WO2011113293A1 - 二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 - Google Patents
二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 Download PDFInfo
- Publication number
- WO2011113293A1 WO2011113293A1 PCT/CN2011/000062 CN2011000062W WO2011113293A1 WO 2011113293 A1 WO2011113293 A1 WO 2011113293A1 CN 2011000062 W CN2011000062 W CN 2011000062W WO 2011113293 A1 WO2011113293 A1 WO 2011113293A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- fluorenyl
- substituted
- aryl
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC[C@]1N(C2CCCC2)c2nc(Nc(cc3)c(*C)cc3C(NC[C@@](C3)(C[C@]4C3CN(C)C4)O)=O)ncc2N(C)C1=O Chemical compound CC[C@]1N(C2CCCC2)c2nc(Nc(cc3)c(*C)cc3C(NC[C@@](C3)(C[C@]4C3CN(C)C4)O)=O)ncc2N(C)C1=O 0.000 description 4
- OZOBVXNSPDPSHA-MRXNPFEDSA-N CC[C@H]1N(C(C)C)c(nc(Nc(ccc(C(C)=O)c2)c2OC)nc2)c2N(C)C1=O Chemical compound CC[C@H]1N(C(C)C)c(nc(Nc(ccc(C(C)=O)c2)c2OC)nc2)c2N(C)C1=O OZOBVXNSPDPSHA-MRXNPFEDSA-N 0.000 description 2
- DQJGEBQIHZAYNL-CQOQZXRMSA-N CC[C@H]1N(C2CCCC2)c(nc(Nc(ccc(C(NC[C@H]2OC[C@@H](CCC3)N3C2)=O)c2)c2OC)nc2)c2N(C)C1=O Chemical compound CC[C@H]1N(C2CCCC2)c(nc(Nc(ccc(C(NC[C@H]2OC[C@@H](CCC3)N3C2)=O)c2)c2OC)nc2)c2N(C)C1=O DQJGEBQIHZAYNL-CQOQZXRMSA-N 0.000 description 2
- UZHVXJZEHGSWQV-PKPIPKONSA-N C(C1)CC2[C@@H]1CNC2 Chemical compound C(C1)CC2[C@@H]1CNC2 UZHVXJZEHGSWQV-PKPIPKONSA-N 0.000 description 1
- BJOVBVSJCSOQTA-XYIZDEMUSA-N CCC(C1)[C@@]1(C1)C[C@@H]2[C@H]1CCN(C)C2 Chemical compound CCC(C1)[C@@]1(C1)C[C@@H]2[C@H]1CCN(C)C2 BJOVBVSJCSOQTA-XYIZDEMUSA-N 0.000 description 1
- QNLSHJVWZNHSED-UHFFFAOYSA-N CCC1N(C(C)C)c(nc(Nc(ccc(C(O)=O)c2)c2OC)nc2)c2N(C)C1=O Chemical compound CCC1N(C(C)C)c(nc(Nc(ccc(C(O)=O)c2)c2OC)nc2)c2N(C)C1=O QNLSHJVWZNHSED-UHFFFAOYSA-N 0.000 description 1
- HLUFIDWHGDEDBN-MRVPVSSYSA-N CC[C@H]1N(C(C)C)c([nH]nc2)c2N(C)C1=O Chemical compound CC[C@H]1N(C(C)C)c([nH]nc2)c2N(C)C1=O HLUFIDWHGDEDBN-MRVPVSSYSA-N 0.000 description 1
- YGGNBCXOMODTPJ-QGZVFWFLSA-N CC[C@H]1N(C2CCCC2)c(nc(Nc(ccc(C(NC)=O)c2)c2OC)nc2)c2N(C)C1=O Chemical compound CC[C@H]1N(C2CCCC2)c(nc(Nc(ccc(C(NC)=O)c2)c2OC)nc2)c2N(C)C1=O YGGNBCXOMODTPJ-QGZVFWFLSA-N 0.000 description 1
- IAOQVXMQCZUFFF-WRQKCLEDSA-N CC[C@H]1N(C2CCCC2)c2nc(Nc(c(OC)c3)ccc3C(NC3=[O][C@@]3(C3)CC4[C@@H]3CN(C)C4)=O)ncc2N(C)C1=O Chemical compound CC[C@H]1N(C2CCCC2)c2nc(Nc(c(OC)c3)ccc3C(NC3=[O][C@@]3(C3)CC4[C@@H]3CN(C)C4)=O)ncc2N(C)C1=O IAOQVXMQCZUFFF-WRQKCLEDSA-N 0.000 description 1
- JEUIYRYQIVXDMU-BBNRHVKASA-N CC[C@H]1N(C2CCCC2)c2nc(Nc(c(OC)c3)ccc3C(NC[C@@H](C3)C[C@@H]4[C@H]3CN(C)C4)=O)ncc2N(C)C1=O Chemical compound CC[C@H]1N(C2CCCC2)c2nc(Nc(c(OC)c3)ccc3C(NC[C@@H](C3)C[C@@H]4[C@H]3CN(C)C4)=O)ncc2N(C)C1=O JEUIYRYQIVXDMU-BBNRHVKASA-N 0.000 description 1
- UDDDOBWLQJDZNN-OAQYLSRUSA-N CN(C)C(N(CC1)C[C@H]2N1c(nc(Nc(c(OC)c1)ccc1C(NC1CCN(C)CC1)=O)nc1)c1N(C)C2=O)=O Chemical compound CN(C)C(N(CC1)C[C@H]2N1c(nc(Nc(c(OC)c1)ccc1C(NC1CCN(C)CC1)=O)nc1)c1N(C)C2=O)=O UDDDOBWLQJDZNN-OAQYLSRUSA-N 0.000 description 1
- JEIPCAYLLQEYIH-PZORYLMUSA-N COc(cc(cc1)C(NCC2OC[C@@H](CCC3)N3C2)=O)c1N Chemical compound COc(cc(cc1)C(NCC2OC[C@@H](CCC3)N3C2)=O)c1N JEIPCAYLLQEYIH-PZORYLMUSA-N 0.000 description 1
- JIAZNRIERWGWTE-GVHYBUMESA-N NCC1OC[C@@H](CCC2)N2C1 Chemical compound NCC1OC[C@@H](CCC2)N2C1 JIAZNRIERWGWTE-GVHYBUMESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5383—1,4-Oxazines, e.g. morpholine ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Definitions
- the present invention relates to a novel dihydroacridone derivative, a process for the preparation thereof, and a pharmaceutical composition containing the same and its use as a therapeutic agent, particularly as a Plk kinase inhibitor. Background technique
- the cyclin-dependent kinase family (Cdks) has long been recognized as the most important regulator of the cell cycle, but as the research progresses, more and more other protein kinases are found to be critical to the cell cycle progression. effect.
- the Polo-like kinase (Plks) family is one of them.
- Plks are a class of serine/threonine kinases that are important in regulating the cell cycle. According to the literature, Plks are involved in the regulation of many steps in the process of mitosis, including the activation of Cdc25C and Cdkl/Cyclin B during G2-M phase transformation, centrosome maturation and spindle formation and assembly. In the later stages of mitosis, Plks are also involved in the separation of sister chromatids, which promote the activation of complex components and the regulation of septiii during cytokinesis.
- Plks family of subtypes
- Plkl is particularly important for the regulation of mitosis (see Glover et al, Dev. 1998, 12:3777-87; Qian et al, ⁇ Biol Cell., 2001, 12: 1791 -9).
- Plkl expression and activity levels It is closely related to the degree of growth of tumor cells (see WO 2004/014899 Al).
- Overexpression of Plk1 has been shown to be associated with a variety of high-proliferation types of tumors, such as non-small cell lung cancer, squamous cell carcinoma, breast cancer, ovarian cancer.
- a number of dihydroacridone derivatives have been disclosed in the prior art as Plkl inhibitors, and such compounds have been reported to have antiproliferative activity.
- the patents WO 03/020722, WO 2004/076454 and WO2008050096 disclose dihydroacridone derivatives, processes for their preparation and their use in pharmaceutical compositions for the treatment of cell cycle kinase activity and are characterized by excessive or Use of diseases with abnormal cell proliferation.
- Patent WO 01/019825 discloses the use of pteridin derivatives as therapeutics for tumors and viral diseases. Due to the resistance of various types of tumors, there is an urgent need to develop new drugs to overcome tumors.
- An object of the present invention is to provide a medicament having Plk1 kinase inhibitory activity which can be used for the treatment of cell proliferation diseases such as cancer, infection, inflammation and autoimmune diseases. Summary of the invention
- R 1 and R 2 are each independently selected from a hydrogen atom or an alkyl group
- R 1 and R 2 together with the atoms to which they are bonded form a 3 to 8 membered ring, wherein the 3 to 8 membered ring contains 0 to 2 N, 0 or 8 (0)
- the 3 to 8 membered ring is optionally further selected from one or more selected from the group consisting of fluorenyl, decyloxy, halogen, hydroxy, cyano, nitro, aryl, heteroaryl, carbonyl, -S(0)ONR 7 R 8, -CONR 7 R 8, -NR 7 R 8, -S (0) oR 9, - COR 9 , or - C (0) oR 9 is substituted with a substituent;
- R 3 is selected from a hydrogen atom, an alkyl group, an alkenyl group, an alkynyl group, a cyclodecyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the fluorenyl group, the alkenyl group, the alkynyl group, the cycloalkyl group, the heterocyclic group Or an aryl or heteroaryl group optionally further selected from one or more selected from the group consisting of alkyl, decyloxy, halogen, hydroxy, cyano, nitro, aryl, -S(0)ONR 7 R 8 , - CONR Substituted by a substituent of 7 R 8 , —NR 7 R 8 , —S(0)OR 9 , —COR 9 or —C(0)OR 9 ;
- R 2 and R 3 together with the atoms to which they are attached form a 3 to 8 membered ring, wherein the 3 to 8 membered ring contains 1 to 2 N, 0 or 8 (0) 111 heteroatoms, and
- the 3 to 8 membered ring is optionally further selected from one or more selected from the group consisting of fluorenyl, alkoxy, halogen, hydroxy, cyano, nitro, aryl, heteroaryl, carbonyl, -S(0)ONR 7 R 8 Substituted with a substituent of -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 , -COR 9 or -C(0)OR 9 ;
- R 4 and R 5 are each independently selected from a hydrogen atom, a fluorenyl group, a halogen, a cyano group or a nitro group;
- L is selected from an alkylene group, optionally further substituted with one or more substituents selected from halogen, cyano, nitro or alkyl, wherein said thiol is optionally further substituted by one or more halogens ;
- R 6 is selected from a fluorenyl group, wherein the thiol group is optionally further substituted with a heterocyclic group;
- R 6 is selected from an unsubstituted alkyl group
- A is selected from a bicycloalkyl group, a heteroheterocyclyl group, a spirocycloalkyl group or a spiroheterocyclyl group, wherein the bicyclononyl group, the biheterocyclyl group, the spirocycloalkyl group Or a spiroheterocyclyl optionally further substituted with one or more R 12 ;
- R 6 is selected from heterocyclyl-substituted indenyl
- A is selected from cyclodecyl, heterocyclyl, bicyclononyl, biheterocyclyl, bridged cycloalkyl, hetero bridged fluorenyl, spirocycloalkyl or Spiroheterocyclyl, wherein the cycloalkyl, heterocyclyl, Bicyclic fluorenyl, bicyclohetero, bridged fluorenyl, heterobridged fluorenyl, spirocycloalkyl or spiroheterocyclyl optionally substituted with one or more R 12 ;
- R 7 and R 8 are each independently selected from a hydrogen atom, a fluorenyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the alkyl group, cyclodecyl group, heterocyclic group, aryl group or heteroaryl group Further optionally further selected from one or more selected from the group consisting of fluorenyl, alkoxy, heterocyclic, aryl, heteroaryl, halogen, hydroxy, cyano, -S(0)OR 9 , -COR 9 , -C Substituting (0) a substituent of OR 9 , -SCOjONR ⁇ R 11 , -CONRWR 11 or -NR 1 Q R";
- R 7 and R 8 together with the N atom to which they are bonded form a 3 to 8 membered heterocyclic group, wherein the 3 to 8 membered heterocyclic group contains one or more N, 0 or S(0) m hetero An atom, and the 3 to 8 membered heterocyclic group is optionally further selected from one or more selected from the group consisting of an alkyl group, a decyloxy group, a heterocyclic group, an aryl group, a heteroaryl group, a halogen, a hydroxyl group, a cyano group, and -S ( 0) 0R 9 , -COR 9 , -C(0)0R 9 , -S(O)ONR 10 U .
- R 9 is selected from a hydrogen atom, an alkyl group, a cycloalkyl group or an aryl group, wherein the cycloalkyl group or aryl group is optionally further substituted with one or more mercapto groups;
- R 1Q and R 11 are selected from a hydrogen atom, a fluorenyl group, a cycloalkyl group or an aryl group;
- R 12 is selected from the group consisting of alkyl, alkoxy, cyclodecyl, halogen, hydroxy, cyano, nitro, carbonyl, -S(0)ONR 7 R 8 , -CON 7 R 8 . -C(0)0R 9 , -0C(0)R 9 , -0(CH 2 ) R C(0)OR 9 , - OC(0)NR 7 R 8 , -S(0) M R 9 , -0S(0)0R 9 , Substituted with a substituent of NHC(0)R 9 or -COR 9 wherein said fluorenyl, decyloxy, cycloalkyl or heterocyclyl is optionally further selected from one or more selected from the group consisting of alkyl, cycloalkane Substituted by a substituent of a heterocyclic group, an aryl group or a heteroaryl group;
- n 0 or 1
- n 0, 1 or 2;
- r is 1, 2 or 3.
- the compounds of the formula (I) are tautomers, racemates, enantiomers, diastereomers, mixtures thereof, and pharmaceutically acceptable Salt: Where:
- R 1 is selected from a hydrogen atom
- R 2 is selected from a thiol group
- R 3 is selected from a decyl group or a cycloalkyl group
- R 2 and R 3 together with the atoms to which they are bonded form a 3 to 8 membered heterocyclic group wherein the 3 to 8 membered heterocyclic group contains 1 to 2 N, 0 or S(0) n heteroatoms.
- the 3 to 8 membered heterocyclic group is optionally further selected from one or more selected from the group consisting of fluorenyl, decyloxy, halogen, hydroxy, cyano, nitro, carbonyl, aryl, -S(0)ONR 7 Substituted by a substituent of R 8 , —CONR 7 R 8 , —NR 7 R 8 , —S(0)0R 9 , —COR 9 or —C(0)0R 9 ;
- R 4 and R 5 are each independently selected from a hydrogen atom, a halogen or a fluorenyl group
- R 6 is selected from a fluorenyl group, wherein the thiol group is optionally further substituted with a heterocyclic group;
- A is selected from a bicycloalkyl group, a biheterocyclyl group, a spirocyclic fluorenyl group or a spiroheterocyclic ring. And wherein said bicycloalkyl, bicyclohetero, bridged fluorenyl, bridged heterocyclyl, spiro fluorenyl or spiroheterocyclyl is optionally substituted with one or more R 12 ;
- R 6 is selected from heterocyclic substituted alkyl
- A is selected from cyclodecyl, heterocyclyl, bicycloalkyl, biheterocyclyl, bridged fluorenyl, bridged heterocyclyl, spiro fluorenyl or spiro a heterocyclic group, wherein said cyclodecyl, heterocyclyl, bicyclononyl, diheterocyclyl, bridged fluorenyl, bridged heterocyclyl, spirocycloalkyl or spiroheterocyclyl is optionally further substituted by one or Substituted by multiple R 1 2 ;
- R 7 and R 8 are each independently selected from a hydrogen atom, a fluorenyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the fluorenyl group, cyclodecyl group, heterocyclic group, aryl group or heteroaryl group Further optionally further selected from one or more selected from the group consisting of fluorenyl, decyloxy, heterocyclyl, aryl, heteroaryl, halogen, hydroxy, cyano, carboxylic acid, carboxylic acid ester, -S(0)OR 9 Substituting -COR 9 , -S(0)ONR 1 Q R ] 1 , - CONR 1 G R" or - NRWR 1 1 ;
- R 7 and R 8 together with the N atom to which they are bonded form a 3 to 8 membered ring, wherein the 3 to 8 membered ring contains one or more N, 0 or S(0) m heteroatoms, and
- the 3 to 8 membered heterocyclic ring is further selected from one or more selected from the group consisting of an anthracenyl group, a decyloxy group, a heterocyclic group, an aryl group, a heteroaryl group, a halogen, a hydroxyl group, a cyano group, a carboxylic acid, a carboxylic acid ester, Substituted by a substituent of a carbonyl group, -S(0)OR 9 , -COR 9 , -S ⁇ ONR'V ⁇ - CONR' GR 11 or -NRWR 1 1 ;
- R 9 is selected from a hydrogen atom, a fluorenyl group, a cycloalkyl group or an aryl group, wherein the cycloalkyl group or the aryl group is optionally further substituted with one or more alkyl groups;
- R 1 Q and R 1 1 are selected from a hydrogen atom, a fluorenyl group, a cyclodecyl group or an aryl group;
- R 1 2 is selected from the group consisting of alkyl, decyloxy, cycloalkyl, hydroxy, cyano, nitro, carbonyl, -S(0)ONR 7 R 8 , - CONR 7 R 8 , -C(0)OR 9 - - OC(0)R 9 , -0(CH 2 ) R C(0)OR 9 , - OC(0)NR 7 R 8 , -S(0) M R 9 , -OS(0)OR 9 Substituted with a substituent of -NHC(0)R 9 or -COR 9 wherein the alkyl, decyloxy, cyclodecyl or heterocyclic group is further optionally further selected from one or more selected from the group consisting of fluorenyl and cyclo Substituted by a substituent of an alkyl group, a heterocyclic group, an aryl group or a heteroaryl group;
- n 0 or 1
- n 0, 1 or 2;
- r is 1, 2 or 3.
- a preferred embodiment of the invention, a compound of the formula (I) or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and a pharmaceutically acceptable compound An acceptable salt, wherein: R 1 and R 2 are each independently selected from a hydrogen atom or an alkyl group;
- R 3 is selected from a hydrogen atom, a decyl group, an alkenyl group, an alkynyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the alkyl group, the alkenyl group, the alkynyl group, the cyclodecyl group, the heterocyclic group Or an aryl or heteroaryl group optionally further selected from one or more selected from the group consisting of alkyl, alkoxy, halogen, hydroxy, cyano, nitro, aryl, -S(0)ONR 7 R 8 , -CONR 7 Substituted by a substituent of R 8 , —NR 7 R 8 , —S(0)0R 9 , —COR 9 or —C(0)OR 9 ;
- R 2 and R 3 together with the atoms to which they are attached form a 3 to 8 membered ring, wherein the 3 to 8 membered ring contains 1 to 2 N, 0 or S(0) m heteroatoms, and 3 ⁇ 8 yuan ring optional further by one or a plurality of selected thiol groups, decyloxy groups, halogens, light groups, cyano groups, nitro groups, aryl groups, heteroaryl groups, carbonyl groups, -S(0)ONR 8 , -CONR 7 R s , -NR 7 R 8 , Substituted by a substituent of -S(0)OR 9 , - COR 9 or -C(0)OR 9 ;
- R 4 and R 5 are each independently selected from a hydrogen atom, an alkyl group, a halogen, a cyano group or a nitro group;
- L is selected from the group consisting of an anthracenylene group, optionally further substituted with one or more substituents selected from halogen, cyano, nitro or alkyl, wherein the alkyl group is optionally further substituted by one or more halogens ;
- R 6 is selected from a thiol group
- A is selected from a bicyclononyl group, a bicyclic heterocyclic group, a spirocyclic fluorenyl group or a spiroheterocyclyl group, wherein the bicyclic fluorenyl group, the bicyclic heterocyclic group, the spiro fluorenyl group or the spiroheterocyclic group is optionally further substituted by one or Substituted by a plurality of R 12 ;
- R 7 and R s are each independently selected from a hydrogen atom, a fluorenyl group, a cycloalkyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the fluorenyl group, cyclodecyl group, heterocyclic group, aryl group or heteroaryl group group optionally further substituted with one or more groups selected from the embankment, embankment group, a heterocyclic group, an aryl group, a heteroaryl group, halogen, hydroxy, cyano, -S (0) 0R 9, -COR 9, C ( 0) 0R 9 , -SiC ONR 1 °R' 1 , -CONR 1 °R' 1 or - NR 1 °R ! 1 substituted by a substituent;
- R 7 and R 8 together with the N atom to which they are bonded form a 3 to 8 membered heterocyclic group, wherein the 3 to 8 membered heterocyclic group contains one or more N, 0 or 8 (0), ⁇ atom, and the 3 to 8 membered heterocyclic group is optionally further selected from one or more selected from the group consisting of an alkyl group, a decyloxy group, a heterocyclic group, an aryl group, a heteroaryl group, a halogen group, a hydroxyl group, a cyano group, Substituted by a substituent of -S(0)0R 9 , -COR 9 , C(0)0R 9 , -S(0)0NR 1 Q R M , -CONI ⁇ R 1 1 or -NR 10 R";
- R 9 is selected from a hydrogen atom, an alkyl group, a cyclodecyl group or an aryl group, wherein the cycloalkyl or aryl group is optionally further substituted with one or more alkyl groups;
- R 1Q and R 11 are selected from a hydrogen atom, a fluorenyl group, a cyclodecyl group or an aryl group;
- R 12 is selected from the group consisting of alkyl, nonyloxy, cyclodecyl, halogen, hydroxy, cyano, nitro, carbonyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -C(0)0R 9 , -OC(0)R 9 , -0(CH 2 ) r C(0)OR 9 , - 0C(0)NR 7 R s , -S(0) m R 9 , -0S(0)0R 9 , Substituted by a substituent of -NHC(0)R 9 or -COR 9 wherein said alkyl, decyloxy, cyclodecyl or heterocyclyl is optionally further selected from one or more selected from the group consisting of alkyl, cyclo Substituted with a substituent of a heterocyclic group, an aryl group or a heteroaryl group;
- n 0 or 1
- n 0, 1 or 2;
- r is 1, 2 or 3.
- a preferred embodiment of the invention, a compound of the formula (I) or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and a pharmaceutically acceptable compound An acceptable salt, wherein: R 1 and R 2 are each independently selected from a hydrogen atom or an alkyl group;
- R 3 is selected from a hydrogen atom, a decyl group, an alkenyl group, an alkynyl group, a cyclodecyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the fluorenyl group, the alkenyl group, the alkynyl group, the cyclodecyl group, the heterocyclic group Or an aryl or heteroaryl group optionally further selected from one or more selected from the group consisting of alkyl, decyloxy, halogen, hydroxy, cyano, nitro, aryl, -S(0)ONR 7 R 8 , -CONR 7 Substituted by a substituent of R 8 , —NR 7 R 8 , —S(0)0R 9 , —COR 9 or —C(0)0R 9 ; Alternatively, R 2 and R 3 together with the atoms to which they are attached form a 3 to 8 membered ring, wherein
- R 4 and R 5 are each independently selected from a hydrogen atom, a fluorenyl group, a halogen, a cyano group or a nitro group;
- L is selected from an alkylene group, optionally further substituted with one or more substituents selected from halogen, cyano, nitro or fluorenyl, wherein said alkyl group is optionally further substituted by one or more halogens Replace
- R 6 is selected from a fluorenyl group, wherein the alkyl group is further substituted with a heterocyclic group;
- R 6 is selected from heterocyclyl-substituted indenyl
- A is selected from cycloalkyl, heterocyclyl, bicyclononyl, biheterocyclyl, bridged fluorenyl, hetero bridged fluorenyl, spiro fluorenyl or a spiroheterocyclyl group, wherein the cyclodecyl, heterocyclyl, bicycloalkyl, biheterocyclyl, bridged cycloalkyl, hetero bridged fluorenyl, spirocycloalkyl or spiroheterocyclyl is optionally substituted Substituted by one or more R 12 ;
- R 7 and R 8 are each independently selected from a hydrogen atom, an alkyl group, a cyclodecyl group, a heterocyclic group, an aryl group or a heteroaryl group, wherein the alkyl group, cyclodecyl group, heterocyclic group, aryl group or heteroaryl group Further optionally further selected from one or more selected from the group consisting of fluorenyl, alkoxy, heterocyclic, aryl, heteroaryl, halogen, hydroxy, cyano, -S(0)OR 9 , -COR 9 , C ( 0) 0R 9 , -S(O)ONR ] 0 R' -CONK ER 1 1 or a substituent of -NRWR 11 substituted;
- R 7 and R 8 together with the N atom to which they are bonded form a 3 to 8 membered heterocyclic group, wherein the 3 to 8 membered heterocyclic group contains one or more N, 0 or 8 (0) faced 1 a hetero atom, and the 3 to 8 membered heterocyclic group is further optionally further selected from one or more selected from the group consisting of fluorenyl, decyloxy, heterocyclic, aryl, heteroaryl, halogen, hydroxy, cyano, -S (0)0R 9 , -COR 9 . C(0)0R 9 , -S(O)ONR 10 R N . Substituted by a substituent of -CONR ⁇ R 1 1 or -NR ⁇ R";
- R 9 is selected from a hydrogen atom, an alkyl group, a cycloalkyl group or an aryl group, wherein the cycloalkyl or aryl group is optionally further substituted with one or more mercapto groups;
- R 1Q and R 1 1 are selected from a hydrogen atom, a fluorenyl group, a cyclodecyl group or an aryl group;
- R 12 is selected from the group consisting of fluorenyl, decyloxy, cyclodecyl, halogen, hydroxy, cyano, nitro, carbonyl, -S(0)ONR 7 R 8 , - CONR 7 R 8 , -C(0)0R 9 -OC(0)R 9 , -0(CH 2 ) R C(0)OR 9 , -OC(0)NR 7 R 8 , -S(0),”R 9 , -0S(0)0R 9 , -NHC (0) R 9 or -COR 9 group substituted with substituents, wherein the embankment group, an alkoxy group, a cycloalkyl group or a heterocyclyl group optionally further substituted with one or more groups selected from alkyl with ring Substituted with a substituent of a fluorenyl, heterocyclyl, aryl or heteroaryl group;
- n 0 or 1
- n 0, 1 or 2;
- r is 1, 2 or 3.
- a preferred embodiment of the invention a compound of the formula (I.) or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and A pharmaceutically acceptable salt, wherein R 1 is selected from a hydrogen atom, R 2 is selected from a thiol group, and R 2 is preferably an ethyl group.
- a preferred embodiment of the invention a compound of the formula (I) or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and a pharmaceutically acceptable compound An acceptable salt wherein R 3 is selected from decyl or cyclodecyl, preferably isopropyl or cyclopentyl.
- R 3 is selected from decyl or cyclodecyl, preferably isopropyl or cyclopentyl.
- the compound of the formula (I) may contain an asymmetric carbon atom and may therefore exist in the form of an optically pure diastereomer, a mixture of diastereomers, a diastereomeric racemate, a mixture of diastereomeric racemates. Or exist as a meso compound.
- the invention includes all of these forms. Mixtures of diastereomeric mixtures, diastereomeric racemates or diastereomeric racemates can be separated by conventional methods, for example by column chromatography, thin layer chromatography and HPLC.
- Typical compounds of the invention include, but are not limited to:
- the present invention provides a compound of the formula (IA) which is an intermediate for the preparation of a compound of the formula (I), wherein:
- G is selected from a leaving group, preferably selected from the group consisting of halogen, methanesulfonyl, p-toluenesulfonyl, trifluoromethanesulfonyl or decyloxy;
- R is selected from a hydrogen atom or a sulfhydryl group
- R 1 is selected from a hydrogen atom or a fluorenyl group
- R 2 and R 3 together with the atoms to which they are attached form a 3 to 8 membered ring, wherein the 3 to 8 membered ring contains! ⁇ 2 N, 0 or S(0) n heteroatoms, and the 3 to 8 membered ring is optionally further further selected from one or more selected from the group consisting of fluorenyl, decyloxy, aryl, halogen, hydroxy, cyano, Substituted by a substituent of a carbonyl group, a carboxylic acid, a carboxylic acid ester, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 or -COR 9 .
- n, R 7 to R 9 are as defined in the general formula (J)
- Typical compounds of formula (IA) include, but are not limited to:
- the present invention provides a process for the preparation of a compound of the formula (I), which comprises:
- the compound of the formula (IA:) is reacted with a compound of the formula (IB) or a salt of the compound of the formula (IB) to give a compound of the formula (I).
- R is selected from a methyl group
- R ⁇ R 3 is as defined for the compound of formula (IA);
- A, n, L, R 4 to R 6 are as defined for the compound of the formula (I).
- Another aspect of the invention relates to a compound of the formula (I) of the invention or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and a pharmaceutically acceptable
- the use of the accepted salt for the preparation of a medicament for a cell proliferative disorder wherein the cell proliferative disorder is cancer, infection, inflammation, and autoimmune disease, and the cancer is cervical cancer or colon cancer.
- Another aspect of the invention relates to a method of treating a cell proliferative disorder, the method comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula (I) or a tautomer, racemate, enantiomer thereof Isomers, diastereomers, mixtures thereof, and pharmaceutically acceptable salts, wherein the cell proliferation diseases are cancer, infection, inflammation, and autoimmune diseases, and the cancer is cervical Cancer or colon cancer.
- Another aspect of the invention relates to a compound of the formula (I) of the invention or a tautomer, a racemate, an enantiomer, a diastereomer, a mixture thereof, and a pharmaceutically acceptable
- the accepted salt is a drug for treating a cell proliferation disease, wherein the cell proliferation disease is cancer, infection, inflammation, and autoimmune disease, and the cancer is cervical cancer or colon cancer.
- the present invention relates to the compound of the formula (I) of the present invention or a tautomer, a racemate, an enantiomer, a diastereomer thereof, a mixture thereof, and a pharmaceutically acceptable form thereof.
- Use of a salt in the preparation of a Plk kinase inhibitor Use of a salt in the preparation of a Plk kinase inhibitor.
- Another aspect of the invention relates to a method of inhibiting Plk kinase comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula (I) or a tautomer, racemate, enantiomer thereof , diastereomers, mixtures thereof, and pharmaceutically acceptable salts.
- the invention further relates to the compounds of the formula (I) or the tautomers, racemates, enantiomers thereof, Diastereomers, mixtures thereof, and pharmaceutically acceptable salts are useful as drugs for inhibiting Plk kinase.
- Another aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention or a tautomer, racemate, enantiomer, diastereomer thereof, and In the form of a mixture, and a pharmaceutically acceptable salt, and a pharmaceutically acceptable carrier or excipient therefor.
- the pharmaceutical composition is a medicament for treating cancer, infection, inflammation, and autoimmune diseases, and the cancer is cervical cancer or colon cancer.
- Another aspect of the invention relates to a method of treating a cell proliferative disorder, comprising administering to a patient in need of treatment a therapeutically effective amount of a compound of formula (I) or a tautomer thereof, a racemate, a pair thereof a pharmaceutical composition of a conjugate, a diastereomer, a mixture thereof, and a pharmaceutically acceptable salt, wherein the cell proliferative disorder is cancer, infection, inflammation, and autoimmune disease,
- the cancer described is cervical cancer or colon cancer.
- the compounds of the invention and their pharmaceutically acceptable salts can be administered orally, dermally or parenterally (e.g., by injection, inhalation, spray, sublingual, rectal or vaginal).
- “Injectable administration” includes intravenous injection, joint injection, intramuscular injection, subcutaneous injection, parenteral injection, and infusion. Dermal administration includes topical or cross-administration. Oral administration is carried out according to methods well known to those skilled in the art, and one or more adjuvants such as diluents, sweeteners, flavoring agents, colorants and preservatives may also be present in such formulations.
- the amount of the pharmaceutically active compound in each case should be in the range of from 0.1 to 90% by weight of the total composition, preferably from 0.5 to 50% by weight, i.e., in an amount sufficient to achieve the metering range provided below. If necessary, multiple specified doses can be provided daily.
- the active ingredient is mixed with non-toxic, pharmaceutically acceptable excipients.
- excipients include: inert diluents (such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate), granulation or disintegrants (such as corn starch, alginic acid) and binders (such as magnesium stearate) , stearic acid, talc).
- the tablets may be uncoated or they may be coated by a known method to alleviate the decomposition and absorption in the gastrointestinal tract to prolong the efficacy, such as glyceryl stearate or glyceryl distearate.
- These compounds can also be prepared in a solid, rapid release mode.
- the active ingredient is mixed with an inert solid diluent such as calcium carbonate, calcium phosphate or china clay; in a soft capsule, the active ingredient is mixed with water or an oil vehicle such as peanut oil, paraffin or olive oil.
- an inert solid diluent such as calcium carbonate, calcium phosphate or china clay
- the active ingredient is mixed with water or an oil vehicle such as peanut oil, paraffin or olive oil.
- the active ingredient is mixed with excipients which are suitable for pharmaceutical use.
- excipients are suspending agents (such as hydroxymethyl cellulose sodium, methyl cellulose, hydroxypropyl-methyl cellulose, sodium alginate, polyvinylpyrrolidone, gum arabic), dispersing agents or wetting agents [ Including naturally occurring phospholipids (such as egg pity) or condensates of alkylenes and fatty acids (such as polyoxyethylene stearate) or condensates of ethylene and long-chain fatty alcohols (such as heptadecylene oxide ten A condensate of hexaol) or ethylene oxide and a partial ester derived from a fatty acid and a hexitol (such as polyethylene sorbitan monooleate):
- the aqueous suspension may also mean a plurality of preservatives (such as ethyl p-hydroxybenzoate or propyl p-hydroxybenzoate); one or more colorants; one or more fragrances and one or A variety of sweeteners (such as sucrose or saccharin:).
- preservatives such as ethyl p-hydroxybenzoate or propyl p-hydroxybenzoate
- colorants such as ethyl p-hydroxybenzoate or propyl p-hydroxybenzoate
- fragrances such as sucrose or saccharin:
- Dispersible powders or granules suitable for the preparation of aqueous suspensions are prepared by admixing water, active ingredient and dispersing or wetting agents, suspending agents and one or more preservatives. Other excipients such as sweeteners, colorants or fragrances may also be added thereto.
- the syrup comprising the active ingredient material of the present invention or a composition thereof may additionally include a sweetener such as saccharin, sweetener, glycerin or sugar, and a flavoring agent.
- a sweetener such as saccharin, sweetener, glycerin or sugar
- a flavoring agent such as sweeteners such as vanilla or orange extracts.
- it may also include a suspending aid or a thickening agent (such as sodium carboxymethylcellulose), a wetting agent (such as a condensation product of a fatty alcohol with ethylene oxime:) or a preservative (such as p-hydroxybenzoic acid). ester;).
- Injection or infusion solutions are prepared in a conventional manner, for example, by adding isotonic agents, preservatives (such as parabens) or stabilizers (such as alkali metal salts of ethylenediaminetetraacetic acid), if necessary, using emulsifiers And/or a dispersing agent, when water is used as a diluent, that is, an organic solvent may be used as a dissolving agent or a co-solvent as needed, and transferred to an injection vial or an ampoule or a perfusion bottle.
- isotonic agents such as parabens
- stabilizers such as alkali metal salts of ethylenediaminetetraacetic acid
- emulsifiers And/or a dispersing agent when water is used as a diluent, that is, an organic solvent may be used as a dissolving agent or a co-solvent as needed, and transferred to an injection vial or an ampoule or a perfusion bottle
- Suitable suppositories are, for example, admixed with a carrier for this purpose, such as a neutral lipid or polyethylene glycol or a derivative thereof.
- Suitable excipients can be, for example, water, in pharmaceutically acceptable organic solvents (such as paraffin, for example, petroleum fractions), vegetable oils (for example, peanut oil or sesame oil), mono- or polyfunctional alcohols (for example, ethanol or glycerol).
- organic solvents such as paraffin, for example, petroleum fractions
- vegetable oils for example, peanut oil or sesame oil
- mono- or polyfunctional alcohols for example, ethanol or glycerol
- carrier such as natural mineral powder (for example, kaolin, clay, talc), synthetic mineral powder (for example, highly dispersible silica and silicate)], sugar (for example, raw sugar, lactose and glucose), emulsifier (eg, lignin, sulfite waste, methylcellulose, starch, and polyvinylpyrrolidone) and lubricants (eg, magnesium stearate, talc, stearic acid, and sodium lauryl sulfate).
- natural mineral powder for example, kaolin, clay, talc
- synthetic mineral powder for example, highly dispersible silica and silicate
- sugar for example, raw sugar, lactose and glucose
- emulsifier eg, lignin, sulfite waste, methylcellulose, starch, and polyvinylpyrrolidone
- lubricants eg, magnesium stearate, talc, stearic acid, and sodium lauryl sulf
- the formulations may be administered in a conventional manner, preferably by the oral or dermal route, preferably orally.
- the tablets may of course include additives (e.g., sodium citrate, calcium carbonate, and dicalcium phosphate) in addition to the above-described carriers, and other additives (e.g., starch, preferably potato starch, gelatin, and the like). It is also possible to use a lubricant to form tablets such as magnesium fatty acid, sodium lauryl sulfate and talc.
- the active substance may be mixed with various flavoring or coloring agents in addition to the above-mentioned excipients. Suitable liquid carrier materials can be used for the solution of the active substance for parenteral use.
- the intravenous dose is from 1 to 1000 mg per hour, preferably from 5 to 500 mg per hour.
- the dosage of the drug depends on a variety of factors including, but not limited to, the following factors: the activity of the particular compound used, the age of the patient, the weight of the patient, the health of the patient, the patient's The route, the patient's diet, the time of administration, the mode of administration, the rate of excretion, the combination of drugs, etc.; in addition, the optimal mode of treatment such as the mode of treatment, the daily dose of the compound of formula (I) or pharmaceutically acceptable
- the type of salt can be verified according to traditional treatment options. ⁇ Detailed description of the invention
- Mercapto refers to a saturated aliphatic hydrocarbon group including straight chain and branched chain groups of 1 to 20 carbon atoms.
- a mercapto group having 1 to 10 carbon atoms such as a methyl group, an ethyl group, a propyl group, a 2-propyl group, a n-butyl group, an isobutyl group, a t-butyl group or a pentyl group or the like is preferable.
- lower fluorenyl groups having 1 to 4 carbon atoms, such as methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl or t-butyl groups and the like.
- the fluorenyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of a decyloxy group, an alkenyl group, an alkynyl group, a halogen, a hydroxyl group, an amino group, a nitro group, and a cyano group.
- alkenyl refers to an alkyl group as defined above consisting of at least two carbon atoms and at least one carbon-carbon double bond. For example, vinyl, 1-propenyl, 2-propenyl, 2-, 2- or 3-butenyl, and the like.
- the alkenyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of an indenyl group, a decyloxy group, an alkynyl group, a halogen group, a hydroxyl group, an amino group, a nitro group, and a cyano group.
- Alkynyl means an alkyl group as defined above consisting of at least two carbon atoms and at least one carbon-carbon triple bond. For example, ethynyl, 1-propynyl, 2-propynyl, 1-, 2- or 3-butynyl, and the like.
- the alkynyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of a decyl group, an alkoxy group, an alkenyl group, a halogen, a hydroxyl group, an amino group, a nitro group, and a cyano group.
- a carboxylic acid or a carboxylic acid ester A carboxylic acid or a carboxylic acid ester.
- Cycloalkyl refers to a non-aromatic monocyclic or polycyclic cyclic system comprising from 3 to 20 carbon atoms, preferably from 3 to 10 carbon atoms, more preferably the cycloalkyl ring contains from 3 to 8 carbon atoms.
- monocyclic cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexane, cyclohexadienyl, cycloheptyl, cycloheptatrienyl Wait.
- Polycyclic fluorenyl groups include spiro, fused, and bridged cycloalkyl groups, and non-limiting examples include 1-naphthoquinone, norbornyl, adamantyl, and the like.
- the cycloalkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of alkyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, Amino, nitro, cyano, carbonyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, bicyclononyl, biheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 , -COR 9 , carboxylic acid or carboxylic acid ester.
- Heterocyclyl means a non-aromatic monocyclic or polycyclic ring system comprising from 3 to 20 ring atoms, wherein one or more ring atoms are selected from nitrogen, oxygen or S(0)n (where n is An integer of 0 to 2) of a hetero atom, but excluding the ring moiety of -0- 0-, -0-S- or -SS-, the remaining ring atoms being carbon.
- the heterocyclic group contains 3 to 10 ring atoms, of which 1 to 4 are hetero atoms, and more preferably the heterocyclic group contains 3 to 8 ring atoms.
- Non-limiting examples of monocyclic heterocyclic groups include pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, homopiperazinyl
- the polycyclic heterocyclic group includes a bicyclic or polycyclic spiro ring, a fused ring, and a bridged heterocyclic group.
- the heterocyclic group may be substituted or unsubstituted.
- the substituent is preferably one or more of the following groups, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, amino, nitro, cyano, carbonyl, naphthenic , heterocyclyl, aryl, heteroaryl, bicycloalkyl, biheterocyclyl, -S(0)ONR 7 R 8 , - CONR 7 R s , -NR 7 R 8 , -S(0)OR 9 , -COR 9 , a carboxylic acid or a carboxylic acid ester.
- groups independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, amino, nitro, cyano, carbonyl, naphthenic , heterocyclyl, aryl, heteroaryl, bicycloalkyl, biheterocyclyl, -S(0)ONR 7
- Bicyclic fluorenyl means a 5 to 14 membered all-carbon fused ring ("fused" ring system means that each ring in the system shares an adjacent pair of carbon atoms with other rings in the system), one of which Multiple rings may contain one or more double one rings with a fully conjugated pi-electron system.
- Bicycloalkyl groups can be substituted or unsubstituted.
- the substituent is preferably one or more, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, amino, nitro, cyano, carbonyl, cyclodecyl, hetero Cyclo, aryl, heteroaryl, bicycloindenyl, biheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 , COR 9 , a carboxylic acid or a carboxylic acid ester.
- “Biheterocyclyl” refers to a 12-membered fused ring ("fused" ring system means each ring in the system shares an adjacent pair of carbon atoms with other rings in the system), one or more of which The ring atoms are selected from nitrogen, oxygen or a hetero atom of S(0)n (where n is an integer from 0 to 2), and the remaining ring atoms are carbon. These can contain one or more double bonds but no ⁇ electronic system. It is preferably 7 to 0 yuan. E.g
- the bicyclic heterocyclic group may be substituted or unsubstituted.
- the substituent is preferably one or more, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, amino, nitro, cyano, carbonyl, cycloalkyl, hetero Cyclo, aryl, heteroaryl, bicycloindenyl, biheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 , COR 9 , carboxylic acid or carboxylic acid ester.
- spirocycloalkyl refers to a polycyclic group of 5 to 14 members, which shares a carbon atom (called a spiro atom) between the monocyclic rings. These may contain one or more double bonds, but none of the rings have a complete conjugation. ⁇ electronic system. It is preferably 7 to 10 yuan.
- the spirocycloalkyl group is divided into a monospirocycloalkyl group, a double according to the number of common spiro atoms between the ring and the ring.
- the spirocyclic fluorenyl or polyspiro is preferably a monospiroindole and a bispirocycloalkyl group.
- it is 4 yuan / 4 yuan, 4 yuan / 5 yuan, 4 yuan / 6 yuan, 5 yuan / 5 yuan or 5 yuan / 6 yuan monospirocycloalkyl.
- the spirocycloalkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy , amino, nitro, cyano, carbonyl, cyclodecyl, heterocyclyl, aryl, heteroaryl, bicyclononyl, biheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 -NR 7 R 8 , - S(0)OR 9 , -COR 9 , a carboxylic acid or a carboxylic acid ester.
- the substituent is preferably one or more of the following groups, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy , amino, nitro, cyano, carbonyl
- spiroheterocyclyl means a polycyclic hydrocarbon of 5 to 4 members, which shares an atom (called a spiro atom) between the monocyclic rings, wherein one or two ring atoms are selected from nitrogen, oxygen or S(0)n (wherein n is an integer 0 to 2) hetero atom, and the remaining ring atoms are carbon. These may contain one or more double bonds, but none of the rings have a fully conjugated ⁇ electron system. It is preferably from 7 to 10.
- the spirocyclic fluorenyl group is classified into a monospirocycloalkyl group, a bispirocyclic fluorenyl group or a polyspirocycloalkyl group, preferably a monospiroheterocyclic group and a bisspiroheterocyclic group, depending on the number of common snail atoms between the ring and the ring. More preferably, it is 4 yuan / 4 yuan, 4 yuan / 5 yuan, 4 yuan / 6 yuan, 5 yuan / 5 yuan or 5 yuan / 6 yuan monospiroheterocyclic group.
- the spiroheterocyclyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy , amino, nitro, cyano, carbonyl, cyclodecyl, heterocyclyl, aryl, heteroaryl, bicyclononyl, diheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 -NR 7 R 8 , -S(0)OR 9 , -COR 9 , carboxylic acid or carboxylic acid ester.
- the substituent is preferably one or more of the following groups, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy , amino, nitro, cyano, carbonyl, cycl
- 3 to 8 membered heterocyclic group means that the number of ring atoms is 3 to 8 yuan, and the atoms constituting the ring contain one or more N, 0 or S(0) n hetero atoms, and the ring may contain 1 to 2
- the double bond is a monocyclic or bicyclic non-aromatic ring group, and when the atom constituting the ring contains a nitrogen atom, a bond may be extended from the nitrogen atom. It is preferably a 4- to 6-membered heterocyclic group, and more preferably 5 to 6 members, such as a pyrrolidinyl group, a piperidinyl group or a piperazinyl group.
- the 3- to 8-membered heterocyclic group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more of the following groups, independently selected from the group consisting of an indenyl group, a decyloxy group, an alkenyl group, an alkynyl group, Halogen, hydroxy, amino, nitro, cyano, carbonyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, bicyclononyl, biheterocyclyl, -S(0)ONR 7 R 8 , -CONR 7 R 8 , -NR 7 R 8 , -S(0)OR 9 , -COR 9 , a carboxylic acid or a carboxylic acid ester.
- the substituent is preferably one or more of the following groups, independently selected from the group consisting of an indenyl group, a decyloxy group, an alkenyl group, an alkynyl group, Halogen,
- a p-membered/q-membered bicycloalkyl group a biheterocyclyl group, a monospirocycloalkyl group or a monospiroheterocyclyl group, which refers to a bicyclononyl group, a biheterocyclic group, a monospirocycloalkyl group or a monospiroheterocyclic group.
- the number of ring atoms of each ring is p and q, respectively, and p or q is selected from an integer of 3 to 8, preferably an integer of 4 to 7.
- Aryl means a 6 to 14 membered all-carbon monocyclic or fused polycyclic ring (ie, a ring that shares a pair of adjacent carbon atoms), a polycyclic ring having a conjugated ⁇ -electron system (ie, having adjacent pairs)
- the ring group of a carbon atom is preferably 6 to 10 members such as a phenyl group, a naphthyl group and an anthracenyl group.
- the aryl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of alkyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy, and independently.
- Heteroaryl refers to a heteroaromatic system containing from 1 to 4 heteroatoms, 5 to 14 ring atoms, wherein the heteroatoms include oxygen, sulfur and nitrogen.
- the heteroaryl group is preferably 5 or 6 members.
- furyl thienyl, pyridyl, pyrrole, N-fluorenylpyrrolyl, pyrimidinyl, pyrazinyl, imidazolyl, tetrazolyl, and the like.
- the heteroaryl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of fluorenyl, alkoxy, alkenyl, alkynyl, halogen, hydroxy. , amino, nitro, cyano, cyclodecyl, heterocyclyl, aryl, heteroaryl, bicyclononyl, biheterocyclyl, -COR 9 , -CONR 9 R 1 Q , -NR 9 R 1Q , Carboxylic acid or carboxylic acid ester.
- Mercaptooxy means -0-(; fluorenyl) and -0-(unsubstituted cycloalkyl). For example, methoxy, ethoxy, propoxy, butoxy, cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy and the like.
- the alkoxy group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of fluorenyl, decyloxy, alkenyl, alkynyl, halogen, hydroxy.
- Aryloxy means -0-aryl and -0-heteroaryl, and aryl and heteroaryl are as defined above. For example, phenoxy, pyridinyloxy, furanyloxy, thienyloxy, pyrimidinyloxy, pyrazinyloxy and the like and derivatives thereof. .
- Halogen means fluoro, chloro, bromo or iodo.
- Amino means -N3 ⁇ 4.
- Neitro means -N0 2 .
- Hydrocarbonyl means -(alkyl)-OH.
- Benzyl refers to -CH 2 - (phenyl).
- heterocyclic group optionally substituted by a thiol group means that a fluorenyl group may be, but is not necessarily, present, including the case where the heterocyclic group is substituted by an alkyl group and the case where the heterocyclic group is not substituted by a thiol group.
- “Pharmaceutical composition” means one or more compounds described herein or a physiologically/pharmaceutically acceptable salt thereof or Mixtures of prodrugs with other chemical components, other components such as physiological/pharmaceutically acceptable carriers and excipients ; the purpose of the pharmaceutical compositions is to facilitate administration of the compounds to the organism.
- the method for preparing the compound of the formula (I) or a salt thereof of the present invention comprises the following steps:
- the compound of the formula (IA) is condensed with a salt of a compound of the formula (IB;) or a compound of the formula (IB;) to give a compound of the formula (I).
- condensation reaction is carried out between an acid and an amine group, under a condensation reagent and basic conditions, and the condensation reagent used is selected from the group consisting of ruthenium, ⁇ -dicyclohexylcarbodiimide, ruthenium, osmium-diisopropyl carbon.
- R is selected from a methyl group
- G, ⁇ 3 is as defined for the compound of formula (IA);
- A, n, L, R 4 to R 6 are as defined for the compound of the formula (I). detailed description
- the structure of the compound is determined by nuclear magnetic resonance (NMR) or/and mass spectrometry (MS).
- NMR shift ( ⁇ ) is given in parts per million (ppm).
- the NMR was measured by a Bmker AVANCE-400 nuclear magnetic apparatus, and the solvent was deuterated chloroform (CDC1 3 ).
- the internal standard was tetramethylsilyl (TMS), and the chemical shift was given in units of 10 _ 0 (ppm).
- the MS was measured using a FINNIGAN LCQAd (ESI) mass spectrometer (manufacturer: Thermo, model: Finnigan LCQ advantage MAX).
- HPLC measurements were performed using an Agilent 1200 DAD high pressure liquid chromatograph (Sunfire C18 150 x 4.6 mm column) and a Waters 2695-2996 high pressure liquid chromatograph (Gimini C18 150 x 4.6 mm column).
- the average inhibition rate of the kinase and the IC 5 o value were determined using a NovoStar plate reader (BMG, Germany).
- the silica gel plate used has a specification of 0.15 mm to 0.2 mm, and the thin layer chromatography separation and purification product has a specification of 0.4 ram-0.5 mm.
- the starting materials of the present invention are known and commercially available from ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc, Dari Chemical Companies such as products may be synthesized by or according to methods known in the art.
- An argon atmosphere or a nitrogen atmosphere means that the reaction flask is connected to an argon or nitrogen balloon having a volume of about 1 L.
- the hydrogen atmosphere means that the reaction flask is connected to a hydrogen balloon of about 1 L volume.
- the pressurized hydrogenation reaction uses a Parr Model 3916EKX hydrogenation apparatus and a clear blue QL-500 hydrogen generator or a HC2-SS type hydrogenation apparatus.
- the hydrogenation reaction is usually evacuated, charged with hydrogen, and operated three times.
- the microwave reaction was carried out using a CEM Discover-S Model 908860 microwave reactor.
- the solution means an aqueous solution.
- reaction temperature is room temperature.
- the optimum temperature for the reaction at room temperature is from 20 ° C to 30 ° C.
- the progress of the reaction in the examples was monitored by thin layer chromatography (TLC).
- TLC thin layer chromatography
- the system used for the reaction was: A: chloroform and methanol system, B: n-hexane and ethyl acetate system, C: petroleum Ether and ethyl acetate system, D: acetone.
- the volume ratio of the solvent is adjusted depending on the polarity of the compound.
- Column chromatography chromatographic eluent systems include: A: chloroform and methanol systems, B: n-hexyl and ethyl acetate systems, C: dichloromethane and acetone systems, D: n-hexyl and acetone systems.
- the volume ratio of the solvent is adjusted depending on the polarity of the compound, and it may be adjusted by adding a small amount of ammonia water and acetic acid.
- EtOAc EtOAc
- EtOAc EtOAc
- EtOAc EtOAc
- EtOAc EtOAc
- EtOAc EtOAc
- EtOAc EtOAc
- the residue was purified by silica gel column chromatography elut elut elut elut elut elut elut elut elut Methyl ester-4-yl-amino)-5-methyl-7,8-dihydro-5H-acridin-6-one lk (550 mg, white solid), yield: 73%.
- N-(l-methyl-(c-pyridin-4-yl)-4-nitro-3-(tetrahydro-furan-3-yl-methoxy)-benzamide 4-nitro-3- (tetrahydro-furan-3-yl-methoxy)-benzoic acid 6d (170 mg, 0.67 mmol), 1-methyl-piperidine-4-yl-amine (77 mg, 0.67 mmol), 0-benzene
- triazole-oxime, hydrazine, hydrazine, ⁇ '-tetramethyluronium tetrafluoroborate (215 mg, 0.67 mmol) and diisopropylethylamine (25 ⁇ g, 1.47 mmol) dissolved in 40 mL In dichloromethane, the reaction was stirred for 2 hours.
- W is) - 8 - cyclopentyl-7-ethyl --5 _-6-oxo - 5, 6, 7, 8 - tetrahydro - pteridin-2-ylamino) - N - (1 - A Base-piperidine
- EtOAc (EtOAc) (EtOAcjjjjjjjj 4-(7-ethyl-8-isopropyl-5-methyl-6-oxo-5,6,7,8-tetrahydro-acridin-2-ylamino)-methoxy)-benzene
- EtOAcjjjjjjj 4-(7-ethyl-8-isopropyl-5-methyl-6-oxo-5,6,7,8-tetrahydro-acridin-2-ylamino)-methoxy)-benzene
- Formic acid 12f (2.24 g, white solid), Yield: 91.8 %.
- 3-methoxy-N-(l-methyl-piperidin-4-yl)-4-nitro-benzamide 3-methoxy-4-nitro-benzoic acid la (9.86 g, 50 Ment) dissolved in 200 mL of dichloromethane, followed by 0-benzotriazole-oxime, hydrazine, ⁇ ', ⁇ '-tetramethyluronium tetrafluoroborate (16.1 g, 50 mmol), Isopropylethylamine (18.2 mL, 110 mmol) and 1-methyl-piperidin-4-yl-amine (5.7 g, 50 mmol), stirred for 2 h, added 200 mL dichloromethane, 1 M aqueous ammonia The organic layer was dried (MgSO4) 10 g, yellow solid), Yield: 68%.
- reaction mixture was concentrated under reduced pressure. ⁇ , triethylamine was added dropwise to the pH of the reaction mixture was 8-9, acetone (0.4 g, 7.1 mmol) was added, the reaction was stirred for 1 hour, sodium triacetoxyborohydride (2.5 g, 11.8 mmol) was added, and the reaction was stirred 12 hour. Add 30 mL of water and dilute with dichloromethane (50 mL x 3).
- EtOAc (EtOAc m. 2-Chloro-5-methyl-8-phenyl-7,8,9,] 0-tetrahydro-5H-pyrazine[2,1 -A]acridine-6(6aH)-one 23k (80 mg , yellow solid), Yield: 78.4%.
- the following in vitro assay was used to determine the proliferation inhibitory activity of the compounds of the present invention against one human cervical cancer cell line Hda in a cell line highly expressing Plk.
- the in vitro cell assay described below can determine the proliferation inhibitory activity of a test compound on tumor cells that express Plk in high, and the activity can be expressed by the IC 5Q value.
- the general protocol for such an assay is as follows: First, Hela cells (purchased in Institute of biochemistry and cell biology) are seeded at a suitable cell concentration (eg 3000 cells/mL medium) on a 96-well culture plate, and then the cells are placed in a carbon dioxide incubator. The culture is carried out, and they are allowed to grow overnight. The medium is changed to a medium containing a series of concentration (usually 7 to 9 concentrations) of the test compound solution, and the plate is returned to the incubator for 72 hours. . After 72 hours, the test compound can be tested for its ability to inhibit cell proliferation using the CCK8 method.
- the IC 5 o value can be calculated from the inhibition values of the test compound for the cells at a range of different concentrations.
- the biochemical activity of the compounds of the present invention was determined by the above test, and the measured IC 5 o values are shown in the following table.
- the compounds of the present invention all have significant proliferation inhibitory activity against Hela cells.
- Test Example 2 Inhibition of proliferation of Plk high expressing cells by the compound of the present invention
- the following in vitro assay was conducted to determine the proliferation inhibitory activity of the compound of the present invention against human colon cancer cell HCT-116 in a cell line highly expressing Plk.
- the in vitro cell assay described below can determine the proliferation inhibitory activity of the test compound on highly proliferating digestive tumor cells, and the inhibitory activity of the compound can be expressed by the IC 5Q value.
- the experimental protocol is briefly described as follows: First, HCT-116 cells (purchased in Institute of biochemistry and cell biology) supplemented with 10% FCS (purchased from Gibco) as DMEM in DMEM, at a suitable cell concentration (eg 3000 cells/mL) The medium was inoculated on a 96-well culture plate, and then cultured overnight in a constant temperature incubator at 37 ° C under 5% CO 2 . After the cells are attached, the medium is replaced with fresh medium containing a gradient of the test compound (typically 7 or 9 concentration points).
- the cell culture plate was continuously cultured for 72 hours under the aforementioned conditions. After 72 hours, the inhibitory activity of the compound on cell proliferation was measured by the method of CCK8 (Cell Counting Kit- 8, Cat. No.: CK04, purchased from Dojindo). The IC 5Q value of the compound can be calculated from the inhibition of cell proliferation by the test compound at various concentrations.
- the biochemical activity of the compound of the present invention was measured by the above test, and the measured IC 5Q value is shown in the following table.
- Test Example 3 Determination of Plkl kinase inhibitory activity by the compound of the present invention
- the method described below can be used to assay the compounds of the present invention, and expressed PIk] ability to inhibit kinase activity by 50 value IC.
- the half-inhibitory concentration of the compound, IC 5Q (the concentration of the compound required to inhibit the enzymatic activity to 50%), is determined by mixing a certain amount of the kinase with a specific substrate and a different concentration of the test compound. To a series of inhibition rates, and then use the calculation tool to calculate.
- the Plk1 kinase used in this experiment was a recombinant human protein, and the reaction system was purchased from Poly-Like kinase 1 Assay/Inhibitor Screening Kit (#CY-]J 63).
- the Pjk] enzyme reacts with the polypeptide substrate and different concentrations of the test compound in the reaction system (25 ° C, 30 minutes), followed by an anti-Anti- Phospho-Serine/Threonine Polyclonal Antibody (PPT-07) and The secondary antibody HRP-conjugated Anti-rabbit IgG labels the phosphorylated substrate and finally measures the A450 nM reading to quantify Plkl kinase activity.
- the biochemical activity of the compound of the present invention was measured by the above test, and the measured IC 5Q value is shown in the following table.
- Example 2 compound, a mixture of Examples 8-1 and 8-2
- the pharmacokinetic parameters of the compounds of the invention are as follows:
- Example 2 The compound of Example 2, the mixture of Examples 8-1 and 8-2, had good pharmacological absorption.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
本发明涉及二氢喋啶酮类衍生物、其制备方法及其在医药上的应用。具体而言,本发明涉及一种通式(Ⅰ)所示的新的二氢喋啶酮类衍生物、其制备方法及含有该衍生物的药物组合物以及其作为治疗剂特别是作为Plk激酶抑制剂的用途,其中通式(Ⅰ)的各取代基与说明书中的定义相同。
Description
二氢喋啶酮类衍生物、 其制备方法及其在医药上的应用 技术领域
本发明涉及一种新的二氢喋啶酮类衍生物、 其制备方法及含有该衍生物的药 物组合物以及其作为治疗剂特别是作为 Plk激酶抑制剂的用途。 背景技术
细胞周期蛋白依赖性激酶家族 (Cdks) 长期以来一直被认为是细胞周期最 主要的调节因子, 但随着研究的深入, 越来越多的其它蛋白激酶被发现对细胞周 期的进程也起着关键作用。 Polo样激酶 (Plks)家族就是其中之一。
Plks是一类在调节细胞周期过程中非常重要的丝氨酸 /苏氨酸激酶。据文献报 道, Plks参与了有丝分裂的过程中许多步骤的调控, 包括在 G2- M期的转化过程 中 Cdc25C和 Cdkl/Cyclin B的活化, 中心体成熟以及纺锤体的形成和组装过程。 在有丝分裂的后期, Plks还参与姐妹染色单体的分离, 细胞分裂后期促进复合物 组分的活化以及胞质分裂期中 septiii的调节过程。
到目前为止, Plks家族中己发现四种亚型, 分别是 Plkl、 Plk2、 k3及 Plk4。 (其中 Plkl对有丝分裂过程的调节尤为重要 (参见 Glover等人, Dev. 1998, 12:3777-87; Qian 等人, Μοϊ Biol Cell., 2001 , 12: 1791 -9)。 Plkl的表达及活性水 平与肿瘤细胞的生长程度密切相关 (参见 WO 2004/014899 Al)。 Plkl的过量表达 巳被证实与多种高增殖类型的肿瘤相关, 如非小细胞肺癌、鳞状细胞癌、乳腺癌、 卵巢癌或乳头状癌以及结肠直肠癌等 (Wolf 等人, Oncogene 1997, 14, 543-549; Knecht等人, Cancer Res. , 1999, 59, 2794-2797; Wolf 等 X, Pathol Res Pract" 2000, 196, 753-759; Weichert 等人, Br: J. Cancer, 2004, 90, 815-821 ; Ito 等人, Br. J. Cancer, 2004, 90, 414-418; Takahashi 等人, Cancer Sci" 2003, 94, 148-152)。
现有技术已公开了许多二氢喋啶酮类衍生物作为 Plkl抑制剂, 文献报道此类 化合物具有抗增殖活性。 例如: 专利 WO 03/020722、 WO 2004/076454和 WO2008050096公开了二氢喋啶酮类衍生物, 其制备方法及其在药物组合物中用 于治疗与细胞周期激酶的活性相关以及特征为过度或异常细胞增殖的疾病的用 途。 专利 WO 01/019825公开了蝶啶酮类衍生物作为治疗肿瘤和病毒疾病的用途。 由于各种不同类型肿瘤的耐药性, 迫切需要研发新的药物来攻克肿瘤。 此外, 还 有一些专利 , 如 WO2004076454 、 WO2006018220、 US20040176380、 WO2007135374、 WO2006018185、 WO2006058876、 WO2006018222、 WO2006018182等还公开了其他一些作为 Plkl抑制剂的化合物。
然而, 尽管已有若干的 Plkl激酶抑制剂被公开, 但是目前其应用还受到药效、 药动等方面的限制, 还未有长效的药物, 因此仍然需要开发安全性、 药代动力学 等性质得到改善的 Plkl激酶抑制剂。
本发明目的在于提供一种具有 Plkl激酶抑制活性的药物, 其可以用于治疗癌 症、 感染、 炎症和自身免疫性疾病等细胞增殖类疾病。 发明内容
为了克服现有技术的不足之处,本发明的目的在于提供一种通式( I )所示的二 氢喋啶酮类衍生物, 以及它们的互变异构体、 外消旋体、 对映异构体、 非对映异 构体、 及其混合物形式、 及药学上可以接受的盐和代谢物或前药:

