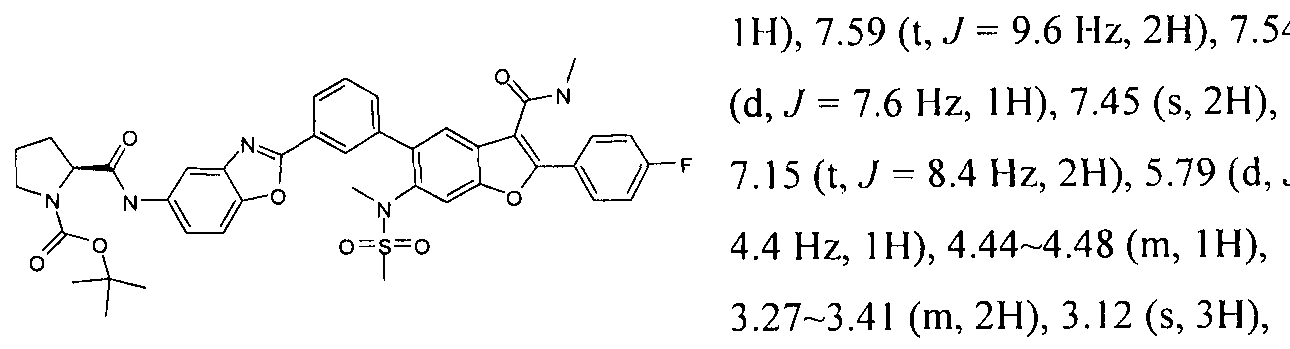
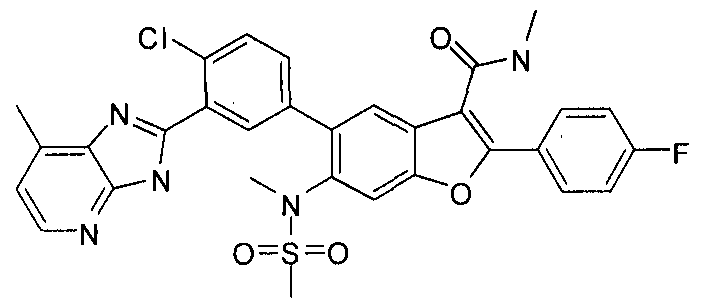
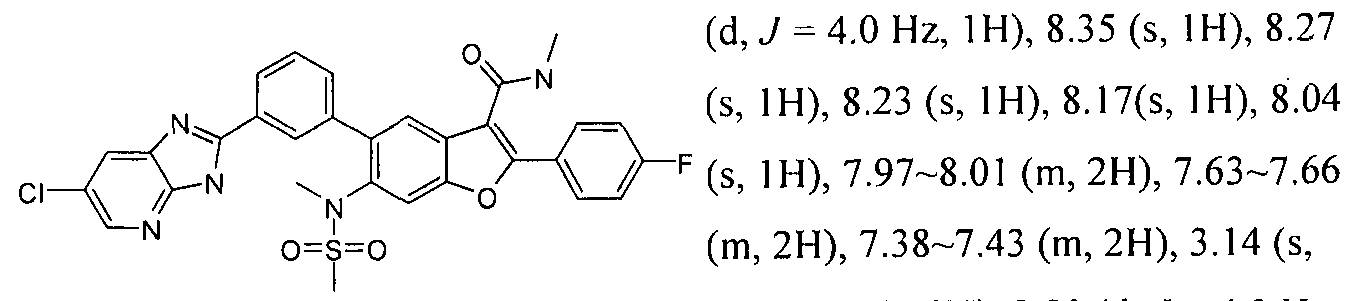
INHIBITORS OF HEPATITIS C VIRUS NS5B POLYMERASE
FIELD OF THE INVENTION
The present disclosure relates to antiviral compounds that are useful as inhibitors of the hepatitis C virus (HCV) NS5B (non-structural protein 5B) polymerase, compositions comprising such compounds, the use of such compounds for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, methods for inhibiting the function of the NS5B polymerase, and methods for inhibiting HCV viral replication and/or viral production.
BACKGROUND OF THE INVENTION
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals. Current treatments for HCV infection include immunotherapy with recombinant interferon-a alone or in combination with the nucleoside analog ribavirin.
Several viral ly-encoded enzymes are putative targets for therapeutic intervention, including a metalloprotease (NS2-3), a serine protease (NS3, amino acid residues 1 -180), a helicase (NS3, full length), an NS3 protease cofactor (NS4A), a membrane protein (NS4B), a zinc metalloprotein (NS5A) and an RNA-dependent RNA polymerase (NS5B).
One identified target for therapeutic intervention is HCV NS5B polymerase. Sven-Erik Behrens et al , Identification and properties of the RNA-dependent RNA polymerase of heptatitis C virus, 15(1) EMBO J. 12-22 (1996). Antagonists of NS5B activity are inhibitors of HCV replication. Steven S. Carroll et al , Inhibition of Hepatitis C Virus RNA Replication by 2'- Modified Nucleoside Analogs, 278(14) J. BIOL. CHEM. 1 1979-84 (2003).
There is a clear and long-felt need to develop effective therapeutics for treatment of HCV infection. Specifically, there is a need to develop compounds that selectively inhibit HCV viral replication and that would be useful for treating HCV-infected patients.
SUMMARY OF THE INVENTION
The present disclosure relates to novel compounds of formula (I) and/or pharmaceutically acceptable salts thereof. These compounds are useful, either as compounds
their pharmaceutically acceptable salts (when appropriate), in the inhibition of HCV (hepatitis C virus) NS5B (non-structural 5B) polymerase, the prevention or treatment of one or more of the symptoms of HCV infection, the inhibition of HCV viral replication and/or HCV viral production, and/or as pharmaceutical composition ingredients. As pharmaceutical composition ingredients, these compounds and their salts may be the primary active therapeutic agent, and, when appropriate, may be combined with other therapeutic agents including but not limited to other HCV antivirals, anti-infectives, immunomodulators, antibiotics or vaccines, as well as the present Standard of Care treatment options for HCV.
More particularly, the present disclosure relates to a compound of formula (I):
(I)
or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently selected from the group consisting of halo, C]-C6 alkyl, -0-(C,-C6 alkyl), -0-(C,-C6 haloalkyl) and -CN;
n is 0, 1 , 2, 3 or 4;
R2 is C(0)NRaRb;
Ra and Rb are independently selected from the group consisting of hydrogen, C|-C6 alkyl, 0(Ci-C6 alkyl) and 5- or 6-membered monocyclic aromatic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S;
R3 is ArA, -C≡C-phenyl or a 15- or 16-membered tetracyclic ring system,
wherein said 15- or 16-membered tetracyclic ring system is substituted by 0, 1 or 2 substitutents independently selected from Ci-C6 alkyl, phenyl, C3-C7 cycloalkyl or 6- membered heteroaryl, and
wherein ArA is an aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and
ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4
heteroatom ring atoms independently selected from the group consisting of N, O or S, and
wherein said ArA is substituted by 0, 1 , 2, 3 or 4 substitutents Rc;
each R° is independently selected from the group consisting of:
a) halogen,
b) OH
c) C,-C6 alkyl,
d) 0(C,-C6 alkyl),
e) CN,
f) (CH2)o-3-ArB, wherein each ArB is an independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and ii) 8-, 9- or 10-membered bicyclic rings, which can be aromatic or non-aromatic, with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S,
g) (CH2)0-3NRdC(O)Re,
h) (CH2)0-3NRdSO2Re,
i) (CH2)0-3C(O)NRdRe,
j) (CH2)0-3SO2Re,
k) -OS02(C,-C6 alkyl), and
1) C2-C6 alkynyl
wherein each Rc c) C|-C6 alkyl, d) 0(C,-C6 alkyl), and
f) (CH2)o-3-ArB is substituted by 0, 1 , 2, 3 or 4 substituents R ; or
wherein any 2 R°. groups on adjacent ring carbon atoms can join to form a group selected from -OC(0)-N-, -OCH2CH20-, -OCH20-, -OCH2CH2-,
each Rd is independently selected from the group consisting of hydrogen and C|-6alkyl;
each Re is independently selected from the group consisting of hydrogen, C|-6alkyl, OCi-6alkyl and 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, wherein each Re C| -6alkyl, OCi-6alkyl and 5- or 6-membered monocyclic rings is substituted by 0, 1 , 2, 3 or 4 substituents independently selected from the group consisting of C|-C6 alkyl, 0(Ci-C6 alkyl), halogen and OH;
each R is independently selected from the group consisting of:
a) halogen,
b) C,-C6 alkyl,
c) 0(C,-C6 alkyl),
d) CN,
e) N( q)2,
OH,
g) C(0)H,
h) NHC(0)Rs,
i) NHS(0)2Rs,
J) C(0)NHRq,
k) C(0)ORq,
1) OS(0)2(C,-C6 alkyl),
m) (CH2)0-3-ArC, wherein each ArC is an independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, wherein each Rf : b) C,-C6 alkyl, c) 0(Ci-C6 alkyl), and m) (CH2)o-3-ArC is substituted by 0, 1 , 2, 3 or 4 substituents Rs;
each Rg is independently selected from the group consisting of halogen, , OH, N(Rq)2, CN, Ci-6alkyl, 0(C,-C6 alkyl), CF3 and C(0)OH;
R4 is selected from the group consisting of NRhR' and 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and
Rh is selected from the group consisting of:
a) hydrogen,
b) C1-6alkyl,
c) C(0)0(Ci-6alkyl), and
d) S02RJ;
RJ is selected from the group consisting of C|. alkyl, C6-io aryl, C3-7 cycloalkyl and NRxRy, where R" and Ry are independently selected from the group consisting of hydrogen and C|.6alkyl;
R1 is selected from the group consisting of:
a) Ci-6alkyl,
b) C2-6alkenyl,
c) C2-6alkynyl,
d) (CH2)0-3(C3-8cycloalkyl),
e) (CH2)o-3(C3-8cycloalkenyl),
f) C(0)Ci.6alkyl, and
g) heterocyclyl,
wherein R1 is substituted by 0, 1 , 2, 3 or 4 Rk groups;
each Rk is independently selected from the group consisting
a) ORL,
b) halogen,
c) CN,
d) NRmRn,
e) OC(0)C,-6alkyl,
f) C(0)OC,-6alkyl, '
g) -P(0)(0-C,-6alkyl)2,
h) -P(0)(OH)(0-C,.6alkyl),
k) -C(0)C(C 1.6alky 1)-NHC(0)-C , .6alkyl,
1) -NHC(0)C(C 1 -6alkyl)-NHC(0)-C , -6alkyl, m) -C(0)OH,
n) (CH2)0-3-ArD, wherein each ArD is an
independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and
ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S,
wherein each Rk e) OC(0)C,.6alkyl, f) C(0)OC,-6alkyl, and n) (CH2)o-3-ArD is substituted by 0, 1 , 2, 3 or 4 R° groups;
RL is selected from the group consisting of hydrogen, Chalky] and phenyl;
Rm is selected from the group consisting of hydrogen, C]-6alkyl, -
CHzCN and (CH2)0-3(phenyl);
R" is selected from the group consisting of hydrogen, Ci.6alkyl, S02(C,-6alkyl), -C(0)H, -C(0)OH, -C(0)0(C,.6alkyl) and C(0)(C,-6alkyl);
or Rm and Rn are taken together with the N to which they are attached to form a 5- to 7-membered ring substituted by 0, 1 , 2 or 3 Rp;
each R° is independently selected from the group consisting of halogen, C,-6alkyl, OC,.6alkyl and C(0)0(Ci-6alkyl);
each Rp is independently selected from the group consisting of halogen, Ci-6alkyl, OCi-6alkyl, oxo and C(0)0(Ci-6alkyl);
each Rq is independently selected from the group consisting of H and C] -6alkyl;
each Rs is independently selected from the group consisting of C |. 6alkyl, heterocyclyl and C6-i0aryl, wherein said heterocyclyl group can be optionally substituted on a ring nitrogen or ring carbon atom with a -C(0)0-(Ci-C6 alkyl) group; and
each Rl is independently selected from the group consisting of C \.
6alkyl and C6-i0aryl;
or Rh and R' are taken together with the N to which they are attached to form a 5- to 7-membered ring. The present invention also includes pharmaceutical compositions containing a compound of the present invention and methods of preparing such pharmaceutical compositions. The present invention further includes methods of treating or reducing the likelihood or severity of HCV infection, methods for inhibiting the activity of the NS5B polymerase, and methods for inhibiting HCV viral replication and/or viral production.
Other embodiments, aspects and features of the present invention are either further described in or will be apparent from the ensuing description, examples and appended claims.
DETAILED DESCRIPTION OF THE INVENTION
The present invention includes compounds of formula (I) above, and
pharmaceutically acceptable salts thereof. The compounds of formula (I) are HCV NS5B polymerase inhibitors.
In a first embodiment of the invention, n is 1. In this embodiment, all other groups are as provided in the general formula above.
In a second embodiment of the invention, the compound is a compound of formula (la):
(la)
or a pharmaceutically acceptable salt thereof. In this embodiment, all other groups are as provided in the general formula above and/or in the first embodiment.
In a third embodiment of the invention, the compound is a compound of formula
(lb):
or a pharmaceutically acceptable salt thereof, wherein:
each R1 is independently selected from the group consisting of halogens;
n is 0, 1 , 2 or 3;
R2 is C(0)NRaRb;
Ra and Rb are independently selected from the group consisting of hydrogen, d-C6 alkyl and 0(d-C6 alkyl);
R3 is ArA, wherein ArA is an aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings, and
ii) 8-, 9- or 10-membered bicyclic rings, and
wherein said ArA is substituted by 0, 1 , 2 or 3 substitutents Rc;
each R° is independently selected from the group consisting of:
a) halogen,
b) OH
c) Ci-C6 alkyl,
d) 0(Ci-C6 alkyl),
e) CN,
f) (CH2)0-3-ArB, wherein each ArB is an independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S,
g) (CH2)0-3NRdC(O)Re,
h) (CH2)0-3NRdSO2Re,
i) (CH2)o-3C(0)NRdRe, and
j) (CH2)0.3SO2Re,
wherein each Rc c) C,-C6 alkyl, d) 0(Ci-C6 alkyl), and
f) (CH2)0-3-ArB is substituted by 0, 1 , 2 or 3 substituents R ;
each Rd is independently selected from the group consisting of hydrogen and C|.6alkyl;
each Re is independently selected from the group consisting of hydrogen, Ci.6alkyl, OCi_6alkyl and 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, wherein each Re Ci-6alkyl, OCi-6alkyl and 5- or 6-membered monocyclic rings is substituted by 0, 1 , 2, 3 substituents independently selected from the group consisting of Ci-C6 alkyl, 0(C]-C alkyl), halogen and OH;
each Rf is independently selected from the group consisting of:
a) halogen,
b) C,-C6 alkyl,
c) 0(C,-C6 alkyl),
d) CN,
e) NH2,
f) (CH2)o-3-ArC, wherein each ArC is an independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S,
wherein each Rf b) C,-C6 alky], c) 0(Ci-C6 alkyl), and
f) (CH2)o-3-ArC is substituted by 0, 1 , 2 or 3 substituents Rg;
each R8 is independently selected from the group consisting of halogen, CN, C,-6alkyl, 0(C,-C6 alkyl), CF3 and C(0)OH;
R4 is selected from the group consisting of NRhR';
Rh is selected from the group consisting of:
a) hydrogen,
b) C,-6alkyl,
c) C(0)0(C,_6alkyl), and
d) S02Rj;
R1 is selected from the group consisting of Ci-6alkyl and NRxRy, where Rx and Ry are independently selected from the group consisting of hydrogen and C |.6alkyl;
R1 is selected from the group consisting of:
a) Ci.6alkyl,
b) C2_6alkenyl,
c) C2-6alkynyl,
d) (CH2)0-3(C3-8cycloalkyl),
e) (CH2)o-3(C3-8cycloalkenyl), and
f) C(0)C,-6alkyl,
wherein R1 is substituted by 0, 1 , 2, 3 or 4 Rk;
each Rk is independently selected from the group consisting of: a) ORL,
b) halogen,
c) CN,
d) NRmRn,
e) OC(0)C,.6alkyl,
f) C(0)OC,-6alkyl,
g) (CH2)o-3-ArD, wherein each ArD is an
independently selected aromatic ring system selected from the group consisting of:
i) 5- or 6-membered monocyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, and
ii) 8-, 9- or 10-membered bicyclic rings with 0, 1 , 2, 3 or 4 heteroatom ring atoms independently selected from the group consisting of N, O or S, wherein each Rk e) OC(0)C1 -6alkyl, f) C(0)OC,.6alkyl, and g) (CH2)0-3-ArD is substituted by 0, 1 , 2 or 3 substituents R°,
RL is selected from the group consisting of hydrogen, C].6alkyl and phenyl;
Rm is selected from the group consisting of hydrogen, Ci-6alkyl and
(CH2)0.3(phenyl);
Rn is selected from the group consisting of hydrogen, C]- alkyl,
S02(C , .6alkyl) and C(0)(C , -6alkyl);
or Rm and Rn are taken together with the N to which they are attached to form a 5- to 7-membered ring substituted by 0, 1 , 2 or 3 Rp;
each R° is independently selected from the group consisting of halogen, Ci-6alkyl, OC1 -6alkyl and C(0)0(C,.6alkyl);
each Rp is independently selected from the group consisting of halogen, C).6alkyl, OC]-6alkyl, oxo and C(0)0(C|-6alkyl);
or Rh and R' are taken together with the N to which they are attached to form a 5- to 7-membered ring.
In a fourth embodiment of the invention, R1 is selected from the group consisting of fluorine, bromine and chlorine. In a first aspect of this third embodiment, R1 is fluorine. In all aspects of this embodiment, all other groups are as provided in the general formula above and/or in the first or second embodiments.
In a fifth embodiment of the invention, Ra is hydrogen. In this embodiment, all other groups are as provided in the general formula above and/or in the first through third embodiments.
In a sixth embodiment of the invention, Rb is selected from the group consisting of -CH3 and -OCH3. In this embodiment, all other groups are as provided in the general formula above and/or in the first through fourth embodiments.
In a seventh embodiment of the invention, ArA is phenyl or pyridyl. In a first aspect of this seventh embodiment, ArA is phenyl, which is optionally substituted with which is substituted by 0, 1 , 2, 3 or 4 substitutents R°. In a second aspect of this seventh embodiment, ArA is pyridyl, which is optionally substituted with which is substituted by 0, 1 , 2, 3 or 4 substitutents Rc.In these embodiments, all other groups are as provided in the general formula above and/or in the first through fifth embodiments.
In an eighth embodiment of the invention, each R° is independently selected from the group consisting of a) fluorine, b) OH, c) C1.3a.kyl, d) OC i.3alkyl, e) CN, f) (CH2)0-i-ArB,
and , g) (CH
2)
0-,N(CH
3)SO
2CH
3, h) (CH
2)
0-,N(H)SO
2CH
3 i) (CH
2)o-iN(CH
3)S0
2phenyl, j) C(0)NHCH
3, k) (CH
2)
0-1N(H)C(O)CH
3, and
1) (CH
2)o_iN(H)C(0)phenyl. In a first aspect of this seventh embodiment each R
c is
independently selected from the group consisting of s , 0 u
and . In all aspects of this embodiment, all other groups are as provided in the general formula above and/or in the first through sixth embodiments.
In a ninth embodiment of the invention, Rh is selected from hydrogen, CH3 and S02CH3. In a first aspect of this eighth embodiment, Rh is S02CH3. In all aspects of this
embodiment, all other groups are as provided in the general formula above and/or in the first through seventh embodiments.
In a tenth embodiment of the invention, R' is selected from the group consisting of Ci-6alkyl and C2-6alkenyl. In this embodiment, all other groups are as provided in the general formula above and/or in the first through eighth embodiments.
In an eleventh embodiment of the invention, Rk is selected from the group consisting of a) ORL, b) halogen, c) CN, d) NRmR", e) OC(0)Ci-6alkyl, and f) C(0)OC)-6alkyl. In this embodiment, all other groups are as provided in the general formula above and/or in the first through ninth embodiments.
In a twelfth embodiment of the invention, RL is selected from the group consisting of Ci_6alkyl. In this embodiment, all other groups are as provided in the general formula above and/or in the first through tenth embodiments.
In a thirteenth embodiment of the invention, Rm is selected from the group consisting of hydrogen and Ci-6alkyl. In this embodiment, all other groups are as provided in the general formula above and/or in the first through eleveth embodiments.
In a fourteenth embodiment of the invention, Rn is selected from the group consisting of C)-6alkyl and S02(Ci_6alkyl). In this embodiment, all other groups are as provided in the general formula above and/or in the first through twelfth embodiments.
In a fifteenth embodiment of the invention, the compound is a compound of formula (Ic):
(Ic)
and pharmaceutically acceptable salts thereof,
wherein:
Z is a phenyl group which is substituted with one R10 group and optionally further substituted with R20;
R10 is an 8- to 10-membered bicyclic heteroaryl group, wherein said 8- to 10- membered bicyclic heteroaryl group is optionally substituted with up to 4 groups, which can be the same or different, and are selected from halo, C,-C6 alkyl, -C(0)H, -(CH2),-N(R,U)2, -(CH2)r OH, -(CH2)rO-(C,-C6 alkyl), -CF3, -NHC(0)-heterocyclyl, -NHC(0)-(C,-C6 alkyl), -C(0)NH- (C,-C6 alkyl), -C(0)OH, -C(0)0-(C,-C6 alkyl), -NHC(0)-aryl, -NHS02-aryl, -NHS02-alkyl, -O- S02-alkyl, -0-(C|-C6 alkyl) and -CN, wherein the heterocyclyl moiety of said -NHC(O)- heterocyclyl group can be optionally substituted on a ring carbon or ring nitrogen atom with a - C(0)0-(Ci-C6 alkyl) group;
R20 represents up to 4 optional substituents, which can be the same or different, and are selected from halo, 8- to 10-membered heteroaryl, Ci-C6 alkyl, -0-(C|-C6 alkyl), -O- (CH2)rOH, -0-(CH2)t-heterocyclyl, -0-(C,-C6 haloalkyl), -0-S02-(C,-C6 alkyl) and -CN;
R30 is H or Ci-C6 alkyl;
R40 is selected from C,-C6 alkyl, C,-C6 haloalkyl, -(CH2)rOH, -(CH2)t- heterocyclyl,-(CH2)rN(R70)2, -(CH2)t-CN, -(CH2)rNHC(0)OR30 and -(CH2)t-NHC(0)R30;
R50 is Ci-C6 alkyl, C6-C,0 aryl or C3-C7 cycloalkyl;
R60 represents up to 4 optional ring substituents, which can be the same or different, and are selected from halo, C|-C6 alkyl, -0-(Ci-C6 alkyl), -0-(Ci-C haloalkyl) and - CN; f
each occurrence of R70 is independently H or Ci-C6 alkyl; and
each occurrence of t is independently an integer ranging from 0 to 6.
In a first aspect of this fifteenth embodiment, Z is:
which can be optionally substituted on the depicted phenyl ring with one or two R groups, which can be the same or different.
In a second aspect of this fifteenth embodiment, Z is selected from:
wherein each occurrence of R is independently CI, F, CN, -OCF3 or -OCH3.
In a third aspect of this fifteenth embodiment, Z is selected from:
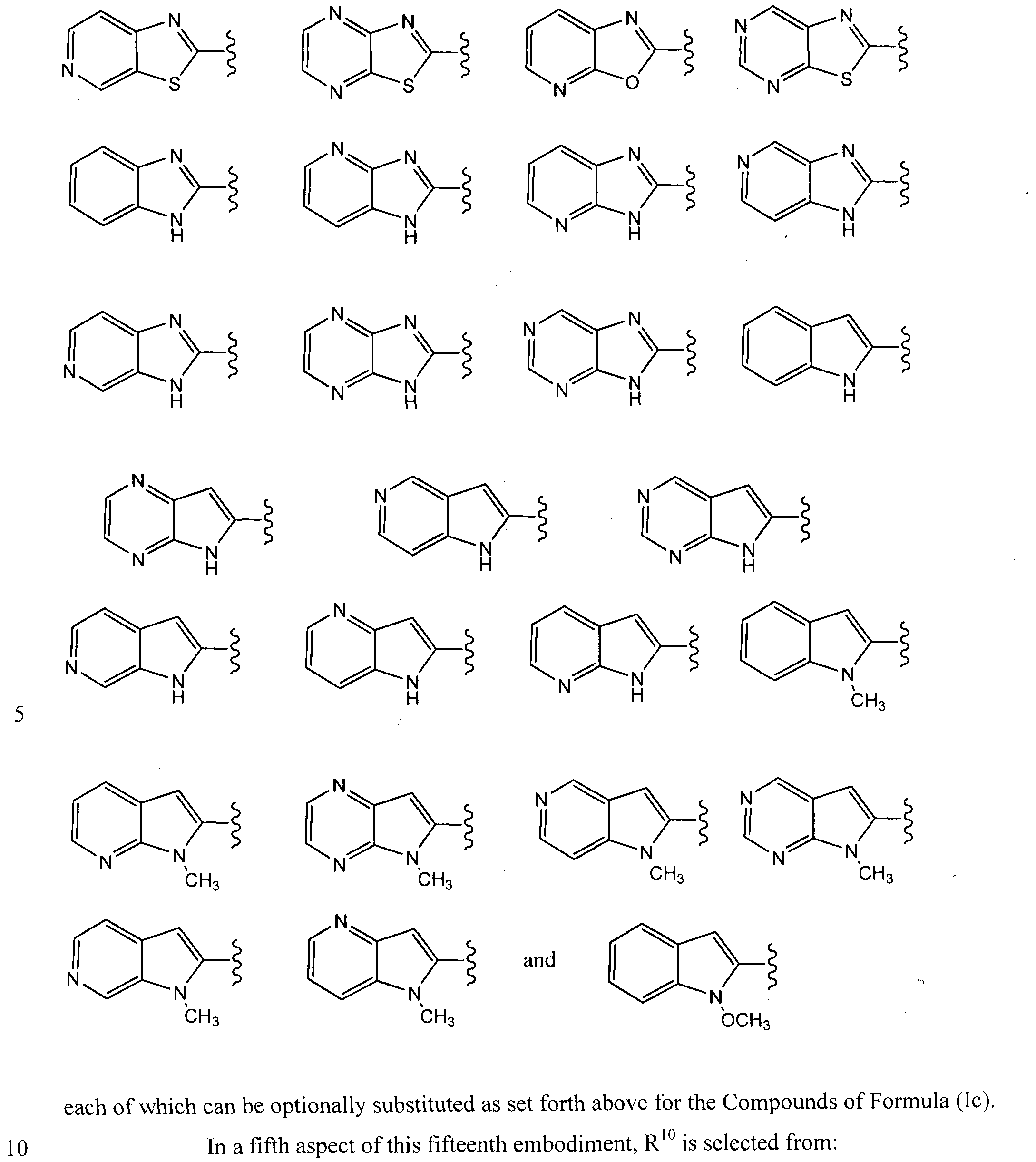
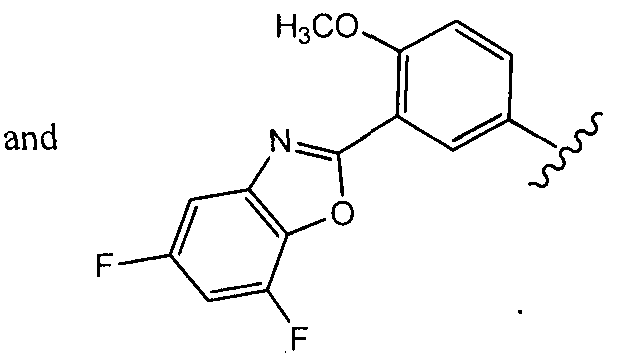
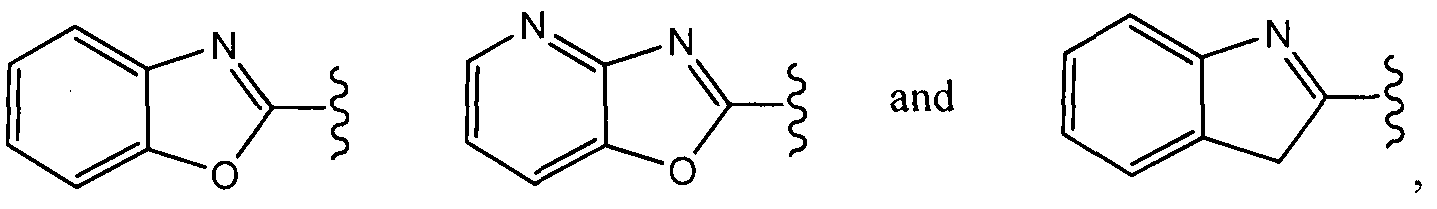
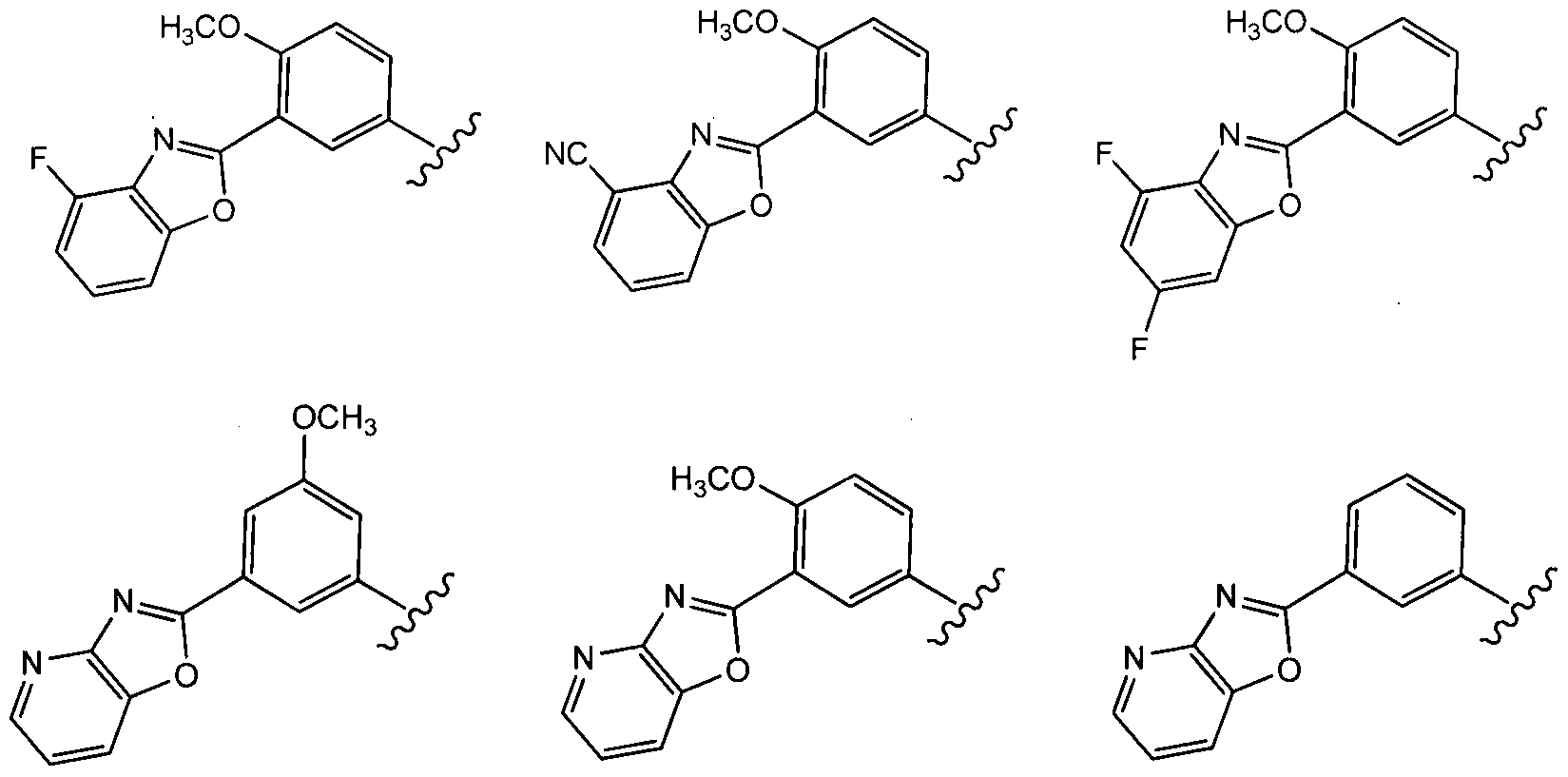
In a fourth aspect of this fifteenth embodiment of the present invention, R10 is selected from:
In a sixth aspect of this fifteenth embodiment, R
10 is:
which can be optionally substituted as set forth above for the Compounds of Formula (Ic).
In a seventh aspect of this fifteenth embodiment, Z is selected from:
wherein each occurrence of R is independently CI, F, CN, -OCF3 or -OCH3; and R10 is selected from:
each of which can be optionally substituted as set forth above for the Compounds of Formula (Ic).
In an eighth aspect of this fifteenth embodiment, Z is selected from:
wherein each occurrence of R20 is independently CI, F, CN, -OCF3 or -OCH3; and R10 is selected from:
In a ninth aspect of this fifteenth embodiment, Z is selected from:
and R10 is selected from:
In a tenth aspect of this fifteenth embodiment, R is -CH3.
In an eleventh aspect of this fifteenth embodiment, R40 is C| -C alkyl, C |-C haloalkyl, -(CH2)t-OH or -(CH2)t-CN, wherein t is an integer ranging from 0 to 6. In a first aspect of this fifth embodiment, R40 is Ci-C6 alkyl. In a second aspect of this fifth embodiment,
Rw is -CH3, -(CH2)3-CN, -CH2CH2F, or -CH2CH2C(CH3)2-OH. In a third aspect of this fifth embodiment, R40 is -CH3.
In a twelfth aspect of this fifteenth embodiment, R50 is Q-C6 alkyl. In a first aspect of this sixth embodiment, R50 is C6-Cio aryl. In a second aspect of this sixth embodiment, R50 is C3-C7 cycloalkyl. In a third aspect of this sixth embodiment, R50 is -CH3, phenyl or cyclopropyl. In a fourth aspect of this sixth embodiment, R50 is -CH3.
In a thirteenth aspect of this fifteenth embodiment, only one R60 group is present. In a first aspect of this seventh embodiment, R60 represents a single halo group. In a second aspect of this seventh embodiment, R60 represents a single F group. In a third aspect of this seventh embodiment, R60 represents a single F group at the para position of the phenyl ring to which it is attached.
In a fourteenth aspect of this fifteenth embodiment, R40 is -CH3, -(CH2)3-CN, -
CH2CH2F or -CH2CH2C(CH3)2-OH, and R5U is -CH3. In a first aspect of this eighth embodiment, R40 and R50 are each -CH3.
In a fifteenth aspect of this fifteenth embodiment, R , R and R are each -CH3.
In a sixteenth aspect of this fifteenth embodiment, R is -CH3, -(CH2)3-CN,
-CH2CH2F or -CH2CH2C(CH3)2-OH; R5U is -CH3; and RbU represents a single F group at the para position of the phenyl ring to which it is attached.
In a seventeenth aspect of this fifteenth embodiment, R30 is -CH3; R40 is -CH3, - (CH2)3-CN, -CH2CH2F or -CH2CH2C(CH3)2-OH; R50 is -CH3; and R60 represents a single F group at the para position of the phenyl ring to which it is attached. In a first aspect of this eleventh embodiment, R30, R40 and R50 are each -CH3 and R60 represents a single F group at the para position of the phenyl ring to which it is attached. -
In a sixteenth embodiment of the present invention, the Compounds of Formula (I) have the formula (Id):
(Id)
and pharmaceutically acceptable salts thereof,
wherein:
Z is:
R is a 9-membered bicyclic heteroaryl group, wherein said 9-membered bicyclic heteroaryl group is optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, C,-C6 alkyl, -(CH2)rN(R70)2, -(CH2)t-OH, -(CH2)t-0-(Ci-C6 alkyl), - CF3, -NHC(0)-heterocyclyl, -NHC(0)-(C,-C6 alkyl), -C(0)NH-(C,-C6 alkyl), -C(0)OH, - C(0)0-(C,-C6 alkyl), -NHC(0)-aryl, -NHS02-aryl, -NHS02-alkyl, -0-S02-alkyl,-0-(C,-C6 alkyl) and -CN, wherein the heterocyclyl moiety of said -NHC(0)-heterocyclyl group can be optionally substituted on a ring carbon or ring nitrogen atom with a -C(0)0-(Ci-C6 alkyl) group;
R20 represents up to 2 optional substituents, which can be the same or different, and are selected from halo, C,-C6 alkyl, -0-(C,-C6 alkyl), -0-(CH2)t-OH, -0-(CH2)t-heterocyclyl, -0-(C,-C6 haloalkyl), -0-S02-(C,-C6 alkyl) and -CN;
R40 is C,-C6 alkyl, C,-C6 haloalkyl, -(CH2)t-OH or -(CH2)t-CN; and each occurrence of t is independently an integer ranging from 0 to 6.
In a first aspect of this sixteenth embodiment, R10 is selected from:
In a third aspect of this sixteenth embodiment, Z is:
fourth aspect of this sixteenth embodiment, Z is
fifth aspect of this sixteenth embodiment, Z is:
a sixth aspect of this sixteenth embodiment, Z
In a seventh aspect of this sixteenth embodiment, Z is:
In a eighth aspect of this sixteenth embodiment, Z is:
In an ninth aspect of this sixteenth embodiment, Z is:
In a tenth aspect of this sixteenth embodiment, R is Ci-C alkyl. In an eleventh aspect of this sixteenth embodiment, R40 is -CH3, -(CH2)3-CN, - CH2CH2F, or -CH2CH2C(CH3)2-OH.
In a twelfth aspect of this sixteenth embodiment, Z is selected from:
Rw is -CH3, -(CH2)3-CN, -CH2CH2F, or -CH2CH2C(CH3)2-OH.
In a thirteenth aspect of this sixteenth embodiment, Z is selected from:
Rw is -CH3.
In a fourteenth aspect of this sixteenth embodiment, Z is:
R4U is -CH3.
In a seventeenth embodiment of the present invention, the Compounds of Formula (I) have the formula (Ie):
(Ie)
and pharmaceutically acceptable salts thereof,
wherein:
Z is a 5- or 6-membered heteroaryl group, which is substituted with one R10 group and optionally substituted with up to two R20 groups;
R10 is a 9-membered bicyclic heteroaryl group, wherein said 9-membered bicyclic heteroaryl group is optionally substituted with up to 2 groups, which can be the same or different, and are selected from halo, C,-C6 alkyl, -(CH2)t-N(R70)2, -(CH2),-OH, -(CH2)(-0-(Ci-C6 alkyl), - CF3, -NHC(0)-heterocyclyl, -NHC(0)-(C,-C6 alkyl), -C(0)NH-(C,-C6 alkyl), -C(0)OH, -
C(0)0-(Ci-C6 alkyl), -NHC(0)-aryl, -NHS02-aryl, -NHS02-alkyl, -0-S02-alkyl,-0-(C,-C6 alkyl) and -CN, wherein the heterocyclyl moiety of said -NHC(0)-heterocyclyl group can be optionally substituted on a ring carbon or ring nitrogen atom with a -C(0)0-(Ci-C alkyl) group;
R20 represents up to 2 optional substituents, which can be the same or different, and are selected from halo, Ci-C6 alkyl, -0-(Ci-C6 alkyl) and -CN;
R40 is C,-C6 alkyl; and
each occurrence of t is independently an integer ranging from 0 to 6.
In a first aspect of this seventeenth embodiment, Z is pyridyl or thiophenyl.
In a second aspect of this seventeenth embodiment, Z is pyridyl, which is optionally substituted with up to 2 groups, each independently selected from methoxy, fluoro or -CN.
In a third aspect of this seventeenth embodiment, R10 is selected from:
each of which can be optionally substituted as set forth above in formula (le).
each of which can be optionally substituted with 1 or 2 groups, independently selected from halo, -CN and -0(C,-C6 alkyl).
:
In a fifth aspect of this seventeenth embodiment, Z is;
In a sixth aspect of this seventeenth embodiment, Z is:
In a seventh aspect of this seventeenth embodiment, Z is:
In a tenth aspect of this seventeenth embodiment, R40 is methyl.
In an eleventh aspect of this seventeenth embodiment, Z is:
In an eighteenth embodiment of the invention, for the compounds of formula (I), variables R1, R2, R3, R4 and n are selected independently of each other.
In a nineteenth embodiment of the invention, the compounds of formula (I) are in isolated and purified form.
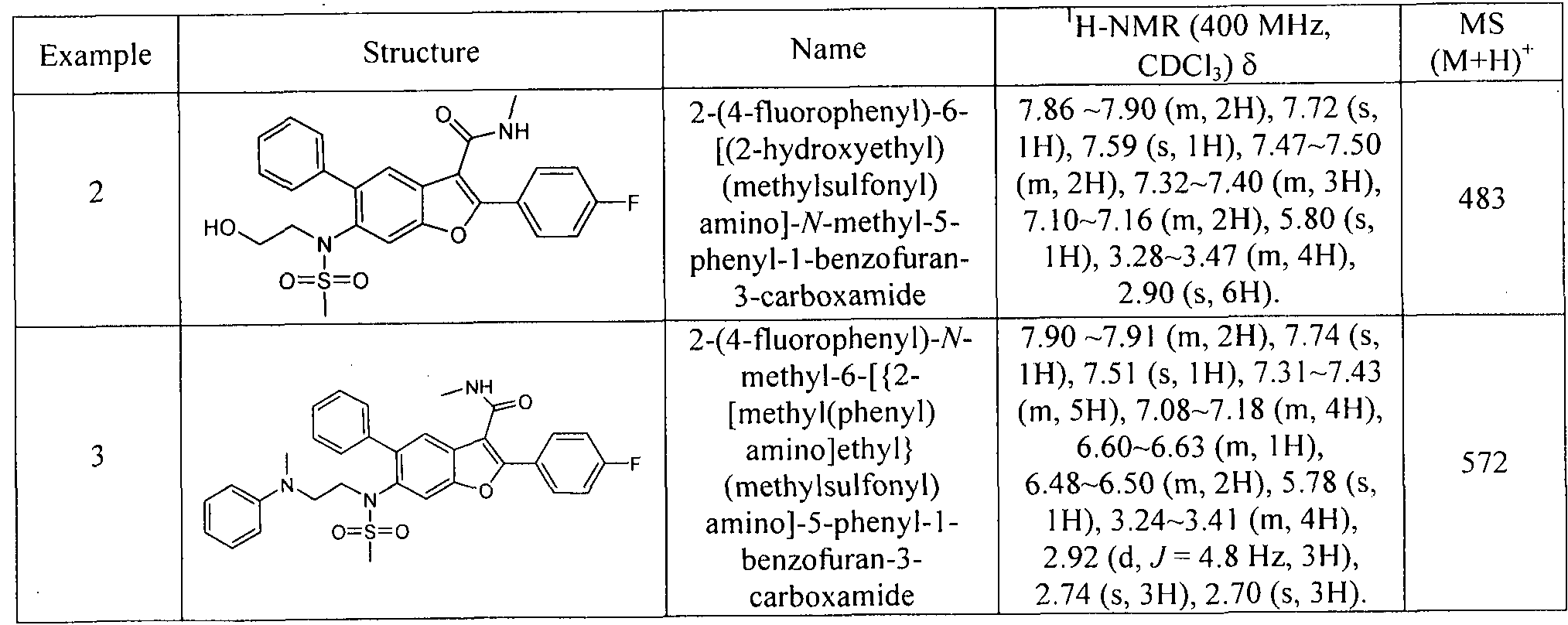
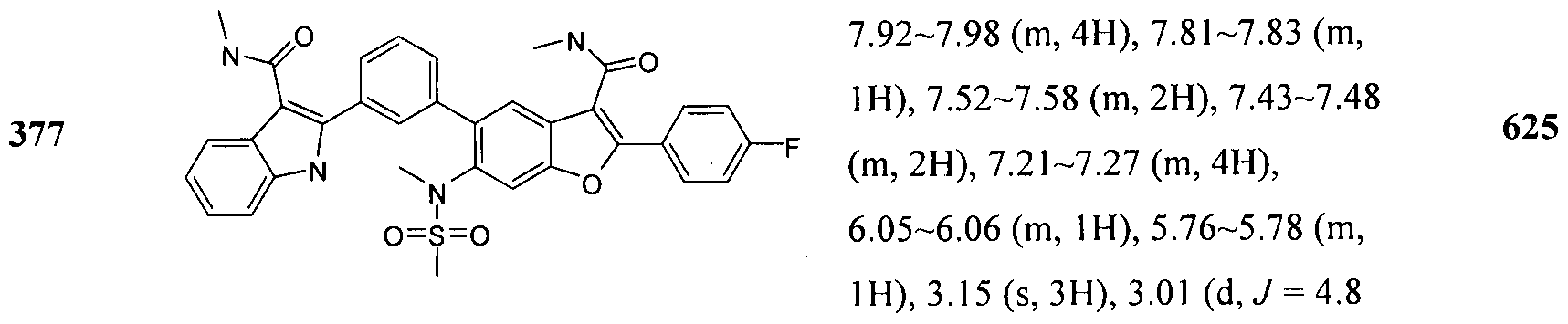
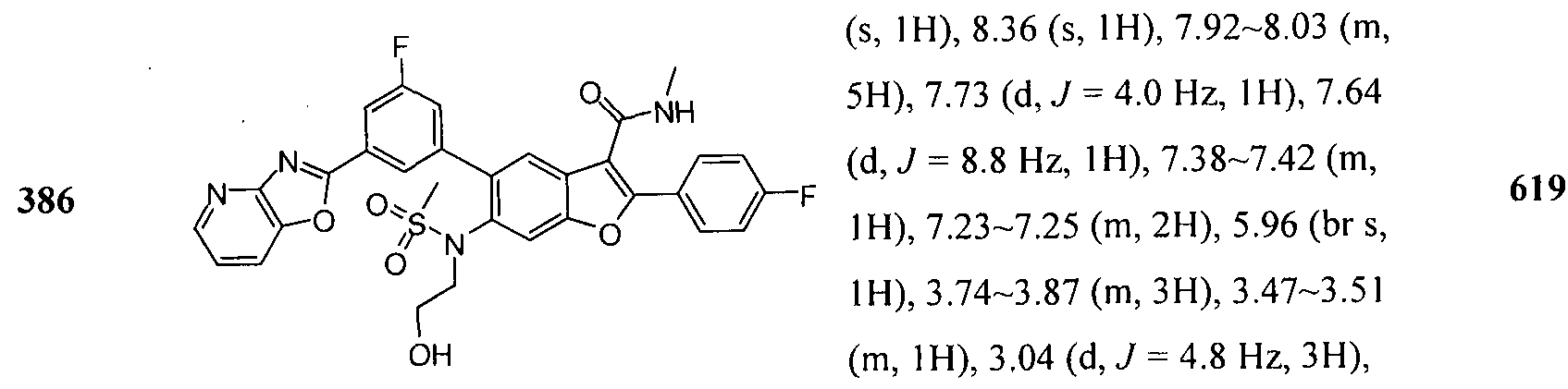
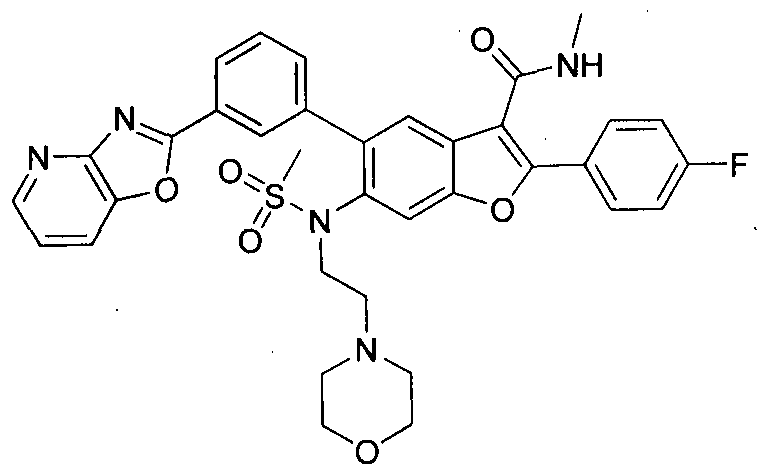
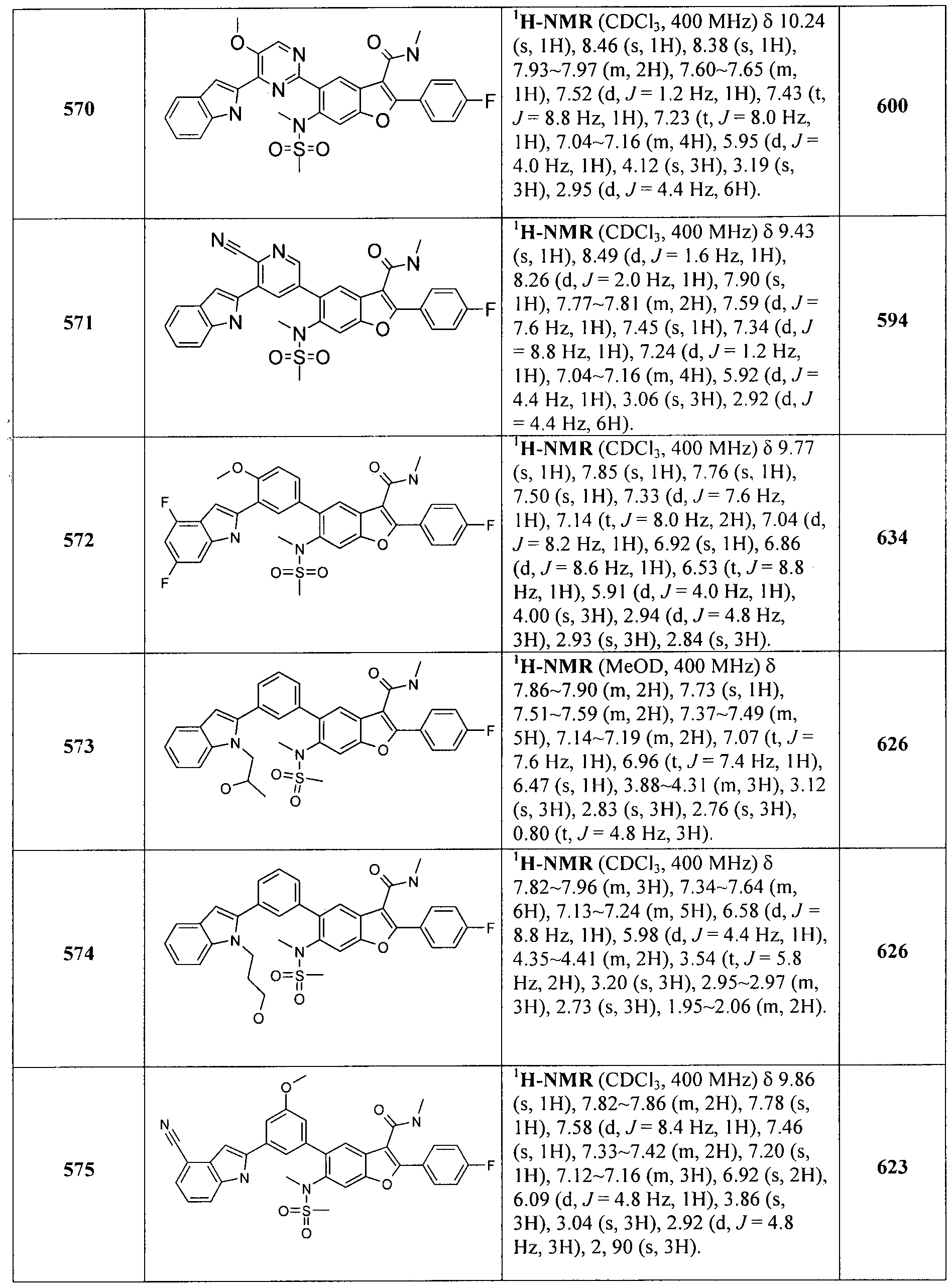
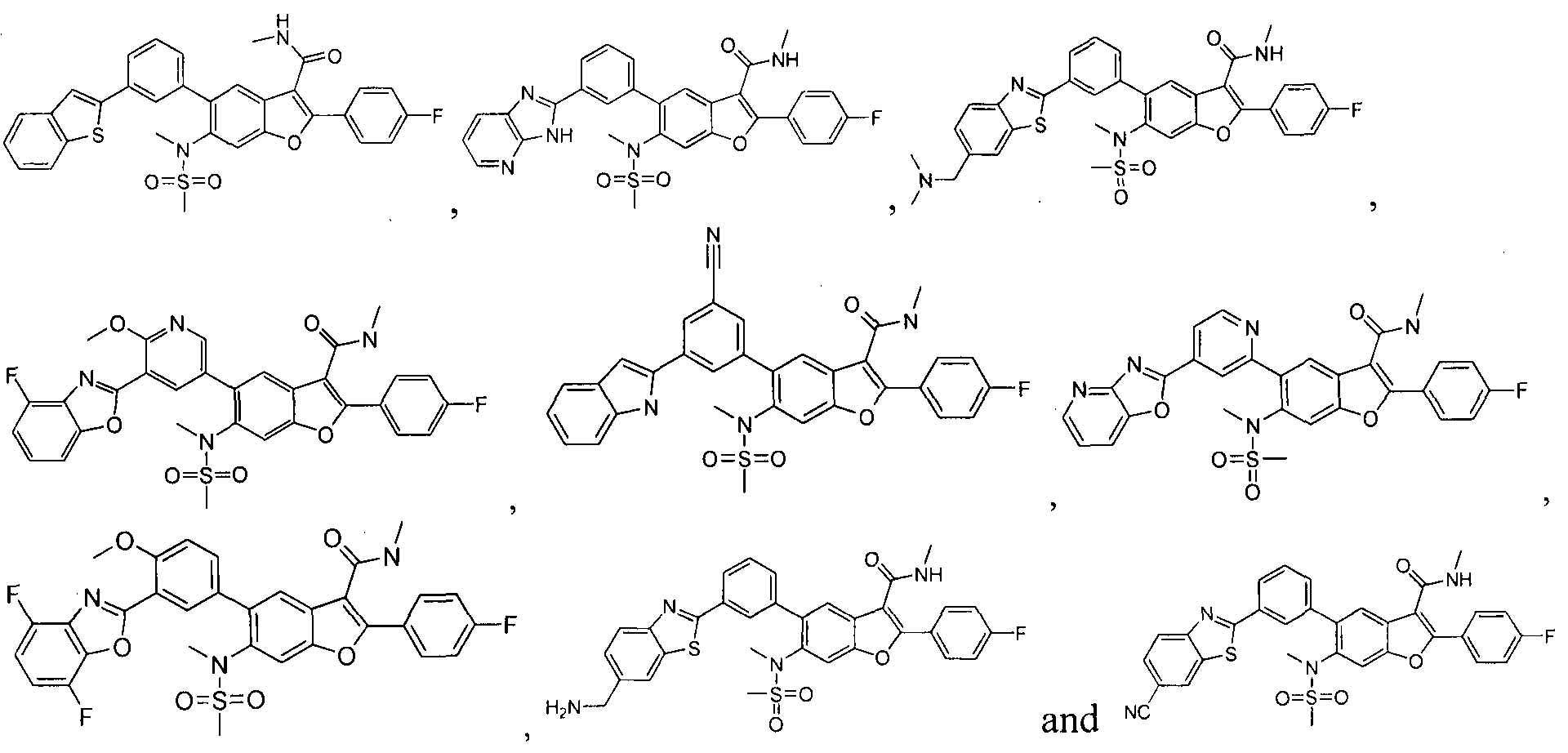
In another embodiment of the invention, the compound of the invention is selected from the exemplary species depicted in Examples 1-880 as shown below, and pharmaceutically acceptable salts thereof.
