたCHO(HPRT−;MAC1,hChr21q22.12−loxP)の13クローンのうち6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ6クローン中、2クローンで75%の割合でMAC1ならびにhChr21q22.12−loxPがCHO細胞に1コピーで導入されていることを確認した(図59)。
以上の結果から、hChr21q22.12−loxPがマウス人工染色体ベクターMAC1含有CHO細胞に導入できたと結論付けた。
[C]CHO(HPRT−;MAC1,hChr21q22.12−loxP)クローンにおけるヒト21番染色体長腕のAP001721よりも遠位の領域12MbのMAC1ベクターへの部位特異的転座
12MbサイズのDNAであるヒト21番染色体長腕のAP001721よりも遠位の領域をマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC1に転座挿入する(図60)
[C.1]トランスフェクションおよびHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC1,hChr21q22.12−loxP)−12,13にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre 3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計19個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(hChr21q22.12−MAC,hChr21−hChr21q22.12))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト21番染色体断片とMAC1上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。

[C.2.2]two−color FISH解析
上記で得られたCHO(hChr21q22.12−MAC,hChr21−hChr21q22.12)の8クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ8クローン中、8クローンで85%以上割合でMAC1上にヒト21番染色体由来によるシグナルが観察されていることを確認した(図61)。
以上の結果から、ヒト21番染色体長腕のAP001721よりも遠位の領域12Mbがマウス人工染色体ベクターMAC1に相互転座によるクローニングができたと結論付けた。
[D]CHO細胞からのhChr21q22.12−MACのマウスES細胞への移入
hChr21q22.12−MACを保持するキメラマウスを作製するために、上記[C]で得られたhChr21q22.12−MACを保持するCHO細胞からマウスES細胞(野性型TT2F)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のhChr21q−MACを保持するCHO細胞(CHO(hChr21q22.12−MAC,hChr21−hChr21q22.12)1,12など)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のマウスES細胞TT2Fをトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4 PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計13個のコロニーを単離し増殖させ、以後の解析を行った。CHO(hChr21q22.12−MAC,hChr21−hChr21q22.12)1からは1クローン、CHO(hChr21q22.12−MAC,hChr21−hChr21q22.12)12からは1クローンがhChr21q22.12−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であった。さらに、上記のうち2クローンについて、ヒトCot−1 DNAならびにマウスminor satellite DNAを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出され、かつマウスの核型が正常なクローンは1クローンであった(図62)。以上のことから、hChr21q22.12−MAC保持TT2F細胞が1クローン得られたと結論付けた。
[E]hChr21q22.12−MACのマウスES細胞における安定性
上記で得られたマウスESクローン(例えばTT2F(hChr21q22.12−MAC)8、上記[D]で取得)について0~50PDLの非選択培養下での長期培養後におけるFISH解析によるhChr21q22.12−MAC保持細胞の割合を計測した結果、50PDLにおいても95%以上の保持率であった。(図63)。
[F]hChr21q22.12−MACを保持するキメラマウスの作製
上記[D]で得られたhChr21q22.12−MAC保持ES細胞クローンを用いてTomizukaらの方法(Nature Genet.16:133,1997)でキメラマウスを作製した。宿主としてはMCH(ICR)(白色、日本クレア社より購入)の雌雄交配により得られる8細胞期胚を用いた。注入胚を仮親に移植した結果生まれる仔マウスは毛色によりキメラであるかどうかを判定できる。hChr21q22.12−MAC保持ESクローン(例えばTT2F(hChr21q22.12−MAC)8,上記[D]で取得)を注入した80個の胚を仮親に移植した結果、43匹のキメラマウス(毛色に濃茶色の部分の認められる)が誕生した。うち3匹は白色の部分がほとんど観察できないキメラ率約100%の個体であった。すなわち、マウス人工染色体ベクターhChr21q22.12−MACを保持するES細胞株(TT2F)はキメラ形成能を保持している、すなわちマウス個体の正常組織に分化する能力を保持していることが示された。
[G]実施例8記載のように、マウス人工染色体ベクターhChr21q22.12−MACを保持するキメラマウスから、hChr21q22.12−MACが子孫伝達されたマウス系統TC(hChr21q22.12−MAC)を作製できる。また、上記TC(hChr21q22.12−MAC)マウス系統を用いて、体細胞におけるhChr21q22.12−MACの安定性を検証できる。また、hChr21q22.12−MAC系統はダウン症候群のモデルマウスとして利用でき、ダウン症候群の症状発症のメカニズム解明や症状改善のための治療薬開発に利用できる。また、TC(hChr21q−MAC)マウス系統とTC(hChr21q22.12−MAC)マウス系統の表現型を比較することで、ダウン症候群の原因遺伝子領域を同定できる。
[実施例13]マウス人工染色体ベクターMAC2の安定性
[A]マウス人工染色体ベクターMAC2のCHO細胞における安定性
上記で得られたCHOクローン(例えばCHO(HPRT−;MAC2)−13,18、上記実施例4で取得)について0~25PDLの非選択培養下での長期培養後におけるFISH解析によるMAC2保持細胞の割合を計測した結果、25PDLにおいても90%以上の保持率であった。一方、Kazukiら報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)を保持するCHO細胞では、25PDLにおいては70%以下の保持率であった。その代表的な結果を図64に示す。
[実施例14]マウス人工染色体ベクターFVIII−MACの構築
マウス人工染色体ベクターMAC2に有用タンパク質をコードする遺伝子の例として、血友病Aの原因遺伝子である第八因子(FVIII)をCre/loxPシステムを用いて挿入し、機能タンパク質の発現と長期安定性を検証する。
[A]マウス人工染色体ベクターMAC2ベクター含有CHO細胞における特定の有用タンパク質をコードする遺伝子(例えばFVIII)のCre/loxPシステムによるマウス人工染色体ベクターMAC2への挿入
マウス人工染色体MACにDNA挿入配列として5’HPRT−loxP−PGKhygタイプのloxP配列を挿入したマウス人工染色体ベクターMAC2においてloxPが稼働し環状DNAが部位特異的に挿入できるかどうか検証する。
[A.1]FVIII挿入ベクターの作製
pCAGGS(大阪大学、岡部博士より分与)のプロモーターとpoly Aの領域をSalIとPstIサイトにて切断し、pBluescript KS(−)(Stratagene)マルチクローニングサイトを改変したpB3のSalIとPstIサイトにクローニングした(pB−CAG)。pKF17Kプラスミド中のBドメイン欠損FVIII cDNA(自治医科大学、坂田教授より分与)をXhoIとSalIサイトにて切断し、pB−CAGにおけるプロモーターとpoly Aの間にあるEcoRIサイトへクローニングした(pB−CAGF8)。pB−CAGF8のCAG−F8−pA領域をSalIとAvrIIサイトにより単離し、pB3ins2上の2つのHS4インスレーター配列にはさまれるようにSalIとAvrIIサイトへクローニングした(pB3−F8ins2)。次にpPAC4(Children’s Hospital Oakland Research Institute(CHORI),BAC/PAC Resources)を3’HPRT−loxP配列を組み込んだベクターへ導入した。pB3−F8ins2上のAscIとFseI領域であるFVIII発現カセット(HS4−CAG−F8−pA−HS4)をpPAC4のAscIとFseIサイトへクローニングし、HPRT再構築系の1コピーFVIII挿入コンストラクトとした(ベクター名:pPAC4 F8ins2 H3−9(1コピーFVIII−PAC))(図65)。AvrIIサイトとNheIサイトのcompatible cohesive endの特徴を利用し、1コピーFVIII−PACにおけるAscIからAvrII1の1発現カセット領域を同ベクターにおけるAscIからNheIサイトへの再クローニングにより2発現カセットをもつ2コピーFVIII−PACを得た。また同様に挿入カセットをAscIとAvrII、ベクター側をAscIとNheIサイトにて再クローニングすることにより2、4、8、16コピーまでのFVIII発現カセットをもつPACベクターを作製した(図66)。
[A.2]トランスフェクションおよびHAT耐性クローンの単離
遺伝子導入はリポフェクション法を用いて行い、90%コンフルエント状態になった6wellの細胞に対して、Cre1μgと1コピーFVIII−PACベクター10μgを市販(インビトロジェン)のプロトコールに従い、上記CHO(HPRT−;MAC2)−13およびKazukiらの報告(Gene Therapy:PMID:21085194,2010)に記載されたCHO(HPRT−;21HAC2)へ導入した(クローン名:CHO(FVIIIx1−MAC)およびCHO(FVIIIx1−HAC))。また、Cre1μgと16コピーFVIII−PACベクター10μgを市販(インビトロジェン)のプロトコールに従い、上記CHO(HPRT−;MAC2)−13へ導入した(クローン名:CHO(FVIIIx16−MAC))。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、4回の導入で得たそれぞれCHO(FVIIIx1−MAC)については46クローン、CHO(FVIIIx16−MAC)については18クローン、CHO(FVIIIx1−HAC)については19クローン、合計83個のコロニーを単離し増殖させ、以後の解析を行った。
[A.3]薬剤耐性クローンの選別
[A.3.1]PCR解析
HAT耐性株のゲノムDNAを鋳型として組換え体を選別するため以下のプライマーを用いてPCRを行い、部位特異的にFVIII遺伝子の挿入が起こっているかを確認した。そのプライマー配列を以下に示す。
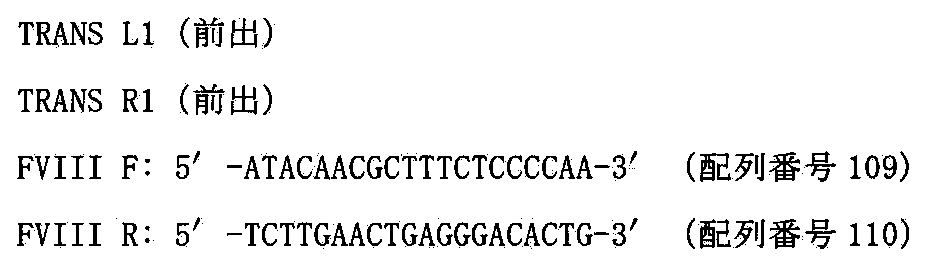
[A.3.2]mono−color FISH解析
上記の結果から7クローンにおいて、mono−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNAまたはヒトcot−1 DNAをプローブにしてFISH解析を行ったところ、CHO(FVIIIx1−MAC)由来については1クローンが90%以上の割合でFVIII−MACが保持され、CHO(FVIIIx1−HAC)由来については2クローンが90%以上の割合でFVIII−HACが保持され、CHO(FVIIIx16−MAC)由来については1クローンが90%以上の割合でFVIII−HACが保持されていた。
[B]CHO細胞におけるFVIII遺伝子の遺伝子発現解析
上記のFVIIIx1−MACが保持されているCHO(FVIIIx1−MAC)1−3およびFVIIIx1−HACが保持されているCHO(FVIIIx1−HAC)1−2およびFVIIIx16−MACが保持されているCHO(FVIIIx16−MAC)16−1,16−2,16−3を用いて、FVIII mRNAが発現しているかどうかを検討するため、上記記載に従い、RNAを抽出し、そのRNAを鋳型としてcDNA合成し、さらに以下のプライマー(前出)を用いてPCRを行った。温度、サイクル条件は94℃10分の熱変性後、94℃30秒、60℃30秒、72℃1分を25サイクル行った。

