WO2011138657A1 - Aryl substituted olefinic compounds as pde10a inhibitors - Google Patents
Aryl substituted olefinic compounds as pde10a inhibitors Download PDFInfo
- Publication number
- WO2011138657A1 WO2011138657A1 PCT/IB2011/000948 IB2011000948W WO2011138657A1 WO 2011138657 A1 WO2011138657 A1 WO 2011138657A1 IB 2011000948 W IB2011000948 W IB 2011000948W WO 2011138657 A1 WO2011138657 A1 WO 2011138657A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- methyl
- prop
- chlorophenyl
- ylmethoxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 ***c(cc1)ccc1C(CO*)=CC1=CC=C/C=C/C=C*(*)=C1 Chemical compound ***c(cc1)ccc1C(CO*)=CC1=CC=C/C=C/C=C*(*)=C1 0.000 description 8
- AZUDJIDLVVUBJT-HYARGMPZSA-N C=Cc1c(C=C)nc(COc(cc2)ccc2/C(/c2nnc[o]2)=C\c2ccc[o]2)cc1 Chemical compound C=Cc1c(C=C)nc(COc(cc2)ccc2/C(/c2nnc[o]2)=C\c2ccc[o]2)cc1 AZUDJIDLVVUBJT-HYARGMPZSA-N 0.000 description 1
- OMZHVELIBXDEQB-ABPOBPNVSA-N CC(OC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c1ccccc1)=N)=N Chemical compound CC(OC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c1ccccc1)=N)=N OMZHVELIBXDEQB-ABPOBPNVSA-N 0.000 description 1
- YVSALFTUYBPYQI-UHFFFAOYSA-N CCc1n[o]c2ccccc12 Chemical compound CCc1n[o]c2ccccc12 YVSALFTUYBPYQI-UHFFFAOYSA-N 0.000 description 1
- WKJJVJQGVVCNSP-JJIBRWJFSA-N CNC(/C(/c(cc1)ccc1OCc1nc2ccccc2cc1)=C/c1ccccc1)=O Chemical compound CNC(/C(/c(cc1)ccc1OCc1nc2ccccc2cc1)=C/c1ccccc1)=O WKJJVJQGVVCNSP-JJIBRWJFSA-N 0.000 description 1
- HXKKLSTXRJBBDM-BWAHOGKJSA-N Clc1ccc(/C=C(/COc2cccnc2)\c(cc2)ccc2OCc2nc(cccc3)c3cc2)cc1 Chemical compound Clc1ccc(/C=C(/COc2cccnc2)\c(cc2)ccc2OCc2nc(cccc3)c3cc2)cc1 HXKKLSTXRJBBDM-BWAHOGKJSA-N 0.000 description 1
- DQXZHDSJTVBSIS-PLRJNAJWSA-N Fc1ccc(/C=C(/CN2CCNCC2)\c(cc2)ccc2OCc2nc(cccc3)c3cc2)cc1 Chemical compound Fc1ccc(/C=C(/CN2CCNCC2)\c(cc2)ccc2OCc2nc(cccc3)c3cc2)cc1 DQXZHDSJTVBSIS-PLRJNAJWSA-N 0.000 description 1
- HRSXJMVXCMBWEH-NHFJDJAPSA-N N#CCCNC(/C(/c1ccc(CCc2nc3ccccc3cc2)cc1)=C/c(cc1)ccc1Cl)=O Chemical compound N#CCCNC(/C(/c1ccc(CCc2nc3ccccc3cc2)cc1)=C/c(cc1)ccc1Cl)=O HRSXJMVXCMBWEH-NHFJDJAPSA-N 0.000 description 1
- UDOVTZQJJZXTED-HYORXLTRSA-O N=COC(/C(/c(cc1)ccc1OCc(cc1)nc2c1[o]cn2)=C/c(cc1)ccc1Cl)=[NH2+] Chemical compound N=COC(/C(/c(cc1)ccc1OCc(cc1)nc2c1[o]cn2)=C/c(cc1)ccc1Cl)=[NH2+] UDOVTZQJJZXTED-HYORXLTRSA-O 0.000 description 1
- ATGYWWKBFYRPSZ-ROVZZSFWSA-O N=COC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(c(F)c1)ccc1Cl)=[NH2+] Chemical compound N=COC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(c(F)c1)ccc1Cl)=[NH2+] ATGYWWKBFYRPSZ-ROVZZSFWSA-O 0.000 description 1
- AMRNGUCGIMSWOK-YTNWSCEGSA-N N=COC(/C(/c1cc(OCc2nc3ccccc3cc2)ccc1)=C/c(cc1)ccc1Cl)=N Chemical compound N=COC(/C(/c1cc(OCc2nc3ccccc3cc2)ccc1)=C/c(cc1)ccc1Cl)=N AMRNGUCGIMSWOK-YTNWSCEGSA-N 0.000 description 1
- KRFDHNQWMPZZTN-ZXVVBBHZSA-N O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)N1CCCC1 Chemical compound O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)N1CCCC1 KRFDHNQWMPZZTN-ZXVVBBHZSA-N 0.000 description 1
- KSBIHPVFGDZWEW-TURZUDJPSA-N O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)NC1CCNCC1 Chemical compound O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)NC1CCNCC1 KSBIHPVFGDZWEW-TURZUDJPSA-N 0.000 description 1
- DFKZEZRBLSZTGH-KOEQRZSOSA-N O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)Nc1ncc[s]1 Chemical compound O=C(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c(cc1)ccc1Cl)Nc1ncc[s]1 DFKZEZRBLSZTGH-KOEQRZSOSA-N 0.000 description 1
- XCTZLMOVKZNLGX-JJIBRWJFSA-N OCCNC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c1ccncc1)=O Chemical compound OCCNC(/C(/c(cc1)ccc1OCc1nc(cccc2)c2cc1)=C/c1ccncc1)=O XCTZLMOVKZNLGX-JJIBRWJFSA-N 0.000 description 1
- OTDGWMABWOZGOL-HVFKXOOMSA-N [N-]=C(/C(/c(cc1)ccc1OCc1ccc2[nH]cnc2c1)=C/c(cc1)ccc1Cl)OC=N Chemical compound [N-]=C(/C(/c(cc1)ccc1OCc1ccc2[nH]cnc2c1)=C/c(cc1)ccc1Cl)OC=N OTDGWMABWOZGOL-HVFKXOOMSA-N 0.000 description 1
- PBPYVSBAPDYZHB-UHFFFAOYSA-O [NH3+]C(C(c(cc1)ccc1OCc1nc(cccc2)c2cc1)=Cc(cc1)ccc1Cl)=CC=N Chemical compound [NH3+]C(C(c(cc1)ccc1OCc1nc(cccc2)c2cc1)=Cc(cc1)ccc1Cl)=CC=N PBPYVSBAPDYZHB-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/12—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D215/14—Radicals substituted by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/28—Radicals substituted by singly-bound oxygen or sulphur atoms
- C07D213/30—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/20—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/10—1,3,4-Oxadiazoles; Hydrogenated 1,3,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Definitions
- the present invention relates to aryl substituted olefinic compounds and their use in treating or preventing diseases, conditions and/or disorders by inhibiting phosphodiesterase 1 OA (PDE1 OA) enzyme.
- PDE1 OA phosphodiesterase 1 OA
- the cyclic nucleotide phosphodiesterases are a class of intracellular enzymes related to a family of phosphohydrolases that selectively catalyze the hydrolysis of the 3' cyclic phosphate bonds of adenosine and/or guanosine 3', 5' cyclic monophosphates (cAMP/cGMP) into their respective 5' monophosphates (5'-AMP/GMP).
- the cyclic nucleotides cAMP and cGMP act as second messengers of intracellular signal transduction in response to extracellular stimuli and are synthesized from ATP and GTP by the catalytic cyclization activity of enzymes adenylyl and guanylyl cyclases, respectively.
- PDEs play a very important role in signal transduction by regulating the cellular levels of these second messengers (cAMP/cGMP) in the way o f controlling their rates of degradation.
- Mammalian PDEs are composed of 21 genes and are categorized into 1 1 families (PDE 1 to PDE1 1 ), with each family typically having several different isoforms and splice variants, based on sequence homology, enzymatic properties, biochemical characteristics and sensitivity to inhibitors. These unique PDEs differ in their three-dimensional structures, kinetic properties, modes of regulation, intracellular localization, cellular expression patterns with different individual isozymes modulating distinct regulatory pathways in the cell. Furthermore, PDEs are di ferentially expressed throughout the body, including in the central nervous system, serving distinct physiological functions. Thus PDEs provide an unique opportunity of selective drug targets for the potential treatment of specific disease states.
- PDEs are also subclassified based on different substrate specificites into cAMP selective (PDE4, 7 and 8), cGMP selective (PDE5, 6 and 9) and cAMP and cGMP dual selective (PDEl , 2, 3, 10 and 1 1 ).
- the human PDEI OA family enzyme was reported essentially at the same time by two different groups (Fujishige K et al., J. Biol. Chem. vol. 274, p.18438- 1 8445, ( 1 999); Loughney K et al., Gene vol. 234, p.
- mice PDEI OA 1 is a 779 amino acid protein that hydrolyzes both cAMP and cGMP to AMP and GMP, respectively.
- PDEI OA hydrolyzes cAMP with a Km of 0.05 ⁇ and cGMP with a Km of 3 ⁇ , suggesting that the affinity of PDEI OA for cAMP is higher than for cGMP.
- Vmax ratio of cGMP/cAMP is 4.7
- PDE 1 OA is a unique cAMP-inhibited cGMP phosphodiesterase [Soderling, S KI et al. ' , Proc. Natl. Acad. Sci. USA vol. 96 p. 7071 -7076, ( 1 999); Fuj ishige et al ., J. Biol. Chem. vol. 274, p. 1 8438- 1 8445, (1999)].
- the PDE I OA family of polypeptides shows a lower degree of sequence homology to previously identified PDE families. These low degrees of sequence homology of PDEI OA family of polypeptide make them insensitive to certain inhibitors that are known to be specific for other known PDE families (US 6,350,603, incorporated herein by reference).
- PDE I OA which is one of the PDE subtypes
- the expression of its mR A has been identified in many tissues and organs such as striatum, testis, kidney, thyroid gland, pituitary gland, thalamus, cerebellum, heart, lungs and placenta, cells such as aortic smooth m-.iscle cells and aortic endothelial cells, cells of cancers such as lung small cell carcinoma, breast cancer and large bowel cancer.
- aortic smooth m-.iscle cells and aortic endothelial cells cells of cancers such as lung small cell carcinoma, breast cancer and large bowel cancer.
- this enzyme is suggested to be involved in, for example, onset or progression of various disorders and diseases related to striatal, basal ganglia related dysfunctions/disorders such as schizophrenia (positive, negative & cognitive symptoms), parkinson's disease, Huntington disease, obsessive compulsive disorders, sleep disorders and disorders of changed circardian rhythm [Siuciak JA et al., Neuropharmacology, vol 51 , p. 374-385, (2006); Threlfell S ef al ., JPET, vol 328, p.
- Neurodegenerative diseases There are very few effective treatments for neurological disorders characterized by progressive cell loss, known as neurodegenerative diseases, as well as those involving acute cell loss, such as stroke and trauma.
- neurodegenerative diseases such as those involving acute cell loss, such as stroke and trauma.
- neurosis which has been linked to altered striatal function relating to changes in expression of the enzyme PDE I OA [J. A. Siuciak, et al. Neuropharmacology, vol . 5 1 , p. 374- 385, (2006)].
- Striatal dysfunction is implicated in a number of CNS disorders including psychosis, schizophrenia, obsessive-compulsive disorders, Parkinson's disease and Huntington's disease.
- PDEI OA inhibitors are useful for treating and/or preventing various diseases caused by enhanced activity of PDE I OA, possibly with reduced side effects (for example, a neural disease such as Parkinson's disease, Huntington disease or Alzheimer's disease, dyskinesia, hypogonadism, diabetes, an ischemic heart disease, hypertension, an inflammatory disease, a disease of the digestive system, an allergic disease, osteoporosis, pain or a malignant tumor).
- a neural disease such as Parkinson's disease, Huntington disease or Alzheimer's disease, dyskinesia, hypogonadism, diabetes, an ischemic heart disease, hypertension, an inflammatory disease, a disease of the digestive system, an allergic disease, osteoporosis, pain or a malignant tumor.
- WO 2003/000269, WO 2003/0141 15, WO 2003/0141 16, WO 2003/0141 1 7, WO 2003/051 877, WO 2006/034491 and WO 2006/034512 describe PDE10 inhibitors for treatment of neurodegenerative diseases, cancer, diabetes and its related disorders.
- WO 2006/072828, WO 2008/084299, WO 2003/093499, WO 2005/082883, WO 2005/120514, WO 2006/01 1 040, WO 2006/070284, WO 2007/077490, WO 2007/085954, WO 2007/096743, WO 2007/1 291 83, WO 2008/001 182, WO 2008/0041 17, WO 2008/020302, WO 2009/070584, WO 2009/068320 ; WO 2009/068246 and WO 2009/036766 describe PDE10 inhibitors for treatment of obesity, diabetes, certain central nervous system disorders, neurodegenerative and psychiatric disorders.
- WO 2009/029214, WO 2009/025839 and WO 2009/025823 describe PDE I O inhibitors lor treatment of obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder and obsessive-compulsive disorder.
- WO 2009/143 178, WO 2009/1 52825, WO ' 2009/1 58393, WO 2009/1 58467, WO 2009/158473, WO 2010/006130, WO 2010/017236, WO 201 0/027097 and WO 201 0/030027 describe PDE10 inhibitors for treatment of anxiety, schizophrenia, drug addiction, movement disorder, certain central nervous system disorders, neurodegenerative and psychiatric disorders.
- the present invention relates to compounds of the formula (I):
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- Y is a bond, or is selected from -(CR 4 R 3 ) P - and -S0 2 -;
- Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
- G ] and G 2 are independently selected from, hydrogen, cyano, nitro, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, haloalkyl, haloalkoxy, cycloalkyl, aryl, heteroaryl , heterocyclyl, -(CR 4 R 5 ) P R 8 , -(CR 4 R 5 ) p C(0)R 8 , -(CR 4 R 5 ) P -NR 9 R 10 , -(CR 4 R 5 ) p -OR' ' , -C(0)R 8 , - C(0)(CR 4 R 5 ) p R 8 , -C(0)NR 9 R 10 , -NR 9 R 10 , -NR 3 C(0)R 8 , -NR 3 C(0)NR 9 R 10 , -N(R 3 )S0 2 R 8 , - OC(0)R 8 and -OC(0)NR 9 R 10 ; with the provis
- R is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR a ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy.
- R 8 is selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl. cyanoalkyl. haloalkoxy, cycloalkyl.
- cycloalkylalkyl cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl, -(CR D R E ) Q R A , -C(0)R A , -C(0)NR B R C , -C(0)OR A , -NR B R C , -NR B C(0)R A , -NR B C(0)NR B R C , -N(R b )S0 2 R 8 , -OC(0)R A , -OC(0)NR B R , - S(0)R A , -S0 2 R A , -SONR B R C , -S0 2 NR B R C and -SR A ;
- R 9 and R 1 0 which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl. hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl .
- R 9 and R 10 together with the nitrogen atom to which they are attached, may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N;
- R " is selected from hydrogen, nitro, substituted or unsubstituted alkyl, alkenyl. al kynyl . alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, cycloalkyl, cycloalkylalkyl, cycloalkenyl.
- R A , R D and R which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl , hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR R , -NR R R G , -C(0)N R L R E J cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R B and R C which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, -C(0)OR F , -C(0)NR F R E , cycloalkyl, aryl, heteroaryl and heterocyclyl : at each occurrence, R f and R 8 are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- 'n' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive;
- 'q' is an integer ranging from 1 to 3, both inclusive;
- the compounds of formula (I) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- Y is a bond, or is selected frorn -(CR 4 R 5 ) p - and -S0 2 -;
- Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, helerocyclvl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
- R ! and R 2 are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl heteroaryl, heteroarylalkyl, -C(0)R A , -C(0)NR B R C , -C(0)OR A , -NR B R C , -NR H C(0)R A , - N R B C(0)NR B R C , -N(R b )S0 2 R A , -OC(0)R A , -OC(0)
- R 3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR A ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)OR A , -NR B R C and -SR A ; or R 4 and R 5 , at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
- R 9 and R 1 0 which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalky L - (CR D R C ) Q R ⁇ -C(0)R A , -C(0)NR B R C , -C(0)OR A , -S(0)R A , -S0 2 R A , -S(0)NR B R C and -S0 2 R B R T: ; or R 9 and R 1 0 together with the nitrogen atom to which they are attached,
- R A , R D and R E which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR F , -NR F R 8 , -C(0)NR F R 8 , cycloalkyl. aryl. heteroaryl and heterocyclyl;
- R B and R C which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, -C(0)0R', -C(0)NR F R , cycloalkyl, aryl, heteroaryl and heterocyclyl; at each occurrence, R f and R s are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- ' ⁇ ' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive;
- 'q' is an integer ranging from 1 to 3, both inclusive.
- R 1 is halogen (e.g., fluorine or chlorine), haloalkyl (e.g., trifluoromethyl), alkoxy (e.g., methoxy) or haloalkoxy (e.g., difluoromethoxy or trifluoromethoxy); and ⁇ ' is 1 .
- halogen e.g., fluorine or chlorine
- haloalkyl e.g., trifluoromethyl
- alkoxy e.g., methoxy
- haloalkoxy e.g., difluoromethoxy or trifluoromethoxy
- R 1 0 is hydrogen, alkyl (e.g., methyl or ethyl), cyanoalkyl (e.g., cyanoethyl), haloalkyl (e.g., trifluoromethyl, trifluoroethyl), hydroxyalkyl (e.g., hydroxyethyl), cycloalkyl (e.g., cyclopropyl), heterocyclyl (e.g., piperidinyl) or heteroaryl (e.g., thiazole).
- alkyl e.g., methyl or ethyl
- cyanoalkyl e.g., cyanoethyl
- haloalkyl e.g., trifluoromethyl, trifluoroethyl
- hydroxyalkyl e.g., hydroxyethyl
- cycloalkyl e.g., cyclopropyl
- heterocyclyl
- R 1 0 is -(CR D R E ) Q R A ; wherein R D and R E are independently alkyl (e.g., methyl or ethyl), hydroxyalkyl (e.g., hydroxymethyl) or phenyl and R A is hydrogen.
- R D and R E are independently alkyl (e.g., methyl or ethyl), hydroxyalkyl (e.g., hydroxymethyl) or phenyl and R A is hydrogen.
- 'q' is 1.
- R 1 0 is -(CR D R E ) Q R A ; wherein both R D and R C are hydrogen and R A is -NR R R E or -C(0)NR'R s .
- R* and R G are independently hydrogen, alkyl (e.g., methyl or ethyl) or -C(0)alkyl (e.g., -C(O)methyl); and 'q' is 1 or 2.
- R 1 0 is -(CR D R E ) Q R A ; wherein both R D and R E are hydrogen or alkoxy (e.g.. methoxy) and R'' is hydrogen.
- 'q' is 2.
- R 1 0 is -(CR D R E ) Q R A ; wherein R A is hydrogen; R D is alkyl (e.g., methyl) and R C is -C(0)OR ⁇
- R 1 is hydrogen or alkyl (e.g., ethyl); and 'q ' is 1 .
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- Y is a bond, or is selected from -(CR 4 R 5 ) P - and -S0 2 -;
- Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl. heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
- R 1 and R 2 which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl . alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl. cycloalkylalkyl .
- cycloalkenyl cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl , heteroaryl, heteroaryl alkyl, -C(0)R A , -C(0)NR B R C , -C(0)OR A , -NR B R C , -NR B C(0)R A , - N R B C(0)NR B R C , -N(R B )S0 2 R A , -OC(0)R A , -OC(0)NR R C , -S(0)R A , -S0 2 R A , -SON R. B R C , - S0 2 NR B R C and -SR";
- R is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, a'kynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR A ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxy] , substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)OR a , -NR b R c and -SR a ; or R 4 and R ' ⁇ at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
- R 9 and R 10 which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryl alkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylal kyl, - (CR d R e ) q R a , -C(0)R a , -C(0)NR R c , -C(0)OR a , -S(0)R a , -S0 2 R ;1 , -S(0)NR b R c and -S0 2 N R h R c ; or R 9 and R
- R a , R d and R e which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl. hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR r , -NR r R g , -C(0) R f R , cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R b and R c which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl , alkoxy, -C(0)OR f , -C(0)NR r R , cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R f and R g are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
- ⁇ ' is an integer ranging from 0 to 5, both inclusive;
- 'n' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive;
- 'q' is an integer ranging from 1 to 3, both inclusive.
- R is halogen (e.g., fluorine or chlorine);
- R 10 is alkyl (e.g., methyl), cyanoalkyl (e.g., cyanoethyl) or -(Cl ⁇ R ⁇ q R 11 ; wherein R d and R c are independently alkyl (e.g., methyl or ethyl) or hydroxyalkyl (e.g.. hydoxymethyl) and R a is hydrogen.
- R d and R c are independently alkyl (e.g., methyl or ethyl) or hydroxyalkyl (e.g.. hydoxymethyl) and R a is hydrogen.
- 'q' is 1 .
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl ;
- Y is a bond, or is selected from -(CR R 5 ) P - and -S0 2 -;
- Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic; al each occurrence, R 1 and R 2 , which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylal kyl, heteroaryl, heteroarylalkyl, -C(0)R A , -C(0)NR B R C , -C
- R 3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR A ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl , aryl, heterocyclyl, heteroaryl, -C(0)OR A , -NR B R C and -SR A ; or R 4 and R 5 , at each occurrence, together with the carbon atom to which they are attached, may form an optionall y substituted cycloalkyl or heterocyclyl ring;
- R 9 and R 1 0 which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl. hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylal kyl, - (CR D R E ) Q R A , -C(0)R A , -C(0)NR B R C , -C(0)OR A , -S(0)R A , -S0 2 R A , -S(0)NR B R C and -S0 2 NR B R C ; or R 9 and R 1 0 together with.
- the nitrogen atom to which they are attached may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N ;
- R A , R D and R E which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR F , -NR F R G , -C(0)NR R R G , cycloalkyl, aryl, heteroaryl and heterocyclyl; at each occurrence, R and R c , which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl. cyanoalkyl, alkoxy, -C(0)OR r , -C(0)NR r R g , cycloalkyl, aryl, heteroaryl and heterocyclyl; .
- R f and R are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- 'n' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive;
- 'q' is an integer ranging from 1 to 3, both inclusive.
- R 1 is halogen (e.g., fluorine or chlorine); and 'm' is 1 or 2.
- R 9 is hydrogen or alkyl, preferably methyl.
- R 10 is alkyl (e.g., methyl), alkynyl (e.g., prop-2-nyl), -C(0)R a , cyanoalkyl (e.g., cyanoethyl) or heterocyclyl (e.g., pyrrolidinyl, pyrrolidin-2-one, piperidinyl, piperidin-2- one or ethyl piperidine- l -carboxylate).
- R" is alkyl (e.g., methyl) or alkoxyalkyl (e.g., -CH 2 -OCH 3 ).
- R 10 is -(CR d R e ) q R a ; wherein both R d and R e are hydrogen and R a is alkoxy (e.g., methoxy) or -NR f R 8 .
- R 1 and R s are independently hydrogen, alkyl (e.g., methyl) or -C(0)alkyl, preferably -C(O)methy) ; and 'q ' is 2.
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- X is a bond, or is selected from -0-, -S-, -NR 3 -, -S(O)-, -S0 2 -, -(CR"R 5 ) P O-
- Z is selected from substituted or unsubstituted alkyl. cycloalkyl. ary 1. helerocycl yl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic:
- Met is selected from heteroaryl and heterocyclyl; wherein said heteroaryl and heterocyclyl may optionally be substituted with atleast one R ;
- R 1 and R 2 which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl.
- R 3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl. alkenyl . alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR A ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro. cyano, hydroxyl, . substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)OR A , -NR B R C and -SR A ; or R" and R 5 . at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
- R 12 is selected from hydrogen, halogen, hydroxyl, cyano, substituted or unsubstituted alkyl, hydroxyalkyl, haloalkyl and alkoxy;
- R a is selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl , haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR F , -NR R R S , -C(0)NR ( R S : cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R B and R C which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl . alkoxy, -C(0)OR F , -C(0)NR , R , cycloalkyl, aryl, heteroaryl and hetei Ocyclyl ; at each occurrence, R f and R ⁇ are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- 'n' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive.
- ring A is aryl, preferably phenyl.
- R 1 is halogen (e.g., fluorine or chlorine), haloalkyl (e.g., trifluoromethyl). alkoxy (e.g., methoxy or ethoxy) or haloalkoxy (e.g., difluoromethoxy or trifluoromethoxy); and 'nf is 1 or 2.
- halogen e.g., fluorine or chlorine
- haloalkyl e.g., trifluoromethyl
- alkoxy e.g., methoxy or ethoxy
- haloalkoxy e.g., difluoromethoxy or trifluoromethoxy
- 'nf is 1 or 2.
- R 2 is halogen (e.g., fluorine or chlorine) or alkoxy (e.g., methoxy or ethoxy); and ' n' is 1 .
- substituted or unsubstituted alkyl e.g., methyl or ethyl
- haloalkyl e.g., trifluoromethyl
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- Y is a bond, or is selected from -(CR 4 R ;, ) p - and -S0 2 -;
- Z is selected from substituted or unsubstituted alky], cycloalkyl, aryl. heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
- Met is selected from heteroaryl and heterocyclyl; wherein said heteroaryl and
- heterocyclyl may optionally be substituted with atleast one R ;
- R 1 and R 2 which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl. alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl. cycloa! ky alky L cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl . heterocyclylalky! .
- R 3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl. alkenyl, alkynyl. alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR a ;
- R 4 , R 5 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy.
- cycloalkyl aryl, heterocyclyl, heteroaryl, -C(0)OR a , -NR b R c and -SR a ; or R 4 and R 5 , at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
- R 12 is selected from hydrogen, halogen, hydroxyl. cyano. substituted or unsubstituted alkyl, hydroxyalkyl, haloalkyl and alkoxy;
- R A is selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR F , -NR R R , -C(0)NR' R 5 ⁇ cycloalkyl, aryl, heteroaryl and heterocyclyl; '
- R B and R C which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl . alkoxy, -C(0)OR F , -C(0)NR R R G , cycloalkyl, aryl, heteroaryl and heterocyclyl ;
- R f and R 8 are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- ' ⁇ ' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive.
- R 1 is halogen, preferably chlorine; and 'm' is 1 .
- A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl
- R 5 ) P (R 6 )C C(R 7 )-;
- Y is a bond, or is selected from -(CR 4 R 3 ) P - and -S0 2 -;
- Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
- R 1 and R 2 which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylal k yL cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl. heterocyclyl. heterocyclylalkyl .
- R 3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)OR a ;
- R 4 , R 3 , R 6 and R 7 which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl. haloalkoxy.
- cycloalkyl aryl, heterocyclyl, heteroaryl, -C(0)OR a , -NR b R c and -SR a ; or R 4 and R 5 , at each occurrence, together with the carbon atom to which they are attached, may form an optionally substituted cycloalkyl or heterocyclyl ring;
- R" is selected from hydrogen, nitro, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, cycloalkyl, cycloalkylalkyl, cycloalkenyL cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl.
- R , R D and R C which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)OR R , -NR'R 8 , -C(0)NR R R 8 S cycloalkyl ; aryl, heteroaryl and heterocyclyl;
- R B and R C which may be the same or different, are independent ly selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoal kyl. alkoxy, -C(0)OR.', -C(0)NR F R G , cycloalkyl, aryl, heteroaryl and heterocyclyl;
- R 1 and R are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
- 'm' is an integer ranging from 0 to 5, both inclusive;
- 'n' is an integer ranging from 0 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive;
- 'q' is an integer ranging from 1 to 3, both inclusive.
- R 1 is halogen, preferably fluorine or chlorine; and 'm' is 1 .
- R 1 1 is aryl, preferably phenyl.
- the present invention also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient, such as a pharmaceutically acceptable carrier or diluent.
- the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein.
- the compounds described in the present patent application may be associated with a pharmaceutically acceptable excipient, such as a carrier or a diluent or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- the compounds and pharmaceutical compositions of the present invention are useful for inhibiting PDE 1 0A, which is related to a variety of disease states.
- the present invention further provides a method of treating a disease, condition or disorder modulated by a PDE10A, in a subject by administering to the subject in need thereof a therapeutically effective amount of a compound of formulas (I) to (If) or a pharmaceutical composition described herein.
- the illustrative examples of the present invention are screened for 'in vivo' PDE 1 OA based efficacy in a rat model of Dizocilpine (MK-801) - induced psychotic behavior.
- Example 91 The effect of Example 91 on MK-801 - induced psychosis behavior in female SD rats as shown in Figure 1 and the effect of Example 1 77 on MK-801 - induced psychosis behavior in female SD rats as shown in Figure 2.
- halogen or halo means fluorine, chlorine, bromine or iodine.
- alkyl refers to a hydrocarbon chain radical that includes solely carbon and hydrogen atoms in the backbone, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n- propyl, 1 -methylethyl (isopropyl), n-butyl, n-pentyl and 1 , 1 -dimethylethyl (t-butyl). Unless set forth or recited to the contrary, all alkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- alkenyl refers to a hydrocarbon chain containing from 2 to 10 carbon atoms and including at least one carbon-carbon double bond. Examples of such alkenyl moiety includes, but are not limited to, ethenyl, 1 -propenyl, 2-propenyl (allyl), wo-propenyl, 2-methyl- l - propenyl, 1 -butenyl and 2-butenyl. Unless set forth or recited to the contrary, all alkenyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- alkynyl refers to a hydrocarbyl radical having at least one carbon-carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 1 0 carbon atoms being preferred).
- alkynyl moiety include, but are not limited to, ethynyl, propynyl and butynyl. Unless set forth or recited to the contrary, all alkynyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- alkoxy refers an alkyl group attached via an oxygen linkage to the rest of the molecule. Examples of such alkoxy-moiety include, but are not limited to, -OCFI3 and -OC2M5. Unless set forth or recited to the contrary, all alkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
- alkoxyalkyl or alkyloxyalkyl refers to an alkoxy or alkyloxy group as defined above directly bonded to an alkyl group as defined above. Example of such alkoxyalkyl moiety includes, but are not limited to, -CH 2 OCH 3 and -CH 2 OC 2 H 5 . Unless set forth or recited to the contrary, all alkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- haloalkyl refers to at least one halo group (selected from F. CI, Br or I), linked to an alkyl group as defined above.
- haloalkyl moiety include, but are not limited to, trifluoromethyl, difluoromethyl. 2,2,2-trifluoroethyl and fluoromethyl groups. Unless set forth or recited to the contrary, all haloalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- haloalkoxy refers to an alkoxy group substituted with one or more halogen atoms.
- haloalkoxy include but are not limited to fluoromethoxy, difluoromethoxy, trifluoromethoxy, 2,2,2-trifiuoroethoxy,. pentafluoroethoxy, pentachloroethoxy, chloromethoxy. dichlorormethoxy, trichloromethoxy and 1 -bromoethoxy.
- all haloalkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
- ' 'hydroxyalkyl refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by hydroxyl groups.
- Examples of hydroxyalkyl moiety include, but are not limited to -CH2OH and -C 2 H 4 OH. Unless set forth or recited to the contrary, all hydroxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- cyanoalkyl refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by cyano groups.
- Examples of cyanoalkyl moiety include, but are not limited to -CH 2 CN and -C 2 H 4 CN. Unless set forth or recited to the contrary, all cyanoalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- cycloalkyl denotes a non-aromatic mono or multicyclic ring system of 3 to about 1 2 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl .
- multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups or sprirobicyclic groups, e.g., sprio(4.4)non-2-yl, spiro
- cycloalkylalkyl refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group.
- the cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- Examples of cycloalkylalkyl moiety include, but are not limited to cyclopropylmethyl, cyclobutylethyl, and eye lopentyl ethyl. Unless set forth or recited to the contrary, all cycloalkylalkyl groups described or claimed herein may be substituted or unsubstituted.
- cycloalkenyl refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cvclopentenyl . Unless set forth or recited to the contrary, all cycloalkenyl groups described or claimed herein may be substituted or unsubstituted.
- cycloalkenylalkyl refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, directly attached to an al kyl group.
- the cycloalkenylalkyl group may be attached to the main structure at any carbon atom in t he alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all cycloalkenylalkyl groups described or claimed herein may be substituted or unsubstituted.
- aryl refers to an aromatic radical having 6 to 14 carbon atoms, including monocyclic, bicyclic and tricyclic aromatic systems, such as phenyl, naphthyl, tetrahydronapthyl, indanyl and biphenyl. Unless set forth or recited to the contrary, all aryl groups described herein may be substituted or unsubstituted.
- aryloxy refers to an aryl group as defined above attached via an oxygen linkage to the rest of the molecule.
- Examples of aryloxy moiety include, but are not l imited to phenoxy and naphthoxy. Unless set forth or recited to the contrary, all aryloxy groups described herein may be substituted or unsubstituted.
- arylalkyl refers to an aryl group as defined above directly bonded to an alkyl group as defined above.
- arylalkyl moiety include, but are not limited to -CH2C6H5 and -C2H4C6H5. Unless set forth or recited to the contrary, all arylalkyl groups described herein may be substituted or unsubstituted.
- heterocyclic ring or “heterocyclyl” unless otherwise speci fied refers to substituted or unsubstituted non-aromatic 3 to 15 membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur.
- the heterocyclic ring radical may be a mono-, bi- or tricyclic ring system, which may- include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sul fur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states.
- the nitrogen atom may be optionally quaternized; also, unless otherwise constrained by the definition the heterocyclic ring or heterocyclyl may optionally contain one or more olefinic bond(s). Examples of such heterocyclic ring radicals include, but are not limited to azepinyl.
- azetidinyl benzodioxolyl, benzodioxanyl, chromanyl, dioxolanyl, dioxaphospholanyl. decahydroisoquinolyl, indanyl, indolinyl, isoindolinyl, isochromanyl.
- phenoxazinyl quinuclidinyl, tetrahydroisquinolyl, tetrahydrofuryl, tetrahydropyranyl, thiazolinyl, thiazolidinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sul f ne. 1 ,4 azathianyl, 7-aza-spiro[3,3]heptanyl, 7-spiro[3,4]octanyl, and 7-aza- spiro[3.4]octanyl .
- heterocyclic ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure.
- al l heterocyclyl groups described herein may be substituted or unsubstituted.
- heterocyclylalkyl refers to a heterocyclic ring radical directly bonded to an alkyl group.
- the heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all heterocyclylalkyl groups described herein may be substituted or unsubstituted.
- heteroaryl refers to substituted or unsubstituted 5 to 1 4 membered aromatic heterocyclic ring radical with one or more heteroatom(s) independently selected from N, O or S.
- the heteroaryl may be a mono-, bi- or tricyclic ring system.
- the heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure. Examples of such heteroaryl ring radicals include, but are not limited to oxazolyl, isoxazolyl, imidazolyl, furyl. indolyl, isoindolyl. pyrrolyl, triazolyl.
- thiazolyl isothiazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, benzofuranyl, benzothiazolyl, benzoxazolyl, benzimidazolyl.
- benzothienyl benzopyranyl, carbazolyl, quinolinyl, isoquinolinyl, quinazolinyl, cinnolinyl, naphthyridinyl, pteridinyl, purinyl, quinoxalinyl, quinolyl, isoquinolyl, thiadiazolyl, indolizinyl, acridinyl, phenazinyl, phthalazinyl, furo[3,2-6]pyridinyl, pyrrolo[3 ,2- >] pyridinyl, thieno
- all heteroaryl groups described herein may be substituted or unsubstituted.
- heteroarylalkyl refers to a heteroaryl ring radical directly bonded to an alkyl group.
- the heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all heteroarylalkyl groups described herein may be substituted or unsubstituted.
- substituted or unsubstituted heterocyclylalkyl ring substituted or unsubstituted heteroarylalkyl . substituted or unsubstituted heterocyclic ring, substituted or unsubstiuted guanidine.
- substituted heterocyclylalkyl ring substituted or unsubstituted heteroarylalkyl, or substituted or unsiibstituted heterocyclic ring.
- the substituents in the aforementioned "substituted” groups cannot be further substituted.
- substituent on “substituted alkyl” is "substituted aryl”
- the substituent on “substituted aryl” cannot be "substituted alkenyl”.
- treating or “treatment” of a state, disorder or condition includes: (a) preventing or delaying the appearance of clinical symptoms of the state, disorder or condition developing in a subject that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition; (b) inhibiting the state, disorder or condition, i.e., arresting or reducing the development of the disease or at least one clinical or subclinical symptom thereof; or (c) relieving the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
- subject includes mammals (especially humans). Other mammals i ncl ude domestic animals (e.g., household pets including cats and dogs) and non-domestic animals (such as wildlife).
- a “therapeutically effective amount” means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment.
- the “therapeutically effective amount” will vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subject to be treated.
- salts forming part of this patent application include salts derived from inorganic bases (such as Li, Na, K, Ca, Mg, Fe, Cu, Zn, and Mn), salts of organic bases (such as N,N'-diacetylethylenediamine, glucamine, triethylamine, choline, hydrox ide, dicyclohexylamine, metformin, benzylamine, trialkylamine, and thiamine), salts of chiral bases (such as alkylphenylamine, glycinol, and phenyl glycinol), salts of natural amino acids (such as glycine, alanine, valine, leucine, isoleucine, norleucine, tyrosine, cystine, cysteine, methionine, proline, hydroxy proline, histidine, ornithine, lysine, arginine, and serine), salts of non-natural amino acids (such as D
- salts include acid addition salts (where appropriate) such as sulphates, nitrates, phosphates, perchlorates, borates, hydrohalides, acetates (such as trifiuoroacetate), tartrates, maleates, citrates, fumarates, succinates. palmoates. methanesulphonates, benzoates, salicylates, benzenesulfonates, ascorbates, glycerophosphates and ketoglutarates.
- acid addition salts such as sulphates, nitrates, phosphates, perchlorates, borates, hydrohalides, acetates (such as trifiuoroacetate), tartrates, maleates, citrates, fumarates, succinates. palmoates. methanesulphonates, benzoates, salicylates, benzenesulfonates, ascorbates, glycerophosphates and ketoglutarates.
- Compounds described herein can comprise one or more asymmetric carbon atoms and thus can occur as racemic mixtures, enantiomers and diastereomers. These compounds can also exist as conformers/rotamers. All such isomeric forms of these compounds are expressly included in the present patent application. Although the specific compounds exemplified in this application may be depicted in a particular stereochemical configuration, compounds having either the opposite stereochemistry at any given chiral centre are envisioned as a part thereof. In addition, compounds of Formulas (I) to (If) can exist in different geometrical isomeric forms. Unless otherwise stated a reference to a particular compound includes al l such isomeric forms, including racemic and other mixtures thereof.
- the pharmaceutical composition of the present patent application comprises one or more compounds described herein and one or more pharmaceutically acceptable excipients, carriers, diluents or mixture thereof.
- the compounds described herein may be associated with one or more pharmaceutically acceptable excipients, carriers, diluents or mixture thereof in the form of capsule, sachet, paper or other container.
- suitable carriers include, but are not limited to, water, salt sol utions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides. pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethyl cellulose and polyvinylpyrrolidone.
- the carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical composition may also include one or more pharmaceutical ly acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmetic pressure, buffers, sweetening agents, flavoring agents, colorants or any combination of the foregoing.
- the pharmaceutical composition of the patent application may be formulated so as to provide quick, sustained or delayed release of the active ingredien after administration to the subject by employing methods known in the art.
- compositions of the present patent application may be prepared by conventional techniques, e.g., as described in Remington: The Science and Practice of Pharmacy, 20 th Ed., 2003 (Lippincott Williams & Wilkins).
- the active compound is mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, sachet, paper or other container.
- the carrier serves as a diluent, it may be a solid, semi-solid or liquid material that acts as a vehicle, excipient or medium for the active compound.
- the active compound is adsorbed on a granular solid contai ner, for example, in a sachet.
- compositions may be in conventional forms, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
- the route of administration may be any route which effectively transports the active compound of the patent application to the appropriate or desired site of action.
- Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal , parenteral, rectal, depot, subcutaneous, intravenous, intraurethral. intramuscular, intranasal, ophthalmic (such as with an ophthalmic solution) or topical (such as with a topical ointment).
- the oral route is preferred.
- Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like arc particularly suitable for oral application. Preferable carriers for tablets, dragees. or capsules include lactose, cornstarch and/or potato starch. A syrup or elixir is used in cases where a sweetened vehicle is employed.
- Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions.
- injectable solutions or suspensions preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil .
- Suitable doses of the compounds for use in treating the diseases and disorders descri bed herein can be determined by those skilled in the relevant art.
- Therapeutic doses are generally identified through a dose ranging study in humans based on preliminary evidence derived from the animal studies. Doses must be sufficient to result in a desired therapeutic benefit without causing unwanted side effects.
- the daily dosage of the PDE 1 0A inhibitors can range from about 0.1 to about 30.0 mg/Kg.
- Mode of administration, dosage forms, suitable pharmaceutical excipients, diluents or carriers can also be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present patent application.
- the present patent application provides a method of treating a disease, condition or disorder modulated by a PDE10A, in a subject by administering to the subject in need thereof a therapeutically effective amount of a compound or a pharmaceutical composition described herein.
- the present patent application further provides a method of treating diseases, disorders or conditions, modulated by a PDE1 0A in mammals including human., of neuropsych iatric. neurodegenerative, neurological, neuroendocrinological nature such as, but not limiting to.
- schizophrenia, psychoses, schizoaffective disorders positive symptoms of schizophrenia including delusions, disordered thoughts and speech, and tactile, auditory, visual, olfactory and gustatory hallucinations, paranoia, paranormal behaviors, negative symptoms of schizophrenia l ike deficits of normal emotional responses or of other thought processes including flat or bl unted affect and emotion, poverty of speech (alogia), inability to experience pleasure (anhedonia), lack of desire to form relationships (asociality), and lack of motivation (avol ition) leading to poor quality of life, functional disabil ities typically regarded as manifestations of psychosis and other comorbidities like cognitive, executive, attention, learning, memory, spatial memory and social cognitive functions, Tic disorders like Tourette's syndrome, autism, autism spectrum disorders, attention deficit hyperactivity disorders (ADHD), pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS), mood disorders, anxiety, depression, major depressive disorders, bipolar disorders, manias, aggression, obsessive compuls
- neurological disorders consisting of movement disorders, ataxias, sensation disorders, cognitive disorders related to multiple sclerosis, amyotrophic lateral sclerosis, abnormalities of brain, spinal cord, nerves leading to symptoms such as paralysis, seizures, catatonias, catalepsies, muscle rigidities, muscle weakness, poor coordination, loss of sensation, confusion, mental suffering, pain and altered levels of consciousness and various other diseases, disorders or conditions related to neuroendocrinological and metabol ic mani festations like change of circardian rhythms, sleep disorders, insomnia, jet lags, eating disorders li ke anorexia nervosa, bulimia nervosa, exercise bulimia or binge eating disorder, aggressive behaviours, obsessive compulsive personality disorders, narcissistic personality disorders, sexual and gender identity disorders, various disorders related to central neurotransmission systems such as dopaminergic, glutamatergic, serotonergic, adrenergic, GABAergic.
- central neurotransmission systems such as dop
- EAA excitatory amino acid
- This patent application also provides a method of treating a disorder or condition comprising as a symptom a deficiency in attention and/or cognition in a mammal, including a human, which method comprises administering to said mammal an amount of a compound of formulas (1) to (If) effective in treating said disorder or condition.
- the phrase "deficiency in attention and/or cognition” as used in the phrase “disorder comprising as a symptom a deficiency in attention and/or cognition” refers to a subnormal functioning in one or more cognitive aspects such as memory, intellect, or learning and logic abi lity, in a particular indiv idual relati ve to other individuals within the same general age population.
- "Deficiency in attention and/or cognition” also refers to a reduction in any particular individual's functioning in one or more cognitive aspects, for example as occur in age-related cognitive decline.
- disorders that comprise as a symptom a deficiency in attention and/or cognition are dementia, for example, Alzheimer's disease, multi-infarct dementia, alcoholic dementia or other drug-related dementia, dementia associated with intracranial tumors or cerebral trauma, dementia associated with Huntington's disease or Parkinson's disease, Multiple sclerosis, Amyotrophic lateral sclerosis.
- Down's syndrome or AIDS-related dementia delirium, amnestic disorder, posttraumatic stress disorder, mental retardation, a learning disorder, for example reading disorder, mathematics disorder, or a disorder of written expression, attention-deficit/hyperactivity disorder and age-related cognitive decline.
- This patent application also provides a method of treating a mood disorder or mood episode in a mammal, including a human, comprising administerin to said mammal an amount of a compound of formulas (I) to (If) effective in treating said disorder or episode.
- This patent application also provides a method of treating a mood disorder or mood episode in a mammal, including a human, comprising administering to said mammal a therapeutically effective amount of a compound of formulas (1) to (If) in inhibiting !' ⁇ )! ⁇ 1 0 ⁇ .
- mood disorders and mood episodes that can be treated according to the present patent application include, but are not limited to, major depressive episode of the mild, moderate or severe type, a manic or mixed mood episode, a hypomanic mood episode; a depressive episode with atypical features; a depressive episode with melancholic features; a depressi ve episode with catatonic features; a mood episode with postpartum onset; post- stroke depression; major depressive disorder; dysthymic disorder; minor depressive disorder; premenstrual dysphoric disorder; post-psychotic depressive disorder of schizophrenia; a major depressive disorder superimposed on a psychotic disorder such as delusional disorder or schizophrenia; a bipolar disorder, for example bipolar I disorder, bipolar II disorder and cyclothymic disorder.
- This patent application further provides a method of treating a neurodegenerative disorder or condition in a mammal, including a human, which method comprises administering lo said mammal a therapeutically effective amount of a compound of the present invention in treating said disorder or condition.
- This patent application further provides a method of treating a neurodegenerative disorder or condition in a mammal, including a human, which method comprises administering to said mammal a therapeutically effective amount of a compound of formulas (1 ) to ( I f) in inhibiting PDE1 0A.
- a neurodegenerative disorder or condition refers to a disorder or condition that is caused by the dysfunction and/or death of neurons in the central nervous system.
- the treatment of these disorders and condi tions can be faci litated by administration of an agent which prevents the dysfunction or death of neurons at risk in these disorders or conditions and/or enhances the function of damaged or healthy neurons in such a way as to compensate for the loss of function caused by the dysfunction or death of at-risk neurons.
- neurotrophic agent refers to a substance or agent that has some or all of these properties.
- neurodegenerative disorders and conditions that can be treated according to the present patent application include, but are not limited to, Parkinson's disease; Huntington's disease; dementia, for example Alzheimer's disease, multi-infarct dementia.
- dementia for example Alzheimer's disease, multi-infarct dementia.
- a ! DS-related dementia, and Pronto temperal Dementia neurodegeneration associated with cerebral trauma; neurodegeneration associated with stroke, neurodegeneration associated with cerebral infarct: hypoglycemia-induced neurodegeneration; neurodegeneration associated with epi leptic seizure: neurodegeneration associated with neurotoxin poisoning; and multi-system atrophy.
- the neurodegenerative disorder or condition comprises neurodegeneration of striatal medium spiny neurons in a mammal, including a human.
- this patent application provides a pharmaceutical composition for treating psychotic disorders, delusional disorders and drug induced psychosis, anxiety disorders, movement disorders, mood disorders, neurodegenerative disorders or drug addiction, comprising a therapeutically effective amount of a compound of the present invention in treating said disorder or condition.
- this patent application provides a method of treating a disorder selected from psychotic disorders, delusional disorders and drug induced psychosis, anxiety disorders, movement disorders, mood disorders, and neurodegenerative disorders, which method comprises administering a therapeutically effective amount of a compound of the present invention in treating said disorder.
- this patent application provides a method of treating the disorders above, where the disorders are selected from the group consisting of: dementia, Alzheimer's disease, multi-infarct dementia, alcoholic dementia or other drug-related dementia, dementia associated with intracranial tumors or cerebral trauma, dementia associated with Huntington ' s disease or Parkinson's disease, or AIDS-related dementia; delirium; amnestic disorder; posttraumatic stress disorder; mental retardation; a learning disorder, for example reading disorder, mathematics disorder, or a disorder of written expression; attention- deficit/hyperactivity disorder; age-related cognitive decline, major depressive episode of the mi ld, moderate or severe type; a manic or mixed mood episode; a hypomanic mood episode; a depressive episode with atypical features; a depressive episode with melancholic features; a depressive episode with catatonic features; a mood episode with postpartum onset; post- stroke depression; major depressive disorder; dysthymic disorder; minor depressive disorder; premenstrual dysphoric disorder; post-psychotic
- a method for preventing, ameliorating or treating a disease or condition selected from obesity or related diseases, conditions; diabetes (including Type 1 and Type II diabetes); diabetic complications; glucose tolerance; hyperinsulinemia; insulin sensitivity or resistance; metabolic syndromes; cardiovascular diseases including, for example, atherosclerosis, lipidemia, dyslipidemia. elevated blood pressure, microalbuminemia.
- hyperuricaemia hypercholesterolemia, hyperlipidemias, hypertriglyceridemias, arteriosclerosis or combination thereof; respiratory diseases or disorders including, for example, sinusitis, asthma, bronchitis or combination thereof; or any combination these diseases, disorders, conditions and/or syndromes thereof; the disease or condition related to serum levels of triglyceride, LDL, HDL, VLDL, total chlolesterol, which method comprises administering to said mammal a therapeutically effective amount of a compound of formulas (I) to (If) in treating said disorder or condition.
- the compounds of formula (la) can be prepared according to Synthetic scheme 1 .
- The. phenyl acetic acid derivative of formula (1 ) is condensed with aromatic aldehyde of formula (2) under suitable Perkin reaction conditions [Journal of Medicinal Chemistry, 1 3, ( 1 970)] to gi ve the acrylic acid of formula (3).
- the regioisomeric amide of formula (lb) can be prepared according to Synthetic scheme 2.
- the acetic acid of formula (6) is condensed with benzaldehyde derivative of formula (5) under suitable Perkin reaction conditions to give the acrylic acid derivative of formula (7).
- Coupling of this acid (7) with amine of formula (4) as described above gives the final compound of formu la (i ).
- the regioiosmeric acrylamide of formula (ib-1 ) where X is an oxygen atom can be prepared as shown in Synthetic scheme 4.
- 4-hydroxybenzaldehyde of the formula ( 1 4) is condensed with an aryl acetic acid derivative of formula (6) under Perkin reaction conditions in the presence of base such as N-methylmorpholine, triethylamine or diisopropylethylamine in acetic anhydride to give acrylic acid of formula (15).
- base such as N-methylmorpholine, triethylamine or diisopropylethylamine in acetic anhydride
- the amine of formula (Ic) can be prepared according to Synthetic scheme 5.
- the ester of formula ( 1 2a) is reduced to corresponding alcohol of formula ( 19) using diisobutyl aluminium hydride, LiAlH or L1BH 4 in a suitable solvent.
- the hydroxyl group of formula (1 9) is converted to an appropriate leaving group such as chloride using thionyl chloride or methane sulphonyl chloride or oxalyl chloride to give compound of formula (20).
- Reaction of Intermediate (20) with an amine of formula (4) in the presence of a base gives the compound of general formula (lc).
- the compound of formula (Ic) can also be prepared from the alcohol of formula ( 1 9) by its oxidation to the corresponding aldehyde of formula (21 ) using an appropriate oxidizi ng agent such as manganese dioxide followed by reductive amination using an appropriate am ine o f formula (4) in presence of sodium triacetoxyborohydride in acetic acid.
- an appropriate oxidizi ng agent such as manganese dioxide
- reductive amination using an appropriate am ine o f formula (4) in presence of sodium triacetoxyborohydride in acetic acid.
- the compounds of formula (lc-1) (wherein R a is substituted or unsubstituted al kyl) can be prepared according to Synthetic scheme 6.
- the chloro compound of formula (20) is treated with sodium azide in the presence of suitable solvent to give azide derivati ve, which on reduction with the suitable reducing agent like triphenylphosphine gives amine derivative of formula (22) via an iminophosphinone intermediate [Tetrahedron letters, 39, p. 3287-3290, ( 1 998)] .
- Intermediate (22) can also be synthesized directly from chloride of formula (20) by treating it with potassium phthalimide followed by hydrazine hydrate in presence of a suitable solvent such as ethanol.
- the amine derivative of formula (22) is then coupled with the carboxylic acid of formula (23a) using an appropriate coupling agent or acid chloride of formula (23b) in the presence of a base to give the amide of formula (Ic-1 ).
- the compounds of formula (Id-1), (Id-2), (Id-3) and (Id-4) can be prepared from the aldehyde derivative (21 ) according to Synthetic scheme 7.
- pyrazole of the formula (Id-1 ) ⁇ Tetrahedron letters, 39, p. 3287-3290, (1998)] is synthesized from aldehyde of formula (21 ) by treating it with diethyl (2- ⁇ 2-[(4-methylphenyl)sulfonyl]hydrazinylidene ⁇ ethyl)phosphonate (24) in the presence of a strong base such as sodium hydride.
- alkylation of the pyrazole of formula (Id-1 ) with suitable alkyl halide of the formula (25) (where X is halogen) in the presence of suitable base such as K 2 CO 3 gives easily separable regioisomeric mixture of products (Id-2) and (Id-3).
- suitable base such as K 2 CO 3
- the aldehyde of formula (21 ) is treated with p-toluenesulphonylmethyl isocyanide (TOSJMI C) in the presence of a suitable base like potassium carbonate in refluxing methanol to give the oxazole derivative of formula (Id-4).
- compound of formula (Id-5) can also be prepared by treating compound of formula (26) with anhydride (27b) or an acid chloride (27c) under suitable conditions.
- Oxadiazolone of formula (Id-6) is synthesized, in good yields, from compound of formula (26) and trichloromethyl chloroformate in presence o a base fol lowed by alkylation ⁇ J. Heterocyclic. chem. 1 9, p. 541 , (1982)] .
- Compound of formula (26) is reacted with carbon disulfide under basic conditions followed by alkylation to give the thio oxadiazole of formula (Id-7) [J. Heterocyclic. chem, 19, p. 541 , (1982)].
- Triazolothione of formula (Id-8) is prepared from Intermediate (26) by its coupling with potassium thiocyanate followed by cyclization under basic conditions [J. Med, Chem. 37, p. 1 25- 1 32, ( 1 994)] .
- Substituted and unsubstituted 1 ,3,4-triazoles derivatives of formula (Id-9) is prepared by the reaction of hydrazide (26) with appropriate amidines of formula (28) under suitable conditions.
- compound of formula (3 1 ) can also be prepared by treating compound of formula (30) with anhydride (27b) under suitable conditions. Deprotection of the carbonate group under basic conditions gives the phenol of formula (32), which on further coupling with alky] halide of formula ( 1 l a) (where L is halogen) under basic conditions or with (l i b) under Mitsunobu reaction conditions gives the final compound of formula (Id-5).
- the isomeric oxadiazole of formula (Id-10) can be prepared as described in Synthetic scheme ( 1 0).
- amide of formula (la) where R 9 and R 10 are H, is condensed with N, N- dimethylformamide dimethyl acetal under reflux conditions to give the imine derivative (33).
- Intermediate (33) on reaction with hydroxyl amine in the presence of a suitable base such as sodium hydroxide gives compound of general formula (Id-10).
- the triazolothione compound of formula (Id-11) and its corresponding sulphur free aromatic compound of formula (Id-12) can be prepared as shown in Synthetic scheme 1 1 .
- intermediate (26) is treated with alkylisothiocyanate of the formula (34) under reflux and the intermediate formed is cyclised under basic conditions to give the triazolothione (Id-11 ) [ . Med. Chein. 37, p. 1 25- 132, ( 1994)] .
- Further oxidative desulphurization of (Id-11 ) by hydrogen peroxide in acetic acid gives the N-substituted triazole derivative (Id-12).
- the lactam derivative of formula (Id-13) can be prepared according to Synthetic scheme 1 2.
- the aldehyde of formula (21 ) undergoes wittig reaction with (carbethoxymethvlene) triphenylphosphorane to give the diene ester of formula (35).
- Michael addition of nitromethane anion, generated by means of suitable base such as 1 , 1 ,2,2-tetramethylguanidine (TMG) gives the adduct of formula (36).
- TMG tetramethylguanidine
- Selective reduction of the nitro group using an appropriate reagent such as iron and ammonium chloride under aqueous conditions gives amine of the formula (37).
- Compound of formula (37) undergoes intramolecular cyclization in refluxing xylene to
- various regioisomeric olefins can be prepared from hydrazide of the isomeric acid (7). Preparation of one such derivative is shown in Synthetic scheme 13.
- the acrylic acid of formula (7) was coupled with hydrazine hydrate to give hydrazide of formula (38) using a suitable coupling agent such as EDCI.
- the oxadiazole derivative (Ie-1) can be prepared as described in Synthetic scheme 1 3 using orthoester of formula (27a) or its equivalent (27b) or (27c) under suitable reaction conditions.
- the ether derivative of formula (If) can be prepared according to Synthetic scheme 1 4.
- chloro compound of formula (20) is coupled with alcohol of formula (39) under basic conditions to give ether of general formula (If).
- final compound of general formula (If) can also be prepared by itsunobu coupling reaction of alcohol ( 1 9) with appropriate aromatic alcohol of formula (39).
- the compound of general formula (Ig) can be prepared as depicted in Synthetic scheme 1 5.
- ethyl (4-iodophenyl)acetate derivative (40) undergoes Sonagashira coupling reaction with ethynyltrimethyl silane followed by desilylation using appropriate reagent such as TBAF.H 2 O, to give the acetylene compound of formula (41 ).
- the coupling reaction of compound of formula (41 ) with 2-bromoquinoline under Sonagashira coupling reaction gives the compound of formula (42).
- Catalytic reduction of the triple bond of compound of formula (42) followed by hydrolysis gives compound of formula (43).
- Perkin reaction of compound of formula (43) with aromatic aldehyde (2) gives acid derivative which on further amide coupli ng with amine of formula (4) gives the final compound of general formaul (I ).
- work-up includes distribution of the reaction mixture between an organic and aqueous phase, separation of layers and drying the organic layer over sodium sulphate, filtration and evaporation of the solvent.
- Purification refers to purification by silica gel chromatographic techniques, in suitable solvents o f a suitable polarity as the mobile phase or crystallization from an appropriate solvent or mixture o f solvents.
- DMSO-i 6 hexadeuterodimethyl su lfoxide
- CDCI3 deuterated chloroform
- J coupling constant in units of Hz
- T or rt room temperature (22-26°C).
- Aq. aqueous; equiv. or eq. : equivalents.
- fhe starling materials represented by the general formula Z-Y-X- used for the preparation intermediates and compounds of invention are in some cases commercially available or can be prepared according to suitable literature procedure or as described below.
- the title compound was synthesized by the reaction of 2-aminothiophenol with glycolic acid using 4 hydrochloric acid as described in the literature [Journal of the Chemical Society, p.
- Step 1 Ethyl 5-amino-6-[2-(trimethylsilyl)ethynyl] pyridine-2-carboxylate:
- Step 2 Ethyl 5-acetamido-6-[2-(trimethylsilyl)ethynyl]pyridine-2-carboxylate: To the well stirred solution of Step 1 intermediate (5.5 g. 20.659 mmol) in DCM (1 00 ml) was added pyridine (3.3 ml) and the reaction mixture was cooled to 0 °C. Acetyl chloride (1 .76 ml. 24.79 mmol) was then added to the reaction mixture drop wise and the reaction was stirred at room temperature for 2 h. The reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (300 ml x 3).
- Step 3 Ethyl l H-pyrrolo[3,2-j]pyridine-5-carboxylate:
- Step 2 intermediate 5.5 g. 1 7.84 mmol
- TH F 50 ml
- tetra-n-butyl ammonium fluoride 4.69 g, 17.59 mmol
- the reaction mixture was refluxed for 8 h.
- the reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (300 ml x 3).
- Step 4 l //-Pyn lo[3,2-£]pyridin-5-ylmethanol :
- Step 3 intermediate 300 g, 1 .578 mmol in THF at 0 °C was added to the well stirred suspension of lithium aluminium hydride (359 g, 9.473 mmol) in dry ' TH F (20 ml) and the reaction mixture was stirred at same temperature for 2 h. The reaction mixture was quenched with saturated sol ution of sodium sulphate, diluted with ethyl acetate and filtered.
- Step 5
- Step 4 Intermediate (400 mg, 2.702 mmol) at 0 °C and the reaction mixture was stirred for 1 0 min.
- the reaction mixture was quenched with water (50 ml) and extracted with chloroform (50 ml x 2).
- the title compound was synthesized by esterification of indazole-6-carboxylic acid using methanol in presence of cone, sulphuric acid followed by reduction of the ester group using lithium aluminium hydride and its subsequent reaction with di-ter/-butyl dicarbonate anhydride;
- Step 1 Ethyl 7-oxo-4,7-dihydropyrazolo[l ,5-a]pyrimidine-5-carboxylate:
- Step 2 Ethyl 7-chloro-4,7-dihydropyrazolo
- Step 1 intermediate (1 g, 4.830 mmol) in phosphoryl chloride (POC1 3 ) (10 ml) was added N,N-dimethylaniline ( 1 ml, 4.83 mmol) and the reaction mixture was refiuxed for 40 min.
- the excess of POCl 3 was distilled under reduced pressure and the residue obtained was poured into crushed ice and extracted with chloroform (2 x 1 00 ml).
- Step 3 7-Chloropyrazolo[ l ,5-a]pyrimidine-5-carbaldehyde:
- Step 2 intermediate (1 g, 4.43 mmol) in dry THF ( 1 5 ml) was added 20 % Diisobutylaluminium hydride (DIBAL-H; 2.22 ml, 13.30 mmol) drop wise at 0°C and the reaction mixture was stirred at the same temperature for 1 h. Water was added to the reaction mixture and it was further stirred for 1 5 min. The precipitate formed was fi ltered and the filtrate was concentrated to yield 1 .3 g of the product: ⁇ N.MR (300 MHz. DMSO-ck) ⁇ 7.29 (s. I H), 7.73 (s, 1 11), 8.56 (s, 1 H), 9.91 (s, I H).
- DIBAL-H Diisobutylaluminium hydride
- Step 4 (7-Chloropyrazolo
- Step 3 intermediate (1 .25 g, 5.449 mmol) in DCM ( 1 0 ml) was added sodium triacetoxyborohydride (1.154 g, 5.449 mmol) and the reaction was stirred at room temperature overnight.
- the reaction mixture was diluted with water and the pH was made neutral with sodium bicarbonate.
- the compound was extracted with chloroform and the organic layer was washed with water, brine, dried, filtered and concentrated to yield the 81 0 mg of the product; ⁇ NMR (300 MHz, DMSO- ⁇ ) ⁇ 4.59 (s, 2H), 5.75 (br s, 1 H), 6.79 (s, 1 H), 7.38 (s, 1T1), 8.3 1 (s, l H).
- Step 5 Pyrazolo[l ,5-o]pyrimidin-5-ylmethanol :
- Step 4 To the well stirred solution of Step 4 Intermediate (800 mg, 4.312 mmol) in a mixture of ethanol ( 12 ml) and ethyl aceatate (27 ml) was added sodium acetate (424 mg, 5. 1 74 mmol) and pal ladium on activated carbon (20 mg) and the reaction mixture was stirred at room temperature under hydrogen atmosphere for 5 h. The reaction mixture was diluted with ethyl acetate fi ltered and washed with saturated solution of sodium bicarbonate, water and brine.
- Step 6 Pyrazolo[l ,5-o]pyrimidin-5-ylmethyl me hanesulfonate:
- Step 5 intermediate 100 mg, 0.662 mmol
- DCM 1 0 ml
- TEA tri ethyl amine
- Methanesulphonyl chloride was added drop wise to the reaction mixture at this temperature and the reaction mixture was stirred for 30 min.
- Step 1 2-[4-(Acetyloxy)phenyl]-3-(4-chlorophenyl)prop-2-enoic acid:
- Step 2 Ethyl -3 -(4-chloro phenyl)-2-(4-hydroxyphenyl)prop-2-enoate:
- Step 2 Intermediate ( 10 g, 33.05 mmol) in dimethyl formamide (60 ml), were added potassium carbonate (6.85 g, 49.57 mmol) and 2-(ch!oromethyl)im idazo
- Step 4 3-(4-Chlorophenyl)-2-[4-(imidazo[l ,2-a]pyridin-2-ylmethoxy)phenyl]prop-2-enoic acid : To a well stirred solution of Step 3 Intermediate (8.5 g, 19.63 mmol) in ethanol (60 ml) was added aqueous solution of 1 N sodium hydroxide and the reaction mixture was stirred for 1 6 h at room temperature. The excess of ethanol was distilled off and the reaction mass was di l uted wi th water (50 ml).
- Step 1 Ethyl -2-[4-(l ,3-benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)prop-2-enoate: To the well stirred solution of ethyl-3-(4-chlorophenyl)-2-(4-hydroxyphenyl)prop-2-enoate (Step 2 of intermediate 1 , 1 .5 g, 4.958 mmol) in dry tetrahydrofuran (30 ml), were added 1 ,3- benzothiazol-2-ylmethanol (81 9 mg, 4.958 mmol) and triphenyl phosphine (,1 .95 g, 7.438 mmol) followed by dropwise addition of diethyl azodicarboxylate ( 1 .01 ml, 6.446 mmol).
- Step 2 2-[4-(l,3-Benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)prop-2-enoic acid:
- Step 1 To a well stirred solution of the Step 1 Intermediate (650 mg, 1.445 mmol) in ethanol (15 ml) was added aqueous solution of IN sodium hydroxide and the reaction mixture was stirred for 16 h at room temperature. The excess of ethanol was distilled off and the reaction mass was diluted with water (25 ml).
- Intemediates 4 to 38 were prepared using appropriate starting materials as described in Intermediate 1. Their structure, names and ⁇ NMR data are given in the Table 1.
- Ethyl-2-(4-hydroxyphenyl)-3-(pyridin-4-yl)prop-2-enoate was synthesized from 4-hydroxy phenylacetic acid and 4-pyridine carboxaldehyde as described in Steps 1 and 2 of Intermediate 1.
- Step 1 Ethyl ⁇ 4-[(trimethylsilyl)ethynyl]phenyl ⁇ acetate:
- Step 1 intermediate (4.9 g, 1 8.82 mmol) in dich loromethane (30 ml) was added tetrabutyl ammonium fluoride hydrate (2.461 g, 9.413 mmol) and the reaction mixture was stirred at room temperature for 10 min.
- the reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (2 x 250 ml). The combined organic layers were washed with water (2 x 50 ml) and brine (50 ml), dried over anhydrous a?S0 4 and concentrated to yield the crude product.
- Step 3 Ethyl [4-(quinolin-2-ylethynyl)phenyl]acetate:
- Step 2 intermediate (1 .44 g, 7.6 mmol) in TEA (20 ml) were added dichlorobis(triphenylphosphine) palladium (II) (54 mg, 0.076 mmol), copper iodide (44 mg, 0.23 mmol) and 2-bromoquinoline (1 .6 g, 7.69 mmol) and the reaction was stirred at room temperature overnight.
- the reaction mixture was diluted with water ( 1 00 ml) and chloroform (300 ml), given charcoal treatment and filtered.
- Step 3 intermediate (1.40 g, 4.44 mmol) in ethanol (30 ml) was added palladium on activated carbon (400 mg) and the reaction was carried for 4 h under 40 psi pressure of hydrogen gas. The reaction mixture was diluted with ethyl acetate and filtered.
- Step 5 ⁇ 4-[2-(Quinolin-2-yl)ethyl]phenyl ⁇ acetic acid:
- Step 4 intermediate 500 mg, 1.566 mmol
- aqueous solution of sodium hydroxide 313 mg, 7.83 mmol
- the reaction mixture was stirred at room temperature overnight.
- the solvent was distilled out and reaction mass was diluted with water and then neutralized with dil. HQ.
- the aqueous layer was extracted with ethyl acetate (2 x 100 ml).
- Step 6 3-(4-Chlorophenyl)-2- ⁇ 4-[2-(quinolin-2-yl)ethyl]phenyl ⁇ prop-2-enoic acid:
- Step 5 intermediate 460 mg, 1.580 mmol
- acetic anhydride 15 ml
- triethylamine 0.33 ml, 2.370 mmol
- 4-chlorobenzaldehyde 222 mg, 1.580 mmol
- the reaction mixture was quenched with water (50 ml) and was further refluxed for half hour after which the aqueous layer was extracted with ethyl acetate.
- Step 1 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -ol :
- Step 2 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enal:
- Step 1 intermediate (1 .1 5 g, 2.287 mmol) in THF (40 ml) was carried out at room temperature and the reaction mixture was further stirred for 4 h.
- Step 1 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]pi p-2-en- 1 -ol :
- Step_2 2-( ⁇ 4-[3-Chloro-l -(4-chlorophenyl)prop- l -en-2-yl]phenoxy ⁇ methyl)quinoline:
- Step 1 intermediate To a well stirred solution of the Step 1 intermediate (580 mg, 1 .03 mmol) in DCM ( 1 0 ml) was added triethylamine (0.28 ml, 2.06 mmol) followed by methanesulfonylchloride (0. 1 2 ml, 1 .558 mmol) at 0 °C and was stirred overnight.
- Step 2 4-( ' l -(4-Chlorophenyl)-3-hydrazinyl-3-oxoprop-l-en-2-yl]phenyl ethyl carbonate:
- Step 1 intermediate 750 mg, 2.73 mmoi
- THF THF
- TEA ethyl chloroformate
- ethyl chloroformate 0.78 ml, 8.19 mmoi
- Step 3 4-
- Step 2 intermediate 700 mg, 1.94 mmoi
- triethyl orlhoformale 15 ml
- PTSA 74 mg, 0.388 mmoi
- the reaction mixture was heated at 90-100 °C for 2 h.
- Step 4 4-J " 2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenol:
- Step 3 intermediate 320 mg, 0.863 mmoi
- ethanol 10 ml
- aqueous solution of sodium hydroxide 173 mg, 4.318 mmoi
- the reaction mixture was stirred at room temperature for 15 min.
- the reaction mixture was diluted with water (25 ml) and the pH was made slightly acidic.
- Intemediates 48 to 82 were prepared using appropriate propenoic acid and hydrazine hydrate as described in Intermediate 47. Their structure, names and H NMR data are given in the Table 3. fable 3 : Structure and characterization data for Intermediates 48 - 82
- Example 36 A / -(2-Cyanoethy])-3-[4-(difluoromethoxy)phenyl]-2-[4-(quinolin-2-ylmethoxy)phenyl] prop-2- enamide
- Example 1 To the well stirred solution of Example 1 (500 mg, 1.206 mmol) in THF (15 ml) was added TEA (0.26 ml, 1.929 mmol) and the reaction mixture was cooled to 0 °C. Trifluoroacetic anhydride was added to this cooled reaction mixture and was further stirred at the same temperature for 2 h.
- Step 1 ter/-Butyl 4-( ⁇ 3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enoyl ⁇ amino)piperidine- 1 -carboxylate:
- Step 2 3-(4-Chlorophenyl)-N-(piperidin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide di (trifluoroacetic acid):
- Step 1 To a well stirred solution of Step 1 Intermediate (170 mg, 0.284 mmol) in dichloromethane (4 ml) was added trifluoroacetic acid (0.5 ml) at 0 °C and the reaction was continued for 4 h. The reaction mixture was concentrated under reduced pressure and dried well under high vacuum.
- Step 1 eri-bu ⁇ y ⁇ [2-( ⁇ 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl ⁇ amino)et ' hyl]carbamale:
- Step 2 N- ⁇ 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl ⁇ ethane- 1 ,2- diamine di(trifluoro acetic acid) salt:
- Step 1 To a well stirred solution of Step 1 Intermediate (115 mg, 0.211 mmol) in dichloromethane (4 ml) was added trifluoroacetic acid (0.5 ml) at 0°C and the reaction was continued for 4 h. The reaction mixture was concentrated under reduced pressure and dried well under high vacuum.
- Step 1 3-(4-ChIorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)pheny]] prop-2-en- 1 -amine:
- Step 2 3-(4-Chlorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -amine dihydrochloride:
- Step 1 2-( ⁇ 4-[l-(4-Fluoi phenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy ⁇ methyl)quinoline: The title compound was prepared from Intermediate 43 (590 mg, 1.302 mmol) and N-BOC piperazine (485 mg, 2.604 mmol) as described in Example 82.
- Step 2 2-( ⁇ 4-[l-(4-Fluorophenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy ⁇ methylquinoline trihydrochloride:
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides aryl substituted olefinic compounds as Phosphodiesterase 1 0A (PDE 1 0A) inhibitors. In particular, compounds described herein are useful for treating or preventing diseases, conditions and/or disorders by inhibiting Phosphodiesterase 1 0A enzyme. Also provided herein are processes for preparing compounds described herein, intermediates used in their synthesis, pharmaceutical compositions thereof.
Description
ARYL SUBSTITUTED OLEFINIC COMPOUNDS AS PDE10A INHIBITORS
Related' applications
This application claims the benefit of Indian Provisional Application Nos. 1 41 8/MUM/201 0 filed on May 04, 201 0; 3006/MUM/2010 filed on October 29, 201 0; and US Provisional application Nos. 61/334,903 filed on May 14, 201 0; 61 /416,498 filed on November 23, 2010 and all of which are hereby incorporated by reference.
Technical Field
The present invention relates to aryl substituted olefinic compounds and their use in treating or preventing diseases, conditions and/or disorders by inhibiting phosphodiesterase 1 OA (PDE1 OA) enzyme.
Background of the Invention
The cyclic nucleotide phosphodiesterases (PDEs) are a class of intracellular enzymes related to a family of phosphohydrolases that selectively catalyze the hydrolysis of the 3' cyclic phosphate bonds of adenosine and/or guanosine 3', 5' cyclic monophosphates (cAMP/cGMP) into their respective 5' monophosphates (5'-AMP/GMP). The cyclic nucleotides cAMP and cGMP act as second messengers of intracellular signal transduction in response to extracellular stimuli and are synthesized from ATP and GTP by the catalytic cyclization activity of enzymes adenylyl and guanylyl cyclases, respectively. PDEs play a very important role in signal transduction by regulating the cellular levels of these second messengers (cAMP/cGMP) in the way o f controlling their rates of degradation.
Mammalian PDEs are composed of 21 genes and are categorized into 1 1 families (PDE 1 to PDE1 1 ), with each family typically having several different isoforms and splice variants, based on sequence homology, enzymatic properties, biochemical characteristics and sensitivity to inhibitors. These unique PDEs differ in their three-dimensional structures, kinetic properties, modes of regulation, intracellular localization, cellular expression patterns with different individual isozymes modulating distinct regulatory pathways in the cell. Furthermore, PDEs are di ferentially expressed throughout the body, including in the central nervous system, serving
distinct physiological functions. Thus PDEs provide an unique opportunity of selective drug targets for the potential treatment of specific disease states.
PDEs are also subclassified based on different substrate specificites into cAMP selective (PDE4, 7 and 8), cGMP selective (PDE5, 6 and 9) and cAMP and cGMP dual selective (PDEl , 2, 3, 10 and 1 1 ). The human PDEI OA family enzyme was reported essentially at the same time by two different groups (Fujishige K et al., J. Biol. Chem. vol. 274, p.18438- 1 8445, ( 1 999); Loughney K et al., Gene vol. 234, p. 109-1 17, ( 1999); Loughney K et al., WO 99/42596) based on the identification of cDNA fragments published in the National Center for Biotechnology Information (NCBI) Expressed Sequence Tags (EST) database. To till date there is only one gene in this family, PDEI OA, although four variants PDE10A 1 -4 have been described. While PDEI OA was found to share homology with known PDEs, no function could be identified for PDEI O. PDE 10 has been identified as a unique family based on primary amino acid sequence and distinct enzymatic activity. The murine homologue has also been cloned [(Soderling, S. et al ., Proc. Natl. Acad. Set USA vol. 96 p. 7071 -7076, (1999)] and N-terminal splice variants of both the rat and human genes have been identified [Kotera, J. et al., Biochem. Biophys. Res. Comm. vol. 261 , p. 551 -557, (1999); Fujishige, K. et al., Eur. J. Biochem. vol. 266, p. 1 1 1 8- 1 1 27, ( 1999)] . There is a high degree of homology across species. The mouse PDEI OA 1 is a 779 amino acid protein that hydrolyzes both cAMP and cGMP to AMP and GMP, respectively. PDEI OA hydrolyzes cAMP with a Km of 0.05 μΜ and cGMP with a Km of 3 μΜ, suggesting that the affinity of PDEI OA for cAMP is higher than for cGMP. However, approximately 5-fold greater Vmax for cGMP over cAMP (Vmax ratio of cGMP/cAMP is 4.7) has lead to the suggestion that PDE 1 OA is a unique cAMP-inhibited cGMP phosphodiesterase [Soderling, S KI et al.', Proc. Natl. Acad. Sci. USA vol. 96 p. 7071 -7076, ( 1 999); Fuj ishige et al ., J. Biol. Chem. vol. 274, p. 1 8438- 1 8445, (1999)].
The PDE I OA family of polypeptides shows a lower degree of sequence homology to previously identified PDE families. These low degrees of sequence homology of PDEI OA family of polypeptide make them insensitive to certain inhibitors that are known to be specific for other known PDE families (US 6,350,603, incorporated herein by reference).
Regarding PDE I OA which is one of the PDE subtypes, the expression of its mR A has been identified in many tissues and organs such as striatum, testis, kidney, thyroid gland,
pituitary gland, thalamus, cerebellum, heart, lungs and placenta, cells such as aortic smooth m-.iscle cells and aortic endothelial cells, cells of cancers such as lung small cell carcinoma, breast cancer and large bowel cancer. Accordingly, the possibility that PDE I OA is involved in diseases related to these cells, tissues and organs has been demonstrated [J. Biol. Chein. vol. 274, p. 1 8438 (1 999), Gene, vol. 234, p. 109 (1999) and WO 01/29199] .
From the view points of strong expression of mRNA of PDE I OA and its enzymatic activity in the striatum, medium spiny striatal neuronal projections, nucleus accumbens, thalamus, pineal & pituitary glands of [Lakics V et al., Neuropharmacology, vol 59, p. 367-374, (201 1 )] this enzyme is suggested to be involved in, for example, onset or progression of various disorders and diseases related to striatal, basal ganglia related dysfunctions/disorders such as schizophrenia (positive, negative & cognitive symptoms), parkinson's disease, Huntington disease, obsessive compulsive disorders, sleep disorders and disorders of changed circardian rhythm [Siuciak JA et al., Neuropharmacology, vol 51 , p. 374-385, (2006); Threlfell S ef al ., JPET, vol 328, p. 785-795, (2009); Grauer SM et al., JPET, vol.33 1 , p.574-590, (2009): Spiwoks-Becker I et al ., Neuroendocrinology, (201 1 )DOI: 1 0.1 1 59/0003271 38] . It has been reported that PDE I OA mRNA expression in the striatum of Huntington disease mouse model is different from that in the striatum of normal mice (WO 01/24781) and chronic suppression of PDE I OA alters striatal expression of genes responsible for neurotransmitter synthesis, neurotransmission and signaling pathways implicated in Huntinton's disease [Kleiman R.1 et al .. JPET, vol .336, p.64-76, (201 1 )] .
There are very few effective treatments for neurological disorders characterized by progressive cell loss, known as neurodegenerative diseases, as well as those involving acute cell loss, such as stroke and trauma. In addition, few effective treatments exist for neurological disorders such as psychosis which has been linked to altered striatal function relating to changes in expression of the enzyme PDE I OA [J. A. Siuciak, et al. Neuropharmacology, vol . 5 1 , p. 374- 385, (2006)]. Striatal dysfunction is implicated in a number of CNS disorders including psychosis, schizophrenia, obsessive-compulsive disorders, Parkinson's disease and Huntington's disease. The results with PDE I OA knock-out mice provide evidence that PDE I OA functions to inhibit striatal output by reducing spiny medium neuron excitability. PDEI OA is selectively expressed in dopamine receptive medium spiny neurons, and considerable data suggests that
cAMP and cGM'P signalling pathways play significant roles in the regulation of medium spiny neuron excitability. Additional studies with papaverine, a potent inhibitor of PDEI OA, confirm that PDEI OA regulates both cAMP and cGMP in vivo in rats [J. A. Siuciak, et al. Neuropharmacology, vol. 51 , p. 386-396, (2006)].
In view of the foregoing, PDEI OA inhibitors are useful for treating and/or preventing various diseases caused by enhanced activity of PDE I OA, possibly with reduced side effects (for example, a neural disease such as Parkinson's disease, Huntington disease or Alzheimer's disease, dyskinesia, hypogonadism, diabetes, an ischemic heart disease, hypertension, an inflammatory disease, a disease of the digestive system, an allergic disease, osteoporosis, pain or a malignant tumor).
WO 2003/000269, WO 2003/0141 15, WO 2003/0141 16, WO 2003/0141 1 7, WO 2003/051 877, WO 2006/034491 and WO 2006/034512 describe PDE10 inhibitors for treatment of neurodegenerative diseases, cancer, diabetes and its related disorders. Also WO 2006/072828, WO 2008/084299, WO 2003/093499, WO 2005/082883, WO 2005/120514, WO 2006/01 1 040, WO 2006/070284, WO 2007/077490, WO 2007/085954, WO 2007/096743, WO 2007/1 291 83, WO 2008/001 182, WO 2008/0041 17, WO 2008/020302, WO 2009/070584, WO 2009/068320; WO 2009/068246 and WO 2009/036766 describe PDE10 inhibitors for treatment of obesity, diabetes, certain central nervous system disorders, neurodegenerative and psychiatric disorders. Also WO 2009/029214, WO 2009/025839 and WO 2009/025823 describe PDE I O inhibitors lor treatment of obesity, non-insulin dependent diabetes, schizophrenia, bipolar disorder and obsessive-compulsive disorder. WO 2009/143 178, WO 2009/1 52825, WO' 2009/1 58393, WO 2009/1 58467, WO 2009/158473, WO 2010/006130, WO 2010/017236, WO 201 0/027097 and WO 201 0/030027 describe PDE10 inhibitors for treatment of anxiety, schizophrenia, drug addiction, movement disorder, certain central nervous system disorders, neurodegenerative and psychiatric disorders.
Summary of the Invention
The present invention relates to compounds of the formula (I):

