WO2011148888A1 - 複素環化合物及びh1受容体拮抗剤 - Google Patents
複素環化合物及びh1受容体拮抗剤 Download PDFInfo
- Publication number
- WO2011148888A1 WO2011148888A1 PCT/JP2011/061734 JP2011061734W WO2011148888A1 WO 2011148888 A1 WO2011148888 A1 WO 2011148888A1 JP 2011061734 W JP2011061734 W JP 2011061734W WO 2011148888 A1 WO2011148888 A1 WO 2011148888A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- ring
- compound
- substituent
- ylmethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- IZDDUTJSMOSXTR-UHFFFAOYSA-N C[n]1nccc1C(c(cc1)ccc1Cl)O Chemical compound C[n]1nccc1C(c(cc1)ccc1Cl)O IZDDUTJSMOSXTR-UHFFFAOYSA-N 0.000 description 1
- ZJSSDJCFSASGNQ-UHFFFAOYSA-N N#Cc1cc(CO)ccn1 Chemical compound N#Cc1cc(CO)ccn1 ZJSSDJCFSASGNQ-UHFFFAOYSA-N 0.000 description 1
- OKQQTGKKYOJTGB-UHFFFAOYSA-N N#Cc1nccc(CN(CC2)CCC2OC(c2ccccc2)c(cc2)ccc2F)c1 Chemical compound N#Cc1nccc(CN(CC2)CCC2OC(c2ccccc2)c(cc2)ccc2F)c1 OKQQTGKKYOJTGB-UHFFFAOYSA-N 0.000 description 1
- MYPPUKQXTQAXPR-UHFFFAOYSA-N OC(c1ccc(CN(CC2)CCC2OC(c2ccccc2)c2ccccc2)[nH]1)=O Chemical compound OC(c1ccc(CN(CC2)CCC2OC(c2ccccc2)c2ccccc2)[nH]1)=O MYPPUKQXTQAXPR-UHFFFAOYSA-N 0.000 description 1
- RDBOVDTWPZSLHG-UHFFFAOYSA-N OC(c1nc(C(N(CC2)CCC2OC(c2ccccc2)c2ccccc2)=O)ccc1)=O Chemical compound OC(c1nc(C(N(CC2)CCC2OC(c2ccccc2)c2ccccc2)=O)ccc1)=O RDBOVDTWPZSLHG-UHFFFAOYSA-N 0.000 description 1
- MWKMXUZXWKTXRH-UHFFFAOYSA-N OC(c1ncc[s]1)c(cc1)ccc1Cl Chemical compound OC(c1ncc[s]1)c(cc1)ccc1Cl MWKMXUZXWKTXRH-UHFFFAOYSA-N 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/38—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D333/40—Thiophene-2-carboxylic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to a heterocyclic compound (or pharmacologically active compound), an antiallergic agent (or H 1 receptor antagonist), and a pharmaceutical composition.
- JP-T-2007-534696 Patent Document 1 describes the formula [AH] -A (AH represents an antihistamine moiety, A represents a linker molecule containing SP and Z, and SP is a spacer molecule. , Z is a drug modulating moiety), a modified antihistamine compound is disclosed.
- This reference also describes ferramine-like compounds and diphenhydramine-like compounds.
- Patent Document 2 discloses a ⁇ opioid receptor agonist represented by the following formula.
- Ar 1 represents an optionally substituted 5- or 6-membered hydrocarbon ring or aromatic heterocyclic ring
- Z represents hydrogen, a carboxyl group or an ester group thereof
- G represents carbon or nitrogen
- R 2 represents hydrogen, a C 1-4 alkyl group, etc .
- R 3 and R 4 represent hydrogen or a C 1-6 alkyl group, R 4 may form a C ⁇ O group
- R 5 represents hydrogen, R 6 represents phenyl, halogen or the like
- R 7 represents hydrogen, C 1-8 alkyl group or the like
- WO 01/046166 Japanese Patent Publication No.
- Patent Document 3 describes compounds (benzhydril) and 6-membered compounds represented by the following formula for treating a condition associated with abnormal N-type calcium channel activity: Piperazine derivatives containing heterocyclic moieties) are disclosed and are also described as being useful in the treatment of stroke, migraine, chronic neuropathic pain and acute pain, hypertension, arrhythmias and the like.
- Z 1 and Z 2 represent N or CH, one of Z 1 and Z 2 is N; n 1 is 1, n 2 is 0 or 1; X 1 and X 2 are linear linkers; Ar represents an aromatic monocyclic ring or an aromatic heterocyclic monocyclic ring, Cy represents an aliphatic cyclic monocyclic ring or a heterocyclic monocyclic ring, and Ya and Yb represent an aromatic monocyclic ring or an aromatic heterocyclic ring. Cyclic monocyclic, aliphatic cyclic monocyclic or heterocyclic monocyclic, etc .; l 1 is 0 or 1; R 1 is C 1-6 alkyl, C 6-10 aryl, C 7-16 Arylalkyl etc.
- Patent Document 3 discloses that Z 1 and Z 2 are N, X 1 is a C 2-6 alkylene group or —C 1-5 alkylene-CO—, X 2 is a methylene group, an ethylene group or an alkenylene group, and Ya is cyclohexyl. Specifically described are compounds in which the group and the phenyl group, Ar is a phenyl group.
- Patent Document 4 discloses a hematopoietic prostaglandin D2 synthase inhibitor represented by the following formula, and has an antiallergic action, antiallergic inflammation, and / or antiasthma. It is also described that it has an action.
- X represents a single bond, a C 1-6 alkylene group
- R 1 represents a C 6-10 aryl group
- R 2 represents a hydrogen atom, a C 1-6 alkyl group, or a C 6-10.
- R 3 represents an aryl group
- R 3 represents an acyl group or a 4- to 10-membered cyclic group
- R 4 and R 5 represent a hydrogen atom
- R 4 and R 5 together represent C 1- 4 represents an alkylene group
- Patent Document 5 discloses a piperazine compound represented by the following formula useful for the treatment of obesity, psychiatric disorders and neurological disorders.
- G is selected from CH and N; R 1 and R 2 independently represent a hydrogen atom, etc .; m and n are independently selected from 1 to 5;
- X is a bond, —C ( L represents a bond, — (CH 2 ) p — or the like; p is selected from 0, 1 and 2;
- R 3 represents C 1-6 alkoxy, —CO 2 R 5 (R 5 represents hydrogen, C 1-6 alkyl, etc.) or a C 6-10 aryl group substituted with a substituent such as; at least one of X and L is not a bond)
- Patent Document 5 specifically describes a compound in which G is N and X is a carbonyl group.
- an object of the present invention is to provide a novel heterocyclic compound, an antiallergic agent (or an antihistamine, an H 1 receptor antagonist, etc.) and a pharmaceutical composition containing these components.
- Another object of the present invention not only have a wide safety margin toxicity (cardiotoxicity, carcinogenicity) no novel heterocyclic compounds, H 1 antiallergic agents having antagonism (or H 1 receptor antagonist) And providing a pharmaceutical composition comprising these components.
- Still another object of the present invention is to provide a novel heterocyclic compound having a high anti-inflammatory action and a little central nervous action, an antiallergic agent (or H 1 receptor antagonist) having an H 1 antagonistic action, and these It is to provide a pharmaceutical composition comprising an ingredient.
- Another object of the present invention is to provide a novel heterocyclic compound, an antiallergic agent, and a pharmaceutical composition containing these components that are effective in the late phase.
- Still another object of the present invention is to provide a method for producing the heterocyclic compound.
- the present inventors have found that a heterostructure having a skeleton in which an aromatic allocyclic or heterocyclic ring having a carboxyl group and a nitrogen atom of the heterocyclic ring are bonded via an alkylene group. It was found that the ring compound has high pharmacological activity (antiallergic action, etc.) and has a wide safety range and low toxicity (cardiotoxicity, carcinogenicity, etc.). The present invention has been completed as a result of further studies based on these findings.
- heterocyclic compound of the present invention is represented by a compound represented by the following formula (1) or a salt thereof.
- ring A represents an allocyclic or heterocyclic ring which may have a substituent
- ring B contains G and nitrogen atom N as constituent atoms of the ring, and may have a substituent.
- R 1 represents a carbonyl group or an alkylene group which may have a substituent;
- R 2a and R 2b are the same or different;
- An alkyl group which may have a substituent, a hydrocarbon ring which may have a substituent (a cycloalkyl group which may have a substituent, an aryl group which may have a substituent, etc.
- a heterocyclic group which may have a substituent a heterocyclic group which may have a substituent
- these rings a homocyclic group or a heterocyclic group
- Substituents may be bonded to each other to form a ring (hydrocarbon ring or heterocycle), and this hydrocarbon ring or heterocycle is substituted
- X represents an oxygen atom or a sulfur atom
- Z represents a hydroxyl group, an alkoxy group, a cycloalkyloxy group, an aryloxy group, an aralkyloxy group, an amino group, or an N-substituted amino group
- R 1 is an alkylene group which may have a substituent.
- ring A may be an aromatic ring optionally having at least one substituent selected from a halogen atom, an alkyl group, a haloalkyl group, an alkoxy group, and a haloalkoxy group. May be an aromatic allocyclic ring or an arene ring (such as a benzene ring) or an aromatic heterocyclic ring (such as a 5-membered or 6-membered heterocyclic ring).
- the ring A may be an aromatic heterocycle containing at least one heteroatom selected from a nitrogen atom, an oxygen atom and a sulfur atom, such as a pyridine ring.
- Ring B may be either an aliphatic or aromatic heterocyclic ring, for example, an aliphatic 4- to 10-membered heterocyclic ring (for example, an aliphatic 5- or 6-membered heterocyclic ring such as a piperidine ring or a piperazine ring). May be.
- R 1 may be a linear or branched alkylene group that may have a halogen atom (for example, a linear or branched C 1-4 alkylene group).
- R 1 may be a linear or branched C 1-3 alkylene group (C 1-2 alkylene group such as methylene group).
- R 2a and R 2b are the same or different and are each a halogen atom, an alkyl group (eg, a C 1-6 alkyl group), a haloalkyl group (eg, a halo C 1-6 alkyl group), an alkoxy group (eg, C 1-6 C 6-10 arene ring (eg benzene ring) or aromatic 5 optionally having at least one substituent selected from an alkoxy group) and a haloalkoxy group (eg a halo C 1-6 alkoxy group)
- R 2a and R 2b are the following formula (2):
- ring C1 and ring C2 are the same or different, a halogen atom, an alkyl group, a haloalkyl group, an optionally C 6-10 optionally having at least one substituent selected from alkoxy groups and haloalkoxy groups
- ring C3 represents a halogen atom, alkyl group, haloalkyl group, hydroxyl group, alkoxy group, haloalkoxy group, carboxyl group, alkoxycarbonyl group, acyl group and carbonyl group
- a 4- to 10-membered hydrocarbon ring or heterocyclic ring optionally having at least one substituent selected from:
- R 2a and R 2b are the same or different and are each a fluorine atom, a chlorine atom, a C 1-3 alkyl group, a halo C 1-3 alkyl group (eg, a fluoro C 1-3 alkyl group), a C 1-3 alkoxy It may be a benzene ring optionally having at least one substituent selected from a group and a halo C 1-3 alkoxy group (for example, a fluoro C 1-3 alkoxy group).
- Z is a hydroxyl group; C 1-6 alkoxy group (eg, C 1-4 alkoxy group); amino group; or C 1-6 alkyl group (eg, C 1-4 alkyl group), C 1-6 alkyl
- An N-substituted amino group in which at least one substituent selected from a carbonyl group (for example, a C 1-4 alkyl-carbonyl group) is substituted with a nitrogen atom may be a hydroxyl group; a C 1-3 alkoxy group (Eg, C 1-2 alkoxy group); amino group; C 1-3 alkyl group (eg, C 1-2 alkyl group), C 1-3 alkyl-carbonyl group (eg, C 1-2 alkyl-carbonyl group) N-substituted amino group in which at least one substituent selected from (1) is substituted with a nitrogen atom may be used.
- G is CH
- n is often 1 and when G is N, n is often 0.
- X is often an oxygen atom.
- Representative compounds represented by the formula (1) include, for example, (4-benzhydryloxypiperidin-1-ylmethyl) pyridinecarboxylic acid, ⁇ 4-[(halophenyl) phenylmethoxy] piperidin-1-ylmethyl ⁇ Pyridine carboxylic acid, ⁇ 4-[(C 1-4 alkylphenyl) phenylmethoxy] piperidin-1-ylmethyl ⁇ pyridine carboxylic acid, ⁇ 4-[(fluoro C 1-4 alkylphenyl) phenylmethoxy] piperidine-1- Ylmethyl ⁇ pyridinecarboxylic acid, ⁇ 4-[(C 1-4 alkoxyphenyl) phenylmethoxy] piperidin-1-ylmethyl ⁇ pyridinecarboxylic acid, ⁇ 4-[(fluoroC 1-4 alkoxyphenyl) phenylmethoxy] piperidine-1 -Ylmethyl ⁇ pyridinecarboxylic acid, ⁇ 4-
- the present invention includes the following reaction process formula
- Y 1 represents a halogen atom
- Z 1 represents an alkoxy group, a cycloalkyloxy group, an aryloxy group or an aralkyloxy group
- Y 2 represents a halogen atom
- R 5 represents an alkyl group or a cycloalkyl group.
- Aryl group and aralkyl group, ring A, ring B, R 1 , G, X, n, R 2a and R 2b are the same as above)
- Z is an alkoxy group, a cycloalkyloxy group, an aryloxy group or an aralkyloxy group in the formula (1), wherein Z is an alkoxy group, a cycloalkyloxy group or an aryl group.
- a heterocyclic compound that is an oxy group or an aralkyloxy group is subjected to hydrolysis or an amide bond forming reaction to produce a heterocyclic compound in which Z is a hydroxyl group, an amino group, or an N-substituted amino group, and Z is an amino group
- Z is a method for producing a heterocyclic compound in which Z is an N-alkylamino group or an N-acylamino group by subjecting the heterocyclic compound to an alkylation or acylation reaction.
- the said compound or its salt of this invention shows pharmacological activity (Antiallergic action, histamine H1 antagonistic action, anti-inflammatory action, etc.). Moreover, it has a wide safety range and is not toxic (genotoxicity such as cardiotoxicity, carcinogenicity, etc.) and is excellent in safety. Furthermore, the central nervous system action is small and does not cause arousal or drowsiness. Therefore, the present invention also includes antiallergic agents, antihistamines (or H 1 receptor antagonists), and anti-inflammatory agents including the above-described compounds or salts thereof (pharmaceutically acceptable salts).
- the present invention also provides an allergic disease (allergic symptoms, bronchial asthma, rhinitis, nasal congestion, atopic dermatitis, urticaria, eczema, skin pruritus) comprising the above compound or a salt thereof (pharmaceutically acceptable salt).
- an allergic disease allergic symptoms, bronchial asthma, rhinitis, nasal congestion, atopic dermatitis, urticaria, eczema, skin pruritus
- a salt thereof pharmaceutically acceptable salt
- the pharmaceutical composition of this invention contains the said compound or its pharmaceutically acceptable salt, and a carrier.
- the compound or a salt thereof includes a prodrug and an active metabolite.
- the heterocyclic compound of the present invention has high pharmacological activity (anti-allergic action, histamine H1 antagonistic action, anti-inflammatory action, etc.) and has a wide safety range as well as toxicity (genotoxicity such as cardiotoxicity and carcinogenicity). There is no safety. Furthermore, the central nervous system action is small and does not cause arousal or drowsiness. Therefore, it is useful as a medicine such as an antiallergic agent.
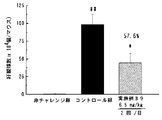
- FIG. 1 is a graph showing the results of the compound of Example 2 in Test Example 6.
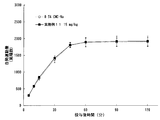
- FIG. 2 is a graph showing the results of the compound of Example 11 in Test Example 6.
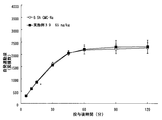
- FIG. 3 is a graph showing the results of the compound of Example 39 in Test Example 6.
- FIG. 4 is a graph showing the results of the compound of Example 2 in Test Example 7.
- FIG. 5 is a graph showing the results of the compound of Example 11 in Test Example 7.
- FIG. 6 is a graph showing the results of the compound of Example 39 in Test Example 7.
- FIG. 7 is a graph showing the results of the compound of Example 2 in Test Example 9.
- FIG. 8 is a graph showing the results of the compound of Example 11 in Test Example 9.
- FIG. 9 is a graph showing the results of the compound of Example 39 in Test Example 9.
- ring A represents an allocyclic ring or a heterocyclic ring, and is usually a 4- to 12-membered ring (preferably a 5- to 10-membered ring, more preferably a 5- or 6-membered ring).
- Ring A may be a condensed ring (such as a condensed ring of an allocyclic ring and a heterocyclic ring), but is usually a single ring.
- Ring A may be an aliphatic ring, but is preferably an aromatic ring in order to enhance pharmacological activity.
- Such an aromatic ring may be an aromatic allocyclic ring, an arene ring or an aromatic heterocyclic ring (such as a 5-membered or 6-membered heterocyclic ring).
- the ring A is usually a heterocyclic ring (for example, an aromatic heterocyclic ring containing at least one heteroatom selected from a nitrogen atom, an oxygen atom and a sulfur atom).
- the heterocycle may contain a plurality of the same or different heteroatoms (for example, two nitrogen atoms, nitrogen and oxygen atoms and / or sulfur atoms).
- Examples of the ring A include aliphatic hydrocarbon rings (C 4-10 cycloalkane rings such as cyclopentane ring and cyclohexane ring), aromatic hydrocarbon rings (C 6-10 arene rings such as benzene ring and naphthalene ring).
- aliphatic heterocycle (5- or 6-membered ring containing an oxygen atom such as pyran as a heteroatom; a condensed ring of a 5- or 6-membered ring containing an oxygen atom such as isochroman or chroman as a heteroatom and a hydrocarbon ring; Pyrrolidine, pyrroline, imidazolidine, imidazoline, pyrazolinidine, pyrazoline, piperidine, piperazine and other nitrogen atoms containing 5 or 6-membered rings as heteroatoms, aromatic heterocycles (including sulfur atoms such as thiophene as heteroatoms 5 or 6 Member ring: condensed ring of a 5- or 6-membered ring containing a sulfur atom as a hetero atom and a hydrocarbon ring (such as a benzene ring) A 5- or 6-membered ring containing an oxygen atom such as furan as a heteroatom; a condensed ring of a 5-
- Preferred ring A is, for example, a benzene ring, a heterocyclic ring (a 5-membered ring such as thiophene, furan, pyrrole, pyrazole, thiazole, oxazole, a 6-membered ring such as pyridine, pyrazine, pyrimidine, pyridazine, etc.), particularly at least a nitrogen atom hetero Includes aromatic heterocycles (pyridine, pyrazine, etc.) contained as atoms.
- a heterocyclic ring a 5-membered ring such as thiophene, furan, pyrrole, pyrazole, thiazole, oxazole, a 6-membered ring such as pyridine, pyrazine, pyrimidine, pyridazine, etc.
- aromatic heterocycles pyridine, pyrazine, etc.
- Ring A may have a substituent.
- substituents include a halogen atom (fluorine atom, chlorine atom, bromine atom, iodine atom), an alkyl group (eg, methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, isobutyl, t-butyl, pentyl, Linear or branched C 1-10 alkyl group such as isopentyl, neopentyl, hexyl group), haloalkyl group [fluoromethyl group (such as trifluoromethyl group), fluoroethyl group (2,2,2-trifluoroethyl) Groups, perfluoroethyl groups, etc.), 3,3,3,2,2-pentafluoropropyl groups, perfluoropropyl groups, etc.), halo C 1-6 alkyls such as chloroalkyl groups corresponding to these fluoroalkyl
- C 6-10 aryl such as benzoyl group - carbonyl group, C 6-10 aryl -C 1-4 alkyl such as benzyl carbonyl group - carbonyl group
- an acyloxy group an acetoxy group, propionyloxy group, like a linear, such as butyryloxy or branched C 2- 6 acyloxy group
- carboxyl group alkoxycarbonyl group [methoxycarbonyl group, ethoxycarbonyl group, n-propoxycarbonyl group, isopropoxycarbonyl group, n-butoxycarbonyl group, isobutoxycarbonyl group, s-butoxycarbonyl group, t
- a linear or branched C 1-6 alkoxy-carbonyl group such as a butoxycarbonyl group]
- a carbamoyl group an N-substituted carbamoyl group [for example, an N-monoC 1-6
- a halogen atom fluorine atom, chlorine atom, etc.
- a linear or branched C 1-6 alkyl group preferably a C 1-4 alkyl group, more preferably a C 1-2 alkyl group
- Linear or branched haloalkyl group [fluoro C 1-6 alkyl group such as trifluoromethyl group, 2,2,2-trifluoroethyl group, perfluoroethyl group, preferably fluoro C 1-4 alkyl Group, more preferably a fluoro C 1-3 alkyl group), a chloroalkyl group corresponding to these fluoroalkyl groups, etc.]
- a linear or branched C 1-6 alkoxy group preferably a C 1-4 alkoxy group
- a linear or branched haloalkoxy group [trifluoromethoxy group, Fluor
- the substitution position of the substituent with respect to ring A is not particularly limited, and may be 2-, 3-, 4-, 5-position, etc. for the homocyclic ring based on the bonding position of R 1. Then, depending on the position of the heteroatom, it may be in the 2-, 3-, 4-, 5-position or the like.
- the substituent may be substituted with a hetero atom (nitrogen atom) in the heterocyclic ring, but is usually substituted with a carbon atom in many cases.
- substitution position of the group —C (O) —Z with respect to ring A may also be the 2-, 3-, 4-, 5-position, etc. of ring A based on the bonding position of R 1 .
- ring A when ring A is a benzene ring, it may be o-, m-, p-position, etc., and when ring A is a pyridine ring, it is based on the nitrogen atom constituting the ring. , 2-, 3-, 4-position, etc.
- Ring B represents a heterocyclic ring containing G and a nitrogen atom N as ring constituent atoms as represented by Formula (1), and G in Ring B represents CH or N.
- Ring B is usually a 4- to 12-membered ring (for example, a 4- to 10-membered ring, preferably a 5- to 10-membered ring, more preferably a 5- to 8-membered ring (for example, a 5-membered or 6-membered ring)). May be.
- Ring B may be either an aliphatic or aromatic heterocycle, and examples of the aliphatic heterocycle include azetidine, pyrrolidine, pyrroline, imidazolidine, imidazoline, pyrazolinidine, pyrazoline, piperidine, piperazine, 1,4
- a 4- to 8-membered ring (for example, a 5-membered or 6-membered ring) containing at least one nitrogen atom as a heteroatom such as diazepan can be exemplified, and examples of the aromatic heterocycle include pyrrole, imidazole, pyrazole, etc.
- 5-, 6-, or 7-membered rings containing at least one nitrogen atom as a heteroatom 5- or 6-membered rings containing a nitrogen atom as a heteroatom such as indole, carbazole, indoline, isoindoline, and a hydrocarbon ring (such as a benzene ring)
- a 5- or 6-membered ring containing a nitrogen atom such as purine as a heteroatom, and Condensed with 5 or 6 membered heterocyclic species or heterologous; and 5 or 6-membered ring containing a nitrogen atom and an oxygen atom, such as morpholine as a hetero atom can be exemplified.
- Preferred examples of the ring B include an aliphatic 5- or 6-membered heterocyclic ring such as a piperidine ring and a piperazine ring.
- Ring B may have a substituent, for example, the same substituent as the ring A (halogen atom, alkyl group, alkoxy group, etc.).
- R 1 represents a carbonyl group or an alkylene group, and this alkylene group may have a substituent.
- the alkylene group include linear or branched C, such as methylene, ethylene, ethane-1,1-diyl, trimethylene, propylene, propane-2,2-diyl, tetramethylene, pentamethylene, and hexamethylene groups. Examples thereof include 1-10 alkylene groups.
- Preferred alkylene groups are C 1-6 alkylene groups, such as linear or branched C 1-4 alkylene groups, especially linear or branched C 1-3 alkylene groups (eg C, such as methylene groups). 1-2 alkylene group), especially a methylene group.
- the substituent of the alkylene group is the same as that of ring A (halogen atom, haloalkyl group, alkoxy group, haloalkoxy group, acyl group, acyloxy group, carboxyl group, alkoxycarbonyl group, carbamoyl group, N-substituted carbamoyl group, Amino group, N-substituted amino group, cyano group, nitro group and the like.
- Preferable substituents include, for example, a halogen atom (fluorine atom, chlorine atom, bromine atom, iodine atom), haloalkyl group [halo C 1-4 alkyl group such as trifluoromethyl group, trichloromethyl group, etc.], alkoxy group (methoxy C 1-4 alkoxy groups such as ethoxy, propoxy, isopropoxy and butoxy groups), haloalkoxy groups [halo C 1-4 alkoxy groups such as trifluoromethoxy group] and the like.
- the alkylene group is usually unsubstituted or has a halogen atom (fluorine atom, chlorine atom, etc.) in many cases.
- R 1 is a substituted (having a substituent) or unsubstituted (having no substituent) alkylene group has higher pharmacological activity than a compound in which R 1 is a carbonyl group. Therefore, R 1 is preferably a substituted or unsubstituted alkylene group (for example, an unsubstituted short chain alkylene group such as a methylene group).
- R 2a and R 2b are the same or different and are an alkyl group, homocyclic ring or hydrocarbon ring group [hydrocarbon ring group [cycloalkyl group (aliphatic homocyclic group)], aryl group (aromatic homocyclic group). ] Or a heterocyclic group (aliphatic or aromatic heterocyclic group).
- alkyl group include linear or branched C 1-10 alkyl groups such as methyl, ethyl, propyl, isopropyl, butyl, s-butyl, isobutyl, t-butyl, pentyl, isopentyl, neopentyl, and hexyl groups. Etc. can be exemplified.
- hydrocarbon ring cycloalkyl group examples include C 4-10 cycloalkyl groups such as cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl groups.
- aryl group aromatic homocyclic group
- aryl groups examples include C 6-10 aryl groups such as a phenyl group and a naphthyl group.
- heterocyclic group examples include the heterocyclic ring exemplified in the above ring A (a 5-membered or 6-membered heterocyclic ring containing at least one heteroatom selected from a nitrogen atom, an oxygen atom, and a sulfur atom, Examples thereof include a group corresponding to a hydrogen ring (such as a condensed ring with a benzene ring), and the heterocyclic group may be an aliphatic heterocyclic group or an aromatic heterocyclic group.
- R 2a and R 2b are aromatic rings such as C 6-10 arene rings (eg benzene rings), or aromatic 5 containing at least one heteroatom selected from a nitrogen atom, an oxygen atom and a sulfur atom.
- a 6-membered heterocyclic ring for example, an aromatic heterocyclic ring such as a pyridine ring containing a nitrogen atom as a hetero atom
- a benzene ring is particularly preferable.
- R 2a and R 2b are preferably at least one is a benzene ring, a combination of both of R 2a and R 2b is a benzene ring, R 2a and R 2b Any one of which is a benzene ring and the other is a cycloalkane ring (for example, a C 5-7 cycloalkane ring such as a cyclohexane ring) or at least one heteroatom selected from a nitrogen atom, an oxygen atom, and a sulfur atom
- a combination that is an aromatic 5-membered or 6-membered heterocyclic ring for example, an aromatic heterocyclic ring containing a nitrogen atom as a heteroatom such as pyrrole, pyrazole, imidazole, pyridine, oxazole, thiazole
- an aromatic 5-membered or 6-membered heterocyclic ring for example, an aromatic heterocyclic ring containing a nitrogen atom as a heteroatom such as
- R 2a and R 2b may have the same or different substituents.
- substituents for R 2a and R 2b include a halogen atom, an alkyl group (for example, a linear or branched C 1-6 alkyl group), and a cycloalkyl group (for example, a C 4-10 cycloalkyl group).
- an aryl group e.g., C 6-10 aryl group
- a haloalkyl group e.g., linear or branched halo-C 1-6 alkyl group
- an alkoxy group e.g., linear or branched C 1- 6 alkoxy group
- haloalkoxy group e.g., linear or branched halo C 1-6 alkoxy group
- acyl group eg, linear or branched C 1-6 alkyl-carbonyl group
- acyloxy group e.g., linear or branched C 2-6 acyloxy group
- a carboxyl group an alkoxycarbonyl group (e.g., straight-chain or branched-chain C 1-6 alkoxy - carbonyl group)
- Carbamoyl group N- substituted carbamoyl group (e.g., C 1-6 alkylcarbamoyl, C 1-6 acyl - such as carbamoy
- these substituents may further include a substituent (a halogen atom such as a fluorine atom, a linear or branched C 1-4 alkyl group, a linear or branched chain, if necessary.
- a substituent a halogen atom such as a fluorine atom, a linear or branched C 1-4 alkyl group, a linear or branched chain, if necessary.
- C 1-4 alkoxy groups linear or branched halo C 1-4 alkyl group, a linear or branched halo C 1-4 alkoxy group, a linear or branched C 1-4 alkyl -
- a carbonyl group, a carboxyl group, a linear or branched C 1-6 alkoxy-carbonyl group, etc. may be substituted to form an alkoxyalkyl group, an alkoxyalkoxy group, a carboxyalkyl group, or the like.
- Preferred substituents include a halogen atom (eg, fluorine atom, chlorine atom), a linear or branched C 1-3 alkyl group, a linear or branched halo C 1-3 alkyl group (eg, fluoro C 1-3 alkyl group), linear or branched C 1-3 alkoxy group, linear or branched halo C 1-3 alkoxy group (for example, fluoro C 1-3 alkoxy group) and the like it can.
- a halogen atom eg, fluorine atom, chlorine atom
- a linear or branched C 1-3 alkyl group eg, a linear or branched halo C 1-3 alkyl group
- linear or branched C 1-3 alkoxy group eg, fluoro C 1-3 alkyl group
- linear or branched halo C 1-3 alkoxy group for example, fluoro C 1-3 alkoxy group
- R 2a and R 2b The types of these substituents may be the same or different in R 2a and R 2b . Moreover, these substituents may be substituted with R 2a and R 2b singly or in combination of two or more. Furthermore, in R 2a and R 2b , the substitution positions of the substituents may be different. For example, the same substituents (halogen atom, alkyl group, haloalkyl group, alkoxy group, haloalkoxy group, etc.) are the same in R 2a and R 2b (for example, 2-, 3-, 4-position etc.
- R 2a may be substituted with a halogen atom, an alkyl group, a haloalkyl group, an alkoxy group, a haloalkoxy group, or the like, and R 2b may be unsubstituted.
- R 2a and R 2b are benzene rings
- the substituent may be substituted at any of the 2-, 3-, and 4-positions.
- 4-position is substituted with a substituent (for example, at least one position selected from 2-, 3-, 4-position of one benzene ring is substituted with one or more substituents), and the other
- the benzene ring may be unsubstituted.
- each of one benzene ring and the other benzene ring may be substituted with different substituents (for example, the substituent is substituted at the 2- or 3-position of one benzene ring, A substituent may be substituted at the 4-position of the other benzene ring).
- R 2a and R 2b are represented by the following formula (2):
- ring C1 and ring C2 are the same or different and are arene rings (eg, C 6-10 arene rings such as benzene rings) or heterocycles (eg, aromatic 5-membered or 6-membered heterocycles) Group or aromatic heterocycle).
- Ring C1 and Ring C2 may be the same or different substituents, for example, a halogen atom (a fluorine atom, a chlorine atom, etc.), an alkyl group (a linear or branched C 1-6 alkyl group, etc.), a haloalkyl group (trifluoromethyl)
- a linear or branched halo C 1-6 alkyl group such as a group
- an alkoxy group such as a linear or branched C 1-6 alkoxy group
- a haloalkoxy group such as a trifluoromethoxy group. It may have at least one substituent selected from a chain or branched halo C 1-6 alkoxy group and the like.
- Ring C1 and ring C2 may be substituted with one or more substituents.
- the substitution site of the substituent is not particularly limited.
- the 1-position to the 4-position for example, the 2-position
- the binding site between ring C3 and ring C1 or ring C2 the binding site on the left side in formula (2).
- it may be located at the 3-position).
- Ring C3 is usually 4- to 10-membered (preferably 5- to 8-membered ring, particularly 6- to 8-membered ring).
- the hydrocarbon ring or heterocyclic ring of ring C3 may be an aliphatic ring or an aromatic ring, respectively, and is usually an aliphatic ring in many cases.
- the hydrocarbon ring is an aliphatic hydrocarbon ring, for example, cycloalkane ring (C 4-10 cycloalkane ring such as cyclohexane ring, cycloheptane ring, cyclooctane ring, etc.), cycloalkene ring (cyclohexene ring, cycloheptene ring, C 4-10 cycloalkene ring such as cyclooctene ring), C 4-10 cycloalkadiene ring (cyclooctadiene ring etc.) and the like, and aromatic hydrocarbon ring (C 6- 10 arene rings).
- cycloalkane ring C 4-10 cycloalkane ring such as cyclohexane ring, cycloheptane ring, cyclooctane ring, etc.
- cycloalkene ring cyclohexene ring, cycloheptene
- the heterocyclic ring contains at least one heteroatom selected from a nitrogen atom, an oxygen atom and a sulfur atom as a constituent atom of the ring.
- the heterocyclic ring may be a ring containing an oxygen atom as a hetero atom (for example, a 6-8 membered heterocyclic ring such as an oxepin ring).
- the heteroatom of ring C3 is from the 1-position to the 3-position (for example, 1-position) from the bond site between ring C3 and ring C1 or ring C2 (the bond site on the left side in formula (2)). -Position or 2-position).
- Ring C3 may have a substituent similar to the substituents of R 2a and R 2b .
- substituents include a halogen atom (a fluorine atom, a chlorine atom, etc.), an alkyl group (for example, a linear or branched C 1-6 alkyl group), and a haloalkyl group (for example, a linear or branched chain).
- Halo C 1-6 alkyl group hydroxyl group, alkoxy group (eg linear or branched C 1-6 alkoxy group), haloalkoxy group (eg linear or branched halo C 1-6 Alkoxy group), carboxyl group, alkoxycarbonyl group (eg, linear or branched C 1-6 alkoxy-carbonyl group), acyl group (eg, linear or branched C 1-6 alkyl-carbonyl group) And at least one substituent selected from a carbonyl group (or an oxo group). That is, one or more substituents may be substituted on ring C3.
- the substitution site of the substituent is not particularly limited.
- the 1-position to the 3-position for example, the 2-position from the binding site between ring C3 and ring C1 or ring C2 (the binding site on the left side in formula (2)).
- 3-position the substitution site of the substituent is not particularly limited.
- the 1-position to the 3-position for example, the 2-position from the binding site
- the bond represented by the following Formula (3) represents a single bond or a double bond, and when n is 0 and G is CH, the bond is a double bond. Indicates.
- X represents an oxygen atom or a sulfur atom, and is usually an oxygen atom.
- alkoxy group represented by Z include the same alkoxy groups as the substituent of the ring A.
- Preferred alkoxy groups are linear or branched C 1-6 alkoxy groups (eg, C 1-4 alkoxy groups).
- Examples of the cycloalkyloxy group include C 4-10 cycloalkyloxy groups such as cyclobutyloxy, cyclopentyloxy, cyclohexyloxy, cycloheptyloxy, and cyclooctyloxy groups.
- Examples of the aryloxy group include C 6-10 aryloxy groups such as phenoxy group and naphthoxy group.
- Examples of the aralkyloxy group include C 6-10 aryl-C 1-4 alkyloxy groups such as benzyloxy and phenethyloxy groups.
- Examples of the N-substituted amino group include the same N-substituted amino groups as those of the ring A.
- N-substituted amino groups include N, N-dimethylamino group, N, N-diC 1-6 alkylamino group such as N, N-diethylamino group, and N—C 1- 1 such as N-acetylamino group. Examples thereof include a 6 alkyl-carbonylamino group and an N, N-diC 1-6 alkyl-carbonylamino group.
- Preferred Z is a hydroxyl group; a C 1-3 alkoxy group (eg C 1-2 alkoxy group); an amino group; or a C 1-3 alkyl group (eg C 1-2 alkyl group) on the nitrogen atom, C 1 A N-substituted amino group (mono- or di-substituted amino group) substituted by a -3 alkyl-carbonyl group (for example, C 1-2 alkyl-carbonyl group) as a substituent.
- Z may form a prodrug or an active metabolite that produces an active compound in vivo.
- N 0 or 1.
- n When G is CH, n is often 1 and when G is N, n is often 0. Further, when G is CH, X is often an oxygen atom.
- R 1 is an alkylene group which may have a substituent.
- R 1 is a carbonyl group, ring A is not a benzene ring but a heterocyclic ring.
- representative compounds can be represented by, for example, the following formula (1a), particularly (1b).
- n is 1 when G is CH
- n is 0 when G is N
- ring C1 and ring C2 are The same or different
- C 6-10 arene ring (for example, benzene ring) or aromatic 5-membered or 6-membered heterocyclic ring
- R 4a and R 4b are the same or different, hydrogen atom, fluorine atom, chlorine atom, etc.
- a halogen atom, a linear or branched C 1-4 alkyl group, a linear or branched halo C 1-4 alkyl group, a linear or branched C 1-4 alkoxy group and a linear or branched halo C 1-4 shows a substituent selected from alkoxy groups, in the ring C1 and ring C2, R 4a and R 4b kinds may be different and be identical .R 4a and R 4b, taken together, hydrocarbon ring of said ring C3 (4 ⁇ 10 membered, or Atom may form a heterocyclic ring) containing as a hetero atom and the ring C3 may have a substituent such as the halogen atom.)
- R 4a and R 4b are the same as above, and ring C 1 And in ring C2, the types of R 4a and R 4b may be the same or different.
- Specific examples of the compound represented by the formula (1a) or (1b) include the following compounds.
- the heterocyclic compound includes a salt of the compound of the formula (1) (such as a salt with a pharmacologically acceptable acid or base).
- Acids that form such salts include inorganic acids (hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, etc.), organic acids (acetic acid, propionic acid, trichloroacetic acid, trifluoroacetic acid, oxalic acid, succinic acid)
- Organic carboxylic acids such as fumaric acid and maleic acid, oxycarboxylic acids such as lactic acid, malic acid, tartaric acid and citric acid, and sulfonic acids such as methanesulfonic acid and toluenesulfonic acid).
- the base examples include inorganic bases (alkali metal hydroxides such as ammonia, sodium hydroxide and potassium hydroxide, alkaline earth metal hydroxides such as alkali metal carbonate, calcium hydroxide and magnesium hydroxide, calcium carbonate) Alkaline earth metal carbonates such as, etc.), organic bases (alkylamines such as triethylamine, alkanolamines such as ethanolamine, polyamines such as alkylenediamine, etc.) and the like. These acids or bases can be used alone or in combination of two or more.
- the heterocyclic compound or a salt thereof may be an anhydride or a hydrate, and may be a solvate (such as a solvate using an organic solvent such as ethanol).
- the heterocyclic compound or a salt thereof includes not only a hydrate or solvate of the compound of the formula (1) or a salt thereof but also an isolated crystal (such as a polymorphic substance).
- the heterocyclic compound of the present invention or a salt thereof is a tautomer of the compound of the formula (1) or a salt thereof, an optically active substance having an asymmetric carbon atom ((R) isomer, (S) isomer, dia Stereomers, etc.), racemates, or mixtures thereof.
- the heterocyclic compound or a salt thereof is a prodrug compound in which the group —C (O) —Z of the compound of the formula (1), the heterocyclic group of ring A, etc. are modified to express the activity in vivo ( Or an active metabolite).
- the prodrug form include compounds that exhibit activity by metabolism such as hydrolysis, oxidation, reduction, transesterification (eg, ester form, ether form, alcohol form, amide form, etc. of the compound of formula (1)). Is mentioned.
- the heterocyclic compound represented by the formula (1) or a salt thereof can be produced according to a conventional method, for example, the following reaction process formula.
- Y 1 represents a halogen atom
- Z 1 represents an alkoxy group, a cycloalkyloxy group, an aryloxy group or an aralkyloxy group
- ring A, ring B, R 1 , G, X, n, R 2a , R 2b is the same as above
- the halogen atom represented by Y 1 represents a fluorine atom, a chlorine atom, a bromine atom or an iodine atom, and is usually a chlorine atom or a bromine atom.
- Examples of the alkoxy group represented by Z 1 include the same alkoxy groups as those described above for Z, and usually a linear or branched C 1-6 alkoxy group (preferably a C 1-4 alkoxy group, more preferably C 1-2 alkoxy group).
- Examples of the cycloalkyloxy group include C 4-10 cycloalkyloxy groups such as a cyclohexyloxy group
- examples of the aryloxy group include C 6-10 aryloxy groups such as a phenoxy group
- examples of the aralkyloxy group include: Examples thereof include C 6-10 aryl-C 1-4 alkyloxy groups such as benzyloxy group and phenethyloxy group.
- Z 1 is usually an alkoxy group or an aralkyloxy group.
- the compound represented by the formula (14) can be generated by the reaction between the compound represented by the formula (12) and the compound represented by the formula (13).
- the reaction can be usually performed in a solvent inert to the reaction in the presence of a base.
- the amount of compound (13) to be used is about 0.8 to 5 mol, preferably 1 to 3 mol, more preferably about 1.1 to 2 mol, per 1 mol of compound (12).
- the solvent examples include hydrocarbons (aliphatic hydrocarbons such as hexane, alicyclic hydrocarbons such as cyclohexane, aromatic hydrocarbons such as toluene), esters (such as ethyl acetate), ketones ( Acetone, methyl ethyl ketone, etc.), ethers (chain ethers such as diethyl ether and diisopropyl ether, cyclic ethers such as dioxane, tetrahydrofuran), nitriles (acetonitrile, propionitrile, benzonitrile, etc.), amides (N, N-dimethylformamide, N, N-dimethylacetamide, etc.), sulfoxides (dimethylsulfoxide, etc.), sulfolane, alcohols (ethanol, isopropanol, etc.) and the like.
- hydrocarbons aliphatic hydrocarbons such as hexane, alicycl
- the base examples include inorganic bases (alkali metal hydroxides such as sodium hydroxide and potassium hydroxide, alkali metal carbonates, alkaline earth metal hydroxides such as calcium hydroxide and magnesium hydroxide, alkaline earth metal carbonates). And organic bases (amines such as trimethylamine, triethylamine, triethanolamine, dimethylaminoethanol, etc.). These bases can be used alone or in combination of two or more.
- the amount of the base used may be an equivalent amount or an excess amount (for example, about 1 to 10 equivalents, preferably about 1.1 to 5 equivalents) relative to 1 mol of the compound (2).
- the reaction can be performed, for example, at a temperature of about 0 to 150 ° C. (preferably 10 to 100 ° C., more preferably room temperature (about 20 to 25 ° C.) to 50 ° C.).
- the reaction can be carried out in air or under an inert gas atmosphere (nitrogen, helium, argon gas, etc.).
- the reaction may be performed under pressure, but is usually performed under atmospheric pressure.
- the resulting compound (14) is separated from the conventional separation or purification (or isolation) method such as filtration, distillation, solvent removal, precipitation, crystallization, recrystallization, decantation, extraction, drying, washing, You may isolate
- a compound in which Z is a hydroxyl group in the formula (1) can be obtained.
- the hydrolysis can be carried out in a conventional manner, for example, in a solvent in the presence of a hydrolysis catalyst (for example, an inorganic base such as sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, potassium hydrogen carbonate).
- a hydrolysis catalyst for example, an inorganic base such as sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, potassium hydrogen carbonate.
- a hydrolysis catalyst for example, an inorganic base such as sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, potassium hydrogen carbonate.
- the solvent for example, various solvents exemplified above (for example, alcohols) can be used, and water may be added to the reaction system.
- the hydrolysis can be carried out at a suitable temperature, for example, 30 to 150 ° C. or under reflux.
- the hydrolysis reaction is carried out by reacting the compound (12) with the compound (13) without
- reaction product After completion of the reaction, the reaction product can be separated or purified (or isolated) in the same manner as described above.
- a compound in which G is N is known, and a compound in which G is CH is a conventional method, for example, the following formula (12a) in the presence of an acid catalyst.
- a compound represented by the formula (12b) can be prepared by dehydration condensation.
- the acid catalyst examples include inorganic acids such as sulfuric acid and organic acids such as p-toluenesulfonic acid, and Lewis acids can also be used.
- the reaction is usually carried out in a solvent, and a hydrophobic solvent such as hydrocarbons (hexane, toluene, etc.), ethers, etc. can be used for dehydration condensation.
- the reaction can be performed, for example, at about 50 to 200 ° C. (for example, 75 to 150 ° C.), and can be performed under reflux of the solvent.
- Y 2 represents a halogen atom
- R 5 represents an alkyl group, a cycloalkyl group, an aryl group, or an aralkyl group
- ring A, ring B, R 1 , G, X, n, R 2a , R 2b Is the same as above
- the halogen atom represented by Y 2 represents a fluorine atom, a chlorine atom, a bromine atom or an iodine atom, and is usually a chlorine atom or a bromine atom.
- Examples of the alkyl group represented by R 5 include an alkyl group corresponding to the same alkoxy group as Z described above, and usually a linear or branched C 1-6 alkyl group (preferably a C 1-4 alkyl group). Group).
- Examples of the cycloalkyl group, aryl group, and aralkyl group represented by R 5 include the same C 4-10 cycloalkyl group, C 6-10 aryl group, and C 6-10 aryl-C 1-4 alkyl group as described above. Can be illustrated.
- a compound represented by formula (17) is obtained by reacting a compound represented by formula (15), a compound represented by formula (16), and carbon monoxide in the presence of a catalyst (carbonylation reaction). Can be generated.
- the reaction is usually performed in a solvent in the presence of a base.
- the amount of compound (16) to be used is equimolar or more per mole of compound (15) (usually an excess mole, for example, 1.5 to 1000 moles, preferably 2 to 500 moles, more preferably 5 to 200 moles). In particular, about 10 to 100 mol).
- the amount of carbon monoxide used is also an excess amount (for example, 2 to 1000 mol, preferably 5 to 500 mol, etc.) relative to 1 mol of compound (15), and is usually in a carbon monoxide atmosphere or carbon monoxide.
- the reaction can be carried out in an air stream or by blowing carbon monoxide into the reaction system.
- the catalyst examples include a carbonylation catalyst such as a palladium catalyst (palladium acetate, palladium sulfate, palladium complex, etc.), a rhodium catalyst, and the like.
- the amount of the catalyst used is, for example, 0.1 to 50 mol%, preferably 0.5 to 20 mol%, more preferably 1 to 10 mol% (for example, relative to the amount of the compound represented by the formula (15)). 3 to 7 mol%).
- phosphines for example, 1,3-bis (diphenylphosphino) propane, triphenylphosphine, etc.
- the amount of the cocatalyst used is, for example, 0.1 to 50 mol%, preferably 0.5 to 20 mol%, more preferably 1 to 10 mol% (based on the amount of the compound represented by the formula (15). For example, it may be about 3 to 7 mol%).
- Solvents include hydrocarbons (aliphatic hydrocarbons such as hexane, alicyclic hydrocarbons such as cyclohexane, aromatic hydrocarbons such as toluene), halogenated hydrocarbons (such as chloroform, dichloromethane, and trichloroethane).
- hydrocarbons aliphatic hydrocarbons such as hexane, alicyclic hydrocarbons such as cyclohexane, aromatic hydrocarbons such as toluene
- halogenated hydrocarbons such as chloroform, dichloromethane, and trichloroethane
- Esters such as ethyl acetate), ketones (such as acetone and methyl ethyl ketone), ethers (chain ethers such as diethyl ether and diisopropyl ether, cyclic ethers such as dioxane and tetrahydrofuran), nitriles (acetonitrile, propio) Nitrile, benzonitrile, etc.), amides (N, N-dimethylformamide, N, N-dimethylacetamide, etc.), sulfoxides (dimethylsulfoxide, etc.), sulfolane, alcohols (ethanol, isopropanol, etc.), etc. It is.
- the reaction can be performed at a temperature of, for example, about 20 to 150 ° C. (preferably 50 to 120 ° C., more preferably 50 to 100 ° C.).
- the reaction can be carried out in air or under an inert gas atmosphere (nitrogen, helium, argon gas, etc.).
- the reaction may be performed under atmospheric pressure or under pressure.
- the produced compound (17) may be separated or purified by the same conventional separation or purification (or isolation) method as described above.
- the compound (17) in which Z is a hydroxyl group can be obtained by hydrolyzing the compound (17). Hydrolysis can be performed in the same manner as the hydrolysis of the compound (14). Further, after completion of the reaction, the reaction product can be separated or purified (or isolated) in the same manner as described above.
- the compound (15) can be prepared by a conventional method, for example, by reacting a compound represented by the following formula (15a) with a compound represented by the formula (15b).
- Y 1 and Y 2 are the same type of halogen atom
- Y 1 preferentially reacts with NH in ring B of compound (15b).
- Preferred Y 1 may be a halogen atom having a higher detachability than Y 2 (for example, if Y 2 is a chlorine atom, Y 1 is a bromine atom).
- the compound (1) contains a basic group (amino group, imino group, heterocyclic basic nitrogen atom, etc.) or an acidic group (carboxyl group, sulfonyl group, etc.), the acid (organic acid and / or inorganic) exemplified above Acids) or bases (organic bases and / or inorganic bases) (eg pharmaceutically acceptable salts) can be readily formed.
- a basic group amino group, imino group, heterocyclic basic nitrogen atom, etc.
- an acidic group carboxyl group, sulfonyl group, etc.
- an amino group represented by Z and an N-alkylamino group (forming a carbamoyl group or an N-alkylcarbamoyl group as a group —C (O) —Z) can be prepared by a conventional amide bond forming method, for example, Z is A compound which is a hydroxyl group, an alkoxy group, a cycloalkyloxy group, an aryloxy group or an aralkyloxy group (a group such as a carboxyl group or an alkoxycarbonyl group is formed as a group -C (O) -Z), ammonia, amines (ethylamine) Primary amines, and secondary amines such as dimethylamine).
- the N-alkylamino group or N-acylamino group represented by Z can be formed by subjecting a heterocyclic compound in which Z is an amino group to an alkylation or acylation reaction.
- a compound in which Z is an amino group and an alkyl halide eg, C 1-6 alkyl halide such as methyl chloride or ethyl chloride
- an acylating agent eg C 1-4 acyl chloride such as acetyl chloride, anhydrous
- Z is an N-alkylamino group or an N-acylamino group by reaction with C 1-3 alkanecarboxylic acid anhydride such as acetic acid, C 6-10 aryl-C 1-4 alkyl chloride such as benzyl chloride
- C 1-3 alkanecarboxylic acid anhydride such as acetic acid, C 6-10 aryl-C 1-4 alkyl chloride such as benzyl chloride
- Heterocyclic compound represented by the formula (1) shows a high pharmacological activity, for example, antiallergic, antihistaminic action (including histamine H 1 antagonism), anti-inflammatory, antipruritic action, etc. Therefore, it is useful as an antiallergic agent, antihistamine (such as histamine H 1 receptor antagonist), anti-inflammatory agent, antipruritic agent and the like for allergic diseases. Moreover, it is useful as a preventive and therapeutic agent for preventing and / or treating allergic diseases.
- allergic diseases include allergic rhinitis, nasal congestion, allergic dermatitis (atopic dermatitis, etc.), allergic inflammation, asthma (bronchial asthma), hives, eczema, cutaneous pruritus, prurigo, pruritus
- allergic rhinitis nasal congestion
- allergic dermatitis atopic dermatitis, etc.
- allergic inflammation asthma (bronchial asthma)
- hives eczema
- cutaneous pruritus prurigo
- pruritus examples include psoriasis vulgaris, allergic conjunctivitis, hypereosinophil syndrome, systemic lupus erythematosus, and rheumatoid arthritis inflammation.
- the heterocyclic compound represented by the formula (1) or a salt thereof has a wide safety range and low toxicity. Furthermore, the heterocyclic compound represented by the formula (1) or a salt thereof has a small central nervous action or hypnotic action such as drowsiness and fatigue.
- the heterocyclic compound may be used alone as a medicine, or may be used as a pharmaceutical composition (or preparation) in combination with a carrier (such as a pharmacologically or physiologically acceptable carrier).
- a carrier such as a pharmacologically or physiologically acceptable carrier.
- the carrier can be selected according to the form (or dosage form), dosage form, use, etc. of the pharmaceutical composition (or preparation).
- the dosage form is not particularly limited, and is a solid preparation (powder, powder, granule (granule, fine granule, etc.), pill, pill, tablet, capsule, dry syrup, suppository, etc., semi-solid preparation (cream) Agents, ointments, gels, gummi, film preparations, sheet preparations, etc.), liquids (solutions, suspensions, emulsions, syrups, elixirs, lotions, injections, etc.) Good. Also included are sprays such as the powders and / or liquids, aerosols and the like.
- the capsule may be a liquid-filled capsule or a capsule filled with a solid agent such as a granule.
- the preparation may be a lyophilized preparation.
- the preparation of the present invention may be a preparation with controlled drug release rate (sustained release preparation, immediate release preparation).
- the aerosol generation method is not particularly limited.
- the same sealed container is filled with a pharmaceutical active ingredient and a propellant (alternative chlorofluorocarbon) and sprayed. It may also be a method in the form of a nebulizer or atomizer using a compressed gas such as carbon dioxide or nitrogen gas filled in a separate container from the active pharmaceutical ingredient.
- the preparation may be an oral administration preparation or a parenteral administration preparation (nasal drops, inhalants, transdermal preparations, etc.).
- the preparation may be a topical preparation (solutions such as injections (aqueous injections, non-aqueous injections, etc.), suspensions, ointments, patches, cataplasms, etc.).
- the preparation of the present invention is often a solid preparation (especially an orally administered preparation).
- the carrier examples include Japanese Pharmacopoeia (Pharmacopoeia), (1) Pharmaceutical Additive Handbook, Maruzen Co., Ltd., (1989), (2) “Pharmaceutical Additives Encyclopedia 2007” (Pharmaceutical Daily Inc., 2007) Issued in July), (3) Pharmacy, revised 5th edition, Nanedo Co., Ltd. (1997), and (4) Pharmaceutical Additives Standard 2003 (Pharmaceutical Daily Inc., August 2003) (For example, an excipient, a binder, a disintegrant, a lubricant, a coating agent, etc.) can be selected according to the administration route and the formulation application.
- a carrier for a solid preparation at least one carrier selected from excipients, binders and disintegrants is often used.
- the pharmaceutical composition may contain a lipid.
- excipient examples include lactose, sucrose, glucose, sucrose, saccharides such as mannitol, sorbitol, and xylitol or sugar alcohols; starch such as corn starch; polysaccharides such as crystalline cellulose (including microcrystalline cellulose); Examples thereof include silicon oxide such as anhydrous silicic acid and synthetic aluminum silicate, or silicate.
- soluble starch such as pregelatinized starch and partially pregelatinized starch; polysaccharides such as agar, gum arabic, dextrin, sodium alginate, tragacanth gum, xanthan gum, hyaluronic acid, sodium chondroitin sulfate; polyvinylpyrrolidone (PVP), polyvinyl Synthetic polymers such as alcohol (PVA), carboxyvinyl polymer, polyacrylic acid polymer, polylactic acid, polyethylene glycol; methyl cellulose (MC), ethyl cellulose (EC), carboxymethyl cellulose (CMC), sodium carboxymethyl cellulose, hydroxyethyl cellulose (HEC) ), Cellulose ethers such as hydroxypropylcellulose (HPC) and hydroxypropylmethylcellulose (HPMC) And others.
- PVP polyvinylpyrrolidone
- PVA polyvinyl Synthetic polymers
- PVA polyvinyl Synthetic polymers
- Disintegrants include calcium carbonate, sodium carboxymethyl starch, carboxymethyl cellulose or salts thereof (carmellose, carmellose sodium, carmellose calcium, croscarmellose sodium, etc.), cross-linked polyvinyl pyrrolidone (crospopidone), low-substituted hydroxypropyl cellulose Etc. can be exemplified. These carriers can be used alone or in combination of two or more.
- the coating agent examples include saccharides, cellulose derivatives such as ethyl cellulose and hydroxymethyl cellulose, polyoxyethylene glycol, cellulose acetate phthalate, hydroxypropyl methylcellulose phthalate, methyl methacrylate- (meth) acrylic acid copolymer, Eudragit (meta Acrylic acid / acrylic acid copolymer).
- the coating agent may be an enteric component such as cellulose phthalate, hydroxypropylmethylcellulose phthalate, methyl methacrylate- (meth) acrylic acid copolymer, or a polymer containing a basic component such as dialkylaminoalkyl (meth) acrylate ( Gastric soluble components composed of Eudragit etc.).
- the preparation may also be a capsule containing these enteric components and gastric components in the skin.
- oily carriers include animal and vegetable oils (vegetable oils such as jojoba oil, olive oil, palm oil, and cottonseed oil; animal oils such as squalane) and mineral oils (liquid paraffin, silicone oil, etc.) Etc. can be exemplified.
- aqueous carrier examples include water (purified or sterile water, distilled water for injection, etc.), physiological saline, Ringer's solution, glucose solution, water-soluble organic solvents [lower aliphatic alcohols such as ethanol and isopropanol; (poly) alkylene glycols ( Ethylene glycol, polyethylene glycol and the like); glycerin and the like], dimethylisosorbide, dimethylacetamide and the like.
- the semi-solid carrier may be selected from the solid pharmaceutical carrier and / or the liquid carrier.
- the carrier of the semisolid agent may contain a lipid.
- Lipids include waxes (beeswax, carnauba wax, lanolin, paraffin, petrolatum, etc.), long chain fatty acid esters (saturated or unsaturated fatty acid alkyl esters, fatty acids and polyhydric alcohols (poly C 2-4 alkylene glycol, glycerin or Polyglycerin, etc.) and esters (such as glycerides), hydrogenated oils, higher alcohols (saturated fatty alcohols such as stearyl alcohol, unsaturated aliphatic alcohols such as oleyl alcohol), higher fatty acids (linoleic acid, linolenic acid, Stearic acid, oleic acid and the like), metal soaps (for example, fatty acid metal salts such as sodium coconut oil fatty acid and calcium stearate) and the like.
- waxes beeswax, carnauba wax, lanolin, paraffin, petrolatum, etc.
- long chain fatty acid esters saturated
- additives can be appropriately used depending on the administration route, dosage form and the like.
- additives include lubricants (for example, talc, magnesium stearate, polyethylene glycol 6000), disintegration aids, antioxidants or antioxidants, and emulsifiers (for example, nonionic surfactants).
- Surfactants dispersants, suspending agents, solubilizers, solubilizers, thickeners (water-soluble polymers such as carboxyvinyl polymer, polyvinyl alcohol, carrageenan, gelatin; cellulose such as carboxymethylcellulose) Ethers, etc.), pH adjusters or buffers (citric acid-sodium citrate buffer, etc.), stabilizers, preservatives or preservatives (parabens, such as methylparaben and butylparaben), fungicides or antibacterial agents (benzoic acid) Benzoic acids such as sodium acid), antistatic agents, flavoring agents or masking agents (for example, Etc.
- Taste agents coloring agents (such as dyes and pigments such as red iron oxide), such as flavoring agents, or perfumes (fragrances), fresheners, defoamers, isotonic agents, soothing agents.
- coloring agents such as dyes and pigments such as red iron oxide
- perfumes fragments
- fresheners such as peppers and peppers
- isotonic agents such as sodium tartrate
- soothing agents such as sodium tartrate, sodium tartrate, sodium tartrate, sodium tartrate, sodium stearate, sodium stearate, sodium stearate, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stevia, sodium stearate, sodium stearate, sodium steacetate
- a solubilizer, a solubilizer, a suspending agent, a buffering agent, a stabilizer, a preservative, etc. are usually used as the additive.
- a solubilizer, a solubilizer, a suspending agent, a buffering agent, a stabilizer, a preservative, etc. are usually used as the additive.
- conventional additives used in powder injection can be used.
- topical administration agents such as inhalants and transdermal absorption agents, dissolution aids, stabilizers, buffers, suspending agents, emulsifiers, preservatives and the like are usually used as the above additives. .
- the pharmaceutical composition of the present invention is prepared by a conventional formulation method, for example, a production method described in the Japanese Pharmacopoeia or the production method described in the 15th revision of the Japanese Pharmacopoeia, using an active ingredient, a carrier component, and additives as necessary. Can be prepared by different methods.
- the heterocyclic compound of the present invention or a salt thereof is a human and non-human animal, usually a mammal (eg, human, mouse, rat, rabbit, Dogs, cats, cows, horses, pigs, monkeys, etc.).
- the dosage of the heterocyclic compound of the present invention or a salt thereof is the subject to be administered, age, weight, sex and condition (general condition, medical condition, presence / absence of complications, etc.), administration time, dosage form, administration method, etc. Depending on the choice.
- the dosage for humans when used as an oral preparation, is usually about 0.01 to 1,000 mg, preferably about 0.1 to 700 mg per day, in a free form of the heterocyclic compound or a salt thereof. More preferably, it is about 0.2 to 500 mg.
- the dosage when used as an injection, is usually about 0.01 to 200 mg, preferably about 0.05 to 100 mg, more preferably per day with the heterocyclic compound or its salt in a free form. Is about 0.1 to 80 mg.
- the dosage when used as a topical agent, is usually about 0.01 to 200 mg, preferably about 0.05 to 100 mg per day with the heterocyclic compound or salt thereof in a free form. Preferably it is about 0.1 to 80 mg.
- Z is a hydroxyl group
- MS mass spectroscopy
- EIMS electron impact mass spectroscopy
- HPLC high performance liquid chromatography
- TFA trifluoroacetic acid
- p-TLC preparative thin layer chromatography
- Production Examples 40, 43, 45, 47 The same operation as in Production Example 38 was carried out except that another known compound was used in place of 2-bromo-4-pyridinecarbaldehyde as the carbaldehyde compound to obtain compound (15) shown in Table 57.
- the title compound was obtained in the same manner as in Production Example 49 except that ethyl 4-formyl-1H-pyrazole-carboxylate was used instead of methyl N-tert-butoxycarbonyl-5-formylpyrrole-2-carboxylate. It was.
- Example 1 except that ethyl 4- (4-benzhydryloxypiperidin-1-ylmethyl) -1H-pyrazole-3-carboxylate synthesized in Production Example 75 was used instead of the compound synthesized in Production Example 2. The same operation was performed to obtain the title compound.
- 4-Chloromethylpyridine synthesized in Preparation Example 88 was prepared by dissolving ( ⁇ ) -4-[(4-fluorophenyl) phenylmethoxy] piperidine (237 mg, 0.83 mmol), a known compound, in N, N-dimethylformamide. -2-Carbonitrile (127 mg, 0.83 mmol) and potassium carbonate (138 mg, 1 mmol) were added and stirred at 70 ° C. overnight. After confirming the completion of the reaction by HPLC, the mixture was allowed to cool to room temperature, water and diethyl ether were added, and the organic layer was washed three times with water and once with saturated brine.
- methyl 2-methoxy-5-methylbenzoate (901 mg, 5 mmol) was dissolved in acetonitrile, N-bromosuccinimide (890 mg, 5 mmol) and benzoyl peroxide (24 mg, 0.1 mmol) were added, Refluxed for 1 hour. After confirming the completion of the reaction by HPLC, the mixture was allowed to cool to room temperature, the solvent was distilled off, the residue was dissolved in ethyl acetate, the organic layer was washed three times with water, and then with saturated brine, magnesium sulfate. And dried.
- Production Examples 137, 140, 143, 146, 149, 152, 203 The same procedure as in Production Example 132 was performed, except that other known compounds were used as the compound (12a) instead of ( ⁇ )-(4-fluorophenyl) phenylmethanol, and piperazine compounds shown in Table 71 (compound (12) )).
- ester compounds (compounds (14) shown in Tables 72 to 81 were used in the same manner as in Production Example 163 except that other known compounds were used in place of ethyl 2-chloromethylisonicotinate as the point of use and / or compound (13). )). Further, the carboxylic acids shown in Tables 72 to 81 were obtained in the same manner as in Example 1 except that this ester compound was used.
- Example 1 except that ethyl 6-((R) -3-benzhydryloxypyrrolidin-1-ylmethyl) pyridine-2-carboxylate synthesized in Production Example 169 was used instead of the compound synthesized in Production Example 2.
- the title compound was obtained in the same manner as in. 1 H-NMR (d 6 -DMSO, ⁇ ): 1.7-2.2 (2H, m), 2.4-2.9 (4H, m), 3.7-4.2 (3H, m ), 5.49 (1H, s), 7.1-7.7 (11H, m), 7.8-8.0 (2H, m) MS (m / z): 387 (M + -1).
- Example 1 except that ethyl 6-((S) -3-benzhydryloxypyrrolidin-1-ylmethyl) pyridine-2-carboxylate synthesized in Production Example 171 was used instead of the compound synthesized in Production Example 2.
- the title compound was obtained in the same manner as in. 1 H-NMR (d 6 -DMSO, ⁇ ): 1.7-2.2 (2H, m), 2.4-2.9 (4H, m), 3.7-3.9 (2H, m ), 4.0-4.2 (1H, m), 5.48 (1H, s), 7.1-7.7 (11H, m), 7.8-8.0 (2H, m) MS (m / z): 387 (M + -1).
- Production Examples 179, 182, 208, 211 The same procedure as in Production Example 175 except that the compound synthesized in Production Examples 178 and 181 or a known compound was used instead of ( ⁇ )-(4-chlorophenyl) (2-methyl-2H-pyrazol-3-yl) methanol. Operation was performed to obtain a compound (12) shown in Table 82.
- Benzaldehyde (8.5 g, 80 mmol) was dissolved in 100 mL of tetrahydrofuran and cooled on an ice bath. Then, a 1.0 M solution of 4-fluorophenyl magnesium bromide in tetrahydrofuran (88 mL, 88 mmol) was added dropwise at 0 ° C. After completion of the dropwise addition, the temperature was raised to room temperature and the completion of the reaction was confirmed by TLC. After completion of the reaction, the mixture was poured into 5% hydrochloric acid and extracted with ethyl acetate. The organic layer was washed with water, dried over sodium sulfate, and the solvent was distilled off.
- the obtained viscous oil was dissolved in dichloromethane (10 mL), and thionyl chloride (10 mL, 140 mmol) was added dropwise at room temperature. After completion of the dropwise addition, the completion of the reaction was confirmed by TLC. After completion of the reaction, toluene was added and unreacted thionyl chloride was distilled off. The obtained pale yellow oil and 1-carboethoxy-4-hydroxypiperidine (2.1 g, 12 mmol) were mixed and stirred overnight at an oil bath temperature of 130 ° C. After confirming the completion of the reaction by TLC, it was allowed to cool. Ethyl acetate was added to the resulting residue, followed by washing with water.
- the organic layer was dried over sodium sulfate, and then the solvent was distilled off to obtain a brown oil.
- Test Example 1 H 1 receptor affinity test H 1 receptor membrane preparations were prepared from rat cerebrum. 50 ⁇ L of each test compound dissolved in incubation buffer, promethazine 800 ⁇ mol / L solution (manufactured by SIGMA-ALDRICH) or incubation buffer was added to 100 ⁇ L of rat H 1 receptor membrane preparation.
- the composition (pH 7.4) of the incubation buffer was disodium monohydrogen phosphate (50 mmol / L; manufactured by Wako Pure Chemical Industries, Ltd.) and potassium dihydrogen phosphate (50 mmol / L; manufactured by Wako Pure Chemical Industries, Ltd.). ).
- Test Example 2 human H 1 receptor affinity test
- a Chinese hamster ovary cell (CHO cell) in which human H 1 receptor was forcibly expressed was purchased and used. 50 ⁇ L of each test compound dissolved in incubation buffer, promethazine 800 ⁇ mol / L solution or incubation buffer was added to 100 ⁇ L of human H 1 receptor membrane preparation.
- the composition (pH 7.4) of the incubation buffer is Tris-hydrochloric acid (50 mmol / L; manufactured by Wako Pure Chemical Industries, Ltd.) and magnesium chloride (5 mmol / L; manufactured by Wako Pure Chemical Industries, Ltd.).
- the compounds of the present invention showed excellent binding inhibitory action on rat histamine H 1 receptor and human histamine H 1 receptor.
- Test Example 3 Rats shaved on the day before the test and fasted overnight were orally administered at a dose of 3 mg / kg with the test compound suspended in 0.5% (W / V) aqueous CMC-Na solution.
- One hour after administration 1% Evans Blue physiological saline (Tokyo Kasei Kogyo) was administered into the tail vein at a volume of 5 mL / kg, and immediately 10 ⁇ g / 0.1 mL / site histamine (manufactured by Wako Pure Chemical Industries, Ltd.) was intradermally. Administered.
- test compounds showed about 40% of the inhibitory effect by oral administration of 3 mg / kg, further low, medium, and set the high dose was determined 50% inhibition dose (ID 50 value). The results are shown in Table C.
- Test Example 4 (mouse skin reaction test) Mice shaved on the day before the test and fasted overnight were orally administered low, medium and high doses of the test compound suspended in 0.5% (W / V) aqueous CMC-Na solution. One hour after administration, 1% Evans blue physiological saline was administered into the tail vein at a volume of 10 mL / kg, and histamine 3 ⁇ g / 50 ⁇ L / site was immediately administered into the dorsal skin. Thirty minutes later, the patient was anesthetized with diethyl ether and euthanized by cervical dislocation. The skin was peeled, the major and minor diameters of the pigment spots exuded into the skin were measured, and the pigment area was calculated. A 50% inhibitory dose (ID 50 value) was determined based on the pigment area. The results are shown in Table C.
- the compounds of the present invention exhibited an excellent antihistaminic action because the compounds of the present invention exhibited an inhibitory effect on histamine-induced vascular permeability enhancement in rats and mice. It is.
- Test Example 5 (Inhibitory effect on mouse IgE-dependent ear edema model) Anti-DNP-IgE antibody (manufactured by SIGMA-ALDRICH) was intravenously administered to mice and sensitized passively. The next day, antigen (DNFB solution; Wako Pure Chemical Industries, Ltd.) was applied to the right auricle to induce dermatitis (auricular swelling). The thickness of the right auricle was measured using a thickness gauge (dial thickness gauge) 1 hour, 24 hours and 7 days after the antigen application. The dose of the test compound was 1/3 times the ID 50 value obtained in the rat histamine skin reaction test, and was orally administered twice a day, 1 hour before and 6 hours after antigen application. The thickness of the right auricle before sensitization was measured in advance, and the suppression rate of swelling in the immediate phase, late phase and very late phase was calculated. The results are shown in Table D.
- the compound of the present invention showed an inhibitory action on the ear edema observed in the immediate phase, late phase, and very late phase, and therefore the compound of the present invention was excellent. It is clear that it exhibits an anti-inflammatory effect.
- Test Example 6 (Effect on mouse antigen-induced lung eosinophil infiltration model) Mice were intraperitoneally administered twice with 0.5 mL of physiological saline containing ovalbumin (OVA) 8 ⁇ g (manufactured by SIGMA-ALDLICH) and aluminum hydroxide gel 2 mg (day 0 (day 1) and day 5 (day 6)). To). One week after the final sensitization, 0.5% OVA physiological saline solution was inhaled twice a day in the morning and in the afternoon using a nebulizer. The dose of test compound was 5 times the ID 50 value obtained in the rat skin reaction test, and was orally administered twice a day 30 minutes before each antigen challenge in the morning and afternoon.
- OVA ovalbumin
- the compound of the present invention showed an inhibitory action on antigen-induced pulmonary eosinophil infiltration, indicating that the compound of the present invention has an excellent anti-inflammatory action.
- the non-challenge group indicates a group that does not inhale 0.5% OVA physiological saline solution (group that does not perform antigen challenge), and the control group includes a 0.5% CMC sodium solution as a test compound. The group which administered is shown.
- the number of eosinophils in the non-challenge group is almost zero.
- Test Example 7 (Effects on mouse locomotor activity) Mice fasted overnight were orally administered test compounds suspended in 0.5% (W / V) aqueous CMC-Na solution. Immediately, 3 mice per group were housed in a polycarbonate cage, and the cumulative locomotor activity was measured every 5 minutes from immediately after administration to 2 hours after administration with a spontaneous exercise mass measuring device. The dose of the test compound was 50 times the ID 50 value obtained in the rat histamine skin reaction test. The results are shown in FIGS.
- the compound of the present invention did not affect the locomotor activity of mice even when 50 times the ID 50 value obtained in the rat histamine skin reaction test was administered. Therefore, the compound of the present invention is a compound having a significantly reduced central nervous action and excellent safety without side effects such as sleepiness.
- Test Example 8 (suppressing effect on mouse IgE-dependent ear edema model)
- Anti-DNP-IgE antibody manufactured by SIGMA-ALDRICH was intravenously administered to mice and sensitized passively.
- antigen DNFB solution; Wako Pure Chemical Industries, Ltd.
- the thickness of the right auricle was measured using a thickness gauge (dial thickness gauge) 1 hour, 24 hours and 7 days after the antigen application.
- the dose of the test compound was 5 or 10 times the ID 50 value obtained in the mouse histamine skin reaction test, and was orally administered twice a day, 1 hour before and 6 hours after antigen application.
- the thickness of the right auricle before sensitization was measured in advance, and the suppression rate of swelling in the immediate phase, late phase and very late phase was calculated. The results are shown in Table E.
- the compounds of the present invention showed a clear inhibitory effect on ear edema observed in the immediate phase, late phase, and very late phase at a low dose. It is clear that this compound exhibits an excellent anti-inflammatory action.
- Test Example 9 (Effect on mouse antigen-induced lung eosinophil infiltration model) Mice were intraperitoneally administered twice with 0.5 mL of physiological saline containing ovalbumin (OVA) 8 ⁇ g (manufactured by SIGMA-ALDLICH) and aluminum hydroxide gel 2 mg (day 0 (day 1) and day 5 (day 6)). To). One week after the final sensitization, 0.5% OVA physiological saline solution was inhaled twice a day in the morning and in the afternoon using a nebulizer. The test compound dose was 30 to 170 times the ID 50 value obtained in the mouse skin reaction test, and was orally administered twice a day 30 minutes before each antigen challenge in the morning and afternoon.
- OVA ovalbumin
- the compound of the present invention since the compound of the present invention exhibits an inhibitory action against antigen-induced pulmonary eosinophil infiltration, the compound of the present invention exhibits an excellent anti-inflammatory action. it is obvious. 7 to 9, the non-challenge group shows a group that does not inhale 0.5% OVA physiological saline solution (group that does not perform antigen challenge), and the control group contains 0.5% CMC sodium solution as a test compound. The group which administered is shown. In addition, the number of eosinophils in the non-challenge group is zero.
- Test Example 10 (Effects on mouse locomotor activity) Test compounds suspended in 0.5% (W / V) CMC-Na aqueous solution in mice fasted overnight (Examples 1, 2, 10, 11, 22, 30, 39, 40, 41, 43, 52, 87, 124, 131, 134, 135 and 136) were administered orally. Immediately, 3 mice per group were housed in a polycarbonate cage, and the cumulative locomotor activity was measured every 5 minutes from immediately after administration to 2 hours after administration with a spontaneous exercise amount measuring device. The dose of the test compound was 300 to 2500 times the ID 50 value obtained in the mouse histamine skin reaction test.
- the compound of the present invention did not affect the locomotor activity of mice even when administered in an amount of 300 times or more the ID 50 value obtained in the rat histamine skin reaction test. Therefore, the compound of the present invention is a compound having a significantly reduced central nervous action and excellent safety without side effects such as sleepiness.
- Formulation example in a tablet 30 mg of the compound of Example 2 Lactose 67mg Corn starch 28mg Crystalline cellulose 15mg Hydroxypropylcellulose 8mg Magnesium stearate 2mg Formulation Example 2 Capsules were obtained in accordance with known methods described in the General Rules for Preparations of JP XIV for the following formulations.
- Formulation example in 1 capsule (total amount 180 mg): 50 mg of the compound of Example 2 Lactose 100mg Corn starch 28mg Magnesium stearate 2mg Formulation Example 3 10 mg of the compound of Example 2 was dissolved in 3 ml of physiological saline, adjusted to pH 7 with a 0.1N aqueous sodium hydroxide solution, and then physiological saline was added to make 5 ml. After dispensing into ampoules, it was heat sterilized to obtain an injection.
- the pharmaceutical composition of the present invention is effective as a preventive and / or therapeutic agent for allergic diseases (for example, bronchial asthma, rhinitis, nasal congestion, dermatitis, etc.).
- allergic diseases for example, bronchial asthma, rhinitis, nasal congestion, dermatitis, etc.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Hydrogenated Pyridines (AREA)
- Pyridine Compounds (AREA)
Abstract
抗アレルギー剤として有用な複素環化合物を提供する。 下記式(1)で表される化合物又はこれらの塩。 (式中、環Aは同素環又は複素環を示し、環BはG及び窒素原子Nを環の構成原子として含む複素環を示し、環BのGはCH又はNを示し、R1はカルボニル基又はアルキレン基を示し、R2a及びR2bはアルキル基、シクロアルキル基、アリール基、又は複素環基を示し、Xは酸素原子又は硫黄原子を示し、Zはヒドロキシル基、アルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アミノ基、N-置換アミノ基を示し、nは0又は1を示す。但し、環Aがベンゼン環であるとき、又は環Bがピペラジン環であるとき、R1は置換基を有していてもよいアルキレン基である)
Description
本発明は、複素環化合物(又は薬理活性化合物)、抗アレルギー剤(又はH1受容体拮抗剤)、及び医薬組成物に関する。
薬理活性を有する化合物として、種々の複素環化合物が知られている。特表2007-534696号公報(特許文献1)には、式[AH]-A(AHは、抗ヒスタミン部分を示し、Aは、SPおよびZを含むリンカー分子を示し、SPはスペーサ分子であり、Zは、薬剤調節部分である)で表される改変型抗ヒスタミン化合物が開示されている。この文献にはフェラミン様化合物、ジフェンヒドラミン様化合物も記載されている。
WO 03/057223(特許文献2)には、下記式で表されるδオピオイド受容体アゴニストが開示されている。