Other embodiments of the present invention include the following: (a) A pharmaceutical composition comprising an effective amount of a compound of formula (I) and a pharmaceutically acceptable carrier.
(b) The pharmaceutical composition of (a), further comprising a second therapeutic agent selected from the group consisting of HCV antiviral agents,
immunomodulators, and anti-infective agents.
(c) The pharmaceutical composition of (b), wherein the HCV antiviral agent is an antiviral selected from the group consisting of direct inhibitors of HCV, including but not limited to NS3 and NS3/4A protease inhibitors, NS5A inhibitors and HCV NS5B polymerase inhibitors.
(d) A pharmaceutical combination that is (i) a compound of formula (I) and (ii) a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound of formula (I) and the second therapeutic agent are each employed in an amount that renders the combination effective for inhibiting HCV NS5B activity, or for inhibiting HCV viral replication, or for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
(e) The combination of (d), wherein the HCV antiviral agents are one or more antiviral agents selected from the group consisting of direct inhibitors of HCV, including but not limited to NS3 and NS3/4A protease inhibitors, NS5A inhibitors and HCV NS5B polymerase inhibitors.
(f) A use of a compound of formula (I) in the preparation of a medicament for inhibiting HCV NS5B activity in a subject in need thereof.
(g) A use of a compound of formula (I) in the preparation of a medicament for preventing and/or treating infection by HCV in a subject in need thereof.
(h) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof, which comprises
administering to the subject an effective amount of a compound of formula (I).
(i) The method of (h), wherein the compound of formula (I) is administered in combination with an effective amount of at least one second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
(j) The method of (i), wherein the HCV antiviral agent is an antiviral selected from the group consisting of direct inhibitors of HCV, including but not limited to NS3 and NS3/4A protease inhibitors, NS5A inhibitors and HCV NS5B polymerase inhibitors.
(k) A method of inhibiting HCV viral replication and/or HCV viral production in a cell-based system, which comprises administering to the subject an effective amount of a
compound of formula (I) in combination with an effective amount of at least one second therapeutic agent selected from the group consisting of HCV antiviral agents,
immunomodulators, and anti-infective agents.
(1) The method of (k), wherein the HCV antiviral agent is an antiviral selected from the group consisting of direct inhibitors of HCV, including but not limited to NS3 and NS3/4A protease inhibitors, NS5A inhibitors and HCV NS5B polymerase inhibitors.
(m) A method of inhibiting HCV NS5B activity in a subject in need thereof, which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
(n) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof, which comprises
administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
In the embodiments of the compounds and salts provided above, it is to be understood that each embodiment may be combined with one or more other embodiments, to the extent that such a combination provides a stable compound or salt and is consistent with the description of the embodiments. It is further to be understood that the embodiments of compositions and methods provided as (a) through (n) above are understood to include all embodiments of the compounds and/or salts, including such embodiments as result from combinations of embodiments.
Additional embodiments of the invention include the pharmaceutical compositions, combinations, uses and methods set forth in (a) through (n) above, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features of the compounds described above. In all of these embodiments, the compound may optionally be used in the form of a pharmaceutically acceptable salt or hydrate as appropriate.
The present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) inhibiting HCV NS5B activity, or (b) inhibiting HCV viral replication, or (c) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, or (d) use in medicine. In these uses, the compounds of the present invention can optionally be employed in combination
with one or more second therapeutic agents selected from HCV antiviral agents, anti-infective agents, and immunomodulators.
As used herein, all ranges are inclusive, and all sub-ranges are included within such ranges, although not necessarily explicitly set forth. In addition, the term "or," as used herein, denotes alternatives that may, where appropriate, be combined; that is, the term "or" includes each listed alternative separately as well as their combination.
As used herein, the term "alkyl" refers to any linear or branched chain alkyl group having a number of carbon atoms in the specified range. Thus, for example, "C |.6 alkyl" (or "C| -C6 alkyl") refers to all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. As another example, "C i -4 alkyl" refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. Alkyl groups may be substituted as indicated.
The term "halogenated" refers to a group or molecule in which a hydrogen atom has been replaced by a halogen. Similarly, the term "haloalkyl" refers to a halogenated alkyl group. The term "halogen" (or "halo") refers to atoms of fluorine, chlorine, bromine and iodine (alternatively referred to as fluoro, chloro, bromo, and iodo).
The term "alkoxy" refers to an "alkyl-O" group. Alkoxy groups may be substituted as indicated.
The term "cycloalkyl" refers to any cyclic ring of an alkane or alkene having a number of carbon atoms in the specified range. Thus, for example, "C3-8 cycloalkyl" (or "C3-C8 cycloalkyl") refers to cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, cycloheptenyl, cyclooctyl, and cyclooctenyl. The term "cycloalkoxy" refers to a "cycloalkyl-O-" group. Cycloalkyl groups may be substituted as indicated.
The term "aryl" (or "aryl ring system") refers to aromatic mono- and poly- carbocyclic ring systems wherein the individual carbocyclic rings in the polyring systems are fused or attached to each other via a single bond. As used herein, the term aryl includes aromatic mono- and poly-carbocyclic ring systems that include from 0 to 4 heteroatoms (non-carbon atoms) that are independently chosen from N, O and S. Suitable aryl groups include phenyl, naphthyl, biphenylenyl, pyridinyl, pyrimidinyl and pyrrolyl, as well as those discussed below. Aryl groups may be substituted as indicated. Aryl ring systems may include, where appropriate, an indication of the variable to which a particular ring atom is attached. Unless otherwise
indicated, substituents to the aryl ring systems can be attached to any ring atom, provided that such attachment results in formation of a stable ring system.
The term "carbocycle" (and variations thereof such as "carbocyclic") as used herein, unless otherwise indicated, refers to (i) a C5 to C7 monocyclic, saturated or unsaturated ring, or (ii) a.Cg to Cjo bicyclic saturated or unsaturated ring system. Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated. Carbocycle groups may be substituted as indicated. When the carbocycles contain one or more heteroatoms independently chosen from N, O and S, the carbocycles may also be referred to as
"heterocycles," as defined below. The carbocycle may be attached to the rest of the molecule at any carbon or nitrogen atom that results in a stable compound. The fused bicyclic carbocycles are a subset of the carbocycles; i. e. , the term "fused bicyclic carbocycle" generally refers to a Cg to Cio bicyclic ring system in which each ring is saturated or unsaturated and two adjacent carbon atoms are shared by each of the rings in the ring system. A fused bicyclic carbocycle in which both rings are saturated is a saturated bicyclic ring system. Saturated carbocyclic rings are also referred to as cycloalkyl rings, e.g. , cyclopropyl, cyclobutyl, etc. A fused bicyclic carbocycle in which one or both rings are unsaturated is an unsaturated bicyclic ring system. Carbocycle ring systems may include, where appropriate, an indication of the variable to which a particular ring atom is attached. Unless otherwise indicated, substituents to the ring systems can be attached to any ring atom, provided that such attachment results in formation of a stable ring system.
Unless indicated otherwise, the term "heterocycle" (and variations thereof such as "heterocyclic" or "heterocyclyl") broadly refers to (i) a stable 5- to 7-membered, saturated or unsaturated monocyclic ring, or (ii) a stable 8- to 10-membered bicyclic ring system, wherein each ring in (ii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, arid the monocyclic ring or bicyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) independently selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the bicyclic ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur heteroatoms is optionally oxidized, and any one or more of the nitrogen heteroatoms is optionally quaternized. Unless otherwise specified, the heterocyclic ring may be attached at any heteroatom or carbon atom, provided that attachment results in the creation of a stable structure. Heterocycle groups may be substituted as indicated, and unless
otherwise specified, the substituents may be attached to any atom in the ring, whether a heteroatom or a carbon atom, provided that a stable chemical structure results. Representative examples include piperidinyl, piperazinyl, azepanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl (or tetrahydrofuranyl). Unless expressly stated to the contrary, the term
"heteroaryl ring system" refers to aryl ring systems, as defined above, that include from 1 to 4 heteroatoms (non-carbon atoms) that are independently chosen from N, O and S. In the case of substituted heteraromatic rings containing at least one nitrogen atom (e.g., pyridine), such substitutions can be those resulting in N-oxide formation. Representative examples of heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl. Representative examples of bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, 2,3-dihydrobenzofuranyl, 2,3-dihydrobenzo- 1 ,4-dioxinyl and benzo-1 ,3-dioxolyl.
Unless otherwise specifically noted as only "substituted", alkyl, cycloalkyl, and aryl groups are not substituted. Preferably, the substituents are selected from the group which includes, but is not limited to, halo, C|-C2o alkyl, -CF3, -NH2, -N(C i-C alkyl)2, -N02, oxo, - CN, -N3, -OH, -0(C,-C6 alkyl), C3-C,0 cycloalkyl, C2-C6 alkenyl, C2-C6 alkynyl, (C0-C6 alkyl) S(0)o-2-, aryl-S(O)0_2-, (C0-C6 alkyl)S(0)o-2(C0-C6 alkyl)-, (C0-C6 alkyl)C(0)NH-, H2N-C( H)- , -0(C,-C6 alkyl)CF3, (C0-C6 alkyl)C(O)-, (C0-C6 alkyl)OC(O)-, (C0-C6alkyl)O(C,-C6 alkyl)-, (C0-C6 alkyl)C(O),-2(C0-C6 alkyl)-, (C0-C6 alkyl)OC(0)NH-, aryl, aralkyl, heteroaryl, heterocyclylalkyl, halo-aryl, halo-aralkyl, halo-heterocycle and halo-heterocyclylalkyl.
As used herein, the term "compound" is intended to encompass chemical agents described by generic formula (I) in all forms, including hydrates and solvates of such chemical agents.
In the compounds of formula (I), the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of formula (I). For example, different isotopic
forms of hydrogen (H) include protium (Ή) and deuterium (2H or D). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
Isotopically-enriched compounds within formula (I) can be prepared without undue
experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
Unless expressly stated to the contrary, all ranges cited herein are inclusive. For example, a heteroaryl ring described as containing from "0 to 3 heteroatoms" means the ring can contain 0, 1 , 2, or 3 heteroatoms. It is also to be understood that any range cited herein includes within its scope all of the sub-ranges within that range. The oxidized forms of the heteroatoms N and S are also included within the scope of the present invention.
When any variable (for example, R1 or R3) occurs more than one time in any constituent or in formula (I) or in any other formula depicting and describing compounds of the invention, its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
Unless expressly stated to the contrary, substitution by a named substituent is permitted on any atom provided such substitution is chemically allowed and results in a stable compound. A "stable" compound is a compound that can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g. , therapeutic or prophylactic administration to a subject).
As a result of the selection of substituents and substituent patterns, certain of the compounds of the present invention can have asymmetric centers and can occur as mixtures of stereoisomers, or as individual diastereomers, or enantiomers. All isomeric forms of these compounds, whether isolated or in mixtures, are within the scope of the present invention.
As would be recognized by one of ordinary skill in the art, certain of the compounds of the present invention can exist as tautomers. For the purposes of the present invention a reference to a compound of formula (I) is a reference to the compound per se, or to any one of its tautomers per se, or to mixtures of two or more tautomers.
The compounds of the present inventions are useful in the inhibition of HCV replication (e.g., HCV NS5B activity), the treatment of HCV infection and/or reduction of the likelihood or severity of symptoms of HCV infection. For example, the compounds of this invention are useful in treating infection by HCV after suspected past exposure to HCV by such means as blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
The compounds of this invention are useful in the preparation and execution of screening assays for antiviral compounds. For example, the compounds of this invention are useful for identifying resistant HCV replicon cell lines harboring mutations within NS5B, which are excellent screening tools for more powerful antiviral compounds. Furthermore, the compounds of this invention are useful in establishing or determining the binding site of other antivirals to the HCV replicase.
The compounds of the present invention may be administered in the form of pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt" refers to a salt that possesses the effectiveness of the parent compound and that is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof). Suitable salts include acid addition salts that may, for example, be formed by mixing a solution of the compound of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid. Many of the compounds of the invention carry an acidic moiety, in which case suitable pharmaceutically acceptable salts thereof can include alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g. , calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts. Also, in the case of an acid (-COOH) or alcohol group being present, pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
The term "administration" and variants thereof (e.g., "administering" a compound) in reference to a compound of the invention mean providing the compound or a prodrug of the compound to the individual in need of treatment. When a compound of the invention is provided in combination with one or more other active agents (e.g., antiviral agents useful for treating HCV infection), "administration" and its variants are each understood to include concurrent and sequential provision of the compound or salt and other agents.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients, as well as any product which results, directly or indirectly, from combining the specified ingredients.
By "pharmaceutically acceptable" is meant that the ingredients of the pharmaceutical composition must be compatible with each other and not deleterious to the recipient thereof.
The term "subject" (alternatively referred to herein as "patient"), as used herein, refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
The term "effective amount" as used herein means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician. In one embodiment, the effective amount is a "therapeutically effective amount" for the alleviation of one or more symptoms of the disease or condition being treated. In another embodiment, the effective amount is a "prophylactically effective amount" for reduction of the severity or likelihood of one or more symptoms of the disease or condition. In another embodiment, the effective amount is a "therapeutically effective amount" for inhibition of HCV viral replication and/or HCV viral production. The term also includes herein the amount of active compound sufficient to inhibit HCV NS5B activity and thereby elicit the response being sought (i.e., an "inhibition effective amount"). When the active compound (i.e., active ingredient) is administered as the salt, references to the amount of active ingredient are to the free acid or free base form of the compound.
For the purposes of inhibiting HCV NS5B polymerase, treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection and inhibiting HCV viral replication and/or HCV viral production, the compounds of the present invention, optionally in the form of a salt, can be administered by any means that produces contact of the active agent with the agent's site of action. They can be administered by one or more
conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice. The compounds of the invention can, for example, be administered by one or more of the following: orally, parenterally
(including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles. Liquid preparations suitable for oral administration (e.g., suspensions, syrups, elixirs and the like) can be prepared according to techniques known in the art and can employ any of the usual media such as water, . glycols, oils, alcohols and the like. Solid preparations suitable for oral administration (e.g., powders, pills, capsules and tablets) can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders,
disintegrating agents and the like. Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids. Injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose. Further description of methods suitable for use in preparing pharmaceutical compositions of the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences, 18th edition (ed. A. R. Gennaro, Mack Publishing Co., 1990).
The compounds of this invention can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses. One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses. Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses. For oral administration, the compositions can be provided in the form of tablets or capsules containing 1 .0 to 500 mg of the active ingredient, particularly 1 , 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, and 500 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated. The specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, HCV viral genotype, viral resistance, and the host undergoing therapy.
As noted above, the present invention also relates to a method of inhibiting HCV NS5B activity, inhibiting HCV viral replication and/or HCV viral production, treating HCV
infection and/or reducing the likelihood or severity of symptoms of HCV infection with a compound of the present invention in combination with one or more therapeutic agents and a pharmaceutical composition comprising a compound of the present invention and one or more therapeutic agents selected from the group consisting of a HCV antiviral agent, an
immunomodulator, and an anti-infective agent. Such therapeutic agents active against HCV include, but are not limited to, ribavirin, levovirin, viramidine, thymosin alpha- 1 , R7025 (an enhanced interferon (Roche)), interferon-β, interferon-α, pegylated interferon-α (peginterferon-a), a combination of interferon-α and ribavirin, a combination of peginterferon-α and ribavirin, a combination of interferon-α and levovirin, and a combination of peginterferon-α and levovirin. The combination of pegylated-interferon and ribaviron represents the current Standard of Care for HCV treatment. The combination of one or more compounds of the present invention with the Standard of Care for HCV treatment, pegylated-interferon and ribaviron is specifically contemplated as being encompassed by the present invention, lnterferon-α includes, but is not limited to, recombinant interferon-a2a (such as ROFERON interferon available from Hoffmann- LaRoche, Nutley, NJ), pegylated interferon-a2a (PEGASYS), interferon-a2b (such as INTRON-A interferon available from Schering Corp., Kenilworth, NJ), pegylated interferon-a2b
(PEGINTRON), a recombinant consensus interferon (such as interferon alphacon- 1 ), albuferon (interferon-α bound to human serum albumin (Human Genome Sciences)), and a purified interferon-α product. Amgen's recombinant consensus interferon has the brand name INFERGEN. Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory activity similar to ribavirin. Viramidine represents an analog of ribavirin disclosed in International Patent Application Publication WO 01/60379. In accordance with the method of the present invention, the individual components of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
For the treatment of HCV infection, the compounds of the invention may also be administered in combination with an antiviral agent NS5B polymerase inhibitor, e.g., R7128 (Roche), valopicitabine (NM-283; Idenix) and 2'-F-2'-beta-methylcytidine (see also WO
2005/003147).
The compounds of the present invention also may be combined for the treatment of HCV infection with antiviral 2'-C-branched ribonucleosides disclosed in Rogers E. Harry-
O'Kuru et al. , A Short, Flexible Route toward -C-Branched Ribonucleosides, 62 J. ORG. CHEM. 1754-59 (1997); Michael S. Wolfe & Rogers E. Harry-O'Kuru, A Concise 2'-C-
Methylribonucleosides, 36(42) TETRAHEDRON LETTERS 761 1 -14 (1995); U.S. Patent No. 3,480,613; and International Patent Application Publications WO 01 /90121 , WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 and WO 04/002422; the entire contents of each of which are incorporated by reference. Such 2'-C-branched ribonucleosides include, but are not limited to, 2'-C-methyl-cytidine, 2'-C-methyl-uridine, 2'-C-methyl-adenosine, 2'-C-methyl- guanosine, and 9-(2-C-methyl- -D-ribofuranosyl)-2,6-diaminopurine, and the corresponding amino acid ester of the ribose C-2', C-3', and C-5' hydroxyls and the corresponding optionally substituted cyclic 1 ,3-propanediol esters of the 5'-phosphate derivatives.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS3 serine protease. HCV NS3 serine protease is an essential viral enzyme and has been described to be an excellent target for inhibition of HCV replication. Exemplary substrate and non-substrate based inhibitors of HCV NS3 protease inhibitors are disclosed in International Patent Application Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/481 16, WO 02/48172, WO 2008/057208 and WO 2008/057209, in British Patent No. GB 2 337 262, and in U.S. Patent Nos. 6,323, 180 and 7,470,664.
Further examples of HCV protease inhibitors useful in the present compositions and methods include, but are not limited to, the following compounds:
40
41
and pharmaceutically acceptable salts thereof.
The compounds of the present invention may also be combined for the treatment of HCV infection with nucleosides having anti-HCV properties, such as those disclosed in International Patent Application Publications WO 02/51425, WO 01/79246, WO 02/32920, WO 02/48165 and WO 2005/003147 (including R1656, (2'7?)-2'-deoxy-2'-fluoro-2'-C- methylcytidine, shown as compounds 3^6 on page 77); WO 01/68663; WO 99/43691 ;
WO 02/18404 and WO 2006/021341 , and U.S. Patent Application Publication US 2005/0038240, including 4'-azido nucleosides such as R1626, 4'-azidocytidine; U.S. Patent Application
Publications US 2002/0019363, US 2003/0236216, US 2004/0006007, US 2004/0063658 and US 2004/01 10717; U.S. Patent Nos. 7,105,499, 7,125,855, 7,202,224; and International Patent Application Publications WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/01 1478, WO 04/013300 and WO 04/028481 ; the content of each is incorporated herein by reference in its entirety.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS5B polymerase. Such HCV NS5B polymerase inhibitors that may be used as combination therapy include, but are not limited to, those disclosed in International Patent Application Publications
WO 02/057287, WO 02/057425, WO 03/068244, WO 2004/000858, WO 04/003138 and
WO 2004/007512; U.S. Patent Nos. 6,777,392, 7,105,499, 7,125,855, 7,202,224 and U.S. Patent
Application Publications US 2004/0067901 and US 2004/01 1071 7; the content of each is incorporated herein by reference in its entirety.
In one embodiment, additional nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS5B inhibitors are selected from the following compounds: 4-amino-7-(2-C-methyl- -D-arabinofuranosyl)-7H-pyrrolo[2,3-iii]pyrimidine; 4- amino-7-(2-C-methyl- -D-ribofuranosyl)-7H-pyrrolo[2,3-(i]pyrimidine; 4-methylamino-7-(2-C- methyl- -D-ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-dimethylamino-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-t/]pyrimidine; 4-cyclopropylamino-7-(2-C-methyl-p-D- ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(2-C-vinyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-d]pyrimidine; 4-amino-7-(2-C-hydroxymethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-(/]pyrimidine; 4-amino-7-(2-C-fluoromethyl- -D-ribofuranosyl)-7H- pyrrolo[2,3-</]pyrimidine; 4-amino-5-methyl-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i pyrimidine; 4-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-cf|pyrimidine-5-carboxylic acid; 4-amino-5-bromo-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-i/]pyrimidine; 4-amino-5-chloro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-t ]pyrimidine; 4-amino-5-fluoro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 2,4-diamino-7-(2-C-methyl-P-D-ribofuranosyl)- 7H-pyrrolo[2,3-<i]pyrimidine; 2-amino-7-(2-C-methyl- -D-ribofuranosyl)-7H- pyrrolo[2,3-t/]pyrimidine; 2-amino-4-cyclopropylamino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-c ]pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-c/]pyrimidin-4(3H)-one; 4-amino-7-(2-C-ethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-t/]pyrimidine; 4-amino-7-(2-C,2-0-dimethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-<i]pyrimidine; 7-(2-C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-( |pyrimidin-4(3H)- one; 2-amino-5-methyl-7-(2-C, 2-0-dimethyl- -D-ribofuranosyl)-7H-pyrrolo[2,3-c/]pyrimidin- 4(3H)-one; 4-amino-7-(3-deoxy-2-C-methyl-P-D-ribofuranosyl)-7H-pyrro]o[2,3-i ]pyrimidine; 4-amino-7-(3-deoxy-2-C-methyl-p-D-arabinofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino- 2-fluoro-7-(2-C-methyl- -D-ribofuranosyl)-7H-pyrrolo[2,3-i |pyrimidine; 4-amino-7-(3-C- methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(3-C-methyl-P-D- xylofuranosyl)-7H-pyrrolo[2,3-t ]pyrimidine; 4-amino-7-(2,4-di-C-methyl-p-D-ribofuranosyl)- 7H-pyrrolo[2,3-<f|pyrimidine; 4-amino-7-(3-deoxy-3-fluoro-2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i/]pyrimidine; and the corresponding 5 '-triphosphates; or a pharmaceutically acceptable salt thereof.
The compounds of the present invention may also be combined for the treatment of HCV infection with non-nucleoside inhibitors of HCV polymerase such as those disclosed in U.S. Patent Applciation Publications US 2006/0100262 and US 2009/0048239; International Patent Application Publications WO 01/77091, WO 01/47883, WO 02/04425, WO 02/06246, WO 02/20497, WO 2005/016927 (in particular JT 003), WO 2004/041201, WO 2006/066079, WO 2006/066080, WO 2008/075103, WO 2009/010783 and WO 2009/010785; the content of each is incorporated herein by reference in its entirety.
In one embodiment, additional non-nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS5B inhibitors are selected from the following compounds: 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8- tetrahydrOindolo[2,l-fl][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2-morpholin- 4-ylethyl)-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine-l l-carboxylic acid; 14- cyclohexyl-6-[2-(dimethylamino)ethyl]-3-methoxy-5,6,7,8-tetrahydroindolo[2,l-a]
[2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-3-methoxy-6-methyl-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; methyl ({[(14-cyclohexyl-3- methoxy-6-methyl-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocin-l 1- yl)carbonyl]amino}sulfonyl)acetate; ({[(14-cyclohexyl-3-methoxy-6-methyl-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocin-l l-yl)carbonyl]amino}sulfonyl)acetic acid; 14- cyclohexyl-N-[(dimethylamino)sulfonyl]-3-methoxy-6-methyl-5,6,7,8-tetrahydroindolo[2,l-a] [2,5]benzodiazocine-l 1-carboxamide; 3-chloro-14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7- oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine 11 -carboxylic acid; N"-(l l-carboxy-14- cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7-yl)-N,N-dimethylethane-l,2- diaminium bis(trifluoroacetate); 14-cyclohexyl-7,8-dihydro-6H-indolo[ 1 ,2-e] [1,5]
benzoxazocine-11-carboxylic acid; 14-cyclohexyl-6-methyl-7-oxo-5,6,7,8-tetrahydroindolo [2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-3-methoxy-6-methyl-7-oxo- 5,6,7,8-tetrahydroindolo[2,l-o][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2- (dimethylamino)ethyl]-3-methoxy-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine- 11-carboxylic acid; 14-cyclohexyl-6-[3-(dimethylamino)propyl]-7-oxo-5,6,7,8-tetrahydroindolo [2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-7-oxo-6-(2-piperidin-l -ylethyl)- 5,6,7,8-tetrahydroindolo[2,l- ][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2- morpholin-4-ylethyl)-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2-(diethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a]
[2,5]benzodiazocine- l 1 -carboxylic acid; 14-cyclohexyl-6-(l -methylpiperidin-4-yl)-7-oxo- 5,6,7,8-tetrahydroindolo[2, l -a][2,5]benzodiazocine-l 1 -carboxylic acid; 14-cyclohexyl-N- [(dimethylamino)sulfonyl]-7-oxo-6-(2-piperidin- l -ylethyl)-5,6,7,8-tetrahydroindolo[2, l -fl] [2,5]benzodiazocine- l 1 -carboxamide; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-V- [(dimethylaminp)sulfonyl]-7-oxo-5,6,7,8-tetrahydroindolo[2, l -a][2,5]benzodiazocine- l 1 - carboxamide; 14-cyclopentyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2, l -(7]
[2,5]benzodiazocine- l 1 -carboxylic acid; 14-cyclohexyl-5,6,7,8-tetrahydroindolo[2, l -a]
[2,5]benzodiazocine- l 1 -carboxylic acid; 6-allyl-14-cyclohexyl-3-methoxy-5,6,7,8- tetrahydroindolo[2, l -tf][2,5]benzodiazocine- l 1 -carboxylic acid; 14-cyclopentyl-6-[2- (dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2, l -a][2,5]benzodiazocine-l 1 -carboxylic acid; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2, l -o][2,5]benzodiazocine- 1 1 -carboxylic acid; 13-cyclohexyl-5-methyl-4,5,6,7-tetrahydrofuro[3',2':6,7][ l ,4]diazocino[l ,8- a]indole- 10-carboxylic acid; 15-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-6,7,8,9- tetrahydro-5H-indolo[2,l -a][2,6]benzodiazonine-12-carboxylic acid; 1 5-cyclohexyl-8-oxo- 6,7,8,9-tetrahydro-5H-indolo[2, l -a][2,5]benzodiazonine-12-carboxylic acid; 13-cyclohexyl-6- oxo-6,7-dihydro-5H-indolo[l ,2-i |[ l ,4]benzodiazepine- 10-carboxylic acid; and pharmaceutically acceptable salts thereof.
In another embodiment, the present HCV NS5B polymerase inhibitors are used in combination with non-nucleoside HCV NS5A inhibitors and pharmaceutically acceptable salts thereof.
The HCV NS5B inhibitory activity of the present compounds may be tested using assays known in the art. The HCV NS5B polymerase inhibitors described herein have activities in a genotype l b replicon assay as described in the Examples. The assay is performed by incubating a replicon harboring cell-line in the presence of inhibitor for a set period of time and measuring the effect of the inhibitor on HCV replicon replication either directly by quantifying replicon RNA level, or indirectly by measuring enzymatic activity of a co-encoded reporter enzyme such as luciferase or β-lactamase. By performing a series of such measurements at different inhibitor concentrations, the effective inhibitory concentration of the inhibitor (EC50 or EC90) is determined. See Jan M. Vrolijk et al , A replicons-based bioassay for the measurement of interferons in patients with chronic hepatitis C, 1 10 J. VlROLOGlCAL METHODS 201 (2003). Such assays may also be run in an automated format for high through-put screening. See Paul
Zuck et al., A cell-based β-lactamase reporter gene assay for the identification of inhibitors of hepatitis C virus replication, 334 ANALYTICAL BIOCHEMISTRY 344 (2004).
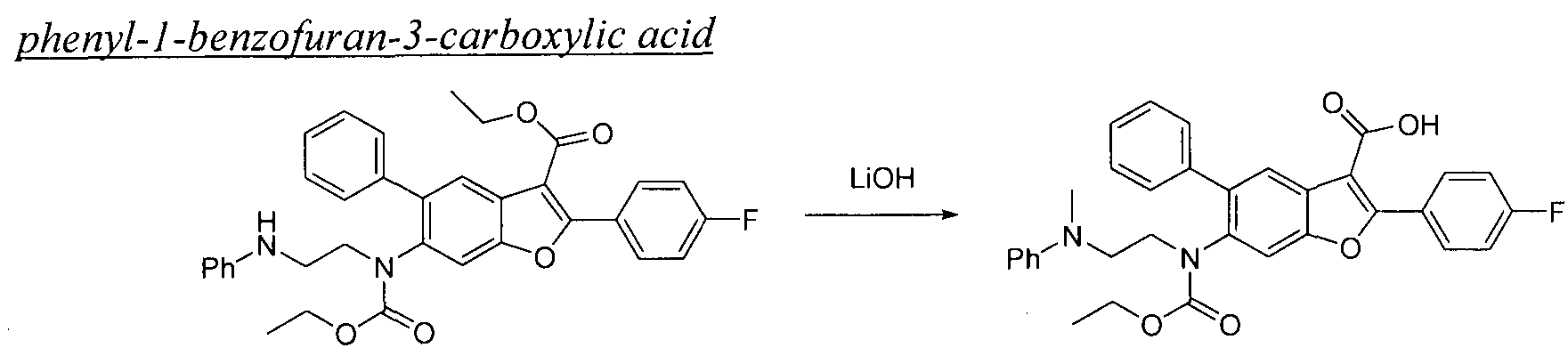
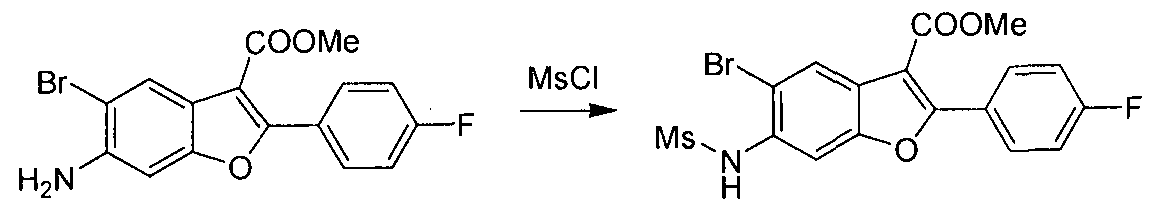
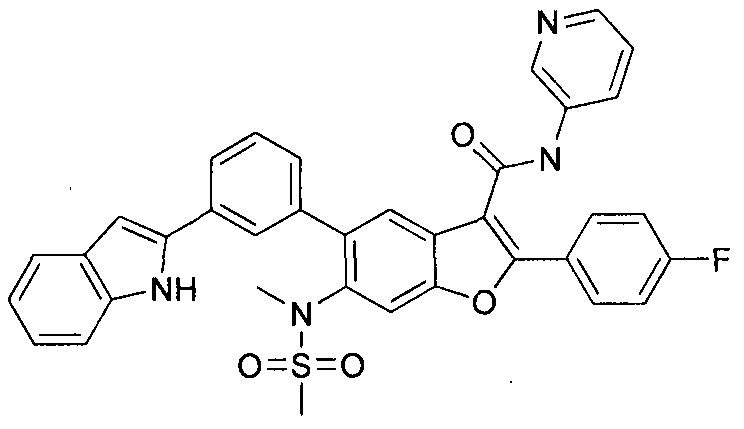
The present invention also includes processes for making Compounds of Formula (I). The compounds of the present invention can be readily prepared according to the following reaction schemes and examples, or modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are themselves known to those of ordinary skill in this art, but are not mentioned in greater detail. Furthermore, other methods for preparing compounds of the invention will be readily apparent to the person of ordinary skill in the art in light of the following reaction schemes and examples. Unless otherwise indicated, all variables are as defined above. The following reaction schemes and examples serve only to illustrate the invention and its practice.
General Schemes
Scheme 1
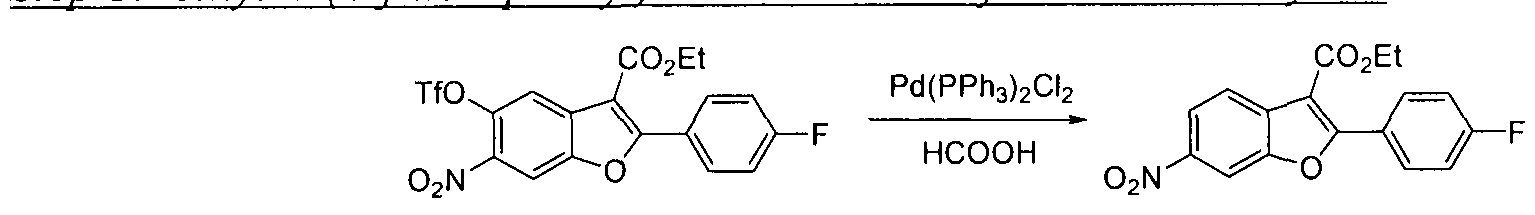
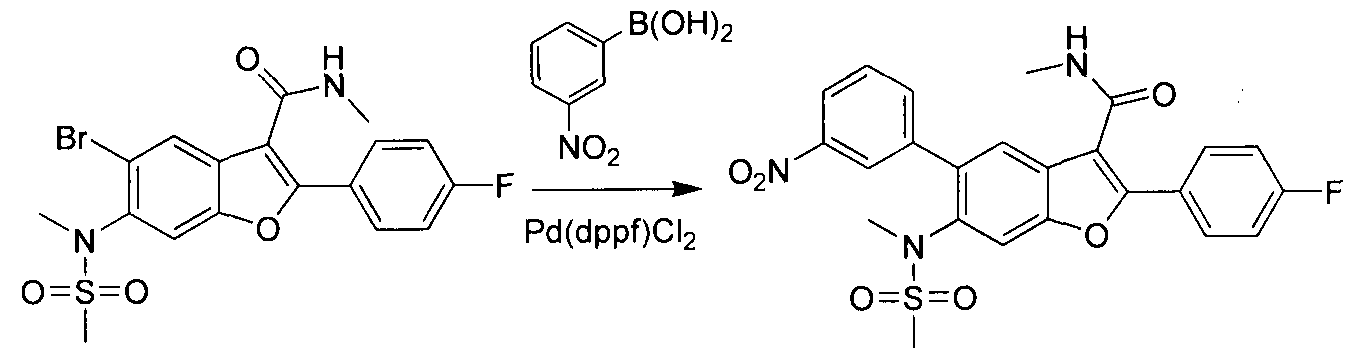
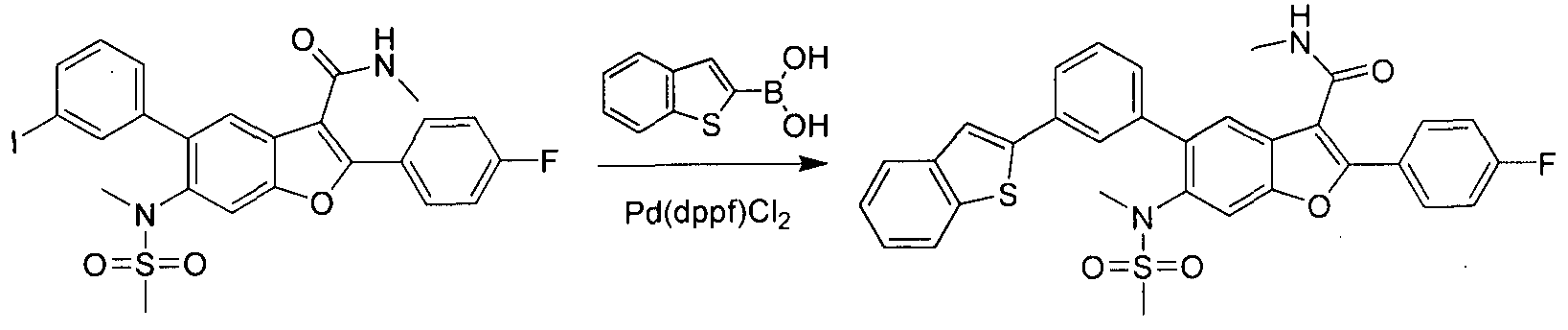
This scheme describes the preparation of compounds with the general structure of G and H. Starting from compound A (obtained according to procedure in WO 2004/041201 A2), coupling with a substituted or unsubstituted phenylboronic acid catalyzed by a transition metal,
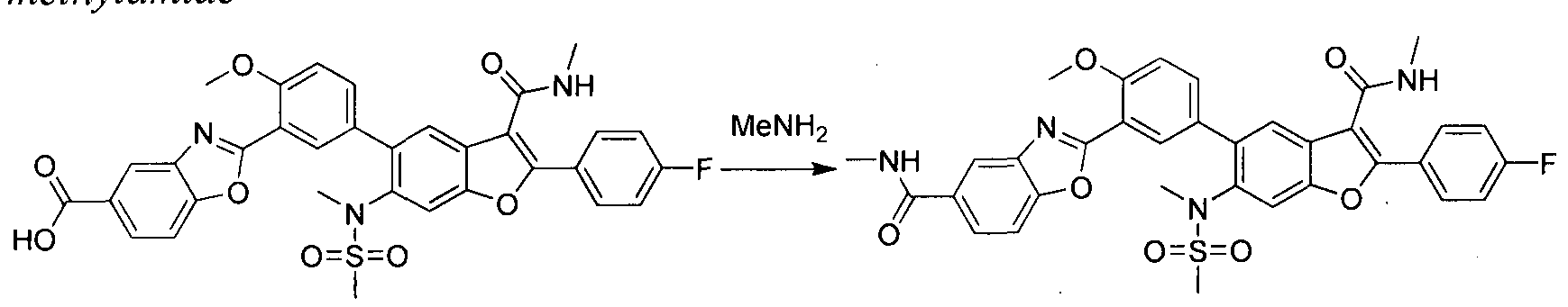
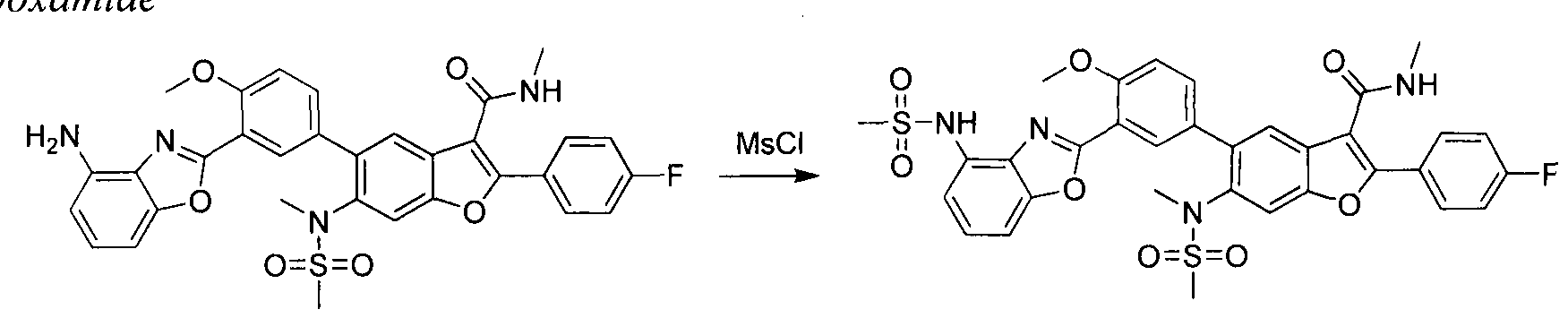
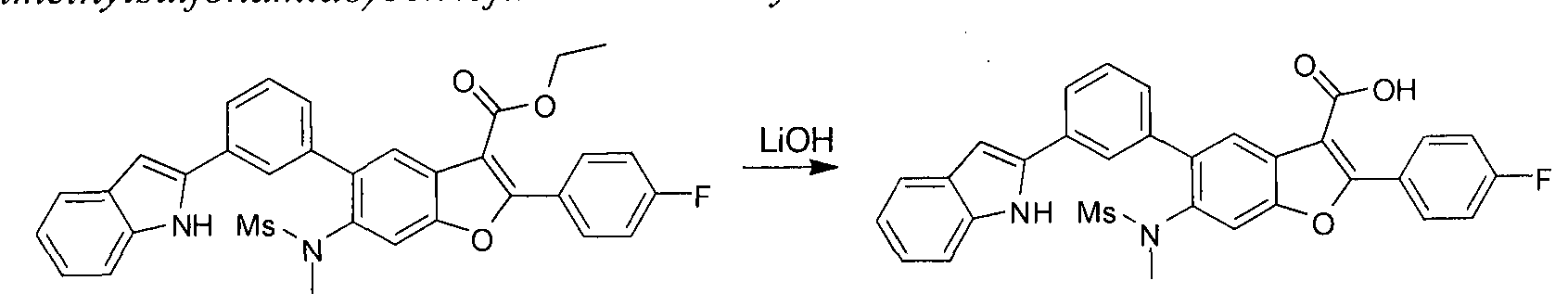
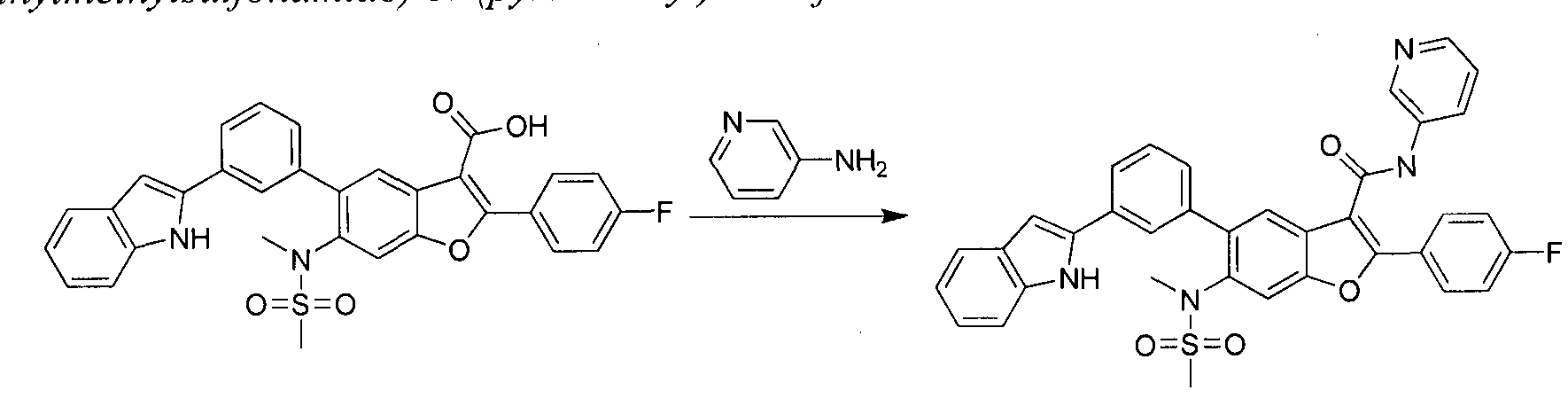
in this case Pd(dppf)Cl2, furnishes compounds of the general structure B. This type of transition- metal-mediated cross-coupling is common and there are numerous conditions that one skilled in the art can use to execute such a transformation. Compounds of type C are next generated by reduction of the nitro group in compound B, which can be accomplished by exposure to common reducing conditions, in this case treatment by Fe in NH4CI solution under reflux. The amino group in compounds C is then sulfonylated with a sulphonyl chloride to give compounds of type D. The sulfonamide D can be coupled with an alkylating agent (an alkyl halide for example) in the presence of a suitable base, such as potassium carbonate, to provide compounds E. The ester functionality in compounds E is readily hydrolyzed by aqueous base to afford compounds F. The carboxylic acid of compound F was condensed with methanamine or
O-methylhydroxylamine using common amide-forming reagents such as EDCI and HOBT to give compounds G or compounds H.
Schem
Compound C can be coupled with an alkylating agent (an alkyl halide for example) in the presence of a suitable base, such as potassium carbonate, to provide compounds I where Z represents an alkylated aniline. Alternatively C may be condensed with substituted carboxylic acid in the presence of coupling reagents, such as EDCI and HOBT, to afford compounds I where Z represents a substituted amide. Compounds J may be obtained from compounds I by further TV-alkylation or 7V-acylation reaction. Compounds of general structure I or J are hydrolyzed by aqueous hydroxide to provide compounds F. The carboxylic acid of
compound F may be condensed with an amine as shown in Scheme 1 to provide target compounds of general structure G and H.
Scheme 3
Compound A may be reduced by a catalyst in the presence of a hydrogen source (for example, Pd in the presence of formic acid) to afford compound K. Further reduction of K provides aniline L. The amino group of compound L is reacted with sulfonyl chloride to afford compound M, which can be further N-alkylated with a wide variety of alkylating agents in the presence of a suitable base, such as potassium carbonate, to provide compound N. Halogenation of compound IN, in this case bromination with FeCl3 and Br2 in anhydrous CC1 gives compound O. Compounds of general structure O are hydrolyzed by aqueous hydroxide to provide compounds P. The carboxylic acid of compound P may be condensed with an amine as shown in Scheme 1 to provide compounds of general structure Q. Transition metal mediated coupling of compounds Q with a boronic acid (alternatively alkyl tin, silicon, or other types of coupling partners may be used) provides the target compounds of general structure G.
Scheme 4
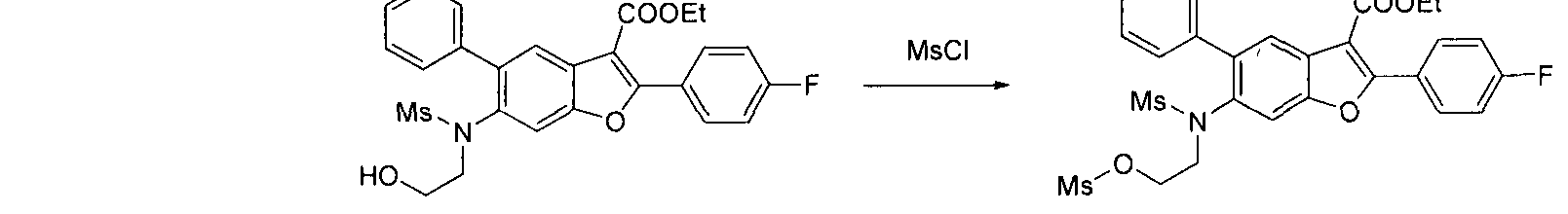
Compounds E that possess a hydroxyl group may be obtained from compounds D by reacting with 2-bromo ethanol. The hydroxyl group E can be converted to a leaving group (by reaction with MsCl for example) to afford compound R. Compound R may be treated with nucleophilic reagents such as an amine in the presence of a suitable base, such as triethylamine, to afford compound S. Compounds T can then be obtained from compound S by further N-alkylation or N-acylation. Compounds of structure T are readily converted to the target structures G following the general procedure described in Scheme 1.
Sche
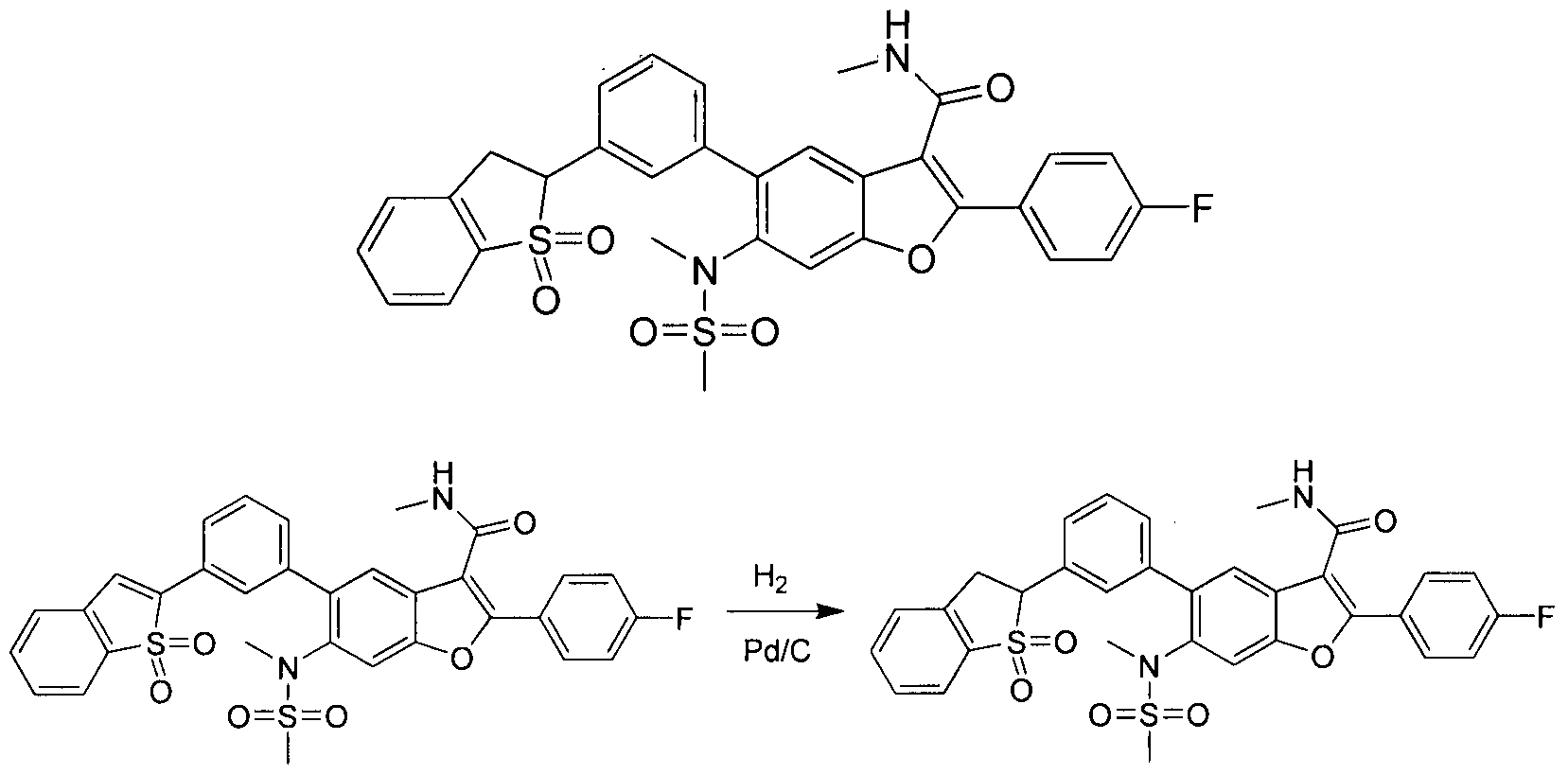
This scheme describes the preparation of compounds with the general structure of M'. Starting from compound A', bromating and esterifying with TBATB in MeOH to afford compound B\ Protecting the phenol group of B' with TBSC1 provides compound C, which can be C-acylated with 4-fluorobenzoyl chloride to give compound D'. After de-protection with TBAF and cyclizing by concentrated HC1, compound D' affords compound E' and F'
sequentially. Compound F' can be converted to compound G' by treated with fuming H 03. Compound H' is generated by reduction of the nitro group in compound G', and the amino group in compound H' is then sulfonylated with MsCl to furnish compound Γ. The sulfonamide Γ can be coupled with Mel in the presence of potassium carbonate to provide compound J'. The ester functionality in compound J1 is readily hydrolyzed by aqueous base to afford compound K'. The carboxylic acid of compound K' was condensed with methanamine using common amide forming reagents such as EDCI and HOBT to give compound L\ Transition metal mediated coupling of compound L' with a meta-heterocycle-substituted phenyl boronic ester (alternatively boronic acid, alkyl tin, silicon, or other types of coupling partners may be used) provides the target compounds of general structure M\
Scheme 6
Coupling compound L' with a substituted or unsubstituted 3-formylphenylboronic acid catalyzed by a transition metal, in this case Pd(dppf)Cl2, furnishes compounds of the general structure '. Compounds of type N' were cyclized with ortho-amino anilines or or ho-amino thiophenols to provide the target compounds of general structure O' or P\
Scheme 7
Τ' U'
This scheme describes a method useful for making the compounds of formula U', which correspond to the Compounds of Formula (II) wherein Het is a heterocyclyl or heteroaryl group; R60 is para-F; and R20, R30, R40 and R50 are defined above for the Compounds of Formula (Π).
