WO2011097496A1 - Selective androgen receptor modulators - Google Patents
Selective androgen receptor modulators Download PDFInfo
- Publication number
- WO2011097496A1 WO2011097496A1 PCT/US2011/023768 US2011023768W WO2011097496A1 WO 2011097496 A1 WO2011097496 A1 WO 2011097496A1 US 2011023768 W US2011023768 W US 2011023768W WO 2011097496 A1 WO2011097496 A1 WO 2011097496A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- tetrahydro
- carbazol
- dichloro
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)(Cc1c2[n]c3c1cccc3)*CCC2(O)[Rh] Chemical compound CC(C)(Cc1c2[n]c3c1cccc3)*CCC2(O)[Rh] 0.000 description 14
- SDYWXFYBZPNOFX-UHFFFAOYSA-N Nc(cc1)cc(Cl)c1Cl Chemical compound Nc(cc1)cc(Cl)c1Cl SDYWXFYBZPNOFX-UHFFFAOYSA-N 0.000 description 2
- LKNZHKDMBMSBOE-UHFFFAOYSA-N CC(CCc1c2[nH]c3ccccc13)C2(O)[Rh] Chemical compound CC(CCc1c2[nH]c3ccccc13)C2(O)[Rh] LKNZHKDMBMSBOE-UHFFFAOYSA-N 0.000 description 1
- KCZDEPKLIPOOPO-UHFFFAOYSA-N CC1(C(O)=C(Cc2ccccc2)C=CC11)Nc2c1c(C)c(C)cc2 Chemical compound CC1(C(O)=C(Cc2ccccc2)C=CC11)Nc2c1c(C)c(C)cc2 KCZDEPKLIPOOPO-UHFFFAOYSA-N 0.000 description 1
- PZBBESSUKAHBHD-UHFFFAOYSA-N COC(C(CCC1)C1=O)=O Chemical compound COC(C(CCC1)C1=O)=O PZBBESSUKAHBHD-UHFFFAOYSA-N 0.000 description 1
- PSFDOLWKHOMXRW-UHFFFAOYSA-N Cc(c1c(cc2)[nH]c(C3=O)c1CCC3(Cc1ccccc1)Br)c2Cl Chemical compound Cc(c1c(cc2)[nH]c(C3=O)c1CCC3(Cc1ccccc1)Br)c2Cl PSFDOLWKHOMXRW-UHFFFAOYSA-N 0.000 description 1
- JINODYMSFZBLPV-UHFFFAOYSA-N N#Cc(cc1c2ccc3)ccc1[nH]c2c3O Chemical compound N#Cc(cc1c2ccc3)ccc1[nH]c2c3O JINODYMSFZBLPV-UHFFFAOYSA-N 0.000 description 1
- LPVQKGRUQLAPJO-UHFFFAOYSA-N N#Cc1ccc2[nH]c(C(C(CC3)Br)=O)c3c2c1 Chemical compound N#Cc1ccc2[nH]c(C(C(CC3)Br)=O)c3c2c1 LPVQKGRUQLAPJO-UHFFFAOYSA-N 0.000 description 1
- UXIHHFHUOJSWII-UHFFFAOYSA-N N#Cc1ccc2[nH]c(C(CCC3)(C(C(F)(F)F)(F)F)O)c3c2c1 Chemical compound N#Cc1ccc2[nH]c(C(CCC3)(C(C(F)(F)F)(F)F)O)c3c2c1 UXIHHFHUOJSWII-UHFFFAOYSA-N 0.000 description 1
- BNDFTXVWHLGWHU-XNTDXEJSSA-N O=C(CCC1)/C1=N/Nc(cc1Cl)ccc1Cl Chemical compound O=C(CCC1)/C1=N/Nc(cc1Cl)ccc1Cl BNDFTXVWHLGWHU-XNTDXEJSSA-N 0.000 description 1
- AQUUJMVARAFDRO-UHFFFAOYSA-N O=C(CCC1)c2c1c(cc(cc1Cl)Cl)c1[nH]2 Chemical compound O=C(CCC1)c2c1c(cc(cc1Cl)Cl)c1[nH]2 AQUUJMVARAFDRO-UHFFFAOYSA-N 0.000 description 1
- OILAIQUEIWYQPH-UHFFFAOYSA-N O=C(CCCC1)C1=O Chemical compound O=C(CCCC1)C1=O OILAIQUEIWYQPH-UHFFFAOYSA-N 0.000 description 1
- FDRMTRSYPSJWQJ-UHFFFAOYSA-N OC(C(CC1)C(F)(F)F)(C(F)(F)F)c2c1c(cc(cc1Cl)F)c1[nH]2 Chemical compound OC(C(CC1)C(F)(F)F)(C(F)(F)F)c2c1c(cc(cc1Cl)F)c1[nH]2 FDRMTRSYPSJWQJ-UHFFFAOYSA-N 0.000 description 1
- KGYITEFEOAWUCU-UHFFFAOYSA-N OC(C(CC1)F)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl Chemical compound OC(C(CC1)F)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl KGYITEFEOAWUCU-UHFFFAOYSA-N 0.000 description 1
- FXBPINRBSNMESY-UHFFFAOYSA-N OC(C(CCC1)C1=O)=O Chemical compound OC(C(CCC1)C1=O)=O FXBPINRBSNMESY-UHFFFAOYSA-N 0.000 description 1
- HVDRWCKUQMRDGT-UHFFFAOYSA-N OC(CC1)(C(F)(F)F)c2c1c(c(Cl)c(cc1)Cl)c1[nH]2 Chemical compound OC(CC1)(C(F)(F)F)c2c1c(c(Cl)c(cc1)Cl)c1[nH]2 HVDRWCKUQMRDGT-UHFFFAOYSA-N 0.000 description 1
- FUIJMEOKOZBXIO-UHFFFAOYSA-N OC(CC1)(C(F)(F)F)c2c1c1c(C3[IH]CC3)c(Cl)ccc1[nH]2 Chemical compound OC(CC1)(C(F)(F)F)c2c1c1c(C3[IH]CC3)c(Cl)ccc1[nH]2 FUIJMEOKOZBXIO-UHFFFAOYSA-N 0.000 description 1
- OZXBEIHRSWLQHT-UHFFFAOYSA-N OC(CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl Chemical compound OC(CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl OZXBEIHRSWLQHT-UHFFFAOYSA-N 0.000 description 1
- AWFSIUCRLQLNFB-UHFFFAOYSA-N OC(CCC1)(C(F)(F)F)c2c1c(cc(cc1Cl)F)c1[nH]2 Chemical compound OC(CCC1)(C(F)(F)F)c2c1c(cc(cc1Cl)F)c1[nH]2 AWFSIUCRLQLNFB-UHFFFAOYSA-N 0.000 description 1
- OZXBEIHRSWLQHT-GFCCVEGCSA-N O[C@@](CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl Chemical compound O[C@@](CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl OZXBEIHRSWLQHT-GFCCVEGCSA-N 0.000 description 1
- OZXBEIHRSWLQHT-LBPRGKRZSA-N O[C@](CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl Chemical compound O[C@](CCC1)(C(F)(F)F)c([nH]c2c3)c1c2cc(Cl)c3Cl OZXBEIHRSWLQHT-LBPRGKRZSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/02—Drugs for genital or sexual disorders; Contraceptives for disorders of the vagina
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/12—Drugs for genital or sexual disorders; Contraceptives for climacteric disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/08—Antiseborrheics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/26—Androgens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/94—[b, c]- or [b, d]-condensed containing carbocyclic rings other than six-membered
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/74—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving hormones or other non-cytokine intercellular protein regulatory factors such as growth factors, including receptors to hormones and growth factors
- G01N33/743—Steroid hormones
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/705—Assays involving receptors, cell surface antigens or cell surface determinants
- G01N2333/72—Assays involving receptors, cell surface antigens or cell surface determinants for hormones
- G01N2333/723—Steroid/thyroid hormone superfamily, e.g. GR, EcR, androgen receptor, oestrogen receptor
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/04—Screening involving studying the effect of compounds C directly on molecule A (e.g. C are potential ligands for a receptor A, or potential substrates for an enzyme A)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2500/00—Screening for compounds of potential therapeutic value
- G01N2500/10—Screening for compounds of potential therapeutic value involving cells
Definitions
- Androgen signaling is mediated through the androgen receptor (AR) and is a nuclear signaling pathway of tremendous importance in mammals.
- AR androgen receptor
- this critical hormone signaling pathway affects a large number of non-sexual tissues including, bone, muscle, CNS, liver, etc.
- testosterone and dihydrotestosterone are the primary ligands that mediate AR- signaling. Both are high affinity ligands for AR, with dihydrotestosterone having somewhat higher affinity.
- Testosterone is converted to dihydrotestosterone through the action of 5ot-reductase enzymes and is converted to 17p-estradiol (potent endogenous estrogen) through the action of P-450 aromatase enzymes.
- AR signaling is mediated by binding of an AR ligand to AR in the cellular cytosol, homodimerization of two AR receptors and nuclear location of the ligand bound dimer to the cell nucleus where the complex associates with various coactivators as well as Androgen Response Elements (palindrome-like sequences of DNA) which serve as activation sites for certain AR-mediated genes.
- both sexual and non-sexual, androgens such as testosterone and dihydrotestosterone have a number of potentially desirable actions as well as non-desirable actions depending on the particular individual's age, sex, therapeutic need, etc.
- certain positive consequences of AR-agonist signaling can be generalized as including increased bone mineral density and a corresponding reduction of risk of bone fractures.
- androgen supplementation can be very valuable in the prevention or treatment of osteoporosis where the osteoporosis might originate from any number of different causes, such as corticosteroid induced osteoporosis and age-related osteoporosis (e.g. postmenopausal).
- males and females respond to agonist supplementation with an increase in muscle mass and very often a decrease in fat mass.
- This is beneficial in a very large number of treatment modalities.
- Other muscle- wasting disorders such as muscular dystrophy in its many forms as well as related disorders might be treated to advantage with androgens.
- the increase in muscle mass with concomitant reduction in fat mass associated with anabolic androgen action has additional health benefits for many men and women including potentially increased sensitivity to insulin.
- Androgen supplementation is also associated with reduction of high triglycerides, though there is a general correlation with androgen use and decreased HDL levels and in some cases, increased LDL levels.
- numerous laudatory benefits have been associated with androgen supplementation including improved sexual desire and functioning, increased cognition, memory, sense of well being and possible decrease in risk of Alzheimer's disease.
- Androgen antagonists have been used in treating prostate cancer, where blockade of androgen signaling is desired whereas some androgens agonists (e.g. dihydrotestosterone) stimulate the hypertrophy of prostate tissue and may be a causative factor in prostate cancer. Androgen agonist activity is often associated with stimulation of benign prostate hyperplasia, a disease characterized by an enlarged prostate often accompanied by discomfort and difficulty in urination due to blockage of the urethra. As a result, androgen antagonists have efficacy in the reduction of the size of the prostate and the corresponding symptoms of benign prostate hyperplasia, though it is much more common to use a 5oc-reductase inhibitor (e.g.