(I)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -SO,-, -(CR4R5)pO-, - (CR4R5)pN(R3)-, -C≡C-, -(CR4R5)PC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-;
Y is a bond, or is selected from -(CR4R3)P- and -S02-;
Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
G ] and G2, are independently selected from, hydrogen, cyano, nitro, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, haloalkyl, haloalkoxy, cycloalkyl, aryl, heteroaryl , heterocyclyl, -(CR4R5)PR8, -(CR4R5)pC(0)R8, -(CR4R5)P-NR9R10, -(CR4R5)p-OR' ' , -C(0)R8, - C(0)(CR4R5)pR8, -C(0)NR9R10, -NR9R10, -NR3C(0)R8, -NR3C(0)NR9R10, -N(R3)S02R8, - OC(0)R8 and -OC(0)NR9R10; with the proviso that at least one of G, or G2 is not hydrogen; at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxy!, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl . cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocycl ylalkyl. heteroaryl, heteroarylalkyl, -C(0)Ra, -C(0)NRbRc, -C(0)ORa, -NRbRc, -NRbC(0)Ra. - NRbC(0)NRbRc, -N(Rb)S02Ra, -OC(0)Ra, -OC(0)NRbRc, -S(0)Ra, -S02Ra 3 -SO RbRc. - S02NRbRc and -SRa;
R is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORa;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy.
cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORA, -NRBRC and -SRA; or R4 and R5, at each occurrence, together with the carbon atom to which they are attached, may form an optionally substituted cycloalkyl or heterocyclyl ring;
R8 is selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl. cyanoalkyl. haloalkoxy, cycloalkyl. cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl, -(CRDRE)QRA, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRBRC, -NRBC(0)RA, -NRBC(0)NRBRC, -N(Rb)S02R8, -OC(0)RA, -OC(0)NRBR , - S(0)RA, -S02RA, -SONRBRC, -S02NRBRC and -SRA;
R9 and R1 0, which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl. hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl . - (CRDRC)QRA, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -S(0)RA, -S02RA, -S(0)NRBRC and -S02 N RBRC; or R9 and R10 together with the nitrogen atom to which they are attached, may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N;
R " is selected from hydrogen, nitro, substituted or unsubstituted alkyl, alkenyl. al kynyl . alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, cycloalkyl, cycloalkylalkyl, cycloalkenyl. cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl, - (CRDRE)QRA, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRV, -NRBC(0)RA, -NRBC(0)NRBRC, - (Rb)S02RA, -S(0)RA, -S02RA, -S(0)NRBRC and -SOZNR ;
at each occurrence, RA, RD and R , which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl , hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORR, -NRRRG, -C(0)N RLRE J cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, RB and RC, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, -C(0)ORF, -C(0)NRFRE, cycloalkyl, aryl, heteroaryl and heterocyclyl :
at each occurrence, Rf and R8 are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive;
'p' is an integer ranging from 1 to 3, both inclusive;
'q' is an integer ranging from 1 to 3, both inclusive;
with the proviso that the compound of formula (I) is not
4-Hydroxy-A/-methyl-3-(pyridine-4-yl)-2-(4-(quinoline-2-ylmethoxy)phenyl but-2-enamide; 4- {4-|'(Quinolin-2-yl)methoxy]phenyl }-3-(phenyl)but-3-en-2-one; and
4- {4-|'(Quinolin-2-yl)methoxy]phenyl} -3-(pyridine-4-yl)but-3-en-2-one.
The compounds of formula (I) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, there is provided a compound of the formula (la):

(la)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein..
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -S02-, -(CR R5)pO-, - (CR4R5)pN(R3)-, -C≡C-, -(CR4R5)pC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-;
Y is a bond, or is selected frorn -(CR4R5)p- and -S02-;
Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, helerocyclvl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
at each occurrence, R! and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl.
alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl heteroaryl, heteroarylalkyl, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRBRC, -NRHC(0)RA, - N RBC(0)NRBRC, -N(Rb)S02RA, -OC(0)RA, -OC(0)NRBRC, -S(0)RA, -S02RA, -SONRBRC, - S02NRBRC and -SRA;
R3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORA;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORA, -NRBRC and -SRA; or R4 and R5, at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
R9 and R1 0, which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalky L - (CRDRC)QR\ -C(0)RA, -C(0)NRBRC, -C(0)ORA, -S(0)RA, -S02RA, -S(0)NRBRC and -S02 RBRT:; or R9 and R1 0 together with the nitrogen atom to which they are attached, may form an optional l y substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or.N;
at each occurrence, RA, RD and RE, which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORF, -NRFR8, -C(0)NRFR8, cycloalkyl. aryl. heteroaryl and heterocyclyl;
at each occurrence, RB and RC. which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, -C(0)0R', -C(0)NRFR , cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, Rf and Rs are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'η' is an integer ranging from 0 to 4, both inclusive;
'p' is an integer ranging from 1 to 3, both inclusive; and
'q' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended lo limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (la) in which A is selected aryl, preferably phenyl.
According to another embodiment, specifically provided are compounds of the formula (la) in which A is heteroaryl selected from a group consisting of

According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 is halogen (e.g., fluorine or chlorine), haloalkyl (e.g., trifluoromethyl), alkoxy (e.g., methoxy) or haloalkoxy (e.g., difluoromethoxy or trifluoromethoxy); and Ίη ' is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (la) in which 'm' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which 'n' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which X is -0-.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which X is bond.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which Y is -(CR4R3)P-; wherein both R4 and R5 are hydrogen and 'p' is 1 or 2.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which Z is substituted or unsubstituted heteroaryl selected from a group consisting of

According to yet another embodiment, specifically provided are compounds of the formula (la) in which R9 is hydrogen.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 0 is hydrogen, alkyl (e.g., methyl or ethyl), cyanoalkyl (e.g., cyanoethyl), haloalkyl (e.g., trifluoromethyl, trifluoroethyl), hydroxyalkyl (e.g., hydroxyethyl), cycloalkyl (e.g., cyclopropyl), heterocyclyl (e.g., piperidinyl) or heteroaryl (e.g., thiazole).
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 0 is -(CRDRE)QRA; wherein RD and RE are independently alkyl (e.g., methyl or ethyl), hydroxyalkyl (e.g., hydroxymethyl) or phenyl and RA is hydrogen. In this embodiment 'q' is 1.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 0 is -(CRDRE)QRA; wherein both RD and RC are hydrogen and RA is -NRRRE or -C(0)NR'Rs. In this embodiment R* and RG are independently hydrogen, alkyl (e.g., methyl or ethyl) or -C(0)alkyl (e.g., -C(O)methyl); and 'q' is 1 or 2.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 0 is -(CRDRE)QRA; wherein both RD and RE are hydrogen or alkoxy (e.g.. methoxy) and R'' is hydrogen. In this embodiment 'q' is 2.
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R1 0 is -(CRDRE)QRA; wherein RA is hydrogen; RD is alkyl (e.g., methyl) and RC is -C(0)OR\ In this embodiment R1 is hydrogen or alkyl (e.g., ethyl); and 'q ' is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (la) in which R9 and R10 together with the nitrogen atom to which they are attached, forms an heterocyclic ring selected from a group consisting of
■N O

According to one embodiment, there is provided a compound of the formula (l b):

(lb)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a Λ'-oxide thereof, wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR.3-, -S(O)-, -S02-, -(CR R5)PO-, - (CR"R5)PN (R3)-, -C≡C-, -(CR4R5)PC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-;
Y is a bond, or is selected from -(CR4R5)P- and -S02-;
Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl. heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl . alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl. cycloalkylalkyl . cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl , heteroaryl, heteroaryl alkyl, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRBRC, -NRBC(0)RA, - N RBC(0)NRBRC, -N(RB)S02RA, -OC(0)RA, -OC(0)NR RC, -S(0)RA, -S02RA, -SON R.BRC, - S02NRBRC and -SR";
R is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, a'kynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORA;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxy] , substituted or
unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORa, -NRbRc and -SRa; or R4 and R'\ at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
R9 and R 10, which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryl alkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylal kyl, - (CRdRe)qRa, -C(0)Ra, -C(0)NR Rc, -C(0)ORa, -S(0)Ra, -S02R;1, -S(0)NRbRc and -S02N RhRc; or R9 and R10 together with the nitrogen atom to which they are attached, may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may- contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N;
at each occurrence, Ra, Rd and Re, which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl. hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORr, -NRrRg, -C(0) RfR , cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, Rb and Rc, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl , alkoxy, -C(0)ORf, -C(0)NRrR , cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, Rf and Rg are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
Ίη' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive;
'p' is an integer ranging from 1 to 3, both inclusive; and
'q' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (l b) in which A is aryl, preferably phenyl.
According to another embodiment, specifically provided are compounds o f the formula
(lb) in which R is halogen (e.g., fluorine or chlorine); and
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which 'n' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which X is -0-.
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which Y is -(CR4R3)P-; wherein both R4 and R3 are hydrogen and ' p' is ] .
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which Z is heteroaryl, preferably quinoliny!.
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which R9 is hydrogen.
According to yet another embodiment, specifically provided are compounds of the formula (lb) in which R10 is alkyl (e.g., methyl), cyanoalkyl (e.g., cyanoethyl) or -(Cl^R^qR11; wherein Rd and Rc are independently alkyl (e.g., methyl or ethyl) or hydroxyalkyl (e.g.. hydoxymethyl) and Ra is hydrogen. In this embodiment 'q' is 1 .
According to one embodiment there is provided a compound of the formula (Ic) :

(Ic)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl ;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -S02-; -(CR4R5)pO-, - (CR4R5)pN(R3)-, -C≡C-, -(CR4R5)pC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-;
Y is a bond, or is selected from -(CR R5)P- and -S02-;
Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
al each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylal kyl, heteroaryl, heteroarylalkyl, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRBRC, - RBC(0)RA, - NRBC(0)NRBRC, -N(RB)S02RA, -OC(0)RA, -OC(0)NRBRC, -S(0)RA, -S02RA, -SONRV, - S02NRBRC and -SRA;
R3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORA;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl , aryl, heterocyclyl, heteroaryl, -C(0)ORA, -NRBRC and -SRA; or R4 and R5, at each occurrence, together with the carbon atom to which they are attached, may form an optionall y substituted cycloalkyl or heterocyclyl ring;
R9 and R1 0, which may be the same or different, are independently selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl. hydroxyalkyl, haloalkyl, cyanoalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylal kyl, - (CRDRE)QRA, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -S(0)RA, -S02RA, -S(0)NRBRC and -S02NRBRC; or R9 and R 1 0 together with. the nitrogen atom to which they are attached, may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N ;
at each occurrence, RA, RD and RE, which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORF, -NRFRG, -C(0)NRRRG, cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, R and Rc, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl. cyanoalkyl, alkoxy, -C(0)ORr, -C(0)NRrRg, cycloalkyl, aryl, heteroaryl and heterocyclyl; .
at each occurrence, Rf and R are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive;
'p' is an integer ranging from 1 to 3, both inclusive; and
'q' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (lc) in v/hich A is aryl, preferably phenyl.
According to another embodiment, specifically provided are compounds of the formula (lc) in which R 1 is halogen (e.g., fluorine or chlorine); and 'm' is 1 or 2.
According to yet another embodiment, specifically provided are compounds of the formula (lc) in which 'm' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (Ic) in which 'n' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (lc) in which X is -0-.
According to yet another embodiment, specifically provided are compounds of the formula (lc) in which Y is -(CR4R5)P-; wherein both R4 and R5 are hyd rogen and p' is 1 .
According to yet another embodiment, specifical ly provided are compounds of the formula (Ic) in which Z is quinolinyl.
According to yet another embodiment, specifically provided are compounds of the formula (Ic) in which R9 is hydrogen or alkyl, preferably methyl.
According to yet another embodiment, specifically provided are compounds of the formula (lc) in which R 10 is alkyl (e.g., methyl), alkynyl (e.g., prop-2-nyl), -C(0)Ra, cyanoalkyl (e.g., cyanoethyl) or heterocyclyl (e.g., pyrrolidinyl, pyrrolidin-2-one, piperidinyl, piperidin-2-
one or ethyl piperidine- l -carboxylate). In this embodiment R" is alkyl (e.g., methyl) or alkoxyalkyl (e.g., -CH2-OCH3).
According to yet another embodiment, specifically provided are compounds of the formula (Ic) in which R10 is -(CRdRe)qRa; wherein both Rd and Re are hydrogen and Ra is alkoxy (e.g., methoxy) or -NRfR8. In this embodiment, R1 and Rs are independently hydrogen, alkyl (e.g., methyl) or -C(0)alkyl, preferably -C(O)methy) ; and 'q ' is 2.
According to yet another embodiment, specifically provided are compounds of the formula (lc) in which R9 and R10 together with the nitrogen atom to which they are attached, forms heterocyclyl or heteroaryl ring selected from,

According to one embodiment, there is provided a compound of the formula (Id):

(Id)
or a pharmaceulically acceptable salt thereof, or a stereoisomer thereof or a yV-oxide thereo f, wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -S02-, -(CR"R5)PO-
(CR4R5)PN(R3)-, -C≡C-, -(CR4R5)PC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-
Y is a bond, or is selected from -(CITRV and -S02-;
Z is selected from substituted or unsubstituted alkyl. cycloalkyl. ary 1. helerocycl yl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic:
Met is selected from heteroaryl and heterocyclyl; wherein said heteroaryl and heterocyclyl may optionally be substituted with atleast one R ;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylalkyl, cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl, heterocyclylalkyl. heteroaryl, heteroarylalkyl, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRV, -NRBC(0)RA, - N RBC(0)NRBRC, -N(RB)S02RA, -OC(0)RA, -OC(0)NRBRC, -S(0)RA, -S02RA, -SONRBRC, - S02 R RC and -SRA;
R3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl. alkenyl . alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORA;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro. cyano, hydroxyl, . substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORA, -NRBRC and -SRA; or R" and R5. at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
at each occurrence, R 12 is selected from hydrogen, halogen, hydroxyl, cyano, substituted or unsubstituted alkyl, hydroxyalkyl, haloalkyl and alkoxy;
Ra is selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl , haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORF, -NRRRS, -C(0)NR(RS : cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, RB and RC, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl . alkoxy, -C(0)ORF, -C(0)NR,R , cycloalkyl, aryl, heteroaryl and hetei Ocyclyl ;
at each occurrence, Rf and R§ are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (Id) in which ring A is aryl, preferably phenyl.
According to another embodiment, specifically provided are compounds of the formula (Id) in which A is heteroaryl selected from a group consisting of

According to yet another embodiment, specifically provided are compounds of the formula (Id) in which R1 is halogen (e.g., fluorine or chlorine), haloalkyl (e.g., trifluoromethyl). alkoxy (e.g., methoxy or ethoxy) or haloalkoxy (e.g., difluoromethoxy or trifluoromethoxy); and 'nf is 1 or 2.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which 'm' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which 'n' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which R2 is halogen (e.g., fluorine or chlorine) or alkoxy (e.g., methoxy or ethoxy); and ' n' is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which X is -0-.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which X is bond.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which Y is -(CR R3)P-; wherein R4 and R'1 are hydrogen and 'p" is 1 or 2.
According to yet another embodiment, specifical ly provided are compounds of the formula (Id) in which Z is aryl, preferably quinolinyl.
According to yet another embodiment, specifically provided are compounds of the formula (Id) in which Z is heteroaryl selected from a group consisting of

According to yet another embodiment, specifically provided are compounds of the

substituted or unsubstituted alkyl (e.g., methyl or ethyl) or haloalkyl (e.g., trifluoromethyl).
According to one embodiment there is provided a compound of the formula (le):
(Ie)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -S02-, -(CR4R5)pO-, - (CR"R5)pN(R3)-5 -C≡C-, -(CR4R5)PC≡C-, -(R6)C=C(R7)- and -(CR4R5)P(R6)C=C(R7)-;
Y is a bond, or is selected from -(CR4R;,)p- and -S02-;
Z is selected from substituted or unsubstituted alky], cycloalkyl, aryl. heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
Met is selected from heteroaryl and heterocyclyl; wherein said heteroaryl and
12
heterocyclyl may optionally be substituted with atleast one R ;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl. alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl. cycloa! ky alky L cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl, heterocyclyl . heterocyclylalky! . heteroaryl, heteroarylalkyl, -C(0)Ra, -C(0)NRbRc, -C(0)ORa, -NRbRc, -NRbC(0)Ra, - N bC(0)NRbRc, -N(Rb)S02Ra, -OC(0)Ra, -OC(0)NRbRc, -S(0)Ra, -S02Ra, -SONRbRc, - S02NRbRc and -SRa;
R3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl. alkenyl, alkynyl. alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORa;
at each occurrence, R4, R5, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy. cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORa, -NRbRc and -SRa; or R4 and R5, at each occurrence, together with the carbon atom to which they are attached, may form an optional ly substituted cycloalkyl or heterocyclyl ring;
at each occurrence, R12 is selected from hydrogen, halogen, hydroxyl. cyano. substituted or unsubstituted alkyl, hydroxyalkyl, haloalkyl and alkoxy;
RA is selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORF, -NRRR , -C(0)NR' R5\ cycloalkyl, aryl, heteroaryl and heterocyclyl; '
at each occurrence, RB and RC, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl . alkoxy, -C(0)ORF, -C(0)NRRRG, cycloalkyl, aryl, heteroaryl and heterocyclyl ;
at each occurrence, Rf and R8 are independently selected from hydrogen, alkyl. alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'η' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (le) in which A is aryl preferably phenyl.
According to another embodiment, specifically provided are compounds of the formula (Ie) in which R 1 is halogen, preferably chlorine; and 'm' is 1 .
According to yet another embodiment, specifical ly provided are compounds of the formula (Ie) in which X is -0-.
According to yet another embodiment, specifical ly provided are compounds of the formula (Ie) in which Y is -(CR4R3)P-; wherein R4 and R5 are hydrogen and ' p: is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (le) in which Z is heteroaryl, preferably quinolinyl.
According to yet another embodiment, specifically provided are compounds of the formula (Ie) in which !n' is 0.
According to yet another embodiment, specifically provided are compounds of the
formula (le) in which Het
 wherein R12 is hydrogen or substituted or unsubstituted alkyl, preferably unsubstituted alkyl, more preferably methyl.
According to one embodiment there is provided a compound of the formula (If):
wherein R12 is hydrogen or substituted or unsubstituted alkyl, preferably unsubstituted alkyl, more preferably methyl.
According to one embodiment there is provided a compound of the formula (If):

(If)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof wherein,
A is selected from cycloalkyl, aryl, heteroaryl and heterocyclyl;
X is a bond, or is selected from -0-, -S-, -NR3-, -S(O)-, -S02-5 -(CR R5)pO:, - (CR R 5)PN(R3)-, -C≡C-, -(CR4R5)PC≡C-, -(R6)C=C(R7)- and -(CR |R5)P(R6)C=C(R7)-;
Y is a bond, or is selected from -(CR4R3)P- and -S02-;
Z is selected from substituted or unsubstituted alkyl, cycloalkyl, aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, cycloalkylal k yL cycloalkenyl, cycloalkenylalkyl, aryl, aryloxy, arylalkyl. heterocyclyl. heterocyclylalkyl . heteroaryl, heteroarylalkyl, -C(0)R", -C(0)NRbRc, -C(0)ORa, -N RbRc, -\ R ViO s!r. - NRbC(0)NRbRc, -N(R )S02Ra, -OC(0)Ra, -OC(0)NRbRc, -S(0)R -S02Ra, -SO RV, - S02NRbRc and -SRa;
R3 is selected from hydrogen, hydroxyl, substituted or unsubstituted alkyl, alkenyl. alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl, haloalkoxy, cycloalkyl, aryl, heterocyclyl, heteroaryl and -C(0)ORa;
at each occurrence, R4, R3, R6 and R7, which may be the same or different, are independently selected from hydrogen, halogen, nitro, cyano, hydroxyl, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxy, alkoxyalkyl, hydroxyalkyl, haloalkyl. haloalkoxy. cycloalkyl, aryl, heterocyclyl, heteroaryl, -C(0)ORa, -NRbRc and -SRa; or R4 and R5, at each
occurrence, together with the carbon atom to which they are attached, may form an optionally substituted cycloalkyl or heterocyclyl ring;
R" is selected from hydrogen, nitro, substituted or unsubstituted alkyl, alkenyl, alkynyl, alkoxyalkyl, hydroxyalkyl, haloalkyl, cyanoalkyl, cycloalkyl, cycloalkylalkyl, cycloalkenyL cycloalkenylalkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl. - (CRDRE)QRA, -C(0)RA, -C(0)NRBRC, -C(0)ORA, -NRBRC, -NRBC(0)RA, -NRBC(0)NRBRC, - .N (Rb)S02R", -S(0)R", -S02RA, -S(0)NRBRC and -S02NRBRC;
at each occurrence, R , RD and RC, which may be the same or different, are independently selected from hydrogen, halogen, cyano, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoalkyl, alkoxy, alkoxyalkyl, -C(0)ORR, -NR'R8, -C(0)NRRR8 S cycloalkyl ; aryl, heteroaryl and heterocyclyl;
at each occurrence, RB and RC, which may be the same or different, are independent ly selected from hydrogen, substituted or unsubstituted alkyl, haloalkyl, hydroxyalkyl, cyanoal kyl. alkoxy, -C(0)OR.', -C(0)NRFRG, cycloalkyl, aryl, heteroaryl and heterocyclyl;
at each occurrence, R1 and R are independently selected from hydrogen, alkyl, alkenyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive;
'p' is an integer ranging from 1 to 3, both inclusive; and
'q' is an integer ranging from 1 to 3, both inclusive.
The embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified.
According to one embodiment, specifically provided are compounds of the formula (I f) in which A is aryl, preferably phenyl.
According to another embodiment, specifically provided are compounds of the formula (If) in which A is heteroaryl, preferably 4-pyridyl.
According to yet another embodiment, specifically provided are compounds of the formula (If) in which R1 is halogen, preferably fluorine or chlorine; and 'm' is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (If) in which em' is 0.
According to yet another embodiment, specifically provided are compounds of the formula (If) in which 'ir is 0.
According to yet another embodiment, specifically provided are compounds of the formula (If) in which X is -0-.
According to yet another embodiment, specifically provided are compounds of the formula (I f) in which Y is -(CR4R3)P-; wherein R4 and R3 are hydrogen and 'p' is 1 .
According to yet another embodiment, specifically provided are compounds of the formula (If) in which Z is heteroaryl, preferably quinolinyl.
According to yet another embodiment, specifically provided are compounds of the formula (If) in which R1 1 is aryl, preferably phenyl.
According to yet another embodiment, specifically provided are compounds of the