[C]CHO細胞におけるFVIII遺伝子の遺伝子機能解析
上記、FVIII mRNAの発現が確認されたCHO(FVIIIx1−MAC)1−3およびCHO(FVIIIx1−HAC)1−2におけるFVIIIタンパク発現が機能的か否かを検証するためClotting assay(コスモバイオ)を添付のプロトコールに従い、行った。0PDLおよび非選択培養で25PDLまで培養した上記細胞を6ウェルディッシュに培養し100%コンフルエント状態から新しいメディウムに交換した。24時間後培養上清を回収しFVIII活性によるFVIIIの活性化度合いを測定した。その結果、0PDLではCHO(FVIIIx1−MAC)1−3およびCHO(FVIIIx1−HAC)1−2に活性の差はほとんど見られなかった。一方、CHO(FVIIIx1−MAC)1−3では25PDLにおいても活性が1.8倍に上昇していたのに対し、CHO(FVIIIx1−HAC)1−2では25PDLにおいては活性が1/6倍に低下していた(図67)。また、CHO(FVIIIx1−MAC)1−3に比べ、CHO(FVIIIx16−MAC)16−1,16−2,16−3の活性は約10倍に上昇しており、コピー数依存的に活性が上昇することが確かめられた(図68)。
以上の実験によりマウス人工染色体MAC2ベクター上にFVIII遺伝子を搭載することにより、FVIII遺伝子の機能的な発現が観察され、さらにFVIII−MACを保持するCHOではFVIII−HAC保持するCHOよりも長期間安定に機能的発現を呈することが確かめられた。また、PACベクターによって、200kb以内の有用タンパク質をコードするDNAが挿入可能である。
[実施例15]複数遺伝子の導入が可能なマウス人工染色体ベクターMI−MACの構築
マウス人工染色体ベクターMAC2に複数の遺伝子を搭載できる例として、5つの部位特異的組換え酵素認識部位(ΦC31 attP、R4 attP、TP901−1 attP、Bxb1 attP、FRT)を持つmulti−integrase platformをCre/loxPシステムを用いて挿入し、複数遺伝子の導入・発現を検証する。
[A.1]複数遺伝子搭載部位(multi−integrase platform)カセットの作製
マウス人工染色体ベクターに複数の遺伝子を導入するための複数遺伝子搭載部位を持つカセットをMultisite−Gateway kit(Invitrogen)を用いて、以下のようにして作製した。まず、PGK−hyg(Clontech)をテンプレートとして、遺伝子導入部位であるΦC31 attP site、R4 attP site、TP901−1 attP site、Bxb1 attP site、FRT siteをfirst PCR(以下の各primer pair F1−R1)によりPGK promoter配列に付加した。PCRは、サーマルサイクラーとしてPerkin−Elmer社製のGene Amp 9600を、Taqポリメラーゼはkodplus(Toyobo)を用い、バッファーやdNTPs(dATP,dCTP,dGTP,dTTP)は添付のものを推奨される条件に従って用いた。温度、サイクル条件は94℃2分の熱変性後、94℃15秒、68℃30秒、72℃90秒を20サイクル行った。PCR産物をプロテネースK(ギブコ)処理した後、CHROMASPIN−TE400(クローンテック)で精製した。


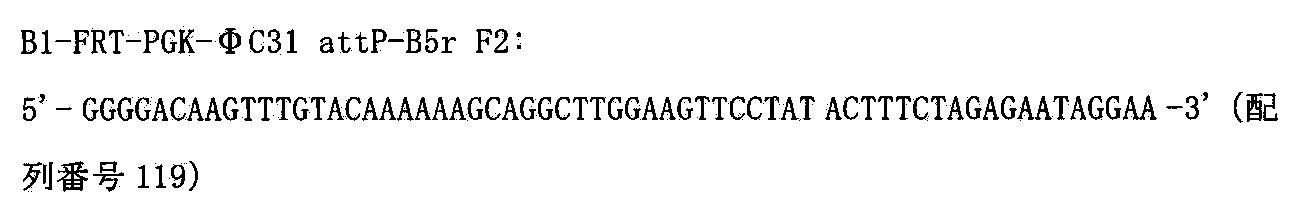

次に、これらの遺伝子導入部位をマウス人工染色体ベクターに導入するために必要な、3’HPRT−loxP配列を組み込んだplasmidを以下の手順で作製した。上記X3.1をテンプレートとして、以下のプライマーを用いて増幅した。PCR条件は、上記の条件(サイクル数25)と同様である。

[A.2]複数遺伝子搭載部位(multi−integrase platform)カセットのマウス人工染色体ベクターへの搭載
複数遺伝子搭載部位(multi−integrase platform)カセットは上述のCHO(HPRT−;MAC2)や下記記載のCHO(HPRT−;MAC4)に上述のようにCre−loxP組み換え後、HAT耐性クローンを取得することでマウス人工染色体ベクターMAC2やMAC4に挿入できる(MI−MACとよぶ)(図71)。
[A.3]遺伝子導入カセットの作製
multi−integrase platformに外来遺伝子を導入するためのカセットベクターを以下のように作製した。まず、薬剤選択のために必要なPromoterlessのNeomycin−resistant geneをpIRES Neo2(Clontech)をテンプレートに、以下のプライマーを用いて増幅した。PCR条件は、上記の条件(サイクル数25)と同様である。

[A.4]部位特異的組換え酵素発現ベクターの作製
各々対応したattB−attP間の組換えを起こす部位特異的組み換え酵素(ΦC31 integrase、R4 integrase、TP901−1 integrase、Bxb1 integrase)(GenBank accession numbers:ΦC31,CAA07153;R4,BAA07372;TP901−1,CAA59475;Bxb1,AAG59740)をde novo合成した(ΦC31:Codon device、他:Invitrogen)。これらのintegraseは哺乳類細胞での高発現を行うよう、codon usageを哺乳類に最適化して合成したものである。この合成したベクターから制限酵素KpnI−XbaIでΦC31 integraseを含むDNA断片を切り出し、制限酵素KpnI−XbaIで切断したpVAX1(Invitrogen)にライゲーションしたpCMV−ΦC31や、制限酵素NheI−XhoIでR4 integrase、あるいはTP901−1 integrase、Bxb1 integraseを含むDNA断片を切り出し、制限酵素NheI−XhoIで切断したpVAX1(Invitrogen)にライゲーションしたpCMV−R4、pCMV−TP901−1、pCMV−Bxb1を作製した(図72)。
[A.5]MI−MACベクターへの遺伝子導入
Cre発現ベクターの代わりに上述の各種部位特異的組換え酵素発現ベクターと、FVIII挿入ベクターの代わりに遺伝子導入カセットを導入することで、複数(1~5個)の遺伝子をマウス人工染色体ベクターMI−MACベクター上へ挿入することができる。また、複数遺伝子搭載部位(multi−integrase platform)カセットをマウス人工染色体MACベクター上に複数搭載することも可能であるので、無制限に遺伝子を挿入することもできる(図72)。
[実施例16]マウス人工染色体ベクターPXR−MACの構築
マウス人工染色体ベクターMAC3に核内受容体であるヒトPXRをCre/loxPシステムを用いて挿入し、PXR−MACを構築する。
[A.1]ヒトPXR挿入ベクターの作製
ヒトPXR遺伝子およびloxP配列を挿入するための基本BACベクターにはヒトPXR遺伝子全長を含むRP11−169N13(CHORI)を用いた。Yamadaら(J Hum Genet.2008;53(5):447−53)の方法に従い、上記BACベクター上のカナマイシン耐性遺伝子領域にマウス人工染色体ベクターMAC3に挿入するためのAmp−5’HPRT−loxP配列を相同組み換えにより挿入した(ベクター名:PXR−loxP)。
Cre/loxPシステムによるHPRT再構築系によるヒトPXR挿入により生じる部位特異的DNA挿入を(図73)に示す。
[A.2]トランスフェクションおよびHAT耐性クローンの単離
遺伝子導入はリポフェクション法を用いて行い、90%コンフルエント状態になった6wellの細胞に対して、Cre1μgとPXR−loxPベクター2μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計11個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(PXR−MAC))。
[A.3]薬剤耐性クローンの選別
[A.3.1]PCR解析
HAT耐性株のゲノムDNAを鋳型として組換え体を選別するため以下のプライマーを用いてPCRを行い、部位特異的にPXR遺伝子の挿入が起こっているかを確認した。そのプライマー配列を以下に示す。