- finasteride as such inhibitors do not decrease androgen signaling systemically to the same extent as a typical anti-androgen (e.g. bicalutamide), but rather reduce androgen drive more site specifically to where testosterone to DHT conversion occurs such as the prostate and scalp.
- Androgen antagonists also find utility in the treatment of hirsutism in women as well as the treatment of acne. Androgens are generally contraindicated in conditions that are treated with androgen antagonists since they can exacerbate the symptoms that are being treated.
- an androgen would retain the benefits of androgen agonists while minimizing the stimulatory effects on the prostate in males as well as some of the other untoward effects of androgens including masculinization of women and increase in acne in both sexes.
- Androgens that demonstrate tissue selective effects compared to the benchmarks testosterone and/or dihydrotestosterone are typically referred to as androgen receptor modulators or more often, selective androgen receptor modulators (SARMs).
- SARMs selective androgen receptor modulators
- this invention describes a compound of Formula (I) or a pharmaceutically acceptable salt thereof:
- X, Y and Z are independently selected from the group consisting of hydrogen, halogen, CN, Ci -4 alkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkyl, N0 2 , N3 ⁇ 4, OCi -3 alkyl and OH; with the proviso that at least one of X, Y and Z is not hydrogen; each R a is independently selected from the group consisting of halogen, OH, NH(CO)C 1-6 alkyl, C 4 alkyl (wherein said C 1-4 alkyl is optionally substituted with from 1-2 substituents each independently selected from the group consisting of CN, OH and OCi -3 alkyl), Ci-s haloalkyl, monocyclic aryl (wherein said monocyclic aryl is optionally substituted with from 1-3 substituents each independently selected from the group consisting of C 1-3 alkyl, Ci-s haloalkyl, CN, halogen, OH and OC 1-3
- R b is independently selected from the group consisting of C 1-4 alkyl (wherein said C 1-4 alkyl is optionally substituted with from 1-2 substituents each
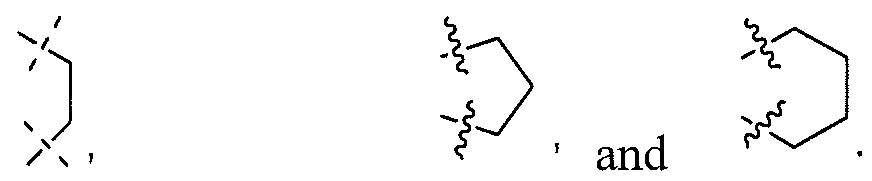
- A is a 2-5 membered carbon alkyl linker selected from the group consisting of
- n 0, 1, 2 or 3.
- this invention describes a compound of Formula (II) a pharmaceutically acceptable salt thereof:
- c 0, 1, 2, or 3;
- R a , X, Y and Z are as defined herein;
- This invention also provides methods of treating a disease, syndrome, illness, or symptom associated with insufficient androgen levels in a mammal in need thereof, wherein said method comprises the administration to said mammal of an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof.
- this invention describes a compound of Formula (I) or a pharmaceutically acceptable salt thereof:
- X, Y and Z are independently selected from hydrogen, halogen, CN, Ci- 4 alkyl, C 1-3 hydroxyalkyl, C 1-3 haloalkyl, N0 2 , NH 2 , Ci -3 alkyl and OH; with the proviso that at least one of X, Y and Z is not hydrogen;
- each R a is independently selected from halogen, OH, NH(CO)C 1-6 alkyl, C 4 alkyl (wherein said C 1-4 alkyl is optionally substituted with from 1-2 substituents each independently selected from CN, OH and OC 1-3 alkyl), C1-5 haloalkyl, monocyclic aryl (wherein said monocyclic aryl is optionally substituted with from 1- 3 substituents each independently selected from Cj -3 alkyl, C 1-5 haloalkyl, CN, halogen, OH and OC 1-3 alkyl), benzyl (wherein the phenyl group of said benzyl is optionally substituted with from 1-3 substituents each independently selected from halogen, d -3 alkyl, S(O) 0-2 Ci -3 alkyl, S(O) 0-2 phenyl, 0-C 1-6 alkyl, and OCF 3 ), C(O)- Ci.io alk l, S0
- R b is independently selected from C 1-4 alkyl (wherein said Ci -4 alkyl is optionally substituted with from 1 -2 substituents each independently selected from CN, OH, OPh (wherein said Ph is optionally substituted with 1 -2 substituents each mdependently selected from the group consisting of halogen, OH, CN and OC 1-3 alkyl), C 1-5 haloalkyl, monocyclic aryl (wherein said monocyclic aryl is optionally substituted with from 1-3 substituents each independently selected from the group consisting of C 1-3 alkyl, 0 1-5 haloalkyl, CN, halogen, OH and OC 1-3 alkyl), and benzyl (wherein the phenyl group of said benzyl is optionally substituted with from 1-3 substituents each independently selected from the group consisting of halogen, C1.3 alkyl, CN, S(O) 0-2 C 1-3 alkyl, S(O) 0-2
- A is a 2-5 membered carbon alkyl linker selected from the group consisting of
- X, Y and Z are independently selected from hydrogen, halogen, Ci -3 haloalkyl and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- X, Y and Z are independently selected from hydrogen, chlorine, fluorine, CF 3 and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- each R a is independently selected from halogen, C 4 alkyl and Ci -5 haloalkyl.
- each R a is independently selected from halogen, C 2 alkyl and C 1-2 haloalkyl.
- each R a is independently selected from chlorine, fluorine, CH 3 , CH 3 CH 2 , CF 3 , and CF 3 CF 2 . In other embodiments of this invention, for the compound of Formula (I), each R a is independently selected from fluorine, CH 3 , and CF 3 .
- R b is C -2 alkyl or Ci ⁇ haloalkyl.
- Rb is CH 3 , CH 3 CH 2 , CF 3 , or CF 3 CF 2 .
- Rb is CH 3 , CF 3 , or CF 3 CF 2 .
- A is a carbon linker selected from the group consisting of:
- n is 0, 1 or 2.
- X and Y are hydrogen; Z is CN; each R a is independently selected from the group consisting of fluorine, CH 3 and CF 3 ; Rb is CH 3 , CF 3 , or CF 3 CF 2 ;
- A is a carbon linker selected from the group consisting of: n is 0, 1 or 2.
- X is hydrogen; Y is CF 3 ; Z is CN; each R a is independently selected from the group consisting of fluorine, CH 3 and CF 3 ; R b is selected from the group consisting of CH 3 , CF 3 , and CF 3 CF 2 ;
- A is:
- n 0, 1 or 2.
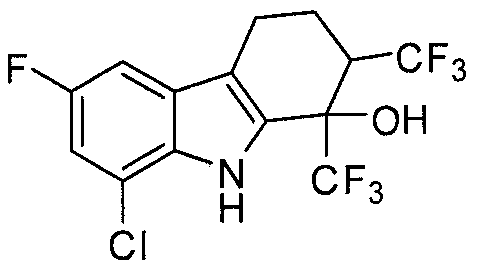
- X is hydrogen; Y and Z are chlorine; each R a is independently selected from the group consisting of fluorine, CH 3 and CF 3 ; R b is selected from the group consisting of CH 3 , CF 3 , and CF 3 CF 2 ;
- n 0, 1 or 2.
- X is hydrogen; Y is chlorine; Z is fluorine; each R a is independently selected from
- A is:
- n 0, 1 or 2.
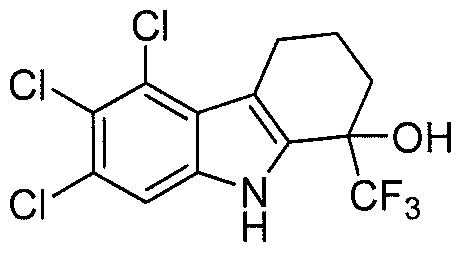
- X, Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 or CF 3 ;
- n 0, 1 or 2.
- this invention describes a compound of Formula (la) or a pharmaceutically acceptable salt thereof:
- X, Y and Z are independently selected from hydrogen, halogen, C 1-3 haloalkyl and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- X, Y and Z are independently selected from hydrogen, chlorine, fluorine, CF 3 and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- each R a is independently selected from halogen, C1-4 alkyl and C 1-5 haloalkyl.
- each R a is independently selected from halogen, C 2 alkyl and Ci -2 haloalkyl.
- each R a is independently selected from chlorine, fluorine, CH 3 , CH 3 CH 2 , CF 3 , and CF 3 CF 2 .
- each R a is independently selected from fluorine, CH 3 , and CF 3 .
- R b is Cj- 2 alkyl or C 1-2 haloalkyl.
- R b is CH 3 , CH 3 CH 2 , CF 3 , or CF 3 CF 2 .
- R b is CH 3 , CF 3 , or CF 3 CF 2 .
- n is 0, 1 or 2.
- X and Y are hydrogen; Z is CN; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is CF 3 ; Z is CN; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 or CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is chlorine; Z is fluorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X, Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 or CF 3 ; R b is CH 3 , CF 3 , and CF 3 CF 2 ; and n is 0, 1 or 2.
- this invention describes a compound of Formula (lb) or a pharmaceutically acceptable salt thereof;
- X, Y, Z, R a , R b and n are as defined for formula I.
- X, Y and Z are independently selected from hydrogen, halogen, C 1-3 haloalkyl and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- X, Y and Z are independently selected from hydrogen, chlorine, fluorine, CF 3 and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- each R a is independently selected from halogen, Ci- 4 alkyl and C 1-5 haloalkyl.
- each R a is independently selected from halogen, Cj- 2 alkyl and Ci -2 haloalkyl.
- each R a is independently selected from chlorine, fluorine, CH 3 , CH 3 CH 2 , CF 3 , and CF 3 CF 2 .
- each R a is independently selected from fluorine, CH 3 , and CF 3 .
- R b is Ci- 2 alkyl or Ci -2 haloalkyl.
- R b is CH 3 , CH 3 CH 2 , CF 3 , or CF 3 CF 2 .
- R b is CH 3 , CF 3 , or CF 3 CF 2 .
- n is 0, 1 or 2.
- X and Y are hydrogen; Z is CN; each R a is independently selected from fluorine, CH 3 or CF 3 ; R b is CH 3 , CF 3 , and CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is CF 3 ; Z is CN; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is chlorine; Z is fluorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X, Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- this invention describes a compound of Formula (Ic) or a pharmaceutically acceptable salt thereof:
- X, Y, Z, R a , R b and n are as defined for formula I .
- X, Y and Z are independently selected from hydrogen, halogen, C 1-3 haloalkyl and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- X, Y and Z are independently selected from hydrogen, chlorine, fluorine, CF 3 and CN; with the proviso that at least one of X, Y and Z is not hydrogen.