(Ar1は置換基を有していてもよい5又は6員炭化水素環又は芳香族複素環を示し;Zは水素、カルボキシル基又はそのエステル基などを示し;Gは炭素又は窒素を示し;R2は水素、C1-4アルキル基など示し;R3,R4は水素又はC1-6アルキル基を示し、R4はC=O基を形成してもよく;R5は水素、アルコキシ基などを示し;R6はフェニル、ハロゲンなどを示し;R7は水素、C1-8アルキル基などを示す)
WO 01/046166(特表2003-518107号公報、特許文献3)には、異常N型カルシウムチャネル活性に関連する状態を処置するための下記式で表される化合物(ベンズヒドリル(benzhydril)及び6員複素環式部分を含有するピペラジン誘導体)が開示され、発作、片頭痛、慢性神経障害性疼痛および急性疼痛、高血圧症、不整脈などの処置に有用であることも記載されている。
WO 01/046166(特表2003-518107号公報、特許文献3)には、異常N型カルシウムチャネル活性に関連する状態を処置するための下記式で表される化合物(ベンズヒドリル(benzhydril)及び6員複素環式部分を含有するピペラジン誘導体)が開示され、発作、片頭痛、慢性神経障害性疼痛および急性疼痛、高血圧症、不整脈などの処置に有用であることも記載されている。

(Z1及びZ2は、N又はCHを示し、Z1及びZ2の一方はNであり;n1は1、n2は0又は1であり;X1及びX2は直鎖リンカーであり;Arは、芳香族単環又は芳香族複素環式単環を表し、Cyは、脂肪族環式単環又は複素環式単環などを表し;Ya及びYbは、芳香族単環又は芳香族複素環式単環、脂肪族環式単環又は複素環式単環などであり;l1は0又は1であり;R1は、C1-6アルキル、C6-10アリール、C7-16アリールアルキルなどを示す)
この特許文献3には、Z1及びZ2がN、X1がC2-6アルキレン基又は-C1-5アルキレン-CO-、X2がメチレン基、エチレン基又はアルケニレン基、Yaがシクロヘキシル基及びフェニル基、Arがフェニル基である化合物が具体的に記載されている。
この特許文献3には、Z1及びZ2がN、X1がC2-6アルキレン基又は-C1-5アルキレン-CO-、X2がメチレン基、エチレン基又はアルケニレン基、Yaがシクロヘキシル基及びフェニル基、Arがフェニル基である化合物が具体的に記載されている。
特開2004-51600号公報(特許文献4)には、下記式で表される造血器型プロスタグランジンD2合成酵素阻害剤が開示され、抗アレルギー作用、抗アレルギー性炎症、及び/又は抗喘息作用を有することも記載されている。

(式中、Xは、単結合、C1-6アルキレン基などを表し、R1はC6-10アリール基を表し、R2は、水素原子、C1-6アルキル基又はC6-10アリール基を表し、R3は、アシル基又は4~10員の環式基を表し、R4及びR5は、水素原子を表すか、又はR4とR5が一緒になってC1-4アルキレン基を表す)
この文献には、Xがカルボニル基、R1及びR2がフェニル基、R3が4-メトキシカルボニルフェニル基である化合物(化合物番号27)も記載されている。
この文献には、Xがカルボニル基、R1及びR2がフェニル基、R3が4-メトキシカルボニルフェニル基である化合物(化合物番号27)も記載されている。
特表2009-504637号公報(特許文献5)には、肥満、精神及び神経障害の治療に有用な下記式で表されるピペラジン化合物が開示されている。
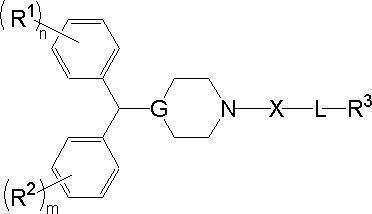
(Gは、CH及びNから選択され;R1及びR2は、独立に、水素原子などを表し;m及びnは、独立して1~5から選択され;Xは、結合、-C(=O)-などを表し;Lは、結合、-(CH2)p-などを表し;pは、0、1及び2から選択され;R3は、C1-6アルコキシ、-CO2R5(R5は、水素、C1-6アルキルなどを示す)などの置換基で置換されたC6-10アリール基などを示し、;X及びLの内の少なくとも1つは結合ではない)
この特許文献5には、GがN、Xがカルボニル基である化合物が具体的に記載されている。
この特許文献5には、GがN、Xがカルボニル基である化合物が具体的に記載されている。
従って、本発明の目的は、新規な複素環化合物、抗アレルギー剤(又は抗ヒスタミン剤、H1受容体拮抗剤など)およびこれらの成分を含む医薬組成物を提供することにある。
本発明の他の目的は、安全域が広いだけでなく毒性(心毒性、癌原性)のない新規な複素環化合物、H1拮抗作用を有する抗アレルギー剤(又はH1受容体拮抗剤)およびこれらの成分を含む医薬組成物を提供することにある。
本発明のさらに他の目的は、高い抗炎症作用を有し、かつ中枢神経作用の少ない新規な複素環化合物、H1拮抗作用を有する抗アレルギー剤(又はH1受容体拮抗剤)およびこれらの成分を含む医薬組成物を提供することにある。
本発明の別の目的は、遅発相に有効な新規な複素環化合物、抗アレルギー剤およびこれらの成分を含む医薬組成物を提供することにある。
本発明のさらに別の目的は、前記複素環化合物の製造方法を提供することにある。
本発明者らは、前記課題を達成するため鋭意検討した結果、アルキレン基を介して、カルボキシル基を有する芳香族同素環又は複素環と、複素環の窒素原子とが結合した骨格を有する複素環化合物が高い薬理活性(抗アレルギー作用など)を有するとともに安全域が広いだけでなく毒性(心毒性、癌原性など)が小さいことを見いだした。本発明はこれらの知見に基づいてさらに検討を加えた結果、完成したものである。
すなわち、本発明の複素環化合物は、下記式(1)で表される化合物又はこれらの塩で表される。
(式中、環Aは置換基を有していてもよい同素環又は複素環を示し;環BはG及び窒素原子Nを環の構成原子として含み、置換基を有していてもよい複素環を示し、環BのGはCH又はN(窒素原子)を示し;R1はカルボニル基又は置換基を有していてもよいアルキレン基を示し;R2a及びR2bは同一又は異なって置換基を有していてもよいアルキル基、置換基を有していてもよい炭化水素環(置換基を有していてもよいシクロアルキル基、置換基を有していてもよいアリール基などの同素環基)又は置換基を有していてもよい複素環基(置換基を有していてもよい複素環基)を示し、これらの環(同素環基又は複素環基)の置換基は互いに結合して環(炭化水素環又は複素環)を形成してもよく、この炭化水素環又は複素環は置換基を有してもよく;Xは酸素原子又は硫黄原子を示し;Zはヒドロキシル基、アルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アミノ基、N-置換アミノ基を示し;nは0又は1を示す。但し、環Aがベンゼン環であるとき、又は環Bがピペラジン環であるとき、R1は置換基を有していてもよいアルキレン基である)
この化合物において、環Aは、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよい芳香族環であってもよく、環Aは芳香族同素環又はアレーン環(ベンゼン環など)又は芳香族複素環(5員又は6員複素環など)であってもよい。さらに、環Aは、窒素原子、酸素原子及び硫黄原子から選択された少なくとも一種のヘテロ原子を含む芳香族複素環、例えば、ピリジン環などであってもよい。
この化合物において、環Aは、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよい芳香族環であってもよく、環Aは芳香族同素環又はアレーン環(ベンゼン環など)又は芳香族複素環(5員又は6員複素環など)であってもよい。さらに、環Aは、窒素原子、酸素原子及び硫黄原子から選択された少なくとも一種のヘテロ原子を含む芳香族複素環、例えば、ピリジン環などであってもよい。
環Bは、脂肪族又は芳香族複素環のいずれであってもよく、例えば、脂肪族4~10員複素環(例えば、ピペリジン環又はピペラジン環などの脂肪族5又は6員複素環)であってもよい。
R1はハロゲン原子を有していてもよい直鎖状又は分岐鎖状アルキレン基(例えば、直鎖状又は分岐鎖状C1-4アルキレン基)であってもよい。また、R1は直鎖状又は分岐鎖状C1-3アルキレン基(メチレン基などのC1-2アルキレン基)であってもよい。
R2a及びR2bは同一又は異なって、ハロゲン原子、アルキル基(例えば、C1-6アルキル基)、ハロアルキル基(例えば、ハロC1-6アルキル基)、アルコキシ基(例えば、C1-6アルコキシ基)およびハロアルコキシ基(例えば、ハロC1-6アルコキシ基)から選択された少なくとも1つの置換基を有していてもよいC6-10アレーン環(例えば、ベンゼン環)又は芳香族5員又は6員複素環であるか、又はR2a及びR2bは下記式(2)

(式中、環C1及び環C2は同一又は異なって、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよいC6-10アレーン環又は芳香族5員又は6員複素環を示し;環C3は、ハロゲン原子、アルキル基、ハロアルキル基、ヒドロキシル基、アルコキシ基、ハロアルコキシ基、カルボキシル基、アルコキシカルボニル基、アシル基およびカルボニル基から選択された少なくとも1つの置換基を有していてもよい4~10員の炭化水素環又は複素環を示し;下記式(3)
で表される結合手は単結合又は二重結合を示し、nが0であり、かつGがCHであるとき、前記結合手は二重結合を示す)
で表される三環式基を形成してもよい。
さらに、R2a及びR2bは同一又は異なって、フッ素原子、塩素原子、C1-3アルキル基、ハロC1-3アルキル基(例えば、フルオロC1-3アルキル基)、C1-3アルコキシ基およびハロC1-3アルコキシ基(例えば、フルオロC1-3アルコキシ基)から選択された少なくとも1つの置換基を有していてもよいベンゼン環であってもよい。
で表される三環式基を形成してもよい。
さらに、R2a及びR2bは同一又は異なって、フッ素原子、塩素原子、C1-3アルキル基、ハロC1-3アルキル基(例えば、フルオロC1-3アルキル基)、C1-3アルコキシ基およびハロC1-3アルコキシ基(例えば、フルオロC1-3アルコキシ基)から選択された少なくとも1つの置換基を有していてもよいベンゼン環であってもよい。
さらに、Zはヒドロキシル基;C1-6アルコキシ基(例えば、C1-4アルコキシ基);アミノ基;又はC1-6アルキル基(例えば、C1-4アルキル基)、C1-6アルキル-カルボニル基(例えば、C1-4アルキル-カルボニル基)から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基であってもよく、ヒドロキシル基;C1-3アルコキシ基(例えば、C1-2アルコキシ基);アミノ基;C1-3アルキル基(例えば、C1-2アルキル基)、C1-3アルキル-カルボニル基(例えば、C1-2アルキル-カルボニル基)から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基であってもよい。
なお、GがCHであるとき、nは1である場合が多く、GがNであるときnは0である場合が多い。また、GがCHであるとき、Xは酸素原子である場合が多い。
前記式(1)で表される代表的な化合物としては、例えば、(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジンカルボン酸、{4-[(ハロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[(C1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[(フルオロC1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[(C1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[(フルオロC1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[ビス(ハロフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、{4-[ビス(ハロフェニル)メチル]ピペラジン-1-イルメチル}ピリジンカルボン酸、(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジンカルボン酸、[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸、[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸などが挙げられる。これらの化合物は、塩、プロドラッグ、又は活性代謝物を形成していてもよい。
本発明は、下記反応工程式