formula (If) in which R1 ' is -heteroaryl, preferably ^— " or
It should be understood that the formulas (I), (la),. (Ib), (Ic), (Id), (le) and (If) structural ly encompasses esters, N-oxide, all tautomers, stereoisomers, including enantiomers and diastereomers, geometrical isomers and pharmaceutically acceptable salts that may be contemplated from the chemical structure of the genera described herein.
The present invention also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient, such as a pharmaceutically acceptable carrier or diluent. Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. The compounds described in the present patent application may be associated with a pharmaceutically acceptable excipient, such as a carrier or a diluent or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
The compounds and pharmaceutical compositions of the present invention are useful for inhibiting PDE 1 0A, which is related to a variety of disease states.
The present invention further provides a method of treating a disease, condition or disorder modulated by a PDE10A, in a subject by administering to the subject in need thereof a therapeutically effective amount of a compound of formulas (I) to (If) or a pharmaceutical composition described herein.
Drawings:
The illustrative examples of the present invention are screened for 'in vivo' PDE 1 OA based efficacy in a rat model of Dizocilpine (MK-801) - induced psychotic behavior.
The effect of Example 91 on MK-801 - induced psychosis behavior in female SD rats as shown in Figure 1 and the effect of Example 1 77 on MK-801 - induced psychosis behavior in female SD rats as shown in Figure 2.
Detailed Description of the Invention
Definitions
The terms "halogen" or "halo" means fluorine, chlorine, bromine or iodine.
The term "alkyl" refers to a hydrocarbon chain radical that includes solely carbon and hydrogen atoms in the backbone, containing no unsaturation, having from one to eight carbon atoms, and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n- propyl, 1 -methylethyl (isopropyl), n-butyl, n-pentyl and 1 , 1 -dimethylethyl (t-butyl). Unless set forth or recited to the contrary, all alkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "alkenyl" refers to a hydrocarbon chain containing from 2 to 10 carbon atoms and including at least one carbon-carbon double bond. Examples of such alkenyl moiety includes, but are not limited to, ethenyl, 1 -propenyl, 2-propenyl (allyl), wo-propenyl, 2-methyl- l - propenyl, 1 -butenyl and 2-butenyl. Unless set forth or recited to the contrary, all alkenyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "alkynyl" refers to a hydrocarbyl radical having at least one carbon-carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 1 0 carbon atoms being preferred). Examples of such alkynyl moiety include, but are not limited to, ethynyl, propynyl and butynyl. Unless set forth or recited to the contrary, all alkynyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "alkoxy" refers an alkyl group attached via an oxygen linkage to the rest of the molecule. Examples of such alkoxy-moiety include, but are not limited to, -OCFI3 and -OC2M5. Unless set forth or recited to the contrary, all alkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "alkoxyalkyl" or "alkyloxyalkyl" refers to an alkoxy or alkyloxy group as defined above directly bonded to an alkyl group as defined above. Example of such alkoxyalkyl moiety includes, but are not limited to, -CH2OCH3 and -CH2OC2H5. Unless set forth or recited to the contrary, all alkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "haloalkyl" refers to at least one halo group (selected from F. CI, Br or I), linked to an alkyl group as defined above. Examples of such haloalkyl moiety include, but are not limited to, trifluoromethyl, difluoromethyl. 2,2,2-trifluoroethyl and fluoromethyl groups. Unless set forth or recited to the contrary, all haloalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "haloalkoxy" refers to an alkoxy group substituted with one or more halogen atoms. Examples of "haloalkoxy" include but are not limited to fluoromethoxy, difluoromethoxy, trifluoromethoxy, 2,2,2-trifiuoroethoxy,. pentafluoroethoxy, pentachloroethoxy, chloromethoxy. dichlorormethoxy, trichloromethoxy and 1 -bromoethoxy. Unless set forth or recited to the contrary, all haloalkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
The term ''hydroxyalkyl" refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by hydroxyl groups. Examples of hydroxyalkyl moiety include, but are not limited to -CH2OH and -C2H4OH. Unless set forth or recited to the contrary, all hydroxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "cyanoalkyl" refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by cyano groups. Examples of cyanoalkyl moiety include, but are not limited to -CH2CN and -C2H4CN. Unless set forth or recited to the contrary, all cyanoalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "cycloalkyl" denotes a non-aromatic mono or multicyclic ring system of 3 to about 1 2 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl . Examples of multicyclic cycloalkyl groups include, but are not limited to, perhydronapththyl, adamantyl and norbornyl groups, bridged cyclic groups or sprirobicyclic groups, e.g., sprio(4.4)non-2-yl,
spiro|"3,3]heptanyl, spiro|"3,4]octanyl and spiro[4,4]heptanyl. Unless set forth or recited to the contrary, all cycloalkyl groups described herein may be substituted or unsubstituted.
The term "cycloalkylalkyl" refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group. The cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Examples of cycloalkylalkyl moiety include, but are not limited to cyclopropylmethyl, cyclobutylethyl, and eye lopentyl ethyl. Unless set forth or recited to the contrary, all cycloalkylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "cycloalkenyl" refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, such as cyclopropenyl, cyclobutenyl, and cvclopentenyl . Unless set forth or recited to the contrary, all cycloalkenyl groups described or claimed herein may be substituted or unsubstituted.
The term "cycloalkenylalkyl" refers to a cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, directly attached to an al kyl group. The cycloalkenylalkyl group may be attached to the main structure at any carbon atom in t he alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all cycloalkenylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "aryl" refers to an aromatic radical having 6 to 14 carbon atoms, including monocyclic, bicyclic and tricyclic aromatic systems, such as phenyl, naphthyl, tetrahydronapthyl, indanyl and biphenyl. Unless set forth or recited to the contrary, all aryl groups described herein may be substituted or unsubstituted.
The term "aryloxy" refers to an aryl group as defined above attached via an oxygen linkage to the rest of the molecule. Examples of aryloxy moiety include, but are not l imited to phenoxy and naphthoxy. Unless set forth or recited to the contrary, all aryloxy groups described herein may be substituted or unsubstituted.
The term "arylalkyl" refers to an aryl group as defined above directly bonded to an alkyl group as defined above. Examples of arylalkyl moiety include, but are not limited to -CH2C6H5 and -C2H4C6H5. Unless set forth or recited to the contrary, all arylalkyl groups described herein may be substituted or unsubstituted.
The term "heterocyclic ring" or "heterocyclyl" unless otherwise speci fied refers to substituted or unsubstituted non-aromatic 3 to 15 membered ring radical which consists of carbon atoms and from one to five heteroatoms selected from nitrogen, phosphorus, oxygen and sulfur. The heterocyclic ring radical may be a mono-, bi- or tricyclic ring system, which may- include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sul fur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states. In addition, the nitrogen atom may be optionally quaternized; also, unless otherwise constrained by the definition the heterocyclic ring or heterocyclyl may optionally contain one or more olefinic bond(s). Examples of such heterocyclic ring radicals include, but are not limited to azepinyl. azetidinyl, benzodioxolyl, benzodioxanyl, chromanyl, dioxolanyl, dioxaphospholanyl. decahydroisoquinolyl, indanyl, indolinyl, isoindolinyl, isochromanyl. isothiazolidinyl, isoxazolidinyl, morpholinyl, oxazolinyl, oxazolidinyl, oxadiazolyl, 2-oxopiperazinyl, 2- oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl, octahydroindolyl, octahydroisoindolyl, perhydroazepinyl, piperazinyl, 4-piperidonyl, pyrrolidinyl, piperidinyl, phenothiazinyl . phenoxazinyl, quinuclidinyl, tetrahydroisquinolyl, tetrahydrofuryl, tetrahydropyranyl, thiazolinyl, thiazolidinyl, thiamorpholinyl, thiamorpholinyl sulfoxide, thiamorpholinyl sul f ne. 1 ,4 azathianyl, 7-aza-spiro[3,3]heptanyl, 7-spiro[3,4]octanyl, and 7-aza- spiro[3.4]octanyl . The heterocyclic ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure. Unless set forth or recited to the contrary, al l heterocyclyl groups described herein may be substituted or unsubstituted.
The term "heterocyclylalkyl" refers to a heterocyclic ring radical directly bonded to an alkyl group. The heterocyclylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all heterocyclylalkyl groups described herein may be substituted or unsubstituted.
The term "heteroaryl" unless otherwise specified refers to substituted or unsubstituted 5 to 1 4 membered aromatic heterocyclic ring radical with one or more heteroatom(s) independently selected from N, O or S. The heteroaryl may be a mono-, bi- or tricyclic ring system. The heteroaryl ring radical may be attached to the main structure at any heteroatom or carbon atom that results in the creation of a stable structure. Examples of such heteroaryl ring radicals include, but are not limited to oxazolyl, isoxazolyl, imidazolyl, furyl. indolyl, isoindolyl.
pyrrolyl, triazolyl. triazinyl, tetrazolyl, thienyl. thiazolyl, isothiazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, benzofuranyl, benzothiazolyl, benzoxazolyl, benzimidazolyl. benzothienyl, benzopyranyl, carbazolyl, quinolinyl, isoquinolinyl, quinazolinyl, cinnolinyl, naphthyridinyl, pteridinyl, purinyl, quinoxalinyl, quinolyl, isoquinolyl, thiadiazolyl, indolizinyl, acridinyl, phenazinyl, phthalazinyl, furo[3,2-6]pyridinyl, pyrrolo[3 ,2- >] pyridinyl, thieno| 3,2-i ] pyridinyl, indazolyl and imidazo[l ,2-a]pyridinyl. Unless set forth or recited to the contrary, all heteroaryl groups described herein may be substituted or unsubstituted.
The term "heteroarylalkyl" refers to a heteroaryl ring radical directly bonded to an alkyl group. The heteroarylalkyl radical may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Unless set forth or recited to the contrary, all heteroarylalkyl groups described herein may be substituted or unsubstituted.
Unless otherwise specified, the term "substituted" as used herein refers to a group or moiety having one or more of the substituents attached to the structural skeleton of the group or moiety, including, but not limited to such substituents as hydroxy, halogen, cyano, nitro, oxo (=0), thio (=S), substituted or unsubstituted alkyl, substituted or unsubstituted haloalkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted haloalkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or ui: substituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl. substituted or unsubstituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl . substituted or unsubstituted heterocyclic ring, substituted or unsubstiuted guanidine. -COOR\ - C(0)R'\ -C(S)R\ -C(0)NRxRy, -C(0)ONRxRy, -NRxCOMRyRz, -N(Rx)SORy ; -N(Rx)S02Rv : - (=N-N(Rx)Ry), -NRxC(0)ORy, -NRxRy, -NRxC(0)Ry, -NRxC(S)Ry, -NRxC(S)NRyRz, -SONRxR , -S02NRxRy, -ORx, -OC(0)ORy, -OC(0)Rx, -OC(0)NRxRy, -SRX, -SORx, -S02Rx, and -ON02; wherein Rx, Ry and Rz are independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl.
substituted heterocyclylalkyl ring, substituted or unsubstituted heteroarylalkyl, or substituted or unsiibstituted heterocyclic ring. The substituents in the aforementioned "substituted" groups cannot be further substituted. For example, when the substituent on "substituted alkyl" is "substituted aryl", the substituent on "substituted aryl" cannot be "substituted alkenyl".
The term "treating" or "treatment" of a state, disorder or condition includes: (a) preventing or delaying the appearance of clinical symptoms of the state, disorder or condition developing in a subject that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition; (b) inhibiting the state, disorder or condition, i.e., arresting or reducing the development of the disease or at least one clinical or subclinical symptom thereof; or (c) relieving the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
The term "subject" includes mammals (especially humans). Other mammals i ncl ude domestic animals (e.g., household pets including cats and dogs) and non-domestic animals (such as wildlife).
A "therapeutically effective amount" means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment. The "therapeutically effective amount" will vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subject to be treated.
Pharmaceutically acceptable salts forming part of this patent application include salts derived from inorganic bases (such as Li, Na, K, Ca, Mg, Fe, Cu, Zn, and Mn), salts of organic bases (such as N,N'-diacetylethylenediamine, glucamine, triethylamine, choline, hydrox ide, dicyclohexylamine, metformin, benzylamine, trialkylamine, and thiamine), salts of chiral bases (such as alkylphenylamine, glycinol, and phenyl glycinol), salts of natural amino acids (such as glycine, alanine, valine, leucine, isoleucine, norleucine, tyrosine, cystine, cysteine, methionine, proline, hydroxy proline, histidine, ornithine, lysine, arginine, and serine), salts of non-natural amino acids (such as D-isomers or substituted amino acids), salts of guanidine, salts of substituted guanidine (wherein the substituents are selected from nitro, amino, alkyl, alkenyl or alkynyl), ammonium salts, substituted ammonium salts and aluminum salts. Other
pharmaceutically acceptable salts include acid addition salts (where appropriate) such as sulphates, nitrates, phosphates, perchlorates, borates, hydrohalides, acetates (such as trifiuoroacetate), tartrates, maleates, citrates, fumarates, succinates. palmoates. methanesulphonates, benzoates, salicylates, benzenesulfonates, ascorbates, glycerophosphates and ketoglutarates. Yet other pharmaceutically acceptable salts include, but are not limited to, quaternary ammonium salts of the compounds of invention with alkyl halides or alkyl sulphates (such as Mel or Me?S04).
Compounds described herein can comprise one or more asymmetric carbon atoms and thus can occur as racemic mixtures, enantiomers and diastereomers. These compounds can also exist as conformers/rotamers. All such isomeric forms of these compounds are expressly included in the present patent application. Although the specific compounds exemplified in this application may be depicted in a particular stereochemical configuration, compounds having either the opposite stereochemistry at any given chiral centre are envisioned as a part thereof. In addition, compounds of Formulas (I) to (If) can exist in different geometrical isomeric forms. Unless otherwise stated a reference to a particular compound includes al l such isomeric forms, including racemic and other mixtures thereof. The various isomeric forms of the compounds o f the present invention may be separated from one another by methods known in the art or a given isomer may be obtained by stereospecific or asymmetric synthesis. Tautomeric forms and mixtures of compounds described herein are also contemplated.
Pharmaceutical Compositions
The pharmaceutical composition of the present patent application comprises one or more compounds described herein and one or more pharmaceutically acceptable excipients, carriers, diluents or mixture thereof. The compounds described herein may be associated with one or more pharmaceutically acceptable excipients, carriers, diluents or mixture thereof in the form of capsule, sachet, paper or other container.
Examples of suitable carriers include, but are not limited to, water, salt sol utions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of
cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides. pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethyl cellulose and polyvinylpyrrolidone.
The carrier or diluent may include a sustained release material, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
The pharmaceutical composition may also include one or more pharmaceutical ly acceptable auxiliary agents, wetting agents, emulsifying agents, suspending agents, preserving agents, salts for influencing osmetic pressure, buffers, sweetening agents, flavoring agents, colorants or any combination of the foregoing. The pharmaceutical composition of the patent application may be formulated so as to provide quick, sustained or delayed release of the active ingredien after administration to the subject by employing methods known in the art.
The pharmaceutical compositions of the present patent application may be prepared by conventional techniques, e.g., as described in Remington: The Science and Practice of Pharmacy, 20th Ed., 2003 (Lippincott Williams & Wilkins). For example, the active compound is mixed with a carrier, or diluted by a carrier, or enclosed within a carrier, which may be in the form of an ampoule, capsule, sachet, paper or other container. When the carrier serves as a diluent, it may be a solid, semi-solid or liquid material that acts as a vehicle, excipient or medium for the active compound. The active compound is adsorbed on a granular solid contai ner, for example, in a sachet.
- The pharmaceutical compositions may be in conventional forms, for example, capsules, tablets, aerosols, solutions, suspensions or products for topical application.
The route of administration may be any route which effectively transports the active compound of the patent application to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, pulmonary, buccal, subdermal, intradermal, transdermal , parenteral, rectal, depot, subcutaneous, intravenous, intraurethral. intramuscular, intranasal, ophthalmic (such as with an ophthalmic solution) or topical (such as with a topical ointment). The oral route is preferred.
Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges. Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the
like arc particularly suitable for oral application. Preferable carriers for tablets, dragees. or capsules include lactose, cornstarch and/or potato starch. A syrup or elixir is used in cases where a sweetened vehicle is employed.
Liquid formulations include, but are not limited to, syrups, emulsions, soft gelatin and sterile injectable liquids, such as aqueous or non-aqueous liquid suspensions or solutions.
For parenteral application, particularly suitable are injectable solutions or suspensions, preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil .
Suitable doses of the compounds for use in treating the diseases and disorders descri bed herein can be determined by those skilled in the relevant art. Therapeutic doses are generally identified through a dose ranging study in humans based on preliminary evidence derived from the animal studies. Doses must be sufficient to result in a desired therapeutic benefit without causing unwanted side effects. For example, the daily dosage of the PDE 1 0A inhibitors can range from about 0.1 to about 30.0 mg/Kg. Mode of administration, dosage forms, suitable pharmaceutical excipients, diluents or carriers can also be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present patent application.
Methods of Treatment
The present patent application provides a method of treating a disease, condition or disorder modulated by a PDE10A, in a subject by administering to the subject in need thereof a therapeutically effective amount of a compound or a pharmaceutical composition described herein.
The present patent application further provides a method of treating diseases, disorders or conditions, modulated by a PDE1 0A in mammals including human., of neuropsych iatric. neurodegenerative, neurological, neuroendocrinological nature such as, but not limiting to. schizophrenia, psychoses, schizoaffective disorders, positive symptoms of schizophrenia including delusions, disordered thoughts and speech, and tactile, auditory, visual, olfactory and gustatory hallucinations, paranoia, paranormal behaviors, negative symptoms of schizophrenia l ike deficits of normal emotional responses or of other thought processes including flat or bl unted affect and emotion, poverty of speech (alogia), inability to experience pleasure
(anhedonia), lack of desire to form relationships (asociality), and lack of motivation (avol ition) leading to poor quality of life, functional disabil ities typically regarded as manifestations of psychosis and other comorbidities like cognitive, executive, attention, learning, memory, spatial memory and social cognitive functions, Tic disorders like Tourette's syndrome, autism, autism spectrum disorders, attention deficit hyperactivity disorders (ADHD), pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS), mood disorders, anxiety, depression, major depressive disorders, bipolar disorders, manias, aggression, obsessive compulsive disorders, Huntington's disease, Alzheimer's disease, Parkinsons disease, rest-less i ·„.·>>. syndrome, various other neurological disorders consisting of movement disorders, ataxias, sensation disorders, cognitive disorders related to multiple sclerosis, amyotrophic lateral sclerosis, abnormalities of brain, spinal cord, nerves leading to symptoms such as paralysis, seizures, catatonias, catalepsies, muscle rigidities, muscle weakness, poor coordination, loss of sensation, confusion, mental suffering, pain and altered levels of consciousness and various other diseases, disorders or conditions related to neuroendocrinological and metabol ic mani festations like change of circardian rhythms, sleep disorders, insomnia, jet lags, eating disorders li ke anorexia nervosa, bulimia nervosa, exercise bulimia or binge eating disorder, aggressive behaviours, obsessive compulsive personality disorders, narcissistic personality disorders, sexual and gender identity disorders, various disorders related to central neurotransmission systems such as dopaminergic, glutamatergic, serotonergic, adrenergic, GABAergic. excitatory amino acid (EAA) mediated signal transduction dysfunctions and diseases related to brain, spinal cord regions, glands and hormones located in the central and peripheral nervous systems related to basal ganglia, limbic system, neostriatum, caudate putamen, striatum, striatal medium spiny neurons, globus pallidus, thalamus, prefrontal cortex, cortex, nucleus accumbens, ventral tegmental area, corpus striatum, substantia nigra, optic chiasm, vomeronasal organ, suprachiasmatic nucleus, hippocampus, amygdala, cerebellum, pineal gland, pituitary gland, hypothalamus, hypothalamo-pituitary-adrenal axis, thyroid, gonads , and trinucleotide repeat expansion diseases of polyglutamine and non-polyglutamine nature.
This patent application also provides a method of treating a disorder or condition comprising as a symptom a deficiency in attention and/or cognition in a mammal, including a human, which method comprises administering to said mammal an amount of a compound of
formulas (1) to (If) effective in treating said disorder or condition. The phrase "deficiency in attention and/or cognition" as used in the phrase "disorder comprising as a symptom a deficiency in attention and/or cognition" refers to a subnormal functioning in one or more cognitive aspects such as memory, intellect, or learning and logic abi lity, in a particular indiv idual relati ve to other individuals within the same general age population. "Deficiency in attention and/or cognition" also refers to a reduction in any particular individual's functioning in one or more cognitive aspects, for example as occur in age-related cognitive decline.
Examples of disorders that comprise as a symptom a deficiency in attention and/or cognition that can be treated according to the present patent application are dementia, for example, Alzheimer's disease, multi-infarct dementia, alcoholic dementia or other drug-related dementia, dementia associated with intracranial tumors or cerebral trauma, dementia associated with Huntington's disease or Parkinson's disease, Multiple sclerosis, Amyotrophic lateral sclerosis. Down's syndrome or AIDS-related dementia; delirium, amnestic disorder, posttraumatic stress disorder, mental retardation, a learning disorder, for example reading disorder, mathematics disorder, or a disorder of written expression, attention-deficit/hyperactivity disorder and age-related cognitive decline. This patent application also provides a method of treating a mood disorder or mood episode in a mammal, including a human, comprising administerin to said mammal an amount of a compound of formulas (I) to (If) effective in treating said disorder or episode. This patent application also provides a method of treating a mood disorder or mood episode in a mammal, including a human, comprising administering to said mammal a therapeutically effective amount of a compound of formulas (1) to (If) in inhibiting !' · )! · 1 0Λ .
Examples of mood disorders and mood episodes that can be treated according to the present patent application include, but are not limited to, major depressive episode of the mild, moderate or severe type, a manic or mixed mood episode, a hypomanic mood episode; a depressive episode with atypical features; a depressive episode with melancholic features; a depressi ve episode with catatonic features; a mood episode with postpartum onset; post- stroke depression; major depressive disorder; dysthymic disorder; minor depressive disorder; premenstrual dysphoric disorder; post-psychotic depressive disorder of schizophrenia; a major depressive disorder superimposed on a psychotic disorder such as delusional disorder or
schizophrenia; a bipolar disorder, for example bipolar I disorder, bipolar II disorder and cyclothymic disorder.
This patent application further provides a method of treating a neurodegenerative disorder or condition in a mammal, including a human, which method comprises administering lo said mammal a therapeutically effective amount of a compound of the present invention in treating said disorder or condition.
This patent application further provides a method of treating a neurodegenerative disorder or condition in a mammal, including a human, which method comprises administering to said mammal a therapeutically effective amount of a compound of formulas (1 ) to ( I f) in inhibiting PDE1 0A. As used herein and unless otherwise indicated, a "neurodegenerative disorder or condition" refers to a disorder or condition that is caused by the dysfunction and/or death of neurons in the central nervous system. The treatment of these disorders and condi tions can be faci litated by administration of an agent which prevents the dysfunction or death of neurons at risk in these disorders or conditions and/or enhances the function of damaged or healthy neurons in such a way as to compensate for the loss of function caused by the dysfunction or death of at-risk neurons. The term "neurotrophic agent" as used herein refers to a substance or agent that has some or all of these properties.
Examples of neurodegenerative disorders and conditions that can be treated according to the present patent application include, but are not limited to, Parkinson's disease; Huntington's disease; dementia, for example Alzheimer's disease, multi-infarct dementia. A ! DS-related dementia, and Pronto temperal Dementia; neurodegeneration associated with cerebral trauma; neurodegeneration associated with stroke, neurodegeneration associated with cerebral infarct: hypoglycemia-induced neurodegeneration; neurodegeneration associated with epi leptic seizure: neurodegeneration associated with neurotoxin poisoning; and multi-system atrophy.
In one aspect of the present patent application, the neurodegenerative disorder or condition comprises neurodegeneration of striatal medium spiny neurons in a mammal, including a human.
In another aspect, this patent application provides a pharmaceutical composition for treating psychotic disorders, delusional disorders and drug induced psychosis, anxiety disorders, movement disorders, mood disorders, neurodegenerative disorders or drug addiction, comprising
a therapeutically effective amount of a compound of the present invention in treating said disorder or condition.
In another aspect, this patent application provides a method of treating a disorder selected from psychotic disorders, delusional disorders and drug induced psychosis, anxiety disorders, movement disorders, mood disorders, and neurodegenerative disorders, which method comprises administering a therapeutically effective amount of a compound of the present invention in treating said disorder.
In another aspect, this patent application provides a method of treating the disorders above, where the disorders are selected from the group consisting of: dementia, Alzheimer's disease, multi-infarct dementia, alcoholic dementia or other drug-related dementia, dementia associated with intracranial tumors or cerebral trauma, dementia associated with Huntington' s disease or Parkinson's disease, or AIDS-related dementia; delirium; amnestic disorder; posttraumatic stress disorder; mental retardation; a learning disorder, for example reading disorder, mathematics disorder, or a disorder of written expression; attention- deficit/hyperactivity disorder; age-related cognitive decline, major depressive episode of the mi ld, moderate or severe type; a manic or mixed mood episode; a hypomanic mood episode; a depressive episode with atypical features; a depressive episode with melancholic features; a depressive episode with catatonic features; a mood episode with postpartum onset; post- stroke depression; major depressive disorder; dysthymic disorder; minor depressive disorder; premenstrual dysphoric disorder; post-psychotic depressive disorder of schizophrenia; a major depressive disorder superimposed on a psychotic disorder comprising a delusional disorder or schizophrenia; a bipolar disorder comprising bipolar I disorder, bipolar IT disorder, cyclothymic disorder, Parkinson's disease; Huntington's disease; dementia, Alzheimer's disease, multi-infarct dementia, AIDS-related dementia, Fronto temperal Dementia; neurodegeneration associated with cerebral trauma; neurodegeneration associated with stroke; neurodegeneration associated wi th cerebral infarct; hypoglycemia-induced neurodegeneration; neurodegeneration associated with epileptic seizure; neurodegeneration associated with neurotoxin poisoning; multi-system atrophy, paranoid, disorganized, catatonic, undifferentiated or residual type; schizophreniform disorder; schizoaffective disorder of the delusional type or the depressive type; delusional disorder; substance-induced psychotic disorder, psychosis induced by alcohol, amphetamine, cannabis,
cocaine, hallucinogens, inhalants, opioids, or phencyciidine; personality disorder of the paranoid type; and personality disorder of the schizoid type.
In another aspect, there is provided a method for preventing, ameliorating or treating a disease or condition selected from obesity or related diseases, conditions; diabetes (including Type 1 and Type II diabetes); diabetic complications; glucose tolerance; hyperinsulinemia; insulin sensitivity or resistance; metabolic syndromes; cardiovascular diseases including, for example, atherosclerosis, lipidemia, dyslipidemia. elevated blood pressure, microalbuminemia. hyperuricaemia, hypercholesterolemia, hyperlipidemias, hypertriglyceridemias, arteriosclerosis or combination thereof; respiratory diseases or disorders including, for example, sinusitis, asthma, bronchitis or combination thereof; or any combination these diseases, disorders, conditions and/or syndromes thereof; the disease or condition related to serum levels of triglyceride, LDL, HDL, VLDL, total chlolesterol, which method comprises administering to said mammal a therapeutically effective amount of a compound of formulas (I) to (If) in treating said disorder or condition.
General Methods of Preparation
The compounds described herein, including compounds of general formulas (I), (la), (l b), (Ic). (Id), (Ie), (If) and specific examples are prepared using techniques known to one skilled in the art through the reaction sequences depicted in schemes 1 - 15. Furthermore, in the fol lowing schemes, where specific acids, bases, reagents, coupling agents, solvents, etc. are mentioned, it is understood that other suitable acids, bases, reagents, coupling agents etc. may be used and are included within the scope of the present invention. Modifications to reaction conditions, for example, temperature, duration of the reaction or combinations thereof, are envisioned as part of the present invention. The compounds obtained by using the general reaction sequences may be of insufficient purity. These compounds can be purified by using any of the methods for purification of organic compounds known to persons skilled in the art, for example, crystallization or silica gel or alumina column chromatography using different solvents i n suitable ratios. All possible geometrical isomers and stereoisomers are envisioned within the scope of this invention.
The starting materials for the below reaction schemes are commercially available or can be prepared according to methods known to one skilled in the art or by methods disclosed herein. I n general, intermediates and compounds of the present invention may be prepared through the reaction scheme as follows, wherein all symbols are as defined in the description.
The compounds of formula (la) can be prepared according to Synthetic scheme 1 . The. phenyl acetic acid derivative of formula (1 ) is condensed with aromatic aldehyde of formula (2) under suitable Perkin reaction conditions [Journal of Medicinal Chemistry, 1 3, ( 1 970)] to gi ve the acrylic acid of formula (3). Coupling reaction of the acid of formula (3) with amine of formula (4) using appropriate reagents such as dicyclohexyl carbodimide (DCC) or AL| 3- (d;.methylamino)piOpyl]-N'-ethylcarbodiimide (EDCI) either alone or in combination with ΓΗ- benzotriazol- 1 -ol (HOBT) gives compounds of formula (la).
Synthetic scheme 1

The regioisomeric amide of formula (lb) can be prepared according to Synthetic scheme 2. The acetic acid of formula (6) is condensed with benzaldehyde derivative of formula (5) under suitable Perkin reaction conditions to give the acrylic acid derivative of formula (7). Coupling of this acid (7) with amine of formula (4) as described above gives the final compound of formu la (i ).
Synthetic scheme 2

(5) (7)
The synthesis of compounds of formula (la), where X is an oxygen atom, is shown in Synthetic scheme 3. Thus, 4-hydroxyphenylacetic acid of the formula (8) is condensed wi th an aldehyde of formula (2) in the presence of a base such as N-methylmorpholine, triethylamine or diisopropylethylamine in acetic anhydride to give acrylic acid of formula (9). Deacetylation fol lowed by esterification in a one pot reaction under acidic conditions gives the phenolic ester of formula (1 0). Alkylation of compound of formula (10) with alkyl halide of formula ( 1 l a) (where L is halogen) under basic conditions or reaction of appropriate alcohol of formula ( l i b) under Mitsunobu reaction conditions gives intermediate ( 1 2). Base hydrolysis of the ester o f formula ( 12) results in the formation of the corresponding acrylic acid derivative ( 1 3) which on coupling with amine (4), using suitable coupling agent such as EDCJ or DCC gives the compound of formula (Ia-1).
Synthetic scheme 3

(8) (9) ( 10)
Z-Y-L (1 1 a), base or
Z-Y-OH (1 1 b), DEAD, PPh3

Synthetic scheme 4

Z-Y-L ( 1 1 a), base or
Z-Y-O H (1 1 b), P Ph3

(Ib-1 ) ( 18) ( 17)
The amine of formula (Ic) can be prepared according to Synthetic scheme 5. The ester of formula ( 1 2a) is reduced to corresponding alcohol of formula ( 19) using diisobutyl aluminium hydride, LiAlH or L1BH4 in a suitable solvent. The hydroxyl group of formula (1 9) is converted to an appropriate leaving group such as chloride using thionyl chloride or methane sulphonyl chloride or oxalyl chloride to give compound of formula (20). Reaction of Intermediate (20) with an amine of formula (4) in the presence of a base, gives the compound of general formula (lc).
The compound of formula (Ic) can also be prepared from the alcohol of formula ( 1 9) by its oxidation to the corresponding aldehyde of formula (21 ) using an appropriate oxidizi ng agent such as manganese dioxide followed by reductive amination using an appropriate am ine o f formula (4) in presence of sodium triacetoxyborohydride in acetic acid.
Synthetic scheme 5

(21 ) (Ic)
The compounds of formula (lc-1) (wherein Ra is substituted or unsubstituted al kyl) can be prepared according to Synthetic scheme 6. Thus, the chloro compound of formula (20) is treated with sodium azide in the presence of suitable solvent to give azide derivati ve, which on reduction with the suitable reducing agent like triphenylphosphine gives amine derivative of formula (22) via an iminophosphinone intermediate [Tetrahedron letters, 39, p. 3287-3290, ( 1 998)] . Alternatively, Intermediate (22) can also be synthesized directly from chloride of formula (20) by treating it with potassium phthalimide followed by hydrazine hydrate in presence of a suitable solvent such as ethanol. The amine derivative of formula (22) is then coupled with the carboxylic acid of formula (23a) using an appropriate coupling agent or acid chloride of formula (23b) in the presence of a base to give the amide of formula (Ic-1 ).
Synthetic scheme 6

(20) (22)
The compounds of formula (Id-1), (Id-2), (Id-3) and (Id-4) can be prepared from the aldehyde derivative (21 ) according to Synthetic scheme 7. Thus, pyrazole of the formula (Id-1 ) {Tetrahedron letters, 39, p. 3287-3290, (1998)] is synthesized from aldehyde of formula (21 ) by treating it with diethyl (2-{2-[(4-methylphenyl)sulfonyl]hydrazinylidene} ethyl)phosphonate (24) in the presence of a strong base such as sodium hydride. Further, alkylation of the pyrazole of formula (Id-1 ) with suitable alkyl halide of the formula (25) (where X is halogen) in the presence of suitable base such as K2CO3 gives easily separable regioisomeric mixture of products (Id-2) and (Id-3). The aldehyde of formula (21 ) is treated with p-toluenesulphonylmethyl isocyanide (TOSJMI C) in the presence of a suitable base like potassium carbonate in refluxing methanol to give the oxazole derivative of formula (Id-4).

Synthetic scheme 8

(Id-6) (Id-7) (Id-8) (ld-9)
An alternate route for the synthesis of oxadiazole (ld-5) is depicted in Synthetic scheme 9. The ester of formula ( 1 0) is hydrolysed to the corresponding acid of formula (29) in presence of aqueous base. The acid group of formula (29) is converted to a hydrazide using hydrazine hydrate in the presence of ethyl chloroformate and a base such as triethylamine. The hydrazide of formula (30) is treated with an ortho ester of formula (27a) in presence of an acid to give compound of formula (31 ) [J. Med. Chem. 44, p.1268- 1285, (2001 )] . Alternatively, compound of formula (3 1 ) can also be prepared by treating compound of formula (30) with anhydride (27b) under suitable conditions. Deprotection of the carbonate group under basic conditions gives the phenol of formula (32), which on further coupling with alky] halide of formula ( 1 l a) (where L is halogen) under basic conditions or with (l i b) under Mitsunobu reaction conditions gives the final compound of formula (Id-5).
Synthetic scheme 9


The isomeric oxadiazole of formula (Id-10) can be prepared as described in Synthetic scheme ( 1 0). Thus, amide of formula (la), where R9 and R10 are H, is condensed with N, N- dimethylformamide dimethyl acetal under reflux conditions to give the imine derivative (33). Intermediate (33) on reaction with hydroxyl amine in the presence of a suitable base such as sodium hydroxide gives compound of general formula (Id-10).
Synthetic scheme 10
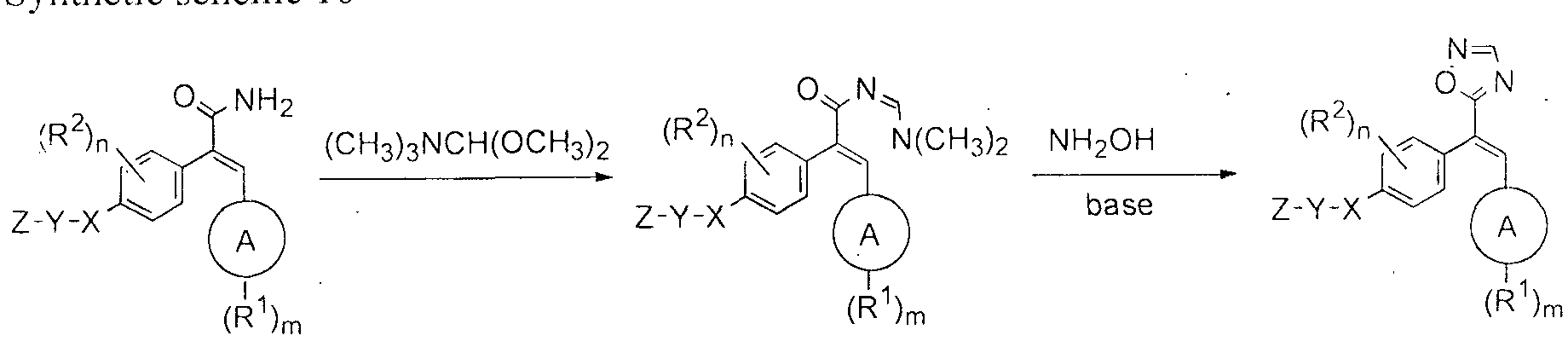
The triazolothione compound of formula (Id-11) and its corresponding sulphur free aromatic compound of formula (Id-12) can be prepared as shown in Synthetic scheme 1 1 . Thus, intermediate (26) is treated with alkylisothiocyanate of the formula (34) under reflux and the intermediate formed is cyclised under basic conditions to give the triazolothione (Id-11 ) [ . Med.
Chein. 37, p. 1 25- 132, ( 1994)] . Further oxidative desulphurization of (Id-11 ) by hydrogen peroxide in acetic acid gives the N-substituted triazole derivative (Id-12).
Synthetic scheme 1 1

The lactam derivative of formula (Id-13) can be prepared according to Synthetic scheme 1 2. Thus, the aldehyde of formula (21 ) undergoes wittig reaction with (carbethoxymethvlene) triphenylphosphorane to give the diene ester of formula (35). Michael addition of nitromethane anion, generated by means of suitable base such as 1 , 1 ,2,2-tetramethylguanidine (TMG) gives the adduct of formula (36). Selective reduction of the nitro group using an appropriate reagent such as iron and ammonium chloride under aqueous conditions gives amine of the formula (37). Compound of formula (37) undergoes intramolecular cyclization in refluxing xylene to give compound of formula (Id-13).
Synthetic scheme 12
Fe/NH4CI
EtOH/ water

Simi lar to the approaches described in synthetic schemes 8 and 1 1 , various regioisomeric olefins can be prepared from hydrazide of the isomeric acid (7). Preparation of one such derivative is shown in Synthetic scheme 13. Thus, the acrylic acid of formula (7) was coupled with hydrazine hydrate to give hydrazide of formula (38) using a suitable coupling agent such as EDCI. The oxadiazole derivative (Ie-1) can be prepared as described in Synthetic scheme 1 3 using orthoester of formula (27a) or its equivalent (27b) or (27c) under suitable reaction conditions.
Synthetic scheme 1 3

The ether derivative of formula (If) can be prepared according to Synthetic scheme 1 4. Thus, chloro compound of formula (20) is coupled with alcohol of formula (39) under basic conditions to give ether of general formula (If). Alternatively, final compound of general
formula (If) can also be prepared by itsunobu coupling reaction of alcohol ( 1 9) with appropriate aromatic alcohol of formula (39).
Synthetic scheme 14

The compound of general formula (Ig) can be prepared as depicted in Synthetic scheme 1 5. Thus, ethyl (4-iodophenyl)acetate derivative (40) undergoes Sonagashira coupling reaction with ethynyltrimethyl silane followed by desilylation using appropriate reagent such as TBAF.H2O, to give the acetylene compound of formula (41 ). The coupling reaction of compound of formula (41 ) with 2-bromoquinoline under Sonagashira coupling reaction gives the compound of formula (42). Catalytic reduction of the triple bond of compound of formula (42) followed by hydrolysis gives compound of formula (43). Perkin reaction of compound of formula (43) with aromatic aldehyde (2) gives acid derivative which on further amide coupli ng with amine of formula (4) gives the final compound of general formaul (I ).
Synthetic scheme 1 5

2. reduction
3. hydrolysis

Unless otherwise stated, work-up includes distribution of the reaction mixture between an organic and aqueous phase, separation of layers and drying the organic layer over sodium sulphate, filtration and evaporation of the solvent. Purification, unless otherwise mentioned, refers to purification by silica gel chromatographic techniques, in suitable solvents o f a suitable polarity as the mobile phase or crystallization from an appropriate solvent or mixture o f solvents. The following abbreviations are used in the text: DMSO-i 6: hexadeuterodimethyl su lfoxide: CDCI3 : deuterated chloroform; J: coupling constant in units of Hz; T or rt: room temperature (22-26°C). Aq. : aqueous; equiv. or eq. : equivalents.
fhe starling materials represented by the general formula Z-Y-X-, used for the preparation intermediates and compounds of invention are in some cases commercially available or can be prepared according to suitable literature procedure or as described below.
Preparation of Intermediates
Preparation of 2-(chloromethyl)imidazo[l 2-o]pyridine

The title compound was prepared by condensation of 2-aminopyridine with 1 ,3-dichloroacetone in presence of dimethoxyethane as described in the literature (WO 2009/1 52825): Ή N MR (300 MHz, CDCI3) δ 4.77 (s, 2H), 6.79 (t, J = 6.9 Hz, 1 H), 7.1 9 (t, J = 7.5 Hz, 1 H), 7.57 (d, J = 93 Hz, 2H), 7.61 (s, 1 H), 8.07 (d, J = 6,3 Hz, 1H).
Preparation of l ,3-benzothiazol-2-ylmethanol

The title compound was synthesized by the reaction of 2-aminothiophenol with glycolic acid using 4 hydrochloric acid as described in the literature [Journal of the Chemical Society, p.
2395. ( 1 928)] ; Ή NMR (300 MHz, CDC13) δ 5.08 (s, 2H), 7.39 (t, J = 7.5 Hz, 1 IT), 7.49 (t, J = 7.2 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1 H).
Preparation of 2-(chloromethyl)- 1 -methyl- 1 H-benzimidazole

Synthesis of the title compound involves conversion of o-phenylenediamine to 1 /-/-benzimidazol- 2-ylmethanol using glycolic acid as described in the literature [Journal of (he Chemical Society. p. 2395, ( 1 928)], followed by alkylation and chlorination; Ή NMR (300 MHz, CDC13) δ 4. 1 4 (s; 3 H), 5.42 (s, 2H), 7.47-7.52 (m, 1 H), 7.56-7.64 (m, 2H), 7.75 (d, J = 8.4 Hz, 1 H ).
Preparation of 2-(l ,2-benzoxazol-3-yl)ethanol
 2)2OH
2)2OH
Reaction of 4-hydroxycoumarin with hydroxylamine hydrochloride in presence of trielhylamine (TEA) gives l ,2-benzoxazol-3-ylacetic acid (WO 02/070495) which after esterification is reduced to the corresponding alcohol, using lithium aluminium hydride, to give the title compound; Ή NMR (300 MHz, CDC13) δ 3.04 (br s, 1 H), 3.24 (t, J = 6.0 Hz, 2H), 4. 1 3 (I, ./ = .5.7 Hz, 2H), 7.30-7.35 (m, 2H), 7.57 (br s, 2H), 7.69 (d, J = 7.8 Hz, 1 H).
Preparation of (1 -methyl- lH-indazol-3-yl)methyl methanesulfonate
The title compound was synthesized by reduction of methyl l -methyl- l H-indazole-3-carboxylate using lithi um aluminium hydride followed by its reaction with methanesulfonyl chloride in the presence of TEA; Ή NMR (300 MHz, CDC13) δ 3.03 (s, 3H), 4.06 (s, 3H), 4.97. (s, 2H), 7. 1 9- 7.24 (m, 1 H), 7.42 (d, J = 7.5 Hz, 2H), 7.82 (d, J = 8.1 Hz, l H).
Preparation of mieno[3,2-/ ]pyridin-5-ylmethyl methanesulfonate
N _.CH2OS02CH3
Reaction of 3-aminothiophene-2-carboxaldehyde with pyruvic acid in presence of sodium hydroxide gave thieno[3,2-6]pyridine-5-carboxylic acid, which was esterified using ethanol and catalytic amount of cone, sulphuric acid to give the corresponding ethyl ester. The ester group
was reduced using lithium aluminium hydride and the alcohol thus obtained was treated with methanesulphonyl chloride to give the title compound (WO 94/22869); Ή NMR (300 MHz, CDCI3) δ 3.10 (s, 3H), 5.46 (s, 2H), 7.46 (d, J= 8.1 Hz, 1H), 7.55 (d, J= 5.4 Hz, 1H), 7.83 (d, J = 5.4 Hz, 1 H), 8.26 (d, J = 5.4 Hz, 1H).
Preparation of 5-(bromomethyl)furo[3,2-£]pyridine
CH2Br
Reaction of 2-iodo-6-methylpyridin-3-ol with ethynyltrimethylsilane under Sonagashira coupling reaction conditions resulted in the formation of 5-methyl-2-(tnmethylsilyl)furo|3.2-/ ]pyridine via C-C coupling and intramolecular cyclization. Desilylation using tetra-n-butyl ammonium fluoride followed by bromination of the methyl group using NBS gives the title compound
(Synlett, 2002, 3, 453 -'457); 1H MR (300 MHz, CDC13) δ 5.12 (s, 2H), 6.93 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.66 (d, J= 7.8 Hz, 1H), 7.81 (s, 1 H).
Preparation of { 1 -[(4-Methylphenyl)sulfonyl]-lH-pyrrolo[3,2-?]pyridin-5-yl}methyl-4-methyl benzenesulfonate

Step 1 : Ethyl 5-amino-6-[2-(trimethylsilyl)ethynyl] pyridine-2-carboxylate:
To the well stirred solution of 5-amino-6-iodopyridine-2-carboxylate (7.5 g, 25.684 mmol) in TEA (40 ml) were added ethynyltrimethylsilane (10.16 ml. 71.915 mmol), bis(triphenylphosphine) palladium(lI)chloride (180 mg, 0.256 mmol) and copper iodide (146 mg, 0.770 mmol) and the reaction mixture was stirred at room temperature overnight. The reaction mixture was quenched with water (100 ml) and extracted with ethyl acetate (300 ml x 3). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 7 g of product as off-white solid; Ή NMR (300 MHz, DMSO-<¾) δ 0.27 (s,
9H), 1.28 (t, J= 6.9 Hz, 3H), 4.24 (q, J= 6.9 Hz, 2H), 6.24 (s, 1H), 6.62 (s, 1H), 7.09-7.14 (m, 1H); 7.74-7.79 (m, 1H).
Step 2: Ethyl 5-acetamido-6-[2-(trimethylsilyl)ethynyl]pyridine-2-carboxylate:
To the well stirred solution of Step 1 intermediate (5.5 g. 20.659 mmol) in DCM (1 00 ml) was added pyridine (3.3 ml) and the reaction mixture was cooled to 0 °C. Acetyl chloride (1 .76 ml. 24.79 mmol) was then added to the reaction mixture drop wise and the reaction was stirred at room temperature for 2 h. The reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (300 ml x 3). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 5.56 g of product as off-white solid; Ή NMR (300 MHz, DMSC ¾ δ 0.30 (s, 9H), 1.32 (t, J = 7.5 Hz, 3H), 2. 16 (s, 3H), 4.33 (q, J = 6.9 I I/. 2H), 8.04 (d, .7 = 8.4 Hz, 1 H), 8.39 (d, J = 8.4 Hz, 1 H), 9.53 (s, l H).
Step 3 : Ethyl l H-pyrrolo[3,2-j]pyridine-5-carboxylate:
To the well stirred solution of Step 2 intermediate (5.5 g. 1 7.84 mmol) in TH F (50 ml) was added tetra-n-butyl ammonium fluoride (4.69 g, 17.59 mmol) and the reaction mixture was refluxed for 8 h. The reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (300 ml x 3). The combined organic layers were washed with water and brine, dried over anhydrous Na2S(Xi and concentrated to yield 2.5 g of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1 .43 (t, J = 6.6 Hz, 3H), 4.51 (q, J = 6.6 Hz, 2H), 6.80 (br s, 1 H), 7.58-7.62 (m3 1 H), 7.80 (d, .7 = 8.1 Hz, 1 H), 8.06 (d, J = 8.4 Hz, 1 H), 9.42 (s, 1 H).
Step 4: l //-Pyn lo[3,2-£]pyridin-5-ylmethanol :
To the well stirred suspension of lithium aluminium hydride (359 g, 9.473 mmol) in dry 'TH F (20 ml) was added a solution of Step 3 intermediate (300 g, 1 .578 mmol) in THF at 0 °C and the reaction mixture was stirred at same temperature for 2 h. The reaction mixture was quenched with saturated sol ution of sodium sulphate, diluted with ethyl acetate and filtered. The filtrate was dried and concentrated under reduced pressure to yield 330 mg of the product; Ή NMR (300 MHz, DMSO-c¾ δ 4.61 (s, 2H), 5.24 (br s, 1 H), 6.46 (s, 1 H), 7.80 (d, J = 8.4 Hz. 1 H), 7.59 (s, l H), 7.74 (d, J = 8.4 Hz, 1H), 1 1 .21 (br s, 1 H).
Step 5 : |T -(Phenylsulfonyl)- l //-pyrrolo[3,2-j]pyridin-5-yl]methyl benzenesulfonate:
To the well stirred suspension of sodium hydride (324 mg, 8. 108 mmol) in dry THF (30 ml) was added Step 4 Intermediate (400 mg, 2.702 mmol) at 0 °C and the reaction mixture was stirred for 1 0 min. The solution of losyl chloride ( 1 .288 g, 6.755 mmol) in THF was added to the reaction mixture at 0 °C and it was further stirred overnight. The reaction mixture was quenched with water (50 ml) and extracted with chloroform (50 ml x 2). The combined organic layers were
washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 290 mg of product as off-white solid; Ή NMR (300 MHz, DMSO-i¾ δ 2.3 1 (s, 3H), 2.34 (s, 3H), 5. 1 8 (br s, 2H), 6.91 (br s, 1 H), 7.36-7.41 (m, 5H), 7.75 (d, J = 7.8 Hz, 2H), 7.91 (d, .7 = 7.5 Hz, 2H), 8.1 8 (br s, 1 H), 8.3 1 (d, .7 = 7.8 Hz, 2H).
Preparation of (1 -methyl- l H-pyrrolo[3,2-6]pyridin-5-yl)methyl methanesulfonate
CH2OMs
H3C
The title compound was synthesized by methylation of ethyl l H-pyrrolo[3,2-i>}pyridine-5- carboxylate using methyl iodide followed by its subsequent reduction and mesylation; Ή NMR (300 M Hz, CDC13) δ 3.03 (s, 3H), 4.02 (s, 3H), 5.09 (s, 2H), 7.05-7.1 1 (m, I H), 7.61 (d, J = 8.4 Hz, 1 H), 7.76 (s, 1 H), 8.30 (d, J = 8.4 Hz, 1 H).
Preparation of tert -butyl 5-(hydroxymethyl)-lH-indazole- l -carboxylate

The title compound was synthesized by esterification of indazole-6-carboxylic acid using methanol in presence of cone, sulphuric acid followed by reduction of the ester group using lithium aluminium hydride and its subsequent reaction with di-ter/-butyl dicarbonate anhydride;
Ή NMR (300 MHz, DMSO-c 6) δ 1.64 (s, 9H), 4.67 (d, J = 5.1 Hz, l H), 5.43 (br s, I H), 7.29 (d, .] = 7.8 Hz, I H), 7.80 (d, J = 8.1 Hz, 1 H), 8.12 (s, 1 H), 8.36 (s, 1 H).
Preparation of ( 1 -methyl- l H-benzimidazol- - l methanol

The title compound was synthesized by esterification of l//-benzimidazole-5-carboxylic acid using methanol in presence of cone, sulphuric acid followed by N-methylation using methyl iodide in presence of potassium carbonate and subsequent reduction of the ester group by lithium
aluminium hydride; Ή NMR (300 MHz, DMSO- 6) δ 3.84 (s, 3H), 4.59 (s, 2H), 5.23 (br s, l H), 7.26 (d, J = 8.4 Hz, 1H), 7.52-7.58 (m, 2H), 8.23-8.31 (m, 1 H).
Preparation of 5-(bromomethyl)[l ,3]oxazolo[4,5-6]pyridine

The title compound was prepared by reaction of 2-amino-6-methylpyridin-3-ol with triethyl orthoformate followed * by bromination of methyl group with N-bromosuccinimide (NBS) in presence of azobisisobutyronitrile (AIBN); 'H NMR (300 MHz, CDC13) δ 5.12 (s, 2H), 7.22 (d, J = 8.4 Hz5 1H), 7.78 (d, J = 8.4 Hz, 1H), 8.29 (s, 1 H).
Preparation of pyrazolo[l ,5-tf]pyrimidin- -ylmethyl methanesulfonate

Step 1 : Ethyl 7-oxo-4,7-dihydropyrazolo[l ,5-a]pyrimidine-5-carboxylate:
To the well stirred solution of lH-pyrazol-3-amine (5 g, 60. 1 75 mmol) in THF (40 ml) was added diethyl acetylene dicarboxylate (7.70 ml, 48.14 mmol) and the reaction mixture was stirred at room temperature for 72 h. The precipitate obtained was filtered, washed with THF and dried to yield 4.5 g of the product; Ή NMR (300 MHz, DMSO-i¾ δ 1 .35 (t, J = 7.2 Hz, 3H), 4.41 (q, J = 6.9 FIz, 2H), 6.27 (s, 2H), 7.95 (s, 1 H).
Step 2: Ethyl 7-chloro-4,7-dihydropyrazolo| l ,5-a]pyrimidine-5-carboxylate:
To the well stirred solution of Step 1 intermediate (1 g, 4.830 mmol) in phosphoryl chloride (POC13) (10 ml) was added N,N-dimethylaniline ( 1 ml, 4.83 mmol) and the reaction mixture was refiuxed for 40 min. The excess of POCl3 was distilled under reduced pressure and the residue obtained was poured into crushed ice and extracted with chloroform (2 x 1 00 ml). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 1 .1 g of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1 .48 (t, J = 7.2 Hz, 3H), 4.54 (q, J = 6.9 Hz, 2H), 7.08 (s, 1 H), 7.75 (s, I H), 8.35 (s, 1 H).
Step 3 : 7-Chloropyrazolo[ l ,5-a]pyrimidine-5-carbaldehyde:
To the well stirred solution of Step 2 intermediate (1 g, 4.43 mmol) in dry THF ( 1 5 ml) was added 20 % Diisobutylaluminium hydride (DIBAL-H; 2.22 ml, 13.30 mmol) drop wise at 0°C
and the reaction mixture was stirred at the same temperature for 1 h. Water was added to the reaction mixture and it was further stirred for 1 5 min. The precipitate formed was fi ltered and the filtrate was concentrated to yield 1 .3 g of the product: Ή N.MR (300 MHz. DMSO-ck) δ 7.29 (s. I H), 7.73 (s, 1 11), 8.56 (s, 1 H), 9.91 (s, I H).
Step 4 : (7-Chloropyrazolo| 1 ,5-fif]pyrimidin-5-yl)methanol :
To the well stirred solution of Step 3 intermediate (1 .25 g, 5.449 mmol) in DCM ( 1 0 ml) was added sodium triacetoxyborohydride (1.154 g, 5.449 mmol) and the reaction was stirred at room temperature overnight. The reaction mixture was diluted with water and the pH was made neutral with sodium bicarbonate. The compound was extracted with chloroform and the organic layer was washed with water, brine, dried, filtered and concentrated to yield the 81 0 mg of the product; Ή NMR (300 MHz, DMSO-^) δ 4.59 (s, 2H), 5.75 (br s, 1 H), 6.79 (s, 1 H), 7.38 (s, 1T1), 8.3 1 (s, l H).
Step 5 : Pyrazolo[l ,5-o]pyrimidin-5-ylmethanol :
To the well stirred solution of Step 4 Intermediate (800 mg, 4.312 mmol) in a mixture of ethanol ( 12 ml) and ethyl aceatate (27 ml) was added sodium acetate (424 mg, 5. 1 74 mmol) and pal ladium on activated carbon (20 mg) and the reaction mixture was stirred at room temperature under hydrogen atmosphere for 5 h. The reaction mixture was diluted with ethyl acetate fi ltered and washed with saturated solution of sodium bicarbonate, water and brine. The organic layer was dried and concentrated to yield 300 mg of the product; Ή NMR (300 MHz, DMSO-c¾ δ 4.57 (s, 2H), 5.66 (br s, 1 H), 6.59 (s, 1 H), 7.09 (d, J = 7.2 Hz. 1 H). 8.1 7 (s. 1 H), 9.05 (d: J = 7.2 Hz, 1 H).
Step 6: Pyrazolo[l ,5-o]pyrimidin-5-ylmethyl me hanesulfonate:
To the wel l stirred solution of Step 5 intermediate (100 mg, 0.662 mmol) in DCM ( 1 0 ml) was added tri ethyl amine (TEA) (0.27 ml, 1 .986 mmol) and the reaction mixture was cooled to 0°C. Methanesulphonyl chloride was added drop wise to the reaction mixture at this temperature and the reaction mixture was stirred for 30 min. The reaction mixture was diluted with water and was extracted with chloroform and the organic layer was washed with water, brine, dried, filtered and concentrated to yield the 150 mg of the product; Ή NMR (300 MHz, DMSO-e¾) δ 2.37 (s: 3H), 5.37 (s, 2H), 6.75 (s, 1 H), 7.1 l (d, J = 7.2 Hz, 1 H), 8.26 (s, 1 H); 9. 1 8 (d, J = 7.5 Hz. 1 H).
The following intermediates were prepared using appropriate methods described in Synthetic schemes 1 to 1 3.
Intermediate 1
3-(4-Chlorophenyl)-2-[4-(imidazo[l ,2-i7]pyridin-2-ylmethoxy)phenyl]prop-2-enoic acid
Step 1 : 2-[4-(Acetyloxy)phenyl]-3-(4-chlorophenyl)prop-2-enoic acid:
To a well stirred solution of 4-hydroxy phenyl acetic acid ( 1 0 g, 65.72 mmol) in acetic anhydride ( 1 00 ml), were added TEA ( 1 3.7 ml, 98.5 1 mmol) and 4-chloro benzaldehyde (9.23 g: 65.66 mmol) and the reaction mixture was refluxed for 3 h. The reaction mixture was quenched with water ( 100 ml) and was further refluxed for 30 mins. Mixture was cooled to room temperature and filtered. The. solid obtained was titured with diethyl ether and dried to yield 1 2 g of off-white solid; Ή NMR (300 MHz, DMSO-c¾ δ 2.28 (s, 3H), 7.06 (d, J = 9.0 Hz, 2H), 7. 1 1 -7.2 1 (m. 4H), 7.27 (d, J = 8.1 Hz, 2H), 7.75 (s, 1 H).
Step 2: Ethyl -3 -(4-chloro phenyl)-2-(4-hydroxyphenyl)prop-2-enoate:
To a well stirred solution of the Step 1 Intermediate ( 1 1 .5 g, 36.33 mmol) in ethanol (70 ml) was added concentrated sulphuric acid (9 ml) and the reaction mixture was refluxed for 1 6 h. The excess of ethanol was distilled off and the reaction mixture was diluted with ethyl acetate (500 ml). The organic layer was washed with water (50 ml x 2) and brine (50 ml), dried over anhydrous Na2S04 and concentrated to yield the 10.5 g of off-white solid; Ή NMR (300 MHz, CDCI3) δ 1 .30 (t, J = 7.5 Hz, 3H), 4.27 (q, J = 7.5 Hz, 2H), 5.17 (br s, 1 H), 6.80 (t, J = 8.4 Hz, 2H), 6.97-7.07 (m, 4H), 7.1 4 (d, J = 8. 1 Hz, 2H), 7.72 (s, 1 H).
S ep 3 : Ethyl-3-(4-ch]orophenyl)-2-[4-(imidazo[l ,2--7]pyridin-2-ylmethoxy)phenyl |piOp-2- enoate:
To a well stirred solution of Step 2 Intermediate ( 10 g, 33.05 mmol) in dimethyl formamide (60 ml), were added potassium carbonate (6.85 g, 49.57 mmol) and 2-(ch!oromethyl)im idazo|' l .2- ajpyridine (5.50 g, 33.05) and the reaction mixture was heated to 60 °C for 1 6 h. The reaction mixture was diluted with water (20 ml) and extracted with ethyl acetate (50 ml x 3). The combined organic layers were washed with water (25 ml x 2) and brine (25 ml), dried over
anhydrous Na2S0 , filtered and concentrated to yield 9 g of off-white solid; Ή NMR (300 MHz. CDCls) δ 1 .30 (t, J = 6.9 Hz, 3H), 4.26 (q, J = 7.5 Hz, 2H), 5.30 (s, 2H), 6.80 (t, J = 6.9 Hz, 1 H), 6.96-7.05 (m, 4H), 7. 12 (d, J = 6.9 Hz, 4H), 7.16-7.23 (m, 1 H), 7.60 (d, J = 9.3 Hz, 1 H), 7.66 (s, I H), 7.72 (s, I H), 8.1 1 (d, J = 6.9 Hz, 1H).
Step 4: 3-(4-Chlorophenyl)-2-[4-(imidazo[l ,2-a]pyridin-2-ylmethoxy)phenyl]prop-2-enoic acid : To a well stirred solution of Step 3 Intermediate (8.5 g, 19.63 mmol) in ethanol (60 ml) was added aqueous solution of 1 N sodium hydroxide and the reaction mixture was stirred for 1 6 h at room temperature. The excess of ethanol was distilled off and the reaction mass was di l uted wi th water (50 ml). It was further acidi fied with 2N HC1 solution and the solid so obtained was filtered and washed with water to yield 7.5 g of product as off-white solid; Ή NMR (300 MHz, D SO-tftf) δ 5.27 (s, 2H), 7.01 - 7.10 (m, 7H), 7.27 (d, J = 8. 1 Hz, 2H), 7.40 (t, J = 7.5 Hz, 1 H), 7.63 (d, J = 8.7 Hz, 1 H), 7.70 (s, 1 H), 8.1 'l (s, 1H), 8.63 (d, J = 6.3 Hz, IH), 13.58 (br s, 1 H, D20 exchangeable).
Intermediate 2
2- 14-( 1 ,3-Benzothiazol-2-ylmethoxy)phenyl]-3-(4-chloiOphenyl)prop-2-enoic acid

Step 1 : Ethyl -2-[4-(l ,3-benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)prop-2-enoate: To the well stirred solution of ethyl-3-(4-chlorophenyl)-2-(4-hydroxyphenyl)prop-2-enoate (Step 2 of intermediate 1 , 1 .5 g, 4.958 mmol) in dry tetrahydrofuran (30 ml), were added 1 ,3- benzothiazol-2-ylmethanol (81 9 mg, 4.958 mmol) and triphenyl phosphine (,1 .95 g, 7.438 mmol) followed by dropwise addition of diethyl azodicarboxylate ( 1 .01 ml, 6.446 mmol). The reaction mixture was stirred at room temperature for 10 mins after which it was heated at 60 °C overnight. The reaction mixture was diluted with water (30 ml) and extracted with ethyl acetate ( 100 ml x 3). The combined organic layers were washed with water (50 ml x 2) and brine (50 ml), dried over anhydrous Na2S04, filtered and concentrated to yield 670 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1 .29 (t, J = 7.5 Hz, 3H), 4.26 (q, J = 7.2 Hz, 2H),
5.52 (s, 2H), 6.96 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8. 1 Hz, 2H), 7.09-7. 1 6 (m, 4H), 7.43 (t, ./ =
7.2 Hz, ΓΗ), 7.52 (t, =7.2 Hz, 1H), 7.73 (s, 1H), 7.93 (d, J - 7.8 Hz, 1H), 8.04 (d: J = 7.8 Hz, IH).
Step 2: 2-[4-(l,3-Benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)prop-2-enoic acid:
To a well stirred solution of the Step 1 Intermediate (650 mg, 1.445 mmol) in ethanol (15 ml) was added aqueous solution of IN sodium hydroxide and the reaction mixture was stirred for 16 h at room temperature. The excess of ethanol was distilled off and the reaction mass was diluted with water (25 ml). It was further acidified with 2N HC1 solution and the solid so obtained was filtered and washed with water to yield 560 mg of product as off-white solid: Ή M (300 MHz, DMSO-i¾) δ 5.63 (s, 2H), 7.04-7.11 (m, 6H), 7.23 (d, .7= 8.4 Hz, 2H), 7.44-7.57 (m, 211), 7.70 (s, 1H), 8.02 (d,J= 8.4 Hz, 1H), 8.14 (d,J= 7.8 Hz, 1 H), 12.75 (br s, 1H).
Intermediate 3
2-{4- 2-(l ,2-Benzoxazol-3-yl)ethoxy]phenyl}-3-(4-chlorophenyl)prop-2-enoic acid

The title compound was prepared by Mitsunobu coupling of ethyI-3-(4-chIorophenyl)-2-(4- hydroxyphenyl)prop-2-enoate and 2-(l,2-benzoxazol-3-yl)ethanol as described in Intermediate 2; Ή NMR (300 MHz, OMSO-d6) δ 3.48-3.53 (m, 2H), 4.44 (t, J = 6.3 Hz, 2H), 6.92 (d, .7= 8.1 Hz, 2H), 7.02-7.08 (m, 4H), 7.26 (d, J = 8.1 Hz, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.63-7.75 (m, 3H), 8.02 (d, J =7.8 Hz, 1H), 12.85 (br s, 1H).
Intemediates 4 to 38 were prepared using appropriate starting materials as described in Intermediate 1. Their structure, names and Ή NMR data are given in the Table 1.
Table 1 : Structure and characterization data for Intermediates 4-38
Molecular Structure and Chemical name and Ή NMR data (δ ppm, 300 MHz)
Intermediate No.