A compound of formula Q' can be coupled with a substituted or unsubstituted 3- nitrophenylboronic acid catalyzed by a transition metal, in this case Pd(dppf)Cl2, to provide the compounds of formula R'. Compounds of formula R' can then be hydrogenated to provide the amino compounds of formula S', which are reacted with i-AmONO / 12, to provide the iodo compounds of formula T'. Transition metal mediated coupling of T with a heterocyclic boronic acid (alternatively boronic ester, alkyl tin, silicon, or other types of coupling partners may be used) provides the target compounds of formula U\
Scheme 8
υ·
This scheme describes an alternate useful for making the compounds of formula U', which correspond to the Compounds of Formula (II) wherein Het is a heterocyclyl or heteroaryl group; R60 is para-F; and R20, R30, R40 and R50 are defined above for the Compounds of Formula (II).
An iodo compound of formula T' can be converted to boronic ester compounds of formula V in the presence of Pd(dppf)Cl2. A compound of formula V can then be coupled with and aryl bromide or heterocyclic bromide to provide the compounds of formula U'. Scheme 9
This scheme describes a method useful for making the the compounds of formula W, which correspond to the Compounds of Formula (II) wherein R10 is indole or other bicyclic
pyrrole derivative; R is para-F; and R , R , R and R are defined above for the Compounds of Formula (II).
A transition metal-mediated coupling of a compound of a bromo compound of formula Q' with a heterocycle substituted phenyl boronic ester (alternatively boronic acid, alkyl tin, silicon, or other types of coupling partners may be used) provides the compounds of formula W. The SEM protecting group of a compound of formula W can subsequently be deproteted using TBAF to provide the compounds of formula X'.
Scheme 1
This scheme describes an alternate method useful for making the compounds of formula U', which correspond to the Compounds of Formula (II) wherein Het is a heterocyclyl or heteroaryl group; R60 is para-F; and R20, R30, R40 and R50 are defined above for the Compounds of Formula (II).
The ester group of a compound of formula Y' can be hydrolyzed using aqueous base to provide a compound of formula Z'. The carboxylic acid moiety of Z' can then be condensed with an amine of formula R30NH2 using common amide forming reagents, such as EDCI and HOBT, to provide the compounds of formula A". The sulfonamide group of A" can then be coupled with a reagent of formula R40X in the presence of potassium carbonate or with a regent of formula R40OH in the presence of PPh3 and DEAD to provide compounds of fomrula B". Transition metal mediated coupling of a compound of formula B" with a heterocycle- substituted phenyl boronic ester (alternatively boronic acid, alkyl tin, silicon, or other types of coupling partners may be used) provides the compounds of formula U'.
U'
This scheme describes yet another alternate method useful for making the compounds of formula U', which correspond to the Compounds of Formula (I) wherein Het is a heterocyclyl or heteroaryl group; R60 is para-F; and R20, R30, R40 and R50 are defined above for the Compounds of Formula (I).
The amino group of a compound of formula FT can be sulfonylated using a reagent of formula R50SO2Cl to provide the sulfonamide compounds of formula C". A compound of formula C" can then be coupled with a reactant of formula R40X in the presence of potassium carbonate to provide the compounds of formula D". The ester moiety of the compounds of formula D" can be readily hydrolyzed using aqueous base to provide the compounds of formula E". The carboxylic acid group of E" is then condensed with an amine of formula R30NH2 using common amide forming reagents, such as EDCI and HOBT, to provide the compounds of formula to F". Transition metal mediated coupling of a compound of formula F" with a heterocycle-substituted phenyl boronic ester (alternatively boronic acid, alkyl tin, silicon, or other types of coupling partners may be used) provides the compounds of formula U'.
List of Abbreviations
AcOH Acetic acid
i-AmONO z'so-Amylnitrite
n-BuLi «-butyllithium
Bu3N Tributylamine
CCI4 Carbon tetrachloride or tetrachloromethane
CDCI3 Deuterated chloroform
MeCN, CH3CN Acetonitrile
MeNH2, CH3NH2 Methylamine
MeONH2, CH3ONH2 Methoxyamine
Cs2C03 Cesium carbonate
DCM Dichloromethane
DEAD Diethylazodicarboxylate
D F Dimethylformamide
DMSO Dimethylsulfoxide
EDCI N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide (also EDC)
Et3N Triethylamine
EtOAc Ethyl acetate
EtOH Ethanol
EtOOCCl, CICOOEt Ethyl chloroformate
HOBT 1 -Hydroxy benzotriazole
Ή-NMR Proton Nuclear Magnetic Resonance
HPLC High Performance Liquid Chromatography
KOAc Potassium acetate
K3P04 Potassium Phosphate
LDA Lithium diisopropylamide
LiHMDS Lithium bis(trimethylsilyl) amide
LiOH H20 Lithium hydroxide monohydrate
MeNH2 Methanamine
MeCN Acetonitrile
MeOD Deuterated methanol
MeOH Methanol
MeONH2 Methoxyamine
MS Mass spectroscopy
Ms Methanesulfonyl (mesyl)
MsCl Methanesulfonyl chloride
NBS N-Bromosuccinimide
NCS N-Chlorosuccinimide
PE Petroleum ether
PPh3 Triphenylphosphine
Pd-C, Pd/C Palladium on carbon
Pd(dppf)Cl2 1 , 1 '-bis(diphenylphosphino)ferrocene-palladium(II)dichloride
Pd(PPh3)2Cl2 1 , 1 '-bis(tetrakis(triphenylphosphine))palladium(II)dichloride
Pd(PPh3)4 Tetrakis(triphenylphospine)palladium(0)
Ph Phenyl
PhB(OH)2 Phenylboronic acid
PhN02 Nitrobenzene
PhS02Cl Benzenesulfonyl chloride
/-PrNH2 Diisopropylamine
Py Pyridine
RT Room temperature, approximately 25°C
SEM 2-(Trimethylsilyl)ethoxymethyl
TBAF Tetrabutyl ammonium fluoride
TBATB Tetrabutylammonium tribromide
TBS Tert-butyldimethylsilyl
TBSC1 Tert-butyldimethylsilylchloride
Tf Trifluoromethanesulfonate (triflate)
THF Tetrahydrofuran
TLC Thin layer chromatography
EXAMPLES
Example 1 : 2-(4-fluorophenyl)-N-methyl-6-fmcthyl(methylsulfonyl)aminol-5-phenyl-l- benzofuran-3-carboxamide
Step 1: ethyl 2-(4-fluorophenyl)-6-nitro-5-phenyl-l-benzofuran-3-carboxylate
Phenylboronic acid (100 mg, 0.8 mmol) and K3P04-3H20 (1 19 mg, 0.8 mmol) were added to a suspension of ethyl 2-(4-fluorophenyl)-6-nitro-5-{[(trifluoromethyl)sulfonyl] oxy}-l -benzofuran-3-carboxylate (obtained according to procedure in WO 2004/041201 A2, 200 mg, 0.4 mmol) in dioxane (2 mL) and DMF (2 mL) under N2 protection. Then, Pd(dppf)Cl2 (5 mg, 0.08 mmol) was added to the mixture under N2 protection. The reaction mixture was heated to 90°C for 30 minutes. After cooling, the mixture was diluted with H20 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-TLC to give pure ethyl 2-(4- fluorophenyl)-6-nitro-5-phenyl-l -benzofuran-3-carboxylate (35 mg, yield: 23%).
Ή-NMR (400 MHz, CDC1
3) δ 7.88-7.98 (m, 2H), 7.62 (s, 1 H), 7.44-7.48 (m, 4H), 7.32-7.38 (m, 1 H), 7.06-7.12 (m, 2H), 6.78 (s, 1 H), 4.29-4.35 (m, 2H), 1.27-1.30 (m, 3H) Step 2: ethyl 6-amino-2-(4-fluorophenyl)-5-phenyl-l-benzofuran-3-carboxylate
A mixture of ethyl 2-(4-fluorophenyl)-6-nitro-5-phenyl-l -benzofuran-3- carboxylate (1 10 mg, 0.27 mmol), Fe (120 mg, 2.16 mmol) and NH4C1 (217 mg, 4.05 mmol) in H20 /MeOH /THF (1 mL /l mL /l mL) was refluxed for 4 hours. Then, ¾0 was added to quench the reaction, and the mixture was extracted with EtOAc. After washing with brine and dried, the solvent was removed by distillation. The pure product of ethyl 6-amino-2-(4- fluorophenyl)-5-phenyl-l -benzofuran-3-carboxylate was obtained (85 mg, yield: 85%) by prep- TLC.
1 H-NMR (400 MHz, CDC13) δ 8.00-8.03 (m, 2H), 7.85 (d, J = 7.2 Hz, 2H), 7.45-7.49 (m, 3H), 7.29-7.32 (m, 2H), 7.10-7.14 (m, 2H), 4.29-4.35 (m, 2H), 1.27-1.30 (m, 3H).
Step 3: ethyl 2-(4-fluorophenyl)-6-[(methylsulfonyl)amino -5-phenyl-l-benzofuran-3- carboxylate
MsCI
MsCl (66 mg, 0.6 mmol) was added to a solution of the product of Step 2 (85 mg, 0.23 mmol) and pyridine (73 mg, 0.92 mmol) in dry DCM (2 mL). The reaction mixture was stirred overnight at RT. After dilution with H
20 and extraction with DCM, the organic layer was washed with brine, dried over Na
2S0
4 and filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by prep-TLC to give ethyl 2-(4-fluorophenyl)- 6-[(methylsulfonyl)amino]-5-phenyl-l -benzofuran-3-carboxylate (90 mg, yield: 86%).
Ή-NMR (400 MHz, CDC13) δ 8.00-8.03 (m, 2H), 7.85 (d, J = 7.2 Hz, 2H), 7.45-7.49 (m, 3H), 7.29-7.32 (m, 2H), 7.10-7.14 (m, 2H), 6.50 (s, 1 H), 4.29-4.35 (m, 2H), 2.80 (s, 3H), 1.27-1.30 (m, 3H).
Step 4: ethyl 2-(4-fluorophenyl)-6-(methyl(methylsulfonyl)aminol-5-phe
NaH (60% in oil, 20 mg, 0.5 mmol) and CH3I (85 mg, 0.6 mmol) were added to a solution of the product of Step 3 (90 mg, 0.2 mmol) in dry DMF under N2 protection. The mixture was stirred overnight at RT, and then ice-cold diluted AcOH was added to the mixture.
After extraction with EtOAc, the organic solvent was washed with brine, dried over Na2S04, filtered and the solvent was evaporated under reduced pressure. The crude product was purified by prep-TLC to give ethyl 2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)amino]-5-phenyl-l - benzofuran-3-carboxylate (78 mg, yield: 84%).
Ή-NMR (400 MHz, CDC13) δ 8.00-8.02 (m, 2H), 7.97-7.98 (m, 1 H), 7.55-7.56
(m, 1 H), 7.39-7.40 (m, 4H), 7.32-7.34 (m, 1H), 7.1 1-7.15 (m, 2H), 4.32 (q, J = 7.2 Hz, 2H),
3.1 1 (s, 3H), 2.45 (s, 3H), 1.26-1.30 (t, J= 6.8 Hz, 3H).
Step 5: 2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)amino]-5^henyl-l-be
The product of Step 4 (78 mg, 0.17 mmol) was dissolved in THF (2 mL) and H20
(2 mL). Then, LiOH (71 mg, 1.7 mmol) was added to the solution, and the mixture was stirred at
RT overnight. After acidification with HC1 (1 N) and extraction with EtOAc, the combined organic phases were washed with brine, dried over Na
2S0 , filtered and evaporated to give the
product of 2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)amino]-5-phenyl-l -benzofuran-3- carboxylic acid (50 mg, yield: 67%). It was used for the next step without further purification. Step 6: 2-(4-fluorophenyl)-N-methyl-6-[methyl(methvhulfonyl)amino]-5^henyl-l-benz 3-carboxamide
The product of Step 5 (50 mg, 0.1 1 mmol), HOBT (24.5 mg, 0.16 mmol) and EDCI (52 mg, 0.27 mmol) were dissolved in dry DMF (2 mL). The resulting solution was stirred for 30 minutes. Then, methanamine (HCl salt, 14 mg, 0.44 mmol) and Et3N (50 mg, 0.47 mmol) were added to the mixture. After stirring overnight, the mixture was diluted with H20 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-TLC to give pure 2-(4-fluorophenyl)-N-methyl-6-[methyl(methylsulfonyl)amino]-5-phenyl-l -benzofuran-3- carboxamide (20 mg, yield: 40%).
Ή-NMR (400 MHz, CDC13) δ 7.92-7.96 (m, 2H), 7.59 (s, 1 H), 7.52-7.54 (m, 1 H), 7.29-7.47 (m, 5H), 7.1 1 -7.18 (m, 2H), 5.84 (s, 1 H), 3.25 (s, 3H), 2.98 (d, J = 7.2 Hz, 3H), 2.61 (s, 3H).
Examples 2 through 6 were prepared according to the general procedu;
Example 7: 2-(4-fluorophenyl)-N-methoxy-6-[methyl(methylsulfonyl)ainino|-5-phenyl-l- benzofuran-3-carboxamide
Steps 1-5
Steps 1 -5 were performed in accordance with Example 1 , Steps 1 -5.
Step 6: 2-(4-fluorophenyl)-N-methoxy-6-fmethyl(methylsulfonyl)amino]-5^henyl-l-bemofuran- 3-carboxamide
The product of Step 5 (50 mg, 0.1 1 mmol), HOBT (24.5 mg, 0.16 mmol) and EDCI (52 mg, 0.27 mmol) were dissolved in dry DMF (2 mL). The resulting solution was stirred for 30 minutes. Then, O-methylhydroxylamine (HCl salt, 36 mg, 0.44 mmol) and Et3N (50 mg, 0.47 mmol) were added to the mixture. After stirred overnight, the mixture was diluted
with H20 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-TLC to give pure product (20 mg, yield: 40%).
1H-NMR (400 MHz, CDC13) δ 8.26-8.27 (m, IH), 7.70-7.87 (m, 2H), 7.56 (s, I H), 7.41 (s, I H), 7.34-7.39 (m, 5H), 7.12-7.16 (m, 2H), 3.78 (s, 3H), 3.10 (s, 3H), 2.45 (s, 3H). MS (M+H)+: 469.
Examples 8-12
Examples 8-12 were prepared according to the general procedures of Example 7.
Example 13: 6-f(cyclohexylmethyl)(methylsulfonyl)ain-noi-2-(4-fluorophenyl)-/V-methyl-5- phenyl-l-benzofuran-3-carboxamide
Steps 1-3
Steps 1 -3 were performed in accordance with Example 1 , Steps 1 -3.
Ste 4: 2-(4-fluoro henyl)-6-[ (meth lsulfonyl)amino]-5-phenyl-l-benzofuran- -carboxylic acid
The compound prepared in Step 3 (1.3 g, 2.74 mmol) was dissolved in 1 ,4- dioxane (7 mL) and H20 (7 mL). Then, LiOH (1.14, 27.4 mmol) was added to the solution, and the mixture was refluxed for 2 hours. After acidified with HCI (1 N) and extracted with EtOAc, the combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated to give the carboxylic acid (990 mg, yield: 85%). It was used for the next step without further purification.
Step 5: 2-(4-fluorophenyl)-N-methyl-6-[ (methylsulfonyl) amino) '-5-phenyl- 1 -benzofuran-3- carboxamide
The carboxylic acid prepared in Step 4 (990 mg, 2.34 mmol), HOBT (631 mg, 4.7 mmol) and EDCI (900 mg, 4.7 mmol) were dissolved in dry DMF (10 mL). The resulting
solution was stirred for 30 minutes. Then, methanamine (HC1 salt, 640 mg, 9.4 mmol) and Et3N (2 mL) were added to the mixture. After stirred overnight, the mixture was diluted with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by column to give pure 2-(4- fluorophenyl)-N-methyl-6-[(methylsulfonyl)amino]-5-phenyl-l -benzofuran-3-carboxamide (900 mg, yield: 88%).
Ή-NMR (400 MHz, CDC13) δ 7.82-7.86 (m, 2H), 7.79 (s, 1H), 7.63 (s, 1 H), 7.41-7.46 (m, 3H), 7.27-7.33 (m, 2H), 7.10-7.44 (m, 2H), 6.51 (br, 1H), 5.84 (br, 1 H), 2.91 (d, J = 4.8 Hz, 3H), 2.80 (s, 3H). MS (M+H)+: 439.
Step 6: 6-f fcvclohexylmethyl)fmethylsulfonyI)amino]-2-f4-fIuorophenyl)-N-methyl-5-phenyl-l- benzofuran-3-carboxamide
The compound prepared in Step 5 (35 mg, 0.08 mmol), (bromomethyl) cyclohexane (21 mg, 0.12 mmol), 2C03 (22 mg, 0.16 mmol), I (2 mg) in DMF (2 mL) was stirred at 90°C for 16 hours under N2. The mixture was concentrated, diluted with DCM, washed with brine, dried over Na2S04, filtered and the solvent was evaporated. The residue was purified by prep-HPLC to give pure product (15 mg, yield: 35%).
Ή-NMR (400 MHz, CDC13) δ 7.97-7.93 (m, 2H), 7.73 (s, 1H), 7.58 (s, 1 H), 7.52-7.50 (m, 2H), 7.44-7.37 (m, 3H), 7.24-7.16 (m, 2H), 5.84 (s, 1H), 3.18-3.13 (m, 1 H), 2.99-2.97 (m, 4H), 2.95 (s, 3H), 1.74-1.58 (m, 1 H),1.54~1.51 (m, 2H), 1.43-1.41 (m, 2H), 1 .04-0.91 (m, 4H), 0.89-0.79 (m, 2H), 0.76-0.56 (m, 1H). MS (M+H)+: 535.
Examples 14-68
Examples 14-68 were prepared according to the general procedures of
3-carboxamide 3H), 2.91 (d, =8.0 Hz, 3H).
Example 69: 2-(4-fluorophenyl)-N-methyl-5-phenyl-6-[(l-phenylethyl)aminol-l- benzofuran-3-carboxamide
Steps 1-3
Steps 1 -3 were performed in accordance with Example 1 , Steps 1 -3.
Step 4: ethyl 2- -fl orophenyl)-5^henyl-6-[(l-phenylethyl)amino]-l-benzofuran-3-carboxylate
A mixture of the product of Step 3 (400 mg, 1.06 mmol), (1-bromoethyl) benzene (197 mg, 1.06 mmol) and Cs2C03 (7.8 g, 24 mmol) in dry DMF (100 mL) was stirred at 140°C for 4 hours. After the mixture was concentrated, the residue was diluted with DCM, washed with water, dried over Na2S04 and concentrated. The residue was purified by prep-TLC to give the product (200 mg, yield: 39%).
Ή-NMR (400 MHz, CDC13) δ 7.90-7.88 (m, 2H), 7.62 (s, 1 H), 7.47-7.45 (m, 2H), 7.45-7.44 (m, 1 H), 7.26-7.25 (m, 5H), 7.18-7.17 (m, 2H), 7.04-7.03 (m, 2H), 6.45 (s. 1 H),
4. 42-4.41 (m, I H), 4 28-4.26 (q, J = 8.0 Hz, 2H), 1.36-1 .34 (d, J = 8.0 Hz, 3H), 1 .26-1 .24 (t, J = 8.0 Hz, 3H). MS (M+H)+: 480.
Step 5: 2-(4-fluorophenyl)-5^henyl-6-f(l^henylethyl)aminol-l-benzofuran-3-carboxylic acid
The product ( 1 10 mg, yield: 58.4%) was prepared in an analogous manner to
Example 1 3 using the general procedure in Example 1 3, Step 4. The crude product was used in the next step without further purification.
1 H-NMR (400 MHz, CDC13) δ 7.93-7.89 (m, 2H), 7.70 (s, I H), 7.46-7.45 (m, 4H), 7.42-7.40 (m, I H), 7.38-7.35 (m, 4H), 7.07-7.03 (m, 3H), 6.50 (s, I H), 4.44-4.39 (m, I H), 1 .37-1 .36 (d, J = 4.0 Hz, 3H). MS (M+H)+: 452.
Step 6: 2-(4-fluorophenyl)-N-methyl-5^henyl~6-[(l^henylethyl)amino]-l-benzofuran-3- carboxamide
Example 69 (20 mg, yield: 48.6%) was prepared according to the general procedure in Example 1 , Step 6.
1 H-NMR (400 MHz, CDC13) δ 7.82-7.78 (m, 2H), 7.45-7.44 (m, 4H),
7.36~7.35(m, 2H), 7.27-7.25 (m, 4H), 7.18-7.16 (m, 2H), 7.05-7.01 (m, 2H), 6.48 (s, I H), 5.72 (s, I H), 4.44-4.39 (m, I H), 2.89-2.87 (s, 3H), 1 .37-1 .35 (d, J = 8.0 Hz, 3H). MS (M+H)+: 465.
Example 70: 2-(4-fluorophenyl)-N-methyl-6-({2-fmethvKphenyl)aminolethvnainino)-5- phenyl-l-benzofuran-3-carboxamide
Example 70 was prepared according to the general procedures of Example 69.
Example 71: 2-f4-fluorophenyl)-N-methyl-6-[methyl(l-phcnylethyl)amino1-5-phenyl-l- benzofuran-3-carboxamide
Steps 1-4
Steps 1 -4 were performed in accordance with Example 69, Steps 1 -4.
Step 5: ethyl 2-f4-fluorophenyl)-6-imethylfJ^henylethyl)aminoJ-5^henyl-l-benzofuran-3- carboxylate
The product of Step 4 (62 mg, 0.13 mmol), CH3I (29 mg, 0.20 mmol), K2C03 (37 mg, 0.27 mmol) in DMF (2 mL) was stirred at 90°C for 16 hours. The mixture was quenched with water, diluted with DCM, dried over Na2S04, filtered, and the solvent was evaporated. The residue was purified by prep-TLC to give pure compound product (49 mg, yield: 77.7%) as a yellow solid.
Ή-NMR (400 MHz, CDC13) δ 7.90-7.88 (m, 2H), 7.62 (s, 1 H), 7.47-7.45 (m,
2H), 7.45-7.44 (m, 1 H), 7.26-7.25 (m, 5H), 7.18-7.17 (m, 2H), 7.04-7.03 (m, 2H), 6.45 (s, 1 H), 4.33-4.28 (q, J = 2.0 Hz, 2H), 4.17-4.12 (m, 1 H), 2.50 (s, 3H), 1 .32-1 .27 (t, J = 2.0 Hz, 3H), 1 .32-1 .34 (d, J = 0.8 Hz, 3H). MS (M+H)+: 494.
Step 6: 2-(4-fiuorophenyl)-6-fmethyl(l-phenylethyl)amino]-5-phenyl-]-benzofuran^
The carboxylic acid (75 mg, yield: 90 %) was prepared in an analogous manner to Example 13 using the general procedure in Example 13, Step 4. The carboxylic acid was used in the next step without further purification.
Ή-NMR (400 MHz, CDC13) δ 7.90-7.88 (m, 2H), 7.62 (s, 1 H), 7.47-7.45 (m,
2H), 7.45-7.44 (m, 1H), 7.26-7.25 (m, 5H), 7.18-7.17 (m, 2H), 7.04-7.03 (m, 2H), 6.45 (s, 1 H), 4. 17-4.12 (m, 1H), 2.50 (s, 3H), 1.32-1.34 (d, J = 0.8 Hz, 3H). MS (M+H)+: 466.
Step 7: 2-(4-fluorophenyl)-N-methyl-6-fmethyl(l-phenylethyl)amino]-5-phenyl-I-benzofuran-3- carboxamide
The product (30 mg, yield: 38.9%) was prepared according to the general procedure in Example 1 , Step 6.
Ή-NMR (400 MHz, CDC13) δ 7.86-7.83 (m, 2H), 7.66 (s, 1 H), 7.50-7.45 (m, 4H), 7.34-7.33 (m, 2H), 7.25-7.20 (m, 2H), 7.17-7.13 (m, 2H), 6.95-6.93 (m, 2H), 6.96 (s, 1 H), 4.55 (m, 1H), 2.93 (s, 3H), 2.85 (s, 3H), 1.35 (s, 3H). MS (M+H)+: 479.
Example 72; ethyl [2-(4-fluorophenyl)-3-(methylcarbamoyl)-5-phenyl-l-benzofuran-6- yll {2-|methyl(phenyl)aminolethyl)carbamate
Steps 1-3
Steps 1 -3 were performed in accordance with Example 1 , Steps 1 -3.
Step 4: ethyl 6-i(ethoxycarbonyl)amino]-2-(4-fluorophenyl)-5^henyl-l-benzofuran-3- carboxylate
A mixture of the product of Step 3 (64 mg, 0.17 mmol), EtOOCCl (22 mg, 0.21 mmol), Py (23 mg, 0.31 mmol) in DCM (3 mL) was stirred at RT for 2 hours. The mixture was quenched with H20, diluted with DCM, dried over Na2S04, filtered, and the solvent was evaporated. The residue was purified by prep-TLC to give pure carbamate (63 mg, yield: 83.3%) as a white solid.
Ή-NMR (400 MHz, CDC13) δ 8.02-8.00 (m, 2H), 7.78 (s, 1 H), 7.49-7.47 (m, 2H), 7.45-7.34 (m, 3H), 7.14-7.09 (m, 2H), 6.67 (m, 1 H), 4.34-4.30 (q, J = 1.6 Hz, 2H),
4.16-4.1 1 (q, J = 2.0 Hz, 2H), 2.18-2.14 (t, J = 1.6 Hz, 3H), 2.13-1 .98 (t, J = 2.0 Hz, 3H). MS (M+H)+: 448.
Step 5: ethyl 6-f(ethoxycarbonyl){2-[methyl(phenyl)amino]ethyl}amino]-2-(4-fluorophenyl)-5- phenyl- -benzofuran-3-carboxylate
The product of Step 4 (474 mg, 1.06 mmol), 2-(methyl(phenyl)amino)ethyl methanesulfonate (243 mg, 1.06 mmol) and Cs2C03 (7.8 g, 24 mmol) in dry DMF (100 mL) was stirred at 140°C for 4 hours. After the mixture was concentrated, the residue was diluted with DCM, washed with water, dried over Na2S04 and concentrated. The residue was purified by prep-TLC to give the desired amino carbamate (335 mg, yield: 54.6%). MS (M+H)+: 581 .
Step 6: 6-f (ethoxycarbonyl)(2-imethyl(phenyl)aminolethyl}amino]-2-(4-fluorophenyl)-5-
The product of Step 5 (25 mg^ yield: 90%) was prepared in an analogous manner to Example 13 using the general procedure in Example 13, Step 4. The carboxylic acid was used directly in the next step without further purification.
Step 7: ethyl [2-(4-fluorophenyl)-3-(methylcarbamoyl)-5-phenyl-l-benzofuran-6-yl]{2- f methyl (phenyl) amino ] ethyl } carbamate
Example 72 ( 1 5 mg, yield: 48.7%) was prepared according to the general procedure in Example 1 , Step 6.
Ή-NMR (400 MHz, CDC13) δ 7.89-7.87 (m, 2H), 7.65 (s, 1 H), 7.40-7.36 (m, 2H), 7.32-7.20 (m, 6H), 7.19-7.1 8 (m, 3H), 7.15-7. 10 (m, 2H), 6.09 (m, 1 H), 4.09-4.04 (m, 2H), 3.35-3.36 (m, 2H), 3.19-3.07 (m, 2H), 2.97-2.89 (m, 6H), 1 .21 -1.10 (m, 3H). MS (M+H)+: 566.
Example 73: 2-(4-fluorophenyl)-N-methyl-6-|(N-methyl-/V-phenylglvcyl)aminol-5-phenyl- l-benzofuran-3-carboxamide
Steps 1-2
Steps 1 -2 were performed in accordance with Example 1 , Steps 1 -2.
Step 3: ethyl 2-(4-fluorophenyl)-6-f(N-methyl-N-phenylslycyl)amino]-5-phenyl-l-benzofuran-3- carboxylate
The amide (75 mg, yield: 50%) was prepared from the product of Step 2 according to the general procedure in Example 1 , Step 6.
1 H-NMR (400 MHz, CDC13) δ 8.41-8.48 (m, 2H), 8.01-8.09 (m, 2H), 7.78 (s, I H), 7.01-7.15 (m, 8H), 6.71-6.75 (m, IH), 6.50 (t, J = 12.0 Hz, 2H), 4.31-4.35 (m, 2H), 3.24 (s, 3H), 2.61 (m, 2H), 1.30-1.33 (t, J = 12.0 Hz, 3H). MS (M+H)+: 523.
Step 4: 2-(4-fluorophenyl)-6-f(N-methyl-N^henylglycyl)aminoJ-5-phenyl-l-benzofuran-3- carboxylic acid
The carboxylic acid (50 mg, yield: 75%) was prepared in an analogous manner to Example 13 using the general procedure in Example 13, Step 4. The carboxylic acid was used in the next step without further purification.
Step 5: 2-(4-fluorophenyl)-N-methyl-6-f (N-methyl-N-phenylglycyl)amino]-5-phenyl-l- benzofuran-3-carboxamide
The amide (35 mg, yield: 78%) was prepared according to the general procedure in Example 1 , Step 6.
1 H-NMR (400 MHz, CDC13) δ 8.85 (s, 3H), 8.71 (s, 3H), 7.81-7.89 (m, 2H), 7.55
(s, I H), 7.23-7.25 (m, 5H), 7.01-7.12 (m, 2H), 6.71-6.75 (m, I H), 6.50 (d, J = 12.0 Hz, 2H), 5.71-5.75 (m, 2H), 3.78 (s, 3H), 2.58 (s, 3H). MS (M+H)+: 508.
Example 74: 2-(4-fluorophenyn-N-incthvI-6-[methyl(N-methyl-N-phenylglvcyl)aminol-5- phenyl-l-benzofuran-3-carboxamide
Steps 1-3
Steps 1 -3 were performed in accordance with Example 73, Steps 1 -3.
Step 4: ethyl 2-(4-fluorophenyl)-6-fmethyl(N-methyl-N-phenylglycyl)amino]-5-phen l-l- benzof ran-3-carboxylate
The alkylated amide (90 mg, yield: 90%) was prepared in an analogous manner to the compound prepared in Example 1 , Step 4.
Ή-NMR (400 MHz, CDC13) δ 8.09 (s, 1 H), 8.01-8.05 (m, 2H), 7.36-7.45 (m, 6H), 7.13-7.18 (m, 2H), 6.96-7.02 (m, 2H), 6.53-6.61 (m, 1 H), 6.53-6.61 (t, J = 4.0 Hz, 2H), 4.31-4.39 (m, 2H), 3.58-3.66 (m, 2H), 3.24 (s, 3H), 2.70 (s, 3H), 1.30-1.33 (t, J= 12.0 Hz, 3H). MS (M+H)+: 537.
Step 5: 2-(4-fluorophenyl)-6-[methyl(N-methyl-N-phenylzlycyl)amino]-5-^
3-carboxylic ac
The carboxylic acid (85 mg, yield: 95%) was prepared in an analogous manner to Example 13 using the general procedure in Example 13, Step 4. The carboxylic acid was used in the next step without further purification.
Step 6: 2-(4-fluorophenyl)-N-methyl-6-[methyl(N-methyl-N^henylglycyl)am
benzofuran-3-carboxamide
The amide was prepared in an analogous manner to Example 1 , Step 6 (25 mg, yield: 68%).
Ή-NMR (400 MHz, CDC13) δ 7.89-7.91 (m, 2H), 7.86 (s, 1 H), 7.39-7.42 (m, 4H), 7.34-7.38 (m, 2H), 7.13-7.18 (m, 2H), 7.00-7.09 (m, 2H), 6.55-6.57 (m, 1 H), 6.16 (d, J = 4.0 Hz, 2H), 5.71-5.73 (m, 1H), 3.48-3.56 (m, 2H), 3.24 (s, 3H), 2.94 (d, J = 8.0 Hz, 3H), 2.69 (s, 3H). MS (M+H)+: 522.
Examples 75-76
Examples 75 and 76 were prepared according to the general procedures of
Example 74.
Example 77: 2-(4-fluorophenyl)-N-methyl-6-f(4S,5R)-4-methyl-2-oxo-5-phenyl-l,3- oxazolidin-3-yl1-5-phenyl-l-benzofuran-3-carboxamide
Steps 1-3
Steps 1-3 were performed in accordance with Example 1 , Steps 1 -3.
Step 4: ethyl 2-( -fluorophenyl)-6-iodo-5-phenyl-l-benzofuran-3-carboxylate
A solution of the product of Step 3 (100 mg, 0.27 mmol) in 30% H2SO4 aqueous solution was cooled at 0°C. Then the solution of NaN02 in 1 mL H20 was added dropwise to amine solution over a period of 1 minute with keeping the temperature at 0°C. The resulting mixture was stirred for an additional 30 minutes at 0°C. An aqueous solution of Kl was added dropwise over 5 minutes. The reaction mixture was stirred for 3 hours at RT, giving a dark
brown solution. The solution was extracted with EtOAc. The organic layer was washed with Na2S03 solution and concentrated to give the crude iodide (40 mg, yield: 31 %).
Ή-NMR (400 MHz, CDC13) δ 8.12 (s, 1 H), 8.06-8.10 (m, 2H), 7.99 (s, 1 H), 7.38-7.48 (m, 5H), 7.17-7.22 (m, 2H), 4.39 (q, J = 7.2 Hz, 2H), 1.35(t, J = 7.2 Hz, 3H). MS (M+H)+: 487.
Step 5: ethyl 2-(4-fluorophenyl)-6-[(4S,5R)-4-methyl-2-oxo-5-phenyl-l,3-oxazolidin-3-yl]-5- phenyl-1 -benzofuran-3-carboxylate
The iodide (30 mg, 0.06 mmol), (4S, 5R)-4-methyl-5-phenyloxazolidin-2-one (17 mg, 0.9 mmol), Cul (15 mg, 0.08 mmol) and K2C03 (20 mg, 0.14 mmol) in dry nitrobenzene (1 mL) was heated to 180°C for 6 hours. When TLC showed the reaction was completed, H20 was added to the mixture and the aqueous phase was extracted by EtOAc. The combined organic phase was washed with brine, dried over Na2S04 and concentrated under reduced pressure. The residue was purified by prep-TLC to give the N-aryl oxizolidinone (10 mg, yield: 30%).
Ή-NMR (400 MHz, CDC13) δ 8.00-8.02 (m, 3H), 7.99 (s, 1 H), 7.39-7.55 (m, 5H), 7.07-7.27 (m, 7H), 5.26 (d, J= 8.0 Hz, 1H), 4.33 (q, J = 7.2 Hz, 2H), 3.61 (br s, 1 H), 1.31 (t, J = 7.2 Hz, 3H), 0.45 (d, J = 6.8 Hz, 3H). MS (M+H)+: 536.
Step 6: 2-(4-fluorophenyl)-6-[(4S,5R)-4-methyl-2-oxo-5-phenyl-l13-oxazolidin-3-yl]-5-phenyl-l- benzofuran- -carboxylic acid
To a stirred solution of ester (40 mg, 0.07 mmol) in dioxane/H20 (1 : 1 , 2 mL) was added LiOH (20 mg, 0.48 mmol), and the mixture was stirred at 100°C for 3 hours. The mixture was concentrated in vacuo. The residue was dissolved in H20, I HCl was added until pH to 3, and the mixture was extracted with EtOAc. The organic solvent was washed with brine, dried over Na2S04 and filtered, and the solvent was evaporated. The solvent was removed by
distillation to provide the crude carboxylic acid (35 mg, yield: 92%). It was used for the next step without further purification.
Step 7: 2-(4-fluorophenyl)-N-methyl-6-[(4S, 5R)-4-methyl-2-oxo-5-phenyl-l , 3-oxazolidin-3-yll- 5 -phenyl- -benzofuran-3 -car boxamide
A solution of carboxylic acid (35 mg, 0.07 mmol), HOBT (40 mg, 0.30 mmol) and EDCI (50 mg, 0.32 mmol) in dry DMF (2 mL) was stirred at RT. After 30 minutes, Et3N (0.2 mL) and CH3NH2 (HC1 salt, 40 mg, 0.59 mmol) was added to the mixture, and the mixture was stirred overnight. After the solvent was removed, H20 was added, and the mixture was extracted with EtOAc. The combined organic layer was washed with H20, brine and
concentrated. The residue was purified by prep-TLC to give the product of Example 77 (20 mg,. yield: 56%).
Ή-NMR (400 MHz, CDC13) δ 7.86-7.89 (m, 2H), 7.74 (s, 1 H), 7.52 (s, 1 H), 7.40-7.42 (m, 5H), 125-1.26 (m, 3H), 7.06-7.14 (m, 4H), 5.84 (br s, 1 H), 5.25 (d, J = 8.0 Hz, l H), 3.62 (br s, 1 H), 2.91 (d, J = 4.8 Hz, 3H), 0.44 (d, J = 6.8 Hz, 3H). MS (M+H)+: 521 .
Example 78: 5-(2-fluorophenvn-2-(4-fluorophenyl)-N-niethyl-6- |niethyl(methylsulfonyl)aininol-l-benzofuran-3-carboxamide
Step 1: ethyl 5-(2-fl orophenyl)-2-(4-fl orophenyl)-6-nitro-l-benzofuran-3-carboxylate
2-Fluorophenylboronic acid (obtained according to procedure in
WO 2004/041201 A2; 283 mg, 2.10 mmol) and Κ3Ρ0 ·3Η20 (556 mg, 2.10 mmol) were added to a suspension of triflate (described in Example 1 ) (500 mg, 1 .05 mmol) in dry DMF (2 mL) under N2. Then Pd(dppf)Cl2 (5 mg, 0.08 mmol) was added to the mixture under N2. The
reaction mixture was heated to 80°C for 6 hours. The mixture was cooled, diluted with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by column to give pure aryl fluoride (250 mg, yield: 55%).
Ή-NMR (400 MHz, CDC13) 8 8.02 (s, 1 H), 8.00-8.01 (m, 3H), 7.31-7.35 (m,
2H), 7.20-7.22 (m, 3Ή), 7.03-7.05 (m, 1 H), 4.30-4.36 (dd, J = 8.0 Hz, 2H), 1.27-1.31 (m, 3H) MS (M+H)+: 424.
A mixture of nitro arene (250 mg, 0.59 mmol), Fe (264 mg, 4.70 mmol) and NH4CI (475 mg, 8.85 mmol) in H20/MeOH/THF (2 mL/2 mL/2 mL) was refluxed for 3 hours. Then, H20 was added to quench the reaction, which was filtered and extracted with EtOAc, washed with brine and dried over Na2S04. The solvent was removed by distillation. After purification by column, the desired aninline was obtained (180 mg, yield: 77%).
Ή-NMR (400 MHz, CDC13) δ 7.94-7.97 (m, 2H), 7.74 (s, 1 H), 7.32-7.35 (m, 2H), 7.05-7.20 (m, 4H), 6.67 (s, 1H), 4.26-4.30 (dd, J = 8.0 Hz, 2H), 1.18-1 .27 (m, 3H). MS (M+H)+: 394.
Step 3: ethyl 5-(2-fluorophenyl)-2-(4-fluorophenyl)-6-[(methylsulfonyl)amino]-l-benzofuran-3- carboxylate
MsCl (65 mg, 0.60 mmol) was added to a solution of aniline (180 mg, 0.50 mmol) and pyridine (79 mg, 1.00 mmol) in dry DCM (2 mL). The reaction mixture was stirred overnight at RT. After diluted with H20 and extracted with DCM, the mixture was washed with brine, dried over Na2S04 and filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by prep-TLC to give sulfonamide (150 mg, yield: 75%).
Ή-NMR (400 MHz, CDC1
3) δ 7.94-7.97 (m, 2H), 7.74 (s, 1 H), 7.71 (s, 1 H), 7.32-7.35 (m, 2H), 7.05-7.20 (m, 4H), 4.26-4.30 (dd, J = 8.0 Hz, 2H), 2.95 (s, 3H), 1 .18-1 .27 (m, 3H). MS (M+H)
+: 472.
Step 4: ethyl 5-(2-fluorophenyl)-2-(4-fluorophenyl)-6-fmethyl(methylsulfonyl)amino]-l- benzofuran-3-carboxylate
I (4 mg, 0.02 mmol), 2C03 (60 mg, 0.40 mmol), and CH3I (1 13 mg, 0.80 mmol) were added to a solution of sulfonamide (100 mg, 0.20 mmol) in dry DMF (5 mL) under N2. The mixture was heated to 80°C overnight. The mixture was cooled, diluted with H20, and extracted with EtOAc; the organic solvent was washed with brine, dried over Na2S04 and filtered; and the solvent was evaporated under reduced pressure. The crude was purified by prep-TLC and the desired alkyl sulfonamide was obtained (90 mg, yield: 87%).
Ή-NMR (400 MHz, CDC13) δ ppm 8.03-8.05 (m, 2H), 8.01 (s, 1 H), 7.63 (s, 1 H), 7.37-7.44 (m, 2H), 7.12-7.27 (m, 4H), 4.34-4.40 (dd, J = 8.0 Hz, 2H), 3.23 (s, 3H), 2.48 (s, 3H), 1.34-1.36 (m, 3H). MS (M+H)+: 486.
Step 5: 5-(2-fluorophenyl)-2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)ami
The ester (90 mg, 0.20 mmol) was dissolved in 1 ,4-dioxane (2 mL) and H20 (2 mL). Then LiOH (84 mg, 2.00 mmol) was added to the solution, and the mixture was refluxed for 2 hours. After acidified with HCl (1 N) and extracted with EtOAc, the combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated to give the carboxylic acid (80 mg, yield: 90%). It was used for the next step without further purification. Step 6: 5-(2-fluorophenyl)-2-(4-fluorophenyl)-N- ethyl-6-[methyl(methyls lfonyl)amino]-l- benzofuran-3-carboxamide
The carboxylic acid (75 mg, 0.16 mmol), HOBT (37 mg, 0.24 mmol) and EDC1 (77 mg, 0.40 mmol) were dissolved in dry DMF (2 mL). The resulting solution was stirred for 30 minutes. Then, methanamine HCl salt (43 mg, 0.64 mmol) and Et3N (73 mg, 0.72 mmol) was
added to the mixture. After stirred overnight, the mixture was diluted with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-TLC to give pure Example 78 (35 mg, yield: 47%).
Ή-NMR (400 MHz, CDCI3) δ 7.88-7.92 (m, 2H), 7.74 (s, 1H), 7.60 (s, 1 H), 7.34-7.40 (m, 2H), 7.10-7.24 (m, 4H), 5.92 (s, 1 H), 3.20 (s, 3H), 2.94-2.95 (d, J = 4.0 Hz, 3H), 2.47 (s, 3H). MS (M+H)+: 471
Examples 79-89
Examples 79-89 were prepared according to the general procedures of
Example 78.
carboxamide
Example 90: 5-(2-fluorophenyl)-2-(4-fluorophenyl)-N-methoxy-6- [methyl(methylsulfonyl)aniino1-l-benzofuran-3-carboxainide
Steps 1-5
Steps 1 -5 were performed in accordance with Example 78, Steps 1 -5.
Step 6: 5-(2-fluorophenyl)-2-(4-fluorophenyl)-N-methoxy-6-[methyl(methylsulfo
Example 90 was prepared using conditions analogous to the coupling reaction described in Example 7, Step 6 (40 mg, yield: 51 %).
Ή-NMR (400 MHz, CDC13) δ 8.43 (s, 1 H), 7.90-7.93 (m, 2Η), 7.74 (s, 1 H), 7.62 (s, 1 H), 7.36-7.38 (m, 2H), 7.13-7.25 (m, 4H), 3.83 (s, 3H), 3.21 (s, 3H), 2.46 (s, 3H). MS (M+H)+: 487.
Examples 91-98
Examples 91 -98 were prepared according to the general procedures of
Example 90.
Ή-NMR (400 MHz, CDC13) MS
Example Structure Name
δ (-M+H)
7.56 (s, 1 H), 7.81 -7.85 (m,
2H), 7.62 (s, 1 H), 7.55 (s,
2-(4-fluorophenyl)-/V-
1 1 H), 7.30-7.35 (m, 1 H),
methoxy-5-(2- 7.2 1-7.24 (m, I H),
methoxyphenyl)-6-
91 7. 10-7. 1 8 (m, I H),
[methyl 450
6.95-7.01 (m, I H),
(methylsulfonyl)amino]
1 6.89-6.91 (d, .7 = 4.0 Hz, o=s=o - l -benzofuran-3- I H), 3.80 (s, 3H), 3.70 (s,
' 1 carboxamide
3H), 3. 1 1 (s, 3H), 2.34 (s,
carboxamide 3H).
Example 99: 2-(4-fluorophenyl)-7V-niethoxy-5-i3-(methoxycarbannovnphenyll-6- [methv methylsulfonyl)amino]-l-benzofuran-3-carboxamide
Steps 1-4: ethyl 5-(3-cyanophenyl)-2-(4-fluorophenyl)-6-[methyl(methyls lfonyl)amino]-l- benzofuran-3-carboxylate
Steps 1 -4 were performed in an analogous manner to Example 1 , Steps 1 -4.
Step 5: 5-(3-cyanophenyl)-2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)aminol-l-benzofuran-3- carboxylic acid and 5-(3-carboxyphenyl)-2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)aminol-l- benzofuran-3 -carboxylic acid
The ester (450 mg, 0.92 mmol) was dissolved in dioxane (5 mL). Then LiOH (96 mg, 4 mmol) was added to the solution, and the mixture was stirred at RT overnight. After acidifing with HCl (1 N) and extracting with EtOAc, the combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated to give the cyano carboxylic acid (300 mg, yield: 50%) and dicarboxylic acid (100 mg, yield: 30%). The crude mixture was used for the next step without further purification.
Step 6: 2-(4-fluorophenyl)-N-methoxy-5-[3-(methoxycarbamoyl)phenyl]-6- [ methyl (methylsulfonyl) amino) '-] ' -benzofuran-3-carboxamide
Example 99 was prepared using condition analogous to the coupling reaction described in Example 7, Step 6 (55 mg, yield: 73%).
1 H-NMR (400 MHz, CDC13) δ 9.49-9.54 (m, 1 H), 8.39 (s, 1 H), 7.86-7.89 (m, 2H), 7.83-7.85 (m, 2H), 7.79 (s, 1H), 7.45-7.51 (m, 3H), 7.13-7.17 (m, 2H), 3.81-3.82 (m, 6H), 2.99 (s, 3H), 2.78 (s, 3H). MS (M+H)+: 542.
Example 100: 2-(4-fluorophenyl)-N-inethyl-5-f3-(methylcarbamovnphenyll-6- [methyl(methylsulfonyl)aminol-l-benzofuran-3-carboxamide
Steps 1-5: 5-(3-carboxyphenyl)-2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)aminol-l- benzofuran-3-carboxylic acid
Steps 1-5 were performed according to the general procedures in Example 99,
Steps 1 -5.
Step 6: 2-(4-fluorophenyl)-N-methyl-5-[3-(methylcarbamoyl)phenyl]-6- f methvHmethylsul fonyl)amino ]-l -benzofuran-3-carboxamide
Example 100 was prepared according to the general procedure in Example 1 ,
Step 6.
1 H-NMR (400 MHz, CDC13) δ 7.86-7.89 (m, 2H), 7.78-7.81 (m, 2H), 7.44-7.51 (m, 3H), 7.12-7.16 (t, J = 12.0 Hz, 2H), 6.72-6.73 (m, 1 H), 5.81-5.82 (m, 1H), 2.92-2.95 (m, 6H), 2.90 (s, 3H), 2.84 (s, 3H). MS (M+H)+: 510.
Example 101 : 5-[3-(aminomethyl)phenyl]-2-(4-fluorophenyl)-7V-methyl-6- |methyl(methylsulfonyl)aminol-l-benzofuran-3-carboxamide
Step 1: 5-[3-(aminomethyl)phenyl -2-(4-fluorophenyl)-N-methyl-6-
Raney-Ni (100 mg) and ammonia (cone. 0.5 mL) were added to a solution of the compound of Example 84 (58 mg, 0.13 mmol) in MeOH (20 mL). And then the mixture was degassed and stirred under 30 psi of H2 overnight at RT. After filtered through CELITE, the filtrate was concentrated to give the desired benzylic amine (50 mg, yield: 85%).
Ή-NMR (400 MHz, CDC13) δ 7.79-7.82 (m, 2H), 7.57 (s, 1 H), 7.29 (d, J = 8.0 Hz, 2H), 7.06-7.10 (t, J = 16.0 Hz, 2H), 6.58-6.59 (m, 3H), 3.98 (s, 2H), 2.93 (s, 3H), 2.71 (d, J = 4.0 Hz, 3H), 2.49 (s, 3H). MS (M+H)+: 482.
Example 102: 2-(4-fluorophenyl)-/V-methyl-6-[methvKinethylsulfonyl)aitiino]-5-(3-
{[(methylsulfonyl)aminolmethyl)phenyl)-l-benzofuran-3-carboxamide
Steps 1-2
Steps 1 -2 were performed according to the general procedures in Example 1 ,
Steps 1 -2.
Step 3: 2-(4-fluorophenyl)-N-methyl-6-[methyl(methylsulfonyl)amino]-5-(3- { [(methylsulfonyl)amino] methyljphenyl)- 1 -benzofuran-3-carboxamide
Example 102 was prepared in an analogous manner to the sulfonamide synthesis described in Example 1, Step 3 (20 mg, yield: 60%).
1 H-NMR (400 MHz, CDC13) δ 7.85-7.88 (m, 2H), 7.76 (s, IH), 7.52 (s, I H), 7.46 (s, IH), 7.36-7.38 (m, I H), 7.28-7.32 (m, 2H), 7.12-7.16 (m, 2H), 5.78-5.79 (m, I H),
4.95-4.96 (m, I H), 4.31 (d, J= 8.0 Hz, 2H), 2.91-2.93 (m, 6H), 2.86 (s, 3H), 2.79 (s, 3H). MS (M+H)+: 560.
Example 103
Example 103 was prepared according to the general procedures of Example 102.
Example 104: 2-(4-fluorophenvn-yV-methyl-6-[methyl(methylsulfonyl)aminol-5-(4- {Kmethylsulfonyl)aminolmethyl|phenyl)-l-benzofuran-3-carboxamide
Step 1: 5-[4-(aminomethyl)phenyll-2-(4-fluorophenyl)-N-methyl-6-
To a solution of the compound of Example 85 (400 mg, 83.8 mmol) in MeOH (10 mL), and Raney-Ni (30 mg) was added. The reaction was degassed and then was shaken under 30 psi H
2 overnight. The reaction mixture was filtered, washed with MeOH. The solvent was evaporated to give the desired benzylic amine (350 mg, yield: 87%).
1 H-NMR (400 MHz, CDC13) 7.82-7.85 (m, 2H), 7.47-7.52 (m, 3H), 7.45 (s, 1 H),
7.31-7.37 (m, 2H), 6.99-7.1 1 (m, 2H), 6.41 (s, 1 H), 4.12 (s, 2H), 2.88 (s, 3H), 2.72 (d, J = 4.0 Hz, 3H), 2.52 (s, 3H). MS (M+H)+: 482.
Step 2: 2-(4-fl orophenyl)-N-methyl-6-[methyl(methylsulfonyl)aminol-5-(4- {[(methylsulfonyl)amino methyl}phenyl)-l-benzof ran-3-carboxamide
Example 104 was prepared in an analogous manner to the sulfonamide prepared in Example 1 , Step 3 (20 mg, yield: 60%).
1 H-NMR (400 MHz, CDC13) 7.85-7.88 (m, 2H), 7.69 (s, 1 H), 7.51 (s, 1 H), 7.37 (s, 4H), 7.14-7.19 (m, 2H), 4.21 (s, 2H), 3.04 (s, 3H), 2.83 (s, 3H), 2.75 (s, 3H) 2.70 (s, 3H). MS (M+H)+: 560.
Examples 105-107
Examples 105-107 were prepared according to the general procedures of
Example 104.
Example 108: 2-(4-fluorophenyl)-N-methyl-6-fmethyl(methYlsulfonyl)ainino]-5-|4- (trifluoromethvnphenyll-l-benzofuran-3-carboxamide
Step 1: ethyl 2-(4-fluorophenyl)-6-nitro-l-benzofuran-3-carboxylate
HCOOH (2.4 g, 71.23 mmol), Bu3N (1 1.6 g, 85.47 mmol) and Pd(PPh3)2Cl2 ( 197 mg, 0.28 mmol) were added to a solution of triflate (obtained according to procedure in WO 2004/041201 A2, 9 g, 28.49 mmol) in DMF (90 mL). The mixture was heated to 1 10°C under N2 protection. After stirred for 0.5 hour, the mixture was diluted with H20 and extracted with ether. The combined organic layers were washed with brine, dried over Na2S04, filtered and the solvent was evaporated. The crude product was purified by column to give pure nitro arene (4.78 g, yield: 51 %).