(I)
其中:
R1和 R2各自独立地选自氢原子或烷基;
或者, R1与 R2与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 0〜2个 N、 0或 8(0)|71杂原子, 并且所述 3〜8元环任选进一步被一个或 多个选自垸基、垸氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 - COR9或- C(0)OR9的取代基所取代;
R3选自氢原子、 烷基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基, 其中 所述垸基、 烯基、 炔基、 环烷基、 杂环基、.芳基或杂芳基任选进一步被一个或多 个选自烷基、垸氧基、 卤素、羟基、氰基、硝基、 芳基、 -S(0)ONR7R8、 - CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9或 -C(0)OR9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 1〜2个 N、 0或 8(0)111杂原子, 并且所述 3〜8元环任选进一步被一个或 多个选自垸基、烷氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 - S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9或 -C(0)OR9的取代基所取代;
R4和 R5各自独立地选自氢原子、 垸基、 卤素、 氰基或硝基;
L选自亚烷基, 任选进一步被一个或多个选自卤素、氰基、硝基或烷基的取代 基所取代, 其中所述的垸基任选进一步被一个或多个卤素所取代;
R6选自垸基, 其中所述的垸基任选进一步被一个杂环基所取代;
当 R6选自未取代的烷基时, A选自双环烷基、 双杂环基、 螺环烷基或螺杂环 基, 其中所述双环垸基、 双杂环基、 螺环烷基或螺杂环基任选进一步被一个或多 个 R12所取代;
当 R6选自杂环基取代的垸基时, A选自环垸基、 杂环基、 双环垸基、 双杂环 基、 桥环烷基、 杂桥环垸基、 螺环烷基或螺杂环基, 其中所述环垸基、 杂环基、
双环垸基、 双杂环基、 桥环垸基、 杂桥环垸基、 螺环烷基或螺杂环基任选进一歩 被一个或多个 R12所取代;
R7和 R8各自独立地选自氢原子、 垸基、 环烷基、 杂环基、 芳基或杂芳基, 其 中所述烷基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 烷氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)OR9、 -COR9 , -C(0)OR9、 -SCOjONR^R11 , -CONRWR11或 -NR1 QR"的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其中所述 3〜8元杂环基内含有一个或多个 N、 0或 S(0)m杂原子, 并且所述 3〜8元杂环基 任选进一步被一个或多个选自烷基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、氰基、 -S(0)0R9、 -COR9, -C(0)0R9、 -S(O)ONR10 U . -CONR ^R1 1或 -NR】0RN 的取代基所取代;
R9选自氢原子、 烷基、 环烷基或芳基, 其中所述环烷基或芳基任选进一步被 一个或多个垸基所取代;
R1Q和 R11选自氢原子、 垸基、 环烷基或芳基;
R12选自烷基、烷氧基、环垸基、卤素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 -CON 7R8. -C(0)0R9、 -0C(0)R9、 -0(CH2)RC(0)OR9、 - OC(0)NR7R8、 -S(0)MR9、 -0S(0)0R9、 -NHC(0)R9或- COR9的取代基所取代, 其中所述的垸基、 垸氧基、 环 烷基或杂环基任选进一步被一个或多个选自烷基、 环烷基、 杂环基、 芳基或杂芳 基的取代基所取代;.
m为 0或 1;
n为 0, 1或 2; 且
r为 1, 2或 3。 本发明的优选方案, 通式(I )所示的化合物其互变异构体、 外消旋体、 对映异 构体、 非对映异构体、 及其混合物形式、 及可药用的盐: 其中:
R1选自氢原子;
R2选自垸基;
R3选自垸基或环烷基;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元杂环基,其中所述 3〜 8元杂环基内含有 1〜2个N、 0或 S(0)n杂原子, 并且所述 3〜8元杂环基任选进 一步被一个或多个选自垸基、 垸氧基、 卤素、 羟基、 氰基、 硝基、 羰基、 芳基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)0R9、 -COR9或 -C(0)0R9的取代基所取 代;
R4和 R5各自独立选自氢原子、 卤素或垸基;
R6选自垸基, 其中所述的垸基任选进一步被一个杂环基所取代;
当 R6选自未取代的垸基时, A选自双环烷基、 双杂环基、 螺环垸基或螺杂环
基, 其中所述的双环烷基、 双杂环基、 桥环垸基、 桥杂环基、 螺环垸基或螺杂环 基任选进一歩被一个或多个 R12所取代;
当 R6选自杂环基取代的烷基时, A选自环垸基、 杂环基、 双环烷基、 双杂环 基、 桥环垸基、 桥杂环基、 螺环垸基或螺杂环基, 其中所述的环垸基、 杂环基、 双环垸基、 双杂环基、 桥环垸基、 桥杂环基、 螺环烷基或螺杂环基任选进一步被 一个或多个 R1 2所取代; .
R7和 R8各自独立地选自氢原子、 垸基、 环烷基、 杂环基、 芳基或杂芳基, 其 中所述垸基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟基、 氰基、 羧酸、 羧酸酯、 -S(0)OR9、 -COR9、 -S(0)ONR1 QR] 1、 - CONR1 GR"或- NRWR1 1的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元环, 其中所述 3〜8 元环内含有一个或多个 N、 0或 S(0)m杂原子, 并且所述 3〜8元杂环上任选进一 步被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟基、 氰基、 羧酸、 羧酸酯、 羰基、 -S(0)OR9、 -COR9 , -S^ONR'V^ - CONR' GR11或 -NRWR1 1 的取代基所取代;
R9选自氢原子、 垸基、 环垸基或芳基, 其中环烷基或芳基任选进一步被一个 或多个烷基所取代;
R1 Q和 R1 1选自氢原子、 垸基、 环垸基或芳基;
R1 2选自烷基、垸氧基、环烷基、 素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 - CONR7R8、 -C(0)OR9、 - OC(0)R9、 -0(CH2)RC(0)OR9、 - OC(0)NR7R8、 -S(0)MR9、 -OS(0)OR9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的烷基、 垸氧基、 环 垸基或杂环基任选进一步被一个或多个选自垸基、 环烷基、 杂环基、 芳基或杂芳 基的取代基所取代;
m为 0或 1;
n为 0, 1或 2; 且
r为 1, 2或 3。 本发明的优选方案, 一种通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中: R1和 R2各自独立地选自氢原子或烷基;
R3选自氢原子、 垸基、 烯基、 炔基、 环烷基、 杂环基、 芳基或杂芳基, 其中 所述烷基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多 个选自烷基、烷氧基、 卤素、羟基、氰基、硝基、芳基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)0R9、 -COR9或 -C(0)OR9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 1〜2个 N、 0或 S(0)m杂原子, 并且所述 3〜8元环任选进一步被一个或
多个选 垸基、垸氧基、卤素、轻基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR 8、 -CONR7Rs、 -NR7R8、 -S(0)OR9、 - COR9或 -C(0)OR9的取代基所取代;
R4和 R5各自独立地选自氢原子、 烷基、 卤素、 氰基或硝基;
L选自亚垸基, 任选进一步被一个或多个选自卤素、氰基、硝基或烷基的取代 基所取代, 其中所述的烷基任选进一步被一个或多个卤素所取代;
R6选自垸基;
A选自双环垸基、 双杂环基、 螺环垸基或螺杂环基, 其中所述双环垸基、 双 杂环基、 螺环垸基或螺杂环基任选进一歩被一个或多个 R12所取代;
R7和 Rs各自独立地选自氢原子、 垸基、 环烷基、 杂环基、 芳基或杂芳基, 其 中所述垸基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 垸氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)0R9、 -COR9, C(0)0R9、 -SiC ONR1 °R' 1、 -CONR1 °R' 1或- NR1 °R! 1的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其中所述 3〜8元杂环基内含有一个或多个 N、 0或 8(0),,^^原子, 并且所述 3〜8元杂环基 任选进一歩被一个或多个选自烷基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、 氰基、 -S(0)0R9、 -COR9, C(0)0R9、 -S(0)0NR1 QRM、 -CONI^R1 1或 -NR10R" 的取代基所取代;
R9选自氢原子、 烷基、 环垸基或芳基, 其中所述环垸基或芳基任选进一步被 一个或多个烷基所取代;
R1Q和 R11选自氢原子、 垸基、 环垸基或芳基;
R12选自烷基、垸氧基、环垸基、卤素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -C(0)0R9、 -OC(0)R9、 -0(CH2)rC(0)OR9、 - 0C(0)NR7Rs、 -S(0)mR9、 -0S(0)0R9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的烷基、 垸氧基、 环 垸基或杂环基任选进一步被一个或多个选自烷基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
m为 0或 1;
n为 0, 1或 2; 且
r为 1, 2或 3。 本发明的优选方案, 一种通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中: R1和 R2各自独立地选自氢原子或烷基;
R3选自氢原子、 垸基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基, 其中 所述垸基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多 个选自烷基、垸氧基、 卤素、羟基、氰基、硝基、 芳基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 - S(0)0R9、 - COR9或 -C(0)0R9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 1〜2个^^、 0或 S C^ ^原子, 并且所述 3〜8元环任选进一步被一个或 多个选自烷基、垸氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)0R9、 -COR9或 -C(0)0R9的取代基所取代;
R4和 R5各自独立地选自氢原子、 垸基、 卤素、 氰基或硝基;
L选自亚烷基, 任选进一步被一个或多个选自卤素、氰基、硝基或垸基的取代 基所取代, 其中所述的烷基任选进一步被- 个或多个卤素所取代;
R6选自垸基, 其中所述的烷基进一步被一个杂环基所取代;
当 R6选自杂环基取代的垸基时, A选自环烷基、 杂环基、 双环垸基、 双杂环 基、 桥环垸基、 杂桥环垸基、 螺环垸基或螺杂环基, 其中所述环垸基、 杂环基、 双环烷基、 双杂环基、 桥环烷基、 杂桥环垸基、 螺环烷基或螺杂环基任选进一歩 被一个或多个 R12所取代;
R7和 R8各自独立地选自氢原子、 烷基、 环垸基、 杂环基、 芳基或杂芳基, 其 中所述烷基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 烷氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)OR9、 -COR9, C(0)0R9、 -S(O)ONR] 0R' -CONK ER1 1或 -NRWR11的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其中所述 3〜8元杂环基内含有一个或多个 N、 0或 8(0)„1杂原子, 并且所述 3〜8元杂环基 任选进一步被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、 氰基、 - S(0)0R9、 -COR9. C(0)0R9、 -S(O)ONR10RN . -CONR^R1 1或 -NR ^R" 的取代基所取代;
R9选自氢原子、 烷基、 环烷基或芳基, 其中所述环垸基或芳基任选进一步被 一个或多个垸基所取代;
R1Q和 R1 1选自氢原子、 垸基、 环垸基或芳基;
R12选自垸基、垸氧基、环垸基、卤素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 - CONR7R8、 -C(0)0R9、 -OC(0)R9、 -0(CH2)RC(0)OR9、 -OC(0)NR7R8、 -S(0),"R9、 -0S(0)0R9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的垸基、 烷氧基、 环 烷基或杂环基任选进一步被一个或多个选自垸基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
m为 0或 1 ;
n为 0, 1或 2; 且
r为 1, 2或 3。 本发明的优选方案, 一种通式 (I.) 所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 R1 选自氢原子, R2选自垸基, R2优选为乙基。
本发明的优选方案, 一种通式 (I) 所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 R3 选自垸基或环垸基, 优选为异丙基或环戊基。 本发明的优选方案, 一种通式 (I) 所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 n 为 0或 1, L选自亚烷基, 优选为亚甲基。 本发明的优选方案, 一种通式 (I)所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 R6 选自杂环基取代的垸基, A选自环垸基。 本发明的优选方案, 一种通式 (I) 所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 A 选自双杂环基。 通式(I )化合物可以含有不对称碳原子, 因此可以以旋光纯的非对映体、 非对 映体混合物、 非对映体外消旋体、 非对映外消旋体的混合物的形式存在或作为内 消旋体化合物存在。 本发明包括所有这些形式。 非对映体混合物、 非对映外消旋 体或非对映外消旋体的混合物可以通过常规方法, 例如通过柱色谱法、 薄层色谱 法和 HPLC等来分离。 本发明的典型化合物包括, 但不限于:
化合物
结构 命 名
编号
4-((i?)- 8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨
1 基) -3-甲氧基 -N-((3i?,8a ?)-六氢-吡 咯并 [2,1 - C][l ,4]-噁嗪 -3-基甲基) -苯