- each R a is independently selected from halogen, Ci- 4 alkyl and C 1-5 haloalkyl.
- each R a is independently selected from halogen, C 2 alkyl and C 1-2 haloalkyl.
- each R a is independently selected from chlorine, fluorine, CH 3 , CH 3 CH 2 , CF 3 , and CF 3 CF 2 .
- each R a is independently selected from fluorine, CH 3 , and CF 3 .
- R b is Ci-2 alkyl or C 1-2 haloalkyl.
- R b is CH 3 , CH 3 CH 2 , CF 3 , or CF 3 CF 2 .
- R b is CH 3 , CF 3 , or CF 3 CF 2 .
- n is 0, 1 or 2.
- X and Y are hydrogen; Z is CN; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is CF 3 ; Z is CN; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 or CF 3 ; R b is CH 3 , CF 3 , and CF 3 CF 2 ; and n is 0, 1 or 2.
- X is hydrogen; Y is chlorine; Z is fluorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- X, Y and Z are chlorine; each R a is independently selected from fluorine, CH 3 and CF 3 ; R b is CH 3 , CF 3 , or CF 3 CF 2 ; and n is 0, 1 or 2.
- selected compound of this invention is selected from the following list.
- the compound names in the list were generated with the assistance of ChemDraw ® versions 8.0, 9.0 and/or 11.0 (CambridgeSoft Corporation, 100 CambridgePark Drive, Cambridge, MA 02140 USA)).
- ChemDraw ® versions 8.0, 9.0 and/or 11.0 (CambridgeSoft Corporation, 100 CambridgePark Drive, Cambridge, MA 02140 USA)
- the stereochemistry at a chiral center is not defined in the compound name this indicates that the sample prepared contained a mixture of isomers at this center.
- this invention describes a compound of Formula (II), or a pharmaceutically acceptable salt thereof:
- c 0, 1, 2, or 3;
- R a , X, Y and Z are as defined for formula I;
- X, Y and Z are independently selected from hydrogen, halogen, C 1-3 haloalkyl and CN; with the proviso that at least two of X, Y and Z are each independently halogen or CN; and provided that two of X, Y and Z are not both Br.
- X, Y and Z are independently selected from hydrogen, chlorine, bromine, CF 3 and CN; with the proviso that at least two of X, Y and Z are each independently halogen, or CN; and provided that two of X, Y and Z are not both Br.
- each R a is independently selected from halogen, Ci- 4 alkyl, C 1-5 haloalkyl and benzyl.
- each R a is independently selected from halogen, Ci- 2 alkyl, Cj -2 haloalkyl and benzyl.
- each R a is independently selected from chlorine, fluorine, bromine, CH 3 and benzyl.
- each R a is independently selected from bromine, CH 3 and benzyl.
- c is 0, or 1.
- this invention describes a compound of Formula (Ila), or a pharmaceutically acceptable salt thereof:
- c 0, 1, 2, or 3;
- R a , X, Y and Z are as defined for formula I;
- X, Y and Z are independently selected from hydrogen, halogen, C 1-3 haloalkyl and CN; with the proviso that at least two of X, Y and Z are each independently halogen or CN; and provided that two of X, Y and Z are not both Br.
- X, Y and Z are independently selected from hydrogen, chlorine, bromine, CF 3 and CN; with the proviso that at least two of X, Y and Z are each independently halogen, or CN; and provided that two of X, Y and Z are not both Br;
- each R a is independently selected from halogen, Q- 4 alkyl, C 1-5 haloalkyl and benzyl.
- each R a is independently selected from halogen, Ci- 2 alkyl, Ci -2 haloalkyl and benzyl.
- each R a is independently selected from chlorine, fluorine, bromine, CH 3 and benzyl.
- each R a is independently selected from bromine, CH 3 and benzyl.
- c is 0, or 1.
- X and Y are hydrogen; Z is CN; each R a is independently selected from bromine and benzyl; c is 0 or 1.
- X and Y are hydrogen; Z is bromine; each R a is independently selected from bromine and benzyl; c is 0 or 1.
- X and Y are chlorine; Z is hydrogen; each R a is independently selected from bromine and benzyl; c is 0 or 1.
- III X and Y are hydrogen; Z is CN; each R a is independently selected from bromine and benzyl; and c is 0 or 1.
- X and Y are hydrogen; Z is bromine; each R a is independently selected from bromine and benzyl; and c is 0 or 1.
- X and Y are chlorine; Z is hydrogen; each R a is independently selected from bromine and benzyl; and c is 0 or 1.
- a compound of this invention is selected from the group below.
- the compound names in the list were generated with the assistance of ChemDraw ® versions 8.0, 9.0 and/or 1 1.0 (CambridgeSoft Corporation, 100 CambridgePark Drive, Cambridge, MA 02140 USA)).
- the invention also relates to pharmaceutical compositions comprising a compound of Formula (I), (la), (lb), (Ic), (II), (Ila) or (III) or any of the structural embodiments described herein and at least one pharmaceutically acceptable excipient.
- the invention also provides a method of modulating an androgen receptor in a cell, comprising the administration of a compound to said cell wherein said compound has structural Formula (I), (la), (lb), (Ic), (II), (Ila) or (III) or any of the structural embodiments described herein, or a pharmaceutically acceptable salt thereof.
- This invention provides a method of identifying a compound capable of modulating an androgen receptor comprising contacting a cell expressing an androgen receptor with a compound according to Formula (I), (la), (lb), (Ic), (II), (Ila) or (III) and monitoring the effect of the compound on the cell.
- This invention also provides a method of treating (e.g., preventing, or ameliorating the symptoms associated with, or reducing the incidence of, reducing the pathogenesis of, facilitating the recovery from or delaying the onset of) a disease, syndrome, illness, or symptom associated with insufficient androgen levels in a mammal in need thereof, wherein said method comprises the administration to said mammal of an effective amount of a compound of Formula (I), (la), (lb), (Ic), (II), (Ila) or (III), or any one of the structural embodiments described herein or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of Formula (I), (la), (lb), (Ic), (II), (Ila) or (III), or one of the structural embodiments described herein, or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
- the mammal is a human.
- this invention provides a method of treating (e.g., preventing, or ameliorating the symptoms associated with, or reducing the incidence of, reducing the pathogenesis of, facilitating the recovery from or delaying the onset of) sarcopenia, frailty, multiple sclerosis, osteoporosis, anemia, cognitive impairment, cachexia, muscular dystrophy, weak appetite, low body weight, anorexia nervosa, acne, seborrhea, polycystic ovarian syndrome, hair loss, AIDs wasting, chronic fatigue syndrome, short stature, low testosterone levels, diminished libido, benign prostate hypertrophy, infertility, erectile dysfunction, vaginal dryness, premenstrual syndrome, postmenopausal symptoms, female hormone replacement therapy, male hormone replacement therapy, depression, Type II diabetes, mood disorders, sleep disorders, memory disorders, neurodegenerative disorders,
- Alzheimer's dementia attention deficit disorder, senile dementia, coronary artery disease, hirsutism, pain, myalgia, myocardial infarction, stroke, clotting disorders, thromboembolisms, congestive heart disorder, low insulin sensitivity, low glucose utilization, high blood sugar, organ transplant, metabolic syndrome, diabetes, glucose intolerance, hyperinsulinemia, insulin resistance, tooth injury, tooth disease, periodontal disease, liver disease, thrombocytopenia, fatty liver conditions, endometriosis, hot flushes, hot flashes, vasomotor disturbance, stress disorders, dwarfism, dyslipidemia, cardiovascular disease, coronary artery disease, renal disease, thin skin disorders, lethargy, osteopenia, dialysis, irritable bowel syndrome, Crohn's disease, Paget's disease, osteoarthritis, connective tissue disease or disorders, injury, burns, trauma, wounds, bone fracture, atherosclerosis, cachexia, cancer cachexia, and obesity, in a mammal in
- this invention describes a method of treating (e.g., preventing, or ameliorating the symptoms associated with, or reducing the incidence of, reducing the pathogenesis of, facilitating the recovery from or delaying the onset of) prostate cancer, breast cancer, endometrial cancer, hepatocellular cancer, lymphoma, multiple endocrine neoplasia, vaginal cancer, renal cancer, thyroid cancer, testicular cancer, leukemia, and ovarian cancer in a mammal in need thereof comprising the administration to said mammal of a compound according to a structure of Formula (I), (la), (lb), (Ic), (II), (Ila) or (III), or one of the structural embodiments described herein, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition comprising a compound of structural Formula (I), (la), (lb), (Ic), (II), (Ila) or (III), or one of the structural embodiments described herein including pharmaceutically acceptable salts thereof and a pharmaceutically
- alkyl refers to both straight and branch chain hydrocarbon radicals, having the number of carbon atoms falling within the specified range.
- C alkyl means that a hydrocarbon radical is attached that may contain anywhere from 1 to 4 carbon atoms with the remaining valence filled in by hydrogen atoms.
- the definition also includes separately each permutation as though it were separately listed.
- C 1- alkyl includes methyl and ethyl.
- C 1-3 alkyl includes methyl, ethyl, propyl and 2-propyl.
- C ⁇ .
- C 4 alkyl includes methyl, ethyl, n-propyl, 2-propyl, n-butyl, 2-butyl, iso-butyl and tert- butyl.
- C 1-5 alkyl includes methyl, ethyl, 2-propyl, n-butyl, 2-methylbutyl, tert-butyl, n-pentyl, pentan-2-yl, pentan-3-yl, and tert-pentyl, iso-pentyl.
- halogen refers to a fluorine, chlorine, bromine or iodine radical.
- haloalkyl refers to an alkyl radical wherein said alkyl radical is the same as defined for the term “alkyl” except that the alkyl radical additionally has from 1 to 5 halogen atoms attached to the alkyl chain.
- C ⁇ haloalkyl includes
- Ci -2 haloalkyl includes -CH 2 F, CHF 2 , CF 3 ,
- C 1-3 haloalkyl is defined to include -CH 2 F, -CHF 2 , -CF 3 , -CH 2 CF 3 , -CHFCF 3 , -CF 2 CF 3 , -CHC1CH 3 , -CH2CH2CI, -CH 2 CH 2 CF 3 , and the like.
- CM haloalkyl is defined to include -CH 2 F, -CHF 2 , -CF 3 , -CH 2 CF 3 , -CHFCF 3 , -CF 2 CF 3 , -CHC1CH 3 , -CH 2 CH 2 C1, -CH 2 CH 2 CF 3 , -CH 2 CH 2 CH 2 CF 3 , CHC1CF 2 CH 2 CH 3 , CF 2 CH 2 CH 2 CHF 2 , CH 2 CH 2 CH 2 CH 2 F, CH2CH2CH2CH2CI, and the like.