[A.3.3]two−color FISH解析
上記から結果から選んだ6クローンにおいて、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNAおよびヒトPXR−BAC由来のDNA(RP11−169N13)(CHORI)をプローブにしてFISH解析を行ったところ、6クローンのうち、5クローンにおいて60%以上の割合でPXR−MACが保持され、さらにPXR−BAC由来のシグナルが現れ、ネガティブコントロールであるPXR−BACを部位特異的挿入する前のMAC3上にはシグナルが検出されなかったことから、部位特異的にヒトPXR遺伝子が挿入されたことが確かめられた(図74)。
以上の実験によりマウス人工染色体MAC3上にヒトPXR遺伝子を搭載することにより、マウス人工染色体ベクターPXR−MACを保持するCHO細胞を得られたことが確認できた。
[B]マウス人工染色体ベクターPXR−MAC含有CHO細胞からマウスES細胞へマウス人工染色体ベクターPXR−MACの導入
PXR−MACを保持するキメラマウスを作製するために、上記[A]で得られたPXR−MACを保持するCHO細胞からマウスES細胞(野性型TT2F)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のPXR−MACを保持するCHO細胞(CHO(PXR−MAC)7,9,10など)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のマウスES細胞TT2Fをトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計34個のコロニーを単離し増殖させ、以後の解析を行った。CHO(PXR−MAC)7からは2クローン、CHO(PXR−MAC)9からは2クローン、CHO(PXR−MAC)10からは12クローンがPXR−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であった。さらに、上記の16クローンについて、ヒトPXR−BAC由来のDNA(RP11−169N13)(CHORI)を用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出されたクローンは16クローンのうち、4クローンであった。以上のことから、PXR−MAC保持TT2F細胞が4クローン得られたと結論付けた(図75)。
[C]実施例8記載のように、マウス人工染色体ベクターPXR−MACを保持するマウスES細胞からキメラマウスを作製し、PXR−MACが子孫伝達されたマウス系統TC(PXR−MAC)を作製できる。また、上記TC(PXR−MAC)マウス系統を用いて、体細胞におけるPXR−MACの安定性を検証できる。また、TC(PXR−MAC)マウス系統はヒトにおける薬物によるCYP遺伝子発現誘導を再現できる。また、上記TC(CYP3A−MAC)マウス系統との交配により、TC(CYP3A−MAC/PXR−MAC)系統を作製でき、医薬品開発における薬効・毒性を調べるためのin vivo試験用のモデルマウスとして利用できる。
[実施例17]マウス人工染色体ベクターMAC4の構築
マウス人工染色体MACにDNA挿入配列としてGFP−5’HPRT−loxP−PGKhygタイプのloxP配列を挿入したマウス人工染色体ベクターMAC4を構築する。5’HPRT−loxP−PGKhygタイプのloxP配列は、Kazukiらの報告(Gene Therapy:PMID:21085194,2010)に記載された21番染色体由来のGFP搭載HACベクター(21HAC2)に挿入されており、HACとMACの遺伝子発現を同じベクターで比較することができる。また、21HAC2への挿入のための遺伝子導入ベクターがベクター作製のステップを経ることなく、そのまま利用可能である。
[A]マウス人工染色体MACへのGFP−5’HPRT−loxP−hygタイプのloxP配列の挿入
[A.1]GFP−5’HPRT−loxP−hygタイプのloxPターゲティングベクターの作成
loxP配列を挿入するための基本プラスミドには上記で作成したpMAC2を用いた。NotIとSalIにより切り出したHS4−CAG−EGFP−HS4(大阪大学、岡部博士及びNIH、Felsenfeld博士より分与)を平滑化し、上記pMAC2をXhoI切断ならびに平滑化後にクローニングした(ベクター名:pMAC4)。ターゲティングベクター、標的配列および相同組換えにより生じる染色体アレルを図76に示す。
[A.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpMAC4を制限酵素NotI(TAKARA)で線状化後、上記で作製したクローンDT40(MAC)にトランスフェクションし、ハイグロマイシン(1.5mg/ml)を含む培地に交換し、96穴培養プレート2枚に分注して約2週間の選択培養を行った。1回のトランスフェクションで得た合計36個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(MAC4))
[A.3]相同組換体の選別
[A.3.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス染色体ベクターMAC上で部位特異的に組換えが起こっているかを確認した。そのプライマー配列を以下に示す。
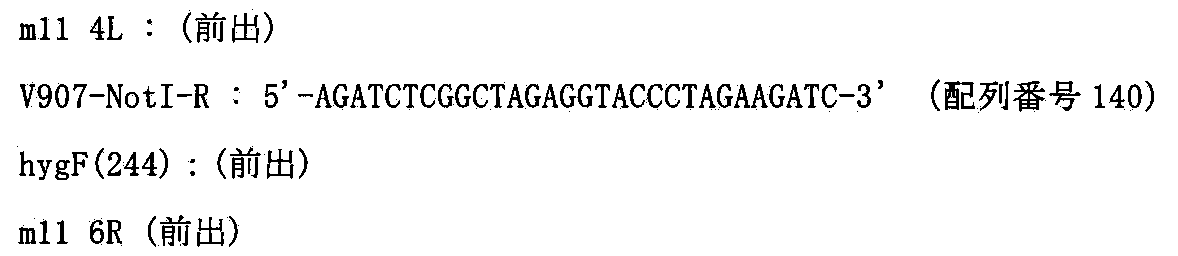
[A.3.2]two−color FISH解析
上記から得られたDT40(MAC4)の5クローンにおいて、two−color FISH解析を松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。マウスcot−1 DNAおよびGFP−5’HPRT−loxP−hygカセットをプローブにしてFISH解析を行ったところ、ネガティブコントロールのターゲティングする前のマウス人工染色体ベクターMACにおいては、プローブ由来のシグナルが検出されなかったのに対し、DT40(MAC4)の5クローンでは、65%以上の割合でプローブ由来のシグナルが検出されたことから、上記5クローンにおいて、部位特異的に組換えが起こったことが視覚的に確かめられた(図77)。これらの結果から、マウス人工染色体ベクターMAC4を保持するDT40細胞クローンが得られたと結論できた。
[B]マウス人工染色体ベクターMAC4含有ニワトリDT40細胞からMAC4のCHO細胞への導入
[B.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(MAC4)−B1−5及びB1−74及びB2−3及びB2−4を用いて、上記と同様にCHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)であるCHO(HPRT−)に微小核細胞融合法を行った。4回の微小核細胞融合で得た合計23個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC4))
[B.2]薬剤耐性クローンの選別
[B.2.1]PCR解析
ハイグロマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、マウス人工染色体ベクターMAC4がCHO細胞に導入できているかを確認した。そのプライマー配列を以下に示す。

[B.2.2]mono−color FISH解析
上記で得られたCHO(HPRT−;MAC4)の7クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNAをプローブにしたFISH解析を行ったところ7クローン中、4クローンで95%以上の割合でCHO細胞にMAC4が導入されていることを確認した(図78)。
以上の結果から、マウス人工染色体ベクターMAC4がCHO細胞に導入できたと結論付けた。
[C]実施例8記載のように、マウス人工染色体ベクターMAC4を保持するマウスES細胞を作製し、そのES細胞を用いてin vitroの安定性を検証できる。また、上記ES細胞からキメラマウスを作製し、MAC4が子孫伝達されたマウス系統TC(MAC4)を作製できる。また、上記TC(MAC4)マウス系統を用いて、体細胞におけるMAC4の安定性を検証できる。
[実施例18]マウス人工染色体ベクターUGT2−MACの構築
マウス人工染色体ベクターMAC4にヒト薬物代謝酵素遺伝子群であるUGT2クラスターをCre/loxPシステムを用いて転座クローニングを行い、実施例3と同様にして、UGT2−MACを構築する。また、UGT2−MACのマウスES細胞での安定性を検証する。
[A]ヒト4番染色体上のAC125239における部位特異的切断
ヒト4番染色体のUGT2遺伝子クラスターから遠位側の遺伝子を削除するために、部位特異的染色体削除であるテロメアトランケーションを行う。
[A.1]ターゲティングベクターpTELpuro−UGT2の作製
ヒト4染色体上のUGT2遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAC125239領域にヒトテロメア配列を挿入するためのターゲティングベクターpTELpuro−UGT2を以下のようにして作製した。まず、AC125239ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
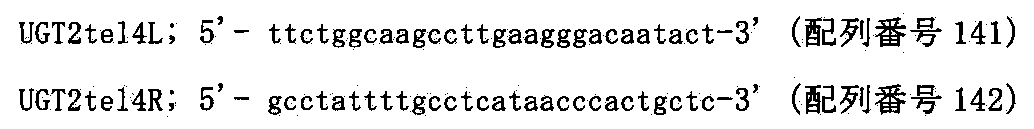
[A.2]トランスフェクションおよび薬剤耐性クローンの単離
Kazuki et al.BBRC 2004に記載された方法で、ヒト4番染色体を保持するA9(KM64−4)(Kugoh et al.DNA research 1999)からヒト4番染色体を保持するニワトリDT40細胞を作製した(クローン名:DT40(hChr4))。次に上述と同様に、上記で作製したターゲティングベクターpTELpuro−UGT2を制限酵素PstI(ニッポンジーン)で線状化後、上記で作製したクローンDT40(hChr4)1にトランスフェクションし、ピューロマイシン(0.3ug/ml)を含む培地に交換し、96穴培養プレート10枚に分注して約2週間の選択培養を行った。4回のトランスフェクションで得た合計96個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr4−tel))。
[A.3]相同組換え体の選別
[A.3.1]PCR解析
ピューロマイシン耐性株のゲノムDNAを鋳型として組換え体を選別するため一次スクリーニングとして切断部位よりテロメア側に位置する以下のプライマーを用いてPCRを行い、部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。


[A.3.2]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち2クローンをヒトcot−1 DNAおよびピューロマイシンDNAをプローブにしてFISH解析を行ったところ、ヒト4番染色体は全てのクローンで宿主染色体に転座することなく、ヒト4番染色体断片末端にピューロマイシン由来のシグナルが検出され、目的部位で切断されていたことから部位特異的に組換えが起こったことが確かめられた(図80)。
以上の結果から、クローンDT40(hChr4−tel)35及び73において、UGT2遺伝子クラスター領域よりテロメア側のAC125239から遠位において切断できたと結論付けた。
[B]ヒト4番染色体のAC074378へのloxP配列の部位特異的挿入
マウス人工染色体ベクターMAC4にloxP配列を介して転座挿入するために、DT40細胞内でhChr4−telのUGT2遺伝子クラスターの近位のAC074378へloxP配列を挿入する。
[B.1]ターゲティングベクターpUGT2loxPneoの作製
ヒト4番染色体上のUGT2遺伝子座のごく近傍かつセントロメア側(約300Kbセントロメア側)に位置するAC074378領域にCre組換え酵素の認識配列loxPを挿入するためのターゲティングベクターpUGT2loxPneoを以下のようにして作製した。まず、AC074378ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
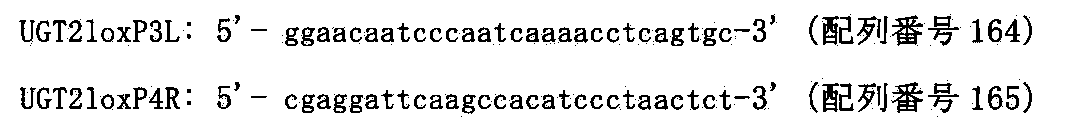
[B.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpUGT2loxPneoを制限酵素NotI(TAKARA)で線状化後、ヒト4番染色体を保持するニワトリDT40細胞(クローンDT40(hChr4−tel)35にトランスフェクションし、ネオマイシン(1.5mg/ml)を含む培地に交換し、96穴培養プレート3枚に分注して約2週間の選択培養を行った。2回のトランスフェクションで得た合計12個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr4−tel−loxP))。
[B.3]相同組換え体の選別
[B.3.1]PCR解析
ネオマイシン耐性クローンからPuregene DNA Isolation Kit(Gentra System社)を用いてゲノムDNAを抽出して、以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。

[B.3.2]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち4クローンをヒトcot−1 DNAおよびネオマイシンDNAをプローブにしてFISH解析を行ったところ、ヒト4番染色体は全てのクローンで宿主染色体に転座することなく、4q13付近にネオマイシン由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた(図82)。以上の結果から、ヒト4番染色体上のAC074378に遺伝子導入部位であるloxP配列が部位特異的に挿入されたと結論づけた。
[C]hChr4−loxP−tel含有DT40からhChr4−loxP−telのMAC4含有CHO細胞への導入。
CHO細胞内でマウス人工染色体ベクターMAC4にloxP配列を介してヒトUGT2遺伝子クラスター領域を転座挿入するために、hChr4−loxP−telをマウス人工染色体ベクターMAC4含有CHO細胞に導入する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(hChr4−loxP−tel)5及び10を用いて、上記と同様にMAC4含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC4)に微小核細胞融合法を行った。3回の微小核細胞融合で得た合計22個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC4,hChr4−loxP−tel))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
ネオマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、ヒト4番染色体断片がMAC4含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。