甲酰胺
4-((i?)-8-环戊基 - 7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨
2 — ΦΊ 。 基) -3-甲氧基 -N-((3a5,5S,6a^)-2-甲
。、 Η ύ 基-八氢-环戊并 [c]吡咯 -5-基) -苯甲 酰胺
4-((/ )-8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢-喋啶 -2-基氨
3 基) -N」(((3a凡 5 & 6a,S)-2-甲基-八氢- 环戊并

4-( )-8-环戊基 -7-乙基- 5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨
4 基) -N-(((3a 5 & 6a )-5-羟基 -2-甲
 基-八氢-环戊并 M吡咯 -5-基) -甲 基)— 3-甲氧基-苯甲酰胺
基-八氢-环戊并 M吡咯 -5-基) -甲 基)— 3-甲氧基-苯甲酰胺
4-((7?)—8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢-喋啶- 2-基氨
5 基) -N-(((3ai?,5 6aS)-5-羟基 -2-甲
H H Ί 基-八氢-环戊并 M吡咯 -5,基) -甲 基) -3-甲氧基-苯甲酰胺
、 0 1 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢-喋啶 -2-基氨
6
基) -N-(l -甲基 -哌啶 -4-基) -3- (四氢- 呋喃 -3-基-甲氧基) -苯甲酰胺
0 1 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢-喋啶 -2-基氨
7
基) -N-(l-甲基 -哌啶 -4-基) -3- 氢-
¾ ώ 呋喃 -2-基 -甲氧基:) -苯甲酰胺
4-((i?)-8-环戊基 - 7-乙基- 5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨-1 基) -N-((4a7?,6i?,7aS -2-甲基 -八氢
-1H-环己垸 [c]吡啶 -6-基) -3-甲氧基
-苯甲酰胺
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨-2 基) -N-((4a5,6 & 7ai?)-2-甲基 -八氢
- η ό 环己垸 M吡啶 -6-基) -3-甲氧基
-苯甲酰胺



或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及 药学上可以接受的盐。 进一步, 本发明提供一种通式(IA )所示的化合物, 其作为制备通式(I )化合 物的中间体, 其中:

(1A)
其中:
G选自离去基团, 优选为选自卤素、 甲磺酰基、 对甲苯磺酰基、 三氟甲磺酰 基或垸氧基;
R选自氢原子或垸基;
R1选自氢原子或垸基;
R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元环内 含有!〜 2个 N、 0或 S(0)n杂原子, 并且所述 3〜8元环任选进一步被一个或多 个选自垸基、垸氧基、芳基、卤素、羟基、氰基、羰基、羧酸、羧酸酯、 -S(0)ONR7R8, -CONR7R8、 -NR7R8、 -S(0)OR9或 -COR9的取代基所取代。
n, R7〜R9的定义如通式 (J)中所述
通式(IA)的典型化合物包括, 但不限于:

进一步, 本发明提供一种通式(I)化合物的制备方法, 该方法包括:
】2

(IA) (IB) ( 1 )
通式( IA:)化合物与通式( IB )化合物或通式( IB >化合物的盐反应,得到通式( I ) 化合物。
其中:
R选自甲基;
G、 R^R3的定义如通式(IA )化合物所述;
A、 n、 L、 R4〜R6的定义如通式 ( I )化合物所述。 本发明的另一方面涉及本发明通式(I )化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐在制备细胞 增殖类疾病的药物中的用途, 其中所述的细胞增殖类疾病为癌症、 感染、 炎症及 自身免疫性疾病, 所述的癌症为宫颈癌或结肠癌。 本发明的另一方面涉及一种治疗细胞增殖类疾病的方法, 该方法包括给予需 要治疗的患者有效治疗量的通式(I )化合物或其互变异构体、 外消旋体、 对映异构 体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中所述的细胞 增殖类疾病为癌症、 感染、 炎症及自身免疫性疾病, 所述的癌症为宫颈癌或结肠 癌。 本发明的另一方面涉及本发明通式(I )化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐作为治疗细 胞增殖类疾病的药物, 其中所述的细胞增殖类疾病为癌症、 感染、 炎症及自身免 疫性疾病, 所述的癌症为宫颈癌或结肠癌。 进一步, 本发明还涉及本发明通式(I )化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐在制备 Plk 激酶抑制剂中的用途。 本发明的另一方面涉及一种抑制 Plk激酶的方法,该方法包括给予需要治疗的 患者有效治疗量的通式(I )化合物或其互变异构体、 外消旋体、 对映异构体、 非对 映异构体、 及其混合物形式、 及药学上可以接受的盐。 本发明还涉及本发明通式(I )化合物或其互变异构体、外消旋体、对映异构体、
非对映异构体、及其混合物形式、及药学上可以接受的盐作为抑制 Plk激酶的药物。 本发明的另一方面涉及一种药物组合物, 其含有治疗有效剂量的本发明化合 物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 及其可药用的载体或赋形剂。 该药物组合物在制备治疗 癌症、 感染、 炎症及自身免疫性疾病的药物中的用途, 所述的癌症为宫颈癌或结 肠癌。该药物组合物在制备 Plk激酶抑制剂的药物中的用途。该药物组合物作为治 疗癌症、 感染、 炎症及自身免疫性疾病的药物, 所述的癌症为宫颈癌或结肠癌。 本发明的另一方面涉及一种治疗细胞增殖类疾病的方法, 该方法包括给予需 要治疗的患者含有有效治疗量的通式(I )化合物或其互变异构体、 外消旋体、 对映 异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐的药物组合物, 其中所述的细胞增殖类疾病为癌症、 感染、 炎症及自身免疫性疾病, 所述的癌症 为宫颈癌或结肠癌。 本发明所得化合物及其可药用的盐可通过口服、 真皮或非肠道 (如通过注射、 吸入、 喷雾、 舌下、 直肠或阴道)给药。 "注射给药"包括静脉注射、 关节注射、 肌 肉注射、 皮下注射、 非肠道注射以及输液。 真皮给药包括局部或交叉给药。 口服 用药是按照本领域技术人员所熟知的方法制备的, 在此类制剂中还可有一种或多 种助剂, 如稀释剂、 甜味剂、 调味剂、 色剂和防腐剂。 在每一种情况中的药物活 性化合物量应该在 0.1〜90重量%的总组合物范围内, 优选 0.5〜50重量%即以充 分达到以下提供的计量范围的量。 若必要, 可以每天提供多次指定的剂量。
在片剂中, 活性成分和无毒、 可药用的赋形剂混合。 这些赋形剂包括: 惰性 稀释剂 (如碳酸钙、 碳酸钠、 乳糖、 磷酸钙或磷酸钠)、 粒化或崩解剂 (如玉米淀粉、 藻酸)和粘合剂 (如硬脂酸镁、 硬脂酸、 滑石)。 片剂可以不包衣, 也可以用己知的 方法包衣来缓解在肠胃道中的分解和吸收从而延长药效时间, 如可以使用硬脂酸 甘油酯或甘油二硬脂酸酯。 这些化合物也可以制备成固态、 快速释放的模式。
在硬胶囊中, 活性组分和惰性固体稀释剂如碳酸钙、 磷酸钙或陶土混合; 在 软胶囊中, 活性成分和水或油媒介如花生油、 石蜡或橄榄油混合。
在水悬浮剂中,活性成分和能适合药用的赋形剂混合。这些赋形剂有悬浮剂 (如 羟基甲级纤维素钠、 甲基纤维素、 羟基丙基-甲基纤维素、 藻酸钠、 聚乙烯吡咯垸 酮、 阿拉伯胶)、 分散剂或湿润剂 [包括自然产生的磷脂 (如卵憐脂)或烯化养和脂肪 酸的缩合物 (如聚氧乙烯硬脂酸酯)或乙烯化养和长链脂肪醇的缩合物 (如十七碳乙 烯氧十六醇)或乙烯氧和由脂肪酸和己糖醇衍生得到的部分酯的缩合物 (如聚乙烯 脱水山梨糖醇单油酸酯): |。 所述水悬浮剂也可以含义中或多种防腐剂 (如对羟基苯 甲酸乙酯、 对羟基苯甲酸丙酯); 一种或多种色剂; 一种或多种芳香剂以及一种或
多种甜味剂 (如蔗糖或糖精:)。
适合制备水悬浮剂的可分散粉末或颗粒是通过将水、 活性组分和分散剂或湿 润剂、 悬浮剂和一种或多种防腐剂混合制备的。 也可以向其中加入其他赋形剂如 甜味剂、 色剂或芳香剂。
包括本发明的活性成分物质或其组合物的糖浆可另外包括甜味剂如糖精、 甜 精、 甘油或糖以及改味剂。 例如, 甜味剂, 如香草或橙萃取液。 此外, 其也可以 包括悬浮助剂或增稠剂 (如羟甲基纤维素钠)、 湿润剂 (例如脂肪醇与环氧乙垸的缩 合产物:)或保存剂 (如对-羟基苯甲酸酯;)。
注射或灌注溶液以常见的方式制备, 例如, 加入等渗剂、 保存剂 (如对羟基苯 甲酸酯)或稳定剂 (如乙二氨四醋酸的碱金属盐类),视需要使用乳化剂及 /或分散剂, 同时使用水作为稀释剂时, 即可视需要使用有机溶剂作为溶解剂或助溶剂, 并转 移至注射小瓶或安瓶或灌注瓶中。
适合的栓剂例如与为此规定的载体 (如中性脂或聚乙二醇或其衍生物)混合而 成。
适合的赋形剂可以是例如水、在可药用的有机溶剂 (如石蜡,例如,石油馏分)、 植物源油 (例如, 花生油或芝麻油)、 单或多官能醇 (例如, 乙醇或甘油)、 载体 [如天 然矿物粉 (例如, 高岭土、 粘土、 滑石粉)、 合成矿物粉 (例如, 高分散性二氧化硅 及硅酸盐)]、 糖 (例如, 粗糖、 乳糖及葡萄糖)、 乳化剂 (例如, 木质素、 亚硫酸盐废 液、 甲基纤维素、 淀粉及聚乙烯吡咯烷酮)及润滑剂 (例如, 硬脂酸镁、 滑石粉、 硬 脂酸及月桂基硫酸钠)。
可将制剂以常见的方式给药, 优选经口或皮肤途径, 优选为口服。 在口服时, 则片剂当然除了上述的载体之外可以包括添加剂 (如柠檬酸钠、碳酸钙及磷酸二钙) 与其他的添加剂 (如淀粉, 优选马铃薯淀粉、 明胶及类似物)。 也可以使用润滑剂形 成药片, 如硬脂肪酸镁、 月桂基硫酸钠及滑石粉。 在水性悬浮液的情况中, 可将 活性物质除了上述的赋形剂之外与各种改味剂或着色剂混合。 对于非肠使用的活 性物质溶液可使用适合的液态载体物质。 静脉注射剂量是以每小时计 1〜1000毫 克, 优选每小时介于 5〜500毫克。
但是, 可视需要偏离上述剂量, 它取决于体重或给药方法, 个体对药物的反 应, 所使用的制剂的性质及给药时间或间隔。 因此, 在一些情况下使用小于以上 指定的最少剂量己足够, 但是在其他的情况下必须超过指定的上限。 在以大的剂 量给药时, 则建议将其分成每天数次的单一剂量。
当然, 如本领域技术人员所熟知的, 药物的给药剂量依赖于多种因素, 包括 但并非限定以下因素: 所用特定化合物的活性、 病人的年龄、 病人的体重、 病人 的健康状况、 病人的行被、 病人的饮食、 给药时间、 给药方式、 排泄的速率、 药 物的组合等; 另外, 最佳的治疗方式如治疗的模式、 通式化合物 (I)的日用量或可 药用的盐的种类可以根据传统的治疗方案来验证。
■ 发明的详细说明
除非有相反陈述, 否则下列用在说明书和权利要求书中的术语具有下述含义。 "垸基 "指饱和的脂族烃基团, 包括 1至 20个碳原子的直链和支链基团。 优 选含有 1至 10个碳原子的垸基, 例如甲基、 乙基、 丙基、 2-丙基、 正丁基、 异丁 基、 叔丁基或戊基等。 更优选的是含有 1至 4个碳原子的低级垸基, 例如甲基、 乙基、丙基、 2-丙基、正丁基、异丁基或叔丁基等。垸基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 环烷基、 杂环基、 芳基、 杂芳基、 双环垸基、 双杂环 基、 -S(0)ONR7R8、 -CONR7Rs、 -NR7R8、 - S(0)OR9、 -COR9, 羧酸或羧酸酯。
"烯基 "指由至少两个碳原子和至少一个碳-碳双键组成的如上述定义的烷基。 例如乙烯基、 1-丙烯基、 2-丙烯基、 〗 -, 2-或 3-丁烯基等。 烯基可以是取代的或未 取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自垸基、 垸氧基、 炔 基、 卤素、 羟基、 氨基、 硝基、 氰基、 磺酰基、 环垸基、 杂环基、 芳基、 杂芳基、 双环垸基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 - COR9、 羧酸或羧酸酯。
"炔基"指至少两个碳原子和至少一个碳-碳三键组成的如上所定义的烷基。 例如乙炔基、 1-丙炔基、 2-丙炔基、 1-, 2-或 3-丁炔基等。 炔基可以是取代的或未 取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自垸基、 烷氧基、 烯 基、 卤素、 羟基、 氨基、 硝基、 氰基、 磺酰基、 环烷基、 杂环基、 芳基、 杂芳基、 双环垸基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9. 羧酸或羧酸酯。
"环垸基"指非芳香族单环或多环环状体系, 其包括 3至 20个碳原子, 优选 包括 3至 10个碳原子, 更优选环烷基环包含 3至 8个碳原子。 单环环烷基的非限 制性实施例包含环丙基、 环丁基、 环戊基、 环戊烯基、 环己烷基、 环己二烯基、 环庚烷基、 环庚三烯基等。 多环环垸基包括螺环、 稠环和桥环的环烷基, 非限制 性实施例包括 1-萘垸基、 降冰片基、 金刚垸基及类似基团。 环垸基可以是取代的 或未取代的, 当被取代时, 取代基优选为一个或多个以下基团, 独立地选自烷基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环烷基、 杂环基、 芳基、杂芳基、双环垸基、双杂环基、 - S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9, 羧酸或羧酸酯。
"杂环基"指非芳香族的单环或多环环状体系, 其包括 3至 20个环原子, 其 中一个或多个环原子选自氮、 氧或 S(0)n (其中 n是整数 0至 2)的杂原子, 但不包 括 -0- 0-、 -0-S-或- S-S-的环部分, 其余环原子为碳。优选杂环基包含 3〜10个环原 子, 其中 1 ~4个是杂原子, 更优选杂环基包含 3〜8个环原子。 单环杂环基的非 限制性实施例包括吡咯烷基、 哌啶基、 哌嗪基、 吗啉基、 硫代吗啉基、 高哌嗪基
等, 多环杂环基包括双环或多环的螺环、 稠环和桥环的杂环基。 杂环基可以是取 代的或未取代的。 当被取代时, 取代基优选为一个或多个以下基团, 独立地选自 垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环烷基、 杂环基、芳基、杂芳基、双环烷基、双杂环基、 -S(0)ONR7R8、 - CONR7Rs、 - NR7R8、 -S(0)OR9、 -COR9, 羧酸或羧酸酯。
"双环垸基"指 5至 14元全碳稠合环 ("稠合"环系意味着系统中的每个环与体 系中的其他环共享毗邻的一对碳原子)基团, 其中一个或多个环可以含有一个或多 个双 一个环具有完全共轭的 π电子系统。 例如

优选为 5元 /5元或 5元 /6元双环烷基。 双环烷基可以是取代的或未取代的。 当被 取代时, 取代基优选为一个或多个, 独立地选自垸基、 垸氧基、 烯基、 炔基、 卤 素、 羟基、 氨基、 硝基、 氰基、 羰基、 环垸基、 杂环基、 芳基、 杂芳基、 双环垸 基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9, 羧酸或 羧酸酯。
"双杂环基" 指 Ί至 12元稠合环 ("稠合"环系意味着系统中的每个环与体系 中的其他环共享毗邻的一对碳原子)基团, 其中一个或多个环原子选自氮、 氧或 S(0)n (其中 n是整数 0至 2)的杂原子, 其余环原子为碳。这些可以含有一个或多个 双键, 但没有一 π电子系统。 优选为 7至 ] 0元。 例如
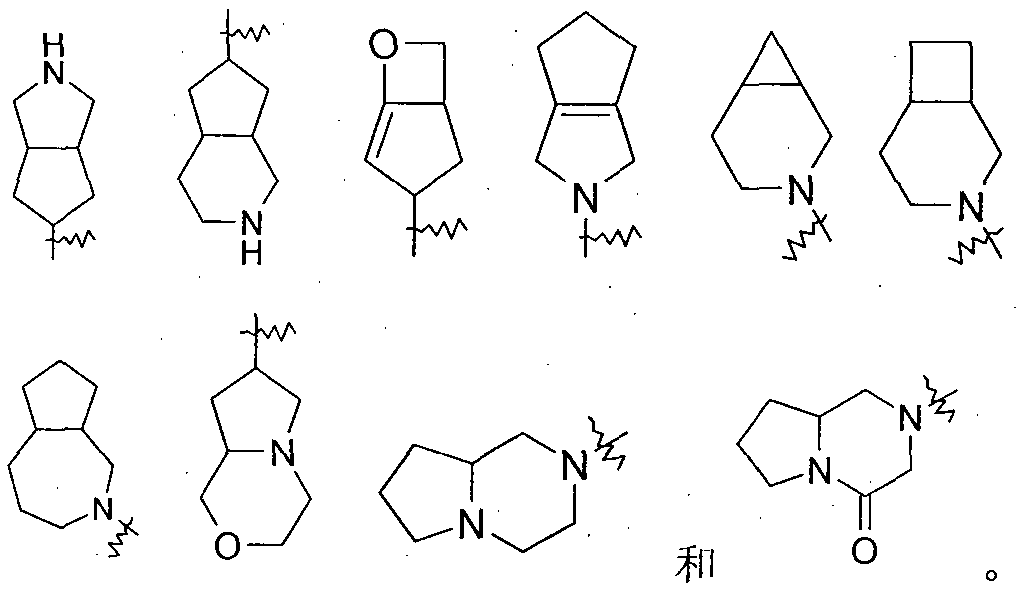
更优选为 5元 /5元或 5元 /6元双杂环基。 双杂环基可以是取代的或未取代的。 当 被取代时, 取代基优选为一个或多个, 独立地选自垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环烷基、 杂环基、 芳基、 杂芳基、 双环 垸基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9、 羧酸 或羧酸酯。
"螺环垸基"指 5至 14元, 单环之间共用一个碳原子 (称螺原子)的多环基团, 这些可以含有一个或多个双键, 但没有一个环具有完全共轭的 π 电子系统。 优选 为 7至 10元。 根据环与环之间共用螺原子的数目将螺环烷基分为单螺环烷基、 双
螺环垸基基或多螺环 优选为单螺环垸基和双螺环烷基。 例如:

更优选为 4元 /4元、 4元 /5元、 4元 /6元、 5元 /5元或 5元 /6元单螺环烷基。 螺环 烷基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个以下基团, 独立地选自垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环垸基、杂环基、芳基、杂芳基、双环垸基、双杂环基、 -S(0)ONR7R8, -CONR7R8、 -NR7R8、 - S(0)OR9、 -COR9, 羧酸或羧酸酯。
"螺杂环基"指 5至 ] 4元, 单环之间共用一个原子 (称螺原子)的多环烃, 其中 一个或两个环原子选自氮、 氧或 S(0)n (其中 n是整数 0至 2)的杂原子, 其余环原 子为碳。 这些可以含有一个或多个双键, 但没有一个环具有完全共轭的 π 电子系 统。 优选为 7至 10 。 例如

根据环与环之间共用螺原子的数目将螺环垸基分为单螺环垸基、 双螺环垸基基或 多螺环烷基, 优选为单螺杂环基和双螺杂环基, 更优选为 4元 /4元、 4元 /5元、 4 元 /6元、 5元 /5元或 5元 /6元单螺杂环基。 螺杂环基可以是取代的或未取代的, 当 被取代时, 取代基优选为一个或多个以下基团, 独立地选自垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环垸基、 杂环基、 芳基、 杂芳基、 双环垸基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9、 羧酸或羧酸酯。
"3〜8元杂环基"指构成环原子的数量为 3〜8元, 构成环的原子中含有一个 或多个 N、 0或 S(0)n杂原子, 环内可以含有 1〜2个双键, 为单环或双环的非芳 香族的环基,构成环的原子中含有氮原子时,可以从氮原子伸出结合键。优选为 4〜 6元杂环基,:更优选为 5〜6元, 例如吡咯烷基、 哌啶基或哌嗪基等。 3〜8元杂环 基可以是取代的或未取代的, 当被取代时, 取代基优选为一个或多个以下基团, 独立地选自垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 羰基、 环烷基、杂环基、芳基、杂芳基、双环垸基、双杂环基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9, 羧酸或羧酸酯。
p元 /q元的双环烷基、 双杂环基、 单螺环垸基或单螺杂环基, 指双环垸基、 双 杂环基、单螺环垸基或单螺杂环基的两个环的环原子数量分别为 p个和 q个, p或 q选自 3〜8的整数, 优选为 4〜7的整数。
"芳基 "指 6至 14元全碳单环或稠合多环 (也就是共享毗邻碳原子对的环)基团, 具有共轭的 π电子体系的多环 (即其带有相邻对碳原子的环)基团, 优选为 6至 10 元, 例如苯基、 萘基和蒽基。 芳基可以是取代的或未取代的, 当被取代时, 取代 基优选为一个或多个, 独立地选自独立地选自烷基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 环垸基、 杂环基、 芳基、 杂芳基、 双环垸基、 双杂环 基、 -S(0)ONR7R8、 -CONR7Rs、 - NR7R8、 -S(0)OR9、 -COR9, 羧酸或羧酸酯。
"杂芳基"指包含 1至 4个杂原子, 5至 14个环原子的杂芳族体系, 其中杂 原子包括氧、 硫和氮。 杂芳基优选为是 5元或 6元。 例如呋喃基、 噻吩基、 吡啶 基、 吡咯、 N-垸基吡咯基、 嘧啶基、 吡嗪基、 咪唑基、 四唑基等。 杂芳基可以是 取代的或未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自独立地 选自垸基、 烷氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 环垸基、 杂 环基、 芳基、 杂芳基、 双环垸基、 双杂环基、 -COR9、 -CONR9R1 Q、 -NR9R1Q、 羧酸 或羧酸酯。
• "垸氧基"指 -0-(;垸基)和 -0-(未取代的环烷基)。例如甲氧基、乙氧基、丙氧基、 丁氧基、 环丙氧基、 环丁氧基、 环戊氧基、 环己氧基等。 烷氧基可以是取代的或 未取代的, 当被取代时, 取代基优选为一个或多个, 独立地选自独立地选自垸基、 垸氧基、 烯基、 炔基、 卤素、 羟基、 氨基、 硝基、 氰基、 环垸基、 杂环基、 芳基、 杂芳基、 双环烷基、 双杂环基、 -S(0)ONR7R8、 -CONR7R8、 - NR7R8、 -S(0)OR9、 -COR9, 羧酸或羧酸酯。
"芳氧基"指 -0-芳基和 -0-杂芳基, 芳基和杂芳基定义同上。 例如苯氧基、 吡 啶氧基、 呋喃氧基、 噻吩氧基、 嘧啶氧基、 吡嗪氧基等及其衍生物。 .
"羟基 "指 -OH基团。 .
"卤素"指氟、 氯、 溴或碘。
"氨基"指 -N¾。
"氰基"指 -CN。
"硝基"指 -N02。
"羰基"指 (基团) -C(=0)- (基团)。
"羟垸基"指- (烷基 )-OH。
"苄基"指 -CH2- (苯基)。
"羧酸"指 (浣基) C(=0)OH。
"羧酸酯"指 (垸基) C(=0)0(垸基)。
"任选 "或"任选地 "意味着随后所描述地事件或环境可以但不必发生,该说明包 括该事件或环境发生或不发生地场合。例如, "任选被垸基取代的杂环基团"意味着 垸基可以但不必须存在, 该说明包括杂环基团被烷基取代的情形和杂环基团不被 垸基取代的情形。
"药物组合物"表示一种或多种本文所述化合物或其生理学上 /可药用的盐或
前体药物与其他化学组分的混合物,其他组分例如生理学 /可药用的载体和赋形剂; 药物组合物的目的是促进化合物对生物体的给药。 本发明化合物的合成方法
为了完成 本发明采用如下技术方案:
本发明通式( I )所述的化合物或其盐的制备方法, 包括以下歩骤:

通式( IA )化合物与通式( IB;)化合物或通式( IB;)化合物的盐, 缩合反应得到通 式(I )化合物。
上述的缩合反应是在酸和胺基之间进行, 在缩合试剂和碱性条件下进行, 所 用缩合试剂选自 Ν,Ν-二环己基碳二亚胺、 Ν,Ν-二异丙基碳二亚、 0-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (TBTU)等,优选为 0-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基 脲四氟硼酸酯 (TBTU); 碱性条件由有机碱或无机碱提供, 有机碱选自如二异丙基 乙胺、 吡啶、 三乙胺、 六氢吡啶、 N-甲基哌嗪、 4-二甲氨基吡啶等, 优选为二异丙 基乙胺; 所用溶剂选自甲苯、 苯、 二氯甲垸、 四氢呋喃、 氯仿或上述溶剂组成的 混合物等, 优选为二氯甲烷; 反应温度控制在 -80°C到 100°C, 优选为 0°C到 60°C ; 反应时间一般控制在 1分钟至 72小时, 优选为 15分钟至 24小时;
其中:
R选自甲基;
G、 Ι^〜Ι 3的定义如通式(IA )化合物所述;
A、 n、 L、 R4〜R6的定义如通式(I )化合物所述。 具体实施方式
以下结合实施例用于进一步描述本发明, 但这些实施例并非限制着本发明的 范围。
实施例
化合物的结构是通过核磁共振 (NMR)或 /和质谱 (MS)来确定的。 NMR位移 (δ) 以百万分之一 (ppm)的单位给出。 NMR的测定是用 Bmker AVANCE-400核磁仪, 测定溶剂为氘代氯仿 (CDC13), 内标为四甲基硅垸 (TMS), 化学位移是以 10_0(ppm) 作为单位给出。
MS的测定用 FINNIGAN LCQAd (ESI)质谱仪 (生产商: Thermo, 型号: Finnigan LCQ advantage MAX)。
HPLC的测定使用安捷伦 1200DAD高压液相色谱仪 (Sunfire C18 150x4.6mm 色谱柱)和 Waters 2695-2996高压液相色谱仪 (Gimini C18 150x4.6mm色谱柱)。
激酶平均抑制率及 IC5o值的测定用 NovoStar酶标仪 (德国 BMG公司)。
薄层层析硅胶板使用烟台黄海 HSGF254或青岛 GF254硅胶板, 薄层色谱法
(TLC)使用的硅胶板采用的规格是 0.15 mm〜0.2 mm, 薄层层析分离纯化产品采用 的规格是 0.4 ram—0.5 mm。
柱层析一般使用烟台黄海硅胶 200~300目硅胶为载体。
本发明的起始原料是已知的,并且可以在市场上购买到,购买自 ABCR GmbH & Co. KG, Acros Organics , Aldrich Chemical Company, 韶远化学禾斗技 (Accela ChemBio Inc) 、 达瑞化学品等公司, 或者可以采用或按照本领域已知的方法来合 成。
氩气氛或氮气氛是指反应瓶连接一个约 1L容积的氩气或氮气气球。
氢气氛是指反应瓶连接一个约 1L容积的氢气气球。
加压氢化反应使用 Parr 3916EKX 型氢化仪和清蓝 QL-500 型氢气发生器或 HC2-SS型氢化仪。
氢化反应通常抽真空, 充入氢气, 反复操作 3次。
微波反应使用 CEM Discover-S 908860型微波反应器。
实施例中无特殊说明, 溶液是指水溶液。
实施例中无特殊说明, 反应的温度为室温。
室温为最适宜的反应温度, 为 20°C〜30°C。
实施例中的反应进程的监测采用薄层色谱法 (TLC), 反应所使用的展开剂的体 系有: A: 二氯甲垸和甲醇体系, B: 正己烷和乙酸乙酯体系, C: 石油醚和乙酸 乙酯体系, D: 丙酮。 溶剂的体积比根据化合物的极性不同而进行调节。
柱层析的洗脱剂的体系包括: A: 二氯甲垸和甲醇体系, B: 正己垸和乙酸乙 酯体系, C: 二氯甲垸和丙酮体系, D: 正己垸和丙酮体系。 溶剂的体积比根据化 合物的极性不同而进行调节, 也可以加入少量的氨水和醋酸等进行调节。 实施例 1
4-(0? -环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )-3-甲氧基 -N-((3i?,8ai?)-六 甲基) -苯甲酰胺