- hydroxyalkyl refers to an alkyl radical wherein said alkyl radical is the same as defined for the term “alkyl” except that the alkyl radical additionally has from 1 or 2 hydroxyl groups attached to the alkyl chain.
- C 2 - 4 hydroxyalkyl includes 2-hydroxyethyl, 2-hydroxypropyl, 2,4-dihydroxybutyl and the like.
- the compounds of this invention may be present as solids and when so present, may be in an amorphous form or they may be crystalline. When the compounds of this invention are in the crystalline form, they might be present as a single polymorph or a mixture of polymorphs or even as a mixture of amorphous material together with one or more distinct polymorphs - the invention is not limited according to any particular solid or liquid state form.
- the compounds of this invention contain at least one stereocenter and therefore, exist in various stereoisomeric forms.
- Stereoisomers are compounds which differ only in their spatial arrangement.
- Enantiomers are pairs of
- the stereochemistry of a disclosed compound is named or depicted by structure
- the named or depicted stereoisomer is at least 60%, 70%, 80%, 90%, 99% or 99.9% by weight pure relative to the other stereoisomers.
- the depicted or named enantiomer is at least 60%, 70%, 80%, 90%, 99% or 99.9% by weight optically pure. Percent optical purity by weight is the ratio of the weight of the enantiomer over the weight of the enantiomer plus the weight of its optical isomer.
- the compounds of the invention may be prepared as individual isomers by incorporating or starting with a specific isomer, isomer-specific synthesis or resolution from an isomeric mixture.
- Conventional resolution techniques include forming the salt of a free base of each isomer of an isomeric pair using an optically active acid (followed by fractional crystallization and regeneration of the free base), forming the salt of the acid form of each isomer of an isomeric pair using an optically active amine (followed by fractional crystallization and regeneration of the free acid), forming an ester or amide of each of the isomers of an isomeric pair using an optically pure acid, amine or alcohol (followed by chromatographic separation and removal of the chiral auxiliary), or resolving an isomeric mixture of either a starting material or a final product using various well known
- acid addition salts can be made and this invention includes such acid addition salts.
- Some representative (non-limiting) acid addition salts include hydrochloride, hydrobromide, hydroiodide, acetate, benzenesulfonate, mesylate, besylate, benzoate, tosylate, citrate, tartrate, sulfate, bisulfate, lactate, maleate, mandelate, valerate, laurate, caprylate, propionate, succinate, phosphate, salicylate, napsylate, nitrate, tannate, resorcinate and the like, including multiprotic salts as well as mixtures of the acid addition salts.
- this invention also embraces quaternized ammonium salts of those amines. It should be appreciated that N-oxides of amines are also embraced within the definition of the compounds of this invention.
- compounds of this invention include one or more acid sites such as carboxylic acids, phenols and the like, basic addition salts can be made and this invention includes such basic addition salts.
- some representative (non-limiting) acidic compounds of this invention may be present as their lithium, sodium, potassium, ammonium, trialkyammonium, calcium, magnesium, barium and the like.
- solvates can also be present as solvates and such solvates are embraced within the scope of this invention even where not explicitly described.
- Such solvates are preferably hydrates but can be solvates comprised of other solvents, preferably where those solvents are considered to be non-toxic or at least acceptable for administration to mammals, preferably humans.
- the solvates can be stoichiometric or non-stoichiometric, singular or in combination.
- Some exemplary solvates include water, ethanol, acetic acid and the like.
- deuterium is optionally included - either at the normal hydrogen to deuterium isotope ratio or, possibly enriched in deuterium up to 100% deuterium at any given position.
- some embodiments of this invention can be prepared so as to include high levels of deuterium (>90%) at one or more positions.
- a structure of Formula (I) is provided wherein A is defined as including cyclohexyl.
- A when A is a cyclohexyl ring, the cyclohexyl will have one or more specified "hydrogens".
- one or more of the hydrogens may be optionally substituted by deuterium and that substitution may range from a very low incorporation to >90% depending on the method used for incorporating the deuterium if a specifc method was used.
- the therapeutic utility of these compounds includes "treating" a mammal, preferably a human where treating is understood to include treating, preventing, or ameliorating the symptoms associated with, or reducing the incidence of, reducing the pathogenesis of, facilitating the recovery from or delaying the onset of the syndrome, illness, malady or condition being considered.
- the compounds of this invention can also be useful in states or conditions where no clear deficit, illness or malady per se is perceived but rather, where a preferred condition, sensation, performance, capability or state is obtainable through therapeutic intervention with a compound of this invention.
- the compounds of this invention when used as therapeutics can be administered by any method known to one of skill in the art such as orally, bucally, intravenously, subcutaneously, intramuscularly, transdermally, intradermally, intravascularly, intranasally, sublingually, intracranially, rectally, intratumorally, intravaginally, intraperitonealy, pulmonary, ocularly and intratumorally.
- the term "effective amount” refers to an amount which, when administered in a proper dosing regimen, is sufficient to treat (therapeutically or prophylactically) the target disorder. For example, and effective amount is sufficient to reduce or ameliorate the severity, duration or progression of the disorder being treated, prevent the advancement of the disorder being treated, cause the regression of the disorder being treated, or enhance or improve the prophylactic or therapeutic effect(s) of another therapy.
- the compounds and compositions of this invention maybe given once daily or with multiple daily doses such as twice per day, three times per day and four times per day.
- a Pharmaceutical composition refers to one or more of the compounds of this invention with one or more pharmaceutically acceptable excipients.
- the compound is administered orally where it can be Formulated for solid dosage administration or liquid dosage administration.
- Solid dosage administration can be in the form of a tablet, granule, capsule, pill, pellet, powder and the like.
- Liquid dosage Formulations include syrups, solutions, gels, suspensions, elixirs, emulsions, colloids, oils, and the like.
- the compounds of this invention may be solids and when present as solids, they maybe of defined particle size. Where the compound of this invention is not particularly water soluble, it is sometimes preferable to administer the compound with a certain particle size - a particle size with a preferred range where the average mean particle size diameter is under 100 microns, or 75 microns, or 50 microns, or 35 microns, or 10 microns or 5 microns.
- Solid dosage Formulations will comprise at least one compound of this invention together with one or more pharmaceutical excipients.
- excipients include, by way of non-limiting example diluents (monosaccharides, disaccharides and polyhydric alcohols including starch, mannitol, dextrose, sucrose, microcrystallme cellulose, maltodextrin, sorbitol, xylitol, fructose and the like), binders (starch, gelatin, natural sugars, gums, waxes and the like), disintegrants (alginic acid, carboxymethylcellulose (calcium or sodium), cellulose, crocarmellose, crospovidone, microcrystallme cellulose, sodium starch glycolate, agar and the like), acidic or basic buffering agents (citrates, phoshphates, gluconates, acetates, carbonates, bicarbonates and the like), chelating agents (edetic), arates, phos
- preservatives benzoic acid, chlorhexidine gluconate, potassium benzoate, potassium sorbate, sorbic acid, sodium benzoate and the like
- glidants and lubricants calcium stearate, oils, magnesium stearate, magnesium trisilicate, sodium fumarate, colloidal silica, zinc stearate, sodium oleate, stearic acid, and the like
- antioxidants and/or preservatives tocopherols, ascorabtes, phenols, and the like
- acidifying agents citric acid, fumaric acid, malic acid, tartaric acid and the like
- coloring agents citric acid, fumaric acid, malic acid, tartaric acid and the like
- the solid dosage Formulations of this invention can be prepared in different forms including most commonly, tablets and capsules.
- the tablets can be
- Formulated by a wide variety of methods known to one of skill in the art including, for example, preparing a dry powder mixture of the drug substance in combination with one or more of the excipients granulating the mixture and pressing to together into a tablet and optionally coating the tablet with an enteric or non-enteric coating.
- the final coat typically includes a light protective pigment such as titanium oxide and a shellac or wax to keep the tablet dry and stable. While not intending to be limited by theory or example, in some instances it might be preferred to prepare the tablets by wet granulating the drug with one or more of the excipients and then extruding the granulated material.
- the solid dosage forms of this invention also include capsules wherein the drug is enclosed inside the capsule either as a powder together with optional excipients or as granules containing usually including one or more excipients together with the drug and wherein the granule in turn can be optionally coated, for example, enterically or non-enterically.
- the solid dosage Formulations of this invention are Formulated in a sustained release Formulation.
- Formulations are known to those of skill in the art and generally rely on the co- Formulation of the drug with one or more matrix forming substances that slow the release of the androgen receptor modulator thus extending the compound's lifetime in the digestive track and thereby extend the compounds half-life.
- matrix forming substances include hydroxypropyl methylcellulose, carbopol, sodium carboxymethylcellulose and the like.
- the compounds are Formulated for delivery other than via a solid oral dosage form.
- a pulmonary route of administration typically means that the compound of this invention is inhaled into the lung where it is absorbed into the circulation.
- Such a route of administration has the advantage of avoiding a first pass liver effect thereby possibly increasing bioavailability as well as decreasing or eliminating undesirable androgen agonist effects on the liver such as increasing liver enzymes and/or decreasing HDL.
- Formulating a compound of the invention for pulmonary delivery can be accomplished by micronizing the compound of the invention to a very fine size particle, typically with a mean average diameter of less than 20 microns, or less than 10 microns or between 2 and 5 microns.
- the powder may then be inhaled by itself or more likely mixed with one or more excipients such as lactose or maltose.
- the powder can then be inhaled in a dry powder inhaling device either once or multiple times per day depending on the particular compound and the patients need.
- Other types of pulmonary dosage forms are also embraced by this invention.
- the compound of this invention may be suspended in an aerosolizing medium and inhaled as a suspension through a meter dosed inhaler or a nebulizer.
- the compounds of this invention can be Formulated for transdermal delivery. Effective advantage of these compounds can be taken through a wide variety of transdermal options.
- the compounds of this invention maybe Formulated for passive diffusion patches where they are preferably embedded in a matrix that allows for slow diffusion of the compound into the treated subject's circulation.
- the compound is preferably dissolved or suspended in solvents including by way of non-limiting examples one or more of ethanol, water, propylene glycol, and Klucel HF.
- a polymer matrix e.g. acrylate adhesive
- the transdermal Formulations maybe designed to be compatible with alternate transdermal delivery technologies.
- some transdermal technologies achieve greater and/or more consistent delivery by creating micropores in the skin using radio frequency, heat, ultrasound or electricity.
- the compounds of this invention can be used with microneedle technology wherein the compound is loaded into very small needles which due not need to penetrate the dermis to be effective.
- the compounds of this invention may be employed alone or in combination with other therapeutic agents.
- the compounds of this invention can be used in combination with anti-lipidemics (statins, fibrates, omega-3 oils, niacinates and the like), bone anti-resorptives (bisphosponates, estrogens, selective estrogen receptor modulators (SERMs), calcitonin, and the like), bone anabolic agents (PTH and fragments e.g teriparatide, PTHRP and analogues e.g. Ba058), anti-diabetics (e.g. insulin sensitizers, glucose absorption and synthesis inhibitors (e.g.