[Intermediate 24] 8.02 (d, J= 8.4 Hz, 2H), 8.40-8.48 (m, 3H), 13.05 (br s, IH).
(t, =

[Intermediate 30] IH).
 3-(3,4-Difliiorophenvl)-2-[4-(quinolin-2-vlmethoxy)pheny | prop-2-enoic acid: (CDCl ) δ 5.44 (s, 2H), 6.78-6.88 (m, 2H), 6.93-6.99 (m, lH), 7.05 (d, ,7 = 8.7 Hz, 2H), 7.16 (d, - 8.1 Hz, 2H), 7.57 (t, J= 7.2 Hz, 1H), 7.71-7.79 (m, 3H)S 7.86 (d, ./ =
3-(3,4-Difliiorophenvl)-2-[4-(quinolin-2-vlmethoxy)pheny | prop-2-enoic acid: (CDCl ) δ 5.44 (s, 2H), 6.78-6.88 (m, 2H), 6.93-6.99 (m, lH), 7.05 (d, ,7 = 8.7 Hz, 2H), 7.16 (d, - 8.1 Hz, 2H), 7.57 (t, J= 7.2 Hz, 1H), 7.71-7.79 (m, 3H)S 7.86 (d, ./ =
 7.8 Hz, 1H), 8.19 (d, J= 8.4 Hz, IH), 8.24 (d, J= 9.0 Hz, lH),
7.8 Hz, 1H), 8.19 (d, J= 8.4 Hz, IH), 8.24 (d, J= 9.0 Hz, lH),
[Intermediate 37] 12.45 (brs, 1H).
3-(4-Chloro-3-fluorophenyl)-2-[4-(quinolin-2-ylmethoxy) phenvl]prop-2-enoic acid: (DMSO-d*) δ 5.41 is, 2H), 6.92 (d, .7 = 9.0 Hz, IH), 7.02-7.10 (m, 5H), 7.42 (t, ./ = 7.8 Hz, IH), 7.60- 7.66 (m, 1H), 7.70 (d, J = 8.7 Hz, 1H), 7.79 (I, J= 7.8 Hz, IH),
CI
8.01 (t, J= 7.5 Hz, 2H), 8.43 (d, J = 8.1 Hz, IH), 12.95 (br s,
[Intermediate 38] IH).
Intermediate 39
3-( 1 -Oxidopyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enoic acid

Ethyl-2-(4-hydroxyphenyl)-3-(pyridin-4-yl)prop-2-enoate was synthesized from 4-hydroxy phenylacetic acid and 4-pyridine carboxaldehyde as described in Steps 1 and 2 of Intermediate 1. TV-oxidation of this intermediate by m-chloroperbenzoic acid gave ethyl-2-(4-hydroxyphenyl)-3- (1 -oxidopyridin-4-yl)prop-2-enoate which was furthur coupled with 2-(chloromethyl)quinoline and hydrolysed as described in Steps 3 and 4 of Intermediate 1 to give the title compound; Ή N R (300 MHz, DMSO-<¾) δ 5.40 (s, 2.H), 7.01 (d, J - 8.1 Hz, 2H), 7.08-7.17 (m, 4H), 7.59- 7.65 (m, 2H), 7.71 (d, J= 8.4 Hz, IH), 7.80 (t, J= 7.5 Hz, IH), 7.99-8.06 (m, 4H), 8.45 (d, J = 8.4 Hz, IH), 12.62 (brs, 1 H).
Intermediate 40
3-(4-Chlorophenyl)-2-{4-[2-(quinolin-2-yl)ethyl]phenyl}prop-2-enoic acid

Step 1 : Ethyl {4-[(trimethylsilyl)ethynyl]phenyl }acetate:
To a well stirred solution of ethyl (4-iodophenyl)acetate (5.5 g, 1 8.96 mmol) in DM'SO (40 ml) were added dichlorobis(triphenylphosphine) palladium (11) ( 1 33 mg, 0.1 8 mmol). copper iodide ( 1 08 mg, 0.568 mmol), triethylamine (3.95 ml, 2.844 mmol) and ethynyltrimethyl silane (2.04 g, 20.85 mmol) and the reaction was stirred at room temperature overnight. The reaction mixture was diluted with water ( 1 00 ml) and ethyl acetate (300 ml), given charcoal treatment and filtered. The organic layer was washed with water (2 x 100 ml), brine (50 ml), dried and concentrated to yield 5 g of the product; Ή NMR (300 MHz, CDC13) δ 0.24 (s, 9H), 1.21 - 1 .27 (m, 3H)5 3.59 (s, 2H), 4.14 (q, J = 6.9 Hz, 2H), 7.03 (d, J = 8.4 Hz, 1 H), 7.25 (d, J = 7.8 Hz, 1 H), 7.4 1 (d, J - 8.4 Hz, 1 H), 7.64 (d, J = 7.8 Hz, 1 H).
Step 2: Ethyl (4-ethynylphenyl)acetate:
To the well stirred solution of Step 1 intermediate (4.9 g, 1 8.82 mmol) in dich loromethane (30 ml) was added tetrabutyl ammonium fluoride hydrate (2.461 g, 9.413 mmol) and the reaction mixture was stirred at room temperature for 10 min. The reaction mixture was quenched with water ( 1 00 ml) and extracted with chloroform (2 x 250 ml). The combined organic layers were washed with water (2 x 50 ml) and brine (50 ml), dried over anhydrous a?S04 and concentrated to yield the crude product. The compound was purified by silica gel column chromatography to yield 2.9 g of the product; Ή NMR (300 MHz, CDC13) δ 1 .25 (t, ,/ - 7.2 Hz, 3H); 3.06 (s, 1 H). 3.61 (s, 2H), 4.1 5 (q, J = 6.9 Hz, 2H), 7.24 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H).
Step 3 : Ethyl [4-(quinolin-2-ylethynyl)phenyl]acetate:
To a well stirred solution of Step 2 intermediate (1 .44 g, 7.6 mmol) in TEA (20 ml) were added dichlorobis(triphenylphosphine) palladium (II) (54 mg, 0.076 mmol), copper iodide (44 mg, 0.23 mmol) and 2-bromoquinoline (1 .6 g, 7.69 mmol) and the reaction was stirred at room temperature overnight. The reaction mixture was diluted with water ( 1 00 ml) and chloroform (300 ml), given charcoal treatment and filtered. The organic layer was washed with water (2 x 100 ml), brine (50 ml), dried and concentrated yield 1 .45 g o f the product; Ή NMR (300 M Hz,
CDCI3) δ 1.26 (t, J = 7.2 Hz, 3H), 3.65 (s, 2H), 4.17 (q, J = 7.2 Hz, 2H), 7.31 (d, J= 7.8 Hz, 2H), 7.56-7.64 (m, 4H), 7.74 (t, J= 7.2 Hz, 1H), 7.81 (d, J= 8.1 Hz, 1H), 8.14 (t, J = 7.8 Hz, 2H). Step 4: Ethyl {4-[2.-(quinolin-2-yl)ethyl] phenyl} acetate:
To the well stirred solution of Step 3 intermediate (1.40 g, 4.44 mmol) in ethanol (30 ml) was added palladium on activated carbon (400 mg) and the reaction was carried for 4 h under 40 psi pressure of hydrogen gas. The reaction mixture was diluted with ethyl acetate and filtered. The organic layer was washed with water and concentrated to yield 1.35 g of the product; Ή NMR (300 MHz, CDCI3) δ 1.25 (t, J= 6.9 Hz, 3H), 3.12-3.17 (m, 2H), 3.29 (d, J = 8.1 Hz, IH), 3.58 (m, 2H), 4.14 (q, J =1.5 Hz, 2H), 7.21 (s, 4H), 7.23 (s, IH), 7.51 (t, J = 7.2 Hz, IH), 7.71 (t, .7 = 7.5 Hz, 1 IT), 7.79 (d, J = 8.1 Hz, IH), 8.04 (d, J = 8.4 Hz, 2H).
Step 5: {4-[2-(Quinolin-2-yl)ethyl]phenyl}acetic acid:
To the well stirred solution of Step 4 intermediate (500 mg, 1.566 mmol) in a mixture of ethanol (15 ml) and tetrahydrofuran (5 ml) was added aqueous solution of sodium hydroxide (313 mg, 7.83 mmol) and the reaction mixture was stirred at room temperature overnight. The solvent was distilled out and reaction mass was diluted with water and then neutralized with dil. HQ. The aqueous layer was extracted with ethyl acetate (2 x 100 ml). 'Hie combined organic layer was washed with water, brine, dried and concentrated under reduced pressure to yield 470 mg of the product; Ή NMR (300 MHz, CDC13) δ 3.02-3.07 (m, 2H), 3.24-3.30 (m, 2H), 3.69 (s, 21T), 7.17 (d, J = 7.8 Hz, 2H), 7.29 (s, 2H), 7.35 (d, J = 8.1 Hz, IH), 7.58 (t, J = 7.5 Hz, IH), 7.76 (t, J = 7.8 Hz, IH), 7.82 (d,J= 7.8 Hz, IH), 8.18 (d,J = 8.1 Hz, IH), 8.30 (d,J= 8.4 Hz, IH).
Step 6: 3-(4-Chlorophenyl)-2-{4-[2-(quinolin-2-yl)ethyl]phenyl}prop-2-enoic acid:
To the well stirred solution of Step 5 intermediate (460 mg, 1.580 mmol) in acetic anhydride (15 ml), were added triethylamine (0.33 ml, 2.370 mmol) and 4-chlorobenzaldehyde (222 mg, 1.580 mmol) and the reaction mixture was refluxed for 5 h. The reaction mixture was quenched with water (50 ml) and was further refluxed for half hour after which the aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with water, brine, dried and concentrated under reduced pressure to yield 650 mg of the compound; Ή NMR (300 MHz, CDCI3) δ 0.88 (br s, 3H), 3.13 (br s, 2H), 6.98 (d, J =8.1 Hz, 2H), 7.08 (d, J= 8.7 Hz, 2H), 7.14- 7.22 (m, 4H), 7.35 (d, J= 8.1 Hz, IH), 7.57 (t, J= 8.7 Hz, IH), 7.76-7.88 (m, 3H), 8.18 (d, = 8:4 Hz, IH), 8.40 (d, J= 7.8 Hz, IH), 13.78 (br s, IH).
Intermediate 41
3-(4-ChloiOphenyl)-2-| 4-(quinolin-2-ylmethoxy)phenyl]prop-2-enal
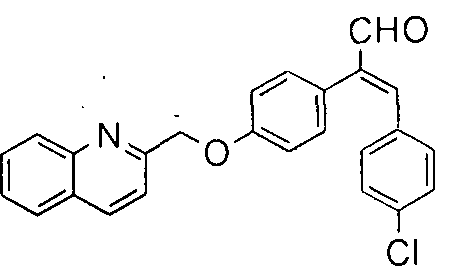
Step 1 : 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -ol :
To a well stirred solution of the ester compound of Intermediate 29 (3.3 g, 7.440 mmol) in THF (40 ml) was added DIBAL( 1 5.85 ml, 22.322 mmol) at -30 to -40 °C and stirred for 3 h at the same temperature. The reaction mixture was quenched with water (15 ml) and stirred for 20 mins after addition of ethyl acetate (200 ml). The precipitate obtained was filtered and the filtrate was dried and concentrated to yield 2.7 g of the product as off-white solid; Ή NMR (300 MHz. CDC13) δ 4.42 (s, 2H),' 5.38 (s, 2H), 6.60 (s, 1 H), 6.91 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 9.0 ! !z; 2H), 7.04-7.14 (m, 4H), 7.54-7.61 (m, 1 H), 7.66-7.74 (m, 2H), 7.84 (d, J = 7.8 Hz, I H), 8.09 (d. J = 8.4 Hz, I H), 8.22 (d, J = 8.7 Hz, 1 H), 12.72 (br s, 1 H).
Step 2: 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enal:
Portion wise addition of Mn02 (2.5 g, 28.737 mmol) to the well stirred solution of Step 1 intermediate (1 .1 5 g, 2.287 mmol) in THF (40 ml) was carried out at room temperature and the reaction mixture was further stirred for 4 h. The reaction mixture was diluted with ethyl acetate (250 ml), filtered, dried and concentrated to yield the crude product which was puri fied by silica gel column chromatography to yield 1 .3 g of the product as off-white solid; Ή NMR (300 M Hz, CDCI3) δ 5.42 (s, 2H), 7.05-7.1 6 (m, 3H), 7.18-7.25 (m, 4H), 7.27 (d, J - 7.8 Hz, 2H), 7.56 (d, J = 7.5 Hz, 1 H), 7.68-7.77 (m, 2H), 7.85 (d, J = 8.4 Hz, 1 H), 8.09 (d, J = 8.4 Hz, 1 H), 8.22 (d, J = 8. 1 Hz, ΓΗ), 9.73 (s, I H).
Intermediate 42
2-( {4-[3-Chloro- l -(4-chlorophenyl)prop- l -en-2-yl]phenoxy} methyI)quinoline
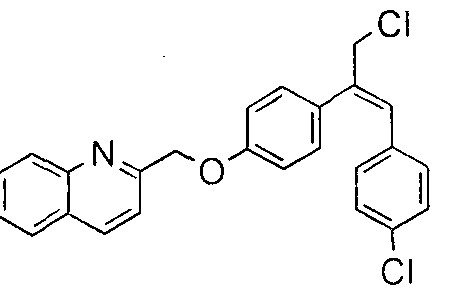
Step 1 : 3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]pi p-2-en- 1 -ol :
To well stirred solution of the ester of Intermediate 29 (4 g, 9.36 mmol) in THF (60 ml) was added 20 % Diisobutylaluminium hydride (23.54 ml, 28.10 mmol) at -30 to -40 °C and stirred for 3 h at the same temperature. The reaction mixture was quenched with water ( 1 ml) and stirred for 20 mins after addition of ethyl acetate (200 ml). The precipitate obtained was fi ltered and the fi lterate was dried and concentrated to yield 3.1 g of the product as off-white solid; Ή MR (300 M Hz, CDC13) δ 4.42 (s, 2H), 4.52 (br s, 1 H), 5.38 (s, 2H), 6.60 (s, 1 H), 6.91 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 9.0 Hz, 2H), 7.05-7.1 3 (m, 4H), 7.56 (t, J = 1.5 Hz, 1 H), 7.6 '-7.7 '4 (m, 21-1), 7.84 (d, J = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1 H), 8.22 (d, J = 8.7 Hz, 1 H).
Step_2: 2-({4-[3-Chloro-l -(4-chlorophenyl)prop- l -en-2-yl]phenoxy}methyl)quinoline:
To a well stirred solution of the Step 1 intermediate (580 mg, 1 .03 mmol) in DCM ( 1 0 ml) was added triethylamine (0.28 ml, 2.06 mmol) followed by methanesulfonylchloride (0. 1 2 ml, 1 .558 mmol) at 0 °C and was stirred overnight. The reaction mixture was quenched with water (20 ml), extracted with ethyl acetate (2 x 25 ml), washed with water (20 ml), brine (20 ml) and dried to yield 590 mg of the product; 1H NMR (300 MHz, CDC13) δ 4.40 (s, 2H), 5.44 (s, 2H), 6.69 (s, 1 .H), 6.88-6.93 (m, 2H), 7.04-7.08 (m, 3H), 7.10-7.1 7 (m, 2H), 7.29 (s, 1 H); 7.59 (t, ,7 = 7.2 Hz, 1 H), 7.72-7.81 (m, 2H), 7.87 (d, J = 8.4 Hz, 1 H), 8.14 (d, J - 8.4 Hz, 1 H), 8.27 (d, J = 8.4 Hz, 1 H). intermediates 43-45 were prepared as described in Intermediate 42 by the reduction of appropriate ester followed by chlorination. Their structure, names and Ή NMR data are given in the Table 2.
Table 2: Structure and characterization data for Intermediates 43 - 45
Molecular Structure and Chemical name and H N MR data (δ ppm, 300 MHz)
Intermediate No.
Molecular Structure and Chemical name and Ή NMR data (δ ppm, 300 MHz) Intermediate No.
2-( i4-|"3-Chloro-l-(4-fluorophenvl)prop-l -εη-2-γΓ|ρ1ιεηοχγ} methvDquinoline: (CDCh) δ 4.40 fs, 2H), 5.41 (s, 2H), 6.70 (s, 1H), 6.81 (t,J = 8.4 Hz, 2H), 6.94-7.03 (m, 4H), 7.16 (d: J = 8.7 Hz, 2H), 7.57 (t, J= 7.2 Hz, 1H), 7.70-7.87 (m, 2H), 7.86 (d, J-
F
7.8 Hz, 1H), 8.11 (d,J=8.1 Hz, 1H), 8.24 (d,J=8.4Hz, 1 H).
[Intermediate 43]
2-({4-|'3-Chloro-l-phenylprop-l -en-2-Yriphenoxy 1· methyl) quinoline: fCDC δ 4.42 fs.2H), 5.38 (s.2H .6.74 fs, 1 H , 7.00 (d,J = 8.1 Hz, 4H), 7.12 (br ss 3H), 7.18 (d..7= 8.4 Hz, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.68-7.76 (m, 2H), 7.84 (d, J= 7.8
[Intermediate 44] Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.21 (d, J= 8.1 Hz, 1H).
CI 2- {4- 3-Chloro-l -f4-chloi -3-fluoropheny])prop-l-en-2- vllphenoxvlmethvDquinoline: (CDCh) δ 4.38 is, 2H), 5.42 fs, 2H), 6.65 (s, 1H), 6.72 (d, J = 9.3 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 7.10-7.16 (m, 3H), 7.57 (t, J= 7.2 Hz, IH), 7.68-7.76 (m; CI
2H), 7.86 (d,J=7.8Hz, 1H), 8.11 (d,J = 9.0 Hz, 1H), 8.24 (d, ./
. [Intermediate 45]
= 8.4 Hz, 1H).
Intermediate 46
4-|'2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol- -yl)ethenyl]phenol

S ep 1 : 3-(4-Chlorophenyl)-2-(4-hydroxyphenyl)prop-2-enoic acid:
To the well stirred solution of ethyl-3-(4-chlorophenyl)-2-(4-hydroxyphenyl)prop-2-enoate (1 g, 3.30 mmol) in ethanol (20 ml) and THF (10 ml) was added aqueous solution of sodium hydroxide (661 mg, 16.52 mmol) and the reaction mixture was stirred at room temperature for 15 min. The reaction mixture was diluted with water (25 ml) and the pH was made acidic. The aqueous layer was then extracted with ethyl acetate (3 x 20 ml), washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 760 mg of the product as off-white solid;
Ή NMR (300 MHz, DMSO-i¾ δ 6.74 (d, J= 8.1 Hz, 2H), 6.94 (d, J= 8.4 Hz, 2H); 7.09 (d. J = 8.7 Hz, 2H), 7.28 (d, J= 8.4 Hz, 2H), 7.64 (s, 1H), 9.54 (br s, 1H), 12.63 (br s, 1H).
Step 2: 4-('l -(4-Chlorophenyl)-3-hydrazinyl-3-oxoprop-l-en-2-yl]phenyl ethyl carbonate:
To the well stirred solution of Step 1 intermediate (750 mg, 2.73 mmoi) in THF (30 ml) was added TEA (1.14 ml, 8.19 mmoi) and the reaction mixture was cooled to 0 °C after which ethyl chloroformate (0.78 ml, 8.19 mmoi) was added to it and the reaction was stirred at the same temperature for 1 hour. Hydrazine hydrate (0.5 ml) was added to the reaction mixture and the reaction mixture was further stirred for lh at 0 °C. The reaction mixture was diluted with water (25 ml) and extracted with ethyl acetate (3 x 20 ml), washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 710 mg of the product as off-white solid; Ή NMR (300 MHz, CDCb) 81.42 (t, J= 7.5 Hz, 3H), 4.35 (q, J= 6.9 Hz, 2H), 6.81 (br s, 1H); 6.92 (d5 J = 8.1 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 7.23-7.30 (m, 4H), 7.81 (s, 111), 9.82 (br s: 2H).
Step 3 : 4-|2-(4-Chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenyl ethyl carbonate:
To the well stirred solution of Step 2 intermediate (700 mg, 1.94 mmoi) in triethyl orlhoformale (15 ml) was added PTSA (74 mg, 0.388 mmoi) and the reaction mixture was heated at 90-100 °C for 2 h. The reaction mixture was diluted with water (25 ml) and extracted with ethyl acetate (3 x 20 ml), washed with water and brine, dried over anhydrous Na2SO^ and concentrated to yield 500 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-c¾ δ 1.30 (t, J = 6.9 Hz, 3H),
4.27 (q, .7= 7.5 ilz.2H), 7.14 (d, .7= 8.4 Hz, 2H), 7.30-7.40 (m, 6H), 7.74 (s, 1H), 7.81 (s, 1H),
9.28 (s, 2H).
Step 4: 4-J"2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenol:
To the well stirred solution of Step 3 intermediate (320 mg, 0.863 mmoi) in ethanol (10 ml) was added aqueous solution of sodium hydroxide (173 mg, 4.318 mmoi) and the reaction mixture was stirred at room temperature for 15 min. The reaction mixture was diluted with water (25 ml) and the pH was made slightly acidic. The aqueous layer was then extracted with ethyl acetate (3 x 20 ml), washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 190 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 6.79 (d, J = 7.8 Hz, 2H), 7.06(t,J=8.4Hz, 2H), 7.15 (d, J= 8.7 Hz, 4H), 7.60 (s, 1H), 8.42 (s, lH), 10.11 (br s, IH).
Intermediate 47
-(4-Chlorophenyl)-2-[4-(imidazo[ l -a]pyridin-2-ylmeth{ \y)phenyl]pi p-2-enehydi

Ethyl chloroformate (0.35 ml, 3.70 mmol) was added drop wise to a well stirred solution of Intermediate 1 ( 1 g, 2.472 mmol) and TEA (0.52 ml, 3.70 mmol) in dry tetrahydrofuran (30 ml) at 0°C under nitrogen atmosphere and continued stirring for 30 min at same temperature. Excess of hydrazine hydrate (2 ml) was then added to the reaction mixture and stirred for further 1 h at 0- 1 0 °C: The reaction mixture was quenched with water (50 ml) and extracted with ethyl acetate ( 100 ml x 2). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 980 mg of product as off-white solid; Ή N MR (300 MHz, CDC13) δ 4.72 (br s, 3H), 5.21 (s, 2H), 6.89 (t, J = 6.3 Hz, 1 H), 7.00-7.08 (m, 5H): 7. 1 8- 7.27 (m, 4H), 7.53 (d, J = 9.0 Hz, I H), 7.68 (s: 1 H), 8.54 (d, J = 6.3 Hz, 1 H). 8.87 (s. 1 H).
Intemediates 48 to 82 were prepared using appropriate propenoic acid and hydrazine hydrate as described in Intermediate 47. Their structure, names and H NMR data are given in the Table 3. fable 3 : Structure and characterization data for Intermediates 48 - 82

CL, NHNH2 3-(4-Chlorophenvl)-2-[4-('pYridin-2-YlmethoxY)phenY ip!Op-2- enehvdrazide: (DMSO-<¾) δ 4.52 (br s, 2H), 5.18 (s, 2H), 6.74 (s, 1H), 6.98-7.06 (m, 6H), 7.24 (d, J= 8.1 Hz, 2H), 7.31-7.39 (m, IH), 7.53 (d, J = 7.8 Hz, 1H), 7.59 (s, IH), 7.82 (i: J =-■ 7.2 CI
Hz, 1H), 8.85 (br s, I H).
[Intermediate 50]
C NHNH2 3-(4-ChlorophenYl)-2-[4-(thieno[3,2-0]pYridin-5-Ylmethoxy)
Dhenvllprop-2-enehvdrazide: (DMSO-d* δ 4.50 (br s.2H), 5.32 (s, 2H), 6.52 (s, IH), 7.06 (br s, 6H), 7.25 (d, J = 8.1 Hz, 2H), 7.51-7.60 (m, 2H), 7.65 (s, IH), 8.19 (d, J = 5.7 Hz, IH), 8.54 CI
(d,J=8.4Hz, IH).
[Intermediate 51]
C NHNH2 3-(4-ChloiOphenYl)-2-[4-(furo[3,2-0]pYridin-5-ylmethoxv)
phenvllprop-2-enehYdrazide: (DMSO-Λ) δ 3.98-4.09 (m, 2H). 5.27 (s, 2H), 7.00-7.06 (m, 6H), 7.15-7.23 (m, 4H), 7.51 (d, J = 8.4 Hz, IH), 8.09 (d, J= 8.7 Hz, IH), 8.36 (s, IH), 8.90 (br s. CI IH).
[Intermediate 52]
C NHNH2 3-(4-Chlorophenyl)-2-{4-r(l-methYl-lH-PYn lo[3,2-/y|pvridin -
5-y] methoxylphenyl}prop-2-enehydrazide: (DMSO-fl ) δ 3.83
(s, 3H), 4.54 (br s, 2H), 5.23 (s, 2H), 6.54 (s, IH), 6.99-7.05 (m: 6H), 7.18 (s, IH), 7.23 (d, J= 8.4 Hz, 21-1), 7.32 (d, .7= 9.0 Hz,- H3C CI
IH), 7.65 (br s, IH), 7.91 (d, .7= 8.4 Hz, IH), 8.87 (s, IH).
[Intermediate 53]
C NHNH2 3-(4-Chlorophenyl)-2-[4-(pYrazolo[l,5-i7]pyrimidin-5-vl
methoxy)phenyl]prop-2-enehydrazide: (DMSO-aM δ 4.01-4.06
(m, 2H), 5.27 (s, 2H), 6.71 (s, IH), 6.99-7.15 (m, 6H), 7.18-7.27 (m, 3H), 8.24 (s, IH), 8.92 (s, IH), 9.15 (d,J= 7.2 Hz, 2H).
CI
[Intermediate 54]
0¾_NHNH2 2-|4-(Ouinolin-2-YlmethoxY)phenYl]-3-(thiophen-2-Yl)prop-2- enehydrazide: (CDCh) δ 3.97 (br s.2H .5.50 (s.2H), 6.90-6.94 (m, IH), 7.18-7.23 (m, 7H), 7.60 (t, J = 6.9 Hz, IH), 7.76-7.81 (m, 2H), 7.88 (d, J= 7.8 Hz, IH), 8.06 (s, IH), 8.16 (d, J= 8.4
[Intermediate 55] Hz, IH), 8.29 (d, J =8.4 Hz, IH).

[Intermediate 61] 7.99-8.05 (m, 2H), 8.44 (d,J= 8.4 Hz, lH), 8.84 (s, 1H).'


 Molecular Structure and Chemical name and Ή NMR data (δ ppm, 300 MHz) Intermediate No.
Molecular Structure and Chemical name and Ή NMR data (δ ppm, 300 MHz) Intermediate No.
[Intermediate 79]
0. NHNH2 3-(3,4-DifluorophenYl)-2-[4-(quinolin-2-vlmethoxY)phenyl] proD-2-enehvdrazide: (CDCM δ 4.01 (br s, 2H), 5.45 (s, 2H), 6.66-6.75 (m, 2H), 6.83 (br s, 1H), 6.95 (q,J= 9.0 Hz, lH), 7.12 (br s, 4H), 7.58 (t, J= 7.2 Hz, 1 H), 7.66-7.73 (m, 3H), 7.86 (d, J F = 8.4Hz, 1H), 8.10 (d, .7=8.4 Hz, 1H), 8.24 (d, ./= 8.1 Hz, 1H).
[Intermediate 80]
. CL NHNH2 3-(4-ChlorophenvlV2-[3-(quinolin-2-vlmethoxy)phenyl] prop-2- enehydrazide
(DMSO-i6) δ 4.59 (br s, 2H), 5.44 (s, 2H), 6.51 (s, 1 H), 6.76 (d, J= 7.8 Hz, 1H), 6.89 (s, 1H), 7.04 (d,J= 8.4 Hz, 2H).7.10 (d../ CI = 8.1 Hz, 1H), 7.18 (d, J = 8.7 Hz, 2H), 7.33 (t, J = 7.8 Hz, 1 H),
[Intermediate 81] 7.70-7.77 (m, 3H), 7.84-7.90 (m, 1H), 8.09 (br s, 2H), 8.59 (br s,
1H).
3-(4-Chlorophenyn-2-{4-[2-(quinolin-2-Yl)ethyr|phenyP, prop-
2-enehvdrazide: (CDC δ 3.20-3.26 fm, 2H), 3.32-3.36 (m. 2H), 3.96 (br s, 2H), 6.73 (s, 1H), 6.88 (d, J= 8.4 Hz, 2H), 7.04- 7.11 (m, 4H), 7.27-7.32 (m, 3H), 7.53 (t, J= 7.8 Hz, IH), 7.70-
CI
7.83 (m, 3H), 8.10 (d, J = 8.4 Hz, 2H).
(Intermediate 82]
Intermediate 83
2-(4-Chlorophenyl)-3-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enoic acid
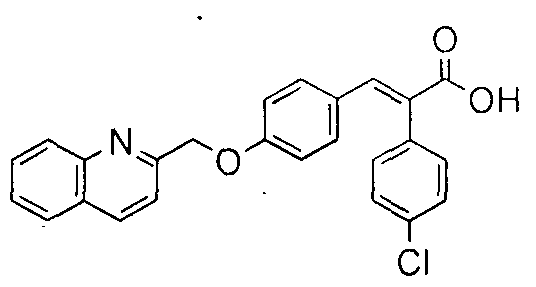
The title compound was prepared from 4-hydroxybenzaldehyde, 4-chlorophenyl acetic acid and 2-(chloromethyl) quinoline in a 4 step procedure as described in Intermediate 1; Ή NMR (300 MHz, D SO-ifc) δ 5.31 (s, 2H), 6.91 (d, J= 8.4 Hz, 2H), 6.98 (d, J= 8.7 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 7.39 (d, ./ = 8.4 Hz, 2H), 7.62 (d, J = 8.7 Hz, 3H), 7.77 (d, J - 7.8 Hz. 1H); 7.97- 8.02 (m, 2H), 8.39 (d, J= 8.4 Hz, 1H), 12.72 (br s, 1H).
Intermediate 84
-Chloropheny])-3-[4-(quinolin-2-ylmethoxy)pheny]]prop-2-enehydrazid

The title compound was prepared using Intermediate 83 and hydrazine hydrate as described in Intermediate 47; Ή NMR (300 MHz DMSO-<¾) δ 4.59 (s, 2H), 5.31 (s, 2H), 6.90-6.97 (m, 4H), 7.16 (d, J= 7.8 Hz, 2H), 7.29 (s, 1H), 7.44 (d, J= 8.4. Hz, 2H), 7.62 (d, J= 8.4 Hz, 2H), 7.77 (t, J =1.2 Hz, 1H), 7.97-8.02 (br s, 2H), 8.40 (d,J= 8.4 Hz, 1H), 8.87 (s, 1H).
Examples
Example 1
3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-enamide
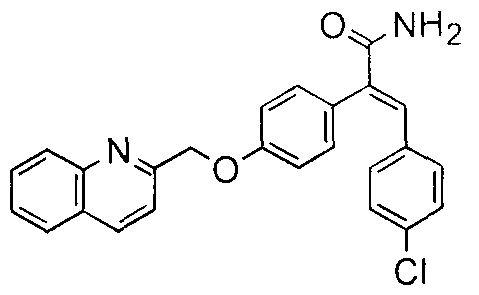
The title compound was prepared by reaction of aqueous ammonia (5 ml) with the mixed anhydride which was prepared from Intermediate 29 (1 g, 2.406 mmol) and ethyl chloroformate (0.39 ml.3.609 mmol) in presence of TEA (0.5 ml, 3.609 mmol) to yield 50 mg of the product as off-white solid as described in Intermediate 47; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2! I).5.50 (br s, 1H), 5.61 (br s, 1H), 6.93 (d, J= 8.7 Hz, 2H), 7.10 (d, .7= 9.0 Hz, 4H), 7.18 (d, .7= 9.011/. 2H), 7.58 (t, J= 7.2 Hz, 1H), 7.71-7.78 (m, 3H), 7.86 (d, .7 = 8.4 Hz, 1H), 8.10 (d, ./ = 9.0 Hz, IH), 8.24 (d, J= 8.1 Hz, 1H); ESI-MS (m/z) 415 (M)+.
Example 2
3-(4-Chlorophenyl)-2-[4-(imidazo[l,2-a]pyridin-2-ylmethoxy)phenyl]-N-methyl-prop-2- enamide

To a well stirred solution of Intermediate 1 (150 mg, 0.37 mmol) in dichloromelhane (10 ml) were added N-[3-(dimethylamino)propyl]-N'-ethylcarbodiimide hydrochloride (EDC1.HC1; 107 mg. 0.55 mmol), lH-benzotriazol-l-ol (HOBT; 75 mg, 0.55 mmol), TEA (0.11 ml, 1.1 mmol) and methylamine hydrochloride (48 mg, 0.74 mmol) and the reaction "mixture was stirred at room temperature for 16 h. The reaction mixture was quenched with water (5 ml) and extracted with ethyl acetate (25 ml x 2). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 110 mg of product as off-white solid; Ή NMR (300 MHz, DMSO-<¾) δ 2.65 (d, J= 4.2 Hz, 3H), 5.22 (s, 2H), 6.90 (t, J =6.3 Hz, 1H), 7.00 (d, J = 9.0 Hz, 2H), 7.05-7.12 (m, 4H), 7.24 (d, J= 9.0 Hz, 2H), 7.28-7.34 (m, 3H), 7.54 (d, J = 9.3 Hz, 1H), 7.56 (s, 1H), 8.55 (d, J= 6.6 Hz, 1H); APCI-MS (m/z) 418 (M+H)+.
Example 3
2-f4-(l .3-Benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)-N-methylprop-2-enamide

The title compound was prepared by coupling Intermediate 2 (150 mg, 0.372 mmol) with methylamine hydrochloride (30 mg, 0.446 mmol) as described in Example 2 to yield 95 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.84 (d, J= 4.5 Hz, 3H), 5.54 (br s, 3H), 6.89 (d, J = 8.4 Hz, 2H), 7.06-7.18 (m, 6H), 7.44 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.2 Hz, 1H), 7.77 (s, 1H), 7.94 (d,J = 7.8 Hz, 1H), 8.05 (d,J = 8.1 Hz, 1H); ESI-MS (m/z) 435 (M+H)+.
Example 4
A/-Methyl-2-{4-[(l -methyl- lH-benzimidazol-2-yl)methoxy]phenyl}-3-(pyridin-3-yl)prop-2- enamide

The title compound was prepared by coupling Intermediate 4 (1 00 mg, 0.259 mmol) with methylamine hydrochloride (35 mg, 0.519 mmol) as described in Example 2 to yield 69 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.85 (d, J = 4.8 Hz, 3H), 3.95 (s, 3H), 5.44 (s, 2H), 5.56 (br s, 1 H), 7.01 -7.06 (m, 1 H), 7.16 (br s, 5H), 7.3 1 -7.40 (m, 3H): 7.79 (br s, 2H), 8.29 (s, 1 H): 8.36 (br s, l H); APCl-MS (m/z) 399 (M+H)+.
Example 5
N-Methyl-2- {4-[ ( 1 -methyl- 1 H-benzimidazol-2-yl)methoxy]phenyl } -3 -(pyridin-4-yl)prop-2- enamide

The title compound was prepared by coupling Intermediate 5 (1 50 mg, 0.389 mmol) with methylamine hydrochloride (52 mg, 0.779 mmol) as described in Example 2 to yield 120 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-i¾ δ 2.65 (d, J = 4.2 Hz, 31-1), 3.88 (s, 3H), 5.45 (s, 2H), 6.91 (d, J = 4.8 Hz, 2H), 7.1 1 (d, J = 8.1 Hz, 2H), 7.1 7 (d, J = .8.4 Hz, H i. 7.23-7.33 (m, 3H), 7.51 (br s, 1H), 7.60 (d, J = 7.8 Hz, 1 H), 7.66 (d, ,7 = 7.8 Hz, l H), 8.35 (d, J = 7.8 Hz, 2H); APCl-MS (m/z) 399 (M+H)+.
Example 6
3-(4-Chlorophenyl)-N-methyl-2-[4-(pyridin-2-ylmethoxy)phenyr|prop-2-enamide

Example 7
A'-Methyl-2-|"4-(quinolin-2-ylmethoxy)phenyl]-3-(thiophen-2-yl)prop-2-enamide

The title compound was prepared by coupling Intermediate 20 ( 1 50 mg, 0.387 mmol) wi th methylamine, hydrochloride (3 1 mg, 0.464 mmol) as described in Example 2 to yield 85 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.83 (d, J = 4.8 Hz, 3H), 5.46 (s, 2H); 5.86 (s, 1 H), 6.91 (t, J = 4.5 Hz, 1 H), 7.12-7.19 (m, 6H), 7.58 (t, .7 = 7.5 Hz, 1 H), 7.74-7.88 (m; 2H), 7.87 (d, J = 7.8 Hz, 1 H), 8.06 (s, 1H), 8.1 1 (d, J = 8.4 Hz, l H), 8.25 (d, J = 8.4 Hz, 1 H); APCI-MS (m/z) 401 (M+H)+.
Example 8
/V-Methyl-3-phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]pi p-2-enamide
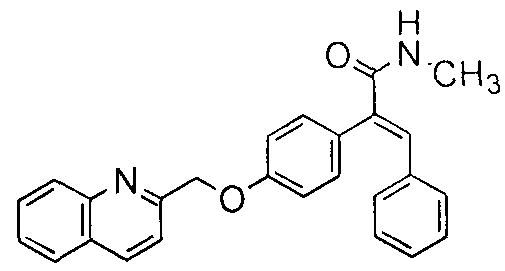
The title compound was prepared by coupling Intermediate 22 (200 mg, 0.524 mmol) with methylamine hydrochloride (53 mg, 0.787 mmol) as described in Example 2 to yield 70 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.84 (d, .7 = 5.1 Hz, 3H), 5.43 (s, 2H), 5.54 (br s, l H), 6.99 (d, J = 6.3 Hz, 2H), 7. 12-7.24 (m, 7H), 7.58 (t, J = 7.5 Hz, 1 H), 7.73- 7.80 (m, 2H), 7.82-7.89 (m, 2H), 8. 1 1 (d, J = 8.4 Hz, l H), 8.25 (d, J - 8.4 Hz, l H); ESl -MS (m/z) 395 (M+H)+.
Example 9
3-(4-Chlorophenyl)-A-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enanu'de

The title compound was prepared by coupling Intermediate 29 (100 mg, 0.24 mmol) with methylamine hydrochloride (32 mg, 0.48 mmol) as described in Example 2 to yield 60 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.85 (br s, 3H), 5.45 (s, 2H), 5.54 (br s, 1H), 6,90 (d, J= 8.1 Hz, 2H), 7.12 (br s, 6H), 7.59 (t, ,1=12 Hz, 1H), 1.12-1.1% (m, 3H), 7.88 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H); APCI-MS (m/z) 429 (M+H)+.
Example 10
3-(4-FluoiOphenyl)-N-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 30 (150 mg, 0.37 mmol) with methyl amine hydrochloride (38 mg, 0.56 mmol) as described in Example 2 to yield 100 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.84.(d, J= 4.5 Hz, 3H), 5.43 (s, 2H), 5.55 (br s, 1H), 6.81 (t,J= 8.7 Hz, 2H), 6.96 (t,J= 8.1 Hz, 2H), 7.09-7.17 (m,.4H), 7.58 (t, J = 7.5 Hz, lH), 7.69-7.80 (m, 3H), 7.86 (d, J= 8.1 Hz, 1H), 8.10 (d, .7- 8.4 Hz, 1H), 8.24 (d, J = 9.0 Hz, l H); APCI-MS (m/z) 413 (M+H)+.
Example 11
yV-Methyl-3-(pyridin-3-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 13 (100 mg, 0.261 mmol) with methylamine hydrochloride (35 mg, 0.523 mmol) as described in Example 2. to yield 60 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.86 (s, 3H), 5.42 (s, 2H), 5.58 (br s, 1H), 7.06-7.13 (m, 6H), 7.60 (d, .7 = 6.9 Hz, 1H), 7.59-7.71 (m, 1H); 7.71-7.80 (m, 2H).7.87 (d, J= 7.2 Hz, 1H), 8.11 (d, J= 8.4 Hz, 1H), 8.26 (d, J= 8.4 Hz, 1H), 8.30-8.38 (m, 2H): ESI- MS im/z) 396 (M+H)+.
Example 12
A-Methyl-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide
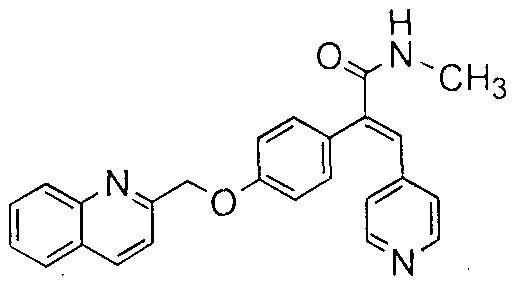
The title compound was prepared by coupling Intermediate 14 (150 mg, 0.392 mmol) with methylamine hydrochloride (26 mg, 0.74 mmol) as described in Example 2 to yield 89 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-<¾ δ 2.66 (d, J = 5.1 Hz, 3H), 5.40 (s; 2H), 6.94 (d, .7-5.1 Hz, 2H), 7.11 (s, 3H), 7.28 (s, 1H), 7.53 (br s, 2H), 7.63 (ts .7 = 6.9 Hz. 111). 7.71 (d, .7= 8.1 Hz, 1 H), 7.80 (t, J = 7.5 Hz, 1 H), 8.02 (d, J = 8.4 Hz, 2H), 8.37 (d, J = 4.8 Hz, 2H), 8.45 (d, .7= 8.7Hz, 1H); APCI-MS (m/z) 396 (.VHIlV.
Example 13
3-(4-Methoxyphenyl)-N-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

Example 14
Benzoxazol-3-yl)ethoxy]phenyl}-3-(4-chl0rophenyl)-N-methylprop-2-enamid

The title compound was prepared by coupling Intermediate 3 (150 mg, 0.357 mmol) with mcthylamine hydrochloride (36 mg, 0.535 mmol) as described in Example 2 to yield 71 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.83 (d, J = 4.8 Hz, 3H), 3.53 (t, = 6.3 Hz, 2H), 4.48 (t, J = 6.3 Hz, 2H), 5.50 (br s, 1H), 6.89-6.99 (m, 4H), 7.10 (d, J = 8.4 Hz, 4H), 7.36 (br s 1H), 7.60 (br s, 2H), 7.76 (s, 1H), 7.82 (d, J = 7.8 Hz, 1H); ESI-MS (m/z) 433 (M)+.
Example 15
A/-Ethyl-3-(pyridin-4-yl)-2-[4-(quinolin-2- lmehox hen l rop-2-enamide

The title compound was prepared by coupling Intermediate 14 (150 mg, 0.392 mmol) with ethylamine hydrochloride (63 mg, 0.784 mmol) as described in Example.2 to yield 135 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI3) δ 1.09 (d, J= 6.9 Hz, 3H), 3.29-3.40 (m, 2H), 5.43 (s, 2H), 5.60 (br s, 1H), 6.82 (d, J= 4.8 Hz, 2H), 7.12 (s, 4H), 7.58 (t, J= 7.2 Hz, 1H),
7.70-7.79 (m, 3H), 7.87 (d, J = 7.8 Hz, 1H), 8.09 (d, J - 8.1 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.37 (d, .1= 5.1 Hz, 2H); APCI-MS (m/z) 410 (M+H)+.
Example 16
3-(4-Chlorophenyl)-2-[4-(pyridin-2-ylmethoxy)phenyl]-N-(2,2,2-trifluoroethyl)piOp-2-enamide

The title compound was prepared by coupling Intermediate 12 (200 mg, 0.547 mmol) and 2,2,2- trifluoroethylamine hydrochloride (148 mg, 1.094 mmo!) as described in Example 2 to yield 109 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.97 (t, .7= 7.8 Hz, 2H), 5.25 (s, 2H).5.78 (br s, 1H), 6.93 (d, J= 7.8 Hz, 2H), 7.07-7.18 (m, 6H), 7.27 (br s, 1 H), 7.56 (d, J - 7.8 Hz, 1H), 7.73-7.82 (m, 2H), 8.63 (d, J= 3.6 Hz, 1H); ESI (m/z) 447 (M)+.