(式中、Y1はハロゲン原子を示し、Z1はアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基を示し、Y2はハロゲン原子を示し、R5はアルキル基、シクロアルキル基、アリール基、アラルキル基を示す。環A,環B,R1,G,X,n,R2a,R2bは前記に同じ)
で表される反応により、前記式(1)においてZがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を製造し、Zがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を加水分解又はアミド結合形成反応に供し、Zがヒドロキシル基、アミノ基又はN-置換アミノ基である複素環化合物を製造し、Zがアミノ基である複素環化合物をアルキル化又はアシル化反応に供し、ZがN-アルキルアミノ基又はN-アシルアミノ基である複素環化合物を製造する方法も包含する。
で表される反応により、前記式(1)においてZがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を製造し、Zがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を加水分解又はアミド結合形成反応に供し、Zがヒドロキシル基、アミノ基又はN-置換アミノ基である複素環化合物を製造し、Zがアミノ基である複素環化合物をアルキル化又はアシル化反応に供し、ZがN-アルキルアミノ基又はN-アシルアミノ基である複素環化合物を製造する方法も包含する。
本発明の前記化合物又はその塩は、薬理活性(抗アレルギー作用、ヒスタミンH1拮抗作用、抗炎症作用など)を示す。しかも、安全域が広いだけでなく毒性(心毒性、癌原性などの遺伝毒性)がなく、安全性に優れている。さらに、中枢神経作用が小さく、覚醒又は眠気を催すことがない。そのため、本発明は、前記化合物又はその塩(薬学的に許容可能な塩)を含む抗アレルギー剤、抗ヒスタミン剤(又はH1受容体拮抗剤)、抗炎症剤も包含する。また、本発明は、前記化合物又はその塩(薬学的に許容可能な塩)を含む、アレルギー疾患(アレルギー症状、気管支喘息、鼻炎、鼻閉、アトピー性皮膚炎、蕁麻疹、湿疹、皮膚そう痒症、痒疹、そう痒を伴う尋常性乾癬、アレルギー性結膜炎、高好酸球症候群、全身性エリテマトーデス、慢性関節リウマチ及び/又は炎症など)の予防治療剤も包含する。さらに、本発明の医薬組成物は、前記化合物又はその薬学的に許容可能な塩と担体とを含んでいる。
なお、本明細書において、前記化合物又はその塩(又は薬学的に許容可能な塩)は、プロドラッグ、活性代謝物も包含する。
本発明の複素環化合物は、高い薬理活性(抗アレルギー作用、ヒスタミンH1拮抗作用、抗炎症作用など)を有するとともに、安全域が広いだけでなく毒性(心毒性、癌原性などの遺伝毒性)がなく、安全性に優れている。さらに、中枢神経作用が小さく、覚醒又は眠気を催すことがない。そのため、抗アレルギー剤などの医薬として有用である。
前記式(1)において、環Aは同素環又は複素環を示し、通常、4~12員環(好ましくは5~10員環、さらに好ましくは5員又は6員環)である。環Aは、縮合環(同素環と複素環との縮合環など)であってもよいが、通常、単環である。環Aは、脂肪族環であってもよいが、薬理活性を高めるためには、芳香族環であるのが好ましい。このような芳香族環は、芳香族同素環又はアレーン環又は芳香族複素環(5員又は6員複素環など)であってもよい。さらに、環Aは、通常、複素環(例えば、窒素原子、酸素原子及び硫黄原子から選択された少なくとも一種のヘテロ原子を含む芳香族複素環)である場合が多い。複素環は同種又は異種の複数のヘテロ原子(例えば、2つの窒素原子、窒素原子と酸素原子及び/又は硫黄原子など)を含んでいてもよい。
環Aとしては、例えば、脂肪族炭化水素環(シクロペンタン環、シクロヘキサン環などのC4-10シクロアルカン環など)、芳香族炭化水素環(ベンゼン環、ナフタレン環などのC6-10アレーン環など)、脂肪族複素環(ピランなどの酸素原子をヘテロ原子として含む5又は6員環;イソクロマン、クロマンなどの酸素原子をヘテロ原子として含む5又は6員環と炭化水素環との縮合環;ピロリジン、ピロリン、イミダゾリジン、イミダゾリン、ピラゾリニジン、ピラゾリン、ピペリジン、ピペラジンなどの窒素原子をヘテロ原子として含む5又は6員環)、芳香族複素環(チオフェンなどの硫黄原子をヘテロ原子として含む5又は6員環;硫黄原子をヘテロ原子として含む5又は6員環と炭化水素環(ベンゼン環など)との縮合環;フランなどの酸素原子をヘテロ原子として含む5又は6員環;イソベンゾフラン、クロメンなどの酸素原子をヘテロ原子として含む5又は6員環と炭化水素環(ベンゼン環など)との縮合環;ピロール、イミダゾール、ピラゾール、ピリジン、ピラジン、ピリミジン、ピリダジンなどの窒素原子をヘテロ原子として含む5又は6員環;インドール、イソインドール、イソキノリン、キノリンなどの窒素原子をヘテロ原子として含む5又は6員環と炭化水素環(ベンゼン環など)との縮合環;プリン、ナフチリジンなどの窒素原子をヘテロ原子として含む5又は6員環と5又は6員複素環との縮合環;チアゾール、イソチアゾールなどの窒素原子及び硫黄原子をヘテロ原子として含む5又は6員環;オキサゾール、イソオキサゾールなどの窒素原子及び酸素原子をヘテロ原子として含む5又は6員環)などが例示できる。
好ましい環Aは、例えば、ベンゼン環、複素環(チオフェン、フラン、ピロール、ピラゾール、チアゾール、オキサゾールなどの5員環、ピリジン、ピラジン、ピリミジン、ピリダジンなどの6員環)、特に少なくとも窒素原子をヘテロ原子として含む芳香族複素環(ピリジン、ピラジンなど)などを含む。
環Aは置換基を有していてもよい。置換基としては、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子)、アルキル基(例えば、メチル、エチル、プロピル、イソプロピル、ブチル、s-ブチル、イソブチル、t-ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル基などの直鎖状又は分岐鎖状C1-10アルキル基)、ハロアルキル基[フルオロメチル基(トリフルオロメチル基など)、フルオロエチル基(2,2,2-トリフルオロエチル基、パーフルオロエチル基など)、3,3,3,2,2-ペンタフルオロプロピル基、パーフルオロプロピル基など)、これらのフルオロアルキル基に対応するクロロアルキル基などのハロC1-6アルキル基]、アルコキシ基(メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、s-ブトキシ、イソブトキシ、t-ブトキシ、ペンチルオキシ、ヘキシルオキシ基などの直鎖状又は分岐鎖状C1-10アルコキシ基など)、ハロアルコキシ基[フルオロメトキシ基(トリフルオロメトキシ基など)、フルオロエトキシ基(2,2,2-トリフルオロエトキシ基、パーフルオロエトキシ基など)、3,3,3,2,2-ペンタフルオロプロポキシ基、パーフルオロプロポキシ基など)、これらのフルオロアルコキシ基に対応するクロロアルコキシ基などのハロC1-6アルコキシ基]、アシル基(ホルミル基、アセチル、プロピオニル、ブチリル基などの直鎖状又は分岐鎖状C1-6アルキル-カルボニル基、シクロヘキシルカルボニル基などのC3-10シクロアルキル-カルボニル基、ベンゾイル基などのC6-10アリール-カルボニル基、ベンジルカルボニル基などのC6-10アリール-C1-4アルキル-カルボニル基など)、アシルオキシ基(アセトキシ基、プロピオニルオキシ基、ブチリルオキシ基などの直鎖状又は分岐鎖状C2-6アシルオキシ基など)、カルボキシル基、アルコキシカルボニル基[メトキシカルボニル基、エトキシカルボニル基、n-プロポキシカルボニル基、イソプロポキシカルボニル基、n-ブトキシカルボニル基、イソブトキシカルボニル基、s-ブトキシカルボニル基、t-ブトキシカルボニル基などの直鎖状又は分岐鎖状C1-6アルコキシ-カルボニル基など]、カルバモイル基、N-置換カルバモイル基[例えば、N-モノC1-6アルキルカルバモイル基、N-C1-6アシル-カルバモイル基、N,N-ジC1-6アルキルカルバモイル基、N,N-ジC1-6アシル-カルバモイル基など]、スルホニル基、スルホン酸アミド基、アミノ基、N-置換アミノ基[例えば、N,N-ジメチルアミノ基、N,N-ジエチルアミノ基などのN-C1-4アルキルアミノ基、N,N-ジC1-4アルキルアミノ基、N-アセチルアミノ基などのN-C1-4アルキル-カルボニルアミノ基、N,N-ジC1-4アルキル-カルボニルアミノ基など]、シアノ基、ニトロ基などが例示できる。これらの置換基は単独で又は二種以上組み合わせて置換していてもよい。
これらの置換基のうち、ハロゲン原子(フッ素原子、塩素原子など)、直鎖状又は分岐鎖状C1-6アルキル基(好ましくはC1-4アルキル基、さらに好ましくはC1-2アルキル基)、直鎖状又は分岐鎖状ハロアルキル基[トリフルオロメチル基、2,2,2-トリフルオロエチル基、パーフルオロエチル基などのフルオロC1-6アルキル基、好ましくはフルオロC1-4アルキル基、さらに好ましくはフルオロC1-3アルキル基)、これらのフルオロアルキル基に対応するクロロアルキル基など]、直鎖状又は分岐鎖状C1-6アルコキシ基(好ましくはC1-4アルコキシ基、さらに好ましくはC1-3アルコキシ基、特にC1-2アルコキシ基など)、直鎖状又は分岐鎖状ハロアルコキシ基[トリフルオロメトキシ基、2,2,2-トリフルオロエトキシ基、3,3,3,2,2-ペンタフルオロプロポキシ基などのフルオロC1-6アルコキシ基、好ましくはフルオロC1-4アルコキシ基、さらに好ましくはフルオロC1-3アルコキシ基)、これらのフルオロアルコキシ基に対応するクロロアルコキシ基など]が好ましい。
環Aに対する置換基の置換位置は、特に限定されず、R1の結合位置を基準にして同素環では、2-,3-,4-,5-位などであってもよく、複素環ではヘテロ原子の位置に応じて、2-,3-,4-,5-位などであってもよい。また、置換基は、複素環ではヘテロ原子(窒素原子)に置換してもよいが、通常、炭素原子に置換する場合が多い。
なお、環Aに対する基-C(O)-Zの置換位置も、R1の結合位置を基準にして環Aの2-,3-,4-,5-位などであってもよい。例えば、環Aがベンゼン環である場合には、o-、m-、p-位などであってもよく、環Aがピリジン環である場合には、環を構成する窒素原子を基準にして、2-,3-,4-位などであってもよい。
環Bは、式(1)で表されるように、G及び窒素原子Nを環の構成原子として含む複素環を示し、環BのGはCH又はNを示す。環Bは、通常、4~12員環(例えば、4~10員環、好ましくは5~10員環、さらに好ましくは5~8員環(例えば、5員又は6員環など))であってもよい。
環Bは、脂肪族又は芳香族複素環のいずれであってもよく、脂肪族複素環としては、例えば、アゼチジン、ピロリジン、ピロリン、イミダゾリジン、イミダゾリン、ピラゾリニジン、ピラゾリン、ピペリジン、ピペラジン、1,4-ジアゼパンなどの少なくとも1つの窒素原子をヘテロ原子として含む4~8員環(例えば、5員又は6員環など)が例示でき、芳香族複素環としては、例えば、ピロール、イミダゾール、ピラゾールなどの少なくとも1つの窒素原子をヘテロ原子として含む5又は6員若しくは7員環;インドール、カルバゾール、インドリン、イソインドリンなどの窒素原子をヘテロ原子として含む5又は6員環と炭化水素環(ベンゼン環など)との縮合環;プリンなどの窒素原子をヘテロ原子として含む5又は6員環と、同種又は異種の5又は6員複素環との縮合環;モルホリンなどの窒素原子及び酸素原子をヘテロ原子として含む5又は6員環などが例示できる。好ましい環Bとしては、脂肪族5又は6員複素環、例えば、ピペリジン環、ピペラジン環などが例示できる。
環Bは、置換基、例えば、前記環Aと同様の置換基(ハロゲン原子、アルキル基、アルコキシ基など)を有していてもよい。
R1はカルボニル基又はアルキレン基を示し、このアルキレン基は置換基を有していてもよい。アルキレン基としては、例えば、メチレン、エチレン、エタン-1,1-ジイル、トリメチレン、プロピレン、プロパン-2,2-ジイル、テトラメチレン、ペンタメチレン、ヘキサメチレン基などの直鎖状又は分岐鎖状C1-10アルキレン基が例示できる。好ましいアルキレン基は、C1-6アルキレン基、例えば、直鎖状又は分岐鎖状C1-4アルキレン基、特に直鎖状又は分岐鎖状C1-3アルキレン基(例えば、メチレン基などのC1-2アルキレン基)、中でもメチレン基である。
アルキレン基の置換基は、環Aと同様の置換基(ハロゲン原子、ハロアルキル基、アルコキシ基、ハロアルコキシ基、アシル基、アシルオキシ基、カルボキシル基、アルコキシカルボニル基、カルバモイル基、N-置換カルバモイル基、アミノ基、N-置換アミノ基、シアノ基、ニトロ基など)などであってもよい。好ましい置換基は、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子)、ハロアルキル基[トリフルオロメチル基、トリクロロメチル基などのハロC1-4アルキル基など]、アルコキシ基(メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ基などのC1-4アルコキシ基など)、ハロアルコキシ基[トリフルオロメトキシ基などのハロC1-4アルコキシ基など]などであってもよい。アルキレン基は、通常、無置換又はハロゲン原子(フッ素原子、塩素原子など)を有している場合が多い。
なお、R1がカルボニル基である化合物に比べてR1が置換(置換基を有する)又は未置換(置換基を有さない)のアルキレン基である化合物は高い薬理活性を示す。そのため、R1は置換又は未置換のアルキレン基(例えば、メチレン基などの未置換の短鎖アルキレン基)であるのが好ましい。
R2a及びR2bは同一又は異なってアルキル基、同素環又は炭化水素環基[炭化水素環基[シクロアルキル基(脂肪族同素環基)、アリール基(芳香族性同素環基)]]、又は複素環基(脂肪族性又は芳香族性複素環基)を示す。アルキル基としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、s-ブチル、イソブチル、t-ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル基などの直鎖状又は分岐鎖状C1-10アルキル基などが例示できる。炭化水素環のシクロアルキル基(脂肪族同素環基)としては、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル基などのC4-10シクロアルキル基などが例示できる。アリール基(芳香族性同素環基)としては、フェニル基、ナフチル基などのC6-10アリール基が例示できる。さらに、複素環基としては、前記環Aで例示の複素環(窒素原子、酸素原子、硫黄原子から選択された少なくとも1つのヘテロ原子を含む5員又は6員複素環、これらの複素環と炭化水素環(ベンゼン環など)との縮合環など)に対応する基が例示でき、複素環基は脂肪族複素環基であってもよく芳香族複素環基であってもよい。
好ましいR2a及びR2bは、芳香族環、例えば、C6-10アレーン環(例えば、ベンゼン環)、又は窒素原子、酸素原子、硫黄原子から選択された少なくとも1つのヘテロ原子を含む芳香族5員又は6員複素環(例えば、ヘテロ原子として窒素原子を含むピリジン環などの芳香族複素環)であり、特にベンゼン環が好ましい。また、R2a及びR2bの組み合わせとしては、R2a及びR2bのうち、少なくとも一方がベンゼン環であるのが好ましく、R2a及びR2bの双方がベンゼン環である組み合わせ、R2a及びR2bのうち、いずれか一方がベンゼン環であり、他方がシクロアルカン環(例えば、シクロヘキサン環などのC5-7シクロアルカン環)又は窒素原子、酸素原子、硫黄原子から選択された少なくとも1つのヘテロ原子を含む芳香族5員又は6員複素環(例えば、ピロール、ピラゾール、イミダゾール、ピリジン、オキサゾール、チアゾールなどのヘテロ原子として窒素原子を含む芳香族複素環)である組み合わせが好ましい。
R2a及びR2bは同一又は異なる置換基を有していてもよい。R2a及びR2bの置換基としては、例えば、ハロゲン原子、アルキル基(例えば、直鎖状又は分岐鎖状C1-6アルキル基)、シクロアルキル基(例えば、C4-10シクロアルキル基)、アリール基(例えば、C6-10アリール基)、ハロアルキル基(例えば、直鎖状又は分岐鎖状ハロC1-6アルキル基)、アルコキシ基(例えば、直鎖状又は分岐鎖状C1-6アルコキシ基)、ハロアルコキシ基(例えば、直鎖状又は分岐鎖状ハロC1-6アルコキシ基)、アシル基(例えば、直鎖状又は分岐鎖状C1-6アルキル-カルボニル基)、アシルオキシ基(例えば、直鎖状又は分岐鎖状C2-6アシルオキシ基)、カルボキシル基、アルコキシカルボニル基(例えば、直鎖状又は分岐鎖状C1-6アルコキシ-カルボニル基)、カルバモイル基、N-置換カルバモイル基(例えば、C1-6アルキルカルバモイル基、C1-6アシル-カルバモイル基など)、アミノ基、N-置換アミノ基(例えば、N,N-ジC1-4アルキルアミノ基、N,N-ジC1-4アルキル-カルボニルアミノ基など)、オキソ基、シアノ基、ニトロ基などが例示できる。置換基の種類に応じて、これらの置換基には、必要により、さらに置換基(フッ素原子などのハロゲン原子、直鎖状又は分岐鎖状C1-4アルキル基、直鎖状又は分岐鎖状C1-4アルコキシ基、直鎖状又は分岐鎖状ハロC1-4アルキル基、直鎖状又は分岐鎖状ハロC1-4アルコキシ基、直鎖状又は分岐鎖状C1-4アルキル-カルボニル基、カルボキシル基、直鎖状又は分岐鎖状C1-6アルコキシ-カルボニル基など)が置換し、アルコキシアルキル基、アルコキシアルコキシ基、カルボキシアルキル基などを形成してもよい。
好ましい置換基としては、ハロゲン原子(例えば、フッ素原子、塩素原子)、直鎖状又は分岐鎖状C1-3アルキル基、直鎖状又は分岐鎖状ハロC1-3アルキル基(例えば、フルオロC1-3アルキル基)、直鎖状又は分岐鎖状C1-3アルコキシ基、直鎖状又は分岐鎖状ハロC1-3アルコキシ基(例えば、フルオロC1-3アルコキシ基)などが例示できる。
これらの置換基の種類は、R2a及びR2bにおいて同じであってもよく異なっていてもよい。また、これらの置換基は、単独で又は二種以上組み合わせてR2a及びR2bに置換していてもよい。さらには、R2a及びR2bにおいて、置換基の置換位置は異なっていてもよい。例えば、R2a及びR2bに同じ置換基(ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基、ハロアルコキシ基など)が同じ位置(例えば、ベンゼン環では、2-,3-,4-位など)に置換していてもよく、R2aにハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基、ハロアルコキシ基などが置換し、R2bは未置換であってもよい。
より具体的には、R2a及びR2bがベンゼン環であるとき、置換基は2-,3-,4-位のいずれに置換していてもよく、一方のベンゼン環の2-,3-,4-位のいずれに置換基が置換し(例えば、一方のベンゼン環の2-,3-,4-位から選択された少なくとも1つの位置に1又は複数の置換基が置換し)、他方のベンゼン環は未置換であってもよい。また、一方のベンゼン環及び他方のベンゼン環のそれぞれに置換位置を異にして置換基が置換していてもよい(例えば、一方のベンゼン環の2-又は3-位に置換基が置換し、他方のベンゼン環の4-位に置換基が置換していてもよい)。
さらに、R2a及びR2bで表される環[前記同素環(炭化水素環)又は複素環]の置換基は、互いに結合して炭化水素環又は複素環を形成してもよく、この炭化水素環又は複素環は置換基を有してもよい。より具体的には、R2a及びR2bは下記式(2)

で表される三環式基を形成してもよい。
式(2)において、環C1及び環C2は同一又は異なってアレーン環(例えば、ベンゼン環などのC6-10アレーン環)又は複素環(例えば、芳香族5員又は6員複素環などの脂肪族又は芳香族複素環)を示す。環C1及び環C2は同一又は異なる置換基、例えば、ハロゲン原子(フッ素原子、塩素原子など)、アルキル基(直鎖状又は分岐鎖状C1-6アルキル基など)、ハロアルキル基(トリフルオロメチル基などの直鎖状又は分岐鎖状ハロC1-6アルキル基など)、アルコキシ基(直鎖状又は分岐鎖状C1-6アルコキシ基など)およびハロアルコキシ基(トリフルオロメトキシ基などの直鎖状又は分岐鎖状ハロC1-6アルコキシ基など)などから選択された少なくとも1つの置換基を有していてもよい。環C1及び環C2には1又は複数の置換基が置換していてもよい。置換基の置換部位は特に制限されず、例えば、環C3と環C1又は環C2との結合部位(式(2)において左側の結合部位)から1-位~4-位(例えば、2-位又は3-位)に位置していてもよい。
環C3は、通常、4~10員(好ましくは5~8員環、特に6~8員環)である。環C3の炭化水素環又は複素環はそれぞれ脂肪族又は芳香族環であってもよく、通常、脂肪族環である場合が多い。炭化水素環は、脂肪族炭化水素環、例えば、シクロアルカン環(シクロヘキサン環、シクロへプタン環、シクロオクタン環などのC4-10シクロアルカン環など)、シクロアルケン環(シクロヘキセン環、シクロヘプテン環、シクロオクテン環などのC4-10シクロアルケン環など)、C4-10シクロアルカジエン環(シクロオクタジエン環など)などであってもよく、芳香族炭化水素環(ベンゼン環などのC6-10アレーン環など)であってもよい。複素環は、窒素原子、酸素原子及び硫黄原子から選択された少なくとも一種のヘテロ原子を環の構成原子として含んでいる。複素環は、ヘテロ原子として酸素原子を含む環(例えば、オキセピン環などの6~8員複素環など)であってもよい。環C3のヘテロ原子は、環C3の員数に応じて、環C3と環C1又は環C2との結合部位(式(2)において左側の結合部位)から1-位~3-位(例えば、1-位又は2-位)に位置していてもよい。
環C3は、前記R2a及びR2bの置換基と同様の置換基を有していてもよい。置換基としては、例えば、ハロゲン原子(フッ素原子、塩素原子など)、アルキル基(例えば、直鎖状又は分岐鎖状C1-6アルキル基)、ハロアルキル基(例えば、直鎖状又は分岐鎖状ハロC1-6アルキル基)、ヒドロキシル基、アルコキシ基(例えば、直鎖状又は分岐鎖状C1-6アルコキシ基)、ハロアルコキシ基(例えば、直鎖状又は分岐鎖状ハロC1-6アルコキシ基)、カルボキシル基、アルコキシカルボニル基(例えば、直鎖状又は分岐鎖状C1-6アルコキシ-カルボニル基)、アシル基(例えば、直鎖状又は分岐鎖状C1-6アルキル-カルボニル基)およびカルボニル基(又はオキソ基)から選択された少なくとも1つの置換基であってもよい。すなわち、環C3には1又は複数の置換基が置換していてもよい。置換基の置換部位は特に制限されず、例えば、環C3と環C1又は環C2との結合部位(式(2)において左側の結合部位)から1-位~3-位(例えば、2-位又は3-位)であってもよい。
なお、式(2)において、下記式(3)で表される結合手は単結合又は二重結合を示し、nが0であり、かつGがCHであるとき、前記結合手は二重結合を示す。
Xは酸素原子又は硫黄原子を示し、通常、酸素原子である。
式(1)において、環Aに置換基-C(O)-Zを導入することにより、薬理活性を大きく向上できる。Zで表されるアルコキシ基としては、前記環Aの置換基と同様のアルコキシ基が例示できる。好ましいアルコキシ基は、直鎖状又は分岐鎖状C1-6アルコキシ基(例えば、C1-4アルコキシ基)である。
シクロアルキルオキシ基としては、例えば、シクロブチルオキシ、シクロペンチルオキシ、シクロヘキシルオキシ、シクロヘプチルオキシ、シクロオクチルオキシ基などのC4-10シクロアルキルオキシ基などが例示できる。アリールオキシ基としては、フェノキシ基、ナフトキシ基などのC6-10アリールオキシ基が例示できる。アラルキルオキシ基としては、ベンジルオキシ、フェネチルオキシ基などのC6-10アリール-C1-4アルキルオキシ基などが例示できる。N-置換アミノ基としては、前記環Aの置換基と同様のN-置換アミノ基が例示できる。好ましいN-置換アミノ基としては、N,N-ジメチルアミノ基、N,N-ジエチルアミノ基などのN,N-ジC1-6アルキルアミノ基、N-アセチルアミノ基などのN-C1-6アルキル-カルボニルアミノ基、N,N-ジC1-6アルキル-カルボニルアミノ基などが例示できる。
好ましいZは、ヒドロキシル基;C1-3アルコキシ基(例えば、C1-2アルコキシ基);アミノ基;又は窒素原子にC1-3アルキル基(例えば、C1-2アルキル基)、C1-3アルキル-カルボニル基(例えば、C1-2アルキル-カルボニル基)が置換基として置換したN-置換アミノ基(モノ又はジ置換アミノ基)である。
なお、Zは、生体内で活性な化合物を産生するプロドラッグ又は活性代謝物を形成してもよい。
nは0又は1を示す。なお、GがCHであるときnは1である場合が多く、GがNであるときnは0である場合が多い。また、GがCHであるとき、Xは酸素原子である場合が多い。
さらに、環Aがベンゼン環であるとき、又は環Bがピペラジン環であるとき、R1は置換基を有していてもよいアルキレン基である。また、R1がカルボニル基であるとき、環Aはベンゼン環ではなく複素環である。
このような化合物のうち代表的な化合物は、例えば、下記式(1a)、特に(1b)で表すことができる。
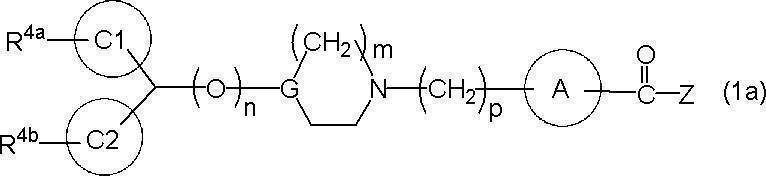
(式中、pは1~4の整数、mは1~5の整数、GがCHであるときnが1であり、GがNであるときnは0であり、環C1及び環C2は同一又は異なって、C6-10アレーン環(例えば、ベンゼン環)又は芳香族5員又は6員複素環を示し、R4a及びR4bは同一又は異なって、水素原子、フッ素原子、塩素原子などのハロゲン原子、直鎖状又は分岐鎖状C1-4アルキル基、直鎖状又は分岐鎖状ハロC1-4アルキル基、直鎖状又は分岐鎖状C1-4アルコキシ基および直鎖状又は分岐鎖状ハロC1-4アルコキシ基から選択された置換基を示し、環C1及び環C2において、R4a及びR4bの種類は同一であってもよく異なってもよい。R4a及びR4bは互いに結合して、前記環C3(4~10員の炭化水素環、又は酸素原子をヘテロ原子として含む複素環など)を形成してもよく、この環C3は前記ハロゲン原子などの置換基を有していてもよい。)

(式中、pは1又は2であり、GがCHであるときnが1であり、GがNであるときnは0であり、R4a及びR4bは前記に同じであり、環C1及び環C2において、R4a及びR4bの種類は同一であってもよく異なってもよい)
前記式(1a)又は(1b)で表される具体的な化合物としては、例えば、以下の化合物が例示できる。
前記式(1a)又は(1b)で表される具体的な化合物としては、例えば、以下の化合物が例示できる。
(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジンカルボン酸[例えば、6-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩;
{4-[(ハロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(C1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル-p-トリルメトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(フルオロC1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル(3-トリフルオロメチルフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(C1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(フルオロC1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル(4-トリフルオロメトキシフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[ビス(ハロフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[ビス(4-フルオロフェニル)メトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[ビス(ハロフェニル)メチル]ピペラジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[ビス(4-フルオロフェニル)メチル]ピペラジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジンカルボン酸[例えば、6-(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸[例えば、6-[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸[例えば、6-[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物)。
{4-[(ハロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(C1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル-p-トリルメトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(フルオロC1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル(3-トリフルオロメチルフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(C1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[(フルオロC1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[フェニル(4-トリフルオロメトキシフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[ビス(ハロフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸[例えば、2-{4-[ビス(4-フルオロフェニル)メトキシ]ピペリジン-1-イルメチル}イソニコチン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
{4-[ビス(ハロフェニル)メチル]ピペラジン-1-イルメチル}ピリジンカルボン酸[例えば、6-{4-[ビス(4-フルオロフェニル)メチル]ピペラジン-1-イルメチル}ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジンカルボン酸[例えば、6-(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸[例えば、6-[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物);
[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸[例えば、6-[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジン-2-カルボン酸など]、又はこれらのエステル若しくは塩(プロドラッグ、活性代謝物)。
前記複素環化合物は、前記式(1)の化合物の塩(薬理学的に許容可能な酸又は塩基との塩など)も含む。このような塩を形成する酸としては、無機酸(塩酸、臭化水素酸、硫酸、硝酸、リン酸など)、有機酸(酢酸、プロピオン酸、トリクロロ酢酸、トリフルオロ酢酸、シュウ酸、コハク酸、フマル酸、マレイン酸などの有機カルボン酸、乳酸、リンゴ酸、酒石酸、クエン酸などのオキシカルボン酸、メタンスルホン酸、トルエンスルホン酸などのスルホン酸など)が例示できる。塩基としては、例えば、無機塩基(アンモニア、水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物、アルカリ金属炭酸塩、水酸化カルシウム、水酸化マグネシウムなどのアルカリ土類金属水酸化物、炭酸カルシウムなどのアルカリ土類金属炭酸塩など)、有機塩基(トリエチルアミンなどのアルキルアミン類、エタノールアミンなどのアルカノールアミン類、アルキレンジアミンなどのポリアミン類など)などが挙げられる。これらの酸又は塩基は、単独で又は二種以上組み合わせて使用できる。
なお、複素環化合物又はその塩は、無水物又は水和物であってもよく、溶媒和物(エタノールなどの有機溶媒などによる溶媒和物など)などであってもよい。また、複素環化合物又はその塩には、式(1)の化合物又はその塩の水和物又は溶媒和物などの他、単離された結晶(結晶多形の物質など)も含まれる。また、本発明の複素環化合物又はその塩は、前記式(1)の化合物又はその塩の互変異性体、不斉炭素原子を有する光学活性体((R)体,(S)体、ジアステレオマーなど)、ラセミ体、又はこれらの混合物なども包含する。特に、Xが酸素原子又は硫黄原子である化合物は、不斉炭素原子を有するため、光学活性体であってもよい。さらに、前記複素環化合物又はその塩は、前記式(1)の化合物の基-C(O)-Z、環Aの複素環基などが修飾され、生体内で活性を発現するプロドラッグ体(又は活性代謝物)であってもよい。プロドラッグ体としては、例えば、加水分解、酸化、還元、エステル交換などの代謝により活性が発現する化合物(例えば、式(1)の化合物のエステル体、エーテル体、アルコール体、アミド体など)などが挙げられる。
[製造方法]
前記式(1)で表される複素環化合物又はその塩は、慣用の方法、例えば、下記の反応工程式に従って製造できる。
前記式(1)で表される複素環化合物又はその塩は、慣用の方法、例えば、下記の反応工程式に従って製造できる。
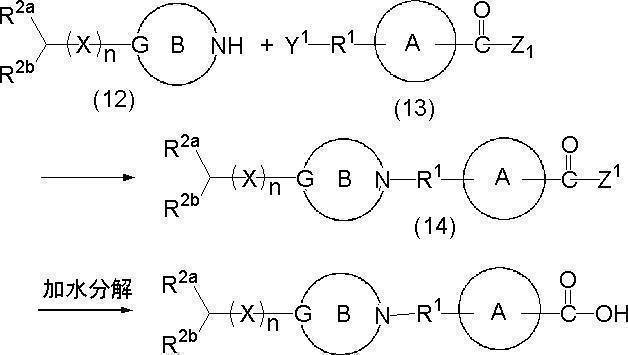
(式中、Y1はハロゲン原子を示し、Z1はアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基を示し、環A,環B,R1,G,X,n,R2a,R2bは前記に同じ)
Y1で表されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子又はヨウ素原子を示し、通常、塩素原子又は臭素原子である。Z1で表されるアルコキシ基としては、前記Zと同様のアルコキシ基が例示でき、通常、直鎖状又は分岐鎖状C1-6アルコキシ基(好ましくはC1-4アルコキシ基、さらに好ましくはC1-2アルコキシ基)である。シクロアルキルオキシ基としては、シクロヘキシルオキシ基などのC4-10シクロアルキルオキシ基が例示でき、アリールオキシ基としては、フェノキシ基などのC6-10アリールオキシ基が例示でき、アラルキルオキシ基として、ベンジルオキシ基、フェネチルオキシ基などのC6-10アリール-C1-4アルキルオキシ基などが例示できる。Z1は、通常、アルコキシ基又はアラルキルオキシ基である。
Y1で表されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子又はヨウ素原子を示し、通常、塩素原子又は臭素原子である。Z1で表されるアルコキシ基としては、前記Zと同様のアルコキシ基が例示でき、通常、直鎖状又は分岐鎖状C1-6アルコキシ基(好ましくはC1-4アルコキシ基、さらに好ましくはC1-2アルコキシ基)である。シクロアルキルオキシ基としては、シクロヘキシルオキシ基などのC4-10シクロアルキルオキシ基が例示でき、アリールオキシ基としては、フェノキシ基などのC6-10アリールオキシ基が例示でき、アラルキルオキシ基として、ベンジルオキシ基、フェネチルオキシ基などのC6-10アリール-C1-4アルキルオキシ基などが例示できる。Z1は、通常、アルコキシ基又はアラルキルオキシ基である。
前記式(12)で表される化合物と式(13)で表される化合物との反応により式(14)で表される化合物を生成できる。反応は、通常、反応に不活性な溶媒中、塩基の存在下で行うことができる。化合物(13)の使用量は、化合物(12)1モルに対して、0.8~5モル、好ましくは1~3モル、さらに好ましくは1.1~2モル程度である。
溶媒としては、例えば、炭化水素類(ヘキサンなどの脂肪族炭化水素類、シクロヘキサンなどの脂環族炭化水素類、トルエンなどの芳香族炭化水素類)、エステル類(酢酸エチルなど)、ケトン類(アセトン、メチルエチルケトンなど)、エーテル類(ジエチルエーテル、ジイソプロピルエーテルなどの鎖状エーテル類、ジオキサン、テトラヒドロフランなどの環状エーテル類)、ニトリル類(アセトニトリル、プロピオニトリル、ベンゾニトリルなど)、アミド類(N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミドなど)、スルホキシド類(ジメチルスルホキシドなど)、スルホラン、アルコール類(エタノール、イソプロパノールなど)などが挙げられる。これらの溶媒は単独で又は二種以上組み合わせて混合溶媒として使用できる。塩基としては、無機塩基(水酸化ナトリウム、水酸化カリウムなどのアルカリ金属水酸化物、アルカリ金属炭酸塩、水酸化カルシウム、水酸化マグネシウムなどのアルカリ土類金属水酸化物、アルカリ土類金属炭酸塩など)、有機塩基(トリメチルアミン、トリエチルアミン、トリエタノールアミン、ジメチルアミノエタノールなどのアミン類など)などが例示できる。これらの塩基は単独で又は二種以上組み合わせて使用できる。塩基の使用量は、化合物(2)1モルに対して当量又は過剰量(例えば、1~10当量、好ましくは1.1~5当量程度)であってもよい。
反応は、例えば、0~150℃(好ましくは10~100℃、さらに好ましくは室温(20~25℃程度)~50℃)程度の温度で行うことができる。また、反応は、空気中又は不活性ガス雰囲気(窒素、ヘリウム、アルゴンガスなど)下で行うことができる。反応は、加圧下で行ってもよいが、通常、大気圧下で行われる。
反応終了後、生成した化合物(14)を、慣用の分離又は精製(あるいは単離)方法、例えば、ろ過、蒸留、溶媒除去、析出、晶析、再結晶、デカンテーション、抽出、乾燥、洗浄、クロマトグラフィー、及びこれらの組み合わせなどにより、分離又は精製してもよい。
化合物(4)を加水分解することにより、式(1)においてZがヒドロキシル基である化合物を得ることができる。加水分解は、慣用の方法、例えば、溶媒中、加水分解触媒(例えば、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素カリウムなどの無機塩基)の存在下で行うことができる。溶媒としては、例えば、前記例示の種々の溶媒(例えば、アルコール類など)を用いることができ、反応系には水を添加してもよい。
加水分解は、適当な温度、例えば、30~150℃又は還流下で行うことができる。なお、加水分解反応は、前記化合物(12)と化合物(13)との反応により生成した化合物(14)を単離又は生成することなく、前記化合物(12)と化合物(13)との反応に引き続いて行うことができる。
加水分解は、適当な温度、例えば、30~150℃又は還流下で行うことができる。なお、加水分解反応は、前記化合物(12)と化合物(13)との反応により生成した化合物(14)を単離又は生成することなく、前記化合物(12)と化合物(13)との反応に引き続いて行うことができる。
反応終了後、反応生成物は、前記と同様にして分離又は精製(あるいは単離)することができる。
なお、前記式(12)で表される化合物において、GがNである化合物は公知であり、GがCHである化合物は、慣用の方法、例えば、酸触媒の存在下、下記式(12a)で表される化合物と式(12b)で表される化合物とを脱水縮合することにより調製できる。
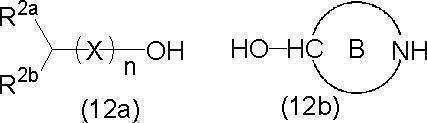
酸触媒としては、例えば、硫酸などの無機酸、p-トルエンスルホン酸などの有機酸が例示でき、ルイス酸なども利用できる。反応は、通常、溶媒中で行われ、溶媒としては、脱水縮合させるため、疎水性溶媒、例えば、炭化水素類(ヘキサン、トルエンなど)、エーテル類などが利用できる。反応は、例えば、50~200℃(例えば、75~150℃)程度で行うことができ、溶媒の還流下で行うことができる。
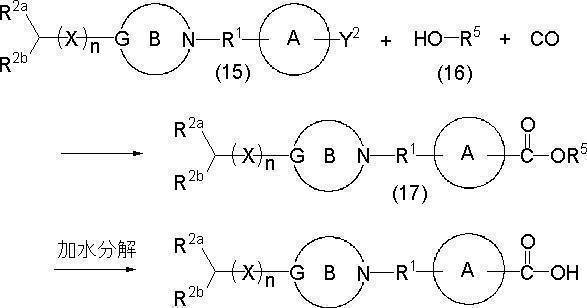
(式中、Y2はハロゲン原子を示し、R5はアルキル基、シクロアルキル基、アリール基、アラルキル基を示し、環A,環B,R1,G,X,n,R2a,R2bは前記に同じ)
Y2で表されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子又はヨウ素原子を示し、通常、塩素原子又は臭素原子である。
Y2で表されるハロゲン原子としては、フッ素原子、塩素原子、臭素原子又はヨウ素原子を示し、通常、塩素原子又は臭素原子である。
R5で表されるアルキル基としては、前記Zと同様のアルコキシ基に対応するアルキル基が例示でき、通常、直鎖状又は分岐鎖状C1-6アルキル基(好ましくはC1-4アルキル基)である。R5で表されるシクロアルキル基、アリール基、アラルキル基としては、前記と同様のC4-10シクロアルキル基、C6-10アリール基、C6-10アリール-C1-4アルキル基などが例示できる。
式(15)で表される化合物と式(16)で表される化合物と一酸化炭素とを、触媒の存在下、反応(カルボニル化反応)させることにより式(17)で表される化合物を生成できる。反応は、通常、溶媒中、塩基の存在下で行われる。化合物(16)の使用量は、化合物(15)1モルに対して等モル以上(通常、過剰モル、例えば、1.5~1000モル、好ましくは2~500モル、さらに好ましくは5~200モル、特に10~100モル程度)である。また、一酸化炭素の使用量も、化合物(15)1モルに対して過剰量(例えば、2~1000モル、好ましくは5~500モルなど)であり、通常、一酸化炭素雰囲気又は一酸化炭素気流中、若しくは反応系への一酸化炭素の吹き込みにより反応を行うことができる。
触媒としては、カルボニル化触媒、例えば、パラジウム触媒(酢酸パラジウム、硫酸パラジウム、パラジウム錯体など)、ロジウム触媒などが例示できる。触媒の使用量は、例えば、式(15)で表される化合物の量に対して0.1~50モル%、好ましくは0.5~20モル%、さらに好ましくは1~10モル%(例えば、3~7モル%)程度であってもよい。
さらに、助触媒として、ホスフィン類(例えば、1,3-ビス(ジフェニルホスフィノ)プロパン、トリフェニルホスフィンなど)などを使用してもよい。助触媒の使用量は、例えば、式(15)で表される化合物の量に対して0.1~50モル%、好ましくは0.5~20モル%、さらに好ましくは1~10モル%(例えば、3~7モル%)程度であってもよい。
塩基の種類と使用量は、前記化合物(12)と化合物(13)との反応と同様である。溶媒としては、炭化水素類(ヘキサンなどの脂肪族炭化水素類、シクロヘキサンなどの脂環族炭化水素類、トルエンなどの芳香族炭化水素類)、ハロゲン化炭化水素類(クロロホルム、ジクロロメタン、トリクロロエタンなど)、エステル類(酢酸エチルなど)、ケトン類(アセトン、メチルエチルケトンなど)、エーテル類(ジエチルエーテル、ジイソプロピルエーテルなどの鎖状エーテル類、ジオキサン、テトラヒドロフランなどの環状エーテル類)、ニトリル類(アセトニトリル、プロピオニトリル、ベンゾニトリルなど)、アミド類(N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミドなど)、スルホキシド類(ジメチルスルホキシドなど)、スルホラン、アルコール類(エタノール、イソプロパノールなど)などが挙げられる。
反応は、例えば、20~150℃(好ましくは50~120℃、さらに好ましくは50~100℃)程度の温度で行うことができる。また、反応は、空気中又は不活性ガス雰囲気(窒素、ヘリウム、アルゴンガスなど)下で行うことができる。反応は、大気圧下又は加圧下で行ってもよい。
反応終了後、生成した化合物(17)を、前記と同様の慣用の分離又は精製(あるいは単離)方法で分離又は精製してもよい。
化合物(17)を加水分解することにより、式(1)においてZがヒドロキシル基である化合物を得ることができる。加水分解は前記化合物(14)の加水分解と同様にして行うことができる。また、反応終了後、反応生成物は、前記と同様にして分離又は精製(あるいは単離)することができる。
なお、前記化合物(15)は慣用の方法、例えば、下記式(15a)で表される化合物と式(15b)で表される化合物とを反応させることにより調製できる。
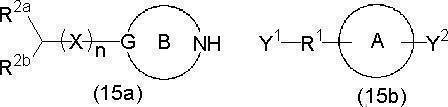
(式中、環A,環B,R1,Y1,Y2,G,X,n,R2a,R2bは前記に同じ)
化合物(15a)と化合物(15b)との反応は、前記化合物(12)と化合物(13)との反応と同様に行うことができる。なお、Y1及びY2が同種のハロゲン原子である場合には、化合物(15b)の環BのNHに対してY1が優先的に反応する。好ましいY1はY2よりも脱離性の高いハロゲン原子(例えば、Y2が塩素原子であれば、Y1は臭素原子)であってもよい。
化合物(15a)と化合物(15b)との反応は、前記化合物(12)と化合物(13)との反応と同様に行うことができる。なお、Y1及びY2が同種のハロゲン原子である場合には、化合物(15b)の環BのNHに対してY1が優先的に反応する。好ましいY1はY2よりも脱離性の高いハロゲン原子(例えば、Y2が塩素原子であれば、Y1は臭素原子)であってもよい。
化合物(1)が塩基性基(アミノ基、イミノ基、複素環の塩基性窒素原子など)、酸性基(カルボキシル基、スルホニル基など)を含む場合、前記例示の酸(有機酸及び/又は無機酸)又は塩基(有機塩基及び/又は無機塩基)との塩(例えば、薬学的に許容可能な塩)を容易に形成できる。
さらに、Zで表されるアミノ基及びN-アルキルアミノ基(基-C(O)-Zとして、カルバモイル基、N-アルキルカルバモイル基を形成)は、慣用のアミド結合形成方法、例えば、Zがヒドロキシル基、アルコキシ基、シクロアルキルオキシ基、アリールオキシ基又はアラルキルオキシ基(基-C(O)-Zとして、カルボキシル基又はアルコキシカルボニル基などを形成)である化合物と、アンモニア、アミン類(エチルアミンなどの一級アミン類、ジメチルアミンなどの二級アミン類)と反応させることにより得ることができる。また、Zで表されるN-アルキルアミノ基又はN-アシルアミノ基は、Zがアミノ基である複素環化合物をアルキル化又はアシル化反応に供することにより形成できる。例えば、Zがアミノ基である化合物と、アルキルハライド(例えば、メチルクロライド、エチルクロライドなどのC1-6アルキルハライドなど)又はアシル化剤(例えば、アセチルクロライドなどのC1-4アシルクロライド、無水酢酸などの無水C1-3アルカンカルボン酸、ベンジルクロライドなどC6-10アリール-C1-4アルキルクロライドなど)との反応によりZがN-アルキルアミノ基又はN-アシルアミノ基である化合物を得ることができる。
[用途及び医薬組成物]
前記式(1)で表される複素環化合物又はその塩は、高い薬理活性、例えば、抗アレルギー作用、抗ヒスタミン作用(ヒスタミンH1拮抗作用を含む)、抗炎症作用、止痒作用などを示すため、アレルギー疾患に対する抗アレルギー剤、抗ヒスタミン剤(ヒスタミンH1受容体拮抗剤など)、抗炎症剤、止痒剤などとして有用である。また、アレルギー疾患を予防及び/又は治療するための予防治療剤として有用である。アレルギー疾患としては、例えば、アレルギー性鼻炎、鼻閉、アレルギー性皮膚炎(アトピー性皮膚炎など)、アレルギー性炎症、喘息(気管支喘息)、蕁麻疹、湿疹、皮膚そう痒症、痒疹、そう痒を伴う尋常性乾癬、アレルギー性結膜炎、高好酸球症候群、全身性エリテマトーデス、慢性関節リウマチ炎症などが例示できる。
前記式(1)で表される複素環化合物又はその塩は、高い薬理活性、例えば、抗アレルギー作用、抗ヒスタミン作用(ヒスタミンH1拮抗作用を含む)、抗炎症作用、止痒作用などを示すため、アレルギー疾患に対する抗アレルギー剤、抗ヒスタミン剤(ヒスタミンH1受容体拮抗剤など)、抗炎症剤、止痒剤などとして有用である。また、アレルギー疾患を予防及び/又は治療するための予防治療剤として有用である。アレルギー疾患としては、例えば、アレルギー性鼻炎、鼻閉、アレルギー性皮膚炎(アトピー性皮膚炎など)、アレルギー性炎症、喘息(気管支喘息)、蕁麻疹、湿疹、皮膚そう痒症、痒疹、そう痒を伴う尋常性乾癬、アレルギー性結膜炎、高好酸球症候群、全身性エリテマトーデス、慢性関節リウマチ炎症などが例示できる。
しかも、前記式(1)で表される複素環化合物又はその塩は、安全域が広く毒性が小さい。さらには、前記式(1)で表される複素環化合物又はその塩は、眠気、倦怠感などの中枢神経作用又は催眠作用が小さい。
前記複素環化合物は単独で医薬として用いてもよく、担体(薬理学的又は生理学的に許容可能な担体など)と組み合わせて医薬組成物(又は製剤)として用いてもよい。本発明の医薬組成物において、担体は、医薬組成物(又は製剤)の形態(又は剤形)、投与形態、用途などに応じて、選択できる。剤形は特に制限されず、固形製剤(粉剤、散剤、粒剤(顆粒剤、細粒剤など)、丸剤、ピル、錠剤、カプセル剤、ドライシロップ剤、座剤など)、半固形製剤(クリーム剤、軟膏剤、ゲル剤、グミ剤、フィルム状製剤、シート状製剤など)、液剤(溶液剤、懸濁剤、乳剤、シロップ剤、エリキシル剤、ローション剤、注射剤など)などであってもよい。また、前記粉剤及び/又は液剤などのスプレー剤、エアゾール剤なども含まれる。なお、カプセル剤は、液体充填カプセルであってもよく、顆粒剤などの固形剤を充填したカプセルであってもよい。また、製剤は凍結乾燥製剤であってもよい。さらに、本発明の製剤は、薬剤の放出速度が制御された製剤(徐放性製剤、速放性製剤)であってもよい。なお、吸入剤などで利用されるエアゾール剤において、エアゾールの発生方法は、特に制限されず、例えば、同一密封容器に医薬有効成分と噴射剤(代替フロンなど)とを充填し、スプレーする方法であってもよく、また、医薬有効成分と別の容器に充填した二酸化炭素や窒素ガスなどの圧縮ガスを用いたネブライザーやアトマイザーなどの形態による方法であってもよい。さらに、製剤は経口投与製剤であってもよく、非経口投与製剤(点鼻剤、吸入剤、経皮投与製剤など)であってもよい。さらに、製剤は局所投与製剤(注射剤(水性注射剤、非水性注射剤など)などの溶液剤、懸濁剤、軟膏剤、貼付剤、パップ剤など)であってもよい。本発明の製剤は固形製剤(特に経口投与製剤)である場合が多い。
前記担体は、例えば、日本薬局方(局方)の他、(1)医薬品添加物ハンドブック、丸善(株)、(1989)、(2)「医薬品添加物事典2007」(薬事日報社、2007年7月発行)、(3)薬剤学、改訂第5版、(株)南江堂(1997)、及び(4)医薬品添加物規格2003(薬事日報社、2003年8月)などに収載されている成分(例えば、賦形剤、結合剤、崩壊剤、滑沢剤、コーティング剤など)の中から、投与経路及び製剤用途に応じて選択できる。例えば、固形製剤の担体としては、賦形剤、結合剤および崩壊剤から選択された少なくとも一種の担体を使用する場合が多い。また、医薬組成物は脂質を含んでいてもよい。
前記賦形剤としては、乳糖、白糖、ブドウ糖、ショ糖、マンニトール、ソルビトール、キシリトールなどの糖類又は糖アルコール類;トウモロコシデンプンなどのデンプン;結晶セルロース(微結晶セルロースも含む)などの多糖類;軽質無水ケイ酸、合成ケイ酸アルミニウムなどの酸化ケイ素又はケイ酸塩などが例示できる。結合剤としては、アルファ化デンプン、部分アルファ化デンプンなどの可溶性デンプン;寒天、アラビアゴム、デキストリン、アルギン酸ナトリウム、トラガントガム、キサンタンガム、ヒアルロン酸、コンドロイチン硫酸ナトリウムなどの多糖類;ポリビニルピロリドン(PVP)、ポリビニルアルコール(PVA)、カルボキシビニルポリマー、ポリアクリル酸系ポリマー、ポリ乳酸、ポリエチレングリコールなどの合成高分子;メチルセルロース(MC)、エチルセルロース(EC)、カルボキシメチルセルロース(CMC)、カルボキシメチルセルロースナトリウム、ヒドロキシエチルセルロース(HEC)、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC)などのセルロースエーテル類などが例示できる。崩壊剤としては、炭酸カルシウム、カルボキシメチルスターチナトリウム、カルボキシメチルセルロース又はその塩(カルメロース、カルメロースナトリウム、カルメロースカルシウム、クロスカルメロースナトリウムなど)、架橋ポリビニルピロリドン(クロスポピドン)、低置換度ヒドロキシプロピルセルロースなどが例示できる。これらの担体は、単独で又は二種以上組み合わせて使用できる。
なお、前記コーティング剤としては、例えば、糖類、エチルセルロース、ヒドロキシメチルセルロースなどのセルロース誘導体、ポリオキシエチレングリコール、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルメタクリレート-(メタ)アクリル酸共重合体、オイドラギット(メタアクリル酸・アクリル酸共重合物)などが用いられる。コーティング剤は、セルロースフタレート、ヒドロキシプロピルメチルセルロースフタレート、メチルメタクリレート-(メタ)アクリル酸共重合体などの腸溶性成分であってもよく、ジアルキルアミノアルキル(メタ)アクリレートなどの塩基性成分を含むポリマー(オイドラギットなど)で構成された胃溶性成分であってもよい。また、製剤は、これらの腸溶性成分や胃溶性成分を剤皮に含むカプセル剤であってもよい。
液剤の担体のうち油性担体としては、動植物系油剤(ホホバ油、オリーブ油、やし油、綿実油などの植物系油剤;スクアランなどの動物系油剤など)、鉱物系油剤(流動パラフィン、シリコーンオイルなど)などが例示できる。水性担体としては、水(精製又は無菌水、注射用蒸留水など)、生理食塩水、リンゲル液、ブドウ糖液、水溶性有機溶媒[エタノール、イソプロパノールなどの低級脂肪族アルコール;(ポリ)アルキレングリコール類(エチレングリコール、ポリエチレングリコールなど);グリセリンなど]、ジメチルイソソルビド、ジメチルアセトアミドなどが挙げられる。また、半固形剤の担体は、前記固形製剤の担体及び/又は液剤の担体から選択してもよい。また、半固形剤の担体は、脂質を含んでいてもよい。
脂質としては、ワックス類(蜜ろう、カルナバろう、ラノリン、パラフィン、ワセリンなど)、長鎖脂肪酸エステル(飽和又は不飽和脂肪酸アルキルエステル、脂肪酸と多価アルコール(ポリC2-4アルキレングリコール、グリセリン又はポリグリセリンなど)とのエステル(グリセライドなど)など)、硬化油、高級アルコール(ステアリルアルコールなどの飽和脂肪族アルコール、オレイルアルコールなどの不飽和脂肪族アルコールなど)、高級脂肪酸(リノール酸、リノレイン酸、ステアリン酸、オレイン酸など)、金属石鹸類(例えば、ヤシ油脂肪酸ナトリウム、ステアリン酸カルシウムなどの脂肪酸金属塩など)などが例示できる。
製剤においては、投与経路や剤形などに応じて、公知の添加剤を適宜使用することができる。このような添加剤としては、例えば、滑沢剤(例えば、タルク、ステアリン酸マグネシウム、ポリエチレングリコール6000など)、崩壊補助剤、抗酸化剤又は酸化防止剤、乳化剤(例えば、非イオン性界面活性剤などの各種界面活性剤など)、分散剤、懸濁化剤、溶解剤、溶解補助剤、増粘剤(カルボキシビニルポリマー、ポリビニルアルコール、カラギーナン、ゼラチンなどの水溶性高分子;カルボキシメチルセルロースなどのセルロースエーテル類など)、pH調整剤又は緩衝剤(クエン酸-クエン酸ナトリウム緩衝剤など)、安定剤、防腐剤又は保存剤(メチルパラベン、ブチルパラベンなどのパラベン類など)、殺菌剤又は抗菌剤(安息香酸ナトリウムなどの安息香酸類など)、帯電防止剤、矯味剤又はマスキング剤(例えば、甘味剤など)、着色剤(ベンガラなどの染顔料など)、矯臭剤又は香料(芳香剤など)、清涼化剤、消泡剤、等張化剤、無痛化剤などが挙げられる。これらの添加剤は単独で又は二種以上組み合わせて使用できる。
例えば、注射剤では、通常、前記添加物として、溶解剤、溶解補助剤、懸濁化剤、緩衝剤、安定剤、保存剤などを使用する場合が多い。なお、投与時に溶解あるいは懸濁して使用するための粉末注射剤では、粉末注射剤で使用される慣用の添加剤が使用できる。
また、吸入剤、経皮吸収剤などの局所投与剤では、上記添加物として、通常、溶解補助剤、安定剤、緩衝剤、懸濁化剤、乳化剤、保存剤などが使用される場合が多い。
本発明の医薬組成物は、有効成分の他、担体成分、必要により添加剤などを用いて、慣用の製剤化方法、例えば、第十五改正日本薬局方記載の製造法又はこの製造方法に準じた方法により調製できる。
本発明の複素環化合物又はその塩(抗アレルギー剤、予防及び/又は治療剤、医薬組成物も含む)は、ヒト及び非ヒト動物、通常、哺乳動物(例えば、ヒト、マウス、ラット、ウサギ、イヌ、ネコ、ウシ、ウマ、ブタ、サルなど)に対して、安全に投与される。本発明の複素環化合物又はその塩の投与量は、投与対象、投与対象の年齢、体重、性別及び状態(一般的状態、病状、合併症の有無など)、投与時間、剤形、投与方法などに応じて、選択できる。
ヒトに対する投与量は、例えば、経口剤として使用する場合、前記複素環化合物又はその塩を遊離の形態で、1日当たり、通常、0.01~1,000mg程度、好ましくは0.1~700mg程度、さらに好ましくは0.2~500mg程度である。また、注射剤として使用する場合、前記投与量は、前記複素環化合物又はその塩を遊離の形態で、1日当たり、通常、0.01~200mg程度、好ましくは0.05~100mg程度、さらに好ましくは0.1~80mg程度である。さらに、局所投与剤として使用する場合、前記投与量は、前記複素環化合物又はその塩を遊離の形態で、1日当たり、通常、0.01~200mg程度、好ましくは0.05~100mg程度、さらに好ましくは0.1~80mg程度である。
以下に、実施例に基づいて本発明をより詳細に説明するが、本発明はこれらの実施例によって限定されるものではない。
なお、以下の製造例及び実施例において、略号は慣用の意味で用い、例えば、下記略号の意味は次の通りである。
NMR:核磁気共鳴スペクトル
MS:マススペクトロスコピー
EIMS:エレクトロンインパクトマススペクトロスコピー
HPLC:高速液体クロマトグラフィ
TFA:トリフルオロ酢酸
p-TLC:分取薄層クロマトグラフィー
また、以下の実施例では、Zがヒドロキシル基である化合物を実施例に記載している。そのため、本発明の化合物(例えば、Zがアルコキシ基である化合物など)であっても、これらの化合物を製造例に記載している場合がある。
MS:マススペクトロスコピー
EIMS:エレクトロンインパクトマススペクトロスコピー
HPLC:高速液体クロマトグラフィ
TFA:トリフルオロ酢酸
p-TLC:分取薄層クロマトグラフィー
また、以下の実施例では、Zがヒドロキシル基である化合物を実施例に記載している。そのため、本発明の化合物(例えば、Zがアルコキシ基である化合物など)であっても、これらの化合物を製造例に記載している場合がある。
製造例1
4-ベンズヒドリルオキシピペリジン
4-ベンズヒドリルオキシピペリジン