第一步
3-甲氧基 -4-硝基 -苯甲酸甲酯
将 3-甲氧基 -4-硝基-苯甲酸 la(5 g, 50 mmol)溶解于 30 mL甲醇中, 滴加二氯 亚砜 (5.50mL, 75mmol), 回流反应 3小时。 减压浓缩反应液, 加入饱和碳酸氫钠 溶液 (50mL), 乙酸乙酯萃取 (50mLx3), 合并有机相, 无水硫酸镁干燥, 过滤, 滤 液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 3- 甲氧基 -4-硝基 -苯甲酸甲酯 lb (5.28 g, 白色固体), 产率: 99.6%
第 ~~ ^步
4-氨基 -3-甲氧基 -苯甲酸甲酯
将 3-甲氧基 -4-硝基 -苯甲酸甲酯 lb(5.28g, 25mmo】)溶解于 40mL甲醇中, 加 入 (600mg, 10%)钯/碳, 氢气氛下搅拌反应 1小时。 过滤, 滤液减压浓縮, 用硅胶 柱色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 4-氨基 -3-甲氧基-苯甲酸 甲酯 lc(4.1g, 淡黄色固体), 产率: 91%。
MS m/z(ESI): 182.1 [M+l]
第三步
(i?)-2-氨基-丁酸甲酯
冰浴下, 将 ((i?)-2-氨基 -丁酸 ld(10 g, 0.096 mol)溶解于 50 mL甲醇中, 滴加 二氯亚砜 (13mL, 0.17 mol), 回流搅拌反应 1小时,冷却至室温,减压浓縮反应液,
得到标题产物 (7?)-2-氨基-丁酸甲酯盐酸盐 le (无色油状物), 直接投入下一步。 MS m/z (ESI):118.0 [ +1]
第四步
. -2-环戊基氨基-丁酸甲酯
将 ((i?)-2-氨基-丁酸甲酯盐酸盐 le(1 1 .24 g, 0.096 mol)和环戊酮 (8.24 g, 0.098 mol)溶解于 150 mL二氯甲垸中,搅拌反应 1.5小时,加入乙酸钠 (8.04 g, 0.098 mol) 和三乙酰氧基硼氢化钠 (30.52 g, 0.14 mol), 搅拌反应 3小时。 将反应液倒入 150 mL10 %碳酸氢钠溶液中, 用二氯甲烷萃取 (100mLx3), 合并有机相, 饱和食盐水 洗涤 (100mLx3), 无水硫酸镁千燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 B纯化所得残余物,得到标题产物 (7?)-2-环戊基氨基-丁酸甲酯 lf(6.04 g,淡 黄色油状物), 产率: 34 %。
MS m/z (ESl): 186.1 [M+1]
第五步
(i -2-[(2-氯- 5-硝基-嘧 I症 -4-基) -环戊基-氨基] -丁酸甲酯 将 ( -2-环戊基氨基-丁酸甲酯 lf(2.50 g, 13.5 mmol)和碳酸氢钠 (4.54 g, 54 nimol)溶解于 ] 00 mL环己烷中, 搅拌 30分钟, 加入 2,4-二氯- 5-硝基-嘧啶 (2.88 g, 14.84 mmol), 60°C下搅拌反应 12小时。 过滤, 滤饼用二氯甲垸洗涤 (50 mL), 滤 液减压浓縮, 用 150 mL乙酸乙酯和正己垸 (V:V=1 :4)混合溶剂重结晶残余物, 得到 标题产物 (ii)- 2-[(2-氯 -5-硝基 -嘧啶 -4-基) -环戊基-氨基] -丁酸甲酯 lg(3.36 g, 浅黄色 固体), 产率: 72.6 %。
MS m/z (ESI):343.1 [M+1]
(i?)-7-乙基 -2-氯 -8-环戊基 -7,8-二氢 -5H-喋啶 -6-酮 将 (i?)-2- [(2-氯 -5-硝基-嘧啶- 4-基) -环戊基-氨基] -丁酸甲酯 lg(l g, 3 mmol)溶解 于 10 mL醋酸中,加入兰尼镍 (0.50 g),氢气氛下, 80°C下搅拌反应 12小时。过滤, 滤饼用二氯甲垸洗涤 (50 mL), 滤液减压浓缩, 加入 100 mL乙酸乙酯, 依次用水 (50mLx3)、 饱和食盐水洗涤 (50mLx3), 合并有机相, 无水硫酸镁干燥, 过滤, 滤 液减压浓縮,用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 (i?)-7- 乙基- 2-氯 -8-环戊基 -7,8-二氢 -5H-喋啶 -6-酮 lh(0.56 g, 白色固体), 产率: 66.7 %。 MS m/z (ESI):281.2 [M+1]
第七步
((7?)-7-乙基 -2-氯 -8-环戊基 -5-甲基- 7,8-二氢 -5H-喋啶 -6-酮 将 (i?)-7-乙基 -2-氯 -8-环戊基 -7,8-二氢 -5H-喋啶 -6-酮 lh(3.50 g, 12.5 mmol, 根 据专利公开号 US2004/176380制备而得)溶解于 80 mL丙酮中, 加入对甲基苯磺酸甲 酉旨 (3.4 g, 18.7 mmol)和碳酸钾 (3.45 g, 25 mmol), 回流搅拌反应 2小时, 冷却至室 温。 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到 标题产物 (i?)-7-乙基 -2-氯 -8-环戊基 -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lj(3.4 g, 白色固
体), 产率: 93 %。
MS m/z (ESI):295.4 [M+l ]
第八歩
( 7-乙基 -8-环戊基 - 2-(3-甲氧基 -苯甲酸甲酯 -4-基-氨基) -5-甲基- 7,8-二氢 喋啶
-6-酮
将 4-氨基 -3-甲氧基-苯屮酸甲酯 lc(305 mg, 1.7 mmol), (i?)-7-乙基 -2-氯 -8-环 戊基- 5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lj(500 mg, 1.7 mmol)和对甲苯磺酸 (500 mg, 25.5 mmol)溶解于 20 mL 4-甲基 -2-戊醇中, 回流反应 2.5小时。 减压浓缩反应液, . 滴加氨水至反应液 pH为 9〜10, 乙酸乙酯萃取 (30 mLx3), 合并有机相, 饱和食盐 水溶液洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以 洗脱剂体系 B纯化所得残余物, 得到标题产物 (7?)-7-乙基 -8-环戊基 -2-(3-甲氧基-苯 甲酸甲酯 -4-基-氨基) -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lk(550 mg, 白色固体), 产率: 73 %。
MS m/z (ESI):440.2 [M+l]
第九歩
(7?)-7-乙基 -8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基) -5-甲基 -7,8-二氢 -5H-喋啶- 6-酮 将 (i?)-7-乙基 -8-环戊基 -2-(3-甲氧基 -苯甲酸甲酯 - 4-基-氨基) -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lk(200 mg, 0.46 mmol)溶解于 20 mL甲醇中, 加入 3 mL 2 M氢氧 化锂 (77 mg, 1.38 mmol)溶液, 60°C下搅拌反应 12小时。 减压浓缩反应液, 乙酸 乙酯萃取 (50 mLx3), 水相加入少量水稀释后, 滴加 1 M盐酸至反应液 pH为 2, 有白色固体析出。 过滤, 烘干滤饼, 得到标题产物 (i?)-7-乙基 -8-环戊基 -2-(3-甲氧 基-苯甲酸—4—基 -氨基 )-5-甲基 -7,8-二氢—5//—喋啶—6—酮!.!^^ !^,白色固体),产率: 71 % o
MS m/z (ESI):426.3 [M+l]
第十步
2-氯 -3-(2-羟基甲基-吡咯烷 -1 -基) -丙腈
氮气氛下, 将吡咯垸 -2-基 -甲醇 ln(200 mg, 2 mmol)溶解于 5 mL无水乙醚中, 加入 2-氯-丙烯腈 (0.16 mL, 2 mmol), 搅拌反应 12小时。 减压浓縮反应液, 得到 标题产物 2-氯 -3-(2-轻基甲基-吡咯垸- 1-基) -丙腈 lo, 直接用于下一步反应。
MS m/z (ESI): 189.1 [M+l]
第十一步
六氢-吡咯并 [2,1-C][1,4]-噁嗪 -3-腈
冰浴下,将 2-氯 -3-(2-羟基甲基-吡咯垸 -1-基) -丙腈 lO(0.38 g, 2 mmol)溶解于 6 mL无水四氢呋喃中, 加入叔丁醇钾 (0.29 g, 2.6 mmol), 室温下搅拌反应 1小时。 过滤, 用 20 mL甲醇洗涤滤饼, 合并滤液, 减压浓缩, 用硅胶柱色谱法以洗脱剂 体系 B纯化所得残余物,得到标题产物六氢-吡咯并 [2,l-c|[l,4]-噁嗦 -3-腈 lp(0.12 g, 无色油状物), 产率: 37.8 %。
MS m/z (ESl):153.2 [M+\]
第十二歩
[(37?,8ai?)-3,4,6,7,8,8a-六氢 -1H-吡咯并 [2,1-ύ][1,4] 噁嗪- 3-基) -甲胺 将六氢-吡咯并 [2,1 - c][l ,4]-噁嗪 -3-腈 lp(0.40 g, 2.63 mmol)溶解于 20 mL甲醇 中, 加入兰尼镍 (0.5 g), 氢气氛下搅拌反应 12小时。 过滤, 滤液减压浓缩, 得到 标题产物 [(3 8a7 -3,4,6,7,8,8a-六氢 -1 H-吡咯并 [2,l-c|[l,4] 噁嗪 -3-基) -甲胺 lq(0.3 g, 无色油状物), 产率: 73 %, 直接用于下一歩反应。
MS m/z (ESI): 157.2 [M+l ]
第十三步
4-((i?)-8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-3-甲氧基
-N- ((3 ?,8aii)-六氢-吡咯并 [2,1- [1,4]-噁嗪 -3-基甲基) -苯甲酰胺 将 [(!K,8ai -3,4,6,7,8,8a-六氢 -1H-吡咯并 [2,l-c][l,4] 噁嗪 -3-基) -甲胺 lq(60 mg, 0.38 mmol), ((7?)-7-乙基- 8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基) -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lm(160 mg, 0.38 mmol), (9-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼 酸酯 (243 mg, 0.38 mmol)和二异丙基乙胺 (180 mg, 1.4 mmol)溶解于 12 mL二氯甲 垸中, 搅拌反应 2小时。加入 20 mL饱和碳酸氢钠溶液, 二氯甲垸萃取 (50 ^1^3), 合并有机相, 饱和食盐水洗漆 (20 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物,得到标题产物 4-((/?)-8-环戊基 -7- 乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )-3-甲氧基 -N-((3i?,8a 0-六氢 -吡咯 并 [2,l -c][l,4]-噁嗪 -3-基甲基) -苯甲酰胺 l(100 mg, 白色固体), 产率: 47 %。
MS m/z (ESI):564.3 [M+l]
'H NMR(400MHz, CDC13i ppm) δ 8.58-8.60 (d, 1H), 7.72 (s, 1H), 7.63 (s, 1H), 7.49 (s, 1 H), 7.32 (d, 1H), 6.53 (t, 1H), 4.34 (m, 1H), 4.24-4.27 (m, 1H), 4.06-4.88 (ra, I H), 4.02 (s, 3H), 3.78-3.8 (m, 2H), 3.47-3.48 (m, 2H), 3.39 (s, 3H), 3.1 1-3.14 (m, 2H); 1.90-2.19 (m, 5H), 1.47-1.92 (m, 12H), 0.90-0.94 (t, 3H) 实施例 2
4— (( ?)-8—环戊基 _7—乙基 _5—甲基 -6—氧代 -5,6,7,8-四氢 -喋啶 _2-基氨基 )-3-甲氧基 -Ν-ί(3 5 & 6 基) -苯甲酰胺


第一步
烯丙基-丙 -2-炔基-胺
冰浴下, 将丙烯胺 2a(225 mL, 3.0 niol)溶解于 100 mL 2 M氢氧化钠溶液中, 滴加溴丙炔 (89.1 mL, 1.0 mol), 升至室温, 搅拌反应 12小时。 减压浓缩反应液, 得到标题产物烯丙基-丙 -2-炔基-胺 2b, 粗产品直接用于下一步反应。
MS m/z (ESI):96.2 [M+l]
第二步
烯丙基-丙 -2-炔基-氨基甲酸叔丁酯
将粗品烯丙基-丙 -2-炔基-胺 2b(90.05 g, 0.95 mol),碳酸钾 (130.75 g, 0.95 mol) 和二碳酸二叔丁酯 (120 g, 0.55 mol)溶解于 200 mL二氯甲垸中,搅拌反应 12小时。 加入 20 mL水,二氯甲垸萃取 (50 mL> 3),合并有机相,饱和食盐水洗涤 (20 mL><3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所 得残余物,得到标题产物烯丙基-丙 -2-炔基-氨基甲酸叔丁酯 2c(76.97 g),产率: 41.7 第三步
5-氧代-六氢-环戊并 [c|吡咯 -2-羧酸叔丁酯 氮气氛下, 将烯丙基-丙 -2-炔基-氨基甲酸叔丁酯 2c(16.7 g, 0.86 mol)溶解于 100 mL乙二醇二甲醚中, 加入八羰基二钴 (29.4 g, 0.86 mol)禾卩 31 mL 7_K, 回流搅 拌反应 3小时。 加入 10 mL水, 减压浓缩反应液, 加入 100 mL乙酸乙酯, 100 mL 水和 50 mL的 1 M盐酸溶解残余物, 用乙酸乙酯萃取 (100 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓縮, 用硅胶柱色 谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 5-酮-六氢-环戊并 [c]吡咯 -2- 羧酸叔丁酯 2d(7.69 g), 产率: 40%。
第四步
(3aS,5/?,6ai0-5-羟基-六氢-环戊并 [c]吡咯 -2-狻酸叔丁酯 将 5-酮-六氢-环戊并 |c]吡咯 -2-羧酸叔丁酯 2d(l .8 g, 8 mmol , 根据现有文献 Tetrahedron, 49(23), 5047-54; 1993制备而得)溶解于 30 四氢呋喃中, 加入硼氢化 钠 (0.6 g, 16 mmol), 搅拌反应 1.2小时。 加入 30 mL饱和碳酸氢钠溶液, 用乙酸乙酯 萃取 (50 mLx3), 合并有机相,饱和食盐水洗涤 (50 mLx3),无水硫酸镁干燥, 过滤, 滤液减压浓^ I, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3aS,5 6ai?)-5-羟基-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 2e(1.64 g,黄色液体),产率: 90%。
MS m/z (ESI):228.1 [M+1 ]
第五步
aS,SR R)-5-甲磺酰基 -六氢 -环戊并 M吡咯 -2-羧酸叔丁酯 冰浴下, 将 (3aS,5 6ai?)-5-羟基-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 2e(1.64 g, 7.2 mmol)溶解于 30 mL二氯甲烷中, 加入甲磺酰氯 (0.85 mL, 11 mmol)和三乙胺 (2 mL, 14.4 mmol), 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯甲烷 萃取 (50 mLx3),合并有机相,饱和食盐水洗涤 (50 mLx3),无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3aS,5 6ai?)-5-甲磺酰基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 2f(1.76 g, 黄色液体), 产率: 80%。
MS m/z (ESI):305.9 [M+1]
(3a5,5 & 6ai?)-5-叠氮基 -六氢 -环戊并 [C]吡咯 -2-羧酸叔丁酯 将 (3a^5i?,6ai -5-甲磺酰基-六氢-环戊并 [c]吡咯- 2-羧酸叔丁酯 2f(] .76 g, 5.76 mmol)溶解于 20 mL N,N-二甲基甲酰胺中, 加入叠氮化钠 (0.94 g, 14.4 mmol), 80 °C下搅拌反应 4小时。 加入 20 mL水, 用乙酸乙酯萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3aS,5 & 6ai?)-5-叠氮基-六氢- 环戊并 M吡咯 -2-羧酸叔丁酯 2g(1.15 g, 白色固体), 产率: 79%。
MS m/z (ESI):253.0 [M+1]
第七步
(3aS,5 & 6ai?)- 5-叠氮基-八氢-环戊并 [c]吡咯盐酸盐 将 (3a5,5 & 6ai?)- 5-叠氮基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 2^0.62 g, 2.44 mmol)溶解于 10 mL二氯甲垸中,加入 10 mL 4 M 氯化氢的 1,4-二氧六环溶液,搅 拌反应 0.5小时。减压浓缩反应液, 得到标题产物 (3a^,5 & 6ai?)-5-叠氮基 - 、氢 -环戊 并 [c]吡咯盐酸盐 2h(0.47 g, 白色固体), 产率: 〗00%。
MS m/z (ESI): 153.1 [M+1]
第八步
(3 5 & 6ai?)-5-叠氮基 -2-甲基-八氢-环戊并 [C]吡咯 冰浴下, 将 (3a&5&6aT -5-叠氮基-八氢-环戊并 [c]吡咯盐酸盐 2h(0.45 g , 2.38 ramol)溶解于 10 mL乙腈中, 加入甲醛 (0.39 niL, 4.76 mmol)和三乙酰氧基硼氢化 钠 (1.5 ] g, 7.14 mmol), 搅拌反应 2小时。滴加饱和碳酸钠溶液至反应液 pH为 10, 用二氯甲烷萃取 (50 mLx3) , 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁 千燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 D纯化所得残余物, 得到标题产物 (3aS,5 & 6ai?)-5-叠氮基 -2-甲基-八氢-环戊并 [c]吡咯 2j(0.28 g, 淡黄色 液体), 产率: 71 %。
MS m/z (ESI): 167.1 [M+ l ]
第九步
(3a5 5 & 6aA)-2-甲基-八氢-环戊并 [c]吡咯 -5-基-胺 将 (3aS,5 & 6ai?)-5-叠氮基 -2-甲基-八氢-环戊并 [c]吡咯 2j(] 50 mg, 0.9 mmol)溶解 于 20 mL甲醇中, 加入 (30 mg, 10%)钯 /碳, 氢气氛下搅拌反应 2小时。 过滤, 滤 饼用甲醇洗涤 (30 mL) , 滤液减压浓缩, 得到标题产物 (3aS,5 & 6ai -2-甲基-八氢-环 戊并 [c]吡咯 -5-基-胺 2k(0.08 g, 淡黄色液体), 产率: 63 %。
MS m/z (ESI): 141.4 [M+l ]
第十步
4-((i?)-8-环戊基 -7-乙基 -5-甲基- 6-氧代 -5,6,7,8-四氢 -喋啶 - 2-基氨基 )-3-甲氧基 -N-((3aS,5 & 6ai?)- 2-甲基-八氢-环戊并 [c]吡咯 -5-基)-苯甲酰胺 将 (7?)-7-乙基 -8-环戊基 -2- (3-甲氧基-苯甲酸- 4-基-氨基) -5-甲基— 7,8-二氢 -5H-喋 啶 -6-酮 lm(242 mg, 0.57 mmol)和(9-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (183 mg, 0.57 mmol)溶解于 20 mL二氯甲烷中, 加入二异丙基乙胺 (0.21 mL, 1.25 mmol)和 10 mL (3aS,5 & 6a ?)-2-甲基-八氢-环戊并 [c]吡咯 -5-基-胺 2k(80 mg, 0.57 mmol)的二氯甲烷溶液, 搅拌反应 3小时。 加入 30 mL饱和碳酸氫钠溶液, 用二氯 甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标 题产物 4-((/?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )-3-甲氧 基 -N-((3aS,5 & 6ai?)- 2-甲基-八氢-环戊并 [c]吡咯 -5-基) -苯甲酰胺 2(193 mg , 白色固 体), 产率: 62 %。
MS m/z (ESI):548.4 [M+ l ]
'Η NMR(400MHz, CDCl3j ppm) δ 8.56-8.59 (d, 1 H), 7.72 (s, 1H), 7.62 (s, 1 H), 7.47 (s, 1 H), 7.23-7.26 (d, 1H), 5.96-5.98 (d, 1 H), 4.53-4.66 (m, 2H), 4.24-4.27 (d, 1H), 4.01 (s, 3H), 3.37 (s, 3H), 2.83 (s, 4H), 2.38 (s, 3H), 2.12-2.32 (m, 3H), 1.70-2.12 (m, 13H), 0.85-0.95 (t, 3H) 实施例 3
4_(( ?)-8-环戊基—7—乙基—5—甲基—6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨
基) -N-((T3a 5S.6a5V2 3-甲氧基-苯甲酰胺

第一步
(3aS,55*,6a/?)-5-氰基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 干冰浴下, 将 5-酮-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 2d(1.9 g, 8.4 mmol)和 2-对甲苯磺酰基 -乙腈 (1.97 g, 10.1 mmol)溶解于 20 mL二氯甲垸中, 滴加 25 mL 叔丁醇钾 (1.88 g, 16.8 mmol)的叔丁醇溶液, 搅拌反应 12小时。 加入 l O mL冰水 饱和碳酸氢钠溶液, 减压浓缩反应液, 用乙酸乙酯萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3a5,5 & 6ai? 5-氰基-六氢-环 戊并 [c]吡咯 -2-羧酸叔丁酯 3a(0.7 g, 黄色液体), 产率: 35 %。
MS m/z (ESI):259.1 [M+23]
第二步
(3a5,5 & 6ai?)-5-氨甲基-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 将 (3aS,5 & 6ai?)-5-氰基-六氢-环戊并 | c]吡咯- 2-羧酸叔丁酯 3a(0.3 g, 1.27 mmol) 溶解于 30 niL甲醇中, 加入兰尼镍 (0.50 g), 氢气氛下搅拌反应 6小时。 过滤, 滤 液减压浓缩, 得到标题产物 (3a&5&6ai?)-5-氨甲基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁 酯 3b(0.2 g, 淡黄色液体), 产率: 65 % , 直接用于下一步反应。
第三步
5-{ [4- )- 8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-3-甲氧基- 苯甲酰胺] -甲基 }-(3 5 & 6ai?)-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 将 (3a5,5 & 6aW)-5-氨甲基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 3b(190 mg, 0.8 mmol), (R)-7-乙基 -8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基) -5-甲基— 7,8-二氢 -5H- 喋啶- 6-酮] m(336 mg, 0.8 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯
(256 rag, 0.8 mmol)和二异丙基乙胺 (228 mg, 1.76 mmol)溶解于 40 mL二氯甲垸中, 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯甲烷萃取 (50 mLx3), 合 并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓縮, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物,得到标题产物 5- {[4-(( -8-环戊 基 -7-乙基 -5-甲基- 6-氧代 _5,6,7,8-四氢-喋啶 -2-基氨基 )-3-甲氧基 -苯甲酰胺] -甲 基 }-(3a&5 & 6a/?)-六氢-环戊并 M吡咯 -2-羧酸叔丁酯 3c(388 mg, 白色固体), 产率: 75 %。
MS m/z (ESI):648.6 [M+1]
第四步
4-((7 -8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨 基) -N-(((3ai?,5 & 6aS)-2-甲基-八氢-环戊并 [C]吡咯- 5-基) -甲基 )-3-甲氧基 -苯甲酰胺 将 5-{[4-((7?)-8-环戊基 -7-乙基 -5-甲基- 6-氧代 -5,6,7,8-四氢 -喋啶 - 2-基氨基 )-3-甲 氧基-苯甲酰胺]-甲基 }-(3aS,5 & 6ai?)-六氢-环戊并 [c]吡咯- 2-羧酸叔丁酯 3c(388 mg, 0.46 mol)溶解于 20 niL二氯甲烷, 加入 20 mL 6 M氯化氢的 1,4-二氧六环溶液, 搅 拌反应 0.5小时, 减压浓缩反应液, 加入甲醛 (0.1 mL, 0.92 mmo】)和两滴乙酸, 搅 拌反应 0.5小时后, 加入三乙酰氧基硼氢化钠 (292 mg, 1.38 mmol), 搅拌反应 2小 时。 加入氨水调节 pH至 9〜10, 用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食 盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以 洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((7?)-8-环戊基 -7-乙基- 5-甲基 -6-氧 代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-(((3ai?,5 & 6a5)-2-甲基-八氢-环戊并 [C]吡咯 -5-基) - 甲基) -3-甲氧基-苯甲酰胺 3(120 mg, 白色固体), 产率: 46.5 %。
MS m/z (ESI):562.5 [M+1]
'H NMR(400MHz, CDCl3;ppm) δ 8.54-8.56 (d, 1H), 7.68 (s, 1H), 7.59 (s, 1H), 7.44-7.45 (d, 1H), 7.25-7.27 (m, 1H), 6.17-6.2 (m, 1H), 4.5-4.54 (m, 1H), 4.2-4.23 (m, 1H), 3.80 (s, 3H), 3.40-3.43 (t, 2H), 3.33 (s, 3H), 2.80 (s, 4H), 2.36-2.42 (m, 4H), 2.26 (s, 2H), 2.13-2.18 (m, 1H), 1.98-2.02 (m, 1H), 1.66-1.89 (m, 10H), 1.50-1.54 (m, 2H), 0.81-0.92 (t, 3H) 实施例 4
4-〔(7?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N-((T3ai?,5 & 6a V5- )-甲基) -3-甲氧基 -苯甲


第一歩
(3a7?,6a5 螺 [l ,3,3a,4,6,6a-六氢环戊并 [c]吡咯- 5,2'-环氧乙烷] -2-羧酸叔丁酯 氮气氛下, 冰浴下, 将三甲基碘化亚砜 (293 mg, 〗.33 mmol)和氢化钠 (60 mg, 1.46 mmol)溶解于 2 mL二甲亚砜, 搅拌 1小时, 滴加 2 mL 5-酮-六氢-环戊并 [c]吡 咯 -2-羧酸叔丁酯 2d 0.3 g, 1.33 mmol)的二甲亚砜溶液, 搅拌 10分钟, 室温搅拌 反应 12小时。 加入 20 mL冰水中, 用乙酸乙酯萃取 (50 niLx3), 合并有机相, 饱 和食盐水洗涤 (50 mLx3), 无水硫酸钠干燥, 过滤, 滤液减压浓縮, 用硅胶柱色谱 法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3a ?,6a -螺 [l ,3,3a,4,6,6a-六氢 环戊并 [c]吡咯 -5,2'-环氧乙烷] -2-羧酸叔丁酯 4a(200 mg,无色油状物),产率: 63 %。 MS m/z (ESI):262.3 [M+23]
第二歩
(3ai?,6aS 5- (氨基 -甲基 )-5-羟基-六氢-环戊并 |c]吡咯 -2-羧酸叔丁酯
(3a ?,6a5)-螺 [l ,3,3a,4,6,6a-六氢环戊并 [c]吡咯 -5,2'-环氧乙烷] -2-羧酸叔丁酯
4a(0.3 g, 1.3 mmol)溶解于 30 mL乙醇中, 加入过量的氨水, 搅拌反应 12小时。 减压浓缩反应液, 得到标题产物 (3a ?,6a5 5- (氨基 -甲基 )-5-羟基-六氢-环戊并 [c]吡 咯 -2-羧酸叔丁酯 4b (黄色油状物), 直接用于下一步反应。
第二步
(3a ?,6aS)-5-{ [4-((i -8-环戊基 - 7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基-氨 基) -3-甲氧基 -苯甲酰胺] -甲基 }-5-羟基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 将 (3a 6aS)-5- (氨甲基) -5-羟基-六氢-环戊并 [c]吡咯- 2-羧酸叔丁酯 4b(193 mg, 0.75 mmol) , (i?)-7-乙基- 8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基) -5-甲基- 7,8-二氢 -5H-喋啶 -6-酮 lm(320 mg, 0.75 mmol) , < -苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼 酸酯 (241 mg, 0.75 mmol)和二异丙基乙胺 (213 mg, 1.65 mmol)溶解于 40 mL二氯 甲垸中, 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯甲垸萃取 (50
mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液 减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A 纯化所得残余物, 得到标题产物 (3a^;6aS)-5-{[4-(( i)-8-环戊基 -Ί-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基 -氨 基) -3-甲氧基 -苯甲酰胺] -甲基 }-5-羟基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 4c(0.42 g, 白色固体), 产率: 84%。
MS m/z (ESI):664.6 [M+l]
第四步
4-(W-8—环戊基— 7—乙基— 5_甲基 _6-氧代 _5,6,7,8—四氢 -噪啶 -2—基-氨基)— N-(((3ai?, 6aS)-5-羟基-八氢-环戊并 [C]吡咯 -5-基)-甲基) -3-甲氧基-苯甲酰胺
将 (3ai?,6aS)-5-{[4- ((i?)- 8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基- 氨基) -3-甲氧基 -苯甲酰胺] -甲基 }-5-羟基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 4c(420 mg, 0.63 mol)溶解于 20 mL二氯甲垸, 加入 20 mL 6 M氯化氢的 1,4-二氧六环溶 液,搅拌反应 0.5小时,滴加氨水至反应液 pH为 8〜9,用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 4-(( -8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基-氨 基 )-Ν- (((3 6Β5 5-羟基-八氢-环戊并 [c]吡咯 -5-基)-甲基) -3-甲氧基-苯甲酰胺 4(1(0.32 g, 白色固体), 产率: 90%。 直接用于下一步反应。
MS m/z (ESI):564.5 [M+l]
第五歩
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基-氨
基) -N-(((3ai?,5 & 6a )-5-羟基 -2-甲基-八氢-环戊并 [C]吡咯 -5-基)-甲基) -3-甲氧基-苯甲.
酰胺
将 4-((i?)- 8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基 -氨 基) -N-(((3ai?,6aS 5-羟基-八氢-环戊并 [C]吡咯 -5-基)-甲基) -3-甲氧基-苯甲酰胺 4d(318 mg, 0.56 mol)溶解于 60 mL二氯甲垸和水 (V:V=1 :1)混合溶剂中, 加入甲醛 (34 mg, 1.13 mmol)和两滴乙酸, 搅拌反应 0.5小时后, 加入三乙酰氧基硼氢化钠 (358 mg, 1.69 mmol), 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯 甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标 题产物 4-((7?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基-氨 基) -N-(((3a ?,5 & 6aS)-5-羟基 -2-甲基-八氢-环戊并 [c]吡咯 -5-基)-甲基) -3-甲氧基 -苯甲 酰胺 4(110 mg, 白色固体), 产率: 34%。
MS m/z (ESI):578.5 [M+l]
'H NMR(400MHz, CDC13, ppm) δ 8.52-8.45 (d, 1H), 7.68 (s, 1H), 7.59 (s, 1H), 7.47 (s: 1H), 7.33-7.35 (d, 1H), 6.74 (s, 1H), 4.49-4.57 (m, 1H), 4.20-4.23 (m, 1H), 3.97 (s, 3H), 3.58-3.59 (d, 2H), 3.53 (s, 3H), 2.79-2.84 (m, 4H), 2.37 (s, 3H), 2.15-2.19 (m, 3H), 1.96-2.02 (m, 3H), 1.66-1.90 (m, 10H), 0.89-0.94 (t, 3H)
实施例 5
4-^)-8-环戊基— 7—乙基— 5-甲基—6 -氧代 -5,6,7,8—四氢 -蝶啶 -2-基氨 基) -N-ffl3aW,5凡 6a6V5-羟基 -2-甲基-八氢-环戊并 W吡咯 -5-基) -甲基 )-3-甲氧基-苯

第一步
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基-氨 基) -N-(((3ai?,5 6a^-5-羟基 -2-甲基 氢-环戊并 M吡咯- 5-基) -甲基 )-3-甲氧基-苯
. 甲酰胺
将 4- ((i?)-8-环戊基 -Ί-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基 -氨 基) -N-(((3ai?,6aS 5-羟基-八氢-环戊并 [C]吡咯 -5-基) -甲基 )-3-甲氧基-苯甲酰胺 4d(318 mg, 0.56 mol)溶解于 60 mL二氯甲垸和水 (V:V=1 :1)混合溶剂中, 加入甲醛 (34 mg, 1.13 mmol)和两滴乙酸, 搅拌反应 0.5小时后, 加入三乙酰氧基硼氢化钠 (358 mg, 1.69 mmol), 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯 甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标 题产物 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基 -氨 基) -N-(((3a/?,5 6aS)-5-羟基 -2-甲基-八氢-环戊并 [C]吡咯 -5-基)-甲基) -3-甲氧基-苯 甲酰胺 5(80 mg, 白色固体), 产率: 25 %。
MS m/z (ESI):578.5 [M+l]
Ή NMR(400MHz, CDC13, ppm) δ 8.59-8.62 (d, 1H), 7.73 (s, 1H), 7.65 (s, 1H), 7.48-7.49 (s, 1H), 7.32-7.33 (d, 1H), 6.62-6.64 (m, 2H), 4.56~4.58 (m, 1H), 4.25-4.28 (m, 1H), 4.02 (s, 3H), 3.71-3.72 (d, 2H), 3.35 (s, 1H), 2.96 (s, 2H), 2.75-2.81 (m, 2H), 2.38-2.25 (m, 5H), 2.19-2.21 (m, 1H), 2.03-2.11 (m, 3H), 1.71-1.93 (m, 11H), 0.89-0.91 (t, 3H) 实施例 6
4-(W)-8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N- (1-甲基 -哌啶
-4-基) -3- (四氢-呋喃 -3-基-甲氧基) -苯甲酰胺

第一歩
3-轻基 -4-硝基-苯甲酸甲酯
氮气氛下, 将 3-羟基 -4-硝基-苯甲酸 6a(3.172 g, 17.32 mmol)溶解于 40 mL无 水甲醇中, 滴加二氯亚砜 (3.09 g, 25.98 mmol), 回流搅拌反应 2小时。 减压浓缩 反应液, 乙酸乙酯萃取 (50 mLx4),合并有机相,依次用饱和碳酸氢钠溶液 (30 mL)、 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产 物 3-羟基 -4-硝基 -苯甲酸甲酯 6b(3.304 g, 黄色固体), 产率: 96.7%。
MS m/z (ESI): 195.8 [M-l]
第二步
4-硝基 -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酸甲酯 氮气氛下, 干冰浴下, 将三苯基膦 (576 mg, 2.2 mmol)溶解于 10 mL无水四氢 呋喃中, 滴加 10 mL偶氮二甲酸二乙酯 (382 mg, 2.2 mmol)的四氢呋喃溶液, 搅拌 反应 30分钟后, 滴加 5 mL 3-羟基 -4-硝基 -苯甲酸甲酯 6b(300 mg, 1.52 mmol)的四 氢呋喃溶液, 搅拌反应 1.5分钟, 加入 (四氢 -呋喃 -3-基) -甲醇 (150 mg, 1.46 mmol), 室温搅拌反应 ]2小时。减压浓缩反应液, 用硅胶柱色谱法以洗脱剂体系 B纯化所 得残余物,得到标题产物 4-硝基 -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酸甲酯 6c(306 mg, 白色固体), 产率: 74.4%。
第三步
4-硝基 -3- (四氢 -呋喃 -3-基 -甲氧基)-苯甲酸
将 4-硝基 -3- (四氢-呋喃- 3-基-甲氧基) -苯甲酸甲酯 6e(240 mg, 0.85 mrnol)溶解 于 5 mL甲醇中, 加入】0 mL 1 M氢氧化锂的四氢呋喃溶液, 搅拌反应 12小时。 减压浓缩反应液, 二氯甲烷萃取 (50 mL), 滴加硫酸氢钾溶液至水相 pH至 2〜3, 乙 酸乙酯萃取 (100 mLx3), 合并有机相, 饱和食盐水洗涤 (30 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 4-硝基 -3- (四氢 -呋喃 -3-基 -甲氧基)-苯甲酸 6d(172 mg, 白色固体), 产率: 75.4 %。
MS m/z (ESI):265.9 [M-l ]
第四步
N-(l-甲基- (截啶 -4-基) -4-硝基 -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 将 4-硝基 -3- (四氢 -呋喃 -3-基 -甲氧基)-苯甲酸 6d(170 mg, 0.67 mmol), 1-甲基- 哌啶- 4-基-胺 (77 mg, 0.67 mmol), 0-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (215 mg, 0.67 mmol)和二异丙基乙胺 (25】 μί, 1.47 mmol)溶解于 40 mL二氯甲烷 中,搅拌反应 2小时。加入 30 mL饱和碳酸氢钠溶液,用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 N-(l-甲基-哌啶 -4- 基)—4-硝基 -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 6e(0.22 g, 淡黄色固体), 产率: 94 %。
MS m/z (ES1):364.3 [M+l]
第五步
4-氨基 - N-(l -甲基 -哌啶 -4-基) -3- (四氢-呋喃 -3-基-甲氧基) -苯甲酰胺 将 N-(l -甲基 -哌啶 -4-基) -4-硝基 -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 6e(220 mg, 0.61 mmol)溶解于 40 mL甲醇中, 加入 (50 mg, 10%)钯 /碳, 氢气氛下搅拌反 应 12小时。过滤,滤液减压浓縮,得到标题产物 4-氨基 -N-(l-甲基 -哌啶 -4-基) -3- (四 氢-呋喃- 3-基-甲氧基)-苯甲酰胺 6f(150 mg, 白色固体), 产率: 73.9%。
MS m/z (ESI):334.3 [M+l]
第六步
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N- (1-甲基 -哌啶
-4-基) -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 将 4-氨基 -N-(l-甲基 -哌啶 -4-基) -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 6f(150 mg, 0.45 mmol), (i?)-7-乙基 -2-氯- 8-环戊基 - 5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lj(132 mg, 0.45 mmol)和对甲苯磺酸 (137 mg, 0.72 mmol)溶解于 20 mL 4-甲基 -2-戊醇中, 回流搅拌反应 2小时。 滴加饱和碳酸氢钠溶液至反应液 pH为 8〜9, 用二氯甲垸 萃取 (50 mLx3),合并有机相,饱和食盐水洗涤 (50 mLx3),无水硫酸镁干燥,过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-(l-甲基 -哌啶
-4-基) -3- (四氢 -呋喃 -3-基-甲氧基) -苯甲酰胺 6(0.08 g, 白色固体), 产率: 30.5 %。 MS m/z (ES1):592.5 [M+l]
Ή NMR(400MHz, CDC】3, ppm) δ 8.10 (d, IH), 7.70 (s, I H), 7.50 (s, IH), 7.39 (s, 1H), 7.25〜7.27 (s, IH), 5.98-6 (d, IH), 4.37-4.47 (m, II- I), 4.19-4.21 (m, IH), 3.74-4.13 (m 7H), 3.33 (s, 3H), 2.91-2.94 (m, 3H), 2.37 (s, 3H), 2.06-2.29 (m, 7H), 1.68-1.91 (m, IH), 0.86-0.90 (t, 3H) 实施例 7
4-(0 -8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-N-(l-甲基-哌啶
-4-基) -3- -呋喃 -2-基-甲氧基) -苯甲酰胺