[C.2.2]two−color FISH解析
上記で得られたCHO(HPRT−;MAC4,hChr4−loxP−tel)の5クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ、1クローンにおいて90%以上のMAC1ならびにhChr4−loxP−telがCHO細胞に1コピーもしくは2コピーで導入されていることを確認した(図83)。
以上の結果から、hChr4−loxP−telがマウス人工染色体ベクターMAC4含有CHO細胞に導入できたと結論付けた。
[D]CHO(HPRT−;MAC4,hChr4−loxP−tel)クローンにおけるヒトUGT2遺伝子クラスター領域周辺(AC074378−ヒトUGT2遺伝子クラスター−AC125239)2MbのMAC4ベクターへの部位特異的転座
2MbサイズのDNAであるヒトUGT2遺伝子クラスターをマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC4に転座挿入する(図84)。
[D.1]トランスフェクションおよびHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC4,hChr4−loxP−tel)8にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計6個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(UGT2−MAC,hChr4−ΔUGT2))。
[D.2]薬剤耐性クローンの選別
[D.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト4番染色体断片とMAC4上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。


[D.2.2]two−color FISH解析
上記で得られたCHO(UGT2−MAC,hChr4−ΔUGT2)の6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でUGT2−BAC(RP11−643N16)(CHORI)DNA及びマウスCot−1 DNAをプローブにしたFISH解析を行ったところ6クローン中、2クローンで50%以上の割合でMAC4上にヒトUGT2由来によるシグナルが観察されていることを確認した(図85)。
以上の結果から、ヒト4番染色体断片上のUGT2クラスター2Mbがマウス人工染色体ベクターMAC4に相互転座によるクローニングができたと結論付けた。
[E]CHO細胞からのUGT2−MACのマウスA9細胞への移入
UGT2−MACを保持するマウスES細を作製するために、上記[D]で得られたUGT2−MACを保持するCHO細胞(CHO(UGT2−MAC,hChr4−ΔUGT2)4,5)からマウスA9細胞へミクロセル形成能の高いマウスA9細胞へ微小核細胞融合法により導入した。4回の微小核細胞融合で得た合計16個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:A9(UGT2−MAC))。その結果、UGT2−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であるクローンが5クローンあった。さらに、UGT2−BAC(RP11−643N16)(CHORI)及びマウスminor satellite DNAプローブを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出されるUGT2−MACの存在が5クローン中、全てのクローンで確認された(図86)。以上のことから、UGT2−MAC保持A9細胞が5クローン得られたと結論付けた。
[F]A9細胞からのUGT2−MACのマウスES細胞への移入
UGT2−MACを保持するキメラマウスを作製するために、上記[E]で得られたUGT2−MACを保持するA9細胞からマウスES細胞(野性型TT2F)へ微小核細胞融合法により導入した。Tomizukaら(Nature Genet.16:133,1997)の方法に従い、約108個のUGT2−MACを保持するA9細胞(A9(UGT2−MAC)13,15など)からミクロセルを精製し、DMEM 5mlに懸濁した。約107個のマウスES細胞TT2Fをトリプシン処理により剥がし、DMEMで3回洗浄後DMEM 5mlに懸濁し、遠心したミクロセルに加え、1250rpmで10分間遠心し、上清を完全に取り除いた。沈殿をタッピングにより十分ほぐし、1:1.4PEG溶液[5g PEG1000(和光純薬)、1ml DMSO(シグマ)をDMEM 6mlに溶解]0.5mlを加え、約1分30秒間、十分攪拌した。その後、DMEM 10mlをゆっくり加え、1250rpmで10分間遠心し、30mlのES培地に懸濁し、予めフィーダー細胞をまいた直径100mmシャーレ(コーニング社)3枚に分注し、培養した。24時間後、300μg/mlのG418を含む培地に交換し、約1週間選択培養した。その結果、合計25個のコロニーを単離し増殖させ、以後の解析を行った。A9(UGT2−MAC)13からは5クローン、A9(UGT2−MAC)15からは4クローンがUGT2−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であった。さらに、上記のうち9クローンについて、UGT2−BAC(RP11−643N16)(CHORI)ならびにマウスminor satellite DNAを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出され、かつマウスの核型が正常なクローンは7クローンであった(図87)。以上のことから、UGT2−MAC保持TT2F細胞が7クローン得られたと結論付けた。
[G]UGT2−MACのマウスES細胞における安定性
上記で得られたマウスESクローン(例えばTT2F(UGT2−MAC)9,10,19、上記[F]で取得)について0~50PDLの非選択培養下での長期培養後におけるFISH解析によるUGT2−MAC保持細胞の割合を計測した結果、50PDLにおいても95%以上の保持率であった。(図88)。
[H]実施例8記載のように、マウス人工染色体ベクターUGT2−MACを保持するマウスES細胞からキメラマウスを作製し、UGT2−MACが子孫伝達されたマウス系統TC(UGT2−MAC)を作製できる。また、上記TC(UGT2−MAC)マウス系統を用いて、体細胞におけるUGT2−MACの安定性を検証できる。また、TC(UGT2−MAC)マウス系統はヒトの薬物代謝を再現できるので、医薬品開発における第二相反応の薬効・毒性を調べるためのin vivo試験用のモデルマウスとして利用できる。
[実施例19]マウス人工染色体ベクターCYP2C−MACの構築
マウス人工染色体ベクターMAC4にヒト薬物代謝酵素遺伝子群であるCYP2CクラスターをCre/loxPシステムを用いて転座クローニングを行い、実施例3と同様にして、CYP2C−MACを構築する。
[A]ヒト10番染色体上のAL157834における部位特異的切断
ヒト10番染色体のCYP2C遺伝子クラスターから遠位側の遺伝子を削除するために、部位特異的染色体削除であるテロメアトランケーションを行う。
[A.1]ターゲティングベクターpTELpuro−CYP2Cの作製
ヒト10染色体上のCYP2C遺伝子座のごく近傍かつテロメア側(約150Kbテロメア側)に位置するAL157834領域にヒトテロメア配列を挿入するためのターゲティングベクターpTELpuro−CYP2Cを以下のようにして作製した。まず、AL157834ゲノム領域を以下のプライマーを用いてPCRにより増幅した。

[A.2]トランスフェクションおよび薬剤耐性クローンの単離
Kazuki et al.BBRC 2004に記載された方法で、ヒト10番染色体を保持するA9(KM32−2)およびA9(KM26−3)(Kugoh et al.DNA research 1999)からヒト10番染色体を保持するニワトリDT40細胞を作製した(クローン名:DT40(hChr10))。次に上述と同様に、上記で作製したターゲティングベクターpTELpuro−CYP2Cを制限酵素PstI(ニッポンジーン)で線状化後、上記で作製したクローンDT40(hChr10)1,42にトランスフェクションし、ピューロマイシン(0.3ug/ml)を含む培地に交換し、96穴培養プレート10枚に分注して約2週間の選択培養を行った。4回のトランスフェクションで得た合計96個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr10−tel))。
[A.3]相同組換え体の選別
[A.3.1]PCR解析
ピューロマイシン耐性株のゲノムDNAを鋳型として組換え体を選別するため一次スクリーニングとして切断部位よりテロメア側に位置する以下のプライマーを用いてPCRを行い、部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。


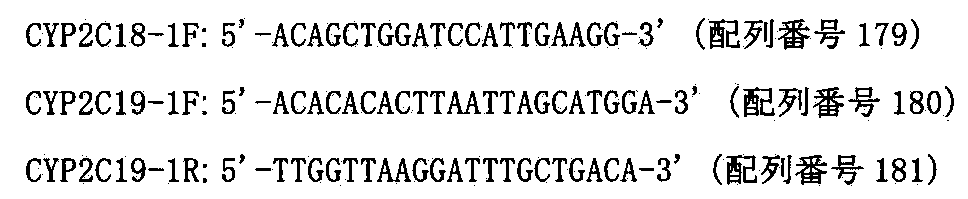
[B.3.2]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認した3クローンをヒトcot−1 DNAおよびピューロマイシンDNAをプローブにしてFISH解析を行ったところ、ヒト10番染色体は全てのクローンで宿主染色体に転座することなく、ヒト10番染色体断片末端にピューロマイシン由来のシグナルが検出され、目的部位で切断されていたことから部位特異的に組換えが起こったことが確かめられた(図90)。
以上の結果から、クローンDT40(hChr10−tel)5及び98及び101において、CYP2C遺伝子クラスター領域よりテロメア側のAL157834から遠位において切断できたと結論付けた。
[B]ヒト10番染色体のAL138759へのloxP配列の部位特異的挿入
マウス人工染色体ベクターMAC4にloxP配列を介して転座挿入するために、DT40細胞内でhChr10−telのCYP2C遺伝子クラスターの近位のAL138759へloxP配列を挿入する。
[B.1]ターゲティングベクターpCYP2CloxPneoの作製
ヒト10番染色体上のCYP2C遺伝子座のごく近傍かつセントロメア側(約300Kbセントロメア側)に位置するAL138759領域にCre組換え酵素の認識配列loxPを挿入するためのターゲティングベクターpCYP2CloxPneoを以下のようにして作製した。まず、AL138759ゲノム領域を以下のプライマーを用いてPCRにより増幅した。
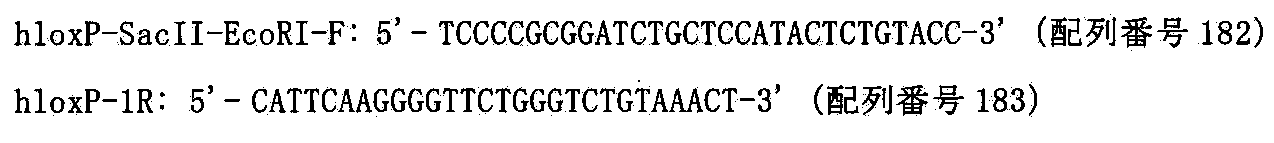
[B.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpCYP2CloxPneoを制限酵素NotI(TAKARA)で線状化後、ヒト10番染色体を保持するニワトリDT40細胞(クローンDT40(hChr10−tel)1−98にトランスフェクションし、ネオマイシン(1.5mg/ml)を含む培地に交換し、96穴培養プレート3枚に分注して約2週間の選択培養を行った。2回のトランスフェクションで得た合計15個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr10−tel−loxP))。
[B.3]相同組換え体の選別
[B.3.1]PCR解析
ネオマイシン耐性クローンからPuregene DNA Isolation Kit(Gentra System社)
を用いてゲノムDNAを抽出して、以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。

[B.3.2]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認したクローンのうち1クローンをヒトcot−1 DNAおよびネオマイシンをプローブにしてFISH解析を行ったところ、ヒト10番染色体は全てのクローンで宿主染色体に転座することなく、10q24付近にネオマイシン由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた。以上の結果から、ヒト10番染色体上のAL138759に遺伝子導入部位であるloxP配列が部位特異的に挿入されたと結論づけた。
[C]hChr10−loxP−tel含有DT40からhChr10−loxP−telのMAC4含有CHO細胞への導入。
CHO細胞内でマウス人工染色体ベクターMAC4にloxP配列を介してヒトCYP2C遺伝子クラスター領域を転座挿入するために、hChr10−loxP−telをマウス人工染色体ベクターMAC4含有CHO細胞に導入する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(hChr10−loxP−tel)7を用いて、上記と同様にMAC4含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC4)に微小核細胞融合法を行った。3回の微小核細胞融合で得た合計8個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC4,hChr10−loxP−tel))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
ネオマイシン耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、ヒト10番染色体断片がMAC4含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。