- the compounds of this invention may be co- Formulated or co-administered wherein said co-administration does not require dosing at exactly the same time but rather indicates that the patient is undergoing treatment with one or more of the additional agents during the timeframe of treatment with the selective androgen modulators of this invention.
- the additional drug(s) for combination treatment can be administered concomitantly, sequentially or separately from the compounds of this invention.
- the compounds of this invention may be administered according to different dosage scheduling and the dosage may be adjusted as deemed necessary by the subject or preferably by the subject in consultation with a qualified practitioner of medicine. Dosing of the compounds of this invention can take place by multiple routes and consequently, the dosing schedule and amounts are dependent not only on the particular subject's weight, sex, age, therapy contemplated, etc but also by the route of the drug chosen.
- the compounds of this invention may be dosed by the oral route in a once daily, twice daily, three times daily or more than three times per day depending on the particular needs of that subject, the
- the dosage will typically be from about 0.01 mg to 500 mg of drug per daily dosage, for example from about 0.1 mg to about 10 mg, such as from about 0.1 mg to about 3 mg, or from about 0.1 mg to about 250 mg of drug per daily dosage, or from about 1 mg to about 150 mg of drug per daily dosage, or from about 5 mg to about 100 mg of drug per daily dosage., or from about 0.1 mg to about 5 mg of drug per daily dosage.
- the amount of compound dosed per day can be administered every day, every other day, every 2 days, every 3 days, every 4 days, every 5 days, etc.
- a 5 mg per day dose can be initiated on Monday with a first subsequent 5 mg per day dose administered on Wednesday, a second subsequent 5 mg per day dose administered on Friday, etc.
- a compound of this invention is dosed once every seven days.
- the compounds of this invention can also be dosed on a monthly basis meaning that administration is done once per month.
- the compounds of this invention can be dosed on a weekly basis (once a week), every other week, every three weeks or every four weeks for a single day or multiple days.
- the compounds of this invention can also be dosed on an as needed or "pro re nata" "prn" schedule, and "on demand".
- the compounds of this invention are administered in a therapeutically effective dose at some time prior to commencement of an activity wherein the therapeutic effect of the compounds of this invention is desirable.
- Administration can be immediately prior to such an activity, including about 0 minutes, about 10 minutes, about 20 minutes, about 30 minutes, about 1 hour, about 2 hours, about 3 hours, about 4 hours, about 5 hours, about 6 hours, about 7 hours, about 8 hours, about 9 hours, or about 10 hours prior to such an activity, depending on the Formulation.
- the compounds of this invention can be prepared by a variety of synthetic routes and techniques known to those of skill in the art.
- the processes disclosed herein should not be construed as limiting the examples or scope of the invention in any way but rather are provided as just some of the representative ways that the compounds of this invention can be or were prepared.
- protective groups are employed in the synthesis of the compounds of this invention and it should be appreciated that there are a diverse array of protective groups and strategies that can be employed in organic synthesis (T.W.Green and P.G.M. Wuts (2006) Greene's Protective Groups in Organic Synthesis, herein incorporated by reference in its entirety) and that where a protective group is referred to generically, any appropriate protective group should be considered.
- leaving groups are employed in the synthesis of compounds of this invention. Where a specific leaving group is referred to, it should be appreciated that other leaving groups might also be used. Leaving groups typically include those groups that can stabilize an anion. In the case of nucleophilic aromatic substitutions, the leaving group may be an anion or a neutrally charged group. In some cases, the leaving group for nucleophilic aromatic substitution may be a group that is not typically considered to be a stabilized anion (e.g. fluoride or hydride).
- nucleophilic leaving groups include halogens, sulfonates (O-mesylates, O-tosylates, etc), hydrides, quaternized amines, nitro, and the like. Additional discussion and examples can be found in leading textbooks on organic chemistry including, for example, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5 th Edition, which is herein incorporated in its entirety.
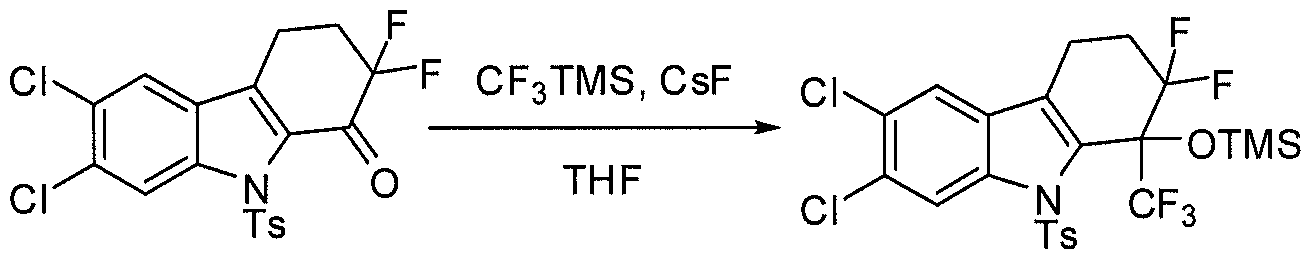
- Scheme 1 gives one embodiment of a general method for preparing compounds of this invention, such as, for example, the mono-substituted compound of Formula E, wherein X, Y, Z and 3 ⁇ 4 are as defined herein and m is 1, 2 or 3.
- the starting material an aryl hydrazine of Formula A
- a 1 ,2,cyclodione of Formula B is coupled with a 1 ,2,cyclodione of Formula B to give the tricyclic keto compound of Formula C using standard coupling reaction conditions known to those of skill in the art.
- a solvent is chosen that can dissolve at least some of each of the components of the reaction.
- EtOH or MeOH can be a very good solvent for these reactions but other solvents such as DMSO, DMF, HMPA, etc, should be considered as well.
- the product C is isolated and carried to the next step. Isolation techniques are well-known to those of skill in the art and include chromatography and/or crystallization.
- the keto product C is converted to the alcohol F using a number of potential reagents well-known to those of skill in the art.
- the keto compound C is converted directly to the product F by reduction of the ketone, such as for example, by reaction with a Grignard reagent, CF 3 TMS or CF 3 CF 3 TMS.
- a Grignard reagent such as for example, by reaction with a Grignard reagent, CF 3 TMS or CF 3 CF 3 TMS.
- the compound D is converted to the substituted alcohol E, wherein P 2 is any alcohol protecting group, known to one skilled in the art, such as, for example, a silyl ether, such as, trimethylsilyl group, a tert-butyldimethylsilyl ether, or a tert-butyldiphenylsilyl ether, but other protecting groups could be useful as well. Removal of the protecting groups Pi and P 2 results in the product F.
- P 2 is any alcohol protecting group, known to one skilled in the art, such as, for example, a silyl ether, such as, trimethylsilyl group, a tert-butyldimethylsilyl ether, or a tert-butyldiphenylsilyl ether, but other protecting groups could be useful as well. Removal of the protecting groups Pi and P 2 results in the product F.
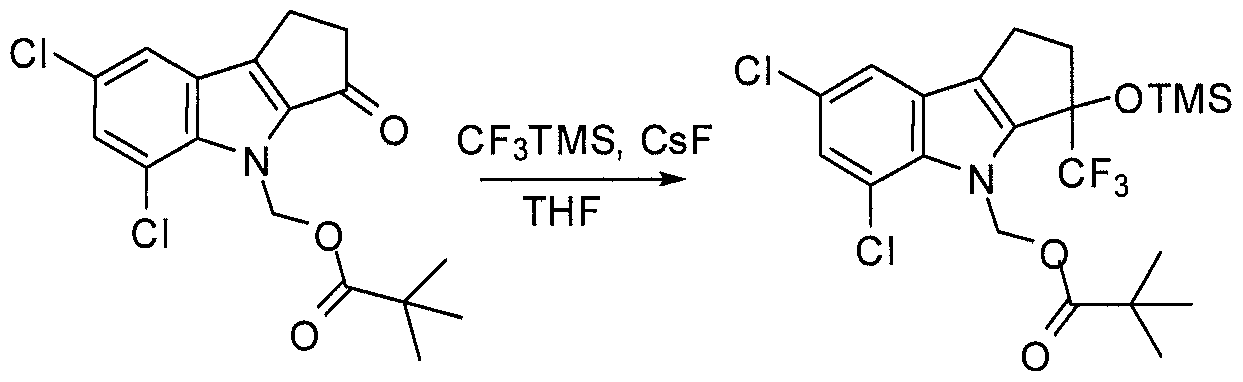
- Scheme 3 shows the modification of Scheme 1 for the preparation of the di- substituted compounds of Formula L wherein X, Y, 3 ⁇ 4 and m are as defined herein.
- the regioisomers for the keto compound of Formula K (corresponding to the compound of Formula C in Scheme 1) are separated using a number of potential methods well-known to those of skill in the art.
- the carbazole amine is converted to the t-butyl carbamate followed by chromatography. Each regioisomer is then separately converted to the compound of Formula L as described in Scheme 1
- an androgen receptor binding assay was performed wherein many of the compounds of this invention are shown to demonstrate significant affinity for the androgen receptor.
- the assay was performed as specified by the manufacturer (Invitrogen, Madison, WI). Briefly, ⁇ of lOmM compound was added to 500 ⁇ 1 of AR screening buffer in a 1.5ml eppendorf tube to make a 2xl0 "5 M stock. 10-fold serial dilutions of the test compounds were prepared ranging in concentration from 10 "5 M to 10 "12 M. Each dilution was added in triplicate to a black 384-microtiter plate. The test compounds will be diluted 2-fold in the final reaction.
- 2x AR-FluormoneTM complex was prepared with 2nM Flourmone AL GreenTM and 30nM AR. 25 ⁇ of 2x complex was aliquoted to each reaction well, such that the final reaction volume was 50 ⁇ 1 per well. The plate was sealed with a foil cover and incubated in the dark at room temperature for 4 h. Polarization values for each well were measured. The polarization values were plotted against the concentration of the test compound. The concentration of the test compound that results in half-maximum shift equals the IC 5 0 of the test compound. As a control, a competition curve for
- R1881(methyltrienolone) was performed for each assay. Curve Fitting was performed using GraphPad Prism® software from GraphPadTM Software Inc.
- Binding data are reported as a single determination if the experiment was run once only and as the average of experiments if the binding experiment was performed two or more times with that compound. Results are set forth in Table 1.
- H and C NMR spectra were typically determined in CDC1 3 or DMSO- ⁇ 3 ⁇ 4, using either a Varian Inova 500 MHz spectrometer or a Varian Gemini 2000 200 MHz spectrometer. Proton chemical shifts ( ⁇ ) are relative to the residual solvent peaks for each deuterated solvent and expressed in ppm. Coupling constants (J) are expressed in hertz. Infra red spectra were obtained in Jasco 460 plus using KBr pellets. Mass Spectra was recorded in ESI & APCI source using AGILENT 6310 Iontrap or Shimadzu LCMS-2010 EV. Chemical reagents were generally commercially available and were used without further purification unless stated otherwise. Chemical reagents were generally commercially available and were used without further purification unless stated otherwise.