' H-NMR (400 MHz, CDCl
3) 6 8.36 (d, J = 2 Hz, 1 H), 8.20-8.23 (m, 1 H), 8.1 1 (d, J = 8.8 Hz, 1 H), 8.03-8.07 (m, 2H), 7.13-7.18 (m, 2H), 4.36-4.41 (m, 2H), 1 .37 (t, J = 7.2 Hz, 3H). MS (M+H)
+: 330.
Step 2: ethyl 6-amino-2-(4-fluorophenyl)- 1 -benzofuran-3-carboxylate
A mixture of the product of Step 1 (4.78 g, 14.5 mmol), Fe (4.06 g, 72.6 mmol) and NH4C1 (6.20 g, 1 16 mmol) in H20/MeOH/THF (50 mL /50 mL /50 mL) was refluxed for 4 hours. Then, H20 was added to quench the reaction, and the mixture was extracted with EtOAc. After washing with brine and dried, the solvent was removed by distillation. The pure aniline was obtained (3.47 g, yield: 80%) by prep-TLC.
H-NMR (400 MHz, CDC13) δ 7.94-7.98 (m, 2H), 7.73 (d, J = 8 Hz, 1 H), 7.08 (t,
J = 8.8 Hz, 2H), 6.77 (s, 1H), 6.68 (d, J= 6.8 Hz, 1H), 4.30-4.35 (m, 2H), 1.34 (t, J = 7.2 Hz, 3H). MS (M+H)+: 300.
Step 3: ethyl 2-(4-fluorophenyl)-6-[(methylsulfonyl)ammo]-l-benzofuran-3-carboxylate
MsCl (122 mg, 1.06 mmol) was added to a solution of aniline (200 mg,
0.67 mmol) and pyridine (107 mg, 1.35 mmol) in dry DCM (2 mL). After stirred overnight at RT, the mixture was diluted with H20 and extracted with DCM. The organic layer was washed with brine, dried over Na2S04 and filtered, and the solvent was evaporated. The crude product was purified by prep-TLC to give the desired sulfonamide (200 mg, yield: 78.5%).
Ή-NMR (400 MHz, CDC13) δ 7.97-8.06 (m, 3H), 7.53-7.54 (m, 1 H), 7.1 1 -7.19 (m, 3H), 6.74 (s, 1 H), 4.30-4.35 (m, 2H), 3.93 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H). MS (M+H)+: 378.
Step 4: ethyl 2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)aminol-l-benzofuran-3-carboxylate
NaH (60 % in oil, 1 1 1 mg, 2.78 mmol) and CH3I (395 mg, 2.78 mmol) were added to a solution of sulfonamide (21 1 mg, 0.56 mmol) in dry DMF (4 mL) under N2. After stirred overnight at RT, ice cold diluted AcOH was added, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered, and the
solvent was evaporated under reduced pressure. The crude product was used for the next step without further purification (210 mg, yield: 96%).
Step 5: ethyl 5-bromo-2-(4-fluorophenyl)-6-[methyl(methylsulfonyl)aminol-l-benzofuran-3- carboxylate
A stirred solution of sulfonamide (500 mg, 1.3 mmol) and FeCl3 (210 mg, 0.78 mmol) in dry CC1 (5 mL) was added Br2 (210 mg, 1.3 mmol) in dry CC14 (2 mL). The mixture was allowed to stir at 50°C for 4 hours. The mixture was cooled, diluted with H20, and extracted with DCM; the organic solvent was washed with brine, dried over Na2S04 and filtered; and the solvent was evaporated under reduced pressure. The crude was purified by column chromatography to give aryl bromide (240 mg, yield: 30%).
Ή-NMR (400 MHz, CDC13) δ 8.25 (s, 1H), 7.91-8.05 (m, 2H), 7.62 (s, 1H), 7.02-7.15 (m, 2H), 4.32-4.46 (m, 2H), 3.37 (s, 3H), 3.02 (s, 3H), 1.35 (t, J = 4.4 Hz, 3H). MS (M+H)+: 470.
Step 6: 5-bromo-2-(4-fl orophenyl)-6-[methyl(methylsulfonyl)amino]-l-benzofurari-3- carboxylic acid
The ester (210 mg, yield: 80%) was hydrolysed in an analogous manner to the general procedure of Example 78, Step 5. The carboxylic acid was used in the next step without further purification.
Step 7: 5-bromo-2-(4-fl orophenyl)-N-methyl-6-[methyl(methylsulfonyl)amm^
The amide was prepared according to the general procedure in Example 1 , Step 6 (180 mg, yield: 75%).
Ή-NMR (400 MHz, CDC13) δ 8.09 (s, 1 H), 7.81-7.85 (m, 2H), 7.63 (s, 1 H), 7.12-7.19 (m, 2H), 5.71 (br, 1 H), 3.27 (s, 3H), 3.02 (s, 3H), 2.93 (d, J = 4.4 Hz, 3H). MS (M+H)+: 455.
Step 8: 2-(4-fluorophenyl)-N-methyl-6-[methyl(methylsulfonyl)amino]-5-[4- (trifluoromethyl)phenyl]-l-benzofuran-3-carboxamide
To a solution of 5-bromo-2-(4-fluorophenyl)-N-methyl-6- [methyl(methylsulfonyl)amino]-l -benzofuran-3-carboxamide (30 mg, 0.066 mmol) in DMF (2 mL) were added 4-hydroxy-phenyl boronic acid (21 mg, 0.13 mmol) and Κ3Ρ04·3Η20 (36.5 mg, 0.13 mmol). Then, Pd(dppf)Cl2 (3.4 mg, 0.004 mmol) was added under N2. The resulting mixture was heated to 90°C for 12 hours. The mixture was cooled to RT, then filtered and purified by prep-HPLC to give 2-(4-fluorophenyl)-5-(4-hydroxyphenyl)-N-methyl-6- [methyl(methylsulfonyl)amino]-l -benzofuran-3-carboxamide. (4.8 mg, Yield; 15.5%).
MS (M+H)+: 469.
Examples 109-122
Examples 109-122 were prepared according to the general procedures of
Example 108.
Example 123: 6-{f2-(benzylaiiiino)ethvn(inethylsulfonyl)aiiiino}-2-(4-fluorophenyl)-N- methyl-5-phenyl-l-benzofuran-3-carboxaniide
Steps 1-4
Steps 1 -4 were performed in an analogous manner to Example 1 , Steps 1 -4.
Step 5: ethyl 2-(4-fluorophenyl)-6-[(methylsulfonyl){2-[(methylsulfonyl)oxylethyl}aminol-5- phenyl-l-benzofuran-3-carboxylate
MsCl (0.2 mL, 3.0 mmol) was added to a solution of alcohol (1 g, 2.0 mmol) and Et3N (0.6 mL, 4.0 mmol) in dry DCM (10 mL), in a manner similar to that of Example 1 , Step 4. The reaction mixture was stirred overnight at RT. After dilution with H20 and extraction with DCM, the mixture was washed with brine, dried over Na2S04 and filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by column to give the mesylate (800 mg, yield: 75%).
Ή-NMR (400 MHz, CDC13) δ 8.00-8.03 (m, 2H), 7.99 (s, 1 H), 7.57 (s, 1 H), 7.48-7.50 (m, 2H), 7.35-7.43 (m, 3H), 7.1 1-7.16 (m, 2H), 4.30-4.35 (dd, J= 8.0 Hz, 2H), 4.02-4.05 (m, 2H), 3.21 -3.83 (m, 2H), 2.98 (s, 3H), 2.90 (s, 3H), 1.27-1.30 (m, 3H). MS (M+H)+: 576.
Step 6: ethyl 6-{f2-(benzylamino)ethyll(methylsulfonyl)amino}-2-(4-fluorophenyl)-5-phenyl-l- benzofuran-3-carboxylate
Benzylamine (0.5 mL, 0.27 mmol)) was added to a solution of mesylate (50 mg, 0.09 mmol) in Et3N (1 mL) and MeCN (1 mL). The reaction mixture was stirred overnight at 60°C. After dilution with H20 and extraction with EtOAc, the mixture was washed with brine, dried over Na2S04 and filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by prep-TLC to give the benzylic amine (30 mg, yield: 58%).
Ή-NMR (400 MHz, CDC13) δ 8.00-8.03 (m, 2H), 7.99 (s, 1 H), 7.57 (s, 1 H), 7.48-7.50 (m, 2H), 7.35-7.43 (m, 7H), 7.1 1-7.16 (m, 3H), 4.30-4.35 (dd, J = 8.0 Hz, 2H), 4.02-4.05 (m, 2H), 3.21 -3.83 (m, 2H), 2.98 (s, 3H), 2.32 (d, J = 8.0 Hz, 2H), 1.27-1 .30 (m, MS (M+H)+: 587.
Step 7: 6-{
' [2-(benzylamino)ethyl] '(methyls lfonyl)amino}-2-(4-fluorophenyl)-5-phenyl-l - benzofuran-3-carboxylic acid
The ester (30 mg, 0.05 mmol) was dissolved in 1 ,4-dioxane (1 mL) and H20 (1 mL). Then LiOH (21 mg, 0.5 mmol) was added to the solution, and the mixture was refluxed for 2 hours. After being acidified with HC1 (1 N) and extracted with EtOAc, the combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated to give the carboxylic acid (22 mg, yield: 79%). The acid was used in the next step without further purification.
Step 8: 6-{[2-(benzylamino)ethyll(methylsulfonyl)amino}-2-(4-fluoropheny
Carboxylic acid (22 mg, 0.04 mmol), HOBT (10 mg, 0.06 mmol) and EDCI (19 mg, 0.10 mmol) were dissolved in dry DMF (1 mL). The resulting solution was stirred for 30 minutes. Then, methanamine HC1 salt (1 1 mg, 0.16 mmol) and Et3N (18 mg, 0.18 mmol) was added to the mixture. After stirred overnight, the mixture was diluted with H20 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-HPLC to give pure amide (Example 124) (20 mg, yield: 70%).
Ή-NMR (400 MHz, CDC13) δ 7.87-7.88 (m, 2H), 7.68 (s, 1 H), 7.44 (s, 1 H), 7.38-7.42 (m, 2H), 7.23-7.25 (m, 6H), 7.13-7.19 (m, 4H), 5.87 (s, 1 H), 3.58-3.61 (m, 2H), 3.51 -3.52 (m, 2H), 3.06 (s, 3H), 2.91 (s, 3H), 2.53-2.59 (m, 2H). MS (M+H)+: 572.
Examples 124-132
Examples 124-132 were prepared according to the general procedures of
Example 123.
Example 133: 2-(4-fluorophenyl)-6-[(2-hvdroxyethyl)finethylsulfonyl)a-ninol-7V-methyl-5- phenyl-l-benzofuran-3-carboxamide
Steps 1-3
Steps 1 -3 were performed in accordance with Example 1 , Steps 1 -3.
Step 4: ethyl 2-(4-fluorophenyl)-6-[(2-hydroxyethyl)(methylsulfonyl)amino]-5-phenyl-l-
I (6 mg, 0.036 mmol), K2C03 (46 mg, 0.33 mmol), and 2-bromo ethanol (80 mg, 0.563 mmol) were added to a solution of ethyl 2-(4-fluorophenyl)-6-[(methylsulfonyl)amino]-5- phenyl-l -benzofuran-3-carboxylate (50 mg, 0.131 mmol) in dry DMF under N2 protection. The mixture was stirred at 60°C overnight. After dilution with H20 and extraction with EtOAc, the organic solvent was washed with brine, dried over Na2S0 and filtered, and the solvent was evaporated under reduced pressure. The crude was purified by prep-TLC to give the desired product of ethyl 2-(4-fluorophenyl)-6-[(2-hydroxyethyl) (methylsulfonyl)amino]-5-pheny 1- 1 - benzofuran-3-carboxylate (60 mg, yield: 91 %).
!H-NMR (400 MHz, CDC13) δ 8.00-8.03 (m, 3H), 7.61 (s, 1 H), 7.51 -7.52 (m, 2H), 7.35-7.44 (m, 3H), 7.1 1-7.16 (m, 2H), 4.30-4.36 (m, 2H), 3.21-3.56 (m, 4H), 2.91 (s, 3H), 1.29 (t, J= 7.2 Hz, 3H).
Step 5: 2-(4-fluorophenyl)-6-f (2-hydroxyethyl) (methylsulfonyl) amino j '-5-phenyl-l -benzofuran- 3-carboxylic aci
To a solution of the product of Step 4 (60 mg, 0.12 mmol) in dioxane (1 mL) was added LiOH H20 (40 mg, 0.952 mmol) and H20 (1 mL), and the resultant solution was stirred for 2 hours at 60°C. H20 was added, and then 2N aqueous HC1 was added to adjust pH = 4-5.
After extraction with EtOAc, the combined organic layer was washed with brine, dried over
Na2SC"4, and evaporated to provide the crude product. The crude was purified by prep-TLC.
The desired product of 2-(4-fluorophenyl)-6-[(2-hydroxyethyl)(methylsulfonyl)amino]-5-phenyl- l -benzofuran-3-carboxylic acid was obtained (50 mg, yield: 88%).
Ή-NMR (400 MHz, CDC13) δ 8.05 (s, 1H), 7.99-8.03 (m, 2H), 7.62 (s, 1H),
7.48-7.49 (m, 2H), 7.38-7.43 (m, 3H), 7.1 1-7.15 (m, 2H), 3.19-3.59 (m, 4H), 2.90 (s, 3H).
Step 6: 2-(4-fluorophenyl)-6- [ (2-hydroxyethyl) (methylsulfonyl)amino]-N-methyl-5-phenyl- 1- benzofuran-3-carboxamide
The product of Step 5 (20 mg, 0.043 mmol), HOBT (12 mg, 0.08 mmol) and EDCI (26 mg, 0.13 mmol) were dissolved in dry DMF (1 mL). The resulting solution was stirred for 30 minutes. Then, methanamine (HC1 salt, 7 mg, 0.22 mmol) and Et3N (25 mg, 0.24 mmol) were added to the mixture. After stirring overnight, the mixture was diluted with H20 and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and evaporated. The crude product was purified by prep-TLC to give pure 2-(4-fluorophenyl)-6-[(2-hydroxyethyl)(methylsulfonyl)amino]-jV-methyl-5-phenyl-l - benzofuran-3-carboxamide (10 mg, yield: 48%).
Ή-NMR (400 MHz, CDC13) δ 7.86-7.90 (m, 2H), 7.72 (s, 1 H), 7.59 (s, 1 H), 7.47-7.50 (m, 2H), 7.32-7.40 (m, 3H), 7.10-7.16 (m, 2H), 5.80 (s, 1 H), 3.28-3.47 (m, 4H), 2.90 (s, 6H).
Example 134: 2-(4-fluorophenyl)- V-methyl-6-K2-[methyl(phenyl)aininolethYl}
(methylsulfonyl)aiiiino1-5-phen -l-benzofuran-3-carboxamide
Steps 1-3
Steps 1 -3 were performed in accordance with Example 1 , Steps 1 -3.
Step 4: ethyl 2-(4-fluorophenyl)-6-[{2-[methyl(phenyl)amino] ethyl} (methylsulfonyl)amino]-5-
Step 4 was performed in an an analogouos manner to Example 133, Step 4. The crude product was purified by prep-TLC to give pure ethyl 2-(4-fluorophenyl)-6-[{2- [methyl(phenyl)amino]ethyl}(methylsulfonyl)amino]-5-phenyl-l -benzofuran-3-carboxylate (60 mg, yield: 77%).
Ή-NMR (400 MHz, CDC13) δ 8.06-8.10 (m, 3H), 7.59 (s, 1 H), 7.49-7.51 (m, 2H), 7.39-7.46 (m, 3H), 7.14-7.22 (m, 4H), 6.66-6.70 (m, 1H), 6.54-6.56 (m, 2H), 4.37-4.42 (m, 2H), 3.23-3.67 (m, 4H), 2.81 (s, 3H), 2.75 (s, 3H), 1 .35 (t, J = 7.2 Hz, 3H).
Step 5: 2-(4-fluorophenyl)-6-[{2-[methyl(phenyl)amino]ethyl}(methylsulfonyl) amino5-yhenyl- 1 -benzofuran- -carboxylic acid
Step 5 was performed in an analogous manner to Example 133, Step 5. The crude product was purified by prep-TLC to give pure 2-(4-fluorophenyl)-6-[{2-[methyl(phenyl)amino] ethyl }(methylsulfonyl) amino5-phenyl- l -benzofuran-3-carboxylic acid (50 mg, yield: 87%).
Ή-NMR (400 MHz, CDC13) δ 7.80-7.89 (m, 3H), 7.50 (s, 1 H), 7.07-7.42 (m,
10H), 6.97-7.01 (m, 2H), 3.41-3.67 (m, 4H), 2.94 (s, 3H), 2.71 (s, 3H).
Step 6: 2-(4-fl orophenyl)-N-methyl-6-[{2-[methyl(phenyl)aminol' 'ethyl} (methylsulfonyl)amino]-' 5 -phenyl- 1 ' -benzofuran-3-carboxamide
Step 6 was performed in an analogous manner to Example 133, Step 6. The crude product was purified by prep-TLC to give pure 2-(4-fluorophenyl)-N-methyl-6-[{2- [methyl(phenyl)amino]ethyl} (methylsulfonyl)amino]-5-phenyl-l -benzofuran-3-carboxamide (13 mg, yield: 42%).
Ή-NMR (400 MHz, CDC13) δ 7.90-7.91 (m, 2H), 7.74 (s, 1H), 7.51 (s, 1H), 7.31-7.43 (m, 5H), 7.08-7.18 (m, 4H), 6.60-6.63 (m, 1 H), 6.48-6.50 (m, 2H), 5.78 (s, 1 H), 3.24-3.41 (m, 4H), 2.92 (d, J - 4.8 Hz, 3H), 2.74 (s, 3H), 2.70 (s, 3H).
Example 135: 5-(3-(benzo[dlthiazol-2-yl)phenvn-2-(4-fluorophenvn-N-methyl-6-(N- methylmethylsulfonainido)-l-benzofuran-3-carboxamide
Step I : Methyl 2-(5-bromo-2-hydroxyphenyl)acetate
2-(2-hydroxyphenyl)acetic acid (100 g, 0.66 mol) was dissolved in MeOH, and then TBATB (320 g, 0.66 mmol) was added to the solution. The resulting mixture was stirred at RT for 18 hours. After evaporation of solvent, the residue was dissolved in diethyl ether. The organic layer was washed with 1 N HC1, 2 M sodium bisulfate, H20 and brine, dried and evaporated to yield methyl 2-(5-bromo-2-hydroxyphenyl)acetate (145 g, yield: 90%).
Ή-NMR (400 MHz, CDC13) δ 7.48 (br s, 1H), 7.20-7.25 (m, 2H), 6.75-6.78 (m, 1 H), 3.74 (s, 3H), 3.62 (s, 2H). MS (M+H)+: 245.
Step 2: Methyl 2-(5-bromo-2-(tert-butyldimethylsilyloxy)phenyl)acetate
To a stirred solution of the product of Step 1 (1 g, 4.1 mmol) in DCM (5 mL) was added imidazole (0.56 g, 8.23 mmol) and TBSC1 (0.93 g, 6.17 mmol) at 0°C. After stirred overnight at RT, the reaction mixture was washed with H20, brine and concentrated in vacuo, the residue was purified by column chromatography to furnish the pure product of methyl 2-(5- bromo-2-(tert-butyldimethylsilyloxy)phenyl)acetate (1 .4 g, yield: 95%).
Ή-NMR (400 MHz, CDC13) δ 7.23 (d, J = 2.4 Hz, 1 H), 7.17 (dd, J, = 8.4 Hz, J2 = 2.4 Hz, 1 H), 6.61 (d, J = 8.4 Hz, 1 H), 3.61 (s, 3H), 3.50 (s, 2H), 0.91 (s, 9H), 0.15 (s, 6H). MS (M+H)+: 359.
Step 3: Methyl 2-(5-bromo-2-(tert-butyldimethylsilyloxy)phenyl)-3-(4-fluorophenyl)-3- oxopropanoate
A solution of the product of Step 2 (500 mg, 1.4 mmol) in THF ( 10 mL) at -78°C was treated dropwise with lithium bis(trimethylsilyl)amide (1.7 mL, 1.7 mmol, 1 N in THF). After stirred 30 minutes, a solution of 4-fluorobenzoyl chloride (250 mg, 1.6 mmol) in THF was added dropwise. The reaction mixture was stirred at -78°C for 1 hour and at 0°C for another 1 hour. The mixture was quenched with 1 N HC1, THF was removed in vacuo, and the residue
was extracted with EtOAc. The organic layer was concentrated and purified by column chromatography to afford the pure product of methyl 2-(5-bromo-2-(tert- butyldimethylsilyloxy)phenyl)-3-(4-fluorophenyl)-3-oxopropanoate (550 mg, yield: 82%).
Ή-NMR (400 MHz, CDC13) δ 7.83-7.87 (m, 2H), 7.28 (d, J = 2.4 Hz, 1 H), 7.16 (dd, J, = 8.4 Hz, J2 = 2.4 Hz, 1H), 6.93-6.98 (m, 2H), 6.63 (d, J = 8.4 Hz, 1 H), 5.86 (s, 1 H), 3.65 (s, 3H), 0.91 (s, 9H), 0.18 (s, 3H), 0.10 (s, 3H). MS (M+H)+: 481.
Step 4: Methyl 2-(5-bromo- -hydroxyphenyl)-3-(4-fluorophenyl)-3-oxopropanoate
To a solution of the product of Step 3 (300 mg, 0.6 mmol) in THF (10 mL), TBAF (500 mg, 1.9 mmol) was added and the mixture was stirred at 0°C for 1 hour. After concentrated in vacuo, the mixture was suspended in H20 and extracted with EtOAc. The organic layer was washed with H20, brine and concentrated. The residue was purified by column chromatography to give the product of methyl 2-(5-bromo-2-hydroxyphenyl)-3-(4- fluorophenyl)-3-oxopropanoate (200 mg, yield: 87%).
Ή-NMR (400 MHz, CDC13) δ 7.99 (m, 2H), 7.33 (s, 1 H), 7.18 (d, J = 8.0 Hz,
1 H), 7.07 (m, 2H), 6.68 (d, J = 8.0 Hz, 1H), 5.93 (s, 1 H), 3.77 (s, 3H). MS (M+H)+: 367.
Step 5: Methyl 5-bromo-2-(4-fluorophenyl)-l-benzofuran-3-carboxylate
To a solution of the product of Step 4 ( 100 mg, 0.3 mmol) in acetone (4 mL) was added concentrated HC1, and the mixture was heated under reflux for 30 minutes. Then, the reaction mixture was concentrated in vacuo, suspended in H20 and extracted with EtOAc. The organic layer was washed with H20, brine and concentrated. The residue was purified by prep- TLC to give pure methyl 5-bromo-2-(4-fluorophenyl)-l -benzofuran-3-carboxylate (70 mg, yield: 73%).
Ή-NMR (400 MHz, CDC13) 5 8.15 (s, 1 H), 8.05 (m, 2H), 7.43 (m, 1 H), 7.37 (m,
1H), 7.16 (m, 2H), 3.94 (s, 3H). MS (M+H)+: 349.
Step 6: Methyl 5-bromo-2-(4-fluorophenyl)-6-nitro-l-benzofuran-3-carboxylate
To a solution of the product of Step 5 (0.5 g, 1.4 mmol) in CHC13 (4 mL), fuming HN03 (1 mL) was added dropwise at RT, and the mixture was stirred for 4 hours. The reaction mixture was poured into ice water and extracted with EtOAc. The organic layer was washed with NaHC03 and brine. The solvent was removed by concentration to provide the crude product of methyl 5-bromo-2-(4-fluorophenyl)-6-nitro-l -benzofuran-3-carboxylate (0.4 g, yield: 70%). It was used for the next step without further purification.
Step 7: Methyl 6-amino-5-bromo-2-(4-fluorophenyl)-l-benzofuran-3-carboxylate
A mixture of the product of Step 6 (200 mg, 0.5 mmol), iron filings (200 mg, 3.58 mmol) and NH4CI (300 mg, 5.61 mmol) in MeOH:THF:H20 (1 : 1 : 1 , 20 mL) was stirred at reflux for 3 hours. After filtered and concentrated in vacuo, the residue was purified by column chromatography to furnish the pure methyl 6-amino-5-bromo-2-(4-fluorophenyl)-l -benzofuran- 3-carboxylate (150 mg, yield: 81 %).
1H-NMR (400 MHz, CDC13) δ 7.99 (s, 1H), 7.96 (m, 2H), 7.05-7.10 (m, 2H), 6.82 (s, 1 H), 4.18 (br s, 2H), 3.86 (s, 3H). MS (M+H)+: 364.
Step 8: Methyl 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido)-l-benzofuran-3-carboxylate
MsCI (60 ί, 0.77 mmol) was added to a solution of the product of Step 7 (150 mg, 0.41 mmol) and pyridine (0.34 mL) in dry DCM (10 mL) at 0°C. After stirring overnight at RT, the mixture was diluted with water, and extracted with DCM. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo, and the residue was purified by prep-TLC to afford the pure product of methyl 5-bromo-2-(4-fluorophenyl)-6- (methylsulfonamido)-l -benzofuran-3-carboxylate (150 mg, yield: 82%)).
Ή-NMR (400 MHz, CDC13) δ 8.21 (s, 1H), 7.99-8.03 (m, 2H), 7.83 (s, 1 H), 7.1 1 -7.16 (m, 2H), 6.82 (br s, 1 H), 3.90 (s, 3H), 2.96 (s, 3H). MS (M+H)+: 442.
Step 9: Methyl 5-bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l-benzofuran-3- carboxylate
CH3I (0.8 mL, 12.85 mmol) was added to a mixture of the product of Step 8 (5.0 g, 1 1.31 mmol), K2C03 (3.2 g, 23.15 mmol) and KI (1.9 mg, 1 1.45 mmol) in DMF (40 mL) under N2 protection. The mixture was stirred at reflux overnight. After filtered and concentrated in vacuo, the residue was purified by column chromatography to give the product of methyl 5- bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l -benzofuran-3-carboxylate (5 g, yield: 96%).
1H-NMR (400 MHz, CDC13) 6 8.32 (s, 1H), 8.05-8.09 (m, 2H), 7.72 (s, 1 H), 7.17-7.22 (m, 2H), 3.96 (s, 3H), 3.35 (s, 3H), 3.10 (s, 3H). MS (M+H)+: 456.
Step 10: 5-bromo-2-(4-fiuorophenyl)-6-(N-methylmethylsulfonamido)-l-benzofuran-3- carboxylic acid
To a solution of the product of Step 9 (5 g, 0.1 1 mol) in dioxane / H20 (1 : 1 , 100 mL) was added LiOH H20 (4.6 g, 0.1 1 mol), and the mixture was stirred at 100°C for 2 hours. After concentration, the residue was dissolved in H20, 1 N HC1 was added until pH reached 3, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed by distillation to provide the crude product of 5-bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l -benzofuran-3- carboxylic acid (4.5 g, yield: 97%). It was used for the next step without further purification. Step 11: 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonam
A solution of the product of Step 10 (5 g, 1 1.31 mmol), HOBT (3.2 g, 23.7 mmol) and EDCI (5.0 g, 26.1 mmol) in dry DMF (100 mL) was stirred at RT. After 30 minutes, Et3N (16 mL) and CH3NH2 (HC1 salt, 3.7 g, 56.5 mmol) was added to the mixture, and the mixture was stirred overnight. After the solvent was removed, H20 was added, and the mixture was extracted with EtOAc. The combined organic layer was washed with H20 and brine and
concentrated. The residue was purified by column chromatography to give the product of 5- bromo-2-(4-fluorophenyl)-N-methyl-6-( -methylmethylsulfonamido)-l -benzofuran-3- carboxamide (4.8 g, yield: 93%).
Ή-NMR (400 MHz, CDC13) δ 8.16 (s, 1H), 7.88-7.92 (m, 2H), 7.70 (s, 1 H), 7.18-7.23 (m, 2H), 5.78 (br s, 1H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J = 4.8 Hz, 3H). MS (M+H)+: 455.
Step 12: 5-(3-(benzofdlihiazol-2- l)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N-
A mixture of Pd(dppf)Cl2 (10 mg), the product of Step 1 1 (50 mg, 0.1 1 mmol), 3PO4 (60 mg, 0.28 mmol) and 2-(3-(4,4,5,5-tetramethyl- l ,3,2-dioxaborolan-2- yl)phenyl)benzo[d]thiazole (100 mg, 0.30 mmol) in DMF (2 mL) was stirred at 100°C under N2 protection overnight. Then, the solvent was removed, and H20 was added. After extracted with EtOAc, the combined organic layer was dried over Na2S04 and evaporated. The residue was purified by prep-HPLC to give the product of 5-(3-(benzo[d]thiazol-2-yl)phenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)- 1 -benzofuran-3 -carboxamide (20 mg, yield: 31 %).
Ή-ΝΜΡν (400 MHz, CDC13) δ 8.19 (s, 1 H), 8.12 (d, J = 7.2 Hz, 1 H), 8.06 (d, J = 8.4 Hz, 1H), 7.91-7.96 (m, 3H), 7.86 (s, 1 H), 7.58-7. 64 (m, 3H), 7.48-7.53 (m, 1 H), 7.38-7.42 (m, 1 H), 7.17-7.22 (m, 2H), 6.03 (br s, 1 H), 3.17 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.71 (s, 3H). MS (M+H)+: 586.
Examples 136-142
Examples 136-142 were prepared according to the general procedures of
Example 135.
Example 143; 2-( -ΑυοΓορΗβηνΙ)-/ν-ηΐ6ΐΗν1-5-ί3-(6-ιη6ί>ινΜ,3-66ηζοίΗί8ζοΙ-2-ν1)ρΗ6ην11-6- [methvKmethylsulfonyl)aminol- -benzofuran-3-carboxamide
Steps 1-11
Steps 1 -1 1 were performed in an analogous manner to Example 135, Steps 1 - 1 1 . Step 12: 2-(4-fluorophenyl)-5-(3-formylphenyl)-N-methyl-6-[methylfmethylsulfonyl)aminoJ-l- benzofuran-3-carboxamide
The aryl aldehyde (45 mg, yield: 73%) was prepared in an analogous manner to Example 136, Step 12.
Step 13: 2-(4-fluorophenyl)-N-methyl-5-[3-(6-methyl-l,3-benzothiazol-2-yl)phenyll-6- [methyl(methylsulfonyl)amino] -1 -benzofuran-3-carboxamide
A mixture of 2-amino-5-methylbenzenethiol (50 mg, 0.10 mmol) and the aryl aldehyde (50 mg, 0.36 mmol) in DMSO was stirred at 200°C for 1 hour. After cooling, 20 mL H20 was added, and the mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed, and the crude product was purified by prep-TLC to give pure 2-(4-fluorophenyl)-N-methyl-5-[3-(6-methyl-l ,3- benzothiazol-2-yl)phenyl]-6-[methyl(methylsulfonyl)amino]-l -benzofuran-3-carboxamide (50 mg, yield: 82%).
Ή-NMR (400 MHz, CDC13) δ 8.19 (s, 1H), 8.12 (d, J = 6.8 Hz, 1H), 7.94-7.99 (m, 3H), 7.87 (s, 1 H), 7.73 (s, 1H), 7.66 (s, 1H), 7.57-7.61 (m, 2H), 7.33 (d, J = 8.4 Hz, 1 H), 7.19-7.24 (m, 2H), 6.05 (br s, 1 H), 3.19 (s, 3H), 3.01 (d, J = 4.8 Hz, 3H), 2.72 (s, 3H), 2.53 (s, 3H). MS (M+H)+: 600.
Examples 144-149
Examples 144-149 were prepared according to the general proced
Example 143.
Example 150: 5-[3-(5-fluoro-lH-benzimidazol-2-yl)phenvn-2-(4-fluorophenvn-N-methyl-6- [tnethv methylsulfonvDamin -l-benzofuran-B-carboxaniide
Steps 1-12
Steps 1-12 were performed in an analogous manner to Example 143, Steps 1 - 12.
Step 13: 5-f3-(5-fluoro-lH-benzimidazol-2-yl)phenyl]-2-(4-fluorophenyl)-N-methyl-6- fmethyl(melhyls \fon l)amino] -1 -benzofuran-3-carboxamide
The aryl aldehyde of Example 143, Step 12 (100 mg, 0.21 mmol) and 4- fluorobenzene-l ,2-diamine (32 mg, 0.25 mmol) were added in PhN02 (4 mL) and the mixture was heated to 120°C and stirred overnight. The mixture was concentrated, and H20 (30 mL) was added. After extraction with EtOAc, the organic layer was washed with brine and concentrated. The residue was purified by prep-HPLC to give pure 5-[3-(5-fluoro-l H-benzimidazol-2- yl)phenyl]-2-(4-fluorophenyl)-N-methyl-6-[methyl(methylsulfonyl)amino]- l -benzofuran-3- carboxamide (30 mg, yield: 41.5%).
Ή-NMR: (400 MHz, CDC13) δ 8.16 (s, 1H), 8.00 (d, J = 6.8 Hz, 1 H), 7.87 (m, 2H), 7.72 (s, 1H), 7.56-7.58 (m, 1H), 7.48-7.50 (m, 1 H), 7.41 (s,l H), 7.28-7.35 (m, 2H), 7.04-7.14 (m, 3H), 6.62-6.68 (m, 1 H), 2.93-2.96 (m, 9H). MS (M+H)+: 587. Examples 151-154
Examples 151 -154 were prepared according to the general procedures of
Example 155: 2-(4-fluorophenyl)-N-methyl-6-(N-methylrnethylsulfonainido)-5-(3- (oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-3-carboxamide
Step 1: Methyl 2-(5-bromo-2-hydroxyphenyl)acetate
2-(2-hydroxyphenyl)acetic acid (484 g, 3.18 mol) was dissolved in methanol, and then tetrabutyl ammonium tribromide (1549 g, 3.18 mol) was added to the solution. The resulting mixture was allowed to stir at room temperature for 18 hours. After evaporation of solvent in vacuo, the residue obtained was dissolved in EtOAc. The organic layer was washed with 1 N HCl, water and brine, dried and concentrated, the residue obtained was purified using flash column chromatography on silica gel (eluted with PE / EtOAc = 10 / 1) to give pure methyl 2-(5-bromo-2-hydroxyphenyl)acetate (750 g, 94%). Ή-NMR (400 MHz, CDC13) δ 7.48 (br s, 1 H), 7.20-7.25 (m, 2H), 6.75-6.78 (m, 1 H), 3.74 (s, 3H), 3.62 (s, 2H). MS (M+H)+: 245.
Methyl 2-(5-brom -2-(tert-butyldimethylsilyloxy)phenyl)acetate
To a stirring solution of methyl 2-(5-bromo-2-hydroxyphenyl)acetate (750 g, 3.06 mol) in dichloromethane (4 L) was added imidazole (416 g, 6.1 mol) and TBSC1 (692 g, 4.6 mol) at 0 °C. After stirred for about 15 hours at room temperature, the reaction mixture was washed with water, brine and concentrated in vacuo, the residue obtained was purified using flash column chromatography on silica gel (eluted with PE / EtOAc = 30 / 1) to furnish pure product of methyl 2-(5-bromo-2-(tertbutyldimethylsilyloxy) phenyl)acetate (880 g, 80%). Ή-NMR (400 MHz, CDC13) 5 7.23 (d, J = 2.4 Hz, 1 H), 7.17 (dd, J, = 8.4 Hz, J2 = 2.4 Hz, 1 H), 6.61 (d, J = 8.4 Hz, 1 H), 3.61 (s, 3H), 3.50 (s, 2H), 0.91 (s, 9H), 0.15 (s, 6H). MS (M+H)+: 359.
Step 3: Methyl 2-(5-bromo-2-(tert-butyldimethylsilyloxy)phenyl)-3-(4-fluorophenyl)-3- oxopropanoate
A solution of methyl 2-(5-bromo-2-(tert-butyldimethylsilyloxy)phenyl)acetate (220 g, 0.62 mol) in THF (1.5 L) at -78 °C was treated dropwise with a THF solution of LDA (0.74 mol, freshly prepared from i-Pr2NH and n-BuLi). After stirred for 1 hour, a solution of 4-
fluorobenzoyl chloride (106 g, 0.68 mol) in THF was added dropwise. The reaction mixture was allowed to stir at -78 °C for 1 hour and at 0 °C for another 1 hours. The mixture was quenched with 1 N HC1, and then THF was removed in vacuo, the residue obtained was extracted with EtOAc. The organic layer was concentrated and purified using flash column chromatography on silica gel (eluted with PE / EtOAc = 10 / 1) to providepure product of methyl 2-(5-bromo-2-(tert- butyldimethylsilyloxy)phenyl)-3-(4-fluorophenyl)-3-oxopropanoate (236 g, 80%). Ή-NMR (400 MHz, CDC13) δ 7.83-7.87 (m, 2H), 7.28 (d, J = 2.4 Hz, 1 H), 7.16 (dd, J, = 8.4 Hz, J2 = 2.4 Hz, 1 H), 6.93-6.98 (m, 2H), 6.63 (d, J = 8.4 Hz, 1H), 5.86 (s, 1H), 3.65 (s, 3H), 0.91 (s, 9H), 0.18 (s, 3H), 0.10 (s, 3H). MS (M+H)+: 481.
Step 4: Methyl 2-(5-bromo-2-hydroxyphenyl)-3-(4-fluorophenyl)-3-oxopropanoate
TBAF (217.5 g, 0.83 mol) was added to a solution of methyl 2-(5-bromo-2-(tert- butyldimethylsilyloxy)phenyl)-3-(4-fluorophenyl)-3-oxopropanoate (267 g, 554.6 mol) in THF (2 L), and the mixture was allowed to stir at 0 °C for 1 hours. The reaction mixture was then concentrated in vacuo and the resulting residue was suspended in H
20 and extracted with ethyl acetate. The organic layer was washed with H
20, brine and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (eluted with PE / EtOAc from 10 / 1 to 5 / 1) to provide methyl 2-(5-bromo-2-hydroxyphenyl)-3-(4-fluorophenyl)-3- oxopropanoate (178.6 g, 88%). Ή-NMR (400 MHz, CDC1
3) δ 7.99 (m, 2H), 7.33 (s, 1 H), 7.18 (d, J = 8.0 Hz, 1 H), 7.07 (m, 2H), 6.68 (d, J = 8.0 Hz, 1 H), 5.93 (s, 1 H), 3.77 (s, 3H). MS (M+H)
+: 367. Step 5: . Methyl 5-bromo-2-(4-fluorophenyl)-l-benzofuran-3-carboxylate
To a solution of methyl 2-(5-bromo-2-hydroxyphenyl)-3-(4-fluorophenyl)-3- oxopropanoate (50 g, 136.1 mmol) in acetone (200 mL) was added concentrated hydrochloric
acid and the mixture was heated to reflux for 1 hour. Then the reaction mixture was concentrated in vacuo, suspended in H20 and extracted with ethyl acetate. The organic layer was washed with aq. NaHC03 and brine. Then the organic layer was concentrated to provide the crude product of methyl 5-bromo-2-(4-fluorophenyl)-l -benzofuran-3-carboxylate. It was used for the next step without further purification. Ή-NMR (400 MHz, CDC13) δ 8.15 (s, 1 H), 8.05 (m, 2H), 7.43 (m, 1 H), 7.37 (m, 1H), 7.16 (m, 2H), 3.94 (s, 3H). MS (M+H)+: 349.
Methyl 5- romo-2-(4-fluorophenyl)-6-nitro-l -benzofuran-3 -carboxylate
To a solution of methyl 5-bromo-2-(4-fluorophenyl)-l -benzofuran-3-carboxylate
(50 g, 143.2 mmol) in CHC13 (300 mL) at room temperature, was added dropwise fuming HN03 (50 mL) and the reaction was allowed to stir for 4 hours. The reaction mixture was poured into ice water and extracted with ethyl acetate. The organic layer was washed with NaHC03 and brine, then concentrated in vacuo to provide methyl 5-bromo-2-(4-fluorophenyl)-6-nitro-l - benzofuran-3-carboxylate, which was used without further purification.
Step 7: Methyl 6-amino- -bromo-2- 4- uoro hen l -l- nzo uran-3- r xylate
A mixture of methyl 5-bromo-2-(4-fluorophenyl)-6-nitro-l-benzofuran-3- carboxylate (100 g, crude), iron filings (100 g, 1.79 mol) and NH
4C1 (200 g, 3.74 mol) in MeOH / THF / H
20 (8 / 8 / 5, 1 L) was heated to reflux and allowed to stir at this temperature for 3 hours. The reaction mixture was then filtered and concentrated in vacuo, the residue obtained was purified using flash column chromatography on silica gel (eluted with PE / EtOAc = 10 / 1 and then with pure dichloromethane) to furnish pure product of methyl 6-amino-5-bromo~2-(4- fluorophenyl)- l -benzofuran-3-carboxylate (41.2 g, 44.5%, 3 steps overall). Ή-NMR (400 MHz, CDC1
3) δ 7.99 (s, 1 H), 7.96 (m, 2H), 7.05-7.10 (m, 2H), 6.82 (s, 1 H), 4.1 8 (br s, 2H), 3.86 (s, 3H). MS (M+H)
+: 364.
Step 8: Methyl 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido)-l-benzofuran-3- carboxylate
MsCI (25.2 g, 219.7 mmol) was added to a solution of methyl 6-amino-5-bromo- 2-(4-fluorophenyl)-l -benzofuran-3-carboxylate (40 g, 109.8 mmol) and pyridine (26.1 g, 329.5 mmol) in dry dichloromethane (300 mL) at 0 °C. After stirred for about 15 hours at room temperature, the mixture was diluted with water, and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2SC>4, filtered and concentrated in vacuo. The residue obtained was crystalized from EtOAc to providethe product of methyl 5-bromo-2-(4- fluorophenyl)-6-(methylsulfonamido)-l -benzofuran-3-carboxylate (38.2 g, 78.6%). Ή-NMR
(400 MHz, CDC13) δ 8.21 (s, 1 H), 7.99-8.03 (m, 2H), 7.83 (s, 1 H), 7.1 1-7.16 (m, 2H), 6.82 (br s, 1 H), 3.90 (s, 3H), 2.96 (s, 3H). MS (M+H)+: 442.
Step 9: Methyl 5-bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l-benzofuran-3- carboxylate
CH3I (3.53 g, 24.9 mmol) was added to a mixture of methyl 5-bromo-2-(4- fluorophenyl)-6-(methylsulfonamido)-l -benzofuran-3-carboxylate (10 g, 22.61 mmol), K2C03 (6.25 g, 45.2 mmol) and KI (1.88 g , \ 1.31 mmol) in DMF (100 mL) under N2 protection. The mixture was allowed to stir at reflux for about 15 hours. After concentrated, H20 was added and the mixture was extracted with dichloromethane. The combined organic layer was washed with H20, brine and concentrated in vacuo. The residue obtained was crystalized from EtOAc to provide methyl 5-bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l -benzofuran-3- carboxylate (9.6 g, 93%). Ή-NMR (400 MHz, CDC13) δ 8.32 (s, 1H), 8.05-8.09 (m, 2H), 7.72 (s, 1 H), 7.17-7.22 (m, 2H), 3.96 (s, 3H), 3.35 (s, 3H), 3.10 (s, 3H). MS (M+H)+: 456.
Step 10 - 5-bromo-2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido)-l-benzofuran-3- car boxy lie acid
To a solution of methyl 5-bromo-2-(4-fluorophenyl)-6-(N- methylmethylsulfonamido)-l -benzofuran-3-carboxylate (20 g, 43.8 mmol) in dioxane / H20 (1 / 1 , 100 mL) was added LiOH H20 (18.39 g, 0.44 mol), and the mixture was heated to reflux for 3 hours, filtered and concentrated in vacuo. The residue obtained was dissolved in H20, 1 N HC1 was added until pH reached 3, and the mixture was extracted with dichloromethane. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed by concentration to provide the crude product of 5-bromo-2-(4-fluorophenyl)-6-(N- methylmethylsulfonamido)-l -benzofuran-3-carboxylic acid (18.2 g, 93.8%). It was used for the next step without further purification.
Step 11 - 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-l-benzofuran- 3-carboxamide (Compound L)
A solution of 5-bromo-2-(4-fluorophenyl)-6-( -methylmethylsulfonamido)-l - benzofuran-3-carboxylic acid (21 g, 47.5 mmol), HOBT (7.06 g, 52.2 mmol) and EDCI (9 g, 47.5 mmol) in dry DMF (200 mL) was allowed to stir at room temperature. After 30 minutes, Et3N (16 mL) and CH3NH2 (HC1 salt, 6.41 g, 95 mmol) was added to the mixture, and the mixture was allowed to stir for about 15 hours. After the solvent was removed, H20 was added and the mixture was extracted with dichloromethane. The combined organic layer was washed with H20, brine and concentrated in vacuo. The residue obtained was crystalized from EtOAc to provide compound L (19.5 g, 90%). Ή-NMR (400 MHz, CDC13) δ 8.16 (s, 1H), 7.88-7.92 (m, 2H), 7.70 (s, 1 H), 7.18-7.23 (m, 2H), 5.78 (br s, 1 H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J = 4.8 Hz, 3H). MS (M+H)+: 455.
Step 12 - 2-(4-fluorophenyl)-N-methyl-6-(N-methylmelhylsulfonamido)-5-(3-(oxazolo[4,5- b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide
To a degassed solution of 2-[3-(4,4,5,5-Tetramethyl-[l ,3,2]dioxaborolan-2-yl)- phenyl]-oxazolo[4,5-b]pyridine (prepared from corresponding bromide, 587 mg, 1.82 mmol) was added a solution of Compound L (635 mg, 1.40 mmol) and K3PO4 (771 mg, 3.64 mmol) in dry DMF (6 raL). To the resulting solution was added Pd(dppf)Cl2 (30 mg) and the reaction mixture was placed under N2 atmosphere, heated to 100 °C and allowed to stir at this temperature for 6 hours. After cooled to room temperature and filtered, the filtrate was washed with H20, brine, and dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column chromatography (PE : EtOAc = 1 : 1 ) to provide the target compound (430 mg, 53.9%) as white solid. 1H-NMR (CDC13, 400 MHz) δ 8.60-8.61 (m, 1 H), 8.39 (s, 1 H), 8.33 (d, J = 6.8 Hz, 1 H), 7.91-7.95 (m, 3H), 7.88 (s, 1H), 7.72 (d, J = 7.6 Hz, 1 H), 7.62-7.66 (m, 2H), 7.35-7.38 (m, 1 H), 7.20 (d, J = 8.8 Hz, 2H), 5.93-5.94 (m, 1 H), 3.18 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.71 (s, 3H). MS (M+H)+: 571. The following compounds of the present invention were prepared using the method described in Example 155 and substituting the appropriate reactants and/or reagents.
MS
Example Structure NM R
(M+H)+
Ή-NMR (CDCI3, 400 MHz) δ
1H), 6.03 (d, = 4.4 Hz, 1H), 3.22 (s, 3H), 3.02 (d, J = 4.8 Hz, 3H), 2.73 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.07
IH), 3.14 (s,3H), 2.93 (d,J=4.8
Hz, 3H), 2.73 (s, 3H).
Ή-NMR (DMSO, 400 MHz) δ 8.54 (s, IH), 8.10-8.11 (d, J = 4.0 Hz,
8
J
(s, 3H), 3.02 (s, 3H), 2.81-2.82 (d, J = 4.4 Hz, 3H).
Ή-NMR (CDCI3, 400 MHz) 58.15 1.6
163 622
5.2 Hz, 3H), 2.82 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 7.93
3H), 2.94 (d, J = 4.8 Hz, 3H), 2.70
(s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.64
2.92 (d, 7=4.8 Hz, 3H), 2.69 (s, 3H).
(s, 3H), 2.49 (s, 3H).
'H-NMR (CDCI3, 400 MHz) δ 8.08
= 4.8 Hz, 3H), 2.71 (s, 3H), 2.52 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.73
2H), 5.31 (s, IH), 3.22 (s, 3H), 3.03 (d, 7= 4.8 Hz, 3H), 2.82 (s, 3H). Ή-NMR (CDCI3, 400 MHz) δ
IH), 3.22 (s, 3H), 2.92 (d,7=4.4 Hz, 3H), 2.55 (s, 3H).
1 H-NMR (DMSO, 400 MHz) δ
8.54-8.55 (d, J = 4.4 Hz, 1 H), 8.25 (s, I H), 8.20-8.22 (d, J = 6.4 Hz,
3H), 2.82 (d, J = 4.8 Hz, 3H), 2.60 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.22
Hz, 3H), 2.60 (s, 3H), 2.44 (s, 3H).
Hz, 3H), 2.77 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.18
3H),2.93 (d, =5.2 Hz, 3H), 2.76
(s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.21
3.12 (s,3H), 2.93 (d, J = 4.8 Hz,
3H), 3.92 (s, 3H), 3.02 (s, 3H), 2.94 (d, J = 4.4 Hz, 3H), 2.87 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.43
8.4 Hz, IH), 5.81 (s, IH), 4.03 (s, 3H), 3.11 (s, 3H), 2.93 (d, J =4.4 Hz, 3H), 2.71 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.11
(s, 1H), 7.83-7.87 (m, 2H), 7.77 (s,
3H), 3.21 (s, 3H), 3.09 (s, 3H), 2.89 (d, .7=4.4 Hz, 3H), 2.65 (s, 3H).
Ή-NMR (CDC13) 400 MHz) 68.12
7 (m,
(m,
Hz, 3H), 2.70 (s, 3H), 2.44 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.22
(s, IH), 7.93-7.94 (m, 2H), 7.87 (s, 1 H), 7.65-7.68 (m, 3H), 7.16-7.25
(m, 5H), 5.91 (d, J =4.4 Hz, IH), 614 4.08 (s, 3H), 3.17 (s, 3H), 3.00 (d, J
= 8.0 Hz, 3H), 2.77 (s, 3H), 2.58 (s, 3H).
3H), 2.94 (d, J = 4.8 Hz, 3H), 2.70
(s, 3H), 1.41 (d, 7=6.0 Hz, 6H).
s, 3H), 2,87 (s, 3H).
Ή-NIVIR (CDCI3, 400 MHz) δ 8.44
3H), 2.92-2.93 (d, J = 4.0 Hz, 3H), 2.68 (s, 3H).
Ή-NMR (CDC13) 400 MHz) δ 8.71
3H), 3.03 (d, J = 5.2 Hz, 3H), 2.78
(s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.27
IH), 3.12 (s,3H), 2.91 (d, .7=5.2
Hz, 3H), 2.67 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.73
3.23 (s, 3H), 3.05 (d, .7=4.4 Hz,
3H), 2.87 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.44 (s, IH), 8.23 (s, 2H), 8.17-8.19 (m, IH), 7.80-7.83 (m, 2H), 7.64 (s,
199 IH), 7.59-7.61 (m, 1 H), 7.41-7.43 623
(m, IH), 7.15 (s,2H), 5.67 (s, IH),
3.10 (s,3H), 2.84 (d, .7=4.8 Hz,
3H), 2.55 (s, 3H).
Ή-N R (CDCI3,400 MHz) δ
3H),2.99 (d, J = 4.8 Hz, 3H),2.81
(s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.43
Hz, 3H), 2.65 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.49 (s, IH), 8.32 (s, 2H), 8.27-8.29 (m, IH), 7.90-7.93 (m, 2H), 7.84 (s,
202 IH), 7.69-7.72 (m, IH), 7.61-7.65 605
(m, 2H), 7.15 (s, 2H), 5.77 (s, IH),
3.13 (s, 3H), 2.94 (d, J =4.8 Hz,
3H), 2.65 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.57
3.15 (s, 3H), 2.94 (d, 7 = 4.8 Hz,
3H), 2.75 (s, 3H).
204 8 605
3H), 3.01 (s, 3H), 2.85 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.75
(s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.85 (s, 3H), 2.68 (s, 3H).
209 .4
3H), 2.78 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.59
3H), 2.99 (d, J = 4.8 Hz, 3H), 2.71 (s, 3H), 2.53 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.25
2H), 5.83 (s, IH), 3.13 (s, 3H), 2.92 (d, .7 =4.8 Hz, 3H), 2.68 (s, 3H). Ή-NMR (CDCI3, 400 MHz) δ 9.21
IH), 3.16 (s,3H), 2.93 (d, = 4.8 Hz, 3H), 2.80 (s, 3H).
'H-NMR (CDCI3, 400 MHz) δ 8.95 8
2H), 5.83 (s, IH), 3.06 (s, 3H), 2.82 (d, J =4.8 Hz, 3H), 2.75 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.53
I H), 3.25 (s, 3H), 3.04 (d, = 4.8 Hz, 3H), 2.82 (s, 3H).
Ή-NMR (CDCIj, 400 MHz) δ 8.27
(s, IH), 8.08 (s, 1H), 7.99 (s, 1H), 7.83-7.92 (m, 4H), 7.57 (s, l'H),
219 7.38(d, .7 = 8.0 Hz, 1H), 7.13-7.17
(m, 2H), 5.83 (s, 1 H), 3.15 (s, 3H),
2.93 (d, J = 4.0 Hz, 3H), 2.73 (s, 3H).
1 H-NMR (CDC13, 400 MHz) δ 8.38
3.03 (d, = 4.8 Hz, 3H), 2.74 (s, 3H).