[C.2.2]two−color FISH解析
上記で得られたCHO(HPRT−;MAC4,hChr10−loxP−tel)の8クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ、2クローンにおいて90%以上の割合でMAC1ならびにhChr10−loxP−telがCHO細胞に1コピーもしくは2コピーで導入されていることを確認した(図92)。
以上の結果から、hChr10−loxP−telがマウス人工染色体ベクターMAC4含有CHO細胞に導入できたと結論付けた。
[D]CHO(HPRT−;MAC4,hChr10−loxP−tel)クローンにおけるヒトCYP2C遺伝子クラスター領域周辺(AL138759−ヒトCYP2C遺伝子クラスター−AL157834)380kbのMAC4ベクターへの部位特異的転座
380kbサイズのDNAであるヒトCYP2C遺伝子クラスターをマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC4に転座挿入する(図93)。
[D.1]トランスフェクションおよびHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC4,hChr10−loxP−tel)1及び5にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計11個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(CYP2C−MAC,hChr10−ΔCYP2C))。
[D.2]薬剤耐性クローンの選別
[D.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト10番染色体断片とMAC4上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。

[D.2.2]two−color FISH解析
上記で得られたCHO(CYP2C−MAC,hChr10−ΔCYP2C)の9クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でCYP2C−BAC(RP11−466J14)(CHORI)DNA及びマウスCot−1 DNAをプローブにしたFISH解析を行ったところ9クローン中、3クローンで50%以上の割合でMAC4上にヒトCYP2C由来によるシグナルが観察されていることを確認した(図94)。
以上の結果から、ヒト10番染色体断片上のCYP2Cクラスター380kbがマウス人工染色体ベクターMAC4に相互転座によるクローニングができたと結論付けた。
[E]CHO細胞からのCYP2C−MACのマウスA9細胞への移入
CYP2C−MACを保持するマウスES細を作製するために、上記[D]で得られたCYP2C−MACを保持するCHO細胞(CHO(CYP2C−MAC,hChr10−ΔCYP2C)2,8,10)からマウスA9細胞へミクロセル形成能の高いマウスA9細胞へ微小核細胞融合法により導入した。4回の微小核細胞融合で得た合計4個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:A9(CYP2C−MAC))。その結果、CYP2C−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であるクローンが4クローンあった。さらに、CYP2C−BAC(RP11−466J14)(CHORI)ならびにマウスminor satellite DNAプローブを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出されるCYP2C−MACの存在が4クローン中、2クローンで確認された。以上のことから、CYP2C−MAC保持A9細胞が2クローン得られたと結論付けた。
[F]実施例8記載のように、マウス人工染色体ベクターCYP2C−MACを保持するマウスES細胞を作製し、そのES細胞を用いてin vitroの安定性を検証できる。また、上記ES細胞からキメラマウスを作製し、CYP2C−MACが子孫伝達されたマウス系統TC(CYP2C−MAC)を作製できる。また、上記TC(CYP2C−MAC)マウス系統を用いて、体細胞におけるCYP2C−MACの安定性を検証できる。また、TC(CYP2C−MAC)マウス系統由来の肝臓ミクロソームは医薬品開発における第一相反応の薬効・毒性を調べるための試料として利用できる。また、TC(CYP2C−MAC)マウス系統はヒトの薬物代謝を再現できるので、医薬品開発における第一相反応の薬効・毒性を調べるためのin vivo試験用のモデルマウスとして利用できる。
[実施例20]マウス人工染色体ベクターMDR1−MACの構築
マウス人工染色体ベクターMAC4にヒト薬物代謝酵素遺伝子群であるMDR1遺伝子をCre/loxPシステムを用いて転座クローニングを行い、実施例3と同様にして、MDR1−MACを構築する。
[A]ヒト7番染色体のAC005045へのloxP配列の部位特異的挿入
マウス人工染色体ベクターMAC4にloxP配列を介して転座挿入するために、DT40細胞内でヒト7番染色体(hChr7)のMDR1遺伝子の近位のAC005045へloxP配列を挿入する。
[A.1]ターゲティングベクターpMDR1loxPbsの作製
ヒト7番染色体上のMDR1遺伝子座のごく近傍かつセントロメア側(約50Kbセントロメア側)に位置するAC005045領域にCre組換え酵素の認識配列loxPを挿入するためのターゲティングベクターpMDR1loxPbsを以下のようにして作製した。まず、AC005045ゲノム領域を以下のプライマーを用いてPCRにより増幅した。

[A.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpMDR1loxPbsを制限酵素NotI(TAKARA)で線状化後、WO01/011951に記載された方法で作製されたヒト7番染色体を保持するニワトリDT40細胞(クローンDT40−#7)にトランスフェクションし、ブラストサイジンS(15μg/ml)を含む培地に交換し、96穴培養プレート3枚に分注して約2週間の選択培養を行った。2回のトランスフェクションで得た合計9個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr7M−loxP))。
[A.3]相同組換え体の選別
[A.3.1]PCR解析
ブラストサイジンS耐性クローンからPuregene DNA Isolation Kit(Gentra System社)を用いてゲノムDNAを抽出して、以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
以下の2組のプライマーを用いたPCRにより相同組換え体の同定を行った。
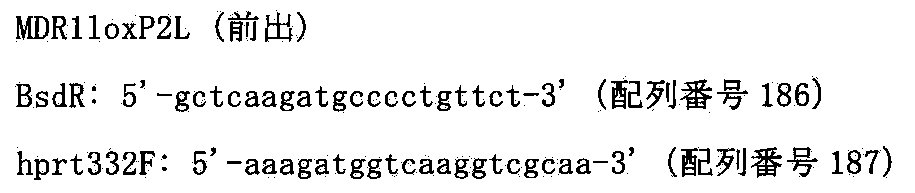
[A.3.3]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認した3クローンをヒトcot−1 DNAおよびブラストサイジンDNAをプローブにしてFISH解析を行ったところ、ヒト7番染色体は全てのクローンで宿主染色体に転座することなく、7q21付近にネオマイシン由来のシグナルが検出されたことから部位特異的に組換えが起こったことが確かめられた。以上の結果から、ヒト7番染色体上のAC005045に遺伝子導入部位であるloxP配列が部位特異的に挿入されたと結論づけた。
[B]ヒト7番染色体上のAC003083における部位特異的切断
ヒト7番染色体のMDR1遺伝子から遠位側の遺伝子を削除するために、部位特異的染色体削除であるテロメアトランケーションを行う。
[B.1]ターゲティングベクターpTELpuro−MDR1の作製
ヒト7染色体上のMDR1遺伝子座のごく近傍かつテロメア側(約50Kbテロメア側)に位置するAC003083領域にヒトテロメア配列を挿入するためのターゲティングベクターpTELpuro−MDR1を以下のようにして作製した。まず、AC003083ゲノム領域を以下のプライマーを用いてPCRにより増幅した。

[B.2]トランスフェクションおよび薬剤耐性クローンの単離
上述と同様に、上記で作製したターゲティングベクターpTELpuro−MDR1を制限酵素EcoRI(ニッポンジーン)で線状化後、上記で作製したクローンDT40(hChr7M−loxP)8,9にトランスフェクションし、ピューロマイシン(0.3ug/ml)を含む培地に交換し、96穴培養プレート10枚に分注して約2週間の選択培養を行った。4回のトランスフェクションで得た合計96個の耐性コロニーを単離し増殖させ、以後の解析を行った(クローン名:DT40(hChr7M−loxP−tel))。
[B.3]相同組換え体の選別
[B.3.1]PCR解析
ピューロマイシン耐性株のゲノムDNAを鋳型として組換え体を選別するため一次スクリーニングとして切断部位よりテロメア側に位置する以下のプライマーを用いてPCRを行い、部位特異的切断が起こっているかを確認した。そのプライマー配列を以下に示す。


次に上記プライマーで検出されなかった3クローンについて以下のプライマーを用いて部位特異的相同組換えが起こっているかをPCRにて確認した。配列は以下のとおりである。

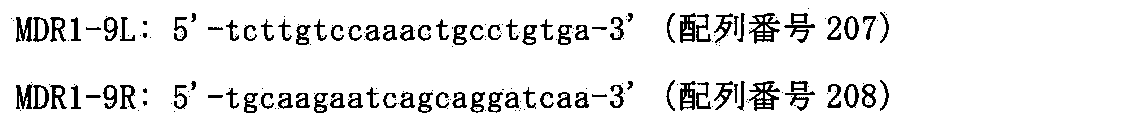
[B.3.2]two−color FISH解析
FISH解析は松原ら(FISH実験プロトコール、秀潤社、1994)に従い行った。上記で組換えを確認した3クローンをヒトcot−1 DNAおよびピューロマイシンDNAをプローブにしてFISH解析を行ったところ、ヒト7番染色体は全てのクローンで宿主染色体に転座することなく、ヒト7番染色体断片末端にピューロマイシン由来のシグナルが検出され、目的部位で切断されていたことから部位特異的に組換えが起こったことが確かめられた(図97)。
以上の結果から、クローンDT40(hChr7M−loxP−tel)10及び12及び70において、MDR1遺伝子領域よりテロメア側のAC003083から遠位において切断できたと結論付けた。
[C]hChr7M−loxP−tel含有DT40からhChr7M−loxP−telのMAC4含有CHO細胞への導入。
CHO細胞内でマウス人工染色体ベクターMAC4にloxP配列を介してヒトMDR1遺伝子領域を転座挿入するために、hChr7M−loxP−telをマウス人工染色体ベクターMAC4含有CHO細胞に導入する。
[C.1]微小核細胞融合と薬剤耐性クローンの単離
レシピエント細胞であるDT40(hChr7M−loxP−tel)10及び70を用いて、上記と同様にMAC4含有CHO hprt欠損細胞(ヒューマンサイエンス研究資源バンクより入手、登録番号JCRB0218)である、CHO(HPRT−;MAC4)に微小核細胞融合法を行った。4回の微小核細胞融合で得た合計15個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:CHO(HPRT−;MAC4,hChr7M−loxP−tel))。
[C.2]薬剤耐性クローンの選別
[C.2.1]PCR解析
ブラストサイジンS耐性株のゲノムDNAを抽出して鋳型として組換え体を選別するため、以下のプライマーを用いてPCRを行い、ヒト7番染色体断片がMAC4含有CHO細胞に導入されているかを確認した。そのプライマー配列を以下に示す。