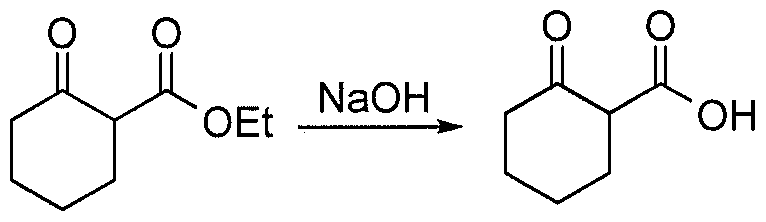
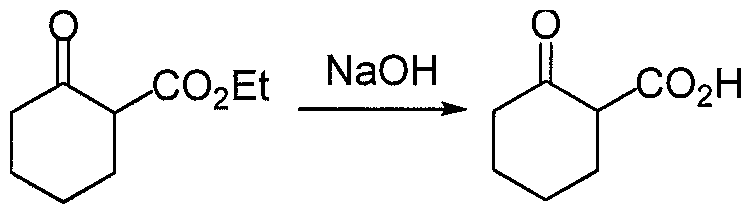
- methyl 2-oxocyclopentanecarboxylate (2.5 g, 17.5 mmol) in 5N NaOH (0.75 g, 18.8 mmol) was stirred for 16 h, cooled to 0 °C, acidified with cone. HC1 (1.1 mL) and added slowly to the diazonium salt prepared above. The reaction mixture was stirred at 0 °C for 10 min. during which time a yellow solid
- reaction mixture was heated to 110 °C for 2 h, cooled to room temperature and quenched with saturated NH 4 C1 (10 mL), extracted with EtOAc (2 x 15 mL) and the combined organic extracts were concentrated in vacuo to give the crude compound which was purified by silica gel chromatography [EtOAc-hexane (1 :9) as eluant] to provide the title compound as light red solid (30 mg, 37%).
- reaction mixture was stirred at 60 °C for 12 h, filtered through a pad of Celite® and the filtrate was concentrated under reduced pressure to give the crude material which was purified by silica gel chromatography [EtOAc-hexane (1 :9) as eluant] to obtain the title compound as a yellow solid (0.48 g, 92%).
- the binding data shown in Table 1 (below) is from the result of a single or multiple determinations based on the same compound. Where multiple data points have been taken, the value reported is the average of the multiple determinations.
- Table 1 Compound AR-Binding Affinity
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurosurgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Reproductive Health (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Obesity (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Gynecology & Obstetrics (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Emergency Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
Abstract
Description






























































































































































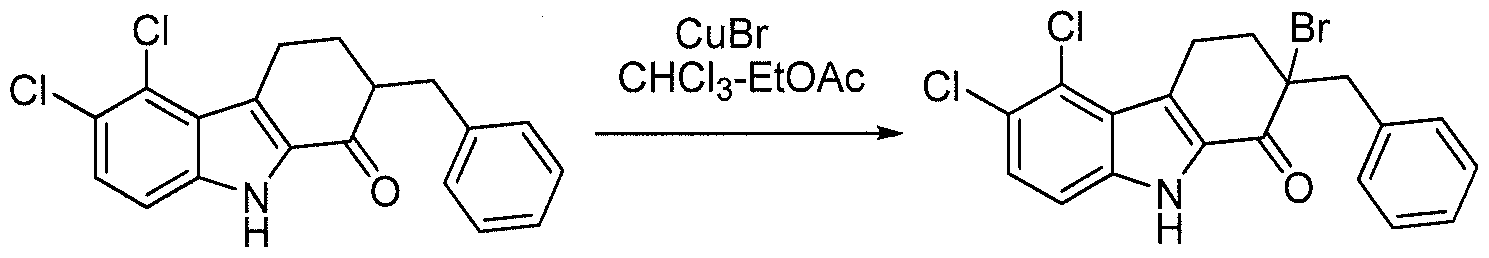







Claims














Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2012008995A MX338831B (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators. |
| BR112012019551A BR112012019551A2 (en) | 2010-02-04 | 2011-02-04 | selective androgen receptor modulators. |
| JP2012552115A JP5964756B2 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulator |
| EP11740437.6A EP2531029B1 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
| CA2788907A CA2788907A1 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
| US13/576,754 US8987319B2 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
| AU2011212813A AU2011212813B2 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
| US14/665,849 US20150266821A1 (en) | 2010-02-04 | 2015-03-23 | Selective androgen receptor modulators |
| US15/330,845 US20170121285A1 (en) | 2010-02-04 | 2016-11-07 | Selective androgen receptor modulators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US30149210P | 2010-02-04 | 2010-02-04 | |
| US61/301,492 | 2010-02-04 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/576,754 A-371-Of-International US8987319B2 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
| US14/665,849 Continuation US20150266821A1 (en) | 2010-02-04 | 2015-03-23 | Selective androgen receptor modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011097496A1 true WO2011097496A1 (en) | 2011-08-11 |
Family
ID=44355809
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/023768 Ceased WO2011097496A1 (en) | 2010-02-04 | 2011-02-04 | Selective androgen receptor modulators |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US8987319B2 (en) |
| EP (1) | EP2531029B1 (en) |
| JP (2) | JP5964756B2 (en) |
| AU (1) | AU2011212813B2 (en) |
| BR (1) | BR112012019551A2 (en) |
| CA (1) | CA2788907A1 (en) |
| MX (1) | MX338831B (en) |
| WO (1) | WO2011097496A1 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103172643A (en) * | 2011-12-26 | 2013-06-26 | 中国医学科学院药物研究所 | Carbazole alkaloids of clausena lansium and preparation method thereof and medical composition and use thereof |
| US8987319B2 (en) | 2010-02-04 | 2015-03-24 | Radius Health, Inc. | Selective androgen receptor modulators |
| US9133182B2 (en) | 2010-09-28 | 2015-09-15 | Radius Health, Inc. | Selective androgen receptor modulators |
| US9555014B2 (en) | 2010-05-12 | 2017-01-31 | Radius Health, Inc. | Therapeutic regimens |
| US10071066B2 (en) | 2014-03-28 | 2018-09-11 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US10385008B2 (en) | 2017-01-05 | 2019-08-20 | Radius Pharmaceuticals, Inc. | Polymorphic forms of RAD1901-2HCL |
| US10420734B2 (en) | 2014-03-28 | 2019-09-24 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| WO2023088388A1 (en) * | 2021-11-19 | 2023-05-25 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole compound, and pharmaceutical composition and use thereof |
| US11771682B2 (en) | 2016-06-22 | 2023-10-03 | Ellipses Pharma Ltd. | AR+ breast cancer treatment methods |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4464384A3 (en) | 2007-08-17 | 2025-01-08 | Purdue Research Foundation | Psma binding ligand-linker conjugates and methods for using |
| US9951324B2 (en) | 2010-02-25 | 2018-04-24 | Purdue Research Foundation | PSMA binding ligand-linker conjugates and methods for using |
| BR112015011118B1 (en) | 2012-11-15 | 2022-12-13 | Endocyte, Inc | CONJUGATE; PHARMACEUTICAL COMPOSITION; AND USE OF A CONJUGATE |
| LT4095130T (en) | 2013-10-18 | 2024-04-25 | Novartis Ag | Labeled inhibitors of prostate specific membrane antigen (psma), their use as imaging agents and pharmaceutical agents for the treatment of prostate cancer |
| BR112016010927A2 (en) | 2013-11-14 | 2017-08-08 | Endocyte Inc | FORMULA CONJUGATE |
| US9682960B2 (en) | 2013-12-19 | 2017-06-20 | Endorecherche, Inc. | Non-steroidal antiandrogens and selective androgen receptor modulators with a pyridyl moiety |
| US10188759B2 (en) | 2015-01-07 | 2019-01-29 | Endocyte, Inc. | Conjugates for imaging |
| JP6757037B2 (en) * | 2016-02-15 | 2020-09-16 | 学校法人近畿大学 | Stress evaluation method |
| US12208102B2 (en) | 2018-04-17 | 2025-01-28 | Endocyte, Inc. | Methods of treating cancer |
| IL279853B2 (en) | 2018-07-04 | 2025-01-01 | Radius Pharmaceuticals Inc | Polymorphic forms of rad 1901-2hcl |
| CN109239214B (en) * | 2018-09-19 | 2020-01-31 | 珠海润都制药股份有限公司 | Method for detecting Shakubiqu isomers in Shakubiqu sodium |
| CN113348163B (en) | 2019-02-12 | 2024-10-08 | 雷迪厄斯制药公司 | Methods and compounds |
| CN114096264B (en) | 2019-05-20 | 2025-03-14 | 因多塞特股份有限公司 | Method for preparing PSMA conjugates |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994027989A1 (en) * | 1993-05-21 | 1994-12-08 | Glaxo Group Limited | Carbazole derivatives with 17,20-lyase-inhibiting activity |
| US6159959A (en) * | 1999-05-06 | 2000-12-12 | American Home Products Corporation | Combined estrogen and antiestrogen therapy |
| US20090042866A1 (en) * | 2004-11-23 | 2009-02-12 | Lennox William | Tetrahydrocarbazoles as Active Agents for Inhibiting VEGF Production by Translational Control |
Family Cites Families (92)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1547758A (en) | 1975-07-29 | 1979-06-27 | Shell Int Research | Herbicidal composition |
| JPH01261381A (en) | 1988-04-12 | 1989-10-18 | Nippon Soda Co Ltd | Oxa(thia)diazole derivative, its production and miticide |
| US5411981A (en) | 1991-01-09 | 1995-05-02 | Roussel Uclaf | Phenylimidazolidines having antiandrogenic activity |
| FR2693461B1 (en) | 1992-07-08 | 1994-09-02 | Roussel Uclaf | New substituted phenylimidazolidines, process for their preparation, their use as medicaments and the pharmaceutical compositions containing them. |
| UA51652C2 (en) | 1995-06-08 | 2002-12-16 | Новартіс Аг | A method of hydrogenation of imines |
| JP2000513362A (en) | 1996-06-27 | 2000-10-10 | リガンド・ファーマスーティカルス・インコーポレーテッド | Androgen receptor modulator compounds and methods |
| US20090264534A1 (en) | 1996-11-27 | 2009-10-22 | Dalton James T | Selective androgen receptor modulators |
| FR2770842B1 (en) | 1997-11-13 | 1999-12-17 | Oreal | NOVEL COMPOUNDS DERIVED FROM N-ARYL 2-HYDROXY ALKYLAMIDES |
| BR0111298A (en) | 2000-06-28 | 2005-05-10 | Bristol Myers Squibb Co | Selective androgen receptor modulators and methods for their identification, design and use |
| EP1401801B1 (en) | 2000-08-24 | 2006-11-02 | The University Of Tennessee Research Corporation | Selective androgen receptor modulators and methods of use thereof |
| WO2003011824A1 (en) | 2001-07-31 | 2003-02-13 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| US8853266B2 (en) | 2001-12-06 | 2014-10-07 | University Of Tennessee Research Foundation | Selective androgen receptor modulators for treating diabetes |