The title compound was prepared by coupling Intermediate 4 (100 mg, 0.259 mmol) with 2,2,2- trifluoroethylamine hydrochloride (70 mg, 0.518 mmol) as described in Example 2 to yield 75 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.89-4.00 (m, 5H), 5.48 (s, 211), 5.79 (br s, lH), 7.02-7.08 (m, 1H), 7.19 (br s, 5H), 7.34-7.44 (m, 3H), 7.78-7.85 (m, 2H), 8.31 (s, 1H), 8.39 (d, J= 3.9 Hz, 1H); APCI-MS {m/z) 467 (M+H)+.
Example 18
2-{4-[(l -Methyl- lH-benzimidazol-2-yl)methoxy]phenyl}-3-(pyridin-4-yl)-/V-(2,2, 2- trifluoroelhyl)prop-2-enarnide

The titl'e compound was prepared by coupling Intermediate 5 (I 50 mg, 0.389 mmol) with 2,2,2- trifluoroethylamine hydrochloride (105 mg, 0.778 mmol) as described in Example 2 to yield 110 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.95 (br s, 5H), 5.47 (s, 2H), 5.84 (br s, 1H), 6.83 (d,J= 5.4 Hz, 2H), 7.12-7.22 (m, 4H), 7.32-7.40 (m, 314), 7.75 (s: 111).7.80 (d, J = 7.8 Hz, 1 H), 8.38 (d, J = 5.4 Hz, 2H); ESI-MS (m/z) 467 (Μ·1Ι)\
Example 19
2-|4-(lmidazo[l,2-o]pyridin-2-y]methoxy)pheny]]-3-(pyridin-4-yl)-N-(2,2,2-trinuoroethy prop- 2-enamide

The title compound was prepared by coupling Intermediate 11 (150 mg, 0.40 mmol) with 2,2,2- trifluoroethylamine hydrochloride (108 mg, 0.80 mmol) as described in Example 2 to yield 80 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.94-4.04 (m, 2H), 5.33 (s. 211), 5.90 (br s, 1H), 6.86 (d, .7= 5.1 Hz, 3H), 7.15 (s, 4H), 7.28 (br s, 1H), 7.65 (d, 7= 8.7 Hz, ll-l), 7.70 (s, 1H), 7.76 (s, 1H), 8.14 (d, J = 6.9 Hz, 1H); 8.41 (d, J= 5.4 Hz, 2H); APC1-MS (m/z) 453 (M+H)+.
Example 20
3-(Pyridin-3-yl)-2-[4-(quinolin-2-ylmethoxy)pheny]]-A/-(2,2,2-trifluoroethyl)prop-2-enami

The title compound was prepared by coupling Intermediate 13 (100 mg, 0.261 mmol) vvith 2,2,2- trifuoroethylamine hydrochloride (70 mg, 0.522 mmol) as described in Example 2 to yield 75 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.92-4.03 (m, 2H), 5.45 (s, 2H), 5.81'(brs, 1H), 7.13-7.23 (m, 6H), 7.60 (t,J=7.5Hz, 1H), 7.76- 7.81 (m, 1H): 7.83-7.9 (m; 3H)S 8.13 (d, J= 7.8 Hz, 1H), 8.26-8.34 (m, 1H), 8.42 (br s, 2H); APC1-MS (m/z) 464 (M -!iV.
Example 21
3-(Pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)ph

The title compound was prepared by coupling Intermediate 14 (100 mg, 0.261 mmol) with 2,2,2- trifluoroethylamine hydrochloride (70 mg, 0.523 mmol) as described in Example 2 to yield 83 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.94 - 4.11 (m, 2H), 5.43 (s, 2H), 5.86 (br s, 1H), 6.84 (d, J= 4.5 Hz, 2H), 7.14 (br s, 4H), 7.58 (t, J =1.5 Hz, 1H), 7.68-7.76 (m, 3H), 7.86 (d, .7 = 7.8 Hz, 1H), 8.09 (d, J = 8.4 Hz, 1 H), 8.25 (d, .7=8.1 Hz, 1 H), 8.39 (d, .7 = 4.5 Hz, 2H); APCI-MS (m/z) 464 (M+H)+.
Example 22
Benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)-N-(2-cyanoethyl)prop-2- enamide

Example 23
3-(4-ChloiOphenyl)-N-(2-cyanoethyl)-2-[4-(imidazo[l,2-a]pyridin-2-ylmethoxy) phenyl]prop-2- enamide

The title compound was prepared by coupling Intermediate 1 (150 mg, 0.37 mmol) with 3- aminopropionitrile fumarate (190 mg, 0.74 mmol) as described in Example 2 to yield 90 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-c¾ δ 2.69 (t, J= 6.3 Hz, 2H), 3.34- 3.39 (m, 2H), 4.20 (br s, 1H), 5.22 (s, 2H), 6.92 (t, J= 6.3 Hz, 1H), 7.02 (d, J= 7.8 Hz, 2H), 7.10 (br s, 4H), 7.25 (d, J= 8.1 Hz, 2H), 7.38 (s, 1H), 7.54 (d, J = 9.3 Hz, 1H), 7.73 (br s, 1H), 8.03 (s, ]H), 8.55 (d, J= 6.3 Hz, 1H); APCI-MS (m/z) 457 (M+H)+.
Example 24
3 -(4-Chlorophenyl)-A'-(2-cyanoethyl)-2-{4-[(l -methyl- lH-indazol-3-yl)methoxy]phenyl} prop- 2-enamide

The title compound was prepared by coupling Intermediate 15 (150 mg, 0.358 mmol) with 3- aminopropionitrile fumarate (110 mg, 0.429 mmol) as described in Example 2 to yield 80 mg of
the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.67 (t, J = 6.0 Hz, 2H), 3.52 (br s; 2H),4.09 (s, 3H), 5.48 (s, 2H), 6.00 (br s, 1H), 6.90 (d,J = 8.1 Hz, 2H), 7.07-7.15 (m, 3H), 7.17- 7.23 (m, 4H), 7.43 (br s, 2H), 775 (s, 1H), 7.87 (d, J= 8.1 Hz, 1H); APC1-MS (m/z) All (M+H)+.
Example 25
3-(4-Chlorophenyl)-N-(2-cyanoethyl -2-[4-(pyridin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 12 (200 mg, 0.547 mmol) with 3- aminopropionitrile fumarate (210 mg, 0.820 mmol) as described in Example 2 to yield 119 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (d, J =5.1 Hz, 2H), 3.54 (q, J = 6.3 Hz, 2H), 5.25 (s, 2H), 5.98 (br s, 1H), 6.93 (d, J= 8.7 Hz, 2H), 7.07-7.19 (m, 6H), 7.29 (br s, 1H), 7.57 (d, J= 7.8 Hz, 1H), 7.74-7.81 (m, 2H), 8.63 (d, J = 4.5 Hz, IH); ESI (m/z) 418 ( I ) .
Example 26
3-(4-Chlorophenyl)-N-(2-cyanoethyl)-2-[4-(thieno[3,2-0]pyridin-5-ylmethoxy)phenyl]pi p-2- enamide

The title compound was prepared by coupling Intermediate 16 (200 mg, 0.474 mmol) with 3- aminopropionitrile fumarate (243 mg, 0.948 mmol) as described in Example 2 to yield 58 mg of product as off-white solid; Ή NMR (30.0 MHz, CDC13) δ 2.68 (t, J= 57 Hz, 2H), 3.53 (q, J = 6.3 Hz, 2H), 5.37 (s, 2H), 5.98 (br s, 1H), 6.92 (d, J= 8.4 Hz, 2H), 7.10-7.18 (m, 6H), 7.54-7.59 (m, 2H), 776 (s, 1H), 7.82 (d, J = 5.4 Hz, 1H), 8.27 (d, J = 8.1 Hz, 1H); APCI-MS (m/z) 474 (M+H)+.
Example 27
N-(2-Cyanoethyl)-2-[4-(quino]in-2-ylmethoxy)phenyl]-3-(thiophen-2-yl)prop-2-enamide

The title compound was prepared by coupling Intermediate 20 ( 1 50 mg, 0.387 mmol) with 3- aminopropionitrile fumarate (1 1 9 mg, 0.464 mmol) as described in Example 2 to yield 90 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.66 (t, .7 = 5.7 Hz, 2H), 3.52 (q, .7 = 6.3 Hz, 2H), 5.47 (s, 2H), 5.87 (br s, 1 H), 6.92 (t, J = 4.5 Hz, 1 H), 7.14-7.21 (m, 6H), 7.58 (t, .7 = 7.2 Hz, 1 H), 7.74-7.79 (m, 2H), 7.87 (d, J = 7.8 Hz, 1 H), 8.06 (s, 1 H), 8. 1 1 (d, J = 9.0 Hz, 1 IT), 8.26 (d, J = 8.1 Hz, 1 H); APCI-MS (m/z) 440 (M+H)+.
Example 28
/V-(2-Cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-(thiophen-3-yl)prop-2-enamide

The title compound was prepared by coupling Intermediate 21 ( 150 mg, 0.387 mmol) with 3 - aminopropionitrile fumarate ( 1 19 mg, 0.464 mmol) as described in Example 2 to give 1 00 mg o f the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.66 (t, J = 6.3 Hz, 2H), 3.49-3.54 (m, 2IT), 5.45 (s, 2H), 5.92 (br s, 1 H), 6.43 (br s, 1 IT), 7.05 (s, 2H), 7.1 5-7.21 (m, 4H), 7.58 (t, .7 = 7.5 Hz, 1 H), 7.72-7.79 (m, 2H), 7.85 (br s, 2H), 8.10 (d, .7 = 8.4 Hz, l H), 8.25 (d, .7 = 8.7 Hz, 1 IT); APCI-MS (m/z) 440 (M)+.
Example 29
A/-(2-Cyanoethyl)-3-phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 22 (200 mg, 0.524 mmol) with 3- aminopropionitrile fumarate (201 mg, 0.728 mmol) as described in Example 2 to yield 50 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J = 6.3 Hz, 2H), 3.53 (d, J = 6.6 Hz, 2H), 5.43 (s, 2H), 5.97 (br s, 1H), 7.05 (d, .7= 6.9 Hz, 2H), 7.14-7.26 (m, 7H), 7.58 (t, J= 7.2 Hz, 1H), 7.71-7.72 (m, 4H), 8.10 (d, J= 8.4 Hz, 1H), 8.25 (d, J= 8.4 Hz, 1H); ES3-MS

Example 30
3-(3-Chlorophenyl)-N-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl ]prop-2-enamide

The title compound was prepared by coupling Intermediate 27 (150 mg, 0.360 mmol) with 3- aminopropionitrile fumarate (130 mg, 0.540 mmol) as described in Example 2 to give 91 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J= 6.3 Hz, 2H), 3.53 (q, - 6.3 Hz, 2H), 5.44 (s, 2H), 5.99 (br s, IH), 6.86 (d, J= 7.8 Hz, 1H), 6.94 (s, IH), 7.04-7.11 (m, 3H), 7.12-7.18 (m, 3H), 7.57 (t, J= 7.8 Hz, 1H), 7.68-7.78 (m, 3H), 7.85 (d, J = 8.1 Hz, IH), 8.09 (d, = 8.4 Hz, IH), 8.23 (d, J= 9.0 Hz, IH); ESI-MS (m/z) 468 (M)+.
Example 31
3-(4-ChloiOphenyl)-7V-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide
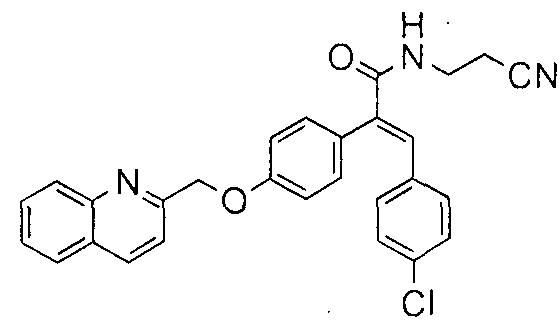
Example 32
/V-(2-Cyanoethyl)-3-(4-fluoi phenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide
The title compound was prepared by coupling Intermediate 30 (150 mg, 0.37 mmol) with 3- aminopropionitrile fumarate (144 mg, 0.56 mmol) as described in Example 2 to yield 103 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.67 (t..7= 6.3 Hz, 2H), 3.53 (q, J - 6.0 Hz, 2H), 5.43 (s, 2H), 5.95 (br s, 1H), 6.82 (t, J= 8.4 Hz, 2H), 7.13-7.20 (m, 6H), 7.58 (t, J = 7.5 Hz, lH), 7.69-7.79 (m, 3H), 7.86 (d, J= 8.4 Hz, 1H), 8.10 (d, J= 8.4 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1 H); APCI-MS (m/z) 452 (M+H)+.
Example 33
N-(2-Cyanoethyl)-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 14 (130 mg, 0.340 mmol) with 3- aminopropionitrile fumarate (174 mg, 0.680 mmol) as described in Example 2 to yield 112 mg of the product as off-white solid; Ή NMR (300 MHz, CDC! ) δ 2.69 (t, J = 6.6 Hz, 2H), 3.49-3.59 (m, 2H), 5.42 (s, 2H), 6.07 (br s, 1H), 6.83 (d, J = 4.5 Hz, 2H), 7.14 (br s, 4H), 7.58 (t, J= 7.5
Hz, 1H), 7.69-7.79 (m, 3H), 7.86 (d, .7- 8.1 Hz, 1H), 8.09 (d, J= 8.4 Hz, 1H), 8.25 (d, .7= 8.4 Hz, 1 H), 8.38 (br s, 2H); APCl-MS (m/z) 435 (M+H)+.
Example 34
7V-(2-Cyanoethyl)-3-(l-oxidopyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 39 (150 mg, 0.376 mmol) with 3- aminopropionitrile fumarate (116 mg, 0.565 mmol) as described in Example 2 to give 38 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J= 6.9 Hz, 2H), 3.55 (q, J = 6.0 Hz, 2H), 5.62 (s, 2H), 6.06 (br s, 1H), 6.84 (d, .7 = 5.4 Hz, 2H), 7.20 (s, 2H), 7.67 (s, 2H), 7.68-7.74 (m, 2H), 7.85-7.97 (m, 5H), 8.37 (d, J= 7.8 Hz, 1H), 8.46 (d, J= 8.4 Hz, 1H); ESi-MS (m/z) 451 (M+H)+.
Example 35
/V-(2-Cyanoethyl)-3-(4-methoxyphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 10 (180 mg, 0.437 mmol) with 3- aminopropionitrile fumarate (168 mg, 0.656 mmol) as described in Example 2 to yield 120 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.66 (t, J= 6.3 Hz, 2H), 3.52 (q, J = 5.7 Hz, 2H), 3.74 (s, 3H), 5.43 (s, 2H), 5.91 (br s, 1H), 6.67 (d, J= 9.0 Hz, 2H), 6.94 (d, J = 8.1 Hz, 2H), 7.12-7.21 (m, 4H), 7.60 (t, J= 8.1 Hz, 1H), 7.73-7.80 (m, 3H), 7.86 (d, J = 8.4 Hz, 1 H), 8.10 (d, J= 8.4 Hz, 1H), 8.25 (d, J= 8.1 Hz, lH); ESI (m/z) 464 (M+H)+.
Example 36
A/-(2-Cyanoethy])-3-[4-(difluoromethoxy)phenyl]-2-[4-(quinolin-2-ylmethoxy)phenyl] prop-2- enamide
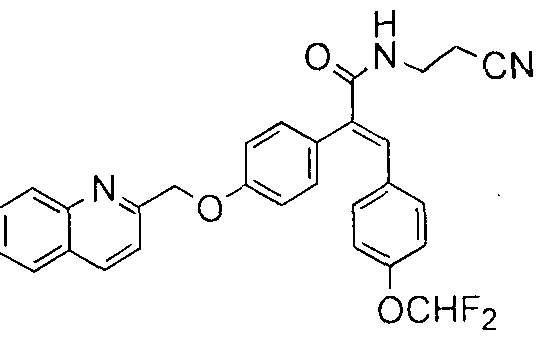
The title compound was prepared by coupling Intermediate 31 (150 mg.0.335 mmol) with 3- aminopropionitrile fumarate (112 mg, 0.435 mmol) as described in Example 2 to give 93 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J = 6.3 Hz, 2H), 3.53 (q. J = 6.3 Hz; 21-1), 5.43 (s, 2H), 5.96 (br s, 1H),.6.45 (t, J = 73.8 Hz, 1H), 6.87 (d; J = 8.1 Hz, 2.H), 6.99 (d, J= 8.4 Hz, 2H), 7.12-7.19 (m, 4H), 7.58 (t, J= 7.2 Hz, l H), 7.70-7.78 (m, 3H), 7.86 (d, .7= 7.8 Hz, ΓΗ), 8.10 (d, J= 8.4 Hz, 1H), 8.25 (d, J= 8.4 Hz, 1H); ES1-MS (m/z) 500 ( -H)'.
Example 37
A-(2-Cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-| -(trifluoromethoxy)phenyl] prop-2- enamide

The title compound was prepared by coupling Intermediate 32 (150 mg, 0.332 mmol) with 3- aminopropionitrile fumarate (1.11 mg, 0.432 mmol) as described in Example 2 to give 70 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J= 6.3 Hz, 2H), 3.50-3.55 (m, 2H), 5.44 (s, 2H), 5.97 (br s, 1H), 6.95-7.03 (m, 4H), 7.15-7.20 (m, 4H), 7.58 (t, J = 7.2 Hz, 1H), 7.71-7.79 (m, 3H), 7.86 (d, J= 8.4 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 8.25 (d, .7= 8.1 Hz, 1 H); ES I -MS {m/z) 518 (M+H)+.
Example 38
/V (2-Cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-[4-(tri:nuoromethyl)p
enamide
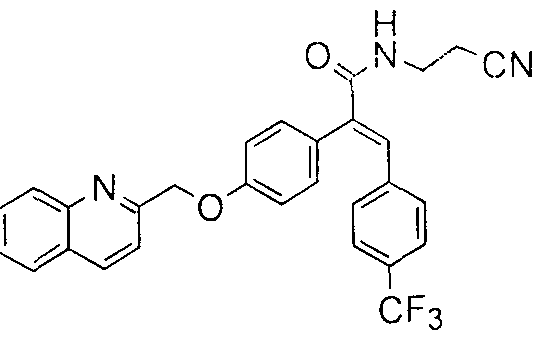
The title compound was prepared by coupling Intermediate 33 ( 100 mg, 0.222 mmol) with 3- aminopropionitrile fumarate (86 mg, 0.333 mmol) as described in Example 2 to yield 50 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.68 (t, J = 6.6 Hz, 2H), 3.55 (q; J - 6.3 Hz, 2H), 5.43 (s, 2H), 6.02 (br s, 1 H), 7.08-7. 1 8 (m, 6H), 7.39 (d, J = 8.4 Hz, 2H), 7.58 (t. J = 8.4 Hz, 1 H), 7.69-7.76 (m, 2H), 7.78-7.85 (m, 2H), 8.09 (d, J = 9.0 Hz, 1 H), 8.24 (d, J = 8.4 Hz, H-i); APCI-MS (m/z) 502 (M+H)+.
Example 39
2- {4-[ 2-(l ,2-Benzoxazol-3-yl)ethoxy]phenyl } -3-(4-chloi phenyl)-N-(2-cyanoethyl) prop-2- enamide
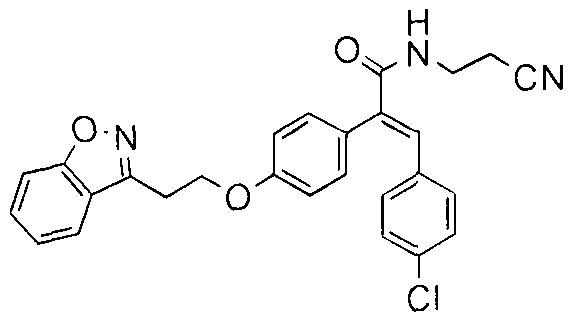
'The title compound was prepared by coupling Intermediate 3 (150 mg, 0.352 mmol) with 3- aminopropionitrile fumarate (138 mg, 0.535 mmol) as described in Example 2 to yield 73 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.67 (t, J = 5.7 Hz, 2H), 3.48-3.56 (m, 4H), 4.48 (t, .7 = 6.3 Hz, 2H), 5.94 (br s, 1 H), 6.91 -7.00 (m, 4H), 7.1 3 (t, J = 7.5 Hz, 4H), 7.38 (br s, I H), 7.60 (br s, 2H), 7.76 (s, 1 H), 7.83 (d, J = 7.8 Hz, 1 H); ESI-MS (m/z) 472 (M+H)+.
Example 40
3-(4-Chloropheny.l)-N-(2-hydroxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide
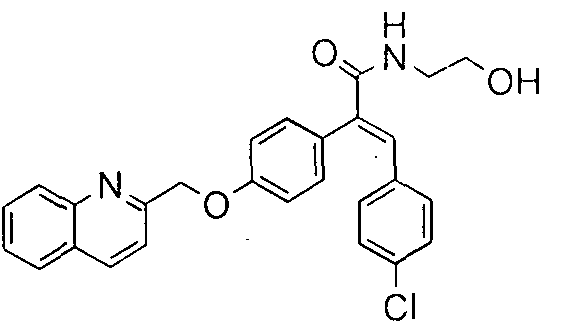
The title compound was prepared by coupling Intermediate 29 (200 mg. 0.481 mmol) with 2- aminoethanol (0.04 ml.0.722 mmol) as described in Example 2 to yield 70 mg of product as off- white solid; Ή NMR (300 MHz, CDC13) δ 2.84 (br s, 1H), 3.43-3.50 (m, 2H), 3.71 (br s, 2H), 5.42 (s, 2H), 5.99 (br s, 1H), 6.44 (d, J= 8.1 Hz, 2H), 7.08-7.17 (m, 6H), 7.58 (t, J = 7.5 Hz, 1H), 7.68-7.79 (m, 3H), 7.86 (d, J= 7.8 Hz, lH), 8.09 (d, J= 8.4 Hz, lH), 8.25 (d, J= 8,4 Hz, 1H); APCI (m/z) 459 (M)+.
Example 41
3- 4-Chloropheiiyl)-A'-|"2-(dimethylamino)ethyl]-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2 enamide

The title compound was prepared by coupling Intermediate 29 (300 mg, 0.721 mmol) with N.N- dimethylethanamine (0.15 ml, 1.44 mmol) as described in Example 2 to. give 52 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.12 (s, 6H), 2.36 (t, J = 6.0 Hz,.2H), 3.38 (q, J = 5.7 Hz, 2H), 5.42 (s, 2H), 6.13 (br s, 1H), 6.91(d, J = 8.7 Hz, 2H), 7.07-7.15 (m, 6H), 7.57 (t, J = 7.2 Hz, 1H), 7.69-7.78 (m, 3H), 7.86 (d, J - 8.1 Hz, 1H), 8.09 (d, .7 = 8.4 Hz, lH), 8.23 (d, J= 8.7 Hz, 1H); ESI-MS (m/z) 486 (M+H)+.
Example 42
3-(4-Clilorophenyl)-A/-[2-(diethylamino)ethyl]-2-[4-(quinolin-2-y]methoxy)phenyl]piOp-2- enamide

The title compound was prepared by coupling Intermediate 29 (300 mg.0.721 mmol) with N,N- diethylethane-1 ,2-diamine (0.202 ml, 1.082 mmol) as described in Example 2 to give 48 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 0.78 (t, J = 6.9 Hz, 6H), 2.34 (d, J = 6.9 Hz, 4H), 2.48 (br s, 2H), 3.34 (d, J = 4.8 Hz, 2H), 5.42 (s, 2H), 6.39 (br s, lH), 6.92 (d, ,7 = 8.4 Hz, 2H), 7.07-7.15 (m, 6H), 7.57 (t, J= 7.2 Hz, 1H), 7.68-7.78 (m, 3H), 7.86 (d, J= 7.8 Hz. H i), 8.09 (d,J= 8.7 Hz, 1H), 8.23 (d,J= 8.7 Hz, 1H); ESI-MS (m/z) 515 (M+H)+.
Example 43
/V-(P!Opan-2-yl)-3-(pyridin-4-yl)-2- -(quino]in-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 14 (150 mg, 0.392 mmol) with isopropylamine hydrochloride (46 mg, 0.784 mmol) as described in Example 2 to yield 90 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI3) 51.10 (d, J = 6.3 Hz, 6H), 4.12-4.2 I Cm, IH), 5.43 (br s, 3H), 6.81 (d, J= 4.5 Hz, 2H), 7.11 (br s, 4H), 7.58 (t, J = 7.511/.. ill).7.67- 7.79 (m, 3H), 7.87 (d, J= 8.1 Hz, 1H), 8.09 (d, J= 8.1 Hz, 1 H), 8.25 (d, .7 = 8.1 Hz, 1 H), 8.37 (d, J = 4.5 Hz, 2H); APCI-MS (m/z) 424 (M+H)+.
Example 44
(±)-3-(4-Chlorophenyl)-N-(] -hydroxypropan-2-y])-2-[4-(quinolin-2-ylmethoxy)phenyJ] prop-2- enamide

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.360 mmol) with (±)-2- aminopropanol (0.03 ml, 0.397 mmol) as described in Example 2 to yield 120 mg of the product as white solid; Ή NMR (300 MHz, DMSO-i¾ δ 1.04 (d, J= 6.6 Hz, 3H), 2.96 (br s: 1H), 3.24- 3.34 (m, 2H), 3.84-3.90 (m, 1H), 4.71 (t, J= 5.4 Hz, 1H), 5.39 (s, 2H), 7.00 (d, J = 8.4 Hz, 2H), 7.15 (s, 4H), 7.21-7.27 (m, 3H), 7.63 (t,J=7.8 Hz, 1H), 7.71 (d,J= 8.1 Hz, 1H), 7.80 (t,J= 7.8 Hz, 1H), 8.02 (d, J =6.9 Hz, 2H), 8.45 (d, .7= 8.4 Hz, 1H); APCl-MS (m/z) 473 (M+H)+.
Example 45
N-[(27i)-l-Hydroxypropan-2-yl]-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide

The title compound was prepared by coupling Intermediate 14 (100 mg, 0.260 mmol) with R- (-) -2-amino- 1 -propanol (0.02 ml, 0.280 mmol) as described in Example 2 to yield 71 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1.11 (d, J= 6.3 Hz, 3H), 3.52 (br s, 2H), 3.65 (br s, 1H), 4.15 (br s, 1H), 5.43 (s, 2H), 5.72 (br s, 1H), 6.84 (br s, 2H), 7.12 (br s, 4H), 7.70 (br s, IH), 7.72-7.78 (m, 3H), 7.87 (d, J = 7.8 Hz, 1H), 8.10 (d, J= 7.8 Hz, lH), 8.25 (d, J= 7.8 Hz, 1H), 8.38 (br s, 2H); APCI-MS (m/z) 440 (M+H)+.
Example 46
3-(4-Chlorophenyl)-AL(l-hydiOxybutan-2-yl)-2-[4-(imidazo[l .2-a]pyridin-2-ylmethoxy) phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 1 (150 mg, 0.370 mmol) with 2- aminobutan-l-ol (0.42 ml, 0.44 mmol) as described in Example 2 to yield 130 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 0.832 (t, J = 7.5 Hz, 3H), 1.29-1.40 (m, 1H), 1.49-1.61 (m, IH), 2.54 (br s, lH), 3.38 (br s, 2H), 3.76 (br s, 1H), 4.65 (br s, 1H), 5.21 (s, 2H), 6.90 (t, ./ = 7.5 Hz, 1 H), 6.98-7.09 (m, 6H), 7.26 (d, .7= 11.7 Hz, 4H), 7.53 (d, J = 9.3 Hz, IH), 8.02 (s, 1H), 8.54 (d, J= 6.9 Hz, 1H); ESI (m/z) 476 (M+H)+.
Example 47
(±)-3-(4-Chlorophenyl)-N-(l-hydroxybutan-2-yl)-2-[4-(pyridin-2-ylmethoxy)phenyl]piOp-2- enamide

The title compound was prepared by coupling Intermediate 12 (200 mg, 0.547 mmol) with 2- aminobutan-l-ol (0.77 ml, 0.820 mmol) as described in Example 2 to give 58 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 0.85 (t, J= 7.5 Hz, 3H), 1.25-1.37 (m, 1 H), 1.50-1.59 (m, 1 H), 1.69 (br s, IH), 3.02 (br s, IH), 3.50-3.58 (m, 1H), 3.70 (br s, 1H), 3.92 (br s, 1H), 5.24 (s, 2H), 5.62 (d, J = 6.6 Hz, IH), 6.92 (d, J = 8.1 Hz, 2H), 7.05-7.13 (m, 3H), 7.15- 7.26 (m, 3H), 7.30 (br s, 1H), 7.56 (d, J= 7.8 Hz, IH), 7.76 (br s, 1 H), 8.63 (d, J= 3.9 Hz, 1 H); ESI-MS (m/z) 437 (M+H)+.
Example 48
3-(4-Chlorophenyl)-N-[(2?)-l-hydroxybutan-2-yl]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2- enamide

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.481 mmol) with (R)-2- aminobutan-l -ol (47 mg, 0.520 mmol) as described in Example 2 to yield 50 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 0.84 (t, .7 = 7.2 Hz, 3H), 1.31-1.41 (m, 1H), 1.46-1.58 (m, 1H), 2.93 (br s, 1H), 3.50-3.56 (m, 1H), 3.64-3.71 (m, 1H)S 3.91 (br s, 1H), 5.42 (s, 2H), 5.60 (d, J= 7.5 Hz, 1H), 6.91 (d, J= 8.7 Hz, 2H), 7.08-7.17 (m, 6H), 7.58 (t, J = 7.2 Hz, 1H), 7.69-7.79 (m, 3H), 7.86 (d, J = 8.1 Hz, 1H), 8.09 (d, J= 8.1 Hz, lH), 8.24 (d, .7-9.0 Hz. lH):ESI(/77/z) 487 (M+H)+.
Example 49
3-(4-Chlorophenyl)-N-[(2S)-l-hydroxybutan-2-yl]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2- enamide

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.481 mmol) with (S)-2- aminobutan-l-ol (47 mg, 0.520 mmol) as described in Example 2 to yield 50 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 0.84 (t. J = 7.2 Hz, 3H), 1.31-1.41 (m, 1 H). 1.46-1.58 (m, 1H), 2.93 (br s, 1H), 3.50-3.56 (m, 1H), 3.64-3.71 (m, 1H), 3.91 (br s, 1H), 5.42 (s, 2H), 5.60 (d, J= 7.5 Hz, 1H), 6.91 (d, J= 8.7 Hz, 2H), 7.08-7.17 (m, 6H), 7.58 (t, J = 7.2 Hz, H I), 7.69-7.79 (m, 3H), 7.86 (d, J= 8.1 Hz, 1H), 8.09 (d, .7= 8.1 Hz, 1H), 8.24 (d, .7 = 9.0 Hz, 1 H); ESI (m/z) 487 (M+H)+.
Example 50
(±)-Λ''-(1 -I-lydroxybutan-2-yl)-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl |prop-2- enamide

The title compound was prepared by coupling Intermediate 14 (100 mg, 0.260 mmol) with 2- amino-1 -butanol (0.03 ml, 0.310 mmol) as described in Example 2 to yield 83 mg of the product as off-white solid; Ή NM (300 MHz, CDC13) δ 0.86 (t, J= 7.2 Hz, 3H), 1.34-1.42 (m, 1H), 1.51-1.60 (m, 1H), 3.54-3.58 (m, 1H), 3.65-3.71 (m, 1H), 3.94 (br s, 1H), 4.23 (br s, IH), 5.42 (s: 2H), 5.69 (d, J= 7.2 Hz, 1H), 6.83 (d, J= 5.1 Hz, 2H), 7.09-7.17 (m, 4H), 7.57 (l, J= 7.2 Hz, IH), 7.70-7.79 (m, 3H), 7.86 (d, J= 8.1 Hz, 1H), 8.09 (d, J= 9.0 Hz, 1H), 8.25 (d, .1 = 8.7 Hz, 1H), 8.38 (d, J = 4.5 Hz, 2H); APCI-MS (mz) 454 (M+H)+.
Example 51
(=i:)-N-(l-HydiOxybutan-2-yl)-3-(4-methoxyphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl J prop-2- enamide

The title compound was prepared by coupling intermediate 10 (200 mg, 0.486 mmol) with 2- aminobutan-1 -ol (47 mg, 0.535 mmol) as described in Example 2 to yield 120 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 0.83 (t, J = 7.2 Hz, 3H), 1.26-1.37 (m, IH), 1.47-1.55 (m, 1H), 3.18 (br s, 1H), 3.54 (br s, 1H), 3.66 (br s, IH), 3.74 (s, 3H), 3.89 (br s, IH), 5.43 (s, 2H), 5.54 (d, J= 6.9 Hz, IH), 6.67 (d, J= 8.7 Hz, 2H), 6.94 (d, J = 9.0 Hz, 2H), 7.12- 7.22 (m, 4H), 7.58 (t, J = 7.2 Hz, IH), 7.70-7.79 (m, 3H), 7.86 (d, J= 8.4 Hz, 1 H), 8.10 (d, .7 = 9.0 Hz, 1 H), 8.25 (d, J = 8.4 Hz, IH); ESI (m/z) 483 (M+H)+.
Example 52
A^-('(l/?)-2-Hydroxy-l -phenylethyl]-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyr| prop-2- enamide

The title compound was prepared by coupling Intermediate 14 (95 mg, 0.240 mmol) with R-(-) - 2-amino-2-phenylethanol (41 mg, 0.290 mmol) as described in Example 2 to yield 61 mg of the product as off-white; Ή NMR (300 MHz, CDC13) δ 3.84 (br s, 3H), 4.26 (br s, 1H); 5.17 (d, J = 6.3 Hz, 1H), 5.43 (s, 2H), 6.34 (d, J= 7.5 Hz, lH), 6.80 (d, J= 4.8 Hz, 2H), 7.10-7.20 (m, 5H), 7.28-7.33 (m, 4H), 7.58 (t, J= 7.2 Hz, 1H), Ί .69-1.19 (m, 2H), 7.86 (d, J = 8.4 Hz, 114), 8.10 (d, J= 8.7 Hz, lH), 8.24 (d, ,7 = 9.0 Hz, 1H), 8.37 (d, ,7 = 4.8 Hz, 2H); ESI (m/z) 502 ί\! Ί I )'.
Example 53
(±)-Ethyl N-{3-(4-chlorophenyl)-2- -(quinolin-2-ylmethoxy)phenyl]prop-2-enoyl}alaninate

The title compound was prepared by . coupling Intermediate 29 (250 mg, 0.601 mmol) with ethyl 2-aminopropanoate (120 mg, 0.783 mmol) as described in Example 2 to yield 170 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 1.24 (t, .7 = 6.9 Hz, 3H), 1.36 (d, .7 = 7.2 Hz, 3H), 4.15 (q,J=7.2 Hz, 2H), 4.62-4.69 (m, 1H), 5.43 (s, 2H), 6.13 (br s, 1H), 6.91 (d, J = 8.7 Hz, 2H), 7.08-7.20 (m, 6H), 7.58 (t, J= 7.2 Hz, 1H), 7.70-7.79 (m, 3H), 7.86 (d, J = 7.8 Hz, 1H), 8.10 (d, J = 8.4 Hz, 1H), 8.25 (d,J= 8.4 Hz, 1H); ESI-MS (m/z) 515 (M)"\
Example 54
(±)-7vr-{3-(4-Ch]orophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-enoyl}alanine
 The title compound was prepared by base hydrolysis of Example 53 (100 mg, 0.194 mmol) to yield 75 mg of the produc as off white solid; Ή NMR (300 MHz, CDC13) δ 1.46 (d, J= 6.6 Hz, 3H), 4.73 (t, J- 6.9 Hz, 1H), 5.49 (br s, 2H), 6.27 (d, J= 6.0 Hz, 1H), 6.91 (d, J = 7.8 Hz, 2H), 7.06-7.13 (m, 311), 7.14-7.21 (m, 3H), 7.59 (t, J= 7.2 Hz, 1H), 7.78 (br s, 3H), 7.87 (d, J= 8.4 Hz, 1H), 8.14 (d, J= 7.8 Hz, 1H), 8.30 (d, J= 6.6 Hz, 1H), 13.24 (br s, 1H); ESI- S (m/z) 487 (M+H)+.
The title compound was prepared by base hydrolysis of Example 53 (100 mg, 0.194 mmol) to yield 75 mg of the produc as off white solid; Ή NMR (300 MHz, CDC13) δ 1.46 (d, J= 6.6 Hz, 3H), 4.73 (t, J- 6.9 Hz, 1H), 5.49 (br s, 2H), 6.27 (d, J= 6.0 Hz, 1H), 6.91 (d, J = 7.8 Hz, 2H), 7.06-7.13 (m, 311), 7.14-7.21 (m, 3H), 7.59 (t, J= 7.2 Hz, 1H), 7.78 (br s, 3H), 7.87 (d, J= 8.4 Hz, 1H), 8.14 (d, J= 7.8 Hz, 1H), 8.30 (d, J= 6.6 Hz, 1H), 13.24 (br s, 1H); ESI- S (m/z) 487 (M+H)+.
Example 55
3-(4-Chlorophenyl)-AL(2,2-dimethoxyelhyl)-2-|"4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamicle

The title compound was prepared by coupling Intermediate 29 (1.0 g, 2.406 mmol) with aminoacetaldehyde dimethylacetal (0.38 ml, 3.610 mmol) as described in Example 2 to yield 900 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.31 (br s, 6H), 3.43 (t, J = 5.7 Hz, 2H), 4.35 (t, J= 5.4 Hz, 1H), 5.43 (s, 2H), 5.75 (br s, 1H), 6.91 (d, J= 8.7 Hz, 2H), 7.08- 7.15 (m, 6H), 7.57 (t, J = 7.5 Hz, 1H), 7.69-7.78 (m, 3H), 7.86 (d, J = 7.8 Hz, 1 H), 8.09 (d, J = 8.1 Hz, 1H), 8.17 (d,J= 8.1 Hz, 1H); ESI-MS (m/z) 502 (M+H)+.
Example 56
A/-(2-Amino-2-oxoethyl)-3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl ]prop-2- enamide

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.480 mmol) and 2- aminoacelamide (80 mg, 0.720 mmol) as described in Example 2 to yield 100 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 3.98 (d, J= 5.4 Hz, 2H), 5.43 (s, 2H), 6.16 (br
s, 1H), 6.24 (br , 2H), 6.92 (d, J= 8.4 Hz, 2H), 7.08-7.19 (m; 6H), 7.57 (t, J =7.2 Hz, 1H).7.70- 7.79 (m, 3H), 7.87 (d,J = 8.4 Hz, 1H), 8.10 (d,J= 8.1 Hz, 1H), 8.25 (d,J= 8.1 Hz, 1 H); APCI- MS (m/z) 472 (M+H)+.
Example 57
A/-|2-(Acetylamiio)ethyj-3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.360 mmol) with Λ'-(2- aminoethyl) acetamide (0.04 ml, 0.433 mmol) as described in Example 2 to yield 90 mg of the product as off white solid; Ή NMR (300 MHz, DMSO-afc) δ 1.76 (s, 3H), 3.10-3.17 (m, 4H), 5.39 (s.2H), 6.99 (d, .7= 8.4 Hz, 2H), 7.10 (s, 4H), 7.23 (d, . = 8.4 Hz, 2H), 7.33 (s, 1H), 7.49- 7.54 (m, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.71 (d, J= 8.4 Hz, lH), 7.80 (t, J= 7.8 Hz, 1H), 7.83- 7.89 (m, 1H), 8.02 (d, J= 7.5 Hz, 2H), 8.45 (d, J= 8.4 Hz, 1H); ES1-MS (m/z) 500 (M+H)+.
Example 58
3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enenitrile

To the well stirred solution of Example 1 (500 mg, 1.206 mmol) in THF (15 ml) was added TEA (0.26 ml, 1.929 mmol) and the reaction mixture was cooled to 0 °C. Trifluoroacetic anhydride was added to this cooled reaction mixture and was further stirred at the same temperature for 2 h. The reaction mixture was diluted with water (50 ml) and extracted with ethyl acetate (3 x 50 ml),, washed with water and brine, dried over anhydrous Na2S0 and concentrated to yield 50 mg of the produc as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.40 (s, 2H), 7.00 (d, J - 9.0 Hz,
2H), 7.10 (d, J= 8.1 Hz, 2H), 7.19 (d, J= 6.3 Hz, 2H), 7.28 (d, J= 8.7 Hz, 3H), 7.57 (t, .7= 6.9
Hz, IH), 7.66 (d, J= 9.0 Hz, 1H), 7.75 (t, J= 7.8 Hz, 1H), 7.85 (d, J
8.1 Hz, IH), 8.22 (d, J = 8.1 Hz, 1H); ESI-MS (m/z) 397 (M+H)+.
Example 59
jV-Cyclopropyl-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 14 (150 mg, 0.39 mmol) with cyclopropylamine hydrochloride (44 mg, 0.78 mmol) as described in Example 2 to yield 120 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 0.43 (br s, 2H), 0.79 (d, J= 6.6 Hz, 2H), 2.79 (br s, IH), 5.42 (s, 2H), 5.65 (br s, IH), 6.81 (d, J= 5.1 Hz, 2H), 7.09 (s, 4H), 7.58 (t, J= 7.2 Hz, IH), 7.71-7.79 (m, 3H), 7.86 (d, J= 7.8 Hz, IH), 8.09 (d, ./= 8.4 Hz, Hi).8.25 (cl, J= 8.7 Hz, IH), 8.37 (d, J= 5.1 Hz, 2H); APCI-MS {m/z) 422 (M+H)+.
Example 60
3-(4-ChloiOphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-A/-(l ,3-thiazol-2-yl)prop-2-enamide

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.481 mmol) and 1,3- thiazol-2-amine as described (130 mg, 0.673 mmol) in Example 2 to yield 50 mg of product as off-white solid; Ή NMR (300 MHz, CDCI3) 55.47 (s, 2H), 6.95 (d, J= 8.4 Hz, 2H), 7.00 (d, J = 3.3 Hz, IH), 7.12-7.23 (m, 6H), 7.40 (d, J= 3.6 Hz, IH), 7.58 (t, J= 7.2 Hz, IH), 7.73-7.79 (m, 2H), 7.87 (d, J= 7.8 Hz, IH), 7.95 (s, IH), 8.11 (d, J= 8.4 Hz, IH), 8.28 (d, J = 8.4 Hz, IH), 8.72 (br s, IH); APCI-MS (m/z) 498 (M+H)+.
Example 61
3-(4-Chlorophenyl)-N-(piperidin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide di (trifluoroacetic acid)

Step 1 : ter/-Butyl 4-({3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enoyl} amino)piperidine- 1 -carboxylate:
The title compound was prepared from Intermediate 29 and 4-amino-l-BOC piperidine as described in Example 2 to give 180 mg of the product as off-white solid; Ή NMR (300 MHz.. CDC13) δ 1.17-1.21 (m, 2H), 1.42 (s, 9H), 1.86 (br s, 2H), 2.87 (br s, 2H), 3.95 (br s, 3H), 5.38 (d, J= 7.8 Hz, 1H), 5.43 (s, 2H), 6.90 (d, J= 8.1 Hz, 2H), 7.07 (br s, 2H), 7.11 (s, 4H), 7.58 (t, J = 7.5 Hz, 1H), 7.70-7.79 (m, 3H), 7.87 (d, J= 7.8 Hz, 1H), 8.10 (d, J= 8.4 Hz, 1H), 8.25 (d, J = 8.7 Hz, 1H).
Step 2: 3-(4-Chlorophenyl)-N-(piperidin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide di (trifluoroacetic acid):
To a well stirred solution of Step 1 Intermediate (170 mg, 0.284 mmol) in dichloromethane (4 ml) was added trifluoroacetic acid (0.5 ml) at 0 °C and the reaction was continued for 4 h. The reaction mixture was concentrated under reduced pressure and dried well under high vacuum. The product was recrystallized from diethyl ether to give 49 mg of an off-white salt; Ή NMR (300 MHz, D SO-flfc) δ 1.66 (br s, 2H), 1.95 (br s, 2H), 2.93-3.02 (rn.2H), 3.29 (br s, 2H), 3.68 (s, IH), 3.92 (s, 1H), 4.31 (br s, 1H), 5.41 (s, 2H), 6.13 (br s, 2H), 7.01 (d, J = 8.4 Hz, 2H), 7.09 (s, 4H), 7.16 (s, 1H), 7.22 (d, J= 7.8 Hz, 1H), 7.74 (d, J= 9.0 Hz, 1H), 7.85 (t, J= 6.9 Hz, 1H), 8.05 (d, J= 6.9 Hz, 2H), 8.52 (d, J= 8.1 Hz, 1H); APCI-MS (m/z) 498 (M+H)+.
Example 62
3-(4-Chlorophenyl)-l -[(3/i)-3-hydroxypyrrolidin-l-yl]-2-[4-(quinoIin-2-ylmethoxy)phenyl] prop-2-en-l-one

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.481 mmol) with (R)- (+)-3-pyrrolidinol (50 mg, 0.570 mmol) as described in Example 2 to yield 70 mg of the product as off' white solid; Ή NMR (300 MHz, CDC13) δ 1.81-1.93 (m, 3H), 3.24 (br s, lH), 3.36-3.41 (m, 1H), 3.63-3.69 (m, 2H), 4.45 (d, .7= 32.7 Hz, 1H), 5.37 (s, 2H), 6.77 (br s, 1H), 6.95 (d, J = 8.4 Hz, 2H), 7.03 (d, J= 8.1 Hz, 2H), 7.13 (d, J= 8.4 Hz, 2H), 7.20 (t, .7= 8.1 Hz, 2H), 7.57 (t, = 7.5 Hz, 1H), 7.68 (d, J= 8.7 Hz, 1H), 7.53 (t, J= 7.2 Hz, 1H), 7.85 (d, .7= 7.8 Hz, 1 H), 8.08 (d, J= 8.4 Hz, 1H), 8.22 (d, J= 8.7 Hz, lH); ESI (m/z) 485 (VP 11)"'.
Example 63
3-(4-Chlorophenyl)-l-[(2R)-2-(hydroxymethyl)pyrrolidin-l-yl]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2-en- 1 -one

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.361 mmol) with (2R)- pyrrolidin-2-ylmethanol (0.53 ml, 0.542 mmol) as described in Example 2 to yield 100 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 1.51-1.58 (m, 2H), 1.65-1.78 (m, HI). 2.08 (br s, 1H), 3.13 (br s, 1H), 3.49 (br s, lH), 3.64-3.73 (m, 2H), 4.28 (d, = 6.0 Hz, 1 IT), 5.00 (br s, 1H), 5.38 (s, 2H), 6.74 (s, 1H), 6.97 (d, ,7= 8.1 Hz, 2H), 7.05 (d, = 7.8 Hz, 2H), 7.14 (d, .7 = 8.1 Hz, 2H), 7.21 (d,J=8.1 Hz, 2H), 7.57 (t,J=7.2 Hz, 1H), 7.66-7.78 (m, 2H), 7.85 (d,J = 7.8 Hz, 1H), 8.08 (d, J = 8.4 Hz, lH), 8,22 (d, J= 8.1 Hz, 1H); ESI-MS (m/z) 499 (M+H)+.
Example 64
3-(4-Chlorophenyl)-l-[(25)-2-(hydroxymethyl)pyrrolidin-l-yl]-2-[4-(quinolin-2-ylmethoxy) pheny]]prop-2-en- 1 -one

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.360 mmol) with (2S)- pyrrolidin-2-ylmethanol (0.053 ml, 0.541 mmol) as described in Example 2 to yield 100 mg of the product as off white solid; Ή NMR (300 MHz, CDCI3) δ 1.63-1.72 (m, 3H), 2.04-2.12 (m, 1H), 3.06-3.16 (m, 1H), 3.49 (br s, 1H), 3.61-3.72 (m, 2H), 4.28 (br s, 1H), 5.01 (br s, 1H), 5.38 (s, 2H), 6.74 (s, 1H), 6.97 (d, J= 8.7 Hz, 2H), 7.05 (d, J= 8.4 Hz, 2H), 7.13-7.22 (m, 4H), 7.57 (t. J= 7.2 Hz, 1H), 7.70-7.78 (m, 2H), 7.85 (d, J= 7.8 Hz, 1 H), 8.09 (d, J = 8.7 Hz, 1 H), 8.22 (d. J - 8.7 Hz, lH); ESI-MS (m/z) 499 (M+H)+.
Example 65
3-(4-Ch]orophenyl)-l-(4-methylpiperazin-l-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l - one

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.36 mmol) with - methyl piperazine (0.06 ml, 0.541 mmol) as described in Example 2 to yield 70 mg of the product as off white solid; Ή NMR (300 MHz, CDCI3) δ 2.18 (br s, 2H), 2.26 (br s, 3H), 2.41 (br s, 2H), 3.51 (br s, 2H), 3.71 (br s, 2H), 5.37 (s, 2H), 6.59 (s, 1H), 6.96 (d, J = 8.4 Hz, 2H), 7.06 (d, J= 8.4 Hz, 2H), 7.14 (d, J= 8.4 Hz, 2H), 7.21 (d, ./= 8.1 Hz, 2H), 7.56 (d, J = 7.2 Hz, 1 H), 7.67 (d, J= 8.1 Hz, 1H), 7.74 (t, J= 7.2 Hz, 1H), 7.84 (d, J= 7.8 Hz, 1H), 8.07 (d, J= 8.7 Hz, 1 H), 8.21 (d, J= 8.7 Hz, 1H); ESI-MS (m/z) 498 (M+H)+.
Example 66
-(4-Chloi phenyl)-2-|"4-(quinolin-2-y]methoxy)phenyl]-l -(thiomorpholin-4-yl)prop-

The title compound was prepared by coupling Intermediate 29 (150 mg, 0.360 mmol) and thiomorpholine (0.041 ml, 0.433 mmol) as described in Example 2 to yield 140 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.22 (br s, 2H), 2.61 (br s, 2H): 3.71 (br s, 2H), 3.91 (br s, 2H), 5.39 (s, 2H), 6.58 (s, 1H), 6.97 (d, J= 8.4 Hz, 2H), 7.05 (d, J= 8.4 Hz, 2H), 7.13-7.22 (m, 4H), 7.57 (t, J= 7.2 Hz, 1H), 7.67 (d, J =9.0 Hz, 1H), 7.75 (t, .7=8.1 Mz; 1H), 7.85 (d, J= 8.4 Hz, 1H), 8.09 (d, J= 8.4 Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H); ES1-MS (m/z) 501 (M+H)+.
Example 67
3-(4-Chlorophenyl)-l-(morpholin-4- -2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-en-l -one
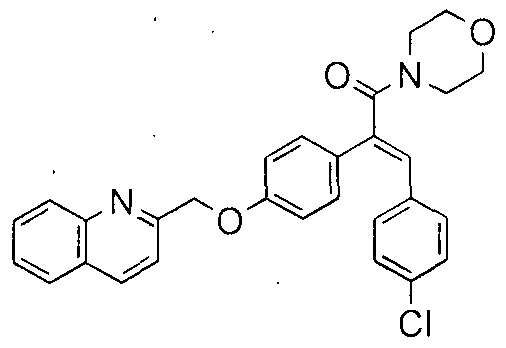
The title compound was prepared by coupling Intermediate 29 (150 mg, 0.361 mmol) with morpholine (0.37 ml, 0.433 mmol) as described in Example 2 to yield 75 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 3.45 (br s, 4H), 3.67 (br s, 4H), 5.38 (s, 2H), 6.6.1 (s, 1H), 6.96 (d, J= 8.7 Hz, 2H), 7.05 (d, J= 8.7 Hz, 1H), 7.12-7.23 (m, 5H), 7.57 (t, J =1.2 Hz, 1H), 7.67 (d, J= 8.7 Hz, 1H), 7.75 (t, J= 7.5 Hz, 1H), 7.85 (d, J= 8.1 Hz, 1H), 8.08 (d, J= 8.4 Hz, HI).8.22 (d, J= 8.4 Hz, 1H); ESI-MS (m/z) 485 (M+H)+.
Example 68
4-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-enoyl}piperazin-2-one

The title compound was prepared by coupling Intermediate 29 (200 mg, 0.481 mmol) with 2- oxopiperazine (72 mg, 0.721 mmol) as described in Example 2 to yield 60 mg of product as off- white solid; Ή NMR (300 MHz, CDC13) δ 3.01 (br s, 1H), 3.41 (br s, IH).3.64 (br sJH), 3.85 (br s, IH), 4.06 (br s, IH), 4.27 (br s, 1H)S 5.37 (s, 2H), 6.09 (br s, 1 H), 6.69 (s51H); 6.97 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.13-7.22 (m, 4H), 7.57 (t, J = 7.2 Hz, IH), 7.67 (d, J = 9.0 Hz, IH), 7.73 (t, J= 7.5 Hz, 1H), 7.85 (d, J= 8.1 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.22 (d, .7 = 8.1 Hz, IH); ESI-MS (m/z) 498 (M+H)+.
Example 69
3-( -Chlorophenyl)-N-(2-cyanoethyl)-2-{4-[2-(quinolin-2-yl)ethyl]phenyl}prop-2-enamide
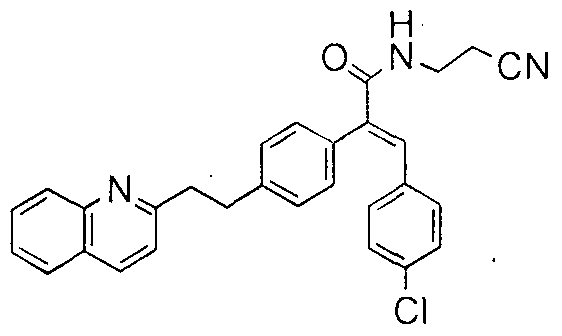
The title compound was prepared by coupling Intermediate 40 (100 mg, 0.241 mmol) with 3- aminopropionitrile fumarate (74 mg, 0.29 mmol) as described in Example 2 to give 65 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI3) δ 2.67 (t, J= 6.3 Hz, 2H); 3.26 (d; J - 7.8 Hz, 2H), 3.35 (d,J= 7.5 Hz, 2H), 3.53 (q,J= 6.3 Hz, 2H), 5.98 (br s, IH), 6.87 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.12 (d, J= 7.8 Hz, 2H), 7.29-7.35 (m, 3H), 7.54 (t, J= 6.6 Hz, .1 H), 7.73-7.83 (m, 3H), 8.09 (br s, 2H); ESI-MS (m/z) 466 (M)+.
Example 70
A/-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-y]methoxy)phenyl]prop-2-en-l-yl}ethane-l ,2-diamine di(trifluoro acetic acid) salt

Step 1 : eri-bu\y\ [2-({3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl} amino)et'hyl]carbamale:
To the well stirred solution of Intermediate 41 (300 mg, 0.750 mmol) in dichloromethane (10 ml) were added N-BOC ethylenediamine (0.12 ml, 0.750 mmol) and sodium triacetoxy borohydride (318 mg, 1.501 mmol) and the reaction mixture was stirred for 30 mins after which acetic acid (0.04 ml, 0.7509) was added to it and it was further stirred overnight. The reaction mixture was quenched with sodium bicarbonate (25 ml), extracted with ethyl acetate (2 x 50ml), washed with water (2 x 25 ml) and dried. The crude product obtained was purified by silica gel column chromatography to yield 125 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI3) δ 1.41 (s, 9H), 2.78 (br s, 21-1), 3.22 (br s, 21T), 3.63 (s, 2H), 4.96 (br s, 1 H), 5.38 (s521 i), 6.20 (br s, 1H), 6.53 (s, 1H), 6.89 (d, J= 9.0 Hz, 2H), 6.98 (d, ,7= 8.4 Hz, 2H), 7.03-7.12 (m, 4H), 7.54 (t. J = 7.2 Hz, 1H), 7.66-7.78 (m, 2H), 7.85 (d, J= 8.4 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1 H), 8.22 (d, ,7 = 8.4 Hz, 1H).
Step 2: N-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl} ethane- 1 ,2- diamine di(trifluoro acetic acid) salt:
To a well stirred solution of Step 1 Intermediate (115 mg, 0.211 mmol) in dichloromethane (4 ml) was added trifluoroacetic acid (0.5 ml) at 0°C and the reaction was continued for 4 h. The reaction mixture was concentrated under reduced pressure and dried well under high vacuum. The product was recrystallized from diethyl ether to give 56 mg of an off-white salt; Ή NMR (300 MHz, CDCI3) δ 3.10-3.18 (m, 4H), 4.06 (s, 2H), 4.32 (br, 2H), 5.40 (s, 2H), 6.80 (s5 1H), 6.95 (d, J= 8.4 Hz, 2H), 7.12 (d, J= 7.8 Hz, 2H), 7.19-7.25 (m, 5H), 7.65-7.71 (m, 2H); 7.82 ft. J= 7.8 Hz, 1H), 8.02 (d, J = 8.1 Hz, 2H), 8.46 (d, J= 8.4 Hz, lH); APC1-MS (m/z) 444 (.V! i !) .
•Example 71
3-(4-Chlorophenyl)-A^-(prop-2-yn-l-yl)-2-[4-(quinolin-2-ylmet'hoxy)phenyl]prop-2-en-l-amine

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and propargyl amine (0.5 1 ml, 0.750 mmol) as described in Step 1 of Example 70 to yield 50 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.24 (s, 1 H), 3.45 (s, 2H), 3.68 (s, 2H), 5.38 (s, 2H), 6.55 (s, I H), 6.71 (d, J = 7.8 Hz, 2H), 6.98 (d, .7 - 8. 1 Hz, 2H), 7.04-7. 14 (m, 5H), 7.56 (t, J = 7.5 Hz, 1 H), 7.67-7.78 (m, 2H), 7.80 (d, J = 7.8 Hz, 1 H), 8.08 (d, J = 8.1 Hz, l H), 8.21 (d, J = 8.4 Hz, 1 H); ESl-MS {m/z) 439 (M+H)+.
Example 72
3-(4-Chlorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -amine dihydrochloride