'H-NMR (CDCI3, 400 MHz) δ
IH), 3.16 (s,3H), 2.97 (d, = 4.8 Hz, 3H), 2.77 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.25-8.30 (m, 3H), 8.01 (t, J = 6.8 Hz, IH), 7.88-7.92 (m, 2H), 7.83 (s, IH), 7.62 (s, IH), 7.26-7.32 (m,
222 2H), 7.15 (t, J = 8.4 Hz, 2H), 5.81
(d, J = 4.8 Hz, IH), 3.23 (s, 3H),
2.95 (d, = 4.8 Hz, 3H), 2.58 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ
IH), 3.14 (s,3H), 2.94 (d, .7 = 4.0
Hz, 3H), 2.64 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.38 (s, IH), 8.34 (d, 7= 7.6 Hz, IH),
8.27 (s, IH), 7.97-8.00 (m, 2H),
224 7.93 (s, IH), 7.67-7.84 (m, 4H), 589
7.25 (t, 7 = 8.4 Hz, 2H), 5.88 (br, s,
IH), 3.24 (s, 3H), 3.03 (d, 7 = 4.8
Hz, 3H), 2.74 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.37
3H), 4.17 (s, 3H), 3.21 (s, 3H), 3.01 (d,7 = 4.0 Hz, 3H), 2.78 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.23
(s, 3H), 2.93 (d, 7 = 4.0 Hz, 3H),
2.72 (s, 3H).
(d, 7= 4.8 Hz, 3H), 2.72 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.43
J =17.6 Hz, 2H), 3.16 (s, 3H), 2.98
(s, 3H), 2.78 (d, = 4.4 Hz, 3H).
Ή-NMR (CDCI3, 400 ΜΗζ)δ 8.42
(d, = 3.6 Hz, IH), 8.34 (s, IH),
3.04 (d, J = 4.8 Hz, 3H), 2.83 (s,
3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.30
3H), 2.93 (d, = 4.8 Hz, 3H), 2.57
(s,3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.56 (s, 1H), 8.43 (s, IH), 7.84-7.90 (m,
4H), 7.71-7.74 (m, IH), 7.59 (s,
231 IH), 7.48-7.50 (m, IH), 7.29-7.31 655
(m, IH), 7.14-7.16 (m, 2H),
5.79-5.80 (m, IH), 3.14 (s, 3H),
2.93-2.94 (m, 3H), 2.72 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.61
1 .2 Hz, 3H), 3.19 (s, 3H), 2.99 (d, J
= 4.4 Hz, 3H), 2.85 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.34
l H), 4.14 (s, 3H), 3.15 (s, 3H), 2.94
(d, = 4.0 Hz, 3H), 2.78 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ
(d, = 4.8 Hz, 3H), 2.94 (s, 3H).
3H), 3.93 (s, 3H), 3.04 (s, 3H), 2.94
(d, .7 = 4.8 Hz, 3H), 2.87 (s, 3H).
H-NMR (CDClj, 400 MHz) δ 8.54
3.15 (br s, 5H), 2.99 (d, J = 4.8 Hz, 3H), 2.93 (brs, 4H), 2.81 (s, 3H).
'H-NMR (CDCI3, 400 MHz) δ 8.55
(s, 3H), 3.00 (d, = 4.8 Hz, 3H),
2.81 (s, 3H).
"H-NMR (CDCl,, 400 MHz) δ 8.53
(s, 3H), 2.94 (d, .7 = 4.8 Hz, 3H),
2.73 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.85 (s, 1 H), 8.54-8.57 (m, 2H), 8.13 (s, IH), 8.02-8.04 (m, 3H), 7.75-7.78
239 (m, IH), 7.62 (s, IH), 7.42-7.45 (m, 635
2H), 5.80 (brs, 1H),4.00 (s3H),
3.15 (s, 3H), 2.99 (s, 3H), 2.80 (d,J
= 4.8 Hz, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.52
(d,J = 2.& Hz, 1H), 8.38 (s, 1H),
3H), 2.93 (d, J= 5.2 Hz, 3H), 2.66
(s, 3H).
'H-NMR (CDCI3, 400 MHz) δ 9.14
3.14 (s, 3H), 2.93 (d, = 4.8 Hz,
3H), 2.69 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.09
1H), 3.15 (s, 3H), 2.94 (d, .7=4.8
Hz, 3H), 2.67 (s, 3H).
Ή-NMR (DMSO, 400 MHz) δ 8.62
3H), 3.05 (s, 3H), 2.83 (d,J = 4.4
Hz, 3H).
1 H-NMR (CDClj, 400 MHz) δ 8.42
IH), 3.22 (s, 3H), 3.01 (d, 7 = 4.8 Hz, 3H), 2.88 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ
2.94 (d, =4.8 Hz, 3H), 2.74 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.51
2H), 5.96 (s, IH), 3.21 (s, 3H), 2.98 (d, 7 = 4.8 Hz, 3H), 2.88 (s, 3H).
IH), 3.29 (s,3H), 3.00 (d,7 = 4.8 Hz, 3H), 2.67 (s, 3H).
248 605
Ή-NMR (CDC13, 400 MHz) δ
1H),4.00 (s, 3H), 3.19 (s,3H), 3.02
(d,J=4.8 Hz, 3H), 2.84 (s, 3H).
Ή-INMR (CDCI3, 400 MHz) δ 9.66 (s, IH), 9.36 (d, = 8.4 Hz, IH),
Hz, 1H),4.99 (s, 3H), 4.32 (s, 3H),
4.12 (d, J = 4.8 Hz, 3H), 3.73 (s,
3H).
Ή-NMR (CDCI3, 400 MHz) 58.18
3H), 3.01 (d,J=4.0 Hz, 3H), 2.72
(s, 3H).
1 H-NMR (CDC13, 400 MHz) δ 8.42
1 H), 3.20 (s, 3H), 3.02 (s, 3 H), 2.62 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.60
(d, .7=6.8 Hz, IH), 8.28 (s, IH),
8.22 (d, J = 7.2 Hz, 1 H), 7.99 (d, J = 9.2 Hz, IH), 7.91-7.94 (m,2H),
257 7.80 (s, I H), 7.68 (d, J= 7.6 Hz, 570
1 H), 7.57-7.64 (m, 3H), 7.14 (X,J =
8.4 Hz, 3H), 6.18 (s, IH), 3.13 (s,
3H), 2.94 (d, = 4.4 Hz, 3H), 2.60
(s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.38
IH), 5.81 (s, IH), 3.15 (s,3H), 2.93
(d, J = 4.0 Hz, 3H), 2.67 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.71
3H), 2.94 (d, J = 4.0 Hz, 3H), 2.68
(s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ
3H), 3.02 (d, J= 4.4 Hz, 3H), 2.84
(s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 8.60
7.15-7.17 (m, 2H), 5.77 (br s, IH), 3.09 (s, 3H), 2.92 (d,J=4.0 Hz, 3H), 2.81 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ
(s, 3H), 3.01 (d, .7=4.8 Hz, 3H),
2.67 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.79 (d, J = 8.0 Hz, IH), 8.63 (d, J = 8.0
Hz, IH), 8.35 (s, IH), 8.24 (d, J =
8.4 Hz, IH), 8.03-8.14 (m, 5H),
266 7.92 (s, IH), 7.83-7.88 (m,2H), 580
7.75 (t, .7 = 7.6 Hz, IH), 7.62 (s,
IH), 7.23 (t, J= 8.4 Hz, 2H), 6.77
(s, IH), 3.13 (s, 3H), 3.06 (d,J = 7.2 Hz, 3H), 2.93 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.24
3.14 (s, 3H), 2.93 (d,J=5.2 Hz,
3H), 2.58 (s, 3H).
Ή-NIMR (CDCI3, 400 MHz) δ 9.56
3H), 3.00 (d, .7= 4.8 Hz, 3H), 2.63
(s, 3H).
'H-NMR (CDC13, 400 MHz) δ 8.68
Hz, 1H), 3.06 (s, 3H), 2.94 (d, 7 = 4.8 Hz, 3H), 2.80 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.55
1H), 5.85 (s, 1H), 3.24 (s, 3H), 2.30 (d, 3H),2.80 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.66
4.19 (s, 3H), 3.19 (s, 3H), 3.03 (d,J = 5.2 Hz, 3H), 2.86 (s, 3H).
Ή-NMR (DMSO, 400 MHz) δ 8.82
2H), 3.17 (s, 3H), 3.02 (s, 3H), 2.83 (d, .7 = 4.4 Hz, 3H).
Ή-NMR (DMSO, 400 MHz) δ 9.27
2.93 (d, .7=8.0 Hz, 3H), 2.70 (s,
3H).
"H-NIMR (MeOD, 400 MHz) δ
(d, = 4.8 Hz, 3H), 2.71 (s, 3H).
Ή NMR: (CDClj, 400 MHz) δ
IH), 2.99 (s, 3H), 2.92 (d, = 4.8
Hz, 3H), 2.60 (s, 3H). ,
277 .66 621
(s, 3H), 2.95 (d, .7=4.8 Hz, 3H),
2.58 (s,3H).
Ή-NMR (CDC13, 400 MHz) δ 8.21
I H), 4.00 (s, 3H), 3.09 (s, 3H), 2.94 (d, .7 = 4.8 Hz, 3H), 2.73 (s, 3H).
1 H-NMR (CDCI3, 400 MHz) δ 8.26
3.15 (s,3H), 2.98 (d, J = 4.8 Hz, 3H), 2.83 (s, 3H).
'H-NMR(CDC13, 400 MHz) δ 8.15
IH), 3.13 (s,3H), 2.94 (d,J=4.8 Hz, 3H), 2.74 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.18
3H), 3.13 (s,3H), 2.97 (s, 3H), 2.78 (d, =8.0 Hz, 3H).
Ή-NMR (CDCI3, 400 MHz) 58.17
3H), 3.01 (d, .7= 4.8 Hz, 3H), 2.82 (s, 3H).
H-NMR (CDClj, 400 MHz) δ 8.26
(s, 1 H), 7.95-7.98 (m, 2H), 7.88 (s,
1 H), 7.74 (d, J = 8.4 Hz, 1 H), 7.65
409 (s, 1 H), 7.20-7.23 (m, 3H),
7.02-7.1 1 (m, 2H), 6.07 (s, 1 H),
4.1 1 (s, 3H), 3.20 (s, 3H), 3.05 (d, J
= 4.8 Hz, 3 H), 2.57 (s, 3H).
Ή-NMR (CDC13, 400 MHz) 5 8. 19
(d, J = 2.0 Hz, 1 H), 7.91 -7.93 (m,
410
Hz, 1 H), 4.07 (s, 3H), 3.17 (s, 3H),
2.99 (d, .7 = 4.0 Hz, 3H), 2.80 (s,
3H).
Example 281 : 2-(4-fluorophenvn-N-methyl-6-(N-methylmethylsulfonamido)-5-(3-(6- (methylsulfonamido)benzofdloxazol-2-yI)phenvnbenzofuran-3-carboxamide
Step 1: 5-(3-(6-aminobenzo[d]oxazol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
Example 195
To a solution of the compound of Example 195 (530 mg, 0.13 mmol) in MeOH (10 mL), Pd/C (10 mg) was added, and the resulting reaction mixture was allowed to stir under 40 psi of H2 atmosphere for 24 hours at 25 °C. The reaction mixture was filtered, concentrated
vacuo and the residue obtained was purified using flash column chromatography (PE : EtOAc = 2 : 1) to provide 5-(3-(6-aminobenzo[d]oxazol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (420 mg, 85%). Ή-NMR (DMSO, 400 MHz) δ 8.55 (s, 1 H), 8.00-8.1 1 (m, 5H), 7.59-7.63 (m, 3H), 7.38-7.40 (m, 3H), 6.80 (s, 1 H), 6.62-6.64 (d, J= 8.4 Hz, 1H), 5.47 (s, 2H), 3.12 (s, 3H), 2.93 (s, 3H), 2.79-2.80 (d, J = 4.0 Hz, 3H). MS (M+H)+: 585.
Step 2: 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(3-(6- (methylsulfonamido)benzo[d]oxazol-2-yl)phenyl)benzofuran-3-carboxamide
To a solution of 5-(3-(6-aminobenzo[d]oxazol-2-yl)phenyl)-2-(4-fluorophenyl)- N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (50 mg, 0.13 mmol) and pyridine (0.2 mL) in 1 mL of dry dichloromethane, MsCI (50 mg, 0.44 mmol) was added dropwise at 0 °C. After stirred at room temperature for 4 hours, the mixture was quenched with 20% aq. NH4C1, then extracted with dichloromethane and washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using preparative HPLC to provide 2-(4-fluorophenyI)-N-methyl-6-(N-methylmethyIsulfonamido)-5-(3-(6- (methylsulfonamido)benzo[d]oxazol^2-yl)phenyl)benzofuran-3-carboxamide (43 mg, 90.1 %). Ή-NMR (CDCI3, 400 MHz) δ 8.17-8.23 (m, 3H), 7.88-7.92 (m, 2H), 7.80 (s, 1 H), 7.55-7.60 (m, 4H), 7.25 (s, 1 H), 7.12-7.14 (m, 2H), 7.06-7.08 (m, 1H), 5.79 (s, 1H), 3.13 (s, 3H), 2.93-2.94 (d, J = 4.8 Hz, 3H), 2.60 (s, 3H), 2.56 (s, 3H). MS (M+H)+: 663.
The following compounds of the present invention were prepared using the method described in Example 281 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-NMR (CDC13, 400 MHz) δ
282 628
283 690
284 726
1.52 (s,9H).
H-NMR (CDCI3, 400 MHz) δ 8.25
(s, 1H), 8.15 (d, J= 8.0 Hz, 1H),
7.87-7.90 (m, 2H), 7.82 (s, 1H),
5.77 (d, .7 = 3.6 Hz, 1H),3.12(s,
3H), 2.92 (d, J = 4.8 Hz, 3H), 2.63
(s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.25
(s, 1H), 8.20 (d, .7 = 8.0 Hz, 1H),
7.91 (t, 7 = 8.0 Hz, 3H), 7.81 (s,
4
2.94 (d, = 5.2 Hz, 3H), 2.62 (s,
3H), 2.50-2.57 (m, 1H), 1.86-1.90
(m, 3H), 1.45 (s, 9H).
Example 288: 5-(3-r6-cvanobenzo|dlthiazol-2-vnphenyl)-2-(4-fluorophenvn-N-methyl-6-(N-
Step 1 : 2-(4-fl orophenyl)-5-(3-formylphenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (Ql)
L Q1
To a degassed solution of 3-formylphenylboronic acid (440 mg, 2.64 mmol) in dry DMF (20 mL) was added Compound L ( l .O g, 2.20 mmol), K3P04 (1.2 g, 4.40 mmol) and Pd(dppf)Cl2 (20 mg). Then the reaction mixture was placed under N2 atmosphere and stirred at 100 °C for 6 hours. After cooled to room temperature and filtered, the filtrate was washed with H20, brine, and dried over Na2S0 , filtered and concentrated in vacuo. The residue obtained was purified using column chromatography (PE : EtOAc = 3: 1) to provide aryl aldehyde Ql (760 mg, 72.1 %) as white solid; 1H-NMR (CDC13, 400 MHz) δ 10.05 (s, 1 H), 7.98-7.88 (m, 4H), 7.82 (s, 1 H), 7.75 (s, 1H), 7.62-7.59 (m, 2H), 7.59-7.16 (m, 2H), 5.96 (s, 1 H), 3.10 (s, 3H), 2.96 (s, 3H), 2.69 (s, 3H). MS (M+H)+: 481.5.
Step 2: 5-(3-(6-cyanobenzo[d]thiazol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
A mixture of the aryl aldehyhyde Ql (150 mg, 0.31 mmol) and 4-amino-3- mercaptobenzonitrile (56 mg, 0.37 mmol) in DMSO (3 mL) was allowed to stir at 200 °C for 2 hours. After cooled, the mixture was diluted with water and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S0 , filtered and concentrated in vacuo. The residue was purified using preparative HPLC to provide the target compound (150 mg, 79%). Ή-NMR (CDC13, 400 MHz) δ 8.27-8.28 (m, 2H), 8.14-8.19 (m, 2H), 7.94-7.99 (m, 3H), 7.76-7.84 (m, 1 H), 7.63-7.72 (m, 3H), 7.23-7.25 (m, 2H), 5.91-5.92 (m, 1 H), 3.19 (s, 3H), 3.20 (d, J= 4.4 Hz, 3H), 2.81 (s, 3H). MS (M+H)+: 61 1.
The following compounds of the present invention were prepared using the method described in Example 288 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-NIVIR (CDC13, 400 MHz) δ
8.01-8.10 (m, 1H), 7.89-7.98 (m,
6H), 7.68 (s, 1H), 7.53-7.57 (m,
289 1 H), 7.43-7.48 (m, 1 H), 7.34-7.37 604
(m, 1H), 7.24 (t, J =8.8 Hz, 2H),
5.97 (brs, IH), 3.21 (s, 3H), 3.04 (d,
7 = 4.8 Hz, 3H), 2.84 (s, 3H).
8.13-8.16 (m, 2H), 8.02-8.05 (m,
IH), 7.90-7.96 (m, 2H), 7.88 (d, J =
4.0 Hz, 2H), 7.67 (s, IH), 7.47-7.51
290 (m, IH), 7.37-7.41 (m, IH), 604
7.26-7.31 (m, IH), 7.17-7.12 (m,
2H), 6.00 (brs, IH), 3.25 (s, 3H),
2.99 (d, .7 = 4.8 Hz, 3H), 2.64 (s,
3H).
78
8.0
(s,
291 719
8
Ή-NMR (CDCI3, 400 MHz) 58.15
(s, IH), 8.04-8.06 (m, IH),
.82 (s, IH),
.66-7.69 (m,
292 Hz, IH), 604
.09-7.17 (m,
(s, 3H), 2.92 (d, =5.2 Hz, 3H),
2.66 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ
Example 297: 5-(3-(6-(aminomethyl)benzo[dlthiazol-2-yl)phenyl)-2-(4-fluorophenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 188
To a solution of the compound of Example 188 (120 mg, 0.20 mmol) and NH4OH (0.5 mL) in MeOH ( 10 mL), was added Raney-Ni ( 100 mg). The resulting solution was degassed and then was shaken under hydrogen gas atmosphere (30 psi) for about 1 5 hours. The reaction mixture was filtered and the collected solid was washed with MeOH. The filtrate and washing were combined and concentrated in vacuo to provide the target compound (80 mg, 66%). Ή-NMR (MeOD, 400 MHz) δ 8.23 (s, 1 H), 8.12-8.14 (m, 1 H), 8.06-8.09 (m, 2H), 7.94-7.97 (m, 2H), 7.82 (s, 1 H), 7.74 (s, 1 H), 7.69 (s, 1 H), 7.57-7.67 (m, 2H), 7.22-7.26 (m, 2H), 4.24 (s, 2H), 3.1 8 (s, 3H), 2.92 (s, 3H), 2.89 (s, 3H). MS (M+H)+: 615.
Example 298: 5-(3-(6-(( dimethylamino)methyl)benzo[dlthiazol-2-yl)phenvn-2-(4- fluorophenyl)-N-methyl-6-(TV-methylmethylsulfonainido)benzofuran-3-carboxainide
Example 197
CF3COOH (0.1 mL) was added to a solution of the compound of Example 197 (50 mg, 0.08 mmol) and paraformaldehyde (5 mg, 0.16 mmol) in MeOH (2 mL). The resulting reaction was allowed to stir at room temperature for 3 hours, then Na(CN)BH3 (10 mg, 0.16 mmol) was added. The reaction mixture was allowed to stir at room temperature for about 15 hours, then was quenched with saturated NH4C1 solution and extracted with EtOAc. The combined organic phases were washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using preparative HPLC to provide the target compound (20 mg, 38%). Ή-NMR (CDC13, 400 MHz) δ 8.14 (s, 1 H), 8.03-8.08 (m, 2H), 7.99 (s, 1 H), 7.87-7.91 (m, 2H), 7.83 (s, 1H), 7.53-7.60 (m, 3H), 7.44-7.46 (m, 1 H), 7.13-7.17 (m, 2H), 5.82-5.83 (m, 1 H), 4.25 (s, 2H), 3.1 1 (s, 3H), 2.92 (d, J = 8.0 Hz, 3H), 2.75 (s, 6H), 2.67 (s, 3H). MS (M+H)+: 643.
Example 299: 5-(3-f3H-iniidazoi4,5-bl pyriditi-2-vi)phenyl)-2-(4-fluorophenyl)-N-methyl-6- (N-inethylmethylsulfonainido)benzofuran-3-carboxamide
A solution of compound Ql (100 mg, 0.385 mmol) in pyridine-2,3-diamine (58 mg, 0.42 mmol) was heated to 160 °C and allowed to stir at this temperature for 2 hours. The
reaction mixture was cooled to room temperature, quenched with water, and extracted with EtOAc. The organic layer was concentrated in vacuo and the resulting residue was purified using prep-TLC (DCM : MeOH = 20 : 1) to provide the target compound (50 mg, 53.7%). Ή- NMR (CDC13, 400 MHz) δ 8.26-8.29 (m, 2H), 8.07 (s, 1 H), 7.74-7.82 (m, 4H), 7.41-7.52 (m, 3H), 7.25-7.27 (m, 1 H), 7.05-7.15 (m, 3H), 3.14 (s, 3H), 2.94 (s, 3H), 2.82 (d, J = 4.8 Hz, 3H). MS (M+H)+: 570.
The following compounds of the present invention were prepared using the method described in Example 299 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure INIMR
(M+H)+ m,
, 3H),
300 ), 587
), 3.15
(s, 3H), 3.04 (d, J = 4.8 Hz, 3H), 2.92
(s, 3H). m,
, 7.43
6.95-7.00 (m, I H), 3.00-3.01 (m,
6H), 2.92 (s, 3H).
3H), 2.99 (d, J = 4.0 Hz, 3H), 2.91
(s, 3H), 2.37 (s, 3H).
1 H-NMR (CDC13, 400 MHz) δ 8.33
3.24 (s, 3H), 2.98 (s. 3H), 2.92 (s, 3H), 2.3 (m, 3H).
Ή-NMR: (CDC)3, 400 MHz) δ 8.54
3.08 (s, 3H),2.94 (d, =4.8 Hz, 3H), 2.80 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ
9.52-9.61 (m, IH), 8.36 (s, IH),
3.01 (s, 3H), 2.96 (d, 7= 4.8 Hz,
3H), 2.86 (s, 3H).
Ή-NMR (MeOD, 400 MHz) δ 8.54
(s, 3H).
Ή-NMR (MeOD, 400 MHz) δ
3.11 (s, 3H), 2.91 (s, 3H), 2.86 (s,
3H).
Ή-NMR (CDClj, 400 MHz) δ 8.32
3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.22
(s, IH), 7.84-7.89 (m, 3H), 7.73 (s,
317 IH), 7.66-7.67 (m, IH), 7.52 (s, IH), 634
7.09-7.14 (m, 4H), 3.87 (s, 3H), 3.04
(s, 3H), 2.87 (s, 3H), 2.77 (s, 3H).
(d,J = 4.0 Hz, IH), 58.26 (d,7 =
8.0 Hz, IH), 8.05 (s, IH), 7.98-8.00 (m, 2H), 7.97 (s, 1 H), 7.90 (s, 1 H), 604 7.74-7.78 (m, 2H), 7.49-7.52 (m,
2H), 7.26-7.30 (m,2H), 3.22 (s,
3H), 2.99 (s, 3H), 2.96 (s, 3H).
Ή-NMR (400 MHz, MeOH) δ
2.96 (s, 3H).
Ή-NMR (MeOD, 400 MHz) δ 8.45 (d, J = 8.0 Hz, IH), 7.96-7.99 (m,
3H), 7.91 (s, IH), 7.76-7.87 (d, J =
8.0 Hz, 3H), 7.50-751 (d, J = 4.0 Hz,
6i
8 IH), 7.36-7.31 (m, 2H), 3.21 (s, 3H),
3.00 (s, 3H), 2.95 (s, 3H), 2.84 (s,
3H).
Ή-NMR (MeOD, 400 MHz) δ
8.42-8.43 (d, J = 4.0 Hz, IH),
8.11-8.12 (d, .7 = 4.0 Hz, IH),
7.97-8.01 (m, 3H), 7.89 (s, IH), 7.79
638 (s, IH), 7.24 (s, 2H), 7.26-7.30 (m,
2H), 3.22 (s, 3H), 2.97 (s, 3H), 2.96
(s, 3H)
Ή-NMR (MeOD, 400 MHz) δ 8.50
= 4.4 Hz, 3H), 2.83 (s, 3H).
Ή NMR (CDCI3, 400 MHz) 5 8.19
(s, I H), 8.15 (d, J = 3.6 Hz, I H),
8.09 (d, J = 7.6 Hz, 1 H), 7.86-7.89
329 (m, 3H), 7.57 (s, 2H), 7.43 (s, I H), 664
7.06-7.1 8 (m, 3H), 6.05 (s, I H),
3.49 (s, 3H), 3.12 (s, 3H), 2.94 (d, J
= 4.8 Hz, 3H), 2.83 (s, 3H).
'H-NMR (CDCh, 400 MHz) δ
2.96 (d, J = 4.4 Hz, 3H), 2.88 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.54
3H), 2.92 (s, 3H), 2.80 (d, J = 4.0 Hz, 3H).
3H), 2.96 (d, J = 4.4 Hz, 3H), 2.88
(s, 3H).
Ή-NMR (MeOD, 400 MHz) δ 9.23 H),
333 01 570
3H), 2.95 (s, 3H), 2.92 (s, 3H).
'H-NMR (CDCIj, 400 MHz) δ 9.78
I H), 3.28 (s, 3H), 3.09 (s, 3H), 2.88
(d, J = 4.8 Hz, 3H).
Ή-NMR (CDCIj, 400 MHz) 9.08
3H), 2.94 (s, 3H), 2.93 (s, 3 H).
Ή-NMR (CDC13, 400 MHz) δ 10.78
(br s, 1 H), 8.54 (s, 1 H), 8.40 (s, 2H),
7.84-7.88 (m, 2H), 7.72-7.74 (m,
338 2H), 7.50 (s, IH), 7.08-7.13 (m, 3H),
6oi 6.68 (s, IH), 4.14 (s,3H), 3.07 (s,
IH), 3.13 (s, 3H), 2.96 (d, J = 4.8 Hz,
3H), 2.82 (s, 3H).
Ή-NMR (DMSO, 400 MHz) δ 9.32 (s, IH), 9.05 (s, IH), 8.20-8.21 (m,
341 1 H), 7.70-7.77 (m, 4H), 7.46-7.48 571
(m, 2H), 7.29-7.31 (m, 3H), 3.10 (s,
3H), 3.01 (s, 3H), 2.85 (s, 3H).
'.H-NMR (MeOD, 400 MHz) δ 9.04
H), 8.89 (s, IH), 8.28-8.3.1 (m,
, 7.87-7.91 (m, 2H), 7.81 (s,
342 , 7.67-7.73 (m, 2H), 7.36-7.41 589
2H), 7.36-7.41 (m, IH), 7.19 (t,
8.8 Hz, 2H), 3.15 (s, 3H), 2.86
(s, 6H).
Ή-NMR (DMSO, 400 MHz) 59.10 (s, IH), 8.95 (s, IH), 8.52-8.53 (m,
IH), 8.15 (s, IH), 7.97-8.08 (m,
4H), 7.70 (s, IH), 7.50-7.52 (m,
589 IH), 7.38-7.42 (m, 2H), 3.18 (s,
3H), 2.98 (s.3H), 2.79-2.80 (m,
3H).
Ή-NMR (CDC13, 400 MHz) δ 9.02
8.55-8.56 (m, 1 H), 8.30-8.3 1 (m,
1 H), 8.04 (s, 1 H), 8.04-7.97 (m,
349 2H), 7.68-7.70 (m, 1 H), 7.59 (s,
lH), 7.40-7.45 (m, 3H), 4.1 1 (br s,
2H), 3.15 (d, J = 4.0 Hz, 6H), 3.00
(s, 3H), 2.80 (d, .7 = 4.8 Hz, 3H).
Example 351 : 5-(3-(benzofblthiophcn-2-yl)phenyl)-2-(4-fluorophenyl)-N-mcthyl-6-(N- methylmethylsu fonainido)benzofuran-3-carboxamide
Step 1: 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(3- nitrophenyl)benzofuran-3-carboxamide
To a degassed solution of Compound L (prepared as described in Example 1 , Step 1 1 , 2.0 g, 4.39 mmol) and 3-nitrophenylboronic acid (880 mg, 5.27 mmol) in dry DMF (1.5 mL) were added Pd(dppf)Cl2 (20 mg) and K3P04 (1.86 g, 8.79 mmol) under N2. The mixture was allowed to stir at 90 °C for about 15 hours. After the mixture was cooled to room temperature, diluted with EtOAc and filtered, the filtrate was washed with H20, brine, and dried over Na2S04. After concentrated, the crude was purified using column chromatography (PE : EtOAc = 3 : 1 ) to provide 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(3-
nitrophenyl)benzofuran-3-carboxamide (1 .78 g, 84%). Ή-NMR (CDC13, 400 MHz) δ 8.24 (s, 1 H), 8.18 (d, J = 8.4 Hz, 1 H), 7.83-7.87 (m, 2H), 7.79 (d, J = 5.6 Hz, 1 H), 7.77 (s, 1 H), 7.58 (s, 1 H), 7.55 (t, J = 4.0 Hz, 1 H), 7.15 (t, J = 8.8 Hz, 2H), 5.83 (d, J = 3.2 Hz, 1 H), 3.09 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H), 2.73 (s, 3H).
Step 2: 5 -(3 -aminophenyl) -2 -(4 -fluorophenyl) -N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
To a solution of 2-(4-fluorophenyl)-N-rnethyl-6-(N-methylmethylsulfonamido)-5-
(3-nitrophenyl)benzofuran-3-carboxamide (1.0 g, 2.01 mmol) in MeOH (30 mL), Pd/C (200 mg) was added and the resulting reaction mixture was allowed to stir under 40 psi of H2 atmosphere for 24 hours at 25 °C. Then the reaction mixture was filtered, and the filtrate was concentrated in vacuo to provide 5-(3-aminophenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (846 mg, 89%). Ή-NMR (DMSO, 400 MHz) δ 8.49 (d, J = 4.8 Hz, 1 H), 7.94-7.97 (m, 2H), 7.84 (s, 1 H), 7.43 (s, 1 H), 7.38 (t, J = 9.2 Hz, 2H), 7.03 (t, J = 8.0 Hz, 1 H), 6.53-6.58 (m, 3H), 5.09 (s, 2H), 3.13 (d, J = 5.6 Hz, 3H), 3.04 (s, 3H), 2.81 (s, 3H). MS (M+H)+: 468. Step 3: 2-(4-fluorophenyl)-5-(3-iodophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
To a stirred solution of 5-(3-aminophenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide ( 1.5 g, 3.21 mmol) in MeCN (20 mL) was added I
2 (488.6 mg, 1 .93 mmol) and Cul (6 mg) at 0 °C, then i-AmONO (394.6 mg, 3.37 mmol) was added dropwise. After the solution was allowed to stir at 25 °C for 6 hours, the mixture was
heated to 90 °C for 1 hour. The mixture was diluted with Na
2S
20
3 and concentrated to remove the organic solvent, and then the residue obtained was extracted with EtOAc. The organic layer was washed with brine, dried over Na
2S0
4 and concentrated in vacuo. The residue obtained was purified using flash column chromatography (PE : EtOAc = 10 : 1 ) to provide 2-(4- fluorophenyl)-5-(3-iodophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide (1.17 g, 65%). Ή-NMR (CDC1
3, 400 MHz) δ 7.85-7.88 (m, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.66 (d, J = 8.0 Hz, 1 H), 7.53 (s, 1 H), 7.38 (d, J = 7.6 Hz, 1 H), 7.14 (t, J = 6.0 Hz, 2H), 5.77 (d, J = 4.0 Hz, 1H), 3.06 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H), 2.61 (s, 3H). MS (M+H)
+: 579. Step 4 - 5-(3-(bemo[b]thiophen-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
197
To a degassed solution of 5-(3-aminophenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (70 mg, 121.0 umol) and
benzo[b]thiophen-2-ylboronic acid (26.1 mg, 145.1 umol) in dry DMF (1.5 mL) were added Pd(dppf)Cl2 (5 mg) and K3P04 (51.4 mg, 171.2 umol) under N2. The mixture was heated to 90 °C for about 15 hours. After the reaction mixture was cooled to room temperature, diluted with EtOAc and filtered, the filtrate was washed with H20, brine, dried over Na2S04. After concentrated, the crude was purified using prep-TLC (PE : EtOAc = 3 : 1 ) to provide 5-(3- (benzo[b]thiophen-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (38 mg, 60%). Ή-NMR (CDC13, 400 MHz) δ 7.95-7.98 (m, 2H), 7.85 (d, J= 12 Hz, 3H), 7.80 (d, J= 7.6 Hz, 1H), 7.76 (d, J = 6.8 Hz, 1 H), 7.64 (t, J= 3.2 Hz, 2H), 7.52 (d, J = 7.6 Hz, 1 H), 7.44 (d, J= 8.0 Hz, 1 H), 7.37 (t, J= 8.8 Hz, 2H), 7.22 (t, J= 8.8 Hz, 2H), 6.04 (d, J = 4.4 Hz, 1 H), 3.20 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.67 (s, 3H). MS (M+H)+: 585.
The following compounds of the present invention were prepared using the method described in Example 351 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-NMR (DMSO, 400 MHz) δ 8.53
(d, = 4.8 Hz, 1H), 8.02 (d,V=6.8
3H), 2.96 (s, 3H), 2.80 (d, J = 4.4
Hz, 3H).
Ή-NMR (CDCI3, 400 MHz) 69.19
(s, IH), 8.71 (d, = 7.2 Hz, 2H),
8.09-8.20 (m, 2H), 7.88-7.91 (m,
353 3H), 7.77-7.82 (m, 3H), 7.53-7.60 580
(m, 3H), 7.11-7.16 (m, 2H), 6.04 (s,
IH), 3.12 (s,3H), 2.93 (d, J = 4.4
Hz, 3H), 2.72 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 9.67
8
(d, .7=4.8 Hz, 3H), 2.96 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.94
(s, IH), 7.89-7.86 (m, 2H), 7.82 (s,
4.0 Hz, IH), 3.78 (s, 3H), 2.91 (d, J
= 6.8 Hz, 9H).
Ή NMR (CDClj, 400 MHz) δ 9.18
(s, IH), 8.35 (s, IH), 8.06 (d,J=8.8
,
Hz, IH), 3.09 (s,3H), 2.91 (d, J =
4.8 Hz, 3H), 2.66 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ
8.85-8.88 (m, IH), 8.05-8.20 (m,
2H), 7.99 (d,J= 1.6 Hz, IH), 7.97
-7.91 (m,
2H), 7.69
358 (s, IH), 580
5.80 (d, J =4.4 Hz, IH), 3.10 (s,
3H), 2.92 (d, J = 4.8 Hz, 3H), 2.64
(s, 3H).
Ή NMR (CDCI3, 400 MHz) δ
7.92- 7.95 (m,4H), 7.83 (s, IH),
2.97 (d, J = 4.8 Hz, 3H), 2.62 (s,
3H).
Ή NMR (CDCI3, 400 MHz) δ 8.92
(s, IH), 7.92-7.96 (m, 2H), 7.87 (s,
IH), 5.84 (s, IH), 5.10 (s, 2H),
2.93- 2.98 (m, 9H).
H-NMR (CDCI3, 400 MHz) δ 9.25
(s, 1 H), 8.46 (d, J = 5.6 Hz, 1 H),
8.16 (s, 1 H), 7.94 (d, J = 8.4 Hz,
=
5.76 (d, = 3.6 Hz, 1 H), 3.85 (s,
3H), 3.09 (s, 3 H), 2.92 (d, J = 4.8
Hz, 3 H), 2.75 (s, 3 H).
Exam le 2:
Example 351
To a solution of the compound of Example 351 (100 mg, 0.38 mmol) in 10 mL of acetic acid was added H202 (2 mL) and the resulting reaction mixture was heated to 65 °C and allowed to stir at this temperature for 3 hours. The reaction was then was quenched with aq. Na2S03 and extracted with EtOAc. The organic phase was washed with H20 and brine, dried over MgS04, filtered and concentrated in vacuo. The residue obtained was purified using preparative HPLC to provide the target compound (45 mg, 28%). Ή NMR: (CDC13, 400 MHz) δ 7.92 (s, 1H), 7.86-7.90 (m, 2H), 7.74-7.76 (s, 2H), 7.69-7.70 (m, 1 H), 7.43-7.56 (m, 5H), 7.34-7.38 (m, 2H), 7.14 (t, J = 8.8 Hz, 2H), 5.84 (s, 1 H), 3.18 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.54 (s, 3H). MS (M+H)+: 617.
Example 362
To a solution of the compound of Example 362 (30 mg, 0.13 mmol) in 10 mL of
MeOH, was added Pd/C (10 mg); and the resulting reaction was placed under H2 atmosphere (40 Psi) and allowed to stir at room temperature for 24 hours. The reaction mixture was then filtered and concentrated in vacuo, and the residue obtained was purified using preparative HPLC to provide Compound 209 (20 mg, 85%). Ή-NMR (CDC13, 400 MHz) δ 7.86-7.90 (m, 2H), 7.72-7.73 (m, 2H), 7.54-7.58 (m, 2H), 7.39-7.46 (m, 6H), 7.1 1-7.16 (m, 2H), 5.77-5.78 (m, 1 H), 4.68 (t, J = 8.2 Hz, 1 H), 3.64 (d, J = 8.2 Hz, 2H), 3.09 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.46 (s, 3H). MS (M+H)+: 619.
Example 364: 2-(4-fluorophenyl)-5-(3-(isoquinolin-6-yl)phenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
Step 1: 2-(4-fluorophenyl)-N- ethyl-6-(N-methylmethylsulfonamido)-5-(3-(4, 4,5,5-ietramethyl- /, 3, 2-dioxaborolan-2-yl)phenyl)benzofuran-3-carboxamide
To a degassed solution of 2-(4-fluorophenyl)-5-(3-iodophenyl)-N-methyl-6-(lSl- methy]methylsulfonamido)benzofuran-3-carboxamide (Prepared as described in Example 7, Step 3, 200 mg, 0.346 mmol) and pinacol diborane (132 mg, 0.519 mmol) in dry DMF (1.5 mL) was added Pd(dppf)Cl2 (10 mg) and KOAc (102 mg, 1 .04 mmol). The mixture was placed under N2 atmosphere, then heated to 90 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature, filtered, and the filtrate was washed with H20, brine, dried over Na2S0 , filtered and concentrated in vacuo to provide 2-(4-fluorophenyl)-N- methyl-6-(N-methylmethylsulfonamido)-5-(3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)phenyl)benzofuran-3-carboxamide (200 mg, 100%), which was used without further purification. Ή-NMR (CDC13, 400 MHz) δ 7.88-7.92 (m, 2H), 7.75-7.78 (m, 2H), 7.72 (s, 1 H), 7.56 (s, 1H), 7.49-7.52 (m, 1H), 7.37-7.41 (m, 1 H), 7.1 1-7.15 (m, 2H), 5.81-5.82 (m, 1 H), 3.05 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.51 (s, 3H), 1.29 (s, 12H). MS (M+H)+: 579. Step 2: 2-(4-fluorophenyl)-5-(3-(isoquinolin-6-yl)phenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5-(3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)phenyl)benzofuran-3-carboxamide (90 mg, 0.189 mmol) and 6-bromo-isoquinoline (51 mg, 0.246 mmol) in dry DMF (1.5 mL) was added Pd(dppf)Cl2 (20 mg) and K3P04 (81 mg, 0.381 mmol) under N2. The mixture was heated to 100 °C for about 15 hours. The reaction mixture was cooled to room temperature and filtered. The filtrate was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep- TLC (PE : EtOAc = 2 : 1 ) to provide 2-(4-fluorophenyl)-5-(3-(isoquinolin-6-yl)phenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (85 mg, 93%). Ή-NMR
(CDCI3, 400 MHz) δ 9.62 (s, 1 H), 8.46 (d, J = 6.0 Hz, 1 H), 8.38 (s, 1 H), 8.31 -8.33 (m, 1 H), 8.21-8.23 (m, 1H), 8.15 (d, J = 6.0 Hz, 1 H), 7.98 (s, 1 H), 7.81-7.85 (m, 3H), 7.71-7.72 (m, 1 H), 7.51-7.60 (m, 3H), 7.12-7.19 (m, 2H), 6.02-6.03 (m, 1 H), 3.02 (s, 3H), 2.89-2.92 (m, 6H). MS (M+H)+: 580.
The following compound of the present invention was prepared using the method described in Example 364 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-NMR (CDCI3, 400 MHz) δ 9.79
(s, 1 H), 8.50 (s, 1 H), 8.3 1 (d, .7 = 8.0
I H), 3.02 (s, 3H), 2.94 (d, 7 = 4.8
Hz, 3H), 2.87 (s, 3H).
Example 366: 5-(3-(lH-indol-2-yl)phenvn-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
Step 1: 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethyls lfonamido)-5-(3-(l-((2- (lrimethylsilyl)ethoxy)methyl)-lH-indol-2-yl)phenyl)benzofuran-3-carboxamide
5-bromo-2-(4-fluorophenyl)-N-methyl-6-( -methylmethylsulfonamido)-l - benzofuran-3-carboxamide (prepared as described in Example 1 , Step 1 1 ) was converted to 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(3-(l -((2- (trimethylsilyl)ethoxy)methyl)-lH-indol-2-yl)phenyl)benzofuran-3-carboxamide (120 mg, 53.4%) using the method described in Example 1 , Step 1. Ή-NMR (CDC1
3, 400 MHz) δ
8.07-8.03 (m, 2H), 7.93 (s, 1H), 7.82-7.80 (m, 2H), 7.74-7.72 (m, 2H), 7.65-7.60 (m, 2H), 7.37-7.35 (m, 2H), 7.32-7.27 (m, 3H), 6.77 (s, 1 H), 6.05 (d, J = 4.4 Hz, 1H), 5.61 (s, 2H), 3.62 (t, J = 8.4 Hz, 2H), 3.31 (s, 3H), 3.08 (d, J = 4.8 Hz, 3H), 2.72 (s, 3H), 0.95 (t, J = 8.4 Hz, 2H), 0.00 (s, 9H). MS (M+H)+: 698.
Step 2: 5-(3-(l H-indol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5-(3 -( 1 -((2-
(trimethylsilyl)ethoxy)methyl)-lH-indol-2-yl)phenyl)benzofuran-3-carboxamide (60 mg, 0.86 mmol) and TBAF (61 AA mg, 2.57 mmol) in DMF (2 mL) was added to a flask, ethylene diamine (25.83 mg, 0.95 mmol) was added. The mixture was purged with nitrogen and heated at 80 °C for about 15 hours. The mixture was diluted with EtOAc and washed with 0.1 M HC1. The phases were separated, and the organic phase was washed with water and brine, dried over
Na2S0 , filtered and concentrated in vacuo. The resulting redisue was purified using preparative TLC to provde 5-(3-(l H-indol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6-( - methylmethylsulfonamido)benzofuran-3-carboxamide (20 mg, 41.4%). Ή-NMR (CDC13, 400 MHz) δ 9.30 (s, 1 H), 7.94 (d, J = 8.8 Hz, 3H), 7.83 (s, 1H), 7.74 (d, J = 8.0 Hz, 1 H), 7.65 (t, J = 7.2 Hz, 1 H), 7.52-7.47 (m, 2H), 7.43 (d, J = 8.0 Hz, 1 H), 7.35 (d, J = 6.8 Hz, 1 H), 7.22-7.17 (m, 3H), 7.14-7.10 (m, 1 H), 6.85 (s, 1H), 6.09 (d, J = 4.4 Hz, 1H), 2.99 (s, 3H), 2.97 (d, J = 4.0 Hz, 3H), 2.92 (s, 3H). MS (M+H)+: 568.
The following compounds of the present invention were prepared using the method described in Example 366 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-N R (CDCIj, 400 MHz) δ 9.10
(s, IH), 7.89-7.84 (m, 4H), 7.66 (d,
7 = 8.0 Hz, 1 H), 7.45 (t, 7 = 5.6 Hz,
2H), 7.33 (d, = 7.2 Hz, 1 H),
367 7.28-7.25 (m, 1 H), 7.17-7.12 (m, 586
3H), 6.88-6.83 (m, 1 H), 6.73 (d, 7 =
I.2 Hz, IH), 5.84 (d, J = 4.4 Hz,
IH), 2.96 (s, 3H), 2.93 (d, J = 4.8
Hz, 3H), 2.88 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ 8.26
(d, .7 = 6.0 Hz, IH), 8.17 (d, 7 = 8.0
IH), 6.52 (s, IH), 2.99-3.99 (m,
9H).
Ή-N!VIR (CDCI3, 400 MHz) δ
14.50 (s, IH), 8.30 (d, 7 = 4.4 Hz,
IH), 8.03-8.05 (m, IH), 7.84-7.95
(m, 4H), 7.81 (s, IH), 7.55-7.86 (m,
369 2H), 7.48-7.50 (m, IH), 7.30-7.32 569
(m, IH), 7.12-7.16 (m,2H), 7.01 (s,
IH), 5.94 (d, 7 = 4.8 Hz, IH), 3.16
(s, 3H), 2.93 (d, 7 = 4.8 Hz, 3H),
2.66 (s, 3H).
Ή-NMR (DMSO, 400 MHz) δ
II.18(s, IH), 8.55 (d,7=4.8 Hz,
(d, 7 = 4.4 Hz, 3H), 2.45 (s, 3H).
Ή-NMR (CDCI3> 400 MHz) 5 9.15
(s, I H), 7.96-8.01 (m, 3H), 7.93 (s,
3H), 2.91 (s, 3H).
Ή-NMR (CDC13, 400 MHz) δ 9.00
Ή-NIVIR (CDCI3, 400 MHz) δ 9.57
(s, IH), 8.07 (d, .7=7.6 Hz, IH),
Hz, 3H), 2.96-2.97 (m, 6H).
Ή-NMR (CDCI3, 400 MHz) δ 8.32
(d,J = 7.6 Hz, IH), 8.01 (d,J= 5.6
Hz, IH), 7.89-7.93 (m, 2H), 7.81 (s, IH), 7.55 (s,2H), 7.41 (s, IH), 7.32
378 (m, IH), 7.12-7.16 (m,2H), 7.15 (s, 599
IH), 7.09 (s, 1Η 6.99 (s, IH), 5.96
(s, IH), 3.91 (s,3H),3.12(s,3H),
2.95 (d, J = 4.8 Hz, 3H), 2.75 (s,
3H).
H-NMR (CDCI3, 400 MHz) δ 9.98
(s, 1 H), 8.54-8.57 (m, 1 H),
(s, 3H).
'H-NMR (CDCI3, 400 MHz) δ 9.75
(s, 1 H), 8.29-8.3 1 (m, 1 H),
8.01 -8.04 (m, 1 H), 7.91 -7.95 (m,
5.84 (d, J = 4.4 Hz, 1 H), 3.02 (s,
3 H), 2.98 (d, 7 = 4.8 Hz, 3 H), 2.97
(s, 3H).
Example 381 : 5-(3-(3-chloro-l H-indol-2-yl)phenvI)-2-(4-fluorophenvn-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
Example 366
To a solution of Example 366 (50 mg, 0.088 mmol) in 2 mL of DMF, was added NCS (15 mg, 0.088 mmol), and the resulting reaction was allowed to stir under N2 atmosphere for 4 hours at 25 °C. The reaction mixture was concentrated in vacuo and the resulting residue
\
was"diluted EtOAc. The resulting solution was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using prep-TLC (PE : EtOAc = 2 1 ) to provide the title compound (20 mg, 50%) as a white solid. Ή-NMR (CDC13, 400 MHz) δ 9.29 (s, 1 H), 7.97 (d, J = 7.6 Hz, 1 H), 7.83-7.86 (m, 2H), 7.78 (s, 1 H), 7.57 (d, J = 8.0 Hz, 1 H), 7.49 (t, J = 7.6 Hz, 1H), 7.41 (s, 1 H), 7.33 (t, J = 5.6 Hz, 2H), 7.17 (d, J = 7.6 Hz, 1 H),
7.09-7.15 (m, 3H), 5.92 (d, J = 4.4 Hz, 1 H), 2.97 (s, 3H), 2.87 (d, J = 4.8 Hz, 3H), 2.85 (s, 3H). MS (M+H)+: 602.
Example 382: 5-(3-(3-bromo-lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)-N-methyl-6- N- methylmethylsulfonamido)benzofuran-3-carboxamide
Example 366
To a solution of the compound of Example 366 (50 mg, 0.088 mmol) in 3 mL of
DMF, was added NBS ( 16 mg, 0.088 mmol) and the resulting reaction was heated to 75 °C and allowed to stir at this temperature for 4 hours. The reaction mixture was cooled to room temperature and concentrated in vacuo. The resulting residue was diluted with EtOAc and the resulting solution was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-TLC (PE : EtOAc = 2 : 1 ) to provide the title compound (40 mg, 89%) as a white solid. Ή-NMR (CDC13, 400 MHz) δ 9.38 (s, 1 H), 8.02 (d, J = 8.0 Hz, 1 H), 7.94 (s, 1 H), 7.88-7.94 (m, 2H), 7.84 (s, 1 H), 7.53 (t, J = 7.6 Hz, 2H), 7.46 (d, J = 4.8 Hz, 1 H), 7.35-7.40 (m, 2H), 7.1 1 -7.1 5 (m, 4H), 5.80 (s, 1 H), 3.04 (s, 3H), 2.94 (d, J = 5.2 Hz, 3H), 2.87 (s, 3H). MS (M+H)+: 646.
Example 383: 2-(4-fluorophenyl)-5-(3-(3-(hvdroxymethyl)-lH-pyrrolo|2,3-b]pyridin-2- yl)phenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 379
To a solution of the compound of Example 379 (50 mg, 0.084 mmol) in MeOH (5 mL) was added NaBH4 (17 mg, 0.5 mmol) and the resulting reacton was allowed to stir at room temperature for 2 hours. The reaction mixture was diluted with water and extracted with dichloromethane and the organic extract was dried over Na2S04, filtered and concentrated in vacuo to provide the title compound (20 mg, 40%). Ή-NMR (CDC13, 400 MHz) δ 10.15-10.25 (m, 1 H), 8.22 (d, J = 3.6 Hz, 1 H), 8.02-8.04 (m, 1H), 7.88-7.91 (m, 3H), 7.82 (s, 1 H),
7.70-7.72 (m, 1 H), 7.50-7.54 (m, 1H), 7.48 (s, 1 H), 7.40-7.42 (m, 1H), 7.12-7.16 (m, 2H), 7.05-7.08 (m, 1 H), 5.93-5.98 (m, 1 H), 4.92 (s, 2H), 2.96 (s, 3H), 2.91-2.93 (m, 6H).
Example 384: 2-(4-fluorophenyl)-6-(N-(3-hvdroxypropyl)methylsulfonamido)-N-inethyl-5- (3-(oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-3-carboxamide
Step 1: 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido)benzofuran-3-carboxylic acid
0
To a solution of methyl 5-bromo-2-(4-fluorophenyI)-6-
(methylsulfonamido)benzofuran-3-carboxylate (prepared as described in Example 1 , Step 8, 0.5 g, 1.13 mmol) in dioxane (3 mL) and water (1 mL) was LiOH H20 (0.24 g, 5.65 mmol). The resulting reaction was heated to 80 °C and allowed to stir at this temperature for 2 hours. The reaction mixture was cooled to room temperature and adjusted to pH = 6-7 using cone. HCI. The resulting solution was extracted with EtOAc, and the organic phase was dried over anhydrous Na2S04, filtered and concentrated in vacuo to provide 5-bromo-2-(4-fluorophenyl)-6- (methylsulfonamido)benzofuran-3-carboxylic acid (0.4 g. 87 %) as a white solid. Ή-NMR (DMSO, 400 MHz) δ 13.49 (s, 1 H), 9.67 (s, 1 H), 8.30 (s, 1 H), 8.12-8.17 (m, 2H), 7.87 (s, 1 H), 7.45-7.50 (m, 2H), 3.16 (s, 3H). MS (M+H)+: 428.
Step 2: 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide
To a solution of 5-bromo-2-(4-fluorophenyl)-6-(methylsulfonamido)benzofuran-
3-carboxylic acid (420 mg, 0.77 mmol) in DMF (10 mL) was added EDCI (295 mg, 1.57 mmol) and HOBT (104 mg, 0.77mmol), and the resulting reaction was allowed to stir at room temperature for 3 hours. CH3NH2 HC1 (102 mg, 1.54 mmol) and Et3N (3 mL) were then added to the reaction mixture and the resulting reaction was allowed to stir at room temperature for an additional 8 hours. The reaction mixture was then concentrated in vacuo and the residue obtained was diluted with EtOAc. The resulting solution was washed with HCI (1 N) and NaOH (1 N), dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-2-(4- fluorophenyl)-N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (400 mg. 87 %).
Ή-NMR (DMSO, 400 MHz) δ 9.55 (br s, 1 H), 8.46-8.48 (m, 1H), 8.12-8.17 (m, 2H), 7.96 (s, 1 H), 7.87 (s, 1 H), 7.45-7.50 (m, 2H), 3.16 (s, 3H), 2.93 (d, J= 8.4 Hz, 3H). MS (M+H)+: 441 .