4-ヒドロキシピペリジン(505mg,5mmol)のトルエン懸濁液にモレキュラーシーブス、ベンズヒドロール(1.1g,5.5mmol)、p-トルエンスルホン酸1水和物(1.0g,5.5mmol)を加え、ディーンスタークコンデンサーを用いて水を除去しながら3時間還流した。HPLCで反応の終了を確認した後、室温まで放冷し、不溶物を除去した後、溶媒を留去した。得られた残渣を酢酸エチルに溶解し、2N水酸化ナトリウム液で3回洗浄した。有機層を硫酸マグネシウムで乾燥した後、溶媒を留去し、表題化合物(676mg,50.6%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.2-1.5(2H,m),1.8-1.9(2H,m),2.3-2.5(2H,m),2.8-3.0(2H,m),3.2-3.4(1H,m),5.64(1H,s),7.1-7.4(10H,m)。
1H-NMR(CDCl3,δ):1.2-1.5(2H,m),1.8-1.9(2H,m),2.3-2.5(2H,m),2.8-3.0(2H,m),3.2-3.4(1H,m),5.64(1H,s),7.1-7.4(10H,m)。
製造例2
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)フラン-2-カルボン酸メチル
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)フラン-2-カルボン酸メチル

製造例1で合成した4-ベンズヒドリルオキシピペリジン401mg、5-クロロメチルフラン-2-カルボン酸メチル288mg、炭酸カリウム352mg、アセトニトリル3.5mLの混合物を18時間撹拌した。反応液を酢酸エチルで希釈した後、水に投入して有機層を分取した。飽和食塩水で有機層を洗った後、硫酸マグネシウムで乾燥した。溶媒を留去した。シリカゲルカラムクロマトグラフィー(クロロホルム/アセトン(容積比)=20:1~10:1)を行い、表題化合物475mgを得た。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.1-2.4(2H,m),2.6-2.9(2H,m),3.3-3.5(1H,m),3.59(2H,s),3.87(3H,s),5.49(1H,s),6.31(1H,d,J=3.5Hz),7.11(1H,d,J=3.5Hz),7.1-7.4(10H,m)
MS(m/z):404(M+-1)。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.1-2.4(2H,m),2.6-2.9(2H,m),3.3-3.5(1H,m),3.59(2H,s),3.87(3H,s),5.49(1H,s),6.31(1H,d,J=3.5Hz),7.11(1H,d,J=3.5Hz),7.1-7.4(10H,m)
MS(m/z):404(M+-1)。
実施例1
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)フラン-2-カルボン酸
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)フラン-2-カルボン酸
製造例2で合成した5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)フラン-2-カルボン酸メチル465mg、エタノール1.7mL、1N水酸化ナトリウム水溶液3.4mLの混合物を40分間加熱還流した。水10mL、1N塩酸3.4mLを加え撹拌した。析出した結晶をろ取し、水洗した。乾燥し表題化合物316mgを得た。
1H-NMR(d6-DMSO,δ):1.4-2.3(6H,m),2.6-2.8(2H,m),3.2-3.4(1H,m),3.53(2H,s),5.61(1H,s),6.44(1H,d,J=3.5Hz),7.12(1H,d,J=3.5Hz),7.1-7.5(10H,m)
MS(m/z):391(M+)。
1H-NMR(d6-DMSO,δ):1.4-2.3(6H,m),2.6-2.8(2H,m),3.2-3.4(1H,m),3.53(2H,s),5.61(1H,s),6.44(1H,d,J=3.5Hz),7.12(1H,d,J=3.5Hz),7.1-7.5(10H,m)
MS(m/z):391(M+)。
製造例3~12、14、15、17~21、23、25~28、30、32、34~35、50、52~61、63、65、67~70、74、76~79、81~83、91、112、113、116~119、121~124、127、128、131、133~135、138、139、141、142、144、145、147、148、150、151、153、155、156、158、159、161、162、189、190、194、197、198、201、202、204、206、207、実施例2~27、36、38~54、56~62、66、83、84、86~93、96、97、100~103、105~121、140~149
化合物(12)として、4-ベンズヒドリルオキシピペリジンの代わりに、製造例16、22、24、29、31、33、62、64、66、80、115、120、132、137、140、143、146、149、152、154、157、160、188、193、200、203、205で合成した化合物又は公知化合物を用いる点、及び/又は化合物(13)として、5-クロロメチルフラン-2-カルボン酸メチルの代わりに、製造例13で合成した化合物又は公知化合物を用いる点を除き、製造例2と同様にして表1~50に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表1~50に示すカルボン酸を得た。
化合物(12)として、4-ベンズヒドリルオキシピペリジンの代わりに、製造例16、22、24、29、31、33、62、64、66、80、115、120、132、137、140、143、146、149、152、154、157、160、188、193、200、203、205で合成した化合物又は公知化合物を用いる点、及び/又は化合物(13)として、5-クロロメチルフラン-2-カルボン酸メチルの代わりに、製造例13で合成した化合物又は公知化合物を用いる点を除き、製造例2と同様にして表1~50に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表1~50に示すカルボン酸を得た。


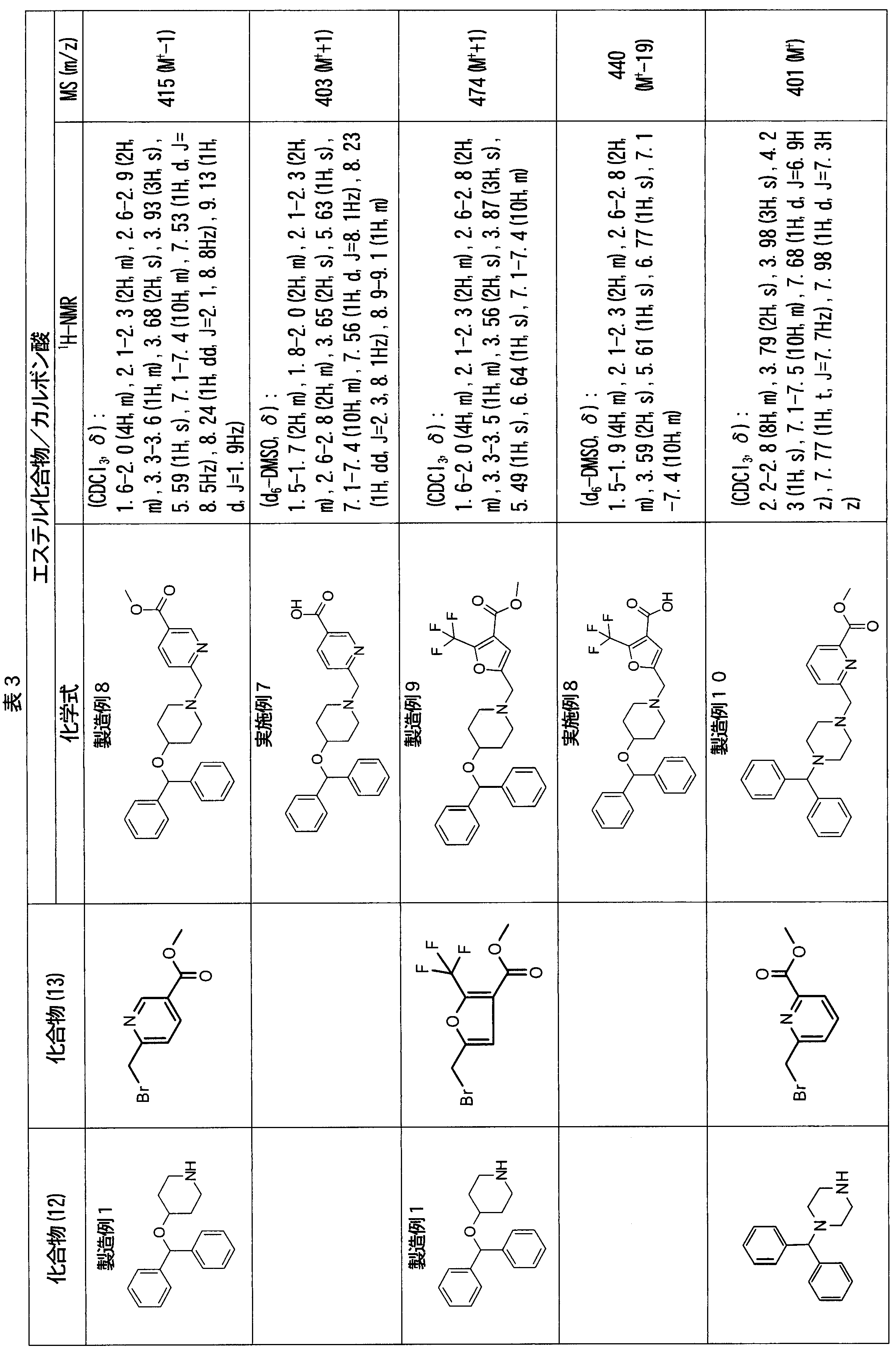

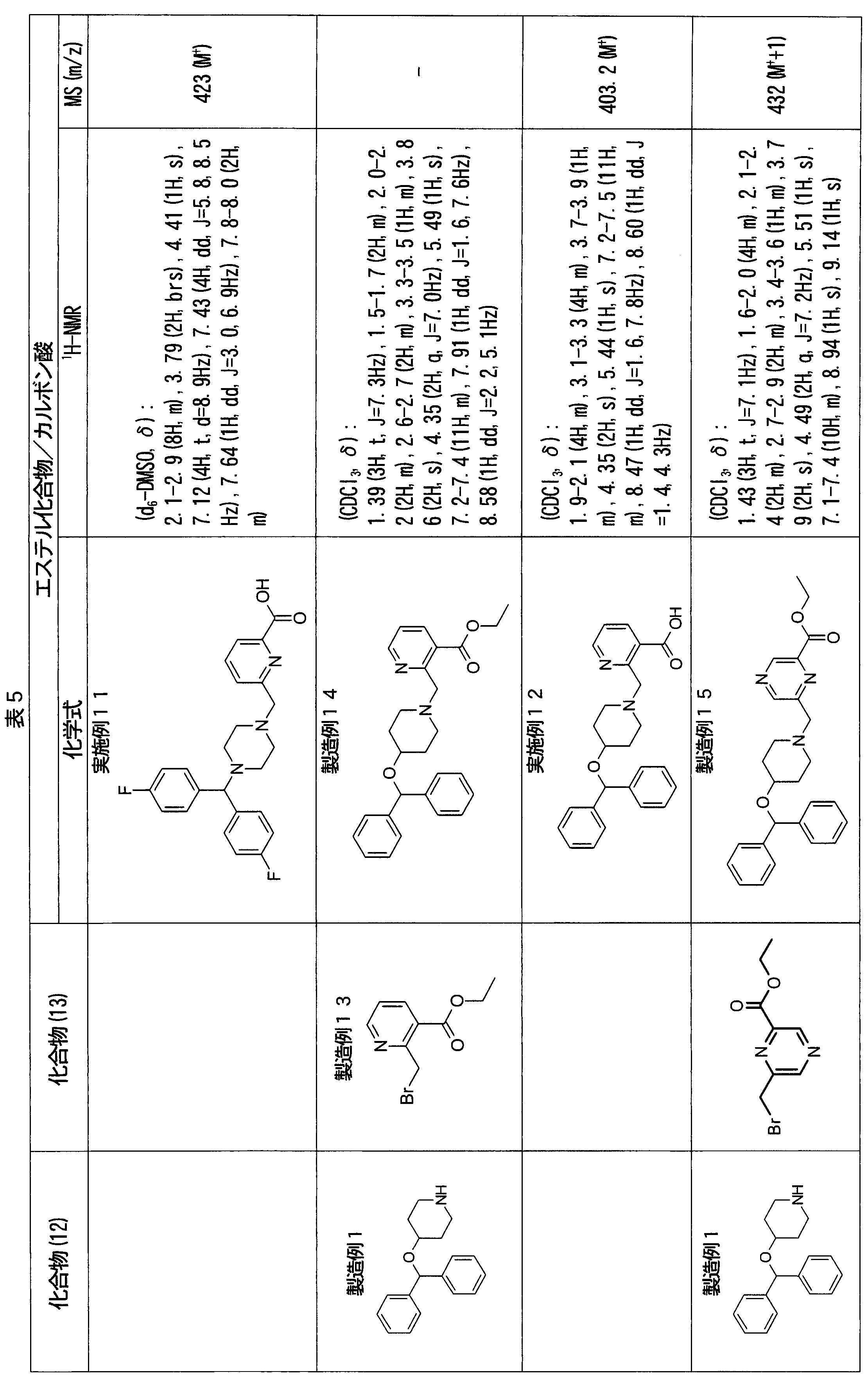




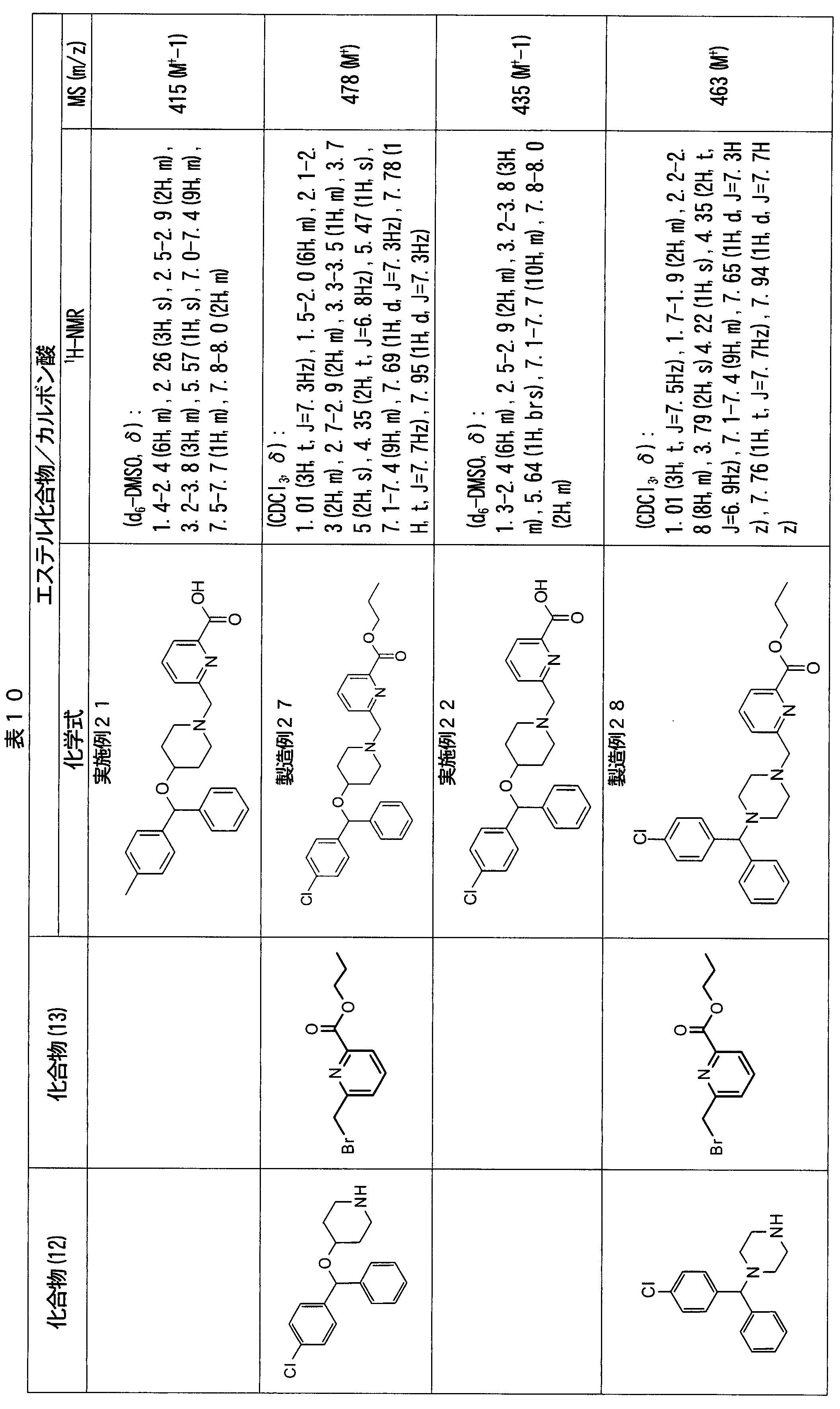

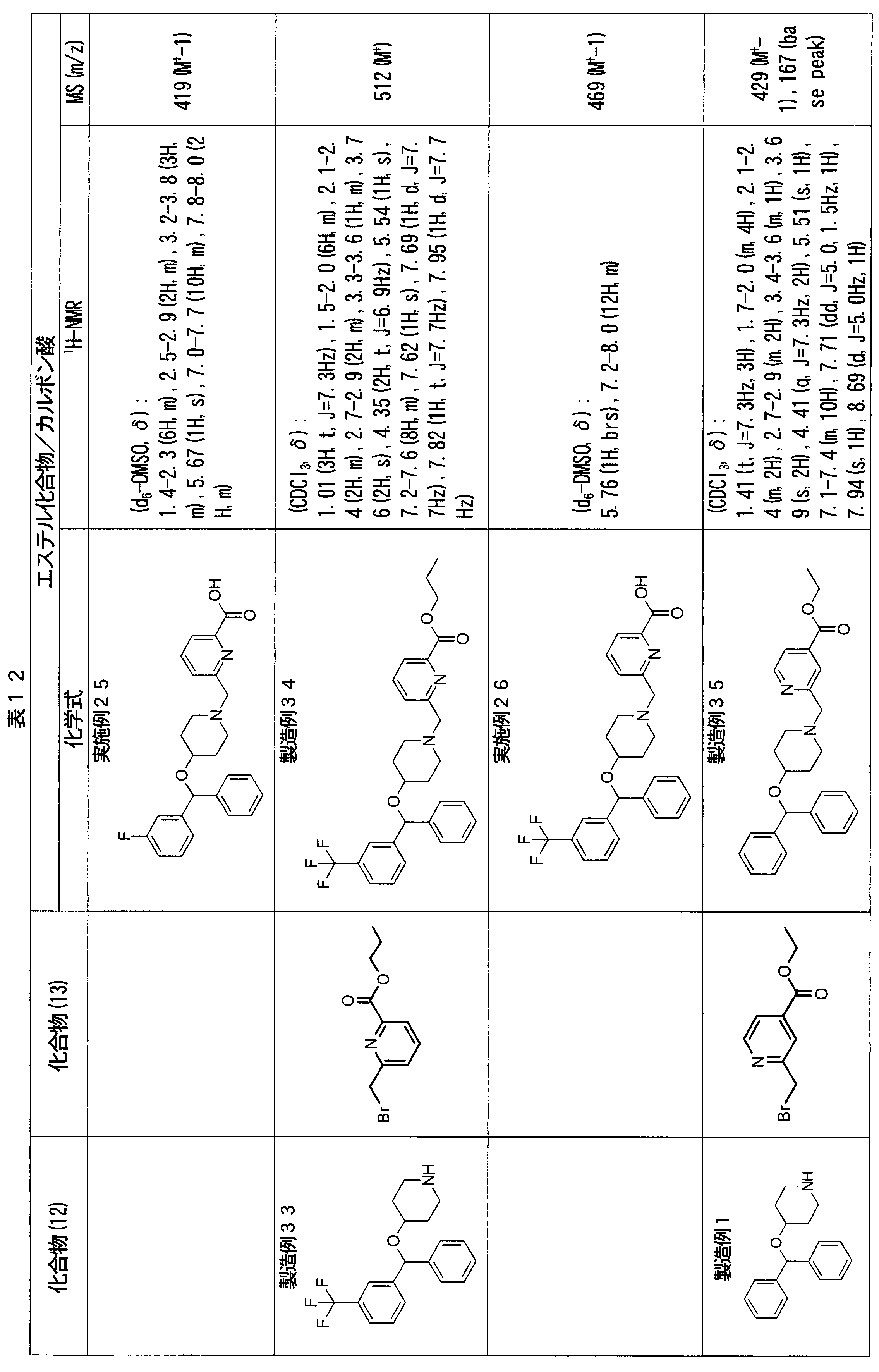





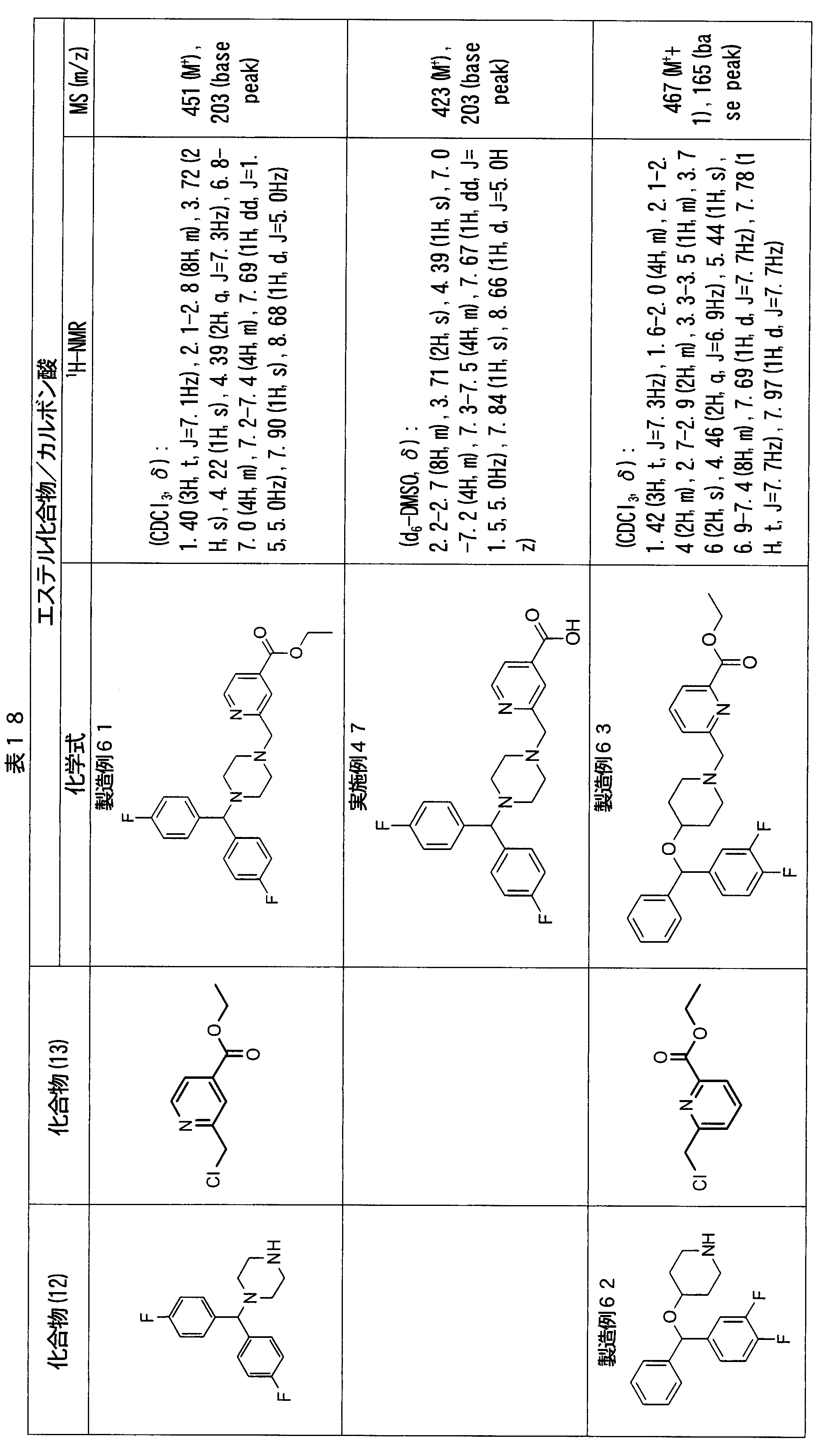
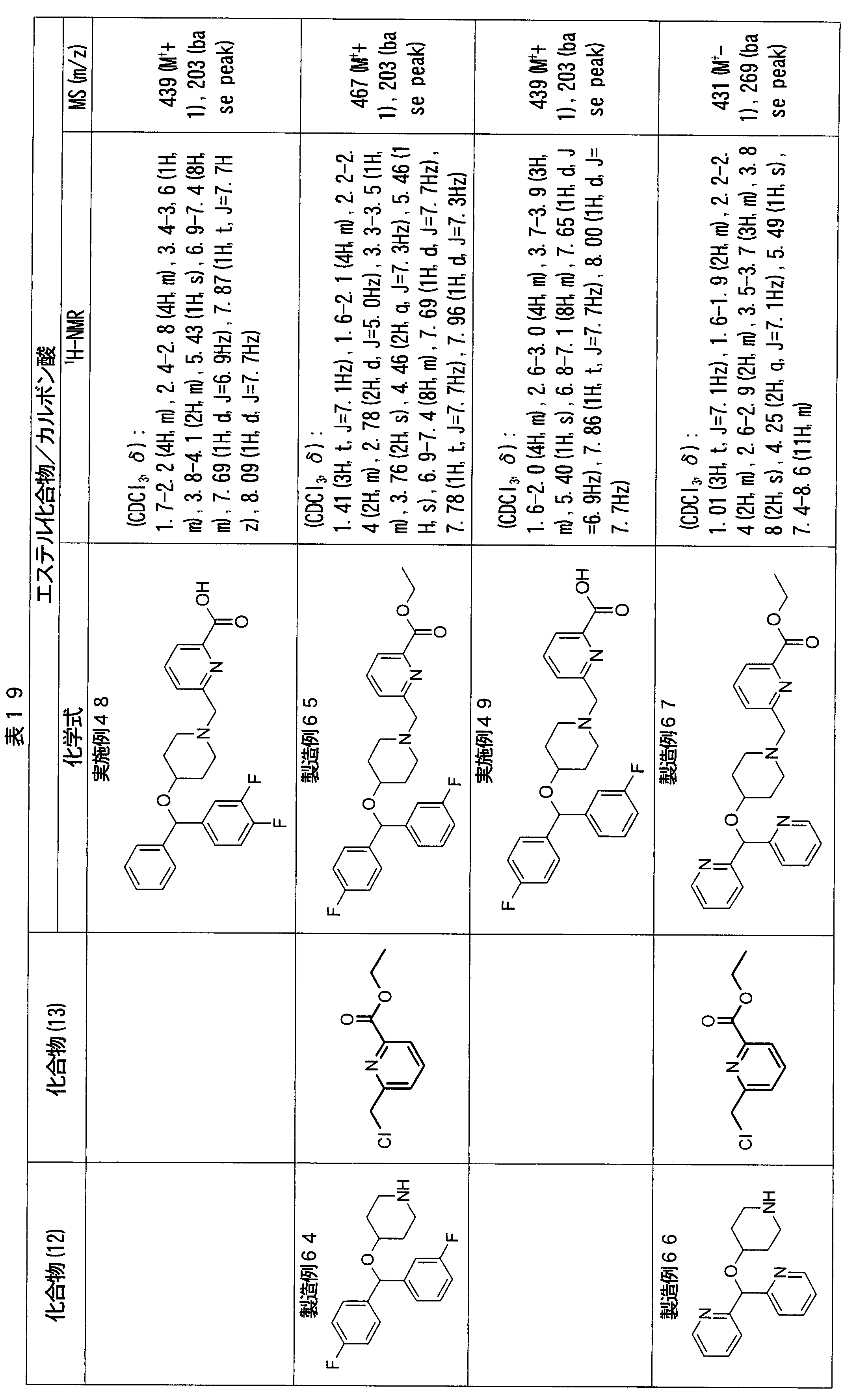