[C.2.2]two−color FISH解析
上記で得られたCHO(HPRT−;MAC4,hChr7M−loxP−tel)の6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でマウスCot−1 DNA及びヒトCot−1 DNAをプローブにしたFISH解析を行ったところ、2クローンにおいて80%以上の割合でMAC1ならびにhChr7M−loxP−telがCHO細胞に1コピーもしくは2コピーで導入されていることを確認した(図98)。
以上の結果から、hChr7M−loxP−telがマウス人工染色体ベクターMAC4含有CHO細胞に導入できたと結論付けた。
[D]CHO(HPRT−;MAC4,hChr7M−loxP−tel)クローンにおけるヒトMDR1遺伝子領域周辺(AC005045−ヒトMDR1遺伝子−AC003083)210kbのMAC4ベクターへの部位特異的転座
210kbサイズのDNAであるヒトMDR1遺伝子をマウス個体内で安定に維持させるために、マウス人工染色体ベクターMAC4に転座挿入する(図99)。
[D.1]トランスフェクションおよびHAT耐性クローンの単離
上記で得られたCHO(HPRT−;MAC4,hChr7M−loxP−tel)7及び15にリポフェクション法を用いて遺伝子導入を行い、90%コンフルエント状態になった6wellの細胞に対して、Cre3μgを市販(インビトロジェン)のプロトコールに従い導入した。HAT選択培養下で2週間培養すると、耐性コロニーが出現し、2回の導入で得た合計10個のコロニーを単離し増殖させ、以後の解析を行った(クローン名:CHO(MDR1−MAC,hChr7−ΔMDR1))。
[D.2]薬剤耐性クローンの選別
[D.2.1]PCR解析
HAT耐性株のゲノムDNAを抽出して鋳型として相互転座クローンを選別するため、以下のプライマーを用いてPCRを行い、ヒト7番染色体断片とMAC4上で染色体相互転座が起こっているかを確認した。そのプライマー配列を以下に示す。