第一步
4-硝基 -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酸甲酯 氮气氛下, 干冰浴下, 将三苯基膦 (694 mg, 2.65 mmol)溶解于 20 mL无水四 氢呋喃中, 依次滴加 10 mL偶氮二甲酸二乙酯 (461 mg, 2.65 mmol)的四氢呋喃溶 液和 10 mL3-羟基 -4-硝基 -苯甲酸甲酯 6b(348 mg, 1.77 mmol)的四氢呋喃溶液, 搅 拌反应 10分钟, 加入 5 mL (四氢 -呋喃 -2-基) -甲醇 (198 mg, 1.94 mmol)的四氢呋喃 溶液, 搅拌反应 30分钟, 室温下搅拌反应 12小时。 减压浓缩反应液, 用硅胶柱 色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 4-硝基 -3- (四氢 -呋喃 -2-基- 甲氧基) -苯甲酸甲酯 7a(317 mg, 黄色固体), 产率: 64%。
MS m/z (ESI):282.1 [M+l]
第二步
4-硝基 -3- (四氢 -呋喃 -2-基 -甲氧基)-苯甲酸
将 4-硝基 -3- (四氢-呋喃 -2-基-甲氧基) -苯甲酸甲酯 7a(317 mg, l . l mmol)溶解于 30 mL甲醇中 , 加入 20 mh】 M氢氧化锂 (370 mg, 6.6 mniol)的四氢呋喃溶液, 搅 拌反应 2小时。加入 10 mL水,用二氯甲烷萃取 (50 mL:),滴加 1 M盐酸至水相 pH 为 3〜4, 乙酸乙酯萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mL), 无水硫 酸镁干燥,过滤,滤液减压浓缩,得到标题产物 4-硝基 -3- (四氢 -呋喃 -2-基-甲氧基) - 苯甲酸 7b(290 mg, 淡黄色固体), 产率: 99%。 直接用于下一歩反应。
第三步 . .
N-(l-甲基 -哌啶 -4-基) -4-硝基 -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 将 4-硝基 -3- (:四氢 -呋喃 -2-基-甲氧基 )-苯甲酸 7b 290 mg, 1.1 mmol), 1-甲基- 哌啶 -4-基-胺 (125 mg, 1 .1 mmol), < -苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (353 mg, 1.1 1¾1¾01)和二异丙基乙胺(313 mg, 2.42 mmol)溶解于 40 mL二氯甲烷中, 搅拌反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯甲烷萃取 (50 mLx3), 合 并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁千燥, 过滤, 滤液减压浓縮, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 N-(】-甲基 -哌啶 -4- 基) -4-硝基 -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7c(0.35 g,淡黄色固体),产率: 87.5
MS m/z (ESI):364.3 [M+l]
第四步
4-氨基 - N-(l-甲基 -哌啶 -4-基)- 3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 将 N-(l-甲基 -哌啶 -4-基)- 4-硝基 -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7c(422 mg, 1. ] 6 1^^01)溶解于40 0^甲醇中, 加入(50 ^¾, ] 0%)钯/碳, 氢气氛下搅拌反 应 12小吋。过滤,滤液减压浓缩,得到标题产物 4-氨基 -N-(l-甲基 -哌啶 -4-基) -3- (四 氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7d(307 nig, 白色固体), 产率 : 79 %。
MS m/z (ESI):334.3 [M+l]
第五步
W)-8—环戊基 -7—乙基 -5_甲基 -6—氧代—5,6,7,8—四氢-喋啶—2—基氨基) -N-(1 -甲基 -哌啶
-4-基) -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 将 4-氨基- N-(l-甲基 -哌啶 -4-基)- 3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7d(150 mg, 0.45 mmol), ((i?)-7-乙基 -2-氯 -8-环戊基 -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 lj(】33 mg, 0.45 mmol)和对甲苯磺酸 (137 mg, 0.72 mmol)溶解于 20 mL 4-甲基 -2-戊醇中, 回流搅拌反应 3小时。 滴加饱和碳酸钠溶液至反应液 pH为 9〜10, 用二氯甲烷萃 取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4- )-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -N- ( 甲基-哌啶 -4-基) -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7(0.12 g, 白色固体), 产率: 45 %。 MS m/z (ESI):592.5 [M+l]
Ή NMR(400MHz, CDC13, ppm) δ 8.57〜8.59 (d, 1H), 7.70-7.72 (d, 2H), 7.46 (s, 1H),
7.30 (s, IH), 5.94-6.03 (d, I H), 4.41-4.50 (m, IH), 4.32-4.38 (m, I H), 4.1 8-4.23 (m, IH), 4.13-4.16 (m, 2H): 3.94-4.03 (m, 2H), 3.80-3.90 (m, IH), 3.86 (s, 3H), 2.83-2.93 (d, 2H), 2.39 (s, 3H), 2.21-2.32 (t, 2B), 1 .96-2.18 (m, 7H), 1 .65-1.94 (m, 11H), 0.86-0.90 (t, 3H) 实施例 8
4-(W)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨 基) -N-(T4a ?,6 7 y)-2-甲基-八氢 -1H-环己垸 W吡啶 -6-基 3-甲氧基-苯甲酰胺 8-1
4— ( R)-8—环戊基 -7—乙基 甲基 -6—氧代— 5,6,7,8—四氢—蝶啶 _2—基氨 基) -N-((4a,6 & 7ai?)-2-甲基 -八氢 -1H-环己烷 kl吡啶 -6-基) -3-甲氧基-苯甲酰胺 8-2


第一步
(3aS,6a5)-2,5-二酮-八氢-并环戊二烯 -1,3,4,6-四羧酸四甲基酯 冰浴下, 将氢氧化钠 (6.4 g, 0.16 mol)溶解于 115 mL甲醇中, 滴加 1,3-二羧酸 二甲酯 -丙酮 (22.6 mL, 0.16 mol), 加热回流至盐全部溶解, 65 °C下快速滴加乙二 8a(12.85 g, 0.088 mol), 冷却至室温, 搅拌反应 12小时, 过滤, 用 50 mL甲醇 洗涤滤饼, 将滤饼溶解于 180 mL二氯甲垸和水 (V:V=5:4)混合溶剂中, 冰浴下滴加 1 M盐酸至反应液 pH为 6, 用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水 洗涤 (50 mLx3),无水硫酸镁干燥,过滤,滤液减压浓缩,得到标题产物 (3aS,6aS)-2,5-
二氧代 -八氢 -并环戊二烯 -],3,4,6-四羧酸四甲基酯 8b(20 g, 白色固体), 产率: 61 MS m/z(ESI):371.3 [M+l]
第二步
cis-四氢 -并环戊二烯 -2,5-二酮
将 (3aS,6a5 2,5-二氧代 -八氢 -并环戊二烯 -1,3,4,6-四羧酸四甲基酯 8b(6.75 g, 0.018 mol)溶解于 3.3 mL醋酸中, 加入 30 mL 1 M盐酸, 回流搅拌反应 3.5小时。 冷却至室温, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 减压浓缩, 所得残余物用 100 mL二氯甲烷溶解, 滴加饱和碳酸氢钠溶液至反应液 pH为 7左右, 无水硫酸 镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 (3a&6a5)-四氢 -并环戊二烯 -2,5-二酮 8c(2g, 白色固体), 产率: 80%。
第三歩
(4a/,7a^)-四氢 -1H-环戊并 [c]吡啶 -3,6-二酮 8d-l
(4a5,7aS 四氢-] H-环戊并 [c]吡啶 -3,6-二酮 8d-2 冰浴下,将 (3a5,6aS)-四氢-并环戊二烯- 2,5-二酮 8c(1.8 g, 13 mmol)溶解于 25 mL 浓盐酸中, 分批加入叠氮化钠 (1.1 g, 16.9 mmol), 室温下搅拌反应 24小时。 滴加 20%的氢氧化钠溶液至反应液 pH为 10〜11, 二氯甲烷萃取 (50 mLx3), 合并有机 相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶 柱色谱法以洗脱剂体系 C纯化所得残余物, 得到标题产物 (4a 7a -四氢 环戊 并 [c]吡啶 -3,6-二酮 8d-l和 Wa^JaS)-四氢- 1H-环戊并 [c]吡啶 -3,6-二酮 8d-2的混合物 (3 g, 白色固体), 产率: 100%。
MS m/z(ESI):154.1 [M+l]
第四步
(4a?,7ai?)-六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊烷] -3(4H)-酮 8c-l
(4a^,7a5 六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊垸] -3(4H)-酮 8e-2
将上述步骤所得的 (4a?,7ai?)-四氢 -1H-环戊并 [c]吡啶 -3,6-二酮 8d-l和 (4aS,7aS)- 四氢 -1H-环戊并 [c]吡啶 -3,6-二酮 8d-2的混合物 (1.2 g, 7.8 mmol), 乙二醇 (1.34 g, 21.5 mmol)和对甲苯磺酸 (24 mg, 0.13 mmol)溶解于 60 mL甲苯中, 回流搅拌反应 8小时。滴加饱和碳酸氢钠溶液至反应液 pH为 7左右, 二氯甲垸萃取 (50mL), 水 相再用二氯甲垸萃取 (50mLx3), 合并有机相, 饱和食盐水洗涤 (50mLx3), 无水硫 酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 C纯化所得残余 物, 得到标题产物 (4aW,7ai?)-六氢螺 [环戊并 [c]吡啶 -6,2'-[1 ,3]二氧戊垸 ]-3(4H)-酮 8e-l和 (4a5,7aS 六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊烷] -3(4H)-酮 8e-2的混合物 (U g, 白色固体), 产率: 70%。
MS m/z(ESI): 198.1 [M+l]
第五步
(4a7?,7ai?)-八氢螺 [环戊并 [c]吡啶 -6,2'-[l ,3]二氧戊垸] 8f-l
(4a&7a -八氢螺 [环戊并 [c|吡啶 -6,2'- [】 ,3]二氧戊烷] 8f-2 氩气氛下, 冰水浴下, 将氢化铝锂 (47 mg, 1.27 ramoj)溶解于 l O mL四氢呋喃 中, 加入上述步骤所得的 〗0 mL(4a7?,7a/?)-六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊 垸] -3(4H)-酮 8c-l 和 (435,7^5)-六氢螺 [环戊并 j c]吡啶 -6,2'-[U]二氧戊烷 ]_3(4H)-酮 8e-2的混合物 020 mg, 0.6 mmol)的四氢呋喃溶液, 搅拌反应 1 小时。 分批加入 0.047 L的水, 0.047 15 %的氢氧化钠溶液, 用二氯甲垸萃取 (50 mL><3), 合并有 机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓縮, 得到 标题产物 (4a^,7ai?)-八氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊垸] 8f-l和 (4a5,7a5 八氢 螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊垸] 8f-2的混合物 (] 50 mg,油状物),产率: 95 %。 直接投入下一歩反应。
、 . 卜
j¾八步
. (4aS,7ai -六氢螺 [环戊并 M吡啶- 6,2'-[1,3]二氧戊烷] -2(1H>羧酸叔丁酯 8g-l (4a/?,7a 六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊烷] -2(】H)-羧酸叔丁酯 8g-2 冰水浴下, 将上述步骤所得的 (4ai?,7a^)-八氢螺 [环戊并 [C]吡啶- 6,2'- [1,3]二氧戊 烷] 8f-l 和 (4aS,7aS 八氢螺 [环戊并 [c]吡啶 -6,2'-[1 ,3]二氧戊垸 ] 8f-2 的混合物 (100 mg, 0.54 mmol)溶解于 15 mL二氯甲烷中, 依次加入三乙胺 (109 mg, 1.08 mmol) 和 5 mL二碳酸二叔丁酯 (130 mg, 0.6 mmol)的二氯甲垸溶液, 搅拌反应 2小时。 滴加饱和碳酸氢钠溶液至反应液 pH为 8〜9, 用二氯甲垸萃取 (50 mLx3), 合并有 机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅 胶柱色谱法以洗脱剂体系 C纯化所得残余物,得到标题产物 (4a^7ai?)-六氢螺 [环戊 并 [c]吡啶 -6,2'-[1,3]二氧戊垸]- 2(1H)-羧酸叔丁酯 8g-l和 (4a/?,7a5 六氢螺 [环戊并 [c] 吡啶 -6,2'-[1,3]二氧戊烷] 羧酸叔丁酯 8g-2的混合物 100 mg, 无色油状物), 产率: 66%。
第七步
(4aS,7aT -6-氧代 -六氢 -1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8h-l
(4aS,7ai?)-6-氧代 -六氢 -1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8h-2
将上述步骤所得的 (4aS,7ai?)-六氢螺 [环戊并 W吡啶 -6,2'-[1,3]二氧戊垸] -2(1H)- 羧酸叔丁酯 8g-l和 (4a ?,7aS 六氢螺 [环戊并 [c]吡啶 -6,2'-[1,3]二氧戊垸] -2(1H)-羧酸 叔丁酯 8g-2的混合物 (620 mg, 2.2 mmol)溶解于 50 mL丙酮中, 加入 20 mL对甲 苯磺酸 (200 mg, 1.1 mmol)的丙酮溶液, 回流搅拌反应 20分钟。 滴加饱和碳酸氢 钠溶液至反应液 pH为 8〜9, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐 水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗 脱剂体系 C纯化所得残余物, 得到标题产物 (4a5,7ai?)-6-氧代 -六氢 -1H-环戊并 [c]吡 啶 -2(3H>羧酸叔丁酯 8h-l 和 (4a5,7a7?)-6-氧代-六氢-】 H-环戊并 [c]吡啶 -2(3H)-羧酸 叔丁酯 8h-2的混合物 (450 mg, 无色油状物), 产率: 85 %。
Ή NMR (400MHz, CDC13, ppra) δ 3.7 3.82 (in, 2H), 3.25 3.29 (d, 1 H), 2.9 3.0] (m, 1H), 2.32-2.40 (m, 4H), 2.08-2.18 (m, 2H), 1 .69-1 .71 (m, 1 H), 1.46-1.66 (s, 9H), 1.27 1.37 (m, 1 H)
第八歩
(4a ,67i,7ai?)-6-甲磺酰基 -六氢 -IH-环戊并 [c]吡啶 -2(3/i)-羧酸叔丁酯 8j-l
. (4ai?,6 & 73^-6-甲磺酰基-六氢-] H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8j-2 将上述步骤所得的 (4a5,7a/?)-6-氧代 -六氢 环戊并 [c]吡啶 -2(3H)-羧酸叔丁 酯 8h(22 mg 0.096 mmol)溶解于 10 mL四氢呋喃中, 加入硼氢化钠(10 mg 0.14 mmol), 搅拌反应 12小时。 减压浓缩反应液, 用 20 二氯甲垸溶液溶解残余物, 加入三乙胺 (40 μί, 0.29 1 ol)和甲磺酰氯 (20 \ih, 0.14 mmol), 搅拌反应 2小时。 加入饱和碳酸氢钠溶液调节 pH至 8~9, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法以洗脱剂体系 C 纯化所得残余物, 得到标题产物 (4aS,6 7ai )-6-甲磺酰基-六 氢 - 1 H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8j-l 和 (4a 6 & 7aS)-6-甲磺酰基-六氢-] H- 环戊并 M吡啶- 2(3H)-羧酸叔丁酯 8j-2的混合物 (28 mg, 无色油状物), 产率: 98
,H NMR(400MHz, CDC13, ppm) δ 5.12-5.18 (m, 1H), 3.61-3.68 (m, 1H), 3.42-3.51 (m, 1H), 3.35-3.48 (m, 1H), 3.1-3.14 (m, 1H), 2.98-3.06 (s, 3H), 2.19-2.26 (m, 2H), 2.07-2.17 (m, 2H), 1.79 1 .86 (m, 2H), 1.6 1.7 (m, 2H), 1.40-1.50 (s, 9H)
第九步
(4a 6 7aS 6-叠氮基-六氢- 1H-环戊并 [c]吡啶 -2(3H>羧酸叔丁酯 8k-l
(4aS,6 & 7a ^-6-叠氮基 -六氢 -1H-环戊并 M吡啶 -2(3H)-羧酸叔丁酯 8k-2 将上述步骤所得的 (4aS 6 ?,7aW)-6-甲磺酰基 -六氢 -1H-环戊并 [c]吡啶 -2(3/ -羧 酸叔丁酯 8j-l 和 (4a/?,6 & 7aS)-6-甲磺酰基 -六氢 -1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁 酯 8j-2的混合物 (32 mg 0.1 mmol)溶解于 10 mL N,N'-二甲基甲酰胺中, 缓慢加入 叠氮化钠 (20 mg 0.25 mmol), 70 80°C下搅拌反应 3.5小时。 滴加饱和碳酸氢钠 溶液至反应液 pH为 7 8, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐水 洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 B纯化所得残余物,得到标题产物 (4ai?,6 7a5)-6-叠氮基 -六氢 -1H-环戊并 [c] 吡啶 -2(3H)-羧酸叔丁酯 8k-l 和 (4a5,6 & 7ai?)-6-叠氮基-六氢-】 H-环戊并 [c]吡啶 -2(3/ )-羧酸叔丁酯 8k-2的混合物 (40 mg, 无色油状物), 产率: 100%
1H NMR(400MHz, CDC13, ppm) δ 4.05-4.08 (m, 1 Η), 3.56-3.61 (m, 1H), 3.39-3.40 (m, 2H), 3.01-3.10 (m, 1H), 2.2-2.29 (m, 2H), 1.7-1.83 (m, 5H), 1.45-1.51 (s, 9H), 1.20-1.31 (m, 1H)
第十步
(4ai?,6i?,7aS 6-氨基-六氢- 1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8m-l
(4a5 6S,7a ?)-6-氨基 -六氢 -1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8m-2
将上述步骤所得的 (4ai?,6 7a5)-6-叠氮基 -六氢-] 环戊并 [c]吡啶 -2(3H>羧酸 叔丁酯 8k-l和 (4a&6 & 7ai?)-6-叠氮基-六氢-] H-环戊并 [C]吡啶 -2(3H)-羧酸叔丁酯 8k-2的混合物 (30 mg, 0. ] 1 mmol)溶解于 ] 2 mL甲醇中, 加入 (20 mg, 10%)钯 /碳, 氢气氛下搅拌反应 2小吋。 过滤, 滤液减压浓缩, 得到标题产物 (4a 6 7aS 6 -氨 基-六氢 环戊并 M吡啶 -2(3H)-羧酸叔丁酯 8m-l和 (4a&6 & 7ai?)-6-氨基 -六氢 -1H- 环戊并 M吡啶- 2(3H>羧酸叔丁酯 8m-2的混合物 (28 mg, 淡黄色油状物), 产率: 100 %
MS m/z (ESI):241 .2 [M+1]
第十一步
(4a/?,6i?,7aS 6-[4-(((7?)- 8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶- 2-基氨 基)] -六氢 环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8n-l (4aS,6 & 7ai?)-6-[4- (((;?)- 8-环戊基 - 7-乙基 -5-甲基 -6-氧代- 5,6,7,8-四氢 -喋啶 - 2-基氨 基)] -六氢 - 1H-环戊并 M吡啶 -2(3//)-羧酸叔丁酯 8n-2 将上述歩骤所得的 (4a 6 ?,7a )-6-氨基-六氢 -1H-环戊并 [C]吡啶 -2(3H)-羧酸叔 丁酯 8m-l和 (4a5,6S,7ai?)-6-氨基 -六氢 -1H-环戊并 [C]吡啶 -2(3H)-羧酸叔丁酯 8m-2 的混合物 (28 mg, 0.12 mmol), ((i?)-7-乙基 -8-环戊基 -2-(3-甲氧基-苹甲酸 -4-基-氨 基) -5-甲基 -7,8-二氢- 5H-喋啶 -6-酮 lm(50 mg, 0.12 mmol), O苯并三氮唑 -Ν,Ν,Ν',Ν'- 四甲基脲四氟硼酸酯 (40 mg, 0.12 mmol)和二异丙基乙胺 (30 mg, 0.23 mmol)溶解 于 12 mL二氯甲烷中, 搅拌反应 2小时。 滴加饱和碳酸钠溶液至反应液 pH为 8〜 9, 用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫 酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余 物, 得到标题产物 (4ai?,6 7aS)-6-[4- (((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8- 四氢 -喋啶 -2-基氨基)] -六氢 环戊并 [C]吡啶 -2(3H)-羧酸叔丁酯 8n-l 和 (4aS 6 & 7ai?)-6-[4-(((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基)] -六氢 环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 811-2的混合物 (36 mg, 白色固体), 产率: 50 %。
MS m/z (ESI):648.6 [M+1]
. 第十二步
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代- 5,6,7,8-四氢-喋啶 -2-基氨 基) -N-((4a/?,6凡 7a¾-八氢 - 1H-环戊并 [c]吡啶 -6-基)- 3-甲氧基-苯甲酰胺 8o-l
4- ((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N-((4a5,6 & 7ai?)-八氢- 1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8o-2 将 (4& 6 ?,735)- 6-[4-(((i?)-8-环戊基 - 7-乙基 -5-甲基 -6-氧代- 5,6,7,8-四氢 -喋啶 -2- 基氨 基)] -六 氢 环 戊 并 [c] 吡 啶 -2(3H)-羧酸 叔 丁 酉旨 8n-l 和 (4aS,6S,7aR)-6-[4-(((R)-S-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨 基)] -六氢 -1H-环戊并 [c]吡啶 -2(3H)-羧酸叔丁酯 8n-2的混合物 (50 mg, 0.077 mmol)
溶解于 5 mL二氯甲垸中, 滴加 4 mL 6 M氯化氢的 ] , 4-二氧六环溶液, 搅拌反应 ]. 小时。 减压浓缩反应液, 得到标题产物 4-((( - 8-环戊基 - 7-乙基 -5-甲基 -6-氧代 - 5,6,7,8-四氢 -喋啶 -2-基氨基) -N-((4a 6 7a 八氢 -1J7-环戊并 [c]吡啶 -6-基) -3-甲氧 基-苯甲酰胺 80-1和 4-(((7?)- 8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基 氨基) -N-((4aS,6 & 7a -八氢 -1 环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8o-2 的混 合物 (80 mg, 白色固体), 产率: ] 00%。 直接投入下一步反应。
第十三步
4-(W-8—环戊基—7—乙基— 5—甲基—6—氧代 -5,6,7,8—四氢—喋啶—2—基氨 基) -N-((4ai?,6 7aS 2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8-1
4-(( - 8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氯 基)—Ν-((4 ;6&7^)- 2-甲基-八氢- 1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8-2 冰浴下, 将 4-(((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N- ((4a7?,6i?,7aS 八氢 - 1 H-环戊并 [C]吡啶- 6-基) -3 -甲氧基-苯甲酰胺 8o-l 和 4-(((7?)-8-环戊基 - 7-乙基 -5- 甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N-((4aS,6 & 7ai?)-八氢 环戊并 [C]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8o-2 的混合 物 (80 mg, 0.15 mmol)溶解于〗0 mL二氯甲烷和乙腈 (V:V=1 :1)的混合液中, 加入 37 %甲醛溶液 (0.023 mL, 0.29 mmol)和三乙酰氧基硼氢化钠(100 mg, 0.45 mmol), 室温下搅拌反应 2小时。 滴加饱和碳酸钠溶液至反应液 pH为 8〜9, 用二氯甲垸 萃取 (50 mLx3),合并有机相,饱和食盐水洗涤 (50 mLx3),无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((i?)-8-环戊基 - 7- 乙基 -5- 甲 基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基)- N-((4a^,6i?,7aS -2-甲基-八氢 -1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8-1和 4-((R)-S-环戊基 -7- 乙基 -5- 甲 基 -6-氧代 -5,6,7,8- 四氢 -喋啶 -2-基氨 基)- N-((4aS,6 & 7a/?)-2-甲基 -八氢 -1H-环戊并 [C]吡啶 -6-基) -3-甲氧基-苯甲酰胺 8-2 的混合物 (30 mg, 白色固体), 产率: 37 %。
MS m/z (ESI):562.5 [M+l]
1H NMR(400MHz, CDC13, ppm) δ 8.52-8.55 (d, 1H), 7.67 (s, 1H), 7.59 (s, 1H), 7.43 (s: 1H), 7.27 (s, 1H), 6.32-6.34 (d, 1H), 4.59-4.61 (m, 1 H), 4.20-4.23 (m, 1 H), 3.97 (s, 3H), 3.33 (s, 3H), 2.21-2.82 (m, 4H), 2.42-2.68 (m, 4H), 2.30-2.33 (m, 3H), 2.02-2.18 (m, 3H), 1.82-2.01 (m, 1H), 1.65-1.78 (m, 11H), 0.85-0.89 (t, 3H) 实施例 9
4-(( ?)-8-环戊基 -7-乙基- 5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-(Y( i?,8aS)-六 氢-吡咯并 r2J-c|「l,41噁嗪- 3-基)-甲基) - 3-甲氧基-苯甲酰胺
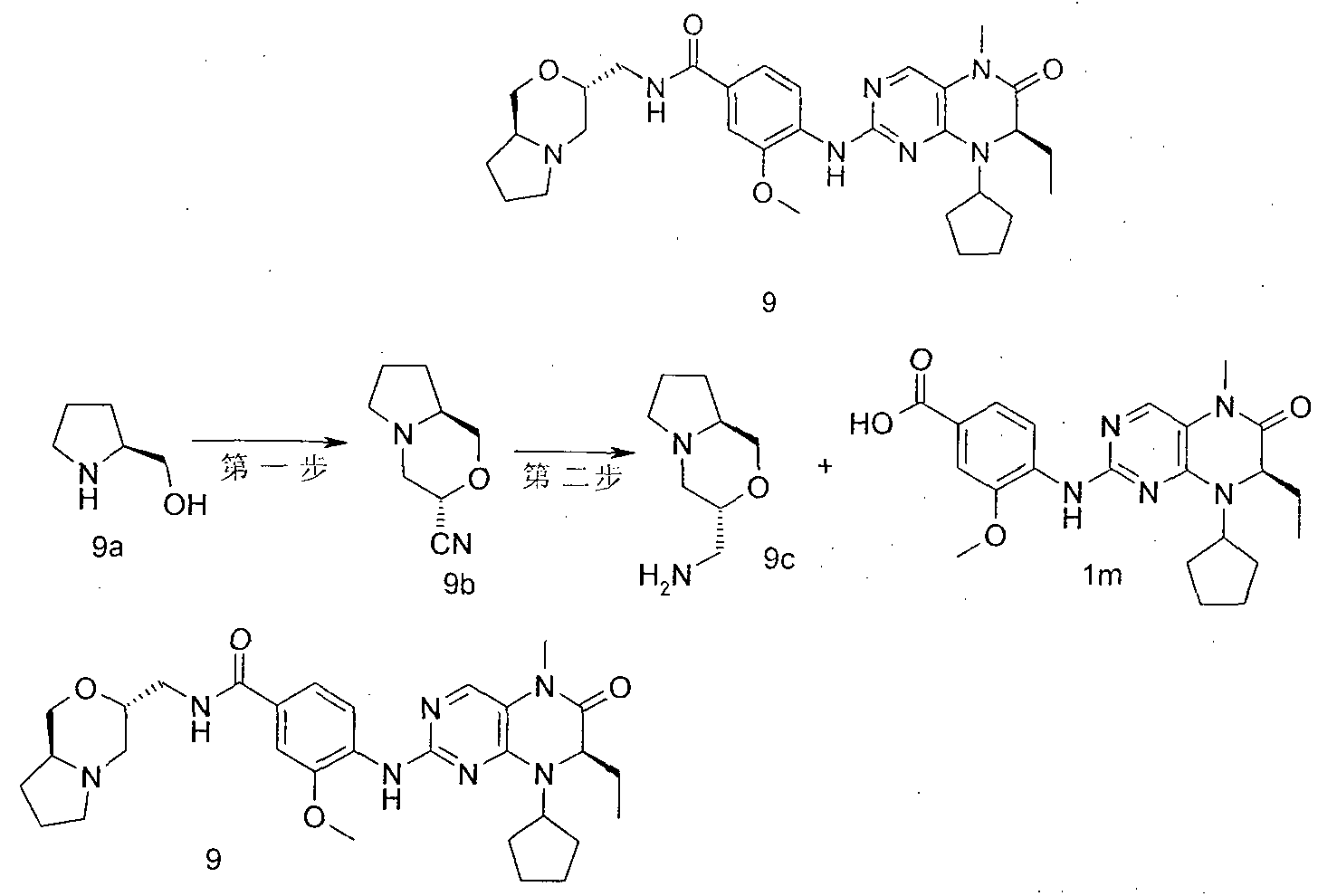
. 第一步
(3 & 8a )-六氢-吡咯并 [2,l-c][l,4]噁嗪 -3-腈 氩气氛下, 将 ((5 吡咯垸 -2-基 -甲醇 9a(1.29 g, 12.7 mmol)溶解于 120 mL四氢 呋喃中, 加入 2-氯-丙烯腈 (1.11 g, 12.7 mmol)和叔丁醇钾 (2.14 g, 19.06 mmol), 搅 拌反应】0分钟。 过滤, 滤液减压浓缩, 加入 50 mL乙酸乙酯, 依次用饱和碳酸氢 钠溶液 (30 mL)、饱和食盐水洗涤 (30 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3 & 8aS 六氢-吡 咯并 [2,l-c][l ,4]噁嗪 -3-腈 9b(357 mg, 白色固体), 产率: 18.5 %。
MS m/z (ESI): 153.1 [M+1]
第二步
((3&8a5 )-六氢 41比咯并 [2J - C][】 ,4]噁嗪 -3-基) -甲胺 将 (3 & 8aS 六氢-吡咯并 [2,1 - C][l,4]噁嗪 -3-腈 9b(391 mg, 2.57 mmol)溶解于 30 mL甲醇中,依次加入一滴氨水和兰尼镍 (0.5 g),氢气氛下搅拌反应 18小时。过滤, 滤液减压浓缩, 得到标题产物 ((3&8a5 )-六氢-吡咯并 [2,1-C][1 ,4]噁嗪 -3-基) -甲胺 9c(200 mg, 无色油状物), 产率: 50%。
MS m/z (ESI): 157.2 [M+1 ]
第三步
4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代- 5,6,7,8-四氢-喋啶 -2-基氨基) -N-(((3 8aS)-六 氢-吡咯并 [2,1-C][1,4]噁嗪 -3-基) -甲基 )- 3-甲氧基-苯甲酰胺 将 ((3&8a5)-六氢-吡咯并 [2,l-c][l,4]噁嗪 -3-基) -甲胺 9c(43 mg, 0.28 mmol)溶解 于 60 mL二氯甲垸中,加入 ((i?)-7-乙基 -8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基)- 5- 甲基 -7,8-二氢- 5H-喋啶 -6-酮 lm(117 mg, 0.28 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四 甲基脲四氟硼酸酯 (88 mg, 0.28 nimol)和二异丙基乙胺 (89 mg, 0.69 mmol), 搅拌 反应 2小时。 加入 30 mL饱和碳酸氢钠溶液, 用二氯甲烷萃取 (50 mLx3), 合并有
机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用二 氯甲垸和正己垸重结晶所得残余物, 得到标题产物 4-(( -8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 - 2-基氨基) -N_(((3 8aS)-六氢 - P比咯并 [2J -c][l,4]ii ;嗪 -3- 基)-甲基) -3-甲氧基 -苯甲酰胺 9(76 mg, 白色固体), 产率: 49%。
S m/z (ESI):564.3 [M+1]
]Η ΝΜΚ(400ΜΗζ, CDC13, ppm) δ 8.60-8.62 (d, 1H), 7.73 (s, 1H), 7.66 (s, 1H), 7.36 (s: 1H), 7.31~7.34 (d, 1H), 6.68-6.77 (s, lH), 4.58-4.60 (m, 1H), 4.54-4.56 (m, 1H), 4.14-4.52 (m, 2H), 4.06 (s, 3H), 3.80-4.03 (d, 1H), 3.72 (m, 2H), 3.37 (s, 3H),
3.17-3.21 (m, 4H), 2.84-2.86 (m, 1H), 2.38 (m, 1H), 1.91-2.19 (m, 5H), 1.76-1.87 (m, 4H), 1.61〜1.74 (m, 4H), 0.92-0.98 (t, 3H) 实施例 10
4-(( -8-环戊基 - 7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )-3-甲氧基