Step 1 : 3-(4-ChIorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)pheny]] prop-2-en- 1 -amine:
The title compound was prepared from Intermediate 41 and 2-methoxyethylamine as described in Step 1 of Example 70.
Step 2: 3-(4-Chlorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -amine dihydrochloride:
Excess of HCl gas in ethyl acetate was added to the Step 1 Intermediate (300 mg, 0.750 mmol) al 0°C and stirred for 2 h. Excess HCl was removed by bubbling nitrogen gas through the reaction mixture and the solid obtained was purified by silica gel column chromatography to yield 25 mg of off-white solid; Ή NMR (300 MHz, CDC13) δ 2.82 (t, J = 4.8 Hz, 2H), 3.3 1 (s, 3H); 3.47-3.5 1 (in, 2H), 3.61 (s, 2H), 5.38 (s, 2H), 6.52 (s, 1 H), 6.88 (d, J = 8. 1 Hz, 2H), 6.98 (d, J = 8. 1 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8. 1 Hz, 2H), 7.56 (t, J = 7.2 Rz, 1H), 7.68-7.77 (m,
3H), 7.84 (d, J = 7.8 Hz, 1H), 8.08 (d, J= 8.1 Hz, lH), 8.21 (d, J = 8.4 Hz, 1H); ESI-M.S (ni/z) 459 (M+H)+.
Example 73
3-(i3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}amino)
propanenitrile dihydrochloride

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and 3- aminopropionitrile (193 mg, 0.750 mmol) as described in Example 72 to yield 25 mg of off- white solid; Ή NMR (300 MHz. DMSO-afc) D20 exchange δ 2.95 (t, J = 6.9 Hz, 2H), 3.27 (t. ,1 = 1.2 Hz, 2H), 3.20 (br s, 1H), 4.03 (br s, 2H), 5.46 (s, 2H), 6.84 (s, 1H), 6.94 (d, J= 8.1 Hz, 2H), 7.12 (d, J= 8.7 Hz, 2H), 7.22 (d, J= 8.4 Hz, 4H), 7.72 (t, J = 7.5 Hz, 1H), 7.79 (d, J - 8.4 Hz, 1H), 7.88 (t, J - 6.9 Hz, 1H), 8.10 (t, J = 7.8 Hz, 2H), 8.60 (d, J - 9.0 Hz, lH); ES1-MS (m/z) 454 (M+H)+.
Example 74
yV-{3-(4-Chk)rophenyl)-2- 4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}-A/'-methyl ethane- 1 ,2-diamine di(trifluoro acetic acid)

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and /e/ -butyl (2- aminoethyl)methylcarbamate (0.13 ml, 0.750 mmol) as described in Example 70 to yield 56 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.63 (s, 3H), 3.33 (br s, 4H), 3.99 (s, 2H), 4.33 (br s, IH), 5.40 (s, 2H), 6.74 (s, 1H), 6.89 (d, J= 8.4 Hz, 2H), 6.98 (d, J= 7.8 Hz, 2H), 7.01-7.13 (m, 5H), 7.61 (t, J = 7.2 Hz, 1H), 7.72 (d, J = 8.1 Hz, 111), 7.79 (t, J = 7.8 Hz, 1 H),
7.88 (d, J= 7.8 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.31 (d, J= 8.4 Hz, 1H); APCI-MS (m/z) 458 (M-l-H)"'".
Example 75
/V-[2-({3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phen.yl]prop-2-en-l -yl}amino)elhyl] acetamide

The title compound was prepared from Intermediate 41 (200 mg. 0.50 mmol) and N-(2- aminoethyl)acetamide (0.04 ml, 0.50 mmol) as described in Step 1 of example 70 to yield 20 mg of product as off white solid; Ή NMR (300 MHz, CDC13) δ 1.86 (br s, 3H), 2.78 (br s, 2H)r 3.30 (br s, 2H), 3.64 (s, 2H), 4.20 (br s, 1H), 5.38 (s, 2H), 6.04 (br s, 1H), 6.52 (s, 11-J), 6.89 (d. J = 7.8 Hz, 2H), 6.95-7.02 (m, 2H), 7.04-7.12 (m, 4H), 7.56 (t, J= 7.5 Hz, 1H), Ί .66-1.11 (m, 2H). 7.85 (d, J = 7.8 Hz, 1H), 8.08 (d, J= 9.0 Hz, 1H), 8.22 (d, J= 8.4 Hz, 1H); ESI-MS (m/z) 486 (\f ll):.
Example 76
(3 )-A/-{3-(4-Chlorophenyl)-2-|"4-(quinolin-2-ylmethoxy)phenyl]piOp-2-en-l -yl} pyrrolidin-3- amine di(trifluoroacetic acid)

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and tert-butyl
(3S)-3-aminopyrrolidine-l -carboxylate (0.13 ml, 0.750 mmol) as described in Example 70 to yield 57 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.07 (br s, 2H), 3.26 (br s, 4H), 3.54 (br s, 1H), 4.05 (br s, 2H), 5.39 (s, 2H), 6.82 (s, 1H), 6.95 (s, 3H), 7.12 (br s, 3H),
7.20 (br s, 4H), 7.69 (br s, 3H), 8.01 (br s, 2H), 8.45 (br s, 1H); ESI-MS (m/z) 470 (M+H)+.
Example 77
(4S)-4-({3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmelhoxy)phenyl]prop-2-en-l -yl} amino) pyrrolidin-2-one

The title compound was prepared from Intermediate 41 (60 mg, 0.439 mmol) and (4S)-4- aminopyrrolidin-2-one hydrochloride (176 mg, 0.439 mmol) as described in Step 1 of Example 70 to yield 40 mg of product as white solid; Ή NMR (300 MHz, CDC13) δ 2.13-2.20 (m: 2H), 2.51-2.63 (m, 1H), 3.13-3.20 (m, 1H), 3.53-3.67 (m, 4H), 5.38 (m, 2H), 5.75 (br s, 1H), 6.52 (s, ill), 6.88 (d, J - 7.8 Hz, 2H), 6.98 (d, J= 8.1 Hz, 2H), 7.04-7.13 (m, 4H), 7.56 (t, J= 7.8 Hz, 1H), 7.66-7.77 (m, 2H), 7.85 (d, J= 8.1 Hz, 1H), 8.08 (d, J= 8.1 Hz, lH), 8.22 (d, J= 8.1 Hz, 1H); APCI-MS (m/z) 484 (M+H)+.
Example 78
A/-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl ]prop-2-en-1 -yl}piperidin-4-aminc di(trifluoroacetic acid) salt

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and ier/-butyl 4- aminopiperidine-1 -carboxylate (150 mg, 0.750 mmol) as described Example 70 to yield 52 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1.76 (br s, 2H), 2.23 (br s; 3H), 2.67 (br s, 1H), 2.95 (br s, 2H), 3.40 (br s, 2H), 4.04 (s, 2H), 5.43 (s, 2H), 5.78 (br s, 1H), 6.81 (s: lH), 6.93 (br s, 2H), 7.11 (br s, 4H), 7.18 (br s, 2H), 7.73 (br s, 2H), 7.88 (br s, 1 H); 8.04 (br s: 2H), 8.51 (d, J= 7.8 Hz, lH); APCI-MS {m/z) 484 (M+H)+.
Example 79
Ethyl 4-({3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl} [amino) piperidine-1 -carboxylate

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and ethyl-4- amino-piperidine carboxylate (0.12 ml, 0.750 mmol) as described in Step 1 of Example 70 to yield 24 mg of product as colourless oil; Ή NMR (300 MHz, CDC13) δ 1.22-1.27 (m, 5H), 1.80 (br s, 21-1), 2.69 (br s, 1H), 2.83 (t, J= 11.1 Hz, 2H), 3.62 (s, 2H), 4.08-4.15 (m, 4H), 4.35 (br s, 1H), 5.38 (s, 2H), 6.54 (s, 1H), 6.88 (d, J= 8.4 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 7.05 (d, ./= 8.7 1-1 z, 2H), 7.10 (d, .7 = 8.4 Hz, 2H), 7.56 (t, J= 7.5 Hz, 1 H), 7.67-7.77 (m, 2H), 7.85 (d, J = 8.4 Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.22 (d, J= 8.4 Hz, 1H); ESl-MS (m/z) 556 (M+H)+.
Example 80
(5S)-5-({3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}amino) piperidin-2-one
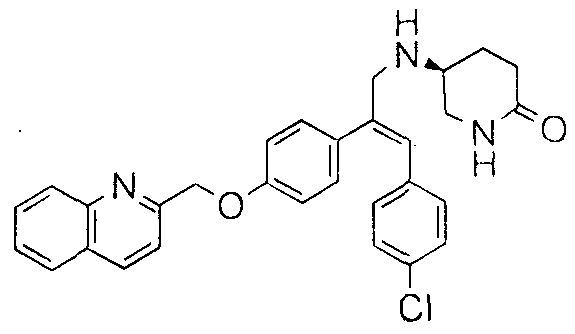
The title compound was prepared from Intermediate 41 (300 mg, 0.751 mmol) and (5S)-5- aminopiperidin-2-one (103 mg, 0.902 mmol) as described in Step 1 of Example 70 to yield 34 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1.25 (s, 2H), 1.79 (br s, 1H), 2.28-2.39 (m, lH), 2.40-2.53 (m, 1H), 3.06 (br s, 2H), 3.45 (br s, 1H), 3.62 (s, 2H), 5.38 (s, 2H), 5.82 (br s, 1H), 6.53 (s, 1H), 6.88 (d, J = 8.4 Hz, 2H), 6.98 (d, J - 8.7 Hz, 2H), 7.05-7.12 (m, 4H), 7.57 (t, J = 7.5 Hz, 1H), 7.60-7.75 (m, 2H), 7.85 (d, ,7 = 7.8 Hz, IH), 8.08 (d, .7 - 8.4 Hz, 1 H), 8.22 (d, J= 8.4 Hz, 1H); ESl-MS (m/z) 498 (M)+.
Example 81
3-(4-Chlorophenyl)-A^N-dimethyl-2-|"4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -amine

The title compound was prepared from Intermediate 41 (200 mg.0.50 mmol) and dimethylamine (41 mg, 0.50 mmol) as described in Step 1 of Example 70 to yield 24 mg of product as white solid; Ή NMR (300 MHz, CDC13) δ 2.29 (s, 6H), 3.30 (br s, 2H), 5.37 (s, 2H), 6.54 (s; 1 H); 6.89 (d, J= 8.1 Hz, 2H), 6.96 (d, J= 8.7 Hz, 2H), 7.06 (d, ./= 8.4 Hz, 2H), 7.12 (d, J= 8.7 ilz. ?.!!). 7.56 (t, J= 7.5 Hz, 1H), 7.67-7.77 (m, 2H), 7.84 (d, J= 8.4 Hz, 1H), 8.08 (d, J = 8.7 Hz, 1H); 8.21 (d..7= 8.1 Hz, 1H); APCI-MS ( /z) 429 (M+H)+.
Example 82
2-({4-| 1 -(4-Chlorophenyl)-3-(pyrrolidin-l- l)prop-l -en-2-yl]phenoxy}methyl)quinoline

Example 83
|X25)-l-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-1-yl}pynOlidin-2- yl methanol

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and (2S)- pyrrolidin-2-ylmethanol (0.07 ml, 0.750 mmol) as described in Step 1 of Example 70 to yield 57 mg of product as off white solid; Ή NMR (300 MHz, CDC13) δ 1.68 (br s, 2H), 1.85 (d, J= 8.4 Hz, 2H), 2.36 (d, J = 7.8 Hz, IH), 2.69 (br s, 2H), 3.1 (br s, 2H), 3.32 (d, J = 10.8 Hz, 1 H), 3.59 (d, J= 9.9 Hz, IH), 3.81 (br s, IH), 5.37 (s, 2H), 6.52 (s, I H), 6.89 (d5 J = 7.8 Hz, 2H), 6.98 (d, J = 8.1 Hz, 2H), 7.04-7.11 (m, 4H), 7.56 (t, J= 7.2 Hz, IH), 7.68-7.76 (m, 2H), 7.85 (d, J = 7.8 Hz, IH), 8.08 (d, J= 8.4 Hz, IH), 8.22 (d, J= 8.4 Hz, IH); APC1-MS (m/z) 485 (M+H)+.
Example 84
2-({4-[Ί -(4-Chlorophenyl)-3-( l/-/-pyrazol- 1 -yl)prop- 1 -en-2-yl]phenoxy } methyl)q

The title compound was prepared from Intermediate 42 (150 mg, 0.352 mmol) and lH-pyrazole (32 mg, 0.462 mmol) as described in Example 82 to yield 45 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.05 (s, 2H), 5.35 (s, 2H), 6.21 (s, IH), 6.32 (s, IH), 6.86-6.98 (m, 6H), 7.05 (d, .7= 8.1 Hz, 2H), 7.31 (s, IH), 7.51-7.60 (m, 2H), 7.67 (d, J - 8.4 Hz, IH).7.74 (t, J= 6.9 Hz, IH), 7.85 (d, J= 8.1 Hz, IH), 8.08 (d, J = 8.1 Hz, IH), 8.21 (d, J = 8.7 Hz, IH): APC1 ( /z) 452 (M+H)+.
Example 85
2-({4-| 1 -(4-Chlorophenyl)-3-(lH-imidazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 42 (225 mg.0.537 mmol) and 1 /-/-imidazole (44 mg, 0.642 mmol) as described in Example 82 to yield 30 mg of product as off-white solid: Ή NMR (300 MHz. CDC13) δ 4.90 (s, 2H), 5.36 (s, 2H), 6.46 (s, IH), 6.88 (d, J = 8.4 Hz, 2H), 6.94 (br s, 5H), 7.08 (d, J = 7.8 Hz, 3H), 7.56 (t, J= 7.2 Hz, 1H), 7.67 (d, = 8.1 Hz, IH), 7.72- 7.77 (m, 2H), 7.85 (d,J= 8.4 Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.22 (d; J= 8.4 Ηζ,ΙΗ); ESI-MS

Example 86
(1 - {3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}-lH-imidazol-4-yl) methanol

The title compound was prepared from Intermediate 42 (500 mg, 1.190 mmol) and 1 /-/-iniidazol - 4-ylmethanol (140 mg, 1.427 mmol) as described in Example 82 to yield 77 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 4.56 (s, 2H), 4.79 (s, 2H), 5.36 (s, 2H), 6.41 (s, 1H), 6.83-6.91 (m, 3H), 6.92-6.98 (m, 5H), 7.04-7.09 (m, 2H), 7.39 (s, 1H), 7.56 (t, J - 7.5 Hz, 1H), 7.66 (d, J= 9.0 Hz, 1H), 7.74 (t, J = 6.9 Hz, 1H), 7.85 (d, J= 7.8 Hz, IH), 8.08 (d, J= 9.0 Hz, 1 H), 8.22 (d, J = 8.4 Hz, IH); ESI-MS (m/z) 482 (M+H)+.
Example 87
2-({4-[ 1 -(4-Chlorophenyl)-3-(lH-l,2,4-triazol-l-yI)prop-l-en-2-yI]phenoxy}methyI)quinoIine
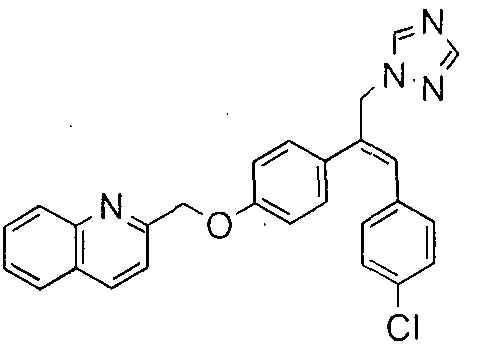
The title compound was prepared from Intermediate 42 (65 mg, 0.154 mmol) and 1/7-1,2,4- triazole (13 mg, 0.185 mmol) as described in Example 82 to yield 20 mg of product as off-while solid; Ή NMR (300 MHz, CDC13) δ 5.23 (s, 2H), 5.32 (s, 2H), 6.61 (s, ΓΗ), 6.94-7.05 (m.6H). 7.19 (d, J = 8.1 Hz, 2H), 7.62-7.69 (m, 2H), 7.79 (t, J = 8.1 Hz, IH), 7.91 (s, 11-1), 8.01 (d, = 9.0 Hz, 2H), 8.36 (s, IH), 8.43 (d, J= 8.4 Hz, IH); ES1-MS (m/z) 453 (M+H)+.
Example 88
4-|"l-(4-ChloiOphenyl)-3-(2H-l, -triazol-2-yl)prop-l-en-2-yl]phenoxy}methy])qu

The title compound was prepared from Intermediate 42 (350 mg, 0.833 mmol) and 2/7-1.2,3- triazole (0.07 ml, 1.249 mmol) as described in Example 82 to yield regioisomeric mixture of products. These regioisomers were separated by silica gel column chromatography and 62 mg of the above less polar isomer was eluted out in 15 % acetone in petroleum ether solution as off- white solid; Ί-TN R (300 MHz, CDC13) δ 5.34 (s, 2H), 5.38 (s, 2H), 6.38 (s, IH), 6.87-6.93 (m, 4H), 7.00-7.07 (m, 4H), 7.52-7.58 (m, 3H), 7.66 (d, J = 8.7 Hz, IH), 7.74 (t, = 7.8 Hz, 1 H), 7.84 (d, ./= 7.8 Hz, IH), 8.07 (d, J = 8.4 Hz, IH), 8.20 (d, J= 8.4 Hz, ΓΗ): ESI-MS (m/z) 453
(M+H) .
Example 89
2-({4-|l-(4-Chlorophenyl)-3-(17/-l,2,3-triazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was the more polar isomer isolated from Example 88 and was eluted out in 20 % acetone in petroleum ether solution to give 70 mg of product as off-white solid; Ή NMR
(300 MHz, CDC13) δ 5.33 (s, 2H), 5.35 (s, 2H), 6.51 (s, 1H), 6.89-7.00 (m, 6H), 7.08 (d, J = 8.4 Hz, 2H), 7.41 (s, 1H), 7.56 (t, J= 7.5 Hz, 1H), 7.62-7.68 (m, 2H), 7.74 (t, J= 7.5 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H), 8.21 (d, J = 8.1 Hz, 1H); ESI-MS (m/z) 453
( il!)'.
Example 90
2-({4-|" 1 -(4-Chlorophenyl)-3-(lH-tetrazol-l -yl)prop-l -en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 42 (400 mg, 0.952 mmol) and 1 /7-tetrazole (4.2 ml, 1.904 mmol) as described in Example 82 to yield regioisomeric mixture of products. These regioisomers were separated by silica gel column chromatography and 78 mg of the above less polar isomer was eluted out in 25 % acetone in petroleum ether solution as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.34 (s, 2H), 5.58 (s, 2H), 6.59 (s, lH), 6.92 (d, J = 8.1 Hz, 4H), 7.02 (d, J= 8.4 Hz, 2H), 7.08 (d, J= 8.4 Hz, 2H), 7.56 (t, J = 7.2 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.74 (t,J= 7.8 Hz, 1H),7.84 (d,J= 8.1 Hz, 1H), 8.08 (d,J= 8.1 Hz, 111), 8.21 (d, J= 8.7 Hz, 1 H), 8.45 (s, 1 H); ESI-MS (m/z) 454 (M+H)+.
Example 91
2-({4-| 1 -(4-Chlorophenyl)-3-(2/7-tetrazol-2-yl)prop- 1 -en-2-yl]phenoxy }methyl)quinoline

The title compound was the more polar isomer isolated from Example 90 and was eluted out in 28 % acetone in petroleum ether solution to give 92 mg of off-white solid; Ή NMR (300 MHz, CDCI3) δ 5.36 (br s, 4H), 6.65 (s, 1H), 6.91-6.96 (m, 6H), 7.10 (d, J= 8.4 Hz, 2H), 7.57 (t, J = 7.2 Hz, 1H), 7.65 (d, J= 8.4 Hz, 1H), 7.75 (t, J= 7.2 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.22 (d, J= 8.4 Hz, 1H), 8.34 (s, 1H); ESI-MS (m/z) 454 (M+H)+.
Example 92
2-({4-[l-(4-Ch]orophenyl)-3-(piperi in-l-yl)prop-l-en-2-yl]phenoxy}methy!)quinoline

The title compound was prepared from Intermediate 42 (5 g, 11.26 mmol) and piperidine (0.52 ml, 0.5711 mmol) as described in Example 82 to yield 16 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 1.58 (br s, 6H), 3.35 (br s, 4H), 4.87 (s, 2H), 5.38 (s, 2H), 6.55 (s, 1H), 6.90-6.99 (m, 4H), 7.04-7.14 (m, 4H), 7.56 (t, J= 7.5 Hz, 1H), 7.67-7.77 (m, 2H), 7.84 (d, J = 8.4 Hz, 1 H), 8.08 (d, J= 8.1 Hz, 1H), 8.21 (d, .7=8.1 Hz, 1H); APCI-MS (m/z) 469 (M+H)+.
Example 93
2-({4-[l-(4-Chlorophenyl)-3-(thiomorpholin-4-yl)prop-l-en-2-yl]phenoxy}methyl)q

Example 94
2-({4-|Ί -(4-Chloropheny])-3-(morpholin-4-yl)prop- l -en-2-yl]phenoxy} methyl)quinoline

The title compound was prepared from Intermediate 42 (240 mg, 0.571 mmol) and morpholine (0.09 ml, 1.142 mmol) as described in Example 82 to yield 103 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.51 (br s, 4H), 3.24 (s, 2H), 3.70 (br s, 4H), 5.38 (s, 2H), 6.5 1 (s, l H), 6.88 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 8.7 Hz, 2H), 7.05 (d, J = 8.1 Hz, 2H), 7.1 3 (d, J = 8.7 Hz, 2H), 7.56 (t, J = 7.5 Hz, 1 H), 7.68-7.77 (m, 2H), 7.85 (d, J = 7.8 Hz, 1 H), 8.08 (d, ./ = 8.4 Hz, 1 H), 8.22 (d, J = 8.1 Hz, 1 H); ESI-MS (m/z) 471 (M+H)"'\
Example 95
2-({4-[l -(4-Chlorophenyl)-3-(piperazin-l -yl)prop- l -en-2-yl]phenoxy}methyl)quinoline di(trifluoro acetic acid)

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and 1 -BOC piperazine ( 140 mg, 0.750 mmol) as described in Example 70 to yield 45 mg of product as off- white solid; Ή NMR (300 MHz, CDC13) δ 3.18 (br s, 4H), 3.33 (br s, 4H), 3.50 (br s, I H), 3.75
(s, 2H), 5.51 (s, 2H), 6.65 (s, 1H), 6.89 (d, J= 7.8 Hz, 2H), 6.98 (d, J - 8.1 Hz, 2H).7.06-7.12 (m, 4H), 7.67 (br s, 1H), 7.83-7.88 (m, 2H), 7.94 (d, J ='8.4 Hz, 1H), 8.24 (d, .7= 8.1 Hz, 1 H), 8.45 (d, J= 8.1 Hz, 1H); ESI-MS (m/z) 470 (M+H)+.
Example 96
2-({4-[l-(4-Fluorophenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline trihydrochloride

Step 1 : 2-({4-[l-(4-Fluoi phenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline: The title compound was prepared from Intermediate 43 (590 mg, 1.302 mmol) and N-BOC piperazine (485 mg, 2.604 mmol) as described in Example 82.
Step 2: 2-({4-[l-(4-Fluorophenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methylquinoline trihydrochloride:
Excess of HCl gas in ethyl acetate was added to Step 1 BOC intermediate at 0°C and stirred for 2 h. HCl was removed by bubbling nitrogen gas through the reaction mixture and the solid obtained was purified by diethyl ether to yield 200 mg of off-white solid; Ή NMR (300 MHz, DMSO-i¾ δ 2.26 (br s, 1H), 3.35 (br s, 8H), 4.23 (br s, 2H), 5.43 (br s, 2H), 6.97 (d, J= 5.4 Hz. 5H), 7.09 (d, J= 7.8 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 7.69-7.79 (m, 2H), 7.87 (br s, 1 H).8.08 (br s, 2H), 8.59 (d, J= 8.4 Hz, 1H); ESI-MS (m/z) 454 (M+H)+.
Example 97
2-({4-| 1 -(4-Chloro-3-fluorophenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl) quinoline trihydrochloride

Example 98
2-({4-|l -(4-Chlorophenyl)-3-(4-methylpiperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl) quinoline

The title compound was prepared from Intermediate 41 (300 mg, 0.750 mmol) and 1 -methyl piperazine (0.083 ml, 0.750 mmol) as described in Step 1 of Example 70 to yield 72 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.30 (s, 3H), 2.47 (br s, 4H), 2.56 (br s, 4H), 3.26 (s, 2H), 5.38 (s, 2H), 6.50 (s, 1H), 6.88 (d, J= 8.1 Hz, 2H), 6.95 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.12 (d, J = 8.7 Hz, 2H), 7.56 (t, J = 7.8 Hz, 1H), 7.68-7.77 (m, 2H), 7.85 (d, J= 8.4 Hz, 1H), 8.08 (d, J= 8.7 Hz, 1H), 8.22 (d, J= 8.1 Hz, 1H); ES1-MS (m/z) 484 (.VI : II)'.
Example 99
4-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}piperazin-2-one

Example 100
1 -{3-(4-Fluorophenyl)-2-[4-(quinoli -2-ylmethoxy)phenyl]prop-2-en-l-yl}pyridin-2(l /J)-one

The title compound was prepared from Intermediate 43 (200 mg, 0.491 mmol) and pyridin- 2(lH)-one (70.70 mg, 0.743 mmol) as described in Example 82 to yield 39 mg of product as off- white solid; Ή NMR (300 MHz, CDCI3) δ 4.91 (s, 2H), 5.35 (s, 2H), 6.06 (t, J= 6.9 Hz. 1 H): 6.45 (s, lH), 6.53 (d, J = 9.3 Hz, lH), 6.76 (t, J= 8.7 Hz, 2H), 6.91-6.97 (m.4H), 7.09 (d, ,7 = 8.7 Hz, 2H), 7.17 (d, J= 6.9 Hz, 1H), 7.23 (s, 1H), 7.56 (t, .7= 7.5 Hz, 1H), 7.67 (d, .7= 8.1 Hz, 1H), 7.74 (t, J - 7.5 Hz, 1H), 7.84 (d, J= 8.1 Hz, 1H), 8.08 (d, J= 9.0 Hz, 1H), 8.21 (d, .7= 9.0 Hz, 1 H); ESI-MS (m/z) 463 (M+H)+.
Example 101
1 -{3-(4-Chlorophenyl)-2-[4-(quinoli -2-ylmethoxy)phenyl]prop-2-en-l-yl}pyrimidin-2(l/J)-one

Example 102
1 -{3-(4-Fluorophenyl)-2-[4-(quinoli -2-ylmethoxy)phenyl]piOp-2-en-l -yl}pyrimidin-2(l/7)-one

The title compound was prepared from Intermediate 43 (200 mg, 0.495 mmol) and pyrimidin- 2(l/7)-one hydrochloride (79 mg, 0.594 mmol) as described in Example 82 to yield 78 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.89 (s, 2H), 5.35 (s, 2H), 6.10-6.14 (m, 1H), 6.66 (s, 1H), 6.79 (t, J= 8.4 Hz, 2H), 6.91-6.97 (m, 4H), 7.06 (d, J= 8.7 Hz, 2H), 7.41- 7.45 (m, 1H), 7.56 (t, J= 7.8 Hz, 1H), 7.67 (d, J= 8.4 Hz, 1H), 7.75 (t, J = 7.5 Hz, IH), 7.85 (d, J= 8.1 Hz, IH), 8.08 (d, J= 8.4 Hz, IH), 8.22 (d, J= 8.7 Hz, IH), 8.48 (br s, IH); APCI-MS (m/z) 464 (M+H)+.
Example 103
3-{3-Phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]pi p-2-en-l-yl}pyrimidin-4(3/-/)-one

The title compound was prepared from Intermediate 44 (200 mg, 0.518 mmol) and pyrimidin- 4(3/-/)-one (60 mg, 0.621 mmol) as described in Example 82 to yield 30 mg of product as off- white solid; Ή NMR (300 MHz, CDC13) δ 4.90 (s, 2H), 5.35 (s, 2H), 6.40 (d, J = 6.3 Hz; IH),
6.63 (s, 1H), 6.90-6.97 (m, 4H), 7.11 (br s, 5H), 7.56 (t, J = 7.8 Hz, 1H), 7.68 (d, J = 9.0 Hz, IH), 7.72-7.80 (m, 2H), 7.84 (d, J= 8.1 Hz, 1H), 7.94 (s, IH), 8.08 (d, J= 8.1 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1 H); APC1-MS (in/z) 446 (M+H)+.
Example 104
3-{3-(4-Fluorophenyl)-2-[4-(quinoli -2-ylmethoxy)phenyl]prop-2-en-l-yl}pyrimidin-4(3/7)-one

The title compound was prepared from Intermediate 43 (300 mg, 0.743 mmol) and pyrimidin- 4(3/7)-one (86 mg, 0.892 mmol) as described in Example 82 to yield 130 mg of product as off- white solid; Ή NMR (300 MHz, CDC13) δ 4.88 (s, 2H), 5.36 (s, 2H), 6.40 (d, J = 6.9 Hz. 1 H), 6.59 (s, IH), 6.79 (t, J= 8.4 Hz, 2H), 6.92-6.98 (m, 4H), 7.07 (d, J= 8.7 Hz, 2H), 7.56 (t, .7-7.2 Hz, IH), 7.67 (d, J= 8.7 Hz, IH), 7.72-7.83 (m, 2H), 7.85 (d, J= 8.4 Hz, IH), 7.91'(s, IH), 8.08 (d, ,]= 8.411/. IH), 8.22 (d, J= 8.7 Hz, IH); ESI-MS (m/z) 464 (M+H)+.
Example 105
2-({4-[l-(4-Fluorophenyl)-3-(lH-tetrazol-l-yl)prop-]-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 43 (400 mg, 0.991 mmol) and lH-tetrazole (4.5 ml, 1.982 mmol) as described in Example 82 to yield 119 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.34 (s, 2H), 5.58 (s, 2H), 6.62 (s, IH), 6.81 (t, J= 8.1 Hz, 2H), 6.91-7.03 (m, 6H), 7.56 (t, J= 7.5 Hz, IH), 7.66 (d, J= 8.4 Hz, IH), 7.74 (t, J = 7.8 Hz, IH), 7.84 (d, J= 7.8 Hz, IH), 8.08 (d, J= 8.4 Hz, IH), 8.21 (d, J= 8.4 Hz, IH), 8.45 (s, IH); APCI- MS (m/z) 438 (M+H)+.
Example 106
2-({4-[l -(4-1 υοΓθρ1ιεηγ1)-3-(2/Υ εΐΓ3Ζθ1-2-γ1)ρΓορ-1-εη-2-γ1]ρΗεηοχγ}ηΊεΐΗγ1)ςυίηο1ίηε

The title compound was prepared from Intermediate 43 (400 mg, 0.991 mmol) and 2/-/-tetrazole (4.5 ml, 1.982 mmol) as described in Example 82 to yield 148 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.35 (s, 4H), 6.68 (s, 1H), 6.83 (t, J = 8.4 Hz, 2H), 6.96 (br s, 6H): 7.57 (t, J =1.5 Hz, 1H), 7.65 (d, J= 8.4 Hz, 1H), 7.75 (t, J= 7.2 Hz, 1H), 7.85 (d, J = 8.4 Hz, IB), 8.08 (d, J = 8.4 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 8.33 (s, lH); APC1-MS (m/z) 438 i -ny.
Example 107
7V-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}acetamide

Step 1 : 2-({4-|3-Azido-l -(4-chlorophenyl)prop-l -en-2-y!]phenoxy}methyl) quinoline:
To the well stirred solution of Intermediate 42 (1.7 g, 4.045 mmol) in dimethylformamide (25 ml) was added sodium azide (1.05 g, 16.18 mmol) and the reaction mixture was heated to 60°C and stirred at the same temperature overnight. The reaction mixture was diluted with water (250 ml) and extracted with ethyl acetate (250 ml x 2). The combined organic layers were washed with water (50 ml x 2) and brine (50 ml), dried over anhydrous Na2S04 and concentrated to yield 1.35 g of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.3,1 (s, 2H), 5.43 (s, 2H), 6.92-7.01 (m, 4H), 7.05-7.15 (m, 4H), 7.47 (d, J = 8.7 Hz, lH), 7.56 (t, J = 7.5 Hz, 1H), 7.67- 7.83 (m, 2H), 7.84 (d, J= 7.8 Hz, 1H), 8.08 (d, .7=8.1 Hz, 1H), 8.21 (d, J= 8.7 Hz, lH).
Step 2: 3-(4-Chloropheny])-2-[4-(quinolin-2-ylmethoxy)pheny]]prop-2-en-l -amine:
To the well stirred solution of Step 1 Intermediate (500 mg. 1 .1 71 mmol) in dry T! 11· ( 1 0 ml) was added triphenylphosphine (338 mg, ] .288 mmol) and the reaction mixture was stirred for 3 h at room temperature. Water was then added to the reaction mixture and was further stirred overnight. The reaction mixture was diluted with water ( 100 ml) and extracted with ethyl acetate ( 150 ml x 2). The combined organic layers were washed with water (50 ml x 2) and brine (50 nil), dried (Na2S0 ) and concentrated to yield 750 mg of product as off-white solid.
Step 3 : 3-(4-Chlorophenyl)-2-f4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -amine
hydrochloride:
To the well stirred solution of Step 2 Intermediate (750 mg, 1 .872 mmol) in acetonitrile ( 1 5 ml) was added TEA (0.52 ml, 3.745 mmol), water (0.5 ml) and BOC anhydride (450 mg, 2.059 mmol) and the reaction mixture was stirred at room temperature for 6 h. The reaction mixture was diluted with water (50 ml) and extracted with ethyl acetate ( 100 ml x 2). The combined organic layers were washed with water (50 ml x 2) and brine (50 ml), dried over anhydrous a2S04 and concentrated. The crude compound was purified by silica gel chromatography to yield 1 50 mg of product. The /<?r/-butyl {3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl] prop-2-en- l -yl }carbamate so formed was deprotected using HQ gas in ethyl acetate to yield 200 mg of its hydrochloride salt.
Step 4: /V-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -yl } acetamide: To the well stirred solution of Step 3 crude Intermediate (200 mg, 0.499 mmol) i n dicholomethane ( 1 0 ml) were added triethlyamine (0.27 ml, 1 .997 mmol) and acetic acid (0.07 m l . 0.749 mmol) and the reaction was stirred at room temperature for 4 h. The reaction mixture was diluted with water (50 ml) and extracted with chloroform ( 100 ml x 2). The combined organic layers were washed with water (20 ml x 2) and brine (20 ml), dried over anhydrous Na2S04 and concentrated to yield 90 mg of product as off-white solid; Ή NMR (300 MHz, DMSO-4i) δ 1.82 (s, 3H), 4.01 (br s, 2H), 5.36 (s, 2H), 6.43 (s, 1 H), 6.91 (d, J = 7.8 Hz, 2H); 7.04-7.09 (m, 4H), 7.16 (d, J = 7.8 Hz, 2H), 7.62-7.70 (m, 2H), 7.75-7.8 1 (m, 1 H), 8.00 (br s, 2H), 8. 1 4 (br s, 1 H), 8.43 (d, J = 8.4 Hz, 1 H); APCI-MS (m/z) 443 (M+H)+.
Example 1 08
N-{3-(4-Chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -yl }-2-methoxy acetamide

The title compound was prepared from 3-(4-chlorophenyI)-2-[4-(quinoIin-2-yImelhoxy) pheny]]prop-2-en- l -amine hydrochloride (Step 3 of Example 107; 1 50 mg, 0.374 mmol) and methoxyacetyl chloride (0.05 ml, 5.617 mmol) as described in Example 1 07 to yield 5 1 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.28 (s, 3H), 3.86 (s, 2H). 4.28 (d„ J = 6.0 Hz, 2H), 5.37 (s, 2H), 6.48 (s, 111), 6.62 (br s, 1 H), 6.89 (d, J = 8.4 Hz, 2H), 6.97 (d, J = 8.7 Hz, 2H), 7.05-7.1 2 (m, 4H), 7.56 (t, J = 7.2 Hz, 1 H), 7.68 (d, ,7 = 8.1 Hz, 1 11), 7.74 (t, J = 7.2 Hz, ΓΗ), 7.84 (d, J = 7.8 Hz, 1 H), 8.08 (d, J = 8.4 Hz, 1 H), 8.2 1 (d, ./ = 9.0 Hz, 1 H); ES1 -MS (m/z) 473 (M+H)+.
Example 109
4- {2-(4-Chlorophenyl)- l -[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}pyrrolidin-2-one

Step 1 : Ethyl-5-(4-chlorophenyl)-4-[4-(quinolin-2-ylmethoxy)phenyl]penta-2,4-dienoate:
To the well stirred solution of Intermediate 42 (1 g, 2.502 mmol) in toluene (30 ml) was added (carbethoxymethylene)triphenylphosphorane (1.04 g, 3.00 mmol) and the reaction mixture was heated overnight at 120 °C. The reaction mixture was quenched with water (50 ml), extracted with ethyl acetate (2 x 1 00 ml), washed with water (2 x 50 ml), brine (25 ml) and dried. The crude product obtained was purified by silica gel column chromatography to yield 900 mg of the product; Ή NMR (300 MHz, CDC13) δ 1 .26 (t, J - 7.5 Hz, 3H), 4.18 (q, J = 7.2 Hz, 2H), 5.42 (s, 21-1), 5.54 (d, J = 15.3 Hz, 1H), 6.82-6.88 (m, 3H), 7.02-7.09 (m, 6H), 7.56 (d, J = 7.2 Hz, 1 H),
7.63 (d, .7= 15.6 Hz, 1H), 7.71-7.78 (m, 2H), 7.86 (d, J= 7.8 Hz, 1 H), 8.10 (d, J = 8.4 Hz, 1 H). 8.24 (d, .7=8.4 Hz, 1H).
Step 2: Ethyl-5-(4-chlorophenyl)-3-(nitromethyl)-4-[4-(quinolin-2-ylmethoxy)phenyl]pent-4- enoate:
To a well stirred solution of Step 2 Intermediate (900 mg, 1.916 mmol) in nitromethane (20 ml) was added 1,1,3,3-tetramethylguanidine (441 mg, 3.833 mmol) and the reaction was heated at 120 °C for 3h. The reaction mixture was quenched with water (50 ml), extracted with ethyl acetate (2 x 100 ml), washed with water (2 x 50 ml), brine (25 ml) and dried. The crude product obtained was purified by silica gel column chromatography to yield 520 mg of the product; Ή NMR (300 MHz, CDC13) δ 1.23 (t, J= 6.9 Hz, 3H), 2.91 (d, J= 6.9 Hz, 2H), 4.07-4.18 (m, 2H), 4.60-4.68 (m, 1H), 4.77-4.86 (m, 1H), 5.39 (br s, 2H), 5.77 (t, J = 6.9 Hz, 1H), 6.73-6.82 (m, 1H), 6.93 (d, J= 8.7 Hz, 1H), 7.01-7.09 (m, 3H), 7.24 (br s, 4H), 7.56 (t, J= 7.5 Hz, 1H), 7.66 (d, J= 9.0 Hz, 1H), 7.75 (t, J= 7.2 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 8.08 (d, J= 9.0 Hz, 1 I ), 8.18-8.25 (m, 1H).
Step 3: Ethy!-3-(aminomethyI)-5-(4-chlorophenyl)-4-[4-(quinolin-2-yhnethoxy)phenyI] pent-4- enoate:
To the well stirred solution of Step 2 Intermediate (500 mg, 0.942 mmol) in ethanol (10 ml) was added aqueous solution of ammonium chloride (505 mg, 9.425 mmol) and the reaction was heated to reflux. At reflux temperature, iron powder (158 mg, 2.827 mmol) was added to the reaction mixture and it was further refluxed for 0.5 h. The reaction mixture was filtered and the filtrate was extracted with ethyl acetate (2 x 100 ml). The combined organic layer was washed with water (25 ml), brine (25 ml), dried over anhydrous Na2SC>4 and concentrated to yield 100 mg of the crude product; Ή NMR (300 MHz, CDC13) δ 1.26 (t, J= 7.2 Hz, 3H), 2.98 (t, J= 7.5 Hz, 2H), 3.98-4.10 (m, 5H), 5.33 (br s, 2H), 5.94 (br s, 2H), 7.10-7.21 (m, 4H), 7.51-7.63 (m, 3H), 7.68-7.80 (m, 4H), 8.05 (d, J= 8.4 Hz, 2H), 8.13-8.21 (m, 2H).
Step 4: 4-{2-(4-Chloi phenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}pyrrolidin-2-one: The Step 3 Intermediate (100 mg, 0.199 mmol) was dissolved in xylene (20. ml) and refluxed for 2 h. The reaction mixture was dried and concentrated to yield the crude product which was purified by silica gel column chromatography to yield 30 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.44-2.50 (m, 2H), 3.45-3.56 (m, 3H), 5.39 (s, 2H), 5.52 (br s,
1H), 6.40 (s, ll-I), 6.80 (d, J= 8.4 Hz, 2H), 6.99-7.06 (m, 6H), 7.59 (t, J= 7.8 Hz, lH), 7.66-7.74 (3), 2H), 7.85 (d, J =7.8 Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.22 (d, J= 8.4 Hz, 1H)S APC1 (m/z) 455 (M+H)+.
Example 110
2-({4-|2-(4-Chlorophenyl)-l-(l,3-ox oxy}methyl)quinoline

To the well stirred solution of Intermediate 41 (100 mg, 0.250 mmol) in methanol (10 ml) was added p-toluenesulfonylmethyl isocyanide (54 mg, 0.275 mmol) followed by potassium carbonate (104 mg, 0.750 mmol) and the reaction mixture was refuxed for 2 h. Methanol was distilled ou under reduced pressure and reaction mass was quenched with water. The compound was extracted with ethyl acetate (2 x 25 ml), washed with water (2 x 25 ml), brine (25 ml), dried and concentrated to yield the crude product. The compound was purified by silica gel column chromatography to yield 78 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI ) δ 5.43 (s, 2H), 6.65 (s, 1H), 6.93 (d, J = 8.1 Hz, 2H), 7.05-7.10 (m, 5H), 7.20 (d, J = 8.4 Hz: 2H); 7.57 (l, J =1.2 Hz, 1H), 7.71-7.78 (m, 2H), 7.83-7.89 (m, 2M), 8.10 (d, J= 8.4 Hz, 111), 8.24 (d, J= 8.1 Hz, 1H); APC1-MS {m/z) 439 (M+H)+.
Example 111
2-({4-|2-(4-Chlorophenyl)-l-(l -methyl- lH-pyrazol-5-yl)ethenyl]phenoxy}methyl)qu

Step \: 2-({4-[2-(4-chlorophenyl)-l-(lH-pyrazol-5-yl)ethenyl]phenoxy}methyl)quinoline: To the well stirred suspension of sodium hydride (100 mg, 2.503 mmol) in dry THF (1 ml) was added a solution of diethyl (2-{2-[(4-methylphenyl)sulfonyl]hydrazinylidene}ethyl)phosphonate
(653 mg, 1 .877 mmol) in THF at 0 °C and the reaction mixture was stirred for 30 minutes at the same temperature. A solution of Intermediate 41 (500 mg, 1 .25 1 mmol) in THF was added to the reaction mixture and it was stirred at room temperature for 1 h. The reaction was quenched with water, neutralized with dilute HCl and extracted with ethyl acetate. The combined organic layers were washed with water, brine, dried and concentrated to yield the crude product. The compound was purified by silica gel column chromatography to yield 595 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.52 (s, 2H), 6.35 (s, 1 H), 6.93-7.01 (m, 2H), 7.05-7. 1 1 (m, 5H), 7.20 (d, J = 8.4 Hz, 3H), 7.53 (br s, 1 H), 7.62 (br s, 1 H), 7.79 (d, = 7.8 Hz, 2H), 7.90 (d, J = 8.1 Hz, 3H), 8.23-8.35 (m, 2H).
Step 2: 2-({4-[2-(4-Chlorophenyl)- l -( l -methyl- l /-pyrazol-5-yl)ethenyl]phenoxy} methyl) quinoline:
To the well stirred solution of Step 1 Intermediate (590 mg, 1.348 mmol) in DMF (20 ml) were added cesium carbonate (527 mg, 1 .618 mmol) followed by methyl iodide (0.21 ml, 3.371 mmol) and the reaction mixture was stirred at room temperature for 16 h. The reaction was quenched with water (50 ml) and extracted with ethyl acetate (2 x 50 ml). The combined organic layers were washed with water (2 x 50 ml), brine (50 ml), dried and concentrated to yield the regioisomeric mixture of products. These regioisomers were separated by silica gel column chromatography and 29 mg of the above less polar isomer was ejuted out in 25 % ethyl acetate in petroleum ether solution as off-white solid; Ή NMR (300 MHz, DMSO-i 6) δ 3.81 (s, 3H), 5.40 (s, 2H), 6.13 (s, 1 H), 6.96 (d, J = 7.8 Hz, 2H), 7.10-7.18 (m, 6H), 7.64 (br s, 1 H), 7.71 -7.80 (m, 2H), 8,01 (br s, 2H), 8.46 (d, J = 7.8 Hz, 1H); ESI-MS (m/z) 452 (M+H)+.
Example 1 1 2
2-({4-[2-(4-Chlorophenyl)- l -(l -methyl- l H-pyrazol-3-yl)ethenyl]phenoxy }methyl)quinoline
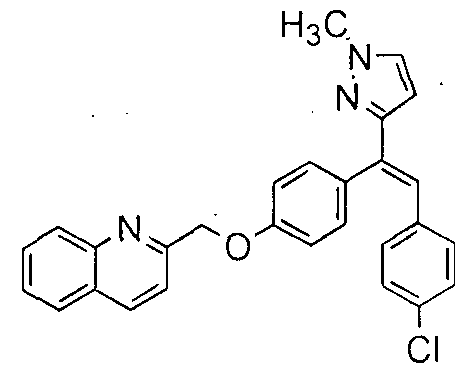
The title compound was the more polar isomer isolated from Example 1 1 1 and was eluted out in
30 % ethyl acetate in petroleum ether solution to give 15 mg of off-white solid; Ή NMR (300
MHz, DMSO-i¾ δ 3.51 (s, 3H), 5.38 (s, 2H), 6.22 (s, Hi), 6.83 (s, 1H), 7.08 (s, 4H), 7.12 (s, 2H), 7.25 (d, J= 8.4 Hz, 2H), 7.40 (s, 1H), 7.63 (t, J= 7.2 Hz, 1H), 7.70 (d, J - 8.4 Hz, 111). 7.79 (t, J =1.5 Hz, 1H), 8.01 (d, J= 8.4 Hz, 2H), 8.44 (d, J= 8.4 Hz, 1H); ES1-MS (m/z) 452 (.V1 -II)'.
Example 113
2-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)vinyl]phenoxy}methyl)imidazo| 1 ,2-a] pyridine

To a well stirred solution of Intermediate 47 (200 mg, 0.477 mmol) in triethyl orthoformate (10 ml) was added p-toluenesulfonic acid (PTSA) (18 mg, 0.095 mmol) and the reaction mixture was heated to 90-100 °C for 3 h. The reaction mixture was diluted with water (10 ml) and extracted with ethyl acetate (25 ml x 2). The combined organic layers were washed with water (20 ml x 2) and brine (15 ml), dried over anhydrous Na2S04 and concentrated to give crude product. The crude product was purified by silica gel column chromatography using 0.5 to 1% methanol in chloroform to yield 25 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.33 (s, 2H), 6.82 (t, J= 6.3 Hz, 1H), 7.07 (d, J= 9.6 Hz, 4H), 7.17 (d, J= 8.7 Hz, 2H), 7.21-7.28 (m, 3H), 7.59-7.68 (m, 3H), 8.12 (d, J= 6.9 Hz, 1H), 8.39 (s, 1H); ESI ( /z) 429 (M+H)+.
Example 114
2-({4-[2-(4-Chlorophenyl)-l-(l , 3, 4-oxadiazol-2-yl)ethenyl]phenoxy} methyl)-! ,3-benzothiazole

The title compound was prepared from Intermediate 48 (250 mg, 0.574 mmol) and excess of triethyl orthoformate (7 ml) as described in Example 113 to yield 70 mg of product as off-white
solid; Ή NMR (300 MHz, CDC13) δ 5.53 (s, 2H), 7.03 (d, J = 8.4 Hz, 2H), 7.12 (q, J = 8.4 Hz, 4H): 7.30 (br s, 2H), 7.43 (t, J= 7.5 Hz, 1H), 7.54 (t, J= 7.2 Hz, 1H), 7.64 (s, 1H), 7.93 (d, J = 8.1 Hz, 1H), 8.05 (d,J = 7.8 Hz, 1H), 8.39 (s, lH); ESI-MS (m/∑) 446 (M+H)+.
Example 115
3-({4 2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l -methyl- 1/-/- indazole

The title compound was prepared from Intermediate 49 (300 mg, 0.693 mmol) and excess of tri ethyl orthoformate (7 ml) as described in Example 113 to yield 25 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.09 (s, 3H), 5.47 (s, 2H), 7.03 (d, J = 8.1 Hz, 2H), 7.11- 7.16 (m, 4H), 7.19-7.25 (m, 3H), 7.42 (br s, 2H), 7.63 (s, 1H), 7.86 (d, J= 8.4 Hz, 1 H), 8.38 (s, ! H); APCI-MS (m/z) 443 ( +H).
Example 116
2-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2- l ethen l]phenoxy}methyl)pyridine

The title compound was prepared from Intermediate 50 (200 mg, 0.527 mmol) and excess of triethyl orthoformate (7 ml) as described in Example 113 to give 56 mg of the product as off- white solid; Ή NMR (300 MHz, CDCI3) δ 5.25 (s, 2H), 7.05 (ds J= 8.4 Hz, 4H)S 7.17 (d, ./ = 8.1 Hz, 2H), 7.24 (br s, 3H), 7.56 (d, J= 7.8 Hz, 1H), 7.63 (s, 1H), 7.75 (t, J= 7.2 Hz, lH), 8.39 (s, l.H), 8.61 (d, J= 3.9 Hz, 1H); APCI (m/z).390 (M+H)+.
Example 117
5-({4-| -(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}rnethyl)thieno[3,2-6] pyridine

The title compound was prepared from Intermediate 51 (300 mg, 0.688 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 78 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.37 (s, 2H), 7.06 (t, J= 8.7 Hz, 4H), 7.16 (d. J = 8.4 Hz, 2H), 7.24 (br s, 2H), 7.53-7.59 (m, 2H), 7.63 (s, 1H), 7.81 (d, J= 5.4 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.39 (s, 1H); APCI-MS (m/z) 446 (M+H)+.
Example 118
5-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}m

The above compound was prepared from Intermediate 52 (400 mg, 0.953 mmol) and excess of triethyl orthoformate (10 ml) as described in Example 113 to yield 55 mg of the product as off- white solid; Ή NMR (300 MHz, DMSO-i/6) 85.31 (s, 2H), 7.10-7.16 (m, 5H), 7.23-7.32 (m, 4H), 7.53 (d, .7 = 8.1 Hz, 1H), 7.66 (s, 1H), 8.10 (d, J = 8.4 Hz, lH), 8.36 (s, 1H), 9.26 (s; 1 H); APC.1-MS (m/z) 430 (M+H)+.
Example 119
5-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-3/-/-pyrrolo[3,2-6] pyridine

Step 1 : 5-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l-[(4- methylphenyl)sulfonyl]-lH-pyrrolo[3,2-0]pyridine:
To the well stirred solution of Intermediate 46 (151 mg, 0.189 mmol) in DMF (20 ml) were added cesium carbonate (246 mg, 0.953 mmol) and {l-[(4-methy'lphenyl)sulfonyl]-lH- pyrrolo|"3,2- 7]pyridin-5-yl}methyl 4-methyl benzenesulfonate (290 mg, 0.635 mmol) and the reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with water (20 ml) and extracted with ethyl acetate (2 x 20 ml), washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 150 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.36 (s, 3H), 5.28 (s, 2H), 6.87 (br s, 1 H), 7.04 (d, J = 8.4 Hz. 4H), 7.16 (d, J= 8.1 Hz, 2H), 7.22-7.28 (m, 4H), 7.52 (d,J= 8.4 Hz, lH), 7.63 (s, 1H).7.77 (d, ./ = 7.8 Hz, 2H), 7.83 (br s, 1H), 8.31 (d, .7= 8.1 Hz, 1H), 8.39 (s, 1H).
Step 2: 5-({4-|~2-(4-Chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l/-/-pyrrolo [3,2-/)]pyridine:
To the well stirred solution of Step 1 Intermediate (150 mg, 0.349 mmol) in methanol (15 ml) was added aqueous sodium hydroxide (140 mg, 3.49 mmol) and the reaction mixture was stirred at room temperature for 15 min. The reaction mixture was diluted with water (20 ml) and extracted with ethyl acetate (2 x 20 ml), washed with water and brine, dried over anhydrous Na2SOi and concentrated to yield 50 mg of the product as off-white solid; Ή NMR (300 MHz, CDCI3) δ 5.36 (s, 21-1), 6.76 (s, 1H), 7.05 (t, J = 9 Hz, 4H), 7.16 (d, J = 8.4 Hz, 2H), 7.22 (br s, 21-1), 7.38 (d, ,7= 8.1 Hz, 1H), 7.48 (br s, 1H), 7.62 (s, 1H), 7.72 (br s, 1H), 8.39 (s, 1H), 8.55 (br s, 1H); ES1-MS (m/z) 429 (M+H)+.
Example 120
5-({4-|2-(4-Chlorophenyl)-l -(1 ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l -methyl-] 77- pyiTo]o[3;2-6]pyridine