| MXPA04007870A (en) | 2002-02-15 | 2005-06-20 | Endorech Inc | Biphenil derivatives and their use as antiandrogenic agents. |
| US7772433B2 (en) | 2002-02-28 | 2010-08-10 | University Of Tennessee Research Foundation | SARMS and method of use thereof |
| TW200407324A (en) | 2002-05-17 | 2004-05-16 | Bristol Myers Squibb Co | Bicyclic modulators of androgen receptor function |
| AU2003248052A1 (en) | 2002-07-12 | 2004-02-02 | Yamanouchi Pharmaceutical Co., Ltd. | N-phenyl-(2r,5s)dimethylpiperadine derivative |
| BR0313405A (en) | 2002-08-12 | 2005-07-12 | Takeda Pharmaceutical | Compounds, methods for preparing a compound and for preventing and / or treating cancer, prodrug, drug, androgen receptor modulator, agent for preventing and / or treating male hypogonadism or climatic disorder, osteoporosis and cancer, and use of a compound. |
| EP1581217A4 (en) | 2002-11-01 | 2007-07-11 | Merck & Co Inc | CARBONYLAMINO-BENZIMIDAZOLE DERIVATIVES AS MODULATORS OF THE ANDROGEN RECEPTOR |
| UA79504C2 (en) | 2002-11-07 | 2007-06-25 | Organon Nv | Indols for treating diseases associated with androgen receptors |
| US7632858B2 (en) | 2002-11-15 | 2009-12-15 | Bristol-Myers Squibb Company | Open chain prolyl urea-related modulators of androgen receptor function |
| EP1603858A2 (en) | 2003-03-11 | 2005-12-14 | NeuroSearch A/S | Kcnq channel modulating compounds and their pharmaceutical use |
| US20060142387A1 (en) | 2003-06-10 | 2006-06-29 | Rodolfo Cadilla | Chemical compounds |
| JP2007500245A (en) | 2003-06-10 | 2007-01-11 | スミスクライン ビーチャム コーポレーション | Compound |
| FI20030958A0 (en) | 2003-06-27 | 2003-06-27 | Orion Corp | New compounds |
| WO2005000309A2 (en) | 2003-06-27 | 2005-01-06 | Ionix Pharmaceuticals Limited | Chemical compounds |
| AU2004272007B2 (en) | 2003-09-10 | 2009-05-28 | Merck Sharp & Dohme Corp. | 17-heterocyclic-4-azasteroid derivatives as androgen receptor modulators |
| US20050124625A1 (en) | 2003-10-21 | 2005-06-09 | Salvati Mark E. | Piperazine derivatives and their use as modulators of nuclear hormone receptor function |
| GB0324551D0 (en) | 2003-10-21 | 2003-11-26 | Karobio Ab | Novel compounds |
| US7256208B2 (en) | 2003-11-13 | 2007-08-14 | Bristol-Myers Squibb Company | Monocyclic N-Aryl hydantoin modulators of androgen receptor function |
| CA2545048A1 (en) | 2003-11-20 | 2005-06-02 | Warner-Lambert Company Llc | Androgen receptor modulators |
| WO2005060956A1 (en) | 2003-12-12 | 2005-07-07 | University Of Maryland, Baltimore | IMMUNOMODULATORY COMPOUNDS THAT TARGET AND INHIBIT THE pY+3 BINDING SITE OF TYROSENE KINASE p56 LCK SH2 DOMAIN |
| KR101536701B1 (en) | 2004-01-07 | 2015-07-14 | 앙도르쉐르슈 인코포레이티드 | Helix 12-oriented type steroid pharmaceutical product |
| US20050182105A1 (en) | 2004-02-04 | 2005-08-18 | Nirschl Alexandra A. | Method of using 3-cyano-4-arylpyridine derivatives as modulators of androgen receptor function |
| US7820702B2 (en) | 2004-02-04 | 2010-10-26 | Bristol-Myers Squibb Company | Sulfonylpyrrolidine modulators of androgen receptor function and method |
| JP4805909B2 (en) | 2004-03-03 | 2011-11-02 | スミスクライン ビーチャム コーポレーション | Aniline derivatives as selective androgen receptor modulators |
| US7696241B2 (en) | 2004-03-04 | 2010-04-13 | Bristol-Myers Squibb Company | Bicyclic compounds as modulators of androgen receptor function and method |
| US7388027B2 (en) | 2004-03-04 | 2008-06-17 | Bristol-Myers Squibb Company | Bicyclic compounds as modulators of androgen receptor function and method |
| US7625923B2 (en) | 2004-03-04 | 2009-12-01 | Bristol-Myers Squibb Company | Bicyclic modulators of androgen receptor function |
| GB0405033D0 (en) | 2004-03-05 | 2004-04-07 | Karobio Ab | Novel pharmaceutical compositions |
| US8519158B2 (en) * | 2004-03-12 | 2013-08-27 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds and methods |
| JP2007223901A (en) | 2004-03-24 | 2007-09-06 | Takeda Chem Ind Ltd | Heterocyclic compound and use thereof |
| CA2562132A1 (en) | 2004-04-08 | 2005-10-27 | Merck & Co., Inc. | 17 beta-acetamide-4-azasteroids as androgen receptor modulators |
| TW200602317A (en) | 2004-04-23 | 2006-01-16 | Akzo Nobel Nv | Novel androgens |
| CN1980917B (en) | 2004-05-03 | 2014-02-12 | 詹森药业有限公司 | Novel indole derivatives as selective androgen receptor modulators (SARMS) |
| CN1980891B (en) * | 2004-05-03 | 2011-03-09 | 詹森药业有限公司 | Indole, benzofuran and benzothiophene derivatives as selective androgen receptor modulators (SARMS) |
| EP1747193A1 (en) | 2004-05-11 | 2007-01-31 | Pfizer Products Incorporated | Benzonitrile derivatives to treat musculoskeletal frailty |
| CA2566942A1 (en) | 2004-05-17 | 2005-12-08 | Acadia Pharmaceuticals Inc. | Androgen receptor modulators and method of treating disease using the same |
| US20080119456A1 (en) | 2004-05-29 | 2008-05-22 | Trond Ulven | Substituted Thiazoleacetic Acid as Crth2 Ligands |
| ATE552235T1 (en) | 2004-06-07 | 2012-04-15 | Univ Tennessee Res Foundation | SELECTIVE ANDROGEN RECEPTOR MODULATOR AND METHOD OF USE THEREOF |
| EP1791821B1 (en) | 2004-09-10 | 2013-06-05 | Janssen Pharmaceutica NV | Novel imidazolidin-2-one derivatives as selective androgen receptor modulators (sarms) |
| ATE514700T1 (en) | 2004-09-20 | 2011-07-15 | Janssen Pharmaceutica Nv | NEW TETRACYCLIC HETEROATOME WITH DERIVATIVES SUITABLE AS SEXUAL STEROID HORMONE RECEPTOR MODULATORS |
| EP1805147B1 (en) | 2004-09-30 | 2014-08-13 | Janssen Pharmaceutica NV | Novel benzimidazole derivatives useful as selective androgen receptor modulators (sarms) |
| US8143425B2 (en) | 2004-10-12 | 2012-03-27 | Bristol-Myers Squibb Company | Heterocyclic aromatic compounds useful as growth hormone secretagogues |
| EP1809275A1 (en) | 2004-10-13 | 2007-07-25 | Smithkline Beecham Corporation | Chemical compounds |
| JP2008518968A (en) | 2004-10-29 | 2008-06-05 | メルク エンド カムパニー インコーポレーテッド | N- (pyridin-3-yl) -2-phenylbutanamide as an androgen receptor modulator |
| CN104803916B (en) | 2004-11-16 | 2017-08-11 | 詹森药业有限公司 | Hete rocyclic derivatives as SARM (SARMS) |
| NZ555974A (en) | 2005-01-10 | 2010-11-26 | Acadia Pharm Inc | Aminophenyl derivatives as selective androgen receptor modulators |
| WO2006113552A2 (en) | 2005-04-15 | 2006-10-26 | Smithkline Beecham Corporation | Cyanoarylamines |
| DK1891038T3 (en) | 2005-05-13 | 2009-01-19 | Lilly Co Eli | Substituted n-arylpyrrolidines as selective androgen receptor modulators |
| EP1888512A2 (en) | 2005-06-06 | 2008-02-20 | Smithkline Beecham Corporation | Chemical compounds |
| US7829589B2 (en) | 2005-06-10 | 2010-11-09 | Elixir Pharmaceuticals, Inc. | Sulfonamide compounds and uses thereof |
| US7709516B2 (en) | 2005-06-17 | 2010-05-04 | Endorecherche, Inc. | Helix 12 directed non-steroidal antiandrogens |
| CN101203491A (en) | 2005-06-24 | 2008-06-18 | 伊莱利利公司 | Tetrahydrocarbazole derivatives (SARMs) useful as androgen receptor modulators |
| WO2007005887A2 (en) | 2005-07-01 | 2007-01-11 | Ligand Pharmaceuticals Incorporated | Androgen receptor modulator compounds, compositions and uses thereof |
| EP1911743B8 (en) | 2005-08-01 | 2013-01-16 | Takeda Pharmaceutical Company Limited | Cyclic amine compound |
| JP2008303145A (en) | 2005-09-22 | 2008-12-18 | Takeda Chem Ind Ltd | Cardiotonic comprising grk inhibitor |
| US7776859B2 (en) | 2005-10-14 | 2010-08-17 | Bristol-Myers Squibb Company | Hexahydroimidazopyrazin-3-one compounds useful as modulators of androgen receptor function |
| TW200730505A (en) | 2005-12-07 | 2007-08-16 | Merck & Co Inc | Polymorphs of an androgen receptor modulator |
| ES2402305T3 (en) | 2006-01-24 | 2013-04-30 | Janssen Pharmaceutica N.V. | New 2-substituted benzimidazoles as selective androgen receptor modulators (SARMS) |
| CA2642598C (en) | 2006-03-03 | 2014-05-27 | Orion Corporation | Selective androgen receptor modulators |
| PT2038252T (en) | 2006-07-12 | 2016-12-16 | Univ Tennessee Res Found | Substituted acylanilides and methods of use thereof |
| CA2658098A1 (en) | 2006-07-19 | 2008-01-24 | Osurf | Selective androgen receptor modulators, analogs and derivatives thereof and uses thereof |
| CN101528214B (en) | 2006-08-24 | 2013-06-05 | 田纳西大学研究基金会 | Substituted N-acylanilides and methods of use thereof |
| ES2452343T3 (en) | 2006-09-29 | 2014-04-01 | Glaxosmithkline Llc | Indole Compounds Substituted |
| WO2008044033A1 (en) | 2006-10-11 | 2008-04-17 | Astrazeneca Ab | Amide derivatives |
| UA98777C2 (en) | 2006-11-20 | 2012-06-25 | Эли Лилли Энд Компани | Tetrahydrocyclopenta[b]indole compounds as androgen receptor modulators |
| WO2008121602A1 (en) | 2007-03-29 | 2008-10-09 | Smithkline Beecham Corporation | Chemical compounds |
| WO2008124000A2 (en) | 2007-04-02 | 2008-10-16 | Ligand Pharmaceuticals Incorporated | Thiazole derivatives as androgen receptor modulator compounds |