第一步.
(3a 5 6a5)-5-苄氨-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 冰浴下, 将 5-酮-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 2d(3.37 g, 15 mmol), 苄 胺 (1.6 g, 15 mmol), 醋酸 (0.9 g, 15 mmol)溶解于 60 mL二氯甲垸中, 搅拌反应 0.5 小时, 加入三乙酰氧基硼氢化钠 (6.4 g, 30 mmol), 室温搅拌反应 12小时。 加入 50 mL饱和碳酸氢钠溶液, 二氯甲垸萃取 (150 mL), 有机层用饱和食盐水洗涤 (30
mLx3), 无水硫酸镁千燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A 纯化所得残余物, 得到标题产物 (3a/i,5凡 6a5 5-苄氨-六氢-环戊并 |c]吡咯- 2-羧酸叔 丁酯: 10a(4.7g, 白色固体), 产率: ]00%。
MSm/z (ESI):317.3 [ +l]
一歩
(3aW,5 6a5)-5-氨基-六氢-环戊并 |c]吡咯 -2-羧酸叔丁酯 将 (3a 5i?,6aS 5-苄氨-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 10a(4.7 g, 14.8 mmol), 醋酸 (l g, 〗4.8 mmol)溶解于 100 mL甲醇中, 加入 (500 mg, 10%)钯 /碳, 氢气氛下搅拌反应 12小时。 过滤, 减压浓缩反应液, 加入 50 mL饱和碳酸氢钠溶 液, 二氯甲垸萃取 (150 mL), 有机层用饱和食盐水洗涤 (30 mLx3), 无水硫酸镁干 燥, 过滤, 滤液减压浓缩, 得到标题产物 (3ai?,5 6aS 5-氨基-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 10b(1.7 g, 白色固体), 产率: 51.5%。
第三步
(3a?,5 ?,6aS 5-苄氧基羰基氨基-六氢-环戊并 [C]吡咯 -2-羧酸叔丁酯 将 (3ai?,5 6∑uS 5-氨基-六氢-环戊并 [c]吡咯- 2-羧酸叔丁酯 10b(1.7 g, 7.5 mmol) 溶解于 60 mL二氯甲垸中, 加入苄氧基酰氯 (1.41 g, 8.26 mmol)和三乙胺 (1.52 g, 15 mmol),搅拌反应 3小时。加入 50 mL饱和碳酸氢钠溶液,二氯甲垸萃取 (]00 mL), 有机层用饱和食盐水洗涤 (30 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用 硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (3ai?,5i?,6aS 5-苄氧 基羰基氨基-六氢-环戊并 M吡咯 -2-羧酸叔丁酯 10c(1.98 g, 无色粘稠液体), 产率: 73.3%。
第四步
(3a 5i?,6a5 (八氢-环戊并 [c]吡咯 -5-基) -羧酸苄酯 将 (3ai?,5 6a5 5-苄氧基羰基氨基-六氢-环戊并 [c]吡咯 -2-羧酸叔丁酯 10^1.96 g, 5.44 mmol)溶解于 10 mL 1,4-二氧六环中, 加入 2M的盐酸 10niL, 50°C搅拌反 应 ί2小时。 减压浓缩反应液, 滴加 2Μ的氢氧化钠溶液至反应液 ρΗ为 12, 二氯 甲烷溶液萃取 (50mL), 饱和食盐水洗涤 (50mLx3), 无水硫酸镁干燥, 过滤, 滤液 减压浓缩,得到标题产物 (3a 5 6a5 (八氢-环戊并 [C]吡咯 -5-基) -羧酸苄酯 10d(635 mg, 淡黄色油状物), 产率: 45%。
MSm/z (ESI):261.2 [M+l]
第五步
3ai?,5凡 6aSH2-甲基-八氢-环戊并 [C]吡咯 -5-基) -羧酸苄酯 冰浴下, 将 (3ai?,5 6aS)-(八氢-环戊并 M吡咯 -5-基) -羧酸苄酯 10d(635 mg, 2.4 mmol)溶解于 20 mL乙腈和水 (V:V=9:1)的混合液中, 加入 37%甲醛溶液 (110 mg, 3.66 mmol)和三乙酰氧基硼氢化钠 (1.52 g, 7.2 mmol), 搅拌反应 2小时。 减压浓缩 反应液, 加入 50 mL饱和碳酸氢钠溶液, 二氯甲垸萃取 (100 mL), 有机层用饱和
食盐水洗涤 (30 mLx3),, 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法 以洗脱剂体系 A纯化所得残余物,得到标题产物 (3ai?,5 6a5 (2-甲基-八氢-环戊并 [c]吡咯 _5 -基) -羧酸苄酯 10e(559 mg, 黄色油状物), 产率: 85 %
1
i¾八少
(3a 5 6a5 2-甲基-八氢-环戊并 [C]吡咯- 5-基-胺 将 (3a 5i?,6aS (2-甲基-八氢-环戊并 [c|吡咯 -5-基) -狻酸苄酯 10e(550 mg 2.0 mmol)溶解于 20 mL甲醇中, 加入 (55 mg 10%)钯 /碳, 氢气氛下搅拌反应 1小时。 过滤, 滤液减压浓缩, 得到标题产物 (3a/?,5 6 〕-2-甲基-八氢-环戊并 [C]吡咯 -5-基- 胺 10f(246 n]g, 无色油状物), 产率: 87.9%
MS m/z (ES1): 141.2 [M+1]
第七步
4-(((/?)- 8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-3-甲氧基 -N-((3a 5 6aS 2-甲基-八氢-环戊并 [c]吡咯 -5-基) -苯甲酰胺 将 ((i -7-乙基 -8-环戊基 -2-(3-甲氧基-苯甲酸 -4-基-氨基) -5-甲基 -7,8-二氢 -5H-喋 啶 -6-酮 lra(165 rag, 0.39 mmol)溶解于 15 mL二氯甲垸中, 加入 (3a ?,5凡 6aS 2-甲 基-八氢-环戊并 [c]P比咯 -5-基-胺 10f(60 mg 0.42 mmol), < -苯并三氮唑 -Ν,Ν,Ν',Ν'- 四甲基脲四氟硼酸酯 (135 mg 0.42 mmol)和二异丙基乙胺 (15】 mg 1.17 mmol), 搅拌反应 2小时。 依次用饱和氯化铰溶液 (30 mL;)、 饱和碳酸氢钠溶液 (30 mL)、 饱 和食盐水洗涤 (30 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱 法以洗脱剂体系 A纯化所得残余物,得到标题产物 4-((( ?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代- 5,6,7 8-四氢 -喋啶 -2-基氨基 )-3-甲氧基 -N-((3a凡 57?,6aS 2-甲基-八氢-环戊并 [c]吡咯 -5-基) -苯甲酰胺: 10(47 mg, 白色固体), 产率: 22.1 %
MS m/z (ES1):548.3 [M+l ]
Ή NMR(400MHz, CDC13, ppm) δ 9.52 (s, 1H), 8.52 8.54 (d, 1H), 7.70 (s, 1H), 7.62 (s: 1 H), 7.57 (s, I H), 7.28-7.29 (d, 1H), 4.65-4.69 (m, 1H), 4.53-4.61 (m, 1H), 4.22-4.25 (m, 1H), 4.00 (s, 3H), 3.34 (s, 3H), 2.84-2.92 (m, 2H), 2.73-2.83 (m, 2H), 2.47 (s, 3H), 2.28-2.3 (m, 2H), 2.1-2.22 (m, 2H), 1.6-2.08 (m, 12H), 0.88-0.91 (t, 3H) 实施例 11
4—(W)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) - N-((4a ,6i?,7a/?V2-甲基 -八氢 -1H-环戊并 W吡啶 -6-基 3-甲氧基-苯甲酰胺 11-1
4-((i?)-8-环戊基 -7-乙基 -5-甲基- 6-氧代 -5,6 7 8-四氢-喋啶 -2-基氨基) - Ν-((4 ? 6<$ 7^-2-甲基-八氢 -1H-环戊并『c|吡啶 -6-基) -3-甲氧基-苯甲酰胺 11-2

第一步
(4aS,6i?,7ai?)-6-苄氨基 -六氢 -1H-环戊并 [c]吡啶 -2-羧酸叔丁酯
(4a 6 & 7a5)-6-苄氨基 -六氢 -1H-环戊并 [C]吡啶- 2-羧酸叔丁酯 lla-2
冰浴下, 将实施例 8 第七步所得的 (4a5,7ai? 6-氧代 -六氢 -1H-环戊并 [c]吡啶 -2(3//)-羧酸叔丁酯.8h-l 和 (4aS",7ai?)-6-氧代-六氢- 1H-环戊并 [c]吡啶- 2(3H)-羧酸叔 丁酯 8h-2的混合物 (1.1 g, 4.6 mniol)和苄胺 (492 mg, 4.6 nimol)溶解于 50 mL二氯 甲烷中, 加入醋酸 (276 mg, 4.6 mmol)和三乙酰氧基硼氢化钠 (1.95 g, 9.2 mmol), 室温搅拌反应 12小时。 滴加饱和碳酸钠溶液至反应液 pH为 9左右, 二氯甲垸萃 取 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂 体系 A纯化所得残余物, 得到标题产物 (4a ,6 7a7?)-6-苄氨基 -六氢 -1H-环戊并 [C] 吡啶 -2-羧酸叔丁酯 l l a-l 和 (4aW,6S,7aS)-6-苄氨基 -六氢 环戊并 [c]吡啶 -2-羧酸 叔丁酯 lla-2 的混合物 (1.25 g, 淡黄色油状物), 产率: 83 %。
MS m/z (ESI):331.3 [M+1]
第二步
(43^,6 7aA)-6-氨基 -六氢 -】H-环戊并 [c]吡啶 -2-羧酸叔丁酯 llb-1 (4a/?,6 & 7a5 6-氨基 -六氢 -1H-环戊并 [c]吡啶 -2-羧酸叔丁酯 llb-2 将上述歩骤所得的 (4a5,6 7ai?)- 6-苄氨基 -六氢 -1H-环戊并 | c]吡啶- 2-羧酸叔丁 酯 ll.a-1和 (4ai?,6 & 7a5)-6-苄氨基 -六氢 - ]H-环戊并 [c]吡啶- 2-羧酸叔丁酯 lla-2 的混 合物 (1 .25 g, 3.78 mmol)溶解于 20 mL甲醇中, 加入 (150 mg, 10%)钯 /碳, 氢气氛 下搅拌反应 12小时。 过滤, 滤液减压浓缩, 得到标题产物 (4a5,6 7a/?)- 6-氨基-六 氫 环戊并 [c]吡啶 -2-羧酸叔丁酯 llb-1和 (4ai?,6S,7a 6-氨基 -六氢 -1 H-环戊并 [c] 吡啶- 2-羧酸叔丁酯 llb-2 的混合物 (910 mg, 淡黄色油状物), 产率: 100%。
MS m/z (ESI):241.2 [M+l]
第三歩
(4a5,6i?,7aii)-6-苄氧基碳基氨基-六氢- 1H-环戊并 [C]吡啶 -2-羧酸叔丁酯 llc-1 (43 6&735)-6-苄氧基碳基氨基-六氢-^-环戊并|^]吡啶-2-羧酸叔丁酯 llc-2 冰浴下,将上述步骤所得的 (4aS,6 7ai?)- 6-氨基 -六氢 -】H-环戊并 [c]吡啶- 2-羧酸 叔丁酯 llb-1 和 (4a 6 & 7aS)-6-氨基 -六氢 环戊并 [c]吡啶 -2-羧酸叔丁酯 llb-2 的混合物 (909 mg, 3.78 mniol)溶解于 25 mL二氯甲垸中,加入三乙胺 (763 mg, 7.56 mmol)和氯甲酸苄酯 (707 mg, 4.16 mmol), 搅拌反应 2小时。 滴加饱和碳酸钠溶液 至反应液 pH为 8〜9, 用二氯甲烷萃取 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液 减压浓縮, 用硅胶柱色谱法以洗脱剂体系 A 纯化所得残余物, 得到标题产物 (4a5,6 7a/ -6-苄氧基碳基氨基 -六氢 -1H-环戊并 M吡啶 -2-羧酸叔丁酯: llc-1和 (4a ?,6 & 7aS)-6-苄氧基碳基氨基-六氢 -1 H-环戊并 [c]吡啶 -2-羧酸叔丁酯 llc-2的混合 物 (520 mg, 无色油状物), 产率: 37 %。
第四步
(4a5,6i?,7ai?)-八氢 环戊并 [c]吡啶 -6-基-羧酸苄酯 盐酸盐 lld-1
(4a/?,6&7aS 八氢 -1H-环戊并 [c]吡啶- 6-基-羧酸苄酯 盐酸盐 lld-2
将上述步骤所得的 (4aS,6i?,7ai?)-6-苄氧基碳基氨基 -六氢 -1H-环戊并 [C]吡啶 -2- 羧酸叔丁酯 llc-1和 (4a 6 & 7α^-6-苄氧基碳基氨基-六氢 -1H-环戊并 [C]吡啶 -2-羧酸 叔丁酯 llc-2的混合物 (513 mg, 1.37 mmol)溶解于 6 mL二氯甲垸中, 加入 6 mL 6 M 氯化氢的 1,4-二氧六环溶液, 搅拌反应 30 分钟。 减压浓缩, 得到标题产物 (4 S,6 ?,7a ?)-八氢 -1H-环戊并 [c]吡啶 -6-基-羧酸苄酯 盐酸盐 lld-1 和 (4a ?,6 & 7aS 八氢 -1H-环戊并 [c]吡啶 -6-基-羧酸苄酯 盐酸盐 lld-2 的混合物 (430 mg, 白色粘 稠液体), 产率: 100%, 直接投入下一步。
MS m/z (ESI):275.2 [M+l]
第五步
(4aS,6 7a -2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基-羧酸苄酯 lle-1
(4a ?,6 & 7a5 2-甲基 -八氢 环戊并 [c]吡啶 -6-基-羧酸苄酯 lle-2
冰浴下, 将上述歩骤所得的 (4a&67?,7a7?)-八氢- 1H-环戊并 [c]吡啶- 6-基-羧酸苄酯 盐 酸盐 lld-1和 (4a/?,6 & 7aS 八氢 环戊并 [C]吡啶- 6-基 -羧酸苄酯 盐酸盐 lld-2的 混合物 (425 mg, 1 .37 mmol)溶解于 20 mL乙腈和水 (V:V=1 :1)的混合液中, 加入三 乙酰氧基硼氢化钠 (871 mg, 4.1 1 mmol), 室温搅拌反应 12小时。 滴加饱和碳酸钠 溶液至反应液 pH为 9左右, 用二氯甲垸萃取 (50 mLx3), 依次用水 (50 mL), 饱和 食盐水洗涤 (30 mLx3) , 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 (4aS,6凡 7ai?)-2-甲基-八氢- 1H-环戊并 [C]吡啶 -6-基-羧酸苄酯 lle-1和 (4a ?,6 & 7a5>2- 甲基-八氢 -1H-环戊并 吡啶 -6-基-羧酸苄酯 lle-2的混合物 (374 mg, 淡黄色油状 物), 产率: 94 %, 直接投入下一歩。
MS m/z (ESI) :289.2 [M+l]
第六步
(4a5,6i?,7aW)- 2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-氨基 llf-1 ( aR,6S aS)-2-甲基 -八氢 - 1 H-环戊并 [c]吡啶 -6-氨基 llf-2 将上述步骤所得的 (4aS,6i?,7a7?)-2-甲基 -八氢 -1H-环戊并 |c]吡啶 -6-基-羧酸苄酯 lle-1和 (4ai?,6 & 7aS)-2-甲基-八氢- 1H-环戊并 [c]吡啶 -6-基-羧酸苄酯 lle-2的混合物 (374 mg, 1.29 ^11101)溶解于25 ^^甲醇中, 加入 (50 mg, 10%)钯 /碳, 氢气氛下搅 拌反应 2小时。 过滤, 滤液减压浓缩, 得到标题产物 (4aS,6凡 7ai? 2-甲基 -八氢 -1H- 环戊并 [c]吡啶 -6-氨基 llf-1 和 (4a ?,6 & 7aS 2-甲基-八氢- 1H-环戊并 [c]吡啶 -6-氨基 llf-2的混合物 (198 mg, 白色固体), 产率: 100 % , 直接投入下一歩。
MS m/z (ESI): 155.2 [M+l]
第七步
4—((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) - N-((4a5,6 7ai?)- 2-甲基-八氢-] H-环戊并 [c]吡啶- 6-基) -3-甲氧基-苯甲酰胺 11-1.
4— ((i?)—8-环戊基 -7-乙基 -5-甲基- 6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) - N- ((4a 6 & 7a5 2-甲基 -八氢 环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 11-2 将上述步骤所得的 (4aS,6i?,7a/ -2-甲基 -八氢 环戊并 [c]吡啶 -6-氨基 llf-1和 (4a/?,6&7a^-2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-氨基 llf-2 的混合物 (60 mg, 0.39 mmol), ((i?)-7-乙基 -8-环戊基 - 2-(3-甲氧基-苯甲酸 -4-基-氨基)- 5-甲基 -7,8-二氢 -5H- 喋啶 -6-酮 lm(182 mg, 0.43 mmol), 0-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (125 mg, 0.39 mmol)和二异丙基乙胺 (150 mg, 1.17 mmol)溶解于 20 mL二氯甲垸 中, 搅拌反应 2小时。 滴加饱和碳酸钠溶液至反应液 pH为 9左右, 用二氯甲垸萃 取 (50 mLx3), 依次用饱和氯化铵溶液 (30 mL)、 饱和碳酸氢钠溶液 (30 mL)、 饱和 食盐水洗涤 (30 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法 以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基:) -N-((4aS,6i?,7ai?)-2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 11-1和 4-((i?)-8-环戊基 -7-乙基 -5-甲基 -6-氧代 -5,6,7,8-四
氢-喋啶 -2-基氨基) -N-((4ai?,6 & 7a5 2-甲基 -八氢 -1H-环戊并 |c]吡啶 -6-基) -3-甲氧基- 苯甲酰胺 ; Π-2的混合物 (70 mg, 白色固体, 混合物), 产率: 32%。
MS m/z (ESI):562.5 [M+l]
IH NMR(400MHz, CDC13, ppm) δ 8.52-8.55 (d, 1H), 7.67 (s, 1H), 7.59 (s, 1H), 7.43 (s: 1H), 7.27 (s, 1H), 6.32-6.34 (d,】H), 4.40-4.60 (m, 2H), 4.20-4.23 (m, 1H), 3.95 (s, 3H), 3.35 (s, 3H), 2.52-2.67 (m, 2H), 2.26-2.4 (m, 6H), 2.11-2.23 (m, 3H), 1.99-2.10 (m, 2H), 1.61-1.9 (m, 11H), 1.45-1.55 (ni, 1H), 0.98-1.06 (t, 3H) 实施例 12
4-07?)- 7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-(T3凡 8aS 六
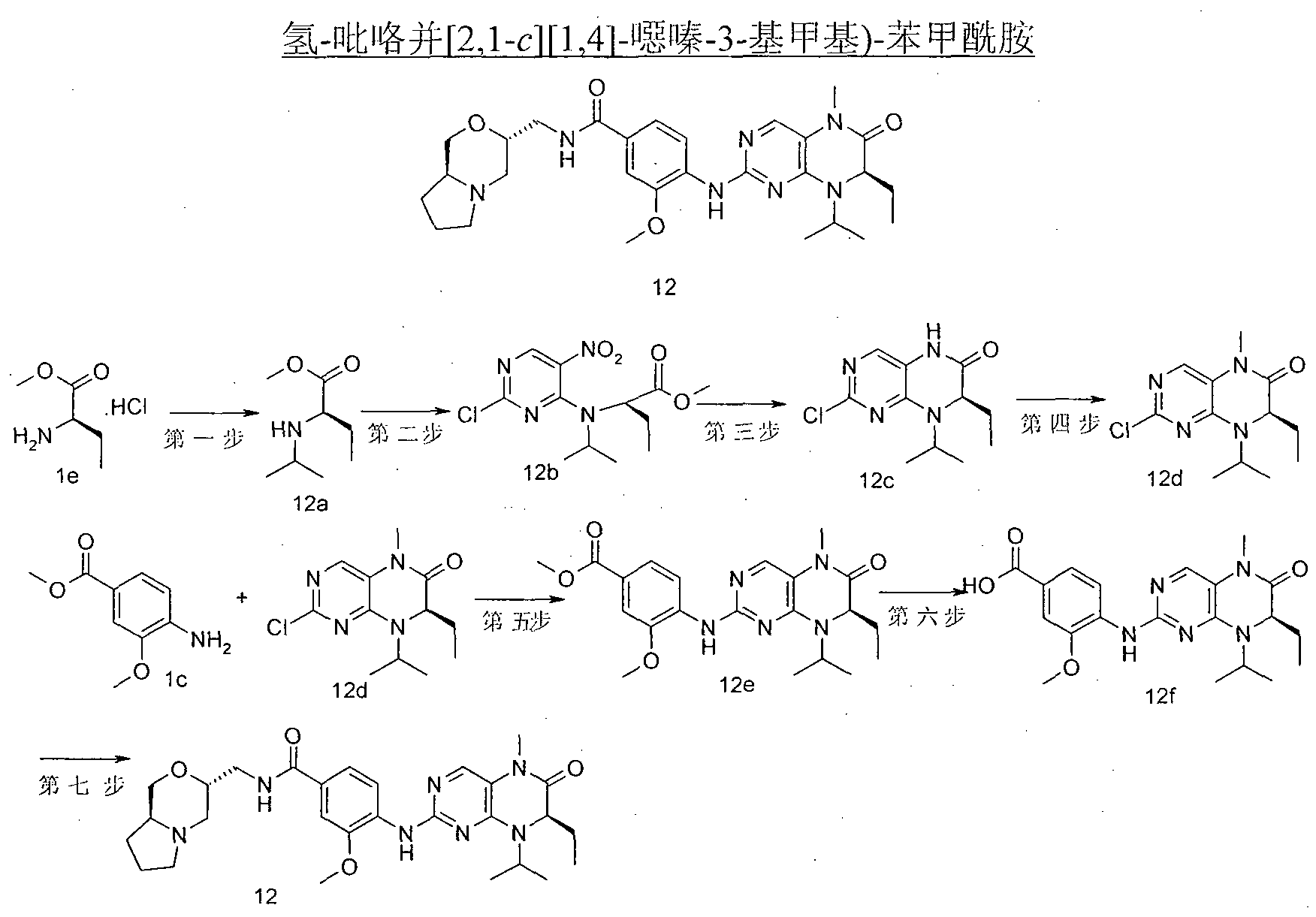
第一步
((i?)-2-异丙基氨基-丁酸甲酯
将 (i?)- 2-氨基-丁酸甲酯盐酸盐 le(28.78 g, 0.19 mol)溶解于 200 mL二氯甲垸 中, 加入丙酮 (11.96 g, 0.21 mol)和醋酸钠 (30.76 g, 0.38 mol), 搅拌 5分钟, 加入 三乙酰氧基硼氢化钠 (59.61 g, 0.281 mol), 搅拌反应 12小时。加入少量 1 M盐酸, 滴加饱和碳酸钠溶液至反应液 pH为 8〜9, 二氯甲垸萃取 (50 mLx3),合并有机相, 饱和食盐水洗涤 (30 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱 法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 (i?)- 2-异丙基氨基-丁酸甲酯 12a(16.4 g, 淡色油状物), 产率: 55 %。
第二步
(i?)-2- [(2-氯 -5-硝基-嘧 - 4-基) -异丙基-氨基] -丁酸甲酯 将 2,4-二氯 -5-硝基 -嘧啶 (19.9 g, 103 mmol)溶解于 300 mL环己垸中,加入 (7?)-2-
异丙基氨基-丁酸甲酯 12a(16.4 g, 103 mmol)和碳酸氢钠 (36.61 g, 412 mmol), 80°C 搅拌反应 4小时。过滤, 滤液减压浓缩, 加入 30 mL水, 二氯甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (30 mL), 无水硫酸镁千燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 (W-2-I 2-氯 -5-硝 基 -(]密啶—4-基) -异丙基-氨基] -丁酸甲酯 12b(20.5 g, 黄色固体), 产率: 62.8 %。
第三步
((i?)-7-乙基- 2-氯 -8-异丙基 - 7,8-二氢 -5H-喋啶 -6-酮 将 (i?)-2-[(2-氯 -5-硝基-嘧啶 -4-基) -异丙基-氨基] -丁基酸甲酯 12b(7.3 g,23 mmol) 溶解于 lOO mL醋酸中, 加入过量兰尼镍, 氢气氛下, 75Ό下搅拌反应 2小时。 过 滤,,滤液减压浓缩, 加入 20 mL冰水, 析出固体, 过滤, 烘干滤饼, 得到标题产 物 ( -7-乙基 -2-氯 -8-异丙基 -7,8-二氢 -5H-喋啶- 6-酮 12c(12.1 g,灰白色固体),产率: 71.7%。 直接投入下一歩反应。
MS m/z (ESl):255.1 [M+1]
第四步
0??)-7-乙基 -2-氯 -8-异丙基 -5-甲基- 7,8-二氢 -5H-喋啶 -6-酮 将 (i?)-7-乙基 -2-氯 -8-异丙基 - 7,8-二氢- 5H-喋啶 -6-酮 12c(12.1 g, 47.5 mmol)溶 解于 180 mL丙酮中, 加入对甲苯磺酸甲酯 (13.28 g, 71 .3 mmol)和碳酸钾 (13.11 g, 95 mmol), 搅拌反应 12小时。 过滤, 滤液减压浓缩, 加入 30 mL水, 二氯甲烷萃 取 (50 niLx3), 合并有机相, 饱和食盐水洗涤 (30 mL), 无水硫酸镁干燥, 过滤, 滤 液减压浓缩,用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物,得到标题产物 ( -7- 乙基 -2-氯 -8-异丙基 -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 12d(10.8 g, 白色固体), 产率: 84.7 %。
第五步
(i?)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -甲氧基) - 苯甲酸甲酯
将 4-氨基 -3-甲氧基 -4-苯甲酸甲酯 lc(l .7 g, 9.56 mmol), (( -7-乙基 -2-氯- 8- 异丙基 -5-甲基 -7,8-二氢 -5H-喋啶 -6-酮 12d(2.4 g, 9.11 mmol)和对甲苯磺酸 (2.8 g, 14.57 mmol)溶解于 50 mL 4-甲基 -2-戊醇中, 回流搅拌反应 2.5小时。 减压浓缩反 应液, 加入 250 mL二氯甲烷, 依次用饱和碳酸氢钠溶液 (50 mL)、 饱和食盐水溶 液洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱 剂体系 B 纯化所得残余物, 得到标题产物 )-4- (7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -甲氧基) -苯甲酸甲酯 12e(1.7 g, 白色固体),产率: 45.5
MS m/z (ESI):414.3 [M+1]
第六步
C/?)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -甲氧基) -苯
甲酸
将 W_4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -甲氧基) - 苯甲酸甲酯 12e(1.7 g, 4.15 mmol)溶解于 40 mL甲醇中,加入 45 mL 1 M氢氧化锂 溶液, 8CTC下搅拌反应 5小时。 减压浓缩反应液, 用乙酸乙酯萃取 (50 mLx3), 加 入固体硫酸氢钾至反应液 pH为 3〜4, 过滤, 滤饼用水和少量乙醇洗涤, 干燥, 得到标题产物 (i?)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) - 甲氧基) -苯甲酸 12f(2.24 g, 白色固体), 产率: 91.8 %。
第七步
4-( )-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基)- N-((3 8aS)-六 氢-吡咯并 [2, l -c][l,4]-噁嗪 -3-基甲基)-苯甲酰胺
将 (3i?,8aS)-六氢-吡咯 [2,1- c][] ,4]噁嗪- 3-基)甲胺 (39 mg, 0.25 mmol , 采用公知 的方法 " CN101392001 "制备而得)溶解于 30 mL二氯甲烷中, 加入 ((7?)-4-(7-乙基 -8-异丙基 -5-甲基- 6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -甲氧基)-苯甲酸 12f(100 mg, 0.25 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (80 mg, 0.25 mmol)和二 异丙基乙胺 (0.1 mL, 0.63 mmol), 搅拌反应 1小时。 加入 30 mL二氯甲烷, 依次 用饱和氯化铵溶液 (30 mL), 饱和食盐水洗涤 (30 mL), 无水硫酸镁千燥, 过滤, 滤 液减压浓缩, 用 HPLC制备分离纯化所得残余物, 得到标题产物 4-((/?)-7-乙基- 8- 异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-N-((3凡 8aS)-六氢 -吡咯并 [2,1- C][l,4]-噁嗪 -3-基甲基) -苯甲酰胺 12(75 mg, 白色固体), 产率: 56 %。
MS m/z (ESI):538.3 [M+l]
1H NMR(400MHz, CDC13, ppm) δ 8.57—8.58 (d, 1 H), 7.63 (s, 1H), 7.46 (s, 1H), 7.34〜7.36 (d, 1H), 6.86 (s, 1 H), 4.69〜4.74 (m, 1H), 4.28〜4.3] (m, 1 H), 4.12—4.19 (m, 2H), 3.90—3.98 (m, 4H), 3.66—3.75 (m, 2H), 3.33〜3.35 (m, 3H), 3.05—3.24 (m, 4H), 2.76〜2.82 (m, 1H), 1.89—2.03 (m, 5H), 1.69〜1.77 (m, 1H), 1.37〜1.45 (m, 3H): L30〜1.36 (m, 3H), 0·87〜0·90 (t, 3H) 实施例 13
4-((iQ-7-乙基 -8-异丙基 - 5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-(i3S,8aS)-六 氢-吡咯 甲酰胺

13

第一歩
4-((i?)-7-乙基- 8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 - 2-基氨基) -N-((3&8aS')-六 氢-吡咯并 [2,1-C][1 ,4]-噁嗪 -3_基甲基) -苯甲酰胺
将 (3 & 8aS)-六氢-吡咯 [2,l-c][l ,4]噁嗪 -3-基)甲胺 (39 mg, 0.25 mmol, 采用公知 的方法 "CN10139200] "制备而得)溶解于 30 mL二氯甲垸中, 加入 ((i?)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代- 5,6,7,8-四氢 -喋啶 -2-基氨基) -甲氧基) -苯甲酸 12f(100 mg, 0.25 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (80 mg, 0.25 mmol)和二 异丙基乙胺 (0.1 mL, 0.63 mmol), 搅拌反应 1小时。 加入 30 mL二氯甲垸, 依次 用饱和氯化铵溶液 (30 mL), 饱和食盐水洗涤 (30 mL), 无水硫酸镁干燥, 过滤, 滤 液减压浓缩, 用 HPLC制备分离纯化所得残余物, 得到标题产物 4-((i?)- 7-乙基 -8- 异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )-N-((3 & 8aS)-六氢 -吡咯并 [2,l-c][l,4]-噁嗪 -3-基甲基) -苯甲酰胺 13(65 mg, 白色固体), 产率: 44.8 %。
MS m/z (ESI):538.5 [M+l]
Ή NMR(400MHz, CDC】3, ppm) δ 8.55〜8.58 (d, 1H), 7.68 (s, 1H), 7.62 (s, 1H), 7.46 (s, 1H), 7.32〜7.45 (d, 1H), 6.5 〜 6.54 (m, 1H), 4.7 〜 4.74 (m, 1H), 4.28〜4.31 (m, 1H), 4.02〜4.06 (m, 1H), 3.97 (s, 3H), 3.73—3.79 (m, 2H), 3.41—3.46 (m, 2H), 3.33 (s: 3H), 3.1 1〜3.14 (m, 2H), 2.15〜2.20 (m, 3H), 1.69—1.93 (m, 6H), 1.40〜1 .45 (m, 3H): 1.36〜1.37 (m, 3H), 0.85〜0.90 (t, 3H) 实施例 14
4-((7?)-7-乙基 -8-异丙基 - 5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-iOQa ?)- ~2,l-c|「l ,4 噁嗪 -3-基甲基) -苯甲酰胺

第一步
4-((i?)-7-乙基- 8-异丙基 - 5-甲基 -6-氧代- 5,6,7,8-四氢 -喋啶 -2-基氨基) -N-((3 8ai?)-六 氢-吡咯并 [2,l -c][l ,4]-噁嗪 -3-基甲基) -苯甲酰胺
将 ((3 8ai?)-六氢-吡咯 [2,1- c][l,4]噁嗪 -3-基)甲胺 (39nig, 0.25 mmol,釆用公知 的方法 "CN] 01392001"制备而得)溶解于 30 mL二氯甲烷中, 加入 ((i?)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -甲氧基)-苯甲酸 12fT100mg, 0.25 mmol), (9-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (80 mg, 0.25 mmol)和二 异丙基乙胺 (0.1 mL, 0.63 mmol), 搅拌反应 1小时。 加入 20 mL二氯甲烷, 依次 用饱和氯化铵溶液 (30mL), 饱和食盐水洗涤 (30mL), 无水硫酸镁干燥, 过滤, 滤 液减压浓缩, 用 HPLC制备分离纯化所得残余物, 得到标题产物 4-((A)-7-乙基 -8- 异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基 )-N-((3i?,8a/?)-六氢 -吡咯并 [2,1-C][1,4]-噁嗪 -3-基甲基) -苯甲酰胺 14(67mg, 白色固体), 产率: 50%。
MSm/z(ESI):538.5 [M+1]
1H NMR(400MHz, CDC13, ppm) δ 8.55〜8.58 (d, 1H), 7.68 (s, 1H), 7.62 (s, 1H), 7.45 (s, 1H), 7.30〜7.32 (d, 1H), 6.49〜6.50 (m, 1H), 4.69〜4.76 (m, 1H), 4.28〜4.30 (m, 1H), 4.02—4.06 (m, 1H), 3.97 (s, 3H), 3.73—3.79 (m, 2H), 3.42—3.47 (m, 2H), 3.33 (s: 3H), 3.01—3.12 (m, 2H), 2.13〜2.18 (m, 3H), 1.68—1.96 (m, 6I-I), 1.43〜1.45 (m, 3H): 1.36〜1.37 (m, 3H), 0.85—0.90 (t, 3H) 实施例 15
4-(0?)-7-乙基- 8-异丙基 - 5-甲基- 6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-"3S,8a/?)-六 氢-吡咯并「2,l-cin,4卜噁嗪 -3-基甲基) -苯甲酰胺

第一歩
4-((i?)-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N-((3S,8ai?)-六 氢-吡咯并 [2,1-C][1,4]-噁嗪 -3-基甲基) -苯甲酰胺 将 ((3S,8ai?)-六氢 -吡咯 [2,l-c][l,4]噁嗪 -3-基)甲胺 (39 mg, 0.25 mmol,釆用公知 的方法 "CN101392001"制备而得)溶解于 30 mL二氯甲垸中, 加入 (( -4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -甲氧基)-苯甲酸 12f(100mg, 0.25 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (80 mg, 0.25 mmol)和二 异丙基乙胺 (0.1 mL, 0.63 mmol), 搅拌反应 1小时。 加入 30 mL二氯甲垸, 依次 用饱和氯化钹溶液 (30mL), 饱和食盐水洗涤 (30mL), 无水硫酸镁干燥, 过滤, 滤
液减压浓缩, 用 HPLC制备分离纯化所得残余物, 得到标题产物 4-((i?)-7-乙基- 8- 异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基 )- N-((3S,8ai?)-六氢-吡咯并 [2,1 -C]n ,4]-噁嗪 -3-基甲基) -苯甲酰胺] 5(62 mg, 白色固体), 产率: 45.5 %。
MS m/z (ESI):538.5 [M+1]
】H NMR(400MHz, CDC13, ppm) δ 8.57—8.59 (d, 1H), 7.67 (s, l H), 7.64 (s, 1H), 7.47 (s, 1 H), 7.40—7.46 (d, 1H), 6.96 (s, 1H), 4.68〜4.75 (m, 1H), 4.28—4.30 (m, 1H), 4.12—4.22 (m, 2H), 3.94—3.98 (m, 4H), 3.62〜3.73 (m, 2H), 3.21 -3.32 (m, 6H), 3.01 -3.12 (m, 1H), 2.76—2,82 (m, 1H), 1.95〜2.01 (m, 4H),】.89〜1.94 (m, 1H), 】.69〜1.88 (m, 1H), ] .55〜1 ·57 (m, 3H), 1.48〜1.53 (m, 3H), 0.85〜0.90 (t, 3H) 实施例 16
4-((iT>-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -
N-((4aR,6R aS/4aS,6S aR)-2-甲基-八 -环戊并 W吡啶 -6-基) -3-甲氧基-苯甲酰