[D.2.2]two−color FISH解析
上記で得られたCHO(MDR1−MAC,hChr7−ΔMDR1)の6クローンについてShinoharaらの報告(Human Molecular Genetics,10:1163−1175,2001)に記された方法でMDR1−BAC(RP11−784L5)(CHORI)DNA及びマウスCot−1 DNAをプローブにしたFISH解析を行ったところ6クローン中、3クローンで60%以上の割合でMAC4上にヒトMDR1由来によるシグナルが観察されていることを確認した(図100)。
以上の結果から、ヒト7番染色体断片上のMDR1遺伝子210kbがマウス人工染色体ベクターMAC4に相互転座によるクローニングができたと結論付けた。
[E]CHO細胞からのMDR1−MACのマウスA9細胞への移入
MDR1−MACを保持するマウスES細を作製するために、上記[D]で得られたMDR1−MACを保持するCHO細胞(CHO(MDR1−MAC,hChr7−ΔMDR1)1,2,4)からマウスA9細胞へミクロセル形成能の高いマウスA9細胞へ微小核細胞融合法により導入した。4回の微小核細胞融合で得た合計7個の耐性コロニーを単離し増殖させ、以降の解析を行った(クローン名:A9(MDR1−MAC))。その結果、MDR1−MAC領域のみを検出する前出のプライマーを用いたPCRで陽性であるクローンが5クローンあった。さらに、MDR1−BAC(RP11−784L5)(CHORI)ならびにマウスminor satellite DNAプローブを用いてFISH解析(Tomizukaら,Nature Genet.16:133,1997)を行った結果、上記プローブにより特異的に検出されるMDR1−MACの存在が5クローン中、3クローンで確認された。以上のことから、MDR1−MAC保持A9細胞が3クローン得られたと結論付けた。
[F]実施例8記載のように、マウス人工染色体ベクターMDR1−MACを保持するマウスES細胞を作製し、そのES細胞を用いてin vitroの安定性を検証できる。また、上記ES細胞からキメラマウスを作製し、MDR1−MACが子孫伝達されたマウス系統TC(MDR1−MAC)を作製できる。また、上記TC(MDR1−MAC)マウス系統を用いて、体細胞におけるMDR1−MACの安定性を検証できる。また、TC(MDR1−MAC)マウス系統はヒトにおける薬物の輸送等を再現できる。また、上記TC(CYP3A−MAC)マウス系統との交配により、TC(CYP3A−MAC/MDR1−MAC)系統を作製できるので、医薬品開発における薬効・毒性を調べるためのin vivo試験用のモデルマウスとして利用できる。
本明細書で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書にとり入れるものとする。
Claims (44)
- マウス染色体由来の天然型セントロメア、セントロメア近傍のマウス染色体長腕の部位から長腕遠位を削除したマウス染色体由来の長腕断片、及びテロメア配列を含むこと、ならびに、哺乳類の細胞及び組織において安定に保持されることを特徴とする、マウス人工染色体ベクター。
- マウス染色体が1~19番染色体のいずれか1つである、請求項1記載のマウス人工染色体ベクター。
- マウス染色体由来の長腕断片が、マウス1~19番染色体のいずれか1つの長腕からその全内在性遺伝子数の少なくとも99.5%が削除された残部からなる、請求項1又は2記載のマウス人工染色体ベクター。
- 寄託細胞株DT40 B6bT−1(FERM BP−11128)に含まれるマウス人工染色体を基本構造として含む、請求項1~3のいずれか1項記載のマウス人工染色体ベクター。
- 哺乳類がげっ歯類である、請求項1~4のいずれか1項記載のマウス人工染色体ベクター。
- げっ歯類がマウス、ラット又はハムスターである、請求項5記載のマウス人工染色体ベクター。
- 1つ又は複数のDNA配列挿入部位をさらに含む、請求項1~6のいずれか1項記載のマウス人工染色体ベクター。
- DNA配列挿入部位が、部位特異的組換え酵素の認識部位である、請求項7記載のマウス人工染色体ベクター。
- DNA配列挿入部位が、loxP配列、FRT配列、φC31attB及びφC31attP配列、R4attB及びR4attP配列、TP901−1attB及びTP901−1attP配列、或いはBxb1attB及びBxb1attP配列である、請求項7又は8記載のマウス人工染色体ベクター。
- レポーター遺伝子、選択マーカー遺伝子又はその両方をさらに含む、請求項1~9のいずれか1項記載のマウス人工染色体ベクター。
- 外来DNA配列をさらに含む、請求項1~10のいずれか1項記載のマウス人工染色体ベクター。
- 外来DNA配列のサイズが200kb以上である、請求項1~11のいずれか1項記載のマウス人工染色体ベクター。
- 外来DNA配列がヒトDNA配列である、請求項11又は12記載のマウス人工染色体ベクター。
- 外来DNA配列が、薬物代謝関連遺伝子のDNA配列である、請求項11~13のいずれか1項記載のマウス人工染色体ベクター。
- 薬物代謝関連遺伝子が、第一相反応又は第二相反応に関わる酵素をコードする遺伝子である、請求項14記載のマウス人工染色体ベクター。
- 第一相反応に関わる酵素が、CYP1A、CYP1B、CYP2A、CYP2B、CYP2C、CYP2D、CYP2E、CYP2J、CYP3A、CYP4A、CYP4B及びそれらのサブファミリーなどのCYP、並びにCES、からなる群より選択される少なくとも1つの酵素をコードするものである、請求項15記載のマウス人工染色体ベクター。
- 第二相反応に関わる酵素が、UGT1及びUGT2からなる群より選択される少なくとも1つの酵素をコードするものである、請求項15記載のマウス人工染色体ベクター。
- 薬物代謝関連遺伝子が、トランスポーターをコードする遺伝子である、請求項14記載のマウス人工染色体ベクター。
- トランスポーターをコードする遺伝子が、MDR1、MDR2、MRP2、OAT、OATP、OCT及びBCRPからなる群より選択される少なくとも1つの遺伝子をコードするものである、請求項18記載のマウス人工染色体ベクター。
- 薬物代謝関連遺伝子が、核内受容体をコードする遺伝子である、請求項14記載のマウス人工染色体ベクター。
- 核内受容体をコードする遺伝子が、PXR、AhR、CAR及びPPARαからなる群より選択される少なくとも1つの遺伝子をコードするものである、請求項20記載のマウス人工染色体ベクター。
- 外来DNA配列が、ヒト染色体の長腕又は短腕のDNA配列である、請求項11~13のいずれか1項記載のマウス人工染色体ベクター。
- 外来DNA配列が、第一相反応に関わる酵素をコードする遺伝子、第二相に関わる酵素をコードする遺伝子、トランスポーターをコードする遺伝子及び核内受容体をコードする遺伝子からなる群より選択される少なくとも2つの遺伝子の配列を含む、請求項11~21のいずれか1項記載のマウス人工染色体ベクター。
- ヒト染色体が、疾患遺伝子の原因領域を含む、ヒト染色体の長腕又は短腕のDNA配列である、請求項22記載のマウス人工染色体ベクター。
- 外来DNA配列が、サイトカイン類、ホルモン類、成長因子類、栄養因子類、造血因子類、血液凝固・溶解因子類、イムノグロブリン類、G蛋白質共役型受容体類、酵素類などのポリペプチド類をコードする遺伝子又はDNAの配列、或いは、腫瘍、筋ジストロフィー、血友病、神経変性疾患、自己免疫疾患、アレルギー性疾患、遺伝性疾患などの疾患に関連する治療用遺伝子又はDNAの配列である、請求項11~13のいずれか1項に記載のマウス人工染色体ベクター。
- 細胞が、肝細胞、腸細胞、腎細胞、脾細胞、肺細胞、心臓細胞、骨格筋細胞、脳細胞、骨髄細胞、リンパ球細胞、巨核球細胞、精子又は卵子である、請求項1~25のいずれか1項記載のマウス人工染色体ベクター。
- 組織が、肝臓、腸、腎臓、脾臓、肺、心臓、骨格筋、脳、骨髄、精巣又は卵巣由来の組織である、請求項1~25のいずれか1項記載のマウス人工染色体ベクター。
- 請求項1~27のいずれか1項記載のマウス人工染色体ベクターを保持する細胞。
- 細胞が、体細胞、非ヒト生殖系列細胞、幹細胞及び前駆細胞からなる群から選択される、請求項28記載の細胞。
- 幹細胞が、胚性幹(ES)細胞又は人工多能性幹(iPS)細胞である、請求項29記載の細胞。
- 細胞が、初代培養細胞、継代細胞又は株化細胞である、請求項28~30のいずれか1項記載の細胞。
- 細胞がヒト抗体産生を可能にする細胞である、請求項28~31のいずれか1項記載の細胞。
- 疾患治療用の外来DNA配列を含むマウス人工染色体ベクターを保持する請求項28~32のいずれか1項記載の細胞を含む医薬組成物。
- 請求項1~27のいずれか1項記載のマウス人工染色体ベクターを保持することを特徴とする、非ヒト動物。
- 疾患モデル動物である、請求項34記載の非ヒト動物。
- 外来のヒト薬物代謝関連遺伝子を発現可能にする動物である、請求項34記載の非ヒト動物。
- ヒト抗体を産生可能にする動物である、請求項34記載の非ヒト動物。
- マウス人工染色体ベクターに含まれる外来DNAに対応する内在遺伝子が破壊されている又は内在遺伝子の発現が低下している、請求項34~37のいずれか1項記載の非ヒト動物。
- 外来DNA配列を含むマウス人工染色体ベクターを保持する請求項28~32のいずれか1項記載の細胞を培養し、産生された該DNAによってコードされるタンパク質を回収することを含む、タンパク質の生産方法。
- ヒト抗体遺伝子を含むマウス人工染色体ベクターを保持する請求項37又は38記載の非ヒト動物を用いてヒト抗体を産生し、該ヒト抗体を回収することを含む、ヒト抗体の製造方法。
- 疾患モデル動物である請求項35記載の非ヒト動物に候補薬剤を投与し、該薬剤の治療効果を評価することを含む、該疾患を治療するのに有効な物質をスクリーニングする方法。
- ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する請求項36又は38記載の非ヒト動物或いは該動物由来の細胞、器官又は組織に、薬物又は食品を投与し、該薬物又は食品の薬理作用及び/又は代謝及び/又は毒性を測定することを含む、薬物又は食品の薬理作用及び/又は代謝及び/又は毒性の試験方法。
- ヒト薬物代謝関連遺伝子を含むマウス人工染色体ベクターを保持する請求項36又は38記載の非ヒト動物から得られたミクロソーム又はミクロソーム画分S9を、培養細胞若しくは細菌と、薬物及び/若しくは食品と共に培養し、薬物又は食品が該細胞又は細菌に及ぼす影響を測定することを含む、薬物又は食品の毒性の試験方法。
- 請求項1~27のいずれか1項記載のマウス人工染色体ベクターを使用して、200kb以上の大サイズの外来DNAをげっ歯類細胞又はげっ歯類個体内で90%以上の保持率で安定に維持させることを含む、細胞又は個体内での大サイズDNAの安定化方法。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HK13105170.6A HK1178206B (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| EP11731868.3A EP2522725B1 (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| KR1020127020182A KR101485840B1 (ko) | 2010-01-06 | 2011-01-06 | 마우스 인공염색체 벡터 |
| AU2011204143A AU2011204143B2 (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| US13/520,754 US8940533B2 (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| JP2011549044A JP5557217B2 (ja) | 2010-01-06 | 2011-01-06 | マウス人工染色体ベクター |
| CA2786659A CA2786659C (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| CN201180012643.2A CN102791857B (zh) | 2010-01-06 | 2011-01-06 | 小鼠人工染色体载体 |
| US14/574,749 US9775331B2 (en) | 2010-01-06 | 2014-12-18 | Rodent comprising mouse artificial chromosome vector |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010001425 | 2010-01-06 | ||
| JP2010-001425 | 2010-01-06 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/520,754 A-371-Of-International US8940533B2 (en) | 2010-01-06 | 2011-01-06 | Mouse artificial chromosome vector |
| US14/574,749 Division US9775331B2 (en) | 2010-01-06 | 2014-12-18 | Rodent comprising mouse artificial chromosome vector |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011083870A1 true WO2011083870A1 (ja) | 2011-07-14 |
Family
ID=44305605
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/050490 Ceased WO2011083870A1 (ja) | 2010-01-06 | 2011-01-06 | マウス人工染色体ベクター |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US8940533B2 (ja) |
| EP (1) | EP2522725B1 (ja) |
| JP (1) | JP5557217B2 (ja) |
| KR (1) | KR101485840B1 (ja) |
| CN (1) | CN102791857B (ja) |
| AU (1) | AU2011204143B2 (ja) |
| CA (1) | CA2786659C (ja) |
| WO (1) | WO2011083870A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013054949A1 (ja) * | 2011-10-13 | 2013-04-18 | 株式会社フェニックスバイオ | ヒト肝細胞を担持するキメラ非ヒト動物 |
| WO2014061829A1 (ja) * | 2012-10-19 | 2014-04-24 | 国立大学法人鳥取大学 | 薬物代謝酵素誘導および細胞毒性の評価方法、ならびにそのためのベクターおよび細胞 |
| JP2015119643A (ja) * | 2013-12-20 | 2015-07-02 | 国立研究開発法人産業技術総合研究所 | 人工染色体ベクター及び形質転換哺乳類細胞 |
| WO2018062392A1 (ja) * | 2016-09-28 | 2018-04-05 | 国立大学法人鳥取大学 | ダウン症モデルラット及びその作製法 |
| WO2018079857A1 (ja) * | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| JP2018196398A (ja) * | 2018-09-25 | 2018-12-13 | 国立研究開発法人産業技術総合研究所 | 人工染色体ベクター及び形質転換哺乳類細胞 |
| JP2019511235A (ja) * | 2016-04-12 | 2019-04-25 | シンプロイド バイオテック エルエルシーSynploid Biotek,Llc | 生合成経路を発現する合成染色体の作成方法及びその使用 |
| WO2019177163A1 (ja) * | 2018-03-16 | 2019-09-19 | 国立大学法人鳥取大学 | マウス人工染色体ベクター及びその使用 |
| WO2020075822A1 (ja) * | 2018-10-10 | 2020-04-16 | 国立大学法人鳥取大学 | 外来染色体を含むヒト人工多能性幹細胞の製造方法 |
| WO2024181584A1 (ja) * | 2023-02-28 | 2024-09-06 | 株式会社Trans Chromosomics | チャイニーズハムスター人工染色体ベクター又はその断片、及びそれを含む哺乳動物由来細胞 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103529059B (zh) * | 2013-10-09 | 2015-10-28 | 辉源生物科技(上海)有限公司 | 人乳腺癌耐药蛋白介导的药物代谢水平评价方法 |
| US11155836B2 (en) | 2015-02-09 | 2021-10-26 | CarryGenes Bioengineering | Compositions and methods for monitoring in real-time construction and bioengineering of mammalian synthetic chromosomes |
| CN104818253B (zh) * | 2015-03-24 | 2018-03-09 | 成都贝爱特生物科技有限公司 | 靶向定点整合cho细胞系的改造及其用途 |
| AU2017250178B2 (en) | 2016-04-12 | 2021-04-15 | CarryGenes Bioengineering | Sequential loadings of multiple delivery vectors using a single selectable marker |
| AU2017250265B2 (en) | 2016-04-12 | 2023-08-17 | CarryGenes Bioengineering | Methods for creating synthetic chromosomes having gene regulatory systems and uses thereof |
| CA3046509A1 (en) | 2016-12-15 | 2018-06-21 | Synploid Biotek, Llc | Methods of cell renewal |
| WO2018169892A1 (en) | 2017-03-15 | 2018-09-20 | Synploid Biotek, Llc | Compositions and methods of chromosomal silencing |
| WO2019050875A1 (en) | 2017-09-05 | 2019-03-14 | Synploid Biotek, Llc | LINEAR REPORTER SYNTHESIS CHROMOSOMES AND METHODS OF USE |
| JP7202573B2 (ja) | 2017-11-02 | 2023-01-12 | 国立大学法人鳥取大学 | 哺乳類人工染色体ベクターを利用するタンパク質の高生産方法 |
| CN109837307B (zh) * | 2017-11-24 | 2023-09-19 | 中国科学院脑科学与智能技术卓越创新中心 | 建立含外源染色体的胚胎干细胞的方法 |
| WO2020075823A1 (ja) | 2018-10-10 | 2020-04-16 | 国立大学法人鳥取大学 | 微小核細胞融合法による目的dnaを含む動物細胞の作製方法 |
| JP7302763B2 (ja) * | 2019-04-12 | 2023-07-04 | ヒューマブ カンパニー リミテッド | 人工組換え染色体およびその使用 |
| CN116209467A (zh) | 2020-09-16 | 2023-06-02 | 国立大学法人鸟取大学 | 针对冠状病毒的抗体 |
| CN115806978B (zh) * | 2021-09-14 | 2026-02-27 | 中国农业大学 | 异源染色体定向转移的方法 |
| JPWO2023090372A1 (ja) | 2021-11-16 | 2023-05-25 | ||
| CN121693564A (zh) | 2023-06-26 | 2026-03-17 | 夏威夷大学 | 进化的整合酶和其用于基因组编辑的方法 |
| CN118370276B (zh) * | 2024-04-12 | 2025-12-09 | 浙江大学 | 可实现条件特异遗传校正唐氏综合征小鼠模型的构建方法及应用 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000010383A1 (fr) | 1998-08-21 | 2000-03-02 | Kirin Beer Kabushiki Kaisha | Procede de modification des chromosomes |
| WO2001011951A1 (fr) | 1999-08-13 | 2001-02-22 | Kirin Beer Kabushiki Kaisha | Souris renfermant un cytochrome p450 humain |
| WO2004031385A1 (ja) | 2002-10-04 | 2004-04-15 | Kirin Beer Kabushiki Kaisha | ヒト人工染色体(hac)ベクター |