Step 3: 5-bromo-2-(4-fluorophenyl)-6-(N-(3-hydroxypropyl)methylsulfonamido)-N- methylbenzofuran-3-carboxamide
To a solution of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-
(methylsulfonamido)benzofuran-3-carboxamide (300 mg, 0.68 mmol) in DMF ( 10 mL) was added 3-bromopropan-l -ol (190 mg, 1.36 mmol), K2C03 (188 mg, 1.36 mmol) and I (1 1 mg, 0.068 mmol). The resulting reaction was heated to 100 °C and allowed to stir at this temperature for 10 hours. The reaction mixture was cooled to room temperature and concentrated in vacuo. The resulting residue was taken up in EtOAc and the resulting solution was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified by flash column chromatography (PE : EtOAc = 2 : 1 ) to provide 5-bromo-2-(4-fluorophenyl)-6- ( -(3-hydroxypropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide (320 mg.,
78.6 %). Ή-NMR (CDC13, 400 MHz) δ 8.12 (s, 1 H), 7.76 (d, J = 8.0 Hz, 2H), 7.65 (s, 1 H), 7.14 (d, J = 8.4 Hz, 2H), 5.78 (br s, 1 H), 3.64-3.67 (m, 2H), 3.55-3.60 (m, 2H), 3.08 (s, 3H), 2.97 (d, J = 4.4 Hz, 3H), 1.72-1.76 (m, 2H). MS (M+H)+: 499. Step 4: 2-(4-fluorophenyl)-6-(N-(3-hydroxypropyl)methylsulfonamido)-N-methyl-5-(3- (oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (Compound 230)
To a degassed solution of 5-bromo-2-(4-fluorophenyl)-6-(N-(3- hydroxypropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide (100 mg, 0.20 mmol) and 2-(3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)oxazolo[4,5-b]pyridine (77 mg,
0.24 mmol) in dioxane / CH3CN / H20 (10 / 1 / 1 , 5 mL) was added Pd(PPh3)4 (2 mg) and K3C03 (100 mg, 0.40 mmol). The reaction was put under N2 atmosphere and heated to 100 °C in microwave for 30 minutes. The reaction mixture was filtered, and the filtrate was diluted with EtOAc, and the resulting solution washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography (PE : EtOAc = 1 : 1 ) 2-(4-fluorophenyl)-6-(N-(3-hydroxypropyl)methylsulfonamido)-N-methyl- 5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (38 mg, 30.9%). Ή-NMR (CDC13, 400 MHz) 5 8.50 (J = 4.4 Hz, 1 H), 8.38-8.41 (m, 1 H), 8.23 (d, J = 8.0 Hz, 1 H), 7.81-7.87 (m, 2H), 7.56-7.58 (m, 3H), 7.25-7.26 (m, 2H), 7.18-7.20 (m, 1 H), 7.1 1-7.15 (m, 2H), 6.07 (br s, 1 H), 3.64-3.67 (m, 2H), 3.41-3.52 (m, 2H), 2.92-2.93 (m, 3H), 2.81 (s, 3H), 1.72-1 .76 (m, 2H). MS (M+H)+: 615.
The following compounds of the present invention were prepared using the method described in Example 384 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure NMR
(M+H)+
Ή-NMR (CDC13, 400 MHz) δ 8.57
(d, .7 = 4.8 Hz, 1 H), 8.46 (s, 1 H),
8.23 (d, J = 8.0 Hz, 1 H), 7.98 (d, J =
8.0 Hz, I H), 7.81 -7.88 (m, 4H),
385 7.58-7.62 (m, 2H), 7.36-7.41 (m, 601
1 H), 7.12-7.17 (m, 2H), 5.98 (br s,
I H), 3.60-3.70 (m„ 3H), 3.38-3.44
(m, I H), 2.93 (d, .7 = 4.4 Hz, 3H),
2.89 (s, 3H).
Ή-NMR (CDC , 400 MHz) δ 8.64
3.03 (s, 3H ).
H-NMR (CDC13, 400 MHz) δ 8.53
(d,J = 4.0 Hz, IH), 8.38 (d, = 4.0
Hz, IH), 8.13-8.15 (m, IH),
7.98-8.00 (m, 2H), 7.94 (d, J = 4.0
Hz, IH), 7.86 (s, IH), 7.73 (s, IH),
387 7.37-7.49 (m, IH), 7.30-7.35 (m, 631
IH), 7.26-7.30 (m,2H), 4.08 (s,
3H), 3.71-3.74 (m, IH), 3.46-3.49
(m, 2H), 3.23 (m, 3H), 3.09-3.14
(m, IH), 2.95 (s, 3H).
Ή-NMR (CDCI3, 400 MHz) δ
8.49-8.50 (m, IH), 8.38-8.41 (m,
388 615
389 645
,
2H), 3.15 (s,3H), 2.95 (s, 3H ),
1.30-1.60 (m, 4H).
Ή-NMR (CDCIj, 400 MHz) δ 8.50
2.22-2.25 (m, 2H), 1 .39-1 .58 (m,
4H).
Ή-NIMR (CDCI3, 400 MHz) δ 8.67
(d, J = 4.4 Hz, 1H), 8.47 (s, 1H),
3.04 (d, .7=4.8 Hz, 3H), 3.02 (s,
3H), 2.84-2.96 (m, 2H), 1.57-1.64
(m, 2H), 1.38 (s, 9H).
Ή-NMR (CDCI3, 400 MHz) δ
8.39-8.42 (m, 2H), 8.09-8.16 (m,
2H), 7.95-8.01 (m, 2H), 7.82-7.85
(m, 2H), 7.62 (t, J = 8.0 Hz, 1H),
396 7.57 (s, 1H), 7.38-7.40 (m, 1H), 614
7.20-7.25 (m, 2H), 6.44 (br s, 1 H),
3.50-3.70 (m, 2H), 3.01 (d, J = 4.8
Hz, 3H), 2.97 (s, 3H), 2.80-2.90 (m,
2H), 1.85-1.95 (m, 2H).
Example 397: 2-(4-fluorophenyl)-N-methyl-6-(N-(2-morpholinoethyl)methylsulfonamido)- 5-(3-(oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-3-carboxamide
Step 1 : 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(2-morpholinoethyl)
methylsulfonamido)benzofuran-3-c rboxamide
Triphenylphosphine (180 mg, 0.69 mmol) and 5-bromo-2-(4-fluorophenyl)-N- methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (200 mg, 0.45 mmol, prepared by taking the product of Example 1 , Step 8 and subjecting it to the methods described in Example 1 , Steps 10 and 1 1 ) were taken up in anhydrous THF (10 mL) and to the resulting suspension was added DEAD (120 mg, 0.69 mmol). The resulting reaction was allowed to stir at room temperature in the dark for 1 hour, then a solution of 2~morpholinoethanol (90 mg, 0.69 mmol) in anhydrous THF was added, and the resulting reaction was allowed to stir in the dark at room temperature for about 15 hours. The reaction mixture was concentrated in vacuo and the resulting residue was purified using flash chromatography (PE : EtOAc = 1 : 1 )to provide 5- bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(2-morpholinoethyl)methylsulfonamido)benzofuran- 3-carboxamide (200 mg, 79%). Ή-NMR (CDC1
3, 400 MHz) δ 8.15 (s, 1H), 7.87-7.91 (m, 2H), 7.73 (s, 1 H), 7.18-7.23 (m, 2H), 5.93 (br s, 1H), 4.04-4.12 (m, 1 H), 3.59-3.66 (m, 5H), 3.1 1 (s, 3H), 2.99 (d, J = 4.4 Hz, 3H), 2.48-2.55 (m, 4H), 2.33-2.37 (m,2H). MS (M+H)
+: 554.
Step 2: 2-(4-fluorophenyl-N-methyl-6-(N-(2-morpholinoethyl)methylsulfonamido)-5-(3- (oxazolo 4, 5-b ]pyridine-2-yl)phenyl)benzofuran-3-carboxamide
5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(2- morpholinoethyl)methylsulfonamido)benzofuran-3-carboxamide (20 mg, 0.04 mmol), 2-(3- (4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)oxazolo[4,5-b]pyridine (12 mg, 0.04 mmol) and K2C03 (10 mg, 0.07 mmol) were taken up in a mixture of dioxane/CH3CN/H20 (10/1/1 , 1 mL total solution volume). To the resulting solution was added Pd(PPh3)4 (2 mg) and the resulting reaction was put under N2 atmosphere and heated to 100 °C using microwave radiation. The reaction was allowed to remain at this temperature under microwave radiation for 20 minutes, then was cooled to room temperature and concentrated in vacuo. The residue obtained was purified using preparative HPLC to provide 2-(4-fluorophenyl)-N-methyl-6-(N-(2- morpholinoethyl)methylsulfonamido)-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-
carboxamide (15 mg, 62%). Ή-NMR (CDC13, 400 MHz) δ 8.56 (br s, 1 H), 8.30 (s, 1 H), 8.20-8.22 (m ,1H), 7.97 (d, J = 8.0 Hz, 1 H), 7.81-7.87 (m, 3H), 7.71 (br s, 1 H), 7.58-7.63 (m, 2H), 7.36-7.40 (m, 1H), 7.14-7.19 (m, 2H), 6.37 (br s, 1H), 3.80-4.05 (m, 6H), 3.42 (br s, 2H), 3.21 (br s, 2H), 2.80-3.10 (m, 8H). MS (M+H)+: 670.
The following compounds of the present invention were prepared using the method described in Example 397 and substituting the appropriate reactants and/or reagents.
MS
Compound Structure N IWR
(M+H)+
Ή-NMR (CDCh, 400 MHz) δ
8.48-8.53 (m, 2H), 8.35 (d, J = 8.0
1 H ), 3.06 (s, 3H), 2.93 (d, .7 = 4.0
Hz, 3H), 2.81 (s, 6H).
Ή-NIVIR (CDCI3, 400 MHz) δ 8.64
(d, 7 = 4.8 Hz, 1 H), 8.42 (s, 1 H),
7H), 2.78-2.87 (m, 7H), 1 .98-2.05
(m, 2H). Example 400: 2-(4-fluorophenyl)-N-methyl-6-(N-inethylphenylsulfonamido)-5-(3- (oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-3-carboxamide
methyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
To a 0 °C solution of methyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3- carboxylate (prepared as described in Example 1 , Step 7, 500 mg, 1.4 mmol) and pyridine (5 mL) in dry dichloromethane (10 mL) was added benzenesulfonyl chloride (1.5 g, 8.5 mmol). The cold bath was removed and the resulting reaction was allowed to stir for about 15 hours at room temperature. The reaction mixture was diluted with water, extracted with dichloromethane and the organic extract was washed with brine, dried (Na
2S0
4), filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography (PE : EtOAc = 5 : 1 ) to provide methyl 5-bromo-2-(4-fluorophenyl)-6-(phenylsulfonamido)benzofuran-3-carboxylate (600 mg, 87%). Ή-NMR (CDC1
3, 400 MHz) δ 8.01-8.03 (m, 2H), 7.93-7.95 (d, 2H),
7.68-7.69 (d, I H), 7.62-7.63 (m, IH), 7.50-7.52 (m, 2H), 7.33-7.37 (m, I H) 7.10-7.16 (m, 2H) 5.23 (s, I H). 3.85-3.89 (d, J= 16.8 Hz, 3H). MS (M+H)+: 504.
Step 2: methyl 5-bromo-2-(4-fluorophenyl)-6-(N-methylphenylsulfonamido)benzofuran-3- carboxylate
A solution of methyl 5-bromo-2-(4-fluorophenyl)-6- (phenylsulfonamido)benzofuran-3-carboxylate (0.6 g, 1.18 mmol) and K2C03 (1.1 g, 8.0 mmol) in DMF (15 mL) was put under N2 atmosphere. CH3I (1.0 mL, 16.0 mmol) was added and the resulting reaction was heated to 40 °C and allowed to stir at this temperature for about 15 hours.
The reaction mixture was then filtered and the filtrate was concentrated in vacuo to provide methyl 5-bromo-2-(4-fluorophenyl)-6-(N-methylphenylsulfonamido)benzofuran-3-carboxylate (500 mg, 81 %) which was used without further purification. Step 3: 5-bromo-2-(4-fluorophenyl)-6-(N-methylphenylsulfonamido)benzofuran-3-carboxy acid
To a solution of methyl 5-bromo-2-(4-fluorophenyl)-6-(N- methylphenylsulfonamido)benzofuran-3-carboxylate (500 mg, 0.96 mmol) in a mixture of dioxane/H20 (1/1 , 10 mL total volume) was added LiOH H20 (90 mg, 2.14 mmol), and the resulting reaction was heated to 100 °C and allowed to stir at this temperature for 2 hours. The reaction mixture was cooled to room temperature, then concentrated in vacuo. The residue obtained was dissolved in H20 and the resulting solution was adjusted to pH 3 using HCl (1 N). The acidific solution was then extracted with EtOAc and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-2-(4- fluorophenyl)-6-(N-methylphenylsulfonamido)benzofuran-3-carboxylic acid (300 mg, 62%), which was used without further purification.
Step 4: 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-methylphenylsulfonamido)benzofuran-3- carboxamide
To a solution of 5-bromo-2-(4-fluorophenyl)-6-(N- methylphenylsulfonamido)benzofuran-3-carboxylic acid (300 mg, 0.59 mmol) in dry DMF (10 mL) was added HOBT (100 mg, 0.74 mmol) and EDCI (100 mg, 0.64 mmol) and the resulting reaction was allowed to stir at room temperature for 1 hour. Et3N (2.0 mL) and CH3NH2 (HCl salt, 100 mg, 1.48 mmol) were then added to the reaction mixture and the resulting reaction was allowed to stir for about 15 hours at room temperature. The reaction mixture was concentrated
in vacuo, the resulting residue was diluted with H20, and the resulting aqueous solution was extracted with ethyl acetate. The organic extract was washed with H20 and brine, then concentrated in vacuo. The residue obtained was purified by flash column chromatography (PE : EtOAc = 2 : 1 ) to provide 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylphenylsulfonamido)benzofuran-3-carboxamide (130 mg, 42%). Ή-NMR (CDC13, 400 MHz) δ 8.02 (s, IH), 7.83-7.86 (m, 2H), 7.75-7.77 (d, 2Η), 7.54-7.56 (m, IH), 7.44-7.48 (m, 2H), 7.36 (s, I H), 7.1 1-7.19 (m, 2H), 5.71 (br s, I H), 3.20 (s, 3H), 2.94 (d, J = 4.8 Hz, 3H). MS (M+H)+: 517. Step 5: 2-(4-fluorophenyl)-N-methyl-6-(N-methylphenylsulfonamido)-5-(3-(oxazolo[4,5~ b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (Compound 246)
5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N- methylphenylsulfonamido)benzofuran-3-carboxamide (30 mg, 0.06 mmol), 2-(3-(4,4,5,5- tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)oxazolo[4,5-b]pyridine (22.5 mg, 0.07 mmol) and K2C03 (16 mg, 0.12 mmol) were taken up in a mixture of dioxane-acetonitrile-water (10: 1 : 1 , 2 mL total volume). To the resulting solution was added Pd(PPh3)4 (5 mg) and the resulting reaction was put under N2 atmosphere and heated to 100 °C using microwave radiation. The reaction was allowed to remain at this temperature under microwave radiation for 20 minutes, then was cooled to room temperature and concentrated in vacuo. The residue obtained was purified using preparative HPLC to provide 2-(4~fluorophenyl)-N-methyl-6-(N- methylphenylsulfonamido)-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (4 mg, 1 1%). Ή-NMR (CDC13, 400 MHz) δ 8.57 (d, IH), 8.30 (m, 2H), 7.86-7.90 (m, 3H), 7.82 (s, I H), 7.68 (d, I H), 7.53-7.58 (m, 3H), 7.47-7.49 (m, IH), 7.36-7.40 (m, 2H), 7.30-7.33 (m, I H), 7.12-7.15 (m, 3H), 5.83 (br s, IH), 3.06 (s, 3H), (d, J = 4.8 Hz, 3H). MS (M+H)+: 633.
The following compound of the present invention was prepared using the method described in Example 400 and substituting the appropriate reactants and/or reagents.
IMS
Compound Structure NMR
(1M+H)+
Ή-NMR (CDClj, 400 MHz) δ
8.57-8.58 (d, J = 4.0 Hz, 1 H), 8.36
(s, 1 H), 8.29-8.3 1 (d, .7 = 8.2 Hz,
1 H), 7.82-7.98 (m, 4H), 7.57-7.60
40] (m, 3H), 7.27-7.29 (m, ) H), 585
7. 1 3-7. 17 (m, 2H), 5.82-5.83 (d, J =
8. 1 Hz, 1 H), 3. 1 5 (s, 3H), 2.93-2.94
(d, J = 5.2 Hz, 3 H), 2.76-2.78 (m,
2H), 1 .09-1 .13 (m, 3 H).
Example 402: 5-(3-(4-fluorobenzo|dloxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Step 1: l-fluoro-3-methoxy-2-nitrobenzene
To a 0 °C solution of l ,3-difluoro-2-nitrobenzene (100 g, 0.63 mol) in MeOH (1 .3 L) was slowly added a solution of MeONa (0.69 mol, in MeOH, freshly prepared from 15.9 g of sodium metal and 200 mL of MeOH). The resulting reaction was allowed to stir for about 15 hours at room temperature, then the reaction mixture was concentrated and diluted with EtOAc. The organic phase was washed sequentially with water and brine, dried over Na
2S0
4, then filtered and concentrated in vacuo to provide l -fluoro-3-methoxy-2-nitrobenzene (98 g, yield: 91.4%), which was used without further purification. Ή-NMR (CDC1
3, 400 MHz) δ 7.38-7.44 (m, 1 H), 6.72-6.88 (m, 2H), 3.95 (s, 3H).
Step 2: 3-fluoro-2-nitrophenol
To a -40 °C solution of 1 -fluoro-3-methoxy-2-nitrobenzene (98 g, 0.57 mol) in dichloromethane (500 mL) was added dropwise a solution of BBr3 ( 1 L, 1 M in dichloromethane The resulting reaction was allowed to stir for about 15 hours at room temperature, then the reaction mixture was slowly poured into ice water (500 mL). The resulting solution was extracted with EtOAc (300 mL x 3), and the combined organic layers were washed with sequentially with 5% aqueous NaHC03 and brine, then dried over Na2S0 , filtered and concentrated in vacuo to provide 3-fluoro-2-nitrophenol (85 g, yield: 95%), which was used without further purification. Ή-NMR (CDC13, 400 MHz) δ 7.43-7.49 (m, 1 H), 6.88 (d, J = 8.0 Hz, 1 H), 6.73-6.78 (m, 1 H). Step 3: 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methyl-6- (N-methylsulfonamido)benzofuran-3-carboxamide
3-fluoro-2-nitrophenol (38 g, 0.24 mol) was dissolved in EtOH and then palladium on carbon (5 g, 10% Pd) was added. The reaction flask was evacuated and the reaction mixture was put under H2 atmosphere (1 atm) and allowed to stir for 3 hours at room temperature. The reaction mixture was then filtered through a short pad of celite and the celite was washed with EtOH. The combined filtrate and washing was concentrated in vacuo to provide 2-amino-3-fluorophenoI (26 g, yield: 85.7%), which was used without further purification. Ή-NMR (DMSO, 400 MHz) δ 9.43 (s, 1 H), 6.42-6.53 (m, 2H), 6.32-6.42 (m, 1 H), 4.34 (s, 2H).
Step 4: 2-(5-bromo-2-methoxyphenyl)-4-fluorobenzo[d]oxazole
To a solution of 2-amino-3-fluorophenol (9 g 70.8 mmol) in 10 mL of PPA was added 5-bromo-2-methoxybenzoic acid (16.3 g, 70.8 mmol), and the resulting reaction was heated to 140 °C and allowed to stir at this temperature for 4 hours. The reaction mixture was then poured into ice water (50 mL), and extracted with EtOAc. The organic extract was concentrated in vacuo and the residue obtained was purified using flash column chromatography on silica gel (petroleum ether/ethyl acetate = 10 / 1 ), to provide 2-(5-bromo-2-methoxyphenyl)- 4-fluorobenzo[d]oxazole (16 g, yield: 82%) as a solid. Ή-NMR (CDC13, 400 MHz) δ 8.29 (d, J = 2.4 Hz, 1 H), 7.57-7.54 (m, 1 H), 7.40 (d, J = 8.0 Hz, 1H), 7.27-7.33 (m, 1H), 7.07 (m, 1 H), 6.96 (d, J = 9.2 Hz, 1 H), 3.99 (s, 3H).
Step 5: 4-fluoro-2-(2-methoxy-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2- yl)phenyl)benzo[d] oxazole
A solution of 2-(5-bromo-2-methoxyphenyl)-4-fluorobenzo[d]oxazole ( 18.4 g,
57.1 mmol) and bis(pinacolato)diboron (17.4 g, 68.5 mmol) in DMF (10 mL) was placed under N2 atmosphere and to the resulting solution was added Pd(dppf)Cl2 (500 mg) and AcOK ( 10 g, 1 14 mmol). The reaction was heated to 80 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was then concentrated in vacuo, the residue obtained was dissolved in dichloromethane, and the resulting solution was filtered through a pad of celite. The organic solution was washed sequentiall wiith ¾0 and brine, then dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography on silica gel (PE / EA = 10 / 1 ) to provide 4-fluoro-2-(2-methoxy-5-(4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)phenyl)benzo[d]oxazole (10 g, yield: 54%) as a solid. Ή-NMR (CDC13, 400
MHz) δ 8.53 (d, J = 1.6 Hz, 1H), 7.85-7.92 (m, 1 H), 7.44 (d, J
6.96-7.05 (m, 2H), 3.97 (s, 3H), 1.29 (s, 12H).
Step 6- 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methyl-6- (N-methylsulfonamido)benzofuran-3-carboxamide
To a solution of Compound L (5 g, 1 1 .0 mmol) and 4-fIuoro-2-(2-methoxy-5- (4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)benzo[d]oxazole (5.27 g, 14.3 mmol) in DMF (150 mL) under N2 atmosphere was added Pd(dppf)Cl2 (200 mg) and 3P04 (4.66 g, 22.0 mmol). The resulting reaction was heated to 100 °C and allowed to stir at this temperature for 1 0 hours, then the reaction mixture was concentrated in vacuo. The residue obtained was dissolved in dichloromethane and filtrated through a short pad of celite. The filtrate was washed sequentially with water and brine, dried over Na2S04, then filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography on silica gel (petroleum ether/ethyl acetate = 4 / 1 to 2 / 1 ) and the product obtained was then recrystallized from dichlormethane/ethyl acetate (5 /l ), to provide 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-2-(4-fluorophenyl)-N-methyl-6-(N-methylsulfonamido)benzofuran-3- carboxamide (3.8 g, yield: 56%) as a white solid. Ή-NMR (CDC13, 400 MHz) δ 8.21 (d, J = 2.0 Hz, 1 H), 7.91-7.95 (m, 2H), 7.83 (s, 1 H), 7.68 (d, J = 2.0 Hz, 1 H), 7.66 (s, 1 H), 7.39 (d, J = 8.0 Hz, 1 H), 7.14-7.27 (m, 4H), 7.06 (t, J= 8.4 Hz, 1 H), 5.95 (br s, 1 H), 4.06 (s, 3H), 3.14 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.77 (s, 3H); MS (M+H)+ 61 8.
Example 403: 5-(3-(4-cvanobenzo[d1oxazol-2-yl)-4-methoxyphenyl)-2- 4-fluorophenyl)-N- methyl-6-fN-methylmethylsulfonamido)benzofuran-3-carboxamide
Step 1: 3-hydroxy-2-nitrobenzonitrile
To a 0 °C solution of NaN03 (4 g, 47 mmol) and H2S04 (aqueous, 3 M, 45 mL) was added a solution of 3-hydroxybenzonitrile (5 g, 42 mmol) in CH2C12 (80 mL). To the resulting solution was added NaN02 (289 mg, 4.2 mmol) and the resulting reaction was allowed to stir for 16 hours. The reaction mixture was then diluted with CH2C12 and the resulting solution was washed sequentially with H20 and brine, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (petroleum ether/ethyl acetate = 40 / 1 ) to provide 3-hydroxy-2-nitrobenzonitrile (1 .7 g, yield: 25%). Ή-
NMR (DMSO, 400 MHz) δ 1 1 .73 (s, 1 H), 8.25 (d, J = 8.4 Hz, 1 H), 7.35 (d, J
7.19 (t, J = 8.4 Hz, 1 H)
Step 2: 2-amino-3-hydroxybenzonitrile
To a solution of 3-hydroxy-2-nitrobenzonitrile (1.7 g, 0.01 mol) in MeOH (30 mL) was added SnCl2 (7.9 g, 4.1 mol). The resulting reaction was heated to 50 °C and allowed to stir at this temperature for 6 hours. The reaction mixture was then concentrated in vacuo and the resulting residue was taken up in EtOAc. To the resulting solution was added saturated aqueous NaHCC"3 solution, which caused a white solid to precipitate out of solution. The resulting suspension was filtered through celite and extracted with EtOAc. The organic layer was dried over MgS04, filtered, and concentrated in vacuo to provide 2-amino-3-hydroxybenzonitrile (1 .1 g, yield: 79.7%), which was used without further purification. Ή-NMR (CDC13, 400 MHz) δ 6.94 (d, J = 8.4 Hz, 1 H), 6.79 (d, J = 8.0 Hz, 1 H), 6.53 (t, J = 8.0 Hz, 1 H), 5.17 (s, 1 H), 4.43 (s, 2H).
Step 3: 5-bromo-N-(2-cyano-6-hydroxyphenyl)-2-methoxybenzamide
A solution of 5-bromo-2-methoxybenzoic acid (1 1.7 g, 50.8 mmol) in SOCl2 (50 mL) was heated to 100 °C and allowed to stir at this temperature for 2 hours. The reaction mixture was then concentrated in vacuo and the resulting residue was dissolved in dry dichloromethane (30 mL). The resulting solution was then added dropwise to a solution of 2- amino-3-hydroxybenzonitrile (6.2 g, 46.22 mmol) in dichloromethane (30mL) and triethylamine (15 mL) at 0 °C under N2. The resulting reaction was allowed to stir for 5 hours at room temperature, then the reaction mixture was poured into ice water (50 mL) and extracted with dichloromethane. The organic phase was washed sequentially with H20 and brine, dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-N-(2-cyano-6-hydroxyphenyl)-2- methoxybenzamide (4.0 g), which was used without further purification.
Step 4: 2-(5-bromo-2-methoxyphenyl)benzo[d]oxazole-4-carbonitrile
A solution of 5-bromo-/V-(2-cyano-6-hydroxyphenyl)-2-heated to reflux and allowed to stir at this temperature for 3 hours using a reflux condenser fitted with a Dean-Stark trap. After the was removed, the residue obtained was dissolved in EtOAc (40 mL). The organic phase was washed sequentially with H20 and brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (PE / EA = 10 / 1) to provide 2-(5-bromo-2-methoxyphenyl)benzo[d]oxazole-4-carbonitrile (2.1 g, yield: 26 % two steps) as solid. Ή-NMR (CDC13, 400 MHz) δ 8.70 (s, 1 H), 8.21 -8.24 (m, 1 H), 7.81-7.83 (m, 1 H), 7.70-7.72 (m, 1 H), 7.46-7.48 (m, 1H), 7.15-7.17 (m, 1 H), 4.14 (s, 3H).
Step 5: 2-(2-methoxy-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2-yl)phenyl)benzo[ dJoxazole-4- carbonitrile
To a solution of 2-(5-bromo-2-methoxyphenyl)benzo[d]oxazole-4-carbonitrile
(2.0 g, 6.08 mmol) and bis(pinacolato)diboron (2.01 g, 7.90 mmol) in toluene (25 mL) under N2 atmosphere, was added Pd(dppf)Cl2 (300 mg) and AcO (1.19 g, 12.15 mmol). The resulting reaction was heated to 80 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was then concentrated in vacuo and the resulting residue was dissolved in
dichloromethane and filtrated through a short pad of celite. The organic phase was washed sequentially with H20 and brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (petroleum ether/ethyl acetate = 10 / 1 ) to provide 2-(2-methoxy-5-(4,4,5, 5-tetramethyl-l , 3,2-dioxaborolan- 2-yl)phenyl)benzo[d]oxazole-4-carbonitrile (1.8 g, yield: 78.6%) as solid, which was used without further purification. Ή-NMR (CDC13, 400 MHz) δ 8.65 (s, 1 H), 8.00-8.02 (m, 1 H),
7.84-7.86 (m, 1 H), 7.68-7.70 (m, 1 H), 7.42-7.46 (m, 1H), 7.10-7.12 (m, 1H), 4.08 (s, 3H), 1 .41 (s, 12H).
Step 6: 5-(3-(4-cyanobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methyl-6- (N-methylsulfonamido)benzofuran-3-carboxamide
To a solution of Compound L (1.21 g, 2.66 mmol) and 2-(2-methoxy-5-(4,4,5,5- tetramethyl- l ,3,2-dioxaborolan-2-yl)phenyl)benzo[d]oxazole-4-carbonitrile ( 1 .20 g, 3.19 mmol) in DMF (12 mL) under N2 atmosphere, was added Pd(dppf)Cl2 (400 mg) and K3P04 (1 .42 g, 5.32 mmol). The resulting reactionwas heated to 100 °C and allowed to stir at this temperature
for 10 hours, then the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue obtained was dissolved in dichloromethane and filtered through a short pad of celite. The filtrate was washed sequentially with water and brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using preparative HPLC to provide the title compound (0.81 g, yield: 50%) as white solid. Ή- MR (CDC13, 400 MHz) δ 8.25 (s, 1H), 7.86-7.89 (m, 2H), 7.76-7.80 (m, 2H), 7.59-7.67 (m, 3H), 7.34-7.38 (m, 1 H), 7.1 1 -7.16 (m, 3H), 5.85 (s, 1 H), 4.02 (s, 3H), 3.10 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.78 (s, 3H); MS (M+H)+ 625. Example 411 : 5-[3-(4-Fluoro-benzooxazol-2-yl)-4-methoxy-phenyll-2- (4-fluoro-phenyl)-6- (methanesulfonYl-methyl-amino)-benzofuran-3-carboxylic acid methylamide
- Synthesis of ethyl 5-bromo-2-(4-fluorophenyl)benzofuran-3-carboxylate
A solution of ethyl 3-(4-fluorophenyl)-3-oxopropanoate (130 g, 0.6 mol), 4- bromophenol (31 1 g, 1.8 mol) and FeCl3-6H20 (19.5 g, 0.09 mol) in DCE (700 mL) was heated to reflux, and then 2-(tert-butylperoxy)-2-methylpropane (193 g, 1 .32 mol) was added drop wise under nitrogen. After 6 hours of refluxing, the mixture was cooled to room temperature and quenched with saturated NaHS03, extracted with dichloromethane. The organic phases were washed with water, brine and dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column chromatography (petroleum ether : dichloromethane = 15 : 1 ) to provide the crude product, which was crystallized from cold MeOH to provide ethyl 5-bromo- 2-(4-fluorophenyl)benzofuran-3-carboxylate (37 g, 14.3%) as a solid. Ή-NMR (CDC13, 400 MHz) δ 8.12 (s, 1 H), 7.97-8.01 (m, 2H), 7.37 (d, J = 4.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.1 1 (t, J = 8.0 Hz, 2H), 4.32-4.38 (m, 2H), 1.36 (t, J = 8.0 Hz, 3H). MS (M+H)+: 363 / 365.
Step 2 - Synthesis of ethyl-5-bromo-2-(4-fluorophenyl)-6-nitrobenzofuran-3
-carboxylate
To a solution of ethyl-5-bromo-2-(4-fluorophenyl)benzofuran-3- carboxylate (50 g, 137.6 mmol) in CHC13 (500 mL), fuming HNO3 (50 mL) was added dropwise at -15 °C and the mixture was stirred for 0.5 hours. The reaction mixture was poured into ice water and extracted with CH2C12. The organic layer was washed with a.q. sat. NaHC03 and brine, after removed the most of solvent, the residue obtained was crystallized with petroleum ether :
dichloromethane = 20 : 1 to provide product of ethyl 5-bromo-2- (4-fluorophenyl)-6- nitrobenzofuran-3-carboxylate (35 g, 66%)
Ή-NMR (CDCI3, 400 MHz) δ 8.36 (s, 1H), 8.02-8.04 (m, 3H), 7.13-7.18 (m, 2H), 4.36-4.41 (m, 2H), 1.37 (t, J = 4.0 Hz, 3H).
Step 2 - Synthesis of ethyl 6-amino-5-bromo-2-(4-fluorophenyl)benzofuran-3 -carboxylate (Compound 411 D)
4U D
A mixture of ethyl 5-bromo-2-(4-fluorophenyl)-6- nitrobenzofuran-3 -carboxylate (52 g, 127 mmol), iron filings (21.3 g, 382.2 mmol) and NH4C1 (41 g, 764.4 mmol) in MeOH / THF / H20 (2: 2: 1 , 500 mL) was allowed to stir at reflux for 3 hours. After being filtered and concentrated in vacuo, the residue obtained was purified using column chromatography
(petroleum ethenEtOAc : dichloromethane = 20 : 1 : 20) to provide the pure ethyl 6-amino-5- bromo-2-(4-fluorophenyl) benzofuran-3 -carboxylate (compound 411D) (40 g, 82%). Ή-NMR (CDC13, 400 MHz) δ 8.01 (s, 1H), 7.94-7.98 (m, 2H), 7.08 (t, J = 8.0 Hz, 2H), 6.83 (s, 1 H), 4.32-4.36 (m, 2H), 4.18 (s, 2H), 1.35 (t, J= 8.0 Hz, 3H). MS (M+H)+: 378 / 380.
Step 3 - Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino- benzofuran-3- carboxylic acid ethyl ester (Compound 41 IE)
MsCI (31.7 g, 277.5 mmol) was added to a solution of ethyl 6-amino-5-bromo-2- (4-fluorophenyl)benzofuran-3-carboxylate (35 g, 92.5 mmol) and pyridine (60 mL) in dry dichloromethane (300 mL) at 0 °C. After stirred for about 15 hours at room temperature, the mixture was diluted with water, and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo, the residue obtained was purified using crystallized with EtOAc to provide the pure product of Compound 4ΠΕ (35 g, 82%). 'H-NMR (CDC13, 400 MHz) δ 8.27 (s, 1H), 8.01-8.05 (m, 2H), 7.87 (s, 1 H), 7.15-7.19 (m, 2H), 6.87 (s, 1H), 4.38-4.43 (m, 2H), 3.00 (s, 3H), 1.40 (t, J = 40 Hz, 3H). MS (M+H)+: 456 / 458.
Step 4 - Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino -benzofuran-3- carboxylic acid (Compound 41 IF)
To a solution of Compound 41 IE (53 g, 0.23 mol) in dioxane / H20 (5: 1 , 600 mL) was added LiOH H20 (25 g, 1.17 mol), and the mixture was allowed to stir at 100 °C and allowed to stir at this temperature for 3 hours. After being concentrated in vacuo, the residue obtained was dissolved in H20, 1 N HCl was added until pH reached 3, and the reaction mixture was extracted with EtOAc. The organic layer was washed with brine, dried over Na2S04 and filtered. The solvent was removed by distillation to provide the crude product of Compound
411F (48 g, 96%). Ή-NMR (400 MHz, DMSO) δ 13.49 (s, 1 H), 9.67 (s, 1 H), 8.30 (s, 1H), 8.12-8.17 (m, 2H), 7.87 (s, 1H), 7.45-7.50 (m, 2H), 3.16 (s, 3H). MS (M+H)+: 428 / 430.
Step 5 - Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-methanesulfonylamino -benzofi
carboxylic acid methylamide (Compound 411G)
A solution of Compound 41 IF (33 g, 77 mmol), HOBT (15.6 g, 1 15.5 mmol) and EDCI (22.2 g, 1 15.5 mmol) in dry DMF (250 mL) was allowed to stir at room temperature. After 2 hours, Et3N (50 mL) and CH3NH2 (HC1 salt, 17.7 g, 231 mmol) was added to the mixture, and the mixture was stirred for about 15 hours. After the solvent was removed, H20 was added and the reaction mixture was extracted with ethyl acetate. The organic extract was washed with H20, brine and concentrated in vacuo and the residue obtained was washed with EtOAc to provide Compound 41 1 G (32 g, 94%). Ή-NMR (400 MHz, DMSO) δ 9.55 (br s, 1 H), 8.46-8.48 (m, I H), 8.12-8.17 (m, 2H), 7.96 (s, IH), 7.87 (s, I H), 7.45-7.50 (m, 2H), 3.16 (s, 3H), 2.93 (d, J = 8.4 Hz, 3H). MS (M+H)+: 441 / 443.
Step 6 - Synthesis of 5-Bromo-2-(4-fluoro-phenyl)-6-(methanesulfonyl-methyl- benzofuran-3-carboxylic acid methylamide (Compound 41 IH)
411G 411H
CH3I (24.3 g, 171 mmol) was added to a mixture of Compound 411 G (25 g, 57.1 mmol), K2C03 (19.8 g, 143 mmol) and I ( 190 mg, 1 .1 mmol) in DMF (1 00 mL) under N2 protection. The reaction was allowed to stir at reflux for about 15 hours, then was concentrated in vacuo and the residue obtained was washed with water and EtOAc to provide Compound
41 IH (24 g, 93%). Ή-NMR (CDC13, 400 MHz) δ 8.16 (s, I H), 7.88-7.92 (m, 2H), 7.70 (s, I H), 7.18-7.23 (m, 2H), 5.78 (br s, 1 H), 3.34 (s, 3H), 3.09 (s, 3H), 3.00 (d, J = 4.8 Hz, 3H). MS (M+H)+: 455 / 457.
Step 7 - Synthesis of l-fluoro-3-methoxy-2-nitrobenzene
To a solution of 1 ,3-difluoro-2-nitrobenzene (100 g, 0.63 mol) in MeOH (1 .3 L) was added a solution of NaOMe (0.69 mol, in MeOH, freshly prepared from 15.9 g of metal Na and 200 mL of MeOH) slowly at 0 °C. The reaction was allowed to stir for about 15 hours at room temperature, then the reaction mixture was concentrated in vacuo and the residue obtained was diluted with EtOAc. The resulting solution was washed with water and brine, dried over Na
2S0
4, filtered and concentrated in vacuo to provide l-fluoro-3-methoxy-2- nitrobenzene (98 g, 91.4%). Ή-NMR (CDC1
3, 400 MHz) δ 7.38-7.44 (m, 1 H), 6.72-6.88 (m, 2H), 3.95 (s, 3H).
Step 8 - Synthesis of 3-Fluoro-2-nitro-phenol
To a solution of l-Fluoro-3-methoxy-2-nitro-benzene (98 g, 0.57 mol) in dichloromethane (500 mL) was added dropwise a solution of BBr3 (1 L, 1 M in dichloromethane) at -40 °C. The reaction was allowed to stir for about 15 hours at room temperature, then the reaction mixture was slowly poured into ice water (500 mL). The mixture was extracted with EtOAc (300 mL x 3), and the combined organic extracts were washed with 5% aqueous NaHC03, brine, dried over Na2S04, filtered and concentrated in vacuo to provide 3-fluoro-2-nitro-phenol (85 g, 95%). Ή-NMR (CDC13, 400 MHz) δ 7.43-7.49 (m, 1H), 6.88 (d, J = 8.0 Hz, 1 H),
6.73-6.78 (m, 1H). Step 9 - Synthesis of 2-Amino-3-fluoro-phenol
3-Fluoro-2-nitro-phenol (38 g, 0.24 mol) was dissolved in EtOH and to the resulting solution was added palladium on carbon (5 g, 10% Pd). The reaction was put under H2 atmosphere (1 atm) and allowed to stir for 3 hours at room temperature. The reaction mixture was filtered and the collected palladium was washed with EtOH. The filtrate and washing was combined and concentrated in vacuo to provide 2-amino- 3-fluoro-phenol (26 g, 85.7%). Ή- NMR (DMSO, 400 MHz) δ 9.43 (s, 1H), 6.42-6.53 (m, 2H), 6.32-6.42 (m, 1 H), 4.34 (s, 2H).
Step 10 - Synthesis of 2-(5-Bromo-2-methoxy-phenyl)-4-fluoro-benzooxazole
To a solution of 2-amino-3-fluoro-phenol (9 g 70.8 mmol) in 10 mL of polyphosphoric acid was added 5-bromo-2-methoxybenzoic acid (16.3 g, 70.8 mmol), and the resulting mixture was heated to 140 °C and allowed to stir at this temperature for 4 hours. The reaction mixture was then poured into ice water (50 mL) and extracted with EtOAc. The organic extract was concentrated in vacuo and the resulting residue was purified using column chromatography on silica gel (petroleum ether:EtOAc = 10 : 1 ) to provide 2-(5-bromo-2- methoxy-phenyl)-4- fluoro-benzooxazole(16 g, 82%) as a solid. Ή-NMR (CDC13, 400 MHz) δ 8.29 (d, J= 2.4 Hz, 1 H), 7.57-7.54 (m, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.27-7.33 (m, l H), 7.07 (m, 1 H), 6.96 (d, J = 9.2 Hz, 1 H), 3.99 (s, 3H).
Step 11 - Synthesis of 4-Fluoro-2-[2-methoxy-5-(4, 4, 5, 5-tetramethyl-[l, 3, 2]dioxaborolan-2- yl) -phenyl] -benzooxazole
To a solution of 2-(5-bromo-2-methoxy-phenyl)-4-fluoro- benzooxazole (18.4 g,
57.1 mmol) in DMF (10 mL), bis(pinacolato)diboron (17.4 g, 68.5 mmol) and AcOK ( 10 g, 1.14 mmol) was added, and the resulting mixture was heated to 80 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was concentrated in vacuo and the residue obtained was dissolved in dichloromethane and filtered through celite. The filtrate was washed with H20 and brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column chromatography (petroleum ether:EtOAc = 10 : 1 ) to provide 4- fluoro-2-[2-methoxy-5-(4, 4, 5, 5-tetramethyl-[l , 3, 2]dioxaborolan-2-yl)-phenyl] -benzooxazole (10 g, 54%) as a solid. Ή-NMR (CDC13, 400 MHz) δ 8.53 (d, J = 1 .6 Hz, 1 H), 7.85-7.92 (m, 1 H), 7.44 (d, J = 8.0 Hz, 1H), 7.20-7.28 (m, 1 H), 6.96-7.05 (m, 2H), 3.97 (s, 3H), 1.29 (s, 12H).
Step 12 - Synthesis of 5-[3-(4-Fluoro-benzooxazol-2-yl)-4-methoxy-phenyl]-2- (4-fluoro-phenyl)- 6-(methanesulfonyl-methyl-amino)-benzofuran-3-carboxylic acid methylamide
411H Example 411
To a solution of 4-fluoro-2-[2-methoxy-5-(4, 4, 5, 5-tetramethyl-[l , 3,
2]dioxaborolan-2-yl)-phenyl]-benzooxazole (5.27 g, 14.3 mmol) and Compound 41 IH (5 g, 1 1 .0 mmol) in DMF (150 mL) was added Pd(dppf)Cl2 (200 mg) and K3P04 (4.66 g, 22.0 mmol) under N2 protection. The resulting mixture was heated to 100 °C and allowed to stir at this temperature for 10 hours, and then the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue obtained was dissolved in dichloromethane and filtered through celite. The filtrate was washed with water, brine, dried over Na2S04 and concentrated in vacuo. The residue obtained was purified using flash column chromatography (petroleum ether:EtOAc = 4: 1 to 2: 1 ) and crystallized from dichloromethane : EtOAc (5 : 1 ) to provide the target compound (3.8 g, 56%) was obtained as white solid. Ή-NMR (CDC1 , 400 MHz) δ 8.21 (d, J = 2.0 Hz, I H), 7.91-7.95 (m, 2H), 7.83 (s, I H), 7.68 (d, J= 2.0 Hz, I H), 7.66 (s, I H), 7.39 (d, J = 8.0 Hz, I H), 7.14-7.27 (m, 4H), 7.06 (t, J = 8.4 Hz, I H), 5.95 (br s, IH), 4.06 (s, 3H), 3.14 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.77 (s, 3H).
Example 412: 5-(5-(4-fluorobenzo[dloxazol-2-yr)thiophen-2-yl)-2-(4- fluorophenyl)- N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Step 1 - Synthesis of2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)- 5-(4, 4, 5, 5- tetramethyl-1, 3, 2-dioxaborolan-2-yl)benzofuran-3-carboxamide (Compound 411J)
To a degassed solution of Compound 411H (1.0 g, 2.20 mmol) and pinacol diborane (2.79 g, 10.98 mmol) in 1 , 4-Dioxane (25 mL) was added OAc (647 mg, 6.59 mmol) under N2 and the resulting reaction was allowed to stir for 4 hours. Pd(dppf)Cl2 (60 mg) was then added and the reaction was stirred for another 30 minutes. The reaction flaski was then put into a pre-heated oil-bath at 130 °C and stirred for another 1 hour under N2. The reaction mixture was cooled to room temperature, then concentrated in vacuo and extracted with EtOAc. The organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column chromatography on silica gel (petroleum ethenEtOAc = 5 : 1 to 2 : 1) to provide Compound 411J as white solid (700 mg, 64%). Ή-NMR (CDC13, 400 MHz) 6 8.17 (s, 1H), 7.87-7.91 (m, 2H), 7.52 (s, 1 H), 7.1 1 (t, J = 7.6 Hz, 2H), 5.81 (d, J= 2.8 Hz, 1 H), 3.30 (s, 3H), 2.97 (d, J = 5.2 Hz, 3H), 2.90 (s, 3H), 1.31 (s, 12H).
Step 2 - Synthesis of 5-(5-(4-fluorobenzo[d]oxazol-2-yl)thiophen-2-yl)-2- (4 fluorophenyl) -N- methyl-6-( -methylmethylsulfonamido)benzofuran-3-carboxamide
Example 412
To a degassed solution of Compound 411J (100 mg, 0.2 mmol) and 2-(5- bromothiophen-2-yl)-4-fluorobenzo[d]oxazole (53 mg, 0.2 mmol, prepared using the methods described in Example 1) in dry DMF (3 mL) was added Pd(dppf)Cl2 (10 mg) and K3PO4 (120 mg, 0.4 mmol) under N2 protection. The reaction was heated to 100 °C and allowed to stir at this temperature for about 15 hours, then was cooled to room temperature and filtered. The filtrate was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the target compound (68 mg, 57.6%). Ή- NMR (CDCI3, 400 MHz) δ 8.04 (s, 1H), 7.86-7.89 (m, 3H), 7.82 (s, 1H), 7.69 (s, 1 H), 7.55 (s, 1 H), 7.23-7.34 (m, 1 H), 7.13 (t, J = 8.0 Hz, 2H), 7.03 (t, J = 8.8 Hz, 1 H), 3.18 (s, 3H), 2.93 (s, 3H), 2.82 (s, 3H). MS (M+H)+: 595.
Example 413: 5-(4-(lH-indol-2-v0pyridin-2-vn-2-(4-fluorophenvn-N-methyl- 6-(N- methylmethylsulfonamido)benzofuran-3-carboxaniide
Step 1 - Synthesis of tert-butyl 1 H-indole-1 -carboxylate
To a solution of indole (1 g, 8.5 mmol) and (Boc)20 (2.2 g, 10.2 mmol) in dichloromethane (10 mL) was added DMAP (100 mg, 0.85 mmol) at room temperature, and the mixture was stirred for 3 hours. Water was added, extracted with dichloromethane and washed with brine, dried over Na2S04. After being concentrated in vacuo, the residue obtained was purified using column chromatography (petroleum ethenEtOAc = 20 : 1) to provide tert-butyl 1 H-indole-1 -carboxylate (1.8 g, 96%). 1H-NMR (CDC13, 400 MHz) δ 8.13 (d, J = 8.0 Hz, 1 H), 7.54-7.59 (m, 2H), 7.20-7.32 (m, 2H), 6.56 (t, J = 1.8 Hz, 1H), 1.67 (s, 9H).
Step 2 - Synthesis of 1 -(tert-butoxycarbonyl)-l H-indol-2-ylboronic acid
To a solution of tert-butyl l H-indole-l-carboxylate (1 g, 4.61 mmol) and B(z- PrO)3 (1.6 1, 6.91 mmol) in THF (7 mL) was added LDA (3.5 mL, 6.91 mmol) at 0 °C, Then warmed up to room temperature and stirred for 30 minutes. 2N HCl was added to acidified the solution until pH = 7, extracted with ethyl acetate and washed with brine, dried over Na2S04. After being concentrated in vacuo, the residue obtained was purified using column
chromatography (petroleum ether:EtOAc = 10: 1 to 2: 1 ) to provide 1 -(tert-butoxycarbonyl)- 1 H- indol-2-ylboronic acid (0.5 g, 45%). Ή-NMR (DMSO, 400 MHz) δ 8.16 (s, 1 H), 8.05 (d, J = 8.8 Hz, 1 H), 7.52 (d, J = 8.8 Hz, 1 H), 7.24 (t, J = 7.2. Hz, 1 H), 7.16 (t, J = 7.2 Hz, 1 H), 6.59 (s, 1 H), 1.57 (s, 9H).
- Synthesis of te -butyl 2-(2-chloropyridin-4-yl)-l H-indole- 1-carboxyh
To a mixture of 1 -(tert-butoxycarbonyl)- l H-indol-2-ylboronic acid (400 mg, 1.56 mmol), 2-Chloro-4-bromopyridine (200 mg, 1.04 mmol) and Κ3Ρ0 ·3Η20 (830 mg, 3.12 mmol) in DMF (6 mL), under nitrogen atmosphere, was added Pd(dppf)Cl2 (60 mg). The reaction was heated to 90 °C and allowed to stir at this temperature for 5 hours. Water was added, the solution was extracted with ethyl acetate and the organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-TLC
(petroleum ether:EtOAc = 5 : 1 ) to provide tert-butyl 2-(2-chloropyridin-4-yl)- 1 H-indole- 1 - carboxylate (300 mg, 88%). MS (M+H)+: 328 / 330.
Step 4 - Synthesis of 2-(2-chloropyridin-4-yl)-l H-indole
To a 0 °C solution of tert-butyl 2-(2-chloropyridin-4-yl)-l H-indole- 1 -carboxylate (328 g, 1.0 mmol) in dichloromethane (5 mL) was added TFA (0.5 mL) dropwise. The reaction was allowed to warm to room temperature with stirring, then was allowed to stir for an additional 1 hour. Water was added and the resulting solution was extracted with dichloromethane and the organic phase was washed with brine, dried over Na S0
4; filtered and concentrated in vacuo to provide 2-(2-chloropyridin-4-yl)- l H-indole (150 mg, 66%) was obtained. Ή-NMR (CDC1
3, 400 MHz) δ 8.48 (s, 1 H), 8.41 (d, J = 4.4 Hz, 1 H), 7.67 (d, J= 8.8 Hz, 1 H), 7.56 (s, 1 H), 7.42-7.46 (m, 2H), 7.30 (d, J = 8.4 Hz, 1 H), 7.17 (t, J = 8.0 Hz, 1 H), 7.06 (s, 1 H). Step 5 - Synthesis of 5-(4-(l H-indol-2-yl)pyridin-2-yl)-2-(4-fluorophenyl)-N-methyl- 6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
A mixture of 2-(2-chloropyridin-4-yl)-l H-indole (34 mg, 0.15 mmol), Compound 41 U (50 mg, 0.1 mmol), Κ3Ρ04·3Η20 (80 mg, 0.3 mmol), Pd2(dba)3 (9.15 mg, 0.01 mmol) and X-Phos (9.50 mg, 0.02 mmol) in 1 ,4-dioxane (2 mL) and H20 (0. 5 mL) was heated to 1 10 °C and allowed to stir at this temperature for 12 hours. Water was added and the reaction mixture was extracted with ethyl acetate. The organic extract was and washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using prep- HPLC to provide the target compound (40 mg, 69%). Ή-NMR (CDC13, 400 MHz) δ 9.48 (s, 1 H), 8.64 (d, J = 5.2 Hz, 1 H), 8.09 (d, J = 2.0 Hz, 1 H), 8.05 (s, 1 H), 7.94-7.97 (m, 2H), 7.64 (d, J =
8.0 Hz, 1 H), 7.55 (d, J = 5.2 Hz, 1H), 7.53 (s, 1 H), 7.43 (d, J = 8.0 Hz, 1 H), 7.1 1 -7.25 (m, 4H),
7.01 (s, 1 H), 6.16 (s, 1 H), 3.09 (d, J = 0.8 Hz, 3H), 3.03 (s, 3H), 2.98 (d, J = 4.8 Hz, 3H). MS (M+H)+: 569. The following compounds of the present invention were made using the methods described above in Examples 41 1 -413 and using the appropriate reactants and/or reagents.
Ή-NMR (CDC1
3, 400 MHz) δ 9.32
(s, 1 H), 7.94 (t, J = 4.6 Hz, 1 H), 1.88
(d, = 4.8 Hz, 1 H), 7.51 (d, .7 = 5.2
Hz, 2H), 7.07-7.27 (m, 6H), 6.94 (s,
605 1 H), 6.89 (s, 1 H), 6.75-6.80 (m, 1 H), 616
5.92 (s, 1 H), 3.93 (d, J = 4.8 Hz,
o=s— 3H), 3.07 (d, J = 4.8 Hz, 3H), 2.99
0
(d, J = 4.8 Hz, 3 H), 2.96 (d, J = 4.8
Hz, 3H).