第一步
(4a^,6 ?, 73^-6-叠氮基 -八氢 -1H-环戊并 [c]吡啶盐酸盐 16a-l
(4a5,6 & 7aW)-6-叠氮基 -八氢 -1//-环戊并 [c]吡啶盐酸盐 16a-2
将实施例 8第九歩所得的 (4a 6 7a5 6-叠氮基 -六氢 -li- /-环戊并 [c]吡啶 -2(3H)- 羧酸叔丁酯 8k-l和 (4a&6 & 7 )-6-叠氮基-六氢- 1 H-环戊并 [c]吡啶 - 2(3H)-羧酸叔丁 酯 8k-2的混合物 (1.15 g, 4.32 mmol)溶解于 5 mL二氯甲垸中, 加入 10 mL 6 M氯 化氢的 1 ,4-二氧六环溶液, 搅拌反应 2 小时。 减压浓缩反应液, 得到标题产物 (4a7?,6 7EUS 6-叠氮基 -八氢 - 1H-环戊并 [c]吡啶盐酸盐 16a-l 和 (4aS,6 & 7a/ -6-叠氮 基 -八氢 -1H-环戊并 M吡啶盐酸盐 16a-2的混合物 (880 mg,无色油状物),产率: 100
%。 直接投入下一步。
MS m/z (ESl): 167.2 [M+l]
第二步
(4a e,6/?,7a5)-6-叠氮基 -2-甲基-八氢 -1H-环戊并 [c]吡啶 16b-l (4a5,6 & 7ai?)-6-叠氮基 -2-甲基 -八氢 -1H-环戊并 [c]吡啶 16b-2 冰水浴下, 将上述歩骤所得的 (4a ?,6 7aS)- 6-叠氮基 -八氢 环戊并 [C]吡啶盐 酸盐 16a-l和 (4a5,6 & 7aW)-6-叠氮基 -八氢 环戊并 [c]吡啶盐酸盐 16a-2的混合物 (880 mg, 4.32 mmo】)溶解于 20 mL乙腈和水 (V:V = 1 : 1)的混合液中, 依次加入 37 %甲醛溶液 (0.71 mL, 8.64 mmol)和三乙酰氧基硼氢化钠 (2.7 g, 〗2.96 mmol), 搅拌 反应 2小时, 加入 10 mL 1 M盐酸, 滴加 15 mL 5 %氢氧化钠溶液至反应液 pH为 9, 搅拌 ] 0 分钟, 用二氯甲垸萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A 纯化所得残余物, 得到标题产物 (4ai?,6 7aS 6-叠氮基 -2-甲基 -八氢 -1H-环戊并 [C] 吡啶 16b-l和 (4a5',6 & 7ai?)-6-叠氮基 -2-甲基 -八氢 -1H-环戊并 [c]吡啶 16b-2的混合物 (650 mg, 无色油状物), 产率: 84%。
MS m/z (ESI): 181.1 [M+l]
第三步
(4a/?,6i?,7aS 2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基-胺 16c-l :
(4aS,6 & 7a ?)-2-甲基-八氢- 1H-环戊并 M吡啶 -6-基-胺 16c-2 将上述步骤所得的 (4a7?,6 7aS)-6-叠氮基 -2-甲基-八氢 -1H-环戊并 [c]吡啶.16b- 1 和 (4a ,6 & 7a/?)-6-叠氮基 - 2-甲基 -八氢 -1H-环戊并 [c]吡啶 16b-2的混合物 (650 mg, 3.6 mmol)溶解于 20 mL甲醇中, 加入 (70 mg, 10%)钯 /碳, 氢气氛下搅拌反应 2小 时, 过滤, 滤液减压浓缩, 得到标题产物 (4a ?,6 7a5)-2-甲基-八氢- 1H-环戊并 [C-] 吡啶 -6-基-胺 16c-l和 (4aS,6S,7ai?)-2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基-胺 16c-2的 混合物 (505 mg, 淡黄色油状物), 产率: 91 %。
MS m/z (ESI): 155.2 [M+l]
第四步
. 4- ((i?)- 7-乙基- 8-异丙基 -5-甲基- 6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) - Ν-((4α^,6 73^-2-甲基 -八氢 -1H-环戊垸 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 16-1
4-(( -7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) - N-((4aS,6 & 7a7?)-2-甲基 -八氢 环戊烷 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 16-2 将上述步骤所得的 (4ai?,6 7a,S 2-甲基 -八氢 -1H-环戊并 |c]吡啶- 6-基-胺 16c-l 和 (4a&6 & 7aW)-2-甲基-八氢-] H-环戊并 [c]吡啶 -6-基-胺 16c-2的混合物 (77 mg, 0.5 mraol) , (i?)—4-(7-乙基- 8-异丙基 -5-甲基 -6-氧代 -5,6/7,8-四氢 -喋啶 -2-基氨基) -甲氧 基)-苯甲酸 12f(199.7 rag, 0.5 mmol), < -苯并三氮唑- Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸 酯 (160 mg, 0.5 mmol)和二异丙基乙胺 (】42 mg, 1.1 mmol), 搅拌反应 2小时。 滴 加氨水至反应液 pH为 9〜10, 加入 50 mL水, 用二氯甲烷萃取 (50 mL><3), 合并 有机相, 饱和食盐水洗涤 (50 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用硅 胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((i?)-7-乙基 -8-异丙 基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基)- N- ((4a 6i?,7a^)- 2-甲基 -八氢 环 戊垸 M吡啶- 6-基) -3-甲氧基-苯甲酰胺 16-1和 4-((i?)- 7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基)- N-((4a5,6&7aW)-2-甲基 -八氢 -1H-环戊垸 [c]吡啶 -6- 基) -3-甲氧基 -苯甲酰胺 16-2的混合物 (60 mg, 白色固体), 产率: 22.3 %。
MS m/z (ESI):536.5 [M+l]
Ή NMR(400MHz, CDC13, ppm) δ 8.48〜8.62 (d, 1H), 7.58〜7.71 (d, 2H), 7.36-7.51 (s, 1H), 7.18-7.31 (m, 1H), 6.12—6.28 (d, 1H), 4.68-4.81 (m, 1 H), 4.55〜4.65 (m, 1H) 4.21—4.45 (m, I H), 3.91—4.05 (s, 3H), 3.26—3.41 (s, 3H), 2.21〜2.59 (m, 1H), 2.48〜2.58 (s, 3H), 2.35〜2.46 (s, 3H), 2.15—2.28 (m, 4H), 1.88—2.02 (m, 2H), 1 ·61〜1 ·75 (m, 4H), 1.43〜1.53 (d, 3H), 1.28〜1.41 (d, 3H), 0.75—0.81 (t, 3H) 实施例 17
4- (W)-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨 基) -Ν-(ϊ437?,6&73^/43^,6凡 7a/D-2-甲基-八氢 环戊并 kl吡啶 -6-基) -3-甲氧基 _苯 甲酰胺 17-1
4-((7?)-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N-(Y4ai?,6S,7aS74¾S,6 7ai?)-2-甲基 -八氢 -1H-环戊并「c|吡啶 -6-基) -3-甲氧基-苯 甲酰胺 17-2

17-1 17-2

第一步
4-((7?)-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N- ((4a 6 & 7aS)-2-甲基 -八氢 -1H-环戊并 [C]吡啶 -6-基)- 3-甲氧基-苯甲酰胺 17-1
4-(( ?)-7-乙基- 8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -N-((4aS,6/?,7ai?)-2-甲基-八氢 -1H-环戊并 [c]吡啶 -6-基) -3-甲氧基-苯甲酰胺 17-2 将实施例 11第六歩中所得的 (4& 6 & 7a5)-2-甲基 -八氢 -1H-环戊并 [c]吡啶 -6-基- 胺 llf-1和 (4aS,6 7a ?)-2-甲基-八氢-] H-环戊并 [6']吡啶 -6-基-胺 llf-2的混合物 (60 mg, 0.39 mmol), (W)-4-(7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨 基) -甲氧基)-苯甲酸 12f(171.6 mg, 0.43 mmol), O-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲 四氟硼酸酯 (125 mg, 0.39 mmol)和二异丙基乙胺 (150 mg, 1.17 mmol), 搅拌反应 2 小时。 滴加饱和碳酸钠溶液至反应液 pH为 8〜9, 用二氯甲烷萃取 (50 mLx3), 合 并有机相, 饱和食盐水洗涤 (50 mL), 无水硫酸钠干燥, 过滤, 滤液减压浓缩, 用 硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((7?)-7-乙基 -8-异 丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -N- ((4ε^,6&735 2-甲基 -八氢 -1H- 环戊并 [c]吡啶 -6-基:) -3-甲氧基-苯甲酰胺 17-1 和 4-((; -7-乙基 -8-异丙基 -5-甲基 -6- 氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基)- N-((4aS,6W,7ai?)- 2-甲基 -八氢 -1H-环戊并 [C]吡啶 -6-基) -3-甲氧基-苯甲酰胺 17-2的混合物 (70 mg, 白色固体), 产率: 34%。
MS m/z (ESI):536.3 [M+l]
1H NMR(400MHz, CDC】3, ppm) δ 8.57-8.59 (d, 1H), 7.76 (s, 1H), 7.71 (s, 1H), 7.48 (s, 1H), 7.29—7.31 (m, 1H), 6.30-6.32 (d, 1H), 4.75〜4.78 (m, 1H), 4.34〜4.56 (m, 1H), 4.32〜4.34 (m, 1H), 4.00 (s, 3H), 3.36 (s, 3H), 2.52〜2.58 (m, 2H), 2.28〜2.44 (m; 9H), 1.92-2.03 (m, 2H), 1.70〜1.81 (m, 4H), 1 ·46〜1 ·53 (d, 3H), 1.31〜1.41 (d, 3H), 0.88—0.92 (m, 3H) 实施例 18
4-((i?)-7-乙基- 8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢 -喋啶 -2-基氨基) -Ν-Π-甲基 -哌啶
-4-基)- 3- (四氧 -呋喃 -2-基-甲氧基) -苯甲酰胺


第一步
4-((i?)-7-乙基 -8-异丙基 -5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶 -2-基氨基) -N-(l-甲基-哌啶
-4-基) -3 - (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 将 4-氨基 -N-(l -甲基 -哌啶 -4-基) -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 7d(150 mg, 0.45 mmol), (i?)-7-乙基 -2-氯- 8-异丙基 -5-甲基- 7,8-二氫 -5H-喋啶 -6-酮 12d(121 mg, 0.45 mmol)和对甲苯磺酸 (137 mg, 0.72 mmol)溶解于 20 mL 4-甲基 -2-戊醇中, 回流搅拌反应 3小时。 滴加饱和碳酸钾溶液至反应液 pH为 9〜】0, 用二氯甲烷萃 取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓縮, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物 4-((i?)-7-乙基 -8-异丙基 - 5-甲基 -6-氧代 -5,6,7,8-四氢-喋啶- 2-基氨基)- N-(l-甲基 -哌啶 -4-基) -3- (四氢 -呋喃 -2-基-甲氧基) -苯甲酰胺 18(100 mg, 白色固体), 产率: 39%。 MS m/z (ESI):566.5 [M+l]
'Η NMR(400MHz, CDC13, ppm) δ 8.45— 8.65 (d, 1H), 7.65-7.75 (d, 2H), 7.40-7.51 (s, 1H), 7.25-7.38 (m, 1H), 5.92-6.05 (m, 1H), 4.36-4.61 (m, 1H), 4.33〜4.46 (m, 1H), 4.21〜4.32 (m, 1H), 4.06〜4.18 (m, 2H), 3.95—4.04 (m, 2H), 3.80—3.92 (m, 1H), 3.25-3.40 (s, 3H), 2.82〜2.96 (d, 2H), 2.30〜2.42 (s, 3H), 2.15〜2.28 (m, 2H), 1.95- 2.06 (m, 6H), 1.63〜 1.75 (m, 4H), 1.42〜1.53 (d, 3H), 1.30〜1.41 (d, 3H), 0.80〜0.95 (t, 3H) 实施例 19
0 ?)-3-甲氧基- 4-(5-甲基 -6-氧代 -8-甲苯磺酰基 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 Γ2 J -W 喋啶 -2-基氨基) -N-(l-甲基哌啶 -4-基)苯甲酰胺

第一步
3-甲氧基 -N-(l -甲基-哌啶 -4-基) -4-硝基-苯甲酰胺 将 3-甲氧基 -4-硝基-苯甲酸 la(9.86 g, 50 mmol)溶解于 200 mL二氯甲垸中, 依次加入 0-苯并三氮唑 -Ν,Ν,Ν',Ν'-四甲基脲四氟硼酸酯 (16.1 g, 50 mmol), 二异丙 基乙胺 (18.2 mL, 110 mmol)和 1-甲基 -哌啶 -4-基-胺 (5.7 g, 50 mmol), 搅拌反应 2 小时, 加入 200 mL二氯甲烷, 用 1 M氨水洗涤, 无水硫酸钠干燥, 过滤, 滤液减 压浓缩, 得到标题产物 3-甲氧基 -N-(l -甲基-哌啶- 4-基)- 4-硝基-苯甲酰胺 19a(10 g, 黄色固体), 产率: 68 %。
MS m/z (ESI):294.2 [M+l]
第二步
4-氨基 -3-甲氧基 -N-(l-甲基 -哌啶 -4-基) -苯甲酰胺 将 3-甲氧基 -N-(l-甲基 -哌啶 -4-基) -4-硝基-苯甲酰胺 19a(5 g, 17 mmol)溶解于 120 mL甲醇中, 加入 (500 mg, 10%)钯 /碳, 氢气氛下搅拌反应 1小时, 过滤, 用 30 mL甲醇洗涤滤饼, 滤液减压浓缩, 得到标题产物 4-氨基 -3-甲氧基 -N-(l-甲基- 哌啶 -4-基) -苯甲酰胺 19b(4.36 g, 白色固体), 产率: 97 %。
MS m/z (ESI):264.3 [M+l]
二歩
((i?)-4- (2-氯 -5-硝基 -嘧啶 -4-基) -哌嗪 -1 ,3-二羧酸 1-叔丁酯 3-甲酯 将 哌嗪 -1,3-二羧酸 1 -叔丁酯 3-甲酯 19c(6 g, 24.6 mmol), 2,4-二氯 -5-硝基
-呃嗪 (4.8 g, 24.8 nimol)和碳酸氢钠 (8.3 g, 98.4 mmol)溶解于 150 mL环己烷中, 65 °C下搅拌反应 0.5 小时, 加入 200 mL 乙酸乙酯, 依次用饱和氯化铵溶液 (60 mLx2)、 饱和食盐水洗涤 (60 mLx2), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得 到标题产物 ((i?)-4-(2-氯 -5-硝基-嘧啶 -4-基) -哌嗪 -1,3-二羧酸 1 -叔丁酯 3-甲酯 19d(8.36 g, 黄色固体), 产率: 84.4%。
MS m/z (ESI):402.2 [M+l ]
第四步
(7?)-2-氯 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1 -/7]喋啶 -8(6H)-羧酸叔丁酯
将 4- (2-氯 -5-硝基 -嘧啶 -4-基) -哌嗪 -1 ,3-二羧酸 1-叔丁酯 3-甲酯】9d(8.36 g, 20.8 mmol)和铁粉 (3.5 g, 62.4 mmol)溶解于 350 mL乙酸中, 70°C下搅拌反应 12小 时, 减压浓缩反应液,.滴加饱和碳酸氢钠溶液至反应液 pH为 8〜9, 二氯甲垸萃 取 (200 mLx3), 饱和食盐水洗涤 (60 mL><2), 无水硫酸镁千燥, 过滤, 滤液减压浓 缩, 依次加入 10 raL二氯甲烷和 50 mL乙酸乙酯, 有白色固体析出, 过滤, 烘干 滤饼,得到标题产物 (i?)-2-氯 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-/7]喋啶 -8(6H>羧酸 叔丁酯 19e(4.55 g, 白色固体), 产率: 64.1 %。
MS m/z (ESI):340.3 [M+l]
第五步 .
(i?)-2-氯 -5-甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2J -/7]喋啶 -8(6H)-羧酸叔丁酯 将 (R)-2-氯 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-/2]喋啶 -8(6H)-羧酸叔丁酯 19e(500 mg, 1.47 mmol)和碳酸钾 (305 mg, 2.2 mmol)溶解于 40 mL碳酸二甲酯中, 回流搅拌反应 12小时, 加入 50 mL二氯甲垸, 依次用饱和氯化胺溶液 (50 mLx2), 饱和食盐水洗涤 (50 mLx2), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色 谱法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 (7?)-2-氯 -5-甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-A]喋啶- 8(6H)-羧酸叔丁酯 19f(0.45 g,白色固体),产率: 79.8 % o
MS ra/z (ESI):354.2 [M+l]
第六步 .
(i?)-2-氯 -5-甲基- 8-甲苯磺酰基 -7,8,9,10-四氢- 5H-吡嗪 [2,1 -A]喋啶 -6(6aH)-酮 将 (i?)- 2-氯- 5-甲基 -6-氧代- 6a,7,9,10-四氢 -5H-吡嗪 [2,1- 喋啶 -8(6H)-羧酸叔丁 酯 19f(415 mg, 1.2 mmol)溶解于 40 mL二氯甲烷中, 加入氯化氢气体,.搅拌反应 2小时, 依次加入三乙胺 (0.24 g, 2.4 mmol)和对甲苯磺酰氯 (0.34 g, 1.8 mmol), 搅 拌反应 2小时。 用饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压 浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标题产物 2-氯 -5- 甲基 -8-甲苯磺酰基 -7,8,9,10-四氢 -5H-吡嗪 [2,1 - 喋啶 -6(6aH)-酮 1.9g(0.14 g,白色固 体), 产率: 27.7%。
MS m/z (ESI) :408.2 [M+l]
第七步
((X)- 3-甲氧基 -4-(5-甲基 -6-氧代 -8-甲苯磺酰基 -6,6a,7,8,9,10-六氢 -5 ^吡嚓并 [2,1 -h] 喋啶 -2-基氨基) -N-(] -甲基哌啶 -4-基)苯甲酰胺 将 4-氨基 -3-甲氧基 -N-(l-甲基 -哌啶 -4-基) -苯甲酰胺 19b(300 mg, 1 .2 mmol), ((7?)-3-氯 -10-甲基 -7- (甲苯基 -4-磺酰基 )-6,7,8,8a-四氢 -5H,10H-2,4,4b,7,10-五氮杂-菲 -9-酮 19g(696 mg, 1.7 mmol)和对甲苯磺酸 (1 . ] 4 g, 6 mmol)溶解于 40 mL 4-甲基 -2- 戊醇中, 回流搅拌反应〗 小时。 滴加三乙胺至反应液 pH为 8〜9, 减压浓缩反应 液, 加入 20 mL饱和碳酸氢钠溶液, 用二氯甲垸萃取 (50 mLx3), 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体 系 A纯化所得残余物,得到标题产物 甲氧基 -4-(5-甲基 -6-氧代 -8-甲苯磺酰基 -6,6a,7,8,9,10-六氢 -5H-吡嗉并 [2,1- h]喋啶 -2-基氨基) -N-(l-甲基哌啶- 4-基)苯甲酰胺 19(212 mg, 黄色固体), 产率: 29.8 %。
MS m/z (ESl):635.3 [M+1]
1H NMR(400MHz, CDC13, ppm) δ 8.39〜8.52 (d, 1 H), 7.72-7.81 (m, 3H), 7·56〜7·71 (s: 1H), 7.42-7.55 (s, 1H), 7.34—7.41 (m, 2H), 7.21-7.32 (m, 1H), 6.03—6.14 (d, 1H), 4.35〜4.49 (m, 1H), 4.16〜4.28 (m,】H), 3.85—4.10 (m, 5H), 3.26—3.39 (s, 3H), 2.96—3.09 (m, 1H), 2.81—2.94 (m, 2H), 2.36〜2.50 (m, 8H), 2.26—2.30 (m, 2H), ] .99〜2.] 2 (m, 4H), 1.6〗〜 1.76 (m, 2H) 实施例 20
W)— 4-(8-异丙基 -5-甲基 -6-氧代 -6,6a,7,8,9,10-六氢 -5H-吡嗉并「2, W喋啶 -2-基氨
-3-甲氧基 -Ν-α -甲基 -哌啶 -4-基)苯甲酰胺

第一步
(i?)-2-氯- 8-异丙基 -5-甲基 -7,8,9,10-四氢- 5H-吡嗪 [2,1- //]喋啶 -6(6aH)-酮 将 2-氯 -5-甲基- 6-氧代 -6a,7,9,10-四氢 -5H-吡嗪并 [2,1-/0喋啶 -8(6H)-羧酸叔丁酯 19f(2.08 g, 5.9 mmol)溶解于 50 mL二氯甲垸中, 加入三氟乙酸 (3.3 g, 29 mmol), 搅拌反应 0.5小时, 减压浓缩反应液, 加入 50 mL二氯甲垸, 滴加三乙胺至反应液 pH为 8〜9, 加入丙酮 (0.4 g, 7.1 mmol), 搅拌反应 1小时, 加入三乙酰氧基硼氢 化钠 (2.5 g, 11.8 mmol),搅拌反应 12小时。加入 30 mL水,二氯甲烷萃取 (50 mLx3),
合并有机相, 用饱和食盐水洗涤 (50 mLx2), 无水硫酸镁干燥, 过滤, 滤液减压浓 缩, 用硅胶柱色谱法以洗脱剂体系 B 纯化所得残余物, 得到标题产物 氯- 8- 异丙基 -5-甲基 -7,8,9,10-四氢 -5H-吡嗪 [2,1 - 喋啶 -6(6aH)-酮 20a(1.32 g, 白色固体), 产率: 25.4 %。
MS m/z (ESI):296.1 [M+l ]
第二步
( ?)-4— ( 异丙基— 5—甲基— 6—氧代 _6,6a,7,8,9,1 0—六氢 -5//—吡嗪并 [ -W喋啶— 2—基氨 基) -3-甲氧基 -N-(l -甲基-哌啶 -4-基)苯甲酰胺 将 4-氨基- 3-甲氧基 -N-(l -甲基 -哌啶 -4-基) -苯甲酰胺 1.9b(170 mg, 0.64 mmol), (7?)-2-氯 -8-异丙基 - 5-甲基 -7,8,9,10-四氢 -5H-吡嗪 [2,1 -/2]喋啶 -6(6aH)-酮 20a(240 mg, 0.81 mmol)和对甲苯磺酸 (0.81 g, 4.3 mmol)溶解于 20 mL4-甲基 -2-戊醇中, 回流搅 拌反应 2小时。 冷至室温, 加入 20 mL饱和碳酸氢钠溶液, 用二氯甲烷萃取 (50 mLx3), 合并有机相, 饱和食盐水洗涤 (50 mLx3), 无水硫酸镁干燥, 过滤, 滤液 减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A 纯化所得残余物, 得到标题产物 (/?)-4-(8-异丙基 -5-甲基 -6-氧代- 6,6a,7,8,9,10-六氢 -5.H-吡嗪 喋啶 -2-基氨基 )-3- 甲氧基 -N-(l -甲基 -哌啶 -4-基)苯甲酰胺 20(61.3 mg, 白色固体), 产率: 18.4 %。 MS i-n/z (ESI):523.4 [M+l]
Ή NMR(400MHz, CDC13, ppm) δ 8.42—8.56 (d, 1H), 7.35〜7.61 (d, 2H), 7.52-7.90 (s, 1H), 7.21-7.33 (m, 1H), 5.99—6.12 (d, 1H), 4.61〜4.72 (d, 1H), 4.1 1〜4.22 (d, 1 H), 3.92—4.05 (m, 4H), 3.42—3.58 (d, 1H), 3.36—3.40 (s, 3H), 2.85〜2.96 (m, 4H), 2.31〜2.42 (m, 4H), 2. ] 6〜2.28 (m, 2H), 1.91—2.06 (m, 6H), 1.02〜 .15 (m, 3H) 实施例 21
(R)-2-(2-甲氧基 -4- (1 -甲基哌啶 -4-基氨甲酰基)苯氨基) -N,N,5-三甲基 -6-氧代
-6a,7,9,10- -5H-吡嗪并「2, 1 -W喋啶 -8(6H)-羧酸酰胺


第一步
(i?)-2-氯 -5-甲基 -7,8,9,] 0-四氢-577-吡嗪并[2,1-/7]喋啶-6(6& /)-酮 将 2-氯 -5-甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪并 [2,l - z]喋啶 -8(6H)-羧酸叔丁酯 19f(2.15 g, 6.08 mmol)溶解于 50 mL二氯甲烷中, 通入氯化氢气体, 搅拌反应 1 小时, 加入 20 mL水, 加入碳酸钾固体至反应液 pH为 9〜10, 二氯甲烷萃取 (50 mLx3), 合并有机相, 用饱和食盐水洗涤 (50 mLx2), 无水硫酸钠干燥, 过滤, 滤 液减压浓缩, 得到标题产物 (i?)-2-氯 -5-甲基 -7,8,9,10-四氢 -5H-吡嗪并 [2,1-h]喋啶 -6(6aHHll 21a(1..05 g, 白色固体), 产率: 68.3 %。
MS m/z (ESI):254.1 [M+1]
第二步
(i?)-2-氯 -N,N,5-三甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-h]喋啶- 8(6H)-羧酸酰胺 冰浴下, 将 (R)-2-氯 -5-甲基 -7,8,9, 10-四氢 -5H-吡嗪并 [2, 1 -h]喋啶 -6(6aH)-酮 21a(0.25 g, 1 mmol)溶解于 15 mL二氯甲烷中, 依次加入三乙胺 (0.3 mL, 2 mmol) 和二甲基氨基甲酰氯 (0.1 mL, 1.1 mmol), 搅拌反应 0.5小时。 撤去冰浴, 室温搅 拌 1小时,加入 50 mL二氯甲垸,用饱和食盐水洗涤 (50 mLx2),无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得到标 题产物 (i?)-2-氯 -N,N,5-三甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-/ |喋啶 -8(6H)-羧 酸酰胺 21b(250 mg, 白色固体), 产率: 78 %。
MS m/z (ESI):325.3 [M+1]
第三步
(7ζ)-2-(2-甲氧基 - 4- (1-甲基哌啶- 4-基氨甲酰基)苯氨基) -N,N,5-三甲基 -6-氧代
-6a,7,9,10-四氢 -5H-吡嗪并 [2,1-/7]喋啶 -8(6H)-羧酸酰胺 将 4-氨基 -3-甲氧基 -N-(l-甲基 -哌啶 -4-基) -苯甲酰胺 19b(0.072 g, 0.27 mmol), (7?)- 2-氯 -N,N,5-三甲基 -6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-/2]喋啶 -8(6H)-羧酸酰胺 21b(100 mg, 0.3 mmol)和对甲苯磺酸 (0.16 g, 0.82 mmol)溶解于 30 mL 4-甲基 -2- 戊醇中, 回流搅拌反应 3小时。 减压浓缩反应液, 加入 100 mL二氯甲垸和 50 mL 5 %饱和碳酸钾溶液,有机相用饱和食盐水洗涤 (50 mLx3),无水硫酸镁干燥,过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 A纯化所得残余物, 得到标题产物
(R)-2-(2-甲氧基 -4-(1-甲基哌啶 -4-基氨甲酰基)苯氨基 )-N,N,5-三甲基 -6-氧代 - 6a,7,9,10-四氢- 5H-吡嗪并 [2J - ?]喋啶- 8(6H)-羧酸酰胺 21(70 mg, 白色固体),产率: 50 %。
MS ra/z (ES1):552.4 [M+l]
!H MR(400MHz, CDC13, ppm) δ 8.42〜8.45 (d, 1H), 7.75 (s, 1 H), 7.5〜7.51 (d, 1 H), 7.48 (s, 1 H), 4.55〜4.58 (m, l H), 4.1-4.1 5 (m, 2H), 4.02-4.03 (m, 1 H), 4.01 (s, 3H), 3.73—3.76 (m, 1H), 3·32〜3·33 (m, 1H), 3.27 (s, 3H), 3.19—3.23 (m, 2H), 2.93-2.98 (m, 2H), 2.9 (s, 6H), 2.57 (s, 3H), 2.62—2.65 (m, 2H), 2.07—2.10 (m, 2H), 1.85〜1.88 (m, 2H) 实施例 22
( )-4-(8-乙酰基 -5-甲基 -6-氧代 -6,6a,7,8,9,10-六氢- 5H-吡嗪并 n -W喋啶 -2-基氨
-3-甲氧基 -Ν-Π -甲基哌啶 -4-基)苯甲酰胺

第一步
(i?)-2-氯 -5-甲基- 6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-Zz]喋啶 -8(6H)-羧酸苄酯 冰浴下, 将 (i?)- 2-氯 -5-甲基 -7,8,9, 10-四氢 -5H-吡嗪并 [2, 1 -h]喋啶 -6(6aH)-酮 21a(1.98 g, 7.8 mmol)溶解于 50 mL二氯甲垸中,依次加入三乙胺 (1.7 mL, 11.7 mmol) 和氯甲酸苄酯 (1 .4 mL, 9.4 mmol), 搅拌反应 0.5小时。 滴加 20 mL饱和碳酸氢钠
溶液至反应液 pH为 8〜9, 二氯甲烷萃取 (40 mLx2), 用饱和食盐水洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所 得残余物,得到标题产物 氯 -5-甲基- 6-氧代 -6a,7,9,10-四氢 -5H-吡嗪 [2,1-/?]喋啶 - 8(6H)-羧酸苄酯 22a(2.25 g, 白色固体), 产率: 74.2 %。
MS m/z (ESI):388.3 [M+l ]
第二步
{R)-2-{2-甲氧基 -4- ( 甲基哌啶 -4-基氨甲酰基)苯氨基 )-5-甲基 -6-氧代 -6a,7,9, 10-四 氢 -5H-吡嗪并 [2,1-W喋啶 -8(6H)-羧酸苄酯 将 4-氨基 -3-甲氧基 -N-0甲基 -哌啶 -4-基)-苯甲酰胺 19b(616 mg, 2.34 mmol)溶 解于 30 mL 4-甲基- 2-戊醇中, 依次加入 (i?)-2-氯- 5-甲基 -6-氧代 -6aJ,9,10-四氢 -5H- 吡嗪 [2,1-A]喋啶 -8(6H)-羧酸苄酯 22a(999 mg, 2.58 mmol)和对甲苯磺酸 (722 mg, 3.75 mmol), 回流搅拌反应 3小时。 减压浓缩反应液, 加入 50 mL水, 滴加饱和碳 酸钾溶液至反应液 pH为 9〜10, 二氯甲垸萃取 (100 mLx2), 有机相用饱和食盐水 洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂 体系 A纯化所得残余物, 得到标题产物 (i?)-2- (2-甲氧基 -4-(1-甲基哌啶 -4-基氨甲酰 基)苯氨基 )- 5-甲基- 6-氧代 -6a,7,9,10-四氢 -5H-吡嗪并 [2,1 -/0喋啶 -8(6H)-羧酸苄酯 22b(404 mg, 白色固体), 产率: 14.2%。
MS m/z (ESl):615.5 [M+l]
第三步
(i?)-3-甲氧基 -4-(5-甲基- 6-氧代 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 [2,] -h]喋啶 -2-基氨 基)- N-(l -甲基哌啶 -4-基)苯甲酰胺
将 (i?)-2-(2-甲氧基 -4-(1 -甲基哌啶 -4-基氨甲酰基)苯氨基 )-5-甲基- 6-氧代 - 6a,7,9,10-四氢 -5/J-吡嗪并 [2,1- /?]喋啶 -8(6H)-羧酸苄酯 22b(500 mg, 0.8 mmol)溶解 于 100 mL二氯甲烷和甲醇 (V:V=] : 1)混合溶液中,依次加入 5滴冰醋酸和 (l OO mg, 10%)钯 /碳, 3个大气压下, 氢气氛下搅拌反应 24小时。 过滤, 滤液减压浓縮, 得 到标题产物 (i?)-3-甲氧基 -4-(5-甲基 -6-氧代 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 [2,1-h]喋 啶 -2-基氨基) -N-(l-甲基哌啶 -4-基)苯甲酰胺 22c(277 mg, 白色固体), 产率: 71 %。 MS m/z (ESI):481.3 [M+l ]
第四步
((/?)—4— (7-乙酰基 -10-甲基 -9-酮 -6,7,8,8a,9,10-六氢 -5H-2,4,4b,7,10-五氮杂 -菲- 3-基氨 基) -3-甲氧基 -N-(l -甲基 -哌啶 -4-基)-苯甲酰胺 冰浴下, 将 ( -3-甲氧基 -4-(5-甲基 -6-氧代 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 [2,1- h] 喋啶 -2-基氨基) -N-(l-甲基哌啶 -4-基)苯甲酰胺 22c(48 mg, 0.1 mmol)溶解于 30 mL 二氯甲烷中, 加入三乙胺 (0.04 mL, 0.3 mmol)和乙酰氯 (0.008 niL, 0.11 mmol), 搅 拌反应 1小时。 加入 l OO mL水, 有机相用饱和食盐水洗搽 (50 mL), 无水硫酸镁 干燥, 过滤, 滤液减压浓缩, 用薄层层析以展开剂体系 A纯化所得残余物, 得到
标题产物 (i?)-4- (8-乙酰基 - 5-甲基 -6-氧代 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 [2,1-h]喋啶 -2-基氨基 )-3-甲氧基 -N-(l-甲基哌啶 -4-基)苯甲酰胺 22(30 mg, 白色固体), 产率: 57%。
MS m/z (ESI):523.2 [M+1]
Ή NMR(400MHz, CDC13, ppm) δ 8.45〜8.47 (d, 1H), 7.79-7.83 (m, 1H), 7.42-7.5 (m 2H), 4.58-4.65 (m, 3H), 4.17-4.21 (m, 1 H), 4.01 (s, 3H), 3.91 -3.92 (m, 1H), 3.31 (s, 3H), 2.95—3.00 (m, 3H), 2.41 (s, 3H), 2.35 (s, 3H), 2.12—2.26 (m, 4H), 1.97-2.01 (m, 2H), 1.71-1.77 (m, 2H) 实施例 23
(70-3-甲氧基 -4-(5-甲基 -6-氧代 -8-苯基 -6,6a,7,8,9,10-六氢- 5H-吡嗪并「2,1-W喋啶 -2- -N-(l-甲基哌啶 -4-基)苯甲酰胺