The title compound was prepared from Intermediate 53 (200 mg, 0.462 mmol) and excees of triethyl orthoformate (10 ml) as described in Example 113 to yield 70 mg of the product as off- white solid; Ή NMR (300 MHz. DMSO-i¾ δ 3.83 (s, 3H), 5.28 (s, 2H), 6.55 (s, 1H), 7.09-7.16 (m, 4H), 7.22 (d, J= 8.1 Hz, 2H), 7.27-7.35 (m, 3H), 7.65 (s, 2H), 7.92 (d, J= 8.4 Hz, Hi), 9.22 (s.11-1); ESI-MS (m/z) 443 (M+H)+.
Example 121
5-({4-|"2-(4-Chlorophenyl)-l-(l ,3, 4-oxadiazol-2-yl)ethenyl]phenoxy} methyl)-! H-indazole
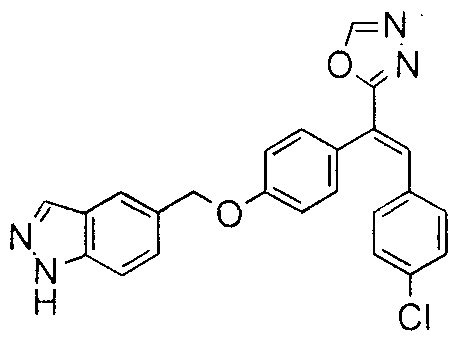
Step 1 : /e/7-Butyl 5-({4-[2-(4-chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy} methyl)- 1 H-indazole- 1 -carboxylate:
To the well stirred solution of Intermediate 46 (250 mg.0.830 mmol) in TMF (1 ml) were added triphenylphosphine (330 mg, 1.25 mmol) and /gr/-butyl 5-(hydroxymethyl)-l H-indazole- 1- carboxylate (208 mg, 0.830 mmol). The reaction was stirred at room temperature for 15 mins followed by drop wise addition of diethylazodicarboxylate (0.19 ml, 1.25 mmol) and the reaction mixture was further stirred for 16 h. The excess solvent was distilled under reduced pressure and purified by silica gel column chromatography to yield 160 mg of the product as oft-white solid;
Ή NMR (300 MHz, DMSO-<¾ δ 1.64 (s, 9H), 5.38 (s, 2H), 7.10-7.17 (m, 3H), 7.24-7.30 (m, 3H), 7.47 (d, J= 7.8 Hz, lH), 7.66 (s, lH), 7.91 (d, J= 8.4 Hz, 1H), 8.22 (s, 1H), 8.42 (s, 1H): 8.99 (s, 1H), 9.26 (s, 1H).
Step 2: 5-({4-[2-(4-Chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}mefhyl)- 1 H- indazole:
To a well stirred solution of the Step 1 Intermediate (180 mg, 0.340 mmol) in dichloromethane (3 ml) was added trifluoroacetic acid (3 ml) at 0 °C and the reaction was continued for 4 h. The reaction mixture was diluted with water and neutralized with sodium bicarbonate. The aqueous layer was extracted with ethyl acetate (3 x 50 ml), washed with water and brine, dried over anhydrous Na2S04 and concentrated under reduced pressure to yield the crude product. The product was purified by silica gel column chromatography to yield 51 mg of an off-white salt; Ή
NMR (300 MHz, OMSO-d6) δ 5.26 (s, 2H), 7.07-7.12 (m, 5H), 7.18-7.25 (m54H), 7.62 (s, 2H). 7.78 (d, .7= 8.1 Hz, 1H), 8.06 (s, 1H), 9.13 (s, l H); APCI-MS (m/z) 429 (M+H)"\ xample 122
5-({4-[2-(4-Chlorophenyl)-l -(1, 3, 4-oxadiazol-2-yl)ethenyl]phenoxy} methyl)-! -methyl- 1/7- indazole

The title compound was prepared from Intermediate 46 (315 mg, 1.055 mmol) and (1-methyl- l/-benziinidazol-5-yl)methanol (190 mg, 1.172 mmol) as described in Step 1 of Example 121 to yield 39 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 3.88 (s, 3H), 5.24 (s, 2H), 7.02-7.07 (m, 4H), 7.12-7.18 (m, 2H), 7.22 (br s, 2H), 7.44 (s, 2H), 7.63 (s, 1H), 7.90 (d, ./ = 8.7 Hz, 2H), 8.38 (s, 1H); APCI-MS (m/z) 443 (M+H)+.
Example 123
5-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl] phenoxy} ethyl)| 1 ,3]oxazolo [4,5-6' pyridine

Example 124
5-({4-[2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)pyrazolo| ~\,5-a] pyrimidine

The title compound was prepared from Intermediate 54 (400 mg, 0.953 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to yield 40 mg of the product as o f- white solid; Ή NMR (300 MHz, CDC13) δ 5.25 (s, 2H), 6.68 (s, 1H), 7.05 (d, J= 7.5 Hz, 4H), 7.12-7.18 (m, 3H), 7.29 (br s, 2H), 7.64 (s, 1H), 8.16 (s, 1H), 8.39 (s, 1H), 8.72 (d, .1=7.2 Hz, lH); APCI-MS (m/z) 430 (M+H)+.
Example 125
2-{2-(4-Chloropheny])-l-[4-(naphthalen-2-ylmethoxy)phenyl]ethenyl}-l ,3,4-oxadiazole

The title compound was prepared from Intermediate 46 (150 mg, 0.502 mmol) and 2- (chloromethyl)napthalene (97 mg, 0.552 mmol) as described in Example 82 to yield 100 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.28 (s, 2H), 7.03-7.09 (m, 3H), 7.i 1-7.17 (m, 3H), 7.24 (br s, 2H), 7.49-7.58 (m, 3H), 7.64 (s, 1H), 7.85-7.91 (m, 4H), 8.39 (s, 1 H); APCI-MS (m/z) 439 (M+H)+.
Example 126
2-({4-|Ί-(1 ,3,4-Oxadiazol-2-yl)-2-(thiophen-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 55 (300 mg, 0.747 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 40 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.46 (s, 2H), 6.95 (br s, 1H), 7.17-7.22 (m, 4H), 7.29- 7.32 (m, 2H), 7.57 (t, J= 7.8 Hz, 1H), 7.74 (d, J= 8.4 Hz, 2H), 7.86 (d, J - 7.8 Hz, 1H), 7.92 (s; IH), 8.10 (d, J = 8.1 Hz, 1H), 8.24 (d, J= 8.4 Hz, 1H), 8.35 (s, 1H); APCI-MS (m/z) 412
(M+H)+.
Example 127
2-({4-[l-(l ,3,4-Oxadiazol-2-yl)-2-(thiophen-3-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 56 (250 mg, 0.622 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 65 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.45 (s, 2H), 6.58 (d, .7= 5.1 Hz, IH), 7.10-7.15 (m, 4H), 7.29-7.35 (m, 3H), 7.57 (t, J= 7.2 Hz, IH), 7.72-7.78 (m, 2H), 7.86 (d, J = 8.4 Hz, IH), 8.09 (d,J= 8.4 Hz, IH), 8.24 (d,J= 8.4 Hz, IH), 8.36 (s, IH); APCI-MS (m/z) 412 (M+H)+.
Example 128
2-({4-[2-(Furan-2-yl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 57 (400 mg, 1.038 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 200 mg of the product as off- white solid; Ή NMR (300 MHz, DMSO-¾) δ 5.46 (s, 2H), 5.95 (d, J= 3.3 Hz, 1H), 6.30 (br s, 1H), 7.13 (d, J= 8.4 Hz, 2H), 7.31-7.38 (m, 3H), 7.54-7.60 (m, 2H), 7.72-7.78 (m, 2H), 7.85 (d, J - 8.4 Hz, 1H), 8.10 (d, J = 9.0 Hz, 1H), 8.24 (d, J= 8.1 Hz, 1H), 8.36 (s, l H); ESI-MS (m/z 396 (M+H)+.
Example 129
2-({4-[2-(Furan-3-yl)-l-(l ,3,4-oxadiazol-2- l ethen l henoxy}methyl)quinoline

The title compound was prepared from Intermediate 58 (450 mg, 1.168 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 150 mg of (he product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.42 (s, 2H), 5.71 (s, 1H), 7.11-7.20 (m, 3H), 7.28 (t. J= 8.7 Hz, 2H), 7.40 (s, 1H), 7.53-7.60 (m, 2H), 7.69-7.78 (m, 2H), 7.85 (d, J - 8.4 Hz, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.23 (d, J= 8.4 Hz, 1H), 8.34 (s, 1H); APCI (m/z) 396 (M+H)+. xample 130
2-({4-| 1 -(1 ,3,4-Oxadiazol-2-yl)-2-(l, -thiazol-5-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 59 (350 mg, 0.870 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 120 mg of the product as off-
white solid; Ή NMR (300 MHz, CDC13) δ 5.46 (s, 2H), 7.29 (q, J= 9.0 Hz, 4H), 7.63 (t, J= 7.5 Hz, IH), 7.73-7.82 (m, 2H), 8.01-8.06 (m, 3H), 8.28 (s, 1H), 8.46 (d, J = 8.4 Hz, 1H), 8.91 (s, 1 H), 9.24 (ss 1 H); ESI-MS (m/z) 413 (M+H)+.
Example 131
2-({4-[2-(2-ChloiO-l,3-thiazol-5-yl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl) quinoline

The title compound was prepared from Intermediate 60 (650 mg, 1.524 mmol) and excess of tri ethyl orthoformate (5 ml) as described in Example 113 to give 250 mg of the product as off- white solid; 'HNMR (300 MHz, DMSO-<¾) δ 5.46 (s, 2H), 7.25 (d, J= 8.4 Hz, 2H), 7.33 (d, = 8.4 Hz, 2H), 7.63 (t, J= 7.5 Hz, 1H), 7.72-7.82 (m, 3H), 8.02 (t, J= 7.2 Hz, 2H), 8.46 (d, J= 8.1 Hz, IH), 8.98 (s, ll-I), 8.27 (s, 1H); ESI-MS (m/z) 447 (M+H)+.
Example 132
2-({4-[l ,-(l,3,4-Oxadiazol-2-yl)-2-phenylethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 61 (300 mg, 0.759 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 120 mg of the product as off- white solid; Ή NMR (300 MHz, CDCI3) δ 5.42 (s, 2H), 7.06-7.13 (m, 4H), 7.19-7.29 (m, 5H), 7.56 (t, J = 7.8 Hz, 1 H), 7.68-7.77 (m, 3H), 7.85 (d, J = 8.4 Hz, 1H), 8.09 (d, J = 8.4 Hz, IH), 8.23 (d, J= 9.0 Hz, 1H), 8.38 (s, IH); ESI-MS (m/z) 406 (M+H)+.
Example 133
2-({2-Chloro-4-[l -(1 ,3,4-oxadiazol-2-yl)-2-phenylethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 62 (300 mg, 0.698 mmol) and excess of triethyl orthoformate as described in Example 113 to give 70 mg of the product as off-white solid: Ή NMR (300 MHz, CDC13) δ 5.50 (s, 2H), 7.06-7.14 (m, 3H), 7.17-7.26 (m, 3H), 7.43 (s, lH), 7.57 (t, ,7= 7.2 Hz, 1H), 7.70 (s, 1H), 7.72-7.78 (m, 2H), 7.84 (d, J= 8.7 Hz, 2H), 8.07 (d, J = 8.7 Hz, lH), 8.25 (d, = 8.1 Hz, 1H), 8.39 (s, lH); APC1-MS (m/z) 440 (M+H)+.
Example 134
2-({4-[2-(3-Chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 63 (300 mg, 0.698 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 26 mg o the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 6.97 (d, J= 7.8 Hz, IH), 7.07-7.13 (m, 4H), 7.16-7.22 (m, 3H), 7.54-7.61 (m, 2H), 7.68-7.77 (m, 2H), 7.85 (d, J= 7.8 Hz, H i), 8.08 (d, ./= 8.1 Hz, 1H), 8.22 (d, J= 9.0 Hz, 1H), 8.39 (s, 1H); APCI-MS (m/z) 440 (M)+.
Example 135
2-({4-|2-(4-Chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

Example 136
2-(|4-|"2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2-y])ethenyl]-2-methoxyphenoxy}methyl) quinoline

The title compound was prepared from Intermediate 65 (300 mg, 0.652 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 35 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 3.84 (s, 3H), 5.51 (s, 2H), 6.79-6.85 (m, 2H), 6.97- 7.04 (m, 3H), 7.15 (d, J= 8.4 Hz, 2H), 7.53-7.62 (m, 2H), 7.74 (d, J = 8.4 Hz, 2H)S 7.85 (d, J = 7.8 Hz, 1H), 8.07 (d, J= 8.7 Hz, lH), 8.22 (d, J= 8.7 Hz, 1H), 8.39 (s, 1H); ESI-MS (m/z) 470 (M+H)+.
Example 137
2-({4-|"2-(4-Fluorophenyl)-l-(l,3,4-oxadiazol-2-y])ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 66 (200 mg, 0.480 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to yield 152 mg of the product as white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 6.88 (d, ./= 8.4 Hz, 2H), 7.09 (d, .7 = 8.1 Hz,
4H), 7.28 (br s, 2H), 7.57 (t, J = 7.5 Ηζ,.ΙΗ), 7.65 (s, 1H), 7.69-7.77 (m, 2H): 7.85 (d, ./ = 8.1
Hz, IH), 8.09 (d, J= 8.4 Hz, IH), 8.23 (d, J = 8.1 Hz, 1H), 8.37 (s, 1H); APCI- S (m/z) 424 ( +H)+.
Example 138
2-({4-[l-(l .3,4-Oxadiazol-2-yl) 2-(pyridin-3-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 67 (500 mg. 1.262 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 81 mg of the product as off- white solid; Ή NMR (300 MHz, DMSO-i¾ δ 5.42 (s, 2H), 7.19 (d, J= 8.7 Hz, 2H), 7.22-7.29 (m, 3H), 7.40 (d: J = 8,4 Hz, IH), 7.65 (t, J= 7.2 Hz, IH), 7.71-7,82 (m, 3H), 8.03 (t, ,/= 7.8 Hz, 2H), 8.41-8.47 (m, 3H), 9.28 (s, IH); ESI-MS (m/z) 407 (M+H)+.
Example 139
2-({4- 2-(6-Methoxypyridin-3-yl)-l- -oxadiazol-2-yl)ethenyl]phenoxy}methyl) quinoline

The title compound was prepared from Intermediate 68 (180 mg. 0.422 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 80 mg of the product as off- white solid; Ή NMR (300 MHz, DMSO-c¾ δ 3.81 (s, 3H), 5.43 (s, 2H); 6.65 (d, J = 8.7 Hz; IH), 7.16-7.29 (m, 5H), 7.60-7.66 (m, 2H), 7.71-7.82 (m, 2H), 8.02 (t, J= 6.3 Hz, 2H), 8.10 (s, I H), 8.45 (d, J= 8.4 Hz, IH), 9.23 (s, IH); ESI-MS (m/z) 437 (M+H)+.
Example 140
2-({4-[2-(6-Ethoxypyndin-3-yl)-l-(l,3,4-oxadiazo]-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 69 (500 mg, 1.10 mmol) and excess of triethyl orthoformate (10 ml) as described in Example 113 to yield 105 mg of the product as off- white solid; Ή N R (300 MHz, CDC13) δ 1.36 (t, J= 6.9 Hz, 3H), 4.32 (q, J = 6.9 Hz, 2H), 5.42 (s, 2H), 6,47 (d, J = 8.7 Hz, 1H), 7.09-7.14 (m, 3H), 7.29 (br s, 2H), 7.54-7.60 (m, 2H), 7.69-7.77 (m.2H), 7.85 (d, J= 7.8 Hz, 1H), 8.04-8.10 (m, 2H), 8.23 (d, J= 8.4 Hz, 1H), 8.36 (s: 1 H); ESl-MS ( /z) 451 (M+H)+.
Example 141
2-({4-[l-(l,3,4-Oxadiazol-2-yl)-2-(pyridin-4-yl)ethenyl]phenoxy}methyl)qi

The title compound was prepared from Intermediate 70 (200 mg. 0.505 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to yield 163 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 6.96 (d, J = 6 Hz, 2H), 7.09 (d, J = 6.9 Hz, 2H), 7.24 (d, J = 6.5 Hz, 2H), 7.54-7.59 (m, 2H), 7.68-7.76 (m, 2H), 7.85 (d, J - 7.5 Hz. 11-1), 8.09 (d, J= 8.4 Hz, 1H), 8.23 (d, J= 8.4 Hz, 1H), 8.40-8.46 (m, 3H); APCI-MS (m/z) 407 (M+H)+.
Example 142
2-({2-Fluoro-4-[l-(l,3,4-oxadiazol-2- -2-(pyridin-4-yl)ethenyl]phenoxy}methyl)quinoline

Example 143
2-({4-|"l-(l ,3,4-Oxadiazol-2-yl)-2-(l -oxidopyridin-4-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 72 (300 mg, 0.724 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 40 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.47 (s, 2H), 6.95 (d, J = 6.3 Hz, 2H), 7.14 (cL J= 8.4 Hz, 2H), 7.29 (s, 2H), 7.53 (s, 1H), 7.59 (t, J = 7.2 Hz, 1H), 7.71-7.80 (m, 2H), 7.88 (d, J= 7.8 Hz, IH), 7.97 (d, J= 6.6 Hz, 2H), 8.14 (d, J= 7.8 Hz, 1H), 8.27 (d. J= 8.4 Hz, 1H), 8.42 (s, lH); APC1-MS (m/z) 423 (M+H)+.
Example 144
2-({4-|2-(4-Methoxyphenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 73 (200 mg, 0.470 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to yield 15 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 3.77 (s, 3H), 5.43 (s, 2H), 6.72 (d, J = 8.4 Hz, 2H), 7.04-7.13 (m, 4H), 7.30 (br s, 2H), 7.57 (t, J= 7.5 Hz, 1H), 7.70-7.78 (m, 2H), 7.85 (d, J = 7.8
Hz, 1H), 8.09 (d, J = 8.4 Hz, 1H), 8.24 (d, J = 8.1 Hz, 1H), 8.35 (s, 1H); ES1-MS (TW/Z) 436 (VI ill)'.
Example 145
2-({4-[2-|4-(DifluoiOmethoxy)phenyl]-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl) quinoline
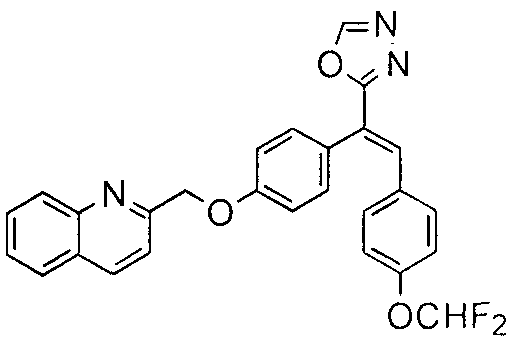
The title compound was prepared from the Intermediate 74 (400 mg, 0.898 mmol) and excess of triethyl orfhoformate (5 ml) as described in Example 113 to give 105 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 6.48 (t, J = 73.2 Hz, lH), 6.93 (d, J = 8.4 Hz, 2H), 7.08-7.13 (m, 4H), 7.28 (br s, 2H), 7.57 (t, J= 7.2 Hz, 1H), 7.65 (s, 1 I ), 7.70-7.78 (m, 2H), 7.86 (d, J = 7.8 Hz, 1H), 8.09 (d, J= 8.4 Hz, 1H), 8.23 (d, J = 8.4 Hz, 1 H), 8.38 (s, 1 H); ES1-MS (m/z) 472 (M+H)+.
Example 146
2-|"(4-{ 1 -(l,3,4-Oxadiazol-2-yl)-2-[4-(trifluo)Omethoxy)phenyl]ethenyl}phenoxy)methyl quinoline
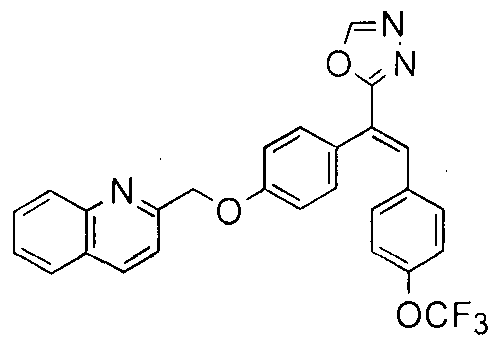
The title compound was prepared from Intermediate 75 (400 mg, 0.835 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 58 mg of the product as off- white solid; Ή NMR (300 MHz, DMSO-i¾) δ 5.43 (s, 2H), 7.16 (d, .7= 8.1 Hz, 2H), 7.20-7.28 (m, 611).7.63 (t, J= 7.2 Hz, 1H), 7.70-7.82 (m, 3H), 8.02 (t, J= 7.2 Hz, 2H), 8.45 (d, J - 8.1 Hz, IH), 9.27 (s, 1H); ES1-MS (m/z) 490 (M+H)+.
Example 147
2-| 4-{l-(l,3,4-Oxadiazol-2-yl)-2-[4-(trifluoromethyl)phenyl]elhenyl}phenoxy)methy]] quinoline
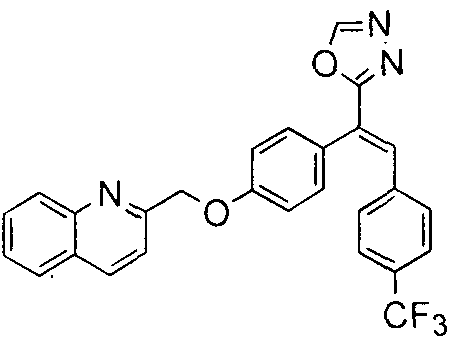
The title compound was prepared from Intermediate 76 (300 mg, 0.647 mmol) and excess of tri ethyl orthoformate (5 ml) as described in Example 113 to give 40 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 7.09 (d, J = 8.4 Hz, 2H); 7.20-7.24 (m, 4H), 7.44 (d, J = 7.8 Hz, 2H), 7.57 (t, J= 7.2 Hz, 1H), 7.69-7.77 (m, 3H), 7.85 (d, J = 7.8 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.23 (d,*J = 8.7 Hz, 1H), 8.41 (s, 1H); APCI-MS (m/z) 474 (M+H)+.
Example 148
2-( {4- 12-(2,4-Di fluorophenyl)-! -(l,3,4-oxadiazol-2-yl)elhenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 77 (280 mg, 0.640 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 40 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.41 (s, 2H), 6.57 (t, J = 8.4 Hz, 1H), 6.84 (q, .7 - 9.0 Hz, 2H), 7.07 (d, J = 8.1 Hz, 2H), 7.27 (br s, 2H), 7.59 (t, .7 = 7.8 Hz, 1H), 7.67-7.78 (m.3H), 7.85 (d, J= 8.4 Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.22 (d, J= 8.7 Hz, 1H), 8.40 (s, 1H); ESl-MS (m/z) 442 (M+H)+.
Example 149
2-({4-[2-(4-Chloro-2-fluoropheny])-l -(1 ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 78 (350 mg, 0.780 mmol) and excess of tri ethyl orthoformate (5 ml) as described in Example 113 to yield 95 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.41 (s, 2H), 6.75-6.81 (m, 2H), 7.03-7.09 (m, 311). 7.24 (br s, 2H), 7.57 (t, J =1.5 Hz, IH), 7.68-7.77 (m, 3H), 7.85 (d5.7= 8.411/. ! IT).8.08 (d, J = 8.4 Hz, 1H), 8.22 (d, .7=8.1 Hz, 1H), 8.41 (s, 1H); ESI-MS (m/z) 458 (M+H)".
Example 150
2-({4-[2-(4-Chloro-3-fluorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 79 (300 mg, 0.670 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 75 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.45 (s, 2H), 6.83-6.89 (m, 2H), 7.10 (d, J = 9.0 Hz, 21-1), 7.19-7.26 (m, 3H), 7.53-7.59 (m, 2H), 7.69-7.78 (m, 2H), 7.86 (d, .7= 8.1 Hz, 1 IT), 8.10 (d, J= 8.4 Hz, IH), 8.24 (d, .7= 8.1 Hz, 1H), 8.40 (s, 1H); ESI-MS (m/z) 458 (M+H)+.
Example 151
2-({4-[2-(3,4-Difluorophenyl)-l-(l,3 -oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

Example 152
2-({3- 2-(4-Chlorophenyl)-l -(1 ,3,4-oxadiazol-2-yl)ethenyl]phenoxy} methyl)quinol ine

The title compound was prepared from Intermediate 8 1 (250 mg, 0.580 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 1 13 to give 103 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 5.36 (s, 2H), 6.92-6.97 (m, 4H), 7.07-7.12 (m, 3H), 7.37 (I, J = 7.8 Hz, 1 H), 7.53-7.65 (m, 3H), 7.73 (t, J = 7.5 Hz, 1 H), 7.84 (d, J = 8.4 Hz, I H), 8.03 (d, .7 = 8.4 Hz, 1 H), 8. 17 (d, J = 8.4 Hz, 1 H), 8.3 1 (s, 1 H); ESI-MS {m/z) 440 (M+H)+
Example 153
2-({4-[ 2-(4-Chlorophenyl)- l -(l ,2,4-oxadiazol-5-yl)ethenyl]phenoxy}methyl)quinoline

Step 1 : 3-(4-Ch]orophenyl)-N-[(Z)-(dimethylamino)methylidene]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2-enamide:
The solution of Example 1 (400 mg, 0.966 mmol) in N, TV-dim ethyl formamide dimethyl acetal (4 ml) was stirred at 120- 130 °C for 3 h. The reaction mixture was concentrated to yield 456 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) 6 3.1 1 (s, 3H), 3.14 (s, 3H), 5:40 (s, 2H),
6.98-7.03 (m, 4H), 7.08-7.15 (m, 4H), 7.55 (t, J= 6.9 Hz, lH), l.lQ-Ί.Ίβ (m, 2H), 7.84 (d, J = 8.4 Hz, 1H), 8 3 (s, 2H), 8.21 (d, J= 8.4 Hz, 1H), 8.46 (s, 1H).
Step 2: 2-({4- 2-(4-Chlorophenyl)-l-(l ,2,4-oxadiazol-5-yl)ethenyl]phenoxy}methyl)quinoline: To the well stirred solution of Step 1 Intermediate (450 mg, 0.958 mmol) in dioxane (5 ml) were added hydroxylamine hydrochloride (134 mg, 1.916 mmol), aqueous sodium hydroxide (5 ml) and acetic acid (4 ml) and the reaction mixture was stirred for 4 h at 110 °C. The reaction mixture was quenched with water (20 ml), neutralized by sodium bicarbonate and was extracted with ethyl acetate (50 ml x 2). The combined organic layers were washed with water and brine, dried over anhydrous Na2S04 and concentrated to yield 24 mg of product as white solid; Ή NMR (300 MHz, CDC13) δ 5.43 (s, 2H), 7.05-7.18 (m, 6H), 7.23 (br s, 2H), 7.57 (t, J = 7.2 Hz, IH), 7.70-7.78 (m, 2H), 7.82-7.87 (m, 2H), 8.09 (d, J= 8.4 Hz, 1 H), 8.23 (d, ./ = 8.1 Hz, 1H); 8.39 (s, 1 H); ESl-MS (m/z) 440 (M+H)+.
Example 154
2-({4-[ l-(5-Methyl'l,3,4-oxadiazol-2-yl)-2-(4-chlorophenyl)ethenyl]phenoxy}methyl)imidazo 11 ,2-a]pyridine

The title compound was prepared from Intermediate 47 (300 mg, 0.710 mmol) and triethyl orthoacetate (15 ml) as described in Example 113 to yield 183 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-<6) 62.53 (s, 3H), 5.24 (s, 2H), 6.09 (I, J= 6.9 Hz, 111).7.10- 7.16 (m, 4H), 7.23 (d, J = 8.7 Hz, 3H), 7.30 (d, J= 8.4 Hz, 2H), 7.55 (d, .7= 9.6 Hz, 2H), 8.03 (s, 1 H), 8.55 (d, J = 9.0 Hz, 1H); APCI-MS (m/z) 443 (M+H)+.
Example 155
2-({4-| -(4-Chlorophenyl)-l-(5-methyl-l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)pyridine

The title compound was prepared from Intermediate 50 (200 mg, 0.527 mmol) and triethyl orthoacetate (7 ml) as described in Example 1 13 to give 86 mg of the product as off-whi le solid; Ή NMR (300 MHz, CDC13) δ 2.57 (s, 3H), 5.24 (s, 2H), 7.00-7.07 (m, 4H), 7. 1 5 (d, .7 = 8. 1 H z, 2H), 7.23 (br s, 3H), 7.52-7.59 (m, 2H), 7.75 (t, J = 7.5 Hz, 1 H), 8.67 (d, J = 3.9 Hz, 1 H); APC1 (m/z) 404 (M+H)+.
Example 156
2-({4-[ l -(5-Methyl-l ,3,4-oxadiazol-2- -2-(thiophen-2-yl)ethenyl]phenoxy}methyl)q

The title compound was prepared from Intermediate 55 (300 mg, 0.747 mmol) and triethvi orthoacetate (5 ml) as described in Example 1 13 to give 45 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.55 (s, 3H), 5.46 (s, 2H), 6.93 (br s, 1 H), 7.12-7.19 (m, 4H), 7.27-7.32 (m, 2H), 7.56 (t, J = 7.8 Hz, 1 H), 7.73-7.81 (m, 3H), 7.85 (d, J = 8.4 Hz, 1 H), 8.09 (d, ./ = 8.4 Hz, 1 H), 8.24 (d, J = 8.1 Hz, 1 H); APCI-MS (m/z) 426 (M+H)+.
Example 1 57
2-({4-| 1 -(5-Methyl-l ,3,4-oxadiazol-2- -2-(thiophen-3-yl)ethenyl]phenoxy} methyl)quinoline

The title compound was prepared from Intermediate 56 (300 mg, 0.747 mmol) and triethyl orthoacetate (5 ml) as described in Example 1 13 to give 60 mg of the product as off-white solid;
Ί ΐ NMR (300 MHz, CDC13) δ 2.56 (s, 3H), 5.44 (s, 2H), 6.56 (d, J - 4.5 Hz, 1H), 7.13 (d, .7 = 8.7 HA 4H), 7.30 (br s, 2H), 7.54-7.61 (m, 2H), 7.72-7.78 (m, 2H), 7.86 (d,J= 7.8 Hz, 1H), 8.09 (d, .7= 8.7 Hz, 1H), 8.24 (d, J= 8.4 Hz, 1H); APCI-MS (m/z) 426 (M+H)+.
Example 158
2-({4-[l-(5-Methyl-l,3,4-oxadiazol-2- -2-phenylethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 61 (300 mg, 0.759 mmol) and tri eth l orthoacetate (10 ml) as described in Example 113 to give 90 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.57 (s, 3H), 5.42 (s, 2H), 7.05-7.12 (m, 4H), 7.18 (d, J = 6.0 Hz, 3H), 7.28 (br s, 2H), 7.50-7.58 (m, 2H), 7.69-7.76 (m, 2H), 7.85 (d, J= 8.4 Hz, lH), 8.08 (d, ./= 8.4 Hz, 1H), 8^22 (d, J= 8.1 Hz, 1H); ES1-MS (m/z) 420 (M+H).
Example 159
2-({4-[l -(5-Methyl- 1,3, 4-oxadiazol-2- -2-(4-chlorophenyl)etheny]]phenoxy} methyl )quinoline

The title compound was prepared from Intermediate 64 (250 mg, 0.582 mmol) and triethyl orthoacetate (5 ml) as described in Example 113 to yield 170 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.56 (s, 3H), 5.44 (s, 2H), 7.01 (d, J - 8.4 Hz, 2H), 7.08 (d, .7= 8.7 Hz, 2H), 7.14 (d, .7= 8.4 Hz, 2H), 7.24 (d,J= 8.7 Hz, 2H), 7.52 (s, 1H), 7.58 (t, J= 7.8 Hz, 1H), 7.71-7.78 (m, 2H), 7.86 (d, J= 7.8 Hz, l H), 8.Ί 2 (d, J = 8.4 Hz, 1H), 8.25 (d, J = 8.1 Hz, 1 H); APCI-MS (m/z) 454 (M+H)+.
Example 160
2-({4-| -(5-MethyI-l,3,4-oxadiazol-2- -2-(4-fluorophenyI)ethenyI]phenoxy}methyI)quinoIine

The title compound was prepared from Intermediate 66 (200 mg, 0.480 mmol) and triethyl orthoacetate (5 ml) as described in Example 113 to give 140 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.56 (s, 3H), 5.42 (s, 2H), 6.86 (t, J= 8.4 Hz, 2H), 7.08 (cL J = 8.7 Hz, 4H), 7.25 (d, J= 6.6 Hz, 2H), 7.53-7.60 (m, 2H), 7.69-7.79 (m, 2H), 7.85 (d. J = 7.8 Hz, lH), 8.09 (d, J= 8.4 Hz, 1H), 8.23 (d, J= 8.7 Hz, 1H); APCI-MS (m/z) 438 (M+H)+.
Example 161
2-({4-[l-(5-Methyl-l ,3,4-oxadia lethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 70 (150 mg, 0.378 mmol) and triethyl orthoacetate (5 ml) as described in Example 113 to yield 63 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.59 (s, 3H), 5.43 (s, 2H), 6.93 (d, J= 6.3 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 7.48 (s, 1H), 7.57 (t, J =6.9 Hz, 1H), 7.68-7.78 (m, 2H), 7.85 (d, .7= 8.1 Hz, 1H), 8.09 (d, .7= 8.1 Hz, 1H), 8.23 (d, 7= 8.7 Hz, 1H), 8.43 (d, J= 5.7 Hz, 2H); APCI-MS (m/z) 421 (M+H)+.
Example 162
2-1 4- {]-(5-Methyl-1.3;4-oxadiazol-2-yl)-2-| -(tTifluoromethy])phenyl]ethenyl}phenoxy) methyl]quinoline

The title compound was prepared from Intermediate 76 (300 mg, 0.647 mmol) and trietbyl orthoacetate (10 ml) as described in Example 113 to give 150 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.58 (s, 3H), 5.44 (s, 2H), 7.08 (d, J= 8.1 Hz, 2H), 7.17- 7.23 (m, 4H), 7.42 (d, J = 8.1 Hz, 2H), 7.59 (br s, 2H), 7.70-7.76 (m, 2H), 7.86 (d, J = 7.8 Hz, 1H), 8.10 (d, J= 8.4 Hz, 1H), 8.24 (d, J= 8.4 Hz, 1H); ESI-MS (m/z) 488 (M-Il) .
Example 163
2-({4-[l-(5-{Trifluoromethyl}-l,3,4-oxadiazol-2-yl)-2-pyridin-4-ylelhenyl ]phenoxy}methyl) quinoline

Trifluoroacetic anhydride (0.1 ml, 0.757 mmol) was added drop wise to a cooled solution of Intermediate 70 (200 mg, 0.505 mmol) in dichloroethane (10 ml) and the reaction mixture was stirred at room temperature for 2 h after which it was further refluxed for 12 h. The reaction mixture was diluted with water (10 ml) and basified with saturated sodium bicarbonate solution. The aqueous layer was extracted with chloroform (25 ml x 2). The combined organic layers were washed with water (20 ml x 2) and brine (15 ml), dried over anhydrous Na2S04 and concentrated to yield 45 mg of product as off-white solid; Ή NMR (300 MHz. CDC13) δ 5.43 (s, 2H), 6.97 (cL J= 5.4 Hz, 2H), 7.10 (d, J= 8.7 Hz, 2H), 7.24 (d, J =1.2 Hz, 2H), 7.57 (t, J = 7.2 Hz, 1H): 7.69- 7.78 (m, 3H), 7.86 (d, J= 8.1 Hz, 1H), 8.09 (d, J - 8.4 Hz, Hi), 8.24 (d, J- 8.4 Hz, 1 H), 8.47 (d; J- 4.5 Hz, 2H); APCI-MS (m/z) 475 (M+H)+.
Example 164
2-({4-[2-(4-Chlorophenyl)- l -(5-methyl-4H- l ^ imidazo [ 1 ,2-a]pyridine

To a well stirred solution of Intermediate 47 (400 mg, 0.955 mmol) in ethanol ( 1 5 ml) were added sodium ethoxide (195 mg, 2.865 mmol) and acetamidine hydrochloride (271 mg. 2.865 mmol). The reaction was refluxed for 16 h after which excess of ethanol was distilled under reduced pressure and the reaction mass was diluted with water ( 1 5 ml). The aqueous layer was extracted with ethyl acetate (25 ml x 2) and the combined organic layers were washed with water (1 5 ml x 2) and brine (1 5 ml), dried over anhydrous Na2S04 and concentrated to yield 43 mg of product as white solid; Ή NMR (300 MHz, CDC13) δ 2.36 (s, 3H), 5.22 (s, 2H), 6.90 (t. J = 6.9 Hz, I H), 7.02-7.1 2 (m, 6H), 7.20-7.27 (m, 3H), 7.46-7:55 (m, 2H), 8.02 (s, 1 H)S 8.55 (d, J = 6.6 Hz, 1 H). 1 3.46 (s, I H); APCI (m/z) 442 (M+H)+.
Example 165
2-({4-[2-(4-Chlorophenyl)-l -(5-methyl-4H- l ,2,4-triazol-3-yl)ethenyl]phenoxy}methyl) quinoline

The title compound was prepared from Intermediate 64 (100 mg, 0.224 mmol) and acetamidine hydrochloride (42 mg, 0.448 mmol) as described in Example 164 to yield 68 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 2.57 (s, 3H), 5.42 (s, 2H), 7.01 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 7.23 (br s, 2H), 1.52-1.59 (m, I H), 7.69- 7.77 (m, 2H), 7.86 (d, J = 7.8 Hz, I H), 8.09 (d, J = 8.4 Hz, I H), 8.23 (d, J = 8.4 Hz, I H), 8.30- 8.38 (m, 2H); APCI-MS (m/z) 454 (M+H)+.
Example 166
5-{2-Phenyl-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-l,3,4-oxadiazol-2(3H)-one

Trichloromethyl chloroformate (0.12 ml, 1.01 mmol) was added drop wise to a well stirred and cooled solution of Intermediate 61 (200 mg, 0.506 mmol) in dioxane (10 ml) and was fefluxed- for 12 h. The reaction mixture was diluted with water (15 ml) and extracted with ethyl acetate (20 ml x 3). The combined organic layers were washed with water (15 ml x 2) and brine (15 ml), dried over anhydrous Na2S04, filtered and concentrated to yield 100 mg of off-white solid; Ή NMR (300 MHz, DMSO-c¾ δ 5.41 (s, 2H), 7.10-7.16 (m, 4H), 7.20-7.32 (m, 5H), 7.32 (s, 1H), 7.63 (t, .7- 6.9 Hz, 1H), 7.71 (d, J= 8.7 Hz, 1H), 7.80 (t, J= 7.2 Hz, 1H), 8.00-8.06 (m, 2 ill 8.45 (d, .7 = 8.4 Hz, 1 H), 12.45 (s, 1H); ES1-MS (m/z) 422 (M+H)+.
Example 167
5-{2-(4-Chlorophenyl)-l-[4-(quinoli -2-ylmethoxy)phenyl]ethenyl}-l,3,4-oxadiazol-2(3/7)-one

The title compound was prepared from Intermediate 64 (200 mg, 0.505 mmol) as described in Example 166 to yield 68 mg of the product as off-white solid; Ή NMR (300 MHz, DMSO-c¼) δ 5.41 (s, 2H), 7.07-7.15 (m, 4H), 7.19 -7.72 (m, 4H), 7.31 (s, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.79 (t, J= 7.8 Hz, 1H), 8.04 (br s, 2H), 8.45 (d, J = 8.7 Hz, 1H), 12.46 (s, 1H); APCI-MS (m/z) 456 (M+H)+.
Example 168
5-{2-(4-Chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-l ,3,4-oxadiazol-2(3/-/)- thione

To a well stirred solution of Intermediate 64 (200 mg.0.465 mmol) in ethanol (10 ml) were added aqueous potassium hydroxide (52 mg, 2.931 mmol) followed by carbon bisulfide (107 mg. 1.396 mmol) and the reaction mixture was refluxed for 12 h. The excess of ethanol was distilled under reduced pressure and the reaction mass was diluted with water (15 ml). The solid so obtained was filtered, dissolved in ethyl acetate (30 ml), dried over anhydrous a?C03 and concentrated to yield 130 mg of the off-white solid; Ή NMR (300 MHz, DMSO-<¾ δ 5.42 (s, 2H), 7.14 (d, .7=7.5 Hz, 4H), 7.23-7.30 (m, 5H), 7.49 (s, 1H), 7.63 (t, J= 5.1 Hz, 1H), 7.71 (d, = 8.7 Hz, 1H), 7.80 (t, J = 7.8 Hz, 1H), 8.01 (br s, 2H), 8.45 (d, J= 8.4 Hz, lH); APCI-MS (m/z) 472 (M+H)+. xample 169
5-{2-(4-Chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-2,4-dihydro
triazole-3-thione

To the well stirred solution of Intermediate 64 (300 mg, 0.698 mmol) in dil.HCl (40 ml) was added potassium thiocyanate (340 mg, 3.505 mmol) and the reaction mixture was refluxed overnight. The crude product obtained was cyclized to give the title compound by refluxing it in IM solution of sodium bicarbonate for 24 h. The compound thus obtained was purified by silica gel column chromatography to yield 200 mg of the product as off-white solid; Ή NMR (300
MHz, DMSO-c/fi) δ 5.40 (s, 2H), 6.98 (d, J= 8.4 Hz, 2H), 7.09 (d, J= 8.7 Hz, 2H), 7.15 (d, J = 9.0 Hz, 2H), 7.25 (d, .7=8.1 Hz, 2H), 7.38 (s, 1H), 7.63 (t, J= 7.8 Hz, 1H), 7.70 (d, = 8.4 Hz,
1H), 7.79 (t, .7= 7.8 Hz, 1H), 8.02 (br s, 2H), 8.45 (d, J= 8.7 Hz, 1H), 13.52 (s, 1H), 13.60 (s, IH); APCI-MS (m/z) 471 (M)+.
Example 170
5-{2-(4-Chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyI]ethenyl}-4-ethyl-2,4-dihydro-3/-/- l,2,4-triazole-3-thione

Step 1 : 2-{3-(4-Chlorophenyl)-2-[4-(quinoliii-2-ylmethoxy)phenyl]prop-2-enoyl}-N-ethyl hydrazinecarbothioamide:
To a well stirred solution of intermediate 64 (500 mg, 1.165 mmol) in dry THF (45 ml) was added ethyl isothiocynate (0.09 ml, 1.04 mmol) and the reaction mixture was refluxed for 2 h and then stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure and recrystallized to give 500 mg of the product as off white solid; Ή NMR (300 MHz, DMSO-40 δ 1.07 (t, J= 6.9 Hz, 3H), 2.50 (br s, 1H), 3.46 (br s, 2H), 5.32 (s, 2H), 6.92-7.00 (m, 4H), 7.26 (d, J= 7.8 Hz, 2H), 7.42-7.51 (m, 3H), 7.58-7.65 (m, 2H), 7.78 (I, .7 = 7.2 Hz, 1 H), 7.99 (d, J = 6.9 Hz, 2H), 8.41 (d, J= 9.0 Hz, 1 H), 9.19 (br s, 1 H), 9.62 (br s, 1 H). Step 2: 5-{2-(4-ChloiOphenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl |ethenyl }-4-ethyl-2.4-dihydro- 37 - 1 ,2,4-triazole-3-thione:
To the Step 1 Intermediate (500 mg, 0.967 mmol) was added saturated solution of sodium bicarbonate (40 ml) and was refluxed for 24 h after which it was filtered as it is. The precipitate obtained was dissolved in chloroform, dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude product thus obtained was purified by silica gel column chromatography to yield 300 mg of the product as off-white solid; Ή NMR (300 MHz, CDC] ) δ
I .00 (t, J = 6.9 Hz, 3H), 3.69 (d, J =1.5 Hz, 2H), 5.41 (s, 2H), 6.89 (d, J = 8.4 Hz, 2H), 7.06 (d, ./= 9.3 Hz, 3H), 7.20 (d, J= 8.4 Hz, 2H), 7.33 (d, J= 8.4 Hz, 2H), 7.57 (t, J= 7.5 Hz, 1H), 7.65 (d, J= 9.0 Hz, 1H), 7.76 (t, J= 7.8 Hz, 1H), 7.84 (d, J= 7.8 Hz, 1H), 8.21 (d, = 8.4 Hz, 2H),
I I .07 (br s, 1 H); APCI-MS (m/z) 499 (M)+.
Example 171
2-({4-[2-(4-Chlorophenyl)-l-(4-ethyl-4H-l,2,4-triazol-3-yl)ethenyl]phenoxy}methyl)qu

To a well stirred solution of Example 170 (100 mg, 0.2 mmol) in glacial acetic acid (5 ml) was added hydrogen peroxide (5 ml) drop wise and was stirred at room temperature for 12 h. The reaction mixture was neutralized with sodium bicarbonate (10 ml) and extracted with ethyl acetate (25 ml x 2). The combined organic layers were washed with water and brine, dried (Na2S04) and concentrated under reduced pressure. The product was purified by silica gel column chromatography to yield 50 mg of product as off-white solid; Ή NMR (300 MHz, DMSO-£¾-61.10 (t, J =7.2 Hz, 3H), 3.66 (q, J = 6.9 Hz, 2H), 5.34 (s, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.997.14 (m, 3H), 7.20 (d, J = 8.4 Hz, 2H), 7.44 (d, J= 8.4 Hz, 2H), 7.607.67 (m, 2H), 7.79 (t, J= 7.2 Hz, 1H), 8.00 (br s, 2H), 8.42 (d, J= 8.7 Hz, 1H), 8.57 (s, lH); APCl-MS (m/z) 467 (M+H)+.
Example 172
2-({4- l-(4-Chlorophenyl)-3-phenoxyprop-l -en-2-yl]phenoxy}methyl)quinoline

To the well stirred solution of the Intermediate 42 (200 mg, 0.476 mmol) in DMF (10 ml) was added phenol (54 mg, 0.571 mmol) followed by potassium carbonate (164 mg, 1.19 mmol) and was stirred at room temperature overnight. The reaction mixture was quenched with water (20 ml), extracted with ethyl acetate (2 x 25 ml), washed with water (20 ml), brine (20 ml) and dried, to yield 69 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.76 (s, 2H), 5.39 (s,2H), 6.71 (s, 1H), 6.92-7.01 (m, 7H), 7.07 (d, J= 8.7 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.30
(d, .7= 7.8 Hz, 2H), 7.56 (t, J =1.2 Hz, 1H).7.67-7.77 (m, 2H), 7.85 (d, = 7.8 Hz, 1H), 8.08 (d, J= 8.4 Hz, 1H), 8.21 (d, J= 8.4 Hz, 1H); ESI-MS (m/z) 478.36 (M+H)+.
Example 173
2-({4-[l-Phenyl-3-(pyridin-3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 44 (200 mg, 0.518 mmol) and 3- hydroxypyridine (98 mg, 1.036 mmol) as described in Example 172 to yield 30 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.86 (s, 2H), 5.41 (s, 2H), 6.75 (s, 1H); 6.98-7.03 (m, 4H), 7.12-7.19 (m, 5H), 7.34 (br s, 2H), 7.57 (t, J = 1.2 Hz, 1H), 7.70-7.78 (m, 2H), 7.85 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 8.24 (d, J= 8.1 Hz, 2H), 8.40 (br s, 1H); A PCI -MS (m/z) 445 (M+H)+.
Example 174
2-({4-[l -(4-ChloiOphenyl)-3-(pyrid -3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 42 (300 mg, 0.714 mmol) and 3- hydroxypyridine (102 mg, 1.071 mmol) as described in Example 172 to yield 26 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.82 (s, 2H), 5.39 (s, 2H), 6.70 (s, 1 H), 6.93 (d,/ = 8.4 Hz, 2H), 7.00 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H), 7.20- 7.27 (m, 2H), 7.56 (t, .1=1.2 Hz, 1H), 7.67-7.75 (m, 2H), 7.85 (d, J = 8.4 Hz, HI).8.08 (d. J = 8.1 Hz, 1H), 8.22 (d, J= 8.4 Hz, 2H), 8.36 (br s, 1H): ESI-MS (m/z) 479 (M+Hf.
Example 175
2-({4-[l -(4-Fluorophenyl)-3-(pyridin-3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 43 (200 mg. 0.495 mmol) and 3- hydroxypyridine (71 mg, 0.743 mmol) as described in Example 172 to yield 38 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 5.09 (s, 2H), 5.30 (s, 2H), 6.76-6.82 (m, 414), 6.90-6.96 (m, 6H), 7.06 (br s, 2H), 7.55 (t, J= 7.2 Hz, 1H), 7.62 (d, J= 9.0 Hz, lH), 7.73 (I, ,7 = 7.5 Hz, 1H), 7.83 (d, J= 7.5 Hz, 2H), 8.06 (d, J= 8.4 Hz, 1H), 8.19 (d, J= 8.4 Hz, 1H); ESl-MS (m/z) 463 (M+H)+.
Example 176
[1 -Phenyl-3-(pyridin-4-y]oxy)prop-l -en-2-yl]phenoxy}methyl)quinoiine

The title compound was prepared from Intermediate 44 (200 mg, 0.518 mmol) and 4- hydroxypyridine (59 mg, 0.621 mmol) as described in Example 172 to yield 91 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.66 (s, 2H), 5.36 (s, 2H), 6.34 (d, J = 7.2 Hz, 2H), 6.58 (s, lH), 6.96 (br s, 5H), 7.10-7.16 (m, 3H), 7.23 (br s, 3H), 7.56 (t, J = 7.2 Hz, 1 H), 7.66 (d, .7= 8.4 Hz, 1H), 7.75 (t, J= 7.2 Hz, 1H), 7.85 (d, J= 7.8 Hz, IH), 8.08 (d, J = 8.1 Hz, 1 H), 8.22 (d, J= 8.7 Hz, 1H); APCI-MS (m/z) 445 (M+H)+.
Example 177
2-({4-| 1 -(4-Chlorophenyl)-3-(pyrid -4-yloxy)prop-l-en-2-yl]phenoxy}methyl)q

The title compound was prepared from Intermediate 42 (300 mg, 0.714 mmol) and 4- hydroxypyridine (102 mg, 1.071 mmol) as described in Example 172 to yield 156 mg of product
as white solid; Ή NMR (300 MHz, CDC13) δ 4.63 (s, 2H), 5.37 (s, 2H), 6.31 (d, J= 7.5 Hz.2H), 6.49 (s, IH), 6.88-6.99 (m, 6H), 7.09 (d, J= 8.1 Hz, 2H), 7.21 (d, J = 7.2 Hz, 2H), 7.57 (t, J = 7.5 Hz, IH), 7.66 (d, J =8.7 Hz, 1H), 7.75 (t, J = 7.2 Hz, IH), 7.85 (d,J=7.8Hz, 1H), 8.08 (d, = 8.1 Hz, IH), 8.22 (d, J= 8.4 Hz, IH); ESI-MS {mz) 479 (M+H)+.
Example 178
2-({4-[]-(4-Fluorophenyl)-3-(pyrid -4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 43 (200 mg, 0.495 mmol) and 4- hydroxypyridine (71 mg, 0.743 mmol) as described in Example 172 to yield 107 mg of product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.64 (s, 2H), 5.37 (s, 2H), 6.34 (d, J =1.2 Hz. 2H), 6.53 (s, 2H), 6.82 (t, J= 9.0 Hz, 2H), 6.92-7.00 (m, 5H), 7.23 (d, J= 7.5 Hz, 2H), 7.57 (t, J = 7.2 Hz, IH), 7.'66 (d, J = 9.0 Hz, IH), 7.75 (t,J=7.2 Hz, IH), 7.86 (d, J = 8.4 Hz, IH), 8.08 (d,J= 8.1 Hz, IH), 8.23 (d,J= 8.4 Hz, IH); APCI-MS {m/z) 463 (M+H)+.
Example 179
2-({4-| l-(Pyridin-4-yl)-3-(pyridin- -yloxy)prop-l-en-2-yl]phenoxy}methyl)qu

Step 1: 3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyr|prop-2-en-l -ol:
To well stirred solution of the ester of Intermediate 14 (2 g, 4.878 mmol) in Tl IF (30 ml) was added 20 % di-isobutylaluminium hydride (DIBAL-H; 13.50 ml, 29.26 mmol) at -30 to -40 °C and stirred for 3 h at the same temperature. The reaction mixture was quenched with water (150 ml) and stirred for 20 mins after addition of ethyl acetate (300 ml). The precipitate obtained was filtered and the filtrate was dried and concentrated to yield 1.1 g of the product as off-white solid; Ή NMR (300 MHz, DMSO-i¾ δ 4.20 (d, J= 5.1 Hz, 2H), 5.38 (s, 2H), 6.60 (s, IH), 6.86
(d, .1=5.1 Hz, 2H), 7.05-7.13 (m, 4H), 7.60-7.70 (m, 2H), 7.79 (t, J= 6.6 Hz, 1H), 7.99-8.03 (m, 3H), 8.27-8.33 (m, 2H), 8.43 (d, J= 8.7 Hz, lH).
Step 2: 2-({4-[l-(Pyridin-4-yl)-3-(pyridin-4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline: To the well stirred solution of Step 1 Intermediate (300 mg, 0.819 mmol) in THF (10 ml) were added triphenylphosphine (322 mg, 0.983 mmol) and 4-hydroxypyridine (86 mg, 0.737 mmol). The reaction was stirred at room temperature for 15 mins followed by drop wise addition of diethylazodicarboxylate (0.19 ml, 1.5 mmol) and the reaction mixture was further stirred for 16h. The excess solvent was distilled under reduced pressure and purified by silica gel column chromatography to yield 34 mg of the product as off-white solid; Ή NMR (300 MHz, CDC13) δ 4.83 (s, 2H), 5.40 (s, 2H), 6.66 (s, 1H), 6.86 (br s, 4H), 7.02 (d, J= 8.4 Hz, 2H), 7.14 (d, .7= 7.8 Hz, 2H), 7.57 (t, J= 7.8 Hz, 1H), 7.67-7.75 (m, 2H), 7.85 (d, J = 7.8 Hz, 1 H), 8.08 (d, J= 7.8 Hz, 1H), 8.22 (d, .7- 8.1 Hz, 1H), 8.35 (d, J= 4.5 Hz, 2H), 8.45 (d, J= 4.8 Hz, 2H); APC1-MS (m/z) 446 (M+H)+.
Example 180
2-(4-Chlorophenyl)-7V-methyl-3-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide

The title compound was prepared by coupling Intermediate 83 (200 mg, 0.481 mmol) with methylamine hydrochloride (49 mg, 0.721 mmol) as described in Example 2 to yield 110 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.84 (d, J= 4.8 Hz, 3H), 5.32 (br s, 3H), 6.80 (d, J= 8.7 Hz, 2H), 6.93 (d, J= 8.1 Hz, 2H), 7.19 (d, .7= 8.1 Hz, 2H), 7.43 (d, J= 8.4 Hz, 2H), 7.52-7.60 (m, 2H), 7.71-7.83 (m, 3H), 8.05 (d, J = 8.4 Hz, lH), 8.16 (d, J= 8.7 Hz, 1 IT): ESI (m/z) 429 (M+H)+.
Example 181
2-(4-ChloiOphenyl)-N-(2-cyanoethyl)-3-[4-(quinolin-2-y]methoxy)phenyl]piOp-2-enamide