Example 606:5-(3-(4-fluorobenzo[dloxazol-2-y-)-4-hydroxyphenv-)-2(4- fluorophenyl) -N-methyl- 6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Example 411 Example 606
A solution of the compound of Example 41 1 (120 mg, 0.19 mmol) in anhydrous CH2C12 (3 mL), was cooled to -30 °C and a solution of BBr3 (142 mg, 0.57 mmol) in
dichloromethane was added dropwise. The reaction was allowed to stir at room temperature for 3 hours, then was quenched with water and extracted with CH2C12. The organic phase was dried over Na2S04, filtered and concentrated in vacuo, and the residue obtained was purified using prep-TLC (petroleum ether:EtOAc = 2 : 1 ) to provide the target compound (1 10 mg, 94%). Ή- NMR (CDCI3, 400 MHz) 8.18 (s, 1 H), 7.94-7.98 (m, 2H), 7.71 (d, J = 2.4 Hz, 1 H), 7.59 (s, 1 H), 7.42-7.43 (m, 2H), 7.33-7.37 (m, 2H), 7.21-7.25 (m, 2H), 7.14 (d, J = 8.8 Hz, 1 H), 5.87 (d, J = 4.4 Hz, 1H), 4.02 (s, 3H), 3.12 (s, 3H), 3.01 (d, J = 4.8 Hz, 3H). MS (M+H)+: 604.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 616: 5-(4-(2,2-difluoroethoxy)-3-f4-fluorobenzofdloxazol-2- yl)phenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)
benzofuran-3- carboxamide
A solution of the compound of Example 606 ( 1 00 mg, 0.16 mmol), 2,2- difluoroethyl methanesulfonate (234 mg, 1 .6 mmol) and K2C03 (43 mg, 0.32 mmol) in DMF (3 mL) was heated to 100 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature and filtered, and the filtrate was concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the target compound (30 mg,
25%). 'H-NMR (CDCI3, 400 MHz) 8.18 (s, I H), 7.82-7.85 (m, 2H), 7.76 (s, I H), 7.59-7.62 (m, I H), 7.52 (s, I H), 7.33 (d, J = 8.0 Hz, I H), 7.22-7.27 (m, I H), 7.06-7.12 (m, 3H), 7.01 (t, J = 1 .8 Hz, I H), 6.06-6.38 (m, 2H), 4.32-4.40 (m, 2H), 3.09 (s, 3H), 2.95 (d, J = 4.8 Hz, 3H), 2.76 (s, 3H). MS (M+H)+: 668.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 631: 2-{5-[2-(4-Fluoro-phenyl)-6-(methanesulfonYl- methylamino)- 3- methylcarbamoyl-benzofuran-5-yll-2-methoxy-phenyl}-benzooxazole-5-carboxylic acid methylamide
Step 1 - Synthesis of 2-{5-[2-(4-Fluoro-phenyl)-6-(methanesulfonyl-methyl-amino)- 3- methylcarbamoyl-benzofuran-5-yl]-2-methoxy-phenyl}-benzooxazole-5-carboxylic acid ester
Compound 411H was converted to methyl 2-(5-(2-(4-fluorophenyl)-3- (methylcarbamoyl)-6-( -methylmethylsulfonamido) benzofuran-5-yl)-2- methoxyphenyl)benzo[d]oxazole-5-carboxylate (95 mg, 58.3%) using the method described in Example 41 1 , Step 12. Ή-NMR (DMSO-d6, 400 MHz) δ 8.51 (d, J = 3.6 Hz, l H), 8.33 (s, I H), 7.97-8.09 (m, 4H), 7.90 (t, J = 8.0 Hz, 1 H), 7.70 (d, J = 6.0 Hz, 1 H), 7.61 (s, 1 H), 7.35-7.41 (m, 3H), 3.98 (s, 3H), 3.87 (s, 3H), 3.1 1 (s, 3H), 2.98 (s, 3H), 2.78 (d, J = 4.4 Hz, 3H). MS (M+H)+: 658.
Step 2 - Synthesis of 2-{5-[2-(4-Fluoro-phenyl)-6-(methanesulfonyl-methyl-amino)- 3- me
Methyl-2-(5-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-6-(N- methylmethylsulfonamido)benzofuran-5-yl)-2-methoxyphenyl)benzo[d]oxazole-5-carboxylate was converted to 2-(5-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-6-(N- methylmethylsulfonamido)benzofuran-5-yl)-2-methoxyphenyl)benzo[d]oxazole-5-carboxylic acid (85 mg, 100%) using the method described in Example 41 1 , Step 4. Ή-NMR (MeOD, 400 MHz) 6 8.03 (d, J = 7.2 Hz, 1H), 7.87-7.90 (m, 2H), 7.59-7.74 (m, 6H), 7.25 (d, J= 8.8 Hz, 1 H), 7.12-7.17 (m, 3H), 3.98 (s, 3H), 3.1 1 (s, 3H), 2.86 (s, 3H), 2.83 (s, 3H). MS (M+H)
+: 644.
Step 3 - Synthesis of 2-{5-[2-(4-Fluoro-phenyl)-6-(methanesulfonyl-methyl-amino)- 3- methylcarbamoyl-benzofuran-5-yl]-2-methoxy-phenyl}-benzooxazole-5-carboxylic acid methylamide
Example 631
2-(5-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-6-( - methylmethylsulfonamido)benzofuran-5-yl)-2-methoxyphenyl)benzo[d]oxazole-5-carboxylic acid was converted to the title compound (35 mg, 35.8%) using the method described in
Example 41 1 , Step 5. Ή-NMR (CDC13, 400 MHz) δ 8.15 (s, 1 H), 8.08 (s, 1 H), 7.87-7.90 (m, 2H), 7.81 (s, 1 H), 7.88 (s, 1 H), 7.63-7.54 (m, 1 H), 7.56 (t, J = 4.0 Hz, 2H), 7.12-7.17 (m, 3H), 6.1 1 (br s, 1 H), 5.80 (d, J = 4.8 Hz, 1 H), 4.03 (s, 3H), 3.10 (s, 3H), 2.99 (d, J = 4.8 Hz, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.73 (s, 3H). MS (M+H)+: 657.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Step 1 - Synthesis of 2-(4-fluorophenyl)-5-(4-methoxy-3-(4~nitrobenzo[d]oxazol-2 -yl) phenyl) -N- methyl- -(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Compound 41 IH was converted to 2-(4-fluoropheny))-5-(4- methoxy-3-(4 nitrobenzo[d]oxazol-2-yl)phenyl)-N- methyl-6-(N- methylmethylsulfonamido)benzofuran-3- carboxamide (2.5 g, 86%) using the method described in Example 41 1 , Step 12. Ή-NMR
(CDCI3, 400 MHz) δ 8.36-8.37 (m, I H), 7.93-7.96 (m, 2H), 7.84 (s, I H), 7.63-7.67 (m, I H), 7.61 (s, IH), 7.32-7.35 (m, 2H), 7.23-7.31 (m, I H), 7.17-7.21 (m, 2H), 6.83 (d, J = 7.6 Hz, I H), 6.05 (d, J = 4.8 Hz, IH), 4.05 (s, 3H), 3.14 (s, 3H), 3.02 (d, J = 4.8 Hz, 3H), 2.80 (s, 3H). Step 2 - Synthesis of 5-(3-(4-aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4- fluorophenyl)- N-meth -6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
A mixture of 2-(4-fluorophenyl)-5-(4-methoxy-3-(4-
nitrobenzo[d]oxazol-2-yl)phenyl)-N-methyl-6-( -methylmethylsulfonamido)benzofuran-3- carboxamide (2.5 g, 3.88 mmol), Fe (0.7 g, 12.5 mmol) and NH4C1 (1 g, 19.4 mmol) in MeOH (10 mL) and H20 (10 mL) THF (5 mL) was allowed to stir at reflux for 3 hours. After being filtered and concentrated in vacuo, the residue obtained was purified using column
chromatography eluted with petroleum ethenEtOAc = 1 : 1 to provide 5-(3-(4- aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-
N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (1.7 g, 72%). Ή-NMR (CDCI3, 400 MHz) δ 8.05 (s, 1H), 7.70-7.78 (m, 2H), 7.48-7.52 (m, 2H), 7.01-7.12 (m, 4H), 6.83-6.88 (m, I H), 6.48-6.53 (m, 1H), 6.02-6.04 (m 1H), 5.25 (s, 1H), 4.05 (s, 3H), 3.14 (s, 3H), 2.70 (m, 3H), 2.65 (s, 3H). MS (M+H)+: 615.
Step 3 - Synthesis of2-(4-fluorophenyl)-5-(4-methoxy-3-(4- (methylsulfonamido)
benzo[d]oxazol-2-yl)phenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzojuran-3- carboxamide
Example 639
5-(3-(4-aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide was converted to the title compound (30 mg, 20%) using the method described in Example 41 1 , Step 3. 1 H-NMR (CDC13, 400 MHz) δ 8.21 (d, J= 2.4 Hz, I H), 7.99 (s, IH), 7.91-7.95 (m, 2H), 7.89 (s, I H), 7.67 (d, J = 8.8 Hz, I H), 7.61 (s, I H), 7.48 (d, J= 7.2 Hz, IH), 7.26-7.35 (m, 2H), 7.17-7.23 (m, 3H), 5.93 (d, J = 4.8 Hz, IH), 4.07 (s, 3H), 3.16 (s, 3H), 3.12 (s, 3H), 2.99 (d, J = 5.2 Hz, 3H), 2.00 (s, 3H). MS (M+H)+: 693.
Example 640: 2-(4-fluorophenyl)-5-(3-(4-(furan-2-yl)benzofdloxazol-2-yl)-4- methoxyphenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Step 1 - Synthesis of2-(4-fluorophenyl)-5-(3-(4-iodobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-N- methyl- -(N-methylmethylsulfonamido)benzofuran-3-carboxamide
A mixture of 5-(3-(4-aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)- 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide (1.5 g, 2.44 mmol), Cul (0.8 g, 4.2 mmol) and h (0.5 g, 1.97 mmol) in CH3CN (10 mL) was allowed to stir at 30 °C and allowed to stir at this temperature for 30 minutes. Then the reaction was cooled to 0 °C and isopentyl nitrite (0.6 g, 5.12 mmol) was added at 0 °C and the reaction was allowed to warm to 30 °C and stir at this temperature for about 15 hours. After being filtered and concentrated in vacuo, the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 2 : 1 to provide 2-(4-fluorophenyl)- 5-(3-(4-iodobenzo[d]oxazol- 2-yl)-4- methoxyphenyl)- N-methyl-6- (N-methylmethylsulfonamido)benzofuran-3 -carboxamide (0.7 g, 40%). Ή-NMR (CDC13, 400 MHz) δ 8.18 (s, 1 H), 7.93-7.96 (m, 2H), 7.84 (s, 1 H), 7.63-7.67 (m, 1 H), 7.61 (s, 1H), 7.32-7.35 (m, 1 H), 7.23-7.31 (m, 1 H), 7.02-7.19 (m, 4H), 5.85 (s, 1 H), 4.00 (s, 3H), 3.10 (s, 3H), 3.93-3.95 (m, 3H), 2.77 (s, 3H). MS (M+H)+: 726.
Step 2 - Synthesis of2-(4-fluorophenyl)-5-(3-(4-(furan-2-yl)benzo[d]oxazol-2-yl)-4- meth
Example 640
2-(4-fluorophenyl)-5-(3-(4-iodobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxarnide was converted to the title compound (40 mg, 44%) using the method described in Example 41 1 , Step 12. Ή-NMR (CDCI3, 400 MHz) δ 8.35 (s, I H), 7.87-7.88 (m, 2H), 7.80 (s, I H), 7.61 -7.70 (m, I H), 7.57-7.60 (m, 2H), 7.48-7.49 (m, 2H), 7.47-7.48 (m, 1 H), 7.28-7.31 (m, 1 H), 7.09-7.13 (m, 3H), 6.50-6.51 (m, I H), 5.84 (d, J = 4.8 Hz, I H), 4.03 (s, 3H), 3.1 1 (s, 3H), 2.92 (d, J = 5.2 Hz, 3H), 2.70 (s, 3H). MS (M+H)+: 666.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 642: (Z)-5-(3-(4-fluorobenzo[dloxazol-2-yl)-5-(l-(hvdroxyimino)ethyl) phenyl) -2-
(4-fluorophenyl)-N-methyl-6- N-methylmethylsulfonamido)
benzofuran-3-carboxamide
Example 642
A mixture of 5-(3-acety 5-(4-fluorobenzo[d]oxazol-2-yl)phenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (120 mg, 0.19 mmol, prepared according to the method described in Example 1), NH2OH HCl (27 mg, 0.38 mmol) and NaHC03 (32 mg, 0.38 mmol) in CH3OH (5 mL) was heated to 50 °C and allowed to stir at this temperature for 5 hours. After the reaction mixture was cooled room temperature and concentrated in vacuo, the residue obtained was washed with CH2C12 and filtered. The filtrate was dried over Na2S04, concentrated in vacuo, and the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 2 : 1 to provide the title compound (100 mg, 80%). 1 H-NMR (CDC13, 400 MHz) 8.46 (s, 1 H), 8.27 (s, 1H), 7.83-7.92 (m, 4H), 7.57 (s, 1 H), 7.34-7.36 (m, 1 H), 7.23-7.29 (m, 3H), 7.00-7.05 (m, 1 H), 5.98 (s, 1 H), 3.06 (s, 3H), 2.94 (t, J = 4.8 Hz, 3H), 2.70 (s, 3H), 2.32 (s, 3H). MS (M+H)+: 645.
Example 643: 5-(3-(l-aininoethyl)-5-(4-fluorobenzo[dloxazol-2-yl)phenyl)- 2-(4- fluorophenyl)-N-methyl-6- N-methylmethylsulfonamido)benzofuraii-3- carboxamide
Example 642 Example 643
To a solution of the compound of Example 642 (40 mg, 0.06 mmol) in CH3OH (3 mL) was added Pd/C (10 mg) and HC1 (IN, 2 drops) and the resulting reaction was put under H2 atmosphere (1 atm) and stirred for 12 hours. The reaction mixture was filtered and the filtrate was concentrated in vacuo to provide the title compound (30 mg, 75%). Ή-NMR (CDC13, 400 MHz) 8.15 (s, 2H), 7.82-7.85 (m, 2H), 7.76 (s, 1H), 7.69 (s, 1 H), 7.37 (s, 1 H), 7.23-7.28 (m, 1 H), 7.1 1 -7.15 (m, 3H), 7.02 (t, J = 8.4 Hz, 1H), 6.44 (d, J = 4.0 Hz, 1H), 4.48-4.51 (m, 1 H), 3.02 (s, 3H), 2.89 (d, J = 4.4 Hz, 3H), 2.75 (s, 3H), 1.67 (d, J = 7.2 Hz, 3H). MS (M+H)+: 631.
Example 644: 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)-5- (3-(4,5,6,7-tetrahvdrofuro[3,2-clpyridin-2-yl)phenvnbenzofuran-3-carboxamide
Example 517 Example 644
A mixture of the compound of Example 517 (166 mg, 0.27 mmol) and NaOH (108 mg, 2.7 mmol) in 9 mL of EtOH:H20 (2: 1 ) was heated to 90 °C under N2 and allowed to stir at this temperature for about 15 hours. Then the reaction mixture was mixture was purified using prep-HPLC to provide the title compound (76 mg, 49.0%). Ή-NMR (DMSO, 400 MHz) 5 9.10 (s, 1 H), 8.50-8.54 (m, 1 H), 7.97-8.00 (m, 3H), 7.74 (s, 1 H), 7.68 (d, J = 7.6 Hz, 1 H), 7.58 (s, 1 H), 7.49 (t, J = 7.6 Hz, 1 H), 7.37-7.43 (m, 3H), 6.92 (s, 1 H), 4.12 (s, 2H), 3.46 (s, 2H), 3.08 (s, 3H), 2.97 (s, 2H), 2.94 (s, 3H), 2.80 (d, J = 4.8 Hz, 3H). MS (M+H)+: 574. Example 645: 5-(3-(4-fluorobenzo[d]oxazol-2-vn-4-(methylsulfonamido)phenyl)- 2-(4-fluorophenyl)-N-methyl-6-rN-methylmethylsulfonamido)benzofuran-3- carboxamide
Step 1 - Synthesis of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4-nitrophenyl)-2-(4- fluorophenyl) -N- methyl- -(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a degassed solution of 2-(5-chloro-2-nitrophenyl)-4-fluorobenzo[d] oxazole (39 mg, 0.13 mmol) and Compound 411J (50 mg, 0.1 mmol) in 1 ,4-dioxane (2.0 mL) was added Pd2(dba)3 (5 mg), X-Phos (5 mg) and K3P04 (42 mg, 0.2 mmol) under N2. The reaction was heated to 100 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was then cooled to room temperature and filtered and the filtrate was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-TLC (petroleum ethenEtOAc = 2 : 1) to provide 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- nitrophenyl)-2- (4-fluorophenyl)-N-methyl-6- (N-methylmethylsulfonamido) benzofuran-3- carboxamide (51 mg, 82%). Ή-NMR (CDC13, 400 MHz) δ 8.18 (s, 1H), 8.05 (d, J = 2.4 Hz, 1 H), 7.80-7.88 (m, 3H), 7.58 (s, 1H), 7.28-7.51 (m, 2H), 7.14 (t, J = 8.4 Hz, 2H), 7.06 (t, J = 8.8 Hz, 1 H), 5.80 (d, J = 4.0 Hz, 1 H), 3.1 1 (s, 3H), 2. 94 (d, J = 4.8 Hz, 3H), 2.89 (s, 3H).
Step 2 - Synthesis of 5-(4-amino-3-(4-fluorobenzo[d]oxazol-2-yl)phenyl)-2-(4 -fluorophenyl) -N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a degassed solution of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- nitrophenyl)-2- (4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (350 mg, 1.3 mmol) in MeOH (9 mL) was added Fe powder (270 mg, 5 mmol) and NH4C1 (395 mg, 7.5 mmol) under N2. The reaction was heated to 70 °C and allowed to stir at this temperature for about 15 hours, then was filtered and the filtrate was concentrated in vacuo to provide 5-(4- amino-3-(4- fluorobenzo[d]oxazol-2-yl)phenyl)-2-(4- fluorophenyl)- N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (300 mg, 94%). Ή-NMR (CDCI3, 400 MHz) 6 8.1 1 (s, 1 H), 8.05 (d, J = 2.4 Hz, 2H), 7.87-7.91 (m, 2H), 7.78 (s, 1 H), 7.36-7.55 (m, 1H), 7.30 (d, J = 7.2 Hz, 1 H), 7.14 (t, J= 8.8 Hz, 3H), 7.09 (t, J = 8.8 Hz, 1 H), 6.82 (d, J = 8.4 Hz, 1 H), 5.80 (d, J = 4.4 Hz, 1 H), 3.12 (s, 3H), 2. 93 (d, J= 4.8 Hz, 3H), 2.68 (s, 3H). MS (Ms+H)+: 603.
Step 3 - Synthesis of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4-(methylsulfonamido) phenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 645
To a degassed solution of 5-(4-amino-3-(4-fluorobenzo[d]oxazol-2- yl)phenyl)-2- (4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (40 mg, 0.07 mmol) and pyridine (1 mL) in dichloromethane (1 mL) was added MsCI (23 mg, 0.2 mmol) at 0 °C under N2 atmosphere. The reaction was allowed stir for 5 hours, then the mixture was concentrated in vacuo and extracted with dichloromethane. The organic phase was washed with brine, dried over Na2S04i filtered and concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 67%). Ή-NMR (CDC13, 400 MHz) δ 8.30 (s, 1 H), 7.85-7.89 (m, 4H), 7.61 (t, J = 6.4 Hz, 1 H), 7.58 (d, J = 5.6 Hz, 1 H), 7.29-7.36 (m, 2H), 7.16 (t, J = 8.4 Hz, 2H), 7.07 (t, J = 8.4 Hz, 1 H), 5.77 (d, J = 4.0 Hz, 1 H), 3.13 (s, 3H), 3.12 (s, 3H), 2. 93 (d, J= 4.8 Hz, 3H), 2.75 (s, 3H). MS (Ms+H)+: 681. Example 646: 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5-(l-hvdroxyethyl) phenyl)- 2-(4- fluorophenyl)-N-methyl-6-rN-methylniethylsulfonainido)benzofuran-3- carboxamide
Step 1 - Synthesis of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5-formylphenyl)-2-(4 -fluorophenyl) -N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a solution of 3-(4-fluorobenzo[d]oxazol-2-yl)-5-(4,4,5,5- tetramethyl-l ,3,2- dioxaborolan-2-yl)benzaldehyde (40 mg, 0.1 1 mmol), Compound 41 1 H (50 mg, 0.10 mmol) and K3P04 (38 mg, 0.20 mmol) in DMF (2 mL) was added Pd(dppf)Cl2 (10 mg) under N2, and then the mixture was heated to 100 °C and allowed to stir at this temperature for 5 hours. Thee reaction mixture was cooled to room temperature and filtered and, the filtrate was concentrated in vacuo. The residue obtained was purified using prep-TLC (petroleum ether:EtOAc = 5 : 1 ) to provide 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5-formylphenyl)-2-(4-fluorophenyl)-N-methyl-6- (N-methylmethylsulfonamido)benzofuran-3-carboxamide (35 mg, 53%). Ή-NMR (CDC13, 400 MHz) 9.86 (s, 1H), 8.24 (s, 1 H), 8.18 (s, 1H), 7.87-7.90 (m, 2H), 7.84 (s, 1 H), 7.74 (s, 1 H), 7.54 (s, 1 H), 7.34-7.36 (m, 1H), 7.23-7.29 (m, 1 H), 7.12 7.17 (m, 2H), 7.01-7.06 (m, 1 H), 5.89 (t, J = 3.2 Hz, 1 H), 3.03 (s, 3H), 2.94 (d, J = 4.8 Hz, 3H), 2.75 (s, 3H). MS (M+H)+: 616.
Step 2 - Synthesis of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5-(l-hydroxyethyl) phenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 646
To a solution of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5- formylphenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (123 mg, 0.2 mmol) in anhydrous THF (5 mL) at 0 °C was added dropwise a solution of
methylmagnesium bromide (0.67 mL, 3 N in ether). The reaction was allowed to stir at room temperature for 3 hours, and then the reaction mixture was quenched with saturated NH4CI, and extracted with CH2C12. The organic phase was dried (Na2S04), filtered and concentrated in vacuo and the residue obtained was purified using column chromatography eluted with petroleum ethenEtOAc = 3: 1 to provide the title compound (100 mg, 70%). Ή-NMR (CDC13, 400 MHz) 8.24 (s, 1H), 8.18 (s, 1 H), 7.87-7.90 (m, 2H), 7.84 (s, 1 H), 7.74 (s, 1 H), 7.54 (s, 1 H), 7.34-7.36 (m, 1 H), 7.23-7.29 (m, 1 H), 7.12-7.17 (m, 2H), 7.01-7.06 (m, 1 H), 5.89 (t, J= 3.2 Hz, 1 H), 5.00-5.05 (m, 1H), 3.03 (s, 3H), 2.94 (d, J = 4.8 Hz, 3H), 2.75 (s, 3H), 1 .55 (d, J = 6.4 Hz, 3H). MS (M+H)+: 632.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 648: 5-(3-(4-fluorobenzo[dloxazol-2-yl)-5-(l-fluoroethyl)phenyl)- 2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Example 648
To a solution of the compound of Example 646 (73 mg, 0.12 mmol) in anhydrous CH2CI2 (3 mL) at 0 °C, was added DAST reagent (0.5 mL, 0.25 mmol) dropwise. The reaction was allowed to stir for 5 hours at room temperature, then the reaction was quenched with water, and extracted with CH2C12. The organic phase was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column
chromatography eluted with petroleum ether:EtOAc = 3: 1 to provide the title compound (35 mg, 50%). Ή-NMR (CDCI3, 400 MHz) 8.25 (s, 2H), 7.87-7.91 (m, 2H), 7.84 (s, 1 H), 7.65 (s, 1 H), 7.58 (s, 1H), 7.29-7.35 (m, 1 H), 7.23-7.28 (m, 1 H), 7.13-7.17 (m, 2H), 7.02-7.06 (m, 1 H), 5.70-5.84 (m, 1H), 5.65-5.76 (m, 1 H), 3.09 (s, 3H), 2.94 (d, J = 4.8 Hz, 3H), 2.69 (s, 3H), 1 .69 (dd, J = 6.4 Hz, 3H). MS (M+H)+: 634.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 650: 3-(4-fluorobenzo[dloxazol-2-yl)-5-(2-(4-fluorophenyl)- 3-(methylcarbamoyl)-6-(N-methylmethylsulfonamido)benzofuran-5-yl)benzoic acid
To a solution of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-5- formylphenyl)-2-(4- fluorophenyl)-N-methyl-6-(TvI-methylmethylsulfonamido)benzofuran-3-carboxamide (360 mg, 0.6 mmol, described in Example 423, step 1 ) in /-BuOH (2 mL) was added 2-methyl-2-butane (0.6 mL) and dioxane (2 mL) and the mixture was cooled to 0 °C. To the cooled mixture was added as solution of NaC102 (600 mg, 6.6 mmol) and NaH2P04 (1 .2 g, 10.8 mmol) in water (3 mL) and the resulting reaction was allowed to stir at room temperature for 2 hours. CH2CI2 was added and the organic phase was separated and washed with water, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (35 mg, 53%). Ή-NMR (CDC13, 400 MHz) 8.01-8.89 (m, 2H), 7.61 -7.98 (m, 2H), 7.44-7.46 (m, 1 H), 7.35-7.39 (m, 1H), 7.1 1-7.19 (m, 4H), 6.94-6.99 (m, 2H), 5.90 (s, 1 H), 3.08 (s, 6H), 2.30 (s, 3H). MS (M+H)+: 632.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 652: 5-(3-carbamoyl-5-(4-fluorobenzo[dloxazol-2-yl)phenyl)- 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
A solution of the compound of Example 650 (160 mg, 0.25 mmol), EDC1 (67 mg, 0.25 mmol) and HOBT (96 mg, 0.25 mmol) in DMF (3 mL) was allowed to stir at room temperature for 3 hours. Et3N (0.6 mL) and the NH4C1 (20 mg, 0.4 mmol) were then added and ther reaction was allowed to stir at room temperature for another 4 hours. The reaction mixture was concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (50 mg, 30%). Ή-NMR (CDC13, 400 MHz) 8.79 (s, I H), 8.52 (s, I H), 8.26 (s, I H), 7.93-7.96 (m, 3H), 7.56 (s, I H), 7.44-7.46 (m, I H), 7.35-7.39 (m, I H), 7.1 1 -7.19 (m, 3H), 6.94-6.99 (m, lH), 6.26 (s, I H), 5.90 (s, I H), 3.08 (s, 6H), 2.30 (s, 3H). MS (M+H)+: 631 .
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 658: 2-(4-fluorophenyl)-5-(4-methoxy-3-(6-(pyrimidin-5-yl)
benzo[dloxazol-2-yl)phenyl)-N-methyl-6-(N-methylmethylsu fonamido)
benzofuran-3-carboxamide
Step 1 - Synthesis of 2-(4-fl orophenyl)-5-(4-methoxy-3-(6-nitrobenzo[d]oxazol-2- yl)phenyl)-N- methyl- -(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Compound 41 1 H was converted to 2-(4-fluorophenyl)-5-(4-methoxy-3-(6- nitrobenzo[d]oxazol-2-yl)phenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide (810 mg, 16%) using the method described in Example 41 1 , Step 12. Ή-NMR (CDC1
3, 400 MHz) δ 8.43 (d, J = 2.0 Hz, 1 H), 8.26 (d, J = 2.0 Hz, 1 H), 8.21 -8.25 (m, 1 H), 7.80-7.88 (m, 4H), 7.67-7.70 (m, 1H), 7.55 (s, 1 H), 7.12-7.17 (m, 3H), 5.81 (d, J = 4.0 Hz, 1 H), 4.04 (s, 3H), 3.1 1 (s, 3H), 2.93 (d, J = 4.0 Hz, 3H), 2.76 (s, 3H).
Step 2 - Synthesis of 5-(3-(6-aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4 -fluorophenyl) - -methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a solution of 2-(4-fluorophenyl)-5-(4-methoxy-3-(6- nitrobenzo[d]oxazol-2- yl)phenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (400 mg, 0.62 mmol) in MeOH (20 mL) was added Pd-C (10 mg) and the resulting reaction was stirred under 40 psi of H2 atmosphere for 24 hours at room temperature. The reaction mixture was filtered and concentrated in vacuo to provide 5-(3-(6-aminobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4- fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (350 mg, 92%), which was used without further purification. MS (M+H)+: 615.
Step 3 - Synthesis of 2-(4-fluorophenyl)-5-(3-(6-iodobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a 0 °C suspension of 5-(3-(6-aminobenzo[d]oxazol-2-yl)-4- methoxyphenyl)- 2-(4-fluorophenyl)-N-methyl-6-( J-methylmethylsulfonamido)benzofuran-3-carboxarnide (620 mg, 1 .01 mmol), I
2 (200 mg, 0.81 mol), Cul (190 mg, 1 .01 mmol) in THF was added t-BuONO dropwise. The reaction was allowed to stir at 0 °C for 1 hour and then stirred at refluxed for about 15 hours. The reaction was then cooled to room temperated, diluted with dichloromethane and filtered. The filtrate was concentrated in vacuo and the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 3 : 1 to provide 2-(4- fluorophenyl)-5-(3-(6-iodobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (360 mg, 53.8%) as a yellow solid. MS (M+H)
+: 726.
Step 4 - Synthesis of2-(4-fluorophenyl)-5-(4-methoxy-3-(6-(pyrimidin-5- yl)benzo[d]oxazol-2- yl)phenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
To a degassed solution of 2-(4-fluorophenyl)-5-(3-(6- iodobenzo[d]oxazol-2-yl)-
4-methoxyphenyl)-N-methyl-6-( -methylmethylsulfonamido)benzofuran-3-carboxamide (100 mg, 0.14 mmol), pyrimidin-5-ylboronic acid (26 mg, 0.21 mmol) and K3P04 (75 mg, 0.28 mmol) in dry DMF (3 mL) was added Pd(dppf)Cl2 (3 mg) under N2. The reaction was heated to 100 °C and allowed to stir at this temperature for 6 hours. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (45 mg, 48.3%). Ή-NMR (CDC13, 400 MHz) δ 9.29 (s, 1 H), 9.13 (s, 2H), 8.30 (s, 1 H), 8.01 (d, J = 8.0 Hz, 1H), 7.95-7.98 (m, 2H), 7.93 (s, 1 H), 7.87 (s, 1 H), 7.76 (d, J = 6.4 Hz, 1 H), 7.64 (d, J = 1 1.2 Hz, 2H), 7.23-7.27 (m, 3H), 5.96 (s, 1 H), 4.14 (s, 3H), 3.21 (s, 3H), 3.03 (d, J = 4.8 Hz, 3H), 2.85 (s, 3H). MS (M+H)+: 678.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 666: 5-(3-(3-((dimethylamino)methyl)-lH-indol-2-yl)phenyl)-
2-(4-fluorophenvI)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Example 552 Example 666
A solution of the compound of Example 552 (50 mg, 0.09 mmol),
polyoxymethylene (3 mg, 0.09 mmol), dimethylamine (41 mg, 0.9 mmol), ZnCl2 (41 mg, 0.27 mmol) in EtOH (2 mL) was heated to 60 °C and allowed to stir at this temperature for 12 hours. The reaction mixture was added to water and then extracted with ethyl acetate and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide Compound 256 (10 mg, 20%). Ή- NMR (CDCI3, 400 MHz) δ 9.63 (s, 1 H), 7.89-7.97 (m, 4H), 7.58-7.67 (m, 3H), 7.47-7.49 (m, 3H), 7.19-7.30 (m, 4H), 6.1 1 (s, 1 H), 4.69 (s, 2H), 3.16 (s, 3H), 2.95-3.00 (m, 6H), 2.55 (s, 6H). MS (M+H)+: 625.
Example 667: 5-f3-f3-qiH-imidazol-l-vnmethvn-lH-indol-2-vnphenyl)-
2-(4-fluorophenvn-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Example 667
A solution of the compound of Example 666 (55 mg, 0.09 mmol) and imidazole (31 mg, 0.45 mmol) in xylenes (1.5 mL) was heated to 120 °C and allowed to stir at this temperature for 1 hour. The reaction mixture was cooled to room temperature and concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 60%). Ή-NMR (CDC13, 400 MHz) δ 9.51 (s, 1 H), 7.93-7.96 (m, 2H), 7.77-7.82 (m, 2H), 7.57 (s, 1 H), 7.38-7.49 (m, 5H), 7.12-7.24 (m, 4H), 6.95-6.98 (m, 2H), 6.32 (s, 1 H), 5.39 (s, 2H), 3.04 (s, 3H), 2.96-2.97 (m, 3H), 2.92 (s, 3H). MS (M+H)+: 648. Example 668; 5-(3-(l-(2-aminoethyl)-lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)- N-methvI-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 562 Example 668
The compound of Example 562 (50 mg, 0.07 mmol) was added to a 0 °C mixture of TFA/dichloromethane (1 : 4, 1 mL). The reaction was allowed to stir at room temperature for 1.5 hours, then saturated aqueous NaHC03 was added to adjust the reaction mixture to pH 7. The reaction mixture was then extracted with EtOAc (30 mL) and the organic extract was washed with saturated aqueous NaHC03 (2x 10 mL), brine (2^20 mL), dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-TLC
(dichloromethane : MeOH = 15 : 1) to provide the title compound (40 mg, 93%). Ή-NMR
(CDC13, 400 MHz) δ 7.92-7.95 (m, 2H), 7.83 (s, 1H), 7.62-7.64 (d, J = 7.6 Hz, 1 H), 7.56 (s, 2H), 7.51-7.52 (d, J = 5.6 Hz, 2H), 7.41-7.44 (m, 2H), 7.12-7.24 (m, 4H), 6.58 (s, 1 H), 6.07-6.08 (d, J = 4.4 Hz, 1 H), 4.30-4.33 (t, d, J = 6.8 Hz, 2H), 3.22 (s, 3H), 2.91 -2.96 (m, 5H), 2.69 (s, 3H). MS (M+H)+: 61 1.
Example 669: - ^fluorophenylVN-methvI^-fN-methylmethylsulfonamido)- 5-(3-(3-
(pyridin-3-yl)-lH-indol-2-yl)phenyl)benzofuran-3-carboxamide
Step 1 - Synthesis of 5-(3-(3-bromo-lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)- N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
Example 552
To a solution of the compound of example 552 (50 mg, 0.09 mmol) in 3 mL of
DMF, was added NBS (16 mg, 0.09 mmol) and the resulting reaction mixture was placed under N2 atmosphere, heated to 75 °C and allowed to stir at this temperature for 4 hours. The reaction mixture was then concentrated in vacuo and the residue obtained was diluted with with EtOAc. The resulting solution was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using prep-TLC (petroleum ether:EtOAc = 2 : 1 ) to provide 5-(3-(3-bromo-lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)-N- methyl-6- (N- methylmethylsulfonamido)benzofuran-3-carboxamide (40 mg, 89%) as white solid. Ή-NMR (CDC13, 400 MHz) δ 9.38 (s, 1 H), 8.02 (d, J = 8.0 Hz, 1H), 7.94 (s, 1 H), 7.88-7.94 (m, 2H), 7.84 (s, 1H), 7.53 (t, J = 7.6 Hz, 2H), 7.46 (d, J = 4.8 Hz, 1H), 7.35-7.40 (m, 2H), 7.1 1-7.1 5 (m, 4H), 5.80 (s, 1 H), 3.04 (s, 3H), 2.94 (d, J= 5.2 Hz, 3H), 2.87 (s, 3H). MS (M+H)+: 646 / 648.
Step 2 - Synthesis of 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)- 5-(3-(3- (pyridin-3-yl)-lH-indol-2-yl)phenyl)benzofuran-3-carboxamide
Example 669
A mixture of 5-(3-(3-bromo-l H-indol-2-yl)phenyl)-2-(4- fluorophenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (100 mg, 0.15 mmol), pyridin-3-ylboronic acid (24 mg, 0.19 mmol), Pd(dppf)Cl2 (12 mg) and Κ3Ρ04·3Η20 (82 mg, 0.31 mmol) in ethanol and water (2.5 mL, 4: 1) was placed in a commercial microwave oven and subjected to microwave irradiation for 30 minutes (120 watts, internal reaction temperature was 100 °C at the conclusion of irradiation). The reaction mixture was cooled to room temperature and then was diluted with water and extracted with EtOAc. The organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound ( 10 mg, 10%). Ή-NMR (MeOD, 400 MHz) δ 8.74 (s, 1 H), 8.60 (d, J= 6.8 Hz, 2H), 7.93-8.00 (m, 3H), 7.84 (s, 1 H), 7.72 (d, J = 8.0 Hz, 1 H), 7.59 (d, J = 4.0 Hz, 2H), 7.54 (d, J - 12.0 Hz, 4H), 7.26-7.30 (m, 3H), 7.20 (t, J = 4.0 Hz, 1H), 3.17 (s, 3H), 2.94 (d, J = 4.0 Hz, 6H). MS (M+H)+: 645. The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 671: 2-(2-(3-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-6-(N- methylmethylsulfonamido)benzofuran-5-yl)phenyl)-lH-indol-l-yl)acetic acid
To the solution of ethyl 2-(2-(3-(2-(4-fluorophenyl)-3- (methylcarbamoyl)-6-(N- methylmethylsulfonamido)benzofuran-5-yl)phenyl)-l H-indol- l -yl)acetate ( 120 mg, 0.1 8 mmol, prepared according to the method described in Example 41 1) in MeOH (1.5 mL) was added a saturated solution of LiOH. The reaction was allowed to stir at room temperature until LCMS indicated that the starting material was consumed. The reaction mixture was extracted with dichloromethane and the organic extract was washed with brine, dried over Na2S0 , filtered and concentrated in vacuo. The residue obtained was purified using HPLC to provide the title compound (1 10 mg, 95.7%). Ή-NMR (CDC13, 400 MHz) δ 7.81-7.91 (m, 3H), 7.64 (d, J = 7.6 Hz, 1 H), 7.56 (s, 1 H), 7.52 (t, J = 6.4 Hz, 4H), 7.26-7.1 (m, 5H), 6.63 (s, lH), 6.19 (s, 1 H), 4.88 (s, 2H), 3.12 (s, 3H), 2.94 (d, J = 4.0 Hz, 3H), 2.70 (s, 3H). MS (M+H)+: 626.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 674: 5-(3-(l-f2-amino-2-ox0ethyl)-lH-indol-2-vI)phenvI)-2-f4- fluorophenyl)-N-inethyl-6-(N--Tiethylmethylsulfonamido)benzofuran-3- carboxamide
To a solution of the compound of Example 671 (50 mg, 0.08 mmol) in MeCN (1 mL) was added EDCI (23 mg, 0.12 mmol) and HOBT (16 mg, 0.12 mmol). The reaction was allowed to stir at room temperature for 2 hours then TEtOAc (16 mg, 0.16 mmol) and NH4C1 (9 mg, 0.16 mmol) were added. The reaction was then stirred at room temperature and monitored using LCMS until the starting material was consumed completely. The reaction mixture was concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (10 mg, 20%). Ή-NMR (CDC13, 400 MHz) δ 8.00 (t, J = 8.0 Hz, 2H), 7.83 (s, 1 H), 7.68 (d, J= 8.0 Hz, 1H), 7.60-7.52 (m, 1H), 7,36 (d, J= 8.0 Hz, 4H), 7.31 (d, J = 7.2 Hz, 2H), 7.20 (t, J= 8.4 Hz, 3H), 6.75 (s, 1H), 6.22 (s, 1H), 5.74 (d, J = 1 1.6 Hz, 1H), 5.57 (s, 1 H), 4.83 (s, 2H), 3.19 (s, 3H), 3.03 (d, J = 4.4 Hz, 3H), 2.72 (s, 3H). MS (M+H)+: 625.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
To a solution of the compound of Example 588 (50 mg, 0.08 mmol) in MeOH (1 mL) was added Pd/C (10 mg), and the mixture was put under H2 atmosphere (50 psi) and allowed to stir for about 15 hours. The reaction mixture was filtered, the filtrate was
concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (20 mg, 40%). Ή-NMR (CDCI3, 400 MHz) δ 7.95-7.92 (m, 2H), 7.84 (s, 1 H), 7.80 (d, J = 2.0 Hz, 1 H), 7.62 (d, J = 10.8 Hz, 2H), 7.42 (d, J = 8.0 Hz, 1 H), 7.34-7.31 (m, 1 H), 7.24-7.1 1 (m, 5H), 6.85 (s, 1 H), 5.94 (d, J = 4.8 Hz, 1 H), 5.82 (d, J = 7.6 Hz, I H), 3.16 (s, 3H), 2.99 (d, J = 5.2 Hz, 3H), 2.71 (s, 3H), 1.58-1.50 (m, 1 H), 0.67-0.57 (m, 4H). MS (M+H)+: 636.
Example 677: 5-(3-ethynylphenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methyimeth\ sulfonamido)benzofuran-3-carboxamide
Step 1 - Synthesis of 2-(4-fluorophenyl)-N-methyl- -(N- methylmethylsulfonamido) -5-(3- ((trimethylsilyl)ethynyl)phenyl)benzofuran-3-carboxamide
411H
To a mixture of trimethyl((3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan- 2- yl)phenyl)ethynyl)silane (480 mg, 1.60 mmol), Compound 41 I H (600 mg, 1 .32 mmol) and
3P04-3H20 (700 mg, 1.99 mmol) in 1 ,4-dioxane (20 mL), was added Pd(dppi)Cl2 (15 mg). The reacton was put under N2 atmosphere, heated to 80 °C and allowed to stir at this temperature for 2 hours. The reaction mixture was then concentrated in vacuo and the residue obtained was diluted with water and extracted with EtOAc. The organic extract was washed with brine, dried (Na2S04), filtered and concentrated in vacuo and the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 3 : 1 to provide 2-(4-fluorophenyl)-N- methyl-6-(N- methylmethylsulfonamido)-5-(3-((trimethylsilyl) ethynyl)phenyl)benzofuran-3- carboxamide (600 mg, 83%). Ή-NMR (CDC13, 400 MHz) δ 7.90-7.94 (m, 2H), 7.75 (s, 1 H), 7.59 (s, I H), 7.47-7.52 (m, 2H), 7.37-7.43 (m, 2H), 7.16-7.21 (m, 2H), 5.93 (br s, I H), 3.13 (s, 3H), 2.98 (d, J = 4.8 Hz, 3H), 2.62 (s, 3H) 0.26 (s, 9H). MS (M+H)+: 549.
Step 2 - Synthesis of5-(3-ethynylphenyl)-2-(4-fluorophenyl)-N-methyl-6 -(N- methylmethylsulfonamido)benzofuran-3 '-carboxamide
To a solution of 2-(4-fluorophenyl)-N-methyl-6-( - methylrnethylsulfonamido)- 5-(3-((trimethylsilyl)ethynyl)phenyl)benzofuran-3-carboxamide (600 mg, 1 .09 mmol) in MeOH was added KF (200 mg, 3.44 mmol) and the reaction was allowed to stir at room temperature for about 15 hours. The reaction mixture was concentrated in vacuo and the residue obtained was diluted with water and extracted with EtOAc. The organic extract was washed with brine, dried (Na2S04), filtered and concentrated in vacuo and the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 3 : 1 to provide the title compound (300 mg, 57%). Ή-NMR (CDC13, 400 MHz) δ 7.91-7.95 (m, 2H), 7.77 (s, I H), 7.61 (s, I H),
7.51 -7.56 (m, 2H), 7.39-7.47 (m, 2H), 7.17-7.23 (m, 2H), 5.87 (br s, I H), 3.14 (s, 3H), 3.1 1 (s, 1 H), 2.99 (d, J = 4.8 Hz, 3H), 2.63 (s, 3H). MS (M+H)+: 477.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 679: 5-(3- 5-bromofuro|2,3-blpyridin-2-yl)phenyl)-2-(4- fluorophenvn-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3- carboxamide
Example 677 Example 679
To a solution of the compound of Example 677 (150 mg, 0.32 mmol) and 5- bromo-3-iodopyridin-2-ol (105 mg, 0.35 mmol) in THF-Et3N (1 : 1 , 4 mL) was added Cul (10 mg) and Pd(PPh3)2Cl2 (20 mg) and the reaction was allowed to stir at room temperature for 3 hours. The reaction mixture was diluted with EtOAc and filtered and the organic phase was washed with NH4C1, water and brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 2 : 1 to provide the title compound (100 mg, 49%). Ή-NMR (CDCI3, 400 MHz) δ 8.29 (s, I H), 8.00
J
(br s, 2H), 7.88-7.94 (m, 3H), 7.82 (s, I H), 7.47-7.61 (m, 3H), 7.15-7.20 (m, 2H), 7.03 (s, I H), 6.07 (br s, I H), 3.12 (s, 3H), 2.97 (d, J = 4.8 Hz, 3H), 2.73 (s, 3H). MS (M+H)
+: 648 / 650. Example 680: 2-(4-nuorophenyl)-5-(3-(furo[2,3-blpyridin-2-vnphenyl)-N- methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 680
A mixture of the compound of Example 679 (30 mg, 0.05 mmol), Pd/C (10 mg, 5%) and Et3N (0.1 mL) in MeOH (5 mL) was put under H2 atmosphere (30 psi) and allowed to stir at room temperature for about 15 hours. The reaction mixture was filtered, the filtrate was concentrated in vacuo and the residue obtained was purified using PTLC to provide the title compound (10 mg, 38%). Ή-NMR (CDC13, 400 MHz) δ 8.30 (d, J= 3.6 Hz, IH), 8.03 (s, I H), 7.91-7.98 (m, 4H), 7.86 (s, I H), 7.63 (s, IH), 7.56 (t, J = 7.6 Hz, IH), 7.49 (d, J= 7.6 Hz, I H), 7.18-7.25 (m, 3H), 7.10 (s, IH), 5.94 (br s, I H), 3.17 (s, 3H), 3.00 (d, J = 4.8 Hz, 3H), 2.70 (s, 3H). MS (M+H)+: 570.
Example 681; 5-(4-fluoro-lH-indol-2-vn-2-(4-fluorophenvn-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide
A solution of Pd(OAc)2 (4 mg) and S-Phos (14 mg, 0.03 mmol) in toluene (2 ml) was stirred for 10 minutes under N2 atmosphere. The reaction mixture was then added to a stirring solution of 2-(2,2-dibromovinyl)-3-fluoroaniline (50 mg, 0.17 mmol), Compound 41 U (126 mg, 0.25 mmol) and K3P04 (108 mg, 0.51 mmol). The resulting reaction was heated to 1 10 °C and allowed to stir at this temperature for 12 hours, then water was added and the solution was extracted with ethyl acetate. The organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using TLC to provide the title compound (30 mg, 35%). MS (M+H)+: 510. Ή-NMR (CDC13, 400 MHz) δ 9.53 (s, I H), 7.99 (s, I H), 7.86-7.87 (m, 2H), 7.48 (s, IH), 7.01-7.19 (m, 4H), 6.19-6.74 (m, 2H), 5.83 (d, J = 4.0 Hz, IH), 3.07 (s, 3H), 3.02 (s, 3H), 2.94 (d, J = 4.8 Hz, 3H). MS (M+H)+: 510.
Example 682: S-O- ^fluorobenzofblthiophen- -vOphenvO-l-^-fluorophenyl)- N-methyl-6-
(N-meth> methylsulfonaniido)benzofuran-3-carboxamidc
Step 1 - Synthesis of 2-(4-fluorophenyl)-N-methyl-6- (N-methylmethylsulfonamido)-5-(3- nitrophenyl)benzofuran-3-carboxamide
To a degassed solution of Compound 41 1 H (2.0 g, 4.39 mmol) and 3- nitrophenylboronic acid (880 mg, 5.27 mmol) in dry DMF (1.5 mL), under nitrogen atmosphere, was added Pd(dppf)Cl2 (20 mg) and K3P04 (1.86 g, 8.79 mmol). The reaction was heated to 90 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature, diluted with EtOAc and filtered, and the filtrate was washed with H20, brine, and dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using flash column chromatography on silica gel (eluted with dichloromethane: EtOAc = 20: 1 ) to provide 2-(4-fluorophenyl)-N-methyl-6- (N-methylmethylsulfonamido)-5-(3-nitrophenyl) benzofuran-3-carboxamide (1.78 g, 84%). Ή-NMR (CDC13, 400 MHz) δ 8.24 (s, 1 H), 8.1 8 (d, J = 8.4 Hz, 1H), 7.83-7.87 (m, 2H), 7.79 (d, J = 5.6 Hz, 1H), 7.77 (s, 1H), 7.58 (s, 1H), 7.55 (t, J = 4.0 Hz, 1 H), 7.15 (t, J= 8.8 Hz, 2H), 5.83 (d, J = 3.2 Hz, 1H), 3.09 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H), 2.73 (s, 3H).
Step 2 - Synthesis of 5-(3-aminophenyl)-2-(4-fluorophenyl)- N-methyl-6- (N- methylmethylsulfonamido)benzofuran-3-carboxamide
To a solution of 2-(4-fluoiophenyl)-N-methyl-6-(N- methylmethylsulfonamido)- 5-(3-nitrophenyl)benzofuran-3-carboxamide (1.0 g, 2.01 mmol) in MeOH (30 mL), Pd/C (200 mg) was added and the resulting reaction mixture was stirred under 40 psi of H
2 atmosphere for 24 h at 25 °C. Then the reaction mixture was filtered, and the filtrate was concentrated in vacuo to provide the crude product of 5-(3-aminophenyl)-2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (846 mg, 89%). Ή-NMR (DMSO, 400 MHz) δ 8.49 (d, J = 4.8 Hz, 1 H), 7.94-7.97 (m, 2H), 7.84 (s, 1 H), 7.43 (s, 1 H), 7.38 (t, J= 9.2 Hz, 2H), 7.03 (t, J = 8.0 Hz, 1H), 6.53-6.58 (m, 3H), 5.09 (s, 2H), 3.13 (d, J = 5.6 Hz, 3H), 3.04 (s, 3H), 2.81 (s, 3H). MS (M+H)
+: 468.
Step 3 - Synthesis of 2-(4-fluorophenyl)-5-(3-iodophenyl)-N- methyl-6- (N- methylmethylsulfonamido)benzofuran-3-carboxamide
a stirred solution of 5-(3-aminophenyl)-2-(4-fluorophenyl)-N- methyl-6-(N- methylmethylsulfonamido)benzofuran-3-carboxamide (1.5 g, 3.21 mmol) in MeCN (20 mL) was added I2 (488.6 mg, 1.93 mmol) and Cul (6 mg) at 0 °C, then i-AmONO (394.6 mg, 3.37 mmol) was added dropwise. The reaction was allowed to stir at 25 °C and allowed to stir at this temperature for 6 hours, then the reaction mixture was heated to 90 °C and allowed to stir at this temperature for 1 hour. The mixture was diluted with Na2S203 and concentrated in vacuo to remove the organic solvent, and then the residue obtained was extracted with EtOAc. The organic layer was washed with brine, dried over Na S04 and concentrated in vacuo. The residue obtained was purified using column chromatography eluted with petroleum ethenEtOAc = 3 : 1 then with pure dichloromethane to provide 2-(4-fluorophenyl)-5-(3- iodophenyl)-N-methyl-6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide (1.17 g, 65%).'H-NMR (CDC13, 400 MHz) δ 7.85-7.88 (m, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.66 (d, J = 8.0 Hz, 1 H), 7.53 (s, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.14 (t, J= 6.0 Hz, 2H), 5.77 (d, J = 4.0 Hz, 1 H), 3.06 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H), 2.61 (s, 3H). MS (M+H)+: 579.
Step 4 - Synthesis of 2-(4-fluorophenyl)-N-methyl-6-(N-methylmethylsulfonamido)- 5-(3-(4, 4,5,5- tetram
To a degassed solution of 2-(4-fluorophenyl)-5-(3-iodophenyl)- N-methyl-6-( - methylmethylsulfonamido)benzofuran-3-carboxamide (200 mg, 0.35 mmol) and pinacol diborane (132 mg, 0.52 mmol) in dry DMF (1.5 mL) was added Pd(dppf)Cl2 (10 mg) and OAc (102 mg, 1.04 mmol) under N2. The mixture was heated to 90 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature and filtered. The filtrate was washed with H20, brine, dried over Na2S04. After being concentrated in vacuo, the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 4 : 1 to provide 2-(4-fluorophenyl)-N-methyl-6-(N- methylmethylsulfonamido)-5- (3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl) phenyl)benzofuran-3-carboxamide (190 mg, 95%). Ή-NMR (CDC13, 400 MHz) δ 7.88-7.92 (m, 2H), 7.75-7.78 (m, 2H), 7.72 (s, 1 H), 7.56 (s, 1 H), 7.49-7.52 (m, 1 H), 7.37-7.41 (m, 1H), 7.1 1-7.15 (m, 2H), 5.81-5.82 (m, 1 H), 3.05 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.51 (s, 3H), 1.29 (s, 12H). MS (M+H)+: 579.
Step 5 - Synthesis of 5-(3-(4-fluorobenzo[b]thiophen-2-yl)phenyl)-2-(4-fluorophenyl)- N-methyl- 6-(N-methylmethylsulfonamido)benzofuran-3-carboxamide
Example 682
To a degassed solution of 2-(4-fluorophenyl)-N-methyl-6- (N- methylmethylsulfonamido)-5-(3-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2- yl)phenyl)benzofuran-3-carboxamide (90 mg, 0.19 mmol) and 4-fluoro-2- iodobenzothiophene (65 mg, 0.25 mmol) in dry DMF (1.5 mL) was added Pd(dppf)Cl2 (20 mg) and K3P04 (81 mg, 0.38 mmol) under N2. The reaction was heated to 100 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature and filtered. The filtrate was washed with H20, brine, dried over Na2S0 . After being concentrated in vacuo, the residue obtained was purified using prep-HPLC to provide the title compound (55 mg, 58.7%). 1 H-NMR (CDCb, 400 ΜΗζ) δ 7.86-7.89 (m, 2H), 7.77 (s, I H), 7.74 (s, I H), 7.64-7.70 (m, 2H), 7.56 (s, 1 H), 7.54 (d, J = 8.0 Hz, 1 H), 7.43-7.47 (m, 1 H), 7.17-7.22 (m, 1 H), 7.13 (t, J = 8.8 Hz, 2H), 6.95-7.98 (m, I H), 5.85 (d, J = 4.4 Hz, I H), 3.12 (s, 3H), 2.91 (d, J = 4.8 Hz, 3H), 2.59 (s, 3H). MS (M+H)+: 603.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 690: 2-(4-fluorophenyl)-6-(N-(3-fluoropropynmethylsulfonamido)- 5-(3-(furo[3,2-blpyridin-2-yl)-4-methoxyphenyl)-N-methylbenzofuran-3-carboxamide
Step 1 - Synthesis of 5-bromo-2-(4-fluorophenyl)-6-(N-(3- fluoropropyl) methylsulfonam methylbenzofur -3-carboxamide
A mixture of 3 -fluoropropyl 4-methylbenzenesulfonate (500 mg, 2.15 mmol) and K2C03 (500 mg, 3.62 mmol) was added to a solution of Compound 41 1 G (500 mg, 1.13 mmol) in DMF (3 mL) under N2. The reaction was heated to 80 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was filtered and concentrated in vacuo, and the residue obtained was purified using column chromatography eluted with petroleum ether:EtOAc = 3 : 1 to provide 5-bromo-2-(4-fluorophenyl)-6-(N-(3- fluoropropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide (500 mg, 88%). Ή- NMR (CDC13, 400 MHz) δ 8.07 (s, IH), 7.78-7.83 (m, 2H), 7.58 (s, IH), 7.10-7.13 (m, 2H), 5.90 (s, IH), 4.40-4.53 (m, 2H), 3.69-3.89 (m, 2H), 3.00 (s, 3H), 2.91 (d, J = 4.8 Hz, 3H), 1.85-1.89 (m, 2H). MS (M+H)+: 501.