第一步
(i?)-哌嗪 -2-羧酸甲酯
将 (7?)-哌嗪 -2-羧酸 23a(10 g, 0.05 mol)溶解于 150 mL甲醇中, 加入 30 mL浓 硫酸, 回流搅拌反应 8小时,冷却至室温,滴加 100 mL三乙胺至反应液 pH为 9〜 10, 减压浓缩反应液, 得到标题产物 (^)-哌嗪 -2-羧酸甲酯 23b, 直接投入下一步。
第二歩
( -哌嗉 -U-二羧酸 3-甲酯 〗-苄酯
冰浴下, 将 ( ?)-哌嗪 -2-羧酸甲酯 23b(7.2 g, 0.05 mol)溶解于 200 mL二氯甲垸 中, 依次加入 20 mL三乙胺和氯甲酸苄酯 (8.5 g, 0.05 mol), 室温搅拌反应 3小时。 减压浓缩反应液, 加入 300 mL乙酸乙酯, 300 mL水, 滴加〗 M盐酸至反应液 pH 为 3〜4, 水层滴加饱和碳酸钠溶液至反应液 pH为 9, 乙酸乙酯萃取 (100 mL>G), 合并有机相, 饱和食盐水洗涤 (50 mL), 无水硫酸镁千燥, 过滤, 滤液减压浓缩, 得到标题产物 ( ?)-哌嗪 -1,3-二羧酸 3-甲酯 1-苄酯 23c(6.6 g, 黄色油状物), 产率: 47 o
MS m/z (ESI):279.1 [M+l ]
第三步
( -哌嗪 -1,2,4-三羧酸 4-苄酯 1-叔丁酯 2-甲酯 冰浴下, 将 (i?)-哌嗪 -1,3-二羧酸 3-甲酯 1-苄酯 23c(6.6 g, 0.023 mol)和三乙胺 (10 mL, 0.07 mol)溶解于 100 mL二氯甲垸中, 加入二碳酸二叔丁酯 (5.4 g, 2.8 mmol), 室温搅拌反应 2小时。 减压浓缩反应液, 加入 200 mL乙酸乙酯, 〗00 mL 水, 滴加 1 M盐酸至反应液 pH为 4〜5, 有机相用饱和食盐水洗涤 (50 mL), 无水 硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 ( ?)-哌嗪 -1,2,4-三羧酸 4-苄酯 1- 叔丁酯 2-甲酯 23d(9 g, 黄色固体), 产率: 100%。
MS nVz (ESI):401.1 [M+23]
第四歩
( ?)-哌嗪 -1 ,2-二羧酸 1-叔丁酯 2-甲酯
将 哌嗪 -1,2,4-三羧酸 4-苄酯 1-叔丁酯 2-甲酯 23d(3 g,8 mmoi:)溶解于 100 mL甲醇中, 依次加入 3滴醋酸和 (300 mg, 10%)钯 /碳, 氢气氛下搅拌反应 12小 时。 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗脱剂体系 B纯化所得残余物, 得 到标题产物 (i?)-哌嗪 -1,2-二羧酸 1-叔丁酯 2-甲酯 23e(l g, 黄色油状物),产率: 52.6 %。
MS m/z (ESI) :245.1 [M+l]
第五步
(i?)-4-苯基 -哌嗪 -1,2-二羧酸 1-叔丁酯 2-甲酯.
氩气氛下, 将叔丁醇钾 (1.45 g, 12.9 mmol), 2- (二叔丁基膦)联苯 (0.1 g, 0.34 mmol)和三 (二亚苄基丙酮)二钯 (0.16 g, 0.17 mmol)溶解于 40 mL甲苯中, 依次加 入 (OR)-哌嗪 -1,2-二羧酸 1-叔丁酯 2-甲酯 23e(2.1 g, 8.6 mmol)和 10 mL溴苯 (1.6 g, 10.3 mmol)的甲苯溶液, 60°C下搅拌反应 2小时, 室温下搅拌反应 12小时。 减压 浓缩反应液, 加入 200 mL乙酸乙酯, 过滤, 滤液减压浓缩, 用硅胶柱色谱法以洗 脱剂体系 B纯化所得残余物, 得到标题产物 (i?)-4-苯基 -哌嗪 -1,2-二羧酸 1-叔丁酯 2-甲酯 23f(500 mg, 黄色固体), 产率: 18.1 %。
MS m/z (ES1):321.2 [M+l]
第六歩
(7?)-4-苯基 -哌嗪 -2-羧酸甲酯盐酸盐
将 ( -4-苯基-哌嗪 -】,2-二羧酸 】-叔丁酯 2-甲酯 23f(0.5 g, 1.5 ramol)溶解于 20 mL二氯甲垸中, 加入 5 mL 6 M氯化氢的 ] ,4-二氧六环溶液, 搅抨反应 2小时。 减压浓缩反应液, 得到标题产物 (i?)-4-苯基 -哌嗪 -2-羧酸甲酯盐酸盐 23g(385 mg, 黄色固体), 产率: 100 %。
MS m/z (ESI):221.2 [M+l]
第七步
(i?)- ] -(2-氯 -5-硝基 -嘧啶 -4-基) -4-苯基-哌嗪 -2-羧酸甲酯 将 4-苯基 -哌嗪 -2-羧酸甲酯盐酸盐 23g(385.05 mg, 1.5 mmol)溶解于 50 mL 环己烷中, 依次加入碳酸氢钠固体 (750 mg, 9 mmol)和 2,4-二氯 -5-硝基 -嘧啶 (290 rag, 1.5 mmol) , 回流搅拌反应 3小时。 冷却至室温, 加入 100 mL水, 100 mL乙 酸乙酯, 水层再用乙酸乙酯萃取 (50 mLx2) , 合并有机相, 用饱和食盐水洗涤 (50 mL) , 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 (i?)-l-(2-氯 -5-硝基- 嘧啶 -4-基) -4-苯基-哌嗪 -2-羧酸甲酯 23h(0.3 g, 黄色固体), 产率: 53.5 %。
MS m/z (ESI):378.2 [M+l]
第八步
(i?)-2-氯 -8-苯基 -7,8,9, 10-四氢 -5H-吡嗪 [2, 1 -h]喋啶 -6(6aH)-酮 将 (7?)-1 -(2-氯- 5-硝基-嘧啶 -4-基) -4-苯基 -哌嗪 -2-羧酸甲酯 23h(300 mg, 0.8 mmol)溶解于 50 mL冰醋酸中, 加入铁粉 (180 mg, 3.2 mmol), 70°C下搅拌反应 3 小时, 减压浓缩反应液, 加入 10 mL水, 二氯甲垸萃取 (50 mLx3), 合并有机相, 用饱和食盐水洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩,. 得到标题产 物 ( -2-氯- 8-苯基 -7,8,9,10-四氢- 5H-吡嗪 [2,1- h]喋啶 -6(6aH)-酮 23j(0.1 g , 黄色固 体), 产率: 40 %。
MS m/z (ES1):316.2 [M+l]
第九步
(i?)-2-氯 -5-甲基 -8-苯基- 7,8,9, 10-四氢 -5H-吡嗪 [2, 1 -/z]喋啶- 6(6aH)-酮 将 (i?)-2-氯 -8-苯基 -7,8,9, 10-四氢 -5H-吡嗪 [2,1-/2]喋啶 -6(6aH)-酮 23j(100 mg, 3.1 mraol),碳酸钾 (87 mg, 6.3 mmol)和对甲苯磺酸甲酉旨 (88 mg, 4.7 mmol)溶解于 50 mL 丙酮中, 回流搅拌反应 3小时。 减压浓缩反应液, 加入 100 mL水和 100 mL二氯 甲烷, 有机相用饱和食盐水洗涤 (50 mL), 无水硫酸镁干燥, 过滤, 滤液减压浓缩, 得到标题产物 (7?)-2-氯 -5-甲基 -8-苯基 -7,8,9,] 0-四氢 -5H-吡嗪 [2,1 -A]喋啶 -6(6aH)-酮 23k(80 mg, 黄色固体), 产率: 78.4 %。
MS m/z (ESI):330.1 [M+l]
第十步
(i?)-3-甲氧基 -4-(5-甲基 -6-氧代 -8-苯基 -6,6a,7,8,9,10-六氢 -5H-吡嗪并 [2, 1-W喋啶 -2- 基氨基) -N-(l -甲基哌啶 -4-基)苯甲酖胺
将 4-氨基 -3-甲氧基 -N-(l -甲基 -哌啶 -4-基) -苯甲酰胺 19b(64 mg, 0.24 mmol),
((/?)- 3-氯 -10-甲基 -7-苯基- 6,7,8,8a-四氢 -5H,10H-2,4,4b,7,10-五氮杂-菲 -9-酮 23k(80 mg, 0.24 mmol)和对甲苯磺酸 (230 mg, 1.2 mmol)溶解于 30 mL4-甲基 -2-戊醇中, 回流搅拌反应 3小时。 减压浓縮反应液, 加入 100 mL饱和碳酸钾溶液, 二氯甲烷 萃取 (100 mLx2), 合并有机相, 用饱和食盐水洗涤 (50 mL), 无水硫酸镁干燥, 过 滤, 滤液减压浓缩, 用薄层层析以展开剂体系 A纯化所得残余物, 得到标题产物 ( )-3-甲氧基 -4-(5-甲基- 6-氧代 -8-苯基 -6,6a,7,8,9, 10-六氢 - 5H-吡嗪并 [2,l -/z]喋啶 -2- 基氨基) -N- (1 -甲基哌啶 -4-基)苯甲酰胺 23(30 mg, 白色固体), 产率: 22.5 %。
MS m/z (ESI):557.4 [M+l ]
Ή NMR(400MHz, CDC13, ppm) δ 8·54〜8.57 (d, 1H), 7.77 (s, 1H), 7.46-7.47 (d, 1H), 7.33-7.37 (m, 3H), 7.06—7.07 (d, 2H), 6.96-7.0 (t, 1H), 4.76〜4.80 (m, 1H),
4.26-4.33 (m, 2H), 4.13〜4J 9 (m, 1H), 4.01 (s, 3H), 3.75-3.78 (m,〗H), 3.38 (s, 3H), 3.】 〜 3.20 (m, 3H), 2.89-2.95 (m, 2H), 2.52-2.57 (ni, 5H), 2.】6〜2.19 (m, 2H), 1.95〜 2.01 (m, 2H) 测试例:
生物学评价 测试例 1、 本发明化合物对 P】k高表达细胞的增殖抑制测定
下面的体外试验是用来测定本发明化合物对高表达 Plk 的细胞株一人宫颈癌 细胞 Hda的增殖抑制活性。
以下所述的体外细胞试验可测定受试化合物对高表达 Plk 的肿瘤细胞的增殖 抑制活性, 其活性可用 IC5Q值来表示。 此类试验的一般方案如下: 首先将 Hela细 胞 (购于 Institute of biochemistry and cell biology)以适宜细胞浓度 (e.g. 3000 个细胞 /mL medium)接种在 96孔培养板上, 然后将细胞在二氧化碳恒温箱内进行培养, 让它们生长至过夜,更换培养基为加有一系列浓度递度 (一般 7到 9个浓度)受试化 合物溶液的培养基, 将培养板重新放回培养箱, 连续培养 72个小时。 72小时后, 可用 CCK8方法进行测试化合物对于抑制细胞增殖活性。 IC5o值可通过一系列不同 浓度下, 受试化合物对于细胞的抑制数值进行计算。
本发明化合物的活性
本发明化合物的生化学活性通过以上的试验进行测定, 测得的 IC5o值见下表。

9 2
10 0.4
11 -1禾 P Π -2的混合物 0.6
16-1和 16-2的混合物 4
17-1和 17-2的混合物 4
结论: 本发明化合物均对 Hela细胞具有明显的增殖抑制活性。 测试例 2、 本发明化合物对 Plk高表达细胞的增殖抑制测定
下面的体外试验是用来测定本发明化合物对高表达 Plk 的细胞株一人结肠癌 细胞 HCT-116的增殖抑制活性。
以下所述的体外细胞实验可测定受试化合物对高增殖的消化系肿瘤细胞的增 殖抑制活性, 化合物的抑制活性可用 IC5Q值来表示。 实验方案简述如下: 首先将 以 DMEM 附加 10% FCS (购于 Gibco)作为完全培养基的 HCT-116 细胞 (购于 Institute of biochemistry and cell biology), 以适宜的细胞浓度 (e.g. 3000 个 /mL medium)接种在 96孔培养板上, 然后在 37°C , 5% C02条件下, 于恒温培养箱内培 养过夜。 待细胞贴壁后, 将培养基更换为含有受试化合物梯度浓度 (一般为 7或 9 个浓度点)溶液的新鲜培养基。此后,将细胞培养板在前述条件下连续培养 72个小 时。 72小时后, 采用 CCK8 (Cell Counting Kit- 8, 货号: CK04, 购于 Dojindo)方法 测定化合物对于细胞增殖的抑制活性。 化合物的 IC5Q值可通过不同浓度下受试化 合物对于细胞增殖的抑制数值计算得出。
本发明化合物的活性
本发明化合物的生化学活性通过以上的试验进行测定, 测得的 IC5Q值见下表。

结论: 本发明化合物均对 HCT-116细胞具有明显的增殖抑制活性。 测试例 3、 本发明化合物对 Plkl激酶抑制活性的测定
体外 Pjkl激酶活性通过以下的方法进行测试。
下面所述的方法可用来测定本发明化合物对 Plk]激酶活性的抑制能力, 并通过 IC50值表示。化合物的半数抑制浓度 IC5Q (将酶活性抑制至 50 %时所需的化合物浓度) 是通过将一定量的激酶与特定的底物及不同浓度的待测化合物混合反应后测定得
到一系列的抑制率,然后运用计算工具计算出的。本实验所用的 Plkl激酶为重组人 源蛋白,反应体系购自 MBL公司 (Po -like kinase 1 Assay/Inhibitor Screening Kit, # CY-]J 63)。Pjk]酶在该反应体系中与多肽底物以及不同浓度的受试化合物共同进行 反应 (25 °C, 30分钟), 随后用一抗 Anti- Phospho-Serine/Threonine Polyclonal Antibody (PPT- 07)和二抗 HRP- conjugated Anti-rabbit IgG对磷酸化底物进行标记, 最后检测 A450 nM下的读数对 Plkl激酶活性进行定量。
本发明化合物的活性
本发明化合物的生化学活性通过以上的试验进行测定, 测得的 IC5Q值见下表。

结论: 本发明化合物均对 P】k-1激酶具有明显的增殖抑制活性。 药代动力学评价 测试例 4、本发明实施例化合物 2和实施例 8-1和 8-2的混合物的药代动力学测试
1、 摘要
以大鼠为受试动物, 应用 LC/MS/MS法测定了大鼠分别静脉注射给予实施例 2 化合物, 实施例 8-1和 8- 2的混合物后不同时刻血浆中的药物浓度, 研究本发明化合 物在大鼠体内的药代动力学行为, 评价其药动学特征。
2、 试验方案:
2.1 试验药品
实施例 2化合物, 实施例 8-1和 8-2的混合物
2.2 试验动物
健康成年 SD大鼠 8只, 雌雄各半, 购自上海西普尔 -必凯实验动物有限公司, 动物生产许可证号: SCXK (沪) 2008-0016。
2.3 药物配制
称取一定量药物, 力口 DMSO 0.5 mL使溶解, 加入 0.1 M HC1 1.5 mL, 生理盐水 稀释至终体积, 使药物浓度为 2.5 mg/mL。
2.4 给药
SD大鼠 8只, 雌雄各半, 平均分成 2组, 禁食过夜后经尾静脉注射给药, 剂量 均为 25 mg/kg。
3、 操作
取给药后各时刻的大鼠血浆 25 L, 加入内标溶液 20 μί, 甲醇 150 L, 涡旋 混合 3 分钟, 离心 10 分钟 (13500 转 /分钟), 血浆样品取上清液 5 μ 进行 LC-MS/MS分析。 主要药代动力学参数采用 DAS 2.0软件计算。
4、 药代动力学参数结果
本发明化合物的药代动力学参数如下:

结论: 实施例 2化合物, 实施例 8-1和 8-2的混合物的药代吸收良好。
Claims
权利要求书: 通式( I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映 异构体、 及其混合物形 及可药用的盐:

R1和 R2各自独立地选自氢原子或烷基;
或者, R1与 R2与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 0〜2个 N、 0或 S(0)m杂原子, 并且所述 3〜8元环任选进一步被一个或 多个选自垸基、烷氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9或 -C(0)OR9的取代基所取代;
R3选自氢原子、 烷基、 烯基、 炔基、 环烷基、 杂环基、 芳基或杂芳基, 其中 所述垸基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多 个选自烷基、垸氧基、 卤素、羟基、氰基、硝基、 芳基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9或 -C(0)OR9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有〗〜2个 N、 0或 8(0),„杂原子, 并且所述 3〜8元环任选进一歩被一个或 多个选自垸基、烷氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)0R9、 -COR9或 -C(0)0R9的取代基所取代;
R4和 R5各自独立地选自氢原子、 烷基、 卤素、 氰基或硝基;
L选自亚烷基, 任选进一歩被一个或多个选自卤素、氰基、硝基或烷基的取代 基所取代, 其中所述的垸基任选进一步被一个或多个卤素所取代;
R6选自烷基, 其中所述的垸基任选进一步被一个杂环基所取代;
当 R6选自未取代的垸基时, A选自双环垸基、 双杂环基、 螺环垸基或螺杂环 基, 其中所述双环垸基、 双杂环基、 螺环垸基或螺杂环基任选进一步被一个或多 个 R12所取代;
当 R6选自杂环基取代的垸基时, A选自环烷基、 杂环基、 双环烷基、 双杂环 基、 桥环烷基、 杂桥环垸基、 螺环烷基或螺杂环基, 其中所述环烷基、 杂环基、 双环垸基、 双杂环基、 桥环垸基、 杂桥环垸基、 螺环垸基或螺杂环基任选进一步 被一个或多个 R12所取代;
R7和 R8各自独立地选自氢原子、 垸基、 环垸基、 杂环基、 芳基或杂芳基, 其 中所述烷基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 烷氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)0R9、 -COR9, C(0)0R9、 -S^ONR^R1 1 , -CONK ER1 1或 -NR' R11的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其屮所述 3〜8元杂环基内含有一个或多个 N、 0或 8(0)|11杂原子, 并且所述 3〜8元杂环基 任选进一步被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、 氰基、 -S(0)0R9、 -C0R9、 C(0)0R9、 -S(O)ONR10R"、 - CONK ER11或 -NR' R11 的取代基所取代;
R9选自氢原子、 烷基、 环垸基或芳基, 其中所述环垸基或芳基任选进一歩被 一个或多个烷基所取代;
R1 Q和 R1 1选自氢原子、 垸基、 环垸基或芳基;
R12选自垸基、垸氧基、环烷基、卤素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -C(0)OR9、 -OC(0)R9、 -0(CH2)RC(0)OR9、 -OC(0)NR7R8、 -S(0)MR9、 -OS(0)OR9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的垸基、 烷氧基、 环 烷基或杂环基任选进一步被一个或多个选自垸基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
■ m为 0或 1 ;
n为 0, 1或 2; 且
r为 1 , 2或 3。
2、 根据权利要求 1所述的通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中: R1选自氢原子;
R2选自烷基;
R3选自垸基或环烷基;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元杂环基,其中所述 3〜 8元杂环基内含有 1〜2个 N、 0或 S(0)n杂原子, 并且所述 3〜8元杂环基任选进 一步被一个或多个选自烷基、 烷氧基、 卤素、 羟基、 氰基、 硝基、 羰基、 芳基、 -S(0)ON 7R8 -CONR7R8、 -NR7R8、 -S(0)OR9、 -COR9或 -C(0)OR9的取代基所取 代;
R4和 R5各自独立选自氢原子、 卤素或垸基;
R6选自烷基, 其中所述的垸基任选进一步被一个杂环基所取代;
当 R6选自未取代的垸基时, A选自双环烷基、 双杂环基、 螺环垸基或螺杂环 基, 其中所述的双环垸基、 双杂环基、 桥环垸基、 桥杂环基、 螺环垸基或螺杂环 基任选进一步被一个或多个 R12所取代;
当 R6选自杂环基取代的垸基时, A选自环垸基、 杂环基、 双环垸基、 双杂环 基、 桥环垸基、 桥杂环基、 螺环垸基或螺杂环基, 其中所述的环烷基、 杂环基、 双环垸基、 双杂环基、 桥环垸基、 桥杂环基、 螺环烷基或螺杂环基任选进一歩被 一个或多个 R12所取代;
R7和 R8各自独立地选自氢原子、 垸基、 环垸基、 杂环基、 芳基或杂芳基, 其 中所述烷基、 环烷基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 烷氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟基、 氰基、 羧酸、 羧酸酯、 -S(0)OR9、 -COR9、 -S(O)ONR10R!\ -CONK ER11或 -NRWR11的取代基所取代;
或者, R7和 R's与其相连接的 N原子一起形成一个 3〜8元环, 其中所述 3〜8 元环内含有一个或多个 N、 0或 S(C m杂原子, 并且所述 3〜8元杂环上任选进一 歩被一个或多个选自垸基、 烷氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟基、 氰基、 羧酸、 羧酸酯、 羰基、 -S(0)0R9、 -COR9, -S(O)ONR,0Rn , -CONFER11或 -NR^R" 的取代基所取代;
R9选自氢原子、 垸基、 环垸基或芳基;
R1 Q和 R11选自氢原子、 垸基、 环垸基或芳基;
R12选自垸基、垸氧基、环垸基、卤素、羟基、氰基、硝基、羰基、 -S(0)ON 7R8, -C0NR7Rs、 -C(0)OR9、 -0C(0)R9、 -0(CH2)rC(0)OR9、 -OC(0)NR7R8、 -S(0)mR9、 -0S(0)0R9, -NHC(0)R9或- COR9的取代基所取代, 其中所述的垸基、 垸氧基、 环 烷基或杂环基任选进一步被一个或多个选自垸基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
ni为 0或 1;
n为 0, 1或 2; 且
r为 1, 2或 3。
3、 根据权利要求 1所述的通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中: R1和 R2各自独立地选自氢原子或垸基;
R3选自氢原子、 烷基、 烯基、 炔基、 环烷基、 杂环基、 芳基或杂芳基, 其中 所述垸基、 烯基、 炔基、 环烷基、 杂环基、 芳基或杂芳基任选进一步被一个或多 个选自烷基、烷氧基、 卤素、羟基、氰基、硝基、芳基、 -S(0)ONR 8、 -CONR7R8、 -NR7R8、 -S(0)0R9, - COR9或 -C(0)0R9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 1〜2个N、 0或 8(0)11、杂原子, 并且所述 3〜8元环任选进一步被一个或 多个选自烷基、垸氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 - NR7R8、 -S(0)0R9、 - COR9或 -C(0)0R9的取代基所取代;
R4和 R5各自独立地选自氢原子、 垸基、 卤素、 氰基或硝基;
L选自亚烷基, 任选进一歩被一个或多个选自卤素、氰基、硝基或垸基的取代 基所取代, 其中所述的垸基任选进一步被一个或多个卤素所取代;
R6选自垸基;
A 选自双环垸基、 双杂环基、 螺环烷基或螺杂环基, 其中所述双环垸基、 双 杂环基、 螺环烷基或螺杂环基任选进一歩被一个或多个 R12所取代;
R7和 R8各自独立地选自氢原子、 烷基、 环垸基、 杂环基、 芳基或杂芳基, 其 中所述垸基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 烷氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)OR9、 -COR9, C(0)OR9, -S(O)ONR10R1 J , -CONK ER1 1或 -NRWR11的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其中所述 3〜8元杂环基内含有一个或多个 N、 0或 8(0)111杂原子, 并且所述 3〜8元杂环基 任选进一步被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、 氰基、 -S(0)0R9、 - C0R9、 C(0)0R9、 -S(O)ONR10RN , -C0NR1 ()R "或 -NR10R" 的取代基所取代;
R9选自氢原子、 垸基、 环垸基或芳基, 其中所述环垸基或芳基任选进一步被 一个或多个烷基所取代;
RI Q和 R1 1选自氢原子、 垸基、 环垸基或芳基;
R12选自垸基、垸氧基、环烷基、卤素、羟基、氰基、硝基、羰基、 -S(0)ONR7R8、 - CONR7R8、 - C(0)0R9、 -0C(0)R9、 -0(C¾)RC(0)0R9、 -OC(0)NR7R8、 -S(0)MR9、 -0S(0)0R9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的垸基、 烷氧基、 环 烷基或杂环基任选进一步被一个或多个选自垸基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
m为 0或 1;
n为 0, 1或 2 ; 且
r为 1, 2或 3。
4、 根据权利要求 1所述的通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中: R1和 R2各自独立地选自氢原子或垸基;
R3选自氢原子、 垸基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基, 其中 所述垸基、 烯基、 炔基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多 个选自垸基、垸氧基、 卤素、羟基、氰基、硝基、 芳基、 -S(0)ONR7R8、 -CONR7R8、 -NR7RS、 -S(0)0R9、 - COR9或 -C(0)0R9的取代基所取代;
或者, R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元 环内含有 1〜2个^^、 0或 8(0)111杂原子, 并且所述 3〜8元环任选进一歩被一个或 多个选自垸基、烷氧基、卤素、羟基、氰基、硝基、芳基、杂芳基、羰基、 -S(0)ONR7R8、 -CONR7R8、 -NR7RS、 -S(0)0R9、 -COR9或 -C(0)0R9的取代基所取代; R4和 R5各自独立地选自氢原子、 烷基、 卤素、 氰基或硝基;
L选自亚垸基, 任选进一歩被一个或多个选自卤素、氰基、硝基或垸基的取代 基所取代, 其中所述的垸基任选进一歩被一个或多个卤素所取代;
R6选自垸基, 其中所述的垸基进一步被一个杂环基所取代;
当 R0选自杂环基取代的垸基时, A选自环垸基、 杂环基、 双环垸基、 双杂环 基、 桥环垸基、 杂桥环垸基、 螺环垸基或螺杂环基, 其中所述环垸基、 杂环基、 双环垸基、 双杂环基、 桥环垸基、 杂桥环垸基、 螺环垸基或螺杂环基任选进一步 被一个或多个 R12所取代;
1 7和1^各自独立地选自氢原子、 垸基、 环垸基、 杂环基、 芳基或杂芳基, 其 中所述烷基、 环垸基、 杂环基、 芳基或杂芳基任选进一步被一个或多个选自垸基、 垸氧基、杂环基、芳基、杂芳基、 卤素、羟基、氰基、 -S(0)OR9、 -COR9, C(0)0R9, -S(O)ONR10Ru , -CONR'V或 -NR1()R"的取代基所取代;
或者, R7和 R8与其相连接的 N原子一起形成一个 3〜8元杂环基, 其中所述 3〜8元杂环基内含有一个或多个 N、 0或 S(0)m杂原子, 并且所述 3〜8元杂环基 任选进一歩被一个或多个选自垸基、 垸氧基、 杂环基、 芳基、 杂芳基、 卤素、 羟 基、 氰基、 -S(0)OR9、 -COR9, C(0)0R9、 -S^ONR^R11 , -CONFER11或 -NI^R' 1 的取代基所取代;
R9选自氢原子、 垸基、 环烷基或芳基, 其中所述环垸基或芳基任选进一歩被 一个或多个烷基所取代;
R1Q和 R11选自氢原子、 垸基、 环烷基或芳基;
R12选自垸基、烷氧基、环烷基、卤素、羟基、氰基、硝基、羰基、 -S(0)0NR7Rs、 -CONR7R8、 - C(0)OR9、 - 0C(0)R9、 -0(C¾)rC(0)0R9、 - OC(0)NR7R8、 -S(0)mR9、 -0S(0)0R9、 -NHC(0)R9或 -COR9的取代基所取代, 其中所述的垸基、 烷氧基、 环 垸基或杂环基任选进一步被一个或多个选自烷基、 环垸基、 杂环基、 芳基或杂芳 基的取代基所取代;
m为 0或 1;
n为 0, 1或 2; 且
r为 1, 2或 3。
5、 根据权利要求 1所述的通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中 R3 选自垸基或环垸基。
6、 根据权利要求 1所述的通式(I )所示的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 其中所 述的化合物包括:



7、 一种通式 (IA)所示的化合物或可药用的盐, 其作为制备通式 (I)化合物的中 间体, 其中: 
(ΙΑ)
其中:
G 选自离去基团, 优选为选自卤素、 甲磺酰基、 对甲苯磺酰基、 三氟甲磺酰 基或垸氧基;
R选自氢原子或烷基;
R1选自氢原子或烷基;
R2和 R3与其相连接的原子一起形成一个 3〜8元环, 其中所述 3〜8元环内含 有 1〜2个 N、 0或 S(0)n杂原子,并且所述 3〜8元环任选进一步被一个或多个选 自垸基、 垸氧基、 芳基、 卤素、 羟基、 氰基、 羰基、 羧酸、 羧酸酯、 -S(0)ONR7R8、 -CONR7R8、 -NR7R8、 -S(0)OR9或 -COR9的取代基所取代;
, R7〜R9的定义如权利要求 1中所述。
8、 根据权利要求 7所述的通式 (IA)所示的化合物或可药用的盐, 其中所述的

9、 一种制备通式 (I)化合物的万法, 包括:

通式 (IA)化合物与通式 (IB)化合物或通式 (IB)化合物的盐反应, 得到通式 (I)化 合物;
其中:
R选自甲基;
G、 Ri〜R3的定义如权利要求 7中所述;
A、 n、 L、 R4〜R6的定义如权利要求 1中所述。
10、 一种药物组合物, 其含有治疗有效剂量的根据权利要求 1〜6任一项所述 的化合物或其互变异构体、 外消旋体、 对映异构体、 非对映异构体、 及其混合物 形式、 及药学上可以接受的盐, 及可药用的载体或赋形剂。
11、 根据权利要求〗〜 6任一项所述的化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 根据权利 要求 10所述的药物组合物在制备治疗细胞增殖类疾病的药物中的用途。
.
12、根据权利要求 11所述的用途, 其中所述的细胞增殖类疾病为癌症、感染、 炎症或自身免疫性疾病。
13、 根据权利要求 12所述的用途, 其中所述的癌症选自非小细胞肺癌、 鳞状 细胞癌、 乳腺癌、 卵巢癌、 宫颈癌、 乳头状癌或结肠直肠癌。
14、 一种治疗细胞增殖类疾病的方法, 该方法包括给予需要治疗的患者有效 治疗量的根据权利要求 1〜6任一项所述的化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 根据权利 要求 10所述的药物组合物, 其中所述的细胞增殖类疾病为癌症、 感染、 炎症及自 身免疫性疾病, 所述的癌症为宫颈癌或结肠癌。
15、 根据权利要求 1〜6任一项所述的化合物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, .根据权利 要求 10所述的药物组合物, 其作为治疗细胞增殖类疾病的药物, 其中所述的细胞 增殖类疾病为癌症、 感染、 炎症及自身免疫性疾病, 所述的癌症为宫颈癌或结肠 癌
16、 根据权利要求 1〜6任一项所述的化^ ·物或其互变异构体、 外消旋体、 对 映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 根据权利 要求 10所述的药物组合物在制备 Plk激酶抑制剂中的用途。
17、 一种抑制 Plk激酶的方法, 该方法包括给予需要治疗的患者有效治疗量的 根据权利要求 1〜6任一项所述的化合物或其互变异构体、外消旋体、对映异构体、 非对映异构体、 及其混合物形式、 及药学上可以接受的盐, 根据权利要求】0所述 的药物组合物。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180002067.3A CN102421779B (zh) | 2010-03-19 | 2011-01-14 | 二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 |
| TW100115019A TW201229050A (en) | 2011-01-14 | 2011-04-28 | Dihydropteridinone derivatives, preparation process and pharmaceutical use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201010131438.5 | 2010-03-19 | ||
| CN2010101314385A CN102190669A (zh) | 2010-03-19 | 2010-03-19 | 二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011113293A1 true WO2011113293A1 (zh) | 2011-09-22 |
Family
ID=44599624
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2011/000062 Ceased WO2011113293A1 (zh) | 2010-03-19 | 2011-01-14 | 二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 |
Country Status (2)
| Country | Link |
|---|---|
| CN (2) | CN102190669A (zh) |
| WO (1) | WO2011113293A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102017005089A1 (de) | 2016-05-30 | 2017-11-30 | Bayer Pharma Aktiengesellschaft | Substitulerte 3,4-Dihydrochinoxalin-2(1H)-one |
| DE102017005091A1 (de) | 2016-05-30 | 2017-11-30 | Bayer Pharma Aktiengesellschaft | Substituierte 3,4-Dihydropyrido[2,3-b]pyrazin-2(1H)-one |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114276349B (zh) * | 2021-12-13 | 2024-04-09 | 复旦大学 | 2-氨基喋啶酮类衍生物或其盐及其制备方法和用途 |
| WO2026050923A1 (zh) * | 2024-09-04 | 2026-03-12 | 上海再启生物技术有限公司 | 一种jak激酶抑制剂关键中间体的合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1745081A (zh) * | 2003-02-26 | 2006-03-08 | 贝林格尔英格海姆法玛两合公司 | 二氢蝶啶酮、其制法及作为药物制剂的用途 |
| CN101039676A (zh) * | 2004-08-14 | 2007-09-19 | 贝林格尔·英格海姆国际有限公司 | 具有长贮存期的二氢蝶啶酮的输注溶液 |
| CN101541800A (zh) * | 2006-10-25 | 2009-09-23 | 色品疗法有限公司 | 用于治疗癌症的作为plk抑制剂的蝶啶衍生物 |
-
2010
- 2010-03-19 CN CN2010101314385A patent/CN102190669A/zh active Pending
-
2011
- 2011-01-14 WO PCT/CN2011/000062 patent/WO2011113293A1/zh not_active Ceased
- 2011-01-14 CN CN201180002067.3A patent/CN102421779B/zh not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1745081A (zh) * | 2003-02-26 | 2006-03-08 | 贝林格尔英格海姆法玛两合公司 | 二氢蝶啶酮、其制法及作为药物制剂的用途 |
| CN101039676A (zh) * | 2004-08-14 | 2007-09-19 | 贝林格尔·英格海姆国际有限公司 | 具有长贮存期的二氢蝶啶酮的输注溶液 |
| CN101541800A (zh) * | 2006-10-25 | 2009-09-23 | 色品疗法有限公司 | 用于治疗癌症的作为plk抑制剂的蝶啶衍生物 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102017005089A1 (de) | 2016-05-30 | 2017-11-30 | Bayer Pharma Aktiengesellschaft | Substitulerte 3,4-Dihydrochinoxalin-2(1H)-one |
| DE102017005091A1 (de) | 2016-05-30 | 2017-11-30 | Bayer Pharma Aktiengesellschaft | Substituierte 3,4-Dihydropyrido[2,3-b]pyrazin-2(1H)-one |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102421779B (zh) | 2015-03-11 |
| CN102421779A (zh) | 2012-04-18 |
| CN102190669A (zh) | 2011-09-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN115192577B (zh) | Kras突变蛋白抑制剂 | |
| WO2024083246A1 (en) | Kras inhibitors | |
| EP4712961A2 (en) | Kras g12s and g12c inhibitors | |
| WO2024032703A1 (en) | Heterocyclic compounds, compositions thereof, and methods of treatment therewith | |
| WO2023150284A2 (en) | Quinazoline pan-kras inhibitors | |
| WO2020146613A1 (en) | Kras g12c inhibitors | |
| WO2022266258A1 (en) | Compounds and methods for the targeted degradation of irak-4 | |
| JP2022530049A (ja) | 統合ストレス応答経路のモジュレーター | |
| TWI828712B (zh) | 作為trk抑制劑的雜環化合物 | |
| WO2020173426A1 (zh) | 基于戊二酰亚胺骨架的含硫化合物及其应用 | |
| KR20170042779A (ko) | Mdm2-p53 억제제로서의 신규 스피로[3h-인돌-3,2'-피롤리딘]-2(1h)-온 화합물 및 유도체 | |
| EP4037677B1 (en) | Antagonists of the muscarinic acetylcholine receptor m4 | |
| JP6110787B2 (ja) | ピリミドジアゼピノン化合物 | |
| JP7840334B2 (ja) | 芳香族複素環式化合物、薬学的組成物、及びその使用 | |
| EP4178964A1 (en) | Benzodiazepine derivatives useful in treating a respiratory syncytial virus infection | |
| EP1515965A1 (de) | Phenylaminopyrimidine und ihre verwendung als rho-kinase inhibitoren | |
| CN112292374A (zh) | 一种新型磷酸肌醇3-激酶抑制剂及其制备方法和用途 | |
| JP2023522725A (ja) | 3-アザビシクロアルキル誘導体およびこれを含む薬剤学的組成物 | |
| WO2022135390A1 (zh) | 己酮糖激酶抑制剂及其用途 | |
| WO2024193464A1 (zh) | 一种含氮三并环衍生物及其在医药上的应用 | |
| TW202428267A (zh) | 用於降解egfr激酶之化合物 | |
| WO2011113293A1 (zh) | 二氢喋啶酮类衍生物、其制备方法及其在医药上的应用 | |
| CN116354932A (zh) | 嘧啶衍生物及其制备方法和用途 | |
| CN121100113A (zh) | 取代的喹喔啉 | |
| WO2023151635A1 (zh) | 基于喹唑啉取代戊二酰亚胺骨架的化合物及其应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180002067.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11755610 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11755610 Country of ref document: EP Kind code of ref document: A1 |