| JP2005230020A (ja) | 2001-05-11 | 2005-09-02 | Kirin Brewery Co Ltd | ヒト抗体λ軽鎖遺伝子を含むヒト人工染色体、および子孫伝達可能な該ヒト人工染色体を含む非ヒト動物 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| JP2007295860A (ja) | 2006-05-01 | 2007-11-15 | Tottori Univ | 内在遺伝子を含まないヒト人工染色体ベクター |
| JP2007306928A (ja) | 2007-06-25 | 2007-11-29 | Chromo Research Inc | 哺乳類人工染色体 |
| WO2008013067A1 (fr) * | 2006-07-07 | 2008-01-31 | Kyowa Hakko Kirin Co., Ltd. | Vecteur de chromosome artificiel humain (cah), et substance pharmaceutique à base de cellules humaines contenant un vecteur de chromosome artificiel humain (cah) |
| JP2008054501A (ja) | 2005-01-26 | 2008-03-13 | Tsuneko Okazaki | 線状哺乳類人工染色体及びその構築方法 |
| WO2009063722A1 (ja) | 2007-11-14 | 2009-05-22 | National University Corporation Tottori University | ヒトチトクロームp450遺伝子(クラスター)を含む哺乳動物人工染色体ベクター及びそれを保持する非ヒト哺乳動物 |
| JP2010001425A (ja) | 2008-06-23 | 2010-01-07 | Toppan Printing Co Ltd | 蛍光インキおよび蛍光印刷物 |
-
2011
- 2011-01-06 WO PCT/JP2011/050490 patent/WO2011083870A1/ja not_active Ceased
- 2011-01-06 US US13/520,754 patent/US8940533B2/en active Active
- 2011-01-06 JP JP2011549044A patent/JP5557217B2/ja active Active
- 2011-01-06 AU AU2011204143A patent/AU2011204143B2/en not_active Ceased
- 2011-01-06 EP EP11731868.3A patent/EP2522725B1/en active Active
- 2011-01-06 CA CA2786659A patent/CA2786659C/en active Active
- 2011-01-06 KR KR1020127020182A patent/KR101485840B1/ko not_active Expired - Fee Related
- 2011-01-06 CN CN201180012643.2A patent/CN102791857B/zh not_active Expired - Fee Related
-
2014
- 2014-12-18 US US14/574,749 patent/US9775331B2/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000010383A1 (fr) | 1998-08-21 | 2000-03-02 | Kirin Beer Kabushiki Kaisha | Procede de modification des chromosomes |
| WO2001011951A1 (fr) | 1999-08-13 | 2001-02-22 | Kirin Beer Kabushiki Kaisha | Souris renfermant un cytochrome p450 humain |
| JP2005230020A (ja) | 2001-05-11 | 2005-09-02 | Kirin Brewery Co Ltd | ヒト抗体λ軽鎖遺伝子を含むヒト人工染色体、および子孫伝達可能な該ヒト人工染色体を含む非ヒト動物 |
| WO2004031385A1 (ja) | 2002-10-04 | 2004-04-15 | Kirin Beer Kabushiki Kaisha | ヒト人工染色体(hac)ベクター |
| JP2008054501A (ja) | 2005-01-26 | 2008-03-13 | Tsuneko Okazaki | 線状哺乳類人工染色体及びその構築方法 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| JP2007295860A (ja) | 2006-05-01 | 2007-11-15 | Tottori Univ | 内在遺伝子を含まないヒト人工染色体ベクター |
| WO2008013067A1 (fr) * | 2006-07-07 | 2008-01-31 | Kyowa Hakko Kirin Co., Ltd. | Vecteur de chromosome artificiel humain (cah), et substance pharmaceutique à base de cellules humaines contenant un vecteur de chromosome artificiel humain (cah) |
| JP2007306928A (ja) | 2007-06-25 | 2007-11-29 | Chromo Research Inc | 哺乳類人工染色体 |
| WO2009063722A1 (ja) | 2007-11-14 | 2009-05-22 | National University Corporation Tottori University | ヒトチトクロームp450遺伝子(クラスター)を含む哺乳動物人工染色体ベクター及びそれを保持する非ヒト哺乳動物 |
| JP2010001425A (ja) | 2008-06-23 | 2010-01-07 | Toppan Printing Co Ltd | 蛍光インキおよび蛍光印刷物 |
Non-Patent Citations (37)
| Title |
|---|
| B. SAUER, METHODS OF ENZYMOLOGY, vol. 225, 1993, pages 890 - 900 |
| CO DO ET AL., CHROMOSOME RES., vol. 8, no. 3, 2000, pages 83 - 91 |
| DIEKEN ET AL., NATURE GENETICS, vol. 12, 1996, pages 1 74 - 182 |
| GENE TARGETING, EXPERIMENTAL MEDICINE, 1995 |
| HOSHIYA ET AL., MOL THER, vol. 17, 2009, pages 309 - 17 |
| HOSHIYA ET AL., MOL THER., vol. 17, no. 2, 2009, pages 309 - 17 |
| J. A. THOMSON ET AL., BIOL. REPROD., vol. 55, 1996, pages 254 - 259 |
| J. A. THOMSON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 7844 - 7848 |
| J. A. THOMSON ET AL., SCIENCE, vol. 282, 1999, pages 1145 - 1147 |
| J. A. THOMSON; V. S. MARSHALL, CURR. TOP. DEV. BIOL., vol. 38, 1998, pages 133 - 165 |
| J. LIAO ET AL., CELL RES., vol. 18, 2008, pages 600 - 603 |
| J. YU ET AL., SCIENCE, vol. 318, 2007, pages 1917 - 1920 |
| K. TAKAHASHI ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| K. TAKAHASHI; S. YAMANAKA, CELL, vol. 126, 2006, pages 663 - 676 |
| KAI ET AL., CELL RES, vol. 19, 2009, pages 247 - 58 |
| KATOH ET AL., BBRC, vol. 321, 2000, pages 280 - 290 |
| KATOH, M ET AL.: "Construction of a novel human artificial chromosome vector for gene delivery.", BIOCHEM BIOPHYS RES COMMUN., vol. 321, no. 2, 20 August 2004 (2004-08-20), pages 280 - 290, XP004522500 * |
| KAZUKI ET AL., BBRC, 2004 |
| KAZUKI ET AL., BBRC, vol. DT40, 2004, pages 21 - 2,3 |
| KAZUKI ET AL., GENE THERAPY, 2010 |
| KOI ET AL., JPN. J. CANCER RES., vol. 80, 1973, pages 413 - 418 |
| KUGOH ET AL., DNA RESEARCH, 1999 |
| KUROIWA ET AL., NAT BIOTECH, vol. 18, 2000, pages 1086 - 1090 |
| KUROIWA ET AL., NATURE BIOTECH., vol. 20, 2002, pages 88 |
| M. J. EVANS; M. H. KAUFMAN, NATURE, vol. 292, 1981, pages 154 - 156 |
| M. NAKAGAWA ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 101 - 106 |
| MATSUBARA ET AL.: "FISH test protocol", 1994, SHUJUNSHA CO., LTD. |
| OMURA ET AL., J. BIOL. CHEM., vol. 239, 1964, pages 2370 |
| S. STEWART ET AL., GENE THERAPY, vol. 9, 2002, pages 719 - 723 |
| See also references of EP2522725A4 * |
| SHINOHARA ET AL., CHROMOSOME RESEARCH, vol. 8, 2000, pages 713 - 725 |
| SHINOHARA ET AL., HMG, vol. 10, 2001, pages 1163 - 75 |
| SHINOHARA ET AL., HUMAN MOLECULAR GENETICS, vol. 10, 2001, pages 1163 - 1175 |
| TAKAHASHI, K. ET AL., CELL, vol. 131, 2007, pages 861 - 872 |
| TOMIZUKA ET AL., NAT GENET, vol. 16, 1997, pages 133 - 143 |
| TOMIZUKA ET AL., NATURE GENET., vol. 16, 1997, pages 133 |
| YAMADA ET AL., J HUM GENET., vol. 53, no. 5, 2008, pages 447 - 53 |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013054949A1 (ja) * | 2011-10-13 | 2013-04-18 | 株式会社フェニックスバイオ | ヒト肝細胞を担持するキメラ非ヒト動物 |
| WO2014061829A1 (ja) * | 2012-10-19 | 2014-04-24 | 国立大学法人鳥取大学 | 薬物代謝酵素誘導および細胞毒性の評価方法、ならびにそのためのベクターおよび細胞 |
| JP2015119643A (ja) * | 2013-12-20 | 2015-07-02 | 国立研究開発法人産業技術総合研究所 | 人工染色体ベクター及び形質転換哺乳類細胞 |
| JP7497139B2 (ja) | 2016-04-12 | 2024-06-10 | キャリージーンズ バイオエンジニアリング | 生合成経路を発現する合成染色体の作成方法及びその使用 |
| JP7601402B2 (ja) | 2016-04-12 | 2024-12-17 | キャリージーンズ バイオエンジニアリング | 生合成経路を発現する合成染色体の作成方法及びその使用 |
| JP2019511235A (ja) * | 2016-04-12 | 2019-04-25 | シンプロイド バイオテック エルエルシーSynploid Biotek,Llc | 生合成経路を発現する合成染色体の作成方法及びその使用 |
| JP2022070950A (ja) * | 2016-04-12 | 2022-05-13 | キャリージーンズ バイオエンジニアリング | 生合成経路を発現する合成染色体の作成方法及びその使用 |
| WO2018062392A1 (ja) * | 2016-09-28 | 2018-04-05 | 国立大学法人鳥取大学 | ダウン症モデルラット及びその作製法 |
| JPWO2018062392A1 (ja) * | 2016-09-28 | 2019-08-22 | 国立大学法人鳥取大学 | ダウン症モデルラット及びその作製法 |
| JP7011228B2 (ja) | 2016-09-28 | 2022-02-10 | 国立大学法人鳥取大学 | ダウン症モデルラット及びその作製法 |
| JPWO2018079857A1 (ja) * | 2016-10-31 | 2019-09-19 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| US12063913B2 (en) | 2016-10-31 | 2024-08-20 | National University Corporation Tottori University | Human antibody-producing non-human animal and method for preparing human antibodies using same |
| WO2018079857A1 (ja) * | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
| JPWO2019177163A1 (ja) * | 2018-03-16 | 2020-08-20 | 国立大学法人鳥取大学 | マウス人工染色体ベクター及びその使用 |
| US12428652B2 (en) | 2018-03-16 | 2025-09-30 | National University Corporation Tottori University | Mouse artificial chromosome vector and use thereof |
| WO2019177163A1 (ja) * | 2018-03-16 | 2019-09-19 | 国立大学法人鳥取大学 | マウス人工染色体ベクター及びその使用 |
| JP2018196398A (ja) * | 2018-09-25 | 2018-12-13 | 国立研究開発法人産業技術総合研究所 | 人工染色体ベクター及び形質転換哺乳類細胞 |
| WO2020075822A1 (ja) * | 2018-10-10 | 2020-04-16 | 国立大学法人鳥取大学 | 外来染色体を含むヒト人工多能性幹細胞の製造方法 |
| JP7079946B2 (ja) | 2018-10-10 | 2022-06-03 | 国立大学法人鳥取大学 | 外来染色体を含むヒト人工多能性幹細胞の製造方法 |
| JPWO2020075822A1 (ja) * | 2018-10-10 | 2021-06-03 | 国立大学法人鳥取大学 | 外来染色体を含むヒト人工多能性幹細胞の製造方法 |
| WO2024181584A1 (ja) * | 2023-02-28 | 2024-09-06 | 株式会社Trans Chromosomics | チャイニーズハムスター人工染色体ベクター又はその断片、及びそれを含む哺乳動物由来細胞 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2786659A1 (en) | 2011-07-14 |
| CN102791857A (zh) | 2012-11-21 |
| JP5557217B2 (ja) | 2014-07-23 |
| CN102791857B (zh) | 2017-03-15 |
| HK1178206A1 (zh) | 2013-09-06 |
| AU2011204143A1 (en) | 2012-08-16 |
| US8940533B2 (en) | 2015-01-27 |
| JPWO2011083870A1 (ja) | 2013-05-16 |
| KR101485840B1 (ko) | 2015-01-23 |
| EP2522725A1 (en) | 2012-11-14 |
| EP2522725B1 (en) | 2016-10-05 |
| US20150096063A1 (en) | 2015-04-02 |
| US9775331B2 (en) | 2017-10-03 |
| US20120272342A1 (en) | 2012-10-25 |
| EP2522725A4 (en) | 2013-09-25 |
| AU2011204143B2 (en) | 2014-09-18 |
| CA2786659C (en) | 2017-08-22 |
| KR20120099300A (ko) | 2012-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5557217B2 (ja) | マウス人工染色体ベクター | |
| JP4318736B2 (ja) | ヒト抗体遺伝子を発現する非ヒト動物とその利用 | |
| AU2008322038B2 (en) | Mammalian artificial chromosome vector comprising human cytochrome P450 gene (cluster) and non-human mammalian animal retaining the same | |
| JP4082740B2 (ja) | 内因性遺伝子が破壊されている分化多能性保持細胞 | |
| JP6868250B2 (ja) | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 | |
| US20250366449A1 (en) | Mouse artificial chromosome vector and use thereof | |
| CN118265449A (zh) | 转基因哺乳动物及其使用方法 | |
| JP2007312792A (ja) | キメラ動物およびその作製法 | |
| HK1178206B (en) | Mouse artificial chromosome vector | |
| WO2022235759A1 (en) | Transgenic rodents expressing chimeric equine-rodent antibodies and methods of use thereof | |
| CN119213132A (zh) | Mhc基因群人源化动物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180012643.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11731868 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 13520754 Country of ref document: US Ref document number: 2011549044 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2786659 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011204143 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 20127020182 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6766/CHENP/2012 Country of ref document: IN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011731868 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011731868 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2011204143 Country of ref document: AU Date of ref document: 20110106 Kind code of ref document: A |