| US9284345B2 (en) | 2007-04-12 | 2016-03-15 | Endorecherche, Inc. | 17alpha-substituted steroids as systemic antiandrogens and selective androgen receptor modulators |
| WO2008128100A1 (en) | 2007-04-13 | 2008-10-23 | The Regents Of The University Of California | Small-molecule inhibitors of the androgen receptor |
| PL2176220T3 (en) | 2007-08-07 | 2013-08-30 | Takeda Pharmaceuticals Co | Pyrrolidin-2-one derivatives as androgen receptor modulators |
| WO2009065600A2 (en) | 2007-11-21 | 2009-05-28 | Technische Universität Dresden | Means for treating myosin-related diseases |
| KR101595238B1 (en) | 2007-12-21 | 2016-02-18 | 리간드 파마슈티칼스 인코포레이티드 | Selective androgen receptor modulators(SARMs) and uses thereof |
| BRPI0821676A2 (en) | 2007-12-21 | 2015-06-16 | Astrazeneca Ab | Compound, pharmaceutical composition, and process for preparing the compounds. |
| ES2488990T3 (en) | 2008-02-22 | 2014-09-01 | Radius Health, Inc. | Selective androgen receptor modulators |
| US8268872B2 (en) | 2008-02-22 | 2012-09-18 | Radius Health, Inc. | Selective androgen receptor modulators |
| WO2009133861A1 (en) | 2008-04-28 | 2009-11-05 | 武田薬品工業株式会社 | Cyclic amine compound |
| BRPI0912394A2 (en) | 2008-05-16 | 2016-07-26 | Lilly Co Eli | androgen receptor modulators based on tetrahydrocyclopenta [b] indole |
| WO2010118287A1 (en) | 2009-04-10 | 2010-10-14 | Radius Health, Inc. | Selective androgen receptor modulators |
| MX338831B (en) | 2010-02-04 | 2016-05-03 | Radius Health Inc | Selective androgen receptor modulators. |
| US8642632B2 (en) | 2010-07-02 | 2014-02-04 | Radius Health, Inc. | Selective androgen receptor modulators |
| US9133182B2 (en) | 2010-09-28 | 2015-09-15 | Radius Health, Inc. | Selective androgen receptor modulators |
-
2011
- 2011-02-04 MX MX2012008995A patent/MX338831B/en active IP Right Grant
- 2011-02-04 AU AU2011212813A patent/AU2011212813B2/en not_active Ceased
- 2011-02-04 JP JP2012552115A patent/JP5964756B2/en not_active Expired - Fee Related
- 2011-02-04 US US13/576,754 patent/US8987319B2/en not_active Expired - Fee Related
- 2011-02-04 WO PCT/US2011/023768 patent/WO2011097496A1/en not_active Ceased
- 2011-02-04 CA CA2788907A patent/CA2788907A1/en not_active Abandoned
- 2011-02-04 BR BR112012019551A patent/BR112012019551A2/en not_active IP Right Cessation
- 2011-02-04 EP EP11740437.6A patent/EP2531029B1/en not_active Not-in-force
-
2015
- 2015-03-23 US US14/665,849 patent/US20150266821A1/en not_active Abandoned
-
2016
- 2016-02-26 JP JP2016035296A patent/JP2016138118A/en active Pending
- 2016-11-07 US US15/330,845 patent/US20170121285A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994027989A1 (en) * | 1993-05-21 | 1994-12-08 | Glaxo Group Limited | Carbazole derivatives with 17,20-lyase-inhibiting activity |
| US6159959A (en) * | 1999-05-06 | 2000-12-12 | American Home Products Corporation | Combined estrogen and antiestrogen therapy |
| US20090042866A1 (en) * | 2004-11-23 | 2009-02-12 | Lennox William | Tetrahydrocarbazoles as Active Agents for Inhibiting VEGF Production by Translational Control |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8987319B2 (en) | 2010-02-04 | 2015-03-24 | Radius Health, Inc. | Selective androgen receptor modulators |
| US9555014B2 (en) | 2010-05-12 | 2017-01-31 | Radius Health, Inc. | Therapeutic regimens |
| US9133182B2 (en) | 2010-09-28 | 2015-09-15 | Radius Health, Inc. | Selective androgen receptor modulators |
| US9920044B2 (en) | 2010-09-28 | 2018-03-20 | Radius Pharmaceuticals, Inc. | Selective androgen receptor modulators |
| CN103172643A (en) * | 2011-12-26 | 2013-06-26 | 中国医学科学院药物研究所 | Carbazole alkaloids of clausena lansium and preparation method thereof and medical composition and use thereof |
| US11779552B2 (en) | 2014-03-28 | 2023-10-10 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US10071066B2 (en) | 2014-03-28 | 2018-09-11 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US11951080B2 (en) | 2014-03-28 | 2024-04-09 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US10420734B2 (en) | 2014-03-28 | 2019-09-24 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US12263142B2 (en) | 2014-03-28 | 2025-04-01 | Duke University | Method of treating cancer using selective estrogen receptor modulators |
| US11771682B2 (en) | 2016-06-22 | 2023-10-03 | Ellipses Pharma Ltd. | AR+ breast cancer treatment methods |
| US12329746B2 (en) | 2016-06-22 | 2025-06-17 | Ellipses Pharma Ltd | AR+breast cancer treatment methods |
| US10385008B2 (en) | 2017-01-05 | 2019-08-20 | Radius Pharmaceuticals, Inc. | Polymorphic forms of RAD1901-2HCL |
| WO2023088388A1 (en) * | 2021-11-19 | 2023-05-25 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole compound, and pharmaceutical composition and use thereof |
| CN118284598A (en) * | 2021-11-19 | 2024-07-02 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole compounds, pharmaceutical compositions and uses thereof |
| CN118284598B (en) * | 2021-11-19 | 2025-06-24 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole compound, pharmaceutical composition and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016138118A (en) | 2016-08-04 |
| AU2011212813A1 (en) | 2012-09-06 |
| JP2013518902A (en) | 2013-05-23 |
| EP2531029A4 (en) | 2013-05-29 |
| US20170121285A1 (en) | 2017-05-04 |
| EP2531029B1 (en) | 2016-10-19 |
| US8987319B2 (en) | 2015-03-24 |
| MX2012008995A (en) | 2012-11-22 |
| US20130041007A1 (en) | 2013-02-14 |
| JP5964756B2 (en) | 2016-08-03 |
| EP2531029A1 (en) | 2012-12-12 |
| AU2011212813B2 (en) | 2014-10-23 |
| BR112012019551A2 (en) | 2016-08-09 |
| MX338831B (en) | 2016-05-03 |
| CA2788907A1 (en) | 2011-08-11 |
| US20150266821A1 (en) | 2015-09-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2531029B1 (en) | Selective androgen receptor modulators | |
| JP7299382B2 (en) | Fused pentacyclic imidazole derivatives as modulators of TNF activity | |
| JP5828848B2 (en) | Beta-secretase inhibitor | |
| WO2010118287A1 (en) | Selective androgen receptor modulators | |
| JP7660040B2 (en) | Solid forms of 3-((1R,3R)-1-(2,6-difluoro-4-((1-(3-fluoropropyl)azetidin-3-yl)amino)phenyl)-3-methyl-1,3,4,9-tetrahydro-2H-pyrido[3,4-b]indol-2-yl)-2,2-difluoropropan-1-ol and methods for preparing fused tricyclic compounds containing substituted phenyl or pyridinyl moieties and methods of using them | |
| AU2006275870B2 (en) | Novel 1-aryl-3-azabicyclo[3.1.0]hexanes: preparation and use to treat neuropsychiatric disorders | |
| JP4377942B2 (en) | Tetrahydroindolone and tetrahydroindazolone derivatives | |
| US20120004270A1 (en) | Selective Androgen Receptor Modulators | |
| JP2022503942A (en) | Isoindoline compounds, methods of preparation thereof, pharmaceutical compositions and uses | |
| EP1902026A2 (en) | Tetrahydrocarbazole derivatives useful as androgen receptor modulators (sarm) | |
| TWI846350B (en) | Tetrahydro-imidazo quinoline compounds as cbp/p300 inhibitors | |
| EP1891038A2 (en) | Substituted n-arylpyrrolidines as selective androgen receptor modulators | |
| TW201406728A (en) | Substituted tricyclic compounds with activity towards EP1 receptors | |
| BG63874B1 (en) | Derivatives of indol-2,3-dion-3-oxime | |
| EP3394050A1 (en) | Fused (hetero)cyclic compounds as s1p modulators | |
| CN102137857A (en) | Process for the preparation of benzimidazol-2-ylpyrimidine derivatives | |
| CN120712253A (en) | Hydroquinazoline derivatives for treating diseases or disorders | |
| CN103429587B (en) | As the azaspiro decanone compound of HSL inhibitor | |
| CN102548996B (en) | Automated radio synthesizes | |
| EP3687533A1 (en) | Novel salts | |
| JP2025521119A (en) | AT2R antagonists and uses thereof | |
| HK1149748A (en) | Azaindole compounds for treatment of central nervous system disorders | |
| HK1143159A (en) | Polycyclic compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11740437 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012552115 Country of ref document: JP Ref document number: MX/A/2012/008995 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2788907 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011212813 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7188/DELNP/2012 Country of ref document: IN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2011740437 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011740437 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2011212813 Country of ref document: AU Date of ref document: 20110204 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13576754 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012019551 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012019551 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120803 |