Th title compound was prepared by coupling Intermediate 83 (200 mg, 0.481 mmol) with 3- aminopropionitrile fumarate (247 mg, 0.962 mmol) as described in Example 2 to yield 120 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 2.67 (t, J = 6.6 Hz, 2H), 3.52 (q, J = 6.3 Hz, 2H), 5.33 (s, 2H), 5.78 (br s, 1H), 6.82 (d, J = 8.7 Hz, 2H), 6.94 (d, J - 8.7 Hz, 2H); 7.20-7.27 (m, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.51-7.61 (m, 2H), 7.71-7.83 (m, 3H), 8.06 (d, ,7 = 8.1 Hz, IH), 8.17 (d,J= 8.1 Hz, 1H); ESI (m/z) 468 (M+H)+.
Example 182
(±)-2-(4-Chlorophenyl)-N-(l-hydroxybutan-2-yl)-3-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide
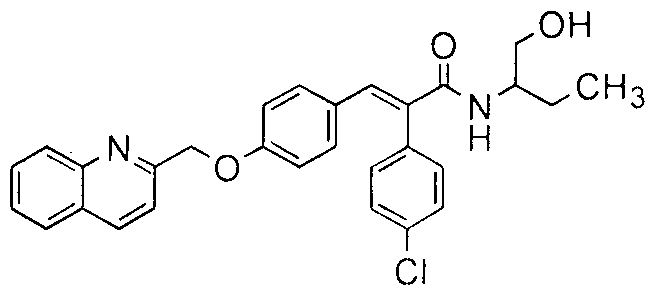
The title compound was prepared by coupling Intermediate 83 (150 mg, 0.360 mmol) with 2- aminobutan-l-ol (39 mg, 0.433 mmol) as described in Example 2 to yield 101 mg of the product as off white solid; Ή NMR (300 MHz, CDC13) δ 0.85 (t, J = 7.5 Hz, 3H), 1.30-1.40 (m: 1 H), 1.48-1.57 (m, IH), 2.92 (br s, 1H), 3.55 (br s, 1H), 3.67 (br s, 1H), 3.92 (br s, IH), 5.33 (s, 2H); 5.42 (d, J = 6.9 Hz, 3H), 6.81 (d, J= 8.7 Hz, 2H), 6.92 (d, J= 8.7 Hz, 2H), 7.19-7.27 (m, 2H), 7.44 (d, J= 7.8 Hz, 2H), 7.54-7.61 (m, 2H), 7.71-7.83 (m, 3H), 8.06 (d, J = 8.4 Hz, IH), 8.17 (d, J= 8.4 Hz, IH); ESI (m/z) 487 (M+H)+.
Example 183
2-({4-[2-(4-Chlorophenyl)-2-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 84 (300 mg, 0.697 mmol) and excess of triethyl orthoformate (5 ml) as described in Example 113 to give 63 mg of the product as off-
white solid; Ή NMR (300 MHz, CDC13) δ 5.36 (s, 2H), 6.86 (d, J= 8.7 Hz, 2H), 7.05 (d, J= 8.7 Hz, 2H), 7.25-7.32 (m, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.52-7.60 (m, 2H), 7.66 (s, 1H), 7.75 (t, J - 7.8 Hz, 1H),7.83 (d, J =8.4 Hz, 1H), 8.07 (d, J = 9.0 Hz, 1H), 8.19 (d, J = 8.1 Hz, IH), 8.37 (s, 1 H); ESI-MS (m/z) 440 (M+H)+.
Example 184
2-({4-[2-(4-Chlorophenyl)-2-(5-methyl-l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline

The title compound was prepared from Intermediate 84 (300 mg, 0.697 mmol) and Iriethyl orthoacetate (15 ml) as described in Example 113 to give 150 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) δ 2.56 (s, 3H), 5.35 (s, 2H), 6.85 (d, J= 9.0 Hz, 2H), 7.03 (d, J= 9.0 Hz, 2H), 7.27 (d, J= 9.6 Hz, 2H), 7.40 (d, J= 8.4 Hz, 2H), Ί .52-1.62 (m, 3H), 7.74 (t, ./ = 7.5 Hz, IH), 7.83 (d, J= 8.4 Hz, IH), 8.07 (d, J= 7.8 Hz, 1H), 8.18 (d, J= 8.7 Hz, IH); ESI-MS (m/z) 454 (M+H)+.
Example 185
2-(2-{4-|"2-(4-Chlorophenyl)-l-(l,3,4-oxadiazol-2- l ethen l]phenyl}ethyl)quinoline

The title compound was prepared from Intermediate 82 (260 mg, 0.607 mmol) and excess of Iriethyl orthoformate (5 ml) as described in Example 113 to give 45 mg of the product as off- white solid; Ή NMR (300 MHz, CDC13) 53.21- 3.26 (m, 2H), 3.36 (d, J= 7.8 Hz, 211 ).6.99 (d, J= 8.4 Hz, 2H), 7.10 (d, J= 8.7 Hz, 2H), 7.21-7.32 (m, 5H), 7.53 (t, J = 7.8 Hz, IH), 7.65 (s, IH), 7.73 (t, .1= 7.8 Hz, IH), 7.82 (d, J= 8.4 Hz, IH), 8.10 (d, J = 7.2 Hz, 2H) 8.39 (s, IH); ESI- MS (m/z) 438 (M+H)+.
Pharmacological activity
Phosphodiesterase 10 enzyme hydrolyses cAMP/cGMP to metabolically inactive 5 ΆΜΡ/5 ΌΜΡ. Inhibition of PDE10 enzyme activity can be quantitated by using a two step radiometric assay procedure (see Sette, C, Iona, S. and Conti, M., J. Biol. Chein., 269 ( 1 2), 1 994, pp 9245-9252). In this assay, PDE10 enzyme converts H-cAMP/3H-cGMP to 3H- 3 3
AMP/ H-GMP which is then converted to H-adenosine/ H-guanosine using snake venom nucleotidase. The radioactivity released in the supernatant liquid is quantitated as an indicator of PDE 1 0 enzyme activity.
ln-vitro screening assay of PDE10A inhibitors
Test compounds or reference compounds such as Dipyridamole, IB MX (Calbiochem) and Papverine (Sigma) were dissolved in pure dimethylsul fox ide (DMSO) to prepare 1 .0 m M stock solution and diluted suitably to afford the desired concentration. Final concentration of DMSO in the assay was 3 % (v/v). Substrate mixture was prepared by mixing 3H-cAMP (GE Healthcare) and 1 .0 mM cold cAMP (Sigma) in order to get 0.5 μθί / mL & 1 μΜ final concentrations of each respectively in the assay buffer. A 1.0 mg/mL of snake venom nucleotidase (Sigma) was prepared in D/w. Dowex (AG1 -X8 from Biorad) slurry was mixed with water and ethanol at 1 .0: 1 .0: 1 .0 ratios. The assay was carried out using suitably diluted PDE 1 0A enzyme preparation (BPS Biosciences) to get around 15-20% substrate hydrolysis to ensure linear reaction kinetics.
PDE 1 0 assay was carried out in 200 reaction volume by addition of assay buffer containing 10.0 mM Tris-HCl (pH 7.4), 0.2 mM MgCl2, test compound at required concentration and diluted enzyme. Reaction mixture was incubated at 30 °C for 30 min. The reaction was stopped by heating the plate in boiling water bath for 5.0 min and then cooling on an ice bath for 1 5 min. This was followed by addition of 50 μΕ of Crotalus atrox snake venom 5 '-nucleotidase and incubation at 30 °C for 30 min. Thereafter 400 μΕ of Dowex was added and incubated on ice bath for 1 5 min. Reaction mixture was centrifuged and supernatant liquid was used for quantifying radioactivity in the samples. Reaction was measured as counts per minute (cpm) using a Packard Biosciences plate reader. An enzyme control without test compounds was run to quantitate maximum PDE10 reaction. Inhibition of enzyme activity was calculated as a percent
of control reaction. The IC50 values were calculated from dose-response curve by nonlinear regression analysis using Graph Pad Prism software.
The compounds prepared were tested using the above assay procedure and the results obtained are given in Table 4. Percentage inhibition of human PDE I OA enzyme at concentrations of 1 .0 μΜ and 10.0 μΜ are given in the table 4 along with IC50 (nM) for selected examples. The IC50 (nM) values of the compounds are set forth in Table 4 wherein "A" refers to an IC50 value of less than 20 nM, "B" refers to an IC50 value in range of 20.01 -50 nM, "C" refers to an IC50 value in range of 50.01 - 100 nM and "D" refers to an IC50 value of more than 1 00 nM.
Table 4: In-vitro screening results (hPDEl OA activity) of compounds of invention
Example Percentage inhibition of hPDE l OA at -C5o (nM)
No. 1 μΜ 10 μΜ
1. 89.45 96.71 C
2. 84.60 98.37 C
42.96 86.79
4. 35.21 73.5 1
5. 47.23 84.58 —
6. 22.09 67,39 -
7. 67.74 99.29 -
8. 55.51 88.00 -
9. 89.01 99.68 B
1 0. 65.82 95.80 D
1 1 . 46.52 87.71 —
1 2. 78.07 96.24 D
■1 3.' 71 .5 1 93.84 D
14. 17.03 1 7.22 . -
15. 79.10 97. 13 D
16. 14.08 67.09 -
17. 49.53 79.34 —
1 8. 55.67 87.74 D
1 9. 38.73 85.93 —
Example Percentage inhibition of hPDElOA at ]Cso(nM) No. 1 μΜ 10 μΜ
20. 55.07 90.54 D
21. 79.80 98.74 C
22. 58.29 91.58 -
23. 90.58 98.62 B
24. 17.31 51.94 -
25. 30.18 77.85 -
26. 94.79 99.49 A
27. 60.31 92.27 -
28. 75.11 97.60 D
29. 64.39 · 94.63 -
30. 45.71 81.87 -
31. 92.07 98.33 A
32. 72.13 95.68 D jj. 90.89 99.02 C
34. 38.63 73.09 -
35. 85.21 96.26 C
36. 70.38 92.15 D
37. 58.79 89.32 D
38. 72.70 95.36 D
39. 02.37 14.02 --
40. 91.18 98.08 C
41. 67.91 94.32 -
42. 44.79 86.43 -
43. 72.72 96.87 D
44. 90.96 98.97 C
45. 83.17 99.71 C
46. 87.83 98.46 D
Example Percentage inhibition of hPDElOA at ICso(nM) No. 1 μΜ . 10 μΜ
47. 07.57 64.63 -
48. 92.74 98.03 B
49. 91.44 98.87 B
50. 86.05 101.33 C
51. 74.96 94.93 D
52. 67.97 93.82 D
53. 73.85 89.23 C
54. 91.42 97.06 C
55. 95.44 99.51 A
56. 90.79 97.93 C
57. 88.60 97.12 C
58. 95.19 97.88 A
59, 65.58 94.50 D
60. 73.01 88.11 -
61. 96.77 97.50 A
62. 66.93 95.44 D
63. 64.67 95.30 D
64. 87.62 97.18 D
65. 46.11 92.14 -
66. 43.03 54.21 -
67. 59.63 91.07 -
68. 83.03 100.78 D
69. 64.95 94.88 -'
70. 94.45 99.90 B
- 71. 99.66 100.00 A
72. 92.99 99.29 C
73. 97.89 99.61 A
Example Percentage inhibition of hPDEl OA at IC30 (nM) No. 1 μΜ 1 0 μΜ
74. 94.01 99.21 13
75. 94.69 99.95 B
76. 95.68 99.56 A
77. 97.39 98.67 A
78. 95.65 97.55 A
79. 99.28 99.82 C
80. 97.82 99.94 A
81 . 95.39 100.52 B
82. 94.52 1 00.00 A
83. 79.15 96.83 D
84. 97.44 97.36 A
85. 101 .16 100.75 A
86. 99.14 98.92 A
87. 100.08 102.00 A
88. 96.67 97.08 A
89. 97.52 98.80 A
90. 97.18 97.87 A
91. 98.19 99.54 A '
92. 86.49 89.58 A
93. 90.67 90.58 B
94. 97.23 99.29 A
95. 97.85 99.86 A
96. 86.80 99.94 C
97. 99.74 100.00 B
98. 96.64 98.26 A
99. 96.84 100.08 A
100. 90.93 98.61 C
Example Percentage inhibition of hPDElOA at IC5o(nM) No. 1 μΜ 10 μΜ
101. 98.13 98.65 A
102. 94.70 98.68 B
103. 85.70 92.54 D
104. 96.99 100.36 B
105. 91.20 97.30 c
106. 95.17 96.21 A
107. 98.97 99.44 A
108. 99.27 98.44 A
109. 93.12 94.16 A
110. 98.11 99.51 A
111. 82.91 94.56 D
112. 84.25 97.65 B
113. 98.81 100.00 A
114. 83.11 96.09 D
115. 73.49 70.69 -
116. 70.96 96.25 D ,
117. 98.14 100.68 A
118. 96.81 99.76 A
119. 99.68 99.27 A'
120. 99.61 99.40 A
121. 25.72 44.03 -
122. 13.07 37.48 -
123. 83.43 96.59 C
124. 93.40 97.34 A
125. 17.89 27.45 -
126. 86.51 98.14 B
127. . 96.90 102.04 B
Example Percentage inhibition of hPDElOA at IC50 (nM) No. 1 μΜ 10 μΜ
128. 94.30 ' 99.94 B
129. 97.52 99.96 B
130. 97.87 102.30 A
131. 92.07 78.21 -
132. 88.70 98.80 D
1 j j. 41.21 83.91 -
134. 85.98 96.78 B
135. 98.55 98.25 A
136. 85.86 95.78 C
137. 95.24 98.26 B
138. 95.31 101.76 B
139. 95.70 99.83 A
140. 87.92 94.96 C
141. 97.64 99.37 A
142. 89.68 98.38 C
143. 76.86 99.78 D
144. 95.18 98.72 A
145. 89.27 97.68 C
146. 05.58 14.63 -
147. 86.24 • 97.93 C
148. 89.03 97.51 c
149. 96.36 99.21 A
150. 100.54 102.21 A
151. 94.72 98.25 B
152. 25.97 57.87
153. 94.37 97.10 B
154. 97.75 98.84 A
Example Percentage inhibition of hPDElOA at lC50(n ) No. 1 μΜ 10 μΜ
155. 48.12 92.05 -
156. 72.89 95.20 D
157. 87.67 97.17 C
158. 81.40 98.40 D
159. 98.88 98.46 A
160. 93.80 99.07 B
161. 96.47 98.26 A
162. 78.61 93.38 D
163. 67.98 80.07 D
164. 84.09 98.62 C
165. 97.62 99.46 A
166. 68.68 93.30 -
167. 98.12 100.43 A
168. 99.91 100.57 A
169. 83.66 97.67 C
170. 13.80 05.78 -
171. 24.79 68.32 -
172. 60.35 65.29 -
173. 77.73 85.74 -
174. 99.64 99.14 A
175. 94.72 99.46 C
176. 98.03 99.06 A
177. 100.00 100.00 A
178. 96.02 97.68 A
179. 98.16 96.97 A
180. 08.38 54.94 -
181. 06.49 09.16 -
Example Percentage inhibition of hPDEl OA at IC50 (nM) No. 1 μΜ 1 0 μΜ
1 82. 1 0.27 56.07 -
1 83. 09.07 1 1 .65 -
1 84. 0.90 06.1 0 -
1 85. 89.69 97.62 B
In vivo efficacy screen for Psychoses:
The illustrative examples of the present invention are screened for 'in vivo'- PDE 1 0 based efficacy in a rat model of Dizocilpine (MK-801 ) - induced psychotic behaviour according to slightly modified procedures described in a Andine. P et al., JPET, 290; p. 1 393- 1 408. ( 1 999)]. The screening of the compounds can also be carried out by some other methods and procedures known to persons skilled in the art.
Dizocilpine (MK-801 ) - induced model of Psychosis in female Sprague-Dawley rats:
Dizocilpine (MK-801), a highly selective non-competitive antagonist of NMD A (N-methyl-d- aspartate) receptor [an excitatory amino acid (glutamate, EAA) receptor] is known to produce psychosis both in rodents and humans. A dysfunction in the main excitatory neurotransmitter system of the brain through the EAA receptors has been well documented in psychoses. On the day of the experiment, rats were acclimatized for 15 min before the administration of 0.2 mg/kg of MK-801 by subcutaneous route, followed by a behavioural observation (three behaviours: locomotion, stereotyped sniffing and ataxia) scoring that started 1 5 min after injection and continued for 1 h (i.e. 15-75 min post MK-801 ) [Andine, P et al ., JPET, 290, p. 1 393- 1 408, ( 1 999)]. The test compounds were administered at appropriate time point prior to the MK-801 injection, based on the route of administration.
Example No. 91 , at a dose of 30 mg/kg (p.o.), administered 60 min prior, potently blocked the MK-801 - induced psychoses in female SD rats as shown in Figure 1 .
Example No. 177, at a dose of 30 mg/kg (i.p.), administered 5 min prior, moderately blocked the MK-801 - induced psychoses in female SD rats as shown in Figure 2.
Claims
WE CLAIM :
! . A compound of the formula

(1)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
G i is selected from hydrogen, cyano, substituted or unsubstituted heteroaryl, heterocyclyl, -(CR4R5)P-NR9R10, -(CR4R5)P-ORN and -C(0)NR9R1 0;
G2 is selected from hydrogen, substituted or unsubstituted heteroaryl, heterocyclyl and - C(0)NR9R10; with the proviso that at least one of G i or G2 is not hydrogen ;
A is aryl or heteroaryl;
X is a bond or -0-;
Y is -(CR4R5)p-;
Z is selected from substituted or unsubstituted aryl, heterocyclyl and heteroaryl, wherein said cyclic ring may be monocyclic, bicyclic or spirocyclic;
at each occurrence, R1 and R2, which may be. the same or different, are independently selected from halogen, substituted or unsubstituted alkoxy, haloalkyl and haloalkoxy;
at each occurrence, both R4 and R3 are hydrogen; .
R9 and R 1 0, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl, alkynyl, cyanoalkyl, hydroxyalkyl, haloalky] , cycloalkyl, heterocyclyl, heteroaryl, -(CRDRE)QRA and -C(0)RA; or R9 and R 10 together with the nitrogen atom to which they are attached, may form an optionally substituted heterocyclyl or heteroaryl ring, wherein said heterocyclic or heteroaryl ring may contain 1 , 2, 3 or 4 hetero atom(s) selected from O, S or N;
R " is substituted or unsubstituted aryl or heteroaryl;
at each occurrence, RA, RD and RE, which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alky], hydroxyalkyl, alkoxy, alkoxyal kyl. aryl, -C(0)NR'R8, -C(0)OR' and -NRFR8; at each occurrence, R and R , which may be the same or different, are independently selected from hydrogen, substituted or unsubstituted alkyl and -C(0)alkyl;
'm' is an integer ranging from 0 to 5, both inclusive;
'ir is an integer ranging from 0 to 4, both inclusive;
'';y is an integer ranging from 1 to 3, both inclusive; and
:q' is an integer ranging from 1 to 3, both inclusive.
A compound selected from
(a)a compound of formula (la

(la)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereol wherein,
A is selected from a group consisting of 
X is a bond or -0-;
Y is -(CH2)P-;
Z is independently selected from a group consisting

R9 is hyd rogen or substituted or unsubstituted alky! ;
R10 is independently selected from a group consisting of hydrogen,
\— CH3 J-CH2CH3 I \— CH(CH3)2 I -CH2CF3 \— (CH2)2CNI \— (CH2)2OH -(CH2)2N(CH3)2 -(CH2)2N(C2H5)2 I \— (CH2)CH(OCH3)2 I ^_(CH2)C(0)NH2 F -(CH2)2NHC(0)CH3J \— <]


' is an integer ranging from 0 to 5, both inclusive;
is an integer ranging from 0 to 4, both inclusive; and
is an integer ranging from 1 to 3, both inclusive.
(b) a compound of the formul
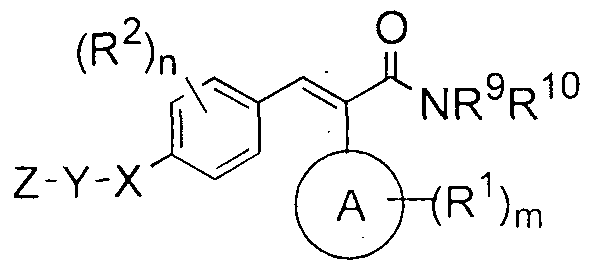
(lb)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-o ide thereof wherein, A is aryl, preferably phenyl;
X is -0-;
Y is -(CH2)p-;
Z is heteroaryl, preferably quinolinyl ;
at each occurrence. R1 and R2, which may be the same or different, are independent! lected from halogen, substituted or unsubstituted alkoxy, haloaikyl and haloalkoxy;
R9 is hydrogen or substituted or unsubstituted alkyl;
R10 is independently selected from a group consisting of hydrogen, 
' m' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive. a compound of the formula Ic)

(lc)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
A is aryl, preferably phenyl;
X is -0-;
y is -(CH2)P-;
Z is heteroaryl, preferably quinolinyl ;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, substituted or unsubstituted alkoxy, haloaikyl and haloalkoxy;
R9 is hydrogen or substituted or unsubstituted alkyl ;
R10 is independently selected from a group consisting of hydrogen, -(CH2)2N H2 i - (CH2)2NHCH3 i }-(CH2)2OCH3 ^--CH2C≡CH \— C(0)CH3 }— C(0)CH2OCH3 i
-(CH2)2CN i i— (CH2)2NHC(0)CH3

and \— \ NC(0)OC2H5 or R , 9 a _„nd_, n Rl O together with the nitrogen atom to which they are attached, forms heterocyclic or heteroaryl ring independently selected from a group consisting of

'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive. compound of the formul (Id)

or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
ring A is selected from a group consisting of 
X is a bond or -0-;

at each occurrence, R and R , which may be the same or different, are independently selected from halogen, substituted or unsubstituted alkoxy, haloalkyl and haloalkoxy;
Het is heteroaryl or heterocyclyl independently selected from a group consisti ng of


'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and 'ρ' is an integer ranging from 1 to 3, both inclusive. (e)a compound of the formula

(le)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
A is aryl preferably phenyl ;
X is -0-;
Y is -(CH2)P-;
Z is heteroaryl, preferably quinolinyl;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, substituted or unsubstituted alkoxy, haloalkyl and haloalkoxy;
Het is heteroaryl selected from
12

'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
(f) a compound of the formula (If) 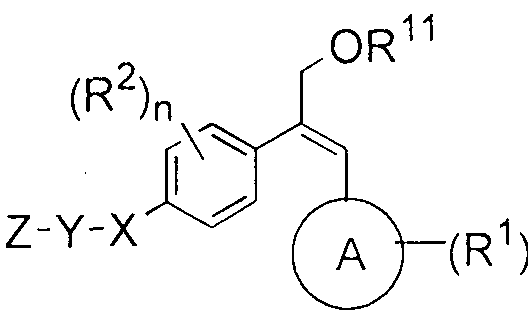
(if)
or a pharmaceutically acceptable salt thereof, or a stereoisomer thereof or a N-oxide thereof, wherein,
ring A is aryl or heteroaryl selected from phenyl and 4-pyridyl;
X is -0-;
Z is heteroaryl, preferably quinolinyl;
at each occurrence, R1 and R2, which may be the same or different, are independently selected from halogen, substituted or unsubstituted alkoxy, haloalkyl and haloalkoxy;
R1 1 is aryl or heteroaryl, selected from phenyl, 3-pyridyl and 4-pyridyl ;
'm' is an integer ranging from 0 to 5, both inclusive;
'n' is an integer ranging from 0 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
3. The compound of claim 1 , wherein G | is cyano.
4. The compound of claim 2, wherein R1 in formula (la) to (If) is haloalkyl.
5. The compound of claim 4, wherein haloalkyl is trifluoromethyl.
6. The compound of claim 2, wherein R1 in formula (la) or (Id) is haloalkoxy.
7. The compound of claim 6, wherein haloalkoxy is difluoromethoxy or trifluoromethoxy.
8. The compound of claim 2, wherein R1 in formula (la) or (Id) is alkoxy.
9. The compound of claim 8, wherein alkoxy is methoxy or ethoxy.
1 0. The compound of claim 2, wherein 'm ' in formula (la) to (If) is 1 .
1 1. The compound of claim 2, wherein 'nT in formula (Ic) or (Id) is 2.
1 2. The compound of claim 2, wherein 'm' in formula (la), (Ic), (Id) or (If) is 0.
1 3. The compound of claim 2, wherein 'n' in formula (la) to (If) is 0.
1 4. The compound of claim 2, wherein 'R2' in formula (Id) is fluorine, chlorine or methoxy and 'n' is 1 .
1 The compound of claim 2, wherein 'p' in formula (la) to (If) is 1 .
16. The compound of claim 2, wherein 'p' in formula (la) or (Id) is 2
1 7. The compound of claim 2, wherein R12 in formula (Id) or (le) is hydrogen.
1 8. The compound of claim 2, wherein R12 in formula (Id) or (Ie) is alkyl.
1 9. The compound of claim 1 8, wherein alkyl is methyl or ethyl .
20. The compound of claim 2, wherein R12 in formula (Id) or (Ie) is haloalkyl .
21 . The compound of claim 20, wherein haloalkyl is trifluoromethyl.
22. The compound of claim 2, wherein
(a) the compound is selected from
3-(4-chlorophenyl)-2-f4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-chlorophenyl)-2-[4-(imidazo[l ,2-tf]pyridin-2-ylmethoxy)phenyl]-N-methyl-prop-2- enamide;
2- |'4-(l ,3-benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)-N-methylprop-2-enamide; N--rnethyl-2- {4-[( l -methyl- lH-benzimidazol-2-yl)methoxy]phenyl } -3 -(pyridin-3-yl)prop-2- enamide;
N-methyl-2- {4-[( 1 -methyl- 1 H-benzimidazol-2-yl)methoxy]pheny 1 } -3 -(pyridin-4-y l)prop-2- enamide;
3 - (4-chlorophenyl)-N-methyl-2-[4-(pyridin-2-ylmethoxy)phenyl]prop-2-enamide;
yV-methyl-2-| 4-(qui.nolin-2-ylmethoxy)phenyl]-3-(thiophen-2-yl)piOp-2-enamide;
A/-methyl-3-phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-chlorophenyl)-N-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl |prop-2-enamide;
3-(4-fluoi phenyl)-N-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
A''-methyl-3-(pyridin-3-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
/V-methyl-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-methoxyphenyl)-N-methyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
2- {4-| 2-( l .2-benzoxazol-3-yl)ethoxy]phenyl}-3-(4-chlorophenyl)-N-methylprop-2-enamide; yV- ethyl-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)pheny]]prop-2-enamide;
3- (4-chlorophenyl)-2-[4-( yridin-2-ylmethoxy)phenyl]-N-(2.2,2-tri'fluoroethyl)prop-2-enamide; 2-{4-[( l -methyl- lH-benzimidazol-2-yl)methoxy]phenyl }-3-(pyridin-3-yl)-N-(2.2.2- trifluoroethyl)prop-2-enamide; 2-{4-| ( l -methyl- l / -benzimidazol-2-yl)methoxy]phenyl}-3-(pyridin-4-yl)-Ai-(2, 2,2- trifiuoroethyl)prop-2-enamide;
2-[4-(imidazo[ l ,2-a]pyridin-2-ylmethoxy)phenyl]-3-(pyridin-4-yl)-N-(2,2,2-trifluoroethy]) prop-
2- enamide;
3- (pyridin-3-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-N-(2,2,2-trifluoroethyl)prop-2-enamide; 3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-N-(2J2,2-trifluoroethyl)pi p-2-enami
2- [4-(l ,3-benzothiazol-2-ylmethoxy)phenyl]-3-(4-chlorophenyl)-N-(2-cyanoet'hyl)prop-2- enamide;
3- (4-chlorophenyl)-AL(2-cyanoethyl)-2-[4-(imidazo[l ,2-£/]pyridin-2-ylmethoxy) phenyl]prop-2- enamide;
3-(4-chlorophenyl)-yV-(2-cyanoethyl)-2-{4-[(] -methyl- ] H-indazol-3-yl)methoxy]phenyl } prop-2- enamide;
3-(4-chlorophenyl)-N-(2-cyanoethyl)-2-[4-(pyridin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-chlorophenyl)-N-(2-cyanoethyl)-2-[4-(thieno[3,2-6]pyridin-5-ylmethoxy)phenyl]prop-2- enamide;
/V-(2-cyanoethyl)-2-| 4-(qiiinolin-2-ylmethoxy)phenyl]-3-(thiophen-2-yl)prop-2-enamide;
N-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-(thiophen-3-yl)prop-2-enamide;
Ar-(2-cyanoethyl)-3-phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(3-chlorophenyl)-N-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-chlorophenyl)-N-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
N-(2-cyanoethyl)-3-(4-fluorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
N-(2-cyanoethyl)-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
N-(2-cyanoethyl)-3-( l -oxidopyridin-4-yl)-2-[4-(quinolin-2-ylrnethoxy)pheny ]prop-2-enamide;
N-(2-cyanoethyl)-3-(4-methoxyphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-enamide;
A/-(2-cyanoethyl)-3-[4-(difluoromethoxy)phenyl]-2-| 4-(quinolin-2-ylmethoxy)phenyl] prop-2- enamide;
N-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-[4-(trifluoromethoxy)phenyl] prop-2- enamide;
A|i-(2-cyanoethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-3-[4-(trifluoromethyl)phenyl]piOp-2- enamid.e; 2- {4-[2-( l ,2-benzoxazol-3-yl)ethoxy]phenyl}-3-(4-chlorophenyl)-N-(2-cyanoethyl) prop-2- enamide;
3- (4-chlorophenyl)-N-(2-hydroxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide; 3-(4-chlorophenyl)-N-[2-(dimethylarnino)ethyl]-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2- enamide;
3-(4-chlorophenyl)-N-[2-(diethylamino)ethyl]-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide;
/ -(piOpan-2-yl)-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
(±)-3-(4-chlorophenyl)-N-(l -hydroxypropan-2-yl)-2-[4-(quinolin-2-ylrnethoxy)phenyl] prop-2- enamide;
N-[(27i)-l -hydroxypropan-2-yl]-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl] piOp-2- enami de;
3-(4-chlorophenyl.)-N-(l -hydroxybutan-2-yl)-2-[4-(i midazo [ l ,2-a]pyrid in-2-yl methox y) phenyl]prop-2-enamide;
(±)-3-(4-chlorophenyl)-/vr-(l -hydroxybutan-2-yl)-2-[4-(pyridin-2-y]methoxy)pheny]]prop-2- enamide;
3-(4-cChlorophenyl)-N-[(27?)-l -hydroxybutan-2-yl]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2- enamide;
3-(4-chloiOphenyl)-N-[(25)- l -hydroxybutan-2-y'l]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2- enamide:
(±)-N-( \ -hydiOxybutan-2-yl)-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide;
(±)-N-(l -hydroxybutan-2-yl)-3-(4-methoxyphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl] pvop-2- enamide;
A'-[(l ii)-2-hydroxy-l -phenylethyl]-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)pheny l] prop-2- enamide;
(±)-ethyl /V- {3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]piOp-2-enoyl } alani nate;
(rJ:)-N- { 3-(4-chlorophenyl)-2-[4-(quino]in-2-ylmethoxy)phenyl]prop-2-enoyl }alanine;
3-(4-chlorophenyl)-N-(2,2-dimethoxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl] piOp-2-enamide; N-(2-arnino-2-oxoethyl)-3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide;
A/-[2-(acetylamino)ethyl]-3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide;
A/-cyclopropyl-3-(pyridin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-N-( l ,3-thiazol-2-yl)prop-2-enamide; 3-(4-chlorophenyl)-N-(piperidin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]pi p-2-enamide di (trifluoroacetic acid);
3-(4-chlorophenyl)-l -[(3i?)-3-hydroxypyrrolidin- l -yl]-2-[4-(quinolin-2-ylmethoxy)phenyl] prop-
2- en- ] -one;
3- (4-chlorophenyl)-l -[(2R)-2-(hydroxymethyl)pyrrolidin- l -yl]-2-[4-(quinolin-2-ylmethoxy) phenyl]prop-2-en- 1 -one;
3-(4-chlorophenyl)- l -[(21S)-2-(hydroxymethyl)pyrrolidin- i -yl]-2-[4-(quinolin-2-ylmethoxy) pheny]]prop-2-en- 1 -one;
3-(4-chlorophenyl)- l -(4-rnethylpiperazin- l -yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l - one;
3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]-l -(thio'morpholin-4-yl)prop-2-en- l -one:
3- (4-chlorophenyl)- l -(morpholin-4-yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -one:
4- { 3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enoyl }piperazin-2-one;
3-(4-chlorophenyl)-A/-(2-cyanoethyl)-2-{4-[2-(quinol in-2-yl)ethyl]phenyl}prop-2-enamide;
(b) the compound is selected from
2-(4-chloi phenyl)-N-methyl-3-[ 4-(quinolin-2-ylmethoxy)pheny]]piOp-2-enamide;
2- (4-chlorophenyl)-N-(2-cyanoethyl)-3-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enamide;
(±)-2-(4-chlorophenyl)-N-(l -hydroxybutan-2-yl)-3-[4-(quinolin-2-ylmethoxy)phenyl]prop-2- enamide;
(c) the compound is selected from
yV-{ 3 -(4-chloiOphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyr|piOp-2-en- l -yl } ethane- 1 ,2 -diamine di(trifluoro acetic acid) salt;
3- (4-chlorophenyl)-N-(prop-2-yn- l -yl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- l -amine; 3-(4-chlorophenyl)-N-(2-methoxyethyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -amine dihydrochloride;
3-({3-(4-chloiOpheny])-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}amino)
propanenitrile dihydrochloride;
A/-{3-(4-chloiOphenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -yl }-N'-methyl ethane- 1 ,2-diamine di(trifluoro acetic acid);
A/- 2-({3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}amino)elhyl| acetamide;
(31S,)-A/-{3-(4-ch]orophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl} pyrrolidin-3- aminc di(trifluoroacetic acid);
(4S)-4-({3-(4-chlorophenyl)-2-[4-(quinohn-2-ylmeihoxy)phenyl]prop-2-en-l-y]}amino) pyrrolidin-2-one;
A'-{3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}piperidin-4-amine di(trifluoroacetic acid) salt;
ethyl 4-({3-(4-chloiOphenyl)-2-|4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}amino) piperidine- 1 -carboxylate;
(5S)-5-({3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}amino) piperidin-2-one;
3-(4-chlorophenyl)-N,N-dimet'hyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -amine: 2-({4-[l -(4-chloropheny])-3-(pyrrolidin-l-yl)prop-l -en-2-yl]phenoxy}methyl)quinoline:
[(25)- 1 - { 3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en- 1 -yl }pyrrolidin-2- yl]methanol;
2-({4-[l -(4-chlorophenyl)-3-(lH-pyrazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline; 2-({4-[l -(4-chlorophenyl)-3-(l/-imidazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline; (1 - {3-(4-chlorophenyl)-2-|4-(quinolin-2-ylmethoxy)phenyl]piOp-2-en-l-yl}-l/7-imidazol-4-yl) methanol;
2- {4- 1 -(4-chlorophenyl)-3-(lH-l,2,4-triazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoIine; 2-({4-[l-(4-chlorophenyl)-3-(2H-l,2,3-triazol-2-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline; 2-({4-[l-(4-chloropheny])-3-(lH-l,2,3-triazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline; 2-({4-[l-(4-chlorophenyl)-3-(lH-tetrazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline; 2-({4-[ -(4-chlorophenyl)-3-(2H-tetrazol-2-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2 {4-|"l-(4-chlorophenyl)-3-(piperidin-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline:
2-({4-[l-(4-chlorophenyl)-3-(thiomorpholin-4-yl)prop-l-en-2-yl]phenoxy}met1iyl)quinoline; 2-({4-[]-(4-chlorophenyl)-3-(morpholin-4-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-[ l-(4-chlorophenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline di(trifluoro acetic acid);
2-({4-[l-(4-fluoiOphenyl)-3-(piperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline trihydrochloride;
2-({4-[l-(4-chloro-3-fluorophenyl)-3-(piperazin-l-yl)prop-l -en-2-yl]phenoxy}meth l) quinoline trihydrochloride;
2- ({4-[l-(4-chloiOphenyl)-3-(4-methylpiperazin-l-yl)prop-l-en-2-yl]phenoxy}methyl) quinoline;
4-{3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}piperazin-2-one; 1 -{3-(4-fluorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}pyridin-2(lH)-one; 1 -{3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}pyrimidin-2(l H)-onc; 1 -{3-(4-fluorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyI]prop-2-en-l -yl}pyrimidin-2(l H)-one:
3- {3-phenyl-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l-yl}pyrimidin-4(3H)-one;
3- {3-(4-fluorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}pyrimidin-4(3//)-one; 2-({4-| 1 -(4-fluorophenyl)-3-(lH-tetrazol-l-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-[l-(4-fluorophenyl)-3-(2H-tetrazol-2-yl)prop-l-en-2-yl]phenoxy}methyl)quinoline;
N-{3-(4-chlorophenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yljacetamide;
N-{3-(4-chloi phenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-en-l -yl}-2-methoxy acetamide;
(d) the compound is selected from
4- {2-(4-chlorophenyl)-l - 4-(quinolin-2-ylmethoxy)phenyl]ethenyl}pyrrolidin-2-one;
2-({4-[2-(4-chlorophenyl)-l-(l ,3-oxazol-5-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4- 12-(4-chlorophenyl)-l-(l -methyl- lH-pyrazol-5-yl)ethenyl]phenoxy}methyl)quinoline; 2-({4-[2-(4-chlorophenyl)-l-(l -methyl- lH-pyrazol-3-yl)ethenyl]phenoxy}methyl)quinoline: 2-({4-|2-(4-chlorophenyl)-l -(1.3,4-oxadiazol-2-yl)vinyl]phenoxy}methyl)imidazo[l ,2-a\ pyridine: 2- ({4-[2-(4-chloiOphenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l,3-benzothiazole;
3- ({4-[2-(4-chloropheny])-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l -methyl- 1H- indazole;
2-({4- 2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)pyndine;
5-({4- 2-(4-chlorophenyl)-l-(1.3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)thieno 3,2-/ | pyridine;
5-({4-[2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)furo[3,2-A]pyridine; 5-({4-[2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-lH-pyrrolo[3,2 ?7] pyridine;
5-({4-[2-(4-chlorophenyl)-l-(l, 3 ,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l -methyl- 1/ - pyrrolo[3,2-6]pyridine;
5-({4- 2-(4-chloiOphenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l //-indazole:
5-({4-[2-(4-chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)-l -methyl- 1 //- indazole;
5-({4-[2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl] phenoxy} ethyl )| l,3]oxazolo [4,5-b] pyridine;
5-({4-[2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)pyrazolo[l ,5-a] pyrimidine;
2-{2-(4-chlorophenyI)-l-[4-(naphthalen-2-ylmethoxy)pheny]]ethenyl}-l,3,4-oxadiazole;
2-({4-[l -(l,3,4-oxadiazol-2-yl)-2-(thiophen-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l -(1 ,3,4-oxadiazol-2-yl)-2-(thiophen-3-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-|2-(furan-2-yl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-|2-(furan-3-yl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-| 1 -(l,3,4-oxadiazol-2-yl)-2-(l,3-thiazol-5-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[2-(2-chloro-l,3-thiazol-5-yl)-l-(l, 3, 4-oxadiazol-2-yl)ethenyl|phenoxy} methyl) quinoline;
2-({4-[l-(l,3,4-oxadiazol-2-yl)-2-phenylethenyl]phenoxy}methyl)quinoline;
2-({2-chloro-4-[l-(l,3,4-oxadiazol-2-yl)-2-phenylethenyl]phenoxy}methyl)quinoline;
2-({4-[2-(3-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[2-(4-chlorophenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline: 2-({4-|"2-(4-chloi phenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]-2-methoxyphenoxy}methyl) quinoline;
2-({4-[2-(4-fluorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l-(l,3,4-oxadiazol-2-yl)-2-(pyridin-3-yl)ethenyl]phenoxy}methyl)quinoline:
2-({4- 2-(6-methoxypyridin-3-yl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}rnethyl)quinoline;
2-({4-|'2-(6-ethoxypyridin-3-yl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l -(1 ,3,4-oxadiazol-2-yl)-2-(pyridin-4-yl)ethenyl]phenoxy}methyl)quinoline;
2-({2-fluoro-4-[l-(l,3,4-oxadiazol-2-yl)-2-(pyridin-4-yl)ethenyl]phenoxy}methy])quinoline;
2-({4-['l -(1 ,3,4-oxadiazol-2-yl)-2-(l-oxidopyridin-4-yl)ethenyl]phenoxy}methyl)quinoline:
2-({4-["2-(4-methoxyphenyl)-l-(l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-['2-[4-(difluoromethoxy)phenyl]-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl) quinoline;
2-|(4-{l-(l,3,4-Oxadiazol-2-yl)-2-[4-(trifluoromethoxy)phenyl]ethenyl}phenoxy)methyl] quinoline;
2-f(4-{ 1 -(1 ,3,4-oxadiazol-2-yl)-2-[4-(trifluoromethyl)phenyl]ethenyl}phenoxy)methyl] quinoline;
2-({4-[2-(2,4-difluorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}.methyl)quinoline:
2-({4-[2-(4-Chloro-2-fluoropheny])-l -(1 ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline: 2-( {4-[2-(4-chloro-3 -fluorophenyl)- 1-(1 ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline: 2-({4-[2-(3,4-di"fluorophenyl)-l-(l,3.4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[2-(4-chlorophenyl)-l-(l,2,4-oxadiazol-5-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-|Ί -(5-methyl-l ,3.4-oxadiazol-2-yl)-2-(4-chlorophenyl)ethenyl]phenoxy}methyl)imidazo 11 ,2-a]pyridine;
2-({4-[2-(4-chlorophenyl)-l-(5-methyl-l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)pyridine;
2-({4-[l -(5-methyl-l, 3, 4-oxadiazol-2-yl)-2-(thiophen-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l -(5-methyl-l, 3,4-oxadiazol-2-yl)-2-(thiophen-3-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l -(5-methyl-l ,3,4-oxadiazol-2-yl)-2-phenylethenyl]phenoxy}methyl)quinoline;
2-( {4- j 1 -(5-methyl-l, 3, 4-oxadiazol-2-yl)-2-(4-chlorophenyl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l-(5-methy]-l,3,4-oxadiazol-2-yl)-2-(4-fluorophenyl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[l-(5-methyl-l,3,4-oxadiazol-2-yl)-2-pyridin-4-ylethenyl]phenoxy}methyl)quinoline; 2-|'(4-{l-(5-methyl-l,3,4-oxadiazol-2-yl)-2-[4-(trifluoromethyl)phenyl]ethenyl}phenoxy) methyljquinoline;
2-({4-[l-(5-{trifluoromehyl}-l ,3,4-oxadiazol-2-yl)-2-pyridin-4-ylethenyl]phenoxy}methyl) quinoline;
2-({.4-[2-(4-chlorophenyl)-l-(5-methyl-4H-l,2,4-triazol-3-yl)ethenyl]phenoxy}methyl) imidazo [],2-a]pyridine;
2-({4-[2-(4-chlorophenyl)-l-(5-methyl-4H-l,2,4-triazol-3-yl)ethenyl]phenoxy}methyl) •quinoline;
5-{2-phenyl-l-f4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-l,3,4-oxadiazol-2(3H)-one;
5-{2-(4-chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-l,3,4-oxadiazol-2(3H)-one; 5-{2-(4-chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-l ,3,4-oxadiazol-2(3/ )- thione;
5-{2-(4-chlorophenyl)-l-[4-(quinolin-2-ylrnethoxy)phenyl]ethenyl}-2,4-dihydro-3/7-] ,2,4- triazole-3-thione;
5-{2-(4-chlorophenyl)-l-[4-(quinolin-2-ylmethoxy)phenyl]ethenyl}-4-ethyl-2,4-dihydro-3H-
1 ,2;4-triazole-3-thione;
2-({4-[2-(4-ch]orophenyl)-l-(4-ethyl-4H-l,2,4-triazol-3-yl)etheny]]phenoxy}melhyl)quinoline; 2-(2-{4-[2-(4-chlorophenyl)-l-(l,3,4-oxadiazol-2-yl)ethenyl]phenyl}ethyl)quinoline;
(e) the compound is selected from
2-({4-[2-(4-chlorophenyl)-2-(l,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinoline;
2-({4-[2-(4-chlorophenyl)-2-(5-methyl-l ,3,4-oxadiazol-2-yl)ethenyl]phenoxy}methyl)quinolinc:
(f) the compound is selected from
2-({4-[Ί -(4-chlorophenyl)-3-phenoxyprop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-[ -phenyl-3-(pyridin-3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-[Ί -(4-chlorophenyl)-3-(pyridin-3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-[] -(4-fluorophenyl)-3-(pyridin-3-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-|"l-phenyl-3-(pyridin-4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-["l-(4-chlorophenyl)-3-(pyridin-4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-|Ί -(4-fluorophenyl)-3-(pyridin-4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
2-({4-|' l-(pyridin-4-yl)-3-(pyridin-4-yloxy)prop-l-en-2-yl]phenoxy}methyl)quinoline;
or a pharmaceutically acceptable salt thereof.
23. The compound of claim 1 , wherein the compound is
3-(4-chloi phenyl)-2-[4-(quinolin-2-ylmethoxy)phenyl]prop-2-enenitrile or a
pharmaceutically acceptable salt thereof.
24. A pharmaceutical composition comprising a compound according to claims 1 to 23either as a free base or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients. carriers, diluents or mixture thereof.
25. A method for preventing, ameliorating or treating a phosphodiesterase 1 OA modulated disease, disorder or syndrome in a subject in need thereof, the method comprising administering to the subject a therapeutically effective amount of a compound according to any one of claims 1 to 23.
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN1418MU2010 | 2010-05-04 | ||
| IN1418/MUM/2010 | 2010-05-04 | ||
| US33490310P | 2010-05-14 | 2010-05-14 | |
| US61/334,903 | 2010-05-14 | ||
| IN3006/MUM/2010 | 2010-10-29 | ||
| IN3006MU2010 | 2010-10-29 | ||
| US41649810P | 2010-11-23 | 2010-11-23 | |
| US61/416,498 | 2010-11-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011138657A1 true WO2011138657A1 (en) | 2011-11-10 |
Family
ID=44903665
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2011/000948 Ceased WO2011138657A1 (en) | 2010-05-04 | 2011-05-03 | Aryl substituted olefinic compounds as pde10a inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2011138657A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012112946A1 (en) | 2011-02-18 | 2012-08-23 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014004064A1 (en) | 2012-06-29 | 2014-01-03 | E. I. Du Pont De Nemours And Company | Fungicidal heterocyclic carboxamides |
| WO2014071044A1 (en) | 2012-11-01 | 2014-05-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014172190A1 (en) | 2013-04-15 | 2014-10-23 | E. I. Du Pont De Nemours And Company | Fungicidal amides |
| EP2940022A1 (en) * | 2014-04-30 | 2015-11-04 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| US9200016B2 (en) | 2013-12-05 | 2015-12-01 | Allergan, Inc. | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| CN107216334A (en) * | 2017-06-29 | 2017-09-29 | 上海吉尔多肽有限公司 | A kind of synthetic method of 6 chlorine furans [3,2 B] pyridine |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| EP3498273A1 (en) * | 2017-12-14 | 2019-06-19 | Universität Wien | Pharmaceutical composition for modulating the response of a gaba-a receptor |
| CN114213424A (en) * | 2021-12-30 | 2022-03-22 | 杭州澳赛诺生物科技有限公司 | Synthetic method of furan [3, 2-b ] pyridine derivative |
| JP2022115888A (en) * | 2017-01-20 | 2022-08-09 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Inhibitors of n-terminal domain of androgen receptor |
| US20230285331A1 (en) * | 2022-01-24 | 2023-09-14 | University Of South Carolina | Tyrosine and resveratrol derivatives as novel modulators of cellular serine-adp-ribosylation |
| WO2025212579A1 (en) * | 2024-04-01 | 2025-10-09 | University Of South Carolina | Methods and compositions for inhibition of tyrosine and phenylalanine-mediated dna damage and repair |
| USRE50799E1 (en) | 2016-10-03 | 2026-02-17 | Hangzhou Highlightll Pharmaceutical Co., Ltd | JAK1 selective inhibitors and uses thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011999A1 (en) * | 1990-02-13 | 1991-08-22 | Merck & Co., Inc. | Angiotensin ii antagonists incorporating a substituted benzyl element |
| EP0669333A1 (en) * | 1994-02-24 | 1995-08-30 | J. URIACH & CIA. S.A. | New imidazopyridine derivatives as angiotensin II antagonists |
| CN1109057A (en) * | 1993-11-23 | 1995-09-27 | 默克专利股份有限公司 | Imidazopyridazine |
-
2011
- 2011-05-03 WO PCT/IB2011/000948 patent/WO2011138657A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991011999A1 (en) * | 1990-02-13 | 1991-08-22 | Merck & Co., Inc. | Angiotensin ii antagonists incorporating a substituted benzyl element |
| CN1109057A (en) * | 1993-11-23 | 1995-09-27 | 默克专利股份有限公司 | Imidazopyridazine |
| EP0669333A1 (en) * | 1994-02-24 | 1995-08-30 | J. URIACH & CIA. S.A. | New imidazopyridine derivatives as angiotensin II antagonists |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9670181B2 (en) | 2011-02-18 | 2017-06-06 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| US8772316B2 (en) | 2011-02-18 | 2014-07-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (PDE10A) |
| WO2012112946A1 (en) | 2011-02-18 | 2012-08-23 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinolinol derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014004064A1 (en) | 2012-06-29 | 2014-01-03 | E. I. Du Pont De Nemours And Company | Fungicidal heterocyclic carboxamides |
| WO2014071044A1 (en) | 2012-11-01 | 2014-05-08 | Allergan, Inc. | Substituted 6,7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (pde10a) |
| WO2014172190A1 (en) | 2013-04-15 | 2014-10-23 | E. I. Du Pont De Nemours And Company | Fungicidal amides |
| US9200016B2 (en) | 2013-12-05 | 2015-12-01 | Allergan, Inc. | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| US9902710B2 (en) | 2013-12-05 | 2018-02-27 | Exonhit Therapeutics, Sa | Substituted 6, 7-dialkoxy-3-isoquinoline derivatives as inhibitors of phosphodiesterase 10 (PDE 10A) |
| EP2940022A1 (en) * | 2014-04-30 | 2015-11-04 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| JP2017513915A (en) * | 2014-04-30 | 2017-06-01 | マサリコヴァ ユニヴェルジタ | Furopyridine as an inhibitor of protein kinases |
| WO2015165428A1 (en) * | 2014-04-30 | 2015-11-05 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| US9902733B2 (en) | 2014-04-30 | 2018-02-27 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| USRE50799E1 (en) | 2016-10-03 | 2026-02-17 | Hangzhou Highlightll Pharmaceutical Co., Ltd | JAK1 selective inhibitors and uses thereof |
| JP2022115888A (en) * | 2017-01-20 | 2022-08-09 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Inhibitors of n-terminal domain of androgen receptor |
| JP7443414B2 (en) | 2017-01-20 | 2024-03-05 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Inhibitor of the N-terminal domain of the androgen receptor |
| CN107216334A (en) * | 2017-06-29 | 2017-09-29 | 上海吉尔多肽有限公司 | A kind of synthetic method of 6 chlorine furans [3,2 B] pyridine |
| EP3498273A1 (en) * | 2017-12-14 | 2019-06-19 | Universität Wien | Pharmaceutical composition for modulating the response of a gaba-a receptor |
| EP3305786A2 (en) | 2018-01-22 | 2018-04-11 | Bayer CropScience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pesticides |
| CN114213424A (en) * | 2021-12-30 | 2022-03-22 | 杭州澳赛诺生物科技有限公司 | Synthetic method of furan [3, 2-b ] pyridine derivative |
| US20230285331A1 (en) * | 2022-01-24 | 2023-09-14 | University Of South Carolina | Tyrosine and resveratrol derivatives as novel modulators of cellular serine-adp-ribosylation |
| WO2025212579A1 (en) * | 2024-04-01 | 2025-10-09 | University Of South Carolina | Methods and compositions for inhibition of tyrosine and phenylalanine-mediated dna damage and repair |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011138657A1 (en) | Aryl substituted olefinic compounds as pde10a inhibitors | |
| US10562915B2 (en) | Bromodomain inhibitors | |
| EP1841757B1 (en) | Heteroaromatic quinoline compounds and their use as pde10 inhibitors | |
| AU2017382185C9 (en) | Pyrazole derivatives as MALT1 inhibitors | |
| AU2015289643B2 (en) | Therapeutic inhibitory compounds | |
| US20090176829A1 (en) | Bicyclic heteroaryl compounds as pde10 inhibitors | |
| JP2009522346A (en) | Bicyclic heteroaryl compounds as PDE10 inhibitors | |
| JP2010524974A (en) | Kinase inhibitors useful for the treatment of myeloproliferative disorders and other proliferative disorders | |
| EP3080100A1 (en) | Inhibitors of lysine specific demethylase-1 | |
| CA2643796A1 (en) | Polycyclic cinnamide derivatives | |
| TWI601724B (en) | Imidazo quinoline derivatives and salts thereof, preparation process and pharmaceutical use thereof | |
| WO2014155300A2 (en) | Substitued pyrimidine amine derivatives as tak-1 inhibitors | |
| WO2011132048A1 (en) | Heteroaryl compounds as pde10a inhibitors | |
| US20130158031A1 (en) | Alkyne-bridged hetero-aromatics and uses thereof | |
| WO2011132051A2 (en) | Tricycle compounds as phosphodiesterase-10 inhibitors | |
| CA2868240A1 (en) | Triazolo compounds as pde10 inhibitors | |
| EP2629616A2 (en) | Substituted amino-triazolyl pde10 inhibitors | |
| WO2014200885A1 (en) | PDE10a INHIBITORS FOR THE TREATMENT OF TYPE II DIABETES | |
| JP6977038B2 (en) | GSK-3 inhibitor | |
| CN104744484A (en) | 3-furan[2,3-b] pyridine-4-indol maleimide derivatives as well as preparation and application thereof | |
| CA3223874A1 (en) | Pyrimidine compounds for use as map4k1 inhibitors | |
| WO2025147320A1 (en) | Stat6 inhibitors and uses thereof | |
| EA048523B1 (en) | PYRIMIDINE COMPOUNDS FOR USE AS MAP4K1 INHIBITORS | |
| HK1113926A (en) | Heteroaromatic quinoline compounds and their use as pde10 inhibitors | |
| HK1167263B (en) | Heteroaromatic quinoline compounds and their use as pde10 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11777333 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11777333 Country of ref document: EP Kind code of ref document: A1 |