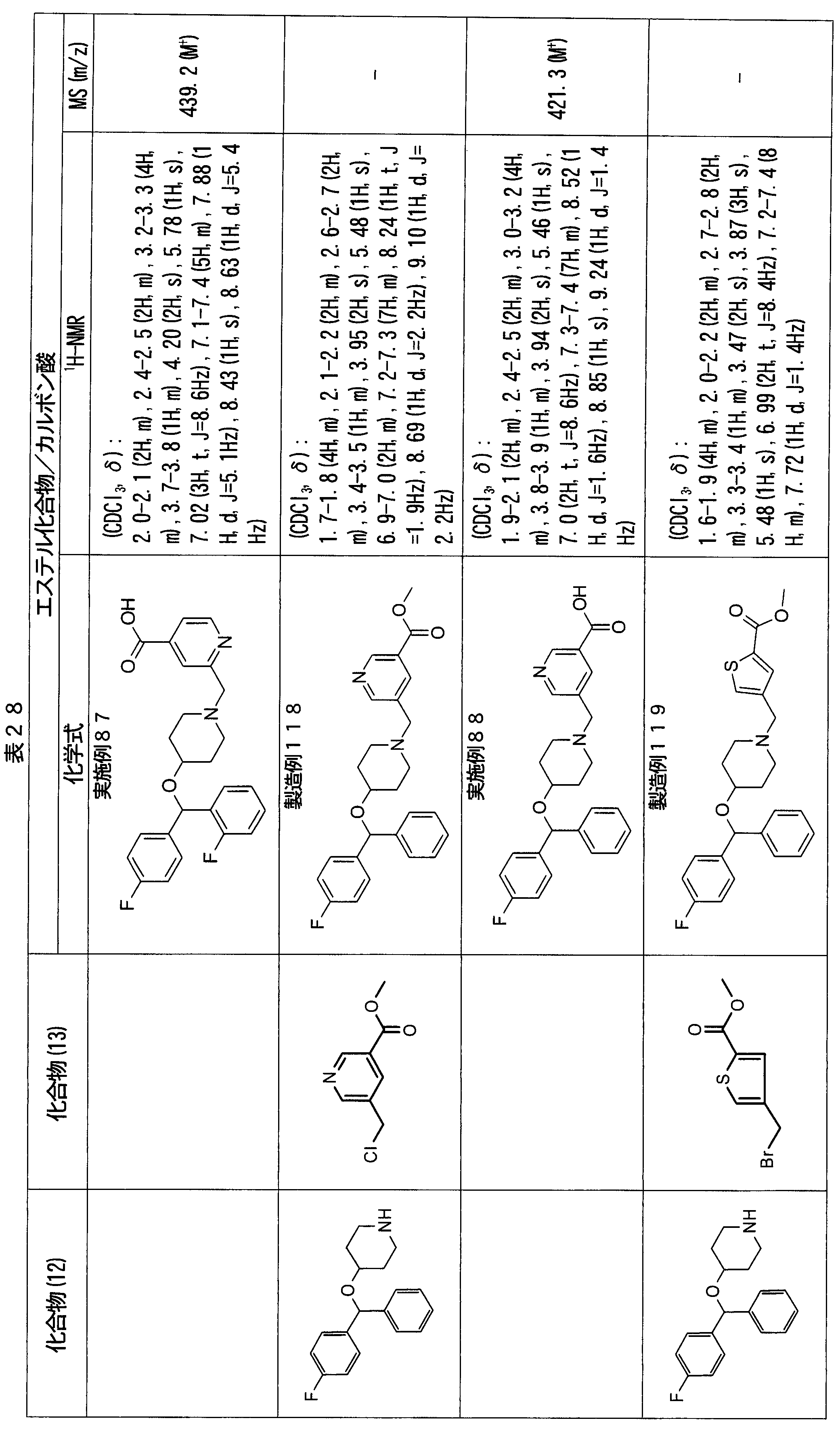



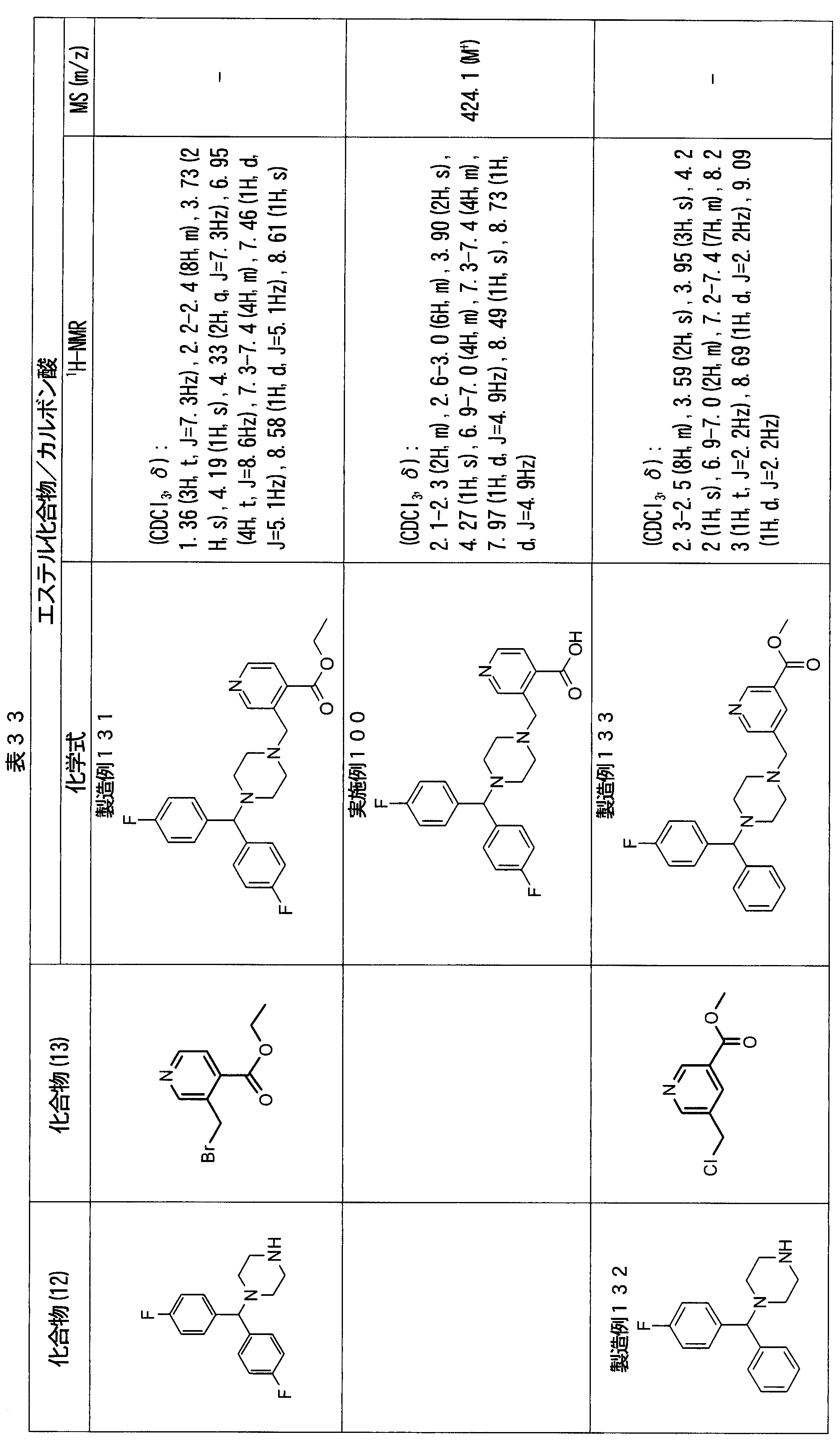
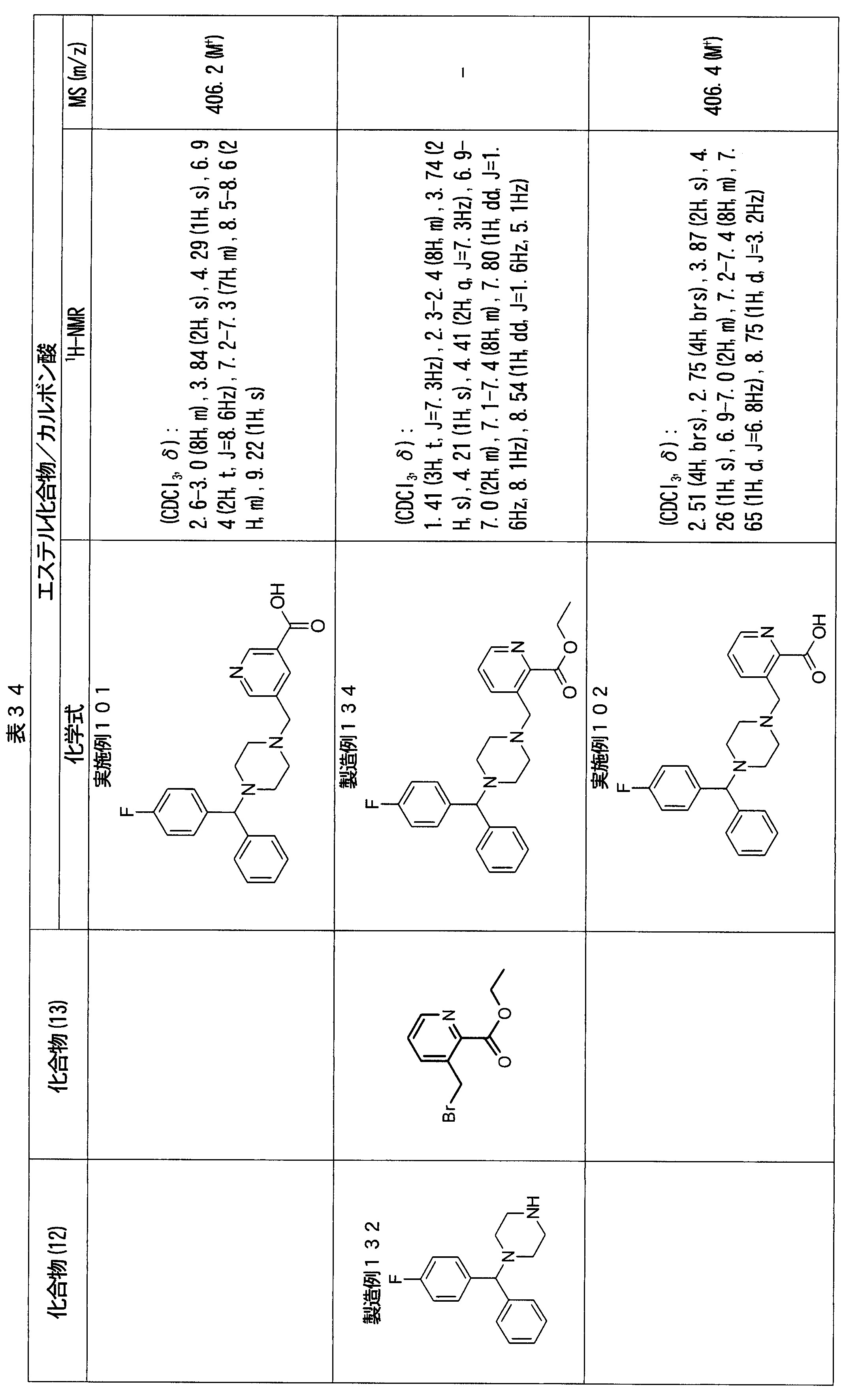
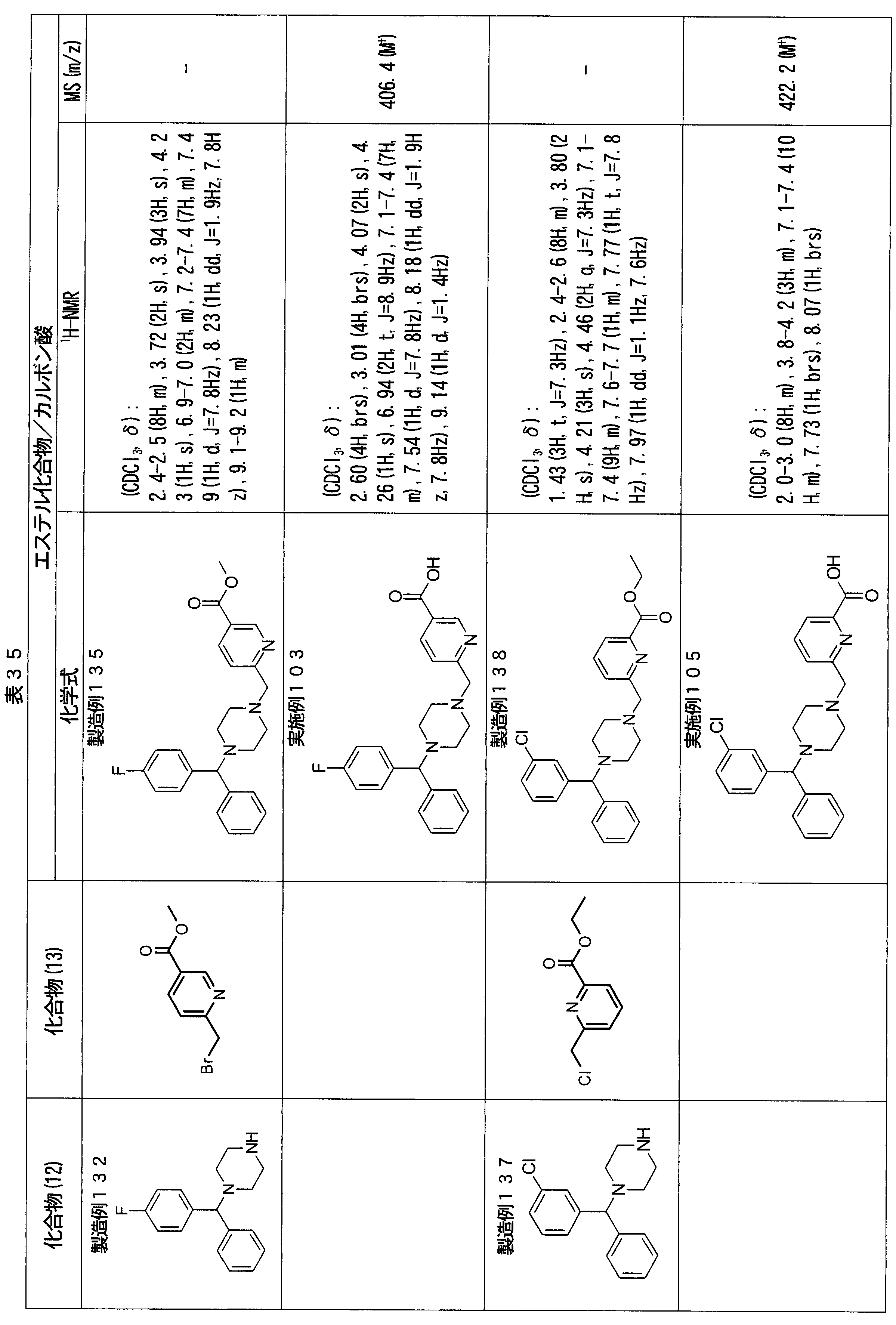










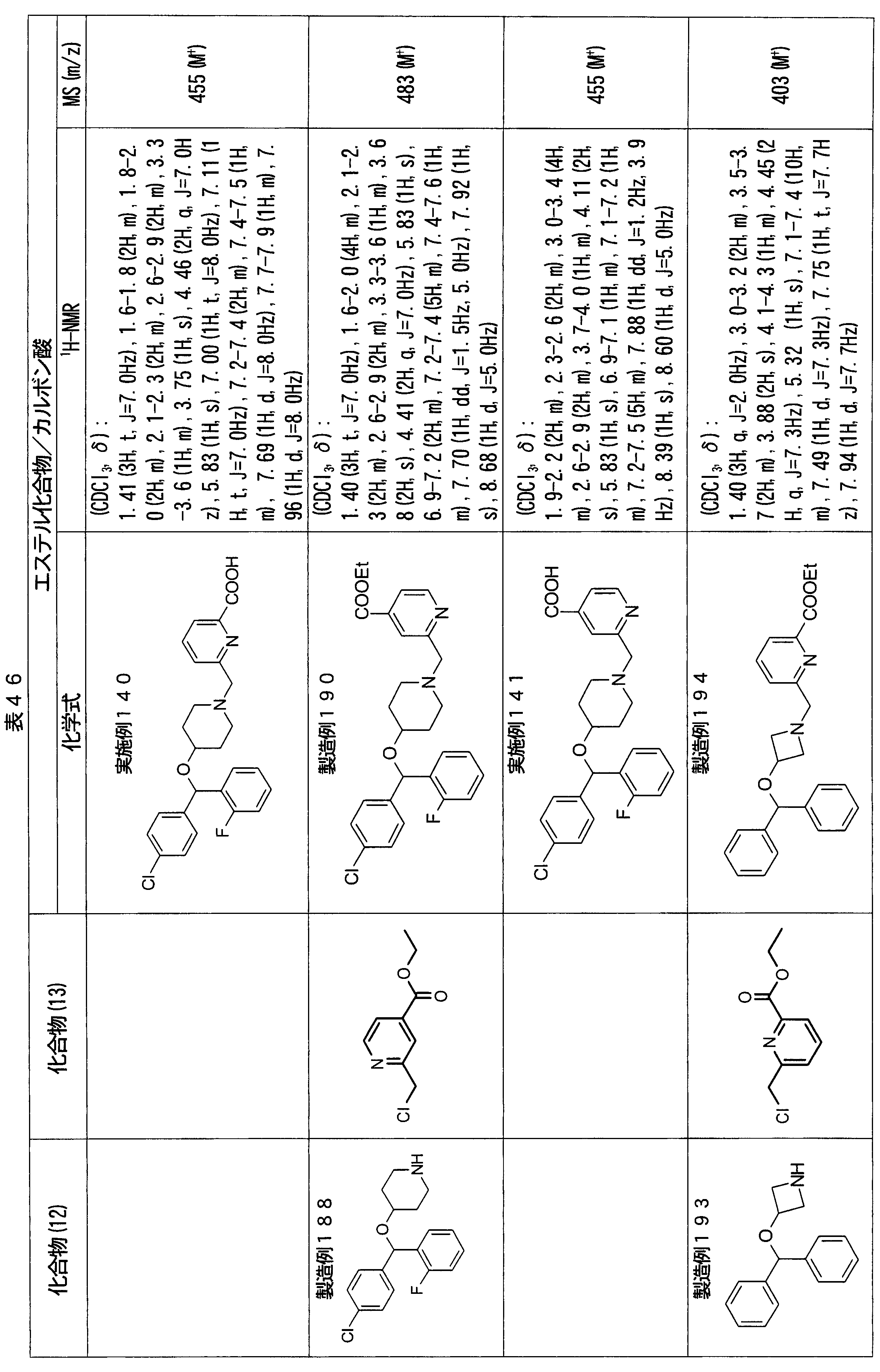
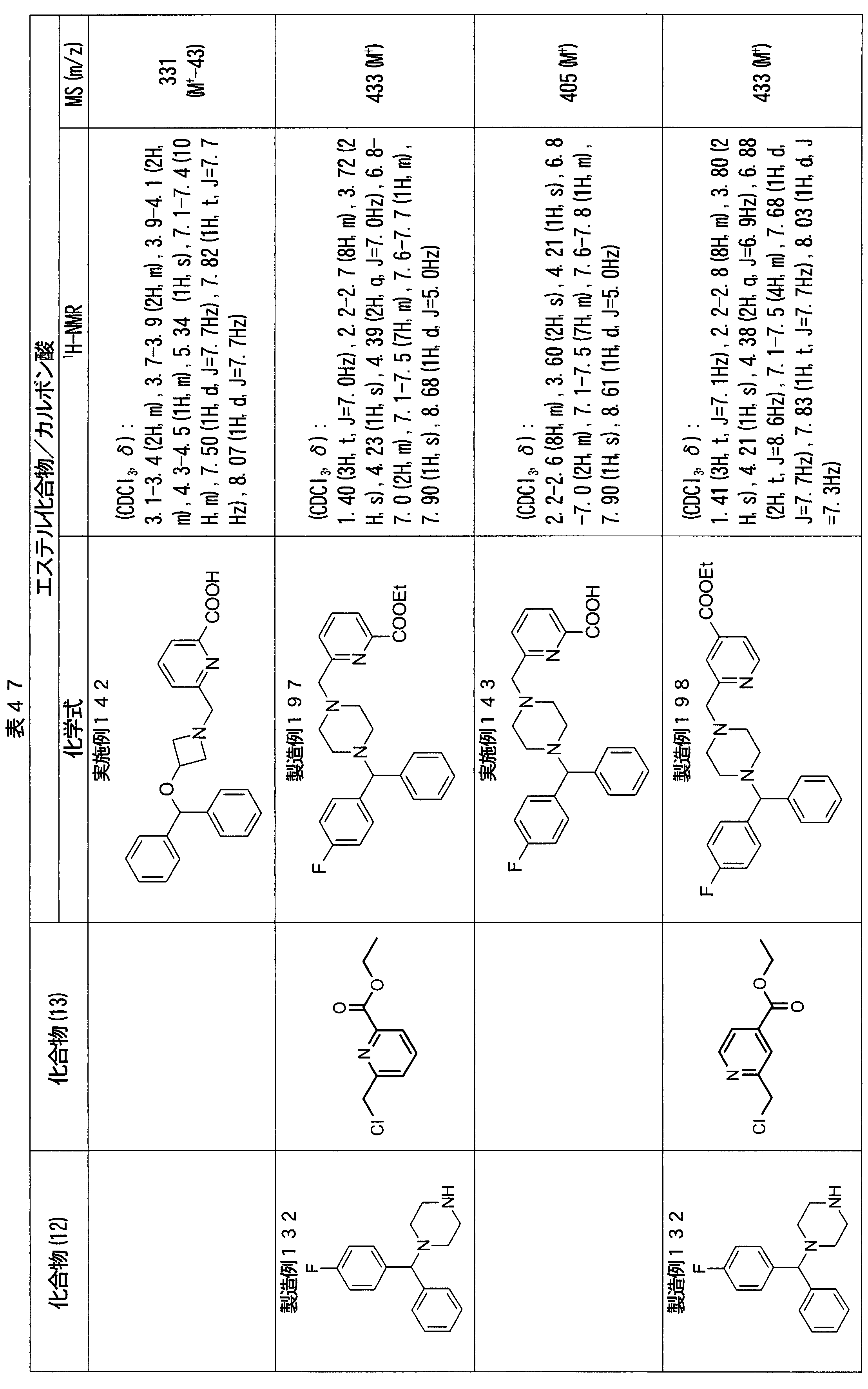


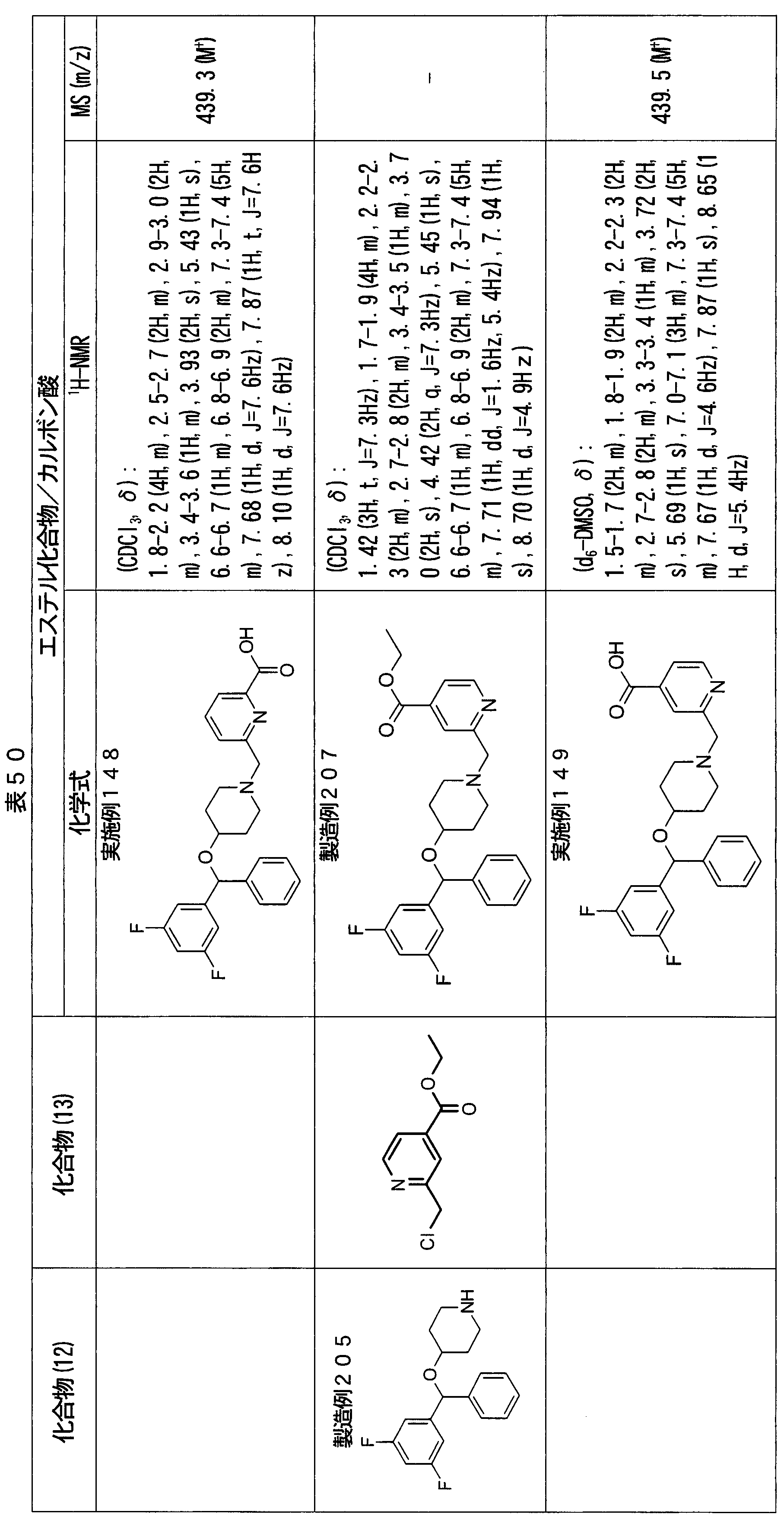
製造例13
2-ブロモメチルニコチン酸エチル
2-ブロモメチルニコチン酸エチル

窒素雰囲気下、2-メチルニコチン酸エチル(1.65g,10mmol)を四塩化炭素に溶解し、N-ブロモこはく酸イミド(2.31g,13mmol)及びアゾイソブチロニトリル(164mg,1mmol)を加え、8時間還流した。HPLCで反応の終了を確認した後、室温まで放冷し、水で有機層を3回、続いて飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した。溶媒を留去した後、得られた残渣をフラッシュカラムクロマトグラフィー(山善,ハイフラッシュカラム,シリカゲル,ヘキサン/酢酸エチル(容積比)=10:1)で精製し、表題化合物(1.45g,59.4%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.3Hz),4.42(2H,q,J=7.3Hz),5.04(2H,s),7.34(1H,app-dd,J=4.6,7.8Hz),8.29(1H,dd,J=1.9,8.1Hz),871(1H,dd,J=1.6,4.6Hz)。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.3Hz),4.42(2H,q,J=7.3Hz),5.04(2H,s),7.34(1H,app-dd,J=4.6,7.8Hz),8.29(1H,dd,J=1.9,8.1Hz),871(1H,dd,J=1.6,4.6Hz)。
製造例16、22、24、29、31、33、62、64、66、115、120、154、168、170、188、214、217
化合物(12a)として、ベンズヒドロールの代わりに他の公知化合物を用いる点、及び/又は化合物(12b)として、4-ヒドロキシピペリジンの代わりに他の公知化合物を用いる点を除き、製造例1と同様の操作を行い、表51~53に示す化合物(12)を得た。
化合物(12a)として、ベンズヒドロールの代わりに他の公知化合物を用いる点、及び/又は化合物(12b)として、4-ヒドロキシピペリジンの代わりに他の公知化合物を用いる点を除き、製造例1と同様の操作を行い、表51~53に示す化合物(12)を得た。
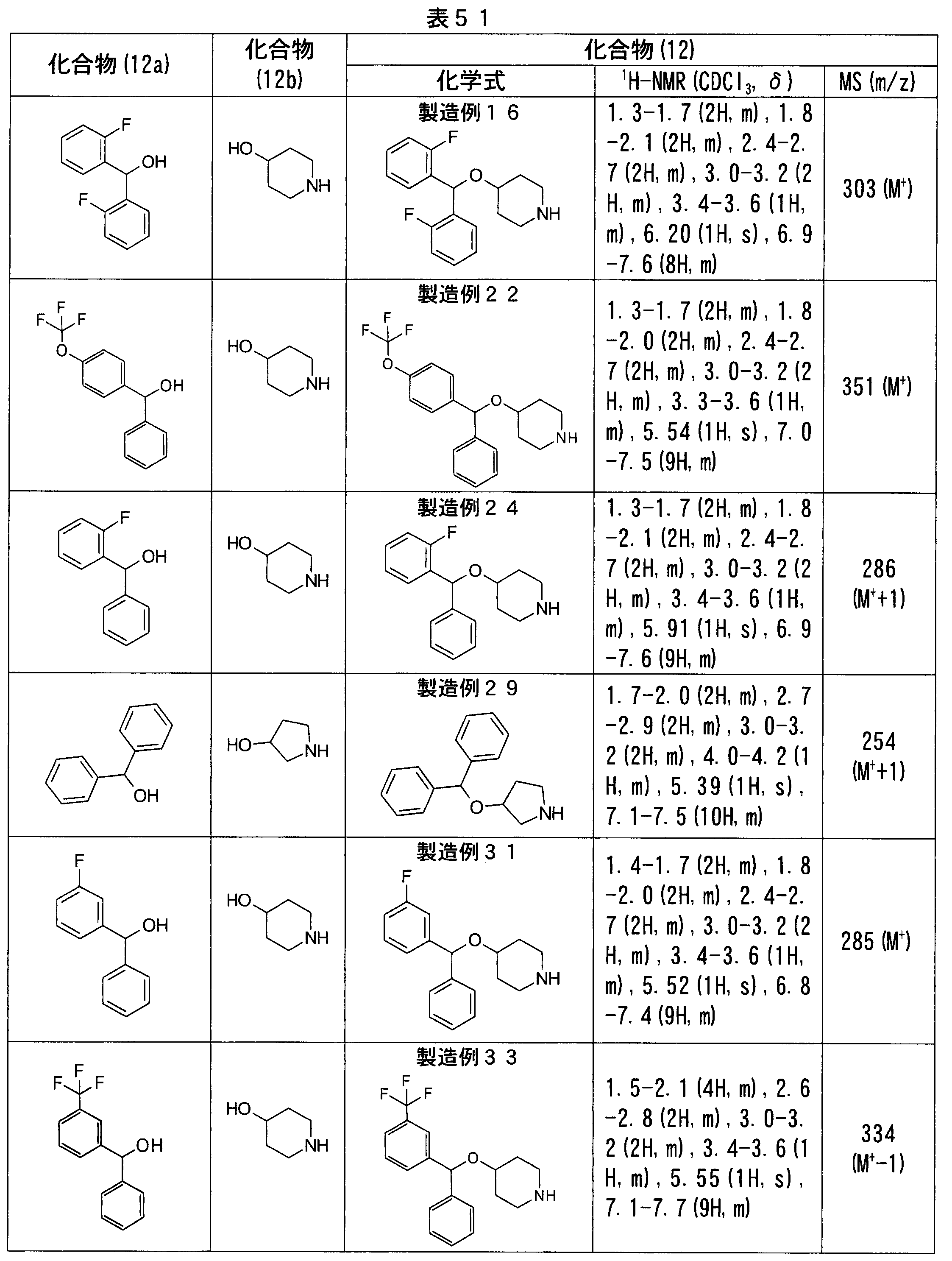
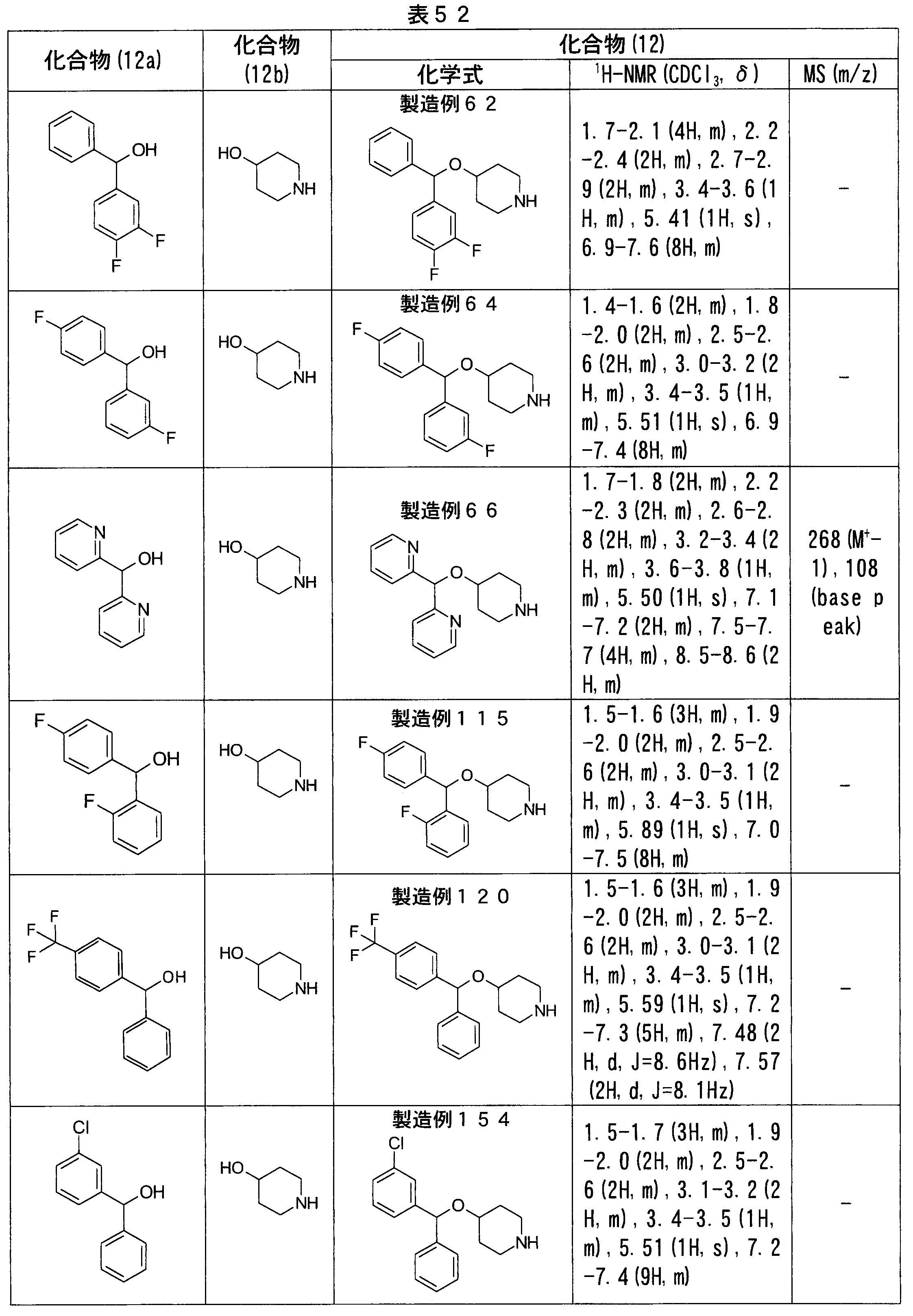

製造例36
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-2-クロロピリジン
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-2-クロロピリジン

5-クロロメチルフラン-2-カルボン酸メチルの代わりに2-クロロ-5-クロロメチルピリジンを用いる以外は、製造例2と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.6-2.3(6H,m),2.6-2.8(2H,m),3.4-3.6(3H,m),5.50(1H,s),7.1-7.4(11H,m),7.63(1H,dd,J=2.3,8.1Hz),8.27(1H,d,J=2.3Hz)
MS(m/z):393(M++1)。
1H-NMR(CDCl3,δ):1.6-2.3(6H,m),2.6-2.8(2H,m),3.4-3.6(3H,m),5.50(1H,s),7.1-7.4(11H,m),7.63(1H,dd,J=2.3,8.1Hz),8.27(1H,d,J=2.3Hz)
MS(m/z):393(M++1)。
製造例37
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸プロピル
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸プロピル

製造例36で合成した5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-2-クロロピリジン721mg、酢酸パラジウム21mg、1,3-ビス(ジフェニルホスフィノ)プロパン41mg、炭酸カリウム380mg、n-プロパノール7.0mL、N,N-ジメチルホルムアミド3.5mLの混合物を一酸化炭素雰囲気下、外温90℃で7時間撹拌した。溶媒を留去した後、酢酸エチルで希釈した。水に投入し有機層を分取した。飽和食塩水で洗い、硫酸マグネシウムで乾燥した。溶媒を留去の後、p-TLCを行い表題化合物569mgを得た。
1H-NMR(CDCl3,δ):1.02(3H,t,J=7.7Hz),1.6-2.0(6H,m),2.0-2.3(2H,m).2.6-2.8(2H,m),3.3-3.7(3H,m),4.36(2H,t,J=6.9Hz),5.50(1H,s),7.1-7.4(10H,m),7.79(1H,d,J=7.7Hz),8.06(1H,d,J=7.7Hz),8.66(1H,d,J=1.5Hz)
MS(m/z):443(M+-1),262(base peak)。
1H-NMR(CDCl3,δ):1.02(3H,t,J=7.7Hz),1.6-2.0(6H,m),2.0-2.3(2H,m).2.6-2.8(2H,m),3.3-3.7(3H,m),4.36(2H,t,J=6.9Hz),5.50(1H,s),7.1-7.4(10H,m),7.79(1H,d,J=7.7Hz),8.06(1H,d,J=7.7Hz),8.66(1H,d,J=1.5Hz)
MS(m/z):443(M+-1),262(base peak)。
実施例28
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸

製造例2で合成した化合物の代わりに製造例37で合成した5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジン-2-カルボン酸プロピルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.5-1.7(2H,m),1.8-1.9(2H,m),2.0-2.2(2H,m),2.5-2.8(2H,m),3.2-3.5(1H,m).3.56(2H,s),5.62(2H,s),7.1-7.5(10H,m),7.86(2H,dd,J=1.9,8.1Hz),7.99(1H,d,J=7.7Hz),8.59(1H,d,J=7.7Hz)
MS(m/z):401(M+-1),220(base peak)。
1H-NMR(d6-DMSO,δ):1.5-1.7(2H,m),1.8-1.9(2H,m),2.0-2.2(2H,m),2.5-2.8(2H,m),3.2-3.5(1H,m).3.56(2H,s),5.62(2H,s),7.1-7.5(10H,m),7.86(2H,dd,J=1.9,8.1Hz),7.99(1H,d,J=7.7Hz),8.59(1H,d,J=7.7Hz)
MS(m/z):401(M+-1),220(base peak)。
製造例38
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-2-ブロモピリジン
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-2-ブロモピリジン

製造例1で合成した4-ベンズヒドリルオキシピペリジン904mg、ジクロロメタン6.4mL、2-ブロモ-4-ピリジンカルバルデヒド629mgの混合物を50分間撹拌した。トリアセトキシヒドロほう酸ナトリウム1.075gを少しずつ加え14時間撹拌した。酢酸エチルで希釈し重曹水に投入した。有機層を分取し、飽和食塩水で洗った。硫酸マグネシウムで乾燥し、溶媒を留去した。シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=2:1~1:1)に付し、表題化合物1.027gを得た。
1H-NMR(CDCl3,δ):1.6-2.3(6H,m),2.6-2.8(2H,m),3.3-3.6(3H,m),5.51(1H,s),7.1-7.4(11H,m),7.46(1H,s),8.27(1H,d,J=5.0Hz)
MS(m/z):437(M+)。
1H-NMR(CDCl3,δ):1.6-2.3(6H,m),2.6-2.8(2H,m),3.3-3.6(3H,m),5.51(1H,s),7.1-7.4(11H,m),7.46(1H,s),8.27(1H,d,J=5.0Hz)
MS(m/z):437(M+)。
製造例39、41、44、46、48、実施例29~30、32~34
化合物(15)として、製造例36で合成した化合物の代わりに、製造例38、40、43、45、47で合成した化合物を用いる以外、製造例37と同様にして表54~56に示すエステル化合物(化合物(17))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表54~56に示すカルボン酸を得た。
化合物(15)として、製造例36で合成した化合物の代わりに、製造例38、40、43、45、47で合成した化合物を用いる以外、製造例37と同様にして表54~56に示すエステル化合物(化合物(17))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表54~56に示すカルボン酸を得た。


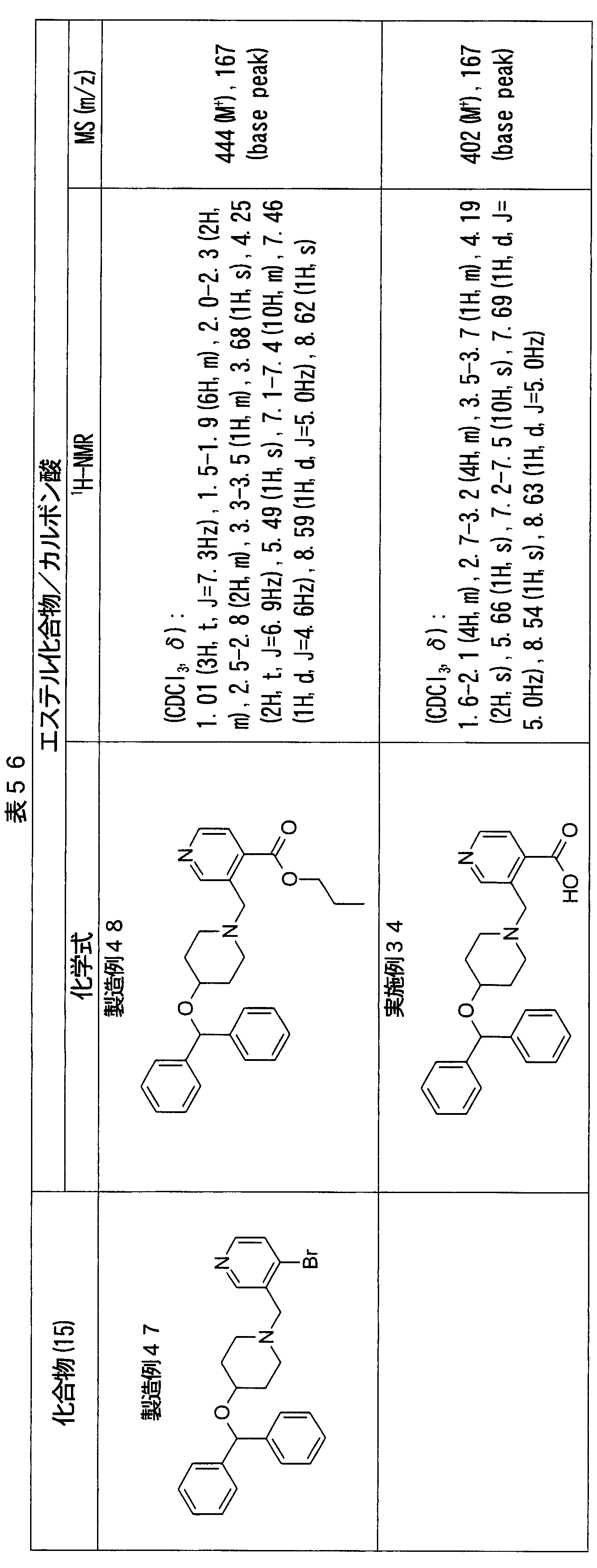
製造例40、43、45、47
カルバルデヒド化合物として、2-ブロモ-4-ピリジンカルバルデヒドの代わりに他の公知化合物を用いる以外は、製造例38と同様の操作を行い、表57に示す化合物(15)を得た。
カルバルデヒド化合物として、2-ブロモ-4-ピリジンカルバルデヒドの代わりに他の公知化合物を用いる以外は、製造例38と同様の操作を行い、表57に示す化合物(15)を得た。

製造例42
6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸メチル
6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸メチル

100mL3口フラスコへ製造例1で合成した4-ベンズヒドリルオキシピペリジン4.0g(15mmol)、テトラヒドロフラン25mLを仕込み内温-10℃に冷却した。そこへ1.6Mn-ブチルリチウム(ヘキサン溶液)11mL(17mmol,1.1eq.)を内温-5℃以下で滴下し同温にて30分間攪拌した。
別に準備した2,6-ピリジンカルボン酸ジメチル2.9g(15mmol)のテトラヒドロフラン懸濁液を-20℃まで冷却し、上記反応物を-10℃以下で滴下した。滴下終了後同温にて5分間熟成し徐々に室温へ昇温し一晩攪拌した。
反応終了を確認後、内容物を濃縮し残渣へ酢酸エチル、水を加えて分液抽出した。有機層を水、飽和食塩水にて洗浄後、硫酸ナトリウムにて乾燥し、溶媒を留去した。得られた残渣をカラムクロマトグラフィー(クロロホルム:酢酸エチル(容積比)=10:1)にて精製し表題化合物3.1g(66%)を得た。
1H-NMR(CDCl3,δ):1.7-2.0(4H,m),3.3-3.5(1H,m),3.6-3.9(4H,m),3.98(3H,s)5.52(1H,s),7.2-7.4(10H,m),7.83(1H,dd,J=1.2,7.3Hz),7.9-8.0(1H,m),8.1-8.2(1H,m)
MS(m/z):430(M+),166(base peak)。
1H-NMR(CDCl3,δ):1.7-2.0(4H,m),3.3-3.5(1H,m),3.6-3.9(4H,m),3.98(3H,s)5.52(1H,s),7.2-7.4(10H,m),7.83(1H,dd,J=1.2,7.3Hz),7.9-8.0(1H,m),8.1-8.2(1H,m)
MS(m/z):430(M+),166(base peak)。
実施例31
6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸
6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸
製造例42で合成した6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸メチル250mg(0.58mmol)を50mLのナス型フラスコへ仕込み、エタノール10mLにて溶解させた。そこへ1N水酸化ナトリウム0.9mLを添加し室温にて反応させた。反応終了を確認後、系内容物を濃縮し残渣へ水を加えて希釈した。そこへ1N塩酸を用いて中和し生じた白色結晶を吸引ろ過し減圧乾燥にて表題化合物200mg(83%)を得た。
1H-NMR(d6-DMSO,δ):1.41(2H,m),1.9-2.1(2H,m),3.3-3.8(5H,m),5.58(1H,s),7.2-7.4(10H,m),7.9(1H,d,J=7.1Hz),8.0(1H,d,J=5.0Hz),8.12(1H,d,J=5.0Hz)
MS(m/z):416(M+),167(base peak)。
1H-NMR(d6-DMSO,δ):1.41(2H,m),1.9-2.1(2H,m),3.3-3.8(5H,m),5.58(1H,s),7.2-7.4(10H,m),7.9(1H,d,J=7.1Hz),8.0(1H,d,J=5.0Hz),8.12(1H,d,J=5.0Hz)
MS(m/z):416(M+),167(base peak)。
製造例49
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピロール-1,2-ジカルボン酸 1-tert-ブチルエステル 2-メチルエステル
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピロール-1,2-ジカルボン酸 1-tert-ブチルエステル 2-メチルエステル

100mL3口フラスコへアルゴン雰囲気下製造例1で合成した4-ベンズヒドリルオキシピペリジン1.1g(4.0mmol)、N-tert-ブトキシカルボニル-5-ホルミルピロール-2-カルボン酸メチル1.0g(4.0mmol)、ジクロロメタン20mLを仕込み内温0℃に冷却した。そこへ水素化トリアセトキシホウ素ナトリウム1.3g(6.1mmol,1.5eq.)を内温-5℃以下で添加し同温にて5分間攪拌した後、室温まで昇温し一晩攪拌した。
反応終了を確認後、5%炭酸水素ナトリウム水溶液にてクエンチし、水を加えて洗浄した。
有機層を水にて再度洗浄後、硫酸ナトリウムにて乾燥させ溶媒を濃縮し黄色オイルを得た。次いでカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=10:1)にて精製し表題化合物1.0g(52%)を得た。
1H-NMR(CDCl3,δ):1.57(9H,s),1.7-1.9(4H,m),2.0-2.2(2H,m),2.6-2.7(2H,m),3.3-3.5(1H,m),3.52(2H,s),3.80(3H,s),5.48(1H,s),6.00(1H,d,J=3.9Hz),6.75(1H,d,J=3.9Hz),7.2-7.4(10H,m)
MS(m/z):505(M++1),57(base peak)。
1H-NMR(CDCl3,δ):1.57(9H,s),1.7-1.9(4H,m),2.0-2.2(2H,m),2.6-2.7(2H,m),3.3-3.5(1H,m),3.52(2H,s),3.80(3H,s),5.48(1H,s),6.00(1H,d,J=3.9Hz),6.75(1H,d,J=3.9Hz),7.2-7.4(10H,m)
MS(m/z):505(M++1),57(base peak)。
実施例35
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピロール-2-カルボン酸
5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピロール-2-カルボン酸
製造例49で合成した5-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピロール-1,2-ジカルボン酸1-tert-ブチルエステル2-メチルエステル790mg(1.5mmol)、メタノール20mLを50mLナス型フラスコへ仕込み室温下攪拌した。次いで1N水酸化ナトリウム水溶液6.0mL(6.0mmol,4.0eq.)を滴下し内温60℃にて終夜攪拌した。
反応終了を確認後、溶媒を留去し水を加えて希釈した。1N塩酸にて中和し生じた白色結晶を吸引ろ過にてろ取し、減圧乾燥後、表題化合物340mg(83%)を得た。
1H-NMR(CDCl3,δ):1.8-2.0(2H,m),2.3-2.6(2H,m),3.0-3.2(4H,m),3.7-3.9(1H,m),4.02(2H,s),5.41(1H,s),6.15(1H,s),6.70(1H,s),7.2-7.4(10H,m),11.78(1H,brs)
MS(m/z):391(M++1),167(base peak)。
1H-NMR(CDCl3,δ):1.8-2.0(2H,m),2.3-2.6(2H,m),3.0-3.2(4H,m),3.7-3.9(1H,m),4.02(2H,s),5.41(1H,s),6.15(1H,s),6.70(1H,s),7.2-7.4(10H,m),11.78(1H,brs)
MS(m/z):391(M++1),167(base peak)。
製造例51
6-[(4-ベンズヒドリルオキシピペリジン-1-イル)ジフルオロメチル]ピリジン-2-カルボン酸メチル
6-[(4-ベンズヒドリルオキシピペリジン-1-イル)ジフルオロメチル]ピリジン-2-カルボン酸メチル