Step 2 - Synthesis of 2-(4fluorophenyl)-6-(N-(3-fluoropropyl) methylsulfonamido)- (furo -b]pyridin-2-yl)-4-methoxyphenyl)-N-methylbenzofuran-3-carboxamide
Example 690
To a solution of 5-bromo-2-(4-fluorophenyl)-6-(N-(3- fluoropropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide (100 mg, 0.2 mmol) in DMF, was added K3P04 (170 mg, 0.8 mmol) and 2-(2-methoxy-5-(4,4,5,5- tetramethyl- 1 ,3,2- dioxaborolan-2-yl)phenyl) furo[3,2-b]pyridine (100 mg, 0.28 mmol) and Pd(dppf)Cl2 (5 mg). The reaction was put under N2 atmosphere, heated to 80 °C and allowed to stir at this
temperature for about 15 hours. The mixture was concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 23%). Ή-NMR (CDC13, 400 MHz) δ 8.46 (d, J = 4.0 Hz, 1 H), 8.09 (d, J = 2.4 Hz, 1H), 7.87-7.90 (m, 2H), 7.79 (s, 1 H), 7.68 (d, J = 8.4 Hz, 1 H), 7.53-7.57 (m, 3H), 7.12-7.19 (m, 3H), 7.06 (d, J = 8.8 Hz, 1 H), 5.91 (d, J = 4.0 Hz, 1 H), 4.01-4.22 (m, 5H), 3.44-3.48 (m, 2H), 2.91 (m, 6H), 1.62-1.77 (m, 2H). MS (M+H)+: 646.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 775: 2-(4-fluorophenyl)-6-(N-(2-hvdroxy-2-methylpropyl)
methylsulfonamido)-N-methyl-5-(3-(oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-3- carboxamide
Step 1 - Synthesis of 5-bromo-2-(4-fluorophenyl)-6-(N- (2-hydroxypropyl)methylsulfonamido)-N- methylbenzofur -3-c rboxamide
To a solution of Compound 411G (8 g, 18.1 mmol), K
2C0
3 (7.5 g, 54.3 mmol) and KI ( 1.5 g, 9.05 mmol) in DMF (150 mL) at 15 °C was added 1 -bromopropan-2-ol (5.03 g, 36.2 mmol, 4.5 mL) dropwise. The reaction was heated to 1 10 °C and allowed to stir at this temperature for 8 hours. The reaction mixture was diluted with water and the resulting solution extracted with EtOAc (500 mL x 5). The combined organic extracts were washed with brine, dried over Na
2S0
4, filtered and concentrated in vacuo, and the residue obtained was purified using column chromatography (dichloromethane / EtOAc = 20: 1 to 15: 1 ) to provide 5-bromo-2- (4-fluorophenyl)-6-(N- (2-hydroxypropyl)methylsulfonamido)- N-methylbenzofuran-3- carboxamide (5 g, 55%) as yellow solid. Ή-NMR (CDC1
3, 400 MHz) δ 8.1 1 (d, J= 4.8 Hz, 1 H), 7.81-7.84 (m, 2H), 7.66-7.71 (m, 1H), 7.15 (t, J = 8.4 Hz, 2H), 5.75 (d, J = 3.6 Hz, 1 H),
3.60-3.92 (m, 2H), 3.46-3.58 (m, 1 H), 3.09 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 1.09-1.13 (m, 3H). MS (M+H)+: 499.
Step 2 - Synthesis of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(2-oxopropyl)
methylsulfonamido)benzofuran-3-carboxamide
To a 0 °C solution of 5-bromo-2-(4-fluorophenyl)-6-(N-(2- hydroxypropyl) methylsulfonamido)-N-methylbenzofuran-3-carboxamide (1.00 g, 2.00 mmol) in
dichloromethane (20 mL) was added DMP (1.19 g, 2.81 mmol) portionwise. The reaction was allowed to stir at 20 °C for 6 hours, then the reaction mixture was diluted with NaHC03 and basified to pH 8, then extracted with dichloromethane (500 mL x 3). The combined organic extracts were washed with Na2S03, brine and dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(2- oxopropyl)methylsulfonamido)benzofuran-3-carboxamide (908 mg, 91 %). Ή-NMR (CDC13, 400 MHz) δ 8.09 (s, 1 H), 7.98 (s, 1 H), 7.81-7.85 (m, 2H), 7.15 (t, J= 8.4 Hz, 2H), 5.76 (d, J = 3.68 Hz, 1H), 4.86-4.89 (m, 1 H), 4.10-4.32 (m, 1 H), 3.09 (s, 3H), 2.93 (d, J = 4.8 Hz, 3H), 2.08 (s, 3H). MS (M+H)+: 497.
Step 3 - Synthesis of 5-bromo-2-(4-fluorophenyl)-6-(N-(2-hydroxy-2 - methylpropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide
oxopropyl)methylsulfonamido)benzofuran-3-carboxamide (1 .00 g, 2.01 mmol) in anhydrous THF (20 mL) was added MeMgBr (3 M, 1 mL) and the reaction was allowed to stir at 20 °C for 3 hours. The reaction mixture was then quenched with aqueous NH4C1, then extracted with EtOAc (100 mL x 3). The combined organic extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-2-(4-fluorophenyl)-6-( -(2- hydroxy-2-methylpropyl) methylsulfonamido)-N-methylbenzofuran-3-carboxamide (910 mg, 91 %). Ή-NMR (CDCI3, 400 MHz) δ 7.77 (d, J= 3.2 Hz, 1 H), 7.67-7.71 (m, 2H), 7.51 (s, 1 H), 7.26 (d, J = 4.4 Hz, 1 H), 6.91 (t, J = 8.4 Hz, 2H), 3.57-3.61 (m, 1 H), 3.42-3.46 (m, 1 H), 2.85 (s, 3H), 2.70 (d, J = 4.8 Hz, 3H), 0.97 (s, 3H), 0.93 (s, 3H). MS (M+H)+: 513. Step 4 - Synthesis of 2-(4-fluorophenyl)-6-(N-(2-hydroxy-2-methylpropyl) methylsulfonamido)-N- meth -5-(3-(oxazolo[4,5-b]pyridi -2-yl)phenyl)benzofuran-3-carboxamide
Example 775
5-bromo-2-(4-fluorophenyl)-6-(N-(2-hydroxy-2- methylpropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide was converted to the title compound (800 mg, 65.6%) using the method described in Example 411, Step 12. Ή-NMR (CDC13,400 MHz)68.52(t,J = 1.6 Hz, 1H), 8.48 (s, 1H), 8.30 (d, J= 8.0 Hz, 1H), 7.88-7.91 (m, 3H), 7.84 (t, J= 6.8 Hz, 1H), 7.82 (s, 1H), 7.79 (s, 1H), 7.59-7.65 (m, 1H), 7.24-7.27 (m, 1H), 7.15 (t,J=8.4 Hz, 2H), 5.83 (d,J=4.8 Hz, 1H), 3.45 (t,J= 12.8 Hz, 1H), 3.20 (s, 3H), 3.05 (d,J= 15.2 Hz, 1H),2.93 (d,J=4.8Hz, 3H), 1.68-1.72 (m, 1H), 0.95 (s, 6H). MS (M+H)+: 629. The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Ή-NMR (CDCI
3, 400 MHz) δ 8.33
(s, 1H), 7.92-7.95 (m,3H),7.81 (s,
1H), 7.67 (s, 1H),7.40 (d,J=8.0
Hz, 1 H), 7.27-7.32 (m, 1 H), 7.18 (t,
779 J= 8.0 Hz, 3H), 7.06 (t, J= 8.8 Hz, 676
1H), 5.96 (s, 1H),4.07 (s, 3H), 3.54
0 (d,J= 15.2 Hz, 1H), 3.29 (s, 3H),
3.10 (d, = 15.2 Hz, 1 H), 2.99 (d, J
= 4.0 Hz, 3H), 1.00 (s,6H).
Ή-NMR (CDC13, 400 MHz) δ 8.31
(s, 1H), 7.92-7.97 (m, 3H), 7.83 (s,
1H), 7.68 (s, 1H), 7.31 (d, =8.8
Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H),
780 6.87-6.92 (m, 1H), 5.84 (d, J = 2.4 694
Hz, 1H),4.09 (s, 3H), 3.54 (d,J =
0 14.8 Hz, lH), 3.29 (s,3H), 3.11 (d, J
= 15.2 Hz, 1H), 2.98 (d,7=4.8 Hz,
3H), 1.01 (d, .7 = 2.8 Hz, 6H).
Example 781: 6-(N-(2,4-dihvdroxybutyl)inethylsulfonamido)-5-(3-(4- fluorobenzo[dloxazol-2-vn-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methv.benzofuran-3-carboxamide
A mixture of the compound of Example 769 (50 mg, 0.06 mmol) and Pd/C (5 mg) in MeOH (5 mL) was placed under hydrogen atmosphere (50 psi) and allowed to stir at room temperature for 5 hours. The reaction mixture was filtered and concentrated in vacuo, and the residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 68%). Ή- NMR (CDCI
3, 400 MHz) δ 8.12-8.17 (m, 1H), 7.72-7.87 (m, 4H), 7.62 (d,J= 3.6 Hz, 1H), 7.00-7.43 (m, 6H), 6.38-6.57 (m, 1H), 4.00 (s, 3H), 3.51-3.72 (m, 4H), 2.81-3.14 (m, 7H), 1.31-1.53 (m,2H). MS (M+H)
+: 692. The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 789: 6-(N-(2-fluoro-2-methylpropyl)methylsulfonainido)-5-(3-(4- fluorobenzo[dloxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methylbenzofuran-3-carboxamide
Step 1 - Synthesis of 5-bromo-6-(N-(2-fluoro-2-methylpropyl) methylsulfonamido)-2-(4- fluorophenyl)-N-methylbenzofuran-3-carboxamide
5-bromo-2-(4-fluorophenyl)-6-(N-(2-hydroxy-2- methylpropyl)methylsulfonamido)-N-methylbenzofuran-3-carboxamide (450 mg, 0.88 mmol) was dissolved in dichloromethane (6 mL) and the solution was put under nitrogen atmosphere and cooled to-70 °C and stirred for 30 minutes. DAST reagent (283 mg, 1.76 mmol) was added dropwise into the mixture and the reaction was stirred for an additional 3 hours. The reaction was diluted with water and extracted with dichloromethane. The combined organic phases were washed with brine, dried over Na2S04, filtered and concentrated in vacuo to provide 5-bromo-6- (N-(2-fluoro-2-methylpropyl) methylsulfonamido)-2-(4-fluorophenyl)-N-methylbenzofuran-3- carboxamide (320 mg, 71%). Ή-NMR (CDC13, 400 MHz) δ 8.08 (s, 1 H), 7.80-7.84 (m, 2H), 7.73 (s, 1H), 7.13 (t, J = 8.0 Hz, 2H), 5.76 (d, J = 4.0 Hz, 1 H), 3.82-4.01 (m, 2H), 2.97 (s, 3H), 2.92 (d, J = 4.8 Hz, 3H), 1 .50 (d, J = 22.0 Hz, 3H), 1 .29 (d, J= 21 .2 Hz, 3H). MS (M+H)+: 515.
Step 2 - Synthesis of 6-(N-(2-fluoro-2-methylpropyl)methylsulfonamido) -5-(3-(4- fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methylbenzofuran-3- carboxamide
Example 789
5-bromo-6-(N-(2-fluoro-2-methylpropyl)methylsulfonamido)-2-(4-fluorophenyl)- N-methylbenzofuran-3 -carboxamide was converted to the title compound (30 mg, 36%) using the method described in Example 41 1 , Step 12. Ή-NMR (CDC13, 400 MHz) δ 8.02 (s, 1 H), 7.85-7.91 (m, 3H), 7.76 (s, 1H), 7.59 (s, 1 H), 7.35 (d, J = 8.4 Hz, 1H), 7.23-7.27 (m, 1 H), 7.12-7.16 (m, 3H), 7.01 (t, J= 8.8 Hz, 1H), 5.84 (d, J = 4.8 Hz, 1H), 4.03 (s, 3H), 3.67-3.74 (m, 1 H), 3.20 (s, 3H), 2.99-3.06 (m, 1H), 2.94 (d, J = 4.8 Hz, 3H), 1.14 (d, J = 21 .6 Hz, 3H), 1 .01 (d, J= 20.8 Hz, 3H). MS (M+H)+: 678.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
F 'H-NMR (CDCI
3, 400 Ηζ)δ 8.54 o. / (s, IH), 8.01 (s, IH), 7.79 (t,J= 14.8
Hz, 4H), 7.54-7.45 (m, 2H), 7.30 (s, IH), 7.19-7.12 (m,2H), 5.97 (s, IH), 4.13 (s, 3H), 3.96-3.39 (m, 2H), 3.23 693 (d, J = 2.2 Hz, 2H), 2.95 (d, J= 3.0
Hz, 3H), 1.68 (d,J = 1.6 Hz, 3H),
1.21 (s, 3H), 1.16 (s, 3H).
F
'H-NMR (MeOD, 400 MHz) δ 8.26
(s, IH), 7.96-8.00 (m,2H),
7.85-7.89 (m, 2H), 7.79 (s, IH),
7.68-7.71 (m, IH), 7.49-7.51 (m,
IH), 7.33-7.42 (m, 2H), 7.24-7.28 690 (m, 2H), 7.13-7.18 (m, 1 H), 4.59 (s, 3H), 4.06 (s, 3H), 3.22 (s, 2H), 2.93
(s, 3H), 1.27 (s, 3H), 0.86-0.88 (m,
IH), 0.32-0.5 (m, 3H).
1 Ή-NMR (CDCI3, 400 MHz) δ 8.21
(s, IH), 7.54-7.91 (m, 5H),
7.22-7.37 (m, 5H), 7.00 (t, J = 8.8
Hz, IH), 5.83 (br s, IH), 4.85-5.00
(m, IH), 4.20-4.52 (m, 2H), 682 3.88-4.03 (m, 4H), 3.18 (s, 2H), 2.90 (d,J = 10.6 Hz, 4H).
1 , 'H-NMR (MeOD, 400 MHz) δ 8.32
(s, IH), 7.87-7.90 (m, 4H), 7.63 (s,
IH), 7.41 (d, .7=8.4 Hz, IH),
7.26-7.31 (m, 2H), 7.15-7.19 (m,
3H), 7.04-7.08 (m, IH), 3.92-3.98 710 (m, 5H), 3.80-3.84 (m, 2H),
3.51-3.55 (m, 1 H), 3.32-3.36 (m,
IH), 3.19 (s,3H), 2.85 (s, 3H), 1.18
F
(s, 3H).
Ή-NMR (MeOD, 400 MHz) δ 8.31 (s, IH), 7.87-7.91 (m, 4H), 7.62 (s,
1 H), 7.1 -7.30 (m, 4H), 6.96-7.01
(m, IH), 3.96-4.03 (m, 5H), 728 3.78-3.84 (m, 2H), 3.51-3.55 (m,
IH), 3.32-3.36 (m, IH), 3.19 (s, 3H),
2.85 (s, 3H), 1.19 (s,3H).
Example 811 : 6-(N-(4-fluoro-2-hvdroxybutyl)methylsulfonamido)-5- 3-(4- fluorobenzo[dloxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methylbenzofuran-3-carboxamide
Step 1 - Synthesis of 4-(N-(5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-2-(4- fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-3-hydroxybutyl 4- methylbenzenesulfonate
Example 781
To a 0 °C solution of the compound of Example 581 (100 mg, 0.16 mmol), DMAP (10 mg) and TEtOAc (0.1 mL) in dichloromethane (1 mL) was added TsCl (30.8 mg, 0.16 mmol) and the reaction was allowed to stir at room temperature for 5 hours. Water was added, and the reaction mixture was extracted with dichloromethane. The combined extracts were washed with brine, dried over Na2S0 , filtered and concentrated in vacuo and the residue obtained was purified using prep-TLC (petroleum efhenEtOAc = 1 : 1) to provide 4-(N-(5-(3-(4- fluorobenzo [d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-3- (methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-3-hydroxybutyl 4- methylbenzenesulfonate (70 mg, 58%). 1H-NMR (CDC13, 400 MHz) δ 8.09-8.31 (m, 1H), 7.92 (s, 2H), 7.99 (d, J = 2.0 Hz, 2H), 7.61-7.72 (m, 3H), 7.03-7.45 (m, 8H), 6.06-6.22 (m, 1 H), 4.14-4.30 (m, 2H), 4.06 (s, 3H), 3.56-3.93 (m, 3H), 2.70-3.06 (m, 6H), 2.33-2.40 (m, 3H), 1.60-1.78 (m, 2H). MS (M+H)+: 846.
Step 2 - Synthesis of 6-(N-(4-fluoro-2-hydroxybutyl)methylsulfonamido)-5-(3- (4- fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methylbenzofuran-3- carboxamide
Example 811
A mixture of 4-(N-(5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-2-(4- fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-3-hydroxybutyl 4- methylbenzenesulfonate (50 mg, 0.06 mmol), CsF (27 mg, 0.12 mmol) in t-BuOH (2 mL) was heated to 80 °C and allowed to stir at this temperature for 5 hours. Then water was added, and the reaction mixture was extracted with dichloromethane. The combined extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo, and the residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 73%). Ή-NMR (CDCl3, 400 MHz) δ 8.16-8.37 (m, 1 H), 7.81-7.93 (m, 4H), 7.72 (d, J = 7.2 Hz, 1 H), 7.66 (s, 1 H), 7.29-7.46 (m, 2H), 7.04-7.20 (m, 4H), 5.89-6.61 (m, 1 H), 4.46-4.85 (m, 2H), 4.05 (s, 3H), 3.57-3.91 (m, 2H), 2.97-3.31 (m, 5H), 2.67 (s, 2H), 1.62-1.69 (m, 2H). MS (M+H)+: 694.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 816: 6-(N-(4-cvano-2-hvdroxybutyl)methy sulfonamido)-5-(3-(4- fluorobenzo[dloxazol-2-vn-4-methoxyphenyl)-2-(4-fluoropheny )-N- methylbenzofuran-3-carboxamide
Example 816
A mixture of 4-(N-(5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4- methoxyphenyl)-2-(4- fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-3-hydroxybutyl 4- methylbenzenesulfonate (50 mg, 0.06 mmol, made as described in Example 444), TMSCN (1 1.7 mg, 0.12 mmol) and TBAF (32.3 mg, 0.12 mmol) in CH3CN (2 mL) was heated to 70 °C and allowed to stir at this temperature for 5 hours. Then water was added and the resulting solution was extracted with dichloromethane. The combined extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo, and the resulting residue was purified using prep- HPLC to provide the title compound (35 mg, 85%). Ή-NMR (CDC13, 400 MHz) δ 8.51 (s, 1 H), 7.61-7.95 (m, 5H), 7.08-7.48 (m, 6H), 5.91 (d, J = 4.8 Hz, 1 H), 3.48-4.1 1 (m, 6H), 2.98 (d, J = 4.8 Hz, 3H), 2.43-2.88 (m, 5H), 1.71-1.73 (m, 2H). MS (M+H)+: 701 .
Example 817: 6-(N-(3-cvano-2-hvdroxypropyl)methylsulfonainido)-5-(3-(4- fluorobenzo|d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N- methylbenzofuran-3-carboxamide
Step 1 - Synthesis of 5-bromo-6-(N-(3-cyano-2-hydroxypropyl) methylsulfonamido)-2-(4- fluorophenyl)-N-methylbenzofuran-3-carboxamide
A mixture of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(N-(oxiran-2- ylmethyl)methylsulfonamido)benzofuran-3-carboxamide (1.05 g, 2.1 mmol, prepared from Compound 411G as described in Example 41 1 , Step 6), TMSCN (837 mg, 8. 5 mmol) and TBAF (2.20 g, 8.5 mmol) in THF (50 mL) was heated to reflux (80 °C) and allowed to stir at this temperature for 2 hours. The reaction was cooled to room temperature, diluted with water and extracted with EtOAc. The organic extract was washed with water and brine, dried over Na
2S0
4, filtered and concentrated in vacuo to provide 5-bromo-6-(N-(3-cyano-2-hydroxypropyl) methylsulfonamido)-2-(4- fluorophenyl)-N-methylbenzofuran-3-carboxamide (1.35 g), which was used without further purification.
Step 2 - Synthesis of 6-(N-(3-cyano-2-hydroxypropyl)methylsulfonamido)-5 -(3-(4- fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2-(4-fluorophenyl)-N-methylbenzofuran-3- carboxamide
Example 817
5-bromo-6-( -(3-cyano-2-hydroxypropyl)methylsulfonamido)-2-(4- fluorophenyl)-N-methylbenzofuran-3-carboxamide was converted to the title compund (18 mg, 10.6%) using the method described in Example 41 1 , Step 12. Ή-NMR (CDC13, 400 MHz) δ 8.08-8.35 (m, 1 H), 7.59-7.91 (m, 5H), 7.03-7.47 (m, 6H), 6.00-6.14 (m, 1 H), 3.83-4.33 (m, 5H), 3.47-3.72 (m, 1H), 2.41-3.03 (m, 8H). MS (M+H)+: 687.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 823: 5-(3- 4-fluorobenzo[dloxazol-2-vI)-4-methoxyphenyl)-2-(4-
fluorophenyl)-6-(N-(2-hvdroxy-3-(lH-imidazol-l-yl)propyl)methylsulfonamido)-N- methylbenzofuran-3-carboxamide
Example 823
To a microwave tube was added 5-bromo-2-(4-fluorophenyl)-N- methyl-6-( -
(oxiran-2-yl-methyl)methylsulfonamido)benzofuran-3-carboxamide (43 mg, 0.07 mmol, prepared according to the method described in Example 440), imidazole ( 1 1 mg, 0.16 mmol), Cs2C03 (53 mg, 0.16 mmol) and 5 mL of DMF. The reaction was placed in a commercial microwave for 30 minutes during which time the reaction temperature reached 120 °C. The reaction mixture was then cooled to RT and water was added and the solution was extracted with EtOAc. The organic extract was washed with H20 and brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using prep-HPLC to provide the title compound (38 mg, 80.3%). Ή-NMR (CDC13, 400 MHz) δ 8.19-8.06 (m, 1H), 7.77-7.60 (m, 5H), 7.49 (s, 1 H), 7.38-7.35 (m, 4H), 7.07-6.98 (m, 1.5H), 6.92-6.67 (m, 1.5H), 5.29 (s, 1 H), 4.13 (s, 1H), 4.00 (t, J = 9.2 Hz, 3H), 3.88-3.55 (m, 4H), 3.1 1 (d, J= 2.4 Hz, 2H), 3.08-2.92 (m, 3H), 2.83 (d, J = 2.0 Hz, 2H). MS (M+H)+: 728.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 829: 2-(4-fluorophenvn-N-methyl-5-(3-(oxazolo[4,5-blpyridin-2- yl)phenyl)-6-(N-(2-(phenylamino)propyl)methylsuIfonamido)benzofuran-3-
carboxamide
Step 1 - Synthesis of l-(N-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-5-(3-(oxazol o[4,5- b]pyridi -2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)propan-2-yl methanesulfonate
To a solution of 2-(4-fluorophenyl)-6-(N-(2-hydroxypropyl) methylsulfonamido)- N-methyl-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (147 mg, 0.24 mmol, prepared according to the method described in Example 440) in dichloromethane ( 1 .5 mL) was added Et3N (30 mg, 0.29 mmol). The reaction was cooled to 0 °C and MsCl (30 mg, 0.26 mmol) was added dropwise. The reaction was warmed to 25 °C and allowed to stir at this temperature for 1 hour, then the reaction mixture was extracted with dichloromethane, and the organic extract was concentrated in vacuo. The residue obtained was purified using prep-TLC (petroleum ethenEtOAc = 2 : 1 ) to provide l -(N-(2-(4-fluorophenyl)-3- (methylcarbamoyl)-5-(3- (oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)propan-2-yl
methanesulfonate (0.1 g, 85%) as yellow solid. Ή-NMR (MeOD, 400 MHz) δ 8.52-8.56 (m, 2H), 8.39-8.40 (m, IH), 8.15-8.17 (m, I H), 7.96-8.03 (m, 4H), 7.71-7.75 (m, 2H), 7.46-7.50 (m, I H), 7.27-7.31 (m, 2H), 4.62 (s, 2H), 4.07-4.12 (m, I H), 3.03 (s, 3H), 2.94 (s, 3H), 2.01 (s, 3H), 2.01 (s, 3H), 1.28 (s, 3H). MS (M+H)+: 693.
Step 2 - Synthesis of 2 -(4 -fluorophenyl) -N -methyl- 5- (3- (oxazolo [4,5 -b]pyridin-2 -yl) phenyl) -6- (N-(2-(phenylamino)propyl)methylsulfonamido)benzofuran-3-carboxamide
To a solution of l -(N-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-5- (3- (oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)propan-2-yl methanesulfonate (100 mg, 0.14 mmol) in MeCN (2 mL) was added Et3N ( 1 mL), PhNH2 ( 130 mg, 0.14 mmol) and DMAP (12 mg) and the mixture was placed in a commercial microwave oven and irradiated for 1 hour, during which time the reaction temperature went to 120 °C. The reaction was cooled to RT, diluted with dichloromethane, and the resulting solution was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 30%) as white solid. 1 H-NMR (MeOD, 400 MHz) δ 8.51 -8.55 (m, 2H), 8.35-8.39 (m, 1 H), 8.14-8.16 (m, 1 H), 7.93-8.00 (m, 4H), 7.68-7.77 (m, 2H), 7.30-7.49 (m, 1 H), 7.25-7.30 (m, 2H), 7.25-7.30 (m, 1 H), 6.26-6.97 (m, 4H), 3.46-3.70 (m, 2H), 3.17-3.19 (m, 4H), 2.92 (s, 3H), 1 .03-1 .05 (m, 1 H), 0.74-0.76 (m, 2H). MS (M+H)+: 689. The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 831 : 2-(4-fluorophenyl)-N-methyl-5-(3-(oxazolo|4,5-blpyridin-2- yl)phenyl)-6-(N-(piperidin-4-ylmethyl)methylsulfonamido)benzofuran-3- carboxamide
Example 831
To a 0 °C solution of tert-butyl 4-((N-(2-(4-fluorophenyl)-3- (methyl carbamoyl)- 5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)methyl)piperidine- 1 -carboxylate (100 mg, 0.13 mmol, prepared according to the method described in Example 440) in dichloromethane (10 mL) was added TFA (75 mg, 0.66 mmol) was added dropwise. The reaction was allowed to stir at 0 °C for 2 hours, and then was diluted with water and basified with aqueous NaHC03 solution. The basified solution was extracted with dichloromethane and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 34.6%). Ή-NMR (CDCb, 400 MHz) δ 8.51-8.56 (m, 2H), 8.32 (d, J = 8.0 Hz, 1 H), 8.15 (d, J = 8.0 Hz, 1 H), 7.95-8.00 (m, 4H), 7.68-7.72 (m, 2H), 7.48 (d, J = 4.0 Hz, 1H), 7.24-7.29 (m, 2H), 3.38-3.40 (m, 2H), 3.18-3.22 (m, 4H), 2.91 -2.97 (m, 4H), 2.75-2.81 (m, 1 H), 2.40-2.44 (m, 1H), 1 .45-1.67 (m, 1 H), 1.30-1.31 (m, 1 H), 1.21-1.27 (m, 1 H), 1.00-1.04 (m, 2H). MS (M+H)+: 654.
Example 832: (S)-methyl l-f4- N-(2-(4-fluorophenyl)-3-(methylcarbamovn-5- (3-
(oxazolo[4,5-blpyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)
tnethyl)pipe"din-l-yl)-3-methyl-l-oxobutan-2-ylcarbainate
A solution of the compound of Example 831 (86 mg, 0.1 3 mmol), HOBT (58 mg, 0.43 mmol) and EDCI (84 mg, 0.43 mmol) in dry DMF (3 mL) was allowed to stir at room temperature for 30 minutes. Triethylamine (0.5 mL) and (S)-2-(methoxycarbonylamino)-3- methylbutanoic acid (70 mg, 0.39 mmol) were then added and the reaction was allowed to stir for about 15 hours. The reaction mixture was concentrated in vacuo and the residue obtained was diluted with water and extracted with ethyl acetate. The organic extract was washed with H
20, brine, dried over Na
2S0
4, filtered and concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (52 mg, 49%). 1H-NMR (CDC1
3, 400 MHz) δ 8.35-8.54 (m, 2H), 8.30-8.35 (m, 1 H), 8.12-8.14 (m, 1 H), 7.90-7.97 (m, 4H),
7.67-7.71 (m, 2H), 7.44 (d, J = 4.0 Hz, 1 H), 7.22-7.26 (m, 2H), 3.91 -4.3 1 (m, 3H), 3.48-3.55 (m, 3H), 3.16-3.1 8 (m, 3H), 2.93 (s, 3H), 2.37-2.85 (m, 2H), 1 .36- 1 .80 (m, 4H), 0.81 -0.99 (m, 2H), 0.49-0.80 (m, 8H). MS (M+H)+: 81 1.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
Example 838: 6-(N-(3-(cyanomethYlamino)propynmethylsulfonainido)-2-(4- fluorophenyl)-N-methyl-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3- carboxamide
Example 838
A mixture of 6-(N-(3-aminopropyl)methylsulfonamido)-2-(4- fluorophenyl)-N- methyl-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl)benzofuran-3-carboxamide (100 mg, 0.16 mmol, prepared according to the method described in Example 449), BrCH2CN (84 mg, 0.71 mmol), K2C03 (97 mg, 0.71 mmol) and KI (27 mg, 0.16 mmol) in dry DMF (2.0 mL) was heated to 100 °C and allowed to stir at this temperature for about 15 hours. The reaction mixture was cooled to room temperature and filtered, then the filtrate was washed with H20, brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (21 mg, 19.8%). Ή-NMR (CDC13, 400 MHz) δ 8.47-8.49 (m, 1 H), 8.32 (s, 1 H), 8.23 (d, J = 7.2 Hz, 1 H), 7.86-7.88 (m, 2H), 7.79-7.83 (m, 2H), 7.71-7.73 (m, 1 H), 7.55-7.57 (m, 2H) 7.22-7.26 (m, 1H), 7.10-7.14 (m, 2H), 6.08 (d, J = 4.4 Hz, 1H), 3.39 (s, 4H), 2.92 (d, J = 5.2 Hz, 3H), 2.84 (s, 3H), 2.38-2.42 (m, 2H), 1.53 (s, 3H). MS (M+H)+: 653.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 840; 3-(N-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-5-(3- (oxazolo[4,5-b1pyridin-2-yl)phenyl)benzofuran-6-yl)inethylsulfonainido)
propanoic acid
Step 1 - Synthesis of2-(4-fluorophenyl)-N-methyl-5-(3-(oxazolo[4,5-b]pyridin- 2-yl)phenyl)-6- (N-(3-oxopropyl)methylsulfonamido)benzofuran-3-carboxamide
A solution of 2-(4-fluorophenyl)-6-(N-(3-hydroxypropyl) methylsulfonamido)-N- methyl-5-(3-(oxazolo[4,5-b]pyridin-2-yl)phenyl) benzofuran-3-carboxamide (350 mg, 0.57 mmol, prepared according to the method described in Example 440) and DMP (1.2 g, 2.8 mmol) in dichloromethane (10 mL) was allowed to stir at room temperature for 2 hours under N
2 atmosphere. The reaction was quenched with saturated aqueous NaHC0
3 and excess Na
2S
20
4 and stirred until all solids were dissolved. The solution was then extracted with dichloromethane and the organic extract was washed with brine, dried over Na
2S0
4, filtered and concentrated in vacuo to provide 2-(4-fluorophenyl)-N-methyl-5-(3- (oxazolo[4,5-b]pyridin-2-yl)phenyl)-6-(N- (3-oxopropyl)methylsulfonamido)benzofuran-3-carboxamide (315 mg, 90.2%), which was used without further purification. Step 2 - Synthesis of 3-(N-(2-(4-fluorophenyl)-3-(methylcarbamoyl)-5- (3-(oxazolo [4,5- b]pyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonamido)propanoic acid
Example 840
To a 0 °C solution of 2-(4-fluorophenyl)-N-methyl-5-(3-(oxazolo [4,5-b]pyridin- 2-yl)phenyl)-6-( J-(3-oxopropyl)methylsulfonamido)benzoftjran-3-carboxamide (300 mg, 0.49 mmol), NaH2P04 (180 mg, 4 mmol) and NH2S03H (72 mg, 0.75 mmol) in dioxane (5 mL) was added a solution of NaC102 (180 mg, 2 mmol) in H20 (2 mL) dropwise. The reaction was allowed to stir for 10 minutes at 0 °C, then the cold bath was removed and the reaction mixture was warmed up to room temperature and stirred for another 15 minutes. The reaction was diluted with water, extracted with dichloromethane and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo to provide the title compound (210 mg, 67.8%). Ή-NMR (DMSO, 400 MHz) δ 8.57 (s, 1 H), 8.28 (s, 1 H), 8.17-8.19 (m, lH), 7.81-7.87 (m, 2H), 7.56-7.58 (m, 3H), 7.25-7.26 (m, 2H), 7.18-7.20 (m, 1 H), 7.1 1-7.15 (m, 2H), 5.98 (s, 1 H), 3.64-3.67 (m, 2H), 2.92-2.93 (m, 3H), 2.81 (s, 3H), 2.51-2.52 (m, 2H). MS (M+H)+: 629. Example 841 : (S)-methyl 2-(3-(N-(2-(4-fluorophenvn-3-(methylcarbamovn-5-(3-
(oxazolo[4,5-b1pyridin-2-yl)phenyl)benzofuran-6-yl)methylsulfonainido)
propanamido)-3-methylbutanoate
Example 840 Example 841
The compound of Example 840 was converted to the title compound (30 mg, 36%) using the method described in Example 41 1 , Step 5. 1 H-NMR (CDC13, 400 MHz) δ 8.57 (s, 1 H), 8.38 (s, 1 H), 8.21 (m, J = 6.4 Hz, 1 H), 8.06 (s, 1 H), 7.88 (s, 2H), 7.78-7.80 (m, 1 H), 7.42-7.59 (m, 3H), 7.1 1-7.15 (m, 3H), 6.21 (s, 2H), 4.29 (s, 1 H), 3.57 (d, J = 4.4 Hz, 3H), 3.25-3.32 (m, 1H), 2.92-2.98 (m, 6H), 2.36 (s, 2H), 1.98-2.01 (m, 2H), 1.1 8 (s, 6H). MS (M+H)+: 742.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Examples 843 and 844: 3-fN-f5-(3-(4-fluorobenzo[d1oxazol-2-yl)-4- methoxyphenyl)-2-(4-fluoronhenyl)-3-(methylcarbamoyl)benzofuran-6-yl)
methylsulfonamido)propylph.osph.onic acid
Example 844
To a solution of the compound of Example 713 (100 mg, 0.13 mmol) in CH3CN (2 mL) was added TMSBr (2.0 g, 9.2 mmol). The reaction was allowed to stir for 16 hours, then was quenched with water and extracted with CH2C12. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified prep-HPLC to provide Example 843 (20 mg, 20%) and Example 844 (10 mg, 10%).
Example 843: 1H-NMR (CDC13, 400 MHz) δ 7.98 (s, 1H), 7.35-7.59 (m, 5H), 7.19-7.23 (s, 1H), 7.12 (s, 1 H), 6.86-6.88 (m, 4H), 3.77 (s, 3H), 3.16-3.33 (m, 2H), 2.65-2.85 (m, 6H), 1.18-1 .51 (m, 4H). MS (M+H)+: 726.
Example 843: Ή-NMR (CDC13, 400 MHz) δ 7.92 (s, 1H), 7.84-7.87 (m, 3H), 7.72 (s, l H), 7.52 (s, 1H), 7.39 (s, 1 H), 7.28-7.29 (m, 1 H), 7.03-7.1 1 (m, 4H), 6.15 (s, 1 H), 3.94 (s, 5H), 3.41 (s, 2H), 3.00 (s, 3H), 2.90 (d, J = 4.4 Hz, 3H), 1.31-1.61 (m, 4H), 1.14-1.16 (m, 3H). MS (M+H)+: 754. Example 845: 5-f3-riH-indoI-2-yl)-4-niethoxyphenyl)-2-(4-fluorophenvn-N- methyl-6-(N-methylmethylsulfonainido)-N-(pyridin-3-yl)benzofuran-3- carboxamide
Example 553 Example 845
A solution of the compound of Example 553 (100 mg, 0.17 mmol), 3-Bromo- pyridine (40 mg, 0.25 mmol) and Cul (3 mg) in toluene (1.5 mL) was put under nitrogen atmosphere and heated to 1 10 °C. The reaction was stirred and monitored using TLC. When the starting material was consumed, the reaction mixture was cooled to RT and the reaction mixture was concentrated in vacuo. The residue obtained was purified using Prep-HPLC to provide the title compound (30 mg, 26.5%). 1H-NMR (CDC13, 400 MHz) δ 8.00 (t, J = 8.0 Hz, 2H), 7.83 (s, 1H), 7.68 (d, J = 8.0 Hz, 1 H), 7.60-7.52 (m, 1 H), 7,36 (d, J= 8.0 Hz, 4H), 7.31 (d, J = 7.2 Hz, 2H), 7.20 (t, J = 8.4 Hz, 3H), 6.75 (s, 1H), 6.22 (s, 1H), 5.74 (d, J = 1 1 .6 Hz, 1 H), 5.57 (s, 1 H), 4.83 (s, 2H), 3.19 (s, 3H), 3.03 (d, J = 4.4 Hz, 3H), 2.72 (s, 3H). MS (M+H)+: 675.
The following compounds of the present invention were made using the method described above and using the appropriate reactants and/or reagents.
8.36-8.37 (m, 2H), 8.16-8.17 (m,
1 H), 7.63-7.66 (m, 2H), 7.56 (s, 1 H),
7.46-7.47 (m, 2H), 7.36-7.38 (m,
2H), 7.22-7.28 (m, 6H), 4.02 (s, 3 H),
3.44 (s, 3H), 3.09 (s, 3H), 2.75 (s,
3H).
Example 851 : 5-(3-(lH-indol-2-yl)phenvt)-2-(4-fluorophenvn-6-(N- methylmethylsulfonamido)-N-(pyridin-3-yl)benzofuran-3-carboxamide
Step 1 - Synthesis of ethyl 5-(3-(l H-indol-2-yl)phenyl)-2-(4-fluorophenyl)-6- (N- methylmethylsulfonamido)benzofuran-3-carboxylate
A mixture of ethyl 5-bromo-2-(4-fluorophenyl)-6-(N- methylmethylsulfonamido)benzofuran-3-carboxylate (884 mg, 1.9 mmol, prepared from
Compound 41 I E with Mel according to the method described in Example 41 1 , step 7), 2-(3- (4,4,5,5-tetramethyl-l ,3,2- dioxaborolan-2-yl)phenyl)-l H-indole (746 mg, 2.3 mmol, prepared from corresponding bromide), K3PO4 (1.03 g, 3.88 mmol) and Pd(dppf)Cl2 (142 mg, 0.19 mmol) in DMF (10 mL) was heated to 100 °C and allowed to stir at this temperature for 8 hour under N2 atmosphere. The reaction was poured into ice water, the resulting solution was filtered and the collected solid was washed with water and dried to provide ethyl 5-(3-(l H-indol-2-yl)phenyl)-2- (4- fluorophenyl)-6-( - methylmethylsulfonamido) benzofuran-3-carboxylate (0.88 g, 79% yield), which was used without further purification. MS (M+H)+: 583.
Step 2 - Synthesis of 5-(3-(lH-indol-2-yl)phenyl)-2-(4- fluorophenyl) -6- (N- methylmethylsulfonamido)benzofuran-3-carboxylic acid
A mixture of ethyl 5-(3-(lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)- 6- (N- methylmethylsulfonamido)benzofuran-3-carboxylate (870 mg, 1.53 mmol), and LiOH (320 mg, 7.65 mmol) in 1 ,4-dioxane/water (1/1 , 40 mL) was heated to 100 °C and allowed to stir at this temperature for 2 hours. The reaction mixture was cooled to RT, concentrated in vacuo and the resulting residue was diluted by water. The resulting solution was adjusted to pH 3 using IN HC1 and the acidified solution was filtered. The collected solid was washed with water and dried to provide 5-(3-(l H-indol-2-yl) phenyl)- 2-(4-fluorophenyl)-6-(N-methylmethylsulfonamido) benzofuran-3- carboxylic acid (0.8 g, 94%). Ή-NMR (DMSO, 400 MHz) δ 13.38 (s, 1 H), 1 1.58 (s, 1 H), 8.13-8.16 (m, 2H), 8.04 (d, J = 9.2 Hz, 2H), 7.94 (s, 1 H), 7.89 (d, J = 7.6 Hz, 1 H), 7.54 (t, J = 7.2 Hz, 2H), 7.38-7.46 (m, 4H), 7.10 (t, J = 8.0 Hz, 1 H), 6.99 (t, J = 8.0 Hz, 1 H), 6.95 (s, 1H), 3.14 (s, 3H), 2.94 (s, 3H). MS (M+H)+: 555.
Step 3 - Synthesis of 5-(3-(lH-indol-2-yl)phenyl)-2-(4-fluorophenyl)-6- (N- methylmethylsulfonamido)-N-(pyridin-3-yl)benzofuran-3-carboxamide
Example 851
A mixture of 5-(3-(l H-indol-2-yl)phenyl)-2-(4-fluorophenyl)-6- (N- methylmethylsulfonamido)benzofuran-3-carboxylic acid (80 mg, 0.14 mmol), pyridin-3 -amine (17 mg, 0.17 mmol), PyBOP (80 mg, 0.17 mmol), and DIPEA (27 mg, 0.21 mmol) in DMF (1 mL) was allowed to stir for 12 hours. Water was added, then the resulting solution was extracted with ethyl acetate and the organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue obtained was purified using prep-HPLC to provide the title compound (30 mg, 33%). Ή-NMR (CDC13, 400 MHz) δ 9.67 (s, I H), 8.78-8:92 (m, 2H), 8.28 (s, I H), 8.08 (s, IH), 7.74 (s, 3H), 7.64-7.67 (m, 3H), 7.47-7.49 (m, I H), 7.36 (s, 2H),
7.14-7.22 (m, 6H), 6.82 (s, IH), 2.91 (s, 6H). MS (M+H)+: 631.
The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Example 853: 5-(3-(4-fluorobenzo[dloxazol-2-yl)-4-methoxyphenyl)-2-(4- fluorophenvI)-N-methyl-6-(2-oxooxazolidin-3-vnbenzofuran-3-carboxamide
Step 1 - Synthesis of ethyl 5-bromo-2-(4-fluorophenyl)-6-(2- oxooxazolidin-3- yl)benzof ran-3- carboxylate
2-Chloroethyl chloroformate (0.38 g, 2.6 mmol) and K2C03 (0.75 g, 7.2 mmol) were added to a solution of Compound 41 ID (0.5 g, 1 .3 mmol, prepared described in Example 1, step 3) in MeCN (10 mL) under N2 atmosphere. The reaction was heated to reflux (80 °C) and allowed to stir at this temperature for about 15 hours. The reaction mixture was then filtered and concentrated in vacuo, and the residue obtained was purified using column chromatography (petroleum ether:EtOAc = 4 : 1) to provide ethyl 5-bromo-2-(4-fluorophenyl)-6-(2- oxooxazolidin-3-yl) benzofuran-3-carboxylate (350 mg, 59%). Ή-NMR (CDC13, 400 MHz) δ 8.35 (s, 1H), 8.04-8.08 (m, 2H), 7.61 (s, 1 H), 7.17-7.21 (m, 2H), 4.59 (t, J = 8.0 Hz, 2H), 4.43 (q, J = 7.2 Hz, 2H), 4.08 (t, J = 8.0 Hz, 2H), 1.42 (t, J= 7.2 Hz, 3H). MS (M+H)+: 448 / 450.
Step 2 - Synthesis of 5-bromo-2-(4-fluorophenyl)-6-(2-oxooxazolidin-3-yl)benzofuran- 3- car boxy lie acid
To a solution of ethyl 5-bromo-2-(4-fluorophenyl)-6-(2- oxooxazolidin-3- yl)benzofuran-3-carboxylate (350 mg, 0.78 mmol) in dioxane (6 mL) and water (6 mL) was added LiOH (187 mg, 7.81 mmol). The reaction was heated to reflux and allowed to stir at this temperature for 3 hours. The reaction mixture was concentrated in vacuo and the resulting residu was was diluted with water. The solution was acidified to pH = 6-7 using 1 N HC1, and extracted with EtOAc. The organic extract was washed with brine, dried over Na
2S0
4, filtered and concentrated in vacuo to provide 5-bromo-2-(4-fluorophenyl)-6-(2-oxooxazolidin-3- yl)benzofuran- 3-carboxylic acid (300 mg, 92%), which was used without further purification. Step 3 - Synthesis of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(2-oxooxazolidin-3 -yl)benzofuran- 3-carboxamide
5-brorno-2-(4-fluorophenyl)-6-(2-oxooxazolidin-3-yl)benzofuran-3-carboxylic acid (300 mg, 0.72 mmol), HOBT (145 mg, 1.07 mmol) and EDCI (166 mg, 1.07 mmol) were taken up in dry DMF (8 mL). The resulting reaction was allowed to stir for 30 minutes, then methanamine HC1 salt (44 mg, 1.43 mmol) and Et3N (1 mL) were added. The reaction was then allowed to stir for about 15 hours, then the reaction mixture was diluted with water and extracted with EtOAc. The organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using prep-TLC (petroleum ether:EtOAc = 2 : 1) to provide pure 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(2-oxooxazolidin- 3-yl) benzofuran-3-carboxamide (200 mg, 66%). Ή-NMR (CDC13, 400 MHz) δ 7.99 (s, 1 H), 7.81 (br s, 2H), 7.46 (s, 1 H), 7.12-7.16 (m, 2H), 6.29 (br s, 1H), 4.55 (t, J= 8.0 Hz, 2H), 4.03 (t, J = 8.0 Hz, 2H), 2.92 (d, J = 4.8 Hz, 3H). MS (M+H)+: 433 / 435.
Step 4 - Synthesis of 5-(3-(4-fluorobenzo[d]oxazol-2-yl)-4-methoxyphenyl)-2- (4-fluorophenyl)- N-methyl-6-(2-oxooxazolidin-3-yl)benzofuran-3-carboxamide
Example 853
To a mixture of 5-bromo-2-(4-fluorophenyl)-N-methyl-6-(2- oxooxazolidin- 3- yl)benzofuran-3-carboxamide (50 mg, 0.12 mmol), 4-fluoro-2- (2-methoxy-5-(4,4,5,5- tetramethyl-l ,3,2-dioxaborolan-2-yl)phenyl)benzo[d]oxazole (51 mg, 0.14 mmol) and
Κ3Ρ04·3Η20 (60 mg, 0.23 mmol) in 1 ,4-dioxane (2 mL), was Pd(dppf)Cl2 (5 mg). The reaction was put under N2 atmosphere, heated to 100 °C and allowed to stir at this temperature for for about 15 hours. The reaction was then cooled to room temperature and concentrated in vacuo and the residue obtained was diluted with water, and extracted with EtOAc. The organic extract was washed with brine, dried over Na2S04, filtered and concentrated in vacuo and the residue obtained was purified using prep-HPLC to provide the title compound (50 mg, 82%). Ή-NMR (CDC13, 400 MHz) δ 8.1 1 (d, J = 2.0 Hz, 1H), 7.79-7.82 (m, 2H), 7.71 (s, 1H), 7.50-7.53 (m, 2H), 7.40 (d, J = 8.0 Hz, 1 H), 7.23-7.32 (m, 1 H), 7.15 (d, J = 8.4 Hz, 1 H), 7.02-7.07 (m, 3H), 6.84 (br s, 1 H), 4.23 (t, J = 8.0 Hz, 2H), 4.09 (s, 3H), 3.47 (t, J = 8.0 Hz, 2H), 3.1 1 (d, J = 4.4 Hz, 3H). MS (M+H)+: 596. The following compound of the present invention was made using the method described above and using the appropriate reactants and/or reagents.
Examples 855-880
The following compounds of the present invention were made using the methods described in the Examples above, and using the appropriate reactants and/or reagents.
877
878
879
880
NA = not available
Example 881: Measuring Compound Inhibitory Potency
Measurement of inhibition by compounds was performed using the HCV replicon system. Several different replicons encoding different HCV genotypes or mutations were used. In addition, potency measurements were made using different formats of the replicon assay, including different ways of measurements and different plating formats. See Jan M. Vrolijk et al., A replicons-based bioassay for the measurement of interferons in patients with chronic
hepatitis C, 1 10 J. ViROLOGlCAL METHODS 201 (2003); Steven S. Carroll et al. , Inhibition of Hepatitis C Virus RNA Replication by 2'-Modified Nucleoside Analogs, 278(14) J. BIOLOGICAL CHEMISTRY 1 1979 (2003). However, the underlying principles are common to all of these determinations, and are outlined below.
Stable neomycin phosphotransferase encoding replicons-harboring cell lines were used, so all cell lines were maintained under G41 8 selection prior to the assay. Potency was deteremined using a cell ELISA assay with an antibody to the replicons encoded NS3/4a protease. See Caterina Trozzi et al. , In Vitro Selection and Characterization of Hepatitis C Virus Serine Protease Variants Resistant to an Active-Site Peptide Inhibitor, 77(6) J. Virol. 3669 (2003). To initiate an assay, replicon cells were plated in the presence of a dilution series of test compound in the absence of G418. Typically, the assays were performed in a 96-well plate formate for manual operation, or a 384-well plate format for automated assay. Replicon cells and compound were incubated for 96 hours. At the end of the assay, cells were washed free of media and compound, and the cells were then lysed. RNA was quantified indirectly through detection of replicon-encoded NS3/4A protein levels, through an ELISA-based assay with an antibody specific for NS3/4A. IC50 determinations were calculated as a percentage of a DMSO control by fitting the data to a four-parameter fit function and the data obtained is provided in the table below.
The activity tables provided below illustrate the observed activity of selected compounds of the present invention:
ND = no data available
176 3.9 252 78.2 345 2.1
177 7.4 260 3.7 346 165.2
178 7.0 261 2.0 347 8.1
179 7.2 262 5.0 348 72.7
180 5.0 264 2.9 349 5.7
181 5.7 271 1.5 350 0.7
182 2.3 272 3.0 353 4.2
183 1.5 273 2.2 356 5.9
185 2.2 278 3.2 360 30.5
186 2.0 279 3.2 366 4.2
187 2.6 281 3.3 367 3.9
188 5.1 282 2.3 368 1.6
189 1.9 283 17.2 369 2.7
190 2.2 284 5.3 370 8.8
191 4.1 285 10.2 371 5.8
192 1.9 286 5.8 372 13.0
193 1 1.6 289 5.4 373 3.9
194 4.2 290 6.9 374 52.4
198 1.4 292 5.9 375 2.7
199 2.9 293 8.6 377 1.2
200 1.6 294 10.0 378 1 .8
201 1.7 295 4.1 380 1.2
202 1.0 296 64.1 381 5.1
203 2.4 297 20.5 382 8.9
204 4.5 298 6.0 383 2.5
205 1 1.9 299 3.3 384 0.9
206 2.1 300 1.5 386 1.3
207 2.4 301 1.9 387 2.3
208 1.8 302 3.1 391 2.4
209 5.5 303 1.8 393 1 .2
210 1.9 304 1.3 394 1.4
211 3.3 305 1.1 395 8.2
212 2.6 306 28.8 396 18.2
213 2.5 307 2.4 397 3.6
214 6.3 308 2.5 398 2.4
215 2.1 309 3.7 399 2.8
216 8.2 310 1.8 400 303.4
217 2.1 311 1.7 401 1.8
218 1.8 312 1.1 402 0.9
219 3.0 314 1 .5 403 3.8
220 1.5 315 4.5 404 2.6
221 1.7 316 2.8 405 4.7
222 3.5 317 2.9 406 15.1
223 10.0 318 18.7 407 2.8
224 2.3 319 13.6 408 3.8
225 3.6 320 13.1 409 2.1
226 4.0 321 6.6 410 0.5
227 2.2 322 19.5
It will be appreciated that various of the above-discussed and other features and functions, or alternatives thereof, may be desirably combined into many other different systems or applications. Also that various presently unforeseen or unanticipated alternatives,
modifications, variations or improvements therein may be subsequently made by those skilled in the art which are also intended to be encompassed by the following claims.