100mL3口フラスコへ製造例42で合成した6-(4-ベンズヒドリルオキシピペリジン-1-カルボニル)ピリジン-2-カルボン酸メチル640mg(1.5mmol)、シクロペンチルメチルエーテル30mLを仕込みアルゴン置換した。次いで室温下オキザリルクロライド190mg(1.5mmol)を滴下し1時間攪拌した。その後加熱還流下終夜攪拌した。反応終了後を確認後、冷却し溶媒を濃縮乾固した。得られた残渣へジメチルイミダゾリジノン20mL、フッ化ナトリウム250mg(6.0mmol)を順次添加し室温下終夜攪拌した。反応終了を確認後、沈殿物をろ別しろ液を濃縮後カラムクロマトグラフィーにて精製(クロロホルム/酢酸エチル(容積比)=10:1)し表題化合物380mg(56%)を得た。
1H-NMR(CDCl3,δ):1.4-1.8(4H,m),3.3-3.9(5H,m),3.94(3H,s),5.50(1H,s),7.2-7.5(10H,m),7.73(1H,d,J=7.7Hz),7.80(1H,t,J=7.7Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):453(M++1),167(base peak)。
1H-NMR(CDCl3,δ):1.4-1.8(4H,m),3.3-3.9(5H,m),3.94(3H,s),5.50(1H,s),7.2-7.5(10H,m),7.73(1H,d,J=7.7Hz),7.80(1H,t,J=7.7Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):453(M++1),167(base peak)。
実施例37
6-[(4-ベンズヒドリルオキシピペリジン-1-イル)ジフルオロメチル]ピリジン-2-カルボン酸
6-[(4-ベンズヒドリルオキシピペリジン-1-イル)ジフルオロメチル]ピリジン-2-カルボン酸

製造例51で合成した6-[(4-ベンズヒドリルオキシピペリジン-1-イル)ジフルオロメチル]ピリジン-2-カルボン酸メチル380mg(0.84mmol)を50mLのナス型フラスコへ仕込み、エタノール10mLにて溶解させた。そこへ1N水酸化ナトリウム水溶液1.7mLを添加し室温にて反応させた。反応終了を確認後、系内容物を濃縮し残渣へ水を加えて希釈した。そこへ1N塩酸を用いて中和しクロロホルムを加えて抽出した。有機層を水、飽和食塩水にて洗浄し硫酸ナトリウムにて乾燥させた。溶媒濃縮後、得られた褐色油状物をカラムクロマトグラフィーにて精製(クロロホルム/メタノール(容積比)=3:1)し表題化合物306mg(83%)を得た。
1H-NMR(d6-DMSO,δ):1.25(2H,t,J=7.1Hz),1.41(2H,t,J=7.1Hz),3.3-3.8(5H,m),5.58(1H,s),7.2-7.5(10H,m),7.71(1H,d,J=7.7Hz),7.78(1H,t,J=7.7Hz),7.94(1H,d,J=7.7Hz)
MS(m/z):439(M++1),167(base peak)。
1H-NMR(d6-DMSO,δ):1.25(2H,t,J=7.1Hz),1.41(2H,t,J=7.1Hz),3.3-3.8(5H,m),5.58(1H,s),7.2-7.5(10H,m),7.71(1H,d,J=7.7Hz),7.78(1H,t,J=7.7Hz),7.94(1H,d,J=7.7Hz)
MS(m/z):439(M++1),167(base peak)。
製造例71
2,5-チオフェンジカルボン酸ジエチル
2,5-チオフェンジカルボン酸ジエチル
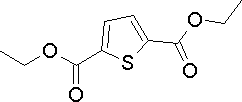
2,5-チオフェンジカルボン酸(861mg,5mmol)をエタノール(10mL)に懸濁し、濃硫酸(1.6mL,30mmol)を加え、一夜加熱還流した。HPLCで反応の終了を確認した後、室温まで放冷し、エタノールを減圧下留去した後、残渣に水と酢酸エチルを加え、酢酸エチルで3回抽出した。有機層を飽和塩化ナトリウム水溶液で洗浄した後、無水硫酸マグネシウムで乾燥し、溶媒を留去して、表題化合物(1.14g,100%)の結晶を得た。
1H-NMR(CDCl3,δ):1.39(6H,t,J=7.3Hz),4.38(4H,q,J=7.3Hz),7.73(2H,s)。
1H-NMR(CDCl3,δ):1.39(6H,t,J=7.3Hz),4.38(4H,q,J=7.3Hz),7.73(2H,s)。
製造例72
5-ヒドロキシメチルチオフェン-2-カルボン酸エチル
5-ヒドロキシメチルチオフェン-2-カルボン酸エチル
製造例71で合成した2,5-チオフェンジカルボン酸ジエチル(1.14g,5mmol)をエタノールに溶解し、テトラヒドロホウ酸ナトリウム(113mg,3mmol)を加え、室温で3日間撹拌した。HPLCで反応の終了を確認した後、水を加え、減圧下濃縮した。残渣を酢酸エチルに溶解し、水及び飽和塩化ナトリウムで洗浄した後、無水硫酸マグネシウムで乾燥し、溶媒を留去した。得られた残渣をフラッシュカラムクロマトグラフィー(山善ハイフラッシュカラム,シリカゲル,ヘキサン/酢酸エチル(容積比)=3:2)で精製し、表題化合物(491mg,52.7%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.37(3H,t,J=7.3Hz),4.34(2H,q,J=7.0Hz),4.86(2H,s),6.99(1H,d,J=3.5Hz),7.67(1H,d,J=3.8Hz)。
1H-NMR(CDCl3,δ):1.37(3H,t,J=7.3Hz),4.34(2H,q,J=7.0Hz),4.86(2H,s),6.99(1H,d,J=3.5Hz),7.67(1H,d,J=3.8Hz)。
製造例73
5-クロロメチルチオフェン-2-カルボン酸エチル
5-クロロメチルチオフェン-2-カルボン酸エチル
製造例72で合成した5-ヒドロキシメチルチオフェン-2-カルボン酸エチルのトルエン懸濁液に、塩化チオニル212μLを滴加し、30分間還流した。室温まで放冷し、1時間撹拌した。TLCにて反応の終了を確認した後、溶媒を留去した。得られた残渣を、精製することなしに次の反応に使用した。
製造例75
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾール-3-カルボン酸エチル
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾール-3-カルボン酸エチル

N-tert-ブトキシカルボニル-5-ホルミルピロール-2-カルボン酸メチルの代わりに4-ホルミル-1H-ピラゾール-カルボン酸エチルを用いる以外は、製造例49と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.38(3H,t,J=7.1Hz)1.6-2.0(4H,m),2.1-2.3(2H,m),2.7-2.9(2H,m),3.3-3.6(1H,m),3.73(2H,s),4.37(2H,q,J=7.3Hz),5.50(1H,s),7.2-7.5(11H,m),7.61(1H,s)
MS(m/z):419(M+),167(base peak)。
1H-NMR(CDCl3,δ):1.38(3H,t,J=7.1Hz)1.6-2.0(4H,m),2.1-2.3(2H,m),2.7-2.9(2H,m),3.3-3.6(1H,m),3.73(2H,s),4.37(2H,q,J=7.3Hz),5.50(1H,s),7.2-7.5(11H,m),7.61(1H,s)
MS(m/z):419(M+),167(base peak)。
実施例55
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾール-3-カルボン酸
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾール-3-カルボン酸

製造例2で合成した化合物の代わりに製造例75で合成した4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾール-3-カルボン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.8-2.2(4H,m),2.9-3.3(4H,m),3.8-4.0(3H,m),5.42(1H,s),7.2-7.4(10H,m),7.54(1H,s)
MS(m/z):390(M+-1),167(base peak)。
1H-NMR(CDCl3,δ):1.8-2.2(4H,m),2.9-3.3(4H,m),3.8-4.0(3H,m),5.42(1H,s),7.2-7.4(10H,m),7.54(1H,s)
MS(m/z):390(M+-1),167(base peak)。
製造例80
(±)-4-[(2,4-ジフルオロフェニル)フェニルメトキシ]ピペリジン
(±)-4-[(2,4-ジフルオロフェニル)フェニルメトキシ]ピペリジン

4-ヒドロキシピペリジン(1.7g,16.8mmol)のトルエン懸濁液にモレキュラーシーブス、(2,4-ジフルオロフェニル)フェニルメタノール(3.69g,16.8mmol)、p-トルエンスルホン酸1水和物(3.84g,20.2mmol)を加え、ディーンスタークコンデンサーを用いて水を除去しながら3時間還流した。HPLCで反応の終了を確認した後、室温まで放冷し、不溶物を除去した後、溶媒を留去した。得られた残渣を酢酸エチルに溶解し、2N水酸化ナトリウム液で3回洗浄した。有機層を硫酸マグネシウムで乾燥した後、溶媒を留去し、表題化合物(2.8g,54.9%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.4-1.7(3H,m),1.9-2.0(2H,m),2.5-2.7(2H,m),3.0-3.6(3H,m),5.86(1H,s),6.7-6.9(2H,m),7.2-7.5(6H,m)。
1H-NMR(CDCl3,δ):1.4-1.7(3H,m),1.9-2.0(2H,m),2.5-2.7(2H,m),3.0-3.6(3H,m),5.86(1H,s),6.7-6.9(2H,m),7.2-7.5(6H,m)。
実施例63
2-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-6-メチルピリジン
2-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-6-メチルピリジン

5-クロロメチルフラン-2-カルボン酸メチルの代わりに2-ブロモメチル-6-メチルピリジンを用いる以外は、製造例2と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.1-2.4(2H,m),2.52(3H,s),2.7-2.9(2H,m),3.3-3.5(1H,m),3.60(1H,s),5.51(1H,s),7.00(1H,d,J=7.7Hz),7.1-7.5(11H,m),7.52(1H,t,J=7.7Hz)
MS(m/z):373(M++1),107(base peak)。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.1-2.4(2H,m),2.52(3H,s),2.7-2.9(2H,m),3.3-3.5(1H,m),3.60(1H,s),5.51(1H,s),7.00(1H,d,J=7.7Hz),7.1-7.5(11H,m),7.52(1H,t,J=7.7Hz)
MS(m/z):373(M++1),107(base peak)。
実施例64
1-[ビス(4-フルオロフェニル)メチル]-4-(6-メチルピリジン-2-イルメチル)ピペラジン
1-[ビス(4-フルオロフェニル)メチル]-4-(6-メチルピリジン-2-イルメチル)ピペラジン
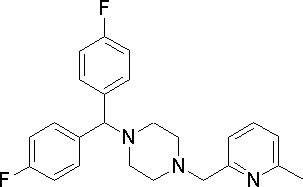
4-ベンズヒドリルオキシピペリジンの代わりに公知化合物である1-[ビス(4-フルオロフェニル)メチル]ピペラジンと5-クロロメチルフラン-2-カルボン酸メチルの代わりに2-ブロモメチル-6-メチルピリジンを用いる以外は、製造例2と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):2.2-2.7(11H,m),3.63(2H,s),4.21(1H,s),6.8-7.1(5H,m),7.18(1H,d,J=7.7Hz),7.2-7.4(4H,m),7.49(1H,t,J=7.7Hz)
MS(m/z):393(M+),107(base peak)。
1H-NMR(CDCl3,δ):2.2-2.7(11H,m),3.63(2H,s),4.21(1H,s),6.8-7.1(5H,m),7.18(1H,d,J=7.7Hz),7.2-7.4(4H,m),7.49(1H,t,J=7.7Hz)
MS(m/z):393(M+),107(base peak)。
製造例84
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン
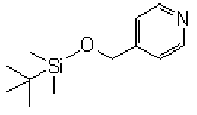
1H-NMR(CDCl3,δ):0.12(6H,s),0.96(9H,s),4.75(2H,s),7.2-7.3(2H,m),8.55(2H,dd,J=1.6,4.6Hz)。
製造例85
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-1-オキシド
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-1-オキシド
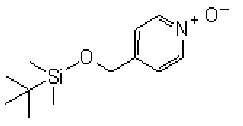
製造例84で合成した4-(tert-ブチルジメチルシリルオキシメチル)ピリジン(1.1g,5mmol)を窒素雰囲気下、ジクロロメタンに溶解し、氷冷下、m-クロロ過安息香酸(1.9g,14.5mmol)をゆっくりと加え、室温までゆっくりと昇温した後、一夜撹拌した。HPLCで反応の終了を確認した後、チオ硫酸ナトリウム液で過剰のm-クロロ過安息香酸を除去し、2N水酸化ナトリウムで3回洗浄した。有機層を硫酸マグネシウムで乾燥し、溶媒を留去して表題化合物(1.2g,100%)を油状物質として得た。
1H-NMR(CDCl3,δ):0.12(6H,s),0.95(9H,s),4.70(2H,s),7.25(2H,d,J=7.3Hz),8.2(2H,d,J=7.0Hz)。
1H-NMR(CDCl3,δ):0.12(6H,s),0.95(9H,s),4.70(2H,s),7.25(2H,d,J=7.3Hz),8.2(2H,d,J=7.0Hz)。
製造例86
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-2-カルボニトリル
4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-2-カルボニトリル

製造例85で合成した4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-1-オキシド(1.2g,5mmol)を窒素雰囲気下、トリエチルアミンに溶解し、トリメチルシリルシアニド(744μL,6mmol)を加え、90℃で3時間撹拌した。反応の終了をHPLCで確認した後、室温まで放冷し、溶媒を留去した。得られた残渣を酢酸エチルに溶解し、水で3回、飽和食塩水で1回洗浄した後、有機層を硫酸マグネシウムで乾燥した。溶媒を留去して表題化合物(260mg,20.9%)を油状物質として得た。
1H-NMR(CDCl3,δ):0.13(6H,s),0.96(9H,s),4.78(2H,s),7.4-7.5(1H,m),7.68(1H,s),8.65(1H,d,J=4.6Hz)。
1H-NMR(CDCl3,δ):0.13(6H,s),0.96(9H,s),4.78(2H,s),7.4-7.5(1H,m),7.68(1H,s),8.65(1H,d,J=4.6Hz)。
製造例87
4-ヒドロキシメチルピリジン-2-カルボニトリル
4-ヒドロキシメチルピリジン-2-カルボニトリル
製造例86で合成した4-(tert-ブチルジメチルシリルオキシメチル)ピリジン-2-カルボニトリル(260mg,1.1mmol)をエタノールに溶解し、濃硫酸(123μL,2.3mmol)を加え、一夜還流した。HPLCで反応の終了を確認した後、室温まで放冷し、溶媒を留去した。得られた残渣を酢酸エチルに溶解し、水及び飽和食塩水で洗浄した後、有機層を硫酸マグネシウムで乾燥し、溶媒を留去して表題化合物(111mg,78.8%)を黄白色の固体として得た。
1H-NMR(CDCl3,δ):4.83(2H,s),7.5-7.6(1H,m),7.74(1H,s),8.68(1H,d,J=5.4Hz)。
1H-NMR(CDCl3,δ):4.83(2H,s),7.5-7.6(1H,m),7.74(1H,s),8.68(1H,d,J=5.4Hz)。
製造例88
4-クロロメチルピリジン-2-カルボニトリル
4-クロロメチルピリジン-2-カルボニトリル
製造例87で合成した4-ヒドロキシメチルピリジン-2-カルボニトリル(111mg,0.83mmol)をトルエンに溶解し、塩化チオニル(73μL)を加え、30分間還流した。その後室温まで放冷し、1時間撹拌した。HPLCで反応の終了を確認した後、溶媒を留去し、得られた残渣を精製することなく次の反応に使用した。
製造例89
(±)-4-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジノ-1-イルメチル}ピリジン-2-カルボニトリル
(±)-4-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジノ-1-イルメチル}ピリジン-2-カルボニトリル

公知化合物である(±)-4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン(237mg,0.83mmol)をN,N-ジメチルホルムアミドに溶解し、製造例88で合成した4-クロロメチルピリジン-2-カルボニトリル(127mg,0.83mmol)及び炭酸カリウム(138mg,1mmol)を加え、70℃で一夜撹拌した。反応の終了をHPLCで確認した後、室温まで放冷し、水及びジエチルエーテルを加え、有機層を水で3回洗浄し、飽和食塩水で1回洗浄した。硫酸マグネシウムで乾燥した後、溶媒を留去し、得られた残渣をフラッシュカラムクロマトグラフィー(山善ハイフラッシュカラム,シリカゲル,ヘキサン/酢酸(容積比)=2:1)で精製し、表題化合物(156mg,46.8%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.7-1.9(4H,m),2.2-2.3(2H,m),2.6-2.8(2H,m),3.4-3.6(3H,m),5.49(1H,s),7.00(2H,t,J=8.6Hz),7.2-7.4(7H,m),7.48(1H,d,J=4.9Hz),7.72(1H,s),8.62(1H,d,J=4.9Hz)。
1H-NMR(CDCl3,δ):1.7-1.9(4H,m),2.2-2.3(2H,m),2.6-2.8(2H,m),3.4-3.6(3H,m),5.49(1H,s),7.00(2H,t,J=8.6Hz),7.2-7.4(7H,m),7.48(1H,d,J=4.9Hz),7.72(1H,s),8.62(1H,d,J=4.9Hz)。
実施例65
(±)-4-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸
(±)-4-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジン-2-カルボン酸

製造例89で合成した(±)-4-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジノ-1-イルメチル}ピリジン-2-カルボニトリル(156mg,0.39mmol)をエタノールに溶解し、50%水酸化ナトリウム液(5mL)を加え、8時間還流した。HPLCで反応の終了を確認した後、室温まで放冷し、溶媒を留去した。残渣を少量の水に溶解し、6N塩酸で液性を中性とした後、析出した固体を集め、温エタノールで洗浄し、表題化合物(90mg,54.9%)を白色の固体として得た。
1H-NMR(d6-DMSO,δ):1.8-2.2(4H,m),2.9-3.3(4H,m),3.67(1H,br),4.41(2H,s),5.67(1H,s),7.1-7.5(8H,m),7.9-8.1(1H,m),8.2-8.4(1H,m),8.77(1H,d,J=4.3Hz)
EIMS:m/e 421.3(M+)
製造例90
5-ブロモメチル-2-メトキシ安息香酸メチル
1H-NMR(d6-DMSO,δ):1.8-2.2(4H,m),2.9-3.3(4H,m),3.67(1H,br),4.41(2H,s),5.67(1H,s),7.1-7.5(8H,m),7.9-8.1(1H,m),8.2-8.4(1H,m),8.77(1H,d,J=4.3Hz)
EIMS:m/e 421.3(M+)
製造例90
5-ブロモメチル-2-メトキシ安息香酸メチル

窒素雰囲気下、2-メトキシ-5-メチル安息香酸メチル(901mg,5mmol)をアセトニトリルに溶解し、N-ブロモこはく酸イミド(890mg,5mmol)及び過酸化ベンゾイル(24mg,0.1mmol)を加え、1時間還流した。HPLCで反応の終了を確認した後、室温まで放冷し、溶媒を留去した後、残渣を酢酸エチルに溶解し、水で有機層を3回、続いて飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した。溶媒を留去した後、得られた残渣をフラッシュカラムクロマトグラフィー(山善,ハイフラッシュカラム,シリカゲル,ヘキサン/酢酸エチル(容積比)=10:1)で精製し、表題化合物(570mg,44.0%)を油状物質として得た。
1H-NMR(CDCl3,δ):1.56(3H,s),3.90(3H,s),4.49(2H,s),6.96(1H,d,J=8.6Hz),7.51(1H,dd,J=2.7,8.6Hz),7.84(1H,d,J=2.7Hz)。
1H-NMR(CDCl3,δ):1.56(3H,s),3.90(3H,s),4.49(2H,s),6.96(1H,d,J=8.6Hz),7.51(1H,dd,J=2.7,8.6Hz),7.84(1H,d,J=2.7Hz)。
製造例92
8-クロロ-11-ピペリジン-4-イリデン-6,11-ジヒドロ-5H-ベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン
8-クロロ-11-ピペリジン-4-イリデン-6,11-ジヒドロ-5H-ベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン

4-(8-クロロ-5,6-ジヒドロ-11H-ベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)-1-ピペリジンカルボン酸エチル(383mg,1mmol)のエタノール溶液に30%水酸化ナトリウム水溶液を加え、一夜還流した。溶媒を留去し、得られた残渣を水に溶解し、濃塩酸で中和した。酢酸エチルで3回抽出し、合わせた有機層を硫酸マグネシウムで乾燥した。溶媒を留去し、表題化合物(290mg,93.3%)を黄白色の固体として得た。
1H-NMR(CDCl3,δ):2.3-2.4(4H,m),2.6-2.9(4H,m)3.0-3.1(2H,m),3.2-3.5(2H,m),7.1-7.2(4H,m),7.43(1H,dd,J=1.4Hz,7.6Hz),8.40(1H,dd,J=1.6Hz,4.9Hz)。
1H-NMR(CDCl3,δ):2.3-2.4(4H,m),2.6-2.9(4H,m)3.0-3.1(2H,m),3.2-3.5(2H,m),7.1-7.2(4H,m),7.43(1H,dd,J=1.4Hz,7.6Hz),8.40(1H,dd,J=1.6Hz,4.9Hz)。
製造例93
3-[4-(8-クロロ-5,6-ジヒドロベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)ピペリジン-1-イルメチル]安息香酸メチル
3-[4-(8-クロロ-5,6-ジヒドロベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)ピペリジン-1-イルメチル]安息香酸メチル
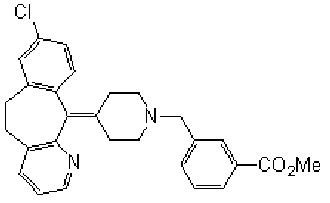
製造例92で得られた化合物(120mg,0.39mmol)をN,N-ジメチルホルムアミドに溶解し、3-ブロモメチル安息香酸メチル(99mg,0.43mmol)及び炭酸カリウム(59mg,0.43mmol)を加え、70℃で一夜撹拌した。溶媒を留去した後、残渣に酢酸エチルを加え、有機層を水で3回、続いて飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した。溶媒を留去した後、得られた残渣をフラッシュカラムクロマトグラフィー(山善,ハイフラッシュカラム,シリカゲル,ヘキサン/酢酸エチル5:1)で精製し、表題化合物(170mg,95.0%)を油状物質として得た。
1H-NMR(CDCl3,δ):2.1-2.5(6H,m),2.7-2.9(4H,m),3.3-3.5(2H,m),3.54(2H,s),3.91(3H,s),7.1-7.2(4H,m),7.3-7.4(2H,m)7.5-7.6(1H,m),7.9-8.0(2H,m),8.39(1H,dd,J=1.9Hz,4.9Hz)。
1H-NMR(CDCl3,δ):2.1-2.5(6H,m),2.7-2.9(4H,m),3.3-3.5(2H,m),3.54(2H,s),3.91(3H,s),7.1-7.2(4H,m),7.3-7.4(2H,m)7.5-7.6(1H,m),7.9-8.0(2H,m),8.39(1H,dd,J=1.9Hz,4.9Hz)。
実施例67
3-[4-(8-クロロ-5,6-ジヒドロベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)ピペリジン-1-イルメチル]安息香酸
3-[4-(8-クロロ-5,6-ジヒドロベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)ピペリジン-1-イルメチル]安息香酸

製造例93で得られた化合物(170mg,0.37mmol)をメタノールに溶解し、2N水酸化ナトリウム水溶液370μLの混合物を2時間加熱還流した。冷後、水3mL、1N塩酸740μLを加え撹拌した。析出した結晶をろ取、水洗、乾燥し表題化合物(104mg、63.2%)を白色の固体として得た。
1H-NMR(d6-DMSO,δ):2.7-2.9(4H,m),3.3-3.4(8H,m),4.36(2H,s),7.1-7.4(4H,m),7.6-7.7(1H,m),7.8-8.2(4H,m),8.43(1H,s),10.74(1H,br)
MS(m/z):445.1(M+)
1H-NMR(d6-DMSO,δ):2.7-2.9(4H,m),3.3-3.4(8H,m),4.36(2H,s),7.1-7.4(4H,m),7.6-7.7(1H,m),7.8-8.2(4H,m),8.43(1H,s),10.74(1H,br)
MS(m/z):445.1(M+)
製造例94、95、97~100、102~107、109~111、実施例68~82
化合物(12)として、製造例92で合成した化合物の代わりに、製造例96、101、108で合成した化合物を用いる点、及び/又は化合物(13)として、3-ブロモメチル安息香酸メチルの代わりに他の公知化合物を用いる点を除き、製造例93と同様にして表58~65に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例67と同様にして表58~65に示すカルボン酸を得た。
化合物(12)として、製造例92で合成した化合物の代わりに、製造例96、101、108で合成した化合物を用いる点、及び/又は化合物(13)として、3-ブロモメチル安息香酸メチルの代わりに他の公知化合物を用いる点を除き、製造例93と同様にして表58~65に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例67と同様にして表58~65に示すカルボン酸を得た。




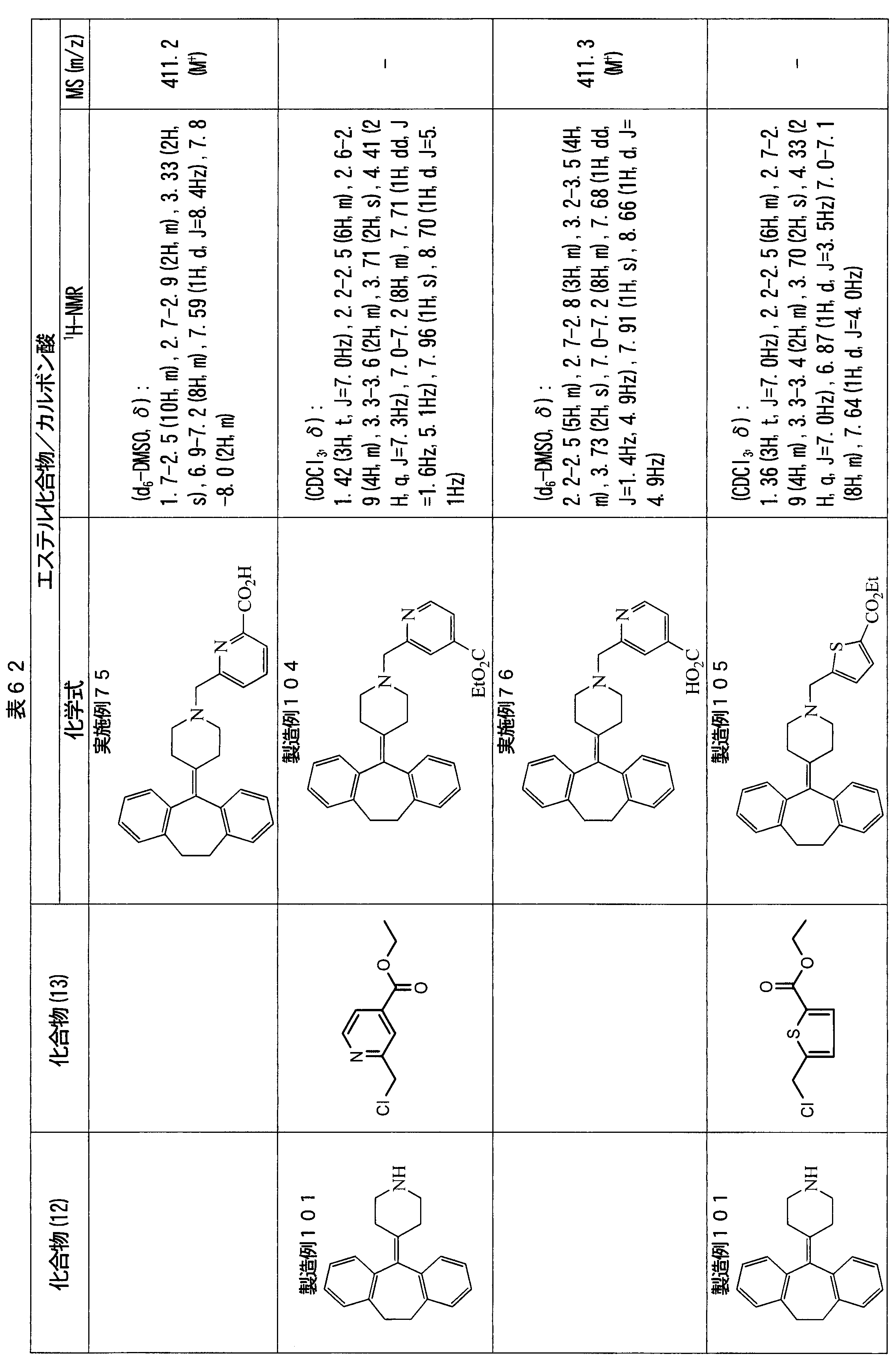



製造例96、101、108、157、160
ピペリジニルエステル化合物として、4-(8-クロロ-5,6-ジヒドロ-11H-ベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)-1-ピペリジンカルボン酸エチルの代わりに他の公知化合物を用いる以外は、製造例92と同様の操作を行い、表66に示すピペリジン化合物(化合物(12))を得た。
ピペリジニルエステル化合物として、4-(8-クロロ-5,6-ジヒドロ-11H-ベンゾ[5,6]シクロヘプタ[1,2-b]ピリジン-11-イリデン)-1-ピペリジンカルボン酸エチルの代わりに他の公知化合物を用いる以外は、製造例92と同様の操作を行い、表66に示すピペリジン化合物(化合物(12))を得た。

製造例114、125、126、129、130、136、実施例85、94、95、98、99、104
化合物(12)として、(±)-4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジンの代わりに、製造例132で合成した化合物又は公知化合物を用いる点、及び/又はカルボニトリル化合物として、4-クロロメチルピリジン-2-カルボニトリルの代わりに、製造例88で合成した化合物又は公知化合物を用いる以外、製造例89と同様にして表67~70に示すシアノ化合物を得た。また、このシアノ化合物を用いる以外、実施例65と同様にして表67~70に示すカルボン酸を得た。
化合物(12)として、(±)-4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジンの代わりに、製造例132で合成した化合物又は公知化合物を用いる点、及び/又はカルボニトリル化合物として、4-クロロメチルピリジン-2-カルボニトリルの代わりに、製造例88で合成した化合物又は公知化合物を用いる以外、製造例89と同様にして表67~70に示すシアノ化合物を得た。また、このシアノ化合物を用いる以外、実施例65と同様にして表67~70に示すカルボン酸を得た。
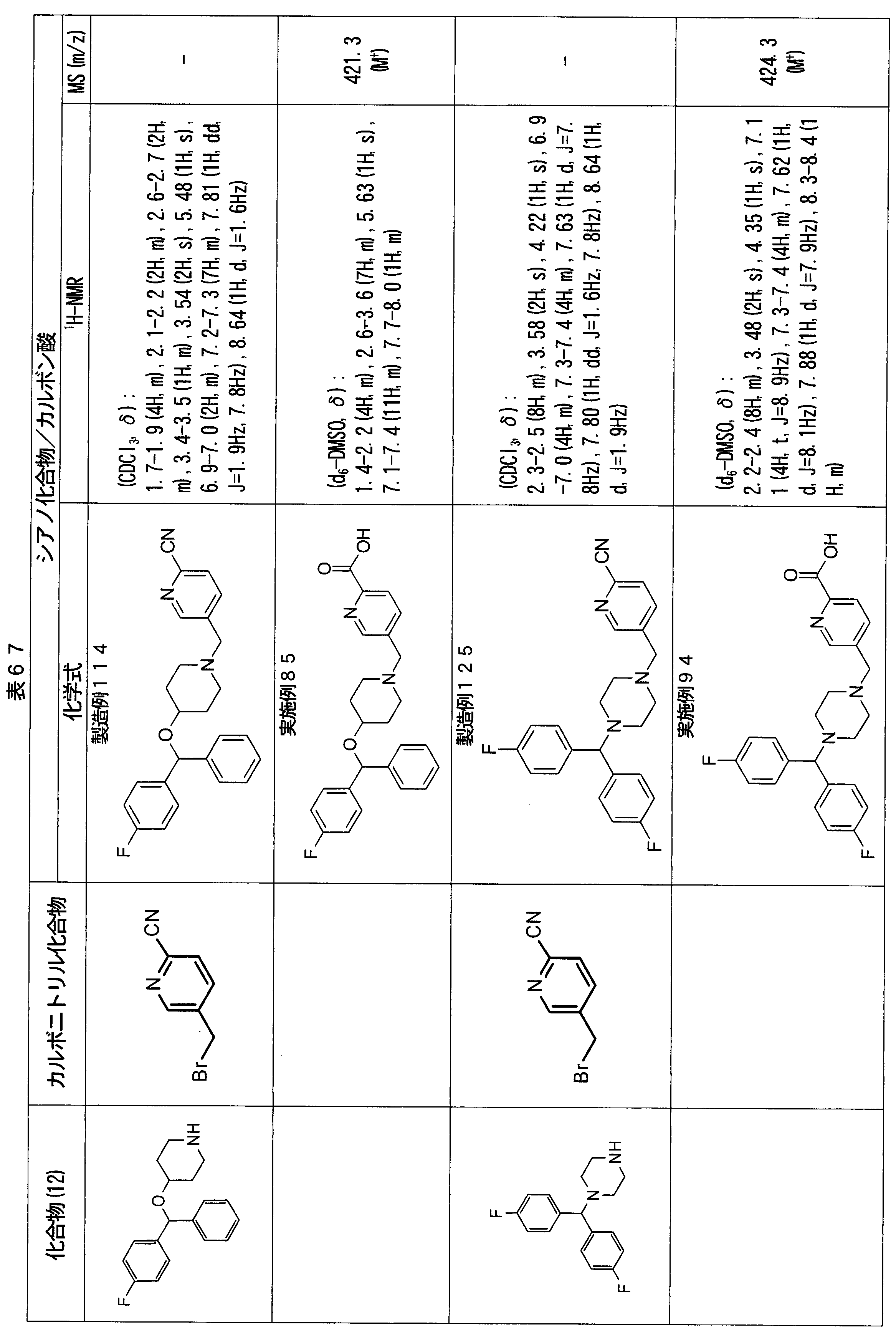



製造例132
(±)-1-[(4-フルオロフェニル)フェニルメチル]ピペラジン
(±)-1-[(4-フルオロフェニル)フェニルメチル]ピペラジン

(±)-(4-フルオロフェニル)フェニルメタノールに塩化チオニルを加え、室温で一夜撹拌した。反応の終了をHPLCで確認した後、酢酸エチルを加え、さらに水を加えた。炭酸水素ナトリウムで中和した後、水で2回洗浄し、硫酸マグネシウムで乾燥し、溶媒を留去した後、残渣をアセトニトリルに溶解し、ピペラジン(1.7g,20mmol)、ヨウ化カリウム(3.3g,20mmol)、炭酸カリウム(3.3g,24mmol)を加え、一夜還流した。HPLCで反応の終了を確認した後、不溶物を除去し、溶媒を留去した。得られた残渣を酢酸エチルに溶解し、水で3回洗浄した。有機層を硫酸マグネシウムで乾燥した後、溶媒を留去し、表題化合物(3.9g,72.7%)を黄白色の固体として得た。
1H-NMR(CDCl3,δ):2.33(4H,brs),2.88(4H,t,J=4.9Hz),4.2-4.3(2H,m),6.95(2H,t,J=8.9Hz),7.1-7.4(7H,m)。
1H-NMR(CDCl3,δ):2.33(4H,brs),2.88(4H,t,J=4.9Hz),4.2-4.3(2H,m),6.95(2H,t,J=8.9Hz),7.1-7.4(7H,m)。
製造例137、140、143、146、149、152、203
化合物(12a)として、(±)-(4-フルオロフェニル)フェニルメタノールの代わりに他の公知化合物を用いる以外は、製造例132と同様の操作を行い、表71に示すピペラジン化合物(化合物(12))を得た。
化合物(12a)として、(±)-(4-フルオロフェニル)フェニルメタノールの代わりに他の公知化合物を用いる以外は、製造例132と同様の操作を行い、表71に示すピペラジン化合物(化合物(12))を得た。

実施例122
(±)-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イル}(4-メチルピリジン-2-イル)メタノン
(±)-{4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン-1-イル}(4-メチルピリジン-2-イル)メタノン

公知化合物である(±)-4-[(4-フルオロフェニル)フェニルメトキシ]ピペリジン2.431g、4-メチルピリジン-2-カルボン酸974mg、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩2.922g、1-ヒドロキシベンゾトリアゾール一水和物1.956g、ピリジン29mL、4-ジメチルアミノピリジン208mgの混合物を1.8時間撹拌した。塩水にあけ酢酸エチルで2回抽出した。飽和食塩水で洗い硫酸マグネシウムで乾燥させた。溶媒を留去の後、シリカゲルカラムクロマトグラフィー(クロロホルム/メタノール(容積比)=20:1~15:1)にて精製し表題化合物1.661g(58%)を得た。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.38(3H,s),3.2-3.4(1H,s),3.5-4.1(4H,m),5.49(1H,s),6.9-7.1(2H,m),7.13(1H,d,J=4.2Hz),7.1-7.4(7H,m),7.40(1H,s),8.41(1H,d,J=5.0Hz)
MS(m/z):404(M+)。
1H-NMR(CDCl3,δ):1.6-2.0(4H,m),2.38(3H,s),3.2-3.4(1H,s),3.5-4.1(4H,m),5.49(1H,s),6.9-7.1(2H,m),7.13(1H,d,J=4.2Hz),7.1-7.4(7H,m),7.40(1H,s),8.41(1H,d,J=5.0Hz)
MS(m/z):404(M+)。
製造例163
(±)-2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸エチル
(±)-2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸エチル

公知化合物である(±)-4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン297mg、公知化合物である2-クロロメチルイソニコチン酸エチル240mg、アセトニトリル2.3mL、トリエチルアミン0.28mLの混合物を一晩撹拌した。塩水にあけ酢酸エチルで抽出した。飽和食塩水で洗い硫酸ナトリウムで乾燥させた。溶媒を留去の後、p-TLCにて精製し表題化合物309mg(67%)を得た。
1H-NMR(CDCl3,δ):1.41(3H,t,J=7.1Hz),1.6-2.0(4H,m),2.1-2.4(2H,m),2.7-2.9(2H,m),3.3-3.6(1H,m),3.68(2H,s),3.77(3H,s),4.40(2H,q,J=7.2Hz),5.47(1H,s),6.7-6.9(2H,m),7.1-7.4(7H,m),7.70(1H,dd,J=1.5Hz,5.0Hz),7.93(1H,s),8.68(1H,dd,J=0.8Hz,5.0Hz)
MS(m/z):461(M++1)。
1H-NMR(CDCl3,δ):1.41(3H,t,J=7.1Hz),1.6-2.0(4H,m),2.1-2.4(2H,m),2.7-2.9(2H,m),3.3-3.6(1H,m),3.68(2H,s),3.77(3H,s),4.40(2H,q,J=7.2Hz),5.47(1H,s),6.7-6.9(2H,m),7.1-7.4(7H,m),7.70(1H,dd,J=1.5Hz,5.0Hz),7.93(1H,s),8.68(1H,dd,J=0.8Hz,5.0Hz)
MS(m/z):461(M++1)。
実施例123
(±)-2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸
(±)-2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸
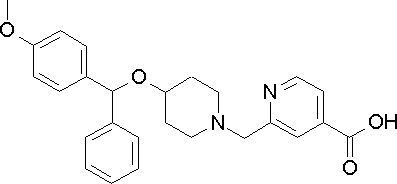
製造例2で合成した化合物の代わりに製造例163で合成した(±)-2-{4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}イソニコチン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.5-2.0(4H,m),2.1-2.4(2H,m),2.7-2.9(2H,m),3.3-3.5(1H,m),3.72(3H,s),3.74(2H,s),5.57(1H,s),6.8-7.0(2H,m),7.1-7.4(7H,m),7.68(1H,dd,J=1.5Hz,5.0Hz),7.88(1H,s),8.65(1H,d,J=4.6Hz)
MS(m/z):431(M+-1)。
1H-NMR(d6-DMSO,δ):1.5-2.0(4H,m),2.1-2.4(2H,m),2.7-2.9(2H,m),3.3-3.5(1H,m),3.72(3H,s),3.74(2H,s),5.57(1H,s),6.8-7.0(2H,m),7.1-7.4(7H,m),7.68(1H,dd,J=1.5Hz,5.0Hz),7.88(1H,s),8.65(1H,d,J=4.6Hz)
MS(m/z):431(M+-1)。
製造例164~167、176、177、180、183~186、209、210、212、213、215、216、218、219、実施例124~127、132~138、150~157
化合物(12)として、(±)-4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジンの代わりに、製造例175、179、182、208、211、214、217で合成した化合物又は公知化合物を用いる点、及び/又は化合物(13)として、2-クロロメチルイソニコチン酸エチルの代わりに他の公知化合物を用いる以外、製造例163と同様にして表72~81に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表72~81に示すカルボン酸を得た。
化合物(12)として、(±)-4-[(4-メトキシフェニル)フェニルメトキシ]ピペリジンの代わりに、製造例175、179、182、208、211、214、217で合成した化合物又は公知化合物を用いる点、及び/又は化合物(13)として、2-クロロメチルイソニコチン酸エチルの代わりに他の公知化合物を用いる以外、製造例163と同様にして表72~81に示すエステル化合物(化合物(14))を得た。また、このエステル化合物を用いる以外、実施例1と同様にして表72~81に示すカルボン酸を得た。
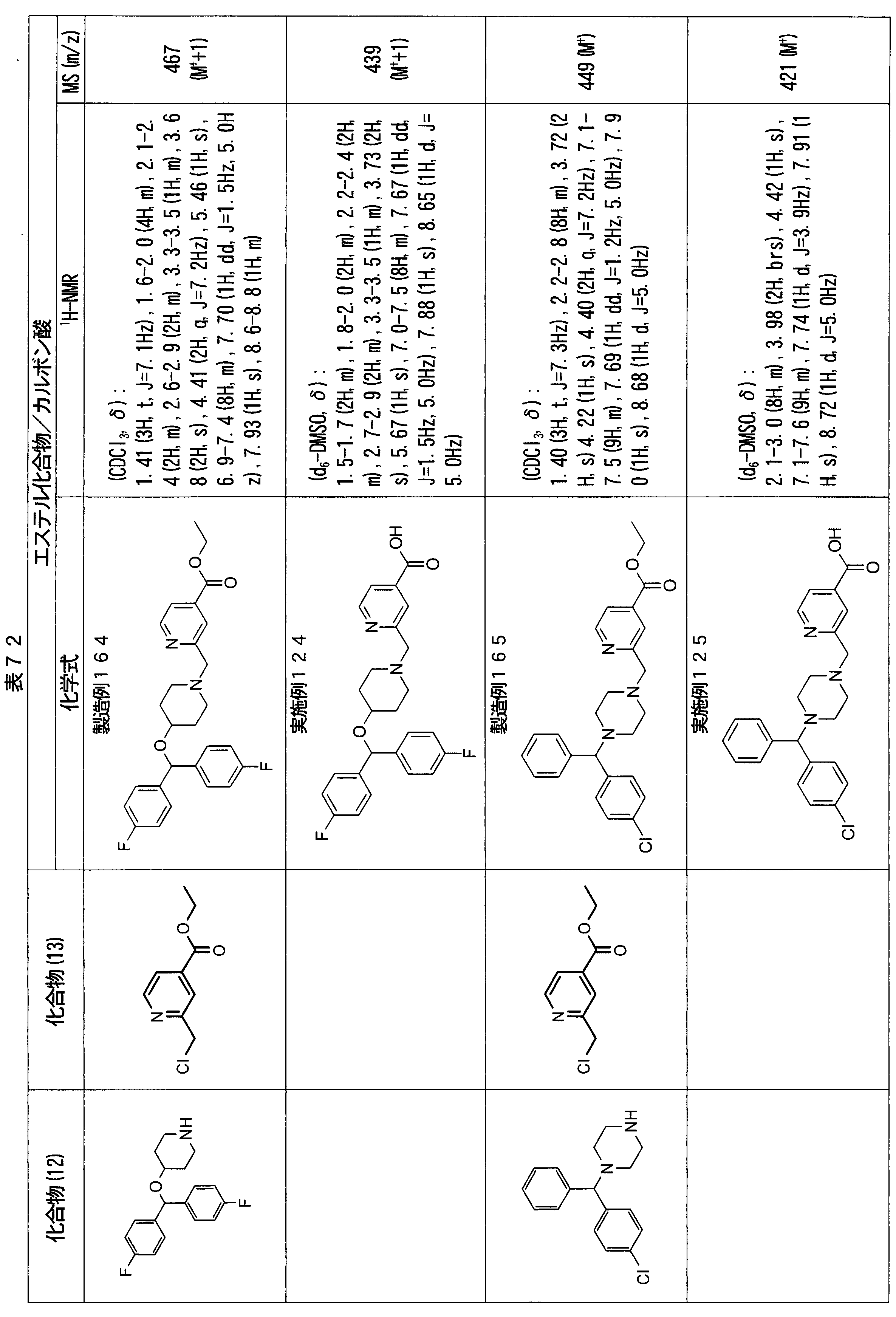
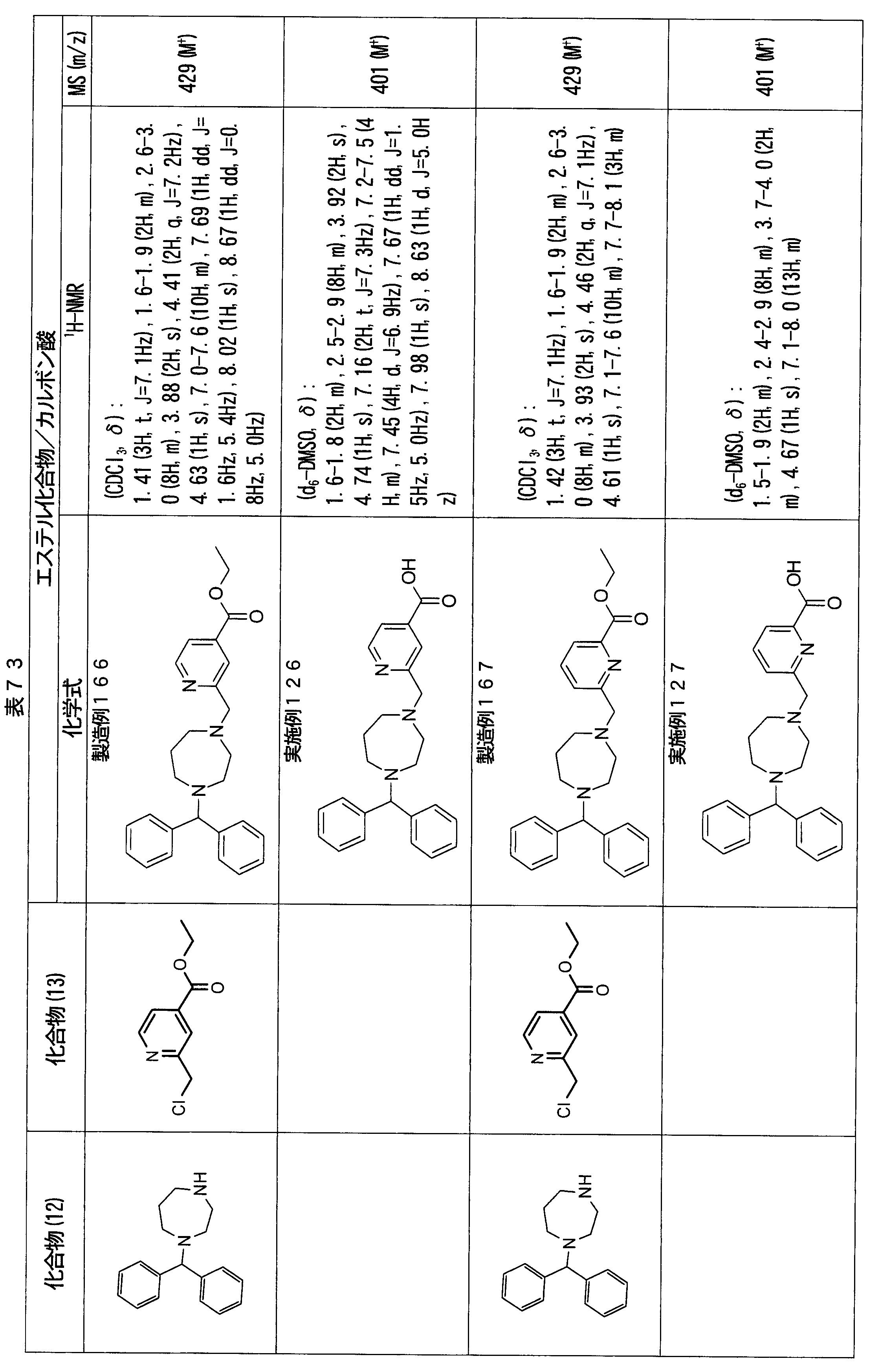
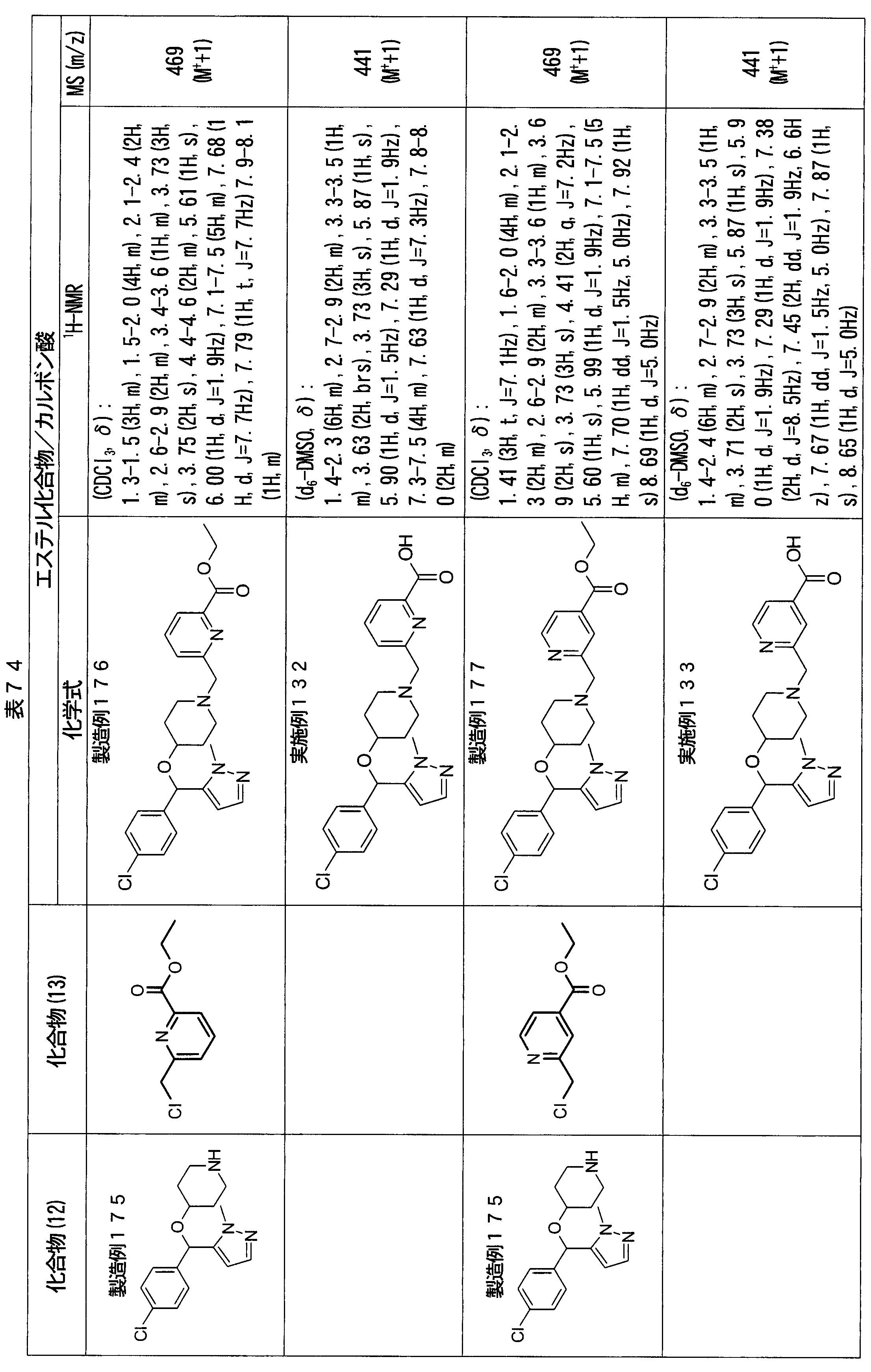
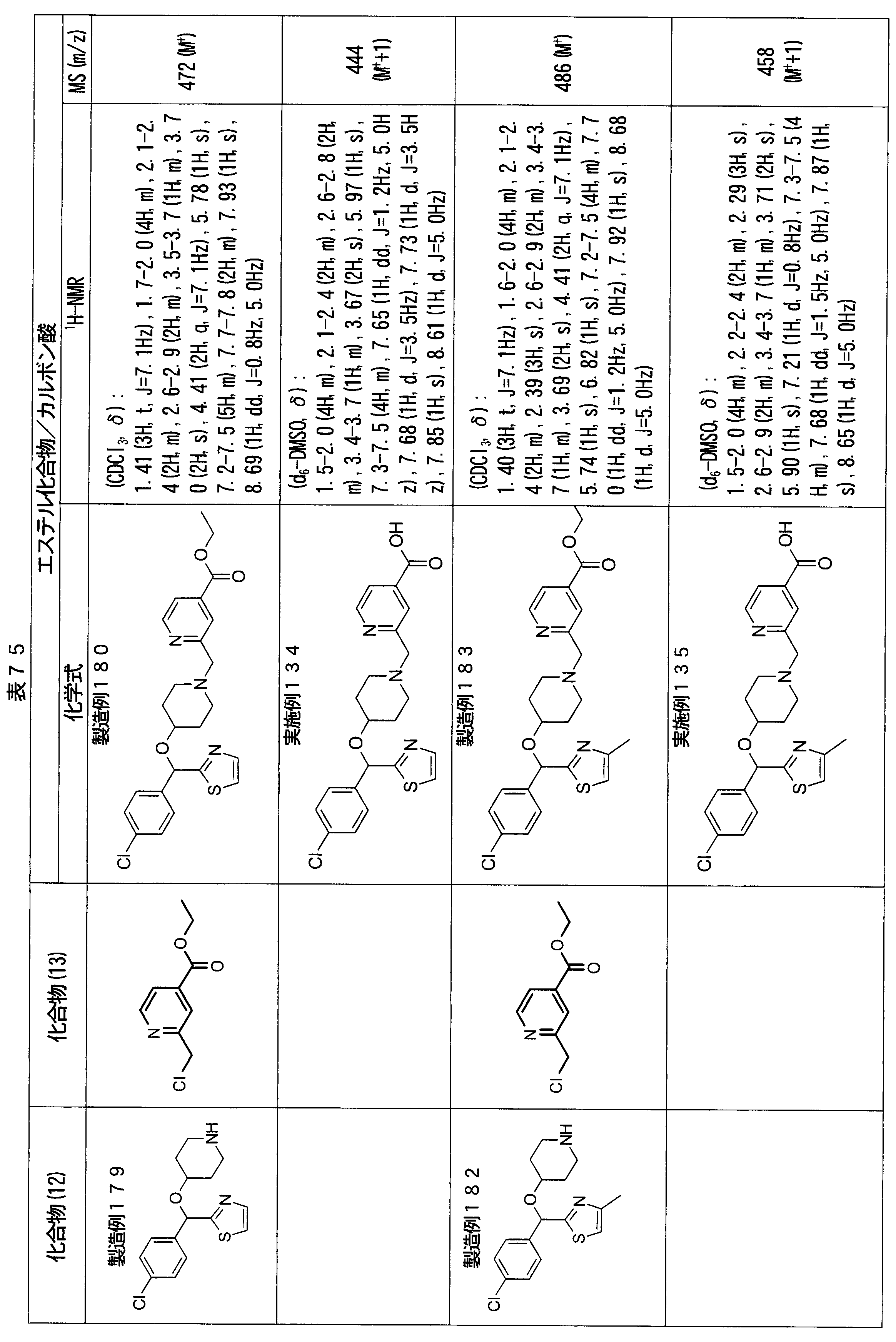


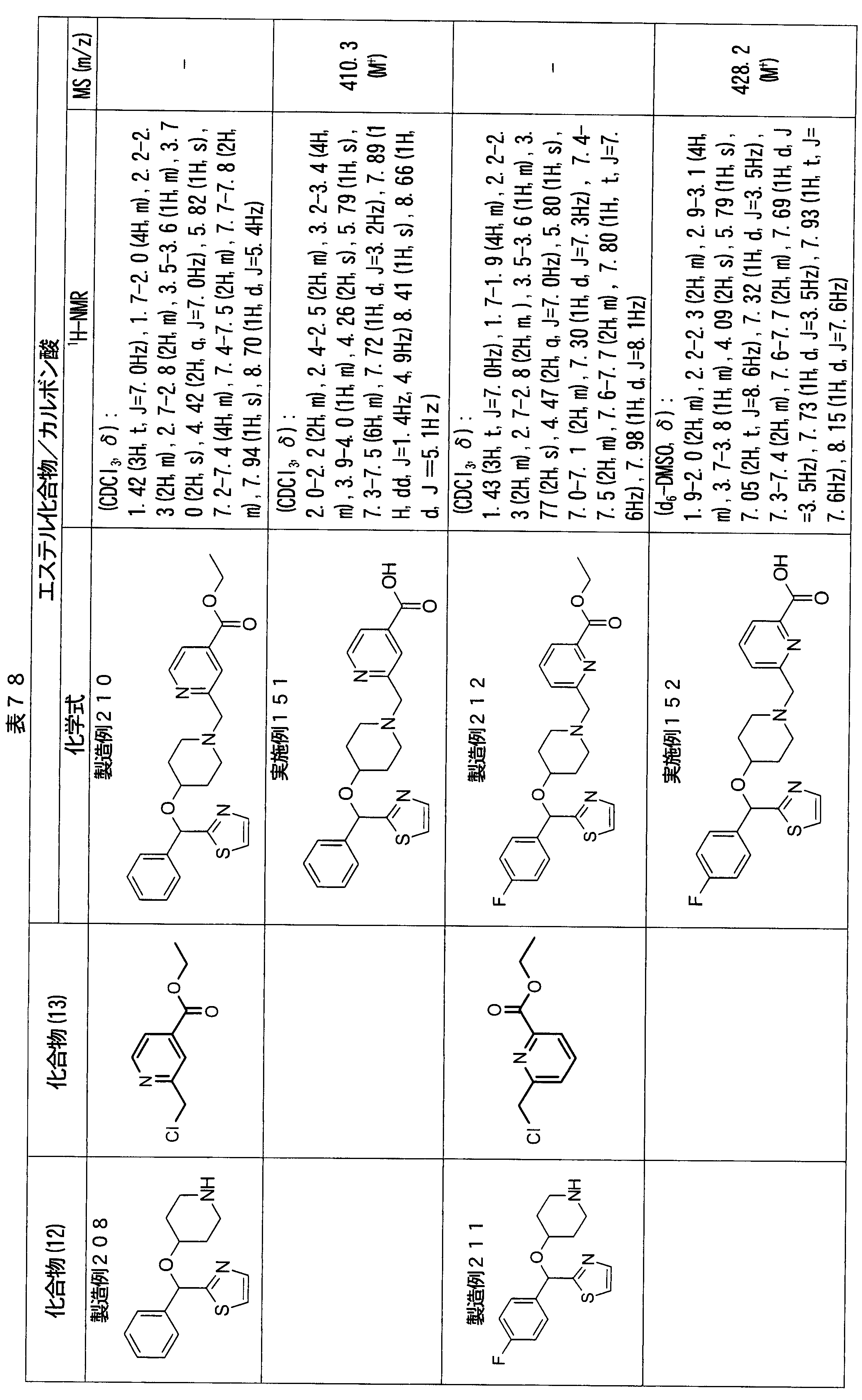

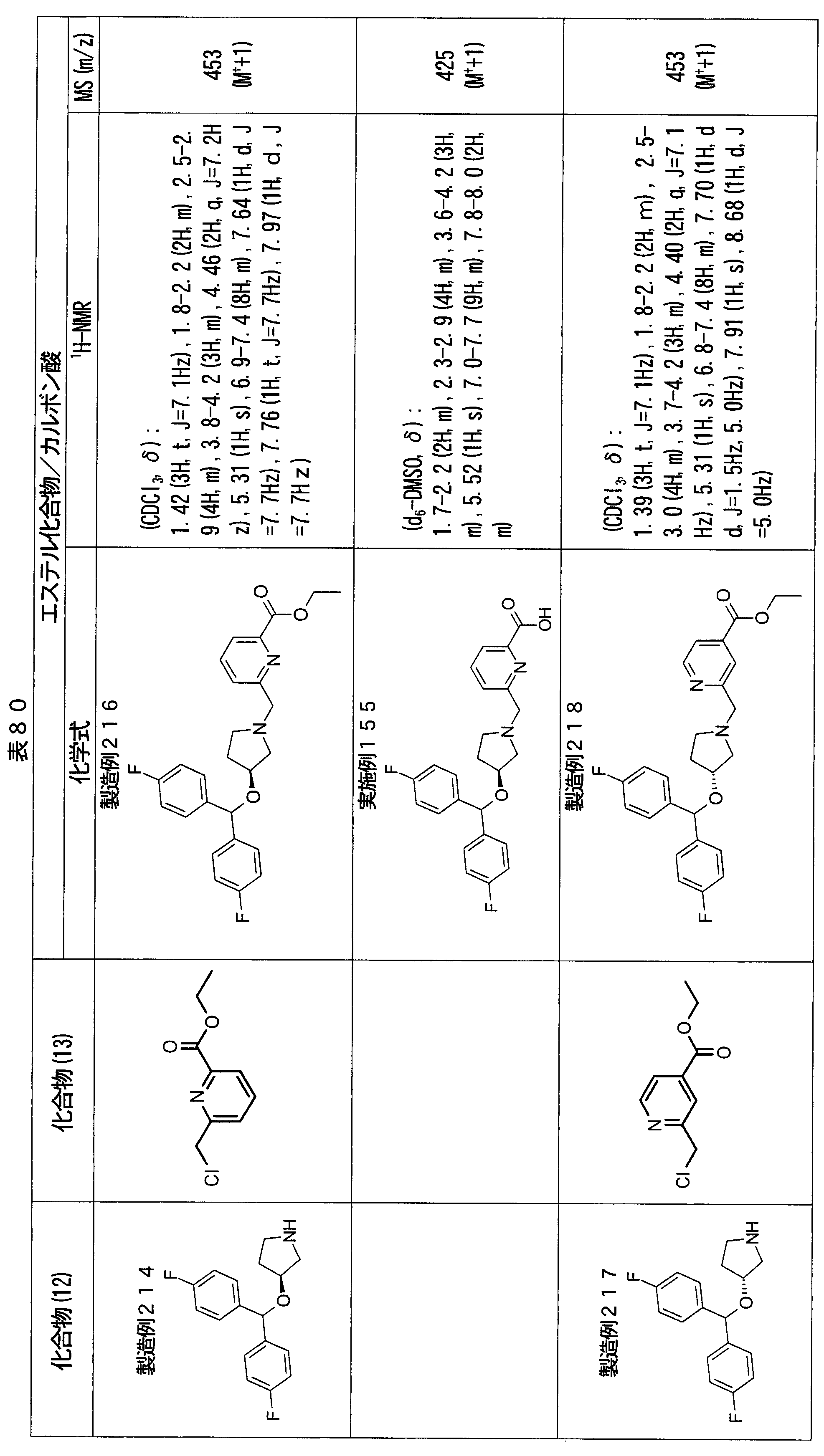

製造例169
6-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチル
6-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチル
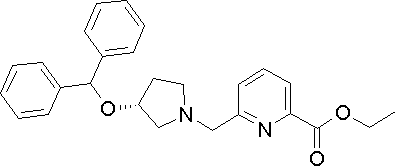
製造例168で合成した(R)-3-ベンズヒドリルオキシピロリジン8.144g、公知化合物である6-クロロメチルピリジン-2-カルボン酸エチル7.701g、アセトニトリル69mL、トリエチルアミン9.0mLの混合物を3時間加熱還流した。放冷の後、酢酸エチルで希釈した。飽和食塩水にあけ有機層を分取した。飽和食塩水で洗い硫酸マグネシウムで乾燥させた。溶媒を留去の後、シリカゲルカラムクロマトグラフィー(クロロホルム/アセトン(容積比)=15:1~10:1)にて精製し表題化合物7.22g(54%)を得た。
1H-NMR(CDCl3,δ):1.42(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-2.9(4H,m),3.8-4.0(2H,m),4.1-4.2(1H,m),4.46(2H,q,J=7.2Hz),5.36(1H,s),7.1-7.4(10H,m),7.65(1H,d,J=7.7Hz),7.76(1H,t,J=7.7Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):416(M+)。
1H-NMR(CDCl3,δ):1.42(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-2.9(4H,m),3.8-4.0(2H,m),4.1-4.2(1H,m),4.46(2H,q,J=7.2Hz),5.36(1H,s),7.1-7.4(10H,m),7.65(1H,d,J=7.7Hz),7.76(1H,t,J=7.7Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):416(M+)。
実施例128
6-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸
6-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸

製造例2で合成した化合物の代わりに製造例169で合成した6-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.7-2.2(2H,m),2.4-2.9(4H,m),3.7-4.2(3H,m),5.49(1H,s),7.1-7.7(11H,m),7.8-8.0(2H,m)
MS(m/z):387(M+-1)。
1H-NMR(d6-DMSO,δ):1.7-2.2(2H,m),2.4-2.9(4H,m),3.7-4.2(3H,m),5.49(1H,s),7.1-7.7(11H,m),7.8-8.0(2H,m)
MS(m/z):387(M+-1)。
製造例171
6-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチル
6-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチル

(R)-3-ベンズヒドリルオキシピロリジンの代わりに製造例170で合成した(S)-3-ベンズヒドリルオキシピロリジンを用いる以外は、製造例169と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.42(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-2.9(4H,m),3.8-4.0(2H,m),4.1-4.2(1H,m),4.46(2H,q,J=7.1Hz),5.36(1H,s),7.1-7.4(10H,m),7.65(1H,d,J=7.7Hz),7.76(1H,t,J=7.9Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):416(M+)。
1H-NMR(CDCl3,δ):1.42(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-2.9(4H,m),3.8-4.0(2H,m),4.1-4.2(1H,m),4.46(2H,q,J=7.1Hz),5.36(1H,s),7.1-7.4(10H,m),7.65(1H,d,J=7.7Hz),7.76(1H,t,J=7.9Hz),7.96(1H,d,J=7.7Hz)
MS(m/z):416(M+)。
実施例129
6-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸
6-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸

製造例2で合成した化合物の代わりに製造例171で合成した6-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジン-2-カルボン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.7-2.2(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.48(1H,s),7.1-7.7(11H,m),7.8-8.0(2H,m)
MS(m/z):387(M+-1)。
1H-NMR(d6-DMSO,δ):1.7-2.2(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.48(1H,s),7.1-7.7(11H,m),7.8-8.0(2H,m)
MS(m/z):387(M+-1)。
製造例172
2-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチル
2-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチル

6-クロロメチルピリジン-2-カルボン酸エチルの代わりに公知化合物である2-クロロメチルイソニコチン酸エチルを用いる以外は、製造例169と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-3.0(4H,m),3.7-4.0(2H,m),4.0-4.3(1H,m),4.40(2H,q,J=6.9Hz),5.36(1H,s),7.1-7.4(10H,m),7.70(1H,d,J=5.0Hz),7.91(1H,s),8.68(1H,d,J=5.4Hz)
MS(m/z):416(M+)。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-3.0(4H,m),3.7-4.0(2H,m),4.0-4.3(1H,m),4.40(2H,q,J=6.9Hz),5.36(1H,s),7.1-7.4(10H,m),7.70(1H,d,J=5.0Hz),7.91(1H,s),8.68(1H,d,J=5.4Hz)
MS(m/z):416(M+)。
実施例130
2-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸
2-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸

製造例2で合成した化合物の代わりに製造例172で合成した2-((R)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.7-2.1(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.49(1H,s),7.1-7.4(10H,m),7.65(1H,dd,J=1.5Hz,5.0Hz),7.84(1H,s),8.5-8.7(1H,m)
MS(m/z):387(M+-1)。
1H-NMR(d6-DMSO,δ):1.7-2.1(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.49(1H,s),7.1-7.4(10H,m),7.65(1H,dd,J=1.5Hz,5.0Hz),7.84(1H,s),8.5-8.7(1H,m)
MS(m/z):387(M+-1)。
製造例173
2-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチル
2-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチル
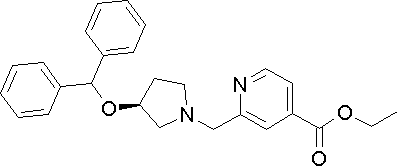
6-クロロメチルピリジン-2-カルボン酸エチルの代わりに公知化合物である2-クロロメチルイソニコチン酸エチルを、(R)-3-ベンズヒドリルオキシピロリジンの代わりに製造例170で合成した(S)-3-ベンズヒドリルオキシピロリジンを用いる以外は、製造例169と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-3.0(4H,m),3.7-4.0(2H,m),4.0-4.3(1H,m),4.40(2H,q,J=7.1Hz),5.36(1H,s),7.1-7.4(10H,m),7.70(1H,dd,J=1.5Hz,5.0Hz),7.91(1H,s),8.68(1H,d,J=5.0Hz)
MS(m/z):417(M++1)。
1H-NMR(CDCl3,δ):1.39(3H,t,J=7.1Hz),1.8-2.2(2H,m),2.5-3.0(4H,m),3.7-4.0(2H,m),4.0-4.3(1H,m),4.40(2H,q,J=7.1Hz),5.36(1H,s),7.1-7.4(10H,m),7.70(1H,dd,J=1.5Hz,5.0Hz),7.91(1H,s),8.68(1H,d,J=5.0Hz)
MS(m/z):417(M++1)。
実施例131
2-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸
2-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸

製造例2で合成した化合物の代わりに製造例173で合成した2-((S)-3-ベンズヒドリルオキシピロリジン-1-イルメチル)イソニコチン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(d6-DMSO,δ):1.7-2.1(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.49(1H,s),7.1-7.4(10H,m),7.65(1H,dd,J=1.5Hz,5.0Hz),7.84(1H,s),8.60(1H,d,J=5.0Hz)
MS(m/z):387(M+-1)。
1H-NMR(d6-DMSO,δ):1.7-2.1(2H,m),2.4-2.9(4H,m),3.7-3.9(2H,m),4.0-4.2(1H,m),5.49(1H,s),7.1-7.4(10H,m),7.65(1H,dd,J=1.5Hz,5.0Hz),7.84(1H,s),8.60(1H,d,J=5.0Hz)
MS(m/z):387(M+-1)。
製造例174
(±)-(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メタノール
(±)-(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メタノール

1-メチルピラゾール6.60gのテトラヒドロフラン150mLの溶液を-60℃に冷却し、窒素雰囲気下、1.6M n-ブチルリチウム/n-ヘキサン溶液60mLを滴下した。1時間撹拌した後、4-クロロベンズアルデヒド11.288gのテトラヒドロフラン50mLの溶液を滴下した。7時間かけて室温に戻し、1N塩化アンモニウム水溶液125mLを加えた。酢酸エチルで2回抽出し飽和食塩水で洗った。硫酸ナトリウムで乾燥した後、溶媒を留去した。粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=1:2=1:3)にて精製し、表題化合物7.898g(44%)を得た。
1H-NMR(CDCl3,δ):2.53(1H,d,J=4.6Hz),3.77(3H,s),5.90(1H,d,J=4.2Hz),6.02(1H,d,J=1.9Hz),7.2-7.5(5H,m)
MS(m/z):222(M+)。
1H-NMR(CDCl3,δ):2.53(1H,d,J=4.6Hz),3.77(3H,s),5.90(1H,d,J=4.2Hz),6.02(1H,d,J=1.9Hz),7.2-7.5(5H,m)
MS(m/z):222(M+)。
製造例175
(±)-4-[(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メトキシ]ピペリジン
(±)-4-[(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メトキシ]ピペリジン
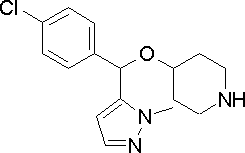
製造例174で合成した(±)-(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メタノール2.074g、4-ヒドロキシピペリジン942mg、p-トルエンスルホン酸一水和物1.949g、トルエン100mLの混合物をディーンスタークで水を除きながら40分間加熱還流した。p-トルエンスルホン酸一水和物1.949gを追加し、さらに4.3時間、水を除きながら加熱還流した。放冷の後、トルエン、1N水酸化ナトリウム水溶液22mLを加え、有機層を分取した。1N水酸化ナトリウム水溶液22mL、飽和食塩水で順に洗った。硫酸ナトリウムで乾燥した後、溶媒を留去し、表題化合物2.487g(87%)を得た。
1H-NMR(CDCl3,δ):1.4-1.6(2H,m),1.8-2.0(2H,m),2.4-2.7(2H,m),2.9-3.2(2H,m),3.3-3.6(1H,m),3.74(3H,s),5.64(1H,s),6.00(1H,d,J=1.9Hz),7.1-7.5(5H,m)
MS(m/z):305(M+)。
1H-NMR(CDCl3,δ):1.4-1.6(2H,m),1.8-2.0(2H,m),2.4-2.7(2H,m),2.9-3.2(2H,m),3.3-3.6(1H,m),3.74(3H,s),5.64(1H,s),6.00(1H,d,J=1.9Hz),7.1-7.5(5H,m)
MS(m/z):305(M+)。
製造例178
(±)-(4-クロロフェニル)(チアゾール-2-イル)メタノール
(±)-(4-クロロフェニル)(チアゾール-2-イル)メタノール

1.6M n-ブチルリチウム/n-ヘキサン溶液23mLのジエチルエーテル20mLの溶液を-30℃に冷却下、2-ブロモチアゾール5.003gのジエチルエーテル20mLの溶液を滴下した。1.1時間撹拌の後、4-クロロベンズアルデヒド5.538gのジエチルエーテル20mLの溶液を滴下した。1.2時間かけて室温に戻し、1N塩酸72mLと氷の混合液へ反応液をあけた。不溶物をろ過で除去した後、有機層を分取した。飽和重曹水、飽和食塩水で順に洗った。硫酸マグネシウムで乾燥した後、溶媒を留去した。粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=2:1~3:2)にて精製し、表題化合物4.425g(64%)を得た。
1H-NMR(CDCl3,δ):3.55(1H,d,J=3.9Hz),6.04(1H,d,J=3.5Hz),7.2-7.5(5H,m),7.73(1H,d,J=3.1Hz)
MS(m/z):225(M+)。
1H-NMR(CDCl3,δ):3.55(1H,d,J=3.9Hz),6.04(1H,d,J=3.5Hz),7.2-7.5(5H,m),7.73(1H,d,J=3.1Hz)
MS(m/z):225(M+)。
製造例179、182、208、211
(±)-(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メタノールの代わりに、製造例178、181で合成した化合物又は公知化合物を用いる以外は、製造例175と同様の操作を行い、表82に示す化合物(12)を得た。
(±)-(4-クロロフェニル)(2-メチル-2H-ピラゾール-3-イル)メタノールの代わりに、製造例178、181で合成した化合物又は公知化合物を用いる以外は、製造例175と同様の操作を行い、表82に示す化合物(12)を得た。
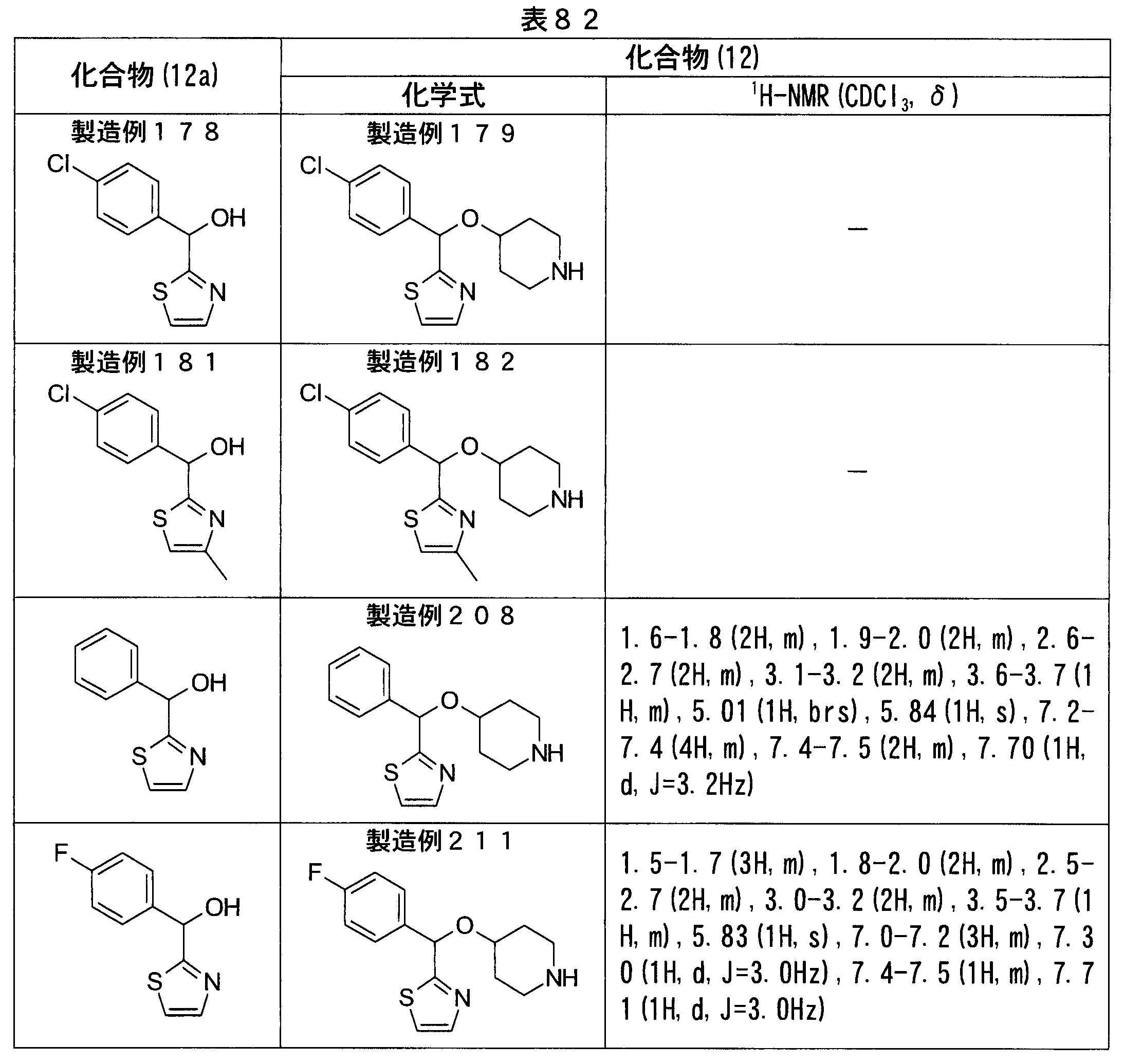
製造例181
(±)-(4-クロロフェニル)(4-メチルチアゾール-2-イル)メタノール
(±)-(4-クロロフェニル)(4-メチルチアゾール-2-イル)メタノール

1.6M n-ブチルリチウム/n-ヘキサン溶液38mLのテトラヒドロフラン6mLの溶液を-60℃に冷却下、4-メチルチアゾール5.949gのテトラヒドロフラン5mLの溶液を滴下した。1.5時間撹拌の後、4-クロロベンズアルデヒド8.434gのテトラヒドロフラン20mLの溶液を滴下した。同温度で1.8時間撹拌の後、2時間かけて室温に戻し、50%エタノール水15mLを加えた。反応液を水にあけジエチルエーテルを加え抽出した。飽和食塩水で洗った後、硫酸マグネシウムで乾燥した。溶媒を留去した後、粗生成物をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=2:1~1:1)にて精製し、表題化合物10.602g(74%)を得た。
1H-NMR(CDCl3,δ):2.40(3H,s),3.68(1H,d,J=3.9Hz),5.98(1H,d,J=3.5Hz),6.83(1H,d,J=0.8Hz),7.2-7.5(4H,m)
MS(m/z):239(M+)。
1H-NMR(CDCl3,δ):2.40(3H,s),3.68(1H,d,J=3.9Hz),5.98(1H,d,J=3.5Hz),6.83(1H,d,J=0.8Hz),7.2-7.5(4H,m)
MS(m/z):239(M+)。
製造例187
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾ-ル-3-カルボン酸エチル
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾ-ル-3-カルボン酸エチル

製造例1で合成した4-ベンズヒドリルオキシピペリジン(1.0g,3.7mmol)、4-ホルミルピラゾ-ル-3-カルボン酸エチル(630mg,3.7mmol)、ジクロロメタン(30mL)を仕込み、アルゴン置換し氷冷した。0℃にてトリアセトキシホウ素ナトリウム(1.2g,5.6mmol)を添加し、室温まで昇温した。TLCにて原料消失を確認後、飽和炭酸水素ナトリウム溶液を加えクエンチした。分液し水層を再びジクロロメタンにて抽出し、先の有機層と合一した。得られた有機層を硫酸ナトリウムにて乾燥し、溶媒留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1)にて精製し、表題化合物(1.3g,83%)を得た。
1H-NMR(CDCl3,δ):1.38(3H,t,J=7.0Hz),1.6-1.8(2H,m),1.8-2.0(2H,m),2.1-2.3(2H,m),2.7-2.9(2H,m),3.3-3.5(1H,m),3.6-3.8(2H,m),4.37(3H,q,J=7.0Hz),5.50(1H,s),7.2-7.4(10H,m),7.60(1H,brs)
MS(m/z):419(M+)。
1H-NMR(CDCl3,δ):1.38(3H,t,J=7.0Hz),1.6-1.8(2H,m),1.8-2.0(2H,m),2.1-2.3(2H,m),2.7-2.9(2H,m),3.3-3.5(1H,m),3.6-3.8(2H,m),4.37(3H,q,J=7.0Hz),5.50(1H,s),7.2-7.4(10H,m),7.60(1H,brs)
MS(m/z):419(M+)。
実施例139
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾ-ル-3-カルボン酸
4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾ-ル-3-カルボン酸

製造例2で合成した化合物の代わりに製造例187で合成した4-(4-ベンズヒドリルオキシピペリジン-1-イルメチル)-1H-ピラゾ-ル-3-カルボン酸エチルを用いる以外は、実施例1と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):1.7-2.2(6H,m),2.9-3.3(4H,m),3.7-4.0(3H,m),5.42(1H,s),7.2-7.4(10H,m),7.54(1H,brs)
MS(m/z):390(M+-1)。
1H-NMR(CDCl3,δ):1.7-2.2(6H,m),2.9-3.3(4H,m),3.7-4.0(3H,m),5.42(1H,s),7.2-7.4(10H,m),7.54(1H,brs)
MS(m/z):390(M+-1)。
製造例191
1-ベンズヒドリルアゼチジン-3-オール
1-ベンズヒドリルアゼチジン-3-オール

ベンズヒドリルアミン(12g,64mmol)、エピクロロヒドリン(5.9g,64mmol)、メタノール30mLをアルゴン下3日間遮光保存した。ついで油浴上3日間加熱還流した。TLCで反応の終了を確認した後、溶媒を留去し得られた残渣にアセトンを加えてろ過した。ろ取物にエーテルを加えて溶解し、6規定水酸化ナトリウム水溶液で洗浄した。有機層を硫酸ナトリウムで乾燥させ、溶媒を留去し、表題化合物(1.3g,83%)を得た。
1H-NMR(CDCl3,δ):2.38(1H,brs),2.8-3.0(2H,m),3.4-3.7(2H,m),4.34(1H,s),4.3-4.5(1H,m),7.1-7.5(10H,m)
MS(m/z):238(M+-1)。
1H-NMR(CDCl3,δ):2.38(1H,brs),2.8-3.0(2H,m),3.4-3.7(2H,m),4.34(1H,s),4.3-4.5(1H,m),7.1-7.5(10H,m)
MS(m/z):238(M+-1)。
製造例192
1-ベンズヒドリル-3-ベンズヒドリルオキシアゼチジン
1-ベンズヒドリル-3-ベンズヒドリルオキシアゼチジン

4-ヒドロキシピペリジンの代わりに製造例191で合成した1-ベンズヒドリルアゼチジン-3-オールを用いる以外は、製造例1と同様の操作を行い、表題化合物を得た。
1H-NMR(CDCl3,δ):2.8-3.0(2H,m),3.3-3.5(2H,m),4.1-4.3(1H,m),4.34(1H,s),5.29(1H,s),7.1-7.5(20H,m)
MS(m/z):483(M+)。
1H-NMR(CDCl3,δ):2.8-3.0(2H,m),3.3-3.5(2H,m),4.1-4.3(1H,m),4.34(1H,s),5.29(1H,s),7.1-7.5(20H,m)
MS(m/z):483(M+)。
製造例193
3-ベンズヒドリルオキシアゼチジン
3-ベンズヒドリルオキシアゼチジン

製造例192で得られた1-ベンズヒドリル-3-ベンズヒドリルオキシアゼチジン(1.8g,4.5mmol)をジクロロメタン30mLに溶解し氷冷した。ついで1-クロロギ酸-1-クロロエチル(1.6g,11mmol)のジクロロメタン溶液を0℃にて滴下した。滴下終了後室温にて一夜攪拌した。TLCで反応の終了を確認した後、反応液を濃縮し、得られた残渣へメタノールを加えて室温にて攪拌した。TLCで反応の終了を確認した後、反応液を濃縮し残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール(容積比)=10:1)にて精製し、表題化合物(0.79g,74%)を得た。
1H-NMR(CDCl3,δ):3.8-4.0(4H,m),4.5-4.7(1H,m),5.32(1H,s),7.1-7.4(10H,m)
MS(m/z):240(M++1)。
1H-NMR(CDCl3,δ):3.8-4.0(4H,m),4.5-4.7(1H,m),5.32(1H,s),7.1-7.4(10H,m)
MS(m/z):240(M++1)。
製造例195
(±)-(4-フルオロフェニル)フェニルメタノール
(±)-(4-フルオロフェニル)フェニルメタノール
ベンズアルデヒド(8.5g,80mmol)をテトラヒドロフラン100mLに溶解し氷浴上で冷却した。ついで4-フルオロフェニルマグネシウムブロミドのテトラヒドロフラン1.0M溶液(88mL,88mmol)を0℃にて滴下した。滴下終了後、室温まで昇温し、TLCで反応の終了を確認した。反応終了後、5%塩酸に注加し、酢酸エチルにて抽出した。有機層を水にて洗浄し、硫酸ナトリウムにて乾燥、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルム/エタノール(容積比)=10:1)にて精製し、表題化合物(13g,81%)を得た。
1H-NMR(CDCl3,δ):2.27(1H,s),5.81(1H,s),7.01(2H,t,J=9.0Hz),7.2-7.5(7H,m)
MS(m/z):202(M+)。
1H-NMR(CDCl3,δ):2.27(1H,s),5.81(1H,s),7.01(2H,t,J=9.0Hz),7.2-7.5(7H,m)
MS(m/z):202(M+)。
製造例196
(±)-1-(クロロフェニルメチル)-4-フルオロベンゼン
(±)-1-(クロロフェニルメチル)-4-フルオロベンゼン

製造例195で合成した(±)-(4-フルオロフェニル)フェニルメタノール(2.1g,10mmol)をジクロロメタン(30mL)に溶解し室温にて塩化チオニル(5.1g,42mmol)を滴下した。滴下終了後加熱還流し、TLCにて反応の終了を確認した。反応終了後濃縮し酢酸エチルを加えて希釈し、水にて洗浄した。有機層を硫酸マグネシウムにて乾燥させ、溶媒を留去し表題化合物(2.1g,94%)を得た。
1H-NMR(CDCl3,δ):6.13(1H,s),6.9-7.1(2H,m),7.2-7.5(7H,m)
MS(m/z):220(M+)。
1H-NMR(CDCl3,δ):6.13(1H,s),6.9-7.1(2H,m),7.2-7.5(7H,m)
MS(m/z):220(M+)。
製造例199
(±)-4-(シクロへキシルフェニルメトキシ)ピペリジン-1-カルボン酸エチル
(±)-4-(シクロへキシルフェニルメトキシ)ピペリジン-1-カルボン酸エチル

シクロヘキシルフェニルケトン(5.0g,27mmol)をエタノール(50mL)に溶解し氷冷した。0℃にて水素化ホウ素ナトリウム(510mg,13.5mmol)を添加した。終了後室温まで昇温し、TLCにて原料消失を確認した。反応液を濃縮後、水を加えて希釈し、1規定塩酸水溶液にて中和した。そこへ酢酸エチルを添加し、抽出後硫酸ナトリウムにて乾燥した。溶媒を留去し、精製をせず次ステップへ用いた。得られた粘性オイルをジクロロメタン(10mL)に溶解し、室温にて塩化チオニル(10mL,140mmol)を滴下した。滴下終了後、TLCにて反応の終了を確認した。反応終了後、トルエンを加えて未反応の塩化チオニルを留去した。得られた淡黄色オイルと1-カルボエトキシ-4-ヒドロキシピペリジン(2.1g,12mmol)を混和し、油浴温130℃にて一夜攪拌した。TLCで反応の終了を確認後、放冷した。得られた残渣に酢酸エチルを加えた後、水にて洗浄した。有機層を硫酸ナトリウムにて乾燥後、溶媒を留去し褐色オイルを得た。得られたオイルをシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル(容積比)=10:1)にて精製し、表題化合物(5.1g,51%)を得た。
1H-NMR(CDCl3,δ):0.8-1.4(3H,m),1.4-2.2(14H,m),3.0-3.3(3H,m),3.3-3.4(1H,m),3.5-3.8(2H,m),3.99(1H,d,J=8Hz),4.10(2H,q,J=7.0Hz),7.1-7.4(5H,m)
MS(m/z):345(M+)。
1H-NMR(CDCl3,δ):0.8-1.4(3H,m),1.4-2.2(14H,m),3.0-3.3(3H,m),3.3-3.4(1H,m),3.5-3.8(2H,m),3.99(1H,d,J=8Hz),4.10(2H,q,J=7.0Hz),7.1-7.4(5H,m)
MS(m/z):345(M+)。
製造例200
(±)-4-(シクロへキシルフェニルメトキシ)ピペリジン
(±)-4-(シクロへキシルフェニルメトキシ)ピペリジン
製造例199で得られた(±)-4-(シクロへキシルフェニルメトキシ)ピペリジン-1-カルボン酸エチル(1.2g,3.5mmol)をエタノール(10mL)に溶解し、30%水酸化ナトリウム水溶液(2.0mL)を加えて加熱還流した。TLCで反応の終了を確認後、放冷した。反応液を濃縮後水にて希釈し、1規定塩酸で中和した。得られた残渣へ酢酸エチルを加えて抽出した。有機層を水にて洗浄した後、有機層を硫酸マグネシウムにて乾燥した。溶媒を留去し、得られた化合物は、精製せずに次の反応に用いた。
1H-NMR(CDCl3,δ):0.8-2.0(14H,m),2.0-2.2(1H,m),2.4-2.6(1H,m),2.9-3.1(2H,m),3.1-3.3(1H,m),3.1-3.3(1H,m),3.9-4.2(2H,m), 7.1-7.4(5H,m)
MS(m/z):273(M+)。
1H-NMR(CDCl3,δ):0.8-2.0(14H,m),2.0-2.2(1H,m),2.4-2.6(1H,m),2.9-3.1(2H,m),3.1-3.3(1H,m),3.1-3.3(1H,m),3.9-4.2(2H,m), 7.1-7.4(5H,m)
MS(m/z):273(M+)。
薬理試験方法及び試験結果
以下に、本発明の複素環化合物又はその塩の有効性及び安全性に関する薬理試験の方法及び結果について説明する。
以下に、本発明の複素環化合物又はその塩の有効性及び安全性に関する薬理試験の方法及び結果について説明する。
試験例1(ラットH
1
受容体親和性試験)
H1受容体膜標品をラット大脳より調製した。ラットH1受容体膜標品100μLにインキュベーション緩衝液、プロメタジン800μmol/L溶液(SIGMA-ALDRICH社製)又はインキュベーション緩衝液に溶解した各試験化合物をそれぞれ50μLずつ添加した。ここで、インキュベーション緩衝液の組成(pH7.4)は、リン酸一水素二ナトリウム(50mmol/L;和光純薬工業社製)及びリン酸二水素カリウム(50mmol/L;和光純薬工業社製)である。これに8nmol/L[3H]-ピリラミン(パーキンエルマー社製)を50μL(最終濃度2nmol/L)加えて混合した。室温にて30分間インキュベーションした後、反応液を、セルハーベスター(モレキュラーデバイス社製)を用いてガラスフィルター上に捕集し、さらにインキュベーション緩衝液によるフィルターの洗浄を行った。ガラスフィルターを切り取り、バイアルに移してシンチレーター(ナショナルダイアグノスティクス社製)5mLを加えて1時間以上振とうした後、放射能濃度を液体シンチレーションカウンターにて測定した。コントロールに対する試験化合物のヒスタミンH1受容体結合阻害率(%)を算出し、50%阻害濃度(IC50)の値を求めた。結果を表A及び表Bに示す。
H1受容体膜標品をラット大脳より調製した。ラットH1受容体膜標品100μLにインキュベーション緩衝液、プロメタジン800μmol/L溶液(SIGMA-ALDRICH社製)又はインキュベーション緩衝液に溶解した各試験化合物をそれぞれ50μLずつ添加した。ここで、インキュベーション緩衝液の組成(pH7.4)は、リン酸一水素二ナトリウム(50mmol/L;和光純薬工業社製)及びリン酸二水素カリウム(50mmol/L;和光純薬工業社製)である。これに8nmol/L[3H]-ピリラミン(パーキンエルマー社製)を50μL(最終濃度2nmol/L)加えて混合した。室温にて30分間インキュベーションした後、反応液を、セルハーベスター(モレキュラーデバイス社製)を用いてガラスフィルター上に捕集し、さらにインキュベーション緩衝液によるフィルターの洗浄を行った。ガラスフィルターを切り取り、バイアルに移してシンチレーター(ナショナルダイアグノスティクス社製)5mLを加えて1時間以上振とうした後、放射能濃度を液体シンチレーションカウンターにて測定した。コントロールに対する試験化合物のヒスタミンH1受容体結合阻害率(%)を算出し、50%阻害濃度(IC50)の値を求めた。結果を表A及び表Bに示す。
試験例2(ヒトH
1
受容体親和性試験)
H1受容体膜標品はチャイニーズハムスター卵巣細胞(CHO細胞)にヒトH1受容体を強制発現させたものを購入し、使用した。ヒトH1受容体膜標品100μLにインキュベーション緩衝液、プロメタジン800μmol/L溶液又はインキュベーション緩衝液に溶解した各試験化合物をそれぞれ50μLずつ添加した。ここで、インキュベーション緩衝液の組成(pH7.4)は、トリス-塩酸(50mmol/L;和光純薬工業社製)及び塩化マグネシウム(5mmol/L;和光純薬工業社製)である。これに8nmol/L[3H]-ピリラミンを50μL(最終濃度2nmol/L)加えて混合した。室温にて3時間インキュベーションした後、反応液をセルハーベスターを用いて0.3%ポリエチレンイミン処理したガラスフィルター上に捕集し、さらに洗浄用緩衝液によるフィルターの洗浄を行った。ここで、洗浄用緩衝液の組成(pH7.4)は、トリス-塩酸(50mmol/L)である。ガラスフィルターを切り取り、バイアルに移してシンチレーター(ナショナルダイアグノスティクス社製)5mLを加えて1時間以上振とうした後、放射能濃度を液体シンチレーションカウンターにて測定した。コントロールに対する試験化合物のヒスタミンH1受容体結合阻害率(%)を算出し、50%阻害濃度(IC50)の値を求めた。結果を表A及び表Bに示す。
H1受容体膜標品はチャイニーズハムスター卵巣細胞(CHO細胞)にヒトH1受容体を強制発現させたものを購入し、使用した。ヒトH1受容体膜標品100μLにインキュベーション緩衝液、プロメタジン800μmol/L溶液又はインキュベーション緩衝液に溶解した各試験化合物をそれぞれ50μLずつ添加した。ここで、インキュベーション緩衝液の組成(pH7.4)は、トリス-塩酸(50mmol/L;和光純薬工業社製)及び塩化マグネシウム(5mmol/L;和光純薬工業社製)である。これに8nmol/L[3H]-ピリラミンを50μL(最終濃度2nmol/L)加えて混合した。室温にて3時間インキュベーションした後、反応液をセルハーベスターを用いて0.3%ポリエチレンイミン処理したガラスフィルター上に捕集し、さらに洗浄用緩衝液によるフィルターの洗浄を行った。ここで、洗浄用緩衝液の組成(pH7.4)は、トリス-塩酸(50mmol/L)である。ガラスフィルターを切り取り、バイアルに移してシンチレーター(ナショナルダイアグノスティクス社製)5mLを加えて1時間以上振とうした後、放射能濃度を液体シンチレーションカウンターにて測定した。コントロールに対する試験化合物のヒスタミンH1受容体結合阻害率(%)を算出し、50%阻害濃度(IC50)の値を求めた。結果を表A及び表Bに示す。


表A及びBから明らかなように、本発明の化合物は、ラットヒスタミンH1受容体及びヒトヒスタミンH1受容体に対して優れた結合阻害作用を示した。
試験例3(ラットヒスタミン誘発皮膚反応試験)
試験前日に剪毛し、一夜絶食したラットに、0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物を3mg/kgの用量で経口投与した。投与1時間後に1%エバンスブルー生理食塩液(東京化成工業)を5mL/kgの容量で尾静脈内投与し、直ちにヒスタミン10μg/0.1mL/site(和光純薬工業社製)を背部皮内に投与した。その30分後にジエチルエーテルにて麻酔後、頚椎脱臼により安楽死させた。皮膚を剥離し、皮内に滲出した色素斑の長径及び短径を測定し、色素面積を算出した。なお、3mg/kgの経口投与により約40%以上の抑制効果を示した試験化合物については、更に低、中、高用量を設定し、50%抑制用量(ID50値)を求めた。結果を表Cに示す。
試験前日に剪毛し、一夜絶食したラットに、0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物を3mg/kgの用量で経口投与した。投与1時間後に1%エバンスブルー生理食塩液(東京化成工業)を5mL/kgの容量で尾静脈内投与し、直ちにヒスタミン10μg/0.1mL/site(和光純薬工業社製)を背部皮内に投与した。その30分後にジエチルエーテルにて麻酔後、頚椎脱臼により安楽死させた。皮膚を剥離し、皮内に滲出した色素斑の長径及び短径を測定し、色素面積を算出した。なお、3mg/kgの経口投与により約40%以上の抑制効果を示した試験化合物については、更に低、中、高用量を設定し、50%抑制用量(ID50値)を求めた。結果を表Cに示す。
試験例4(マウス皮膚反応試験)
試験前日に剪毛し、一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物の低、中、高用量を経口投与した。投与1時間後に1%エバンスブルー生理食塩液を10mL/kgの容量で尾静脈内投与し、直ちにヒスタミン3μg/50μL/siteを背部皮内に投与した。その30分後にジエチルエーテルにて麻酔後、頚椎脱臼により安楽死させた。皮膚を剥離し、皮内に滲出した色素斑の長径及び短径を測定し、色素面積を算出した。色素面積を基に50%抑制用量(ID50値)を求めた。結果を表Cに示す。
試験前日に剪毛し、一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物の低、中、高用量を経口投与した。投与1時間後に1%エバンスブルー生理食塩液を10mL/kgの容量で尾静脈内投与し、直ちにヒスタミン3μg/50μL/siteを背部皮内に投与した。その30分後にジエチルエーテルにて麻酔後、頚椎脱臼により安楽死させた。皮膚を剥離し、皮内に滲出した色素斑の長径及び短径を測定し、色素面積を算出した。色素面積を基に50%抑制用量(ID50値)を求めた。結果を表Cに示す。

表Cから明らかなように、本発明の化合物について、ラット及びマウスにおけるヒスタミン誘発血管透過性亢進の抑制作用が認められたことから、本発明の化合物が、優れた抗ヒスタミン作用を示すことは明らかである。
試験例5(マウスIgE依存性耳介浮腫モデルに対する抑制効果)
マウスに抗DNP-IgE抗体(SIGMA-ALDRICH社製)を0.25mL静脈内投与し、受動感作した。その翌日、抗原(DNFB液;和光純薬工業社製)を右耳介に塗布することにより皮膚炎(耳介の腫脹)を惹起した。抗原塗布の1時間後、24時間後及び7日後に、右耳介の厚さを厚み測定装置(dial thickness gauge)を用いて測定した。なお、試験化合物の投与用量はラットヒスタミン皮膚反応試験で得られたそれぞれのID50値の1/3倍量とし、1日2回、抗原塗布の1時間前及び6時間後に経口投与した。感作前の右耳介の厚さを予め測定しておき、即時相、遅発相及び超遅発相での腫脹の抑制率を算出した。結果を表Dに示す。
マウスに抗DNP-IgE抗体(SIGMA-ALDRICH社製)を0.25mL静脈内投与し、受動感作した。その翌日、抗原(DNFB液;和光純薬工業社製)を右耳介に塗布することにより皮膚炎(耳介の腫脹)を惹起した。抗原塗布の1時間後、24時間後及び7日後に、右耳介の厚さを厚み測定装置(dial thickness gauge)を用いて測定した。なお、試験化合物の投与用量はラットヒスタミン皮膚反応試験で得られたそれぞれのID50値の1/3倍量とし、1日2回、抗原塗布の1時間前及び6時間後に経口投与した。感作前の右耳介の厚さを予め測定しておき、即時相、遅発相及び超遅発相での腫脹の抑制率を算出した。結果を表Dに示す。

表Dから明らかなように、本発明の化合物は、即時相、遅発相、超遅発相に見られる耳介浮腫に対して抑制作用を示したことから、本発明の化合物が、優れた抗炎症作用を示すことは明らかである。
試験例6(マウス抗原誘発肺好酸球浸潤モデルに対する効果)
マウスにオボアルブミン(OVA)8μg(SIGMA-ALDLICH社製)及び水酸化アルミニウムゲル2mgを含む生理食塩液0.5mLを2回腹腔内投与した(day0(1日目)及びday5(6日目)に投与)。最終感作1週間後に午前・午後の2回に0.5%OVA生理食塩溶液をネブライザーを用いて1回あたり1時間吸入させた。なお、試験化合物の投与用量はラット皮膚反応試験で得られたそれぞれのID50値の5倍量とし、1日2回、午前・午後の各抗原チャレンジの30分前に経口投与した。抗原チャレンジから約48時間後にペントバルビタール深麻酔により安楽死させ、肺胞洗浄液(BALF)採取を行った。BALF中の総細胞数を測定した後、スメア標本を作製してメイグリュンワルド・ギムザ染色を行い、単核球、好酸球及び好中球数を測定することにより基礎データとしての細胞構成比を算出し、更に好酸球数を算出した。結果を図1~図3に示す。
マウスにオボアルブミン(OVA)8μg(SIGMA-ALDLICH社製)及び水酸化アルミニウムゲル2mgを含む生理食塩液0.5mLを2回腹腔内投与した(day0(1日目)及びday5(6日目)に投与)。最終感作1週間後に午前・午後の2回に0.5%OVA生理食塩溶液をネブライザーを用いて1回あたり1時間吸入させた。なお、試験化合物の投与用量はラット皮膚反応試験で得られたそれぞれのID50値の5倍量とし、1日2回、午前・午後の各抗原チャレンジの30分前に経口投与した。抗原チャレンジから約48時間後にペントバルビタール深麻酔により安楽死させ、肺胞洗浄液(BALF)採取を行った。BALF中の総細胞数を測定した後、スメア標本を作製してメイグリュンワルド・ギムザ染色を行い、単核球、好酸球及び好中球数を測定することにより基礎データとしての細胞構成比を算出し、更に好酸球数を算出した。結果を図1~図3に示す。
図1~図3から明らかなように、本発明の化合物は、抗原誘発による肺好酸球浸潤に対して抑制作用を示したことから、本発明の化合物が、優れた抗炎症作用を示すことは明らかである。なお、図1~3において、非チャレンジ群は、0.5%OVA生理食塩溶液を吸入させない群(抗原チャレンジを行わない群)を示し、コントロール群は、試験化合物として0.5%CMCナトリウム溶液を投与した群を示す。また、非チャレンジ群の好酸球数は殆ど0である。
試験例7(マウス自発運動量に及ぼす影響)
一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物を経口投与した。直ちにポリカーボネートケージに1群3匹ずつ収容し、自発運動量測定装置にて、投与直後から投与2時間後まで5分毎に累積的な自発運動量を測定した。なお、試験化合物の投与用量はラットヒスタミン皮膚反応試験で得られたそれぞれのID50値の50倍量とした。結果を図4~図6に示す。
一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物を経口投与した。直ちにポリカーボネートケージに1群3匹ずつ収容し、自発運動量測定装置にて、投与直後から投与2時間後まで5分毎に累積的な自発運動量を測定した。なお、試験化合物の投与用量はラットヒスタミン皮膚反応試験で得られたそれぞれのID50値の50倍量とした。結果を図4~図6に示す。
図4~図6から明らかなように、本発明の化合物はラットヒスタミン皮膚反応試験で得られたID50値の50倍量を投与してもマウス自発運動量に影響を及ぼさなかった。従って、本発明の化合物は、中枢神経作用が明らかに軽減されており、眠気等の副作用のない安全性に優れた化合物である。
試験例8(マウスIgE依存性耳介浮腫モデルに対する抑制効果)
マウスに抗DNP-IgE抗体(SIGMA-ALDRICH社製)を0.25mL静脈内投与し、受動感作した。その翌日、抗原(DNFB液;和光純薬工業社製)を右耳介に塗布することにより皮膚炎(耳介の腫脹)を惹起した。抗原塗布の1時間後、24時間後及び7日後に、右耳介の厚さを厚み測定装置(dial thickness gauge)を用いて測定した。なお、試験化合物の投与用量はマウスヒスタミン皮膚反応試験で得られたそれぞれのID50値の5又は10倍量とし、1日2回、抗原塗布の1時間前及び6時間後に経口投与した。感作前の右耳介の厚さを予め測定しておき、即時相、遅発相及び超遅発相での腫脹の抑制率を算出した。結果を表Eに示す。
マウスに抗DNP-IgE抗体(SIGMA-ALDRICH社製)を0.25mL静脈内投与し、受動感作した。その翌日、抗原(DNFB液;和光純薬工業社製)を右耳介に塗布することにより皮膚炎(耳介の腫脹)を惹起した。抗原塗布の1時間後、24時間後及び7日後に、右耳介の厚さを厚み測定装置(dial thickness gauge)を用いて測定した。なお、試験化合物の投与用量はマウスヒスタミン皮膚反応試験で得られたそれぞれのID50値の5又は10倍量とし、1日2回、抗原塗布の1時間前及び6時間後に経口投与した。感作前の右耳介の厚さを予め測定しておき、即時相、遅発相及び超遅発相での腫脹の抑制率を算出した。結果を表Eに示す。

表Eから明らかなように、本発明の化合物は、低用量において、即時相、遅発相、超遅発相に見られる耳介浮腫に対して明らかな抑制作用を示したことから、本発明の化合物が、優れた抗炎症作用を示すことは明らかである。
試験例9(マウス抗原誘発肺好酸球浸潤モデルに対する効果)
マウスにオボアルブミン(OVA)8μg(SIGMA-ALDLICH社製)及び水酸化アルミニウムゲル2mgを含む生理食塩液0.5mLを2回腹腔内投与した(day0(1日目)及びday5(6日目)に投与)。最終感作1週間後に午前・午後の2回に0.5%OVA生理食塩溶液をネブライザーを用いて1回あたり1時間吸入させた。なお、試験化合物の投与用量はマウス皮膚反応試験で得られたそれぞれのID50値の30~170倍量とし、1日2回、午前・午後の各抗原チャレンジの30分前に経口投与した。抗原チャレンジから約48時間後にペントバルビタール深麻酔により安楽死させ、肺胞洗浄液(BALF)採取を行った。BALF中の総細胞数を測定した後、スメア標本を作製してメイグリュンワルド・ギムザ染色を行い、単核球、好酸球及び好中球数を測定することにより基礎データとしての細胞構成比を算出し、更に好酸球数を算出した。結果を図7~図9に示す。
マウスにオボアルブミン(OVA)8μg(SIGMA-ALDLICH社製)及び水酸化アルミニウムゲル2mgを含む生理食塩液0.5mLを2回腹腔内投与した(day0(1日目)及びday5(6日目)に投与)。最終感作1週間後に午前・午後の2回に0.5%OVA生理食塩溶液をネブライザーを用いて1回あたり1時間吸入させた。なお、試験化合物の投与用量はマウス皮膚反応試験で得られたそれぞれのID50値の30~170倍量とし、1日2回、午前・午後の各抗原チャレンジの30分前に経口投与した。抗原チャレンジから約48時間後にペントバルビタール深麻酔により安楽死させ、肺胞洗浄液(BALF)採取を行った。BALF中の総細胞数を測定した後、スメア標本を作製してメイグリュンワルド・ギムザ染色を行い、単核球、好酸球及び好中球数を測定することにより基礎データとしての細胞構成比を算出し、更に好酸球数を算出した。結果を図7~図9に示す。
図7~図9から明らかなように、本発明の化合物は、抗原誘発による肺好酸球浸潤に対して抑制作用を示すことから、本発明の化合物が、優れた抗炎症作用を示すことは明らかである。なお、図7~9において、非チャレンジ群は、0.5%OVA生理食塩溶液を吸入させない群(抗原チャレンジを行わない群)を示し、コントロール群は、試験化合物として0.5%CMCナトリウム溶液を投与した群を示す。また、非チャレンジ群の好酸球数は0である。
試験例10(マウス自発運動量に及ぼす影響)
一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物(実施例1、2、10、11、22、30、39、40、41、43、52、87、124、131、134、135及び136)を経口投与した。直ちにポリカーボネートケージに1群3匹ずつ収容し、自発運動量測定装置にて、投与直後から投与2時間後まで5分毎に累積的な自発運動量を測定した。なお、試験化合物の投与用量はマウスヒスタミン皮膚反応試験で得られたそれぞれのID50値の300~2500倍量とした。
一夜絶食したマウスに0.5%(W/V)のCMC-Na水溶液に懸濁した試験化合物(実施例1、2、10、11、22、30、39、40、41、43、52、87、124、131、134、135及び136)を経口投与した。直ちにポリカーボネートケージに1群3匹ずつ収容し、自発運動量測定装置にて、投与直後から投与2時間後まで5分毎に累積的な自発運動量を測定した。なお、試験化合物の投与用量はマウスヒスタミン皮膚反応試験で得られたそれぞれのID50値の300~2500倍量とした。
試験例7と同様に、本発明の化合物はラットヒスタミン皮膚反応試験で得られたID50値の300倍量以上を投与してもマウス自発運動量に影響を及ぼさなかった。従って、本発明の化合物は、中枢神経作用が明らかに軽減されており、眠気等の副作用のない安全性に優れた化合物である。
製剤例1
下記処方について日局XIVの製剤総則記載の公知方法に従って錠剤を得た。
下記処方について日局XIVの製剤総則記載の公知方法に従って錠剤を得た。
錠剤1錠中の処方例(全量150mg):
実施例2の化合物 30mg
乳糖 67mg
トウモロコシデンプン 28mg
結晶セルロース 15mg
ヒドロキシプロピルセルロース 8mg
ステアリン酸マグネシウム 2mg
製剤例2
下記処方について日局XIVの製剤総則記載の公知方法に従ってカプセル剤を得た。
実施例2の化合物 30mg
乳糖 67mg
トウモロコシデンプン 28mg
結晶セルロース 15mg
ヒドロキシプロピルセルロース 8mg
ステアリン酸マグネシウム 2mg
製剤例2
下記処方について日局XIVの製剤総則記載の公知方法に従ってカプセル剤を得た。
カプセル剤1カプセル中の処方例(全量180mg):
実施例2の化合物 50mg
乳糖 100mg
トウモロコシデンプン 28mg
ステアリン酸マグネシウム 2mg
製剤例3
実施例2の化合物10mgを生理食塩水3mlに溶解し、0.1Nの水酸化ナトリウム水溶液でpH7に調整した後、さらに生理食塩水を加えて5mlとした。アンプルに分注した後、加熱滅菌し注射剤を得た。
実施例2の化合物 50mg
乳糖 100mg
トウモロコシデンプン 28mg
ステアリン酸マグネシウム 2mg
製剤例3
実施例2の化合物10mgを生理食塩水3mlに溶解し、0.1Nの水酸化ナトリウム水溶液でpH7に調整した後、さらに生理食塩水を加えて5mlとした。アンプルに分注した後、加熱滅菌し注射剤を得た。
本発明の医薬組成物は、アレルギー性疾患(例えば、気管支喘息、鼻炎、鼻閉、皮膚炎など)の予防及び/又は治療剤として有効である。
Claims (12)
- 下記式(1)

(式中、環Aは置換基を有していてもよい同素環又は複素環を示し;環BはG及び窒素原子Nを環の構成原子として含み、置換基を有していてもよい複素環を示し、環BのGはCH又はNを示し;R1はカルボニル基又は置換基を有していてもよいアルキレン基を示し;R2a及びR2bは同一又は異なって置換基を有していてもよいアルキル基、置換基を有していてもよいシクロアルキル基、置換基を有していてもよいアリール基、又は置換基を有していてもよい複素環基を示し、これらの環の置換基は互いに結合して炭化水素環又は複素環を形成してもよく、この炭化水素環又は複素環は置換基を有してもよく;Xは酸素原子又は硫黄原子を示し;Zはヒドロキシル基、アルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基、アミノ基、N-置換アミノ基を示し;nは0又は1を示す。但し、環Aがベンゼン環であるとき、又は環Bがピペラジン環であるとき、R1は置換基を有していてもよいアルキレン基である)
で表される複素環化合物又はその塩。 - 環Aが、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよい芳香族環であり、
環Bが脂肪族又は芳香族複素環であり、
R1がハロゲン原子を有していてもよい直鎖状又は分岐鎖状アルキレン基であり、
R2a及びR2bが同一又は異なって、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよいC6-10アレーン環又は芳香族5員又は6員複素環であるか、又はR2a及びR2bは下記式(2)
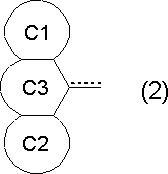
(式中、環C1及び環C2は同一又は異なって、ハロゲン原子、アルキル基、ハロアルキル基、アルコキシ基およびハロアルコキシ基から選択された少なくとも1つの置換基を有していてもよいC6-10アレーン環又は芳香族5員又は6員複素環を示し;環C3は、ハロゲン原子、アルキル基、ハロアルキル基、ヒドロキシル基、アルコキシ基、ハロアルコキシ基、カルボキシル基、アルコキシカルボニル基、アシル基およびカルボニル基から選択された少なくとも1つの置換基を有していてもよい4~10員の炭化水素環又は複素環を示し;下記式(3)

で表される結合手は単結合又は二重結合を示し、nが0であり、かつGがCHであるとき、前記結合手は二重結合を示す)
で表される三環式基を形成し、
Zがヒドロキシル基;C1-6アルコキシ基;アミノ基;又はC1-6アルキル基、C1-6アルキル-カルボニル基から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基であり、
GがCHであるときnは1であり、GがNであるときnは0である
請求項1記載の複素環化合物又はその塩。 - 環Aがベンゼン環又は芳香族5員又は6員複素環であり、
環Bが脂肪族4~10員複素環であり、
R1が直鎖状又は分岐鎖状C1-4アルキレン基であり、
R2a及びR2bが同一又は異なって、ハロゲン原子、C1-6アルキル基、ハロC1-6アルキル基、C1-6アルコキシ基およびハロC1-6アルコキシ基から選択された少なくとも1つの置換基を有していてもよいベンゼン環であり、
Zがヒドロキシル基;C1-4アルコキシ基;アミノ基;又はC1-4アルキル基、C1-4アルキル-カルボニル基から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基であり、
GがCHであるとき、Xが酸素原子であるとともにnが1であり、GがNであるとき、nは0である
請求項1又は2記載の複素環化合物又はその塩。 - 環Aが、窒素原子、酸素原子及び硫黄原子から選択された少なくとも一種のヘテロ原子を含む芳香族複素環であり、
環Bが脂肪族5又は6員複素環であり、
R1が直鎖状又は分岐鎖状C1-3アルキレン基であり、
R2a及びR2bが同一又は異なって、フッ素原子、塩素原子、C1-3アルキル基、ハロC1-3アルキル基、C1-3アルコキシ基およびハロC1-3アルコキシ基から選択された少なくとも1つの置換基を有していてもよいベンゼン環であり、
Zがヒドロキシル基;C1-3アルコキシ基;アミノ基;C1-3アルキル基、C1-3アルキル-カルボニル基から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基である
請求項1~3のいずれかに記載の複素環化合物又はその塩。 - 環Aがピリジン環であり、
環Bがピペリジン環又はピペラジン環であり、
R1がC1-2アルキレン基であり、
R2a及びR2bが同一又は異なって、フッ素原子、塩素原子、C1-3アルキル基、フルオロC1-3アルキル基、C1-3アルコキシ基およびフルオロC1-3アルコキシ基から選択された少なくとも1つの置換基を有していてもよいベンゼン環であり、
Zがヒドロキシル基;C1-2アルコキシ基;アミノ基;C1-2アルキル基、C1-2アルキル-カルボニル基から選択された少なくとも一種の置換基が窒素原子に置換したN-置換アミノ基である
請求項1~4のいずれかに記載の複素環化合物又はその塩。 - (4-ベンズヒドリルオキシピペリジン-1-イルメチル)ピリジンカルボン酸、
{4-[(ハロフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[(C1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[(フルオロC1-4アルキルフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[(C1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[(フルオロC1-4アルコキシフェニル)フェニルメトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[ビス(ハロフェニル)メトキシ]ピペリジン-1-イルメチル}ピリジンカルボン酸、
{4-[ビス(ハロフェニル)メチル]ピペラジン-1-イルメチル}ピリジンカルボン酸、
(3-ベンズヒドリルオキシピロリジン-1-イルメチル)ピリジンカルボン酸、
[4-(10,11-ジヒドロジベンゾ[a,d]シクロヘプテン-5-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸、及び
[4-(6H-ジベンゾ[b,e]オキセピン-11-イリデン)ピペリジン-1-イルメチル]ピリジンカルボン酸
又はこれらの塩、プロドラッグ、活性代謝物から選択された請求項1記載の複素環化合物又はその塩。 - (I)下記式(12)
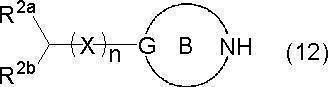
(式中、環B,R1,G,X,n,R2a,R2bは前記に同じ)
で表される化合物と、下記式(13)
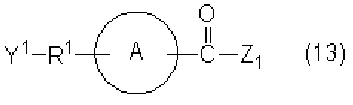
で表される化合物との反応、又は
(II)下記式(15)

(式中、Y2はハロゲン原子を示し、環A,環B,R1,G,X,n,R2a,R2bは前記に同じ)
で表される化合物と、下記式(16)
(式中、R5はアルキル基、シクロアルキル基、アリール基、アラルキル基を示す)
で表される化合物と、一酸化炭素との反応
により、請求項1記載の式(1)においてZがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を製造し、
Zがアルコキシ基、シクロアルキルオキシ基、アリールオキシ基、アラルキルオキシ基である複素環化合物を加水分解又はアミド結合形成反応に供し、Zがヒドロキシル基、アミノ基又はN-置換アミノ基である複素環化合物を製造し、又は
Zがアミノ基である複素環化合物をアルキル化又はアシル化反応に供し、ZがN-アルキルアミノ基又はN-アシルアミノ基である複素環化合物を製造する方法。 - 請求項1~6のいずれかに記載の化合物又はその薬学的に許容可能な塩を含む抗アレルギー剤。
- 請求項1~6のいずれかに記載の化合物又はその薬学的に許容可能な塩を含む抗ヒスタミン剤。
- 請求項1~6のいずれかに記載の化合物又はその薬学的に許容可能な塩を含む抗炎症剤。
- 請求項1~6のいずれかに記載の化合物又はその薬学的に許容可能な塩を含む、アレルギー疾患の予防及び/又は治療剤。
- 請求項1~6のいずれかに記載の化合物又はその薬学的に許容可能な塩と担体とを含む医薬組成物。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012517249A JP5841529B2 (ja) | 2010-05-27 | 2011-05-23 | 複素環化合物及びh1受容体拮抗剤 |
| EP11786587.3A EP2578569B1 (en) | 2010-05-27 | 2011-05-23 | Heterocyclic ring compound and h1 receptor antagonist |
| US13/699,892 US9365553B2 (en) | 2010-05-27 | 2011-05-23 | Heterocyclic compound and H1 receptor antagonist |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-122125 | 2010-05-27 | ||
| JP2010122125 | 2010-05-27 | ||
| JP2010-165134 | 2010-07-22 | ||
| JP2010165134 | 2010-07-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011148888A1 true WO2011148888A1 (ja) | 2011-12-01 |
Family
ID=45003878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/061734 Ceased WO2011148888A1 (ja) | 2010-05-27 | 2011-05-23 | 複素環化合物及びh1受容体拮抗剤 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9365553B2 (ja) |
| EP (1) | EP2578569B1 (ja) |
| JP (1) | JP5841529B2 (ja) |
| WO (1) | WO2011148888A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150063517A (ko) * | 2012-10-02 | 2015-06-09 | 에피테라퓨틱스 에이피에스 | 히스톤 탈메틸효소의 저해제 |
| JP2017512804A (ja) * | 2014-03-31 | 2017-05-25 | ギリアード サイエンシーズ, インコーポレイテッド | ヒストン脱メチル化酵素の阻害剤 |
| EP4333821A4 (en) * | 2021-05-04 | 2025-07-09 | Texas A & M Univ Sys | SARS-COV-2 INHIBITORS |
| EP4508043A4 (en) * | 2022-04-14 | 2026-04-01 | Mayo Found Medical Education & Res | TREATMENT METHODS FOR OCULAR FIBROSIS DISEASES |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2901022C (en) | 2013-02-27 | 2021-05-04 | Epitherapeutics Aps | Substituted pyridine compounds as inhibitors of histone demethylases |
| SG11201701182VA (en) * | 2014-08-27 | 2017-03-30 | Gilead Sciences Inc | Compounds and methods for inhibiting histone demethylases |
| CN113880808B (zh) * | 2020-07-03 | 2024-12-31 | 合肥医工医药股份有限公司 | 一类三唑类化合物、制备方法及其医药用途 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61282359A (ja) * | 1985-06-05 | 1986-12-12 | ジ−.デイ.サ−ル アンド コンパニ− | N−ベンジル−ピペリジン化合物およびそれを含有する医薬組成物 |
| JPH05345765A (ja) * | 1992-04-16 | 1993-12-27 | Hokuriku Seiyaku Co Ltd | ジベンズアゼピン誘導体 |
| JPH07224038A (ja) * | 1994-02-14 | 1995-08-22 | Mitsui Toatsu Chem Inc | ピコリン酸誘導体 |
| WO2001046166A2 (en) | 1999-12-20 | 2001-06-28 | Neuromed Technologies, Inc. | Partially saturated calcium channel blockers |
| WO2003057223A1 (en) | 2002-01-02 | 2003-07-17 | Ardent Pharmaceuticals, Inc. | Method of treating sexual dysfunctions with delta opioid receptor agonist compounds |
| JP2004051600A (ja) | 2002-07-24 | 2004-02-19 | Iyaku Bunshi Sekkei Kenkyusho:Kk | 造血器型プロスタグランジンd2合成酵素阻害剤 |
| JP2006516281A (ja) * | 2003-01-02 | 2006-06-29 | アーデント ファーマスーティカルズ,インコーポレイテッド | 心臓保護的デルタオピオイド受容体アゴニストおよびその使用法 |
| JP2007534696A (ja) | 2004-04-23 | 2007-11-29 | ヒプニオン, インコーポレイテッド | Cns標的モジュレーターを使用する処置またはcns診断 |
| JP2009504637A (ja) | 2005-08-08 | 2009-02-05 | アストラゼネカ・アクチエボラーグ | 治療薬 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4002629A (en) | 1970-11-11 | 1977-01-11 | A. Christiaens Societe Anonyme | Certain pyridine carboxamide derivatives |
| GB8816365D0 (en) * | 1988-07-08 | 1988-08-10 | Pfizer Ltd | Therapeutic agents |
| JP2664238B2 (ja) * | 1989-03-01 | 1997-10-15 | 日清製粉株式会社 | ニコチン酸またはそのエステル誘導体 |
| US4994456A (en) * | 1989-03-01 | 1991-02-19 | Nisshin Flour Milling Co., Ltd. | Pyridinecarboxylic acid amide derivatives and pharmaceutical compositions comprising same |
| JP2843632B2 (ja) * | 1989-03-01 | 1999-01-06 | 日清製粉株式会社 | ピリジンカルボン酸アミド誘導体 |
| US4992629A (en) * | 1990-03-15 | 1991-02-12 | Morais Fortunato R | Cable shock absorbing apparatus |
| US5877177A (en) * | 1997-06-17 | 1999-03-02 | Schering Corporation | Carboxy piperidylacetamide tricyclic compounds useful for inhibition of G-protein function and for treatment of proliferative diseases |
| WO1999038515A1 (en) * | 1998-01-29 | 1999-08-05 | Akzo Nobel N.V. | Optic papillary circulation improving agents |
-
2011
- 2011-05-23 EP EP11786587.3A patent/EP2578569B1/en active Active
- 2011-05-23 JP JP2012517249A patent/JP5841529B2/ja active Active
- 2011-05-23 WO PCT/JP2011/061734 patent/WO2011148888A1/ja not_active Ceased
- 2011-05-23 US US13/699,892 patent/US9365553B2/en active Active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS61282359A (ja) * | 1985-06-05 | 1986-12-12 | ジ−.デイ.サ−ル アンド コンパニ− | N−ベンジル−ピペリジン化合物およびそれを含有する医薬組成物 |
| JPH05345765A (ja) * | 1992-04-16 | 1993-12-27 | Hokuriku Seiyaku Co Ltd | ジベンズアゼピン誘導体 |
| JPH07224038A (ja) * | 1994-02-14 | 1995-08-22 | Mitsui Toatsu Chem Inc | ピコリン酸誘導体 |
| WO2001046166A2 (en) | 1999-12-20 | 2001-06-28 | Neuromed Technologies, Inc. | Partially saturated calcium channel blockers |
| JP2003518107A (ja) | 1999-12-20 | 2003-06-03 | ニューロメド テクノロジーズ, インコーポレイテッド | 部分飽和カルシウムチャネルブロッカー |
| WO2003057223A1 (en) | 2002-01-02 | 2003-07-17 | Ardent Pharmaceuticals, Inc. | Method of treating sexual dysfunctions with delta opioid receptor agonist compounds |
| JP2004051600A (ja) | 2002-07-24 | 2004-02-19 | Iyaku Bunshi Sekkei Kenkyusho:Kk | 造血器型プロスタグランジンd2合成酵素阻害剤 |
| JP2006516281A (ja) * | 2003-01-02 | 2006-06-29 | アーデント ファーマスーティカルズ,インコーポレイテッド | 心臓保護的デルタオピオイド受容体アゴニストおよびその使用法 |
| JP2007534696A (ja) | 2004-04-23 | 2007-11-29 | ヒプニオン, インコーポレイテッド | Cns標的モジュレーターを使用する処置またはcns診断 |
| JP2009504637A (ja) | 2005-08-08 | 2009-02-05 | アストラゼネカ・アクチエボラーグ | 治療薬 |
Non-Patent Citations (6)
| Title |
|---|
| "Handbook of Pharmaceutical Excipients", 1989, MARUZEN COMPANY, LTD. |
| "Japanese Pharmaceutical Excipients Dictionary 2007", July 2007, YAKUJI NIPPO LTD. |
| "Japanese Pharmaceutical Excipients", August 2003, YAKUJI NIPPO LTD. |
| "Pharmaceutics", 1997, NANKODO, CO., LTD. |
| CARCELLER, E. ET AL.: "[(3-Pyridylalkyl) piperidylidene]benzocycloheptapyridine Derivatives as Dual Antagonists of PAF and Histamine", JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 17, 1994, pages 2697 - 2703, XP003005099 * |
| See also references of EP2578569A4 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150063517A (ko) * | 2012-10-02 | 2015-06-09 | 에피테라퓨틱스 에이피에스 | 히스톤 탈메틸효소의 저해제 |
| CN104981458A (zh) * | 2012-10-02 | 2015-10-14 | Epi生物医疗有限公司 | 组蛋白去甲基化酶抑制剂 |
| JP2015532295A (ja) * | 2012-10-02 | 2015-11-09 | エピセラピューティックス・エイ・ピー・エス | ヒストン脱メチル化酵素の阻害剤 |
| CN104981458B (zh) * | 2012-10-02 | 2017-07-21 | 吉利德科学公司 | 组蛋白去甲基化酶抑制剂 |
| US10189787B2 (en) | 2012-10-02 | 2019-01-29 | Gilead Sciences, Inc. | Inhibitors of histone demethylases |
| US10221139B2 (en) | 2012-10-02 | 2019-03-05 | Gilead Sciences, Inc. | Inhibitors of histone demethylases |
| KR102160320B1 (ko) * | 2012-10-02 | 2020-09-28 | 에피테라퓨틱스 에이피에스 | 히스톤 탈메틸효소의 저해제 |
| JP2017512804A (ja) * | 2014-03-31 | 2017-05-25 | ギリアード サイエンシーズ, インコーポレイテッド | ヒストン脱メチル化酵素の阻害剤 |
| EP4333821A4 (en) * | 2021-05-04 | 2025-07-09 | Texas A & M Univ Sys | SARS-COV-2 INHIBITORS |
| EP4508043A4 (en) * | 2022-04-14 | 2026-04-01 | Mayo Found Medical Education & Res | TREATMENT METHODS FOR OCULAR FIBROSIS DISEASES |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2578569A1 (en) | 2013-04-10 |
| US20130085127A1 (en) | 2013-04-04 |
| EP2578569B1 (en) | 2015-10-28 |
| US9365553B2 (en) | 2016-06-14 |
| JPWO2011148888A1 (ja) | 2013-07-25 |
| EP2578569A4 (en) | 2014-01-15 |
| JP5841529B2 (ja) | 2016-01-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5841529B2 (ja) | 複素環化合物及びh1受容体拮抗剤 | |
| AU2012305212B2 (en) | Piperazine- substituted benzothiophene derivatives as antipsychotic agents | |
| RU2167152C2 (ru) | N-замещенные азагетероциклические карбоновые кислоты или их фармацевтически приемлемые соли, способ их получения, фармацевтическая композиция на их основе и способ ингибирования нейрогенного воспаления | |
| DE69132827T2 (de) | Ringamide-Derivate | |
| JP5932779B2 (ja) | シグマ受容体阻害剤としてのピラゾール化合物 | |
| DE69330823T2 (de) | Thiacyclische piperidinylderivate | |
| US4791112A (en) | N-heterocyclic-N-(4-piperidyl)amides and pharmaceutical compositions and methods employing such compounds | |
| WO1997013766A1 (en) | Substituted heteroaromatic derivatives | |
| TWI775723B (zh) | 嗎啡喃衍生物 | |
| JPH05279357A (ja) | アザヘテロサイクリルメチル−クロマン類 | |
| WO2000053600A1 (en) | Novel piperidine derivatives | |
| JPWO2013129622A1 (ja) | 複素環化合物およびその用途 | |
| US9745323B2 (en) | Compound and pharmaceutical composition for neuropsychological disorder or malignant tumor | |
| AU2005335298A1 (en) | N-phenyl-2-pyrimidine-amine derivatives and process for the preparation thereof | |
| JP2008535782A (ja) | mGluR5アンタゴニストとして使用するための、ピロリジンおよびピペリジンアセチレン誘導体 | |
| JP3162523B2 (ja) | ピペリジルメチル−置換クロマン誘導体 | |
| US20100168099A1 (en) | 1h-quinolin-4-one compounds, with affinity for the gaba receptor, processes, uses and compositions | |
| EP0719773B1 (en) | Imidazolidinone derivative, acid-addition salt thereof, and remedy for senile dementia | |
| JP3306827B2 (ja) | 新規ニコチン酸エステル | |
| EP2064185B1 (en) | 1h-quinolin-4-one compounds, with affinity for the gaba receptor, processes, uses and compositions | |
| JP5442711B2 (ja) | Hdl−コレステロール上昇剤としての2−トリフルオロメチルニコチンアミド誘導体 | |
| JPWO2000023436A1 (ja) | キナゾリノン誘導体 | |
| JP5165578B2 (ja) | イソキノリンおよびベンゾ[h]イソキノリン誘導体、調製およびヒスタミンh3受容体のアンタゴニストとしてのこの治療上の使用 | |
| JPH1171350A (ja) | ヒドロキシピペリジン化合物およびその剤 | |
| KR102607510B1 (ko) | 무스카린 m1 수용체 양성 알로스테릭 조절제로서의 피롤로-피리다진 유도체 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11786587 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012517249 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011786587 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13699892 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |